practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations.laws. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant administrative, civil, and/or criminal and administrative penalties, including, without limitation, damages, fines, disgorgement, individual imprisonment, exclusion from participation in government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations or other sanctions.operations. If any of the physicians or other healthcare providers or entities with whom we expect to do business isare found to be not in compliance with applicable laws, and regulations, itthey also may be subject to criminal,administrative, civil, and/or administrativecriminal sanctions, including exclusions from participation in government funded healthcare programs.
FailureLegal Proceedings
From time to buildtime we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our finance infrastructure and improve our accounting systems and controls could impair our ability to comply with the financial reporting and internal controls requirements for publicly traded companies.
As a U.S. public company, we operate in an increasingly demanding regulatory environment that requires us to comply with, among things, the Sarbanes-Oxley Act of 2002, or “SOX” as from December 31, 2016, and related rules and regulationsbusiness. Regardless of the SEC’s substantial disclosure requirements, accelerated reporting requirementsoutcome, litigation can have an adverse impact on us because of defense and complex accounting rules. Company responsibilities required by the Sarbanes-Oxley Act include establishing corporate oversightsettlement costs, diversion of management resources and adequate internal control over financial reporting and disclosure controls and procedures. Effective internal controls are necessary for us to produce reliable financial reports and are important to help prevent financial fraud.other factors.
C. Organizational Structure.
We have limited accounting personnel and other resources to address our internal controls and procedures. Our independent registered public accounting firm has not conducted an audit of our internal control over financial reporting. However, in connection with the audit of our consolidated financial statements as of and for the year ended December 31, 2016, we identified a material weakness in our internal control over financial reporting. A material weakness is a deficiency,subsidiaries, or combination of deficiencies, such that there is reasonable possibility that a material misstatement of our annual or interim consolidated financial statements will not be prevented or detected on a timely basis by our employees. The material weakness identified is related to a lack of segregation of duties given the size of our finance and accounting team.
As of December 31, 2014, we reported three material weaknesses. Since then, we have taken several remedial actions to address these material weaknesses. In particular, we have hired in 2015 additional staff for the finance and legal departments, including a corporate controller and legal advisor. Also, the Group, has worked in 2016 with an independent firm to better define, formalize and upgrade our internal controls and processes with the goal of being compliant with the SOX regulation by end of 2016. Under the supervisionis made of the Company’s management, an independent firm tested our internal controls and processes respectively in September 2016 and January 2017 in light of the SOX regulation. No additional material weaknesses were identified other than those related to the segregation of duties. Management will be addressing the recommendations of the independent firm in view of a continuous improvement of our processes.
As a result of these actions, two out of three material weaknesses identifiedfollowing entities as of December 31, 2014 are no longer valid.2018. The lack of accounting resources to fulfill the reporting requirements of IFRSfollowing diagram illustrates our corporate structure.
| | | | | | | | | | | | | | | | | | |
Name | | Country of
Incorporation
and Place of
Business | | | Nature of
Business | | Proportion of
ordinary shares
directly held by
parent (%) | | | Proportion of
ordinary
shares held
by the group
(%) | | | Proportion of
ordinary
shares held by
non-controlling
interests (%) | |
Celyad SA | | | BE | | | Biopharma | | | Parent company | | | | | | | | | |
Celyad Inc. | | | US | | | Biopharma | | | 100 | % | | | 100 | % | | | 0 | % |
Biological Manufacturing Services SA | | | BE | | | Manufacturing | | | 100 | % | | | 100 | % | | | 0 | % |
CorQuest Medical, Inc. | | | US | | | Medical Device | | | 100 | % | | | 100 | % | | | 0 | % |
See “Item 4.A.—History and the lack of comprehensive knowledge of IFRS accounting policies are no longer identified as material weaknesses as of December 31, 2016.
Furthermore, we believe it is possible that if our independent registered public accounting firm had performed an audit of our internal control over financial reporting, other material weaknesses may have been identified. Section 404Development of the Sarbanes-Oxley Act, or Section 404, requires that we includeCompany.”
D. Property, Plants and Equipment.
We rent a report of management on our internal control over financial reporting2,284 square meter office space from the Axis Parc developer located at the Axis Parc in our annual report on Form20-F beginning with our annual report for the fiscal year ending December 31, 2016. However, until we ceaseMont-Saint-Guibert pursuant to be an “emerging growth company,”a lease agreement dated October 15, 2015 as that term is defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, our independent registered public accounting firm will not be required to attest to and report on the effectiveness of our internal control over financial reporting.
Our management may conclude that our internal control over financial reporting is not effective. Moreover, even if our management concludes that our internal control over financial reporting is effective, our independent registered public accounting firm, after conducting its own independent testing, may issue a report that is qualified if it is not satisfied with our internal controls or the level at which our controls are documented, designed, operated or reviewed, or if it interprets the relevant requirements differently from us. In addition, after we become a public company, our reporting obligations may place a significant strain on our management, operational and financial resources and systems for the foreseeable future. We may be unable to timely complete our evaluation, testing and any required remediation.
In addition, if we fail to maintain the adequacy of our internal control over financial reporting, as these standards are modified, supplemented or amended from time to time, we may not be able to concludewhich expires on an ongoing basis that we have effective internal control over financial reporting in accordance with Section 404. If we fail to achieveSeptember 30, 2025. We also rent a 1,120 square meter office and maintain an effective internal control environment, we could experience material misstatements in our consolidated financial statements and fail to meet our reporting obligations, which would likely cause investors to lose confidence in our reported financial information. This could in turn limit our access to capital markets, harm our results of operations, and leadlaboratory space from the Axis Parc developer pursuant to a decline in the trading price of our ordinary shares. Additionally, ineffective internal control over financial reporting could expose us to increased risk of fraud or misuse of corporate assets and subject us to potential delisting from The NASDAQ Global Market, or NASDAQ, regulatory investigations and civil or criminal sanctions. We may also be required to restate our financial statements for prior periods.
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems and those of our current and future CROs and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we have not experienced any such material system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development and commercialization of our product candidates could be delayed.
Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses.
Our operations, and those of our third-party research institution collaborators, CROs, CMOs, suppliers, and other contractors and consultants, could be subject to earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics, and other natural orman-made disasters or business interruptions. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses. Damage or extended periods of interruption to our corporate, development or research facilities due to fire, natural disaster, power loss, communications failure, unauthorized entry or other events could cause us to cease or delay development of some or all of our drug product candidates. Although we maintain property damage and business interruption insurance coverage on these facilities, our insurance might not cover all losses under such circumstances and our business may be seriously harmed by such delays and interruption.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our drug product candidates.
We face an inherent risk of product liabilitylease agreement dated November 11, 2017, as a result of the clinical testing of our drug product candidates and will face an even greater risk if we commercialize any products. For example, we may be sued if our drug product candidates cause or are perceived to cause injury or are found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our drug product candidates.
Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
decreased demand for our products;
injury to our reputation;
withdrawal of clinical trial participants and inability to continue clinical trials;
initiation of investigations by regulators;
costs to defend the related litigation;
a diversion of management’s time and our resources;
substantial monetary awards to trial participants or patients;
product recalls, withdrawals or labeling, marketing or promotional restrictions;
loss of revenue;
exhaustion of any available insurance and our capital resources;
the inability to commercialize any drug product candidate; and
a decline in our share price.
Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of products we develop, alone or with collaborators. Although our clinical trials are currently covered by a clinical trial insurance, the amount of such insurance coverage may not be adequate, we may be unable to maintain such insurance, or we may not be able to obtain additional or replacement insurance at a reasonable cost, if at all. Our insurance policies may also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We may have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts. Even if our agreements with any future corporate collaborators entitle us to indemnification against losses, such indemnification may not be available or adequate should any claim arise.
Our international operations subject us to various risks, and our failure to manage these risks could adversely affect our results of operations.
We face significant operational risks as a result of doing business internationally, such as:
fluctuations in foreign currency exchange rates;
potentially adverse and/or unexpected tax consequences, including penalties due to the failure of tax planning or due to the challenge by tax authorities on the basis of transfer pricing and liabilities imposed from inconsistent enforcement;
potential changes to the accounting standards, which may influence our financial situation and results;
becoming subject to the different, complex and changing laws, regulations and court systems of multiple jurisdictions and compliance with a wide variety of foreign laws, treaties and regulations;
reduced protection of, or significant difficulties in enforcing, intellectual property rights in certain countries;
difficulties in attracting and retaining qualified personnel;
restrictions imposed by local labor practices and laws on our business and operations, including unilateral cancellation or modification of contracts; and
rapid changes in global government, economic and political policies and conditions, political or civil unrest or instability, terrorism or epidemics and other similar outbreaks or events, and potential failure in confidence of our suppliers or customers due to such changes or events; and tariffs, trade protection measures, import or export licensing requirements, trade embargoes and other trade barriers.
We are subject to certain covenants as a result of certainnon-dilutive financial support we have received to date.
We have receivednon-dilutive financial support totaling €23.1 million as of December 31, 2016, to support various research programs from the Walloon Region, or the Region. The support has been granted in the form of recoverable cash advances, or RCAs, and subsidies.
In the event we decide to exploit any discoveries or products from the research funded by under an RCA, the relevant RCA becomes refundable, otherwise the RCA is not refundable. We own the intellectual property rights which result from the research programs partially funded by the Region, unless we decide not to exploit, or cease to exploit, the results of the research in which case the results and intellectual property rights are transferred to the Region. Subject to certain exceptions, however, we cannot grant to third parties, by way of license or otherwise, any right to use the results without the prior consent of the Region. We also need the consent of the Region to transfer an intellectual property right resulting from the research programs or a transfer or license of a prototype or installation. Obtaining such consent from the Region could give rise to a review of the applicable financial terms. The RCAs also contain provisions prohibiting us from conducting research for any other person which would fall within the scope of a research program of one of the RCAs. Most RCAs provide that this prohibition is applicable during the research phase and the decision phase but a number of RCAs extend it beyond these phases.
Subsidies received from the Region are dedicated to funding research programs and patent applications and are not refundable. We own the intellectual property rights which result from the research programs or with regard to a patent covered by a subsidy. Subject to certain exceptions, however, we cannot grant to third parties, by way of license, transfer or otherwise, any right to use the patents or research results without the prior consent of the Region. In addition, certain subsidies require that we exploit the patent in the countries where the protection was granted and to make an industrial use of the underlying invention. In case of bankruptcy, liquidation or dissolution, the rights to the patents covered by the patent subsidies will be assumed by the Region by operation of law unless the subsidy is reimbursed. Furthermore, we would lose our qualification as a small ormedium-sized enterprise, the patent subsidies will terminate and no additional expenses will be covered by such patent subsidies.
We may be exposed to significant foreign exchange risk.
We incur portions of our expenses, and may in the future derive revenues, in currencies other than the euro, in particular, the U.S. dollar. As a result, we are exposed to foreign currency exchange risk as our results of operations and cash flows are subject to fluctuations in foreign currency exchange rates. We currently do not engage in hedging transactions to protect against uncertainty in future exchange rates between particular foreign currencies and the euro. Therefore, for example, an increase in the value of the euro against the U.S. dollar could be expected to have a negative impact on our revenue and earnings growth as U.S. dollar revenue and earnings, if any, would be translated into euros at a reduced value. We cannot predict the impact of foreign currency fluctuations, and foreign currency fluctuations in the future may adversely affect our financial condition, results of operations and cash flows.
The requirements of being a U.S. public company may strain our resources and divert management’s attention.
We are required to comply with various corporate governance and financial reporting requirements under the Sarbanes-Oxley Act of 2002, the Securities and Exchange Act of 1934, as amended or the Exchange Act, and the rules and regulations adopted by the SEC and the Public Corporation Accounting Oversight Board. Further, compliance with various regulatory reporting requires significant commitments of time from our management and our directors, which reduces the time available for the performance of their other responsibilities. Our failure to track and comply with the various rules may materially adversely affect our reputation, ability to obtain the necessary certifications to financial statements, lead to additional regulatory enforcement actions, and could adversely affect the value of the ADSs or the ordinary shares.
The investment of our cash and cash equivalents may be subject to risks that may cause losses and affect the liquidity of these investments.
As of December 31, 2016, we had cash and cash equivalents of €48.4 million and short term investments of €34.2 million. We historically have invested substantially all of our available cash and cash equivalents in corporate bank accounts. Pending their use in our business, we may invest the net proceeds of the global offering in investments that may include corporate bonds, commercial paper, certificates of deposit and money market funds. These investments may be subject to general credit, liquidity, and market and interest rate risks. We may realize losses in the fair value of these investments or a complete loss of these investments, which would have a negative effect on our financial statements.
Risks Related to Ownership of our Ordinary Shares and ADSs
The market price for the ADSs may be volatile or may decline regardless of our operating performance.
The trading price of the ADSs has fluctuated, and is likely to continue to fluctuate, substantially. The trading price of the ADSs depends on a number of factors, many of which are beyond our control and may not be related to our operating performance, including, among others:
actual or anticipated fluctuations in our financial condition and operating results;
actual or anticipated changes in our growth rate relative to our competitors;
competition from existing products or new products that may emerge;
announcements by us, our partners or our competitors of significant acquisitions, strategic partnerships, joint ventures, collaborations, or capital commitments;
failure to meet or exceed financial estimates and projections of the investment community or that we provide to the public;
issuance of new or updated research or reports by securities analysts;
fluctuations in the valuation of companies perceived by investors to be comparable to us;
additions or departures of key management or scientific personnel;
disputes or other developments related to proprietary rights, including patents, litigation matters, and our ability to obtain patent protection for our technologies;
changes to coverage policies or reimbursement levels by commercial third-party payors and government payors and any announcements relating to coverage policies or reimbursement levels;
announcement or expectation of additional debt or equity financing efforts;
sales of the ADSs or ordinary shares by us, our insiders or our other shareholders; and
general economic and market conditions.
These and other market and industry factors may cause the market price and demand for the ADSs to fluctuate substantially, regardless of our actual operating performance, which may limit or prevent investors from readily selling their ADSs and may otherwise negatively affect the liquidity of our ADSs shares. In addition, the stock market in general, and biotechnology and biopharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies.
Fluctuations in the exchange rate between the U.S. dollar and the euro may increase the risk of holding the ADSs.
Our shares currently trade on Euronext Brussels and Euronext Paris in euros, while the ADSs trade on NASDAQ in U.S. dollars. Fluctuations in the exchange rate between the U.S. dollar and the euro may result in differences between the value of the ADSs and the value of our ordinary shares, which may result in heavy trading by investors seeking to exploit such differences. In addition, as a result of fluctuations in the exchange rate between the U.S. dollar and the euro, the U.S. dollar equivalent of the proceeds that a holder of the ADSs would receive upon the sale in Belgium of any ordinary shares withdrawn from the depositary upon calculation of the corresponding ADSs and the U.S. dollar equivalent of any cash dividends paid in euros on our ordinary shares represented by the ADSs could also decline.
Holders of the ADSs are not treated as shareholders of our company.
Holders of the ADSs are not treated as shareholders of our company, unless they cancel the ADSs and withdraw our ordinary shares underlying the ADSs. The depositary (or its nominee) is the shareholder of the ordinary shares underlying the ADSs. Holders of ADSs therefore do not have any rights as shareholders of our company, other than the rights that they have pursuant to the deposit agreement.
If securities or industry analysts do not publish research or publish inaccurate research or unfavorable research about our business, the price of the ordinary shares and the ADSs and trading volume could decline.
The trading market for the ordinary shares and the ADSs depends in part on the research and reports that securities or industry analysts publish about us or our business. If no or few securities or industry analysts cover our company, the trading price for the ordinary shares and the ADSs would be negatively impacted. If one or more of the analysts who covers us downgrades the ordinary shares or the ADSs or publishes incorrect or unfavorable research about our business, the price of the ordinary shares and the ADSs would likely decline. If one or more of these analysts ceases coverage of our company or fails to publish reports on us regularly, or downgrades the ordinary shares or the ADSs, demand for the ADSs and ordinary shares could decrease, which could cause the price of the ADSs and ordinary shares or trading volume to decline.
We have no present intention to pay dividends on our ordinary shares in the foreseeable future and, consequently, your only opportunity to achieve a return on your investment during that time is if the price of the ADSs or the ordinary shares increases.
We have no present intention to pay dividends in the foreseeable future. Any recommendation by our board of directors to pay dividends will depend on many factors, including our financial condition (including losses carried-forward), results of operations, legal requirements and other factors. Furthermore, pursuant to Belgian law, the calculation of amounts available for distribution to shareholders, as dividends or otherwise, must be determined on the basis of ournon-consolidated statutory accounts prepared in accordance with Belgian accounting rules. In addition, in accordance with Belgian law and our articles of association, we must allocate each year an amount of at least 5% of our annual net profit under ournon-consolidated statutory accounts to a legal reserve until the reserve equals 10% of our share capital. Therefore, we are unlikely to pay dividends or other distributions in the foreseeable future. If the price of the ADSs or the ordinary shares declines before we pay dividends, you will incur a loss on your investment, without the likelihood that this loss will be offset in part or at all by potential future cash dividends.
Our shareholders residing in countries other than Belgium may be subject to double withholding taxation with respect to dividends or other distributions made by us.
Any dividends or other distributions we make to shareholders will, in principle, be subject to withholding tax in Belgium at a rate of 25%, except for shareholders which qualify for an exemption of withholding tax such as, among others, qualifying pension funds or a company qualifying as a parent company within the meaning of the Council Directive (90/435/EEC) July 23, 1990, known as the Parent-Subsidiary Directive, or that qualify for a lower withholding tax rate or an exemption by virtue of a tax treaty. Various conditions may apply and shareholders residing in countries other than Belgium are advised to consult their advisers regarding the tax consequences of dividends or other distributions made by us. Our shareholders residing in countries other than Belgium may not be able to credit the amount of such withholding tax to any tax due on such dividends or other distributions in any other country than Belgium. As a result, such shareholders may be subject to double taxation in respect of such dividends or other distributions. Belgium and the United States have concluded a double tax treaty concerning the avoidance of double taxation, or the U.S.-Belgium Tax Treaty. The U.S.-Belgium Tax Treaty reduces the applicability of Belgian withholding tax to 15%, 5% or 0% for U.S. taxpayers, provided that the U.S. taxpayer meets the limitation of benefits conditions imposed by the U.S.-Belgium Tax Treaty. The Belgian withholding tax is generally reduced to 15% under the U.S.-Belgium Tax Treaty. The 5% withholding tax applies in case where the U.S. shareholder is a company which holds at least 10% of the shares in the company. A 0% Belgian withholding tax applies when the shareholder is a company which has held at least 10% of the shares in the company for at least 12 months, or is, subject to certain conditions, a U.S. pension fund. The U.S. shareholders are encouraged to consult their own tax advisers to determine whether they can invoke the benefits and meet the limitation of benefits conditions as imposed by the U.S.-Belgium Tax Treaty.
We believe that we were a passive foreign investment company (a “PFIC”) for the 2015 taxable year and in prior taxable years, but that we likely were not a PFIC for our 2016 taxable year. It is uncertain whether we will be a PFIC in future taxable years. Our status as a PFIC is a fact intensive determination and we cannot provide any assurances regarding our PFIC status for past, current or future taxable years. U.S. holders of the ADSs may suffer adverse tax consequences if we are characterized as a PFIC for any taxable year.
Generally, if, for any taxable year, at least 75% of our gross income is passive income, or at least 50% of the value of our assets is attributable to assets that produce passive income or are held for the production of passive income, including cash, we would be characterized as a passive foreign investment company, or PFIC, for U.S. federal income tax purposes. For purposes of these tests, passive income includes dividends, interest, and gains from the sale or exchange of investment property and rents and royalties other than rents and royalties which are received from unrelated parties in connection with the active conduct of a trade or business. If we are characterized as a PFIC, U.S. holders of the ADSs may suffer adverse tax consequences, including having gains realized on the sale of the ADSs treated as ordinary income, rather than capital gain, the loss of the preferential rate applicable to dividends received on the ADSs by individuals who are U.S. holders, and having interest charges apply to distributions by us and the proceeds of sales of the ADSs.
Our status as a PFIC will depend on the composition of our income, including the receipt of milestones, and the composition and value of our assets (which may be determined in large part by reference to the market value of the ADSs and ordinary shares, which may be volatile) from time to time.
We believe we were a PFIC in our 2015 taxable year and in prior taxable years, but that we likely were not a PFIC for our 2016 taxable year. With respect to our 2017 taxable year and possibly future taxable years, it is uncertain whether we will be a PFIC based upon the expected value of our assets, including any goodwill, and the expected composition of our income and assets. Our status as a PFIC is a fact intensive determination made on an annual basis and we cannot provide any assurances regarding our PFIC status for past, current or future taxable years.
Future Sales Of Ordinary Shares Or ADSs By Existing Shareholders Could Depress The Market Price Of The ADSs.
If our existing shareholders sell, or indicate an intent to sell, substantial amounts of ordinary shares or ADSs in the public market, the trading price of the ADSs could decline significantly. In the future we may file one or more registration statements with the SEC covering ordinary shares available for future issuance under our equity incentive plans. Upon effectiveness of such registration statements, any shares subsequently issued under such plans will be eligible for sale in the public market, except to the extent that they are restricted by thelock-up agreements referred to above and subject to compliance with Rule 144 in the case of our affiliates. Sales of a large number of the shares issued under these plans in the public market could have an adverse effect on the market price of the ADSs and the ordinary shares.
We are a Belgian public limited liability company, and shareholders of our company may have different and in some cases more limited shareholder rights than shareholders of a U.S. listed corporation.
We are a public limited liability company incorporated under the laws of Belgium. Our corporate affairs are governed by Belgian corporate and securities law. The rights provided to our shareholders under Belgian corporate law and our articles of association differ in certain respects from the rights that you would typically enjoy as a shareholder of a U.S. corporation under applicable U.S. federal and state laws. Under Belgian corporate law, other than certain information that we must make public and except in certain limited circumstances, our shareholders may not ask for an inspection of our corporate records, while under Delaware corporate law any shareholder, irrespective of the size of its shareholdings, may do so. Shareholders of a Belgian corporation have more limited rights to initiate a derivative action, a remedy typically available to shareholders of U.S. companies, in order to enforce a right of our Company, in case we fail to enforce such right ourselves.
A liability action can be instituted for our account by one or more of our shareholders who, individually or together, hold securities representing at least 1.0% of the votes or a part of the capital worth at least €1.25 million and have not approved of the discharge from liability that was granted to the directors. If the court orders the directors to pay damages, they are due to us, though the amounts advanced by the minority shareholders (for example attorney’s fees) are to be reimbursed by us. If the action is disallowed, the minority shareholders may be ordered to pay the costs, and, should there be grounds therefor, to pay damages to the directors, for example for having conducted provocative and reckless legal proceedings.
In addition, a majority of our shareholders present or represented at our meeting of shareholders may release a director from any claim of liability we may have, provided that the financial position of the company is accurately reflected in the annual accounts. This includes a release from liability for any acts of the directors beyond their statutory powers or in breach of the Belgian Company Code, provided that the relevant acts were specifically mentioned in the convening notice to the meeting of shareholders deliberating on the discharge. In contrast, most U.S. federal and state laws prohibit a company or its shareholders from releasing a director from liability altogether if he or she has acted in bad faith or has breached his or her duty of loyalty to the company. Finally, Belgian corporate law does not provide any form of appraisal rights in the case of a business combination. As a result of these differences between Belgian corporate law and our articles of association, on the one hand, and the U.S. federal and state laws, on the other hand, in certain instances, you could receive less protection as an ADS holder of our company than you would as a shareholder of a listed U.S. company.
Takeover provisions in the national law of Belgium may make a takeover difficult.
Public takeover bids on our shares and other voting securities, such as warrants or convertible bonds, if any, are subject to the Belgian Act of April 1, 2007 on public takeover bids, as amended and implemented by the Belgian Royal Decree of April 27, 2007, or Royal Decree, and to the supervision by the Belgian Financial Services and Markets Authority, or FSMA. Public takeover bids must be made for all of our voting securities, as well as for all other securities that entitle the holders thereof to the subscription to, the acquisition of or the conversion into voting securities. Prior to making a bid, a bidder must issue and disseminate a prospectus, which must be approved by the FSMA. The bidder must also obtain approval of the relevant competition authorities, where such approval is legally required for the acquisition of our company. The Belgian Act of April 1, 2007 provides that a mandatory bid will be required to be launched for all of our outstanding shares and securities giving access to ordinary shares if a person, as a result of its own acquisition or the acquisition by persons acting in concert with it or by persons acting on their account, directly or indirectly holds more than 30% of the voting securities in a company that has its registered office in Belgium and of which at least part of the voting securities are traded on a regulated market or on a multilateral trading facility designated by the Royal Decree. The mere fact of exceeding the relevant threshold through the acquisition of one or more shares will give rise to a mandatory bid, irrespective of whether or not the price paid in the relevant transaction exceeds the current market price.
There are several provisions of Belgian company law and certain other provisions of Belgian law, such as the obligation to disclose important shareholdings and merger control, that may apply to us and which may make an unfriendly tender offer, merger, change in management or other change in control, more difficult. These provisions could discourage potential takeover attempts that third parties may consider and thus deprive the shareholders of the opportunity to sell their shares at a premium (which is typically offered in the framework of a takeover bid).
Holders of ADSs do not have the same voting rights as holders of our ordinary shares.
Holders of ADSs may exercise voting rights with respect to the ordinary shares represented by the ADSs only in accordance with the provisions of the deposit agreement. The deposit agreement provides that, upon receipt of notice of any meeting of holders of our ordinary shares, the depositary will fix a record date for the determination of ADS holders who shall be entitled to give instructions for the exercise of voting rights. Upon timely receipt of notice from us, if we so request, the depositary shall distribute to the holders as of the record date (1) the notice of the meeting or solicitation of consent or proxy sent by us and (2) a statement as to the manner in which instructions may be given by the holders. You may instruct the depositary of your ADSs to vote the ordinary shares underlying your ADSs. Otherwise, you will not be able to exercise your right to vote, unless you withdraw the ordinary shares underlying the ADSs you hold. However, you may not know about the meeting far enough in advance to withdraw those ordinary shares. We cannot guarantee you that you will receive the voting materials in time to ensure that you can instruct the depositary to vote your ordinary shares or to withdraw your ordinary shares so that you can vote them yourself. In addition, the depositary and its agents are not responsible for failing to carry out voting instructions or for the manner of carrying out voting instructions. This means that you may not be able to exercise your right to vote, and there may be nothing you can do if the ordinary shares underlying your ADSs are not voted as you requested.
Holders of ADSs may be subject to limitations on the transfer of their ADSs and the withdrawal of the underlying ordinary shares.
ADSs are transferable on the books of the depositary. However, the depositary may close its books at any time or from time to time, when it deems expedientwhich expires on September 30, 2020. In January 2016, we entered into asix-year lease agreement for our U.S. corporate offices located in connection with the performance of its duties. The depositary may refuse to deliver, transfer or register transfers of your ADSs generally when our books or the books of the depositary are closed, or at any time if we or the depositary think it is advisable to do so because of any requirement of law, government or governmental body, or under any provision of the deposit agreement, or for any other reason, subject to your right to cancel your ADSs and withdraw the underlying ordinary shares. Temporary delays in the cancellation of your ADSs and withdrawal of the underlying ordinary shares may arise because the depositary has closed its transfer books or we have closed our transfer books, the transfer of ordinary shares is blocked to permit voting at a shareholders’ meeting or we are paying a dividend on our ordinary shares.
In addition, you may not be able to cancel your ADSs and withdraw the underlying ordinary shares when you owe money for fees, taxes and similar charges and when it is necessary to prohibit withdrawals in order to comply with any laws or governmental regulations that apply to ADSs or to the withdrawal of ordinary shares or other deposited securities.
We are an “emerging growth company” and are availing ourselves of reduced disclosure requirements applicable to emerging growth companies, which could make the ADSs or the ordinary shares less attractive to investors.Boston, Massachusetts.
We are an “emerging growth company,” as defined in the JOBS Act, and we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find the ADSs or the ordinary shares less attractive because we may rely on these exemptions. If some investors find the ADSs or the ordinary shares less attractive as a result, there may be a less active trading market for the ADSs or the ordinary shares and the price of the ADSs or the ordinary shares may be more volatile. We may take advantage of these reporting exemptions until we are no longer an emerging growth company. We would cease to be an emerging growth company upon the earliest to occur of (1) the last day of the fiscal year in which we have more than $1.0 billion in annual revenue; (2) the date we qualify as a “large accelerated filer,” with at least $700 million of equity securities held bynon-affiliates; (3) the issuance, in any three-year period, by our company of more than $1.0 billion innon-convertible debt securities held bynon-affiliates; and (4) December 31, 2020. We may choose to take advantage of some but not all of these exemptions.
Even after we no longer qualify as an emerging growth company, we may still qualify as a “smaller reporting company” which would allow us to take advantage of many of the same exemptions from disclosure requirements, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act and reduced disclosure obligations regarding executive compensation. We cannot predict if investors will find our ordinary shares less attractive because we may rely on these exemptions. If some investors find our ordinary shares less attractive as a result, there may be a less active trading market for our ordinary shares and our share price may be more volatile.
As a foreign private issuer, we are exempt from a number of rules under the U.S. securities laws and are permitted to file less information with the SEC than a U.S. company. This may limit the information available to holders of ADSs or ordinary shares.
We are a “foreign private issuer,” as defined in the SEC’s rules and regulations and, consequently, we are not subject to all of the disclosure requirements applicable to public companies organized within the United States. For example, we are exempt from certain rules under the Exchange Act, that regulate disclosure obligations and procedural requirements related to the solicitation of proxies, consents or authorizations applicable to a security registered under the Exchange Act, including the U.S. proxy rules under Section 14 of the Exchange Act. In addition, our officers and directors are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and related rules with respect to their purchases and sales of our securities. Moreover, while we currently make annual and semi-annual filings with respect to our listing on Euronext Brussels and Euronext Paris, we will not be required to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. domestic issuers and will not be required to file quarterly reports on Form10-Q or current reports on Form8-K under the Exchange Act. Accordingly, there will be less publicly available information concerning our company than there would be if we were not a foreign private issuer.
As a foreign private issuer, we are permitted to adopt certain home country practices in relation to corporate governance matters that differ significantly from NASDAQ corporate governance listing standards. These practices may afford less protection to shareholders than they would enjoy if we complied fully with corporate governance listing standards.
As a foreign private issuer listed on NASDAQ, we are subject to corporate governance listing standards. However, rules permit a foreign private issuer like us to follow the corporate governance practices of its home country. Certain corporate governance practices in Belgium, which is our home country, may differ significantly from corporate governance listing standards. For example, neither the corporate laws of Belgium nor our articles of association require a majority of our directors to be independent and we could includenon-independent directors as members of our Nomination and Remuneration Committee, and our independent directors would not necessarily hold regularly scheduled meetings at which only independent directors are present. Currently, we intend to follow home country practice to the maximum extent possible. Therefore, our shareholders may be afforded less protection than they otherwise would have under corporate governance listing standards applicable to U.S. domestic issuers.
We may lose our foreign private issuer status in the future, which could result in significant additional cost and expense.
While we currently qualify as a foreign private issuer, the determination of foreign private issuer status is made annually on the last business day of an issuer’s most recently completed second fiscal quarter and, accordingly, the next determination will be made with respect to us on June 30, 2017. In the future, we would lose our foreign private issuer status if we to fail to meet the requirements necessarycommitted to maintain our foreign private issuer status asheadquarters and registered office in the Walloon region of the relevant determination date. For example, if more than 50%Belgium and all of our securities are held by U.S. residents and more than 50% ofexisting activities will continue to be performed in the members of our executive management team or members of our board of directors are residents or citizens of the United States, we could lose our foreign private issuer status.
The regulatory and compliance costs to us under U.S. securities laws as a U.S. domestic issuer may be significantly more than costs we incur as a foreign private issuer. If we are not a foreign private issuer, we will be required to file periodic reports and registration statements on U.S. domestic issuer forms with the SEC, which are more detailed and extensive in certain respects than the forms available to a foreign private issuer. We would be required under current SEC rules to prepare our financial statements in accordance with U.S. generally accepted accounting principles, or U.S. GAAP, rather than IFRS, and modify certain of our policies to comply with corporate governance practices associated with U.S. domestic issuers. Such conversion of our financial statements to U.S. GAAP will involve significant time and cost. In addition, we may lose our ability to rely upon exemptions from certain corporate governance requirements on U.S. stock exchanges that are available to foreign private issuers such as the ones described above and exemptions from procedural requirements related to the solicitation of proxies.
It may be difficult for investors outside Belgium to serve process on, or enforce foreign judgments against, us or our directors and senior management.
We are a Belgian public limited liability company. Less than a majority of the members of our board of directors and members of our executive management team are residents of the United States. All or a substantial portion of the assets of suchnon-resident persons and most of our assets are located outside the United States. As a result, it may not be possible for investors to effect service of process upon such persons or on us or to enforce against them or us a judgment obtained in U.S. courts. Original actions or actions for the enforcement of judgments of U.S. courts relating to the civil liability provisions of the federal or state securities laws of the United States are not directly enforceable in Belgium.
The United States and Belgium do not currently have a multilateral or bilateral treaty providing for reciprocal recognition and enforcement of judgments, other than arbitral awards, in civil and commercial matters. In order for a final judgment for the payment of money rendered by U.S. courts based on civil liability to produce any effect on Belgian soil, it is accordingly required that this judgment be recognized or be declared enforceable by a Belgian court in accordance with Articles 22 to 25 of the 2004 Belgian Code of Private International Law. Recognition or enforcement does not imply a review of the merits of the case and is irrespective of any reciprocity requirement. A U.S. judgment will, however, not be recognized or declared enforceable in Belgium if it infringes upon one or more of the grounds for refusal that are exhaustively listed in Article 25 of the Belgian Code of Private International Law. Actions for the enforcement of judgments of U.S. courts might be successful only if the Belgian court confirms the substantive correctness of the judgment of the U.S. court and is satisfied that:Walloon region.
the effect of the enforcement judgment is not manifestly incompatible with Belgian public policy;
the judgment did not violate the rights of the defendant;
the judgment was not rendered in a matter where the parties transferred rights subject to transfer restrictions with the sole purpose of avoiding the application of the law applicable according to Belgian international private law;
the judgment is not subject to further recourse under U.S. law;
the judgment is not compatible with a judgment rendered in Belgium or with a subsequent judgment rendered abroad that might be recognized in Belgium;
a claim was not filed outside Belgium after the same claim was filed in Belgium, while the claim filed in Belgium is still pending;
the Belgian courts did not have exclusive jurisdiction to rule on the matter;
the U.S. court did not accept its jurisdiction solely on the basis of either the nationality of the plaintiff or the location of the disputed goods; and
the judgment submitted to the Belgian court is authentic.
In addition to recognition or enforcement, a judgment by a federal or state court in the United States against us may also serve as evidence in a similar action in a Belgian court if it meets the conditions required for the authenticity of judgments according to the law of the state where it was rendered. The findings of a federal or state court in the United States will not, however, be taken into account to the extent they appear incompatible with Belgian public policy.
We may be subject at an increased risk of securities class action litigation.
Historically, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies have experienced significant share price volatility in recent years. If we were to be sued, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
ITEM 4 –INFORMATION ON THE COMPANY
A. History And Development Of The Company
Our legal and commercial name is Celyad SA. Prior to May 5, 2015, our corporate name was Cardio3 Biosciences SA. We are a limited liability company incorporated in the form of anaamloze vennootschap/société anonyme under Belgian law. We are registered with the Register of Legal Entities (RPM Nivelles) under the enterprise number 891.118.115. We were incorporated in Belgium on July 24, 2007 for an unlimited duration. Our fiscal year ends December 31.
Our principal executive and registered offices are located at rue Edouard Belin 2, 1435 Mont-Saint-Guibert, Belgium and our telephone number is +32 10 394 100. Our agent for service of process in the United States is CT Corporation System. We also maintain a website at www.celyad.com. The reference to our website is an inactive textual reference only and the information contained in, or that can be accessed through, our website is not a part of this Annual Report.
Our actual capital expenditures for the years ended December 31, 2014,2015 and 2016 amounted to €0.6 million, €0.8 million and €1.8 million, respectively. These capital expenditures primarily consisted of the acquisition of laboratory equipment and industrial tools, the refurbishment of our research and development laboratories and leasehold improvements of our corporate offices located in Belgium and the United States. We expect our capital expenditures to increase in absolute terms in the near term as we continue to advance our research and development programs and grow our operations. We anticipate our capital expenditure in 2017 to be financed mostly from finance leases and partly with proceeds of our global offering.
B. Business Overview
Overview
We are a leader in engineered cell therapy treatments with clinical programs currently targeting indications in immune-oncology.
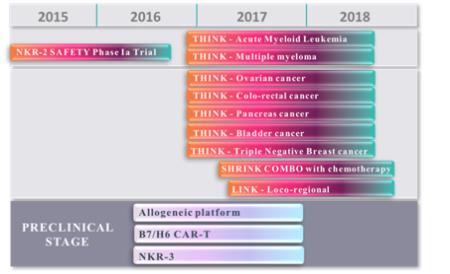
Our lead product candidate in oncology isCAR-TNKR-2, an autologous chimeric antigen receptor, or CAR, using NKG2D, an activating receptor of Natural Killer, or NK, cells transduced onT-lymphocytes. We successfully completed in 2016 our first clinical trial for this product candidate, called theCM-CS1 trial. TheCM-CS1 trial was a Phase 1 dose escalation study that was conducted at the Dana Farber Cancer Institute in Boston, Massachusetts. It enrolled patients with refractory or relapsed acute myeloid leukemia, or AML, or multiple myeloma, or MM. The outcome of theCM-CS1 trial was presented at the 2016 American Society of Hematology, or ASH, Annual Meeting in December 2016. No treatment-related safety concerns were reported at the four doses tested. First signals of activity were also identified and reported.
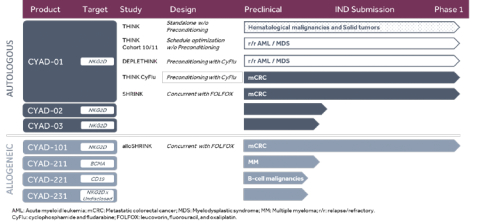
In December 2016, a second Phase 1 clinical trial was initiated. The THINK trial (THerapeutic Immunotherapy withCAR-TNKR-2) is a multinational (EU/US) open-label Phase 1b trial to assess the safety and clinical activity of multiple administrations of autologousCAR-TNKR-2 cells in seven refractory cancers, including five solid tumors (colorectal, ovarian, bladder, triple-negative breast and pancreatic cancers) and two hematological tumors (AML and MM). The THINK trial will test three dose levels adjusted to body weight: up to 3x108, 1x109 and 3x109CAR-TNKR-2 cells. At each dose, the patients will receive three successive administrations, two weeks apart, ofCAR-TNKR-2 cells. The dose escalation part of the study will enroll up to 24 patients while the extension phase would enroll 86 additional patients. The dose escalation part is expected to be completed in the last quarter of 2017.
In July 2016, we announced the signing of an exclusive licensing agreement with the Japanese immuno-oncology company ONO Pharmaceutical Co. Ltd., or ONO, for the development and commercialization of our allogeneicCAR-TNKR-2 immunotherapy. The license agreement with ONO grants them the exclusive right to develop and commercialize our allogeneicCAR-TNKR-2T-Cell immunotherapy in Japan, Korea and Taiwan. In exchange for receiving a license in these countries, ONO will pay us up to $311.5 million in development and commercial milestones, including an upfront payment of $12.5 million plus double digit royalties on net sales in ONO territories.
Our lead drug product candidate in cardiovascular disease is calledC-Cure, an autologous cell therapy for the treatment of patients with ischemic heart failure, or HF.C-Cure was evaluated inCHART-1, a Phase 3 trial conducted in Europe and Israel with 290 patients suffering from advanced ischemic HF. Topline results from theCHART-1 trial were reported in June 2016. Results indicated that the trial was overall neutral but with a positive trend, consistent across all parameters tested. Although the primary endpoint of the randomized trial was not met among the entireCHART-1 patient population, a significant
subpopulation representing more than 60% of the overall trial patients, defined by their Left Ventricular End Diastolic Volume, did meet the trial primary endpoint with a P value of 0.015. Based on the results of theCHART-1 trial, a U.S. trial, orCHART-2, has been designed to exclusively enroll the subset of patients that met the primary endpoint of theCHART-1 trial. We are currently seeking partners to further develop and commercializeC-Cure.
Strategy
Our goal is to become a leader inCAR-T cell therapies dedicated to the treatment of cancer. The key elements of this strategy are as follows:
| • | | Rapidly advanceCAR-TNKR-2 clinical development for the treatment of hematological and solid indications targeted in our THINK trial. We are currently enrolling patients with refractory or relapsed AML, MM, colorectal, bladder, ovarian, pancreatic and triple negative breast cancers in a Phase 1b clinical trial ofCAR-TNKR-2 in Belgium and in the United States. We intend to progressCAR-TNKR-2 in further stages of development in the indications that demonstrate clinical activity. |
| • | | Explore the potentially synergisticeffect of the combination ofCAR-TNKR-2 with standard chemotherapy in first or second line metastatic colorectal cancer patients. We believe there is an opportunity to study the properties of increased ligand expression in tumor cells exposed to aggressive chemotherapy regime, with the administration ofCAR-TNKR-2 in between the chemotherapy cycles. |
| • | | Explore the potential ofCAR-TNKR-2 administered loco regionally.We believe there is an opportunity to study the safety and effectiveness of administration ofCAR-TNKR-2 in the liver in colorectal cancer patients with liver metastasis. |
| • | | Develop our allogeneicCAR-TNKR-2 clinical program. We are developing a proprietary approach to inhibitT-cell receptor, or TCR, expression inCAR-T cells that allows us to use donor cells as opposed to the patient’s own T lymphocytes, which may make the administration ofCAR-T therapies feasible in patients lacking sufficient T lymphocytes, and, potentially, provide an approach to the administration ofCAR-T cells in certain conditions where the resident immune system can be temporarily inhibited. |
| • | | Make manufacturing and logistics of autologousCAR-T therapies feasible to address large indications. While current autologous approaches may be suited for relatively small indications, and allogeneic approaches have yet to demonstrate their equivalent effectiveness innon-hematological malignancies, we believe it is important to find manufacturing and supply chain solutions that allow the administration ofCAR-T cells in large indications feasible, cost effective and safe. Building on our experience in autologous cell therapy from ourC-Cure program, we, with external partners, are developing a fully automated point of care manufacturing solution that will may provide a paradigm shift in the field of autologous cell therapy. |
| • | | Leverage our expertise and knowledge of engineered-cell therapies to expand ourNKR-T cell therapy drug product candidate pipeline. In addition, we are continuing preclinical studies of our other preclinical product candidates, including NKp30, which is another activating receptor of NK cells, and B7H6, which is a NKp30 ligand expressed on cancer cells. We expect that B7H6 will enter clinical development in 2018 for the treatment of a pediatric rare and serious malignancy neuroblastoma. |
| • | | Find a partner to continue the development of our cardiovascular assets, in particularC-Cure. Based on the results from our previous trials, we believe thatC-Cure is a promising candidate for the treatment of ischemic HF. We are actively seeking a partner that could leverage that data and conductCHART-2 for registration ofC-Cure in the United States and in Europe. |
Drug product candidates
We currently hold worldwide rights to develop and commercialize all of our drug product candidates.
Our most advanced product candidates are autologous cell therapy treatments. In autologous procedures, a patient’s cells are harvested, selected, reprogrammed and expanded, and then infused back into the same patient. A benefit of autologous therapies is that autologous cells are not recognized as foreign by patients’ immune systems. We believe that we are well positionned to effectively advance autologous cell therapy treatments for cancer and other indications as a result of the expertise andknow-how that we have acquired through our development ofC-Cure.
Oncology
Cancer is the second leading cause of death in the United States after cardiovascular diseases, according to the U.S. Centers for Disease Control and Prevention. According to the American Cancer Society, in 2014, there were an estimated 1.6 million new cancer cases diagnosed and over 550,000 cancer deaths in the United States alone. In the past decades, the cornerstones of cancer therapies have been surgery, chemotherapy and radiation therapy. Since 2001, molecules that specifically target cancer cells have emerged as standard treatments for a number of cancers. For example, Gleevec is marketed by Novartis AG for the treatment of leukemia, and Herceptin is marketed by Genentech, Inc. for the treatment of breast and gastric cancer. Although targeted therapies have significantly improved the outcomes for certain patients with these cancers, there is still a high unmet need for the treatment of these and many other cancers.
Below are the statistics regarding certain forms of solid and hematological cancers and their estimated death rates in the United States for 2017:
| | | | | | | | |
| | | 2017 estimates for the US | |
| | | New cases | | | Deaths | |
Acute myeloid leukemia | | | 21,380 | | | | 11,960 | |
Multiple myeloma | | | 30,280 | | | | 12,790 | |
Colorectal cancer | | | 135,430 | | | | 50,260 | |
Pancreatic cancer | | | 53,670 | | | | 43,090 | |
Urinary bladder cancer | | | 79,030 | | | | 16,870 | |
Ovary cancer | | | 22,440 | | | | 14,080 | |
Triple negative breast cancer | | | 30,620 | | | | >>4,900 | |
Source: SEER, American Cancer Society
CART-Cell Therapy
The immune system has a natural response to cancer, as cancer cells express antigens that can be recognized by cells of the immune system. Upon recognition of a cancer antigen, activatedT-cells release substances that kill cancer cells and attract other immune cells to assist in the killing process. However, cancer cells can develop the ability to release inhibitory factors that allow them to evade immune response, resulting in the formation of cancers.
CART-cell therapy is a new technology that broadly involves engineering patients’ ownT-cells to express CARs so that thesere-engineered cells recognize and kill cancer cells, overcoming cancer cells’ ability to evade the immune response. CARs are comprised of the following elements:
binding domains that encode proteins, such as variable fragments of antibodies that are expressed on the surface of aT-cell and allow theT-cell to recognize specific antigens on cancer cells;
intracellular signaling domains derived fromT-cell receptors that activate the signaling pathways responsible for the immune response following binding to cancer cells. This allows the T cell to trigger the killing activity of the target cancer cell once it is recognized; and
costimulatory and adaptor domains, which enhance the effectiveness of theT-cells in their immune response.
Once activated, CART-cells proliferate and kill cancer cells directly through the secretion of cytotoxins that destroy cancer cells, and these cytokines attract other immune cells to the tumor site to assist in the killing process.
The CART-cell manufacturing process starts with collecting cells from a patient’s blood.T-cells are then selected, following which the CAR is introduced into theT-cells using vectors. The CART-cells are then expanded prior to injection back into the patient.
Current Investigational Treatments of Cancer using CART-Cells
CAR-T cell therapy is an emerging approach for the treatment of some cancers, such asB-cell malignancies.
CAR CD19 is the most studied CAR. CAR CD19 has an antigen binding domain that recognizes the CD19 antigen that is present on all B lymphocytes. This means that if a cancer originates from B lymphocytes, such as Acute Lymphoblastic Leukemia (ALL), then a CAR bearing the CD19 antibody could potentially recognize it and destroy it. Indeed, results of a clinical trial reported in the New England Journal of Medicine in October 2014 demonstrated that CAR CD19 CAR therapy was effective in treating patients with relapsed and refractory ALL. Treatment was associated with a complete remission rate of 90% and sustained remissions of up to two year after treatment. Despite its promise, CAR CD19 therapy is inherently limited to the treatment ofB-cell malignancies. CAR CD19 also targets normal B lymphocytes leading to the need to treat those patients with gamma globulins.
Our Approach to CART-Cell Therapy
Our approach toCAR-T cell therapy has the potential to treat a broader range of cancers than CD19 CARs as we employ natural receptors that target multiple ligands, and not just CD 19. Our most advanced CAR technologies use activating receptors of NK cells, which are lymphocytes of the innate immune system that kill diseased cells directly and indirectly, by secreting cytokines that attract other immune cells to assist in the killing process. The receptors of NK cells used in our therapies target the ligands that are activated in cancer cells, but are absent or expressed at very low levels in normal cells.
CAR-TNKR-2
Our leadCAR-TNKR-T cell drug product candidate isCAR-TNKR-2.CAR-TNKR-2 uses the native sequence of NKG2D in the CAR construct. InCAR-TNKR-2, the human natural sequence of NKG2D is expressed outside the T lymphocyte and bound to an intra cellular domain called CD3 Zeta. This intracellular domain is the same as the one used in other CARs and is responsible for the activation of theT-cell once NKG2D recognizes and binds to its target. In addition, the complex NKG2D CD3 Zeta binds to DAP 10 which is aco-stimulatory molecule present onT-cells, which means that the activation triggered by the primary stimulatory chain CD3 Zeta is further strengthened by DAP 10, a secondary orco-stimulatory domain.
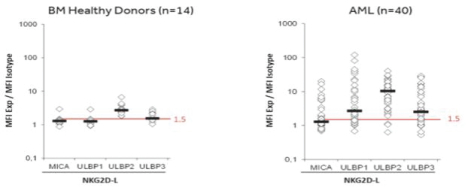
Ligands for the NKG2D receptor are expressed at the surface of cancer cells, allowing for the targeting of multiple tumor types. NKG2D receptor ligands, such asULBP1-6, MICA and MICB, are expressed in numerous solid tumors and blood cancers, including ovarian, bladder, breast, lung and liver cancers, as well as leukemia, lymphoma and myeloma. Within a given cell population, we have observedin vitro effectiveness ofCAR-TNKR-2 even when as few as 7% of the cancer cells expressed a NKG2D receptor ligand.
Preclinical Development
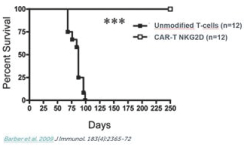
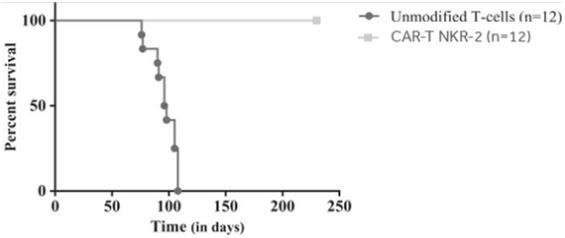
CAR-TNKR-2 has been tested in preclinical models of solid and blood cancers, including lymphoma, ovarian cancer, melanoma and myeloma. In preclinical studies, treatment withCAR-TNKR-2 significantly increased survival. In studies, 100% of treated mice survived through thefollow-up period of the applicable study, which in one study was 325 days. All untreated mice died during thefollow-up period of the applicable study.
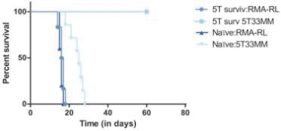
In one representative study, as shown in the figure below, the treatment withCAR-TNKR-2 completely prevented tumor development in mice injected with ovarian cancer cells and followed over a period of 225 days. In contrast, all mice injected with ovarian cancer cells that were treated with unmodifiedT-cells developed cancerous tumors and died during that period.
Our leadCAR-TNKR-T cell drug product candidate isCAR-TNKR-2.CAR-TNKR-2 uses the native sequence of NKG2D in the CAR construct. InCAR-TNKR-2, the human natural sequence of NKG2D is expressed outside the T lymphocyte and bound to an intra cellular domain called CD3 Zeta. This intracellular domain is the same as the one used in other CAR and is responsible for the activation of the T cell once NKG2D recognizes and binds to its target. In addition, the complex NKG2D CD3 Zeta binds to DAP 10 which is a co stimulatory molecule present on T cell, which means that the activation triggered by the primary stimulatory chain CD3 Zeta is further strengthened by DAP 10 a secondary or co stimulatory domain.
Ligands for the NKG2D receptor are expressed at the surface of cancer cells, allowing for the targeting of multiple tumor types. NKG2D receptor ligands, such asULBP1-6, MICA and MICB, are expressed in numerous solid tumors and blood cancers, including ovarian, bladder, breast, lung and liver cancers, as well as leukemia, lymphoma and myeloma. Within a given cell population, we have shownin vitro effectiveness ofNKR-2 even when as few as 7% of the cancer cells expressed a NKG2D receptor ligand.
Preclinical Development
CAR-TNKR-2 has been tested in preclinical models of solid and blood cancers, including lymphoma, ovarian cancer, melanoma and myeloma. In preclinical studies, treatment withNKR-2 significantly increased survival. In studies, 100% of treated mice survived through thefollow-up period of the applicable study, which in one study was 325 days. All untreated mice died during thefollow-up period of the applicable study.
In one representative study, as shown in the figure below, the treatment withNKR-2 completely prevented tumor development in mice injected with ovarian cancer cells and followed over a period of 225 days. In contrast, all mice injected with ovarian cancer cells that were treated with unmodifiedT-cells developed cancerous tumors and died during that period.
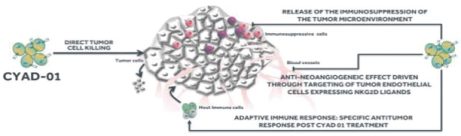
Our preclinical models have shown that administration ofCAR-TNKR-2 is followed by changes in a tumor’s micro-environment resulting from the local release of chemokines, a family of small cytokines.
In a preclinical study, mice that had been injected with 5T33MM cancer cells (a myeloma cancer) and treated withCAR-TNKR-2 were rechallenged, either with the 5T33MM cancer cells or a different tumor type (RMA lymphoma cells). The mice that were rechallenged with the same tumor type survived, while the mice that were challenged with a different tumor type died, as shown in the figure below. Of note, at the time of there-challenge of the surviving animals, noCAR-TNKR-2 were detected in the animals, hence the protection against the original tumor is linked to an adaptive immunity mechanism.
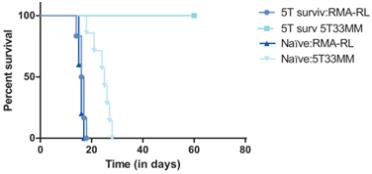
We do not believe that this effect has been observed with other CARs.
Moreover, preclinical studies have suggested thatCART-TNKR-2 could potentially have a direct effect on tumor vasculature. Tumor vessels express ligands for the NKG2D receptor that are not generally expressed by normal vessels. We believe that this expression may be linked to genotoxic stress, hypoxia andre-oxygenation in tumors and therefore thatCART-TNKR-2 could potentially inhibit tumor growth by decreasing tumor vasculature, which enhances the activity through a virtuous circle of anoxia of tumor cells and increased ligand expression of tumor cells.
Preclinical studies also suggest thatCART-TNKR-2 is active without lymphodepletion conditioning, which is the destruction of lymphocytes andT-cells, normally by radiation. We believe this absence of apre-conditioning regimen may significantly expand the range of patients eligible for CART-cell treatment, reduce costs, reduce toxicity and thereby improve patient experience and acceptance.
No significant toxicology findings were reported from preclinical multiple-dose studies at dose levels below 107CART-TNKR-2 per animal. Some temporary weight loss was noted in animals treated withCART-TNKR-2 at doses of 2x107 per animal, a dose practically unattainable in human equivalents.
Clinical Development
CM-CS1 Phase 1a trial
On June 9, 2014, we filed an IND with the FDA in order to conduct a clinical trial for ourCAR-TNKR-2 product candidate. This trial was calledCM-CS1. The purpose of the trial was to evaluate the safety and feasibility of administering a single intravenous dose ofCAR-TNKR-2 to patients with AML and MM. The trial was designed to start at low doses in order to evaluate the potential “on target/off tumor” toxicity, meaning the undue targeting of healthy tissues.
All patients enrolled in theCM-CS1 trial were patients with (i) AML who are not in remission and for which standard therapy options are not available or (ii) relapsed or refractory progressive MM. All patients received a single-dose intravenous administration ofCAR-TNKR-2T-cells without prior lymphodepletion conditioning.CM-CS1 was a dose escalation trial to test four different dose levels. Patients received doses from 1×106 up to 3×107CAR-TNKR-2 in a single intravenous injection. For each dose level, three patients, one with AML, one with MM, and one with either AML or MM, were recruited.
This trial was conducted at the Dana Farber Cancer Institute in the United States. The outcome of the trial was:
• | | Among AML/MDS and MM patients, a single dose ofCAR-TNKR-2 cells without lymphodepletion was feasible and well tolerated without dose limiting toxicities, or DLTs, over a range of 1×106 to 3×107 T cells. |
Cases of unexpected survival and/or improvement in hematologic parameters were noted in both AML and MM patients, some with and some without subsequent therapy.
NKG2DCAR-T-specific activity against autologous tumor-containing cells was observed in vitro in the patients tested.
TheCMCS-1 trial results that were presented at ASH in December 2016 and paved the way for studies of multiple infusions and higher doses ofCAR-TNKR-2 cells, closer to a potential therapeutic range, in numerous malignancies.
THINK Phase 1b Trial
In December 2016, we initiated THINK, a second clinical trial with ourCAR-TNKR-2 product candidate.
THINK (THerapeutic Immunotherapywith NKr-2) is a multinational (EU/US) open-label Phase Ib study to assess the safety and clinical activity of multiple administrations of autologousCAR-TNKR-2 cells in seven cancers, including five solid tumors (colorectal, ovarian, bladder, triple-negative breast and pancreatic cancers) and two hematological tumors (acute myeloid leukemia and multiple myeloma) in patients who did not respond to or relapsed after first line therapies.
The trial will test three dose levels adjusted to body weight: up to 3x108, 1x109 and 3x109CAR-TNKR-2 cells. At each dose, the patients will receive three successive administrations of the specified dose, two weeks apart. The dose escalation part of the study will enroll up to 24 patients while the extension phase is planned to enroll a maximum of 86 additional patients.
Other CART-Cell Development
Additional Autologous Programs
We also have two additional autologousNKR-T cells programs that are in preclinical development. One program involves the use of aNKR-T expressing NKp30, another activated receptor of NK cells.NKR-T cells expressing NKp30 target ligands, which are expressed on many types of cancer cells, including lymphoma, leukemia and gastrointestional stromal tumors. The primary ligand of NKp30 is B7H6. Previous preclinical studies performed at Dartmouth College and reported in the Journal of Immunology in 2012 demonstrated thatNKR-T cells expressing NKp30 were able to kill cancer cells expressing NKp30 ligands both in vitro and in vivo. Another program involves the specific targeting of B7H6 to kill cancer cells that express B7H6. This program is a more canonical CAR using B7H6 as a single chain variable fragement antibody associated with primary CD3 Zeta andco-stimulatory CD 28 domains, and with the lymphodepletion to allow in vivo proliferation. Previous preclinical studies performed at Dartmouth College and reported in the Journal of Immunology in 2015 demonstrated that therapy targeting B7H6 decreased tumor burden of melanoma- and ovarian cancer-bearing mice. We intend to initiate clinical development ofCAR-T B7H6 in Neuroblastoma, a devastating pediatric malignancy, in early 2018.
Allogeneic Platform
AutologousCAR-T cells have yielded impressive results in B cell malignancies. B cell malignancies represent a small proportion of cancer patients. Addressing larger indications such as some solid tumors make autologous treatments using the current centralized manufacturing paradigm a challenging option both from a cost and logistical perspectives. Finding alternative approaches for addressing those high unmet medical needs is therefore of paramount importance. An allogeneic approach must deal with two issues, one related to the rejection of the patient tissues by the grafted cells, ie, the graft versus host disease, or GvHD, and another related to the rejection of the graft by the host immune system, the Transplant Rejection. A truly off the shelf approach must deal with those two issues. We are developing an allogeneic approach that may render some of those challenges more addressable. We have developed a mechanism to prevent the GvHD by silencing theT-Cell Receptor, or TCR, complex on T Lymphocytes. This silencing suppresses the GvHD. In hematological malignancies using the canonicalCAR-T cells, lymphodepletion is used which reduces or eliminates transplant rejection during the time the transplant is alive and the patient’s immune system is dampened. While this may indeed be feasible in this setting, expanding this to other type of cancers requires other approaches that we are currently investigating. We hope to enter clinical development of our allogeneic program beginning of 2018.
Comparison of ourNKR-T cell therapy approach to canonical CAR Therapy.
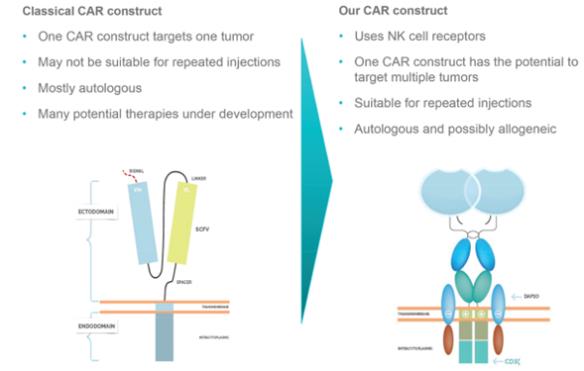
Cardiovascular Diseases
Cardiovascular diseases, which are diseases of the heart and blood vessels, are the largest cause of mortality in the world and, in 2012, approximately 31% of all global deaths were attributable to cardiovascular diseases, according to the World Health Organization. A subset of cardiovascular diseases, cardiac diseases, which are diseases of the heart, represent the single largest cause of death in the cardiovascular diseases population, according to the American Heart Association. If left untreated, cardiac diseases can lead to heart failure, or HF, a condition in which the heart is unable to pump enough blood to meet the body’s metabolic needs.
Current Treatments of Heart Failure
Although existing therapies have been somewhat effective in the treatment of HF, there is still great unmet medical need. In particular, in the case of ischemic HF, which is caused by insufficient oxygen to the heart, current treatments fail to address the decrease in the number of functional myocytes in the heart that result from this lack of oxygen. Over time this functional decrease modifies the dynamics of cardiac contractions leading to tissue remodeling and loss of cardiac function. We believe that cellular therapies have the potential to repair or replace thenon-functioning myocytes of ischemic HF patients.
C-Cure
Our drug product candidate,C-Cure, is an autologous cell therapy that we believe has the potential to treat moderate to severe HF. We have developedC-Cure based predominantly on technology that we licensed from the Mayo Clinic.
To guide cardiac tissue formation, ourC-Cure therapy reprograms multipotent stem cells harvested from a patient’s bone marrow into cardio reparative cells using naturally occurring cytokines and growth factors that mimic the signaling that occurs in embryonic heart tissue development. In theC-Cure process, stem cells are collected from an ischemic HF patient through bone marrow aspiration. The stem cells are then harvested, selected, expanded and differentiated into cardio reparative cells at our manufacturing facility, yielding a homogeneous and pure cell population. The cells are thenre-injected into the heart of the ischemic HF patient with ourC-Cathezcell injection catheter.
Clinical Development Status
CHART-1
CHART-1 has been conducted in Europe and Israel and was first authorized in November 2012.CHART-1 is a290-patient prospective controlled randomized double-blinded trial, including NYHA Class III and IV ischemic HF patients. The primary endpoint of this trial is an improvement in the composite hierarchical endpoint using the Finkelstein-Schoenfeld statistical method. The elements of this endpoint are, in hierarchical order, mortality, morbidity, quality of life, six minutes’ walk distance test, left ventricular end systolic volume (LVESV) and left ventricular ejection fraction (LVEF). Each patient in theC-Cure treatment group will be compared to each patient in the control group and a comprehensive score will be derived to compare one group against the other.
Outcome ofCHART-1
TheCHART-1 trial is the largest cardiovascular regenerative medicine trial to date addressing the effect of cardiopoiesis-based cell therapy in ischemic HF patients with moderate to severe symptoms. In thisat-risk population with limited therapeutic options, the trial was neutral regarding the primary endpoint, a hierarchical composite encompassingall-cause mortality, worsening heart failure events, Minnesota Living with Heart Failure Questionnaire score,6-min walk distance, LVESV and LVEF assessed at 39 weeks. Exploration of the primary composite endpoint according to baseline heart failure severity revealed a clinically relevant population that appeared to benefit from cardiopoietic cell therapy and in which the primary endpoint was met (p=0.015). This sizable target population (the responder population), representing 60% of the whole study cohort, was characterized by severe heart enlargement (baseline LVEDV 200–370 mL). Patients displaying a lower (< 200 mL) or greater (> 370 mL) LVEDV did not appear to respond to cell therapy in this study. We aim to confirm the present findings in subsequent studies with broader representation of women andnon-Caucasian racial groups.
CHART-2
On January 27, 2012, we filed our Investigational New Drug Application, or IND, for the use ofC-Cure inCHART-2 with the FDA (NCT02317458). The purpose of the IND trial is to evaluate the efficacy and safety of bone marrow-derived mesenchymal cardiopoietic cells for improving exercise capacity in subjects with advanced chronic ischemic HF.CHART-2 is expected to be conducted in the United States and Europe.CHART-2 has subsequently been amended and is currently a400-patient prospective controlled randomized double-blinded trial, for which the patient population is limited to the responder population identified inCHART-1. The primary efficacy endpoint ofCHART-2 is the change in Mortality, Worsening Heart Failure Event and Quality of Life frompre-procedure to 12 months. We are looking for a partner to runCHART-2 and continue the regulatory and commercial development ofC-Cure.
Our Complementary Devices
We developedC-Cathez, which isCE-marked, with the goal of overcoming limitations of existing cell injection devices that we discovered during our development ofC-Cure. Due to continuous heart movements, we believe that injecting cells into the heart requires a stable needle that anchors into the tissue during injection. In addition, excess pressure on cells during injection has an adverse impact on cell retention. To respond to these challenges,C-Cathez features a curved needle that provides stability during the injection and multiple holes along the needle that increase the exit surface, reducing the pressure exerted on cells during the injection procedure. In preclinical studies, we obtained a higher retention rate of injected cells through the use ofC-Cathez as compared to other commercially available catheters. In a preclinical study ofC-Cathez, use ofC-Cathez did not cause myocardial perforation or clinically relevant increases in the blood levels of cardiac enzymes were observed in pigs or dogs. We also market C Cathez in the European Union as a stand-alone medical device for delivery of diagnostic and therapeutic agents indicated for delivery into the heart to research laboratories and clinical-stage companies only.
In addition, we believe our heart access technology will enable cardiologists to take a unique access route directly to the patient’s left atrium, which may potentially enable the deployment of catheters or other necessary instruments for use in the treatment of various indications such as mitral valve disorders and structural heart diseases, conditions often linked to HF. This heart access technology includes the heart access sheath, mitral valveneo-chordae, and closure device. These devices are either in the discovery phase or preclinical development.
Licensing and Collaboration Agreements
Academic and clinical collaborations
We have core relationships and collaborations with the Mayo Foundation for Medical Education and Research, or the Mayo Clinic, and the Trustees of Dartmouth College, or Dartmouth.
Dartmouth College and Celdara
In January 2015, we entered into a stock purchase agreement, or the Celdara Agreement, with Celdara Medical, LLC, or Celdara, pursuant to which we purchased all of the outstanding membership interests of OnCyte, LLC, or OnCyte, for a $10.0 million upfront payment to Celdara, $6.0 million of which was paid in cash and $4.0 million of which was paid in the form of 93,087 of our ordinary shares. After this transaction we, Celdara and OnCyte entered into an asset purchase agreement, or OnCyte APA, pursuant to which Celdara sold to OnCyte, data, protocols, regulatory documents and intellectual property, including the rights and obligations under license agreements between Celdara and Dartmouth College, related to ourNKR-T cell therapy programs, or the Transferred Assets. Pursuant to the OnCyte APA, we are obligated to make development-based milestone payments to Celdara of $40.0 million for clinical products and of $36.5 million for preclinical products, as well as sales-based milestone payments of up to $80.0 million for products based on the Transferred Assets, or CAR Products. The OnCyte APA also requires us to make tiered single-digit royalty payments to Celdara in connection with the sales of CAR Products. Such royalties are payable on a CARProduct-by CAR Product andcountry-by-country basis until the later of (i) the last day that at least one valid patent claim covering the CAR Product exists, or (ii) the tenth anniversary of the day of the first commercial sale of the CAR Product in such country. Under the OnCyte APA, we can opt out of the development of any CAR Product if the data does not meet the scientific criteria of success. We may also opt out of development of any CAR Product for any other reason upon payment of a termination fee of $2.0 million to Celdara.
2010 Dartmouth License Agreement
Under the exclusive license agreement with Dartmouth College entered into in April 2010 and amended in February 2012, July 2013 and January 2015, Dartmouth College granted us (as successor in interest to Celdara) an exclusive, worldwide, royalty-bearing license to certainknow-how and patent rights to make, have made, use, and/or sell any product or process for human therapeutics, the manufacture, use or sale of which, is covered by such patent rights. Dartmouth College reserves the right to use the licensed patent rights and licensedknow-how, in the same field, for education and research purposes only. The patent rights covered by this agreement are related, in part, to methods for treating cancer involving chimeric NKG2D and NKP30 receptor targeted therapeutics and T cell receptor-deficientT-cell compositions in treating tumor, infection, GvHD, transplant and radiation sickness.
In consideration for the rights granted to us under the agreement, we are required to pay to Dartmouth College an annual license fee of $20,000 as well as a low single-digit royalty based on annual net sales of the licensed products by us and by our permitted sublicenses, with certain minimum net sales obligations beginning April 30, 2024 and continuing for each year of sales thereafter. We are also obligated to pay to Dartmouth College a certain tiered percentage of sublicensing income ranging from themid-single digits to themid-teens based on the development stage of the technology at the time the sublicense is granted. We are not required to pay sublicensing income on transactions in which we form a newspin-off entity and transfer at least a portion of our assets. Additionally, the agreement requires that we exploit the licensed products, and we have agreed to meet certain developmental and regulatory milestones. Upon successful completion of such milestones, we are obligated to pay to Dartmouth College certain milestone payments up to an aggregate amount of $1.5 million. We are responsible for all expenses in connection with the preparation, filing, prosecution and maintenance of the patents covered under the agreement.
After April 30, 2024, Dartmouth College may terminate the license if we fail to meet the specified minimum net sales obligations for any year, unless we pay to Dartmouth College the royalty we would otherwise be obligated to pay had we met such minimum net sales obligation. Dartmouth College may also terminate the license if we fail to meet a milestone within the specified time period, unless we pay the corresponding milestone payment. Either party may terminate the agreement in the event the other party defaults or breaches any of the provisions of the agreement, subject to 30 days’ prior notice and opportunity to cure. In addition, the agreement automatically terminates in the event we become insolvent, make an assignment for the benefit of creditors or file, or have filed against us, a petition in bankruptcy. Absent early termination, the agreement will continue until the expiration date of the last to expire patent right included under the agreement in the last to expire territory. We expect that the last to expire patent right included under this agreement will expire in 2033, absent extensions or adjustments.
2014 Dartmouth License Agreement
Under the exclusive license agreement with Dartmouth College entered into in June 2014 and amended in January 2015, Dartmouth College granted us (as successor in interest to Celdara) an exclusive, worldwide, royalty-bearing license under certainknow-how and patent rights to make, have made, use, modify, exploit,
distribute, and/or sell any product or process for human therapeutics, the manufacture, use or sale of which, is covered by such patent rights. Our license is subject to any rights that may be required to be granted to the government of the United States, and Dartmouth College reserves the right to use the licensed patent rights and licensedknow-how, in the same field, for education and research purposes only. The patent rights covered by this agreement are related, in part, toanti-B7-H6 antibody, fusion proteins and methods of using the same.
In consideration for the rights granted to us under the agreement, we are required to pay to Dartmouth College a license maintenance fee of $10,000 upon the first anniversary of the agreement and an annual license maintenance fee of $20,000 thereafter. We are also required to pay to Dartmouth College a low single-digit royalty based on annual net sales of the licensed products by us and by our permitted sub licensees, with a specified minimum royalty payment for each year of sales. We are obligated to pay to Dartmouth College a certain tiered percentage of sublicensing income ranging from themid-single digits to themid-teens based on the time or development stage of the technology at the time the sublicense is granted. We are not required to pay sublicensing income on transactions in which we form a newspin-off entity and transfer at least a portion of our assets. Additionally, the agreement requires that we exploit the licensed products, and we have agreed to meet certain developmental and regulatory milestones. Upon successful completion of such milestones for each licensed product, we are obligated to pay to Dartmouth College certain milestone payments up to an aggregate amount of $1.6 million. We are responsible for all expenses in connection with the preparation, filing, prosecution and maintenance of the patents covered under the agreement.
Dartmouth College may, at its option, terminate the license, upon thirty days written notice, if we fail to pay at least the minimum royalty payment amount or make such minimum payment within suchthirty-day period. In addition, Dartmouth College has the right to terminate if we fail to meet a milestone within the specified time period or fail to make the corresponding milestone payment, subject to 30 days’ prior written notice and opportunity to cure. We may unilaterally terminate the agreement by giving three months advance written notice to Dartmouth College and paying a termination fee of $2,500. Either party may terminate the agreement in the event the other party defaults or breaches any of the provisions of the agreement, subject to 30 days’ prior notice and opportunity to cure. In addition, the agreement automatically terminates in the event we become insolvent, make an assignment for the benefit of creditors or file, or have filed against us, a petition in bankruptcy. Absent early termination, the agreement will continue until the expiration date of the last to expire patent right included under the agreement in the last to expire territory. We expect that the last to expire patent right included under this agreement will expire in 2033, absent extensions or adjustments.
Mayo Clinic
C-Cure is based on technology discovered at the Molecular Pharmacology and Experimental Therapeutics lab at the Mayo Clinic, led by Dr. Andre Terzic. Under our technology license agreement with the Mayo Clinic entered into in June 2007 and amended in July 2008 and October 2010, or Mayo License, we were granted an exclusive, worldwide license to make, use, modify, enhance, promote, market and/or sell the “Cardiogenic Cocktail for the production of Cardiac Cells” and “Stem Cell Based Therapy forNon-ischemic Cardiomyopathic Heart Failure” within the field of cardiovascular regeneration or protection, including certain related patents. In addition, we were granted anon-exclusive, worldwide license to licensedknow-how in connection with the licensed inventions within the same field. The exclusive license is subject to the Mayo Clinic’s right to make and use the licensed inventions and licensedknow-how within its affiliates’ own programs, and to the rights, if any, of the United States government. The Mayo Clinic has the right to purchase quantities of licensed invention from us at cost to meet its and its affiliates’ internal needs.
In consideration for the rights granted to us under the Mayo License, we were required to pay an upfront fee to Mayo Clinic of €9,500,000 upon the initial agreement and $3,193,125 upon the execution of the second amendment, which were subsequently converted into our share capital. We also paid the Mayo Clinic $337,000 for the purchase of equipment for research purposes. Additionally, we are required to pay to the Mayo Clinic a low single-digit royalty on net commercial sales by us or by our permitted sublicensees from the commercialization of licensed products, on a licensedproduct-by-licensed product basis, beginning on the date of the first commercial sale of the relevant licensed product and extending until the earlier of (i) the 15 year anniversary of the first commercial sale of such licensed product, (ii) the date on which the licensed product is no longer covered by a valid claim of a licensed patent in the applicable territory, or (iii) termination of the Mayo License. The Mayo License permits a reduction of these royalties, not to exceed a specified floor, for amounts payable to third parties as required toin-license necessary third-party technology.
Under the Mayo License, we are responsible for the development, manufacture and commercialization of the licensed inventions. We committed to provide the Mayo Clinic with $500,000 of directed research funding per year for the years 2012 through 2014. Any results of this research will automatically be included as licensed inventions under the Mayo License. We will also fund research at the Mayo Clinic in the amount of $1,000,000 per year for four years in the area of regeneration or protection for cardiovascular applications. Such payments will begin once we have achieved both commercial sale of a licensed product and a positive cash flow from operations in the previous financial year. We will have an exclusive right of first negotiation to acquire an exclusive license to inventions that are the direct result of work carried out under these grants, in accordance with the mechanism described in the Mayo License. The Mayo Clinic provided us with directed research and conducted a dose finding study for us at no additional cost. Subject topre-existing obligations, until October 18, 2015, we also have an exclusive right of first negotiation to obtain an exclusive license from the Mayo Clinic on any guided cardiopoiesis technology developed by Dr. Andre Terzic or developed orco-developed by Dr. Atta Behfar, the senior investigator involved in the discovery of the cardiopoiesis technology. With respect to both of the foregoing rights of first negotiation, if we and the Mayo Clinic do not reach agreement for a license for the applicable invention within the prescribed negotiation period or permitted extension, the Mayo Clinic is prohibited from entering into a license agreement for such invention with a third party for a period of nine months.
The Mayo License will continue until the later of ten years or as long as the Mayo Clinic has any rights to any part of the licensed inventions. The Mayo Clinic may terminate the license on aproduct-by-product basis or licensedinvention-by-licensed invention basis if we default in making payment when due and payable or under other circumstances specified in the Mayo License, subject to 120 days’ prior written notice and opportunity to cure. The Mayo Clinic may also terminate the Mayo License if we deliberately make false statements in reports delivered to Mayo Clinic. Additionally, Mayo Clinic may convert the license tonon-exclusive or terminate the license upon final decision of an arbitral tribunal that we breached our diligence obligations under the Mayo License. Further, Mayo Clinic may terminate the agreement immediately for our insolvency or bankruptcy, as described in the Mayo License.
In November 2014, we entered into a Preferred Access Agreement with the Mayo Clinic. Pursuant to this agreement, the Mayo Clinic will review with us, on a quarterly basis, technologies arising from the Mayo Center for Regenerative Medicine, and we may review certain other technologies upon request. If, as a result of such reviews, we and the Mayo Clinic decide to advance a certain technology, we will enter into a separate exclusive license agreement with respect to such technology. This agreement remains in effect until December 2017, and may be extended by mutual agreement.
ONO Pharmaceuticals
On July 11, 2016, we entered into a license and collaboration agreement with ONO Pharmaceuticals Co., Ltd., or ONO, in connection with which we granted ONO an exclusive license for the development, manufacture and commercialization of allogenic products incorporating ourNKR-T cell technology in Japan, Korea and Taiwan. Under the terms of the collaboration, ONO is solely responsible for and bears all costs incurred in the research, development and commercialization of such products in its geographies. In addition, we granted ONO an exclusive option to obtain an exclusive license to develop, manufacture and commercialize autologous products incorporating our autologousCAR-TNKR-2 cell technology in Japan, Korea and Taiwan.
In consideration for the rights granted to ONO under the agreement, we received anup-front payment in the amount of 1.25 billion JPY ($12.5 million) and we are eligible to receive additional milestones for up to 30.075 billion JPY ($299 million) in development and commercial milestones. In addition, we are entitled to receive double digit royalties on nets sales of licensed products in licensed territories.
Intellectual Property
Patents and patent applications
Patents, patent applications and other intellectual property rights are important in the sector in which we operate. We consider on acase-by-case basis filing patent applications with a view to protecting certain innovative products, processes, and methods of treatment. We may also license or acquire rights to patents, patent applications or other intellectual property rights owned by third parties, academic partners or commercial companies which are of interest to us.
Our patent portfolio includes pending patent applications and issued patents in the United States and in foreign countries. These patents and applications generally fall into four broad categories:
patents and patent applications licensed from Dartmouth that relate to ourNKR-T platform;
patents and pending patent applications relating to cardiopoiesis, a subset of which are licensed from the Mayo Clinic;
patents and pending applications we own that relate to cardiac injection catheter technology;
and patent applications owned by our subsidiary Corquest that relate to cardiac medical device technology.
The term of a U.S. patent may be eligible for patent term extension under the Hatch-Waxman Act to account for at least some of the time the drug or device is under development and regulatory review after the patent is granted. With regard to a drug or device for which FDA approval is the first permitted marketing of the active ingredient, the Hatch-Waxman Act allows for extension of the term of one U.S. patent. The extended patent term cannot exceed the shorter of five years beyond thenon-extended expiration of the patent or 14 years from the date of the FDA approval of the drug or device. Some foreign jurisdictions have analogous patent term extension provisions that allow for extension of the term of a patent that covers a device approved by the applicable foreign regulatory agency.
NKR-T cell Platform Patents
As of February 28, 2017, our CART-cell portfolio includes four patent families exclusively licensed to us by Dartmouth. This portfolio includes four issued U.S. patents; six pending U.S. patent applications; and 13 foreign patent applications pending in jurisdictions including Australia, Brazil, Canada, China, Europe, Hong Kong, India, Japan, Mexico, and Russia. These patents and patent applications relate to particular chimeric antigen receptors and toT-cell receptor-deficientT-cells, and will be further detailed below.
A first patent family relates to chimeric NK receptors and methods for treating cancer. There are two granted US patents in this family and a further pending US application. The scope of this patent family includes chimeric natural killer cell receptors (NKR CARs), T cells with such receptors (NKR CAR T cells) and methods of treating cancer with these NKR CAR T cells. Patents in this family will begin to expire in 2025, absent any adjustments or extensions. We expect that any patents that eventually issue from currently pending applications in this family will begin to expire in 2025, absent any adjustments or extensions.
A second patent family relates to T cell receptor-deficient T cell compositions. Its scope envisages human T cells that express a chimeric receptor and have impaired or reduced expression of the endogenous T cell receptor (TCR). Such cells are particularly useful in allogeneic cell therapy. The family includes members that relate to the concept (irrespective of the way the T cell is made T cell receptor deficient), as well as members describing specific ways of making the cellsTCR-deficient. There are two granted US patents, as well as three further pending US applications and ten applications in other jurisdictions. Claim 1 of patent US9,181,527 was challenged by an anonymous third party in anex parte reexamination procedure, but the USPTO has determined that claim 1 is patentable as amended. Patents in this family will begin to expire in 2030, absent any adjustments or extensions. We expect that any patents that eventually issue from currently pending applications in this family will begin to expire in 2030, absent any adjustments or extensions.
A third patent family is entitled NKp30 receptor targeted therapeutics and describes a specific NKR CAR based on the NKp30 receptor. It is pending in the US. We expect that any patents that eventually issue from currently pending applications in this family will begin to expire in 2032, absent any adjustments or extensions.
A fourth family relates to ananti-B7-H6 antibody, CARs and BiTE molecules containing such antibody, CAR T cells, and methods of treating cancer with theCAR-T cells. Applications are pending in China, Europe, Japan and the US. We expect that any patents that eventually issue from currently pending applications in this family will begin to expire in 2033, absent any adjustments or extensions.
Cardiopoiesis Platform Patents
As of February 1, 2017, our cardiopoiesis platform portfolio includes five living patent families, four of which are owned by the Mayo Clinic or jointly by Mayo and Celyad, which we refer to as the Mayo Cardiopoiesis Patents and are exclusively licensed to us, and one family that is owned by us, which we refer to as our Cardiopoiesis Patents.
The Mayo Cardiopoiesis Patents include three issued U.S. patents; 26 foreign patents issued in jurisdictions including Australia, China, Europe, Hong Kong, Israel, Japan, Mexico, New Zealand, Russia, Singapore, South Africa, the Republic of Korea and Taiwan; three pending U.S. patent applications; and 17 foreign patent applications pending in jurisdictions including Europe, Australia, Brazil, Canada, China, Hong Kong, Israel, India, Japan, Mexico, Russia, Singapore, South Korea, and Thailand. These patents and patent applications relate to compositions and methods for inducing cardiogenesis in embryonic stem cells, methods of identifying cardiopoietic stem cells, and methods of using cardiopoietic stem cells to treat cardiovascular tissue. The Mayo Cardiopoiesis Patents will begin to expire in 2025, absent any adjustments or extensions. We expect that any patents that eventually issue from currently pending applications in the Mayo Cardiopoiesis patent portfolio will begin to expire in 2025, absent any adjustments or extensions.
Our Cardiopoiesis Patents include one issued U.S. patent and granted patents in each of Australia, Europe, Hong Kong, Japan, Mexico, New Zealand, Russia, Singapore, South Korea and Taiwan; a pending U.S. patent application; and 7 foreign patent applications pending in jurisdictions including Brazil, Canada, China, India, Israel and Thailand. An anonymous third party has filed an opposition to the granted European patent, this is currently being examined by the EPO. Oral proceedings were held on 6 March 2017 during the opposition procedure, which resulted in revocation of the patent. This decision still needs to be confirmed in writing, and can be appealed. These patents and patent applications relate to pharmaceutical compositions containing cardiopoietic stem cells and methods of their production. Our Cardiopoiesis Patents will begin to expire in 2030, absent any adjustments or extensions. We expect that any patents that eventually issue from currently pending applications in our Cardiopoiesis patent portfolio will begin to expire in 2030, absent any adjustments or extensions.
Cardiac Injection Catheter Technology Patents
As of February 1, 2017, our cardiac injection catheter technology portfolio includes two patent families we own. This portfolio includes one issued U.S. patent and two pending U.S. patent applications; nine patents issued in jurisdictions including Australia, Canada, Europe, Hong Kong, Israel, Japan, Mexico, New Zealand and Russia; and nine foreign patent applications pending in jurisdictions including Australia, Brazil, Canada, China, Europe, India, Israel, Japan and Singapore. These patents and patent applications relate to injection catheters and processes for their use. Patents in this portfolio will begin to expire in 2029, absent any adjustments or extensions. We expect that any patents that eventually issue from currently pending applications in the Cardiac Injection Catheter Technology patent portfolio will begin to expire in 2030, absent any adjustments or extensions.
Heart Access Technology Patents
As of February 1, 2017, our heart access technology portfolio includes five patent families owned by Corquest, our wholly owned subsidiary. This portfolio includes eight pending U.S. patent applications and twenty-four foreign patent applications pending in various jurisdictions including Australia, Canada, Europe, Israel, Japan, and Mexico. These patent applications relate to devices, assemblies and methods for treating cardiac injuries and defects. Patents in this portfolio, when eventually issued, will begin to expire in 2032.
Trade secrets
In addition to our patents and patent applications, we keep certain of our proprietary information as trade secrets, which we seek to protect by confidentiality agreements with our employees and third parties, and by fragmentingknow-how between different individuals, in accordance with standard industry practices.
Competition
The industry in which we operate is subject to rapid technological change. We face competition from pharmaceutical, biopharmaceutical and medical devices companies, as well as from academic and research institutions. Some of these competitors are pursuing the development of medicinal products and other therapies that target the same diseases and conditions that we are targeting.
Some of our current or potential competitors, either alone or with their collaboration partners, have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical, biotechnology and gene therapy industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. The key competitive factors affecting the success of all of our programs are likely to be their efficacy, safety and convenience.
Many of our competitors have substantially greater financial, technical and other resources
CART-Cell Therapy
Early results from clinical trials have fueled continued interest in CART-cell therapies and our competitors as of the date of this Annual Report include Bellicum Pharmaceuticals, Inc., bluebird bio, Inc., Cellectis S.A., Juno Therapeutics, Inc., Kite Pharma Inc., Novartis AG, NantKwest Inc and Ziopharm Oncology, Inc.
C-Cure
We have identified several companies that are active in cardiac cell therapy as of the date of this Annual Report, including Capricor Inc., Mesoblast Ltd, Biocardia Inc. and Vericel Corporation.
Government Regulation
Government authorities in the United States at the federal, state and local level and in other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of drug and biological products, or biologics, such as our drug product candidates. Generally, before a new drug or biologic can be marketed, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific for each regulatory authority, submitted for review and an application for marketing authorization must be approved by the regulatory authority.
Certain products may be comprised of components that are regulated under separate regulatory authorities and by different centers at the FDA. These products are known as combination products. A combination product is comprised of a combination of a drug and a device; a biological product and a device; a drug and a biological product; or a drug, a device, and a biological product. Under regulations issued by the FDA, a combination product includes:
a product comprised of two or more regulated components that are physically, chemically, or otherwise combined or mixed and produced as a single entity;
two or more separate products packaged together in a single package or as a unit and comprised of drug and device products, device and biological products, or biological and drug products;
a drug, device, or biological product packaged separately that according to its investigational plan or proposed labeling is intended for use only with an approved individually specified drug, device or biological where both are required to achieve the intended use, indication, or effect and where upon approval of the proposed product the labeling of the approved product would need to be changed, e.g., to reflect a change in intended use, dosage form, strength, route of administration, or significant change in dose; or
any investigational drug, device, or biological packaged separately that according to its proposed labeling is for use only with another individually specified investigational drug, device, or biological product where both are required to achieve the intended use, indication, or effect.
One of our drug product candidates is a combination product that is comprised of a biologic and a device (an endocardial injection catheter) that is used for delivery of the biologic. Under the FDCA, the FDA is charged with assigning a center with primary jurisdiction, or a lead center, for review of a combination product. That determination is based on the “primary mode of action” of the combination product, which means the single mode of action that provides the most important therapeutic action of the combination product, i.e., the mode of action expected to make the greatest contribution to the overall intended therapeutic effects of the combination product. Thus, if the primary mode of action of a device-biologic combination product is attributable to the biologic product, that is, if it acts by means of a virus, therapeutic
serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, or analogous product, the FDA center responsible for premarket review of the biologic product (the Center for Biologics Evaluation and Research, or CBER) would have primary jurisdiction for the combination product. CBER is the agency component with primary jurisdiction for the premarket review and regulation for ourC-Cure investigational product. BecauseC-Cure utilizes a catheter as a delivery system to the heart, CBER may consult or collaborate with the agency center that is responsible for the premarket review of that device, the Center for Devices and Radiological Health, or CDRH.
U.S. Biological Product Development
In the United States, the FDA regulates biologics under the Federal Food, Drug, and Cosmetic Act, or FDCA, and the Public Health Service Act, or PHSA, and their implementing regulations. Biologics are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include, among other actions, the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, product recalls or withdrawals from the market, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
Our drug product candidates must be approved by the FDA through the Biologics License Application, or BLA, process before they may be legally marketed in the United States. The process required by the FDA before a biologic may be marketed in the United States generally involves the following:
completion of extensive nonclinical, sometimes referred to as preclinical laboratory tests, preclinical animal studies and formulation studies in accordance with applicable regulations, including the FDA’s Good Laboratory Practice, or GLP, regulations;
submission to the FDA of an IND, which must become effective before human clinical trials may begin;
performance of adequate and well-controlled human clinical trials in accordance with applicable IND regulations, good clinical practices, or GCPs, and other clinical trial-related regulations to establish the safety and efficacy of the proposed drug product candidate for its proposed indication;
submission to the FDA of a BLA;
satisfactory completion of an FDApre-approval inspection of the manufacturing facility or facilities where the product is produced to assess compliance with the FDA’s current good manufacturing practice, or cGMP, requirements to assure that the facilities, methods and controls are adequate to preserve the product’s identity, strength, quality, purity and potency;
potential FDA audit of the preclinical and/or clinical trial sites that generated the data in support of the BLA; and
FDA review and approval of the BLA prior to any commercial marketing or sale of the product in the United States.
The data required to support a BLA is generated in two distinct development stages: preclinical and clinical. The preclinical development stage generally involves laboratory evaluations of drug chemistry, formulation and stability, as well as studies to evaluate toxicity in animals, which support subsequent clinical testing. The conduct of the preclinical studies must comply with federal regulations, including GLPs. The sponsor must submit the results of the preclinical studies, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, as well as other information, to the FDA as part of the IND. An IND is a request for authorization from the FDA to administer an investigational drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for human trials. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions regarding the proposed clinical trials and places the IND on clinical hold within that30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin.
The FDA may also impose clinical holds on a drug product candidate at any time before or during clinical trials due to safety concerns,non-compliance, or other issues affecting the integrity of the trial. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that could cause the trial to be suspended or terminated. Where a trial is conducted at, or sponsored by, institutions receiving NIH funding for recombinant DNA research, prior to the submission of an IND to the FDA, a protocol and related documentation is submitted to and the trial is registered with the NIH Office of Biotechnology Activities, or OBA, pursuant to the NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules, or NIH Guidelines. Compliance with the NIH Guidelines is mandatory for investigators at institutions receiving NIH funds for research involving recombinant DNA, however many companies and other institutions not otherwise subject to the NIH Guidelines voluntarily follow them. The NIH is responsible for convening the Recombinant NDA Advisory Committee, or RAC, a federal advisory committee, which discusses protocols that raise novel or particularly important scientific, safety or ethical considerations at one of its quarterly public meetings. The OBA will notify the FDA of the RAC’s decision regarding the necessity for full public review of a gene therapy protocol. RAC proceedings and reports are posted to the OBA web site and may be accessed by the public.
The clinical stage of development involves the administration of the drug product candidate to healthy volunteers and patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control, in accordance with GCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety and assess efficacy. Each protocol, and any subsequent amendments to the protocol, must be submitted to the FDA as part of the IND. Further, each clinical trial must be reviewed and approved by an independent institutional review board, or IRB, at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed.
There are also requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries. Sponsors of certain clinical trials ofFDA-regulated products, including biologics, are required to register and disclose certain clinical trial information, which is publicly available at www.clinicaltrials.gov. Information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to discuss the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed until the new product or new indication being studied has been approved. However, there are evolving rules and increasing requirements for publication of trial-related information, and it is possible that data and other information from trials involving biologics that never garner approval could in the future require disclosure. In addition, publication policies of major medical journals mandate certain registration and disclosures as apre-condition for potential publication, even if not currently mandated as a matter of law. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs.
Clinical trials are generally conducted in three sequential phases, known as Phase 1, Phase 2 and Phase 3, and may overlap. Phase 1 clinical trials generally involve a small number of healthy volunteers who are initially exposed to a single dose and then multiple doses of the drug product candidate. The primary purpose of these clinical trials is to assess the metabolism, pharmacologic action, side effect tolerability and safety of the drug product candidate and, if possible, to gain early evidence on effectiveness. Phase 2 clinical trials typically involve studies in disease-affected patients to determine the dose required to produce the desired benefits. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected, as well as identification of possible adverse effects and safety risks and preliminary evaluation of efficacy. Phase 3 clinical trials generally involve large numbers of patients at multiple sites, in multiple countries, and are designed to provide the data necessary to demonstrate the efficacy of the product for its intended use, its safety in use, and to establish the overall benefit/risk relationship of the product and provide an adequate basis for product approval. Phase 3 clinical trials may include comparisons with placebo and/or other comparator treatments. The duration of treatment is often extended to mimic the actual use of a product during marketing. Generally, two adequate and well-controlled Phase 3 clinical trials are required by the FDA for approval of a BLA.
Post-approval trials, sometimes referred to as Phase IV clinical trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, FDA may condition approval of a BLA on the sponsor’s agreement to conduct additional clinical trials to further assess the biologic’s safety and effectiveness after BLA approval.
Progress reports detailing the results of the clinical trials, among other information, must be submitted at least annually to the FDA, and written IND safety reports must be submitted to the FDA and the investigators for serious and unexpected suspected adverse events, findings from other studies suggesting a significant risk to humans exposed to the biologic, findings from animal or in vitro testing that suggest a significant risk for human subjects, and any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA, the IRB, or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether or not a trial may move forward at designated intervals based on access to certain data from the trial. We may also suspend or terminate a clinical trial based on evolving business objectives and/or competitive climate. Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the drug product candidate as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug product candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug product candidate does not undergo unacceptable deterioration over its shelf life.
BLA and FDA Review Process
Following trial completion, trial data are analyzed to assess safety and efficacy. The results of preclinical studies and clinical trials are then submitted to the FDA as part of a BLA, along with proposed labeling for the product and information about the manufacturing process and facilities that will be used to ensure product quality, results of analytical testing conducted on the chemistry of the drug product candidate, and other relevant information. The BLA is a request for approval to market the biologic for one or more specified indications and must contain proof of safety, purity, potency and efficacy, which is demonstrated by extensive preclinical and clinical testing. The application may include both negative or ambiguous results of preclinical and clinical trials as well as positive findings. Data may come from company-sponsored clinical trials intended to test the safety and efficacy of a use of a product, or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of the investigational product to the satisfaction of the FDA. FDA approval of a BLA must be obtained before a biologic may be marketed in the United States.
Under the Prescription Drug User Fee Act, or PDUFA, as amended, each BLA must be accompanied by a significant user fee, which is adjusted on an annual basis. PDUFA also imposes an annual product fee for human drugs and an annual establishment fee on facilities used to manufacture prescription drugs. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business.
Once a BLA has been accepted for filing, which occurs, if at all, sixty days after the BLA’s submission, the FDA’s goal is to review BLAs within 10 months of the filing date for standard review or six months of the filing date for priority review, if the application is for a product intended for a serious or life-threatening condition and the product, if approved, would provide a significant improvement in safety or effectiveness. The review process is often significantly extended by FDA requests for additional information or clarification.
After the BLA submission is accepted for filing, the FDA reviews the BLA to determine, among other things, whether the proposed drug product candidate is safe and effective for its intended use, and whether the drug product candidate is being manufactured in accordance with cGMP to assure and preserve the drug
product candidate’s identity, strength, quality, purity and potency. The FDA may refer applications for novel drug product candidates or drug product candidates which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. The FDA will likelyre-analyze the clinical trial data, which could result in extensive discussions between the FDA and us during the review process. The review and evaluation of a BLA by the FDA is extensive and time consuming and may take longer than originally planned to complete, and we may not receive a timely approval, if at all.
Before approving a BLA, the FDA will conduct apre-approval inspection of the manufacturing facilities for the new product to determine whether they comply with cGMPs. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. In addition, before approving a BLA, the FDA may also audit data from clinical trials to ensure compliance with GCP requirements. After the FDA evaluates the application, manufacturing process and manufacturing facilities, it may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application will not be approved in its present form. A Complete Response Letter usually describes all of the specific deficiencies in the BLA identified by the FDA. The Complete Response Letter may require additional clinical data and/or an additional pivotal Phase III clinical trial(s), and/or other significant and time-consuming requirements related to clinical trials, preclinical studies or manufacturing. If a Complete Response Letter is issued, the applicant may either resubmit the BLA, addressing all of the deficiencies identified in the letter, or withdraw the application. Even if such data and information is submitted, the FDA may ultimately decide that the BLA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive, and the FDA may interpret data differently than we interpret the same data.
There is no assurance that the FDA will ultimately approve a product for marketing in the United States, and we may encounter significant difficulties or costs during the review process. If a product receives marketing approval, the approval may be significantly limited to specific populations, severities of allergies, and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling or may condition the approval of the BLA on other changes to the proposed labeling, development of adequate controls and specifications, or a commitment to conduct post-market testing or clinical trials and surveillance to monitor the effects of approved products. For example, the FDA may require Phase IV testing which involves clinical trials designed to further assess the product’s safety and effectiveness and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized. The FDA may also place other conditions on approvals including the requirement for a Risk Evaluation and Mitigation Strategy, or REMS, to assure the safe use of the product. If the FDA concludes a REMS is needed, the sponsor of the BLA must submit a proposed REMS. The FDA will not approve the NDA without an approved REMS, if required. A REMS could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Product approvals may be withdrawn fornon-compliance with regulatory standards or if problems occur following initial marketing.
Expedited Development and Review Programs
The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and nonclinical or clinical data demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a new drug or biologic may request the FDA to designate the drug or biologic as a Fast Track product concurrently with, or at any time after, submission of an IND, and the FDA must determine if the product qualifies for Fast Track designation within 60 days of receipt of the sponsor’s request. Unique to a Fast Track product, the FDA may consider for review sections of the marketing application on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the application, the FDA agrees to accept sections of the application and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the application.
Any product submitted to the FDA for marketing, including under a Fast Track program, may be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. Any product is eligible for priority review, or review within asix-month timeframe from the date a complete BLA is accepted for filing, if it has the potential to provide a significant improvement in safety and effectiveness compared to available therapies. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug or biological product designated for priority review in an effort to facilitate the review.
Additionally, a product may be eligible for accelerated approval. An investigational drug may obtain accelerated approval if it treats a serious or life-threatening condition and generally provides a meaningful advantage over available therapies and demonstrates an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, or IMM, that is reasonably likely to predict an effect on IMM or other clinical benefit. As a condition of approval, the FDA may require that a sponsor of a drug or biological product receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials. If the FDA concludes that a drug shown to be effective can be safely used only if distribution or use is restricted, it will require such post-marketing restrictions as it deems necessary to assure safe use of the drug, such as:
distribution restricted to certain facilities or physicians with special training or experience; or
distribution conditioned on the performance of specified medical procedures.
The limitations imposed would be commensurate with the specific safety concerns presented by the product. In addition, the FDA currently requires as a condition for accelerated approvalpre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product. Fast Track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
Breakthrough Designation
The Food and Drug Administration Safety and Innovation Act, or FDASIA, amended the FDCA to require the FDA to expedite the development and review of a breakthrough therapy. A product can be designated as a breakthrough therapy if it is intended to treat a serious or life-threatening condition and preliminary clinical evidence indicates that it may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. A sponsor may request that a drug product candidate be designated as a breakthrough therapy concurrently with, or at any time after, the submission of an IND, and the FDA must determine if the drug product candidate qualifies for breakthrough therapy designation within 60 days of receipt of the sponsor’s request. If so designated, the FDA shall act to expedite the development and review of the product’s marketing application, including by meeting with the sponsor throughout the product’s development, providing timely advice to the sponsor to ensure that the development program to gather preclinical and clinical data is as efficient as practicable, involving senior managers and experienced review staff in a cross-disciplinary review, assigning a cross-disciplinary project lead for the FDA review team to facilitate an efficient review of the development program and to serve as a scientific liaison between the review team and the sponsor, and taking steps to ensure that the design of the clinical trials is as efficient as practicable.
Accelerated Approval for Regenerative Advanced Therapies
As part of the 21st Century Cures Act, Congress recently amended the FD&C Act to create an accelerated approval program for regenerative advanced therapies, which include cell therapies, therapeutic tissue engineering products, human cell and tissue products, and combination products using any such therapies or products. Regenerative advanced therapies do not include those human cells, tissues, and cellular and tissue based products regulated solely under section 361 of the Public Health Service Act and 21 CFR Part 1271. The new program is intended to facilitate efficient development and expedite review of regenerative advanced therapies, which are intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition. A drug sponsor may request that FDA designate a drug as a regenerative advanced therapy concurrently with or at any time after submission of an IND. FDA has 60 calendar days to determine whether the drug meets the criteria, including whether there is preliminary clinical evidence indicating that the drug has the potential to address unmet medical needs for a serious or life-threatening disease or condition. A new drug application or BLA for a regenerative advanced therapy may be eligible
for priority review or accelerated approval through (1) surrogate or intermediate endpoints reasonably likely to predict long-term clinical benefit or (2) reliance upon data obtained from a meaningful number of sites. Benefits of such designation also include early interactions with FDA to discuss any potential surrogate or intermediate endpoint to be used to support accelerated approval. A regenerative advanced therapy that is granted accelerated approval and is subject to postapproval requirements may fulfill such requirements through the submission of clinical evidence, clinical studies, patient registries, or other sources of real world evidence, such as electronic health records; the collection of larger confirmatory data sets; or postapproval monitoring of all patients treated with such therapy prior to its approval.
Pediatric Trials
Under the Pediatric Research Equity Act, or PREA, a BLA or supplement to a BLA must contain data to assess the safety and efficacy of the product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. FDASIA requires that a sponsor who is planning to submit a marketing application for a drug or biological product that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration submit an initial Pediatric Study Plan, or PSP, within sixty days of anend-of-Phase II meeting or as may be agreed between the sponsor and FDA. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. FDA and the sponsor must reach agreement on the PSP. A sponsor can submit amendments to an agreed-upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from nonclinical studies, early phase clinical trials, and/or other clinical development programs. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of data or full or partial waivers.
Post-Marketing Requirements
Following approval of a new product, a manufacturer and the approved product are subject to continuing regulation by the FDA, including, among other things, monitoring and recordkeeping activities, reporting to the applicable regulatory authorities of adverse experiences with the product, providing the regulatory authorities with updated safety and efficacy information, product sampling and distribution requirements, and complying with promotion and advertising requirements, which include, among others, standards fordirect-to-consumer advertising, restrictions on promoting products for uses or in patient populations that are not described in the product’s approved labeling (known as“off-label use”), limitations on industry-sponsored scientific and educational activities, and requirements for promotional activities involving the internet. Although physicians may prescribe legally available drugs and biologics foroff-label uses, manufacturers may not market or promote suchoff-label uses.
Modifications or enhancements to the product or its labeling or changes of the site of manufacture are often subject to the approval of the FDA and other regulators, which may or may not be received or may result in a lengthy review process. Prescription drug promotional materials must be submitted to the FDA in conjunction with their first use. Any distribution of prescription drug products and pharmaceutical samples must comply with the U.S. Prescription Drug Marketing Act, or the PDMA, a part of the FDCA.
In the United States, once a product is approved, its manufacture is subject to comprehensive and continuing regulation by the FDA. The FDA regulations require that products be manufactured in specific approved facilities and in accordance with cGMPs. The cGMP requirements for constituent parts of cross-labeled combination products that are manufactured separately and notco-packaged are the same as those that would apply if these constituent parts were not part of a combination product. For single-entity andco-packaged combination products, there are two ways to demonstrate compliance with cGMP requirements, either compliance with all cGMP regulations applicable to each of the constituent parts included in the combination product, or a streamlined approach demonstrating compliance with either the drug/biologic cGMPs or the medical device quality system regulation rather than demonstrating full compliance with both, under certain conditions. These conditions include demonstrating compliance with specified provisions from the other of these two sets of cGMP requirements. Because theC-Cure device comprises a biologic and a catheter that are notco-packaged, we need to comply with the cGMPs requirements for each constituent part. We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our products in accordance with cGMP regulations. cGMP regulations require, among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation and the obligation to investigate and correct any deviations from cGMP.
Manufacturers and other entities involved in the manufacture and distribution of approved products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance. These regulations also impose certain organizational, procedural and documentation requirements with respect to manufacturing and quality assurance activities. BLA holders using contract manufacturers, laboratories or packagers are responsible for the selection and monitoring of qualified firms, and, in certain circumstances, qualified suppliers to these firms. These firms and, where applicable, their suppliers are subject to inspections by the FDA at any time, and the discovery of violative conditions, including failure to conform to cGMP, could result in enforcement actions that interrupt the operation of any such facilities or the ability to distribute products manufactured, processed or tested by them. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved BLA, including, among other things, recall or withdrawal of the product from the market.
The FDA also may require post-approval testing, sometimes referred to as Phase IV testing, risk minimization action plans and post-marketing surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product. Discovery of previously unknown problems with a product or the failure to comply with applicable FDA requirements can have negative consequences, including adverse publicity, judicial or administrative enforcement, untitled or warning letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties, among others. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
Other Regulatory Matters
Manufacturing, sales, promotion and other activities following product approval are also subject to regulation by numerous regulatory authorities in addition to the FDA, including, in the United States, the Centers for Medicare & Medicaid Services, or CMS, other divisions of the Department of Health and Human Services, the Drug Enforcement Administration, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency and state and local governments. In the United States, sales, marketing and scientific/educational programs must also comply with federal and state fraud and abuse laws, data privacy and security laws, transparency laws, and pricing and reimbursement requirements in connection with governmental payor programs, among others. The handling of any controlled substances must comply with the U.S. Controlled Substances Act and Controlled Substances Import and Export Act. Products must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. Manufacturing, sales, promotion and other activities are also potentially subject to federal and state consumer protection and unfair competition laws.
The distribution of pharmaceutical products is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
The failure to comply with regulatory requirements subjects firms to possible legal or regulatory action. Depending on the circumstances, failure to meet applicable regulatory requirements can result in criminal prosecution, fines or other penalties, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of product approvals, or refusal to allow a firm to enter into supply contracts, including government contracts. In addition, even if a firm complies with FDA and other requirements, new information regarding the safety or efficacy of a product could lead the FDA to modify or withdraw product approval. Prohibitions or restrictions on sales or withdrawal of future products marketed by us could materially affect our business in an adverse way.
Changes in regulations, statutes or the interpretation of existing regulations could impact our business in the future by requiring, for example: (i) changes to our manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of our products; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of our business.
U.S. Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of the FDA approval of our drug product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generallyone-half the time between the effective date of an IND and the submission date of a BLA plus the time between the submission date of a BLA and the approval of that application, except that the review period is reduced by any time during which the applicant failed to exercise due diligence. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The U.S. PTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we may apply for restoration of patent term for our currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant BLA.
An abbreviated approval pathway for biological products shown to be biosimilar to, or interchangeable with, anFDA-licensed reference biological product was created by the Biologics Price Competition and Innovation Act of 2009, or BPCI Act, which was part of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively the ACA. This amendment to the PHSA attempts to minimize duplicative testing. Biosimilarity, which requires that the biological product be highly similar to the reference product notwithstanding minor differences in clinically inactive components and that there be no clinically meaningful differences between the product and the reference product in terms of safety, purity, and potency, can be shown through analytical studies, animal studies, and a clinical trial or trials. Interchangeability requires that a biological product be biosimilar to the reference product and that the product can be expected to produce the same clinical results as the reference product in any given patient and, for products administered multiple times, that the product and the reference product may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biological product. However, complexities associated with the larger, and often more complex, structure of biological products as compared to small molecule drugs, as well as the processes by which such products are manufactured, pose significant hurdles to implementation that are still being worked out by the FDA.
A reference biological product is granted twelve years of exclusivity from the time of first licensure of the product, and the FDA will not accept an application for a biosimilar or interchangeable product based on the reference biological product until four years after first licensure. “First licensure” typically means the initial date the particular product at issue was licensed in the United States. This does not include a supplement for the biological product or a subsequent application by the same sponsor or manufacturer of the biological product (or licensor, predecessor in interest, or other related entity) for a change that results in a new indication, route of administration, dosing schedule, dosage form, delivery system, delivery device, or strength, unless that change is a modification to the structure of the biological product and such modification changes its safety, purity, or potency. Whether a subsequent application, if approved, warrants exclusivity as the “first licensure” of a biological product is determined on acase-by-case basis with data submitted by the sponsor.
Pediatric exclusivity is another type of regulatory market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. Thissix-month exclusivity, which attaches to the twelve-year exclusivity period for reference biologics, may be granted based on the voluntary completion of a pediatric trial in accordance with anFDA-issued “Written Request” for such a trial.
European Union Drug Development
In the European Union, our future drug product candidates will also be subject to extensive regulatory requirements. As in the United States, medicinal products can only be marketed if a marketing authorization, or MA, from the competent regulatory agencies has been obtained.
Clinical Trials
Similar to the United States, the various phases of preclinical and clinical research in the European Union are subject to significant regulatory controls. Although the EU Clinical Trials Directive 2001/20/EC has sought to harmonize the European Union clinical trials regulatory framework, setting out common rules for the control and authorization of clinical trials in the European Union, the European Union Member States
have transposed and applied the provisions of the Directive differently. This has led to significant variations in the Member State regimes. To improve the current system, a new Regulation No. 536/2014 on clinical trials on medicinal drug product candidates for human use, which repealed Directive 2001/20/EC, was adopted on April 16, 2014, and published in the European Official Journal on May 27, 2014. The new Regulation aims at harmonizing and streamlining the clinical trials authorization process, simplifying adverse event reporting procedures, improving the supervision of clinical trials, and increasing their transparency. The new Regulation entered into force on June 16, 2014, and is applicable since May 28, 2016. Until then the Clinical Trials Directive 2001/20/EC will still apply. In addition, the transitory provisions of the new Regulation offer the sponsors the possibility to choose between the requirements of the Directive and the Regulation for one year from the entry into application of the Regulation.
Under the current regime, before a clinical trial can be initiated it must be approved in each of the European Union countries where the trial is to be conducted by two distinct bodies: the National Competent Authority, or NCA, and one or more Ethics Committees, or ECs. More specifically, a clinical trial may not be started until the relevant EC has issued a favorable opinion, and the NCA has not informed the Sponsor of the trial of any grounds fornon-acceptance or confirmed that no such grounds exist. Approval will only be granted if satisfactory information demonstrating the quality of the investigational agent and itsnon-clinical safety has been provided, together with a study plan that details the manner in which the trial will be carried out.
ECs determine whether the proposed clinical trial will expose participants to unacceptable conditions of hazards, while considering, among other things, the trial design, protocol, facilities, investigator and supporting staff, recruitment of clinical trial subjects, the Investigator’s Brochure, or IB, indemnity and insurance, etc. The EC also determines whether clinical trial participants have given informed consent to participate in the trial. Following receipt of an application (which must be submitted in the national language), ECs must deliver their opinion within 60 days (or sooner if the Member State has implemented a shorter time period). For clinical trials of gene therapy, somatic cell therapy, and all medicinal products containing genetically modified organisms, this timeline may be extended (with an additional 120 days).
Similarly, a valid request for authorization (in the national language) must be submitted to the NCA of each Member State where the trial will be conducted. Sponsors must be notified of the decision within 60 days of receipt of the application (unless shorter time periods have been fixed), in the absence of which, the trial is considered approved. However, for clinical trials of gene therapy, somatic cell therapy, and all medicinal products containing genetically modified organisms, a written authorization by the competent NCA is required. Similar timeline extensions as for ECs exist.
Studies must comply with ethical guidelines and Good Clinical Practice (GCP) guidelines. Monitoring of adverse reactions that occur during clinical trials, including, where applicable, notification of the same to the competent NCA and ECs, is also required. Trials can be terminated early if a danger to human health is established or continuing the trial would be considered unethical. Consequently, the rate of completion of clinical trials may be delayed by many factors, including slower than anticipated patient enrollment or adverse events occurring during clinical trials.
Drug Review and Approval
In the European Economic Area, or EEA (which is comprised of the 28 Member States of the European Union plus Norway, Iceland and Liechtenstein), medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. There are two types of marketing authorizations:
The Centralized MA, which is issued by the European Commission through the Centralized Procedure, based on the opinion of the Committee for Medicinal Products for Human Use, or CHMP, of the European Medicines Agency, or EMA, and which is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, advanced-therapy medicines such as gene-therapy, somatic cell-therapy or tissue-engineered medicines, and medicinal products containing a new active substance indicated for the treatment of HIV, AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and other immune dysfunctions, and viral diseases. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the European Union.
National MAs, which are issued by the competent authorities of the Member States of the EEA and only cover their respective territory, are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in other Member State(s) through the Mutual Recognition
Procedure, or MRP. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure, or DCP. Under the DCP an identical dossier is submitted to the competent authorities of each of the Member States in which the MA is sought, one of which is selected by the applicant as the Reference Member State, or RMS. The competent authority of the RMS prepares a draft assessment report, a draft summary of product characteristics, or SmPC, and a draft of the labeling and package leaflet, which are sent to the other Member States (referred to as the Concerned Member States, or CMSs) for their approval. If the CMSs raise no objections, based on a potential serious risk to public health, to the assessment, SmPC, labeling, or packaging proposed by the RMS, the product is subsequently granted a national MA in all the relevant Member States (i.e. in the RMS and the CMSs).
Under the above described procedures, before granting the MA, the EMA or the competent authorities of the Member States of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
Marketing Authorization Application
Following positive completion of clinical trials, pharmaceutical companies can submit a MA application. The MA application shall include all information that is relevant to the evaluation of the medicinal products, whether favorable or unfavourable. The application dossier must include, among other things, the results of pharmaceutical (physico-chemical, biological, or microbiological) tests, preclinical (toxicological and pharmacological) tests, and clinical trials, including the therapeutic indications, contra-indications, and adverse reactions, and the recommended dosing regimen or posology.
In addition to demonstrating the safety and efficacy of the medicinal product, pharmaceutical companies are required to guarantee the consistent quality of the product. Therefore, the conditions for obtaining a MA include requirements that the manufacturer of the product complies with applicable legislation including Good Manufacturing Practice, or GMP, related implementing measures and applicable guidelines that involve, amongst others, ongoing inspections of manufacturing and storage facilities.
Early Access Mechanisms
Several schemes exist in the EU to support earlier access to new medicines falling within the scope of the Centralized Procedure, in particular (i) accelerated assessment; (ii) conditional MAs ; and (iii) MAs granted under exceptional circumstances.
For a medicine, which is of “major public health interest” (in particular, in terms of therapeutic innovation), accelerated assessment can be requested, taking up to 150 days instead of the usual period of up to 210 days. There is no single definition of what constitutes major public health interest. This should be justified by the applicant on acase-by-case basis. The justification should present the arguments to support the claim that the medicinal product introduces new methods of therapy or improves on existing methods, thereby addressing to a significant extent the greater unmet needs for maintaining and improving public health.
Conditional MAs may be granted on the basis of less complete data than usual in order to meet unmet medical needs of patients and in the interest of public health, subject to specific obligations with regard to further studies and intended to be replaced by a full unconditional MA once the missing data is provided. A conditional MA is valid for one year on a renewable basis.
Medicines for which the MA applicant can demonstrate that the normally required comprehensive efficacy and safety data cannot be provided (for example because the disease which the medicine treats is extremely rare) may be eligible for a MA under exceptional circumstances. These are medicines for which it is never intended that a full MA will be obtained. MAs under exceptional circumstances are reviewed annually to reassess the risk-benefit balance.
Supplementary Protection Certificates and data/market exclusivity
In Europe, the extension of effective patent term to compensate originator pharmaceutical companies for the period between the filing of an application for a patent for a new medicinal product and the first MA for such product, has been achieved by means of a Supplementary Protection Certificate (SPC) which can be applied for by the originator pharmaceutical company within 6 months from the granting of the first MA
and comes into effect on expiry of the basic patent. Such SPC attaches only to the active ingredient of the medicinal product for which the MA has been granted. The SPC for an active ingredient has a single last potential expiry date throughout the EEA, and cannot last for more than five years from the date on which it takes effect (i.e., patent expiry. Furthermore, the overall duration of protection afforded by a patent and a SPC cannot exceed 15 years from the first MA. The duration of a medicinal product SPC can be extended by a singlesix-month period, or pediatric extension, when all studies in accordance with a pediatric investigation plan, or PIP, have been carried out.
Innovative medicines benefit from specific data and marketing exclusivity regimes. These regimes are intended to provide general regulatory protection to further stimulate innovation. The current rules provide for (i) an8-year data protection (from the MA of an innovative medicine) against the filing of an abridged application for afollow-on product, referring to the data supporting the MA of the innovative medicine (data exclusivity); and (ii) a10-year protection against the marketing of afollow-on product (marketing exclusivity), with a possible extension by 1 year if, during the first 8 years, a new therapeutic indication (which is considered to bring a significant clinical benefit in comparison with existing therapies) is approved. This protection is often referred to as the “eight, plus two, plus one” rule. Additional reward mechanisms exist, most notably a10-year orphan medicines’ marketing exclusivity, and a1-year data exclusivity for developing a new indication for an old substance and for switch data supporting a change in prescription status.
The current rules also provide for a system of obligations and rewards and incentives intended to facilitate the development and accessibility of pediatric medicinal products, and to ensure that such products are subject to high quality ethical research. Pursuant to such rules, pharmaceutical companies are often required to submit a Pediatric Investigation Plan, or PIP, at a relatively early stage of product development, which defines the pediatric studies to be completed before a MA application can be submitted. Upon completion of the studies in the agreed PIP, the company may be entitled to a “reward”,i.e., the afore-mentioned6-month pediatric extension of the SPC fornon-orphan medicinal products; or atwo-year extension of the10-year marketing exclusivity period for orphan medicines.
Post-marketing and pharmacovigilance requirements
When granting a MA, competent authorities (i.e., the EMA or the relevant NCAs) may impose an obligation to conduct additional clinical testing, sometimes referred to as Phase IV clinical trials, or other post-approval commitments, to monitor the product after commercialization. Additionally, the MA may be subjected to limitations on the indicated uses for the product.
Also, after a MA has been obtained, the marketed product and its manufacturer and MA holder will continue to be subject to a number of regulatory obligations, as well as to monitoring/inspections by the competent authorities.
Under applicable pharmacovigilance rules, pharmaceutical companies must, in relation to all their authorized products, irrespective of the regulatory route of approval, collect, evaluate and collate information concerning all suspected adverse reactions and, when relevant, report it to the competent authorities. This information includes both suspected adverse reactions signaled by healthcare professionals, either spontaneously or through post-authorization studies, regardless of whether or not the medicinal product was used in accordance with the authorized SmPC and/or any other marketing conditions, and suspected adverse reactions identified in worldwide-published scientific literature. To that end, a MA holder must have (permanently and continuously) at its disposal an appropriately qualified person responsible for pharmacovigilance and establish an adequate pharmacovigilance system. All relevant suspected adverse reactions, including suspected serious adverse reactions, which must also be reported on an expedited basis, should be submitted to the competent authorities in the form of Periodic Safety Update Reports, or PSURs. PSURs are intended to provide an update for the competent authorities on the worldwide safety experience of a medicinal product at defined time points after authorization. PSURs must therefore comprise a succinct summary of information together with a critical evaluation of the risk/benefit balance of the medicinal product, taking into account any new or changing information. The evaluation should ascertain whether any further investigations need to be carried out, and whether the SmPC or other product information needs to be modified.
To ensure that pharmaceutical companies comply with pharmacovigilance regulatory obligations, and to facilitate compliance, competent authorities will conduct pharmacovigilance inspections. These inspections
are either routine (i.e. aimed at determining whether the appropriate personnel, systems, and resources are in place) or targeted to companies suspected of beingnon-compliant. Reports of the outcome of such inspections will be used to help improve compliance and may also be used as a basis for enforcement action.
Other regulatory matters
Advertising of medicines is subject to tighter controls than general consumer goods and specific requirements are set forth in Directive 2001/83/EC, which apply in addition to the general rules. In general, advertising of unapproved medicinal products or of unapproved uses of otherwise authorized medicinal products (e.g.,off-label uses) is prohibited, and advertising for prescription medicinal products must be directed only towards health care professionals (i.e., advertising of these products to the general public is prohibited). Member States have implemented the advertising rules differently and the requirements vary significantly depending on the specific country. Advertising of medicinal products in an online setting, including social media, can be particularly challenging given the strict rules in place.
Pricing &Reimbursement
United States
Sales of our products will depend, in part, on the extent to which our products, once approved, will be covered and reimbursed by third-party payors, such as government health programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly reducing reimbursements for medical products and services. The process for determining whether a third-party payor will provide coverage for a drug product, including a biologic, typically is separate from the process for setting the price of a drug product or for establishing the reimbursement rate that a payor will pay for the drug product once coverage is approved. Third-party payors may limit coverage to specific drug products on an approved list, also known as a formulary, which might not include all of the approved drugs for a particular indication.
In order to secure coverage and reimbursement for any drug product candidate that might be approved for sale, we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the drug product candidate, in addition to the costs required to obtain FDA or other comparable regulatory approvals. Whether or not we conduct such studies, our drug product candidates may not be considered medically necessary or cost-effective. A third-party payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage, and adequate reimbursement, for the product. Third party reimbursement may not be sufficient to enable us to maintain price levels high enough to realize an appropriate return on our investment in product development.
The containment of healthcare costs has become a priority of federal and state governments, and the prices of drugs, including biologics, have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Decreases in third-party reimbursement for our drug product candidate or a decision by a third-party payor to not cover our drug product candidate could reduce physician usage of the drug product candidate and have a material adverse effect on our sales, results of operations and financial condition.
For example, the ACA, enacted in March 2010, has had, a significant impact on the health care industry. The ACA has expanded coverage for the uninsured while at the same time containing overall healthcare costs. With regard to pharmaceutical products, among other things, the ACA expanded and increased industry rebates for drugs covered under Medicaid programs and made changes to the coverage requirements under the Medicare Part D program.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. On August 2, 2011, the Budget Control Act of 2011 among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was
unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, started in April 2013 and will stay in effect through 2025 unless additional Congressional action is taken. On January 2, 2013, then President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Some of the provisions of ACA have yet to be fully implemented, while certain provisions have been subject to judicial and Congressional challenges. In January 2017, Congress voted to adopt a budget resolution for fiscal year 2017, that while not a law, is widely viewed as the first step toward the passage of legislation that would repeal certain aspects of ACA. Further, on January 20, 2017, President Trump signed an Executive Order directing federal agencies with authorities and responsibilities under ACA to waive, defer, grant exemptions from, or delay the implementation of any provision of ACA that would impose a fiscal burden on states or a cost, fee, tax, penalty or regulatory burden on individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices. Congress also could consider subsequent legislation to replace elements of ACA that are repealed. Thus, the full impact of ACA, any law replacing elements of it, or the political uncertainty related to any repeal or replacement legislation on our business remains unclear.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country.
European Union
In Europe, pricing and reimbursement for pharmaceutical products are not harmonized and fall within the exclusive competence of the national authorities, provided that basic transparency requirements (such as maximum timelines) defined at the European level are met as set forth in the EU Transparency Directive 89/105/EEC. A Member State may approve a specific price for a medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. For example, in France, effective access to the market assumes that our future products will be reimbursed by social security. The price of medications is negotiated with the Economic Committee for Health Products, or CEPS.
As a consequence, reimbursement mechanisms by public national healthcare systems, or private health insurers also vary from country to country. In public healthcare systems, reimbursement is determined by guidelines established by the legislator or a competent national authority. In general, inclusion of a product in reimbursement schemes is dependent upon proof of the product efficacy, medical need, and economic benefits of the product to patients and the healthcare system in general. Acceptance for reimbursement comes with cost, use and often volume restrictions, which again vary from country to country.
The pricing and reimbursement level for medicinal products will depend on the strength of the clinical data set and, as for most novel therapies, restrictions may apply. In most countries, national competent authorities ensure that the prices of registered medicinal products sold in their territory are not excessive. In making this judgment, they usually compare the proposed national price either to prices of existing treatments and/or to prices of the product at issue in other countries –so-called “international reference pricing” – also taking into account the type of treatment (preventive, curative or symptomatic), the degree of innovation, the therapeutic breakthrough, volume of sales, sales forecast, size of the target population and/or the improvement (including cost savings) over comparable treatments. Given the growing burden of medical treatments on national healthcare budgets, reimbursement and insurance coverage is an important determinant of the accessibility of medicines.
The various public and private plans, formulary restrictions, reimbursement policies, patient advocacy groups, and cost-sharing requirements may play a role in determining effective access to the market of our product candidates. The national competent authorities may also use a range of policies and other initiatives intended to influence pharmaceutical consumption. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our drug product candidates. Historically, products launched in the European Union do not follow price structures of the United States and generally tend to be priced at a significantly lower level.
Other Healthcare Laws and Compliance Requirements
Our business operations in the United States and our arrangements with clinical investigators, healthcare providers, consultants, third-party payors and patients may expose us to broadly applicable federal and state fraud and abuse and other healthcare laws. These laws may impact, among other things, our research, proposed sales, marketing and education programs of our drug product candidates that obtain marketing approval. The laws that may affect our ability to operate include, among others:
the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering or paying remuneration (including any kickback, bribe or rebate), directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, lease, order, or recommendation of, an item, good, facility or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs;
federal civil and criminal false claims laws and civil monetary penalty laws, which impose penalties and provide for civil whistleblower or qui tam actions against individuals and entities for, among other things, knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payers that are false or fraudulent, or making a false statement or record material to payment of a false claim or avoiding, decreasing, or concealing an obligation to pay money to the federal government, including for example, providing inaccurate billing or coding information to customers or promoting a productoff-label;
the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created federal criminal statutes that prohibit, among other things, executing a scheme to defraud any healthcare benefit program, obtaining money or property of the health care benefit program through false representations, or knowingly and willingly falsifying, concealing or covering up a material fact, making false statements or using or making any false or fraudulent document in connection with the delivery of or payment for healthcare benefits or services.
the federal Physician Payments Sunshine Act, enacted as part of the ACA, which requires applicable manufacturers of covered drugs, devices, biologics and medical supplies to track and annually report to CMS payments and other transfers of value provided to physicians and teaching hospitals and certain ownership and investment interest held by physicians or their immediate family members in applicable manufacturers and group purchasing organizations;
HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH, and its implementing regulations, which imposes certain requirements on covered entities and their business associates relating to the privacy, security and transmission of individually identifiable health information; and
state law equivalents of each of the above federal laws, such as state anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers, state marketing and/or transparency laws applicable to manufacturers that may be broader in scope than the federal requirements, state laws that require biopharmaceutical companies to comply with the biopharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect as HIPAA, thus complicating compliance efforts.
The ACA broadened the reach of the federal fraud and abuse laws by, among other things, amending the intent requirement of the federal Anti-Kickback Statute and certain applicable federal criminal healthcare fraud statutes. Pursuant to the statutory amendment, a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. In addition, the ACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act or the civil monetary penalties statute.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving
applicable fraud and abuse or other healthcare laws. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant administrative, civil, and/or criminal penalties, damages, fines, disgorgement, individual imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If the physicians or other healthcare providers or entities with whom we expect to do business are found to be not in compliance with applicable laws, they also may be subject to administrative, civil, and/or criminal sanctions, including exclusions from government funded healthcare programs.
Legal Proceedings
From time to time we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our business. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
On July 8, 2016, following the unsuccessful outcome of the conciliation procedure organized under Swiss laws, a Swiss company named AtonRâ Partners SA formalized its claim against us before the Tribunal of First Instance of Geneva (Switzerland), or the Tribunal. AtonRâ Partners SA claims the payment of respectively 95.250 EUR and 300.300 USD as alleged broker intermediary commissions in the context of our fund raising of March 3 2015 and our Initial Public Offering (IPO) on the NASDAQ on June 18, 2015. We fully contest the merits of the claim and the jurisdiction of the Tribunal as Atonrâ was not a party of the bank syndicate of these two placements. The procedure is pending and the Tribunal has not fixed the judgment date. The decision is subject to appeal in accordance with Swiss laws.
C. Organizational Structure.
The Company and its subsidiaries, or the Group, is made of the following entities as of the end of December 2016. The following diagram illustrates our corporate structure.
| | | | | | | | | | | | | | | | |
Name | | Country of
Incorporation
and Place of
Business | | Nature of
Business | | Proportion of
ordinary
shares directly
held by parent
(%) | | | Proportion of ordinary
shares held by the
group (%) | | | Proportion of ordinary
shares held by non-
controlling interests (%) | |
Celyad SA | | BE | | Biopharma | | | Parent company | | | | | | | | | |
Celyad Inc | | US | | Biopharma | | | 100 | % | | | 100 | % | | | 0 | % |
Biological Manufacturing Services SA | | BE | | Biopharma | | | 100 | % | | | 100 | % | | | 0 | % |
Oncyte LLC | | US | | Biopharma | | | 100 | % | | | 100 | % | | | 0 | % |
CorQuest | | US | | Medical Device | | | 100 | % | | | 100 | % | | | 0 | % |
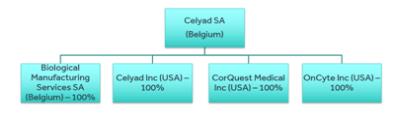
D. Property, Plants and Equipment.
We rent a 2,284 square meter office space from the Axis Parc developer located at the Axis Parc in Mont-Saint-Guibert pursuant to a lease agreement dated June 24, 2015 as amended from time to time, which expires on June 30 2025. We also rent a 1,120 square meter office and laboratory space from the Axis Parc developer pursuant to a lease agreement dated October 31, 2007, as amended from time to time, which expires on September 30, 2017. In January 2016, we entered into a6-year lease agreement for our US corporate offices located in Boston, Massachussets.
We plan to identify additional facilities in the Flemish region of Belgium to construct our contemplated future European manufacturing plant. We have committed to maintain our headquarters and registered office in the Walloon region of Belgium and all of our existing activities will continue to be performed in the Walloon region
| ITEM 4A. | UNRESOLVED STAFF COMMENTS. |
Not applicable.
| ITEM 5. | OPERATING AND FINANCIAL REVIEW AND PROSPECTS |
We are a leaderclinical-stage biopharmaceutical company focused on the development of specialized cell-based therapies. We utilize our expertise in engineered cell therapy treatments with clinical programs currently targeting indications in immune-oncology.
engineering to target cancer. Our lead drug product candidate,