5
• | our ability to contract with third-party suppliers and manufacturers and their ability to perform adequately; |
our ability to build our commercialization organization;
regulatory developments in the United States and foreign countries; and
You should refer to the section of this Annual Report on Form20-F titled “Item 3.D—Risk Factors” for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Annual Report on Form20-F will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame or at all. We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
You should read this Annual Report on Form20-F and the documents that we reference in this Annual Report on Form20-F and have filed as exhibits to this Annual Report on Form20-F completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this Annual Report on Form 20-F, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete. Our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and investors are cautioned not to unduly rely upon these statements.
This Annual Report on Form20-F contains market data and industry forecasts that were obtained from industry publications. These data involve a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. We have not independently verified any third-party information. While we believe the market position, market opportunity and market size information included in this Annual Report on Form20-F is generally reliable, such information is inherently imprecise.
6
Item 1. Identity of Directors, Senior Management and Advisers.
Not applicable.
Item 2. Offer Statistics and Expected Timetable.
Not applicable.
We have derived the selected consolidated statements of comprehensive loss for the years ended December 31, 2016, 20152019, 2018, and 20142017 presented below and the selected consolidated balance sheet data as of December 31, 20162019 and 20152018 presented below from our audited consolidated financial statements included elsewhere in this Annual Report on Form20-F. The selected consolidated statement of comprehensive loss for the years ended December 31, 2016 and 2015 and the selected consolidated balance sheet data as of December 31, 2014 has2017, 2016 and 2015 have been derived from our audited consolidated financial statements not included in this Annual Report on Form20-F. This data should be read together with, and is qualified in its entirety by reference to, “Item 5. Operating and Financial Review and Prospects” as well as our financial statements and notes thereto appearing elsewhere in this Annual Report on Form20-F. Our historical results are not necessarily indicative of the results to be expected in the future.
We present the audited consolidated financial statements in U.S. dollars and in accordance with IFRS. On January 1, 2019, IFRS 16 Leases became effective and as a result of this new adoption, we recognized right-of-use assets and lease liabilities of $2.7 million, as further detailed in note 2.3 of our audited consolidated financial statements included elsewhere in this Annual Report on Form 20-F. The adoption of IFRS 16 Leases did not have a material impact on our net loss after tax or on our loss per share for the year ended December 31, 2019.
| Year ended December 31, |
| Year ended December 31, |
| |||||||||||||||||||||||||||||
| 2016 | 2015 | 2014 |
| 2019 |
|
| 2018 |
|
| 2017 |
|
| 2016 |
|
| 2015 |
| |||||||||||||||
| (in thousands, except share and per share data) |
| (in thousands, except share and per share data) |
| |||||||||||||||||||||||||||||
Consolidated Statements of Comprehensive Loss: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Other operating income | $ | 22 | $ | 17 | $ | 45 | ||||||||||||||||||||||||||
Operating income other than revenue |
| $ | 16 |
|
| $ | 15 |
|
| $ | 16 |
|
| $ | 22 |
|
| $ | 17 |
| ||||||||||||
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||
Research and development expenses | (23,711 | ) | (16,892 | ) | (11,402 | ) |
|
| (88,053 | ) |
|
| (62,872 | ) |
|
| (54,912 | ) |
|
| (23,711 | ) |
|
| (16,892 | ) | ||||||
General and administrative expenses | (6,452 | ) | (2,954 | ) | (1,560 | ) |
|
| (19,058 | ) |
|
| (14,297 | ) |
|
| (12,568 | ) |
|
| (6,452 | ) |
|
| (2,954 | ) | ||||||
|
|
| ||||||||||||||||||||||||||||||
Total operating expenses | (30,163 | ) | (19,846 | ) | (12,962 | ) |
|
| (107,111 | ) |
|
| (77,169 | ) |
|
| (67,480 | ) |
|
| (30,163 | ) |
|
| (19,846 | ) | ||||||
|
|
| ||||||||||||||||||||||||||||||
Operating loss | (30,141 | ) | (19,829 | ) | (12,917 | ) |
|
| (107,095 | ) |
|
| (77,154 | ) |
|
| (67,464 | ) |
|
| (30,141 | ) |
|
| (19,829 | ) | ||||||
Finance income | 36 | — | 109 |
|
| 854 |
|
|
| 393 |
|
|
| 590 |
|
|
| 36 |
|
|
| — |
| |||||||||
Finance expense | (97 | ) | (38 | ) | — |
|
| (2,482 | ) |
|
| — |
|
|
| (1 | ) |
|
| (97 | ) |
|
| (38 | ) | |||||||
|
|
| ||||||||||||||||||||||||||||||
Net loss before tax | (30,202 | ) | (19,867 | ) | (12,808 | ) |
|
| (108,723 | ) |
|
| (76,761 | ) |
|
| (66,875 | ) |
|
| (30,202 | ) |
|
| (19,867 | ) | ||||||
Income tax expense | — | — | — | |||||||||||||||||||||||||||||
|
|
| ||||||||||||||||||||||||||||||
Income tax (expense) / benefit |
|
| (67 | ) |
|
| 45 |
|
|
| (51 | ) |
|
| — |
|
|
| — |
| ||||||||||||
Net loss | (30,202 | ) | $ | (19,867 | ) | $ | (12,808 | ) |
| $ | (108,790 | ) |
| $ | (76,716 | ) |
| $ | (66,926 | ) |
| $ | (30,202 | ) |
| $ | (19,867 | ) | ||||
|
|
| ||||||||||||||||||||||||||||||
Net loss per share, basic and diluted(1) | $ | (1.40 | ) | $ | (1.87 | ) | $ | (1.41 | ) |
| $ | (2.49 | ) |
| $ | (1.91 | ) |
| $ | (2.25 | ) |
| $ | (1.40 | ) |
| $ | (1.87 | ) | |||
|
|
| ||||||||||||||||||||||||||||||
(1) | See Note |
7
Consolidated Balance Sheet Data: Cash and cash equivalents Working capital(1) Total assets Total liabilities Share capital Accumulated losses Total shareholders’ equity As of December 31, 2016 2015 2014 $ 25,508 $ 54,275 $ 4,008 22,054 50,266 2,566 45,525 71,590 9,279 9,484 6,915 3,292 1,740 1,694 764 (39,599 ) (39,437 ) (18,214 ) 36,041 64,675 5,987
|
| As of December 31, |
| |||||||||||||||||
(in thousands) |
| 2019 |
|
| 2018 |
|
| 2017 |
|
| 2016 |
|
| 2015 |
| |||||
Consolidated Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
| $ | 69,370 |
|
| $ | 138,640 |
|
| $ | 110,841 |
|
| $ | 25,508 |
|
| $ | 54,275 |
|
Working capital(1) |
|
| 55,305 |
|
|
| 128,311 |
|
|
| 103,684 |
|
|
| 22,054 |
|
|
| 50,266 |
|
Total assets |
|
| 103,943 |
|
|
| 167,440 |
|
|
| 135,235 |
|
|
| 45,525 |
|
|
| 71,590 |
|
Total liabilities |
|
| 54,988 |
|
|
| 20,524 |
|
|
| 12,584 |
|
|
| 9,484 |
|
|
| 6,915 |
|
Share capital |
|
| 3,499 |
|
|
| 3,420 |
|
|
| 2,864 |
|
|
| 1,740 |
|
|
| 1,694 |
|
Accumulated losses |
|
| (297,411 | ) |
|
| (183,927 | ) |
|
| (106,667 | ) |
|
| (39,599 | ) |
|
| (39,437 | ) |
Total shareholders’ equity |
|
| 48,955 |
|
|
| 146,916 |
|
|
| 122,651 |
|
|
| 36,041 |
|
|
| 64,675 |
|
(1) | We define working capital as current assets less current liabilities. See our audited consolidated financial statements included elsewhere in this Annual Report on Form20-F for further details regarding our current assets and current liabilities. |
B. Capitalization and Indebtedness
Not applicable.
C. Reasons for the Offer and Use of Proceeds
Not applicable.
Our business faces significant risks. You should carefully consider all of the information set forth in this Annual Report and in our other filings with the United States Securities and Exchange Commission, or the SEC, including the following risk factors which we face and which are faced by our industry. Our business, financial condition or results of operations could be materially adversely affected by any of these risks. This report also contains forward-looking statements that involve risks and uncertainties. Our results could materially differ from those anticipated in these forward-looking statements, as a result of certain factors including the risks described below and elsewhere in this Annual Report and our other SEC filings. See “Special Note Regarding Forward-Looking Statements” above.
Risks Related to Our Financial Position and Need for Additional Capital
We have incurred significant operating losses since inception and anticipate that we will continue to incur substantial operating losses for the foreseeable future and may never achieve or maintain profitability.
Since our inception, we have incurred significant operating losses. Our net loss was $30.2$108.8 million, $19.9$76.7 million and $12.8$66.9 million for the years ended December 31, 2016, 20152019, 2018 and 2014,2017, respectively. As of December 31, 2016,2019, we had accumulated losses of $70.2$328.0 million, out of which $30.6 million were offset with share premium. We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. We have devoted substantially all of our efforts toin-licensing and developing our product candidates, OBE2109,linzagolix, OBE022 and nolasiban, and OBE022, as well as capital raising, and building our management team. It could be several years, if ever, before we have a commercialized product. The net losses we incur may fluctuate significantly from quarter to quarter and year to year. We anticipate that our expenses will increase substantially if and as we:
conduct nonclinical studies required for the continued development and regulatory approval of our existing clinical programs, including an environmental assessment for linzagolix;
initiate preclinical studies and clinical trials for any additional indications for our current product candidates and any future product candidates that we may pursue;
8
prepare for the commercialization of certain product candidates;
hire additional clinical, regulatory, scientific, commercial and accounting personnel; and
To become and remain profitable, we must develop and eventually commercialize one or more product candidates with significant market potential. This will require us to be successful in a range of challenging activities, including completing clinical trials of OBE2109,linzagolix, OBE022 and nolasiban, and OBE022, developing commercial scale manufacturing processes, obtaining marketing approval, manufacturing, marketing and selling any current and future product candidates for which we may obtain marketing approval, and satisfying any post-marketing requirements. We are only in the preliminary stages of most of these activities and, in some cases, have not yet commenced certain of these activities. We may never succeed in any or all of these activities and, even if we do, we may never generate sufficient revenue to achieve profitability.
Because of the numerous risks and uncertainties associated with product development, we are unable to accurately predict the timing or amount of expenses or when, or if, we will obtain marketing approval to commercialize any of our product candidates. If we are required by the U.S. Food and Drug Administration, or FDA, or other regulatory authorities such as the European Medicines Agency, or EMA, to perform studies and trials in addition to those currently expected, or if there are any delays in the development, or in the completion of any planned or future preclinical studies or clinical trials of our current or future product candidates, our expenses could increase and profitability could be further delayed.
Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would decrease the value of our company and could impair our ability to raise capital, maintain our research and development efforts, expand our business or continue our operations. A decline in the value of our company also could cause you to lose all or part of your investment.
We have a limited operating history and have never generated any revenue from product sales, which may make it difficult to evaluate the success of our business to date and to assess our future viability.
We commenced operations in 2012, and our operations to date have been largely focused onin-licensing and developing our product candidates, including conducting preclinical studies and clinical trials, raising capital, and building our management team and infrastructure. We have not yet demonstrated an ability to successfully complete later-stage clinical trials,development of a product candidate, obtain regulatory approvals, manufacture products on a commercial scale, or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful commercialization. Additionally, the markets for our product candidates are competitive, complex and have characteristics that differ by geography. Consequently, any predictions you make about our future success or viability may not be as accurate as they could be if we had a longer operating history or a history of successfully developing and commercializing products.
We expect our financial condition and operating results to continue to fluctuate from quarter to quarter and year to year due to a variety of factors, many of which are beyond our control. We will need to eventually transition from a company with a research and development focus to a company capable of undertaking commercial activities. We may encounter unforeseen expenses, difficulties, complications and delays, and may not be successful in such a transition.
We will need substantial additional funding to pursue our business objectives. If we are unable to raise capital when needed or on terms favorable to us, we could be forced to curtail our planned operations and the pursuit of our growth strategy.
Identifying potential product candidates and conducting preclinical studies and clinical trials is a time-consuming, expensive and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain regulatory approval and achieve product sales. We expect our expenses to increaseremain significant in connection with our ongoing activities, particularly as we continue to develop our product candidates. Our expenses could increase beyond our current expectations if the FDA, EMA or other foreign regulatory agencies require us to perform clinical trials and other studies in addition to those that we currently anticipate. In addition, our product candidates, if approved, may not achieve commercial success. Our revenue, if any, will be derived from sales of products that we domay not expect to bebecome commercially available for a
9
number of years, if at all. Additionally, if we obtain marketing approval for our product candidates, we expect to incur significant expenses related to manufacturing, marketing, sales and distribution. We also expect to incur additional costs associated with operating as a public company.
As of December 31, 2016,2019, our cash and cash equivalents was $25.5 million, and we$69.4 million. We raised approximately $86.8$88.5 million in net proceeds from our initial public offering.offering in January 2017, $56.3 million in net proceeds from our private placement in October 2017 and $72.4 million in net proceeds from our underwritten public offering in June 2018. In addition, in 2018 and 2019, we sold treasury shares from our “at the market” (ATM) program, generating net proceeds of $22.9 million. In August 2019, we borrowed $25.0 million under our senior secured term loan credit facility. We expect our existing cash and cash equivalents will enable us to fund our operating expenses and capital expenditure requirements into the first quarter of 2019.2021. This estimate is based on assumptions that may prove to be wrong, and we could use our available capital resources sooner than we expect. Changes may occur beyond our control that would cause us to consume our available capital before that time, including changes in and progress of our development activities and changes in regulation. Our future capital requirements will depend on many factors, including:
We will require additional capital to complete our planned clinical development programs for our current product candidates to seek regulatory approval.approval and may determine to engage in equity or debt financings or enter into credit facilities for other reasons. If we receive regulatory approval for any of our product candidates, we expect to incur significant commercialization expenses related to product manufacturing, sales, marketing and distribution. Any additional capital raising efforts may divert our management from theirday-to-day activities, which may adversely affect our ability to develop and commercialize our current and future product candidates, if approved.
In addition, we cannot guarantee that futuremay not be able to timely secure debt or equity financing will be available on a timely basis, in sufficient amountsfavorable terms or on terms acceptable to us, if at all. Moreover,Any debt financing obtained by us in the termsfuture could involve restrictive covenants relating to our capital raising activities and other financial and operational matters, which may make it more difficult for us to obtain additional capital and to pursue business opportunities. If we raise additional funds through further issuances of any financing may adversely affect the holdingsequity, convertible debt securities or the rightsother securities convertible into equity, our existing shareholders could suffer significant dilution in their percentage ownership of our shareholderscompany, and the issuanceany new equity securities we issue could have rights, preferences and privileges senior to those of additional securities, whether equity or debt, by us, or the possibilityholders of such issuance, may cause the market price of our common shares to decline.shares. Further, as a Swiss corporation we have less flexibility to raise capital, particularly in a quick and efficient manner. As a result, once we are a listed company in the United States, we may not be able to access the capital markets as frequently as comparable U.S. companies. See the Risk Factor entitled “Our status as a Swiss corporation means that our shareholders enjoy certain rights that may limit our flexibility to raise capital, issue dividends and otherwise manage ongoing capital needs” for additional information related to our ability to timely raise capital. If we are unable to obtain funding on a timely basis on acceptable terms, we may be required to significantly curtail, delay or discontinue one or more of our research or development programs or the commercialization of any product candidates, if approved, or be unable to expand our operations or otherwise capitalize on our business opportunities, as desired.
The incurrence of debt may impact our financial position and subject us to additional financial and operating restrictions.
10
On August 7, 2019, we entered into a $75.0 million senior secured term loan credit facility, or the 2019 Facility, with Oxford Finance LLC, or Oxford, which is subject to funding in three tranches, of which $50.0 million is available to be drawn. Upon entry into the 2019 Facility, we borrowed $25.0 million. The second tranche of $25.0 million is not eligible to be drawn, due to adverse nolasiban clinical trial results. The third tranche of $25.0 million will become available upon positive results in the Phase 3 PRIMROSE 1 clinical trial of linzagolix, expected in the second quarter of 2020. Our overall leverage and certain covenants and obligations contained in the related documentation could adversely affect our financial health and business and future operations by, among other things:
making it more difficult to satisfy our obligations, including under the terms of the 2019 Facility;
limiting our ability to refinance our debt on terms acceptable to us or at all;
limiting our flexibility to plan for and adjust to changing business and market conditions and increasing our vulnerability to general adverse economic and industry conditions;
limiting our ability to use our available cash flow to fund future acquisitions and to make dividend payments; and
limiting our ability to obtain additional financing for working capital, to fund growth or for general corporate purposes, even when necessary to maintain adequate liquidity.
Furthermore, substantially all of our assets, including our intellectual property, secure the 2019 Facility. If an event of default under the 2019 Facility occurs and is continuing, Oxford may request the acceleration of the related debt and foreclose on the underlying security interests.
The LIBOR calculation method may change and LIBOR is expected to be phased out after 2021.
Interest on the outstanding principal balance of the loans under the 2019 Facility is calculated based on one-month LIBOR, plus an applicable margin. On July 27, 2017, the U.K. Financial Conduct Authority, or the FCA, announced that it will no longer require banks to submit rates for the calculation of LIBOR after 2021. In the meantime, actions by the FCA, other regulators or law enforcement agencies may result in changes to the method by which LIBOR is calculated. At this time, it is not possible to predict the effect of any such changes or any other reforms to LIBOR that may be enacted in the United Kingdom or elsewhere.
Raising additional capital may cause dilution to our shareholders, restrict our operations or require us to relinquish rights to our intellectual property or future revenue streams.
Until such time as we can generate substantial product revenue, if ever, we expect to finance our operations through a combination of equity offerings, royalty financing, debt financings, and license and development agreements in connection with any future collaborations. WeOther than the 2019 Facility, we do not have any committed external source of funds. In the event we seek additional funds, we may raise additional capital through the sale of equity or convertible debt securities. In such an event, our shareholders may experience substantial dilution, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of holders of our common shares. Royalty financing, if available, may only provide future payments contingent upon development, regulatory or commercial milestones and royalty payments as a percentage of our future sales. Debt financing, if available, could result in increased fixed payment obligations and may involve agreements that include restrictive covenants, such as limitations on our ability to incur additional debt, make capital expenditures, acquire, sell or license intellectual property rights or declare dividends, and other operating restrictions that could hurt our ability to conduct our business.
Further, if we raise additional capital through collaborations, strategic alliances, or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our intellectual property future revenue streams, research programs or product candidates, or grant licenses on terms that may not be favorable to us.
Fluctuations in exchange rates may adversely affect our results of operations.
Our reporting and functional currency is in U.S. dollars, but the results of operations and the financial position of our operations in Switzerland are reported in Swiss francs and then translated into U.S. dollars for reporting purposes. Our financial results are, thus, impacted primarily by currency fluctuations between U.S. dollars and Swiss francs. Moreover, adollars. A change in the concentration of our business activities could result in an increased effect of exchange rates on our financial position and results of operations. Although we do currently hedge against certain currency risks, see “Item 11— Quantitative and Qualitative Disclosures about Market Risks” for more information regarding our exposure to currency fluctuations. There is no assurance that we will, in the future, be successful in fully or even adequately hedging our currency risk.
11
Legal, political and economic uncertainty surrounding the planned exit of the United Kingdom from the European Union may be a source of instability in international markets, create currency fluctuations and pose additional risks to our business operations and financial condition.
The United Kingdom’s referendum to leave the European Union, or “Brexit,” has caused and may continue to cause disruptions to capital and currency markets worldwide. The full impact of the Brexit decision, however, remains uncertain. A process of negotiation will determine the future terms of the United Kingdom’s relationship with the European Union. During this period of negotiation and afterwards, our results of operations and access to capital may be negatively affected by interest rate, exchange rate and other market and economic volatility, as well as political uncertainty. Brexit may have a limited detrimental effect on the timing of our research and development activities, which may, in turn, adversely affect our development and commercialization of certain product candidates.
Risks Related to the Development of Our Product Candidates
We depend entirely on the success of a limited number of product candidates, which are in clinical development and none of which have completed a pivotal trial.development. If we do not obtain regulatory approval for and successfully commercialize one or more of our product candidates or we experience significant delays in doing so, we may never become profitable.
We do not have any products that have received regulatory approval and may never be able to develop marketable product candidates. We expect that a substantial portion of our efforts and expenses over the next few years will be devoted primarily to OBE2109, nolasibanlinzagolix and OBE022, and as a result, our business currently depends heavily on the successful development, regulatory approval and commercialization of these product candidates. We cannot be certain that our product candidates will receive regulatory approval or will be successfully commercialized even if they receive regulatory approval. The research, testing, manufacturing, safety, efficacy, labeling, approval, sale, marketing and distribution of our product candidates are, and will remain, subject to comprehensive regulation by the FDA, EMA and comparable foreign regulatory agencies. Failure to obtain regulatory approval for our product candidates in the United States, the European Union or other jurisdictions will prevent us from commercializing and marketing our product candidates. The success of our product candidates will depend on several additional factors, including:
need for additional clinical trials necessitated by future interactions with regulatory authorities;
receiving marketing approvals from applicable regulatory authorities;
Many of these factors are beyond our control, including the time needed to adequately complete clinical testing, the regulatory submission process, potential threats to our intellectual property rights and changes in the competitive landscape. It is possible that none of our product candidates will ever obtain regulatory approval, even if we expend substantial time and resources seeking such approval. If we do not achieve one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully complete clinical trials, obtain regulatory approval or, if approved, commercialize our product candidates, which would harm our business, financial condition and results of operations.
12
Clinical trials are very expensive, time-consuming and difficult to design and implement and involve uncertain outcomes. Furthermore, results of earlier preclinical studies and clinical trials may not be predictive of results of future preclinical studies or clinical trials.
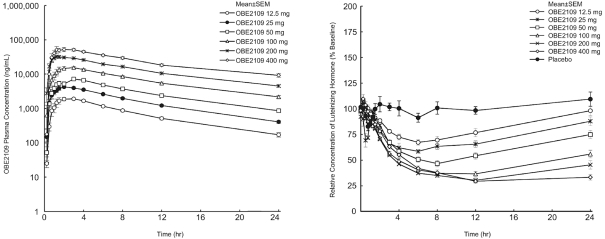
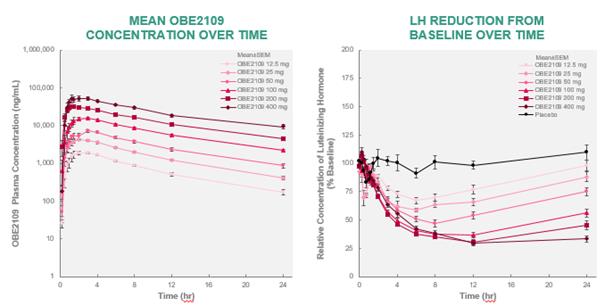
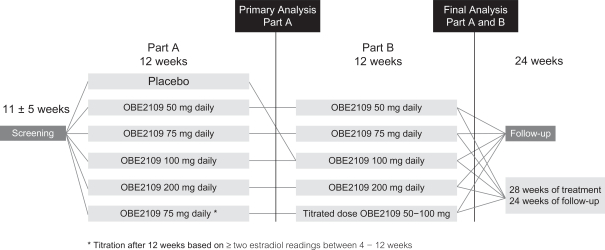
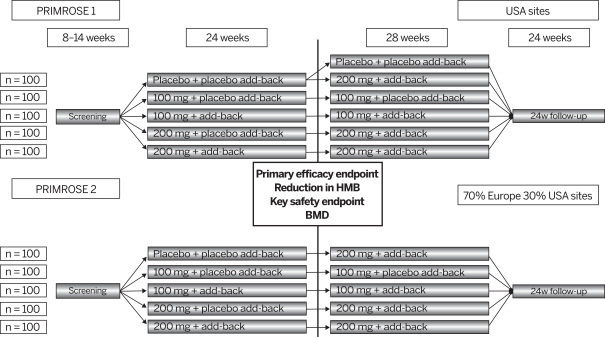
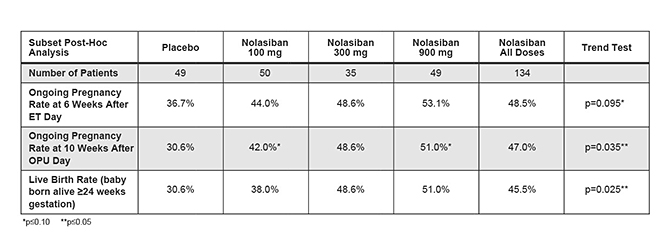
The risk of failure for our product candidates is high. It is impossible to predict when or if any of our product candidates will prove effective or safe in humans or will receive regulatory approval. To obtain the requisite regulatory approvals to market and sell any of our product candidates, we must demonstrate through extensive preclinical studies and clinical trials that our product candidates are safe and effective in humans. Clinical testing is expensive and can take many years to complete, and the outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. For example, in our Phase 2 clinical trial of nolasiban in women undergoing in vitro fertilization, or IVF, nolasiban did not achieve the primary endpoint of a statistically significant increase in pregnancy rate after six weeks and, as a result, we are required to conduct additional clinical trials and analyses which may ultimately delay the development of nolasiban. Additionally, we are planning to conduct a pharmacokinetic, or PK, and pharmacodynamic, or PD, study to confirm theadd-back dosage to be utilized in our Phase 3 PRIMROSE clinical trials for OBE2109’s uterine fibroid indication. If the results of the PK and PD study do not support our dosing assumptions, we may need to adapt ouradd-back drug dosing in the Phase 3 PRIMROSE clinical trials.
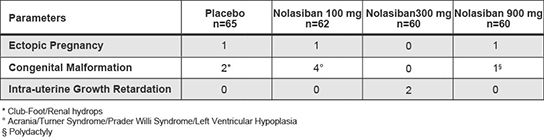
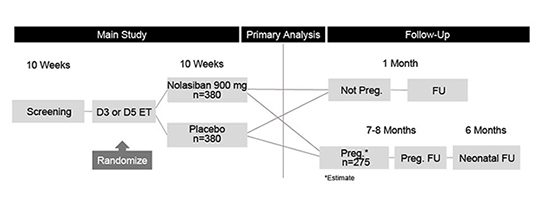
In addition, the results of preclinical studies and earlier clinical trials may not be predictive of the results of later-stage clinical trials. The results generated to date in preclinical studies or clinical trials for our product candidates do not ensure that later preclinical studies or clinical trials will demonstrate similar results. Further, we have limited clinical data for each of our product candidates and have not completed Phase 3 clinical trials for any of our product candidates. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through preclinical and earlier stage clinical trials. For example, we announced on November 7, 2019 that the IMPLANT 4 Phase 3 clinical trial of nolasiban in women undergoing embryo transfer following in-vitro fertilization, or IVF, did not meet the primary endpoint of an increase in ongoing pregnancy rate at 10 weeks. Based on these results, we discontinued our previously ongoing nolasiban IVF program. A number of companies in the biopharmaceutical industry have suffered significant setbacks in later-stage clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier trials, and we cannot be certain that we will not face similar setbacks.setbacks for our other product candidates. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval of their products.
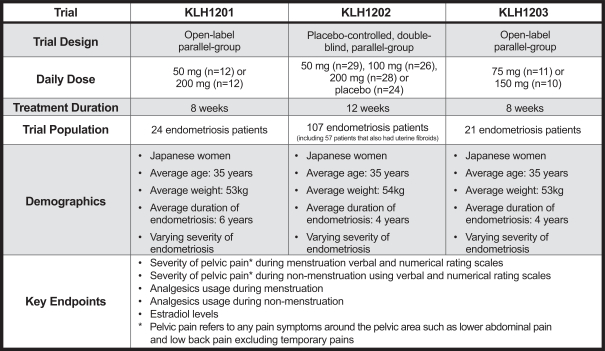
In some instances, there can be significant variability in safety or efficacy results between different clinical trials of the same product candidate due to numerous factors, including changes in clinical trial procedures set forth in protocols, differences in the size and type of the patient populations, adherence to the dosing regimen and other clinical trial protocols, and the rate of dropout among clinical trial participants. Specifically, the three Phase 2 clinical trials of OBE2109 only enrolled women of Japanese descent, and therefore the effects of OBE2109 on women of European descent in our Phase 2b clinical trial of OBE2109 in women with pain associated with endometriosis or our two planned Phase 3 clinical trials of OBE2109, which will be conducted in Europe and in the United States, in women with heavy menstrual bleeding associated with uterine fibroids may not be consistent with the results of the Japanese Phase 2 clinical trials. Additionally, in the case of our late-stage clinical product candidates, results may differ in general on the basis of the larger number of clinical trial sites and additional countries and languages involved in Phase 3 clinical trials. Different countries have different standards of care and different levels of access to care for patients. These differences may, in part, drive the heterogeneity of the patient populations that enroll in our studies.studies and that may affect clinical trial results.
In addition, because wein-licensed OBE2109 linzagolix from Kissei Pharmaceutical Co., Ltd., or Kissei,and OBE022 and nolasiban and OBE022 from Ares Trading S.A., an affiliate of Merck Serono, or Merck Serono, we were not involved in and had no control over the preclinical and clinical development of these product candidates prior to entering into thesein-license agreements. In addition, we are relying on Kissei and Merck Serono to have conducted such research and development in accordance with the applicable protocol, legal, regulatory and scientific standards, having accurately reported the results of all clinical trials conducted prior to our acquisition of OBE2109,linzagolix, OBE022 and nolasiban, and OBE022, and having correctly collected and interpreted the data from these studies and trials. To the extent any of these has not occurred, expected development time and costs may be increased which could adversely affect the marketing approval for and any future revenue from these product candidates.
The European Phase 3 clinical trial for nolasiban based on observations from ourpost-hoc analysis of the failed Phase 2 clinical trial may not achieve statistically significant results or otherwise meet its targeted endpoint.
Following the failure to achieve the primary endpoint of a statistically significant increase in pregnancy rate after six weeks in our Phase 2 clinical trial for nolasiban, we completed apost-hoc analysis of the Phase 2 data. In thepost-hoc analysis, which excluded patients with progesterone levels in the top quartile of the patient pool, we identified a statistically significant result for the primary endpoint. Based on these results, we intend to initiate a European Phase 3 clinical trial. There may be greater risk to the ultimate success of this clinical trial given the fact that we are basing our European Phase 3 clinical trial design onpost-hoc analysis and this trial may not achieve in statistically significant results or otherwise meet its primary endpoint.
Clinical trials may be delayed, suspended or terminated for many reasons, which will increase our expenses and delay the time it takes to develop our product candidates.
We may experience delays in our ongoing or future preclinical studies or clinical trials, and we do not know whether future preclinical studies or clinical trials will begin on time, need to be redesigned, enroll an adequate number of patients on time or be completed on schedule, if at all. The completion of clinical trials for our clinical product candidates may be delayed, suspended or terminated as a result of many factors, including:
13
delays in patient enrollment and variability in the number and types of patients available for clinical trials;
We could also encounter delays if a clinical trial is suspended or terminated by us, by the IRBs or Ethics Committees of the institutions in which such trials are being conducted, by the Data Safety Monitoring Board for such trial, or by the FDA or other regulatory authorities. Such authorities may suspend or terminate a clinical trial due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial.
Further, conducting clinical trials in foreign countries, aswhich we plan to doare doing for our product candidates, presents additional risks that may delay completion of our clinical trials. These risks include the failure of enrolled patients in foreign countries to adhere to the clinical protocol as a result of differences in healthcare services or cultural customs, managing additional administrative burdens associated with foreign regulatory schemes, as well as political and economic risks relevant to such foreign countries. Similarly, conducting clinical trials in the United States, which we are doing for our product candidates, presents risks that may delay completion of our clinical trials. These risks include the failure of enrolled patients in the United States to adhere to the clinical protocol, managing additional administrative burdens associated with United States regulatory schemes, as well as economic risks relevant to the United States.
If we experience delays in the completion, or termination, of any clinical trial of our product candidates, the commercial prospects of our product candidates may be harmed, and our ability to generate product revenue from sales of any of these product candidates will be delayed or not realized at all.
14
We do not know whether any of our preclinical studies or clinical trials will begin as planned, will need to be restructured or will be completed on schedule, or at all. Any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenue from product sales. Any of these occurrences may significantly harm our business, financial condition and prospects. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates. Significant preclinical study or clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our product candidates.
The regulatory approval process of the FDA, EMA or any comparable foreign regulatory agency may be lengthy, time-consuming and unpredictable.
Our future success is dependent upon our ability to successfully develop, obtain regulatory approval for and then successfully commercialize one or more of our product candidates. The time required to obtain approval by the FDA and comparable foreign authorities is unpredictable but typically takes many years following the commencement of clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions. We have not obtained regulatory approval for any product candidate and it is possible that none of our existing product candidates or any product candidates we may seek to develop in the future will ever obtain regulatory approval. Neither we nor any future collaborator is permitted to market any of our product candidates in the United States or abroad until we receive regulatory approval of a New Drug Application, or NDA, from the FDA or approval from the EMA or other applicable foreign regulatory agency.
Prior to obtaining approval to commercialize a product candidate in any jurisdiction, we or our collaborators must demonstrate with substantial evidence from well-controlled clinical trials, and to the satisfaction of the FDA, EMA or any comparable foreign regulatory agency, that such product candidates are safe and effective for their intended uses. Results from preclinical studies and clinical trials can be interpreted in different ways. The FDA, EMA or any comparable foreign regulatory agency can delay, limit or deny approval of our product candidates or require us to conduct additional preclinical or clinical testing or abandon a program for many reasons, including:
15
Any of our current or future product candidates could take a significantly longer time to gain regulatory approval than expected or may never gain regulatory approval. This could delay or eliminate any potential product revenue by delaying or terminating the potential commercialization of our product candidates.
Of the large number of drugs in development, only a small percentage successfully complete the FDA or foreign regulatory approval processes and are commercialized. The lengthy approval process as well as the unpredictability of future clinical trial results may result in our failing to obtain regulatory approval to market our product candidates, which would significantly harm our business, financial condition, results of operations and prospects.
We have previously sought, and intend to seek in the future, formal advice and guidance from the FDA or other applicable foreign regulatory agencies prior to advancing our product candidates into further studies andor pivotal clinical trials. If
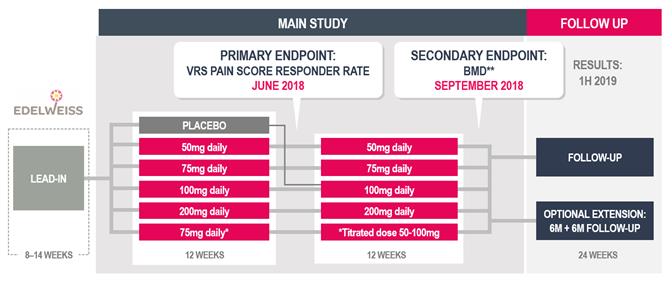
In December 2018, we sought guidance from the feedback we receive is different from what we currently anticipate, this could delay the development and regulatory approval process for these product candidates. For example, we do not believeFDA on our subjective11-point numeric pain rating scale we are using in our Phase 2b clinical trial of OBE2109 in women with pain associated with endometriosis will be sufficient, on its own, to support a suitable endpoint for future Phase 3 clinical trials. Asdevelopment program for linzagolix at an End of Phase 2 meeting. Based upon FDA feedback, we initiated a result, we believe we will be required to validate an additional assessment of pain for our Phase 3 program. We will have limited experience using this new pain assessment rating scaleclinical development program for linzagolix for the endometriosis indication which includes the EDELWEISS 2 and EDELWEISS 3 clinical trials in May 2019.
In July 2019, we sought guidance from the FDA on our uterine fibroid Phase 3 clinical development program for linzagolix at a clinical setting, which, as a result, increasesType C meeting regarding key secondary endpoints for the risk that we may have unforeseen, or variabilityongoing Phase 3 PRIMROSE 1 and PRIMROSE 2 trials to support labeling claims in results from this clinical trial.an NDA. Our statistical analysis plan was updated based upon FDA feedback.
We generally plan to seek regulatory approval to commercialize our product candidates in the United States, the European Union and other key global markets. With regard to each of our product candidates, we may experience delays or encounter issues in the development program in the relevant jurisdictions, including imposition of a clinical hold, failed studies, inconclusive or hard-to-interpret results, safety or efficacy issues, refusal to file the application, or refusal to approve it. To obtain regulatory approval in other countries, we must comply with numerous and varying regulatory requirements of such other countries regarding safety, efficacy, chemistry, manufacturing and controls, clinical trials, commercial sales, pricing and distribution of our product candidates. Even if we are successful in obtaining approval in one jurisdiction, we cannot ensure that we will obtain approval in any other jurisdiction. Failure to obtain approval in one jurisdiction may negatively impact our ability to obtain approval elsewhere. Failure to obtain marketing authorization for our product candidates will result in our being unablethe inability to market and sell such products. If we fail to obtain approval in any jurisdiction, the geographic market for our product candidates could be limited. Similarly, regulatory agencies may not approve the labeling claims that are necessary or desirable for the successful commercialization of our product candidates or may grant approvals for more limited patient populations than requested.
Even if we eventually complete clinical testing and receive approval of an NDA or foreign marketing application for our product candidates, the FDA or the applicable foreign regulatory agency may grant approval contingent on the performance of costly additional clinical trials, including Phase 4 clinical trials or the implementation of a Risk Evaluation and Mitigation Strategy, or REMS, which may be required to ensure safe use of the drug after approval. Any delay in obtaining, or inability to obtain, applicable regulatory approval would delay or prevent commercialization of that product candidate and would adversely impact our business and prospects.
Our clinical trials may fail to demonstrate the safety and efficacy of our product candidates, or serious adverse or unacceptable side effects may be identified during the development of our product candidates, which could prevent or delay regulatory approval and commercialization, increase our costs or necessitate the abandonment or limitation of the development of some of our product candidates.
Before obtaining regulatory approvals for the commercial sale of our product candidates, we must demonstrate through lengthy, complex and expensive preclinical testing and clinical trials that our product candidates are both safe and effective for use in each target indication, and failures can occur at any stage of testing. Clinical trials, such as our initial Phase 2 clinical trial for nolasiban and our IMPLANT 4 Phase 3 clinical trial of nolasiban, often fail to demonstrate definitive efficacy or safety of the product candidate studied for the target indication.
Moreover, undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or comparable foreign regulatory authorities. Results of our clinical trials could reveal a high and unacceptable severity and prevalence of side effects or unexpected characteristics. Accordingly, we may need to abandon their development or limit
16
development to certain uses orsub-populations in which such side effects are less prevalent, less severe or more acceptable from a risk-benefit perspective. Many compounds that initially showed promise in preclinical or early-stage testing have later been found to cause side effects that restricted their use and prevented further development of the compound for larger indications.
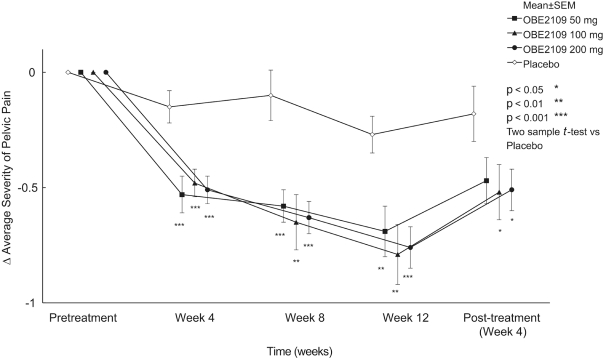
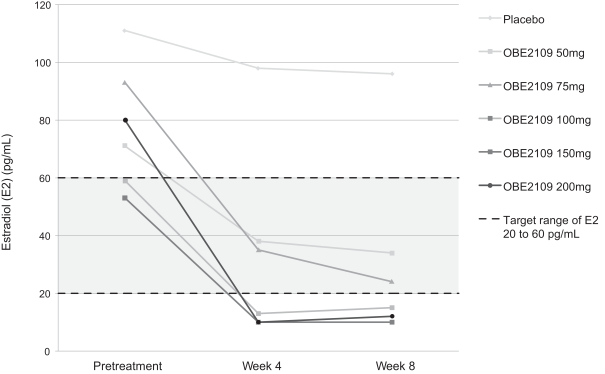
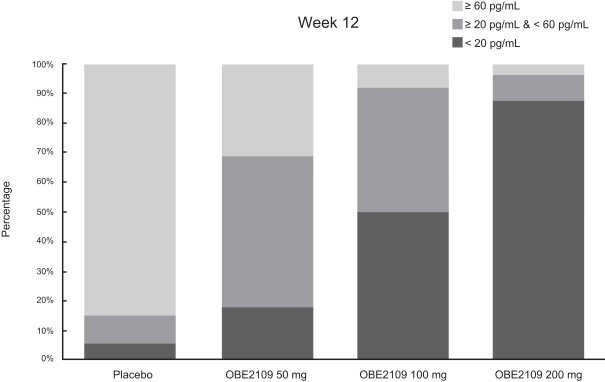
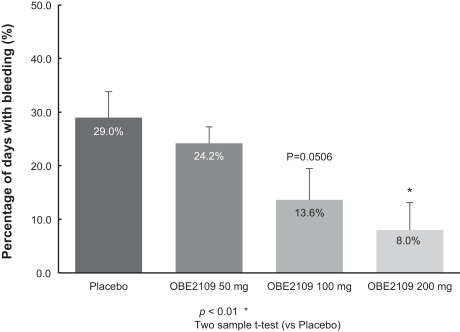
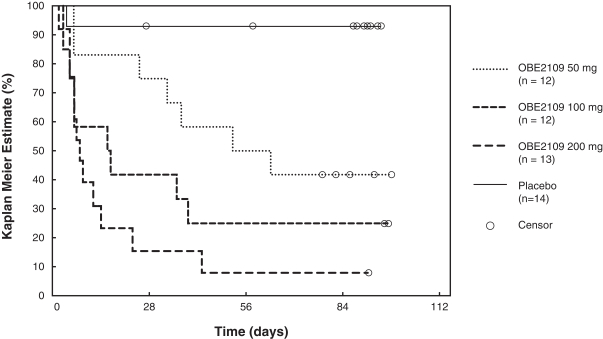
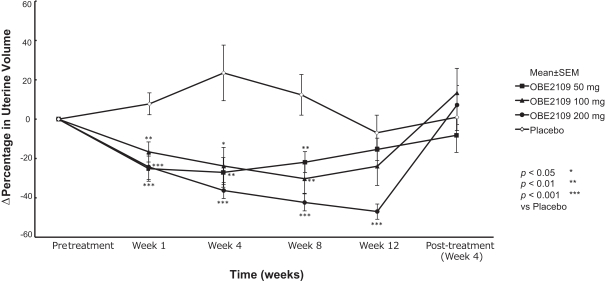
For example, in evaluation of OBE2109linzagolix to date, patients have experienced adverse events consistent with the suppression of estradiol, including hot flashes and abnormalirregular uterine bleeding. Three patients experienced hot flashes that were moderate in severity. Occurrence of serious treatment-related side effects could impede subject recruitment and clinical trial enrollment or the ability of enrolled patients to complete the trial, require us to halt the clinical trial, and prevent receipt of regulatory approval from the FDA, EMA or any comparable foreign regulatory agency. They could also adversely affect physician or patient acceptance of our product candidates or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly.
Additionally, if one or more of our product candidates receives regulatory approval, and we or others later identify undesirable side effects caused by these product candidates, a number of potentially significant negative consequences could result, including:
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and could significantly harm our business, financial condition, and results of operations.
We depend on enrollment of patients in our clinical trials for our product candidates. If we are unable to enroll patients in our clinical trials, our research and development efforts could be adversely affected.
Identifying and qualifying patients to participate in clinical trials of our product candidates is critical to our success. Successful and timely completion of clinical trials will require that we enroll a sufficient number of patients who remain in the study until its conclusion. If patients are unwilling to participate in our clinical trials because of a lack of familiarity with our approach to the treatment of reproductive health conditions, negative publicity from adverse events in the reproductive health field or for other reasons, including competitive clinical trials for similar patient populations and general social or cultural stigmas and sensitivities towards reproductive health, our timelines for recruiting patients, conducting clinical trials and obtaining regulatory approval of potential products may be delayed. These delays could result in increased costs, delays in advancing our product development, delays in testing the effectiveness of our technology or termination of our clinical trials altogether.
We cannot predict how successful we will be at enrolling patients in future clinical trials. Patient enrollment is affected by other factors including:
17
the efficiency with which our external vendor, or contract research organization (CRO), manages the logistics of patient recruitment, randomization, and follow-up within the clinical trial.
In addition, our clinical trials will compete with other clinical trials for product candidates that are in the same therapeutic areas as our product candidates, and this competition will reduce the number and types of patients available to us, because some patients who might have opted to enroll in our trials may instead opt to enroll in a trial being conducted by one of our competitors. We expect to conductthat some oftrial sites that participate in our clinical trials at the samemay also participate in clinical trial sites that some of our competitors use,trials being conducted to develop competitive compounds, which will reduce the number of patients who are available for our clinical trials inat such clinical trial site.
For example, enrollment in our Phase 3 clinical trial of linzagolix for the treatment of uterine fibroids and endometriosis may be impacted by other compounds being developed for those indications, as well as the fact that a competitive oral GnRH receptor antagonist received FDA approval for treating endometriosis associated pain in 2018 and is now commercially available. In particular,addition, for the clinical trials for nolasiban and OBE022, we will need to enroll women undergoing IVF andare enrolling pregnant women respectively. We believepresenting with pre-term labor, a patient population that these patient populations may be reluctant to enroll in clinical trials, given the sensitivity of reproductive health issues, particularly for women who are undergoing IVF or are experiencing spontaneous preterm labor. In addition, to date, pregnant women have not been extensively evaluated in clinical trials.around pregnancy. As a result, enrollment in our planned clinical trials for OBE022 is difficult to predict and may take longer or cost more than we anticipate.
Delays in the completion of any clinical trial of our product candidates will increase our costs, slow down our product candidate development and approval process, and delay or potentially jeopardize our ability to commence product sales and generate revenue. In addition, many of the factors that may lead to a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
We may not be successful in our efforts toin-license or acquire additional product candidates for other serious conditions compromising women’s reproductive health and pregnancy.
A significant element of our strategy is to further build and expand our pipeline of product candidates throughin-licensing or acquiring additional product candidates for other serious conditions compromising women’s reproductive health and pregnancy. Currently, we do not have the internal expertise, nor do we intend to develop the internal expertise, necessary to discover new chemical entities for therapeutic purposes. As a result, if we are not able to identify and acquire additional product candidates, we will not be able to expand our pipeline. Even if we are successful in continuing to build our pipeline throughin-licensing or acquisitions, the potential product candidates that wein-license or acquire may not be suitable for clinical development, including as a result of being shown to have harmful side effects or other characteristics that indicate that they are unlikely to be drugs that will receive marketing approval and achieve market acceptance.
18
We may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
We have limited financial and managerial resources. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. For example, a substantial portion of our efforts and expenses have been devoted to nolasiban, and we announced on November 7, 2019 that the IMPLANT 4 Phase 3 clinical trial of nolasiban in women undergoing embryo transfer following IVF did not meet the primary endpoint of an increase in ongoing pregnancy rate at 10 weeks. Based on these results, we discontinued our previously ongoing nolasiban IVF program. Our spending on current and future research and development programs and product candidates for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate.
We may become exposed to costly and damaging liability claims, either when testing our product candidates in the clinic or at the commercial stage, and our product liability insurance may not cover all damages from such claims.
We are exposed to potential product liability and professional indemnity risks that are inherent in the research, development, manufacturing, marketing, and use of pharmaceutical products. We currently have no products that have been approved for commercial sale. However, the current and future use of product candidates by us in clinical trials, and the sale of any approved products in the future, may expose us to liability claims. These claims might be made by patients that use the product, healthcare providers, pharmaceutical companies, or others selling such products. Any claims against us, regardless of their merit, could be difficult and costly to defend, and could compromise the market acceptance of our product candidates or any prospects for commercialization of our product candidates, if approved.
Although the clinical trial process is designed to identify and assess potential side effects, it is always possible that a drug, even after regulatory approval, may exhibit unforeseen side effects. If any of our product candidates were to cause adverse side effects during clinical trials or after approval of the product candidate, we may be exposed to substantial liabilities. Physicians and patients may not comply with any warnings that identify known potential adverse effects and patients who should not use our product candidates.
Although we maintain standard product liability insurance coverage, such insurance may not be adequate to cover all liabilities that we may incur. We may need to increase our insurance coverage each time we commence a clinical trial and if we successfully commercialize any product candidate. As the expense of insurance coverage is increasing, we may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise. If a successful product liability claim or series of claims is brought against us for uninsured liabilities or in excess of insured liabilities, our assets may not be sufficient to cover such claims and our business operations could be impaired.
Risks Related to Commercialization of Our Product Candidates
We have never commercialized a product candidate and we may lack the necessary expertise, personnel and resources to successfully commercialize any of our products that receive regulatory approval on our own or together with collaborators.
We have never commercialized a product candidate. Our operations to date have been limited to organizing and staffing our company, business planning, raising capital,in-licensing or acquiring our product candidates and undertaking preclinical studies and clinical trials of our product candidates. We currently have no sales force, marketing or distribution capabilities. To achieve commercial success of our product candidates, if any are approved, we will have to develop our own sales, marketing and supply capabilities or outsource these activities to a third party. While we hired a Chief Commercial Officer in July 2018 to lead the strategic and logistical planning of these activities, including market access, the success of such activities once undertaken may be influenced by several factors outside of our control.
Factors that may affect our ability to commercialize our product candidates on our own include recruiting and retaining adequate numbers of effective sales and marketing personnel, obtaining access to or persuadingand educating an adequate numbers of physicians as to prescribethe benefits of our product candidates and other unforeseen costs associated with creating an independent sales and marketing organization. Developing a sales and marketing organization requires significant investment, is time-consuming and could delay the launch of our product candidates. We may not be able to build an effective sales and marketing organization in the United States, the European Union or other key global markets. If we are unable to build our
19
own distribution and marketing capabilities or to find suitable partners for the commercialization of our product candidates, we may have difficulties generating revenue from them.
We operate in a highly competitive and rapidly changing industry.
Biopharmaceutical product development is highly competitive and subject to rapid and significant technological advancements. Our success is highly dependent upon our ability toin-license, acquire, develop and obtain regulatory approval for new and innovative products on a cost-effective basis and to market them successfully. In doing so, we face and will continue to face intense competition from a variety of businesses, including large, fully integrated, well-established pharmaceutical companies who already possess a large share of the market, specialty pharmaceutical and biopharmaceutical companies, academic institutions, government agencies and other private and public research institutions in the United States, the European Union and other jurisdictions.
With respect to OBE2109, there are nolinzagolix, in 2018, the first compound from the oral gonadotropin-releasing hormone, or GnRH, antagonists currently approvedreceptor antagonist class, elagolix, received regulatory approval in the United States for the treatment of pain associated with endometriosis orendometriosis. AbbVie Inc. has been commercializing elagolix, brand named Orilissa, in the United States since August 2018. In August 2019, AbbVie submitted an NDA to the FDA for elagolix for the management of heavy menstrual bleeding associated with uterine fibroids. However, weWe are aware that AbbVie Inc.,of one other oral GnRH receptor antagonist product candidate being developed in Phase 3 clinical trials for endometriosis and uterine fibroids indications, relugolix from Myovant Sciences, Inc. In 2019, Myovant reported positive 6-month results for the two Phase 3 trials in the fibroid indication (LIBERTY 1 and Astellas Pharma Inc. are developing GnRH antagonist product candidates for2) and stated it would file a MAA and a NDA on the basis of 52-week treatment data in the first quarter of symptoms associated with endometriosis or uterine fibroids.2020 and in April 2020, respectively. We also anticipate competing with GnRH receptor agonists, including Lupron (leuprolide acetate), marketed by AbbVie Inc. and Takeda Pharmaceuticals, the progestin Visanne (dienogest), which is approved for the treatment of endometriosis outside the United States and marketed by Bayer, and ulipristal acetate, a Selective Progesterone Receptor Modulator, or SPRM, which is approved for the treatment ofmoderate-to-severe symptoms of uterine fibroids outside the United States and marketed by Gedeon Richter in Europe and other regions, and by Actavis (Allergan)Allergan in Canada. Actavis (Allergan) has stated that it expectsUlipristal acetate, experienced severe label restrictions of usage in 2018 due to submitpost marketing liver safety issues. Allergan had submitted an NDA for ulipristal acetate withbut disclosed receipt of a complete response letter from the FDA in 2017.August 2018 that the NDA is not approvable in its current form and requesting additional information. Enrollment in Phase 3 clinical trials of vilaprisan, another SPRM developed by Bayer Schering for the treatment of uterine fibroids and endometriosis, was halted by Bayer Schering after long-term toxicology studies in rodents indicated a potential problem. In addition, oral contraceptives and nonsteroidal anti-inflammatory drugs, or NSAIDs, are routinely used as a first-line therapy for the treatment of symptoms associated with endometriosis and uterine fibroids and have a meaningful success rate at mitigating the symptoms associated with these conditions.
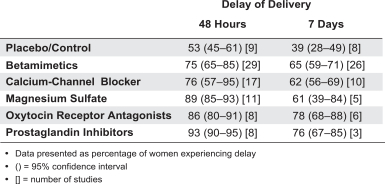
With respect to OBE022, Tractotile (atosiban) is approved to delay preterm birth outside of the United States, and we anticipate potential competition as a single agent, if not used in combination with OBE022 given their different mechanisms of action. In terms of clinical development, it is our understanding that GlaxoSmithKline terminated the in-house development of retosiban, an oxytocin receptor antagonist, designed to delay preterm birth. Currently available prostaglandin synthesis inhibitors, such as NSAIDs may also represent competitive therapies, some of which may be used off-label as standard of care, despite risk of serious side effects for the neonates. Another potential competitive therapy frequently used off-label are calcium channel blockers, such as nifedipine. Makena, which is registered in the USA for preventing preterm delivery in high risk patients, is seen as a complement rather than a competitor for OBE022, due to its mechanism of action and regime of administration (bi-weekly administration to be initiated between week 16 and 20 of gestation).
With respect to nolasiban, there are no other oxytocin receptor antagonists approved either for oral administration or for use in connection with IVF. However, we are awareit is our understanding that Ferring Pharmaceuticals Inc. has been developing barusiban in its development pipeline, an oxytocin receptor antagonist, to be administered subcutaneously, that may be developed for use in connection with IVF. Nevertheless, to our knowledge, no new clinical trial activity has been publicly announced since completion of a Phase 2 trial in 2015. Ferring Pharmaceuticals’ atosiban, an oxytocin receptor antagonist, to be administered by continuous infusion, has been used off-label in investigator initiated trials in connection with IVF outside the United States.
With respect to OBE022, we anticipate competing with atosiban, which has been approved to delay preterm birth outside of the United States, as well as currently available prostaglandin inhibitors, such as NSAIDs. We are also aware that GlaxoSmithKline is developing retosiban, an oxytocin receptor antagonist, to delay preterm birth.
We may also compete with other companies acquiring and developing or marketing drug therapies or products for women’s reproductive health diseases.
Many of the companies against which we are competing or against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved drugs than we do. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our
20
programs. Mergers and acquisitions in the biopharmaceutical industry could result in even more resources being concentrated among a small number of our competitors.
Competition may further increase as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our competitors may succeed in developing, acquiring or licensing, on an exclusive basis, products that are more effective or less costly than any product candidate that we may develop.
Established biopharmaceutical companies may invest heavily to accelerate discovery and development of novel compounds or toin-license novel compounds that could make our product candidates less competitive. In addition, any new product that competes with an approved product must demonstrate compelling advantages in efficacy, convenience, tolerability and safety in order to overcome price competition and to be commercially successful. Accordingly, our competitors may succeed in obtaining patent protection, discovering, developing, receiving FDA, EMA or any comparable foreign regulatory agency approval for or commercializing drugs before we do, which would have an adverse impact on our business and results of operations.
The availability of our competitors’ products could limit the demand and the price we are able to charge for any product candidate we commercialize, if any. The inability to compete with existing or subsequently introduced drugs would harm our business, financial condition and results of operations.
The successful commercialization of certain of our product candidates will depend in part on the extent to which governmental authorities and health insurers establish coverage and adequate coverage, reimbursement levels, andas well as pricing policies. Failure to obtain or maintain adequate coverage and adequate reimbursement for our product candidates, if approved, could limit our ability to market those products and decrease our ability to generate revenue.
The availability and adequacy of coverage and adequate reimbursement by governmental healthcare programs, such as Medicare and Medicaid, private health insurers and other third-party payors, are essential for most patients to be able to afford products such as our product candidates, if approved. Our ability to achieve acceptable levels of coverage and reimbursement for products by governmental authorities, private health insurers and other organizations will have an effect on our ability to successfully commercialize, and attract additional collaboration partners to invest in the development of our product candidates. Coverage under certain government programs, such as Medicare, Medicaid and Tricare, may not be available for certain of our product candidates. Assuming we obtain coverage for a given product by a third-party payor, the resulting reimbursement payment rates may not be adequate or may requireco-payments that patients find unacceptably high. We cannot be sure that coverage and reimbursement in the United States, the European Union or elsewhere will be available for any product that we may develop, and any reimbursement that may not be adequate to make our products affordable for patients or profitable for us and may become available, may be decreased or eliminated in the future.
Third-party payors increasingly are challenging prices charged for pharmaceutical products and services, and many third-party payors may refuse to provide coverage and reimbursement for particular drugs when an equivalent generic drug or a less expensive therapy is available. It is possible that a third-party payor may consider our product candidates and other therapies as substitutable and only offer to reimburse patients for the less expensive product. Even if we show improved efficacy or improved convenience of administration with our product candidates, pricing of existing drugs may limit the amount we will be able to charge for our product candidates. These payors may deny or revoke the reimbursement status of a given product or establish prices for new or existing marketed products at levels that are too low to enable us to realize an appropriate return on our investment in product development. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize our product candidates, and may not be able to obtain a satisfactory financial return on products that we may develop.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. In the United States, third-party payors, including private and governmental payors, such as the Medicare and Medicaid programs, play an important role in determining the extent to which new drugs and biologics will be covered. The Medicare and Medicaid programs increasingly are used as models for how private payors and other governmental payors develop their coverage and reimbursement policies for drugs and biologics. Some third-party payors may requirepre-approval of coverage for new or innovative devices or drug therapies before they will reimburse health care providers who use such therapies. It is difficult to predict at this time what third-party payors will decide with respect to the coverage and reimbursement for our product candidates.
Obtaining and maintaining reimbursement status is time-consuming and costly. No uniform policy for coverage and reimbursement for products exists among third-party payors in the United States. Therefore, coverage and reimbursement for
21
products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. Furthermore, rules and regulations regarding reimbursement change frequently, in some cases at short notice, and we believe that changes in these rules and regulations are likely.
Outside the United States, international operations are generally subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis on cost-containment initiatives in Europe, Canada, and other countries has and will continue to put pressure on the pricing and usage of our product candidates. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. Other countries allow companies to fix their own prices for medical products, but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our product candidates. Accordingly, in markets outside the United States, the reimbursement for our products may be reduced compared with the United States and may be insufficient to generate commercially reasonable revenue and profits.
In the European Union, for example, the main legal instrument at the European Union level governing the pricing and reimbursement of medicinal products is Council Directive 89/105/EEC, or the Price Transparency Directive. The aim of the Price Transparency Directive is to ensure the transparency of measures established by European Union countries to control the pricing and reimbursement of medicinal products. It defines a series of procedural requirements designed to verify that national pricing and reimbursement decisions do not create obstacles to the pharmaceutical trade within the European Union’s Internal Market. The Price Transparency Directive does not, however, provide any guidance concerning the specific criteria on the basis of which pricing and reimbursement decisions are to be made in individual EU member states, except as far as is necessary to achieve the level of transparency required by the Price Transparency Directive. The national authorities of the individual EU member states are free to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices and/or reimbursement of medicinal products for human use. Some individual EU member states adopt policies according to which a specific price or level of reimbursement is approved for the medicinal product. Other EU member states adopt a system of reference pricing, basing the price or reimbursement level in their territory either, on the pricing and reimbursement levels in other countries, or on the pricing and reimbursement levels of medicinal products intended for the same therapeutic indication. Furthermore, some EU member states impose direct or indirect controls on the profitability of the company placing the medicinal product on the market.
Moreover, increasing efforts by governmental and third-party payors in the United States and abroad to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for newly approved products and, as a result, they may not cover or provide adequate payment for our product candidates. We expect to experience pricing pressures in connection with the sale of any of our product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations, and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
Even if we obtain regulatory approval for OBE2109,linzagolix, OBE022, nolasiban OBE022 or future product candidates, they will remain subject to ongoing regulatory oversight.
Even if we obtain any regulatory approval for OBE2109,linzagolix, OBE022, nolasiban OBE022 or future product candidates, they will be subject to extensive and ongoing regulatory requirements for manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, sampling and record-keeping. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with current good manufacturing practices, or cGMP, regulations and good clinical practices, or GCPs, for any clinical trials that we conduct post-approval, all of which may result in significant expense and limit our ability to commercialize such products. In addition, any regulatory approvals that we receive for OBE2109,linzagolix, OBE022, nolasiban OBE022 or future product candidates may also be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the product candidate. The FDA may also require a REMS as a condition of approval of our product candidates, which could include requirements for a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools.
The FDA’s and other regulatory authorities’ policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent
22
of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability. Moreover, if there are changes in the application of legislation or regulatory policies, or if problems are discovered with a product or our manufacture of a product, or if we or one of our distributors, licensees orco-marketers fails to comply with regulatory requirements, the regulators could take various actions. These include imposing fines on us, imposing restrictions on the product or its manufacture and requiring us to recall or remove the product from the
market. The regulators could also suspend or withdraw our marketing authorizations, requiring us to conduct additional clinical trials, change our product labeling or submit additional applications for marketing authorization. If any of these events occurs, our ability to sell such product may be impaired, and we may incur substantial additional expense to comply with regulatory requirements, which could adversely affect our business, financial condition and results of operations.
Off-label use is common in the indications for which our product candidates are under development, which may result in enforcement actions by the FDA and other regulatory agencies for violations of the laws and regulations prohibiting the promotion ofoff-label uses.
Although physicians, in the practice of medicine, may prescribe approved drugs for unapproved indications, pharmaceutical companies are prohibited from marketing or promoting their drug products for uses outside the approved label, a practice known asoff-label promotion. Certain of our product candidates, including OBE022 and nolasiban, are under development for indications for whichoff-label use is common. For example, nifedipine isand NSAIDS are prescribedoff-label for the treatment of preterm labor, although it isthey are not approved for this use. Similarly, the anticipated market for OBE2109linzagolix is characterized by the use of oral contraceptives as a first-line therapy, which have been prescribedoff-label for the treatment of a variety of indications. To the extent the price of our product candidates, if approved, is significantly higher than the prices of commercially available products that are frequently prescribedoff-label, physicians may recommend and prescribe these commercial alternatives instead of writing prescriptions for our products. Either of these outcomes may adversely impact our results of operations by limiting how we price our product and increasing our competition.
In addition, if any of our product candidates are approved, our product labeling, advertising and promotional materials would be subject to regulatory requirements and continuing review by the FDA, Department of Justice, Department of Health and Human Services’ Office of Inspector General, state attorneys general, members of Congress and the public. If we are found to have improperly promotedoff-label uses of our product candidates, if approved, we may become subject to significant liability. Such enforcement has become more common in the industry. If we are found to have promoted our products for any suchoff-label uses, the federal government could levy civil, criminal or administrative penalties, and seek fines against us. The FDA or other regulatory authorities could also requestrequire that we enter into a consent decree or a corporate integrity agreement, or seek a permanent injunction against us under which specified promotional conduct is monitored, changed or curtailed. If we cannot successfully manage the promotion of our product candidates, if approved, we could become subject to significant liability, which would materially adversely affect our business and financial condition.
In the United States, engaging in the impermissible promotion of our products, following approval, foroff-label uses can also subject us to false claims litigation under federal and state statutes, which can lead to civil and criminal penalties and fines, agreements with governmental authorities that materially restrict the manner in which we promote or distribute drug products through, for example, corporate integrity agreements, and debarment, suspension or exclusion from participation in federal and state healthcare programs. These false claims statutes include, the, among others, the federal civil False Claims Act, which allows any individual to bring a lawsuit against a pharmaceutical company on behalf of the federal government alleging submission of false or fraudulent claims, or causing others to present such false or fraudulent claims, for payment by a federal program such as Medicare or Medicaid. If the government decides to intervene and prevails in the lawsuit, the individual will share in the proceeds from any fines or settlement funds. If the government declines to intervene, the individual may pursue the case alone. These false claims lawsuits against pharmaceutical companies have increased significantly in volume and breadth, leading to several substantial civil and criminal settlements regarding certain sales practices promotingoff-label drug uses. This growth in litigation has increased the risk that a pharmaceutical company will have to defend a false claim action, pay settlement fines or restitution, as well as criminal and civil penalties, agree to comply with burdensome reporting and compliance obligations, and be excluded from Medicare, Medicaid and other federal and state healthcare programs. If we do not lawfully promote our approved products, if any, we may become subject to such litigation and, if we do not successfully defend against such actions, those actions may have an adverse effect on our business, financial condition, results of operations and prospects.
23
Even if any of our product candidates receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success.
Even if the FDA, EMA or any comparable foreign regulatory agency approves the marketing of any product candidates that we develop, physicians, patients, third-party payors or the medical community may not accept or use them. Efforts to educate the medical community and third-party payors on the benefits of our product candidates may require significant resources and may not be successful. If OBE2109,linzagolix, OBE022, nolasiban OBE022 or any future product candidate that we develop does not achieve an adequate level of acceptance, we may not generate significant product revenue or any profits from operations. The degree of market acceptance of OBE2109,linzagolix, OBE022, nolasiban OBE022 or any of our future product candidates that are approved for commercial sale will depend on a variety of factors, including:
Our efforts to educate physicians, patients, third-party payors and others in the medical community on the benefits of our products, if approved, may require significant resources and may never be successful. Such efforts may require more resources than are typically required due to the complexity and uniqueness of our product candidates. Because we expect sales of our product candidates, if approved, to generate substantially all of our product revenue for the foreseeable future, the failure of our product candidates to find market acceptance would harm our business and could require us to seek additional financing.
In addition, the potential market opportunity for OBE2109,linzagolix, OBE022, nolasiban OBE022 or any other product candidate we may develop is difficult to estimate precisely. Our estimates of the potential market opportunity are predicated on several key assumptions such as industry knowledge and publications, third-party research reports and other surveys. While we believe that our internal assumptions are reasonable, these assumptions may be inaccurate. If any of the assumptions proves to be inaccurate, then the actual market for OBE2109,linzagolix, OBE022, nolasiban OBE022 or our future product candidates could be smaller than our estimates of the potential market opportunity. If the actual market for OBE2109,linzagolix, OBE022, nolasiban OBE022 or our future product candidates is smaller than we expect, or if the products fail to achieve an adequate level of acceptance by physicians, health care payors and patients, our revenue from product sales may be limited and we may be unable to achieve or maintain profitability.
We currently have no marketing, sales or distribution infrastructure. If we are unable to develop sales, marketing and distribution capabilities on our own or through collaborations, or if we fail to achieve adequate pricing or reimbursement we will not be successful in commercializing our product candidates, if approved.
We currently have no marketing, sales and distribution capabilities and our product candidates are still in clinical development. If any of our product candidates are approved, we intend either to establish a sales and marketing organization with technical expertise and supporting distribution capabilities to commercialize our product candidates, or to outsource this these functions to a third party. Either of these options would be expensive and time-consuming. These costs may be incurred in advance of any approval of our product candidates. In addition, we may not be able to hire a sales force that is sufficient in size or has adequate expertise in the medical markets that we intend to target. Any failure or delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of our products.
24
To the extent that we enter into collaboration agreements with respect to marketing, sales or distribution, our product revenue may be lower than if we directly marketed or sold any approved products. In addition, any revenue we receive will depend in whole or in part upon the efforts of these third-party collaborators, which may not be successful and are generally not within our control. If we are unable to enter into these arrangements on acceptable terms or at all, we may not be able to successfully commercialize any approved products. If we are not successful in commercializing any approved products, either on our own or through collaborations with one or more third parties, our future product revenue will suffer and we may incur significant additional losses.
Our business and operations would suffer in the event of computer system failures, cyber-attacks or a deficiency in our cyber-security.
Despite the implementation of security measures, our internal computer systems, and those of third parties on which we rely, are vulnerable to damage from computer viruses, malware, natural disasters, terrorism, war, telecommunication and electrical failures, cyber-attacks or cyber-intrusions over the Internet, attachments to emails, persons inside our organization, or persons with access to systems inside our organization. The risk of a security breach or disruption, particularly through cyber-attacks or cyber-intrusion, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our product development programs. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach was to result in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur material legal claims and liability, damage to our reputation, and the further development of our product candidates could be delayed.
If we fail to comply with data protection laws and regulations, we could be subject to government enforcement actions (which could include civil or criminal penalties), private litigation, increased compliance costs and/or adverse publicity, which could negatively affect our operating results and business.
We are subject to data protection laws and regulations (i.e., laws and regulations that address privacy and data security). In the United States, numerous federal and state laws and regulations, including state data breach notification laws, state health information privacy laws, and federal and state consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act), govern the collection, use, disclosure, and protection of health-related and other personal information. Failure to comply with data protection laws and regulations could result in government enforcement actions and create liability for us, including civil and/or criminal penalties, private litigation and/or adverse publicity that could negatively affect our operating results and business. In addition, we may obtain health information from third parties (e.g., healthcare providers who prescribe our products) that are subject to privacy and security requirements under the U.S. federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by HITECH. Although we are not directly subject to HIPAA—other than potentially with respect to providing certain employee benefits—we could be subject to criminal penalties if we knowingly obtain or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA. HIPAA generally requires that healthcare providers and other covered entities obtain written authorizations from patients prior to disclosing protected health information of the patient (unless an exception to the authorization requirement applies). If authorization is required and the patient fails to execute an authorization or the authorization fails to contain all required provisions, then we may not be allowed access to and use of the patient’s information and our research efforts could be delayed. Furthermore, use of protected health information that is provided to us pursuant to a valid patient authorization is subject to the limits set forth in the authorization (e.g., for use in research and in submissions to regulatory authorities for product approvals). In addition, HIPAA does not replace federal, state, international or other laws that may grant individuals even greater privacy protections.
On June 28, 2018, California enacted the California Consumer Privacy Act, or CCPA, which took effect on January 1, 2020. The CCPA gives California residents expanded rights to access and delete their personal information, opt out of certain personal information sharing, and receive detailed information about how their personal information is used. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. The CCPA may increase our compliance costs and potential liability. Some observers have noted that the CCPA could mark the beginning of a trend toward more stringent state privacy legislation in the U.S., which could increase our potential liability and adversely affect our business.
In the European Union, the General Data Protection Regulation, or GDPR, took effect on May 25, 2018, introducing sweeping new data protection requirements that carry potential fines of up to the greater of 20 million Euros or 4% of annual global revenue. The GDPR introduces strict requirements for processing personal data, including potentially burdensome
25
documentation requirements, more stringent requirements for obtaining valid consent (where applicable), obligations to honor expanded rights of individuals to control the use and retention of their personal data, and requirements to notify regulators and affected individuals of certain personal data breaches. The GDPR also imposes heightened restrictions on processing of special categories of personal data, such as health and genetic personal data. In addition, the GDPR prohibits the transfer of personal data to countries outside of the EEA, such as the United States, which are not considered by the European Commission to provide an adequate level of data protection. Switzerland has adopted similar restrictions. Although there are legal mechanisms to allow for the transfer of personal data from the EEA and Switzerland to the United States, they are subject to pending legal challenges that, if successful, could invalidate these mechanisms, restrict our ability to process personal data of European residents outside of Europe and adversely impact our business. The GDPR has increased our responsibility and potential liability in relation to personal data that we process, exposes us to substantial potential fines in the event of violations, increases our compliance costs and could restrict our operations in Europe.
Risks Related to Our Dependence on Third Parties
If we fail to comply with our obligations under our existing and any future intellectual property licenses with third parties, we could lose license rights that are important to our business.
We are party to several license agreements under which wein-license patent rights and other intellectual property related to orour business, including a license and supply agreement with Kissei, under which we were granted an exclusive license relating to OBE2109linzagolix and license agreements with Merck Serono, pursuant to which we were granted exclusive worldwide licenses relating to nolasibanOBE022 and OBE022.nolasiban. We may enter into additional license agreements in the future. Our license agreements impose, and we expect that future license agreements will impose, various diligence, milestone payment, royalty, insurance and other obligations on us. Any uncured, material breach under these license agreements could result in our loss of rights to practice the patent rights and other intellectual property licensed to us under these agreements, and could compromise our development and commercialization efforts for OBE2109,linzagolix, OBE022 and nolasiban, and OBE022, or any future product candidates. See “Item 4.B—Business Overview” for a more detailed description of our current license agreements.
We may be required to make significant payments in connection with our license and supply agreement with Kissei.
We acquired exclusive rights to OBE2109linzagolix pursuant to our license and supply agreement with Kissei in November 2015. Under the terms of the Kissei license and supply agreement, we are obligated to cover substantialshare certain development costs for OBE2109, andwith Kissei in addition to our own direct development costs. Additionally, we may make significant payments in connection with certain milestones and the sale of resulting products. If these obligations become due under the terms of the Kissei license and supply agreement within the next eighteen months, our development efforts may be delayed and/or we may have to seek additional funding earlier than currently planned.
Our intellectual propertyin-licenses with in-licensed from third parties may be subject to disagreements over contract interpretations, which could narrow the scope of our rights to the relevant intellectual property or technology or increase our financial or other obligations to our licensors.
The agreements under which we currentlyin-license intellectual property or technology from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could harm our business, financial condition, results of operations and prospects. If any of our current or future licenses or material relationships or anyin-licenses upon which our current or future licenses are based are terminated or breached, we may:
If we experience any of the foregoing, it could harm our business, financial condition and results of operations.
26
We rely on third parties to conduct our preclinical studies and clinical trials and if these third parties perform in an unsatisfactory manner, our business could be substantially harmed.
We currently do not have the ability to independently conduct preclinical studies that comply with the regulatory requirements known as good laboratory practice, or GLP, requirements. We also do not currently have the ability to independently conduct any clinical trials. We have relied upon and plan to continue to rely upon medical institutions, clinical investigators, contract laboratories and other third parties, such as CROs, to conductGLP-compliant preclinical studies andGCP-compliant clinical trials on our product candidates properly and on time, and may not currently have all of the necessary contractual relationships in place to do so. Once we have established contractual relationships with such third-party CROs, we will have only limited control over their actual performance of these activities.
We and our CROs and other vendors are required to comply with cGMP, GCP and GLP, which are regulations and guidelines enforced by the FDA, the Competent Authorities of the Member States of the European UnionEMA and any comparable foreign regulatory authorities for all of our product candidates in preclinical and clinical development. Regulatory authorities enforce these regulations through periodic inspections of trial sponsors, principal investigators, clinical trial sites and other contractors. Although we rely on CROs to conduct any current or plannedGLP-compliant preclinical studies andGCP-compliant clinical trials and have limited influence over their actual performance, we remain responsible for ensuring that each of our preclinical studies and clinical trials is conducted in accordance with its investigational plan and protocol and applicable laws and regulations, and our reliance on the CROs does not relieve us of our regulatory responsibilities. If we or any of our CROs or vendors fail to comply with applicable regulations, the data generated in our preclinical studies and clinical trials may be deemed unreliable and the FDA, EMA or any comparable foreign regulatory agency may require us to perform additional preclinical studies and clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory agency, such regulatory agency will determine that all of our clinical trials comply with GCP regulations. In addition, our clinical trials must be conducted with products produced under cGMP requirements. Our failure to comply with these requirements may require us to repeat clinical trials, which would delay the regulatory approval process.
While we will have agreements governing their activities, our CROs will not be our employees, and we will not be able to control whether or not they devote sufficient time and resources to our future preclinical and clinical programs. These CROs may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials, or other drug development activities which could harm our business. We face the risk of potential unauthorized disclosure or misappropriation of our intellectual property by CROs, which may reduce our trade secret protection and allow our potential competitors to access and exploit our proprietary technology. If our CROs do not successfully carry out their contractual duties or obligations, fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for any other reason, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize any product candidate that we develop. As a result, our financial results and the commercial prospects for any product candidate that we develop would be harmed, our costs could increase, and our ability to generate revenue could be delayed.
If our relationship with these CROs terminates, we may not be able to enter into arrangements with alternative CROs or do so on commercially reasonable terms. Switching or adding additional CROs involves substantial cost and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which can negatively impact our ability to meet our desired clinical development timelines. Though we intend to carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a negative impact on our business and financial condition.
27
We currently rely on third parties forour third-party manufacturers to source the production of our clinical supply of the materials for our product candidates and, we intendin the case of linzagolix, Kissei is the exclusive supplier of API. The inability to continue to rely on third partiesobtain supply of the materials for our clinicalproduct candidates or the failure of, or loss of, our exclusive supplier to supply us with the API for linzagolix would materially and commercial supply.adversely affect our business.
We currently rely on and expect to continue to rely on third parties for the manufacturing and supply of chemical compounds for the clinical trials of our product candidates and, if approved, our commercial supply. Further, Kissei has the exclusive right to supply us with the active pharmaceutical ingredient, or API, for OBE2109linzagolix for our clinical trials and commercial supply,supplies, if approved, subject to limited specified exceptions within the control of Kissei. Reliance on third-party suppliers may expose us to different risks than if we were to manufacture product candidates ourselves. The facilities used by our contract manufacturers to manufacture our product candidates must be approved by the FDA or other regulatory authorities pursuant to inspections that will be conducted after we submit our NDA or comparable marketing application to the FDA or other regulatory agency. Although we have auditing rights with all our manufacturing counterparties, we do not have control over a supplier’s or manufacturer’s compliance with these laws, regulations and applicable cGMP standards and other laws and
regulations, such as those related to environmental health and safety matters. For example, a contract
To meet our projected needs for clinical supplies and support our activities through regulatory approval and commercial manufacturing, organization,the CMOs with whom we currently work will need to increase scale of production, or we will need to secure alternate suppliers. With respect to linzagolix, we have entered into an exclusive agreement for the supply of the linzagolix API with Kissei. A CMO that Kissei is using to supply the linzagolix API for OBE2109 received a warning letter from the FDA in November 2016 citing deviations from cGMP requirements with respect to its drug manufacturing facility. WeKissei has now obtained linzagolix cGMP supply from two additional suppliers, both of which are alsodifferent from the supplier who received the warning letter from the FDA in the process of qualifying additional suppliers. IfNovember 2016. For OBE022 and nolasiban, we obtain supply on a purchase order basis from a different single source. However, we believe that there are multiple potential sources for our contract manufacturers cannot successfully manufacture material that conformsmanufacturing for OBE022 and nolasiban. We have started engaging alternative suppliers for the cGMP manufacturing of nolasiban. We have not started engaging alternate suppliers for the manufacturing of OBE022. Additionally, any damage to our specifications and the strict regulatory requirements of the FDA or others, they will not be able to secure or maintain regulatory approval for their manufacturing facilities. In addition, we have no control over the abilitydestruction of our contract manufacturersor our third-party manufacturers’ or suppliers’ facilities or equipment may significantly impair our ability to maintain adequate quality control, quality assurance and qualified personnel. If the FDA or a comparable foreign regulatory agency does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved. Any failure to achieve and maintain compliance with these laws, regulations and standards could subject us to the risk that we may have to suspend the manufacturing of our product candidates or that obtained approvals could be revoked, which would adversely affect our business and reputation. Furthermore, third-party providers may breach agreements they have with us because of factors beyond our control. They may also terminate or refuse to renew their agreements because of their own financial difficulties or business priorities, potentially aton a time that is costly or otherwise inconvenient for us. If we were unable to find adequate replacement or another acceptable solution in time, our clinical trials could be delayed or our commercial activities could be harmed.
In addition, the fact that we are dependent on third parties for the manufacture, storage and distribution of our product candidates means that we are subject to the risk that our product candidates and, if approved, commercial products may have manufacturing defects that we have limited ability to prevent or control. The sale of products containing such defects could result in recalls or regulatory enforcement action that could adversely affect our business, financial condition and results of operations.
We rely on our third-party manufacturers to source the supply of the materials for our product candidates and, in the case of OBE2109, a single and exclusive supplier for its API. The inability to obtain supply of the materials for our product candidates or the failure of, or loss of, our sole and exclusive supplier to supply us with the API for OBE2109 would materially and adversely affect our business.timely basis.
We rely on our manufacturers to purchase from third-party suppliers the materials necessary to produce our product candidates for our clinical trials, and we expect to continue to depend on third-party suppliers for the foreseeable future. There are a limited number of suppliers for raw materials that we use to manufacture our drugs and there may be a need to assess alternate suppliers to prevent a possible disruption of the manufacture of the materials necessary to produce our product candidates for our clinical trials, and if approved, ultimately for commercial sale. We do not have any control over the process or timing of the acquisition of these raw materials by our manufacturers. Moreover, we currently do not have any agreements for the commercial production of these raw materials. Any significant delay in the supply of a product candidate, or the raw material components thereof, for an ongoing clinical trial due to the need to replace a third-party manufacturer could considerably delay completion of our clinical trials, product testing and potential regulatory approval of our product candidates. If our manufacturers or we are unable to purchase these raw materials after regulatory approval has been obtained for our product candidates, the commercial launch of our product candidates would be delayed or there would be a shortage in supply, which would impair our ability to generate revenue from the sale of our product candidates.
Other than in respect of our agreement with Kissei, we have not yet entered into a long-term agreement with any alternate supplier of API for our product candidates. We may be unable to enter into long-term arrangements with alternative suppliers or do so on commercially reasonable terms, which could have a material adverse impact upon our business. With respect to OBE2109, we have entered into an exclusive agreement for the supply of the API for OBE2109 with Kissei. A CMO that Kissei is using to supply the API for OBE2109 received a warning letter from the FDA in November 2016 citing deviations from cGMP requirements with respect to its drug manufacturing facility. In the event of a disruption to Kissei or to a CMO Kissei elects to utilize for this source of supply, we will have no other means of producing OBE2109 until Kissei restores the affected facilities, selects an alternate CMO or we or it procures alternative manufacturing facilities. Additionally, any damage to or destruction of our or our third-party manufacturers’ or suppliers’ facilities or equipment may significantly impair our ability to manufacture our product candidates on a timely basis.
We rely on our manufacturers and other subcontractors to comply with and respect the proprietary rights of others in conducting their contractual obligations for us. If our manufacturers or other subcontractors fail to acquire the proper licenses or otherwise infringe third-party proprietary rights in the course of completing their contractual obligations to us, we may have to find alternative manufacturers or defend against claims of infringement, either of which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved.
If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or others, they will not be able to secure or maintain regulatory approval for their manufacturing facilities. In addition, we have no control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If the FDA or a comparable foreign regulatory agency does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved. Any failure to achieve and maintain compliance with these laws, regulations and standards could subject us to the risk that we may have to suspend the manufacturing of our product candidates or that obtained approvals could be revoked, which would adversely affect our business and reputation. Furthermore, third-party providers may breach agreements they have with us because of factors beyond our control. They may also terminate or refuse to renew their agreements because of their own financial difficulties or business priorities,
28
potentially at a time that is costly or otherwise inconvenient for us. If we were unable to find adequate replacement or another acceptable solution in time, our clinical trials could be delayed or our commercial activities could be harmed.
In addition, the fact that we are dependent on third parties for the manufacture, storage and distribution of our product candidates means that we are subject to the risk that our product candidates and, if approved, commercial products may have manufacturing defects that we have limited ability to prevent or control. The sale of products containing such defects could result in recalls or regulatory enforcement action that could adversely affect our business, financial condition and results of operations.
We may in the future enter into collaborations with third parties to develop our product candidates. If these collaborations are not successful, our business could be harmed.
We may potentially enter into collaborations with third parties in the future. We will face, to the extent that we decide to enter into collaboration agreements, significant competition in seeking appropriate collaborators. Moreover, collaboration arrangements are complex and time-consuming to negotiate, document, implement and maintain. We may not be successful in our efforts to establish and implement collaborations or other alternative arrangements should we so chose to enter into such arrangements. The terms of any collaborations or other arrangements that we may establish may not be favorable to us.
Any future collaborations that we enter into may not be successful. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborations are subject to numerous risks, including:
29
If any such potential future collaborations do not result in the successful development and commercialization of product candidates, or if one of our future collaborators terminates its agreement with us, we may not receive any future research funding or milestone or royalty payments under the collaboration. If we do not receive the funding we expect under these agreements, the development of our product candidates could be delayed and we may need additional resources to develop our product candidates. In addition, if one of our future collaborators terminates its agreement with us, we may find it more difficult to attract new collaborators and the perception of us in the business and financial communities could be adversely affected. All of the risks relating to product development, regulatory approval and commercialization apply to the activities of our potential future collaborators.
If we are not able to establish or maintain collaborations, we may have to alter some of our future development and commercialization plans.
Our product development programs and the potential commercialization of our product candidates will require substantial additional capital to fund expenses. For some of our product candidates, we may decide to collaborate with pharmaceutical and biotechnology companies for the future development and potential commercialization of those product candidates, particularly in Asia. For example, in January 2020, we announced our entrance into a sublicense agreement with Hangzhou Yuyuan BioScience Technology Co., Ltd., or Yuyuan, to develop and commercialize nolasiban for improving clinical pregnancy and live birth rates in women undergoing embryo transfer following IVF in the People’s Republic of China, or PRC. Under the terms of the sublicense agreement, Yuyuan has the exclusive rights to develop and commercialize nolasiban in the PRC. We are also exploring various alternatives for the future potential commercialization of linzagolix, including through a collaboration with a third party. Furthermore, we may find that our programs require the use of proprietary rights held by third parties, and the growth of our business may depend in part on our ability to acquire,in-license or use these proprietary rights.
We face significant competition in seeking appropriate collaborators, and a number of more established companies may also be pursuing strategies to license or acquire third-party intellectual property rights that we may consider attractive. These established companies may have a competitive advantage over us due to their size, financial resources and greater clinical development and commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. Whether we reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA, EMA or similar foreign regulatory authorities, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge, and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such a collaboration could be more attractive than the one with us for our product candidate. We may also be restricted under existing license agreements from entering into agreements on certain terms with potential collaborators. Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
We may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. Even if we are able to obtain a license to intellectual property of interest, we may not be able to secure exclusive rights, in which case others could use the same rights and compete with us. If we are unable to successfully obtain rights to required third party intellectual property
30
rights or maintain the existing intellectual property rights we have, we may have to curtail the development of such product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our product candidates or bring them to market and generate product revenue.
Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
Because we rely on third parties to develop and manufacture our product candidates, we must, at times, share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements or other similar agreements with our collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, such as trade secrets. Despite these contractual agreements with third parties, sharing trade secrets and other confidential information increases the risk that such trade secrets become known by our competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on ourknow-how and trade secrets, a competitor’s discovery of our trade secrets or other unauthorized use or disclosure would impair our competitive position and may harm our business.
In addition, these agreements typically restrict the ability of our advisors, employees, third-party contractors and consultants to publish data potentially relating to our trade secrets, although our agreements may contain certain limited publication rights. Despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of our agreements with third parties, independent development or publication of information by any of our third-party collaborators. A competitor’s discovery of our trade secrets would impair our competitive position and have an adverse impact on our business.
Risks Related to Regulatory Compliance
Enacted and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates and may affect the prices we may set.
In the United States, the European Union, and other foreign jurisdictions, there have been, and we expect there will continue to be, a number of legislative and regulatory changes and proposed changes to the healthcare system that could affect our future results of operations. In particular, there have been and continue to be a number of initiatives at the United States federal and state levels that seek to reduce healthcare costs and improve the quality of healthcare. For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively the ACA, was enacted, which substantially changes the way healthcare is financed by both governmental and private insurers. Among the provisions of the ACA, those of greatest importance to the pharmaceutical and biotechnology industries include:
31
There have beenremains judicial and Congressional challenges to certain aspects of the ACA, as well as efforts by the Trump administration to repeal or replace certain aspects of the ACA. Since January 2017, President Trump has signed two Executive Orders and we expect there will be additional challenges and amendmentsother directives designed to delay the ACA in the future. The current administration and Congress will likely continue to seek legislative and regulatory changes, including repeal and replacementimplementation of certain provisions of the ACA or otherwise circumvent some of the requirements for health insurance mandated by the ACA. In January 2017, President Trump signed an Executive Order directing federal agencies with authoritiesConcurrently, Congress has considered legislation that would repeal or repeal and responsibilitiesreplace all or part of the ACA. While Congress has not passed comprehensive repeal legislation, two bills affecting the implementation of certain taxes under the ACA have been signed into law. Legislation enacted in 2017, informally titled the Tax Cuts and Jobs Act, or Tax Act, includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to waive, defer, grant exemptionsmaintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate”. In addition, the 2020 federal spending package permanently eliminates, effective January 1, 2020, the ACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, also eliminates the health insurer tax. The Bipartisan Budget Act of 2018, or the BBA, among other things, amends the ACA, effective January 1, 2019, to close the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole”. In December 2018, CMS published a new final rule permitting further collections and payments to and from or delaycertain ACA qualified health plans and health insurance issuers under the implementationACA risk adjustment program in response to the outcome of any provisionfederal district court litigation regarding the method CMS uses to determine this risk adjustment. On December 14, 2018, a Texas U.S. District Court Judge ruled that the ACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress as part of the Tax Act. Additionally, on December 18, 2019, the U.S. Court of Appeals for the 5th Circuit upheld the District Court ruling that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA that would impose a fiscal or regulatory burden on states, individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices. In March 2017, following the passage of the budget resolution for fiscal year 2017, the U.S. House of Representatives introduced legislation knownare invalid as the American Health Care Act, which, if enacted, would have amended or
repealed significant portions of the ACA. However, consensus over the scope of the American Health Care Act could not be reached by its proponents in the U.S. House of Representatives. Thus, the proposed legislation has been withdrawnwell. It is unclear how this decision, future decisions, subsequent appeals, and the prospects for legislative action on this bill are uncertain. Congress could consider other legislationefforts to repeal orand replace certain elements of the ACA. We continue to evaluate the effect thatACA will impact the ACA and its possible repeal and replacement has on our business. At this time, the full effect that the ACA would have on our business remains unclear.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. On August 2, 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of 2% per fiscal year. These reductions went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute, including amendments by the BBA, will remain in effect through 20252029 unless additional Congressional action is taken. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other health care funding, which could have an adverse effect on our customers and accordingly, our financial operations.
Moreover, payment methodologies may be subject to changes in healthcare legislation and regulatory initiatives. For example, CMS may develop newthe Medicare Access and CHIP Reauthorization Act of 2015 ended the use of the statutory formula, also referred to as the Sustainable Growth Rate, for clinician payment and delivery models, suchestablished a quality payment incentive program, also referred to as bundled payment models. The U.S. Department of Healththe Quality Payment Program. This program provides clinicians with two ways to participate, including through the Advanced Alternative Payment Models, or APMs, and Human Services,the Merit-based Incentive Payment System, or HHS, setMIPS. In November 2019, CMS issued a goal of moving 30% of Medicare payments to alternative payment models tiedfinal rule finalizing the changes to the Quality Payment Program. At this time, it is unclear how the introduction of the quality payment program will impact overall physician reimbursement under the Medicare program. Any reduction in reimbursement from Medicare or value of services by 2016 and 50% of Medicareother government programs may result in a similar reduction in payments into these alternative payment models by the end of 2018. In March, HHS announced that it has achieved its goal for 2016. from private payors.
In addition, recently there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, including several recent Congressional inquiries and proposed billsand enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing
32
and manufacturer patient programs, reduce the cost of drugs under Medicare and reform government program reimbursement methodologies for products. At the federal level, the Trump administration’s budget proposal for fiscal year 2020 contains further drug price control measures that could be enacted during the budget process or in other future legislation, including, for example, measures to permit Medicare Part D plans to negotiate the price of certain drugs under Medicare Part B, to allow some states to negotiate drug prices under Medicaid, and to eliminate cost sharing for generic drugs for low-income patients. Additionally, the Trump administration released a “Blueprint” to lower drug prices and reduce out of pocket costs of drugs that contains additional proposals to increase manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products and reduce the out of pocket costs of drug products paid by consumers. HHS has solicited feedback on some of these measures and implemented others under its existing authority. For example, in May 2019, CMS issued a final rule to allow Medicare Advantage plans the option to use step therapy for Part B drugs beginning January 1, 2020. This final rule codified CMS’s policy change that was effective January 1, 2019. While a number of these and other measures may require additional authorization to become effective, Congress and the Trump administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs. We expect that additional U.S. federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that the U.S. federal government will pay for healthcare products and services, which could result in reduced demand for our product candidates or additional pricing pressures.
IndividualAt the state level, individual states in the United States have also become increasingly aggressive in passingpassed legislation and implementingimplemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions could harm our business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce the ultimate demand for our products or put pressure on our product pricing, which could negatively affect our business, results of operations, financial condition and prospects.
In the European Union, similar political, economic and regulatory developments may affect our ability to profitably commercialize any of our product candidates, if approved. In addition to continuing pressure on prices and cost containment measures, legislative developments at the European Union or member state level may result in significant additional requirements or obstacles that may increase our operating costs. The delivery of healthcare in the European Union, including the establishment and operation of health services and the pricing and reimbursement of medicines, is almost exclusively a matter for national, rather than European Union, law and policy. National governments and health service providers have different priorities and approaches to the delivery of health care and the pricing and reimbursement of products in that context. In general, however, the healthcare budgetary constraints in most EU member states have resulted in restrictions on the pricing and reimbursement of medicines by relevant health service providers. Coupled with ever-increasing European Union and national regulatory burdens on those wishing to develop and market products, this could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to commercialize any products for which we obtain marketing approval. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various EU member states, and parallel trade, i.e., arbitrage between low-priced and high-priced member states, can further reduce prices. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any products, if approved in those countries. In international markets, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies.
33
We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we or our collaborators are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or our collaborators are not able to maintain regulatory compliance, our product candidates may lose any regulatory approval that may have been obtained and we may not achieve or sustain profitability, which would adversely affect our business.
Our business operations and current and future relationships with investigators, health care professionals, consultants, third-party payors and customers willmay be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws, health information privacy and security laws, and other healthcare laws and regulations. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
Although we do not currently have any products on the market, if we obtain FDA approval for OBE2109, nolasiban, OBE022 or any future product candidates, and begin commercializing those products in the United States, our operations may be directly, or indirectly through our prescribers,health care professionals, consultants, customers and third-party payors, subject to various U.S. federal and state healthcare laws and regulations, including, without limitation, the U.S. federal Anti-Kickback Statute, the U.S. federal civil and criminal false claims laws and Physician Payments Sunshine Act and regulations. Healthcare providers, physicians and others play a primary role in the recommendation and prescription of any products for which we obtain marketing approval. These laws may impact, among other things, our current business operations, including our clinical research activities, and proposed sales, marketing and education programs and constrain the business of financial arrangements and relationships with healthcare providers, physicians and other parties through which we market, sell and distribute our products for which we obtain marketing approval. In addition, we may be subject to patient data privacy and security regulation by both the U.S. federal government and the states in which we conduct our business. Finally, we may be subject to additional healthcare, statutory and regulatory requirements and enforcement by foreign regulatory authorities in jurisdictions in which we conduct our business. The laws that may affect our ability to operate include:
34
analogous state laws and regulations, including: state anti-kickback and false claims laws, which may apply to our business practices, including, but not limited to, research, distribution, sales and marketing arrangements and claims involving healthcare items or services reimbursed by any third-party payor, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the U.S. federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws and regulations that require drug manufacturers to file reports relating to pricing and marketing information, which requires tracking gifts and other remuneration and items of value provided to healthcare professionals and entities; state and local laws that require the registration of sales representatives in the jurisdiction; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts; and
Ensuring that our internal operations and future business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations, agency guidance or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of the laws described above or any other governmental laws and regulations that may apply to us, we may be subject to significant penalties, including civil, criminal and administrative penalties, damages, fines, exclusion from U.S. government funded healthcare programs, such as Medicare and Medicaid, or similar programs in other countries or jurisdictions, disgorgement, individual imprisonment, contractual damages, reputational harm, diminished profits, and corporate integrity agreements, which impose, among other things, rigorous operational and monitoring requirements on companies, and the curtailment or restructuring of our operations. Further, defending against any such actions can be costly, time-consuming and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired. If any of the physicians or other providers or entities with whom we expect to do business is found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs and imprisonment. If any of the above occur, it could adversely affect our ability to operate our business and our results of operations.
Risks Related to Our Intellectual Property
If we fail to comply with our obligations under our existing and any future intellectual property licenses with third parties or if such licenses are subject to a disagreement over contract interpretation, we could lose license rights that are important to our business or be subject to a narrowing of the scope of our rights to the relevant intellectual property or technology or an increase of our financial or other obligations to our licensors.
We are party to several license agreements under which wein-license patent rights and other intellectual property related to our business, and we may enter into additional license agreements in the future. See “Risk Factors—Risks Related to Our Dependence on Third Parties” for a more detailed description of risks related to current and future license agreements.
If we are unable to obtain and maintain patent protection for our technology and product candidates, or if the scope of the patent protection obtained is not sufficiently broad, we may not be able to compete effectively in our markets.
We rely upon a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to our development programs and product candidates. Our success depends in large part on our ability to obtain and maintain patent protection in the United States and other countries with respect to our current and future product candidates. We have sought to protect our proprietary position by filing andin-licensing patent applications in the United States and abroad related to our development programs and product candidates. The patent prosecution process is expensive and time-consuming, and we or our licensors may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner or in all jurisdictions.
35
It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. Moreover, in some circumstances, we do not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the patents, covering technology that we have licensed from third parties. We prosecute and maintain thecertain patent rights for OBE022our product candidates and rely on our licensors Kissei and Merck Serono to prosecute and maintain theother relevant patent rights for OBE2109linzagolix, OBE022 and nolasiban. Therefore, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business. The patent applications that we own orin-license may fail to result in issued patents with claims that cover our current and future product candidates in the United States or in other foreign countries. Our patent applications cannot be enforced against third parties practicing the technology claimed in such applications unless, and until, a patent issues from such applications, and then only to the extent the issued claims cover the technology.
If the patent applications we hold or havein-licensed with respect to our development programs and product candidates fail to issue, if their breadth or strength of protection is threatened, or if they fail to provide meaningful exclusivity for our current and future product candidates, it could dissuade companies from collaborating with us to develop product candidates, and threaten our ability to commercialize, products. Any such outcome could have a negative effect on our business.
The patent position of biopharmaceutical companies generally is highly uncertain, involves complex legal and factual questions and has in recent years been the subject of much litigation. In addition, the laws of foreign countries may not protect our rights to the same extent as the laws of the United States. For example, EU patent law restricts the patentability of methods of treatment of the human body more than U.S. law does. Publications of discoveries in scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions remain confidential for a period of time after filing, and some remain so until issued. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in our owned or licensed patents or pending patent applications, or that we were the first to file for patent protection of such inventions. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain. There is no assurance that all potentially relevant prior art relating to our patents and patent applications has been found, and such prior art could potentially invalidate a one or more of our patents or prevent a patent from issuing from a one or more of our pending patent applications. There is also no assurance that there is not prior art of which we are aware, but which we do not believe affects the validity or enforceability of a claim in our patents and patent applications, which may, nonetheless, ultimately be found to affect the validity or enforceability of a claim. Even if patents do successfully issue and even if such patents cover our current and future product candidates, third parties may challenge their validity, enforceability or scope, which may result in such patents being narrowed, invalidated or held unenforceable, which could allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights. Any successful opposition to these patents or any other patents owned by or licensed to us could deprive us of rights necessary for the successful commercialization of any product candidates that we may develop. Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property, provide exclusivity for our product candidates, prevent others from designing around our claims or provide us with a competitive advantage. Any of these outcomes could impair our ability to prevent competition from third parties. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection.
We, independently or together with our licensors, have filed several patent applications covering various aspects of our product candidates. We cannot offer any assurances about which, if any, patents will issue, the breadth of any such patent, or whether any issued patents will be found invalid and unenforceable or will be challenged by third parties. Any successful opposition to these patents or any other patents owned by or licensed to us after patent issuance could deprive us of rights necessary for the successful commercialization of any product candidates that we may develop. Further, if we encounter delays in regulatory approvals, the period of time during which we could market a product candidate under patent protection could be reduced.
36
We may not identify relevant third-party patents or may incorrectly interpret the relevance, scope or expiration of a third-party patent which might adversely affect our ability to develop and market our product candidates.
We cannot guarantee that any of our or our licensors’ patent searches or analyses, including the identification of relevant patents, the scope of patent claims or the expiration of relevant patents, are complete or thorough, nor can we be certain that we have identified each and every third-party patent and pending application in the United States and abroad that is relevant to or necessary for the commercialization of our product candidates in any jurisdiction. For example, U.S. applications filed before November 29, 2000 and certain U.S. applications filed after that date that will not be filed outside the United States remain confidential until patents issue. Patent applications in the United States and elsewhere are published approximately 18 months after the earliest filing for which priority is claimed, with such earliest filing date being commonly referred to as the
priority date. Therefore, patent applications covering our product candidates could have been filed by others without our knowledge. Additionally, pending patent applications that have been published can, subject to certain limitations, be later amended in a manner that could cover our product candidates or the use of our product candidates. The scope of a patent claim is determined by an interpretation of the law, the written disclosure in a patent and the patent’s prosecution history. Our interpretation of the relevance or the scope of a patent or a pending application may be incorrect, which may negatively impact our ability to market our product candidates. We may incorrectly determine that our product candidates are not covered by a third-party patent or may incorrectly predict whether a third party’s pending application will issue with claims of relevant scope. Our determination of the expiration date of any patent in the United States or abroad that we consider relevant may be incorrect, which may negatively impact our ability to develop and market our product candidates. Our failure to identify and correctly interpret relevant patents may negatively impact our ability to develop and market our product candidates.
If we fail to identify and correctly interpret relevant patents, we may be subject to infringement claims. We cannot guarantee that we will be able to successfully settle or otherwise resolve such infringement claims. If we fail in any such dispute, in addition to being forced to pay damages, we may be temporarily or permanently prohibited from commercializing any of our product candidates that are held to be infringing. We might, if possible, also be forced to redesign product candidates that we no longer infringe the third-party intellectual property rights. Any of these events, even if we were ultimately to prevail, could require us to divert substantial financial and management resources that we would otherwise be able to devote to our business.
We are aware of issued patents and pending patent applications, in the United States and abroad, relating to methods of improving embryo implantation outcomes in patients undergoing an embryo transfer procedure. If any such patent, or a patent that issues from any such application, were to be asserted against us, we believe that we have defenses against any such action, including that these patents would not be infringed by our product candidates and/or that these patents are not valid. However, if these patents were asserted against us and our defenses to such an action were unsuccessful, unless we obtain a license to these patents, which may not be available on commercially reasonable terms, or at all, we could be liable for damages and precluded from commercializing any product candidates that were ultimately held to infringe these patents, which could have a material adverse effect on our business, financial condition, cash flows or results of operations.
Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S.non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering OBE2109,linzagolix, OBE022 and nolasiban and OBE022 are obtained, once the patent life has expired for a product candidate, we may be open to competition from competitive medications, including biosimilar or generic medications. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such product candidates might expire before or shortly after such product candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing product candidates similar or identical to ours.
Depending upon the timing, duration and conditions of FDA marketing approval of our product candidates, one or more of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Amendments, and similar legislation in the European Union. The Hatch-Waxman Amendments permit a patent term extension of up to five years for a patent covering an approved product as compensation for effective patent term lost during product development and the FDA regulatory review process. However, we may not receive an extension if we fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. Moreover, the length of the extension could be less than we request. If we are unable to obtain patent term extension or the term of any such extension is less than we request, the period
37
during which we can enforce our patent rights for that product will be shortened and our competitors may obtain approval to market competing products sooner. As a result, our revenue from applicable products could be reduced. Further, if this occurs, our competitors may take advantage of our investment in development and trials by referencing our clinical and preclinical data and launch their product earlier than might otherwise be the case.
Intellectual property rights do not necessarily address all potential threats to our business.
Once granted, patents may remain open to opposition, interference,re-examination, post-grant review, inter partes review, nullification or derivation action in court of before patent offices or similar proceedings for a given period after allowance or grant, during which time third parties can raise objections against such grant. In the course of such proceedings, which may continue for a protracted period of time, the patent owner may be compelled to limit the scope of the allowed or granted claims thus attacked, or may lose the allowed or granted claims altogether. In addition, the degree of future protection afforded by our intellectual property rights is uncertain because even granted intellectual property rights have limitations, and may not adequately protect our business. The following examples are illustrative:
Changes in patent laws or patent jurisprudence could diminish the value of patents in general, thereby impairing our ability to protect our product candidates.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involve both technological complexity and legal complexity. Therefore, obtaining and enforcing biopharmaceutical patents is costly, time-consuming and inherently uncertain. In addition, the America Invents Act, or the AIA, was signed into law on September 16, 2011, and many of the substantive changes became effective on March 16, 2013.
An important change introduced by the AIA is that, as of March 16, 2013, the United States transitioned to a“first-to-file” “first-to-file” system for deciding which party should be granted a patent when two or more patent applications are filed by different parties claiming the same invention. A third party that files a patent application in the U.S. Patent and Trademark Office, or USPTO, after that date but before us could therefore be awarded a patent covering an invention of ours even if we had made the invention before it was made by the third party. This will require us to be cognizant going forward of the time from invention to filing of a patent application, but circumstances could prevent us from promptly filing patent applications on our inventions.
38
Among some of the other changes introduced by the AIA are changes that limit where a patentee may file a patent infringement suit and providing opportunities for third parties to challenge any issued patent in the USPTO. This applies to all of our U.S. patents, even those issued before March 16, 2013. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in U.S. federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use the USPTO procedures to invalidate our patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. The AIA and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
The USPTO has developed, in the last few years, regulations and procedures to govern administration of the AIA, and many of the substantive changes to patent law associated with the AIA, and, in particular, the first to file provisions, only became effective on March 16, 2013. Accordingly, it is not clear what, if any, impact the AIA will have on the operation of our business. However, the AIA and its implementation could increase the uncertainties and costs surrounding the prosecution of our or our licensors’ or collaboration partners’ patent applications and the enforcement or defense of our or our licensors’ or collaboration partners’ issued patents, all of which could have an adverse effect on our business and financial condition.
Additionally, the U.S. Supreme Court has ruled on several patent cases in recent years, such as Association forMolecular Pathology v. Myriad Genetics, Inc.(Myriad I),Mayo Collaborative Services v. Prometheus Laboratories, Inc., andAlice Corporation Pty. Ltd. v. CLS Bank International, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future. Similarly, the complexity and uncertainty of European patent laws has also increased in recent years. For example, the April 2010 amendment of the European Patent Convention, which limited the time permitted for filing divisional applications, was subsequently abrogated. This amendment and subsequent abrogation illustrates the uncertainty involved in the prosecution of European patent laws. In addition, the European patent system is relatively stringent in the type of amendments that are allowed during prosecution. These changes could limit our ability to obtain new patents in the future that may be important for our business.
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could have a negative impact on the success of our business.
Our commercial success depends, in part, upon our ability, and the ability of our future collaborators, to develop, manufacture, market and sell OBE2109,linzagolix, OBE022 and nolasiban OBE022 and any future product candidates, if approved, and use our proprietary technologies without alleged or actual infringement, misappropriation or other violation of the patents and proprietary rights of third parties. There have been many lawsuits and other proceedings involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries, including patent infringement lawsuits, interferences, oppositions andre-examination proceedings before the USPTO, and corresponding foreign patent offices. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are developing product candidates. Some claimants may have substantially greater resources than we do and may be able to sustain the costs of complex intellectual property litigation to a greater degree and for longer periods of time than we could. In addition, patent holding companies that focus solely on extracting royalties and settlements by enforcing patent rights may target us. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our product candidates may be subject to claims of infringement of the intellectual property rights of third parties.
We may in the future become party to, or be threatened with, adversarial proceedings or litigation regarding intellectual property rights with respect to OBE2109,linzagolix, OBE022 and nolasiban OBE022 and any future product candidates and technology, including interference or derivation proceedings, post grant review and inter partes review before the USPTO or similar adversarial proceedings or litigation in other jurisdictions. Similarly, we or our licensors or collaborators may initiate such proceedings or litigation against third parties, including to challenge the validity or scope of intellectual property rights controlled by third parties. Third parties may assert infringement claims against us based on existing patents or patents that may be granted in the future, regardless of their merit. There is a risk that third parties may choose to engage in litigation with us to enforce or to otherwise assert their patent rights against us. Even if we believe such claims are without merit, a court of competent jurisdiction could hold that these third-party patents are valid, enforceable and infringed, and the holders
39
of any such patents may be able to block our ability to commercialize such product candidate unless we obtained a license under the applicable patents, or until such patents expire or are finally determined to be invalid or unenforceable. Similarly, if any third-party patents were held by a court of competent jurisdiction to cover aspects of our compositions, formulations, or methods of treatment, prevention or use, the holders of any such patents may be able to block our ability to develop and commercialize the applicable product candidate unless we obtained a license or until such patent expires or is finally determined to be invalid or unenforceable. In either case, such a license may not be available on commercially reasonable terms, or at all. Even if we were able to obtain a license, it could benon-exclusive, thereby giving our competitors access to the same technologies licensed to us. Furthermore, even in the absence of litigation, we may need to obtain licenses from third parties to advance our research or allow commercialization of our product candidates, and we have done so from time to time. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In such an event, we would be unable to further practice our technologies or develop and commercialize any of our product candidates at issue, which could harm our business significantly.
Parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize one or more of our product candidates, if approved. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. Third parties making such claims may have the ability to dedicate substantially greater resources to these legal actions than we or our licensors or collaborators can. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, pay royalties, redesign our infringing products or obtain one or more licenses from third parties, which may be impossible or require substantial time and monetary expenditure.
We may become involved in lawsuits to protect or enforce our patents, the patents of our licensors or our other intellectual property rights, which could be expensive, time-consuming and unsuccessful.
Competitors may infringe or otherwise violate our or our licensors’ patents or misappropriate or otherwise violate our or our licensor’s other intellectual property rights. To counter infringement or unauthorized use, we may be required to file legal claims, which can be expensive and time-consuming. Our agreements with Merck Serono give Merck Serono the first right to control such claims. Therefore, these patents and applications may not be enforced in a manner consistent with the best interests of our business. Our or our licensors’ adversaries in these proceedings may have the ability to dedicate substantially greater resources to prosecuting these legal actions than we or our licensors can. In addition, in an infringement proceeding, a court may decide that a patent of ours or our licensors is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing. The initiation of a claim against a third party may also cause the third party to bring counter claims against us such as claims asserting that our patents are invalid or unenforceable or claims challenging the scope of the intellectual property rights we own or control. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness,non-enablement or lack of statutory subject matter. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant material information from the USPTO, or made a materially misleading statement, during prosecution. Third parties may also raise similar validity claims before the USPTO in post-grant proceedings such as ex partere-examinations, inter partes review, or post-grant review, or oppositions or similar proceedings outside the United States, in parallel with litigation or even outside the context of litigation. The outcome following legal assertions of invalidity and unenforceability is unpredictable. We cannot be certain that there is no invalidating prior art, of which we, our licensors and the patent examiner were unaware during prosecution. For the patents and patent applications that we have licensed, we may have limited or no right to participate in the defense of any licensed patents against challenge by a third party. Our agreements with Kissei and Merck Serono give our licensors the first right to defend such validity challenges. Therefore, these patents and applications may not be defended in a manner consistent with the best interests of our business. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of any future patent protection on our current or future product candidates. Such a loss of patent protection could harm our business. In addition, if the breadth or strength of protection provided by our or our licensors’ patents and patent applications is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates.
We may not be able to prevent, alone or with our licensors, infringement or misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States. Our business could be harmed if in litigation the prevailing party does not offer us a license on commercially reasonable terms. Any
40
litigation or other proceedings to enforce our intellectual property rights may fail, and even if successful, may result in substantial costs and distract our management and other employees.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have an adverse effect on the price of our common shares.
We may not be able to protect our intellectual property rights throughout the world, which could negatively impact our business.
Filing, prosecuting and defending patents covering OBE2109,linzagolix, OBE022, nolasiban OBE022 and any future product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Further, licensing partners may not prosecute patents in certain jurisdictions in which we may obtain commercial rights, thereby precluding the possibility of later obtaining patent protection in these countries. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions
where we have not obtained patent protection to develop their own products and may also export infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our product candidates, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions, whether or not successful, could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license. Furthermore, while we intend to protect our intellectual property rights in our expected significant markets, we cannot ensure that we will be able to initiate or maintain similar efforts in all jurisdictions in which we may wish to market our product candidates. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate, which may have an adverse effect on our ability to successfully commercialize our product candidates in all of our expected significant foreign markets.
Additionally, the requirements for patentability may differ in certain countries, particularly developing countries. For example, unlike other countries, China has a heightened requirement for patentability, and specifically requires a detailed description of medical uses of a claimed drug. In India, unlike the United States, there is no link between regulatory approval of a drug and its patent status. Furthermore, generic or biosimilar drug manufacturers or other competitors may challenge the scope, validity or enforceability of our or our licensors’ patents, requiring us or our licensors to engage in complex, lengthy and costly litigation or other proceedings. Generic or biosimilar drug manufacturers may develop, seek approval for, and launch biosimilar versions of our products. In addition to India, certain countries in Europe and developing countries, including China, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In those countries, we and our licensors may have limited remedies if patents are infringed or if we or our licensors are compelled to grant a license to a third party, which could materially diminish the value of those patents. This could limit our potential revenue opportunities. Accordingly, our and our licensors’ efforts to enforce intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we own or license.
41
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of their former employers or other third parties.
We may employ individuals who were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees, consultants and independent contractors do not use the proprietary information orknow-how of others in their work for us, and we are not currently subject to any claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties, we may in the future be subject to such claims. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Such intellectual property rights could be awarded to a third party, and we could be required to obtain a license from such third party to commercialize our technology or products. Such a license may not be available on commercially reasonable terms or at all. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
We may be subject to claims challenging the inventorship of our patents and other intellectual property.
Although we are not currently experiencing any claims challenging the inventorship of our patents or ownership of our intellectual property, we may in the future be subject to claims that former employees, collaborators or other third parties have an interest in our patents or other intellectual property as an inventor orco-inventor. While it is our policy to require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who, in fact, conceives or develops intellectual property that we regard as our own. For example, the assignment of intellectual property rights may not be self-executing or the assignment agreements may be breached, or we may have inventorship disputes arise from conflicting obligations of consultants or others who are involved in developing our product
candidates. Litigation may be necessary to defend against these and other claims challenging inventorship. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Intellectual property litigation could cause us to spend substantial resources and distract our personnel from their normal responsibilities.
Litigation or other legal proceedings relating to intellectual property claims, with or without merit, is unpredictable and generally expensive and time-consuming and is likely to divert significant resources from our core business, including distracting our technical and management personnel from their normal responsibilities. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common shares. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities.
We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing upon, misappropriating or successfully challenging our intellectual property rights. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have an adverse effect on our ability to compete in the marketplace.
Our inability to protect our confidential information and trade secrets would harm our business and competitive position.
In addition to seeking patents for some of our technology and products, we also rely on trade secrets, including unpatentedknow-how, technology and other proprietary information, to maintain our competitive position. We seek to protect these trade secrets, in part, by entering intonon-disclosure and confidentiality agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants. We also seek to preserve the integrity and confidentiality of our data, trade secrets andknow-how by maintaining physical security of our premises and physical and electronic security of our information technology systems. Monitoring unauthorized uses and disclosures is difficult, and we do not know whether the steps we have taken to protect our
42
proprietary technologies will be effective. We cannot guarantee that our trade secrets and other proprietary and confidential information will not be disclosed or that competitors will not otherwise gain access to our trade secrets. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts both within and outside the United States may be less willing or unwilling to protect trade secrets. If a competitor lawfully obtained or independently developed any of our trade secrets, we would have no right to prevent such competitor from using that technology or information to compete with us, which could harm our competitive position.
Trade secrets andknow-how can be difficult to protect as trade secrets andknow-how will over time be disseminated within the industry through independent development, the publication of journal articles, and the movement of personnel skilled in the art from company to company or academic to industry scientific positions. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent such competitor from using that technology or information to compete with us, which could harm our competitive position. If we are unable to prevent material disclosure of the intellectual property related to our technologies to third parties, we will not be able to establish or maintain a competitive advantage in our market, which could adversely affect our business, results of operations and financial condition. Even if we are able to adequately protect our trade secrets and proprietary information, our trade secrets could otherwise become known or could be independently discovered by our competitors. Competitors could purchase our products and attempt to replicate some or all of the competitive advantages we derive from our development efforts, willfully infringe our intellectual property rights, design around our protected technology or develop their own competitive technologies that fall outside of our intellectual property rights. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, in the absence of patent protection, we would have no right to prevent them, or those to whom they communicate, from using that technology or information to compete with us.
We may not be able to prevent misappropriation of our intellectual property, trade secrets or confidential information, particularly in countries where the laws may not protect those rights as fully as in the United States. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common shares.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated fornon-compliance with these requirements.
Periodic maintenance and annuity fees on any issued patent are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction.Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits,non-payment of fees and failure to properly legalize and submit formal documents. If we or our licensors fail to maintain the patents and patent applications covering our products, our competitors might be able to enter the market, which would harm our business.
Risks Related to Our Business Operations, Employee Matters and Managing Growth
Our future growth and ability to compete depends on retaining our key personnel and recruiting additional qualified personnel.
We are highly dependent on the management, development, clinical, financial and business development experience of Ernest Loumaye, our Chief Executive Officer, Tim Adams, our Chief Financial Officer, Jean-Pierre Gotteland, our Chief Scientific Officer Elke Bestel,and Head of R&D, Wim Souverijns, our Chief Commercial Officer, and Elizabeth Garner, our Chief Medical Officer and Head of Pharmacovigilance, Ben T.G. Tan, our Vice President of Commercial and Business Development, and Fabien Lefebvre de Ladonchamps, our Vice President of Finance.Officer. Each of these officers may currently terminate their employment with us at any time.on short notice. We do not maintain “key person” insurance for any of our executives or employees.
43
Laws and regulations on executive compensation, including legislation in our home country, Switzerland, may restrict our ability to attract, motivate and retain the required level of qualified personnel. In Switzerland, legislation affecting public companies has been passed that, among other things, (1) imposes an annual binding shareholders’ “say on pay” vote with respect to the compensation of executive management, including executive officers and the board of directors, (2) generally prohibits severance, advances, transaction premiums and similar payments to members of our executive management and board of directors, (3) imposes other restrictive compensation practices and (4) requires companies to specify various compensation-related matters in their articles of association, thus requiring them to be approved by a shareholders’ vote. In addition, the competition for qualified personnel in the biopharmaceutical field is intense, and our future success depends upon our ability to attract, retain and motivate highly-skilled scientific, technical and managerial employees. Because the Swiss legislation affecting public companies will apply to operations in the United States and are more onerous and restrictive than comparable laws and regulations applying to U.S. domiciled companies, recruiting and retaining employees in the United States will be even more difficult as compared to companies in the United States. We face competition for personnel from other companies, universities, public and private research institutions and other organizations. If our recruitment and retention efforts are unsuccessful in the future, it may be difficult for us to implement business strategy, which could harm our business.
In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us. If we are unable to continue to attract and retain high quality personnel, our ability to pursue our growth strategy will be limited.
Our future growth depends, in part, on our ability to penetrate foreign markets, where we would be subject to additional regulatory burdens and other risks and uncertainties.
Our future profitability will depend, in part, on our ability to commercialize our product candidates in markets outside of the United States and the European Union. If we commercialize our product candidates in foreign markets, we will be subject to additional risks and uncertainties, including:
Foreign sales of our products could also be adversely affected by the imposition of governmental controls, political and economic instability, trade restrictions and changes in tariffs.
44
The novel coronavirus outbreak could impact our business.
In December 2019, a novel strain of coronavirus was reported in China. This virus has now spread to numerous other countries, including Switzerland and the United States. While we do not currently have significant operations in geographical locations where the coronavirus was initially reported to be most prevalent, we recently entered into a sublicense agreement with Yuyuan, which is based in China. We cannot reasonably estimate at this time the impact, if any, that the coronavirus may have on our collaboration with Yuyuan, our business or operations. The extent to which the coronavirus impacts our business will depend on future developments, which are highly uncertain and cannot be predicted, including new information which may emerge concerning the severity of the coronavirus and the actions to contain the coronavirus or treat its impact including on financial markets or otherwise.
We expect to expand our development and regulatory capabilities and potentially implement sales, marketing and distribution capabilities, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
As of December 31, 2016,2019, we had 2753 employees. As our clinical development progresses, we expect tomay experience growth in the number of our employees and the scope of our operations, particularly in the areas of clinical operations, regulatory affairs and, if any of our product candidates receives marketing approval, sales, marketing and distribution. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Due to our limited financial resources and the limited experience of our management team in managing a company with such anticipated growth, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
Our employees, independent contractors, consultants, commercial collaborators, principal investigators, CROs and vendors may engage in misconduct or other improper activities, includingnon-compliance with regulatory standards and requirements.
We are exposed to the risk that our employees, independent contractors, consultants, commercial collaborators, principal investigators, CROs and vendors may engage in fraudulent conduct or other illegal activity. Misconduct by these parties could include intentional, reckless or negligent conduct or unauthorized activities that violates (1) the laws and regulations of the FDA, the EMA and other similar regulatory authorities, including those laws requiring the reporting of true, complete and accurate information to such authorities, (2) manufacturing standards, (3) federal and state data privacy, security, fraud and abuse and other healthcare laws and regulations in the United States and abroad and (4) laws that require the true, complete and accurate reporting of financial information or data. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-
dealingself-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Misconduct by these parties could also involve the improper use of individually identifiable information, including information obtained in the course of clinical trials, creating fraudulent data in our preclinical studies or clinical trials or illegal misappropriation of product candidates, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a code of business conduct and ethics, but it is not always possible to identify and deter misconduct by employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. Additionally, we are subject to the risk that a person or government could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant civil, criminal and administrative penalties, including damages, fines, disgorgement, individual imprisonment, exclusion from participation in government healthcare programs, such as Medicare and Medicaid, additional integrity oversight and reporting obligations, contractual damages, reputational harm and the curtailment or restructuring of our operations.
45
Risks Related to Our Common Shares
The price of our common shares may be volatile and may fluctuate due to factors beyond our control.
The share price of our common shares has fluctuated, and is likely to continue to fluctuate substantially. The market price of our securities depends on a number of factors, including those described in this “Risk Factors” section, many of which are beyond our control and may not be related to our operating performance.
The market price of our securities may fluctuate significantly in response to numerous factors, many of which are beyond our control, including
changes in the structure of healthcare payment systems;
general market or regulatory conditions in the pharmaceutical industry or in the economy as a whole; or
These and other market and industry factors may cause the market price and demand for our securities to fluctuate substantially, regardless of our actual operating performance, which may limit or prevent investors from selling their common shares at or above the price paid for the shares and may otherwise negatively affect the liquidity of our common shares. In addition, the stock market in general, and biopharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies.
Some companies that have experienced volatility in the trading price of their shares have been the subject of securities class action litigation. Any lawsuit to which we are a party, with or without merit, may result in an unfavorable judgment. We also may decide to settle lawsuits on unfavorable terms. Any such negative outcome could result in payments of substantial damages or fines, damage to our reputation or adverse changes to our offerings or business practices. Defending against litigation is costly and time-consuming, and could divert our management’s attention and resources. Furthermore, during the course of litigation, there could be negative public announcements of the results of hearings, motions or other interim proceedings or developments, which could have a negative effect on the market price of our common shares.
46
Concentration of ownership of our common shares among our existing executive officers, directors and principal shareholders may prevent new investors from influencing significant corporate decisions.
Based upon our common shares outstanding as of December 31, 2016, upon the closing of our initial public offering in January 2017,2019, our executive officers, directors and shareholders who owned more than 5% of our outstanding common shares beneficially own, in the aggregate, approximately 73%31% of our outstanding common shares. This beneficial ownership percentage does not include the number of common shares that our 5% shareholders and their affiliated entities purchased in our initial public offering. These shareholders, acting together, will be able to significantly influence all matters requiring shareholder approval, including the election and removal of directors and approval of any merger, consolidation or sale of all or substantially all of our assets.
Some of these persons or entities may have interests different than our other shareholders. For example, because many of these shareholders purchased their shares at prices substantially below the price at which shares were sold in our initial public offering and have held their shares for a longer period, they may be more interested in selling our company to an acquirer than other investors, or they may want us to pursue strategies that deviate from the interests of other shareholders.
Future sales, or the possibility of future sales, of a substantial number of our common shares could adversely affect the price of our common shares.
As of December 31, 2016, 23,181,2622019, 48,567,605 common shares were issued and outstanding. Such share number does not include the 6,450,000 common shares that were sold in connection with our initial public offering in January 2017. Sales ofIf a substantial number of our commonthese shares or the perception that such sales will occur, could cause a decline in the market price of our common shares. Approximately 94% of the common shares outstanding as of December 31, 2016 are held by existing shareholders. A significant portion of these common shares are subject to certainlock-up agreements entered in connection with our initial public offering. If, after the end of suchlock-up agreements, these shareholders sell substantial amounts of common shareswere sold in the public market, or the market perceives that such sales may occur, the market price of our common shares and our ability to raise capital through an issue of equity securities in the future could be adversely affected.
We have also entered into a registration rights agreement pursuant to which we have agreed under certain circumstances to file a registration statement to register the resale of the common shares held by certain of our existing shareholders, as well as to cooperate in certain public offerings of such common shares. Once we register these common shares, they can be freely sold in the public market upon issuance, subject to volume limitations applicable to affiliates and thelock-up agreements.
In addition, we have adopted a newan omnibus equity incentive plan, under which we have the discretion to grant a broad range of equity-based awards to eligible participants. We have filed a registration statement with the SEC to register the common shares that may be issued under our equity incentive plan. The common shares subject to outstanding options under our equity incentive plan, common shares reserved for future issuance under our equity incentive plan and common shares subject to outstanding warrants will become eligible for sale in the public market in the future, subject to certain legal and contractual limitations. Sales of a large number of the shares issued under these plans in the public market could have an adverse effect on the market price of our securities.
We do not expect to pay dividends in the foreseeable future.
We have not paid any dividends since our incorporation. Even if future operations lead to significant levels of distributable profits, we currently intend that any earnings will be reinvested in our business and that dividends will not be paid until we have an established revenue stream to support continuing dividends. The proposal to pay future dividends to shareholders will in addition effectively be at the discretion of our board of directors and shareholders after taking into account various factors including our business prospects, cash requirements, financial performance and new product development. In addition, payment of future dividends is subject to certain limitations pursuant to Swiss law or by our articles of association. See “Item 10.B—Memorandum and Articles of Association.” Accordingly, investors cannot rely on dividend income from our common shares and any returns on an investment in our common shares will likely depend entirely upon any future appreciation in the price of our common shares.
We are a Swiss stock corporation. The rights of our shareholders may be different from the rights of shareholders in companies governed by the laws of U.S. jurisdictions.
We are a Swiss stock corporation. Our corporate affairs are governed by our articles of association and by the laws governing companies incorporated in Switzerland. The rights of our shareholders and the responsibilities of members of our board of directors may be different from the rights and obligations of shareholders and directors of companies governed by the U.S. laws. In the performance of its duties, our board of directors is required by Swiss law to consider the interests of our company, our shareholders, our employees and other stakeholders, in all cases with due observation of the principles of reasonableness and fairness. It is possible that some of these parties will have interests that are different from, or in addition to, your interests as a shareholder. Swiss corporate law limits the ability of our shareholders to challenge resolutions made or other actions taken by our board of directors in court. Our shareholders generally are not permitted to file a suit to reverse a decision or an action taken by our board of directors but are instead only permitted to seek damages for breaches of fiduciary duty. As a matter of Swiss law, shareholder claims against a member of our board of directors for breach of fiduciary duty would have to be brought in Geneva, Switzerland, or where the relevant member of our board of directors is domiciled. In addition, under Swiss law, any claims by our shareholders against us must be brought exclusively in Geneva, Switzerland. Class actions and derivative actions as such are not available under Swiss law. In addition, Swiss corporation law restricts our
47
ability to implement rights plans or U.S.-style “poison pills.” Since the Swiss takeover rules do not apply to us, our ability to resist an unsolicited takeover attempt may be limited. See “Item 10.B—Memorandum and Articles of Association.” Also, there can be no assurance that Swiss law will not change in the future or that it will serve to protect investors in a similar fashion afforded under corporate law principles in the United States, which could adversely affect the rights of investors.
Our common shares are not listedtraded on more than one market and this may result in Switzerland,price variations and adversely affect the liquidity and value of our home jurisdiction. As a result, certain Swiss law provisions designed to protect shareholders in the event of a public takeover offer or change of control transaction will not apply.common shares.
The Swiss rules that require investors to disclose their interest in our company if they reach, exceed or fall below certain ownership thresholds only applies to issuers that have a listing for their equity securities in Switzerland. Since ourOur common shares are listed exclusively on The NASDAQthe Nasdaq Global Select Market a U.S. market,and the disclosure obligations generally applicable to holders of significant interestsSIX Swiss Exchange. Trading in our common shares on these markets takes place in different currencies (U.S. dollars on the Nasdaq Global Select Market and Swiss corporations do not apply to us. Likewise,Francs on the SIX Swiss takeover regime only applies to issuers that have a listing for their shares in Switzerland. This regime does consequently not apply to us or our shareholders. In particular, the Swiss rules that oblige any person or group of persons that acquires more than one third of a company’s voting rights to submit a cash offer for all the outstanding listed equity securities of the relevant companyExchange), and at a minimum price do not apply to us. Since Swiss corporation law restricts our ability to implement rights plans or U.S.-style “poison pills,” our ability to protect minority shareholdersdifferent times (resulting from different time zones, different trading days and different public holidays in the eventUnited States and Switzerland). The trading prices of our common shares on these two markets may differ due to these and other factors. Any decrease in the price of our common shares on the SIX Swiss Exchange could cause a changedecrease in the trading price of control transaction may be limited.our common shares on the Nasdaq Global Select Market. Investors could seek to sell or buy our common shares to take advantage of any price differences between the markets through a practice referred to as arbitrage. Any arbitrage activity could create unexpected volatility in both our share prices on one exchange and the common shares available for trading on the other exchange.
U.S. shareholders may not be able to obtain judgments or enforce civil liabilities against us or our executive officers or members of our board of directors.
We are a Swiss stock corporation, and our jurisdiction of incorporation is Geneva, Switzerland. Moreover, a number of our directors and executive officers are not residents of the United States, and all or a substantial portion of the assets of such persons are located outside the United States. As a result, it may not be possible for investors to effect service of process within the United States upon us or upon such persons or to enforce against them judgments obtained in U.S. courts, including judgments in actions predicated upon the civil liability provisions of the federal securities laws of the United States. We have been advised by our Swiss counsel that there is doubt as to the enforceability in Switzerland of original actions, or in actions for enforcement of judgments of U.S. courts, of civil liabilities to the extent predicated upon the federal and state securities laws of the United States. Original actions against persons in Switzerland based solely upon the U.S. federal or state securities laws are governed, among other things, by the principles set forth in the Swiss Federal Act on International Private Law. This statute provides that the application of provisions ofnon-Swiss law by the courts in Switzerland shall be precluded if the result is incompatible with Swiss public policy. Also, mandatory provisions of Swiss law may be applicable regardless of any other law that would otherwise apply.
Switzerland and the United States do not have a treaty providing for reciprocal recognition and enforcement of judgments in civil and commercial matters. The recognition and enforcement of a judgment of the courts of the United States in Switzerland is governed by the principles set forth in the Swiss Federal Act on Private International Law. This statute provides in principle that a judgment rendered by anon-Swiss court may be enforced in Switzerland only if:
Our status as a Swiss stock corporation means that our shareholders enjoy certain rights that may limit our flexibility to raise capital, issue dividends and otherwise manage ongoing capital needs.
Swiss law reserves for approval by shareholders certain corporate actions over which a board of directors would have authority in some other jurisdictions. For example, the payment of dividends and cancellation of treasury shares must be approved by shareholders. Swiss law also requires that our shareholders themselves resolve to, or authorize our board of directors to, increase our share capital. While our shareholders may authorize share capital that can be issued by our board of directors without additional shareholder approval, Swiss law limits this authorization to 50% of the issued share capital at the time of the authorization. The authorization, furthermore, has a limited duration of up to two years and must be renewed by the shareholders from time to time thereafter in order to be available for raising capital. Additionally, subject to specified
48
exceptions, including exceptions explicitly described in our articles of association, Swiss law grantspre-emptive rights to existing shareholders to subscribe for new issuances of shares. Swiss law also does not provide as much flexibility in the various rights and regulations that can attach to different classes of shares as do the laws of some other jurisdictions. These Swiss law requirements relating to our capital management may limit our flexibility, and situations may arise where greater flexibility would have provided benefits to our shareholders. See “Item 10.B—Memorandum and Articles of Association.”
We are a foreign private issuer and, as a result, are not subject to U.S. proxy rules and are subject to Exchange Act reporting obligations that, to some extent, are more lenient and less frequent than those of a U.S. domestic public company.
We report under the Securities Exchange Act of 1934, as amended, or the Exchange Act, as anon-U.S. company with foreign private issuer status. Because we qualify as a foreign private issuer under the Exchange Act and although we are subject to Swiss laws and regulations with regard to such matters and intend to furnish quarterly financial information to the SEC, we are exempt from certain provisions of the Exchange Act that are applicable to U.S. domestic public companies, including: (1) the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act; (2) the sections of the Exchange Act requiring insiders to file public reports of their share ownership and trading activities and liability for insiders who profit from trades made in a short period of time; and (3) the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form10-Q containing unaudited financial and other specified information, or current reports on Form8-K, upon the occurrence of specified significant events. In addition, foreign private issuers are not required to file their annual report on Form20-F until four months after the end of each financial year, while U.S. domestic issuers that are accelerated filers are required to file their annual report on Form10-K within 75 days after the end of each fiscal year. Foreign private issuers are also exempt from the Regulation Fair Disclosure, aimed at preventing issuers from making selective disclosures of material information. As a result of the above, you may not have the same protections afforded to shareholders of companies that are not foreign private issuers.
As a foreign private issuer and as permitted by the listing requirements of NASDAQ,Nasdaq, we will have the option to follow certain home country governance practices rather than the corporate governance requirements of NASDAQ.Nasdaq.
We are a foreign private issuer. As a result, in accordance with NASDAQNasdaq Listing Rule 5615(a)(3), we may choose to comply with home country governance requirements and certain exemptions thereunder rather than complying with certain of the corporate governance requirements of NASDAQ.Nasdaq.
Swiss law does not require that a majority of our board of directors consist of independent directors. Our board of directors therefore may include fewer independent directors than would be required if we were subject to NASDAQNasdaq Listing Rule 5605(b)(1). In addition, we are not subject to NASDAQNasdaq Listing Rule 5605(b)(2), which requires that independent directors regularly have scheduled meetings at which only independent directors are present.
Although Swiss law also requires that we set up a compensation committee, we may follow home country requirements with respect to such committee.
Our articles of association provide for an independent proxy elected by our shareholders, who may represent our shareholders at a general meeting of shareholders, and we must provide shareholders with an agenda and other relevant documents for the general meeting of shareholders. However, Swiss law does not have a regulatory regime for the solicitation of proxies and company solicitation of proxies is prohibited for public companies in Switzerland, thus our practice may vary from the requirement of NASDAQNasdaq Listing Rule 5620(b), which sets forth certain requirements regarding the solicitation of proxies. Furthermore, in accordance with Swiss law and generally accepted business practices, our articles of association do not provide quorum requirements generally applicable to general meetings of shareholders. Our practice thus varies from the requirement of NASDAQNasdaq Listing Rule 5620(c), which requires an issuer to provide in its bylaws for a generally applicable quorum, and that such quorum may not be less thanone-third of the outstanding voting stock.
For an overview of our corporate governance principles, see “Item 10.B—Memorandum and Articles of Association.” As a result of the above, you may not have the same protections afforded to shareholders of companies that are not foreign private issuers.
We may lose our foreign private issuer status, which would then require us to comply with the domestic reporting requirements of the Exchange Act and cause us to incur significant legal, accounting and other expenses.
We are currently a foreign private issuer and therefore, are not be required to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act applicable to U.S. domestic issuers. In order to maintain our current
49
status as a foreign private issuer, either (1) a majority of our common shares must be either directly or indirectly owned of record bynon-residents of the United States or (2)(a) a majority of our executive officers or directors may not be United States citizens or residents, (b) more than 50% of our assets cannot be located in the United States and (c) our business must be administered principally outside the United States. If we lost this status, we would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. We may also be required to make changes in our corporate governance practices in accordance with various SEC and stock exchange rules. The regulatory and compliance costs to us under U.S. securities laws if we are required to comply with the reporting requirements applicable to a U.S. domestic issuer may be significantly higher than the cost we would incur as a foreign private issuer. As a result, we expect that a loss of foreign private issuer status would increase our legal and financial compliance costs and would make some activities highly time-consuming and costly. If we lose our foreign private issuer status and are unable to devote adequate funding and the resources needed to maintain compliance with U.S. securities laws, while continuing our operations, we could be forced to deregister with the SEC. A deregistration would substantially reduce or effectively terminate the trading of our securities in the United States. We also expect that if we were required to comply with the rules and regulations applicable to U.S. domestic issuers, it would make it more difficult and expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These rules and regulations could also make it more difficult for us to attract and retain qualified members of our board of directors.
WhileIf we do not expect to be are a “passive foreign investment company,” or PFIC, for U.S. federal income tax purposes for our current year and future years, we maythere could be or become a PFIC, which could result in adverse U.S. federal income tax consequences to U.S. holders.Holders.
AlthoughBased on our analysis of our income, assets, activities and market capitalization for our taxable year ending December 31, 2019, we do not expect to bebelieve that we were a PFIC for our taxable year ending December 31, 2016,2019. However, our operations generate very limited amounts of non-passive income. Until we generate sufficient revenue from active licensing and other non-passive sources, there is a risk that we will be a PFIC under the PFIC income test. Moreover, we hold a substantial amount of cash and cash equivalents. Because the calculation of the value of our assets may be based in part on the value of our common shares, the value of which may fluctuate considerably, our PFIC status may change from year to year and it is difficult to predict whether we will be a PFIC under the PFIC asset test for the current year or any future year. Therefore, we have not yet made any determination as to our expected PFIC status for the current year. Even if we determine that we are not a PFIC after the close of a taxable year, there can be no assurance that the Internal Revenue Service, or IRS, will agree with our conclusion. Because PFIC status is a fact specific determination, and generally cannot be made until the close of the taxable year in question, no assurance can be given that we will not be a PFIC for our current taxable year and that we will not be a PFIC in future taxable years. Our United States counsel expresses no opinion with respect to our PFIC status for prior years, we could bethe current taxable year or become a PFIC. any future years.
Under the Internal Revenue Code of 1986, as amended, we will be a PFIC for any taxable year in which (1) 75% or more of our gross income consists of passive income or (2) 50% or more of the average quarterly value of our assets consists of assets that produce, or are held for the production of, passive income. For purposes of the above calculations, anon-U.S. corporation that directly or indirectly owns at least 25% by value of the shares of another corporation is treated as if it held its proportionate share of the assets and received directly its proportionate share of the income of such other corporation. Passive income generally includes dividends, interest, certain rents and royalties, and capital gains.
If we are a PFIC for any taxable year during which a U.S. holderHolder (as defined in “Item 10.E—Taxation”) holds our shares, the U.S. holderHolder may be subject to adverse tax consequences, including (1) the treatment of all or a portion of any gain on the disposition of our common shares as ordinary income, (2) the addition of an interest charge to the tax on such gain and (3) the obligation to comply with certain reporting requirements.
Each U.S. holderHolder is strongly urged to consult its tax advisor regarding these issues. For further discussion of the adverse U.S. federal income tax consequences in the event we are classified as a PFIC, see “Item 10.E—Taxation.”
If a United States person is treated as owning at least 10% of our common shares, such holder may be subject to adverse U.S. federal income tax consequences.
If a U.S. Holder is treated as owning (directly, indirectly or constructively) at least 10% of the value or voting power of our common shares, such U.S. Holder may be treated as a “United States shareholder” with respect to each “controlled foreign corporation” in our group (if any). Because our group includes at least one U.S. subsidiary (ObsEva USA Inc.), our Irish subsidiary (ObsEva Ireland Limited) and any other non-U.S. subsidiaries we form or acquire in the future, may be treated as controlled foreign corporations (regardless of whether ObsEva SA is treated as a controlled foreign corporation). A United States shareholder of a controlled foreign corporation may be required to report annually and include in its U.S. taxable income its pro rata share of “Subpart F income,” “global intangible low-taxed income” and investments in U.S. property by
50
controlled foreign corporations, regardless of whether we make any distributions. An individual that is a United States shareholder with respect to a controlled foreign corporation generally would not be allowed certain tax deductions or foreign tax credits that would be allowed to a United States shareholder that is a U.S. corporation. Failure to comply with these reporting obligations may subject you to significant monetary penalties and may prevent the statute of limitations from starting with respect to your U.S. federal income tax return for the year for which reporting was due. We cannot provide any assurances that we will assist investors in determining whether any of our non-U.S. subsidiaries are treated as a controlled foreign corporation or whether such investor is treated as a United States shareholder with respect to any of such controlled foreign corporations. Further, we cannot provide any assurances that we will furnish to any United States shareholders information that may be necessary to comply with the aforementioned reporting and tax payment obligations. U.S. Holders should consult their tax advisors regarding the potential application of these rules to their investment in our common shares.”
As a result of changes in tax laws, treaties, rulings, regulations or agreements, or their interpretation, of Switzerland or any other country in which we operate, the loss of a major tax dispute or a successful challenge to our operating structure, intercompany pricing policies or the taxable presence of our key subsidiaries in certain countries, or other factors, our effective income tax rates may increase in the future, which could adversely affect our net income and cash flows.
We operate in multiple jurisdictions and our profits are taxed pursuant to the tax laws of these jurisdictions. Our effective income tax rate may be affected by changes in or interpretations of tax laws, treaties, rulings, regulations or agreements in any given jurisdiction, utilization of net operating loss and tax credit carryforwards, changes in geographical allocation of income and expense, and changes in management’s assessment of matters such as the realizability of deferred tax assets. In the past, we have experienced fluctuations in our effective income tax rate. Our effective income tax rate in a given fiscal year reflects a variety of factors that may not be present in the succeeding fiscal year or years. There is no assurance that our effective income tax rate will not change in future periods.
We file Swiss andnon-Swiss tax returns. We are frequently subject to tax audits, examinations and assessments in various jurisdictions. If any tax authority successfully challenges our operational structure, intercompany pricing policies or the taxable presence of our key subsidiaries in certain countries, if the terms of certain income tax treaties are interpreted in a manner that is adverse to our structure, or if we lose a material tax dispute in any country, our effective income tax rate could increase. A material assessment by a governing tax authority could adversely affect our profitability. If our effective income tax rate increases in future periods, our net income and cash flows could be adversely affected.
Future changes to tax laws could materially adversely affect our company and reduce net returns to our shareholders.
New income, sales, use or other tax laws, statutes, rules, regulations or ordinances could be enacted at any time, which could adversely affect our business operations and financial performance. Further, existing tax laws, statutes, rules, regulations or ordinances could be interpreted, changed, modified or applied adversely to us. For example, legislation enacted in 2017 informally titled the Tax Cuts and Jobs Act or (the “Tax Act”) enacted many significant changes to the U.S. tax laws. Future guidance from the Internal Revenue Service and other tax authorities with respect to the Tax Act may affect us, and certain aspects of the Tax Act could be repealed or modified in future legislation. In addition, it is uncertain if and to what extent various states will conform to the Tax Act or any newly enacted federal tax legislation. Changes in corporate tax rates, the realization of net deferred tax assets relating to our operations, the taxation of foreign earnings, and the deductibility of expenses under the Tax Act or future reform legislation could have a material impact on the value of our deferred tax assets, could result in significant one-time charges, and could increase our future U.S. tax expense.
On May 19, 2019, the Swiss Federal Act on Tax Reform and AHV Financing, or TRAF, was approved by a public vote. The main part of the TRAF provisions entered into force, at the Swiss federal level, on January 1, 2020. The TRAF, among other things, contains significant changes to corporate taxation, including (i) the elimination of certain preferential tax regimes at both the federal and cantonal levels with certain transitional rules, (ii) the introduction of a mandatory cantonal patent box in line with the standards of the OECD, (iii) the introduction of R&D super deductions for R&D costs incurred in Switzerland, (iv) the introduction of an optional notional interest deduction in cantons with higher tax rates, (v) the introduction of a maximum relief limitation and (vi) the introduction of a limitation of the ability of Swiss resident corporations listed on a Swiss stock exchange to distribute dividends out of capital contributions exempt from withholding tax (these companies can only pay 50% of their annual dividend distributions out of capital contribution reserves). In addition, the cantons will implement new tax rates. The measures should enter into force in 2020. As a result, we expect that the standard combined (federal, cantonal, communal) effective corporate income tax rate, except for dividend income for which we could claim a participation exemption, will be approximately 14% for 2020 in the canton of Geneva. However, the standard effective corporate income tax rate in the canton of Geneva can change from time to time.
51
Our ability to use our net operating loss carryforwards to offset future taxable income may be subject to certain limitations.
Our U.S. net operating loss, or NOL, carryforwards generated in tax years ending on or prior to December 31, 2017, are only permitted to be carried forward for 20 years under applicable U.S. tax law. Under the Tax Act, our federal NOLs generated in tax years ending after December 31, 2017, may be carried forward indefinitely, but the deductibility of such federal NOLs generated in tax years beginning after December 31, 2017, is limited. It is uncertain if and to what extent various states will conform to the Tax Act. In addition, under Sections 382 and 383 of the Code and corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50% change, by value, in its equity ownership over a three-year period, the corporation’s ability to use its pre-change NOL carryforwards and other pre-change U.S. tax attributes to offset its post-change income or taxes may be limited. We may experience ownership changes in the future as a result of subsequent shifts in our stock ownership.
As a result, our pre-2018 NOL carryforwards may expire prior to being used, and our NOL carryforwards generated in 2018 and thereafter will be subject to a percentage limitation. In addition, it is possible that we have in the past undergone, and in the future may undergo, additional ownership changes that could limit our ability to use all of our pre-change NOLs and other pre-change tax attributes to offset our post-change income or taxes. Similar provisions of state tax law may also apply to limit our use of accumulated state tax attributes. In addition, at the state level, there may be periods during which the use of NOLs is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed. As a result, we may be unable to use all or a material portion of our NOLs and other tax attributes, which could adversely affect our future cash flows.
Our NOL carryforwards are only permitted to be carried forward for seven years under applicable Swiss tax law. As a result, our NOL carryforwards may expire prior to being used and we may be unable to use all or a material portion of our NOLs, which could adversely affect our future cash flows.
We are an “emerging growth company,” and we cannot be certain if the reduced reporting requirements applicable to “emerging growth companies” will make our common shares less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. For as long as we continue to be an “emerging growth company,” we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies,” including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. As an “emerging growth company,” we are required to report only two years of financial results and selected financial data compared to three and five years, respectively, for comparable data reported by other public companies. We may take advantage of these exemptions until we are no longer an “emerging growth company.” We could be an “emerging growth company” for up to five years, although circumstances could cause us to lose that status earlier, including if the aggregate market value of our common shares held bynon-affiliates exceeds $700 million as of any June 30 (the end of our second fiscal quarter) before that time, in which case we would no longer be an “emerging growth company” as of the following December 31 (our fiscal year end). We cannot predict if investors will find our common shares less attractive because we may rely on these exemptions. If some investors find our common shares less attractive as a result, there may be a less active trading market for our common shares and the price of our common shares may be more volatile.
52
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. As a result, shareholders could lose confidence in our financial and other public reporting, which would harm our business and the trading price of our common shares.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. In addition, any testing by us conducted in connection with Section 404, or any subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses or that may require prospective or retroactive changes to our financial statements or identify other areas for further attention or improvement. Inferior internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common shares.
We will beare required to disclose changes made in our internal controls and procedures on a quarterly basis and our management will beis required to assess the effectiveness of these controls annually. However, for as long as we are an “emerging growth company” under the JOBS Act, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal controls over financial reporting pursuant to Section 404. We could be an “emerging growth company” for up to five years. An independent assessment of the effectiveness of our internal controls could detect problems that our management’s assessment might not. Undetected material weaknesses in our internal controls could lead to financial statement restatements and require us to incur the expense of remediation.
If equity research analysts do not publish research or reports, or publish unfavorable research or reports, about us, our business or our market, our share price and trading volume could decline.
The trading market for our common shares is influenced by the research and reports that equity research analysts publish about us or our business, our market and our competitors. As a newlynew public company, we have only limited research coverage by equity research analysts. Equity research analysts may elect not to initiate or continue to provide research coverage of our common shares, and such lack of research coverage may adversely affect the market price of our common shares. Even if we have equity research analyst coverage, we will not have any control over the analysts or the content and opinions included in their reports. The price of our shares could decline if one or more equity research analysts downgrade our shares or issue other unfavorable commentary or research. If one or more equity research analysts ceases coverage of our company or fails to publish reports on us regularly, demand for our shares could decrease, which in turn could cause our share price or trading volume to decline.
Item 4. Information on the Company.
A. History and Development of the Company
Our legal and commercial name is ObsEva SA. We are a Swiss stock corporation (société anonyme) organized under the laws of Switzerland. We were formed in 2012 with an indefinite duration. We are currently registered inPlan-les-Ouates, Geneva, Switzerland. Our principal executive offices are located at Chemin des Aulx, 12, 1228Plan-les-Ouates, Geneva, Switzerland. Our telephone number is +41 22 552 38 40. Investors should contact us for any inquiries through the address and telephone number of our principal executive office.office, or via our U.S. office at 1 Financial Center in Boston, MA, telephone number +1 (857) 972-9366. We maintain a web sitewebsite atwww.obseva.com. The reference to our website is an inactive textual reference only and the information contained in, or that can be accessed through, our web site is not a part of this Annual Report on Form 20-F. The SEC maintains a website at 20-F.www.sec.gov that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC.
Our actual capital expenditures for the years ended December 31, 2016, 20152019, 2018 and 20142017 amounted to $45$5.0 million, $105 thousand $10.0 million, and $0.1$5.3 million respectively. These capital expenditures primarily consisted of purchase of furniture and office equipment, except forand payments of $5.0 million each to Kissei in 2019 and 2017 upon execution of the year 2015 when $10.0 million were used to acquire the OBE2109 license from Kissei Pharmaceutical.linzagolix licensing agreement. We expect our capital expenditures to increase in absolute terms in the near term in line with our previous expenditures, as we continue to advance our drug development programs and grow our operations. We anticipate our capital expenditures in 20172020 to be financed from our existing cash and cash equivalents, including the net proceeds from our initial public offering.equivalents.
We are a clinical-stage biopharmaceutical company focused on the development and commercialization of novel therapeutics for serious conditions that compromise a woman’s reproductive health and pregnancy. We are advancing a pipeline of orally-administeredorally-
53
administered innovative new chemical entities, or NCEs, for the treatment of symptoms associated with endometriosis and uterine fibroids, treatment of preterm labor and improvement of clinical pregnancy and live birth rates in women undergoing IVF and treatment of preterm labor.IVF. We have assembled a strong management team with extensive experience in successfully developing and commercializing therapeutics in our target market. Our goal is to build the leading women’s reproductive health and pregnancy company focused on conditions where current treatment options are limited and significant unmet needs exist.
We were founded in November 2012 by former executives of PregLem SA, or PregLem, a Swiss-based specialty biopharmaceutical company dedicated to the development and commercialization of innovative drugs for women’s reproductive medicine. While at PregLem, our senior management team collaborated in the clinical development and commercialization of several women’s reproductive health therapeutics, including Esmya (ulipristal acetate) for the treatment of uterine fibroids. PregLem was subsequently acquired by Gedeon Richter in 2010. We believe we will be able to leverage our senior management team’s long-standing experience working together and with key opinion leaders, patient groups, payors, reproductive health networks, fertility clinics, obstetricians and gynecologists, or OB/GYNs, nurses and pharmacists to identify,in-license or acquire, develop and commercialize product candidates. We are merging our passion for, and extensive experience in, the field of women’s reproductive health and pregnancy, to develop therapeutics that can help women lead healthier and more healthy and fulfilling lives.
We are focused on providing therapeutic solutions for women between the ages of 15 and 49 who suffer from reproductive health conditions that affect their quality of life, ability to conceive or that complicate pregnancy and the health of newborns. There are millions of women of reproductive age affected by conditions such as endometriosis, uterine fibroids and preterm labor, or that require IVF to conceive. We believe the efficacy of current treatment options is limited and creates a significant unmet need for improved therapeutics for these women. The graphic below depicts the segments and associated characteristics of the therapeutic market for women’s reproductive health products:
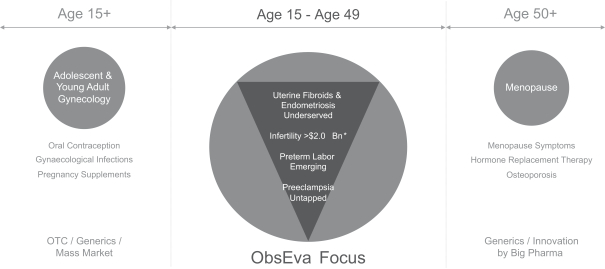
Our portfolio currently consists of threein-licensed NCEs in clinical development for four indications intended to address areas that we believe present significant unmet medical needs:
Linzagolix for the treatment of pain associated with endometriosis and HMB associated with uterine fibroids. We are developing linzagolix as a novel, oral GnRH receptor antagonist, for the treatment of pain associated with endometriosis and HMB associated with uterine fibroids in pre-menopausal women. Endometriosis is an often painful disorder in which the tissue that normally lines the inside of the uterus, called the endometrium, grows outside of the uterus, causing monthly bleeding and chronic inflammatory reactions inside the abdomen that may result in ovarian cyst formation, scar tissue and adhesions. The symptoms of endometriosis include significant pain during menstrual periods (dysmenorrhea), chronic pelvic pain, pain during intercourse (dyspareunia), pain during defecation (dyschezia), excessive menstrual bleeding and infertility. These symptoms can impact general physical, mental and social well-being. We believe that approximately 5 million women in the United States were diagnosed and being treated annually for endometriosis and that the majority of those women experience significant pain during menstrual periods as well as non-menstrual pelvic pain that is not associated with their menstrual periods. Uterine fibroids are common non-cancerous tumors that develop in the muscular wall of the uterus and have disabling symptoms such as HMB and pain. According to a study published in the American Journal of Obstetrics & Gynecology in 2003, uterine fibroids affect an estimated 20 to 40% of women over the age of 30 in the United States based on clinical cases and women who undergo treatment.
In previous early stage Phase 1 and Phase 2 clinical trials, OBE2109linzagolix was observed to have a linear PK profile, a predictable dose-dependent suppression of estradiol and a dose range that was well-tolerated and provided symptom relief. Aimed at addressing the need of the largest possible population in each indication, our clinical trials for both of these indications are designed to assess and potentially support the registration of two regimens of administrations for OBE2109linzagolix i.e. (i) a moderate dose of OBE2109linzagolix without hormonal add-back therapy and (ii) a high dose of OBE2109linzagolix with hormonal add-back therapy. Add-back therapy (ABT) consists of co-administering estrogen and progestin with a high dose of GnRH receptor antagonist to compensate for the severe depletion of estrogen levels and thus prevent the side effects of full estrogen suppression such as hot flushes, vaginal dryness, and loss of bone mineral density (BMD).
We are currently conductinghave completed a multiple-dose, placebo-controlled Phase 2b clinical trial of OBE2109linzagolix in approximately 330 patients with endometriosis, the EDELWEISS 1 trial, which met its primary endpoint at Month 3 and showed maintenance or increase of the effect of linzagolix on endometriosis-associated pain and favorable safety up to 52 weeks of treatment and up to 24 weeks after end of treatment. The results were presented at the 75th American Society of Reproductive Medicine (ASRM) Scientific Congress & Expo in October 2019.
We believe the BMD results support our plan to pursue further development of two doses of linzagolix for the treatment of endometriosis, including a 75 mg once daily dose without ABT, and a 200 mg once daily dose in
54
combination with ABT (1mg E2 / 0.5mg NETA, or Activella). Based on the results of our EDELWEISS 1 trial, we believe nearly three out of four patients with moderate to severe endometriosis-associated pain may achieve significant symptom relief with linzagolix 75 mg once daily with no need for ABT to mitigate BMD loss. Following the positive results of EDELWEISS 1 and the End of Phase 2 meeting with the U.S. Food and Drug Administration (FDA) in December 2018, the EDELWEISS 2 and EDELWEISS 3 Phase 3 clinical trials were initiated in May 2019. These Phase 3 trials will each enroll approximately 450 patients with endometriosis associated pain, with a target enrollmentco-primary endpoint of 330 patients, which we refer to as the EDELWEISS trial. We expect to report data from the first24-week evaluation period of the EDELWEISS trialdysmenorrhea (menstrual pain) and non-menstrual pain (NMPP). Both trials include a 75 mg once daily dose without ABT option, and a 200 mg once daily dose in the first half of 2018. combination with ABT option.
For the uterine fibroids indication, we plan to performhave completed a Phase 1 PK and PK/PD clinical trial to assess two different doses ofadd-back therapy ABT in patients receiving 100 mg and 200 mg doses of OBE2109linzagolix over six weeks. We expect that theThe results of this clinical trial, which we expect to receivewere announced in June 2017, support the first half of 2017, will confirm the selection of theadd-back therapyABT dose or doses planned(1mg E2 /0.5mg NETA) being utilized in the two randomized, placebo-controlled Phase 3 clinical trials that we intend to commencecommenced in the first half of 2017.
We refer to these Phase 3 clinical trials of OBE2109linzagolix in patients with heavy menstrual bleedingHMB associated with uterine fibroids as the PRIMROSE 1 (conducted in the US) and PRIMROSE 2 (conducted in Europe and in the US) clinical trials. In December 2019, we announced positive Phase 3 trial results from the PRIMROSE 2 trial of linzagolix for the treatment of heavy menstrual bleeding (HMB) due to uterine fibroids. The PRIMROSE clinical trials will each have2 trial enrolled 535 women with uterine fibroids. The trial was conducted in Europe and the US, and evaluated the efficacy and safety of once daily oral linzagolix, including 100 mg and 200 mg doses, both with and without hormonal ABT. The primary efficacy endpoint was the reduction in HMB at 24 weeks; responders were defined as patients with menstrual blood loss volume of ≤ 80 mL and a target enrollment50 percent or greater reduction from baseline in menstrual blood loss volume, measured using the alkaline hematin method. BMD was measured centrally via Dual Energy X-ray Absorptiometry (DEXA) scan at baseline and 24 weeks. The responder rate was 93.9% (p < 0.001) for patients receiving 200 mg with ABT and 56.7% for patients receiving 100 mg without ABT (p < 0.001), compared to 29.4% in the placebo group. Both doses achieved significant rates of approximately 500 patients. amenorrhea (p< 0.001), reduction in pain (p < 0.001), and improvement in quality of life (p < 0.001). Additionally, significant improvement (p< 0.001) in Hb levels, a reduction in number of days of bleeding and reduction in uterine volume were observed. A significant reduction in fibroid volume was also observed for the 200 mg dose with ABT (p = 0.008). The overall safety profile was in line with expectations. The most frequently observed adverse events (occurring in > 5% of patients) were headache, hot flushes, and anemia. Mean percentage change from baseline in BMD was consistent with the previous trial.
We expect to report databoth the primary endpoint results following 24 weeks of treatment from the PRIMROSE clinical trials1 trial conducted in the first halfU.S. and the 52 weeks of treatment results from PRIMROSE 2 trial in the second quarter of 2020. A Marketing Authorization Application (MAA) submission to the European Medicines Agency (EMA) and a New Drug Application (NDA) submission to the FDA are planned by the fourth quarter of 2020 and the first quarter of 2021 respectively, pending PRIMROSE 1 positive results and feedback from regulatory agencies.
We believe OBE2109,linzagolix, if approved in either indication,or both indications, has the potential to be abest-in-class oral GnRH receptor antagonist based on its favorable PK and PD profiles, and its expected balance between safety and efficacy. We expect OBE2109linzagolix to potentially reduce endometriosis-associated pain symptoms associated with endometriosis and heavy menstrual bleeding associated with uterine fibroids, while mitigating bone mineral density loss and other adverse effects associated with excessivefull estradiol suppression. Further, we believe that OBE2109linzagolix has the potential to offer personalizedflexible dosing alternatives to achieve either partial or full estrogen suppression that can be tailoredadministered with or without hormonal add-back therapy. Our intent is to a patient’s individual response.demonstrate that the majority of endometriosis patients may be able to experience significant symptomatic relief by utilizing our partial estrogen suppression linzagolix dose of 75 mg with no ABT. For uterine fibroids, we believe our 100 mg linzagolix dose without ABT is the only oral GnRH dosing regimen being developed for this indication without the use of ABT. Finally, we believe OBE2109linzagolix has certain advantageous characteristics including the absence of food effect, high bioavailability, low volume of distribution, no induction of liver enzymes known as cytochrome P450 3A4, or CYP3A4, no active transport into the liver by organic-anion-transporting polypeptide 1B1 and 1B3 or OATP1B1/1B3, and low PK and PD variability. We believe these characteristics could be key product differentiators compared to other oral GnRH receptor antagonists in clinical development. We are also exploring various alternatives for the future potential commercialization of linzagolix, including through a collaboration with a third party.
55
34 weeks gestational age, or GA. PGF |
To date, only treatments with limited efficacy and/or restrictive safety issues are available to treat preterm labor. In the United States, no drugs are approved for acute treatment of preterm labor and recommended off-label tocolytic treatments (medications that inhibit labor) include beta-adrenergic receptor agonists, calcium channel blockers, or NSAIDs, which are used for short-term prolongation of pregnancy (up to 48 hours) to allow for the administration of antenatal steroids (e.g. betamethasone). Magnesium sulfate, used for fetal neuroprotection can also be used (up to 48 hours) to inhibit acute preterm labor, but has limited efficacy. Approved tocolytic treatments in Europe include beta-adrenergic agonists, which carry severe maternal cardiovascular risks, and intravenous infusions of atosiban (an oxytocin receptor antagonist).
While prostaglandin synthesis inhibitors, a sub-group of NSAIDs, have been shown to be effective for inhibiting preterm labor, use of such drugs is limited, due to the threat of serious and sometimes life-threatening side effects in the fetus. In nonclinical studies, ObsEva has observed that OBE022 markedly reduces spontaneous and induced uterine contractions in pregnant rats without causing the fetal side effects seen with NSAIDS, such as indomethacin.
Through specific antagonism of the PGF2α receptor, OBE022 is designed to control preterm labor by reducing inflammation, decreasing uterine contractions and preventing cervical changes and fetal membrane rupture. Based on its PK profile and efficacy observed in animal models, we believe OBE022 has the potential to become a first-in-class therapy to suppress preterm labor and delay or avoid preterm birth, without significant safety concerns for the fetus. In February 2017, we completed a Phase 1 clinical trial assessing the safety, tolerability and PK profile of OBE022 in healthy post-menopausal female volunteers after single doses of 10 mg to 1,300 mg and multiple doses between 100 mg per day and 1,000 mg per day over 7 consecutive days. In this trial, OBE022 was observed to have a favorable PK profile, no clinically significant food effect, a favorable safety profile and to be well-tolerated at doses up to 1,300 mg after single dose administration and up to 1,000 mg per day after multiple dose administration over 7 days, each of which are above the estimated clinical effective dose. In March 2017, we completed a set of drug-drug interaction, or DDI, Phase 1 clinical pharmacology studies investigating the safety, tolerability and PK profile of OBE022 when combined with magnesium sulfate, atosiban, nifedipine or betamethasone (medications typically used in patients with preterm labor) in pre-menopausal female volunteers. OBE022 in combination with those drugs was observed to have a favorable safety profile and to be well-tolerated up to 1,100 mg per day, which was the highest tested dose. In December 2017, we announced the initiation of our Phase 2a proof-of-concept trial of OBE022, referred to as the PROLONG clinical trial. PROLONG is a proof-of-concept Phase 2a trial conducted in two parts: Part A and Part B. In this trial, OBE022 is orally administered daily for 7 days, to pregnant women, who are already receiving standard of care therapy for preterm labor, atosiban infusion for 48 hours. This clinical trial will enroll up to 120 pregnant women presenting with spontaneous preterm labor at gestational ages between 24 and 34 weeks.
56
In December 2018, we announced the completion of the open label Part A of the PROLONG trial in nine patients assessing OBE022 safety and pharmacokinetic (PK) profile. OBE022 was observed to be well tolerated by the mothers and their fetuses and we were able to demonstrate that the pharmacokinetics of OBE022 were similar to those previously observed in non-pregnant women. Also, 8 of the 9 treated women did not deliver during the 7 days of treatment. Based on these data, we began the randomized Part B of the trial assessing efficacy in delaying childbirth in women at 24 to 34 weeks gestation who are experiencing symptoms of preterm labor and potentially preterm delivery. We announced in July 2019 that the trial Independent Data Monitoring Committee (IDMC) completed the unblinded review of data from the first 30 subjects randomized in Part B of the PROLONG trial and recommended to continue the trial without modifications. We subsequently announced in January 2020 that the IDMC completed the unblinded review of data from the first 60 subjects and again recommended to continue the trial without modifications. Pending enrollment of the remaining 60 subjects, final PROLONG trial results are anticipated in the second half of 2020.
Nolasiban to improve embryo transfer outcomes after IVF. We have been developing nolasiban, an oral oxytocin receptor antagonist, to improve clinical pregnancy and live birth rates in women undergoing embryo transfer (ET) following an IVF cycle. In 2018, we reported positive results for the primary endpoint of ongoing pregnancy 10 weeks post embryo transfer and the secondary endpoint of live birth rate from the European Phase 3 clinical trial in 778 women undergoing IVF, or the IMPLANT 2 clinical trial. Patients receiving nolasiban prior to either Day 3 or Day 5 ET experienced an approximate 7% absolute or 25% relative increase in live birth rate over placebo. The Day 5 ET only population experienced an approximate 12% absolute or 35% relative increase in live birth rate over placebo.
In November 2019, we announced that the IMPLANT 4 trial did not meet the primary endpoint of an increase in ongoing pregnancy rate at 10 weeks, (39.1 % placebo vs 40.5 % nolasiban) (p = 0.745). As these results did not confirm the prior positive Phase 3 IMPLANT 2 trial findings, we have discontinued our previously ongoing development of nolasiban for IVF, and are exploring potential repositioning of the compound, such as through higher dose levels and earlier and longer exposure of nolasiban, as well as focusing on subjects with a high uterus contraction rate at the time of ET. In connection with this potential repositioning, in January 2020, we and Hangzhou Yuyuan BioScience Technology Co., Ltd. (Yuyuan) entered into a sublicense agreement to develop and commercialize nolasiban for improving clinical pregnancy and live birth rates in women undergoing embryo transfer as part of an IVF cycle in the People's Republic of China (PRC). Under the terms of the agreement, Yuyuan has the exclusive rights to develop and commercialize nolasiban in the PRC. They will fund all development and registration activities in the PRC, starting with the obligation to fund and conduct a Phase 1 trial and a Phase 2 proof-of-concept trial in China. We and Yuyuan will collaborate on the global development of nolasiban. We retain all rights to the product outside of PRC.
The following table summarizes key information regarding our current product candidates:
57

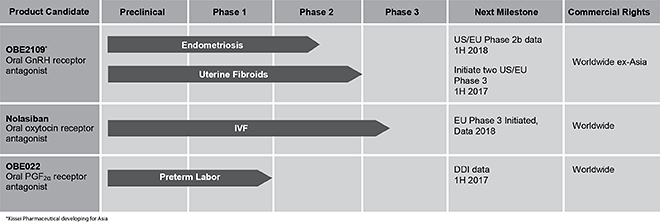
We are also evaluating additional indications for our current product candidates as well as opportunities toin-license or acquire additional product candidates in our therapeutic field.
Our executive team has substantial experience in developing and commercializing pharmaceutical products in this field. For example, Ernest Loumaye, M.D., Ph.D., OB/GYN, our Chief Executive Officer andco-founder, is a board certified and academically trained OB/GYN with extensive experience developing therapeutics for women’s health and over 90 publications in peer-reviewed journals. Most recently he was the Chief Executive Officer andCo-Founder of PregLem. Prior to PregLem, Dr. Loumaye spent nine years as Head of Clinical Development for Reproductive Health at Serono, now Merck Serono, where he led the worldwide clinical development and contributed to the worldwide registration ofGonal-F, Luveris and Ovidrel.
In addition, Jean-Pierre Gotteland, Ph.D., our Chief Scientific Officer and Elke Bestel M.D., our Chief Medical OfficerHead of R&D, held the same roles at PregLem where theyhe worked with Dr. Loumaye for six years and successfullyin-licensed, developed and registered afirst-in-class product, Esmya (ulipristal acetate), for the treatment of uterine fibroids.
Wim Souverijns, our Chief Commercial Officer is based at our headquarters in Geneva, Switzerland and is responsible for leading our transition from a development company to a commercial company. Mr. Souverijns brings nearly 20 years of experience in the pharmaceutical industry and recently served as Corporate Vice President, Global Marketing, Hematology & Oncology within Celgene out of Summit, New Jersey. Previously, Mr. Souverijns developed his pharmaceutical experience through various international assignments at PwC Consulting and in different market access leadership roles at Amgen, both in Europe and the U.S.
In July 2019, Elizabeth Garner joined us as our Chief Medical Officer, after spending many years in the women’s health industry at Agile Therapeutics Inc., Myriad Genetics Laboratories, Abbott Laboratories, and Merck. Dr. Garner has several years of experience in academic clinical practice, research and teaching at Harvard Medical School. Dr. Garner holds M.D. and M.P.H. degrees from the Harvard Medical School and Harvard School of Public Health, Boston, and received board certification in both general Obstetrics and Gynecology and Gynecologic Oncology.
58
Collectively, our management team has led the clinical development or contributed to the worldwide registration of three market-leading fertility products at Serono,Gonal-F, Luveris and Ovidrel, as well as other products including Esmya, Puregon Pen, Implanon, NuvaRing and Evamist. In addition, members of our management team bring pharmaceutical development, regulatory approval, manufacturing, reimbursement and commercialization experience from other pharmaceutical and biotechnology companies, including Merck Serono, PregLem, Organon, Allergan, Pierre Fabre, Novartis Pharma AG, Roche, SmithKline Beecham, Shire, Galderma, Speedel, Evolva SA and Acrux.
We have demonstrated an ability to successfully execute on the first part of our strategy by leveraging our extensive network in the field of women’s reproductive health and pregnancy toin-license OBE2109 linzagolix from Kissei and nolasiban and OBE022 from Merck Serono. Additionally, we have raised $93.2$334.2 million in equity financing from inception to December 31, 20162019 from leading healthcare investors, including HBM Healthcare Investments, New Enterprise Associates, Novo A/S, OrbiMed, Sofinnova Partners and Sofinnova Ventures.as well as $25.0 million from the issuance of a debt instrument.
Our Strengths
We believe our clinical and product development experience in the field of women’s reproductive health and pregnancy provides us with the following strengths:
Our Strategy
Our goal is to build the leading women’s reproductive health and pregnancy company focused on conditions where current treatment options are limited and significant unmet needs exist. The key elements of our strategy include the following:
Continue to advance each of our current product candidates in their respective indications.
Develop a targeted commercialization strategy for any approved product candidates. We have obtained worldwide commercial rights to our lead product candidates, except for certain countries in Asia with respect to linzagolix and for the PRC with respect to nolasiban. As we move our product candidates through development toward regulatory approval we will evaluate several options for each product candidate’s commercialization strategy. These options include building our own internal sales force, entering into a joint marketing partnership with another pharmaceutical or biotechnology company, or out-licensing the product to another pharmaceutical or biotechnology company. We are also exploring various alternatives for the future potential commercialization of linzagolix, including through a collaboration with a third party.
Pursue additional indications for our current product candidates. We believe each of our current product candidates have application outside the indications we are currently developing and we plan to pursue additional indications for our existing product candidates in the near future.
Leverage our international product development experience and extensive network of clinical experts and pharmaceutical industry executives within women’s reproductive health and pregnancy to in-license or acquire novel product candidates. We are focused on identifying, and in-licensing or acquiring, additional clinical-stage product candidates that we believe have the potential to become best-in-class or first-in-class products for the treatment of serious conditions in women’s reproductive health and pregnancy, if approved. We intend to focus on product candidates that we believe will be efficient from a capital-management standpoint, and we are exploring additional needs in our therapeutic field, such as premenstrual syndrome, fibrocystic breast disease, post-menopausal hot flashes, preeclampsia, dysmenorrhea and menopause-related auto-immune diseases.
59
OBE2109:Linzagolix: Investigational GnRH Receptor Antagonist for Symptoms Associated with Endometriosis and Uterine Fibroids
We are developing OBE2109linzagolix as an oral GnRH receptor antagonist, which we have observed in our clinical trials to induce a dose-dependent reduction of estradiol levels. Through that mechanism, we expect linzagolix to be indicated for the treatment of endometriosis-associated pain associated with endometriosis and heavy menstrual bleeding associated with uterine fibroids. We believe OBE2109,linzagolix, if approved, has the potential to be abest-in-class oral GnRH receptor antagonist based on its favorable PK and PD profiles, and its potential to provide targeted estradiol suppression to reduce pain symptoms associated with endometriosis and heavy menstrual bleedingHMB associated with uterine fibroids, while mitigating bone mineral density loss and other adverse effects that are typically associated with excessivefull estradiol suppression. We believe that OBE2109linzagolix has the potential to offer personalizedflexible dosing that can be tailoredalternatives to a patient’s individual response and that it will beaddress the symptoms of the broad patient population, supported by key differentiating product characteristics, including absence of food effect, high bioavailability, low volume of distribution, no CYP3A4 induction or OATP1B1/B3 interaction, and low PK and PD variability. We believe these characteristics are key product differentiators compared to other GnRH receptor antagonists in development.
In 2015, wein-licensed OBE2109 linzagolix from Kissei. Kissei completed three Phase 2a clinical trials in Japan of OBE2109linzagolix in patients with endometriosis, including one double blind placebo-controlled trial with a subgroup of patients diagnosed with both endometriosis and uterine fibroids.
Aimed at addressing the needneeds of the largest possible population in each indication, our clinical trials for both of these indications are designed to assess and potentially support the registration of two regimens of administrations for OBE2109linzagolix i.e. (i) a moderate dose of OBE2109linzagolix withoutadd-back therapy hormonal ABT and (ii) a high dose of OBE2109linzagolix withadd-back therapy. hormonal ABT. We are currently conducting thehave completed a 330-patient multiple-dose, placebo-controlled Phase 2b EDELWEISS 1 clinical trial of OBE2109linzagolix in endometriosis patients across 4670 sites in the United States and 15 sites in Central and Eastern Europe. We expectThis prospective, dose finding, randomized, parallel-group, double-blind, placebo-controlled Phase 2b study was designed to report data frominvestigate the first24-week evaluation periodefficacy and safety of this triala range of doses of linzagolix in the first halftreatment of 2018. We intendwomen with endometriosis associated pain. The 24-week treatment period (primary endpoint after 12 weeks of treatment) was followed either by a 24-week post treatment follow-up (PTFU) or an optional treatment extension phase with a 24-week PTFU.
The EDELWEISS 1 clinical trial successfully met its primary endpoint, a statistically significant patient response rate vs. placebo following 12 weeks of treatment. Patient response was measured by a reduction of at least 30% in combined menstrual and non-menstrual pelvic pain on a verbal rating scale (VRS) of 0 (no pain) through 3 (severe pain). Observed response rates were 34.5% for placebo, 61.5% for 75mg linzagolix, and 56.3% for 200mg linzagolix. Respective p values were 0.003 and 0.034. The efficacy in reducing pelvic pain, including dysmenorrhea and non-menstrual pelvic pain, as well as improvements in dyspareunia, dyschezia, and quality of life measures observed after 12 weeks of treatment were further improved or maintained up to commence our twoWeek 52, with the greatest treatment benefit observed at dose levels of 75 mg and above. The EDELWEISS 1 trial also demonstrated that linzagolix is well tolerated and has clinical benefits when administered continuously for up to 52 weeks.
After an End of Phase 3 PRIMROSE clinical trials2 meeting with the FDA in patients with heavy menstrual bleeding associated with uterine fibroids inDecember 2018, we announced the first halfinitiation of 2017. The PRIMROSE clinical trials will each have a target enrollment of approximately 500 patientsthe EDELWEISS 2 and will be conducted in the United States and in Europe. We expect to report data from theseEDELWEISS 3 Phase 3 clinical trials in May 2019. These Phase 3 trials will each enroll approximately 450 patients with endometriosis associated pain, with a co-primary endpoint of patients’ response on both dysmenorrhea (menstrual pain) and non-menstrual pain. Both trials include a 75 mg once daily dose without hormonal ABT option, and a 200 mg once daily dose in combination with ABT (1mg E2 / 0.5mg NETA) option.
In addition, we are conducting two Phase 3 clinical trials of linzagolix in patients with HMB associated with uterine fibroids, the PRIMROSE 1 (conducted in the US) and PRIMROSE 2 (conducted in Europe and in the US) clinical trials.
In December 2019, we announced positive Phase 3 trial results from the PRIMROSE 2 trial of linzagolix for the treatment of HMB due to uterine fibroids. The PRIMROSE 2 trial enrolled 535 women with uterine fibroids. The trial was conducted in Europe and the US, and evaluated the efficacy and safety of once daily oral linzagolix, including 100 mg and 200 mg doses, both with and without hormonal ABT. The primary efficacy endpoint was the reduction in HMB at 24 weeks; responders were defined as patients with menstrual blood loss volume of ≤ 80 mL and a 50 percent or greater reduction from baseline in menstrual blood loss volume, measured using the alkaline hematin method. Bone mineral density (BMD) was measured centrally via Dual Energy X-ray Absorptiometry (DEXA) scan at baseline and 24 weeks. The responder rate was 93.9% (p < 0.001) for patients receiving 200 mg with ABT and 56.7% for patients receiving 100 mg without ABT (p < 0.001), compared to 29.4% in the placebo group. Both doses achieved significant rates of amenorrhea (p< 0.001), reduction in pain (p < 0.001), and improvement in quality of life (p < 0.001). Additionally, significant improvement (p< 0.001) in Hb levels, a reduction in number of days of bleeding and reduction in uterine volume were observed. A significant reduction in fibroid volume was
60
also observed for the 200 mg dose with ABT (p = 0.008). The overall safety profile was in line with expectations. The most frequently observed adverse events (occurring in > 5% of patients) were headache, hot flushes, and anemia. Mean percentage change from baseline in BMD was consistent with previous clinical data.
We expect to report both the primary endpoint results following 24 weeks of treatment from the PRIMROSE 1 trial conducted in the U.S. and the 52 weeks of treatment results from PRIMROSE 2 trial in the second quarter of 2020. A MAA submission to the EMA and a NDA submission to the FDA are planned by the fourth quarter of 2020 and the first halfquarter of 2020.2021 respectively, pending PRIMROSE 1 positive results and feedback from regulatory agencies.
Background ofon Endometriosis and Uterine Fibroids
Endometriosis is a painful disorder in which the endometriumendometrial tissue grows outside of the uterus, typically on the lining of the pelvis, on the ovaries, in the rectovaginal septum, on the bladder, and inon the bowels. Endometriosis causes pain with monthly bleeding and chronic inflammatory reactions in the abdomen that may result in ovarian cyst formation, scar tissue and adhesions. The symptoms of endometriosis include significant pain during menstrual periods, chronic pelvic pain, pain during intercourse, excessive menstrual bleeding and infertility which in turn can impact general physical, mental and social well-being. Often the pain associated with endometriosis is cyclical in nature and reflects the response to reproductive hormones circulating throughout the body, particularly estrogen. Endometriosis is also one of the leading causes of infertility. In many instances, endometriosis is only diagnosed when women seek treatment for such infertility.
According to the World Endometriosis Research Foundation, as of 2014, endometriosis affects an estimated one in ten women during their reproductive years, totaling approximately 176 million women globally between the ages of 15 and 49. As of 2014, weWe believe that approximately 2.55 million women in the United States were diagnosed and treated annually for endometriosis, and the majority of those women experience significant pain during menstrual periods.
Uterine fibroids are commonnon-cancerous tumors that develop in the muscular wall of the uterus. Uterine fibroids can vary in size from a few millimeters to more than 20 centimeters, and in number from a single fibroid to several dozen fibroids. The main symptoms of uterine fibroids are heavy menstrual bleeding, anemia, abdominal pressure, abdominal pain, bloating, increased urinary frequency and reproductive dysfunction. Heavy menstrual blood loss is the most frequent disabling symptom of uterine fibroids.fibroids which often lead to anemia. Uterine fibroids also carry an increased risk of pregnancy complications such as infertility, miscarriage, placental abruption and premature onset of labor.
According to a study published in the American Journal of Obstetrics & Gynecology in 2003, uterine fibroids affect an estimated 20 to 40% of women over the age of 30 in the United States based on clinical cases and women who undergo treatment. We believe that more thanapproximately four million women in the United States are diagnosed and being treated for uterine fibroids.
The Role of GnRH
The exact causes of endometriosis and uterine fibroids are not currently understood. However, several factors can contribute to their development and progression, including the rise and fall of hormones, particularly estrogen, mainly in the form of
estradiol. The production of estrogen in the body is regulated by GnRH. GnRH is responsible for stimulating the synthesis and release of luteinizing hormone, or LH, and follicle stimulating hormone, or FSH, by the pituitary gland. LH and FSH in turn drive estrogen production through stimulation of the ovaries. Estradiol is the hormone that, among other effects, causes the endometrium inside the uterus to thicken during the menstrual cycle. Similarly, estradiol has been determined to promote the growth of endometriosis lesions and uterine fibroids. Various pharmacological treatments directed at addressing endometriosis and uterine fibroids attempt to regulate the production of estrogen, particularly estradiol, by controlling the activity of GnRH.
Limitations of Current Therapies for Endometriosis and Uterine Fibroids
Current treatment options for endometriosis and uterine fibroids are either pharmacological or surgical.
61
For endometriosis, the treatment selected is based on the severity of pain and the extent of the disease. Endometriosis treatments aim first to alleviate pain, then to remove or decrease the size and number of endometrial lesions, and possibly improve fertility. Oral contraceptives, progestins and NSAIDs are generally first-line treatments for women experiencing pain. Following the failure of first-line therapies, current treatment options are limited to intra-muscular or subcutaneous GnRH agonist injections and GnRH agonists nasal spray pumps or surgerypumps. In July 2018, AbbVie Inc announced that their GnRH antagonist elagolix (Orilissa) received regulatory approval in the U.S. for the treatment of moderate-to-severe endometriosis-associated pain.
Surgery may be performed for the most symptomatic cases.
Surgery However, in most cases conservative surgery can provide short-term relief by excising theand/or ablating endometrial lesions, but often does not prevent the endometrial lesions and associated symptoms from recurring. Surgery requires general anesthesia, and has a risk of scar tissue and adhesion formation in the pelvis, which couldcan lead to infertility, makeworsened pain, worse, requirerequirement for additional surgeries, orand damage to other pelvic structures. Surgical treatments for endometriosis range from laparoscopy to more complex open abdominal surgery. If a woman has not responded to other medical or surgical treatments, a radical hysterectomy, which is the removal of all or part of the uterus, andmay be performed. Depending on the woman, removal of the ovaries may also be required, resulting in definitive infertility and immediate menopause.
The World Endometriosis Research Foundation through its EndoCost study estimated the aggregate annual cost of endometriosis to be approximately $80 billion in the United States and approximately $60 billion in Germany, the UK, France and Italy in 2012 based on current exchange rates.
Uterine Fibroids
ForCurrent treatment options for heavy menstrual bleeding associated with uterine fibroids current treatment options are limited and generally consist of oral contraceptives, GnRH agonist injections or surgery. Oral contraceptives are generally used as the first-line therapy.therapy, but are often not effective in reducing heavy bleeding. Upon failure of a first-line therapy or contraindication to oral contraceptives, surgical intervention is generally the next treatment option. Hysterectomy is the most commonly performed surgical treatment option. Other less invasive procedures include (1) myomectomy, which is a selective removal of the fibroidfibroids, typically performed by laparoscopy, whichlaparoscopy; this usually preserves fertility, (2) uterine artery embolization, which is a procedure to obstruct the arteries nurturingfeeding the fibroid, performed by arterial catheterization, and (3) if the dominant symptom is bleeding, endometrial ablation, which is a procedure to remove the inner layerlining of the uterus performed by thermic or ultrasonic process.
According to a study published in the American Journal of Obstetrics & Gynecology in 2012, approximately 200,000300,000 hysterectomies and 30,000 myomectomies are performed annually for the treatment of uterine fibroids in the United States as of 2003. Hysterectomies are major surgeries and, according to the National Uterine Fibroids Foundation, approximately 660 women die each year in the United States from complications from a hysterectomy. Hysterectomies can be both physically and psychologically damaging, not only resulting in a woman becoming infertile,loss of fertility, but they also can be perceived by some women as impairing their feminine integrity.sense of femininity. Surgery also carries a risk of scar tissue and adhesions, which couldcan lead to infertility, makeworsening of pain, worse,damage to other pelvic structures, and may require additional surgeries or damage other pelvic structures.surgical management.
Treating uterine fibroids is expensive, as surgery constitutes a significant cost burden. Patients who do not undergo surgery often require medical management, hospitalization and additional outpatient physician visits, which further increase the annual costs of the disease. According to a systematic review of literature published in the American Journal of Obstetrics &Gynecology in 2012, direct and indirect costs associated with uterine fibroids were estimated in 2010 to be up to $34.4 billion annually in the United States.
Mechanism of Action and Limitations of GnRH Agonists
GnRH agonists are a standard pharmaceutical therapy for estrogen dependent conditions such as endometriosis and uterine fibroids. However, GnRH agonistsfibroids as they have significant drawbacks and limitations.
been demonstrated to reduce estradiol levels. GnRH agonists act by overstimulating the GnRH receptors which desensitizesinitially may worsen the symptoms for several weeks (the flair effect). Subsequently the pituitary cells are desensitized, resulting in reduced secretion of LH and FSH, and severely reduced production of estrogen, a contributing factor to endometriosis and uterine fibroids.estrogen. This leads to a state referred to as pseudo-menopause, in which patients experience menopausal symptoms before ultimately experiencing symptom relief. While GnRH agonists may be effective at treating the symptoms of endometriosis and uterine fibroids, they can be accompanied with serious drawbacks and limitations including: