| • | | PKU GOLIKE® in phenylketonuria (PKU). We intend to maximize the commercial potential of the PKU GOLIKE® family of products in all major territories either directly or through distribution partners
PKU GOLIKE® in phenylketonuria (PKU). We recently announced a license and supply relationship with Eton Pharmaceuticals, Inc. to ensure the commercialization of PKU GOLIKE in the United States. We further intend to maximize the commercial potential of the PKU GOLIKE® family of products in all major territories through licencees in order to create a steady and growing positive cash flow; PKU GOLIKE® is an approved, and fully reimbursed line of patented and differentiated medical food products for the dietary management of PKU. | • | | RLF-OD032 in PKU. The Company acquired worldwide rights (except in the United Kingdom) to RLF-OD032 in 2022. We intend to complete clinical studies of this product candidate in 2024 after which time we expect to file a 505(b)(2) NDA with the FDA. If approved, we intend to divest or out-license this product. |
| | • | | OLPRUVA™® in urea cycle disorders (UCDs). Pursuant toThe Company in-licensed the FDA approval grantedrights for OLPRUVA in December 2022, we are expectingEurope and funded part of the development of OLPRUVA™ to become commercially available in the U.S. from mid-June 2023 as a new prescription optionby Acer Therapeutics, Inc. (Acer). The FDA approved OLPRUVA for the treatment of UCDs;UCDs in parallel, together with our collaboration partner AcerDecember 2022. Zevra Therapeutics Inc. (Acer), we are currently pursuing approval for use(Zevra) acquired Acer in 2023 and went through a full commercial launch of OLPRUVA™the product in Europe. |
| • | | January 2024. Relief is entitled to receive a 10% continuing royalty on the net sales of OLPRUVA™ in maple syrup urine disease (MSUD). Togetherthe U.S. up to a cumulative amount of USD 45 million. We plan to continue working with our collaboration partner Acer, we intendZevra to pursuemaximize the developmentcommercial potential of OLPRUVA™ for the treatment of MSUD in the U.S. and Europe, especially considering this disease currently has no approved treatments available.to identify partnership opportunities for Europe. |
| • | | RLF-OD032 in PKU. We intend to complete the development of this prescription drug candidate intended as a new, patented and highly differentiated treatment option for PKU; we anticipate the completion of the development and the filing of a 505(b)(2) NDA before the FDA in 2024 and anticipate an expected approval sometime in Q3/2025.
|
| • | | RLF-TD011 in epidermolysis bullosa (EB). We intend to advance the development of this drug candidate and to complete the ongoing proof of concept, investigator initiated clinical trial sometime before the end of 2023 subject to patient enrolment pace.
|
| • | | RLF-TD011 in cutaneous T-cell lymphoma (CTCL). Subject to additional financing, we intend to conduct a proof of concept, investigator initiated clinical trial on CTCL patients at Northwestern University in the U.S. where clinical protocol has been already approved.
|
| • | | Rare Respiratory Diseases: |
In 2023, we completed the development of a shelf stable formulation of RLF-100® (aviptadil acetate) for both injectable intravenous (IV) and inhaled use.
The IV RLF-100® formulation is specifically designed and indicated for all COVID or non-COVID related acute respiratory distress syndromes (ARDSs) because (i) it allows a prompt and efficient delivery of the drug in patients with severe conditions (e.g., patients in ICUs) while (ii) keeping under control the possible side effects because administered in a hospital setting.
The Inhaled RLF-100® formulation is specifically designed and indicated for all the rare chronic lung diseases (CLDs) we are targeting because (i) it can maximize the clinical efficacy directly in the target organs (the lungs), (ii) while reducing the side effects of the drug (vasodilator and hypotension) because not systemically absorbed.
| | • | | RLF-100 in pulmonary indications. We intend to explore partnership opportunities with seasoned respiratory biopharmaceutical companies to advance RLF-100® in ARDSs. Subject(aviptadil acetate) for both injectable intravenous and inhaled use, and to additional financing, we intend to advancemaximize the developmentpotential value of this drug candidate in ARDSs by entering into Phase 2 development.the program. |
Additionally, we will continue to evaluate business development opportunities that expand our portfolio in the rare dermatological therapeutic area. We will consider partnerships with, or acquisitions of, companies that have late-stage clinical assets with strong safety and efficacy profiles where we can bring to bear our development expertise and platform technologies to quickly and capital efficiently develop and commercialize these product candidates. | • | | RLF® in CLDs. Subject to additional financing, we intend to advance the development of this drug candidate in the targeted CLDs, namely sarcoidosis, berylliosis and checkpoint inhibitor-induced pneumonitis (CIP) by entering into Phase 2 development.
|
3630
| • | | Expansion of our pipeline:
|
We intend to leverage our strength and experience to identify, acquire and bring to market drug candidates for therapeutic areas that align with our areas of focus.
Product Portfolio and Development TimelinePipeline Relief Therapeutics’ portfolio offersconsists of a balanced mix of marketed, revenue-generating products our proprietary,and globally patented Physiomimic™ and TEHCLO™ drug delivery platform technologies and a highly targeted clinical developmenttechnologies. Our pipeline consisting of risk-mitigated assets focused inspans three core therapeutic areas: rare metabolicdermatological disorders, rare skin diseasesmetabolic disorders, and rare respiratory diseases where the company can best leverage its internal research and development (R&D) capabilities and track record. In addition, Relief Therapeutics is commercializing several legacy products via licensing and distribution partners.diseases.
 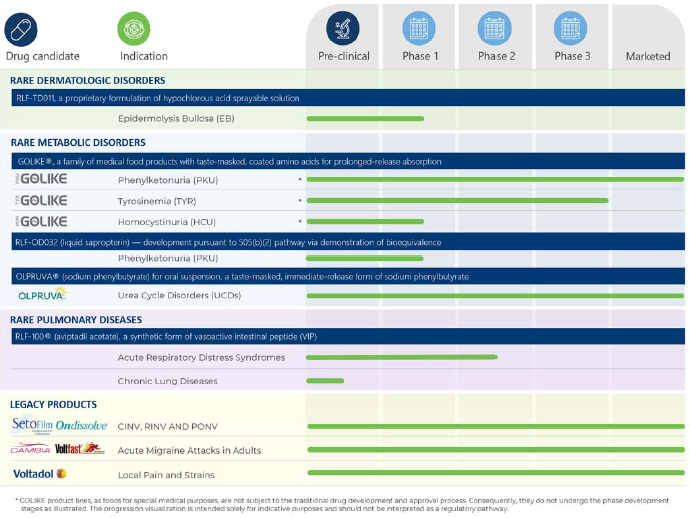
Rare Dermatologic Disorders 37
DRUG DELIVERY PLATFORM TECHNOLOGIESThe Company is committed to developing product candidates in rare dermatological disorders which it believes is an area of high unmet need where its developmental expertise and platform technology offer the opportunity to improve patient outcomes and quality of life. RLF-TD011 is being studied for the treatment of epidermolysis bullosa where patients have relatively few optimal treatment options.
Our drug delivery platform technologies enable us to optimize the therapeutic potential of established products with proven efficacy, known safety profiles or where proof-of-concept exists. These platforms have utility for development in other specialty or rare disease therapeutic areas, partnerships and out-licensing.
TEHCLO NANOTECHNOLOGY™ Our TEHCLO Nanotechnology™ Nanotechnology
The TEHCLO Nanotechnology platform (TEHCLO™) consists(TEHCLO), used in the development of RLF TD011, is our proprietary, globally patent-protected electrode with nanocoating,patented technology. Characterized by its nanocoated electrodes, this technology enables the method for preparing and makingproduction of a highly stable aqueous solutionselectrolytic water resulting in a hypochlorous acid solution that is low in pH, isotonic, and our device for the electrolytic treatment of a fluid. TEHCLO™ was used to develop RLF-TD011, Nexodyn and some of our legacy products.oxidizing. Our TEHCLO™ intellectual property portfolio consists of fourthree patent families. The first threetwo families include 10740 granted patents world-wideworldwide directed to systems and methods for generating APR’sour hypochlorous acid solution, compositions comprising APR’s hypochlorous acid solution and methods for treating ocular disorders.solutions. These patents expire between October 2026 and JuneFebruary 2030, exclusive of any patent term adjustments or extensions, or any form of potential exclusivity. IfA third patent family will cover certain medical uses, and if granted, additional patents wouldwill expire no earlier than July 2040. PHYSIOMIMIC TECHNOLOGY™Epidermolysis Bullosa (EB)
Our PhysiomimicEpidermolysis bullosa (EB) is a group of rare, genetic skin disorders which cause the skin to blister and tear from minimal contact or friction. There are several genetic and symptomatic variations of the disease, yet all types have the symptom of exceedingly fragile skin. Individuals born with EB have skin of such fragility they are often referred to as ‘butterfly children’, a metaphor that highlights the extreme delicacy of their skin, akin to the wings of a butterfly.
31
In patients with EB, painful open wounds and sores form where the skin has been damaged. Moreover, in some cases, the disease can severely impact internal linings and organs. Complications commonly result due to secondary infections and extensive scarring. Individuals with milder forms of EB may still live long, productive lives. However, the more severe forms of EB require multiple medical interventions to treat manifestations and complications, which may lead to disfigurement, disability, and in some cases, premature mortality. The National Epidermolysis Bullosa Registry (NEBR) reports, based on 16 years of data, that the incidence of EB in the U.S. is 19.57 per 1 million live births, with prevalence rate of 11.07 per 1 million population. Globally, EB affects approximately 500,000 individuals. The current classification for EB includes four subtypes defined by the level of cleavage at the dermal/epidermal junction, as detailed hereafter (Fine et al. 2008). | | | EB Subtype | | Characteristics | | Genetically Inherited | | | | EB simplex (EBS) | | Blistering occurs in the upper layer of the skin (the epidermis). This is the most common type of EB, accounting for 70% of cases, and tends to be milder than the other types. | | | | Dystrophic EB (DEB) | | Blistering occurs below the basement membrane zone in the upper part of the dermis. DEB accounts for approximately 25% of cases and can manifest as either recessive (RDEB) or dominant (DDEB) | | | | Junctional EB (JEB) | | Blistering occurs at the junction between the epidermis and the dermis (lower layer of the skin) in a layer of skin known as the basement membrane zone. JEB accounts for around 5% of cases and is usually considered the most severe type of EB. | | | | Kindler Syndrome (KS) | | An extremely rare, recessively inherited disorder characterized by blistering in infancy, followed by poikiloderma and photosensitivity in childhood (Burch et al. 2006). Blistering may occur within any layer of the skin. | | | Non-Genetically Inherited | | | | Acquired EB (Epidermolysis Bullosa Acquisita, EBA) | | Blistering occurs at the basal derma. This chronic autoimmune condition is caused by antibodies targeting type VII collagen, the major component of anchoring fibrils that connect the basement membrane to dermal structures. It is a very rare form and is not genetic (Kasperkiewicz et al. 2016). |
The four major forms of EB (EBS, DEB, JEB, and KS) are inherited genetically. The cause of inherited EB involves at least 20 distinct genes, with over 1,000 mutations identified that affect proteins for the adherence of the epidermis to the underlying dermis (Denyer et al. 2007). These molecular anomalies not only alter the structure of epidermis and dermis but can also interfere with the functional and structural integrity of the basal membrane zone (BMZ). The BMZ, a highly specialized interface between epithelial cells and the underlying matrix, is crucial for cell adhesion, proliferation, differentiation, tissue repair, and barrier function. Consequently, these disruptions lead to cell and tissue dehiscence, severely impacting the skin’s ability to perform its protective and regenerative functions (Laimer et al. 2015). Figure: Cross-section diagram of the skin 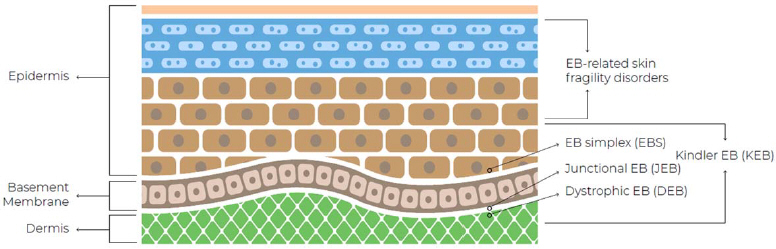
32
Considerable genetic heterogeneity and complex genotype-phenotype correlations are observed across the EB subtype and are attributed to several factors. These include the type of the mutation (homozygosity versus heterozygosity), the number of genes involved (monogenic, digenic inheritance), the location of mutations within each gene, and the range of resulting alterations in protein expression. Beyond the primary structural-functional defects, secondary epigenetic factors (e.g. the differentially regulated expression of a myriad of other genes involved in the maintenance and function of this microenvironment, as well as induction of inflammatory cascades) and environmental factors further contribute to the highly variable phenotype of EB (Laimer et al. 2015, Küttner et al. 2013). In most cases, symptoms of EB are apparent from birth or shortly thereafter. A physician may suspect EB from the appearance of the affected skin. To confirm the diagnosis, a limited number of laboratory tests are available, which may include a skin biopsy for immunofluorescent mapping or a genetic testing. The signs and symptoms of this disorder vary greatly among the different EB subtypes and individuals affected. In milder cases, blistering mainly affects the hands and feet, whereas severe forms of EB are characterized by generalized skin fragility and devastating blistering from minor trauma, severely impairing the quality of life of affected patients. Some EB types may also affect the eyes, tongue, and esophagus, leading to mutilating scarring and disabling musculoskeletal deformities. Complications in EB may include infections, fusion of fingers and joint changes, nutritional problems, dental and oral issues, skin cancer, and premature death. VYJUVEK®, was approved by the FDA on May 19, 2023, for the treatment of DEB, and the company Krystal Biotech, Inc. (NASDAQ: KRYS) subsequently initiated the U.S. commercial launch. VYJUVEK is the first medicine approved by the FDA for the treatment of DEB. VYJUVEK® is a re-dosable, off-the-shelf gene therapy designed to deliver two copies of the COL7A1 gene when applied topically, directly onto an open wound. Unlike the previous standard of care, VYJUVEK treats DEB at the molecular level by providing patient’s skin cells the template to produce normal COL7 protein, thereby addressing the fundamental disease-causing mechanism. FILSUVEZ® was approved by the FDA on December 19, 2023. It is a topical gel indicated for the treatment of partial thickness wounds in patients six months and older with JEB and DEB. FILSUVEZ contains a dry extract from two species of birch bark consisting of naturally occurring substances known as triterpenes, including betulin, betulinic acid, erythrodiol, lupeol and oleanolic acid. The topical gel is applied on the wound and covered by a wound dressing. The current standard of care for EB patients includes wound management to prevent infection, pain management to reduce discomfort, and nutritional support to promote healing. This involves careful wound cleaning and disinfection to minimize the risk of infection. Gentle cleansing of the affected areas with mild, non-irritating solutions helps remove bacteria and other pathogens from the wound surface. Antibiotics may be used to prevent and treat infections, while analgesics are prescribed for pain relief. Management of bioburden in EB patients involves antiseptics and often requires the use of antibiotics. However, topical antibiotics should be used sparingly in EB due to the risk of promoting antibiotic-resistant bacteria strains and potential wound sensitization. The emergence of antibiotic-resistant strains of bacteria, such as methicillin-resistant Staphylococcus aureus and increasingly ciprofloxacin-resistant Pseudomonas, is a significant challenge, potentially compromising the efficacy of current treatments. These resistant strains are frequently isolated from EB wounds (Singer et al. 2018). While both infection and inflammation can impair wound healing, no specific product has been developed for EB wounds that can simultaneously control infection and bioburden while reducing inflammation. Wound care management for EB patients and their care givers is a complex and time-consuming process. Nurses and families often find their lives overwhelmed by the continuous routine of wound management and medication administration. Care involves piercing, draining, and dressing blisters, with bathing and dressing changes alone requiring more than three hours. Pain medications and antibiotics must be administered regularly. Additionally, a significant amount is dedicated to frequent visits to doctors, clinics and support groups (Denyer et al. 2007). The goal of developing RLF-TD011 is to efficiently control bioburden while reducing the need for antibiotics and alleviating inflammation and pain associated with EB. Additionally, the method of administration may reduce the complexity and time required to treat EB thereby consuming less healthcare and care giver resources. RLF-TD011 for the Potential Treatment of Epidermolysis Bullosa We are developing RLF-TD011 as a differentiated acid oxidizing solution of hypochlorous acid (HCIO) that combines strong antimicrobial action with anti-inflammatory properties, thereby allowing for infection control, reduction of wound colonization and improved wound healing. We believe RLF TD011, if approved, may be a fast, easy to use, and effective treatment for EB wound care management. Importantly, RLF-TD011 could also enhance the efficacy and usability of newly developed EB treatments given its unique properties. Developed with our proprietary, patent-protected TEHCLO Nanotechnology, RLF-TD011 employs an exclusive combination of four physio-chemical properties—high-purity HCIO, hypotonic, low pH and high oxidation-reduction potential, which we believe can support a faster physiological healing of wounds by creating a favorable wound microenvironment. HCIO is well known as a broad-spectrum, fast acting antimicrobial agent, which reinforced by low pH and high ORP contributes to the prevention and treatment of skin infections. RLF-TD011 is a self-administered, sprayable solution enabling targeted application while avoiding skin contact and cross-contamination. Wound care remains the cornerstone of treatment for patients with EB, potentially facilitating a rapid and natural wound healing, while minimizing or preventing infections (and thereby reducing the reliance on antibiotics), and avoiding or limiting the chronicization of wounds. 33
RLF-TD011 is intended to prevent or reduce infections and inflammation by modulating the wound microenvironment as it exhibits characteristics conducive to accelerated natural wound healing, including: | • | | Antimicrobial activity: HClO is well-known as a broad-spectrum, fast acting antimicrobial substance naturally produced by our body as part of the innate immune system’s response to infections. We believe using active chlorine compounds such as HClO constitute a viable solution with a lower risk of developing resistance. Pure HClO has been described to be 80-100 times more potent as a germicide than the hypochlorite anion (ClO-). Since the cytoplasmic pH of bacteria is generally higher than that of the external environment, the acid dissociates and releases a proton, thus leading to acidification of the cytoplasm. The combination of low pH and high ORP in RLF-TD011 is expected to reinforce the known antimicrobial activity of HClO that fosters the wound healing process (Mellerio 2010). |
| • | | Anti-inflammatory activity: HClO inhibits NF-kB (Nuclear Factor kappa-light-chain-enhancer of activated B cells), blocking the activation of the inflammatory pathway. Low pH reinforces the anti-inflammatory activity by inhibition of the alkaline-pH-dependent MMP’s activity. |
Skin repair is a complex sequence of events, orchestrated by molecular interactions among different cell populations at the wound site, ensuring the effective restoration of skin homeostasis. However, repeated injury before completion healing leads to excessive inflammation, disruption of the regenerative processes, altered extracellular matrix (ECM) architecture, pathological scarring, fibrosis, and subungual squamous cell carcinoma (SCC) (Nyström and Bruckner-Tuderman 2018). In EB-affected skin, critical wound colonization or infection could trigger an acute inflammatory response. Certain findings also indicate the presence of an intrinsic pro-inflammatory state in RDEB skin (Cianfarani et al. 2017) RLF-TD011 could also be effective in reducing skin inflammation by (i) inhibiting the NF-kB pro-inflammatory pathway, and (ii) irreversibly inactivating the main pro-inflammatory proteases MMP- 2. In 2019, RLF-TD011 was granted Orphan Drug Designation (ODD) by the FDA for the treatment of EB, which qualifies the sponsor of the treatment for certain development incentives, including seven-year marketing exclusivity after FDA marketing approval is received. In February 2023, we announced the first three patients were enrolled in a proof-of-concept, investigator-initiated study to evaluate RLF-TD011 as a treatment for EB (NCT05533866). The primary aim of this study is to assess changes in the skin microbiome before, during and after treatment with RLF TD011. Patients with dystrophic or junctional EB whose wounds are colonized by staphylococcus aureus, pseudomonas aeruginosa or commensal organisms, were treated with RLF-TD011 for eight weeks followed by discontinuation of treatment for four weeks with assessment of their wound microbiome at each stage. As of the date of this annual report, the study has completed enrollment and treatment of patients. The results are expected in mid- to late-2024. Subject to a positive outcome, we intend to engage in consultations with the Food and Drug Administration (FDA). These discussions will aim to finalize and validate our development and regulatory plan, ensuring an efficient path to market approval. Rare Metabolic Disorders PHYSIOMIMIC Technology™, The Physiomimic Technology, used in the PKUdevelopment and manufacturing of our GOLIKE® product line, is our proprietary globally patented proprietary method to engineer amino acids to modify theirtechnology. Through a complex coating process, this technology alters the release and absorption to mimicprofile of amino acids, mimicking the physiological absorption of natural dietary proteins. This technology provides extended-release,unique approach reduces the inherent taste and odor maskingof amino acids and increased absorption.increases their nutritional value compared to standard free amino acids available on the market. Our PKU GOLIKE®Physiomimic Technology intellectual property portfolio consists of two patent families including 3414 pending applications and 5036 granted patents world-wide.worldwide. Patents resulting from these families, if granted, will expire no earlier than 2036 and 2038, respectively, exclusive of any patent term adjustments or extensions or any form ofand other potential market exclusivity. RARE METABOLIC DISORDERS
Phenylketonuria (PKU) Phenylketonuria (PKU) is a rare metabolic disorder that hinders the body’s ability to break downmetabolize the amino acid phenylalanine (Phe), resulting. This deficiency results in a dangerous build-uptoxic accumulation of Phe when patients eat foods containing protein or aspartame. Highto toxic levels, of the Phe can lead topotentially inducing severe symptoms,systemic damage, with some being irreversible, including: permanent cognitive disorders and intellectual disability; behavioral, emotional and social problems, and psychiatric disorders; a musty odor in the breath, skin or urine, caused by too much Phe in the body;urine; neurological problems, which may include seizures; 34
fair skin and blue eyes, because phenylalanine can’t transform into melanin, the pigment responsible for hair and skin tone;
abnormally small head (microcephaly); and hyperactivity,hyperactivity.
In PKU patients, the enzyme needed to convertSince Phe is missing or severely reduced, which can resultfound in high build-upa wide array of Phe foods, including chicken, meat, eggs, dairy products, nuts, grains, and severe systemic damage, mainly in the brain.legumes, individuals diagnosed with PKU are prescribed a special diet. Treatment guidelines for these patientsPKU require a life-long strictlifelong, stringent, and very limited,restrictive low protein diet combineddiet. This regimen is supplemented with a Phe-free mix of(or Phe low content) amino acid (AA) supplementation (whichmix, which can representconstitute up to 75 percent of the total daily protein intake).
A further reduction ofintake. While PKU is not curable, an affected newborn can grow up with a normal brain development if diagnosed early enough by managing and controlling Phe levels is important before conception: pregnant women with PKU, including those with less severe forms of the disease, may place their unborn children at risk by not following the PKUthrough a strict diet. Children of woman with untreated PKU may have an unusually small head (microcephaly), congenital heart disease, developmental abnormalities or facial abnormalities. There is a strong relationship between the severity of these symptoms and high Phe levels in the mother.
38
Diagnosis and Incidence
Diagnosis of PKU in Europe and the U.S. is systematically conducted on each and everyall newborns through mandatory newborn through the new-bornscreening (NBS) programs which are mandatory in those countries. When diagnosed,programs. Diagnosed newborns are referred to specialized referral centers/hospitalscenters trained to deal with suchat managing rare metabolic disorders. According to a study published in August 2020 in the American Journal of Human Genetics,, approximately 450,000 people suffer from PKU worldwide. In the U.S., approximately 17,500 people are living with PKU and, annually, about 350 newborns are diagnosed with this condition. Current Treatment Options for PKU
While PKU is not curable, if diagnosed early enough, an affected newborn can grow up with normal brain development by managing and controlling Phe levels through a strict diet combined with AA supplementation or with available medications. Diet is composed by few amounts of natural food (based on severity of the disease) supplemented with AA mix with absence or low Phe content plus low protein foods. Diet is recommended for the entire life since it has been demonstrated that high Phe levels has an impact not only during growing but also in adulthood. In 2018, the FDA approved an enzyme substitute called pegvaliase, sold by BioMarin Pharmaceuticals under the brand name PALYNZIQ®(pegvaliase-pqpz). PALYNZIQ®is a derivative of the enzyme Phe ammonia-lyase that metabolizes Phe to reduce its blood levels (but it is not able to produce tyrosine as the natural enzyme, which still needs to be supplemented). Tetrahydrobiopterin (BH4), a cofactor for the oxidation of Phe, when taken by mouth, is also thought to reduce blood levels of Phe in some people. Along with PALYNZIQ®, BioMarin Pharmaceuticals also markets KUVAN® (sapropterin dihydrochloride) for the treatment of PKU. There are also other amino acid products, sold both by prescription and over the counter, that are marketed toward PKU patients.
PKU GOLIKE® for the Dietary Management of PKU Patients with PKU require supplementation of amino acid (AA)-basedAA-based foods for special medical purposes (FSMPs or medical formula)Medical Foods) to prevent protein deficiency and optimize metabolic control. Many of these FSMPs canHowever, Medical Foods may result in poor dietary compliance due to their taste and odor. Further, the unpleasantoften-unpleasant odor and aftertaste of current AA supplements can become a barrier to social interaction for PKU patients. In addition, proteins needed for normal growth and coming from natural food sources are broken down during the digestion process and are gradually absorbed, keeping blood AA levels sufficient stable over time. However, free-AA mix administered to PKU patients in the form of FSMPs are not comparable to natural proteins because they do not need to be broken down before they are absorbed, resulting in a rapid peak of absorption and rapid decrease of their concentration into the bloodstream.
This rapid peak in blood amino acids following ingestion of FSMPs impairs the ability of the body to process them properly and incorporate them into the body’s own tissue, through a process known as anabolism, resulting in a portion of unprocessed amino acids which are then oxidated and eliminated. This rapid elimination of unprocessed AAs that is associated with traditional FSMPs represents a fundamental unmet need for PKU patients especially during any prolonged fasting period.
In order to compensate for the low levels of AAs during fasting periods, the body is forced to initiate a process called catabolism, where the body will break down lean muscle mass to obtain the AAs or energy it requires.
PKU GOLIKE® products are Phe-free FSMPs (or low Phe content) Medical Foods for both children and adults. Developed with the Relief Therapeutics proprietary, patent-protected Physiomimic Technology™ drug delivery platform, PKU GOLIKE® productsadults and are the first prolonged-release AA FSMPs,Medical Food. They are characterized by a special coating that ensures a physiological absorption of the AAs mirroring that of natural proteins.proteins’ absorption profile. The special coating also masks the unpleasant taste, odor and aftertaste of the AAs. PKU GOLIKE® granules are flavorless and can be mixed with many foods. foods. PKU GOLIKE® products contain all 19 amino acids that people with PKU patients need to maintain neurological and muscular health and isare fortified with 27 essential vitamins and minerals, including ones normally found in protein-rich foods like iron, calcium and vitamin B12.B12 which are normally contained in protein-rich foods. In 2023, Relief released pre-clinical and clinical data on PKU GOLIKE, demonstrating the product’s ability to decrease catabolic events and lower blood Phe levels. Additionally, the data indicated a reduction in gastrointestinal discomfort among patients with phenylketonuria (PKU). We are currently conducting two sponsored, randomized, controlled, studies in PKU patients to demonstrate additional benefits in Phe fluctuations with PKU GOLIKE versus standard free AA products (study numbers GLK-IT-2023 and GLK-UK-2021) and expect to report the results in 2024. We believe these results, if positive, may allow for increased utilization of our PKU GOLIKE products. The PKU GOLIKE® line of products has a life cycle management plan aimed at increasing the variety of available formulations. Today our products are available in convenient packets of flavorless granules (PKU GOLIKE Plus®for ages3-16and ages 16+), medical food bars (PKU GOLIKE BAR®)BAR) and tablets to be chewed (PKU GOLIKE KRUNCH®)KRUNCH). Relief Therapeutics plans to expand theWe are also developing PKU GOLIKE® commercial infrastructure beyond products in additional solid and liquid forms based on the current countries and aims to strengthen the commercial activities to increase and accelerate future growth.same technology. PKU GOLIKE® is currently promoted and marketed by a direct sales and marketing infrastructure in U.S., Germany, Italy, Switzerland and Austria; in addition, the product is marketed in the UK, Spain, Israel and Ecuador by local distributors under contract with APR. 39
PKU GOLIKE® products have been commercially available in Europe since 2018. Relief Therapeutics launched2018 and in the PKUU.S. since October 2022.
Following our business-to-business strategy, we granted Eton Pharmaceuticals, Inc. an exclusive license on March 21, 2024, for the commercialization of the GOLIKE® family of products in the U.S.United States. We are actively pursuing similar licensing arrangements for the commercialization of GOLIKE in late October 2022, with its recently assembled commercial infrastructure and team. In early 2023, the company announced the U.S. and EU availability of the new red fruit and tropical fruit flavored PKU GOLIKE BAR®, which contain real fruit flavors, from natural ingredients. More flavors of the bars and other forms of PKU GOLIKE® are currently in development. On August 23, 2022, APR was issued U.S. patent number 11,419,837, which covers certain formulations of PKU GOLIKE® and supplements the PKU GOLIKE® intellectual property portfolio, which includes U.S. patent number 10,500,180, which was issued on December 10, 2019. The patents will expire no earlier than September 27, 2036, with the payment of all prescribed maintenance fees.
In March 2023, Relief Therapeutics presented the findings from pre-clinical research evaluating the metabolic impact of PKU GOLIKE® on nitrogen balance, muscle strength and glucose in a poster session at the Society for Inherited Metabolic Disorders (SIMD) 44th Annual Meeting. The poster summarized the acute and long-term metabolic effects of PKU GOLIKE® supplementation on the utilization of AAs and glucose metabolism in a pre-clinical rat model using biomarkers for muscle metabolism, functional muscle performance and a glucose tolerance test. Due to the prolonged release of the AAs, beneficial effects were observed on AA oxidation, muscle metabolism, grip strength and glucose tolerance in healthy rats. BUN (blood urine nitrogen test) was significantly lower in the acute treatment with PKU GOLIKE® indicating the potential to improve AA utilization in PKU patients resulting in a reduction of catabolic episodes. The results from this pre-clinical research demonstrate the important body composition benefits of the physiological absorption of our prolonged-release AA supplement PKU GOLIKE®. Detailed results from this study are available on the Relief Therapeutics website.
Relief Therapeutics plans to expand the PKU GOLIKE® commercial infrastructure beyond the current countries to increase and accelerate future growth. This will be supported by newer formulations of PKU GOLIKE®.
In the U.S., PKU GOLIKE® was granted ODD by the FDA for its development as a prescription drug. We have decided not to pursue this program and will continue to market our PKU GOLIKE® line of products solely as FSMPs.key European markets.
RLF-OD032 (formerly known as APR-OD32) for the Treatment of PKU ThroughRLF-OD032 is a definitive agreementnovel liquid formulation of a Sapropterin dihydrochloride product in oral suspension to reduce blood phenylalanine (Phe) levels in adult and pediatric PKU patients. If approved, RLF-OD032 would be the first and only liquid formulation of a Sapropterin dihydrochloride product.
Sapropterin dihydrochloride is a pharmaceutical version of the tetrahydrobiopterin (BH4) molecule. It enhances phenylalanine hydroxylase (PAH) enzyme activity in Sapropterin-responsive PKU patients and, in conjunction with Meta Healthcare Ltd. (Meta), dietary management, helps lower blood Phe concentrations. It has been widely demonstrated that increased Phe tolerance and reduced Medical Food requirement improves patients’ stress of a strict diet and quality of life. We believe there remains a significant unmet need to provide additional benefits to PKU patients. The large volume of solid products needed to be consumed daily by patients and the need to tailor treatment quantities based on patient’s weight render the treatment challenging, especially in the pediatric population, thereby affecting patient compliance. If approved, our liquid suspension product may improve patients’ acceptance and compliance by reducing the amount of drug product that must be consumed compared to other generic versions of Sapropterin dihydrochloride. Low volume and no mixing requirement make RLF-OD032 a more convenient administration form compared to the existing dosage forms and would be administered orally via a metered syringe, thereby offering significant improvement in the management of PKU in newborns, children and adults. 35
Relief Therapeutics has acquired theRLF-OD032 worldwide rights, title and interest, except in the UK, from Meta Healthcare Ltd in 2022. We have since developed RLF-OD032’s formulation for a novel dosage formclinical and potential commercial use and are preparing the initiation of a prescriptionPilot PK Trial in mid-2024. Upon completion of a Pivotal PK Trial, we expect to file an 505(b)(2) NDA with the FDA. If approved, we intend to divest or out-license this product. Tyrosinemia and Homocystinuria Tyrosinemia (TYR) TYR is a genetic disease that affects the metabolism of Tyrosine. It is classified into three distinct forms, each caused by deficiencies in different enzymes involved in the metabolism of tyrosine: Type I (TYR1): This form results from a deficiency in the enzyme fumarylacetoacetate hydrolase, leading to liver failure and hepatocellular carcinoma. The worldwide incidence is 1:100,000, with screening available only in some countries. Type II (TYR2): Caused by a deficiency in tyrosine aminotransferase, this form may result in mental retardation, herpetiform corneal ulcers, and skin hyperkeratotic lesions. Its incidence is less than 1: 250,000, with screening available only in some countries. | • | | Type III (TYR3): This extremely rare form results from a deficiency in 4-hydroxyphenylpyruvate dioxygenase, and its symptoms include intermittent ataxia, without hepatorenal involvement, corneal ulcers, or skin lesions. |
In 2002, the orphan drug alreadyNitisinone (NTBC) was approved byin EU and the FDA and intendedU.S. for the treatment of patientsTYR1. While NTBC treatment significantly increases plasma tyrosine concentrations, it requires a complementary diet restricted in Tyrosine and Phe and should not be administered on its own. Importantly, this drug is not effective for TYR2 and TYR3, for which a low-protein diet remains the standard of care with a protein intake similar to those for PKU. Homocystinuria (HCU) Classical homocystinuria is an inherited genetic disorder resulting from mutations in the cystathionine beta synthase (CBS) gene, impairing the body’s ability to metabolize the amino acid homocysteine (Hcy), crucial for several metabolic processes. This improved productdeficiency in the CBS enzyme leads to elevated levels of Hcy. Thus, affected individuals may manifest symptoms ranging from mild to severe, impacting the ocular, skeletal, vascular and central nervous systems. Its prevalence is expectedapproximately 1 in 200,000 to increase patient acceptance335,000 worldwide, and compliance as well as provide easy metered dosing and dispensing. Meta will provide the technology transfer package, while Relief Therapeutics will conduct clinical studies, manufacturing, regulatory submission and commercialization1 in 100,000 to 200,000 in the U.S. However, its prevalence may be higher due to poor detection rates in newborn screening. The treatment for HCU varies based on the patient’s responsiveness to pyridoxine (vitamin B6), leading to its classification into two main types: (i) Pyridoxine responsive homocystinuria, and EU.(ii) Pyridoxine non-responsive homocystinuria. For the latter, dietary management is similar to those for PKU patients and is based on a low protein diet supplemented with a methionine-free amino acid mix. AccordingGOLIKE® for the Dietary Management of TYR and HCU
TYR and HCU require lifelong diets with significant compliance challenges, often due to the termspoor palatability of AAs and the purchase agreement, Meta will transfersuboptimal nutritional value from the fast absorption of standard products. Given the limited range of products available for these rarer diseases, GOLIKE can offer substantial benefits to Relief Therapeutics allpatients. TYR GOLIKE products are Phe-free and Tyrosine-free (or Phe and Tyrosine low content) Medical Foods. data, know-how, asHCU-GOLIKE well as any intellectual property asproducts are Methionine (Meth) free (or Meth low content) Medical Foods. Both product lines are developed or generated so far by Meta. Relief Therapeutics will only be responsible for fundingwith our Physiomimic Technology™ drug delivery platform and intend to address both children and adults’ dietary needs. We anticipate the remaining development work as well as for filing and prosecuting NDA (or equivalent thereof) in all countries worldwide exceptregulatory completion of GOLIKE for the UK, where we will grant a license back to Meta, enabling Meta to market the productdietary management of TYR in that country. Other than the initial acquisition payment2025 and low double-digit royalty payments on net profits of the productHCU in the various countries, we2026. If approved, these products will be under no obligation to fund or pay any other amount to Meta.commercialized through licensees. Relief Therapeutics anticipates filing for registration approval in the U.S. and Europe through a 505(b)(2) NDA sometime in the second half of 2024.
ACER-001 / OLPRUVA™ (SODIUM PHENYLBUTYRATE) FOR ORAL SUSPENSION
In March 2021, Relief Therapeutics signed a collaboration and license agreement with Acer Therapeutics Inc. (Acer) for the worldwide development and commercialization of ACER-001 (sodium phenylbutyrate) for the treatment of various inborn errors of metabolism, urea cycle disorders (UCDs) and maple syrup urine disease (MSUD).
40
ACER-001 is a proprietary, coated powder formulation of sodium phenylbutyrate (NaPB) designed to be both taste-masked and immediate release. ACER-001 was developed using a multiple coating process, and the microparticles consist of an inert core center, a coated layer of active drug and a final taste-masking coating that quickly dissolves in the stomach to avoid a bitter taste while still allowing for rapid systemic absorption. ACER-001’s taste-masked formulation is designed to improve the palatability of NaPB and could make it a compelling alternative to existing NaPB-based treatments, as the unpleasant taste associated with NaPB is cited as a major impediment to patient compliance with those treatments. Additionally, bioequivalence trials have shown ACER-001 to have similar relative bioavailability to BUPHENYL® under both fasted and fed conditions, along with significantly lower projected pricing compared to RAVICTI®.
On December 22, 2022, the FDA approved ACER-001 under the brand name OLPRUVA™ (sodium phenylbutyrate) for oral suspension as a prescription medicine as an adjunctive treatment for use in certain patients with UCDs.
Urea Cycle Disorders (UCDs) UCDs are a group of rare, genetic disorders that can cause harmful ammonia to build up in the blood, potentially resulting in brain damage and neurocognitive impairments, if ammonia levels are not controlled. Any increase in ammonia over time is serious. Therefore, it is important to adhere to any dietary protein restrictions and have alternative medication options to help control ammonia levels. The urea cycle is a series of biochemical reactions that occur primarily in the liver, which converts toxic ammonia produced by the breakdown of protein and other nitrogen-containing molecules in the human body into urea for excretion. Primary hyperammonemia is a term to describe an elevation of ammonia in blood or plasma due to a defect within the urea cycle, which is the pathway responsible for ammonia detoxification and arginine biosynthesis. Urea cycle disorders (UCDs)UCDs are rare diseases caused by genetic defects affecting any of the six enzymes or two transporters that are directly involved in the urea cycle function. The clinical situation is variable and largely depends on the time of onset. Newborns who 36
are often affected by hyper-ammonaemic encephalopathy carry a potential risk of severe brain damage, which may lead to death. Outside the neonatal period, symptoms are very unspecific but most often neurological (with wide variability), psychiatric and/or gastrointestinal. Early identification of patients is extremely importantessential to start effective treatment modalities immediately. The acute management includes detoxification of ammonia, which often requires extracorporeal means such as hemodialysis, and the use of intravenous drugs that work as nitrogen scavengers. Long-term management of patients with UCDs consists of a low-protein diet, which needs to be balanced and supplemented to avoid deficiencies of essential amino acids, trace elements or vitamins and the use of nitrogen scavengers. In cases where dietary management or medication is not effective, patients with UCD may require a liver transplant. Diagnosis and Incidence
The diagnosis of UCDs is based on clinical observations, confirmed by biochemical and molecular genetic testing. A plasma ammonia concentration of 150 µmol/L or higher associated with a normal anion gap and a normal plasma glucose concentration is an indication for the presence of UCDs. Plasma quantitative amino acid analysis and measurement of urinary orotic acid can distinguish between the various types of UCDs. A definitive diagnosis of UCDs depends on either molecular genetic testing or measurement of enzyme activity. Molecular genetic testing is possible for all urea cycle defects. Studies suggest the incidence of UCDs in the U.S. and Europe is 1 in 35,000 live births. Approximately 2,1001 in 100,000 people have UCD, and there are an estimated 800 patients suffer from UCDswho are actively treated in the U.S.
OLPRUVA offers benefits over other UCD treatments by eliminating issues with palatability, offering improved portability with its single-dose envelopes, and it comes in a dosage that is personalized to the patient based on weight. OLPRUVA® (sodium phenylbutyrate)for oral suspension OLPRUVA is a proprietary and novel formulation of sodium phenylbutyrate powder, packaged in pre-measured single-dose envelopes, that has shown bioequivalence to existing sodium phenylbutyrate powder but with a pH-sensitive polymer coating that is designed to minimize dissolution of the coating for up to five minutes after preparation. OLPRUVA was approved in the U.S by the Food and Drug Administration in December 2022 as an adjunctive therapy for the long-term management of UCDs involving deficiencies of carbamylphosphate synthetase (CPS), ornithine transcarbamylase (OTC), orargininosuccinic acid synthetase (AS). OLPRUVA is currently marketed in the U.S. by Acer Therapeutics, Inc. (Acer), a wholly owned subsidiary of Zevra Therapeutics, Inc. (NASDAQ: ZVRA). Under the terms of our contractual agreement with Acer, we are entitled to a 10% continuing royalty on the net sales of OLPRUVA in the U.S., up to a cumulative amount of USD 45 million. Additionally, we hold exclusive development and commercialization rights of OLPRUVA within Europe. The commercialization of OLPRUVA in Europe through partnerships is contingent on evidence of commercial viability and the performance of a bridging PK study for regulatory purposes. Current Treatment Options for UCDs The current treatment of UCDs consists of dietary management to limit ammonia production in conjunction with medications that provide alternative pathways for the removal of ammonia from the bloodstream. Dietary protein must be carefully monitored, and some restriction is necessary; too much dietary protein causes excessive ammonia production. However, if protein intake is too restrictive or insufficient calories are consumed, the body will break down lean muscle mass to obtain the amino acids or energy it requires, which can also lead to excessive ammonia in the bloodstream. Dietary management may also include supplementation with special AA formulas developed specifically for UCDs, which can be prescribed to provide approximately 50 percent of the daily dietary protein allowance. Some patients may also require individual branched-chain amino acid supplementation.
Medications for UCDs primarily comprise nitrogen scavenger drugs, which are substances that provide alternative metabolic excretion pathways for nitrogen, thereby bypassing the urea cycle. The use of these alternative pathways for nitrogen removal is important for the management of acute episodes of hyperammonemia and are also included as part of a long-term treatment regime for UCD patients. Current nitrogen scavenger treatments for UCDs are based on sodium benzoate or phenylbutyrate, which conjugate with glycine and glutamine, respectively, allowing for urinary excretion of nitrogen as hippurate and phenylacetylglutamine, respectively.
41
According to a 2016 study by Shchelochkov et al., published in Molecular Genetics and Metabolism Reports, while nitrogen scavenging medications are effective in helping to manage UCD, non-compliance with treatment is common. Reasons given for non-compliance include the unpleasant taste associated with available medications, the frequency with which medication must be taken and the high cost of the medication.
Sodium phenylbutyrate (NaPB) is currently approved in the U.S. and the EU to treat patients with UCDs, which is marketed as BUPHENYL® (sodium phenylbutyrate) Tablets, BUPHENYL® (sodium phenylbutyrate) Powder and RAVICTI® (glycerol phenylbutyrate) Oral Liquid. While a study provided by Horizon Therapeutics, Inc. in the RAVICTI® package insert involving 46 adults with UCD demonstrated that BUPHENYL® and RAVICTI® were similarly effective in controlling the blood level of ammonia over a 24-hour period, many patients who take their medicine orally prefer RAVICTI®, as it is significantly more palatable than BUPHENYL®. However, the very high annual treatment cost of RAVICTI®, based on patient weight, is often prohibitive. RAVICTI® and BUPHENYL® are registered trademarks owned by or licensed to Horizon Therapeutics plc. Phenylburate is also marketed in the U.S., Europe, Australia and New Zealand under the trade name PHEBURANE® (sodium phenylbutyrate) Oral Pellets. AMMONAPS (sodium phenylbutyrate) Tablets, another formulation of NaPB that claims to be tasteless and odor free is approved and marketed in Europe. Rationale for OLPRUVA™ (formerly ACER-001) Treatment in UCDs
Two Phase 1 bridging studies that evaluated the bioavailability and bioequivalence of OLPRUVA™ (sodium phenylbutyrate) compared to sodium phenylbutyrate (BUPHENYL®) powder showed the bioequivalence of OLPRUVA™ administered as a suspension with sodium phenylbutyrate (BUPHENYL®) powder administered as a solution in healthy adult volunteers after a single dose under fasting and fed conditions (Steiner 2022).
Additionally, two Phase 1, open-label, repeated measures, taste assessment studies of polymer-coated sodium phenylbutyrate (OLPRUVA™) and sodium phenylbutyrate (BUPHENYL®) powder, conducted on healthy panelists who were required to complete a training program for a minimum of six months that educated panelists on the identification, description and quantification of sensory attributes of products, concluded that OLPRUVA™ had overall lower flavor intensity scores than sodium phenylbutyrate (BUPHENYL®) powder when administered within five minutes of preparation (Cedarbaum 2022).
Currently approved therapies for UCDs, including BUPHENYL® and RAVICTI®, are required to be administered with food. BUPHENYL is required to be administered in a fed state due to its aversive odor and taste, with side effects including nausea, vomiting and headaches, which can lead to discontinuation of treatment. Additionally, prescribing information states that the BUPHENYL food effect is unknown. RAVICTI PK and pharmacodynamic (PD) properties were determined to be indistinguishable in fed or fasted states. OLPRUVA™ is uniquely formulated with its multi-particulate, taste-masked coating to allow for administration in a fasted state, while still allowing for rapid systemic release.
Based on the results from the food effect study within the first OLPRUVA™ BE trial, Acer commissioned Rosa & Co. LLC to create a PhysioPD PK model to evaluate the potential food effect on exposure, tolerability and efficacy of OLPRUVA™ in UCDs patients. Results from this in silico model suggested that administration of OLPRUVA™ in a fasted state required approximately 30 percent less PBA to achieve comparable therapeutic benefit to that in a fed state. In addition, the model predicted that administration of OLPRUVA ™ in a fasted state compared to administration of BUPHENYL® or RAVICTI® (same amounts of PBA) in their required fed states would be expected to result in higher peak blood PBA, PAA and PAGN concentrations, which should achieve a 43 percent increase in urinary PAGN levels (a negative correlation between blood ammonia area under the curve and 24-hour urinary PAGN amount has been demonstrated).
42
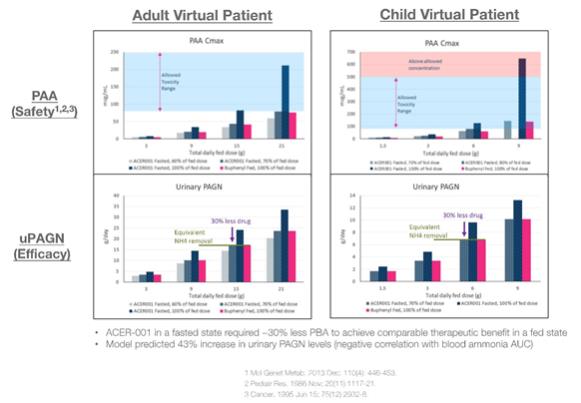
In February 2021, Acer announced topline results from its bioequivalence trial in which OLPRUVA™ (formerly ACER-001) showed similar relative bioavailability to BUPHENYL® (sodium phenylbutyrate) under fed conditions. The single-center, single-blind, randomized, single-dose crossover trial evaluated BE of OLPRUVA™ compared to BUPHENYL® when administered under fed conditions in 36 healthy adults. The topline data from this trial showed OLPRUVA™ to have similar PK profiles for both PBA and PAA compared to BUPHENYL® under fed conditions.
Registration Plan and FDA Approval of OLPRUVA™ (Sodium Phenylbutyrate) for Oral Suspension for UCDs
On December 22, 2022, the FDA approved ACER-001 under the brand name OLPRUVA™ (sodium phenylbutyrate) for oral suspension as a prescription medicine for use with certain therapy, including changes in diet, for the long-term management of adults and children weighing 44 pounds (20 kg) or greater and with a body surface area (BSA) of 1.2 m2 or greater, with UCDs, involving deficiencies of carbamylphosphate synthetase (CPS), ornithine transcarbamylase (OTC) or argininosuccinic acid synthetase (AS). OLPRUVA™ is not used to treat rapid increase of ammonia in the blood (acute hyperammonemia), which can be life-threatening and requires emergency medical treatment. Please see Important Safety Information and full Prescribing Information, including Patient Information.
On March 15, 2023, Acer and Relief Therapeutics announced that, in support of a planned launch for commercial OLPRUVA™ in the third quarter of 2023, that Acer is actively adding resources to establish its commercial and medical affairs presence in the U.S. Acer has recently introduced its patient support service, OLPRUVA™ Navigator by Acer Therapeutics, designed to support UCD patients with support, access, education and patient adherence to treatment. Representatives are expected to begin accepting prescriptions late in the second quarter of 2023. Acer also reported that it is actively engaged in negotiations regarding access for OLPRUVA™ with the major commercial payers and state Medicaid organizations.
According to Acer, it has established a pricing strategy reflecting its commitment to deliver innovative treatments that are responsibly priced and accessible to those in need. Acer intends to price OLPRUVA™ competitively, at a significant discount to the currently available commercial product RAVICTI®, while implementing predictable pricing that will not increase beyond the rate of inflation. Acer indicated that it also plans to invest a portion of OLPRUVA™ revenue back into additional solutions aimed at improving outcomes for UCD patients.
43
We intend to seek approval from the European Medicines Agency (EMA) in the EU and potentially other territories outside the U.S. Because the FDA has approved an NDA for BUPHENYL®, which is referred to as the reference listed drug (RLD), we intend to rely on the RLD’s preclinical and clinical safety and efficacy data, while supplementing the data with a bridging study that shows similar relative bioavailability of OLPRUVA™ to BUPHENYL®. In parallel or after initial potential FDA approval for administration under fed conditions, and subject to additional capital, we also plan to evaluate potential development of OLPRUVA™ for administration under fasted (pre-meal) conditions, which will likely require additional nonclinical and clinical studies to provide the necessary evidence of safety and efficacy of OLPRUVA™ to be considered for FDA approval for administration under fasted (pre-meal) conditions.
Maple Syrup Urine Disease (MSUD)
Maple syrup urine disease (MSUD) is a rare inherited disorder caused by defects in the mitochondrial branched-chain ketoacid dehydrogenase complex, which results in elevated blood levels of the branched-chain amino acids (BCAA), leucine, valine and isoleucine, as well as the associated branched-chain ketoacids (BCKA) in a patient’s blood. Left untreated, this can result in neurological damage, mental disability, coma, or death. The most severe presentation of MSUD, known as “classic” MSUD, accounts for 80 percent of cases and can result in neonatal onset with encephalopathy and coma. Although metabolic management of the disease is possible via a highly restrictive diet, the outcome is unpredictable, and a significant portion of affected individuals are mentally impaired or experience neurological complications.
Diagnosis and Incidence of MSUD
MSUD is typically diagnosed at birth via newborn screening. Studies indicate that MSUD affects an estimated 1 in 185,000 infants worldwide. The disorder occurs more frequently in the Old Order Mennonite population, with an estimated incidence of about 1 in 380 newborns, and the Ashkenazi Jewish population, with an estimated incidence of 1 in 26,000. Approximately 3,000 patients suffer from MSUD worldwide, of whom approximately 1,000 are located in the U.S.
Current Treatment Options in MSUD
There are currently no approved pharmacologic therapies in the U.S. or the EU for MSUD. Treatment of MSUD consists primarily of a severely restricted diet to limit the intake of BCAA, with aggressive medical interventions when blood-levels of BCAA or BCKA become elevated.
Rationale for OLPRUVA™ Treatment in MSUD
Therapy with NaPB in UCD patients has been associated with a selective reduction in BCAA despite adequate dietary protein intake.
Based on this clinical observation, investigators at Baylor College of Medicine (BCM) explored the potential of NaPB treatment to lower BCAA and their corresponding BCKA in patients with MSUD. The investigators found that BCAA and BCKA were both significantly reduced following NaPB therapy in control subjects and in patients with MSUD, although there was no simple correlation between the patients’ levels of residual enzymatic activity with the response of plasma BCAA and their BCKA to NaPB. NaPB showed a statistically significant reduction of BCAA leucine, in all three healthy subjects and in three out of the five MSUD patients who participated in the trial. The reduction in leucine, the most toxic of the BCAAs, in the three responsive MSUD patients ranged between 28-34 percent, which is considered by clinicians to be a clinically meaningful response.
Investigators at BCM further explored the mechanistic rationale for NaPB lowering BCAA/BCKA levels. NaPB was found to be an allosteric inhibitor of the branched-chain keto acid dehydrogenase complex kinase (BCKD-kinase), and enzyme that regulates the activity of the branched-chain keto acid dehydrogenase complex (BCKDC) enzyme that is responsible for the normal metabolism of BCKAs. By inhibiting the BCKD-kinase, the BCKDC is constitutively activated, thus the increased activity results in a reduction in the plasma levels of BCAA and BCKA in all people, including those with MSUD, suggesting that NaPB may be an effective treatment for people with MSUD, who experience elevated BCAA levels.
In November 2020, study results evaluating the effect of NaPB in the management of acute MSUD attacks in pediatric patients (n=10) were published in the Journal of Pediatric Endocrinology and Metabolism showing a significant reduction in leucine levels in MSUD patients experiencing an acute attack. However, verifying this outcome would require additional validation in a controlled trial. If OLPRUVA™ is approved for the treatment of chronic MSUD, we believe patients will not be required to interrupt their therapy in the event of an acute crisis.
44
Registration Plan for MSUD
Acer has submitted an investigational new drug (IND) application to the FDA to evaluate the safety and efficacy of OLPRUVA™ for the treatment of MSUD, and although there can be no assurance, we expect that Acer will begin such study during the first quarter of 2023. Given its regulatory status with regard to UCDs, there is no requirement for Phase 1 studies in healthy volunteers. The timing of a Phase 2 clinical trial, if such a trial occurs, would be subject to completion of a commercial assessment of the opportunity, including, but not limited to, a possible pre-IND meeting with the FDA and/or the EMA, with an objective of validation and agreement on the primary and secondary clinical trial end-points, which remain important given that there are presently no approved treatment options for this disease nor are there guidelines to assess efficacy and safety of an investigational drug in this disease. If a successful clinical trial for OLPRUVA™ in MSUD is completed, along with our partner Acer, we plan to seek FDA approval to market OLPRUVA™ for the treatment of MSUD as an added indication in the U.S. by submitting a supplemental NDA (sNDA) incorporating the efficacy and safety data from the MSUD population, assuming OLPRUVA™ is approved for the treatment of UCDs prior to sNDA submission. We also intend to seek approval in the EU and other territories outside the U.S. after the sNDA for treatment of MSUD is filed, or simultaneously with the U.S. filing.
Royalty Sharing for OLPRUVA™
The companies will split net profits from OLPRUVA™ from Acer’s territories (consisting of the U.S., Canada, Turkey, Japan and Brazil) 60 percent to 40 percent in favor of Relief Therapeutics. In addition, Relief Therapeutics has licensed the rights for the rest of the world, where Acer will receive from Relief Therapeutics a 15 percent royalty on all net revenues received in the Relief Therapeutics territories.
RARE PULMONARY DISEASES
RLF-100® (AVIPTADIL ACETATE) (Aviptadil acetate) Aviptadil acetate is a synthetic form of vasoactive intestinal peptide (VIP) consisting of 28 amino acids which was first discovered in 1970. That same year, Nature published a short report entitled “Potent It is an abundant biologically active peptide endogenous in humans as well as in other species. It is produced by neurons in the peripheral and splanchnic vasodilatorcentral nervous system, by endocrine cells like the pituitary lactotrophs, cells of the endocrine pancreas as well as T-lymphocytes, and B-lymphocytes. This natural peptide from normal gut,” authored by two young scientists working atis one of the Karolinska Institute (Said, Mutt, 1970)signal molecules of the neuroendocrine-immune network comprising anti-inflammatory, immunosuppressive, anti-proliferative, and vasodilating features (Mukherjee et al, 2021). Although named (or mis-named) for the intestinal tissue in which it was first isolated, human VIPIt is now known to be produced by neuroendocrine cells throughout the body by T-lymphocytes, B-lymphocytes and macrophages, primarily concentrated in the lungs. VIP is highlypredominantly localized in the lung (Leys 1986, Virgolini 1995) but islungs and a widely distributed that showed, in various models, effects in hemodynamics and coronary circulation (Feliciano 1998, Frase 1987, Henning 2001), kidney (Dimaline 1983, Calam 1983 and 1988), immune system (Gonzales-Rey 2007, Ganea 2015, Li 2013), intestinal tract (Iwasaki 2019) and reproduction (Fredericks 1983, Fraccaroli 2012). VIP has a multimodal mechanismvast body of action: decrease of inflammatory cytokines release leadingexperimental, pharmacological as well as clinical evidence suggests Aviptadil to prevention of cytokine storm syndrome and viral replication, immunomodulating effect, vasodilating and bronchodilating effects and prevention of surfactant depletion. Seventy percent of VIP in the body is bound to a less common type of cell in the lung, the alveolar epithelial type II (AT2) cell, which is critical to the absorption of oxygen into the body. Five decades of subsequent research documented VIP’s rolebe an attractive candidate as a potent natural anti-cytokine that has unique capability to block pathways of cell death in ATII cells – the cell targeted by the SARS-CoV-2 virus.treatment option for pulmonary disorders.
Since RLF-100’s mechanism of action is not restricted to the protection of AT2 cells, we believe that its beneficial effects could extend to other types of acute lung injury (ALI) as supported by pre-clinical and preliminary clinical data in sepsis-induced ALI. It is our objective to establishWe have developed our proprietary and patent-protected formulation of aviptadil,stable Aviptadil formulations (codenamed RLF-100RLF-100),® intended for intravenous (IV) and inhaled administration as the standard of care for the prevention and treatment of respiratory failure and its complications in both the acute intensive care and chronic ambulatory settings.
In April 2023, Relief Therapeutics announced positive 12-month Based on the latest Aviptadil stability data for the liquidresults, we filed a US Patent Application and lyophilized preparations of RLF-100®, intended for intravenous (IV) and inhaled administration. RLF-100® is the company’s proprietary, patent protected investigational formulation of aviptadil acetate. The data from the stability study showed that both inhaled and IV RLF-100® demonstrated high purity levels at 12 months at all temperatures tested, including refrigerated and room temperature environments. The results are consistent with prior data observed at three- and six-month intervals. The stability testing study will continue to determine the maximum shelf life of RLF-100®.a PCT Patent Application.
4537
Based on the latest results, Relief Therapeutics intendsAs we continue to amend its previously filed provisional patent application for RLF-100®explore Aviptadil’s applicability, with the new findings. If granted, this patent could provide exclusivity for RLF-100® at least until 2042, without considering Hatch-Waxman extensions or other patent term adjustments. The Hatch-Waxman Act permits a patent extension term of up to five years as compensation for patent term lost during the FDA regulatory review process.
Relief Therapeutics intendsboth IV and inhaled formulations, we see opportunity to develop the intravenous (IV) formulation of RLF-100® for targeted COVID or non-COVIDthis product in acute respiratory distress syndromes (ARDSs) because (i) it allows a prompt and efficient delivery of the drug in severe conditions (e.g., patients in intensive care units (ICUs)) while (ii) keeping under control the possible side effects because administered in a hospital setting and (iii) it remains a dosage form easy to handle by healthcare professionals (HPCs).
At the same time, Relief Therapeutics intends to develop the inhaled formulation of RLF-100®for targetedcertain chronic lung diseases (CLDs) such as, including sarcoidosis, berylliosis and checkpoint inhibitor-induced pneumonitis (CIP) because (i) it can.
It is anticipated that the IV formulation would ensure an efficient and acute delivery of the active compound in severe conditions while the inhalation route would allow to maximize the clinical efficacy directly inbenefit to the target organs (the lungs), (ii)affected organ (lung) while reducingminimizing the sidepotential adverse effects related to the systemic activity of the drug (vasodilator and hypotension) because it is not systemically absorbed and (iii) itAviptadil offering a home-based treatment. ARDS ARDS is a patient friendly dosage form as those CLDs are primarily treated at home and drug administered directly by the patients. COVID-19 ARDS
Acute respiratory failure is the primary cause of death in COVID-19. In some cases, the injury is attributed to cytokine storm – i.e., a massive release of inflammatory cytokines that then cause destruction of pulmonary epithelium cells. However, the cytokine storm is only produced after the SARS-CoV2 virus enters the ATII cell through binding of its spike protein to angiotensin converting enzyme 2 (ACE2) surface receptors (Mason 2020). ACE2 is not present on type I alveolar cells, which comprise 95 percent of the pulmonary epithelium hence those cells are not infected by the coronavirus. Similarly, only the ATII cell expresses the VPAC1 receptor to which VIP binds. VIP is shown to prevent their apoptosis in models of lung injury (Ao 2011, Pakbaz 1993). Hence, VIP represents a highly specific approach to rescuing the lung from the overwhelming failure of oxygenation seen in COVID-19.
Pulmonary drugs are difficult to develop, given regulatory requirements for long-term toxicology studies in multiple species, including primates (Tepper 2016). The FDA has asserted that these preclinical toxicology requirements must be observed in the case of candidate drugs to treat COVID-19. VIP, on the other hand, have successfully completed four species toxicology and safety pharmacology studies in both intravenous (IV) and inhaled dosages. Phase 2 trials in sarcoidosis (Prasse 2010), pulmonary hypertension (Petkov 2003, Leuchte 2008), pulmonary fibrosis (unpublished data) and asthma (Bundgaard 1983, Morice 1983 and 1986, Altiere 1984, Barnes 1984, Crimi 1988, Morice 1986) document that VIP has no major toxicities when inhaled at doses of 300µg/day or infused at dose of 6 pmol/kg/min. VIP was first proposed as a modulator of lung inflammation by Said (Said 1988, 1991). It has demonstrated positive effects indevastating clinical trials of sepsis-related acute respiratory distress syndrome (ARDS) (Youssef 2020 preprint) and sarcoidosis (Prasse 2010).
In March 2020, at the beginning of the first wave of the pandemic in the U.S., our former collaboration partner, NeuroRx Inc. (NeuroRx), submitted an IND application to the FDA for a Phase 2b/3 trial of intravenous (IV)aviptadil for the treatment of patients with critical COVID-19 respiratory failure. Within 24 hours, the FDA issued a “Study May Proceed” letter and the first patients were treated in April 2020 at Thomas Jefferson University Hospital in Philadelphia.
In late 2020, a Phase 2b/3 clinical study with aviptadil acetate IV in patients with COVID-19-induced acute respiratory distress syndrome (ARDS) was completed in the U.S. by NeuroRx. In its press release reporting those results, NeuroRx announced that across all patients and sites, the aviptadil acetate IV treated cohort met the primary endpoint for successful recovery from respiratory failure at days 28 (p=0.14) and 60 (p=0.13) and had a meaningful survival benefit after controlling for ventilation status and clinical site. However, they also reported that the trial did not demonstrate a statistically significant difference on the study’s primary endpoint without statistical adjustment for these pre- specified covariates. Based on these findings, NeuroRx announced on June 1, 2021, the company applied to the FDA for emergency use authorization (EUA) for aviptadil acetate IV for the treatment of acute respiratory failure due to criticalwith progressive arterial hypoxemia, dyspnea, and a marked increase in the work of breathing with a need for mechanical ventilation. It has a rapid onset COVID-19(7-10 and that it planned to submit an NDAdays) with the FDA. On November 5, 2021, NeuroRx announced the FDA declined its application for EUA of aviptadil IV for the treatment of acute respiratory failure due to critical COVID-19. Subsequent applications filed by NeuroRx with the FDA seeking EUA for more limited usea progressive malfunction of the product for the treatment of COVID-19 and for breakthrough therapy designation for the product were also denied in the first half of 2022.
46
In March 2021, NeuroRx announcedlungs that aviptadil acetate IV was included in a National Institutes of Health (NIH)-sponsored Phase 3 ACTIV-3b/TESICO clinical trial in severely ill patients with COVID-19. In May 2022, Relief Therapeutics learned that the ACTIV-3b/TESICO trial was discontinued by its Data Safety Monitoring Board (DSMB) based on futility.
Relief Therapeutics intends to obtain and review all available clinical data, including data from the NIH-sponsored trial to better understand the results observed, up to and including the point at which the study was discontinued.
While regulatory approval for aviptadil acetate IV to treat COVID-19-induced ARDS has not been granted in the U.S., an unrelated pharmaceutical company received approval for this indication in India in early 2022 for their formulation of aviptadil, thereby substantiating Relief Therapeutics’ original hypothesis.
Inhaled RLF-100® is being evaluated in an investigator-initiated trial at a site in Switzerland for the treatmentquickly evolves into respiratory failure. The pathophysiology of ARDS is complex and is associated with COVID-19 (Leuppi/NCT04536350). While the study is in an advanced stage of recruitment, changing disease patterns have hindered the completion of patient recruitment. The lead investigator has reported that top-line data is now expected in the fourth quarter of 2023.
Non-COVID-19 ARDS
Infectious acute respiratory distress syndrome (ARDS) is a potentially life-threatening condition in which the lungs become severely inflamed, leading to buildupextensive lung inflammation and accumulation of fluid in the lungs, preventing oxygenalveoli (air sacs) severely affecting the lung gas exchanging ability. Global awareness of ARDS was heightened during the COVID-19 pandemic due to a sharp increase in its incidence, but ARDS is defined more broadly as a heterogeneous syndrome resulting from getting to the bloodstreamvarious direct or indirect pulmonary insults. ARDS is widely recognized as a major clinical problem worldwide: it globally affects approximately 3 million patients annually, accounting for 10% of intensive care unit (ICU) admissions, and the rest24% of the body. Infectious ARDS results from an injury or an infection (such as pneumonia, severe flu, sepsis, etc.) of the air sacspatients receiving mechanical ventilation in the lung. PlansICU with an estimated mortality rate of approximately 40-60%for depending on disease severity (Battaglini et al. 2021). No approved drug treatment is currently available despite the completion of several clinical trials and ongoing research efforts.
Aviptadil was granted U.S. Food and Drug Administration (FDA) Fast Track Designation for treating critical COVID-19-induced ARDS. It has been recently tested in IV or inhaled forms in several clinical trials: | • | | Phase 2 and 3 studies were conducted with Aviptadil during the COVID pandemic with mixed results A Phase 2b/3 multicenter study did not reach its primary end point but demonstrated a statistically significant two-fold decrease in mortality and a significant improvement in respiratory distress ratio (Youssef et al. 2022). This finding was deemed “hypothesis generating” by the US FDA and insufficient to warrant Emergency Use Authorization. Aviptadil was further evaluated for improving treatment of severely and critically ill COVID-19 patients in the I-SPY COVID-19 trial and the TESICO trial but was withdrawn from these two studies before completion. |
| • | | A Phase 3 randomized, multicentric, double-blind, placebo-controlled, comparative clinical trial (150 participants) with severe COVID-19-induced ARDS conducted in India by an unrelated third-party on a different formulation of Aviptadil reported that Aviptadil was safe and effective in improving the resolution of respiratory failure, shortening the time to recovery, decreasing respiratory distress, and preventing death in respiratory failure patients (Dewan and Shinde, 2022). In comparison to placebo, patients on Aviptadil demonstrated a 2.1-fold increase (p=0.0410) of being free of respiratory failure (no oxygen requirement) at day 3 and a 2.6-fold increase (p=0.0035) at day 7. While this was not our study, its results support the proposition that Aviptadil may be an effective treatment for treating ARDS. |
| • | | A retrospective observational study evaluating Aviptadil in severe viral-related ARDS demonstrated an improvement of clinical outcomes (Sampley et al. 2023). Six patients who developed ARDS after viral pneumonias, have been treated with 3 days of infusion. Mean oxygen saturation significantly improved from 87.86% before the first Aviptadil dose to 93.43% afterward. Similarly, PaO2 values rose from 54.3 to 68.4 post-therapy (p-value < 0.004) and the SpO2/FiO2 ratio from 149 to 336 post-therapy (p-value < 0.003). |
| • | | A Phase 1 trial showed promising results in treating sepsis-related ARDS (JP Youssef et al. 2020). Eight patients under mechanical ventilation were treated with IV Aviptadil for 12 hours. Seven demonstrated a successful course during intensive care and were successfully removed from mechanical ventilation and discharged from intensive care. Of those who were discharged from the ICU, six demonstrated successful 30-day survival and serum levels of TNF decreased in five patients. Hypotension was seen in association with two infusions and diarrhea in association with one but did not necessitate cessation of therapy. |
Additionally, Aviptadil has showed promising results in a recent case series of RLF-100severe ARDS cases with rapid deterioration of clinical conditions (Mehta et al. 2024). There can be no assurance that Aviptadil will ever be approved for commercialization in the treatment of infectious ARDS are in development.ARDS. RLF-100Chronic lung diseases (CLDs)® (AVIPTADIL ACETATE) IN CHRONIC LUNG DISEASES (CLDs)
InhaledWe continue to assess the development of inhaled RLF-100® is under development for targeted CLDs, including pulmonary sarcoidosis, checkpoint inhibitor-induced pneumonitis (CIP) and chronic berylliosis.
Pulmonary sarcoidosis, chronic berylliosis and CIP These indications are generally classified as granulomatous chronic lung diseases because they all havedue to their similar pathogenesis which lead toresulting in the formation of lung granulomas. It is a process driven by an exaggerated immune response, wherewherein the activation of CD4+ Th1 and Th17 cells causesleads to the development of pro-inflammatory cytokine storms and lung granuloma formation.
4738
InhaledWe believe inhaled RLF-100®, which is can bind to the synthetic form of VIP, is supposed to bind its receptor VPAC1 on CD4+ Th1 and Th17 immune cells, thus inhibiting NfKB. ThanksNF-kB (Martinez et al., 2019). NF-kB (Nuclear Factor kappa-light-chain-enhancer of activated B cells) is a protein complex that plays a crucial role in regulating the immune response to infection. It is involved in cellular responses to stimuli such as stress, cytokines, free radicals, ultraviolet irradiation, oxidized LDL, and bacterial or viral antigens.
Due to this specific mechanism of action, RLF-100® is ableexpected to reduce the pro-inflammatory cytokines and increase anti-inflammatory cytokines, thereby preventing granuloma formation and allowingfacilitating disease resolution as detailed in the illustration below. 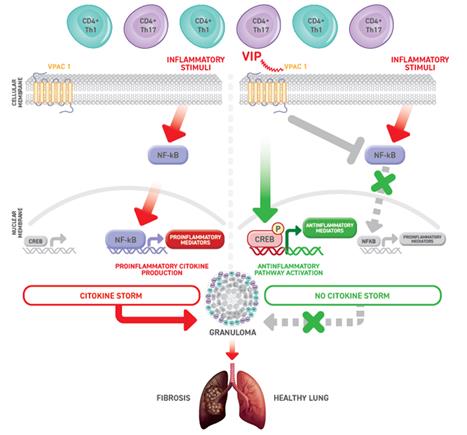 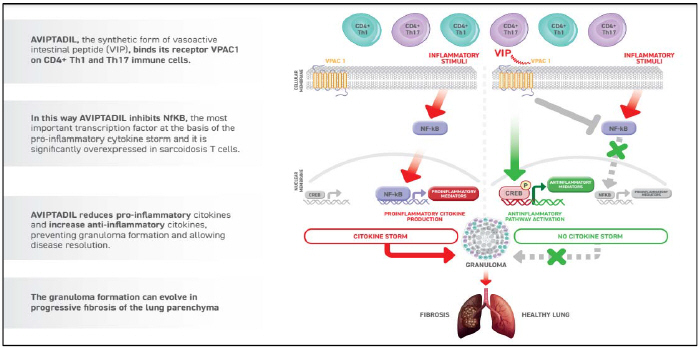
48
Pulmonary Sarcoidosis
Sarcoidosis isWhile these assumptions are pending validation in clinical trials, preliminary observations provide partial support. Notably, clinical evidence on sarcoidosis has been generated in a study on 20 patients treated for four weeks (Prasse et al. 2010). In addition, a Named Patient Program in Germany has yielded encouraging results. Lastly, a case involving an inflammatory disease characterized by the formation of granulomas—tiny clumps of inflammatory cells that can develop in any part of the body. When the disease occurs in the lungs, it is called pulmonary sarcoidosis and is a form of interstitial lung disease (ILD) which are a group of immune-mediated disorders that cause progressive fibrosis of the lung interstitium (the extravascular and extracellular space between cells in tissue).
The granulomas disrupt the intake of oxygen and can cause scarring on the lungs, preventing the lungs stretching fully, and therefore limiting their capacity. The prognosis for patientselderly patient with pulmonary sarcoidosis ranges from benign and self-limiting to chronic, debilitating disease and death. Despite increasing advances in research, pulmonary sarcoidosis remains difficult to diagnose with limited treatment options to manage symptoms and no known cure. According to the Foundation for Sarcoidosis Research, approximately 200,000 Americans live with pulmonary sarcoidosis. Relief Therapeutics was granted ODD by the FDA for inhaled RLF-100® for the treatment of pulmonary sarcoidosis in August 2021.
Checkpoint Inhibitor-Induced Pneumonitis (CIP)
Checkpoint inhibitor-induced pneumonitis (CIP) is a rare, potentially fatal form of lung inflammationCIP demonstrated positive outcome following treatment with immune checkpoint inhibitors (ICIs). ICIs are a type of immune therapy used to treat cancer. CIP can result in cough, dyspnea, fever, chest pain, and in severe cases, lack of oxygen in the lungs (hypoxia) and respiratory distress. The use of inhaled RLF-100® for this indication will be further evaluated to explore whether such use could enhance compliance with chemotherapy and improve outcomes for cancer patients. Relief Therapeutics received a Swiss method-of-use patent protection related to thean inhaled formulation of RLF-100® for the potential treatment of CIP extending into at least 2039.
Berylliosis / Chronic Beryllium Disease (CBD)
Chronic beryllium disease (CBD) is an orphan lung disease caused by the inhalation of beryllium particles, dust or fumes in the workplace, resulting in severe inflammation of the lungs, coughing and increasing breathlessness (dyspnea). CBD is a clinical phenocopy of sarcoidosis. Currently there are no treatments approved for berylliosis. The ex-vivo effect of RLF-100® on mononuclear cells in the setting of CBD is currently being evaluated. Together with the results from the Phase 2b sarcoidosis trial, these results would justify the therapeutic use of inhaled RLF-100® in CBD, providing a rationale for the clinical trial design in this indication.
SENTINOX
Sentinox, is a novel, acid-oxidizing solution containing hypochlorous acid in a nasal spray formulation that was developed by APR. Sentinox was certified in Europe on February 16, 2021, as a Class III medical device (certificate number EPT 0477.MDD21/4200.1). Sentinox is intended for irrigation, cleansing and moistening of the nasal cavities and is indicated to reduce the risk of infections caused by bacteria and viruses, including SARS-CoV-2, by lowering the nasal microbial load; symptomatic nasal care; and nasal care in cases of minor lesions/alterations of the nasal mucosa.
On October 27, 2021, we reported positive interim results from our randomized, controlled clinical trial designed to evaluate the safety and efficacy of Sentinox in reducing viral load in the upper respiratory airways in recently SARS-CoV-2 infected patients. We also reported that data from the study suggest Sentinox could potentially be effective in reducing the SARS-CoV-2 viral load at the level of the nasal mucosa.
On March 17, 2022, Relief Therapeutics and APR reported the final data from APR’s clinical trial of nasal spray, Sentinox, in SARS-CoV-2 infected patients. The post-market, interventional, randomized, controlled clinical study (NCT04909996, clinicaltrials.gov) enrolled 57 patients who were randomized to receive Sentinox treatment 0.5 ml into each nostril, performed three times/day or five times/day for five days as add-on to the standard therapy, vs. no Sentinox treatment group. The study was designed to assess the efficacy and safety of Sentinox spray in terms of viral load reduction, negativization and infectivity in recently infected SARS-CoV-2 individuals. It was conducted by the Hygiene Unit of IRCCS Policlinico San Martino Hospital in Genoa, Italy, and coordinated by Professor Giancarlo Icardi.
Considering the small sample size and the high variability in the baseline viral load observed within study groups, the primary endpoint was not reached; however, the results of the study suggest the potential efficacy of Sentinox in the reduction of the nasal viral load, negativization and infectivity and confirmed its safety and tolerability.
49
Additional analyses have been conducted in patients stratified according to baseline value of RT-PCR cycles: in the subgroup with medium (Ct 20-30) viral load, the use of Sentinox significantly reduced the viral load of 1.9761 Log10 (p=0.0178) at day five compared to the control group, suggesting a positive trend in the treatment effect. Further efficacy analyses on the ITT population showed that negativization in the Sentinox three times/day group started at day four; at day six patients with negative swab were almost two-fold compared to the control group (47 percent in Sentinox group versus 22 percent in no treatment group) (p=0.0005). Similar results were obtained in the analysis conducted in the 20-30 RT PCR cycles subpopulation.
Analysis on infectivity data was also conducted in the ITT population: patients were considered “not infectious” (patient likely not be able to spread virus to others) when the cycle threshold value of >35 cycles was achieved (Carrouel et al. 2021; Jang et al. 2021; Iwanami et al. 2021; ChoudhuriAviptadil (Frye et al. 2020). More recently, a long-term treatment of granulomatosis with inhaled Aviptadil was published in a poster at the 2022 European Respiratory Society (ERS) congress.
We recently developed a new Aviptadil drug product for inhaled administration, based on our stable formulation. In alignment with our development strategy and regulatory requirements, we anticipate completing certain pre-clinical In the three times/day Sentinox group, 71 percent of patients were non-infectious versus 44 percent in the control group at day six (p<0.0001). Overall safety data monitored through clinical examination showed a good safety profile for Sentinox. This has been confirmed also by VAS and LIKERT scale results. Relief Therapeutics initiated a confirmatory, controlled clinical trial in the prevention of viral and bacterial airborne infections in the fourth quarter of 2022. Completion of the clinical trial is subject to availability of capital.
RARE CONNECTIVE TISSUE DISORDERS
NEXODYN® Acid-Oxidizing Solution (AOS)
Nexodyn® Acid-Oxidizing Solution (AOS) was developed using APR’s proprietary, patent protected TEHCLO Nanotechnology® and is a solution of highly pure and stabilized hypochlorous acid (HClO >95% of free chlorine species), acidic pH (2.5 – 3.0) with high reduction-oxidation potential (ORP 1.000 – 1.200 mV). The product is a self-administered sprayable solution with ancillary antimicrobial properties intended for use in the debridement, irrigation, cleansing and moistening of acute and chronic wounds (e.g., diabetic foot ulcers, pressure ulcers and vascular ulcers), post-surgical wounds, burns and other lesions. The product is certified in the EU as a Class III medical device and is certified as a 510(k) medical device in the U.S.
Nexodyn® AOS is proven to restart healing in chronic wounds by creating an ideal microenvironment to sustain the physiological healing process. A wealth of evidence and real-world experience has consistently shown accelerated wound closure with reduced infection rates and less wound-associated pain.
The anti-microbial and anti-inflammatory properties of Nexodyn® AOS, along with its tolerability, could make this an attractive treatment candidate for the management of wounds in epidermolysis bullosa (EB), with the potential to be the only product approved for the control of wound infection in this disease, thereby reducing long term antibiotic use, while assisting wound healing and decreasing wound related pain, all of which would significantly benefit quality of life in patientsdemonstrations with this genetic disorder.
RLF-TD011 (formerly referred to as APR-TD011 or APR-TM011)
RLF-TD011 is a differentiated acid oxidizing solution of hypochlorous acid (HCIO) that combines strong antimicrobial action with anti-inflammatory properties, thereby allowing for infection control, reduction of wound colonization, alleviation of pain and itching and improved wound healing.
Developed with APR’s proprietary, patent-protected TEHCLO Nanotechnology®, RLF-TD011 employs an exclusive combination of three physio-chemical properties—high-purity HCIO, hypotonic low pH and high oxidation-reduction potential (ORP), which is believed to support a faster physiological healing of wounds by creating a favorable wound microenvironment. HCIO is well known as a broad-spectrum, fast acting antimicrobial agent, which reinforced by low pH and high ORP contributes to the prevention and treatment of skin infections.
RLF-TD011 for the Potential Treatment of Epidermolysis Bullosa
RLF-TD011 is an investigational drug candidate with the potential to treat wounds in epidermolysis bullosa (EB) as it is a self-administered, sprayable solution enabling targeted application while avoiding skin contact and cross-contamination. EB, also known as “Butterfly Skin,” is a group of rare, genetic, life-threatening connective tissue disorders characterized by skin fragility and blistering, which may appear in response to minor injury, even from heat, rubbing or scratching. These widely distributed, painful, chronic wounds can easily become infected, resulting in an elevated risk of sepsis and death. A crucial element of patient management involves rigorous and timely wound care.
50
There are four main types of EB, which are classified based on the depth, or level, of blister formation: EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB) and Kindler syndrome. In severe cases, the blisters may develop into chronic wounds or occur inside the body, such as the lining of the mouth or stomach. Patients with JEB and DEB are at increased risk for serious complications, including aggressive squamous cell carcinoma. The National Epidermolysis Bullosa Registry (NEBR) reports, based on 16 years of data, that the incidence of EB in the U.S. is 19.57 per 1 million live births and the prevalence is 11.07 per 1 million population. Worldwide, EB impacts 500,000 lives.
Currently there is no cure or approved treatments for EB in the U.S. RLF-TD011 could represent the first product specifically indicated for EB patients that provides a comprehensive solution to prevent or reduce wound colonization and infection. This, along with its anti-inflammatory action, could provide symptom relief and wound healing. The Company estimates the global market opportunity for EB to exceed USD 1.0 billion.
Subject to clinical demonstration of efficacy and safety in clinical trials, RLF-TD011 could play an important role in the reduction of inflammation by inhibiting the NF-kB pro-inflammatory pathway and, at the same time, may offer a faster wound healing in EB patients and by reducing the itching and pain linked to infections and inflammation. In a preliminary clinical trial, EB patients who administered RLF-TD011 demonstrated improvement in skin blistering and tissue repair within just two weeks of treatment, and the product candidate was shown to be well tolerated with a favorable safety profile.
In late 2019, RLF-TD011 was granted ODD by the FDA for the treatment of EB, which qualifies the sponsor of the treatment for certain development incentives, including seven-year marketing exclusivity after FDA marketing approval is received. Relief Therapeutics intends to seek qualified infectious disease product (QIDP) designation status for RLF-TD011, which may confer up to an additional five years of market exclusivity regardless of patent protection status. Good Manufacturing Practice (GMP) grade product is being prepared forbefore initiating clinical development under an FDA-authorized IND.in CLDs.
In February 2023, Relief Therapeutics announcedAs the first three patients were enrolled in a proof-of-concept, investigator-initiated study to evaluate RLF-TD011 as a treatment for EB (NCT05533866). The primary aim of this studyCompany is focusing on rare dermatological disorders, we will be to assess changes in the skin microbiome before, during and after treatment with RLF-TD011. Patients with dystrophicseeking partnerships or junctional EB whose wounds are colonized by staphylococcus aureus, pseudomonas aeruginosa or commensal organisms will be treated with RLF-TD011 for eight weeks followed by discontinuation of treatment for four weeks with assessment of their wound microbiome at each stage. All study participants will have the optioncollaborations to continue treatment in a six-month open label study extension. Results of this study are expected sometime between the fourth quarter of 2023 and the first quarter of 2024 depending on the enrollment and treatment pace.
RLF-TD011 for Potential Treatment in Oncology Supportive Care
RLF-TD011 is currently approved in Europe as a Class III medical device for the treatment of skin lesions and toxicities induced by cancer treatments, including anti-epidermal growth factor receptors (anti-EGFR) monoclonal antibodies, such as Cetuximab. The use of anti-EGFR inhibitors causes papulopustular manifestations due to their interference of epidermal growth factor receptor (EGFR) signaling in the skin with a high risk of secondary infections.
Following commercial assessment, the company is planning to conduct a follow-on clinical study to renew product approval in Europe as a Class III medical device beyond 2024, when the new EU device regulations will apply. This clinical study will be a multi-center, post-market, double-blinded, placebo-controlled trial to evaluate the efficacy, safety and tolerability of RLF-TD011 in the management of skin lesions and reactions resulting from anti-EGFR monoclonal antibodies and/or radiotherapy treatments in oncology patients. This study would follow a preliminary, proof of concept study completed by the company on 15 patients with head and neck cancer treated for 8-12 weeks with Cetuximab and showing a mean reduction of 94 percent of the lesion area compared to the standard of care
In January 2023, Relief Therapeutics announced that an independent institutional review board (IRB) approved the protocol of an investigator-initiated trial to evaluate RLF-TD011 as an adjunctive treatment for patients diagnosed with cutaneous t-cell lymphoma (CTCL) (NCT05728879). The study will evaluate the effect of RLF-TD011, on the microbiome of CTCL skin lesions and determine tolerability, symptom improvement, and potential for reducing lesion size and skin disease activity.
51
Cutaneous t-cell lymphoma (CTCL) is a rare, heterogeneous group of non-Hodgkin’s lymphomas characterized by abnormal accumulation of malignant t-cells in the skin that can result in the development of rashes, plaques and tumors. Because CTCL is rare and often looks like eczema or another common skin disease, itour formulations for Aviptadil. However, there can be difficult to diagnose. Advanced CTCL lesions harbor staphylococcus aureus, which release toxinsno assurance that stimulate malignant cells and drive disease progression. This often leads to recurrent skin infections with a high riskAviptadil will ever be approved for sepsis and death. Treatment of advanced CTCL remains a challenge, with five-year disease-specific survival rates ranging from 70 percent for early stage to 24 percent for advanced disease, with the greatest mortality stemming from bacterial infections.commercialization.
While there are many types of CTCLs, the most common diagnoses are mycosis fungoides, primary CTCL and primary cutaneous anaplastic large cell lymphoma. The overall incidence rate of CTCL was 8.55 per 1 million with MF being the subtype with the highest incidence, at 5.42 per 1 million. The overall incidence of CTCL in the U.S. and Europe has increased, a reflection of better diagnostic tools and increased awareness among physicians and patients, which has led to improved disease detection.
According to Fortune Business Insights, the North American CTCL therapeutics market size is projected to reach an annual valuation of USD 587.4 million by 2028, registering a 13.6 percent compound annual growth rate (CAGR) in the 2021-2028 period. The market value was estimated to be worth USD 225.9 million in 2020 and reached USD 240.9 million in 2021. The increasing burden of CTCL in the region is slated to increase the demand for novel CTCL therapeutics solutions. Cleveland Clinic reports that more than 3,000 new CTCL patients are diagnosed in the U.S. each year and about 16,000-20,000 individuals suffer from mycosis fungoides, the most common form of CTCL that is linked to skin-localized immune cell stimulation.
LEGACY PRODUCTS Our legacy products are revenue-generating, approved products marketed in various countries and regions of the world including the U.S. and Europe, originally developed and patented by APRRelief and subsequently licensed to third parties for commercialization in the different territories. CAMBIA® Diclofenac potassium is an off-patent, potent non-steroidal anti-inflammatory drug (NSAID) widely used for treating inflammatory conditions and pain management. By applying our patented Dynamic Buffering Technology (DBT), we developed the first and only NSAID approved by the FDA for the treatment of acute migraine attacks with or without aura in adults. CAMBIA is currently available in the form of a powder packed into a single dose envelope to be poured and dissolved in water before administration. The rightsproduct is marketed in the U.S. as CAMBIA by Assertio Therapeutics Inc. (Nasdaq: ASRT) and in Canada by Aralez Pharmaceuticals Canada Inc. 39
CAMBIA is protected by a family of four patents listed in the FDA Orange Book, all expiring in 2026. In 2023, based on litigation settlements between Assertio Therapeutics Inc. and specific generic filers, generic versions at of CAMBIA became available in the legacy products were acquired by Relief Therapeutics as part of the 2021 acquisition of APR.U.S., significantly reducing our royalty income from CAMBIA. SETOFILM/ONDISSOLVE SETOFILM / ONDISSOLVE SETOFILM® is the first prescription-only medicine approved in Europe and Canada, developed as an orodispersible film (ODF) formulation. The product is available in 4 mg and 8 mg doses. Once placed on the tongue, it dissolves in a few seconds and is swallowed with saliva without the need for water. The innovative ODF form may reduce the patient pill burden and enable patients to take their medication virtually anywhere.
The product is indicated for radiotherapy induced nausea and vomiting (RINV), chemotherapy induced nausea and vomiting (CINV) as well as postoperative induced nausea and vomiting (PONV) in both adults and children 6 months of age or older. The product has been formulated and developed using the RapidFilm drug delivery technology and is the form of a soluble film to be placed on the tongue where it dissolves in a few seconds thus greatly improving patient compliance and avoiding possible risks of suffocation in kids. The product is approvedmarketed in Europe and Canada as prescription drug and it is marketed by Norgine B.V.B.V and Takeda Pharmaceuticals respectively under license from APR. CAMBIA™
Diclofenac potassium is an off-patent, potent non-steroidal anti-inflammatory drug (NSAID) widely used for treating inflammatory conditions and pain management. By applying its patented Dynamic Buffering Technology (DBT), APR developed the first and only NSAID approved by the FDA for the treatment of acute migraine attacks with or without aura in adults. The product is currently marketed as CAMBIATM by Assertio Therapeutics Inc. (Nasdaq: ASRT) in the U.S. and Miravo Healthcare (formerly Nuvo Pharmaceuticals Inc.) in Canada under an exclusive, royalty-bearing license agreement with APR.by Takeda Pharmaceuticals.
In January 2022, APR received a notice of allowance from the U.S. Patent and Trademark Office (USPTO) for patent application number 16/713,052 entitled, “Ready to Use Diclofenac Packs” with an expiration date in 2039.
52
On February 28, 2022, Unimedica Laboratories Pvt. Ltd., India, sent APR a Notice of Certification under the Federal Food, Drug, and Cosmetic Act (FFDCA) related to the filing of an abbreviated new drug application (ANDA) for CAMBIA. While there can be no assurance, it is unlikely that Unimedica will get accelerated approval, and we reserve the right to seek to enforce our patents.
DBT and CAMBIA are currently protected by a family of four patents listed in the FDA Orange Book, all expiring in 2026. In 2023, based on litigation settlements between Assertio and specific generic filers, generic versions at Cambia may become available. CAMBIA is currently available in the form of a dry powder packed into a single dose envelope to be poured and dissolved in water before administration.
VOLTADOL® Developed by APR using awith our patented matrix patch technology, Voltadol is a topical, locally applied and locally acting patch delivering diclofenac sodium, an off-patent, potent non-steroidal anti-inflammatory drug (NSAID) for the local treatment of painful, acute conditions such as muscle and joint strains. Unlike heat plaster, the patch contains an anti-inflammatory. It penetrates deep to the source of pain to provide powerful pain relief. The medicated patch provides up to two times more powerful deep pain relief, compared to a non-medicated, non-heated placebo patch. The patch also provides 12 hours continuous release of the active ingredient (diclofenac) to the site of pain. This means the patch only needs to be applied once in the morning and once in the evening to provide effective pain relief. The product is marketed in various countries as an over-the-counter medicine by GlaxoSmithKline (GSK) which recently spun-off the rights to Haleon. Discontinued Products In 2023, the development of Sentinox was discontinued due to a decrease in market needs as the COVID-19 pandemic subsided. Commercialization efforts for Nexodyn®, a hypochlorous acid-based spray solution for wound management, have also been discontinued. Neither Sentinox nor Nexodyn significantly contributed to the Company’s revenue. Third-Party Agreements We are party to certain agreements with third parties relating to licensing, collaboration, or other matters that are material to our business and performance. OLPRUVA™ License On January 25, 2021, we entered into an option agreement with Acer Therapeutics, Inc. (Acer) providing exclusivity for the right to negotiate a potential collaboration and license agreement for worldwide development and commercialization for ACER-001 (now OLPRUVA™) for the treatment of UCDs, MSUD, and other potential indications. Under the terms of the option agreement, we paid Acer a USD 1 million non-refundable payment in return for exclusivity until June 30, 2021, to negotiate and enter into a collaboration and license agreement for the development of ACER-001. Further, in connection with entering into the option agreement, we made a USD 4 million secured loan to Acer. On March 19, 2021, we executed the definitive collaboration and license agreement (the March 2021 CLA). Under this agreement, Acer received a $10 million USD cash payment (originally USD 14 million, offset by repayment of the USD 4 million outstanding balance of the prior loan, plus interest, to Acer). We also paid Acer in 2021 and 2022 approximately USD 20 million in U.S. development and commercial launch costs of OLPRUVA®. In December 2022, ACER-001 was approved in the U.S by the Food and Drug Administration. under the trademark OLPRUVA®, for the treatment of Urea Cycle Disorders involving deficiencies of carbamylphosphate synthetase, ornithine transcarbamylase, or argininosuccinic acid synthetase. On August 30, 2023, Relief and Acer terminated the March 2021 CLA. Concurrently, the parties entered into a new exclusive license agreement (the ELA). Pursuant to the ELA, Relief holds exclusive development and commercialization rights for OLPRUVA in the European Union, Liechtenstein, San Marino, Vatican City, Norway, Iceland, Principality of Monaco, Andorra, Gibraltar, Switzerland, United Kingdom, Albania, Bosnia, Kosovo, Montenegro, Serbia and North Macedonia (Geographical Europe). Under the terms of the termination agreement and the ELA, Relief is entitled to receive from Acer a 10% continuing royalty on net sales of OLPRUVA® in the Acer territory 40
(worldwide, excluding Geographical Europe), and 20% of any value received by Acer from licensing or divestment transactions relating to OLPRUVA®, up to a cumulative amount of USD 45 million. Relief committed to paying Acer a variable, continuing royalty up to a maximum of 10% of potential future net sales of OLPRUVA® in Geographical Europe and 20% of any value received by Relief from sublicensing transactions relating to OLPRUVA®. Furthermore, Relief returned to Acer development and commercialization rights for non-US-territories, excluding Geographical Europe where Relief retained these rights. Relief received from Acer upon execution of the termination agreement and the ELA a non-refundable USD 10 million upfront payment in cash and is due to receive an additional non-contingent cash payment of USD 1.5 million in August 2024. On November 17, 2023, Zevra Therapeutics, Inc. (NASDAQ: ZVRA) (Zevra), completed its acquisition of Acer. Zevra confirmed its assumption of Acer’s obligations under the termination agreement referenced above and the ELA. As of the date of this annual report, Acer is a wholly owned subsidiary of Zevra. License and Supply Agreement with Eton Pharmaceuticals, Inc. On March 21, 2024, we entered into a license and supply agreement granting the exclusive right to Eton Pharmaceuticals, Inc. (Nasdaq: ETON) (Eton) for the commercialization of GOLIKE® family of products in the United States. As part of the agreement, Eton also received U.S. rights to Relief’s GOLIKE medical food line extensions under development for the management of other inherited rare metabolic diseases such as tyrosinemia and homocystinuria. For an initial term of six years, Relief committed to manufacture and supply GOLIKE products for the U.S., with renewals possible for three-year periods. Under the terms of the agreement, we received from Eton an upfront payment of USD 2.2 million and are eligible to receive up to USD 2 million in additional sales milestones, consisting of one-time USD 500,000 payments when U.S. net sales over a year reach USD 4 million, USD 8 million, USD 15 million, and USD 20 million. We will also receive a royalty of 30% of U.S. net sales, which will include the cost of the products we supply to Eton. We anticipate this royalty, net of our cost of goods, will range between 13% and 18% of net sales based on our current cost of goods. Acquisition and license agreement with Meta Healthcare Ltd On July 11, 2022, we acquired from Meta Healthcare Ltd. (Meta) the worldwide rights, except for the United Kingdom, of RLF-OD032, a liquid formulation of a Sapropterin dihydrochloride product intended for the treatment of patients with phenylketonuria. Under the terms of the agreement, we paid Meta approximately CHF 0.3 million prior to certain subsequent price adjustments and may issue additional payments of approximately CHF 0.3 million contingent to pre-specified development milestones. We committed to pay Meta a 10% royalty on possible future net commercialization profit from RLF-OD032. Share Subscription Facility with GEM On January 20, 2021, we signed a binding agreement with our largest shareholder, GEM Global Yield LLC SCS and GEM Yield Bahamas Limited (GEM) for the implementation of a new share subscription facility (SSF) in the amount of up to CHF 50 million for a duration of up to three years. On February 27, 2024, the SSF agreement was extended until January 20, 2027. Under the terms of the SSF, we have the right to periodically, during the timeframe of the agreement, issue and sell shares to GEM. Under the facility, GEM undertakes to subscribe to or acquire our ordinary registered shares upon our exercise of a drawdown notice. In accordance with the customary terms of the SSF agreement, we control the timing and maximum amount of any drawdown and retain the right, not the obligation, to draw down on the full commitment amount. Future subscription prices under the SSF will correspond to 90 percent of the average of the closing bid prices on the SIX Swiss Exchange during the reference period, which corresponds to fifteen trading days following Relief’s drawdown notice. As of the date of this annual report, no amounts have been drawn on this facility. Under the terms of the agreement dated January 20, 2021, we were required to pay GEM a commitment fee of CHF 1.25 million, which remained outstanding as an interest-bearing loan. Pursuant to the extension agreement dated February 27, 2024, GEM agreed to forgive the commitment fee and accrued interests. In consideration for GEM’s capital commitment and this debt waiver, Relief committed to issuing GEM warrants to purchase up to 3.35 million ordinary shares at a purchase price of CHF 1.70 per share, exercisable from the issuance date, and expiring on January 20, 2027. The issuance of these warrants was contingent upon shareholder approval for a reduction in the nominal value of the Company’s ordinary shares. At the extraordinary general meeting held on April 26, 2024, such reduction was approved, and the warrants will be formally issued upon the registration of the reduction in nominal value with the commercial register of Geneva, Switzerland. Acquisition of APR Applied Pharma Research SA On June 28, 2021, we signed and closed a definitive agreement for Relief to acquire all outstanding shares of APR Applied Pharma Research SA (APR), a privately held Swiss company with over 25 years of experience in identifying developing and commercializing known molecules engineered with drug delivery systems in niche and rare diseases. 41
Under the terms of the agreement, APR’s former shareholders received from Relief CHF 21.5 million in cash and 516,967 Relief shares for a value of CHF 45 million when the consideration shares were granted. The former APR shareholders were also eligible to receive possible future contingent milestone payments in the aggregate maximum amount of up to CHF 35 million, upon achievement of pre-agreed objectives involving (i) the execution of a definitive agreement for the commercialization of Sentinox™, (ii) the launch of Sentinox in the first of France, Germany, Spain, Italy, and the UK, (iii) the launch of PKU GOLIKE® in the U.S., and (iv) the launch of RLF-TD011 in the first of France, Germany, Spain, Italy and the UK. The launch of PKU GOLIKE® in the U.S. on October 10, 2022, marked the completion of the third milestone, for which Relief issued a cash payment of CHF 2.8 million as well as 375,500 shares. The remaining milestones remained outstanding as of the date of this annual report. Acquisition of AdVita Lifescience GmbH On July 28, 2021, we announced the closing of a definitive agreement to acquire all the outstanding shares of AdVita Lifescience GmbH (AdVita). AdVita was founded in 2019 for the purpose of developing products and strategies to improve the therapy and diagnosis of rare lung diseases. Among AdVita’s assets were intellection property rights that could cover RLF-100 inhaled formulation specifications and the potential application of inhaled Aviptadil in the treatment of Acute Respiratory Distress Syndrome, Checkpoint Inhibitor-induced Pneumonitis and Sarcoidosis. Under the terms of the agreement, AdVita’s former shareholders received 339,353 of our ordinary shares, representing EUR 25 million in value based on the then 60-day volume weighted average price of our ordinary shares and were also eligible to receive additional contingent payments in cash. In April 2022, we issued a cash payment of EUR 5 million upon completion of the first milestone pertaining to the issuance by the Swiss Patent Office IPI to Relief of a patent entitled Vasoactive Intestinal Peptide (VIP) for the Use in the Treatment of Drug- induced Pneumonitis. As of the date of this annual report, AdVita’s former shareholders may receive up to EUR 10 million upon achievement of pre-agreed milestones involving (i) the approval in the U.S. or Europe of the inhaled form of aviptadil for the treatment of sarcoidosis or berylliosis, and (ii) the conduct of a phase II clinical study for the inhaled form of aviptadil in the treatment of checkpoint inhibitor-induced pneumonitis. Collaboration Agreement with InveniAI LLC On November 24, 2021, we announced that we had entered into a collaboration agreement with InveniAI LLC (InveniAI), a U.S. based company that has pioneered the application of artificial intelligence and machine learning across biopharma and other industries, in order to identify promising drug candidates to treat rare and specialty diseases. Under the terms of the agreement, we paid InveniAI an initial up-front fee of USD 500,000 and will be required to pay success milestones for any products brought to us in connection with this agreement as well as royalties on any such product if commercialized. We are currently not developing any product brought to us by InveniAI, nor are we actively seeking to identify new product candidates or concepts in collaboration with them. NeuroRx Collaboration Agreement On September 18, 2020, we entered into a binding collaboration agreement (the Collaboration Agreement) with NeuroRx. The Collaboration Agreement established the terms under which we will collaborate and assist with NeuroRx in order to maximize revenues in our respective territories from the sale of RLF-100® for intravenous and inhaled use primarily in the treatment of COVID-19 related conditions. The NeuroRx territory included the U.S., Canada, and Israel. The Relief territory comprised the rest of the world and includes the EU, Switzerland, Iceland, Norway, the UK, the Channel Islands, Liechtenstein, Monaco, Andorra, San Marino and Vatican City. The Collaboration Agreement provided that we would fund the costs associated with the clinical trials and development of RLF-100® (aviptadil acetate) in the U.S., which development would be conducted and managed by NeuroRx. NeuroRx was responsible for ensuring that the costs of the clinical trials and development activities for RLF-100 IV did not exceed the budget contemplated by the parties by more than 30 percent. The Collaboration Agreement also provided options for the parties to treat health conditions outside COVID-19 and for the commercialization of RLF-100® outside of the above-described territories. Dispute, Litigation and LitigationSettlement with NeuroRx On October 7, 2021, because of breaches of the Collaboration Agreement by NeuroRx, among others, we filed a lawsuit against NeuroRx and its Chief Executive Officer,then chief executive officer, Dr. Jonathan Javitt, for multiple breaches of the Collaboration Agreement (Complaint). The Complaint was filed in the Supreme Court in the State of New York in Manhattan. The suit alleged, among other matters, breaches of the covenant of good faith and fair dealing and tortious interference with prospective economic advantage. The Complaint, among other remedies, sought damages, an order compelling NeuroRx to comply with multiple provisions of the Collaboration Agreement, and a declaration directing NeuroRx to deliver the entire data set from the Phase 2b/3 clinical trial of intravenously administering aviptadil to Relief. On January 10, 2022, NeuroRx, through its parent, NRx Pharmaceuticals, Inc. (NRx), filed a complaint against Relief. In the complaint, NeuroRx claimed financial damages in excess of $185 million and sought a ruling that the Collaboration Agreement iswas void. It also included a count for defamation. 42
On November 14, 2022, Relief and NRx issued a press release announcingannounced that the parties had entered into a definitive settlement agreement to resolve all matters relating to the pending litigation. At the closing of this settlement, which was held on December 19, 2022, (i) NRx transferred to Relief all of the assets that it previously used in its aviptadil development program, including its regulatory filings, patent applications, clinical data, and the formulation of the aviptadil product it was previously developing, (ii) Relief hashad the exclusive right and control going forward to develop and commercialize an aviptadil product, (iii) Relief has agreed to use commercially reasonable efforts to continue the existing Right to Try Program for aviptadil in the U.S. for at least two years, (iv) Relief will pay NRx milestone payments if it can successfully obtain commercial approval of an aviptadil product (whether for COVID-19 or any other indication), (v) Relief will pay NRx royalties based on a percentage of future sales of an aviptadil product (whether for COVID-19 or any other indication), up to a maximum of $30 million in the aggregate, (vi) NRx Pharmaceuticals has agreed not to compete in the development of an aviptadil product in the future, and (vii) at the closing, Relief and NRx Pharmaceuticals dismissed their pending litigation. Further, the Collaboration Agreement was cancelled. Finally, as part of the settlement, the parties exchanged releases of all claims that could have been brought in the lawsuits between the parties. We have agreed to use commercially reasonable efforts to develop, market, and commercialize aviptadil. In September 2023, we received a copy of a complaint filed in Israel by Jonathan Javitt, former chief executive officer and chief scientist of NRx, against Relief and certain of its current and former directors, officers, and consultants. The complaint alleged, among other matters, that statements made by Relief and other defendants regarding Dr. Javitt were defamatory, causing material harm to Dr. Javitt. Dr. Javitt also appeared to be seeking financial damages, an injunction against future alleged defamatory statements, and for Relief to turn over global rights to NRx’s version of Aviptadil to Dr. Javitt. We reported denying all allegations in the complaint, considering them to be without merit and lacking both factual and legal foundation. After we objected to the service of process, the complaint was dismissed in March 2024. 53
Patents and LicensesIntellectual Property
Our success depends significantly on our ability to develop, obtain and maintain intellectual property rights for our product candidates, technology and know-how, to operate without infringing intellectual property rights of others and to prevent others from infringing our intellectual property rights. We seek to protect our proprietary position by, among other methods, filing patent applications in Europe, the U.S. and other relevant jurisdictions related to our proprietary technology, inventions and improvements that are vital to the development of our business, where patent protection is available. We also rely on trade secrets, know-how and in licensingin-licensing opportunities to develop and maintain our proprietary position. OLPRUVA™ License
We in-licensed from Acer the rights to commercialize OLPRUVA™ for the treatment of UCD and MSUD. Under the terms of our collaboration agreement, Acer received approximately $10 million cash payment (originally $14 million, offset by repayment of the $4 million outstanding balance of the prior loan, plus interest, from Relief to Acer). Relief will also pay Acer up to $20 million ($15 million which has been paid to-date) in U.S. development and commercial launch costs for the UCDs and MSUD indications. Acer will retain development and commercialization rights in the U.S., Canada, Brazil, Turkey, and Japan. The companies will split net profits from Acer’s territories 60 percent to 40 percent favor of Relief. In addition, Relief has licensed the rights for the rest of the world, where Acer will receive from Relief a 15 percent royalty on all revenues received in Relief’s territories. Acer may also receive a total of $6 million in development milestone payments following the first EU marketing approvals for UCDs and MSUD. Acer intends to submit the patent for listing by the FDA in the Approved Drug Products with Therapeutic Equivalence Evaluations, or Orange Book.
In parallel with Acer’s actions, Relief and Acer are pursuing similar claims in the European Patent Office to cover OLPRUVA™ as Relief continues to execute on its plan to submit a Marketing Authorization Application (MAA) for OLPRUVA™ for the treatment of patients with UCDs in Europe. There can be no assurance that Relief and Acer will be successful in those endeavors.
Acer maintains its own intellectual property portfolio. In August 2014, Acer was granted ODD by the FDA to sodium phenylbutyrate (OLPRUVA™) for the treatment of Maple Syrup Urine Disease.
As of the date of this annual report, Acer’s patent portfolio for ACER-001(OLPRUVA™) consists of three patent families. The first family includes 41 granted patents world-wide directed towards novel sodium phenylbutyrate particle formulations and methods of use. These patents have an expiration date of October 2036, exclusive of any patent term adjustments or extensions, or any form of potential exclusivity. If granted, additional patents, would expire no earlier than October 2036. Acer’s patent portfolio further includes PCT/US2021/040760 and PCT/US2022/040082. Patents granting from applications claiming priority to PCT/US2021/040760 will expire in July 2041, excluding any patent term adjustments or extensions, or any form of potential exclusivity. Patents granted from applications claiming priority to PCT/US2022/040082 will expire in April 2042, excluding any patent term adjustments or extensions, or any form of potential exclusivity.
54
OLPRUVA™’s patent applications worldwide are as follows:
| | | | | Summary Description of Patent or Patent Application
| | United States or Foreign Jurisdiction
| | Expiration Date
| Palatable Compositions Including Sodium Phenylbutyrate and Uses Thereof | | Granted: United States (Patent Nos. 11,154,521, 11,202,767, and 11,433,041), Albania, Austria, Belgium, Bulgaria, Switzerland, Cyprus, Czechia, Germany, Denmark, Estonia, European Patent Convention, Spain, Finland, France, United Kingdom, Greece, Croatia, Hungary, Ireland, Iceland, Italy, Lithuania, Luxembourg, Latvia, Monaco, North Macedonia, Malta, Mexico, Netherlands, Norway, Poland, Portugal, Romania, Serbia, Sweden, Slovenia, Slovakia, San Marino
Pending: Bahrain, Brazil, Canada, European Patent Convention, Israel*, Japan, Republic of Korea, Kuwait, Mexico, New Zealand*, Oman, Qatar, Saudi Arabia, United Arab Emirates
| | October 17, 2036. Expiration of pending patents to be determined upon grant. | | | | Administration of Sodium Phenylbutyrate in a Fasted State to Treat Urea Cycle Disorders | | Pending: Patent Cooperation Treaty | | Applications claiming priority to this PCT application, if granted, will expire no earlier than July 7, 2041. | | | | Dosage Form for Improving Palatability of Drug Substance | | Granted: China
Pending: China, Patent Cooperation Treaty
| | August 24, 2031 (China). Applications claiming priority to the PCT application, if granted, will expire no earlier than April 12, 2042. |
Aviptadil Acetate/RLF-100® Patents As of the date of this annual report, Relief haswe have two groups of patent families covering formulations of aviptadilAviptadil acetate. The first familygroup includes a U.S. patent that will expire in the U.S. valid until at least July 2029, with extension opportunities up to five years, as well as patents in several countries in Europe and the rest of the world valid until at least 2026, excluding extension opportunities comparable to the U.S. The first family applications were filed in 2006 and granted between 2011 and 2012. The second family includes an unpublisheda pending U.S. provisional and a PCT application. Relief’s aviptadilAviptadil patents and patent applications worldwide are as follows: | | | | | Summary Description of Patent or Patent Application | | United States or Foreign Jurisdiction | | Expiration Date | | Patent Family 1 | | | | | | | | | Formulation for Aviptadil | | Granted: United States (No. 8,178,489), China, European Patent Convention, Mexico, India, Austria, Denmark, Switzerland/Lichtenstein, Germany, Spain, United Kingdom, Ireland, Netherlands, Turkey | | July 3, 2029 (United States), March 7, 2026 (all other jurisdictions) | | | | **Unpublished**Patent Family 2 | | | | | | | | | Stable Aviptadil Formulation | | Pending: United States (provisional), PCT/IB2023/060966 | | Expiration of any potential applications claiming priority to this provisional application and/or PCT application to be determined upon grant. |
AdVita
As ofIn the date of this annual report, AdVita hassecond group, we have three patent families in various stages of prosecution. The first family includes 1one granted patent and 15four applications world-wide claiming priority to PCT/EP2020/062420. Patents grantingThe patent and any patents granted from the pending applications claiming priority to PCT/EP2020/062420 will expire in May 2040, excluding any patent term adjustments or extensions, or any form of potential exclusivity. The second family includes 15 applications world-wide claiming priority to PCT/EP2021/052151. Patents granted from applications claiming priority to PCT/EP2021/052151 will expire in January 2041, excluding any patent term adjustments or extensions, or any form of potential exclusivity. The last family includes an a PCT application ,three applications claiming priority to PCT/IB2022/053709. Patents granted from applications claiming priority to PCT/IB2022/053709 will expire in April 2042, excluding any patent term adjustments or extensions, or any form of potential exclusivity. Each family of applications is directed to novel uses and/or formulations of Aviptadil for treating various conditions such as drug-induced pneumonitis. AdVita’s patents and
43
Our Aviptadil patent applications worldwide are as follows: 55
| | | | | Summary Description of Patent Application | | United States or Foreign Jurisdiction | | Expiration Date | | Patent Family 1 | | | | | | | | | Vasoactive Intestinal Peptide (VIP) for Use in the Treatment of Drug-Induced Pneumonitis | | Granted: Switzerland Pending: United States (Application No. 17/595,025), Australia, Brazil, Canada, Switzerland, China, European Patent Convention, Hong Kong, Israel, Japan, Republic of Korea, Mexico, New Zealand, Russian Federation, Singapore, South Africa | | May 5, 2040 (Switzerland) Applications, Other applications, if granted, will expire no earlier than May 5, 2040. | | | | | Patent Family 2 | | | | | | | | | Human Anti-Inflammatory Peptides for the Inhalatory Treatment of Inflammatory Pulmonary Diseases | | Pending: United States (Application No. 17/759,559), Australia, Brazil, Canada, China, European Patent Convention, Hong Kong, Israel, India, Japan, Republic of Korea, Mexico, New Zealand, Singapore, South Africa | | Applications claiming priority to this PCT application, if granted, will expire no earlier than Jan 29, 2041. | | | | | Patent Family 3 | | | | | | | | | Use of Aviptadil Alone or in Combination with Alpha Lipoic Acid as a Therapeutic Medicament for Post-Viral Infection Syndrome | | Pending United States (Application No. 18/556,073), Canada and European Patent Cooperation TreatyConvention. | | Expiration of any potential applications claiming priority to this PCT application to be determined upon grant.Applications, if granted, will expire no earlier than April 20, 2042. |
Partner Patents and Licenses
TECHLO Technology™ As of the date of this annual report, our TEHCLOTM™ portfolio consists of fourthree patent families. The first threetwo families include 10740 granted patents world-wideworldwide directed to systems and methods for generating hypochlorous acid solution and compositions comprising Relief’sour hypochlorous acid solution, and methods for treating ocular disorders.solution. These patents expire between October 2026 and JuneFebruary 2030, exclusive of any patent term adjustments or extensions, or any form of potential exclusivity. If granted, additional patents of the third family would expire no earlier than July 2040. 56
| | | | | Summary Description of Patent or Patent Application | | United States or Foreign Jurisdiction | | Expiration Date | | Patent Family 1 | | | | | | | | | Electrolytic Water Treatment Device Having Sintered Nanoparticle Coated Electrode and Method for Making Acid or Basic Water Therewith | | Granted: United States (Patent No. 8,277,634) | | August 23, 2029 | | | | | Device Comprising an Electrode with Nanocoating for Preparing a Highly Stable Aqueous Solution and Method for Making this Aqueous Solution | | Austria, Belgium, Bulgaria,Granted: Switzerland, Switzerland, Cyprus, Czechia, Germany, Denmark, Estonia, European Patent Convention, Spain, Finland, France, United Kingdom, Greece, Hungary, Ireland, Iceland, Italy, Lithuania, Luxembourg, Latvia, Monaco, Netherlands, Poland, Portugal, Romania, Sweden, Slovenia, Slovakia Turkey | | October 24, 2026 (Luxembourg), October 23, 2026 (all other jurisdictions) | | | | | New Highly Stable Aqueous Solution, Electrode with Nanocoating for Preparing the Solution and Method for Making this Electrode | | Australia,Granted: Canada, China, Israel, Mexico, New Zealand, Republic of Korea, Russian Federation, Singapore, South Africa | | October 22, 2026 (China), October 23, 2026 (all other jurisdictions) | | | | A Device for the Electrolytic Treatment of a Fluid | | India | | October 23, 2026 | | | | | Patent Family 2 | | | | | | | | | Highly Stable Electrolytic Water with Reduced NMR Half Line Width | | Granted: United States (Patent Nos. 8,709,495, 9,402,192, and 9,889,153), Austria, Australia, Belgium, Bulgaria, Brazil, Canada, Switzerland, Cyprus, Czechia, Germany*, Denmark, Estonia, European Patent Convention*, Spain*, Finland, France*, United Kingdom*, Greece, Croatia, Hungary, Ireland, Iceland, Italy*, Japan, Republic of Korea, Lithuania, Luxembourg, Latvia, Monaco, Malta, Mexico, Netherlands, Norway, New Zealand, Poland*, Portugal, Romania, Russian Federation, Sweden, Singapore, Slovenia, Slovakia, Turkey*, South Africa *Two patentsPatents | | February 7, 2030 (United States Patent No. 8,709,495), April 24, 2028 (one United Kingdom patent), April 26, 2028 (Greece), April 25, 2028 (all other patents and jurisdictions)patents) | | | | Electrolytic Acid Water | | India | | April 25, 2028 | | | | | Patent Family 3 | | | | | | | | Methods of Treating Outer Eye Disorders Using High ORP Acid Water and Compositions Thereof | | United States (Patent No. 8,691,289), Germany, European Patent Convention, Spain, France, United Kingdom, Italy, South Africa | | June 15, 2030 (United Kingdom), March 13, 2032 (United States), June 16, 2030 (all other jurisdictions) | | | | Patent Family 4 | | | | | | | | Therapeutic Uses of OxidisingOxidizing Hypotonic Acid Solutions | | Pending: United States (Application No. 17/597,220), United Arab Emirates, Australia, Brazil, Canada, China, Colombia, Egypt, European Patent Convention, Israel, Japan, Korea, Kuwait, Qatar, Russian Federation | | Applications, if granted, will expire no earlier than July 2, 2040. |
We were granted worldwide licenses for TECHLOTM to numerous regional and national pharmaceutical firms. None of the licenses, either individually or as a whole, currently represent a material amount of the revenues of the consolidated company.
5744
The above-mentioned patents and applications intend to protect RLF-TD011. Physiomimic Technology™ - PKU GOLIKE® As of the date of this annual report, the PKU GOLIKE® portfolio consists of two patent families including 3414 pending applications and 5036 granted patents world-wide.worldwide. Patents resulting from these families, if granted, will expire no earlier than 2036 and 2038, respectively, exclusive of any patent term adjustments or extensions, or any form of potential exclusivity. | | | | | Summary Description of Patent or Patent Application | | United States or Foreign Jurisdiction | | Expiration Date | | | | | Patent Family 1 | | | | | | | | | Modified Release Orally Administered Amino Acid Formulations | | Granted: United States (Nos.(Patents Nos. 10,500,180 and 11,419,837), Armenia, Austria, Australia, Azerbaijan,Australia*, Belgium, Bulgaria, Belarus,Brazil, Switzerland, China,China*, Colombia, Czechia, Germany, Denmark, Eurasian Patent Convention, European Patent Convention, Spain, Finland, France, United Kingdom, Gulf Cooperation Council Greece, Croatia, Hungary, Indonesia,Hong Kong, Ireland, Israel, Italy, Jordan, Kyrgyzstan, Kazakhstan, Lebanon, Lithuania, Macao, Malta, Mexico, Malaysia, Norway, Netherlands, Poland, Portugal, Romania, Russian Federation, Sweden, Slovenia, Slovakia, Tajikistan, Turkmenistan,Saudi Arabia ,Sweden, Turkey, Taiwan South Africa Pending: United States (Application No. 17/660,999), Argentina, Australia, Brazil, Canada, Chile, China, Egypt, European Patent Convention, Gulf Cooperation Council, Hong Kong*, Israel, Iraq, Malaysia, Philippines, Pakistan, Saudi Arabia, Uruguay, Venezuela, Vietnam Kong * Two ApplicationsPatents | | September 25, 2036 (Jordan), September 28,29, 2036 (Taiwan), September 27, 2036 (all other jurisdictions). Applications, if granted, will expire no earlier than September 27, 2036. | | | | | Patent Family 2 | | | | | | | | | Methods of Normalizing Markers of Amino Acid Metabolism | | Granted: IraqUnited States (Patent No. 11,701,335), Canada Pending: United States (Application No. 18/326,259), Australia, Brazil, Chile, China, European Patent Convention, Hong Kong, Israel, Saudi Arabia, Taiwan | | August 29,16, 2039 (Iraq)(United States Patent 11,701,335 and Canada) Applications, if granted, will expire no earlier than August 16, 2039. |
RLF-OD032 – Sapropterine suspension As of the date of this annual report, the RLF-OD032 portfolio consists of one Patent Family: | | | | | Summary Description of Patent Application | | United States or Foreign Jurisdiction | | Expiration Date | | | | | Sapropterine Formulation | | Pending: United States (Application No. 16/543,437), Australia, Brazil, Canada, Chile, China, Colombia, European Patent Convention, Hong Kong, Israel, Pakistan, Saudi Arabia, Taiwan18/163,748) and PCT/IB2023/050932 | | Expiration of pending patentsapplications to be determined upon grant. |
We were granted licenses for PKU GOLIKE® in Spain, Portugal, the UK, Ireland, Brazil, Israel, Gaza territories and West Bank, Ecuador, Greece and Cyprus.. None of the licenses, either individually or as a whole, currently represent a material amount of the revenues of the consolidated company.
Dynamic Buffer Technology -– Diclofenac As of the date of this annual report, our diclofenac patent portfolio consists of multiple patent families comprising 4944 granted patents world- wide,worldwide, with expiration dates in either betweenranging from 2026 andto 2040, exclusive of any patent term adjustments or extensions, or any form of potential exclusivity. The portfolio further includes 148 pending applications directed to new diclofenac formulations and methods of use.applications. If granted, patents resulting from these pending applications will expire between 2026 and 2041, exclusive of any patent term adjustments or extensions, or any form of potential exclusivity. 58
| | | | | Summary Description of Patent or Patent Application | | United States or Foreign Jurisdiction | | Expiration Date | | | | | Patent Family 1 | | | | | | | | | Diclofenac Formulations and Methods of Use | | Granted: United States (Nos. 7,759,394, 8,097,651, 8,927,604 and 9,827,197), Australia, Canada*, Switzerland*, Germany***, European Patent Convention***, Spain**, France**, United Kingdom**, Greece*, Indonesia, Italy***, Jordan, Republic of Korea, Lebanon, Malta, Mexico, Norway, New Zealand, Pakistan, Poland, Portugal, Russian Federation, Turkey, South Africa Pending: United States (Application No. 16/716,511), China, Egypt, Gulf Cooperation Council*, Hong Kong, Thailand
* Two Patents
** Three Patents
*** Four Patents
| | June 16, 2026 (all United States Patents), patents) June 8, 2026 (Lebanon), June 14, 2026 (Malta), June 15, 2026 (United Kingdom), June 16, 2026 (all other jurisdictions). Expiration of pending applications to be determined upon grant. |
45
| | | | | | | | | Diclofenac FormulationsJordan, Lebanon, Malta, New Zealand, Pakistan, Poland, Russian Federation, Thailand, South Africa Pending: Egypt, Gulf Cooperation Council* * Two patents or applications ** Three patents *** Four patents | | | Granted: Germany, Spain, France, United Kingdom, Italy, | June 15, 2026 (United Kingdom), June 16, 2026 (All other jurisdictions) | | | | | Patent Family 2 | | | | | | | | Moisture Resistant Container Systems for Rapidly Bioavailable Dosage Forms | | Granted: United States (Nos. 7,700,125 and 8,097,267) | | February 7, 2026 (No. 8,097,267), October 11, 2026 (No. 7,700,125) | | | | Patent Family 3 | | | | | | | | | Substantially Sodium Free Diclofenac Potassium Oral Solutions | | Granted: United States (No. 11,127,318) Pending: United States (Application No. 17/463,154), European Patent Convention
| | January 27, 2038 (United States Patent No. 11,127,318). Expiration of pending patents to be determined upon grant. | | | | Patent Family 43 | | | | | | | | | Ready to Use Diclofenac Stick Packs | | Granted: United States (No. 11,260,026) Pending: United States (Application No. 17/584,212), European Patent Convention Hong Kong | | February 22, 2040 (United States Patent No. 11,260,026). Expiration of pending applications to be determined upon grant. Applications, if granted, will expire no earlier than February 22, 2040. | | | | Patent Family 54 | | | | | | | | | Bioavailable Sugar-Based Diclofenac Formulations | | Pending: United States (Application No. 18/001,313), European Patent Cooperation TreatyConvention, Canada | | Expiration of pending application to be determined upon grant.Applications, if granted, will expire no earlier than June 8, 2041. |
We licensed Diclofenac Potassium 50 mg Powder for Oral Solution to Assertio Therapeutics (Cambia®) and to Novartis (Voltaren®/Voltfast®). 59Oral Disposable Film—Ondansetron
As of the date of this annual report, our Ondansetron patent portfolio consists of one patent family. | | | | | Summary Description of Patent Application | | United States or Foreign Jurisdiction | | Expiration Date | | Patent Family 1 | | | | | | Non-Mucoadhesive Film Dosage Forms | | Granted: Canada | | October 2, 2027 |
APR granted a license right on the Canadian patent to Takeda. In-licensed OLPRUVA™ Patents Pursuant to our exclusive license agreement with Acer Therapeutics Inc., we hold exclusive development and commercialization rights for OLPRUVA in Geographical Europe (European Union, Liechtenstein, San Marino, Vatican City, Norway, Iceland, Principality of Monaco, Andorra, Gibraltar, Switzerland, United Kingdom, Albania, Bosnia, Kosovo, Montenegro, Serbia and North Macedonia). We have a license/sublicense on the following patents: Title: Palatable Compositions Including Sodium Phenylbutyrate and Uses Thereof Assignee: Acer Therapeutics Inc. Priority Application Number: U.S. Prov. Appl. No. 62/308,614 Priority Date: March 15, 2026 Title: Taste Masked Phenylbutyrate and Compositions Therefore Assignee: Acer Therapeutics Inc. Priority Application Numbers: U.S. Prov. Appl. Nos. 63/048,892 & 63/065,272 Priority Dates: July 7, 2020 (August 13, 2020) Title: Dosage Form for Improving Palatability of Drug Substance Assignee: Acer Therapeutics Inc. Priority Application Number: 63/232,011 Priority Date: August 11, 2021 46
We licensed Diclofenac to Assertio Therapeutics for its Cambia® productTitle: Method of Modulation of Branch Chained Acid and to Novartis for its Voltaren®/Voltfast® product. We entered into a partnership agreement with Fidia Farmaceutici S.p.A. for diclofenac patches. We also entered into license and supply agreements with Actavis Group PTC ehf (MerckleGmbH) and Zentiva k.s..Uses Thereof
We sold the IT Patent for Diclofenac to the Neilos s.r.l. (an affiliateAssignee: Baylor College of Shedir Pharma Group S.p.A) but retained a Medicine
Priority Application Number: 61/228,485 Priority Date: July 24, 2009 non-exclusiveOther In-licensed and perpetual license right on such patent for the production in the country of Italy of drops solution for oral administration containing Diclofenac Potassium as sole active ingredient in a concentration of 5 percent. The sale of this patent does not have an effect on the license and supply agreements described in this section.Patents
We received a sublicense right in the territory of U.S. and China from Fidia Farmaceutici in relation to the following patents owned by IBSA Farmaceutici on Diclofenac transdermal patch: Chinese Patent No. CN101001616B; U.S. Patent No. 10,328,034. We do not believe that this license agreement our material to its business.
Oral Disposable Film—Ondansetron
As of the date of this annual report, our Ondansetron patent portfolio consists of two patent families comprising two pending applications and six granted patents with expiration dates ranging from 2027 to 2031, exclusive of any patent term adjustments or extensions, or any form of potential exclusivity.
| | | | | Summary Description of Patent or Patent Application
| | United States or Foreign Jurisdiction
| | Expiration Date
| | | | Patent Family 1 | | | | | | | | Non-Mucoadhesive Film Dosage Forms | | United States (Patent Nos. 8,580,830 and 9,682,037), Canada, Republic of Korea | | November 22, 2029 (United States Patent No. 8,580,830), October 2, 2027 (All other patents) | | | | Patent Family 2 | | | | | | | | Fast Dissolving Drug Delivery Systems | | Granted: Russian Federation, South Africa
Pending: Brazil, Egypt
| | March 23, 2031. Expiration of pending patents to be determined upon grant. |
APR has granted a license right on the abovementioned patents and patent applications to Takeda in Canada. This license does not represent a material amount of our revenues.
Other APR IP In addition to the patents and applications described above, APR has several other pending applications and granted patents:we also have: U.S. Patent No. 8,039,024, entitled “Device and composition for the delivery of a preservative-free balsamic cream” and patents in Canada, Russia and Ukraine entitled “Adhesive Label with Bittering Agent and Fluidifying Agents for Natural Airway Secretions” claim and cover a preservative-free, OTC decongestant stick pack which is no longer marketed.
I.T. patent 102020000021223 and pending unpublished applicationsA sublicense right in the U.S. and China from Fidia Farmaceutici in relation to the following patents owned by IBSA Farmaceutici on Diclofenac transdermal patch: Chinese Patent No. CN101001616B; U.S. Patent No. 10,328,034.
A patent in Italy and applications in the United States (Application No. 18/044,358), Canada, related to dermal compositions, entitledand the European Patent Convention (all titled “Dermal Compositions Replicating the Vernix Caseosa”,) that cover and claim OTC formulations targeting atopic dermatitis as well as other moderate skin disorders. Commercialization 60We maintained an internal marketing and sales infrastructure in Switzerland, the U.S., Italy and Germany, dedicated to the direct commercialization of PKU GOLIKE. For the commercialization of our other commercially available products, as well as PKU GOLIKE outside of these key markets, we entered into licensing or distribution agreements with third parties. In December 2023, we decided to transition progressively from our direct marketing and sales infrastructure to a partnership-based model for PKU GOLIKE to enable more efficient patient access through leveraging external expertise and infrastructure.
On March 21, 2024, we entered into a license and supply agreement granting the exclusive right to Eton Pharmaceuticals, Inc. (Nasdaq: ETON) for the commercialization of GOLIKE® family of products in the United States. Consequently, we terminated our U.S. marketing and sales infrastructure. As of the date of this annual report, we are in active discussions with potential licensees for the commercialization of PKU GOLIKE in certain European countries.
As we move our product candidates through development toward regulatory approval, we intend to enter into strategic marketing partnerships with third parties, including other pharmaceutical or biotechnology companies. We believe this approach is tailored to leverage both our internal capabilities and strategic partnerships to ensure efficient and widespread patient access to our future therapies. Manufacturing and Supply We do not own or operate facilities for the manufacture, packaging, labeling, storage or distribution of preclinical or clinical supplies of any of our drug candidates. We instead contract with and rely on third-party CMOs to manufacture, package, label, store test and distribute all preclinical development and clinical supplies of our drug candidates, and we plan to continue to do so for the foreseeable future. APR maintains laboratories for the testing of its products. Such laboratories are also used to develop new formulations. Compliance with governing rules and quality requirements The facilities used by our collaboration partners and CMOs to manufacture our product candidates are systematically audited by local authorities and occasionally inspected by competent authorities where the clinical studies are ongoing. The facilities where the commercial productions are performed must be approved by the FDA or other relevant regulatory authorities, pursuant to inspections that are conducted after we submit our NDA or comparable marketing applications. We perform periodic quality audits of the manufacturing facilities and CMOs to monitor their compliance with the regional laws, regulations and applicable cGMP standards and other laws and regulations, such as those related to environmental health and safety matters. The scope of our audits also involves monitoring the ability of our providers to maintain adequate QCs and QA systems including personnel qualification. 47
After manufacturing, our products are submitted to extensive characterization and QC testing plans performed by using properly developed analytical methods that are qualified or validated; this ensures the accuracy of the results generated and provides evidence of the quality of our products. In addition, our products are submitted to detailed and standardized stability programs aimed at demonstrating product stability during the storage period; this, in addition to guaranteeing the safety of the products, supports the definition of a suitable supply chain that may encompass the distribution of the products in different continents. Contractual framework We have established, with CMOs supplying drug substances or drug products under cGMP, quality agreements and master service agreements. Quality agreements define the quality standards required to develop, produce and supply the product, and also define the responsibilities related to the collaboration with regards to the quality related aspects. Manufacturing service agreements define the commercial and financial framework under which product manufacturing under cGMP is performed. Any failure to achieve and maintain compliance with the laws, regulations and standards, suspension of the manufacturing of our product candidates or revoke of cGMP permissions, which would adversely affect our business and reputation, are defined in the master service agreements and quality agreements. The risk that any third-party providers may breach the agreements they have with us because of factors beyond our control and the possibility that they may also terminate or refuse to renew their agreements because of their own financial difficulties or business priorities, potentially at a time that is costly or otherwise inconvenient for us, is managed by us with constant investments toward maintaining reserve stocks and in-depth process know-how. Interaction with collaboration partners and CMOs Finally, our partnership with CMOs is managed through an efficient project management platform in which teams are formed with the representatives of each key function from both parties. Meetings occur either through telephone conferences aimed at updating short-term actions or or face-to-face conferences conferences when mid- to long-term development plans are discussed. Acquisition of APR Applied Pharma Research SA
On April 30, 2021, we entered into a binding term sheet with the then current shareholders of APR Applied Pharma Research SA, a privately held Swiss company with over 25 years of experience in identifying developing and commercializing known molecules engineered with drug delivery systems in niche and rare diseases, to acquire all of the outstanding shares of APR. On June 28, 2021, the former shareholders of APR and Relief signed and closed a definitive agreement for Relief to acquire all outstanding shares of APR. Under the terms of the agreement APR’s shareholders have received from Relief CHF 21.5 million in cash and 206,786,784 Consideration Shares at a value of CHF 45 million when the Consideration Shares were issued and listed. The APR shareholders were also eligible to receive possible future contingent milestone payments in the aggregate maximum amount of up to CHF 35 million, upon achievement of pre-agreed objectives involving (i) the execution of a definitive agreement for the commercialization of Sentinox (as such product is defined below), (ii) the launch of Sentinox in the first of France, Germany, Spain, Italy, and the UK, (iii) the launch of PKU GOLIKE® in the U.S., and (iv) the launch of RLF-TD011 (as such product is described below) in the first of France, Germany, Spain, Italy and the UK. The launch of PKU GOLIKE® in the U.S. on October 10, 2022 marked the completion of the third milestone, for which Relief issued a cash payment of CHF 2.8 million as well as 150,200,120 shares.
61
Acquisition of AdVita Lifescience GmbH
On July 28, 2021, we announced the closing of a definitive agreement to acquire all of the outstanding shares of AdVita Lifescience GmbH. Under the agreement, the stockholders of AdVita received 135,741,063 of our ordinary shares, representing EUR 25 million (approximately CHF 27.4 million) in value based on a 60-day volume weighted average price (VWAP) of our ordinary shares and were also eligible to receive additional contingent payments in cash. In April 2022, we made an initial milestone payment of EUR 5 million (approximately CHF 5 million) upon completion of the first milestone.
As of the date of this annual report, the former shareholders of AdVita may receive up to EUR 10 million (approximately CHF 10 million) upon achievement of pre-agreed milestones involving (i) the approval in the U.S. or Europe of the inhaled form of aviptadil for the treatment of sarcoidosis or berylliosis, and (ii) the conduct of a phase II clinical study for the inhaled form of aviptadil in the treatment of checkpoint inhibitor-induced pneumonitis.
AdVita was founded in 2019 for the purpose of developing products and strategies to improve the therapy and diagnosis of rare lung diseases. Among AdVita’s assets are intellection property rights that may cover RLF-100 inhaled formulation specifications and the potential application of inhaled Aviptadil in the treatment of Acute Respiratory Distress Syndrome, Checkpoint Inhibitor-induced Pneumonitis and Sarcoidosis.
Collaboration Agreement with InveniAI LLC
On November 24, 2021, we announced that we had entered into a collaboration agreement with InveniAI LLC (InveniAI), a U.S. based company that has pioneered the application of artificial intelligence and machine learning across biopharma and other industries, in order to identify promising drug candidates to treat rare and specialty diseases (the InveniAI Collaboration Agreement).
Under the terms of the InveniAI Collaboration Agreement, InveniAI will use its proprietary platform for the identification of potential pharmaceutical product opportunities using its Pharma Big Innovation Data Lab, consisting of (i) its proprietary AlphaMeld platform, a cloud-based artificial intelligence platform that uses its proprietary machine learning and deep learning based neural networks to identify product opportunities in therapeutic areas, (ii) its cross-functional teams at its Integrated Center of Excellence, and (iii) domain expertise, to generate novel pharmaceutical opportunities and the related development pathway for the development of such concepts.
In the collaboration it is expected that InveniAI will use its platform to navigate the volume of data for all regulatory agency approved drugs and their associated active ingredients to identify potential rate and specialty disease indications for development and commercialization by us (product concepts). InveniAI will seek to prioritize top product concepts, associated diseases, scientific packages and evidence to support the potential drug development opportunities by us. We anticipate that InveniAI’s platform will complement APR’s existing capabilities in research and development and in drug reformulation. Based on product leads developed by InveniAI, we hope to develop proprietary versions of existing drugs, and to protect those drugs with long-lived intellectual property and defensible product claims.
Under the terms of the InveniAI Collaboration Agreement, we paid InveniAI an initial up-front fee of $500,000. We will be required to pay success milestones for any products brought to us in connection with the InveniAI Collaboration Agreement ranging from $200,000 per product candidate for which we exercise our option to acquire IP rights to $50 million for any required product reaching $1 billion per year in net sales. We will also be required to pay royalties on any such commercialized product in certain countries a royalty of approximately three percent.
We are not currently developing any product brought to us by InveniAI, and there can be no assurance that our collaboration with InveniAI will result in the development of new product candidates or product concepts.
62
Regulation in the United States The Company assumes that some of its product candidates will be submitted under an NDA and that approval of not only the products but also their manufacture is required before starting to market them. According to the definition of the U.S. Code of Federal Regulations, a drug product is approved only after demonstrating that it meets standards that assure the product’s safety, purity, effectiveness and potency. The design, pre-clinical and clinical study, manufacture, labeling, packaging, storage, holding, sale, distribution, marketing, and promotion of pharmaceutical products – including biologic products – are subject to extensive and rigorous government regulation. The Federal Food, Drug, and Cosmetic Act (FFDCA) and other federal and state statutes and regulations govern or influence these activities. Non-compliance with applicable requirements can result in fines, recall or seizure of products, total or partial suspension of production and/or distribution, refusal of the government to enter into supply contracts or to approve NDAs, civil penalties and criminal prosecution. Product Approval Process Pharmaceutical products are subject to extensive regulation by the FDA. The FFDCA, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending NDAs, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution. Pharmaceutical product development for a new product or certain changes to an approved product in the U.S.United States typically involves preclinical laboratory and animal tests, the submission to the FDA of an IND, which must become effective before clinical testing may commence, and adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug for each indication for which FDA approval is sought. Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease. Preclinical tests include laboratory evaluation of product chemistry, formulation and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with federal regulations and requirements, including good laboratory practices. The results of preclinical testing are submitted to the FDA as part of an IND along with other information, including information about product chemistry, manufacturing and controls, and a proposed clinical trial protocol. Long term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted. The FDA’s Center for Drug Evaluation and Research fosters early communications between sponsors and new drug review divisions to provide guidance on the data necessary to warrant IND submission, and a 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has neither commented on nor questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin. 48
Clinical trials involve the administration of the IND to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted: (i) in compliance with federal regulations; (ii) in compliance with good clinical practice, or GCP, an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators, and monitors; as well as (iii) under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to the FDA as part of the IND. The FDA may order the temporary or permanent discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The study protocol and informed consent information for patients in clinical trials must also be submitted to an institutional review board, or IRB, for approval. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions. 63
Clinical trials to support NDAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap. In Phase 1, after the initial introduction of the drug into healthy human subjects or patients, the drug is tested to assess metabolism, pharmacokinetics, pharmacological actions, side effects associated with increasing doses, and, if possible, early evidence on effectiveness. Phase 2 usually involves trials in a limited patient population to determine the effectiveness of the drug for a particular indication, dosage tolerance, and optimum dosage, and to identify common adverse effects and safety risks. If a compound demonstrates evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to obtain the additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug and to provide adequate information for the labeling of the drug. In most cases the FDA requires two adequate and well-controlled Phase 3 clinical trials to demonstrate the efficacy of the drug. A single Phase 3 trial with other confirmatory evidence may be sufficient in instances where the study is a large multicenter trial demonstrating internal consistency and a statistically persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible. In addition, the manufacturer of an investigational drug in a Phase 2 or Phase 3 clinical trial for a serious or life-threatening disease is required to make available, such as by posting on its website, its policy on evaluating and responding to requests for expanded access. After completion of the required clinical testing, an NDA is prepared and submitted to the FDA. FDA approval of the NDA is required before marketing of the product may begin in the U.S.. The NDA must include the results of all preclinical, clinical and other testing and a compilation of data relating to the product’s pharmacology, chemistry, manufacture and controls. The cost of preparing and submitting an NDA exceeds USD 3,000,000. The FDA has 60 days from its receipt of an NDA to determine whether the application will be filed based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. If the NDA submission is filed, the FDA reviews the NDA to determine, among other things, whether the proposed product is safe and effective for its intended use. The FDA has agreed to certain performance goals in the review of NDAs. Most such applications for standard review drug products are reviewed within ten to twelve months; most applications for priority review drugs are reviewed in six to eight months. Priority review can be applied to drugs that the FDA determines offer major advances in treatment or provide a treatment where no adequate therapy exists. The review process for both standard and priority review may be extended by the FDA for three additional months to consider certain late-submitted information, or information intended to clarify information already provided in the submission. The FDA may also refer applications for novel drug products, or drug products that present difficult questions of safety or efficacy, to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. After the FDA evaluates the NDA and the manufacturing facilities, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing, or information, in order for the FDA to reconsider the application. If, or when, those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require a risk evaluation and mitigation strategy, or REMS, to help ensure that the benefits of the drug outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the drug. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing. 6449
Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA supplement before the change can be implemented. An NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing NDAs. Quality Assurance The FDA regulates the facilities, processes and procedures used to manufacture and market pharmaceutical products in the U.S. Manufacturing facilities, including those located outside the U.S., must be registered with the FDA and all products made in such facilities must be manufactured in accordance with cGMP regulations enforced by the FDA. Compliance with cGMP regulations requires the dedication of substantial resources and requires significant expenditures. These cGMP standards are particularly stringent for biologic products. The FDA periodically inspects manufacturing facilities and procedures to assure compliance. The FDA may cause a suspension or withdrawal of product approvals if regulatory standards are not maintained. In the event an approved manufacturing facility is required by the FDA to curtail or cease operations, or otherwise becomes inoperable, or a third party contract manufacturing facility faces manufacturing problems, obtaining the required FDA authorization to manufacture at the same or a different manufacturing site could result in production delays, which could adversely affect the Company’s business, results of operations, financial condition and cash flow. The FDA conducts pre-approval inspections of facilities engaged in the development, manufacture, processing, packing, testing and holding of the products subject to INDs. If the FDA concludes that the facilities to be used do not or did not meet cGMP, GLP or GCP requirements, it will not approve an IND application. Corrective actions to remedy the deficiencies must be performed and are usually verified in a sub-sequent inspection. In addition, manufacturers of both pharmaceutical products and active pharmaceutical ingredients (APIs) used to formulate the product also ordinarily undergo a pre-approval inspection, although the inspection can be waived when the manufacturer has had a passing cGMP inspection in the immediate past. Failure of any facility to pass a pre-approval inspection will result in delayed approval and would have a material adverse effect on the Company’s business, results of operations, financial condition and cash flows. The FDA also conducts periodic inspections of facilities to assess their cGMP status. If the FDA were to find serious cGMP non-compliance during such an inspection, it could take regulatory actions that could adversely affect the Company’s business, results of operations, financial condition and cash flows. Imported API and other components needed to manufacture products could be rejected by U.S. Customs, usually after conferring with the FDA. In respect to domestic establishments, the FDA could initiate product seizures or request product recalls and seek to enjoin a product’s manufacture and distribution. In certain circumstances, violations could support civil penalties and criminal prosecutions. In addition, if the FDA concludes that a company is not in compliance with cGMP requirements, sanctions may be imposed that include classifying that company as an “unacceptable supplier”, thereby disqualifying that company from selling products to federal agencies. Marketing Companies that market pharmaceutical products in the U.S. are subject to various federal and state laws pertaining to healthcare fraud and abuse, including prohibitions on the offer of payment or acceptance of kickbacks or other remuneration for the purchase of products, such as inducements to potential patients to request the company’s products. Specifically, the federal Anti-Kickback Statute prohibits persons or entities from knowingly and willfully soliciting, receiving, offering or providing remuneration, directly or indirectly, to induce either the referral of an individual, or the furnishing, recommending, or arranging for a good or service, for which payment may be made under a federal healthcare program, such as the Medicare and Medicaid pro-grams. Due to legislative changes, violations of the Anti-Kickback Statute also carry potential federal False Claims Act liability. Because of the sweeping language of the federal Anti-Kickback Statute, many potentially beneficial business arrangements would be prohibited if the statute were strictly applied. To avoid this outcome, the U.S. Department of Health and Human Services’ Office of Inspector General has published regulations—known as “safe harbors”—that identify exceptions or exemptions to the statute’s prohibitions. Arrangements that do not fit within the safe harbors are not automatically deemed to be illegal but must be evaluated on a case-by-case basis for compliance with the statute. Additionally, many states have adopted laws similar to the federal Anti-Kickback Statute. Some of these state prohibitions apply to referral of patients for healthcare items or services reimbursed by any third party payer, not only the Medicare and Medicaid programs, and do not contain identical safe harbors. 65
The Company is unaware of any violations of these laws. However, due to the breadth of the statutory provisions and the absence of uniform guidance in the form of regulations or court decisions, there can be no assurance that its practices will not be challenged under anti-kickback or similar laws. Violations of such restrictions may be punishable by civil and/or criminal sanctions, including fines and civil monetary penalties, as well as the possibility of exclusion from participation in U.S. federal and state healthcare programs (including Medicaid and Medicare). Any liability from such a violation could have a material adverse effect on our business, financial condition, results of operations and cash flows. 50
In addition, the FDA has the authority to regulate the claims made by a manufacturer in marketing its products to ensure that such claims are true, not misleading, supported by scientific evidence and consistent with the products approved or cleared labeling. Failure to comply with FDA requirements in this regard could result in, among other things, suspensions or withdrawal of approvals, product seizures, injunctions against the manufacture, holding, distribution, marketing and sale of a product, civil and criminal sanctions. Also, the federal False Claims Act prohibits persons from knowingly filing, or causing to be filed, a false claim to, knowingly concealing or knowingly and improperly avoiding or decreasing an obligation to pay or transmit money or property to, or the knowing use of false statements to obtain payment from, the government. When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties for each separate false claim. Various states have also enacted laws modeled after the federal False Claims Act. Federal and state authorities and private whistleblower plaintiffs have brought actions against pharmaceutical product manufacturers alleging that the manufacturers’ activities constituted causing healthcare providers to submit false claims, alleging that the manufacturers themselves made false or misleading statements to the federal government, or alleging that the manufacturers improperly promoted their products for “off-label” uses not approved by the FDA, or offered inducements to referral sources that are prohibited by the federal Anti-Kickback Statute. To the extent the Company becomes the subject of any such investigations or litigation, it could be time-consuming and costly to the Company and could have a material adverse effect on its business. In addition, if its activities are found to violate federal or state False Claims Act statutes, it could have a material adverse effect on its business, financial conditions, results of operations and cash flows. Product Liability There are potential liability risks that arise from the testing, manufacturing, marketing and sale of pharmaceutical products. In addition to direct expenditures for damages, settlement and defense costs, there is a possibility of adverse publicity as a result of product liability claims. Some plaintiffs have received substantial damage awards in some jurisdictions against pharmaceutical companies based upon claims for injuries allegedly caused by the use of their products. In addition, it may be necessary for the Company to voluntarily or mandatorily recall or withdraw products that do not meet approved specifications, or which subsequent data demonstrate may be unsafe or ineffective, which would also result in adverse publicity as well as in costs connected to the recall and loss of revenue. Health Information Privacy and Security There are potential liability risks that arise from the testing, manufacturing, marketing and sale of pharmaceutical products. In addition to direct expenditures for damages, settlement and defense costs, there is a possibility of adverse publicity as a result of product liability claims. Some plaintiffs have received substantial damage awards in some jurisdictions against pharmaceutical companies based upon claims for injuries allegedly caused by the use of their products. In addition, it may be necessary for the Company to voluntarily or mandatorily recall or withdraw products that do not meet approved specifications or which subsequent data demonstrate may be unsafe or ineffective, which would also result in adverse publicity as well as in costs connected to the recall and loss of revenue. Legislative and regulatory initiatives at the state and federal levels address concerns about the privacy and security of health information. HITECH expands the health information privacy and security protections under HIPAA and imposes new obligations to notify individuals and the U.S. Department of Health and Human Services Office for Civil Rights (OCR), of breaches of certain unsecured health information. Compliance with these laws and regulations may require the Company to spend substantial sums, including, but not limited to, purchasing new information technology, which could negatively impact financial results. Additionally, if the Company fails to comply with the HIPAA privacy, security and breach notification standards, it could suffer civil penalties of up to USD 1,500,000 per calendar year for violations of an identical standard and criminal penalties of up to USD 250,000 and 10 years in prison for offenses committed with the intent to sell, transfer, or use individually identifiable health information for commercial advantage, personal gain or malicious harm. In addition, healthcare providers will continue to remain subject to any state laws that are more restrictive than the federal privacy regulations. These privacy laws vary by state and could impose additional penalties. 66
The provisions of HIPAA criminalize situations that previously were handled exclusively civilly through repayments of overpayments, offsets and fines by creating new federal healthcare fraud crimes. Further, as with the federal laws, general state criminal laws may be used to prosecute healthcare fraud and abuse. A violation could subject the Company to penalties, fines and/or possible exclusion from Medicare or Medicaid. Such sanctions could significantly reduce its financial results. Future healthcare legislation and regulation or other changes in the administration of or interpretation of existing legislation or regulations regarding governmental healthcare pro-grams could have an adverse effect on the Company’s business the results of its operations. Regulation in the European Union Product development, the regulatory approval process, and safety monitoring of medicinal products and their manufacturers in the EU proceed in much the same manner as they do in the U.S.. Therefore, many of the issues discussed above apply similarly in the context of the EU. In addition, drugs are subject to the extensive price and reimbursement regulations of the various EU Member States. 51
In the EEA, which is comprised of the 27 Member States of the EU plus Norway, Iceland and Liechtenstein, medicinal products can only be commercialized after obtaining a marketing authorization. There are two types of marketing authorization: the Community Marketing Authorization, which is issued by the EC through the Centralized Procedure based on the opinion of the Committee for Medicinal Products for Human Use (CHMP), a body of the EMA, and which is valid throughout the entire territory of the EEA; and the National Marketing Authorization, which is issued by the competent authorities of the Member States of the EEA and authorizes marketing only in that Member State’s national territory and not the EEA as a whole. The Centralized Procedure is compulsory for human medicines for the treatment of human immunodeficiency virus or acquired immune deficiency syndrome (AIDS), cancer, diabetes, neurodegenerative diseases, autoimmune and other immune dysfunctions, and viral diseases; for veterinary medicines for use as growth or yield enhancers; for medicines derived from biotechnology processes, such as genetic engineering; for advanced-therapy medicines, such as gene-therapy, somatic cell-therapy or tissue-engineered medicines; and for officially designated ‘orphan medicines’ (medicines used for rare human diseases). The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation, or for products that are in the interest of public health in the EU. The National Marketing Authorization is for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National Marketing Authorization can be recognized in another Member State through the Mutual Recognition Procedure. If the product has not received a National Marketing Authorization in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure. Under the Decentralized Procedure, an identical dossier is submitted to the competent authorities of each of the Member States in which the marketing authorization is sought, one of which is selected by the applicant as the Reference Member State (RMS). If the RMS proposes to authorize the product, and the other Member States do not raise objections, the product is granted a National Marketing Authorization in all the Member States in which the authorization was sought. Before granting the marketing authorization, the EMA or the competent authorities of the Member States of the EEA assesses the risk–benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy. Clinical Studies As is the case in the U.S., the various phases of preclinical and clinical research in the EU are subject to significant regulatory controls. The EU Clinical Trial Regulation (EU) No. 536/2014 (Clinical(“Clinical Trials Relation)Relation”) on clinical trials and medicinal products for human use, repealed Directive 2001/20/EC. The Clinical Trials Regulation entered into application on January 31, 2022 and is intended to simplify the current rules for clinical trial authorization and standards of performance. For instance, there will be a streamlined application procedure via a single entry point, a EU portal and database. The new clinical trial portal and database will be maintained by the EMA in collaboration with the European Commission and the EU Member States. The objectives of the Clinical Trials Regulation include consistent rules for conducting trials throughout the EU, consistent data standards and adverse events listing, and consistent information on authorization status. Additionally, information on the conduct and results of each clinical trial carried out in the EU will be made publicly available. 6752
Marketing Approval Marketing approvals under the EU regulatory system may be obtained through a centralized or decentralized procedure. The centralized procedure results in the grant of a single marketing authorization, which is valid for all (currently 27) EU Member States and the three European Free Trade Association (EFTA) members (Norway, Iceland and Liechtenstein). Pursuant to Regulation (EC) No. 726/2004, as amended, the centralized procedure is mandatory for drugs developed by means of specified biotechnological processes, advanced-therapy medicinal products, drugs for human use containing a new active substance for which the therapeutic indication is the treatment of specified diseases, including but not limited to AIDS, neurodegenerative disorders, auto-immune diseases and other immune dysfunctions, as well as drugs designated as orphan drugs. The CHMP also has the discretion to permit other products to use the centralized procedure if it considers them sufficiently innovative or they contain a new active substance. In the marketing authorization application, the applicant has to properly and sufficiently demonstrate the quality, safety and efficacy of the drug. Under the centralized approval procedure, the CHMP, possibly in conjunction with other committees, is responsible for drawing up the opinion of the EMA on any matter concerning the admissibility of the files submitted in accordance with the centralized procedure, such as an opinion on the granting, variation, suspension or revocation of a marketing authorization, and pharmacovigilance. The CHMP and other committees are also responsible for providing guidelines and have published numerous guidelines that may apply to our product candidates. These guidelines provide additional guidance on the factors that the EMA will consider in relation to the development and evaluation of drug products and may include, among other things, the preclinical studies required in specific cases, the manufacturing and control information that should be submitted in a marketing authorization application, and the post-approval measures required to monitor patients and evaluate the long-term efficacy and potential adverse reactions. Although these guidelines are not legally binding, we believe that our compliance with them is likely to be necessary to gain approval for any of our product candidates. The maximum timeframe for the evaluation of a marketing authorization application by the CHMP under the centralized procedure is 210 days after receipt of a valid application. This period will be suspended until such time as the supplementary information requested by the CHMP has been provided by the applicant. Likewise, this time limit will be suspended for the time allowed for the applicant to prepare oral or written explanations. When an application is submitted for a marketing authorization in respect of a drug that is of major interest from the viewpoint of public health and in particular therapeutic innovation, the applicant may request an accelerated assessment procedure. If the CHMP accepts such a request, the time limit of 210 days will be reduced to 150 days, but it is possible that the CHMP can revert to the standard time limit for the centralized procedure if it considers that it is no longer appropriate to conduct an accelerated assessment. If the CHMP concludes that the quality, safety and efficacy of the product are sufficiently proven, it adopts a positive opinion. This is sent to the EC, which drafts a decision within approximately 67 days following the CHMP opinion. After consulting with the Member States, the EC adopts a decision and grants a marketing authorization, which is valid for the whole of the EEA. The marketing authorization may be subject to certain conditions, which may include, without limitation, the performance of post-authorization safety and/or efficacy studies. The EMA has various programs, including accelerated assessment, conditional approval and Priority Medicines (PRIME), which are intended to increase agency interactions, expedite or facilitate the process for reviewing drug candidates, and/or provide for initial approval on the basis of surrogate endpoints. One or more of our product candidates may qualify for some of these expedited development and review programs. However, even if a drug candidate qualifies for one or more of these programs, the EMA may later decide that the drug candidate no longer meets the conditions for qualification. Eligibility to the PRIME scheme is limited to products considered to offer a major therapeutic advantage in populations with high unmet need. PRIME is a voluntary scheme aimed at enhancing interaction and early dialogue with developers of promising medicines through achieving the early appointment of the Rapporteur for the product, optimizing development plans and speeding up evaluation so these medicines can reach patients earlier. Products benefiting from PRIME can expect to be eligible for accelerated assessment at the time of application for a marketing authorization application. 68
EU legislation also provides for a system of regulatory data and market exclusivity. According to Article 14(11) of Regulation (EC) No. 726/2004, as amended, and Article 10(1) of Directive 2001/83/EC, as amended, upon receiving marketing authorization, new chemical entities approved on the basis of a complete independent data package benefit from 8 years of data exclusivity and an additional 2 years of market exclusivity. Data exclusivity prevents regulatory authorities in the EU from referencing the innovator’s data to assess a generic (abbreviated) application. During the additional 2-year period of market exclusivity, a generic marketing authorization can be submitted, and the innovator’s data may be referenced, but no generic medicinal product can be marketed until the expiration of the market exclusivity. The overall 10-year period will be extended to a maximum of 11 years if, during the first 8 years of those 10 years, the marketing authorization holder (MAH) obtains an authorization for one or more new therapeutic indications that, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. Even if a compound is considered to be a new chemical entity and the innovator can gain the period of data exclusivity, another company nevertheless could also market another version of the drug if such company obtained marketing authorization based on a marketing authorization application with a completely independent data package of pharmaceutical test, preclinical tests and clinical studies. However, products designated as orphan medicinal products enjoy, upon receiving marketing authorization, a period of 10 years of orphan market exclusivity. See also “Orphan drug regulation” below. Depending upon the timing and duration of the EU marketing authorization process, products may be eligible for an SPC of up to five years, pursuant to Regulation (EC) No. 469/2009. Such SPCs extend the rights under the basic patent for the drug. 53
In the EU, the pediatric regulation (Regulation (EC) No 1901/2006, as amended) requires sponsors to submit a pediatric investigation plan at the end of Phase 1. This plan will provide the details of the quality, non-clinical and clinical studies required to support the authorization of a pediatric indication. Additional rules apply to medicinal products for pediatric use under Regulation (EC) No. 1901/2006. Potential incentives include a six-month extension of any supplementary protection certificate granted pursuant to Regulation (EC) No. 469/2009, but not in cases in which the relevant product is designated as an orphan medicinal product pursuant to Regulation (EC) No. 141/2000, as amended. Instead, a medicinal product designated as an orphan medicinal product may enjoy an extension of the 10-year market exclusivity period granted under Regulation (EC) No. 141/2000 to 12 years subject to the conditions applicable to orphan drugs. Orphan Drug Regulation In the EU, Regulation (EC) No. 141/2000, as amended, states that a drug will be designated as an orphan drug if its sponsor can establish: that it is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting not more than 5 in 10,000 persons in the EU when the application is made, or that it is intended for the diagnosis, prevention or treatment of a life- threatening, seriously debilitating or serious and chronic condition in the EU and that without incentives it is unlikely that the marketing of the drug in the EU would generate sufficient return to justify the necessary investment; and that there exists no satisfactory method of diagnosis, prevention or treatment of the condition in question that has been authorized in the EU or, if such method exists, that the drug will be of significant benefit to those affected by that condition. Regulation (EC) No. 847/2000 sets out further provisions for implementation of the criteria for designation of a drug as an orphan drug. An application for the designation of a drug as an orphan drug must be submitted at any stage of development of the drug before filing of a marketing authorization application. If a EU-wide community marketing authorization in respect of an orphan drug is granted or if all the EU Member States have granted marketing authorizations in accordance with the procedures for mutual recognition, the EU and the Member States will not, for a period of 10 years, accept another application for a marketing authorization, or grant a marketing authorization or accept an application to extend an existing marketing authorization, for the same therapeutic indication, in respect of a similar drug. This period may, however, be reduced to 6 years if, at the end of the fifth year, it is established, with respect to the drug concerned, that the criteria for orphan-drug designation are no longer met; in other words, when it is shown on the basis of available evidence that the product is sufficiently profitable not to justify maintenance of market exclusivity. Notwithstanding the foregoing, a marketing authorization may be granted, for the same therapeutic indication, to a similar drug if: the holder of the marketing authorization for the original orphan drug has given its consent to the second applicant; the holder of the marketing authorization for the original orphan drug is unable to supply sufficient quantities of the drug; or 69
the second applicant can establish in the application that the second drug, although similar to the orphan drug already authorized, is safer, more effective or otherwise clinically superior. Other incentives available to orphan drugs in the EU include financial incentives such as a reduction of fees or fee waivers and protocol assistance. ODD does not shorten the duration of the regulatory review and approval process. Manufacturing and Manufacturers’ License Pursuant to Directive 2003/94/EC, as transposed into the national laws of the Member States, the manufacturing of investigational medicinal products and approved drugs is subject to a separate manufacturer’s license and must be conducted in strict compliance with cGMP requirements, which mandate the methods, facilities and controls used in manufacturing, processing and packing of drugs to assure their safety and identity. Manufacturers must have at least one qualified person permanently and continuously at their disposal. The qualified person is ultimately responsible for certifying that each batch of finished product released onto the market has been manufactured in accordance with cGMP and the specifications set out in the marketing authorization or investigational medicinal product dossier. cGMP requirements are enforced through mandatory registration of facilities and inspections of those facilities. Failure to comply with these requirements could interrupt supply and result in delays, unanticipated costs and lost revenues, and subject the applicant to potential legal or regulatory action, including but not limited to warning letters, suspension of manufacturing, seizure of product, injunctive action, or possible civil and criminal penalties. 54
Wholesale Distribution and License Pursuant to Directive 2001/83/EC, the wholesale distribution of medicinal products is subject to the possession of an authorization to engage in activity as a wholesaler in medicinal products. Possession of a manufacturing authorization includes authorization to distribute by wholesale the medicinal products covered by that authorization. The distribution of medicinal products must comply with the principles and guidelines of cGDP. Advertising In the EU, the promotion of prescription medicines is subject to intense regulation and control, including EU and national legislation as well as self- regulatory codes (industry codes). Advertising legislation inter alia includes a prohibition on direct-to-consumer advertising. All advertising of prescription medicines must be consistent with the product’s approved Summary of Product Characteristics, and must be factual, accurate, balanced and not misleading. Advertising of prescription medicines pre-approval or off-label is not allowed. Some jurisdictions require that all promotional materials for prescription medicines be subjected to prior review and approval, either internal or regulatory. Other Regulatory Requirements A Marketing Authorization Holder (MAH) for a medicinal product is legally obliged to fulfill a number of obligations by virtue of its status as an MAH. The MAH can delegate the performance of related tasks to third parties, such as distributors or marketing partners, provided that this delegation is appropriately documented and the MAH maintains legal responsibility and liability. An MAH for a medicinal product is legally obliged to fulfill a number of obligations by virtue of its status as an MAH. The MAH can delegate the performance of related tasks to third parties, such as distributors or marketing partners, provided that this delegation is appropriately documented and the MAH maintains legal responsibility and liability. The obligations of an MAH include the following: | | • | | Manufacturing and batch release. MAHs should guarantee that all manufacturing operations comply with relevant laws and regulations, applicable GMPs, and the product specifications and manufacturing conditions set out in the marketing authorization, and that each batch of product is subject to appropriate release formalities. |
| | • | | Availability and continuous supply. Pursuant to Directive 2001/83/EC, as transposed into the national laws of the Member States, the MAH for a medicinal product and the distributors of the said medicinal product actually placed on the market in a Member State shall, within the limits of their responsibilities, ensure appropriate and continued supplies of that medical product to pharmacies and persons authorized to supply medicinal products so that the needs of patients in the Member State in question are covered. |
70
| | • | | Advertising and promotion. MAHs remain responsible for all advertising and promotion of their products, including promotional activities by other companies or individuals on their behalf, and in some cases must conduct internal or regulatory pre-approval of promotional materials. Regulation in this area also covers interactions with healthcare practitioners and/or patient groups, and in some jurisdictions legal or self-regulatory obligations to disclose such interactions exist. |
| | • | | Medical affairs/scientific service. MAHs are required to disseminate scientific and medical information on their medicinal products to healthcare professionals, regulators and patients. |
| | • | | Legal representation and distributor issues. MAHs are responsible for regulatory actions or inactions of their distributors and agents. |
| | • | | Preparation, filing and maintenance of the application and subsequent marketing authorization. MAHs must maintain appropriate records, comply with the marketing authorization’s terms and conditions, fulfill reporting obligations to regulators, submit renewal applications and pay all appropriate fees to the authorities. We may hold any future marketing authorizations granted for our product candidates in our own name or appoint an affiliate or a collaboration partner to hold marketing authorizations on our behalf. Any failure by an MAH to comply with these obligations may result in regulatory action against an MAH and ultimately threaten our ability to commercialize our products. |
55
International Regulation In addition to regulations in the U.S. and Europe, a variety of foreign regulations govern clinical trials, commercial sales and distribution of product candidates. The approval process varies from country to country and the time to approval may be longer or shorter than that required for FDA or EMA approval. Pharmaceutical Coverage, Pricing and Reimbursement In both domestic and foreign markets, our or our collaboration partners’ sales of any approved products will depend in part on the availability of coverage and adequate reimbursement from third-party payors. Third-party payors include government authorities, managed care providers, private health insurers and other organizations. Patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Patients are unlikely to use our products, if approved, unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our products. Sales of our products will therefore depend substantially, both domestically and abroad, on the extent to which the costs of our products will be paid by third-party payors. These third-party payors are increasingly focused on containing healthcare costs by challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the coverage and reimbursement status of newly approved healthcare product candidates. The market for our product candidates for which we may receive regulatory approval will depend significantly on access to third-party payors’ drug formularies or lists of medications for which third-party payors provide coverage and reimbursement. The industry competition to be included in such formularies often lead to downward pricing pressures on pharmaceutical or biopharmaceutical companies. Additionally, third-party payors may refuse to include a particular branded drug in their formularies or otherwise restrict patient access to a branded drug when a less costly generic equivalent or another alternative is available. Because each third-party payor individually approves coverage and reimbursement levels, obtaining coverage and adequate reimbursement is a time-consuming, costly and sometimes unpredictable process. We may be required to provide scientific and clinical support for the use of any product to each third-party payor separately with no assurance that approval would be obtained, and we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our products. This process could delay the market acceptance of any product and could have a negative effect on our future revenues and operating results. We cannot be certain that our product candidates will be considered cost-effective. Because coverage and reimbursement determinations are made on a payor-by-payor basis, obtaining acceptable coverage and reimbursement from one payor does not guarantee we will obtain similar acceptable coverage or reimbursement from another payor. If we are unable to obtain coverage of, and adequate reimbursement and payment levels for, our product candidates from third-party payors, physicians may limit how much or under what circumstances they will prescribe or administer them and patients may decline to purchase them. This in turn could affect our ability to successfully commercialize our products and impact our profitability, results of operations, financial condition and future success. 71
In the EU, the pricing and reimbursement mechanisms by private and public health insurers vary largely by country and even within countries. The public systems reimbursement for standard drugs is determined by guidelines established by the legislator or responsible national authority. The approach taken varies by Member State. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. Other Member States allow companies to fix their own prices for medicines but monitor and control company profits and may limit or restrict reimbursement. The downward pressure on healthcare costs in general, particularly prescription drugs, has become very intense. As a result, increasingly high barriers to the entry of new products are being erected and some EU countries require the completion of studies that compare the cost-effectiveness of a particular product candidate with that of currently available therapies in order to obtain reimbursement or pricing approval. Special pricing and reimbursement rules may apply to orphan drugs. Inclusion of orphan drugs in reimbursement systems tend to focus on the medical usefulness, need, quality and economic benefits to patients and the healthcare system as for any drug. Acceptance of any medicinal product for reimbursement may come with cost, use and often volume restrictions, which again can vary by country. In addition, results based rules of reimbursement may apply. Environmental, Health, and Safety Laws and Regulations We are subject to numerous environmental, health and safety laws and regulations and permitting requirements, including those governing laboratory procedures, decontamination activities, and the handling, transportation, use, remediation, storage, treatment, and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, and the risk of injury, contamination or noncompliance with environmental, health and safety requirements cannot be eliminated. Although compliance with such laws and regulations and permitting requirements has not had a material effect on our capital expenditures, earnings or competitive position, environmental, health and safety laws, and regulations and permitting requirements have tended to become increasingly stringent and, to the extent that legal or regulatory changes may occur in the future, they could result in, among other things, increased costs to us or the impairment of our research, development or production efforts. 56
Competition The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies and intense competition. While we believe that our knowledge, experience and scientific resources provide us with competitive advantages, we face potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions and governmental agencies and public and private research institutions, which may in the future develop products to treat those diseases that we currently or, in the future, seek to treat. Any product candidates that we successfully develop and commercialize may compete with existing therapies and new therapies that may become available in the future. Many of our competitors have far greater marketing and research capabilities than us. All of these companies and institutions may have product candidates in development that are or may become superior to our product candidates. Our commercial opportunity would be reduced significantly if our competitors develop and commercialize products that are safer, more effective, more convenient, have fewer side effects or are less expensive than our product candidates. | C. | ORGANIZATIONAL STRUCTURE |
We are a Swiss stock corporation (société anonyme). We were originally formed in 2013, with our registered office and domicile in Geneva, Switzerland. Our Swiss enterprise identification number is CHE-113.516.874. We are located in the Canton of Geneva, City of Geneva,headquartered at Avenueavenue de Sécheron 15, 1202 Genève,Geneva, Switzerland. As of the date of this annual report, we, have the following direct subsidiaries: | | | | | | | | Name | | Domicile | | Percent
Owned | | Relief Therapeutics International SA | | Switzerland | | | 100 | | Relief Therapeutics US, Inc. | | Connecticut (U.S.) | | | 100 | | Relief Therapeutics, Inc. | | Delaware (U.S.) | | | 100 | | APR Applied Pharma Research SA | | Switzerland | | | 100 | | APR Applied Pharma Research Holding SA | | Switzerland | | | 100 | | APR Applied Pharma Research – Italy s.r.l. | | Italy | | | 100 | | APR Applied Pharma Research Deutschland GmbH | | Germany | | | 100 | | AdVita Lifescience GmbH | | Germany | | | 100 | | AdVita Lifescience AG | | Switzerland | | | 100 | | AdVita Lifescience, Inc. | | New York (U.S.) | | | 100 | |
| D. | PROPERTY, PLANT AND EQUIPMENT |
The Group leases approximately 1,800 square feet of office, lab space and representative offices located in Geneva and Balerna (Switzerland), Freiburg im Breisgau and Offenbach am Main (Germany), and RomeMonza (Italy). Our headquarters are at Avenue de Sécheron 15, Geneva, Switzerland. We aredo not aware ofown any environmental issues or other constraints that would materially impact the intended use of our facilities.real property. While we may require additional space and facilities our business expands, we believe that our current facilities are suitable and adequate space will be available to meet our current needs. 72
| ITEM 4A | UNRESOLVED STAFF COMMENTS |
None. | ITEM 5 | OPERATING AND FINANCIAL REVIEW AND PROSPECTS |
We are a company developing drugs via participationYou should read the following discussion and analysis of our financial condition and results of operations together with our audited consolidated financial statements, including the notes thereto, included in active entities that have obtained intellectual properties through their own research activities, or via in-licensing, or via internal research and development activities. Historically, our development has focused primarily on clinical-stage projects based on molecules of natural origin (peptides and proteins) with a history of clinical testing and use in human patients or a strong scientific rationale.
this Annual Report. The following discussion contains referencesis based on our financial information prepared in accordance with IFRS as issued by the IASB, which might differ in material respects from generally accepted accounting principles in other jurisdictions. The following discussion includes forward-looking statements that involve risks, uncertainties and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of many factors, including but not limited to the financial statements of Relief Therapeutics Holding SAthose described under “Item 3. Key information—D. Risk factors” and its consolidated subsidiaries (also referred to as the Company). These financial statements consolidate the Company’s subsidiaries and include the Company’s interest in investments held at fair value. Subsidiaries are those entities over which the Company retains control. Where we have neither control nor significant influence for financial accounting purposes, we recognize our holding in such entity at fair value. For additional information regarding the accounting treatment of these entities, see Note 1 of our consolidated financial statements includedelsewhere in this annual report. For additional information regarding our operating structure, see “Basis of Presentation and Consolidation” below.Annual Report. Overview We are a Swiss, commercial-stage biopharmaceutical company committed to delivering innovative treatment options with the potential for transformative outcomes to benefit those suffering from rare debilitating conditions that have no or limited treatment options. Our cost-effective, capital-efficient approach to drug development and commercialization is focused on rare metabolic disorders, rare skin diseases, rare respiratory diseases and rare monogenetic diseases. OurHistorically, we have had a diversified portfolio offers a balanced mix of marketed revenue-generating products and developmental product candidates many of which use our proprietary globally patented drug delivery platform technologies that have utility for development in other specialty or rare disease therapeutic areas and a highly targeted clinical development pipeline consisting of risk-mitigated assets that have been engineeredwhich we believe allow for improvements in efficacy, safety or convenience to benefit the lives of patients. In addition, the Company is commercializing several legacy products via licensingFollowing a comprehensive review of our commercial and distribution partners.
Weclinical product portfolio, we are actively pursuing a strategy to diversifyof refocusing our business on products addressing rare conditions in the dermatological therapeutic area where we believe there is considerable unmet need. For our other commercial and development stage products currently in our portfolio, we are actively seeking potential partners through out-licensing, divestitures or other collaboration transactions.
57
In accordance with this strategy, on March 21, 2024, we entered into an exclusive license and supply agreement with Eton Pharmaceuticals, Inc. for the commercialization of the GOLIKE® family of products in the United States. As we transition to a business-to-business strategy for selected commercial and development stage products, we are focusing our efforts on advancing RLF-TD011, an acid oxidizing solution of hypochlorous acid that is self-administered, sprayable, and is being studied for the treatment of wounds in epidermolysis bullosa. RLF-TD011 was developed using the Company’s proprietary, patented TEHCLO™ Nanotechnology platform. Our Strategy Our mission is to provide therapeutic relief to those suffering from debilitating rare diseases that have limited or no treatment options to help them live their best possible lives and achieve their full potential. We target established products with a proven history of safety and efficacy and either initial human therapeutic activity, proof-of-concept or a strong scientific rationale, allowing for relatively short, capital-efficient clinical trials with clear endpoints. Our research and development resources are directed toward optimizing the therapeutic potential of these assets to deliver improvements in efficacy, safety and convenience through the ongoing evaluationapplication of our proprietary platform technologies, drug delivery systems or novel dosage forms. Our strategy plans leverage our clinical trial and business development expertise to establish and grow our business in rare dermatologic disorders with high unmet need. We intend to use our product portfolio in rare metabolic and respiratory diseases through partnerships and collaborations to generate cash flow in support of investments in the rare dermatological therapeutic area. Rare Dermatologic Disorders: | • | | RLF-TD011 in epidermolysis bullosa (EB). We are currently studying RLF-TD011 in a 12-subjectproof-of-concept (POC) investigated initiated trial (IIT) where enrollment and treatment have been completed and the results are expected in mid to late-2024. Based on these results, we intend to initiate pre-IND discussions with the FDA by the end of 2024, after which time we expect to file an IND and initiate a Phase 2 trial shortly thereafter. If approved, we would evaluate additional indications for RLF-TD011 and expand our portfolio in rare dermatological therapies through internal or business development activities. |
Rare Metabolic Disorders: PKU GOLIKE® in phenylketonuria (PKU). We recently announced a license and supply relationship with Eton Pharmaceuticals, Inc. to ensure the commercialization of PKU GOLIKE in the United States. We further intend to maximize the commercial potential in-licensing opportunities. To bring treatmentsof the PKU GOLIKE® family of products in all major territories through licencees in order to patients as quickly as possible,create a steady and growing positive cash flow; PKU GOLIKE is an approved, and fully reimbursed line of patented and differentiated medical food products for the dietary management of PKU. | • | | RLF-OD032 in PKU. The Company acquired worldwide rights (except in the United Kingdom) to RLF-OD032 in 2022. We intend to complete clinical studies of this product candidate in 2024 after which time we expect to file a 505(b)(2) NDA with the FDA. If approved, we intend to divest or out-license this product. |
| • | | OLPRUVA® in urea cycle disorders (UCDs). The Company in-licensed the rights for OLPRUVA in Europe and funded part of the development of OLPRUVA in the U.S. by Acer Therapeutics, Inc. (Acer). The FDA approved OLPRUVA for the treatment of UCDs in December 2022. Zevra Therapeutics Inc. (Zevra) acquired Acer in 2023 and went through a full commercial launch of the product in January 2024. Relief is entitled to receive a 10% continuing royalty on the net sales of OLPRUVA in the U.S. up to a cumulative amount of USD 45 million. We plan to continue working with Zevra to maximize the commercial potential of OLPRUVA in the U.S. and to identify partnership opportunities for Europe. |
Rare Respiratory Diseases: | • | | RLF-100 in pulmonary indications. We intend to explore partnership opportunities with seasoned respiratory biopharmaceutical companies to advance RLF-100® (aviptadil acetate) for both injectable intravenous and inhaled use, and to maximize the potential value of the program. |
Additionally, we are seekingwill continue to evaluate business development opportunities that expand our portfolio in the rare dermatological therapeutic area. We will consider partnerships with, or acquisitions of, companies that have late-stage clinical moleculesassets with a strong human safety profile, allowing for relatively short, capital-efficient clinical trials with clear endpoints. We are also evaluating prospective opportunities that fit withinand efficacy profiles where we can bring to bear our genetic medicine initiative for devastating, as-yet-unaddressed, rare monogenetic diseases. We are led by a provendevelopment expertise and seasoned management team of business leaders with significant experience in discovering, developingplatform technologies to quickly and commercializing important new medicines, delivering them to marketcapital efficiently develop and maximizing shareholder value. Collectively, the members of our management team have overseen research and development of products supporting regulatory approvals as well as commercial launches of marketed products.
In March 2021, we signed a Collaboration and License Agreement with Acer Therapeutics, Inc. (Acer) for the worldwide development and commercialization of ACER-001 (now OLPRUVA™) for the treatment of urea cycle disorders (UCDs) and maple syrup urine disease (MSUD). OLPRUVA™ is a proprietary powder formulation of sodium phenylbutyrate (NaPB) designed to be both taste-masked and immediate release. In August 2021, Acer submitted an NDA for ACER-001 to the FDA for use as a treatment of UCD, which submission was accepted for filing in November 2021 with a PDUFA decision date of June 5, 2022. However, on or about the PDUFA decision date, Acer received a complete response letter stating that a satisfactory inspection of Acer’s third-party contract manufacturer would be required before its NDA for ACER-001 could be approved. On December 27, 2022, Relief and Acer announced that, after resubmission, the FDA had approved ACER-001, under the trade name OLPRUVA™, for the treatment of UCDs. In addition, Acer has filed an IND to investigate OLPRUVA™ for the treatment of MSUD.
In June 2021, we signed and closed a definitive agreement to acquire all outstanding shares of APR Applied Pharma Research SA (APR), a privately held Swiss pharmaceutical company with over 25 years’ experience in identifying, developing and globally commercializing known molecules engineered with drug delivery systems in niche and rare diseases.commercialize these product candidates.
7358
In July 2021, Product Portfolio and Pipeline
Relief acquired AdVita Lifescience GmbH (AdVita),Therapeutics’ portfolio consists of a Germany-based privately held pharmaceutical company developingbalanced mix of marketed, revenue-generating products for the treatment and diagnosis ofglobally patented drug delivery platform technologies. Our pipeline spans three core therapeutic areas: rare lung diseases. AdVita’s capabilities helped the Company to further progress the development of RLF-100® for a range of lungdermatological disorders, rare metabolic disorders, and rare respiratory diseases. In July 2022, we executed a definitive agreement with Meta Healthcare Ltd. (Meta), acquiring the worldwide rights, except for the UK, for a novel dosage form of a prescription drug already approved by the FDA and intended for the treatment of patients with PKU. This improved product is expected to increase patient acceptance and compliance as well as enable easier, self or caregiver administered metered dosing and dispensing. According to the terms of the agreement, Meta shall transfer to Relief all data, know-how, as well as any intellectual property as developed or generated so far by Meta. Relief shall only be responsible for funding the remaining development work as well as for filing and prosecuting an NDA in all countries worldwide except for the UK where Relief Therapeutics shall grant a license back to Meta, enabling Meta to directly promote and commercialize the product in such country. Other than the initial acquisition payment and low double-digit royalty payments on net profit of the product in the various countries, Relief shall be under no obligation to fund or pay any other amount to Meta.
Previously, Relief partnered with NeuroRx, Inc. (NeuroRx) to seek to
| A. | OPERATING RESULTSdevelop RLF-100® as a treatment of COVID-19. In September 2020, Relief entered into a binding collaboration agreement with NeuroRx (the Collaboration Agreement). The Collaboration Agreement established the terms under which we and NeuroRx collaborate and assist each other to maximize the revenues in our respective territories from the sale of aviptadil for intravenous and inhale use primarily for the treatment of COVID-19 related conditions. Based on numerous breaches of the Collaboration Agreement by NeuroRx, in October 2021, we filed a lawsuit against NeuroRx and its then CEO, Dr. Jonathan Javitt, for multiple breaches of the Collaboration Agreement. The complaint was filed in the Supreme Court of the State of New York in Manhattan. On January 10, 2022, NeuroRx filed a complaint against Relief alleging that we were in breach of the Collaboration Agreement and have thus repudiated and cancelled the Collaboration Agreement. Additionally, NeuroRx’s claims included a count for defamation.
On November 14, 2022, Relief and NRx issued a press release announcing that the parties had entered into a definitive settlement agreement to resolve all matters relating to the pending litigation. At the closing of this settlement, held on December 19, 2022, (i) NRx transferred to Relief all of the assets that it previously used in its aviptadil development program, including its regulatory filings, patent applications, clinical data, and the formulation of the aviptadil product it was previously developing, (ii) Relief has the exclusive right and control going forward to develop and commercialize an aviptadil product, (iii) Relief has agreed to use commercially reasonable efforts to continue the existing Right to Try Program for aviptadil in the U.S. for at least two years, (iv) Relief will pay NRx milestone payments if it can successfully obtain commercial approval of an aviptadil product (whether for COVID-19 or any other indication), (v) Relief will pay NRx royalties based on a percentage of future sales of an aviptadil product (whether for COVID-19 or any other indication), up to a maximum of $30 million in the aggregate, (vi) NRx Pharmaceuticals has agreed not to compete in the development of an aviptadil product in the future, and (vii) at the closing, Relief and NRx Pharmaceuticals have dismissed their pending litigation. Further, as part of the settlement, the parties cancelled the Collaboration Agreement and exchanged mutual releases with respect to matters that were the subject of the litigation.
Material Agreements
We are party to certain agreements with third parties relating to licensing, collaboration, or other matters that are material to our business and performance.
NeuroRx
In September 2020, we entered into a collaboration agreement with NeuroRx for the global commercialization of RLF-100® (aviptadil acetate) and the selection of commercial partners. This partnership was designed to rapidly advance RLF-100® through clinical development so that it reaches COVID-19 patients worldwide as soon as possible. Under the agreement, NeuroRx was to lead commercialization in the U.S., Canada, and Israel, while we were to lead commercialization in Europe and the rest of the world.
On December 19, 2022, we finalized a settlement with NeuroRx by which the collaboration agreement was cancelled. For additional information, “Item 4. Information on the Company – B. Business Overview.”
74
Acer Therapeutics
On January 25, 2021, we entered into an option agreement with Acer Therapeutics Inc providing exclusivity for the right to negotiate a potential collaboration and license agreement (CLA) for worldwide development and commercialization for ACER-001 (now OLPRUVA™).
Under the terms of the option agreement, we paid Acer a $1 million USD non-refundable payment in return for exclusivity until June 30, 2021 to negotiate and enter into a definitive collaboration and license agreement for the development of ACER-001. Further, in connection with entering into the option agreement, we made a $4 million USD secured loan to Acer.
On March 22, 2021, both companies announced the execution of the CLA. Acer since received a $10 million USD cash payment (originally $14 million USD, offset by repayment of the $4 million USD outstanding balance of the prior loan, plus interest, to Acer). We also committed to pay Acer up to $20 million USD ($15 million which has been paid to-date) in U.S. development and commercial launch costs of the molecule for the treatment of the UCD and MSUD indications. Acer will retain development and commercialization rights in the U.S., Canada, Brazil, Turkey and Japan. The companies will split net profits from Acer’s territories 60 percent to 40 percent in our favor. In addition, we have licensed the rights for the rest of the world, where Acer will receive from us a 15 percent royalty on all revenues received in our territories. Acer may also receive a total of $6 million USD in development milestone payments following the first EU marketing approvals for UCDs and MSUD.
GEM Global Yield LLC SCS
On January 20, 2021, we signed a binding agreement with our largest shareholder, GEM Global Yield LLC SCS (GEM) for the implementation of a new share subscription facility (SSF) in the amount of up to CHF 50 million.
Under the terms of the SSF, we have the right to periodically, during a timeframe of up to three years, issue and sell shares to GEM. Under the facility, GEM undertakes to subscribe to or acquire our ordinary registered shares upon our exercise of a drawdown notice. In accordance with the customary terms of the SSF agreement, we control the timing and maximum amount of any drawdown and retains the right, not the obligation, to draw down on the full commitment amount. Future subscription prices under the SSF will correspond to 90 percent of the average of the closing bid prices on the SIX Swiss Exchange during the reference period, which corresponds to fifteen trading days following Relief’s drawdown notice.
Under the terms of the SSF, we are required to pay GEM a commitment fee of CHF 1.25 million, which is currently outstanding as an interest bearing loan pursuant to the SSF. As of the date of this annual report, no amounts have been drawn on this facility.
APR Applied Pharma Research SA
On April 30, 2021, we entered into a binding term sheet with the then current shareholders of APR Applied Pharma Research SA, a privately held Swiss company with over 25 years of experience in identifying developing and commercializing known molecules engineered with drug delivery systems in niche and rare diseases, to acquire all of the outstanding shares of APR. On June 28, 2021, the former shareholders of APR and Relief signed and closed a definitive agreement for Relief to acquire all outstanding shares of APR. Under the terms of the agreement APR’s shareholders have received from Relief CHF 21.5 million in cash and 206,786,784 Consideration Shares at a value of CHF 45 million when the Consideration Shares were issued and listed. The APR shareholders were also eligible to receive possible future contingent milestone payments in the aggregate maximum amount of up to CHF 35 million, upon achievement of pre-agreed objectives involving (i) the execution of a definitive agreement for the commercialization of Sentinox (as such product is defined below), (ii) the launch of Sentinox in the first of France, Germany, Spain, Italy, and the UK, (iii) the launch of GOLIKE in the U.S., and (iv) the launch of RLF-TD011 (as such product is described below) in the first of France, Germany, Spain, Italy and the UK. The launch of PKU GOLIKE® in the U.S. on October 10, 2022 marked the completion of the third milestone, for which Relief issued a cash payment of CHF 2.8 million as well as 150,200,120 shares.
AdVita Lifescience GmbH
On July 28, 2021, we announced the closing of a definitive agreement to acquire all of the outstanding shares of AdVita Lifescience GmbH. Under the agreement, the stockholders of AdVita received 135,741,063 of our ordinary shares, representing EUR 25 million (approximately CHF 27.4 million) in value based on a 60-day Volume Weighted Average Price of our ordinary shares and were also eligible to receive additional contingent payments in cash. In April 2022, we made an initial milestone payment of EUR 5 million (approximately CHF 5 million) upon completion of the first milestone.
75
As of the date of this annual report, the former shareholders of AdVita may receive up to EUR 10 million (approximately CHF 10 million) upon achievement of pre-agreed milestones involving (i) the approval in the U.S. or Europe of the inhaled form of aviptadil for the treatment of sarcoidosis or berylliosis, and (ii) the conduct of a phase II clinical study for the inhaled form of aviptadil in the treatment of checkpoint inhibitor-induced pneumonitis.
AdVita was founded in 2019 for the purpose of developing products and strategies to improve the therapy and diagnosis of rare lung diseases. Among AdVita’s assets are intellection property rights that may cover RLF-100® inhaled formulation specifications and the potential application of inhaled aviptadil in the treatment of acute respiratory distress syndrome, checkpoint inhibitor-induced pneumonitis and sarcoidosis.
InveniAI LLC Collaboration Agreement
On November 24, 2021, we announced that we had entered into a collaboration agreement with InveniAI LLC (InveniAI), a U.S. based company that has pioneered the application of artificial intelligence and machine learning across biopharma and other industries, in order to identify promising drug candidates to treat rare and specialty diseases (the InveniAI Collaboration Agreement).
Under the terms of the InveniAI Collaboration Agreement, InveniAI will use its proprietary platform for the identification of potential pharmaceutical product opportunities using its Pharma Big Innovation Data Lab, consisting of (i) its proprietary AlphaMeld platform, a cloud-based artificial intelligence platform that uses its proprietary machine learning and deep learning based neural networks to identify product opportunities in therapeutic areas, (ii) its cross-functional teams at its Integrated Center of Excellence, and (iii) domain expertise, to generate novel pharmaceutical opportunities and the related development pathway for the development of such concepts.
In the collaboration it is expected that InveniAI will use its platform to navigate the volume of data for all regulatory agency approved drugs and their associated active ingredients to identify potential rate and specialty disease indications for development and commercialization by us (“product concepts”) . InveniAI will seek to prioritize top product concepts, associated diseases, scientific packages and evidence to support the potential drug development opportunities by us. We anticipate that InveniAI’s platform will complement APR’s existing capabilities in research and development and in drug reformulation. Based on product leads developed by InveniAI, we hope to develop proprietary versions of existing drugs, and to protect those drugs with long-lived intellectual property and defensible product claims.
Under the terms of the InveniAI Collaboration Agreement, we paid InveniAI an initial up-front fee of $500,000. We will be required to pay success milestones for any products brought to us in connection with the InveniAI Collaboration Agreement ranging from $200,000 per product candidate for which we exercise our option to acquire IP rights to $50 million for any required product reaching $1 billion per year in net sales. We will also be required to pay royalties on any such commercialized product in certain countries a royalty of approximately three percent.
We are not currently developing any product brought to us by InveniAI, and there can be no assurance that our collaboration with InveniAI will result in the development of new product candidates or product concepts.
Basis of Preparation and Consolidation
Our consolidated financial statements have been prepared in accordance with IFRS, as issued by the IASB, and comply with Swiss law. They have been prepared under the historical cost convention, as modified by the revaluation of financial instruments at fair value, are presented in Swiss Francs (CHF), and all values are rounded to the nearest thousand (TCHF), except when otherwise indicated.
The consolidated financial statements comprise the financial statements of us and our subsidiaries as of December 31, 2022. Control is achieved when we are exposed, or have rights, to variable returns from our involvement with the investee and have the ability to affect those returns through our power over the investee.
Specifically, we control an investee if and only if we have:
power over the investee (i.e., existing rights that give us the current ability to direct the relevant activities of the investee);
exposure, or rights, to variable returns from our involvement with the investee; and
76
the ability to use our power over the investee to affect our returns.
When we have less than a majority of the voting or similar rights over an investee, we consider all relevant facts and circumstances in assessing whether we have power over an investee, including:
any contractual arrangement over the other vote holders of the investee;
rights arising from other contractual arrangements; and
our voting rights and potential voting rights.
We reassess whether or not we control an investee if facts and circumstances indicate that there are changes to one or more of the three elements of control. Consolidation of a subsidiary begins when we obtain control over the subsidiary and ceases when we lose control of the subsidiary. Assets, liabilities, income and expenses of a subsidiary acquired or disposed of during the year are included in the statement of comprehensive income from the date we gain control until the date we cease to control the subsidiary.
Profit or loss and each component of other comprehensive income are attributed to the equity holders of RELIEF THERAPEUTICS Holding SA and to the non-controlling interests, even if this results in the non-controlling interests having a deficit balance. When necessary, adjustments are made to the financial statements of subsidiaries to bring their accounting policies into line with our accounting policies. Inter-company transactions, balances and unrealized gains/losses on transactions between us and our subsidiaries are eliminated. The accounting policies of subsidiaries are consistent with the policies adopted by RELIEF THERAPEUTICS Holding SA.
|
Components of Our Results of Operations Revenue and Other Gains Revenue is primarily derived from our portfolio of marketed products and the provision of R&D services to third parties. We generate revenue from product sales, licensing fees, and royalties since the date of acquisition of APR in June 2021. Prior to thethis acquisition, Reliefwe did not generate any revenue from commercial activities. To date, our revenue has been substantially less than our operating expenses and does not significantly contribute to our cash needs.expenses. Accordingly, we rely primarily on our available cash and external funding to continue operations and fund our clinical and commercial development plan. We expect the expansion of our PKU GOLIKE® franchise as a medical food in the U.S. and other territories, as well as the commercialization of OLPRUVA™ by Acer (for which we may receive royalty repayments in the amount of 60 percent of net profits) will contribute to increases in future revenues. We do not expect to generate revenue from product candidates unless and until we complete their development and obtain regulatory approvals. Other Gains Other gains generally consist of gains on disposal of intangible assets, write-offs of liabilities and adjustments in fair value of certain assets and liabilities. Raw Materials and Consumables Expenses Raw materials and consumables expenses are comprised of expendituresinclude costs incurred with third parties in relation to the purchasefor purchasing and manufacturing ofmedical food and drug products for sale, as well as laboratory supplies in connection withfor R&D services provided to customers. External Selling and Distribution Expenses External selling and distribution expenses are comprised of expendituresinclude costs incurred with third parties in relation tofor advertising, marketing, sales promotion, shipping, distribution, and commission on sales, for the sale ofdistributing our products and R&D services. External Research and Development Expenses External research and development expenses include costs associated with outsourced clinical research organization activities, sponsored research studies, clinical trial costs, process development, drug candidate manufacturing expenses, license fees, and investigator-sponsored trials, including licensing fees and milestone payments charged by licensors or collaboration partners, as well as expenses related to laboratory supplies and materials. 77
Costs for certain development activities, such as clinical trials, are recognized based on an evaluation of the progress to completion of specific tasks using information from the clinical sites and our vendors. Costs associated with the development activity under collaboration agreements are recognized based on actual expenses reported by our collaboration partners. Personnel Expenses Personnel expenses consist of employee-related expenses, including salaries, benefits, share-based compensation, and other related costs. Our benefits under our defined benefit (pension) plan are determined using the projected unit credit method, pursuant to which costs are recognized immediately in the statement of financial position with a corresponding debit or credit to retained earnings through other comprehensive income in the period in which they occur.
Past service costs are recognized in profit or loss on the earlier of: the date of the plan amendment or curtailment, or the date that the restructuring- related costs are recognized.
The following changes in the net defined benefit obligation are recognized under ‘personnel expense’ in the consolidated statement of comprehensive income:
| • | | service costs comprising of current service costs, past-service costs, gains and losses on curtailments and non-routine settlements; and
|
net interest expense or income.
The cost of share-based compensation is recognized over the period in which the performance and/or service conditions are fulfilled. The cumulative expense recognized for such transactions at each reporting date reflects the extent to which the performance and/or service conditions underlying the share award are earned. The financial statements in any period represent the movement in cumulative expense recognized at the beginning and the end of such period. No expense is recognized for awards that do not ultimately vest, except in certain cases.
Other Administrative Expenses Other administrative expenses consist primarily of corporate facility costs, fees for legal and audit services, insurance costs, and consulting fees not otherwise included in research and development expenses. Financial Income Financial income consists mainly of interest on term deposits and foreign exchange net result, when positive. Foreign exchange net result is allocated to financial expense when negative. We use the Swiss francs (CHF) as our reporting currency and conduct business on a global basis in various currencies. As a result, the Group is exposed to foreign currency exchange movements, primarily in European and U.S. market currencies. 59
Financial Expense Financial expense consists mainly of interest expense associated with the discounting over timeovertime of provisions for contingent payments measured at fair value. The commitment fee that became due upon execution of our current share subscription facility agreement with GEM in January 2021 is expensed over the three-year period of effectiveness of the instrument. In addition, we incurred negative interest charge on our Swiss franc and Euro cash deposits until year-end 2022. Income Taxes We are subject to corporate income taxation in Switzerland, the U.S., Italy, and Germany. We are also subject to corporate capital tax for our parent company and subsidiaries located in Switzerland. Unless and until the Group becomes profitable in certain tax jurisdictions, we expect income tax losses and gains will primarily arise from variations of deferred tax assets and liabilities. 78
Relief Results of Operations
The following table, which hasnumbers below have been derived from our audited consolidated financial statements included elsewhere in this Annual Report. The discussion below should be read along with these consolidated financial statements and it is qualified in its entirety by reference to them. The following table presents our consolidated results of operations for the years ended December 31, 2023 and 2022 and 2021, together with changes from year to year.(in thousands CHF): | | | | December 31, | | | Change | | | December 31, | | | Change | | Statement of Operations Data | | 2022 | | | 2021 | | | (2022 to 2021) | | | 2023 | | | 2022 | | | (2023 to 2022) | | Revenue | | | 6,081 | | | | 3,321 | | | | 2,760 | | | | 6,033 | | | | 6,081 | | | | (48 | ) | Other gains | | | 9,921 | | | | 1,171 | | | | 8,750 | | | | 295 | | | | 1,029 | | | | (734 | ) | Total Income | | | 16,002 | | | | 4,492 | | | | 11,510 | | | | | | | | | | | | | | | | | | | | Total income | | | | 6,328 | | | | 7,110 | | | | (782 | ) | Raw materials and consumables expenses | | | (1,250 | ) | | | (750 | ) | | | (500 | ) | | | (1,736 | ) | | | (1,250 | ) | | | (486 | ) | External selling and distribution expenses | | | (3,307 | ) | | | (365 | ) | | | (2,942 | ) | | | (2,200 | ) | | | (3,307 | ) | | | 1,107 | | External research and development expenses | | | (12,393 | ) | | | (19,024 | ) | | | 6,631 | | | | (1,328 | ) | | | (12,393 | ) | | | 11,065 | | Personnel expenses | | | (12,998 | ) | | | (9,121 | ) | | | (3,877 | ) | | | (11,838 | ) | | | (12,998 | ) | | | 1,160 | | Other administrative expenses | | | (7,747 | ) | | | (6,750 | ) | | | (997 | ) | | | (5,391 | ) | | | (7,747 | ) | | | 2,356 | | Other losses | | | (63 | ) | | | (752 | ) | | | 689 | | | | (48 | ) | | | (63 | ) | | | 15 | | | | | | | | | | | | | | | | | | | | EBITDA | | | (21,756 | ) | | | (32,270 | ) | | | 10,514 | | | | (16,213 | ) | | | (30,648 | ) | | | 14,435 | | Reversal of impairment losses on intangible assets | | | (26,424 | ) | | | — | | | | (26,424 | ) | Change in fair value of contingent consideration | | | | 4,782 | | | | 8,892 | | | | (4,110 | ) | Impairment expense | | | | (96,079 | ) | | | (26,424 | ) | | | (69,655 | ) | Amortization and depreciation expense | | | (3,860 | ) | | | (2,036 | ) | | | (1,824 | ) | | | (3,318 | ) | | | (3,860 | ) | | | 542 | | | | | | | | | | | | | | | | | | | | Operating loss | | | (52,040 | ) | | | (34,306 | ) | | | (17,734 | ) | Operating result | | | | (110,828 | ) | | | (52,040 | ) | | | (58,788 | ) | Financial income | | | 18 | | | | 97 | | | | (79 | ) | | | 93 | | | | 18 | | | | 75 | | Financial expenses | | | (2,294 | ) | | | (1,316 | ) | | | (978 | ) | Financial expense | | | | (949 | ) | | | (2,294 | ) | | | 1,345 | | | | | | | | | | | | | | | | | | | | Result before income taxes | | | (54,316 | ) | | | (35,525 | ) | | | (18,791 | ) | Net loss before taxes | | | | (111,684 | ) | | | (54,316 | ) | | | (57,368 | ) | Income taxes | | | 3,526 | | | | 820 | | | | 2,706 | | | | 13,503 | | | | 3,526 | | | | 9,977 | | | | | | | | | | | | | | | | | | | | Results for the period | | | (50,790 | ) | | | (34,705 | ) | | | (16,085 | ) | | | | | | | | | | | Net loss for the period | | | | (98,181 | ) | | | (50,790 | ) | | | (47,391 | ) |
Comparison of the Years Ended December 31, 2023 and 2022 60
Revenue The following table details our revenue figures for 2023 and 2022. | | | | | | | | | | | | | | in CHF thousands | | 2023 | | | 2022 | | | Change | | Product sales | | | 4’256 | | | | 2’525 | | | | 1’731 | | Royalties | | | 1’411 | | | | 2’482 | | | | (1’071 | ) | Licensing fees | | | 19 | | | | 380 | | | | (361 | ) | Revenue from research and development services | | | 347 | | | | 694 | | | | (347 | ) | | | | | | | | | | | | | | Total revenue | | | 6’033 | | | | 6’081 | | | | (48 | ) | | | | | | | | | | | | | |
Product sales revenue increased by CHF 1.7 million, mainly driven by sales of PKU GOLIKE following its launch in the U.S. in October 2022 and 2021the roll-out of PKU GOLIKE bars in 2023. Royalty revenue fell by CHF 1.1 million as generic competitors to our out-licensed Diclofenac-based products entered the U.S. market in 2023. Revenue and otherOther gains
In 2022, we generated CHF 6.1 million in revenue from product sales, licensing fees, royalties and contract services, compared to CHF 3.3 million in 2021. Relief began generating revenue following the acquisition of APR at the end of June 2021, which generated net sales of CHF 3.2 million in the six-month period from July 1, 2021 to December 31, 2021. On an annualized basis, revenue decreased by 8.4 percent mainly due to a decrease in non-recurring license fees and in contract services revenue.
Other gains were CHF 9.90.3 million in 2022,2023, compared to CHF 1.21.0 million in 2021.2022. In the current period, other gains consisted mainlywere primarily constituted by a divestment gain related to the renegotiation of the license agreement with Acer and by income from sublease facilities. Other gains from prior year were primarily constituted by reversals of impairment on financial assets, contributing CHF 0.7 million. Raw materials and consumables expenses Raw materials and consumables expenses increased by 39% in 2023 compared to the prior year. This correlates with the 69% increase in product sales revenue. The difference is attributable to a change in the product mix, with a higher proportion of sales stemming from higher-margin products. Raw materials and consumables expenses include costs incurred with third parties for purchasing and manufacturing medical food and drug products for sale, as well as laboratory supplies for R&D services provided to customers. External selling and distribution expenses External selling and distribution expenses decreased to CHF 2.2 million in 2023, from CHF 3.3 million in 2022, a decrease of CHF 1.1 million mainly due to scaled-back marketing activities in 2023, following the preparation and launch of PKU GOLIKE in the U.S. in 2022. External selling and distribution expenses include costs incurred with third parties for advertising, marketing, and distributing our products and R&D services. External research and development expenses External research and development expenses decreased to CHF 1.3 million in 2023, from CHF 12.4 million in 2022, a decrease of CHF 11.1 million. The decrease is mainly attributable to the prior year’s CHF 9.9 million expenses under our former license and collaboration agreement with Acer for the development and premarketing activities of OLPRUVA™. In 2023, our external development expenses were mainly directed to the development of RLF-OD032, RLF-TD011 and RLF-100®, alongside the continued development of our GOLIKE® product franchise. External research and development expenses include costs associated with outsourced clinical research organization activities, sponsored research studies, clinical trial costs, process development, drug candidate manufacturing expenses, license fees, as well as expenses related to laboratory supplies and materials. Personnel expenses Personnel expenses decreased to CHF 11.8 million in 2023, compared to CHF 13.0 million in 2022, a decrease of CHF 1.2 million mainly due to lower expense recognized from the granting of stock options. Specifically, non-cash expenses resulting from stock option grants were CHF 0.8 million in 2023, compared to CHF 2.2 million in 2022. As of December 31, 2023, Relief employed 49 full-time equivalents, a decrease from 69 full-time equivalents on December 31, 2022. 61
Other administrative expenses Other administrative expenses decreased to CHF 5.4 million in 2023, compared to CHF 7.7 million in 2022, a decrease of CHF 2.3 million. The decrease is mainly attributable to a CHF 2 million reduction in legal and regulatory affairs expenses, driven by non-recurring events in 2022. In addition, we achieved a CHF 1.2 million reduction in other professional fees across various administrative activities. These savings were partially offset by an increase of CHF 0.8 million for directors and officers’ insurance coverage. Change in fair value of provisions for contingent liabilities (CHF 8.9 million) and an impairment reversal (CHF 0.5 million) following the repayment of a loan issued to NeuroRx in 2020 and for which we had recorded a complete impairment allowance. In the comparative period, other gains were mainly related to write-offs of liabilities.consideration The 2022 one-off gain of CHF 8.9 million resulted from unfavorable changes in the estimated market potential and development programs for certain clinical stage assets acquired inUnder the APR and AdVita business combinations. Under the acquisition agreements, Relief agreed to pay additional consideration upon completion of specific milestones. The fair value of the contingent consideration is recorded as a liability on our balance sheet and adjusted at the end of each reporting period based on the estimated probability of occurrence and the time factor. Any changes in fair value of the contingent liability due to assumption adjustments are recorded as ‘Other gains’ or ‘Other losses’. Refer to notes 7 and 18 of our consolidated financial statements for further information.in the income statement.
79
Raw materials and consumables expenses, and external selling and distribution expenses
Raw materials and consumables expenses, and external selling and distribution expenses, were, respectively,In 2023, a CHF 1.34.8 million and CHF 3.3 million in 2022. We did not incur any such expenses prior to the acquisition of APR and its marketing activities. In addition, premarketing and marketing activitiesgain was recognized in relation to the launchpostponement of PKU GOLIKE® in the U.S. in October 2022 accounted for CHF 2.5 million in 2022.
External Researchexpected completion dates of development and Development Expenses
External research and development expenses were primarily driven by development activitiescommercial milestones for RLF-100®, RLF-TD011 and expenses incurred by Acer for theSentinox™ development and intended commercialization of ACER-001. The decrease ofprograms. In 2022, similar unfavorable changes resulted in a CHF 6.68.9 million to CHF 12.4 million in 2022 from CHF 19.0 million in 2021, was primarily due to a reduction of CHF 6.7 million in development expenses associated with RLF-100®. Expenditures associated with other in-process programs did not vary materially.
Contingent upon availability of funds, we plan to further increase our research and development expenses for the foreseeable future as we commence additional clinical trials and pursue discovery and development of new product candidates.gain.
Personnel ExpensesImpairment expense
Personnel expenses increased to CHF 13.0 million in 2022, compared to CHF 9.1 million in 2021,We conducted an increase of CHF 3.9 million mainly due to an increase in employee headcount resulting from the acquisitions of APR and AdVita and the establishmentimpairment test of our U.S. sales force. Non-cash remuneration in the form of stock options amounted to CHF 2.2 millionintangible assets and CHF 1.1 million in 2022 and 2021, respectively. Asgoodwill as of December 31, 2022, Relief had 69 full-time equivalents on its payroll.
Other Administrative Expenses
Other administrative expenses increased to CHF 7.7 million in 2022, compared to CHF 6.7 million in 2021, an increase of CHF 1.0 million. The main drivers of this increase were higher expenses related to the maintenance and prosecution of intellectual property, travel of Company’s staff, and IT infrastructure and software primarily due to the addition of APR and AdVita. Legal fees remain flat as non-recurring costs incurred as part of Relief’s effort to list its securities on Nasdaq were offset by a reduction in costs incurred for other legal and regulatory matters.
Other Losses
Other losses were not material in 2022, compared to CHF 0.8 million in 2021. Other losses for the comparative period were mainly constituted by an impairment loss of CHF 0.4 million on the loan issued to NeuroRx in 2020.
Impairment Expense
We conducted impairment tests of intangible assets as of June 30 and December 31, 2022,2023, and concluded that the carrying amount of certain assets, mainly intangible assets and goodwill associated with PKU GOLIKERLF-100®, RLF-TD011 and Sentinox™, exceeded their recoverable amount.were impaired. As a result, we recognized a non-cash impairment charge on intangible assets of CHF 26.496.1 million in the current period. The impairment charge reflects a reductionreduced projected returns and their delayed realization, attributable to adjustments to our development and commercialization strategies. Refer to note 7 of estimated future net cash flows from PKU GOLIKE® following changes in market assumptions,our consolidated financial statements for further information on intangible assets and for Sentinox™, a one-year delay in the estimated launch date.related impairment.
Amortization and Depreciation Expensedepreciation expenses Amortization and depreciation expenses were CHF 3.3 million in 2023, compared to CHF 3.9 million in 2022, compared to CHF 2.0 million in 2021. These2022. Amortization and depreciation expenses are mainly relatedpredominantly pertain to the amortization of our intangible assets. Prior to the acquisition of APR in June 2021, we did not have amortizable intangible assets nor material property, plant, and equipment assets on our balance sheet. Financial Incomeincome Financial income was not material in 2022, comparedincreased to CHF 0.1 million in 2021. Financial2023, compared to CHF 0.02 million in 2022. In 2023, financial income for the comparative period was mainlyprimarily constituted by interest income from cash deposits and the recognition of interest on a net foreign exchange gain on monetary assets and liabilities and transactions denominateddeferred payment expected from Acer in U.S. dollars and Euros. In 2022, the net foreign exchange result was a loss recorded in financial expense.2024. 8062
Financial Expensesexpenses Financial expenses increaseddecreased to CHF 0.9 million in 2023, compared to CHF 2.3 million in 2022, compareda decrease of CHF 1.4 million. This reduction was primarily due to the decrease in interest costs, from CHF 1.3 million in 2021, an increase ofto CHF 1.0 million. The cost of0.3 million, associated with the unwinding of the time discount ofon provisions for contingent consideration forconsiderations, in line with the acquisition of APR and AdVita increased to CHF 1.3 milliondecrease in 2022 from CHF 0.7 million in 2021 as the period that elapsed was 12 months in 2022, compared to 6 months in 2021. Financial expenses also increased as the Company incurred a net foreign exchange loss of CHF 0.4 million in 2022, compared to a net foreign exchange gain in 2021.these provisions. Income Taxestaxes Income taxes were a gain of CHF 3.513.5 million in 2022,2023, compared to a gain of CHF 0.83.5 million in 2021.2022. The income tax gaingains resulted mainly from the amortization and impairment of intangible assets and a corresponding reduction in the temporary difference between the carrying amount of these assets and their tax base. In 2022,Unless and until the Company also wrote offGroup becomes profitable in certain tax jurisdictions, we expect income tax losses and gains will primarily arise from variations of deferred tax assets for a total amount of CHF 1.5 million as the criteria for the recognition of these assets were not met as of December 31, 2022. The write-off charge was offset by a reduction in deferred tax liabilities of CHF 5.0 million.and liabilities. Summary of criticalCritical accounting judgements and key sources of estimation uncertainty
Our operating and financial review and prospects were derived from our consolidated financial statements in conformity with IFRS as issued by the IASB. The preparation of the consolidated financial statements in conformity with IFRS requires management to make estimates and assumptions that affect the application of policies and reported amounts of assets, liabilities, income, expenses and related disclosures. The estimates and underlying assumptions are based on historical experience and various other factors that are believed to be reasonable under the circumstances, the results of which form the basis for making the judgments about carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates. The estimates and assumptions that have a significant risk of causing a material adjustment to the carrying amounts of assets and liabilities within the next financial year are described below. Critical accounting judgements and key sources of estimation uncertainty are described in note 4 of our 20222023 consolidated financial statements included in this annual report. | B. | LIQUIDITY AND CAPITAL RESOURCES |
Sources of Liquidity To date, we have funded our operations primarily through private placements, at-the-market sales of treasury shares and equity offerings and loans from our largest shareholder, GEM.offerings. We have never been profitable and have incurred operating losses in each year since inception. We have an accumulated deficit of CHF 119.6218.3 million as of December 31, 20222023, and expect tomay incur further losses over the foreseeable future as we develop our business. We have spent, and expect to continue to spend, a substantial amount of funds in connection with implementing our business strategy, including our planned product development and commercialization efforts. As Relief continues to incur significant operating losses, our ability to pursue and finance our operations and our intended development plans depends on our ability to continue to raise additional financing. Our primary uses of capital are R&D expenses, personnel compensation expenses, outsourced R&D expenses, and administrative expenses. We expect to continue to incur substantial expenses in connection with our product candidates at various stages of development and for working capital requirements. We expect to continue to raise financing through the sale of equity and debt financing.equity. We intend to use future expected proceeds, together with cash on hand, to finance our development and commercial activities and the diversification of our pipeline, as well as to fund our outstanding liabilities and other commitments. We expect our expenses to increase in connection with our ongoing activities, particularly as we continue to advance our portfolio of product candidates, initiate further clinical trials, and seek marketing approval for our product candidates. In addition, if we obtain marketing approval for any of our product candidates, we expect to incur additional commercialization expenses related to program sales, marketing, manufacturing, and distribution to the extent that such sales, marketing and distribution are not the responsibility of potential partners. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. 81
Going Concern
As of December 31, 2022,2023, we had cash and cash equivalents of CHF 19.214.6 million. Based on current operating plans,financial projections and available cash, we expectbelieve that we have sufficient resources to fund operations into the third quarter of 2023. This raises substantial doubt about our ability to continue as a going concern. Please refer to note 4.1 ‘going concern’ of our consolidated financial statements included in2026. We have based this annual report. Our consolidated financial statements do not include any adjustments or classificationsestimate on assumptions that may result fromprove to be wrong, and we could utilize our possible inability to continue as a going concern. available capital resources sooner than we currently expect. Our future capital requirements will depend on many factors, including: the scope, progress, results and costs of our ongoing and planned preclinical studies and clinical trials; the number and development requirements of other product candidates that we may pursue; 63
the costs, timing and outcome of regulatory review of our product candidates; the timing amount of milestone payments we may have to pay in relation to the acquisitions of APR and AdVita; the timing and amount of milestone and royalty payments we may receive under our license agreements; | | • | | the extent to which we in-license or acquire other product candidates and technologies; |
the costs and timing of future commercialization activities, including drug manufacturing, marketing, sales and distribution, for any of our product candidates for which we receive or have received marketing approval; the timing of repayment of the Relief’s borrowings; and
the funding necessary to sustain our commercial operations until we attain the breakeven point. We willmay need to raise additional capital to fund continued operations beyond the third quarter of 2023. We2026 and may not be successful in our efforts to raise additional funds or achieve profitable operations. We continueintend to explore potential opportunities and alternatives to obtain the additional resources that will be necessary to support our ongoing operations, beyond the third quarter of 2023, including raising additional capital through either private or public equity or debt financing, or additional program collaborations or non-dilutive funding, as well as using our treasury share sales program or our shares subscription facility with GEM. If we are unable to obtain additional funding to support our current or proposed activities and operations, we may not be able to continue our operations as proposed, which may require us to suspend or terminate any ongoing development activities, modify our business plan, curtail various aspects of our operations, cease operations, or seek relief under applicable bankruptcy laws. In such event, our stockholders may lose a substantial portion or even all of their investment. 82
Cash Flows The following table summarizes our cash flows for each of the periods presented. | | | | December 31, | | | December 31, | | In thousands | | 2022 | | | 2021 | | | 2023 | | | 2022 | | Cash flow from operating activities | | | (24,126 | ) | | | (35,718 | ) | Cash flow from investing activities | | | (7,999 | ) | | | (30,262 | ) | Cash flow used in operating activities | | | | (17,612 | ) | | | (24,126 | ) | Cash flow from (used in) investing activities | | | | 8,695 | | | | (7,999 | ) | Cash flow from financing activities | | | 6,417 | | | | 67,689 | | | | 4,050 | | | | 6,417 | | Net (decrease) increase in cash and cash equivalents | | | (25,708 | ) | | | 1,709 | | | | (4,867 | ) | | | (25,708 | ) | Cash and cash equivalents at end of period | | | 19,237 | | | | 44,761 | | | | 14’556 | | | | 19,237 | |
Operating Activities Net cash used in operating activities consistconsists of the net operating loss adjusted for changes in net working capital and for non-cash items, such asincluding impairment and depreciation, fair value adjustments, the value of share-based servicesservice payments, and changes in post-employment benefits.benefit obligations. NetIn 2023, net cash used in operating activities was CHF 17.6 million, primarily as the result of our negative EBITDA of CHF 16.2 million, and an increase of CHF 1.0 million in net working capital. The increase in net working capital was mainly due to a decrease in outstanding payables.
In 2022, net cash used in operating activities was CHF 24.1 million, in 2022, compared toprimarily as the result of our negative EBITDA of CHF 35.730.6 million, partially offset by a reduction of CHF 5.6 million in 2021.net working capital. The decrease in cash used in operating activities of CHF 11.6 million was due to an increase in net loss of CHF 16.1 million, before adjustments for non-operating transactions, offset by an increase in non-cash items of CHF 19.0 million and an increase in changereduction in net working capital of CHF 10.8 million.was mainly due to a decrease in year-end prepayments. Investing Activities NetIn 2023, net cash provided by investing activities was CHF 8.7 million and consisted mainly of a payment from Acer under the termination and revised license agreements executed in August 2023.
In 2022, net cash used in investing activities was CHF 8.0 million in 2022, compared to CHF 30.3 million in 2021. In 2022, net cash used in investing activitiesand consisted primarilymainly of payments to the former shareholders of APR and AdVita in relation to the completion of contractual milestones.
In 2021, net cash used in investing activities consisted primarily in payments for the acquisition of APR and ACER 001 license.
64
Financing Activities NetIn 2023, net cash from financing activities was CHF 4.0 million and was derived mainly from a CHF 4.5 million private placement, net of transaction costs.
In 2022, net cash from financing activities was CHF 6.4 million in 2022, compared to CHF 67.7 million in 2021. In 2022, net cash from financing activities consisted primarily of CHF 7.1 million gross proceedsand was derived mainly from the offer of treasuryour shares into the trading market partially offset by issuance costs of CHF 0.2 million and by debt repayments of CHF 0.5 million.
In 2021, net cash from financing activities consisted primarily of CHF 50.9 million gross proceeds from the offer of treasury shares into the trading market and private placements of CHF 25 million, partially offset by issuance costs of CHF 2.8 million and by debt repayments of CHF 5.6 million.through our direct share placement program.
Quantitative and Qualitative Disclosure about Financial RisksMarket Risk We are exposed to various financialFor qualitative and quantitative disclosures about market risks such as creditincluding foreign currency risk, interest rate risk, liquidity risk and market risk (including interest rate and currency risk). The following is an overview of the extent of such risks and our processes employed to handle those risks.
Credit Risk
Credit risk refers to the risk that a party will default on its contractual obligations to us, resulting in financial losses. We do not currently have any revenue and as a result we do not have credit risk, with relationsee note 34 to our customers. Our financial assets consist of mainly cash, for which the risk is minimal as such deposits are at well-known banks in Switzerland with an “A” rating as per Standard & Poors.
83
Liquidity Risk
Liquidity risk implicates our ability to maintain sufficient cash and cash equivalents to meet our financial obligations. Our management monitors our net liquidity through rolling forecasts of projected cash flows.
Interest Rate Risk
We are exposed to interest risk in respect of the Company’s cash deposits, bank loans and other interest-bearing liabilities. We deem the interest rate risk as low on its performance and its equity.
Currency Risk
We operate internationally and are exposed to currency risk arising from various exposures, primarily with respect to the Swiss francs, Euros and U.S. dollars. Currency risk arises from future transactions, recognized assets and liabilities and net investments in foreign operations. To manage such risk, we monitor our exposure by periodically assessing future spending needs in foreign currencies and maintain foreign currency cash balances to cover anticipated future requirements. We do not enter into any forward currency transactions.
While we consider our current exposure to foreign currency risk to be low, adverse changes in the value of the Swiss franc could still have a significant negative impact on our financial condition, results of operations, and future prospects.
JOBS Act Exemptions and Foreign Private Issuer Status
We qualify as an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012 (JOBS Act) in the U.S. An emerging growth company may take advantage of specified reduced reporting and other requirements that are otherwise applicable generally to public companies. This includes an exemption from the auditor attestation requirement in the assessment of our internal control over financial reporting pursuant to the Sarbanes-Oxley Act of 2002. We may take advantage of this exemption for up to five years or such earlier time that we are no longer an emerging growth company. We will cease to be an emerging growth company if we have more than $1.07 billion in total annual gross revenue, have more than $700 million in market value of our ordinary shares held by non-affiliates or issue more than $1 billion of nonconvertible debt over a three- year period. We may choose to take advantage of some but not all of these provisions that allow for reduced reporting and other requirements.
Upon completion of the U.S. listing to which this annual report relates, we will report under the Securities Exchange Act of 1934, as amended, or the Exchange Act, as a non-U.S. company with foreign private issuer status. Even after we no longer qualify as an emerging growth company, as long as we qualify as a foreign private issuer under the Exchange Act, we will be exempt from certain provisions of the Exchange Act that are applicable to U.S. domestic public companies, including:
the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act;
sections of the Exchange Act requiring insiders to file public reports of their stock ownership and trading activities and liability for insiders who profit from trades made in a short period of time;
| • | | the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form 10-Q containing unaudited financial and other specified information, or current reports on Form 8-K, upon the occurrence of specified significant events; and
|
Regulation FD, which regulates selective disclosures of material information by issuers.
C. | RESEARCH AND DEVELOPMENT, PATENTS AND LICENSES, ETC.
|
See “Item 4. Information on the Company – B. Business Overview” and “Item 5. Operating and Financial Review and Prospects – A. Operating Results”.
See “Item 5. Operating and Financial Review and Prospects – A. Operating Results and B. Liquidity and Capital Resources”.
84
E. | OFF-BALANCE SHEET ARRANGEMENTS
|
As of the date of this annual report, the Company had the contingent liabilities listed in note 36 of the 2022 consolidated financial statements included in this annual report. There were no other material off-balance sheet arrangements.
Material Cash Requirements from contractual Obligations F. | TABULAR DISCLOSURE OF CONTRACTUAL ARRANGEMENTS
|
The following table summarizes our contractual commitments as of December 31, 2022:2023: | | (TCHF) | | Less than
1 year | | | 1 to 3
years | | | 4 to 5
years | | | More than 5 years | | | Total | | | Less than
1 year | | | 1 to 3
years | | | 4 to 5
years | | | More than 5
years | | | Total | | Trade payable, accrued expenses, and other payables | | | | 4,838 | | | | — | | | | — | | | | — | | | | 4,838 | | Lease liabilities (1) | | | 340 | | | | 309 | | | | 140 | | | | — | | | | 789 | | | | 427 | | | | 526 | | | | 16 | | | | — | | | | 969 | | Financial borrowings (2) | | | 1’652 | | | | 12 | | | | 3 | | | | — | | | | 1’667 | | Financial Borrowings | | | | 1,692 | | | | 9 | | | | — | | | | — | | | | 1,701 | | Total | | | | | | | | | | | | | 6,957 | | | | 535 | | | | 16 | | | | — | | | | 7,508 | |
| (1) | Represents the minimum lease payments due under our operating leases for offices, laboratory space, company cars, and office and labs material. |
(2) | Bank loans are classified as short term due to the absence of contractual repayment date that renders their repayment date uncertain and dependent on the creditor’s demand.
|
Further, as described elsewhere in this document, we have committed to pay royalties on any revenue derived from the sale of aviptadil and OLPRUVA™, to NeuroRx and Acer Therapeutics, respectively, as well as royalties on net commercialization profit of a low double-digit percentage to Meta if we commercialize or license RLF-OD32. We currently do not generate any revenue from these three compounds and therefore have no current payment obligations. We have not included any contingent payment obligation, such as milestone payments and royalties, in the table above as there is no current obligation to issue payments and the timing and likelihood of such payments are not known. We enter into contracts in the normal course of business with CROs, contract manufacturing organizations and other third parties for clinical trials, preclinical studies, manufacturing and other services and products for operating purposes. These contracts generally provide for termination upon notice, and therefore we believe that our non-cancelable obligations under these agreements are not material. This annual report contains forward-looking statements within the meaning of Section 27AAs of the Securities Act and Section 21Edate of the Exchange Act,discussion and analysis and during the period presented, we did not have, and we do not currently have, any off-balance sheet arrangements, as defined in the Privaterules and regulations of the U.S. Securities Litigation Reform Act of 1995. and Exchange Commission
| C. | RESEARCH AND DEVELOPMENT, PATENTS AND LICENSES, ETC. |
See “Special Note with Respect to Forward Looking Statements” included elsewhere in this annual report.“Item 4.B. Business Overview” and “Item 5A. Operating Results”. See “Item 5.A. Operating Results” and “Item 5.B. Liquidity and Capital Resources”. | E. | CRITICAL ACCOUNTING ESTIMATES |
Not applicable. | ITEM 6 | DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES |
| A. | DIRECTORS AND SENIOR MANAGEMENT |
For information aboutThe following table sets forth the names, dates of birth and positions of our directors and senior management see “Item 1. Identityas of the date of this annual report. Unless otherwise stated, the business address for our directors and executive officers is c/o RELIEF THERAPEUTICS Holding SA, Av. de Sécheron 15, 1202 Geneva, Switzerland. In principle, our directors are appointed for one-year terms, which expire on the occasion of each annual general meeting. Accordingly, the terms of the directors set forth below will expire on the date of our next annual general meeting of shareholders expected in June 2024.
65
Directors | | | | | Name | | Date of Birth | | Position | | Raghuram Selvaraju | | 11/25/1978 | | Chairman | | Michelle Lock | | 11/21/1968 | | Director | | Peter de Svastich | | 09/03/1943 | | Director | | Gregory Van Beek | | 05/19/1969 | | Director | | Tommy Elzinga | | 03/19/1997 | | Director | | | | | Senior Management (Executive Committee) | | | | | | | | Name | | Date of Birth | | Position | | Michelle Lock | | 11/21/1968 | | Interim Chief Executive Officer | | Paolo Galfetti | | 05/03/1965 | | Chief Operating Officer | | Jeremy Meinen | | 02/21/1989 | | Chief Financial Officer and Treasurer |
Current Directors Raghuram (Ram) Selvaraju, Ph.D., MBA, serves as Chairman of our Board of Directors. Dr. Selvaraju Managing Director of Equity Research at H.C. Wainwright & Co., LLC whose research focuses on the healthcare sector. Dr. Selvaraju has over 16 years of experience on Wall Street and previously was a pharmaceutical researcher at Serono in Switzerland. In addition, Dr. Selvaraju has appeared numerous times on Bloomberg, CNBC, Business News Network and BTV where he discussed drug development trends, healthcare reform policy, and pharma and biotech M&A. Prior to joining Wainwright, Dr. Selvaraju held Senior Research positions at MLV & Co., Aegis Capital Corp. – Head of Healthcare Equity Research and Director of Equity Research, Hapoalim Securities U.S.A. and Rodman & Renshaw LLC. Dr. Selvaraju became the youngest-ever recipient of the Serono Pharmaceutical Research Institute’s Inventorship Award for exceptional innovation and creativity in 2003. Dr. Selvaraju earned his Ph.D. in cellular immunology and molecular neuroscience and an M.S. in molecular biology from the University of Geneva in Switzerland on the basis of his drug development research. He also holds an M.B.A. from the Cornell University accelerated one-year program for scientists and engineers and a B.S. in biological sciences and technical writing from Carnegie Mellon University. Michelle Lock is a member of our Board of Directors since January 2022 and became Relief’s Interim Chief Executive Officer in November 2023. She has nearly 30 years of biopharmaceutical strategic, operational and commercialization experience. Previously, Ms. Lock served as Chief Operating Officer of Covis Pharma Group, a Switzerland-based global specialty pharmaceutical company that markets therapeutic solutions for patients with life-threatening conditions and chronic illnesses. Ms. Lock’s broad biopharmaceutical industry experience spans nearly 30 years and includes leadership roles in commercialization across various therapeutic areas including oncology, hematology, cardiovascular and metabolic disease, liver disease, immunology, virology and neuroscience. Previously, Ms. Lock served as the Senior Vice President and Head of International organization at Acceleron Pharma Inc, a biopharmaceutical company dedicated to the discovery, development, and commercialization of therapeutics to treat serious and rare diseases. Before that, she was a consultant to biotechnology companies, providing leadership, guidance, and strategic support to managements seeking to establish or improve their international businesses based in Switzerland. Earlier, Ms. Lock was Senior Vice President & Head of International at Sage Therapeutics, a clinical-stage biopharmaceutical company committed to discovering, developing, and commercializing novel medicines to transform the lives of patients with life- altering central nervous system (CNS) disorders. During her career, Ms. Lock also spent 24 years with Bristol-Myers Squibb (BMS) in positions of increasing responsibility in sales, commercial, general management, regional leadership and business strategy. In her most recent role at BMS, she served as Vice President and General Manager for EU Country Clusters & Global Capabilities Hub leadership, Switzerland, driving the company’s leadership efforts in immuno-oncology. She has served as Honorary Ambassador between Switzerland and the U.S. since 2018, as well is a past member of the board of directors of the Swiss American Chamber of Commerce and the Interpharma Switzerland Pharmaceutical Industry. She earned a degree in Science/Nursing at Royal Melbourne University, Australia and studied General Management and Advisers – A.Internal General Management at CEDEP, France. Peter de Svastich is a Managing Director at GEM. He has served on the Company’s Board from May 2016 to December 2020. Mr. de Svastich has deep expertise in the areas of commercial banking, investment banking and alternative investments. For four decades he built banking and financial businesses in the U.S., Brazil, Chile, Spain, and France. He also founded WestHem International Group, a privately held investment management and financial services company. He has formed joint ventures in banking and alternative investments with N.M. Rothschild & Sons (Spain), Banco Internacional y de Comercio Exterior (Chile), Banque Française de Commerce Extérieur (France), and Banque Nationale de Paris (Brazil). Mr. de Svastich obtained a Bachelor of Arts Degree in Art and Archeology from Princeton University, an LLB/JD from The Yale Law School, and was the recipient of a Latin American Teaching Fellowship - Fellow in International Law - from The Fletcher School of Law and Diplomacy at Tufts University. 66
Gregory Van Beek is a Managing Director at GEM, focusing on special situation investments in both public capital markets and private opportunities. He has 25 years of experience in private equity, portfolio management, investment research and strategy. Previously, Mr. Van Beek was Senior Vice President at Franklin Templeton Investments. Prior to that, he was Director, Strategy at Temasek International Ltd., the Singaporean sovereign investor. He was also Director and Investment Officer for the firm with transactional responsibilities in various sectors and markets, both emerging and developed. Mr. Van Beek holds degrees in Russian and Business from Southern Methodist University, and an MBA from the American Graduate School of International Management. Tommy Elzinga is an Investment Associate at GEM. He is responsible for evaluating both public and private investment opportunities, as well as managing portfolio businesses. Prior to GEM, Mr. Elzinga was a Senior Associate for the Boston Consulting Group (BCG). He began his career as a consultant for Ernst & Young. Mr. Elzinga holds a Bachelor of Science in Business Administration from the Olin Business School at Washington University in Saint Louis. Directors until April 26, 2024 Thomas Plitz served as Vice Chairman of our Board of Directors and Senior Management”.was chairperson of the Nomination and Compensation Committee of the Board. He is the CEO of Precirix, a biotechnology company developing precision radiopharmaceuticals in oncology. He recently served as Chief Executive Officer of Chord Therapeutics SA, a privately held biopharmaceutical firm based in Geneva, Switzerland, which was acquired by Merck KGaA in January 2022. Prior to Chord Therapeutics, Dr. Plitz worked as Chief Scientific Officer of the rare disease company, Wilson Therapeutics, which was acquired for USD 855 million by Alexion Pharmaceuticals in April 2018. Dr. Plitz’s previous assignments include senior roles at Serono, Merck, and Shire Pharmaceuticals, where he worked across multiple therapeutic areas including neuroinflammatory, metabolic, and rare diseases, completing more than two decades of experience in pharmaceutical R&D. Mr. Plitz holds a Ph.D. from Technical University of Munich, Germany. Patrice Jean was a member of our Board of Directors and was chairperson of the Audit and Finance Committee and of the Corporate Governance Committee. She is the Chair of the Life Sciences Practice at Hughes Hubbard & Reed, an international law firm based in New York City. She has over a decade of experience counselling and leading startup pharmaceutical, chemical and biotechnology companies in all areas of intellectual property law including asserting and defending patent rights underlying core technologies and innovations. Dr. Jean serves as Vice-President of the New York Intellectual Property Law Education Foundation and is a board member of the New York Intellectual Property Law Association. She currently does not hold, and has not held in the past, any management positions or significant business connections with the Company. Ms. Jean holds a Ph.D. in molecular biology from Princeton University, a J.D. from Columbia University School of Law, and a B.A. in biochemistry from Xavier University. Executive Officers Michelle Lock. See biographical information above. Paolo Galfetti is the Chief Operating Officer of Relief. He has more than thirty years of management experience in the pharmaceutical sector including in the areas of business development and licensing, operational strategic management, clinical research and pharmaceutical discovery and development. Mr. Galfetti joined APR in 1995 as head of licensing and business development and was appointed Chief Executive Officer in 2002. Prior to joining APR, he was a founding partner, Chief Executive Officer and board member of the Institute for Pharmacokinetic and Analytical Studies AG (IPAS), a Swiss contract research organization focusing on Phase I and II clinical trials, as well as Chief Executive Officer and board member of Farma Resa s.r.l., an Italian contract research organization dedicated to Phase III and IV clinical trial on a contract basis. Mr. Galfetti is a Chartered Financial Analyst (CFA) and has a bachelor’s degree in economics from the Commercial University Bocconi, Milan, Italy. Jeremy Meinen became our Chief Financial Officer and Treasurer in December 2022. Previously Mr. Meinen had served as our Vice President Finance and Administration since October 2020 and our Chief Accounting Officer since December 2021. He joined Relief as ad-interim Chief Financial Officer in April 2020. Prior to joining Relief, Jeremy provided financial consulting, controlling and auditing services to companies in various industries. He began his career in an international audit firm, where he held positions of increasing responsibility and scope over more than six years. Mr. Meinen holds a Master of Science in finance from Bocconi University and a Bachelor of Arts degree in Business Administration from the University of Geneva. He is a Swiss certified public accountant and former licensed audit expert. Family Relationships There are no family relationships among any of our executive officers or directors. Compensation in General We are subject to the Directive on Information Relating to Corporate Governance of the SIX Swiss Exchange (Corporate Governance Directive) and the Swiss Ordinance Against Excessive Compensation in Public Companies of November 20, 2013, as amended from time to time (Compensation Ordinance), and the Swiss Code of Obligations.
In line with the requirements of the Compensation Ordinance,Swiss Code of Obligations, the Company’s Articles of Association and the Organizational RegulationRegulations include provisions on the following governance and compensation-related matters: 67
number of permissible mandates in the supreme governing bodies of other legal entities; maximum terms of employment contracts and maximum notice period for members of the Executive Committee; 85
principles of compensation applicable to the Board of Directors and Executive Committee; shareholders’ binding vote on compensation of the Board of Directors and Executive Committee; additional amount for members of the Executive Committee hired after the vote on compensation by the Annual General Meeting; and loans, credit facilities and post-employment benefits for members of the Board of Directors and of the Executive Committee. Role of the Compensation Committee The Nomination and Compensation Committee (NCC)(“NCC”) assists the Board of Directors in all nomination and compensation matters. As detailed in the Organizational RulesRegulations of the Company, the NCC is responsible for ensuring the best possible leadership and management talent for the company and an appropriate compensation policy. In particular, the NCC is responsible for the following activities: identification of suitable candidates for positions on the Board of Directors and on the Executive Committee; recommendation and proposal of compensation principles and programs, including share-based compensation plans; and recommendation and proposal of the compensation for the members of the Board of Directors and Executive Committee; and recommendation and proposal of specific compensation packages for other members of management.Committee.
The decision-making authorities in compensation matters are summarized in the table below: | | | | | | | | | | | | | | | | | | | | | Level of Authority CEO* | | CEO
CEO* | | | Nomination
Compensation
Committee | | | Board of
Directors | | | Annual
General
Meeting | | Compensation policy including share-based plans | | | | | | | proposes | | | | approves | | | | | | Aggregate compensation of the Board of Directors | | | | | | | proposes | | | | reviews | | | | approves | | Individual remuneration of the Board of Directors members | | | | | | | proposes | | | | approves | | | | | | Aggregate compensation of the Executive Committee | | | | | | | proposes | | | | reviews | | | | approves | | Individual compensation of the CEO | | | | | | | proposes | | | | approves | | | | | | Individual compensation of Executive Committee members | | | proposes | | | | reviews | | | | approves | | | | | | Compensation report | | | | | | | proposes | | | | approves | | | | | |
The NCC consists of a minimum of one member of the Board. The members of the NCC are elected individually and annually by the Annual General Meeting (AGM) for the period until the following AGM. At the AGM 2022,2023, Thomas Plitz (NCC Chairman) and Raghuram Selvaraju were elected members of the NCC. The NCC meets as often as the business requires, but at least once a year. The NCC Chairman may invite the Chairman of the Board, the CEO or other members of the Executive Committee to join the meeting in an advisory capacity. However, the executives do not take part in the meeting, or parts of meeting, during which their own compensation is discussed. The NCC Chairman reports to the Board on the activities of the committee after each meeting. The NCC may retain external advisors to obtain support in fulfilling its duties. Role of Shareholders and Say-On-Pay Vote At the AGM to be held by the endWe hold an annual general meeting of June 2023, ashareholders (AGM) each year. A binding vote on the compensation amount of the Board and Executive Committee will beis conducted. The AGM will votevotes on the maximum compensation amount of the Board for the period of office until the following AGM and on the maximum compensation amount of the Executive Committee for the next financial year. The prospective voting structure provides the Company and its management with the necessary level of planning certainty to operate efficiently.
86
The AGM held on May 31, 2022, approved a maximum2023 did not approve the proposed compensation amount of CHF 2’500’000 for the Boardenvelope for the period from the AGM 20222023 to the AGM 2023 and2024. This led the Board to suspend all compensation payments from June 2023. Subsequently, the Extraordinary General Meeting held on April 26, 2024 (the “EGM”) approved a maximum compensation amountenvelope of CHF 5’000’000500,000. This approval allowed the Board to resume payments, including compensation for the Executive Committee for the financial year 2023.period from June 2023 to April 2024. 68
Method of Determination of Compensation Based on the recommendation of the NCC, the Board of Directors decides upon the compensation of the Board of Directors and Executive Committee at its own discretion, which is ultimately approved by the AGM. When preparing the compensation proposals, the NCC takes the following factors into consideration:
affordability and overall situation of the Company;
business financial results and individual performance; and
level of compensation paid by other companies that are deemed to be comparable in terms of industry (where they compete for talent) and complexity (defined by their size and geographic scope).
The compensation of the Board of Directors and Executive Committee is reviewed annually on the basis of those factors; however, the review does not necessarily lead to any adjustment.
Based on the recommendation of the NCC, the Board decides on the compensation of the Board and Executive Committee at its own discretion, which is prospectively approved by the AGM. When preparing the compensation proposals, the NCC takes the following factors into consideration: Affordability and overall situation of the Company; Achievement of corporate goals and individual objectives; and Level of compensation paid by comparable companies in the biotech and pharmaceutical industry (where they compete for talent) and complexity (defined by their size and geographic scope). The compensation of the Board and Executive Committee is reviewed annually on the basis of those factors. However, the review does not necessarily lead to adjustments. Executive Compensation In accordance with the laws of Switzerland, we do not report executive compensation on an individualized basis. The disclosure below includes all forms of compensation given by us in exchange for services rendered in 20222023 and 20212022 by members of the Executive Committee. In 2022 and 2021, membersCompensation of the Executive Committee received totalfor the 2023 Calendar Year, in CHF
| | | | | | | | | | | | | | | | | | | | | | | | | | | | Fixed
Compensation | | | Cash
Bonus | | | Pension
Benefits | | | Other
Benefits | | | Options (2) | | | Total (3) | | Total Executive Committee (1) | | | 1,461,412 | | | | 85,500 | | | | 90,315 | | | | 486,892 | | | | 246,059 | | | | 2,370,178 | |
| (1) | The highest paid member of the Executive Committee in 2023 was the Company’s former Chief Executive Officer, Jack Weinstein, who received CHF 409,005 of fixed compensation and CHF 476,466 in other benefits. Other benefits include an additional 12-month remuneration of CHF 465,119, in accordance with pre-existing contractual terms, pursuant to Mr. Weinstein’s post-employment non-compete and non-solicitation obligation. |
| (2) | Reflects the value of share-based payments in accordance with IFRS 2 at grant date independently of the vesting schedule. Such stock option values are theoretical values at grant date and do not reflect taxable income nor realized income. These options were not yet granted as of December 31, 2023, but committed by the Company, contingent upon the Company meeting specific technical requirements. For accounting purposes, in accordance with IFRS 2, these were treated as granted options |
| (3) | Does not include the Company’s mandatory contribution to social security of CHF 115,991. |
During the 2023 fiscal year, remuneration as follows:to the executive committee amounted to CHF 2,370,178. This was within the limit of CHF 5,000,000 approved by the AGM 2022. Compensation of the Executive Committee for the 2022 Calendar Year, in CHF | | | | | | | | | | | | | | | | | | | | | | | | | | | | Fixed
Compensation | | | Cash
Bonus | | | Pension
Benefits | | | Other
Benefits | | | Options | | | Total (3) | | Total Executive Committee (1) | | | 1,635,477 | | | | 160,130 | | | | 89,087 | | | | 35,068 | | | | — | | | | 1,919,762 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | Fixed
Compensation | | | Cash
Bonus | | | Pension
Benefits | | | Other
Benefits | | | Options | | | Total (2) | | Total Executive Committee (1) | | | 1,635,477 | | | | 160,130 | | | | 89,087 | | | | 35,068 | | | | — | | | | 1,919,762 | |
| (1) | The highest paid member of the Executive Committee in 2022 was our former Chief Financial Officer and later Chief Executive Officer, Jack Weinstein, who received CHF 481,521 of fixed compensation, CHF 50,130 of variable cash compensation, and CHF 10,929 in other benefits. |
| (2) | Does not include the Company’s mandatory contribution to social security (AHV) of CHF 128,696. |
87
Compensation of the Executive Committee for the 2021 Calendar Year, in CHF
| | | | | | | | | | | | | | | | | | | | | | | | | | | | Fixed
Compensation | | | Cash
Bonus | | | Pension
Benefits | | | Other
Benefits | | | Options (2) | | | Total (3) | | Total Executive Committee (1) | | | 1,763,451 | | | | 231,340 | | | | 30,151 | | | | — | | | | 1,947,634 | | | | 3,972,476 | |
(1) | The highest paid member of the Executive Committee in 2021 was our then Chief Financial Officer, Jack Weinstein, who received CHF 460,614 of fixed compensation, CHF 175,000 of variable cash compensation and CHF 1,096,799 of options.
|
(2) | Reflects the value of share-based payments in accordance with IFRS 2 at grant date independently of the vesting schedule. Such stock option values are theoretical values at grant date and do not reflect taxable income nor realized income.
|
(3) | Does not include the Company’s mandatory contribution to social security (AHV) of CHF 82,953
|
During the 2022 fiscal year, remuneration to the executive committee amounted to CHF 1,919,762. This was within the limit of CHF 5,000,000 approved by the AGM 2022.
Employment Arrangements We have entered into employment letters with each of our executive officers. These agreements provide for a base salary and annual incentive cash and option bonus opportunities as well as payments upon termination.opportunities. Agreements with our executive officers may be terminated upon twothree to twelve months’ advance notice. Principles and Compensation Architecture Our compensation principles are aligned with the Company’s strategy of becoming profitable by growing its business and increasing revenue. The compensation principles are as follows: 69
Balance between competitiveness and affordability: within the Company’s financial ability, compensation levels are competitive and aligned with market practice for similar functions in comparable companies in the biotech and pharmaceutical industry; Pay for performance: part of compensation is directly linked to the performance of the business and to the achievement of individual objectives; and Alignment with shareholders’ interests: part of compensation is delivered in the form of stock options and thus is directly tied to the Company’s long-term share performance. The compensation of the members of the Executive Committee consists of fixed and variable remunerations. Fixed remuneration comprises the base salary and other benefits. Variable remuneration may comprise a performance-based cash bonus and equity grants. Compensation Model for Executive Officers | | | | | | | | | | | | Vehicle | | Purpose | | Drivers | | Performance | | Fixed Base Salary | | Monthly Cash | | Attract & Retain | | Market practice | | — | | Performance bonus | | Cash bonus | | Pay for performance | | Business and individual performance | | Company’s profitability,goals, individual performance | | Employee Participation Program | | Share options | | Align to shareholders’ interests | | Level of the roleresponsibility | | Share price | | Benefits | | Pension/insurance plans | | Protect against risk | | Market practice | | — |
Fixed Base Salary. The fixed base salary pays for the function and depends on our financial ability, the market value of the function and the profile of the individual in terms of qualifications and skill set. Performance Bonus. The performance bonus rewards the effective and successful conduct of the business and the achievement of individual objectives. The target performance bonus is generally expressed as a percentage of the fixed base salary. At the discretion of the Board and the NCC, decision to grant a bonus may be taken. The bonus amount effectively paid out is then determined by the Board, based upon the proposal of the NCC. The performance bonus is usually paid in cash or options, usually at the end of the financial year. 88
Employee Participation Program. The Employee Participation Program provides an incentive for management to make significant contributions towards our long-term success and aligns their interests to those of our shareholders. The Board of Directors determines the individual allocation of stock options at its own discretion, taking into account the level of the role and economic considerations. The value of the options for financial reporting purpose is calculated according to the Black-Scholes valuation model. Benefits. Executive Officers participate in the regular pension and retirement plans applicable to all employees in their country of employment. The provisions of those pension and retirement plans are in line with local regulations and prevailing market practice. Further, executive officers may be entitled to benefits in kind, in line with local market practice, such as a company car or other benefits. Contractual Provisions.Provisions. The employment contracts of our executive officers aremembers of the Executive Committee may be concluded for ana definite or indefinite period. The duration of definite employment contracts shall not exceed one year. Renewal is possible. The termination notice period and without a notice period. Theyof indefinite employment contracts may not exceed 12 months. The Company may enter into non-compete agreements with members of the Executive Committee for the time after termination of the employment agreement. Any non-compete provision for the period after termination of employment shall not exceed one year with the maximum compensation for such period not exceeding the average annual compensation of the last three years. Their employment contracts do not contain any agreement onprovision for severance provisions.payments. 8970
NON-EMPLOYEENon-Employee DIRECTOR COMPENSATIONDirector Compensation
The disclosure of compensation below includes all forms of compensation given by us in exchange for services rendered by members of our Board of Directors in 20222023 and 2021,2022, in CHF. | | | | | | | | | | | | | | | | | | | | | | | | | Board of Directors | | Cash Fee
2022 | | | Cash Fee
2021 | | | Options 2022
(1) | | | Options 2021
(1) | | | Total 2022
(2) | | | Total 2021 (2) | | Raghuram Selvaraju Chairman since May 25, 2016 | | | 500,000 | | | | 475,000 | | | | — | | | | 248,470 | | | | 500,000 | | | | 723,470 | | Thomas Plitz Member since December 17, 2020 | | | 150,000 | | | | 125,000 | | | | — | | | | 266,103 | | | | 150,000 | | | | 391,103 | | Patrice Jean Member since June 18, 2021 | | | 150,000 | | | | 76,998 | | | | — | | | | 16,330 | | | | 150,000 | | | | 93,329 | | Paolo Galfetti (3) Member since June 18, 2021 | | | 150,000 | | | | 79,722 | | | | — | | | | — | | | | 150,000 | | | | 79,722 | | Michelle Lock Member since January 28, 2022 | | | 92,473 | | | | — | | | | — | | | | — | | | | 92,473 | | | | — | | Thomaz Burckhardt Member until 8 February 2021 | | | — | | | | 7,500 | | | | — | | | | — | | | | — | | | | 7,500 | | Total | | | 1,042,473 | | | | 764,220 | | | | — | | | | 530,903 | | | | 1,042,473 | | | | 1,295,123 | |
| | | | | | | | | | | | | | | | | | | | | | | | | Board of Directors | | Cash Fee
2023 | | | Cash Fee
2022 | | | Options
2023 | | | Options
2022 | | | Total
2023 (1) | | | Total
2022 (1) | | Raghuram Selvaraju Chairman since May 25, 2016 | | | 248,260 | | | | 500,000 | | | | — | | | | — | | | | 248,260 | | | | 500,000 | | Thomas Plitz Member till April 26, 2024 | | | 133,213 | | | | 150,000 | | | | — | | | | — | | | | 133,213 | | | | 150,000 | | Patrice Jean Member till April 26, 2024 | | | 133,213 | | | | 150,000 | | | | — | | | | — | | | | 133,213 | | | | 150,000 | | Paolo Galfetti (3) Member till June 30, 2023 | | | 70,833 | | | | 150,000 | | | | — | | | | — | | | | 70,833 | | | | 150,000 | | Michelle Lock (2) Member since January 28, 2022 | | | 100,000 | | | | 92,473 | | | | — | | | | — | | | | 100,000 | | | | 92,473 | | Total | | | 685,519 | | | | 1,042,473 | | | | — | | | | — | | | | 685,519 | | | | 1,042,473 | |
| (1) | Reflects value of share-based payments in accordance with IFRS 2 at grant date independently of the vesting schedule. Such stock option values are theoretical values at grant date and do not reflect taxable income nor realized income.
|
(2) | Does not include the Company’s mandatory contribution to social security of CHF 35,515 (2021: 18,628)27,779 (2022: 35,515). |
| (2) | For her executive role, Ms. Lock received in 2023 an additional remuneration of CHF 71,750 in cash, CHF 246,059 in options subject to a vesting schedule, and CHF 7,534 in the form of pension contribution. Her executive compensation is reported as part of the compensation of the Executive Committee in the section above. |
| (3) | For his executive role during his 2023 tenure on the Board, Mr. Galfetti received an additional remuneration of CHF 442’990171,300 in cash and CHF 64’63627,697 in the form of pension contribution for the 2022 calendar year.contribution. His executive compensation is reported as part of the compensation of the Executive Committee. Committee in the section above. |
The figures in the table above cover the 2022 calendar year, as required by Swiss law. They differ fromFor the period authorized by the AGM, which runs from AGM 2023 to AGM (the Authorization Period). Differences between calendar years and Authorization periods are shown in the tables below.
90
During the current Authorization Period,2024, members of the Board of Directors are expected to earn a total compensation of CHF 1,141,667.500,000. This is within the limit of CHF 2,500,000500,000 approved atby the EGM 2022.
| | | | | | | | | | | | | | | | | | | | | | | | Calendar Year 2022 | | | Authorization period 2023/2022 | | | | | Compensation (CHF) | | | Period | | | | Amount | | | | Period | | | | Amount | (1) | | | Approved | | Cash Fee | |
| January 2022-
December 2022 |
| | | 1,042,473 | | |
| July 2022-June
2023 |
| | | 1,141,667 | | | | | | Options | |
| January 2022-
December 2022 |
| | | — | | |
| July 2022-June
2023 |
| | | — | | | | | | | | | | | | | | | | | | | | | | | | | | | Total | | | | | | | 1,042,473 | | | | | | | | 1,141,667 | | | | 2,500,000 | | (1) As this period is not yet ended as of the date of this report, the amount includes actual to date and an estimate of the compensation to be earned over the remaining period to the AGM 2023. | |
| | | | | | | | | | | | | | | | | | | | | | | | Calendar Year 2021 | | | Authorization period 2022/2021 | | | | | Compensation (CHF) | | | Period | | | | Amount | | | | Period | | | | Amount | (1) | | | Approved | | Cash Fee | |
| January 2021-
December 2021 |
| | | 764,220 | | |
| July 2021-
June 2022 |
| | | 922,110 | | | | | | Options | |
| January 2021-
December 2021 |
| | | 530,903 | | |
| July 2021-
June 2022 |
| | | 264,800 | | | | | | | | | | | | | | | | | | | | | | | | | | | Total | | | | | | | 1,295,123 | | | | | | | | 1,186,911 | | | | 2,500,000 | |
In 2022 and 2021, no compensation was granted to former members of the Board of Directors or related parties.held on April 26, 2024.
Option Grants to Members of the Board of Directors and Executive Committee During 2023 and 2022, and 2021, the followingno options to purchase shares of the Company’s ordinary stock as listed on the SIX Swiss Exchange were granted to members of the Board of Directors and Executive Committee:Committee. However, the Company had committed to granting 320,000 options to Ms. Lock, interim executive chief officer, contingent upon the Company meeting certain technical requirements, including the availability of conditional capital and a reduction in the nominal value of the Company’s share capital. Such conditions were met on April 26, 2024. The options are subject to a vesting schedule, have an exercise price of CHF 2.00, and will expire between May 2029 and May 2030. | | | | | | | | | | | Date of Grant | | Options Granted | | | Strike Price (in
CHF) | | | Expiration Date | January 1, 2021 | | | 100,000 | | | | 0.2690 | | | December 31, 2025 | May 30, 2021 | | | 8,500,000 | | | | 0.0100 | | | 1/2 on each of May 29, 2027 and 2028 | September 12, 2021 | |
| 1,900,000
3,200,000 |
| |
| 0.0100
0.1540 |
| | 1/3 on each of September 12, 2027, 2028 and 2029 | December 20, 2021 | | | 3,000,000 | | | | 0.0100 | | | December 27, 2027 | December 30, 2021 | |
| 15,500,000
11,000,000 |
| |
| 0.0610
0.0100 |
| | 1/3 on each of December 27, 2027, 2028 and 2029 |
Our Board of Directors currently consists of five members. As a foreign private issuer, we are not required to have independent directors on our Board of Directors, except that our audit committee is required to consist fully of independent directors, subject to certain phase-in schedules. 91
Overview The Board of Directors is self-constituting and determines our internal organization. The Chairman convenes the Board as often as our affairs require and presides (or in his absence, another Board member specifically designated by the majority of the other members present at the meeting) over the Board meetings. Each Board member is entitled to request to the Chairman, in writing, a meeting of the Board by indicating the grounds for such a request. The Chairman decides on the agenda items and motions. Every Director is entitled to request to the Chairman, in writing, the inclusion of a specific agenda item by indicating the grounds for such a request. 71
To pass a valid resolution, the majority of the Board members has to attend the meeting. Meetings may also be held by telephone or video conference, to which all the Board members are invited. No quorum is required for confirmatory resolutions and adaptations of the Articles in connection with capital increases. The Board of Directors passes its resolutions by way of simple majority. The members of the Board may only vote in person, not in proxy. In the event of a tie vote, the Chairman has the deciding vote. Minutes of deliberations and resolutions are kept and signed by the Chairman and the designated Secretary. The Board has established the following permanent committees to further strengthen our corporate governance structure. Audit and Finance Committee The Audit and Finance Committee (AFC) advises the Board of Directors in the performance of its supervisory duties. In particular, the AFC reviews the financial reporting to shareholders and the general public as well as the relationship with the external auditors; satisfies itself that our financial risk management and our internal controls are of an appropriate standard; ensures that its activities are consistent and compliant with the Organizational Regulations; assesses adherence to the relevant ‘best practice’ corporate governance provisions, to the extent such practice has effect on the activities and the functions of the AFC; satisfies itself that our overall fraud prevention procedures are of an appropriate standard and ensures that appropriate procedures to enable employees to confidentially and anonymously submit their concerns regarding accounting, internal controls or auditing matters are in place. AsUntil April 26, 2024, the AFC was composed of the date of this annual report,Patrice Jean (Chair) and Thomas Plitz. From April 27, 2024, the AFC is composed of Patrice JeanGregory Van Beek (Chair), Michelle Lock and Thomas Plitz.Elzinga.
Nomination and Compensation Committee The Nominating and Compensation Committee (NCC) advises the Board of Directors in the performance of its supervisory duties related to nomination and compensation matters. It is responsible for ensuring the best possible leadership and management of the Company and for determining compensation policies, including share-based incentive programs, for the Company’s top management and BoardBoard. Until April 26, 2024, the NCC was composed of Directors. As of the date of this annual report,Thomas Plitz (Chair) and Raghuram Selvaraju. From April 27, 2024, the NCC is composed of Thomas PlitzPeter de Svastich (Chair) and Raghuram Selvaraju.
Corporate Governance Committee The Corporate Governance Committee (CGC) advises the Board on all matters of corporate governance. It is responsible for carrying out in-depth analysis of specific corporate governance-related matters and monitors compliance with corporate governance principles and policies. AsUntil April 26, 2024, the CGC was composed of the date of this annual report,Patrice Jean (Chair) and Michelle Lock. From April 27, 2024, the CGC is composed of Paolo GalfettiRaghuram Selvaraju (Chair) and Patrice Jean.
Other Corporate Governance Matters
The Sarbanes-Oxley Act of 2002, as well as related rules subsequently implemented by the SEC, requires foreign private issuers, including our company, to comply with various corporate governance practices. In addition, Nasdaq rules provide that foreign private issuers may follow home country practice in lieu of the Nasdaq corporate governance standards, subject to certain exceptions and except to the extent that such exemptions would be contrary to U.S. federal securities laws.
92
We intend to take all actions necessary for us to maintain compliance as a foreign private issuer under the applicable corporate governance requirements of the Sarbanes-Oxley Act of 2002, the rules adopted by the SEC and Nasdaq’s listing standards.
Because we are a foreign private issuer, our directors and senior management are not subject to short-swing profit and insider trading reporting obligations under Section 16 of the U.S. Securities Exchange Act of 1934, as amended, or the Exchange Act. They will, however, be subject to the obligations to report changes in share ownership under Section 13 of the Exchange Act and related SEC rules.Michelle Lock.
Indemnification of Officers and Directors Under Swiss law, a corporation may indemnify its officers and directors against losses and expenses, except for such losses and expenses arising from willful misconduct or negligence (although some legal scholars advocate that at least gross negligence be required), including attorney’s fees, judgments, fines, and settlement amounts actually and reasonably incurred in a civil or criminal action, suit or proceeding by reason of having been the representative of, or serving at the request of, the corporation. Subject to Swiss law, our articles of association provide for indemnification of the exiting and former members of our board of directors,Board, executive management, and the heirs, executors and administrators, against liabilities arising in connection with the performance of their duties in such capacity, and permits us to advance the expenses of defending any act, suit or proceeding to members of our board of directorsBoard and executive management. In addition, under general principles of Swiss employment law, an employer may be required to indemnify an employee against losses and expenses incurred by such employee in the proper execution of their duties under their employment agreement with the company. We have entered into indemnification agreements with each of the members of our board of directorsBoard and with our executive officers. 72
AtAs of December 31, 2022,2023, we had a total of 6949 employees. Our relationshipNone of our employees are represented by any collective bargaining agreements. We believe that we maintain good relations with our employees is good and no employees are covered by a labor union. We did not employ any temporary employees during 2022.employees.
For information about share ownership by our directors and senior management, see “Item 7. Major Shareholders and Related Party Transactions – A. Major Shareholders”. | F. | DISCLOSURE OF A REGISTRANT’S ACTION TO RECOVER ERRONEOUSLY AWARDED COMPENSATION |
Not applicable. | ITEM 7 | MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS |
The following table sets forth information with respect to the beneficial ownership of our ordinary shares as of April 30, 2023the date of this annual report by: each of our executive officers; each of our directors; and each person, or group of affiliated persons, who is known by us to beneficially own more than 3 percent of our outstanding ordinary shares. These share holding numbers have not been adjusted for the 400:1 reverse split which occurred on May 5, 2023.
93
Beneficial ownership is determined in accordance with the rules and regulations of the SEC and includes voting or investment power with respect to our ordinary shares. Ordinary shares subject to options that are currently exercisable or exercisable within 60 days after calculation date are considered outstanding and beneficially owned by the person holding the options for the purpose of calculating the percentage ownership of that person but not for the purpose of calculating the percentage ownership of any other person. Except as otherwise noted, the persons and entities in this table have sole voting and investment power with respect to all of the ordinary shares beneficially owned by them, subject to community property laws, where applicable. Except as otherwise set forth below, the address of the beneficial owner is c/o the Company. The information in the table below is based on information known to us or ascertained by us from public filings made by the shareholders. We have also set forth below information known to us regarding any significant change in the percentage ownership of our ordinary shares by any major shareholders during the past three years. The major shareholders listed below do not have voting rights with respect to their ordinary shares that are different from the voting rights of other holders of our ordinary shares. | | Name | | Shares | | | Option | | | Percentage | | | Shares | | | Option * | | Percentage ** | | 5 Percent Stockholders | | | | | | | GEM Global Yield LLC SCS† | | | 1,155,899,126 | | | | 0 | | | | 20.58 | %* | 3 Percent Stockholders | | GEM Global Yield LLC SCS (a) | | | | 2,889,747 | | | | 3,350,000 | (c) | | | 49.76 | % | Armistice Capital Master Fund Ltd. (b) | | | | 688,000 | | | | 1,500,000 | | | | 17.45 | % | Executive Officers and Directors | | | | | | | Jack Weinstein | | | 185,000 | | | | 18,600,000 | | | | * | * | Michelle Lock | | | | 0 | | | | 320,000 | (d) | | | 1.28 | % | Paolo Galfetti | | | | 78,093 | | | | 31,250 | | | | * | ** | Jeremy Meinen | | | 140,655 | | | | 1,100,000 | | | | * | * | | | 351 | | | | 2,750 | | | | * | ** | Marco Marotta | | | 0 | | | | 1,500,000 | | | | * | * | Raghuram Selvaraju | | | 0 | | | | 8,963,197 | | | | * | * | | | 0 | | | | 22,408 | | | | * | ** | Thomas Plitz | | | 500,000 | | | | 1,500,000 | | | | * | * | Patrice Jean | | | 140,000 | | | | 200,000 | | | | * | * | Paolo Galfetti | | | 31,237,437 | | | | 12,500,000 | | | | * | * | Michelle Lock | | | 0 | | | | 0 | | | | * | * | Peter de Svastich (e) | | | | 7,001 | | | | 0 | | | | * | ** | Gregory Van Beek (e) | | | | 0 | | | | 0 | | | | * | ** | Thomas Elzinga (e) | | | | 0 | | | | 0 | | | | * | ** |
†(a) | As set forth in a filing with the SIX Swiss Exchange on April 7, 2023. Pursuant to such filing, Christopher Brown, a resident of the State of New York, is the person who can exercise the voting rights at his own discretion. |
*(b) | Calculated atAs set forth in a filing with the date of notification basedSIX Swiss Exchange on the number of outstanding shares registered at the Commercial Register of Geneva.April 12, 2024.
|
| (c) | Of which 3,350,000 are expected to be issued by the Company within 60 days of April 30, 2024, and immediately exercisable at a price per share of CHF 1.70. |
| (d) | Of which 160,000 are expected to be issued by the Company within 60 days of April 30, 2024, and immediately exercisable at a price per share of CHF 2.00. |
| (e) | Although they are affiliated with GEM, each of these directors expressly disclaim beneficial ownership over the shares of the Company’s stock owned by GEM. |
| * | Unless stated otherwise, all options are either already exercisable or will become exercisable within 60 days of April 30, 2024. |
73
| ** | Percentage ownership calculations are based on 12,540,439 shares issued and outstanding, excluding 1,500,398 treasury shares, plus, in accordance with SEC rules on disclosure of beneficial ownership, shares that each securityholder has the ability to acquire within 60 days of April 30, 2024, due to outstanding equity interests becoming vested or exercisable. |
| *** | Less than one percent. |
We are not aware of any arrangement, the operation of which may result in a change of control of the Company. Except for significant shareholders reported in the table above, the Company is not owned or controlled by another corporation, by any foreign government or by any other natural or legal person. | B. | RELATED PARTY TRANSACTIONS |
The following is a description of related party transactions we have entered into with the beneficial owners of 10 percent or more of our ordinary shares, which are our only voting securities, senior management and members of our Board of Directors, from January 1, 2021,2023, to December 31, 2022.the date of this annual report. Director and Executive Officer Compensation See “Item 6.B—Compensation of Directors and Executive Officers” for information regarding compensation of directors and executive officers. Indemnification of directors and executive management We entered into indemnification agreements with our executive officers and directors. Share Subscription Facility with GEM Global Yield LLC SCS On January 20, 2021, we signed a binding agreement with our largest shareholder, GEM Global Yield LLC SCS and GEM Yield Bahamas Limited (GEM) for the implementation of a new share subscription facility (SSF) in the amount of up to CHF 50 million.million for a duration of up to three years. On February 27, 2024, the SSF agreement was extended until January 20, 2027. Under the terms of the SSF, we have the right to periodically, during athe timeframe of up to three years,the agreement, issue and sell shares to GEM. Under the facility, GEM undertakes to subscribe to or acquire our ordinary registered shares of the Company upon the Company’sour exercise of a drawdown notice. In accordance with the customary terms of the SSF agreement, the Company controlswe control the timing and maximum amount of any drawdown and retainsretain the right, not the obligation, to draw down on the full commitment amount. Future subscription prices under the SSF will correspond to 90 percent of the average of the closing bid prices on the SIX Swiss Exchange during the reference period, which corresponds to fifteen trading days following Relief’s drawdown notice. Under the terms of the SSF, we are required to pay GEM a commitment fee of CHF 1.25 million, which is currently outstanding as an interest bearing loan pursuant to the SSF. As of the date of this annual report, no amounts have been drawn on this facility.
94
ManagementUnder the terms of the agreement dated January 20, 2021, we were required to pay GEM a commitment fee of CHF 1.25 million, which remained outstanding as an interest-bearing loan. Pursuant to the extension agreement dated February 27, 2024, GEM agreed to forgive the commitment fee and Boardaccrued interests to date. In consideration for GEM’s capital commitment and this debt waiver, Relief committed to issuing GEM warrants to purchase up to 3.35 million ordinary shares at a purchase price of Directors
CHF 1.70 per share, exercisable from the issuance date, and expiring on January 20, 2027. The following additional transactions were carried outissuance of these warrants was contingent upon shareholder approval for a reduction in the nominal value of the Company’s ordinary shares. At the extraordinary general meeting held on April 26, 2024, such reduction was approved and the warrants will be formally issued upon the registration of the reduction in nominal value with our executive officers and membersthe commercial register of our board of directorsGeneva, Switzerland. | | | | | | | | | | Key Management Compensation, in TCHF | | 2022 | | | 2021 | | Fees, salaries and other short-term employee benefits | | | 2,873 | | | | 2,759 | | Post-employment benefits | | | 89 | | | | 30 | | Share-based compensation | | | — | | | | 814 | | Total compensation for key management | | | 2,962 | | | | 3,603 | |
| C. | INTERESTS OF EXPERTS AND COUNSEL |
None.Not applicable..
| ITEM 8 | FINANCIAL INFORMATION |
| A. | CONSOLIDATED STATEMENTS AND OTHER FINANCIAL INFORMATION |
Consolidated Financial Statements Our audited financial statements for the fiscal years ended December 31, 20222023 and 20212022 are included in Item 1817 of this annual report. 74
Legal Proceedings From time to time, we may become involved in litigation or other legal governmental or arbitration proceedings or be subjectrelating to claims arising infrom the ordinary course of business. There are currently no claims or actions pending against us that, in the opinion of our business. Except as set forth thereinmanagement, are likely to have a material adverse effect on our results of operations, financial condition or elsewherecash flows. A description of and update on our previously reported and terminated litigation with NeuroRx can be found in this Annual Report, we are not presently a party to any legal, governmental or arbitration proceeding. Regardless of“Item 4. Information on the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.Company - 4.B. Business Overview.” – Material Contracts” Dividend Distribution PolicyBoard of Directors
| | Cash Fee
2023 | | | Cash Fee
2022 | | | Options
2023 | | | Options
2022 | | | Total
2023 (1) | | | Total
2022 (1) | |

