We may experience delays or setbacks in our ongoing clinical trials, and we do not know whether future clinical trials will begin on time, need to be redesigned, enroll an adequate number of patients on time or be completed on schedule, if at all. Clinical trials can be delayed or terminated for a variety of reasons, including delay or failure to:
generate sufficient preclinical, toxicology, or other in vivo or in vitro data to support the initiation or continuation of clinical trials
obtain regulatory approvalauthorization to commence a trial, if applicable;trial;
reach agreement on acceptable terms with prospective contract research organizations, or CROs, and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
obtain Ethics Committee, institutional review board, or IRB, or ethics committee approval at each site;
manufacture, test, release, validate or import sufficient quantities of drug product for use in a trial;
recruit, screen and enroll suitable patients to participate in a trial;
have patients complete a trial or return forpost-treatmentfollow-up;post-treatment follow-up;
ensure that clinical sites observe trial protocol or continue to participate in a trial;
address any patient safety concerns that arise during the course of a trial;
address any conflicts with new or existing laws or regulations; or
initiate or add a sufficient number of clinical trial sites; orsites.
manufacture sufficient quantities of product candidate for use in clinical trials.
There is alsoThe novel Coronavirus (COVID-19) pandemic has had, and may continue to have an evolving impact of the novel Coronavirus(COVID-19) pandemic on the conduct of clinical trials of investigational therapeutic candidates, and any challenges which may arise, for example, from quarantines, site closures, travel limitations, interruptions to the supply chain for our product candidates, or other considerations if site personnel or trial subjects become infected withCOVID-19, which may lead to difficulties in meeting protocol-specified procedures, including administering or using the therapeutic candidate or adhering to protocol-mandated visits and laboratory/diagnostic testing, unavoidable protocol deviations due toCOVID-19 illness and/orCOVID-19 control measures, which will likely vary depending on many factors, including the nature of disease under study, the trial design, and in what region(s) the study is being conducted.
Patient enrollment is a significant factor in the timing of clinical trials and is affected by many factors, including the size and nature of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, competing clinical trials and clinicians’ and patients’ perceptions as to the potential advantages of the drug being studied in relation to other available therapies, including any new drugs or treatments that may be approved for the indications we are investigating.
We could also encounter delays if a clinical trial is suspended or terminated by us our collaboration partner for a product candidate, by the Ethics Committee or IRBs of the institutions in which such trials are being conducted, by an independent data safety monitoring board, or DSMB, for such trial or by European Economic Area, or EEA, Competent Authorities, the FDA or similar regulatory authorities. Such authorities, or we, may suspend or terminate a clinical trial due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by EEA Competent Authorities, the FDA or similar regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial.
Further, we are conducting, phase 3 studiesand plan to conduct, clinical trials in sites outside of TransCon hGH across clinical sites located in North America, Europe, the Middle East, and Oceania (Australia/New Zealand).United States. Conducting clinical trials in foreign countries presents additional risks that may delay completion of clinical trials. These risks include the failure of physicians or enrolled patients in foreign countries to adhere to clinical protocol as a result of differences in healthcare services or cultural customs, managing additional administrative burdens associated with foreign regulatory schemes, as well as political and economic risks relevant to such foreign countries. In addition, the EMA or the FDA may determine that the clinical trial results obtained in foreign subjects do not representestablish the safety and efficacy of a product candidate when administered in EEA or U.S. patients, and are thus not supportive of an application for a marketing authorization in
the EEA or of a New Drug Application, or NDA, or Biologics License Application, or BLA, approval in the United States. As a result, the EMA or the FDA may not accept data from clinical trials conducted outside the EEA or the United States, respectively, and may require that we conduct additional clinical trials or obtain additional data before we can submit an NDA or BLA in the United States or a marketing authorization application in the EEA. The EMA or the FDA may even require conducting additional clinical trials in the EEA or the United States, respectively.
If there are delays in the completion of, or termination of, any clinical trial of our product candidates or if we are required to conduct additional clinical trials in addition to those we have currently planned, the commercial prospects of our product candidates may be harmed, and our ability to generate revenue from commercial product sales from any of these product candidates will be delayed. In addition, any delays in completing the clinical trials will increase costs, slow down our product candidate development and approval process and jeopardize the ability to commence product sales and generate revenue from commercial product sales. Any of these occurrences may significantly harm our business, financial condition and prospects. Clinical trial delays may also allow our competitors to bring products to market before we do, which could impair our ability to obtain orphan exclusivity for our products that potentially qualify for orphan drug designation. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
We depend on certain collaboration partners to develop and conduct clinical studies with, obtain regulatory approvals for, market and sell our collaboration product candidates, and if such collaboration partners fail to perform as expected, or are unable to obtain the required regulatory approvals for such product candidates, the potential for us to generate future revenue fromof such product candidates would be significantly reduced and our business would be significantly harmed.
We rely on our collaboration partners to conduct certain clinical studies. For example, in November 2018, we announced the formation of VISEN Pharmaceuticals, or Visen,VISEN, a company established to develop, manufacture, and commercialize our endocrinology rare disease therapies in Greater China. In connection with the formation of Visen,VISEN, we granted VisenVISEN exclusive rights to develop and commercialize our rare disease endocrinology products based on our proprietary TransCon technologies, including TransCon hGH, TransCon PTH and TransCon CNP, in Greater China for use in all human indications, subject to certain exceptions. We may also enter into collaboration agreements with other parties in the future relating to our other product candidates. Under our existing collaboration agreements, our collaboration partners are responsible for completing preclinical and/or clinical development and obtaining and maintaining regulatory approval for the applicable product candidates from the EMA, the FDA, the National Medical Product Administrations of the People’s Republic of China, or NMPA, and similar regulatory authorities. Ultimately, if such product candidates are advanced through clinical trials and receive marketing approval from the EMA, the FDA, the NMPA or similar regulatory authorities, such partners will be responsible for commercialization of these collaboration products. The potential for us to obtain future development milestone payments and, ultimately, generate revenue from royalties on sales of such collaboration products depends entirely on successful development, regulatory approval, marketing and commercialization by our collaboration partners.
If our collaboration partners do not perform in the manner we expect or fulfill their responsibilities in a timely manner, or at all, if our agreements with them terminate or if the quality or accuracy of the clinical data they obtain is compromised, the clinical development, regulatory approval and commercialization efforts related to our collaboration product candidates could be delayed or terminated and it could become necessary for us to assume the responsibility at our own expense for the clinical development of such product candidates. In that event, we would likely be required to limit the size and scope of efforts for the development and commercialization of such product candidate, to seek additional financing to fund further development, or to identify alternative collaboration partners, and our potential to generate future revenue from royalties and milestone payments from such product candidate would be significantly reduced or delayed and our business would be harmed. Our existing collaborations and any future collaboration arrangements that we may enter into with third partiesthird-parties may not be scientifically or commercially successful. In addition to the risks inherent in the development of a drug product candidate, factors that may affect the success of our collaborations include the following:
our collaboration partners have the unilateral ability to choose not to develop a collaboration product for one or more indications for which such product has been or is currently being evaluated, and our collaboration partners may choose to pursue an indication that is not in our strategic best interest or to forego an indication that they believe does not provide significant market potential even if clinical data is supportive of further development for such indication;
our collaboration partners may choose not to develop and commercialize our collaboration products in certain relevant markets;
our collaboration partners may take considerably more time advancing our product candidates through the clinical and regulatory process than we currently anticipate, which could materially delay the achievement of milestones and, consequently the receipt of milestone payments from our collaboration partners;
our collaboration partners have substantial discretion under their respective agreements regarding how they structure their efforts and allocate resources to fulfill their obligations to diligently develop, obtain regulatory approval for and commercialize our collaboration products;
our collaboration partners control all aspects of commercialization efforts under their respective license agreements and may change the focus of their development and commercialization efforts or pursuehigher-priority programs and, accordingly, reduce the efforts and resources allocated to their collaborations with us;
our collaboration partners are solely responsible for obtaining and maintaining all regulatory approvals and we or our collaboration partners may fail to develop a commercially viable formulation or manufacturing process for our product candidates, and we or our collaboration partners may fail to manufacture or supply sufficient drug substance for commercial use, if approved, which could result in lost revenue under such collaborations;
our collaboration partners may not comply with all applicable regulatory requirements or may fail to report safety data in accordance with all applicable regulatory requirements;
if any of our agreements with our collaboration partners terminate, we will no longer have any rights to receive potential revenue under such agreement, in which case we would need to identify alternative means to continue the development, manufacture and commercialization of the affected product candidates, alone or with others;
our collaboration partners have the discretion to sublicense their rights with respect to our collaboration technology in connection with collaboration product candidates to one or more third partiesthird-parties without our consent; and
our collaboration partners may be pursuing alternative technologies or developing alternative products, either on their own or in collaboration with others, that may be competitive with products on which they are collaborating with us or which could affect our collaboration partners’ commitment to the collaboration; and
if our collaboration partners receive approval for any of the collaboration product candidates, reductions in marketing or sales efforts or a discontinuation of marketing or sales of our product candidates by our collaboration partners would reduce any royalties we could be entitled to receive, which are based on the sales of our product candidates by our collaboration partners.collaboration.
In addition, the collaboration agreements provide our collaboration partners with rights to terminate such agreements and licenses under various conditions, which if exercised would adversely affect our product development efforts, make it difficult for us to attract new partners and adversely affect our reputation in the business and financial communities. Our collaboration partners have the right to terminate their respective collaboration agreements with us, upon advance written notice, in the event of our uncured material breach of the agreement and for convenience. In addition, Visenparticular, VISEN may terminate in the event of our bankruptcy or insolvency.
The timing and amount of any milestone and royalty payments we may receive under our agreements with our collaboration partners and the value of any equity we own in our collaboration partners (such as the equity we own in Visen)VISEN) will depend on, among other things, the efforts, allocation of resources, and successful development and commercialization of our product candidates by our collaboration partners. We cannot be certain that any of the development and regulatory milestones will be achieved or that we will receive any future milestone payments under these agreements.agreements we may enter into with collaboration partners. In addition, in certain circumstances we may believe that we have achieved a particular milestone and the applicable collaboration partner may disagree with our belief. In that case, receipt of that milestone payment may be delayed or may never be received, which may require us to adjust our operating plans. We also cannot be certain that any equity we own in our collaboration partners (such as the equity we own in Visen)VISEN) will maintain its value or grow in value.
We may form additional strategic collaborations in the future with respect to our proprietary programs, but we may not realize the benefits of such collaborations.
We may form strategic collaborations, create joint ventures or enter into licensing arrangements with third partiesthird-parties with respect to our independent programs that we believe will complement or augment our existing business. We have historically engaged, and intend to continue to engage, in partnering discussions with a range of biopharmaceutical companies and could enter into new collaborations at any time. For example, in November 2018, we announced the formation of Visen,VISEN, a company established to develop, manufacture, and commercialize our endocrinology rare disease therapies in Greater China. In connection with the formation of Visen,VISEN, we granted VisenVISEN exclusive rights to develop and commercialize our rare disease endocrinology products based on our proprietary TransCon technologies, including TransCon hGH, TransCon PTH and TransCon CNP, in Greater China for use in all human indications, subject to certain exceptions. We face significant competition in seeking appropriate strategic partners, and the negotiation process to secure appropriate terms istime-consuming and complex. Any delays in identifying suitable development partners and entering into agreements to develop our product candidates could also delay the commercialization of our product candidates, which may reduce their competitiveness even if they reach the market. Moreover, we may not be successful in our efforts to establish such a strategic partnership for any future product candidates and programs on terms that are acceptable to us, or at all. This may be for a number of reasons. For example, under our collaboration with Visen, VisenVISEN, VISEN has a right of first negotiation to develop certain of our endocrinology product candidates in Greater China, so our ability to negotiate such a collaboration with suitable third partiesthird-parties may be hampered by such rights we granted to Visen.VISEN. Additionally, our product candidates and programs may be deemed to be at too early of a stage of development for collaborative effort, our research and development pipeline may be viewed as insufficient, and/or third partiesthird-parties may not view our product candidates and programs as having sufficient potential for commercialization, including the likelihood of an adequate safety and efficacy profile. Even if we are successful in entering into a strategic alliance or license arrangement, there is no guarantee that the collaboration will be successful, or that any future collaboration partner will commit sufficient resources to the development, regulatory approval, and commercialization of our product candidates, or that such alliances will result in us achieving revenues that justify such transactions.
OurCertain of our product candidates other than TransCon hGH, TransCon PTH and TransCon CNP, are in various stages of preclinical development and we may not be successful in our efforts to successfully develop these products or expand our pipeline of product candidates.
A key element of our strategy is to expand our pipeline of product candidates utilizing our proprietary TransCon technologies, and to advance such product candidates through clinical development, either ondevelopment. Certain of our own or in conjunction with strategic collaboration partners. Our other product candidates are in preclinical development and may require significant time and additional research and development before we can file INDINDs or equivalent foreign regulatory filings with regulatory authorities to begin clinical studies. Of the large number of drugs in development, only a small percentage of such drugs successfully complete the EMA or FDA regulatory approval process and are commercialized. Accordingly, even if we are able to continue to fund such development programs, our product candidates may not be advanced to clinical studies or be successfully developed or commercialized. In addition, our preclinical product candidates may not demonstrate the advantages we expect from application of our TransCon technologies in preclinical studies. In such event, we may decide not to progress any such product candidates into clinical trials.
Research programs to identify product candidates require substantial technical, financial and human resources, whether or not any product candidates are ultimately identified. Although our research and development efforts to date have resulted in several development programs, we may not be able to develop product candidates that are safe and effective. Our research programs may initially show promise in identifying potential product candidates, yet fail
to yield product candidates for clinical development or commercialization for many reasons, including the following:
the research methodology used and our TransCon technologies may not be successful in creating potential product candidates;
competitors may develop alternatives that render our product candidates obsolete or less attractive;
product candidates we develop may nevertheless be covered by third parties’third-parties’ intellectual property rights or other types of exclusivity and we may not be able to obtain a license from such third partythird-party or the license terms may not be acceptable to us;
the market for a product candidate may change during our program or we may discover that such market was smaller than initially expected so that such a product may become financially unfeasible to continue to develop;
a product candidate may be demonstrated to have harmful side effects or not to be effective, or otherwise not to meet other requirements for regulatory approval;
a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and
a product candidate may not be accepted as safe and effective by patients, the medical community orthird-party payors, or reimbursable bythird-party payors, if applicable.
Even if we are successful in continuing to expand our pipeline, through our own research and development efforts or by pursuingin-licensing or acquisition of product candidates, the potential product candidates that we identify or acquire may not be suitable for clinical development, including as a result of being shown to have harmful side effects or other characteristics that indicate that they are unlikely to receive marketing approval and achieve market acceptance. If we do not successfully develop and commercialize a product pipeline, we may not be able to generate revenue from commercial product sales in future periods or achieve or sustain profitability.
Interim, and/or“topline” and preliminary data from our clinical trials that we have announced, or that we may announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publish interimpublicly disclose preliminary or preliminarytop-line data from our preclinical studies and clinical studies. For example, in January 2020, we announcedtrials, which is based on a preliminary analysis of then-available data, from our phase 2 study of TransCon PTH. Interim data forand the trials we may completeresults and related findings and conclusions are subject to change following a more comprehensive review of the riskdata related to the particular study or trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the top-line or preliminary results that onewe report may differ from future results of the same studies, or more of clinical outcomesdifferent conclusions or considerations may materially change as patient enrollment continues and/or more patientqualify such results, once additional data become available. Preliminaryhave been received and fully evaluated. Top-line data would also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, any interim and preliminarytop-line data should be viewed with caution until the final data are available. Material adverse changes in
From time to time, we may also disclose interim data from our preclinical studies and clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available or as patients from our clinical trials continue other treatments for their disease. Adverse differences between preliminary or interim data and final data could significantly harm our business prospects.
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our company in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and others may not agree with what we determine is material or otherwise appropriate information to include in our disclosure. If the interim, top-line, or preliminary data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize, our product candidates may be harmed, which could harm our business, operating results, prospects or financial condition.
We may expendBy expending our limited resources to pursue a particular product candidate or indicationcandidates and areas of focus we may fail to capitalize on product candidates or indicationsareas of focus that may beare more profitable or for which there is a greater likelihood of success.
We have focused on research programs and product candidates that utilize our proprietary TransCon technologies.on the endocrinology and oncology therapeutic areas of focus. As a result, we may forego or delay pursuit of opportunities with other product candidates or forin other indicationsfocus areas that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and product candidates for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate.
We rely on third partiesthird-parties to conduct our nonclinical studies and clinical trials. If these third partiesthird-parties do not successfully carry out their contractual duties or meet expected deadlines, we may be unable to obtain regulatory approval for, or commercialize, our product candidates.
We do not currently have the ability to independently conduct clinical trials or nonclinical studies. We rely on medical institutions, clinical investigators, contract laboratories, collaboration partners and other third parties,third-parties, such as CROs, to conduct clinical trials of our product candidates. The third partiesthird-parties with whom we contract for execution of our clinical trials play a significant role in the conduct of these trials and the subsequent collection and analysis of data. However, these third partiesthird-parties are not our employees, and except for contractual duties and obligations, we control only certain aspects of their activities and have limited ability to control the amount or timing of resources that they devote to our programs. Although we rely on these third partiesthird-parties to conduct our nonclinical studies and our clinical trials, we remain responsible for ensuring that each of our nonclinical studies and clinical trials is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards and our reliance on third partiesthird-parties does not relieve us of our regulatory responsibilities. We and these third partiesthird-parties are required to comply with current good laboratory practices, or GLPs, for nonclinical studies, and good clinical practices, or GCPs, for clinical studies. GLPs and GCPs are regulations and guidelines enforced by the Competent Authorities of the Member States of the European Economic Area, or EEA, the FDA and comparable foreign regulatory authorities for all of our products in nonclinical and clinical development, respectively. Regulatory authorities enforce GCPs through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of our third partythird-party contractors fail to comply with applicable regulatory requirements, including GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the EMA, the FDA, or similar regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot be certain that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP regulations. In addition, our clinical trials must be conducted with products produced under cGMP regulations. The failure of our contract manufacturers to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process.
Even if our product candidates obtain regulatory approval, they may never achieve market acceptance or commercial success, which will depend, in part, upon the degree of acceptance among physicians, patients, patient advocacy groups,third-party payors and the medical community.
Even if our product candidates obtain EMA, FDA or other regulatory approvals, and are ultimately commercialized, our product candidates may not achieve market acceptance among physicians, patients,third-party payors, patient advocacy groups and the medical community. The degree of market acceptance, if any, for our most advanced product candidates for which marketing approval is obtained will depend on a number of factors, including:
the efficacy of the products as demonstrated in clinical trials;
the prevalence and severity of any side effects and overall safety profile of the product;
the perceived safety of the TransCon technologies;
the convenience and features of theauto-injector or drug delivery device used to administer the drug;
the clinical indications for which the product is approved;
acceptance by physicians, major operators of clinics and patients of the product as a safe and effective treatment and their willingness to pay for them;
relative convenience and ease of administration of our products;
the potential and perceived advantages of our product candidates over current treatment options or alternative treatments, including future alternative treatments;
the availability of supply of our products and their ability to meet market demand;
marketing and distribution support for our product candidates;
the quality of our relationships with patient advocacy groups; and
coverage and reimbursement policies of government and otherthird-party payors.
If our product candidates that obtain regulatory approval do not achieve significant market acceptance or commercial success, this could harm our business, results of operations and prospects, and the value of our shares or ADSs.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following regulatory approval, if any. If any of our product candidates receives marketing approval and subsequently causes undesirable side effects, the ability to market the product candidates could be compromised.
Undesirable side effects caused by TransCon hGH,lonapegsomatropin, TransCon PTH, TransCon CNP, or our other product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the EMA, the FDA or similar authorities. In the event that trials conducted by us or ourany collaboration partners, or trials we conduct with our unlicensed product candidates, reveal a high and unacceptable severity and prevalence of side effects, such trials could be suspended or terminated and the EMA, the FDA or similar regulatory authorities could order ourany collaboration partners or us to cease further development of or deny approval of our product candidates for any or all targeted indications. Thedrug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly.
Additionally, if we successfully develop a product candidate and it receives marketing approval, the FDA could require us to adopt a Risk Evaluation and Mitigation Strategy, or REMS, to ensure that the benefits of treatment with such product candidate outweigh the risks for each potential patient, which may include, among other things, a communication plan to health care practitioners, patient education, extensive patient monitoring or distribution systems and processes that are highly controlled, restrictive and more costly than what is typical for the industry. For example, in June 2020, we submitted a BLA to the FDA for approval of lonapegsomatropin for the treatment of pediatric GHD. In our discussions with the FDA relating to this BLA submission to date, the FDA has indicated that it has not identified significant safety issues and a REMS is not currently being considered for lonapegsomatropin, if approved; however, the FDA’s review of this BLA submission is ongoing and the FDA may still require us to adopt a REMS for lonapegsomatropin for the treatment of GHD.
In addition, in the event that any of our product candidates receives regulatory approval and we or others later identify undesirable side effects caused by one of our products, a number of potentially significant negative consequences could occur, including:
regulatory authorities may withdraw their approval of the product or seize the product;
we, or ourany collaboration partners, may be required to recall the product;
additional restrictions may be imposed on the marketing of the particular product or the manufacturing processes for the product or any component thereof, including the imposition of a REMS or requirements for similar actions, such as patient education, certification of health care professionals or specific monitoring;
we, or ourany collaboration partners, may be subject to fines, injunctions or the imposition of civil or criminal penalties;
regulatory authorities may require the addition of labeling statements, such as a “black box” warning or a contraindication;
we could be sued and held liable for harm caused to patients;
the product may become less competitive; and
our reputation may suffer.
Any of the foregoing events could prevent us, or ourany collaboration partners, from achieving or maintaining market acceptance of a particular product candidate, if approved, and could result in the loss of significant revenue to us, which would harm our results of operations and business.
Competition in the biotechnology and pharmaceutical industries is intense and our competitors may discover, develop or commercialize products faster or more successfully than us. If we are unable to compete effectively our business, results of operations and prospects will suffer.
The markets in which we intend to compete are undergoing, and are expected to continue to undergo, rapid and significant technological changes. Some of our product candidates are for fields in which competitive products already exist and are established. We expect competition to intensify as technological advances are made or new drugs and biotechnology products are introduced. New developments by competitors may render our current or future product candidates and/or technologiesnon-competitive, obsolete or not economical. Our competitors’ products may be more efficacious or marketed and sold more effectively than any of our product candidates.
We are aware of several pharmaceutical and biopharmaceutical companies that have commenced clinical studies of products or have successfully commercialized products addressing areas that we are targeting. WhileTo our knowledge, as of the date of this report, there are currently no commercially available long-acting growth hormone treatment options available in the United States or Europe, aEurope. A permanently PEGylated long-acting growth hormone developed by GeneScience Pharmaceuticals Co., Ltd. is available in China and the Somatropin Biopartners product (LB03002), is available in Korea. On August 28, 2020, the FDA granted Novo Nordisk Inc. approval of somapacitan for replacement of endogenous growth hormone in adult patients with GHD but as of the date of this report to our knowledge Novo Nordisk has not commercially launched the product in the United States. In additionJanuary 2021, Novo Nordisk received a positive opinion from the Committee for Medicinal Products for Human Use, under the EMA for once-weekly somapacitan, recommending marketing authorisation for use in adult patients with GHD. Pfizer (in collaboration with OPKO Health Inc.) has submitted to the currently approved and marketed dailyFDA, a Biologics License Application, or BLA, for somatrogon, a long-acting growth hormone therapies, there are a varietyfor the treatment of pediatric patients with GHD. In January 2021, Pfizer (in collaboration with OPKO Health) announced the FDA has accepted the regulatory submission and set the PDUFA goal date in October 2021. Other experimental growth hormone therapies based on permanent modification are in different stages of clinical development by various companies, including GeneScience Pharmaceuticals Co., Ltd., Genexine Inc,Inc., I-MAB, and JCR Pharmaceuticals Co., Ltd., Novo Nordisk A/S, and OPKO Health, Inc. (in collaboration with Pfizer Inc.). In addition, Shire plc owns the rights to NATPARA,Natpara, a treatment for hypoparathyroidism. NATPARANatpara was voluntarily recalled in September 2019 in the U.S. and is now only available to a limited number of seriously-ill patients through a Special Use Program offered by its manufacturer, Takeda Pharmaceutical Company. In addition, we are aware of several academic groups and companies working on making longer actinglonger-acting agonists of the PTH receptor, or PTH1R. Other companies and groups are developing or commercializing therapies for hypoparathyroidism, including Shire, Chugai Pharmaceutical Co., Ltd., Entera Bio, Extend Biosciences, Massachusetts General Hospital, AlizéAmolyt Pharma, Bridgebio and Eli Lilly and Company. Other companies are developing therapies for achondroplasia, including BioMarin, Pfizer, QED Therapeutics and BioClin Therapeutics. BioMarin Pharmaceutical, Inc. is developing vosoritide for the treatment of achondroplasia, and other companies thatTherachon and BioClin Therapeutics, Inc. are developing therapiescompounds for achondroplasia include Pfizer, QEDachondroplasia. Other companies are developing toll like receptor agonists for cancer immunotherapy including: Nektar Therapeutics, CureVac N.V., Seven and BioClinEight Biopharmaceuticals Inc., Idera Pharmaceuticals, Inc., Checkmate Pharmaceuticals, Inc., Exicure, Inc., Bolt Therapeutics, Inc., and Silverback Therapeutics, Inc. In addition to product basedproduct-based competition, our TransCon technologies face technology basedtechnology-based competition as we believe other companies are developing or evaluating enhanced drug delivery and sustained release technologies. In particular, we believe Nektar Therapeutics, OPKO Health, Inc., ProLynx LLC and Serina Therapeutics, Inc. are developing technologies that use reversible linkers and that may be competitive with our TransCon technologies.
It is also possible that our competitors will commercialize competing drugs or treatments before we or our collaboration partners can launch any products developed from our product candidates. We also anticipate that we will face increased competition in the future as new companies enter into our target markets.
Furthermore, to the extent we are developing TransCon product candidates that incorporate already approved drugs, we face competition from the pharmaceutical companies which are currently marketing such approved products. These pharmaceutical companies can generally be expected to seek to delay the introduction of competing products through a variety of means including:
filing new formulation patent applications on drugs whose original patent protection is about to expire;
filing an increasing number of patent applications that are more complex and costly to challenge;
filing suits for alleged patent infringement that automatically delay FDA approval;
developing patentedcontrolled-release or other“next-generation” products, which may compete with TransCon product candidates;
establishing exclusive contracts with third partythird-party payors; or
changing product claims and product labeling.
Any one of these strategies may increase the costs and risks associated with our efforts to introduce any of our product candidates and may delay or altogether prevent such introduction.
Many of our competitors have:
significantly greater name recognition, financial, marketing, research, drug development and technical and human resources than we have at every stage of the discovery, development, manufacturing and commercialization process and additional mergers and acquisitions in the biotechnology industries may result in even more resources being concentrated in our competitors;
more extensive experience in commercializing drugs, conducting preclinical testing, conducting clinical studies, obtaining regulatory approvals, challenging patents and in manufacturing and marketing pharmaceutical products;
products that have been approved or are in late stages of development; and
collaboration arrangements in our target markets with leading companies and research institutions.
If we successfully develop and obtain approval for our product candidates, we will face competition based on many different factors, including:
the safety and effectiveness of our product candidates;
the timing of and specific circumstances relating to regulatory approvals for these product candidates;
the availability and cost of manufacturing, marketing and sales capabilities;
the effectiveness of our marketing and sales capabilities;
the price of our product candidates;
the availability and amount ofthird-party reimbursement for our product candidates; and
the strength of our patent position.
In addition, academic institutions, government agencies, and other public and private organizations conducting research may seek patent protection with respect to potentially competitive products or technologies. These organizations may also establish exclusive collaborative or licensing relationships with our competitors.
Our competitors may develop or commercialize products with significant advantages in regard to any of these factors. Our competitors may therefore be more successful in commercializing their products than we are, which could adversely affect our business, results of operations and prospects, and the value of our shares or ADSs.
For additional information regarding the competitive landscape for our product candidates, see “Item 4 B. Information on the Company – Business Overview – TransCon Product Candidates.”
Our proprietary TransCon technologies include a new approach to extending the residence time and duration of action of a variety of drug products and may not result in any products of commercial value.
Our TransCon technologies have been developed to improve the delivery of a variety of drug products. However, we cannot be certain that our TransCon technologies will be deemed safe or efficacious, nor that any aspects of our TransCon technologies will yield additional product candidates that could be commercially valuable. Further, one of our two carrier systems, the TransCon hydrogel carrier system, has never been used in humans. As a result, our TransCon hydrogel carriers, when dosed in humans, may fail to perform as we expect. Failure of any of our product candidates to be successfully developed and approved may result in our TransCon technologies being viewed as an ineffective approach to developing drug products which would harm our business and prospects.
We apply our TransCon technologies to both approved and unapproved parent drugs to extend the life of such drugs in the body, and to enhance the overall benefit of a given therapy. Even when applied to approved parent drugs, we have generated limited clinical data on our product candidates using our systemic TransCon technologies with respect to safety and efficacy forlong-term treatment in humans. Thelong-term safety and efficacy of our TransCon
technologies and the extended life in the body of our product candidates utilizing TransCon technologies compared to currently approved products is unknown, and it is possible that our product candidates may have an increased risk of unforeseen reactions following extended treatment relative to other currently approved products. If extended treatment with product candidates utilizing TransCon in our ongoing or future clinical trials results in any concerns about the safety or efficacy of our TransCon technologies, we may be unable to successfully develop or commercialize our product candidates.
Product candidates created utilizing the TransCon technologies are new chemical entities that employ novel technologies that have not yet been approved by the FDA, EMA or other regulatory authorities. These regulatory authorities have limited experience in evaluating our technologies and product candidates.
Our TransCon technologies allow for the creation of new molecular entities through the transient conjugation of parent drug molecules to our soluble and microparticle TransCon carrier molecules via our TransCon linkers. We and our collaboration partners are developingdevelop product candidates based on these novel technologies, and we intend to work closely with our collaboration partners to understand and deliver the requisite demonstration of safety and efficacy that the FDA, the EMA and other regulatory authorities may seek for the approval of product candidates that incorporate the TransCon technologies. It is possible that the regulatory approval process may take significant time and resources and require deliverables from independent third partiesthird-parties not under our control. For some of our product candidates, the regulatory approval path and requirements may not be clear, which could add significant delay and expense. Delays or failure to obtain regulatory approval of any of the products that we or ourany collaboration partners develop using our novel technologies would adversely affect our business.
We have limited clinical data on product candidates utilizing theour TransCon technology platformtechnologies to indicate whether they are safe or effective forlong-term use in humans.
Our product candidates transiently link a parent drug molecule to select TransCon carriers via our TransCon linkers. Once injected, we believe that our prodrugs predictably release the unmodified parent drug molecule over time, thus preserving the parent drug’s original mode of action, and, we believe, the parent drug’s original safety and efficacy profile. We believe that our TransCon carriers remain bound to our TransCon linkers and that they are cleared from the body predominantly by renal filtration and biliary transport with fecal excretion. We have limited clinical data on product candidates utilizing the systemic TransCon technologies to indicate whether they are safe or effective forlong-term use in humans, including the safety of any degradation products that may result after the TransCon carrier and TransCon linker are cleaved from the parent drug molecule. As an example, our TransCon prodrugs utilize polyethylene glycol, or PEG, and hydrogels incorporatingPEG-based polymers as TransCon carriers. Although the safety and efficacy of PEG and permanently PEGylated proteins has been demonstrated within their respective
indications by the approval of drugs such as PegIntron®, PegaSys®, Neulasta®, Somavert®, Cimzia®, Krystexxa®, Adynovate® and Rebinyn® and we are not aware of any evidence forPEG-related safety issues with PEGylated proteins in the clinic, health authorities, including the EMA, have historically posed general questions relating to the distribution, elimination, and the potential for PEG accumulation to pharmaceutical companies involved in the development of PEGylated drug products. If treatment with any of our product candidates in our clinical trials results in concerns about their safety or efficacy, we and ourany collaboration partners may be unable to successfully develop or commercialize any or all of our TransCon technologies based product candidates or enter into collaborations with respect to our product candidates.
We have limited clinical data on TransCon PTH and TransCon CNP and no clinical data on our other preclinical product candidates, to indicate whether they are safe or effective forlong-term use in humans.
We have generated limited clinical data on TransCon PTH and TransCon CNP. It is unknown whetherlong-term repeated administration of TransCon PTH or TransCon CNP could result in issues that may adversely affect safety. In addition, we have generated no clinical data on our preclinical product candidates. If extended treatment with TransCon PTH, TransCon CNP, or any of our preclinical product candidates, in our clinical trials, results in any safety or efficacy concerns, we may be unable to successfully develop or commercialize our product candidates or enter into collaborations with respect to our product candidates.
We may seek orphan drug designation for some of our product candidates and we may be unsuccessful, or may be unable to maintain the benefits associated with orphan drug designation, including the potential for market exclusivity, for product candidates for which we obtain orphan drug designation.
Regulatory authorities in some jurisdictions, including the United States, may designate drugs or biologics intended to treat relatively small patient populations as orphan drug products. Under the Orphan Drug Act, the FDA may designate a drug or biologic as an orphan drug if it is intended to treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. In the European Union, the EMA’s, Committee for Orphan Medicinal Products grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting not more than five in 10,000 persons in the European Union. Orphan drug designation must be requested before submitting a BLA or NDA in the United States or an MAA in Europe.
If a drug or biologic with an orphan drug designation subsequently receives the first marketing approval for the indication for which it has such designation, the drug or biologic is entitled to a period of marketing exclusivity, which precludes the FDA from approving another marketing application for the same drug and indication for that time period, except in limited circumstances. If our competitors are able to obtain orphan drug exclusivity prior to us, for products that constitute the “same drug” and treat the same indications as our product candidates, we may not be able to have competing products approved by the applicable regulatory authority for a significant period of time. The applicable period is seven years in the United States. The applicable exclusivity period is ten years in the European Union (“EU”), but such exclusivity period can be reduced to six years if a product no longer meets the criteria for orphan designation or if the product is sufficiently profitable so that market exclusivity is no longer justified.
As part of our business strategy, we intend to pursue orphan drug designation for certain of our product candidates. For example, in June 2018 we were granted orphan drug designation by the FDA for TransCon PTH andfor the treatment of hypoparathyroidism, in February 2019, we were granted orphan drug designation by the FDA for TransCon CNP.CNP for the treatment of achondroplasia, and in April 2020, we were granted orphan drug designation by the FDA for lonapegsomatropin for the treatment of GHD. Additionally, in August 2020, we were granted orphan designation by the European Commission for TransCon CNP for the treatment of achondroplasia and in October 2020, we were granted orphan designation by the European Commission for TransCon PTH for Treatment of hypoparathyroidism. In October 2019, we were granted orphan designation by the European Commission for lonapegsomatropin for GHD. However, we may be unsuccessful in obtaining orphan drug designation or orphan designation for other product candidates, and may be unable to maintain the benefits associated with orphan drug designation.
Even if we obtain orphan drug exclusivity for any of our product candidates, that exclusivity may not effectively protect those product candidates from competition because different drugs can be approved for the same condition, and orphan drug exclusivity does not prevent the FDA from approving the same or a different drug in another indication. Even after an orphan drug is granted orphan exclusivity and approved, the FDA can subsequently approve a later application for the same drug for the same condition before the expiration of the seven-year exclusivity period if the FDA concludes that the later drug is clinically superior in that it is shown to be safer in a substantial portion of the target populations, more effective or makes a major contribution to patient care. In addition, a designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. Moreover,orphan-drug-exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if we are unable to manufacture sufficient quantities of the product to meet the needs of patients with the rare disease or condition. Orphan drug designation neither shortens the development time or regulatory review time of a drug nor gives the drug any advantage in the regulatory review or approval process.
Any biological product for which we intend to seek approval may face competition sooner than anticipated.
The Affordable Care Act, or the ACA, includes a subtitle called the Biologics Price Competition and Innovation Act of 2009, or BPCIA, which created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with anFDA-licensed reference biological product. Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until twelve years from the date on which the reference product was first licensed. During thistwelve-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing the sponsor’s own preclinical data and data from adequate andwell-controlled clinical trials to demonstrate the safety, purity and potency of its product. The law is complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation and meaning are subject to uncertainty, and any processes adopted by the FDA to implement the BPCIA could have a material adverse effect on the future commercial prospects for our biological products.
We believe that any of our future biological product candidates approved under a BLA should qualify for thetwelve-year period of exclusivity. However, there is a risk that this exclusivity could be shortened due to Congressional action or otherwise, or that the FDA will not consider our product candidates to be reference products for competing products, potentially creating the opportunity for generic competition sooner than anticipated. Other
aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation. Moreover, the extent to which a biosimilar, once approved, could be substituted for any one of our reference products in a way that is similar to traditional generic substitution fornon-biological products will depend on a number of marketplace and regulatory factors that are still developing.
We have limited direct sales and distribution capabilities and no sales experience with any of our own product candidates and we may not be able to successfully commercialize any of our product candidates.
We have limited direct sales and distribution capabilities and no sales experience with any of our own product candidates. Except for our license agreements with VisenVISEN for Greater China, we have no sales, marketing or distribution agreements for TransCon hGH, TransCon PTH, TransCon CNP, or our other product candidates. We may enter into arrangements with third partiesthird-parties to market and sell certain of our other product candidates in one or multiple geographies. We may not be able to enter into such marketing and sales arrangements with others on acceptable terms, if at all. To the extent that we enter into marketing and sales arrangements with other companies, our revenues, if any, will depend on the terms of any such arrangements and the efforts of others. These efforts may turn out not to be sufficient.
We currently have a limited sales organization and have no sales experience with any of our own product candidates. To commercialize any of our product candidates, we or ourany collaboration partners must build and/or maintain marketing, sales, distribution, managerial and othernon-technical capabilities or make arrangements with third partiesthird-parties to perform these services, and we or ourany collaboration partners may not be successful in doing so. If one or more of our product candidates receives regulatory approval, we may establish a specialty sales organization with technical expertise and supporting distribution capabilities toco-promote and/or commercialize our product candidates, which will be expensive and time consuming. As a company, we have no prior experience in the sale and distribution of pharmaceutical products and there are significant risks involved in building and managing a sales organization, including our ability to hire, retain, and incentivize qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel, comply with regulatory requirements applicable to the marketing and sale of drug products and effectively manage a geographically dispersed sales and marketing team. Any failure or delay in the development of our internal sales, marketing and distribution capabilities with respect to anon-licensed product candidate would adversely impact the commercialization of such product candidate.
We may choose to work with third partiesthird-parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems. If we are unable to enter into such arrangements on acceptable terms or at all, we may not be able to successfully commercialize our product candidates.
We rely on third partiesthird-parties to manufacture our preclinical and clinical drug supplies, and we intend to rely on third partiesthird-parties to produce commercial supplies of any approved product candidate and device.
We do not own facilities for manufacturing our products and product candidates for the potential pivotal clinical studies and/or commercial manufacturing of our products and product candidates. We depend on our collaboration partners and other third partiesthird-parties to manufacture and provide analytical services with respect to our most advanced product candidates and device.
In addition, if our product candidates are approved, to produce the quantities necessary to meet anticipated market demand, we and/or ourany collaboration partners will need to secure sufficient manufacturing capacity withthird-party manufacturers. If we and/or ourany collaboration partners are unable to produce our product candidates in sufficient quantities to meet the requirements for the launch of the product or to meet future demand, our revenues and gross margins could be adversely affected. For example, public health epidemics or pandemics, such as the novel coronavirus disease(COVID-19) currently impacting multiple jurisdictions worldwide may impact the ability of our existing or future manufacturers to perform their obligations under our manufacturing agreements with such parties. Such failure or substantial delay could materially harm our business. To be successful, our product candidates must be manufactured in commercial quantities in compliance with regulatory requirements and at acceptable costs. We and/or ourany collaboration partners will regularly need to secure access to facilities to manufacture some of our product candidates commercially. All of this will require additional funds and inspection and approval by the Competent Authorities of the Member States of the EEA, the FDA and other regulatory authorities. If we and/or our
any collaboration partners are unable to establish and maintain a manufacturing capacity within our planned time and cost parameters, the development and sales of our products and product candidates as well as our business, results of operations and prospects, and the value of our shares or ADSs could be adversely affected.
We and/or ourany collaboration partners may encounter problems with aspects of manufacturing our collaboration products and product candidates, including the following:
production yields;
quality control and assurance;
shortages of qualified personnel;
compliance with FDA and EEA regulations;
production costs; and
development of advanced manufacturing techniques and process controls.
We evaluate our options for clinical study supplies and commercial production of our product candidates on a regular basis, which may include use ofthird-party manufacturers, or entering into a manufacturing joint venture relationship with a third party. We are aware of only a limited number of companies on a worldwide basis who operate manufacturing facilities in which our product candidates can be manufactured under cGMP regulations, a requirement for all pharmaceutical products. We cannot be certain that we or our collaboration partners will be able to contract with any of these companies on acceptable terms, if at all, all of which could harm our business, results of operations and prospects, and the value of our shares or ADSs.
In addition, we, or our collaboration partners, as well as anythird-party manufacturer, will be required to register such manufacturing facilities with the FDA (and have a U.S. agent for the facility, if outside the United States), the Competent Authorities of the Member States of the EEA, and other regulatory authorities. The facilities will be subject to inspections confirming compliance with the FDA, the Competent Authorities of the Member States of the EEAs, or other regulatory authority cGMPs requirements. We do not control the manufacturing process of our product candidates, and other than with respect to our collaboration product candidates, we are dependent on our contract manufacturing partners for compliance with cGMPs regulations for manufacture of both active drug substances and finished drug products. If we or our collaboration partners or anythird-party manufacturer fails to maintain regulatory compliance, our business, financial condition and results of operations may be harmed, and the FDA, the Competent Authorities of the Member States of the EEA, or other regulatory authorities can impose regulatory sanctions that range from a warning letter to withdrawal of approval to seeking product seizures, injunctions and, where appropriate, criminal prosecution.
Under our collaborationagreements with Visen,VISEN, we are obligated to use commercially reasonable efforts to supply clinical trial material for VisenVISEN to conduct certain clinical trials, therefor, and will negotiate in good faith with VisenVISEN the terms and conditions governing our commercial supply of relevant products to Visen.VISEN. In turn, we currently rely on third partythird-party manufacturers in fulfilling our supply obligations to Visen. For additional information regarding the risks of our dependence on our collaboration partners, see the risk factors above “Item 3 D. Risk Factors—We are substantially dependent on the success of our product candidates, which may not be successful in nonclinical studies or clinical trials, receive regulatory approval or be successfully commercialized” and “Item 3 D. Risk Factors—We depend on collaboration partners to develop and conduct clinical studies with, obtain regulatory approvals for, and market and sell our collaboration product candidates, and if such collaboration partners fail to perform as expected, or are unable to obtain the required regulatory approvals for such product candidates, the potential for us to generate future revenue from such product candidates would be significantly reduced and our business would be significantly harmed.”VISEN.
If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or similar regulatory authorities, they will not be able to secure and/or maintain regulatory approval for their manufacturing facilities. In addition, we have no control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If the FDA, the Competent Authorities of the Member States of the EEA, or a similar regulatory authority does not
approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved.
We rely on our manufacturers to purchase fromthird-party suppliers the materials necessary to produce our product candidates for our clinical studies. Any significant delay or discontinuation in the supply of such materials would delay completion of our clinical studies or clinical studies conducted by our collaboration partners who rely on us for supply, and harm our business.
There are a limited number of suppliers for raw materials that we use to manufacture our drugs, and there may be a need to identify alternate suppliers to prevent a possible disruption of the manufacture of the materials necessary to produce our product candidates for our clinical studies, and, if approved, ultimately for commercial sale. We do not have any control over the process or timing of the acquisition of these raw materials by our manufacturers. Although we generally do not begin a clinical study unless we believe we have on hand, or will be able to manufacture, a sufficient supply of a product candidate to complete such study, and we currently envision that Visen,VISEN, who relies on us for clinical supply of our product candidates, would do the same, any significant delay or discontinuity in the supply of a product candidate, or the raw material components thereof, for a clinical study due to the need to replace athird-party manufacturer could considerably delay completion of our or Visen’sVISEN’s clinical studies, product testing, and potential regulatory approval of our product candidates, which could harm our business and results of operations.
Any inability to obtain suppliers, including an inability to obtain, or delay in obtaining, approval of a supplier from the Competent Authorities of the Member States of the EMA, the FDA or other regulatory authorities, would delay or prevent the clinical development and commercialization of our product candidates, and could impact our ability to meet supply obligations to collaboration partners for the development of, or future marketing and sale, of our product candidates.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
Our business exposes us to potential product liability risks which are inherent in research and development, preclinical and clinical studies, manufacturing, marketing and use of our product candidates. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability, and a breach of warranties. Claims could also be asserted under state consumer protection acts. Product liability claims may be expensive to defend and may result in judgmentsjudgements against us which are potentially punitive. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
decreased demand for our product candidates;
injury to our reputation;
withdrawal of clinical trial participants;
costs to defend the related litigation;
a diversion of management’s time and our resources;
substantial monetary awards to trial participants or patients;
regulatory investigations, product recalls or withdrawals, or labeling, marketing or promotional restrictions;
loss of revenue; and
the inability to commercialize orco-promote our product candidates.
It is generally necessary for us to secure certain levels of insurance as a condition for the conduct of clinical studies. We believe that our product liability insurance for clinical studies is sufficient to cover claims. We currently maintain liability insurance with certain specified coverage limits. We cannot be certain that the insurance policies will be sufficient to cover all claims that may be made against us. Our inability to obtain and maintain sufficient product liability insurance at an acceptable cost and scope of coverage to protect against potential product liability claims could prevent or inhibit the commercialization of any products we develop. We currently carry product liability insurance covering use in our clinical trials in the amount of $20 million in the aggregate on our primary insurance policy and $40 million in the aggregate on our excess insurance policy. Any claim that may be brought against us could result in a court judgmentjudgement or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various, limits, exclusions and deductibles, and given these various limits, exclusions and deductibles, we may be subject to a product liability claim for which we have no coverage. We will have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts. Moreover, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses. Product liability insurance is expensive, difficult to obtain and may not be available in the future on acceptable terms.
We will need to significantly increase the size of our organization and we may have difficulties in managing our growth and expanding our operations successfully.
As of December 31, 2019,2020, we had 330482 full-time employees worldwide, with key facilities in Denmark, Germany, and the United States. As we and/or our collaboration partners advance our product candidates through the development and commercialization process, we will need to expand managerial, operational, financial and other resources to manage our operations, preclinical and clinical trials, research and development activities, regulatory filings, manufacturing and supply activities, and any marketing and commercialization activities or contract with other organizations to provide these capabilities for us. As operations expand, we expect that we will need to manage additional relationships with various collaboration partners, suppliers and other organizations. Our ability to manage our operations and growth requires us to continue to improve our operational, financial and management controls, reporting systems and procedures across a global organization. Such growth could place a strain on our administrative and operational infrastructure. We may not be able to make improvements to our management information and control systems in an efficient or timely manner and may discover deficiencies in existing systems and controls. Our management, personnel, systems and facilities currently in place may not be adequate to support this future growth. Our need to effectively execute our growth strategy requires that we either internally, together with our collaboration partners or through third partythird-party contractors, as applicable:
expand our general and administrative functions;
identify, recruit, retain, incentivize and integrate additional employees;
manage our internal development efforts effectively while carrying out our contractual obligations to third parties;third-parties;
establish and build a marketing and commercial organization; and
continue to improve our operational, legal, financial and management controls, reporting systems and procedures.
If we are not able to attract, retain and motivate necessary personnel to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy.
We incur significant costs as a result of operating as a public company, and our management devotes substantial time to compliance initiatives. We may fail to comply with the rules that apply to public companies, including Section 404 of theSarbanes-Oxley Act of 2002, which could result in sanctions or other penalties that would harm our business.
We incur significant legal, accounting and other expenses as a public company, including costs resulting from public company reporting obligations under the Securities Exchange Act of 1934, as amended, or the Exchange Act, and regulations regarding corporate governance practices. Our senior management and other personnel need to devote a substantial amount of time to ensure that we maintain compliance with all of these requirements. Moreover, the reporting requirements, rules and regulations increase our legal and financial compliance costs and make some activities more time consuming and costly. Any changes we make to comply with these obligations may not be sufficient to allow us to satisfy our obligations as a public company on a timely basis, or at all. These reporting requirements, rules and regulations, coupled with the increase in potential litigation exposure associated with being a public company, could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors or board committees or to serve as members of our senior management, or to obtain certain types of insurance, including directors’ and officers’ insurance, on acceptable terms.
We are subject to Section 404 of TheSarbanes-Oxley Act of 2002, or Section 404, and the related rules of the Securities and Exchange Commission, or SEC, which generally require our senior management and independent registered public accounting firm to report on the effectiveness of our internal control over financial reporting. Beginning with the year ended December 31, 2015, Section 404 requiredrequires an annual management assessment of the effectiveness of our internal control over financial reporting, and beginning with the year ended December 31, 2018, we are required to include an opinion from our independent registered public accounting firm on the effectiveness of our internal controls over financial reporting.
As we grow our business and enter into new activities, and as the reporting requirements increase, we may identify deficiencies and be unable to remediate them before we must provide the required reports. Furthermore, if we have a material weakness in our internal controls over financial reporting, we may not detect errors on a timely basis and our consolidated financial statements may be materially misstated. We may not be able to conclude on an ongoing basis that we have effective internal control over financial reporting, which could harm our operating results, cause investors to lose confidence in our reported financial information and cause the trading price of the ADSs to fall. In addition, as a public company we are required to file accurate and timely annual reports with the SEC under the Exchange Act. Any failure to report our financial results on an accurate and timely basis could result in sanctions, lawsuits, delisting of the ADSs from theThe Nasdaq Global Select Market or other adverse consequences that would harm our business.
Our operating results may vary significantly from period to period and these variations may be difficult to predict.
Our potential future revenues and operating results are expected to vary significantly from period to period due to a number of factors. Many of these factors are outside of our control. These factors include:
the timing of regulatory approvals, if any, for our most advanced product candidates;
the initiation of intellectual property litigation by third partiesthird-parties or by us;
the amount and timing of operating costs and capital expenditures relating to the expansion of our business operations and facilities;
the timing of the commencement, completion or termination of collaboration agreements;
the timing and amount of payments to us under our collaboration agreements, if any;
the introduction of new products and services by us, our collaboration partners or our competitors;
delays in preclinical testing and clinical studies;
changes in regulatory requirements for clinical studies;
costs and expenses associated with preclinical testing and clinical studies; and
payment of license fees for the right to usethird-party proprietary rights.rights, if any.
Our revenues in any particular period may be lower than we anticipate and, if we are unable to reduce spending in that period, our operating results will be harmed.
We may engage in strategic transactions that could impact our liquidity, increase our expenses and present significant distractions to our management.
We may consider strategic transactions, such as acquisitions of companies, asset purchases, andin-licensing orout-licensing of products, product candidates or technologies. Additional potential transactions that we may consider include a variety of different business arrangements, includingspin-offs, strategic partnerships, joint ventures, restructurings, divestitures, business combinations and investments. Any such transaction may require us to incurnon-recurring or other charges, may increase ournear- andlong-term expenditures and may pose significant integration challenges or disrupt our senior management or business, which could adversely affect our operations and financial results. For example, these transactions may entail numerous operational and financial risks, including:
up-front, milestone and royalty payments, equity investments and financial support of new research and development candidates including increase of personnel, all of which may be substantial;
exposure to unknown liabilities, including potential indemnification claims from a potentialspin-off orout-license of certain of our intellectual property rights;
disruption of our business and diversion of our management’s time and attention to develop acquired products, product candidates or technologies;
incurrence of substantial debt or dilutive issuances of equity securities to pay for acquisitions;
higher-than-expected acquisition and integration costs;
lower-than-expected benefits, fromout-licensing or selling our technology, intellectual property or any of our subsidiaries or, fromin-licensing intellectual property or purchasing assets;
write-downs of assets or goodwill or impairment charges;
difficulty and cost in combining or separating the operations and personnel of any acquired or sold businesses with our existing operations and personnel;
impairment of relationships with key suppliers or customers of any acquired or sold businesses due to changes in our senior management and ownership; and
inability to retain key employees of any acquired businesses.
Accordingly, although we cannot be certain that we will undertake or successfully complete any transactions of the nature described above, any transactions that we do complete may be subject to the foregoing or other risks, and could harm our business, results of operations, financial condition and prospects.
Exchange rate fluctuations or abandonment of the euro currency may harm our results of operations and financial condition.
Due to the international scope of our operations, fluctuations in exchange rates, particularly between the euro,Euro, the Danish krone and the U.S. dollar,Dollar, may adversely affect us. Although we are based in Denmark, we source research and development, manufacturing, consulting and other services from several countries. In addition, our arrangements with our collaboration partners are denominated in euros and U.S. dollars. Further, potential future revenue may be derived from abroad, including from the United States. We currently attempt to limit our exposure to exchange rate risks by maintaining cash positions in the currencies in which we expect to incur the majority of our future expenses; however, for a variety of reasons we may be unable to maintain cash positions in the currencies in which we expect to incur the majority of our future expenses and we may fail to predict the currency of our future
expenses, accurately or at all. As a result, our business and the price of the ADSs may be affected by fluctuations in foreign exchange rates between the euroEuro and these other currencies, which may also have a significant impact on our reported results of operations and cash flows from period to period. We currently do not enter into foreign exchange contracts to cover our exposure to exchange rate fluctuations, or any other form of exchange rate hedging arrangements. If we fail to manage foreign exchange risk adequately our business, results of operations and prospects, and the value of our shares or ADSs may be adversely affected.
In addition, the possible abandonment of the euroEuro by one or more members of the European UnionEU could harm our business in the future. Despite measures taken by the European Union to provide funding to certain E.U.EU member states in financial difficulties and by a number of European countries to stabilize their economies and reduce their debt burdens, it is possible that the euroEuro could be abandoned in the future as a currency by countries that have adopted its use. This could lead to there-introduction of individual currencies in one or more E.U.EU member states. The effects on our business of a potential dissolution of the European Union, the exit of one or more E.U.EU. member states from the European Union or the abandonment of the euroEuro as a currency, are impossible to predict with certainty, and any such events could harm our business, financial condition and results of operations.
The United Kingdom’s withdrawal from the European Union may have a negative effect on global economic conditions, financial markets and our business.
Following a national referendum and enactment of legislation by the government of the United Kingdom, the United Kingdom formally withdrew from the European Union on January 31, 2020 and entered intoratified a transition period during which it will continuetrade and cooperation agreement governing its ongoing and complex negotiationsfuture relationship with the European Union. The agreement, which was applied provisionally from January 1, 2021 until it is ratified by the European Parliament and the Council of the European Union, relating toaddresses trade, economic arrangements, law enforcement, judicial cooperation and a governance framework including procedures for dispute resolution, among other things. Because the future trading relationshipagreement merely sets forth a framework in many respects and will require complex additional bilateral negotiations between the parties. SignificantUnited Kingdom and the European Union as both parties continue to work on the rules for implementation, significant political and economic uncertainty remains about whetherhow the precise terms of the relationship between the parties will differ materially from the terms before withdrawal, as well as about the possibility that aso-called “no deal” separation will occur if negotiations are not completed by the end of the transition period. withdrawal.
These developments, or the perception that any of themrelated developments could occur, have had and may continue to have a material adverse effect on global economic conditions and the stability of global financial markets, and may significantly reduce global market liquidity, and restrict the ability of key market participants to operate in certain financial markets.markets or restrict our access to capital. Any of these factors could depress economic activity and restrict our access to capital, which could have a material adverse effect on our business, financial condition and results of operations and reduce the price of the ADSs.
Risks associated with our international operations, including seeking and obtaining approval to commercialize our product candidates in foreign jurisdictions, could harm our business.
We engage extensively in international operations, which include seeking marketing approval for certain of our product candidates in foreign jurisdictions. We expect that we are or will be subject to additional risks related to entering into these international business markets and relationships, including:
different regulatory requirements for drug approvals in foreign countries;
differing U.S. andnon-U.S. drug import and export rules;
reduced protection for intellectual property rights in foreign countries;
unexpected changes in tariffs, trade barriers and regulatory requirements;
different reimbursement systems, and different competitive drugs;
economic weakness, including inflation, or political instability in particular foreign economies and markets;
compliance with tax, employment, immigration and labor laws for employees living or traveling abroad;
foreign taxes, including withholding of payroll taxes;
foreign currency fluctuations, which could result in increased operating expenses and reduced revenues, and other obligations incident to doing business in another country;
workforce uncertainty in countries where labor unrest is more common than in the United States;
production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad;
potential liability resulting from work conducted by these distributors;
regulatory and compliance risks that relate to maintaining accurate information and control over sales and activities that may fall within the purview of the FCPA, its books and records provisions, or its anti-bribery provisions; and
business interruptions resulting from geopolitical actions, including war and terrorism, or natural disasters.
The manufacture of our TransCon product candidates is dependent upon third partythird-party manufacturers that are based in other parts of the world, including Europe and Japan. This manufacturing process requires that the components used in our product candidates are transported long distances, through multiple countries, which increases the risk that issues in the global supply chain or other disruptions to the international marketplace could harm our business.
The parent drug, drug substance, drug product and other components of our product candidates are currently acquired fromsingle-source suppliers. The loss of these suppliers, or their failure to supply could materially and adversely affect our business.
Our growth hormone parent drug as well as our TransCon hGHlonapegsomatropin drug substance are supplied by Fujifilm Diosynth Biotechnologies UK Limited, or Fujifilm, pursuant to our agreement with Fujifilm. TransCon hGHlonapegsomatropin drug product in vials is manufactured by Vetter Pharma Fertigung, or Vetter, pursuant to our agreement with Vetter. TransCon hGHlonapegsomatropin drug product in dual chamber cartridges will be supplied by Vetter for use in our drug delivery device made by Philips Medisize A/S (formerly Medicom Innovation Partner A/S). The intermediates of our proprietary TransCon linkers are made by CARBOGEN AMCIS AG under an agreement with CARBOGEN AMCIS AG and accompanying purchase orders. For products that utilize soluble TransCon carriers, NOF Corporation (Japan), or NOF, supplies PEGs. Furthermore, NOF is responsible for coupling the TransCon linker used for TransCon hGHlonapegsomatropin to methoxy PEG, or mPEG, under manufacturing agreements and accompanying purchase orders. Our PTH as well as our TransCon PTH drug substance is supplied by Bachem, Switzerland, pursuant to our agreement with Bachem. TransCon PTH drug product in vials is manufactured by Baccinex, SA, Switzerland in collaboration with Bachem. We expect Vetter to manufacturemanufactures the TransCon PTH drug product in cartridges and assembleassembles the cartridges with a drug delivery device made by Ypsomed AG. Intermediate for TransCon CNP is supplied by Corden Pharma, Switzerland and CNP drug substance is supplied by Wacker Biotech, Germany. Our TransCon CNP drug product in vials is manufactured by Vetter pursuant to our agreement with Vetter. WeWith the exception of Lonza, who will be supplying Transcon hGH drug substance, we do not currently have any other suppliers for the drug substance, drug product or other components of our product candidates for TransCon hGH,lonapegsomatropin, TransCon PTH and TransCon CNP, although we believe that there are alternate sources of supply that could satisfy our clinical and commercial requirements, we cannot provide assurance that identifying alternate sources and establishing relationships with such sources would not result in significant delays in the development of our product candidates. Additionally, we may not be able to enter into supply arrangements with alternative suppliers on commercially reasonable terms or at all. A delay in the development of our product candidates or having to enter into a new agreement with a different third partythird-party on less favorable terms than we have with our current suppliers could have a material adverse impact upon on our business.
We may not be successful in our efforts to identify additional product candidates based on our TransCon technologies.
An important element of our strategy is to develop new products and product candidates based on our TransCon technologies. Research programs to identify new product candidates require substantial technical, financial and human resources. These research programs may initially show promise in identifying potential product candidates, yet fail to yield product candidates for clinical development for a number of reasons, including that:
the research methodology used may not be successful in identifying potential product candidates; or
potential product candidates may, on further study, be shown to have inadequate efficacy, harmful side effects or other characteristics suggesting that they are unlikely to be effective or safe products, or that they may not be sufficiently differentiated or offer substantial improvement over the currently available treatment options or standard of care in a given therapeutic category.
If we are unable to develop suitable product candidates through internal research programs or otherwise, we will not be able to increase our revenues in future periods, which could harm our business, results of operations and prospects, and the value of our shares or ADSs.
We are highly dependent on the services of our President and Chief Executive Officer, Jan Møller Mikkelsen, and if we are not able to retain this member of our senior management or recruit additional management, clinical and scientific personnel, our business will suffer.
Our success depends in part on our continued ability to attract, retain and motivate highly qualified personnel. We may not be able to attract or retain qualified management and scientific and clinical personnel in the future due to the intense competition for qualified personnel among biotechnology, pharmaceutical and other businesses. Our industry has experienced a high rate of turnover of management personnel in recent years. If we are not able to attract, retain and motivate necessary personnel to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy.
In particular, we are highly dependent upon Jan Møller Mikkelsen, our President and Chief Executive Officer. The loss of services of this individual could result in delays in product development and harm our business.
We may have difficulties in attracting and retaining key personnel, and if we fail to do so our business may suffer.
We are highly dependent on the principal members of our senior management and scientific staff, the loss of whose services could adversely affect the achievement of planned development objectives. AlthoughIn addition, we have not historically experienced uniquecould experience difficulties attracting and retaining qualified employees we could experience such problems in the future. For example, competition for qualified personnel in the biotechnology and pharmaceuticals field is intense due to the limited number of individuals who possess the skills and experience required by our industry. This is particularly true in Heidelberg, Germany where we operate our research and development activities. As such, we could have difficulty attracting experienced personnel to our company and may be required to expend significant financial resources in our employee recruitment and retention efforts.
For us to further expand our product development plans, we will need to hire additional qualified scientific personnel to perform research and development. We will also need to hire personnel with expertise in clinical testing, government regulation, sales and marketing, and finance, and might need to hire personnel with expertise in manufacturing. We may not be able to attract and retain personnel on acceptable terms, given the competition for such personnel among biotechnology, pharmaceutical and healthcare companies, universities andnon-profit research institutions. Although we may be successful in attracting and retaining suitably qualified scientific personnel, there can be no assurance that we will be able to attract and retain such personnel on acceptable terms given the competition for experienced scientists from numerous pharmaceutical and chemical companies, specialized biotechnology firms, universities and other research institutions. Our failure to do so could adversely affect our business, results of operations and prospects, and the value of our shares or ADSs.
Our internal computer systems, or those of our CROs or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs and other critical business functions.
Despite the implementation of security measures, our internal computer systems and those of our CROs and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war, telecommunication and electrical failures,failures. Attacks upon information technology systems are increasing in their frequency, levels of persistence, sophistication and public health epidemicsintensity, and are being conducted by sophisticated and organized groups and individuals with a wide range of motives and expertise. As a result of the COVID-19 pandemic, we may also face increased cybersecurity risks due to our reliance on internet technology and the number of our employees who are working remotely, which may create additional opportunities for cybercriminals to exploit vulnerabilities. Furthermore, because the techniques used to obtain unauthorized access to, or pandemics, such as the novel coronavirus disease(COVID-19) currently impacting multiple jurisdictions worldwide. Whileto sabotage, systems change frequently and often are not recognized until launched against a target, we have not experienced any such system failure, accidentmay be unable to anticipate these techniques or implement adequate preventative measures. We may also experience security breach to date, ifbreaches that may remain undetected for an extended period.If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our programs. For example, the loss of
clinical trial data from completed or ongoing clinical trials for any of our product candidates could result in delays in our regulatory approval efforts, and the loss of research data could result in delays of our research and development efforts and it would be expensive to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of our product candidates could be delayed.
The global pandemic caused byCOVID-19 could materially adversely impact our business, including our clinical trials.trials, supply chain operation, regulatory timelines and commercial activities.
In December 2019, a novel strain of coronavirus,COVID-19, was reported to have surfaced in Wuhan, China. Since then, theCOVID-19 coronavirus has been declared by WHO to be a worldwide pandemic. As a result of the rapidly growing spread ofCOVID-19 throughout the areas we operate, we may experience disruptions that could severely impact our business and clinical trials, including:
delays or difficulties in enrolling and retaining patients in our clinical trials;trials, which could potentially have a negative impact on clinical trial timelines;
delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff;
��
significant increases in expenses required to manage impacts to our business to complete our planned operations within our projected timelines;
diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials;
interruption of key clinical trial activities, such as clinical trial site monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers and others;
limitations in employee resources that would otherwise be focused on the conduct of our clinical trials, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people;
delays in receiving approval from local regulatory authorities to initiate our planned clinical trials;
delays in clinical sites receiving the supplies and materials needed to conduct our clinical trials;
interruption in global shipping that may affect the transport of clinical trial materials, such as comparator drugs used in certain of our clinical trials;
interruptions in our global supply chain with regards to clinical trial and potential commercial grade material;
changes in local regulations as part of a response to theCOVID-19 outbreak which may require us to change the ways in which our clinical trials are conducted, which may result in unexpected costs, or to discontinue the clinical trials altogether;
delays in necessary interactions with local regulators, ethics committees and other important agencies and contractors due to limitations in employee resources or forced furlough of government employees; and
refusal of regulatory authorities to accept data from clinical trials in these affected geographies.
in conducting our clinical trials, suppliers may experience delays in providing necessary equipment, consumables and services, which potentially could cause temporary delays in clinical trial activities;
global demand for COVID-19 vaccines could result in contract manufactures not having sufficient capacity to meet scheduled manufacturing. In addition, sourcing of certain types of raw materials, consumables and equipment could result in scheduled manufacturing being delayed or postponed;
travel restrictions and local outbreaks of COVID-19 could restrict authorities from performing site inspections in connection with their review procedures of marketing applications for lonapegsomatropin, which could potentially delay the commercial launch; and
our commercial launch strategy, including for lonapegsomatropin, could be negatively impacted by patients not being able to see their physicians, and similarly, our commercial team not being able to meet with physicians, which could both have a negative impact on the commercial launch strategy.
In addition, the pandemic has caused, and is likely to cause further, disruption to global financial markets. This may reduce our ability to access capital on favorable terms or to access capital at all. Furthermore, sustained adverse market events (such as a recession or depression) resulting from the pandemic could materially and adversely affect our business and the price of our ADSs.
The global outbreak ofCOVID-19 continues to rapidly evolve. The extent to which theCOVID-19 coronavirus further impacts our business and clinical trials will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the speed and extent of geographic spread of the disease, the duration of the outbreak, travel restrictions and social distancing in the affected areas, business closures or business disruptions and the effectiveness of actions taken in the affected areas to contain and treat the disease.
Risks Related to Government Regulatory and Legal Requirements
The regulatory approval processes of the EMA, the FDA and comparable authorities are lengthy, time consuming, and inherently unpredictable. If we are ultimately unable to obtain regulatory approval for our product candidates, our business will be substantially harmed.
The research, testing, manufacturing, labeling, approval, selling, import, export, marketing and distribution of drug products are subject to extensive regulation by the FDA, E.U.EU legislative bodies and other regulatory authorities in the United States, the EEA and other jurisdictions, which regulations differ from country to country. Neither we nor any of our collaboration partners isWe are not permitted to market any drug product in the United States until we receive marketing approval from the FDA. Equally, neither we nor any of our collaboration partners isare not permitted to market any drug product in the EEA until we receive a marketing authorization from the EMA or EEA Member State Competent Authorities. In September 2020, the FDA filed our BLA for lonapegsomatropin for the treatment for pediatric GHD for substantive review and set a target action date of June 25, 2021, and in September 2020, we submitted a marketing authorization application to the EMA for lonapegsomatropin for the treatment of pediatric hormone growth deficiency. We have not submitted an application or obtained marketing approval for any of our other product candidates anywhere in the world.
Obtaining regulatory approval of an NDA or BLA, can be a lengthy, expensive and uncertain process. In addition, failure to comply with FDA and other applicable U.S., EEA and foreign regulatory requirements may subject us to administrative or judicially imposed sanctions or other actions, including:
warning letters;
civil and criminal penalties;
injunctions;
withdrawal of regulatory approval of products;
product seizure or detention;
product recalls;
total or partial suspension of production; and
refusal to approve pending NDAs or BLAs, marketing authorization applications, or supplements to approved NDAs or BLAs or extensions or variations to marketing authorizations.
Prior to obtaining approval to commercialize a drug or biological product candidate in the United States, the EEA or other regions, we or our collaboration partners must demonstrate with substantial evidence fromwell-controlled clinical trials, and to the satisfaction of the EMA, the FDA or other similar regulatory authorities, that such drug candidates are safe and effective for their intended uses. The number of nonclinical studies and clinical trials that will be required for FDA, or EMA approval varies depending on the product candidate, the disease or condition that the product candidate is designed to address, and the regulations applicable to any particular product candidate. Results from nonclinical studies and clinical trials can be interpreted in different ways. Even if we believe the nonclinical or clinical data for our product candidates are promising, such data may not be sufficient to support approval by the EMA, the FDA and other regulatory authorities. Administering drug or biological product candidates to humans may produce undesirable side effects, which could interrupt, delay or halt clinical trials and result in the EMA, the FDA or other regulatory authorities denying approval of a product candidate for any or all targeted indications.
The time required to obtain approval by the EMA, the FDA and comparable authorities is unpredictable, typically takes many years following the commencement of clinical studies, and depends upon numerous factors. The EMA, the FDA and comparable authorities have substantial discretion in the approval process and we may encounter
matters with the EMA, the FDA or such comparable authorities that requires us to expend additional time and resources and delay or prevent the approval of our product candidates. For example, the FDA or EMA may require us to conduct additional studies or trials for drug or biological product candidates either prior to orpost-approval, such as additionaldrug-drug interaction studies or safety or efficacy studies or trials, or it may object to elements of our clinical development program such as the number of subjects in our current clinical trials from the United States or Europe. In addition, approval policies, regulations or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions, which may cause delays in the approval or result in a decision not to approve an application for regulatory approval. Despite the time and expense exerted, failure can occur at any stage. Applications for our product candidates could fail to receive regulatory approval for many reasons, including but not limited to the following:
the EMA, the FDA or other comparable foreign regulatory authorities may disagree with the design or implementation of our, or ourany collaboration partners’, clinical studies;
the population studied in the clinical program may not be sufficiently broad or representative to assure safety in the full population for which approval is sought;
the EMA, the FDA or comparable foreign regulatory authorities may disagree with the interpretation of data from preclinical studies or clinical studies;
the data collected from clinical studies of our product candidates may not be sufficient to support the submission of aan NDA or BLA, marketing authorization application, or other submission or to obtain regulatory approval in the United States, the EEA or elsewhere;
we, or ourany collaboration partners, may be unable to demonstrate to the EMA, the FDA or comparable foreign regulatory authorities that a product candidate’srisk-benefit ratio for its proposed indication is acceptable;
the EMA, the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes, test procedures and specifications, or facilities ofthird-party manufacturers responsible for clinical and commercial supplies; and
the approval policies or regulations of the EMA, the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval.
This lengthy approval process, as well as the unpredictability of the results of clinical studies, may result in our failure to obtain regulatory approval to market any of our product candidates, which would significantly harm our business, results of operations, and prospects. Additionally, if the EMA, the FDA or comparable foreign regulatory authorities require that we conduct additional clinical studies, place limitations on our label, delay approval to market our product candidates or limit the use of our products, our business and results of operations may be harmed.
In addition, even if we were to obtain approval, regulatory authorities may approve any of our product candidates for fewer or more limited indications than we request, may not approve the price we intend to charge for our products, may grant approval contingent on the performance of costlypost-marketing clinical trials, may impose a REMS, or may approve a product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. Any of the foregoing scenarios could harm the commercial prospects for our product candidates.
We do not have and may never obtain the regulatory approvals we need to market our product candidates.
We have not yet received any regulatory approvals required for the commercial sale of TransCon hGH,lonapegsomatropin, TransCon PTH, TransCon CNP, or any of our other product candidates in the United States, the EMA or in any other jurisdiction. Furthermore,In June 2020, we submitted a BLA with the FDA for lonapegsomatropin, for the treatment for pediatric GHD. In September 2020, the FDA filed our BLA for lonapegsomatropin for substantive review and set a target action date of June 25, 2021, and in September 2020, we submitted a marketing authorization application to the EMA for lonapegsomatropin for the treatment of pediatric hormone growth deficiency We have yet to submit an NDA or BLA to the FDA for TransCon PTH, TransCon CNP, or any of our other product candidates. We have yet to submit a Marketing Authorization Application, or MAA, to the EMA, national regulatory authorities in Europe or to any international regulatory authorities for TransCon PTH, TransCon CNP, or any of our other product candidates. We have only limited experience in filing and pursuing applications
necessary to obtain regulatory approval or licensure, and we cannot be certain that any of our product candidates will be approved or licensed for marketing. The process of applying for regulatory approval is expensive, often takes many years and can vary substantially based upon the type, complexity and novelty of the product candidates involved. If any or all of our product candidates are not approved, this could harm our business, results of operations and prospects, and the value of our shares or ADSs.
If we are unable to file an MAA for approval to the EMA for any of our other product candidates, or if we are required to generate additional data related to safety and efficacy, to obtain approval from the FDA for any of our product candidates, we may be unable to meet our anticipated development and commercialization timelines.
While we have an active IND with the FDA for TransCon hGH, and we have completed substantive discussions with the FDA regarding the development of TransCon hGH in pediatric growth hormone deficiency, and we believe we are satisfying the necessary criteria, there is no guarantee that FDA requirements will not change between now and the time of our filing in the United States. We have not yet filed an MAA with the EMA for any of our product candidates. Depending on the data that may be required by the EMA for approval, we may be required to conduct substantial new research and development activities beyond those in which we currently plan to engage to obtain approval of our product candidates. Such additional new research and development activities would be costly and time consuming.
We have developed anauto-injector to facilitate the administration of the product byend-users and additional time may be required to obtain regulatory approval for ourauto-injector.
We have developed anauto-injector with Phillips Medisize A/S (formerly Medicom Innovation Partner A/S) to facilitate the administration of TransCon hGHlonapegsomatropin by patients. In addition, we are developinghave developed a drug delivery device with Ypsomed to facilitate the administration of TransCon PTH by patients. We anticipate the EMA, the FDA and other similar regulatory authorities may require approval of ourauto-injector and TransCon PTH drug delivery device as part of the approval of TransCon hGHlonapegsomatropin and TransCon PTH. Because of ourauto-injector and TransCon PTH drug delivery device, the FDA’s review of TransCon hGHlonapegsomatropin and/or TransCon PTH may include the participation of both the FDA’s Center for Drug Evaluation and Research and the FDA’s Center for Devices and Radiological Health, which may complicate or prolong the review, and in the EEA the EMA’s review may require the involvement of an EU Notified Body. As a result, we may experience delays for ourauto-injector and TransCon hGHlonapegsomatropin and/or our drug delivery device of TransCon PTH and TransCon PTH.
Safety issues with the parent drugs or other components of our product candidates, or with approved products of third partiesthird-parties that are similar to our product candidates, could give rise to delays in the regulatory approval process.
Our product development portfolio consists of prodrugs that are new molecular entities that incorporate existing parent drug molecules, many of which have been previously approved by the EMA, the FDA or other foreign regulatory authorities. Discovery of previously unknown problems with any of the parent drugs that we use in our TransCon product candidates may result in restrictions on its permissible uses, including withdrawal of the product from the market.
Additionally, problems with approved parent drugs marketed by third partiesthird-parties that utilize the same therapeutic target as the parent drug we use in our TransCon product candidates could adversely affect the development of our product candidates.
Any failure or delay in commencing or completing clinical trials or obtaining regulatory approvals for our product candidates would delay commercialization of the product candidates and severely harm our business and financial condition.
We are subject to extensive and costly government regulation. If we fail to obtain or maintain governmental approvals, we will not be able to commercialize our product candidates and our business will suffer.
Pharmaceutical products, including product candidates employing our TransCon technologies, are subject to extensive and rigorous government regulation. The FDA, the EMA and other regulatory authorities regulate the development, testing, manufacture, safety, efficacy,record- keeping, recordkeeping, labeling, storage, approval, advertising,
promotion, sale and distribution of pharmaceutical products. If products employing our TransCon technologies are marketed in countries outside of the European Union and the United States, they will also be subject to extensive regulation by other governments. The regulatory review and approval or licensing process, including preclinical testing and clinical studies of each product candidate, is lengthy, expensive and uncertain. Securing marketing approval requires the submission of extensive preclinical and clinical data and supporting information to the FDA, EMA and/or EEA Competent Authorities for each indication to establish the candidate’s safety and efficacy. The approval process takes many years, requires substantial resources, involvespost-marketing surveillance, and may involve ongoingpost-marketing studies. While clinical studies are designed with scientific advice from regulatory authorities, such plans must often be put in place years in advance of application for marketing approval. At the time of such application, the clinical and regulatory environment may have changed significantly as a result of new scientific discoveries, competitor product evaluations, changes in medical health care policies, new technical standards and other factors beyond our control.
Regulators can refuse marketing approval, or can require us or our collaboration partners to repeat previous clinical studies or conduct further clinical studies. Apre-approval inspection of manufacturing facilities by regulatory authorities may need to be completed before marketing approval can be obtained, and such facilities will be subject to periodic inspections that could prevent or delay marketing approval, or require the expenditure of financial or other resources to address. In addition, as part of a review process, the FDA may refer an application for a novel drug or biologic to an advisory committee to review, evaluate and provide a recommendation as to whether the application should be approved and
under what conditions. At the time of our mid-cycle review with the FDA relating to our BLA submitted to the FDA for approval of lonapegsomatropin for the treatment of pediatric GHD, the FDA indicated that it had no plans for an Advisory Committee Meeting for our BLA submission; however, there can be no assurance that the FDA will not convene an Advisory Committee Meeting relating to our BLA submission. If we or our collaboration partners do not succeed in obtaining regulatory approval, or succeed only after delays, this could have a material effect on our ability to generate revenues. Delays in obtaining regulatory approvals may:
adversely affect the successful commercialization of any product that we or our collaboration partners develop;
impose costly procedures on us or ourany collaboration partners;
diminish any competitive advantages in the market placemarketplace that we or ourany collaboration partners may attain; and
adversely affect our receipt of revenues or royalties.
Material changes to an approved product, such as manufacturing changes or additional labeling claims, require further FDA and EMA and/or EEA Competent Authorities review and approval before marketing. Once obtained, any approvals may be withdrawn or revoked because of unforeseen safety, effectiveness or potency concerns or failure to comply with governmental regulations. Further, if we, ourany collaboration partners or our contract manufacturers fail to comply with applicable FDA, EMA, and/or EEA Competent Authorities regulatory requirements at any stage during the regulatory process, the FDA, EMA, and/or EEA Competent Authorities and other regulatory authorities may impose sanctions, including:
delays;
warning letters;
fines;
importation restrictions;
product recalls or seizures;
injunctions;
refusal of the FDA, EMA or other regulatory authorities to review pending market approval applications or supplements to approval applications;
total or partial suspension of production;
suspension or debarment from sellingFDA-regulated products to the U.S. government for periods of time that vary depending on the cause of such suspension or debarment;
civil penalties;
withdrawal or revocation of previously approved marketing applications or licenses; and
criminal prosecutions.
Even if we receive regulatory approval for a product candidate, we will be subject to ongoing regulatory obligations and review, which may result in significant additional expense. Additionally, any product candidates, if approved, could be subject to labeling and other restrictions and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our products.
The governmental regulation of the development of products and product candidates extends beyond clinical studies to approval required for their sale and monitoring of such products after sale. This regulation, approval and monitoring is the responsibility of numerous authorities in Denmark, the United States, the European Union and authorities in other territories. Following any regulatory approval of a product candidate, we, ourany collaboration partners and the manufacturers of our products will be subject to continuing regulatory obligations, including safety reporting requirements, regulatory oversight of product promotion and marketing, and cGMP requirements. Furthermore, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These regulations cover all aspects of manufacturing, testing, quality control and record keepingrecordkeeping of our products. If we or ourany collaboration partners or manufacturers fail to comply with applicable regulatory requirements, we may be subject to fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution. These requirements include submissions of safety and otherpost-marketing information and reports, registration, as well as continued compliance with cGMPs and GCPs for any clinical trials that we conductpost-approval. As such, we and our third partythird-party contract manufacturers will be subject to continual review and periodic inspections to assess compliance with regulatory requirements. Accordingly, we and others with whom we work must continue to expend time, money, and effort in all areas of regulatory compliance, including manufacturing, production, and quality control. Regulatory authorities may also impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costlypost-marketing studies. Furthermore, any new legislation addressing drug safety issues could result in delays or increased costs to assure compliance.
In the United States, advertising and promotional materials must comply with FDA rules in addition to other potentially applicable U.S. laws. In particular, the promotional claims that we would be permitted to make for our products would be limited to those supported by (or, under FDA guidance, consistent with) the approved product labeling. In addition, under the Federal Food, Drug, and Cosmetic Act, particular restrictions are placed on the distribution of human growth hormone products, potentially including TransCon hGH.lonapegsomatropin. The distribution of product samples to physicians must also comply with the requirements of the Prescription Drug Marketing Act. Manufacturing facilities remain subject to FDA inspection and must continue to adhere to International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use and the FDA’s cGMP requirements. Application holders must obtain FDA approval for many product and manufacturing changes, depending on the nature of the change. Sales, marketing, and scientific/educational grant programs must comply with the U.S.Anti-Kickback Statute, the False Claims Act, as amended, the privacy regulations promulgated under the Health Insurance Portability and Accountability Act, or HIPAA, and similar state laws. Certain payments and other transfers of value to U.S. licensed physicians (as defined under statute) and teaching hospitals must be reported under the Physician Payments Sunshine Act. Pricing and rebate programs must comply with the Medicaid Drug Rebate Program requirements of the Omnibus Budget Reconciliation Act of 1990, as amended, and the Veterans Health Care Act of 1992, as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. All of these activities are also potentially subject to U.S. consumer protection and unfair competition laws.
We will also be required to report certain adverse reactions and production problems, if any, to the FDA, and to comply with requirements concerning advertising and promotion for our products. Promotional communications with respect to prescription pharmaceutical products are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s approved label. As such, we may not promote our products for indications or uses for which they do not have FDA approval.
Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with ourthird-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
warning letters, fines or holds on clinical trials;
restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market or voluntary or mandatory product recalls;
injunctions or the imposition of civil or criminal penalties;
suspension or revocation of existing regulatory approvals;
suspension of any of our future or ongoing clinical trials;
refusal to approve pending applications or supplements to approved applications submitted by us;
restrictions on our or our contract manufacturers’ operations; or
product seizure or detention, or refusal to permit the import or export of products.
Any government investigation of alleged violations of law could require us to expend significant time and resources in response, and could generate negative publicity. Any failure to comply with ongoing regulatory requirements may significantly and adversely affect our ability to commercialize our product candidates. If regulatory sanctions are applied or if regulatory approval is withdrawn, the value of our company and our operating results will be adversely affected.
In addition, the FDA’s policies may change and additional government laws or regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. For example, the results of the 2020 U.S. Presidential Election may impact our business and industry. Namely, the Trump administration took several executive actions, including the issuance of a number of Executive Orders, that could impose significant burdens on, or otherwise materially delay, the FDA’s ability to engage in routine oversight activities such as implementing statutes through rulemaking, issuance of guidance, and review and approval of marketing applications. It is difficult to predict whether or how these orders will be implemented, or whether they will be rescinded and replaced under the Biden administration. The policies and priorities of the new administration are unknown and could materially impact the regulations governing our product candidates. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval thatbe subject to enforcement action and we may have obtained, which would adversely affect our business, prospects and ability tonot achieve or sustain profitability.
Within the European Union, once a Marketing Authorization is obtained, numerouspost-approval requirements also apply, and as in the United States,off-label promotion of medicinal products is not permitted. Furthermore, advertising to the general public of medicinal products which are available on medical prescription only is prohibited. The requirements are regulated by both E.U.EU regulations (such as advertising of medicinal products and reporting of adverse events) as well as national applicable regulations (namely related to prices and promotional activities).
The regulatory requirements relating to the manufacturing, testing, marketing and sale of pharmaceutical products are subject to periodic change. This may impact our ability and the ability of our collaboration partners to conduct clinical studies in the European Union. Changes in the regulations governing us could increase costs and adversely affect our business.
Furthermore, companies developing pharmaceutical products are facing increased demands to publish clinical trial results. Any such publication by us may, in addition to the additional cost of the publication, lead to investors misinterpreting the published data due to its technical and scientific nature, which, in turn, may adversely affect our business, results of operations and prospects and the value of our shares or ADSs.
Changes in funding forDisruptions at the FDA and other government agencies caused by funding shortages or global health concerns could hinder their ability to hire, and retain or deploy key leadership and other personnel, or otherwise prevent new or modified products and services from being developed, approved or commercialized in a timely manner or at all, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, statutory, regulatory, and policy changes, the FDA’s ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes.other events that may otherwise affect the FDA’s ability to perform routine functions. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs, medical devices and biologics or modifications to approved drugs, and biologics to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, including for 35 days beginning on December 22, 2018, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA, have had to furlough critical FDA employees and stop critical activities.
Separately, in response to the COVID-19 pandemic, on March 10, 2020 the FDA announced its intention to postpone most inspections of foreign manufacturing facilities, and on March 18, 2020, the FDA temporarily postponed routine surveillance inspections of domestic manufacturing facilities. Subsequently, on July 10, 2020 the FDA announced its intention to resume certain on-site inspections of domestic manufacturing facilities subject to a risk-based prioritization system. The FDA intends to use this risk-based assessment system to identify the categories of regulatory activity that can occur within a given geographic area, ranging from mission critical inspections to resumption of all regulatory activities. Regulatory authorities outside the United States may adopt similar restrictions or other policy measures in response to the COVID-19 pandemic. If a prolonged government shutdown occurs, or if global health concerns continue to prevent the FDA or other regulatory authorities from conducting their regular inspections, reviews, or other regulatory activities, it could significantly impact the ability of the FDA or other regulatory authorities to timely review and process our regulatory submissions, which could have a material adverse effect on our business. For example, in connection with the FDA’s review of our BLA submission for approval of lonapegsomatropin for the treatment of pediatric GHD, the FDA has indicated a desire to conduct an on-site inspection of certain manufacturing facilities in the United Kingdom. We have discussed with the FDA alternatives for inspection if restrictions imposed due to the COVID-19 pandemic prevent an on-site inspection of the facility in the United Kingdom. However, if the FDA determines it needs to conduct an on-site inspection of certain manufacturing facilities, such as the manufacturing site in the United Kingdom, to approve lonapegsomatropin for the treatment of pediatric GHD, the COVID-19 pandemic could delay approval until an on-site inspection is completed.
Third-party payor coverage and reimbursement status ofnewly-approved products is uncertain. Failure to obtain or maintain adequate coverage and reimbursement for our product candidates could limit our ability to market those products and decrease our ability to generate revenue.
The availability and adequacy of coverage and reimbursement by governmental healthcare programs such as Medicare and Medicaid, private health insurers and otherthird-party payors are essential for most patients to be able to afford treatments such as ours, assuming approval. Our ability to achieve acceptable levels of coverage and reimbursement for drug treatments by governmental authorities, private health insurers and other organizations will have an effect on our ability to successfully commercialize, and attract additional collaboration partners to invest in the development of our product candidates. We cannot be sure that coverage and reimbursement in the United States, the European Union or elsewhere will be available for any product that we may develop, and any reimbursement that may become available may be decreased or eliminated in the future.Third-party payors increasingly are challenging prices charged for pharmaceutical products, medical devices and services, and manythird-party payors may refuse to provide coverage and reimbursement for particular drugs when an equivalent generic drug is available. It is possible that athird-party payor may consider our product candidate and the generic parent drug as substitutable and only offer to reimburse patients for the generic drug. Even if we show improved efficacy or improved convenience of administration with our product candidate, pricing of the existing parent drug may limit the amount we will be able to charge for our product candidate. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize our product candidates, and may not be able to obtain a satisfactory financial return on products that we may develop.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. In the United States,third-party payors, including private and governmental payors, such as the Medicare and Medicaid programs, play an important role in determining the extent to which new drugs, biologics and medical devices will be covered. The Medicare and Medicaid programs increasingly are used as models for how private payors and other governmental payors develop their coverage and reimbursement policies for drugs, biologics and medical devices. It is difficult to predict at this time whatthird-party payors will decide with respect to the coverage and reimbursement for our product candidates.
Outside the United States, international operations are generally subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis oncost-containment initiatives in Europe, Canada, and other countries has and will continue to put pressure on the pricing and usage of our product candidates, if approved, and on related parent drugs. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. Many countries, including the European Union member
states, established complex and lengthy procedures to obtain price approvals, coverage and reimbursement. These procedures vary from country to country but are commonly initiated after grant of the related marketing authorization. Other countries allow companies to fix their own prices for medical products, but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our product candidates. Accordingly, in markets outside the United States, the
reimbursement for our products may be reduced compared with the United States and may be insufficient to generate commercially reasonable revenue and profits. As an example, many EU member states review periodically their decisions concerning the pricing and reimbursement of medicinal products. The outcome of these reviews cannot be predicted and could have adverse effects on the pricing and reimbursement of our medicinal products in the EU member states.
Moreover, increasing efforts by governmental andthird-party payors in the United States and abroad to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for new products approved and, as a result, they may not cover or provide adequate payment for our product candidates. We expect to experience pricing pressures in connection with the sale of any of our product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations, and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs, medical devices and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
We and our collaboration partners and contract manufacturers are subject to significant regulation with respect to manufacturing our product candidates. The manufacturing facilities on which we rely may not continue to meet regulatory requirements or may not be able to meet supply demands.
We depend on third partiesthird-parties to manufacture products employing our TransCon technologies. Components of a finished therapeutic product approved for commercial sale or used inlate-stage clinical studies must be manufactured in accordance with cGMP. These regulations govern manufacturing processes and procedures (including record keeping) and the implementation and operation of quality systems to control and assure the quality of investigational products and products approved for sale. All entities involved in the preparation of product candidates for clinical studies or commercial sale, including our existing contract manufacturers for our product candidates, are subject to extensive regulation. Manufacturing facilities are subject topre-approval and ongoing periodic inspection by the FDA, EEA Competent Authorities and other corresponding governmental authorities, including unannounced inspections, and must be licensed before they can be used in commercial manufacturing of products employing our TransCon technologies. After regulatory approvals or licensure are obtained, the subsequent discovery of previously unknown manufacturing, quality control or regulatory documentation problems or failure to maintain compliance with the regulatory requirements may result in restrictions on the marketing of a product, revocation of the license, withdrawal of the product from the market, seizures, injunctions, or criminal sanctions. Poor control of production processes can lead to the introduction of contaminants or to inadvertent changes in the properties or stability of our product candidates that may not be detectable in final product testing. We our collaboration partners, or our contract manufacturers must supply all necessary documentation in support of an NDA, BLA, MAA or comparable regulatory filing on a timely basis and must adhere to cGMP regulations enforced by the FDA, EEA Competent Authorities and other regulatory authorities through their facilities inspection programs. Some of our contract manufacturers have never produced a commercially approved pharmaceutical product and therefore have not obtained the requisite regulatory authority approvals to do so. Although we oversee the contract manufacturers, we cannot control the manufacturing process of, and are completely dependent on, our contract manufacturing partners for compliance with the regulatory requirements. If these facilities do not pass apre-approval plant inspection, regulatory approval of the products may not be granted or may be substantially delayed until any violations are corrected to the satisfaction of the regulatory authority, if ever. In addition, we have no control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel.
The regulatory authorities also may, at any time following approval of a product for sale, audit the manufacturing facilities of our collaboration partners andthird-party contractors. If any such inspection or audit identifies a failure to comply with applicable regulations or if a violation of our product specifications or applicable regulations occurs independent of such an inspection or audit, we or the relevant regulatory authority may require remedial measures that may be costly and/or time consuming for us or a third partythird-party to implement, and that may include the temporary or permanent suspension of a clinical study or commercial sales or the temporary or permanent suspension of production or closure of a facility. Any such remedial measures imposed upon us or third partiesthird-parties with whom we contract could harm our business.
If we our collaboration partners, or any of ourthird-party manufacturers fail to maintain regulatory compliance, the FDA or other applicable regulatory authority can impose regulatory sanctions including, among other things, refusal to approve a pending application for a new pharmaceutical product, withdrawal of an approval, or suspension of production. As a result, our business, financial condition, and results of operations may be harmed.
Additionally, if supply from one approved manufacturer is interrupted, an alternative manufacturer would need to be qualified through an NDA or BLA, a supplemental NDA or BLA, a marketing authorization variation application or equivalent foreign regulatory filing, which could result in further delay. The regulatory authorities may also require additional studies if a new manufacturer is relied upon for commercial production. Switching manufacturers may involve substantial costs and is likely to result in a delay in our desired clinical and commercial timelines. Furthermore, interruption or delay in supplies from one contract manufacturer may cause delays further down the supply chain, as certain contract manufacturers may rely on delivery of materials from other contract manufacturers.
These factors could cause us to incur higher costs and could cause the delay or termination of clinical studies, regulatory submissions, required approvals, or commercialization of our product candidates. Furthermore, if our suppliers fail to meet contractual requirements and we are unable to secure one or more replacement suppliers capable of production at a substantially equivalent cost, our clinical studies may be delayed, or we could lose potential revenue.
Our operations involve hazardous materials and we and third partiesthird-parties with whom we contract must comply with environmental laws and regulations, which can be expensive and restrict how we do business.
As a pharmaceutical company, we are subject to environmental and safety laws and regulations, including those governing the use of hazardous materials. The cost of compliance with health and safety regulations is substantial. Our business activities involve the controlled use of hazardous materials. Our research and development activities involve the controlled storage, use and disposal of hazardous materials, including the components of our product candidates and other hazardous compounds. We and manufacturers and suppliers with whom we may contract are subject to laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. In some cases, these hazardous materials and various wastes resulting from their use are stored at our and our manufacturers’ facilities pending their use and disposal. We cannot eliminate the risk of accidental contamination or injury from these materials, which could cause an interruption of our commercialization efforts, research and development efforts and business operations, environmental damage resulting in costlyclean-up and liabilities under applicable laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. We cannot guarantee that the safety procedures utilized bythird-party manufacturers and suppliers with whom we may contract will comply with the standards prescribed by laws and regulations or will eliminate the risk of accidental contamination or injury from these materials. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources and European, U.S. federal and state or other applicable authorities may curtail our use of certain materials and/or interrupt our business operations. Furthermore, environmental laws and regulations are complex, change frequently and have tended to become more stringent. We cannot predict the impact of such changes and cannot be certain of our future compliance. We do not currently carry biological or hazardous waste insurance coverage. In the event of an accident or environmental discharge, we may be held liable for any consequential damage and any resulting claims for damages, which may exceed our financial resources and may materially adversely affect our business, results of operations and prospects, and the value of our shares or ADSs.
If we fail to comply or are found to have failed to comply with EEA, FDA and other regulations related to the promotion of our products for unapproved uses, we could be subject to criminal penalties, substantial fines or other sanctions and damage awards.
The regulations relating to the promotion of products for unapproved uses are complex and subject to substantial interpretation by the EEA Competent Authorities, the FDA and other regulatory authorities, as well as courts. If any of our product candidates receives marketing approval, we and any collaboration partner will be restricted from marketing the product outside of its approved labeling, also referred to as promotion. However, physicians may nevertheless lawfully prescribe an approved product to their patients in a manner that is inconsistent with the approved label, which is anoff-label use.
We intend to implement compliance and training programs designed to ensure that our sales and marketing practices comply with applicable regulations regardingoff-label promotion and other illegal promotional activities. Notwithstanding these programs, the EEA Competent Authorities, the FDA or other government authorities may allege or find that our practices constitute prohibited promotion of our product candidates for unapproved uses. We also cannot be sure that our employees will comply with company policies and applicable regulations regarding the promotion of products.
Over the past several years, a significant number of pharmaceutical and biotechnology companies have been the target of inquiries and investigations by various U.S. federal and state regulatory, investigative, prosecutorial and administrative entities in connection with the promotion of products for unapproved uses and other sales practices, including the Department of Justice and various U.S. Attorneys’ Offices, the Office of Inspector General of the Department of Health and Human Services, the FDA, the Federal Trade Commission and various state Attorneys General offices. These investigations have alleged violations of various U.S. federal and state laws and regulations, including claims asserting antitrust violations, violations of the Food, Drug and Cosmetic Act, the False Claims Act, the Prescription Drug Marketing Act,anti-kickback laws, and other alleged violations in connection with the promotion of products for unapproved uses, pricing and Medicare and/or Medicaid reimbursement. Many of these investigations originate as “qui tam” actions under the False Claims Act. Under the False Claims Act, any individual can bring a claim on behalf of the government alleging that a person or entity has presented a false claim, or caused a false claim to be submitted, to the government for payment. The person bringing a qui tam suit is entitled to a share of any recovery or settlement. Qui tam suits, also commonly referred to as “whistleblower suits,” are often brought by current or former employees. In a qui tam suit, the government must decide whether to intervene and prosecute the case. If it declines, the individual may pursue the case alone.
If the FDA or any other governmental agency initiates an enforcement action against us or if we are the subject of a qui tam suit and it is determined that we violated prohibitions relating to the promotion of products for unapproved uses, we could be subject to substantial civil or criminal fines or damage awards and other sanctions such as consent decrees and corporate integrity agreements pursuant to which our activities would be subject to ongoing scrutiny and monitoring to ensure compliance with applicable laws and regulations. Any such fines, awards or other sanctions would have an adverse effect on our revenue, business, financial prospects and reputation.
If approved, our product candidates may cause or contribute to adverse medical events that we are required to report to regulatory authorities and if we fail to do so we could be subject to sanctions that would harm our business.
Some participants in clinical trials of our product candidates have reported adverse events. As with all clinical trials, serious or severe adverse events may occur which may compromise the program. The FDA, EEA, and foreign regulatory authority regulations require that we report certain information about adverse medical events if those products may have caused or contributed to those adverse events, both during their development and after commercialization, if approved. The timing of our obligation to report is triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events we become aware of within the prescribed timeframe. We may also fail to appreciate that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the FDA, EEA Competent Authorities, or a foreign regulatory authority could take action, including criminal prosecution, the imposition of civil monetary penalties, seizure of our products or delay in approval or clearance of future products.
Our employees, independent contractors, principal investigators, CROs, consultants, vendors and collaboration partners may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk that our employees, independent contractors, principal investigators, CROs, consultants, vendors and collaboration partners may engage in fraudulent conduct or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or unauthorized activities that violate: (1) FDA regulations, including those laws that require the reporting of true, complete and accurate information to the FDA; (2) manufacturing standards; (3) U.S. federal and state fraud and abuse and other healthcare laws and regulations; or (4) laws that require the reporting of true and accurate financial information and data. Specifically, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks,self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. These activities also include the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter misconduct by employees and other third parties,third-parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in
protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. Additionally, we are subject to the risk that a person or government could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other U.S. federal healthcare programs, individual imprisonment, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Additionally, our business activities may be subject to the Foreign Corrupt Practices Act, or FCPA, and similar anti-bribery or anti-corruption laws, regulations or rules of other countries in which we operate. The FCPA generally prohibits offering, promising, giving, or authorizing others to give anything of value, either directly or indirectly, to a non-U.S. government official in order to influence official action, or otherwise obtain or retain business. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect the transactions of the corporation and to devise and maintain an adequate system of internal accounting controls. Our business is heavily regulated and, therefore, involves significant interaction with public officials, including officials of non-U.S. governments. Additionally, in many other countries, the health care providers who prescribe pharmaceuticals are employed by their government, and the purchasers of pharmaceuticals are government entities; therefore, our dealings with these prescribers and purchasers are subject to regulation under the FCPA.
There is no certainty that all of our employees, agents, contractors, or collaborators, or those of our affiliates, will comply with all applicable laws and regulations, particularly given the high level of complexity of these requirements. We have adopted a code of conduct, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may be ineffective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these requirements. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business.
Failure to obtain regulatory approvals innon-U.S. jurisdictions would prevent us from marketing our products outside of the United States.
In the EEA, medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. There are two types of MA:
The Community MA, which is issued by the European Commission through the centralized procedure, based on the opinion of the Committee for Medicinal Products for Human Use (CHMP) of the EMA, is valid throughout the entire territory of the EEA. The centralized procedure is mandatory for certain types of products, such as medicinal products derived from biotechnology processes, orphan medicinal products, and medicinal products containing a new active substance indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes andauto-immune and viral diseases. The centralized procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the European Union.
National MAs, which are issued by the Competent Authorities of the Member States of the EEA and only cover their respective territory, are available for products not falling within the mandatory scope of the centralized procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in other Member States through the mutual recognition procedure. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the decentralized procedure.
Under the above described procedures, before granting the MA, the EMA or the Competent Authorities of the Member States of the EEA make an assessment of therisk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
In the EEA, the EMA’s Committee for Orphan Medicinal Products grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment oflife-threatening or chronically debilitating conditions affecting not more than five in 10,000 persons in the E.U. Community and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized (or the product would be a significant benefit to those affected). Additionally, designation is granted for products intended for the diagnosis, prevention, or treatment of alife-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in the European Union would be sufficient to justify the necessary investment in developing the medicinal product. An E.U. orphan drug designation entitles a party to financial incentives such as reduction of fees or fee waivers and 10 years of market exclusivity is granted following medicinal product approval. This period may be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. We have as of December 31, 2019 received orphan drug designation for TransCon hGH in the European Union and for TransCon PTH and TransCon CNP in the United States.
In the EEA, marketing authorization applications for new medicinal products not authorized in the EU will only be regarded as valid, if they include one of the following: (i) the results of all studies performed and details of all information collected in compliance with a paediatric investigation plan; PIP, agreed with the EMA’s Pediatric
Committee, or the PDCO, (ii) a decision of the EMA granting a waiver from the obligation to provide the results of studies in the paediatric population in accordance with a PIP, or (iii) a decision by the EMA agreeing to a deferral of the initiation or completion of some or all of the measures set out in the PIP. The PIP sets out the timing and measures proposed to generate data to support a pediatric indication of the drug for which marketing authorization is being sought. The PDCO can grant a deferral of the obligation to implement some or all of the measures of the PIP until there are sufficient data to demonstrate the efficacy and safety of the product in adults. Further, the obligation
to provide pediatric clinical trial data can be waived by the PDCO when these data is not needed or appropriate because the product is likely to be ineffective or unsafe in children, the disease or condition for which the product is intended occurs only in adult populations, or when the product does not represent a significant therapeutic benefit over existing treatments for pediatric patients. Once the marketing authorization is obtained in all Member States of the European Union and study results are included in the product information, even when negative, the product is eligible for six months’ supplementary protection certificate extension. Fororphan-designated medicinal products, the10-yearten-year period of market exclusivity is extended to 12twelve years. At this time, although other long-acting products in development have received PIP waivers, we are currently in discussions with PDCO and have not agreed to a PIP or received a PIP waiver for TransCon hGH.
Outside the U.S. and the EEA, approval procedures vary among countries and can involve additional clinical testing, and the time required to obtain approval may differ from that required to obtain FDA or EEA approval. Clinical trials conducted in one country may not be accepted by regulatory authorities in other countries. Approval by the FDA, EMA, or EEA Competent Authorities does not ensure approval by regulatory authorities in other countries, and approval by one or more foreign regulatory authorities does not ensure approval by regulatory authorities in other foreign countries or by the FDA, EMA or EEA Competent Authorities. However, a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval, EMA, or EEA Competent Authority. We may not be able to file for regulatory approvals or to do so on a timely basis, and even if we do file, we may not receive necessary approvals to commercialize our products in any market.
We may be subject to healthcare laws, regulation and enforcement; our failure to comply with these laws could harm our results of operations and financial conditions.
Although we do not currently have any products on the market, once we begin commercializing our products, we may be subject to additional healthcare, statutory and regulatory requirements and enforcement by the U.S. federal government and the states and foreign governments in which we conduct our business. The laws that may affect our ability to operate as a commercial organization include:
the U.S.Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under U.S. federal healthcare programs such as the Medicare and Medicaid programs. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the U.S. federalAnti-Kickback Statute constitutes a false or fraudulent claim for purposes of the false claims statutes;violation;
U.S. false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or otherthird-party payors that are false or fraudulent;fraudulent. In addition, the government may assert that a claim including items or services resulting from a violation of the U.S. federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act;
U.S. federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. Similar to the U.S. federalAnti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation;
HIPAA, which imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement, in connection with the delivery of, or payment for, healthcare benefits, items or services; similar to the U.S. Health Insurance Portability and Accountability Actfederal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act, which governs the conduct of certain electronic healthcare transactions and protects the security and privacy of protected health information;statute or specific intent to violate it in order to have committed a violation;
the U.S. federal physician sunshine requirements under the Affordable CarePhysician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics, and medical supplies to report annually to the CMS information related to payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain other healthcare providers beginning in 2022, and teaching hospitals, and ownership and investment interests held by physicians (as defined under statute) and their immediate family members;
state law equivalents of each of the above U.S. federal laws, such asanti-kickback and false claims laws which may apply to items or services reimbursed by anythird-party payor, including commercial insurers;
state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the U.S. federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources;
state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures and pricing information;
state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways, thus complicating compliance efforts. For example, California recently enacted legislation, the California Consumer Privacy Act, or CCPA, effective January 1, 2020. The CCPA, among other things, creates new data privacy obligations for covered companies and provides new privacy rights to California residents, including the right to opt out of certain disclosures of their information. The CCPA also creates a private right of action with statutory damages for certain data breaches, thereby potentially increasing risks associated with a data breach. Although the law includes limited exceptions, including for “protected health information” maintained by a covered entity or business associate, it may regulate or impact our processing of personal information depending on the context; and
European and other foreign law equivalents of each of the laws, including regulation regarding advertising of medicinal products and reporting requirements detailing interactions with and payments to healthcare providers.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. The risk of our activities being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations.
Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. If our operations are found to be in violation of any of the laws described above or any other governmental laws and regulations that apply to us, we may be subject to significant penalties, including civil and criminal penalties, damages, fines, the curtailment or restructuring of our operations, the exclusion from participation in U.S. federal and state and/or EEA healthcare programs and imprisonment, any of which could adversely affect our ability to market our products and adversely impact our financial results.
We are subjectChanges in and failures to governmental regulationcomply with U.S. and other legal obligations, particularly related toforeign privacy and data protection laws, regulations and information security,standards may adversely affect our business, operations and financial performance.
The global data protection landscape is rapidly evolving, and we are or may become subject to consumer protectionnumerous state, federal and foreign laws, that regulate our marketing practices and prohibit unfair or deceptive acts or practices. Our actual or perceived failure to comply with such obligations could harm our business.
We are subject to diverse lawsrequirements and regulations relating to data privacygoverning the collection, use, disclosure, retention and security including, in the United States, HIPAA and certain state lawsof personal data, such as information that we may collect in connection with clinical trials. Implementation standards and enforcement practices are likely to remain uncertain for the foreseeable future, and we cannot yet determine the impact future laws, regulations, standards, or perception of their requirements may have on our business. This evolution may create uncertainty in our business, affect our ability to operate in certain jurisdictions or to collect, store, transfer use and share personal information, necessitate the Stateacceptance of California and,more onerous obligations in the EU and the European Economic Area,our contracts, result in liability or EEA, Regulation 2016/679, known as the General Data Protection Regulation, or GDPR. New privacy rules are being enacted in the United States and globally, and existing ones are being updated and strengthened.impose additional costs on us. Complying with these numerous, complex and often changing regulations is expensive and difficult, and any failure or perceived failure to comply with any data privacy laws or data security laws, our policies and procedures, our contracts governing our processing of personal information or any security incident or breach involving the misappropriation, loss or other unauthorized use or disclosure of sensitive or confidential patient or consumer information, whether by us, one of our business associatespartners or anotherthird-party, could adversely affect our business, financial condition and results of operations, including but not limited to: investigation costs,and could result in negative publicity, government investigations and enforcement actions, claims by third-parties and damage to our reputation, any of which could have a material finesadverse effect on our operations, financial performance and penalties; compensatory, special, punitive,business.
As our operations and statutory damages; litigation; consent orders regarding ourbusiness grow, we may become subject to or affected by new or additional data protection laws and regulations and face increased scrutiny or attention from regulatory authorities. In the U.S., HIPAA imposes, among other things, certain standards relating to the privacy, security, transmission and breach reporting of individually identifiable health information. Certain states have also adopted comparable privacy and security practices;laws
and regulations, some of which may be more stringent than HIPAA. Such laws and regulations will be subject to interpretation by various courts and other governmental authorities, thus creating potentially complex compliance issues for us and our future customers and strategic partners. In addition, California enacted the California Consumer Privacy Act, or CCPA, which went into effect on January 1, 2020. The CCPA creates individual privacy rights for California consumers and increases the privacy and security obligations of entities handling certain personal information. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. The CCPA may increase our compliance costs and potential liability, and many similar laws have been proposed at the federal level and in other states. Further, the California Privacy Rights Act, or the CPRA, recently passed in California. The CPRA will impose additional data protection obligations on covered businesses, including additional consumer rights processes, limitations on data uses, new audit requirements for higher risk data, and opt outs for certain uses of sensitive data. It will also create a new California data protection agency authorized to issue substantive regulations and could result in increased privacy and information security enforcement. The majority of the provisions will go into effect on January 1, 2023, and additional compliance investment and potential business process changes may be required. In the event that we provide notices, credit monitoring services and/are subject to or credit restoration servicesaffected by HIPAA, the CCPA, the CPRA or other relevant servicesdomestic privacy and data protection laws, any liability from failure to impacted individuals; adverse actions againstcomply with the requirements of these laws could adversely affect our licenses to do business; and injunctive relief. Furthermore, these rules are constantly changing; for example,financial condition.
In Europe, the General Data Protection Regulation, or the GDPR came into force in May 2018 changingimposes strict requirements for processing the European regime. Before that,personal data of individuals within theUS-EU Safe Harbor framework was declared invalid in 2015 and replaced with theUS-EU Privacy Shield framework which, along with other methods which permit transfer under European privacy law, are under ongoing review and subject to challenge.
The privacy laws in the EU have been significantly reformed. On May 25, 2018, the GDPR entered into force and became directly applicable in all EU member states. The GDPR implements more stringent operational requirements than its predecessor legislation. EEA, including clinical trial data. For example, the GDPR requires us to make more detailed disclosures to data subjects, requires disclosure of the legal basis on which we can process personal data, makes it harder for us to obtain valid consent for processing and in other cases prevents the use of consent as legal basis for processing of personal data, will requirerequires the appointment of data protection officers when sensitive personal data, such as health data, is processed on a large scale, provides more robust rights for data subjects, introducesimposes mandatory data breach notification through the EU and EEA, imposes additional obligations on us when contracting with service providers and requires us to adopt appropriate privacy governance including policies, procedures, training and data audit. If we do not comply with our obligations under the GDPR, we could be exposed to fines of up to the greater of €20 million or up to 4% of our total global annual revenue in the event of a significant breach. In addition, we may be the subject of litigation and/or adverse publicity, which could adversely affect our business, results of operations and financial condition.
The law in this area is also developing rapidly. For example, in July 2020, the Court of Justice of the EU invalidated the Privacy Shield, limiting how organizations could lawfully transfer personal data from the EEA to the U.S. Relatedly, following the United Kingdom’s withdrawal from the EEA and the EU, and the expiry of the transition period, companies have to comply with both the GDPR and the GDPR as incorporated into United Kingdom national law, the latter regime having the ability to separately fine up to the greater of £17.5 million or 4% of global turnover. The relationship between the United Kingdom and the European Union in relation to certain aspects of data protection law remains unclear, for example around how data can lawfully be transferred between each jurisdiction, which exposes us to further compliance risk. We cannot assure you that ourthird-party service providers with access to our or our customers’, suppliers’, trial patients’, and employees’ personally identifiable and other sensitive or confidential information in relation to which we are responsible will not breach contractual obligations imposed by us, or that they will not experience data security breaches or attempts thereof, which could have a corresponding effect on our business including putting us in breach of our obligations under privacy laws and regulations and/or which could in turn adversely affect our business, results of operations and financial condition. While we attempt to address the associated risks by performing security assessments and detailed due diligence, we cannot assure you that these contractual measures and our own privacy andsecurity-related safeguards will protect us from the risks associated with thethird-party processing, storage and transmission of such information.
Legislative or regulatory healthcare reforms in the United States may make it more difficult and costly for us to obtain regulatory clearance or approval of our product candidates in the United States and to produce, market and distribute our products in the United States after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in U.S. Congress that could significantly change the statutory provisions governing the regulatory clearance or approval, manufacture, and marketing of regulated products or the reimbursement thereof. In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. In addition, the Trump administration has taken several executive actions, including the issuance of a number of Executive Orders, that could impose significant burdens on, or otherwise materially delay, the FDA’s ability to engage in routine oversight activities such as implementing statutes through rulemaking, issuance of guidance, and review and approval of marketing applications. It is difficult to predict how these requirements will be interpreted and implemented, and the extent to which they will impact the FDA’s ability to exercise its regulatory authority. If these executive actions
impose restrictions on the FDA’s ability to engage in oversight and implementation activities in the normal course, our business may be negatively impacted. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of our product candidates. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require:
additional clinical trials to be conducted prior to obtaining approval;
changes to manufacturing methods;
recall, replacement, or discontinuance of one or more of our products; and
additional record keeping.
Each of these would likely entail substantial time and cost and could harm our business and our financial results. In addition, delays in receipt of or failure to receive regulatory clearances or approvals for any future products would harm our business, financial condition and results of operations.
In addition, the trend toward managed healthcare in the United States and the changes in health insurance programs, as well as legislative proposals to reform healthcare or reduce government insurance programs, may result in lower prices for pharmaceutical products, including any product that may be offered by us. In addition, any future regulatory change regarding the healthcare industry orthird-party coverage and reimbursement may affect demand for any products that we may develop and could harm our sales and profitability. For example, in the United States, the ACA was enacted in 2010 with a goal of reducing the cost of healthcare and substantially changing the way healthcare is financed by both government and private insurers. The ACA, among other things, increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extended the rebate program to individuals enrolled in Medicaid managed care organizations, addressed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, established annual fees and taxes on manufacturers of certain branded prescription drugs and medical devices, and created a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50%point-of-sale discounts, which, through subsequent legislative amendments, was increased to 70%, starting in 2019, off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D.
We expect that the current presidential administrationSince its enactment, there have been judicial, executive and U.S. Congress will likely continueCongressional challenges to seek to modify, repeal, or otherwise invalidate all, or certain provisions of, the ACA. For example, the Tax Cuts and Jobs Act, enacted on December 22, 2017, removes penalties for not complying with the ACA’s individual mandate to carry health insurance. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas, ruled that because the individual mandate is a critical and inseverable feature of the ACA, the remaining provisions of the ACA were invalid as well. Upon appeal, the U.S. Court of Appeals for the Fifth Circuit affirmed that the individual mandate was unconstitutional but remanded the case back to the U.S. District Court to determine what portions of the ACA, if any, might continue to be valid. On January 21, 2020, theThe U.S. Supreme Court declined a motion byis currently reviewing the U.S. House of Representatives and others seeking expedited review of the case. Itcase, although it is unclear how these decisions, subsequent appeals andthe Supreme Court will rule. It is also unclear how other efforts to challenge, repeal or replace the ACA, if any, will impact the law. We cannot predict the ultimate content, timing or effect of any healthcare reform legislation or the impact of potential legislation on us.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted, including reductions in Medicare payments to providers, capped at 2% per fiscal year, which went into effect on April 1, 2013. These reductions, extended by subsequent legislation will stay in effect through 20292030, with the exception of a temporary suspension from May 1, 2020 through March 31, 2021, unless additional Congressional action is taken, further reductions totaken. Further, the American Taxpayer Relief Act of 2012 reduced Medicare payments to several types of providers, including hospitals, and an increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Recently, there has also been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed bills designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for pharmaceutical products. The current presidential administration has offered multiple proposals and plans as means to lower drug costs. Congress and the current administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs. The likelihood of implementation of any of these reform initiatives is uncertain, particularly in light of the new Presidential administration. Individual states in the United States have also become increasingly
active in passing legislation and implementing regulations designed to control pharmaceutical product pricing, including through constraints on reimbursement, imposition of mandatory discounts, discounts, restrictions on access to certain products, transparency measures, and programs for importation from other countries or bulk purchasing.
We expect that additional U.S. local and national healthcare reform measures will be adopted within and outside the United States in the future, any of which could limit the amounts that governments will pay for healthcare products and services, which could result in reduced demand for our product candidates or additional pricing pressures. The continuing efforts of the U.S. government, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare may adversely affect the demand for any drug products for which we may obtain regulatory approval, our ability to set a price that we believe is fair for our products, our ability to obtain coverage and reimbursement approval for a product, our ability to generate revenues and achieve or maintain profitability, and the level of taxes that we are required to pay.
Risks Related to Our Intellectual Property
If our intellectual property related to our product candidates is not adequate, we may not be able to compete effectively in our market.
Our success depends in part on our ability to:
protect our trade secrets;
apply for, obtain, maintain and enforce patents; and
operate without infringing upon the proprietary rights of others.
We will be able to protect our proprietary technologies from unauthorized use by third partiesthird-parties only to the extent that such proprietary rights are covered by valid and enforceable patents or are effectively maintained as trade secrets. Anynon-confidential disclosure to or misappropriation by third partiesthird-parties of our confidential or proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, thus eroding our competitive position in our market. Where we elect to pursue patent protection on our proprietary technologies, we file, prosecute and maintain international and other national patent applications covering such technologies, including in the United States, Europe, China, and other jurisdictions.
As of December 31, 2019,twenty-four2020, 28 patents have been issued to us in the United States. 1617 patents are directed to our TransCon technologies and five are directed to TransCon hGH. In addition, as of December 31, 2019,2020, we have approximately 139170 issued patents in jurisdictions outside of the United States, at least 7489 of which are directed to our TransCon technologies, and 3543 of which are directed to our product candidates. As of December 31, 2019,2020, our TransCon hGH is covered by seven different patent families and an additional nine patent families covering theauto-injector auto injector device, our TransCon PTH is covered by sevennine different patent families and our TransCon CNP is covered by 11eleven different patent families. Most members of these families are applications in an early stage, so it is impossible to make any statements regarding whether or not they will be granted. We are not aware of any challenge to our issued patents, in the United States, Europe or in any other jurisdiction.
The patent application process, also known as patent prosecution, is expensive andtime-consuming, and we and our current or future licensors and licensees may not be able to prepare, file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we or our current licensors, or any future licensors or licensees, will fail to identify patentable aspects of inventions made in the course of development and commercialization activities before it is too late to obtain patent protection on them. Therefore, these and any of our patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business. It is possible that defects of form in the preparation or filing of our patents or patent applications may exist, or may arise in the future, for example with respect to proper priority claims, inventorship, etc., although we are unaware of any such defects. If we or our current licensors or licensees, or any future licensors or licensees, fail to establish, maintain or protect such patents and other intellectual property rights, including due to the impact of the COVID-19 pandemic on our or our licensors’ business operations, such rights may be reduced or eliminated. If our current licensors or licensees, or any future licensors or licensees, are not fully cooperative or disagree with us as to the prosecution, maintenance or enforcement of any patent rights, such patent
rights could be compromised. If there are material defects in the form or preparation of our patents or patent applications, such patents or applications may be invalid and unenforceable. Any of these outcomes could impair our ability to prevent competition from third parties,third-parties, which may harm our business.
The strength of patents in the biotechnology and pharmaceutical field involves complex legal and scientific questions and can be highly uncertain. The patent applications that we own or license may fail to result in issued patents in the United States or in other countries. Even if patents do issue on such patent applications, third partiesthird-parties may challenge the validity, enforceability or scope thereof, which may result in such patents being narrowed, invalidated or held unenforceable. For example, U.S. patents can be challenged by any person before the USPTO Patent Trial and Appeals Board at any time within theone-year period following that person’s receipt of an allegation of infringement of the patents. Patents granted by the European Patent Office may be similarly opposed by any person within nine months from the publication of the grant. Similar proceedings are available in other jurisdictions, and in the United States, Europe and other jurisdictions third partiesthird-parties can raise questions of validity with a patent office even before a patent has granted. Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property or prevent others from designing around our claims. For example, a third partythird-party may develop a competitive product that provides therapeutic benefits similar to one or more of our product candidates but that has a different composition that falls outside the scope of our patent protection. If the breadth or strength of protection provided by the patents and patent applications we hold or pursue with respect to our product candidates is successfully challenged, then our ability to commercialize such product candidates could be negatively affected, and we may face unexpected competition that could have harm our business. Further, ifpatents have a limited lifespan. Although various extensions may be available, the life of a patent, and the protection it affords, is limited. If we encounter delays in our clinical trials, the period of time during which we or our collaboration partners could market our product candidates under patent protection would be reduced. If we do not have sufficient patent life to protect our products, our business and results of operations will be adversely affected.
The degree of future protection of our proprietary rights is uncertain. Patent protection may be unavailable or severely limited in some cases and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
we might not have been the first to invent or the first to file the inventions covered by each of our pending patent applications and issued patents;
others may independently develop similar or alternative technologies or duplicate any of our technologies;
the patents of others may have an adverse effect on our business;
any patents we obtain or ourin-licensed issued patents may not encompass commercially viable products, may not provide us with any competitive advantages or may be challenged by third parties;third-parties;
any patents we obtain or ourin-licensed issued patents may not be valid or enforceable; or
we may not develop additional proprietary technologies that are patentable.
If we or our current licensors or licensees, or any future licensors or licensees, fail to prosecute, maintain and enforce patent protection for our product candidates, our ability to develop and commercialize our product candidates could be harmed and we might not be able to prevent competitors from making, using and selling competing products. This failure to properly protect the intellectual property rights relating to our product candidates could harm our business, financial condition and operating results. Moreover, our competitors may independently develop equivalent knowledge, methods andknow-how.
Even where laws provide protection, costly andtime-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and the outcome of such litigation would be uncertain. If we or one of our collaboration partners were to initiate legal proceedings against a third partythird-party to enforce a patent covering the product candidate, the defendant could counterclaim that our patent is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness ornon-enablement. Patents may be unenforceable if someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during
prosecution. The outcomes of proceedings involving assertions of invalidity and unenforceability are unpredictable. It is possible that prior art of which we and the patent examiner were unaware during prosecution exists, which would render our patents invalid. Moreover, it is also possible that prior art may exist that we are aware of, but that we do not believe are relevant to our current or future patents, that could nevertheless be determined to render our patents invalid. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability of our patents covering one of our product candidates, we would lose at least part, and perhaps all, of the patent protection on such product candidate. Such a loss of patent protection would harm our business. Moreover, our competitors could counterclaim in any suit to enforce our patents that we infringe their intellectual property. Furthermore, some of our competitors have substantially greater intellectual property portfolios, and resources, than we do.
We license intellectual property rights from third-parties. Such licenses may be subject to early termination if we fail to comply with our obligations in our licenses with third-parties, which could result in the loss of rights or technology that are material to our business.
We are or may become a party to licenses that give us rights to third-party intellectual property or technology that is necessary or useful for our business, and we may enter into additional licenses in the future. Under these license agreements, we are or may become obligated to pay the licensor fees, which may include annual license fees, milestone payments, royalties, a percentage of revenues associated with the licensed technology and a percentage of sublicensing revenue. These fees may be significant, which could make it difficult for us to achieve or maintain profitability. In addition, under certain of such agreements, we are or may become required to diligently pursue the development of products using the licensed technology. If we fail to comply with these obligations, including due to the impact of the COVID-19 pandemic on our business operations or our use of the intellectual property licensed to us in an unauthorized manner, and fail to cure our breach within a specified period of time, the licensor may have the right to terminate the applicable license, in which event we could lose valuable rights and technology that are material to our business, harming our ability to develop, manufacture and/or commercialize our platform or product candidates.
In addition, the agreements under which we license intellectual property or technology to or from third-parties can be complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations and prospects.
The licensing and acquisition of third-party intellectual property rights is a competitive practice, and companies that may be more established, or have greater resources than we do, may also be pursuing strategies to license or acquire third-party intellectual property rights that we may consider necessary or attractive in order to commercialize our product candidates. More established companies may have a competitive advantage over us due to their larger size and cash resources or greater clinical development and commercialization capabilities. There can be no assurance that we will be able to successfully complete such negotiations and ultimately acquire the rights to the intellectual property surrounding the additional product candidates that we may seek to acquire. If we fail to obtain in-license any compositions, methods of use, processes or other third-party intellectual property rights at a reasonable cost or on reasonable terms, we could harm our business. If we fail to obtain licenses to necessary third-party intellectual property rights, we may need to cease use of the compositions or methods covered by such third-party intellectual property rights. Furthermore, we may need to seek to develop alternative approaches that do not infringe on such intellectual property rights which may entail additional costs and development delays, even if we were able to develop such alternatives, which may not be feasible. Even if we are able to obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. In that event, we may be required to expend significant time and resources to develop or license replacement technology.
If we are unable to prevent disclosure of our trade secrets or other confidential information to third parties,third-parties, our competitive position may be impaired.
In addition to patents, we rely on trade secrets and proprietaryknow-how. We seek protection, in part, through confidentiality and proprietary information clauses in agreements with our collaboration partners, employees, consultants, outside scientific collaboration partners and sponsored researchers and other advisors. Although we
generally require all of our employees, consultants, advisors and any third partiesthird-parties who have access to our proprietaryknow-how, information or technology to assign or grant similar rights to their inventions to us, and endeavor to execute confidentiality agreements with all such parties, we cannot be certain that we have executed such agreements with all parties who may have contributed to our intellectual property or who had access to our proprietary information, nor can we be certain that our agreements with such parties will not be breached. These agreements may not effectively prevent disclosure of confidential and proprietary information and may not provide an adequate remedy in the event of unauthorized use or disclosure of confidential and proprietary information. We cannot guarantee that our trade secrets and other confidential proprietary information will not be publicly disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position. Furthermore, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad. The failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
If we are sued for infringing intellectual property rights of third parties,third-parties, it will be costly and time consuming, and an unfavorable outcome in that litigation could harm our business.
Our commercial success depends significantly on our ability to operate without infringing, violating or misappropriating the patents and other proprietary rights of third parties.third-parties. Our own technologies may infringe, violate or misappropriate the patents or other proprietary rights of third parties,third-parties, or we may be subject tothird-party claims of such infringement. Numerous U.S. and foreign issued patents and pending patent applications owned by third partiesthird-parties exist in the fields in which we are developing our product candidates. For example, we are aware of several issued patents related toauto-injection devices that may be relevant to ourauto-injection device under development with Phillips Medisize A/S (formerly Medicom Innovation Partner A/S); however, we believe that these (i) will expire prior to our anticipated product launch, (ii) are invalid, and/or (iii) do not and will not cover our product or device. Additionally, we are aware of an allowed patent application owned by a competitor related to macromolecules capable of releasing CNP variants and methods of treating various disorders including achondroplasia using such macromolecules. Although we believe that these allowed claims are invalid, we could be wrong in our assessment. We cannot be certain that our product candidates will not infringe these or other existing or future patents. BecauseThe scope of a patent claim is determined by an interpretation of the law, the written disclosure in a patent and the patent’s prosecution history. Our interpretation of the relevance or the scope of a patent or a pending application may be incorrect, which may negatively impact our ability to market our products. Additionally, because patent applications can take many years to issue and may be confidential for 18 months or more after filing, and because pending patent claims can be revised before issuance, there may be applications now pending which may later result in issued patents that may be infringed by the manufacture, use or sale of our product candidates or our TransCon technologies. We may not be aware of patents that have already issued that a third partythird-party might assert are infringed by our product candidates. It is also possible that patents of which we are aware, but which we do not believe are relevant to our product candidates, could nevertheless be found to be infringed by our product candidates. Moreover, we may face patent infringement claims from non-practicing entities that have no relevant product revenue and against whom our own patent portfolio may thus have no deterrent effect. Nevertheless, we are not aware of any valid issued patents that we believe would prevent us from marketing our product candidates, if approved. Moreover, we may face patent infringement claims fromnon-practicing entities that have no relevant product revenue and against whom our own patent portfolio may thus have no deterrent effect.
In addition, we and our collaboration partners may face costly andtime-consuming intellectual property litigation with the NDA holders, BLA holders and Orange Book patentees of the products in respect of which we seek to obtain FDA approval. Companies that produce branded pharmaceutical products for which there are listed patents in the FDA’s Orange Book routinely bring patent infringement litigation against applicants seeking FDA approval to manufacture and market branded and/or generic forms of their products. Accordingly, we may face patent litigation as a result of our submission of NDA and BLA applications to the FDA or as a result of submitting an MAA with the EMA.
Intellectual property litigation involves many risks and uncertainties, and there is no assurance that we will prevail in any lawsuit brought against us. Third partiesThird-parties making claims against us for infringement, violation or misappropriation of their intellectual property rights may seek and obtain injunctive or other equitable relief, which
could effectively block our ability to further develop and commercialize our product candidates. Further, if a patent infringement suit were brought against us, we could be forced to stop or delay research, development, manufacturing or sales of the product or product candidate that is the subject of the suit. Defense of these claims, regardless of their merit, would cause us to incur substantial expenses and, would be a substantial diversion of resources from our business. In the event of a successful claim of any such infringement, violation or misappropriation, we may need to obtain licenses from such third partiesthird-parties and we and our collaboration partners may be prevented from pursuing product development or commercialization and/or may be required to pay damages. We cannot be certain that any licenses required under such patents or proprietary rights would be made available to us, or that any offer to license would be made available to us on commercially reasonable terms. If we cannot obtain such licenses, we and our collaboration partners may be restricted or prevented from manufacturing and selling products employing our technologies. These adverse results, if they occur, could adversely affect our business, results of operations and prospects, and the value of our shares or ADSs.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time consuming and unsuccessful.
The biotechnology and pharmaceutical industries have been characterized by extensive litigation regarding patents and other intellectual property rights. The defense and prosecution of contractual or intellectual property lawsuits, USPTO interference or derivation proceedings, European Patent Office oppositions and related legal and administrative proceedings in the United States, Europe and other countries, involve complex legal and factual questions. As a result, such proceedings may be costly andtime-consuming to pursue and their outcome is uncertain.
Litigation may be necessary to:
protect and enforce our patents and any future patents issuing on our patent applications;
enforce or clarify the terms of the licenses we have granted or may be granted in the future;
protect and enforce trade secrets,know-how and other proprietary rights that we own or have licensed, or may license in the future; or
determine the enforceability, scope and validity of the proprietary rights of third partiesthird-parties and defend against alleged patent infringement.
Competitors may infringe our intellectual property. As a result, we may be required to file infringement claims to stopthird-party infringement or unauthorized use. This can be expensive, particularly for a company of our size, andtime-consuming. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings and some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. In addition, in an infringement proceeding, a court may decide that a patent of ours is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patent claims do not cover its technology or that the factors necessary to grant an injunction against an infringer are not satisfied. An adverse determination of any litigation or other proceedings could put one or more of our patents at risk of being invalidated, interpreted narrowly, or amended such that they do not cover our product candidates. Moreover, such adverse determinations could put our patent applications at risk of not issuing, or issuing with limited and potentially inadequate scope to cover our product candidates or to prevent others from marketing similar products.
Interference, derivation or other proceedings brought at the USPTO, may be necessary to determine the priority or patentability of inventions with respect to our patent applications or those of our licensors or potential collaboration partners. Litigation or USPTO proceedings brought by us may fail or may be invoked against us by third parties.third-parties. Even if we are successful, domestic or foreign litigation or USPTO or foreign patent office proceedings may result in substantial costs and distraction to our management. We may not be able, alone or with our licensors or potential collaboration partners, to prevent misappropriation of our proprietary rights, particularly in countries where the laws may not protect such rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or other proceedings, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation or other proceedings. In addition, during the course of this kind of litigation or proceedings, there could be public announcements of the results of hearings, motions or other interim proceedings or developments or public access to related documents. If investors perceive these results to be negative, the market price for the ADSs could be significantly harmed.
Changes to the patent law in the United States and other jurisdictions could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity and is therefore costly, time consuming and inherently uncertain. Patent reform legislation in the United States and other countries, including theLeahy-Smith America Invents Act, orLeahy-Smith Act, signed into law on September 16, 2011, could increase those uncertainties and costs. TheLeahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted, redefine prior art and provide more efficient andcost-effective avenues for competitors to challenge the validity of patents. In addition, theLeahy-Smith Act has transformed the U.S. patent system into a “first to file” system. Thefirst-to-file provisions, however, only became effective on March 16, 2013. Accordingly, it is not yet clear what, if any, impact theLeahy-Smith Act will have on the operation of our business. However, theLeahy-Smith Act and its implementation could make it more difficult to obtain patent protection for our inventions and increase the uncertainties and costs surrounding the prosecution of our or our collaboration partners’ patent applications and the enforcement or defense of our or our collaboration partners’ issued patents, all of which could harm our business, results of operations and financial condition.
The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. Additionally, there have been recent proposals for additional changes to the patent laws of the United States and other countries that, if adopted, could impact our ability to obtain patent protection for our proprietary technologies or our ability to enforce our proprietary technologies. Depending on future actions by the U.S. Congress, the U.S. courts, the USPTO and the relevantlaw-making bodies in other countries, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
Certain of our employees and patents are subject to German law.
As of December 31, 2019, 802020, 96 of our employees work in Germany and are subject to German employment law. Ideas, developments, discoveries and inventions made by such employees are generally subject to the provisions of the German Act on Employees’ Inventions, which regulates the ownership of, and compensation for, inventions made by employees. Under this act, we face the risk that we may be required to pay additional compensation for assigned patent rights and disputes can occur between us and our employees orex-employees pertaining to allegednon-adherence to the provisions of this act that may be costly to defend and consume our management’s time and efforts whether we prevail or fail in such dispute. In addition, under the German Act on Employees’ Inventions, certain employees may have retained rights to patents they invented orco-invented before October 2009. Although substantially all of these employees have assigned their interest in these patents to us, to the extent permitted by law, there is a risk that the compensation we provided to them may be deemed to be insufficient and we may be required under German law to increase the compensation due to such employees for the use of the patents. In those cases where employees have not assigned their interests to us, we may need to pay compensation for the use of those patents. If we are required to pay additional compensation or face other disputes under the German Act on Employees’ Inventions, our results of operations could be adversely affected.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated fornon-compliance with these requirements.
The USPTO and various foreign patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions to maintain patent applications and issued patents. Noncompliance with theseAlthough an inadvertent lapse, including due to the effect of the COVID-19 pandemic on us or our patent maintenance vendors, can in many cases
be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance with these requirements can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Losing our patent rights could enable competitors to enter the market earlier than would otherwise have been the case.
Failure to secure trademark registrations for a commercial trade name for any of our product candidates in the United States or elsewhere could adversely affect our business.
We use various trademark rights in our business, including, Ascendis, and our trade name TransCon. Ascendis and TransCon are our only registered trademarks in the United States. Trademark applicationsA trademark application for a number of commercial trade name candidates for TransCon hGH havelonapegsomatropin has been filed. Infiled in the United States and the European UnionUnion. However, our current or future trademarks and trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks, and we have registered both Ascendis, TransCon and a number of commercial trade name candidates for TransCon hGH. We may not be able to obtain trademark protection in other territories that we consider of significant importance to us. Furthermore, we have not yet registered trademarks for a commercial trade name for any other of our product candidates in the United States or elsewhere. During trademark registration proceedings, our trademark applications may be rejected. Although we are given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the USPTO and in comparable agencies in many foreign jurisdictions, third partiesthird-parties can oppose pending trademark applications and seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. In the event that our trademarks are successfully challenged, we could be forced to rebrand our products, which could result in loss of brand recognition and could require us to devote resources to advertising and marketing our products under new brands. We may license our trademarks and trade names to third-parties, such as distributors. Though these license agreements may provide guidelines for how our trademarks and trade names may be used, a breach of these agreements or misuse of our trademarks and tradenames by our licensees may jeopardize our rights in or diminish the goodwill associated with our trademarks and trade names.
As a result of the United Kingdom’s referendum on exitingexit from the European Union, our trademark istrademarks in the European Union are likely to require some form ofre-registration in the UK. While this is assumed to be a purely administrative act, we may accidentally not perform all required steps in time which may lead to a lapse of our trademarktrademarks in the UK.
Moreover, any name we propose to use with our product candidates in the United States or any other country must be approved by the FDA, EMA or any other relevant health authority regardless of whether we have registered it, or applied to register it, as a trademark. The FDA as well as EMA typically conducts a review of proposed product names, including an evaluation of potential for confusion with other product names. If the FDA, EMA or any other relevant approval authority objects to any of our proposed proprietary product names, we may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third partiesthird-parties and be acceptable to the FDA, EMA or any other relevant approval authority.
We may not be able to enforce our intellectual property rights throughout the world.
Filing,Patents are of national or regional effect, and filing, prosecuting and defending patents on our product candidates in all countries throughout the world would be prohibitively expensive. The requirements for patentability may differ in certain countries, particularly in developing countries. Moreover, our ability to protect and enforce our intellectual property rights may be adversely affected by unforeseen changes in foreign intellectual property laws. Additionally, laws of some countries outside of the United States and Europe do not afford intellectual property protection to the same extent as the laws of the United States and Europe. For example, patents with claims directed to dry pharmaceutical formulations of TransCon hGH have issued in the United States, Europe, and other jurisdictions, but related claims were rejected in China. This decision is currently on appeal, and we intend to vigorously defend the patentability of these claims. However, we may be unsuccessful, and our patent protection for TransCon hGH may expire sooner in China than in other jurisdictions. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, including India, China and othercertain developing countries, do not favor the enforcement of patents and other intellectual property rights. This could make it difficult for us to stop the infringement of our patents or the misappropriation of our other intellectual property rights. For example, many foreign countries have compulsory
licensing laws under which a patent owner must grant licenses to third parties.third-parties. Consequently, we may not be able to prevent third partiesthird-parties from practicing our inventions in certain countries outside the United States and many countries in Europe. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, if our ability to enforce our patents to stop infringing activities is inadequate. These products may compete with our products, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Proceedings to enforce our patent rights in foreign jurisdictions, whether or not successful, could result in substantial costs and divert our efforts and resources from other aspects of our business. Furthermore, while we intend to protect our intellectual property rights in major markets for our products, we cannot ensure that we will be able to initiate or maintain similar efforts in all jurisdictions in which we may wish to market our products. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate.
We may be subject to claims that we or our employees have misappropriated the intellectual property, includingknow-how or trade secrets, of a third party,third-party, or claiming ownership of what we regard as our own intellectual property.
Many of our employees, consultants and contractors were previously employed at or engaged by other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Some of these employees, consultants and contractors, executed proprietary rights,non-disclosure andnon-competition agreements in connection with such previous employment. Although we try to ensure that our employees, consultants and contractors do not use the intellectual property, proprietary information,know-how or trade secrets of others in their work for us, we may be subject to claims that we or these employees, consultants and contractors have used or disclosed such intellectual property, includingknow-how, trade secrets or other proprietary information. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel, or access to consultants and contractors. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
In addition, while we typically require our employees, consultants and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own, which may result in claims by or against us related to the ownership of such intellectual property. If we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights. Even if we are successful in prosecuting or defending against such claims, litigation could result in substantial costs and be a distraction to our senior management and scientific personnel.
We may be subject to claims challenging the inventorship or ownership of our patents and other intellectual property.
We may be subject to claims that former employees, collaborators, or other third-parties have an interest in our patents or other intellectual property as an inventor or co-inventor. The failure to name the proper inventors on a patent application can result in the patents issuing thereon being unenforceable. Inventorship disputes may arise from conflicting views regarding the contributions of different individuals named as inventors, the effects of foreign laws where foreign nationals are involved in the development of the subject matter of the patent, conflicting obligations of third-parties involved in developing our product candidates or as a result of questions regarding co-ownership of potential joint inventions. For example, we may have inventorship disputes arise from conflicting obligations of consultants or others who are involved in developing our product candidates. Alternatively, or additionally, we may enter into agreements to clarify the scope of our rights in such intellectual property. Litigation may be necessary to defend against these and other claims challenging inventorship. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
We or our licensors may have relied on third-party consultants or collaborators or on funds from third-parties, such as national governments, such that we or our licensors are not the sole and exclusive owners of the patents we in-licensed. If other third-parties have ownership rights or other rights to our patents, including in-licensed patents, they may be able to license such patents to our competitors, and our competitors could market competing products and technology. This could have a material adverse effect on our competitive position, business, financial conditions, results of operations, and prospects.
Risks Relatedrelating to Our Ordinary Sharesour ordinary shares and ADSs
The price of ourthe ADSs may be volatile and ADSthe holders of the ADSs may not be able to resell our ADSs at or above the price they paid.
The trading price of ourthe ADSs could be highly volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. These factors include those discussed in this “Risk Factors” section of this annual report and others such as:include:
results from, or any delays in, clinical trial programs relating to our product candidates, including clinical trials for TransCon hGH,lonapegsomatropin, TransCon PTH and TransCon CNP;
the effects on our business, operating results, prospects and financial condition of the worldwide COVID-19 pandemic;
our ability to apply our TransCon technologies to therapeutic areas other than endocrinology, including the therapeutic area of oncology;
our ability to commercialize or obtain regulatory approval for our product candidates, or delays in commercializing or obtaining regulatory approval;
announcements of regulatory approval or a complete response letter to our product candidates, or specific label indications or patient populations for its use, or changes or delays in the regulatory review process;
announcements relating to current or future collaborations or joint ventures, including decisions regarding the exercise by our collaboration partners of their options, if any, or any termination by them of their collaborations with us;
timing and amount of payments to us under our collaborations or joint ventures, if any;ventures;
announcements of therapeutic innovations or new products by us or our competitors;
announcements regarding the parent drugs that we use in developing our product candidates;
adverse actions taken by regulatory authorities with respect to our clinical trials, manufacturing supply chain or sales and marketing activities;
changes or developments in laws or regulations applicable to our product candidates;
any adverse changes to our relationship with any manufacturers or suppliers;
the success of our testing and clinical trials;
the success of our efforts to acquire, license or discover additional product candidates;
any intellectual property infringement actions in which we may become involved;
announcements concerning our competitors or the pharmaceutical industry in general;
achievement of expected product sales and profitability;
manufacture, supply or distribution shortages;
actual or anticipated fluctuations in our operating results;
European Medicines Agency, or EMA, FDA or other similar regulatory actions affecting us or our industry or other healthcare reform measures in the European Union, United States or in other markets;
changes in the structure of healthcare payment systems;
changes in financial estimates or recommendations by securities analysts;
trading volume of the ADSs;
sales of ordinary shares and/or ADSs by us, our senior management and board members, holders of the ADSs or our shareholders in the future;
general economic and market conditions and overall fluctuations in the United States and international equity markets; and
the loss of any of our key scientific or senior management personnel.
In addition, the stock markets in general, and the markets for pharmaceutical, biopharmaceutical and biotechnology stocks in particular, have experienced extreme volatility that may have been unrelated to the operating performance of the issuer. These broad market fluctuations may adversely affect the trading price or liquidity of our ADSs. In the past, when the market price of a stock has been volatile, holders of that stock have sometimes instituted securities class action litigation against the issuer. If any of the holders of ordinary shares or ADSs were to bring such a lawsuit against us, we could incur substantial costs defending the lawsuit and the attention of our senior management would be diverted from the operation of our business, which could seriously harm our financial position. Any adverse determination in litigation could also subject us to significant liabilities.
ADS holders do not directly hold our ordinary shares and do not have the rights of a holder of our ordinary shares.
ADS holders are not treated as our shareholders and do not have the rights of a holder of our ordinary shares. Danish law governs shareholder rights. Our depositary, Bank of New York Mellon, is the holder of the ordinary shares underlying our ADSs. The deposit agreement among us, the depositary, and all other persons directly and indirectly holding ADSs, sets out ADS holder rights as well as the rights and obligations of the depositary. In addition, our depositary charges certain fees to holders of our ADSs as set forth in “Item 12 D. Description of Securities Other than Equity Securities – American Depositary Shares.”ADSs.
ADS holders may not be able to exercise their right to vote the ordinary shares underlying their ADSs.
Holders of ADSs may exercise voting rights with respect to the ordinary shares represented by the ADSs only in accordance with the provisions of the deposit agreement and not as a direct shareholder in the Company. The deposit agreement provides that, upon receipt of notice of any meeting of holders of our ordinary shares, the depositary will fix a record date for the determination of ADS holders who shall be entitled to give instructions for the exercise of voting rights. Upon timely receipt of notice from us, if we so request, the depositary shall distribute to the holders as of the record date (1) the notice of the meeting or solicitation of consent or proxy sent by us and (2) a statement as to the manner in which instructions may be given by the holders. However, we may not request the depositary to distribute this information which could effectively limit the ability of ADS holders to direct voting of the ordinary shares underlying their ADSs.
ADS holders may instruct the depositary of their ADSs to vote the ordinary shares underlying their ADSs. Otherwise, ADS holders are not able to exercise their right to vote, unless they withdraw the ordinary shares underlying the ADSs they hold. However, they may not know about the meeting far enough in advance to withdraw those ordinary shares. If we ask for instructions from ADS holders, the depositary, upon timely notice from us, will notify the ADS holders of the upcoming vote and arrange to deliver our voting materials to the ADS holders. We cannot guarantee that ADS holders will receive the voting materials in time to ensure that such holders can instruct the depositary to vote the ordinary shares underlying their ADSs or to withdraw the ordinary shares underlying their ADSs so that they can vote such shares directly. If the depositary does not receive timely voting instructions from an ADS holder, the depositary may give a proxy to a person designated by us to vote the ordinary shares underlying ADSs. In addition, the depositary and its agents are not responsible for failing to carry out voting instructions or for the manner of carrying out voting instructions. This means that ADS holders may not be able to exercise any right to vote, and there may be nothing an ADS holder can do if the ordinary shares underlying their ADSs are not voted as they requested.
An ADS holder may be subject to limitations on the transfer of their ADSs and the withdrawal of the underlying ordinary shares.
ADSs, which may be evidenced by American Depositary Receipts, or ADRs, are transferable on the books of the depositary. However, the depositary may close its books at any time or from time to time when it deems expedient in connection with the performance of its duties. The depositary may refuse to deliver, transfer or register transfers of ADSs generally when our books or the books of the depositary are closed, or at any time if we or the depositary think it is advisable to do so because of any requirement of law, government or governmental body, or under any provision of the deposit agreement, or for any other reason subject to an ADS holders’ right to cancel their ADSs and withdraw the underlying ordinary shares. Temporary delays in the cancellation of ADSs and withdrawal of the underlying ordinary shares may arise because the depositary has closed its transfer books or we have closed our transfer books, the transfer of ordinary shares is blocked to permit voting at a shareholders’ meeting or we are paying a dividend on our ordinary shares. In addition, an ADS holder may not be able to cancel their ADS and withdraw the underlying ordinary shares when such holder owes money for fees, taxes and similar charges and when it is necessary to prohibit withdrawals to comply with any laws or governmental regulations that apply to ADSs or to the withdrawal of ordinary shares or other deposited securities.
If securities or industry analysts do not publish research or reports about our business, or if they issue an adverse or misleading opinion regarding our ordinary shares or ADSs, the price of the ADSs and trading volume could decline.
The trading market for the ADSs may be influenced by the research and reports that industry or securities analysts publish about us or our business. If any of the analysts who cover us issue an adverse or misleading opinion regarding us, our business model, our intellectual property or our stock performance, or if our clinical trials and operating results fail to meet the expectations of analysts, the price of our ADSs would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, we could lose visibility in the financial markets, which in turn could cause the price of our ADSs or trading volume to decline.
If we issue shares or ADSs in future financings, shareholders or holders of ADSs may experience immediate dilution and, as a result, the price of our ADSs may decline.
We may from time to time issue additional shares or ADS at a discount from the trading price of our ADSs. As a result, our shareholders and holders of ADSs would experience immediate dilution upon the issuance of ADSs at such discount. In addition, as opportunities present themselves, we may enter into financing or similar arrangements in the future, including the issuance of debt securities, preference share, ADSs or ordinary shares. If we issue shares or securities convertible into shares of our share capital, our ordinary shareholders and holders of ADSs would experience additional dilution and, as a result, the price of our ADSs may decline.
Sales of a substantial number of our ordinary shares or ADSs in the public market could cause the price of the ADSs to fall.
If our existing shareholders or holders of ADSs sell, or indicate an intention to sell, substantial amounts of our ordinary shares or ADSs representing our ordinary shares in the public market, the trading price of our ADSs could decline. Based upon the number of sharesIf our outstanding as of March 1, 2020, we have outstanding a total of 47,985,837 ordinary shares. Of those shares, approximately 6,776,094 were owned by current board members, members of our senior management and their respective affiliates, or may otherwise be subject to Rule 144 under the Securities Act. In addition, pursuant to a registration statement on Form F-3 filed in February 2016, we registered for resale by shares owned by certain selling shareholders, including shareholders that are affiliated with members of our board of directors.
As of December 31, 2019, there were 5,820,211 warrants outstanding. If these warrants are exercised an additional 5,820,211 ordinary shares or ADSs will become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules and Rule 144 and Rule 701 under the Securities Act. If these additional ordinary shares or ADSs are sold, or if it is perceived that they will be sold, in the public market, the trading price of the ADSs could decline. Any sales of securities by these security holders could have a negative effect on the trading price of the ADSs.
Our principal shareholders and senior management own a significant percentage of our shares and are able to exert significant control over matters subject to shareholder approval.
As of March 1, 2020,2021, our senior management, board members, holders of 5% or more of our share capital and their respective affiliates beneficially own approximately 60.5%68.6% of our outstanding voting securities. See “Item 7 A. Major Shareholders” for information relating to the determination of the number of shares beneficially owned by an entity or a person. As a result, these security holders have the ability either alone or voting together as a group to determine and/or significantly influence the outcome of matters submitted to our shareholders for approval, including the election and removal of board members, payment of dividends, amendments to our articles of association, including changes to our share capital or any mergers, demergers, liquidations and similar transactions. This may prevent or discourage unsolicited acquisition proposals or offers for our ordinary shares or ADSs that our shareholders or ADS holders may feel are in their best interest as a shareholder or holder of ADSs. In addition, this group of shareholders may have the ability to control our management and affairs. Such control and concentration of ownership may affect the market price of the ADSs and may discourage certain types of transactions, including those involving actual or potential change of control of us (whether through merger, consolidation,take-over or other business combination), which might otherwise have a positive effect on the market price of the ADSs.
The rights of our shareholders may be different from the rights of shareholders in companies governed by the laws of U.S. jurisdictions.
Our corporate affairs are governed by our articles of association and by the laws governing companies incorporated in Denmark, including the Danish Companies Act. The rights of shareholders and the responsibilities of members of our board of directors may be different from the rights and obligations of shareholders in companies governed by the laws of U.S. jurisdictions. In the performance of its duties, our board of directors is required by Danish law to consider the interests of our company, its shareholders and its creditors. It is possible that some of these parties will have interests that are different from, or in addition to, the interests of our shareholders or the holders of our ADS.shareholders.
Claims of U.S. civil liabilities may not be enforceable against us.
We are incorporated under the laws of Denmark. Substantially all of our assets are located outside the United States. A significant portion of our board members and employees reside outside the United States. As a result, it may not be possible for investors to effect service of process within the United States upon such persons or to enforce against them or us in U.S. courts, including judgmentsjudgements predicated upon the civil liability provisions of the U.S. securities laws of the United States.
The United States and Denmark currently do not have a treaty providing for the reciprocal recognition and enforcement of judgments,judgements, other than arbitration awards, in civil and commercial matters. Consequently, a final judgmentjudgement for payment given by a court in the United States, whether or not predicated solely upon U.S. securities laws, would not automatically be recognized or enforceable in Denmark. To obtain a judgmentjudgement which is enforceable in Denmark, the party in whose favor a final and conclusive judgmentjudgement of the U.S. court has been rendered will be required to file its claim with a court of competent jurisdiction in Denmark. Such party may submit to the Danish court the final judgmentjudgement rendered by the U.S. court. If and to the extent that the Danish court finds that the jurisdiction of the U.S. court has been based on grounds which are internationally acceptable and that proper legal procedures have been observed, the Danish court should, in principle, give binding effect to the judgmentjudgement of the U.S. court, unless such judgmentjudgement contravenes principles of public policy of Denmark. Danish courts are likely to deny the recognition and enforcement of punitive damages or other awards. Moreover, a Danish court may reduce the amount of damages granted by a U.S. court and recognize damages only to the extent that they are necessary to compensate actual losses or damages. Enforcement and recognition of judgmentsjudgements of U.S. courts in Denmark are solely governed by the provisions of the Danish Civil Procedure Code.Administration of Justice Act.
Based on the lack of a treaty as described above, U.S. investors may not be able to enforce against us or members of our board of directors, our executive board, our senior management or certain experts named herein who are residents of Denmark or countries other than the United States any judgmentsjudgements obtained in U.S. courts in civil and commercial matters, including judgmentsjudgements under the U.S. federal securities laws.
We qualify as a foreign private issuer and, as a result, we will not be subject to U.S. proxy rules and will be subject to Exchange Act reporting obligations that, to some extent, are more lenient and less frequent than those of a U.S. domestic public company.
We report under the Exchange Act, as anon-U.S. company with foreign private issuer status. Because we qualify as a foreign private issuer under the Exchange Act and although we are subject to Danish laws and regulations with regard to such matters and intend to furnish quarterly financial information to the SEC, we are exempt from certain provisions of the Exchange Act that are applicable to U.S. domestic public companies, including (i) the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act; (ii) the sections of the Exchange Act requiring insiders to file public reports of their stock ownership and trading activities and liability for insiders who profit from trades made in a short period of time; and (iii) the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form10-Q containing unaudited financial and other specified information, or current reports on Form8-K, upon the occurrence of specified significant events. In addition, foreign private issuers are not required to file their annual report on Form20-F until four months after the end of each fiscal year, while U.S. domestic issuers that are large accelerated filers are required to file their annual report on Form10-K within 60 days after the end of each fiscal year. Foreign private issuers are also exempt from the Regulation Fair Disclosure, aimed at preventing issuers from making selective disclosures of material information. As a result of the above, our shareholders and the holders of our ADS may not have the same protections afforded to shareholders of companies that are not foreign private issuers.
Our status as a “foreign private issuer” allows us to adopt International Financial Reporting Standards, or IFRS, accounting principles, which are different than accounting principles under U.S. Generally Accepted Accounting Principles, or U.S. GAAP.
We have adopted and presented our consolidated financial statements in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board.Board and as adopted by the European Union. IFRS is an internationally recognized body of accounting principles that are used by many companies outside of the United States to prepare their financial statements; and the SEC permits foreign private issuers such as our company to prepare and file their financial statements in accordance with IFRS rather than U.S. GAAP. IFRS accounting principles are different from
those of U.S. GAAP, and SEC rules do not require us to provide a reconciliation of IFRS accounting principles to those of U.S. GAAP. Investors who are not familiar with IFRS may misunderstand certain information presented in our consolidated financial statements. Accordingly, we suggest that readers of our consolidated financial statements familiarize themselves with the provisions of IFRS accounting principles to better understand the differences between these two sets of principles.
As a foreign private issuer and as permitted by the listing requirements of theThe Nasdaq Global Select Market, we rely on certain home country governance practices rather than the corporate governance requirements of theThe Nasdaq Global Select Market.
We qualify as a foreign private issuer. As a result, in accordance with the listing requirements of theThe Nasdaq Global Select Market, we rely on home country governance requirements and certain exemptions thereunder rather than relying on the corporate governance requirements of theThe Nasdaq Global Select Market. For instance, the Listing Rules for theThe Nasdaq Stock Market, or theThe Nasdaq Listing Rules, for domestic U.S. issuers require listed companies to have, among other things, a majority of their board members be independent, and to have independent director oversight of executive compensation, nomination of board members and corporate governance matters. As a foreign private issuer, however, while we intend to comply with these requirements, we are permitted to follow home country practice in lieu of the above requirements. Danish law does not require that a majority of our board consist of independent directors or the implementation of a remuneration committee or nominating and corporate governance committee, and our board may thus in the future not include, or include fewer, independent directors than would be required if we were subject to theThe Nasdaq Listing Rules, or they may decide that it is in our interest not to have a remuneration committee or nominating and corporate governance committee, or have such committees governed by practices that would not comply with The Nasdaq Listing Rules. Since a majority of our board of directors may not consist of independent directors if we decide to rely on the foreign private issuer exemption to theThe Nasdaq Listing Rules, our board’s approach may, therefore, be different from that of a board with a majority of independent directors, and as a result, the management oversight of our company could, in the future, be more limited than if we were subject to theThe Nasdaq Listing Rules. We intend to follow home country practice with regard to, among other things, quorum requirements generally applicable to general meetings of shareholders.
Furthermore, Danish law does not have a regulatory regime for the solicitation of proxies and the solicitation of proxies is not a generally accepted business practice in Denmark, thus our practice varies from the requirement of Nasdaq Listing Rule 5620(b). In addition, our shareholders have authorized our board of directors to issue securities including in connection with certain events such as the acquisition of shares or assets of another company, the establishment of or amendments toequity-based compensation plans for employees, a change of control of us, rights issues at or below market price, certain private placements and issuance of convertible notes. To this extent, our practice varies from the requirements of Nasdaq Rule 5635, which generally requires an issuer to obtain shareholder approval for the issuance of securities in connection with such events. Accordingly, our shareholders and holders of our ADS may not have the same protections afforded to shareholders of companies that are subject to these Nasdaq requirements.
We may lose our foreign private issuer status, which would then require us to comply with the Exchange Act’s domestic reporting regime and cause us to incur significant legal, accounting and other expenses.
We qualify as a foreign private issuer and therefore we are not required to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act applicable to U.S. domestic issuers. We may no longer be a foreign private issuer as of June 30, 2020,2021, which would require us to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act applicable to U.S. domestic issuers as of January 1, 2021.2022. To maintain our current status as a foreign private issuer, either (a) a majority of our ordinary shares or ADSs must be either directly or indirectly owned of record bynon-residents of the United States or (b)(i) a majority of our executive officers or directors may not be U.S. citizens or residents, (ii) more than 50% of our assets cannot be located in the United States and (iii) our business must not be administered principally inside the United States. If we lost this status, we would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. We may also be required to make changes in our corporate governance practices in accordance with various SEC and Nasdaq rules. The regulatory and compliance costs to us under U.S. securities laws if we are required to comply with the reporting requirements applicable to a U.S. domestic issuer may be significantly higher than the cost we would incur as a foreign private issuer. As a result, we expect that a loss of foreign private issuer status
would increase our legal and financial compliance costs and would make some activities highly time consuming and costly. We also expect that if we were required to comply with the rules and regulations applicable to U.S. domestic issuers, it would make it more difficult and expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These rules and regulations could also make it more difficult for us to attract and retain qualified members of our board of directors.
We may be a “passive foreign investment company” for U.S. federal income tax purposes for our current taxable year and future taxable years, which could result in adverse U.S. federal income tax consequences to U.S. investors.
Under the Internal Revenue Code of 1986, as amended, the determination of passive foreign investment company, or PFIC, status isfact-specific, and generally cannot be made until after the close of the taxable year in question. Based on the market price of our ADSs, the value of our assets and the composition of our income and assets, we do not believe we were a PFIC for U.S. federal income tax purposes for our taxable year ended December 31, 2019. However, we cannot assure you we will not be a PFIC for any taxable year.
Anon-U.S. corporation will be considered a PFIC for any taxable year if either (1) at least 75% of its gross income for such year is passive income or (2) at least 50% of the value of its assets (based on an average of the quarterly values of the assets during such year) is attributable to assets that produce or are held for the production of passive income. A separate determination must be made each taxable year as to whether we are a PFIC (after the close of each such taxable year). If we are a PFIC for any taxable year during which a U.S. Holder (as defined in “Item 10 E. Additional Information—Taxation—Material U.S. Federal Income Tax Consequences to U.S. Holders”) holds ordinary shares or ADSs, the U.S. Holder may be subject to adverse tax consequences, including (i) the treatment of all or a portion of any gain on disposition as ordinary income, (ii) the application of an interest charge with respect to such gain and certain dividends and (iii) compliance with certain reporting requirements. Each U.S. Holder is strongly urged to consult its tax advisor regarding these issues. See “Item 10 E. Additional Information—Taxation—Material U.S. Federal Income Tax Consequences to U.S. Holders.”
If a United States person is treated as owning at least 10% of our ordinary shares or ADSs, such holder may be subject to adverse U.S. federal income tax consequences.
If a U.S. Holder (as defined in “Item 10 E. Additional Information—Taxation—Material U.S. Federal Income Tax Consequences to U.S. Holders”) is treated as owning (directly, indirectly or constructively) at least 10% of the value or voting power of our ordinary shares or ADSs, such U.S. Holder may be treated as a “United States shareholder” with respect to each “controlled foreign corporation” in our group (if any). Because our group includes one or more U.S. subsidiaries, certain of ournon-U.S. subsidiaries could be treated as “controlled foreign corporations” (regardless of whether we are treated as a “controlled foreign corporation”). A “United States shareholder” of a “controlled foreign corporation” may be required to report annually and include in its U.S. taxable income its pro rata share of “Subpart F income,” “global intangiblelow-taxed income” and investments in U.S. property by “controlled foreign corporations,” regardless of whether we make any distributions. An individual that is a “United States shareholder” with respect to a “controlled foreign corporation” generally would not be allowed certain tax deductions or foreign tax credits that would be allowed to a “United States shareholder” that is a U.S. corporation. Failure to comply with these reporting obligations may subject you to significant monetary penalties and may prevent the statute of limitations from starting with respect to your U.S. federal income tax return for the year for which reporting was due. We cannot provide any assurances that we will assist investors in determining whether any of ournon-U.S. subsidiaries are treated as a “controlled foreign corporation” or whether such investor is treated as a “United States shareholder” with respect to any of such “controlled foreign corporations.” Further, we cannot provide any assurances that we will furnish to any “United States shareholders” information that may be necessary to comply with the aforementioned reporting and tax payment obligations. U.S. Holders should consult their tax advisors regarding the potential application of these rules to their investment in our ordinary shares or ADSs.
We do not currently intend to pay dividends on our ordinary shares or ADSs, and, consequently, our shareholders’ and ADS holders’ ability to achieve a return on their investment will depend on appreciation in the price of the ADSs or our ordinary shares.
We do not currently intend to pay any cash dividends on our ordinary shares for the foreseeable future. We currently intend to invest our future earnings, if any, to fund our growth. Therefore, our shareholders and ADS holders are not likely to receive any dividends on their investment for the foreseeable future. Because we do not intend to pay
dividends, our shareholders’ and ADS holders’ ability to receive a return on their investment will depend on any future appreciation in the market value of our ADSs. There is no guarantee that our ordinary shares or ADSs will appreciate or even maintain the price at which our holders have acquired them.
Investors should be aware that the rights provided to our shareholders and holders of ADSs under Danish corporate law and our articles of association differ in certain respects from the rights that would typically be provided to a shareholder of a U.S. company under applicable U.S. federal and state laws.
Under Danish corporate law, except in certain limited circumstances (which require as a minimum that a proposal for inspection has been supported by a minimum of 25% of the shareholders voting and being present at a general meeting), our shareholders may not ask for an inspection of our corporate records, while under Delaware corporate law any shareholder, irrespective of the size of such shareholder’s shareholdings, may do so. Shareholders of a Danish limited liability company are also unable to initiate a derivative action, a remedy typically available to shareholders of U.S. companies, to enforce a right of our company, in case we fail to enforce such right ourselves, other than in certain cases of board member/management liability under limited circumstances. In addition, a majority of our shareholders may release a board member or manager from any claim of liability we may have, including if such board member or manager has acted in bad faith or has breached his/her duty of loyalty and only if a minority of at least 10% of the shareholders represented at the relevant general meeting have opposed the decision, may a shareholder bring a derivative action on behalf of our company. In contrast, most U.S. federal and state laws prohibit a company or its shareholders from releasing a board member from liability altogether if such board member has acted in bad faith or has breached such board member’s duty of loyalty to our company. Additionally, distribution of dividends from Danish companies to foreign companies and individuals can be eligible fornon-refundable withholding tax, and not all receiving countries allow for deduction. Also, the rights as a creditor may not be as strong under Danish insolvency law, as under U.S. law or other insolvency law, and consequently creditors may recover less in the event our company is subject to insolvency compared to a similar case including a U.S. debtor. In addition, the use of the tax asset consisting of the accumulated tax deficit requires that we are able to generate positive taxable income and can be restricted by future amendments to Danish tax law. Finally, Danish
corporate law may not provide appraisal rights in the case of a business combination equivalent to those generally afforded a shareholder of a U.S. company under applicable U.S. laws. As a result of these differences between Danish corporate law and our articles of association, on the one hand, and U.S. federal and state laws, on the other hand, in certain instances, shareholders and ADS holders could receive less protection as an equity holder of our company than they would as a shareholder of a U.S. company.
Holders of our ordinary shares or ADSs may not be able to exercise theirpre-emptive subscription rights and may suffer dilution of their equity holding in the event of future issuances of our shares.
Under the Danish Companies Act, our shareholders benefit from apre-emptive subscription right on the issuance of ordinary shares for cash consideration only and not in the event of issuance of shares againstnon-cash contribution or debt conversion. Even the shareholders’pre-emptive subscription rights in the event of issuances of shares against cash payment may be disapplied by a resolution of the shareholders at a general meeting of our shareholders and/or the shares or ADSs may be issued on the basis of an authorization granted to the board of directors pursuant to which the board may disapply the shareholders’pre-emptive subscription rights. Such shares or ADSs may be issued above, or at market value as well as by way of incorporation of available reserves (including premium). In addition, a shareholder may not be able to exercise the shareholder’spre-emptive right on a timely basis or at all, unless the shareholder complies with the Danish Companies Act and applicable laws in the jurisdiction in which the shareholder is resident. Furthermore, the use ofpre-emptive subscription rights in relation to future capital increases in our company can be restricted for U.S. residents according to U.S. securities law. As a result, the shareholding or holders of ADSs of such shareholders or ADS holders may be materially diluted in the event shares or ADSs are issued in the future. Shares or ADSs may be issued at a discount to market price in rights offerings provided that the resolution is approved bytwo-thirds of the votes cast and the share capital represented at the general meeting and in these cases a restriction on the ability to exercisepre-emptive rights may materially dilute the value of the ordinary shares or ADSs held by the shareholder or ADS holder in question. Rights issues may also be carried out by the board of directors according to valid authorizations in our articles of association.
However, our ADS holders in the United States are not entitled to exercise or sell suchpre-emptive subscription rights related to the ordinary shares, which they represent unless we register thepre-emptive subscription rights and the securities to which thepre-emptive subscription rights relate under the Securities Act or an exemption from the
registration requirements is available. In addition, the deposit agreement provides that the depositary will not make rights available to ADS holders unless the distribution to ADS holders or both the rights and any related securities are either registered under the Securities Act or exempted from registration under the Securities Act. Further, if we offer holders of our ordinary shares the option to receive dividends in either cash or shares, under the deposit agreement the depositary may require satisfactory assurances from us that extending the offer to holders of ADSs does not require registration of any securities under the Securities Act before making the option available to holders of ADSs. We are under no obligation to file a registration statement with respect to any such rights or securities or to endeavor to cause such a registration statement to be declared effective. Moreover, we may not be able to establish an exemption from registration under the Securities Act. Accordingly, ADS holders may be unable to participate in our rights offerings or to elect to receive dividends in shares and may experience dilution in their holdings. In addition, if the depositary is unable to sell rights that are not exercised or not distributed or if the sale is not lawful or reasonably practicable, it will allow the rights to lapse, in which case our shareholders and ADS holders will receive no value for these rights.
If securities or industry analysts do not publish research or reports about our business, or if they issue an adverse or misleading opinion regarding our ordinary shares or ADSs, the price of the ADSs and trading volume could decline.
The trading market for the ADSs may be influenced by the research and reports that industry or securities analysts publish about us or our business. If any of the analysts who cover us issue an adverse or misleading opinion regarding us, our business model, our intellectual property or performance of the ADSs, or if our clinical trials and operating results fail to meet the expectations of analysts, the price of the ADSs would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, we could lose visibility in the financial markets, which in turn could cause the price of the ADSs or trading volume to decline.
We may be a “passive foreign investment company” for U.S. federal income tax purposes for our current taxable year and future taxable years, which could result in adverse U.S. federal income tax consequences to U.S. investors.
Under the Internal Revenue Code of 1986, as amended, the determination of passive foreign investment company, or PFIC, status is fact-specific, and generally cannot be made until after the close of the taxable year in question. Based on the market price of our ADSs, the value of our assets and the composition of our income and assets, we do not believe we were a PFIC for U.S. federal income tax purposes for our taxable year ended December 31, 2020. However, the application of the PFIC rules is subject to uncertainty in several respects, and we cannot assure you we will not be a PFIC for any taxable year.
A non-U.S. corporation will be considered a PFIC for any taxable year if either (1) at least 75% of its gross income for such year is passive income or (2) at least 50% of the value of its assets (generally based on an average of the quarterly values of the assets) during such year is attributable to assets that produce or are held for the production of passive income. A separate determination must be made each taxable year as to whether we are a PFIC (after the close of each such taxable year). If we are a PFIC for any taxable year during which a U.S. Holder (as defined in “Taxation—Material U.S. federal income tax consequences to U.S. holders”) holds ordinary shares or ADSs, the U.S. Holder may be subject to adverse tax consequences, including (i) the treatment of all or a portion of any gain on disposition as ordinary income, (ii) the application of an interest charge with respect to such gain and certain dividends and (iii) compliance with certain reporting requirements. Although we do not believe we were a PFIC for U.S. federal income tax purposes for our taxable year ended December 31, 2020, the application of the PFIC rules is subject to uncertainty in several respects. Whether we will be a PFIC in any year depends on the composition of our income and assets, and the relative fair market value of our assets from time to time, which we expect may vary substantially over time. Among other things, because (i) we currently own a significant amount of passive assets, including cash, and (ii) the value of our assets (including our intangible assets) that generate non-passive income for PFIC purposes is uncertain and may vary substantially over time, we cannot assure you we will not be a PFIC for any tax year. Each U.S. Holder is strongly urged to consult its tax advisor regarding these issues. See “Item 10 E. Additional Information—Taxation—Material U.S. Federal Income Tax Consequences to U.S. Holders.”
If a United States person is treated as owning at least 10% of our ordinary shares or ADSs, such holder may be subject to adverse U.S. federal income tax consequences.
If a U.S. Holder (as defined in “Item 10 E. Additional Information—Taxation—Material U.S. Federal Income Tax Consequences to U.S. Holders.”) is treated as owning (directly, indirectly or constructively) at least 10% of the value or voting power of our ordinary shares or ADSs, such U.S. Holder may be treated as a “United States shareholder” with respect to each “controlled foreign corporation” in our group (if any). Because our group includes one or more U.S. subsidiaries, certain of our non-U.S. subsidiaries could be treated as “controlled foreign corporations” (regardless of whether we are treated as a “controlled foreign corporation”). A “United States shareholder” of a “controlled foreign corporation” may be required to report annually and include in its U.S. taxable income its pro rata share of “Subpart F income,” “global intangible low-taxed income” and investments in U.S. property by “controlled foreign corporations,” regardless of whether we make any distributions. Failure to comply with these reporting obligations may subject a “United States shareholder” to significant monetary penalties and may prevent the statute of limitations from starting with respect to such shareholder’s U.S. federal income tax return for the year for which reporting was due. Further, an individual that is a “United States shareholder” with respect to a “controlled foreign corporation” generally would not be allowed certain tax deductions or foreign tax credits that would be allowed to a “United States shareholder” that is a U.S. corporation. We cannot provide any assurances that we will assist investors in determining whether any of our non-U.S. subsidiaries are treated as a “controlled foreign corporation” or whether such investor is treated as a “United States shareholder” with respect to any of such “controlled foreign corporations.” Further, we cannot provide any assurances that we will furnish to any “United States shareholders” information that may be necessary to comply with the aforementioned reporting and tax payment obligations. U.S. Holders should consult their tax advisors regarding the potential application of these rules to their investment in our ordinary shares or ADSs.
| Item 4 | Information on the Company |
A. History and Development of the Company |
We were organized under the laws of the Kingdom of Denmark in September 2006 as a private limited liability company (Anpartsselskab, or ApS) and then transformed into a public limited liability company (Aktieselskab, or A/S), effective December 17, 2007. In connection with this conversion, our legal name changed from Ascendis Pharma ApS to Ascendis Pharma A/S. We commenced operations in December 2007 in connection with the acquisition of the company that invented our TransCon technologies, Complex Biosystems GmbH.
Our registered office and principal executive offices are located at Tuborg Boulevard 12,DK-2900 Hellerup, Denmark and our telephone number is +45 70 22 22 44. Our agent for service of process in the United States is Ascendis Pharma, Inc. Our corporate website address is www.ascendispharma.com. The information on, or that can be accessed through, our website is not part of and should not be incorporated by reference into this annual report or any other report we file or furnish to the SEC. We have included our website address as an inactive textual reference only. Our ADSs are traded on The Nasdaq Global Select Market under the symbol ASND.“ASND”.
The SEC maintains an internet site at www.sec.gov that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC.
For additional information relating to the development of our company, see “Item 4 B. Information on the Company – Business Overview.” For additional information relating to the Company’s capital expenditures, see “Item 5 A. Operating Results.”
B. Business Overview |
Overview
We are applying our innovative TransCon technologies to build a leading, fully integrated biopharmaceutical company and develop a pipeline of product candidates with potentialbest-in-class profiles to address unmet medical needs. We have created a portfolio of potentialbest-in-class rare disease endocrinology product candidates to address unmet medical needs by utilizing our TransCon technologies with clinically validated parent drugs. We currently have three product candidates in clinical development in rare endocrine diseases and we are advancing multiple preclinicaltwo product candidates identified in oncology, our second therapeutic area of focus. We are also working to apply our TransCon technology platformtechnologies in additional therapeutic areas to address unmet patient needs.
Our most advanced investigational product candidate, TransCon Growth Hormone, or TransCon hGH (adoptedor lonapegsomatropin (the adopted nonproprietary name lonapegsomatropin)for TransCon hGH), is currently under regulatory review by the Food and Drug Administration, or the FDA in developmentthe U.S. and European Medicines Agency in Europe as a once-weekly long-acting prodrug of recombinant human growth hormone, also referred to as somatropin or hGH, as a potential treatment for pediatric and adult growth hormone deficiency, or GHD. Our phase 3 pediatric program for TransCon hGH consists of the heiGHt, fliGHt and enliGHten Trials. Our results from the pivotal, phase 3 heiGHt Trial demonstrated a statistically significant increase in annualized height velocity, or AHV, compared to daily hGH at 52 weeks, and showed a safety profile comparable to that of daily hGH in pediatric subjects who weretreatment-naïve.
Nearly all subjects who completed the heiGHt or fliGHt Trials have enrolled in the open-label extension study, or the enliGHten Trial, which is designed to provide long-term safety data to support the planned regulatory filings for TransCon hGH. We initiated the enliGHten Trial in 2017 as the first subjects began to roll over from the heiGHt Trial, and we have enrolled approximately 300 subjects, which we believe will form a safety database consistent with input received from regulatory authorities.
In September 2019, we completed the last subject visit forming thetwo-year follow up for the TransCon hGH phase 3 program in pediatric GHD. These data will form the safety database to support submission of a Biologics License Application, or BLA, to the U.S. Food and Drug Administration, or the FDA, for TransCon hGH to treat pediatric GHD, which we expect to submit in the second quarter of 2020, as well as submission of a Marketing Authorisation Application to the European Medicines Agency expected in the fourth quarter of 2020.
In October 2019, we received Orphan Designation from the European Commission for TransCon hGH for pediatric GHD. Orphan Designation is granted to therapies aimed at the treatment, prevention or diagnosis of a disease that is life-threatening or chronically debilitating, affects no more than five in 10,000 persons in the European Union, or EU, and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized (or the product would provide significant additional benefit over existing therapies).
We believe that TransCon hGH, if approved, may offer a once-weekly therapy for pediatric and adult GHD with the potential to improve outcomes compared to currently approved daily hGH. If approved, we believe TransCon hGH may reduce the burden of daily treatment by requiring significantly fewer injections, which we believe may improve compliance and treatment outcomes. After receiving feedback from the FDA, we have filed an IND amendment to initiate a global, phase 3 trial in subjects with adult GHD and we intend to pursue other indications for TransCon hGH consistent with our strategy to create sustainable growth.
We are also using our TransCon technology platformtechnologies to develop other product candidates to address rare endocrine diseases. These product candidates include TransCon PTH, an investigationalonce-dailylong-acting prodrug of parathyroid hormone, or PTH as a potential treatment for adult hypoparathyroidism a rare endocrine disorder of calcium and phosphate metabolism. We completed a phase 1 trial in healthy subjects in 2018, the results of which were consistent with our target product profile for TransCon PTH as a “true” replacement therapy. In this trial, TransCon PTH showed the predicted pharmacokinetic and pharmacodynamic response, suggesting the ability to normalize serum and urinary calcium levels in patients with hypoparathyroidism.
Our ongoing phase 2 PaTH Forward Trial is evaluating the safety, tolerability and efficacy of three fixed doses of TransCon PTH using aready-to-use prefilled pen device. The goal of PaTH Forward is to identify a starting dose (15, 18, or 21 µg per day) for a pivotal phase 3 trial, establish a titration regimen for complete withdrawal of standard of care (i.e., active vitamin D and calcium supplements), and evaluate TransCon PTH control of serum and urinary calcium. In February 2020, we completed enrollment of the trial with 59 subjects and we expect to reporttop-line data from the one-month blinded portion of the PaTH Forward Trial inmid-April 2020. Following evaluation of Phase 2 data from the PaTH Forward Trial, we expect and plan to initiate a global phase 3 program for TransCon PTH in the fourth quarter of 2020, including trial sites in the United States, Canada, Europe and Asia-Pacific, including Japan.
In June 2018, we were granted Orphan Drug Designation, or ODD, by the FDA for TransCon PTH. ODD is provided to drugs that are intended for the safe and effective treatment, diagnosis, or prevention of rare diseases or disorders that affect fewer than 200,000 people in the United States. We believe TransCon PTH, if approved, may provide patients suffering from hypoparathyroidism with a PTH replacement therapy that is designed to address both the short-term symptoms and long-term complications of the disease.
We are also developing TransCon CNP, an investigationallong-acting prodrug ofC-type natriuretic peptide, or CNP as a potential therapeutic option for achondroplasia, the most common form of dwarfism. TransCon CNP is designed to provide continuous CNP exposure with the goal of optimizing efficacy with a safe and convenient once-weekly dose. Currently, there are no medical therapies for achondroplasia approved by the FDA. In November 2018, we reported preliminary results from a phase 1 trial in healthy adult subjects, which supported our target product profile for TransCon CNP. In February 2019, we were granted ODD by the FDA for TransCon CNP. Following successful submission of an IND application in July 2019, we initiated the phase 2 ACcomplisH Trial to evaluate safety and efficacy of TransCon CNP in children (ages2-10 years) with achondroplasia. The company continues to work towards escalating sequential dose cohorts throughout the year, while ensuring the safety of subjects during the current pandemic and access to physicians for future monitoring visits. Our goal is to develop TransCon CNP as a safe and effective therapeutic option for achondroplasia and potentially other related growth disorders.
In addition to our pipeline of candidates in rare endocrine disorders,diseases, in January 2019, we establisheddisclosed oncology as our second independent therapeutic area of focus for our TransCon technologies. In June 2019, we announced three of our oncology product candidates and reported preclinical data supporting their development rationale. Our goal is to improve treatment efficacy while limiting or reducing toxicity by applying our TransCon technologies to clinically validated drugs, using our unique algorithm for product innovation. We are conducting preclinical studies within the field of oncology to explore multiple potential product candidates and evaluate systemic as well as localized delivery systems using our TransCon platform.
In November 2018, we announced the formation of VISEN Pharmaceuticals, or Visen, a company established to develop and commercialize our endocrinology rare disease therapies in the People’s Republic of China, Hong Kong, Macau and Taiwan, or Greater China. In connection with the formation of Visen, we granted Visen exclusive rights to develop and commercialize certain product candidates based on our proprietary TransCon technologies, including TransCon hGH, TransCon PTH and TransCon CNP, in Greater China for use in all human indications, subject to certain exceptions. As consideration for the rights granted to Visen, we received 50% ownership in the outstanding shares of Visen and concurrently with the rights we granted to Visen, entities affiliated with Vivo Capital and Sofinnova Ventures purchased shares in Visen for an aggregate purchase price of $40 million in cash. We believe Visen supports our strategy to extend our endocrinology rare disease portfolio globally and establish a presence in China in partnership with collaborators who have significant experience and knowledge of the biopharmaceutical opportunity in China. In part because Visen was established in China, we believe Visen will be able to effectively develop and, if approved, market our innovative technologies to address the needs of the local markets in Greater China.
We believe that the effectiveness of our TransCon technologies is supported by data from our preclinical research and the ongoing clinical programs, including our TransCon hGH, TransCon PTH and TransCon CNP programs, as well as findings from our ongoing development of other product candidates. We have applied the TransCon technologies in combination with a clinically validated parent drug or pathway using our algorithm for creating products with the potential to bebest-in-class in endocrinology rare diseases, and we will continue to apply this algorithm for product selection in new therapeutic areas. We believe this approach may reduce the risks associated with traditional drug development.
In addition to our pipeline of product candidates in endocrinology, we are conducting preclinical studies in the field of oncology to explore multiple potential product candidates and evaluate systemic as well as localized delivery systems using our TransCon technologies. In December 2020, we submitted an investigational new drug application, or IND, to the FDA to initiate the clinical program of TransCon TLR7/8 Agonist, a long-acting prodrug of resiquimod, a small molecule agonist of Toll-like receptors (TLR) 7 and 8. TransCon IL-2b/g, our second oncology product candidate, is currently in preclinical development.
We believe that the effectiveness of our TransCon technologies is supported by data from our preclinical research and the ongoing clinical programs, including our lonapegsomatropin, TransCon PTH and TransCon CNP programs, as well as findings from our ongoing development of other product candidates. We have applied our TransCon technologies in combination with a clinically validated parent drug or pathway using our algorithm for creating products with the potential to be best-in-class in endocrinology rare diseases, and we plan to apply this algorithm for product selection in new therapeutic areas. We believe this approach may reduce the risks associated with traditional drug development.
TransCon Product Candidate Pipeline

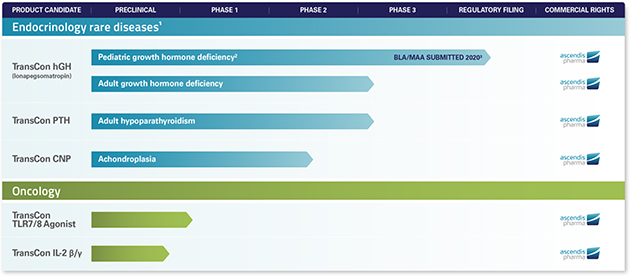
| 1. | Excludes rights granted to VISEN Pharmaceuticals in Greater China |
| 2. | In phase 3 development for pediatric growth hormone deficiency in Greater China through VISEN Pharmaceuticals |
When we apply our TransCon technologies to already approved drug compounds, we may benefit from established clinical safety and efficacy data, which we believe increases the probability of success compared to traditional drug development.
| 3. | U.S. PDUFA user fee goal date June 25, 2021 |
We maintain an intellectual property portfolio comprising over 160198 issued patents and approximately 335385 patent applications as of December 31, 20192020 with claims directed to composition of matter, process, formulation and/ormethods-of-use for our product candidates, including aproduct-specific device and core TransCon technologies. In addition, Sanofi has granted us rights that enable us to freely commercialize all improvements to the TransCon technologies developed by Sanofi outside of the field of diabetes. Other than the rights we have granted to Sanofi relating to the field of diabetes and to VisenVISEN as noted above,in this report, we hold worldwide rights to our TransCon technologies and owe nothird-party royalty or milestone payment obligations with respect to our TransCon technologies or any of our product candidates. While our TransCon prodrugs may incorporate already approved parent drugs, each of our product candidates is a new molecular entity and is therefore eligible to be granted new intellectual property rights, including new composition of matter patents.
Our Strategy
Our goal is to build a fully integrated global biopharmaceutical company by applying our TransCon technology platformtechnologies to create a pipeline of proprietary products. When we apply our TransCon technologies to already approved drug compounds, we may benefit from established clinical safety and efficacy data, which we believe increases the probability of success compared to traditional drug development. Our algorithm for product innovation focuses on identifying indications that have an unmet medical need, have a clinically validated parent drug or pathway, are suitable to our TransCon technologies, have a clearly differentiated product, have a potential established development pathway and have a large potentially addressable market.
Using this approach for our endocrinology rare disease franchise, we have obtained positive clinical data for all three of our TransCon product candidates. We are working towards regulatory approval of these candidates in three high value indications, and we are exploring label expansion opportunities. We expect ournear-term therapeutic focus on endocrinology will provide important synergies and a strong foundation for building our commercial infrastructure, including expertise in endocrinology, a concentrated prescriber base, apatient-centric support system, reimbursement and payor expertise and distribution networks.
For the longer term, our aim is to utilize our product innovation algorithm to advance into new therapeutic areas and create sustainable growth through multiple approaches. We have establisheddisclosed oncology as our second therapeutic area of focus and intend to select a third independent therapeutic area as part of our Vision 3x3 strategic roadmap through 2025, which was introduced in January 2019.2025.
The key elements of Vision 3x3 include to:
obtain regulatory approval for three independent endocrinology rare disease products: TransCon hGH for pediatric GHD, TransCon PTH for adult hypoparathyroidism and TransCon CNP for achondroplasia;
create further growth of the company’sCompany’s endocrinology rare disease pipeline through establishing global clinical reach either directly or through partnerships, pursuit of 9 total indications, and, label optimization and lifecycle management activities;activities, and through new endocrinology products;
establish a global commercial presence for the endocrinology rare disease franchise by building an integrated commercial business in North America and select European countries, and establishing relationshipsfurther global presence through partners with local expertise and infrastructure in other geographic areas; and
creation ofcreate a third independent therapeutic areasarea with a diversified pipeline built on our TransCon technology platformtechnologies and the company’sour unique algorithm for product innovation.
In November 2018, we announced the formation of VISEN Pharmaceuticals, or VISEN, a company established to develop and commercialize our endocrinology rare disease therapies in the People’s Republic of China, Hong Kong, Macau and Taiwan, or Greater China. In connection with the formation of VISEN, under three exclusive license agreements, effective November 7, 2018 and as amended January 4, 2021, or the Rights Agreements, we granted VISEN exclusive rights to develop and commercialize certain product candidates based on our proprietary TransCon technologies, including TransCon hGH, TransCon PTH and TransCon CNP, in Greater China for use in all human indications, subject to certain exceptions. As consideration for the rights granted to VISEN, we received 50% ownership in the outstanding shares of VISEN and concurrently with the rights we granted to VISEN, entities affiliated with Vivo Capital and Sofinnova Ventures purchased shares in VISEN for an aggregate purchase price of $40 million in cash. In January 2021, we invested additional $12.5 million in VISEN as part of VISEN’s $150 million Series B financing. Following the Series B financing, we retained approximately 44% of VISEN’s issued and outstanding shares. We believe our investment in VISEN supports our strategy to extend our endocrinology rare disease portfolio globally and establish a presence in China in partnership with collaborators who have significant experience and knowledge of the biopharmaceutical opportunity in China. In part because VISEN was established in China, we believe VISEN will be able to effectively develop and, if approved, market our innovative technologies to address the needs of the local markets in Greater China.
TransCon Technologies
Overview
Our TransCon technologies are designed to solve the fundamental limitations of previous approaches applied to extend duration of a drug’s action in the body, and to enhance the overall benefit of a given therapeutic. Many drugs suffer from suboptimal pharmacokinetics, short residence time in the body, poor tolerability at the administration site and/or systemic side effects that result from initial drug concentrations that are too high. Frequent administration and poor tolerability negatively impact patient compliance, potentially leading to suboptimal treatment outcomes. To address these issues, several approaches are currently being applied to improve drug characteristics, such as prodrug and sustained release technologies.
Our TransCon technologies combine the benefits of conventional prodrug and sustained release technologies to create new therapies with potentially optimized therapeutic effect, including efficacy, safety and dosing frequency. We believe the technologies can be applied broadly to a protein, peptide, antibody or small molecule in multiple therapeutic areas. TransCon molecules have three components: an existing parent drug, an inert carrier that protects it, and a linker that temporarily binds the two. When bound, the carrier inactivates and shields the parent drug from clearance. When injected into the body, physiologic pH and temperature conditions initiate the release of the active, unmodified parent drug in a predictable release manner. Because the parent drug is unmodified, its original mode of action is expected to be maintained. Depending upon the type of TransCon carrier we employ, we can design our TransCon prodrugs to act systemically or locally in areas that are difficult to treat with conventional therapies. In addition to retaining the original mode of action of the unmodified parent drug, we believe this predictable release may improve the likelihood of clinical development success. We refer to our systemic and localized applications of TransCon as individual technologies.
We believe that our TransCon technologies enable multiple potential therapeutic, drug development and intellectual property benefits:
Efficacy Benefits
Same mode of action as parent drug
Predictable release of unmodified parent drug supporting dosing frequency from daily to up to six months or more
Designed to enable sustained localized or systemic drug exposure
Designed to reduce dosing frequency to improve patient adherence and improve overall treatment outcomes
Dosing and release tailored to desired pharmacokinetic profile and potentially optimizing effects of parent drug
Safety and Tolerability Benefits
Same predicted safety profile as parent drug with potential enhancements or improvements due to application of TransCon technologies
May enable comparable alternative to continuous infusions or subcutaneous injections
TransCon localized delivery platform may offer improved safety profile by maintaining high local concentrations of drug while minimizing systemic exposure
Immunogenic potential, or the ability of a substance to provoke an immune response, comparable to parent drug
Development Benefits
Potentially higher drug development success rate when investigating clinically validated parent drugs and mechanisms by leveraging existing knowledge
Intellectual Property Benefits
New composition of matter patents
TransCon Technology Components
Our TransCon prodrug product candidates consist of three components: the TransCon linker, the TransCon carrier and a parent drug.
Our broad selection of TransCon linkers, in combination with our systemic and localized carriers, provides us with a powerful and flexible technology platform that we leverage to design potentiallybest-in-class therapeutics to address unmet medical needs.
TransCon Linkers
Our TransCon linkers are reversible linkers that enable the transient conjugation of a broad range of therapeutics, including proteins, peptides and small molecules, to our TransCon carriers. We have a large library of TransCon linkers that are applicable to various types of parent drugs, and that can be tailored to achievehalf-life extension enabling daily, weekly, monthly andhalf-yearly dosing, and customize the pharmacokinetic, or PK, profile for each individual product candidate to potentially optimize therapeutic effect. TransCon linkers areself-cleaving through a process calledintra-molecular assisted cleavage, which causes the linker to release the unmodified parent drug. We can tailor the release properties of the linker to a given therapeutic indication and parent drug by modifying the linker structures. We believe theself-cleaving process of our linker avoids many of the shortcomings of conventional prodrug technologies, which often depend on metabolic processes, such as enzymatic degradation, to convert the prodrug into the active drug. The rate of metabolic conversion of prodrugs in these types of processes may differ between patients, and even within different tissues in the same patient. As a result, conventional prodrugs do not always offer predictable release of the parent drug. Our TransCon linkers predictably release an unmodified active parent drug at predetermined rates governed by physiological pH and temperature conditions, which are tightly regulated in the body. Consequently, we can design our prodrugs to release the unmodified parent drug at predictable rates.
TransCon Carriers
Our TransCon technologies incorporate two carrier platforms that can be used for providing localized or systemic drug exposure. These biocompatible carrier platforms include our TransCon systemic carriers and our proprietary TransCon localized carriers, which areself-eliminating hydrogels. Our carriers inactivate and protect the drug through a shielding effect, which prevents rapid excretion and degradation of the parent drug and may enable benefits that include improved injection site tolerability, reduced systemic adverse effects and low immunogenicity.
Our TransCon systemic carriers are used for providing systemic drug exposure and are based on soluble compounds such as polyethylene glycol, or PEG, or other natural or synthetic polymers. Prodrugs created using our systemic carriers are readily absorbed into the bloodstream after administration, thus minimizing exposure of the subcutaneous tissue to active drug, which we believe may improve injection site tolerability. Our most advanced product candidates, TransCon hGH,lonapegsomatropin, TransCon PTH and TransCon CNP, utilize PEG as a carrier molecule. PEG is widely used to improve the pharmacokinetic or pharmacodynamic properties of marketed therapeutics. Below is an illustration of our systemic carrier:
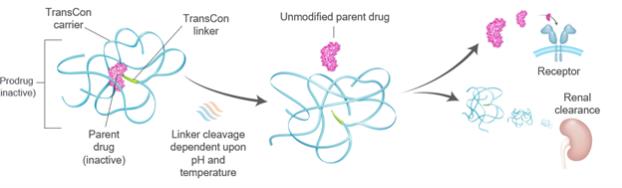
TransCon carrier Transcon linker Parent drug (active) Prodrug (Inactive) Parent drug (inactive) Linker cleavage dependent upon pH and temperature Receptor Renal clearance
Our TransCon localized carriers are being developed to provide either localized or systemic parent drug exposure. Our TransCon hydrogels may be based on PEG, hyaluronic acid or other biopolymers. Our TransCon hydrogel is designed toself-eliminate to soluble, biocompatible molecules after the drug payload has been released. With our current and future collaboration partners, we are developing certain of the TransCon hydrogel carriers to provide both systemic and localized parent drug delivery applications. When applied for localized delivery, the TransCon hydrogel enables the release of parent drug at high local concentrations while minimizing systemic exposure. We believe this may widen the therapeutic window for parent drugs that suffer from significant systemic side effects and facilitate the development of highly efficacious product opportunities that have improved safety and tolerability profiles. Below is an illustration of our hydrogel carrier:

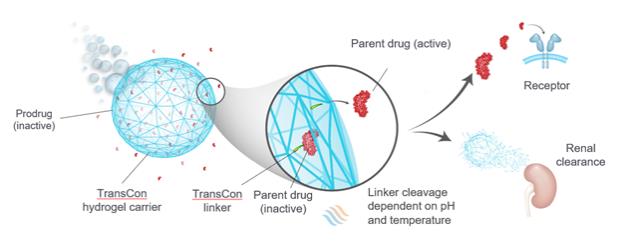
Parent drug (active) Prodrug (inactive) Receptor Transcon hydrogel carrier TransCon linker Parent drug (inactive) Linker cleavage dependent on pH and temperature Renal clearance
Parent Drug
Our TransCon technologies are applicable across a broad range of therapeutic classes and are currently used to create potentiallybest-in-classlong-acting product candidates based on proteins, peptides and small molecules. By primarily focusing on biological targets that have been clinically validated, we can leverage available knowledge regarding a target’s activity. Based on this selective approach, we know what drug levels must be maintained in the body for optimal efficacy and safety, and we can design the releasehalf-life and dosing frequency of our TransCon prodrugs to maintain these levels to achieve the desired pharmacological effect. We move a product candidate into development after it demonstrates a superior profile to such medicines or drugs in animal models that we believe correlate to human
clinical experience. Furthermore, based on the established translational relationships between preclinical animal models and clinical efficacy, we believe experimental results generated in validated animal models are highly predictive of clinical results and reduce the development risk of our TransCon prodrugs. This strategy is designed to reduce risk and increase productivity.
This approach has enabled us to generate a pipeline of product candidates to address significant unmet medical needs and to become potential sources of significant revenue for our company. Because our TransCon technologies
release an unmodified drug with established clinical safety and efficacy, we believe we may benefit from a higher development and regulatory success rate as compared to development of drug compounds without established biology.
TransCon Product Candidates - Endocrinology
TransCon Growth Hormone (hGH)
Market Opportunity in GHD
GHD is a serious orphan disease that affects both children and adults. Children with GHD are characterized by short stature, metabolic abnormalities, cognitive deficiencies and poor quality of life. GHD in adults is associated with increased adiposity, or fat mass, as well aspsychiatric-cognitive, cardiovascular, muscular, metabolic and skeletal abnormalities. The current standard of care for GHD is daily subcutaneous injections of somatropin (hGH). Daily therapy with hGH has been shown to increase growth in children, and improve metabolic effects, including reducing adiposity and improving cardiovascular health in both children and adults. These daily hGH therapies have been shown to be safe andwell-tolerated.
In both therapy-compliant children and adults with GHD, daily subcutaneous injections of hGH have resulted in improved body composition parameters, bone density, cardiovascular outcomes and quality of life. Growthhormone-deficient children who are fully adherent with their daily hGH treatment regimen may achieve a height in adulthood that is comparable to that of their family members and national norms. In boththerapy-compliant children and adults with GHD, daily subcutaneous injections of hGH have resulted in improved body composition parameters, bone density, cardiovascular outcomes and quality of life.
Despite the demonstrated benefits of hGH therapy, adherence continues to be a challenge, as patients treated with daily hGH typically receive thousands of injections over the course of many years. Published studies have shown that the majority of patients on a daily hGH regimen are not fully adherent with their daily dosing schedule, and therefore fail to achieve expected treatment outcomes. According to a 2018 study (Graham et al), as many as 7seven out of 10ten pediatric patients arenon-adherent to daily growth hormone therapy, which may result in significant reductions in the degree of growth and suboptimal outcomes.
Reducing injection frequency is associated with better adherence and thus may improve height velocity (HV) for pediatric patients experiencing poor adherence with daily hGH injections. As shown in the figure below, for patients missing two or more injections per week, there was a clinically relevant reduction in their change in HV standard deviation score, or HVSDS, compared tohigh-compliance patients. A greater HVSDS indicates more rapid growth:
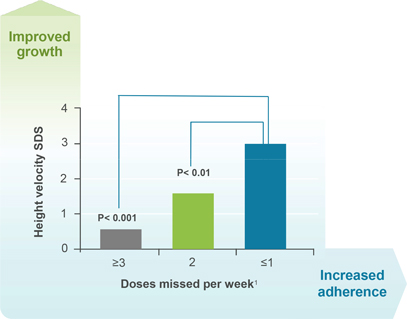
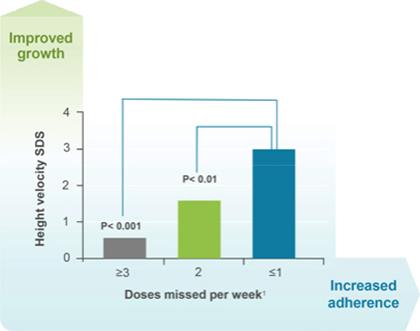
Improved growth Height velocity SDS P<0.001 P<0.01 Doses missed per week1 Increased adherence 0 1 2 3 4 >3 2 <1
Figure 1. Negative impact of poor compliance on growth response. Patients missing two or more injections per week havehad a statistically significant reduction in height velocity. A result is considered statistically significant when the p-value, representing the probability that random chance could explain the result, is lower than 0.05. (Cutfield et al, 2011.)
Since the introduction of hGH in 1981, a number of the world’s largest pharmaceutical companies have developed and now market daily hGH products. All currently marketed hGH products in the United States, Norditropin® (Novo Nordisk A/S), Humatrope® (Eli Lilly and Company), Nutropin AQ® (Genentech), Genotropin® (Pfizer Inc.), Saizen® (Merck Serono S.A.), Zomacton® (Ferring Pharmaceuticals, Inc.) and Omnitrope® (Sandoz GmbH), contain unmodified somatropin (hGH) and are administered by daily subcutaneous injections. The global market for daily hGH products is dominated by Novo Nordisk, Pfizer, Eli Lilly, Sandoz, Merck KGaA and Roche, which together account for most of the global market share.
Primary indications for hGH in children are GHD, idiopathic short stature, chronic kidney disease,Prader-Willi syndrome, small for gestational age and Turner syndrome. In adults, primary indications for hGH include GHD andAIDS-induced weight loss. Pediatric indications comprise up to 90% of the total hGH market, of which approximately half is for GHD.
Global annual sales from currently marketed hGH products are currently estimated at approximately $4 billion. We believe a significant market opportunity exists for along-acting version of hGH with comparable efficacy, safety and tolerability as daily hGH products. We are developing TransCon hGHlonapegsomatropin as aonce-weekly somatropin (hGH) therapy with a target profile designed to have comparable efficacy, safety and tolerability to daily hGH, but with a dosing regimen that could improve adherence and overall health.
Competitive Landscape forLong-Acting Growth Hormone Therapies
Since the 1990s, the pharmaceutical industry has employed various approaches to developlong-acting growth hormone products to reduce the patient burden of daily injections and increase patient compliance with the dosing regimen. These approaches generally fall into two categories: unmodified somatropin (hGH) and permanent modification of growth hormone:
| • | Unmodified somatropin (hGH): Twolong-acting growth hormone products using encapsulation technologies have previously received regulatory approval in U.S. and Europe and were subsequently discontinued due to commercial challenges. These include Nutropin Depot®, formerly marketed by Genentech, and Somatropin Biopartners, developed by LG Life Sciences and Biopartners GmbH. Nutropin Depot was approved in 1999 and later withdrawn; Somatropin Biopartners (LB03002), was approved by the European Medicines Agency, |
Permanent modification of growth hormone: Modification technologies prolong activity in the body by creating analogs of growth hormone through permanent modification of the growth hormone molecule. This modification may alter the molecular size and interaction with the growth hormone receptor and/or change the natural association affinity to endogenous proteins, as well as the distribution in the body. These changes may alter and reduce the efficacy of these drugs compared to unmodified daily somatropin (hGH) and may also negatively impact the drug’s safety.
ThereTo our knowledge, as of the date of this report, there are currently no commercially available long-acting growth hormone treatment options available in the United States or Europe. A permanently PEGylatedlong-acting growth hormone developed by GeneScience Pharmaceuticals Co., Ltd. is available in China and the Somatropin Biopartners product (LB03002), is available in Korea.
On August 28, 2020, the FDA granted Novo Nordisk Inc. approval of somapacitan for replacement of endogenous growth hormone in adult patients with GHD but as of the date of this report to our knowledge Novo Nordisk has not commercially launched the product in the United States. In additionJanuary 2021, Novo Nordisk received a positive opinion from the Committee for Medicinal Products for Human Use, under the EMA for once-weekly somapacitan, recommending marketing authorisation for use in adult patients with GHD. Pfizer (in collaboration with OPKO Health Inc.) has submitted to the currently approved and marketed dailyFDA, a Biologics License Application, or BLA, for somatrogon, a long-acting growth hormone therapies, there are a varietyfor the treatment of pediatric patients with GHD. In January 2021, Pfizer (in collaboration with OPKO Health) announced the FDA has accepted the regulatory submission and set the PDUFA goal date in October 2021.
Other experimental growth hormone therapies based on permanent modification are in different stages of clinical development by various companies, including GeneScience Pharmaceuticals Co., Ltd., Genexine Inc,Inc., I-MAB, and JCR Pharmaceuticals Co., Ltd., Novo Nordisk A/S, and OPKO Health, Inc. (in collaboration with Pfizer Inc.).
Our Solution: TransCon hGHLonapegsomatropin
TransCon hGHLonapegsomatropin is an investigationallong-acting prodrug of somatropin (hGH) that is being developed for the treatment of GHD. It is designed to maintain the same mode of action as daily therapies by releasing the same growth hormone molecule, somatropin, as daily hGH therapy. TransCon hGHlonapegsomatropin is composed of an unmodified somatropin that is transiently bound to a carrier and proprietary linker.
We believe ouronce-weekly TransCon hGH Lonapegsomatropin has the same mode of action and distribution into key growthhormone-responsive tissues, such as brain, bone, muscle, liver and fat tissue, as the hGH administered from daily injections and endogenous growth hormone. We use daily growth hormone as an active comparator in our clinical studies, allowing us to directly compare the activity of TransCon hGHlonapegsomatropin to daily growth hormone in an identical clinical setting.
In October 2019, we received Orphan Designation from the European Commission for lonapegsomatropin for pediatric GHD. Orphan Designation is granted to therapies aimed at the treatment, prevention or diagnosis of a disease that is life-threatening or chronically debilitating, affects no more than five in 10,000 persons in the European Union, or EU, and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized (or the product would provide significant additional benefit over existing therapies). We received Orphan Drug Designation, or ODD, from the FDA for lonapegsomatropin as a treatment for GHD in April 2020.
Clinical Development of TransCon hGHLonapegsomatropin for Pediatric GHD
Our phase 3 pediatric program for TransCon hGHlonapegsomatropin consists of the heiGHt, fliGHt and enliGHten Trials. In March 2019, we reportedThe top-linetwo-year follow-up resultsdata for the heiGHt Trial,phase 3 program for pediatric GHD provides the safety database to support our BLA submission to the U.S. FDA in which TransCon hGH was observedJune of 2020 and our Marketing Authorisation Application, or MAA, to have superior efficacy (as demonstrated by a statistically significant increasethe EMA in AHV at 52 weeks) and comparable safety and tolerability to thatSeptember of daily hGH.2020.
Results from the Phase 3 heiGHt Trial in Pediatric Subjects with GHD
The heiGHt Trial was a randomized,open-label,active-controlled phase 3 registrational trial that enrolled 161 children with GHD who had not previously been treated. Subjects received eitheronce-weekly TransCon hGH lonapegsomatropin (0.24 mg/kg/week) or daily injections of Genotropin® at 34 µg/kg/day (0.24 mg/kg/week) with a 2:1 randomization. The primary endpoint was annualized height velocity (AHV) at 52 weeks, with anon-inferiority analysis comparing the difference between the two treatment groups.groups, followed by a test of superiority if non-feriority was met. Two subjects, one from each arm, withdrew from the trial prior to the final visit.
Top-line resultsResults showed thatonce-weekly TransCon hGH lonapegsomatropin was superior toonce-daily hGH on the primary endpoint of AHV at 52 weeks. In the primary analysis of theintent-to-treat population using least squared mean (LS Mean) results from an ANCOVA TransCon hGHmodel, lonapegsomatropin was associated with an AHV of 11.2 cm/year compared to 10.3 cm/year for the daily hGH. The treatment difference was 0.86 cm/year with a 95 percent confidence interval of 0.22 to 1.50 cm/year. The AHV for TransCon hGHlonapegsomatropin was significantly greater than the daily hGH (p=0.0088).
The AHV was greater for TransCon hGHlonapegsomatropin than for the daily hGH at each visit, with the treatment difference reaching statistical significance from and including week 26 onward. The incidence of subjects with AHV < 8 cm/year at 52 weeks, referred to as poor responders (AHV < 8.0 cm/year) was 4 percent4% and 11 percent11% in the TransCon hGHlonapegsomatropin and daily hGH arms, respectively. All sensitivity analyses completed from the trial support the primary outcome, indicating the robustness of these results.
Results from the trial indicate that TransCon hGHlonapegsomatropin was generally safe andwell-tolerated, with adverse events consistent with the type and frequency observed with daily hGH therapy and comparable between arms of the trial. No serious adverse events related to study drug were observed in either arm. Notreatment-emergent adverse events leading to discontinuation of study drug were observed in either arm.
Additional analyses from the heiGHt Trial showed:
No incidence of treatment emergent anti-hGHneutralizing antibodies detected, and post-baseline low level oflow-titernon-neutralizinganti-hGHlow-titer non-neutralizing anti-hGH binding antibodies was similar between the two arms (7 [6.7%(6 [5.7%] in TransCon hGHlonapegsomatropin vs. 2 [3.6%] in Genotropin arm).
Height SDS at 52 weeks increased over baseline by 1.10 for TransCon hGHlonapegsomatropin and by 0.96 for the daily hGH, with the treatment difference increasing at each visit over 52 weeks.
Body Mass Index (BMI) was in the normal range over 52 weeks in both the TransCon hGHlonapegsomatropin group (with a mean increase in BMI SDS from -0.32 at baseline to -0.03 at Week 52) and in the daily hGH group (with a mean BMI SDS decrease from -0.14 at baseline to -0.40 at week 52).
Mean hemoglobin A1c values were generally stable over the course of the trial and remained within the normal range for both arms.
Model-derived mean average insulin-like growth insulin-like growthfactor-1(IGF-1)factor-1 (IGF-1) SDS values were 0.610.72 for TransCon hGHlonapegsomatropin and 0.01-0.02 for the daily hGH at week 52.
Adverse events leading to dose reduction(IGF-1 levels or clinical symptoms) occurred twice in the TransCon hGHlonapegsomatropin arm (representing 1.9 percent) and once in the daily hGH arm (representing 1.8 percent).
Observed change in bone age over 52 weeks from baseline was 1.36 years for the TransCon hGHlonapegsomatropin arm and 1.35 years in the daily arm.
Additional Clinical Trials of TransCon hGH in Pediatric Subjects with GHD
The results presented for the heiGHt Trial are consistent with findings from the phase 2 trial, anactive-controlled trial which found that three different doses of TransCon hGH were comparable to daily hGH in 53treatment-naïve,pre-pubertal children with GHD. The phase 2 trial was conducted with a bioequivalent predecessor molecule and designed to evaluate the safety, tolerability, pharmacokinetics, pharmacodynamics and efficacy of TransCon hGH. This6-monthmulti-center,6-month multi-center, randomized,open-label trial compared three dose levels of TransCon hGH (0.14; 0.21; and 0.30 mg GH/kg/week), administered once per week, to Genotropin at 30 µg/kg/day (0.21 mg GH/kg/week), administered as a daily injection. The primary efficacy endpoint was annualized mean height velocity at six months. Mean annualized height velocities among the three dosing levels administered weekly were 11.9 cm for the 0.14 mg/kg/week dose, 12.9 cm for the 0.21 mg/kg/week dose, and 13.9 cm for the 0.30 mg/kg/week dose, which were comparable to 11.6 cm for the active comparator, daily injections of Genotropin at a 0.21 mg/kg/week dose.
Additionally, in May 2019, we announced preliminary data from theThe single arm phase 3 fliGHt Trial of TransCon hGH, whichlonapegsomatropin was designed to create additional exposures and expand the safety database for TransCon hGHlonapegsomatropin in pediatric subjects. In this single armopen-label trial, 146 patients were enrolled, and subjects switched from daily hGH to weekly TransCon hGHlonapegsomatropin with a primary objective of evaluating the safety and tolerability of TransCon hGHlonapegsomatropin in subjects with pediatric GHD. TheResults of the trial enrolled 146 subjects, and indicated treatment with TransCon hGHlonapegsomatropin was safe and well-tolerated in those with pediatric GHD who were previously treated with commercially-available daily growth hormone therapies. In the trial, theThe safety profile of TransCon hGHlonapegsomatropin was similar to the published safety profile of daily growth hormone therapies. We believe the results from the fliGHt Trial support switching to once-weekly TransCon hGHlonapegsomatropin from a daily hGH, and also provide evidence supporting the tolerability of TransCon hGHlonapegsomatropin in subjects under three years of age.
Nearly all subjects who completed the heiGHt or fliGHt Trials have enrolled in the open-label extension study, the enliGHten Trial, which is designed to provide long-term safety data to support the planned regulatory filings for TransCon hGH.lonapegsomatropin. We initiated the enliGHten Trial in 2017, and approximately 300 pediatric subjects enrolled. We expect dataData from enliGHten will form aformed the long-term safety database consistent with the input we have received from regulatory authorities.
In September 2019, we completed the last subject visit forming thetwo-year follow up for the TransCon hGH phase 3 program in pediatric GHD. These data, including results from approximately 300 subjects treated with TransCon hGH (approximately 300 for six months, 120 for 12 months and 45 for 24 months), will form the safety database to supportsupporting our BLA submission of a BLA to the FDA for TransCon hGHlonapegsomatropin for the treatment of pediatric GHD expectedwhich occurred in the second quarter ofJune 2020, as well as submission of a Marketing Authorisation ApplicationMAA to the EMA which occurred in September 2020. The PDUFA user fee goal date is set for June 25, 2021. If approved by the FDA, we expect to commercially launch lonapegsomatropin for the treatment of pediatric GHD in the U.S. in the third quarter of 2021. In addition, in July 2020, we received approval of our proposed Paediatric Investigation Plan covering ages 6 months to less than 18 years of age from the EMA for lonapegsomatropin and we anticipate the European Medicines Agency, expectedCommission to issue a decision on our MAA in the fourth quarter of 2020.2021.
Additionally, in January 2020,2021, we presented 78-weekannounced 104-week analysis of data from the ongoing enliGHten Trial, includingfollow-up on subjects from heiGHt who continued into enliGHten. The data showed
maintenance of a treatment advantage in subjects initially treated with TransCon hGHlonapegsomatropin beyond the first year of therapy. The safety results, which were comparable to Genotropin in the phase 3 heiGHt Trial, were consistent across the phase 3 clinical trials.
In September 2020, we filed a Clinical Trial Notification with the Pharmaceuticals and Medical Device Agency in Japan to initiate the company’s phase 3 riGHt Trial of lonapegsomatropin for the treatment for pediatric GHD. The primary objective of the riGHt Trial is to evaluate and compare the annualized height velocity of 40 Japanese prepubertal treatment naïve children with GHD treated with weekly lonapegsomatropin to that of a commercially available daily hGH formulation at 52 weeks.
VISEN, our strategic investment partner, is conducting a clinical study in China evaluating lonapegsomatropin for the treatment of pediatric GHD.
ProprietaryAuto-injector Development
As part of our effort to improve treatment compliance among children with GHD, we are developing astate-of-the-artauto-injector for the administration of TransCon hGH.lonapegsomatropin. In May 2019, we introduced the auto-injector into the enliGHten Trial and we expect to make it available to patients in conjunction with a potential commercial launch. As of January 2020, overOver 160 subjects have used auto-injector and dual-chamber cartridges in the enliGHten extension trial, which we believe meets our objective of collecting required usability data to support inclusion of the auto-injector as part of our initial BLA submission.
The auto-injector provides for room temperature storage, includes anempty-all design and is expected to last for at least four years. With simple operation, the device has a single,low-volume injection for the majority of patients of less than 0.6 mL and requires a small,31-gauge needle that is only 4 millimeters in length, which is comparable to needles used to administer daily hGH. We are also working on strategies that will enable theauto-injector to integrate with the digital healthcare system, including Bluetooth connectivity features to allow for easy tracking of dosing complianceadherence over time.
Figure 2. Ourstate-of-the-artauto-injector is designed to improve treatment compliance for children with GHD.
Clinical Development of TransCon hGHLonapegsomatropin in Adults
We have successfully completed a phase 2, European,multi-center, multiple dose,open-label,active-controlled, study to examine the safety, tolerability, pharmacokinetics and pharmacodynamics in 37 adult male and female subjects with GHD. We have also completed several phase 1 trials in healthy adult subjects.
Results from the Phase 2 Trial in Adults with GHD
In September 2011, we reported data from aFollowing our phase 2 European,multi-center, multiple dose,open-label,active-controlled,study to examine the safety, tolerability, pharmacokinetics and pharmacodynamics in 37 adult male and female subjects with GHD.
In this study, serum levels of somatropin (hGH) increased proportionallydiscussions with the administered dose and the maximum serum concentration of somatropin released from TransCon hGH was comparable to the levels achieved by daily hGH injections.
TransCon hGH also elicited anIGF-1 response that was similar to theIGF-1 response of the same cumulative dose of hGH administered as daily injections over a week. Importantly, theIGF-1 response at Week 1 and Week 4 were similar and without significant accumulation.
In addition to demonstrating similar maximum hGH and resultingIGF-1 concentrations when administered at the same cumulative weekly dose, the exposure to hGH andIGF-1 over one week, as judged by AUC, orArea-Under-the-Curve, was similar between TransCon hGH and daily hGH.
In this study, adverse events were comparable to the incidence and type generally expected when hGH is administered to adults with GHD. Only mild and transient injection site reactions were observed across all treatment groups with no difference between treatment groups, including daily hGH.
Notreatment-emergentanti-hGH antibody formation was observed during thismultiple-dose study. We did not observe any injection site lipoatrophy following repeated injections of TransCon hGH. We believe the PK and pharmacodynamics, or PD, data gathered in our phase 2multi-dose study in adult subjects supports the desiredonce-weekly dosing regimen and confirms the favorable safety profile of TransCon hGH previously observed in phase 1 studies.
Future Development Plans
Following discussions with FDA, we have submitted an amendment to our IND amendment to initiate the foresiGHt Trial, a global phase 3 foresiGHt Trial in adult GHD. The foresiGHt Trial is expected to begin enrollment later this year, and aimsstudy with the aim to demonstrate the metabolic benefits of TransCon hGHlonapegsomatropin in adults, with the primary objective to evaluate change in trunk fat percentage. The three arms of the study include patients treated with once-weekly lonapegsomatropin, once-weekly placebo, and daily hGH with patients randomized in a 1:1:1 ratio. The primary endpoint of the study is a change from baseline in percentage trunk fat at 38 weeks. Following the 38-week main period, all patients will receive once-weekly lonapegsomatropin during the 52-week open-label extension. We expect to complete enrollment of foresiGHt by the end of 2021 or early 2022.
In addition, we
Future Development Plans
We are currently evaluating clinical development plans for TransCon hGHlonapegsomatropin in additional pediatric indications, and we intend to initiatefollow-up clinical trials in the future. We are also considering other potential indications for TransCon hGHlonapegsomatropin where along-acting hGH therapy may offer abest-in-class option for patients with rare growth disorders.
As part of our strategy to establish global reach, we also intend to initiate a phase 3 trial in pediatric GHD in Japan in fourth quarter of 2020.
TransCon PTH
Market Opportunity in Hypoparathyroidism
Hypoparathyroidism, or HP, is a rare endocrine disorderdisease characterized by deficient or absent parathyroid hormone, or PTH, affecting approximately 200,000 patients in the U.S., Europe, Japan and South Korea. The most common cause in approximately 75% of cases is inadvertent removal or damage to the parathyroid tissue during neck surgery. Patients with HP cannot adequately regulate calcium and phosphate metabolism and suffer from low calcium and elevated phosphate levels in the blood. The condition results in a diverse range of physical, cognitive and emotional symptoms. Short term symptoms include weakness, severe muscle cramps (tetany), abnormal sensations such as tingling, burning and numbness (paresthesia), memory loss, impaired judgmentjudgement and headache. Over the long term, treatment with the current standard of care, (SoC)or SoC, can increase risk of major complications, such as extraskeletal calcium depositions occurring within the brain, lens of the eye, and kidneys, which can lead to impaired renal function. Patients often experience decreased quality of life.
PTH controls serum calcium via several mechanisms. PTH acts to release calcium from the skeleton by regulating bone turnover, and in the kidney to reabsorb calcium from the urine. In addition, PTH facilitates the conversion of 25 hydroxyvitamin D to the active form, which in turn acts on the intestines to increase calcium absorption. Through these primary pathways, calcium homeostasis is maintained.
Current SoC for HP patients primarily consists of oral calcium and active vitamin D and oral calcium supplementation. However, since PTH is not present at the kidney to facilitate calcium reabsorption from the urine, the goal of SoC is to maintain serum calcium (sCa) levels just below or within the lower part of the normal range and thereby limit as much as possible the damage from excess urinary calcium. Nonetheless, SoC frequently leads to significant sCa fluctuations accompanied by symptomatichyper- or hypocalcemia. SoC with calcium and active vitamin D and calcium have been shown to contribute to the risk of renal disease.
HP also poses a high burden on the healthcare system despite current standard of care.SoC. For example, one survey of 374 patients showed that 72% experienced more than 10ten symptoms in the preceding 12twelve months, with symptoms experienced for a mean of 13 ± 9 hours a day. Other studies showed that 79% of HP cases require hospitalizations and that patients with the disorderdisease results have afour-fold increase in the risk of renal disease compared to healthy controls. Patients often experience decreased quality of life. We conducted a survey of 42 patients which found that 100 percent of subjects reported negative psychological impacts, interference with daily life and impact on physical functioning from HP, and that 76 percent were either no longer able to work or experienced interference with work productivity.
An effective PTH replacement therapy that fully addresses the condition is not widely available to patients with HP.HP in the U.S. In 2015, NATPARA,PTH(1-84), was approved foronce-daily subcutaneous injection as an adjunct to vitamin D and calcium in patients with hypoparathyroidism. NATPARA was voluntarily recalled in September 2019 in the U.S. and is now only available to a limited number ofseriously-ill patients through a Special Use Program offered by its manufacturer, Takeda Pharmaceutical Company. We are also aware of several academic groups and companies working on making longer-acting agonists of the PTH receptor, or PTH1R. In addition, other companies and groups are developing or commercializing therapies for hypoparathyroidism, including Shire, Chugai Pharmaceutical Co., Ltd., Entera Bio, Extend Biosciences, Massachusetts General Hospital, AlizéAmolyt Pharma, Prolynx Inc., and Eli Lilly and Company.
Teriparatide,PTH(1-34), approved since 2002 for the treatment of osteoporosis, has sometimes been used for treatment of hypoparathyroidism using multiple daily injections, despite not being approved for this indication. Clinical research conducted by the NIH of subjects receiving continuous exposure toPTH(1-34), administered by an infusion pump, has demonstrated simultaneous normalization of sCa and urinary calcium, as well as normalization of bone turnover.
Our Solution: TransCon PTH
TransCon PTH is an investigational long-acting prodrug of parathyroid hormone, or PTH, that is designed as a novel replacement therapy for parathyroid hormone, or PTH dosedonce-daily to achieve and maintain a steady concentration of PTH in the bloodstream within the normal range, at levels similar to those observed in healthy individuals. TransCon PTH is designed to restore physiologic levels of PTH 24 hours per day, thereby more fully addressing all aspects of the disease including normalizing serum and urinary calcium and serum phosphate levels. Pharmacokinetic data from our phase 1 trial of TransCon PTH in healthy subjects demonstrated ahalf-life of approximately 60 hours, supporting aninfusion-like profile with daily administration. Thishalf-life is a substantial increase compared toPTH(1-34) andPTH(1-84), both of which havehalf-lives of only a few hours after subcutaneous administration to humans.
Withonce-daily dosing, we believe this substantialhalf-life extension of PTH could more closely reflect the physiological levels of PTH observed in healthy individuals thereby maintaining blood calcium levels and normalizing urinary calcium excretion. Pharmacokinetic data from multiple ascending dose (MAD) cohorts in our phase 1 trial of TransCon PTH in healthy subjects demonstrated aninfusion-like profile of free PTH. By providing steady levels of PTH in the physiological range, we believe TransCon PTH can address the fundamental limitations of short-acting PTH molecules such as NATPARA, orPTH(1-84), and Teriparatide, orPTH(1-34) and become a highly differentiated therapy for HP.
Clinical Development of TransCon PTH for Adult Hypoparathyroidism
Our ongoing phase 2 PaTH Forward Trial is evaluating the safety, tolerability and efficacy of three fixed doses (15, 18, or 21 µg per day) of TransCon PTH compared to placebo over a four-week double-blinded period, followed by a range of doses intended to cover the range of individual requirements for hormone replacements in a long-term open-label extension, or OLE, using a ready-to-use prefilled pen device. The goal of PaTH Forward is to evaluate TransCon PTH control of serum and urinary calcium, identify a starting dose (15, 18, or 21 µg per day) for a pivotal phase 3 trial, and establish a titration regimen for complete withdrawal of standard of care (i.e., active vitamin D and calcium supplements), and evaluate TransCon PTH control of serum and urinary calcium.SoC.
Previously, in May 2018, we completed a phase 1 trial to evaluate the safety tolerability, pharmacodynamics and pharmacokinetics of TransCon PTH in healthy adults. Primary objectives of the trial included assessing the safety and tolerability of single and 10ten multiple daily doses of TransCon PTH in healthy adults. Secondary objectives of this trial included evaluation of pharmacodynamics, including serum calcium, down regulation of endogenousPTH(1-84), and bone markers; pharmacokinetics following single and multiple daily doses of TransCon PTH; assessment of whether TransCon PTH treatment affects fractional excretion of urinary calcium; and, incidence ofanti-PTH andanti-PEG antibodies.
Results of the trial showed that TransCon PTH led to sustained anddose-dependent elevations of serum calcium with lowinter-subject variability. Thisdose-dependent response and lowinter-subject variability suggests the ability to titrate and individualize dosing in patients. Following 10ten repeated doses, free PTH exhibited a flat,infusion-like profile with lowinter-patient variability. profile. TransCon PTH was also observed to have the expected effects on renal calcium reabsorption basedas evaluated by on fractional excretion and down regulation of endogenousPTH(1-84).
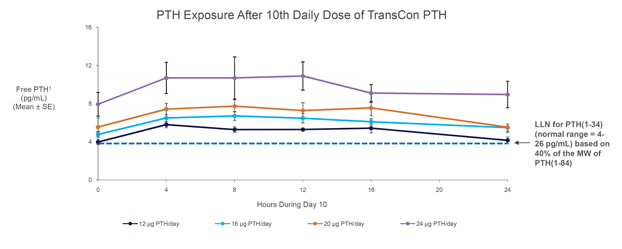
Figure 3. TransCon PTH daily dosing for 10 days of 12, 16, 20 and 24 µg per day. TransCon PTH daily dosing for 10 days provided a flatinfusion-like profile with a low PTHpeak-to-trough ratio at day 10.
In addition, the TransCon PTH PK translated into a predictable sCa response, suggesting the ability to titrate patients with HP into the normal calcemic range, consistent with preclinical data.
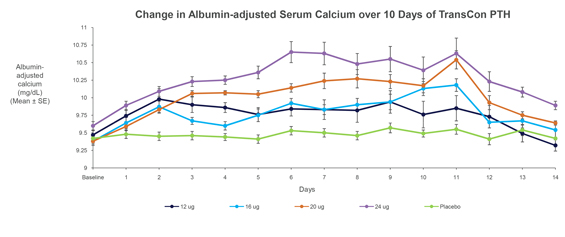
Figure 4. Serum calcium over 10 days of TransCon PTH. TransCon PTH daily dosing for 10 days provided adose-dependent increase of serum calcium.
TransCon PTH also demonstrated the expected effect on renal calcium reabsorption, and maintained a normal fractional excretion of calcium, even in the presenceand also down regulation of hypercalcemia, predicting control of both serum and urine calcium.
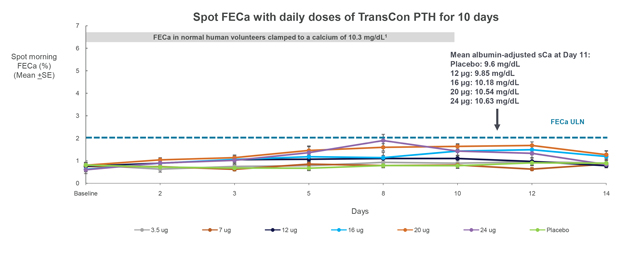
Figure 5. Control of urinary calcium with multiple doses of TransCon PTH.
The final results of the phase 1 trial were consistent with our target product profile for TransCon PTH, showing the predicted pharmacokinetic and pharmacodynamic response, and suggesting the ability to normalize serum and urinary calcium levels in patients with hypoparathyroidism. We believe TransCon PTH may provide patients suffering from hypoparathyroidism with a PTH replacement therapy that is designed to address both the short-term symptoms and long-term complications of HP.endogenous PTH(1-84) secretion.
In June 2018,April 2020, we were granted Orphan Drug Designation, or ODD, byannounced top-line data from the FDA, for TransCon PTH. The FDA may designatefour-week fixed dose, double-blinded portion of PaTH Forward, a drug or biologic as an orphan drug if it is intended to treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States.
We initiated theglobal phase 2 PaTH Forward Trial in adult subjects with hypoparathyroidism in the first quarter of 2019. PaTH Forward isstudy evaluating the safety, tolerability and efficacy of threeTransCon PTH in adult subjects with hypoparathyroidism. A total of 59 subjects were randomized in a blinded manner to receive fixed doses of TransCon PTH at 15, 18 or 21 µg/day or placebo for four weeks using aready-to-use prefilled pen device. The goalinjector planned for commercial presentation. All doses of PaTH Forward isTransCon PTH were well-tolerated, and no serious or severe treatment-related adverse events, or TEAEs, were observed at any point. No treatment-emergent adverse events led to identify a startingdiscontinuation of study drug, and the overall incidence of TEAEs was comparable between TransCon PTH and placebo. Additionally, there were no drop-outs during the four-week fixed dose (15, 18, or 21 µg per day) for a pivotal phase 3 trial, establish a titration regimen for complete withdrawal ofperiod. In the modified full analysis set (n=57), TransCon PTH eliminated standard of care (i.e., off active vitamin D and £ 500 mg per day of calcium supplements), in 100 percent of subjects in the highest dose arm (21 µg/day) and evaluate TransCon PTH control82 percent of serum and urinary calcium. Followingsubjects across all dosage arms. Fifty-eight subjects reached the one-month blindedsix-month analysis in the OLE portion of PaTH Forward, subjects entered an open-label extensionthe trial, where they will receive a customized maintenance dose of TransCon PTH (6 to 30 µg per day) titrated to optimize their calcium control and evaluated on the primary composite endpoint, both as planned for phase 3..
On November 14, 2019, we announced an expansion of the patient population in the ongoing PaTH Forward Trial to facilitate enrollment of subjects affected by the NATPARA® recall, a parathyroid hormone. Previously, patients treated with NATPARA were required to undergo a long washout period prior to entering screening. In response to the recall of NATPARA in the United States, we evaluated opportunities to help enroll patients affected by the recall. Following the protocol addendum, patients previously treated with NATPARA in the United States were able to utilize an expedited process to enroll in PaTH Forward. In February 2020, we completed enrollment of the expanded trial with 59 subjects and we expect to reportPreliminary top-linesix-month dataresults from the one-month blinded portion of the PaTH Forward Trial inOLE demonstrated
91 percent of all subjects eliminated standard of care (defined as (1) off active vitamin D and (2) mid-April£500 mg per day of calcium supplements), including 76 percent who eliminated all supplements.
86 percent of all subjects normalized or reduced by 50 percent 24-hour 2020, with six-month dataurine calcium.
| • | All mean summary and subdomain SF-36® Health Survey scores normalized despite all mean scores starting below norms at baseline including subjects randomized to placebo who switched to TransCon PTH group at week four. Importantly, subjects randomized to TransCon PTH demonstrated continued improvements from week four to month six. |
Bone mineral density mean Z-scores trended towards normalization at week 26.
All doses of TransCon PTH were well-tolerated, and no treatment-related serious or severe adverse events were observed at any point. No subjects had PTH treatment-emergent adverse events related to hyper- or hypocalcemia leading to emergency visit, urgent care visit, or hospitalization.
Adherence to daily injections of TransCon PTH was 99.8 percent.
As of March 10, 2021, fifty-eight out of the open-label extension phase expected to be reported during the third quarter of 2020. Final enrollment of PaTH Forward included 17 subjects previously treated with NATPARA.
On January 12, 2020, we reported preliminary data from patient diaries on the first 8 subjects completing 4 weeks offollow-upfifty-nine patients continued in the open-label extension portion which showed that all subjects were completely off current standard of care. All 8 subjects no longer required active vitamin D, and 7 of 8 subjects no longer required calcium supplements (one subject taking < 500 mg calcium). 59 subjects completed the blinded portion, and 58 subjects continued in the open-label extension, with one subject withdrawing for reasons unrelated to safety or efficacy of the study drug.
Following evaluation of Phase 2 data from the PaTH Forward Trial,In September 2020, we expect and plansubmitted an amendment to our IND to initiate aPaTHway, our global phase 3 clinical trial forevaluating the safety, tolerability and efficacy of TransCon PTH in adults with HP following discussions with FDA and European regulatory authorities. The double-blind, placebo-controlled trial is expected to enroll approximately 76 subjects at sites in North America and Europe in order to obtain 68 evaluable subjects. We expect topline results from this trial in the fourth quarter of 2020, including trial sites2021. In addition, we are planning to conduct a phase 3 study in Japan designed to evaluate the safety, tolerability, and efficacy of TransCon PTH. We anticipate filing a Clinical Trial Notification for this proposed phase 3 study in the United States, Canada, Europe and Asia-Pacific, including Japan.second quarter of 2021.
In June 2018, we were granted ODD by the FDA, for TransCon PTH for the treatment of for hypoparathyroidism. In October 2020, we were granted Orphan Designation by the European Commission for TransCon PTH for the treatment of hypoparathyroidism.
TransCon CNP
Market Opportunity in Achondroplasia
Achondroplasia is the most common form of dwarfism, occurring in about 1one in 10,000 to 30,000 newborns or approximately 250,000 worldwide. Achondroplasia results in severe skeletal complications and comorbidities, including spinal stenosis due to premature fusion of the foramen magnum, sleep apnea, and chronic ear infections.
Patients often face multiple surgeries to alleviate its many complications. There are currently noFDA-approved pharmacological treatments for achondroplasia.
Achondroplasia is caused by an autosomal dominant activating mutation in fibroblast growth factor receptor 3, or FGFR3, that leads to an imbalance in the effects of the FGFR3 andC-type natriuretic peptide (CNP) signaling pathways. In achondroplasia, mutations in FGFR3 result in constitutive activation, suppressing the proliferation and differentiation of chondrocytes resulting in improper cartilage to bone conversion in the growth plate.
Preclinical and clinical data show that the CNP pathway stimulates growth and increased CNP helps to counteract the effects of the FGFR3 mutation downstream. Administration of a CNP analog to patientschildren with achondroplasia and in animal models of achondroplasia has been found to stimulate growth.
BioMarin Pharmaceutical, Inc. is developing vosoritide for the treatment of achondroplasia, and other companies that are developing therapies for achondroplasia include Pfizer, QED Therapeutics and BioClin Therapeutics, Inc.
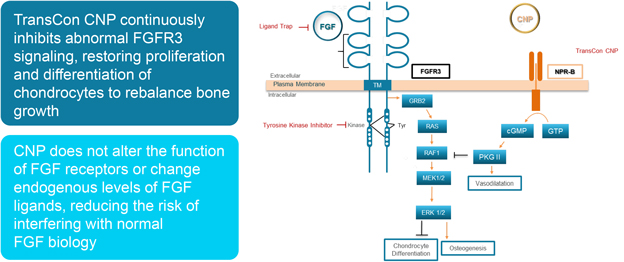
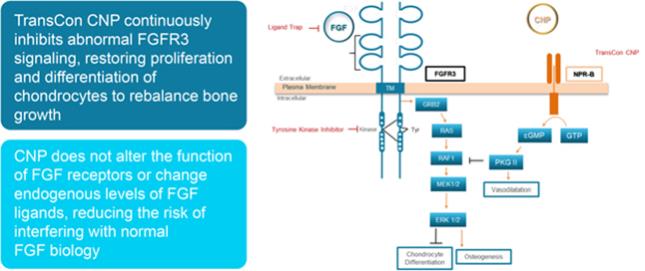
TransCon CNP continuously inhibits abnormal FGFR3 signaling, restoring proliferation and differentiation of chondrocytes to rebalance bone growth CNP Does not alter the function of fgf receptors or change endogenous levels of fgf ligands, reducing the risk of interfering with normal fgf biology LIGAND TRAGS fgf fgfr3 npr-8 transcos CNP pkg 11 mek 1/7 erk 1/2 GTPFGFR3 NRP-Bchondrocyte differentationplasma membrane12790
Adapted from Current Opin Pediatrics 2010;22:516-523. |
Figure 6.3. The role of a defect in the FGFR3 signaling pathway in the development of achondroplasia is well understood.
Our Solution: TransCon CNP
Endogenous CNP has an extremely shorthalf-life of only approximately two minutes. Vosoritide, a CNP analogue, has been developed to provide better stability of CNP, and has ahalf-life of about 20 minutes. In a phase 3 trial, vosoritide also demonstrated a statistically significant change in growth velocity from baseline over one year in children treated with vosoritide compared to placebo.
However, in addition to limited efficacy,short-acting CNP and CNP analogues that result in high Cmax levels may cause adverse cardiovascular events. As achondroplasia is caused by an FGFR3 mutation that chronically inhibits growth, we expect a more constant CNP exposure at lower Cmax to correlate with better therapeutic outcomes, with lower cardiovascular risk. TransCon CNP is an investigationallong-acting prodrug ofC-type natriuretic peptide designed to provide continuous CNP exposure at therapeutic levels with awell-tolerated and convenientonce-weekly dose. It is being developed for the treatment of children with achondroplasia. TransCon CNP is designed to provide effective shielding of CNP from neutral endopeptidase degradation in subcutaneous tissue and the blood compartment, minimize binding of CNP to theNPR-C receptor to decrease clearance, reduce binding of CNP to theNPR-B receptor in the cardiovascular system to avoid hypotension, and release unmodified CNP, which is small enough in size to allow effective penetration into growth plates. We believe TransCon CNP offers advantages over short-acting CNP and CNP analogs in development that result in high Cmax levels which may cause adverse cardiovascular events. In addition, we expect a more constant CNP exposure at lower Cmax to correlate with better therapeutic outcomes.
Clinical Development of TransCon CNP for Achondroplasia
TransCon CNP is currently being evaluated in a global phase 2 trial, known as the ACcomplisH Trial, which is designed to evaluate the safety and efficacy of TransCon CNP in children (ages2-10 two to ten years) with achondroplasia.
In November 2018, we reported preliminary resultsResults from aour phase 1 trial of TransCon CNP in healthy adult subjects which supported our target product profile for TransCon CNP. In this phase 1,double-blind, randomized,placebo-controlled trial, 45 healthy adult subjects were enrolled. Five doses of TransCon CNP were tested sequentially, beginning with the lowest dose: 3, 10, 25, 75 and 150 µg/kilogram. Up to 10ten subjects in each dose cohort were randomized to receive TransCon CNP or placebo in a 4:1 ratio. After each cohort completed dosing, a Data Safety Monitoring Board reviewed the blinded data to approve escalation to the next higher dose. The primary endpoint was frequency of adverse events after administration of TransCon CNP. Secondary endpoints included additional safety parameters, tolerability and pharmacokinetics.
The results showed TransCon CNP provided continuous exposure to CNP with a pharmacokinetic profile designed to provide efficacy withonce-weekly dosing. No serious adverse events were reported in the trial and TransCon CNP was generally well tolerated at doses up to 150 µg/kilogram. Mean orthostatic changes in vital signs appeared unrelated to TransCon CNP exposure and were consistent between placebo and TransCon CNP cohorts. Mean resting blood pressure and heart rate were unchanged frompre-dose at all time points, in all cohorts. Injections were well tolerated in all dose cohorts. Noanti-CNP antibodies were detected in any subjects.
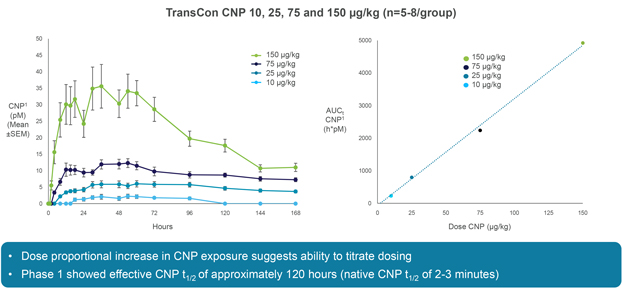
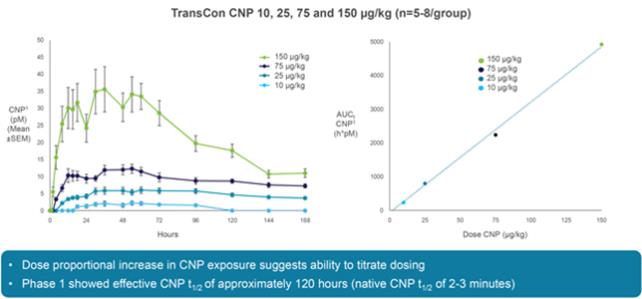
Transcon cnp 10, 25 , 75 and 150 ug/kg (n=5-8/group) cnp1 (pm) (Mean a SEM)50 , 40 , 35, 30, 25, 20, 15, 10 , 5,0,24,40,72,96,120,144,163 HOURS 150 UG/KG75 ug/kg25 ug/kg10 ug/kgAUC2 CNP1 (h*pm) HOURS DOSE proportional increase in cnp exposure suggests ability to titrate dosingphase 1 showed effective CNP T1/12 of approximately 120 hours ( native CNP T1/2 OF 2-3 MINUTES)5000 4000 3000 2000 1000 0 25 50 75 100 125 150 DOSE CNP (UG/KG)150 UG/KG75 UG/KG25 UG/KG10 UG/KG
| 1 | CNP measured asCNP-38. |
Figure 7.4. Pharmacokinetic results for TransCon CNP 10, 25, 75 and 150 µg/kg administered as aonce-weekly subcutaneous injection are presented.
Following completion of the phase 1 trial, and a successful submission of an IND in July 2019, we initiated the phase 2 ACcomplisH Trial, a randomized, double-blind, placebo-controlled, sequential rising dose trial to evaluate the safety and efficacy of TransCon CNP in approximately 60 children with achondroplasia (ages 2two to 10ten years). Subjects will be randomized to receive either TransCon CNP or placebo in a 3:1 ratio. The primary efficacy endpoint is annualized height velocity at 12twelve months. Key secondary and additional endpoints include body proportionality and change in BMI, both evaluated after 12twelve months of weekly TransCon CNP treatment, and patient reported outcome (PRO) measures. The company continuesWe continue to work towards escalating sequential dose cohorts throughout this year.
In collaboration with VISEN, we are sponsoring the ACcomplisH China Trial, a randomized, double-blind, placebo-controlled, phase 2 dose expansion trial to evaluate the safety and efficacy of TransCon CNP in subjects with achondroplasia. The primary endpoint is to evaluate the safety of treatment and its effect on 12-month annualized height velocity. In January 2021, China Center for Drug Evaluation (CDE) of National Medical Products Administration (NMPA) approved VISEN’s IND application to conduct the ACcomplish China Trial.
In parallel, we are conducting the ACHieve Study, a multi-center natural history study designed to gain insight into the experience of pediatric patientssubjects with achondroplasia. ACHieve will study growth velocity, body proportionality, and comorbidities over time of children with achondroplasia up to 8eight years old. No study medication will be administered.
We plan to provide a TransCon CNP clinical program update in the fourth quarter of 2021.
In February 2019, we were granted ODD by the FDA for TransCon CNP for the treatment of achondroplasia. Our goal is to developIn August 2020, we received Orphan Designation from the European Commission for TransCon CNP as a safe and effective therapeutic option for achondroplasia and potentially other related growth disorders.the of achondroplasia.
TransCon Product Candidates - Oncology
Market Opportunity in Oncology
Cancer remains a major unmet medical need and the incidence of many cancer types continues to rise. Improved understanding of the cellular and molecular mechanisms involved inanti-tumor immune responses has fueled the rapid growth ofimmuno-oncology therapeutics. In particular, immune checkpoint inhibitors, such asanti-PD-1,anti-PD-L1 andanti-CTLA-4 antibodies, have provided new therapeutic options for patients who did not respond to previous therapies. Supported by these and other advancements, oncology is now the largest therapy class in terms of revenue in pharmaceutical industry, with worldwide prescription drug andover-the-counter sales of $124 billion in 2018 with projected growth to $237 billion in 2024.
Despite these recent advances, there is still a high unmet medical need for new treatment options, as many patients do not respond to current therapies and the efficacy is often limited by toxicities that result in dose reductions or treatment discontinuations. One approach to minimizing adverse events while retaining or improving efficacy is to create product candidates with longerhalf-lives, allowing for more consistent circulating drug levels and less frequent administration than the corresponding parental molecules. Another approach is intratumoral injection, which is to directly administer a drug to the tumors. Early clinical results for intratumoral treatment of cutaneous tumors are encouraging, but the frequent administration ofshort-acting molecules todifficult-to-access tumors is often impractical. Increased and prolonged therapeutic activity in tumors has the potential to improve outcomes in patients with suboptimal responses to current therapies.
Our Solution: TransCon Technologies for Oncology
Building from the success of our programs in endocrinology, our vision in oncology is to createbest-in-class therapeutics by applying both systemic and sustained localized TransCon technologies for clinically validated pathways. By applying our unique algorithm for product innovation, we believe we can improve outcomes in oncology currently limited by suboptimal efficacy and systemic toxicity.
We believe TransCon is particularly well-suited to oncology because of the large number of validated targets with known limitations. We are working to not only prolong the activity of approved drugs at efficacious levels, but to extend the exposure times without reaching high toxic levels that often complicate oncology therapeutic regimens. By prolonging therapeutic levels, we believe that our technologies have the potential to increase the efficacy of small molecules, peptides and proteins without increasing toxicity – addressing a long-standing challenge in oncology.
Our TransCon product candidates in oncology are designed for sustained systemic or intratumoral administration to provide durable and potentanti-tumor effects. TransCon product candidates can be designed to facilitate all the critical steps of the cancer immunity cycle that lead to eradication of malignant cells. We believe these product candidates have the potential to optimize the efficacy of clinically validated therapies while limiting adverse effects.
For example, many therapies are administered at the maximum tolerated dose, and the ability to deliver therapies directly to the tumor may improve both tolerability and efficacy, as high local drug concentration may be achieved, while maintaining low systemic exposure. In addition, TransCon product candidates are designed for sustained release and, hence, may allow for reduced frequency of dosing, enabling treatment of tumor types that cannot be easily accessed for frequent injection.
We have conducted nonclinical studies that have found that our oncology product candidates can slowly releaseimmuno-oncology agents, highlighting their potential to enhance the immune system to attack malignant tumor cells.
Development of TransCon Product Candidates in Oncology
Our goal is to improve treatment efficacy while limiting or reducing toxicity by applying TransCon technologies to clinically validated pathways, using our unique algorithm for product innovation. We are conducting nonclinical studies within the field of oncology to explore multiple potential product candidates and evaluate systemic as well as localized delivery systems using our TransCon technologies.
In June 2019, we reported nonclinical data for three of our oncology programs:
TransConIL-2 ß/g is designed for prolonged exposure of anIL-2 variant that selectively activates theIL-2Rß/g with low binding toIL-2Rα. ThisWe are currently advancing two product candidate is designed to provide potent anti-tumor activity and to have reduced risk of toxicity, such as vascular leak syndrome.candidates:
TransCon TLR7/8 Agonist is designed for sustained release of TLR7/8 agonist, resiquimod, and intended for intratumoral administration. This product candidate is designed to provide potent activation of the innate immune system in the tumor and draining lymph nodes and to have low risk of systemic toxicity. In December 2020, we filed an IND with the U.S. Food and Drug Administration to initiate the clinical program of TransCon TLR7/8 Agonist with the transcendIT-101 Trial. We expect initial results from the first part of transcendIT-101, monotherapy dose escalation, in the fourth quarter of 2021. We plan to initiate the second part of transcendIT-101, dose escalation of TransCon TLR7/8 Agonist in combination with a checkpoint inhibitor, in the second quarter of 2021.
TransConVEGF-TKIIL-2b/g is designed for sustained releaseprolonged exposure of aan VEGF-TKIIL-2 molecule, axitinib, and intended for intratumoral treatment. Thevariant that selectively activates the IL-2Rb/g with minimal binding to IL-2Rα. This product candidate is designed to enable high intratumoral concentrationsprovide potent anti-tumor activity and to have reduced risk of axitinib that are not achievable via oral route, potentially improving efficacy and avoiding toxicitiestoxicity, such as vascular leak syndrome. We plan to submit an IND or similar for TransCon IL-2b/g in the third quarter of oral treatments that often result in treatment interruptions and discontinuations.2021.
We are evaluating multipleadditional TransCon product candidates in nonclinical research studies for the treatment a variety of tumor types. Examples of TransCon product candidates under evaluation include stimulators of innate and adaptive immunity, as well as modulators of the tumor environment. We are exploring systemic and intratumoral administration both as a monotherapy and as a component of combination regimens.
We believe these programs have potential to target multiple steps of the immunity cycle that drives the immune response against tumor cells. Our goal is to file an IND or equivalent in oncology in the fourth quarter of 2020.
Strategic Collaborations
We also engage in strategic collaborations to further leverage our TransCon technology platformtechnologies in certain geographies withmarket-leading biopharmaceutical companies. These collaborations aim to further monetize both our TransCon technology platformtechnologies and our internal product candidates, particularly into therapeutic areas where we believe a partner may have more expertise, capability and capital.
In addition, we may choose to pursue a collaboration to develop and market our internal,wholly-owned product candidates in geographic markets outside our core focus of the United States and Europe.
Strategic Investment
VISEN Pharmaceuticals
In November 2018, we announced the formation of VISEN Pharmaceuticals, or Visen,VISEN, a company established to develop and commercialize our endocrinology rare disease therapies in the People’s Republic of China, Hong Kong, Macau, and Taiwan, or Greater China. In connection with the formation of Visen,VISEN, we granted VisenVISEN exclusive rights to develop and commercialize certain product candidates based on our proprietary TransCon technologies, including TransCon hGH, TransCon PTH and TransCon CNP, in Greater China for use in all human indications, subject to
certain exceptions. As consideration for the rights granted to Visen,VISEN, we received 50% ownership in the outstanding shares of VisenVISEN and concurrently with the rights we granted to Visen,VISEN, entities affiliated with Vivo Capital and Sofinnova Ventures purchased shares in VisenVISEN for an aggregate purchase price of $40 million in cash. OnIn January 2021, we invested additional $12.5 million in VISEN as part of VISEN’s $150 million Series B financing. Following the Series B financing, we retain approximately 44% of VISEN’s issued and outstanding shares. In October 25, 2019, VisenVISEN received approval of a clinical trial application for TransCon hGH from the Center for Drug Evaluation in China and initiated a phase 3 trial in pediatric growth hormone deficiency. In addition, Visen expectsJanuary 2021, China’s CDE approved VISEN’s application to initiateconduct the ACcomplish China Trial, a randomized, double-blind, placebo-controlled, phase 2 dose expansion trial to evaluate the safety and efficacy of TransCon CNP in childrensubjects with achondroplasia during the fourth quarter of 2020.ACH.
Market Opportunity in China
China is the second largest pharmaceutical market in the world after the United States and represents one of the fastest growing pharmaceutical markets worldwide. In recent years, the Chinese government has initiated a number of regulatory reforms that are expected to accelerate drug development, as well as drive growth and demand for new therapeutics in China. In addition to joining an international organization that standardizes regulations for clinical development, the National Medical Products Administration (NMPA) has introduced initiatives such as fast track review for drugs for unmet medical needs and adopted new rules that streamline the drug approval process in China for global companies.
We believe VisenVISEN supports our strategy to extend our endocrinology rare disease portfolio globally and establish a presence in China in partnership with collaborators who have significant experience and knowledge of the biopharmaceutical opportunity in China. In part because VisenVISEN was established in China, we believe VisenVISEN will be able to effectively develop and, if approved, market our innovative technologies to address the needs of the local markets in Greater China.
Rights Agreements
Under the Rights Agreements, VisenVISEN must use diligent efforts to develop and commercialize licensed products in Greater China. Additionally, we and VisenVISEN will conduct certain research and development activities allocated to the respective party under a research and technical development plan, and VisenVISEN will reimburse us for costs of conducting such activities, including costs of our personnel committed to performing such activities in Greater China.
We will provide product supply to VisenVISEN for use in conducting clinical trials in Greater China pursuant to separate clinical supply agreements entered into concurrently with the Rights Agreements in accordance with the terms and conditions set forth therein. Additionally, we and VisenVISEN will negotiate in good faith the terms and conditions governing commercial supply of licensed product to VisenVISEN on the terms and conditions set forth in the Rights Agreements.
Under the Rights Agreements, we agreed not to research, develop, or commercialize competing products in Greater China, and VisenVISEN agreed not to grant certain rights under its interest in any inventions or intellectual property arising out of the activities conducted under the Rights Agreements to third parties,third-parties, in each case, under the terms and conditions specified in the Rights Agreements. We will have the right to exploit inventions and intellectual property arising out of the activities conducted under the Rights Agreements outside of Greater China. Additionally, we granted VisenVISEN a right of first negotiation to develop and commercialize certain of its endocrinology products in Greater China.
The Rights Agreements continue in effect for as long as a valid claim of a licensed patent exists in Greater China. VisenVISEN may terminate a Rights Agreement for convenience, for uncured material breach by us of a Rights Agreement and for our bankruptcy orinsolvency-related events. We may terminate a Rights Agreement for certain specified material breaches thereof by Visen,VISEN, in the event VisenVISEN undergoes a change of control in favor of a competitor, if VisenVISEN challenges the validity of any of the licensed patents and for Visen’sVISEN’s bankruptcy orinsolvency-related events.
Amended and Restated Shareholders Agreement
In connection with the formation of Visen,Company’s investment in VISEN, on January 8, 2021, the Company entered into an Amended and Restated Shareholders Agreement (the “Amended Shareholders Agreement”), amending and restating the Shareholders Agreement dated November 7, 2018, we entered into a Shareholders Agreementbetween the Company and the parties set forth therein (the “Shareholders Agreement”) providing for certain. In addition to rights and obligations of Visen and its shareholders. Pursuant topreviously granted under the Shareholders Agreement, Visen andunder the Visen shareholders agreed to certain negotiated information and inspection rights, rights relating to registration of shares held by shareholders, pro rata rights to participate in future offerings by Visen of certain securities of Visen subject to certain limited exceptions, drag along provisions relating to a change of control of Visen, rights of first refusal andco-sale with respect to proposed sales (if any) by shareholders of Visen (including sales by us).
Pursuant to theAmended Shareholders Agreement, we havethe Company has the right to designate an individualtwo individuals for election to the board of directors of VisenVISEN, which individuals are initially Jan Møller Mikkelsen and VisenMichael Wolff Jensen. In addition, VISEN has agreed that certain specified events (including a certain liquidation events) shall require the approval of (i) shareholders of VisenVISEN holding at least 50% of VISEN’s Series B preferred shares, (ii) shareholders of VISEN holding at least 60% of Visen’sVISEN’s Series A preferred shares and/or (ii)(iii) certain members of theVISEN’s board of directors. Under theThe Amended Shareholders Agreement and in connection with the formation of Visen, we have agreed to refrain from carrying out, or engaging in, the research, development, manufacture or commercialization of certain competing products in Greater China.
The Shareholders Agreement terminatescan be terminated by written agreement between us and an entity affiliated with Vivo Capital, and automatically terminates uponamong the dissolution of Visen. In addition, holders of a specified percentageat least 60% of VISEN’s Series A preferred shares in Visen can terminate the Shareholders Agreement by written notice to Visen upon the occurrenceand at least 50% of certain events set forth in the Shareholders Agreement.
Strategic Collaboration with Sanofi
In December 2010, we entered into a strategic collaboration agreement with Sanofi under which we assigned to Sanofi certain diabetes-related patent rights, and granted to Sanofi an exclusive, worldwide, royalty-free license to research, develop, make and commercialize (1) products based on the TransCon technologies and any combination ofglucagon-like-peptide-1, orGLP-1, glucagon and insulin to treat any diseases in humans or animals, or (2) any other product developed by Sanofi incorporating our TransCon technologies, other technology covered by the assigned patents or other improvements to our TransCon technologies or the foregoing products, to treat diabetes in humans or animals. During the term of the agreement, we are prohibited from engaging in any research, development or commercialization activities related to certain specified products. In addition, we granted Sanofi anon-exclusive, royalty-free license to research, develop, manufacture and commercialize products other than those based on the TransCon technologies and any combination ofGLP-1, glucagon and insulin that are developed by Sanofi incorporating our TransCon technologies, other technology covered by the assigned patents or other improvements to our TransCon technologies or the foregoing products for the treatment of certain diabetes-related metabolic disorders and obesity in humans and animals, so long as, for any such products that are peptides, Sanofi first develops them for diabetes or obesity in humans and the first application for regulatory approval for such products is for diabetes or obesity in humans in a major country, and for any such products that are not peptides, Sanofi first develops such products for diabetes in humans and animals and the first application for marketing approval is for diabetes in humans in a major country. This license will become exclusive, on apeptide-by-peptide basis, for any licensed product containing a peptide that isnon-proprietary to Sanofi and is designated by Sanofi if certain specified conditions are met. Under the agreement, Sanofi has granted us anon-exclusive, royalty-free license (with the right to grant sublicenses) under Sanofi’s rights in any improvements generated in connection with the collaboration, to research, develop, make or commercialize products outside the scope of the collaboration and outside the field of diabetes.
In consideration for these licenses to the TransCon technologies and as payment for the assignment of specific diabetes-related product patents, Sanofi provided an aggregate of €25 million innon-refundable,up-front payments to us. Sanofi also committed to fund our development activities for a fixed amount over the first three years of the collaboration, in accordance with an agreed upon development plan. For the first two products developed under the Sanofi collaboration, we are also eligible to receive up to an aggregate of €170 million upon Sanofi’s achievement of specified clinical development and regulatory approval milestones and up to an aggregate of €100 million upon Sanofi’s achievement of certain sales-related milestones.
The term of the agreement expires upon the expiration of the last to expire of the patents licensed or assigned to Sanofi under the agreement and we currently expect thelast-to-expire licensed or assigned patent will expire in October 2030. We may terminate the agreement upon 30 days’ prior written notice if Sanofi fails to remit any undisputed sum it must pay to us. Each party may terminate the agreement upon 60 days’ prior written notice for the other party’s uncured material breach. Sanofi has the right to terminate the agreement in its entirety for convenience upon 90 days’ prior written notice. Either party may terminate the agreement by written notice to the other party if the other party institutes a lawsuit or proceeding allegingnon-infringement, invalidity or unenforceability with respect to any patent licensed to such other party under the agreement. Upon any such termination by us or byVISEN’s Series B preferred shares.
Sanofi for convenience, all licenses granted to Sanofi would terminate and, if such termination is by Sanofi for convenience prior to IND approval of a product under the agreement, we may require Sanofi to assign back to us the assigned patent rights upon payment of a specified amount. Our collaboration agreement with Sanofi remains in effect; however, in December 2019 Sanofi announced it plans to end its investments in the field of diabetes.
Manufacturing
As we do not maintain the capability to manufacture finished drug products, we utilize contract manufacturers to manufacture finished drug product of our proprietary TransCon product candidates intended for clinical or commercial use. We source starting materials for our manufacturing activities from one or more suppliers. For the starting materials necessary for our proprietary TransCon product candidate development, we have agreements for the supply of such starting materials with drug manufacturers or suppliers that we believe have sufficient capacity to meet our demands. However, from time to time, we source critical raw materials and services from one or a limited number of suppliers and there is a risk that if such supply or services were interrupted, it would materially harm our business. In addition, we typically order raw materials and services on a purchase order basis and do not enter intolong-term dedicated capacity or minimum supply arrangements. We utilize the services of contract manufacturers to manufacture drug substance required for later phases of clinical development and eventual commercialization for us under all applicable laws and regulations.regulations and are subject to long term forecasting obligations and certain minimum purchase requirements for all parts of the commercial supply chain.
We have analytical and process development capabilities in our own facility. We generally perform analytical and process development for our proprietary TransCon product candidates internally and manufacture internally our TransCon product candidates necessary to conduct thenon-GLP preclinical studies thereof. However, we occasionally outsource the manufacture of research anddevelopment-stage TransCon product candidates. Occasionally our collaboration partners may manufacture the research anddevelopment-stage TransCon product candidates for which they are licensed. Each of our collaboration partners have granted us rights that enable us to freely commercialize all improvements to the TransCon prodrug technologies and manufacturing process developed by our collaboration partners outside of the fields of use and/or territories (as applicable) licensed to such collaboration partners under the relevant collaboration agreements with such partners.
We do not have, and we do not currently plan to acquire or develop, the facilities or capabilities to manufacture bulk drug substance or filled drug product for use in human clinical trials. We rely onthird-party manufacturers to produce the bulk drug substances required for our clinical trials and expect to continue to rely on third partiesthird-parties to manufacture and test clinical trial drug supplies for the foreseeable future.
Our contract suppliers manufacture drug substance and finished drug product for our TransCon product candidates for clinical trial use in compliance with cGMP and applicable local regulations. cGMP regulations include requirements relating to organization of personnel; buildings and facilities; equipment; control of components and drug product containers and closures; production and process controls; packaging and labeling controls; holding and distribution; laboratory controls; records and reports; and returned or salvaged products. The manufacturing facilities for our products must be in compliance with cGMP requirements, and for device and device components, the Quality System Regulation, or QSR, requirements, before any product is approved. We ensure cGMP compliance of our suppliers through regular quality inspections performed by our Quality Assurance group. Ourthird-party manufacturers may also be subject to periodic inspections of facilities by the FDA, the Competent Authorities of the Member States of the European Economic Area, and other authorities, including reviews of procedures and operations used in the testing and manufacture of our products to assess our compliance with applicable regulations. Failure to comply with statutory and regulatory requirements subjects a manufacturer to possible legal or regulatory action, including warning letters, the seizure or recall of products, injunctions, consent decrees placing significant restrictions on or suspending manufacturing operations and civil and criminal penalties. These actions could have a material impact on the availability of our products. In addition, contract manufacturers often encounter difficulties involving production yields, quality control and quality assurance, as well as shortages of qualified personnel.
We also contract with additional third partiesthird-parties for the filling, labeling, packaging, testing, storage and distribution of our TransCon product candidates. We employ personnel with the significant scientific, technical, production, quality and project management experience required to oversee our network ofthird-party suppliers and to manage manufacturing, quality data and information for regulatory compliance purposes.
NOF Manufacturing and Supply Agreement
On December 21, 2017, we entered into amulti-year Manufacturing and Supply Agreement (the “NOF Agreement”) with NOF Corporation (“NOF”). Under the NOF Agreement, NOF has agreed to manufacture and supply the mPEG Linker (the “NOF Product”) for our TransCon hGH product candidate. We have agreed to purchase certain quantities of NOF Product. We may purchase NOF Product from other manufacturers and are not obligated to purchase NOF Product from NOF, other than certain quantities that have been forecasted by us in accordance with a mandatory rolling forecast that we must deliver to NOF from time to time.
The NOF Agreement is effective as of December 21, 2017. The initial term of the NOF Agreement terminates on December 31, 2025 unless earlier terminated. The parties may extend the initial term of NOF Agreement pursuant to a written agreement until five years following the finalization of NOF’s capacity expansion. After the expiration of the initial term of the NOF Agreement, the NOF Agreement continues until it is terminated. The NOF Agreement may be terminated (i) by either party for the other party’s assignment for the benefit of creditors, insolvency, bankruptcy, liquidation, dissolution, or the taking of any action by the other party under an act for relief from creditors, (ii) by either party for the other party’s uncured material breach, (iii) by us after the initial term of the NOF Agreement with one year written notice, or (iv) by mutual agreement of the parties. In addition, the NOF Agreement may be terminated by us in the event of a change of fifty percent or more of the direct or indirect ownership of NOF or manufacturing facilities relevant to the NOF Agreement, if such ownership goes to a third partythird-party materially involved in the treatment of growth related disorders in humans. The NOF Agreement may also be terminated by either party for a continuing event of force majeure.
The NOF Agreement contains, among other provisions customary representations and warranties by us and NOF, grants certain limited license rights related to either party’s intellectual property in connection with the manufacturing and supply of NOF Product, provides for certain indemnification rights in favor of both parties and customary confidentiality provisions.
Carbogen Manufacturing and Supply Agreement
On October 26, 2018, we entered into amulti-year Manufacturing and Supply Agreement (the “Carbogen Agreement”) with Carbogen Amcis AG (“Carbogen”). Under the Carbogen Agreement, Carbogen has agreed to manufacture and supply the C13 Linker (the “Carbogen Product”) for our TransCon hGH product candidate. We may purchase C13 Linker from other manufacturers and are not obligated to purchase Carbogen Product from Carbogen, other than certain quantities that have been forecasted by us in accordance with a mandatory rolling forecast that we must deliver to Carbogen from time to time.
The Carbogen Agreement is effective as of October 26, 2018. The initial term of the Carbogen Agreement expires five years after the first commercial launch of our TransCon hGH product candidate (the “Carbogen Initial Term”) unless earlier terminated. After the expiration of the Carbogen Initial Term of the Carbogen Agreement, the Carbogen Agreement continues until it is terminated. The Carbogen Agreement may be terminated (i) by either party for the other party’s assignment of the Carbogen Agreement for the benefit of creditors, insolvency, bankruptcy, dissolution, or taking of any action under an act for relief from creditors, (ii) by either party for the other party’s uncured material breach, (iii) by us after the Carbogen Initial Term of the Carbogen Agreement with one year written notice, (iv) by Carbogen after the Carbogen Initial Term of the Carbogen Agreement with four years written notice (subject to Carbogen’s technology transfer obligation to an alternate supplier) or (iv)(v) by mutual agreement of the parties. In addition, the Carbogen Agreement may be terminated by us in the event of a change of fifty percent or more of the direct or indirect ownership of Carbogen, if such ownership goes to a third partythird-party materially involved in the treatment ofgrowth-related disorders in humans. The Carbogen Agreement may also be terminated by either party for a continuing event of force majeure.
The Carbogen Agreement contains, among other provisions, certain representations and warranties by us and Carbogen, grants certain rights to intellectual property relating to, or inventions made in connection with, the manufacturing and supply of Carbogen Product, provides for certain indemnification rights in favor of both parties and includes confidentiality provisions.
Philips Medisize (formerly B&O Medicom and Medicom Innovation Partner)
On January 12, 2017, we entered into amulti-year Manufacturing and Supply Agreement (the “Medicom Agreement”) with Medicom Innovation Partner (“Medicom”). Under the Medicom Agreement, Medicom has agreed to exclusively manufacture and supply the auto injector injection device (the “Medicom Product”) for our TransCon hGH product candidate. We are obligated to purchase certain quantities that have been forecasted by us in accordance with a mandatory rolling forecast that we must deliver to Medicom from time to time.
The Medicom Agreement is effective as of January 12, 2017. The term of the Medicom Agreement terminates on June 30, 2025 (“Initial Term”) unless earlier terminated or unless extended unilaterally by us, with notice of extension to be given no later than June 30, 2024, by five years until June 30, 2030 (“Extended Term”) after which date it shall continue indefinitely unless terminated. The Medicom Agreement may be terminated (i) by either party for the other party��sparty’s bankruptcy orinsolvency-related events, (ii) by either party for the other party’s uncured material breach, (iii) by us by not extending the Initial Term into the Extended Term, (iv) by Medicom after the Extended Term of the Medicom Agreement with two year’s advance written notice or by us after the Extended Term of the Medicom Agreement with one year’s advance notice, or (v) by Medicom if we purchase less than an agreed volume of the Medicom Product (provided that we may avoid such termination by paying Medicom’s lost profits up to such agreed minimum volume). In addition, the Medicom Agreement may be terminated by us in the event of a change of control of Medicom, if such control goes to a third partythird-party materially involved in the treatment of certain defined endocrinology disordersdiseases in humans. In all events of termination Medicom is obligated to support a tech transfer of manufacture of Medicom Product to an alternate supplier.
The Medicom Agreement contains, among other provisions certain representations and warranties by us and Medicom, grants certain limited license rights related to either party’s intellectual property in connection with the manufacturing and supply of Medicom Product, provides for certain indemnification rights in favor of both parties and includes confidentiality provisions.
Vetter Pharma International GmbH
On December 14, 2018, we entered into amulti-year Supply Agreement (the “Vetter Agreement”) with Vetter Pharma International (“Vetter”). Under the Vetter Agreement, Vetter has agreed to manufacture andfill-and-finish drug product indual-chamber cartridges (the “Ascendis Product”) for our TransCon hGH product candidate. Vetter has agreed to supply in accordance with along-term forecast in addition to a rolling forecast with a binding part that we must deliver to Vetter from time to time.
The Vetter Agreement is effective as of January 1, 2019. The term of the Vetter Agreement expires on thefive-year anniversary of the date of first regulatory approval of the TransCon hGH product (the “Initial Term”) after which term it shall be automatically renewed for subsequenttwo-year terms unless terminated. The Vetter Agreement may be terminated (i) by either party for the other party’s uncured material breach, including certain enumerated events constituting material breach such as bankruptcy orinsolvency-related events, (ii) by us with two years’ notice, with effect no earlier than two years after expiry of the Initial Term or (iii) by either party if the other party is taken over by our or a Vetter competitor, as applicable.
The Vetter Agreement contains, among other provisions, certain representations and warranties by us and Vetter, grants certain limited license rights in connection with Vetter’s manufacturing and supply, and our sale, distribution and other use, of Ascendis Product, provides for certain indemnification rights in favor of both parties and includes confidentiality provisions.
Fujifilm Commercial Supply Agreement
On January 9, 2019, we entered into amulti-year Commercial Supply Agreement (the “Fujifilm Agreement”) with Fujifilm Diosynth Biotechnologies UK Ltd. (“Fujifilm”). Under the Fujifilm Agreement, Fujifilm has agreed to manufacture and supply TransCon hGH Drug Substance (the “Fujifilm Product”) for our TransCon hGH product candidate. We may purchase TransCon hGH Drug Substance from other manufacturers and are not obligated to purchase Fujifilm Product from Fujifilm, other than a total of 6six batches each year in 2020 and 2021.
The Fujifilm Agreement is effective as of January 9, 2019. The initial term of the Fujifilm Agreement expires on December 31 in the year of the five-year anniversary of the first commercial sale of our TransCon hGH product candidate (the “Fujifilm Initial Term”) unless earlier terminated. After the expiration of the Fujifilm Initial Term of the Fujifilm Agreement, the Fujifilm Agreement continues until it is terminated. The Fujifilm Agreement may be terminated (i) by either party for the other party’s bankruptcy orinsolvency-related events, (ii) by either party for the other party’s uncured material breach or material breach that is not capable of remedy, (iii) by us after the Fujifilm Initial Term of the Fujifilm Agreement with two years written notice, or (iv) by Fujifilm after the Fujifilm Initial Term of the Fujifilm Agreement with five years written notice. We are entitled to terminate the Fujifilm Agreement with regards to the manufacture of recombinant hGH after one year following launch with two years written notice. In addition, the Fujifilm Agreement may be terminated by us in the event of a change in control of Fujifilm, where the new controlling entity is our competitor. The Fujifilm Agreement may also be terminated by either party for a continuing event of force majeure.
The Fujifilm Agreement contains, among other provisions, certain warranties by us and Fujifilm, grants certain limited license rights related to either party’s intellectual property in connection with the manufacturing and supply of Fujifilm Product, provides for certain indemnification rights in favor of both parties and includes confidentiality provisions.
Lonza Tech Transfer and Manufacturing Agreement
On December 12, 2019, we entered into a multi-year commercial supply agreement (the “Lonza Agreement”) with Lonza Ltd (“Lonza”). Under the Lonza Agreement, Lonza has agreed to manufacture and supply drug substance for our TransCon hGH product candidate (the “TransCon hGH Drug Substance”). We may purchase TransCon hGH Drug Substance from other manufacturers but have granted Lonza status as primary supplier of TransCon hGH Drug Substance and are obligated to purchase a certain minimum annual quantity of TransCon hGH Drug Substance per year starting in 2023.
The Lonza Agreement secures us a certain capacity of TransCon hGH Drug Substance per year. For requirements above such capacity, we are free to have manufactured and purchase TransCon hGH Drug Substance from other suppliers, including but not limited to, subject to certain restrictions, transferring the manufacturing process of TransCon hGH Drug Substance to ourselves.
The Lonza Agreement is effective as of December 12, 2019. The initial term of the Lonza Agreement expires seven years after first approval of a drug product manufactured using the TransCon hGH Drug Substance (the “Lonza Initial Term”) unless earlier terminated. During the first five years of the Lonza Initial Term, we may decide, in our sole discretion, to extend the term of the Lonza Agreement by two years. The Lonza Agreement may be terminated (i) by either party for the other party’s bankruptcy or insolvency-related events, (ii) by either party for the other party’s uncured material breach, (iii) by either party for a continuing event of force majeure, (iv) by either party upon written notice after a specified time period in the event of our change of control, and (v) by either party in the event of the occurrence of certain conditions related to the manufacturing of the TransCon hGH Drug Substance as more fully described in the Lonza Agreement.
The Lonza Agreement contains, among other provisions, certain warranties by us and Lonza, grants certain limited license rights related to either party’s intellectual property in connection with the manufacturing and supply of TransCon hGH Drug Substance, provides for certain indemnification rights in favour of both parties and includes confidentiality provisions.
Sharp Corporation Packaging and Supply Agreement
On December 1, 2019 we entered into a multi-year packaging agreement (the “Sharp Agreement”) with Sharp Corporation (“Sharp”). Under the Sharp Agreement, Sharp will package, assemble, and label TransCon hGH for commercial use in certain territories, including the United States and the European Union. We arenon-exclusive to Sharp and may engage other manufacturers to package, assemble, and label TransCon hGH but we are obligated to meet certain minimum spend requirements for TransCon hGH during the first12-month period after first shipment of TransCon hGH for commercial sale after regulatory approval thereof.
The Sharp Agreement is effective as of December 1, 2019. The initial term of the Sharp Agreement expires on December 31, 2025 and will be automatically extended for additionaltwo-year periods unless earlier terminated. The Sharp Agreement may be terminated (i) by either party upon mutual consent, (ii) by either party for the other party’s uncured material breach, (iii) by either party for the other party’s bankruptcy or insolvency-related events, (iv) by either party for a continuing event of force majeure, (v) by either party after the initial term of the Sharp Agreement has been completed.
The Sharp Agreement contains, among other provisions, certain warranties by Sharp, provides for certain indemnification rights in favor of both parties and includes confidentiality provisions.
Competition
The pharmaceutical industry is very competitive and subject to rapid and significant innovation. Our potential competitors include major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical and generic drug companies, universities, and other research institutions. Many of our competitors have greater resources, as well as larger research and development functions and more experienced marketing and manufacturing organizations. As a result, these companies may obtain regulatory approval more rapidly than we are able to and may be more effective in selling and marketing their products.
Smaller orearly-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Our competitors may succeed in developing, acquiring or licensing technologies and drug products that are superior to, or more effectively marketed than, the product candidates that we are currently developing or that we may develop, which could render our products obsolete and noncompetitive. For additional information regarding the companies that may be competitive with our product candidates currently in development, please see the descriptions of our current product candidates included above under the caption “TransCon Product Candidates.”
In addition, many of our competitors have greater experience than we do in conducting preclinical and clinical trials and obtaining FDA and other regulatory approvals. Accordingly, our competitors may succeed in obtaining FDA or other regulatory approvals for drug candidates more rapidly than we do. Companies that complete clinical trials, obtain required regulatory authority approvals and commence commercial sale of their drugs before their competitors may achieve a significant competitive advantage. Drugs resulting from our research and development efforts or from our joint efforts with collaboration partners therefore may not be commercially competitive with our competitors’ existing products or products under development.
We are aware that other companies are developing or evaluating enhanced drug delivery and sustained release technologies, which may be competitive with our TransCon technologies. In particular, we believe Nektar Therapeutics, OPKO Health, Inc., ProLynx LLC and Serina Therapeutics, Inc. are developing technology platforms in the areas of enhanced drug delivery and reversible linkers that may be competitive with our TransCon technologies. We also expect that technological developments will occur at a rapid rate and that competition is likely to intensify as various enhanced delivery and sustained released technologies may achieve similar advantages.
Intellectual Property
We actively seek to protect the intellectual property and proprietary technology that we believe is important to our business, which includes seeking and maintaining patents covering our technology, i.e., TransCon linkers and carriers, specific lead candidate structures, broad product concepts, proprietary processes and any other inventions that are commercially and/or strategically important to the development of our business. We also rely on trade secrets that may be important to the development of our business and actively seek to protect the confidentiality of such trade secrets.
Our success will depend on our ability to obtain and maintain patent and other proprietary protection for commercially important technology, inventions andknow-how related to our business, defend and enforce our patents, preserve the confidentiality of our trade secrets and operate without infringing the valid and enforceable patents and proprietary rights of third parties.third-parties. For more information, please see “Risk Factors—Risks Related to Our Intellectual Property.”
As of December 31, 2019,2020, we own a total of 8087 patent families, of which 2629 are currently in their priority year or international phase and we own several granted patents in the United States (24)(28), Europe (12)(17), Australia (24)(26), Canada (11)(19), China (7), Israel (11)(13), Indonesia (2), India (3)(4), Korea (3), Malaysia (3)(5), New Zealand (4), Japan (15)(17), Mexico (10)(13), Singapore (6)(7), Russia (8)(10), the United Arab Emirates (1) and South Africa (19)(21) and have approximately 366389 pending national/regional applications in a total of 19 jurisdictions (excluding the member states of the European Patent Convention in which our European patents were validated).
So far none of our granted patents has been subject to opposition proceedings, appeals or similar actions aiming at revoking or restricting the scope of a granted patent.
The patent portfolios for the fields containing our most advanced product candidates as of December 31, 20192020 are summarized below and the expected expiration dates included in the summary below do not give effect to patent term extensions that may be available.
TransCon hGH
Our patent portfolio related to TransCon hGH includes seven patent families relating to different aspects of TransCon hGH and an additional nine patent families covering various aspects of theauto-injector device for administration of TransCon hGH. The first of these patent families is a composition of matter patent family directed to the particular stoichiometry of TransCon hGH and a related TransCon carrier. As of December 31, 2019,2020, this patent family included patents granted in Europe and the United States. We expect any patents granted in this patent family to expire in October 2024.
The second of these patent families is a composition of matter patent family directed to a TransCon linker used in TransCon hGH. As of December 31, 2019,2020, this patent family included patents granted in the United States, Europe, Australia, Brazil, Canada, Japan and Mexico and included patent applications in Europe, the United States, and Brazil. We expect any patents granted in this patent family to expire in March 2025.
The third of these patent families is a composition of matter patent family directed to a broad class of TransCon hGH lead candidate structures. As of December 31, 2019,2020, this patent family included patents granted in the United States, Europe, Australia, Canada, China, Israel, India, Japan, Mexico, Russia and South Africa and included patent applications in Europe, the United States, Brazil, Canada, India, Japan, Mexico and Russia. We expect any patents granted in this patent family to expire in April 2029.
The fourth of these patent families is a composition of matter patent family directed to specific dry pharmaceutical compositions comprising TransCon hGH. As of December 31, 2019,2020, this patent family included patents granted in the United States, Europe, Australia, Canada, India, Israel, Mexico, Singapore and South Africa and included patent applications in the United States Brazil, and India.Brazil. We expect any patents granted in this patent family to expire in December 2030.
The fifth of these patent families is a composition of matter patent family directed to a broad class of TransCon hGH lead candidate structures. As of December 31, 2019,2020, this patent family included a patentpatents granted in the United States, Japan, Russia and South Africa and patent applications in the United States, Europe, Australia, Brazil, Canada, Israel, Japan, South Korea, Mexico, New Zealand, Russia and Singapore. We expect any patents granted in this patent family to expire in November 2035.
The sixth of these patent families is directed to a particular dosage regimen forlong-acting growth hormone formulations. As of December 31, 2019,2020, this patent family included patent applications in the United States and in Europe. We expect any patents granted in this patent family to expire in November 2035.
The seventh of these patent families is directed to potential superior efficacy achieved with TransCon hGH treatment. This patent family is currently in its priority yearPCT phase and any patents granted thereof are expected to expire in March 2040.
Seven of the nine patent families covering theauto-injector device with a filing date of December 29, 2016, includesinclude patent applications in the United States, Europe, Australia, Canada, Japan and New Zealand and one granted patent in the United States as of December 31, 2019.2020. We expect any patents granted from these patent families to expire in December 2036. As of December 31,
2019 2020 the other two patent families covering the auto-injector device with a filing date of May 23, 2018 and June 29, 2018, respectively, include patent applications in the United States, Europe, the United Arab Emirates, Australia, Brazil, Canada, China, Indonesia, Israel, India, Japan, South Korea, Mexico, Malaysia, New Zealand, Russia, Singapore and South Africa and one granted patent in South Africa. We expect any patents granted from these patent families to expire in March and June 2038, respectively.
TransCon PTH
Our patent portfolio related to TransCon PTH includes sevennine patent families relating to different aspects of TransCon PTH. The first of these patent families is a composition of matter patent family directed to the TransCon linker used in TransCon PTH. As of December 31, 2019,2020, this patent family included granted patents in the United States, Europe, the United Arab Emirates, Australia, Canada, China, Israel, Japan, Mexico, Russia and South Africa and included patent applications in Europe, the United States, Brazil and Russia. We expect any patents granted in this family to expire in January 2029.
The second of these patent families is a composition of matter patent family directed to a broad class of TransCon PTH candidate structures. As of December 31, 2019,2020, this patent family included patent applications in the United States, Europe, Australia, Brazil, Canada, China, Indonesia, Israel, India, Japan, South Korea, Mexico, Malaysia, New Zealand, Russia, Singapore and Thailand and a granted patent in South Africa. We expect any patents granted in this patent family to expire in February 2037.
The third and fourth of these patent families are method of treatment patent families directed to a particular dosage regimen. As of December 31, 2019,2020, one of these patent families includes patent applications in the United States, Europe, Australia, Brazil, Canada, China, Israel, Indonesia, Japan, South Korea, Mexico, Malaysia, New Zealand, Russia, Singapore and Thailand and a granted patent in South Africa. The other one of these patent families includes patent applications in the United States, Europe, Australia, Canada, China and Japan. We expect any patents granted in this patent family to expire in September 2037.
The fifth of these patent families is a composition of matter family directed to PTH compounds exhibiting a beneficial pharmacokinetic profile. As of December 31, 2019,2020, this patent family includes patent applications in the United States, Europe, Australia, Brazil, Canada, China, Israel, Japan, South Korea, Mexico, New Zealand, Russia and Singapore and a granted patent in South Africa. We expect any patents granted in this patent family to expire in September 2037.
The sixth patent family relates to a starting dose for treatment with reversible PTH conjugates. As of December 31, 2019,2020, this patent family consists of an international application.includes patent applications in the United States, Europe, Australia, Brazil, Canada, China, Indonesia, Israel, Japan, South Korea, Mexico, Malaysia, New Zealand, Russia, Singapore, Thailand and South Africa. We expect any patents granted from this patent family to expire in May 2039.
The seventh patent family relates to a pharmaceutical composition comprising reversible PTH conjugates. As of December 31, 2019,2020, this patent family consists of an priorityinternational application. We expect any patents granted from this patent family to expire in February 2040.
The eighth and ninth patent families relate to a method of titrating hypoparathyroidism patients off of standard of care within four weeks from the beginning of daily treatment with a PTH compound and the treatment of the physical and mental well-being of hypoparathyroidism patients, respectively. As of December 31, 2020, these patent families are in their priority year. We expect any patents granted from these patent families to expire in January 2041 and September 2041, respectively.
TransCon CNP
Our patent portfolio related to TransCon CNP includes eleven patent families relating to different aspects of TransCon CNP. The first of these patent families is a composition of matter patent family directed to the particular stoichiometry of TransCon CNP and a related TransCon carrier. As of December 31, 2019,2020, this patent family included patents granted in Europe and the United States and a patent application in Europe. We expect any patents granted in this patent family to expire in October 2024.
The second of these patent families is a composition of matter patent family directed to the TransCon linker used in TransCon CNP. As of December 31, 2019,2020, this patent family included granted patents in the United States, Europe, the United Arab Emirates, Australia, Brazil, Canada, China, Israel, Japan, Mexico and South Africa and included patent applications in Europe, the United States, Brazil, Mexico and Russia. We expect any patents granted in this family to expire in January 2029.
The third of these patent families is a composition of matter patent family directed to a broad class of TransCon CNP candidate structures. As of December 31, 2019,2020, this patent family included patent applications in the United States, Europe, Australia, Brazil, Canada, China, Israel, India, Japan, South Korea, Mexico, Malaysia, New Zealand, Russia, Singapore and Thailand and a granted patent in Russia, Singapore and South Africa. We expect any patents granted in this patent family to expire in January 2036.
The fourth to the ninth patent familyfamilies are composition of matter patent families directed various CNP compounds having beneficial properties. As of December 31, 2019,2020, the first one of these six patent families included patent applications in the United States, Europe, Australia, Canada, Japan, Mexico and New Zealand and a granted patent in the United States and South Africa. As of December 31, 2019,2020, the second one included patent applications in the United States, Europe, Australia, Canada, Japan and New Zealand and a granted patent in South Africa. As of December 31, 2019,2020, the third one included patent applications in the United States, Europe, Australia, Brazil, Canada, China, Indonesia, Israel, India, Japan, South Korea, Mexico, Malaysia, New Zealand and Singapore and a granted patent in South Africa. As of December 31, 2019,2020, the fourth one included patent applications in the United States, Europe, Australia, Canada, Israel and New Zealand. As of December 31, 2019,2020, the fifth one included patent application in the United States, Europe, Australia, Brazil, Canada, China, Israel, South Korea, New Zealand and Singapore. As of December 31, 2019,2020, the sixth one included patent applications in the United States, Europe, Australia, Canada, Israel and New Zealand. We expect any patents granted in thisthese patent familyfamilies to expire in January 2037.
The tenth patent family covers a combination therapy of TransCon CNP. As of December 31, 2019,2020, this patent family includes patent applications in the United States, Europe, Australia, Brazil, Canada, China, Indonesia, Israel, Japan, South Korea, Mexico, Malaysia, New Zealand, Russia, Singapore and Thailand and a granted patent in South Africa. We expect any patents granted from this patent family to expire in September 2037.
The eleventh patent family relates to a pharmaceutical composition comprising reversible CNP conjugates. As of December 31, 2019,2020, this patent family consists of an EP priorityinternational application. We expect any patents granted from this patent family to expire in February 2040.
General Field of Oncology
As of December 31, 2019,2020, our oncology-related patent portfolio includes eleventhirteen patent families relating to various TransCon oncology product candidates, all of which, except for one, all are currently in their priority year or in international phase. We expect any patents granted in these patent families to expire in or after March 2039.
TransCon TLR7/8
As of December 31, 2020, our patent portfolio related to TransCon TLR7/8 includes six patent families. The first patent family relates to hydrogels, which are first synthesized and subsequently loaded with drug-linker conjugates. As of December 31, 2020, this patent family included granted patents in the United States and in Europe and a patent application in Europe. We expect any patents granted in these patent families to expire in July 2025.
The second patent family relates to a specific class of PEG-based hydrogels. As of December 31, 2020, this patent family included granted patents in Europe, Australia, Brazil, Canada, China, Indonesia, Israel, India, Japan, South Korea, Mexico, Malaysia, New Zealand, Russia, Singapore and South Africa and patent applications in the United States, Europe and Thailand. We expect any patents granted in this patent family to expire in July 2030.
The third patent family relates to a broad class of TransCon TLR7/8 candidate structures. As of December 31, 2020, this patent family consist of an international application and patent applications in Argentina and Taiwan. The fourth to sixth patent families relate to TransCon TLR7/8 compounds having beneficial properties. As of December 31, 2020, these patent families consist of one international application each. We expect any patents granted in all four of these patent families to expire in January 2040.
TransCon Technologies
Our patent portfolio also includes patents and patent applications generally relating to our TransCon technologies, including TransCon linkers, TransCon carriers and certain soluble conjugates. We own an aggregate of 1315 patent families relating to TransCon linkers, the material components of which are described above. We own an aggregate of 11 patent families relating to TransCon carriers, the material components of which are described above. Finally, we own a composition of matter patent family that is directed to soluble conjugates in which one drug molecule is connected to one TransCon carrier molecule. As of December 31, 2019,2020, this patent family included patents in Europe and the United States and a patent application in Europe. We expect any patents granted in this patent family to expire in October 2024.
Laws and Regulations Regarding Patent Terms
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the earliest date of filing anon-provisional patent application. In the United States, a patent term may be shortened if a patent is terminally disclaimed over another patent or if there are delays in patent prosecution by the patentee. A patent’s term may be lengthened by a patent term adjustment, which compensates a patentee for administrative delays by the USPTO in granting a patent. The patent term of a European patent is 20 years from its filing date, which, unlike in the United States, is not subject to patent term adjustments.
The term of a patent that covers anFDA-approved drug or biologic may also be eligible for patent term extension, which permits patent term restoration as compensation for the patent term lost during the FDA regulatory review
process. The Drug Price Competition and Patent Term Restoration Act of 1984, or theHatch-Waxman Act, permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the drug or biologic is under regulatory review. Patent extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved drug may be extended. Similar provisions are available in Europe and other jurisdictions to extend the term of a patent that covers an approved drug. In the future, if and when our products receive FDA approval, we expect to apply for patent term extensions on patents covering those products. We anticipate that some of our issued patents may be eligible for patent term extensions.
Government Regulation and Product Approval
Government authorities in the United States, at the federal, state and local level, and in other countries extensively regulate, among other things, the research, development, testing, manufacture, including any manufacturing changes, safety surveillance, efficacy, quality control, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, sale, import, export and the reporting of safety and otherpost-market information of pharmaceutical and medical device products such as those we are developing. A new drug must be approved by the FDA through the NDA process and a new biologic must be licensed by the FDA through the BLA process before it may be legally marketed in the United States. Similarly, new drugs and biologics must be approved by the EMA through the marketing authorization application, or MAA, process before they may be legally marketed in Europe. Our product candidates will be subject to similar requirements in other countries prior to marketing in those countries. The processes for obtaining regulatory approvals in the United States, the EEA and in foreign countries, along with subsequent compliance with appropriate federal, state, local and foreign statutes and regulations, require the expenditure of substantial time and resources.
U.S. Government Regulation
In the United States, we are subject to extensive regulation by the FDA, which regulates drugs under the Federal Food, Drug, and Cosmetic Act, or the FDCA, and in the case of biologics, also under the Public Health Service Act, or PHSA, and their implementing regulations, and other federal, state, and local regulatory authorities. The FDCA, PHSA and their implementing regulations set forth, among other things, requirements for the research, testing, development, manufacture, quality control, safety, effectiveness, approval, labeling, storage, record keeping, reporting, distribution, import, export, advertising and promotion of our products. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA’s refusal to approve pending NDAs or BLAs, withdrawal of an approval, imposition of a clinical hold on clinical studies, issuance of warning letters or other notices of violation, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties.
The process required by the FDA before a drug or biologic may be marketed in the United States generally involves the following:
completion of preclinical laboratory tests, animal studies and formulation studies in compliance with the FDA’s Good Laboratory Practice regulations;
submission to the FDA of an IND which must become effective before human clinical trials may begin;
approval by an independent institutional review board, or IRB, at each clinical site before each trial may be initiated;
performance of adequate andwell-controlled human clinical trials in accordance with good clinical practice, or GCP, requirements to establish the safety and efficacy of the proposed drug or biological product for each indication;
submission to the FDA of an NDA or BLA;BLA after completion of all pivotal clinical trials;
satisfactory completion of an FDA advisory committee review, if applicable;
satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current good manufacturing practice, or cGMP, requirements and to assure that the facilities, methods and controls are adequate to preserve the drug’s or biologic’s identity, strength, quality and purity;purity and of selected clinical investigation sites to assess compliance with GCP; and
FDA review and approval of the NDA or BLA.
We are developing anauto-injector with Philips Medisize A/SBLA to facilitate the administration of TransCon hGH byend-users. A medical device (such as an auto-injector) marketed together with a drug or biologic is considered a combination product. Typically, the FDA’s Office of Combination Products assigns a combination product to a specific Agency Center as the lead reviewer after an applicant submits a Request for a Designation, although submission of a Request for Designation is not mandatory. The FDA determines which Center will lead a product’s review based upon the product’s primary mode of action. Depending on the type of combination product, its approval, clearance or licensure may often be obtained through the submission of a singlepermit commercial marketing application. However, the FDA sometimes will require separate marketing applications for individual constituent parts of the combination product which may require additional time, effort, and information. Even when a single marketing application is required for a combination product, such as an NDAparticular indications for a combination pharmaceutical and device product, both the FDA’s Center for Drug Evaluation and Research and the FDA’s Center for Devices and Radiological Health will participateuse in the review. An applicant may also need to discuss with the Agency how to apply certain premarket requirements andpost-marketing regulatory requirements, including conduct of clinical trials, adverse event reporting and good manufacturing practices, to their combination product.United States.
Nonclinical Studies and Investigational New Drug Applications
Nonclinical studies include laboratory evaluations of product chemistry, toxicity and formulations, as well as animal studies to assess safety and efficacy. A USAn IND is a request for authorization from the FDA to administer an investigational pharmaceutical product to humans. A sponsor must submit the results of the nonclinical tests, together with chemistry, manufacturing & control information, and any available clinical data or literature, to the FDA as part of an IND. Some nonclinical testing may continue after the IND is submitted. An IND automatically becomes effective and a clinical trial proposed in the IND may begin 30 days after the FDA receives the IND, unless during this30-day waiting period, the FDA raises concerns or questions related to one or more proposed clinical trials and places the clinical trial on a clinical hold. In such a case, the sponsor must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence. The FDA may impose clinical holds on a product candidate at any time before or during clinical trials due to safety concerns ornon-compliance. If the FDA imposes a clinical hold, trials may not recommence without FDA authorization and then only under terms authorized by the FDA.

