
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 20-F
(Mark One)
| REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
| ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31 2018, 2021
OR
| TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
| SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Date of event requiring this shell company report: Not applicable
For the transition period from______ to _______
Commission file number 001-36581
Vascular Biogenics Ltd.
(Exact name of registrant as specified in its charter)
N/A
(Translation of Registrant’s name into English)
Israel
Israel
(Jurisdiction of incorporation or organization)
8 HaSatat St
Modi’in
Israel7178106
(Address of principal executive offices)
Dror Harats, Chief Executive Officer
8 HaSatat St.
Modi’in
Israel7178106
Tel: +972-8-9935000+972-8-9935000
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act.
Securities registered pursuant to Section 12(b) of the Act:
| Title of | Trading symbol(s) | Name of | ||
| Ordinary Shares, par value NIS 0.01 each | VBLT | The |
Securities registered or to be registered pursuant to Section 12(g) of the Act. None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act. None
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report.
As of December 31, 2018,2021, the Registrant had 35,881,128 Ordinary Shares outstanding.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes [ ] ☐ No [X] ☒
If this report is an annual report or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. Yes [ ] ☐ No [X] ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [X] ☒ No [ ]☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files) Yes [X] ☒ No [ ]☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer, or an emerging growth company.
See definition of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | Accelerated filer | Non-accelerated filer | ||
| Emerging Growth Company |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act. [ ]☐
† The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☒
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
| U.S. GAAP | International Financing Reporting Standards as issued | Other ☐ | ||
| by the International Accounting Standards Board |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow.
Item 17 [ ]☐ Item 18 [ ]☐
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes [ ] ☐ No [X] ☒
| 2 |
General Matters
In this Annual Report on Form 20-F, (“or the Annual Report”),Report, unless the context indicates otherwise, references to “NIS” are to the legal currency of Israel, “U.S. dollars,” “$” or “dollars” are to United States dollars, and the terms “we,” “us,” “our company,” “our,” “VBL,” and “Vascular Biogenics” refer to Vascular Biogenics Ltd.
Throughout this Annual Report, we refer to various trademarks, service marks and trade names that we use in our business. The “Vascular Biogenics” design logo, “VBL Therapeutics,” “Vascular Targeting System,” “VTS,” “VB-111,” “VB-601,” the “OVAL” design logo and other trademarks or service marks of Vascular Biogenics Ltd. appearing in this Annual Report are the property of Vascular Biogenics Ltd. We have several other registered trademarks, service marks and pending applications relating to our products. Although we have omitted the “®” and trademark designations for such marks in this Annual Report, all rights to such trademarks are nevertheless reserved. Other trademarks and service marks appearing in this Annual Report are the property of their respective holders.
Cautionary Note Regarding Forward-Looking Statements
This Annual Report contains forward-looking statements that relate to future events or our future financial performance, which express the current beliefs and expectations of our management. Such statements involve a number of known and unknown risks, uncertainties and other factors that could cause our actual future results, performance or achievements to differ materially from any future results, performance or achievements expressed or implied by such forward-looking statements. Forward-looking statements include all statements that are not historical facts and can be identified by words such as, but not limited to, “believe,” “expect,” “anticipate,” “estimate,” “intend,” “plan,” “targets,” “likely,” “will,” “would,” “could,” and similar expressions or phrases. We have based these forward-looking statements largely on our management’s current expectations and future events and financial trends that we believe may affect our financial condition, results of operations, business strategy and financial needs. Forward-looking statements include, but are not limited to, statements about:
| ● | the initiation, timing, progress and results of our |
| ● | our expectations about the availability of data from our clinical trials; |
| ● | our ability to advance product candidates into, and successfully complete, clinical trials; |
| ● | our plans for future clinical trials; |
| ● | our ability to manufacture our product candidates in sufficient quantities for clinical |
| ● | the timing or likelihood of regulatory filings and |
| ● | the commercialization of our product candidates, if approved; |
| ● | potential advantages of our product candidates; |
| ● | the pricing and reimbursement of our product candidates, if approved; |
| ● | our ability to develop and commercialize additional product candidates based on our platform technologies; |
| ● | our business strategy; |
| ● | the implementation of our business model, strategic plans for our business, product candidates and technology; |
| ● | the scope and duration of protection we are able to establish and maintain for intellectual property rights covering our product candidates and technology; |
| estimates of our expenses, future revenues, capital requirements and our needs for additional financing; | |
| ● | our ability to establish and maintain collaborations and the benefits of such collaborations; |
| ● | our ability to maintain our level of grant funding or obtain additional grant funding; |
| ● | developments relating to our competitors and our industry; |
| ● | our anticipated loss of foreign private issuer status, and |
| ● | other risks and uncertainties, including those listed under the caption “Risk Factors.” |
| 3 |
All forward-looking statements involve risks, assumptions and uncertainties. You should not rely upon forward-looking statements as predictors of future events. The occurrence of the events described, and the achievement of the expected results, depend on many events, some or all of which are not predictable or within our control. Actual results may differ materially from expected results. See the sections “Item 3. Key Information-D. Risk Factors,” “Item 5. Operating and Financial Review and Prospectus” and elsewhere in this Annual Report for a more complete discussion of these risks, assumptions and uncertainties and for other risks and uncertainties. These risks, assumptions and uncertainties are not necessarily all of the important factors that could cause actual results to differ materially from those expressed in any of our forward-looking statements. Other unknown or unpredictable factors also could harm our results.
All of the forward-looking statements we have included in this Annual Report are based on information available to us on the date of this Annual Report. We undertake no obligation, and specifically decline any obligation, to update publicly or revise any forward-looking statements, whether as a result of new information, future events or otherwise. In light of these risks, uncertainties and assumptions, the forward-looking events discussed in this Annual Report might not occur.
The audited financial statements for the years ended December 31, 2018, 20172021, 2020 and 20162019 in this Annual Report have been prepared in accordance in accordance with U.S. GAAP.
Summary of Risk Factors
Investing in our common shares involves a high degree of risk. You should carefully consider the risks summarized below and other risks that we face, a detailed discussion of which can be found under “Item 3. Key Information-D. Risk Factors” below, together with other information in this annual report on Form 20-F and our other filings with the International Financial Reporting Standards (“IFRS”) as issued bySecurities and Exchange Commission, or SEC. This summary list of risks is not exhaustive of the International Accounting Standards Board (“IASB”).factors that may affect any of our forward-looking statements and our business and financial results. If any of these risks actually occur, our business, financial condition and financial performance would likely be materially adversely affected. In such case, the trading price of our common shares would likely decline and you may lose part or all of your investment. Below is a summary of some of the principal risks we face:
| ● | We have incurred significant losses since our inception and anticipate that we will continue to incur significant losses for the foreseeable future. |
| ● | We have never generated any revenue from product sales and may never be profitable. |
| ● | We may need to raise additional funding, which may not be available on acceptable terms, or at all. Failure to obtain this necessary capital when needed may force us to delay, limit or terminate our product development efforts or other operations. |
| ● | We have received and may continue to receive Israeli or other governmental grants to assist in the funding of our research and development activities. If we lose our funding from these research and development grants, we may encounter difficulties in the funding of future research and development projects and implementing technological improvements, which would harm our operating results. |
| ● | We have been selected for €17.5 million of funding from the Horizon Europe EIC Accelerator Program, which funding is subject to a lengthy process, including finalization of agreements, prior to receipt, which we may not successfully achieve. |
| ● | We are highly dependent on the success of ofra-vec in oncology applications, and our platform technologies in general, and we cannot be certain that any of them will receive regulatory approval or be commercialized. Any failure to successfully develop, obtain regulatory approval for and commercialize ofra-vec for cancer indications or any other product candidates, independently or in cooperation with a third party collaborator, or the experience of significant delays in doing so, would compromise our ability to generate revenue and become profitable. |
| ● | Our product candidates are based on novel technologies, which makes it difficult to predict the time and cost of product candidate development and potential regulatory approval. |
| ● | We may find it difficult to enroll patients in our clinical trials, and patients could discontinue their participation in our clinical trials, which could delay or prevent clinical trials of our product candidates. |
| ● | We may encounter substantial delays in our clinical trials or we may fail to demonstrate safety and efficacy to the satisfaction of applicable regulatory authorities. |
| ● | The results from our clinical trials may not be sufficiently robust to support the submission for marketing approval for our product candidates. Before we submit our product candidates for marketing approval, the U.S. Food and Drug Administration, or FDA, and the European Medicines Agency, or EMA, may require us to conduct additional clinical trials, or evaluate subjects for an additional follow-up period. |
| ● | Legislative and regulatory activity may exert downward pressure on potential pricing and reimbursement for any of our product candidates, if approved, that could materially affect the opportunity to commercialize. |
| ● | We expect to rely on third parties to conduct some or all aspects of our product manufacturing, protocol development, research and preclinical and clinical testing, and these third parties may not perform satisfactorily. |
| ● | We intend to at least partially rely on third-party manufacturers to produce commercial quantities of any of our product candidates that receives regulatory approval, but we have not entered into binding agreements with any such manufacturers to support commercialization. Additionally, these manufacturers do not have experience producing our product candidates at commercial levels and may not pass regulatory inspections or achieve the necessary regulatory approvals or produce our product candidates at the quality, quantities, locations and timing needed to support commercialization. |
| ● | Legislative and regulatory activity may exert downward pressure on potential pricing and reimbursement for any of our product candidates, if approved, that could materially affect the opportunity to commercialize. |
| ● | Our future success depends on our ability to retain key employees, consultants, and advisors and to attract, retain and motivate qualified personnel. |
| ● | Pandemics, such as the ongoing COVID-19 pandemic, could have an adverse impact on our developmental programs and our financial condition. |
| ● | We depend on our license agreement with Janssen Vaccines & Prevention B.V. and if we cannot meet requirements under such license agreement, we could lose the rights to our products, which could have a material adverse effect on our business. |
| ● | The market price of our ordinary shares may be highly volatile, and you may not be able to resell your shares at the purchase price. |
| ● | We are currently a “foreign private issuer” and intend to follow certain home country corporate governance practices, and our shareholders may not have the same protections afforded to shareholders of companies that are subject to all Nasdaq corporate governance requirements. Additionally, we cannot be certain if the reduced disclosure requirements applicable to our status as a foreign private issuer, will make our ordinary shares less attractive to investors. | |
| ● | We expect to lose our foreign private issuer status, which will require us to comply with the Exchange Act’s domestic reporting regime and cause us to incur significant legal, accounting and other expenses, even if we are able to qualify as a “smaller reporting company.” |
| 4 |
PART I
Item 1. Identity of Directors, Senior Management and Advisers
Not applicable.
Item 2. Offer Statistics and Expected Timetable
Not applicable.
A. Selected Financial Data[Reserved]
The following table summarizes our financial data. We have derived the summary statements of operations data for the years ended December 31, 2018, 2017, and 2016, and the balance sheet data as of December 31, 2018 and 2017 from our audited financial statements included elsewhere in this Annual Report. The statements of operations data for the years ended December 31, 2015 and 2014, and the balance sheet data as of December 31, 2016, 2015 and 2014 is derived from audited financial statements not included in this Annual Report.
The summary of our financial data set forth below should be read together with our audited financial statements and the related notes, as well as the section entitled “Item 5. Operating and Financial Review and Prospects,” included elsewhere in this Annual Report.
| (in thousands, except share and per-share data) | 2018 | 2017 | 2016 | 2015 | 2014 | |||||||||||||||
| Statements of operations data: | ||||||||||||||||||||
| Revenues | $ | 585 | $ | 13,864 | $ | - | $ | - | $ | - | ||||||||||
| Cost of revenues | (235 | ) | (340 | ) | - | - | - | |||||||||||||
| Gross Profit | $ | 350 | $ | 13,524 | $ | - | $ | - | $ | - | ||||||||||
| Research and development expenses, net | $ | 15,940 | $ | 17,770 | $ | 12,447 | $ | 11,198 | $ | 10,974 | ||||||||||
| Marketing expenses | 397 | 562 | - | - | - | |||||||||||||||
| General and administrative expenses | 5,220 | 5,847 | 3,828 | 3,673 | 3,804 | * | ||||||||||||||
| Operating loss | 21,207 | 10,655 | 16,275 | 14,871 | 14,778 | |||||||||||||||
| Financial income | (908 | ) | (544 | ) | (285 | ) | (100 | ) | (15 | ) | ||||||||||
| Financial expenses: | ||||||||||||||||||||
| Loss from change in fair value of convertible loan | - | - | - | - | 2,342 | |||||||||||||||
| Other financial expenses | 159 | 27 | 12 | 117 | 302 | |||||||||||||||
| Financial (income) expenses, net | (749 | ) | (517 | ) | (273 | ) | 17 | 2,629 | ||||||||||||
| Other comprehensive loss (income) | (25 | ) | 24 | 5 | (6 | ) | (10 | ) | ||||||||||||
| Comprehensive loss | 20,433 | $ | 10,162 | $ | 16,007 | 14,882 | $ | 17,397 | ||||||||||||
| Loss per ordinary share, basic and diluted | 0.62 | $ | 0.37 | $ | 0.64 | $ | 0.73 | $ | 3.09 | |||||||||||
| Weighted average ordinary shares outstanding, basic and diluted | 32,969,094 | 27,398,169 | 24,970,585 | 20,309,596 | 5,627,324 | |||||||||||||||
* Includes a one-time expense related to the IPO grant of options to our Chief Executive Officer of $2.2 million.
| December 31, | ||||||||||||||||||||
| 2018 | 2017 | 2016 | 2015 | 2014 | ||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Statements of financial position data: | ||||||||||||||||||||
| Cash and cash equivalents and short-term bank deposits | $ | 50,482 | $ | 54,729 | $ | 45,254 | $ | 37,146 | $ | 36,783 | ||||||||||
| Total assets | 60,678 | 65,689 | 47,274 | 39,238 | 38,138 | |||||||||||||||
| Total liabilities | 7,585 | 9,789 | 4,874 | 4,231 | 3,036 | |||||||||||||||
| Ordinary shares | 73 | 57 | 50 | 38 | 32 | |||||||||||||||
| Total equity | 53,093 | 55,900 | 42,400 | 35,007 | 35,102 | |||||||||||||||
| 5 |
B. Capitalization and Indebtedness
Not applicable.
C. Reasons for the Offer and Use of Proceeds
Not applicable.
D. Risk Factors
You should consider carefully the risks and uncertainties described below, together with all of the other information in this Annual Report, including the financial statements and the related notes included elsewhere in this Annual Report.Report and “Item 5. Operating and Financial Review and Prospects.” The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties that we are unaware of, or that we currently believe are not material, may also become important factors that adversely affect our business. If any of the following risks actually occurs, our business, financial condition, results of operations, and future prospects could be materially and adversely affected.
Risks Related to Our Financial Condition and Capital Requirements
We have incurred significant losses since our inception and anticipate that we will continue to incur significant losses for the foreseeable future.
We are a clinical-stage biotechnology company, and we have not yet generated any regular revenue streams. We have incurred losses in each year since our inception in 2000, including net losses of $20.5$29.9 million, $10.1$24.2 million and $16.0$19.4 million for the years ended December 31, 2018, 20172021, 2020 and 2016,2019, respectively. As of December 31, 2018,2021, we had an accumulated deficit of $188.6$262.1 million.
We have devoted most of our financial resources to research and development, including our clinical and pre-clinicalpreclinical development activities. To date, we have financed our operations primarily through the sale of equity securities and convertible debt and, to a lesser extent, through grants from governmental agencies. The amount of our future net losses will depend, in part, on the rate of our future expenditures and our ability to obtain funding through equity or debt financings, strategic collaborations or additional grants. We have completed only a single pivotal clinical trial for our product candidates, which did not meet the primary endpoint in such trial and it will be a few years, if ever, before we have a product candidate ready for commercialization. Even if our current Phase 3 trial or future clinical trials are successful and we obtain regulatory approval to market a product, candidate, our future revenues will depend upon the size of any markets in which oursuch product candidates have receivedreceives approval, and our ability to achieve sufficient market acceptance, reimbursement from third-party payorspayers and adequate market share for ourany approved product candidates in those markets.
We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. We anticipate that our expenses will increase substantially if and as we:
| ● | continue our research, |
| ● | expand the scope of our current clinical trials for our product candidates; |
| ● | initiate additional |
| ● | seek regulatory and marketing approvals for any of our product candidates that successfully complete clinical trials; |
| ● | further develop the manufacturing process for our product candidates; |
| ● | |
| ● | change or add additional manufacturers or suppliers; |
| ● | establish a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain marketing approval; |
| ● | expand our operations in the United States; |
| ● | seek to identify and validate additional product candidates; |
| ● | acquire or in-license other product candidates and technologies; |
| ● | make milestone or other payments under any in-license or other intellectual property related agreements, including our agreement with Tel Hashomer-Medical Research, Infrastructure and Services Ltd. and our license from Janssen Vaccines & Prevention B.V., or Janssen (formerly known as Crucell Holland B.V.), and any other licensing arrangements we may enter into the future; |
| ● | maintain, protect and expand our intellectual property portfolio; |
| ● | attract and retain skilled personnel; |
| 6 |
| ● | |
| create additional infrastructure to support our operations as a public company; | |
| ● | transition from being a foreign private issuer to a U.S. reporting company; and |
| ● | experience any delays or encounter issues with any of the above. |
The net losses we incur may fluctuate significantly from quarter to quarter and year to year, such that a period-to-period comparison of our results of operations may not be a good indication of our future performance. In any particular quarter or quarters, our operating results could be below the expectations of securities analysts or investors, which could cause our share price to decline.
We have never generated any revenue from product sales and may never be profitable.
Our ability to generate revenue and achieve profitability depends on our ability, alone or with strategic collaboration partners, to successfully complete the development of, obtain the regulatory approvals of, and commercialize our product candidates. We do not anticipate generating revenues from product sales for the foreseeable future, if ever. Our ability to generate future revenues from product sales depends heavily on our success in:
| ● | completing research, |
| ● | |
| ● | obtaining regulatory and marketing approvals for product candidates for which we complete successful clinical trials; |
| ● | developing a sustainable, scalable, reproducible, and transferable manufacturing process for our product candidates; |
| ● | establishing and maintaining supply and manufacturing relationships with third parties that can provide products and services adequate, in amount and quality, to support clinical development and the market demand for our product candidates, if approved; |
| ● | |
| ● | launching and commercializing any product candidates for which we obtain regulatory and marketing approval, either by collaborating with a partner or, if launched independently, by establishing a sales, marketing and distribution infrastructure; |
| ● | obtaining market acceptance of any product candidates that receive regulatory approval as viable treatment options; |
| ● | addressing any competing technological and market developments; |
| ● | implementing additional internal systems and infrastructure, as needed; |
| ● | identifying and validating new product candidates; |
| ● | negotiating favorable terms in any collaboration, licensing or other arrangements into which we may enter; |
| ● | maintaining, protecting and expanding our portfolio of intellectual property rights, including patents, trade secrets and know-how; and |
| ● | attracting, hiring and retaining qualified personnel. |
Even if one or more of our product candidates isare approved for commercial sale, we anticipate incurring significant costs associated with commercializing any approved product candidate. Our expenses could increase beyond expectations if we are required by the FDA, the European Medicines Agency, or the EMA, or other regulatory agencies, domestic or foreign, to perform clinical and other studies in addition to those that we currently anticipate. Even if we are able to generate revenues from the sale of any approved products, we may not become profitable and may need to obtain additional funding to continue operations.
We may need to raise additional funding, which may not be available on acceptable terms, or at all. Failure to obtain this necessary capital when needed may force us to delay, limit or terminate our product development efforts or other operations.
We are currently advancing VB-111ofra-vec (ofranergene obadenovec, also known as VB-111) for solid cancer indications.oncology indications, and VB-601 for inflammatory applications. We intend to advance this current clinicalthese product candidatecandidates through clinical development and other product candidates through pre-clinicalpreclinical and clinical development. Developing pharmaceutical products is expensive, and we expect our research and development expenses to increase substantially in connection with our ongoing activities, particularly as we advance our product candidates in clinical trials.
As of December 31, 2018,2021, our cash and cash equivalents and short-term bank deposits were $50.5$53.5 million. As of December 31, 2018,the date of this Annual Report, we estimate that our existingthe balance of cash, cash equivalents and short-term bank deposits at December 31, 2021 will be sufficient to fund our operations for aboutat least twelve months from the date of the readout of top-line PFS data from the Phase 3 years.OVAL trial (data we anticipate receiving in the second half of 2022). However, our operating plan may change as a result of many factors, currently unknown to us, and we may need to seek additional funds sooner than planned through public or private equity or debt financings, government or other third-party funding, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements or a combination of these approaches. In any event, we might require additional capital to obtain regulatory approval for our product candidates, and we will require additional capital to commercialize and market any products that receive regulatory approval.approval, including full pre-commercialization activities. Raising funds in the current economic environment may present additional challenges. Global health concerns resulting from the outbreak of the coronavirus and worldwide macroeconomic turmoil may have long-term lasting effects on our ability to raise capital, many of which are difficult for us to predict at this time. Even if we believe we have sufficient funds for our current or future operating plans, we may seek additional capital if market conditions are favorable or if we have specific strategic considerations.
| 7 |
Any additional fundraising efforts may divert our management from their day-to-day activities, which may compromise our ability to develop and commercialize our product candidates. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights of our shareholders, and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our ordinary shares to decline. The sale of additional equity or convertible securities would dilute all of our shareholders. The incurrence of indebtedness would result in increased fixed payment obligations and we may be required to agree to certain restrictive covenants such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. We could also be required to seek funds through arrangements with collaborative partners or otherwise at an earlier stage than otherwise would be desirable, and we may be required to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail, delay or discontinue one or more of our research or development programs or the commercialization of any product candidates, and we may be unable to expand our operations or otherwise capitalize on our business opportunities, as desired.
We have received and may continue to receive Israeli governmental grants to assist in the funding of our research and development activities. If we lose our funding from these research and development grants, we may encounter difficulties in the funding of future research and development projects and implementing technological improvements, which would harm our operating results.
Through December 31, 20182021 we had received an aggregate of $22.6$29.2 million in the form of grants from the Israeli Office of the Chief Scientist, or OCS, which has later transformed to the Israeli Innovation Authority, or IIA. The requirements and restrictions for such grants are found inUnder the Israel Encouragement of Research and Development in Industries, or the Research Law. Under the Research Law, royalties of 3% to 3.5% on the revenues derived from sales of products or services developed in whole or in part using these IIA grants are payable to the Israeli government. We developed both ofthe VTS platform technology from which our platform technologies,product candidate, ofra-vec, is derived, at least in part, with funds from these grants, and accordingly we would be obligated to pay these royalties on sales of any of our product candidates derived from the VTS technology developed using these IIA grants that achieve regulatory approval.approval, such as ofra-vec. We also developed another technology utilizing IIA funds, which we do not expect to be able to commercialize. The maximum aggregate royalties paid for each technology or program separately, generally cannot exceed 100% of the grants made to us for such technology or program, plus annual interest equal to the 12-month LIBOR applicable to dollar deposits, as published on the first business day of each calendar year.interest. As of December 31, 2018,2021, the balance of the principal and interest in respect of our commitments for future payments to the IIA for both programs combined totaled approximately $27.8$37.6 million. As partTo date, we have paid the IIA in relation to our licenses agreement royalties of funding our current and planned product development activities, we submitted follow-up grant application.approximately $0.6 million.
These grants have funded some of our personnel, development activities with subcontractors and other research and development costs and expenses. However, if these awards are not funded in their entirety or if new grants are not awarded in the future, due to, for example, IIA budget constraints or governmental policy decisions, our ability to fund future research and development and implement technological improvements would be impaired, which would negatively impact our ability to develop our product candidates.
The Israeli government grants we have received for research and development expenditures restrict our ability to manufacture productsproduct candidates and transfer technologies outside of Israel and require us to satisfy specifiedcertain conditions. If we fail to satisfy these conditions, we may be required to refund grants previously received together with interest and penalties.
Our research and development efforts have been financed, in part, through the grants that we have received from the IIA. We, therefore, must comply with the requirements of the Research Law.
Under the Research Law, we are required to manufacture the major portionmajority of each of our productsproduct candidates developed using these grants in the State of Israel or otherwise ask for special approvals. We may not receive the required approvals for any proposed transfer of manufacturing activities.activities outside of Israel. Even if we do receive approval to manufacture productsproduct candidates developed with government grants outside of Israel, the royalty rate may be increased and we may be required to pay up to 300% of the grant amounts plus interest, depending on the manufacturing volume that is performed outside of Israel. This restriction may impair our ability to outsource manufacturing or engage in our own manufacturing operations for those productsproduct candidates or technologies. See “Item 5. Operating and Financial Review and Prospects-Financial Overview-Research and Development Expenses” for additional information.
Additionally, under the Research Law, we are prohibited from transferring, including by way of license, the IIA-financed technologies and related intellectual property rights and know-how outside of the State of Israel, except under limited circumstances and only with the approval of the IIA Research Committee. We may not receive the required approvals for any proposed transfer and, even if received, we may be required to pay the IIA a portion, to be set by the IIA upon their approval of such transaction, of the consideration or milestone and royalties payments that we receive upon any sale or out licensing of such technology to a non-Israeli entity, up to 600% of the grant amounts plus interest. The scope of the support received, the royalties that we have already paid to the IIA, the amount of time that has elapsed between the date on which the know-how or the related intellectual property rights were transferred and the date on which the IIA grants were received and the sale price and the form of transaction will be taken into account in order to calculate the amount of the payments to the IIA. ApprovalFor Israeli entities, approval of the transfer of technology to residents of the State of Israel is required, and may be granted in specific circumstances only if the recipient abides by the provisions of applicable laws, including the restrictions on the transfer of know-how and the obligation to pay royalties. No assurance can be made that approval to any such transfer, if requested, will be granted.
These restrictions may impair our ability to sell our technology assets or to perform or outsource manufacturing outside of Israel, engage in change of control transactions or otherwise transfer our know-how outside of Israel and may require us to obtain the approval of the IIA for certain actions and transactions and pay additional royalties and other amounts to the IIA. In addition, any change of control and any change of ownership of our ordinary shares that would make a non-Israeli citizen or resident an “interested party,” as defined in the Research Law, requires prior written notice to the IIA, and our failure to comply with this requirement could result in criminal liability.
| 8 |
These restrictions will continue to apply even after we have repaid the full amount of royalties on the grants. For the years ended December 31, 2018, 20172021, 2020 and 2016,2019, we recorded grants totaling $2.0$0.5 million, $2.7$1.5 million and $1.7$2.7 million from the IIA, respectively. The grants represented 11.22%an approximately 2%, 14%,7% and 12%15%, respectively, of our gross research and development expenditures for the years ended December 31, 2018, 20172021, 2020 and 2016.2019. If we fail to satisfy the conditions of the Research Law, we may be required to refund certain grants previously received together with interest and penalties, and may become subject to criminal charges.
We have been selected for €17.5 million of funding from the Horizon Europe EIC Accelerator Program, which funding is subject to a lengthy process, including negotiation and finalization of documentation, prior to receipt.
On December 20, 2021, VBL announced that it had been selected for €17.5 million of blended funding by the European Innovation Council, or EIC, Accelerator. The funding is comprised of a €2.5 million grant and an additional €15 million direct equity investment by the EIC. The funding process can be lengthy, including establishing and arranging for implementation of the investment and finalization of documentation, and we have yet to receive either the grant or the equity funding. The funding is also subject to meeting the specific requirements of the program and there can be no assurance that we meet and will continue to meet these requirements in order to receive the funding.
Risks Related to the Discovery and Development of Our Product Candidates and Platform Technologies
We have plannedare highly dependent on the future success of ofra-vec in oncology applications, and our lead product candidate, VB-111,platform technologies in general, and we cannot be certain that missed the primary end points in the Phase 3 study and continue to advance it for other indications.any of them will receive regulatory approval or be commercialized. Any failure to successfully develop, obtain regulatory approval for and commercialize VB-111ofra-vec for cancer indications or any other product candidates, independently or in cooperation with a third party collaborator, or the experience of significant delays in doing so, would compromise our ability to generate revenue and become profitable.
We have invested a significant portion of our effortsspent time, money and financial resources ineffort on the development of VB-111 for rGBMour platform technologies and VB-201 for psoriasis and ulcerative colitis for whichproduct candidates, particularly ofra-vec. To date, we have completed clinical trials in which they did not meet their primary endpoints. received regulatory approval for any of our product candidates. Positive results obtained during early development do not necessarily mean later development will succeed or that regulatory approvals will be obtained.
Our ability to generate product revenue from our product candidatecandidates depends heavily on the successful development and commercialization of our products,product candidates, which, in turn, depends on several factors, including the following:
| ● | our ability to continue and support the Vascular Targeting System, or VTS, platform technology and its lead candidate |
| ● | successfully enrolling and completing our ongoing and future trials of |
| ● | our ability to raise additional funding sufficient to conduct future clinical |
| ● | demonstrating that |
| ● | operating our facility to manufacture commercial quantities of our product candidates, if approved; |
| ● | manufacturing our product candidates in large scale and qualifying such processes in compliance with the regulatory requirements for clinical and commercial supply; |
| ● | establishing successful manufacturing arrangements with third-party manufacturers that are compliant with current good manufacturing practices, or cGMP, to ensure adequate supply of our product candidates for clinical development and |
| ● | |
| ● | maintaining an acceptable safety and efficacy profile for our products; |
| ● | the availability of coverage and reimbursement to patients from healthcare payers for our products, if approved; and |
| ● | other risks described in these “Risk Factors.” |
| 9 |
Our product candidates are based on novel technologies, which makes it difficult to predict the time and cost of product candidate development and potential regulatory approval.
We have concentrated our product research and development efforts on our three distinct platform technologies, and our future success depends on the successful development of these technologies. We could experience development problems in the future related to our technologies, which could cause significant delays or unanticipated costs, and we may not be able to solve such development problems. We may also experience delays in developing a sustainable, reproducible and scalable manufacturing processprocesses or transferring that processthose processes to commercial partners, if we decide to do so, which may prevent us from completing our clinical trials or commercializing our products, if approved, on a timely or profitable basis, if at all. If an issue is identified in one of our platform technologies, it may cause us to cease development of the product candidates that utilize the underlying technology.
In addition, the clinical trial requirements of the FDA, the EMA and other regulatory agencies and the criteria these regulators use to determine the safety and efficacy of a product candidate vary substantially according to the type, complexity, novelty and intended use and market of the potential products. The regulatory approval process for novel product candidates such as ours can be more expensive and take longer than for other, better known or extensively studied pharmaceutical or other product candidates. Approvals by the FDA may not be indicative of what the EMA or other regulatory agencies may require for approval, and vice versa.
Certain of our product candidates are based on novel adenovirus technology with which there is limited clinical or regulatory experience to date, which makes it difficult to predict the time and cost of product candidate development and subsequently obtaining regulatory approval. Regulatory requirements governing pharmaceuticalvirus-based products have changed frequently and may continue to change in the future. Also,For example, in addition to the submission of an Investigational New Drug, or IND, application to the FDA before initiation of a clinical trial can begin at an institution fundedin the United States, certain human clinical trials involving recombinant or synthetic nucleic acid molecules are subject to oversight by institutional biosafety committees, or IBCs, as set forth in the U.S. National Institutes of Health, or NIH, Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules, or NIH Guidelines. Under the NIH Guidelines, recombinant and synthetic nucleic acids are defined as: (i) molecules that institution’sare constructed by joining nucleic acid molecules and that can replicate in a living cell (i.e., recombinant nucleic acids); (ii) nucleic acid molecules that are chemically or by other means synthesized or amplified, including those that are chemically or otherwise modified but can base pair with naturally occurring nucleic acid molecules (i.e., synthetic nucleic acids); or (iii) molecules that result from the replication of those described in (i) or (ii). Specifically, under the NIH Guidelines, supervision of human gene transfer trials includes evaluation and assessment by an IBC, a local institutional review board,committee that reviews and oversees research utilizing recombinant or IRB, and its Institutional Biosafety Committee will have to review the proposed clinical trial to assesssynthetic nucleic acid molecules at that institution. The IBC assesses the safety of the research and identifies any potential risk to the public health or the environment, and such review may result in some delay before initiation of a clinical trial. While the NIH Guidelines are not mandatory unless the research in question is being conducted at or sponsored by institutions receiving NIH funding of recombinant or synthetic nucleic acid molecule research, many companies and other institutions not otherwise subject to the NIH Guidelines voluntarily follow them.
In addition, adverse developments in clinical trials of pharmaceutical products conducted by others may cause the FDA or other regulatory bodies to change the requirements for approval of any of our product candidates.
These regulatory agencies and review committees and the new requirements and guidelines they promulgate may lengthen the regulatory review process, require us to perform additional studies, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of these treatment candidates or lead to significant post-approval limitations or restrictions. As we advance our product candidates, we will be required to consult with these regulatory groups, and comply with applicable requirements and guidelines. If we fail to do so, we may be required to delay or discontinue development of our product candidates. Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a potential product candidate to market could impair our ability to generate product revenue and to become profitable.
We may find it difficult to enroll patients in our clinical trials, and patients could discontinue their participation in our clinical trials, which could delay or prevent clinical trials of our product candidates.
Identifying and qualifying patients to participate in clinical trials of our product candidates is critical to our success. The timing of our clinical trials depends on the speed at which we can recruit patients to participate in testing our product candidates. We have experienced delays in some of our clinical trials, and we may experience similar delays in the future. If patients are unwilling to participate in our clinical trials because of negative publicity from adverse events in the biotechnology or pharmaceutical industries or for other reasons, including competitive clinical trials for similar patient populations, the timeline for recruiting patients, conducting trials and obtaining regulatory approval of potential products may be delayed. These delays could result in increased costs, delays in advancing our product development, delays in testing the effectiveness of our technology or termination of the clinical trials altogether.
We may not be able to identify, recruit and enroll a sufficient number of patients, or those with required or desired characteristics to achieve diversity in a trial, to complete our clinical trials in a timely manner. Patient enrollment is affected by factors including:
| ● | severity of the disease under investigation; |
| ● | design of the trial protocol; |
| ● | size of the patient population; |
| ● | eligibility criteria for the trial in question; |
| 10 |
| ● | perceived risks and benefits of the product candidate under study, and specifically in reference to studies in other indications, with the same product; |
| ● | proximity and availability of clinical trial sites for prospective patients; |
| ● | availability of competing therapies and clinical trials; |
| ● | efforts to facilitate timely enrollment in clinical trials; |
| ● | patient referral practices of physicians; and |
| ● | ability to monitor patients adequately during and after treatment. |
In particular, VB-111some of the indications we may develop our candidates for ovarian cancer is intendedmay be for a rare disorderdisorders with limited patient pools from which to draw for clinical trials. The eligibility criteria of our clinical trials will further limit the pool of available trial participants. Additionally, the process of finding and diagnosing patients may prove costly.
We plan to seek initial marketing approval in Europe and Japan, in addition to the United States. We may not be able to initiate or continue clinical trials if we cannot enroll a sufficient number of eligible patients to participate in the clinical trials required by the EMA or other foreign regulatory agencies. Our ability to successfully initiate, enroll and complete a clinical trial in any foreign country is subject to numerous risks unique to conducting business in foreign countries, including:
| ● | difficulty in establishing or managing relationships with contract research organizations, or CROs, and physicians; |
| ● | different standards for the conduct of clinical trials; |
| ● | our inability to locate qualified local consultants, physicians and partners; and |
| ● | the potential burden of complying with a variety of foreign laws, medical standards and regulatory requirements, including the regulation of pharmaceutical and biotechnology products and treatment. |
If we have difficulty enrolling a sufficient number of patients to conduct our clinical trials as planned, we may need to delay, limit or terminate ongoing or planned clinical trials.
In addition, patients enrolled in our clinical trials may discontinue their participation at any time during the trial as a result of a number of factors, including withdrawing their consent or experiencing adverse clinical events, which may or may not be judged related to our product candidates under evaluation. The discontinuation of patients in any one of our trials may cause us to delay or abandon our clinical trial, or cause the results from that trial not to be positive or sufficient to support a filing for regulatory approval of the applicable product candidate.
We may encounter substantial delays in our clinical trials or we may fail to demonstrate safety and efficacy to the satisfaction of applicable regulatory authorities.
We are currently in a Phase 3 clinical trial for VB-111evaluating ofra-vec for ovarian cancer and expect to launch a coupleare supporting two clinical trials of Investigator Initiated trials oneofra-vec for recurrent glioblastoma multiforme, or rGBM, and the other for GI tumorscolorectal cancer in combination with an immune-oncology drug. Before obtaining marketing approval from regulatory authorities for the sale of our product candidates, we must conduct extensive clinical trials to demonstrate the safety and efficacy of the product candidates in humans. Clinical testing is expensive, time-consuming and uncertain as to outcome. We cannot guarantee that any clinical trials will be conducted as planned or completed on schedule, if at all.all, and that the trial will result in a positive outcome. Our Phase 3 clinical trial has two individual primary endpoints, progression-free survival, or PFS, and overall survival, or OS. We cannot guarantee that successfully meeting PFS, or interim OS data, will be sufficient to support FDA approval or that successfully meeting PFS will be indicative of OS results. We also cannot guarantee that we will receive regulatory approval if we achieve statistical significance absent clinically meaningful benefit. A failure of one or more clinical trials can occur at any stage of testing. Events that may prevent successful or timely completion of clinical development include:
| ● | delays in reaching a consensus with regulatory agencies on trial design; |
| ● | delays in reaching agreement on acceptable terms with prospective CROs and clinical trial sites; |
| ● | delays in obtaining required Institutional Review Board, or IRB, or ethics committee approval at each clinical trial site; |
| ● | delays in recruiting suitable patients to participate in our clinical trials including in particular for those trials for rare diseases such as ovarian cancer; |
| ● | delays in clinical trial supply, due to manufacturing delays or other issues, including as a result of FDA technical reviews (such as what occurred in June 2021 when we had to temporarily suspend supply from our Modi’in facility for the U.S. trial sites); |
| ● | imposition of a clinical hold by regulatory agencies, including due to safety reasons with either our product candidate or other product candidates in the same class or after an inspection of our clinical trial operations or trial sites; |
| ● | failure by our CROs, other third parties or us to adhere to clinical trial requirements; |
| ● | failure to perform in accordance with the FDA’s good clinical practices, or GCP, or applicable regulatory requirements in other countries; |
| 11 |
| ● | |
| delays in the testing, validation, manufacturing and delivery of our product candidates to the clinical sites; | |
| ● | delays in having patients complete participation in a trial or return for post-treatment follow-up; |
| ● | clinical trial sites or patients dropping out of a trial; |
| ● | occurrence of serious adverse events associated with the product candidate that are viewed to outweigh its potential benefits; |
| ● | changes in regulatory requirements and guidance that require amending or submitting new clinical trial |
| ● |
Any inability to successfully complete pre-clinicalpreclinical and clinical development could result in additional costs to us or impair our ability to generate revenue from product sales. In addition, if we make manufacturing or formulation changes to our product candidates, we may need to conduct additional studies to bridge our modified product candidates to earlier versions. Clinical trial delays could also shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates.
If the results of our clinical trials are inconclusive or if there are safety concerns or adverse events associated with our product candidates, we may:
| ● | fail to obtain, or be delayed in obtaining, marketing approval for our product candidates; |
| ● | obtain approval for indications or patient populations that are not as broad as intended or desired; |
| ● | obtain approval with labeling that includes significant use or distribution restrictions or safety warnings; |
| ● | need to change the way the product is administered; |
| ● | be unable to compete with other approved products; |
| ● | be required to perform additional clinical trials to support approval or be subject to additional post-marketing testing requirements; |
| ● | have regulatory authorities withdraw their approval of the product or impose restrictions on its distribution or use in the form of a risk evaluation and mitigation strategy, or REMS, or modified REMS; |
| ● | be subject to the addition of labeling statements, such as warnings or contraindications; |
| ● | be sued; or |
| ● | experience damage to our reputation. |
Any of these events could prevent us from achieving or maintaining market acceptance of our product candidates and impair our ability to commercialize our product candidates.
Side effects may occur following treatment with our product candidates, which could make it more difficult for our product candidates to receive regulatory approval.
Treatment with our product candidates may cause side effects or adverse events. In addition, sincebecause our product candidates are in some cases administered in combination with other therapies, patients or clinical trial participants may experience side effects or other adverse events that are unrelated to our product candidate, but may still impact the success of our clinical trials. Additionally, our product candidates could potentially cause other unforeseen adverse events that have not yet been predicted.we cannot predict. The inclusion of critically ill patients in our clinical trials may result in deaths or other adverse medical events due to other therapies or medications that such patients may be using or the severity of the medical condition treated. The experience of side effects and adverse events in our clinical trials could make it more difficult to achieve regulatory approval of our product candidates, or, if approved,at all, or could negatively impact the market acceptance of such products.products, if approved.
Success in early and prior clinical trials may not be indicative of results obtained in later trials.
There is a high failure rate for drugs and biologics proceeding through clinical trials. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in later stage clinical trials even after achieving promising results in earlier stage and prior clinical trials. The results of nonclinical and preclinical studies and clinical trials may not be predictive of the results of later-stage clinical trials, and interim results of a clinical trial do not necessarily predict final results. The results of preclinical studies and clinical trials in one set of patients or disease indications may not be predictive of those obtained in another. In some instances, there can be significant variability in safety or efficacy results between different clinical trials of the same product candidate due to numerous factors, including changes in trial procedures and timing of such procedures as set forth in protocols, differences in the size and type of the patient populations, changes in and adherence to the dosing regimen and the rate of dropout among clinical trial participants, among other factors. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy profile despite having progressed through nonclinical studies and initial clinical trials. For example, ofra-vec did not meet the primary endpoint in a Phase 3 trial in rGBM. We believe that this lack of efficacy was due to a significant change in the treatment regimen between the Phase 2 and Phase 3 trials to administer Avastin in combination with ofra-vec rather than ofra-vec monotherapy priming, and the mechanistic incompatibility of ofra-vec and Avastin, however, we cannot conclusively confirm this hypothesis prior to generating additional clinical data. Data obtained from pre-clinicalpreclinical and clinical activities are subject to varying interpretations, which may delay, limit or prevent regulatory approval. In addition, regulatory delays or rejections may be encountered as a result of many factors, including changes in regulatory policy during the period of product development.
| 12 |
The results from our clinical trials may not be sufficiently robust to support the submission for marketing approval for our product candidates. Before we submit our product candidates for marketing approval, the FDA and the EMA may require us to conduct additional clinical trials, or evaluate subjects for an additional follow-up period.
It is possible that, even if we achieve favorable results in our clinical trials, the FDA or the EMA may require us to conduct additional clinical trials, possibly involving a larger sample size or a different clinical trial design, particularly if the FDA or the EMA does not find the results from our completed clinical trials to be sufficiently persuasive to support a Biologics License Application, or BLA, or a New Drug Application, or NDA. For example, because the dose we used in our Phase 2 trial was limited by our production capacity, the doseachieving statistical significance is no guarantee of VB-111 that we intend to use in our Phase 3 potential registration trial may not be the maximum efficacious dose. The FDA might require data on higher doses of VB-111, this will likely delay development. The FDA may also require that we conduct a longer follow-up period of subjects treated with our product candidates prior to accepting our BLA or NDA.approval if there is no clinically meaningful benefit.
It is also possible that the FDA or the EMA may not consider the results of our clinical trials to be sufficient for approval of our product candidates for their target indications. If the FDA or the EMA requires additional studies for any reason, we would incur increased costs and delays in the marketing approval process, which may require us to expend more resources than we have available. In addition, it is possible that the FDA and the EMA may have divergent opinions on the elements necessary for a successful BLA or NDA and Marketing Authorization Application, which is the equivalent of a BLA, respectively, which may cause us to alter our development, regulatory or commercialization strategies.
Even if we complete the necessary pre-clinicalpreclinical studies and clinical trials, we cannot predict when or if we will obtain regulatory approval to commercialize a product candidate or the approval may be for a more narrow indication than we expect.
We cannot commercialize a product until the appropriate regulatory authorities have reviewed and approved the product candidate. Even if our product candidates demonstrate safety and efficacy in clinical trials, the regulatory agencies may not complete their review processes in a timely manner, or we may not be able to obtain regulatory approval. Additional delays may result if an FDA Advisory Committee or other regulatory authority recommends non-approval or restrictions on approval.approval, or if the FDA is unable to conduct a timely inspection of our or our third party manufacturing facility. In addition, we may experience delays or rejections based upon additional government regulation from future legislation or administrative action, or changes in regulatory agency policy during the period of product development, clinical trials and the review process. Regulatory agencies also may approve a treatment candidate for fewer or more limited indications than requested or may grant approval subject to the performance of post-marketing studies. In addition, regulatory agencies may not approve the labeling claims that are necessary or desirable for the successful commercialization of our treatment candidates.
A fast track designation by the FDA may not actually lead to a faster development or regulatory review or approval process.
If a drug is intended for the treatment of a serious or life-threatening disease or condition and the drug demonstrates the potential to address unmet medical needs for this disease or condition, the drug sponsor may apply for FDA fast track designation. If fast track designation is obtained, the FDA may initiate review of sections of a new drug application,an NDA or NDA,BLA, before the application is complete. This “rolling review” is available if the applicant provides, and the FDA approves, a schedule for submission of the individual sections of the application.
We have received fast track designation from the FDA for VB-111ofra-vec for prolongation of survival in patients with glioblastoma that has recurred following treatment with temozolomide, a chemotherapeutic agent commonly used to treat newly diagnosed glioblastoma, and radiation. We may seek fast track designation for other product candidates and other indications. Even though we have received fast track designation, we may not experience a faster development process, review or approval compared to conventional FDA procedures. The FDA may withdraw fast track designation if it believes that the designation is no longer supported by data from our clinical development program. Our fast track designation does not guarantee that we will qualify for or be able to take advantage of the expedited review procedures or that we will ultimately obtain regulatory approval of VB-111.ofra-vec.
Even though we have obtained orphan drug designation for VB-111ofra-vec for treatment of ovarian cancer in Europe, and the treatment of malignant glioma in the United States and glioma in Europe, and for the treatment of ovarian cancer in Europe, we may not be able to obtain orphan drug exclusivity for this drug or for any of our other product candidates.
Regulatory authorities in some jurisdictions, including the United States and Europe, may designate drugs for relatively small patient populations as orphan drugs. Under the Orphan Drug Act, the FDA may designate a product as an orphan drug if it is a drug intended to treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals annually in the United States, or a patient population of more than 200,000 in the United States where there is no reasonable expectation that the cost of developing the product will be recovered from sales in the United States. For VB-111,ofra-vec, we have obtained orphan drug designation from the FDA for the treatment of malignant glioma and from the EMAEuropean Commission for the treatment of gliomaovarian cancer and ovarian cancer,glioma, and we may seek orphan drug designation for other product candidates or indications, as appropriate.
Similarly, in Europe, the European Commission, upon the recommendation of the EMA’s Committee for Orphan Medicinal Products, grants orphan drug candidates.designation to promote the development of drugs that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than 5 in 10,000 persons in Europe and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized (or the product would be a significant benefit to those affected). Additionally, designation is granted for drugs intended for the diagnosis, prevention, or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in Europe would be sufficient to justify the necessary investment in developing the drug.
| 13 |
Generally, if a product with an orphan drug designation subsequently receives the first marketing approval for the indication for which it has such designation, the product is entitled to a period of marketing exclusivity, which precludes the EMA or the FDA from approving another marketing application for the same drug for the same use or indication for that time period. The applicable period is seven years in the United States and ten years in Europe. The European exclusivity period can be reduced to six years if a drug no longer meets the criteria for orphan drug designation or if the drug is sufficiently profitable so that market exclusivity is no longer justified. Orphan drug exclusivity may be lost if the FDA or EMA determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the drug to meet the needs of patients with the rare disease or condition. In addition, a designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation.
Even if we obtain orphan drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different drugs can be approved for the same condition. Even after an orphan drug is approved, the FDA can subsequently approve the same drug for the same condition if the FDA concludes that the later drug is clinically superior in that it is shown to be safer, more effective or makes a major contribution to patient care. Orphan drug designation neither shortens the development time or regulatory review time of a product nor gives the product any advantage in the regulatory review or approval process. While we may seek orphan drug designation for our product candidates, we may never receive such designations. Even if we do receive such designations, there is no guarantee that we will enjoy the benefits of those designations.
Even if we obtain regulatory approval for a product candidate, our products will remain subject to regulatory scrutiny.
Even if we obtain regulatory approval in a jurisdiction, the regulatory authority may still impose significant restrictions on the indicated uses or marketing of our product candidates, or impose ongoing requirements for potentially costly post-approval studies or post-market surveillance. For example, the holder of an approved BLA is obligated to monitor and report adverse events and any failure of a product to meet the specifications in the BLA. The FDA may also impose a REMS which could entail requirements for a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The holder of an approved BLA must also submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling or manufacturing process. Advertising and promotional materials must comply with FDA rules and are subject to FDA review, in addition to other potentially applicable federal and state laws.
In addition, product sponsors and their manufacturers and theirmanufacturing facilities are subject to payment of user fees and continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP and other regulatory requirements, such as product tracking and tracing, and adherence to commitments made in the BLA or NDA as the case may be. If we or a regulatory agency discover previously unknown problems with a product such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions relative to that product or the manufacturing facility, including requiring recall or withdrawal of the product from the market or suspension of manufacturing.
If we fail to comply with applicable regulatory requirements following approval of any of our product candidates, a regulatory agency may:
| ● | issue a warning letter asserting that we are in violation of the law; |
| ● | seek an injunction or impose civil or criminal penalties or monetary fines; |
| ● | suspend or withdraw regulatory |
| ● | suspend any ongoing clinical trials; |
| ● | refuse to approve a pending BLA or NDA or supplements to a BLA or NDA submitted by us for other indications or new drug products; |
| ● | impose restrictions on the marketing or manufacturing of our products; |
| ● | seize our product; or |
| ● | refuse to allow us to enter into supply contracts, including government contracts. |
Any government investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. The occurrence of any event or penalty described above may inhibit our ability to commercialize our product candidates and generate revenues.
We have only limited experience in regulatory affairs and intend to rely on consultants and other third parties for regulatory matters, which may affect our ability or the time we require to obtain necessary regulatory approvals.
We have limited experience in filing and prosecuting the applications necessary to gain regulatory approvals for drug and biologicsinvestigational product candidates. Moreover, the product candidates that are likely to resultderived from our development programs are based on new technologies that have not been extensively tested in humans. The regulatory requirements governing these types of product candidates may be less well defined or more rigorous than for conventional products. As a result, we may experience a longer regulatory process in connection with obtaining regulatory approvals of any productsproduct candidates that we develop. We intend to rely on independent consultants for purposes of our regulatory compliance and product development and approvals in the United States and elsewhere. Any failure by our consultants to properly advise us regarding, or properly perform tasks related to, regulatory compliance requirements could compromise our ability to develop and seek regulatory approval of our product candidates.
| 14 |
In addition to the level of commercial success of our product candidates, if approved, our future prospects are also dependent on our ability to successfully develop a pipeline of additional product candidates, and we may not be successful in our efforts in using our platform technologies to identify or discover additional product candidates.
The success of our business depends primarily upon our ability to identify, develop and commercialize products based on our two platform technologies. Our research programs may fail to identify other potential product candidates for clinical development for a number of reasons. Our research methodology may be unsuccessful in identifying potential product candidates or our potential product candidates may be shown to have harmful side effects or may have other characteristics that may make the products unmarketable or unlikely to receive marketing approval.
If any of these events occur, we may be forced to abandon our development efforts for a program or programs. Research programs to identify new product candidates require substantial technical, financial and human resources. We may focus our efforts and resources on potential programs or product candidates that ultimately prove to be unsuccessful.
Risks Related to Our Reliance on Third Parties
We expect to rely on third parties to conduct some or all aspects of our product manufacturing, protocol development, research and pre-clinicalpreclinical and clinical testing, and these third parties may not perform satisfactorily.
We do not expect to independently conduct all aspects of our product manufacturing, protocol development, research and pre-clinicalpreclinical and clinical testing. We currently rely, and expect to continue to rely, on third parties with respect to these items. In addition, we may pursue further clinical development of VB-111and indication expansion for thyroid cancer or other indicationsofra-vec with a strategic partner.
We do not have the ability to independently conduct clinical trials. We rely and expect to continue to rely on medical institutions, clinical investigators, contract laboratories and other third parties, such as CROs, to conduct or otherwise support clinical trials for our product candidates. We may also rely on academic and private non-academic institutions to conduct and sponsor clinical trials relating to our product candidates. We will not control the design or conduct of the investigator-sponsored trials, and it is possible that the FDA or non-U.S. regulatory authorities will not view these investigator-sponsored trials as providing adequate support for future clinical trials, whether controlled by us or third parties, for any one or more reasons, including elements of the design or execution of the trials or safety concerns or other trial results. Such arrangements will likely provide us certain information rights with respect to the investigator-sponsored trials, including access to and the ability to use and reference the data, including for our own regulatory filings, resulting from the investigator-sponsored trials. However, we would not have control over the timing and reporting of the data from investigator-sponsored trials, and we may not own the data from certain investigator-sponsored trials. If we are unable to confirm or replicate the results from the investigator-sponsored trials or if negative results are obtained, we would likely be further delayed or prevented from advancing further clinical development of our product candidates. Further, if investigators or institutions breach their obligations with respect to the clinical development of our product candidates, or if the data proves to be inadequate compared to the first-hand knowledge we might have gained had the investigator-sponsored trials been sponsored and conducted by us, then our ability to design and conduct any future clinical trials ourselves may be adversely affected.
Any of these third parties may terminate their engagements with us at any time. If we need to enter into alternative arrangements, it could delay our product development activities. Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibility to ensure compliance with all required regulations and study protocols. For example, for product candidates that we develop and commercialize on our own, we will remain responsible for ensuring that each of our Investigational New Drug, or IND enabling studies and clinical trials are conducted in accordance with the study plan and protocols. For any violations of laws and regulations during the conduct of our clinical trials, we could be subject to warning letters or enforcement action that may include civil penalties up to and including criminal prosecution.
If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our studies in accordance with regulatory requirements or our stated study plans and protocols, we will not be able to complete, or may be delayed in completing, the pre-clinicalpreclinical studies and clinical trials required to support future IND submissions and approval of our product candidates.
Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured the product candidates ourselves, including:
| ● | the inability to negotiate manufacturing agreements with third parties under commercially reasonable terms; |
| ● | reduced control as a result of using third-party manufacturers for all aspects of manufacturing activities; |
| ● | termination or nonrenewal of manufacturing agreements with third parties in a manner or at a time that is costly or damaging to us; and |
| ● | disruptions to the operations of our third-party manufacturers or suppliers caused by conditions unrelated to our business or operations, including the bankruptcy of the manufacturer or supplier. |
Any of these events could lead to clinical trial delays or failure to obtain regulatory approval, or impact our ability to successfully commercialize future products. Some of these events could be the basis for FDA action, including injunction, recall, seizure or total or partial suspension of production.
We and our contract manufacturers are subject to significant regulation with respect to manufacturing our product candidates. The manufacturing facilities on which we rely may not continue to meet regulatory requirements and have limited capacity.
We currently have relationships with a limited number of suppliers for the manufacturing of our product candidates. Each supplier may require licenses to manufacture components of our product candidates or to utilize certain processes for the manufacture of our product candidates. If such components or licenses are not owned by the supplier or in the public domain, we may be unable to transfer or sublicense the intellectual property rights we may have with respect to such activities.
| 15 |
All entities involved in the preparation of therapeutics for clinical trials or commercial sale, including our existing contract manufacturers for our product candidates, are subject to extensive regulation. Components of a finished therapeutic product approved for commercial sale or used in late-stage clinical trials must be manufactured in accordance with cGMP. These regulations govern manufacturing processes and procedures (including record keeping) and the implementation and operation of systems to control and assure the quality of investigational products and products approved for sale. Poor control of production processes can lead to the introduction of contaminants, or to inadvertent changes in the properties or stability of our product candidates that may not be detectable in final product testing. We or our contract manufacturers must supply all necessary documentation in support of a BLA or NDA, as applicable, on a timely basis and must adhere to the FDA’s good laboratory practices, or GLP, and cGMP regulations enforced by the FDA through its facilities inspection program. OurInformation requests from the FDA or failure to meet FDA requirements can result in delays in clinical trials, and any future commercial supply. For example, in June 2021, we voluntarily paused recruitment of patients into the OVAL trial in the United States while the FDA reviewed comparability data for new ofra-vec batches manufactured at our Modi’in facility for clinical use in the United States and the FDA has since provided clearance for us to use batches of ofra-vec produced in our Modi’in facility in the OVAL trial. We and our contract manufacturer for VB-111 hasofra-vec have not produced a commercially approved product based on viral vectors and therefore hashave not yet obtained the requisite FDA approvals to do so. Our facilities and controls and the facilities and controls of some or all of our third-party contractors must pass a pre-approval inspection for compliance with the applicable regulations as a condition of regulatory approval of our product candidates or any of our other potential products. In addition, the regulatory authorities may, at any time, audit or inspect a manufacturing facility involved with the preparation of our product candidates or our other potential products or the associated controls for compliance with the regulations applicable to the activities being conducted. If these facilities do not pass a pre-approval plant inspection, FDA or other regulatory authority approval of the productsproduct candidates will not be granted.
The regulatory authorities also may, at any time following approval of a product for sale, audit our manufacturing facilities or those of our third-party contractors. If any such inspection or audit identifies a failure to comply with applicable regulations or our product specifications, or if a violation of applicable regulations, including a failure to comply with the product specifications, occurs independent of such an inspection or audit, we or the relevant regulatory authority may require remedial measures that may be costly and/or time-consuming for us or a third party to implement and that may include the temporary or permanent suspension of a clinical trial or commercial sales or the temporary or permanent closure of a facility.
If we or any of our third-party manufacturers fail to maintain regulatory compliance, the FDA can impose regulatory sanctions including, among other things, refusal to approve a pending application for a new drug product or biologic product, or revocation of a pre-existing approval.
If any manufacturer with whom we contract fails to perform its obligations, we may be forced to manufacture the materials ourselves, for which we may not have the capabilities or resources, or enter into an agreement with a different manufacturer, which we may not be able to do on reasonable terms, if at all. In either scenario, our clinical trials supply could be delayed significantly as we establish alternative supply sources. In some cases, the technical skills required to manufacture our products or product candidates may be unique or proprietary to the original manufacturer and we may have difficulty, or there may be contractual restrictions prohibiting us from, transferring such skills to a back-up or alternate supplier, or we may be unable to transfer such skills at all. In addition, if we are required to change manufacturers for any reason, we will be required to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations. We will also need to verify, such as through a manufacturing comparability study, that any new manufacturing process will produce our product candidate according to the specifications previously submitted to the FDA or another regulatory authority. The delays associated with the verification of a new manufacturer could negatively affect our ability to develop product candidates or commercialize our products in a timely manner or within budget. In addition, changes in manufacturers often involve changes in manufacturing procedures and processes, which could require that we conduct bridging studies between our prior clinical supply used in our clinical trials and that of any new manufacturer. We may be unsuccessful in demonstrating the comparability of clinical supplies which could require the conduct of additional clinical trials. Additionally, if any of our product candidates receive regulatory approval and supply from one approveda manufacturer is interrupted, there could be a significant disruption in commercial supply. An alternative manufacturer would need to be qualified through a BLA or NDA supplement which could result in further delay. The regulatory agencies may also require additionalcomparability studies if a new manufacturer is relied upon for commercial production. Switching manufacturers may involve substantial costs and is likely to result in a delay in our desired clinical and commercial timelines.
In addition, there are risks associated with large scale manufacturing for clinical trials or commercial scale including, among others, supplier delays, cost overruns, potential problems with process scale-up, process reproducibility, stability issues, compliance with cGMP, lot consistency and timely availability of raw materials. Even if we obtain marketing approval for any of our current product candidates or any future product candidates, there is no assurance that we or our manufacturers will be able to manufacture the approved product to specifications acceptable to the FDA or other comparable foreign regulatory authorities, to produce it in sufficient quantities to meet the requirements for the potential commercial launch of the product or to meet potential future demand. If we or our manufacturers are unable to produce sufficient quantities for clinical trials or for commercialization, our development and commercialization efforts would be impaired, which would have an adverse effect on our business, financial condition, results of operations and growth prospects.
These factors could cause the delay of clinical trials, regulatory submissions, required approvals or commercialization of our product candidates, cause us to incur higher costs and prevent us from commercializing our products successfully. Furthermore, if our suppliers fail to meet contractual requirements, and we are unable to secure one or more replacement suppliers capable of production at a substantially equivalent cost, our clinical trials and potential commercialization may be delayed or we could lose potential revenue.
We expect to rely on third parties to conduct, supervise and monitor our clinical trials, and if these third parties perform in an unsatisfactory manner, it may harm our business.
We expect to rely on CROs and clinical trial sites, including clinical investigators, to ensure our clinical trials are conducted properly and on time. While we will have agreements governing their activities, we will have limited influence over their actual performance. We will control only some aspects of our CROs’ activities. Nevertheless, we will be responsible for ensuring that each of our clinical trials is conducted in accordance with the applicable protocol, legal, regulatory and scientific requirements and standards, and our reliance on the CROs does not relieve us of our regulatory responsibilities.
| 16 |
We and our CROs are required to comply with the FDA’s GCPs for conducting, recording and reporting the results of IND-enabling studies and clinical trials to assure that the data and reported results are credible and accurate and that the rights, integrity and confidentiality of clinical trial participants are protected. The FDA enforces these GCPs through periodic inspections of study sponsors, principal investigators and clinical trial sites. If we or our CROs fail to comply with applicable GCPs, the clinical data generated in our future clinical trials may be deemed unreliable and the FDA may require us to perform additional clinical trials before approving any marketing applications. Upon inspection, the FDA may determine that our clinical trials did not comply with GCPs. In addition, our future clinical trials will require a sufficient number of test subjects to evaluate the safety and effectiveness of our product candidates. Recruitment in rare diseases may be challenging in the event of rare diseases and may require the performance of trials in a significant number of sites which may be harder to monitor. Accordingly, if our CROs fail to comply with these regulations or fail to recruit a sufficient number of patients, we may be required to repeat such clinical trials, which would result in significant additional costs and delay the regulatory approval process.
Our CROs are not our employees, and we are therefore unable to directly monitor whether or not they devote sufficient time and resources to our clinical and nonclinical programs. These CROs may also have relationships with other commercial entities, including parties developing potentially competitive products, for whom they may also be conducting clinical trials or other drug development activities that could harm our competitive position. If our CROs do not successfully carry out their contractual duties or obligations, fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements, or for any other reason, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize our product candidates. As a result, the commercial prospects for our product candidates would be harmed, our costs could increase, and our ability to generate revenues could be delayed.
We also expect to rely on other third parties to store and distribute our product candidates for any clinical trials that we may conduct. Any performance failure on the part of our distributors could delay clinical development or marketing approval of our product candidates or commercialization of our products,product candidates, if approved, producing additional losses and depriving us of potential product revenue.
Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
Because we rely on third parties to manufacture our product candidates, and because we collaborate with various organizations and academic institutions on the advancement of our technology, we must, at times, share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements or other similar agreements with our collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, such as trade secrets. Despite these contractual provisions, the need to share trade secrets and other confidential information increases the risk that such trade secrets become known by potential competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and trade secrets, discovery by a third party of our trade secrets or other unauthorized use or disclosure would impair our intellectual property rights and protections in our product candidates.
In addition, these agreements typically restrict the ability of our collaborators, advisors, employees and consultants to publish data potentially relating to our trade secrets. Our academic collaborators typically have rights to publish data, provided that we are notified in advance and may delay publication for a specified time in order to secure our intellectual property rights arising from the collaboration. In other cases, publication rights are controlled exclusively by us, although in some cases we may share these rights with other parties. Despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of these agreements, independent development or publication of information including our trade secrets in cases where we do not have proprietary or otherwise protected rights at the time of publication.
Risks Related to Commercialization of Our Product Candidates
We intend to fully rely orat least partially rely on third-party manufacturers to produce commercial quantities of any of our product candidates that receives regulatory approval, but we have not entered into binding agreements with any such manufacturers to support commercialization. Additionally, these manufacturers do not have experience producing our product candidates at commercial levels and may not pass regulatory inspections or achieve the necessary regulatory approvals or produce our product candidates at the quality, quantities, locations and timing needed to support commercialization.
We have not yet secured manufacturing capabilities for commercial quantities of our product candidates to fully support world-wide commercialization of our product candidates. Although we intend to partially rely on third-party manufacturers for commercialization, in addition to our internal manufacturing, we have onlynot yet entered into agreements with such manufacturers to assist in the scaling up of the manufacturing process of VB-111.third party manufacturers. We may be unable to negotiate binding agreements with the manufacturers to support our commercialization activities on commercially reasonable terms, which agreements will further be required to comply with the restrictions imposed under the Research Law.
We may encounter technical or scientific issues related to manufacturing or development that we may be unable to resolve in a timely manner or with available funds. Although we have established a company site in which we are planningplan to apply ause for commercial scale manufacturing, the available capacity to manufacture our product candidates on a commercial scale is still limited. In addition, our product candidates are novel, and no manufacturervery few manufacturers currently hashave the experience or ability to produce our product candidates at commercial levels. If we are unable to produce or engage manufacturing partners to produce our product candidates on a larger scale on reasonable terms, our commercialization efforts will be harmed.
Even if we timely complete the development of a manufacturing process and successfully transfer it to the third- party manufacturers of our product candidates, if we or such third-party manufacturers are unable to produce the necessary quantities of our product candidates, or in compliance with cGMP or with pertinent regulatory requirements, and within our planned time frame and cost parameters, the development and sales of our product candidates, if approved, may be impaired.
| 17 |
In addition, any significant disruption in our supplier relationships could harm our business. We source key materials from third parties, either directly through agreements with suppliers or indirectly through our manufacturers who have agreements with suppliers. During the COVID-19 pandemic, global supply chain disruptions have been seen, particularly with raw materials and supplies used in viral production. For example, we have seen significant delays obtaining certain materials and equipment needed to manufacture our product candidates. There are a small number of suppliers, and in some cases a single supplier for certain key materials that are used to manufacture our product candidates. Such suppliers may not sell these key materials to us or our manufacturers at the times we need them or on commercially reasonable terms. We do not have any control over the process or timing of the acquisition of these key materials by our manufacturers. Moreover, we currently do not have any agreements for the commercial production of these key materials.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell any of our product candidates that obtain regulatory approval, we may be unable to generate any revenue.
We have no experience selling and marketing our product candidates or any other products. To successfully commercialize any products that may result from our development programs and obtain regulatory approval, we will need to develop these capabilities, either on our own or with others. We may seek to enter into collaborations with other entities to utilize their marketing and distribution capabilities, but we may be unable to do so on favorable terms, if at all. If any future collaborative partners do not commit sufficient resources to commercialize our future products, if any, and we are unable to develop the necessary marketing capabilities on our own, we will be unable to generate sufficient product revenue to sustain our business. We will be competing with many companies that currently have extensive and well-funded marketing and sales operations. Without ansufficient internal teamcapability or the support of a third party to perform marketing and sales functions, we may be unable to compete successfully against these more established companies or successfully commercialize any of our product candidates.
We face intense competition and rapid technological change and the possibility that our competitors may develop therapies that are more advanced or effective than ours, which could impair our ability to successfully commercialize our product candidates.
We are engaged in pharmaceutical development, which is a rapidly changing field. We have competitors both in the United States and internationally, including majorlarge multinational pharmaceutical companies, biotechnology companies and universities and other research institutions.
Many of our potential competitors have substantially greater financial, technical and other resources, such as larger research and development staff and experienced marketing and manufacturing organizations. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our potential competitors may succeed in developing, acquiring or licensing on an exclusive basis, products that are more effective, safer, or less costly than any product candidate that we may develop, or achieve earlier patent protection, regulatory approval, product commercialization and market penetration than us. Additionally, technologies developed by others may render our potential product candidates uneconomical or obsolete, and we may not be successful in marketing our product candidates against competitors.
In particular, VB-111ofra-vec may face competition from currently approved drugs and drug candidates under development by others to treat rGBMovarian cancer or ovarian cancer.rGBM. In May 2009, the FDA granted accelerated approval to Avastin (bevacizumab)bevacizumab (Avastin®), which is an angiogenesis inhibitor, to treat patients with rGBM at progressionGBM with progressive disease following prior therapy. Bevacizumab also received FDA approval for platinum-resistant ovarian cancer in 2014, and for newly diagnosed patients after standard first-line therapy.their initial surgery in 2018. In addition to bevacizumab, a number of companies are conducting late-stage clinical trials to test targeted drugs focused on angiogenesis inhibition for the treatment of ovarian cancer, including, among others, Amgen’s trebananib, Boehringer Ingelheim’s nintedanib, AstraZeneca’s cediranib and Novartis’s Votrient.cancer. The expansion of poly adenosine diphosphate-ribose polymerase, or PARP, inhibitors (such as olaparib)olaparib, niraparib and veliparib) for ovarian cancer, and clinical studies evaluating the potential use of drug candidates such as checkpoint inhibitors, antibody-drug conjugates (including mirvetuximab soravtansine), bispecific antibodies, GAS6/AXL inhibitors, WEE1 inhibitors, CDK4/6 inhibitors or tumor treating fields medical device for ovarian cancer may also affect the prior lines of therapy used before ofra-vec, or the segment of patient population who will seek treatment with VB-111.ofra-vec.
Even if we are successful in achieving regulatory approval to commercialize a product candidate faster than our competitors, we may face competition from biosimilars. In the United States, the Biologics Price Competition and Innovation Act of 2009 created an abbreviated approval pathway for biological products that are demonstrated to be “highly similar,” or biosimilar, to or “interchangeable” with an FDA-approved biological product. This pathway could allow competitors to reference data from biological products already approved after 12 years from the time of approval. In Europe, the European Commission has granted marketing authorizations for several biosimilars pursuant to a set of general and product class-specific guidelines for biosimilar approvals issued over the past few years. In Europe, a competitor may reference data from biological products already approved, but will not be able to market a biosimilar until ten years after the time of approval. This 10-year period will be extended to 11 years if, during the first eight of those 10 years, the marketing authorization holder obtains an approval for one or more new therapeutic indications that bring significant clinical benefits compared with existing therapies. In addition, companies may be developing biosimilars in other countries that could compete with our products. If competitors are able to obtain marketing approval for biosimilars referencing our products, our products may become subject to competition from such biosimilars, with the attendant competitive pressure and consequences. Expiration or successful challenge of our applicable patent rights could also trigger competition from other products, assuming any relevant exclusivity period has expired.
In addition, although VB-111 has been granted orphan drug status by the FDA and EMA for a specified indication, there are limitations to the exclusivity. In the United States, the exclusivity period for orphan drugs is seven years, while pediatric exclusivity adds six months to any existing patents or exclusivity periods. In Europe, orphan drugs may be able to obtain 10 years of marketing exclusivity and up to an additional two years on the basis of qualifying pediatric studies. However, orphan exclusivity may be reduced to six years if the drug no longer satisfies the original designation criteria. Additionally, a marketing authorization holder may lose its orphan exclusivity if it consents to a second orphan drug application or cannot supply enough drug. Orphan drug exclusivity also can be lost when a second applicant demonstrates its drug is “clinically superior” to the original orphan drug.
Finally, as a result of the expiration or successful challenge of our patent rights, we could face more litigation with respect to the validity or scope of patents relating to other parties’ products. The availability of other parties’ products could limit the demand, and the price we are able to charge, for any products that we may develop and commercialize.
| 18 |
SinceBecause some of our product candidates are aimed fortargeting rare diseases, loss of exclusivity or competition as described above may be veryhave a significant impact on our business in light of the limited size of the relevant market.
The commercial success of any current or future product candidate, if approved, will depend upon the degree of market acceptance by physicians, patients, third-party payorspayers and others in the medical community.
Even if we obtain the requisite regulatory approvals, the commercial success of our product candidates will depend in part on the medical community, patients, and third-party payorspayers accepting our product candidates as medically useful, cost-effective, and safe. Any product that we bring to the market may not gain market acceptance by physicians, patients, third-party payorspayers and others in the medical community. If these products do not achieve an adequate level of acceptance, we may not generate significant product revenue and may not become profitable. The degree of market acceptance of these product candidates, if approved for commercial sale, will depend on a number of factors, including:
| ● | the potential efficacy and potential advantages over alternative treatments; |
| ● | the prevalence and severity of any side effects, including any limitations or warnings contained in a product’s approved labeling; |
| ● | the prevalence and severity of any side effects resulting from the procedure by which our product candidates are administered; |
| ● | relative convenience and ease of administration; |
| ● | the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies; |
| ● | the strength of marketing and distribution support and timing of market introduction of competitive products; |
| ● | ability to adhere to the handling and storage requirements of our product candidates; |
| ● | publicity concerning our products or competing products and treatments; and |
| ● | sufficient third-party insurance coverage or reimbursement. |
Even if a potential product displays a favorable efficacy and safety profile in pre-clinicalpreclinical studies and clinical trials, market acceptance of the product will not be known until after it is launched. Our efforts to educate the medical community and third-party payorspayers on the benefits of the product candidates may require significant resources and may never be successful. Such efforts to educate the marketplace may require more resources than are required by conventional technologies.
A variety of risks associated with international operations could hurt our business.
If any of our product candidates are approved for commercialization, it is our current intention to market them on a worldwide basis, either alone or in collaboration with others. In addition, we conduct development activities in various jurisdictions throughout the world. We expect that we will be subject to additional risks related to engaging in international operations, including:
| ● | different regulatory requirements for approval of drugs and biologics in foreign countries; |
| ● | reduced protection for intellectual property rights; |
| ● | unexpected changes in tariffs, trade barriers and regulatory requirements; |
| ● | economic weakness, including inflation, or political instability in particular foreign economies and markets; |
| ● | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; |
| ● | foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; |
| ● | workforce uncertainty in countries where labor unrest is more common than in the United States and Israel; |
| ● | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
| ● | business interruptions resulting from geopolitical actions, including war and terrorism, global outbreaks of disease, or natural disasters including earthquakes, typhoons, floods and fires. |
We have not undertaken a systematic analysis of the potential consequences to our business as a result of any such natural disaster, public health crisis or pandemic diseases and do not have an applicable recovery plan in place. In addition, if any of our third-party contract manufacturers are affected by natural disasters, such as earthquakes, power shortages or outages, floods, wildfire, public health crises, such as pandemics and epidemics, terrorism or other events outside of our control, our business and operating results could suffer. For example, in December 2019, a novel coronavirus was identified and caused, and continues to cause, massive global business interruptions. More recently, in late February 2022, Russian military forces launched significant military action against Ukraine, and sustained conflict and disruption in the region is likely. The impact to Ukraine, as well as actions taken by other countries, including new and stricter sanctions by Canada, the United Kingdom, the European Union, the United States and other countries and organizations against officials, individuals, regions, and industries in Russia, Ukraine and Belarus, and each country’s potential response to such sanctions, tensions, and military actions could have an adverse effect on our operations. We carry only limited business interruption insurance that would compensate us for actual losses from interruption of our business that may occur and any losses or damages incurred by us in excess of insured amounts could cause our business to materially suffer.
| 19 |
Legislative and regulatory activity may exert downward pressure on potential pricing and reimbursement for any of our product candidates, if approved, that could materially affect the opportunity to commercialize.
The United States and several other jurisdictions are considering, or have already enacted, a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell any of our product candidates profitably, if approved. Among policy-makers and payers in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access to healthcare. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives. There have been, and likely will continue to be, legislative and regulatory proposals at the federal and state levels directed at broadening the availability of healthcare and containing or lowering the cost of healthcare. We cannot predict the initiatives that may be adopted in the future.
The continuing efforts of the government, insurance companies, managed care organizations and other payers of healthcare services to contain or reduce costs of healthcare may adversely affect:
| ● | the demand for any of our product candidates, if approved; |
| ● | the ability to set a price that we believe is fair for any of our product candidates, if approved; |
| ● | our ability to generate revenues and achieve or maintain profitability; |
| ● | the level of taxes that we are required to pay; and |
| ● | the availability of capital. |
We expect that the as amended by the Health Care and Education Reconciliation Act of 2010, or, collectively, the ACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our product candidates.
Moreover, increasing efforts by governmental and third-party payers in the United States and abroad to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for newly approved products and, as a result, they may not cover or provide adequate payment for our product candidates. There has been increasing legislative and enforcement interest in the United States with respect to specialty drug pricing practices. Specifically, there have been several recent U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs.
We expect that the healthcare reform measures that have been adopted and may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product and could seriously harm our future revenues. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our products.
Our relationships with customers and third-party payers may be subject, directly or indirectly, to applicable anti-kickback laws, fraud and abuse laws, false claims laws, health information privacy and security laws and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm, administrative burdens and diminished profits and future earnings.
We are subject to additional healthcare statutory and regulatory requirements and enforcement by the federal government and the states and foreign governments in which we conduct our business. Healthcare providers, physicians and third-party payers play a primary role in the recommendation and prescription of any of our approved drugs and drug candidates for which we obtain marketing approval. Our current and future arrangements with third-party payers and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute any products for which we obtain marketing approval. In addition, we may be subject to health information privacy and security regulation of the European Union, the United States and other jurisdictions in which we conduct our business. For example, the laws that may affect our ability to operate include:
| ● | the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, either the referral of an individual, or the purchase or recommendation of an item or service for which payment may be made under a federal healthcare program, such as the Medicare and Medicaid programs. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act or federal civil money penalties statute (as discussed below); |
| ● | U.S. federal civil and criminal false claims laws and civil monetary penalty laws, including the federal False Claims Act, which impose criminal and civil penalties against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, including the Medicare and Medicaid programs, claims for payment that are false or fraudulent, making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government, or knowingly concealing or knowingly and improperly avoiding or decreasing such an obligation; |
| ● | the U.S. federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, and its implementing regulations, which created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and prohibit knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statements in connection with the delivery of or payment for healthcare benefits, items or services; |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and their respective implementing regulations, which impose certain obligations, including mandatory contractual terms, on covered healthcare providers, health plans, and healthcare clearinghouses, as well as their business associates, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| ● | the U.S. federal Physician Payments Sunshine Act which requires certain manufacturers of drugs, devices, biologics, and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services information related to payments or transfers of value made to physicians (currently defined to include doctors, dentists, optometrists, podiatrists and chiropractors), physician assistants, nurse practitioners, clinical nurse specialists, certified registered nurse anesthetists and teaching hospitals, as well as information regarding ownership and investment interests held by the physicians described above and their immediate family members; |
| ● | federal government price reporting laws, which require us to calculate and report complex pricing metrics in an accurate and timely manner to government programs; |
| ● | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and |
| ● | analogous state and laws and regulations in other jurisdictions, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payers, including private insurers, and state and laws in other jurisdiction governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform. Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. Ensuring that our internal operations and future business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations, agency guidance or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of the laws described above or any other governmental laws and regulations that may apply to us, we may be subject to significant penalties, including administrative, civil and criminal penalties, damages, fines, disgorgement, the exclusion from participation in federal and state healthcare programs, individual imprisonment, reputational harm, and the curtailment or restructuring of our operations, as well as additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws. Further, defending against any such actions can be costly and time consuming, and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired. If any of the physicians or other providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs and imprisonment. If any of the above occur, our ability to operate our business and our results of operations could be adversely affected.
| 20 |
The insurance coverage and reimbursement status of newly approved products is uncertain. Failure to obtain or maintain adequate coverage and reimbursement for any of our product candidates that are approved could limit our ability to market those products and compromise our ability to generate revenue.
The availability of reimbursement by governmental and private payorspayers is essential for most patients to be able to afford expensive treatments. Sales of our product candidates will depend substantially, both in the U.S. and abroad, on the extent to which the costs of our product candidates will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities, private health coverage insurers and other third-party payors.payers. If reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize our product candidates. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize a sufficient return on our investment.
Third-party payers decide which drugs and treatments they will cover and the amount of reimbursement. Reimbursement by a third-party payer may depend upon a number of factors, including, but not limited to, the third-party payer’s determination that use of a product is:
| ● | a covered benefit under its health plan; |
| ● | safe, effective and medically necessary; |
● | appropriate for the specific patient; |
| ● | cost-effective; and |
| ● | neither experimental nor investigational. |
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. In the United States, the principal decisions about reimbursement for new medicines are typically made by the Centers for Medicare & Medicaid Services, or CMS, an agency within the U.S. Department of Health and Human Services, as CMS decides whether and to what extent a new medicine will be covered and reimbursed under Medicare. Private payorspayers tend to follow CMS to a substantial degree. It is difficult to predict what CMS will decide with respect to reimbursement for fundamentally novel products such as ours, as there is no body of established practices and precedents for these new products. Reimbursement agencies in Europe may be more conservative than CMS. For example, a number of cancer drugs have been approved for reimbursement in the United States and have not been approved for reimbursement in certain European countries.
The intended
Obtaining coverage and reimbursement of a product from a government or other third-party payer is a time consuming and costly process that could require us to provide to the payer supporting scientific, clinical and cost-effectiveness data for the use of our products. Even if we obtain coverage for a druggiven product, byif the resulting reimbursement rates are insufficient, hospitals may not approve our product for use in their facility or third-party payers may require co-payments that patients find unacceptably high. Patients are unlikely to use our product candidates unless coverage is provided, and reimbursement is adequate to cover a physician can also affect pricing. For example, CMS could initiate a National Coverage Determination administrative procedure, by whichsignificant portion of the agency determines which usescost of a therapeuticour product would and wouldcandidates. Separate reimbursement for the product itself may or may not be reimbursable under Medicare. This determination process can be lengthy, thereby creating a long period during whichavailable. Instead, the future reimbursement for a particular producthospital or administering physician may be uncertain.reimbursed only for providing the treatment or procedure in which our product is used. Further, from time to time, CMS revises the reimbursement systems used to reimburse health care providers, including the Medicare Physician Fee Schedule and Outpatient Prospective Payment System, which may result in reduced Medicare payments. In some cases, private third-party payers rely on all or portions of Medicare payment systems to determine payment rates. Changes to government healthcare programs that reduce payments under these programs may negatively impact payments from private third-party payers and reduce the willingness of physicians to use our product candidates.
Outside the United States, international operations are generally subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis on cost-containment initiatives in Europe, Canada, and other countries is likely to put pressure on the pricing and usage of any of our product candidates that are approved for marketing. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. In general, the prices of medicines under such systems are substantially lower than in the United States. Other countries allow companies to fix their own prices for medicines, but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our product candidates. Accordingly, in markets outside the United States, the reimbursement for our products may be reduced compared with the United States and may be insufficient to generate commercially reasonable revenue and profits.
Moreover, increasing efforts by governmental and third-party payors,payers, in the United States and abroad, to cap or reduce healthcare costs, resulting in legislation and reforms such as the Patient Protection and Affordable Care Act of 2010, may cause such organizations to limit both coverage and level of reimbursement for new products approved and, as a result, they may not cover or provide adequate payment for our product candidates. We expect to experience pricing pressures in connection with the sale of any of our product candidates, due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
The marketability of any product candidates for which we receive regulatory approval for commercial sale may suffer if government and other third-party payers fail to provide coverage and adequate reimbursement. We expect downward pressure on pharmaceutical pricing to continue. Further, coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
The prescription for or promotion of off-label uses of our products by physicians could adversely affect our business.
Any regulatory approval of our products iswill be limited to thosethe specific diseases and indications for which ourthe products have been deemed safe and effectiveapproved by the FDA or similar authorities in other jurisdictions. In addition, any new indication for an approved product also requires regulatory approval. If we produce an approved therapeuticobtain approval for a product candidate, we will rely on physicians to prescribe and administer it as we have directed and for the indications described on the labeling. It is not, however, uncommon for physicians to prescribe medication for unapproved, or “off-label,” uses or in a manner that is inconsistent with the manufacturer’s directions. To the extent such off-label uses and departures from our administration directions become pervasive and produce results such as reduced efficacy or other adverse effects, the reputation of our products in the marketplace may suffer. In addition, off-label uses may cause a decline in our revenue or potential revenue, to the extent that there is a difference between the prices of our product for different indications.
Furthermore, while physicians may choose to prescribe our drugsproducts, if approved, for off-label uses, our ability to promote the products is limited to those indications that are specifically approved by the FDA or other regulators. Although regulatory authorities generally do not regulate the behavior of physicians, they do restrict communications by companies with respect to off-label use. If our promotional activities fail to comply with these regulations or guidelines, we may be subject to warnings from, or enforcement action by, these authorities. In addition, failure to follow FDA rules and guidelines relating to promotion and advertising can result in the FDA’s refusal to approve a product, the suspension or withdrawal of an approved product from the market, product recalls, fines, disgorgement of money, operating restrictions, injunctions or criminal prosecution.
Due to the small target patient populations for some of our product candidates, we face uncertainty related to pricing and reimbursement for these product candidates.
Some of ourthe target patient populations for our initial product candidates are relatively small, as a result of which the pricing and reimbursement of our product candidates, if approved, must be adequate to support commercial infrastructure. If we are unable to obtain adequate levels of reimbursement, our ability to successfully market and sell our product candidates will be adversely affected. Inadequate reimbursement for such services may lead to physician resistance and adversely affect our ability to market or sell our products.
| 21 |
Risks Related to Our Business Operations
Our future success depends on our ability to retain key employees, consultants and advisors and to attract, retain and motivate qualified personnel.
We are highly dependent on principal members of our executive team listed under “Management” in this report, including Prof. Dror Harats, our chief executive officer, the loss of whose services may adversely impact the achievement of our objectives. While we have entered into employment agreements with each of our executive officers, any of them could leave our employment at any time, as all of our employees are “at will” employees. Recruiting and retaining other qualified employees, consultants and advisors for our business, including scientific and technical personnel, will also be critical to our success. There is currently a shortage of skilled executives in our industry, which is likely to continue. As a result, competition for skilled personnel is intense and the turnover rate can be high. We may not be able to attract and retain personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for individuals with similar skill sets.sets and greater financial resources than us. In addition, failure to succeed in pre-clinicalpreclinical studies or clinical trials may make it more challenging to recruit and retain qualified personnel. The inability to recruit or loss of the services of any executive, key employee, consultant or advisor may impede the progress of our research, development and commercialization objectives.
Our collaborations with outside scientists and consultants may be subject to restriction and change.
We work with medical experts, chemists, biologists and other scientists at academic and other institutions, and consultants who assist us in our research, development and regulatory efforts, including the members of our scientific advisory board. In addition, these scientists and consultants have provided, and we expect that they will continue to provide, valuable advice regarding our programs and regulatory approval processes. These scientists and consultants are not our employees and may have other commitments that would limit their future availability to us. If a conflict of interest arises between their work for us and their work for another entity, we may lose their services. In addition, we are limited in our ability to prevent them from establishing competing businesses or developing competing products. For example, if a key scientist acting as a principal investigator in any of our clinical trials identifies a potential product or compound that is more scientifically interesting to his or her professional interests, his or her availability to remain involved in our clinical trials could be restricted or eliminated.
We will need to expand our organization and external resources, and we may experience difficulties in managing this growth, which could disrupt our operations.
As of March 1, 2019,2022, we had 3941 employees. As we matureWe continue to expand our infrastructure and undertakeexternal resources to support the activities required to advance our product candidates into later stagethrough clinical development and topotential commercialization as well as operate as a public company, wecompany. We expect to expand our full-time employee base and to hire more consultants and contractors.contractors to support the company’s activities. Our management may need to divert a disproportionate amount of its attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. We may not be able to effectively manage the expansion of our operations, which may result in weaknesses in our infrastructure, operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. Our expected growth could require significant capital expenditures and may divert financial resources from other projects, such as the development of additional product candidates. If our management is unable to effectively manage our growth, our expenses may increase more than expected, our ability to generate or grow revenue could be compromised, and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize product candidates and compete effectively will depend, in part, on our ability to effectively manage any future growth.
Our employees, principal investigators, consultants and commercial partners may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of fraud or other misconduct by our employees, principal investigators, consultants and commercial partners. Misconduct by these parties could include intentional failures to comply with the regulations of the FDA and non-U.S. regulators, provide accurate information to the FDA and non-U.S. regulators, comply with healthcare fraud and abuse laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Such misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and cause serious harm to our reputation. We have adopted a code of conduct applicable to all of our employees, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Pandemics, such as the ongoing COVID-19 pandemic, could have an adverse impact on our developmental programs and our financial condition.
In December 2019, an outbreak of a novel strain of coronavirus, or the SARS-CoV-2 coronavirus, which results in COVID-19, had ripple effects to businesses around the world, negatively impacted activity and operations, including extended shutdowns of certain businesses, in many countries, including the United States, European countries and Israel, where our operations are. The list of countries and regions affected by the coronavirus outbreak and new variants are constantly changing and our clinical trial sites and third party collaborators may be located in regions currently being afflicted by the COVID-19 pandemic. Any outbreak of contagious diseases, or other adverse public health developments, could have a material and adverse effect on our business operations. These could include disruptions or restrictions on our ability to travel, pursue partnerships and other business transactions, conduct clinical trials, make shipments of biologic materials, as well as be impacted by the temporary closure of the facilities of suppliers and clinical trial sites. Any disruption of suppliers, clinical trial sites or access to patients would likely impact our ability to complete the development of our product candidates in a timely manner and access capital through the financial markets.
Additionally, timely enrollment in clinical trials is reliant on clinical trial sites that may be adversely affected by the COVID-19 pandemic and other global health matters. Some factors from the coronavirus outbreak that we believe may adversely affect enrollment in our trials include:
| ● | the diversion of healthcare resources away from the conduct of clinical trial matters to focus on pandemic concerns, including the attention of infectious disease physicians serving as our clinical trial investigators, hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials; |
| ● | limitations on travel that interrupt key trial activities, such as clinical trial site initiations and monitoring; |
| 22 |
| ● | increased rates of patients withdrawing from our clinical trials following enrollment as a result of contracting COVID-19, being forced to quarantine or not accepting home health visits; |
| ● | interruption in global shipping affecting the transport of clinical trial materials, such as investigational drug product and comparator drugs used in our trials; and |
| ● | employee furlough days that delay necessary interactions with local regulators, ethics committees and other important agencies and contractors. |
These and other factors arising from the ongoing COVID-19 pandemic could worsen in countries that are already afflicted with the virus or could continue to spread to additional countries, each of which may further adversely impact our clinical trials. The global outbreak of the SARS-CoV-2 coronavirus continues to evolve and the conduct of our trials may continue to be adversely affected, despite efforts to mitigate this impact.
For more information on the extent that the ongoing COVID-19 pandemic has impacted our development programs to date, please refer to the related section in the Overview.
The ongoing COVID-19 pandemic could also interrupt the business of our subcontractors, vendors and external laboratories. For example, since the beginning of the ongoing COVID-19 pandemic, three vaccines for COVID-19 have received Emergency Use Authorization by the FDA and two of those later received marketing approval. Additional vaccines may be authorized or approved in the future. The resultant demand for vaccines and potential for manufacturing facilities and materials to be commandeered under the Defense Production Act of 1950, or equivalent foreign legislation, may make it more difficult to obtain materials or manufacturing slots for the products needed for our clinical trials, which could lead to delays in these trials. For example, the COVID-19 pandemic has, at least in part, caused supply chain disruptions which have led to significant delays receiving materials and equipment required to manufacture our product candidates. Continued or future similar supply chain disruptions may result in delays in our clinical trials or commercial manufacturing.
The extent to which the coronavirus impacts our business will depend on future developments, which are highly uncertain and cannot be predicted, including new information which may emerge concerning the severity of the coronavirus, the identification of new variants of the virus, and the actions to contain the coronavirus or treat its impact, among others.
Inadequate funding for the FDA, the SEC and other government agencies, including from government shut downs, or other disruptions to these agencies’ operations, could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory and policy changes. Average review times at the agency have fluctuated in recent years as a result. Disruptions at the FDA and other agencies may also slow the time necessary for new product candidates to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities, is subject to the political process, which is inherently fluid and unpredictable.
Separately, since March 2020 when foreign and domestic inspections of facilities were largely placed on hold due to the COVID-19 pandemic, the FDA has been working to resume routine surveillance, bioresearch monitoring and pre-approval inspections on a prioritized basis. Since April 2021, the FDA has conducted limited inspections and employed remote interactive evaluations, using risk management methods, to meet user fee commitments and goal dates. Ongoing travel restrictions and other uncertainties continue to impact oversight operations both domestic and abroad and it is unclear when standard operational levels will resume. The FDA is continuing to complete mission-critical work, prioritize other higher-tiered inspectional needs (e.g., for-cause inspections), and carry out surveillance inspections using risk-based approaches for evaluating public health. Should FDA determine that an inspection is necessary for approval and an inspection cannot be completed during the review cycle due to restrictions on travel, and the FDA does not determine a remote interactive evaluation to be adequate, the agency has stated that it generally intends to issue, depending on the circumstances, a complete response letter or defer action on the application until an inspection can be completed. During the COVID-19 public health emergency, a number of companies announced receipt of complete response letters due to the FDA’s inability to complete required inspections for their applications. Regulatory authorities outside the U.S. may adopt similar restrictions or other policy measures in response to the ongoing COVID-19 pandemic and may experience delays in their regulatory activities. If a prolonged government shutdown or other disruption occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Future shutdowns or other disruptions could also affect other government agencies such as the SEC, which may also impact our business by delaying review of our public filings, to the extent such review is necessary, and our ability to access the public markets.
We face potential product liability, and, if successful claims are brought against us, we may incur substantial liability and costs. If the use of our product candidates harms patients, or is perceived to harm patients even when such harm is unrelated to our product candidates, our regulatory approvals could be revoked or otherwise negatively impacted and we could be subject to costly and damaging product liability claims.
The use of our product candidates in clinical trials and the sale of any products for which we obtain marketing approval exposes us to the risk of product liability claims. Product liability claims might be brought against us by consumers, healthcare providers, pharmaceutical companies or others selling or otherwise coming into contact with our product candidates. There is a risk that our product candidates may induce adverse events. If we cannot successfully defend against product liability claims, we could incur substantial liability and costs. In addition, regardless of merit or eventual outcome, product liability claims may result in:
| ● | impairment of our business reputation; |
| ● | withdrawal of clinical trial participants; |
| ● | costs due to related litigation; |
| ● | distraction of management’s attention from our primary business; |
| ● | substantial monetary awards to patients or other claimants; |
| 23 |
| ● | |
| the inability to commercialize our product candidates; | |
| ● | decreased demand for our product candidates, if approved for commercial sale; and |
| ● | impairment of our ability to obtain product liability insurance coverage. |
We carry combined public and products liability (including human clinical trials extension) insurance of $5.0 million per occurrence and $5.0 million aggregate limit, with extension to $10.0 million for the prior Phase 3 studytrial of ofra-vec in rGBM and for the ongoing Phase 3 studytrial of ofra-vec in ovarian cancer. We believe our product liability insurance coverage is sufficient in light of our current clinical programs; however, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. If we obtain marketing approval for any product candidates, we intend to expand our insurance coverage to include the sale of commercial products, but we may not be able to obtain product liability insurance on commercially reasonable terms or in adequate amounts. On occasion, large judgments have been awarded in class action lawsuits based on drugs or medical treatments that had unanticipated adverse effects. A successful product liability claim or series of claims brought against us could cause our share price to decline and, if judgments exceed our insurance coverage, could materially and adversely affect our financial position.
Patients with the diseases targeted by some of our product candidates are often already in severe and advanced stages of disease and have both known and unknown significant pre- existingpre-existing and potentially life-threatening health risks. During the course of treatment, patients may suffer adverse events, including death, for reasons that may be related to our product candidates. Such events could subject us to costly litigation, require us to pay substantial amounts of money to injured patients, delay, negatively impact or end our opportunity to receive or maintain regulatory approval to market our products, or require us to suspend or abandon our commercialization efforts. Even in a circumstance in which we do not believe that an adverse event is related to our product candidate, the investigation into the circumstance may be time-consuming or inconclusive. These investigations may harm our reputation, delay our regulatory approval process, limit the type of regulatory approvals our product candidates receive or maintain, and compromise the market acceptance of any of our product candidates that receive regulatory approval. As a result of these factors, a product liability claim, even if successfully defended, could hurt our business and impair our ability to generate revenue.
If our existing or future manufacturing facility is damaged or destroyed, or production at any of those facilities is otherwise interrupted, our business and prospects would be negatively affected.
We have a manufacturing facility that is currently manufacturing product for our Phase 3 OVAL trial and that we expect to use for commercial scale production. If our existing or future manufacturing facilities, or the equipment in it, is damaged or destroyed, we likely would not be able to quickly or inexpensively replace our manufacturing capacity and possibly wouldmay not be able to replace it at all.all, which would increase our reliance on third party manufacturers. Any new facility needed to replace our existing or future manufacturing facility or any new third party manufacturer would need to comply with the necessary regulatory requirements, and be tailored to our manufacturing requirements and processes. We would need FDA approvalauthorization before using any product candidates manufactured at a new facility or by a new manufacturer in clinical trials or selling any products that are ultimately approved. Such an event could delay our clinical trials or, if any of our product candidates are approved by the FDA, reduce or eliminate our product sales.
See “-We and our contract manufacturers are subject to significant regulation with respect to manufacturing our product candidates. The manufacturing facilities on which we rely may not continue to meet regulatory requirements and have limited capacity” and “-We intend to at least partially rely on third-party manufacturers to produce commercial quantities of any of our product candidates that receives regulatory approval, but we have not entered into binding agreements with any such manufacturers to support commercialization. Additionally, these manufacturers do not have experience producing our product candidates at commercial levels and may not pass regulatory inspections or achieve the necessary regulatory approvals or produce our product candidates at the quality, quantities, locations and timing needed to support commercialization” under this Item for more information.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials or other work-related injuries, this insurance may not provide adequate coverage against potential liabilities. In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
If our shipping capabilities become unavailable due to an accident, an act of terrorism, a labor strike or other similar event, our supply, production and distribution processes could be disrupted.
Some of our raw materials for the manufacturing of VB-111,ofra-vec, and VB-111ofra-vec itself, must be transported at a temperature controlled cold chain at temperatures varying between -4 degrees Celsius to -70 degrees Celsius (25 to -94 degrees Fahrenheit) to ensure their quality and vitality. Not all shipping or distribution channels are equipped to transport at these temperatures. If any of our shipping or distribution channels become inaccessible because of a serious accident, an act of terrorism, global health pandemic, a labor strike or other similar event, we may experience disruptions in our continued supply of raw materials, delays in our production process or a reduction in our ability to distribute our therapeutics toproduct candidates for our customers.clinical trials.
We may use our financial and human resources to pursue a particular research program or product candidate and fail to capitalize on programs or product candidates that may be more profitable or for which there is a greater likelihood of success.
Because we have limited resources, we may forego or delay pursuit of opportunities with certain programs or product candidates or for indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs for product candidates may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through strategic collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate, or we may allocate internal resources to a product candidate in a therapeutic area in which it would have been more advantageous to enter into a collaboration arrangement.
| 24 |
We will continue to incur significant increased costs as a result of operating as a public company, and our management will continue to be required to devote substantial time to new compliance initiatives.
As a public company, we will continue to incur significant legal, accounting and other expenses. In addition, the Sarbanes-Oxley Act, as well as rules subsequently implemented by the Securities and Exchange Commission, or SEC and The NASDAQthe Nasdaq Global Market have imposed various requirements on public companies. Recent legislation permits smaller “emerging growth companies” to implement manyAs noted herein, it is expected that as a result of these requirements over a longer period and up to five years from the pricingcomposition of our offering. We intend to take advantageboard of this new legislation but cannot guarantee thatdirectors, management team and current shareholder base, we will notcease to be a “foreign private issuer” on January 1, 2023 and will be required to implement thesecomply with U.S. reporting requirements sooner than budgeted or planned and thereby incur unexpected expenses.as well as become subject to additional obligations due to our anticipated change in status. Shareholder activism, the current political environment and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact the manner in which we operate our business in ways we cannot currently anticipate. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly. For example, we expect these rules and regulations, as well as the increase in the number of class actions and other securities litigation filed against publicly traded life sciences companies, to make it more difficult and more expensive for us to obtain director and officer liability insurance and we may be required to incur substantial costs to maintain our current levels of such coverage. While compliance with these additional requirements and the transition from being a foreign private issuer will result in increased costs to us, we cannot accurately predict or estimate at this time the amount of additional costs we may incur as a public company under both U.S. and Israeli laws.
Additionally, we are no longer an “emerging growth company,” as defined in the JOBS Act, and are now required to comply with additional disclosure and reporting requirements. These additional reporting requirements may increase our legal and financial compliance costs and cause management and other personnel to divert attention from operational and other business matters to devote substantial time to these public company requirements. Further, we are now an accelerated filer based on our public float as of June 30, 2021 and must comply with Section 404(b) of the Sarbanes-Oxley Act, which requires us to include an attestation report issued by our independent registered public accounting firm on our management’s assessment of our internal control over financial reporting. If we identify material weaknesses in our internal control over financial reporting, if we are unable to assert that our internal control over financial reporting is effective, or if our independent registered public accounting firm is unable to express an opinion or issues an adverse opinion in its attestation as to the effectiveness of our internal control over financial reporting required by Section 404(b), investors may lose confidence in the accuracy and completeness of our financial reports and the trading price of our ordinary shares could be negatively affected. We could also become subject to investigations by the stock exchange on which our securities are listed, the SEC or other regulatory authorities, which could require additional financial and management resources.
Irrespective of compliance with Sections 404(a) and 404(b) of the Sarbanes-Oxley Act, any failure of our internal control could have a material adverse effect on our stated results of operations and harm our reputation. In order to implement changes to our internal control over financial reporting triggered by a failure of those controls, we could experience higher than anticipated operating expenses, as well as higher independent auditor fees during and after the implementation of these changes.
We are subject to foreign currency exchange risk, and fluctuations between the U.S. dollar and the NIS, the Euro and other non-U.S. currencies may negatively affect our earnings and results of operations.
We operate in a number of different currencies. While the dollar is our functional and reporting currency and investments in our share capital have been denominated in dollars, our financial results may be adversely affected by fluctuations in currency exchange rates as a significant portion of our operating expenses, including our salary-related and manufacturing expenses are denominated in the NIS, and a significant portion of our clinical trials and manufacturing expenses are denominated in euros.NIS.
We are exposed to the risks that the NIS may appreciate relative to the dollar, or, if the NIS instead devalues relative to the dollar, that the inflation rate in Israel may exceed such rate of devaluation of the NIS, or that the timing of such devaluation may lag behind inflation in Israel. In any such event, the dollar cost of our operations in Israel would increase and our dollar- denominated results of operations would be adversely affected. We cannot predict any future trends in the rate of inflation in Israel or the rate of devaluation (if any) of the NIS against the dollar. For example, the average exchange rate of the dollar against the NIS decreased in 2018, in 2017 and in 2016.late 2021 strengthened to levels that were multi-year highs. Market volatility and currency fluctuations may limit our ability to cost- effectively hedge against our foreign currency exposure and, in addition, our ability to hedge our exposure to currency fluctuations in certain emerging markets may be limited. Hedging strategies may not eliminate our exposure to foreign exchange rate fluctuations and may involve costs and risks of their own, such as devotion of management time, external costs to implement the strategies and potential accounting implications. Foreign currency fluctuations, independent of the performance of our underlying business, could lead to materially adverse results or could lead to positive results that are not repeated in future periods.
Under applicable employment laws, we may not be able to enforce covenants not to compete.
We generally enter into non-competition agreements with our employees. These agreements prohibit our employees, if they cease working for us, from competing directly with us or working for our competitors or clients for a limited period. We may be unable to enforce these agreements under the laws of the jurisdictions in which our employees work and it may be difficult for us to restrict our competitors from benefitting from the expertise our former employees or consultants developed while working for us. For example, Israeli labor courts have required employers seeking to enforce non-compete undertakings of a former employee to demonstrate that the competitive activities of the former employee will harm one of a limited number of material interests of the employer which have been recognized by the courts, such as the protection of a company’s trade secrets or other intellectual property. We have recently expanded operations into the United States and entered into U.S. employment agreements. As employment law varies from U.S. state to state, we may not be able to enforce non-compete rights in such agreements if U.S. employees reside in states that do not recognize such rights.
Risks Related to Our Intellectual Property
We depend on our license agreement with Janssen Vaccines & Prevention B.V. and if we cannot meet requirements under such license agreement, we could lose the rights to our products, which could have a material adverse effect on our business.
VB-111Ofra-vec incorporates an adenoviral vector as theits delivery vehicle, which is manufactured based on our rights under a license agreement within-licensed from Janssen Vaccines & Prevention B.V., or Janssen. If we fail to meet our obligations under this license agreement, including various diligence, milestone payment, royalty and other obligations, Janssen Vaccines & Prevention B.V. has the right to terminate our license, and upon the effective date of such termination, our right to use the licensed technology would terminate. We may enter into additional agreements in the future with Janssen Vaccines & Prevention B.V. that may impose similar obligations on us. While we would expect to exercise all rights and remedies available to us, including attempting to cure any breach by us, and otherwise seek to preserve our rights under the patents and other technology licensed to us, we may not be able to do so in a timely manner, at an acceptable cost or at all. Any uncured, material breach under the license agreement could result in our loss of rights and may lead to a complete termination of our product development and any commercialization efforts for the applicable product candidates sincebecause there are currently no significantsignificantly similar alternatives on the market.market that we would use to produce these candidates including ofra-vec.
| 25 |
If we are unable to obtain or protect intellectual property rights related to our product candidates, we may not be able to obtain exclusivity for our product candidates or prevent others from developing similar competitive products.
We rely upon a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to our product candidates. The strength of patents in the biotechnology and pharmaceutical field involves complex legal and scientific questions and can be uncertain. The patent applications that we own or in-license may fail to result in issued patents with claims that cover our product candidates in the United States or in other foreign countries. There is no assurance that all of the potentially relevant prior art relating to our patents and patent applications has been found, which can invalidate a patent or prevent a patent from issuing from a pending patent application. Even if patents do successfully issue and even if such patents cover our product candidates, third parties may challenge their validity, enforceability or scope, which may result in the patent claims being narrowed or invalidated. Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property, provide exclusivity for our product candidates or prevent others from designing around our claims. Any of these outcomes could impair our ability to prevent competition from third parties.parties and materially affect our operations and financial condition.
If the patent applications we hold or have in-licensed with respect to our programs or product candidates fail to issue, if the breadth or strength of our patent protection is threatened, or if our patent portfolio fails to provide meaningful exclusivity for our product candidates, it could dissuade companies from collaborating with us to develop product candidates and threaten our ability to commercialize future products. Several patent applications covering our product candidates have been filed recently. We cannot offer any assurances about which, if any, applications will issue as patents, the breadth of any such issued patent claims or whether any issued claims will be found invalid and unenforceable or will be threatened by third parties. Any successful opposition to these patents or any other patents owned by or licensed to us could deprive us of rights necessary for the successful commercialization of any product candidates that we may develop. Further, if we encounter delays in regulatory approvals, the period of time during which we could market a product candidate under patent protection could be reduced. SinceBecause patent applications in the United States and most other countries are confidential for a period of time after filing, and some remain so until issued, we cannot be certain that we or our licensors were the first to file any patent application related to a product candidate. Furthermore, if third parties have filed such patent applications, an interference proceeding in the United States can be initiated by a third party to determine who was the first to invent any of the subject matter covered by the patent claims of our applications. In addition, patents have a limited lifespan. In the United States, the natural expiration of a patent is generally 20 years after it is filed. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our product candidates are obtained, once the patent life has expired for a product, we may be open to competition from generic medications. This risk is material in light of the length of the development process of our products and lifespan of our current patent portfolio.
In addition to the protection afforded by patents, we rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable or that we elect not to patent, processes for which patents are difficult to enforce and any other elements of our product candidate discovery and development processes that involve proprietary know- how,know-how, information or technology that is not covered by patents. However, trade secrets can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with our employees, consultants, scientific advisors and contractors. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. Security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. Although we expect all of our employees and consultants to assign their inventions to us, and all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology to enter into confidentiality agreements, we cannot provide any assurances that all such agreements have been duly executed or that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. Misappropriation or unauthorized disclosure of our trade secrets could impair our competitive position and may have a material adverse effect on our business. Additionally, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating the trade secret. In addition, others may independently discover our trade secrets and proprietary information. For example, the FDA, as part of its Transparency Initiative, is currently considering whether to make additional information publicly available on a routine basis, including information that we may consider to be trade secrets or other proprietary information, and it is not clear at the present time how the FDA’s disclosure policies may change in the future, if at all.
Further, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad. If we are unable to prevent material disclosure of the non-patented intellectual property related to our technologies to third parties, and there is no guarantee that we will have any such enforceable trade secret protection, we may not be able to establish or maintain a competitive advantage in our market.
Third-party claims of intellectual property infringement may prevent or delay our development and commercialization efforts.
Our commercial success depends in part on our avoiding infringement of the patents and proprietary rights of third parties. There is a substantial amount of litigation, both within and outside the United States, involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries, including patent infringement lawsuits, interferences, oppositions and inter partes review proceedings before the U.S. Patent and Trademark Office, or U.S. PTO, and corresponding foreign patent offices. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are pursuing development candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our product candidates may be subject to claims of infringement of the patent rights of third parties.
Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our product candidates. Because patent applications can take many years to issue, there may be currently pending patent applications which may later result in issued patents that our product candidates may be accused of infringing. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. If any third-party patents were held by a court of competent jurisdiction to cover the manufacturing process of any of our product candidates, any molecules formed during the manufacturing process or any final product itself, the holders of any such patents may be able to block our ability to commercialize such product candidate unless we obtained a license under the applicable patents, or until such patents expire. Similarly, if any third-party patents were held by a court of competent jurisdiction to cover aspects of our formulations, processes for manufacture or methods of use, the holders of any such patents may be able to block our ability to develop and commercialize the applicable product candidate unless we obtained a license or until such patent expires. In either case, such a license may not be available on commercially reasonable terms or at all.
| 26 |
Parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize one or more of our product candidates. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, pay royalties, redesign our infringing products or obtain one or more licenses from third parties, which may be impossible or require substantial time and monetary expenditure.
We may not be successful in obtaining or maintaining necessary rights to pharmaceutical product components and processes for our development pipeline through acquisitions and in- licenses.in-licenses.
Presently we have rights to the intellectual property, through licenses from Janssen Vaccines & Prevention B.V. and under patents that we own, to develop our product candidates. Because our programs may involve additional product candidates that may require the use of proprietary rights held by third parties, the growth of our business may depend in part on our ability to acquire, in-license or use these proprietary rights. In addition, our product candidates may require specific formulations to work effectively and efficiently and these rights may be held by others. We may be unable to acquire or in-license any compositions, methods of use, processes or other third- party intellectual property rights from third parties that we identify. The licensing and acquisition of third-party intellectual property rights is a competitive area, and a number of more established companies are also pursuing strategies to license or acquire third-party intellectual property rights that we may consider attractive. These established companies may have a competitive advantage over us due to their size, cash resources and greater clinical development and commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire third-party intellectual property rights on terms that would allow us to make an appropriate return on our investment.
We may enter into license agreements with third parties, and if we fail to comply with our obligations in such agreements under which we license intellectual property rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose license rights that are important to our business.
We may need to obtain licenses from third parties to advance our research or allow commercialization of our product candidates, and we have done so from time to time. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In that event, we may be required to expend significant time and resources to develop or license replacement technology. If we are unable to do so, we may be unable to develop or commercialize the affected product candidates.
In many cases, patent prosecution of our in-licensed technology is controlled solely by the licensor. If our licensors fail to obtain and maintain patent or other protection for the proprietary intellectual property we license from them, we could lose our rights to the intellectual property or our exclusivity with respect to those rights, and our competitors could market competing products using the intellectual property. In some cases, we control the prosecution of patents resulting from licensed technology. In the event we breach any of our obligations related to such prosecution, we may incur significant liability to our licensing partners. Licensing of intellectual property is of critical importance to our business and involves complex legal, business and scientific issues and is complicated by the rapid pace of scientific discovery in our industry. Disputes may arise regarding intellectual property subject to a licensing agreement, including:
| ● | the scope of rights granted under the license agreement and other interpretation-related issues; |
| ● | the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement; |
| ● | the sublicensing of patent and other rights under any collaboration relationships we might enter into in the future; |
| ● | our diligence obligations under the license agreement and what activities satisfy those diligence obligations; |
| ● | the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and |
| ● | the priority of invention of patented technology. |
If disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates.
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time-consuming and unsuccessful.
Competitors may infringe our patents or the patents of our licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours or our licensors is not valid, is unenforceable or is not infringed, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
Interference proceedings provoked by third parties or brought by us may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our licensors. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our defense of litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent, alone or with our licensors, misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
| 27 |
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the trading price of our ordinary shares.
Recent patentPatent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
Patent reform legislation (the Leahy-Smith Act) enacted in 2013 continues to increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. The Leahy-Smith Act introduced a number of significant changes to U.S. patent law, including provisions that affect the way patent applications are prosecuted and patent litigation is conducted. The U.S. PTO continues to develop regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Act,therewith, in particular, the Inter Partes Review (IPR)inter partes review proceedings. It remains to be seen what impact the Leahy-Smith Act will have on the operation of our business. However, the Act and its implementation increases the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business and financial condition.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties or that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
Certain of our key employees and personnel are or were previously employed at universities, medical institutions or other biotechnology or pharmaceutical companies, including our competitors or potential competitors.
Although we try to ensure that our employees, consultants and independent contractors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of any of our employee’s former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees. Furthermore, universities or medical institutions who employ some of our key employees and personnel in parallel to their engagement by us may claim that intellectual property developed by such person is owned by the respective academic or medical institution under the respective institution intellectual property policy or applicable law.
We may become subject to claims for remuneration or royalties for assigned service invention rights by our employees, which could result in litigation and adversely affect our business.
A significant portion of our intellectual property has been developed by our employees in the course of their employment for us. Under the Israeli Patent Law, 5727-1967, or the Patent Law, inventions conceived by an employee during the term and as part of the scope of his or her employment with a company are regarded as “service inventions,” which belong to the employer, absent a specific agreement between the employee and employer giving the employee service invention rights. The Patent Law also provides that if there is no such agreement between an employer and an employee, the Israeli Compensation and Royalties Committee, or the Committee, a body constituted under the Patent Law, shall determine whether the employee is entitled to remuneration for his inventions. Recent decisions by the Committee (which have been upheld by the Israeli Supreme Court on appeal) have created uncertainty in this area, as it held that employees may be entitled to remuneration for their service inventions despite having specifically waived any such rights. Further, the Committee has not yet determined the method for calculating this remuneration nor the criteria or circumstances under which an employee’s waiver of his right to remuneration will be disregarded. We generally enter into assignment-of-invention agreements with our employees pursuant to which such individuals assign to us all rights to any inventions created in the scope of their employment or engagement with us. Although our employees have agreed to assign to us service invention rights, we may face claims demanding remuneration in consideration for assigned inventions. As a consequence of such claims, we could be required to pay additional remuneration or royalties to our current or former employees, or be forced to litigate such claims, which could negatively affect our business.
We may be subject to claims challenging the inventorship or ownership of our patents and other intellectual property.
We may be subject to claims that former employees, collaborators or other third parties have an ownership interest in our patents or other intellectual property. We may have to in the future, ownership disputes arising, for example, from conflicting obligations of consultants or others who are involved in developing our product candidates. Litigation may be necessary to defend against these and other claims challenging inventorship or ownership. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and applications will be due to be paid to the U.S. PTO and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents and applications. The U.S. PTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. There are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction.
| 28 |
Issued patents covering our product candidates could be found invalid or unenforceable if challenged in court.
If we or one of our licensing partners initiated legal proceedings against a third party to enforce a patent covering one of our product candidates, the defendant may contend that the patent covering our product candidate is invalid, unenforceable or fails to cover the product candidate or the infringing product. In patent litigation in the United States, defendants commonly allege that asserted patent claims are invalid and unenforceable. Grounds for a validity challenge could be an alleged failure to meet one or more of several statutory requirements, including lack of novelty, obviousness, lack of written description, indefiniteness and non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the U.S. PTO, or made a misleading statement, during prosecution. Third parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, post grant review, and equivalent proceedings in foreign jurisdictions, such as opposition proceedings. Such proceedings could result in revocation, amendments to our patent claims or statements being made on the record such that our claims may no longer be construed to cover our product candidates. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity, unenforceability or non-infringement, we would lose at least part, and perhaps all, of the patent protection on our product candidates. Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses, and could distract our technical and management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments, and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the market price of our ordinary shares. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other biotechnology companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biotechnology industry involve both technological and legal complexity, and is therefore is costly, time- consumingtime-consuming and inherently uncertain. In addition, the United States has recently enacted and is currently implementing wide-ranging patent reform legislation. Recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in some situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the U.S. PTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
We have not yet registered trademarks for a commercial trade name for some of our product candidates and failure to secure such registrations could adversely affect our business.
We have not yet registered trademarks for a commercial trade name for some of our product candidates. During trademark registration proceedings, we may receive rejections. Although we would be given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the U.S. PTO and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. Moreover, any name we propose to use with our product candidates in the United States must be approved by the FDA, regardless of whether we have registered it, or applied to register it, as a trademark. The FDA typically conducts a review of proposed product names, including an evaluation of potential for confusion with other product names. If the FDA objects to any of our proposed proprietary product names, we may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Potential competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our product candidates, if approved, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
| 29 |
Under applicable employment laws, we may not be able to enforce covenants not to compete.
We generally enter into non-competition agreements with our employees. These agreements prohibit our employees, if they cease working for us, from competing directly with us or working for our competitors or clients for a limited period. We may be unable to enforce these agreements under the laws of the jurisdictions in which our employees work and it may be difficult for us to restrict our competitors from benefitting from the expertise our former employees or consultants developed while working for us. For example, Israeli labor courts have required employers seeking to enforce non-compete undertakings of a former employee to demonstrate that the competitive activities of the former employee will harm one of a limited number of material interests of the employer which have been recognized by the courts, such as the protection of a company’s trade secrets or other intellectual property.
Risks Related to Ownership of Our Ordinary Shares
The market price of our ordinary shares may be highly volatile, and you may not be able to resell your shares at the purchase price.
An active trading market for our ordinary shares may not be available. You may not be able to sell your shares quickly or at the market price if trading in our ordinary shares is not active.
The market price of our ordinary shares has been and is likely to remain volatile. Our share price could be subject to wide fluctuations in response to a variety of factors, including the following:
| ● | adverse results or delays in |
| ● | reports of adverse events in other similar products or clinical trials of such products; |
| ● | inability to obtain additional |
| ● | any delay in filing an IND or BLA for any of our product candidates and any adverse development or perceived adverse development with respect to the FDA’s review of that IND or BLA; |
| ● | failure to develop successfully and commercialize our product candidates for the proposed indications and future product candidates for other indications or new candidates; |
| ● | failure to maintain our licensing arrangements or enter into strategic collaborations; |
| ● | failure by us or our licensors and strategic collaboration partners to prosecute, maintain or enforce our intellectual property rights; |
| ● | changes in laws or regulations applicable to future products; |
| ● | inability |
| ● | adverse regulatory decisions, including by the IIA under the Research Law; |
| ● | introduction of new products, services or technologies by our competitors; |
| ● | |
| failure to meet or exceed the |
| ● | the perception of the pharmaceutical industry by the public, legislatures, regulators and the investment community; |
| ● | announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our competitors; |
| ● | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| ● | additions or departures of key scientific or management personnel; |
| ● | significant lawsuits, including patent or shareholder litigation; |
| ● | changes in the market valuations of similar companies; |
| ● | general economic and market conditions and overall fluctuations in the U.S. equity market; |
| ● | any identified material weakness in our internal control over financial reporting; |
| ● | changes in the Nasdaq listing of our stock; |
| ● | recommendations of equity analysts covering our stock; |
| ● | sales of our ordinary shares by us or our shareholders in the future; and |
| ● | trading volume of our ordinary shares. |
| 30 |
In addition, companies trading in the stock market in general, and biopharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our ordinary shares, regardless of our actual operating performance.
There has been limited trading volume for our ordinary shares.
Even though our ordinary shares have been listed on the NASDAQNasdaq Global Market, there has been limited liquidity in the market for the ordinary shares, which could make it more difficult for holders to sell their ordinary shares. There can be no assurance that an active trading market for our ordinary shares will be sustained. In addition, the stock market generally has experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of listed companies. Broad market and industry factors may negatively affect the market price of our ordinary shares, regardless of our actual operating performance. The market price and liquidity of the market for our ordinary shares that will prevail in the market may be higher or lower than the price you pay and may be significantly affected by numerous factors, some of which are beyond our control.
Our principal shareholders and management own a significant percentage of our shares and will be able to exert significant control over matters subject to shareholder approval.
As of December 31, 2018,2021, to the best of our information, our executive officers, key management, directors, five percent shareholders and their affiliates beneficially owned approximately 42.0%36.1% of our voting shares. Therefore, these shareholders have the ability to control us through their ownership positions. These shareholders may be able to determine all matters requiring shareholder approval. For example, these shareholders, if they were to act together, may be able to control elections of directors, amendments of our organizational documents, or approval of any merger, sale of assets, or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our ordinary shares that you may believe are in your best interest as one of our shareholders.
We are an “emerging growth company” and a “foreign private issuer,” and we cannot be certain if the reduced reporting requirements applicable to emerging growth companies and foreign private issuers will make our ordinary shares less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes- Oxley Act of 2002, or the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in this prospectus and our periodic reports and proxy statements and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We could be an emerging growth company for up to five years, although circumstances could cause us to lose that status earlier, including if the market value of our ordinary shares held by non-affiliates exceeds $700.0 million as of any June 30 before that time or if we have total annual gross revenue of $1.07 billion or more during any fiscal year before that time, in which cases we would no longer be an emerging growth company as of the following December 31 or, if we issue more than $1.0 billion in non-convertible debt during any three-year period before that time, we would cease to be an emerging growth company immediately. For more details please refer to item 4. below.
Furthermore, as a foreign private issuer, we are not subject to the same requirements that are imposed upon U.S. domestic issuers by the SEC. Under the Securities Exchange Act of 1934, or the Exchange Act, we will be subject to reporting obligations that, in certain respects, are less detailed and less frequent than those of U.S. domestic reporting companies. For example, we will not be required to issue quarterly reports, proxy statements that comply with the requirements applicable to U.S. domestic reporting companies, or individual executive compensation information that is as detailed as that required of U.S. domestic reporting companies. We will also have four months after the end of each fiscal year to file our annual reports with the SEC and will not be required to file current reports as frequently or promptly as U.S. domestic reporting companies. Furthermore, our officers, directors and principal shareholders will be exempt from the requirements to report transactions in our equity securities and from the short-swing profit liability provisions contained in Section 16 of the Exchange Act. These exemptions and leniencies will reduce the frequency and scope of information and protections to which you may otherwise have been eligible in relation to a U.S. domestic reporting companies. See “Item 16G. Corporate Governance” for more information.
We cannot predict if investors will find our ordinary shares less attractive because we may rely on these reduced requirements. If some investors find our ordinary shares less attractive as a result, there may be a less active trading market for our ordinary shares and our share price may be more volatile.
Under the JOBS Act, emerging growth companies can also delay adopting new or revised accounting standards until such time as those standards apply to private companies. We are electing to not take advantage of the extended transition period afforded by the JOBS Act for the implementation of new or revised accounting standards, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision to not take advantage of the extended transition period for complying with new or revised accounting standards is irrevocable.
Our ordinary shares are subject to substantial dilution in their book value.
As of December 31, 2018, options and warrants to purchase 12,211,676 ordinary shares at a weighted average exercise price of $3.50 per share were outstanding. The exercise of any of these options and warrants would result in additional dilution.
Sales of a substantial number of our ordinary shares in the public market could cause our share price to fall.
If our existing shareholders sell, indicate an intention to sell or the market perceives that they intend to sell, substantial amounts of our ordinary shares in the public market, the market price of our ordinary shares could decline significantly. As of December 31, 2018, we had outstanding a total of 35,881,128 ordinary shares. Substantially all of the shares are available for sale in the public market.
As of December 31, 2018, 10,467,971 ordinary shares are held by directors, executive officers and other affiliates (holders of more than 10% of the Company’s share capital) and are subject to Rule 144 under the Securities Act of 1933, as amended, or the Securities Act.
In addition, as of March 1, 2019, an aggregate of 9,784,907 ordinary shares that are either subject to outstanding options, reserved for future issuance under our 2014 Plan or subject to outstanding warrants will or may become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules and Rule 144 and Rule 701 under the Securities Act. If these additional ordinary shares are sold, or if it is perceived that they will be sold, in the public market, the market price of our ordinary shares could decline.
Future sales and issuances of our ordinary shares or rights to purchase ordinary shares, including pursuant to our equity incentive plans, could result in additional dilution of the percentage ownership of our shareholders and could cause our share price to fall.
Additional capital will be needed in the future to continue our planned operations. To the extent we raise additional capital by issuing equity securities, our shareholders may experience substantial dilution. We may sell ordinary shares, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time.time, including pursuant to the at-the-market facility with Jefferies LLC, or the Jefferies ATM , or the equity investment component of our European Innovation Commission award. If we sell ordinary shares, convertible securities or other equity securities in one or more than one transaction,transactions, investors may be materially diluted by subsequent sales. These sales may also result in material dilution to our existing shareholders, and new investors could gain rights superior to our existing shareholders.
Pursuant to our Employee Share Ownership and Option Plan (2014), orWe also have equity plans that provide for the 2014 Plan, our management is authorized to grant of share options and other equity-based awards to our employees, directors and consultants. Currently, we plan to registerconsultants, and have issued warrants. The exercise of any of these options and warrants would result in additional share issuances and may be dilutive. As these securities are registered, many are available for resale into the increasedpublic market. Sales of a substantial number of shares available for issuance underof our ordinary shares by our existing stockholders in the 2014 Plan each year. Ifpublic market, or the perception that these sales might occur, could depress the market price of our boardcommon stock and could impair our ability to raise capital through the sale of directors electsadditional equity securities. We are unable to increasepredict the numbereffect that such sales may have on the prevailing market price of shares available for future grant by the maximum amount each year, our shareholders may experience additional dilution, which could cause our share price to fall.ordinary shares.
We could be subject to securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because pharmaceutical companies have experienced significant share price volatility in recent years. For example,Certain milestones we expect to achieve in 2022, including the pricetop-line PFS results from the OVAL Phase 3 trial, have the potential to increase the volatility of our ordinary shares, which reached its high record of $17.02 per share at the close of the trading on January 27, 2015, decreased as low as $2.8 per share at the close of the trading on February 1, 2016 a drop of about 84%. In addition, the price of our ordinary shares, which was $6.80 per share at the close of the trading on March 7, 2018, decreased to $2.65 per share at the close of the trading on March 8, 2018 and to its low record of $0.6 per share at the close of the trading on December 21, 2018.shares. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
We do not intend to pay dividends on our ordinary shares in the foreseeable future, so any returns will be limited to the value of our shares.
We have never declared or paid any cash dividends on our share capital. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. Any return to shareholders will therefore be limited to the appreciation of their shares. In addition, Israeli law limits our ability to declare and pay dividends, and may subject our dividends to Israeli withholding taxes. Furthermore, our payment of dividends (out of tax- exempttax-exempt income) may retroactively subject us to certain Israeli corporate income taxes, to which we would not otherwise be subject.
If equity research analysts do not publish research reports about our business or if they issue unfavorable commentary or downgrade our ordinary shares, the price of our ordinary shares could decline.
The trading market for our ordinary shares relies in part on the research and reports that equity research analysts publish about us and our business. The price of our ordinary shares could decline if we do not obtain research analyst coverage, or one or more securities analysts downgrade our ordinary shares, or if those analysts issue other unfavorable commentary or expectations that we are unable to meet, or cease publishing reports about us or our business.
| 31 |
Risks Related to Our Incorporation and Operations in Israel
We are currently a “foreign private issuer” and intend to follow certain home country corporate governance practices, and our shareholders may not have the same protections afforded to shareholders of companies that are subject to all NASDAQNasdaq corporate governance requirements. Additionally, we cannot be certain if the reduced disclosure requirements applicable to our status as a foreign private issuer, will make our ordinary shares less attractive to investors.
As a foreign private issuer, we are permitted, and intend, to follow certain home country corporate governance practices instead of those otherwise required under the NASDAQNasdaq Stock Market for domestic U.S. issuers. For instance, we intend to follow home country practice in Israel with regard to the quorum requirement for shareholder meetings. As permitted under the Israeli Companies Law, 5759-1999, or the Companies Law, our articles of association provide that the quorum for any meeting of shareholders shall be the presence of at least two shareholders present in person, by proxy or by a voting instrument, who hold at least 25% of the voting power of our shares instead of the 33 1/3% of the issued share capital requirement. We may in the future elect to follow home country practices in Israel (and consequently avoid the requirements that would otherwise apply to a U.S. company listed on The NASDAQthe Nasdaq Global Market) with regard to other matters, as well, such as the formation of compensation, nominating and governance committees, separate executive sessions of independent directors and non-management directors and the requirement to obtain shareholder approval for certain dilutive events (such as for the establishment or amendment of certain equity-based compensation plans, issuances that will result in a change of control of the company, certain transactions other than a public offering involving issuances of a 20% or more interest in the company and certain acquisitions of the stock or assets of another company). Following our home country governance practices as opposed to the requirements that would otherwise apply to a U.S. company listed on The NASDAQthe Nasdaq Global Market may provide less protection to you than what is accorded to investors under the NASDAQNasdaq Stock Market rules applicable to domestic U.S. issuers. See “Item 16G. Corporate Governance” for more information.
In addition, as a foreign private issuer, we are currently exempt from the rules and regulations under the Exchange Act related to the furnishing and content of proxy statements, including with regards to compensation of executive officers and our officers, directors and principal shareholders we are exempt from the requirement for an emerging growth companyreporting and the short-swing profit recovery provisions contained in Section 16 of the Exchange Act. We are also permitted to disclose thelimited compensation of the chief executive officer and other two highest compensatedinformation for our executive officers on an individual rather than aggregate, basis.basis and we are generally exempt from filing quarterly reports with the SEC under the Exchange Act. A recent amendment to regulations under the Israeli Companies Law requires us to disclose in the notice for our annual meeting of shareholders, the annual compensation of our five most highly compensated officers on an individual, rather than aggregate, basis. However, this disclosure is not as extensive as that required of a U.S. domestic issuer.
Further, we are not currently required under the Exchange Act to file annual and current reports and financial statements with the SEC as frequently or as promptly as U.S. domestic companies whose securities are registered under the Exchange Act. Moreover, we are not required to comply with Regulation FD, which restricts the selective disclosure of material nonpublic information to, among others, broker-dealers and holders of a company’s securities under circumstances in which it is reasonably foreseeable that the holder will trade in the company’s securities on the basis of the information. These exemptions and leniencies reduce the frequency and scope of information and protections to which you may otherwise have been eligible in relation to a U.S. domestic issuer.
We would lose our foreign private issuer status if a majority of our directors or executive officers are U.S. citizens or residents and we fail to meet additional requirements necessary to avoid loss of foreign private issuer status.status, which we assess annually on the last business day of our most recently completed fiscal quarter. As noted elsewhere, we currently anticipate that we will lose foreign private issuer status at our next assessment and cease to be a foreign private issuer as of January 1, 2023. Although we have elected to comply with certain U.S. regulatory provisions, our anticipated loss of foreign private issuer status would make such provisions mandatory. The regulatory and compliance costs to us under U.S. securities laws as a U.S. domestic reporting company may be significantly higher. IfOnce we are notno longer a foreign private issuer, we will be required to file periodic reports and registration statements on U.S. domestic reporting company forms with the SEC, which are more detailed and extensive than the forms available to a foreign private issuer. We will have to change our reporting from IFRS to US GAAP accounting standards and we may also be required to modify certain of our policies to comply with accepted governance practices associated with U.S. domestic reporting companies. Such conversion and modifications will involve additional costs. In addition, we may lose our ability to rely upon exemptions from certain corporate governance requirements on U.S. stock exchanges that are available to foreign private issuers.
We cannot predict if investors will find our ordinary shares less attractive because we may rely on these reduced requirements. If some investors find our ordinary shares less attractive as a result, there may be a less active trading market for our ordinary shares and our share price may be more volatile.
We expect to lose our foreign private issuer status, which will require us to comply with the Exchange Act’s domestic reporting regime and cause us to incur significant legal, accounting and other expenses, even if we are able to qualify as a “smaller reporting company.”
We currently qualify as a foreign private issuer. As a foreign private issuer, we are permitted by the SEC to file an annual report on Form 20-F and copies of certain home country materials on Form 6-K in lieu of filing annual, quarterly and current reports on Forms 10-K, 10-Q and 8-K. We are exempt from SEC proxy statement requirements and certain SEC tender offer requirements and our affiliates are exempt from Section 16 of the Exchange Act.
We currently expect that due to the composition of our board of directors, our management team and our shareholder base, we will cease to be a foreign private issuer and cease to be eligible for the foregoing exemptions and privileges effective January 1, 2023. When we cease to be a foreign private issuer, we will be required to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act applicable to U.S. domestic issuers as of January 1, 2023, which are more detailed and extensive than the requirements for foreign private issuers. We may also be required to make changes in our corporate governance practices in accordance with various SEC and Nasdaq rules. Although we already report under U.S. GAAP and voluntarily publish quarterly financial information, the regulatory and compliance costs to once we are required to comply with the reporting requirements applicable to a U.S. domestic issuer may be significantly higher than the cost we incur as a foreign private issuer even if we are able to qualify as a “smaller reporting company.”
As a result, we expect that a loss of foreign private issuer status will increase our legal and financial compliance costs and may make some activities highly time consuming and costly. It will also impose additional burdens on holders of our securities, which may make an investment in our company less attractive. We expect that complying with the rules and regulations applicable to United States domestic issuers may make it more difficult and expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These rules and regulations could also make it more difficult for us to attract and retain qualified members of our board of directors.
Potential political, economic and military instability in the State of Israel, where the majority of our senior management and our research and development facilities are located, may adversely affect our results of operations.
We are incorporated under Israeli law and our offices and core operations are located in the State of Israel.Israel, with a smaller operational base in the United States. In addition, most of our key employees and officers and all but twothree of our directors are residents of Israel. Accordingly, political, economic and military conditions in Israel directly affect our business. Since the State of Israel was established in 1948, a number of armed conflicts have occurred between Israel and its neighboring countries.
Any hostilities involving Israel or the interruption or curtailment of trade between Israel and its present trading partners, or a significant downturn in the economic or financial condition of Israel, could affect adversely our operations. Since October 2000, there have been increasing occurrences of terrorist violence. Ongoing and revived hostilities or other Israeli political or economic factors could harm our operations, product development and results of operations.
Although Israel has entered into various agreements with Egypt, Jordan and the Palestinian Authority, there has been an increase in unrest and terrorist activity, which began in October 2000 and has continued with varying levels of severity. The establishment in 2006 of a government in the Palestinian Authority by representatives of the Hamas militant group has created additional unrest and uncertainty in the region. In 2006, a conflict between Israel and the Hezbollah in Lebanon resulted in thousands of rockets being fired from Lebanon up to 50 miles into Israel. Starting in December 2008, for approximately three weeks, Israel engaged in an armed conflict with Hamas in the Gaza Strip, which involved missile strikes against civilian targets in various parts of Israel and negatively affected business conditions in Israel. In November 2012, for approximately one week, Israel experienced a similar armed conflict, resulting in hundreds of rockets being fired from the Gaza Strip and disrupting most day-to-day civilian activity in southern Israel. Beginning in July 2014, for approximately seven weeks, Israel experienced additional armed conflict between Israel and Hamas, which included rocket strikes against civilian targets in various parts of Israel. If renewed, these hostilities may negatively affect business conditions in Israel. In addition, Israel faces threats from more distant neighbors, in particular, Iran. Our insurance policies do not cover us for the damages incurred in connection with these conflicts or for any resulting disruption in our operations. The Israeli government, as a matter of law, provides coverage for the reinstatement value of direct damages that are caused by terrorist attacks or acts of war; however, the government may cease providing such coverage or the coverage might not be enough to cover potential damages. In the event that hostilities disrupt the ongoing operation of our facilities or the airports and seaports on which we depend to import and export our supplies and products, our operations may be materially adversely affected.
In addition, since the end of 2010, numerous acts of protest and civil unrest have taken place in several countries in the Middle East and North Africa, many of which involved significant violence. The civil unrest inviolence, including Egypt and Syria, which borders Israel, resulted in the resignation of its president Hosni Mubarak, and to significant changes to the country’s government. In Syria, also bordering Israel, a civil war is continuing to take place.border Israel. The ultimate effect of these developments on the political and security situation in the Middle East and on Israel’s position within the region is not clear at this time. Such instability may lead to deterioration in the political and trade relationships that exist between the State of Israel and certain other countries.
Popular uprisings in various countries in the Middle East and North Africa are affecting the political stability of those countries. Such instability may lead to deterioration in the political and trade relationships that exist between the State of Israel and these countries. Several countries, principally in the Middle East, still restrict doing business with Israel and Israeli companies, and additional countries may impose restrictions on doing business with Israel and Israeli companies if hostilities in Israel or political instability in the region continues or increases. Any hostilities involving Israel or the interruption or curtailment of trade between Israel and its present trading partners, or significant downturns in the economic or financial condition of Israel, could adversely affect our operations and product development and adversely affect our share price. Similarly, Israeli companies are limited in conducting business with entities from several countries. For instance, in 2008, the Israeli legislature passed a law forbidding any investments in entities that transact business with Iran.
Our operations may be disrupted by the obligations of personnel to perform military service.
As of March 1, 2019,2022, we had 3941 employees, all39 of whom were based in Israel. Some of our employees may be called upon to perform up to 36 days (and in some cases more) of annual military reserve duty until they reach the age of 40 (and in some cases, up to 45 or older) and, in emergency circumstances, could be called to immediate and unlimited active duty. In the event of severe unrest or other conflict, individuals could be required to serve in the military for extended periods of time. Since September 2000, in response to increased tension and hostilities, there have been occasional call-ups of military reservists including in connection with the 2006 conflict in Lebanon, and the December 2008 and November 2012 conflicts with Hamas, and it is possible that there will be additional call-ups in the future. Our operations could be disrupted by the absence of a significant number of our employees related to military service or the absence for extended periods of one or more of our key employees for military service. Such disruption could materially adversely affect our business and results of operations. Additionally, the absence of a significant number of the employees of our Israeli suppliers and contractors related to military service or the absence for extended periods of one or more of their key employees for military service may disrupt their operations.
The tax benefits that are available to us if and when we generate taxable income require us to meet various conditions and may be prevented or reduced in the future, which could increase our costs and taxes.
If and when we generate taxable income, we would be eligible for certain tax benefits provided to “Benefited Enterprises” under the Israeli Law for the Encouragement of Capital Investments, 1959, as amended, or the Investment Law. In order to remain eligible for the tax benefits for “Benefited Enterprises” we must continue to meet certain conditions stipulated in the Investment Law and its regulations, as amended. In addition, we informed the Israeli Tax Authority of our choice of 2012 as a “Benefited Enterprise” election year, all under the Investment Law. The benefits available to us under this tax regulation are subject to the fulfillment of conditions stipulated in the regulation. Further, in the future these tax benefits may be reduced or discontinued. If these tax benefits are reduced, cancelled or discontinued, our Israeli taxable income would be subject to regular Israeli corporate tax rates. The standard corporate tax rate for Israeli companies is 25% for 2016, 24% for 2017 and 23% for 2018 and thereafter. Additionally, if we increase our activities outside of Israel through acquisitions, for example, our expanded activities might not be eligible for inclusion in future Israeli tax benefit programs. See “Item 10E. Taxation-Israeli Tax Considerations and Government Programs-Law for the Encouragement of Capital Investments, 5719-1959.”
| 33 |
It may be difficult to enforce a U.S. judgment against us, our officers and directors and the Israeli experts named in this prospectusdocument in Israel or the United States, or to assert U.S. securities laws claims in Israel or serve process on our officers and directors and these experts.
We were incorporated in Israel, and our corporate headquarters and substantially all of our operations are located in Israel. AllMost of our executive officers and all but twothree of our directors, and the Israeli experts named in this prospectus,document, are located in Israel. The majority of our assets and the assets of these persons are located outside the United States. Therefore, it may be difficult for an investor, or any other person or entity, to enforce a U.S. court judgment based upon the civil liability provisions of the U.S. federal securities laws against us or any of these persons in a U.S. or Israeli court, or to effect service of process upon these persons in the United States. Additionally, it may be difficult for an investor, or any other person or entity, to assert U.S. securities law claims in original actions instituted in Israel. Israeli courts may refuse to hear a claim based on an alleged violation of U.S. securities laws against us or our officers and directors on the grounds that Israel is not the most appropriate forum in which to bring such a claim. Even if an Israeli court agrees to hear a claim, it may determine that Israeli law and not U.S. law is applicable to the claim. If U.S. law is found to be applicable, the content of applicable U.S. law must be proved as a fact, which can be a time-consuming and costly process. Certain matters of procedure will also be governed by Israeli law. There is little binding case law in Israel addressing the matters described above.
Your rights and responsibilities as our shareholder will be governed by Israeli law, which may differ in some respects from the rights and responsibilities of shareholders of U.S. corporations.
SinceBecause we are incorporated under Israeli law, the rights and responsibilities of our shareholders are governed by our articles of association and Israeli law. These rights and responsibilities differ in some material respects from the rights and responsibilities of shareholders of U.S. corporations. In particular, a shareholder of an Israeli company has a duty to act in good faith and in a customary manner in exercising its rights and performing its obligations towards the company and other shareholders and to refrain from abusing its power in the company, including, among other things, in voting at the general meeting of shareholders on certain matters, such as an amendment to the company’s articles of association, an increase of the company’s authorized share capital, a merger of the company and approval of related party transactions that require shareholder approval. A shareholder also has a general duty to refrain from discriminating against other shareholders. In addition, a controlling shareholder or a shareholder who knows that it possesses the power to determine the outcome of a shareholder vote or to appoint or prevent the appointment of an officer of the company has a duty to act in fairness towards the company with regard to such vote or appointment. However, Israeli law does not define the substance of this duty of fairness. There is limited case law available to assist us in understanding the nature of this duty or the implications of these provisions. These provisions may be interpreted to impose additional obligations and liabilities on our shareholders that are not typically imposed on shareholders of U.S. corporations. See “Item 6. Directors, Senior Management and Employees-Approval of Related Party Transactions Under Israeli Law-Shareholders’ Duties.Law.”
Provisions of Israeli law and our amended and restated articles of association could make it more difficult for a third party to acquire us or increase the cost of acquiring us, even if doing so would benefit our shareholders.
Israeli law regulates mergers, requires tender offers for acquisitions of shares above specified thresholds, requires special approvals for transactions involving directors, officers or significant shareholders and regulates other matters that may be relevant to such types of transactions. For example, a tender offer for all of a company’s issued and outstanding shares can only be completed if the acquirer receives positive responses from the holders of at least 95% of the issued share capital. Completion of the tender offer also requires approval of a majority of the offerees that do not have a personal interest in the tender offer, unless at least 98% of the company’s outstanding shares are tendered. Furthermore, the shareholders, including those who indicated their acceptance of the tender offer (unless the acquirer stipulated in its tender offer that a shareholder that accepts the offer may not seek appraisal rights), may, at any time within six months following the completion of the tender offer, petition an Israeli court to alter the consideration for the acquisition. See “Item 10B. Memorandum and Articles of Association-Acquisitions under Israeli Law” for additional information.
Further, Israeli tax considerations may make potential transactions undesirable to us or to some of our shareholders whose country of residence does not have a tax treaty with Israel granting tax relief to such shareholders from Israeli tax. For example, Israeli tax law does not recognize tax-free share exchanges to the same extent as U.S. tax law. With respect to mergers, Israeli tax law allows for tax deferral in certain circumstances but makes the deferral contingent on the fulfillment of a number of conditions, including, in some cases, a holding period of two years from the date of the transaction during which sales and dispositions of shares of the participating companies are subject to certain restrictions. Moreover, with respect to certain share swap transactions, the tax deferral is limited in time, and when such time expires, the tax becomes payable even if no disposition of the shares has occurred.
Certain U.S. shareholders may be subject to adverse tax consequences if we are characterized as “Controlled Foreign Corporation.”
Each “Ten Percent Shareholder” in a non-U.S. corporation that is classified as a “controlled foreign corporation,” or a CFC, for U.S. federal income tax purposes generally is required to include in income for U.S. federal tax purposes such Ten Percent Shareholder’s pro rata share of the CFC’s “Subpart F income” and investment of earnings in U.S. property, even if the CFC has made no distributions to its shareholders. A non-U.S. corporation generally will be classified as a CFC for U.S. federal income tax purposes if Ten Percent Shareholders own, directly or indirectly, more than 50% of either the total combined voting power of all classes of stock of such corporation entitled to vote or of the total value of the stock of such corporation. A “Ten Percent Shareholder” is a U.S. person (as defined by the U.S. Internal Revenue Code of 1986, as amended), who owns or is considered to own 10% or more of the total combined voting power of all classes of stock entitled to vote of such corporation. The determination of CFC status is complex and includes attribution rules, the application of which is not entirely certain.
We do not believe that we were a CFC for the taxable year ended December 31, 20182021 or that we are currently a CFC. It is possible, however, that a shareholder treated as a U.S. person for U.S. federal income tax purposes will acquire, directly or indirectly, enough shares to be treated as a Ten Percent Shareholder after application of the constructive ownership rules and, together with any other Ten Percent Shareholders of our company, cause us to be treated as a CFC for U.S. federal income tax purposes. We believe that certain of our shareholders are Ten Percent Shareholders for U.S. federal income tax purposes. Holders should consult their own tax advisors with respect to the potential adverse U.S. federal income tax consequences of becoming a Ten Percent Shareholder in a CFC.
| 34 |
We might be classified as a passive foreign investment company in future years, and our U.S. shareholders may suffer adverse tax consequences as a result.
Generally, if, for any taxable year, at least 75% of our gross income is passive income, or at least 50% of the value of our assets is attributable to assets that produce passive income or are held for the production of passive income, including cash, we would be characterized as a passive foreign investment company, or PFIC, for U.S. federal income tax purposes. For purposes of these tests, passive income includes dividends, interest, and gains from the sale or exchange of investment property and rents and royalties other than rents and royalties which are received from unrelated parties in connection with the active conduct of a trade or business. If we are characterized as a PFIC, our U.S. shareholders may suffer adverse tax consequences, including having gains realized on the sale of our ordinary shares treated as ordinary income, rather than capital gain, the loss of the preferential rate applicable to dividends received on our ordinary shares by individuals who are U.S. holders, and having interest charges apply to distributions by us and the proceeds of share sales. See “Item 10E. Taxation-Certain Material U.S. Federal Income Tax Considerations-Passive Foreign Investment Company Considerations.”
SinceBecause PFIC status depends on the composition of our income and the composition and value of our assets (which may be determined in part by reference to the market value of our ordinary shares, which may be volatile) from time to time, there can be no assurance that we will not be considered a PFIC for any taxable year. We had no revenue-producing operations until and including table year 2016. We believe that we were not a PFIC for our 2017, 2018, 2019, 2020 and 20182021 taxable years. In addition, unless and until we generate sufficient revenue from active licensing and other non-passive sources and otherwise satisfy the asset test above, we might be treated as a PFIC in future taxable years.
Item 4. Information on the Company
Corporate Information
The legal name of our company is Vascular Biogenics Ltd., and we conduct business under the name VBL Therapeutics. We were incorporated in Israel on January 27,31, 2000 as a company limited by shares under the name Medicard Ltd. In January 2003,February 2002, we changed our name to Vascular Biogenics Ltd. Our registered and principal office is located 8 HaSatat St., Modi’in, Israel 7178106.7178106 and our telephone number is 972-8-9935000. We also have a wholly owned U.S. subsidiary, VBL Inc., with an office located at 1 Blue Hill Plaza, Suite 1509, Pearl River, NY 10965. Our service agent in the United States is located at c/o CT Corporation System, 111 8thPuglisi and Associates, 850 Library Avenue, New York, New York 10011 and our telephone number is 972-8-9935000. Throughout this prospectus, we refer to various trademarks, service marks and trade names that we use in our business. The “Vascular Biogenics” design logo, “VBL Therapeutics,” “Vascular Targeting System,” “VTS,” “Lecinoxoids,” “VB-111,” “VB-201,” the “OVAL” design logo and other trademarks or service marks of Vascular Biogenics Ltd. appearing in this prospectus are the property of Vascular Biogenics Ltd. We have several other registered trademarks, service marks and pending applications relating to our products. Although we have omitted the “® ” and “” trademark designations for such marks in this prospectus, all rights to such trademarks are nevertheless reserved. Other trademarks and service marks appearing in this prospectus are the property of their respective holders.Suite 204, Newark, Delaware 19711.
Emerging Growth Company
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act, or the JOBS Act. Thus, we may take advantage of certain exemptions from various reporting requirements that are applicable to public companies generally. For example, we have elected not to have our independent registered public accounting firm provide an attestation report on the effectiveness of our internal control over financial reporting, as would otherwise be required by Section 404(b) of the Sarbanes-Oxley Act, or SOX.
We will cease to be an “emerging growth company” upon the earliest of:
The JOBS Act also provides that an “emerging growth company” can utilize the extended transition period provided in Section 7(a)(2)(B) of the Securities Act, for complying with new or revised accounting standards. However, we have chosen to “opt out” of such extended transition period, and, as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for companies that are not “emerging growth companies.” Section 107 of the JOBS Act provides that our decision to opt out of the extended transition period for complying with new or revised accounting standards is irrevocable.
Capital Expenditures
For a discussion of our capital expenditures, see “Item 5. Operating and Financial Review and Prospects-Liquidity and Capital Resources.”
Business OverviewSummary
We are a clinical stage biopharmaceutical company committed to developing next-generation, targeted medicines for difficult-to-treat medical conditions. Using our novel platform technologies we have created a pipeline of therapeutics to uniquely address cancer and immune-inflammatory diseases with the goal of significantly improving patient outcomes and overcoming the limitations of currently approved treatments.
Our product candidates are built off of our two platform technologies: VTS, a gene-based technology targeting newly formed blood vessels, and Monocyte Targeting Technology, or MTT, an antibody-based technology able to specifically inhibit monocyte migration for immune-inflammatory applications.
We are currently evaluating our lead candidate, ofra-vec, in a clinical-stage biopharmaceutical company focused onPhase 3 registration-enabling trial in platinum resistant ovarian cancer, for which we anticipate PFS primary endpoint data in the discovery, developmentsecond half of 2022. We are also supporting Phase 2 trials in rGBM and commercializationmetastatic colorectal cancer, or mCRC where we expect preliminary data in 2022. Our second program, VB-601, is an investigational proprietary monoclonal antibody that binds MOSPD2, which we call the “mono-walk”, receptor, and is engineered to specifically prevent monocytes from exiting the blood stream and traveling to inflamed tissues, and is expected to begin a first-in-human clinical trial in the second half of first-in-class treatments for cancer. 2022.
| 36 |
Our program is based on our proprietary Platform Technologies
Vascular Targeting System
Our proprietary VTS technology is designed to enable systemic administration of our gene-based investigational candidates to either destroy or VTS, platform technology, which utilizes genetically targeted therapy to destroy newly formed, orpromote angiogenic (newly formed) blood vessels, and be both tissue- and condition-specific. By harnessing nature and using our body’s molecular machinery, it allows for targeted gene expression in endothelial cells, the thin layer of cells that lines the interior surface of blood vessels undergoing angiogenesis, without detectable activation or expression in other cells, thereby limiting the potential for off-target effects.
The platform is made up of three components: a viral vector, novel promoter and therapeutic gene. The viral vector delivers the genetic code into cells. The novel promoter imparts specificity for angiogenic cells, and the therapeutic gene executes the desired biological activity. We can use different combinations and modifications of these components to custom tailor the attributes of a VTS-based candidate to enhance its profile for a specific indication. We are currently developing the VTS technology for oncology applications.
Monocyte Targeting Technology
We have also developed a second platform technology, MTT, based on the internal discovery of a novel target, MOSPD2. This novel target, which we call the “mono-walk” receptor, is selectively expressed on the surface of monocytes and controls their ability to migrate (or “walk” to) inflamed tissues. Monocytes are an important cell implicated in the chronicity of disease in inflammatory indications and previous attempts by others to specifically target this cell type and prevent its migration to sites of inflammation have been unsuccessful. We believe will allow usthat our approach can address this gap in being able to develop product candidates for multipleoptimally address chronic inflammation and we are utilizing antibody technology to specifically inhibit this target with high potency.
Product Pipeline
Utilizing our platform technologies, we have created a pipeline of novel and differentiated programs addressing difficult to treat oncology indications.and inflammatory applications:
Ofra-vec - Lead VTS Candidate
Our lead product candidate utilizing the VTS platform is ofra-vec (also known as VB-111), which is currently being evaluated in the OVAL Phase 3 registration-enabling clinical trial in platinum resistant ovarian cancer (NCT03398655) and Phase 2 trials in rGBM (NCT04406272) and mCRC (NCT04166383). Ofra-vec’s mechanism of action is designed to combine the blockade of tumor microvasculature (the blood vessels required for tumor growth) with immune recruitment resulting in an anti-tumor immune response and a highly differentiated potential treatment for solid tumors. As an investigational drug engineered to work through the body’s molecular machinery, ofra-vec is designed to be activated specifically when and where it is needed. To date, over 300 patients have been dosed with ofra-vec in completed clinical trials.
Ofra-vec is a custom designed therapeutic comprised of several components and characteristics. Each individual component brings its own unique potential advantages as it relates to difficult to treat solid tumors:
| 1. | Viral vector (Adenovirus Type 5) – Non-integrating, and non-replicating modified virus designed to deliver the gene construct to target cells and create a localized immune response in the tumor microenvironment. Unlike challenges seen with other approved and investigational therapeutics using an adeno-associated virus, our adenovirus can be re-dosed chronically. | |
| 2. | Promoter (PPE 1-3x) - Promoter designed to impart specificity for angiogenic endothelial cells and engineered to contain the expression of the therapeutic gene and anti-angiogenic effect to the tumor microvasculature without affecting healthy vasculature or other tissues. | |
| 3. | Therapeutic gene - Chimeric pro-apoptotic death receptor comprised of TNFR1 (extracellular part) and FAS (intracellular part) (TNF-Induced death receptor). This genetic sequence is designed to take advantage of high tumor necrosis factor TNF-alpha levels in tumors to enhance activity. Once the promoter expresses the gene in the target cells and the chimeric TNF-Induced death receptor is engaged in the tumor microenvironment, it initiates a self-death process in the tumor microvasculature (blood vessels), potentially leading to vascular disruption, tumor starvation and immune recruitment. |
We believe these three components which comprise ofra-vec result in a unique and highly differentiated dual mechanism of action:

| 37 |
Our lead product candidate, VB-111 (ofranergene obadenovec),Unlike anti-vascular endothelial growth factor, or anti-VEGF agents (such as Avastin/ bevacizumab) or tyrosine-kinase inhibitors, or TKIs, ofra-vec does not aim to block a specific pro-angiogenic pathway; instead, it is designed to use our proprietary angiogenesis-specific promoter to specifically induce cell death in angiogenic endothelial cells in the tumor microenvironment. This mechanism has the potential to retain activity regardless of baseline tumor mutations or the identity of the pro-angiogenic factors secreted by the tumor and has demonstrated clinical activity even after failure of prior treatment with other anti-angiogenic agents. Nevertheless, as ofra-vec expression is induced by VEGF, it should not be co-administrated with anti-VEGF agents such as Avastin.
Overview of Ofra-vec Clinical Programs
Ofra-vec’s dual mechanism of action targeting the vasculature and recruiting the immune system into tumors has the potential to treat a gene-based biologic that we are developing forbroad range of solid tumor indications,tumors without relying on specific genetic drivers or receptors to enhance and enrich its activity. Most solid tumors create vasculature to support growth and cloak themselves from the immune system, both of which we have advancedbelieve ofra-vec may be able to programs for recurrent glioblastoma, or rGBM, an aggressive form of brain cancer,address and thereby provide a more effective therapeutic solution and be potentially applicable to a broader patient population than current therapies.
We are evaluating ofra-vec in multiple clinical trials including the ongoing Phase 3 registration-enabling OVAL trial in platinum resistant ovarian cancer, and thyroid cancer.have seen encouraging data from previous Phase 2 oncology trials demonstrating a dose response and the potential to increase survival, particularly in patients with limited treatment options and poor prognosis. In our clinical trials to date, ofra-vec was generally well-tolerated, and the most common adverse event was transient mild-moderate fever. We have received orphan drug designation for ofra-vec for the treatment of malignant glioma by the FDA and for the treatment of ovarian cancer and the treatment of glioma by the European Commission. We have also obtained fast track designation for VB-111ofra-vec in the United States for prolongation of survival in patients with glioblastoma that has recurred following treatment with standard chemotherapy and radiation. We have also received orphan drug designationan open IND for GBMofra-vec with the Office of Tissues and Advanced Therapies within FDA’s Center for Biologics Evaluation and Research.
Ofra-vec Clinical Program in bothOvarian Cancer
Ovarian cancer is the leading cause of gynecologic cancer death in the United States affecting approximately 21,400 women annually. In patients with platinum-resistant disease, the addition of the anti-angiogenic agent Avastin to chemotherapy has resulted in significantly improved PFS and Europe. VB-111 has also receivedresponse rates. However, the addition of Avastin did not result in a significant improvement of OS and the overall outcomes and prognosis for these patients remains poor. Other approved agents such as PARP inhibitors are being used in this indication but their utility is somewhat limited by genetic factors and so far they have failed to demonstrate an orphan designation forincrease in survival. Given the treatmentlimitations of current therapies, there is an unmet need to make significant improvements in the outcomes of patients with recurrent platinum-resistant ovarian cancer byfollowing first line therapy. We believe that ofra-vec’s unique dual mechanism of action specifically targeting the European Medicines Agency. In September 2015,tumor vasculature and immune recruitment has the potential to improve outcomes and we reported completeare actively pursuing this application.
A Phase 1/2 study of ofra-vec in recurrent platinum-resistant ovarian cancer was initially conducted and final results from our Phase 2 trialwere published in a peer-reviewed publication (Arend et al., Gynecol Oncol. 2020). The data demonstrated a median OS of VB-111498 days in rGBM, demonstrating a statistically-significant benefitthe ofra-vec therapeutic-dose arm, versus 172.5 days in overall survival and favorable response rate inthe low-dose arm (p=0.03). Of the evaluable patients treated with VB-111the therapeutic dose of ofra-vec, 58% had a Gynecologic Cancer Intergroup, or GCIG, CA-125 response. Ofra-vec activity signals were seen despite unfavorable prognostic characteristics (48% platinum refractory disease and 52% previous treatment with anti-angiogenics). There was a trend for favorable survival in patients who had CA-125 decrease >50% in the ofra-vec therapeutic-dose arm (808 vs. 351 days; p=0.067) implicating CA-125 as a potentially valuable biomarker for response with ofra-vec. An immunotherapeutic effect was also observed in biopsies taken from patients. In addition, hematoxylin and eosin and immunohistochemistry staining showed regions of apoptotic cancer cells and infiltration of cytotoxic CD8 T-cells following treatment with ofra-vec.
After an end-of-Phase 2 meeting with the FDA to discuss the clinical path of ofra-vec in ovarian cancer, we aligned with the FDA on our clinical plan to proceed to a Phase 3 registration-enabling trial of ofra-vec in platinum-resistant patients and initiated the OVAL Phase 3 trial. We have since completed enrollment for this global Phase 3 randomized, multi-center, placebo-controlled registration-enabling clinical trial evaluating the combination of ofra-vec and paclitaxel versus placebo and paclitaxel, in patients with platinum-resistant ovarian cancer. A total of 409 patients have been enrolled in the OVAL trial at approximately 75 clinical sites in the United States, Europe, Israel and Japan, with the majority having previously failed Avastin and/or PARP inhibitor therapies. Patients were randomized 1:1 to ofra-vec (1x1013 viral particles, or VPs, once every eight weeks) in combination with bevacizumab. Our pivotal Phase 3 GLOBE study in rGBM began in August 2015 and was comparing a combination of VB-111 and bevacizumabchemotherapy (80mg/m2 paclitaxel once weekly), or to bevacizumab alone.placebo with chemotherapy. The study which enrolled a total of 256 patients in the US, Canadaincludes two individual primary endpoints, PFS and Israel was conducted under a special protocol assessment, or SPA, agreement with the U.S. Food and Drug Administration, or FDA, with full endorsement by the Canadian Brain Tumor Consortium (CBTC). On March 8, 2018,OS. Based on regulatory guidance, we announced top-line results from the GLOBE study, which showed that the study did not meet its pre-specifiedbelieve successfully meeting either primary endpoint of overall survival (OS). No new safety concerns associated with VB-111 have been identified in the GLOBE study. In November 2018, full data from the GLOBE trial were presented at the 2018 Society for Neuro-Oncology Annual Meeting. Analyses pointed to the regimen in GLOBE trial as the potential reason for its negative outcome, indicating that priming with VB-111 without bevacizumab may be critical for the immune and vascular-disruptive/anti-angiogenic mechanism of VB-111 in rGBM. We do not think that results of the GLOBE study in rGBM will necessarily have implications on the prospects for VB-111 in other regimens or tumor types. Accordingly, following independent MRI analyses at UCLA which validated the overall clinical and survival benefit of VB-111 among patients who were primed with VB-111 in our prior Phase 2 study in rGBM,sufficient to support a new investigator-sponsored Phase 2BLA submission. The OVAL trial evaluating VB-111 in rGBM patients is expected to be launched in the first half of 2019. Our OVAL phase 3 potential registration study of VB-111 in platinum resistant ovarian cancer was launched in December 2017 and isbeing conducted in collaboration with the GOG Foundation, Inc., a leading organization for research excellence in the field of gynecologic malignancies. An
We conducted the first pre-planned interim analysis of the OVAL trial after the first 60 enrolled subjects were evaluable for CA-125 response, an important biomarker of disease in ovarian cancer. The interim analysis was utilized to analyze futility and confirm the initial activity of ofra-vec in the OVAL trial. The independent Data Safety Monitoring Committee, or DSMC, reviewed unblinded data and assessed CA-125 response, measured according to the GCIG criteria, and confirmed that the study is expectedmet the interim pre-specified efficacy criterion, of an absolute percentage advantage of 10% or higher CA-125 response rate for the ofra-vec treatment arm, and recommended that the study continue as planned. The cumulative CA-125 response rate in year-end 2019. Inthe first 60 randomized evaluable patients was 53%. Assuming a balanced randomization, the blinded CA-125 response rate in the treatment arm (ofra-vec in addition a collaborative Phase 2 study evaluating VB-111 in combination with a checkpoint-inhibitor in gastrointestinal tumors is expectedto weekly paclitaxel) was estimated to be launched58%. Ofra-vec was well tolerated and the most common adverse event was transient mild-moderate fever. Results of the interim analysis were published in the second half of 2019.
We also have been conducting a program targeting anti-inflammatory diseases,peer-reviewed publication (Arend et al., Gynecol Oncol. 2021). The response rate was markedly higher than what would be expected based on the use of our Lecinoxoid platform technology. Lecinoxoids are a novel class of small molecules we developed that are structurally and functionally similar to naturally occurring molecules known to modulate inflammation. The lead product candidate from this program, VB-201, is a Phase 2-ready molecule that demonstrated efficacy in reducing vascular inflammation in a Phase 2 sub-study in psoriatic patients with cardiovascular risk. Due to business limitations associated with the heavy burden of developing medications for cardiovascular diseases, we chose to test it in psoriasis and ulcerative colitis; however, as we reported in February 2015, VB-201 failed to meet the primary endpoint in Phase 2 clinical trials for psoriasis and for ulcerative colitis. As a result, we have terminated our development of VB-201 in those indications. Nevertheless, based on recent pre-clinical studies, we believe that VB-201 and some second generation molecules such as VB-703 may be applicable for NASH and renal fibrosis. Since the company intends to focus its efforts and resources on advancing our oncology program, we will seek to advance our Lecinoxoid assets via strategic deals. Accordingly, in March 2019, we announced a strategic exclusive option license agreement with one of the world-leading European animal health companies, for the development of VB-201 for veterinary use. We retain the VB-201 rights for treatment of humans, worldwide.
We are also conducting a research program exploring the potential of targeting of MOSPD2 for immuno-oncology applications. In January 2017, we reported that targeting of MOSPD2 inhibits chemotaxis of monocytes and neuropils. In 2018, we published a manuscript showing that MOSPD2 can play a major role in breast cancer cell migration and metastasis, and that targeting MOSPD2 may be employed to prevent the spreading of breast cancer cells. We also presented proof-of-concept data demonstrating antibody-mediated killing of MOSPD2-expressing cancer cells. We believe that targeting of MOSPD2 may have several therapeutic applications, including inhibition of monocyte migration in chronic inflammatory conditions, inhibition of tumor cell metastases and targeting of MOSPD2-expressing tumor cells. We are developing our “VB-600 series” of pipeline candidates towards inflammatory and oncology applications.
We are developing our lead oncology product candidate, VB-111, for solid tumor indications, with current clinical programs in rGBM, thyroid cancer and ovarian cancer. In interim analyses of data from our ongoing open-label Phase 2 clinical trial of VB-111 in rGBM, we observed dose-dependent attenuation of tumor growth and an increase in median overall survival, which is the time interval from initiation of treatment to the patient’s death. The U.S. Food and Drug Administration, or FDA, has granted VB-111 fast track designation for prolongation of survival in patients with glioblastoma that has recurred following treatment with temozolomide, a chemotherapeutic agent commonly used to treat newly diagnosed glioblastoma, and radiation. On July 1, 2014, the FDA concurred with the design and planned analyses of our Phase 3 pivotal trial of VB-111 in rGBM pursuant to an SPA. While VBL's request was to use the same regimen of the Phase 2 clinical trial in rGBM for the Phase 3 study, the agency requested that the Phase 3 study design would be revised. Accordingly, whereasweekly paclitaxel control arm performance in the Phase 2historical Avastin registration study the experimental arm received VB-111 monotherapy and upon disease progression to continue VB-111 in combination with bevacizumab, the SPA language defined that subjects in the Phase 3 experimental arm were to receive VB-111 in combination with bevacizumab, from start. At the time, commencement of the trial was subject to our providing the agency with more information regarding our potency release assay for the trial. We developed this assay and submitted initial information to the FDA on May 26, 2014. On February 5, 2015 the FDA has found our data satisfactory and removed the partial hold.(AURELIA Study).
We began our Phase 3 pivotal trial of VB-111 in rGBM in August 2015 and completed patient enrollment for the study in December 2016, five months ahead of our initial plan. Following positive safety reviews announced in December 2016, in April 2017 and the third and final safety review that was announced in October 2017, the GLOBE trial continued to completion. On March 8, 2018, we announced top-line results from the GLOBE study, which showed that the study did not meet its pre-specified primary endpoint of overall survival (OS). In November 2018, full data from the GLOBE trial were presented at the 2018 Society for Neuro-Oncology Annual Meeting by Dr. Timothy Cloughesy, MD, Professor of Clinical Neurology and Director of the Neuro-Oncology Program, UCLA School of Medicine and principal investigator of the GLOBE trial. Thorough analyses of the baseline risk factors of the Phase 2 and the Phase 3 treatment groups did not reveal any differences. Therefore, patient selection or different patient populations could not explain the difference between the results of the two studies. The analyses pointed to the regimen in GLOBE trial as the potential reason for its failure, indicating that priming with VB-111 without bevacizumab may be critical for the immune and vascular-disruptive/anti-angiogenic mechanism of VB-111 in rGBM. Following independent MRI analyses at UCLA which validated the overall clinical and survival benefit of VB-111 among patients who were primed with VB-111 in our prior Phase 2 study in rGBM, a new investigator-sponsored trial evaluating VB-111 in rGBM patients is expected to be launched in the first half of 2019.
| 38 |
VB-111 was also being studied in a Phase 2 trial for recurrent platinum-resistant Ovarian Cancer and in a Phase 2 study in recurrent, iodine-resistant differentiated Thyroid Cancer. In a Phase 2 trial for recurrent platinum-resistant ovarian cancer, VB-111 demonstrated a statistically significant increase in overall survival and about 60% durable response rate (as measured by reduction in CA-125), approximately twice the historical response with bevacizumab plus chemotherapy in ovarian cancer. In December 2016, we had an end-of-Phase-2 meeting with the FDA, in which we received approval from the FDA to advance VB-111 for a Phase 3 study in platinum-resistant ovarian cancer, which we launched in December 2017. The OVAL study is conducted in collaboration with the Gynecologic Oncology Group (GOG) Foundation, Inc., a leading organization for research excellence in the field of gynecologic malignancies. An interim analysis in the OVAL study is expected to occur in year-end 2019. In March 2019, at the Society of Gynecologic Oncology conference, we presented data from of ovarian cancer patients, showing the potential of VB-111 to stimulate the immune system and drive immune cells to infiltrate the tumor microenvironment.
In February 2017, we reported full data from our exploratory Phase 2 study of VB-111 in recurrent, iodine-resistant differentiated thyroid cancer. The primary endpoint of the trial, defined as 6-month progression-free-survival (PFS-6) of 25%, was met with a dose response. Forty-seven percent of patients in the therapeutic-dose cohort reached PFS-6, versus 25% in the sub-therapeutic cohort, both groups meeting the primary endpoint. An overall survival benefit was seen, with a tail of more than 40% at 3.7 years for the therapeutic-dose cohort, similar to historical data for pazopanib (Votrient®), a tyrosine kinase inhibitor; however, most patients in the VB-111 study had tumors that previously had progressed on pazopanib or other kinase inhibitors. As of December 31, 2016, we had studied VB-111 in over 200 patients and have observed it to be well-tolerated. In December 2015, we have been granted a US composition of matter patents that provides intellectual property protection for VB-111 in the US until October 2033 before any patent term extension.
In October 2017, we announced the opening of our new gene therapy manufacturing plant in Modiin, Israel. This plant can be the commercial facility for production of VB-111, if approved. The Modiin facility is the first commercial-scale gene therapy manufacturing facility in Israel and currently one of the largest gene-therapy designated ones in the world (20,000 sq. ft.).
In November 2017, we signed an exclusive license agreement with NanoCarrier Co., Ltd. (TSE Mothers:4571) for the development, commercialization, and supply of VB-111 in Japan. VBL retains rights to VB-111 in the rest of the world. Under terms of the agreement, VBL has granted NanoCarrier an exclusive license to develop and commercialize VB-111 in Japan for all indications. VBL will supply NanoCarrier with VB-111, and NanoCarrier will be responsible for all regulatory and other clinical activities necessary for commercialization in Japan. In exchange, we received an up-front payment of $15 million, and are entitled to receive greater than $100 million in development and commercial milestone payments. VBL will also receive tiered royalties on net sales in the high-teens.
Based on support from pre-clinical data, which we presented at the American Society of Gene & Cell Therapy (ASGCT) conference in May 2017, and the histological data in ovarian cancer showing ability of VB-111 to turn an immunologically “cold” to “hot” tumor, an exploratory Phase 2 study for VB-111 in combination a checkpoint inhibitor, is planned in gastrointestinal tumors. Launch of this trial is expected in theA second half of 2019.
Our Strategy
Our goal is to become a leading biopharmaceutical company focused on discovering, developing and commercializing innovative therapeutics that leverage our proprietary technologies for oncology indications. We intend to achieve this goal by pursuing the following strategies:
We believe VB-111 has the potential for applications in various solid tumors, and that the outcome of the GLOBE study in rGBM will not necessarily have implications on the prospects for VB-111 in other regimens or tumor types. A new investigator-sponsored Phase 2 trial evaluating VB-111 in rGBM patients is expected to be launched in the first half of 2019.
We have conducted Phase 2 clinical trials of VB-111 in both ovarian and thyroid cancer, with positive results. We intend to continue development of VB-111 for platinum-resistant ovarian cancer, and launched a Phase 3 study for this indication in December 2017. Anpre-planned interim analysis in the OVAL trial is expectedwas conducted and the DSMC reviewed unblinded OS data from the first 100 enrolled subjects with a follow-up of at least three months. The committee also looked at the CA-125 response rate and safety information. The DSMC recommended that the study continue as planned. Additional DSMC meetings have been conducted in year-end 2019.2021 and 2022, after the randomization of 200, 300, and 370 patients, respectively, and the committee found no safety issues with the trial and recommended its continuation as planned. We intendexpect to advance VB-111 to additional cancer indications, either independently or through investigator-sponsored studies or strategic collaborations. Accordingly, the launch of an exploratory Phase 2 study for VB-111 in combination a checkpoint inhibitor in gastrointestinal tumors is expectedread out topline PFS primary endpoint results in the second half of 2019.
As we advance our pipeline of anti-cancer product candidates, we will evaluate opportunities to selectively form collaborative alliances for our non-oncology assets to expand our capabilities2022 and accelerate the development and commercialization of our oncology products. Accordingly, in March 2019, we announced a strategic exclusive option license agreement with one of the world-leading European animal health companies, for the development of VB-201 for veterinary use. We retain the VB-201 rights for treatment of humans, worldwide, as well as the global rights for other Lecinoxoids and VB-600 candidates. We engage in conversations with third parties to evaluate such potential collaborations on an ongoing basis.
We previously manufactured clinical quantities of VB-111 at our facility in Or-Yehuda, Israel and through a third party in the United States. In October 2017, we announced the opening of our new gene therapy manufacturing plant in Modiin, Israel. This plant can be the first commercial facility for production of VB-111 if it receives regulatory approval. On the longer term, we intend to have more than one manufacturing site for VB-111, if regulatory approved.
Our Product Candidates and Technology
The following table summarizes the status of pipeline:

Our VTS Platform
Overview
Our innovative, proprietary VTS platform technology enables systemic administration of gene therapy to either destroy or promote angiogenic blood vessels. VTS is both tissue- and condition-specific, allowing for targeted and limited gene expression in endothelial cells, the thin layer of cells that lines the interior surface of blood vessels undergoing angiogenesis.
Our VTS platform technology comprises three components, a viral vector, a promoter and a transgene:
1. Viral vector-a modified virus that is used as a delivery vehicle to distribute the promoter and the transgene throughout the body.
2. Promoter-our proprietary, genetically modified promoter, called PPE-1-3X, that specifically targets the endothelial cells of angiogenic blood vessels. When present in these cells, the promoter initiates the expression of the transgene.
3. Transgene-a genetic sequence designed to yield a specific biologic effect, the expression of which is directed by PPE-1-3X. The particular transgene will vary depending on the therapeutic objectives of the product candidate.
Once the gene therapy has reached the angiogenic blood vessels, the PPE-1-3X promoter activates expression of the transgene to produce a desired RNA or protein in the endothelial cells of those vessels. For oncology applications, the transgene selected is designed to destroy angiogenic blood vessels that feed solid tumors. For other potential applications, such as the treatment of ischemia, a different transgene can be selected that is designed to promote the development of angiogenic blood vessels instead of their destruction.
VB-111 (ofranergene obadenovec)
VB-111 is a unique biologic agent that uses a dual mechanism to target solid tumors. Its mechanism combines blockade of tumor vasculature with an anti-tumor immune response.
Based on a non-integrating, non-replicating, Adeno 5 vector, VB-111 utilizes VBL’s proprietary Vascular Targeting System (VTS) to target the tumor vasculature for cancer therapy. We designed VB-111 to address oncology indications, specifically solid tumors, by selectively targeting the blood vessels required for tumor growth and encouraging the programmed cell-death process, or apoptosis, of cells in those blood vessels. VB-111 is administered intravenously. PPE-1-3X is activated specifically in angiogenic endothelial cells and regulates a transgene consisting of a combination of two gene sequences known as Fas and TNFR1. When expressed, the transgene produces a unique pro-apoptotic protein, the Fas-TNFR1 chimera, that interacts with a native inflammatory molecule, Tumor Necrosis Factor, or TNF- alpha, and results in the destruction of newly formed or immature blood vessels. When activated by PPE-1-3X, specifically in angiogenic endothelial cells, this combination enables VB-111 to reduce tumor growth in a highly targeted manner.
In addition, VB-111 induces a specific anti-tumor immune response. In 2004, we published preclinical data, which suggested that there is an immune inflammatory response to the presence of the viral vector and the Fas-TNFR1 chimera. Further support for a potential role of the immune system as part of VB-111’s mechanism of action came from an observation that patients who developed fever as a response to VB-111 administration, at least once along the treatment course, had a survival benefit over those who did not experience post-dosing fever. Moreover, an immunotherapeutic effect was also observed in biopsies taken from ovarian cancer patients. Immunohistochemistry staining showed regions of apoptotic cancer cells and infiltration of cytotoxic CD8 T-cells following treatment with VB-111.
In November 2017, at the Society for Neuro-Oncology conference, we presented data, which support the relationship between VB-111’s novel dual immuno-oncology and vascular targeting mechanisms of action to overall survival, and show that molecular and radiographic biomarkers may serve as predictors of clinical benefit.
VB-111’s mechanism of action is illustrated below:
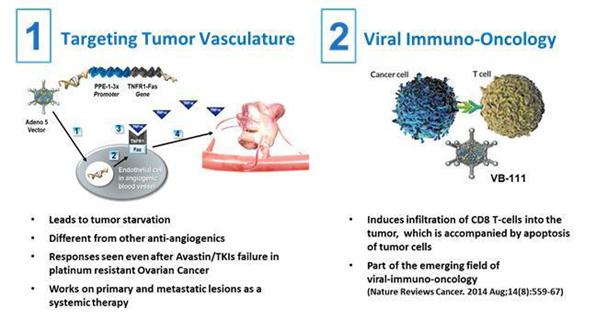
Unlike anti-VEGF agents (such as Avastin®) or tyrosine-kinase inhibitors (TKIs), VB-111 does not aim to block a specific pro-angiogenic pathway; instead, it uses an angiogenesis-specific sensor (VBL’s PPE-1-3x proprietary promoter) to specifically induce cell death in angiogenic endothelial cells in the tumor milieu. This mechanism may retain activity regardless of baseline tumor mutations or the identity of the pro-angiogenic factors secreted by the tumor and shows activity even after failure of prior treatment with other anti-angiogenics. Moreover, VB-111 induces specific anti-tumor immune response, which is accompanied by recruitment of CD8 T-cells and apoptosis of tumor cells. We believe that this mode of action makes VB-111 less susceptible to resistance and, therefore, potentially applicable for a broader patient population than current therapies.
We have conducted pre-clinical studies in animal models of lung cancer, colon cancer, thyroid cancer, rGBM and melanoma. Based on those studies, and clinical results to date, we believe that VB-111 has anti-tumoral activity that may hold clinical promise and may be suitable for treatment of some solid tumors. We initially decided to focus on rGBM as our first indication because we expected the rapid kinetics of this disease would enable us to accumulate clinicalOS primary endpoint data in a short time, but we also advanced VB-111 to a randomized-controlled Phase 3 study in platinum-resistant ovarian cancer.2023.
VB-111 Clinical Programs- Overview
We initially studied VB-111 in a Phase 1 “all comers” trial involving patients with multiple types of advanced metastatic cancer types, including thyroid cancer, neuroendocrine cancer, renal cell carcinoma and lung cancer. In that trial, VB-111 was well-tolerated and showed a dose- dependent extension in median overall survival across a range of tumor types. Based on these results, we decided to proceed with the development of VB-111 for the lead indication of rGBM, as well as to investigate VB-111 as a monotherapy for the treatment of thyroid cancer and, in combination with chemotherapy, for ovarian cancer. We have an open IND for VB-111 with the Office of Cellular, Tissue, and Gene Therapeutics within FDA’s Center for Biologics Evaluation and Research.
VB-111Ofra-vec Clinical Program in GBMrGBM
Glioblastoma, or GBM, is a brain cancer that affects approximately 12,000 to 13,000 newly diagnosed people each year in the United States. It is a devastating, rapidly progressing tumor, with a median time from diagnosis to the patient’s death of 12 to 15 months. In recurrent glioblastoma,GBM, or rGBM, treatment consists of both symptomatic and palliative therapies. However, with currently available therapies, glioblastoma typically remains fatal within a very short period of time.
We conducted an open-label Phase 2 trial in rGBM, which was originally initiated as an adaptive Phase 1/2 trial. The trial was intended to evaluate the safety and efficacy of VB-111, both by itself and in combination with bevacizumab, an anti-angiogenesis agent approved by the FDA for use in rGBM. In this trial, patients were initially dosed with VB-111 alone. After disease progression on VB-111 alone, they receive either bevacizumab alone as standard of care, or, in a second cohort, a combination of VB-111 and bevacizumab. Disease progression was defined as a worsening of the patient’s cancer with an increase of at least 25% in the overall mass of measurable tumors, the appearance of new tumors, the worsening of non-measurable tumors since beginning of treatment, a need for an increased dose of corticosteroids or clinical deterioration.
Our Phase 2 trial results include 46 patients with rGBM treated with VB-111; upon disease progression, 23 patients were treated with VB-111 in combination with Avastin® , and 22 received Avastin® alone. One patient, who achieved a complete response, is still stable on VB-111 alone for more than 5 years as of January 2019, and was included in the continuous exposure cohort. The median number of bi-monthly VB-111 doses was four for the cohort, which was treated with VB-111 through progression, versus one in the limited exposure cohort (average of 4.7 vs. 2.2, respectively). Continuous exposure to VB-111 demonstrated significant improvement in overall survival, with median overall survival of 59.1 weeks (414 days), compared to 31.9 weeks (223 days) in patients on limited VB-111 exposure (p=0.043), meeting the primary endpoint of the trial. Two complete responses and five partial responses were seen in the VB-111 continuous exposure cohort (n=24), compared to only two partial responses in VB-111 limited exposure cohort (n=22). VB-111 was found to be well tolerated.
Trial data also showed that VB-111 may induce an immuno-therapeutic effect. Of the 46 patients who received VB-111, 25 patients experienced a fever post-dosing of VB-111 at least once, while 21 patients did not. Patients with fevers demonstrated increased overall survival of 16 months, compared to patients without fevers, who had a median overall survival of 8.5 months (p=0.03). Additional biomarkers analyses presented at the SNO conference in November 2017 have demonstrated that in addition to fever, VB-111 is also associated with immune-mediated responses, including secretion of immune-stimulatory cytokines that correlate with OS, further supporting a role of the immune system as part of VB-111’s dual mechanism of action.
In June 2016 at the ASCO conference, we presented clinical data that demonstrate a significant overall survival benefit in rGBM patients receiving VB-111 compared with historical Avastin® meta-analysis data. In the Phase 2 VB-111 trial, the median overall survival of patients who received continuous exposure of VB-111 in combination with Avastin was 59.1 weeks. This is compared to 32.1 weeks in the pooled data from the 8 studies in the meta-analysis (p=0.0295; Hazard Ratio 0.62, 95% CI: 0.40-0.96). Median survival ranged from 26.0 weeks to 45.7 weeks in the meta-analysis. Overall survival at 12 months for patients on continuous exposure of VB-111 was 57%, compared with 24% overall survival (range 16%-38%) for the pooled Avastin® treated rGBM data (p=0.03).period.
In 62 patients with rGBM, the most frequent toxicity was self-limited fever, starting several hours post therapy and usually resolving within 24 hours and controlled with anti-pyretics. There were 22 adverse events classified as grade 3 or higher, of which 7 were considered possibly related to VB-111 including asthenia, pyrexia, brain edema, depressed consciousness, pulmonary embolism, or PE (in a patient with PE prior to enrollment in the trial) and hypertension. Safety results were reviewed five times by the trial Data and Safety Monitoring Board, as well as by the FDA, without safety concerns. Based on interim Phase 2 data of VB-111 instudy for rGBM, the FDA has allowed VBL to launch a Phase 3 study of VB-111 in rGBM patients even prior to the completion of the Phase 2 trial.
In June 2016 at the ASCO conference, we presented clinical data that demonstrate a significant overall survival benefit in rGBM patients receiving VB-111 compared with historical Avastin® meta-analysis data. In the Phase 2 VB-111 trial, the median overall survival of patients who received continuous exposure of VB-111 in combination with Avastin was 59.1 weeks. This is compared to 32.1 weeks in the pooled data from the 8 studies in the meta-analysis (p=0.0295; Hazard Ratio 0.62, 95% CI: 0.40-0.96). Median survival ranged from 26.0 weeks to 45.7 weeks in the meta-analysis. Overall survival at 12 months for patients on continuous exposure of VB-111 was 57%, compared with 24% overall survival (range 16%-38%) for the pooled Avastin® treated rGBM data (p=0.03).
VBL’s pivotal Phase 3 GLOBE study was conducted under a Special Protocol Assessment (SPA) granted by the FDA, with full endorsement by the Canadian Brain Tumor Consortium (CBTC). Enrollment in the study, 256 patients in total, started in August 2015 and has been completed in December 2016, five months ahead of schedule.
Three safety reviews were conducted during the GLOBE trial, by the independent Data Safety Monitoring Committee (DSMC). The DSMC is an independent multidisciplinary group that conducts detailed review of un-blinded study data, discusses potential safety concerns and provides recommendations regarding trial continuation. In December 2016, we announced that the independent Data Safety Monitoring Committee (DSMC) met to conduct its first safety review of the Phase 3 GLOBE Study investigating ofranergene obadenovec (VB-111) in recurrent glioblastoma (rGBM). The committee reviewed the GLOBE safety data collected through a cutoff date in September 2016, and did not find any adverse events that would be cause for concern. As a result, the DSMC recommended that the study continue as planned. In April 2017, we announce that the committee reviewed the GLOBE safety data collected through a cutoff date in March 2017 and unanimously recommended that the study continue as planned. The Third and final DSMC review took place in September 2017. The committee reviewed the GLOBE safety data, including mortality data, collected through a cutoff date in August 2017 and stated that they did not identify any safety concerns. The DSMC confirmed that no additional follow up will be necessary. Accordingly, the DSMC unanimously recommended that the study continue as planned, to completion.
On March 8, 2018 we announced top-line data from the GLOBE study. These results show that the GLOBE study did not meet its pre-specified primary endpoint of overall survival (OS), or the secondary endpoint of progression-free-survival (PFS). No new safety concerns associated with VB-111 have been identified in the GLOBE study.
In November 2018, full data from the GLOBE trial were presented at the 2018 Society for Neuro-Oncology Annual Meeting by Dr. Timothy Cloughesy, MD, Professor of Clinical Neurology and Director of the Neuro-Oncology Program, UCLA School of Medicine and principal investigator of the GLOBE trial. Thorough analyses of the baseline risk factors of the Phase 2 and the Phase 3 treatment groups did not reveal any differences. Therefore, patient selection or different patient populations could not explain the difference between the results of the two studies. The analyses pointed to the regimen in GLOBE trial as the potential reason for its failure, indicating that priming with VB-111 without bevacizumab may be critical for the immune and vascular-disruptive/anti-angiogenic mechanism of VB-111 in rGBM. Following independent MRI analyses at UCLA which validated the overall clinical and survival benefit of VB-111 among patients who were primed with VB-111ofra-vec monotherapy that was continued after progression with the addition of Avastin showed a significantly longer OS (414 vs 223 days; HR 0.48; p=0.043) and PFS advantage (90 vs 60 days; HR 0.36; p=0.032) compared to a cohort of patients that had limited exposure to ofra-vec (monotherapy until progression). Full study results were published in a peer reviewed publication (Brenner et al., Neuro Oncol. 2019). Objective radiographic responses were seen during the ofra-vec monotherapy phase and responders exhibited specific imaging characteristics related to ofra-vec’s mechanism of action. A survival advantage was also seen in comparison to historic controls, with the percentage of patients living more than one year almost doubling from 24% to 57%.
We conducted the Phase 3 GLOBE study in rGBM comparing upfront concomitant administration of ofra-vec, without priming, and Avastin to Avastin monotherapy. The treatment did not improve OS and PFS outcomes in rGBM, which conflicted with the results seen in our prior Phase 2 study where an ofra-vec monotherapy priming regimen was used. The full study results were published in a peer-reviewed publication (Cloughesy et al. Neuro Oncol. 2019). We, and the paper’s authors, attribute the contradictory outcomes between the Phase 2 and Phase 3 trials as being related to the lack of ofra-vec monotherapy priming in the GLOBE study, providing clinical, mechanistic and radiographic support for this hypothesis. In-vivo preclinical data demonstrate that Avastin appears to neutralize the effect of ofra-vec by inactivating the angiogenic process that ofra-vec depends on.
A new Phase 2 clinical trial investigating ofra-vec for the treatment of rGBM has since been initiated. The Phase 2 trial, sponsored by Dana-Farber Cancer Institute in collaboration with a new investigator-sponsored trial evaluating VB-111group of top neuro-oncology medical centers in the United States, is investigating neo-adjuvant and adjuvant treatment with ofra-vec in rGBM patients is expectedundergoing a second surgery and looks to be launchedreplicate the Phase 2 results in rGBM, utilizing the ofra-vec monotherapy priming regimen that was not used in the first half of 2019.Phase 3 GLOBE study. Enrollment in this study is ongoing and we expect preliminary data from this study in 2022. We do not believe that the results and confounding factors from the GLOBE study will necessarily have implications on the prospects for ofra-vec in other regimens or tumor types.
VB-111 ClinicalOfra-vec Program in OvarianColorectal Cancer
In addition to GBM, based on observations from early clinical trials, we have advanced VB-111 into tumor specific, repeat-dose trials,Ofra-vec is also being evaluated in ovarian cancer and thyroid cancer.
Ovarian cancer was diagnosed in approximately 22,000 American women in 2013, according to the National Cancer Institute. In ovarian cancer, clinical trials of bevacizumab, which, like VB-111, is an anti-angiogenic agent, demonstrated some improvement in progression free survival in women with high-risk advanced ovarian cancer. Therefore, we conducted a Phase 1/2 clinical trial in ovarian cancer using VB-111 in combination with paclitaxel,Opdivo (nivolumab), an immune checkpoint inhibitor, for the treatment of mCRC, under a common chemotherapeutic agent.
This trial was designed as a Phase 1/2 dose escalation study. The primary objectives were to evaluate the safetycooperative research and tolerability and identify dose limiting toxicity in combination of VB-111 and weekly paclitaxel; and explore the efficacy in an expanded cohort of the optimally tolerated dose of combination VB-111 and weekly paclitaxel, based on RECIST response, CA-125 response, progression free survival (PFS) and overall survival (OS) in patients with recurrent platinum-resistant ovarian cancer.
Twenty one patients with recurrent platinum-resistant Müllerian/ovarian cancer were enrolled at Massachusetts General Hospital and Dana Farber Cancer Institute, and received up to 7 doses of treatment. Patients were treated in two consecutive cohorts: Low Dose Treatment (n=4, 3x1012 VPs + 40mg/80 mg paclitaxel) or a Therapeutic Dose (n=17, 1x1013 VPs + 80 mg paclitaxel). All patients had measurable disease, with a grade at diagnosis of: 1A (1, 5%), 1B (1, 5%), 1C (1, 5%); IIIC (12, 57%); or IV (6, 29%). The patients included in the study were of particularly adverse prognosis as 48% of the patients were primary platinum refractory and 52% had tumors that failed to respond to prior anti-angiogenic agents, including Avastin.
Trialresults showed a significant increase in overall survival at the therapeutic dose of VB-111 vs. the low dose level (498 vs. 172 days, p=0.03). About 60% of patients on the therapeutic dose had a response, as defined by a 50% reduction in CA-125. Durable RECIST responses and disease stabilizations were seen. This represents an approximate doubling in response rate, compared to historical data with ovarian cancer patients treated with a combination of Avastin® and chemotherapy in the AURELIA trial which reported CA-125 response in 11.6% of patients treated with chemotherapy and 31.8% CA-125 response in ovarian cancer patients treated with a combination of chemotherapy and Avastin®.
An immunotherapeutic effect was also observed in biopsies taken from patients. H&E and immunohistochemistry staining showed regions of apoptotic cancer cells and infiltration of cytotoxic CD8 T-cells following treatment with VB-111. VB-111 was found to be safe and well tolerated. Toxicity was similar to what would be expected with antiangiogenics and taxanes in this patient population. Eight serious adverse events were reported, 2 were considered by the investigator to be possibly related to be VB-111. No dose limiting toxicities were reported at any dose level.
In December 2016 we had an end-of-Phase 2 meeting with the FDA to discuss the clinical path of VB-111 in ovarian cancer. We reacheddevelopment agreement with the FDA on our clinical plan to proceed to a Phase 3 potential registration study of VB-111 in platinum-resistant patients. We intend to advance VB-111U.S. National Cancer Institute, or NCI. NCI serves as the sponsor for this patient population, for which most current therapies failtrial. The open label exploratory Phase 2 trial will investigate if priming with ofra-vec can recruit immune cells into the tumor and turn the colorectal tumors from being immunologically “cold” to prolong patient survival, and in December 2017 announce the enrollment“hot.” The purpose of the first patientstudy is to determine whether the immune recruitment seen in other organ tumors from previous ofra-vec clinical trials can be replicated in the OVAL potential registration Phase 3 studygut immune system which is in continuous contact with viruses, bacteria and foreign proteins and may behave differently than the rest of VB-111the body. Preliminary data from this trial are expected in platinum-resistant ovarian cancer. The OVAL study is conducted in collaboration with the Gynecologic Oncology Group (GOG) Foundation, Inc., a leading organization for research excellence in the field of gynecologic malignancies.2022 and will determine whether we should further explore this indication.
In March 2019, at the Society of Gynecologic Oncology conference, we presented data from of ovarian cancer patients treated with VB-111 and paclitaxel, showing the potential of VB-111 to stimulate the immune system and drive immune cells to infiltrate the tumor microenvironment.
The randomized, controlled, double-blind, Phase 3 OVAL study in recurrent platinum-resistant ovarian cancer has been designed to enroll up to 400 adult patients at approximately 75 clinical sites in the United States and Israel. Patients are randomized 1:1 to VB-111 (1x1013 VPs once every 8 weeks) in combination with chemotherapy (80mg/m2 paclitaxel once weekly), or to placebo with chemotherapy. The primary endpoint is overall survival. Additional endpoints include objective response rate (ORR), progression free survival (PFS), CA-125, RECIST 1.1 response and patient reported outcome measures. Given the GLOBE trial results, we added an interim analysis to the OVAL study, which is expected to occur in year-end 2019.
VB-111Ofra-vec Program in Thyroid Cancer
We conducted an exploratory Phase 2 clinical trial to evaluate the safety and efficacy of VB-111ofra-vec in advanced thyroid cancer. According to the National Cancer Institute, there are an estimated 535,000Thyroid cancer affects approximately 44,000 newly diagnosed people currently living with thyroid cancereach year in the United States, with an estimated 60,000 new cases of thyroid cancer each year.States. Most cases can be treated by surgery and radioactive iodine. If radioactive iodine is ineffective, other treatments are prescribed, such tyrosine kinase inhibitorsas TKIs and systemic chemotherapy. However, if such treatments are unsuccessful, the therapeutic options for patients are currently very limited. There are an estimated 2,000 annual deaths in the U.S. as a result of the disease. This subset of patients has ana significant unmet need for novel therapeutic options such as VB-111.options. The estimated number of U.S. deaths of thyroid cancer in 2021 was 2,200.
OurWe conducted a Phase 2 open-label, dose-escalating studytrial which enrolled patients with advanced, recently-progressive, differentiated thyroid cancer that was unresponsive to radioactive iodine, in two cohorts. Most patients had tumors that had not responded to multiple therapies prior to enrollment, including radiation and kinase inhibitors. In the first cohort, thirteen patients received a single intravenous infusion of VB-111ofra-vec at a sub-therapeutic dose of 3X1012 viral particles (VPs).3X10e12 VP. The second cohort included seventeen patients, who received VB-111ofra-vec at a therapeutic dose of 1013 VPs10e13 VP every two months until disease progression. One patient proceeded from a single low dose to later receive later multiple high doses at progression and was included in both groups (for PFS analysis only).
On February 21, 2017 we announced full data from this trial. The primary endpoint of the trial was defined as 6-month progression-free-survival (PFS-6) of 25%, was met with a dose response.PFS. Forty-seven percent (47%; 8/17) of patients in the therapeutic-dose cohort reached PFS-6, versus 25% (4/12) in the sub-therapeutic cohort, both groups meeting the primary endpoint.demonstrating a dose response. Reduction in tumor measurement after the first dose was seen in 44% (7/16) of patients in the therapeutic-dose cohort, compared to 9% (1/11) in the sub-therapeutic-dose cohort. An overall survivalOS benefit was seen with a tail of more than 40% at 3.7 years for the therapeutic-dose cohort (mOS(median OS of 684 days). This is similar to historical data for pazopanib (Votrient® ),(Votrient), a tyrosine kinase inhibitor;TKI; however, most patients in the VB-111 studyPhase 2 trial had tumors that previously had progressed on pazopanib or other kinase inhibitors. VB-111 was observed to be well-tolerated in this study, with no signs of clinically significant safety issues.
| 39 |
We see these positiveencouraging data as additional proof-of-concept for VB-111ofra-vec in another advanced solid tumor, particularly important for investigating the therapeutic potential of VB-111ofra-vec even as monotherapy. Our primary focus continues to be the advancement of VB-111ofra-vec towards commercialization, if approved, in ovarian cancer. Further clinical development of VB-111ofra-vec for thyroid cancer may also be pursued, potentially with a strategic partner, or via an investigator-sponsored study.trial.
Additional VB-111 ProgramOfra-Vec in Other Solid Tumor Indications
We believe ofra-vec has potential to treat a wide range of solid tumors provided they contain either a vascular component, a lack of or minimal immune infiltration, or both. In addition to the encouraging data we have observed in our Phase 2 studies, we also conducted a Phase 1 “all comers” trial involving patients with multiple types of advanced metastatic cancer types, including medullary thyroid cancer, neuroendocrine cancer, renal cell carcinoma and lung cancer which showed a dose-dependent extension in median OS. We believe this Phase 1 trial provides a road map for potential indication expansion in new oncology indications.
Ofra-vec Partnership in Japan
In November 2017, we signed an exclusive license agreement with NanoCarrier Co., Ltd. (TSE Mothers:4571), or NanoCarrier, for the development, commercialization, and supply of ofra-vec in Japan. We retained rights to ofra-vec in the rest of the world. Under terms of the agreement, we granted NanoCarrier an exclusive license to develop and commercialize ofra-vec in Japan for all indications. We will supply NanoCarrier with ofra-vec, and NanoCarrier will be responsible for all regulatory and other clinical activities necessary for commercialization in Japan. Under the agreement, we are entitled to receive greater than $100 million in development and commercial milestone payments, in addition to tiered royalties on net sales in the high-teens.
VB-601 Program- Lead MTT Candidate
Our lead MTT candidate, VB-601, is an investigational proprietary monoclonal antibody that binds the MOSPD2 receptor, which we call the “mono-walk” receptor, and is engineered to specifically prevent monocytes from exiting the blood stream and traveling to inflamed tissues. Monocytes are one of the key cells types in inflammation and particularly implicated in being responsible for the chronicity of disease. VB-601 is designed to offer a novel and differentiated approach in the landscape of current anti-inflammatory agents, most of which target pro-inflammatory molecules and work through T and B lymphocytes but are not targeted to the monocyte cells.
We have conducted various ex-vivo and in-vivo pharmacology studies that demonstrate VB-601’s activity against a broad range of prevalent chronic inflammatory indications:
Based on support from pre-clinicalour preclinical in-vivo and human ex-vivo data, which we presented atbelieve VB-601 has potential utility in a wide range of immune-inflammatory diseases, such as multiple sclerosis (relapsing-remitting (RRMS) and progressive (PMS), rheumatoid arthritis (RA), psoriatic arthritis (PsA), non-alcoholic steatohepatitis, inflammatory bowel disease (including Crohn’s disease (CD) and ulcerative colitis (UC)) and other immune-inflammatory diseases. We had a successful pre-IND meeting with the American SocietyFDA regarding our development plan and have since completed IND-enabling toxicology studies that demonstrated a favorable tolerability profile that supports moving VB-601 into the clinic. We are conducting a cGMP manufacturing run of Gene & Cell Therapy (ASGCT) conferenceVB-601 and expect to initiate a first in May 2017, and the recently presented histological data in ovarian cancer showing ability of VB-111 to turn an immunologically “cold” to “hot” tumor, an exploratory Phase 2 study for VB-111 in combination a checkpoint inhibitor, is planned in gastrointestinal tumors. Launch of thishuman clinical trial is expected in the second half of 2019.2022.
| 40 |
Our Strategy
Our goal is to provide safe, effective and life-improving medicines to people living with cancer and immune-inflammatory diseases. We intend to achieve this goal by pursuing the following strategies:
| ● | Pursue regulatory approval for our lead oncology drug candidate, ofra-vec |
We believe ofra-vec has the potential for applications in various solid tumors. Currently, our focus is on the development of ofra-vec for platinum-resistant ovarian cancer. We recently completed enrollment in the OVAL Phase 3 registration-enabling trial and expect the topline readout of the PFS primary endpoint in the second half of 2022. If positive, we believe OVAL has the potential to support BLA submission, which we expect to file in the first half of 2023.
Additional VTS Pipeline candidatesOfra-vec is also being studied in Phase 2 clinical trials in rGBM and mCRC, with preliminary data expected in 2022. We also conducted a Phase 2 clinical trial of ofra-vec in thyroid cancer, with encouraging results. We intend to advance ofra-vec in additional cancer indications, either independently or through investigator-sponsored studies or strategic collaborations.
| ● | Selectively enter into licensing and collaboration arrangements to supplement our internal development capabilities and commercialize our products, if approved |
We will evaluate opportunities to selectively form collaborative alliances for our assets to expand our capabilities and accelerate their development and commercialization. We engage in conversations with third parties to evaluate such potential collaborations on an ongoing basis.
| ● | Expand our manufacturing capacity to support clinical trials and possible commercialization of ofra-vec |
We have an in-house gene therapy manufacturing plant in Modi’in, Israel. Our facility was certified by a European Union, or EU, Qualified Person, or QP, as being in compliance with EU Good Manufacturing Practices, or GMP. The facility is currently being used to manufacture product for our Phase 3 OVAL trial. We believe this plant can be the first commercial facility for production of ofra-vec if we receive regulatory approval. We intend to bring on at least one additional manufacturing site to support long term scale up and label expansion activities for ofra-vec.
Governmental Regulation
The FDA and other regulatory authorities at federal, state and local levels, as well as in foreign countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, recordkeeping, approval, advertising, promotion, marketing, post-approval monitoring and post-approval reporting of drugs and biologics. We, along with our vendors, CROs, clinical investigators and contract development and manufacturing organizations, or CDMOs, will be required to navigate the various preclinical, clinical, manufacturing and commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval of our product candidates. The process of obtaining regulatory approvals of drugs and biologics and ensuring subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources.
In the United States, the FDA regulates drug products under the Federal Food, Drug, and Cosmetic Act, or FD&C Act, and biologics under the FD&C Act and the Public Health Service Act, or PHSA, as amended, and their implementing regulations. Both drugs and biologics are also subject to other federal, state and local statutes and regulations. If we fail to comply with applicable FDA or other requirements at any time with respect to product development, clinical testing, approval or any other regulatory requirements relating to product manufacture, processing, handling, storage, quality control, safety, marketing, advertising, promotion, packaging, labeling, export, import, distribution, or sale, we may become subject to administrative or judicial sanctions or other legal consequences. These sanctions or consequences could include, among other things, the FDA’s refusal to approve pending applications, issuance of clinical holds for ongoing studies, suspension or revocation of approved applications, warning or untitled letters, product withdrawals or recalls, product seizures, relabeling or repackaging, total or partial suspensions of manufacturing or distribution, injunctions, fines, civil penalties or criminal prosecution.
Our VTS platform technology enables systemicproduct candidates must be approved for therapeutic indications by the FDA before they may be marketed in the United States. For drug product candidates regulated under the FD&C Act, FDA must approve an NDA. For biologic product candidates regulated under the FD&C Act and PHSA, FDA must approve a BLA. The process is similar and generally involves the following:
| ● | completion of extensive preclinical studies in accordance with applicable regulations, including studies conducted in accordance with good laboratory practice, or GLP, requirements; | |
| ● | completion of the manufacture, under cGMP conditions, of the drug substance and drug product that the sponsor intends to use in human clinical trials along with required analytical and stability testing; | |
| ● | submission to the FDA of an IND which must become effective before clinical trials may begin and must be updated annually and when certain changes are made; | |
| ● | approval by an institutional review board (IRB or independent ethics committee at each clinical trial site before each trial may be initiated; | |
| ● | performance of adequate and well-controlled clinical trials in accordance with applicable IND regulations, good clinical practice, or GCP, requirements and other clinical trial-related regulations to establish the safety and efficacy of the investigational product for each proposed indication; |
| 41 |
| ● | preparation and submission to the FDA of an NDA or BLA; | |
| ● | a determination by the FDA within 60 days of its receipt of an NDA or BLA to file the application for review; | |
| ● | satisfactory completion of one or more FDA pre-approval or pre-license inspections of the manufacturing facility or facilities where the product will be produced to assess compliance with cGMP requirements to assure that the facilities, methods and controls are adequate to preserve the drug or biological product’s identity, strength, quality and purity; | |
| ● | satisfactory completion of FDA audit of the clinical trial sites that generated the data in support of the NDA or BLA; | |
| ● | payment of user fees for FDA review of the NDA or BLA; and | |
| ● | FDA review and approval of the NDA or BLA, including, where applicable, consideration of the views of any FDA advisory committee, prior to any commercial marketing or sale of the product in the United States. |
Preclinical Studies and Clinical Trials for Drugs and Biologics
Before testing any drug or biologic in humans, the product candidate must undergo rigorous preclinical testing. Preclinical studies include laboratory evaluations of product chemistry, formulation and stability, as well as in vitro and animal studies to assess safety and in some cases to establish the rationale for therapeutic use. The conduct of preclinical studies is subject to federal and state regulation and requirements, including GLP requirements for safety/toxicology studies. The results of the preclinical studies, together with manufacturing information and analytical data, must be submitted to the FDA as part of an IND.
An IND is a request for authorization from the FDA to administer an investigational product to humans and must become effective before clinical trials may begin. The central focus of an IND submission is on the general investigational plan and the protocol(s) for clinical studies. The IND also includes the results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls, or CMC, information; and any available human data or literature to support the use of the investigational product. Some long-term preclinical testing may continue after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the conduct of the clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks, and imposes a full or partial clinical hold. FDA must notify the sponsor of the grounds for the hold and any identified deficiencies must be resolved before the clinical trial can begin. Submission of an IND may result in the FDA not allowing clinical trials to commence or not allowing clinical trials to commence on the terms originally specified in the IND. A clinical hold can also be imposed once a trial has already begun, thereby halting the trial until the deficiencies articulated by FDA are corrected.
In addition to the submission of an IND to the FDA before initiation of a clinical trial in the United States, certain human clinical trials involving recombinant or synthetic nucleic acid molecules are subject to oversight of IBC as set forth in the NIH Guidelines. Under the NIH Guidelines, recombinant and synthetic nucleic acids are defined as: (i) molecules that are constructed by joining nucleic acid molecules and that can replicate in a living cell (i.e., recombinant nucleic acids); (ii) nucleic acid molecules that are chemically or by other means synthesized or amplified, including those that are chemically or otherwise modified but can base pair with naturally occurring nucleic acid molecules (i.e., synthetic nucleic acids); or (iii) molecules that result from the replication of those described in (i) or (ii). Specifically, under the NIH Guidelines, supervision of human gene transfer trials includes evaluation and assessment by an IBC, a local institutional committee that reviews and oversees research utilizing recombinant or synthetic nucleic acid molecules at that institution. The IBC assesses the safety of the research and identifies any potential risk to public health or the environment, and such review may result in some delay before initiation of a clinical trial. While the NIH Guidelines are not mandatory unless the research in question is being conducted at or sponsored by institutions receiving NIH funding of recombinant or synthetic nucleic acid molecule research, many companies and other institutions not otherwise subject to the NIH Guidelines voluntarily follow them.
The clinical stage of development involves the administration of genethe product candidate to healthy volunteers or patients under the supervision of qualified investigators, who generally are physicians not employed by or under the trial sponsor’s control, in accordance with GCP requirements, which include the requirements that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameters and criteria to be used in monitoring safety and evaluating effectiveness. Each protocol, and any subsequent amendments to the protocol, must be submitted to the FDA as part of the IND. Furthermore, each clinical trial must be reviewed and approved by an IRB for each institution at which the clinical trial will be conducted to ensure that the risks to individuals participating in the clinical trials are minimized and are reasonable compared to the anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. The FDA, the IRB, or the sponsor may suspend or discontinue a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a DSMC. This group provides authorization for whether or not a trial may move forward at designated intervals based on access to certain data from the trial. There also are requirements governing the reporting of ongoing clinical trials and completed clinical trials to public registries. Information about clinical trials, including results for clinical trials other than Phase 1 investigations, must be submitted within specific timeframes for publication on www.ClinicalTrials.gov, a clinical trials database maintained by the National Institutes of Health.
A sponsor who wishes to conduct a clinical trial outside of the United States may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. If a foreign clinical trial is not conducted under an IND, FDA will nevertheless accept the results of the study in support of an NDA if the study was conducted in accordance with GCP requirements, and the FDA is able to validate the data through an onsite inspection if deemed necessary.
| 42 |
Clinical trials to evaluate therapeutic indications to support NDAs or BLAs for marketing approval are typically conducted in three sequential phases, which may overlap.
| ● | Phase 1—Phase 1 clinical trials involve initial introduction of the investigational product in a limited population of healthy human volunteers or patients with the target disease or condition. These studies are typically designed to test the safety, dosage tolerance, absorption, metabolism, distribution and excretion of the investigational product in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence of effectiveness. | |
| ● | Phase 2—Phase 2 clinical trials typically involve administration of the investigational product to a limited patient population with a specified disease or condition to evaluate the drug’s potential efficacy, to determine the optimal dosages and dosing schedule and to identify possible adverse side effects and safety risks. | |
| ● | Phase 3—Phase 3 clinical trials typically involve administration of the investigational product to an expanded patient population to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the investigational product and to provide an adequate basis for product approval and physician labeling. Generally, two adequate and well-controlled Phase 3 trials are required by the FDA for approval of an NDA or BLA. |
Post-approval trials, sometimes referred to as Phase 4 clinical trials or post-marketing studies, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication and are commonly intended to generate additional safety data regarding use of the product in a clinical setting. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of NDA or BLA approval.
Progress reports detailing the results of the clinical trials, among other information, must be submitted at least annually to the FDA. Written IND safety reports must be submitted to the FDA and the investigators fifteen days after the trial sponsor determines the information qualifies for reporting for serious and unexpected suspected adverse events, findings from other studies or animal or in vitro testing that suggest a significant risk for human volunteers and any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must also notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction as soon as possible but in no case later than seven calendar days after the sponsor’s initial receipt of the information.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the product candidate and finalize a process for manufacturing the drug product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and manufacturers must develop, among other things, methods for testing the identity, strength, quality and purity of the final drug product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
U.S. Marketing Approval for Drugs and Biologics
Assuming successful completion of the required clinical testing, the results of the preclinical studies and clinical trials, together with detailed information relating to the product’s CMC and proposed labeling, among other things, are submitted to the FDA as part of an NDA or BLA. An NDA is a request for approval to market a new drug for one or more specified indications and must contain proof of the drug’s safety and efficacy for the requested indications. A BLA is a request for approval to market a new biologic for one or more specified indications and must contain proof of the biologic’s safety, purity and potency for the requested indications. The marketing application is required to include both negative and ambiguous results of preclinical studies and clinical trials, as well as positive findings, together with detailed information relating to the product’s CMC, and proposed labeling, among other things. Data may come from company-sponsored clinical trials intended to test the safety and efficacy of a product’s use or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of the investigational drug, or the safety, purity and potency of the investigational biologic, to the satisfaction of the FDA. FDA must approve an NDA or BLA before a drug or biologic may be marketed in the United States.
The FDA reviews all submitted NDAs and BLAs to ensure they are sufficiently complete to permit substantive review before it accepts them for filing and may request additional information rather than accepting the NDA or BLA for filing. The FDA must make a decision on accepting an NDA or BLA for filing within 60 days of receipt, and such decision could include a refusal to file by the FDA. Once the submission is accepted for filing, the FDA begins an in-depth substantive review of the NDA or BLA. The FDA reviews an NDA or BLA to determine, among other things, whether the product is safe and effective for the indications sought and whether the facility in which it is manufactured, processed, packaged or held meets standards designed, including cGMP requirements, designed to assure and preserve the product’s continued identity, strength, quality and purity. Under the goals and polices agreed to by the FDA under the Prescription Drug User Fee Act, or PDUFA, the FDA targets ten months, from the filing date, in which to complete its initial review of a new molecular entity NDA or BLA and respond to the applicant, and six months from the filing date of a new molecular entity NDA or BLA for priority review. The FDA does not always meet its PDUFA goal dates for standard or priority NDAs or BLAs, and the review process is often extended by FDA requests for additional information or clarification.
Further, under PDUFA, as amended, each NDA or BLA must be accompanied by a substantial user fee. The FDA adjusts the PDUFA user fees on an annual basis. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on NDAs or BLAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
The FDA also may require submission of a REMS if it believes that a risk evaluation and mitigation strategy is necessary to ensure that the benefits of the drug outweigh its risks. A REMS can include use of risk evaluation and mitigation strategies like medication guides, physician communication plans, assessment plans, and/or elements to assure safe use, such as restricted distribution methods, patient registries, special monitoring or other risk-minimization tools.
| 43 |
The FDA may refer an application for a novel drug or biologic to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, which reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA or BLA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and are adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA or BLA, the FDA may inspect one or more clinical trial sites to assure compliance with GCP and other requirements and the integrity of the clinical data submitted to the FDA.
After evaluating the NDA or BLA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a Complete Response Letter. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter generally contains a statement of specific conditions that must be met in order to secure final approval of the NDA or BLA, except that where the FDA determines that the data supporting the application are inadequate to support approval, the FDA may issue the Complete Response Letter without first conducting required inspections, testing submitted product lots, and/or reviewing proposed labeling. In issuing the Complete Response Letter, the FDA may require additional clinical or preclinical testing or recommend other actions, such as requests for additional information or clarification, that the applicant might take in order for the FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications.
Even if the FDA approves a product, depending on the specific risk(s) to be addressed it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess a product’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes, and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Orphan Drug Designation and Exclusivity
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug or biologic intended to treat a rare disease or condition, which is a disease or condition with either a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 individuals in the United States when there is no reasonable expectation that the cost of developing and making the product available in the United States for the disease or condition will be recovered from sales of the product. Orphan drug designation must be requested before submitting an NDA or BLA. After the FDA grants orphan drug designation, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process, though companies developing orphan products are eligible for certain incentives, including tax credits for qualified clinical testing and waiver of application fees.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to a seven-year period of marketing exclusivity during which the FDA may not approve any other applications to market the same therapeutic agent for the same indication, except in limited circumstances, such as a subsequent product’s showing of clinical superiority over the product with orphan exclusivity or where the original applicant cannot produce sufficient quantities of product. Competitors, however, may receive approval of different therapeutic agents for the indication for which the orphan product has exclusivity or obtain approval for the same therapeutic agent for a different indication than that for which the orphan product has exclusivity. Orphan product exclusivity could block the approval of one of our products for seven years if a competitor obtains approval for the same therapeutic agent for the same indication before we do, unless we are able to demonstrate that our product is clinically superior. If an orphan designated product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan exclusivity. Further, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or the manufacturer of the approved product is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
Expedited Development and Review Programs for Drugs and Biologics
The FDA maintains several programs intended to facilitate and expedite development and review of new drugs and biologics to address unmet medical needs in the treatment of serious or life-threatening diseases or conditions. These programs include fast track designation, breakthrough therapy designation, priority review and accelerated approval, and the purpose of these programs is to either destroyexpedite the development or review of important new drugs and biologics to get them to patients more quickly than standard FDA review timelines typically permit.
A new drug or biologic is eligible for fast track designation if it is intended to treat a serious or life-threatening disease or condition and demonstrates the potential to address unmet medical needs for such disease or condition. Fast track designation applies to the combination of the product candidate and the specific indication for which it is being studied. Fast track designation provides increased opportunities for sponsor interactions with the FDA during clinical development, in addition to the potential for rolling review once a marketing application is submitted. Rolling review means that the FDA may review portions of the marketing application before the sponsor submits the complete application.
| 44 |
In addition, a new drug or biologic may be eligible for breakthrough therapy designation if it is intended to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug or biologic, alone or in combination with one or more other drugs or biologics, may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Breakthrough therapy designation provides all the features of fast track designation in addition to intensive guidance on an efficient product development program beginning as early as Phase 1, and FDA organizational commitment to expedited development, including involvement of senior managers and experienced review staff in a cross-disciplinary review, where appropriate.
Any product submitted to the FDA for approval, including a product with fast track or breakthrough therapy designation, may also be eligible for additional FDA programs intended to expedite the review and approval process, including priority review designation and accelerated approval. A product is eligible for priority review, once an NDA or BLA is submitted, if the product that is the subject of the marketing application has the potential to provide a significant improvement in safety or effectiveness in the treatment, diagnosis or prevention of a serious disease or condition. Under priority review, the FDA’s goal date to take action on the marketing application is six months compared to ten months for a standard review. Products are eligible for accelerated approval if they can be shown to have an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or an effect on a clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality, which is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments.
Accelerated approval is usually contingent on a sponsor’s agreement to conduct, in a diligent manner, adequate and well-controlled additional post-approval confirmatory studies to verify and describe the product’s clinical benefit. The FDA may withdraw approval of a drug or an indication approved under accelerated approval if, for example, the confirmatory trial fails to verify the predicted clinical benefit of the product. In addition, for products being considered for accelerated approval, the FDA generally requires, unless otherwise informed by the Agency, that all advertising and promotional materials intended for dissemination or publication within 120 days of marketing approval be submitted to the agency for review during the pre-approval review period. After the 120-day period has passed, all advertising and promotional materials must be submitted at least 30 days prior to the intended time of initial dissemination or publication.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or the time period for FDA review or approval may not be shortened. Furthermore, fast track designation, breakthrough therapy designation, priority review and accelerated approval do not change the scientific or medical standards for approval or the quality of evidence necessary to support approval, though they may expedite the development or review process.
Pediatric Information and Pediatric Exclusivity
Under the Pediatric Research Equity Act, or PREA, as amended, certain NDAs and BLAs and certain NDA and BLA supplements must contain data that can be used to assess the safety and efficacy of the product candidate for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of pediatric data or full or partial waivers. The FD&C Act requires that a sponsor who is planning to submit a marketing application for a product candidate that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration submit an initial Pediatric Study Plan, or PSP, within 60 days of an end-of-Phase 2 meeting or, if there is no such meeting, as early as practicable before the initiation of the Phase 3 or Phase 2/3 study. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. The FDA and the sponsor must reach an agreement on the PSP. A sponsor can submit amendments to an agreed-upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from preclinical studies, early phase clinical trials and/or other clinical development programs. Unless otherwise required by regulation, PREA does not apply to a drug or biologic for an indication for which orphan designation has been granted, except that PREA will apply to an original NDA or BLA for a new active ingredient that is orphan-designated if the drug or biologic is a molecularly targeted cancer product intended for the treatment of an adult cancer and is directed at a molecular target that FDA determines to be substantially relevant to the growth or progression of a pediatric cancer.
A drug or biologic can also obtain pediatric market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study.
U.S. Post-Approval Requirements for Drugs and Biologics
Drugs and biologics manufactured or distributed pursuant to FDA approvals are subject to continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, reporting of adverse experiences with the product, complying with promotion and advertising requirements, which include restrictions on promoting products for unapproved uses or patient populations (known as “off-label use”) and limitations on industry-sponsored scientific and educational activities. Although physicians may prescribe approved products for off-label uses, manufacturers may not market or promote angiogenic blood vessels. Beyond VB-111, wesuch uses. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, including not only by company employees but also by agents of the company or those speaking on the company’s behalf, and a company that is found to have generated additional product candidates which utilize the same vector and promoter as in VB-111, yet comprise alternative functional transgenes. VB-511 is an anti-angiogenic candidate, while VB-211 and VB-411 are pro-angiogenic candidates thatimproperly promoted off-label uses may be employedsubject to significant liability, including investigation by federal and state authorities. Failure to comply with these requirements can result in, among other things, adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties, including liabilities under the False Claims Act where products are obtain reimbursement under federal health care programs. Promotional materials for ischemic conditions like peripheral vascular disease. All three candidates have demonstrated pre-clinical efficacyapproved drugs and biologics must be submitted to the FDA in conjunction with their first use or first publication. Further, if there are ready for toxicology.any modifications to the drug or biologic, including changes in indications, labeling or manufacturing processes or facilities, the applicant may be required to submit and obtain FDA approval of a new NDA or BLA or NDA or BLA supplement, which may require the development of additional data or preclinical studies and clinical trials.
| 45 |
Our Lecinoxoid Platform TechnologyThe FDA may impose a number of post-approval requirements as a condition of approval of an NDA or BLA. For example, the FDA may require post-market testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization. In addition, manufacturers and their subcontractors involved in the manufacture and distribution of approved drugs and biologics are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including cGMPs, which impose certain procedural and documentation requirements on sponsors and their CMOs. Changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting requirements upon us and any third party manufacturers that a sponsor may use. Manufacturers and manufacturers’ facilities are also required to comply with applicable product tracking and tracing requirements. Accordingly, manufacturers must continue to expend time money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance. Failure to comply with statutory and regulatory requirements may subject a manufacturer to possible legal or regulatory action, such as warning letters, suspension of manufacturing, product seizures, injunctions, civil penalties or criminal prosecution. There is also a continuing, annual program user fee for any marketed product.
Our proprietary Lecinoxoid platform technology comprisesThe FDA may withdraw approval of a familyproduct if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of orally administered small molecules designedpreviously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to modulatecomply with regulatory requirements, may result in revisions to the body’s inflammatory response. Lecinoxoids are compoundsapproved labeling to add new safety information, requirements for post-market studies or clinical trials to assess new safety risks, or imposition of distribution or other restrictions under a REMS. Other potential consequences include, among other things:
| ● | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; | |
| ● | the issuance of safety alerts, Dear Healthcare Provider letters, press releases or other communications containing warnings or other safety information about the product; | |
| ● | fines, warning letters or holds on post-approval clinical trials; | |
| ● | refusal of the FDA to approve applications or supplements to approved applications, or suspension or revocation of product approvals; | |
| ● | product seizure or detention, or refusal to permit the import or export of products; | |
| ● | injunctions or the imposition of civil or criminal penalties; and | |
| ● | consent decrees, corporate integrity agreements, debarment or exclusion from federal healthcare programs; or | |
| ● | mandated modification of promotional materials and labeling and issuance of corrective information. |
United States Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA approval of our product candidates, some of our United States patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit restoration of the patent term of up to five years as compensation for patent term lost during the FDA regulatory review process. Patent term restoration, however, cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date and only those claims covering such approved drug product, a method for using it or a method for manufacturing it may be extended. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA or BLA plus the time between the submission date of an NDA or BLA and the approval of that are structurallyapplication, except that the review period is reduced by any time during which the applicant failed to exercise due diligence. Only one patent applicable to an approved drug is eligible for the extension and functionally similarthe application for the extension must be submitted prior to naturally occurring molecules, known as oxidized phospholipids, which possess immune modulating anti-inflammatory properties, modifiedthe expiration of the patent. The U.S. PTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we may apply for restoration of patent term for our currently owned or licensed patents to enhance stabilityadd patent life beyond a patent’s current expiration date, depending on the expected length of the clinical trials and activity. We believe that Lecinoxoids hold significant promiseother factors involved in their abilitythe filing of the relevant NDA or BLA.
Marketing exclusivity provisions under the FD&C Act also can delay the submission or the approval of certain applications. The FD&C Act provides a five-year period of non-patent marketing exclusivity within the United States to treatthe first applicant to gain approval of an NDA for a range of chronic immune-based inflammatory diseases.
The inflammatory responsenew chemical entity. A drug is a complex physiologic process balancing both pro- and anti- inflammatory componentsnew chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an Abbreviated New Drug Application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FD&C Act also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that interact intimatelywere conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions of use associated with the body’s immune system. Oxidized phospholipids are instrumental innew clinical investigations and does not prohibit the interplayFDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of these components that maintain equilibrium. Whena full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the inflammatory response is not adequately balanced, excess inflammation resultspreclinical studies and may cause both acuteadequate and chronic disease states.well-controlled clinical trials necessary to demonstrate safety and effectiveness.
The Lecinoxoid platform seeks to harness the ability of oxidized phospholipids to regulate and attenuate key immune-inflammatory signaling. We believe that our approach-identifying naturally occurring anti-inflammatory compounds and modifying them to enhance stability and activity-may lead to more physiologically balanced responses than other available anti-inflammatory therapies.
| 46 |
VB-201Biosimilars and Exclusivity
Our lead Lecinoxoid-based compound, VB-201,
The ACA includes a subtitle called the Biologics Price Competition and Innovation Act, or BPCIA, which created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-licensed reference biological product. The FDA has issued several guidance documents outlining an approach to review and approval of biosimilars in the United States. Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency, can be shown through analytical studies, animal studies, and a clinical study or studies. Interchangeability requires that a product is biosimilar to the reference product and the product must demonstrate that it can be expected to produce the same clinical results as the reference product in any given patient and, for products that are administered multiple times to an individual, the biologic and the reference biologic may be alternated or switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic.
Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was designed as an oral agentfirst licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the controlcompeting product containing that applicant’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of chronic inflammatory disorders. It was clinically developedits product. The BPCIA also created certain exclusivity periods for psoriasisbiosimilars approved as interchangeable products. At this juncture, it is unclear whether products deemed “interchangeable” by the FDA will, in fact, be readily substituted by pharmacies, which are governed by state pharmacy law.
Other Regulatory Matters
Following product approval, where applicable, the manufacturing, sales, promotion and ulcerative colitis, however,other activities around product candidates and/or commercialization are also subject to regulation by numerous regulatory authorities in the United States in addition to the FDA. Regulatory agencies with authority over product candidates may include, and are not limited to, the Centers for Medicare & Medicaid Services, other divisions of the U.S. Department of Health and Human Services, the Department of Justice, the Drug Enforcement Administration, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency and state and local governments and governmental agencies.
If any products that we may develop are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. Products must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. Manufacturing, labeling, packaging, distribution, sales, promotion and other activities also are potentially subject to federal and state consumer protection and unfair competition laws, among other requirements to which we may be subject.
The distribution of pharmaceutical products is subject to additional requirements and regulations, including extensive recordkeeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
The failure to comply with any of these laws or regulatory requirements may subject firms to legal or regulatory action. Depending on the circumstances, failure to meet applicable regulatory requirements can result in criminal prosecution, fines or other penalties, injunctions, exclusion from federal healthcare programs, requests for recall, seizure of products, total or partial suspension of production, denial or withdrawal of product approvals, relabeling or repackaging, or refusal to allow a firm to enter into supply contracts, including government contracts. Any claim or action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our recent Phase 2 data do not support further developmentmanagement’s attention from the operation of our business. Prohibitions or restrictions on marketing, sales or withdrawal of future products marketed by us could materially affect our business in an adverse way.
Changes in regulations, statutes or the interpretation of existing regulations could impact our business in the future by requiring, for these indications. We believe that VB-201example: (i) changes to our manufacturing arrangements; (ii) additions or modifications to product labeling or packaging; (iii) the recall or discontinuation of our products; or (iv) additional recordkeeping requirements. If any such changes were to be imposed, they could adversely affect the operation of our business.
Other Healthcare Laws
If we obtain regulatory approval of our products, we may still have potentialbe subject to various federal and state laws targeting fraud, waste, and abuse in the healthcare industry. These laws may impact, among other disordersthings, our proposed sales, marketing, and education programs. In addition, we may be subject to patient privacy regulation by both the federal government and the states in which TLR-2 and TLR-4 or monocytes play a role. In this regard, in April 2016 we announced a scientific publication of preclinical findings demonstratingconduct our business. The U.S. laws that VB-201, and its next-generation derivative VB-703, can inhibit Non-Alcoholic Steatohepatitis, or NASH, and liver fibrosis in a murine NASH model.may affect our ability to operate include:
| ● | the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, either the referral of an individual, or the purchase or recommendation of an item or service for which payment may be made under a federal healthcare program, such as the Medicare and Medicaid programs. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act or federal civil money penalties statute (as discussed below); |
Non-alcoholic fatty liver disease is the most common liver disease in the western world. This pathological condition is characterized by lipid accumulation in hepatocytes and ranges from the non-progressive form termed steatosis to NASH, the progressive form that is prone to the development of cirrhosis, liver cancer, and liver failure. NASH is characterized by fatty liver inflammation. Several studies have implicated TLR4 and its co-receptor CD14 in NASH and fibrosis. In addition, studies have emphasized the crucial role of infiltrating monocytes/macrophages for the progression of liver inflammation and fibrosis in experimental mouse models and in patients with liver cirrhosis. It has become clear that the macrophage compartment of the liver, traditionally called Kupffer cells, is greatly augmented by an overwhelming number of infiltrating monocytes upon acute or chronic liver injury.
| ● | U.S. federal civil and criminal false claims laws and civil monetary penalty laws, including the federal False Claims Act, which impose criminal and civil penalties against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, including the Medicare and Medicaid programs, claims for payment that are false or fraudulent, making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government, or knowingly concealing or knowingly and improperly avoiding or decreasing such an obligation; |
| ● | the U.S. federal Health Insurance Portability and Accountability Act of 1996, or HIPAA and its implementing regulations, which created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and prohibit knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statements in connection with the delivery of or payment for healthcare benefits, items or services; |
VB-201 inhibits the CD-14/TLR4 and TLR2 pathways as well as monocyte migration. Using an external CRO, we studied VB-201 in a mouse model of NASH and found that while treatment with VB-201 did not reduce steatosis, it significantly decreased liver lobular inflammation and liver fibrosis compared to vehicle treated mice. Furthermore, in April 2017 VBL presented findings of a post-hoc, hypothesis-driven analysis of data from completed Phase 2 studies with VB-201, indicating that VB-201 may reduce certain liver enzymes in blood samples from treated patients, in a time-dependent and dose-dependent manner.
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and their respective implementing regulations, which impose certain obligations, including mandatory contractual terms, on covered healthcare providers, health plans, and healthcare clearinghouses, as well as their business associates, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| ● | the U.S. federal Physician Payments Sunshine Act which requires certain manufacturers of drugs, devices, biologics, and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services information related to payments or transfers of value made to physicians (currently defined to include doctors, dentists, optometrists, podiatrists and chiropractors), physician assistants, nurse practitioners, clinical nurse specialists, certified registered nurse anesthetists and teaching hospitals, as well as information regarding ownership and investment interests held by the physicians described above and their immediate family members; |
Beyond VB-201, we have developed second and third generations of Lecinoxoid product candidates. Our results highlight the potential of some of these molecules, such as VB-703, a third generation candidate whose IP life-cycle can extend to the mid-2030s. In May 2015 at the DDW conference, we presented that VB-703 inhibits liver fibrosis by blocking TLR4 signaling pathways.
| ● | federal government price reporting laws, which require us to calculate and report complex pricing metrics in an accurate and timely manner to government programs; |
| ● | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and |
Recent preclinical studies which we performed also demonstrate efficacy of VB-201 and VB-703 in a rat model of renal fibrosis. Renal fibrosis is a direct consequence of the kidney’s limited capacity to regenerate after injury. Renal scarring results in a progressive loss of renal function, ultimately leading to end-stage renal failure and a requirement for dialysis or kidney transplantation.
| ● | analogous state and laws and regulations in other jurisdictions, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payers, including private insurers, and state and laws in other jurisdiction governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
Our lead Lecinoxoid molecule, VB-201, is a Phase-2 ready oral molecule which has demonstrated safety in > 600 patients and proof-of-concept in a Phase 2 study for vascular inflammation. Our next-generation molecule VB-703, offers long IP lifecycle with demonstrated efficacy in liver and renal fibrosis models.
| 47 |
We believeMany U.S. states have adopted laws similar to the federal Anti-Kickback Statute and False Claims Act, and may apply to our business practices, including, but not limited to, research, distribution, sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental payers, including private insurers. In addition, some states have passed laws that Lecinoxoidsrequire pharmaceutical companies to comply with the April 2003 Office of Inspector General Compliance Program Guidance for Pharmaceutical Manufacturers and/or the Pharmaceutical Research and Manufacturers of America’s Code on Interactions with Healthcare Professionals. Several states also impose other marketing restrictions or require pharmaceutical companies to make marketing or price disclosures to the state and require the registration of pharmaceutical sales representatives.
At the state level, legislatures have therapeutic potentialincreasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in disorderssome cases, designed to encourage importation from other countries and bulk purchasing.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, in which TLR-2the event we obtain regulatory approval for any one of our products, it is possible that some of our business activities could be subject to challenge and TLR-4 or monocytes play a role, such as atherosclerosis, NASH/Liver fibrosis and renal fibrosis. In spite of that potential, we have decided strategicallyfound to focus our efforts and resources on development of novel anti-cancer therapies, such as VB-111 and MOSPD2. Therefore, we intend to seekviolate one or more strategic partnersof such laws, regulations, and guidance. Law enforcement authorities are increasingly focused on enforcing fraud and abuse laws, and it is possible that some of our practices may be challenged under these laws. Violations of these laws can subject us to administrative, civil and criminal penalties, damages, fines, disgorgement, the exclusion from participation in federal and state healthcare programs, individual imprisonment, reputational harm, and the curtailment or restructuring of our operations, as well as additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws. Efforts to ensure that our current and future business arrangements with third parties, and our business generally, will comply with applicable healthcare laws and regulations will involve substantial costs.
Insurance Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any drug products for which we may obtain regulatory approval. In the United States, sales of any products for which we may receive regulatory approval for commercial sale will depend in part on the availability of coverage and reimbursement from third-party payers. Third-party payers include government programs such as Medicare and Medicaid, managed care providers, private health insurers, and other organizations. The process for determining whether a payer will provide coverage for a drug product may be separate from the process for setting the reimbursement rate that the payer will pay for the drug product. In the United States, the principal decisions about reimbursement for new medicines are typically made by CMS, an agency within HHS. CMS decides whether and to what extent a new medicine will be covered and reimbursed under Medicare and private payers tend to follow CMS to a substantial degree. Third-party payers may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA-approved drugs for a particular indication. Moreover, a payer’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development. Third-party payers are increasingly challenging the price and examining the medical necessity and cost- effectiveness of medical products and services, in addition to their safety and efficacy. In order to obtain coverage and reimbursement for any product that might be approved for sale, we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of any products, in addition to the costs required to obtain regulatory approvals. Factors payers consider in determining reimbursement are based on whether the product is:
| ● | a covered benefit under its health plan; |
| ● | safe, effective and medically necessary; |
| ● | appropriate for the specific patient; |
● | cost-effective; and |
| ● | neither experimental nor investigational. |
Our product candidates may not be considered medically necessary or cost-effective. If third-party payers do not consider a product to be cost-effective compared to other available therapies, they may not cover the product after approval as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow a company to sell its products at a profit.
| 48 |
The U.S. government and state legislatures have shown significant interest in implementing cost containment programs to limit the growth of government-paid health care costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs. For example, the ACA contains provisions that may reduce the profitability of drug products, including, for example, increased rebates for drugs reimbursed by Medicaid programs, extension of Medicaid rebates to Medicaid managed care plans, mandatory discounts for certain Medicare Part D beneficiaries, and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Adoption of government controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceuticals.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payers fail to provide adequate coverage and reimbursement. In addition, an increasing emphasis on cost containment measures in the United States has increased and we expect will continue to increase the pressure on pharmaceutical pricing. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Current and Future Healthcare Reform Legislation
The United States and many foreign jurisdictions have enacted or proposed legislative and regulatory changes affecting the healthcare system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any product for which we obtain marketing approval. Changes in regulations, statutes or the interpretation of existing regulations could impact our business in the future by requiring, for example: (i) changes to our manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of our products; or (iv) additional record-keeping requirements.
In March 2010, the ACA was enacted, which includes measures that have or will significantly change the way health care is financed by both U.S. governmental and private insurers. Among the provisions of the ACA of greatest importance to the pharmaceutical industry are the following:
| ● | The Medicaid Drug Rebate Program requires pharmaceutical manufacturers to enter into and have in effect a national rebate agreement with the Secretary of the Department of HHS as a condition for states to receive federal matching funds for the manufacturer’s outpatient drugs furnished to Medicaid patients. The ACA made several changes to the Medicaid Drug Rebate Program, including increasing pharmaceutical manufacturers’ rebate liability by raising the minimum basic Medicaid rebate on most branded prescription drugs from 15.1% of average manufacturer price, or AMP, to 23.1% of average manufacturer price, or AMP, and adding a new rebate calculation for “line extensions” (i.e., new formulations, such as extended release formulations) of solid oral dosage forms of branded products, as well as potentially impacting their rebate liability by modifying the statutory definition of AMP. The ACA also expanded the universe of Medicaid utilization subject to drug rebates by requiring pharmaceutical manufacturers to pay rebates on Medicaid managed care utilization and by expanding the population potentially eligible for Medicaid drug benefits. In addition, the ACA provides for the public availability of retail survey prices and certain weighted average AMPs under the Medicaid program. |
| ● | In order for a pharmaceutical product to receive federal reimbursement under the Medicare Part B and Medicaid programs or to be sold directly to U.S. government agencies, the manufacturer must extend discounts to entities eligible to participate in the 340B drug pricing program. The required 340B discount on a given product is calculated based on the AMP and Medicaid rebate amounts reported by the manufacturer. The ACA expanded the types of entities eligible to receive discounted 340B pricing, although, under the current state of the law, with the exception of children’s hospitals, these newly eligible entities will not be eligible to receive discounted 340B pricing on orphan drugs when used for the orphan indication. In addition, as 340B drug pricing is determined based on AMP and Medicaid rebate data, the revisions to the Medicaid rebate formula and AMP definition described above could cause the required 340B discount to increase. |
| 49 |
| ● | Expansion of the eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for certain individuals with income at or below 133% of the Federal Poverty Level, thereby potentially increasing manufacturers’ Medicaid rebate liability. |
| ● | The ACA imposed a requirement on manufacturers of branded drugs to provide a 70% discount off the negotiated price of branded drugs dispensed to Medicare Part D patients in the coverage gap (increased from 50% pursuant to the Bipartisan Budget Act of 2018, effective as of 2019). |
| ● | The ACA imposed an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs, apportioned among these entities according to their market share in certain government healthcare programs, although this fee would not apply to sales of certain products approved exclusively for orphan indications. |
| ● | A Patient-Centered Outcomes Research Institute was established pursuant to the ACA to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. The research conducted by the Patient-Centered Outcomes Research Institute may affect the market for certain pharmaceutical products. |
| ● | The ACA established the Center for Medicare and Medicaid Innovation within CMS, which is charged with testing new, innovative payment and service delivery models. |
Since its enactment, there have been judicial, Congressional and executive challenges to certain aspects of the ACA. On June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the ACA brought by several states without specifically ruling on the constitutionality of the ACA. Prior to the Supreme Court’s decision, President Biden issued an executive order to initiate a special enrollment period from February 15, 2021 through August 15, 2021 for purposes of obtaining health insurance coverage through the ACA marketplace. The executive order also instructed certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA. It is unclear how other healthcare reform measures of the Biden administration or other efforts, if any, to challenge, repeal or replace the ACA will impact our business.
In addition, other legislative and regulatory changes have been proposed and adopted in the United States since the ACA was enacted:
| ● | On August 2, 2011, the U.S. Budget Control Act of 2011, among other things, included aggregate reductions of Medicare payments to providers of 2% per fiscal year. These reductions went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute, will remain in effect through 2030, with the exception of a temporary suspension from May 1, 2020 through March 31, 2022 due to the COVID-19 pandemic. Following the temporary suspension, a 1% payment reduction will occur beginning April 1, 2022 through June 30, 2022, and the 2% payment reduction will resume on July 1, 2022. |
| ● | On January 2, 2013, the U.S. American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers. |
| ● | On April 13, 2017, CMS published a final rule that gives states greater flexibility in setting benchmarks for insurers in the individual and small group marketplaces, which may have the effect of relaxing the essential health benefits required under the ACA for plans sold through such marketplaces. |
| ● | On May 30, 2018, the Right to Try Act, was signed into law. The law, among other things, provides a federal framework for certain patients to access certain investigational new drug products that have completed a Phase 1 clinical trial and that are undergoing investigation for FDA approval. Under certain circumstances, eligible patients can seek treatment without enrolling in clinical trials and without obtaining FDA permission under the FDA expanded access program. There is no obligation for a pharmaceutical manufacturer to make its drug products available to eligible patients as a result of the Right to Try Act. |
| 50 |
| ● | On May 23, 2019, CMS published a final rule to allow Medicare Advantage Plans the option of using step therapy for Part B drugs beginning January 1, 2020. |
| ● | On December 20, 2019, former President Trump signed into law the Further Consolidated Appropriations Act (H.R. 1865), which repealed the Cadillac tax, the health insurance provider tax, and the medical device excise tax. It is impossible to determine whether similar taxes could be instated in the future. |
Additionally, there has been increasing legislative and enforcement interest in the United States with respect to drug pricing practices. Specifically, there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, and review the relationship between pricing and manufacturer patient programs. At a federal level, President Biden signed an Executive Order on July 9, 2021 affirming the administration’s policy to (i) support legislative reforms that would help uslower the prices of prescription drug and biologics, including by allowing Medicare to exploitnegotiate drug prices, by imposing inflation caps, and, by supporting the potentialdevelopment and market entry of lower-cost generic drugs and biosimilars; and (ii) support the enactment of a public health insurance option. Among other things, the Executive Order also directs HHS to provide a report on actions to combat excessive pricing of prescription drugs, enhance the domestic drug supply chain, reduce the price that the Federal government pays for drugs, and address price gouging in the industry; and directs the FDA to work with states and Indian Tribes that propose to develop section 804 Importation Programs in accordance with the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, and the FDA’s implementing regulations. FDA released such implementing regulations on September 24, 2020, which went into effect on November 30, 2020, providing guidance for states to build and submit importation plans for drugs from Canada. On September 25, 2020, CMS stated drugs imported by states under this rule will not be eligible for federal rebates under Section 1927 of the Social Security Act and manufacturers would not report these drugs for “best price” or Average Manufacturer Price purposes. Since these drugs are not considered covered outpatient drugs, CMS further stated it will not publish a National Average Drug Acquisition Cost for these drugs. If implemented, importation of drugs from Canada may materially and adversely affect the price we receive for any of our Lecinoxoid assets.product candidates. Further, on November 20, 2020 CMS issued an Interim Final Rule implementing the Most Favored Nation, or MFN, Model under which Medicare Part B reimbursement rates would have been be calculated for certain drugs and biologicals based on the lowest price drug manufacturers receive in Organization for Economic Cooperation and Development countries with a similar gross domestic product per capita. However, on December 29, 2021 CMS rescinded the Most Favored Nations rule. Additionally, on November 30, 2020, HHS published a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers. Pursuant to court order, the removal and addition of the aforementioned safe harbors were delayed and recent legislation imposed a moratorium on implementation of the rule until January 1, 2026. Although a number of these and other proposed measures may require authorization through additional legislation to become effective, and the Biden administration may reverse or otherwise change these measures, both the Biden administration and Congress have indicated that they will continue to seek new legislative measures to control drug costs.
We expect that additional U.S. federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that the U.S. Federal Government will pay for healthcare drugs and services, which could result in reduced demand for our drug candidates or additional pricing pressures.
Other U.S. Environmental, Health and Safety Laws and Regulations
We may be subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. From time to time and in the future, our operations may involve the use of hazardous and flammable materials, including chemicals and biological materials, and may also produce hazardous waste products. Even if we contract with third parties for the disposal of these materials and waste products, we cannot completely eliminate the risk of contamination or injury resulting from these materials. In the event of contamination or injury resulting from the use or disposal of our hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
We maintain workers’ compensation insurance to cover us for costs and expenses that we may incur due to injuries to our employees, but this insurance may not provide adequate coverage against potential liabilities. However, we do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. Current or future environmental laws and regulations may impair our research, development or production efforts. In addition, failure to comply with these laws and regulations may result in substantial fines, penalties or other sanctions.
| 51 |
Accordingly, in March 2019, we announced a strategic exclusive option license agreement with oneGovernment Regulation of world-leading European animal health companies, for the development of VB-201 for veterinary use. Under termsDrugs and Biologics Outside of the agreement, VBL has granted an exclusive option licenseUnited States
In addition to explore the potential of VB-201 for animal health indications. In exchange, VBL is receiving an undisclosed up-front payment, and is entitled to receive additional development milestone payments. Upon exercising the option to license, VBL will receive additional milestones and royalties on net sales, which may exceed €50 million over the course of the license term. We retain the VB-201 rights for treatment of humans, worldwide.
Expanding our Pipeline-The VB-600 series of monoclonal antibodies targeting MOSPD2 for Oncology and Inflammation
We are conducting two parallel drug development programs that are exploring the potential of MOSPD2, a protein which we identified as a key regulator of cell motility, as a therapeutic target for inflammatory diseases and cancer.
MOSPD2 is a membrane protein whose function was unknown. We discovered new biological findings, which seem to position MOSPD2 as a critical pathway controlling monocyte migration and as a key regulator of disease pathogenesis in different inflammatory autoimmune settings. Our data show that inhibition of MOSPD2 by either knockdown, silencing or proprietary antibodies, results in a significant reductionregulations in the abilityUnited States, we will be subject to a variety of monocytes to migrate, regardlessregulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of the inflammatory signals employed to attract them.
Sinceour products. The cost of establishing a regulatory compliance system for numerous varying jurisdictions can be very significant. Although many of the receptors involved in regulation of immune cell migration are also utilized by cancer cellsissues discussed above with respect to the United States apply similarly in the context of the European Union and in other jurisdictions, the approval process varies between countries and jurisdictions and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries and jurisdictions might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the regulatory process in others.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of disseminationclinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application much like the IND prior to the commencement of human clinical trials. In the European Union, for example, a request for a clinical trial authorization, or CTA, must be submitted to each country’s national health authority and an independent ethics committee, much like the FDA and the IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed.
The European Union adopted the new Clinical Trials Regulation (EU) No 536/2014 in April 2014, which replaced the Clinical Trials Directive 2001/20/EC on January 31, 2022. The transitory provisions of the new Regulation offer sponsors the possibility to choose between the requirements of the previous Directive and the new Regulation if the request for authorization of a clinical trial is submitted in the year after the new Regulation became applicable. If the sponsor chooses to submit under the Directive, the clinical trial continues to be governed by the previous Directive until three years after the new Regulation became applicable. If a clinical trial continues for more than three years after the Regulation became applicable, the new Regulation will at that time begin to apply to the clinical trial. The new Regulation overhauls the system of approvals for clinical trials in the European Union. Specifically, it is directly applicable in all Member States (meaning that no national implementing legislation in each Member State is required), and aims at simplifying and streamlining the approval of clinical trials in the European Union. The main characteristics of the new Regulation include: a streamlined application procedure via a single-entry point through the bodyClinical Trials Information System; a single set of documents to be prepared and formation of metastases, we asked whether MOSPD2 plays a role in this setting as well. We found that MOSPD2 was detected insubmitted for the majority of cancerous organs, including colon, esophagus, liver and breast. In a manuscript published in International Journal of Cancerapplication as well as in scientific conferences, we showed that MOSPD2 is requiredsimplified reporting procedures for clinical trial sponsors; and a harmonized procedure for the migrationassessment of applications for clinical trials, which is divided in two parts (Part I contains scientific and invasionmedicinal product documentation and Part II contains the national and patient-level documentation). Part I is assessed by a coordinated review by the competent authorities of breast cancer cellsall European Union Member States in vitro,which an application for authorization of a clinical trial has been submitted (Concerned Member States) of a draft report prepared by a Reference Member State. Part II is assessed separately by each Concerned Member State. Strict deadlines have also been established for the assessment of clinical trial applications.
The requirements and process governing the conduct of clinical trials, product approval or licensing, pricing and reimbursement vary from country to country. In all cases, the clinical trials are conducted in accordance with GCP, the applicable regulatory requirements and the ethical principles that it promotes breast cancer cell metastasishave their origin in vivo. Given the specificityDeclaration of MOSPD2 expression and its highly elevated expressionHelsinki.
To obtain regulatory approval of a medicinal product under European Union regulatory systems, we must submit a marketing authorization application. The application required in tumors, we believe MOSPD2 can serve as a novel mechanism for targetingthe European Union is similar to an NDA or BLA in the United States, with the exception of, tumor cells.
Accordingly, we are developing our VB-600 platform of monoclonal antibodies targeting MOSPD2 for oncology and inflammatory indications.
Bi-specific mAbs for Oncology Indications
We are developing bi-specific antibodies aimed to kill tumor cells, based on MOSPD2 as a target whose expression is inducedamong other things, country-specific document requirements. Marketing approvals in multiple tumors.European Union Member States may be obtained through a centralized, mutual recognition or decentralized procedure. The centralized procedure results in the grant of a single marketing authorization that is valid throughout the European Union Member States, as well as the additional Member States of the European Economic Area (Norway, Iceland and Liechtenstein).
MOSPD2 can be found in many typesPursuant to Regulation (EC) No. 726/2004, as amended, the centralized procedure is mandatory for certain products, including those developed by means of solid tumors,specified biotechnological processes, advanced therapy medicinal products (gene-therapy, somatic-cell therapy, and it seems to be highly expressed on tumor cells when they start invading tissues or creating metastatic lesions. This correlates withtissue-engineered products), products for human use containing a new active substance for which the role we identified for MOSPD2 as a key regulator of cell motility in tumor cells. Our preliminary data indicate that knock-out of MOSPD2 in tumor cells may reduce metastasis by 95% in some pre-clinical settings.
In July 2018, we published a manuscript demonstrating, for the first time, that MOSPD2 can play a major role in breast cancer cell migration and metastasis. Histological analysis of human specimens shows that MOSPD2 levels were correlated with the stage of tumor invasiveness, and were profoundly elevated in invasive and metastatic breast cancer.
Based on these findings, our approachtherapeutic indication is to utilize MOSPD2 as a target for attacking the tumor cells in the treatment of late-stage breastspecified diseases, including AIDS, HIV, cancer, diabetes, neurodegenerative disorders, auto-immune diseases and other tumor types. To this end, we are developing bi-specific antibodies that aimimmune dysfunctions and viral diseases, as well as products designated as orphan medicinal products. The Committee for Medicinal Products for Human Use, or CHMP, of the EMA also has the discretion to induce killingpermit other products to use the centralized procedure if it considers them sufficiently innovative or they contain a new active substance or they may be of MOSPD2-positive tumors cells through binding and activation of T-cells. We have presented proof-of-concept for this approachbenefit to public health at the AACR conferenceEuropean Union level.
Under the centralized procedure in April 2018 usingthe European Union, the maximum timeframe for the evaluation of a BiTE antibody, and are currently advancing our lead bi-specific candidates towards an IND filing,marketing authorization application by the EMA is 210 days, excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP. Clock stops may extend the timeframe of evaluation of a marketing authorization application considerably beyond 210 days. Where the CHMP gives a positive opinion, it provides the opinion together with supporting documentation to the European Commission, who makes the final decision to grant a marketing authorization, which is issued within 67 days of receipt of the EMA’s recommendation. Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of major interest from the point of view of public health and in particular from the viewpoint of therapeutic innovation. If the CHMP accepts such request, the time limit of 210 days will be reduced to 150 days, excluding clock stops, but it is possible that the CHMP can revert to the standard time limit for the centralized procedure if it considers that it is no longer appropriate to conduct an accelerated assessment.
Now that the UK (which comprises Great Britain and Northern Ireland) has left the European Union, Great Britain will no longer be covered by centralized marketing authorizations (under the Northern Ireland Protocol, centralized marketing authorizations will continue to be recognized in Northern Ireland). All medicinal products with a current centralized marketing authorization were automatically converted to Great Britain marketing authorizations on January 1, 2021. For a period of two years from January 1, 2021, the Medicines and Healthcare products Regulatory Agency the United Kingdom’s medicines regulator, may rely on a decision taken by the European Commission on the approval of a new marketing authorization in the second halfcentralized procedure, in order to more quickly grant a new Great Britain marketing authorization. A separate application will, however, still be required.
| 52 |
The European Union also provides opportunities for market exclusivity. For example, in the European Union, upon receiving marketing authorization, new chemical innovative medicinal products generally receive eight years of 2020.data exclusivity and an additional two years of market exclusivity. If granted, data exclusivity prevents applicants for authorization of generics or biosimilars of these innovative products in the European Union from referencing the innovator’s preclinical and clinical trial data when applying for a generic or biosimilar marketing authorization in the European Union, during a period of eight years from the date on which the reference product was first authorized in the European Union. During the additional two-year period of market exclusivity, a generic marketing authorization can be submitted, and the innovator’s data may be referenced, but no generic product can be marketed until the expiration of the market exclusivity. The innovator may obtain an additional one year of market exclusivity if the innovator obtains an additional authorization during the initial eight year period for one or more new indications that demonstrate significant clinical benefit over currently approved therapies. Even if a product is considered to be an innovative medicinal product so that the innovator gains the prescribed period of data exclusivity, another company may market another version of the product if such company obtained a marketing authorization based on a marketing authorization application with a completely independent data package of pharmaceutical tests, preclinical tests and clinical trials. However, there is no guarantee that a product will be considered by the European Union’s regulatory authorities to be a new chemical entity, and products may not qualify for data exclusivity. Similar exclusivity periods are available for new biologics.
Classical mAbsA product can be designated as an orphan medicinal product by the European Commission if its sponsor can establish: that (1) the product is intended for Inflammatory Indicationsthe diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition; (2) either (a) such condition affects no more than five in 10,000 persons in the European Union when the application is made, or (b) it is unlikely that the product, without the benefits derived from orphan status, would generate sufficient return in the European Union to justify the necessary investment in its development; and (3) there exists no satisfactory method of diagnosis, prevention or treatment of such condition authorized for marketing in the European Union or, if such method exists, the product will be of significant benefit to those affected by that condition. Orphan medicinal products are eligible for financial incentives such as reduction of fees or fee waivers. The application for orphan designation must be submitted before the application for marketing authorization. The applicant will receive a fee reduction for the marketing authorization application if the orphan designation has been granted, but not if the designation is still pending at the time the marketing authorization is submitted. Orphan designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
Our data show, that MOSPD2Orphan medicinal products in the European Union are eligible for 10-year market exclusivity during which time no “similar medicinal product” may be placed on the market. A “similar medicinal product” is defined as a medicinal product containing a similar active substance or substances as contained in an authorized orphan medicinal product, and which is predominantly expressedintended for the same therapeutic indication. This 10-year market exclusivity may be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria for orphan designation, for example, if the product is sufficiently profitable not to justify maintenance of market exclusivity. Additionally, marketing authorization may be granted to a similar product for the same indication at any time if:
| ● | the second applicant can establish that its product, although similar, is safer, more effective or otherwise clinically superior; | |
| ● | the marketing authorization holder for the authorized orphan product consents to a second orphan medicinal product application; or | |
| ● | the marketing authorization holder for the authorized orphan product cannot supply enough orphan medicinal product. |
The aforementioned European Union rules are generally applicable in the European Economic Area, which consists of the European Union Member States, plus Norway, Liechtenstein and Iceland.
On June 23, 2016, the electorate in the United Kingdom voted in favor of leaving the European Union, commonly referred to as Brexit, and the UK formally left the European Union on January 31, 2020. There was a transition period during which European Union pharmaceutical laws continued to apply to the United Kingdom, which expired on December 31, 2020. However, the European Union and the United Kingdom have concluded a trade and cooperation agreement, or TCA, which was provisionally applicable since January 1, 2021 and has been formally applicable since May 1, 2021. The TCA includes specific provisions concerning pharmaceuticals, which include the mutual recognition of GMP, inspections of manufacturing facilities for medicinal products and GMP documents issued, but does not foresee wholesale mutual recognition of United Kingdom and European Union pharmaceutical regulations. At present, Great Britain has implemented European Union legislation on the surfacemarketing, promotion and sale of human monocytes,medicinal products through the Human Medicines Regulations 2012 (as amended) (under the Northern Ireland Protocol, the European Union regulatory framework will continue to apply in Northern Ireland). The regulatory regime in Great Britain therefore broadly aligns with current European Union regulations, however it is essentialpossible that these regimes will diverge in future now that Great Britain’s regulatory system is independent from the European Union and the TCA does not provide for their migration. By inhibiting this protein,mutual recognition of United Kingdom and European Union pharmaceutical legislation.
If we seekfail to block this migrationcomply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of monocytes to sitesregulatory approvals, product recalls, seizure of inflammation,products, operating restrictions and accordingly to reduce inflammation and tissue damage.criminal prosecution.
At the ECTRIMS 2018 meeting, we presented the critical role of MOSPD2 in the development of multiple sclerosis, and its potential as a novel target for treatment of inflammation in the Central Nervous System (or CNS) and other organs. One of the key cell types that causes inflammation in MS, is the monocyte. In MS, monocytes that circulate in the peripheral blood infiltrate into the CNS and play a key role in the inflammatory process, particularly through damaging the myelin coating which protects the nerve fibers, therefore leading to acute neurological symptoms. Using MOSPD2 knockout mice, our data show that MOSPD2 was critical for the development of the disease in the experimental autoimmune encephalomyelitis (or EAE) model for MS, as knockout mice essentially do not develop the disease. Furthermore, we developed proprietary monoclonal antibodies against MOSPD2 that successfully prevented development of EAE, and were also effective in treatment of the animals after the neurological symptoms had already appeared. These data suggest that MOSPD2 is a critical path in MS.
In February 2019, we presented additional data implicating the potential of our VB-600 platform of antibodies targeting MOSPD2 for treatment of NASH and RA.
Data presented at Keystone symposia in February 2019 showed that mice in which the MOSPD2 gene was knocked out, had minimal or no disease in the collagen antibody-induced arthritis model for RA. Knockout of MOSPD2 also lead to significant reduction in liver fibrosis in a high-fat-high-carbohydrate model for NASH. Collectively, these data point to MOSPD2 as a key pathway through which the body is recruiting monocytes to specific sites of inflammation. Accordingly, we believe that antibodies targeting MOSPD2 have potential for treatment of various inflammatory indications, and are advancing lead candidates towards an IND submission which is expected in the first half of 2020.
Intellectual Property
Our success depends, at least in part, on our ability to protect our proprietary technology and intellectual property, and to operate without infringing or violating the proprietary rights of others. We rely on a combination of patent, trademark, trade secret and copyright laws, know- how,know-how, intellectual property licenses and other contractual rights, including confidentiality and invention assignment agreements to protect our intellectual property rights.
See “Item 3. Key Information-D. Risk Factors-Risks Related to Our Intellectual Property” for additional discussion on our intellectual property and associated risks.
| 53 |
Patents
As of January 30, 2019,2022, we had 234have more than 200 granted patents and 3445 applications pending worldwide for our oncology program and VTS platform technologytechnology. Our lead VTS asset, ofra-vec, is covered by a US granted patent with an expiration date in 2033, prior to any patent term extension. We also have pending patent applications covering ofra-vec diagnostic methods, the use of ofra-vec for enhancing antitumor immune responses, and 120ofra-vec combination therapy with immune checkpoint inhibitors that, if granted, may extend protection into 2040. We also have more than 25 granted patents and 26 patentmore than 25 applications pending worldwide for our anti-inflammatoryVB-601 program and Lecinoxoid family of compounds. Our lead VTS asset, VB-111, is covered by US granted patent extending to 2033 before any extensions. Our lead Lecinoxoid, VB-201, is protected by US granted composition-of-matter patent extending to 2027 before any extensions. In addition, we have pending patent applications covering use of VB-201, VB-703 and additional Lecinoxoid for NASH and fibrosis indications that may extend, if granted, to the 2030s. For MOSPD2, there are 19 applications pending worldwide.MTT.
Trademarks
We rely on trade names, trademarks and service marks to protect our name brands. Our trademarks and registered trademarks in several countries include the following: “VTS,” “VBL THERAPEUTICS,” “VASCULAR TARGETING SYSTEM VTS,” “VBL,” “V VBL THERAPEUTICS & Design,” “VASCULAR BIOGENICS,” “VASCULAR THERAPEUTICS,” “V & Design,” “GLOBE & Design,” and “OVAL & Design”.
Trade Secrets and Confidential Information
In addition to patented technology, we rely on our unpatented proprietary technology, trade secrets, processes and know-how. We rely on, among other things, confidentiality and invention assignment agreements to protect our proprietary know-how and other intellectual property that may not be patentable, or that we believe is best protected by means that do not require public disclosure. For example, we require our employees to execute confidentiality agreements in connection with their employment relationships with us, and to disclose and assign to us inventions conceived in connection with their services to us. However, there can be no assurance that these agreements will be enforceable or that they will provide us with adequate protection.
We may be unable to obtain, maintain and protect the intellectual property rights necessary to conduct our business, and may be subject to claims that we infringe or otherwise violate the intellectual property rights of others, which could materially harm our business. For a more comprehensive summary of the risks related to our intellectual property, see “Risk Factors.”“Item 3. Key Information-D. Risk Factors “
Sales and Marketing
We have not yet established sales, marketing or product distribution operations because our lead candidates are still in early clinical development. We recently appointed a Chief Commercial Officer and we intend to expand our pre-commercialization activities as we prepare for the readout of the PFS primary endpoint from our Phase 3 OVAL trial in the second half of 2022 which we believe has the potential to support a BLA filing and, if approved, subsequent commercialization.
Manufacturing
We generallycurrently perform process development, characterization and manufacturing for our drug substance candidates and manufacture quantities of our drug candidates necessary to conduct pre-clinical studies and clinical trials of our drug candidates.lead candidate ofra-vec in-house. We rely on third-party manufacturers to produce bulk drug substance required for our clinical trials and expect to continue to rely on third parties to manufacture clinical trial drug supplies for the foreseeable future.other drug candidates. We also contract with additional third parties for the formulating, labeling, packaging, storage and distribution of the final drug products.
VB-111
Until late 2017, we manufactured the active pharmaceutical ingredient and the formulated drug product of VB-111ofra-vec for the clinical development at our small-scale cGMP-compliant production facility in Or-Yehuda, Israel and pursuant to an arrangement with a third party in the United States.
In October 2017, we announced the opening of our new gene therapy manufacturing plant in Modiin,Modi’in, Israel. ThisWe expect this plant willto be the commercial facility for production of the Company’sour lead product candidate, ofranergene obadenovec (VB-111),ofra-vec, if approved. The Modi’in facility is a commercial-scale gene therapy manufacturing facility in Israel (20,000 sq. ft.). The site design enables modular expansion of the manufacturing capacity, to supply growing demand following commercialization. The ModiinWe believe the Modi’in facility shallwill also enable us to comply with the restrictions of the Research Law and our undertaking to the OCS that an essential portion of our VB-111ofra-vec production, and in any event not less than the majority, of VB-111 production, will remain in Israel. In July 2019, our facility was certified by a EU QP as being in compliance with EU GMP. In November 2019 our facility was awarded by the Israeli Ministry of Health the Certificate of GMP Compliance of a Manufacturer. Ofra-vec batches produced in our commercial-scale facility were authorized by the FDA for use in our clinical trials in the United States.
Employees
As of March 1, 2019,2022, we employed 3941 employees, including 3130 in research and development, and 811 in general and administrative positions, and of which 1312 employees have either MDsM.D.s or PhDs. AllPh.D.s. 39 of our employees are located in Israel.Israel and 2 in the U.S. We believe our employee relations are good.
Israeli labor laws govern the length of the workday, minimum wages for employees, procedures for hiring and dismissing employees, determination of severance pay, annual leave, sick days, advance notice of termination of employment, equal opportunity and anti- discrimination laws and other conditions of employment. Subject to specified exceptions, Israeli law generally requires severance pay upon the retirement, death or dismissal of an employee, and requires us and our employees to make payments to the National Insurance Institute, which is similar to the U.S. Social Security Administration. Our employees have defined benefit pension plans that comply with the applicable Israeli legal requirements.
None of our employees currently work under any collective bargaining agreements.
Property
None of our employees currently work under any collective bargaining agreements.
Property
Our corporate headquarters and research facilities are currently located in Modi’in, Israel, where we lease an aggregate of approximately 21,500 square feet of office and laboratory space, pursuant to a lease agreement that expires in June 2023.May 2024. This facility additionally houses our clinical development, clinical operations, regulatory and management functions, as well as our local biological drugs manufacturing facility. We also have a U.S. office located at 1 Blue Hill Plaza, Suite 1509, Pearl River, NY 10965.
Organizational Structure
In September 2021, we incorporated VBL, Inc, a Delaware corporation and wholly owned subsidiary of VBL. We do not have any subsidiaries.conduct U.S. operations from of this entity.
Legal Proceedings
From time to time, we may become involved in litigation or other legal proceedings relating to claims arising from the ordinary course of business. We are currently not a party to any legal proceedings.proceedings that are likely to have a material adverse effect on our results of operations, financial condition or cash flows.
| 55 |
Item 4A. Unresolved Staff Comments
Not applicable.
Item 5. Operating and Financial Review and Prospects
The following discussion of our financial condition and results of operations should be read in conjunction with “Item 3. Key Information-Selected Financial Data” and our financial statements and the related notes to those statements included elsewhere in this Annual Report. In addition to historical financial information, the following discussion and analysis contains forward-looking statements that involve risks, uncertainties and assumptions. See “Cautionary Note Regarding Forward-Looking Statements.” Our actual results and timing of selected events may differ materially from those anticipated in these forward-looking statements as a result of many factors, including those discussed under “Item 3. Key Information-D. Risk Factors” and elsewhere in this Annual Report.
The audited financial statements for the years ended December 31, 2018, 20172021, 2020 and 20162019 and as of December 31, 20182021 and 20172020 in this Annual Report have been prepared in accordance with IFRS as issued by the IASB. None of the financial information in this Annual Report has been prepared in accordance with U.S. GAAP.
Overview
We are a clinical stage biopharmaceutical company committed to developing next-generation, targeted medicines for difficult-to-treat medical conditions. Using our novel platform technologies, we have created a pipeline of therapeutics to uniquely address cancer and immune-inflammatory diseases with the goal of significantly improving patient outcomes and overcoming the limitations of currently approved treatments.
Our product candidates are built off of our two platform technologies: VTS, a gene-based technology targeting newly formed blood vessels, and Monocyte Targeting Technology, or MTT, an antibody-based technology able to specifically inhibit monocyte migration for immune-inflammatory applications.
We are a clinical-stage biopharmaceutical company focused on the discovery, development and commercialization of first-in-class treatments for cancer. Our program is based oncurrently evaluating our proprietary Vascular Targeting System, or VTS, platform technology, which utilizes genetically targeted therapy to destroy newly formed, or angiogenic, blood vessels, and which we believe will allow us to develop product candidates for multiple oncology indications.
Our lead product candidate, VB-111 (ofranergene obadenovec), isofra-vec, in a gene-based biologic that we are developing for solid tumor indications, and which we have advanced to programs for recurrent glioblastoma, or rGBM, an aggressive form of brain cancer, ovarian cancer and thyroid cancer. We have obtained fast track designation for VB-111 in the United States for prolongation of survival in patients with glioblastoma that has recurred following treatment with standard chemotherapy and radiation. We have also received orphan drug designation for GBM in both the United States and Europe. VB-111 has also received an orphan designation for the treatment of ovarian cancer by the European Medicines Agency. In September 2015, we reported complete results from our Phase 2 trial of VB-111 in rGBM, demonstrating a statistically-significant benefit in overall survival and favorable response rate in patients treated with VB-111 in combination with bevacizumab. Our pivotal Phase 3 GLOBE study in rGBM began in August 2015 and was comparing a combination of VB-111 and bevacizumab to bevacizumab alone. The study, which enrolled a total of 256 patients in the US, Canada and Israel was conducted under a special protocol assessment, or SPA, agreement with the U.S. Food and Drug Administration, or FDA, with full endorsement by the Canadian Brain Tumor Consortium (CBTC). On March 8, 2018, we announced top-line results from the GLOBE study, which showed that the study did not meet its pre-specified primary endpoint of overall survival (OS). No new safety concerns associated with VB-111 have been identified in the GLOBE study. In November 2018, full data from the GLOBEregistration-enabling trial were presented at the 2018 Society for Neuro-Oncology Annual Meeting. Analyses pointed to the regimen in GLOBE trial as the potential reason for its negative outcome, indicating that priming with VB-111 without bevacizumab may be critical for the immune and vascular-disruptive/anti-angiogenic mechanism of VB-111 in rGBM. We do not think that results of the GLOBE study in rGBM will necessarily have implications on the prospects for VB-111 in other regimens or tumor types. Accordingly, following independent MRI analyses at UCLA which validated the overall clinical and survival benefit of VB-111 among patients who were primed with VB-111 in our prior Phase 2 study in rGBM, a new investigator-sponsored Phase 2 trial evaluating VB-111 in rGBM patients is expected to be launched in the first half of 2019. Our OVAL phase 3 potential registration study of VB-111 in platinum resistant ovarian cancer, was launched in December 2017 and is conducted in collaboration with the GOG Foundation, Inc., a leading organization for research excellence in the field of gynecologic malignancies. An interim analysis in the OVAL study is expected in year-end 2019. In addition, a collaborative Phase 2 study evaluating VB-111 in combination with a checkpoint-inhibitor in gastrointestinal tumors is expected to be launchedwhich we anticipate PFS primary endpoint data in the second half of 2019.
We also have been conducting a program targeting anti-inflammatory diseases, based on the use of our Lecinoxoid platform technology. Lecinoxoids are a novel class of small molecules we developed that are structurally and functionally similar to naturally occurring molecules known to modulate inflammation. The lead product candidate from this program, VB-201, is a Phase 2-ready molecule that demonstrated efficacy in reducing vascular inflammation in a Phase 2 sub-study in psoriatic patients with cardiovascular risk. Due to business limitations associated with the heavy burden of developing medications for cardiovascular diseases, we chose to test it in psoriasis and ulcerative colitis; however, as we reported in February 2015, VB-201 failed to meet the primary endpoint in Phase 2 clinical trials for psoriasis and for ulcerative colitis. As a result, we have terminated our development of VB-201 in those indications. Nevertheless, based on recent pre-clinical studies, we believe that VB-201 and some second generation molecules such as VB-703 may be applicable for NASH and renal fibrosis. Since the company intends to focus its efforts and resources on advancing our oncology program, we will seek to advance our Lecinoxoid assets via strategic deals. Accordingly, in March 2019, we announced a strategic exclusive option license agreement with one of the world-leading European animal health companies, for the development of VB-201 for veterinary use. We retain the VB-201 rights for treatment of humans, worldwide.
2022. We are also conducting a research program exploring the potential of targeting of MOSPD2 for immuno-oncology applications. In January 2017, we reported that targeting of MOSPD2 inhibits chemotaxis of monocytes and neuropils. In 2018, we published a manuscript showing that MOSPD2 can play a major role in breast cancer cell migration and metastasis, and that targeting MOSPD2 may be employed to prevent the spreading of breast cancer cells. We also presented proof-of-concept data demonstrating antibody-mediated killing of MOSPD2-expressing cancer cells. We believe that targeting of MOSPD2 may have several therapeutic applications, including inhibition of monocyte migration in chronic inflammatory conditions, inhibition of tumor cell metastases and targeting of MOSPD2-expressing tumor cells. We are developing our “VB-600 series” of pipeline candidates towards inflammatory and oncology applications.
We are developing our lead oncology product candidate, VB-111, for solid tumor indications, with current clinical programssupporting Phase 2 trials in rGBM thyroidand metastatic colorectal cancer, or mCRC, where we expect preliminary data in 2022. Our second program, VB-601, is an investigational proprietary monoclonal antibody that binds MOSPD2, which we call the “mono-walk” receptor, and ovarian cancer. In interim analyses of datais engineered to specifically prevent monocytes from our ongoing open-label Phase 2 clinical trial of VB-111 in rGBM, we observed dose-dependent attenuation of tumor growthexiting the blood stream and an increase in median overall survival, which is the time interval from initiation of treatmenttraveling to the patient’s death. The U.S. Foodinflamed tissues, and Drug Administration, or FDA, has granted VB-111 fast track designation for prolongation of survival in patients with glioblastoma that has recurred following treatment with temozolomide, a chemotherapeutic agent commonly used to treat newly diagnosed glioblastoma, and radiation. On July 1, 2014, the FDA concurred with the design and planned analyses of our Phase 3 pivotal trial of VB-111 in rGBM pursuant to an SPA. At the time, commencement of the trial was subject to our providing the agency with more information regarding our potency release assay for the trial. We developed this assay and submitted initial information to the FDA on May 26, 2014. On February 5, 2015 the FDA has found our data satisfactory and removed the partial hold.
We began our Phase 3 pivotal trial of VB-111 in rGBM in August 2015 and completed patient enrollment for the study in December 2016, five months ahead of our initial plan. Following positive safety reviews announced in December 2016, in April 2017 and the third and final safety review that was announced in October 2017, the GLOBE trial continued to completion. On March 8, 2018, we announced top-line results from the GLOBE study, which showed that the study did not meet its pre-specified primary endpoint of overall survival (OS). In November 2018, full data from the GLOBE trial were presented at the 2018 Society for Neuro-Oncology Annual Meeting by Dr. Timothy Cloughesy, MD, Professor of Clinical Neurology and Director of the Neuro-Oncology Program, UCLA School of Medicine and principal investigator of the GLOBE trial. Thorough analyses of the baseline risk factors of the Phase 2 and the Phase 3 treatment groups did not reveal any differences. Therefore, patient selection or different patient populations could not explain the difference between the results of the two studies. The analyses pointed to the regimen in GLOBE trial as the potential reason for its failure, indicating that priming with VB-111 without bevacizumab may be critical for the immune and vascular-disruptive/anti-angiogenic mechanism of VB-111 in rGBM. Following independent MRI analyses at UCLA which validated the overall clinical and survival benefit of VB-111 among patients who were primed with VB-111 in our prior Phase 2 study in rGBM, a new investigator-sponsored trial evaluating VB-111 in rGBM patients is expected to be launched in the first half of 2019.
VB-111 was also being studied inbegin a Phase 2first-in-human clinical trial for recurrent platinum-resistant Ovarian Cancer and in a Phase 2 study in recurrent, iodine-resistant differentiated Thyroid Cancer. In a Phase 2 trial for recurrent platinum-resistant ovarian cancer, VB-111 demonstrated a statistically significant increase in overall survival and about 60% durable response rate (as measured by reduction in CA-125), approximately twice the historical response with bevacizumab plus chemotherapy in ovarian cancer. In December 2016, we had an end-of-Phase-2 meeting with the FDA, in which we received approval from the FDA to advance VB-111 for a Phase 3 study in platinum-resistant ovarian cancer, which we launched in December 2017. The OVAL study is conducted in collaboration with the Gynecologic Oncology Group (GOG) Foundation, Inc., a leading organization for research excellence in the field of gynecologic malignancies. An interim analysis in the OVAL study is expected to occur in year-end 2019.
In March 2019, at the Society of Gynecologic Oncology conference, we presented data from of ovarian cancer patients, showing the potential of VB-111 to stimulate the immune system and drive immune cells to infiltrate the tumor microenvironment.
In February 2017, we reported full data from our exploratory Phase 2 study of VB-111 in recurrent, iodine-resistant differentiated thyroid cancer. The primary endpoint of the trial, defined as 6-month progression-free-survival (PFS-6) of 25%, was met with a dose response. Forty-seven percent of patients in the therapeutic-dose cohort reached PFS-6, versus 25% in the sub-therapeutic cohort, both groups meeting the primary endpoint. An overall survival benefit was seen, with a tail of more than 40% at 3.7 years for the therapeutic-dose cohort, similar to historical data for pazopanib (Votrient® ), a tyrosine kinase inhibitor; however, most patients in the VB-111 study had tumors that previously had progressed on pazopanib or other kinase inhibitors. As of December 31, 2016, we had studied VB-111 in over 200 patients and have observed it to be well-tolerated. In December 2015, we have been granted a US composition of matter patents that provides intellectual property protection for VB-111 in the US until October 2033 before any patent term extension.
In October 2017, we announced the opening of our new gene therapy manufacturing plant in Modiin, Israel. This plant can be the commercial facility for production of VB-111, if approved. The Modiin facility is the first commercial-scale gene therapy manufacturing facility in Israel and currently one of the largest gene-therapy designated ones in the world (20,000 sq. ft.). It is capable of manufacturing in large-scale capacity of 1,000 liters and is scalable to 2,000 liters.
In November 2017, we signed an exclusive license agreement with NanoCarrier Co., Ltd. (TSE Mothers:4571) for the development, commercialization, and supply of VB-111 in Japan. VBL retains rights to VB-111 in the rest of the world. Under terms of the agreement, VBL has granted NanoCarrier an exclusive license to develop and commercialize VB-111 in Japan for all indications. VBL will supply NanoCarrier with VB-111, and NanoCarrier will be responsible for all regulatory and other clinical activities necessary for commercialization in Japan. In exchange, we received an up-front payment of $15 million, and are entitled to receive greater than $100 million in development and commercial milestone payments to the extent they are achieved. VBL will also receive tiered royalties on net sales in the high-teens.
Based on support from pre-clinical data, which we presented at the American Society of Gene & Cell Therapy (ASGCT) conference in May 2017, and the histological data in ovarian cancer showing ability of VB-111 to turn an immunologically “cold” to “hot” tumor, an exploratory Phase 2 study for VB-111 in combination a checkpoint inhibitor, is planned in gastrointestinal tumors. Launch of this trial is expected in the second half of 2019.2022.
We commenced operations in 2000, and our operations to date have been limited to organizing and staffing our company, business planning, raising capital, developing our VTS and Lecinoxoid platform technologies and developing our product candidates, including conducting pre-clinicalpreclinical studies and clinical trials of VB-111ofra-vec and VB-201.VB-601, and programs we are no longer pursuing. To date, we have funded our operations through private sales of preferred shares, a convertible loan, public offeringofferings, revenues from licensing agreements and grants from the Israeli Office of Chief Scientist, or OCS, which has later transformed to the Israeli Innovation Authority, or IIA under the Israel Encouragement of Research and Development in Industry, or the Research Law. We have no products that have received regulatory approval and accordingly have never generated regular revenue streams.streams from sales of our products. Since our inception and through December 31, 2018,2021, we had raised an aggregate of $248.9$325.7 million to fund our operations, of which $113.4 million was from sales of our equity securities, $40.5 from our initial public offering, or IPO, $15 million from a November 3, 2015 underwritten offering, approximately $24.0 million from a June 7, 2016 registered direct offering, $17.9 million from a November 16, 2017 underwritten offering, $15.5 million from a June 27, 2018 registered direct offering and $22.6including $29.2 million from IIA grants.
Since inception, we have incurred significant losses. For the years ended December 31, 2018, 20172021, 2020 and 2016,2019, our loss was $20.5$29.9 million, $10.1$24.2 million and $16.0$19.4 million, respectively. We expect to continue to incur significant expenses and losses for at least the next several years.years and increased expenses related to our development programs, including expenses related to pre-commercialization activities for ofra-vec and the initiation of new clinical trials. As of December 31, 2018,2021, we had an accumulated deficit of $188.6$262.1 million. Our losses may fluctuate significantly from quarter to quarter and year to year, depending on the timing of our clinical trials, the receipt of payments under any future collaborations we may enter into, and our expenditures on other research and development activities.
As of December 31, 2018,2021, we had cash, cash equivalents, and short-term bank deposits and restricted bank deposits of $50.5$53.5 million. To fund further operations and obtain regulatory approval for our product candidates we may need to raise additional capital.capital, and we will require additional capital to commercialize and market any products that receive regulatory approval, including full pre-commercialization activities. We may seek to raise more capital to pursue additional activities, which may be through a combination of private and public equity offerings, government grants, strategic collaborations and licensing arrangements. Additional financing may not be available when we specifically need it or may not be available on terms that are favorable to us. As of March 1, 2019,2022, we had 3941 employees.
Financial Overview
Revenues and Cost of Revenues
To date,Since inception, we have generated cumulative revenues of approximately $14.5$16.7 million revenueprimarily from an exclusive license agreementagreements for the development, commercialization, and supply of ofranergene obadenovec (“VB-111”)ofra-vec in Japan for all indications for a $15.0 millionindications. The generated revenues comprise upfront payment, in addition to a $2.0 million incurredand milestone payment.payments. The cost of revenues associated with these payments was approximately $0.3 million to Tel Hashomer for a 2% consideration that was received for granting a license or similar rights to this intellectual property.$1.5 million. We do not expect to receive any other revenue from any product candidates that we develop unless and until we obtain regulatory approval and commercialize our products, meet regulatory milestones in relation to our existing collaborative agreements, or enter into new collaborative agreements with third parties. For more detail on our revenue recognition treatment under U.S. GAAP, refer to Note 1.m. in the Notes to the Financial Statements.
Research and Development Expenses
Research and development expenses consist of costs incurred for the development of both of our platform technologies and our product candidates. Those expenses include:
| ● | employee-related expenses, including salaries and share-based compensation expenses for employees in research and development functions; |
| ● | expenses incurred in operating our laboratories and |
| ● | expenses incurred under agreements with CROs and investigative sites that conduct our clinical trials; |
| ● | expenses |
| ● | supply, development and manufacturing costs relating to clinical trial materials; |
| ● | maintenance of facilities, depreciation and other expenses, which include direct and allocated expenses for rent and insurance; and |
| ● | costs associated with |
Research and development activities are the primary focus of our business. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. Our research and development expenses may increase in absolute dollars in future periods as we continue to invest in research and development activities related to the development of our platform technologies and product candidates. In particular, our research and development expenses may increase as we develop VB-111ofra-vec beyond rGBM,ovarian cancer, and continue its clinical development in ovarian cancer and thyroid cancer.other oncology indications. In addition, our research and development expenses may increase as we develop our VB-600 series ofVB-601 product candidates.candidate into clinical development.
Research expenses are recognized as incurred. An intangible asset arising from the development of our product candidates is recognized if certain capitalization conditions are met. As of December 31, 2018,2021, we did not have any capitalized development costs.
Costs for certain development activities are recognized based on an evaluation of the progress to completion of specific tasks using information and data provided to us by our vendors and clinical sites. Nonrefundable advance payments for goods or services to be received in future periods for use in research and development activities are deferred and capitalized. The capitalized amounts are then expensed as the related goods are delivered and the services are performed.
We have received grants from the IIA as part of the research and development programs for our VTS and Lecinoxoid platform technologies.technology. The requirements and restrictions for such grants are found in the Research Law. These grants are subject to repayment through future royalty payments on any products resulting from these research and development programs, including VB-111 and VB-201.ofra-vec. Under the Research Law, royalties of 3% to 3.5% on the revenues derived from sales of products or services developed in whole or in part using these IIA grants are payable to the Israeli government. The maximum aggregate royalties paid generally cannot exceed 100% of the grants made to us, plus annual interest generally equal to the 12-month LIBOR applicable to dollar deposits, as published on the first business day of each calendar year. The total gross amount of grants actually received by us from the IIA, including accrued LIBOR interest as of December 31, 20182021 and 2017,2020, totaled $27.8$37.6 million and $26.9$36.0 million, respectively.
The Research Law is targeted at maintaining the intellectual property and manufacturing rights relating to IIA-funded projects in Israel. Under certain circumstances, where the above is not followed, the royalty rate might be higher and accordingly calculated to a formula based on the ratio of the participation by the State in the project to the total project costs incurred us.
| 57 |
In addition to paying any royalty due, we must abide by other restrictions associated with receiving such grants under the Research Law that continue to apply following repayment to the IIA. These restrictions may impair our ability to outsource manufacturing, engage in change of control transactions or otherwise transfer our know-how outside of Israel, and may require us to obtain the approval of the IIA for certain actions and transactions and pay additional royalties and other amounts to the IIA. In addition, any change of control and any change of ownership of our ordinary shares that would make a non-Israeli citizen or resident an “interested party,” as defined in the Research Law, requires prior written notice to the IIA. If we fail to comply with the Research Law, we may be subject to criminal charges.
Under applicable accounting rules, the grants income from the IIA have been accounted for as an off-set against the related research and development expenses in our financial statements. As a result, our research and development expenses are shown on our financial statements net of the IIA grants.
General and Administrative Expenses
General and administrative expenses consist principally of salaries and related costs for personnel in executive and finance functions such as salaries, benefits and share-based compensation. Other general and administrative expenses include facility costs not otherwise included in research and development expenses, communication expenses, and professional fees for legal services, patent counselingcounsel and portfolio maintenance, consulting, auditing and accounting services.
Marketing Expenses
Marketing expenses consists principally of salaries and related cost for personnel in marketing and commercialization functions such as salaries, benefits and share-based compensation, in addition to commercialization consulting services.
Financial Expenses (Income), Net
Financial income is comprised of interest income generated from interest earned on our cash, cash equivalents and short-term bank deposits and gains and losses due to fluctuations in foreign currency exchange rates mainly in the appreciation and depreciation of the NIS exchange rate against the U.S. dollar.
Financial expenses primarily consist of gains and losses due to fluctuations in foreign currency exchange rates.
Taxes on Income
We have not generated taxable income since our inception, and hadhave carry forward tax losses as of December 31, 20182021 of $164.9$222.0 million. We anticipate that we will be able to carry forward these tax losses indefinitely to future tax years. Accordingly, we do not expect to pay taxes in Israel until we have taxable income after the full utilization of our carry forward tax losses.
We recognize deferred tax assets on losses for tax purposes carried forward to subsequent years if utilization of the related tax benefit against a futurefull valuation allowance because we do not expect taxable income is expected. We have not created deferred taxes on our tax loss carry forward since their utilization is not expected in the foreseeable future.income.
Critical Accounting Policies and Significant Judgments and Estimates
This management’s discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with IFRS.U.S. GAAP. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported expenses incurred during the reporting periods. Estimates and judgments are continually evaluated and are based on historical experience and other factors, including expectations of future events that are believed to be reasonable under the circumstances.
We make estimates and assumptions concerning the future. The resulting accounting estimates will, by definition, seldom equal the related actual results. The estimates and assumptions that have a significant risk of causing a material adjustment to the carrying amounts of assets and liabilities within the next financial year are discussed below.
Revenue recognition
InThe recognition of revenue under our November 2017 license agreement with NanoCarrier requires the Company signed a license and supply agreement as more fully described in note 8. In determining the amounts to be recognized as revenue, the Company used its judgementexercise of judgment by management. Notably, our management exercises judgment in the following main issues:areas:
Identifying the performance obligations in the agreement and determining whether the license provided is distinct - based on the Company’sour analysis, the license is distinct as the licensee is able to benefit from the license on its own at its current stage (inter alia, due to sublicensing rights, rights and responsibility for development in the territory, etc.).
Allocation of the transaction price - the Companywe estimated the standalone selling prices of the services to be provided based on expected cost plus a margin and used the residual approach to estimate the standalone selling price of the license as the Company haswe have not yet established a price for the license, and it has not previously been sold on a standalone basis.
Variable consideration consists of potential future milestone payments. We determined that all such variable consideration shall be allocated to the license (the satisfied performance obligation).
See also Note 1.m in the Notes to the Financial Statements.
Share-Based Compensation
We operate a number of equity-settled, share-based compensation plans for employees (as defined in IFRS 2 “Share-Based Payments”), directors and service providers. As part of the plans, we grant employees, directors and service providers, from time to time and at our discretion, options to purchase our ordinary shares. The fair value of the services received in exchange for the grant of the options is recognized as an expense in our statements of comprehensive loss and is carried to additional paid in capital in our statements of financial position. The total amount is recognized as an expense ratably over the vesting period of the options, which is the period during which all vesting conditions are expected to be met.
Share-Based Compensation
We estimate
With respect to grants to employees, the value is measured on the date of grant based on the fair value of our share-based awardsthe equity instruments granted to employees, directors and service providersthe employees. We determine grant date fair value using the Black-Scholes option pricing model, which requires the input of highly subjective assumptions, including (a)management to make significant estimates and judgments. See Note 9 in the expected volatility of our shares, (b) the expected term of the award, (c) the risk-free interest rate, and (d) expected dividends. DueNotes to the lack of a public marketFinancial Statements for the trading of our shares until October 2014 and a lack of company-specific historical and implied volatility data, we have based our estimate of expected volatility on the historic volatility of a group of similar companies that are publicly traded. For options granted since 2015, the expected volatility was calculated using weighted average and was based on the stock price volatility of the Company since October 1st, 2014 (IPO date) and the remaining years on the stock price volatility of similar companies.
We will continue to apply this process until a sufficient amount of historical information regarding the volatility of our own share price becomes available.various assumptions used.
We are also required to estimate forfeitures at the time of grant, and revise those estimates in subsequent periods if actual forfeitures differ from the estimates. Vesting conditions are included in assumptions about the number of options that are expected to vest. At the end of each reporting period, we revise our estimates
The value of the number of options that are expected to vest based ontransactions, measured as aforesaid, is expensed over the nonmarket vesting conditions. We recognizeperiod during which the impactright of the revisionemployees and non-employees to original estimates, if any, in profitexercise or loss,receive the underlying equity instruments vests; commensurate with every periodic recognition of the expense, a corresponding adjustmentincrease is recorded to additional paid in capital.capital, included under our equity (see also Note 9 in the Notes to the Financial Statements).
The following table summarizes the weighted average assumptions we have used in our Black-Scholes calculations for the years ended December 31, 2018, 2017 and 2016:
| Year ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Employee share options: | ||||||||||||
| Risk-free interest rate | 2.46%-2.86 | % | 2.15%-2.44 | % | 1.64%-1.78 | % | ||||||
| Expected dividend yield | 0 | % | 0 | % | 0 | % | ||||||
| Expected volatility | 97.0% -100 | % | 97.0 | % | 86%-97.0 | % | ||||||
| Expected term (years) | 11 | 11 | 11 | |||||||||
The following table summarizes the allocation of our share compensation expense:
| Year ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| (in thousands) | ||||||||||||
| Research and development | $ | 2,255 | $ | 2,027 | $ | 900 | ||||||
| General and administrative | 1,541 | 1,977 | 520 | |||||||||
| Marketing | 71 | 148 | - | |||||||||
| Total | $ | 3,867 | $ | 4,152 | $ | 1,420 | ||||||
Share-based compensation expense for the year ended December 31, 2018 were $3,867 thousand, compared to $4,152 thousand for the year ended December 31, 2017, a decrease of $285 thousand.
Clinical trial accruals
Clinical trial expenses are charged to research and development expense as incurred. The Company accruesWe accrue for expenses resulting from obligations under contracts with clinical research organizations (CROs).CROs. The financial terms of these contracts are subject to negotiations, which vary from contract to contract and may result in payment flows that do not match the periods over which materials or services are provided. The Company’s objective is toWe reflect the appropriate trial expense in the financial statements by matching the appropriate expenses with the period in which services and efforts are expended. As of December 31, 2018,2021, we had approximately $1.9 million of clinical trial accruals.
Lease
Under applicable accounting rules, accounting for our leases require the company had clinical accrualsexercise of significant judgment and estimates by management. We recognize a liability at the present value of the lease payments to be made over the lease term, and concurrently recognize a right of use, or ROU, asset at the same amount of the liability, adjusted for any prepaid or accrued lease payments, plus initial direct costs incurred in respect of the lease. Determination of the lease term by management requires the exercise of significant judgment and estimates. In determining the lease term, we consider all facts and circumstances that create an economic incentive to exercise an extension option, or not exercise a termination option. Extension options are only included in the amountlease term if the lease is reasonably certain to be extended. At initial recognition of approximately $1.2 million.lease liability, we used the incremental borrowing rate, which is the rate that the lessee would have to pay to borrow the funds necessary to obtain an asset of similar value in a similar economic environment with similar terms and conditions (see also Notes 1.p and 5 in the Notes to the Financial Statements).
Results of Operations
Comparison of Years Ended December 31, 2018, 20172021, 2020 and 20162019 (in thousands)
2021 Increase | 2020 Increase | |||||||||||||||||||
| Year ended December 31, | (Decrease) | (Decrease) | ||||||||||||||||||
| 2021 | 2020 | 2019 | $ | $ | ||||||||||||||||
| Revenues | $ | 768 | 922 | 562 | $ | (154 | ) | $ | 360 | |||||||||||
| Cost of Revenues | (365 | ) | (394 | ) | (222 | ) | 29 | (172 | ) | |||||||||||
| Gross profit | 403 | 528 | 340 | (125 | ) | 188 | ||||||||||||||
| Expenses: | ||||||||||||||||||||
| Research and development, gross | $ | 23,206 | $ | 21,125 | $ | 17,460 | $ | 2,081 | $ | 3,665 | ||||||||||
| Government grants | (511 | ) | (1,469 | ) | (2,746 | ) | 958 | 1,277 | ||||||||||||
| Research and development, net | $ | 22,695 | $ | 19,656 | $ | 14,714 | $ | 3,039 | $ | 4,942 | ||||||||||
| General and administrative | 7,704 | 5,355 | 5,708 | 2,349 | (353 | ) | ||||||||||||||
| Marketing | - | - | - | - | - | |||||||||||||||
| Operating loss | 29,996 | 24,483 | 20,082 | 5,513 | 4,401 | |||||||||||||||
| Financial expense (income), net | (76 | ) | (258 | ) | (686 | ) | 182 | 428 | ||||||||||||
| Loss for the year | $ | 29,920 | $ | 24,225 | $ | 19,396 | $ | 5,695 | $ | 4,829 | ||||||||||
Revenues
| Year ended December 31, | 2018Increase (Decrease) | 2017Increase (Decrease) | ||||||||||||||||||||||||||
| 2018 | 2017 | 2016 | $ | % | $ | % | ||||||||||||||||||||||
| (in thousands) | ||||||||||||||||||||||||||||
| Revenues | $ | 585 | 13,864 | - | $ | (13,279 | ) | -96 | % | 13,864 | 100 | % | ||||||||||||||||
| Cost of Revenues | (235 | ) | (340 | ) | 105 | -31 | % | (340 | ) | 100 | % | |||||||||||||||||
| Gross profit | 350 | 13,524 | - | (13,174 | ) | -97 | % | 13,524 | 100 | % | ||||||||||||||||||
| Expenses: | ||||||||||||||||||||||||||||
| Research and development, gross | $ | 17,955 | $ | 19,959 | $ | 14,147 | $ | (2,004 | ) | -10 | % | $ | 5,812 | 41 | % | |||||||||||||
| Government grants | (2,015 | ) | (2,189 | ) | (1,700 | ) | 174 | -8 | % | (489 | ) | 29 | % | |||||||||||||||
| Research and development, net | $ | 15,940 | $ | 17,770 | $ | 12,447 | $ | (1,830 | ) | -10 | % | $ | 5,323 | 43 | % | |||||||||||||
| General and administrative | 5,220 | 5,847 | 3,828 | (627 | ) | -11 | % | 2, 019 | 53 | % | ||||||||||||||||||
| Marketing | 397 | 562 | - | (165 | ) | -29 | % | 562 | 100 | % | ||||||||||||||||||
| Operating loss | 21,207 | 10,655 | 16,275 | 10,552 | 99 | % | (5,620 | ) | -35 | % | ||||||||||||||||||
| Financial expense (income), net | (749 | ) | (517 | ) | (273 | ) | (232 | ) | 45 | % | (244 | ) | 89 | % | ||||||||||||||
| Loss | $ | 20,458 | $ | 10,138 | $ | 16,002 | $ | (10,320 | ) | 102 | % | $ | (5,864 | ) | -37 | % | ||||||||||||
Revenues.
On November 3, 2017 the Company entered into an exclusive license agreement with NanoCarrier Co., Ltd.Revenues for the development, commercialization, and supply of ofranergene obadenovec (“VB-111”)year ended December 31, 2021 were $0.8 million, compared to $0.9 million for the year ended in Japan for all indications. In exchange, the Company received an up-front and a milestone payment of $17.0 million in aggregate, of which $13.9 million was recognized in 20172020 and $0.6 million was recognizedfor the year ended December 31, 2019, a decrease of $0.1 million and an increase of $0.4 million, respectively.
Cost of revenues for the year ended December 31, 2021 were $0.4 million compared to $0.4 million for the year ended in 2018. We expect2020 and $0.2 million for the year ended December 31, 2019. The cost of revenues is attributed to recognize the balancelabor costs and other expenses related to the performance obligations that were delivered during the coming 2-3 years.period.
Research and development expenses, net.
Research and development expenses are shownpresented net of IIA grants. Research and development expenses, net, for the year ended December 31, 20182021 were $15.9$22.7 million, compared to $17.8$19.7 million for the year ended December 31, 20172020 and $12.4$14.7 million for the year ended December 31, 2016, a decrease2019, an increase of $1.8$3.0 million or 10% and an increase of $5.3$4.9 million, or 43%, respectively. The decrease in research and development expenses, net, in 2018 was comprised of approximately $4.3 million decrease in costs incurred for the Phase 3 pivotal trial of VB-111 in rGBM that was at its peak activity in 2017 and ended in 2018 and of approximately $1.5 million in contract manufacturing, partially offset by increased internal manufacturing and facility expenses at our own new manufacturing facility of approximately $3.3 million for operations (approximately $0.6 million), utilities and maintenance (approximately $0.8 million), materials (approximately $0.9 million) and depreciation (approximately $1.0 million). In addition, costs incurred for the Ovarian Phase 3 trial that commenced in 2017 increased by approximately $0.5 million in 2018.
The increase in research and development expenses, net, in 20172021 was mainly duerelated to increased expenses for the VB-111 subcontractors and consultants of $4.0 million asincrease in activity in the Phase 3 pivotalOVAL trial and ofra-vec CMC development towards anticipated BLA submission of VB-111approximately $2.0 million and a decrease of $1.0 million in rGBM continued with the completionIIA grants received in 2021 compared to 2020.
The increase in research and development expenses, net, in 2020 was comprised of patient recruitmentan increase of VB-601 development and Phase 3 clinical trial progression,activity for approximately $4.0 million in addition to costs incurred for the Ovarian Phase 3 trial that commenceda decrease in 2017, and an increase of payroll related costs due to an overall increase of share based compensation of approximately $1.0 million. This was offset by an increase in the amounts of IIA grants received of $0.5approximately $1.3 million, in 2017 due to the realizationoffset mainly by payroll related costs for share-based compensation expense of the 2016 approved grant for the GBM program.approximately $0.3 million.
General and administrative expenses.expenses
General and administrative expenses for the year ended December 31, 20182021 were $5.2$7.7 million, compared to $5.8$5.4 million for the year ended December 31, 20172020 and $3.8$5.7 million for the year ended December 31, 2016,2019, an increase of $2.3 million, and a decrease of $0.6$0.4 million, or 11%, and anrespectively.
This increase of $2.0 million or 53%, respectively. The decrease in 20182021 is mainly dueattributed to lower share-based compensation of approximately $0.6 million and lower bonuses (approximately $0.3 million, partially offset by increase in facility and other costs, mostly due to the move of the Company to the new site in Modiin. The increase in 2017 is mainly due to payroll relatedhigher premium costs for managementour directors’ and officers’ insurance, share-based compensation expense of approximately $1.0 million,and U.S. operational and professional costs compared to 2020.
This decrease in addition to an increase2020 is mainly attributed to share-based compensation expense for options granted to independent directors of approximately $600 thousand.
Marketing expenses
Marketing expenses for the year ended December 31, 2018 were $0.4 millionand financial advisory costs as compared to $0.6 million for the year ended December 31, 2017 a decrease of $0.2 million or 29% respectively.2019.
Financial expense (income), net.net
Financial expense (income), net for the year ended December 31, 20182021 was ($749) thousand,$0.1 million, compared to ($517) thousand$0.3 million for the year ended December 31, 2017,2020, and ($273) thousand$0.7 million for the year ended December 31, 2016,2019, a decrease of $232 thousand$0.2 million in income, and $244 thousand or 45% and 89%,a decrease of $0.4 million in income, respectively. The decrease in 2018 and 2017 was2021 in mainly related to higher interest received due to more favorablelower interest ratesincome compared to 2020, and favorable exchange rates each yearthe decrease in comparison2020 is mainly due lower interest income compared to its preceding year.2019.
Liquidity, and Capital Resources, and Financial Requirements
Since our inception and through December 31, 2018,2021, we have raised a totalan aggregate of $113.4$325.7 million from sales ofto fund our equity securities before the initial public offering, $40.5 million gross in our initial public offering ($34.9 million net), $15.0 million from a November 3, 2015 underwritten offering ($13.6 million net), $24.0 million from a June 7, 2016 registered direct offering ($21.9 million net), $17.9 million from a November 16, 2017 underwritten offering, $15.5 million from a June 27 registered direct offering and $22.6operations, including $29.2 million from IIA grants. Our primary uses of cash have been to fund working capital requirements and research and development, and we expect these will continue to represent our primary uses of cash. We intend to use our cash resources, together with the proceeds from theour previous offerings, described above, to advance clinical programs, working capital, certain pre-commercialization activities, and other general corporate purposes. We expect that our cash resources as of
During the year ended December 31, 2018 would provide sufficient funding for2021, we received $26.4 million in net proceeds from the sale of ordinary shares and pre-funded warrants in an underwritten public offering and an aggregate of $23.1 million in gross proceeds from warrant exercises, sales under our operations through 2021.
at-the-market facility with Oppenheimer & Co. Inc., or the Oppenheimer ATM, and direct shares sales under the ordinary share purchase agreement.
In December 2016,On February 11, 2022, we terminated the Oppenheimer ATM and entered into separate Equity Distribution Agreementsthe Jefferies ATM pursuant to an Open Market Sale AgreementSM with JMP SecuritiesJefferies LLC or Jefferies, providing for the offer and Chardan Capital Markets, LLC, as sales agents, to implement an “at the market offering” ("ATM") program under which we,sale from time to time may offer and sellof our ordinary shares having an aggregate offering price of up to $20.0$50.0 million. Sales agents were entitled to a fixed commission of 3.0% of the aggregate gross proceeds. For the year ended
On December 31, 2017, we have sold an aggregate of 224,695 ordinary shares under the ATM, the total consideration for which amounted to $1,322 thousand, net of issuance costs. In the year ended December 31, 2018 we have not sold any shares under the ATM. The ATM expired on October 18, 2018.
Funding Requirements
At December 31, 2018,2021, we had cash, cash equivalents, and short-term bank deposits and restricted bank deposits of $50.5$53.5 million and working capital of $47.0$44.9 million. We expect that our cash and cash equivalents and short-term bank deposits will enable us to fundwould provide sufficient funding for our current operating expenses and capital expenditure requirements through 2021.plans for at least the next twelve months from the date of the readout of top-line PFS data from the Phase 3 OVAL trial (data we anticipate receiving in the second half of 2022). We are unable to estimate the amounts of increased capital outlays and operating expenses associated with completing the development and commercialization, if approved, of VB-111ofra-vec and our other product candidates. Our future capital requirements will depend on many factors, including:
| ● | the costs, timing and outcome of regulatory review of |
| ● | the costs of future development activities, including clinical trials, for |
| ● | the costs of pre-commercialization and commercialization activities for ofra-vec, if approved; |
| ● | the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending intellectual property-related claims; |
| ● | the extent to which we acquire or in-license other products and technologies; and |
| ● | our ability to establish any future collaboration arrangements on favorable terms, if at all. |
Until such time, if ever, as we can generate substantial product revenue, we expect to finance our cash needs through a combination of equity offerings, debt financings, collaborations, strategic alliances and licensing arrangements. In any event, we might require additional capital to obtain regulatory approval for our product candidates, and we will require additional capital to commercialize and market any products that receive regulatory approval, including full pre-commercialization activities. We do not currently have any committed external source of funds. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interest of our shareholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a holder of our ordinary shares. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams or research programs or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market VB-111ofra-vec and any other product candidates that we would otherwise prefer to develop and market ourselves.
Cash Flows
The following table sets forth the primary sources and uses of cash for each of the periods set forth below:
| Year ended December 31, | ||||||||||||
| 2021 | 2020 | 2019 | ||||||||||
| (in thousands) | ||||||||||||
| Cash used in operating activities | $ | (24,984 | ) | $ | (23,378 | ) | $ | (13,089 | ) | |||
| Cash generated from (used in) investing activities | (15,489 | ) | 9,891 | (6,100 | ) | |||||||
| Cash provided by (used in) financing activities | 49,109 | 17,067 | (359 | ) | ||||||||
| Net increase (decrease) in cash and cash equivalents and restricted cash | $ | 8,636 | $ | 3,580 | $ | (19,548 | ) | |||||
| 61 |
| Year ended December 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| (in thousands) | ||||||||||||
| Cash used in operating activities | $ | (15,680 | ) | $ | (3,821 | ) | $ | (13,412 | ) | |||
| Cash generated from (used in) investing activities | 24,733 | (20,840 | ) | (4,091 | ) | |||||||
| Cash provided by financing activities | 13,671 | 19,510 | 21,980 | |||||||||
| Net (decrease) increase in cash and cash equivalents | $ | 22,724 | $ | (5,151) | $ | (4,477 | ) | |||||
Operating Activities
Cash used in operating activities for the year ended December 31, 20182021 was $15.7$25.0 million and consisted primarily of primarilya net loss of $20.5$29.9 million arising primarily from research and development activities, and partially offset by a $1.7 million in net reduction ofincrease in working capital and an aggregate of $0.2$3.2 million in non-cash charges.
Cash used in operating activities for the year ended December 31, 2020 was $23.4 million and consisted of primarily a net loss of $24.2 million arising primarily from research and development activities in addition to working capital changes of $2.1 million, partially offset by net aggregate non-cash charges of $4.2$2.9 million, comprised mostly of share basedshare-based compensation at fair value and depreciation.
Cash used in operating activities for the year ended December 31, 20172019 was $3.8 million and consisted primarily of net loss of $10.1 million arising primarily from research and development activities, partially offset by net reduction in working capital of $2.3 million and net aggregate non-cash charges of $3.7 million, comprised mostly of share based compensation at fair value and depreciation, partially offset by deferred revenues.
Cash used in operating activities for the year ended December 31, 2016 was $13.4$13.1 million and consisted of primarily a net loss of $16.0$19.4 million arising primarily from research and development activities, partially offset by a net reduction of working capital of $1.1$1.6 million and net aggregate non-cash charges of $1.3$3.7 million, comprised mostly of share basedshare-based compensation at fair value and depreciation.
Investing Activities
Net cash used in investing activities was $15.5 million for the year ended December 31, 2021. This was primarily due to $51.1 million investments in short-term bank deposits, offset by the maturation of $37.1 in short-term bank deposits.
Net cash generated from investing activities was $24.7$9.9 million for the year ended December 31, 2018.2020. This was primarily due to the maturity of short-term bank deposits and the purchases of Property Plant & Equipment in relation to the new Modiin facility.
Net cash used in investing activities was $20.8 million for the year ended December 31, 2017. This was primarily due to the purchases of short-term bank deposits and the purchases of Property Plant & Equipment in relation to the new Modiin facility.
deposits.
Net cash used in investing activities was $4.1$6.1 million for the year ended December 31, 2016.2019. This was primarily due to the purchases of short-term bank deposits.
Financing Activities
Net cash provided by financing activities was $13.7$49.1 million for the year ended December 31, 20182021 and was mainly the result of the proceeds from the April underwritten public offering of ordinary shares and pre-funded warrants, as well as the sales of shares pursuant to the Oppenheimer ATM and direct sales under an agreement with an institutional investor.
Net cash provided by financing activities was $17.1 million for the year ended December 31, 2020 was mainly the result of the net receipt of $13.7$16.4 million from the issuance of ordinary shares per the closing of the June 28, 2018 underwritten offering.
securities purchase agreements on May 7, 2020 and May 11, 2020.
Net cash provided byused in financing activities was $19.5$0.4 million for the year ended December 31, 2017 was mainly the result of the net receipt of $17.9 million from the issuance of ordinary shares per the closing of November 16, 2017 securities offering.
Net cash provided by financing activities was $22.0 million for the year ended December 31, 20162019 was the result of the net receipt of $21.9 million from the issuance of ordinary shares per the closing of June 7, 2016 securities offering.lease payments.
Contractual Obligations and Commitments
The following tables summarize our contractual obligations and commitments as of December 31, 2018 that will affect our future liquidity:
| Total | Less than 1 Year | 1-3 Years | 3-5 Years | More than 5 Years | ||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Licenses | $ | 345 | $ | 115 | $ | 230 | $ | - | $ | - | ||||||||||
| Operating Leases | 2,100 | 516 | 811 | 640 | 133 | |||||||||||||||
| Total | $ | 2,445 | $ | 631 | $ | 1,041 | $ | 640 | $ | 133 | ||||||||||
We also have obligations to make future payments to third parties that become due and payable on the achievement of certain development, regulatory and commercial milestones, such as the start of a clinical trial, filing of an NDA, approval by the FDA or product launch, or royalties upon sale of products. We have not included these commitmentsSee Note 8 of the Notes to the Financial Statements for further details on our statements of financial position or in the table above because the achievement and timing of these milestones is not fixed and determinable. These potential future commitments include:commitments.
|
There are no limits or caps on the amount of potential royalties. Pursuant to the agreement, the Company has the right to terminate the agreement by giving Janssen Vaccines & Prevention B.V. three months’ written notice.
|
Off-Balance Sheet Arrangements
Since our inception, we have not engaged in any off-balance sheet arrangements, as defined in the rules and regulations of the SEC, such as relationships with unconsolidated entities or financial partnerships, which are often referred to as structured finance or special purpose entities, established for the purpose of facilitating financing transactions that are not required to be reflected on our statements of financial position.
Recently Issued and Adopted Accounting Pronouncements
The recent accounting pronouncements are set forth in Note 2 to our audited financial statements beginning on page F-1 of this Annual Report.
JOBS Act
On April 5, 2012, the JOBS Act was signed into law. The JOBS Act contains provisions that, among other things, reduce certain reporting requirements for an “emerging growth company.” As an “emerging growth company,” we are electing to not take advantage of the extended transition period afforded by the JOBS Act for the implementation of new or revised accounting standards, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision to not take advantage of the extended transition period for complying with new or revised accounting standards is irrevocable.
Safe Harbor
See “Cautionary Note Regarding Forward-Looking Statements” in the introduction to this Annual Report.
Item 6. Directors, Senior Management and Employees
Executive Officers, Key Employees and Directors
The following table sets forth certain information relating to our executive officers, key employees and directors, including their ages as of February 1, 2019.2022. Unless otherwise stated, the address for our directors, and executive officers and key employees is c/o Vascular Biogenics Ltd., 8 Hasatat St. Modiin,Modi’in, Israel.
| Name | Age | Position | ||
| Executive Officers, Key Employees, and Executive Director | ||||
| Dror Harats | Chief Executive Officer and Director | |||
| Chief Financial Officer | ||||
| Eyal Breitbart | 55 | Senior Vice President, Research and | ||
| Erez Feige | Senior Vice President, Business Operations | |||
| Tami Rachmilewitz | Senior Vice President, Clinical Development | |||
| Naamit Sher | Senior Vice President, Drug Development & Regulatory Affairs | |||
Non-Executive Directors | ||||
| Marc Kozin (1)(3)(4)(6) | 60 | Chairman and Director | ||
| Ruth | Director | |||
| Shmuel (Muli) Ben Zvi (1)(2) | Director | |||
| Ron Cohen (4)(5)(6) | 66 | Director | ||
| Alison Finger (4)(5)(6) | 58 | Director | ||
| David Hastings (2)(6) | 60 | Director | ||
| Michael Rice (1)(6) | 57 | Director | ||
| Bennett M. Shapiro (3)(5)(6) | 82 | Director |
| (1) | Member of the compensation committee. |
| (2) | Member of the audit committee. |
| (3) | Member of the nominating and corporate governance committee. |
| (4) | Member of the commercialization and business committee. |
| (5) | Member of the scientific committee. |
| (6) | Independent director under the rules of the |
Executive Officers and Key Employees
Prof. Dror Harats, M.D. founded our company in 2000 and has served as our chief executive officer since our inception.January 2001. He has been a member of our board of directors since January 2001. Prof. Harats is the Chairman of the Bert W. Strassburger Lipid Center and chair of the R&D division at the Chaim Sheba Medical Center at Tel Hashomer and chairman of its Institute Review Board. Prof. Harats received his M.D. from Hadassah Medical School at the Hebrew University of Jerusalem, Israel, following which he conducted post-doctoral work at the University of California, San Francisco. Prof. Harats has also served as a visiting scientist at Syntax Discovery Research. Prof. Harats has more than 30 years of both research in the field of medicine and biotechnology as well as a professional and experienced consultant specializing in the biotechnology & pharmaceutical industry for healthcare organizations and companies. Prof. Harats currently serves on the board of directors of Art Healthcare Ltd, and as a part time chair of the R&D division at the Chaim Sheba Medical Center at Tel Hashomer and as chair of its Institute Review Board. Prof. Harats is also a Professor of Medicine in the Departments of Internal Medicine and Biochemistry at the Sackler Faculty of Medicine of Tel-Aviv University, Israel. Prof. Harats has also served as a visiting scientist at Syntax Discovery Research. Prof. Harats currently serves as an observer on the board of directors of Art Healthcare Ltd. We believe Prof. Harats is qualified to serve on our board of directors because of his extensive technical and industry experience, as well as his knowledge of our company.
Amos RonSam Backenroth has served as our chief financial officer since May 2011.October 2021. Prior to joining our company, from July 20082019 to April 2011,2021, Mr. RonBackenroth was the chief financial officer at NeuBase Therapeutics, a novel genetic medicine platform company focused on rare genetic diseases and oncology. Prior to that, from 2010 to 2019, Mr. Backenroth was the chief financial officer of Atlantium Technologies Ltd.Ohr Pharmaceutical, where he was instrumental in the company’s growth from startup to a public market capitalization of several hundred million and helped move its lead program from preclinical into late-stage clinical development. He is also a founder of Orphion Therapeutics, a company focused on one-time gene therapy treatments for ocular and central nervous system manifestations of ultra-rare diseases, and DepYmed, Inc., a privately held start-uppharmaceutical company focused on a novel phosphatase inhibition technology platform for rare diseases and cancer. From 2008 to 2010, he was an investment banker with The Benchmark Company LLC, where he raised capital and provided advisory services for biotechnology companies. Mr. Backenroth holds a B.Sc. degree in the field of clean-tech. Prior to that, Mr. Ronfinance from Touro College in New York.
Eyal Breitbart, Ph.D. has served as the chief financial officerour senior vice president, research and chief operating officer of Medical Compression Systems,operations since February 2022, and prior to that, Mr. Ronhe served as the chief financial officerour vice president, research and operations from 2013 to 2022. Prior to that, from 2006 to 2013, Dr. Breitbart served as our vice president, research. Prior to that, Dr. Breitbart served as head of Interpharm Laboratories Group,research from 2002 to 2006 and prior to that as project manager from 2001 to 2002. Dr. Breitbart holds a wholly owned subsidiaryB.Sc., M.Sc. and Ph.D. from Bar-Ilan University, Israel, and completed a post-doctoral fellowship at Tufts University School of Serono S.A. Mr. Ron holds an M.Sc. (Honors) in Chemical Technology Management from the Hebrew UniversityMedicine.
Erez Feige, Ph.D. has served as our senior vice president of Jerusalem, a B.Sc. in Business Administration, Empire State College (SUNY) (Jerusalem Branch)business operations February 2022 and a B.Sc. in Chemistry from the Hebrew University of Jerusalem.
Dr. Erez Feige hasprior to that, he served as our vice president of business operations since January 2014.from 2014 to 2022. Prior to that, from 2012 to 2014, Dr. Feige served as our director of business development and, from 2006 to 2012, Dr. Feige served as our head of biochemistry. Dr. Feige holds a B.Sc., and M.B.A. and a Ph.D. from Bar-Ilan University, Israel and completed a post-doctoral fellowship at the Dana-Farber Cancer Institute and Harvard Medical School.
Dr. Tami Rachmilewitz, M.D. has served as our senior vice president of clinical development since February 2022 and prior to that, she served as our vice president of clinical development since June 2018.from 2018 to 2022. Prior to joining our company, from 2016 to 2018,Dr. Tami Rachmilewitz served as Medical Director and Head of Pharmacovigilance for NeuroDerm, holding responsibility for all development aspects of clinical phase projects. Prior to that, from 2013 to 2016, she acted as Clinical Program Leader for Teva, leading a pivotal phase III trial in Multiple Sclerosis. From 2009 to 2013 she was a Clinical Development Medical Advisor for Novartis with expertise in Immunology. Dr. Rachmilewitz holds an M.D. from the Hadassah Medical School at the Hebrew University in Jerusalem, which is where she did her internship and residency in Psychiatry.
Dr. Eyal BreitbartNaamit Sher, Ph.D. has served as our senior vice president researchof drug development and operationsregulatory affairs since January 2014. Prior to that, from 2006 to 2013, Dr. Breitbart served as our vice president, research. Prior to that, Dr. Breitbart served as head of research from 2002 to 2006February 2022 and prior to that, as project manager from 2001 to 2002. Dr. Breitbart holds a B.Sc., M.Sc. and Ph.D. from Bar-Ilan University, Israel, and completed a post-doctoral fellowship at Tufts University School of Medicine.
Dr. Naamit Sher hasshe served as our vice president of drug development and regulatory affairs since 2006.from 2006 to 2022. Prior to joining our company, from 2005 to 2006, Dr. Sher was head of QC laboratories, operations division at Teva Pharmaceutical Industries Ltd. From 1992 to 2005, Dr. Sher acted as quality control/quality assurance director at InterPharm, a subsidiary of Ares-Serono. Dr. Sher holds a B.Sc., M.Sc. and Ph.D. from the Hebrew University of Jerusalem, Israel. She completed post-doctoral fellowships at each of the Hebrew University, Jerusalem, Israel, and Rutgers University.
Matt Trudeau joined our company in January 2022. Prior to joining our company, from 2016 to 2021, Mr. Trudeau served at bluebird bio in senior commercialization roles, most recently as head of the U.S. business team where he led design and implementation of their commercial operating model and was accountable for all aspects of U.S. commercialization for an innovative gene therapy portfolio. Prior to that, from 2010 to 2016 Matt served at Biogen, where he led Asia-Pacific business operations for a portfolio of biologics targeting neurologic and hematologic diseases. At Biogen, he also led global strategic design and implementation of the company’s first companion diagnostic platform. Matt started his career at Genzyme where, between 1996 to 2010, he advanced through sales and marketing roles of increasing impact in the diagnostics and surgical oncology segments. He holds a M.Sc. degree from Northeastern University and a B.A. degree from Colby College.
Non-Executive Directors
Adv. Ayelet HornMarc Kozin joined our board of directors in November 2020 as vice chairman and was appointed to chairman in July 2021. Mr. Kozin has three decades of industry expertise advising biopharmaceutical, life sciences and medtech companies. He is currently the chairman of the strategy advisory board of HealthCare Royalty Partners (HCR), a leading investment firm in healthcare, providing royalty monetization and senior debt, a position he has held since 2013. Previously, Mr. Kozin was a career strategy consultant, having served as president of L.E.K. Consulting’s North American practice from 1997 to 2012 and as senior advisor from 2012 to 2018. He began his career at L.E.K. in 1987 by helping establish the Boston office and led the development of L.E.K.’s industry-leading life science strategic planning practice. Mr. Kozin has served on more than a dozen boards in a variety of roles and on all committees. He serves as director and serves on the compensation committee of UFP Technologies (Nasdaq: UFPT). Previously, he served as director for Dicerna Pharmaceuticals (Nasdaq: DRNA), prior to its acquisition by Novo Nordisk. He also served on the board of Endocyte (Nasdaq: ECYT), and was also a board member of Dyax (Nasdaq: DYAX), which was acquired by Shire Plc in 2015. He also served on the boards of directors of Brandwise, Inc., Lynx Therapeutics, Inc., Assurance Medical, Inc., Medical Simulation Corporation, Advizex, and CrunchTime! Information Systems. Mr. Kozin has served as director of The Greenlight Fund, a non-profit focused on improving the lives of inner city children in families, since 2017. He was also on the board of governors at New England Medical Center and the board of DukeEngage for several years. Mr. Kozin received a B.S. degree in economics from Duke University in Durham, N.C. and a M.B.A. in finance from The Wharton School of the University of Pennsylvania in Philadelphia. We believe Mr. Kozin is qualified to serve on our general counsel since our inception in 2000,board of directors because of his extensive industry and has served as our company secretary between 2007 to 2016. Adv. Horn holds an LL.B from Tel-Aviv University, Israel, and an M.B.A. from Herriot Watt University, Edinburgh, Scotland.business background.
Non-Executive Directors
Ruth Alon has served on our board of directors since March 2010. Ms. Alon is currently the founder and chief executive officer of Medstrada, an advisory and consultancy firm in the healthcare and foodtech sectors. From 1997 to 2016, Ms. Alon has served as a general partner in Pitango Venture Capital, where she headed the life sciences activities and has led several of its portfolio companies to successful acquisitions, among them Disc-O-Tech, Colbar, Ventor and Optonol. Prior to her tenure at Pitango, Ms. Alon held senior analyst positions with Montgomery Securities from 1981 to 1987, Kidder Peabody & Co. from 1987 to 1993 and Genesis Securities, LLC from 1993 to 1996, and managed her own independent consulting business in San Francisco in the medical devices industry from 1995 to 1996. Ms. Alon was the founder and chairperson of Israel Life Science Industry, a not-for-profit organization representing the mutual goals of the then approximately 1000 Israeli life science companies. She was also the co-founder of the Israeli Advanced Technology Industries, or IATI, an umbrella organization of the hi-tech and life sciences industries in Israel, which includes venture capital funds, research and development centers of multinational corporations and others. Ms. Alon is also a board member of Moringa Acquisition Corp (Nasdaq: MACA) and of several privately held companies, including Treos Bio, Phoska Biopharma and Blue Tree Technologies. She is the chairperson of Brainsgate (privately held). Ms. Alon has a B.A. in Economics from the Hebrew University of Jerusalem, Israel, an M.B.A. from Boston University, and an M.Sc. from the Columbia University School of Physicians and Surgeons. We believe Ms. Alon is qualified to serve on our board of directors because of her extensive business and industry background, as well as her experience as a seasoned investor.
Shmuel (Muli) Ben Zvi, Ph.D. has served on our board of directors since September 2018. Dr. Ben Zvi is currently a board member at Bank Leumi, the second largest bank in Israel, and a member of its credit, technology and strategy committees. Dr. Ben Zvi is also a board member of SOL-GEL Technologies (Nasdaq: SLGL) and a member of the audit and compensation committees. From 2004 to 2014, Dr. Ben Zvi held various managerial positions at Teva Pharmaceuticals Industries Ltd., dual listed on Nasdaq and the TASE, including as vice president of finance and vice president of strategy. From 2000 to 2004, Dr. Ben Zvi was the financial advisor to the chief of general staff of the Israel Defense Forces and head of the Defense Ministry budget department. Dr. Ben Zvi holds a Ph.D. in economics from Tel-Aviv University, Israel and participated in the Harvard Business School Advanced Management Program (AMP). We believe Dr. Ben Zvi is qualified to serve on our board of directors because of his extensive finance and industry background.
Ron Cohen, M.D. has served on our board of directors since February 2015. In addition to serving on our board of directors, Dr. Cohen has served as president, chief executive officer, founder and director of Acorda Therapeutics, Inc. (Nasdaq: ACOR), since 1995. Previously he was a principal in the startup and an officer of Advanced Tissue Sciences, Inc., a biotechnology company engaged in the growth of human organ tissues for transplantation, from 1986 to 1992. Dr. Cohen is a member of the board of the Biotechnology Innovation Organization (BIO) and previously served as chairman. He served as a member of the board of Dyax Corporation (Nasdaq: DYAX) until 2016, and also previously served as Director and Chair of the New York Biotechnology Association. He is a recipient of the NY CEO Lifetime Achievement Award and the Ernst & Young Entrepreneur of the Year Award for the New York Metropolitan Region, and has been recognized by PharmaVOICE Magazine as one of the 100 Most Inspirational People in the Biopharmaceutical Industry. Dr. Cohen received his B.A. with honors in Psychology from Princeton University, and his M.D. from the Columbia College of Physicians & Surgeons. He completed his residency in Internal Medicine at the University of Virginia Medical Center, and is Board Certified in Internal Medicine. We believe Dr. Cohen is qualified to serve on our board of directors because of his extensive business and industry background.
Alison Finger joined our board in July 2021. Ms. Finger has nearly three decades of biotech and pharmaceutical leadership experience building and optimizing brands and portfolios in the areas of genetic medicine, cell therapy, oncology, neurology, virology and metabolics. Ms. Finger has served as a Principal at Auburn House Consulting LLC since June 2021. Ms. Finger previously served as chief commercial officer at bluebird bio, where she served in senior marketing and commercialization roles from 2015 until January 2021 and built the commercial infrastructure for Europe and the United States in advance of bluebird’s first gene and cell therapy product launches. Prior to bluebird, Ms. Finger served as vice president global marketing at Bristol-Myers Squibb, or BMS, from 2005 until 2014, leading the hematology/oncology, neurology, and virology franchises. In these roles, she led portfolio planning, brand and franchise commercial strategy, and supported research and development and corporate business development decisions. From 2007 until 2009, Ms. Finger also served as managing director of BMS Australia/New Zealand. Previously, she served in various marketing positions in BMS, from 1993 until 2004. Ms. Finger currently serves on the board of Decibel Therapeutics (Nasdaq: DBTX) and as member of its audit committee. Ms. Finger earned her B.A. from St. Lawrence University and an M.B.A. from Duke University’s Fuqua School of Business. We believe Ms. Finger is qualified to serve on our board of directors because of her extensive marketing and commercialization background.
David Hastings has served on our board of directors since January 2018. Mr. Hastings has more than 20 years of finance, accounting and operations experience in the bio-pharmaceutical industry. Mr. Hastings joined Arbutus BioPharma in June 2018 and currently serves as its chief financial officer. Mr. Hastings previously served as the chief financial officer and executive vice president of Incyte Corporation from 2003 until 2014. During this time, Mr. Hastings oversaw all financial aspects as Incyte transitioned from research and development to commercialization, following the launch of Jakafi(ruxolitinib). Mr. Hastings also previously served as vice president, chief financial officer and treasurer of ArQule Inc. During his tenure at ArQule, he played an important role in ArQule’s transition into a drug discovery and development organization, and in two strategic acquisitions, including the purchase of Cyclis Pharmaceuticals Inc. Prior to that, Mr. Hastings was with Genzyme Corporation as its vice president and corporate controller, and with Sepracor, Inc. where he was director of finance. Most recently, Mr. Hastings served as the chief financial officer and senior vice president of Unilife Corporation (a medical device company) from 2015 to 2017 and as its chief accounting officer and treasurer from 2016 to 2017. He is a member of the Board Director of SCYNEXIS, Inc. (Nasdaq: SCYX) and Entasis, Inc. (Nasdaq: ETTX) and chairs their Audit Committees. We believe Mr. Hastings is qualified to serve on our board of directors because of his extensive financial and business background.
| 65 |
Michael Rice joined our board in July 2021. Mr. Rice has deep experience in portfolio management, investment banking, and capital markets. Mr. Rice is a founding partner of LifeSci Advisors LLC, a life sciences investor relations consultancy, since 2010 and of LifeSci Capital LLC, a research-driven investment bank, since 2013. Previously, Mr. Rice was the co-head of Health Care Investment Banking at Canaccord Adams, where he was involved in debt and equity financing. Mr. Rice was also a managing director at Think Equity Partners, where he was responsible for managing Healthcare Capital Markets, which included structuring and executing numerous transactions. Prior to that, he served as a managing director at Bank of America serving large hedge funds and private equity healthcare funds while working closely with Investment Banking. Previously, he was a managing director at JP Morgan/Hambrecht & Quist. Mr. Rice graduated from the University of Maryland with a degree in Economics and currently sits on the board of 9 Meters Biopharma Inc. (Nasdaq: NMTR) and Navidea Biopharmaceuticals, Inc. (NYSE: NAVB). We believe Mr. Rice is qualified to serve on our board of directors because of his extensive banking and industry background.
Bennett M. Shapiro, M.D.has served on our board of directors since September 2004 and as Chairman since 2007.chairman from 2007 until 2021. In addition to serving on our board of directors, Dr. Shapiro has been a senior partner at Puretech Ventures, an innovation enterprise, since 2004, and as chairman from 2009-2015; he now continuescontinued as a Non-Executive Directornon-executive director of PureTech HealthPLC-PRTC.HealthPLC-PRTC until 2020. From 1990 to 2003, Dr. Shapiro served as executive vice president, Merck Research Laboratories. Prior to that, from 1970 to 1990, Dr. Shapiro was a professor of the Department of Biochemistry at the University of Washington and served as chairman from 1985 to 1990. Prior to joining the University of Washington, from 1965 to 1970 Dr. Shapiro served as a research associate, then section head, in the Laboratory of Biochemistry of the National Heart Institute of the U.S. National Institutes of Health. Dr. Shapiro has served as an external director on the board of directors of Momenta Pharmaceuticals from 2003-2016, various private companies, and the Drugs for Neglected Diseases Initiative, an independent, non-profit drug development partnership. Dr. Shapiro previously served on the board of directors of Celera Corporation prior to its acquisition by Quest Diagnostics Inc. Dr. Shapiro received his B.S. in chemistry from Dickinson College and his M.D. from Jefferson Medical College. Dr. Shapiro has been a Guggenheim Fellow, a fellow of the Japan Society for the Promotion of Science and a visiting professor at the University of Nice. WeDr. Shapiro received his B.S. in chemistry from Dickinson College and his M.D. from Jefferson Medical College.We believe Dr. Shapiro is qualified to serve on our board of directors because of his extensive technical and industry background, and his experience serving on boards of directors of companies in our industry, including public companies.
Prof. Ruth Arnon has served on our board of directors since August 2007. Prof. Arnon is an immunologist with the Weizmann Institute of Science in Israel. Prof. Arnon joined the staff of the Weizmann Institute in 1960, and served as vice president of the Institute from 1988 to 1997. Prof. Arnon is a member of the Israel Academy of Sciences, and from 2010 until 2015 served as its president. Prof. Arnon is also an elected member of the European Molecular Biology Organization. She has served as president of the European Federation of Immunological Societies, and as secretary-general of the International Union of Immunological Societies. Her awards and honors include the Robert Koch Prize in Medical Sciences, Spain’s Jimenez Diaz Memorial Award, France’s Legion of Honor, the Hadassah World Organization’s Women of Distinction Award, the Wolf Prize for Medicine, the Rothschild Prize for Biology, and the Israel Prize. Prof. Arnon earned her M.Sc. in Chemistry from the Hebrew University, Jerusalem, Israel, and her Ph.D. from the Hebrew University. We believe Prof. Arnon is qualified to serve on our board of directors because of her extensive technical and industry background.
Ruth Alon has served on our board of directors since March 2010. Ms. Alon is currently the founder and CEO of Medstrada. Since 1997 and until December 24, 2016, Ms. Alon has served as a general partner in Pitango Venture Capital. Prior to her tenure at Pitango, Ms. Alon held senior positions with Montgomery Securities from 1981 to 1987, Genesis Securities, LLC from 1993 to 1996, and Kidder Peabody & Co. from 1987 to 1993, and managed her own independent consulting business in San Francisco in the medical devices industry from 1995 to 1996. Ms. Alon is the chairperson of Israel Life Science Industry, a not-for-profit organization representing the mutual goals of approximately 700 Israeli life science companies. Ms. Alon has a B.A. in Economics from the Hebrew University of Jerusalem, Israel, an M.B.A. from Boston University, and an M.S. from the Columbia University School of Physicians and Surgeons. We believe Ms. Alon is qualified to serve on our board of directors because of her extensive business and industry background, as well as her experience as a seasoned investor.
Dr. Ron Cohen, M.D. joined our board in February 2015. In addition to serving on our board of directors, Dr. Cohen has served as President, Chief Executive Officer and founder of Acorda Therapeutics, Inc., since 1995. Previously he was a principal in the startup and an officer of Advanced Tissue Sciences, Inc., a biotechnology company engaged in the growth of human organ tissues for transplantation, from 1986 to 1992. Dr. Cohen received his B.A. with honors in Psychology from Princeton University, and his M.D. from the Columbia College of Physicians & Surgeons. He completed his residency in Internal Medicine at the University of Virginia Medical Center, and is Board Certified in Internal Medicine. Dr. Cohen is a Director on the Board of the Biotechnology Innovation Organization (BIO) and previously served as Chair of the Board. He served as a Director of Dyax Corporation until the end of 2015, and also previously served as Director and Chair of the New York Biotechnology Association. He is a recipient of the NY CEO Lifetime Achievement Award and the Ernst & Young Entrepreneur of the Year Award for the New York Metropolitan Region, and has been recognized by PharmaVOICE Magazine as one of the 100 Most Inspirational People in the Biopharmaceutical Industry. We believe Dr. Cohen is qualified to serve on our board of directors because of his extensive business and industry background.
Dr. Susan L. Kelley, M.D. joined our board in January 2018. Dr. Kelley is a medical oncologist with extensive experience in drug development and commercialization. Dr. Kelley worked with Bristol-Myers Squibb in Oncology and Immunology drug development from 1987 to 2001. From 2001 to 2008, Dr. Kelley worked with Bayer Healthcare Pharmaceuticals as Vice President, Global Clinical Development and Therapeutic Area Head - Oncology. From 2008 to 2011, she was Chief Medical Officer of the Multiple Myeloma Research Consortium, Dr. Kelley served as a member of the Board of Directors of Alchemia from 2013-2015. She is currently a Director at ArQule, Immune Design, and Daré Bioscience (formerly Cerulean Pharmaceuticals), all publicly-traded, US-based biotechnology companies. Susan Kelley received her M.D. from Duke University School of Medicine and completed oncology training at the Dana-Farber Cancer Institute in Boston. She was also a Fellow in Medical Oncology and Pharmacology at Yale University School of Medicine. We believe Dr. Kelley is qualified to serve on our board of directors because of her extensive industry background.
David Hastings joined our board in January 2018. Mr. Hastings has more than 18 years of finance, accounting and operations experience in the bio-pharmaceutical industry. Mr. Hastings joined Arbutus BioPharma in June 2018 and currently serves as its Chief Financial Officer. Mr. Hastings previously served as the Chief Financial Officer and Executive Vice President of Incyte Corporation from 2003 until 2014. During this time, Mr. Hastings oversaw all financial aspects as Incyte transitioned from research and development to commercialization, following the launch of Jakafi®(ruxolitinib). Mr. Hastings also previously served as Vice President, Chief Financial Officer and Treasurer of ArQule Inc. During his tenure at ArQule, he played an important role in ArQule’s transition into a drug discovery and development organization, and in two strategic acquisitions, including the purchase of Cyclis Pharmaceuticals Inc. Prior to that, Mr. Hastings was with Genzyme Corporation as its Vice President and Corporate Controller, and with Sepracor, Inc. where he was Director of Finance. Most recently, Mr. Hastings served as the Chief Financial Officer and Senior Vice President of Unilife Corporation (a medical device company) from 2015 to 2017 and as its Chief Accounting Officer and Treasurer from 2016 to 2017. He is a member of the Board Director of SCYNEXIS, Inc. and Entasis, Inc. and chairs their Audit Committees. We believe Mr. Hastings is qualified to serve on our board of directors because of his extensive financial and business background.
Dr. Shmuel (Muli) Ben Zvi joined our board in September 2018. Dr. Ben Zvi is currently a board member at Bank Leumi, the second largest bank in Israel, and a member of its audit, risk management, technology and strategy committees. Dr. Ben Zvi is also a board member of SOL-GEL Technologies (NASDAQ SLGL) and a member of the audit and compensation committees. From 2004 to 2014, Dr. Ben Zvi held various managerial positions at Teva Pharmaceuticals Industries Ltd., dual listed on Nasdaq and the TASE, including VP Finance and VP Strategy. From 2000 to 2004, Dr. Ben Zvi was the financial advisor to the Chief of General Staff of the Israel Defense Forces and head of the Defense Ministry budget department. Dr. Ben Zvi holds a Ph.D. in economics from Tel-Aviv University, Israel and participated in the Harvard Business School Advanced Management Program (AMP).
Arrangements Concerning Election of Directors; Family Relationships
Our current board of directors consists of eightnine directors.
We are not a party to, and are not aware of, any voting agreements among our shareholders. In addition, there are no family relationships among our executive officers and directors.
Advisory BoardsScientific Advisors
We established an advisory board with specific expertise in oncology. In addition we have an advisory board comprised ofWe also consult with medical key opinion leaders and industry experts with significant experience in the pharmaceutical industry.
Head of Scientific Advisory Board-Rachel W. Humphrey,Committee
Ruth Arnon, Ph.D., Weizmann Institute of Science
Timothy Cloughesy, M.D., UCLA
Jonathan A. Ledermann, M.D., UCL Cancer Institute, UK
Oncology ExpertsBradley J. Monk, M.D., FACS, FACOG, Univ. of Arizona & Creighton Univ.
Patrick Y. Wen, M.D., Dana-Farber Cancer Institute
Ovarian Cancer Advisors
Rebecca C. Arend, M.D., University of Alabama at Birmingham
Antonio Casado Herraez, M.D., Ph.D., Hospital Clínico San Carlos, Madrid, Spain
Thomas Herzog, M.D., University of Cincinnati Cancer Institute
Jonathan A. Ledermann, M.D., UCL Cancer Institute, UK
Bradley J. Monk, M.D., FACS, FACOG, Univ. of Arizona & Creighton Univ.
Kathleen Moore, M.D., University of Oklahoma Health Sciences Center
Richard T. Penson, M.D., MRCP, Massachusetts General Hospital
Ronnie Shapira-Frommer, M.D., Sheba Medical Center
Krishnansu S. Tewari, M.D., University of California
Glioblastoma (GBM) Advisors
Andrew J. Brenner, MD, PhD,M.D., Ph.D., The University of Texas Health Science Center
Nicholas Butowski, MD,M.D., University of California
Timothy Cloughesy, MD,M.D., UCLA
Patrick Y. Wen, MD,M.D., Dana-Farber Cancer Institute
Ovarian Cancer
Rebecca C. Arend, MD, University of Alabama at Birmingham
Robert A. Burger , MD, University of Pennsylvania
Thomas Herzog , MD, University of Cincinnati Cancer Institute
Bradley J. Monk , MD, FACS, FACOG, Univ. of Arizona& Creighton Univ.
Kathleen Moore , MD, University of Oklahoma Health Sciences Center
Richard T. Penson , MD, MRCP, Massachusetts General Hospital
Ronnie Shapira-Frommer, MD, Sheba Medical Center
Thyroid Cancer
Keith Bible, MD, PhD, Mayo Clinic Cancer Center
Additional Experts
Ronald Goldblum, M.D.
Bonnie Goldman, M.D.
Melanie Hartsough, PhD
John Konz, Ph.D.
David Smolin, Ph.D.
Compensation of Executive Officers, Key Employees, and Directors
The aggregate compensation paid by us to our current directors, and executive officers, and key employees, including share basedshare-based compensation, for the year ended December 31, 2018,2021, was $4.3$3.9 million. This amount includes any amounts set aside or accrued to provide pension, severance, retirement, annual leave and recuperation or similar benefits or expenses. It does not include any business travel, relocation, professional and business association dues and expenses reimbursed to office holders,expenses, and other benefits commonly reimbursed or paid by companies in Israel. The above also includes the provision for bonuses for the year ended December 31, 20182021 in the amount of $0.4$0.5 million. As of December 31, 2018,2021, options and RSU’s to purchase an aggregate of 3,408,5876,065,642 ordinary shares granted to our directors, and executive officers, and key employees were outstanding under the Employee Share Ownership and Option Plan (2000), or the 2000 Plan, and the Employee Share Ownership and Option Plan (2011), or the 2011 Plan, and the Employee Share Ownership and Option Plan (2014), or the 2014 Plan at a weighted average exercise price of $2.81$2.19 per share.
At our annual general meeting held on October 19, 2021, our shareholders approved the terms of the Non-Employee Directors New Compensation Scheme, effective as of the date of the said annual general meeting. The New Compensation Scheme includes the following:
| ● | Cash Compensation |
| ○ | Annual cash compensation of $35,000 to each Non-Employee director, other than the Chairman of the Board and the former Chairman of the Board. |
| ○ | Audit Committee: Additional annual cash compensation of $15,000 to the Chairman of the Audit Committee and $7,500 to each member of the Audit Committee other than the Chairman. |
| ○ | Compensation Committee: Additional annual cash compensation of $12,000 to the Chairman of the Compensation Committee and $6,000 to each member of the Compensation Committee other than the Chairman. |
| ○ | Nominating and Corporate Governance Committee and other committees: Additional annual cash compensation of $8,000 to the Chairman of the Nominating and Corporate Governance Committee or other committees and $4,000 to each member of the Nominating and Corporate Governance Committee or other committees other than the Chairman. |
| ○ | Proration: Pro rata cash compensation of the annual cash compensation amounts set forth above shall be made, as applicable, to (i) any director who ceases to be a director, Chairman of the Board or member or chairman of any committee of the Board and (ii) any new Non-Employee director who is appointed by the Board, any independent director who is appointed to the position of Chairman of the Board or chairman of any such committee of the Board or any independent director who is appointed to serve on any such committee of the Board, for their services rendered as a director and/or committee member, for the portion of the year in which such director so served. |
| ● | Equity Compensation |
| ○ | Initial Equity Grant: One-time equity grant upon initial appointment or election to the Board equal to 0.1% of the Company’s capital on a fully diluted basis as of the date of grant, which shall vest upon and in the manner approved by the Compensation Committee and the Board of Directors, but not less than 2 years until full vesting. |
| ○ | Annual Equity Grant: Annual equity grants of 0.067% of the Company’s share capital on a fully diluted basis as of the date of grant to each continuing director, which shall vest upon and in the manner approved by the Compensation Committee and the Board of Directors, but not less than 2 years until full vesting. |
The New Compensation Scheme added to and replaced the respective provisions of our 2019 Compensation Policy.
| 67 |
Board of Directors
Under the Israeli Companies Law, 5759-1999, or the Companies Law, the management of our business is vested in our board of directors. Our board of directors may exercise all powers and may take all actions that are not specifically granted to our shareholders or to management. Our executive officers are responsible for our day-to-day management and have individual responsibilities established by our board of directors. Our chief executive officer is appointed by, and serves at the discretion of, our board of directors, subject to the employment agreement that we have entered into with him. All other executive officers are also appointed by our board of directors, and are subject to the terms of any applicable employment agreements that we may enter into with them.
Under our amended and restated articles of association, our board of directors must consist of at least three and not more than nine directors, including the external directors. Our board of directors currently consists of seven directors, including two directors who were formerly defined as externalnine directors. Our amended and restated articles of association further provides that external directors are elected according to the special election requirements under the Companies Law and two ofFollowing our directors were nominated as external directors in compliance with the Companies Law. Following the adoption by the Company of certain reliefs provided under the Companies Law, the Company iswe are exempt from the requirement to appoint external directors and the individuals formerly appointed as external directors continue to serve as part of our board of directors until the end of their term and may be removed from office in the same manner as any other director. We have only one class of directors.
The following of our directors were elected in accordance with the terms of our articles of association in effect prior to the initial public offering of our shares on NASDAQNasdaq and are nominated for re-election by our shareholders at any consecutive annual general meeting:
| ● | Dr. Shapiro was appointed as an industry expert by a majority of the other directors, a majority that included representatives of our major shareholders. |
| ● | Prof. Harats was entitled to be a board member for so long as Prof. Harats is either (i) the chief executive officer of our company; or (ii) a holder of 3% or more of our issued and outstanding share capital; |
| ● | Ms. Alon was originally appointed by persons affiliated with Pitango Venture Capital; Since 2017 Ms. Alon is not related to |
Upon the adoption of our amended and restated articles of association upon the closing of our initial public offering,IPO, the rights set forth in the previous articles were terminated and no additional agreements exist with respect to the nomination of our board members.
On February 11, 2015, at a general meeting of our shareholders, each of Dr. Ron Cohen and Mr. Philip Serlin was appointed as an external director by a majority of the shareholders who have no personal interest in his nomination for a period of three years. In accordance with the exemption available to certain Israeli public companies, whose shares are traded on NASDAQ,Nasdaq, we chose as of November 7, 2016 not to follow the requirements of Companies Law with regard to the appointment of “external directors” as defined in the Companies Law, and instead, to follow the NASDAQNasdaq rules applicable to USU.S. domestic companies with respect to the appointment of independent directors. Dr. Ron Cohen and Mr. Philip Serlin shall continue to serve as part of our board of directors until the end of their term and may be removed from office in the same manner as any other director. As long as we follow such reliefs,these requirements, any reference to the election of our external directors in our amended articles of association shall have no actual expression.
We comply with NASDAQNasdaq rules that a majority of our directors are independent. Our board of directors has determined that with the exception of Prof.Dror Harats, all of our directors are independent under such rules.
In accordance with the exemption available to foreign private issuers under NASDAQNasdaq rules, we do not intend to follow the requirements of NASDAQNasdaq rules with regard to the process of nominating directors, and instead, will follow Israeli law and practice, in accordance with which our board of directors (or a committee thereof) is authorized to recommend to our shareholders director nominees for election. See “Item 16G. Corporate Governance” for more information.
Under the Companies Law and our amended and restated articles of association, nominees for directors may also be proposed by any shareholder holding at least 1% of our outstanding voting power. However, any such shareholder may propose a nominee only if a written notice of such shareholder’s intent to propose a nominee has been given to our company secretary (or, if we have no such company secretary, our chief executive officer).secretary. Any such notice must include certain information, including, among other things, a description of all arrangements between the nominating shareholder and the proposed director nominee(s) and any other person pursuant to which the nomination(s) are to be made by the nominating shareholder, the consent of the proposed director nominee(s) to serve as our director(s) if elected and a declaration signed by the nominee(s) declaring that there is no limitation under the Companies Law preventing their election, and that all of the information that is required under the Companies Law to be provided to us in connection with such election has been provided.
| 68 |
In addition, our amended and restated articles of association allow our board of directors to appoint directors to fill vacancies on our board of directors, for a term of office equal to the remaining period of the term of office of the director(s) whose office(s) have been vacated.
Under the Companies Law, our board of directors must determine the minimum number of directors who are required to have accounting and financial expertise (as defined in the Companies Law). In accordance with the exemption available to certain Israeli public companies, whose shares are traded on NASDAQ,Nasdaq, our board of directors elected not to follow the requirements of Companies Law with regard to the appointment of directors with accounting and financial expertise as defined in the Companies Law, and instead, to follow the NASDAQNasdaq rules applicable to USU.S. domestic companies with respect to the financial expertise of the directors. The exemption applies as long as the Company haswe have no controlling shareholder, isare in compliance with applicable USU.S. law and regulations and compliescomply with the NASDAQNasdaq rules applicable to USU.S. domestic companies with respect to the appointment of independent directors and to the composition of the compensation and audit committees. Our board may further resolve at any time that we shall no longer follow the reliefs and in such event, we shall be required to appoint directors with accounting and financial expertise as defined in the Companies Law. Our board of directors has determined that the minimum number of directors who are required to have accounting and financial expertise is one.
External Directors
Under the Companies Law, a public company is required to have at least two directors who qualify as external directors. In accordance with the exemption available to certain Israeli public companies, whose shares are traded on NASDAQNasdaq and which do not have a controlling shareholder (the “Exemption to Foreign-listed Israeli Companies”), our board of directors elected not to follow the requirements of Companies Law with regard to the appointment of “external directors” as defined in the Companies Law, and instead, to follow the NASDAQNasdaq rules applicable to USU.S. domestic companies with respect to the appointment of independent directors. The exemption applies as long as the Company haswe have no controlling shareholder, isare in compliance with applicable USU.S. law and regulations and compliescomply with the NASDAQNasdaq rules applicable to USU.S. domestic companies with respect to the appointment of independent directors and to the composition of the compensation and audit committees. Our board may further resolve at any time that we shall no longer follow the reliefsor determine that we are no longer in compliance with the requirements of such exemption, and in such event, we shall be required to appoint two directors as external directors.
Role of Board in Risk Oversight Process
Risk assessment and oversight are an integral part of our governance and management processes. Our board of directors encourages management to promote a culture that incorporates risk management into our corporate strategy and day-to-day business operations. Management discusses strategic and operational risks at regular management meetings, and conducts specific strategic planning and review sessions during the year that include a focused discussion and analysis of the risks facing us. Throughout the year, senior management reviews these risks with the board of directors at regular board meetings as part of management presentations that focus on particular business functions, operations or strategies, and presents the steps taken by management to mitigate or eliminate such risks.
Leadership Structure of the Board
In accordance with the Companies Law and our amended and restated articles of association, our board of directors is required to appoint one of its members to serve as chairman of the board of directors. Our board of directors has appointed Dr. ShapiroMarc Kozin to serve as chairman of the board of directors.
Committees of the Board of Directors
We have the following statutory required committees: an audit committee, a compensation committee and a nominatingnomination and corporate governance committee. We have adopted a charter for each of these committees. In addition, we also have commercialization, business, and scientific committees.
Audit Committee
Under the Companies Law, we are required to appoint an audit committee. The audit committee must be comprised of at least three directors, including all of the external directors, one of whom must serve as chairman of the committee. In accordance with the exemption available to certain Israeli public companies, whose shares are traded on NASDAQ,Nasdaq, we chose as of November 7, 2016 and for as long the required conditions precedent are met and unless otherwise decided by our board of directors, not to follow the requirements of Companies Law with regard to the composition of the audit committee, and instead, will follow the NASDAQNasdaq rules applicable to USU.S. domestic companies with respect to the appointment and composition of the audit committee.
Under the NASDAQNasdaq listing requirements, we are required to maintain an audit committee consisting of at least three independent directors, all of whom are financially literate and at least one of whom has accounting or related financial management expertise. Our audit committee consists of Mr. David Hastings, Ms. Ruth Alon and Dr. Shmuel (Muli) Ben Zvi and is chaired by Mr. Hastings. Mr. Hastings and Dr. Ben Zvi are the audit committee financial experts as defined by the Securities and Exchange CommissionSEC rules and all of the members of our audit committee have the requisite financial literacy as defined by the NASDAQNasdaq Stock Market rules. All the members of our audit committee are “independent” as such term is defined in Rule 10A-3(b)(1) under the Exchange Act and under the listing standards of NASDAQ.Nasdaq.
| 69 |
Our board of directors has adopted an audit committee charter setting forth the responsibilities of the audit committee consistent with the rules of the SecuritiesSEC and Exchange Commission and NASDAQNasdaq rules as well as the requirements for such committee under the Companies Law, including the following:
| ● | oversight of our independent registered public accounting firm and recommending the engagement, compensation or termination of engagement of our independent registered public accounting firm to the board of directors in accordance with Israeli law; |
| ● | recommending the engagement or termination of the person filling the office of our internal auditor; and |
| ● | recommending the terms of audit and non-audit services provided by the independent registered public accounting firm for pre-approval by our board of directors. |
Our audit committee provides assistance to our board of directors in fulfilling its legal and fiduciary obligations in matters involving our accounting, auditing, financial reporting, internal control and legal compliance functions by pre-approving the services performed by our independent accountants and reviewing their reports regarding our accounting practices and systems of internal control over financial reporting. Our audit committee also oversees the audit efforts of our independent accountants and takes those actions that it deems necessary to satisfy itself that the accountants are independent of management.
Under the Companies Law, our audit committee is responsible for:
| ● | determining whether there are deficiencies in our business management practices, including in consultation with our internal auditor or the independent auditor, and making recommendations to the board of directors to improve such practices; |
| ● | determining whether to approve certain related party transactions (including transactions in which an office holder has a personal interest) and whether such transaction is extraordinary or material under the Companies Law (see “-Approval of Related Party Transactions Under Israeli Law”); |
| ● | where the board of directors approves the work plan of the internal auditor, to examine such work plan before its submission to the board and propose amendments thereto; |
| ● | establishing the approval process for certain transactions with a controlling shareholder or in which a controlling shareholder has a personal interest; |
| ● | examining our internal controls and internal auditor’s performance, including whether the internal auditor has sufficient resources and tools to dispose of its responsibilities; |
| ● | examining the scope of our independent auditor’s work and compensation and submitting a recommendation with respect thereto to our board of directors or shareholders, depending on which of them is considering the appointment of our auditor; and |
| ● | establishing procedures for the handling of employees’ complaints as to deficiencies in the management of our business and the protection to be provided to such employees. |
Our audit committee may not approve any actions requiring its approval (see “-Approval of Related Party Transactions Under Israeli Law”), unless at the time of approval a majority of the committee’s members are present, which majority consists of unaffiliated directors including at least one external director.directors.
Compensation Committee
Our compensation committee consists of Dr. Ben Shapiro, Dr. CohenMarc Kozin, Shmuel (Muli) Ben-Zvi, and of Dr. Ruth Arnon. Dr. Ben ShapiroMichael Rice. Mr. Kozin serves as the chairman of the compensation committee. The members of our compensation committee are independent under the NASDAQNasdaq listing requirements.
Under the Companies Law, the board of directors of a public company must appoint a compensation committee. In accordance with the exemption available to certain Israeli public companies, whose shares are traded on NASDAQ,Nasdaq, we chose as of November 7, 2016 and for as long the required conditions precedent are met and unless otherwise decided by our board of directors, not to follow the requirements of Companies Law with regard to the composition of the compensation committee, and instead, will follow the NASDAQNasdaq rules applicable to USU.S. domestic companies with respect to the appointment and composition of the compensation committee.
The duties of the compensation committee include the recommendation to our board of directors of a policy regarding the terms of engagement of office holders, to which we refer as a compensation policy. That policy must be adopted by the company’s board of directors, after considering the recommendations of the compensation committee, and will need to be brought every three years for approval by the company’s shareholders, which approval requires what we refer to as a special majority. A special majority approval requires shareholder approval by a majority vote of the shares present and voting at a meeting of shareholders called for such purpose, provided that either: (a) such majority includes at least a majority of the shares held by all shareholders who are not controlling shareholders and do not have a personal interest in such compensation arrangement; or (b) the total number of shares of non-controlling shareholders and shareholders who do not have a personal interest in the compensation arrangement and who vote against the arrangement does not exceed 2% of the company’s aggregate voting rights. On May 27, 2015December 30, 2019, our shareholders approved our compensation policy and we intend to extend the compensation policy for an additional three-year term.term and on October 19, 2021, our shareholders approved our New Compensation Scheme, and described in “Item 6: Compensation of Executive Officers, Key Employees, and Directors.”
Our compensation policy must serve as the basis for decisions concerning the financial terms of employment or engagement of office holders, including exculpation, insurance, indemnification or any monetary payment or obligation of payment in respect of employment or engagement. The compensation policy must relate to certain factors, including advancement of the company’s objectives, the company’s business plan and its long term strategy, and creation of appropriate incentives for office holders. It must also consider, among other things, the company’s risk management, size and nature of its operations. The term office holder is defined under the Companies Law as the general manager, chief executive officer, chief business manager, deputy general manager, vice general manager, any other person assuming the responsibilities of any of these positions regardless of that person’s title, a director, or a manager directly subordinate to the general manager. The compensation policy must furthermore consider the following additional factors:
| ● | the knowledge, skills, expertise, and accomplishments of the relevant office holder; |
| ● | the office holder’s roles and responsibilities and prior compensation agreements with him or her; |
| ● | the relationship between the terms offered and the average compensation of the other employees of the company, including those employed through manpower companies; |
| ● | the impact of disparities in salary upon work relationships in the company; |
| ● | the possibility of reducing variable compensation at the discretion of the board of directors; |
| ● | the possibility of setting a limit on the exercise value of non-cash variable equity-based compensation; and |
| ● | as to severance compensation, the period of service of the office holder, the terms of his or her compensation during such service period, the company’s performance during that period of service, the person’s contributions towards the company’s achievement of its goals and the maximization of its profits, and the circumstances under which the person is leaving the company. |
The compensation policy must also include the following principles:
| ● | the link between variable compensation and long term performance and measurable criteria; |
| ● | the relationship between variable and fixed compensation, and the ceiling for the value of variable compensation; |
| ● | the conditions under which an office holder would be required to repay compensation paid to him or her if it was later shown that the data upon which such compensation was based was inaccurate and was required to be restated in the company’s financial statements; |
| ● | the minimum holding or vesting period for variable, equity-based compensation; and |
| ● | maximum limits for severance compensation. |
The compensation committee is responsible for (a) recommending the compensation policy to a company’s board of directors for its approval (and subsequent approval by its shareholders) and (b) duties related to the compensation policy and to the compensation of a company’s office holders as well as functions previously fulfilled by a company’s audit committee with respect to matters related to approval of the terms of engagement of office holders, including:
| ● | recommending whether a compensation policy should continue in effect, if the then- current policy has a term of greater than three years (approval of either a new compensation policy or the continuation of an existing compensation policy must in any case occur every three years); |
| ● | recommending to the board of directors periodic updates to the compensation policy; |
| ● | assessing implementation of the compensation policy; and |
| ● | determining whether the compensation terms of the chief executive officer of the company need not be brought to approval of the shareholders. Our board of directors has adopted a compensation committee charter setting forth the responsibilities of the committee, which include: |
Our board of directors has adopted a compensation committee charter setting forth the responsibilities of the committee, which include:
| ● | the responsibilities set forth in the compensation policy; |
| ● | reviewing and approving the granting of options and other incentive awards to the extent such authority is delegated by our board of directors; and |
| ● | reviewing, evaluating and making recommendations regarding the compensation and benefits for our non-employee directors. |
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee consists of Dr.Bennett Shapiro, Mr. Gonczarowski, Ms.Marc Kozin, and Ruth Alon and Dr. Arnon, and is chaired by Dr. Shapiro.Mr. Kozin. Each of the members of our nominating and corporate governance committee are independent under the listing requirements of The NASDAQthe Nasdaq Global Market.
Our board of directors has adopted a nominating and governance committee charter sets forth the responsibilities of the nominating and governance committee which include:
| ● | overseeing and assisting our board in reviewing and recommending nominees for election as directors; |
| ● | assessing the performance of the members of our board; and |
| ● | establishing and maintaining effective corporate governance policies and practices, including, but not limited to, developing and recommending to our board a set of corporate governance guidelines applicable to our company. |
Internal AuditorScientific Committee
Our scientific committee consists of Bennett Shapiro, Ron Cohen, Allison Finger, Dror Harats and Ruth Arnon as an advisor to the committee, and is chaired by Dr. Shapiro.
Commercial and Business Committee
Our commercial and business committee consists of Allison Finger, Marc Kozin and Ron Cohen, and is chaired by Ms. Finger.
Internal Auditor
Under the Companies Law, the board of directors of a public company must appoint an internal auditor based on the recommendation of the audit committee. The role of the internal auditor is to examine, among other things, our compliance with applicable law and orderly business procedures. The audit committee is required to oversee the activities and to assess the performance of the internal auditor as well as to review the internal auditor’s work plan. Our internal auditor is Mr. Zachi Refaeli from Ernst & Young Israel.
An internal auditor may not be:
| ● | a person (or a relative of a person) who holds more than 5% of the company’s outstanding shares or voting rights; |
| ● | a person (or a relative of a person) who has the power to appoint a director or the general manager of the company; |
| ● | an office holder or director of the company; or |
| ● | a member of the company’s independent accounting firm, or anyone on its behalf. |
| 72 |
Approval of Related Party Transactions Under Israeli Law
Fiduciary Duties of Directors and Executive Officers
The Companies Law codifies the fiduciary duties that office holders owe to a company. Each person listed in the table under “Management-Executive Officers, Senior Management and Directors” isand management members of at least a VP level are considered an office holder under the Companies Law.
An office holder’s fiduciary duties consist of a duty of care and a duty of loyalty. The duty of care requires an office holder to act with the level of care with which a reasonable office holder in the same position would have acted under the same circumstances. The duty of loyalty requires that an office holder act in good faith and in the best interests of the company.
The duty of care includes a duty to use reasonable means to obtain:
| ● | information on the advisability of a given action brought for his or her approval or performed by virtue of his or her position; and |
| ● | all other important information pertaining to these actions. |
The duty of loyalty includes a duty to:
| ● | refrain from any conflict of interest between the performance of his or her duties to the company and his or her other duties or personal affairs; |
| ● | refrain from any activity that is competitive with the company; |
| ● | refrain from exploiting any business opportunity of the company to receive a personal gain for himself or herself or others; and |
| ● | disclose to the company any information or documents relating to the company’s affairs which the office holder received as a result of his or her position as an office holder. |
Disclosure of Personal Interests of an Office Holder and Approval of Certain Transactions
The Companies Law requires that an office holder promptly disclose to the company any personal interest that he or she may be aware of and all related material information or documents concerning any existing or proposed transaction by the company. An interested office holder’s disclosure must be made promptly and in any event no later than the first meeting of the board of directors at which the transaction is considered. An office holder is not obliged to disclose a personal interest if it derives solely from the personal interest of his or her relative in a transaction that is not considered as an extraordinary transaction.
A “personal interest” is defined under the Companies Law to include a personal interest of any person in an act or transaction of a company, including the personal interest of such person’s relative or of a corporate body in which such person or a relative of such person is a 5% or greater shareholder, director or general manager or in which he or she has the right to appoint at least one director or the general manager, but excluding a personal interest stemming from one’s ownership of shares in the company.
A personal interest furthermore includes the personal interest of a person for whom the office holder holds a voting proxy or the personal interest of the office holder with respect to his or her vote on behalf of a person for whom he or she holds a proxy even if such shareholder has no personal interest in the matter. An office holder is not, however, obliged to disclose a personal interest if it derives solely from the personal interest of his or her relative in a transaction that is not considered an extraordinary transaction.
Under the Companies Law, an extraordinary transaction is defined as any of the following:
| ● | a transaction other than in the ordinary course of business; |
| ● | a transaction that is not on market terms; or |
| ● | a transaction that may have a material impact on the company’s profitability, assets or liabilities. |
If it is determined that an office holder has a personal interest in a transaction, approval by the board of directors is required for the transaction, unless the company’s articles of association provide for a different method of approval. Further, so long as an office holder has disclosed his or her personal interest in a transaction, the board of directors may approve an action by the office holder that would otherwise be deemed a breach of duty of loyalty. However, a company may not approve a transaction or action that is adverse to the company’s interest or that is not performed by the office holder in good faith. An extraordinary transaction in which an office holder has a personal interest requires approval first by the company’s audit committee and subsequently by the board of directors. The compensation of, or an undertaking to indemnify or insure, an office holder who is not a director requires approval first by the company’s compensation committee, then by the company’s board of directors, and, if such compensation arrangement or an undertaking to indemnify or insure is inconsistent with the company’s stated compensation policy or if the office holder is the chief executive officer (apart from a number of specific exceptions), then such arrangement is subject to a special majority approval. Arrangements regarding the compensation, indemnification or insurance of a director require the approval of the compensation committee, board of directors and shareholders by ordinary majority, in that order, and under certain circumstances, a special majority approval. If shareholders of a company do not approve the compensation terms of office holders, other than directors, but including the chief executive officer, the compensation committee and board of directors may override the shareholders’ decision, subject to certain conditions.
| 73 |
Generally, a person who has a personal interest in a matter which is considered at a meeting of the board of directors or the audit committee may not be present at such a meeting or vote on that matter unless the chairman of the relevant committee or board of directors (as applicable) determines that he or she should be present in order to present the transaction that is subject to approval. If a majority of the members of the audit committee or the board of directors (as applicable) has a personal interest in the approval of a transaction, then all directors may participate in discussions of the audit committee or the board of directors (as applicable) on such transaction and the voting on approval thereof, but shareholder approval is also required for such transaction.
Disclosure of Personal Interests of Controlling Shareholders and Approval of Certain Transactions
Pursuant to Israeli law, the disclosure requirements regarding personal interests that apply to directors and executive officers also apply to a controlling shareholder of a public company. See “-Major Shareholders and Related Party Transactions” for a definition of controlling shareholder. In the context of a transaction involving a shareholder of the company, a controlling shareholder also includes a shareholder who holds 25% or more of the voting rights in the company if no other shareholder holds more than 50% of the voting rights in the company. For this purpose, the holdings of all shareholders who have a personal interest in the same transaction will be aggregated. The approval of the audit committee, the board of directors and a special majority, in that order, is required for (a) extraordinary transactions with a controlling shareholder or in which a controlling shareholder has a personal interest, (b) the engagement with a controlling shareholder or his or her relative, directly or indirectly, for the provision of services to the company, (c) the terms of engagement and compensation of a controlling shareholder or his or her relative who is not an office holder or (d) the employment of a controlling shareholder or his or her relative by the company, other than as an office holder.
To the extent that any such transaction with a controlling shareholder is for a period extending beyond three years, approval is required once every three years, unless, with respect to certain transactions, the audit committee determines that the duration of the transaction is reasonable given the circumstances related thereto.
Arrangements regarding the compensation, indemnification or insurance of a controlling shareholder in his or her capacity as an office holder require the approval of the compensation committee, board of directors and shareholders by a special majority and the terms thereof may not be inconsistent with the company’s stated compensation policy.
Pursuant to regulations promulgated under the Companies Law, certain transactions with a controlling shareholder or his or her relative, or with directors, that would otherwise require approval of a company’s shareholders may be exempt from shareholder approval upon certain determinations of the audit committee and board of directors. Under these regulations, a shareholder holding at least 1% of the issued share capital of the company may require, within 14 days of the publication of such determinations, that despite such determinations by the audit committee and the board of directors, such transaction will require shareholder approval under the same majority requirements that would otherwise apply to such transactions.
Employment Agreements with Executive Officers, Key Employees, and Directors
We have entered into written employment agreements with each of Dror Harats Erez Feige, Amos Ron, Tami Rachmilewitz, Eyal Breitbart and Naamit Sher.other senior members of management located in Israel. All such agreements contain provisions regarding non-competition, confidentiality of information and assignment of inventions. The non-competition provisions apply for a period of 24 months following termination of the respective officer’s or key employee’s employment. In addition, we are required to provide notice of between three and sixnine months prior to terminating thetheir employment of such executive officers other than in the case of a termination for cause. Other than with respect to Prof. Harats, these agreements do not provide for benefits upon the termination of these executives’ respective employment with us, other than payment of salary and benefits during the required notice period for termination of these agreements, which varies under these individual agreements. Prof. Harats’s agreement provides for six months of severance in the event Prof. Harats’s employment is terminated by us without cause or terminated by Prof. Harats for good reason. Pursuant to his employment agreement, “Cause” means breach by Prof. Harats’s of any of the material terms or conditions of his employment agreement; Prof. Harats’s conviction of any felony related to our business,crime involving moral turpitude or dishonesty or the commission of a serious breach of trust by Prof. Harats, including theft, embezzlementcriminal offense, or fraud against us or any of our funds, self-dealing, prohibited disclosure of confidentialsubsidiaries or proprietary information andaffiliates; Prof. Harats’s engagement in any prohibited business competitivewillful refusal to our own, Prof. Harats’s disregard ofperform the lawful instructions of our board of directors with respectpertaining to the Prof. Harats’ employment under his employment agreement; any breach of Prof. Harats’s fiduciary duties or duties of care to our company or any of our subsidiaries or affiliates (except for conduct taken in good faith); any conduct (other than conduct in good faith) materially detrimental to us following notice, or Prof. Harats’s willful failure to perform any of his fundamental functionsour subsidiaries or duties.affiliates; or circumstances that deny Prof. Harats to severance payment under any applicable law or under any judicial decision of a competent tribunal authority. Pursuant to his employment agreement, “Good reason” means a material reduction in Prof. Harats’s status, title, positionduties, responsibilities or responsibilities,authority, a material reduction in Prof. Harats’s salaryoverall compensation package which is not part of a general reduction inpolicy pursuant to which the salary applicable toof all of our employees,other senior officers is reduced in a failure by us to continue any material compensation or benefit plan, program or practice in which Prof. Harats is participating, orcomparable manner, a material breach by us of any provision of Prof. Harats’s employment agreement.agreement, or if Prof. Harats resigns when such resignation is considered as a discharge or constructive dismissal which entitles him to severance payment under Israeli law.
We have also entered into written employment agreements and Employee Confidentiality, Assignment, Non-solicitation and Non-competition Agreements, or the Confidentiality Agreements, through our wholly owned U.S. subsidiary, VBL Inc., with Sam Backenroth as Chief Financial Officer and Matthew Trudeau as Chief Commercial Officer. Both Confidentiality Agreements contain provisions regarding non-competition, confidentiality of information and assignment of inventions. The non-competition provisions apply for a period of 12 months following the last day of employment of the respective officer. Under certain conditions, these employment agreements provide for benefits upon the termination of these executives’ respective employment with us, other than payment of salary and benefits. Such benefits include (i) continuation of the base salary as of the date of termination for the 12-month period in Mr. Backenroth’s case and nine months in Mr. Trudeau’s case; (ii) continuation of group health plan benefits under certain terms; and (iii) any vested equity rights and vested retirement benefits, if any, to which such officers may be entitled under our employee benefit plans as of the termination date.
In addition, we have entered into compensation agreements with certain of our directors. The amounts payable pursuant to these arrangements have been approved by our board of directors and shareholders.
Our directors do not receive compensation for their service as our directors or otherwise, unless such compensation is approved by our compensation committee, and then by the board of directors followed by the shareholders. The compensation of our directors may be fixed, as an all-inclusive payment or as payment for participation in meetings, or as a combination thereof. In addition, such compensation may include: (i) in the case of a director who is also an officer, a salary or other compensation in respect of his or her work as an officer, as may be agreed upon by the director and us; and (ii) reimbursement of expenses, including travel expenses, expended in connection with his or her duties as a member of the board of directors.
| 74 |
Employees
As of MarchFebruary 1, 2019,2022, we employed 3941 employees, including 3130 in research and development, and 811 in general and administrative positions, and of which 1312 employees have either MDsM.D.s or PhDs. AllPh.D.s. 39 of our employees are located in Israel.Israel and two are located in the United States. We believe our employee relations are good.
Israeli labor laws govern the length of the workday, minimum wages for employees, procedures for hiring and dismissing employees, determination of severance pay, annual leave, sick days, advance notice of termination of employment, equal opportunity and anti- discrimination laws and other conditions of employment. Subject to specified exceptions, Israeli law generally requires severance pay upon the retirement, death or dismissal of an employee, and requires us and our employees to make payments to the National Insurance Institute, which is similar to the U.S. Social Security Administration. Our employees have defined benefit pension plans that comply with the applicable Israeli legal requirements.
None of our employees currently work under any collective bargaining agreements.
Share Ownership
For information regarding the share ownership of our directors and executive officers, please refer to “-Equity“Equity Compensation Plans” below and “Item 7. Major Shareholders and Related Party Transactions-MajorTransactions- Significant Shareholders.”
As of March 1, 2019, our directors and executive officers hold, in the aggregate, options, warrants and RSU’s outstanding for 3,408,587 ordinary shares. These options have an average exercise price of $2.81 per share and have expiration dates generally twenty years after the grant date of the option.
1,846,317options and warrants are exercisable as of February 1, 2019 and have a weighted average exercise price of $3.06 per share.
Equity Compensation Plans
The 2000 Plan, the 2011 Plan and the 2014 Plan, allow us to grant options to purchase our ordinary shares to our directors, officers, employees, consultants, advisers and service providers. The 2022 Inducement Plan allows us to grant options to purchase our ordinary shares to new hires in the U.S.A, upon recruitment. The option plans are intended to enhance our ability to attract and retain desirable individuals by increasing their ownership interests in us. We no longer intend to grant options under the 2000 Plan or the 2011 Plan, and the remaining shares reserved for future grants under the option plans will constitute the initial share reserve for the 2014 Plan. Additionally, upon the expiration of options granted under the 2000 Plan or the 2011 Plan, the ordinary shares underlying such expired options will increase the pool reserved for allocation under the 2014 Plan. As of March 1, 2019, we had reserved an aggregate of 6,955,287 ordinary shares under the option plans. As of March 1, 2019, options to purchase an aggregate of 5,024,981 ordinary shares were outstanding and options to purchase 650,993 ordinary shares had been exercised.
The plans are designed to reflect the provisions of the Israeli Income Tax Ordinance [New Version]-1961, as amended, mainly Sections 102 and 3(i), of the Ordinance, which affords certain tax advantages to Israeli employees, officers and directors that are granted options in accordance with its terms.
Section 102 of the Ordinance allows employees, directors and officers, who are not controlling shareholders and who are Israeli residents, to receive favorable tax treatment for compensation in the form of shares or options. Section 102 of the Ordinance includes two alternatives for tax treatment involving the issuance of options or shares to a trustee for the benefit of the grantees and also includes an additional alternative for the issuance of options or shares directly to the grantee. Section 102(b)(2) of the Ordinance, which provides the most favorable tax treatment for grantees, permits the issuance to a trustee under the “capital gains track.” In order to comply with the terms of the capital gains track, all options granted under a specific plan and subject to the provisions of Section 102 of the Ordinance, as well as the shares issued upon exercise of such options and other shares received following any realization of rights with respect to such options, such as share dividends and share splits, must be registered in the name of a trustee selected by the board of directors and held in trust for the benefit of the relevant employee, director or officer. The trustee may not release these options or shares to the relevant grantee before the second anniversary of the registration of the options in the name of the trustee. However, under this track, we are not allowed to deduct an expense with respect to the issuance of the options or shares. Section 3(i) does not provide for a similar tax benefits.benefit.
The plans may be administered by our board of directors either directly or upon the recommendation of a committee appointed by our board of directors.
The compensation committee recommends to the board of directors, and the board of directors determines or approves the eligible individuals who receive options under the plans, the number of ordinary shares covered by those options, the terms under which such options may be exercised, and other terms and conditions of the options, all in accordance with the provisions of the plans. Option holders may not transfer their options except in the event of death or if the compensation committee determines otherwise. Our compensation committee or board of directors may at any time amend or terminate each of the plans; however, any amendment or termination may not adversely affect any options or shares granted under such plan prior to such action.
| 75 |
The option exercise price is determined by the compensation committee and specified in each option award agreement. In general, the option exercise price is the fair market value of the shares on the date of grant as determined in good faith by our board of directors.
Inducement Plan (2022)
On January 27, 2022, our board of directors adopted the Vascular Biogenic Ltd. Inducement Plan (2022), or the Inducement Plan. The purpose and intent of the Inducement Plan is to enable us to grant equity awards to induce highly qualified prospective officers and employees who are Eligible Employees to accept employment and to provide them with a proprietary interest in our company, pursuant to Rule 5635(c)(4) or 5635(c)(3), if applicable, of the Marketplace Rules of the Nasdaq and the related guidance under Nasdaq IM 5635-1. An “Eligible Employee” under the Inducement Plan is an individual who was not previously an employee or a non-employee director or of any of our affiliates (or who has had a bona fide period of non-employment), who is hired as a full or part-time employee by us or one of our affiliates.
The maximum number of shares that may be issued under the Inducement Plan is 2,000,000 of our ordinary shares, and the types of awards that may be granted under the Inducement Plan, are: nonqualified options, restricted shares; RSU awards and other share or share-based awards
Employee Share Ownership and Option Plan (2014)
In June 2014, we adopted and obtained shareholder approval for our 2014 Plan and the U.S. Appendix thereto. The 2014 Plan provides for the grant of options, restricted shares, restricted share units and other share- based awards to our directors, employees, officers, consultants, advisors and service providers, among others and to any other person whose services are considered valuable to us. Following the approval of the 2014 Plan by the Israeli tax authorities, we will only grant options or other equity incentive awards under the 2014 Plan, although previously-granted options and awards will continue to be governed by our 2000 Plan and 2011 Plan. The initial reserved pool under the 2014 Plan was 928,288 ordinary shares, and was adjusted as set forth in the 2014 Plan, including an automatic annual increase on January 1 of each year such that the number of shares issuable under the 2014 Plan will equal 4% of our issued and outstanding share capital on a fully diluted basis on each such January 1, or a lesser number of shares determined by the board of directors. As of March 1, 2019, the outstanding reserved pool under the 2014 Plan stands on 1,279,486.
The 2014 Plan is administered by our board of directors or by a committee designated by the board of directors, which shall determine, subject to Israeli law, the grantees of awards and the terms of the grant, including, exercise prices, vesting schedules, acceleration of vesting and the other matters necessary in the administration of the 2014 Plan. The 2014 Plan enables us to issue awards under various tax regimes including, without limitation, pursuant to Sections 102 and 3(i) of the Ordinance, and under Section 422 of the Code. Options granted under the 2014 Plan to U.S. residents may qualify as “incentive stock options” within the meaning of Section 422 of the Code, or may be non-qualified. The exercise price for “incentive stock options” must not be less than the fair market value on the date on which an option is granted, or 110% of the fair market value if the option holder holds more than 10% of our share capital.
We currently intend to grant awards under the 2014 Plan only to our employees, directors and officers who are not controlling shareholders, and are considered Israeli residents, under the capital gains track of Section 102(b)2 of the Ordinance.
Awards under the 2014 Plan may be granted until June 8, 2034, 20 years from the date on which the 2014 Plan was approved by our board of directors, provided that awards granted to any U.S. participants may be granted until June 8, 2024, 10 years from the date on which the 2014 Plan was approved by our board of directors.
ThePrior to January 2022, options granted under the 2014 Plan generally vest over four years commencing on the date of grant such that 25% vest on the first anniversary of the date of grant and an additional 6.25% vest at the end of each subsequent three-month periodquarterly thereafter for 36 months.three years such that vested in full on the four-year anniversary of the grant date. Beginning January 2022, options granted under the 2014 Plan generally vest over three years commencing on the date of grant such that 25% vest on the first anniversary of the date of grant and then quarterly thereafter for two years, such that it is vested in full on the three-year anniversary of the grant date. Options, other than certain incentive share options, that are not exercised within 20 years from the grant date expire, unless otherwise determined by our board of directors or its designated committee, as applicable. Share options that qualify as “incentive stock options” granted to a person holding more than 10% of our voting power under the U.S. appendix to the 2014 Plan will expire within five years from the date of the grant and any other options granted under the U.S. appendix to the 2014 Plan will expire within 10 years from the date of grant. Except as otherwise determined by the board of directors or as set forth in an individual’s award agreement, in the event of termination of employment or services for reasons of disability or death, or retirement, the grantee, or in the case of death, his or her legal successor, may exercise options that have vested prior to termination within a period of one year from the date of disability or death, or within 180 days following retirement. If we terminate a grantee’s employment or service for cause, all of the grantee’s vested and unvested options will expire on the date of termination. If a grantee’s employment or service is terminated for any other reason, the grantee may exercise his or her vested options within 90 days of the date of termination. Any expired or unvested options return to the pool for reissuance.
In the event of a merger or consolidation of our company, or a sale of all, or substantially all, of our shares or assets or other transaction having a similar effect on us, then without the consent of the option holder, our board of directors may determine, at its absolute discretion, whether outstanding awards held by or for the benefit of any grantee and which have not yet vested, is to be assumed or substituted and whether acceleration of such awards will be available.
Employee Share Ownership and Option Plan (2011)
In April 2011, we adopted the 2011 Plan. The term of the 2011 Plan is twenty20 years. Each option granted under the 2011 Plan entitles the grantee to purchase our ordinary shares. The options granted under the 2011 Plan generally vest during a four-year period following the date of the grant in 13 installments: 25% of the options vest one year following the grant date, and additional 1/16 of the options vest at the end of each subsequent quarter over the course of the following three years. The options expire twenty20 years after the date of grant if not exercised earlier.
In the case of certain changes in our share capital structure, such as a consolidation or share split or dividend, appropriate adjustments will be made to the numbers of shares and exercise prices under outstanding options. Unless otherwise determined by the board of directors, upon the consummation of certain kinds of transactions, such as a liquidation, a merger, reorganization or sale of all or substantially all of our assets, any unexercised outstanding options shall expire, provided that in case of merger or consolidation or the sale, transfer or exchange of all or substantially all our assets or shares, the surviving corporation does not assume the options or substitute them with appropriate options in the surviving corporation.
| 76 |
In general, when an option holder’s employment or service with us terminates, his or her option will no longer continue to vest following termination, and the holder may exercise any vested options for a period of 90 days following termination without cause. If an option holder’s employment with us terminates due to disability (as determined by the board of directors) or if the termination of employment results from his or her death, then the option holder or his or her estate (as applicable) has twelve months to exercise the option. If an option holder retires from our company, then, with the approval of the board of directors, the option holder or his or her estate (as applicable) has six months to exercise the option. If termination of employment results from cause, his or her outstanding options will expire upon termination. No option may be exercised after its scheduled expiration date.
Employee Share Ownership and Option Plan (2000)
In February 2000, we adopted the 2000 Plan, which was amended and restated in 2003 due to changes in applicable tax law. The original term of the 2000 Plan was ten years. In 2013, the terms of outstanding options were extended by 10 years.
Each option granted under the 2000 Plan entitles the grantee to purchase one of our ordinary shares. The options granted under the 2000 Plan generally vest during a four-year period following the date of the grant in three installments: 50% of the options vest two years following the grant date, 25% of the options vest three years following the grant date and the remaining 25% of the options vest four years following the grant date. The options under the plan expire ten years after the date of grant if not exercised earlier.
In the case of certain changes in our share capital structure, such as a consolidation or share split or dividend, appropriate adjustments will be made to the numbers of shares and exercise prices under outstanding options. In the event of certain transactions, such as an acquisition, or a merger or reorganization or a sale of all or substantially all of our assets, there shall be an acceleration of exercise of unvested options, immediate or otherwise, which depends on, among other things, the nature of such transaction, and provided that in case of merger or consolidation the surviving corporation does not assume the options or substitute them with appropriate options in the surviving corporation.
In general, when an option holder’s employment or service with us terminates, his or her option will no longer continue to vest following termination, and the holder may exercise any vested options for a period of 90 days following termination without cause. If an option holder’s employment with us terminates due to disability (as determined by the board of directors) or if the termination of employment results from his or her death or due to retirement after age 60, then with the approval of the board of directors, the option holder or his or her estate (as applicable) has twelve months to exercise the option; however, the option may not be exercised after its scheduled expiration date. If termination of employment results from cause, his or her outstanding options will expire upon termination.
Item 7. Major Shareholders and Related Party Transactions
Major Shareholders
The following table sets forth information with respect to the beneficial ownership of our ordinary shares as of FebruaryMarch 1, 2019:2022:
| ● | each person or entity known by us to own beneficially more than 5% of our outstanding ordinary shares; |
| ● | each of our executive officers and directors individually; and |
| ● | all of our executive officers and directors as a group. |
The beneficial ownership of our ordinary shares is determined in accordance with the rules of the SEC and generally includes any shares over which a person exercises sole or shared voting or investment power, or the right to receive the economic benefit of ownership. For purposes of the table below, we deem ordinary shares issuable pursuant to options that are currently exercisable or exercisable within 60 days of FebruaryMarch 1, 20192022 to be outstanding and to be beneficially owned by the person holding the options for the purposes of computing the percentage ownership of that person, but we do not treat them as outstanding for the purpose of computing the percentage ownership of any other person. The percentage of ordinary shares beneficially owned is based on 35,881,12869,337,312 ordinary shares outstanding as of FebruaryMarch 1, 2019.2022.
According to our transfer agent, as of February 23, 2019March 1, 2022, there were 12 record holders of our ordinary shares, of which twothree record holders were located in the United States. None of our shareholders hashave different voting rights from other shareholders.
Except as described in the footnotes below, we believe each shareholder has voting and investment power with respect to the ordinary shares indicated in the table as beneficially owned. Unless otherwise indicated, the address of each beneficial owner is c/o Vascular Biogenics Ltd., 8 HaSatat St., Modi’in, Israel 7178106.
| 77 |
| Number of | ||||||||
| Ordinary Shares | Percentage of | |||||||
| Name | Beneficially Owned | Ownership | ||||||
| 5% Shareholders | ||||||||
| Thai Lee (1) | 17,361,793 | 19.99 | % | |||||
| Aurum Ventures M.K.I. Ltd (2) | 6,839,059 | 9.86 | % | |||||
| David M. Slager (3) | 4,203,082 | 6.01 | % | |||||
| Victor Leo (4) | 3,619,048 | 5.22 | % | |||||
| Executive Officers and Directors | ||||||||
| Dror Harats (5) | 2,250,475 | 3.18 | % | |||||
| Sam Backenroth† | - | * | ||||||
| Matthew Trudeau† | - | * | ||||||
| Bennett M. Shapiro | - | * | ||||||
| Marc Kozin | - | * | ||||||
| Ruth Alon | - | * | ||||||
| Shmuel (Muli) Ben Zvi | - | * | ||||||
| Ron Cohen | �� | - | * | |||||
| Alison Finger | - | * | ||||||
| David Hastings | - | * | ||||||
| Michael Rice | - | * | ||||||
| Erez Feige | - | * | ||||||
| Eyal Breitbart | - | * | ||||||
| Naamit Sher | - | * | ||||||
| Tamar Rachmilewitz | - | * | ||||||
All directors, executive officers, and key employees as a group (15 individuals total)(6) | 4,455,006 | 6.13 | % | |||||
| Name | Number of Ordinary Shares Beneficially Owned | Percentage of Ownership | ||||||
| 5% Shareholders | ||||||||
| Thai Lee Family Trust | 5,447,811 | 15.18 | % | |||||
| Aurum Ventures M.K.I. Ltd (1) | 4,254,778 | 11.86 | % | |||||
| Sabby Management LLC | 2,373,768 | 6.61 | % | |||||
| The Estate of Mr. Jecheskiel Gonczarowski (2) | 2,168,337 | 6.03 | % | |||||
| Executive Officers and Directors | ||||||||
| Dr. Bennett M. Shapiro (3) | 314,122 | 0.87 | % | |||||
| Prof. Dror Harats (4) | 1,731,436 | 4.69 | % | |||||
| Prof. Ruth Arnon | - | * | ||||||
| Ms. Ruth Alon | - | * | ||||||
| Dr. Ron Cohen | - | * | ||||||
| Dr. Susan L. Kelley | - | * | ||||||
David Hastings | - | * | ||||||
| Dr. Shmuel (Muli) Ben Zvi | - | * | ||||||
| Mr. Amos Ron | - | * | ||||||
| Dr. Erez Feige | - | * | ||||||
| Dr. Eyal Breitbart | - | * | ||||||
| Dr. Naamit Sher | - | * | ||||||
| Dr. Corinne Epperly | - | * | ||||||
| Adv. Ayelet Horn | - | * | ||||||
| All directors and executive officers as a group (5) | 2,705,833 | 7.09 | % | |||||
* Less than 1%
† Address is 1 Blue Hill Plaza, Suite 1509, Pearl River, NY 10965.
Consists of (i) 1,000,000 ordinary shares held directly by Thai Lee, (ii) 6,001,531 ordinary shares held by the Thai Lee Family Trust, or the Family Trust, (iii) 2,814,262 ordinary shares held by the Thai Lee 2008 DE Trust, or the 2008 Trust, (iv) 146,000 shares held in UTMA accounts for Ms. Lee’s child, over which Ms. Lee disclaims beneficial ownership and (v) 7,400,000 pre-funded warrants to purchase ordinary shares exercisable as of March 1, 2022. Such pre-funded warrants are only exercisable to the extent that Ms. Lee, together with her affiliates, would beneficially own no more than | |
| Consists of | |
| (3) | Consists of (i) 1,812,913 ordinary shares held by Regals Capital Management LP, (ii) 1,740,169 ordinary shares held directly by David M. Slager and (iii) 650,000 pre-funded warrants held by Regals Fund LP. Mr. Slager may be deemed to have beneficial ownership over our shares and warrants held by Regals Capital Management LP and Regals Fund LP. The address of Regals Capital Management LP is 152 West 57th Street, 9th Floor, New York, NY 10019. |
| Consists of | |
Consists of (a) |
| Consists of (a) 1,078,160 outstanding shares; and (b) 3,376,846 shares underlying options |
Related Party Transactions
The following is a description of the material terms of those transactions with related parties to which we are party since January 1, 2018.2021.
Underwritten Public Offering
In April 2021, we completed the underwritten public offering of 5,150,265 of our ordinary shares and, to certain investors in lieu thereof, pre-funded warrants to purchase 8,050,000 ordinary shares, at a price to the public of $1.90 per ordinary share and $1.89 per pre-funded warrant. Thai Lee and David M. Slager, major shareholders, participated in the public offering and acquired an aggregate of 7,400,000 and 650,000 pre-funded warrants, respectively, on the same terms as other investors purchasing pre-funded warrants in the offering.
Warrant Exercises
In April 2021, Prof. Harats exercised warrants to acquire 31,932 ordinary shares at an exercise price of NIS 0.01 per share.
In November 2021, Thai Lee, a major shareholder, exercised warrants to acquire 1,904,762 ordinary shares at an exercise price of $1.45 per ordinary share for cash.
In November 2021, Victor Leo, a major shareholder, exercised warrants to acquire 1,809,524 ordinary shares at an exercise price of $1.45 per ordinary share for cash.
In November 2021, Aurum Ventures M.K.I. Ltd, a major shareholder, exercised warrants to acquire 1,269,841 ordinary shares at an exercise price of $1.45 per ordinary share for cash.
Employment Arrangement
On May 27, 2015 our shareholders approved our written compensation policyOctober 4, 2021 and December 20, 2021, we intend in to extend the compensation policy for anentered into employment agreements with Mr. Sam Backenroth as Chief Financial Officer and Mr. Matthew Trudeau as Chief Commercial Officer, respectively. For additional three-year term. We have adopted a written policy which provides that the approval of the audit committee is required to effect specified actions and transactions with our directors, executive officers and controlling shareholders, or in which such persons have an interest. Seeinformation, see “Item 6. Directors, Senior Management and Employees-ApprovalEmployees—Employment Agreements with Executive Officers and Directors.”
On October 19, 2021, our shareholders approved our New Compensation Scheme, see “Item 6. Directors, Senior Management, and Employees-Compensation of Executive Officers, Key Employees and Directors”.
Service Provider Agreements
In September 2021, VBL Therapeutics entered into an Administrative Services Agreement with VBL Inc., its US based wholly owned subsidiary, whereby VBL Inc. will provide administrative and other services to VBL Therapeutics, as needed. VBL Therapeutics will reimburse VBL Inc. for services provided plus five percent.
In October 2021, pursuant to the review and approval of the audit committee, we entered into an executive search agreement with an affiliate of one of our directors. The agreement provided for an initial $30,000 retainer and total fees equal to 30% of the candidate’s first year estimated compensation, subject to a $150,000 fee cap.
In December 2021, pursuant to the review and approval of the audit committee, we entered into an investor relations services agreement with an affiliate of one of our directors. The term of the agreement is six months, automatically renewed for additional 6 month terms unless otherwise terminated, and incurs monthly fees of $20,000 plus third party expense reimbursement.
See “Item 6 Directors, Senior Management and Employees—Approval of Related Party Transactions Under Israeli Law.” The term “controlling shareholder” means a shareholder withLaw” for more information on the ability to direct the activities of our company, other than by virtue of being an executive officer or director. A shareholder is presumed to be a controlling shareholder if the shareholder holds 50% or more of the voting rights in a company or has the right to appoint the majority of the directors of the company or its general manager. For the purpose of approving transactions with controlling shareholders, as well as corporate approval of executive compensation, the term also includes any shareholder (or two or more shareholders having a personal interest in the same matter being broughtprocess for approval) that holds 25% or more of the voting rights of a company if the company has no shareholder that owns more than 50% of its voting rights. The transactions described below were entered into prior to the effectiveness of this policy.these agreements.
Indemnification Agreements
We have in place indemnification agreements with each of our executive officers exculpating them from a breach of their duty of care to us to the fullest extent permitted by law, subject to limited exceptions, and undertaking to indemnify them to the fullest extent permitted by Israeli law, subject to limited exceptions, including with respect to liabilities resulting from the initial public offering to the extent such liabilities are not covered by insurance.
Employment Agreements
We have entered into employment agreements with our executive officers and key employees. The employment agreements contain standard provisions, including assignment of invention provisions and non-competition clauses. See “Item 6. Directors, Senior Management and Employees-Employment Agreements with Executive Officers.”
Registration Rights Agreement
Our investor rights agreement entitles our preferred shareholders to certain registration rights following the closing of our initial public offering. In accordance with this agreement, and subject to conditions described below, the following executives, directors and entities, which as of the date of the prospectus relating to the initial public offering beneficially owned more than 5% of our ordinary shares are entitled to registration rights: the estate of Jecheskiel Gonczarowski and entity affiliated therewith, Thai Lee Family Trust, Aurum Ventures and Pitango Ventures.
Form F-1 Demand Rights. Upon the request of the holders of more than 50% of the shares held by our former preferred shareholders given more than 180 days after the effective date of the registration statement related to our initial public offering, we are required to file a registration statement on Form F-1 in respect of the ordinary shares held by our former preferred shareholders. Following a request to effect such a registration, we are required to give notice of the request to the other holders of registrable securities and offer them an opportunity to include their shares in the registration statement. We are not required to effect more than two registrations on Form F-1 in the aggregate and not more than one registration in any 12 month period and we are only required to do so if the aggregate proceeds from any such registration are estimated in good faith to be in excess of $6.0 million.
Form F-3 Demand Rights. Upon filing an F-3 and at the request of the holders of more than 20% of the shares held by our former preferred shareholders, we are required to file a registration statement on Form F-3 in respect of the ordinary shares held by our former preferred shareholders.Following a request to effect such a registration, we are required to give notice of the request to the other holders of registrable securities and offer them an opportunity to include their shares in the registration statement. We are not required to effect a registration on Form F-3 more than twice in any 12 month period and are only required to do so if the aggregate proceeds from any such registration are estimated in good faith to be in excess of $2.0 million.
Piggyback Registration Rights. Following our initial public offering, shareholders holding registrable securities also have the right to request that we include their registrable securities in any registration statement filed by us in the future for the purposes of a public offering for cash, subject to specified exceptions.
Cutback. In the event that the managing underwriter advises the registering shareholders that marketing factors require a limitation on the number of shares that can be included in a registered offering, the shares will be included in the registration statement in an agreed order of preference among the holders of registration rights. The same preference also applies in the case of a piggyback registration, but we have first preference and the number of shares of shareholders that are included may not be less than 30% of the total number of shares included in the offering.
Termination. All registration rights granted to holders of registrable securities terminate on the fifth anniversary of the closing of our initial public offering and, with respect to any of our holders of registrable securities when the shares held by such shareholder can be sold within a 90 day period under Rule 144.
Expenses. We will pay all expenses in carrying out the foregoing registrations other than selling shareholders’ underwriting discounts and transfer taxes.
Financial statements are set forth under Item 18.
We have never declared or paid any cash dividends to our shareholders. We currently anticipate that we will retain all of our future earnings, if any, for use in the operation of our business. Additionally, our ability to pay dividends on our ordinary shares is limited by restrictions under the terms of the agreements governing our indebtedness and under Israeli law.
Our ordinary shares are quoted on the Nasdaq Global Market under the symbol “VBLT.”
Nasdaq Global Market
The following table sets forth, for the periods indicated since October 1, 2014, which was the date on which ourOur ordinary shares began trading on the Nasdaq Global Market under the symbol “VBLT,” the high and low sales prices of our ordinary shares as reported by the Nasdaq Global Market.“VBLT” on October 1, 2014.
| Price Per Ordinary Share | ||||||||
| High | Low | |||||||
| Annual: | ||||||||
| 2014 | $ | 7.56 | $ | 4.65 | ||||
| 2015 | 17.02 | 3.09 | ||||||
| 2016 | 7.58 | 2.76 | ||||||
| 2017 | 9.05 | 3.90 | ||||||
| 2018 | 8.50 | 0.60 | ||||||
| Quarterly: | ||||||||
| First Quarter 2017 | $ | 6.50 | $ | 4.20 | ||||
| Second Quarter 2017 | $ | 6.70 | $ | 4.35 | ||||
| Third Quarter 2017 | $ | 7.25 | $ | 3.90 | ||||
| Fourth Quarter 2017 | $ | 9.05 | $ | 5.60 | ||||
| First Quarter 2018 | 8.50 | 2.30 | ||||||
| Second Quarter 2018 | 2.80 | 2.10 | ||||||
| Third Quarter 2018 | 2.175 | 1.55 | ||||||
| Fourth Quarter 2018 | 1.72 | 1.26 | ||||||
| First Quarter 2019 (through March 11, 2019) | $ | 1.64 | $ | 0.94 | ||||
| Most Recent Six Months: | ||||||||
| September 2018 | 1.70 | 1.55 | ||||||
| October 2018 | $ | 1.72 | $ | 1.25 | ||||
| November 2018 | $ | 1.43 | $ | 1.10 | ||||
| December 2018 | $ | 1.26 | $ | 0.60 | ||||
| January 2019 | $ | 1.29 | $ | 0.939 | ||||
| February 2019 | $ | 1.63 | $ | 1.26 | ||||
On March 11, 2019,21, 2022, the last reported sale price of our ordinary shares on the Nasdaq Global Market was $1.64$1.49 per share.
Item 10. Additional Information
A. Share Capital
Not applicable.
B. Memorandum and Articles of Association
Ordinary Shares
Voting
All ordinary shares will have identical voting and other rights in all respects.
Transfer of Shares
Our fully paid ordinary shares are issued in registered form and may be freely transferred under our amended and restated articles of association, unless the transfer is restricted or prohibited by another instrument, applicable law or the rules of a stock exchange on which the shares are listed for trade. The ownership or voting of our ordinary shares by non-residents of Israel is not restricted in any way by our amended and restated articles of association or the laws of the State of Israel, except for ownership by nationals of some countries that are, or have been, in a state of war with Israel.
Election of Directors
Our ordinary shares do not have cumulative voting rights for the election of directors. As a result, the holders of a majority of the voting power represented at a shareholders meeting have the power to elect all of our directors, subject to the special approval requirements for external directors described under “Item 6. Directors, Senior Management and Employees-Board of Directors.”
Under our amended and restated articles of association, our board of directors must consist of not less than three, not including two external directors, but no more than nine directors (including the external directors). Pursuant to our amended and restated articles of association, other than the external directors, for whom special election requirements apply under the Companies Law, the vote required to appoint a director is a simple majority vote of holders of our voting shares, participating and voting at the relevant meeting. Each director will serve until his or her successor is duly elected and qualified or until his or her earlier death, resignation or removal by a vote of the majority voting power of our shareholders at a general meeting of our shareholders or until his or her office expires by operation of law, in accordance with the Companies Law. In addition, our amended and restated articles of association allow our board of directors to appoint directors to fill vacancies on the board of directors to serve for a term of office equal to the remaining period of the term of office of the directors(s) whose office(s) have been vacated. External directors are elected for an initial term of three years, may be elected for additional terms of three years each under certain circumstances, and may be removed from office pursuant to the terms of the Companies Law. See “Item 6. Directors, Senior Management and Employees-Board of Directors.”
Dividend and Liquidation Rights
We may declare a dividend to be paid to the holders of our ordinary shares in proportion to their respective shareholdings. Under the Companies Law, dividend distributions are determined by the board of directors and do not require the approval of the shareholders of a company unless the company’s articles of association provide otherwise. Our amended and restated articles of association do not require shareholder approval of a dividend distribution and provide that dividend distributions may be determined by our board of directors.
Pursuant to the Companies Law, the distribution amount is limited to the greater of retained earnings or earnings generated over the previous two years, according to our then last reviewed or audited financial statements, provided that the date of the financial statements is not more than six months prior to the date of the distribution, or we may otherwise only distribute dividends that do not meet such criteria only with court approval. In each case, we are only permitted to distribute a dividend if our board of directors and the court, if applicable, determines that there is no reasonable concern that payment of the dividend will prevent us from satisfying our existing and foreseeable obligations as they become due.
In the event of our liquidation, after satisfaction of liabilities to creditors, our assets will be distributed to the holders of our ordinary shares in proportion to their shareholdings. This right, as well as the right to receive dividends, may be affected by the grant of preferential dividend or distribution rights to the holders of a class of shares with preferential rights that may be authorized in the future.
Shareholder Meetings
Under Israeli law, we are required to hold an annual general meeting of our shareholders once every calendar year that must be held no later than 15 months after the date of the previous annual general meeting. All meetings other than the annual general meeting of shareholders are referred to in our amended and restated articles of association as extraordinary general meetings. Our board of directors may call extraordinary general meetings whenever it sees fit, at such time and place, within or outside of Israel, as it may determine. In addition, the Companies Law provides that our board of directors is required to convene an extraordinary general meeting upon the written request of (i) any two of our directors or one- quarter of the members of our board of directors or (ii) one or more shareholders holding, in the aggregate, either (a) 5% or more of our outstanding issued shares and 1% of our outstanding voting power or (b) 5% or more of our outstanding voting power. One or more shareholders, holding 1% or more of the outstanding voting power, may ask the board to add an item to the agenda of a prospective meeting, if the proposal merits discussion at the general meeting.
Subject to the provisions of the Companies Law and the regulations promulgated thereunder, shareholders entitled to participate and vote at general meetings are the shareholders of record on a date to be decided by the board of directors, which may be between four and 40 days prior to the date of the meeting. Furthermore, the Companies Law requires that resolutions regarding the following matters must be passed at a general meeting of our shareholders:
| ● | amendments to our articles of association; |
| ● | appointment or termination of our auditors; |
| ● | appointment of external directors; |
| ● | approval of certain related party transactions; |
| ● | increases or reductions of our authorized share capital; |
| ● | a merger; and |
| ● | the exercise of our board of director’s powers by a general meeting, if our board of directors is unable to exercise its powers and the exercise of any of its powers is required for our proper management. |
The Companies Law and our amended and restated articles of association require that a notice of any annual general meeting or extraordinary general meeting be provided to shareholders at least 21 days prior to the meeting and if the agenda of the meeting includes the appointment or removal of directors, the approval of transactions with office holders or interested or related parties, or an approval of a merger, notice must be provided at least 35 days prior to the meeting.
Under the Companies Law and our amended and restated articles of association, shareholders are not permitted to take action via written consent in lieu of a meeting.
Quorum Requirements
Pursuant to our amended and restated articles of association, holders of our ordinary shares have one vote for each ordinary share held on all matters submitted to a vote before the shareholders at a general meeting. As a foreign private issuer, the quorum required for our general meetings of shareholders consists of at least two shareholders present in person, by proxy or written ballot who hold or represent between them at least 25% of the total outstanding voting rights. A meeting adjourned for lack of a quorum is generally adjourned to the same day in the following week at the same time and place or to a later time or date if so.so specified in the notice of the meeting. At the reconvened meeting, any two or more shareholders present in person or by proxy shall constitute a lawful quorum.
Vote Requirements
Our amended and restated articles of association provide that all resolutions of our shareholders require a simple majority vote, unless otherwise required by the Companies Law or by our amended and restated articles of association. Under the Companies Law, each of (i) the approval of an extraordinary transaction with a controlling shareholder and (ii) the terms of employment or other engagement of the controlling shareholder of the company or such controlling shareholder’s relative (even if not extraordinary) requires, the approval described above under “Management-Approval“Item 6. Directors, Senior Management and Employees-Approval of Related Party Transactions Under Israeli Law-Disclosure of Personal Interests of Controlling Shareholders and Approval of Certain Transactions.” Under our amended and restated articles of association, the alteration of the rights, privileges, preferences or obligations of any class of our shares requires a simple majority vote of the class so affected (or such other percentage of the relevant class that may be set forth in the governing documents relevant to such class), in addition to the ordinary majority vote of all classes of shares voting together as a single class at a shareholder meeting. An exception to the simple majority vote requirement is a resolution for the voluntary winding up, or an approval of a scheme of arrangement or reorganization, of the company pursuant to Section 350 of the Companies Law, which requires the approval of holders of 75% of the voting rights represented at the meeting, in person, by proxy or by voting deed and voting on the resolution.
Access to Corporate Records
Under the Companies Law, shareholders are provided access to: minutes of our general meetings; our shareholders register and principal shareholders register, articles of association and financial statements; and any document that we are required by law to file publicly with the Israeli Companies Registrar or the Israel Securities Authority. In addition, shareholders may request to be provided with any document related to an action or transaction requiring shareholder approval under the related party transaction provisions of the Companies Law. We may deny this request if we believe it has not been made in good faith or if such denial is necessary to protect our interest or protect a trade secret or patent.
Acquisitions Under Israeli Law
Full Tender Offer
A person wishing to acquire shares of an Israeli public company and who would as a result hold over 90% of the target company’s issued and outstanding share capital is required by the Companies Law to make a tender offer to all of the company’s shareholders for the purchase of all of the issued and outstanding shares of the company. A person wishing to acquire shares of a public Israeli company and who would as a result hold over 90% of the issued and outstanding share capital of a certain class of shares is required to make a tender offer to all of the shareholders who hold shares of the relevant class for the purchase of all of the issued and outstanding shares of that class. If the shareholders who do not accept the offer hold less than 5% of the issued and outstanding share capital of the company or of the applicable class, and more than half of the shareholders who do not have a personal interest in the offer accept the offer, all of the shares that the acquirer offered to purchase will be transferred to the acquirer by operation of law. However, a tender offer will also be accepted if the shareholders who do not accept the offer hold less than 2% of the issued and outstanding share capital of the company or of the applicable class of shares.
| 81 |
Upon a successful completion of such a full tender offer, any shareholder that was an offeree in such tender offer, whether such shareholder accepted the tender offer or not, may, within six months from the date of acceptance of the tender offer, petition an Israeli court to determine whether the tender offer was for less than fair value and that the fair value should be paid as determined by the court. However, under certain conditions, the offeror may include in the terms of the tender offer that an offeree who accepted the offer will not be entitled to petition the Israeli court as described above.
If (a) the shareholders who did not respond or accept the tender offer hold at least 5% of the issued and outstanding share capital of the company or of the applicable class or the shareholders who accept the offer constitute less than a majority of the offerees that do not have a personal interest in the acceptance of the tender offer, or (b) the shareholders who did not accept the tender offer hold 2% or more of the issued and outstanding share capital of the company (or of the applicable class), the acquirer may not acquire shares of the company that will increase its holdings to more than 90% of the company’s issued and outstanding share capital or of the applicable class from shareholders who accepted the tender offer.
Special Tender Offer
The Companies Law provides that an acquisition of shares of an Israeli public company must be made by means of a special tender offer if as a result of the acquisition the purchaser would become a holder of 25% or more of the voting rights in the company. This requirement does not apply if there is already another holder of at least 25% of the voting rights in the company. Similarly, the Companies Law provides that an acquisition of shares in a public company must be made by means of a special tender offer if, as a result of the acquisition, the purchaser would become a holder of more than 45% of the voting rights in the company, provided that there is no other shareholder of the company who holds more than 45% of the voting rights in the company, subject to certain exceptions.
A special tender offer must be extended to all shareholders of a company, but the offeror is not required to purchase shares representing more than 5% of the voting power attached to the company’s outstanding shares, regardless of how many shares are tendered by shareholders. A special tender offer may be consummated only if (i) outstanding shares representing at least 5% of the voting power of the company will be acquired by the offeror and (ii) the number of shares tendered in the offer exceeds the number of shares whose holders objected to the offer (excluding the purchaser, controlling shareholders, holders of 25% or more of the voting rights in the company or any person having a personal interest in the acceptance of the tender offer). If a special tender offer is accepted, then the purchaser or any person or entity controlling it or under common control with the purchaser or such controlling person or entity may not make a subsequent tender offer for the purchase of shares of the target company and may not enter into a merger with the target company for a period of one year from the date of the offer, unless the purchaser or such person or entity undertook to effect such an offer or merger in the initial special tender offer.
Merger
The Companies Law permits merger transactions if approved by each party’s board of directors and, unless certain requirements described under the Companies Law are met, by a majority vote of each party’s shareholders, and, in the case of the target company, a majority vote of each class of its shares, voted on the proposed merger at a shareholders meeting.
For purposes of the shareholder vote, unless a court rules otherwise, the merger will not be deemed approved if a majority of the votes of shares represented at the shareholders meeting that are held by parties other than the other party to the merger, or by any person (or group of persons acting in concert) who holds (or hold, as the case may be) 25% or more of the voting rights or the right to appoint 25% or more of the directors of the other party, vote against the merger. If, however, the merger involves a merger with a company’s own controlling shareholder or if the controlling shareholder has a personal interest in the merger, then the merger is instead subject to the same special majority approval that governs all extraordinary transactions with controlling shareholders (as described under “Item 6. Directors, Senior Management and Employees-DisclosureEmployees-Approval of Related Party Transactions Under Israeli Law- Disclosure of Personal Interests of Controlling Shareholders and Approval of Certain Transactions”).
If the transaction would have been approved by the shareholders of a merging company but for the separate approval of each class or the exclusion of the votes of certain shareholders as provided above, a court may still approve the merger upon the request of holders of at least 25% of the voting rights of a company, if the court holds that the merger is fair and reasonable, taking into account the value of the parties to the merger and the consideration offered to the shareholders of the target company.
Upon the request of a creditor of either party to the proposed merger, the court may delay or prevent the merger if it concludes that there exists a reasonable concern that, as a result of the merger, the surviving company will be unable to satisfy the obligations of the merging entities, and may further give instructions to secure the rights of creditors.
In addition, a merger may not be consummated unless at least 50 days have passed from the date on which a proposal for approval of the merger was filed by each party with the Israeli Registrar of Companies and at least 30 days have passed from the date on which the merger was approved by the shareholders of each party.
Anti-takeover Measures
The Companies Law allow us to create and issue shares having rights different from those attached to our ordinary shares, including shares providing certain preferred rights with respect to voting, distributions or other matters and shares having preemptive rights. No preferred shares are currently authorized under our amended and restated articles of association. In the future, if we do authorize, create and issue a specific class of preferred shares, such class of shares, depending on the specific rights that may be attached to it, may have the ability to frustrate or prevent a takeover or otherwise prevent our shareholders from realizing a potential premium over the market value of their ordinary shares. The authorization and designation of a class of preferred shares will require an amendment to our amended and restated articles of association, which requires the prior approval of the holders of a majority of the voting power attaching to our issued and outstanding shares at a general meeting. The convening of the meeting, the shareholders entitled to participate and the majority vote required to be obtained at such a meeting will be subject to the requirements set forth in the Companies Law as described above in “Voting Rights”.
Tax Law
Israeli tax law treats some acquisitions, such as stock-for-stock swaps between an Israeli company and a foreign company, less favorably than U.S. tax law. For example, Israeli tax law may subject a shareholder who exchanges ordinary shares in an Israeli company for shares in a non-Israeli corporation to immediate taxation unless such shareholder receives authorization from the Israeli Tax Authority for different tax treatment.
Modification of Class Rights
Under the Companies Law and our amended and restated articles of association, the rights attached to any class of share, such as voting, liquidation and dividend rights, may be amended by adoption of a resolution by the holders of a majority of the shares of that class present at a separate class meeting, or otherwise in accordance with the rights attached to such class of shares, as set forth in our amended and restated articles of association.
Establishment
Our registration number with the Israeli Registrar of Companies is 51-289976-6. Our purpose as set forth in our amended and restated articles of association is to engage in any lawful activity.
Transfer Agent and Registrar
The transfer agent and registrar for our ordinary shares is American Stock Transfer & Trust Company, LLC.
C. Material Contracts
We have not entered into any material contracts other than in the ordinary course of business and other than those described in “Item 4. Information on the Company”,Company,” “Item 6. Directors, Senior Management and Employees” or elsewhere in this Annual Report.
D. Exchange Controls
There are currently no Israeli currency control restrictions on remittances of dividends on our ordinary shares, proceeds from the sale of the shares or interest or other payments to non- residents of Israel, except for shareholders who are subjects of countries that are, or have been, in a state of war with Israel.
In 1998, Israeli currency control regulations were liberalized significantly, so that Israeli residents generally may freely deal in foreign currency and foreign assets, and non-residents may freely deal in Israeli currency and Israeli assets. There are currently no Israeli currency control restrictions on remittances of dividends on the ordinary shares or the proceeds from the sale of the shares provided that all taxes were paid or withheld; however, legislation remains in effect pursuant to which currency controls can be imposed by administrative action at any time.
Non-residents of Israel may freely hold and trade our securities. Neither our articles of association nor the laws of the State of Israel restrict in any way the ownership or voting of ordinary shares by non-residents, except that such restrictions may exist with respect to citizens of countries which are in a state of war with Israel. Israeli residents are allowed to purchase our ordinary shares.
E. Taxation [Note: subject to the review of company’s tax advisors]
The following description is not intended to constitute a complete analysis of all tax consequences relating to the acquisition, ownership and disposition of our ordinary shares. You should consult your own tax advisor concerning the tax consequences of your particular situation, as well as any tax consequences that may arise under the laws of any state, local, foreign or other taxing jurisdiction.
| 83 |
Israeli Tax Considerations and Government Programs
The following is a brief summary of the material Israeli tax laws applicable to us, and certain Israeli Government programs that may benefit us. This section also contains a discussion of material Israeli tax consequences concerning the ownership and disposition of our ordinary shares purchased by investors. This summary does not discuss all the aspects of Israeli tax law that may be relevant to a particular investor in light of his or her personal investment circumstances or to some types of investors subject to special treatment under Israeli law. Examples of such investors include residents of Israel or traders in securities who are subject to special tax regimes not covered in this discussion. Because parts of this discussion are based on new tax legislation that has not yet been subject to judicial or administrative interpretation, we cannot assure you that the appropriate tax authorities or the courts will accept the views expressed in this discussion. The discussion below is subject to change, including due to amendments under Israeli law or changes to the applicable judicial or administrative interpretations of Israeli law, which change could affect the tax consequences described below.
General Corporate Tax Structure in Israel
Israeli companies are generally subject to corporate tax, currently at the rate of 23% of a company’s taxable income. However, the effective tax rate payable by a company that derives income from an Approved Enterprise, a Benefited Enterprise, a Preferred Enterprise or a Preferred Technology Enterprise (as discussed below) may be considerably less. Capital gains derived by an Israeli company are generally subject to tax at the prevailing corporate tax rate.
Law for the Encouragement of Industry (Taxes), 5729-1969
The Law for the Encouragement of Industry (Taxes), 5729-1969, generally referred to as the Industry Encouragement Law, provides several tax benefits for “Industrial Companies.”
The Industry Encouragement Law defines an “Industrial Company” as a company incorporated and resident in Israel, of which 90% or more of its income in any tax year, other than income from defense loans, is derived from an “Industrial Enterprise” owned by it that is located in Israel. An “Industrial Enterprise” is defined as an enterprise whose principal activity in a given tax year is industrial production.
The following corporate tax benefits, among others, are available to Industrial Companies:
| ● | amortization over an eight-year period of the cost of patents and rights to use patents and know-how which were purchased in good faith and are used for the development or advancement of the Industrial Enterprise; |
| ● | under certain conditions, an election to file consolidated tax returns with related Israeli Industrial Companies; and |
| ● | expenses related to a public offering are deductible in equal amounts over three years. |
There is no assurance that we qualify as an Industrial Company or that the benefits described above are currently available to us or will be available to us in the future.
Law for the Encouragement of Capital Investments, 5719-1959
The Law for the Encouragement of Capital Investments, 5719-1959, generally referred to as the Investment Law, provides certain incentives for capital investments in productive assets, such as production facilities, by “Industrial Enterprises” (as defined under the Investment Law).
The Investment Law was significantly amended effective April 1, 2005, (the “2005 Amendment”),or the 2005 Amendment, and further amended as of January 1, 2011, (the “2011 Amendment”)or the 2011 Amendment, and as of January 1, 2017, (the “2017 Amendment”).or the 2017 Amendment. Pursuant to the 2005 Amendment, tax benefits granted in accordance with the provisions of the Investment Law prior to its revision by the 2005 Amendment remain in force but any benefits granted subsequently are subject to the provisions of the 2005 Amendment. Similarly, the 2011 Amendment introduced new benefits to replace those granted in accordance with the provisions of the Investment Law in effect prior to the 2011 Amendment. However, companies entitled to benefits under the Investment Law as in effect prior to January 1, 2011 were entitled to choose to continue to enjoy such benefits, provided that certain conditions are met, or elect instead, irrevocably, to forego such benefits and have the benefits of the 2011 Amendment apply. Finally, the 2017 Amendment provided another benefits track, which represents an alternative to the tracks available under the 2005 Amendment and the 2011 Amendment. We have examined the possible effect, if any, of these provisions of the 2011 Amendment and the 2017 Amendment on our financial statements and have decided, at this time, not to opt to apply the new benefits under the 2011 Amendment or the 2017 Amendment.
Tax Benefits Prior to the 2005 Amendment
An investment program that is implemented in accordance with the provisions of the Investment Law prior to the 2005 Amendment, referred to as an “Approved Enterprise,” is entitled to certain benefits. A company that wished to receive benefits as an Approved Enterprise must have received approval from the Investment Center of the Israeli Ministry of the Economy (formerly the Ministry of Industry, Trade and Labor), or the Investment Center. Each certificate of approval for an Approved Enterprise relates to a specific investment program in the Approved Enterprise, delineated both by the financial scope of the investment and by the physical characteristics of the facility or the asset.
| 84 |
In general, an Approved Enterprise is entitled to receive a grant from the Government of Israel or an alternative package of tax benefits, known as the alternative benefits track. The tax benefits from any certificate of approval relate only to taxable income attributable to the specific Approved Enterprise. Income derived from activity that is not integral to the activity of the Approved Enterprise does not enjoy tax benefits.
In addition, a company that has an Approved Enterprise program is eligible for further tax benefits if it qualifies as a Foreign Investors’ Company, (“FIC”),or FIC, which is a company with a level of foreign investment, as defined in the Investment Law, of more than 25%. The level of foreign investment is measured as the percentage of rights in the company (in terms of shares, rights to profits, voting and appointment of directors), and of combined share capital and loans, that are owned, directly or indirectly, by persons who are not residents of Israel. The determination as to whether a company qualifies as an FIC is made on an annual basis.
If a company elects the alternative benefits track and distributes a dividend out of income derived by its Approved Enterprise during the tax exemption period it will be subject to corporate tax in respect of the amount of the dividend (grossed-up to reflect the pre-tax income that it would have had to earn in order to distribute the dividend) at the corporate tax rate which would have been applicable without the tax exemption under the alternative benefits track. In addition, dividends paid out of income attributed to an Approved Enterprise are generally subject to withholding tax at source at the rate of 15% or such lower rate as may be provided in an applicable tax treaty.
The Investment Law also provides that an Approved Enterprise is entitled to accelerated depreciation on its property and equipment that are included in an Approved Enterprise program during the first five years in which the equipment is used.
The benefits available to an Approved Enterprise are subject to the fulfillment of conditions stipulated in the Investment Law and its regulations and the criteria in the specific certificate of approval. If a company does not meet these conditions, it would be required to repay the amount of tax benefits, as adjusted by the Israeli consumer price index, and interest.
We do not have Approved Enterprise programs.
Tax Benefits Subsequent to the 2005 Amendment
The 2005 Amendment applies to new investment programs commencing after 2004, but does not apply to investment programs approved prior to April 1, 2005. The 2005 Amendment provides that terms and benefits included in any certificate of approval that was granted before the 2005 Amendment became effective (April 1, 2005) will remain subject to the provisions of the Investment Law as in effect on the date of such approval.
The 2005 Amendment provides that a certificate of approval from the Investment Center will only be necessary for receiving cash grants. As a result, it was no longer necessary for a company to obtain an Approved Enterprise certificate of approval in order to receive the tax benefits previously available under the alternative benefits track. Rather, a company may claim the tax benefits offered by the alternative benefits track directly in its tax returns, provided that it meets the criteria for tax benefits set forth in the amendment. In order to receive the tax benefits, the 2005 Amendment states,inter alia, that a company must make an investment which meets all of the conditions, including a minimum qualifying investment in certain productive assets as specified in the Investment Law. Such investment, along with the fulfillment of certain export requirements, allows a company to receive “Benefited Enterprise” status, and may be made over a period of no more than three years culminating with the end of the Benefited Enterprise election year.
The extent of the tax benefits available under the 2005 Amendment to qualifying income of a Benefited Enterprise depends on, among other things, the geographic location in Israel of the Benefited Enterprise. The location will also determine the period for which tax benefits are available. Such tax benefits include an exemption from corporate tax on undistributed income generated by the Benefited Enterprise for a period of between two to ten years, depending on the geographic location of the Benefited Enterprise in Israel, and a reduced corporate tax rate of between 10% to 25% for the remainder of the benefits period, depending on the level of foreign investment in the company in each year. The benefits period is limited to 12 years from the beginning of the Benefited Enterprise election year. With respect to an establishment Benefited Enterprise plan located in certain specific locations, the benefits period is limited to 14 years from the beginning of the Benefited Enterprise election year, depending on the location of the Benefited Enterprise. We informed the Israeli Tax Authority of our choice of 2012 as a Benefited Enterprise election year. A company qualifying for tax benefits under the 2005 Amendment which pays a dividend out of income derived by its Benefited Enterprise during the tax exemption period will be subject to corporate tax in respect of the amount of the dividend (grossed-up to reflect the pre-tax income that it would have had to earn in order to distribute the dividend) at the corporate tax rate which would have otherwise been applicable. Dividends paid out of income attributed to a Benefited Enterprise are generally subject to withholding tax at source at the rate of 15% or such lower rate as may be provided in an applicable tax treaty.
The benefits available to a Benefited Enterprise are subject to the fulfillment of conditions stipulated in the Investment Law and its regulations. If a company does not meet these conditions, in a given tax year during the benefits period, it would generally not be eligible for tax benefits during such tax year; however, the company’s eligibility for tax benefits in prior and future years should not be impacted.
| 85 |
We currently have one Benefited Enterprise program under the Investments Law, which, we believe, may entitle us to certain tax benefits. The tax benefit period for this program has not yet commenced but is expected to end no later than the end of tax year 2023. During the benefits period, which shall commence with the year we will first earn taxable income relating to such enterprise, subject to the 12 years limitation described above, and shall run for a period of up to 10 years (assuming FIC status), a corporate tax exemption is expected to apply with respect to the taxable income from our Benefited Enterprise program (once generated) generated during the first two years of the benefits period (so long as it remains undistributed) and reduced corporate tax rates are expected to apply to such taxable income generated in the remaining years of the benefits period.
There is no assurance that our future taxable income will qualify as Benefited Enterprise income or that the benefits described above will be available to us in the future.
Tax Benefits Under the 2011 Amendment
The 2011 Amendment canceled the availability of the benefits granted to companies under the Investment Law prior to 2011, subject to certain exceptions, and, instead, introduced new benefits for income generated by a “Preferred Company” through its “Preferred Enterprise” (as such terms are defined in the Investment Law) as of January 1, 2011. The definition of a Preferred Company includes a company incorporated in Israel that is not wholly-owned by a governmental entity, and that has, among other things, Preferred Enterprise status and is controlled and managed from Israel. Pursuant to the 2011 Amendment, in 2014 and thereafter a Preferred Company is entitled to a reduced corporate tax rate of 16% with respect to its income derived by its Preferred Enterprise unless the Preferred Enterprise is located in development zone A, in which case the rate will be 9%. This latter rate was reduced to 7.5% as of January 1, 2017. It should be noted that the classification of income generated from the provision of usage rights in know-how or software that were developed in the Preferred Enterprise, as well as royalty income received with respect to such usage, as Preferred Enterprise income may be subject to the issuance of a pre-ruling from the Israel Tax Authority stipulating that such income is associated with the productive activity of the Preferred Enterprise in Israel.
Dividends paid out of income attributed to a Preferred Enterprise are generally subject to withholding tax at source at the rate of 20% or such lower rate as may be provided in an applicable tax treaty. However, if such dividends are paid to an Israeli company, no tax is required to be withheld (although, if such dividends are subsequently distributed to individuals or a non-Israeli company, withholding tax at a rate of 20% or such lower rate as may be provided in an applicable tax treaty will apply).
The 2011 Amendment also provided transitional provisions to address companies that may be eligible for tax benefits under the Approved Enterprise or Benefited Enterprise regimes. These transitional provisions provide, among other things, that unless an irrevocable request is made to apply the provisions of the Investment Law as amended in 2011 with respect to income to be derived as of January 1, 2011: (1) the terms and benefits included in any certificate of approval that was granted to an Approved Enterprise which chose to receive grants before the 2011 Amendment became effective will remain subject to the provisions of the Investment Law as in effect on the date of such approval, and subject to certain other conditions, (2) terms and benefits included in any certificate of approval that was granted to an Approved Enterprise which had participated in an alternative benefits track before the 2011 Amendment became effective will remain subject to the provisions of the Investment Law as in effect on the date of such approval, provided that certain conditions are met, and (3) a Benefited Enterprise can elect to continue to benefit from the benefits provided to it before the 2011 Amendment came into effect, provided that certain conditions are met.
We have examined the potential Israeli tax implications associated with the adoption and implementation of the provisions of the 2011 Amendment and have decided, at this time, not to apply the new benefits under the 2011 Amendment. There is no assurance that our future taxable income will qualify as Preferred Enterprise income or that the benefits described above will be available to us in the future.
The termination or substantial reduction of any of the benefits available under the Investment Law could materially increase our tax liabilities.
Tax Benefits Under the 2017 Amendment
The 2017 Amendment introduced new benefits for income generated by a “Preferred Company” (as defined above) through its “Preferred Technology Enterprise” (as defined in the Investment Law) as of January 1, 2017. Pursuant to the 2017 Amendment, in 2017 and thereafter a Preferred Company is entitled to a reduced corporate tax rate of 12% with respect to its income derived by its Preferred Technology Enterprise unless the Preferred Enterprise is located in development zone A, in which case the rate will be 7.5%. It should be noted that the calculation of a Preferred Company’s Preferred Technology Enterprise income is based on a complex formula and the income not classified as such may be classified as Preferred Enterprise income or ordinary income depending on the circumstances. In addition, a Preferred Company must generally fulfill certain conditions to be eligible for Preferred Technology Enterprise status including,inter alia, an R&D expenses level of at least 7% of total revenues or NIS 75 million per year.
Dividends paid out of Preferred Technology Enterprise income are generally subject to withholding tax at source at the rate of 20% or such lower rate as may be provided in an applicable tax treaty. However, subject to the fulfillment of certain conditions, to the extent that the dividends are paid to a direct foreign parent company holding at least 90% of the shares of the Preferred Company, a reduced withholding tax rate of 4% shall apply. Notwithstanding the above, if such dividends are paid to an Israeli company, no tax is required to be withheld (although, if such dividends are subsequently distributed to individuals or a non-Israeli company, withholding tax at a rate of 20% or such lower rate as may be provided in an applicable tax treaty will apply).
| 86 |
We have examined the potential Israeli tax implications associated with the adoption and implementation of the provisions of the 2017 Amendment and have decided, at this time, not to apply the new benefits under the 2017 Amendment. There is no assurance that our future taxable income will qualify as Preferred Technology Enterprise income or that the benefits described above will be available to us in the future.
The termination or substantial reduction of any of the benefits available under the Investment Law could materially increase our tax liabilities.
Taxation of Our Shareholders
This discussion does not address the tax consequences applicable to shareholders that own, or have owned at any time, directly or indirectly, 10% or more of our shares, (“or Controlling Shareholders”),Shareholders, and such shareholders should consult their tax advisers as to the tax consequences of owning or disposing of our shares.
Capital Gains Taxes Applicable to Non-Israeli Resident Shareholders
A non-Israeli resident who derives capital gains from the sale of shares in an Israeli resident company that were purchased after the Company was listed for trading on a stock exchange outside of Israel will be exempt from Israeli tax so long as,inter alia, such capital gains were not attributable to a permanent establishment that the non-resident maintains in Israel.
However, non-Israeli resident corporations will not be entitled to the foregoing exemption if the Israeli residents: (i) have a controlling interest, directly or indirectly, alone, together with another (i.e., together with a relative, or together with someone who is not a relative but with whom, according to an agreement, there is regular cooperation in material matters of the company, directly or indirectly), or together with another Israeli resident, of more than 25% in one or more of the means of control in such non-Israeli resident corporation, or (ii) Israeli residents are the beneficiaries of, or are entitled to, 25% or more of the revenues or profits of such non-Israeli resident corporation, whether directly or indirectly.
Additionally, a sale of securities by a non-Israeli resident may be exempt from Israeli capital gains tax under the provisions of an applicable tax treaty. For example, under the United States- Israel Tax Treaty, the disposition of shares by a shareholder who (1) is a U.S. resident (for purposes of the treaty), (2) holds the shares as a capital asset, and (3) is entitled to claim the benefits afforded to such person by the treaty, is generally exempt from Israeli capital gains tax. Such exemption will not apply if: (1) the capital gain arising from the disposition can be attributed to a permanent establishment in Israel, (2) the shareholder holds, directly or indirectly, shares representing 10% or more of the voting power of the company during any part of the 12-month period preceding the disposition, subject to certain conditions, or (3) such U.S. resident is an individual and was present in Israel for 183 days or more during the relevant taxable year. In such case, the sale, exchange or disposition of our ordinary shares would be subject to Israeli tax, to the extent applicable; however, under the United States-Israel Tax Treaty, the taxpayer would be permitted to claim a credit for such taxes against the U.S. federal income tax imposed with respect to such sale, exchange or disposition, subject to the limitations under U.S. law applicable to foreign tax credits. The United States-Israel Tax Treaty does not relate to U.S. state or local taxes.
In some instances where our shareholders may be liable for Israeli tax on the sale of their ordinary shares, the payment of the consideration may be subject to the withholding of Israeli tax at source. Shareholders may be required to demonstrate that they are exempt from tax on their capital gains in order to avoid withholding at source at the time of sale.
Taxation of Non-Israeli Shareholders on Receipt of Dividends
Non-Israeli residents are generally subject to Israeli withholding tax on the receipt of dividends paid on our ordinary shares at the rate of 25%, unless relief is provided in a treaty between Israel and the shareholder’s country of residence, subject to receipt of a valid certificate from the Israeli Tax Authority allowing for such reduced rate. With respect to a person who is a “substantial shareholder” at the time of receiving the dividend or at any time during the preceding twelve months, the applicable withholding tax rate is 30%. Furthermore, an additional 3% tax might be applicable to individual shareholders if certain conditions are met. A “substantial shareholder” is generally a person who alone or together with such person’s relative or another person who collaborates with such person on a permanent basis, holds, directly or indirectly, at least 10% of any of the “means of control” of the corporation. “Means of control” generally include the right to vote, receive profits, nominate a director or an executive officer, receive assets upon liquidation, or order someone who holds any of the aforesaid rights how to act, regardless of the source of such right. Notwithstanding the above, dividends paid to a non-Israeli resident “substantial shareholder” on publicly traded shares, like our ordinary shares, which are held via a “nominee company” (as defined under the Securities Law, 1968), are generally subject to Israeli withholding tax at a rate of 25%, unless a different rate is provided under an applicable tax treaty, provided that a certificate from the Israeli Tax Authority allowing for a reduced withholding tax rate is obtained in advance. Under the United States-Israel Tax Treaty, the maximum rate of tax withheld at source in Israel on dividends paid to a holder of our ordinary shares who is a U.S. resident (for purposes of the United States- Israel Tax Treaty) is 25%. Unless a reduced tax rate is provided under an applicable tax treaty, a distribution of dividends to non-Israeli residents is subject to withholding tax at source at a rate of 15% if the dividend is distributed from income attributed to an Approved Enterprise or a Benefited Enterprise, while a 20% rate applies if the dividend is distributed from Preferred Enterprise income or Preferred Technology Enterprise income (unless the dividend is paid to a foreign parent company directly holding at least 90% of the shares of the Preferred Company, in which case a 4% withholding tax rate shall apply). We cannot assure you that in the event we declare a dividend we will designate the income out of which the dividend is paid in a manner that will reduce shareholders’ tax liability.
If the dividend is attributable partly to Approved Enterprise income, Benefited Enterprise income, Preferred Enterprise income or Preferred Technology Enterprise income, and partly to other sources of income, the withholding rate will be a blended rate reflecting the relative portions of the two types of income. U.S. residents who are subject to Israeli withholding tax on a dividend may be entitled to a credit or deduction for Untied States federal income tax purposes in the amount of the taxes withheld, subject to detailed rules contained in U.S. tax legislation.
| 87 |
Estate and Gift Tax
Israeli law presently does not impose estate or gift taxes.
Certain Material U.S. Federal Income Tax Considerations
The following is a description of the material U.S. federal income tax considerations relating to the ownership and disposition of our ordinary shares by a U.S. Holder (as defined below). This description addresses only the U.S. federal income tax considerations to U.S. Holders that will hold such ordinary shares as capital assets. This description does not address tax considerations applicable to U.S. Holders that may be subject to special tax rules, including, without limitation:
| ● | banks, financial institutions or insurance companies; |
| ● | real estate investment trusts, regulated investment companies or grantor trusts; |
| ● | brokers, dealers or traders in securities, commodities or currencies; |
| ● | tax exempt entities or organizations, including an “individual retirement account” or “Roth IRA” as defined in Section 408 or 408A of the Code (as defined below), respectively; |
| ● | certain former citizens or long term residents of the United States; |
| ● | persons that received our shares as compensation for the performance of services; |
| ● | persons that will hold our shares as part of a “hedging,” “integrated” or “conversion” transaction or as a position in a “straddle” for U.S. federal income tax purposes; |
| ● | partnerships (including entities classified as partnerships for U.S. federal income tax purposes) or other pass-through entities, or holders that will hold our shares through such an entity; |
| ● | S corporations; |
| ● | persons that acquire ordinary shares as a result of holding or owning our preferred shares; |
| ● | persons whose “functional currency” is not the U.S. dollar; or |
| ● | persons that own directly, indirectly or through attribution 10% or more of the voting power or value of our shares. |
Moreover, this description does not address the U.S. federal estate, gift, or alternative minimum tax considerations, or any U.S. state, local or non-U.S. tax considerations of the ownership and disposition of our ordinary shares.
This description is based on the U.S. Internal Revenue Code of 1986, as amended, or the Code, changes to the code based on the U.S. tax reform (as described below) existing, proposed and temporary U.S. Treasury Regulations promulgated thereunder and administrative and judicial interpretations thereof, in each case as in effect and available on the date hereof. All the foregoing is subject to change, which change could apply retroactively, and to differing interpretations, all of which could affect the tax considerations described below. There can be no assurances that the U.S. Internal Revenue Service, or the IRS, will not take a different position concerning the tax consequences of the ownership and disposition of our ordinary shares or that such a position would not be sustained. Holders should consult their own tax advisers concerning the U.S. federal, state, local and foreign tax consequences of owning and disposing of our ordinary shares in their particular circumstances.
For purposes of this description, the term “U.S. Holder” means a beneficial owner of our ordinary shares that, for U.S. federal income tax purposes, is (i) a citizen or resident of the United States, (ii) a corporation (or entity treated as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States, any state thereof, or the District of Columbia, (iii) an estate the income of which is subject to U.S. federal income tax regardless of its source, or (iv) a trust with respect to which a court within the United States is able to exercise primary supervision over its administration and one or more U.S. persons have the authority to control all of its substantial decisions.
| 88 |
If a partnership (or any other entity treated as a partnership for U.S. federal income tax purposes) holds our ordinary shares, the U.S. federal income tax consequences relating to an investment in our ordinary shares will depend in part upon the status of the partner and the activities of the partnership. Such a partner or partnership should consult its tax advisor regarding the U.S. federal income tax considerations of acquiring, owning and disposing of our ordinary shares in its particular circumstances.
As indicated below, this discussion is subject to U.S. federal income tax rules applicable to a “passive foreign investment company,” or a PFIC.
Persons considering an investment in our ordinary shares should consult their own tax advisors as to the particular tax consequences applicable to them relating to the ownership and disposition of our ordinary shares, including the applicability of U.S. federal, state and local tax laws and non-U.S. tax laws.
Distributions
Subject to the discussion under “-Passive“Passive Foreign Investment Company Considerations,” below, if you are a U.S. Holder, the gross amount of any distribution made to you with respect to our ordinary shares before reduction for any Israeli taxes withheld therefrom, other than certain distributions, if any, of our ordinary shares distributed pro rata to all our shareholders, generally will be includible in your income as dividend income to the extent such distribution is paid out of our current or accumulated earnings and profits as determined under U.S. federal income tax principles. To the extent that the amount of any distribution by us exceeds our current and accumulated earnings and profits as determined under U.S. federal income tax principles, it will generally be treated first as a return of your adjusted tax basis in our ordinary shares and thereafter as either long-term or short-term capital gain depending upon whether the U.S. Holder has held our ordinary shares for more than one year as of the time such distribution is received. We do not expect to maintain calculations of our earnings and profits under U.S. federal income tax principles. Therefore, U.S. Holders should expect that the entire amount of any distribution generally will be reported as dividend income. Non-corporate U.S. Holders may qualify for the preferential rates of taxation with respect to dividends on ordinary shares applicable to long-term capital gains (i.e., gains from the sale of capital assets held for more than one year) applicable to qualified dividend income (as discussed below). The Company,company, which is incorporated under the laws of the State of Israel, believes that it qualifies as a resident of Israel for purposes of, and is eligible for the benefits of, the Convention between the Government of the United States of America and the Government of the State of Israel with Respect to Taxes on Income, signed on November 20, 1975, as amended and currently in force, or the U.S.-Israel Tax Treaty, although there can be no assurance in this regard. Further, the IRS has determined that the U.S.-Israel Tax Treaty is satisfactory for purposes of the qualified dividend rules and that it includes an exchange-of-information program. Therefore, subject to the discussion under “-Passive Foreign Investment Company Considerations,” below, if the U.S.-Israel Tax Treaty is applicable, such dividends will generally be “qualified dividend income” in the hands of individual U.S. Holders, provided that certain conditions are met, including holding period and the absence of certain risk reduction transaction requirements are met. The dividends will not be eligible for the dividends received deduction generally allowed to corporate U.S. Holders.
On December 22, 2017, President Trump signed into law the Tax Cuts and Jobs Act, or the TCJA. The TCJA provides a 100% deduction for the foreign-source portion of dividends received after January 1, 2018 from “specified 10-percent owned foreign corporations” by U.S. corporate holders, subject to a one-year holding period. No foreign tax credit, including Israeli withholding tax (or deduction for foreign taxes paid with respect to qualifying dividends) would be permitted for foreign taxes paid or accrued with respect to a qualifying dividend. Deduction would be unavailable for “hybrid dividends.” The dividend received deduction enacted under the TCJA may not apply to dividends from a passive foreign investment company.PFIC.
U.S. Holders, other than certain U.S. Holder’s that are U.S. corporations, generally may claim the amount of Israeli withholding tax withheld either as a deduction from gross income or as a credit against U.S. federal income tax liability. However, the foreign tax credit is subject to numerous complex limitations that must be determined and applied on an individual basis. Generally, the credit cannot exceed the proportionate share of a U.S. Holder’s U.S. federal income tax liability that such U.S. Holder’s “foreign source” taxable income bears to such U.S. Holder’s worldwide taxable income. In applying this limitation, a U.S. Holder’s various items of income and deduction must be classified, under complex rules, as either “foreign source” or “U.S. source.” In addition, this limitation is calculated separately with respect to specific categories of income. The amount of a distribution with respect to the ordinary shares that is treated as a “dividend” may be lower for U.S. federal income tax purposes than it is for Israeli income tax purposes, potentially resulting in a reduced foreign tax credit for the U.S. Holder. Each U.S. Holder should consult its own tax advisors regarding the foreign tax credit rules.
In general, the amount of a distribution paid to a U.S. Holder in a foreign currency will be the dollar value of the foreign currency calculated by reference to the spot exchange rate on the day the U.S. Holder receives the distribution, regardless of whether the foreign currency is converted into U.S. dollars at that time. Any foreign currency gain or loss a U.S. Holder realizes on a subsequent conversion of foreign currency into U.S. dollars will be U.S. source ordinary income or loss. If dividends received in foreign currency are converted into U.S. dollars on the day they are received, a U.S. Holder generally should not be required to recognize foreign currency gain or loss in respect of the dividend.
| 89 |
Sale, Exchange or Other Taxable Disposition of Our Ordinary Shares
Subject to the discussion below under “-Passive Foreign Investment Company Considerations,” if you are a U.S. Holder, you generally will recognize gain or loss on the sale, exchange or other taxable disposition of our ordinary shares equal to the difference between the amount realized on such sale, exchange or other taxable disposition and your adjusted tax basis in our ordinary shares, and such gain or loss will be capital gain or loss. The adjusted tax basis in an ordinary share generally will be equal to the cost of such ordinary share. If you are a non-corporate U.S. Holder, capital gain from the sale, exchange or other taxable disposition of ordinary shares is generally eligible for a preferential rate of taxation applicable to capital gains, if your holding period determined at the time of such sale, exchange or other taxable disposition for such ordinary shares exceeds one year (i.e., such gain is long-term capital gain). The deductibility of capital losses for U.S. federal income tax purposes is subject to limitations under the Code. Any such gain or loss that a U.S. Holder recognizes generally will be treated as U.S. source income or loss for foreign tax credit limitation purposes.
For a cash basis taxpayer, units of foreign currency paid or received are translated into U.S. dollars at the spot rate on the settlement date of the purchase or sale. In that case, no foreign currency exchange gain or loss will result from currency fluctuations between the trade date and the settlement date of such a purchase or sale. An accrual basis taxpayer, however, may elect the same treatment required of cash basis taxpayers with respect to purchases and sales of our ordinary shares that are traded on an established securities market, provided the election is applied consistently from year to year. Such election may not be changed without the consent of the IRS. For an accrual basis taxpayer who does not make such election, units of foreign currency paid or received are translated into U.S. dollars at the spot rate on the trade date of the purchase or sale. Such an accrual basis taxpayer may recognize exchange gain or loss based on currency fluctuations between the trade date and the settlement date. Any foreign currency gain or loss a U.S. Holder realizes will be U.S. source ordinary income or loss.
Passive Foreign Investment Company Considerations
If we are classified as a PFIC in any taxable year, a U.S. Holder would be subject to special rules generally intended to reduce or eliminate any benefits from the deferral of U.S. federal income tax that a U.S. Holder could derive from investing in a non-U.S. company that does not distribute all of its earnings on a current basis.
A non-U.S. corporation is classified as a PFIC for U.S. federal income tax purposes in any taxable year in which, after applying certain look-through rules with respect to the income and assets of subsidiaries, either (i) at least 75% of its gross income is “passive income” or (ii) at least 50% of the average quarterly value of its total gross assets (which, assuming we are not a CFC for the year being tested, would be measured by fair market value of the assets, and for which purpose the total value of our assets may be determined in part by the market value of our ordinary shares, which is subject to change) is attributable to assets that produce “passive income” or are held for the production of passive income.
Passive income for this purpose generally includes dividends, interest, royalties, rents, gains from commodities and securities transactions, the excess of gains over losses from the disposition of assets which produce passive income, and includes amounts derived by reason of the temporary investment of funds raised in offerings of our ordinary shares. If a non-U.S. corporation owns directly or indirectly at least 25% by value of the stock of another corporation, the non-U.S. corporation is treated for purposes of the PFIC tests as owning its proportionate share of the assets of the other corporation and as receiving directly its proportionate share of the other corporation’s income. If we are classified as a PFIC in any year with respect to which a U.S. Holder owns our ordinary shares, we will continue to be treated as a PFIC with respect to such U.S. Holder in all succeeding years during which the U.S. Holder owns our ordinary shares, regardless of whether we continue to meet the tests described above.
We must determine our PFIC status annually based on tests which are factual in nature, and our status will depend on our income, assets and activities each year.
However, weWe believe that we were not a PFIC for our 20182021 taxable year, butyear. However, we expect that unless and until we generate sufficient revenue from active licensing and other non-passive sources and otherwise satisfy the asset test above, we will be treated as a PFIC in future taxable years.
If we are a PFIC, and you are a U.S. Holder, then unless you make one of the elections described below, a special tax regime will apply to both (a) any “excess distribution” by us to you (generally, your ratable portion of distributions in any year which are greater than 125% of the average annual distribution received by you in the shorter of the three preceding years or your holding period for our ordinary shares) and (b) any gain realized on the sale or other disposition of the ordinary shares. Under this regime, any excess distribution and realized gain will be treated as ordinary income and will be subject to tax as if (a) the excess distribution or gain had been realized ratably over your holding period, (b) the amount deemed realized in each year had been subject to tax in each year of that holding period at the highest marginal rate for such year (other than income allocated to the current period or any taxable period before we became a PFIC, which would be subject to tax at the U.S. Holder’s regular ordinary income rate for the current year and would not be subject to the interest charge discussed below), and (c) the interest charge generally applicable to underpayments of tax had been imposed on the taxes deemed to have been payable in those years. In addition, dividend distributions made to you will not qualify for the lower rates of taxation applicable to long-term capital gains discussed above under “Distributions.”
Certain elections may potentially be used to reduce the adverse impact of the PFIC rules on U.S. Holders (“qualifying electing fund” (“QEF”)fund,” or QEF, and “mark-to-market” elections), but these elections may accelerate the recognition of taxable income and may result in the recognition of ordinary income.
The rules described above for excess distributions would not apply to a U.S. Holder if the U.S. Holder makes a timely QEF election for the first taxable year of the U.S. Holder’s holding period for ordinary shares and we comply with specified reporting requirements. A timely QEF election for a taxable year generally must be made on or before the due date (as may be extended) for filing the taxpayer’s U.S. federal income tax return for the year. A U.S. Holder who makes a QEF election generally must report on a current basis a pro rata share of our ordinary earnings and net capital gain for any taxable year in which we are a PFIC, whether or not those earnings or gains are distributed. A U.S. Holder who makes a QEF election must file a Form 8621 with its annual income tax return. We have not historically provided the information necessary for U.S. Holders to make qualified electing fund elections. However, beginning with our 2016 taxable year, for U.S. Holders who seek to make a QEF election with respect to our ordinary shares, we intend to make available an information statement that will contain the necessary information required for making a QEF election and permit such U.S. Holders access to certain information in the event of an audit by the U.S. tax authorities.
If a U.S. Holder does not make a QEF election for the first taxable year of the U.S. Holder’s holding period for ordinary shares during which we are a PFIC, the QEF election will not be treated as timely and the adverse tax regime described above would apply to dispositions of or excess distributions on the ordinary shares. In such case, a U.S. Holder may make a deemed sale election whereby the U.S. Holder would be treated as if the U.S. Holder had sold the ordinary shares in a fully taxable sale at fair market value on the first day of such taxable year in which the QEF election takes effect. Such U.S. Holder would be required to recognize any gain on the deemed sale as an excess distribution and pay any tax and interest due on the excess distribution when making the deemed sale election. The effect of such further election would be to restart the U.S. Holder’s holding period in the ordinary shares, subject to the QEF regime, and to purge the PFIC status of such ordinary shares going forward.
If a U.S. Holder makes the mark-to-market election, the U.S. Holder generally will recognize as ordinary income any excess of the fair market value of the ordinary shares at the end of each taxable year over their adjusted tax basis, and will recognize an ordinary loss in respect of any excess of the adjusted tax basis of the ordinary shares over their fair market value at the end of the taxable year (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). If a U.S. Holder makes the election, the U.S. Holder’s tax basis in the ordinary shares will be adjusted to reflect these income or loss amounts. Any gain recognized on the sale or other disposition of ordinary shares in a year when we are a PFIC will be treated as ordinary income and any loss will be treated as an ordinary loss (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). The mark-to-market election is available only if we are a PFIC and our ordinary shares are “regularly traded” on a “qualified exchange.” Our ordinary shares will be treated as “regularly traded” in any calendar year in which more than a de minimis quantity of the ordinary shares are traded on a qualified exchange on at least 15 days during each calendar quarter (subject to the rule that trades that have as one of their principle purposes the meeting of the trading requirement as disregarded). The NASDAQNasdaq Global Market is a qualified exchange for this purpose and, consequently, if the ordinary shares are regularly traded, the mark-to-market election will be available to a U.S. Holder.
U.S. Holders should consult their tax advisors to determine whether any of these elections would be available and if so, what the consequences of the alternative treatments would be in their particular circumstances.
If we are a PFIC, the general tax treatment for U.S. Holders described in this section would apply to indirect distributions and gains deemed to be realized by U.S. Holders in respect of any of our subsidiaries that also may be determined to be PFICs.
If a U.S. Holder owns ordinary shares during any year in which we are a PFIC and the U.S. Holder recognizes gain on a disposition of our ordinary shares or receives distributions with respect to our ordinary shares, the U.S. Holder generally will be required to file an IRS Form 8621 (Information Return by a Shareholder of a Passive Foreign Investment Company or Qualified Electing Fund) with respect to the company, generally with the U.S. Holder’s federal income tax return for that year. If our company were a PFIC for a given taxable year, then you should consult your tax advisor concerning your annual filing requirements.
The U.S. federal income tax rules relating to PFICs are complex. Prospective U.S. investors are urged to consult their own tax advisers with respect to the ownership and disposition of our ordinary shares, the consequences to them of an investment in a PFIC, any elections available with respect to our ordinary shares and the IRS information reporting obligations with respect to the ownership and disposition of our ordinary shares.
Medicare Tax
Certain U.S. Holders that are individuals, estates or trusts may be required to pay an additional 3.8% Medicare tax on all or a portion of their “net investment income,” which may include all or a portion of their dividend income and net gains from the disposition of ordinary shares. U.S. Holders will likely not be able to credit foreign taxes against the 3.8% Medicare tax. Each U.S. Holder that is an individual, estate or trust is urged to consult its tax advisors regarding the applicability of the Medicare tax to its income and gains in respect of its investment in our ordinary shares.
| 91 |
Backup Withholding Tax and Information Reporting Requirements
U.S. backup withholding tax and information reporting requirements may apply to certain payments to certain shareholders. Information reporting generally will apply to payments of dividends on, and to proceeds from the sale or redemption of, our ordinary shares made within the United States, or by a U.S. payorpayer or U.S. middleman, to a holder of our ordinary shares, other than an exempt recipient (including a payee that is not a U.S. person that provides an appropriate certification and certain other persons). A payorpayer may be required to withhold backup withholding tax from any payments of dividends on, or the proceeds from the sale or redemption of, ordinary shares within the United States, or by a U.S. payorpayer or U.S. middleman, to a holder, other than an exempt recipient, if such holder fails to furnish its correct taxpayer identification number or otherwise fails to comply with, or establish an exemption from, such backup withholding tax requirements. Any amounts withheld under the backup withholding rules should generally be allowed as a credit against the beneficial owner’s U.S. federal income tax liability, if any, and any excess amounts withheld under the backup withholding rules may be refunded, provided that the required information is timely furnished to the IRS.
Foreign Asset Reporting
Certain U.S. Holders who are individuals may be required to report information relating to an interest in our ordinary shares, subject to certain exceptions (including an exception for shares held in accounts maintained by U.S. financial institutions) by filing IRS Form 8938 (Statement of Specified Foreign Financial Assets) with their federal income tax return. U.S. Holders are urged to consult their tax advisors regarding their information reporting obligations, if any, with respect to their ownership and disposition of our ordinary shares.
Foreign Account Tax Compliance Act
The Foreign Account Tax Compliance Act, (“FATCA”)or FATCA, encourages foreign financial institutions to report information about their U.S. account holders (including holders of certain equity interests) to the IRS. Foreign financial institutions that fail to comply with the withholding and reporting requirements of FATCA and certain account holders that do not provide sufficient information under the requirements of FATCA are subject to a 30% U.S. withholding tax on certain payments they receive, including foreign passthru payments (which may include payments made by us with respect to our ordinary shares). The term “foreign passthru payment” is not currently defined in U.S. Treasury Regulations, and therefore, the future application of FATCA withholding tax on foreign pass-thru payments to holders of ordinary shares is uncertain. If a holder of ordinary shares is subject to withholding, there will be no additional amounts payable by way of compensation to the holder of such securities for the deducted amount. Holders of ordinary shares should consult their own tax advisors regarding this legislation in light of such holder’s particular situation.
THE DISCUSSION ABOVE IS A GENERAL SUMMARY. IT DOES NOT COVER ALL TAX MATTERS THAT MAY BE OF IMPORTANCE TO A PROSPECTIVE INVESTOR. EACH PROSPECTIVE INVESTOR IS URGED TO CONSULT ITS OWN TAX ADVISOR ABOUT THE TAX CONSEQUENCES TO IT OF AN INVESTMENT IN ORDINARY SHARES IN LIGHT OF THE INVESTOR’S OWN CIRCUMSTANCES.
F. Dividends and Paying Agents
Not applicable.
G. Statement by Experts
Not applicable.
H. Documents on Display
You may inspect our securities filings, including this Annual Report and the exhibits and schedules thereto, without charge at the offices of the SEC at 100 F Street, N.E., Washington, D.C. 20549. You may obtain copies of all or any part of the Annual Report from the Public Reference Section of the SEC, 100 F Street, NE, Washington, D.C. 20549 upon the payment of the prescribed fees. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains a website at www.sec.gov that contains reports, proxy and information statements and other information regarding registrants like us that file electronically with the SEC. You can also inspect the Annual Report on thisthe SEC’s website.
A copy of each document (or a translation thereof to the extent not in English) concerning our company that is referred to in this Annual Report is available for public view (subject to confidential treatment of certain agreements pursuant to applicable law) at our principal executive offices.offices at 8 HaSatat St., Modi’in, Israel 7178106.
I. Subsidiary Information
Not applicable.
| 92 |
Item 11. Quantitative and Qualitative Disclosures About Market Risk
We are exposed to market risks in the ordinary course of our business. Market risk represents the risk of loss that may impact our financial position due to adverse changes in financial market prices and rates. Our market risk exposure is primarily a result of foreign currency exchange rates. Approximately 39%38% of our expenses in 20182021 were denominated in New Israeli Shekels. Changes of 5% in the US$/NIS exchange rate will increase or decrease the operating expenses by up to 1%2%.
Foreign Currency Risk
Fluctuations in exchange rates, especially the NIS against the U.S. dollar, may affect our results, as some of our assets are linked to NIS, as are some of our liabilities. In addition, the fluctuation in the NIS exchange rate against the U.S. dollar may impact our results, as a portion of our operating costs are NIS denominated.
The following table presents information about the changes in the exchange rates of the NIS against the U.S. dollar at year end:
| Period | % | |||
| Year ended December 31, | % | |||
| Year ended December 31, | ( | )% | ||
| Year ended December 31, | ( | )% | ||
Inflation Risk
We do not believe that inflation had a material effect on our business, financial condition or results of operations in the last three fiscal years. If our costs were to become subject to significant inflationary pressures, we may not be able to fully offset such higher costs through hedging transactions. Our inability or failure to do so could harm our business, financial condition and results of operations.
Item 12. Description of Securities Other Than Equity Securities
Not applicable.
PART II
Item 13. Defaults, Dividend Arrearages and Delinquencies
Not applicable.
Item 14. Material Modifications to the Rights of Security Holders and Use of Proceeds
Use of Proceeds from Initial Public OfferingNot applicable.
On October 6, 2014, we completed an initial public offering in the United States on the NASDAQ Global Market of our ordinary shares, par value NIS 0.01 per share. Pursuant to the initial public offering, we sold a total of 6,760,418 ordinary shares (including the shares sold pursuant to the over-allotment option) at a price of $6.00 per share. The aggregate offering price of the shares sold (including the over-allotment option) was approximately $40.5 million. The net proceeds we received from the offering (including the over-allotment option) were approximately $34.9 million.
As of December 31, 2018, we had used all of the net proceeds of our initial public offering. None of the proceeds from our initial public offering were used for direct or indirect payments to our directors, officers or their associates, or to persons owning 10% or more of our equity securities, or to our affiliates.
Item 15. Controls and Procedures
Disclosure Controls and Procedures
We have performed an evaluation of the effectiveness of our disclosure controls and procedures that are designed to ensure that the material financial and non-financial information required to be disclosed to the SEC is recorded, processed, summarized and reported timely. Based on our evaluation, our management, including the Chief Executive Officer, or CEO, and the Chief Financial Officer, or CFO, has concluded that our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934, as amended) as of the end of the period covered by this report are effective. Notwithstanding the foregoing, there can be no assurance that our disclosure controls and procedures will detect or uncover all failures of persons within the Companyour company to disclose material information otherwise required to be set forth in our reports.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rule 13a-15(f) promulgated under the Exchange Act. Our internal control system was designed to provide reasonable assurance to our management and board of directors regarding the reliability of financial reporting and the preparation and fair presentation of published financial statements for external purposes in accordance with generally accepted accounting principles. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation and may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with policies or procedures may deteriorate.
Our management, including our CEO and CFO, conducted an evaluation, pursuant to Rule 13a-15(c) promulgated under the Exchange Act, of the effectiveness, as of the end of the period covered by this Annual Report, of the Compay’sour internal control over financial reporting based on the framework in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013). Based on the results of this evaluation, management concluded that our internal control over financial reporting was effective as of December 31, 2018.2021.
Attestation Report of the Registered Public Accounting Firm
See report of PricewaterhouseCoopers International Ltd., independent registered public accounting firm, included under “Item 18. Financial Statements” on page F-1.
Report of the Independent Accounting Firm
This annual report does not include an attestation report of the Company’s registered public accounting firm because management’s report was not subject to attestation by our independent registered public accounting firm as emerging growth companies are exempt from this requirement.
Changes in Internal Control over Financial Reporting
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures or our internal controls will prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within the Company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the control. The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Over time, controls may become inadequate because of changes in conditions, or the degree of compliance with the policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
There were no changes in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) that occurred during the yearfourth fiscal quarter ended December 31, 20182021 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 16A. Audit committee financial expert
All members of our audit committee meet the requirements for financial literacy under the applicable rules and regulations of the Securities and Exchange CommissionSEC and the NASDAQNasdaq corporate governance rules. Our board of directors has determined that Mr. David Hastings and Dr. Shmuel (Muli) Ben Zvi are the audit committee financial experts as defined by the Securities and Exchange CommissionSEC rules, has the requisite financial experience and is independent as defined by the NASDAQNasdaq corporate governance rules.
Item 16B. Code of Ethics
We have adopted a Code of Business Conduct and Ethics applicable to all of our directors and employees, including our Chief Executive Officer, Chief Financial Officer,CEO, CFO, controller or principal accounting officer, or other persons performing similar functions, which is a “code of ethics” as defined in this Item 16B of Form 20-F promulgated by the SEC. The full text of the Code of Business Conduct and Ethics is posted on our website atwww.vblrx.com Information contained on, or that can be accessed through, our website does not constitute a part of this Form 20-F and is not incorporated by reference herein. If we make any amendment to the Code of Business Conduct and Ethics or grant any waivers, including any implicit waiver, from a provision of the code of ethics, we will disclose the nature of such amendment or waiver on our website to the extent required by the rules and regulations of the SEC.
Item 16C. Principal Accountant Fees and Services
The following table sets forth, for each of the years indicated, the fees billed by Kesselman & Kesselman, a member firm of PricewaterhouseCoopers International Ltd., our independent registered public accounting firm:
| Year Ended December 31, | Year Ended December 31, | |||||||||||||||
| 2018 | 2017 | 2021 | 2020 | |||||||||||||
| (in thousands) | (in thousands) | |||||||||||||||
| Service rendered | ||||||||||||||||
| Audit Fees (1) | $ | 135.0 | $ | 145.0 | $ | 330 | $ | 225 | ||||||||
| Audit-Related Fees (2) | - | - | - | - | ||||||||||||
| Tax Fees (3) | 33.0 | 31.0 | 5 | 8 | ||||||||||||
| All Other Fees | - | - | - | - | ||||||||||||
| Total | $ | 168.0 | $ | 176.0 | $ | 335 | $ | 233 | ||||||||
| (1) | Audit fees consist of services that would normally be provided in connection with statutory and regulatory filings or engagements, including services that generally only the independent accountant can reasonably provide, including work regarding the public listing or |
| (2) | Audit related services relate to reports to the IIA. |
| (3) | Tax fees relate to tax compliance, planning and advice. |
Our board of directors reviews and pre-approves all audit services and permitted non-audit services (including the fees and other terms) to be provided by our independent auditors.auditors pursuant to pre-approval policies and procedures established by the audit committee, which are detailed as to the particular service and the audit committee is informed of each service. The pre-approval policies and procedures do not delegate audit committee responsibilities under the Securities Exchange Act of 1934 to management.
Item 16D. Exemptions from the Listing Standards for Audit Committees
Not applicable.
Item 16E. Purchase of Equity Securities by the Issuer and Affiliated Purchasers
In the year ended December 31, 2018, neither we nor any2021, the following equity securities were purchased by affiliated purchaser (as defined in the Exchange Act) purchased any of our ordinary shares.purchasers:
| Period | Total Number of Shares Purchased | Average Price Paid per Share | Total Number of Shares Purchased as Part of Publicly Announced Plans or Programs | Maximum Number of Shares that May be Purchased Under the Plans or Programs | ||||||||||
| April 2021 | 31,932 | NIS | 0.01 | 31,932 | ||||||||||
| April 2021 | 8,050,000 | $ | 0.01 | |||||||||||
| November 2021 | 4,984,127 | $ | 1.45 | |||||||||||
| December 2021 | 14,000 | $ | 2.21 | |||||||||||
| Total | 13,080,059 | $ | 0.56 | 31,923 | ||||||||||
| 95 |
Item 16F. Change in Registrant’s Certifying Accountant
None.
Item 16G. Corporate Governance
As a foreign private issuer whose shares are listed on the NASDAQNasdaq Global Market, we have the option to follow certain Israeli corporate governance practices rather than those of NASDAQ,Nasdaq, except to the extent that such laws would be contrary to U.S. securities laws and provided that we disclose the practices we are not following and describe the home country practices we follow instead. We rely on this “foreign private issuer exemption” with respect to the following NASDAQNasdaq requirements:
| ● | Quorum requirement. Under our articles of association and as permitted under the Companies Law, a quorum for any meeting of shareholders shall be the presence of at least two shareholders present in person, by proxy or by a voting instrument, who hold at least 25% of the voting power of our shares instead of 3313% of the issued share capital required under Nasdaq requirements. |
Except as stated above, we comply with the rules generally applicable to U.S. domestic companies listed on NASDAQ, subject to certain exemptions the JOBS Act provides to emerging growth companies.Nasdaq. We may in the future elect to follow home country practices in Israel with regard to other matters, including the formation of compensation, nominating and corporate governance committees, separate executive sessions of independent directors and non-management directors and the requirement to obtain shareholder approval for certain dilutive events (such as for the establishment or amendment of certain equity-based compensation plans, issuances that will result in a change of control of the company, certain transactions other than a public offering involving issuances of a 20% or more interest in the company and certain acquisitions of the stock or assets of another company).
Following our home country governance practices, as opposed to the requirements that would otherwise apply to a company listed on NASDAQ,Nasdaq, may provide less protection than is accorded to investors under NASDAQNasdaq listing requirements applicable to domestic issuers. For more information, see “Item 3. Key Information-D. Risk Factors-We are an ‘emerging growth company’ and a ‘foreign private issuer,’ and we cannot be certain if the reduced reporting requirements applicable to emerging growth companies and foreign private issuers will make our ordinary shares less attractive to investors” and “Risk Factors-WeFactors -We are a ‘foreign“foreign private issuer’issuer” and intend to follow certain home country corporate governance practices, and our shareholders may not have the same protections afforded to shareholders of companies that are subject to all NASDAQNasdaq corporate governance requirements. Additionally, we cannot be certain if the reduced disclosure requirements applicable to our status as a foreign private issuer, will make our ordinary shares less attractive to investors.” We will also be required to comply with Israeli corporate governance requirements under the Companies Law applicable to Israeli public companies such as us whose shares are also listed for trade on an exchange outside Israel.
Item 16H. Mine Safety Disclosure
Not applicable.
Item 16I. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
Financial Statements are set forth under Item 18.
Our Financial Statements beginning on pages F-1 through F-8,F-29, as set forth in the following index, are incorporated herein by reference. These Financial Statements are filed as part of this Annual Report.
| F-1 |


Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders of Vascular Biogenics Ltd.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the board of directors and shareholders of VASCULAR BIOGENICS LTD.
OpinionOpinions on the Financial Statements and Internal Control over Financial Reporting
We have audited the accompanying statements of financial positionconsolidated balance sheets of Vascular Biogenics Ltd. and its subsidiary (the “Company”) as of December 31, 20182021 and 2017,2020, and the related consolidated statements of net loss and comprehensive loss, changes in shareholders’ equity and cash flows for each of the three years in the period ended December 31, 2018,2021, including the related notes (collectively referred to as the “financial“consolidated financial statements”). We also have audited the Company’s internal control over financial reporting as of December 31, 2021, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 20182021 and 2017,2020, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 20182021 in conformity with International Financial Reporting Standardsaccounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2021, based on criteria established in Internal Control - Integrated Framework (2013) issued by the International Accounting Standards Board.
COSO.
Basis for OpinionOpinions
TheseThe Company’s management is responsible for these consolidated financial statements, are the responsibilityfor maintaining effective internal control over financial reporting, and for its assessment of the Company’s management.effectiveness of internal control over financial reporting, included in Management’s Report on Internal Control over Financial Reporting appearing under Item 15. Our responsibility is to express an opinionopinions on the Company’s consolidated financial statements and on the Company’s internal control over financial reporting based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits of these financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the auditaudits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding offraud, and whether effective internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.was maintained in all material respects.
Our audits of the consolidated financial statements included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinion.opinions.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Critical Audit Matters
Critical audit matters are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that (i) relate to accounts or disclosures that are material to the consolidated financial statements and (ii) involved our especially challenging, subjective, or complex judgments. We determined there are no critical audit matters.
/s/ Kesselman & Kesselman C.P.A.s
Certified Public Accountants (Isr.)
A member of PricewaterhouseCoopers International Limited
Tel Aviv, Israel
March 23, 2022
We have served as the Company’s auditor since 2001.
|
Kesselman & Kesselman, Derech Menachem Begin 146 Tel Aviv-Yafo 6492103 Israel,
P.O Box 7187 Tel-Aviv 6107120 Telephone: +972 -3- 7954555, Fax:+972 -3- 7954556, www.pwc.com/il]
| F-2 |
VASCULAR BIOGENICS LTD.
CONSOLIDATED BALANCE SHEETS
STATEMENTS OF FINANCIAL POSITION
| 2021 | 2020 | |||||||
| December 31 | ||||||||
| 2021 | 2020 | |||||||
| U.S. dollars in thousands | ||||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 21,986 | $ | 13,184 | ||||
| Restricted bank deposits | - | 151 | ||||||
| Short-term bank deposits | 31,164 | 17,110 | ||||||
| Trade receivables | - | 129 | ||||||
| Other current assets | 1,697 | 1,419 | ||||||
| Total current assets | 54,847 | 31,993 | ||||||
| Non-current assets: | ||||||||
| Restricted bank deposits | $ | 362 | 362 | |||||
| Long-term prepaid expenses | 182 | 241 | ||||||
| Funds in respect of employee rights upon retirement | 415 | 354 | ||||||
| Property, plant and equipment, net | 6,847 | 6,632 | ||||||
| Operating lease right-of-use assets | 2,008 | 2,124 | ||||||
| Total non-current assets | 9,814 | 9,713 | ||||||
| Total assets | $ | 64,661 | $ | 41,706 | ||||
| LIABILITIES AND SHAREHOLDERS’ EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable: | ||||||||
| Trade | $ | 4,331 | $ | 1,960 | ||||
| Other | 4,408 | 4,275 | ||||||
| Deferred revenue | 658 | 725 | ||||||
| Current maturity of operating leases | 529 | 393 | ||||||
| Current maturity of finance lease liability | - | 106 | ||||||
| Total current liabilities | 9,926 | $ | 7,459 | |||||
| Non-current liabilities: | ||||||||
| Liability for employee rights upon retirement | 546 | 474 | ||||||
| Deferred revenue | - | 704 | ||||||
| Operating lease liability | 1,823 | 2,029 | ||||||
| Other non-current liability | 188 | 123 | ||||||
| Total non-current liabilities | 2,557 | 3,330 | ||||||
| Commitments (Note 8) | ||||||||
| Total liabilities | $ | 12,483 | $ | 10,789 | ||||
| Ordinary shares subject to possible redemption, shares at redemption value (Note 9) | 1,598 | - | ||||||
| Shareholders’ equity: | ||||||||
| Ordinary shares, NIS par value; Authorized as of December 31, 2021 and 2020, and shares, respectively; issued and outstanding as of December 31, 2021 and 2020, and shares, respectively (excluding and -- shares subject to possible redemption, as of December 31, 2021 and December 31, 2020, respectively) | 171 | 108 | ||||||
| Additional paid in capital | 309,355 | 252,561 | ||||||
| Warrants | 3,127 | 10,401 | ||||||
| Accumulated deficit | (262,073 | ) | (232,153 | ) | ||||
| Total equity | 50,580 | 30,917 | ||||||
| Total liabilities and equity | $ | 64,661 | $ | 41,706 | ||||
| December 31 | ||||||||||||
| Note | 2018 | 2017 | ||||||||||
| U.S. dollars in thousands | ||||||||||||
| Assets | ||||||||||||
| CURRENT ASSETS: | ||||||||||||
| Cash and cash equivalents | $ | 29,347 | $ | 6,694 | ||||||||
| Short-term bank deposits | 5 | 21,135 | 48,035 | |||||||||
| Trade receivables | 8 | - | 2,000 | |||||||||
| Other current assets | 12a | 1,227 | 1,729 | |||||||||
| TOTAL CURRENT ASSETS | 51,709 | 58,458 | ||||||||||
| NON-CURRENT ASSETS: | ||||||||||||
| Property and equipment, net | 6 | 8,921 | 7,128 | |||||||||
| Long-term prepaid expenses | 48 | 103 | ||||||||||
| TOTAL NON-CURRENT ASSETS | 8,969 | 7,231 | ||||||||||
| TOTAL ASSETS | $ | 60,678 | $ | 65,689 | ||||||||
| Liabilities and equity | ||||||||||||
| CURRENT LIABILITIES- | ||||||||||||
| Accounts payable: | ||||||||||||
| Trade | $ | 1,193 | $ | 3,058 | ||||||||
| Other | 12b | 2,944 | 3,465 | |||||||||
| Deferred revenue | 8 | 290 | 1,046 | |||||||||
| Lease liability | 347 | - | ||||||||||
| TOTAL CURRENT LIABILITIES | 4,774 | 7,569 | ||||||||||
| NON-CURRENT LIABILITIES- | ||||||||||||
| Severance pay obligations, net | 7 | 99 | 128 | |||||||||
| Deferred revenue | 8 | 2,263 | 2,092 | |||||||||
| Lease liability | 449 | - | ||||||||||
| TOTAL NON-CURRENT LIABILITIES | 2,811 | 2,220 | ||||||||||
| TOTAL LIABILITIES | 7,585 | 9,789 | ||||||||||
| COMMITMENTS | 9 | |||||||||||
| EQUITY: | 10 | |||||||||||
Ordinary shares, NIS 0.01 par value; Authorized as of December 31, 2018 and 2017, 70,000,000 shares; issued and outstanding as of December 31, 2018 and 2017, 35,881,128 and 29,879,323 shares, respectively | 73 | 57 | ||||||||||
| Accumulated other comprehensive income | 41 | 16 | ||||||||||
| Additional paid in capital | 233,721 | 221,055 | ||||||||||
| Warrants | 7,904 | 2,960 | ||||||||||
| Accumulated deficit | (188,646 | ) | (168,188 | ) | ||||||||
| TOTAL EQUITY | 53,093 | 55,900 | ||||||||||
| TOTAL LIABILITIES AND EQUITY | $ | 60,678 | $ | 65,689 | ||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-3 |
VASCULAR BIOGENICS LTD.
CONSOLIDATED STATEMENTS OF NET LOSS AND COMPREHENSIVE LOSS
| Year ended December 31 | ||||||||||||||||
| Note | 2018 | 2017 | 2016 | |||||||||||||
| U.S. dollars in thousands | ||||||||||||||||
| REVENUES | 8 | 585 | 13,864 | - | ||||||||||||
| COST OF REVENUES | 9c | (235 | ) | (340 | ) | - | ||||||||||
| GROSS PROFIT | 350 | 13,524 | - | |||||||||||||
| RESEARCH AND DEVELOPMENT EXPENSES, net | 12c | $ | 15,940 | $ | 17,770 | $ | 12,447 | |||||||||
| MARKETING EXPENSES | 12e | 397 | 562 | - | ||||||||||||
| GENERAL AND ADMINISTRATIVE EXPENSES | 12d | 5,220 | 5,847 | 3,828 | ||||||||||||
| OPERATING LOSS | 21,207 | 10,655 | 16,275 | |||||||||||||
| FINANCIAL INCOME | 14 | (908 | ) | (544 | ) | (285 | ) | |||||||||
| FINANCIAL EXPENSES | 14 | 159 | 27 | 12 | ||||||||||||
| FINANCIAL (INCOME), net | (749 | ) | (517 | ) | (273 | ) | ||||||||||
| LOSS FOR THE YEAR | 20,458 | 10,138 | 16,002 | |||||||||||||
| OTHER COMPREHENSIVE LOSS (INCOME)- | ||||||||||||||||
| Items that will not be reclassified to profit or loss- | ||||||||||||||||
| Re-measurements of post-employment benefit obligation | (25 | ) | 24 | 5 | ||||||||||||
| COMPREHENSIVE LOSS | $ | 20,433 | $ | 10,162 | $ | 16,007 | ||||||||||
| U.S. dollars | ||||||||||||||||
| LOSS PER ORDINARY SHARE | 13 | |||||||||||||||
| Basic and diluted | $ | 0.62 | $ | 0.37 | $ | 0.64 | ||||||||||
| Number of shares | ||||||||||||||||
| WEIGHTED AVERAGE ORDINARY SHARES OUTSTANDING- | ||||||||||||||||
| Basic and diluted | 32,969,094 | 27,398,169 | 24,970,585 | |||||||||||||
(U.S. dollars in thousands, except share and per share amounts)
| 2021 | 2020 | 2019 | ||||||||||
| Year ended December 31 | ||||||||||||
| 2021 | 2020 | 2019 | ||||||||||
| U.S. dollars in thousands | ||||||||||||
| Revenues | $ | 768 | $ | 922 | $ | 562 | ||||||
| Cost of revenues | (365 | ) | (394 | ) | (222 | ) | ||||||
| Gross profit | 403 | 528 | 340 | |||||||||
| Research and development expenses, net | 22,695 | 19,656 | 14,714 | |||||||||
| General and administrative expenses | 7,704 | 5,355 | 5,708 | |||||||||
| Operating loss | 29,996 | 24,483 | 20,082 | |||||||||
| Financial income | (120 | ) | (363 | ) | (870 | ) | ||||||
| Financial expenses | 44 | 105 | 184 | |||||||||
| Financial income, net | (76 | ) | (258 | ) | (686 | ) | ||||||
| Net loss and comprehensive loss | $ | 29,920 | $ | 24,225 | $ | 19,396 | ||||||
| U.S. dollars | ||||||||||||
| Loss per ordinary share | ||||||||||||
| Basic and diluted | $ | 0.45 | $ | 0.55 | $ | 0.54 | ||||||
| Number of shares | ||||||||||||
| Weighted average ordinary shares outstanding | ||||||||||||
| Basic and diluted | 66,346,506 | 43,668,155 | 35,881,256 | |||||||||
The accompanying notes are an integral part of the consolidated financial statements.
| F-4 |
(Continued)-1
VASCULAR BIOGENICS LTD.
CONSOLIDATED STATEMENTS OF CHANGES IN SHAREHOLDERS’ EQUITY
Number of ordinary shares | Ordinary shares | Accumulated other comprehensive income | Additional paid in capital | Warrants | Accumulated deficit | Total equity | ||||||||||||||||||||||
| U.S. dollars in thousands | ||||||||||||||||||||||||||||
| BALANCE AT JANUARY 1, 2016 | 22,470,321 | $ | 38 | $ | 45 | $ | 174,012 | 2,960 | $ | (142,048 | ) | $ | 35,007 | |||||||||||||||
| CHANGES DURING THE YEAR ENDED DECEMBER 31, 2016: | ||||||||||||||||||||||||||||
| Comprehensive loss | - | - | (5 | ) | - | - | (16,002 | ) | (16,007 | ) | ||||||||||||||||||
| Employee stock options exercised | 72,873 | * | - | 121 | - | - | 121 | |||||||||||||||||||||
| Issuance of ordinary Shares, net of issuance costs of $2,117 thousand | 4,359,091 | 12 | - | 21,847 | - | - | 21,859 | |||||||||||||||||||||
| Share based payments to employees and non-employees services | - | - | - | 1,420 | - | - | 1,420 | |||||||||||||||||||||
| BALANCE AT DECEMBER 31, 2016 | 26,902,285 | $ | 50 | $ | 40 | $ | 197,400 | $ | 2,960 | $ | (158,050 | ) | $ | 42,400 | ||||||||||||||
| CHANGES DURING THE YEAR ENDED DECEMBER 31, 2017 | ||||||||||||||||||||||||||||
| Comprehensive Loss | - | - | (24 | ) | - | - | (10,138 | ) | (10,162 | ) | ||||||||||||||||||
| Employee stock options exercised | 252,343 | * | - | 479 | - | - | 479 | |||||||||||||||||||||
| Issuance of ordinary shares, net of issuance costs in amount of $288 thousand | 2,724,695 | 7 | - | 19,024 | - | - | 19,031 | |||||||||||||||||||||
| Share based payments to employees and non-employees services | - | - | - | 4,152 | - | - | 4,152 | |||||||||||||||||||||
| BALANCE AT DECEMBER 31, 2017 | 29,879,323 | $ | 57 | $ | 16 | $ | 221,055 | 2,960 | $ | (168,188 | ) | $ | 55,900 | |||||||||||||||
| ordinary | shares | APIC | Warrants | AD | Total | Shares | Amount | ||||||||||||||||||||||||
Number of ordinary | Ordinary | Additional paid in | Accumulated | Total | Ordinary shares subject to possible | ||||||||||||||||||||||||||
shares | shares | capital | Warrants | deficit | equity | redemption | |||||||||||||||||||||||||
| Shares | Amount | ||||||||||||||||||||||||||||||
| U.S. dollars in thousands | |||||||||||||||||||||||||||||||
| Balance at January 1, 2019 | 35,881,128 | $ | 73 | $ | 233,721 | $ | 7,904 | $ | (188,532 | ) | $ | 53,166 | - | - | |||||||||||||||||
| Changes during the year ended December 31, 2019: | |||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | (19,396 | ) | (19,396 | ) | - | - | |||||||||||||||||||||
| Exercise of options by employees | 97,043 | -* | - | - | - | - | - | - | |||||||||||||||||||||||
| Issuance of ordinary shares and warrants, net of issuance costs | |||||||||||||||||||||||||||||||
| Issuance of ordinary shares and warrants, net of issuance costs, shares | |||||||||||||||||||||||||||||||
| Issuance of ordinary shares, net of issuance costs | |||||||||||||||||||||||||||||||
| Issuance of ordinary shares, net of issuance costs, shares | |||||||||||||||||||||||||||||||
| Issuance of ordinary shares | 1,800 | - | 2 | - | - | 2 | - | - | |||||||||||||||||||||||
| Exercised Warrants | |||||||||||||||||||||||||||||||
| Exercised Warrants, shares | |||||||||||||||||||||||||||||||
| Expired warrants | |||||||||||||||||||||||||||||||
| Share-based compensation | - | - | 2,251 | - | - | 2,251 | - | - | |||||||||||||||||||||||
| Balance at December 31, 2019 | 35,882,928 | 73 | 235,974 | 7,904 | (207,928 | ) | 36,023 | - | - | ||||||||||||||||||||||
| Changes during the year ended December 31, 2020: | |||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | (24,225 | ) | (24,225 | ) | - | - | |||||||||||||||||||||
| Issuance of ordinary shares and warrants, net of issuance costs of $1.7 million | 12,304,535 | 35 | 13,110 | 4,313 | - | 17,458 | - | - | |||||||||||||||||||||||
| Expired warrants | - | - | 1,816 | (1,816 | ) | - | - | - | - | ||||||||||||||||||||||
| Share-based compensation | - | - | 1,661 | - | - | 1,661 | - | - | |||||||||||||||||||||||
| Balance at December 31, 2020 | 48,187,463 | 108 | 252,561 | 10,401 | (232,153 | ) | 30,917 | - | - | ||||||||||||||||||||||
| Balance | 48,187,463 | 108 | 252,561 | 10,401 | (232,153 | ) | 30,917 | - | - | ||||||||||||||||||||||
| Changes during the year ended December 31, 2021: | |||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | (29,920 | ) | (29,920 | ) | - | - | |||||||||||||||||||||
| Issuance of ordinary shares | 28,334 | -* | * | - | - | - | - | - | - | ||||||||||||||||||||||
| Issuance of ordinary shares, net of issuance costs of $2.2 million | 8,971,790 | 27 | 30,925 | - | - | 30,952 | - | - | |||||||||||||||||||||||
| Exercised Warrants | 11,523,997 | 36 | 20,974 | (4,347 | ) | - | 16,663 | - | - | ||||||||||||||||||||||
| Expired warrants | - | - | 2,927 | (2,927 | ) | - | - | - | - | ||||||||||||||||||||||
| Issuance of ordinary shares subject to possible redemption | - | - | - | - | - | - | 615,366 | $ | 1,598 | ||||||||||||||||||||||
| Share-based compensation | - | - | 1,968 | - | - | 1,968 | - | - | |||||||||||||||||||||||
| Balance at December 31, 2021 | 68,711,584 | $ | 171 | $ | 309,355 | $ | 3,127 | $ | (262,073 | ) | $ | 50,580 | 615,366 | $ | 1,598 | ||||||||||||||||
| Balance | 68,711,584 | $ | 171 | $ | 309,355 | $ | 3,127 | $ | (262,073 | ) | $ | 50,580 | 615,366 | 1,598 | |||||||||||||||||
*
| * | Amount less than $1 thousand |
The accompanying notes are an integral part of the consolidated financial statements.
| F-5 |
(Concluded)-2
VASCULAR BIOGENICS LTD.
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITYCASH FLOWS
| Number of ordinary shares | Ordinary shares | Accumulated other comprehensive income | Additional paid in capital | Warrants | Accumulated deficit | Total equity | ||||||||||||||||||||||
| U.S. dollars in thousands | ||||||||||||||||||||||||||||
| BALANCE AT DECEMBER 31, 2017 | 29,879,323 | $ | 57 | $ | 16 | $ | 221,055 | 2,960 | $ | (168,188 | ) | $ | 55,900 | |||||||||||||||
| CHANGES DURING THE YEAR ENDED DECEMBER 31, 2018: | ||||||||||||||||||||||||||||
| Comprehensive income (loss) | - | - | 25 | - | - | (20,458 | ) | (20,433 | ) | |||||||||||||||||||
| Employee stock options exercised | 97,043 | - | - | 34 | - | - | 34 | |||||||||||||||||||||
| Issuance of ordinary shares and warrants, net of issuance costs in an amount of $1,775 thousand | 5,904,762 | 16 | - | 8,765 | 4,944 | - | 13,725 | |||||||||||||||||||||
| Share based payments to employees and non-employees services | - | - | - | 3,867 | - | - | 3,867 | |||||||||||||||||||||
| BALANCE AT DECEMBER 31, 2018 | 35,881,128 | $ | 73 | $ | 41 | $ | 233,721 | 7,904 | $ | (188,646 | ) | $ | 53,093 | |||||||||||||||
(U.S. dollars in thousands)
* Amount less than $1 thousand
| 2021 | 2020 | 2019 | ||||||||||
| Year ended December 31, | ||||||||||||
| 2021 | 2020 | 2019 | ||||||||||
| U.S. dollars in thousands | ||||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||||||
| Net loss | $ | (29,920 | ) | $ | (24,225 | ) | $ | (19,396 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
| Depreciation | 1,256 | $ | 1,194 | 1,219 | ||||||||
| Interest expense (income) | (31 | ) | 48 | 61 | ||||||||
| Net changes in operating leases | 46 | 174 | 241 | |||||||||
| Interest expenses on leases | (2 | ) | 9 | 54 | ||||||||
| Exchange gains on cash and cash equivalents | (15 | ) | (175 | ) | (143 | ) | ||||||
| Changes in accrued liability for employee rights upon retirement | 11 | 12 | 6 | |||||||||
| Share-based compensation | 1,968 | 1,661 | 2,251 | |||||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Increase in other current assets and long-term prepaid expenses | (219 | ) | (119 | ) | (381 | ) | ||||||
| Decrease (increase) in trade receivables | 129 | (129 | ) | - | ||||||||
| Increase (decrease) in accounts payable: | ||||||||||||
| Trade | 2,371 | (1,370 | ) | 2,136 | ||||||||
| Other (including other non-current liability) | 193 | 222 | 1,307 | |||||||||
| Decrease in deferred revenue | (771 | ) | (680 | ) | (444 | ) | ||||||
| Net cash used in operating activities | $ | (24,984 | ) | $ | (23,378 | ) | $ | (13,089 | ) | |||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||||||
| Purchase of property and equipment | (1,465 | ) | (51 | ) | (73 | ) | ||||||
| Investment in short-term bank deposits | (51,109 | ) | (41,085 | ) | (63,027 | ) | ||||||
| Maturity of short-term bank deposits | 37,085 | 51,027 | 57,000 | |||||||||
| Net cash provided by (used in) investing activities | $ | (15,489 | ) | $ | 9,891 | $ | (6,100 | ) | ||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||||||
| Proceeds from issuance of ordinary shares and warrants | 33,155 | 19,132 | 2 | |||||||||
| Issuance costs | (2,202 | ) | (1,674 | ) | - | |||||||
| Proceeds from issuance of ordinary shares subject to possible redemption | 1,598 | - | - | |||||||||
| Proceeds from exercised warrants | 16,662 | - | - | |||||||||
| Finance lease payments | (104 | ) | (391 | ) | (361 | ) | ||||||
| Net cash provided by (used in) financing activities | $ | 49,109 | $ | 17,067 | $ | (359 | ) | |||||
| INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS AND RESTRICTED CASH | 8,636 | 3,580 | (19,548 | ) | ||||||||
| CASH AND CASH EQUIVALENTS AND RESTRICTED CASH AT BEGINNING OF YEAR | 13,697 | 9,942 | 29,347 | |||||||||
| EFFECT OF EXCHANGE RATE ON CASH AND CASH EQUIVALENTS AND RESTRICTED CASH | 15 | 175 | 143 | |||||||||
| CASH AND CASH EQUIVALENTS AND RESTRICTED CASH AT END OF YEAR | $ | 22,348 | $ | 13,697 | $ | 9,942 | ||||||
| SUPPLEMENTARY INFORMATION ON INVESTING AND FINANCING ACTIVITIES NOT INVOLVING CASH FLOWS: | ||||||||||||
| Non cash activity - Purchase of property and equipment in payables | $ | 6 | - | - | ||||||||
| Right of use assets obtained in exchange for new operating lease liabilities | $ | 240 | $ | 230 | $ | 28 | ||||||
| RECONCILIATION OF CASH, CASH EQUIVALENTS, AND RESTRICTED CASH REPORTED IN THE STATEMENT OF FINANCIAL POSITION | ||||||||||||
| Cash and cash equivalents | 21,986 | 13,184 | 9,436 | |||||||||
| Restricted bank deposits | - | 151 | - | |||||||||
| Restricted bank deposits included in non-current assets | 362 | 362 | 506 | |||||||||
| Total cash, cash equivalents, and restricted cash shown in the statement of cash flows | 22,348 | 13,697 | 9,942 | |||||||||
| SUPPLEMENTARY DISCLOSURE ON CASH FLOWS | ||||||||||||
| Interest received | $ | 141 | $ | 416 | $ | 927 | ||||||
| Interest paid | $ | (2 | ) | $ | (9 | ) | $ | (20 | ) | |||
The accompanying notes are an integral part of the consolidated financial statements.
| F-6 |
VASCULAR BIOGENICS LTD.
| Year ended December 31 | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| U.S. dollars in thousands | ||||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||||||
| Loss for the year | $ | (20,458 | ) | $ | (10,138 | ) | $ | (16,002 | ) | |||
| Adjustments required to reflect net cash used in operating activities (see Appendix A) | 3,951 | 5,993 | 2,340 | |||||||||
| Interest received | 849 | 324 | 250 | |||||||||
| Interest paid | (22 | ) | - | - | ||||||||
| Net cash used in operating activities | (15,680 | ) | (3,821 | ) | (13,412 | ) | ||||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||||||
| Purchase of property and equipment | (2,229 | ) | (6,482 | ) | (491 | ) | ||||||
| Proceeds from sale of property and equipment | 4 | - | - | |||||||||
| Investment in short-term bank deposits | (21,000 | ) | (81,332 | ) | (3,600 | ) | ||||||
| Maturity of short-term bank deposits | 47,958 | 66,974 | - | |||||||||
| Net cash generated from (used in) from investing activities | 24,733 | (20,840 | ) | (4,091 | ) | |||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||||||
| Exercise of employees stock options | 34 | 479 | 121 | |||||||||
| Issuance of ordinary shares and warrants, net | 13,725 | 19,031 | 21,859 | |||||||||
| Principal elements of finance lease payments | (88 | ) | - | - | ||||||||
| Net cash generated from financing activities | 13,671 | 19,510 | 21,980 | |||||||||
| NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS | 22,724 | (5,151 | ) | 4,477 | ||||||||
| CASH AND CASH EQUIVALENTS AT BEGINNING OF THE YEAR | 6,694 | 11,585 | 7,090 | |||||||||
| EXCHANGE GAINS (LOSSES) ON CASH AND CASH EQUIVALENTS | (71 | ) | 260 | 18 | ||||||||
| CASH AND CASH EQUIVALENTS AT END OF THE YEAR | $ | 29,347 | $ | 6,694 | $ | 11,585 | ||||||
| APPENDIX A: | ||||||||||||
| Adjustments required to reflect net cash used in operating activities: | ||||||||||||
| Depreciation | $ | 1,156 | $ | 156 | $ | 130 | ||||||
| Interest income | (908 | ) | (331 | ) | (263 | ) | ||||||
| Interest paid | 22 | - | - | |||||||||
| Loss from sale of property and equipment | 47 | - | - | |||||||||
| Exchange gains on lease liability | (60 | ) | - | - | ||||||||
| Exchange losses (gains) on cash and cash equivalents | 71 | (260 | ) | (18 | ) | |||||||
| Net changes in severance pay obligations | (4 | ) | 17 | 8 | ||||||||
| Share based payments | 3,867 | 4,152 | 1,420 | |||||||||
| 4,191 | 3,734 | 1,277 | ||||||||||
| Changes in working capital: | ||||||||||||
| Decrease (increase) in other current assets | 502 | (409 | ) | 126 | ||||||||
| Decrease (increase) in trade receivables | 2,000 | (2,000 | ) | - | ||||||||
| Decrease (increase) in long term prepaid expenses | 55 | (90 | ) | 307 | ||||||||
| Increase (decrease) in accounts payable: | ||||||||||||
| Trade | (1,691 | ) | 421 | 472 | ||||||||
| Other | (521 | ) | 1,199 | 158 | ||||||||
| Increase (decrease) in deferred revenue | (585 | ) | 3,138 | - | ||||||||
| (240 | ) | 2,259 | 1,063 | |||||||||
| $ | 3,951 | $ | 5,993 | $ | 2,340 | |||||||
| APPENDIX B: | ||||||||||||
| Non cash activity- Purchase of property and equipment in payables | $ | 796 | 115 | |||||||||
The accompanying notes are an integral part of the financial statements.
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS
NOTE 1-GENERAL INFORMATION:1 – SIGNIFICANT ACCOUNTING POLICIES:
| a. | General |
Vascular Biogenics Ltd. (the “Company” or VBL) was incorporated on January 27, 2000. The CompanyVBL is a late-stage clinical stage biopharmaceutical company focused on the discovery, development and commercialization of first-in-class treatmentscommitted to developing next-generation, targeted medicines for cancer.difficult-to-treat medical conditions. Using its novel platform technologies, VBL has also developedcreated a proprietarypipeline of therapeutics to uniquely address cancer and immune-inflammatory diseases with the goal of significantly improving patient outcomes and overcoming the limitations of currently approved treatments.
VBL’s product candidates are built off of our two platform of small molecules, Lecinoxoids,technologies: Vascular Targeting System (VTS™), a gene-based technology targeting newly formed blood vessels, and Monocyte Targeting Technology (MTT), an antibody-based technology able to specifically inhibit monocyte migration for the treatment of chronic immune-related indications, and is also conducting a research program exploring the potential of targeting of MOSPD2 for immuno-oncology and anti-inflammatoryimmune-inflammatory applications.
VB-111 (ofranergene obadenovec),We are currently evaluating our lead candidate, ofra-vec, in a Phase 3 drug candidate, is the Company’s lead product candidateregistration-enabling trial in platinum resistant ovarian cancer, for which we anticipate PFS primary endpoint data in the Company’ssecond half of 2022. We are also supporting Phase 2 trials in rGBM and metastatic colorectal cancer, program. VB-201,or mCRC where we expect preliminary data in 2022. Our second program, VB-601, is an investigational proprietary monoclonal antibody that binds MOSPD2, which we call the “mono-walk”, receptor, and is engineered to specifically prevent monocytes from exiting the blood stream and traveling to inflamed tissues, and is expected to begin a Phase 2-ready drug candidate, isfirst-in-human clinical trial in the Company’s lead Lecinoxoid-based product candidate. The Company’s “VB-600 series” for targetingsecond half of MOSPD2 is at pre-clinical stage.2022.
In November 2017, theThe Company entered intohas an exclusive license agreement with NanoCarrier Co., Ltd. (hereinafter - “The License Agreement”) for the development, commercialization, and supply of ofranergene obadenovec (“VB-111”)ofra-vec”, also known as VB-111) in Japan for all indications, see notes 2(m)1(m) and 8.7.
Since inception, the CompanyVBL has incurred significant losses, and it expects to continue to incur significant expenses and losses for at least the next several years. As of December 31, 2018,2021, the Company had an accumulated deficit of $188.6$262.1 million. The Company’sVBL’s losses may fluctuate significantly from quarter to quarter and year to year, depending on the timing of its clinical trials, the receipt of payments under any future collaboration agreements it may enter into, and its expenditures on other research and development activities.
As of December 31, 2018,2021, the Company had cash, cash equivalents, and short-term bank deposits and restricted cash of $50.5 $53.5 million. The CompanyBased on its current cash resources, VBL believes its current cash will be sufficient to fund our operations for at least twelve months from the date of the filing of these financial statements. VBL may seek to raise moreadditional capital to pursue additional activities. The Company may seek these fundsactivities through a combination of private and public equity offerings, government grants, strategic collaborations and licensing arrangements. Additional financing may not be available when the CompanyVBL needs it or may not be available on terms that are favorable to the Company.
In September 2021, the Company established VBL Inc., a U.S. based subsidiary of VBL, and began U.S. operations from this entity in the fourth quarter of 2021.
On December 20, 2021, the Company announced that it was awarded €17.5 million of blended funding from the Horizon Europe Innovation Council (EIC) Accelerator Program, which will be comprised of a €2.5 million grant and an additional €15 million direct equity investment by the EIC. The award is subject to the terms of the program and entering into definitive documentation. The Company has not yet received this funding.
| b. |
These financial statements were approved by the Board of Directors on March 27, 2019.
NOTE 2-SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES:
| Basis of preparation of the financial statements |
The Company’s financial statements of the Company have been prepared in accordance with International Financial Reporting Standardsgenerally accepted accounting principles in the United States of America (“IFRS”) as issued by the International Accounting Standards Board (“IASB”U.S. GAAP”).
| F-7 |
The significant accounting policies described below have been applied consistently in relation to all the periods presented, unless otherwise stated.VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
The financial statements have been prepared under the historical cost convention except of severance pay obligation, net.
NOTE 1 – SIGNIFICANT ACCOUNTING POLICIES (continued):
| c. | Use of estimates in the preparation of financial statements |
The preparation of financial statements in conformity with IFRS requires the use of certain critical accounting estimates. It alsoU.S. GAAP requires management to exercise its judgment inmake estimates and assumptions that affect the processreported amounts of applyingassets and liabilities, the Company’s accounting policies. The areas involving a higher degreedisclosure of judgment or complexity, or areas where assumptions and estimates are significant tocontingent liabilities at the date of the financial statements are disclosed in note 3.
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 2-SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued):
and the reported amounts of expenses during the reporting period. Actual results couldmay differ from those estimates and assumptions.estimates.
| Functional and presentation currency: |
| 1) | Functional and presentation currency |
Items included in the financial statements of the Company are measured usingThe U.S. dollar (“dollar”) is the currency of the primary economic environment in which the entity operates (the “functional currency”). The financial statementsoperations of the Company are conducted. Accordingly, the functional currency of the Company and its U.S. subsidiary is the dollar.
| 2) | Transactions and balances |
Transactions and balances originally denominated in dollars are presented at their original amounts. Balances in U.S. dollar ($), which is the Company’s functional and presentation currency.
Foreign currency transactionsnon-dollar currencies are translated into the functional currencydollars using historical and current exchange rates prevailing at the dates of the transactions. Foreign exchange gainsfor non-monetary and losses resulting from the settlement of suchmonetary balances, respectively. For non-dollar transactions and fromother items in the translation at year-endstatements of operations (indicated below), the following exchange rates of monetary assetsare used: (i) for transactions - exchange rates at transaction dates or average rates; and liabilities denominated in foreign currencies are recognized in the income statement.(ii) for other items (derived from non-monetary balance sheet items such as depreciation and amortization, etc.) - historical exchange rates.
All foreign exchange gains and losses are presented in the statements of comprehensive lossoperations within financial income or expenses.
| Cash, |
Cash andThe Company considers all short-term, highly liquid investments, to be a cash or cash equivalents, include cash on hand andwhich includes short-term bank deposits (withwith original maturities of three months or less)less from the date of purchase that are not restricted as to withdrawal or use and are therefore consideredreadily convertible to known amounts of cash, in addition to restricted cash required to be cash equivalents.set aside for operating lease contractual agreements recorded in non-current assets on the balance sheet.
| Property, plant and |
| 1) | All property and equipment (including leasehold improvements) are stated at |
Repairs and maintenance are charged torecorded in the income statement of comprehensive loss during the period in which they are incurred.
| 2) | The assets are depreciated using the straight-line method to allocate their cost over their estimated useful lives. Annual rates of depreciation are as follows: |
Annual rates of depreciation are as follows:
SCHEDULE OF ESTIMATED USEFUL LIVE
Years | ||||
| Laboratory equipment | ||||
| Computers | ||||
| Office furniture and equipment | ||||
Leasehold improvements in buildings under operating leases are depreciated using the straight-line method over the shorter of the term of the lease or the estimated useful life of the improvements.
| F-8 |
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 1 – SIGNIFICANT ACCOUNTING POLICIES (continued):
| 3) | Gains and losses on disposals are determined by comparing proceeds with the associated carrying amount. These are included in the statements of |
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 2-SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued):
| Impairment of |
Assets that are subject to depreciation are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount may not be recoverable. AnIf the sum of expected future cash flows (undiscounted and without interest charges) of the assets is less than the carrying amount of such assets, an impairment loss is recognized forwould be recognized. The assets would be written down to their estimated fair values, calculated based on the amount by which the asset carrying amount exceeds its recoverable amount. The recoverable amount is the higherpresent value of an assetexpected future cash flows (discounted cash flows), or some other fair value less costs to dispose and its value in use. For the purposes of assessing impairment, assets are grouped at the lowest levels for which there are separately identifiable cash flows (cash-generating units).measure.
Through December 31, 2018, no2021, 0 impairment has been recognized.
As of January 1, 2018, the Company adopted IFRS 9“Financial Instruments”.
From 1 January 2018, the Company classifies its financial assets as measured at amortized cost. The classification depends on the entity’s business model for managing the financial assets and the contractual terms of the cash flows.
Financial assets at amortized cost of the Company are included in trade receivables, other current assets and short term bank deposits in the Statements of Financial Position.
Regular way purchases and sales of financial assets are recognised on trade-date, the date on which the group commits to purchase or sell the asset. Financial assets are derecognised when the rights to receive cash flows from the financial assets have expired or have been transferred and the Company has transferred substantially all the risks and rewards of ownership.
At initial recognition, the Company measures a financial asset at its fair value plus, in the case of a financial asset not at fair value through profit or loss (FVPL), transaction costs that are directly attributable to the acquisition of the financial asset.
Assets that are held for collection of contractual cash flows where those cash flows represent solely payments of principal and interest are measured at amortised cost. Interest income from these financial assets is included in finance income using the effective interest rate method. Any gain or loss arising on derecognition is recognised directly in profit or loss and presented in the Statement of Comprehensive Loss under “Financial Expenses (Income), net” together with foreign exchange gains and losses. Impairment losses are presented as separate line item in the statement of profit or loss.
From 1 January 2018, the company assesses on a forward looking basis the expected credit losses associated with its debt instruments carried at amortised cost. The impairment methodology applied depends on whether there has been a significant increase in credit risk.
For trade receivables, the company applies the simplified approach permitted by IFRS 9, which requires expected lifetime losses to be recognised from initial recognition of the receivables.
Prior to the effective date and adoption of IFRS 9, the financial assets of the Company were classified as loans and receivables. The classification depended on the purpose for which the financial assets were acquired, also, prior to the adoption of IFRS 9, the Company assessed at December 31, 2017 whether there is any objective evidence that a financial asset or group of financial assets was impaired.
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 2-SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued):
Accounts payable
Accounts payable are initially measured at fair value. In subsequent periods, the other financial liabilities are presented at amortized cost. Any difference between the consideration and the redemption value is accreted to profit or loss over the term of the liability, using the effective interest method.
Financial liabilities are classified as current liabilities, unless the Company has an unconditional right to defer settlement of the liability for at least 12 months after the end of the reporting period, in which case they are classified as noncurrent liabilities.
Ordinary Shares are classified as equity. Incremental costs directly attributable to the issue of new shares or options are included in equity as a deduction from the proceeds.
| Deferred income tax |
Deferred taxes are recognized using the asset and liability method on temporary differences arising between the tax bases of assets and liabilities and their carrying amounts in the financial statements.
Deferred income tax assets areA valuation allowance is recognized only to the extent that it is probablemore likely than not that future taxablethe deferred taxes will not be realized in the foreseeable future.
Given the Company’s losses, the Company has provided a full valuation allowance with respect to its deferred tax assets.
| i. |
The Company follows a two-step approach in recognizing and measuring uncertain tax positions. The first step is to evaluate the tax position for recognition by determining if the available evidence indicates that it is more likely than not that the position will be available against whichsustained based on technical merits. If this threshold is met, the temporary differences can be utilized. Deferred incomesecond step is to measure the tax is determined using tax rates and lawsposition as the largest amount that have been enacted or substantively enacted by the balance sheet date and are expected to apply when the related deferred income tax asset ishas more than a 50% likelihood of being realized or the deferred income tax liability is settled.upon ultimate settlement.
Due to absence of expectation of taxable income in the future, no deferred tax assets have been recorded in the Company’s financial statements.
| j. | Employee |
a. Post-employment benefit obligation
Israeli labor laws and the Company’s agreements require the Company to pay retirement benefits to employees terminated or leaving their employment in certain other circumstances. Most of the Company’s employees are covered by a defined contribution plan under Section 14 of the Israel Severance Pay Law from the beginning of their employment with the Company.
With respect to the remaining employees, which are not covered by a defined contribution plan under Section 14 of the Israel Severance Pay Law only from January 1, 2010, the Company records a liability in its statement of financial position for defined benefit plans that represents the present value of the defined benefit obligation as of the statement of financial position date, net of the fair value of plan assets.balance sheet.
The defined benefit obligation is calculated annually by independent actuaries using the projected unit credit method. The present value of the defined benefit obligation is determined by discounting the estimated future cash outflows using interest rates of highly rated corporate bonds that are denominated in the currency in which the benefits will be paid (NIS) and that have terms to maturity approximating the terms of the related liability.
Remeasurement gains and losses arising from adjustments to reflect actual experience and changes in actuarial assumptions are charged or credited to equity in other comprehensive loss (income) in the period in which they arise.
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 2-SUMMARY OF1 – SIGNIFICANT ACCOUNTING POLICIES(continued):
| Vacation and recreation pay |
Under Israeli law, each employee is entitled to vacation days and recreation pay, both computed on an annual basis. The entitlement is based on the length of the employment period. The Company recognizes a liability and an expense for vacation and recreation pay based on the entitlement of each employee.
| k. | Share-based |
The Company operates a number of equity-settled,accounts for employees’ and directors’ share-based compensation plans to employees (as defined in IFRS 2 “Share-Based Payments”), directors and service providers. As part ofpayment awards classified as equity awards using the plans, the Company grants employees, directors and service providers, from time to time and at its discretion, options and RSU’s to purchase Company shares. grant-date fair value method.
The fair value of the employee and service provider services received in exchange for the grant of the options and RSU’s are recognized as an expense in profit or loss andshare-based payment transactions is recorded to Additional paid in capital within equity. The total amount recognized as an expense over the requisite service period.
The Company elected to recognize compensation costs for awards conditioned only on continued service that have a graded vesting periodschedule using the accelerated method over the related service period.
Share based payments to employees and directors were measured by reference to the fair value of the options (the period during which all vesting conditions areand restricted share (hereinafter “RSUs”) granted at date of grant.
The Company calculates the fair value of stock-based option awards on the date of grant using the Black-Scholes option pricing model. This option pricing model requires estimates as to the option’s expected to be met) was determined as follows:term and the price volatility of the underlying stock.
Service conditions and performanceThe Company measures compensation expense for the restricted stock units based on the market value of the underlying stock at the date of grant.
Performance vesting conditions are included in assumptions about the number of options and RSU’s that are expected to vest.
The total expense is recognized over the vesting period, which is the period over which all of the specified vesting conditions are to be satisfied.
At the end of each reporting period, the Company revises its estimates of the number of options and RSU’s that are expected to vest based on the vesting conditions. The Company recognizes the impact of the revision to original estimates, if any, in profit or loss, with a corresponding adjustment to Additional paid in capital.
When options are exercised, the Company issues new shares, with proceeds less directly attributable transaction costs recognized as share capital (par value) and additional paid in capital.
The Company has elected to recognize forfeitures as they occur.
| l. |
Provisions are recognized whenCertain conditions may exist as of the date of the financial statements, which may result in a loss to the Company, hasbut which will only be resolved when one or more future events occur or fail to occur. If the assessment of a present legal or constructive obligation as a result of past events andcontingency indicates that it is probable that an outflow of resources will be required to settle the obligation. Provisions are measured by discounting the future cash outflow at a pretax interest rate that reflects current market assessments of the time value of moneymaterial loss has been incurred and the risks specific to the obligation. The carrying amount of the provisionliability can be estimated, then the estimated liability is adjusted in each reporting period in order to reflect the passage of time and the changesrecorded as accrued expenses in the carrying amountsCompany’s financial statements. If the assessment indicates that a potential material loss contingency is not probable but is reasonably possible, or is probable but cannot be estimated, then the nature of the contingent liability, together with an estimate of the range of possible loss if determinable and material are carried to the statement of comprehensive loss.disclosed.
As of December 31, 2018, no provisions2021, 0 contingent liabilities have been recognized.
| F-10 |
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 1 – SIGNIFICANT ACCOUNTING POLICIES (continued):
| m. | Revenue from contracts with |
General
The Company recognizes revenues from the License Agreement according to ASC 606, “Revenues from Contracts with Customers”.
In November 2017,determining the appropriate amount of revenue to be recognized as the Company signed an exclusive license agreement with NanoCarrier Co. Ltd (hereinafter - “The License Agreement”), for the development, commercialization, and supplyfulfills its obligations under each of VB-111 in Japan (see note 8 for further details).
As of January 1, 2017,its agreements, the Company early adopted IFRS 15, with full retrospective application. Sinceperforms the Company has not generated revenues until 2017, the adoption of IFRS 15 did not have an effect on accumulated deficits as of January 1, 2015 nor on 2015’s and 2016’s comparatives.following steps:
IFRS 15 introduces a five-step model for recognizing revenue from contracts with customers, as follows:
| 1. | identify the contract with a customer; | |
| 2. | identify the performance obligations in the contract; | |
| 3. | determine the transaction price; | |
| 4. | allocate the transaction price to the performance obligations in the contract; | |
| 5. | recognize revenue when (or as) the entity satisfies a performance obligation. |
Revenues from licensing agreement
According to IFRS 15,ASC 606, a performance obligation is a promise to provide a distinct good or service or a series of distinct goods or services. Goods and services that are not distinct are bundled with other goods or services in the contract until a bundle of goods or services that is distinct is created. A good or service promised to a customer is distinct if the customer can benefit from the good or service either on its own or together with other resources that are readily available to the customer and the entity’s promise to transfer the good or service to the customer is separately identifiable from other promises in the contract.
The Company has identified two performance obligations in The License Agreement: (1) Grant of the license and use of its IP; and (2) Company’s participation and consulting assistance services. In addition, there is a potential performance obligation regarding future manufacturing.
IFRS 15ASC 606 defines the ‘Transaction Price’ as the amount of consideration to which the entity expects to be entitled in exchange for transferring the promised goods or services to a customer. The Company allocates the transaction price to each performance obligation identified based onestimates the standalone selling prices of the goods or services beingto be provided based on expected cost-plus margin approach and uses the residual approach to estimate the customer.selling price of the license.
The Grant of the license and use of its IP performance obligation considered to be a right to use IP in accordance with IFRS 15.ASC 606. Therefore, revenue is recognized at a point in time, upon transfer of control over the license to the licensee.
The Company’s participation and consulting assistance services performance obligation is recognized as revenue over the service period, based on input method, which is costs incurred and labor hours expended.
Revenue from achieving additional milestones is recognized only when it is highly probable that a significant reversal of cumulative revenues will not occur, usually upon achievement of the specific milestone.
The transaction price contains variable considerationsconsideration contingent upon the licensee achieving certain milestones, as well as sales-based royalties, in accordance with the relevant agreement.
Variable payments, contingent on achieving additional milestones, are included in the transaction price based on most likely amount method. Amounts included in the transaction price are recognized only when it is highly probable that a significant reversal of cumulative revenues will not occur, usually upon achievement of the specific milestone, in accordance with the relevant agreement.
Sales-based royalties are not included in the transaction price. Rather, they are recognized as the related sale occurs, due to the specific exception of IFRS 15ASC 606 for sales-based royalties in licensing of intellectual properties.
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 2-SUMMARY OF1 – SIGNIFICANT ACCOUNTING POLICIES(continued):
| n. | Research and development expenses: |
Research and development expenses are chargedinclude costs directly attributable to profit or loss as incurred. An intangible asset arising from developmentthe conduct of the Company’s products is recognized if all of the following conditions are met:
Other development costs that do not meet the above criteria are recognized as expenses as incurred. Development costs previously recognized as an expense are not recognized as an asset in a subsequent period.
As of December 31, 2018, the Company has not yet capitalized any development costs.
Government grants, which are received from the Israeli Innovation Authority or IIA (formerly known as the Israeli Office of Chief Scientist, or the “OCS”) by way of participation in research and development that is conducted byprograms, including the Company, fall within the scopecost of “forgivable loans,” as set forth in International Accounting Standard 20 “Accounting for Government Grantsclinical trials, clinical trial supplies, salaries, share-based compensation expenses, payroll taxes and Disclosure of Government Assistance” (“IAS 20”).
As approved by the IIA, the grants are received in installments as the program progresses. The Company recognizes each forgivable loan on a systematic basis at the same time the Company records, as an expense, the relatedother employee benefits, lab expenses, consumable equipment and consulting fees. All costs associated with research and development costs for which the grant is received, provided that there is reasonable assurance that (a) the Company complies with the conditions attached to the grant, and (b) it is probable that the grant will be received (usually upon receipt of approval notice). The amount of the forgivable loan is recognized based on the participation rate approved by the IIA; thus, a forgivable loan is recognizeddevelopments are expensed as a receivable when approved research and development costs have been incurred before grant funds are received.incurred.
Since at the time of grant approval there is reasonable assurance that the Company will comply with the forgivable loan conditions attached to the grant, and it is reasonably assured that the Company will not pay royalties to IIA, grant income is recorded against the related research and development expenses in the statements of comprehensive loss.
If forgivable loans are initially carried to income, as described above, and in subsequent periods it is no longer reasonably assured that royalties will not be paid to the IIA, the Company recognizes a liability that is measured based on the Company’s best estimate of the amount required to settle the Company’s obligation at the end of each reporting period.
Leases in which a significant portion of the risks and rewards of ownership are retained by the lessor are classified as operating leases. Payments made under operating leases are charged to the statements of comprehensive loss on a straight-line basis over the period of the lease.
Leases of property, plant and equipment where the Company, as lessee, has substantially all the risks and rewards of ownership are classified as finance leases. Finance leases are capitalised at the lease’s inception at the fair value of the leased property or, if lower, the present value of the minimum lease payments. The corresponding rental obligations, net of finance charges, are included in short-term lease liability and long-term lease liability. Each lease payment is allocated between the liability and finance cost. The finance cost is charged to the profit or loss over the lease period so as to produce a constant periodic rate of interest on the remaining balance of the liability for each period. The property, plant and equipment acquired under finance leases is depreciated over the asset’s useful life or over the shorter of the asset’s useful life and the lease term if there is no reasonable certainty that the Company will obtain ownership at the end of the lease term.
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 2-SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued):
Operating segments are reported in a manner consistent with the internal reporting provided to the chief operating decision-maker, who is responsible for allocating resources and assessing performance of the operating segments. The Company operates in one operating segment.
Basic loss per share is calculated by dividing the loss attributable to equity holders of the Company by the weighted average number of Ordinary Shares issued and outstanding during the year.
The diluted loss per share is calculated by adjusting the weighted average number of outstanding Ordinary Shares, assuming conversion of all dilutive potential shares.
The Company’s dilutive potential shares consist of options and RSU’s granted to employees and service providers and warrants.
The dilutive potential shares were not taken into account in computing loss per share in 2018, 2017, and 2016 as their effect would not have been dilutive.
s.Trade receivables
Trade receivables are amounts due from customers for goods sold or services performed in the ordinary course of business. If collection of the amounts is expected in one year or less, they are classified as current assets. The Company’s impairment for trade and other receivables are outlined in note 2(f)(3).
|
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 3-SIGNIFICANT ACCOUNTING ESTIMATES AND JUDGEMENTS
Estimates and judgments are continually evaluated and are based on historical experience and other factors, including expectations of future events that are believed to be reasonable under the circumstances.
The Company makes estimates and assumptions concerning the future. The resulting accounting estimates will, by definition, seldom equal the related actual results. The estimates and assumptions that have a significant risk of causing a material adjustment to the carrying amounts of assets and liabilities within the next financial year are discussed below:
Revenue
In 2017, the Company signed a license and supply agreement as more fully described in note 8. In determining the amounts to be recognized as revenue, the Company used its judgement in the following main issues:
Identifying the performance obligations in the agreement and determining whether the license provided is distinct - based on the Company’s analysis, the license is distinct as the licensee is able to benefit from the license on its own at its current stage (inter alia, due to sublicensing rights, rights and responsibility for development in the territory, etc.).
Allocation of the transaction price - the Company estimated the standalone selling prices of the services to be provided based on expected cost plus a margin and used the residual approach to estimate the standalone selling price of the license as the Company has not yet established a price for the license, and it has not previously been sold on a standalone basis.
Variable consideration consists of potential future milestone payments. The Company determined that all such variable consideration shall be allocated to the license (the satisfied performance obligation).
Share-based payments
With respect to grants to employees, the value of the labor services received from them in return is measured on the date of grant based on the fair value of the equity instruments granted to the employees. For options granted prior to the Company’s IPO in order to measure the fair value of the labor service received, the Company first measured the share value by using the income approach method, which is an analysis that involves forecasting the appropriate cash flow stream over an appropriate period and then discounting it back to a present value at an appropriate discount rate. This discount rate should consider the time value of money, inflation, and the risk inherent in ownership of the asset or security interest being valued. Once the share value was estimated, the Company used the Black-Scholes model to value the equity instrument. Since the Company was not publicly traded, it looked for comparable companies in the healthcare sector to set the volatility assumption and estimated the equity instrument’s life. For options granted since 2015, the expected volatility was calculated using weighted average and was based on the stock price volatility of the Company since October 1st, 2014 (IPO date) and the remaining years on the stock price volatility of similar companies.
The Company’s management estimates the fair value of the options granted to consultants based on the value of services receivable over the vesting period of the applicable options.
The value of the transactions, measured as aforesaid, is expensed over the period during which the right of the employees and non-employees to exercise or receive the underlying equity instruments vests; commensurate with every periodic recognition of the expense, a corresponding increase is recorded to additional paid in capital, included under the Company’s equity (see also note 10).
Clinical trial accruals
Clinical trial expenses are charged to research and development expense as incurred. The Company accrues for expenses resulting from obligations under contracts with clinical research organizations (CROs). The financial terms of these contracts are subject to negotiations, which vary from contract to contract and may result in payment flows that do not match the periods over which materials or services are provided. The Company’s objective is to reflect the appropriate trial expense in the financial statements by matching the appropriate expenses with the period in which services and efforts are expended. As
| o. | Government grants |
Government grants, which are received from the Israeli Innovation Authority or IIA (formerly known as the Israeli Office of Chief Scientist, or the “OCS”) by way of participation in research and development that is conducted by the Company, are received in installments as the program progresses based on qualified research spending. Grants received are recognized when the grant becomes receivable, provided there was reasonable assurance that the Company will comply with the conditions attached to the grant and there was reasonable assurance the grant will be received.
The grant is deducted from the research and development expenses as the applicable costs are incurred. Research and development expenses, net, for the years ended December 31, 2018, the company had clinical accruals2021, 2020 and 2019, include participation in research and development expenses in the amount of approximately $1.2 million.$0.5 million, $1.4 million and $2.7 million, respectively.
NOTE 4-FINANCIAL RISK MANAGEMENT:
The Company’s activities expose itCompany determines if an arrangement is a lease at inception. Balances related to a variety of financial risks: market risk (including foreign exchange riskoperating leases are included in operating lease right-of-use (“ROU”) assets, other current liabilities, and price risk), credit and interest risks and liquidity risk. The Company’s overall risk management program focuses onoperating lease liabilities in the unpredictability of financial markets and seeks to minimize potential adverse effects on the Company’s financial performance.
Risk management is performed by the Chief Financial Officer of the Company, who identifies and evaluates financial risks in close cooperation with the Company’s Chief Executive Officer.consolidated balance sheets.
The Company’s finance department is responsible for carrying out risk management activitiesCompany also elected to combine lease and non-lease components and to keep leases with an initial term of 12 months or less off the balance sheet and recognize the associated lease payments in accordance with policies approved by its Boardthe consolidated statements of Directors. The Board of Directors provides guidelines for overall risk management, as well as policies dealing with specific areas such as exchange rate risk, interest rate risk, credit risk, use of financial instruments, and investment of excess cash. In order to minimize exposure to market risk and credit risk,income on a straight-line basis over the Company invested the majority of its cash balances in highly-rated banks.lease term.
ROU assets represent the Company’s right to use an underlying asset for the lease term, and lease liabilities represent the Company’s obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized as of the commencement date based on the present value of lease payments over the lease term. The Company’s lease terms may include options to extend or terminate the lease when it is reasonably certain that the Company will exercise that option. The discount rate for the lease is the rate implicit in the lease unless that rate cannot be readily determined. As the Company’s leases do not provide an implicit rate, the Company’s uses its estimated incremental borrowing rate based on the information available at the commencement date in determining the present value of lease payments. Lease expense for lease payments is recognized on a straight-line basis over the lease term (see also note 5).
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 4-FINANCIAL RISK MANAGEMENT1 – SIGNIFICANT ACCOUNTING POLICIES (continued):
An operating segment is defined as a component that engages in business activities whose operating results are reviewed by the chief operating decision maker for the purpose of assessing performance and allocating resources and for which discrete financial information is available. The Company has 1 operating segment.
| r. | Loss per Ordinary Share |
VBL complies with accounting and disclosure requirements of FASB ASC Topic 260, Earnings Per Share. Basic loss per share of common stock is computed by dividing the net loss by the weighted average number of ordinary shares(including fully vested RSUs and PSUs) outstanding during the period. Due to the existence of Ordinary shares subject to possible redemption, the Company follows the two-class method in calculating loss per share. In computing diluted earnings per share, basic earnings per share are adjusted to take into account the potential dilution that could occur upon the exercise of options and non-vested RSUs and PSUs, using the treasury stock method.
Accretion associated with the ordinary shares subject to possible redemption is excluded from loss per ordinary share.
Potentially dilutive securities have been excluded from VBL’s computation of dilutive loss per share as such securities would have been anti-dilutive. There were , , ordinary shares underlying outstanding options and warrants at December 31, 2021, 2020, and 2019, respectively.
| s. | Concentration of credit risks |
Credit and interest risk arisesarise from cash and cash equivalents and deposits with banks. A substantial portion of the liquid instruments of the Company are invested in short-term deposits in a leading Israeli bank. The Company estimates that since the liquid instruments are mainly invested for the short-term and with a highly-ratedhighly rated institution, the credit and interest risk associated with these balances is immaterial.
Prudent liquidity risk management implies maintaining sufficient cashIn November 2021, the FASB issued ASU 2021-10 “Government Assistance (Topic 832)”, which requires annual disclosures that increase the transparency of transactions involving government grants, including (1) the types of transactions, (2) the accounting for those transactions, and (3) the availabilityeffect of funding throughthose transactions on an adequate amount of committed credit facilities.
Management monitors rolling forecasts of the Company’s liquidity reserve (comprising cash and cash equivalents and deposits). This is generally carried out based on the expected cash flowsentity’s financial statements. The amendments in accordance with practice and limits set by the management of the Company.
this update are effective for financial statements issued for annual periods beginning after December 15, 2021. The Company is in a research stage. It is therefore exposedcurrently evaluating this guidance to liquidity risk, taking into considerationdetermine the forecasts of cash flows required to financeimpact it may have on its investments and other activities.consolidated financial statements.
The Company might be exposed to foreign exchange risk as a result of making payments to employees or service providers and investment of some liquidity in currencies other than the U.S. dollar (the functional currency of the Company). The Company manages the foreign exchange risk by aligning the currencies for holding liquidity with the currencies of expected expenses, based on the expected cash flows of the Company. Had the dollar been stronger by 5% against the New Israeli Shekel (“NIS”), assuming all other variables remained constant, the Company would have recognized an additional expense of $62 thousand, $51 thousand and $13 thousand in profit or loss for the years ended December 31, 2018, 2017 and 2016, respectively.
The Company’s objectives when managing capital are to safeguard the Company’s ability to continue as a going concern in order to provide returns for shareholders and to maintain an optimal capital structure to reduce the cost of capital. It should be noted that the Company is in the research and development stage and does not yet generate regular revenue streams. (See also note 1a).VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 2 – FAIR VALUE MEASUREMENTS
The different levels of valuation of financial instruments are defined as follows:
| Level 1 | Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs. | |
| Level 2 | Observable prices that are based on inputs not quoted on active markets, but corroborated by market data or active market data of similar or identical assets or liabilities. | |
| Level 3 |
The fair value of financial instruments traded in active markets is based on quoted market prices at the dates of the statements of financial position.
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 4-FINANCIAL RISK MANAGEMENT (continued):
A market is regarded as active if quoted prices are readily and regularly available from an exchange, dealer, broker, industry group, pricing service, or regulatory agency, and those prices represent actual and regularly occurring market transactions on an arm’s length basis. These instruments are included in level 1.
The fair value of financial instruments that are not traded in an active market is determined by using valuation techniques. These valuation techniques maximize the use of observable market data where it is available and rely as little as possible on entity specific estimates. If all significant inputs required to fair value an instrument are observable, the instrument is included in level 2.
If one or more of the significant inputs is not based on observable market data, the instrument is included in level 3.
As of December 31, 20182021, 2020 and 2017,2019, the fair value of financial instruments (cash and cash equivalents, short term bank deposits, other current assets trade receivables and accounts payable) areis approximate to their carrying value.
NOTE 3 – SHORT-TERM BANK DEPOSITS
The bank deposits in 2021 of $31.2 million are for terms of three months to one year and carry interest at annual rates of 0.65%-0.85%. The bank deposits in 2020 of $17.1 million are for terms of three months to one year and carry interest at annual rates of 0.01%-0.75%.
NOTE 4 – PROPERTY AND EQUIPMENT
SCHEDULE OF PROPERTY AND EQUIPMENT
| December 31 | ||||||||
| 2021 | 2020 | |||||||
| (in thousands) | ||||||||
| Cost: | ||||||||
| Laboratory equipment* | $ | 6,005 | $ | 4,705 | ||||
| Computers | 328 | 304 | ||||||
| Office furniture and equipment | 200 | 198 | ||||||
| Leasehold improvements | $ | 6,707 | 6,653 | |||||
| $ | 13,240 | $ | 11,860 | |||||
| Less: | ||||||||
| Accumulated depreciation* | $ | 6,393 | $ | 5,228 | ||||
| Property and Equipment, net | $ | 6,847 | $ | 6,632 | ||||
| * | Laboratory equipment includes the finance lease (see Note 5) with a cost of $1.1 million as of December 31, 2021 and 2020. The related accumulated depreciation for the finance lease as of December 31, 2021 and 2020 was $0.6million and $0.5million, respectively. |
Depreciation expense totaled $1.3 million, $1.2 million and $1.2 million for the years ended December 31, 2021, December 31, 2020 and December 31, 2019, respectively.
During the years ended December 31, 2021, the Company disposed of $0.1 million of fixed assets. During the years ended December 31, 2020 and December 31, 2019, the Company did not dispose of any fixed assets.
| F-14 |
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 5 – LEASES
Operating leases
| 1) | In October 2016, the Company entered into a long-term lease contract for approximately $2.2 million over 7 years commencing May 2017 for a new facility in Modi’in, Israel with the option to extend for an additional two periods of three years each. The facility houses the Company’s local biological drugs manufacturing facility, headquarters, discovery research and clinical development. The lease includes a security deposit of $0.4 million which is included in non-current assets on the balance sheet. |
| 2) | The Company maintains operating lease agreements for vehicles it uses. The lease periods are generally for three years. |
Finance Lease
In July 2017, the Company entered into a long-term lease contract for approximately $1.1 million over 3 years commencing April 2018 for a laboratory water purification system used in our manufacturing process.
The following table sets forth data regarding the Company’s leases:
SCHEDULE OF LEASES
| Year ended December 31, | ||||||||||||
| 2021 | 2020 | 2019 | ||||||||||
| (in thousands) | ||||||||||||
| Lease cost | ||||||||||||
| Finance lease cost: | ||||||||||||
| Amortization of right-of-use assets | $ | 168 | $ | 168 | $ | 168 | ||||||
| Interest on lease liabilities | 1 | 9 | 20 | |||||||||
| Operating lease cost | 595 | 535 | 554 | |||||||||
| Other information | ||||||||||||
| Cash paid for amounts included in the measurement of lease liabilities: | ||||||||||||
| Financing cash flows from finance leases | $ | 104 | $ | 391 | $ | 361 | ||||||
| Operating cash flows from operating leases | $ | 586 | $ | 530 | $ | 506 | ||||||
| Financing cash flows from finance leases | $ | 1 | $ | 9 | $ | 20 | ||||||
| Right-of-use assets obtained in exchange for new operating lease liabilities | $ | 368 | $ | 230 | $ | 200 | ||||||
| F-15 |
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 5 – LEASES (continued):
December 31, | ||||||||||||
| 2021 | 2020 | 2019 | ||||||||||
| Weighted-average discount rate - finance leases | 3.0 | % | 3.0 | % | 3 | % | ||||||
| Weighted-average discount rate - operating leases | 4.0 | % | 4.1 | % | 4.2 | % | ||||||
| Weighted-average remaining lease term – finance lease | - | 0.25 | 1.25 | |||||||||
| Weighted-average remaining lease term - operating leases | 4.80 | 5.92 | 7.01 | |||||||||
Future minimum lease payments under non-cancellable leases as of December 31, 2021 were as follows:
SCHEDULE OF FUTURE MINIMUM LEASE PAYMENTS
| Operating Leases | ||||
| (Dollars in thousands) | ||||
| Year ending December 31, | ||||
| 2022 | $ | 615 | ||
| 2023 | 548 | |||
| 2024 | 429 | |||
| 2025 | 435 | |||
| 2026 | 435 | |||
| Thereafter | 145 | |||
| Total future minimum lease payments | 2,607 | |||
| Less imputed interest | (255 | ) | ||
| Total | $ | 2,352 | ||
| F-16 |
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 6 – SEVERANCE PAY OBLIGATIONS
Israeli law generally requires payment of severance pay upon dismissal of an employee or upon termination of employment in certain other circumstances. The Israel pension and severance pay liability to employees are covered mainly by regular deposits with recognized pension and severance pay funds under the employees’ names and through the purchase of insurance policies.
Most of the Company’s employees are covered by a defined contribution plan under Section 14 of the Israel Severance Pay Law. According to the plan, the Company regularly makes payments to severance pay or pension funds without having a legal or constructive obligation to pay further contributions if the funds do not hold sufficient assets to pay all employees in the plan the benefits relating to employee service in the current and prior periods. Neither severance pay liability nor severance pay funds under Section 14 for such employees is recorded on the Company’s balance sheet as the Company is relieved of its obligation upon contribution.
For certain Israeli employees, the Company accrues severance pay liability, calculated pursuant to Israeli Severance Pay Law based on the most recent salary of the employees multiplied by the number of years of employment as of the balance sheet date (the “Shut-Down method”). The liability is recorded as if it was payable at each balance sheet date on an undiscounted basis.
The Company’s liability with respect to Israeli employees’ is covered by monthly deposits with severance pay funds. The value of the deposited funds is based on the cash surrender value of these policies and includes profits (or loss) accumulated through the balance sheet date. The deposited funds may be withdrawn only upon the fulfillment of the obligations pursuant to Israeli Severance Pay Law or labor agreements. The amounts funded are presented separately in the balance sheet as funds in respect to employees’ rights upon retirement.
During the five-year period following December 31, 2021, the Company expects to pay future benefits to two employees upon each such employee’s normal retirement age. The Company anticipates that the benefits payable will be approximately $0.1 million.
The amounts of severance pay expenses were approximately $0.3 million, $0.2 million, and $0.2 million for each of the years ended December 31, 2021, 2020 and 2019, respectively, which were substantially made up of company payments to the Contribution Plans. Gains on amounts funded in respect of employee rights upon retirement for the years ended December 31, 2021, 2020 and 2019 was immaterial.
The Company expects to contribute approximately $0.3 million in the year ending December 31, 2022 to insurance companies in connection with its severance liabilities for its operations for that year, approximately all of which will be contributed to one or more Contribution Plans.
The above amounts were determined based on the employees’ current salary rates and the number of years’ service that will have been accumulated at their retirement date. These amounts do not include amounts that might be paid to employees that will cease working with the Company before reaching their normal retirement age.
| F-17 |
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 4-FINANCIAL RISK MANAGEMENT (continued):
The composition of financial instruments by currency:
As of December 31, 2018:
| Dollars | NIS | Pound sterling | Euro & SEK | Total | ||||||||||||||||
| U.S. dollars in thousands | ||||||||||||||||||||
| Assets: | ||||||||||||||||||||
| Cash and cash equivalents | $ | 26,340 | $ | 2,827 | $ | 95 | $ | 85 | $ | 29,347 | ||||||||||
| Short term bank deposits | 21,135 | - | - | - | 21,135 | |||||||||||||||
| Trade receivables | - | - | - | - | - | |||||||||||||||
| Other current assets (except for prepaid expenses) | - | 521 | - | - | 521 | |||||||||||||||
| 47,475 | 3,348 | 95 | 85 | 51,003 | ||||||||||||||||
| Liabilities- | ||||||||||||||||||||
| Accounts payable and accrued expenses | $ | 1,966 | $ | 2,109 | $ | - | $ | 62 | $ | 4,137 | ||||||||||
| Net assets | $ | 45,509 | $ | 1,239 | $ | 95 | $ | 23 | $ | 46,866 | ||||||||||
As of December 31, 2017:
| Dollars | NIS | Pound sterling | Euro & SEK | Total | ||||||||||||||||
| U.S. dollars in thousands | ||||||||||||||||||||
| Assets: | ||||||||||||||||||||
| Cash and cash equivalents | $ | 4,781 | $ | 1,546 | $ | 187 | $ | 180 | $ | 6,694 | ||||||||||
| Short term bank deposits | 48,035 | - | - | - | 48,035 | |||||||||||||||
| Trade receivables | 2,000 | - | - | - | 2,000 | |||||||||||||||
| Other current assets (except for prepaid expenses) | - | 1,447 | - | - | 1,447 | |||||||||||||||
| 54,816 | 2,993 | 187 | 180 | 58,176 | ||||||||||||||||
| Liabilities- | ||||||||||||||||||||
| Accounts payable and accrued expenses | $ | 4,492 | $ | 1,973 | $ | 45 | $ | 13 | $ | 6,523 | ||||||||||
| Net assets | $ | 50,324 | $ | 1,020 | $ | 142 | $ | 167 | $ | 51,653 | ||||||||||
NOTE 5-SHORT-TERM BANK DEPOSITS:
The bank deposits in 2018 of $21,135 thousand are for terms of three months to one year and carry interest at annual rates of 2.44%-2.64%.The bank deposits in 2017 of $48,035 thousand are for terms of three months to one year and carry interest at annual rates of 1.56%-1.81%.
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 6-PROPERTY AND EQUIPMENT
Composition of assets, grouped by major classifications, and changes therein is as follows:
| Cost | Accumulated depreciation | |||||||||||||||||||||||||||||||||||
Balance at beginning of year | Additions during the year | Disposals during the year | Balance at end of year | Balance at beginning of year | Additions during the year | Disposals during the year | Balance at end of year | Net book value | ||||||||||||||||||||||||||||
| U.S. dollars in thousands | ||||||||||||||||||||||||||||||||||||
| Year ended December 31, 2018: | ||||||||||||||||||||||||||||||||||||
| Laboratory equipment | $ | 3,016 | $ | 1,627 | $ | (4 | ) | $ | 4,639 | $ | 1,501 | $ | 432 | $ | (4 | ) | $ | 1,929 | $ | 2,710 | ||||||||||||||||
| Computers | 238 | 37 | - | 275 | 154 | 50 | - | 204 | 71 | |||||||||||||||||||||||||||
| Office furniture and equipment | 111 | 85 | - | 196 | 20 | 15 | - | 35 | 161 | |||||||||||||||||||||||||||
| Leasehold improvements | 5,675 | 1,251 | (300 | ) | 6,626 | 237 | 659 | (249 | ) | 647 | 5,979 | |||||||||||||||||||||||||
| $ | 9,040 | $ | 3,000 | (304 | ) | $ | 11,736 | $ | 1,912 | $ | 1,156 | $ | (253 | ) | $ | 2,815 | $ | 8,921 | ||||||||||||||||||
| Year ended December 31, 2017: | ||||||||||||||||||||||||||||||||||||
| Laboratory equipment | $ | 2,024 | $ | 1,186 | $ | (194 | ) | $ | 3,016 | $ | 1,605 | $ | 90 | $ | (194 | ) | $ | 1,501 | $ | 1,515 | ||||||||||||||||
| Computers | 400 | 95 | (257 | ) | 238 | 368 | 43 | (257 | ) | 154 | 84 | |||||||||||||||||||||||||
| Office furniture and equipment | 67 | 83 | (39 | ) | 111 | 57 | 2 | (39 | ) | 20 | 91 | |||||||||||||||||||||||||
| Leasehold improvements | 752 | 5,233 | (310 | ) | 5,675 | 526 | 21 | (310 | ) | 237 | 5,438 | |||||||||||||||||||||||||
| $ | 3,243 | $ | 6,597 | (800 | ) | $ | 9,040 | $ | 2,556 | $ | 156 | $ | (800 | ) | $ | 1,912 | $ | 7,128 | ||||||||||||||||||
| Cost | Accumulated depreciation | |||||||||||||||||||||||||||||||
Balance at beginning of year | Additions during the year | Disposals during the year | Balance at end of year | Balance at beginning of year | Additions during the year | Balance at end of year | Net book value | |||||||||||||||||||||||||
| U.S. dollars in thousands | ||||||||||||||||||||||||||||||||
| Year ended December 31, 2016: | ||||||||||||||||||||||||||||||||
| Laboratory equipment | $ | 1,683 | $ | 341 | - | $ | 2,024 | $ | 1,528 | $ | 77 | $ | 1,605 | $ | 419 | |||||||||||||||||
| Computers | 374 | 26 | - | 400 | 336 | 32 | 368 | 32 | ||||||||||||||||||||||||
| Office furniture and equipment | 66 | 1 | - | 67 | 54 | 3 | 57 | 10 | ||||||||||||||||||||||||
| Leasehold improvements | 629 | 123 | - | 752 | 508 | 18 | 526 | 226 | ||||||||||||||||||||||||
| $ | 2,752 | $ | 491 | - | $ | 3,243 | $ | 2,426 | $ | 130 | $ | 2,556 | $ | 687 | ||||||||||||||||||
NOTE 7-SEVERANCE PAY OBLIGATIONS, net:
| Year ended December 31 | ||||||||
| 2018 | 2017 | |||||||
| U.S. dollars in thousands | ||||||||
| Severance pay obligations | $ | 391 | $ | 469 | ||||
| Fair value of plan assets | 292 | 341 | ||||||
| Liability in the statements of financial position | $ | 99 | $ | 128 | ||||
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 7-SEVERANCE PAY OBLIGATIONS, net (continued):
Amounts charged to other comprehensive loss (income) for the years ended December 31, 2018, 2017 and 2016 were $(25) thousand, $24 thousand and $5 thousand, respectively.
The principal actuarial assumptions used for December 31, 2018 and 2017 were as follows:
| 2018 | 2017 | |||||||
| Discount rate | 3.41 | % | 2.8 | % | ||||
| Future salary increases | 4.0 | % | 4.5 | % | ||||
NOTE 8-LICENSE7 – LICENSE AND SUPPLY AGREEMENTAGREEMENTS
In November 2017, the Company signed an exclusive license agreement with NanoCarrier Co., Ltd. for the development, commercialization, and supply of VB-111ofra-vec in Japan. VBL retains rights to VB-111 in the rest of the worldofra-vec globally except for Japan (“The License Agreement”). Under the terms of the agreement, VBL has granted NanoCarrier an exclusive license to develop and commercialize VB-111ofra-vec in Japan for all indications. VBL will supply NanoCarrier with VB-111,ofra-vec, and NanoCarrier will be responsible for all regulatory and other clinical activities necessary for commercialization in Japan. In exchange, the Company received an up-front nonrefundable payment of $15.0 $15.0 million, and is entitled to receive greater than $100.0$100.0 million in additional payments only if certain development orand commercial milestones are achieved. VBL will also receive tiered royalties on net sales. In addition, in caseif NanoCarrier will enterenters into a sublicense agreement, the Company willwould be entitled to receive royalties from the sublicense income received by NanoCarrier.
In December 2017,March 2019, the Company metentered into exclusive option license agreement (hereafter- Agreement) with an animal health company, for the first milestone relatingdevelopment of VB-201 for veterinary use. Under the Agreement, the Company granted a right to use intellectual property and transfer materials. In addition, the commencementCompany granted an option to obtain an exclusive worldwide, royalty-bearing, transferable license under the Company’s intellectual property and materials to research, develop and sell the product worldwide.
As part of the Ovarian Phase 3 trial; and therefore, it recognized $2.0 million in trade receivables, which was received in January 2018.
As of December 2017, and in accordance with IFRS 15,Agreement, the Company concluded that it has satisfied the performance obligation for the grant of the licensereceived an immaterial non-refundable and use of its IP andnon-creditable upfront payment recognized revenue in the amount of $11.9 million.as revenues during 2019. In addition, the Company is entitled to receive an immaterial amount upon the commencementachievement of the Ovarian Phase 3 triala milestone the Company recognized additional variable revenue in the amount of $2.0 million.event.
The performance obligation relating to the Company’s participation and consulting assistance services during the development period is recognized over the service period. During 20182021, 2020 and 2019 the Company recognized revenue in an amount of $0.6$0.8 million, $0.9 million and $0.6 million, respectively related to the Company’s participation and consulting assistance services.services of ofra-vec in Japan for all indications and from the option to license agreement for the development of VB-201 for animal healthcare worldwide. Out of the consideration received in the License Agreement, as of December 31, 20182021, the Company has deferred revenue in the amount of $2.6$0.7 million ($0.3 millionin 2021 that is classified within current liabilities, and $2.3 million within non-current liabilities, which will be recognized until 2021).liabilities.
All revenueRevenues recognized in 2017 was related to the license2021, 2020 and use of the Company’s IP. All revenues recognized in 20182019 were related to the Company’s participation and consulting assistance services.services from the License Agreement and from the option to license agreement for the development of VB-201 for animal healthcare worldwide. All of revenues recognized in 20182021 were included in the opening balance of the deferred revenue in the statements of financial position.balance sheets.
| F-18 |
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 9-COMMITMENTS:8 – COMMITMENTS:
| a. | In April 2011, the Company executed a Commercial License Agreement withJanssen Vaccines & Prevention B.V. (“Janssen”), for incorporating the adenovirus 5 in |
| ● | an annual license fee of | |
| ● | a milestone payment of | |
| ● | royalties of |
There are no limits or caps on the amount of potential royalties. Pursuant to the agreement, the Company has the right to terminate the agreement by giving Janssen three months’ written notice.
| b. |
|
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 9-COMMITMENTS (continued):
The Company entered into operating lease agreements for vehicles it uses. The leases will expire in 2021. The expected lease payments for the years ending December 31, 2019, 2020 and 2021 are approximately $196 thousand, $144 thousand and $27 thousand, respectively.
| In February 2013, the Company entered into an agreement with Tel Hashomer-Medical Research, Infrastructure and Services Ltd. (“Tel Hashomer”). The agreement with Tel Hashomer provides that the Company will pay |
| c. |
| The Company is committed to pay royalties to the Government of Israel on proceeds from sales of products in the research and development of which the Government participates by way of grants. At time the grants were received, successful development of the related project was not assumed. In the case of failure of the project that was partly financed by the Government of Israel, the Company is not obligated to pay any such royalties. Under the terms of the Company’s funding from the Israeli Government, royalties of |
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 9-COMMITMENTS8 – COMMITMENTS (continued):
In addition, under the Research Law, the Company is prohibited from transferring, including by way of license, the IIA-financed technologies and related intellectual property rights and know-how outside of the State of Israel, except under limited circumstances and only with the approval of the IIA Research Committee. The Company may not receive the required approvals for any proposed transfer and, even if received, may be required to pay the IIA a portion of the consideration that it receives upon any sale of such technology to a non-Israeli entity up to 600% of the grant amounts plus interest.
NOTE 9 – SHARE CAPITAL:
| a. | ||
NOTE 10-SHARE CAPITAL:
| Number of shares | ||||||||
| December 31 | ||||||||
| 2018 | 2017 | |||||||
| Authorized: | ||||||||
| Ordinary Shares | 70,000,000 | 70,000,000 | ||||||
| Issued: | ||||||||
| Ordinary Shares (1) | 35,881,128 | 29,879,323 | ||||||
| The Ordinary Shares confer upon their holders the following rights: (i) the right to vote in any general meeting of the Company; (ii) the right to receive dividends; and (iii) the right to receive upon liquidation of the Company a sum equal to the nominal value of the share, and if a surplus remains, to receive such surplus. |
On May 17, 2019, the Company entered into an Equity Distribution Agreement with Oppenheimer & Co. Inc., or Oppenheimer to offer and sell from time to time its ordinary shares, NIS par value, having an aggregate offering price of up to $15,000,000 through Oppenheimer acting as its agent and/or principal. For the year-ended December 31, 2021, the Company sold an aggregate of ordinary shares under its at-the-market (“ATM”) equity facility. The total gross consideration amounted to approximately $3.5 million.
The Company failed to file a prospectus supplement specifying details of the share sales under the ATM. This may have constituted a violation of Section 5 of the U.S. Securities Act of 1933, as amended (the “Securities Act”) and may give rise to liability under Section 12 of the Securities Act (which generally provides a rescission remedy for offers and sales of securities in violation of Section 5) as well as potential liability under the anti-fraud provisions of federal and state securities laws and state rescission laws. In such event, anyone who acquired such ordinary shares would have a right to rescind the purchase. If all the shareholders who acquired ordinary shares demanded rescission, the maximum that VBL would be obligated to repay would be approximately $million, plus interest. Out of the approximately $million of sales, one identified buyer purchased approximately $million of the Company’s ordinary shares. Such identified buyer has agreed to waive any rescission rights and has signed a waiver evidencing such agreement. The Securities Act generally requires that any claim brought for a violation of Section 5 of the Securities Act be brought within one year of the violation. Additionally, if it is determined that such sales did in fact violate the Securities Act, VBL may become subject to fines and penalties imposed by the SEC and state securities agencies. Based on consultation with its counsel and management assessment, VBL did not recognize any provision related to this uncertainty.
VBL analyzed the classification of the ordinary shares. Based on ASC 480-10-S99-3A(f), VBL determined that since the redemption obligation is outside of its control, the ordinary shares should be considered ordinary shares subject to possible redemption, and $1.6 million should classified as temporary equity as ordinary shares subject to possible redemption, as reflected in the balance sheet, see also note 13.
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 10-SHARE9 – SHARE CAPITAL(continued):
| b. |
| On |
|
| The fair value of the | ||
| As of December 31, 2021, all 11,492,065 warrants issued pursuant to these securities purchase agreements were exercised for gross proceeds of approximately $16.7 million. | ||
| c. | On July 29, 2020, at the general meeting of the shareholders of the Company, such shareholders approved the increase of the authorized share capital of the Company to ordinary shares, par value NIS per share. | |
| d. | On January 14, 2021, the Company entered into an ordinary share purchase agreement (Agreement) of up to $ million of VBL’s ordinary shares, par value NIS per share, with an institutional investor. The ordinary shares may be sold from time to time based on our notice to the investor over the 30-month term of the purchase Agreement. As of December 31, 2021, the Company issued shares under the Agreement for gross proceeds of approximately $3.0 million. | |
| e. | On April 9, 2021, VBL entered into an underwriting agreement pursuant to which the Company issued(a) of its ordinary shares to certain investors at a price of $ per ordinary share and (b) pre-funded warrants to purchase 8,050,000 ordinary shares at price of $1.89 per pre-funded warrant with an exercise price of each pre-funded warrant equal to $ per share. In addition, the underwriters exercised an option to purchase additional shares and purchased additional ordinary shares. Net proceeds from the issuance and sale of the ordinary shares and 8,050,000 pre-funded warrants was approximately $26.4 million, after deducting the underwriting discounts and commissions and the estimated offering expenses. | |
| f. | On May 6, 2021, 1,250,000five year warrants issued in the Company’s November 6, 2015 registered direct offering expired. |
| F-21 |
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 9 – SHARE CAPITAL (continued):
| g. |
In February 2000, the Company’s boardBoard of directorsDirectors approved an option plan (the “Plan”) as amended through 2008. Under the Plan, the Company reserved up to Ordinary Shares of NIS par value of the Company for allocation to employees and non-employees.
In April 2011, the Company’s board of directors approved a new option plan (the “New Plan”). Under the New Plan, the Company reserved up to Ordinary Shares (of which Ordinary Shares shall be taken from the unallocated pool reserved under the Plan) for allocation to employees and non-employees.
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 10-SHARE CAPITAL (continued):
In September 2014, the Company’s shareholders approved the adoption of the Employee Share Ownership and Option Plan (2014) (“2014 Plan”) effective as of the closing of the public offering. Under the 2014 Plan, the Company reserved up to Ordinary Shares (of which Ordinary Shares shall be taken from the unallocated pool reserved under the New Plan). The Ordinary Shares to be issued upon exercise of the options confer the same rights as the other Ordinary Shares of the Company, immediately upon allotment. Any option granted under the Plan that is not exercised within ten years from the date upon which it becomes exercisable, will expire. .
Option exercise prices and vesting periods shall be asoption grants are determined by the board of directors of the Company on the date of the grant.
The options granted to employees through December 31, 2002 are subject to the terms stipulated by section 102 of the Israeli Income Tax Ordinance (the “Ordinance”). Among other things, the Ordinance provides that the Company will be allowed to claim as an expense for tax purposes the amounts credited to the employees as a benefit upon sale of the shares allotted under the plans at a price exceeding the exercise price, when the related tax is payable by the employee.
The options granted to employees after December 31, 2002, are subject to the terms stipulated by section 102(b)(2) of the Ordinance. According to these provisions, the Company will not be allowed to claim as an expense for tax purposes the amounts credited to the employees as a capital gain benefit in respect of the options granted.
Options granted to related parties or non-employees of the Company are governed by Section 3(i) of the Ordinance. The Company will be allowed to claim as an expense for tax purposes the amounts equal to the expenses it recorded in the financial statements in the year in which the related parties or non-employees exercised the options into shares.
In March 2017, the Board of Directors approved the increase of 1,027,911 Ordinary Shares to the number of shares available for issuance under the 2014 Plan.
In January 2018, the Board of Directors approved the increase of 1,402,385 Ordinary Shares to the number of shares available for issuance under the 2014 Plan
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 10-SHARE CAPITAL (continued):
Options and Restricted Stock Units (“RSUs”) granted in 2016:
Number of options and RSUs granted according to option plan of the Company | Exercise price per Ordinary | The fair value of options and RSUs | ||||||||||||||||||
| Date of grant | Other than directors | To directors | Total | Share ($) | of grant (in thousands) | |||||||||||||||
| 1) February 2016 | - | 20,000 | 20,000 | $ | 3.48 | $ | 51 | |||||||||||||
| 2) May and June 2016 | 70,000 | - | 70,000 | $ | 3.30 - $3.40 | $ | 230 | |||||||||||||
| 3) March 2016 | 114,129 | - | 114,129 | $ | $ | 412 | ||||||||||||||
| 4) November 2016 | 725,000 | - | 725,000 | $ | 5.08 | $ | 3,232 | |||||||||||||
| 5) November 2016 | 100,000 | - | 100,000 | $ | $ | 492 | ||||||||||||||
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 10-SHARE CAPITAL (continued):
Options and Restricted Stock Units (“RSUs”) granted in 2017:
Number of options granted according to option plan of the Company | Exercise price per Ordinary | The fair value of options on date | ||||||||||||||||||
| Date of grant | Other than directors | To directors | Total | Share ($) | of grant (in thousands) | |||||||||||||||
| 1) February 2017 | - | 20,000 | 20,000 | $ | 5.22 | $ | 100 | |||||||||||||
| 2) March 2017 | 10,000 | - | 10,000 | $ | 5.43 | $ | 30 | |||||||||||||
| 3) March 2017 | - | 65,000 | 65,000 | $ | 5.71 | $ | 337 | |||||||||||||
| 4) June 2017 | 100,000 | - | 100,000 | $ | 5.39 | $ | 437 | |||||||||||||
| 5) June 2017 | 36,000 | - | 36,000 | $ | $ | 175 | ||||||||||||||
| 6) October 2017 | 700,000 | - | 700,000 | $ | 5.99 | $ | 4,113 | |||||||||||||
| 7) October 2017 | 140,000 | - | 140,000 | $ | $ | 903 | ||||||||||||||
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
Options granted in 2018:2019, 2020 and 2021:
SCHEDULE OF STOCK OPTION ACTIVITY
Number of options granted according to option plan of the Company | Exercise price per Ordinary | The fair value of options on date | ||||||||||||||||||
| Date of grant | Other than directors | To directors | Total | Share ($) | of grant (in thousands) | |||||||||||||||
| 1) January 2018 | - | 128,000 | 128,000 | $ | 6.9 | $ | 838,470 | |||||||||||||
| 2) June 2018 | 50,000 | - | 50,000 | $ | 2.22 | $ | 119,264 | |||||||||||||
| 3) September 2018 | - | 30,000 | 30,000 | $ | 1.78 | $ | 45,574 | |||||||||||||
| 4) December 2018 | 935,000 | 370,000 | 1,305,000 | $ | 1.22 | $ | 1,299,867 | |||||||||||||
| Number of | ||||||||||||
| options granted | The fair | |||||||||||
| according to | Exercise | value of | ||||||||||
| option plan of | price per | options on date | ||||||||||
| the company | Ordinary Share | of grant (in | ||||||||||
| Date of grant | Total | ($) | thousands) | |||||||||
| December 19, 2019 | 1,346,000 | $ | 1.22 | $ | 1,411 | |||||||
| November 24, 2020 | 125,000 | $ | 1.17 | $ | 135 | |||||||
| December 8, 2020 | 1,343,000 | $ | 1.22 | $ | 1,753 | |||||||
| July 20, 2021 | 125,000 | $ | 2.38 | $ | 276 | |||||||
| October 4, 2021 | 307,500 | $ | 2.22 | $ | 530 | |||||||
| October 19, 2021 | 174,000 | $ | 2.20 | $ | 340 | |||||||
| December 7, 2021 | 1,188,287 | $ | 2.31 | $ | 2,258 | |||||||
|
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 10-SHARE9 – SHARE CAPITAL(continued):
The fair value of the options on the date of grant was computed using the Black-Scholes model. Fair value of the options was estimated using the expected volatility. The risk-free interest rate was determined based on rates of return on maturity of unlinked treasury bonds with time to maturity that equals the average life of the options.
SCHEDULE OF FAIR VALUE OF STOCK OPTIONS AND RSUs GRANTED
| 2021 | 2020 | 2019 | ||||||||||
| Value of one ordinary share | $-$ | $-$ | $ | |||||||||
| Expected stock price volatility | % | % | % | |||||||||
| Expected term (in years) | ||||||||||||
| Risk free interest rate | %- | % | %- | % | % | |||||||
| Dividend yield | - | - | - | |||||||||
| h. | Changes in the number of options and RSUs and weighted average exercise prices are as follows: |
SCHEDULE OF CHANGES IN NUMBER OF OPTIONS AND RSUS AND WEIGHTED AVERAGE EXERCISE PRICES
| Year ended December 31 | ||||||||||||||||||||||||||||||||||||||||||||||||
| 2021 | 2020 | 2019 | ||||||||||||||||||||||||||||||||||||||||||||||
| Weighted | Weighted | Weighted | ||||||||||||||||||||||||||||||||||||||||||||||
| Year ended December 31 | Number | average | Number | average | Number | average | ||||||||||||||||||||||||||||||||||||||||||
| 2018 | 2017 | 2016 | of | exercise | of | exercise | of | exercise | ||||||||||||||||||||||||||||||||||||||||
Number of options | Weighted average exercise price | Number of options | Weighted average exercise price | Number of options | Weighted average exercise price | options | price | options | price | options | price | |||||||||||||||||||||||||||||||||||||
| Outstanding at beginning of year | 4,036,095 | $ | 3.88 | 3,241,535 | $ | 3.41 | 2,304,179 | $ | 3.17 | 7,569,626 | $ | 2.53 | 6,373,331 | $ | 2.91 | 5,056,914 | $ | 3.36 | ||||||||||||||||||||||||||||||
| Granted | 1,513,000 | 1.74 | 1,071,000 | 4.91 | 1,029,129 | 3.87 | 1,794,787 | 2.29 | 1,468,000 | 1.22 | 1,346,000 | 1.22 | ||||||||||||||||||||||||||||||||||||
| Exercised | (97,042 | ) | 0.33 | (252,343 | ) | 1.91 | (72,873 | ) | 1.66 | (60,265 | ) | 0.01 | - | - | - | - | ||||||||||||||||||||||||||||||||
| Forfeited | (395,140 | ) | 3.31 | (24,097 | ) | 4.18 | (18,900 | ) | 6.92 | |||||||||||||||||||||||||||||||||||||||
| Outstanding at end of year | 5,056,914 | $ | 3.36 | 4,036,095 | $ | 3.88 | 3,241,535 | $ | 3.41 | |||||||||||||||||||||||||||||||||||||||
| Forfeited and expired | (65,500 | ) | 3.01 | (271,705 | ) | 4.35 | (29,583 | ) | 3.30 | |||||||||||||||||||||||||||||||||||||||
| Outstanding at end of year (1) | 9,238,648 | $ | 2.5 | 7,569,626 | $ | 2.53 | 6,373,331 | $ | 2.91 | |||||||||||||||||||||||||||||||||||||||
| Exercisable at end of year | 2,478,796 | $ | 3.70 | 1,844,283 | $ | 2.97 | 1,718,713 | $ | 2.16 | 5,308,234 | $ | 3.1 | 4,149,359 | $ | 3.43 | 3,294,647 | $ | 3.73 | ||||||||||||||||||||||||||||||
| Includes RSUs of , and for the years ended December 31, 2021, 2020 and 2019, respectively |
| F-23 |
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 9 – SHARE CAPITAL (continued):
| i. | The following is information about exercise price and remaining contractual life of outstanding options and RSUs at year-end: |
| December 31, 2018 | December 31, 2017 | December 31, 2016 | ||||||||||||||||||||||||||||||||
| Number of options outstanding at end of year | Exercise price | Weighted average of remaining contractual life | Number of options outstanding at end of year | Exercise Price | Weighted average of remaining contractual life | Number of options outstanding at end of year | Exercise price | Weighted average of remaining contractual life | ||||||||||||||||||||||||||
| 521,509 | $ | 0.002 | 11.34 | 758,928 | $ | 0.002 | 8.29 | 620,970 | $ | 0.002 | 10.73 | |||||||||||||||||||||||
| 72,990 | $ | 1.21 | 5.72 | 98,657 | $ | 1.21 | 6.78 | 117,990 | $ | 1.21 | 7.65 | |||||||||||||||||||||||
| 1,898,969 | $ | 1.22-$2.47 | 15.54 | 513,969 | $ | 2.47 | 10.45 | 713,282 | $ | 2.47 | 1.12 | |||||||||||||||||||||||
| 559,871 | $ | 3.30 - $3.48 | 13.96 | 584,871 | $ | 3.30 - $3.48 | 14.96 | 588,023 | $ | 3.30 - $ 3.48 | 15.96 | |||||||||||||||||||||||
| 60,000 | $ | 6.03 | 16.13 | 60,000 | $ | 6.03 | 17.13 | 60,000 | $ | 6.03 | 18.13 | |||||||||||||||||||||||
| 116,000 | 6.9 | 19.2 | ||||||||||||||||||||||||||||||||
| 372,470 | $ | 7.52 | 16.88 | 409,670 | $ | 7.52 | 17.88 | 416,270 | $ | 7.52 | 18.88 | |||||||||||||||||||||||
| 1,455,105 | $ | 5.08 - $ 5.99 | 18.38 | 1,610,000 | $ | 5.08 - $ 5.99 | 19.38 | 725,000 | $ | 5.08 | 19.87 | |||||||||||||||||||||||
| 5,056,914 | 4,036,095 | 3,241,535 | ||||||||||||||||||||||||||||||||
| December 31, 2021 | December 31, 2020 | December 31, 2019 | ||||||||||||||||||||||||||||||||
| Number of | Weighted | Number of | Weighted | Number of | Weighted | |||||||||||||||||||||||||||||
| options | average of | options | average of | options | average of | |||||||||||||||||||||||||||||
| outstanding | remaining | outstanding | remaining | outstanding | remaining | |||||||||||||||||||||||||||||
| at end of | Exercise | contractual | at end of | Exercise | contractual | at end of | Exercise | contractual | ||||||||||||||||||||||||||
| year | Price | life | year | price | life | year | Price | life | ||||||||||||||||||||||||||
| 448,911 | $ | 0.002 | 509,176 | $ | 0.002 | 509,176 | $ | 0.002 | ||||||||||||||||||||||||||
| 125,000 | $ | 1.17 | 125,000 | 1.17 | - | - | - | |||||||||||||||||||||||||||
| 72,990 | $ | 1.21 | 72,990 | $ | 1.21 | 72,990 | $ | 1.21 | ||||||||||||||||||||||||||
| 6,241,406 | $ | - | 4,491,494 | $ | - | 3,244,969 | $ | - | ||||||||||||||||||||||||||
| 538,871 | $ | - | 538,871 | $ | - | 559,871 | $ | - | ||||||||||||||||||||||||||
| $ | 6.03 | 30,000 | $ | 6.03 | 60,000 | $ | 6.03 | |||||||||||||||||||||||||||
| 86,000 | $ | 6.90 | 106,625 | $ | 6.90 | 116,000 | $ | 6.90 | ||||||||||||||||||||||||||
| 342,470 | $ | 7.52 | 342,470 | $ | 7.52 | 372,470 | $ | 7.52 | ||||||||||||||||||||||||||
| 1,353,000 | $ | - | 1,353,000 | $ | - | 1,437,855 | $ | - | ||||||||||||||||||||||||||
| 9,238,648 | 7,569,626 | 6,373,331 | ||||||||||||||||||||||||||||||||
The aggregate intrinsic value for the options outstanding as of December 31, 2021, 2020 and 2019 was $million, $million and, $million, respectively.
| Expenses for share based compensation recognized in statements of comprehensive loss were as follows: |
SCHEDULE OF SHARE BASED COMPENSATION
Year ended December 31 | Year ended December 31 | |||||||||||||||||||||||
| 2018 | 2017 | 2016 | 2021 | 2020 | 2019 | |||||||||||||||||||
U.S. dollars in thousands | U.S. dollars in thousands | |||||||||||||||||||||||
| Research and development expenses | $ | 2,255 | $ | 2,027 | $ | 900 | $ | 774 | $ | 834 | $ | 1,236 | ||||||||||||
| Administrative and general expenses | 1,541 | 1,977 | 520 | 1,194 | 827 | 1,015 | ||||||||||||||||||
| Marketing expenses | 71 | 148 | - | |||||||||||||||||||||
| $ | 1,968 | $ | 1,661 | $ | 2,251 | |||||||||||||||||||
| $ | 3,867 | $ | 4,152 | $ | 1,420 | |||||||||||||||||||
The remaining unrecognized compensation expenses as of December 31, 20182021 are $3,481 thousand;$million; The unrecognized compensation cost is expected to be recognized over a weighted average period of 1 year.years.
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 11-TAXES10 – TAXES ON INCOME
The Company is taxed according to Israeli tax laws:
| a. | Measurement of results for tax purposes |
The Company as a “foreign-investment company” measures its results for tax purposes in dollar based on Income Tax Regulations (Bookkeeping Principles of Foreign Invested Companies and of Certain Partnerships and the Determination of Their Taxable Income), 1986.
| The Company as a “foreign-investment company” measures its results for tax purposes in dollar based on Income Tax Regulations (Bookkeeping Principles of Foreign Invested Companies and of Certain Partnerships and the Determination of Their Taxable Income), 1986. | |
| b. | Tax rates |
The income of the Company, other than income from Benefitted Enterprises (see c below), is subject to the regular corporate tax rate. In January 2016, the Law for the Amendment of the Income Tax Ordinance (No. 216) was published, enacting a reduction of corporate tax rate beginning in 2016 and thereafter, from 26.5% to 25%.
In December 2016, the Economic Efficiency Law (Legislative Amendments for Implementing the Economic Policy for the 2017 and 2018 Budget Year) was published, introducing a gradual reduction in corporate tax rate from 25% to 23%. As a result, the corporate tax rate in 2017 is 24% and in 2018 and thereafter reduced to 23%.
The corporate tax rate for 2018, 2017 and 2016 were 23%, 24% and 25%, respectively.
| The Company is taxed according to Israeli tax laws. The taxable income of the Company, other than income from Benefited Enterprises (see c below), is subject to the regular Israeli corporate tax rate, which is currently 23%. | |
| c. | Tax benefits under the Law for the Encouragement of Capital Investments, 1959 |
Under the Investment Law, including Amendment No. 60 to the Investment Law that was published in April 2005, by virtue of the Benefited Enterprise program for certain of its production facilities, the Company may be entitled to various tax benefits.
The main benefit arising from such status is the reduction in tax rates on income derived from a Benefited Enterprise. The extent of such benefits depends on the location of the enterprise. Since the Company’s facilities are not located in “national development zone A,” income derived from Benefited Enterprises will be tax exempt for a period of two years and then have a reduced tax rate for a period of up to an additional eight years.
The period of tax benefits, as described above, is limited to 12 years from the beginning of the Benefited Enterprise election year (2012). As of December 31, 2018,2021, the period of benefits has not yet commenced.
In the event of distribution or deemed distribution of dividends from income which was tax exempt as above, the amount distributed will be subject to the tax rate it was exempted from.
The Company is entitled to claim accelerated depreciation in respect of equipment used by the approved enterprisesBenefited Enterprises during five tax years.
Entitlement to the above benefits is conditioned upon the Company fulfilling the conditions stipulated by the Investment Law and regulations published thereunder.
In the event of failure to comply with these conditions, the benefits may be canceled and the Company may be required to apply the regular tax depreciation rates and pay tax on the income in question at the regular corporate tax rates with the addition of linkage differences to the Israeli consumer price index and interest.
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 11-TAXES ON INCOME (continued):
The Investment Law was amended as part of the Economic Policy Law for the years 2011-2012 (the “Amendment”), which became effective on January 1, 2011.
The Amendment sets alternative benefit tracks to the ones currently in place under the provisions of the Investment Law, including a reduced corporate tax rate. Tax rate for “Preferred Enterprise” income of companies not located in national development zone A is 16%16% for fiscal year 2014 and thereafter.
| F-25 |
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 10 – TAXES ON INCOME (continued):
The benefits are granted to companies that qualify under criteria set forth in the Investment Law; for the most part, those criteria are similar to the criteria that have existed in the Investment Law prior to its amendment and the benefit period is unlimited in time. However, in accordance with the Amendment, the classification of licensing income as Preferred income may be subject to the issuance of a pre-ruling by the Israel Tax Authority.
Additional amendments to the Investment Law became effective in January 2017 (the “2017 Amendment”). Under the 2017 Amendment, and provided the conditions stipulated therein are met, income derived by Preferred Companies from ‘Preferred Technological Enterprises’ (“PTE”) (as defined in the 2017 Amendment), would be subject to reduced corporate tax rates of 7.5% in Development Zone “A” and 12% elsewhere, or 6% in case of a ‘Special Preferred Technological Enterprise’ (“SPTE”) as defined in the 2017 Amendment) regardless of the company’s geographical location within Israel. A Preferred Company distributing dividends from income derived from its PTE or SPTE, would subject the recipient to a 20% tax (or lower, if so provided under an applicable tax treaty). The 2017 Amendment further provides that, in certain circumstances, a dividend distributed to a corporate shareholder who is not an Israeli resident for tax purposes would be subject to a 4% tax (inter alia, if the amount of foreign investors in the distributing company exceeds 90%). Such taxes would generally be withheld at source by the distributing company.
On June 14, 2017, the Encouragement of Capital Investments Regulations (Preferred Technology Income and Capital Profits for a Technological Enterprise), 2017 (the “Regulations”) were published, which adopted Action 5 under the base erosion and profit shifting (“BEPS”) regulations. The Regulations describe, inter alia, the mechanism used to determine the calculation of the benefits under the PTE and under the SPTE Regime and determine certain requirements relating to documentation of intellectual property for the purpose of the PTE. According to these provisions, a company that complies with the terms under the PTE regime may be entitled to certain tax benefits with respect to income generated during the company’s regular course of business and derived from the preferred intangible asset (as determined in the Investments Law), excluding income derived from intangible assets used for marketing and income attributed to production activity. In the event that intangible assets used for marketing purposes generate over 10% of the PTE’s income, the relevant portion, calculated using a transfer pricing study, would be subject to regular corporate income tax. If such income does not exceed 10%, the PTE will not be required to exclude the marketing income from the PTE’s total income. The Regulations set a presumption of direct production expenses plus 10% with respect to income related to production, which can be countered by the results of a supporting transfer pricing study. Tax rates applicable to such production income expenses will be similar to the tax rates under the Preferred Enterprise regime, to the extent such income would be considered as eligible. In order to calculate the preferred income, the PTE is required to take into account the income and the research and development expenses that are attributed to each single preferred intangible asset. Nevertheless, it should be noted that the transitional provisions allow companies to take into account the income and research and development expenses attributed to all of the preferred intangible assets they have. Under the Regulations, the Company’s corporate tax rate is expected to be between 12% to 16%.
Under the transitional provisions of the Investment Law, a company is allowed to continue to enjoy the tax benefits available under the Investment Law prior to its amendment until the end of the period of benefits, as defined in the Investment Law.
In each year during the period of benefits of its Benefited Enterprise, the Company will be able to opt for application of the Amendment, thereby making available to itself the tax rate described above. The Company’s election to apply the Amendment is irrevocable.
As of December 31, 2018,2021, the Company’s management decided not to adopt the application of the Amendment.
There is no assurance that future taxable income of the Company will qualify as Benefited, Preferred or Preferred Technological income or that the benefits described above will be available to the Company in the future.
The balance of carry forward losses as of December 31, 2018 and 2017 are $164.9 million and $144.9 million, respectively.
Under Israeli tax laws, carryforward tax losses have no expiration date.
Deferred tax assets on losses for tax purposes carried forward to subsequent years are recognized if utilization of the related tax benefit against a future taxable income is expected.
The Company has not created deferred taxes on its carry forward losses since their utilization is not expected in the foreseeable future.
The Company has tax assessments that are considered to be final through tax year 2013.
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 12-SUPPLEMENTARY FINANCIAL INFORMATION:10 – TAXES ON INCOME (continued):
| December 31 | ||||||||
| 2018 | 2017 | |||||||
U.S. dollars in thousands | ||||||||
| a. Other current assets: | ||||||||
| Institutions - VAT | $ | 287 | $ | 865 | ||||
| Prepaid expenses | 706 | 282 | ||||||
| Government grants receivable | 234 | 482 | ||||||
| Other | - | 100 | ||||||
| $ | 1,227 | $ | 1,729 | |||||
| b. Accounts payable-other: | ||||||||
| Accrued expenses | $ | 2,434 | $ | 2,956 | ||||
| Employee-related accrued expenses | 281 | 317 | ||||||
| Provision for vacation | 229 | 192 | ||||||
| $ | 2,944 | $ | 3,465 | |||||
| d. | Losses for tax purposes carried forward to future years |
| The balance of carry forward losses of the Company as of December 31, 2021 is $222.0 million. | |
| Under Israeli tax laws, carryforward tax losses have no expiration date. | |
| Deferred tax assets on losses for tax purposes carried forward to subsequent years are recognized if utilization of the related tax benefit against a future taxable income is expected. | |
| As the achievement of required future taxable income is not likely, the Company recorded a full valuation allowance. | |
| e. | Tax assessments |
| The Company has tax assessments that are considered to be final through tax year 2016. | |
| f. | Deferred Taxes |
The following table presents summary of information concerning the Company’s deferred taxes as of the periods ending December 31, 2021 and December 31, 2020.
| Year ended December 31 | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
U.S. dollars in thousands | ||||||||||||
| c. Research and development expenses, net: | ||||||||||||
| Payroll and related expenses | $ | 5,182 | $ | 4,636 | $ | 2,921 | ||||||
| Subcontractors and consultation | 7,158 | 12,450 | 8,894 | |||||||||
| Materials | 1,681 | 768 | 556 | |||||||||
| Patent expenses | 780 | 797 | 752 | |||||||||
| Depreciation | 1,086 | 106 | 88 | |||||||||
| Office rent and maintenance | 1,364 | 721 | 397 | |||||||||
| Other | 704 | 481 | 539 | |||||||||
| 17,955 | 19,959 | 14,147 | ||||||||||
| Government grants (see note 9e) | (2,015 | ) | (2,189 | ) | (1,700 | ) | ||||||
| $ | 15,940 | $ | 17,770 | $ | 12,447 | |||||||
| d. Administrative and general expenses: | ||||||||||||
| Payroll and related expenses | $ | 1,838 | $ | 2,681 | $ | 1,499 | ||||||
| Management and professional fees | 2,131 | 2,212 | 1,614 | |||||||||
| Foreign travel | 340 | 279 | 259 | |||||||||
| Depreciation | 70 | 50 | 42 | |||||||||
| Other | 841 | 625 | 414 | |||||||||
| $ | 5,220 | $ | 5,847 | $ | 3,828 | |||||||
| e. Marketing expenses | ||||||||||||
| Payroll and related expenses | $ | 355 | 346 | - | ||||||||
| Consultation | 42 | 216 | - | |||||||||
| $ | 397 | 562 | - | |||||||||
SCHEDULE OF DEFERRED TAXES
| 2021 | 2020 | |||||||
| December 31 | ||||||||
| 2021 | 2020 | |||||||
| U.S. dollars | ||||||||
| in thousands | ||||||||
| In respect of: | ||||||||
| Net operating loss carry forwards | 51,070 | 45,553 | ||||||
| Research and development expenses | 4,310 | 3,244 | ||||||
| Other timing differences | 309 | 375 | ||||||
| Less – valuation allowance | (55,690 | ) | (49,172 | ) | ||||
| Net deferred tax assets | - | - | ||||||
Deferred taxes are computed using the tax rates expected to be in effect when those differences reverse.
The changes in valuation allowance are comprised as follows:
SCHEDULE OF CHANGES IN VALUATION ALLOWANCE
| 2021 | 2020 | |||||||
| Year ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| (U.S. dollars in thousands) | ||||||||
| Balance at the beginning of year | $ | 49,172 | $ | 43,770 | ||||
| Additions during the year | 6,158 | 5,402 | ||||||
| Balance at end of year | $ | 55,690 | $ | 49,172 | ||||
Losses for tax purposes carried forward to future years:
The main reconciling item between the statutory tax rate of the Company and the effective rate is the provision for a full valuation allowance in respect of tax benefits from carry forward tax losses due to the uncertainty of the realization of such tax benefits and the Company’s three year cumulative loss position (see above).
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 13-LOSS11 – SUPPLEMENTARY FINANCIAL INFORMATION:
SCHEDULE OF SUPPLEMENTARY FINANCIAL INFORMATION
| 2021 | 2020 | |||||||
| December 31 | ||||||||
| 2021 | 2020 | |||||||
| U.S. dollars in thousands | ||||||||
a. Other current assets: | ||||||||
| Institutions - VAT | $ | 280 | $ | 187 | ||||
| Prepaid expenses | 1,217 | 1,215 | ||||||
| Government grants receivable | 185 | 6 | ||||||
| Other | 15 | 11 | ||||||
| Other current assets | $ | 1,697 | $ | 1,419 | ||||
| b. Accounts payable-other: | ||||||||
| Accrued expenses | $ | 3,611 | $ | 3,632 | ||||
| Employee-related accrued expenses | 489 | 337 | ||||||
| Provision for vacation | 308 | 306 | ||||||
| Accounts payable-other | $ | 4,408 | $ | 4,275 | ||||
Basic and diluted loss per share:
Basic
Basic loss per share is calculated by dividing the result attributable to equity holders of the Company by the weighted average number of Ordinary Shares in issue during the year.
Diluted
All Ordinary Shares underlying outstanding options, RSU’s and warrants have been excluded from the calculation of the diluted loss per share for the years ended December 31, 2018, 20172021, 2020 and 20162019 since their effect was anti-dilutive. The total number of options, RSU’s and warrants excluded from the calculations of diluted loss per share were – 12,211,676, 5,286,095,, and 4,491,535 for the years ended December 31, 2018, 20172021, 2020 and 2016,2019, respectively.
| Year ended December 31 | Year ended December 31 | |||||||||||||||||||||||
| 2018 | 2017 | 2016 | 2021 | 2020 | 2019 | |||||||||||||||||||
| U.S. dollars in thousands, except per share data | U.S. dollars in thousands, except per share data | |||||||||||||||||||||||
| Basic and diluted: | ||||||||||||||||||||||||
| Loss attributable to equity holders of the Company | $ | 20,458 | $ | 10,138 | $ | 16,002 | $ | 29,920 | $ | 24,225 | $ | 19,396 | ||||||||||||
| Weighted average number of ordinary shares in issue | 32,969,094 | 27,398,169 | 24,970,585 | 66,346,506 | 43,668,155 | 35,881,256 | ||||||||||||||||||
| Loss per ordinary share | $ | 0.62 | $ | 0.37 | $ | 0.64 | $ | 0.45 | $ | 0.55 | $ | 0.54 | ||||||||||||
NOTE 14-FINANCIAL (INCOME) EXPENSES, net:
| Year ended December 31 | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
U.S. dollars in thousands | ||||||||||||
| Financial income: | ||||||||||||
| Interest from deposits | $ | 908 | $ | 335 | $ | 263 | ||||||
| Exchange differences | - | 209 | 22 | |||||||||
| 908 | 544 | 285 | ||||||||||
| Financial expenses: | ||||||||||||
| Bank fees | 37 | 27 | 12 | |||||||||
| Exchange differences | 122 | - | - | |||||||||
| 159 | 27 | 12 | ||||||||||
| TOTAL FINANCIAL (INCOME) EXPENSES, net | $ | (749 | ) | $ | (517 | ) | $ | (273 | ) | |||
VASCULAR BIOGENICS LTD.
NOTES TO THE FINANCIAL STATEMENTS (continued)
NOTE 15-RELATED PARTIES-TRANSACTIONS AND BALANCES:13 – SUBSEQUENT EVENTS:
Key management personnel include membersa. On February 11, 2022, the Company entered into an Open Market Sale AgreementSM with Jefferies LLC, or Jefferies, to offer and sell from time to time its ordinary shares, NIS par value, having an aggregate offering price of up to $50.0 million. As of March 23, 2022, no shares were sold under this ATM facility.
b. Effective February 13, 2022, the board of directors of Vascular Biogenics Ltd. (VBL) approved the adoption of the BoardInducement Plan (2022) to reserve an additional two million () of Directors,VBL’s ordinary shares, NIS par value per ordinary share, to be exclusively for grants of awards to individuals who were not previously employees or non-employee directors of VBL (or following a bona fide period of non-employment with VBL), as an inducement material to each such individual’s entry into employment with VBL within the Chief Executive Officer and all Vice Presidentsmeaning of Rule 5635(c)(4) of the CompanyNasdaq Listing Rules (Rule 5635(c)(4)). The Inducement Plan (2022) was approved by the board of directors without shareholder approval pursuant to Rule 5635(c)(4).
c. In February 2022, the shares that were classified as redeemable shares in 2021 were no longer subject to redemption and companies controlled by them.classified as shareholders’ equity in the first quarter of 2022, see note 9.
| Year ended December 31 | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| U.S. dollars in thousands | ||||||||||||
| Key management compensation: | ||||||||||||
| Labor cost and related expenses | $ | 2,267 | $ | 2,202 | $ | 1,737 | ||||||
| Share-based payments | 1,866 | 2,075 | 817 | |||||||||
| Other | 427 | 406 | 420 | |||||||||
| $ | 4,560 | $ | 4,683 | $ | 2,974 | |||||||
| December 31 | ||||||||
| 2018 | 2017 | |||||||
U.S. dollars in thousands | ||||||||
| Key management- | ||||||||
| Payables and accrued expenses - for salary and related expenses | $ | 484 | $ | 409 | ||||
| Severance pay obligations | $ | 76 | $ | 88 | ||||
| Provision for vacation | $ | 116 | $ | 97 | ||||
NOTE 16-SUBSEQUENT EVENT:
In March 2019, the Company executed an exclusive option license agreement with an animal health company, for the development of the Company’s proprietary anti-inflammatory molecule, VB-201, for veterinary use. The Company retains VB-201 rights for treatment of humans, worldwide. Under the terms of the agreement, the Company has granted an exclusive option license to explore the potential of VB-201 for animal health indications. In consideration, the Company receiving an undisclosed up-front payment, and is entitled to receive additional development milestone payments. Upon exercising the option to license, the Company will receive additional milestones and royalties on net sales.
| † | Portions of this exhibit have been omitted pursuant to a grant of confidential treatment by the Securities and Exchange Commission and the non-public information has been filed separately with the Securities and Exchange Commission. |
| # | English summary of original Hebrew document. |
| * | Filed herewith |
| ** | The certifications furnished in Exhibit 13.1 hereto are deemed to accompany this Annual Report on Form 20-F and will not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended. Such certifications will not be deemed to be incorporated by reference into any filings under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, except to the extent that the Registrant specifically incorporates it by reference. |
SIGNATURES
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
VASCULAR BIOGENICS LTD.
| /s/ Dror Harats | ||
Dror Harats | ||
| Chief Executive Officer | ||
Date: March 28, 201923, 2022
| 99 |