false2021FY0001757064 | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | |
(1): Of which Financial income incurred by renegotiating the convertible bond debt OCEANE | | | | — | | — | | 35,578 |
0.88290.81490.89029,5858,785P2Y
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 20-F
(Mark One)
| | | | | |
| o | REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
| | | | | |
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 20212023
OR
| | | | | |
| o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
| | | | | |
| o | SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| Date of event requiring this shell company report |
Commission File Number 001-38844
GENFIT S.A.
(Exact name of Registrant as specified in its charter and translation of Registrant’s name into English)
France
(Jurisdiction of incorporation or organization)
Parc Eurasanté
885, avenue Eugène Avinée
59120 Loos, France
(Address of principal executive offices)
Pascal Prigent
Chief Executive Officer
GENFIT S.A.
Parc Eurasanté
885, avenue Eugène Avinée
59120 Loos, France
Tel: +33 (0)3 2016 4000 / Fax: +33 (0)3 2016 4001
(Name, Telephone, Email and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act.
| | | | | | | | |
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
| American Depositary Shares, each representing one ordinary share, nominal value €0.25 per share | GNFT | The Nasdaq Global Select Market |
| Ordinary shares, nominal value €0.25 per share* | * | The Nasdaq Global Select Market* |
*Not for trading, but only in connection with the registration of the American Depositary Shares.
Securities registered or to be registered pursuant to Section 12(g) of the Act. None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act. None
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report. Ordinary shares: 49,815,48949,834,983 shares outstanding as of December 31, 20212023
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ☐ Yes ☒ No
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. ☐ Yes ☒ No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ☒ Yes ☐ No
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). x Yes o☒Yes ☐ No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | | | | | | | |
Large accelerated filer o | Accelerated filer x | Non-accelerated filer o | Emerging growth company x |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act .☐
| | | | | |
| † | The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012. |
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant's executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
U.S. GAAP ☐
International Financial Reporting Standards as issued by the International Accounting Standards Board ☒ Other ☐
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow. ☐ Item 17 ☐ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). ☐ Yes ☒ No
TABLE OF CONTENTS
| | | | | | | | |
| | | Page |
| PART I | |
| Item 1. | Identity of Director, Senior Management and Advisers. | |
| Item 2. | Offer Statistics and Expected Timetable. | |
| Item 3. | Key Information. | |
| Item 4. | Information on the Company. | |
| Item 5. | Operating and Financial Review and Prospects. | |
| Item 6. | Directors, Senior Management and Employees. | |
| Item 7. | Major Shareholders and Related Party Transactions. | |
| Item 8. | Financial Information. | |
| Item 9. | The Offer and Listing. | |
| Item 10. | Additional Information. | |
| Item 11. | Quantitative and Qualitative Disclosures About Market Risk. | |
| Item 11C. | Interim Periods. | |
| Item 11D. | Safe Harbor | |
| Item 12. | Description of Securities Other than Equity Securities. | |
| PART II | |
| Item 13. | Defaults, Dividend Arrearages and Delinquencies. | |
| Item 14. | Material Modifications to the Rights of Security Holders and Use of Proceeds. | |
| Item 15. | Disclosure Controls and Procedures. | |
| Item 16A. | Audit Committee Financial Expert. | |
| Item 16B. | Code of Business Conduct and Ethics. | |
| Item 16C. | Principal Accountant Fees and Services. | |
| Item 16D. | Exemptions from the Listing Standards for Audit Committees. | |
| Item 16E. | Purchases of Equity Securities by the Issuer and Affiliated Purchasers. | |
| Item 16F. | Change in Registrant’s Certifying Accountant. | |
| Item 16G. | Corporate Governance. | |
| Item 16H. | Mine Safety Disclosure. | |
| Item 16I. | Disclosure Regarding Foreign Jurisdictions that Prevent Inspections. | |
| Item 16J. | Insider trading policies | |
| Item 16K. | Cybersecurity | |
| PART III | |
| Item 17. | Financial Statements. | |
| Item 18. | Financial Statements. | |
| Item 19. | Exhibits. | |
INTRODUCTION
Unless otherwise indicated, “GENFIT,” “the company,” “our company,” ‘the group,” “we,” “us” and “our” refer to GENFIT S.A. and its consolidated subsidiaries.
“GENFIT,”GENFIT”, the GENFIT logo, “RESOLVE-IT”“RESOLVE-IT®”, “NIS4”"UNVEIL-IT®", “ELATIVE”“NIS4®”, “The NASH Education Program”"NIS2+®", “The NASH Epidemiology Institute”“ELATIVE®”, "NASHnext""NASHNext®", and other trademarks or service marks of GENFIT S.A. appearing in this Annual Report on Form 20-F, or annual report, are the property of GENFIT S.A. or its subsidiaries. Solely for convenience, the trademarks, service marks and trade names referred to in this annual report are listed without the ® and ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their right thereto. All other trademarks, trade names and service marks appearing in this annual report are the property of their respective owners. We do not intend to use or display other companies’ trademarks and trade names to imply any relationship with, or endorsement or sponsorship of us by, any other companies.
Our audited consolidated financial statements have been prepared in accordance with International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board, or IASB.IASB, and in accordance with IFRS as adopted by the European Union. Our financial statements included in this annual report are presented in euros and, unless otherwise specified, all monetary amounts are in euros. All references in this annual report to “$,” “US$,” “U.S.$,” “U.S. dollars,” “dollars” and “USD” mean U.S. dollars and all references to “€” and “euros,” mean euros, unless otherwise noted. Throughout this annual report, references to ADSs mean ADSsAmerican Depositary Shares or ordinary shares represented by such ADSs, as the case may be.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 20-F, or annual report, contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, that are based on our management’s beliefs and assumptions and on information currently available to our management. All statements other than present and historical facts and conditions contained in this annual report, including statements regarding our future results of operations and financial positions, business strategy, plans and our objectives for future operations, are forward-looking statements. When used in this annual report, the words “anticipate,” “believe,” “can,” “could,” “estimate,” “expect,” “intend,” “is designed to,” “may,” “might,” “plan,” “potential,” “predict,” “objective,” “should,” or the negative of these and similar expressions identify forward-looking statements. Forward-looking statements include, but are not limited to, statements about:
•ourpotential regulatory approval and commercialization of elafibranor, as well as future plans to developfor development and commercialize elafibranor,commercialization of tests powered by our NIS4NIS4® technology or its improvements and our other drug candidates;
•the initiation, timing, progress and results of our preclinical studies and clinical trials, including the timing of availability of data from our clinical trials;
•our ability to successfully expand and advance our pipeline of drug candidates, including through in-licensing agreements;
•our and our collaborators' ability to expand the research, clinical and commercial use of diagnostics incorporating our NIS4 technology;NIS4® technology or its improvements;
•the timing of our planned regulatory filings;
•the timing of and our ability to obtain and maintain regulatory approvals;
•the clinical utility and market acceptance of our drug candidates and tests powered by our NIS4 technology;NIS4® technology or its improvements;
•the potential clinical utility of our product candidates and their potential advantages over existing therapies as well as those in development;
•our ability to establish and maintain manufacturing and supply arrangements for our product candidates;
•our ability to build our commercial organization in the event we elect to directly commercialize any approved products;
•the ability of third parties with whom we contract to successfully conduct, supervise and monitor clinical trials for our product candidates;
•the potential benefits of strategic collaboration agreements and our ability to enter into strategic arrangements;
•the effects of increased competition as well as innovations by new and existing competitors in our industry;
•our ability to maintain, protect and enhance our intellectual property rights and proprietary technologies and to operate our business without infringing the intellectual property rights and proprietary technology of third parties;
•our estimates regarding future milestone payments and royalties, cash consumption, revenues, expenses and needs for additional financing, including our ability to fund our existing programs and execute our strategy based on our current financial position;
•the impact of the COVID-19 pandemic on our business and operations; and
•other risks and uncertainties, including those listed in this annual report under the caption “Risk Factors.”
You should refer to the section of this annual report titled “Item 3.D—Risk Factors” for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this annual report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame or at all. We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law. You should read this annual report and the documents that we reference in this annual report and have filed as exhibits to this annual report completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
This annual report contains market data and industry forecasts that were obtained from industry publications. These data involve a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. We have not independently verified any third-party information. While we believe the market position, market opportunity and market size information included in this annual report are generally reliable, such information is inherently imprecise.
SUMMARY RISK FACTORS
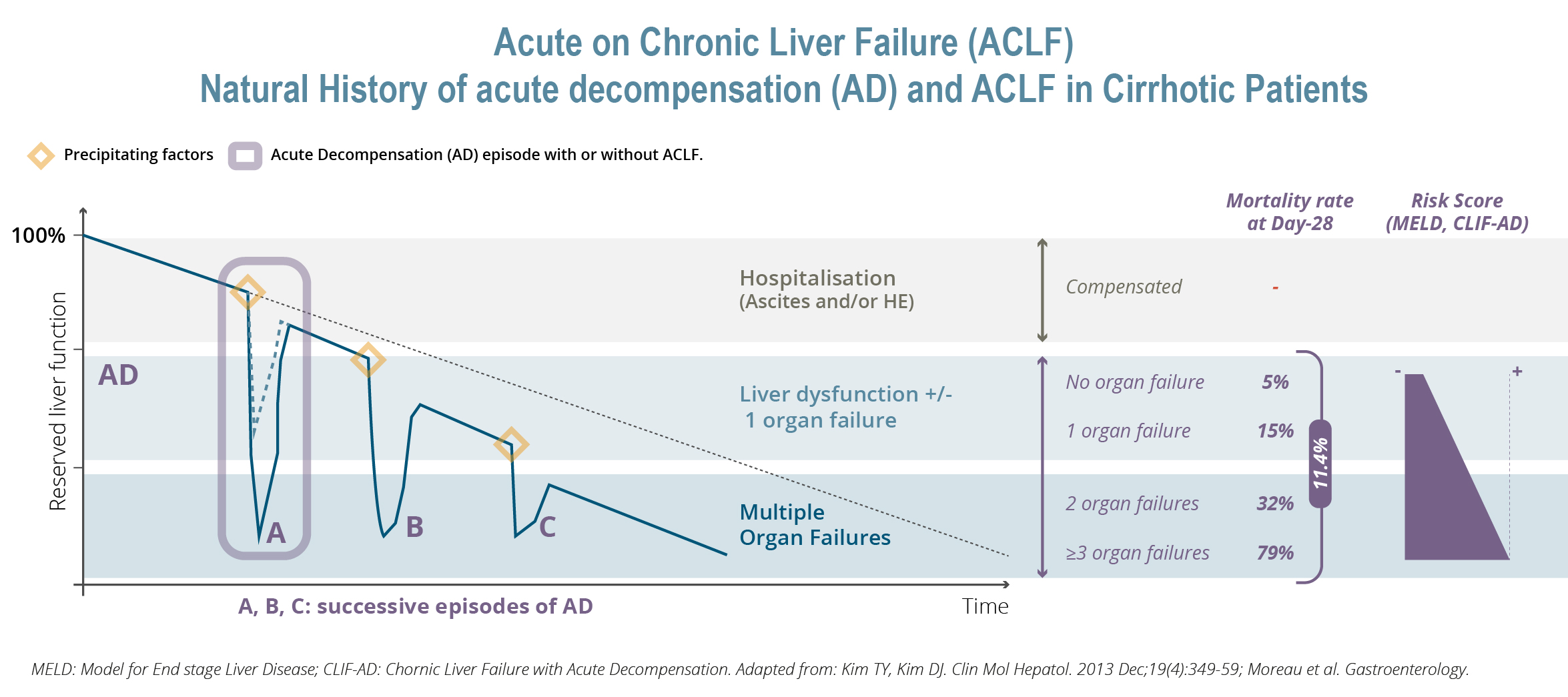
Investing in our shares involves numerous risks, including the risks described in “Item 3.D—3.D - "Risk Factors”" of this annual report. Below are some of our principal risks, any one of which could materially adversely affect our business, financial condition, results of operations, and prospects: •Our drug candidate development activities are focused primarily on the development of our drug candidate elafibranor in PBC as well as on other drug candidates for which development is less advanced. Drug development is subject to a number of risks and the Group is highly exposed to the occurrence of any one of these inherent risks. Our activities in this area are all the more risky as many of our drug candidates are being evaluated in ACLF, a new therapeutic area, are at an early development stage and, for some of them, we were not involved in the initial research and discovery work, and may be less familiar with their mechanisms of action.
•ClinicalDevelopment failure can occur at any stage of preclinical or clinical development, as was the case with our Phase 3 RESOLVE-IT trial of elafibranor in NASH.development. The results of earlier preclinical studies or clinical trials are not necessarily predictive of future results and elafibranor in PBC or any otherof product candidatecandidates that we or our collaborators advance through preclinical studies or clinical trialstrials. We may not have favorable results in later clinical trials, which may delay, limit or prevent our ability to receive regulatory approval or marketing authorization.
•Delays in the commencement enrollment and completion of preclinical studies and clinical trials, and in enrollment of patients for clinical trials, including our Phase 3 ELATIVE trial of elafibranor in PBC,ongoing clinical trials, could result in increased costs to us and delay or limit our ability and that of Ipsen and Terns Pharmaceuticals, our partners for elafibranor, and that of any future collaborators, to obtain regulatory approval for elafibranor and our other drug candidates. Such delays and costs could impair our financing capacity, and these events may limit or compromise our ability to continue development and to eventually commercialize our drug candidates.
•We cannot be certain that elafibranor or any of our other product candidates, even if they meet clinical and regulatory requirements, will receive regulatory approval or certification, as applicable, and without regulatory approval or certification, we will not be able to market our product candidates.
•WeEven though we have obtained breakthrough therapyorphan drug designation from the U.S. Food and Drug Administration or FDA for elafibranor infor the treatment of PBC in both the US and EEA, we, or Ipsen, may through our partnershipnot be able to obtain or maintain the benefits associated with Ipsen, seek to avail ourselves of such mechanisms to expedite the development or approval of elafibranor for another indication or in combination in the future or in order toorphan drug status, including market exclusivity. To accelerate the development, approval or approvalfuture commercialization of some of our other drug candidates, we, or our current or future collaborators, may seek to use certain regulatory pathways, but such mechanisms may not actually lead to a faster development or regulatory review or approval process, and it may not increase the likelihood that elafibranorour drug candidates will receive marketing approval for this indication.approval.
•Due toOur near and medium-term future capital resources depend in large part on the regulatory approval of elafibranor in PBC. Because our limited resources and access to capital,alternative financing is limited, failure in PBC could impact our strategic decisions with respect to the development of certainour other product candidates and may affect the development or timing of our business prospects.
•The development of our NIS4 technology and tests powered by this technology requires access to clinical trials, data and clinical samples in NASH patients and therefore our development is subject to the risks related to these trials.prospects
•We intendwill require substantial additional funding to develop and market an in-vitro diagnosticcommercialize our products, if approved, as well as to reinforce our pipeline, which may not be available to us, or IVD powered by NIS4 as a clinical diagnosticto our current or future partners on acceptable terms, or at all, and, as such, NIS4 remains a product in development subjectif not so available, may require us or them to the hazards of diagnostic product development. In addition, there is no assurance that we will be able to receive the necessary regulatory approvals (including CE Certificate of Conformity) to market an IVD, powered by NIS4 technologydelay, limit, reduce or achieve commercialization of this product candidate forcease our intended market.operations.
•Even if approved, our product candidates may not achieve broad market acceptance among physicians, patients and healthcare payors, and as a result our revenues generated from their sales may be limited.
•If we, or our current and future collaborators are unable to establish sales, marketing and distribution capabilities for elafibranor or our other product candidates, we may not be successful in commercializing those product candidates if and when they are approved.
•Government restrictions on pricing and reimbursement, as well as other healthcare payor cost-containment initiatives, may negatively impact our ability or that of our current or future collaborators to generate revenues even if we or they obtain regulatory approval to market a product candidate.
•We have entered, and may in the future enter into, collaboration, licensing or co-marketing agreements with third parties for the development and eventual commercialization of our product candidates and NIS4NIS4® diagnostic technology or its improvements and may not generate revenues from these agreements.
•We depend on third-party contractors for a substantial portion of our operations, namely contract research organizations or CROs for our preclinical studies and clinical trials and contract manufacturing organizations or CMOs for manufacturing of our active ingredients and therapeutic units and may not be able to control their work as effectively as if we performed these functions ourselves.
•We rely entirely on third parties for the manufacturing of our drug candidates and the future manufacturing of an in-vitro diagnostic, or IVD, powered by NIS4NIS4® or its improvements for use as a clinical diagnostic including one manufacturer for the active ingredient in elafibranor and another manufacturer for the therapeutic units of elafibranor used in our clinical trials.diagnostic. Our business could be harmed if those third parties fail to provide us with sufficient quantities of drug product or tests, or fail to do so at acceptable quality levels or prices.
•Starting in mid-2020 and into 2021, we embarked on a significant strategic reorientation which resulted in a significant changes to our organization and workforceworkforce. As a result, we may encounter difficulties in managing development of our product candidate pipeline, which could disrupt our operations.
•The outbreakWe have recently acquired and may in the future acquire, products or businesses or form new strategic alliances, and we may not realize the benefits of the novel coronavirus disease, COVID-19, has adversely impactedsuch partnerships or acquisitions.
•Our internal information technology systems and those of our current or future collaborators or those of our third-party contractors or consultants, may fail or suffer security breaches, any of which could continue to adversely impactresult in a material disruption of our business, including our preclinical studiesproduct development and clinical trials.commercialization programs.
•If we are unable to obtain and maintain sufficient patent protection for our product candidates, or if the scope of the patent protection is not sufficiently broad, our competitors could develop and commercialize products similar or identical to ours, and our ability or that of a potential future partner to commercialize our product candidates successfully may be adversely affected.
•Currently, besides NASHnextNASHNext® commercialized by our partner, Labcorp, we have no products approved for commercial sale, and to date we have not generated any significant recurring revenue from product sales. As a result, our ability to sustainably reduce our losses, reach lasting profitability, as a result of such types of revenue, and reach sustainable profitability and rebuildmaintain our shareholders equity on our own is unproven, and we may never achieve or sustain profitability.
•Our ability to be profitable in the future will depend on our ability and that of our current or future collaborators to obtain marketing approval for and commercialize our product candidates, particularly our lead product candidate, elafibranor, and an LDT or IVD powered by NIS4 for clinical care.
•We will require substantial additional funding to develop and commercialize our products, if approved, which may not be available to us, or our current or future collaborators on acceptable terms, or at all, and, if not so available, may require us or them to delay, limit, reduce or cease our operations.elafibranor.
•Our stock price may never reach a price at which certain bondholders will deem conversion economically viable, in which case we would need to repay the nominal amount at maturity in October 2025. The terms of our convertible bonds require us to meet certain operating covenants, and if we fail to comply with those covenants the bondholders would be able to accelerate our repayment obligations. Additionally, the conversion of some or all of our bonds into ordinary shares would dilute the ownership interests of existing shareholders
•The market price of our equity securities is particularly volatile and may decline regardless of our operating performance.
•The dual listing of our ordinary shares and our ADSs may adversely affect the liquidity and value of our ordinary shares and ADSs.
•The purported securities class action litigation against us is currently on appeal and we may become subject to additional litigation, which could harm our business and financial condition.
•The rights of shareholders in companies subject to French corporate law differ in material respects from the rights of shareholders of corporations incorporated in the United States.
PART I
Not applicable.
| | | | | |
| Item 2. | Offer Statistics and Expected Timetable. |
Not applicable.
| | | | | |
| B. | Capitalization and Indebtedness |
Not applicable.
| | | | | |
| C. | Reasons for the Offer and Use of Proceeds |
Not applicable.
Our business faces significant risks. You should carefully consider all of the information set forth in this annual report and in our other filings with the United States Securities and Exchange Commission, or the SEC, including the following risk factors which we face and which are faced by our industry. Our business, financial condition or results of operations could be materially adversely affected by any of these risks. This report also contains forward-looking statements that involve risks and uncertainties. Our results could materially differ from those anticipated in these forward-looking statements, as a result of certain factors including the risks described below and elsewhere in this annual report and our other SEC filings. See “Special Note Regarding Forward-Looking Statements” above. Risks Related to the Discovery and Development of and Obtaining Regulatory Approval for Our Product Candidates
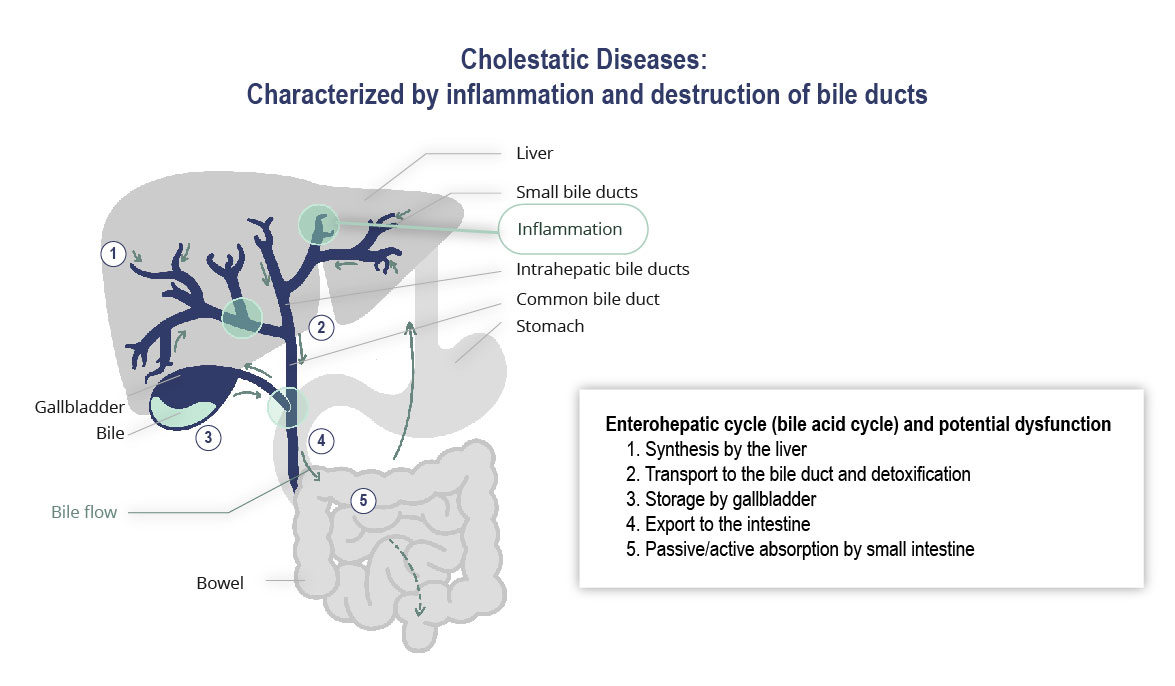
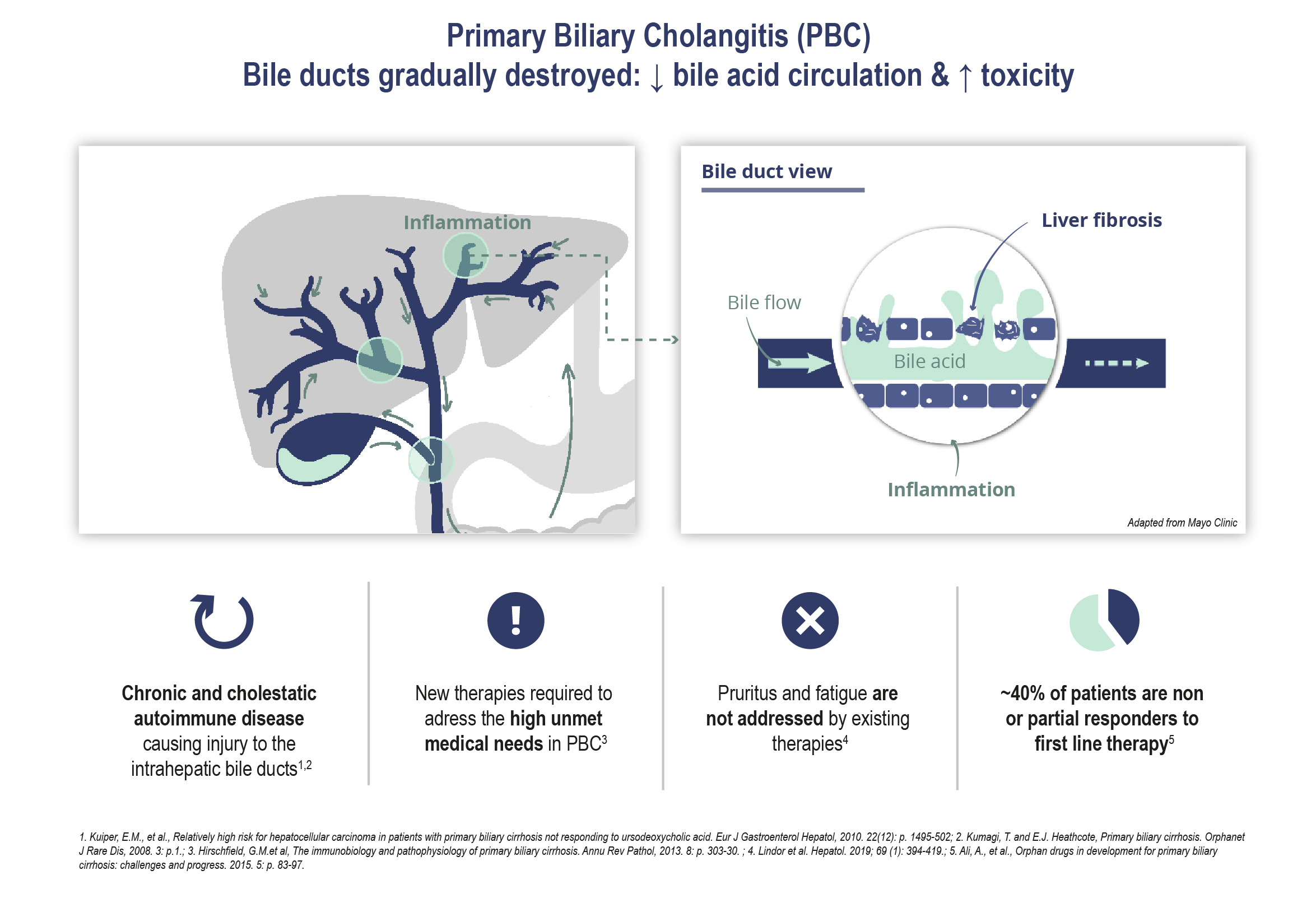
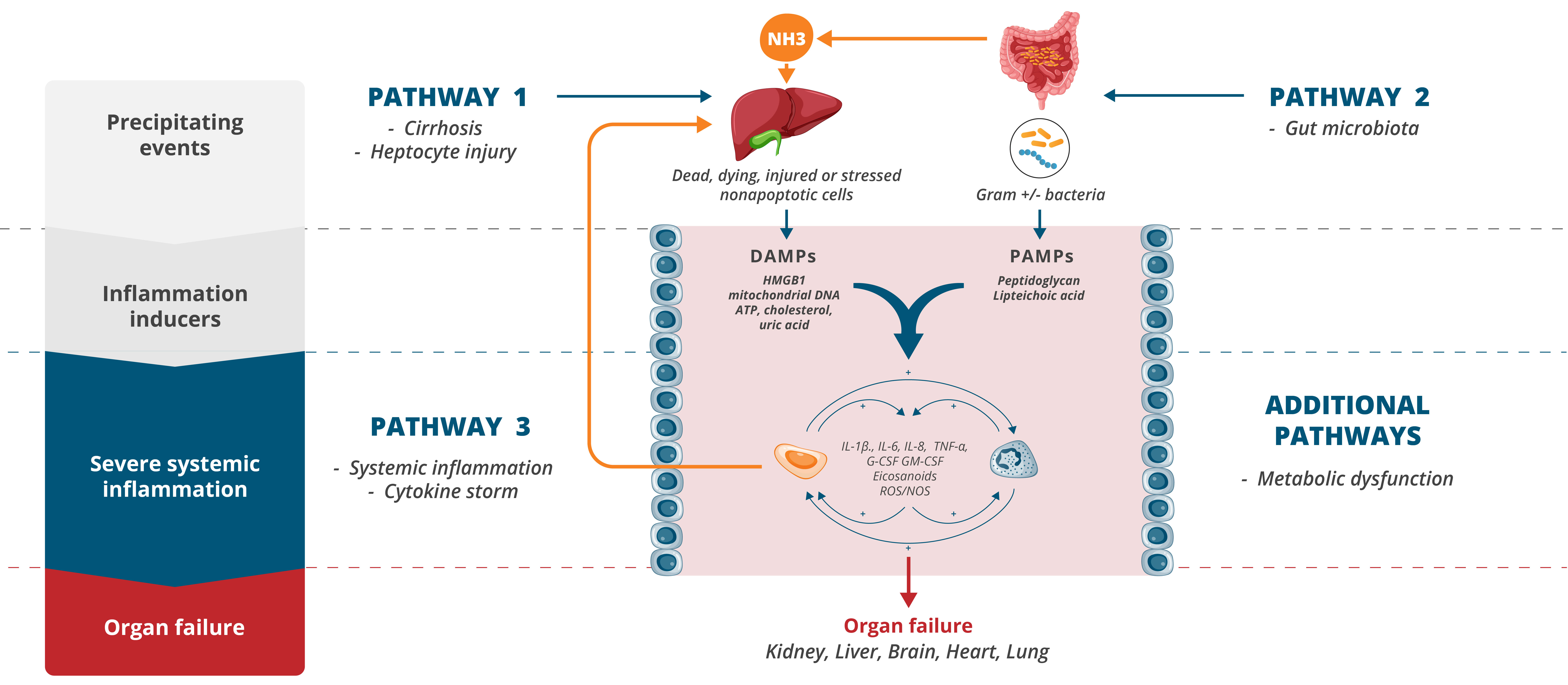
Our drug candidate development activities are focused primarily on the development of our drug candidate elafibranor in PBC as well as on other drug candidates for which development is less advanced. Drug development is subject to a number of risks and the Group is highly exposed to the occurrence of any one of these inherent risks. Our activities in this area are all the more risky as many of our drug candidates are being evaluated in ACLF, a new therapeutic area, are at an early development stage and, for some of them, we were not involved in the initial research and discovery work, and may be less familiar with their mechanisms of action.
Drug development is a long, costly and uncertain process, aimed at demonstrating the therapeutic benefit of a drug candidate that competes with existing products and standards of care or other drug candidates in development.
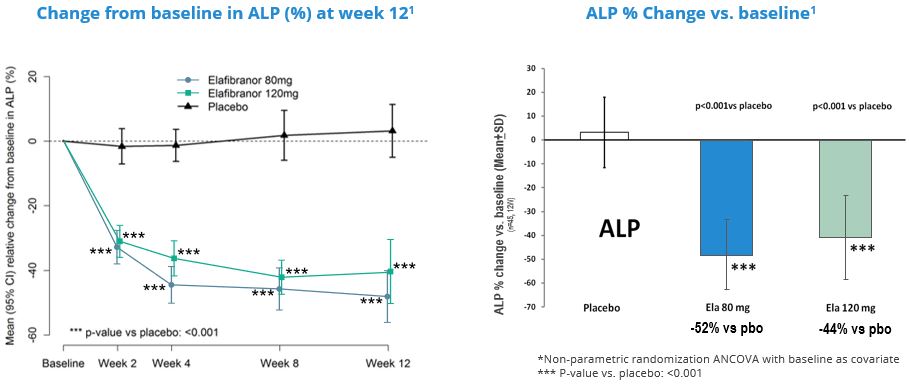
In 2019,June 2023, we entered into aannounced positive interim results from the Phase 3 ELATIVE® trial for our drug candidate elafibranor in PBC following clinical development carried out under the licensing and collaboration agreementagreements we signed with Terns Pharmaceuticals for elafibranorin 2019 in Greater China, and Ipsen in December 2021 the remaining worldwide rights to elafibranor in all indications were licensed to Ipsen. As part of the collaboration with Ipsen, elafibranor,other major pharmaceutical markets. Following these results, our most advanced drug candidate, is currently being evaluated in a Phase 3 ELATIVE clinical trial in primary biliary cholangitis or PBC. Pursuant to this agreement, we remain responsible for the conduct of the phase 3 ELATIVE study until the end of the double-blind period.
Only two treatments are currently approved and marketed in this indication and do not meet the medical needs of all patients. A limited number of treatments are therefore approved for the management of this disease and we have little experience with drug development in this disease area. The development and approvalproduct pipeline now composed of drug candidates whose development is much less advanced and therefore inherently more risky. These drug candidates, even if they have demonstrated promising initial preclinical or clinical results, have yet to treat PBC may therefore presentobtain their preclinical and/or clinical proof-of-concept in the indications for which they are intended.
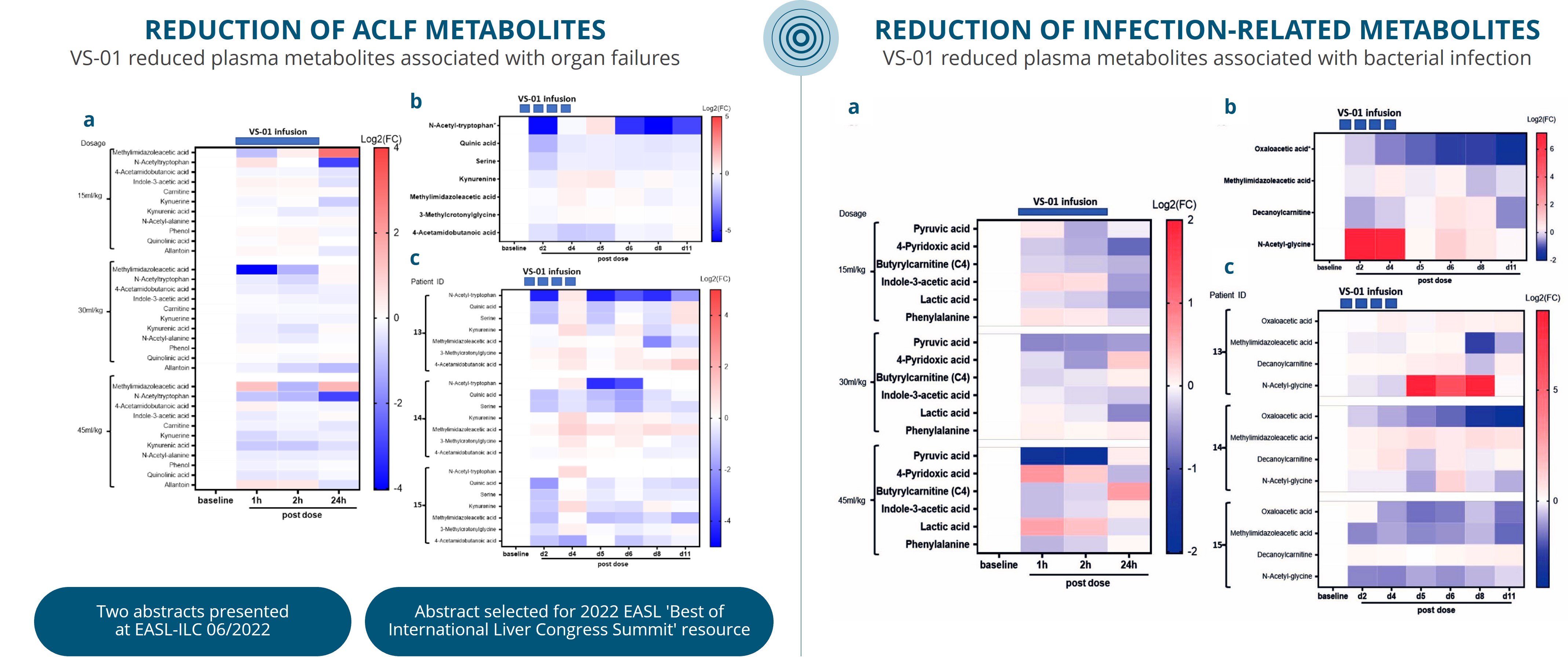
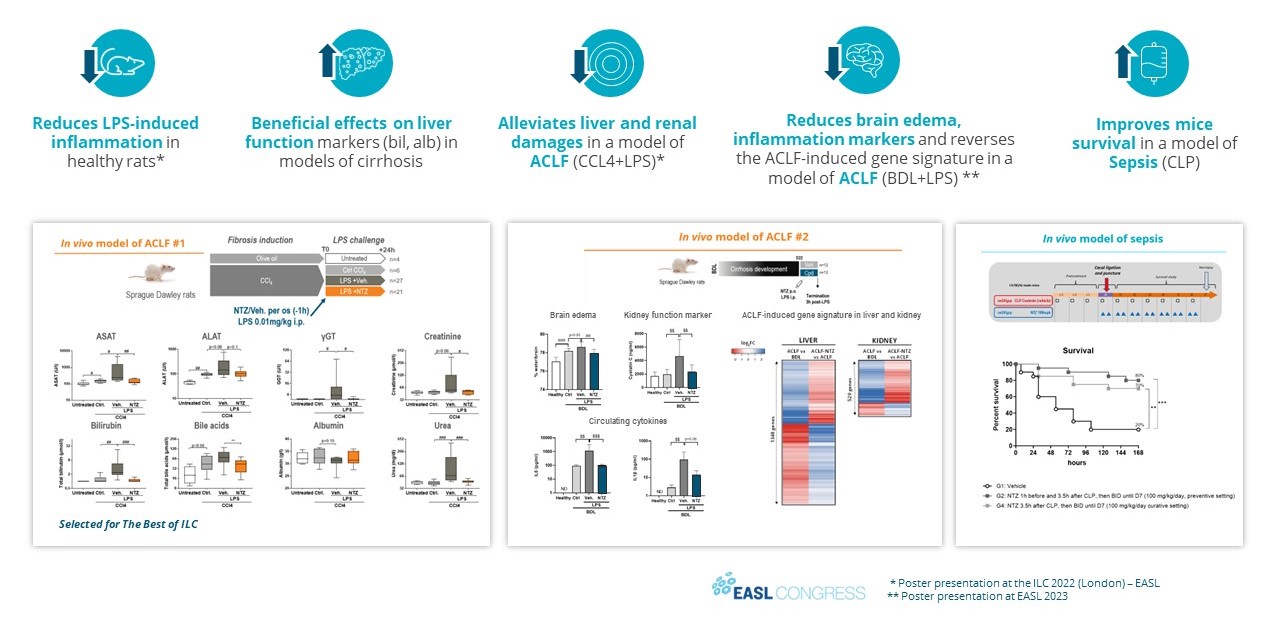
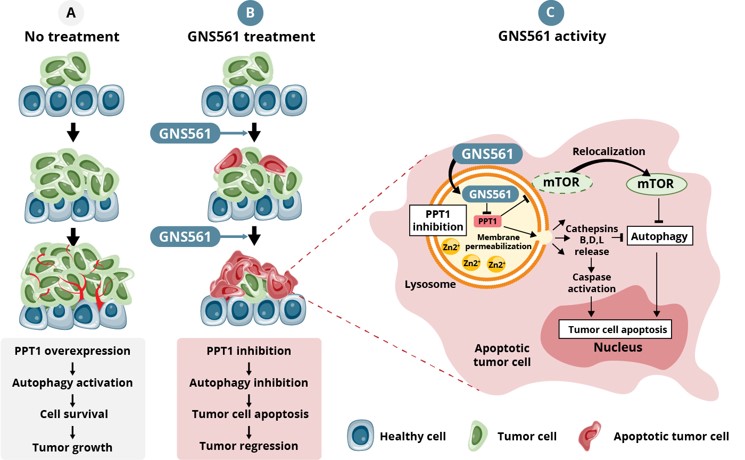
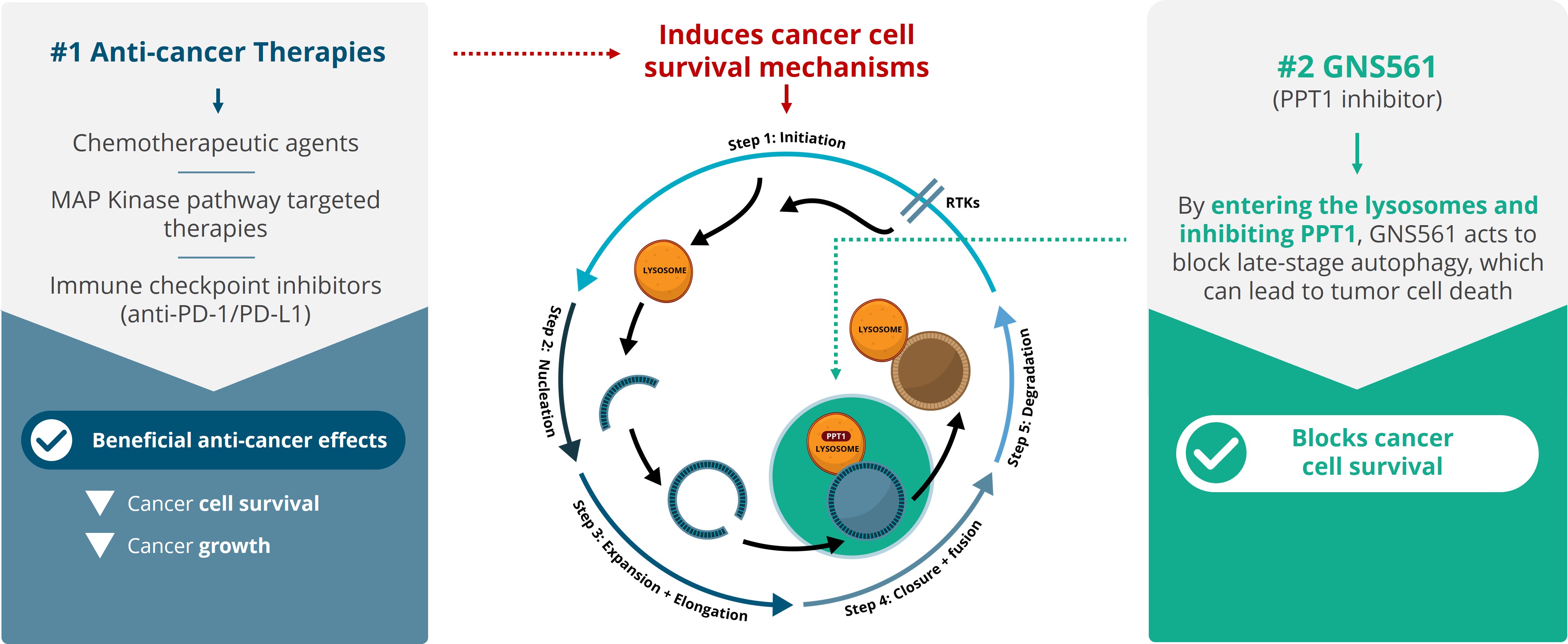
For example, in the second half of 2023, our drug candidates VS-01 in ACLF and GNS561 in CCA have just entered Phase 2 and Phase 1b/2 respectively, in order to provide clinical proof-of-concept.
Our other drug candidates are at an even higher level of risk than in other indications.
As a result, it is possible that the ELATIVEearlier stage, since they have either obtained initial Phase 1 clinical trial results (NTZ), or other clinical trials of elafibranorhave never been administered in other indications,humans (SRT-015, VS-01 in UCD/OAs, VS-02 in HE and our other ongoing or future clinical trialsCLM-022), in general, fail to meet their primary endpoints, as was the case with our Phase 3 RESOLVE-IT trial evaluating elafibranortherapeutic areas in non-alcoholic steatohepatitis or NASH in 2020, orwhich we are delayed, additional development is necessary or, despite a favorable outcome in clinical trials, the regulatory authorities consider that the clinical results of these trials are insufficient to grant or maintain a marketing authorization. These different risks are further described below.developing them.
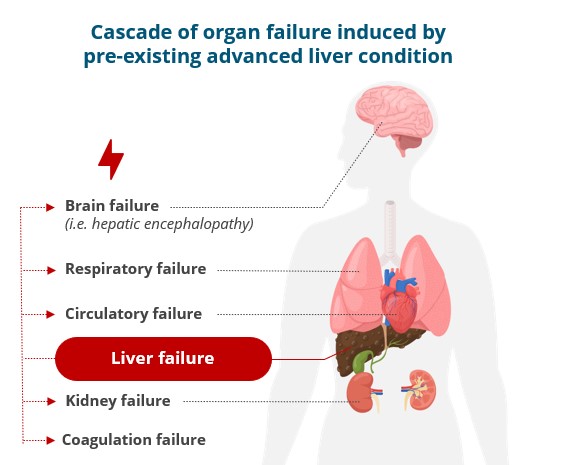
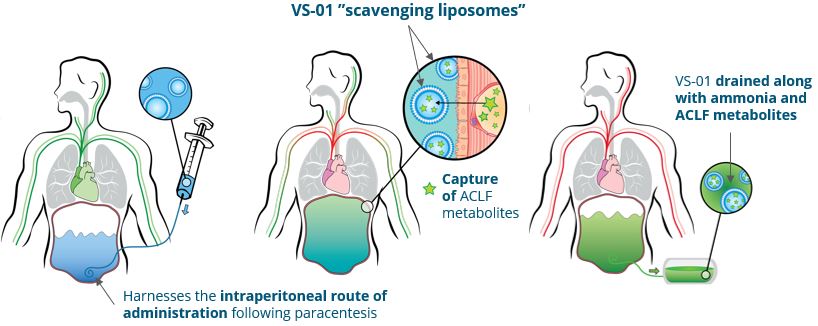
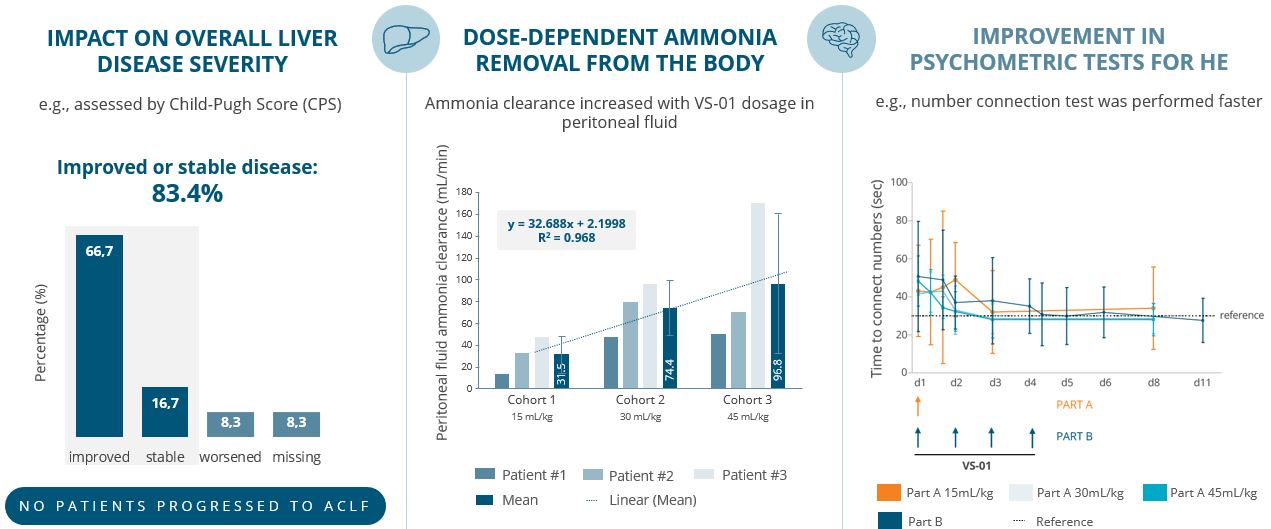
Our other development programsMany of these drug candidates are atbeing developed to treat ACLF (VS-01, NTZ, SRT-015, CLM-022), a much earlier stage of development. Nitazoxanide or NTZ,condition for which is being repositioned in Acute-on Chronic Liver Failure or ACLF, is currently being evaluated inwe have little experience and for which no treatment has yet been approved. As a Phase 1 trialresult, we are more exposed to evaluate pharmacokineticsthe risks associated with the preclinical and safety in individuals with hepatic impairment, and another Phase 1 study is planned to be conducted in renal impairment. We have also just begunclinical development of our programdrug candidates than companies operating in cholangiocarcinoma or CCA with GNS561, licensed from Genoscience Pharma in December 2021. Clinical developmentbetter-explored therapeutic areas, while being, like them, still exposed to the risk of not being able to demonstrate that our drug candidates provide sufficient therapeutic benefit. Some of these product candidates faces similarare also intended to treat diseases for which we have limited experience with drug development, which creates further risks and challenges as our development of elafibranor in PBC.their development.
A clinical failure of elafibranor in PBC, a delay orFinally, the failurerecent addition to receive marketing authorization would therefore have a negative impact, even more so since it would impact our primary program and the most advanced in our portfolio of drug candidates. As a result,some of the programs we could be forced to discontinueare developing (GNS561, VS-01 and VS-02, SRT-015 and CLM-022) results either from the recent acquisition of licensing rights from other companies (Genoscience, Seal Rock Therapeutics and Celloram), or from our Group's acquisition of Versantis AG. Despite due diligence and evaluation procedures we have carried out on the quality of previous results obtained by these companies, the development in PBC, one of our mainthese programs which could significantly affectis riskier than if we had developed them ourselves from the future of our Group.outset.
ClinicalDevelopment failure can occur at any stage of preclinical or clinical development, as was the case with our Phase 3 RESOLVE-IT trial of elafibranor in NASH.development. The results of earlier preclinical studies or clinical trials are not necessarily predictive of future results for elafibranor in PBC or any otherof product candidate, including NTZ or GNS561,candidates that we or our collaborators advance through preclinical studies or clinical trials. Results fromWe may not have favorable results in later clinical trials, may not be favorable, which may delay, limit or prevent our ability to receive regulatory approval or marketing authorization.
ClinicalDevelopment failure can occur at any stage of our preclinical or clinical development or those of our current partner or a future partner. ClinicalPreclinical studies or clinical trials may produce negative or inconclusive results, and we or our collaborators may decide, or regulators may require us, to conduct additional clinical trials or preclinical studies. In addition, data obtained from trials and studies are susceptible to varying interpretations, including interim data, and regulators may not interpret our data as favorably as we or our collaborators do, which may delay, limit or prevent regulatory approval or marketing authorization.
Success in preclinical studies and early clinical trials, or positive interim clinical results, does not ensure that final clinical results or subsequent clinical trials will generate the same or similar results or otherwise provide adequate data to demonstrate the efficacy and safety of a product candidate. A number of companies in the pharmaceutical industry, including those with greater resources and experience than us or our current and potential future collaborators, have suffered significant setbacks in later-stage trials, including Phase 3 clinical trials and at other stages of preclinical and clinical development, for example in particular in NASH and PBCMASH, even after seeing promising results in earlier clinical trials.
For example, in May 2020, we published the topline results of the interim analysis of our Phase 3 RESOLVE-ITRESOLVE-IT® trial of elafibranor in NASH.Metabolic dysfunction associated steatohepatitis or MASH. Elafibranor did not demonstrate a statistically significant effect on the primary surrogate efficacy endpoint of NASHMASH resolution without worsening of fibrosis noror on the key secondary endpoints. These results led us to stop development of elafibranor in NASH in 2020MASH due to lack of efficacy but not due to safety reasons.
In addition, the design of a preclinical study or clinical trial can determine whether its results will support approval of a product and flaws in the design of a clinical trial may not become apparent until the clinical trial is well-advanced. We or our collaborators may be unable to design and execute a preclinical study or clinical trial to support regulatory approval. Further, clinical trials of potential products often reveal that it is not practical or feasible to continue development efforts. If elafibranor or our other drug candidates are found to be unsafe or lack efficacy for any indication, we or our collaborators will not be able to obtain regulatory approval for them, and our prospects and business may be materially and adversely affected. For example, if the results of our Phase 3 ELATIVE trial of elafibranor in PBC does not achieve the primary efficacy endpoints or demonstrate an acceptable safety profile, the prospects for approval of elafibranor in PBC would be materially and adversely affected.
In some instances, there can be significant variability in safety and/or efficacy results between different trials of the same product candidate due to numerous factors, including changes or differences in trial protocols, patient distribution by clinical investigator site, standards of care across sites, differences in composition of the patient populations, adherence to the dosing regimen and other trial protocols and the rate of dropout among clinical trial participants. We do not know whether any Phase 2, Phase 3 or otherSuch instances undermine the readability and acceptability of the results, both for the clinical trials we or any of our collaborators may conduct will demonstrate consistent or adequate efficacytrial sponsor and safety to obtain regulatory approval to market our product candidates. If we or our collaborators are unable to bring any of our current or future product candidates to market, or to acquire any marketed, previously approved products,authorities, and our ability to create long-term shareholder value, will be limited.and could lead to halting the development of the product candidate.
Delays in the commencement enrollment and completion of preclinical studies and clinical trials, and in enrollment of patients for clinical trials, including our Phase 3 ELATIVE trial of elafibranor in PBC,ongoing clinical trials, could result in increased costs to us and delay or limit our ability and that of Terns Pharmaceuticals or Ipsen, our partners for elafibranor and that of any future collaborators, to obtain regulatory approval for elafibranor and our other drug candidates. Such delays and costs could impair our financing capacity, and these events may limit or compromise our ability to continue development and to eventually commercialize our drug candidates.
Preclinical and clinical development of a drug candidate is a long, costly and uncertain process, aimed at demonstrating the therapeutic benefit of a drug candidate that competes with existing products and standards of care or those currently conducting our Phase 3 ELATIVE trialunder development.
At the preclinical stage, we may not be able to generate and complete the preclinical, toxicological, in vivo or in vitro data needed to support the launch of elafibranor in PBC forclinical trials with regulatory authorities, or such data may be obtained later than anticipated, which the first patient was enrolled in September 2020 and is currently enrolling patients as of the date of this annual report. Delays in the commencement, enrollment and completion of our clinical trials or those of our partners Terns Pharmaceuticals or Ipsen or any future collaboratorlatter case could increase our product development costs, ordelay the subsequent phase of clinical development, and potentially limit our ability to obtain regulatory approval of our drug candidates. In the past, we have experienced some delays in enrollment in our clinical trials, including in our RESOLVE-IT clinical trial in NASH. We have also experienced, and may continue to experience delays and challenges in enrollment in clinical trials due to the ongoing COVID-19 pandemic.
The results from these trials may not be available when we expect or we or our collaborators may be required to conduct additional clinical trials or preclinical studies not currently planned in order to receive approval for our product candidates, including elafibranor.elafibranor in PBC. In addition, our clinical programs and those of our partners Ipsen and Terns Pharmaceuticals are subject to a number of variables and contingencies, such as the results of other trials, patient enrollments or regulatory interactions that may result in a change in timing. As such, we do not know whether any future trials or studies in elafibranor or our other product candidates will begin on time or will be completed on schedule, if at all.contingencies.
The commencement, enrollment and completion of clinical trials can be delayed or suspended for a variety of reasons, including:
•inability to demonstrate sufficient safety and efficacy to obtain regulatory approval to commence a clinical trial;
•inability to validate test methods to support quality testing of the drug substance and drug product;
•inability to determine dosing and clinical trial design;
•inability to obtain sufficient funds required for a clinical trial or lack of adequate funding to continue the clinical trial due to unforeseen costs or other business decisions;decisions of the Group, or its current or future partners;
•our inability to enter into collaborations relating to the development and commercialization of our product candidates;
•inability to reach agreements on acceptable terms with prospective contract research organizations, or CROs, and trial sites and contract manufacturing organizations or CMOs, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs, trial sites and trial sites;CMOs;
•clinical holds, other regulatory objections to commencing or continuing a clinical trial or the inability to obtain regulatory approval to commence a clinical trial in countries that require such approvals;
•discussions with the FDA, European Medicines Agency or EMA, the competent authorities of European Economic Area, or EEA, countries or other non-U.S. regulators regarding the scope or design of our clinical trials, which may occur at various times, including subsequent to the initiation of the clinical trial;
•governmental or regulatory delays and changes in regulatory requirements, policy and guidelines, including mandated changes in the scope or design of clinical trials or requests for supplemental information with respect to clinical trial results;
•varying interpretations of our data, and regulatory commitments and requirements by the FDA, EMA, European Commission (EC) and similar foreign regulatory agencies;authorities;
•inability to identify and maintain a sufficient number of trial sites, many of which may already be engaged in other clinical trial programs, including some that may be for the same indications targeted by our product candidates;
•the delay in receiving results from or the failure to achieve the necessary results in other clinical trials;
•inability to obtain approval from institutional review boards, or IRBs, or positive opinions from Ethics Committees, to conduct a clinical trial at their respective sites;
•lack of effectiveness of product candidates during clinical trials;
•suspension or termination by a data and safety monitoring board, or DSMB, that is overseeing the clinical trial;
•changes in the standard of care on which a clinical development plan was based, which may require new or additional trials;
•failure to conduct clinical trials in accordance with regulatory requirements;
•severe or unexpected drug-related adverse effects experienced by patients, death of a patient during a trial or any determination that a clinical trial presents unacceptable health risks;
•a breach of the terms of any agreement with, or termination for any other reason by, current or future collaborators that have responsibility for the clinical development of any of our product candidates, or investigators leading clinical trials on our product candidates;
•inability to timely manufacture or deliver sufficient quantities of the product candidate, or other consumables required for preclinical studies or clinical trials;
•difficulty identifying, recruiting and enrolling patients to participate in clinical trials for a variety of reasons, including meeting the enrollment criteria for our trial, the rarity of the disease or condition (for example PBC, ACLF and CCA), the rarity of the characteristics of the population being studied (for example PBC(as is the case for the profile of patients enrolled in our Phase 1b/2 trial evaluating GNS561 and ACLF and CCA),our Phase 2 trial evaluating VS-01) the nature of the protocol, the risks ofor technological difficulties related to procedures that may be required as part of the trial (related to, for example, to the intravenous administration of some of our drug candidates such as a liver biopsy,VS-01 or SRT-015), the availability of effective treatments for the relevant disease and the eligibility criteria for the clinical trial, insufficient human resources or organizational difficulties within clinical investigation centers, and competition from other clinical trial programs for the same indications or with products with the same mechanism of action as our product candidates;
•global health pandemics such as COVID-19, armed conflicts, warnatural disasters or natural disasters;pandemics; and
•inability to retain enrolled patients after a clinical trial is underway.
For example, our RESOLVE-ITRESOLVE-IT® trial was a large and complex Phase 3 clinical trial in a disease without any approved therapies at the time and the diagnosis of which generally involves invasive procedures such as liver biopsies. These specificities led us to face significant competition for patient enrollment, and to delay the publication date of our topline interim analysis.
As we engageDelays in other largethe commencement, enrollment and complicated trials and trials in advanced disease populations, including our ongoing Phase 3 ELATIVE trial evaluating elafibranor in PBC, we may experience a number of complications that may negatively affect our plans or our development programs. The ELATIVE trial evaluating elafibranor in PBC in particular is made complex by the fact that it is an orphan disease with a small number of patients and the fact that one of our competitor’s product is the only one to have recently received market approval in this indication, and another Phase 3 trial in PBC is enrolling patients at the same time as ours which may compromise our ability to retain or recruit patients or complete the trial on time. Potential discussions with the FDA, the EMA or other regulatory authorities outside the United States or European Economic Area or EEA regarding the scope or designcompletion of our clinical trials may also happen at any time.
More broadly, changes in the treatment of PBC, such as the approval of a drug therapycould significantly increase our product development costs, which could impair our financing capacity or limit our ability to obtain regulatory approvals required for the treatmentcontinued development of PBC by one of our competitors, could result in difficulties retainingother drug candidates and future commercialization, or enrolling patients in our clinical trials and those of our current or future collaborators. Any difficulty retaining patients may delay or produce negative or inconclusive results from our clinical trials, and we or our collaborators may decide, or regulators may require us, to conduct additional clinical trials or preclinical studies. Any delay or compromises with respect to our clinical trials may have a material adverse effectimpact on our business or diminish our competitivefinancial position, relativecommercial prospects and ability to other biotechnology or pharmaceutical companies.generate revenues.
We cannot be certain that elafibranor or any of our other product candidates, even if they meet preclinical, clinical and regulatory requirements, will receive regulatory approval or certification, as applicable, and without regulatory approval or certification, we or our collaborators will not be able to market our product candidates.
We currently have no products approved for sale and we cannot guarantee that we or any of our current or future collaborators will ever have marketable products. Our business and financial situation, including future revenues and financing capacity, currently depends substantially on the successful development, regulatory approval and commercialization of elafibranor in PBC.PBC by our partner Ipsen, and to a lesser extent, on the clinical development and future commercialization by Terns Pharmaceuticals in Greater China. Our ability to generate near-term revenue related toderived from product sales will depend on the successful development andIpsen’s ability to obtain regulatory approval of elafibranor in the indications we and our collaborators are developing or will developPBC in the United States, the European UnionEEA and other countries. Our ability to generate substantial revenue is also dependent on the future of the development and marketing of an IVD test using our NIS4 technology.
The development of drug candidates and NIS4 technology and issues relating to their approval and marketing are subject to extensive regulation by the FDA in the United States, and EMA and European Commission (EU) in the EU and regulatory authorities in other countries, with regulations differing from country to country.as well as successful commercialization.
We or our current or future collaborators will not be permitted to market our drug candidates in the United States or the EEA until we receive approval of a New Drug Application, or NDA, from the FDA or a marketing authorization, or MA, from the EC (based on the positive opinion of the EMA), as applicable. The same is true for other countries, including the United Kingdom since Brexit. We have not submitted at this time any marketing applications for any of our product candidates and neither have Ipsen nor Terns Pharmaceuticals, our development partners for elafibranor, for its products. NDAs, marketing authorization applications or MAAs and MAs in other countries must include extensive preclinical and clinical data and supporting information to establish the drug candidate’s safety and effectiveness for each desired indication. These marketing applications must also include significant information regarding the chemistry, manufacturing and controls for the drug. Obtaining approval of a NDA, MA or other marketing authorization is a lengthy, expensive and uncertain process, and we may not be successful in obtaining approval.
We cannot predict whether our ongoing or planned future trials and studies will be successful or whether regulators will agree with our conclusions regarding the preclinical studies and clinical trials we have conducted to date, or for ongoing trials, with our interim results.
RegulatoryObtaining marketing authorization is therefore a long and costly process, with an uncertain outcome, and these applications may fail.
Even if a drug is approved (whether conditional approval or final approval),the FDA, EMA, or competent authorities in other countries outside ofmay limit the United States and EEA also have requirementsindications for which the drug can be marketed, require a comprehensive warning to appear on the drug's label, packaging and/or package insert, or make approval of drug candidates and diagnostics with which we and our collaborators must comply prior to marketing in those countries. Obtainingconditional on additional clinical trials or costly and/or time-consuming reports, or post-marketing studies. In some cases, authorization may be withdrawn after it has been granted. In some cases, regulatory approval or certification for any of our product candidates may be withdrawn.
Finally, obtaining regulatory approval or certification for marketing of a drug candidate or diagnostic in one country does not ensure that we will be able to obtain regulatory approval or certification in any other country. In addition, delays
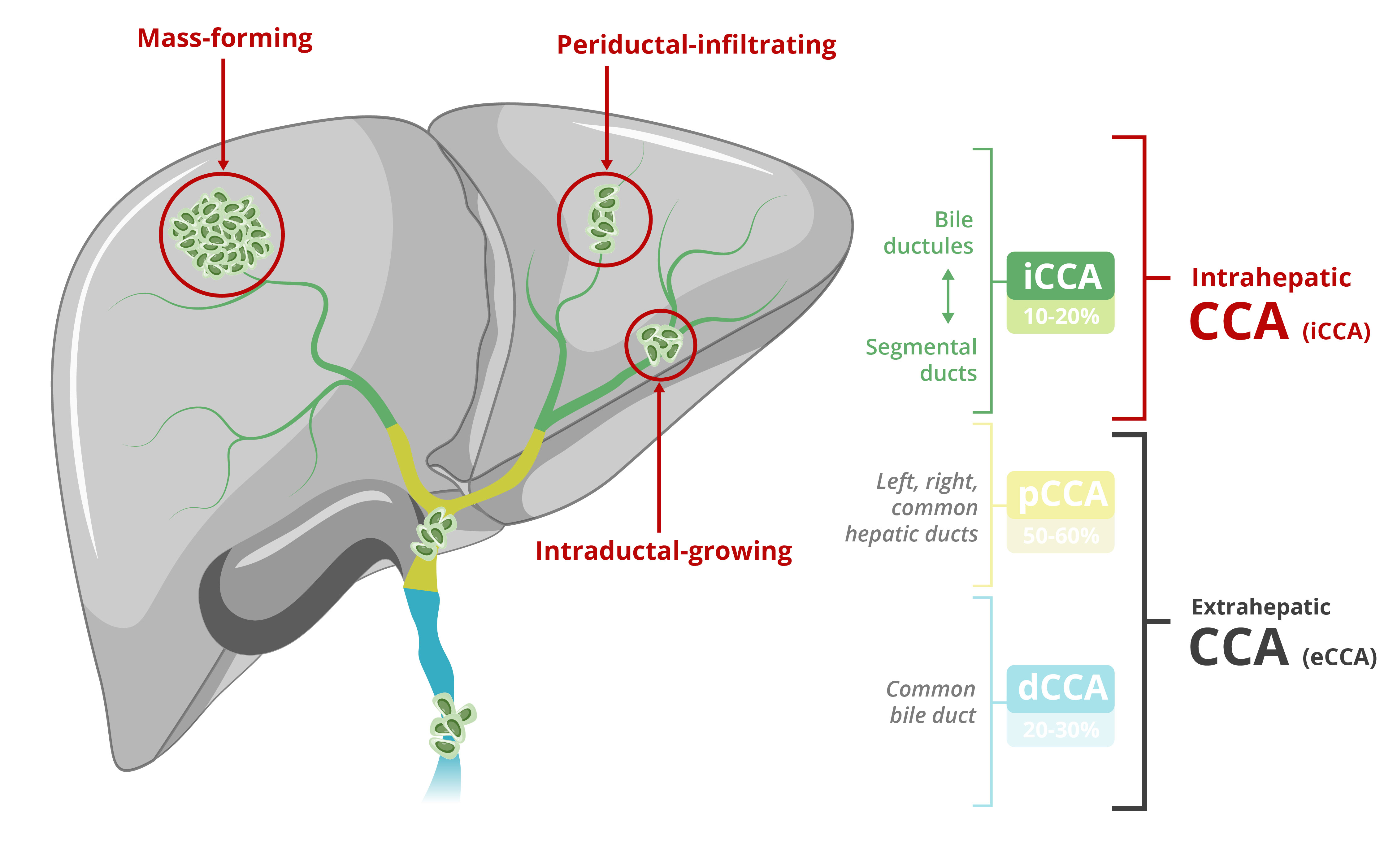
We are currently developing GNS561 in approvals or rejections of marketing applicationscombination with another treatment which is not proprietary to GENFIT, and may pursue other combination programs in the United States, EEAfuture, which present additional risks in comparison with single drug programs.
We are currently developing GNS561 in Cholangiocarcinoma in a Phase 1b/2 trial with trametinib, an MEK-targeting protein kinase inhibitor. We may also assess in the future, as part of some of our other current programs or future programs, the potential combinations of some of our drug candidates in combination with other treatments or other countries may be based upon many factors, including regulatory requests for additional analyses, reports, data, preclinical studies and clinical trials, regulatory questions regarding different interpretations of data and results, changes in regulatory policy during the period of product development and the emergence of new information regarding our product candidates or other products. Also, regulatory approval for any of our product candidatesdrug candidates.
Patients enrolled in this and future trials may be withdrawn.
If we, our collaborators Ipsen and Terns Pharmaceuticals or a future partner are unable to obtain approval from the FDA, the EC or other regulatory agencies for elafibranor, an IVD using NIS4 technology and our other product candidates, or if, subsequent to approval, we, our collaborators Ipsen or Terns Pharmaceuticals or a future partner are unable to successfully commercialize elafibranor, an IVD using NIS4 technology or our other product candidates, we will not be able to generate sufficient revenuetolerate these drug candidates in combination with other treatments. Even if any drug candidate in development were to become profitablereceive marketing approval or be marketed for use in combination with other existing treatments, we would still be exposed to continue our operations.
We have obtained breakthrough therapy designation fromthe risks that the FDA, EMA or other regulatory authorities may withdraw approval of the treatment used in combination with our drug candidate or that safety, efficacy, manufacturing or supply issues arise with such existing treatments. Combination treatments are commonly used for elafibranor in the treatment of PBCcancers and we would be exposed to similar risks if we developed another of our drug candidates for use in combination with other treatments for indications other than cancer. This could result in our own products, if approved, being taken off the market or being less commercially successful.
We may seekalso evaluate our current drug candidates or any other future drug candidates in combination with other treatments that have not yet been approved for marketing by the FDA, EMA or other regulatory authorities. We or potential current or future partners would not be able to avail ourselves of various designation mechanisms (such as orphancommercialize and sell these drug designation, Fast Track and breakthrough therapy designation) tocandidates if, in the end, these associated treatments do not obtain marketing approval.
To accelerate the development, approval or approvalfuture commercialization of some of our other drug candidates, including GNS561 in CCAwe, or our current or future collaborators, may seek to use certain regulatory pathways, but such mechanisms may not actually lead to a faster development or regulatory review or approval process, and it may not increase the likelihood that elafibranorour drug candidates will receive marketing approval for this indication.
In 2019, the FDA granted breakthrough therapy designation for elafibranor for the treatment of PBC. A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. For drugs that are designated as breakthrough therapies, interaction and communication between the FDA and the sponsor can help to identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens.
Designation as a breakthrough therapy is within the discretion of the FDA. Accordingly, even if we believe a drug candidate meets the criteria for designation as a breakthrough therapy, the FDA may disagree and instead determine not to make such designation. In any event, the receipt of a breakthrough therapy designation for a drug candidate may not result in a faster development process, review or approval compared to conventional FDA procedures and does not assure ultimate approval by the FDA.
In addition, even if one or more drug candidatecandidates qualifies as a breakthrough therapy, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened. We may also seek various other designation mechanisms (such as Fast Track designation from the FDA, or orphan drug designationdesignation) for our product candidates in the future, and even if granted, these designations may not lead to accelerated regulatory and approval, or approval at all.
For the development and eventual commercialization of elafibranor in PBC, we and our current partner in the territories concerned (Ipsen) may also be able to benefit from two other regulatory approval procedures. These are accelerated approval by the FDA and conditional marketing authorization by the EMEA.
The advantage of these procedures is that it is possible to obtain marketing authorization on the basis of surrogate endpoints (a marker, laboratory measurement, physical sign or other measure, which is thought to predict clinical benefit but which is not itself a measure of clinical benefit).
As is customary, the benefit of these procedures for the development and eventual marketing of elafibranor in PBC has been subject to our partner Ipsen's commitment to diligently conduct post-authorization studies to verify, describe and confirm the clinical benefit of the drug. Elafibranor, if approved in this context for the second-line treatment of PBC, would therefore be subject to strict compliance requirements after its eventual marketing, such as the performance of Phase 4 trials or post-authorization clinical trials by our partner Ipsen in order to confirm the effect on the clinical endpoint. In the absence of post-marketing studies or confirmation of clinical benefit by such post-marketing studies, the FDA and the EMA or regulatory authorities in other countries may initiate proceedings to withdraw approval of the drug in question. B
More generally, accelerated FDA approval is possible if the drug candidate (1) represents a treatment for a serious disease, (2) offers a real benefit compared to other existing therapies, and (3) demonstrates an effect on an endpoint that provides reasonable assurance of clinical benefit. Conditional marketing authorization by the EMEA is possible if (1) the benefit/risk ratio of the drug candidate is positive, (2) it is likely that the applicant will be able to provide the required comprehensive clinical trial data, (3) the drug candidate corresponds to an unmet medical need, and (4) the public health interest in the immediate availability of the drug candidate on the market outweighs the risks associated with the fact that additional data still need to be provided.
We are also studying the possibility of benefiting from the two regulatory approval procedures described above for the development of GNS561 in Cholangiocarcinoma and VS-01 in ACLF. In view of the significant unmet medical needs in these indications, the Orphan Drug Designation granted by the FDA for GNS561 and VS-01 could make these programs eligible for the various accelerated regulatory pathways proposed by the health authorities. However, the processes described above entail decisions which are at the discretion of the EMEA, the FDA or any other competent authority, and no guarantee can be given that they will be obtained.
Even though we have obtained orphan drug designation for elafibranor for the treatment of PBC in both the US and EU,EEA, we, or Ipsen, may not be able to obtain or maintain the benefits associated with orphan drug status, including market exclusivity. We have also received and may alsocontinue to seek the sameorphan drug designation for other drugof our product candidates, but we may not be able to obtain it or maintain the benefits associated.
Regulatory authorities in some jurisdictions, including the United States and the European Union,EEA, may designate drugs for relatively small patient populations as orphan drugs. Generally, if a drug with an orphan drug designation subsequently receives the first marketing approval for the indication for which it has such designation, the drug may be entitled to a period of marketing exclusivity, which precludes the FDA or the EC from approving another marketing application for the same drug for that time period.
Elafibranor received orphan drug designation for the treatment of PBC in both the US and the EEA in 2019. GNS561 also received orphan drug designation in the United States for the treatment of CCA, and VS-01 received orphan drug designation in both the USUnited States and the EUEEA for elafibranor for the treatment of PBCACLF and in 2019, and Ipsenthe United States for treatment of hyperammonemic crisis. We may request thealso seek orphan drug designation for elafibranor in another indication or for other drugfuture product candidates that we may develop in the EU and/or the United States.and indications.
However, we or our partners may not receive such designation for other drug candidates that we or our partners may develop in Europethe EEA and/or the United States or for any other drug candidate in any other jurisdiction, or for elafibranor, VS-01 or GNS561 in any other indication. Even if we or our partners successfully receive the orphan drug designation, the orphan drug designation does not necessarily guarantee market exclusivity on a given market. Even if we or our partners successfully obtain the exclusivity pertaining to the orphan drug designation for any of our drug candidates, this exclusivity may not protect the product efficiently as exclusivity may be suspended under certain circumstances. In the United States, even after a drug is granted orphan exclusivity and approved, the FDA can subsequently approve another drug for the same condition if the FDA concludes that the later drug is clinically superior in that it is shown to be safer, more effective or makes a major contribution to patient care. In the European Union,EEA, the exclusivity pertaining to the orphan drug designation will not prevent the marketing approval of a similar drug for the same condition if the later drug is shown to be safer, more effective or otherwise clinically superior to the first drug, or if the owner of the market approval of the first product does not have the capacity to deliver sufficient quantities of the product. In addition, if another orphan designated product receives marketing approval and exclusivity for the same condition as the one for which we or a future partner seek to develop a drug candidate, we or our partner may not be able to receive approval of our drug candidate by the relevant regulatory authorities for a significant period of time.
If the FDA does not conclude that certain of our product candidates satisfy the requirements for the Section 505(b)(2) regulatory approval pathway, or if the requirements for such product candidates under Section 505(b)(2) are not as we expect, the approval pathway for those product candidates may likely take significantly longer, cost significantly more and entail significantly greater complications and risks than anticipated, and in either case may not be successful.
We are currently conducting a clinical-stage program based on drug repositioning to develop the drug candidate NTZ for ACLF, for which we may seek FDA approval through the Section 505(b)(2) regulatory pathway. The Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act, added Section 505(b)(2) to the FDCA. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from trials that were not conducted by or for the applicant and for which the applicant has not obtained a right of reference. Section 505(b)(2), if applicable to us under the FDCA, would allow an NDA we submit to the FDA to rely in part on data in the public domain or the FDA’s prior conclusions regarding the safety and effectiveness of approved compounds, which could expedite the development program for our product candidates by potentially decreasing the amount of clinical data that we would need to generate in order to obtain FDA approval. NTZ is approved in another indication in the United States, and a previously-conducted Phase 2 investigator-initiated clinical trial of NTZ in NASH-inducedMASH-induced fibrosis was allowed based on the existing FDA evaluations of safety in the currently-approved indication, which is a hallmark of the Section 505(b)(2) regulatory pathway. As we progress the NTZ clinical program in ACLF, we plan to initiate such discussions with the FDA. If the FDA does not allow us to pursue the Section 505(b)(2) regulatory pathway as we anticipated, we may need to conduct additional clinical trials, provide additional data and information and meet additional standards for regulatory approval. Even if we are allowed to pursue the Section 505(b)(2) regulatory pathway, we cannot assure you that our product candidates will receive the requisite approvals for commercialization.
In addition, the pharmaceutical industry is highly competitive, and Section 505(b)(2) NDAs are subject to special requirements designed to protect the patent rights of sponsors of previously approved drugs that are referenced in a Section 505(b)(2) NDA. These requirements may give rise to patent litigation and mandatory delays in approval of our NDAs for up to 30 months or longer depending on the outcome of any litigation. It is not uncommon for a manufacturer of an approved product to file a citizen petition with the FDA seeking to delay approval of, or impose additional approval requirements for, pending competing products. If successful, such petitions can significantly delay, or even prevent, the approval of the new product. However, even if the FDA ultimately denies such a petition, the FDA may substantially delay approval while it considers and responds to the petition. In addition, even if we or a future partner are able to utilize the Section 505(b)(2) regulatory pathway, there is no guarantee this would ultimately lead to accelerated product development or earlier approval.
Moreover, even if our product candidates are approved under Section 505(b)(2), the approval may be subject to limitations on the indicated uses for which the products may be marketed or to other conditions of approval, or may contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the products.
The EEA and third countries have equivalent laws and obligations that could equally impact the approval of our product candidates.
Our near and medium-term future capital resources depend in large part on the success of developmentregulatory approval of elafibranor in PBC. Because our access to alternative financing is limited, failure in PBC could impact our strategic decisions with respect to the development of our other product candidates and may affect the development or timing of our business prospects.
Our near and mid-term future capital resources depend in large part on the success of developmentfuture potential regulatory approval of elafibranor in PBC.PBC in the territories covered by our licensing agreement with our partner Ipsen, the confirmation of its therapeutic benefit after this eventual marketing and the success of its eventual commercialization in this indication and in these territories. Because we have limited access to capital to fund our operations, failurea delay or the refusal of the PBC programmarketing authorization, unsuccessful post-marketing studies or limited commercial success in this indication could significantly negatively affect our resources available to allocate to research, collaboration, management and financial resources toward particular compounds, programs, product candidates or therapeutic areas. We may be restricted in the opportunities we can pursue, and we may be required to collaborate with third parties to advance a particular product candidate at terms that are less than optimal to us.
Because of our limited resources, we may also have to decline to pursue opportunities that may otherwise prove to be profitable. Furthermore, any failure (or in some cases delay) in the successful development of elafibranor in PBC would result in the non-payment of milestones and/or lower royalties negotiated under our partnership agreement with Ipsen. To a lesser extent, development failure of elafibranor in Greater China through Terns Pharmaceuticals could result in similar outcomes.
Our product candidates may have undesirable side effects which may require us to stop their development, including a clinical trial or which may delay or prevent marketing approval, or, if approval is received, require our product candidates to be taken off the market, require them to include safety warnings or otherwise limit their sales.
Unforeseen side effects from any of our product candidates could arise either during clinical development, forcing us to potentially stop or terminate preclinical development or a clinical trial, or, if approved or CE marked, after the approved or CE marked product has been marketed. If severe side effects were to occur, or if elafibranor or one of our other product candidates is shown to have other unexpected characteristics, we or our current or future collaborators may need to either restrict our use of such product to a smaller population or abandon our or their development.
In addition, our product candidates are being developed as potential treatments for severe, life-threatening diseases and, as a result, our trials will necessarily be conducted in a patient population that will be more prone than the general population to exhibit certain disease states or adverse events. For example,Patients with PBC, patientsACLF or CCA may suffer from other co-morbidities such as osteoporosis that may increase the likelihood of certain adverse events. It may be difficult to discern whether certain events or symptoms observed during our trials were due to our product candidates or some other factor, resulting in our company and our development programs being negatively affected even if such events or symptoms are ultimately determined to be unlikely related to our drug candidates. We cannot assure youensure that additional or more severe adverse side effects with respect to elafibranor, NTZ, GNS561, VS-01 or any other drug candidate will not develop in current or future preclinical studies or clinical trials or commercial use, which could delay or preclude their regulatory approval, limit their commercial use or require them to be taken off the market. However, DSMBs are set up in our main clinical trials to evaluate side effects observed during our studies at regular intervals defined in our study protocols, and to issue recommendations concerning their continuation or the conditions for their continuation, although they may not be effective.
If we or others later identify undesirable or unacceptable side effects caused by our products or product candidates:
•regulatory authorities may require the addition of labeling statements, specific warnings, a contraindication or field alerts to physicians and pharmacies;
•we or current or future collaborators may be required to change instructions regarding the way the product is administered, conduct additional clinical trials or change the labeling of the product;
•we may be subject to limitations on how we may promote the product;
•sales of the product may decrease significantly;
•regulatory authorities may require us or current or future collaborator(s) to take our approved or CE marked product off the market;
•we or current or future collaborators may be subject to litigation or product liability claims; and
•our reputation or that of our current or future collaborators may suffer.
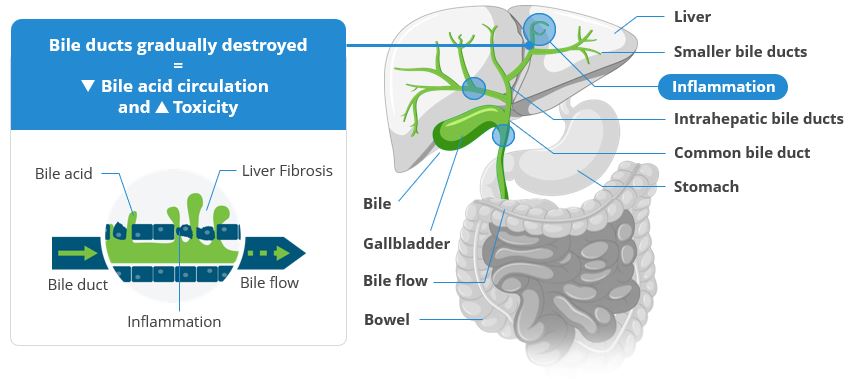
Risks Related to the Discovery and Development of, and Obtaining Regulatory Approval or CE Certificates of Conformity for, our Diagnostic TestTechnology
The development of our NIS4NIS4® technology and its variations and improvements, including NIS2+®, and tests powered by this technology requires access to clinical trials, data and clinical samples in NASHMASH patients and therefore our development is also subject to the risks related to these trials.
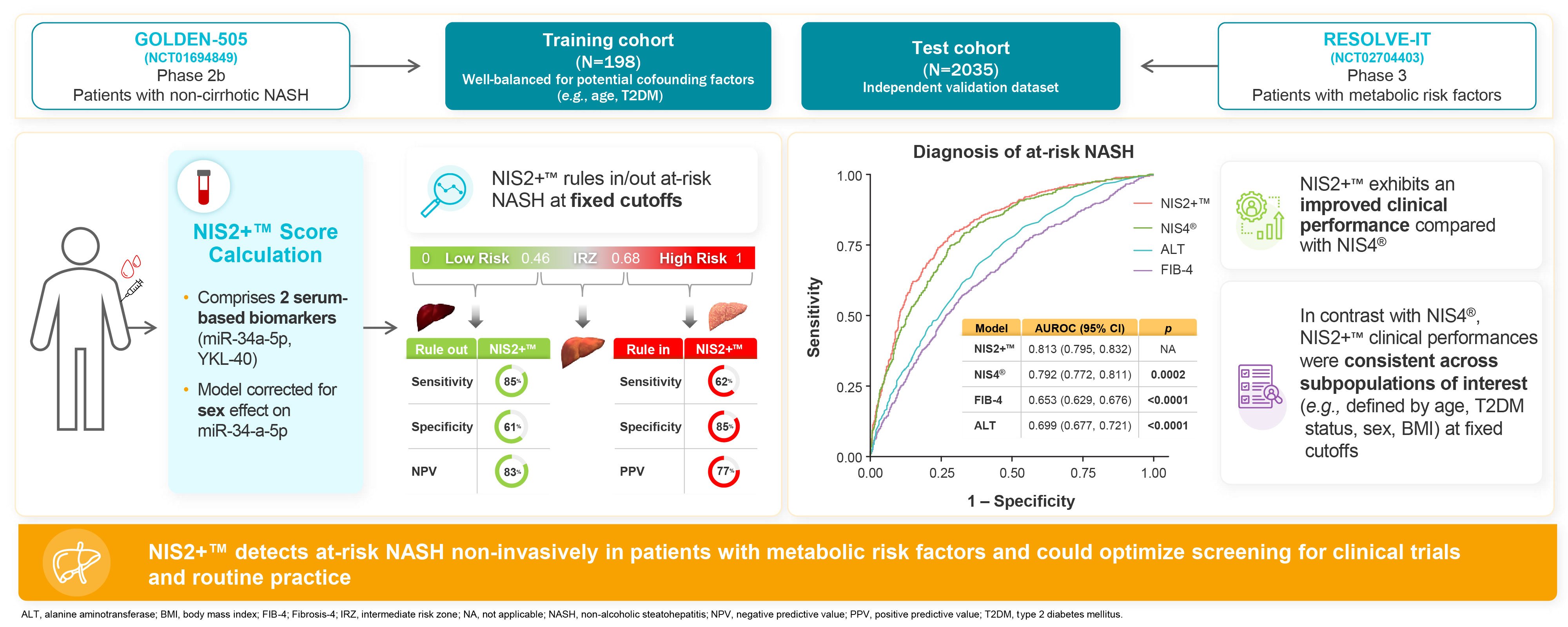
In support of the development of our drug candidates, we conduct research and development programs to identify new, innovative diagnostic strategies, in particular to determine the population of patients to be treated. We initially developed NIS4® diagnostic technology and have sought to continually make improvements, with the primary objective of making it easier to identify patients with MASH who are eligible for therapeutic intervention. Our NIS2+® technology is one of the improvements on NIS4® and carries with it the same objective.
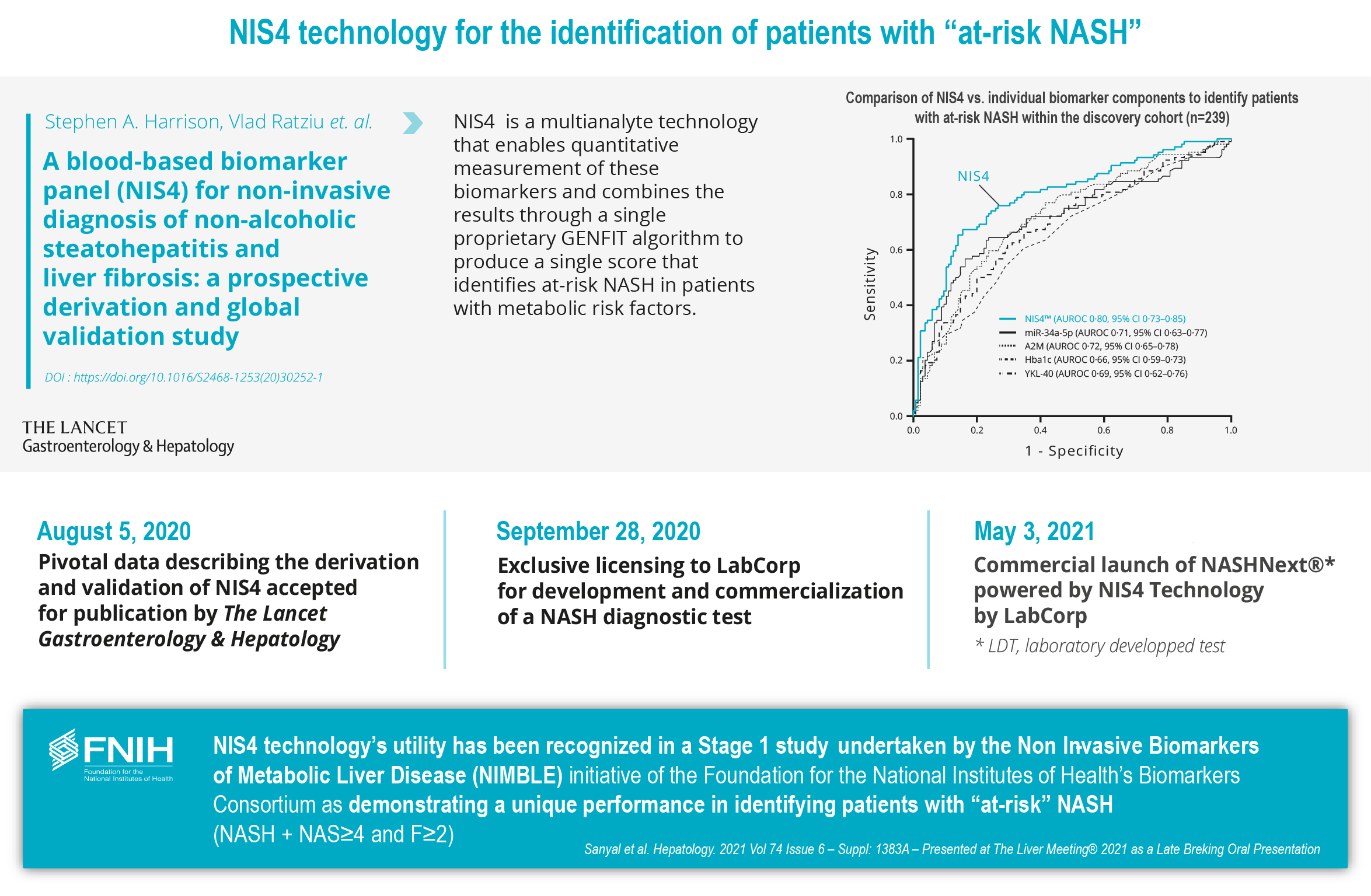
In January 2019, we entered into a license agreement withToday, NIS4® technology is out-licensed to Labcorp and Q Squared Solutions LLC or Q2 to allow them to develop and deploy a test powered by NIS4NIS4® technology in the clinical research space. We believe that leveraging the capabilities of a large diagnostic company such as Labcorp, through its Covance laboratory network, will allow for early adoption of NIS4 technology and result in third party publications. In SeptemberSince 2020, we entered into a five-year exclusive licensing agreement for NIS4have also out-licensed to Labcorp the rights to develop NIS4® technology with Labcorp. As part of the agreement, Labcorp will developas an LDT and commercialize a blood-based molecular diagnostic test powered by NIS4 technology throughout the U.S. and Canada enabling widespread access to healthcare providers. In order to reinforce use of our NIS4 technology, we also entered into an agreement in May 2021 with Q Squared Solutions LLC or Q2, to broaden the availability of the NIS4 technology in the clinical research field. In April 2021, Labcorp launched NASHnext,NASHNext®, an LDT powered by NIS4NIS4® technology to provide broad clinical availability of the test to specialty and primary care physicians across the U.S. and Canada and to identify patients with significant fibrosis or at-risk of NASH. Labcorp is leveragingMASH.
Further development of our NIS4® technology and its deep experience in commercializing innovative diagnostics to educate providers on NASH and the importance of non-invasive testing. We believe this agreement will enable broader test availability to support evidence generation, demonstration of clinical utility, and favorable market access of the test powered by NIS4. We intend to benefit from these advantages to support the next step of the development, clearance, and commercialization of an in vitro diagnostic or IVD powered by NIS4 to enable even broader availability of the clinical diagnostic outside of the central lab setting.
Development ofimprovements as an IVD will nevertheless require us or our future partners to keep gathering clinical data within the framework of trials or observational studies in which NIS4NIS4® is currently being evaluated or within the framework of potential additional clinical trials or observational studies to come.
In these trials or observational studies, we will continue to use human samples. Even though we have preferred access to the samples collected during the clinical development of elafibranor in NASH,MASH, we may be unable to access a sufficient quantity of samples or samples of a sufficient quality or usability, in which case the continuation of the development of NIS4NIS4® and its improvements could be slowed down or even interrupted. In order to have access to samples, we may be required to enter into partnership agreement with hospitals or other third parties, and we may not be able to enter into these agreements under satisfactory conditions or within the desired timeframes, if at all.
The strength of NIS4NIS4® technology initially identified on a relatively limited number of samples could turn out to not be sufficient during potential future validation studies on larger target populations, and notably not display sufficient levels of accuracy, sensitivity or specificity in order to allow for the development of a competitive test for clinical care that would be adopted by the medical community.
Despite the care applied to the development of NIS4 technology, we could discover, after the development phase, inherent defects in the product or technology that were undetectable or inconspicuous defects based on the existing technical and scientific knowledge during the development. A failure may occur at any time during one of these clinical developments. The results of earlier clinical trials or studies does not allow predicting future results and NIS4NIS4® technology may not obtain favorable results in ongoing or future clinical studies. Results for additional clinical trials may not validate earlier positive results from other trials, which could call into question NIS4NIS4® technology's utility and medico-economic benefit. It is possible, in particular, that an LDT or IVD powered by NIS4, at
Developing the timefull medical and commercial potential of NIS4® and its launch on the market for clinical care, will not replace the currentderivatives, and of diagnostic tests and medical examinations. In that case, the place of a test powered by NIS4, initially or as a complement or substitute of certain examinations would have to be assessed through additional clinical studies that would allow evaluating its medico-economic benefit often required to obtain reimbursement. The results ofusing these studies may not support the use of a test using NIS4 technology within the standard of care in a way that meets the needs of clinical practitioners or demonstrates a favorable economic outcome. With such results, a test powered by NIS4 may not obtain reimbursement, especially in European countries, which could materially affect product sales.
Moreover, the data gathered during these trials and studies aretechnologies, remains subject to different interpretations, andthe risks associated with diagnostic product development, requires regulatory authorities may not interpret our data as favorably as us or our collaborators,approval which may delay, limit or prevent the regulatory authorization for the use of an IVD powered by NIS4 as a diagnostic tool for clinical care. In addition, the design of these trials may determine if their results can support the application for marketing approval and procedural defects of a trial may not be visible beforeobtained, and the trial reaches an advanced stage. We or our collaborators may not be able to design and conduct a clinical trial sufficient to support a regulatory market approval of an IVD powered by NIS4drugs to treat for clinical care, which may have a significant unfavorable impact on our prospects and activities.
Changes in regulatory requirements or guidelines issued by the regulatory authorities, or unforeseen events occurring during these trials may force us or our collaborators to alter the protocol or impose new requirements within the framework of these trials or studies, which may result in higher costs and delays in the development schedule of NIS4 technology. If delays occurred in the completion of these clinical trials, or if they were terminated, or if additional clinical trials or studies were required besides the planned ones, this would impact the commercial prospects of an IVD powered by NIS4 and our ability to generate direct or indirect commercial revenue from this product would be delayed.
We intend to develop and market an IVD powered by NIS4 technology as a clinical diagnostic and as such, NIS4 remains a product in development subject to the hazards of diagnostic product development. In addition, there is no assurance that we will be able to receive the necessary regulatory approvals to market an IVD, powered by NIS4 technology or achieve commercialization of this product candidate for our intended market.
In order to reach the largest number of NASHMASH patients possible, we intendor our future partners need to develop an IVD powered by NIS4NIS4® technology or its improvements to identify patients with NASHMASH and fibrosis who may be eligible for therapeutic interventions in a field where no NASH-specific non-invasive test has been approved nor commercialized for clinical care to date and for which clinical experience is currently limited. Our development approach relies therefore on new methodologies. It is thus possible that, in this context, our clinical trials do not meet a favorable outcome or that, despite a favorable outcome, regulatory authorities determine that the results of our clinical trials or those of our collaborators are insufficient to grant market approval for an IVD test using the NIS4 technology for clinical care.intervention.
In order to be allowed to directly market and sell an IVD powered by NIS4NIS4® or its improvements in the European Union and/or Norway, Iceland or Liechtenstein (collectively EEA) andEEA, IVD manufacturers must demonstrate compliance of their products through a conformity assessment procedure, which, depending on the risk classification of the product, may involve a Notified Body. The Notified Body issues a CE Certificate of Conformity following successful completion of a conformity assessment procedure. The successful completion of the conformity assessment procedure is a prerequisite to being able to affix the CE mark to products, allowing manufacturers to market IVDs in the EEA. In the United States, the product must achieve CE marking from an accredited Notified Body in Europe and FDA approval/clearance in the United States.clearance. Other relevant regulatory requirements must be met to market in other countries.
In the United States, IVD tests are regulated as medical devices. Therefore, to be commercially distributed for clinical care, an IVD diagnostic product must demonstrate, depending on its regulatory classification, either its safety and efficiency through a pre-market approval, or its substantial equivalence to a previously FDA-approved medical device through clearance of a 501(k) premarket notification. This regulatory classification may not be obtained. A clinical trial is almost always required to support a pre-market approval or PMA application and is sometimes required for 510(k) clearance. All clinical studies of medical devices must be conducted in compliance with any applicable FDA and Institutional Review Board requirements.
Alternatively, the product may be marketed as an LDT, which does not require FDA approval, but requires the laboratory conducting the test to have been certified under the Clinical Laboratory Improvement Amendments of 1988 Act or CLIA and certain state laboratory licenses. Both testing services by Labcorp and Covance are currently conducted within the framework of CLIA, which establishes quality standards that must be followed in laboratory testing in order to ensure accuracy, reliability and speed of patient test results wherever the test is conducted. This law has instated an accreditation program for clinical laboratories, which Labcorp and Covance have received.
We currently do not have any IVD approved, cleared or clearedCE marked test that has been approved for marketing through such a regulatory process and we cannot guarantee that we or potential collaborators will ever develop marketable IVD tests. We have not submitted any marketing applications for any IVD test with the FDA, nor submitted any application for certification with any Notified Body in the EEA, and, in particular, we have not submitted any marketing application for NIS4.NIS4®.
As with approval of our drug candidates, the process for obtaining marketing authorization of diagnostic candidatesThe NIS4® technology and its improvements have been developed in a field where no MASH-specific non-invasive test has been approved or CE marked nor commercialized for clinical care to date, and in an area where clinical experience is lengthy, uncertain and expensive. Incurrently limited. Our development approach relies therefore on new methodologies. It is thus possible that, in this context, our diagnostic development does not meet a favorable outcome or that, despite a favorable outcome, regulatory authorities determine that the United States, IVD testsresults of our clinical trials or those of our collaborators are regulated as medical devices. The Federal Food, Drug, and Cosmetic Act,insufficient to grant market approval or the FDCA, and its implementing regulations, and other federal and state statutes and regulations govern, among other things, medical device design and development, preclinical and clinical testing, premarket clearance or approval, registration and listing, manufacturing, labelling, storage, advertising and promotion, sales and distribution, export and import, and post-market surveillance.
Concurrently with evaluating the FDA approval processCE Certificates of Conformity for ouran IVD test we are collecting data to obtain CE Certificateusing the NIS4® technology for clinical care of Conformity and to affix the CE mark to the IVD test in the key EEA markets subsequent market authorization in the key European markets. Like the U.S. approval process, the conformity assessment process preceding the CE Certificate of Conformity and CE marking process in the EEA may be lengthy and expensive, and the exact date of a CE Certificate of Conformity, if achieved at all, remains hard to predict.MASH patients.
Each regulatory authority may indeed refuse to issue approval or certification, impose its own conditions to such issuance, or require additional data prior to issuance, even when such approval or certification would have been already granted by regulatory authorities in other jurisdictions. Regulatory authorities may also modify their approval or certification policies, particularly by adding new or additional conditions to grant approval.approval or certification. As an example, Regulation (EU) 2017/746 (IVDR) will entergoverning IVDs in the EEA entered into application on May 26, 2022. The Regulation will introduce a new classification procedure2022 includes stricter requirements for an IVD medical devicesmanufacturers of IVDs to obtain the CE Certificate of Conformity and amplify the requirements which must be fulfilled by IVD manufacturers before they can CE mark and market theircommercialize IVDs in the EEA. We are also required to provide clinical data in the form of a performance evaluation report as part of the conformity assessment process prior to CE marking and in post marketing clinical follow-up activities. Fulfillment of the obligations imposed by the IVDR may cause us to incur substantial costs. We may be unable to fulfil these obligations, or our Notified Body, where applicable, may consider that we have not adequately demonstrated compliance with our related obligations to merit a CE Certificate of Conformity on the basis of the IVDR.
We or our potential collaborators may be subject to delays in obtaining the CE Certificate of Conformity required to affix the CE Mark to our IVD and market a test using NIS4NIS4® or its improvements for clinical care, or even not be successful in receiving approval,certification, due to the entry into force of new IVD medical device regulationsthe IVDR in Europe.the EEA. Such delay or failure may have an unfavorable impact on our ability to market a test using NIS4NIS4® technology or its improvements and our ability to generate direct or indirect revenue from this activity.
Once these authorizations have been obtained, the deployment of the IVD test will also depend to a large extent on the approval of treatment solutions for MASH, such as the recent approval of Madrigal Therapeutic’s product Rezdiffra.
Even after regulatory approval or CE Certificates of Conformity have been granted or declarations of commercialization have been filed with regulatory authorities, IVD tests remains subject to materiovigilance and market-surveillance obligations concerning incidents and risks of incidents related to their use. Even though such incidents may occur and lead regulatory authorities to suspend, vary or even revoke the market authorization or CE Certificates of Conformity of such products. Regulatory authorities may also conclude that procedures put in place by us or our collaborators are insufficient in order to identify and handle incidents, and could suspend commercialization of the products until these procedures are considered sufficient.
It is possible, in particular, that an LDT or IVD powered by NIS4® or its variations, at the time of its launch on the market for clinical care, will not replace the current tests and medical examinations. In that case, the place of a test powered by NIS4® or its variations, initially or as a complement or substitute of certain examinations would have to be assessed through additional clinical studies that would allow evaluating its medico-economic benefit often required to obtain reimbursement. The results of these studies may not support the use of a test using NIS4® technology within the standard of care in a way that meets the needs of clinical practitioners or demonstrates a favorable economic outcome. With such results, a test powered by NIS4® or its variations may not obtain reimbursement, especially in European countries, which could materially affect product sales.Risks Related to the Commercialization of Our Drug Candidates and Diagnostic TestTechnology
Even if approved, our product candidates may find themselves at a competitive disadvantage or not achieve broad market acceptance among physicians, patients and healthcare payors, and as a result our revenues generated from their sales may be limited.
The commercial success of elafibranor as a potential treatment for PBC or in other indications, our other drug candidates or an LDT or IVD powered by NIS4NIS4® or our other drug candidates,its improvements, if approved or cleared, will depend upon their acceptance among the medical community, including physicians, healthcare payors and patients. Given that there are a limited number of products approved for the treatment of PBC, and no products approved for treatment of ACLF, we do not know the degree to which elafibranor or our other product candidates would be accepted as a therapy, if approved. Additionally, we cannot be assured that NASHnext,NASHNext®, or IVD powered by NIS4NIS4® or its improvements will be accepted by the medical community as a means of identifying patients with NASHMASH or fibrosis who may be appropriate candidates for therapeutic intervention, and even if an LDT or IVD powered by NIS4NIS4® or its improvements is used, a physician may still require additional testing (e.g. liver biopsy) to confirm diagnosis. diagnosis using a test based on our technologies. The competitive intensity represented by future treatments (such as seladelpar for the treatment of PBC, which at the date of this Annual Report has not been reviewed by the FDA) and future diagnostic solutions could very significantly influence this adoption.
The degree of market acceptance of elafibranor NASHnext or IVD powered by NIS4 and any of our other drug candidates, that mayor NASHNext® or IVD using our diagnostic technologies, if and when they would be approved will depend on a number of factors, including:
•changes in the standard of care or availability of alternative therapies at similar or lower costs (including generics) or with better reimbursement rates for the targeted indications for any of our product candidates, such as competitors’ product candidates that are in development for the treatment of PBC, or other cholestatic diseases like ACLF or CCA, or an alternative to liver biopsy for the diagnosis of NASHMASH and fibrosis;
•limitations in the approved clinical indications or patient populations for our product candidates;
•demonstrated clinical safety and efficacy compared to other products;
•limitations or warnings, including boxed warnings, contained in our drug candidates’ FDA- or EC-approved labeling, if and when approved;
•in the case of elafibranor, our ability and that of our partners, Ipsen and Terns Pharmaceuticals or of a potential future collaborator to access the PBC market or in other future indications;
•for an LDT powered by NIS4, the ability of our partner, Labcorp or of a potential future collaborator to access the clinical research or clinical diagnostic market;
•for an IVD powered by NIS4, our ability to develop, obtain regulatory approval and commercialize an IVD test for clinical care;
•lack of significant adverse side effects;
•sales, marketing and distribution support;support for our products and those of our competitors;
•availability of coverage and adequate reimbursement from managed care plans and other third-party payors;
•timing of market introduction and perceived effectiveness of competitive products;
•the degree of cost-effectiveness;
•availability of alternative therapies or diagnostic solutions at similar or lower cost, including generics and over-the-counter products;
•the extent to which our product candidates are approved for inclusion on formularies of hospitals and managed care organizations;
•whether our drug or diagnostic candidates are designated under physician diagnostic and treatment guidelines for the treatment of the indications for which we, our partners Ipsen and Terns Pharmaceuticals or a potential future partner have received regulatory approval;
•adverse publicity about our product candidates or favorable publicity about competitive products;
•convenience and ease of administration of our product candidates; and
•potential product liability claims.
The following could also have a negative impact on sales:
•if they were subject to intellectual property rights held by third parties;
•if we or our current or future partners had no stock, or if we or our current or future partners were unable to have stock of our authorized products manufactured; and
•if we or our current or future partners fail to obtain regulatory approval for the manufacture of our products.
If our product candidates are approved, but do not achieve an adequate level of acceptance by physicians, patients, the medical community and healthcare payors, sufficient revenue may not be generated from these products and we may not become or remain profitable. In addition, efforts to educate the medical community and third-party payors on the benefits of our product candidates may require significant resources and may never be successful.
If we, or our current orand future collaborators are unable to establish sales, marketing and distribution capabilities for elafibranor or our other product candidates, we may not be successful in commercializing those product candidates if and when they are approved.
We have no sales, marketing or distribution experience and if we are unable to establish sales, marketing and distribution capabilities, we may not be successful in commercializing our product candidates if and when they are approved. To develop internal sales, distribution and marketing capabilities, we have already begunwould need to invest significant amounts of financial and management resources, and we may continue to do so, even prior to any confirmation that our product candidates will be approved. DevelopmentWorldwide development and commercialization rights for elafibranor, our most advanced drug candidate, are licensed exclusively to Ipsen in PBC and in all other indications, with the exception of rights licensed to Terns Pharmaceuticals for the treatmentdevelopment and commercialization of NASHelafibranor in MASH and PBC in mainland China, Hong Kong, Macau and Taiwan (Greater China) and to Ipsen in PBC and other indications in the rest of the world. Additionally, in connection with the development of NIS4 technology, we entered into a license agreement with Labcorp and Q2 to allow them to develop and deploy a test powered by NIS4 in the clinical research space. In September 2020, we entered into a five-year exclusive licensing agreement for NIS4 technology with Labcorp. As part of the agreement, Labcorp will develop and commercialize a blood-based molecular diagnostic test powered by NIS4 technology throughout the U.S. and Canada enabling widespread access to healthcare providers. We believe this agreement with Labcorp will provide broad clinical availability of a LDT powered by NIS4 technology to specialty and primary care physicians across the U.S. and Canada.Greater China. We are therefore heavily dependent on the sales, marketing and distribution capabilities of our partners, Terns,and Ipsen, and Labcorp.in particular.
If we decide to market any of our products ourselves, we would need to develop our own sales and marketing capabilities. For any product candidates where we decide to perform sales, marketing and distribution functions ourselves or through third parties, we could face a number of additional risks, including:
•we or our third-party sales collaborators may not be able to attract and build an effective marketing or sales force;
•our sales personnel may be unable to obtain access to physicians or persuade adequate numbers of physicians to prescribe any future products;
•the cost of securing or establishing a marketing or sales force may exceed the revenues generated by any products; and
•our direct sales and marketing efforts may not be successful.successful or less successful than those of our competitors.
If we are unable to establish our own sales, marketing and distribution capabilities and decide to enter into arrangements with third parties to perform these services for the products on the markets or indications that are not already subject to licensing agreements, our revenue and our profitability, if any, are likely to be lower than if we were to sell, market and distribute any products that we develop ourselves. Additionally, such collaboration agreements with current or potential collaborators may limit our control over the marketing of our products and expose us to a number of risks, including the risk that the partner will not prioritize the marketing of the product candidate or diagnostic test candidate or does not provide sufficient resources for its commercialization.
We have entered into, and may continue to seek and form, strategic alliances or enter into licensing or co-marketing arrangements to commercialize our approved drugs or diagnostic products, and we may not realize the benefits of such arrangements.
We may enter into licensing arrangements with third parties that we believe will complement or augment our commercialization efforts, particularly with respect to the diagnostic use of NIS4 for clinical care or our other drug candidates. For example, we have entered into an exclusive licensing and collaboration agreement with Ipsen to develop and commercialize elafibranor for the treatment of PBC and other indications worldwide, with the exceptions of Greater China which is licensed to Terns Pharmaceuticals. We have also entered into a license agreement with Labcorp to allow them to deploy an LDT powered by NIS4 in the clinical research and clinical diagnostics spaces. Any of these relationships may require us to incur costs, increase our near and long-term expenditures, issue securities that dilute our existing shareholders or disrupt our management and business. If we enter into any such arrangements with any third parties, we will likely have limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of elafibranor or any other product candidate. Our ability to generate revenues from these arrangements will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements. We cannot be certain that, following a strategic transaction or license, we will achieve the revenue or specific net income that justifies such transaction.
Collaborations involving elafibranor, an LDT or IVD powered by NIS4 or any of our other drug candidates pose the following risks to us:
•collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations;
•collaborators may not perform their obligations as expected;
•collaborators may not pursue commercialization or may elect not to continue or renew commercialization programs based on changes in the collaborator’s strategic focus or available funding or external factors such as an acquisition that diverts resources or creates competing priorities;
•collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidate if the collaborators believe that competitive products are more likely to be successfully commercialized under terms that are more economically attractive than ours;
•a collaborator with marketing and distribution rights to one or more product candidates may not commit sufficient resources to the marketing and distribution of any such product candidate;
•collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our proprietary information or expose us to potential litigation;
•collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability;
•disputes may arise between the collaborators and us that result in the delay or termination of the commercialization of our product candidate or that result in costly litigation or arbitration that diverts management attention and resources;
•we may lose certain valuable rights under circumstances identified in our collaborations, including if we undergo a change of control;
•collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates;
•collaborators may learn about our discoveries and use this knowledge to compete with us in the future;
•there may be conflicts between different collaborators that could negatively affect those collaborations and potentially others;
•the number and type of our collaborations could adversely affect our attractiveness to future collaborators or acquirers;
•collaboration agreements may not lead to commercialization of our product candidate in the most efficient manner or at all. If a present or future collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our commercialization program under such collaboration could be delayed, diminished or terminated; and
•collaborators may be unable to obtain the necessary marketing approvals.
If current or future collaboration partners fail to develop or effectively commercialize elafibranor, an LDT or IVD powered by NIS4 or any other drug candidate for any of these reasons, such product candidate may not be cleared for sale and our sales of such product candidate, if approved, may be limited, which would have an adverse effect on our operating results and financial condition.
Any of our product candidates for which we or our collaborators obtain marketing approval or CE Certificates of Conformity will be subject to ongoing regulation and could be subject to post-marketing restrictions or withdrawal from the market. Furthermore, we or our collaborators may be subject to substantial penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our products following approval.approval or receipt of CE Certificates of Conformity.
Even if we or our collaborators receive regulatory approval or CE Certificates of Conformity for a product candidate, this approval or certification may carry conditions that limit the market for the product or put the product at a competitive disadvantage relative to alternative therapies or diagnostic solutions. For instance, a regulatory approval may limit the indicated uses for which we or our collaborators can market a product or the patient population that may utilize the product, or may be required to carry a warning, such as a boxed warning, in its labelling and on its packaging. Products with boxed warnings are subject to more restrictive advertising regulations than products without such warnings. These restrictions could make it more difficult to market any product candidate effectively.
Additionally, any of our product candidates for which we or our collaborators obtain regulatory approval or certification, as well as the manufacturing processes, post-approval studies and measures, labelling, advertising and promotional activities for such products, among other things, will be subject to continual requirements of and review by the EMA, competent authorities of EEA countries, FDA, other regulatory authorities, and notified bodies.Notified Bodies, as applicable. These requirements include submissions of safety and other post-marketing information and reports, registration and listing requirements, requirements relating to manufacturing, quality control, quality assurance and corresponding maintenance of records and documents, requirements regarding the distribution of samples to physicians, and recordkeeping.
Approved drugs that are manufactured or distributed in the United States pursuant to FDA approvals are subject to pervasive and continuing regulation by the EC and EMA following approval by the EC, or national regulatory authorities in EEA countries and the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, drug sampling and distribution, advertising and promotion and reporting of adverse experiences with the drug.
After approval, most changes to the approved drug, such as adding new indications or other labelling claims and some manufacturing and supplier changes are subject to prior FDA, EC or national regulatory authorities of the EEA countries review and approval. There also are continuing, annual program user fee requirements for marketed drugs, as well as new application fees for certain supplemental applications. Once approval is granted, the FDA, or other comparable foreign regulatory authorities, may issue enforcement letters or withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the drug reaches the market. Corrective action could delay drug distribution and require significant time and financial expenditures. Later discovery of previously unknown problems with a drug, including adverse effects of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labelling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a risk evaluation and mitigation strategy, or REMS. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use. Elements to assure safe use can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS can be costly to establish and can materially affect the potential market and profitability of the drug.
Depending on the outcome, the FDA, EC, or national regulatory authorities of the EEA countries could revoke, suspend or vary the previously granted approval.
Other potential consequences include, among other things:
•restrictions on the marketing or manufacturing of the drug, under a risk evaluation and mitigation strategy, or REMS, or comparable foreign strategy, suspension of the approval, complete withdrawal of the drug from the market or product recalls;
•revisions to the approved labelling to add new safety information;
•fines, warning letters or holds on post-approval clinical trials;
•refusal of the FDA, EC, or national regulatory authorities of the EEA countries to approve applications or supplements to approved applications, or suspension, variation or revocation of drug approvals;
•drug seizure or detention, or refusal to permit the import or export of drugs; or
•injunctions or the imposition of civil or criminal penalties.
Corrective action could delay drug distribution and require significant time and financial expenditures. The requirement for a REMS or comparable foreign strategies can be costly to establish and can materially affect the potential market and profitability of the drug.
The FDA and other comparable foreign regulatory authorities strictly regulate marketing, labelling, advertising and promotion of drugs that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies,comparable national authorities and industry associations activelyforeign regulatory authorities enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including civil, criminal and administrative penalties. Industry associations may also actively supervise promotional activities and report any non-compliance to the competent authorities. However, physicians may, in their independent medical judgment, prescribe legally available products for off-label uses. The FDA and other comparable foreign regulatory authorities do not regulate the behavior of physicians in their choice of treatments but the FDA and other comparable foreign regulatory authorities do restrict manufacturer’s communications on the subject of off-label use of their products.
Similarly,EEA countries' legislation may also restrict or impose limitations on our ability to advertise our products directly to the general public. In addition, voluntary EU and national industry Codes of Conduct provide guidelines on the advertising and promotion of our products to the general public and may impose limitations on our promotional activities with healthcare professionals, which could negatively impact our business, operating results and financial condition.
In addition, if we are able to affix the CE mark to an IVD powered by NIS4 is authorizedNIS4® for marketing for clinical care in the United States, the test will be subject to quality system regulation, or QSR, labelling regulations, registration and listing, the Medical Device Reporting regulation which requires that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur and the Reports of Corrections and Removals regulation which requires manufacturers to report recalls and field actions to the FDA if initiated to reduce a risk to health posed by the device or to remedy a violation of the FDCA. The FDA enforces these requirements by inspection and market surveillance. If the FDA finds a violation, it can institute a wide variety of enforcement actions, ranging from an untitled or public warning letter to more severe sanctions such as fines, injunctions and civil penalties; recall or seizure of products; operating restrictions and partial suspension or total shutdown of production; refusing requests for 510(k) clearance or PMA approval of new products; withdrawing 510(k) clearance or PMAs already granted; and criminal prosecution.
Similarly, in the EEA, IVDs are strictly regulatedwe may be required to conduct costly post-market testing and surveillance to monitor the safety or effectiveness of such products in the EEA. We would also be required comply with IVD reporting requirements, including the reporting of adverse events and malfunctions related to our IVDs will be subjectproducts. Later discovery of previously unknown problems with our products, including unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturing problems, or failure to vigilance, post-market surveillance, quality management systems and many othercomply with regulatory requirements imposed bymay result in changes to labeling, restrictions on such products or manufacturing processes, withdrawal of the IVDR. products from the market, voluntary or mandatory recalls, a requirement to repair, replace or refund the cost of any IVD we would manufacture or distribute, fines, suspension, variation or withdrawal of CE Certificates of Conformity, product seizures, injunctions or the imposition of civil or criminal penalties which would adversely affect our business, operating results and prospects. All manufacturers placing IVDs on the market in the EEA are legally bound to report incidents within strict deadlines and trends involving devices they produce or sell to the regulator authority, in whose jurisdiction the incident occurred. Malfunction of our products could result in future voluntary corrective actions, such as recalls, including corrections, or customer notifications, or regulatory action, such as inspection or enforcement actions. If malfunctions do occur, we may be unable to correct the malfunctions adequately or prevent further malfunctions, in which case we may need to cease manufacture and distribution of the affected products, initiate voluntary recalls, and redesign the products.
In addition, any significant changes made to CE marked IVDs placed on the EEA market, or substantial changes to the related quality assurance system affecting the IVD, must be notified to the Notified Body having delivered the related CE Certificate of Conformity. Obtaining variation of existing CE Certificates of Conformity or a new CE Certificate or Conformity can be a time-consuming process, and delays in obtaining required future clearances or approvals would adversely affect our ability to introduce new or enhanced products in a timely manner, which in turn would harm our future growth.
If a regulatory authority of an EEA country finds a violation of the IVDR obligations for which we are considered to be responsible we may be subject to a wide variety of enforcement actions, ranging from warning letters, injunction letters, ordering recalls, fines, seizing affected products, civil penalties and criminal prosecution.
Accordingly, assuming we or our current or future collaborators receive regulatory approval or certification for one or more of our product candidates, we and our collaborators will continue to expend time, money and effort in all areas of regulatory compliance.
Government restrictions on pricing and reimbursement, as well as other healthcare payor cost-containment initiatives, may negatively impact our ability or that of our current or future collaborators to generate revenues even if we or they obtain regulatory approval to market a product.product candidate.
Our ability to successfully commercialize any of our product candidates or that of our current or future collaborators, if approved, also will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from third-party payors, including government authorities, such as Medicare and Medicaid in the United States, private health insurers and health maintenance organizations. These third-party payors determine which medications they will cover and establish reimbursement levels. Assuming we or our current or future collaborators obtain coverage for a given product by a third-party payor, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. Patients who are prescribed medications for the treatment of their conditions, and their prescribing physicians, generally rely on third-party payors to reimburse all or part of the costs associated with their prescription drugs. Patients are unlikely to use our products unless coverage is provided and reimbursement is adequate to cover all or a significant portion of the cost of our products. Therefore, coverage and adequate reimbursement is critical to new product acceptance. Coverage decisions may depend upon clinical and economic standards that disfavor new drug products when more established or lower cost therapeutic alternatives are already available or subsequently become available.
Third-party payors are developing increasingly sophisticated methods of controlling healthcare costs, such as by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices as a condition of coverage, are using restrictive formularies and preferred drug lists to leverage greater discounts in competitive classes, and are challenging the prices charged for medical products. In addition, in the United States, federal programs impose penalties on drug manufacturers in the form of mandatory additional rebates and/or discounts if commercial prices increase at a rate greater than the Consumer Price Index-Urban, and these rebates and/or discounts, which can be substantial, may impact our or our collaborators’ ability to raise commercial prices. Further, Moreover, no uniform policy requirement for coverage and reimbursement for drug products exists among third-party payors in the United States. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement rates, but also have their own methods and approval process apart from Medicare determinations. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us or our collaborators to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance.
The continuing efforts of third-party payors of healthcare costs to contain or reduce costs of healthcare may negatively affect our or our collaborators commercialization prospects, including:
•the ability to set a price we believe is fair for our or our collaborators’ products, if approved;
•the ability to obtain and maintain market acceptance by the medical community and patients;
•the ability to generate revenues and achieve profitability; and
•the availability of capital.
Our or our collaborators’ ability to obtain an acceptable reimbursement rate for our drugs from third-party payors will be determined in the coming years, in particular by our collaborators' Ipsen and Terns Pharmaceuticals with respect to PBC. We cannot be sure that coverage and reimbursement will be available for any potential product candidate that we or our collaborators may commercialize and, if reimbursement is available, what the level of reimbursement will be. Since few drugs have been commercialized in PBC, we cannot predict the conditions of elafibranor’s future reimbursement. However, because negotiations with the payors are traditionally based on the results (intermediate, or otherwise) of Phase 3 clinical trials, we have only had preliminary discussions with the organizations concerned. Coverage and reimbursement may impact the demand for, or the price of, any product candidate for which we or our collaborators obtain marketing approval. If coverage and reimbursement are not available or reimbursement is available only to limited levels, we may not successfully commercialize any product candidate for which we obtain marketing approval.
In the United States,We expect that the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, ACA, is significantly impacting the provision of, and payment for, healthcare. With regard to pharmaceutical products specifically, the ACA, among other things, expanded and increased industry rebates for drugs covered under Medicaid programs and made changes to the coverage requirements under the Medicare prescription drug benefit. Some of the provisions of the ACA have yet to be implemented, and there have been executive, judicial and Congressional challenges to certain aspects of the ACA. For example, on June 17, 2021, the U.S. Supreme Court dismissed a challenge on procedural grounds that argued the ACA is unconstitutional in its entirety because the individual mandate was repealed by the U.S Congress. Thus the ACA will remain in effect in its current form. Moreover, prior to the U.S. Supreme Court ruling, , on January 28, 2021, President Biden issued an executive order to initiate a special enrollment period for purposes of obtaining health insurance coverage through the ACA marketplace. The executive order also instructs certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA. It is possible that the ACA will be subject to additional judicial or Congressional challenges in the future. It is unclear how the Supreme Court ruling, other such litigation and the health reform measures of the Biden administration will impact the ACA and our business.
In addition, both the Budget Control Act of 2011 and the American Taxpayer Relief Act of 2012 have instituted, among other things, mandatory reductions in Medicare payments to certain providers which went into effect on April 2013 and will remain in effect through 2031 unless additional Congressional action is taken. .Additional legislative proposals to reform healthcare and government insurance programs, along with the trend toward managed healthcare in the United States, could influence the purchase of medicines and reduce coverage and/or reimbursement of our product candidates, if approved. Further, Congress is considering additional health reform measures.
Moreover, recently, there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products. Such scrutiny has resulted in several recent Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the costs of drugs under Medicare and reform government program reimbursement methodologies for drug products. At the state level, legislatures are increasingly passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
We expect that the ACA, as well as other healthcare reform and cost-containment measures that may be adopted in the future, at both the federal and state levels in the United States, as well as internationally, may result in more rigorous coverage criteria and lower reimbursement from both government funded programs as well as private payors, and in additional downward pressure on the price that we or our partners receive for any approved product candidate. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our drugs. Moreover, we cannot predict what healthcare reform initiatives may be adopted in the future. We expect that additional state and federal healthcare reform measures will be adopted in the future. Further, it is possible that additional governmental action is taken in response to the COVID-19 pandemic.
In some non-U.S. countries, the proposed pricing and reimbursement conditions for a drug must be approved by relevant authorities before it may be lawfully marketed. Reimbursement may in some cases be unavailable. The requirements governing drug pricing and reimbursement vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. Non U.S. countries may approve a specific price for the medicinal product, may refuse to reimburse a product at the price set by the manufacturer or may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for biopharmaceutical products will allow favorable reimbursement and pricing arrangements for elafibranor or any of our other product candidates that may be approved.
Failures to reimburse an LDT or IVD powered by NIS4,NIS4® or its variations, if commercialized for clinical care, or changes in reimbursement rates by third-party payors and variancesvariations in reimbursement rates could materially and adversely affect our revenues and could result in significant fluctuations in our revenues.
Our ability or that of a potential future collaboratorscollaborator to successfully commercialize an LDT or IVD powered by NIS4 alsoNIS4® or its variations will depend in parton the availability of an approved drug to treat MASH and also on the extent to which coverage and adequate reimbursement for this test will be available from third-party payors, such as government health administration authorities, private health insurers and other organizations. Insurance coverage and reimbursement rates for diagnostic tests are uncertain, subject to change and particularly volatile during the early stages of a newly commercialized diagnostic test. As of the date of this annual report, NASHnextNASHNext® has not obtained reimbursement status in the countries where it is commercialized by Labcorp. It is uncertain as to what extent third-party payors will provide coverage for NASHnext,NASHNext®, another LDT or IVD powered by NIS4,NIS4® or its variations, if commercialized for clinical care. We will also likely experience volatility in the coverage and reimbursement of NASHnext,NASHNext®, another LDT or IVD test due to contract negotiation with third-party payors and implementation requirements.
The reimbursement amounts we receive from third-party payors will vary from payor to payor, and, in some cases, the variation is material. Third-party payors have increased their efforts to control the cost, utilization and delivery of healthcare services. These measures have resulted in reduced payment rates and decreased utilization for the diagnostic test industry. From time to time, Congress has considered and implemented changes to the Medicare fee schedules in conjunction with budgetary legislation, and pricing for tests covered by Medicare is subject to change at any time. Reductions in the reimbursement rate provided by third-party payors may occur in the future. Reductions in the price at which NASHnext,NASHNext®, another LDT or IVD powered by NIS4NIS4® or its variations is reimbursed could have a material adverse effect on our revenues. If we and our potential future collaborators are unable to establish and maintain broad coverage and adequate reimbursement for NASHnext,NASHNext®, another LDT or IVD powered by NIS4NIS4® or its variations or if third-party payors change their coverage or reimbursement policies with respect to NASHnext,NASHNext®, another LDT or IVD test, our revenues could be materially and adversely affected.
Our future growth depends, in part, on our or our collaborators’ ability to penetrate international markets, where we or they would be subject to additional regulatory burdens and other risks and uncertainties.
Our future profitability will depend on our or our collaborators’ (Ipsen, Terns Pharmaceuticals, Labcorp/Covance, Q2) ability to commercialize our product candidates in the United States, EuropeEEA and other territories around the world. If we or our collaborators commercialize our product candidates in international markets, we would be subject to additional risks and uncertainties, including:
•economic weakness, including inflation;
•political instability, armed conflict or war in particular economies and markets;markets, such as in Ukraine;
•global pandemics like COVID-19;
•the burden of complying with complex and changing non-U.S. regulatory, tax, accounting and legal requirements, many of which vary between countries;
•different medical practices and customs in non-U.S. countries affecting acceptance in the marketplace;
•governmental controls, export controls, tariffs and other trade barriers;barriers and modifications thereto;
•other trade protection measures, import or export licensing requirements or other restrictive actions by U.S. or other governments;
•longer accounts receivable collection times;
•longer lead times for shipping;
•compliance with tax, employment, immigration and labor laws for employees living or traveling abroad;
•workforce uncertainty in countries where labor unrest is common;
•language barriers for technical training;
•reduced protection of intellectual property rights in some countries outside the United States, and related prevalence of generic alternatives to therapeutics;
•foreign currency exchange rate fluctuations and currency controls;
•differing reimbursement landscapes globally;
•uncertain and potentially inadequate reimbursement of our products; and
•the interpretation of contractual provisions governed by laws outside the United States in the event of a contract dispute.
Sales of our products outside the United States could also be adversely affected by the imposition of governmental controls, political and economic instability, trade restrictions and changes in tariffs.
Adverse market and economic conditions may exacerbate certain risks associated with commercializing our product candidates.
Future sales of our product candidates, if they are approved, will be dependent on purchasing decisions of and reimbursement from government health administration authorities, distributors and other organizations. As a result of adverse conditions affecting the global economy and credit and financial markets, including disruptions due to political instability, armed conflict, such as in Ukraine, wars, the COVID-19 pandemicglobal pandemics or otherwise, these organizations may defer purchases, may be unable to satisfy their purchasing or reimbursement obligations, or may delay payment for elafibranor, NASHNextNASHNext® or another LDT or IVD powered by NIS4NIS4® or its improvements or any of our product candidates that are approved for commercialization in the future. In addition, the increase of inflation rates following the COVID-19 pandemic erain recent years and the current armed conflictconflicts in Ukraine or Israel may additionally affect the commercialization of our products and product candidates.
Risks Related to the Dependency on Third Parties
We depend on third-party contractors for a substantial portion of our operations, namely contract research organizations or CROs for our preclinical studies and clinical trials and contract manufacturing organizations or CMOs for manufacturing of our active ingredients and therapeutic units and may not be able to control their work as effectively as if we performed these functions ourselves.
Under our supervision, we outsource substantial portions of our operations to third-party service providers, including preclinical studies and clinical trials, collection and analysis of data and manufacturing of our drug candidates and the realization of certain analyses performed under our agreements with Labcorp and Q2 pertaining to an LDT or IVD powered by NIS4NIS4® technology or its variations for use in the clinical research and clinical diagnostics markets. In particular, we subcontract certain elements of the design and/or conduct of our preclinical studies and clinical trials to CROs, as well as the manufacturing of our active ingredients and therapeutic units to CMOs, especially with regard to our Phase 3 ELATIVE trial evaluating elafibranor in PBC.CMOs.
We also contract with external investigators and other specialized services providers, for example with respect to certain statistical analyses, to perform services such as carrying out and supervising, and collecting, analyzing and formatting of data for our trials. Although we are involved in the design of the protocols for these trials and in monitoring them, we do not control all the stages of test performance and cannot guarantee that the third parties will fulfil their contractual and regulatory obligations. In particular, a contractor’s failure to comply with protocols or regulatory constraints, or repeated delays by a contractor, could compromise the development of our products or result in liability for us, including our contractual liability resulting from provisions in agreements we have signed with Ipsen and Terns Pharmaceuticals for the development of elafibranor.elafibranor in Greater China. Such events could also inflate the product development costs borne by us.
This strategy means that we do not directly control certain key aspects of our product development, such as:
•the quality of the product manufactured;
•the delivery times for therapeutic units (pre-packaged lots specifically labeled for a given clinical trial);
•the clinical and commercial quantities that can be supplied; and
•compliance with applicable laws and regulations.regulations; and
•the quality or accuracy of the data obtained by third parties.
Additionally, our development activities or clinical trials conducted in reliance on third parties may be delayed, suspended, or terminated if:
•the third parties do not devote a sufficient amount of time or effort to our activities or otherwise fail to successfully carry out their contractual duties or to meet regulatory obligations or expected deadlines; or
•we replace a third party; or
•the quality or accuracy of the data obtained by third parties is compromised due to their failure to adhere to pre-clinical and clinical protocols, regulatory requirements, or for other reasons.
We may not be able to control the performance of third parties in their conduct of development activities. In the event of a default, bankruptcy or shutdown of, or a dispute with, a third party, we may be unable to enter into a new agreement with another third party on commercially acceptable terms. Further, third-party performance failures may increase our development costs, delay our ability to obtain regulatory approval, and delay or prevent the commercialization of our product candidates. In addition, our third-party agreements usually contain a clause limiting such third party’s liability, such that we may not be able to obtain full compensation for any losses we may incur in connection with the third party’s performance failures. While we believe that there are numerous alternative sources to provide these services, in the event that we seek such alternative sources, we may not be able to enter into replacement arrangements without incurring delays or additional costs.
We rely entirely on third parties for the manufacturing of our drug candidates and the future manufacturing of an IVD powered by NIS4NIS4® or its variations for use as a clinical diagnostic including one manufacturer for the active ingredient in elafibranor and another manufacturer for the therapeutic units of elafibranor used in our clinical trials and those of our collaborators.diagnostic. Our business could be harmed if those third parties fail to provide us with sufficient quantities of drug product or tests, or fail to do so at acceptable quality levels or prices.
We do not currently and do not intend in the future to manufacture the drug products, nor future test kits related to an IVD powered by NIS4NIS4® or its variations, that we or our collaborators plan to sell if approved, or successfully complete the conformity assessment procedure for use as a clinical diagnostic.
We currently have agreements with a contract manufacturermanufacturers for the production of the active pharmaceutical ingredients and the formulation of sufficient quantities of drug product for our preclinical studies and clinical trials in elafibranor that we plan to conduct prior to and after seeking regulatory approval and, if applicable, for the manufacturing of the first commercial lots of the product. We rely on one supplier for the active ingredient in elafibranor and another manufacturer for the therapeutic units of elafibranor used in our clinical trials and, if applicable, for the provision of the first commercial lots. In addition, we rely on Genoscience Pharma for the provision of drug product for the development and future commercialization of GNS561 and external third parties for the supply of NTZ.trials. If any of these suppliers should cease to provide services to us, or our collaborators, for any reason, we likely would experience delays in advancing our clinical trials and, if applicable, for the commercial launch while we or our collaborators identify and qualify one or more replacement suppliers and we may be unable to obtain replacement supplies on terms that are favorable to us.
While we believe that our current inventory and drugs in production at various levels of the production chain are sufficient for our needs on a short-term basis, awe and Ipsen rely on one supplier for the active ingredient in elafibranor and another manufacturer for the therapeutic units of elafibranor used in our clinical trials and, if applicable, for the provision of the first commercial lots. A failure at both of the storage sites of the therapeutic units used for the ongoing ELATIVEELATIVE® Phase 3 study evaluating elafibranor in PBC would be detrimental to our and Ipsen's clinical development plan.
For example, we have had to face the temporary closing of one of these units for a duration of 15 days due to a suspected case of COVID-19, even though this unit has indicated to us that this would not affect the provision of future clinical lots. However, in case of failure of these units, we may not be able to enter into additional long-term commercial supply agreements for elafibranor with other third-party manufacturers on terms sufficiently advantageous to us.
We do not have agreements for long-term supplies of any of our other product candidates. We currently obtain these suppliesWith regard to VS-01, we are also dependent on several CMOs to cover the supply of therapeutic units and services, such asother materials required for the ongoing Phase 2 trial in ACLF. Concerning NTZ, we use the already commercialized formulation in our clinical trials, which is available to purchase from pharmaceutical wholesalers and are until we are able to finalize our third-party contract manufacturers on a purchase order basis and can bereformulation process are therefore subject to market fluctuations in priceavailability and availability. With respect toprice. Regarding the supply of GNS561, we must enter intodepend on our partner Genoscience Pharma with whom we have signed a supply agreement withto cover the needs of the Phase 1b/2 trial evaluating GNS561 in Cholangiocarcinoma. We also depend on our partner, Genoscience Pharma.partners Seal Rock Therapeutics and Celloram to cover the supply needs linked to the first preclinical developments of SRT-015 and CLM-022.
Additionally, the facilities used by any contract manufacturer to manufacture elafibranor or any of our other product candidates must be the subject of a satisfactory inspection before the FDA, the national competent authority of the EU member states, or the regulators in other jurisdictions that approve the product candidate manufactured at that facility. We are completely dependent on these third-party manufacturers for compliance with the requirements of U.S. and non-U.S. regulators for the manufacture of our finished products. If our manufacturers cannot successfully manufacture material that conform to our specifications and current good manufacturing practice requirements of any governmental agency whose jurisdiction to which we are subject, our products or product candidates will not be approved or, if already approved, may be subject to recalls or other enforcement action.
Reliance on third-party manufacturers entails risks to whichIn the event of a default, bankruptcy or liquidation of a subcontractor, a service provider (CRO or CMO) or a collaborator, such as Genoscience, with whom we wouldhave entered into a supply agreement, or Seal Rock Therapeutics or Celloram, or a dispute with one of these collaborators or service providers, we may not be subject if we manufactured the products or product candidates, including:
•the possibility that we are unableable to enter into or renew a manufacturing agreementnew contract with a third party to manufacture elafibranordifferent subcontractor or service provider on commercially acceptable terms. In addition, failures of our subcontractors, collaborators or service providers in the course of their work could increase our development costs, delay obtaining regulatory approval or prevent the commercialization of our product candidates;
•the possible breach of the manufacturing agreements by the third parties because of factors beyond our control; and
•the possibility of termination or nonrenewal of the agreements by the third parties before we are able to arrange for a qualified replacement third-party manufacturer.
candidates. Any of these factors could cause the delaydelays in launch or completion of our clinical trials, or of approval or disruption of commercialization of our products or product candidates, cause us to incur higher costs, prevent us or our potential future collaborators from commercializing our products and product candidates successfully or disrupt the supply of our products after commercial launch. Furthermore, if any of our partners, such as Genoscience Pharma, or contract manufacturers failfails to deliver the required clinical or commercial quantities of finished product on acceptable commercial terms and we or our current or future collaborators are unable to find one or more replacement manufacturers capable of production at substantially equivalent cost, volume and quality and on a timely basis, we would likely be unable to meet demand for our products and could lose potential revenue. It may take several years to establish an alternative source of supply and to have any such new source approved by the government agencies that regulate our products.
We have entered, and may in the future enter into, collaboration, licensing or co-marketing agreements with third parties for the development and eventual commercialization of our product candidates and NIS4NIS4® diagnostic technology or its variations, and may not generate revenues from these agreements.
We have limited experience in product development and marketing and may seek to enter into collaborations with third parties for the development and potential commercialization of our product candidates including those at an early and preclinical stage, particularly those candidates outside of our main therapeutic areas of interest. We have entered into an exclusive licensing and collaboration agreement with Ipsen to develop and commercialize elafibranor for the treatment of PBC and other indications worldwide, with the exception of Greater China which is licensed to Terns Pharmaceuticals. Our NIS4NIS4® technology isand its improvements are licensed to two partners, both to Labcorp to allow them to deploy an LDT powered by NIS4NIS4® technology in the clinical research and clinical diagnostics spaces and also to Q2 in the clinical research space. Should we seek to collaborate with additional third parties with respect to our development programs, we may not be able to locate a suitable collaborator and may not be able to enter into an agreement on commercially reasonable terms or at all.
We also signed licensing agreements with Genoscience to develop and commercialize GNS561 in CCA, with Seal Rock Therapeutics to develop and commercialize an injectable formulation of SRT-015 in acute liver disease, and with Celloram to develop and commercialize CLM-022 in liver disease.
Any new collaboration may require additional expenditures, increase our short and long term investments, require us to issue new shares and dilute our existing shareholders or disrupt our management team or activities. With our current agreements, or even if we succeedsucceeded in securing collaborators for the development and commercialization of elafibranor, our NIS4NIS4® technology, the NASHnextNASHNext® LDT or our other product candidates, we have limited control over the amount and timing that our collaborators may dedicate to the development or commercialization of our product candidates.
These collaborations and licensing agreements pose a number of risks, including:
•the means and resources used within the framework of these agreements remain, for the most part, at the discretion of the partner;partner, and they may not allocate sufficient resources to carrying out development and commercialization;
•the partner might not fulfill its contractual obligations;
•the partner might interrupt the development or commercialization or decide to interrupt or not renew the development or commercialization programs due to a change in strategic orientation, a lack of financing or external factors such as an acquisition that would reallocate resources or induce different priorities;
•the partner might develop, independently or with the assistance of third parties, products, in the case of pharmaceuticals or in-vitro tests, in the case of diagnostic technologies that are in direct or indirect competition with our product candidates or future IVD powered by NIS4NIS4® or its variations if it believes that it is easier to successfully commercialize competing products under more attractive economic conditions than ours;
•the partner, as holder of the commercialization and distribution rights on a product candidate or technology for a set time period or a specific territory or territories, might not allocate sufficient resources to these activities;
•the partner might not protect or defend our intellectual property rights in an appropriate manner or might use exclusive information that belongs to us in a manner resulting in disputes that may compromise or discredit our exclusive information or expose us to potential disputes;
•the partner might not respect the property rights of third parties, which might expose us to litigation and potentially involve our liability;
•disputes might arise between us and the partner, which could result in delays or suspension of the commercialization of the product candidate, or legal action or costly procedures that would monopolize resources as well as divert management’s attention;
•we might lose certain important rights obtained through these partnerships, notably in the case of change of control of our company;
•the collaboration might be terminated and, in such case, require additional financing to further develop or market the product candidate licensed to it;
•the partner has access to our discoveries and might use this information to develop future competing products;
•there may be conflicts between different partners that could negatively affect those partnerships and potentially others;
•the collaboration, due to its nature, might have a negative impact on our attractiveness for collaborators or potential acquirers;
•the collaboration might not result in the development and commercialization of the product candidate(s) in an optimal fashion or never fulfill its objectives; and
•if the partner were to take part in a merger, the continuity of advancement and the central nature of our commercialization program might be delayed, reduced or suspended by it. it; and
•the partner may be unable to obtain or maintain the necessary marketing approvals.
Thus, collaboration agreements may not lead to development or commercialization of product candidates in the most efficient manner or at all. For example, although we have entered into a license agreement with Labcorp to enable them to develop and commercialize an LDT powered by NIS4NIS4® or its variations for clinical research and clinical diagnostic purposes, there is no guarantee that our collaboration with Labcorp will result in widespread clinical or commercial use of NASHnext,NASHNext®, an LDT powered by NIS4NIS4® technology for clinical care. Commercial launch of NASHnextNASHNext® in 2021 was slowed by COVID-19 and also impacted by the lack of approved treatment for NASH.MASH. Similarly, although we have entered into a collaboration and license agreement with Ipsen for the treatment of PBC and other indication worldwide, with the exception of Greater China which is licensed to Terns Pharmaceuticals, there is no guarantee that our partnership with Ipsen or Terns Pharmaceuticals will successfully result in a generalized clinical or commercial use of elafibranor for these indications and in those jurisdictions. Finally, the conclusion of licensing-out agreements, such as those we have signed with Ipsen, Terns Pharmaceuticals, Labcorp and Q2, necessarily implies that part of the value of the product candidates concerned is transferred to the partner. This reduces our ability to generate revenues and profits, without necessarily being fully offset by the source of financing represented by the payments received on signature or on reaching development milestones, or in the form of royalties.
Some collaboration agreements may be terminated without cause on short notice. Once a collaboration agreement is signed, it may not lead to commercialization of a product candidate. We also face competition in seeking out collaborators. If we are unable to secure new collaborations that achieve the collaborator’s objectives and meet our expectations, we may be unable to advance our product candidates and may not generate meaningful revenues.
Licensing-in agreements, such as those we have signed with Genoscience, Sealrock Therapeutics and Celloram, provide for payments to partners in the event of scientific and regulatory milestones being met, and royalties in the event of commercialization. These agreements may also impair our ability to generate profits if we fail to achieve the expected direct or indirect commercial benefits.
If the manufacturing facilities of our third-party manufacturers of drug candidates as well as the central testing laboratories of Labcorp fail to comply with applicable regulations or maintain these approvals, our business will be materially harmed.
We do not currently and do not intend in the future to manufacture the drug candidates we or our collaborators intend to sell. We outsource the manufacturing of our products to third parties, who are, in turn, subject to ongoing regulation and periodic inspection by the national regulatory authorities of the EEA countries, FDA and other regulatory bodies to ensure compliance with current Good Manufacturing Practices, or cGMP. Any failure to follow and document their adherence to such cGMP regulations or other regulatory requirements may lead to significant delays in the availability of products for commercial sale or clinical trials, may result in the termination of or a hold on a clinical trial, or may delay or prevent filing or approval of marketing applications for our products.product candidates, may lead to the shutdown of the third-party vendor or invalidation of drug product lots or processes and in some cases, a product recall may be warranted or required, which would materially affect our ability to supply and market our product candidates.
Failure to comply with applicable regulations could also result in the national regulatory authorities of the EEA countries, FDA or other applicable regulatory authorities taking various actions, including:
•levying fines and other civil penalties;
•imposing consent decrees or injunctions;
•requiring us or our current or future collaborators to suspend or put on hold one or more of our clinical trials;
•suspending, varying or withdrawing regulatory approvals;
•delaying or refusing to approve pending applications or supplements to approved applications;
•requiring us or our current or future collaborators or our third-party manufacturers to suspend manufacturing activities or product sales, imports or exports;
•requiring us or our current or future collaborators to communicate with physicians and other customers about concerns related to actual or potential safety, efficacy, and other issues involving our products;
•mandating product recalls or seizing products;
•imposing operating restrictions; and
•seeking criminal prosecutions.
Any of the foregoing actions could be detrimental to our reputation, business, financial condition or operating results. Furthermore, our key suppliers may not continue to be in compliance with all applicable regulatory requirements, which could result in our failure or that of our current or future collaborators to produce our products on a timely basis and in the required quantities, if at all. In addition, before any additional products would be considered for marketing approval in the United States, EEA or elsewhere, our suppliers will have to pass an audit by the applicable regulatory agencies.authorities. We are dependent on our suppliers’ cooperation and ability to pass such audits, and the audits and any audit remediation may be costly. Failure to pass such audits by us or any of our suppliers would affect our ability or that of our current or future collaborators to commercialize our product candidates in the United States, Europe or elsewhere.
The deployment of an LDT powered by NIS4NIS4® or its variations depends on the ability of the central laboratories of our partner Labcorp that conduct the diagnostic test to retain its CLIA certification or other regulatory authorizations or operating licenses, which certification sets quality standards that must be followed in laboratory testing in order to ensure accuracy, reliability and speed of test results for the patients wherever the testing is conducted. We do not plan on manufacturing the test kits that we plan on marketingwould market and that will be associated with an IVD powered by NIS4NIS4® or its variations if it were to be approved or CE marked on the market of routine care; and the manufacturing sites of the contractor that we or our potential collaborators may choose for their production would also be subject to significant authorizations, inspections and regulations.
Risks Related to Our Operations
Starting in mid-2020 and into 2021, we embarked on a significant strategic reorientation which resulted in a significant changes to our organization and workforceworkforce. As a result, we may encounter difficulties in managing development of our product candidate pipeline, which could disrupt our operations.
In mid-2020 we terminated our development program of elafibranor in NASHMASH and redefined our strategic priorities with respect to our product candidate pipeline. As a result, we implemented a multi-year cost reduction program and workforce reduction program that had a significant impact on our organization, infrastructure and operations. In 2021, given that our access to market financing was limited, we chose to enter into licensing and collaboration agreements to support the development and commercialization of certain of our product candidates, and elafibranor in particular, as well as the in-licensing of a product candidate developed by a third party, for which we need to develop our expertise.
In particular, this strategy of acquiring new product candidates developed by third-parties was realized in September 2022 with the acquisition of Versantis AG and its programs, and the in-licensing of drug candidates from Genoscience, Seal Rock Therapeutics and Celloram in 2021 and 2023, respectively. We may undertake a similar type of transaction or additional in-licensing projects in the future. In the context of these significant changes in our organization, the focus of our resources on managing the success of these partnerships and new programs could result in weaknesses in our infrastructure (including our internal control over financial reporting), give rise to operational mistakes, loss of business opportunities, loss of employees and reduced productivity among employees. In particular, running so many programs simultaneously could lead to a work overload and an inappropriate dispersion of our resources, and negatively impact their development. This overload could, conversely, force us to make choices that might not prove to be advantageous. These changes in our organization may lead to significant costs and may divert financial resources from other projects, such as the development of our other product candidates. If our management is unable to effectively manage these changes efficiently, our expenses may increase more than expected, our ability to generate or increase our revenue could be impacted and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize our other product candidates, if approved, and compete effectively will depend, in part, on our ability to effectively manage the changes related to the significant strategic reorientation we have undertaken.
We depend on qualified management personnel and our business could be harmed if we lose key personnel and cannot attract new personnel.
Our success depends to a significant degree upon the technical and management skills of our co-founders, scientific advisers, senior management team, including, in particular, Pascal Prigent, our chief executive officer, Jean-François Mouney, our chairman, and Dean Hum, our chief scientific officer and Pascal Caisey, our chief operating officer. The loss of the services of Messrs. Prigent, Mouney, Hum or HumCaisey would likely have a material adverse effect on us. Our success also will depend upon our ability to attract and retain additional qualified scientific, management, marketing, technical, and sales executives and personnel, in particular in the new therapeutic areas where we need to build up our experience, despite the workforce reduction plan we implemented in 2020.. We compete for key personnel against numerous companies, including larger, more established companies with significantly greater financial resources than we possess. In addition, there is risk of departures or difficulties in hiring qualified personnel following the announcement of disappointing clinical results such as those we announced in May 2020 regarding our Phase 3 RESOLVE-IT trial and our recentor the implementation of a workforce reduction plan. There can be no assurance that we will be successful in attracting or retaining such personnel, and the failure to do so could harm our operations and our growth prospects.
We may use hazardous chemicals and biological materials in our business. Any claims relating to improper handling, storage or disposal of these materials could be time-consuming and costly.
Our research and development processes for our product candidates involve the controlled use of hazardous materials, including chemicals and biological materials. We cannot eliminate the risk of accidental contamination or discharge and any resultant injury from these materials. During their work, our researchers come into contact with a number of potentially dangerous substances, including in particular (1) genetically modified organisms, or GMO, the safety of which is overseen in France by the Ministry in charge of Research with the assistance of High Council for Biotechnologies (or the Haut Conseil des Biotechnologies), (2) animals used for experimentation, the authorization of which is overseen by the local Préfet with the assistance of the local Department for the Protection of People, or DDPP (for Direction départementale de la protection des populations) and (3) human samples. This research is subject to application for authorization from the competent authorities, in particular the National Drug and Health Product Authority, or ANSM (for Autorité Nationale de Sécurité du Médicament et des produits de santé) to assess the usefulness of the research, ensure that patients have been properly informed, and assess the management of information obtained from the sampling.
We may be subject to fines or sued for any injury or contamination resulting from our use or the use by third parties of these materials, and our liability may exceed any insurance coverage and our total assets, and we may also suffer reputational harm. European, French and U.S. federal, state, local or foreign laws and regulations govern the use, manufacture, storage, handling and disposal of these hazardous materials and specified waste products, as well as the discharge of pollutants into the environment and human health and safety matters. Compliance with health, safety and/or environmental laws and regulations may be expensive and may impair our research and development efforts. If we fail to comply with these requirements, we could incur substantial costs, including civil or criminal fines and penalties, clean-up costs or capital expenditures for control equipment or operational changes necessary to achieve and maintain compliance. Furthermore, we could face the rejection, suspension or withdrawal of regulatory approval for our drugs candidates or an IVD powered by NIS4NIS4® or its variations if they had received market approval. In addition, we cannot predict the impact on our business of new or amended health, safety and/or environmental laws or regulations or any changes in the way existing and future laws and regulations are interpreted and enforced.
We have recently acquired and may in the future acquire, products or businesses or form new strategic alliances, and despite due diligence and evaluation procedures, we may not realize the benefits of such partnerships or acquisitions.
As part of our growth strategy, we have sought and intend to seek opportunities to in-license rights to drug candidates in clinical development. This could also include the acquisition of companies or technologies facilitating or enabling us to access to new medicines, new research projects, or new geographical areas, or enabling us to express synergies with our existing operations. If such acquisitions occur in the future, we may not be able to identify appropriate targets or make acquisitions under satisfactory conditions, in particular, satisfactory price conditions. In addition, we may be unable to obtain the financing for these acquisitions on favorable terms, which could require us to finance these acquisitions using our existing cash resources that could have been allocated to other purposes. If we acquire businesses with promising markets or technologies, we may not be able to realize the benefit of acquiring such businesses or the expected synergies if we are unable to successfully integrate them with our existing operations and company culture.
In December 2021, we licensed the exclusive rights from Genoscience Pharma to develop and commercialize the investigational treatment GNS561 in CCA in the United States, Canada and Europe, including the United Kingdom and Switzerland. As CCA is a new therapeutic area for us, and despite our due diligence, or in the event we are unable to collaborate efficiently, we may not be successful in realizing the full potential of the GNS561 program.
We also acquired Versantis AG in September 2022 to strengthen our product candidate pipeline, including the drug candidates VS-01-ACLF, VS-01-HAC and VS-02 that we are developing respectively in ACLF, UCD and OA, and HE. As these three therapeutic areas are relatively or totally new to us, despite our due diligence and our evaluation of the potential of these programs, we may be unsuccessful in integrating the company or realizing the full potential of these programs and potential synergies. The anticipated benefits and synergies of this acquisition are based on projections and assumptions, not actual experience, and assume a successful integration.
Finally, in May 2023, we announced that we had entered into a licensing agreement with Seal Rock Therapeutics for exclusive worldwide rights to the ASK1 inhibitor SRT-015, with a view to developing an injectable formulation for use in acute liver disease and ACLF in particular; and in July 2023, we entered into a licensing agreement with Celloram for the worldwide rights to the inflammasome inhibitor CLM-022, to develop and exploit it in liver diseases and ACLF in particular; in return, both companies are eligible for potential clinical, regulatory and commercial development milestone payments, as well as royalties if the products are commercialized. As ACLF is a new therapeutic area for us, it is possible that despite the due diligence and evaluation procedures carried out, or in the event of less-than-efficient collaboration with these two companies, we may not be able to realize the full potential of these two programs.
Our internal information technology systems and those of our current or future collaborators or those of our third-party supplies, contractors or consultants, may fail or suffer security breaches, any of which could result in a material disruption of our product development and commercialization programs.
Despite the implementation of security measures, our internal information technology systems and those of our current or future collaborators, or third-party contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our programs.
In the ordinary course of our business, we collect and store sensitive data, including, among other things, legally protected patient health information, personally identifiable information about our employees, intellectual property and proprietary business information. We manage and maintain our applications and data utilizing on-site systems and outsourced vendors. These applications and data encompass a wide variety of business critical information, including research and development information, commercial information and business and financial information. Because information systems, networks and other technologies are critical to many of our operating activities, shutdowns or service disruptions at our company or vendors that provide information systems, networks or other services to us pose increasing risks. Such disruptions may be caused by events such as computer hacking, phishing attacks, ransomware, dissemination of computer viruses, worms and other destructive or disruptive software, denial of service attacks and other malicious activity and cyberattacks, as well as power outages, natural disasters (including extreme weather), terrorist attacks or other similar events. Such events could have an adverse impact on us and our business, including loss of data and damage to equipment and data. In addition, system redundancy may be ineffective or inadequate, and our disaster recovery planning may not be sufficient to cover all eventualities. Any of these developments could result in a disruption of our operations, damage to our reputation and our credibility or a loss of revenues. In addition, we may not have adequate insurance coverage to compensate for any losses associated with such events. For example, the loss of clinical trial data for our product candidates could result in delays in our regulatory approval efforts or those of our current or collaborators and significantly increase our costs because we could be required to repair or replace information systems or networks and recover or reproduce the lost data.
We could be subject to risks caused by misappropriation, misuse, leakage, falsification or intentional or accidental release or loss of information maintained in the information systems and networks of our company and our vendors, including personal information of our employees and patients, and company and vendor confidential data, as could information stored in the networks or systems of our current or future collaborators. In addition, outside parties may attempt to penetrate our systems, those of our current or future collaborators or those of our vendors or fraudulently induce our personnel or the personnel of our current or future collaborators or our vendors to disclose sensitive information in order to gain access to our data and/or systems.
We may experience threats to our data and systems, including malicious codes and viruses, phishing and other cyber-attacks.
The number and complexity of these threats continue to increase over time. If a material breach of our information technology systems or those of our vendors occurs, the market perception of the effectiveness of our security measures could be harmed and our reputation and credibility could be damaged. We could be required to expend significant amounts of money and other resources to repair or replace information systems or networks.harmed. In addition, we could be subject to regulatory actions and/or claims made by individuals and groups in private litigation involving privacy issues related to data collection and use practices and other data privacy laws and regulations, including claims for misuse or inappropriate disclosure of data, as well as unfair or deceptive practices. Although we develop and maintain systems and controls designed to prevent these events from occurring, and we have a process to identify and mitigate threats, the development and maintenance of these systems, controls and processes is costly and requires ongoing monitoring and updating as technologies change and efforts to overcome security measures become increasingly sophisticated. Moreover, despite our efforts, the possibility of these events occurring cannot be eliminated entirely. As we outsource more of our information systems to vendors, engage in more electronic transactions with payors and patients, and rely more on cloud-based information systems, the related security risks will increase and we will need to expend additional resources to protect our technology and information systems. In addition, there can be no assurance that our internal information technology systems, those of our collaborators or our third-party contractors, or our consultants’ efforts to implement adequate security and control measures, will be sufficient to protect us against breakdowns, service disruption, data deterioration or loss in the event of a system malfunction, or prevent data from being stolen or corrupted in the event of a cyberattack, security breach, industrial espionage attacks or insider threat attacks which could result in financial, legal, business or reputational harm.
UseThe spread of rumors and false information, particularly through social medianetworks, and their inappropriate use, may materially and adversely impact our reputation.
We use social media to relay our official financial communications and participation in scientific congresses and other events. Unauthorized communications, such as press releases or posts on social media, purported to be issued by us, may contain information that is false or otherwise damaging and could have an adverse impact on the price of our securities. Negative or inaccurate posts or comments about us, our research and development programs, and our directors or officers could seriously damage our reputation. Tools using artificial intelligence have made disinformation easier and less costly to generate and spread, and made such information seemingly more credible.
In addition, our employees and collaborators and other third parties with whom we have business relationships may use social media and mobile technologies inappropriately, for which we may be held liable, or which could lead to breaches of data security, loss of trade secrets or other intellectual property or public disclosure of sensitive information. Such uses of social media and mobile technologies could have a material adverse effect on our reputation, business, financial condition and results of operations.
We are exposed to a number of regulatory and commercial risks related to the United Kingdom leaving the European Union.
The United Kingdom left the European Union on January 31, 2020, a development commonly known as Brexit. Given the lack of precedent in the history of the European Union, the financial, commercial, regulatory and legal consequences of the withdrawal of the United Kingdom from the European Union are unclear. The United Kingdom and the European Union have signed a EU-UK Trade and Cooperation Agreement, or TCA, which entered into force on May 1, 2021. This agreement provides details on how some aspects of the United Kingdom and European Union’s relationship will operate going forwards, however there are still many uncertainties.
Our clinical trials in the United Kingdom are subject to the requirements of the Medicines and Healthcare products Regulatory Agency or MHRA and the regulations of the EMA. For example, we plan to open new investigation sites in the United Kingdom for our ELATIVE Phase 3 trial evaluating elafibranor in PBC and potentially other clinical trials. As it relates to marketing authorizations, Great Britain will have a separate regulatory submission process, approval process and a separate national marketing authorization granted by the UK competent authorities in order to place medicinal products on the market in Great Britain. Northern Ireland will, however, continue to be covered by the marketing authorizations granted by the European Commission (EC). For example, the scope of a marketing authorization for a medicinal product granted by the EC or by the competent authorities of EU Member States will no longer encompass Great Britain (England, Scotland and Wales). If we or our potential future collaborators obtain market approval within the European Union, this market approval may not allow us to commercially market our product candidates in the United Kingdom and we or our potential future collaborators may not be in a position to obtain the required approval from the British regulatory authority. If we or our potential collaborators need to obtain additional approvals in the United Kingdom, we will have to bear additional costs which could be considerable.
The outbreak of the novel coronavirus disease, COVID-19, has adversely impacted and could continue to adversely impact our business, including our preclinical studies and clinical trials.
In December 2019, a novel strain of coronavirus disease, SARS-CoV-2, identified as COVID-19, was identified in Wuhan, China. This virus has since spread globally, including throughout the United States, across Europe and in France, where we are headquartered, and in countries where we or our current and future partners have planned or ongoing clinical trials, or where our important subcontractors – for clinical research and manufacturing of our API and drug product for elafibranor and NTZ, in particular, are located. The initial outbreak, subsequent outbreaks resulting from new variants of the disease such as Delta and Omicron, and government measures taken in response have also had a significant impact, both direct and indirect, on businesses and commerce, as worker shortages have occurred; supply chains have been disrupted; facilities have been closed and production have been suspended; and demand for certain goods and services, such as medical services and supplies, has spiked, while demand for other goods and services, such as travel, has fallen.
Strict confinement measures have been taken by the governments in the majority of countries where there has been a COVID-19 outbreak. Although as of the date of this Annual Report, some confinement measures have been lifted in some countries, there is no guarantee that governments will not take additional measures in the event there is a new outbreak of the disease or variants thereof in certain regions.
In response to the spread of COVID-19, in 2020, we made several changes to our operations, including
•temporarily suspending our planned Phase 3 study of elafibranor in PBC;
•suspending enrollment of patients or putting certain other clinical trials on hold;
•enacting remote working for certain of our employees, including most of our general administrative and finance personnel, and applying social distancing and other safety measures for employees who continue to work at our offices and in the laboratories; and
•strictly limiting business travel to that which is considered absolutely critical to our operations.
As of the date of this annual report, although most employees have returned to our offices and business travel has recommenced, the COVID-19 pandemic continues to impact operations. As the result of measures implemented in consultation with our CRO, including virtual appointments, biological evaluations performed by local laboratories and delivery of the drug candidate to the patients’ homes, to ensure the safety of participants in the ELATIVE study, the ELATIVE Phase 3 clinical trial of elafibranor in PBC was able to enroll its first patient in September 2020.
At the start of the ELATIVE trial, and considering the pandemic situation, we had estimated that enrollment in the ELATIVE study would take approximately 18 months and so far, enrollment has been broadly in-line with this estimate. However, the recent rapid expansion of the highly contagious Omicron strain of COVID has created additional complications for us in enrolling patients and in clinical trial operations generally. The rate of infection, as well as the containment measures put in place to control its growth have led to patients postponing site visits or having to be re-screened because they had fallen outside the screening window. This recent worsening of the COVID pandemic has also created significant additional administrative backlogs at sites and regulatory agencies, due to the combination of continued high volume of trials and staffing shortages. This has disproportionately impacted regions where there were already significant delays, such as Latin America. We continue to work with our CRO, trial sites and investigators to regularly revise our program execution estimations to take into account the evolution of the pandemic situation and its impact on our activities. Although we currently do not anticipate these recent complexities to substantially change the guidance related to availability of the ELATIVE topline results, we continue to assess the impacts of COVID-19 for all of our ongoing and planned clinical trials. In 2021, the COVID-19 pandemic also impacted the timing of Labcorp’s commercial launch of an LDT powered by NIS4 in the clinical care space in the United States and as a result, impacted net sales in 2021. More generally, we have observed that the COVID-19 pandemic has diverted our collaborators’ resources towards the prevention, diagnosis and treatment of COVID-19 patients, to the detriment of other activities, including our programs.
As a result of the COVID-19 pandemic, we have experienced and may continue to experience disruptions, some of which could severely impact our business, preclinical studies and clinical trials, including:
•delays or difficulties in manufacturing active pharmaceutical ingredients or drug products used in clinical trials of our product candidates, including interruption in global shipping that may affect the transport of clinical trial materials, such as investigational drug product used in clinical trials of our product candidates;
•delays or difficulties in enrolling patients in our clinical trials;
•delays or difficulties in clinical site initiation, including initiation of their activities, in particular for newly launched trials or trials in preparation, difficulties in recruiting clinical site investigators and clinical site staff;
•diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials or those of our current or future partners;
•interruption of key clinical trial activities, such as clinical trial site monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers and others;
•limitations in employee resources that would otherwise be focused on the conduct of our clinical trials or those of our current or future partners,, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people;
•delays in receiving approval from local regulatory authorities to initiate our or those of our current or future partners, planned clinical trials;
•delays in clinical sites receiving the supplies and materials needed to conduct our, or those of our current or future partners, clinical trials;
•changes in local regulations as part of a response to the COVID-19 coronavirus outbreak which may require us to change the ways in which our, or those of our current or future partners', clinical trials are conducted, which may result in unexpected costs, or to discontinue the clinical trials altogether;
•delays in necessary interactions with local regulators, in particular the FDA and EMA, other regulatory agencies, ethics committees and other important agencies and contractors due to limitations in employee resources or forced furlough of government employees;
•delay in the timing of interactions with the FDA due to absenteeism by federal employees or by the diversion of their efforts and attention to approval of other therapeutics or other activities related to COVID-19; and
•refusal of the FDA or EMA or other regulatory agencies to accept data from clinical trials in affected geographies.
In addition, the outbreak of COVID-19 could disrupt our operations or those of our partners for a significant period of time, due to absenteeism or inability to work from home by infected or ill members of management or other employees, or absenteeism by members of management and other employees who elect not to come to work due to the illness affecting others in our or their office or laboratory facilities, or due to mandated quarantines. COVID-19 could also impact members of our board of directors, resulting in absenteeism from meetings of the directors or committees of directors, and making it more difficult to convene the quorums of the full board of directors or its committees needed to conduct meetings for the management of our affairs.
The global outbreak of COVID-19 continues to rapidly evolve, in particular as a result of new variants. The extent to which COVID-19 may impact our or our partners' businesses, clinical trials and financial situation will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the ultimate geographic spread of the disease, variations in the virus, the duration of the outbreak, travel restrictions and social distancing in France, the United States and other countries, business closures or business disruptions and the effectiveness of actions taken around the world to contain and treat the disease, including the vaccination efforts currently underway in some countries. In addition, the world economy has been strongly impacted by the epidemic and many economists, governments and business leaders predict a severe impact on gross world product. We cannot predict the extent of the impact of this epidemic on the financial markets or on our stock price and as a result, on our ability to obtain additional funding if we should seek to raise additional funding.
Risks Related to Intellectual Property
If we are unable to obtain and maintain sufficient patent protection for our product candidates, or if the scope of the patent protection is not sufficiently broad, our competitors could develop and commercialize products similar or identical to ours, and our ability or that of a potential future partner to commercialize our product candidates successfully may be adversely affected.
Our success depends in large part on our ability to obtain and maintain patent protection in the United States and other countries with respect to our proprietary product candidates. If we do not adequately protect our intellectual property, competitors may be able to erode or negate any competitive advantage we may have, which could harm our business and ability to achieve profitability. To protect our proprietary position, we file patent applications in the United States and abroad related to our novel product candidates that are important to our business. The patent application and approval process is expensive and time-consuming. We may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner.
•we may not have been the first to make the inventions covered by pending patent applications or issued patents;
•we may not have been the first to file patent applications for our product candidates or the compositions we developed or for their uses;
•others may independently develop identical, similar or alternative products or compositions and uses thereof;
•our disclosures in patent applications may not be sufficient to meet the statutory requirements for patentability;
•any or all of our pending patent applications may not result in issued patents;
•we may not seek or obtain patent protection in countries that may eventually provide us a significant business opportunity;
•any patents issued to us may not provide a basis for commercially viable products, may not provide any competitive advantages, or may be successfully challenged by third parties;
•our compositions and methods may not be patentable;
•others may design around our patent claims to produce competitive products which fall outside of the scope of our patents; or
•others may identify prior art or other bases which could invalidate our patents.
Our pending patent applications cannot be enforced against third parties practicing the technology claimed in such applications unless and until patent issues. Because the issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, our patents or pending patent applications may be challenged in the courts or patent offices in the United States and abroad. For example, we may be subject to a third party preissuance submission of prior art to the U.S. Patent and Trademark Office, or USPTO, or become involved in post-grant review procedures, oppositions, derivations, reexaminations, inter partes review or interference proceedings, in the United States or elsewhere, challenging our patent rights or the patent rights of others. An adverse determination in any such challenges may result in loss of exclusivity or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. In addition, given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized.
For example, on May 15, 2019, Nashpharm, a French company, brought before the Paris High Court (Tribunal Judiciaire de Paris) a nullity action against the French part of European patent EP 2 504 005 related to the use of the drug candidate elafibranor. After the filing by Nashpharm of desist conclusions in December 2021, this action has been definitively closed by the Judge on January 11, 2022. Even if this action did not result in any limitation or nullity of our patent, it incurred nevertheless some costs and was time-consuming.
Obtaining and maintaining a patent portfolio entails significant expense and resources. Part of the expense includes periodic maintenance fees, renewal fees, various other official fees on patents and/or applications due in several stages over the lifetime of patents and/or applications, as well as the cost associated with complying with numerous procedural provisions during the patent application examination proceedings. We may not choose to pursue or maintain protection for particular inventions. In addition, there are situations in which failure to make certain payments or noncompliance with certain requirements in the patent process can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. If we choose to forgo patent protection or allow a patent application or patent to lapse purposefully or inadvertently, our competitive position or that of our current of future collaborators could suffer.
Even if our patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner. Our competitors may also seek approval to market their own products similar to or otherwise competitive with our products. Alternatively, our competitors may seek to market generic versions of any approved products by submitting Abbreviated New Drug Applications, or ANDAs, to the FDA, in which they claim that patents owned or licensed by us are invalid, unenforceable or not infringed. In these circumstances, we may need to defend or assert our patents, or both, including by filing lawsuits alleging patent infringement. In any of these types of proceedings, a court or other agency with jurisdiction may find our patents invalid or unenforceable, or that our competitors are competing in a non-infringing manner. Thus, even if we have valid and enforceable patents, these patents still may not provide protection against competing products or processes sufficient to achieve our business objectives or those of our current of future collaborators.
Legal actions to enforce our patent rights can be expensive and may involve the diversion of significant management time. In addition, these legal actions could be unsuccessfulgiven the amount of time required for the development, testing and could also result in the invalidationregulatory review of our patents or a finding that they are unenforceable. We may or may not choose to pursue litigation or other actions against those that have infringed or are currently infringing our patent rights, or used them without authorization, due to the associated expense and time commitment of monitoring these activities. If we fail to protect or to enforce our intellectual property rights successfully, our competitive position or that of our current or future collaborators could suffer, which could harm our results of operations.
Even if we have or obtain patents covering ournew product candidates, patents protecting such candidates might expire before or compositions, we may still be prevented from making, using, selling, offering for sale, or importing our productshortly after such candidates or technologies because of the patent rights of others. Others may have filed, and in the future may file, patent applications covering compositions or products that are similar or identical to ours. These filings could materially affect our ability or that of current or future collaborators to develop our product candidates or sell our products if they are approved. Because patent applications can take many years to issue and are not published for a period of time after filing, there may be currently pending applications unknown to us that may later result in issued patents that our product candidates or compositions may infringe. These patent applications may have priority over patent applications filed by us.commercialized.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time consuming and unsuccessful and issued patents covering our product candidates could be found invalid or unenforceable if challenged in court. An unfavorable outcome could harm our business.
If we initiate legal proceedings against a third party to enforce a patent covering one of our product candidates or technologies, the defendant could counterclaim that the patent covering one of our product candidates or technologies is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and unenforceability of an asserted patent or patents are common. Grounds for a validity challenge include alleged failures to meet any of several statutory requirements, including lack of novelty, obviousness, insufficient written description or non-enablement. Grounds for unenforceability assertions include allegations that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. Third parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, post-grant review or PGR and/or inter partes review and equivalent proceedings in foreign jurisdictions, such as opposition proceedings. Such proceedings could result in revocation or amendment of our patents in such a way that they no longer cover our product candidates or competitive products. Similarly, we may initiate proceedings before the Patent Trial and Appeal Board, or PTAB, of the USPTO, such as post grant review, or PGR, derivation, or inter partes review, against patents granted to third parties. This may delay us from obtaining issued patents with similar claims in the U.S.United States and may prompt additional proceedings in the USPTO against such patent or against other third party applications or patents or may consider the need or benefit of entering into a license agreement with such third party or parties in order to exploit such patent alone or together with such other third party or parties. In the event that we do not prevail or the settlement terms with the adverse party are unfavorable, or we are unable to reach an agreement on terms sufficiently favorable to us, our ability to market our product candidates may be affected or delayed. The outcome following legal assertions of invalidity and unenforceability in the PTAB or the federal courts is unpredictable. With respect to validity, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our product candidates.
Even if we establish infringement, the court may decide not to grant an injunction against further infringing activity and instead award only monetary damages, which may or may not be an adequate remedy. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, in particular, in the United States, there is a risk that some of our confidential information could be compromised by disclosure during litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our ADSs or ordinary shares. Moreover, there can be no assurance that we will have sufficient financial or other resources to file and pursue such infringement claims in the federal courts, which typically last for years before they are concluded. Even if we ultimately prevail in such claims, the monetary cost of such litigation and the diversion of the attention of our management and scientific personnel could outweigh any benefit we receive as a result of the proceedings.
In addition, if one of our patents is revoked or abandoned as a result of an adverse court decision or a settlement, we may face the risk that government, private third party payers or purchasers of pharmaceuticals products may claim damages alleging that they have over-reimbursed or overpaid for a drug. Biopharmaceutical patents and patent applications involve highly complex legal and factual questions, which, if determined adversely to us, could negatively impact our patent position.
The patent positions of biopharmaceutical companies can be highly uncertain and involve complex legal and factual questions. Typically, the development, manufacture, sale and distribution of biopharmaceutical compositions is complicated by third-party intellectual property rights to a greater extent than for the development, manufacture, sale and distribution of small molecule drugs. The interpretation and breadth of claims allowed in some patents covering biopharmaceutical compositions may be uncertain and difficult to determine, and are often affected materially by the facts and circumstances that pertain to the patented compositions and the related patent claims. The standards of the USPTO are evolving and could change in the future. Consequently, we cannot predict the issuance and scope of patents with certainty. Patents, if issued, may be challenged, invalidated or circumvented. U.S. patents and patent applications may also be subject to derivation or interference proceedings, and U.S. patents may be subject to reexamination proceedings, post-grant review and/or inter partes review at the USPTO. Foreign patents may be subject also to opposition or comparable proceedings in the corresponding foreign patent office, which could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such interference, reexamination, post-grant review, inter partes review and opposition proceedings may be costly. Accordingly, rights under any issued patents may not provide us with sufficient protection against competitive products or processes.
In addition, changes in or different interpretations of patent laws in the United States and foreign countries may permit others to use our discoveries or to develop and commercialize our technology and products without providing any compensation to us, or may limit the number of patents or claims we can obtain. The laws of some countries do not protect intellectual property rights to the same extent as U.S. laws and those countries may lack adequate rules and procedures for defending our intellectual property rights.
If we fail to obtain and maintain patent protection and trade secret protection for our product candidates, we could lose our competitive advantage and the competition we face would increase, reducing any potential revenues and adversely affecting our ability to attain or maintain profitability.
If we are sued for infringing intellectual property rights of third parties, such litigation could be costly and time consuming and could prevent or delay us from developing or commercializing our product candidates.
Our commercial success depends, in part, on our ability to develop, manufacture, market and sell our product candidates and use our technologies without infringing the intellectual property and other proprietary rights of third parties. If any third-party patents or patent applications are found to cover our product candidates or their methods of use, we may not be free to manufacture or market our product candidates as planned without obtaining a license, which may not be available on commercially reasonable terms, or at all.
There is a substantial amount of intellectual property litigation in the biotechnology and pharmaceutical industries, and we may become party to, or threatened with, litigation or other adversarial proceedings regarding intellectual property rights with respect to our product candidates, including interference proceedings before the USPTO. Third parties may assert infringement claims against us based on existing or future intellectual property rights. The outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance. The pharmaceutical and biotechnology industries have produced a significant number of patents, and it may not always be clear to industry participants, including us, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If we were sued for patent infringement, we would need to demonstrate that our product candidates, products or methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid or unenforceable, and we may not be able to do this. Proving invalidity is difficult. For example, in the United States, proving invalidity requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents. Even if we are successful in these proceedings, we may incur substantial costs and the time and attention of our management and scientific personnel could be diverted in pursuing these proceedings, which could significantly harm our business and operating results. In addition, we may not have sufficient resources to bring these actions to a successful conclusion.
If we are found to infringe a third party’s intellectual property rights, we could be forced, including by court order, to cease developing, manufacturing or commercializing the infringing product candidate or product. Alternatively, we may be required to obtain a license from such third party in order to use the infringing technology and continue developing, manufacturing or marketing the infringing product candidate. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. In addition, we could, in certain circumstances, be found liable for monetary damages, including treble damages and attorneys’ fees if we are found to have willfully infringed a patent. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could materially harm our business. Claims may also be made that we have misappropriated the confidential information or trade secrets of third parties, which could have a similar negative impact on our business.
Developments in patent law in the United States and in other jurisdictions could have a negative impact on our business.
From time to time, the U.S. Supreme Court, other federal courts, the U.S. Congress, the USPTO or similar foreign authorities may change the standards of patentability and any such changes could have a negative impact on our business. In addition, the Leahy-Smith America Invents Act, or the America Invents Act, which was signed into law in 2011, includes a number of significant changes to U.S. patent law. These changes include a transition from a “first-to-invent” system to a “first-to-file” system, changes to the way issued patents are challenged, and changes to the way patent applications are disputed during the examination process. In certain areas, these changes may favor larger and more established companies that have greater resources to devote to patent application filing and prosecution. The USPTO has developed new regulations and procedures to govern the full implementation of the America Invents Act, and many of the substantive changes to patent law associated with the America Invents Act, and, in particular, the first-to-file provisions, became effective on March 16, 2013. Substantive changes to patent law associated with the America Invents Act, or any subsequent U.S. legislation regarding patents, may affect our ability to obtain patents, and if obtained, to enforce or defend them.
Furthermore, recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances for diagnostic method claims and gene patents.
In view of these and other U.S. federal appellate cases, we cannot guarantee that our efforts to seek patent protection for our tools and biomarkers will be successful.
In May 2023, the European Commission proposed the creation of a unitary Supplementary Protection Certificate (SPC), valid in all EU countries. If this project is accepted in the future, it would enable third parties to bring a single legal action to try and obtain a decision invalidating the SPC valid in all member countries. The European Commission has also proposed a revision of pharmaceutical legislation to reduce the duration of regulatory data protection and market exclusivity for orphan drugs. If these proposals are accepted, they could reduce the duration of regulatory protection for our products.
If we do not obtain protection under the Hatch-Waxman Amendments and similar non-U.S. legislation for extending the term of patents covering each of our product candidates, our business may be materially harmed.
Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. We expect to seek extensions of patent terms for certain patents in the United States and, if available, in other countries where we are prosecuting patents and seeking approval of various products. Depending upon the timing, duration and conditions of FDA marketing approval of our product candidates, one or more of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Amendments; similarly, selected patents outside the U.S., may be eligible for supplementary protection certificate, or SPC, under corresponding legislation in the EEA and several other countries.
Depending upon the circumstances, the Hatch-Waxman Amendments permit a patent term extension of up to five years for a patent covering an approved product as compensation for effective patent term lost during product development and the FDA regulatory review process. However, we may not receive an extension if we fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. Moreover, the length of the extension could be less than we request. If we are unable to obtain patent term extension or the term of any such extension is less than what we request, the period during which we can enforce our patent rights for that product will be shortened. If this occurs, our competitors may be able to take advantage of our investment in development and clinical trials by referencing our clinical and preclinical data and launch their product earlier than might otherwise be the case.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to patent protection, because we operate in the highly technical field of development of therapies, we rely in part on trade secret protection in order to protect our proprietary technology and processes. However, trade secrets are difficult to protect. We have entered into confidentiality and intellectual property assignment agreements with our employees, consultants, outside scientific collaborators, sponsored researchers, and other advisors. These agreements generally require that the other party keeps confidential and does not disclose to third parties all confidential information developed by the party or made known to the party by us during the course of the party’s relationship with us. These agreements also generally provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us.
In addition to contractual measures, we try to protect the confidential nature of our proprietary information using physical and technological security measures. Such measures may not, for example, in the case of misappropriation of a trade secret by an employee or third party with authorized access, provide adequate protection for our proprietary information.
For example, in 2021 we filed a complaint in the U.S. District Court for the Northern District of California against CymaBay Therapeutics, Inc. (“CymaBay”). The suit alleged that CymaBay misappropriated our ELATIVE® Phase 3 clinical trial Protocol synopsis for our drug candidate elafibranor in PBC (the “Protocol synopsis”). In February 2023, we reached a settlement agreement. The settlement agreement, which is confidential, reflects that CymaBay improperly received, reviewed and circulated our Protocol synopsis upon receipt, but also that CymaBay is not using any of our trade secrets in its clinical trials. CymaBay has not admitted legal liability and we and CymaBay have agreed to resolve the litigation completely.
This example shows that the remedies we would then pursue against this type of misconduct may not be sufficient to fully protect our interests, or those of our current partners, or those of potential future partners
Our security measures may not prevent an employee or consultant from misappropriating our trade secrets and providing them to a competitor, and recourse we take against such misconduct may not provide an adequate remedy to protect our interests fully. Although we try to ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, and no such claims against us are currently pending, we may be subject to claims that we or our employees, consultants or independent contractors have used or disclosed intellectual property, including trade secrets or other proprietary information, of a former employer or other third parties. Either we or these individuals may be subject to allegations of trade secret misappropriation or other similar claims as a result of prior affiliations.
Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive, and time-consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets. Trade secrets may be independently developed by others in a manner that could prevent legal recourse by us. If any of our confidential or proprietary information, such as our trade secrets, were to be disclosed or misappropriated, or if any such information was independently developed by a competitor, our competitive position could be harmed.
We will not seek to protect our intellectual property rights in all jurisdictions throughout the world and we may not be able to adequately enforce our intellectual property rights even in the jurisdictions where we seek protection.
Filing, prosecuting and defending patents on our product candidates in all countries and jurisdictions throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States and Europe could be less extensive than those in the United States and Europe, assuming that patent rights are obtained in the United States. Competitors may use our technologies in jurisdictions where we do not pursue and obtain patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States and Europe. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing. Even if we pursue and obtain issued patents in particular jurisdictions, our patent claims or other intellectual property rights may not be effective or sufficient to prevent third parties from so competing.
In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as the federal and state laws in the United States. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, particularly in developing countries, do not favor the enforcement of patents and other intellectual property rights, especially those relating to biopharmaceuticals or biotechnologies. This could make it difficult for us to stop the infringement of our patents, if obtained, or the misappropriation of our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties for certain products. In addition, many countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries.
Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly, could put our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. In addition, changes in the law and legal decisions by courts in the United States and foreign countries may affect our ability to obtain adequate protection for our technology and the enforcement of intellectual property. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Third parties may assert ownership or commercial rights to inventions we develop.
Third parties may in the future make claims challenging the inventorship or ownership of our intellectual property. We have written agreements with collaborators that provide for the ownership of intellectual property arising from our collaborations. These agreements provide that we must negotiate certain commercial rights with collaborators with respect to joint inventions or inventions made by our collaborators that arise from the results of the collaboration. In some instances, there may not be adequate written provisions to clearly address the resolution of intellectual property rights that may arise from collaboration. If we cannot successfully negotiate sufficient ownership and commercial rights to the inventions that result from our use of a third-party collaborator’s materials where required, or if disputes otherwise arise with respect to the intellectual property developed with the use of a collaborator’s samples, we may be limited in our ability to capitalize on the market potential of these inventions. In addition, we may face claims by third parties that our agreements with employees, contractors, or consultants obligating them to assign intellectual property to us are ineffective, or in conflict with prior or competing contractual obligations of assignment, which could result in ownership disputes regarding intellectual property we have developed or will develop and interfere with our ability to capture the commercial value of such inventions. Litigation may be necessary to resolve an ownership dispute, and if we are not successful, we may be precluded from using certain intellectual property, or may lose our exclusive rights in that intellectual property. Either outcome could have an adverse impact on our business.
A dispute concerning the infringement or misappropriation of our proprietary rights or the proprietary rights of others could be time-consuming and costly, and an unfavorable outcome could harm our business.
There is significant litigation in the biopharmaceutical industry regarding patent and other intellectual property rights. We may be exposed to future litigation by third parties based on claims that our product candidates, technologies or activities infringe the intellectual property rights of others. If our development activities are found to infringe any such patents, we may have to pay significant damages or seek licenses to such patents. A patentee could prevent us from using the patented drugs or compositions. We may need to resort to litigation to enforce a patent issued to us, to protect our trade secrets, or to determine the scope and validity of third-party proprietary rights. For example, in 2020 we received an anonymous whistleblower allegation that CymaBay Therapeutics, Inc. (“CymaBay”) had improperly acquired and disclosed the protocol synopsis (“Protocol”) for our Phase 3 ELATIVE clinical trial of elafibranor in PBC. We subsequently filed a Complaint on January 15, 2021 against CymaBay in the U.S. District Court for the Northern District of California alleging that CymaBay, among other things, violated the U.S. federal Defend Trade Secrets Act and the California Uniform Trade Secrets Act when it misappropriated the Protocol. On the same day that we filed the Complaint, we sought a temporary restraining order (“TRO”) against CymaBay, and on March 12, 2021 the Court granted the TRO (which has since been converted into a preliminary injunction), finding in relevant part that we are likely to succeed on the merits of our trade secret claims. We subsequently filed two Amended Complaints with additional allegations against CymaBay, and following CymaBay’s Motion to Dismiss the Second Amended Complaint, the Court declined to dismiss the trade secret claims that are based on the Protocol as a whole, but dismissed certain other claims. Accordingly, the case will now proceed on the federal and California trade secret misappropriation claims based on the Protocol as a whole, and is currently in the discovery phase. While the outcome of the litigation still remains uncertain, we intend to pursue it vigorously.
From time to time, we may hire scientific personnel or consultants formerly employed by other companies involved in one or more areas similar to the activities conducted by us. Although we try to ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, and no such claims against us are currently pending, we may be subject to claims that we or our employees, consultants or independent contractors have used or disclosed intellectual property, including trade secrets or other proprietary information, of a former employer or other third parties. Either we or these individuals may be subject to allegations of trade secret misappropriation or other similar claims as a result of prior affiliations.
If we become involved in litigation, it could consume a substantial portion of our managerial and financial resources, regardless of whether we win or lose. We may not be able to afford the costs of litigation. Any adverse ruling or perception of an adverse ruling in defending ourselves against these claims could have a negative impact on our cash position. Any legal action against us or our collaborators could lead to:
•payment of damages, potentially treble damages, if we are found to have willfully infringed a party’s patent rights;
��•injunctive or other equitable relief that may effectively block our ability to further develop, commercialize, and sell products; or
•us having to enter into license arrangements that may not be available on commercially acceptable terms, if at all.
Any of these outcomes could hurt our cash position and financial condition and our ability to develop and commercialize our product candidates.
Moreover, our commercial success depends, in part, on our ability to develop, manufacture, market and sell our product candidates and use our technologies without infringing the intellectual property and other proprietary rights of third parties. If any third-party patents or patent applications are found to cover our product candidates or their methods of use, we may not be free to manufacture or market our product candidates as planned without obtaining a license, which may not be available on commercially reasonable terms, or at all.
If our trademarks and trade names are not adequately protected, we may not be able to build name recognition in our markets of interest.
Our registered or unregistered trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names, which we will need to build name recognition by potential collaborators or customers in our markets of interest. Over the long term, if we are unable to establish name recognition based on our trademarks and trade names, we may not be able to compete effectively.
Risks Related to Legal and Other Compliance Matters
We are subject to transparency, ethics and healthcare laws and regulations that may require substantial compliance efforts and could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings, among other penalties.
Healthcare providers and others in the healthcare and pharmaceutical sector will play a primary role in the clinical development and potential regulatory approval or certification of our product candidates and their recommendation and prescription, if approved.approved or CE marked. Our arrangements with them and third party payors as well as our activities expose us to broadly applicable federal and state fraud and abuse and other healthcare laws, which may restrict these arrangements and relations through which we research and develop our products, and if approved or CE marked, we or our current or future collaborators will market and distribute them. These laws may thus impact, among other things, our research, development, proposed sales, marketing and education programs of our product candidates that obtain marketing approval. Restrictions under applicable U.S. federal, state and non-U.S. healthcare laws and regulations include, but are not limited to, the following:
•the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowinglyfraud and willfully soliciting, offering, receiving or providing remuneration, including any kickback, bribe or rebate, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase or lease, order or recommendation of, any item, good, facility or service, for which payment may be made under federal healthcare programs such as Medicare and Medicaid;
•U.S. federal civil and criminal false claimsabuse laws, including the civil False Claims Act, which can be enforced through civil whistleblower or qui tam actions,federal anti-kickback and civil monetary penaltiesfalse claims laws; healthcare data privacy and security laws, impose penalties, against individuals or entities for, among other things, knowingly presenting, or causing to be presented, claims for payment that are false or fraudulent or making a false statement to avoid, decrease, or conceal an obligation to pay money to the federal government;
•such as the U.S. federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created additional federal criminal statutes that impose criminalHIPAA; and civil liability for, among other things, executing or attemptingtransparency laws related to execute a scheme to defraud any healthcare benefit program or knowingly and willingly falsifying, concealing or covering up a material fact or making false statements relating to healthcare matters;
•HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and its implementing regulations, which impose certain requirements on certain covered healthcare providers, health plans, and healthcare clearinghouses and their respective business associates and covered subcontractors that perform functions or activities that involve HIPAA Protected Health Information on their behalf, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information;
•U.S. federal transparency requirements under the Physician Payments Sunshine Act, enacted as part of the ACA, that require applicable manufacturers of covered drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, with specific exceptions, to track and annually report to the CMS payments andand/or other transfers of value providedmade to physicians (defined to include doctors, optometrists, podiatrists and chiropractors)other healthcare professionals and teaching hospitals, including the federal Physician Payments Sunshine Act. Many states have similar laws that may differ from each other and certain ownership and investment interests held by physicians or their immediate family members. Beginningfederal law in 2022, applicable manufacturers also will be required to report information regarding payments and transfers of value provided during the previous year to physician assistants, nurse practitioner, clinical nurse specialists, anesthesiologist assistants, certified nurse anesthetists and certified nurse-midwives;
•analogous state or non-U.S. laws and regulations, such as statesignificant ways, thus complicating compliance efforts. For example, states have anti-kickback and false claims laws that may be broader in scope than analogous federal laws and may apply regardless of payor. In addition, state data privacy laws that protect the security of health information may differ from each other and may not be preempted by federal law. Moreover, several states have enacted legislation requiring pharmaceutical manufacturers to, among other things, establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales and marketing activities, report information related to drug pricing, require the registration of sales representatives, and prohibit certain other sales and marketing practices.
Outside the United States, interactions between pharmaceutical companies and health care professionals are also governed by strict laws, such as national anti-bribery laws of European countries, national sunshine rules, regulations, industry self-regulation codes of conduct and physicians’ codes of professional conduct. These laws may include the French “Bertrand Law”, French Ordinance n° 2017-49 of January 19, 2017 and Decree No. 2020-730 of June 15, 2020 relating to benefits offered by persons manufacturing or marketing health products or services, and the UK’s Bribery Act 2010, which may apply to items or services reimbursed by any third-party payor, including commercial insurers, state marketing and/or transparency laws applicable to manufacturers or any company providing services related to their products that may be broader in scope than the federal requirements, laws that require biopharmaceutical companiesrequirements. Failure to comply with the biopharmaceutical industry’s voluntary compliance guidelines, laws requiring manufacturers to declare information related to payment and other gratification to physicians and other healthcare providers or to publicly divulge the expenses related to marketing products and communicate information on their price, and laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect as HIPAA, thus complicating compliance efforts;
•the Foreign Corrupt Practices Act, or FCPA, prohibits any U.S. individual or business from paying, offering, or authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party, or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also requires companies whose securities are listed in the United States to comply with accounting provisions requiring the company to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations. Activities that violate the FCPA, even if they occur wholly outside the United States, canthese requirements could result in criminal and civilreputational risk, public reprimands, administrative penalties, fines imprisonment, disgorgement, oversight, and debarment from government contracts. The FCPA presents particular challenges for the pharmaceutical industry since, in many countries, hospitals are managed by the government, and their physicians and other employees are considered foreign public agents. As such, some payments to hospitals related to clinical trials and other work have been regarded as irregular payments to foreign agents and lead to enforcement action on the basis of the FCPA; and.
•the equivalent anticorruption laws in foreign countries, such as the French law of December 9 2016 or the UK Bribery Act of 2010 that may also be invoked under similar circumstances related to corrupt practices.imprisonment.
Ensuring that our business arrangements with third parties comply with applicable healthcare laws and regulations will likely be costly. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations were found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, disgorgement, imprisonment, possible exclusion from government funded healthcare programs, such as Medicare and Medicaid or comparable foreign programs, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could substantially disrupt our operations. If the physicians or other providers or entities with whom we expect to do business are found not to be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.programs and their professional orders. Although an effective compliance program can mitigate the risk of investigation and prosecution for violations of these laws, the risks cannot be entirely eliminated. Moreover, achieving and sustaining compliance with applicable federal and state privacy, security, and fraud laws, and foreign equivalents, may prove costly. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business.
We are subject to laws and regulations related to data privacy, both in the United States and the European Union whose breach might have a significant negative impact on our activities.
We, and our service providers, receive, process, store and use personal information and other data about our clinical trial participants, employees, partners and others. We, and our service providers, must comply with numerous foreign and domestic laws and regulations regarding privacy and the storing, sharing, use, processing, disclosure, security, and protection of personal information and other data, such as information that we collect about patients and healthcare providers in connection with clinical trials in the EEA, the United States and elsewhere. Third parties (principally CROs during clinical trials) manage on our behalf a significant part of the personal data we may use.
For example, HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and its respective implementing regulations imposes certain requirements on covered entities relating to the privacy, security, and transmission of certain individually identifiable health information, known as protected health information. Among other things, HITECH, through its implementing regulations, makes HIPAA’s security standards and certain privacy standards directly applicable to covered subcontractors and business associates, defined as a person or organization, other than a member of a covered entity’s workforce, that creates, receives, maintains, or transmits protected health information on behalf of a covered entity for a function or activity regulated by HIPAA. HITECH also strengthened the civil and criminal penalties that may be imposed against covered entities, business associates, and individuals, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions. In addition, other federal and state laws may govern the privacy and security of health and other information in certain circumstances, many of which differ from each other in significant ways and may not be preempted by HIPAA, thus complicating compliance efforts.
In May 2018 the European Union General Data Protection Regulation (EU) 2016/679, or GDPR, went into effect in the EEA. The GDPR imposes stringent data protection requirements for processing the information of individuals in (i) the EEA and (ii) the United Kingdom as the GDPR continues to form part of law in the United Kingdom, or the UK GDPR.EEA. The GDPR increases our obligations with respect to clinical trials conducted in Europe (including the EEA United Kingdom and Switzerland) by expressly expanding the definition of personal data to include “pseudonymized” or key-coded data and requiring changes to informed consent practices and more detailed notices for clinical trial subjects and investigators.
The GDPR also provides for more robust regulatory enforcement and greater penalties for noncompliance than previous data protection laws, including fines of up to €20 million or 4% of global annual revenue of any noncompliant company for the preceding financial year, whichever is higher. In addition to administrative fines, a wide variety of other potential enforcement powers are available to competent supervisory authorities in respect of potential and suspected violations of the GDPR, including extensive audit and inspection rights, and powers to order temporary or permanent bans on all or some processing of personal data carried out by non-compliant actors. The GDPR also confers a private right of action on data subjects and consumer associations to lodge complaints with supervisory authorities, seek judicial remedies, and obtain compensation for damages resulting from violations of the GDPR.
European Union data protection laws, including the GDPR, generally restrict the transfer of personal data from Europe, including the EEA United Kingdom and Switzerland, to the United States and most other countries unless the parties to the transfer have implemented specific safeguards to protect the transferred personal data. The current mechanisms that may be used to transfer personal data from the EEA to the United States in compliance with law are subject to legal challenges, and there is no assurance that we can satisfy or rely on these measures to lawfully transfer personal data to the United States.
If there is no lawful manner for us to transfer personal data from the EEA, the UK or other jurisdictions to the United States, or if the requirements for a legally-compliant transfer are too onerous, we could face significant adverse consequences, including the interruption or degradation of our operations, the need to relocate part of or all of our business or data processing activities to other jurisdictions at significant expense, increased exposure to regulatory actions, substantial fines and penalties, the inability to transfer data and work with partners, vendors and other third parties, and injunctions against our processing or transferring of personal data necessary to operate our business. Additionally, companies that transfer personal data out of the EEA and by virtue of the UK GDPR into other jurisdictions, particularly to the United Kingdom, in a broadly uniform manner. However,States, are subject to increased scrutiny from regulators, individual litigants and activist groups. Some European regulators have ordered certain companies to suspend or permanently cease certain transfers out of Europe for allegedly violating the GDPR’s cross-border data transfer limitations.
The GDPR provides that EEA countries may make their own further laws and regulations to introduce specific requirements related to the processing of “special categories of personal data,” including personal data related to health, biometrichealth. In addition, in France, the conduct of clinical trials is subject to compliance with specific provisions, which may include the filing of compliance undertakings with “reference methodologies” (such as the MR-001) adopted by the French data used for unique identification purposes and genetic information– in the United Kingdom, the United Kingdom Data Protection Act 2018 complements the UK GDPR in this regard.protection authority. This fact could expose us to twomultiple parallel regimes or may lead to, greater divergence on the law that applies to the processing of such data types across the EEA and/or United Kingdom, compliance with which, as and where applicable, may increase our costs and could increase our overall compliance risk. Such country-specific regulations could also limit our ability to collect, use and share data and/or could cause our compliance costs to increase, ultimately having an adverse impact on our business, and harming our business and financial condition.
Additionally, other countries outside of the EEA, including Switzerland, the UK and China, have enacted or are considering enacting similar cross-border data transfer restrictions and laws requiring local data residency, which could increase the cost and complexity of delivering our services and operating our business.
The global data protection landscape is rapidly evolving, and we expect that there will continue to be newNew and proposed laws, regulations, andpolicies, codes of conduct, industry standards and legal obligations concerning privacy, data protection and information security, and we cannot yet determine the impact that such future laws, regulations and standards may have on our business. We strive to comply with all applicable requirements and obligations. However, new laws, policies, codes of conduct and legal obligations may arise, continue to evolve, be interpreted and applied in a manner that is inconsistent from one jurisdiction to another and conflict with one another. Moreover, we cannot yet determine the impact that they will have on our business.
Any failure or perceived failure by us or third parties working on our behalf to adequately comply with applicable laws and regulations, any privacy and data security obligations pursuant to contract or pursuant to our stated privacy or security policies or obligations to third parties may result in governmental enforcement actions (including fines, penalties, judgments, settlements, imprisonment of company officials and public censure), civil claims, litigation, damage to our reputation and loss of goodwill, any of which could have a material adverse effect on our business, operations and financial performance. With substantial uncertainty over the interpretation and application of these laws, regulations and other obligations, we may face challenges in addressing their requirements and making necessary changes to our policies and practices, and may incur significant costs and expenses in our efforts to do so.
We cannot assure that our CROs or other third-party service providers with access to our or our suppliers’, manufacturers’, trial participants’ and employees’ sensitive data in relation to which we are responsible will not experience data security incidents, which could have a corresponding adverse effect on our business, financial condition, results of operations and prospects, including putting us in breach of our obligations under privacy laws and regulations. Any actual or perceived failure by us to comply with federal, state or foreign laws, rules or regulations, industry standards, contractual or other legal obligations, or any actual, perceived or suspected cybersecurity incident, whether or not resulting in unauthorized access to, or acquisition, release or transfer of personal data, may result in enforcement actions and prosecutions, private litigation, significant fines, penalties and censure, claims for damages by customers and other affected individuals, regulatory inquiries and investigations or adverse publicity and could cause our customers to lose trust in us, any of which could adversely affect our business, financial condition, results of operations and prospects.Our employees may engage in misconduct or other improper activities, including violating applicable regulatory standards and requirements or engaging in insider trading, which could significantly harm our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with legal requirements or the requirements of FDA, EMA and other government regulators, provide accurate information to applicable government authorities, comply with fraud and abuse and other healthcare laws and regulations in the United States and abroad, report financial information or scientific and medical data accurately or disclose unauthorized activities to us.
In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the falsification or improper use of, including trading on, information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We strive to maintain an ethical corporate culture and have adopted a Code of Business Conduct and Ethics and have a training program in place, but it is not always possible to identify and deter employee misconduct, and the precautions we take to train employees and detect and prevent this activity may be ineffective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Product liability and other lawsuits could divert our resources, result in substantial liabilities, reduce the commercial potential of our product candidates and harm our reputation.
The risk that we may be sued on product liability claims is inherent in the development and commercialization of biopharmaceutical and diagnostic products that are intended to be tested and evaluated on humans in an initial phase, then commercialized. Side effects of, or manufacturing defects in, products that we develop could result in the deterioration of a patient’s condition, injury or even death. This risk is particularly important where patients suffer from life-threatening illnesses, such as ACLF. For example, our liability or that of our current or future collaborators could be sought after by patients participating in the clinical trials in the context of the development of the therapeutic or diagnostic products tested and unexpected side effects resulting from the administration of these products.
Once a product is approved for sale and commercialized, the likelihood of product liability lawsuits increases. Criminal or civil proceedings might be filed against us by patients, regulatory authorities, biopharmaceutical companies and any other third party using or marketing our products. These actions could include claims resulting from acts by our collaborators, licensees, service providers and subcontractors, over which we have little or no control. These lawsuits may divert our management from pursuing our business strategy and may be costly to defend. In addition, if we are held liable in any of these lawsuits, we may incur substantial liabilities and may be forced to limit or forgo further commercialization of the affected products, which may harm our reputation. Patients may not follow warnings identifying potential known side effects, including some patients who should not be using our drug candidates.
We maintain product liability insurance coverage for our clinical trials at levels which we believe are appropriate for our clinical trials and at levels granted by insurers to biopharmaceutical companies like us. Nevertheless, our insurance coverage may be insufficient to reimburse us for any expenses or losses we may suffer. In addition, insurance coverage has become more and more expensive, and in the future, we may not be able to obtain or maintain sufficient insurance coverage at an acceptable cost or for sufficient amounts to otherwise protect against potential product or other legal or administrative liability claims by us or our current or potential collaborators. A successful liability claim against our products may lower the value of our stock, and if the decision awards damages that exceed our insurance coverage, might reduce our available funds and have an unfavorable effect on our activities. It could notably prevent or inhibit the commercial production and sale of any of our product candidates that receive regulatory approval. Product liability claims could also harm our reputation, which may adversely affect our ability to commercialize our products successfully.
Risks Related to our Financial Position and Capital Needs
Currently, we have no products approved for commercial sale, and to date we have not generated any significant recurring revenue from product sales. As a result, our ability to sustainably reduce our losses, reach lasting profitability, as a result of such types of revenue, and maintain our shareholders equity on our own is unproven, and we may never achieve or sustain profitability.
Last year, weWe recorded a net profitloss of €67,259€28,894 thousand for the year ended December 31, 2021, mainly due to operating income resulting from the one-time upfront payment received from Ipsen as part of the license2023 and collaboration agreement signed in December 2021 and the recognition of a redemption bonus resulting from the renegotiation of our convertible bond loan in January 2021. However, we have regularly recorded losses during previous financial years, in particular a net loss of €101,221€23,719 thousand for the year ended December 31, 2020, due in particular to2022. Other than the low amountyear ended December 31, 2021, we have a history of our operating income
recorded losses during prior years.We have never generated any profits from the sale of approved products and we do not expect to become profitable from such sales in the foreseeable future. In 2020, in particular,Although the disappointing intermediate results of the RESOLVE-IT trial make profitability even less likely in the foreseeable future. More recently, although thecollaboration and license and collaboration agreement entered into with Ipsen in 2021 includes the prospect of receiving milestones and royalties in the event of, among other things, the success of the ELATIVE trial and the marketing of elafibranor in PBC and future commercial success, there is no assurance that this will occur.occur on the timelines we expect or ever.
In recent years, our most significant revenue has resulted from one-time upfront payments received in 2019 under our license agreement with Terns Pharmaceuticals and in 2021 and 2023 under our license agreement and our transition service agreements with Ipsen. To these are added, to a lesser extent, the reimbursements of our research tax credit or CIR, which alone have the character of significant recurring operating income, although our ability to continue to benefit from the CIR depends on our ability to continue to meet the criteria and decisions of French policy makers with respect to the scope or rate of the CIR benefit (see Note 11- "Income Tax" to the financial statements for the year ended December 31, 2023). Revenues from our agreements with Labcorp/Covance and Q2 for the use of our NIS4NIS4® diagnostic technology and its improvements have so far been insignificant. Their eventual growth will depend on many external factors, including the market availability and commercialization of a treatment for NASH, which remains uncertain. MASH. However, these revenues will never be of the same order as those that could result from the eventual commercialization of our drug candidates, and will never enable us to be profitable on their own.
Historically, we have also received funding from co-research alliances with other pharmaceutical companies, although we do not currently have any such alliances in place.
At the same time, we plan to continue to incur significant expenses for the development of some of our existing product candidates and new product candidates for which we acquire licensing rights, or preparation of the marketing of such products. We have devoted almost all of our resources to our research and development projects related to our drug candidates, to our NIS4 program, and at to a lesser extentproportion to our NIS4® program and to providing general and administrative support for our operations, protecting our intellectual property and engaging in activities to prepare for the potential commercialization of our drug candidates and an IVD powered by NIS4.NIS4® or its variations. In addition, during the regulatory development process for some of our drug candidates and for IVD tests using our NIS4NIS4® technology or its variations, our operating costs may increase, particularly if the FDA, EMA or European CommissionEC requires studies or preclinical studies or clinical trials additional to those already planned, or, if a delay occurs in the realization of our preclinical studies or clinical trials or, more generally, in the development of one of our products.
As a result, we expect to continue to incur losses for the foreseeable future, and we expect these losses to increase as we continue our development of, and seek regulatory approvals with our current or future partners, as the case may be, for elafibranor in PBC and an IVD powered by NIS4.NIS4® or its variations.
One of the potential consequences of such losses, and which we experienceexperienced at December 31, 2020, is the inability to maintain the amount of our equity at a level at least half of our share capital. As a result, and in accordance with Article L.225-248 of the French Commercial Code, we were required to submit to our June 30, 2021 general meeting a resolution to decide to continue our activities. This resolution was approved by our shareholders in June 2021, and we were able to reconstitute positive shareholders' equity at least equal to half of the share capital at June 30, 2021 and further reinforce our share capital at December 31, 2021 due to the agreement signed with Ipsen and their equity investment in December 2021, and therefore a third party is no longer able to sue to dissolve the company on these grounds. However, we could still face this situation again in the future depending on the development of our product candidates, in particular if elafibranor does not receive regulatory approval in PBC, and we are unable to realize expected revenues from the Phase 3 ELATIVE trial is unsuccessful.potential success of elafibranor in PBC.
Our ability to be profitable in the future will depend on our ability and that of our current or future collaborators to obtain marketing approval for and commercialize our product candidates, particularly our lead product candidate, elafibranor, and the NASHnext LDT or an IVD powered by NIS4 for clinical care.elafibranor.
Our ability to be profitable in the future will depend on our ability and that of our current or future collaborators to obtain marketing approval for and commercialize our product candidates, particularly our lead product candidate, elafibranor and the NASHnextelafibranor. The success of NASHNext® LDT commercialized by Labcorp powered by NIS4NIS4® technology, or anby Q2, or a future IVD powered by NIS4NIS4® or its improvements for clinical care.care will not on their own enable us to be profitable. We or our partners may not be successful in our or their efforts to obtain such approval and to commercialize the products.
Obtaining marketing approval will require us or our current or future collaborators to be successful in a range of challenging activities, including:
•obtaining positive results in preclinical studies and clinical trials;
•regulatory bodies determining that clinical data are sufficient, without further clinical data, to support an application for approval, whether or not conditional or accelerated;
•obtaining approval to market elafibranor;elafibranor and our other product candidates;
•obtaining additional positive results in our or our partners’ formal validation studies required to commercialize a test powered by NIS4NIS4® or its improvements for clinical care;care that would allow an IVD test to be developed and approved for diagnosing MASH patients;
•expanding our manufacturing of commercial supply for elafibranor;our licensed product candidates;
•establishing sales, marketing and distribution capabilities to effectively market and sell elafibranor and NASHnext or IVD powered by NIS4 in the United States, Europe and in other territories;our drug candidates;
•market acceptance by patients and the medical community of elafibranor;elafibranor and our other product candidates;
•market acceptance by patients and the medical community of an LDT or IVD powered by NIS4NIS4® as a diagnostic complement to liver biopsy for clinical care; and
•negotiating and securing coverage and adequate reimbursement from third-party payors for elafibranor and an LDT or IVD powered by NIS4;NIS4® or its improvements and our other product candidates.
•expanding our contract manufacturingWe may also carry out preparatory activities for the commercial supplyfuture commercialization of some of our product candidates, in order to gain a better understanding of how doctors treat and diagnose their patients, without deriving any benefit from them, particularly in the manufacturing under licenseabsence of subsequent approval. Furthermore, as most of the therapeutic areas for which we are targeting our product candidates are characterized by medical needs that remain largely unsatisfied, there is considerable uncertainty as to the level of adoption of future treatments and diagnostic kit accompanying the potential commercialization of an IVD poweredtools by NIS4 for clinical care.patients and healthcare professionals, as well as third-party payers.
Even if we or our collaborators receive marketing approvals for our product candidates and commence our commercial launch, we may not be able to generate significant revenues in the near term. We cannot foresee if our product candidates will ever be accepted as a therapytherapies in PBCtheir designated indications eventually resulting in sustained revenues and it may take the passage of a significant amount of time to generate significant sustained revenues even if elafibranor becomesour product candidates become accepted as a therapytherapies in PBC.their designated indications.
MASH is currently an under-diagnosed disease, and we believe that an LDT or IVD powered by NIS4NIS4® or its improvements will facilitate the identification of patients with NASHMASH and fibrosis who may be eligible for therapeutic intervention. However, NASHMASH is also a disease with no approved drug therapy. As such, there is significant uncertainty in the degree of market acceptance that future treatments or diagnostic tools will have among NASHMASH patients and their healthcare providers as well as third-party payors. If an IVD powered by NIS4NIS4® or its improvements does not obtain marketing authorization or is unable to be commercialized, we, or our collaborators, may not be able to generate sufficient test volume to generate significant revenues. Even if an IVD powered by NIS4® or its improvements were approved, revenues from that IVD alone would not be sufficient alone for us to be profitable.
If elafibranor, NASHnextNASHNext® or an IVD powered by NIS4NIS4® or its improvements or any of our other product candidates fails in preclinical studies or clinical trials or do not gain regulatory approval, or do not achieve market acceptance, we may never become profitable. Our net losses have had, and will continue to have, an adverse effect on our shareholders’ equity and working capital. Because of the numerous risks and uncertainties associated with pharmaceutical and diagnostic product development and commercialization, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. The amount of future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues, including from licensing agreements with current or future partners.
We will require substantial additional funding to develop and commercialize our products,drug candidates, if approved, as well as to reinforce our pipeline, which may not be available to us, or to our current or future partners on acceptable terms, or at all, and, if not so available, may require us or them to delay, limit, reduce or cease our operations.
We are currently advancing elafibranor through clinical development in PBC and our otherOur drug candidates throughare in preclinical or clinical or preclinical development. Additionally, we are also planning formal validation studies of an IVD powered by NIS4 in preparation for submitting the test for marketing authorization for clinical care. Developing pharmaceutical and diagnostic products, including conducting preclinical studies and clinical trials, along with obtaining necessary validation, is expensive.
Subject to obtaining regulatory approval of any of our drug candidates or an IVD powered by NIS4,NIS4® or its improvements, we or our current or future collaborators expect to incur significant pre-marketing and commercialization expenses for product sales, marketing, manufacturing and distribution. We anticipate incurring significant expenses in connection with our planned commercialization of an IVD powered by NIS4, along with an increase in our product development, scientific, commercial and administrative personnel and expansion of our facilities and infrastructure in the United States, France and other countries. We also expect to incur additional costs associated with operating as a public company in the United States and further plan on expanding our operations in the United States, Europe and in other territories. We could continue to require substantial additional capital in connection with our continuing operations, in particular to expand our pipeline, and to continue our clinical development and pre-commercialization activities.
We could therefore still have significant needs in terms of additional funds to pursue our activities, particularly if the revenues we expect to receive under and pursuant to our licensing-out agreements are lower than expected, or if we no longer receive any, and/or if we further strengthen our current portfolio of product candidates and programs, and consequently our preclinical and clinical development activities and, where applicable, pre-commercialization and commercialization.
In addition, access, in particular under acceptable conditions, to necessary financing is subject to contextual factors affecting the financial markets, investors and potential lenders.lenders including certain unfavorable geopolitical circumstances impact by the conflict between Russia and Ukraine, which are deteriorating and could further deteriorate such access and conditions. In addition, our convertible bond contract initially issued on October 16, 2017 contains customary restrictive covenants, some of which limit, but generally do not exclude, the creation of new guarantees on our assets and the incurring of additional indebtedness.
Because successful development of our drug candidates and diagnostic program is uncertain, we are unable to estimate the actual funds required to complete the research and development and commercialization of our products under development.
Our stock price may never reach a price at which certain bondholders will deem conversion economically viable, in which case we would need to repay the nominal amount at maturity in October 2025. The terms of our convertible bonds require us to meet certain operating covenants, and if we fail to comply with those covenants the bondholders would be able to accelerate our repayment obligations. Additionally, theThe conversion of some or all of our bonds into ordinary shares would dilute the ownership interests of existing shareholders.
On January 29, 2021, we amended the terms and conditions of our convertible bonds initially issued in October 2017, mainly to extend the maturity by an additional three years, from October 16, 2022 to October 16, 2025, and increase the conversion ratio from one (1) share per bond to 5.5 shares for one bond, i.e., an implicit conversion price of €5.38 per share instead of €29.60. In addition, we carried out a partial repurchase of 2,895,260 convertible bonds, representing 48% of the outstanding bonds.bonds, resulting in €94.3 million nominal amount of bonds remaining outstanding on January 29, 2021 (compared to €180 million nominal amount initially). Following the closing of the transaction, we received conversion requests covering 1,262,159 convertible bonds. As of the date of this annual report, 1,923,662 convertible bonds are outstanding, representing a nominal amount of €56,940 thousand (versus €180,000 thousand initially). We cannot guarantee that additional conversion will take place, or that only part of the remaining bonds will be converted, before the maturity of this loan. As of the date of this Annual Report, our stock price remains below €5.38, which is the theoretical conversion price of the OCEANEs. It is possible that if our stock price does not reach a price at which the bondholders will deem conversion economically viable, we will be required to repay the nominal amount at maturity in October 2025.
In addition, in 2021 we contracted three bank loans, for a total nominal amount of €15,250 thousand, including two loans guaranteed up to 90% by the French State (PGE) subscribed respectively in June and July 2021 ( initial(initial maturities of one year with options to stagger repayments up to six years), supplemented by a subsidized loan taken out in November 2021 (repayable in six years).
Our ability to repay these loans at maturity, and in particular our convertible bond due October 2025, depends in part on our future performance, which is subject to the success of our research and development programs, the ability of our partners and future partners to successfully commercialize our products, and future operations, as well as on economic, financial and competitive factors that are beyond our control. In addition, we may be required to incur additional debt in the future to meet our additional financing needs. . Even if we are permitted by the terms and conditions of the convertible bonds, or our other bank loans, to incur additional debt or to take other measures with regard to incurring new debt, the terms of these loan could reduce our ability to repay new debts at maturity.
The agreement governing the bonds contains customary negative covenants and events of default. The negative covenants include restrictions on creating other liens on our assets, incurring certain additional indebtedness and engaging in certain mergers or acquisitions. If we default under the agreement governing the bonds, the bondholders may accelerate all of our repayment obligations, which would significantly harm our business and prospects and could cause the price of our ordinary shares to decline.
Finally, the conversion of some or all of our currently outstanding convertible bonds into ordinary shares would dilute the ownership interests of existing shareholders, including holders of our ADSs. Any sales in the public market of the ordinary shares issuable upon such conversion or any anticipated conversion of our convertible bonds into ordinary shares could adversely affect prevailing market prices of our ordinary shares or ADS and limit our ability to raise funds through capital raises. In addition, since 2016, we have set up several stock option plans, free allocation of free shares and stock warrants, many of which are still outstanding. We may in the future allocate or issue new equity-linked instruments, including convertible bonds or equity-linked compensation, the vesting and/or exercise of which could further dilute the ownership interests of shareholders, including holders of ADSs.
We have carried out a specific review of our liquidity risk and consider that we will be able to meet our maturities for the next 12 months. As of December 31, 2021, the Group has €263.22023, we had €77.8 million, in cash and cash equivalents and other financial assets (as(€136.0 million as of December 31, 2020: €172.5 million)2022). In view of these amounts as of December 31, 2021,2023, and in light of the renegotiation of the convertible bonds in January 2021, including the extension of their maturity, we do not consider that we are exposed to a short-term liquidity risk. In particular, we believe that the amount of cash, cash equivalents and other current financial instrumentsassets and future revenues we may receive from our licensing agreements is sufficient to ensure our financing, in view of its projects and current obligations, over the next twelve months.
Our failure to maintain certain tax benefits applicable to French biopharmaceutical companies may adversely affect our results of operations.
As a French biopharmaceutical company, we have benefited from certain tax advantages, including, for example, the French Research Tax Credit, or CIR (Crédit d'Impôt Recherche), which is a French tax credit aimed at stimulating research and development. The CIR can be offset against French corporate income tax due and the portion in excess, if any, may be refunded. The CIR is calculated based on our claimed amount of eligible research and development expenditures in France and was €5.3€5.6 million for the year ended December 31, 2021.2023. We believe, due to the nature of our business operations, that we will continue to be eligible to receive the CIR tax credit. However, if the French Parliament decides to eliminate, or to reduce the scope or the rate of, the CIR benefit, either of which it could decide to do at any time, our results of operations could be adversely affected.
Risks Related to Ownership of Our Ordinary Shares and ADSs and Our Status as a Non-U.S. Company with Foreign Private Issuer Status
The market price of our equity securities is particularly volatile and may decline regardless of our operating performance.
The trading price for our ADSs and ordinary shares has fluctuated, and is likely to continue to fluctuate, substantially. The stock market in general and the market for biopharmaceutical companies in particular have experienced extreme volatility that has often been unrelated to the operating performance ofcof particular companies. As a result of this volatility, investors may not be able to sell their ADSs or ordinary shares at or above the price originally paid for the security. The market price for our ADSs and ordinary shares may be influenced by many factors, including:
•announcements of clinical trial results;
•actual or anticipated fluctuations in our financial condition and operating results;
•actual or anticipated changes in our growth rate relative to our competitors;
•competition from existing products or new products that may emerge;
•announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures, collaborations, or capital commitments;
•failure to meet or exceed financial estimates and projections of the investment community or that we provide to the public;
•issuance of new or updated research or reports by securities analysts;
•fluctuations in the valuation of companies perceived by investors to be comparable to us;
•share price and volume fluctuations attributable to inconsistent trading volume levels of our shares;
•additions or departures of key management or scientific personnel;
•lawsuits threatened or filed against us, including securities litigation, disputes or other developments related to proprietary rights, including patents, litigation matters, and our ability to obtain patent protection for our technologies;
•changes to coverage policies or reimbursement levels by commercial third-party payors and government payors and any announcements relating to coverage policies or reimbursement levels;
•announcement or expectation of additional debt or equity financing efforts;projects;
•sales of our ordinary shares or ADSs by us, our insiders or our other shareholders; and
•general economic and market conditions.
These and other market and industry factors may cause the market price and demand for our ordinary shares and ADSs to fluctuate substantially, regardless of our actual operating performance, which may limit or prevent investors from readily selling their ordinary shares or ADSs and may otherwise negatively affect the liquidity of the trading market for our ordinary shares and ADSs.
The dual listing of our ordinary shares and our ADSs may adversely affect the liquidity and value of our ordinary shares and ADSs.
Our ADSs are listed on the Nasdaq Global Select Market, and our ordinary shares trade on Euronext Paris. We cannot predict the effect of this dual listing on the value of our ADSs and ordinary shares. However, the dual listing of our ADSs and ordinary shares may dilute the liquidity of these securities in one or both markets and may adversely affect the trading market or price for our ADSs and ordinary shares. In the past, there been less liquidity for our ADSs trading on the Nasdaq Global Select Market as compared to trading for our ordinary shares trading on Euronext Paris.
We are currentlyhave been the subject of a securities class action litigation and may become subject to additional litigation, which could harm our business and financial condition.
Historically, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies have experienced significant share price volatility in recent years. We may have actions brought against us by shareholders relating to past transactions, changes in our stock price or other matters. For example, in May 2020, following our announcement that elafibranor had not achieved the primary or key secondary endpoints of the RESOLVE-ITRESOLVE-IT® trial, a purported shareholder class action complaint was filed in state court in the Commonwealth of Massachusetts, naming us, our board of directors and certain members of our senior management as defendants, alleging that we made materially misleading statements about the development of elafibranor in connection with our U.S. initial public offering in violation of U.S. federal securities laws. In October 2020, the plaintiff voluntarily dismissed the Commonwealth of Massachusetts action, but in December 2020, the same plaintiff filed a purported shareholder class action complaint in state court in the State of New York, alleging claims substantially similar to those in the previous complaint against the same defendants, as well as the underwriters of our U.S. initial public offering. In March 2021, we and the other defendants filed a motion to dismiss before the state court of New York. In August 2021, the court granted our motion andSupreme Court of the State of New York, New York County, dismissed the complaint with prejudice. In September 2021, theThe plaintiff filed a notice of appeal toappealed, and in December 2022, the Supreme Court, Appellate Division, First Department and perfectedaffirmed the dismissal of the complaint, except that it deleted the phrase “with prejudice” from the Supreme Court’s judgment. The time to appeal on March 9, 2022. We intend to vigorously defend this action. However, this and future actionsthe decision of the Appellate Division has expired. Future litigation could give rise to substantial damages, and thereby have a material adverse effect on our financial position, liquidity, or results of operations. Even if this action issuch actions are not resolved against us, the uncertainty and expense associated with shareholder actions could harm our business, financial condition and reputation. Litigation can be costly, time-consuming and disruptive to business operations. The defense of lawsuits could also result in diversion of our management's time and attention away from business operations, which could harm our business.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, the price of our ordinary shares and ADSs and their trading volume could decline.
The trading market for our ADSs and ordinary shares depends in part on the research and reports that securities or industry analysts publish about us or our business. If no or few securities or industry analysts cover our company, the trading price for our ADSs and ordinary shares would be negatively impacted. If one or more of the analysts who covers us downgrades our equity securities or publishes incorrect or unfavorable research about our business, the price of our ordinary shares and ADSs would likely decline. If one or more of these analysts ceases coverage of our company or fails to publish reports on us regularly, or downgrades our securities, demand for our ordinary shares and ADSs could decrease, which could cause the price of our ordinary shares and ADSs or their trading volume to decline.
We do not currently intend to pay dividends on our securities and, consequently, your ability to achieve a return on your investment will depend on appreciation in the price of our ordinary shares and ADSs. In addition, French law may limit the amount of dividends we are able to distribute.
We have never declared or paid any cash dividends on our ordinary shares and do not currently intend to do so for the foreseeable future. We currently intend to invest our future earnings, if any, to fund our growth. Therefore, you are not likely to receive any dividends on your ordinary shares or ADSs for the foreseeable future and the success of an investment in ordinary shares or ADSs will depend upon any future appreciation in its value. Consequently, investors may need to sell all or part of their holdings of ordinary shares or ADSs after price appreciation, which may never occur, as the only way to realize any future gains on their investment. There is no guarantee that the ordinary shares or ADSs will appreciate in value or even maintain the price at which our shareholders have purchased them. Investors seeking cash dividends should not purchase our ADSs or ordinary shares.
Further, under French law, the determination of whether we have been sufficiently profitable to pay dividends is made on the basis of our statutory financial statements prepared and presented in accordance with accounting standards applicable in France. In addition, payment of dividends may subject us to additional taxes under French law. Therefore, we may be more restricted in our ability to declare dividends than companies not based in France.
In addition, exchange rate fluctuations may affect the amount of euros that we are able to distribute, and the amount in U.S. dollars that our shareholders receive upon the payment of cash dividends or other distributions we declare and pay in euros, if any. These factors could harm the value of our ADSs, and, in turn, the U.S. dollar proceeds that holders receive from the sale of our ADSs.
Future sales, or the possibility of future sales, of a substantial number of our ADSs or ordinary shares could adversely affect the price of our ADSs and ordinary shares.
As of April 13, 2021,1, 2024, we had 4981548949,860,983 ordinary shares issued and outstanding. Sales of a substantial number of our ADSs or ordinary shares, or the perception that such sales will occur, could cause a decline in the market price of our securities and could impair our ability to raise capital through the sale of additional equity securities. A substantial number of our ordinary shares and ADSs are now generally freely tradable, subject, in the case of sales by our affiliates, to the volume limitations and other provisions of Rule 144 under the Securities Act of 1933, as amended, or the Securities Act. If holders of these shares sell, or indicate an intent to sell, substantial amounts of our securities in the public market, or if we issue additional shares or securities, the trading price of our securities could decline significantly.
The rights of shareholders in companies subject to French corporate law differ in material respects from the rights of shareholders of corporations incorporated in the United States.
We are a French company with limited liability. Our corporate affairs are governed by our bylaws and by the laws governing companies incorporated in France. The rights of shareholders and the responsibilities of members of our board of directors are in many ways different from the rights and obligations of shareholders in companies governed by the laws of U.S. jurisdictions. For example, in the performance of its duties, our board of directors is required by French law to consider the interests of our company, its shareholders, its employees and other stakeholders, rather than solely our shareholders and/or creditors. It is possible that some of these parties will have interests that are different from, or in addition to, your interests as a shareholder or holder of ADSs. See the sections of this annual report titled “Item 6. 6. C —"Directors, Senior Management and Employees—Board Practices”Practices” and the documents referenced in “Item 10. 10. B —"Additional Information—Memorandum and Articles of Association.Association”. U.S. investors may have difficulty enforcing civil liabilities against our company and directors and senior management and the experts named in this annual report.
CertainThe vast majority of the members of our board of directors and senior management and certain experts named in this annual report are non-residents of the United States, and all or a substantial portion of our assets and the assets of such persons are located outside the United States. As a result, it may not be possible to serve process on such persons or us in the United States or to enforce judgments obtained in U.S. courts against them or us based on civil liability provisions of the securities laws of the United States. Additionally, it may be difficult to assert U.S. securities law claims in actions originally instituted outside of the United States. Courts outside the United States may refuse to hear a U.S. securities law claim because non-U.S. courts may not be the most appropriate forums in which to bring such a claim. Even if a court outside the United States agrees to hear a claim, it may determine that the law of the jurisdiction in which the non-U.S. court resides, and not U.S. law, is applicable to the claim. Further, if U.S. law is found to be applicable, the content of applicable U.S. law must be proved as a fact, which can be a time-consuming and costly process, and certain matters of procedure would still be governed by the law of the jurisdiction in which the non-U.S. court resides. In particular, there is some doubt as to whether French courts would recognize and enforce certain civil liabilities under U.S. securities laws in original actions or judgments of U.S. courts based upon these civil liability provisions. In addition, awards of punitive damages in actions brought in the United States or elsewhere may be unenforceable in France. An award for monetary damages under the U.S. securities laws would be considered punitive if it does not seek to compensate the claimant for loss or damage suffered but is intended to punish the defendant. French law provides that a shareholder, or a group of shareholders, may initiate a legal action to seek indemnification from the directors of a corporation in the corporation’s interest if it fails to bring such legal action itself. If so, any damages awarded by the court are paid to the corporation and any legal fees relating to such action may be borne by the relevant shareholder or the group of shareholders.
The enforceability of any judgment in France will depend on the particular facts of the case as well as the laws and treaties in effect at the time. The United States and France do not currently have a treaty providing for recognition and enforcement of judgments, other than arbitration awards, in civil and commercial matters.
Our bylaws and French corporate law contain provisions that may delay or discourage a takeover attempt.
Provisions contained in our bylaws and French corporate law could make it more difficult for a third party to acquire us, even if doing so might be beneficial to our shareholders. In addition, provisions of our bylaws impose various procedural and other requirements, which could make it more difficult for shareholders to effect certain corporate actions. These provisions include the following:
•under French law, the owner of 90% of voting rights of a public company listed on a regulated market in a Member State of the European Union or in a state party to the European Economic Area, or EEA, Agreement, including from the main French stock exchange, has the right to force out minority shareholders following a tender offer made to all shareholders;
•under French law, certain foreign investments in companies incorporated under French laws are subject to the prior authorization from the French Minister of the Economy, where all or part of the target’s business and activity relate to a strategic sector, such as energy, transportation, public health, telecommunications, etc.;, or constitutes a critical technology, such as biotechnologies;
•a merger (i.e., in a French law context, a share for share exchange following which our company would be dissolved into the acquiring entity and our shareholders would become shareholders of the acquiring entity) of our company into a company incorporated in the European Union would require the approval of our board of directors as well as a two-thirds majority of the votes held by the shareholders present, represented by proxy or voting by mail at the relevant meeting;
•a merger of our company into a company incorporated outside of the European Union would require 100% of our shareholders to approve it;
•under French law, a cash merger is treated as a share purchase and would require the consent of each participating shareholder;
•our shareholders have granted and may grant in the future our board of directors broad authorizations to increase our share capital or to issue additional ordinary shares or other securities, such as warrants, to our shareholders, the public or qualified investors, including as a possible defense following the launching of a tender offer for our shares;
•our shareholders have preferential subscription rights on a pro rata basis on the issuance by us of any additional securities for cash or a set-off of cash debts, which rights may only be waived by the extraordinary general meeting by a two-thirds majority vote of our shareholders or on an individual basis by each shareholder;
•our board of directors has the right to appoint directors to fill a vacancy created by the resignation or death of a director, subject to the approval by the shareholders of such appointment at the next shareholders’ meeting, which prevents shareholders from having the sole right to fill vacancies on our board of directors;
•our board of directors can be convened by our chairman, including upon request from our managing director,chief executive officer, if any, or, when no board meeting has been held for more than two consecutive months, from directors representing at least one-third of the total number of directors;
•our board of directors meetings can only be regularly held if at least half of the directors attend either physically or by way of videoconference or teleconference enabling the directors’ identification and ensuring their effective participation in the board’s decisions;
•our shares are registered or bearer, if the legislation so permits, according to the shareholder’s choice;
•approval of at least a majority of the votes held by shareholders present, represented by a proxy, or voting by mail at the relevant ordinary shareholders’ general meeting is required to remove directors with or without cause;
•advance notice is required for nominations to the board of directors or for proposing matters to be acted upon at a shareholders’ meeting, except that a vote to remove and replace a director can be proposed at any shareholders’ meeting without notice;
•our bylaws can be changed in accordance with applicable French laws and regulations;
•transfers of shares shall comply with applicable insider trading rules and regulations and, in particular, with the Market Abuse Directive and Regulation dated April 16, 2014; and
•pursuant to French law, the sections of our Bylaws relating to the number of directors and election and removal of a director from office, may only be modified by a resolution adopted by two-thirds of the votes of our shareholders present, represented by a proxy or voting by mail at the meeting.
You may not be able to exercise your right to vote the ordinary shares underlying your ADSs.
Holders of ADSs may exercise voting rights with respect to the ordinary shares represented by the ADSs only in accordance with the provisions of the deposit agreement. The deposit agreement provides that, upon receipt of notice of any meeting of holders of our ordinary shares, the depositary will fix a record date for the determination of ADS holders who shall be entitled to give instructions for the exercise of voting rights. Upon timely receipt of notice from us, if we so request, the depositary shall distribute to the holders as of the record date (1) the notice of the meeting or solicitation of consent or proxy sent by us and (2) a statement as to the manner in which instructions may be given by the holders.
A holder of ADSs may instruct the depositary of the ADSs to vote the ordinary shares underlying his or her ADSs. Otherwise, such holder will not be able to exercise voting rights unless he or she withdraws the ordinary shares underlying the ADSs that he or she holds. However, a holder of ADSs may not know about the meeting far enough in advance to withdraw those ordinary shares. If we ask for a holder of ADSs’ instructions, the depositary, upon timely notice from us, will notify him or her of the upcoming vote and arrange to deliver our voting materials to him or her. We cannot guarantee to any holder of ADSs that he or she will receive the voting materials in time to ensure that he or she can instruct the depositary to vote his or her ordinary shares or to withdraw his or her ordinary shares so that he or she can vote them. If the depositary does not receive timely voting instructions from a holder of ADSs, it may give a proxy to a person designated by us to vote the ordinary shares underlying his or her ADSs. In addition, the depositary and its agents are not responsible for failing to carry out voting instructions or for the manner of carrying out voting instructions. This means that a holder of ADSs may not be able to exercise his or her right to vote, and there may be nothing he or she can do if the ordinary shares underlying his or her ADSs are not voted as he or she requested.
Holders of ADSs are not holders of our ordinary shares.
A holder of ADSs is not treated as one of our shareholders and does not have direct shareholder rights. French law governs our shareholder rights. The depositary is the holder of the ordinary shares underlying ADSs. The deposit agreement among us, the depositary and all persons directly and indirectly holding ADSs sets out ADS holder rights, as well as the rights and obligations of the depositary.
A double voting right is attached to each registered share which is held in the name of the same shareholder for at least two years. However, the ordinary shares underlying our ADSs will not be entitled to double voting rights as the depositary will hold the shares underlying our ADSs in bearer form.
The right as a holder of ADSs to participate in any future preferential subscription rights or to elect to receive dividends in shares may be limited, which may cause dilution to the holdings of ADS holders.
Under French law, if we issue additional securities for cash, current shareholders will have preferential subscription rights for these securities on a pro rata basis unless they waive those rights at an extraordinary meeting of our shareholders by a two-thirds majority vote or individually by each shareholder. However, ADS holders will not be entitled to exercise or sell such rights unless we register the rights and the securities to which the rights relate under the Securities Act or an exemption from the registration requirements is available. In addition, the deposit agreement provides that the depositary will not make rights available to purchasers of ADSs unless the distribution to ADS holders of both the rights and any related securities are either registered under the Securities Act or exempted from registration under the Securities Act. Further, if we offer holders of our ordinary shares the option to receive dividends in either cash or shares, under the deposit agreement the depositary may require satisfactory assurances from us that extending the offer to holders of ADSs does not require registration of any securities under the Securities Act before making the option available to holders of ADSs. We are under no obligation to file a registration statement with respect to any such rights or securities or to endeavor to cause such a registration statement to be declared effective. Moreover, we may not be able to establish an exemption from registration under the Securities Act. Accordingly, ADS holders may be unable to participate in our rights offerings or to elect to receive dividends in shares and may experience dilution in their holdings. In addition, if the depositary is unable to sell rights that are not exercised or not distributed or if the sale is not lawful or reasonably practicable, it will allow the rights to lapse, in which case you will receive no value for these rights.
Holders of ADSs may be subject to limitations on the withdrawal of the underlying ordinary shares.
Temporary delays in the cancellation of ADSs and withdrawal of the underlying ordinary shares may arise because the depositary has closed its transfer books or we have closed our transfer books, the transfer of ordinary shares is blocked to permit voting at a shareholders’ meeting or we are paying a dividend on our ordinary shares. In addition, a holder of ADSs may not be able to cancel his or her ADSs and withdraw the underlying ordinary shares when he or she owes money for fees, taxes and similar charges and when it is necessary to prohibit withdrawals in order to comply with any laws or governmental regulations that apply to ADSs or to the withdrawal of ordinary shares or other deposited securities.
ADSs holders may not be entitled to a jury trial with respect to claims arising under the deposit agreement, which could result in less favorable outcomes to the plaintiffs in any such action.
The deposit agreement governing the ADSs representing our ordinary shares provides that, to the fullest extent permitted by law, ADS holders waive the right to a jury trial of any claim they may have against us or the depositary arising out of or relating to our shares, the ADSs or the deposit agreement, including any claim under the U.S. federal securities laws.
If we or the depositary opposed a jury trial demand based on the waiver, the court would determine whether the waiver was enforceable based on the facts and circumstances of that case in accordance with the applicable state and federal law. To our knowledge, the enforceability of a contractual pre-dispute jury trial waiver in connection with claims arising under the federal securities laws has not been finally adjudicated by the United States Supreme Court. However, we believe that a contractual pre-dispute jury trial waiver provision is generally enforceable, including under the laws of the State of New York, which govern the deposit agreement, by a federal or state court in the City of New York, which has non-exclusive jurisdiction over matters arising under the deposit agreement. In determining whether to enforce a contractual pre-dispute jury trial waiver provision, courts will generally consider whether a party knowingly, intelligently and voluntarily waived the right to a jury trial. We believe that this is the case with respect to the deposit agreement and the ADSs. It is advisable that you consult legal counsel regarding the jury waiver provision before entering into the deposit agreement.
If you or any other holders or beneficial owners of ADSs bring a claim against us or the depositary in connection with matters arising under the deposit agreement or the ADSs, including claims under federal securities laws, you or such other holder or beneficial owner may not be entitled to a jury trial with respect to such claims, which may have the effect of limiting and discouraging lawsuits against us and the depositary. If a lawsuit is brought against either or both of us and the depositary under the deposit agreement, it may be heard only by a judge or justice of the applicable trial court, which would be conducted according to different civil procedures and may result in different outcomes than a trial by jury would have, including results that could be less favorable to the plaintiffs in any such action.
Nevertheless, if this jury trial waiver provision is not permitted by applicable law, an action could proceed under the terms of the deposit agreement with a jury trial. No condition, stipulation or provision of the deposit agreement or ADSs serves as a waiver by any holder or beneficial owner of ADSs or by us or the depositary of compliance with U.S. federal securities laws and the rules and regulations promulgated thereunder.
As a foreign private issuer, we are exempt from a number of rules under the U.S. securities laws and are permitted to file less information with the SEC than a U.S. company. This may limit the information available to holders of ADSs and our ordinary shares.
We are a foreign private issuer, as defined in the SEC’s rules and regulations and, consequently, we are not subject to all of the disclosure requirements applicable to public companies organized within the United States. For example, we are exempt from certain rules under the Securities Exchange Act of 1934, as amended, or the Exchange Act, that regulate disclosure obligations and procedural requirements related to the solicitation of proxies, consents or authorizations applicable to a security registered under the Exchange Act, including the U.S. proxy rules under Section 14 of the Exchange Act. In addition, our officers and directors are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and related rules with respect to their purchases and sales of our securities. Moreover, while we currently make annual and semi-annual filings with respect to our listing on Euronext Paris and have filed, and expect to continue to file, financial reports on an annual and semi-annual basis, we willare not be required to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. public companies and willare not be required to file quarterly reports on Form 10-Q or current reports on Form 8-K under the Exchange Act. Accordingly, there is, and will continue to be, less publicly available information concerning our company than there would be if we were not a foreign private issuer.
As a foreign private issuer, we are permitted and we expect to follow certain home country practices in relation to corporate governance matters that differ significantly from Nasdaq’s corporate governance standards. These practices may afford less protection to shareholdersADS holders than they would enjoy if we complied fully with the corporate governance standards of the Nasdaq Global Select Market.
As a foreign private issuer listed on the Nasdaq Global Select Market, we are subject to Nasdaq’s corporate governance standards. However, Nasdaq rules provide that foreign private issuers are permitted to follow home country corporate governance practices in lieu of Nasdaq’s corporate governance standards as long as notification is provided to Nasdaq of the intention to take advantage of such exemptions. We have relied, and expect to continue to rely, on exemptions for foreign private issuers and follow French corporate governance practices in lieu of Nasdaq’s corporate governance standards, to the extent possible. Certain corporate governance practices in France, which is our home country, may differ significantly from Nasdaq corporate governance standards. For example, as a French company, neither the corporate laws of France nor our bylaws require a majority of our directors to be independent and we can include non-independent directors as members of our remunerationcompensation committee, and our independent directors are not required to hold regularly scheduled meetings at which only independent directors are present.
We are also exempt from provisions set forth in Nasdaq rules which require an issuer to provide in its bylaws for a generally applicable quorum, and that such quorum may not be less than one-third of the outstanding voting stock. Consistent with French law, our bylaws provide that a quorum requires the presence of shareholders having at least (1) 20% of the shares entitled to vote in the case of an ordinary shareholders’ general meeting or at an extraordinary shareholders’ general meeting where shareholders are voting on a capital increase by capitalization of reserves, profits or share premium, or (2) 25% of the shares entitled to vote in the case of any other extraordinary shareholders’ general meeting. If a quorum is not present, the meeting is adjourned. There is no quorum requirement when an ordinary general meeting is reconvened, but the reconvened meeting may consider only questions which were on the agenda of the adjourned meeting. When an extraordinary general meeting is reconvened, the quorum required is 20% of the shares entitled to vote, except where the reconvened meeting is considering capital increases through capitalization of reserves, profits or share premium. For these matters, no quorum is required at the reconvened meeting. If a quorum is not present at a reconvened meeting requiring a quorum, then the meeting may be adjourned for a maximum of two months.
As a foreign private issuer, we are required to comply with Rule 10A-3 of the Exchange Act, relating to audit committee composition and responsibilities. Under French law, the audit committee may only have an advisory role and appointment of our statutory auditors, in particular, must be decided by the shareholders at our annual meeting. Therefore, our shareholders may be afforded less protection than they otherwise would have under Nasdaq’s corporate governance standards applicable to U.S. domestic issuers. For an overview of our corporate governance practices, see "“Item 6. 6. C - "Directors, Senior Management and Employees—Board Practices.Practices”. We are an “emerging growth company” under the JOBS Act and are able to avail ourselves of reduced disclosure requirements applicable to emerging growth companies, which could make our ADSs less attractive to investors.
We are an “emerging growth company,” as defined in the U.S. Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and we intend to continue to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies,” including not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. In addition, Section 107 of the JOBS Act also provides that an emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended, or the Securities Act, for complying with new or revised accounting standards. We have not taken advantage of, and do not intend to take advantage of, the extended transition period provided under Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. Since IFRS makes no distinction between public and private companies for purposes of compliance with new or revised accounting standards, the requirements for our compliance as a private company and as a public company are the same.
We cannot predict if investors will find our ADSs less attractive because we may rely on these exemptions. If some investors find our ADSs less attractive as a result, there may be a less active trading market for our ADSs and the price of our ADSs may be more volatile. We may take advantage of these reporting exemptions until we are no longer an emerging growth company. We will remain an emerging growth company until the earliest of (1) the last day of the fiscal year in which we have total annual gross revenue of $1.07$1.235 billion or more; (2) December 31, 2024; (3) the date on which we have issued more than $1.0 billion in nonconvertible debt during the previous three years; and (4) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
We may lose our foreign private issuer status in the future, which could result in significant additional cost and expense.
While we currently qualify as a foreign private issuer, the determination of foreign private issuer status is made annually on the last business day of an issuer’s most recently completed second fiscal quarter and, accordingly, the next determination will be made with respect to us on June 30, 2022.2023. In the future, we would lose our foreign private issuer status if we fail to meet the requirements necessary to maintain our foreign private issuer status as of the relevant determination date. We will remain a foreign private issuer until such time that more than 50% of our outstanding voting securities are held by U.S. residents and any of the following three circumstances applies: (1) the majority of our executive officers or directors are U.S. citizens or residents; (2) more than 50% of our assets are located in the United States; or (3) our business is administered principally in the United States.
The regulatory and compliance costs to us under U.S. securities laws as a U.S. domestic issuer may be significantly more than costs we incur as a foreign private issuer. If we are not a foreign private issuer, we will be required to file periodic reports and registration statements on U.S. domestic issuer forms with the SEC, which are more detailed and extensive in certain respects than the forms available to a foreign private issuer. We would be required under current SEC rules to prepare our financial statements in accordance with U.S. GAAP, rather than IFRS, and modify certain of our policies to comply with corporate governance practices associated with U.S. domestic issuers. Such conversion of our financial statements to U.S. GAAP would involve significant time and cost. In addition, we may lose our ability to rely upon exemptions from certain corporate governance requirements on U.S. stock exchanges that are available to foreign private issuers such as the ones described herein and exemptions from procedural requirements related to the solicitation of proxies.
Changes to U.S. and non-U.S. tax laws could materially adversely affect our company.
Our tax treatment is subject to the enactment of, or changes in, tax laws, regulations and treaties, or the interpretation thereof, tax policy initiatives and reforms under consideration and the practices of tax authorities in jurisdictions in which we operate, including those related to the Organization for Economic Co-Operation and Development’s or OECD, Base Erosion and Profit Shifting or BEPS, Project, the European Commission’sEC’s state aid investigations and other initiatives. Such changes may include (but are not limited to) the taxation of operating income, investment income, dividends received or (in the specific context of withholding tax) dividends paid. We are unable to predict what tax reform may be proposed or enacted in the future or what effect such changes would have on our business, but such changes, to the extent they are brought into tax legislation, regulations, policies or practices, could affect our financial position and overall or effective tax rates in the future in countries where we have operations, reduce post-tax returns to our shareholders, and increase the complexity, burden and cost of tax compliance.
Further, existing tax laws, statutes, rules, regulations or ordinances could be interpreted, changed, modified or applied adversely to us. For example, U.S. federalin the United States, the Inflation Reduction Act of 2022 imposes, among other rules, a 15% minimum tax legislation enacted in 2017 informally titledon the Tax Cutsbook income of certain large corporations and Jobs Act or the Tax Act enacted many significant changes to the U.S.a 1% excise tax laws. Future guidance from the U.S. Internal Revenue Service, or IRS, and other tax authorities with respect to the Tax Act may affect us, andon certain aspects of the Tax Act could be repealed or modified in future legislation. For example, the Coronavirus Aid, Relief, and Economic Security Act, or CARES Act, modified certain provisions of the Tax Act. In addition, it is uncertain if and to what extent various states will conform to the Tax Act, CARES Act or any newly enacted federal tax legislation.corporate stock repurchases. Changes in corporate tax rates, the realization of net deferred tax assets relating to our operations, the taxation of foreign earnings, and the deductibility of expenses under the Tax Act or future reform legislation could have a material impact on the value of our deferred tax assets, could result in significant one-time charges, and could increase our future U.S. tax expense.
IfAlthough not free from doubt, we aredo not believe we were a passive"passive foreign investment company, there could" or PFIC, for U.S. federal income tax purposes for the taxable year ended December 31, 2023. However, we cannot assure you that we will not be classified as a PFIC for current taxable year or any future taxable year, which may result in adverse U.S. federal income tax consequences to U.S. holders.
Although the matter is not free from doubt, based on our analysis of our income, assets, activities and market capitalization for our taxable year ended December 31, 2021,2023, we do not believe that we were classified as a passive foreign investment company, or PFIC for the taxable year ended December 31, 2021.2023. Whether we are a PFIC for any taxable year will depend on our assets and income (including whether we receive certain non-refundable grants or subsidies, and whether such amounts along with reimbursements of certain refundable research tax credits and certain intercompany service payments will constitute gross income for purposes of the PFIC income test) in each year, and because this is a factual determination made annually after the end of each taxable year there can be no assurance that we will not be considered a PFIC in any taxable year. In addition, we hold a substantial amount of cash and cash equivalents.equivalents, which are generally treated as a passive asset for purposes of determining PFIC status. Because the calculation of the value of our assets may be based in part on the value of our ordinary shares or ADSs, the value of which may fluctuate considerably, our PFIC status may change from year to year and it is difficult to predict whether we will be a PFIC for the current year or any future year. Therefore, we have not yet made any determination as to our expected PFIC status for the current taxable year. However, we could continue to be considered a PFIC for the current taxable year or a future taxable year if the current percentage of our passive assets compared to our total assets remains the same or increases. Even if we determine that we are not a PFIC after the close of a taxable year, thereThere can be no assurance that the IRS will agree with our conclusion.conclusion with respect to any taxable year that we were not a PFIC for such taxable year. Our U.S. counsel expresses no opinion regarding our conclusions or our expectations regarding our PFIC status.
Under the Internal Revenue Code of 1986, as amended, or the Code, a non-U.S. company will be considered a PFIC for any taxable year in which (1) 75% or more of its gross income consists of passive income or (2) 50% or more of the average quarterly value of its assets consists of assets that produce, or are held for the production of, passive income. For purposes of these tests, passive income includes dividends, interest, gains from the sale or exchange of investment property and certain rents and royalties. In addition, for purposes of the above calculations, a non-U.S. corporation that directly or indirectly owns at least 25% by value of the shares of another corporation is treated as if it held its proportionate share of the assets and received directly its proportionate share of the income of such other corporation. If we are a PFIC for any taxable year during which a U.S. holder (as defined below under “Item 10. E -"Additional Information—Taxation”Information—Taxation”) holds our ordinary shares or ADSs, we will continue to be treated as a PFIC with respect to such U.S. holder in all succeeding years during which the U.S. holder owns the ordinary shares or ADSs, regardless of whether we continue to meet the PFIC test described above for a particular year, unless the U.S. holder makes a specified election once we cease to be a PFIC. If we are classified as a PFIC for any taxable year during which a U.S. holder holds our ordinary shares or ADSs, the U.S. holder may be subject to adverse tax consequences regardless of whether we continue to qualify as a PFIC, including ineligibility for any preferred tax rates on capital gains or on actual or deemed dividends, interest charges on certain taxes treated as deferred, and additional reporting requirements. For further discussion of the PFIC rules, and the adverse U.S. federal income tax consequences in the event we are classified as a PFIC and the availability of elections that may mitigate such adverse consequences, see the section of this annual report titled “Item 10. AdditionalE - "Additional Information—Taxation.Taxation”.
If a United States person is treated as owning at least 10% of our ordinary shares, such holder may be subject to adverse U.S. federal income tax consequences.
If a U.S. holder is treated as owning, directly, indirectly or constructively, at least 10% of the value or voting power of our ordinary shares or ADSs, such U.S. holder may be treated as a “United States shareholder” with respect to each “controlled foreign corporation” in our group, if any. Because our group currently includes one U.S. subsidiary, our non-U.S. subsidiarysubsidiaries (and any other non-U.S. subsidiaries we form or acquire in the future) could be treated as controlled foreign corporations, regardless of whether we are treated as a controlled foreign corporation. A United States shareholder of a controlled foreign corporation may be required annually to annually report and include in its U.S. taxable income its pro rata share of “Subpart F income,” “global intangible low-taxed income” and investments in U.S. property by controlled foreign corporations, regardless of whether we make any distributions. An individual that is a United States shareholder with respect to a controlled foreign corporation generally would not be allowed certain tax deductions or foreign tax credits that would be allowed to a United States shareholder that is a U.S. corporation. Failure to comply with controlled foreign corporation reporting obligations may subject a United States shareholder to significant monetary penalties. We cannot provide any assurances that we will furnish to any United States shareholder information that may be necessary to comply with the reporting and tax paying obligations applicable under the controlled foreign corporation rules of the Code. U.S. holders should consult their tax advisors regarding the potential application of these rules to their investment in our ordinary shares or ADSs.
We must maintain effective internal control over financial reporting, and if we are unable to do so, the accuracy and timeliness of our financial reporting may be adversely affected, which could hurt our business, lessen investor confidence and depress the market price of our securities.
As a public company, we must maintain effective internal control over financial reporting in order to accurately and timely report our results of operations and financial condition. In addition, as a public company listed in the United States, the Sarbanes-Oxley Act requires, among other things, that our management assesses the effectiveness of our internal control over financial reporting beginning with this Annual Report.
The rules governing the standards that must be met for our management to assess our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act are complex and require significant documentation, testing and possible remediation. These stringent standards require that our audit committee be advised and regularly updated on management’s review of internal control over financial reporting. To comply with this obligation, we must maintain an extensive framework of internal control over financial reporting, that we need to regularly update and test. This process is time-consuming, costly, and complicated. In addition, our independent registered public accounting firm will be required to attest to the effectiveness of our internal controls over financial reporting beginning with our annual report following the date on which we are no longer an “emerging growth company,” which may be through December 31, 2024. Our management may not be able to effectively and timely implement controls and procedures that adequately respond to the increased regulatory compliance and reporting requirements that are now applicable to us as a public company listed in the United States.
Management identified no material weakness as of December 31, 2021.2023. See “Item 15—15 - "Disclosure Controls and Procedures” of this Annual Report for further discussion of management’s assessment of the effectiveness of our internal control over financial reporting. Assessing our procedures to improve our internal control over financial reporting is an ongoing process. In connection with the audit of our consolidated financial statements for the year ended December 31, 2018, our independent registered public accounting firmWe have identified a control deficiencymaterial weaknesses in our internal control over financial reporting andin the material weakness was remediated. Wepast, which were remediated and can provide no assurance that we will not have material weaknesses in the future. Any additional material weaknesses we identify could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our consolidated financial statements. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. If material weaknesses occur which we are unable to remediate and we conclude that our internal control over financial reporting is ineffective, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of the ADSs could decline, and we could be subject to sanctions or investigations by the NASDAQ Stock Market, the SEC or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
The outbreak of any new public health crisis could adversely impact our business, including our preclinical studies and clinical trials.
In December 2019, COVID-19 spread across the world, including to countries where our facilities are located, where our product candidates are being evaluated in ongoing or future clinical trials, and where our CROs and CMOs are located, which had a significant impact on our activities.
No assurance can be given that new, restrictive measures will not be adopted by governments, and it is not possible to predict with certainty the economic impact and the extent of the possible recovery from the pandemic or the emergence of a new pandemic. However, a long-lasting pandemic recovery accompanied by the implementation of new restrictive measures could lead to an economic slowdown in one or several markets in which the Group operates, or have disruptions that could have a very significant impact on our activities (including the development and, pre-marketing and marketing activities), our operations and those of our current or future partners, our clinical trials, and in particular:
•delays or difficulties manufacturing active ingredients and therapeutic units to be sent to our clinical investigation sites;
•delays or difficulties in enrolling patients in clinical trials in which our product candidates are being evaluated;
•delays or difficulties in recruiting new clinical investigation sites and in starting their activities, including difficulties in recruiting physician investigators and personnel assigned to trials of the clinical investigation site. In particular, the delays in the launch and in enrollment of patients for the Phase 3 ELATIVE® trial evaluating elafibranor in PBC which led us to have to revise our forecasts with regard to obtaining clinical results;
•reallocations of resources normally dedicated to the conduct of clinical trials, including the resources of hospitals hosting clinical investigation sites and hospital staff involved in the conduct of our clinical trials or those of our current partners or potential future partners that made conducting trials technically difficult or impossible;
•disruptions to key clinical trial-related activities, such as monitoring clinical investigation sites;
•limitations if management, members of the Board of Directors and/or employees are unable to work due to illness or unable to work remotely, or in case of the Board of Directors, unable to meet and specifically in the human resources that would usually be concentrated on the conduct of our clinical trials, or those of our current or future partners;
•additional costs related to the implementation of specific protocols within the framework of our ongoing or future clinical trials, or those of our current or future partners;
•delays in obtaining authorizations from the regulatory authorities necessary to start clinical or preclinical studies that we, or our current partners, have planned to launch;
•delays in receipt by the clinical investigation sites of the supplies and equipment needed to carry out these clinical trials;
•disruptions in global trade that may affect the transportation of clinical trial materials such as our therapeutic units required in our clinical trials;
•changes in local regulations imposed by a resumption of the pandemic that could require us or our current partners to modify the terms of our clinical trials, which could result in unexpected costs, or lead to the interruption of our clinical trials;
•delays in necessary interactions with local regulatory agencies, particularly the FDA and EMA, Ethics Committees and other important agencies and contractors due to limited human resources or the unavailability or forced leave of public officials or due to the concentration of their efforts on the examination of other treatments or other activities related to the pandemic; and
•refusals by the FDA or the EMA to accept clinical trial data collected in geographical areas affected by the COVID-19 pandemic.
In addition, the extent of the negative impact of this possible pandemic recovery on the financial markets, on our share price and therefore on our ability to obtain additional financing is unknown at this time. Disaster recovery, business continuity or restructuring plans may be inadequate or insufficient in these circumstances.
| | | | | |
| Item 4. | Information on the Company. |
| | | | | |
| A. | History and Development of the Company |
GENFIT is a late-stage biopharmaceutical companygroup conducting late stage clinical trials dedicated to improving the lives of patients with metabolicliver diseases with high unmet medical needs, with a special focus on rare, life-threatening and chronic liver diseases.acute pathologies. Our legal name is "GENFIT SA," or a French société anonyme, and our principal executive office is located at Parc Eurasanté 885, avenue Eugène Avinée 59120 Loos, France. Our telephone number at our principal executive office is +33 (0)3 2016 4000. Our agent for service of process in the United States is Corporation Service Company, located at 19 West 44th Street, Suite 200, New York, NY 10036.
With its rich scientific heritage spanning more than two decades, GENFITthe Group is a pioneer in the discovery and development of drugs basedfor liver diseases. Our pipeline encompasses a total of ten programs. The main franchise focuses on nuclear receptors. GENFIT's integrated approachAcute on Chronic Liver Failure (ACLF) and includes five therapeutic programs at different development stages (preclinical, Phase 1, Phase 2): VS-01-ACLF, nitazoxanide (NTZ), SRT-015, CLM-022 and VS-02-HE. A second franchise includes two therapeutic programs targeting other life-threatening liver diseases: GNS561 in managing patients with liver diseases has led itCholangiocarcinoma (CCA) and VS-01-HAC in Urea Cycle Disorder (UCD) and Organic Acidemia (OA). In addition, in 2021, GENFIT successfully out-licensed to develop drug candidatesIpsen a proprietary program, elafibranor, which had been developed internally up to and including Phase 3. Our pipeline also includes a diagnostic franchise including NIS2+® in Metabolic dysfunction-associated steatohepatitis (MASH, previously known as NASH, for the treatment of Primary Biliary Cholangitis (PBC), cholangiocarcinoma, ACLFNonalcoholic Steatohepatitis) and an innovative non-invasive diagnostic technology to identify patients with "at-risk" NASH. All these patients are exposed to an increased risk of progressing to severe complications, hence the reason forTS-01 focusing our efforts on these therapeutic areas for which high unmet medical needs remain.blood ammonia levels.
Our company, a French société anonyme, or S.A.,GENFIT was co-foundedfounded and incorporated in 1999 by Jean-François Mouney, now Chairman of the Board of Directors. Our Chief Executive Officer, Pascal Prigent, took his position on September 16, 2019, following the recommendation of Jean-François Mouney.Mouney and board of directors' approval. In 2003, GENFIT created GENFIT CORP., our subsidiary in Massachusetts, United States. In 2006, GENFIT was listed on the Alternext Market, ofmanaged by Euronext Paris, and transferred in 2014 onto the Euronext Market in Paris (compartment B - ISIN : FR0004163111). In March 2019, GENFIT SA listed its American Depositary Shares on the Nasdaq Global Select Market in the United States under the symbol "GNFT". On September 29, 2022, GENFIT completed the acquisition of Versantis AG, a Swiss-based clinical stage biotechnology company focused on providing solutions for increasing unmet medical needs in liver diseases, which has since then become its wholly-owned subsidiary. In 2023, we in-licensed two additional investigational drugs in ACLF. SRT-015 is an ASK1 inhibitor in-licensed from Seal Rock Therapeutics in acute liver diseases and CLM-022 is a small molecule inhibitor targeting the NLRP3 inflammasome in-licensed from Celloram.
We are led by an executive team and board of directors with deep experience at leading biotech companies, large pharmaceutical companies and academic institutions.
The chair of our scientific advisory board, Bart Staëls,Staels, is the other co-founder of our company and a world-renowned expert in metabolic & inflammatory disorders, and nuclear receptors. Our Scientific Advisory Board is composed of world-renowned key opinion leaders in metabolic and inflammatory diseases with a particular focus on hepatic and gastroenterological diseases.
The Group's workforce is spread over 3 sites: Lille and Paris (France), and Cambridge (Massachusetts, United States). In total, there were 122 employees in early 2022.
Today, our R&D efforts are focused on bringing to the market therapeutic solutions targeting metabolic, inflammatory, autoimmune or fibrotic diseases that mostly affect the liver (like PBC) but also the gastroenterology sphere more broadly. The Company rolls out new approaches combining new treatments and biomarkers. Elafibranor, the Company's most advanced molecule, is being evaluated in a Phase 3 clinical trial in PBC.
Throughout our company’s history, we have carried out numerous R&D programs through consortiums and co-research agreements with large pharmaceutical companies, and experts from the academic world. The experience and expertise we’ve gained have fueled our own research and development efforts, including the discovery of new therapeutic targets, the development of novel technologies and the identification of drug candidates that have demonstrated potential therapeutic efficacy in clinical trials.
The Group's workforce is spread over 4 sites: Lille and Paris (France), Zurich (Switzerland) and Cambridge (Massachusetts, United States). As of December 31, 2023, we had a total of 159 employees.
Our capital expenditures in the years ended December 31, 2023, 2022, and 2021 totaled €0.4 million, €44.9 million, and €0.6 million respectively. The 2022 amount is primarily related to our acquisition of Versantis. Investments in software and scientific equipment primarily account for amounts in 2021 and 2023. (Note that in 2023 we acquired licence rights totaling €2.1 million not included in the figure above.) We expect our capital expenditures in 2024 to be primarily financed from our existing cash.
We maintain a corporate website at www.genfit.com. We intend to post our annual report on our website promptly following it being filed with the SEC. Information contained on, or that can be accessed through, our website does not constitute a part of this annual report. We have included our website address in this annual report solely as an inactive textual reference.
The SEC maintains an internet site at http://www.sec.gov that contains reports and other information regarding issuers that file electronically with the SEC.
i. Our Purpose
GENFIT is a late-stage biopharmaceutical company dedicated to improving the lives of patients affected by severe chronicrare and life-threatening liver diseases that are characterized by high unmet medical needs.
Our purpose supports our long-term commitment with regardsregard to the role we want to play in society, not only as an economic player seeking to create long-term value for our ecosystem and partners but also as an innovative biotechnology company working to improve people'spatients' quality of life, and finally as a civic company striving to promote professional and personal development for its employees.
We intend to create general public benefit by generating a positive and significant social, societal and environmental impact through our activities. As part of this approach, our Board of Directors commits to taking into consideration (i) the social, societal and environmental consequences of its decisions on all of the Company's stakeholders, and (ii) the consequences of its decisions on the environment. As part of this commitment, we have created a dedicated ESG Committee of the Board of Directors which meets at least bi-annually, to measure and track our extra-financial performance and communicate to the public through an annual extra-financial performance report.
ii. Our Vision
Our ambition is to capitalize on our scientific, clinical and regulatory expertise acquired during more than two decades in the field of liver disease to build and expand a pipeline of innovative therapeutic and diagnostic solutions targeting severe chronicrare and life-threatening liver diseases with high unmet medical needs, and representing a significant market potential in order to generate revenuesfinance innovation to enable us to sustain excellence in medical innovation, research and development.development over time.
iii. Our Mission
Our mission is to act asremain a pioneer in clinical research,the field of liver diseases, i.e. identify high potential assets andto bring them from discovery or early stages up to late development stages, typically the end of Phase 3,3. Subject to successful development and marketing approval, and depending on the point at whichnature of our collaboration and licensing agreements, we can decidewould either commercialize the assets ourselves, capitalize on the know-how of our commercialization strategy: either building-up a marketing and sales force ourselves, or leveraging our global partnership with our main commercial partnercurrent partners, such as Ipsen, or partneringenter into additional distribution agreements with another strategic partner.new partners.
iv. Our Founding Values and Principles
Our employees are driven by common principles that shape their actions:
–Innovation to serve patients: We are deeply committed to improving the health and quality of life of patients affected by severe chronicrare and life-threatening liver diseases.diseases characterized by high unmet medical needs. We seek new ways to advance science and medicine, with the goal of optimizing care for patients. With a strong desire to leverage our agility and responsiveness, we and our employees are striving to move our scientific and medical approaches forward, and improve patient management in terms of diagnostics, prevention and care.
–Respect and diversity: We bring together talented employees with unique perspectives and experiences, we recognize and value diversity as a great strength, and ensure that all employees and third parties are treated fairly, with dignity and respect.
–Ethics: We deliver true and accurate information to our partners and stakeholders and build our business relationships with honesty and transparency. We demand of ourselves and others the highest ethical standards and we conduct our business in a socially and environmentally sustainable manner.
Overviewv. Our Sustainability Journey
In May 2021,GENFIT considers Corporate Social Responsibility, or CSR, a key driver for success, in that extra-financial performance can be considered as closely associated with financial performance. Although we announced, followingare not yet subject to significant CSR reporting regulations, we strive to be as proactive and transparent as possible, and publish an Extra-Financial Performance Report, or EFPR, on an annual basis.
Our CSR journey pursues several objectives. First is our desire as a company to uphold the termination of all development of elafibranor in NASH and the redefinitionprinciples of our clinical products portfolio,code of ethics and our internal policies. Secondly, we seek to manage risks that could potentially affect our business activity, and to seize opportunities that could potentially contribute to our growth. Third, we had redeployedengage with key stakeholders in our R&D effort across three franchises covering therapeutic areas where patients have little or no treatment and/or diagnostics options: cholestatic diseases, Acute on Chronic Liver Failure (ACLF),ecosystem (doctors, patient associations, investors, talents, employees, etc.) in order to capture, understand and diagnostics.address challenges that are material for them and for us. Finally, we attempt to anticipate future regulations that may apply to our organization in the coming years.
•Cholestatic diseases franchise
Chronic cholestatic diseases are characterized by defective bile acid transport from the liver to the intestine, which is caused by primary damage to the biliary epitheliumWith this in most cases.
Atmind, at the end of 2021, this franchise includes one Phase 3 programour Board of Directors created a dedicated environmental, social and governance, or ESG, Committee, which meets at least twice per year and makes recommendations to the Board of Directors. This committee reviews in PBC, with investigational drug candidate elafibranor,particular the annual ESG roadmap (specific actions and one Phase 2 program in Cholangiocarcinoma (CCA)initiatives conducted or to be launched), with investigational drug candidate GNS561. Elafibranor's global rights are out-licensed to Ipsen, with the exception of Greater China where Terns Pharmaceuticals has licensedand is involved in the rightsdrafting and review of the annual EFPR. This report describes our philosophy, our priorities and the nature of our engagement in terms of (1) policies, (2) actions and (3) performance indicators, including criteria related to elafibranor.(1) the environment, (2) social and societal topics and (3) governance matters.
Internally, our CSR approach involves stakeholders at all levels of the Company. At the top of the organization, beyond the ESG Committee, the Audit Committee and the Nomination and Compensation Committee play a key role. The Economic and Social Council, or Works Council, a council that is statutorily required in France and composed of employee representatives, also plays a significant role. In December 2021,addition, each functional department is responsible for ensuring that E- and/or S- and/or G-related matters are properly addressed. Then at the bottom of the organization, a group of ESG volunteers - or ESG champions - is making sure that CSR remains at the heart of our organization.
In 2023, the independent rating agency Gaïa Research by EthiFinance SAS modified our rating from Bronze to Gold and ranked our company in second place out of 75 companies in our sector. We also obtained a Prime Status label from Institutional Shareholder Services (ISS). In addition, ODDO Research classified GENFIT as best-in-class for ESG in the Biotech sector, and GENFIT was the sole biotechnology company chosen by the LEEM ("Les Entreprises du Médicament", the professional organization representing pharmaceutical companies operating in France) as one of the four finalists in the "ESG governance" category of their ESG trophies. Since 2022, we acquiredalso engaged in a series of self-evaluation processes, as part of our dedication to continuous improvement, based on sector-specific tools developed around the IS026000 standard as well as the 17 Sustainable Development Goals (SDGs) from the United Nations, and with regards to environmental standards we referred to ADEME (Agency for the Environment and Energy Management), Science Based Targets initiative (SBTi) and Greenhouse gases (GhG).
In 2024 and beyond, we are committed to further enhancing our ESG strategy, building upon a robust foundation established through a formal materiality assessment initiated during the second half of 2023 in collaboration with key stakeholders. This strategy aligns with evolving regulations and underscores our dedication to responsible and sustainable business practices relevant to our business.
vi. Overview of our main programs
Over the past few years, GENFIT has made a strategic pivot towards Acute On-Chronic Liver Failure (ACLF) and other life-threatening liver conditions, broadening its research pipeline to include promising drug candidates that aim to meet the urgent and unmet needs of this challenging condition:
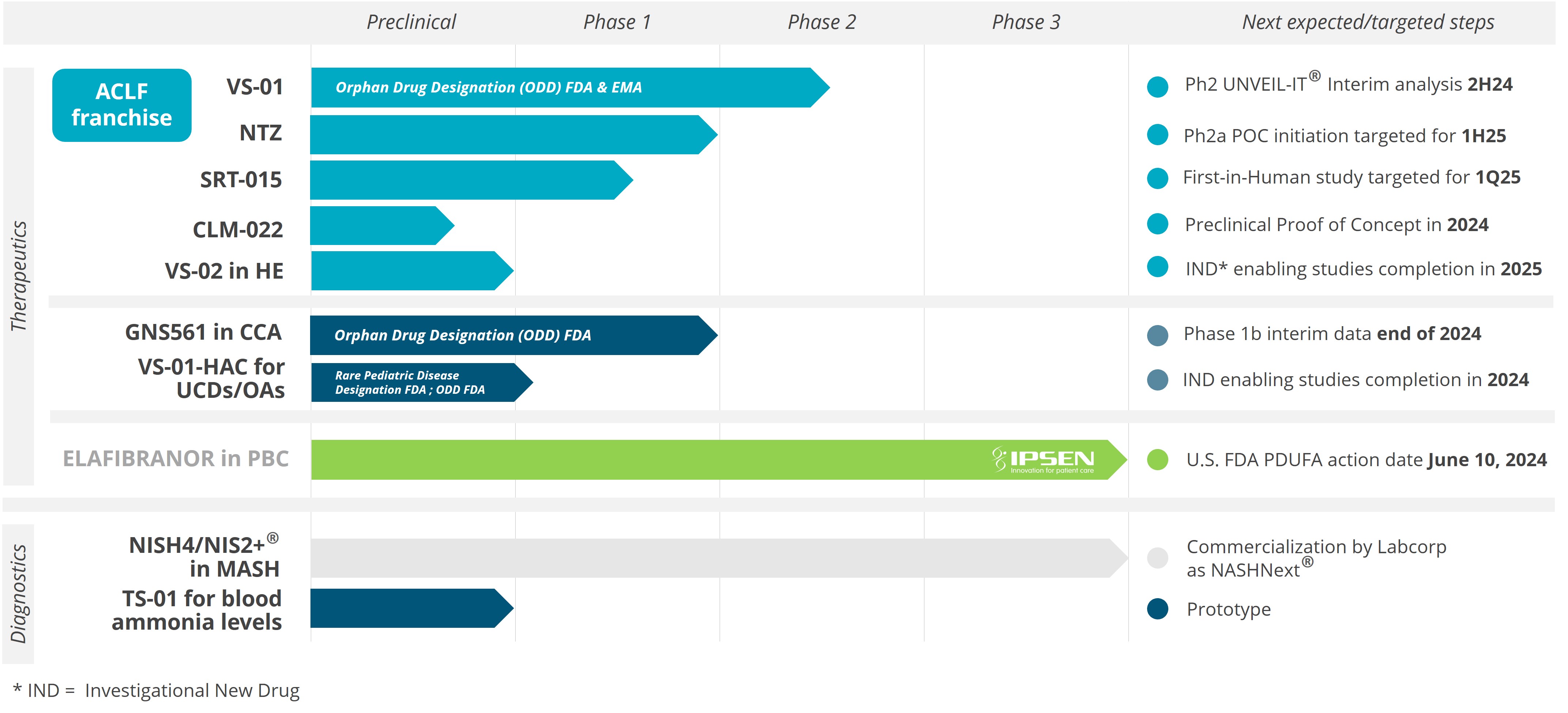
Upcoming milestones, data announcements and launch dates are anticipated and subject to change. PBC: Primary Biliary Cholangitis ; ACLF: Acute- on- Chronic Liver Failure. CCA: Cholangiocarcinoma; HAC: Hyperammonemic Crises; UCD = Urea Cycle Disorders ; OA = Organic Acidemias ; HE: Hepatic Encephalopathy; MASH: Metabolic dysfunction-Associated Steatohepatitis; NTZ: Repositioned molecule (Nitazoxanide); All drugs under development are investigational compounds that have not been reviewed nor been approved by a regulatory authority in targeted indications. Ipsen has global rights to develop and commercialize GNS561elafibranor in PBC (including open-label extension, confirmatory PBC study and life cycle management), with the exception of China, Hong Kong, Taiwan, and Macau (Greater China) where Terns Pharmaceuticals holds the exclusive license to develop and commercialize elafibranor. GENFIT has licensed the exclusive worldwide rights of ASK1 Inhibitor SRT-015 (injectable formulation in acute liver disease) from Seal Rock Therapeutics. GENFIT licensed the exclusive worldwide rights of CLM-022, a potential first-in-class inflammasome inhibitor, from Celloram Inc. GENFIT has in-licensed the exclusive rights for GNS651 in Cholangiocarcinoma in the United States, Canada and Europe, (includingincluding the United-Kingdom and Switzerland, from Genoscience Pharma. Labcorp has a five-year exclusive license for the development and commercialization of NIS4® technology to power a next-generation MASH diagnostic laboratory-developed test (LDT) to identify patients with at-risk MASH in the United KingdomStates and Switzerland) from Genoscience Pharma, for CCA.
On April 7, 2022, we announced that we had completed recruitment for the double-blind part of ELATIVE, which is the part to be used to support accelerated approval. We reaffirm our commitment to delivering ELATIVE topline data in the second quarter 2023. Our Phase 1b/2 clinical trial in CCA is expected to start in the fourth quarter of 2022.
•ACLF franchise
ACLFCanada. NIS2+® is a serious syndrome associated with chronic liver diseases and is defined by an acute episode of hepatic decompensation in patients with cirrhosis that progresses to one or more extra-hepatic organ failures, including the brain, kidneys, heart and/or lungs.
The first program launched in this franchise aims at developing the repurposed drug nitazoxanide (NTZ). A Phase 1 open-label, non-randomized, 2-center, repeated-dose, parallel-group study is expected to provide preliminary insight into NTZ pharmacokinetics or PK and safety in the setting of hepatic impairment in the second half of 2022. A second Phase 1 study to evaluate NTZ PK and safety in the setting of renal impairment is planned to initiate in the first half of 2022. The results of these studies will inform the potential need for dose adjustment in future studies to be conducted in patients with cirrhosis and hepatic impairment and/or renal impairment.
•Diagnostics franchise
This franchise is currently exclusively focused on NASH and the main program aims at developing our non-invasive, blood-based diagnosticnext-generation technology called NIS4, and identifying patients with NASH (NAS≥4) and significant advanced fibrosis (F>2), also referred to as "at-risk" NASH. Following two licensing agreements signed with Labcorp in 2019 and 2020, NIS4 technology is available today for use in clinical research, and also commercialized in the U.S. and Canada as a Laboratory Developed Test or LDT for use in the clinic under the name "NASHnext, powered by NIS4 technology"derived from NIS4®.
In 2021, we signed an additional licensing agreement with Q Squared Solutions, LLC or Q2 to strengthen the availability of NIS4 technology for use in the field of clinical research.
| | | | | | | | | | | |
Cholestatic Diseases Franchise | ACLF Franchise | Diagnostic Franchise |
Primary Biliary Cholangitis | Cholangiocarcinoma |
elafibranor - Phase 3 ELATIVE | GNS561 - Phase 1b/2 | nitazoxanide (NTZ) - Phase 1 | NASH: NIS4 Technology |
A note about the evolving COVID-19 pandemic and its potential consequences on our business
The unprecedented spread of COVID-19 – characterized as a pandemic by the World Health Organization on March 11, 2020 – is impacting the global health and business ecosystem, GENFIT included. During this evolving crisis, our priorities continue to be to ensure the safety and well-being of our employees, of the patients and healthcare professionals involved in our clinical trials, as well as the integrity of our ongoing clinical trials. We remain committed to ensuring business continuity and have been monitoring the situation closely.
In light of our priorities and in accordance with guidance documents issued by the FDA, EMA and other national regulatory authorities, we have worked with our contract research organizations or CROs, trial sites and investigators to critically reassess all our existing programs. We regularly revise our program execution estimations to take into account the evolution of the pandemic situation and its impact on our activities. As a result of measures implemented in consultation with our CRO we were able to minimize disruption to our ELATIVE Phase 3 clinical trial of elafibranor in PBC, which enrolled its first patient in September 2020. At the start of the trial, and considering the pandemic situation, we had estimated that enrollment in the ELATIVE study would take approximately 18 months and so far, enrollment has been broadly in-line with this estimate. However, the recent rapid expansion of the highly contagious Omicron strain of COVID has created additional complications for us in enrolling patients and in clinical trial operations generally. The rate of infection, as well as the containment measures put in place to control its growth have led to patients postponing site visits or having to be re-screened because they had fallen outside the screening window. This recent worsening of the COVID pandemic has also created significant additional administrative backlogs at sites and regulatory agencies, due to the combination of continued high volume of trials and staffing shortages. This has disproportionately impacted regions where there were already significant delays, such as Latin America. Although we currently do not anticipate these recent complexities to substantially change the guidance related to availability of the ELATIVE topline results, we continue to assess the impacts of COVID-19 for all of our ongoing and planned clinical trials.
vii. Our Strengths
We believerely on our strengths listed below, provide the foundation that will allow us to successfully expandaccelerate our activitiesresearch and development efforts over the coming years in both drug and diagnostic research and development:years.
•–A recognized expertise in bringing earlyearliest stage assets into latelater development stages
Over the years, we have brought two early stageGENFIT has demonstrated its capacity to develop assets upfrom the earliest stages to the pre-commercialization stage, first with elafibranor in the Phase 3 RESOLVE-IT trial in NASH and also the Phase 3 ELATIVE trial in PBC.stage. This track record illustrates GENFIT’swas materialized by the development of elafibranor from discovery to Phase 3 in MASH, and then in PBC, leveraging GENFIT's expertise in several fields: research (target identification, understanding of molecular mechanisms of action, establishing a network of experts, etc.), clinical development (KOL management and Ad boards, study(study design and protocol definition, KOL coordination and Advisory Boards, clinical trial execution from site activation and patient recruitment to data readout and statistical analysis), regulatory (FDA/EMA(U.S. Food and Drug Administration (FDA)/European Medicines Agency (EMA) interactions for INDInvestigational New Drug (IND) submissions, Breakthrough Therapy/Fast Track/Orphan designations, accelerated pathways such as Subpart H, NDA presubmission, etc.) and pre-commercialization (disease awareness, patient engagement, pre-marketing, forecasting, sales force sizing, market-access, ,etc.etc.).
•–A portfolio rationalized in 2021 to focuspipeline focused on disease areas with high unmet needs and high market potential
Our current portfolio is well-diversified, with earlyGENFIT's pipeline has become widely diversified, expanding from a single asset (elafibranor) and a single indication (PBC) to late stage programs evaluating differenta much larger pipeline. The wide range of mechanisms of actions in different disease areas with high unmet medical needs. action and indications we are targeting allow us to distribute risk over several programs. The distribution of these programs across several development stages provides a dynamic and diverse potential news flow over the coming months and years. Given their positioning and potential, some programs have received special designations from regulatory agencies:
| | | | | | | | |
| Program | Designation |
| Elafibranor in PBC | Orphan Drug Designation (FDA, EMA) | Breakthrough Therapy Designation (FDA) |
| VS-01-ACLF | Orphan Drug Designation (FDA, EMA*) | |
| GNS561 in CCA | Orphan Drug Designation (FDA) | |
| VS-01-HAC** | Orphan Drug Designation (FDA) | Rare Pediatric Disease Designation (FDA) |
*In addition, the existing body of evidence with our assets forms the rationale to support our programs. In PBC, the Phase 2 trial results give us confidence in elafibranor’s potential to succeed in this indication, and our collaboration with Ipsen - a large pharmaceutical company with proven experience in the field - further supports this potential. In ACLF, where systemic inflammationEU Orphan Drug Designation is a major driver for poor patient outcomes, several KOLs support the clinical evaluation of NTZ, which has demonstrated notable anti-inflammatory effects in a preclinical model of ACLF. In CCA, we also believe in the potential of GNS561, a Phase 2-ready asset which has completed preclinical studies and a Phase 1b trial. Last, in NASH diagnostics, the performance and utility of NIS4 technology have been recognized by the Non-Invasive Biomarkers of Metabolic Liver Disease (NIMBLE) initiative of the FoundationALF
**VS-01-HAC is potentially eligible for the National Institutes of Health’s Biomarkers Consortium or FNIH, a large biomarker consortium of well-respected experts using an independent approach and a robust methodology. Feedback received from the field via our partner Labcorp also confirms the potential of our non-invasive technology once the first anti-NASH treatments are approved and come to market.Priority Review Voucher (PRV) upon approval (FDA)
•–Partners with a strong commercial track-record
Ipsen became an 8% shareholder of GENFIT at the end of 20212021. The strategic partnership also provides Ipsen with access to our research capabilities and also becameother clinical programs through rights to first negotiation, therefore becoming a potential natural partner for GENFIT to commercialize any late stage asset successfully developed in the future. Ipsen’s world-class development capabilities, well-established global commercial footprint and excellent track record in delivering therapies to patient populations with unmet medical need indeed makes it an ideal partner for GENFIT. We have also developed other partnerships with other stakeholders, creating potential avenues to generate revenues in the future. In 2019, wethe Company signed a licensing and collaboration agreement with Terns Pharmaceuticals for the development and commercialization of elafibranor in Greater China, and another onealso has agreements with Labcorp, to commercialize NIS4NIS4® technology in the USU.S. and Canada as an LDT.a Laboratory Developed Test, as well as with Q2 lab in the clinical research space.
•–A robust financial situationExpected future milestone and royalty payments for elafibranor in PBC to support future development, subject to approval by applicable regulatory authorities
As of December 31, 2023, the Company’s cash and cash equivalents amounted to €77.8 million compared with €140.2 million as of December 31, 2022. As of September 30, 2023, cash and cash equivalents totaled €93.9 million. This amount does not include the receipt in February 2024 of a strong€13.3 million milestone payment from Ipsen, which was invoiced in December 2023, triggered by the acceptance of the New Drug Application (NDA) filing by the US Food and Drug Administration (FDA) and Marketing Authorization Application (MAA) by the European Medicines Agency (EMA) for accelerated approval of elafibranor in Primary Biliary Cholangitis (PBC) in December 2023.
In 2024, GENFIT expects to receive total milestone payments of approximately €89 million (including the €13.3 milestone already received in February 2024), subject to the approval and commercialization of elafibranor in PBC by Ipsen.
We expect that our cash positionand cash equivalents will enable us to fund our operating expenses and capital expenditure requirements until approximately the fourth quarter of 2025, taking into account our expectations to receive future milestone revenue in 2024, subject to approval by applicable regulatory authorities, and the US and European commercial launches of elafibranor in PBC by Ipsen. This estimation is based on management’s reasonable, current assumptions and programs, and does not include exceptional events.
As of December 31, 2021, our cash and cash equivalents amounted to €258.8 million. The long-term strategic partnership for global collaboration signed with Ipsen in December 2021 considerably strengthened our financial position, with a €120 million upfront, €28 million investment in equity and up to €360 million in potential milestones payments. In addition, we are eligible for tiered double-digit royalties up to 20%. In January 2021, we successfully completed the renegotiation of our OCEANE convertible debt, decreasing the outstanding nominal amount to €0.1 million as of the date of this annual report, approximately one-third of the original outstanding nominal amount of €180 million, and extending the maturity by 3 years until October 2025. This financial situation provides us with the flexibility and reactivity which are necessary to seize strategic opportunities and choose the most adequate means of growing our pipeline with the right assets, in the right indications, at the right time, be it through the acceleration of our in-house R&D effort or through in-licensing of new innovative assets.
In 2022, we intend to capitalize on all strengths described above to grow and expand our business prospects. We are now well-positioned to execute on our existing programs, to initiate and/or acquire new ones, and have a solid track record for negotiating partnerships that recognizes the potential of these programs.
viii. Our Strategy
GENFIT's goalstrategy is to make the most of our strengths to become a world leader in the development of innovative therapies and diagnostics in severe chronic liver-relatedlife-threatening liver diseases, prioritizing rare diseases. To do so, we intendThis strategy is designed to leverageserve our unique expertise acquired over the last two decades in the development of therapeutic and diagnostic assets from the early stages (screening, preclinical stages) to pre-marketing (Phase 3), to grow and diversify our pipeline.
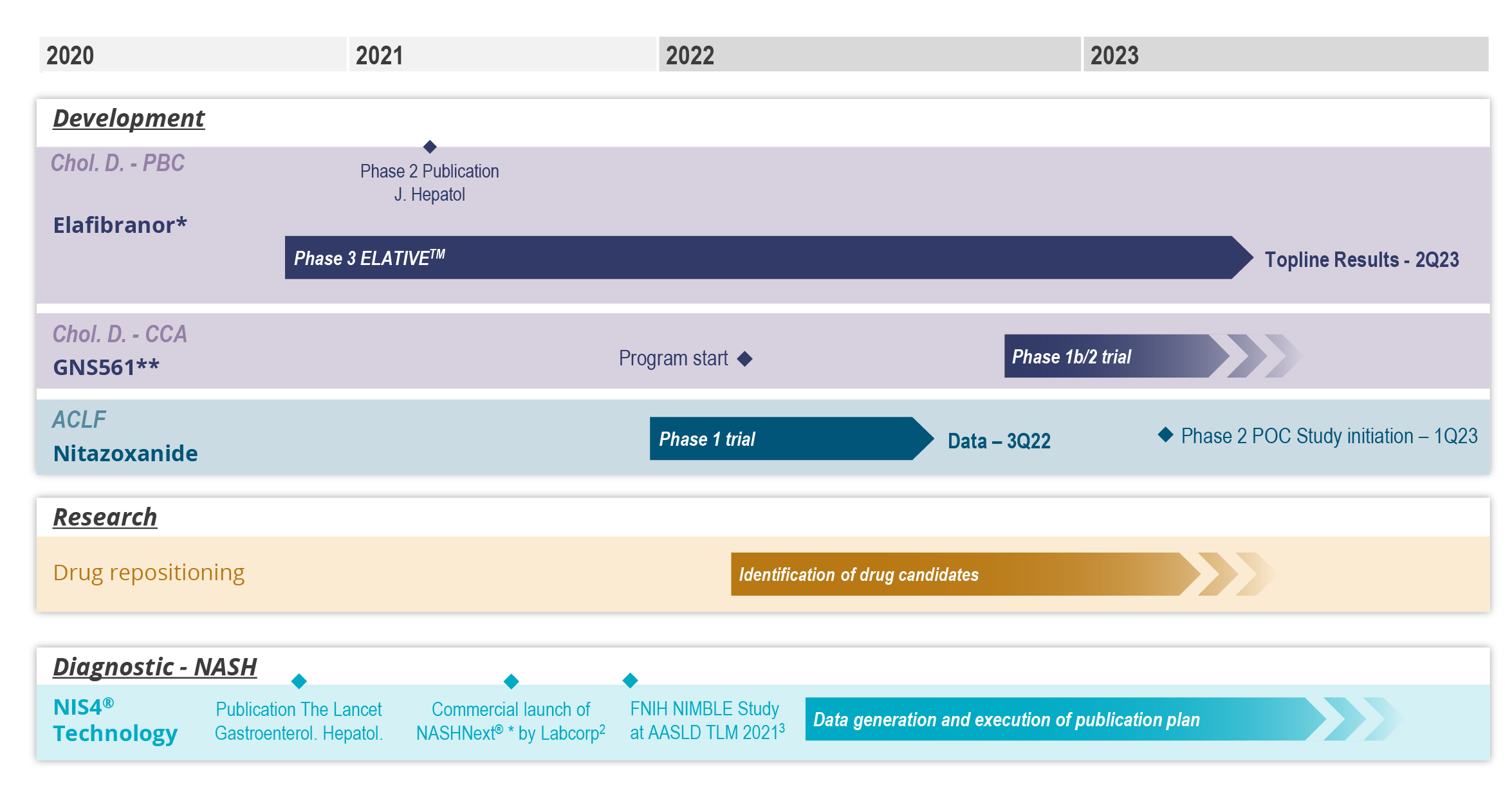
Upcoming milestones, data announcements and launch dates are anticipated and subject to change. ACLF: Acutepurpose, focused on Chronic Liver Failure. CCA: Cholangiocarcinoma. NASH: Non-Alcoholic Steatohepatitis. PBC: Primary Biliary Cholangitis. POC: Proof-of-Concept. *Elafibranor is an investigational compound and has not been approved by any regulatory authority in any indication. Ipsen has global rights to develop and commercialize elafibranor in primary biliary cholangitis (including open-label extension, confirmatory PBC study and life cycle management), with the exception of China, Hong Kong, Taiwan, and Macau where Terns Pharmaceuticals holds the exclusive license to develop and commercialize elafibranor. **GNS561 is an investigational compound and has not been approved by any regulatory authority in any indication. GENFIT holds the exclusive rights for GNS651 in Cholangiocarcinoma in the United States, Canada and Europe. 1. 1. Exclusive licensing to Labcorp for development and commercialization of NIS4 Technology to power a next-generation NASH diagnostic test. 2. Sanyal et al. Hepatology. 2021 Vol 74 Issue 6 – Suppl: 1383A.improving patients' lives.
•–Targeted therapeutic areas
In drug development, GENFIT’s strategic focus is on severe orThe relevance of our positioning in rare, life-threatening liver diseases withfor which unmet needs remain high unmet medical needs. In 2021, GENFIT has defined two specificis threefold:
•It allows us to act, as a pioneer, for the benefit of patients whose lives are in danger, and who have few, if any, therapeutic areas of interest with these criteria: cholestatic diseasesoptions;
•It allows us to apply our know-how, our expertise and ACLF. Givenexperience to try to bring patients satisfactory solutions thanks to the current landscape, standard of care, lack of marketed optionsadvances enabled by our innovation work in the preclinical and KOLs opinions, we see a possibilityclinical fields and;
•Finally, it allows us to shorten development and approval timelines and we intend to interact with regulatory agencies to investigateconsider potential accelerated paths to approval. In 2022 the Company will continue to explore opportunities to evaluate new assets in these indications, as well as opportunities to expand focus on other severe chronic diseases.
In diagnostics, GENFIT's strategic focus remains primarily on NASH, where we intend to further establish NIS4 technology, and we also intend to explore diagnostic opportunities in new indications that are integrated into our pipeline.approval processes.
•–Our approach to generate value
In terms of drug development, our goal is to capitalize onfocus our expertiseefforts in one specific area - rare and life-threatening liver diseases - for greater operational efficiency, and to expand our pipelinedistribute the risk across different programs with drug candidates with high potential that act via different mechanisms of action. To achieve thisaction, with the goal to improve our chances of success.
Our goal is also to reduce development timelines, and we follow a dual-track approach based on:therefore favor two approaches to strengthen our portfolio:
–•Repurposing of molecules approved in other indications (e.g. NTZ, an antiparasitic drug, in ACLF); and
–•In-licensing and/or acquisition of molecules developed by other companies (e.g. GNS561, from Genoscience Pharma, in CCA)CCA, and in ACLF VS-01-ACLF from Versantis AG, SRT-015 from Seal Rock Therapeutics and CLM-022 from Celloram Inc.).
OurGENFIT's ambition is to develop drug candidates from earlythe earliest stages up to the latest stages, including Phase 3 and pre-marketing.3. Depending on predefined criteria such as therapeuticthe targeted indication or competitive environment, weor potential opportunities in terms of partnerships, GENFIT will then choose what we consider to be the best option to commercialize our most promising assets:assets for which the company has not yet licensed the rights:
–•Build our own marketing and sales forces to commercialize the asset on our own, or
–•Leverage the existing relationship with our preferred commercial partner Ipsen which provides a natural path to commercialization, or
–•Commercialize via another partner.
In diagnostics,We consider the patient journey as a whole and are also looking to continue to be present in the diagnostic field, specifically to determine which populations to treat within the therapeutic areas we are targeting with our goal is to expand our footprint and extend awareness about our technology, leveraging our data, expertise, understanding of the medical needs, and KOL network.drug candidates.
•–Our corporate priorities in 20222024
We have defined three corporate priorities for 2022.
The first priority consists of growing and diversifying our pipeline of innovative products, with two possible avenues: repurposing and in-licensing.
The second priority is to accelerateIn 2024, GENFIT will prioritize the execution of existing programs:its clinical development programs, as well as research programs focused on pre-clinical/non-clinical development.
–Elafibranor in PBC: Phase 3 ELATIVE clinical trial topline data readout in the second quarter of 2023;
–NTZ in ACLF: Phase 1 data readout by third quarter of 2022 ;
–GNS561 in CCA: Finalize development plan and start Phase 2 trial by the second half of 2022;
–NASH Diagnostics: Further establish NIS4 technology.
The third priority is to further strengthen our internal organization, to ensure our ability to meet our corporate ambitions for the coming years.
ix. Our Drug Candidates and Diagnostic Development Programs
Cholestatic DiseasesOur pipeline includes our most advanced asset elafibranor in PBC (out-licensed to Ipsen) and two therapeutic franchises: one focused on ACLF with five proprietary or in-licensed assets, and one focused on other life threatening diseases with two proprietary assets. We also have developed two programs in a diagnostic franchise.
Chronic cholestatic diseases are1.Elafibranor in Primary Biliary Cholangitis (PBC) - out-licensed to Ipsen
•About PBC
PBC is a rare, chronic, progressive liver disease of autoimmune etiology, characterized by defectiveinjury of the intrahepatic bile ducts that, in untreated patients or non-responders to existing therapies, may progress to hepatic fibrosis, cirrhosis, hepatic decompensation, and death unless they receive a liver transplant. PBC disproportionately affects women versus men (approximately 10:1) and is typically diagnosed in patients between 40 years to 60 years of age. The incidence and prevalence rates for PBC in Europe, North America, Asia, and Australia are reported as ranging from 0.33 to 5.8 per 100,000 inhabitants and 1.91 to 40.2 per 100,000 inhabitants, respectively. It is estimated that there were 47,000 prevalent cases of PBC in the United States white population and that approximately 3500 new cases are diagnosed each year. Over 60% of the newly diagnosed cases are asymptomatic. The majority of asymptomatic patients become symptomatic within 10 years and the estimates for developing symptoms at 5 and 20 years are 50% and 95%, respectively. Patients with PBC progress at varying rates, some experiencing liver decompensation over a period of several years while others experience liver decompensation over decades. PBC is one of the leading indications for liver transplantation. Despite its rarity, PBC remains an important cause of morbidity in the Western world. PBC has also been identified as an important risk factor for hepatocellular carcinoma.
PBC is characterized by cholestasis caused by autoimmune destruction of biliary ducts with progressive impairment of bile flow in the liver. This results in increased hepatocellular bile acid transport from the liverconcentrations, which are toxic to the intestine, whichliver. Such hepatocellular injury is caused byassociated with a local inflammatory response resulting early on in an abnormal elevation of serum alkaline phosphatase (ALP) levels, a hallmark of the disease. Antimitochondrial antibody and IgM are specific immunological hallmarks of PBC, and antimitochondrial antibody is a diagnostic marker of the disease in approximately 90% of patients. Liver biopsy, while confirmatory, is no longer the standard of care.
ALP is also routinely used to clinically monitor the disease and serves as a leading indicator of disease progression. ALP increases with disease progression as bilirubin starts to decline in more advanced disease (as the excretory function starts to decline), and both have been shown to be highly predictive of long-term clinical outcomes (e.g., transplant-free survival). There is a near log-linear correlation of both elevated ALP and bilirubin after 1 year of follow-up with long-term liver transplant-free survival.
The most common symptoms of PBC are fatigue and pruritus. The mechanisms underlying these symptoms are not well elucidated and neither correlates with disease stage or clinical outcomes.
The following diagram depicts where and how bile ducts are destroyed.
–Limitations of Current Treatment Options
Ursodeoxycholic acid (UDCA), an epimer of the primary damagehuman bile acid, was the only medicine approved to the biliary epitheliumtreat PBC until May 2016. UDCA has been shown to improve ALP and bilirubin, and to delay histological progression, thereby increasing liver transplant-free survival. While UDCA has had a marked impact on clinical outcomes in most cases.PBC, a large proportion of patients have an inadequate response. It is estimated that up to 40% of UDCA-treated patients have a suboptimal response to UDCA. ALP levels remain elevated in up to 70% of patients who are currently being treated or are intolerant to UDCA. Such patients remain at risk of disease progression and longer term adverse clinical outcomes.
Elafibranor for the Potential Treatment of PBC
About PBC
PBC is an autoimmune disease resulting from progressive destruction of the small bile ducts inside the liver. When liver bile ducts are destroyed, the bile which normally would travel to the small intestines to aid in digestion and elimination of waste instead accumulates in the liver, contributing to inflammation and fibrosis. PBC is believed to be an autoimmune disease in which a person’s immune system is overactive and attacks normal, healthy bile duct cells. The following diagram depicts the distinction between normal bile ducts and those that have been destroyed.
PBC is a disease with a global prevalence of approximately 40 cases per 100,000. In the United States, the prevalence of PBC increased from 21.7 to 39.2 per 100,000 from 2006 through 2014. Women are much more likely to be affected by PBC than men, and the incidence increases after the age of 50.
The initial symptoms of PBC are general fatigue and pruritus, which is itchy skin; other potentially associated symptoms include dry eyes, dry mouth and jaundice. However, approximately 60% of patients are asymptomatic when the disease is diagnosed. PBC is diagnosed based on blood tests revealing the presence of anti-mitochondrial antibodies, or AMAs, and high levels of the liver enzyme ALP (alkaline phosphatase). Although cirrhosis occurs in advanced stages of the disease, most patients do not have cirrhosis at the time of PBC diagnosis.
Left untreated, PBC typically leads to cirrhosis, liver failure and the need for liver transplantation. In the absence of treatment, the 10-year survival of asymptomatic patients is estimated to be between 50 and 70%, with a median survival of 16 years. Among symptomatic patients, median survival in the absence of treatment is only seven to eight years. PBC is believed to be responsible for 2-3% of deaths by cirrhosis.
Limitations of Current Treatment Options
There is currently no cure for PBC, although there are medications that work to slow its progression. For many years, ursodeoxycholic acid, or UDCA, was the only drug approved by the FDA for the treatment of PBC. UDCA is a naturally occurring bile acid that is normally produced in the liver by healthy cells. UDCA, administered orally, is designed to help move bile through the liver and into the intestines. Although UDCA is effective in more than 50% of patients, up to 40% of patients do not respond or respond poorly to treatment and an additional 5-10% of patients are unable to tolerate the drug.
In May 2016, the FDA approved obeticholic acid, marketed as Ocaliva™ by Intercept Pharmaceuticals, Inc., for the treatment of PBC in combination with UDCA in adults with an inadequate response to UDCA, or as a single therapymonotherapy in adults unable to tolerate UDCA. In September 2017, following the death of 19 PBC patients being treated with Ocaliva,™, the FDA published a safety announcement for Ocaliva,™, indicating that some patients with moderate to severe decreases in liver function had been incorrectly dosed, resulting in an increased risk of serious liver injury and death. The FDA also indicated that Ocaliva™ may also be associated with liver injury in some patients with mild disease who are receiving the correct dose. In February 2018, the FDA issued a Boxed Warning added to the Ocaliva™ label, the most severe warning required to be included in labeling by the FDA. Concerns remain over pruritus and serious liver injury or liver death caused by administration of Ocaliva™.Ocaliva. In its Phase 3 clinical trial, severe pruritus was reported in 23% of patients in the Ocaliva™ 10 mg dose cohort and in 19% of patients in the Ocaliva™ titration cohort, in which dosing was initiated at 5 mg and titrated up to 10 mg based on clinical response, compared to 7% of patients in the placebo group. In May 2021, the FDA issued a drug safety communication restricting the use of Ocaliva™ in patients with PBC having advanced cirrhosis. The use of Ocaliva™ is now contraindicated in advanced cirrhosis due to the risk of liver failure, which may require liver transplant.
Accordingly, we believe there is still a significant medical need for new therapies, as current treatments either are ineffective for a large portion of PBC patients, cause significant side effects or include safety risks.
•Our Solution:Program: Elafibranor for the Potential Treatment of PBC
We believe that elafibranor has the potential to offer a therapeutic solution that can be effective in treating PBC while also maintaining a favorable tolerability profile and lacksafety profile.
–Elafibranor in PBC: rationale and mechanism of demonstrated safety concerns.action
Elafibranor mechanism of action targets PPARα and PPARδ. Targeting PPAR receptors has shown multiple beneficial activities,effects, including the reduction of bile acid synthesis, improved detoxification of bile in the bile duct and anti-inflammatory activity. In third-party clinical trials, drugs targeting PPAR receptors resulted in a significant decrease in serum alkaline phosphatase (ALP) and improved biochemical profiles and pruritus in PBC patients. Patients with PBC often have elevated ALP, a marker of cholestasis, and studies have shown a correlation between elevated ALP levels and increased risk of adverse patient outcomes.
We have observed elafibranor’s effect in loweringreducing ALP levels and markers of inflammation in our Phase 2 clinical trial in patients with PBC.
Our Clinical Program for ElafibranorPhase 2: positive Phase 2 results published in the Treatment of PBC
Phase 3 ELATIVE triala renowned scientific journal
Positive results from our Phase 2 clinical trial of elafibranor in PBC whichformed the rationale to launch the ELATIVE® Phase 3 trial previously described. These results were announced in December 2018 and then presented in April 2019 at the International Liver Congress 2019 organized by EASL (European Association for the Study of the Liver), formed the rationale to move the program intoand then published in The Journal of Hepatology in 2021.
The Phase 3 and to launch the ELATIVE Phase 32 clinical trial for the evaluation of elafibranor in this indication.
ELATIVE is an international Phase 3 double-blind randomized placebo-controlled study with an open-label long-term extension (LTE) evaluating the efficacy and safety of 80 mg elafibranor once daily versus placebo in patients with PBC and inadequate response or intolerance to UDCA. In the double-blind treatment period, patients will be randomized inwas a 2:1 ratio to receive 80 mg elafibranor (n=100) or placebo (n=50) once daily.
After the variable double-blind treatment period (52 - 104 weeks), all patients will receive elafibranor at 80 mg per day for five years at most during the LTE.
The primary endpoint is the response to treatment at week 52 defined as defined by biochemical parameters: ALP < 1.67 x ULN and total bilirubin ≤ ULN and ALP decrease ≥ 15%. Secondary endpoints include response to treatment based on ALP normalization at week 52 and change from baseline in pruritus through week 52 on PBC Worst Itch NRS score.
Due to the COVID-19 pandemic, we announced in March 2020 a delay in the initiation of the ELATIVE trial. In September 2020, we announced the first patient first visit in the ELATIVE trial. Appropriate measures have been implemented, including virtual appointments, biological evaluations performed by local laboratories and delivery of the drug candidate to the patients’ homes, to ensure the safety of participants in the study. Enrollment is expected to be completed in the second quarter of 2022 for the double-blind part of ELATIVE, which is to be used to support accelerated approval. We remain committee to delivering ELATIVE topline data in the second quarter of 2023.
Following publication of the interim Phase 3 topline results, Ipsen will lead the further clinical and commercial development of elafibranor in PBC. See below "Partnering with elafibranor - Strategic Collaboration with Ipsen" for additional information. In addition to the ELATIVE Phase 3 trial that will be used to support and seek regulatory approval under the accelerated approval pathway, led by Ipsen, the program will also include a confirmatory study based on hard clinical endpoints, which will be conducted by Ipsen and which is required in order to obtain full marketing approval.
Phase 2
In December 2018, we announced positive preliminary results, including achievement of the primary endpoint and the composite endpoint, from our Phase 2 multi-center, double-blind, randomized, placebo-controlled clinical trial to evaluateevaluating the efficacy and safety of elafibranor after 12 weeks of treatment in patients with PBC and inadequate response to UDCA. The trial was conducted at multiple clinical centers in the United States and in three European countries and enrolled a total of 45 patients. The patients were randomized into one of three treatment arms, receiving either elafibranor 80 mg, elafibranor 120 mg or placebo.
The primary objective of the trial was to determine the effect of daily oral administration of elafibranor on ALP in these patients, based on relative change from baseline in serum ALP levels compared to placebo. In addition to assessing the tolerability and safety of elafibranor in patients with PBC, secondary endpoints included assessment of elafibranor 80 mg and 120 mg as compared to placebo on several outcome measures, including:
•composite endpoint composed of ALP and bilirubin, with response defined as (1) ALP less than 1.67 times the upper limit of normal, or ULN, (2) total bilirubin within normal limits and (3) a reduction of ALP of more than 15%;
•changes in patients’ risk scores as measured by several PBC risk scoring systems (Paris I and II, Toronto I and II and UK-PBC);
•change from baseline in pruritus, as measured by a 5-D itch scale and visual analogue scale; and
•change from baseline in quality of life, as measured by PBC-40, a patient-derived questionnaire.
In the preliminary results published in December 2018, weWe observed that the mean decreasechanges from baseline in ALP in both of the elafibranor treatment groups showed statistically significant improvementdecreases compared to placebo. In the elafibranor 80 mg and 120 mg treatment groups mean decreases in ALP were 48% (n=15) and 41% (n=14), respectively, whereas the mean ALP increased by 3% (n=15) in the placebo group. When adjusted for placebo, the treatment effect of the elafibranor 80 mg and 120 mg treatment groups was a mean decrease in ALP of 52% (p<0.001) and 44% (p<0.001), respectively. Based on these results, elafibranor achieved the primary endpoint of the trial with high statistical significance.
| | | | | | | | |
| | |
| (1) Schattenberg et al. J. of Hepatol. 2021, Vol. 74, Issue 6:1344-1354; | |
Elafibranor also achieved high statistical significance on the composite endpoint of ALP and bilirubin, with response defined as (1) ALP less than 1.67 times the ULN, (2) total bilirubin within normal limits and (3) a reduction of ALP of more than 15%. The elafibranor 80 mg and 120 mg treatment groups achieved mean response rates of 67% (p=0.001) and 79% (p<0.001), respectively, as compared to 6.7% in the placebo group. This composite endpoint was the primary endpoint in the Phase 3 clinical trial of Ocaliva™Ocaliva that led to its FDA marketing approval. In a three-month Phase 2 clinical trial of Ocaliva™,Ocaliva, treatment with Ocaliva™10 mg of Ocaliva resulted in a mean response rate of 23%, compared to a placebo response rate of 10%, on this composite endpoint.
Patients treated with elafibranor showed improvement in other PBC markers such as gamma-glutamyl transferase (γGT), markers of inflammation, and metabolic markers such as total cholesterol, low-density lipoprotein-C, and triglycerides. γGT level remained stable throughout the treatment period in placebo treated patients (+0.2±26%), while significant reductions were observed in both elafibranor-treated groups (at week-12: -37.1±25.5%; p<0.001 vs placebo with 80 mg and -40.0±24.1%; p<0.01 vs placebo with 120 mg). The γGT change over time was similar to the changes in ALP observed in the elafibranor-treated groups. Additionally, a reduction of 5’-nucleotidase at both doses of elafibranor vs placebo was observed at week 12. Finally, significant decreases in the elafibranor-treated groups relative to placebo patients were observed in IgM and inflammatory markers including C-reactive protein and haptoglobin. As expected, patients had features of PBC-related dyslipidemia, notably high HDL-cholesterol at baseline. As compared to placebo, elafibranor-treated groups showed decreases in total cholesterol, LDL-cholesterol and triglycerides. Finally, circulating levels of the bile acid precursor C4 were decreased in the elafibranor-treated groups, but not in the placebo group.
Elafibranor treatment did not induce or exacerbate pruritus. In contrast, a favorable trend was evidenced by a reduction of the virtual analogue scale or VAS score in patients that reported pruritus (VAS ≥0 mm) at baseline. A similar trend was observed in the pruritus domain of the PBC-40 QoL questionnaire with a median change from baseline of -25% and -21% in the 80 mg and 120 mg group, compared to placebo, which remained unchanged. This apparent improvement in pruritus is particularly impressive considering that it was observed in this trial of a duration of 3-months. Considering the burden that pruritus has on the quality of life in a significant proportion of patients with PBC, it will be importantwe designed our ELATIVE® Phase 3 trial with several secondary endpoints designed to confirmmeasure the benefitpotential benefits that elafibranor may have in the phase 3 study, which will be of longer duration.alleviating this symptom.
Treatment with elafibranor was generally well tolerated, with a similar number of patients experiencing adverse events in the drug treatment and placebo arms of the trial, with the most common adverse events being of a gastrointestinal nature and of mild or moderate intensity, and included nausea, fatigue and headache. Two patients experienced serious adverse events, of which only one was considered as possibly drug-related. The latter patient suffered from two preexisting auto-immune diseases (PBC and myasthenia gravis) and during the trial presented with a third auto-immune disease (auto-immune hepatitis, or AIH). This diagnosis was made in a patient with poly-auto-immune diseases, and AIH consecutive to PBC or AIH-PBC overlap syndrome are not uncommon, occurring in up to 2.5% and 14% of PBC patients, respectively. While this factor and/or other concomitant medications could be considered as confounding factors, a causal relationship to study drug could not be excluded. The other patient experienced a serious adverse event or SAE deemed unrelated to treatment with elafibranor and withdrew from the trial after only one daily dose.
In April 2019, the FDA granted elafibranor Breakthrough Therapy Designation, based on the Phase 2 data, for treatment of PBC in adults with inadequate response to UDCA and in July 2019, both the FDA and EMA granted elafibranor Orphan Drug Designation in PBC.
–Phase 3 ELATIVE® trial: topline data announced in June 2023
On June 30, 2023 we and Ipsen announced positive 52-week interim topline data from the pivotal ELATIVE® Phase 3 trial. In November 2023, Ipsen provided additional details in a late breaking oral presentation during the American Association for the Study of Liver Diseases congress in Boston, Massachusetts, USA, and published detailed results in the New England Journal of Medicine.
The first part of the trial assessed the efficacy and safety of elafibranor, an investigational dual α,δ PPAR agonist, in the treatment of patients with the rare cholestatic liver disease, PBC, who have an inadequate response or intolerance to the current standard of care therapy, UDCA. Results position elafibranor as a potentially important new treatment option, where there is still high unmet need.
Results showed statistically significant improvements in biomarkers of disease progression across key endpoints with a significant treatment benefit achieved in the primary composite endpoint, demonstrating a 47% placebo-adjusted difference (P<0.001) between patients on elafibranor 80mg (51%) compared with patients on placebo (4%) achieving a biochemical response. In the trial, a biochemical response is defined as alkaline phosphatase (ALP) <1.67 x upper limit of normal (ULN), an ALP decrease ≥ 15 percent and total bilirubin (TB) ≤ ULN at 52 weeks. ALP and bilirubin are important predictors of PBC disease progression. Reductions in levels of both can indicate reduced cholestatic injury and improved liver function.
Only patients receiving elafibranor achieved normalization of ALP (upper limit of normal 104 U/L in females and 129 U/L in males) at Week 52 (15% vs 0% placebo, P=0.002), a key secondary endpoint of the trial. The significant biochemical effect of elafibranor measured by ALP reduction was further supported by data demonstrating reductions from baseline in ALP levels were rapid, seen as early as Week 4 in the elafibranor group, and were sustained through Week 52, with a decrease in ALP of 41% on elafibranor compared with placebo.
Additional details covered the effect of treatment with elafibranor on pruritus (severe itch) across three separate patient-reported outcome measures. On the key secondary endpoint using the PBC Worst Itch NRS score, the reduction of pruritus observed for elafibranor versus placebo was not statistically significant (LS mean, –1.93 versus –1.15; difference, –0.78; 95% CI, –1.99 to 0.42; P=0.20). Two other secondary patient-reported outcome measures were used to assess itch, and greater reductions in pruritus were observed with elafibranor compared with placebo at Week 52, according to the itch domain of PBC-40 quality of life questionnaire (LS mean difference -2.3; 95% CI, -4.0 to -0.7) and 5-D Itch total score (LS mean difference, -3.0; 95% CI, -5.5 to -0.5).
In the study, elafibranor was generally well tolerated with a safety profile consistent with that observed in previously reported studies.
Ipsen will assume responsibility for all additional clinical development, including completion of the long-term extension period of the ELATIVE® trial, and global commercialization (outside of Greater China, where elafibranor is licensed to Terns).
On December 7, 2023, Ipsen confirmed U.S. FDA granted priority review for New Drug Application for elafibranor for the treatment of PBC, with a PDUFA date set for June 10, 2024. The European Medicines Agency (EMA) has also validated Ipsen’s Marketing Authorization Application (MAA) for elafibranor and the review of the submission to the EMA’s Committee for Medicinal Products for Human Use (CHMP) began on October 26, 2023. Furthermore, a third simultaneous regulatory filing of elafibranor has been validated for review by the UK Medicines and Healthcare products Regulatory Agency (MHRA).
–Next milestones
Acceptance of filings in the US and Europe have triggered the first milestone payment for GENFIT in December 2023. In case of favorable decision by the authorities, we would receive additional milestone payments after US and European commercial launches by Ipsen, and royalties on the sales of elafibranor by Ipsen. For more information about the financial terms of the agreement, including the milestone payment received in 2023, see Note 2.1.1 - "Major Events in the Period and Events after the Period - Positive Results from Phase 3 ELATIVE® trial" to our consolidated financial statements included in this annual report. 2. Our therapeutic franchise in ACLF
GENFIT’s ACLF franchise is now comprised of five assets (VS-01-ACLF, NTZ, SRT-015, CLM-022, VS-02-HE) based on differentiated mechanisms of action leveraging complementary pathways.
•About ACLF
ACLF is a rare, life-threatening, but potentially reversible condition of varied etiology. ACLF is a syndrome, globally defined by multi-organ dysfunction and failure in patients with chronic liver disease or liver cirrhosis and high short-term mortality within a period of 28 to 90 days. Today, hepatologists recognize ACLF to be a medical entity as a whole.
Patients with cirrhosis may initially be compensated. With progression, many patients will go on to have acute decompensation of cirrhosis characterized by the rapid development of complications such as ascites, HE, gastrointestinal hemorrhage, or bacterial infection, which are very common causes of hospitalization. On admission, approximately 30% of these patients will have or develop liver and/or other organ failure(s) (i.e, brain, kidneys, cardiovascular and respiratory) and will be considered as having ACLF.
ACLF is an underserved medical condition associated with high short-term mortality (23% to 74% mortality at 28 days, depending on severity grade). Currently, no drugs have been approved in ACLF. In 2021, the prevalence of ACLF is estimated to be approximately 294 thousand across the U.S., EU4 (France, Germany, Italy, and Spain) and UK. This market is expected to grow to approximately 300 thousand patients by 2036 due to an aging population and a higher prevalence of non-alcoholic fatty liver disease (MAFLD)/MASH, diabetes, obesity, alcohol consumption and drug induced liver injuries.
Rising alcohol consumption has already impacted China, the United States, and Denmark, all of which have documented a doubling in alcoholic liver disease hospitalizations over a 10-year period.
In the U.S., there are over 600,000 hospitalizations per year for decompensated cirrhosis. With a 10-30% ACLF prevalence in this population the annual number of ACLF hospitalizations in the U.S. is estimated to be between 60,000 and 180,000. In the five major European countries, there are about 800,000 hospitalizations for decompensated cirrhosis. With a 20-30% prevalence in this population, the annual number of ACLF hospitalization is estimated to be between 160,000 and 240,000.
Cirrhosis and ACLF represent a substantial health and economic burden. For example, in the United States in 2011, the total inpatient costs for cirrhosis with and without ACLF was estimated to be more than $10 billion. In the same study, the cost per hospitalization was 3.5-fold higher for patients with ACLF than for patients with cirrhosis who did not have ACLF.
Such high hospitalization costs for critically ill patients with ACLF as compared to patients with cirrhosis without ACLF can be easily explained by higher rates of hospitalization in the ICU and, most importantly, by 2-3-fold longer hospital stays: average of 16 days for patients with ACLF versus 7 days for patients with cirrhosis who did not have ACLF. Complications are the key drivers impacting the length of patient's hospital stays with renal and infectious complications being associated with the longest hospital stays.
–A high unmet medical need
There are no approved therapies currently available for patients with ACLF other than treatment of precipitating events, when identified, and organ failure support (e.g., renal replacement therapy in the case of kidney failure). The only definitive treatment option is liver transplantation. Due to the emergency setting, limited access to compatible liver donors and, in some cases, no accessible liver transplant capabilities, approximately 15-30% of patients die while awaiting liver transplant.
Patients with acute decompensated cirrhosis are generally hospitalized in the regular hepatology ward. Within one week, patients may progress to ACLF and are usually transferred to an intensive care unit where organ support and general care can most effectively be provided. Despite intense efforts to improve the standard of care, the current high short-term mortality rate highlights the critical medical need of new therapies to help patients to rapidly recover and survive an ACLF episode without liver transplantation or bridge them to liver transplant, when appropriate.
The mean survival time in patients with ACLF is 3-5 years. In a study of 1,343 hospitalized patients with cirrhosis and acute decompensation, 303 had ACLF when the study began, 112 developed ACLF, and 928 did not have ACLF. The 28-day mortality rate among patients who had ACLF when the study began was 33.9%, among those who developed ACLF was 29.7%, and among those who did not have ACLF was 1.9%. In general, a greater number of organ failures is associated with higher short-term mortality. For example, the 28-day mortality rate for patients having 3 or more organ failures approaches 80%.
–Bedside management of ACLF patient hospitalized in Intensive Care Unit
| | | | | | | | |
| 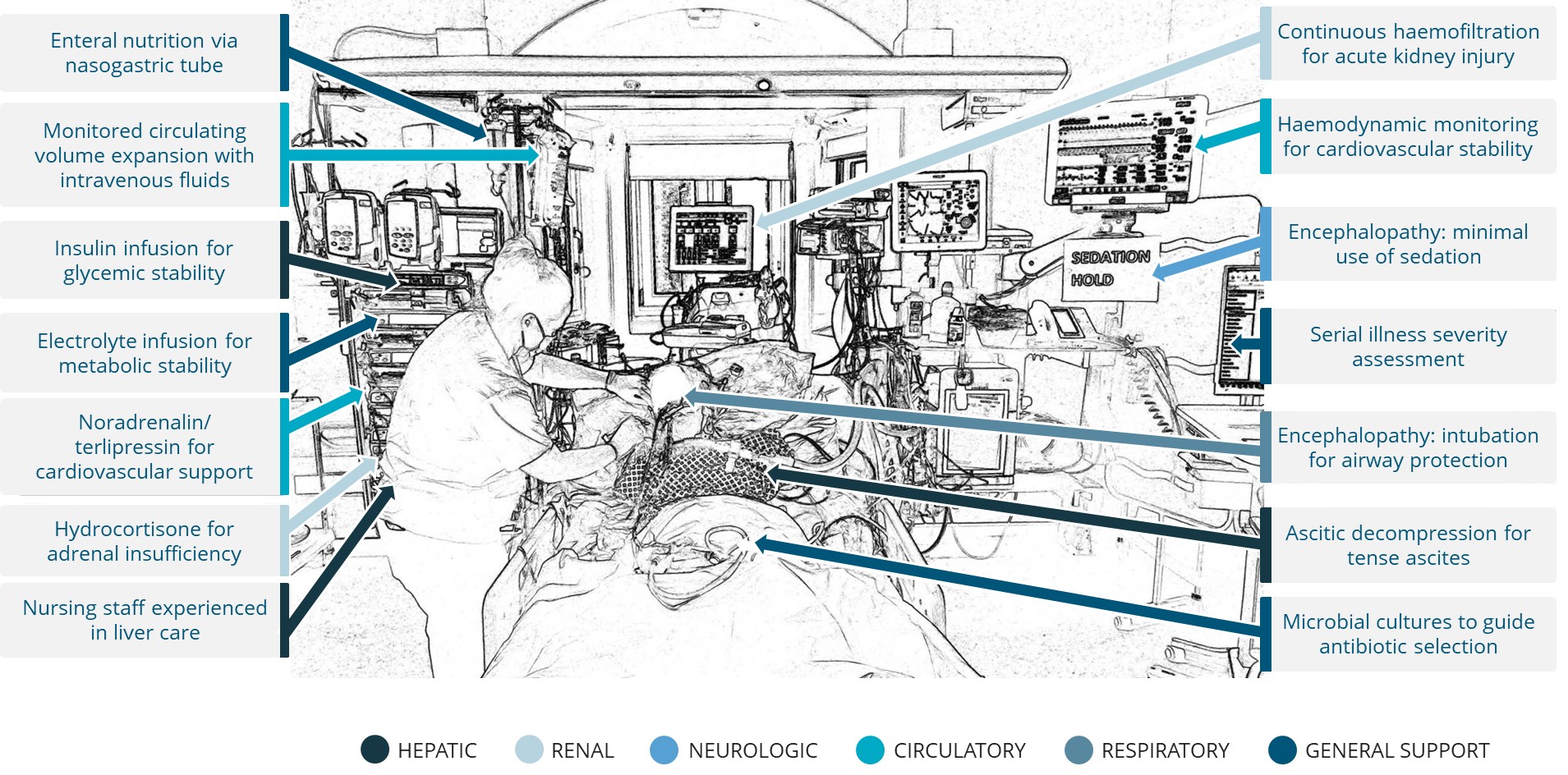 Bernal W. et al, J Hepatol 2021 Bernal W. et al, J Hepatol 2021 | |
•Our first program: VS-01-ACLF for enhancing the systemic elimination of ammonia and other ACLF-related metabolites
–Rationale and mechanism of action
VS-01-ACLF is an innovative, potential first-in-class investigational drug candidate based on a proprietary scavenging liposomal technology. It is administered directly into the peritoneal (abdominal) cavity following drainage (paracentesis) of ascites, one of the most common complications in patients with ACLF. VS-01-ACLF was granted the Orphan Drug Designation in ACLF by the FDA.
In the setting of ACLF, toxic metabolites build up in the bloodstream due to organ failures. VS-01-ACLF is designed to enhance the clearance of ACLF-related metabolites by extracting them from the blood into the peritoneal cavity by passive diffusion. Toxic metabolites, either captured by the liposomes or in the surrounding fluid, are then drained from the body.
VS-01-ACLF is in clinical development as a potential first-line therapy for the timely resolution of ACLF. The identification of the toxic metabolites extracted by VS-01 and associated clinical outcomes are being further investigated in the ongoing proof of concept Phase 2a study. Preclinical and clinical pharmacodynamic and metabolomic studies have shown that VS-01-ACLF could be the first drug to use the intraperitoneal route to:
•Simultaneously support the liver, kidney and brain, the organs that most often fail in patients with cirrhosis, ACLF, and ascites; and
•Reduce inflammation, which is a key driver of ACLF.
More specifically, VS-01-ACLF liposomes are designed to trap bacterial endotoxins and mediators of inflammation as well as ammonia, one of the main toxins associated with Hepatic Encephalopathy and brain failure. Overall, we believe VS-01-ACLF will enhance the clearance of hepatic and uremic toxins to support liver, kidney and brain function.
Thus, VS-01-ACLF may be well suited as a treatment for patients with ACLF, with the potential to improve survival, to increase the probability of success for liver transplant in selected patients, and to reduce healthcare costs.
–Evidence supporting further development
◦Non-clinical evidence
Non-clinical studies evaluated the efficacy of VS-01-ACLF in small and large animal models. VS-01-ACLF was shown to extract kidney and liver toxins (185 extracted metabolites, including ACLF-related metabolites and uremic toxins) as well as inflammation mediators (28 lipophilic compounds identified including fatty acids and bile acids). Moreover, VS-01-ACLF efficiently captured ammonia. In healthy rats, VS-01-ACLF was shown to remove 20 times more ammonia than a control solution without liposomes. The extraction of ammonia in the peritoneal space led to a decrease in ammonemia in rats and pigs and to a decrease in brain edema in a model of bile duct ligated rats.
In rats, VS-01-ACLF was shown to be safe and well tolerated during a prolonged intraperitoneal dwell time (>4h) and during single and multiple doses.
Based on safety pharmacology studies and a GLP repeated dose toxicity study in minipigs receiving a daily session for 10 days, VS-01-ACLF was found to be safe and well tolerated. No immune reactions were observed in pigs which are known to be highly sensitive to colloidal formulation and prone to the so-called complement activation-related pseudoallergy (CARPA) reaction following single and daily administration for 10 days.
◦Clinical evidence
A Phase 1b first-in-human (FIH) open-label study has been completed in 12 patients with cirrhosis, ascites, and covert Hepatic Encephalopathy. As a primary objective, the study assessed the safety and tolerability of VS-01-ACLF following intraperitoneal administrations of single-ascending doses and multiple doses on top of standard of care (SOC). VS-01-ACLF pharmacokinetics and efficacy profile were assessed as secondary objectives. VS-01-ACLF was generally well tolerated. Importantly, >80% of patients demonstrated improvement or stabilization of the severity of their liver disease (as assessed by Child-Pugh score). There was a trend towards dose related increases in the clearance of ammonia removed from the peritoneal cavity as well as improvement in cognitive assessments used in the evaluation of patients with Hepatic Encephalopathy. Taken together, the benefit risk profile of VS-01-ACLF is supportive of ongoing clinical investigation in patients with ACLF having ascites. The main outcomes of the Phase 1b FIH study were presented at the AASLD 2021.
Effect of VS-01-ACLF on metabolites reduction presented at EASL 2022 (2 abstracts):
•Abstract 1 (metabolites associated with organ failure)
•Abstract 2 (metabolites associated with bacterial infection
–Next milestones
An international Phase 2a, open-label, randomized, controlled, multi-center, proof of concept study (UNVEIL-IT®) is ongoing and aims to assess the efficacy, safety and tolerability of VS-01 in addition to standard of care (SOC), compared to SOC alone, in approximately 60 adult patients with ACLF grades 1 and 2 and ascites.
The primary objective is to evaluate efficacy as measured by the CLIF-C ACLF (Chronic Liver Failure Consortium Acute on Chronic Liver Failure score) at Day 7, which is highly correlated with mortality in patients with ACLF. Secondary objectives include 90-day mortality, 28-day mortality, time to death, change in ACLF grade, transplant-free survival, and safety and tolerability.
Interim data from the UNVEIL-IT® trial are targeted for the second half of 2024.
•Our second program: nitazoxanide (NTZ)
–Rationale and mechanism of action
The identification of NTZ is the result of our research program initially designed to discover novel anti-fibrotic molecules with a priority given to liver fibrosis.
During further research we have also discovered that NTZ and its circulating metabolite, tizoxanide (TZ), have additional anti-inflammatory effects through the inhibition of inflammatory cell activation. In our preclinical research, the apparent beneficial effects we have observed with NTZ may be explained in part by the anti-bacterial properties of NTZ (acting on intestinal microbiota dysbiosis/overgrowth and improving the intestinal barrier), and direct dose-dependent anti-inflammatory effects on immune cells (macrophages).
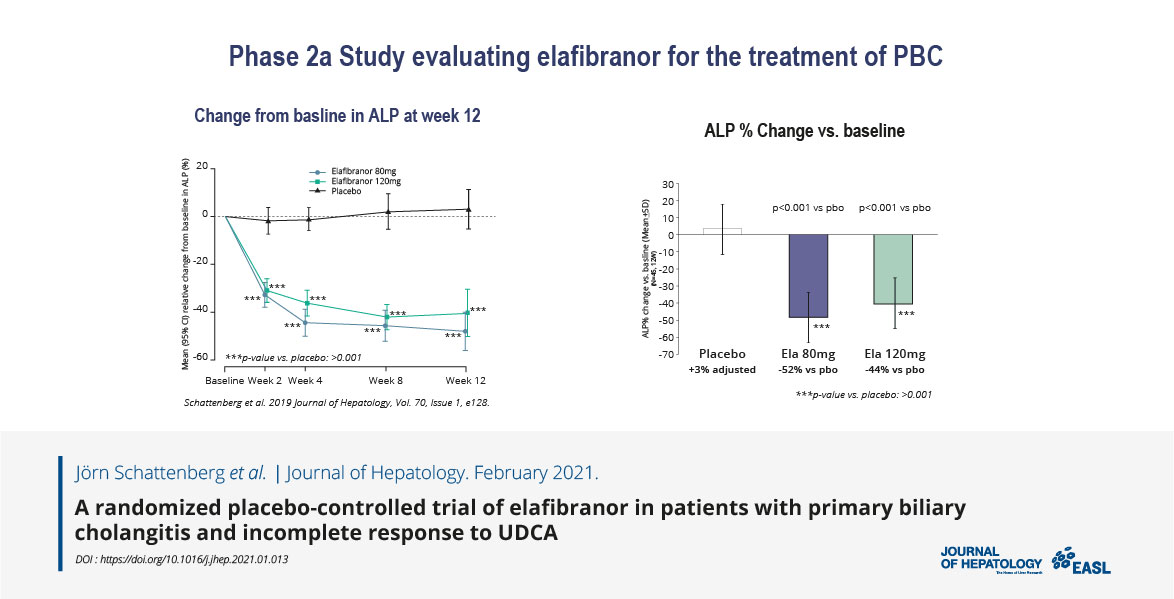
Partnering
◦Preclinical evidence
As part of our preclinical program, we have studied NTZ in cell and animal disease models.
NTZ and TZ, its active circulating metabolite, have a wide anti-infectious spectrum acting on bacteria, viruses and parasites commonly encountered in human intestinal flora. Thus, an oral treatment with elafibranorNTZ is expected to improve bacteria overgrowth and dysbiosis and possibly preserve the intestinal barrier in patients with ACLF. We also observed that, in cultured human liver cells, TZ inhibits a key pathway of programmed cell death (apoptosis) in a dose dependent manner.
•NTZ reduces LPS-induced inflammation in healthy rats: our research has demonstrated that an oral administration of NTZ concomitant with intraperitoneal injection of LPS significantly reduced the LPS-induced rise in circulating cytokines and inflammatory markers;
•NTZ showed beneficial effects on liver function markers (bilirubin, albumin) in two distinct rat models of ACLF (cirrhotic rats given LPS), we found that NTZ has hepatoprotective effects by reducing ALT and AST while totally preventing LPS-induced rise in GGT and total bilirubin, as well as two markers in renal function in plasma (cystatin C and creatinine);
•NTZ reduced brain edema in a model of ACLF (rats with bile duct ligation);
•NTZ reduced inflammation markers in a model of ACLF (rats with bile duct ligation); and
•NTZ improved survival in treatment models of Sepsis (cecal ligation puncture, or CLP): the mortality rates in NTZ treated vs vehicle treated group were 53% vs 90% at 72 hours and 67% vs 100% 5 days after CLP surgery.
◦Clinical evidence
Two Phase 1 studies were conducted to evaluate the safety and pharmacokinetics of NTZ metabolites in the setting of hepatic impairment or renal impairment. These studies were completed in the fourth quarter of 2022 and the first quarter of 2023, respectively, and are supportive of future investigation in patients with ACLF. The data for the hepatic impairment study were presented in a poster presentation during Digestive Disease Week® (DDW) 2023, taking place May 6-9, 2023, at McCormick Place in Chicago, IL, and online.
–Next milestones
A new formulation of NTZ is currently under development to permit greater dosing flexibility and to optimize dose-response in patients with ACLF, who are known to have varying degrees of organ impairment or failure. The new formulation will also permit the optimization of the benefit/risk profile in this patient population.
The reformulation and Phase 2 is under preparation in 2024 with a proof-of-concept study initiation targeted for the first half of 2025.
•Our third program SRT-015 (injectable formulation): an ASK1 inhibitor with multi-system benefits
–Rationale and mechanism of action
SRT-015 is an ASK1 inhibitor in-licensed from Seal Rock Therapeutics in acute liver diseases. It targets the inhibition of cellular apoptosis, inflammation and fibrosis.
ASK1 triggers the activation of several pathways, most notably two key ones: the p38 MAPK pathway and the JNK pathway. This activation contributes to increased inflammation, cell death and fibrosis.
ASK1 inhibition has shown several potentially beneficial effects that may be relevant in ACLF, such as blocking LPS (lipopolysaccharide) associated hyperinflammatory response, reducing the ROS (Reactive Oxygen Species)-related immune response, reducing apoptosis, reducing release of the proinflammatory cytokines, reducing fibrosis, and protecting macrophage mitochondrial function.
–Evidence supporting further development
Preclinical and clinical evidence support ASK1 inhibition as a relevant therapeutic strategy in multi-system disorders such as Acute-on-Chronic Liver Failure (ACLF). Multi-organ activities of ASK1 inhibitors have been observed in several animal models and clinical trials. SRT-015 alleviates hepatic injury in a model of drug-induced liver injury, in association with reduction of phosphorylation of both JNK and ASK1. In addition, SRT-015 alleviates hepatic injury in a model of alcoholic hepatitis.
–Next Milestones
A First-in-Human study with an intravenous formulation of SRT-015 is planned in the first quarter of 2025.
•Our fourth program CLM-022: a potential first-in-class NLRP3 inflammasome inhibitor
–Rationale and mechanism of action
CLM-022 is a small molecule inhibitor targeting the NLRP3 inflammasome. This pathway is particularly relevant to ACLF (Acute-on-Chronic Liver Failure). The significance of this pathway is underscored by several studies, including one which observed a key difference in NLRP3 inflammasome activity between patients with chronic HBV infection alone and those with both HBV infection and ACLF. Thus, inhibiting the NLRP3 pathway is a promising strategy for treating ACLF.
Activation of the NLRP3 pathway leads to proinflammatory cell death, known as pyroptosis, and also initiates the synthesis and maturation of pro-inflammatory cytokines, particularly IL-1β and IL-18.
–Evidence supporting further development
Primarily in animal models of liver injury and inflammation studies have shown promise for NLRP3 inflammasome inhibitors in reducing liver damage and inflammation.
It was shown to:
• Block the production of inflammatory cytokines in a LPS-induced endotoxemia model;
• Block ASC speck formation at nanomolar concentrations;
• Decreases IL-1β secretion by macrophages stimulated with LPS and nigericin; and
• Protect against nigericin-induced pyroptosis in LPS-primed macrophages.
–Next Milestones
Preclinical Proof of Concept is expected to be obtained by end-2024.
•Our fifth program VS-02-HE: a urease inhibitor
VS-02-HE is being developed in HE, one of the major complications of advanced liver disease and portal hypertension.
–About HE
HE is a central nervous system disorder representing a diverse spectrum of neurologic symptoms (sleep-wake cycle disturbance, fatigue, concentration difficulty, personality changes, tremor, cognitive deficits, and, in severe cases, coma), and typically occurs in patients with advanced chronic liver disease or porto-systemic shunting. In chronic liver disease, toxins, including ammonia, accumulate in the systemic circulation and can cross the blood-brain barrier. Excess ammonia induces accumulation of glutamine in astrocytes causing osmotic stress and alteration of cell metabolism and can result in brain edema or swelling. As many as 45% of patients with cirrhosis will experience at least one episode of HE. In the U.S. only, two million patients are believed to be at risk of developing HE and 200,000 patients are hospitalized yearly. In Europe, incidence is close to one million patients. Patients with ACLF and HE have higher mortality rates compared to patients who have ACLF only. The prevalence of overt HE at the time of cirrhosis diagnosis is approximately 10–14%. The estimated annual economic burden associated with HE in the US was $7.2 billion in 2009 and around $12 billion in 2014.
–A high unmet medical need
HE is largely underdiagnosed and undertreated and is associated with poor quality of life. Due to its neurotoxic effect, ammonia has been the main target for HE therapy. Current treatment options for HE focus on either reducing ammonia production and absorption (e.g., non-absorbable disaccharides) or on promoting its elimination by eliminating ammonia-producing colonic bacteria (e.g., antibiotics). Non-absorbable disaccharides such as lactulose, however, exhibit various limitations such as persistent side effects leading to poor compliance which indirectly affects overall efficacy. Additionally, antibiotics (e.g., rifaximin), according to the approved label for rifaximin as of the date of this annual report, are limited to the reduction of overt HE recurrence rather to the treatment of overt HE.
•VS-02-HE : Our Program to Reduce Hyperammonemia and Stabilize Ammonia Levels in the Blood
–Rationale and mechanism of action
VS-02-HE is a urease inhibitor designed to inhibit ureases by binding to nickel atoms in their active site. As urease-producing bacteria in the gut are one of the main sources of circulating ammonia in humans, urease-inhibitors may represent a promising therapeutic approach for HE.
We are developing VS-02, a urease inhibitor currently in preclinical stage. VS-02 is a hydroxamic acid (HA) derivative, which is designed to inhibit ureases by binding to nickel atoms in their active site. Inspired by earlier studies, the in vitro activity of a series of novel hydroxamic acid (HA) derivatives was investigated on rat caecum content. The lead candidate, VS-02, showed a potency largely exceeding that of HA derivatives tested in former clinical trials. It was further found that VS-02 was neither cytotoxic nor mutagenic at up to 1 mM, which makes it an ideal candidate for development as a novel treatment for HE via a colonic formulation.
–Evidence supporting further development
In vivo efficacy studies showed that VS-02-HE (30 mg/kg) was able to reduce ammonia blood levels in bile-duct ligated (BDL) rats. Additionally, in vivo 1H MRS measurements performed at 9.4T in the cerebellum (SPECIAL sequence, TE=2.8ms, VOI=2.5x2.5x2.5mm3) showed a significant decrease in brain glutamine levels after 5 days of treatment compared to non-treated BDL rats confirming the therapeutic effects of VS-02-HE. In summary, we believe VS-02-HE represents a promising oral candidate for further evaluation in the treatment of HE.
–Next milestones
We intend to develop VS-02-HE as a unique oral formulation designed to act where ammonia is primarily produced, minimizing systemic absorption of ammonia while reducing glutamine levels in the brain. The treatment goal is to reduce/stabilize the accumulation of ammonia in the blood and prevent rehospitalization.
Investigational New Drug-enabling nonclinical studies starting in 2024 with completion expected in 2025.
3. Other life-threatening diseases franchise
•GNS561 in Cholangiocarcinoma (CCA)
•About Cholangiocarcinoma
Biliary tract cancer (BTC) is the second most common primary liver malignancy diagnosed globally. Cholangiocarcinoma (CCA) is a type of BTC and represents approximately 15% of all primary liver tumors and 3% of gastrointestinal cancers.
| | | | | | | | |
| | |
| Adapted from Nature Reviews Gastroenterology & Hepatology volume 17, p. 557–588 | |
CCA is comprised of a heterogeneous group of cancers with pathologic features of biliary tract differentiation and is presumed to arise from the intra- or extrahepatic biliary tract. Gallbladder cancer is distinct from Cholangiocarcinoma in epidemiology, pathophysiology, clinical presentation, and management and is considered as a different type of biliary tract cancer. Based on its anatomical origin, CCA is best classified anatomically as intrahepatic (iCCA) or extrahepatic (eCCA), which is comprised of perihilar (pCCA) and distal (dCCA) CCA. The incidence of iCCA appears to be increasing and may be as high as 2.1 per 100,000 person years in Western countries.
CCA may occur in normal livers or in the setting of underlying liver disease, and in these cases, it appears as a mixed type hepatocellular-cholangiocarcinoma instead of traditional adenocarcinoma. Several risk factors of chronic inflammatory damage and increased cellular turnover have been established, such as hepatobiliary flukes (Opistorchis viverrini and Clonorchis sinensis), primary sclerosing cholangitis, biliary tract cysts, hepatolithiasis and toxins. Cirrhosis, chronic hepatitis B and C, obesity, diabetes mellitus and alcohol-related liver disease are also emerging as risk factors for CCA.
The clinical presentation of CCA is non-specific and most often insufficient to establish a diagnosis. Early diagnosis is a major challenge as most patients with early-stage disease do not have symptoms due to limited biliary obstruction. Rather, patients characteristically manifest symptoms related to their underlying cirrhosis, a condition present in some patients with CCA.
Taken together, the majority of patients with CCA are diagnosed with advanced disease, often precluding potentially curative therapies. Once symptomatic, CCA is often associated with non-specific complaints, including right upper abdominal or epigastric pain or discomfort, jaundice, weight loss, malaise, hepatomegaly or a palpable abdominal mass. The onset of ascites, encephalopathy, jaundice or variceal bleeding in patients with previously compensated cirrhosis also increases the clinical suspicion for liver tumor. Tumor-related fever may rarely occur, although night sweats are common in advanced disease. CCA should be considered in patients with underlying hepatolithiasis or primary sclerosing cholangitis (PSC) with worsening performance status, unexplained loss of weight or failure to thrive.
–A high unmet medical need
There are limited therapeutic options for this aggressive disease. The 5-year survival rates drop to 5-15% in the advanced and unresectable settings. The only potentially curative treatment remains surgical resection. Unfortunately, at time of first diagnosis, only about 25% of the patients are eligible for surgery. Moreover, even after curative intent surgery, the clinical outcomes are disappointing, with 5-year survival rates of 7% to 20%. The role of adjuvant therapies, including systemic chemotherapy and radiotherapy, remains poorly defined yielding only a modest survival benefit. Around 60% to 70% of patients are diagnosed with advanced disease, which is defined as unresectable or metastatic disease. For these patients, palliative treatment with systemic chemotherapy is the only treatment option. Patients progressing on first line chemotherapy often have a rapidly worsening performance status, and only a small number of patients may be suitable for further treatment. The estimated median survival for these patients is 3.7 months.
In the advanced setting, the standard of care for first line therapy is a combination of gemcitabine and platinum-based chemotherapy; other gemcitabine- or fluoropyrimidines-based regimens are also commonly used. At time of relapse, patients whose tumor displays fibroblast growth factor receptor 2 (FGFR2) or isocitrate dehydrogenase 1 (IDH-1) alterations may receive approved therapies that target these specific alterations. All other patients are offered second line chemotherapy. The most efficacious regimen is currently a combination of cytotoxics (folic acid, 5-FU/fluorouracil, and liposomal irinotecan (FOLFIRI)) yielding a median overall survival of 8.6 months.
•Our Program: GNS561
To address the significant unmet need in patients diagnosed with CCA, GENFIT is developing GNS561 to prolong the overall survival of patients who present with iCCA and eCCA. GNS561 is a Palmitoyl Protein Thioesterase-1 (PPT-1) inhibitor that blocks autophagy, which GENFIT in-licensed in 2021 from Genoscience (See Item 4.B - "Information on the Company-Out-licensing partnerships"). –GNS561: rationale and mechanism of action
Autophagy is activated in tumor cells as a survival mechanism in a nutrient poor environment, due to tumor cell growth in advanced cancers. One of the key cellular organelles implicated in the autophagy process is the lysosome. By decreasing the activity of PPT1 in lysosomes, GNS561 may have an important inhibiting activity on late-stage autophagy, which leads to tumor cell death.
–Evidence supporting development
Lysosomal function is an essential element in autophagy, and GNS561 is a lysosomotropic small molecule which inhibits PPT1, a lysosomal enzyme required to maintain lysosome-autophagy function. PPT1 expression is high in most cancer cell lines, increased in tumors compared with paired normal tissue, and in metastases versus primary tumors, and high levels of PPT1 have been associated with shorter overall survival. Thus, these findings, along with the role of PPT1 in maintaining lysosome-autophagy function, establishes the potential of PPT1 inhibition as a strategy in cancer therapy. In addition to its inhibition of PPT1, studies with GNS561 showed that it has high liver tropism when administered orally, significantly reduced cell viability in human iCCA cell lines and induced apoptosis. GNS561-mediated cell death was correlated with inhibition of late-stage autophagy and induction of a dose-dependent build-up of dysfunctional lysosomes. GNS561 was also efficient in vivo against a human intrahepatic CCA cell line in a chicken chorioallantoic membrane xenograft model, with a good tolerance at doses high enough to induce an antitumor effect in this model.
In a first-in-human Phase 1 study in patients with advanced primary (HCC and iCCA) and secondary liver cancer (metastasis from distant carcinomas), GNS561 was observed to have good tolerability, exposure, and preliminary signal of activity. Taken together, the results generated with GNS561 highlight its potential to provide benefit in prolonging survival of patients diagnosed with CCA. In particular, we believe that GNS561, as an inhibitor of autophagy, could potentially be beneficial in combination therapy, including combinations with inhibitors of the MAP kinase pathway or immunotherapy/checkpoint inhibitors.
Cytotoxic chemotherapy drugs as well as multiple targeted therapies such as kinase inhibitors have been proposed to induce autophagy as a survival mechanism in cancer cells. In 2019, the results of two major studies showed that, in the context of a cancer with the KRAS mutation (active RAS leading to activation of the MAP kinase pathway), inhibitors of the MAP kinase pathway can induce autophagy in pancreatic cancer, and combinations of MAP kinase pathway inhibitors with autophagy inhibition can enhance tumor cell killing. Importantly, a significant proportion of CCA patients have mutations including KRAS. Therefore, the combination of therapies targeting the MAP kinase pathway with GNS561 to inhibit autophagy is a potential therapeutic strategy to treat CCA patients.
–Next milestones
GNS561 received orphan drug designation for CCA from the FDA in September 2022. Given the high unmet need in this indication and the Orphan Drug Designation obtained from the FDA for GNS561, we believe that the program should qualify for some of the expedited regulatory pathways provided by health authorities.
The GNS561 Phase 1b/2a clinical trial.is currently ongoing. Phase 1b/2a clinical trial is currently ongoing. In Phase 1b of this study, patients with advanced KRAS mutated CCA, who have previously failed a standard of care first line therapy,, will be enrolled to evaluate the safety and tolerability of GNS561 when given in combination with trametinib, a MEK inhibitor, and to identify the recommended doses of the combination to be administered in Phase 2a. In Phase 2a, the safety and efficacy of the combination will be assessed in patients with advanced KRAS mutated CCA who have otherwise failed standard-of-care for first line therapy and who do not have an actionable mutation. Preliminary data from Phase 1b is targeted by the end of 2024.
•VS-01-HAC in Urea Cycle Disorders (UCD) and Organic Acidemias (OA)
•About Hyperammonemic Crisis (HAC) in UCDs and OAs
Hyperammonemia is defined as plasma ammonia levels above 80 µmol/L in newborns up to 1 month of age and above 55 µmol/L in older children. In the mammalian organism, the hepatic urea cycle is the main pathway to detoxify ammonia. Hyperammonemic crisis occurs whenever the load of waste nitrogen exceeds the detoxification capacity. Plasma ammonia levels in HAC can reach up to 1000 µmol/L.
Inborn errors of metabolism causing HAC comprise a group of hereditary disorders in which a single gene defect results in a clinically significant block of the urea cycle responsible for the metabolic clearance of ammonia from the bloodstream. The accumulation of ammonia, which is continuously produced by the breakdown of protein and other nitrogen-containing molecules, rapidly leads to cerebral edema and the related signs of lethargy, anorexia, hyperventilation or hypoventilation, hypothermia, seizures, neurologic posturing, and coma.
| | | | | | | | |
| | |
| Adapted from Rupesh Raina et al., Nature 2020 | |
Hyperammonemia in Inborn Errors of Metabolism (IEM) is classified as follows:
•Primary hyperammonemia, when the urea cycle is directly affected by a defect of any of the involved enzymes or transporters, defining UCDs; and
•Secondary hyperammonemia, when enzymes of the urea cycle are inhibited due to accumulating metabolites or substrate deficiencies. The most relevant group of disorders associated with secondary hyperammonemia is called Organic Acidemias, or OAs.
Regardless of the underlying genetic disorder, the clinical characteristics, outcome, prognosis and treatment of HACs associated with IEM are similar.
Patients are usually diagnosed shortly after birth via universal newborn screening tests. The clinical presentation of patients with HAC caused by IEM may start as early as the first days of life and as late as adulthood. The most severe cases present in the first week after birth with unspecific symptoms like feeding refusal and vomiting, loss of thermoregulation, neurologic posturing, seizures, hyperventilation and then hypoventilation, and irritability that progress rapidly to somnolence, lethargy, coma, multi-organ failure and death.
While these conditions are ultra-rare with 1,900 acute hyperammonemic crisis in the U.S. and the five major European countries per year, the mortality rate is as high as 75%. Most patients will die after 5 years, and survivors will often have severe brain injuries. Patients with HAC associated to IEM must be transferred to specialized tertiary centers to be treated which increases the costs on the healthcare system.
–A high unmet medical need
The treatment of hyperammonemic crisis typically involves prompt management of the elevated ammonia levels in the blood. This may involve hospitalization, administration of medications such as sodium benzoate and phenylacetate, and intravenous fluids to help remove excess ammonia from the bloodstream. In severe cases, hemodialysis may be necessary to help remove ammonia from the blood. In centers where hemodialysis is not available, hemofiltration or other forms of dialysis should be used.
In practice, pediatric patients presenting HAC must be transferred in highly specialized tertiary centers having devices adapted to their size. Consequently, dialysis in IEM HAC is often initiated late when ammonia levels are above 1000 µmol /L and this may contribute to poor outcomes. Moreover, neonatal hemodialysis is risky, highly invasive and widely unavailable. As many as 45% of UCD patients remain untreated, and no drug is currently approved for treatment of OA.
•Our Program: VS-01-HAC for Ammonia Clearance and Prevention of HAC
–VS-01-HAC: rationale and mechanism of action
We are developing VS-01-HAC, a potential first-line lifesaving treatment for acute hyperammonemic crisis associated with IEMs.
To reduce high mortality and morbidity associated with HAC in IEMs, early diagnosis and immediate start of treatment are thought to improve the prognosis. Indeed, coma duration and levels of ammonia blood concentration are the main factors for determining mortality and neurologic outcome.
Therefore, a new drug using the peritoneal route with optimized ammonia clearance and a quick implementations time, would allow for the initiation of efficient dialysis immediately after HAC is confirmed and could help in overcoming the crises. Moreover, as the peritoneal route of administration is well adapted to pediatric patients, this treatment could be safely feasible in the hospital setting. Speed of implementation and safety represent tremendous improvements over neonatal hemodialysis, which is only possible in specialized centers and is a long and risky procedure in pediatric patients.
Use of a new treatment before transferring the patient to a tertiary center would save costs to the healthcare system as well as reduce burden on pediatric patients and their parents.
In addition to Orphan Drug Designation for the treatment of hyperammonemia in inborn errors of metabolism, Rare Pediatric Disease Designation (RPDD) has been granted to VS-01-HAC by the FDA for treatment of Urea Cycle Disorders (UCD) indication. GENFIT is potentially eligible to receive a Priority Review Voucher upon approval of an NDA by the FDA.
–Evidence supporting further development
An in vivo feasibility study was performed with OTC-deficient mice (homozygous females (Otcspf-ash/spf-ash) and hemizygous males (Otcspf-ash/Y)), a gold standard model which develops hyperammonemia and presents many characteristics of the human disorder. The results showed that ammonia extracted from blood into the peritoneal cavity was significantly (p < 0.0006) higher following single intraperitoneal injection of VS-01 compared to the control solution at all timepoints during the dwell time and led to a significant decrease in blood ammonia.
Our non-clinical and first-in-human clinical data showed that ammonia clearance in the peritoneal fluid increased proportionally with the volume of fluid infused and ranged between 5 and 95 mL/min following treatment with 0.3 L and 3 L VS-01, respectively. These values are in the same range as those reported in UCD patients treated with different extra corporal dialysis modalities.
–Next milestones
Following completion of the non-clinical feasibility study, we plan to develop formulation optimization for specific pediatric implementation and conduct IND-enabling nonclinical studies with a target to complete such studies in 2024.
4. Our diagnostics franchise
–NIS2+®, a next-generation technology derived from NIS4® for the identification of patients with at-risk NASH/MASH
•About NASH/MASH
At EASL Congress in June 2023 it was announced that nonalcoholic steatohepatitis (NASH) would now be referred to as Metabolic dysfunction-Associated Steatohepatitis (MASH). Nonalcoholic fatty liver disease (NAFLD) will now be referred to as metabolic dysfunction-associated steatotic liver disease (MASLD).
MASH, the most severe form of metabolic dysfunction-associated steatotic liver disease (MASLD) is characterized by the presence of hepatocyte ballooning and inflammation, in addition to steatosis. MASH can progress silently towards cirrhosis, precluding the opportunity for clinicians to diagnose and intervene therapeutically prior to the development of severe liver complications, and constitutes a growing cause of cirrhosis, liver failure, and liver cancer globally. Furthermore, MASH is projected to become the leading cause of liver transplantation in the United States—it already is the primary cause among women and the secondary cause overall. Given this clinical scenario, there is a pressing need to identify patients at higher risk of disease progression, who could be considered for therapeutic intervention with existing options or when potentially promising agents currently in late-stage clinical development obtain regulatory approval.
•Today’s Challenges in Diagnosing MASH
Liver biopsy is the reference standard for the diagnosis of MASH among patients with clinical risk factors for this disease, such as metabolic disorders in the absence of alternative causes for steatosis. The implementation of this diagnostic approach, however, is limited in routine clinical practice by its invasive procedure, cost, attendant risks, variability in interpretation, and the restricted number of professionals able to perform and interpret the test, among other factors. These limitations preclude liver biopsies from being broadly used as the primary diagnostic in such a prevalent disease. Providing a non-invasive alternative to liver biopsy will therefore be critical to facilitate improved patient diagnosis, management, and future treatment access in routine clinical practice, and may eventually reduce the morbidity and mortality associated with this disease.
At the end of 2022, Madrigal Pharmaceuticals announced positive data in its pivotal Phase 3 MAESTRO-NASH clinical trial of resmetirom for the treatment of MASH and liver fibrosis. On March 14, 2024, Madrigal Pharmaceuticals announced FDA approval of Rezdiffra™ (resmetirom) in conjunction with diet and exercise for the treatment of adults with noncirrhotic NASH with moderate to advanced liver fibrosis. Rezdiffra™ is thus the first-ever approved drug for the treatment of MASH, which should increase the focus on diagnosis over the coming years. The treatment of MASH is a pressing public health challenge and there is a large unmet need for a widely available, non-invasive test, or NIT, to identify patients with at-risk MASH as an alternative to liver biopsy. The availability of such a test would help address the under diagnosis of MASH by supporting physicians in identifying patients with at-risk MASH, who are at higher risk for clinical outcomes and would be eligible for therapeutic intervention.
•Our Program: NIS Technology Comprising Our Proprietary Biomarker Algorithms
As part of our strategy to address unmet needs in MASH, we have an advanced diagnostic program based on the identification of specific biomarkers that are expressed at different levels in patients with MASH and significant fibrosis (F≥2) as compared to patients with less severe disease. This discovery kicked off a multi-year effort that has resulted in the development of NIS4® technology, a blood-based molecular technology for the identification of patients with MASH (NAS≥4) and significant fibrosis (F≥2), also referred to as “at-risk” MASH, who are at higher risk of disease progression and may be appropriate candidates for therapeutic intervention.
Our first biomarker technology, NIS4®, integrated the outputs of four MASH-associated biomarkers (alpha-2-macroglobulin, YKL-40, hemoglobin A1c, and miR-34a-5p) through an algorithm to produce a single score that can be utilized to rule in and rule out at-risk MASH, while minimizing the number of indeterminate test results.
•In August 2020, we announced that pivotal data describing the derivation and validation of NIS4® technology was accepted for publication by The Lancet Gastroenterology & Hepatology.
•In November 2021, NIS4® technology’s utility was demonstrated in a biomarker qualification Phase 1 study undertaken by the NIMBLE consortium with a strong performance for identifying patients with “at-risk” MASH and the components of "at-risk MASH (MASH, NAS ≥ 4 and fibrosis stage ≥ 2). In September 2023, these data have been published in the prestigious scientific journal Nature Medicine.
We out-licensed our NIS4® technology to Labcorp in 2019 and 2020 in the field of clinical research and for the development of a laboratory-developed test (LDT), respectively. In 2021, we also signed a non-exclusive license with Q Squared Solutions LLC, or Q2, with the objective to broaden access to our NIS4® technology in the clinical research space. See Item 4.B - "Commercialization perspectives—Out-licensing partnerships”. In October 2022, we announced the development of NIS2+®, a next-generation technology for the diagnosis of at-risk MASH, designed as a robust optimization of NIS4®. Since then, NIS2+® performances were detailed and presented in four different manuscripts, accepted and published in important scientific journals.
•The first paper, published in the Journal of Hepatology in May 2023, highlighted the development and validation of NIS2+® as an optimization of NIS4® technology for identifying at-risk MASH. NIS2+® demonstrated strong clinical performance in detecting at-risk MASH, while its composite scores were not impacted by the status of important subpopulations such as Type-2 diabetes, age and sex, addressing an important unmet need. In addition, the increased robustness and simplicity of NIS2+® technology (from a 4 to a 2-biomarker panel) should allow for a wider and easier application in clinical settings.
•The second paper, published in conjunction with Labcorp in Hepatology Communications in August 2023, highlighted the high clinical performances of NIS2+® in a population of older adults (≥65 years of age), which were superior to other well-known tests for the diagnosis of at-risk MASH. These data support the clinical value of this blood-based technology for the diagnosis of at-risk MASH in older adults who would benefit from intensive lifestyle or therapeutic interventions, and is expected to greatly assist with US Centers for Medicare & Medicaid Services (CMS) reimbursement efforts.
•The third paper, published in the Journal of Hepatology in December 2023, positioned NIS2+® as a powerful tool for improving the recruitment of at-risk MASH patients into clinical trials, reducing the high inclusion failure rates based on liver biopsy results (>60%) and thus the overall cost associated with recruitment, without significantly increasing the number of patients to be examined or introducing bias into the recruited patient population.
•The last in date paper, published in JHep Reports in January 2024, provided a global analysis of the impact of age on different NITs concluding that, unlike other usual NITs such as FIB-4 or ELF, NIS2+® was not impacted by this factor, allowing physicians to use and interpret NIS2+® results irrespective of patients' age.
Besides these publications in scientific journals, further data on NIS2+® (diagnostic, screening, prognostic, subpopulations analyses) were presented in 10 posters and three oral presentations at five international congresses.
Our agreements with Labcorp and Q2 also provide access to NIS2+®.
–Next milestones
We believe the future of NIS2+® is through an IVD test as a standalone diagnostic with the potential to enable a non-invasive, accessible and validated alternative to the liver biopsy to benefit patients, improve overall clinical care and greatly reduce barriers to entry for innovative therapies.
We continue to explore the possibility of initiating and completing validation studies necessary to obtain regulatory approval and CE Certificates of Conformity, alone or with a development and commercial partner, to release an IVD powered by NIS2+® technology on the U.S. and European markets. In the meantime, we will continue to seek the most appropriate ways to optimize on the potential of NIS2+®.
–TS-01 as a point of care (POC) device for measuring ammonia in blood
Approximately 90% of hyperammonemia cases in adults are in people who have cirrhosis of the liver. Cirrhosis is the end stage of every chronic liver disease and is the 11th leading cause of death worldwide. Globally, an estimated 112 million people suffer from compensated cirrhosis, claiming more than 1.3 million lives in 2017. Complications of cirrhosis are marked by liver metabolic dysfunctions and the development of clinical signs, of which the most frequent is HE. HE is a serious neurologic condition caused when ammonia accumulates in blood, eventually affecting the brain. Elevated ammonia concentration in blood and brain (hyperammonemia) is associated with high mortality and is the mainstay for pathogenesis and treatment of HE. In patients with cirrhosis, fully symptomatic overt HE leads to hospitalizations and readmissions. HE-related hospitalizations generated charges of approximately US $11.9 billion per year in the United States, a 46% cost increase from 2010 to 2014. Costs are expected to further increase due to disease progression, requiring more complex health care efforts.
Overt HE occurs in 30-45% of patients with cirrhosis, leading to approximately 1 million cases considering 2,828,000 cases of cirrhosis worldwide. There is a need for a reliable point of care devise to measure ammonia in the blood in patients with HE, so that there can be a repeated quantification of ammonia levels to test the efficacy of ammonia-lowering treatments. Furthermore, ammonia levels can predict the onset of new episodes of HE even with mild hyperammonemia, but there are currently logistical challenges to accurately measure ammonia in the blood.
We believe that the ammonia POC diagnosis would complement both VS-01 and VS-02 product candidates and is in line with our business strategy to improve the management of severe liver diseases globally. We believe combining diagnostics with therapeutics under one umbrella synergistically multiplies the value of each product.
–A high unmet medical need
When patients with altered mental status are admitted to the emergency department, HE should be diagnosed as fast as possible to initiate further diagnostic tests, especially in the emergency department, where resources of medical staff and time are limited. Since many of the symptoms of HE also occur in people with other types of brain disease or damage (e.g., stroke, brain tumor, or bleeding inside the skull), an ideal bedside test for fast, precise and accurate ammonia measurements would:
–Allow for the rapid diagnosis of HE. A high ammonia level increases the probability of HE especially in patients who have known liver disease.
–Trigger other diagnostic steps to explore other etiologies of altered mental status (a low ammonia level reduces the probability of HE) or to rule out potential gastrointestinal bleeding if HE confirms (e.g., endoscopy).
–Initiate specific medical treatment (e.g., lactulose/antibiotic therapy). Especially in the emergency department, where resources of medical staff and time are limited.
In addition, self-monitoring of ammonia with an accurate and user-friendly POC ("Point of Care") device offers the opportunity for early identification of severe HE episodes, timely therapeutic management, and therefore decreasing hospital visits, long-term risks of complications, and global burden on public health. Moreover, close follow-up of the ammonia offers the possibility to better tailor current therapies for HE, which are unfortunately associated with poor compliance due to their side effects. Adapting treatment dose and schedule, can increase compliance and hence reduce occurrence of severe episodes. Finally, HE impacts daily functioning by altering fitness to drive, attention, memory, mood, and psychomotor speed. A tighter control of the disease is expected to increase the quality of life of patients and their families.
Today, serum ammonia testing and interpretation remain logistically challenging. After the sample is collected, erythrocyte and platelet metabolism persist in vitro, and ammonia concentrations increases at room temperature. Therefore, it is recommended that samples are kept on ice and immediately processed after collection, which increases the overall burden on staff.
Despite these challenges, the literature indicate that serum ammonia testing is increasing. Future improved ammonia testing may enhance value-based use of ammonia in patients with cirrhosis and HE. A POC device for ammonia is expected to save time, efforts, and expenses to the health care professionals while supporting caregivers, and family members.
Currently, the only marketed POC device for ammonia measurement is the PocketChem. It is mainly used in research because of its narrow quantification range (7-286 µmol/L), its interference issues and underestimation of ammonia levels in comparison to enzymatic assays.
•TS-01 for at-home monitoring of ammonia in liver disease patients to help detect HE
TS-01 is a device based on a "transmembrane pH-gradient polymersome" technology designed to easily measure ammonia levels at home.
The underlying technology behind the Transmembrane pH-gradient polymersomes for ammonia quantification in blood consist of vesicles made of non-biodegradable polymers that form a bi-layer membrane. The aqueous core of the vesicles is loaded with a pH-sensitive dye in an acidic buffer. An alkaline buffer on the outside generates the pH-gradient across the polymersomes’ membrane. Uncharged ammonia in blood samples can easily diffuse across the polymeric membrane into the core of the polymersomes, where it is protonated due to the acidic environment. Generated ammonium ions cannot diffuse back due to their charge. Accumulation of protonated ammonia inside the core of the vesicles triggers an increase in pH and consequently an increase in fluorescence intensity of the pH-sensitive dye. The increase correlates with the ammonia concentrations in the sample. When an equilibrium state is reached, fluorescence can be easily measured and thus ammonia concentrations in blood derived. We believe this unique mechanism will allow us to scale polymersome technology from high throughput to single measurements in a POC.
The polymersomes technology was developed and validated by the Federal Institute of Technology Zurich (ETH Zurich) and we hold an exclusive worldwide license to develop and commercialize this technology in all fields, with an option to purchase the intellectual property subject to certain conditions.
–Next milestones
The development of TS-01 based on the polymersome technology is performed in collaboration with ZHAW School of Engineering with expertise in optoelectronics as well as in the development of demanding biomedical instrumentation. The lab-bench prototype has been miniaturized to allow portability of the device. The next steps include validation of the test in blood and further miniaturization of the device.
–Strategic Partnerships
•Out-Licensing Partnerships
◦Strategic Collaboration with Ipsen
In December 2021, we entered into a long-term strategic partnership for global collaboration with Ipsen Pharma SAS, or Ipsen, a global, mid-sized biopharmaceutical company focused on transformative medicines in oncology, rare disease and neuroscience. The agreement gives Ipsen an exclusive worldwide (excluding Greater China which is licensed to Terns, see below) license to develop, manufacture and commercialize our investigational treatment elafibranor, for people living with PBC, and in other indications. The partnership also gives Ipsen access to future clinical programs led by GENFIT through rights to first negotiation and combines GENFIT’s scientific expertise and proprietary technologies in liver disease with Ipsen’s development and commercialization capabilities.
GENFIT remainsremained responsible for the Phase III ELATIVE3 ELATIVE® trial untilthrough the completion of the double-blind treatment period. Ipsen will assumehas assumed responsibility for all additional clinical development, including completion of the long-term, open-label extension period of the ELATIVEELATIVE® trial, and global (excluding Greater China) commercialization. This newly established strategic partnership will also provideAt the date of this report, the responsibility of the pursuit of the trial is almost entirely transferred to Ipsen with access(only one clinical investigation site remains to GENFIT’s research capabilities and other clinical programs through rights to first negotiation.be transferred).
Under the agreement, Ipsen will pay GENFIT up to €480m,€480 million, comprising an upfront cash payment of €120m,€120 million received in 2021, as well as regulatory, commercial, and sales-based milestone payments up to €360m,€360 million, plus tiered double-digit royalties of up to 20%. In addition, to underscore its long-term commitment, Ipsen also became our largest shareholder through the purchase of 3,985,239 newly issued shares representing 8% of GENFIT S.A after issuance, via a €28m€28 million investment. The new shares are subject to a lock-up period ending on the earlier of the date on which the EMA makes a formal recommendation to the European Commission for the marketing authorization of elafibranor in PBC, the date on which the FDA grants approval of elafibranor in PBC (PDUFA action date: June 10, 2024) or in the event the ELATIVEELATIVE® trial does not meet its primary endpoint. For more information about the financial terms of the agreement, including milestones received to date, see Note 7 - "Revenues and other income" as well as Note 29 - "Commitments and contingent liabilities" to our consolidated financial statements included in this annual report. This agreement will remain in force until the later of either a 10-year period after the first sale of a licensed product in the territory or the expiration of the last patent concerning such a licensed product in the relevant country (determined on a per-country basis). For more information, see "
◦Item 10.C – Collaboration and License Agreement with Ipsen Pharma SAS".
Agreement with Terns Pharmaceuticals
In June 2019, we announced the signing of a licensing and collaboration agreement with Terns Pharmaceuticals, a global biopharmaceutical company based in the U.S. and China with a focus on developing novel and combination therapies to treat liver disease. Under the agreement, Terns has been granted the exclusive rights to develop, register and commercialize elafibranor in Greater China (mainland China, Hong Kong, Macau, and Taiwan), for the treatment of NASHMASH and PBC.
Under the terms of the license agreement, GENFIT has received an initial payment of $35 million from Terns and may receive up to $193 million in additional payments upon completion of clinical, regulatory and commercial milestones. At commercial launch of elafibranor in Greater China, GENFIT may receive mid-teen percentage royalties from Terns based on the sales in this territory. As part of the agreement, GENFIT and Terns will also undertake joint R&D projects in liver disease.
The preparation of the inception of clinical trials with elafibranor in PBC in China is underway, and its timeline will be determined by the resolution of the COVID-19 crisis and discussions with regulatory authorities.
This agreement will remain in force until the later of either a 10-year period after the first sale of a licensed product in the territory or the expiration of the last patent concerning such a licensed product in the relevant territory (determined on a per-territory basis). For more information, see "
◦Item 10.C – CollaborationAgreements with LabCorp and License Agreement with Terns Pharmaceuticals, Inc".Q2
Cholangiocarcinoma (CCA)
About Cholangiocarcinoma
CCA isIn January 2019, we entered into a type of cancer that forms in the bile ducts that carry the digestive fluid bile and is the second most common primary hepatic malignancy comprising approximately 15% of all primary liver tumors. Cases of CCA are usually asymptomatic in early stages, and are therefore often diagnosed when the disease is already in advanced stages. The silent presentation of these tumors combinedworldwide, non-exclusive license agreement with their highly aggressive nature and being refractory to chemotherapy contribute to poor prognosis and high mortality, representing ~2% of all cancer-related deaths worldwide yearly.
Although CCA is a rare cancer, its incidence (0.3–6 per 100,000 inhabitants per year) and mortality (1–6 per 100,000 inhabitants per year), have been increasing in the past few decades worldwide, representingLabcorp, a global life sciences leader specializing in health problem.
CCA constitutes a diverse group of heterogeneous biliary malignant tumors that can arise at any point of the biliary tree. CCAs are divided into two subtypes depending on their anatomical site of origin into intrahepatic CCA (iCCA)improvement and extrahepatic CCA (eCCA). Lesions involving the second-order bile ducts are defined as iCCApatient treatment decision support, to enable them to further develop and account for up to 50% of all CCAs. On the other hand, eCCAs are divided into perihilar CCAs (pCCAs), when they arise in the right or left hepatic duct or at their junction (50-60% of eCCAs), and distal CCAs (dCCAs), when they arise in the common bile duct (approx. 20-30% of eCCAs).
Several risk factors have been linked to CCA. However, a common characteristic amongst many of these risk factors is the association with chronic inflammation of the biliary epithelium and bile stasis, which are features of cholestatic diseases. In particular, primary sclerosing cholangitis has been identified as a risk factor linked to CCA. Several recognized risk factors have increased globally over recent decades and could be contributing to increasing CCA rates. These factors include high alcohol consumption, tobacco smoking, viral infections (hepatitis B virus and hepatitis C virus), as well as the global obesity pandemic, the metabolic syndrome and nonalcoholic fatty liver disease, which have been reported to increase the risk of CCA.
Although surgery is a potential curative option for CCA, most patients are diagnosed at late stages due to lack of specific symptoms. The majority of patients with CCA have metastatic or locally advanced (that is, unresectable) disease at presentation, and only ∼25% are eligible for resection. When disease is unresectable, the current first-line treatment is chemotherapy with gemcitabine + cisplatin. After progression on first-line chemotherapy, the second-line treatment includes FOLFOX chemotherapy, along with several targeted therapies including pemigatinib, dabrafenib + trametinib, and ivosidenib. However, despite the treatment options presently available, the unmet need remains high due to limited benefits on survival, and numerous programs are ongoing to develop additional first-line and second-line therapies in CCA.
GNS561 in CCA
To address the significant unmet need in patients diagnosed with CCA, GENFIT is developing GNS561 to prolong the overall survival of patients who present with iCCA and eCCA. GNS561 is a PPT-1 (Palmitoyl Protein Thioesterase-1) inhibitor that blocks autophagy. Autophagy is activated in tumor cells as a survival mechanism in a nutrient poor environment, due to tumor cell growth in advanced cancers. One of the key cellular organelles implicated in the autophagy process is the lysosome. By decreasing the activity of PPT1 in lysosomes, GNS561 may have an important inhibiting activity on late-stage autophagy, which leads to tumor cell death.
Autophagy is induced by starvation to capture and degrade intracellular proteins and organelles in lysosomes, which recycles intracellular components to sustain metabolism and survival. Autophagy also plays a major homeostatic role in controlling protein and organelle quality and quantity, and through these processes play a role in protecting cells from undergoing programmed cell death. Therefore, autophagy confers the ability to adapt to environmental stresses, preventing cellular damage, and promoting cell survival. Autophagy has opposing roles in cancer, preventing tumor initiation in healthy tissues, but favoring cancer progression once the tumor is formed. Although autophagy may play a role to prevent tumor initiation, it often promotes tumor cell proliferation and survival in more advanced cancers, where it is able to help cancer cells cope with the harsh tumor environments characterized by nutrient depletion, hypoxia, and other stresses such as chemotherapy. As such, autophagy has been described as a pro-survival mechanism present in most advanced tumors, facilitating tumor adaptation to different stresses, thus mediating tumor progression. Accordingly, many types of advanced cancers show higher basal autophagic activity than normal tissues, and some have been described as “autophagy-dependent” tumors, such as pancreatic cancer or tumors with active RAS (leading to active MAP kinase pathway). This basal autophagy also facilitates cancer cell adaptation to therapy-induced stresses, provoking therapy resistance in these tumors, which is one of the major challenges in the clinic.
Beyond the variable basal levels of autophagy, many general cytotoxic chemotherapy drugs as well as multiple targeted therapies such as kinase inhibitors have been proposed to induce autophagy as a protective measure in cancer cells. In 2019, the results of two major studies showed that,deploy NIS4® in the context of a cancer with the KRAS mutation (active RAS leadingclinical research. We believe this agreement will provide expanded access to, activationand further validation of the MAP kinase pathway), inhibitors of the MAP kinase pathway can induce autophagy in pancreatic cancer,an LDT powered by NIS4®. The license agreement permits Labcorp, through its subsidiary Covance, to market and combinations of these kinase inhibitors with autophagy inhibition can enhance tumor cell killing (Bryant et al., 2019; Kinsey et al., 2019). Importantly, a significant proportion of CCA patients have mutations including KRAS. Therefore, the combination of therapies targeting the MAP kinase pathway with GNS561 to inhibit autophagy is a potential therapeutic strategy to treat CCA patients.
Lysosomal function issell an essential element in autophagy (macroautophagy) and GNS561 is a lysosomotropic small molecule which inhibits PPT1, a lysosomal enzyme required to maintain lysosome-autophagy function. PPT1 expression is high in most cancer cell lines, increased in tumors compared with paired normal tissue, and in metastases versus primary tumors. As well,LDT powered by NIS4® test in the TCGA database, stage IVcontext of clinical research studies. Covance processes samples and provides test results to clinical trial sponsors. Covance has made significant progress in the deployment of NIS4® in several clinical trials conducted by leading players in the pharmaceutical industry. Covance is permitted and accredited, and will be responsible for submitting any validation that may be required under applicable state and federal laws.
In September 2020 we and Labcorp announced the signature of a five-year exclusive license agreement for our NIS4® technology, which seeks to enable easier identification of patients with multiple tumor types had significantly shorter overall survival if they exhibited high expression of PPT1. Thus, these findings along withat-risk MASH. Under the role of PPT1 in maintaining lysosome-autophagy function establishes the potential of PPT1 inhibition aslicense agreement, Labcorp will commercialize a strategy in cancer therapy. In addition to its inhibition of PPT1, studies with GNS561 showed that it has high liver tropism when administered orally, significantly reduced cell viability in two human iCCA cell lines and induced apoptosis. GNS561-mediated cell death was correlated with inhibition of late-stage autophagy and induction of a dose-dependent build-up of dysfunctional lysosomes. GNS561 was also efficient in vivo against a human intrahepatic CCA cell line in a chicken chorioallantoic membrane xenograft model, with a good tolerance at doses high enough to induce an antitumor effect in this model.
Basedblood-based molecular test based on the current knowledge of the tumor-promoting role of autophagy, numerous pharmacological approaches aimed at inhibiting autophagy at different stages of the pathway are being developed, the majority of which are in preclinical stage. However, the lysosomal inhibitor CQ and its derivative hydroxychloroquine (HCQ) are the only drugs that are currently used in patients with the deliberate goal of targeting autophagy (Towers and Thorburn, 2016). The first wave of clinical trials with CQ/HCQ alone or in combination with other therapies showed promising results, but more recent trial results have been less positive, which may be due to insufficient drug concentrations being used, leading to inconsistent results. Retinopathy and cardiac toxicity (QT prolongation) are major toxicities reported with CQ and HCQ. It has been suggested that novel PPT1 inhibitors which do not cross the blood brain barrier could have an advantage to avoid this retinal toxicity. In comparison, in preclinical studies, GNS561 has been observed to have a 10-fold higher potency than CQ/HCQ to decrease the viability of CCA cell lines (GNS communication). As well, a whole body tissue distribution study of GNS561NIS4® technology in the rat showed very limited exposureUnited States and Canada, thereby making it more widely accessible to health professionals. Leveraging the NIS4® technology, in April 2021, Labcorp launched the LDT "NASHNext®".
In May 2021, we signed a worldwide, non-exclusive license agreement with Q2 to broaden the availability of NIS4® technology in the brain and eyes. GNS561 has been evaluated in a Phase 1b trial which included patients diagnosed with CCA and HCC, and did not detect any neurological or retinal toxicity, cardiac toxicity, or any dose limiting toxicity.
GNS561 is an inhibitor of PPT1, which has been observed to induces lysosomal dysregulation, inhibit autophagy, induce CCA cell killing, and has demonstrated anti-tumor activity in an in vivo allograft model. Taken together, the compelling results generated with GNS561 highlight its potential to provide benefit in prolonging survival of patients diagnosed with CCA. In particular, we believe that GNS561, as an inhibitor of autophagy, is well positioned to be beneficial in combination therapy, including combinations with inhibitors of the MAP kinase pathway or immunotherapy/checkpoint inhibitors.
GNS561 is an investigational compound and has not been registered by any regulatory authority. It is a candidate for breakthrough therapy designation, orphan drug designation and accelerated approval.
We plan to start the Phase 2 clinical trial by the fourth quarter of 2022.
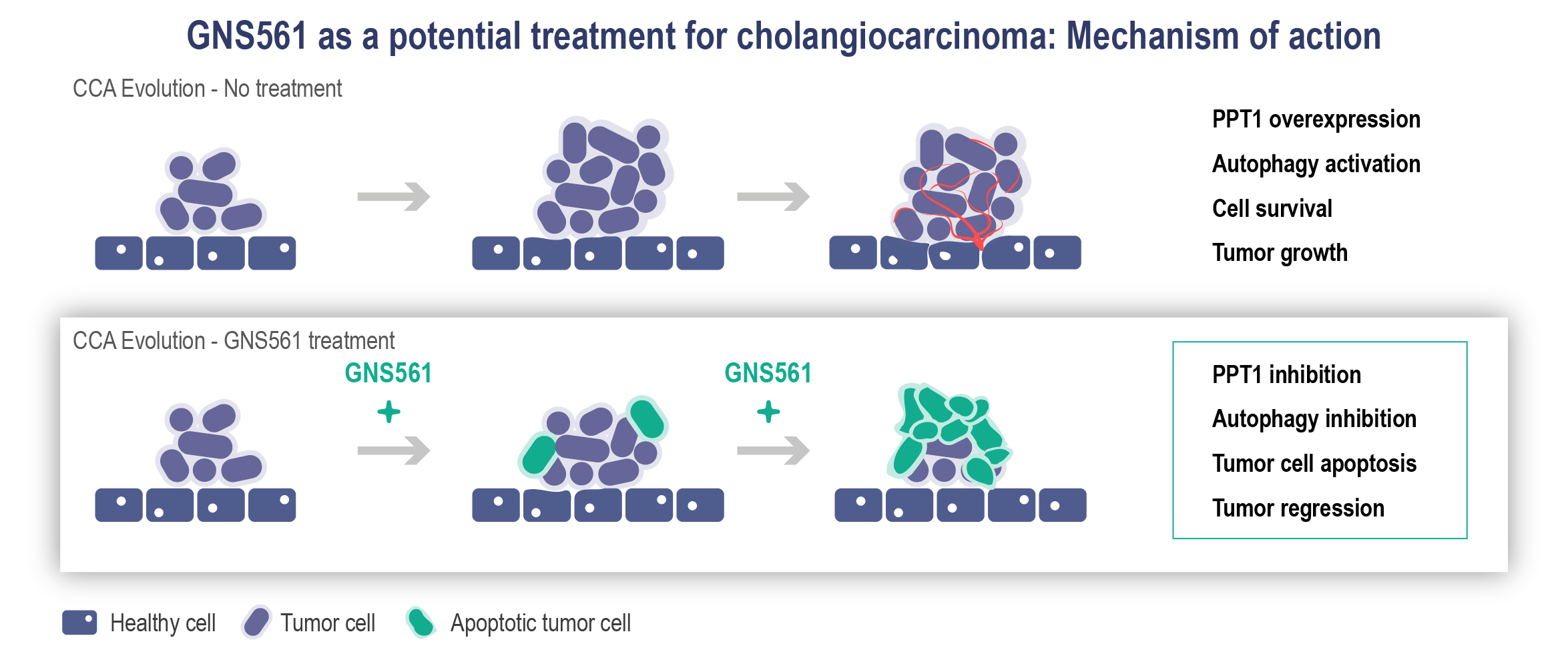
◦License and Development Agreement with Genoscience Pharma
On December 16, 2021, we signed an exclusive license from Genoscience Pharma to develop and commercialize the investigational treatment GNS561 in CCA in the United States, Canada and Europe, including the United Kingdom and Switzerland. Genoscience Pharma is a French clinical-stage biotechnology company developing novel lysosomotropic therapeutics to establish a new standard of care against cancer, autoimmune and infectious diseases.
Under the agreement, Genoscience Pharma is eligible for clinical and regulatory milestone payments of up to €50 million and tiered royalties. The first payable milestone is contingent on positive Phase 2 clinical trial results.results, and may result in payments of up to €20 million.
In addition, we also have a right of first negotiation with respect to any license or assignment, or option for a license or an assignment, with any third party to develop or commercialize other Genoscience Pharma assets in the field of CCA, to the extent Genoscience Pharma is looking to partner the asset with a third party or receives a spontaneous offer for collaboration.
For the period commencing on the date of the agreement until the first regulatory approval of GNS561 for commercialization, Genoscience Pharma has the right to repurchase the license to GNS561 in CCA at a pre-determined price in the event that Genoscience Pharma receives an offer from a third party to acquire or obtain a license to GNS561 in all indications, provided that GENFIT shall first have the opportunity to negotiate the acquisition or license to GNS561 in all indications.
The agreement shall remain in force, on a country by country basis in the territory until the later of (i) the date on which the last patent rights included in the licensed patents expires, or is otherwise cancelled, withdrawn or abandoned, in such country, or (ii) upon the regulatory approval of a generic product with respect to the licensed product in such country or (iii) the tenth anniversary of the first commercial sale of the licensed product in such country.
GENFIT also purchased a 10% equity stake in Genoscience Pharma through the subscription of new ordinary shares for a totaltotal amount of approximately €3.1€2.3 million.
◦License Agreement with Seal Rock Therapeutics
In May 2023, GENFIT licensed the exclusive worldwide rights of ASK1 Inhibitor SRT-015 (injectable formulation in acute liver disease) from Seal Rock Therapeutics, a Seattle, Washington (USA) based clinical stage company.
Under the terms of the agreement, Seal Rock Therapeutics is eligible for payments of up to €100 million, including regulatory, clinical and commercial milestones, as well as tiered royalties. All of these payments would only begin to become due following positive Phase 2 results, which would not occur before 2026, according to our best estimates. For more information about the financial terms of the agreement, see Note 2.52 - "Major Events in the Period and Events after the Period - Major events in the period" "Signature of a Licensing to our consolidated financial statements included in this annual report ◦License Agreement with Genoscience Pharma"Celloram Acute on Chronic Liver FailureOn July 28 2023, GENFIT licensed the exclusive worldwide rights to CLM-022, a first-in-class inflammasome inhibitor in liver disease, from Celloram Inc., a Cleveland-based biotechnology company.
Regardless of their etiology, chronic liver diseases, or CLD are invariably associated with progressive accumulation of fibrosis inUnder the liver culminating in cirrhosis. At a compensated stage, cirrhosis is a silently evolving disease with no specific symptoms. In the absence of an approved anti-fibrotic drug and if the underlying CLD is not treated, fibrosis further invades the liver, which is ultimately no longer able to efficiently function. This transition from compensated to decompensated cirrhosis occurs when anyterms of the followingagreement, Celloram is eligible for payments of up to €160 million, including regulatory, clinical hallmark events occur: presence of ascites, variceal hemorrhage and/or hepatic encephalopathy.
Patients with CLD, and particularly patients with compensated and decompensated cirrhosis, are highly sensitive to acute insults triggering a sudden deterioration of liver function, which may consequently jeopardize normal functioning of other organs. Such an acute decompensation (AD) event is typically the result of a precipitating event, such as bacterial infection, acute alcoholic hepatitis, relapse of viral hepatitis or drug-induced liver injury, although as many as 40% of patients with AD have no identifiable precipitating event.
Acute on Chronic Liver Failure or ACLF syndrome is generally recognized as an episode of AD associated with one or more failures affecting vital organ systems including the liver, kidneys, brain, coagulation, circulation and/or respiratory systems.
Due to associated organ failures, patients with ACLF syndrome are at very high risk for short-term mortality, and globally the mean survival time in this population is 3 – 5 years. In a study of 1,343 hospitalized patients with cirrhosis and AD, 303 had ACLF when the study began, 112 developed ACLF, and 928 did not have ACLF. The 28-day mortality rate among patients who had ACLF when the study began was 33.9%, among those who developed ACLF was 29.7%, and among those who did not have ACLF was 1.9%. In general, a greater number of organ failures is associated with higher short-term mortality. For example, the 28-day mortality rate for patients having 3 or more organ failures approaches 80%.
Management of patients with ACLF
There are no specific therapies currently available for patients with ACLF other than treatment of precipitating events, when identified, and organ failure support (e.g., hemodialysis in the case of kidney failure). The only definitive treatment option is liver transplantation but due to emergency setting, limited access to compatible liver donors and in some case no accessible liver transplant capabilities, a vast majority of patients experiencing a life-threatening ACLF episode do not have access to transplant surgery.
Patients with AD are generally hospitalized in the regular hepatology ward, although an intensive care unit where organ support can most effectively be provided generally provides optimal care of patients with ACLF.
Despite intense efforts to improve the standard of care, the current high short-term mortality rate highlights the critical medical need of new therapies to help patients to rapidly recover and survive an ACLF episode without liver transplantation.
ACLF is a rare life-threatening condition with significant costs for health care systems
Worldwide, there are about 10 million hospitalizations per year for decompensated cirrhosis. With a 35% ACLF prevalence in patients hospitalized for decompensated cirrhosis, there are approximately 3-4 million hospitalizations for ACLF each year.
In the US, there are over 600,000 hospitalizations per year for decompensated cirrhosis. With a 10-30% ACLF prevalence in this population the annual number of ACLF hospitalizations in the US is estimated to be between 60,000 and 180,000. In the 5 major European countries, there are about 800,000 hospitalizations for decompensated cirrhosis. With a 20-30% prevalence in this population, the annual number of ACLF hospitalization is estimated to be between 160,000 and 240,000.
Cirrhosis and ACLF represent a substantial health and economic burden. For example, in the United States in 2011, the total inpatient costs for cirrhosis with and without ACLF was estimated to be more than $10 billion. In the same study, the cost per hospitalization was 3.5-fold higher for ACLF patients with two or more organ failures than for patients with cirrhosis who did not have ACLF.
Such high hospitalization costs for critically ill ACLF patients as compared to cirrhotic patients without ACLF can be easily explained by higher rates of hospitalization in the ICU and, most importantly, by 2-3-fold longer hospital stays: average of 16 days for ACLF patient versus 7 days for patients with cirrhosis who did not have ACLF.
ACLF pathophysiological mechanisms and GENFIT research strategies
In the absence of available treatments for the management of ACLF, we believe R&D efforts should focus on scientific strategies targeted on mechanisms leading to rapid development of organ failures after an acute insult. Although the pathophysiological processes driving ACLF development are not fully understood, some events appear as particularly important. For instance, there is now a large body of evidence implying that an uncontrolled and generalized inflammatory storm (so called Systemic Inflammatory Response Syndrome or SIRS) plays a key role in ACLF development. This may at least partly result from rapid translocation of intact bacteria and bacterial products (pathogen-associated molecular patterns or PAMPs) crossing the intestinal barrier, reaching the circulation and peripheral organs. About 30% of patients with ACLF have sepsis, which confirms this hypothesis.
GENFIT believes that the ideal treatment must simultaneously target all components of the pathological process.
In ACLF, GENFIT is assessing two classes of compounds, NTZ , on the one hand, an antiparasitic drug which we are repositioning, and compounds within the PPAR agonist therapeutic class with optimized metabolic and anti-inflammatory effects, on the other hand.
Preclinical and Clinical Development Program
The identification of NTZ is the result of our research program initially designed to discover novel anti-fibrotic molecules with a priority given to liver fibrosis.
During further research,we have also discovered that NTZ and its circulating metabolite, tizoxanide or TZ, have additional anti-inflammatory effects through the inhibition of inflammatory cell activation. In our preclinical research, we have observed that NTZ's anti-infectious properties may act on intestinal microbiota dysbiosis/overgrowth and improve the intestinal barrier and direct dose-dependent anti-inflammatory effects on immune cells (macrophages and polymorphonuclear leukocytes).
As part of our preclinical program, we have studied NTZ in in vitro and in vivo disease models .
In disease models, NTZcommercial milestones, as well as its active circulating metabolite, have a wide anti-infectious spectrum acting on bacteria, viruses and parasites commonly encountered in human intestinal flora. Thus, an oral treatment with NTZ is expectedtiered royalties. Almost all of these payments would only begin to improve bacteria overgrowth and dysbiosis and possibly preservebecome due following positive Phase 2 results, which would not occur before 2028, according to our best estimates. For more information about the intestinal barrier in patients with ACLF. We also observed that, in cultured human liver cells, TZ inhibits a key pathwayfinancial terms of programmed cell death (apoptosis) in a dose dependent manner.
Our research has demonstrated that in healthy rats, an oral administration of NTZ concomitant with intraperitoneal injection of LPS significantly reduced the LPS-induced rise in circulating cytokines and inflammatory markers.
In two distinct rat models of ACLF, we found that NTZ has hepatoprotective effects by reducing ALT and AST while totally preventing LPS-induced rise in GGT and total bilirubin. NTZ also significantly reduced LPS induced brain edema and LPS-induced rise in inflammatory markers. Treatment with NTZ also prevented plasma increases in two renal function markers : cystatin C and creatinine.
In addition, we are currently researching the effects of NTZ on sepsis, with encouraging preliminary results. In the sepsis model, the mortality rates in NTZ treated vs vehicle treated group were 53% vs 90% at 72 hours and 67% vs 100% 5 days after CLP surgery.
agreement, see
NTZ clinical program
NIS4 Technology to Power the Identification of Patients with NASH and Fibrosis
As part of our strategy to address unmet needs in NASH, we have advanced a diagnostic program based on the identification of specific biomarkers that are expressed at different levels in patients with NASH and significant fibrosis (F≥2) as compared to patients with less severe disease. This discovery kicked off a multi-year effort that has resulted in the development of NIS4 technology, a blood-based molecular technology for the identification of patients with NASH (NAS≥4) and significant fibrosis (F≥2), also referred to as “at-risk” NASH, who are at higher risk of disease progression and may be appropriate candidates for therapeutic intervention. In January 2019, we entered into a license agreement with Labcorp to allow them to develop, market, and sell a test powered by NIS4 in the clinical research space. In September 2020, we signed a five-year exclusive license agreement with Labcorp to allow them to develop and commercialize a Laboratory Developed Test or LDT powered by NIS4 technology for use in routine clinical diagnostic testing in the United States and Canada. In late April 2021, Labcorp launched its LDT NASHnext powered by NIS4 technology. Its commercial launch is however still constrained by the lack of approved therapeutic options in NASH.
In May 2021, we signed a worldwide, non-exclusive license agreement with Q Squared Solutions LLC or Q2, to broaden the availability of NIS4 technology in the clinical research field.
Today’s Challenges in Diagnosing NASH
NASH, the most severe form of NAFLD, is characterized by the presence of hepatocyte ballooning and inflammation, in addition to steatosis. NASH can progress silently towards cirrhosis, precluding the opportunity for clinicians to diagnose and intervene therapeutically prior to the development of severe liver complications, and constitutes a growing cause of cirrhosis, liver failure, and liver cancer globally. Furthermore, NASH is projected to become the leading cause of liver transplantation in the United States—it already is the primary cause among women and the secondary cause overall. Given this clinical scenario, there is a pressing need to identify patients at higher risk of disease progression, who could be considered for therapeutic intervention with existing options or when potentially promising agents currently in late-stage clinical development obtain regulatory approval.
The main histological determinants of the risk for long-term severe liver outcomes are NASH activity and fibrosis stage (F). NASH activity is assessed by the NAFLD activity score (NAS), a composite index derived from the sum of the scores for macrovesicular steatosis, hepatocellular injury (i.e., ballooning), and lobular inflammation. In a study with paired liver biopsies, steatohepatitis was associated with liver-related outcomes, and a higher NAS at baseline was associated with a high probability of fibrosis stage increase after ≥1 year, suggesting an association between increased NASH activity and fibrosis progression. Furthermore, a Phase 2b clinical trial in NASH demonstrated higher rates of spontaneous disease regression in both treated and untreated patients with milder NASH severity (NAS=3) compared with patients with higher activity (NAS ≥4) at baseline. Additionally, multiple studies have shown that fibrosis stage reflects the extent of disease progression toward cirrhosis—in particular, that F ≥2 (significant fibrosis) increases the risk of liver-related clinical outcomes. Given that the overall disease state is described by the combination of NASH activity and fibrosis stage, this is the rationale for inclusion of patients with NASH, a NAS ≥4 and a F ≥2 (referred to as “at-risk NASH”) in pharmacological intervention clinical trials.
Liver biopsy is the clinical reference standard for the diagnosis of NASH among patients with clinical risk factors for this disease, such as metabolic disorders (with or without abnormal liver biochemistries) in the absence of alternative causes for steatosis. The implementation of this diagnostic approach, however, is limited in routine clinical practice by its invasiveness, cost, attendant risks, variability in interpretation, and the restricted number of professionals able to perform and interpret the test, among other factors. These limitations preclude liver biopsies from being broadly used as the primary diagnostic in such a prevalent disease. Providing a non-invasive alternative to liver biopsy will therefore be critical to facilitate improved patient diagnosis, management, and future treatment access in routine clinical practice, and may eventually reduce the morbidity and mortality associated with this disease.
Currently, there are few non-invasive diagnostics specifically designed to identify at-risk NASH. Existing tests used in a NAFLD or NASH clinical context can be generally characterized as either repurposed or not optimized for the identification of this condition. Multiple algorithm or imaging-based tests used today (e.g., Fibrosis-4 [FIB-4] score, aspartate aminotransferase [AST]-to- Platelet Ratio Index [APRI], Enhanced Liver Fibrosis [ELF] score, vibration-controlled transient elastography ([VCTE] also known as FibroScan) were originally designed for use in mixed liver aetiologies (e.g., hepatitis C virus, hepatitis B virus), and have since been repurposed for use in NAFLD or NASH. Limitations associated with many of these tests have been reported, including performance (area under the receiver operating characteristics curve [AUROC] <0∙80) for the identification of NASH or fibrosis stage ≥2, or both, in individuals with type 2 diabetes. Additionally, several NAFLD-focused or NASH-focused tests (e.g., body-mass index [BMI], AST/alanine aminotransferase [ALT] ratio and diabetes [BARD] score, and NAFLD Fibrosis Sore [NFS]) were developed to identify advanced liver fibrosis (i.e., fibrosis stage ≥3), and might therefore not be optimized for the identification of at-risk NASH. Even widely used imaging-based techniques for liver disease management, such as VCTE, have been shown to be influenced by a number of clinical features, including the presence of type 2 diabetes, dyslipidaemia, elevated waist circumference, elevated AST concentrations, and elevated systolic blood pressure at the time of examination. Limitations or confounders of non-invasive tests are of importance to clinicians to help them assess the right NASH diagnostic tests to use in their patients. We aimed to develop and validate a blood-based diagnostic multivariate index test that is specifically designed to rule in and rule out at-risk NASH.
The treatment of NASH being a pressing public health challenge, there is a large unmet need for a widely available, non-invasive tool to identify patients with at-risk NASH as an alternative to liver biopsy. The availability of such a test would help address the under diagnosis of NASH by supporting physicians in identifying patients with at-risk NASH, who are at higher risk for clinical outcomes and would be eligible for therapeutic intervention. We believe a test powered by our NIS4 technology, if validated and approved for marketing, may directly address this clinical gap.
Circulating Biomarkers and miRNA
Biomarkers are characteristics of the body that are objectively measured and have the potential to correlate to a specific biological state or disease condition. Circulating biomarkers are biological molecules, such as proteins, DNA or RNA, found in body fluids such as cerebrospinal fluid, blood or urine that modulate with disease. A single circulating biomarker or a panel of markers has the potential to be used to not only identify but also monitor the progression, regression, or stability of disease.
MicroRNAs or miRNA represent an emerging class of small non-coding RNA whose principal function is the regulation of the expression of target genes by acting on the stability and the translation of their messenger RNA, or mRNA. miRNAs play an essential role in many cell functions, such as development, proliferation, differentiation, cell-cycle arrest and apoptosis, or cell death. Multiple studies have shown a close association between circulating levels of miRNA and the development and progression of several cancers and have highlighted an important role for miRNAs in the regulation of human liver development and pathophysiology. Because miRNAs are released from cells in response to stress, they can be detected in most biological fluids, including blood.
Our Solution: NIS4 Technology Comprising Our Proprietary Biomarker Algorithm
Aware of the challenges associated with diagnosing at-risk NASH, we initiated a research program to combine our technical expertise in informatics, machine learning and molecular biology with access to our extensive NASH clinical biobank, including cohorts from our GOLDEN-505 and RESOLVE-IT clinical trials, in addition to cohorts from academic partnerships, to pursue the discovery of novel biomarkers that may hold the key in developing novel diagnostic tests or technologies in NASH. In 2015, we reached a key milestone with the discovery that two miRNA biomarkers, miR-200a and miR-34a-5p, that were expressed at higher levels in patients with at-risk NASH as compared to patients with less severe disease.
Since then, we have further refined our science which has uncovered four unique biomarkers that we believe provide the best overall diagnostic performance to identify patients with at-risk NASH. Our lead technology, NIS4, integrates the outputs of four independent NASH-associated biomarkers [alpha-2-macroglobulin, YKL-40, hemoglobin A1c, and miR-34a-5p] through an algorithm to produce a single score that can be utilized to rule in and rule out at-risk NASH, while minimizing the number of indeterminate test results. We intend to market an IVD-powered by NIS4 technology, if it receives FDA/CE marketing authorization, as a standalone diagnostic with the potential to enable a non-invasive, accessible and validated alternative to the liver biopsy to benefit patients, improve overall clinical care and greatly reduce barriers to entry for innovative therapies.
Development and validation of NIS4 technology
Blood samples, clinical data, and liver biopsy results from three independent cohorts with suspected NAFLD were used to develop and validate the NIS4 non-invasive blood-based diagnostic technology. Derivation was done in the discovery cohort, which comprised 239 prospectively recruited patients with biopsy-confirmed NASH (NAFLD NAS ≥3; fibrosis stage 0‒3) from the international GOLDEN-505 phase 2b clinical trial. The overall diagnostic performance of NIS4 was externally validated in two independent cohorts: RESOLVE-IT diag and Angers. The RESOLVE-IT diag cohort comprised the first 475 patients screened for potential inclusion into the randomized, double-blind, placebo-controlled, multicenter, international RESOLVE-IT phase 3 clinical trial. ANGERS was a retrospective cohort of 227 prospectively recruited patients with suspected NAFLD and clinical risk factors for NASH or fibrosis stage 2 or more according to abnormal elastography results or abnormal liver biochemistry. Clinical cutoffs were established within the discovery cohort to optimize both rule out and rule in clinical performance while minimizing indeterminate results. NIS4 was validated in the RESOLVE-IT diag cohort (AUROC 0∙83, 95% CI 0∙79‒0∙86) and the Angers cohort (AUROC 0∙76, 0∙69‒0∙82). The diagnostic performance of NIS4 within the external validation cohorts was not influenced by age, sex, BMI, or aminotransferase concentrations.
In the pooled validation cohort, NIS4 significantly outperformed other non-invasive NASH or fibrosis diagnostics, including FIB-4, NFS, ELF, APRI, and BARD for the identification of “at-risk” NASH (all p<0∙010, figure below). The overall performance of NIS4 and VCTE was not statistically different. In addition, although NIS4 was not developed to specifically identify the subpopulation of “at-risk” NASH with fibrosis stage ≥3, performance was significantly better than FIB-4, NFS, BARD and APRI (all p<0∙05), with performance not significantly different to VCTE and ELF.
Subpopulation analyses were done in the pooled validation cohort to assess the overall performance of NIS4 compared with other diagnostics among specific subpopulations of clinical relevance in NASH (figure below). The overall diagnostic performance of NIS4 was the highest across non-invasive tests evaluated, and was neither dependent on (i.e., included as variables in the NIS4 algorithm) nor statistically affected by patient age within the range studied, sex, BMI, or transaminase concentrations. The clinical performance of NFS was significantly better in patients 55 years or older than in those younger than 55 years (AUROC 0∙69 vs 0∙59; p=0∙013), whereas FIB-4 showed higher performance in females than in males (0∙75 vs 0∙67; p=0∙039). Similarly, APRI showed higher performance in females than in males (0∙79 vs 0∙68; p=0∙0040). ELF however showed higher performance in patients with a BMI of 30 kg/m2 or less than in those with a BMI of more than 30 kg/m2 (0∙84 vs 0∙74; p= 0∙029), and in females than in males (0∙81 vs 0∙71; p= 0∙019). BARD showed consistent results across the categories explored, and VCTE— although not significant—had directionally higher performance in patients without type 2 diabetes than in those with type 2 diabetes (AUROC 0∙80 vs 0∙65; p= 0∙056).
We believe that an LDT or IVD powered by NIS4 technology can provide an effective way to noninvasively rule in or rule out at-risk NASH in patients with metabolic risk factors and suspected disease. Use of an LDT or IVD powered by NIS4 in clinical trials or in the clinic has the potential to greatly reduce unnecessary liver biopsies in patients with lower risk of disease progression, achieve straightforward integration into clinical care pathways, and be more cost-effective, accessible, and acceptable for patients than liver biopsy. In doing so, an LDT or IVD powered by NIS4 could also help improve the accuracy of NASH diagnosis in patients with suspected disease, and help healthcare providers identify those most in need of therapeutic intervention.
In August 2020, we announced that pivotal data describing the derivation and validation of NIS4 technology has been accepted for publication by The Lancet Gastroenterology & Hepatology. This published study details NIS4 algorithm development and clinical validation against the liver biopsy reference standard in two independent populations comprised of data from over 700 patients. In addition to the high overall performance in identifying patients with at-risk NASH, NIS4 technology also provided consistent results in critical sub-populations (i.e. diabetic vs. non-diabetic, men vs. women) as compared to other non-invasive tests evaluated in the same individuals.
In November 2020, we announced that data relating to the NIS4 technology and the final results of the RESOLVE-IT Phase 3 clinical trial could be consulted on five posters in the context of the Liver Meeting Digital Experience, the annual conference of the American Association for the Study of Liver Diseases, which was held virtually on 13, 14 and 15 November 2020.
In June 2021, we presented new NIS4 data at the International Liver Congress™ organized by the European Association for the Study of the Liver (EASL), and the 81st Scientific Sessions of the American Diabetes Association (ADA). The data highlights the clinical performance of NIS4 technology in diagnosing at-risk NASH in patients with type 2 diabetes compared to other non-invasive tests. It shows the potential of NIS4 technology to be a valuable clinical tool either alone or in sequential combination with other blood-based non-invasive tests in identifying at-risk NASH in patients with and without type 2 diabetes.
In November 2021, NIS4 technology’s utility was recognized in a study undertaken by NIMBLE as demonstrating a unique performance in identifying patients with “at-risk” NASH. This study highlighted the importance for the diagnosis of “at risk” NASH patients (defined as NASH + NAS ≥4 and fibrosis ≥2) as a subpopulation at greater risk of liver-related outcomes. Of the five blood-based biomarker panels which were assessed in this study, only NIS4 technology produced results showing the capacity to identify these “at-risk” NASH patients.
The study also provided evidence that NIS4 technology – with a sensitivity of 82.3 and a specificity of 79.9 – had the best results among the other biomarkers tested for the diagnosis of fibrosis stage ≥ 2, a critical component of “at-risk” NASH. The presentation concluded that NIS4 technology met the a priori criteria established for Stage 1 by the NIMBLE Circulating Biomarkers Workstream (CWS) for:
• Diagnosis of NAS≥4
• Fibrosis stage 2 or higher.
Regulatory• and Commercial StrategyCompetitive Landscape
We began communications with the FDA in 2017 to discuss potential regulatory pathways for an IVD powered by NIS4 technology. Based on these discussions,Because we will be using blood samples and liver biopsy results from non-treated patients enrolled in our clinical trials conducted to date in order to provide support for the potential validation of our test. By calculating NIS4 from a patient’s blood sample, and then comparing the test score to that patient’s liver biopsy result, we can assess whether an IVD powered byNIS4 is accurate in diagnosing patients with at-risk NASH, who are at higher risk of disease progression and may be considered for therapeutic intervention. We are conducting physician, payer, and patient primary market research to best understand clinical performance goals that would meet the needs of the evolving NASH marketplace. Based on these insights, we will finalize the analytical and clinical study designs, which are required prior to initiating formal validation studies for both the FDA and Notified Body submissions.
We continue to explore the possibility of obtaining regulatory approval and CE Certificates of Conformity to release an IVD powered by NIS4 technology on the US and European markets.
In January 2019, we entered into a license agreement with Labcorp, a global life sciences leader specializing in health improvement and patient treatment decision support, to enable them to further develop and deploy NIS4 in the context of clinical research. We believe this agreement will provide expanded access to, and further validation of an LDT powered by NIS4. Initially, we will enable Labcorp through its subsidiary Covance to market and sell an LDT powered by NIS4 test in the context of clinical research studies. Covance will process samples and provide test results to clinical trial sponsors. Covance is permitted and accredited, and will be responsible for submitting any validation that may be required under applicable state and federal laws.
In September 2020 we and Labcorp announced the signature of a five-year exclusive license agreement for our NIS4 technology, which seeks to enable easier identification of patients with at-risk NASH. Under the license agreement, Labcorp will commercialize a blood-based molecular test based on NIS4 technology in the United States and Canada, thereby making it more widely accessible to health professionals.
In May 2021, we announced that Labcorp had launched NASHnext, a novel, noninvasive diagnostic test powered by GENFIT’s NIS4 technology for the identification of NASH patients with significant fibrosis – also known as “at-risk” NASH. The test, offered by Labcorp exclusively in the U.S. and Canada, aims to identify at-risk NASH in patients with at least one metabolic risk factor. The clinical pertinence as well as the market potential for this test have been confirmed through its use in clinical trials and the new data presented in the first half of 2021. Nonetheless, the COVID-19 pandemic, the lack of approved therapeutic options in NASH and the current lack of reimbursement for NASHnext have contributed to commercial launch that remains modest at the time of this annual report.
Our Clinical Program for Elafibranor in the Potential Treatment of NASH
RESOLVE-IT—Pivotal Phase 3 Clinical Trial in NASH
In May 2020, we announced the topline results of the interim analysis of the RESOLVE-IT Phase 3 clinical trial evaluating the efficacy of the daily administration of elafibranor 120 mg in adults with NASH.
The RESOLVE-IT Phase 3 clinical trial evaluated the effect of elafibranor compared to placebo in 1,070 patients (ITT population) with biopsy proven NASH as defined by NAFLD activity score (NAS) greater than or equal to 4, fibrosis stage 2 or 3. Patients were randomized 2:1 to receive elafibranor 120mg or placebo once daily, with a follow-up liver biopsy at week 72 to evaluate histologic endpoints (resolution of NASH without worsening of fibrosis or fibrosis improvement of at least one stage).
Resolution of NASH is defined by a ballooning score of 0 and an inflammation score of 0 or 1, and the non-worsening of fibrosis corresponds to a fibrosis score that does not increase.
The trial did not meet the predefined primary endpoint of NASH resolution without worsening of fibrosis in the ITT population. In the ITT population, 19.2% of patients who received elafibranor (N=138) achieved NASH resolution without worsening of fibrosis compared to 14.7% of patients in the placebo arm (N=52) (p=0.07).
On the key secondary endpoint of fibrosis improvement of at least one stage, 24.5% of patients who received elafibranor (N=176) achieved fibrosis improvement of at least one stage compared to 22.4% (N=79) in the placebo arm (p=0.445).
Elafibranor was generally well tolerated over the 72 weeks of exposure, which is consistent with previously conducted studies and with DSMB conclusions based upon their review of RESOLVE-IT safety data throughout the study. The safety, including histological assessment, and tolerability profile of elafibranor continues to be supportive of ongoing clinical investigation, which is encouraging for the ongoing Phase 3 trial evaluating elafibranor in PBC.
On July 22, 2020, following the detailed review of the full RESOLVE-IT interim efficacy dataset, we determined that the investment needed to continue the trial was not justified, as it was unlikely to provide results that would be sufficient to support elafibranor for registration in NASH in the United States and Europe.
Competition
We focus on therapeutic areas with high unmet medical needs, i.e. areas characterized by a lack of diagnostic or treatment options. As a result,options, there are relatively few companies with approved products compared with other therapeutic or diagnostic areas where several options are already approved from a regulatory standpoint, and available for healthcare providers and patients.
We however operate in a competitive sector. Several companies are working on technologies, therapeutic targets or drug or biomarker candidates that aim to treat or diagnose the same diseases or identify the same patient population as our product candidates. While we believe that our drug candidates and diagnostic solutions, combined with our expertise and know-how, provide us with competitive advantages, we face potential competition from various sources, including pharmaceutical and biotechnology companies, as well as from academic institutions, governmental agencies and public and private research institutions. We anticipate that we will face intense and increasing competition as new drugs and therapies enter the market and advanced technologies become available.
Cholestatic Diseases In some indications, off-label use of non-approved drugs can also be considered as competition.
•◦PBC
Only two drugs are approved in this indication. UDCA, was approved by the FDA to treat PBC in 1997, and remained the only approved treatment for PBC until 2016, when Ocaliva™Ocaliva was approved by the FDA and ECEuropean Medicines Agency for the treatment of PBC in combination with UDCA in adults with an inadequate response to UDCA, or as monotherapy in adults unable to tolerate UDCA. Although approved as a second-line treatment, Ocaliva™ is the subject of continued safety concerns (with respect to pruritus and serious liver injury or death, leading to the FDA issuing a Black Box Warning in 2018). If approved, elafibranor would compete with these drugs already approved for the treatment of PBC.
The other molecule that could become a direct competitor of elafibranor is seladelpar, developed by the American company CymaBay, which announced at the endreadout of March 2021 that it had commenced recruitmenttopline data for its new Phase 3 (RESPONSE) trial. We know thattrial early September 2023 and NDA submission mid-December 2023. In February 2024, Gilead announced the acquisition of CymaBay Therapeutics.
Other companies are developing other companies develop otherless advanced drug candidates to treat PBC, and therefore may also become competitors. The latest IQVIA study made available to the Company however indicates that these molecules are in relatively early stages of clinical development compared to elafibranor, and would capture together – if approved – less than 20% of market shares. This includedFor instance, Calliditas Therapeutics which announced on February 15,in 2022 that the first patient was randomizedenrolled in its Phase 2b/3 TRANSFORM study evaluating setanaxib in patients with PBC. In addition
◦ACLF
No drugs have been approved in this indication so far and the only therapeutic option currently available is liver transplantation. Some companies, such as Cellaïon, are investigating the potential of certain technologies, but given known challenges in the space, we believe those would likely become complementary to these approved drugs and drug candidates in development, we also may compete with approved drugs in other indications which could be used off-label for the treatment of PBC.what GENFIT is developing rather than serve as direct competitors.
•◦CCA
There is a high unmet need in CCA patients without actionable alterations as currentCurrent treatment options are limited to chemotherapy. The current pipeline of drugs in development includes anti-PD-(L)1 combinations, FGFR2 and PARP inhibitors. FGFR2 and PARP inhibitors are limited to patients with specific alterations, while the expectations from anti-PD-(L) to work in CCA are currently low. A combination of atezolizumab and cobimetinib (anti-PD-(L)1 and MEKi) is being evaluated but preliminary data do not show a major benefit.
•◦ACLFHAC in UCD and OA
There is also a high unmet needNo drugs have been approved for HAC. However, Buphenyl and Ravicti are ammonia scavengers approved in ACLF, a serious condition for whichUCD in the only therapeutic option is liver transplantation. There is therefore a strong need forU.S. and in the development of a promising therapyUS and Europe, respectively.
◦HE
Standard-of-care therapeutics include lactulose (with various brands) and rifaximin (Xifaxan approved in the U.S. and EU, and Rifxima approved in Japan), both oral treatments aiming to reduce ammonia. LOLA (Hepa-Merz approved in the morbidity and mortalityEU) is a third option, but not approved in these patients, as well as to provide healthcare professionals with non-surgical therapeutic options. Some companies, such as Versantis or Promethera, are investigating the potential of certain technologies that may become complementary to what GENFIT is developing, rather than direct competitors.U.S.
•◦NASHMASH Diagnostics
With respect to NIS4, our technology that usesNo blood-based biomarkersdiagnostic solution is approved to identify patients with NASH"at-risk" MASH.
◦At-home ammonia monitoring
The international state of the art of ammonia quantification in blood is enzymatic assays that are implemented in extremely costly large automatic analyzer machines usually only available at central or hospital clinical laboratories. Considering that ammonia blood samples should be collected on ice and fibrosis who we believe could benefit from therapeutic intervention, there are a number of clinical diagnostic tools available foranalyzed within the management of chronic liver disease patients, but none are validated for NASH. In November 2021, NIS4 technology’s utility was recognized in a Stage 1 study undertaken by NIMBLE as demonstrating a unique performance in identifying patients with “at-risk” NASH. The report highlightedhour, these limitations may delay the importance forresults and may add uncertainties to the diagnosis of “at risk” NASH patients (defined as NASH + NAS ≥4HE.
These main limitations of the current gold standard can be resolved with a reliable point of care device at the patients' bedside. The current point of care device commercially available (Arkray’s PocketChem BA analyzer) is however limited by its narrow quantification range (7-286 µmol/L), its interference issues and fibrosis ≥2) asits underestimation of ammonia levels in comparison to enzymatic assays.
Therefore, the need of a subpopulation at greater riskfast, accurate, and precise point of liver-related outcomes, and of all blood-based biomarkers which were assessed in this study (FIB4, ALT, OWL, NIS4, ELF, PROC3, FM-VCTE), only NIS4 technology produced results showing the capacity to identify these “at-risk” NASH patients.care device has not yet been achieved satisfactorily.
◦Other considerations
We believe that elafibranor’s differentiated mechanism of action in targeting PPARα and PPARδ, and the favorable tolerability profile observed to date suggest the potential for elafibranor to have competitive advantages over approved drugs and drug candidates in development by our competitors. NTZ also provided encouraging preclinical data.
However, manyMany of our competitors, either alone or with their strategic collaborators, have substantially greater financial, technical and human resources than we do. Accordingly, our competitors may be more successful than we are in obtaining approval for their drug candidates and achieving widespread market acceptance and may render our drug candidates, such as elafibranor, obsolete or non-competitive. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical study sites and patient registration for clinical studies, as well as in acquiring technologies complementary to, or necessary for, our programs.
We anticipate that we will face intense and increasing competition as new drugs and therapies enter the market and advanced technologies become available. We expect any drugs that we develop and commercialize to compete on the basis of, among other things, efficacy, safety, delivery, price and the availability of reimbursement from government and other third-party payors.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize drugs that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive or better reimbursed than any drugs that we may commercialize. Our competitors also may obtain FDA, EMA or other regulatory approval for their drugs more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position for either the product or a specific indication before we are able to enter the market.
The same considerations apply in the NASH diagnostic field.
–Manufacturing and Supply
We do not have any manufacturing facilities or personnel. We currently rely, and expect to continue to rely, on third parties for the manufacturing of our drug candidates for preclinical and clinical testing, as well as for commercial manufacturing if our drug candidates receive marketing approval.
With respect to our lead drug candidate, elafibranor, we use one supplier for theour remaining active ingredient and another manufacturer for the therapeutic units used in ourstocks were sold to Ipsen to meet their short term clinical trials. Although we could use a substitute company in the event of failure or breach of these two manufacturers, we may face challenges in finding new suppliers within an acceptable timeframe or under commercially reasonable conditions. To mitigate this risk, we have performed an evaluation of the expectedneeds. Ipsen is responsible for managing its clinical and commercial needs for elafibranor manufacturing delays and costs in the event of a disaster at the supplier of the active ingredient or at the manufacturer of therapeutic units. Based on the results of this evaluation, we believe that given the current inventory and drugs in production at various levels of the production chain, which is sufficient to supply our ongoing clinical trials, the short-term failure of one of these manufacturers would not be critical.directly.
Pursuant to our agreement with Genoscience Pharma, Genoscience Pharma will supply our clinical and commercial requirements for GNS561.
NTZ is already approved and commercialized in several jurisdictions in various indications and we therefore purchasepurchased our supply of NTZ for clinical purposes in the market through pharmaceutical wholesalers. A new formulation of NTZ to be used for our upcoming clinical trials is in development. We will use one supplier for the active substance NTZ and another CDMO for the manufacture of the therapeutic units.
VS-01 contains citric acid anhydrous as active ingredient for which a supply agreement covering clinical trials is in place with a third party GMP supplier. VS-01 is a kit containing three intermediate products, supplied by different GMP suppliers, each responsible for the production of VS-01 lipid blend, citric acid solution, liposomal aqueous suspension, xylitol alkaline solution and the kitting activities. The supply of VS-01 kits to clinical sites is managed by an external supplier in accordance with the GDP. The kits are to be reconstituted at the pharmacy hospital based on the instructions provided in the pharmacy manual and prior administration to patients.
With respect to our NIS4NIS4® technology, we have entered into two license agreements with Labcorp to further develop and manufacture a test using NIS4NIS4® technology for clinical research as well as to allow them to develop and commercialize an LDT named NASHNext® and powered by our NIS4NIS4® technology in routine clinical care in the USU.S. and Canada, respectively.Canada.
x. Intellectual Property
Our intellectual property is critical to our business, which we strive to protect by obtaining and maintaining patent protection in territories throughout the world for our drug and biomarker candidates, innovative methods and tools, production methods and other inventions that are important to our business. We also rely on trade secrets to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection.
Our commercial success depends in part upon obtaining and maintaining patent protection and trade secret protection of our current and future drug and biomarker candidates and the methods used to develop and manufacture them, as well as successfully defending these patents against third-party challenges. Our ability to stop third parties from making, using, selling, offering for sale in the United Sates or importing into the United States, our products depends on the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities. We cannot guarantee that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed by us in the future, nor can we guarantee that any of our existing patents or any patents that may be granted to us in the future will be commercially useful in protecting our drug and biomarker candidates, discovery programs and processes from competitors. Furthermore, our patents may be challenged, circumvented, or invalidated by third parties. Because patent applications in the United States and certain other jurisdictions are maintained in secrecy for 18 months or potentially even longer, and since publication of discoveries in the scientific or patent literature often lags behind actual discoveries, we cannot be certain of the priority of inventions covered by our pending patent applications. For this and more comprehensive risks related to our intellectual property, please see “Item 3D - "Risk Factors—Risks Relating to Our Intellectual Property”.”
We monitor our competitors and seek to challenge patent infringements when such infringements would negatively impact our business. We also seek to challenge validity of our competitors’ patents when we think that these patents do not fulfill patentability or validity requirements.
–Patents
As of March 31, 2022,2024, we own or have rights to 4349 issued U.S. patents, over 440500 issued foreign patents in force, and 2432 pending U.S. applications, and over 319324 pending foreign patent applications. Our patent portfolio contains 6073 different patent families, which are made up of over 760824 patents and patents applications. Twenty-twoTwenty-five of our patent families relate to our lead product candidate, elafibranor. Following the acquisition of Versantis AG, three patent families (including three U.S. patent applications and two issued U.S. patents) were integrated into our patent portfolio.
•Elafibranor
Our patent portfolio for elafibranor, a molecule synthesized by us, includes issued patents and pending patent applications directed to compositions of matter, manufacturing methods, and methods of use. As of March 31, 2022,2024, we own threeone U.S. patentspatent directed to the composition of matter of elafibranor, which are expected towill expire in September 2024 without any significant impact on the monopoly of elafibranor in the treatment of PBC, protected in several later patents, without taking patent term extensions into account. We also have counterpart patents in various countries and regions, including Australia, Brazil, Canada, China, Europe, Israel and Japan.
In addition, we own sixfour U.S. granted patents and ten U.S. patent applications (some of them derivable from PCT applications) directed to the treatment of cholestatic diseases, in particular PBC, which, if issued, are expected to expire infrom 2037 andto 2041, without taking patent term extensions into account. We also have counterpart pending patent applications in various countries or regions, including Australia, Canada, Europe, Israel, China, and Japan.
In addition, we own two U.S. patents directed to the method of preparing elafibranor, which are expected to expire in 2024 without any significant impact for the business of elafibranor, and 2031. We also have counterpart patents granted in various countries and regions, including Canada, China, Europe, and Israel.
In addition to these patents and pending applications, we are also pursuing additional patents directed to specific forms of elafibranor, and combinations with other pharmaceutical compounds.
Repurposing of moleculesWe also filed in 2023 three new priority patent applications on elafibranor for treating PBC, thus reinforcing and further protecting our lead product.
All the U.S. patent applications and patents are under license to IPSEN.
•ACLF franchise
We are pursuingfurther developing patent protection directed to our repositioning of nitazoxanide for treating ACLF/sepsis. As of March 31, 2024, two U.S. patent applications are pending. These patent applications, if granted, would be expected to expire in 2041 and 2042 (excluding any patent term extension). In addition, we maintain protection directed to nitazoxanide for treating cholestatic and fibrotic diseases. As of March 31, 2022,2024, six U.S. patents have been granted to us for the use of NTZ in the treatment of different fibrotic diseases and onediseases. Three U.S. patent application is pending. One U.S. patent haspatents have been granted for combination of NTZ with other therapeutic agents in the treatment of different fibrotic diseases and one other U.S. patent application is pending in this family.. These patents and patent applications, if granted, would be expected to expire infrom 2037 to 2038 (excluding any patent term extension).
We also filed in 2021 four priority2023 one international patent applicationsapplication for the use of nitazoxanide and some other proprietary molecules in the treatment of ACLF / sepsis.
Diagnostic Tools and Biomarkerssepsis, reinforcing our patent portfolio on this compound.
As of March 31, 2022,2024, we also own fivetwo U.S. patent applications directed to proprietary compounds for treating ACLF/sepsis. These patent applications, if granted, would be expected to expire in 2042 (excluding any patent term extension). We also filed in 2023 one international patent application for the use of other compounds in the treatment of ACLF/sepsis.
Acquisition of VERSANTIS has allowed us to reinforce our patent portfolio for the treatment of ACLF. As of March 31, 2023, we own three U.S. granted patents and one U.S. patent application (some of them derivable from PCT applications) directed to VS-01 for the treatment of ACLF. These patent applications, if granted, would be expected to expire from 2033 to 2036 (excluding any patent term extension).
We also filed in 2023 one international patent application for the use of VS-01 in the treatment of UCD (Urea Cycle Disorder) reinforcing our patent portfolio on this compound.
We also own one U.S. patent application directed to VS-02 in the treatment of UCD or Hepatic Encephalopathy.
•Diagnostic Franchise
As of March 31, 2024, we own nine U.S. patent applications and one U.S. granted patent directed to the diagnosis of NASH,MASH, in particular our NIS4NIS2+® and NIS4® diagnostic technology, using certain biomarkers. The U.S. applications, if issued, would be expected to expire between 2036 and 2041.
We also have filed several US patent applications covering some other NIS4 diagnostic tools and protecting some other research tools. We filed in 2023 an international patent application and four priority patent applications to reinforce our protection on this diagnostic technology.
As of March 31, 2024, we also own one U.S. patent applications and one U.S. granted patent directed to the diagnosis of MASH, using other biomarkers. The U.S. applications, if issued, would be expected to expire from 2038 to 2040. We also filed in 2023 two international patent applications on methods and devices for the diagnosis of advanced liver fibrosis or liver cirrhosis.
•In-licensed compounds
The licensing agreements signed in 2023 have reinforced our leadership position in the treatment of ACLF, with the intellectual property rights attached to the candidate drugs SRT-015 and CLM-022. The licensing agreements have also reinforced our intellectual property rights of the diagnostic franchise with TS-01 and in oncology with GNS561.
As of March 31, 2024, two U.S. patent applications and one U.S. granted patent are directed to SRT-015. The U.S. applications, if issued, would be expected to expire from 2038 and 2044. An international patent application was also filed in 2023 for CLM-022 in relation with the treatment of ACLF.
As of March 31, 2024, three U.S. patent applications and one U.S. granted patent are directed to TS-01, a diagnostic device for hyperammonemia. The U.S. applications, if issued, would be expected to expire from 2037 to 2038.
One U.S. patent application has been filed in relation with GNS561. This U.S. application, if issued, would be expected to expire in 2035. One international patent application and one priority patent application were also filed in 2023 to further develop the protection on this compound.
•Patent Term Extension (PTE)
In the United States, the term of a patent covering an FDA-approved drug may be eligible for a patent term extension, or PTE, under the Hatch-Waxman Act as compensation for the reduction of patent monopoly time during the FDA regulatory review process. This extended coverage period, PTE, can only be obtained provided we apply for and receive a marketing authorization for a product. The period of extension may be up to five years beyond the normal expiration of the patent, but cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval. Only one patent among those eligible for an extension may be extended. In Europe, Supplementary Protection Certificates, or SPCs, may also be available to patents, which would be available by application to the member states. However, there is no guarantee that the applicable authorities, including the FDA, will agree with our assessment of whether such extensions should be granted, and even if granted, the length of such extensions. We will use the procedures established to compensate regulatory delays via Patent Term Extension in the USU.S. and via Supplementary Protection Certificates in the EU as soon as Health authorities grant NDA in the USU.S. or MA in the EU for our products.
•Trademarks
Our candidate products are protected and will be sold around the world under trademarks that we consider to be of material importance.
Our trademarks will help to identify our products and services and will protect the sustainability of our growth.
It is our policy to file and protect our trademarks with a strategy adapted to each product or service, depending on the countries where the product will be commercialized or where the service will be proposed. Basically our trademarks are protected worldwide for our products and services.
We own close to 500more than 530 registered or filed trademarks worldwide.
The protection offered by trademark varies country by country. In most of the countries, trademark right may only be obtained through the filing and registration of a trademark application at the corresponding Patent and Trademark Office. Registrations are granted for a fixed term (usually ten years) and can be renewed indefinitely, except in certain countries where use of the trademark needs to be demonstrated at renewal time.
In most of the countries, protection of the trademark applies to the products and services designated in the registration certificate.
We monitor our trademarks and defend them against competing trademarks by filing oppositions, observations when appropriate. Similarly, we may enter into coexistence agreement when a third party owns a potentially conflicting or confusing trademark with some of our products or services.
It is also our policy to defend our trademarks against infringement, counterfeiting and/or unfair competition.
•Domain names
It is our policy to file domain names for communicating or giving information on our products or services to patients, prescribers or payers. We own today more than 210close to 170 domain names.
•Know-How and Trade Secrets
In addition to patent protection, we also rely on trade secret protection of our proprietary information that is not amenable to, or that we do not consider appropriate for, patent protection. However, trade secrets can be difficult to protect. Although we take steps to protect our proprietary information, including restricting access to our premises (we seek to preserve the integrity and confidentiality of our data, trade secrets and know-how by maintaining physical security of our premises and physical and electronic security of our information technology systems) and our confidential information, as well as entering into agreements with our employees, consultants, advisors, and potential collaborators, that prohibit the disclosure of confidential information, and require disclosure and assignment to us of ideas, developments, discoveries and inventions important to our business.
xi. Government Regulation
Our drug candidates must be approved by the FDA through the NDA process before they may be legally marketed in the United States and by the European Commission following a positive opinion provided by the EMA through the MAA process for a drug falling within the scope of the Centralized procedure before they may be legally marketed in the European Economic Area or by aone of the procedures administered by the national Competent Authority through other MAA processesAuthorities of EEA countries (National Procedure, Mutual Recognition or Decentralized procedure) before they may be legally marketed in the European Union. respective country/countries.
Our drug candidates will be subject to similar requirements in other countries prior to marketing in those countries. The process of obtaining regulatory approvals and the compliance with applicable federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
–United States Government Regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or the FDCA, and its implementing regulations. The process of obtaining regulatory approvals and compliance with appropriateapplicable federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S.legal requirements at any time during the drug development process, approval process or afterpost approval may subject an applicant and/or sponsor to a variety of administrative or judicial sanctions,other enforcement proceedings, including imposition of a clinical hold, refusal by the FDA to approve applications, withdrawal of an approval, import/export delays, issuance of warning letters and other types of enforcement letters,actions, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement of profits, or civil or criminal investigations and penalties broughtpenalties. These actions may be instituted or prosecuted by a variety of governmental entities, such as the FDA, and the U.S. Department of Justice, state attorneys general or other governmental entities.entities and, in certain cases, by private parties.
The clinical testing, manufacturing, labeling, storage, distribution, record keeping, advertising, promotion, import, export and marketing, among other things, of our drug candidates are governed by extensive regulation by governmental authorities in the United States and other countries. The FDA, under the FDCA, regulates pharmaceutical products in the United States. The steps required before a drug may be approved for marketing in the United States generally include:
•completion of preclinical laboratory tests, animal studies and formulation studies in compliance with the FDA’s good laboratory practice, or GLP, regulations;
•the submission to the FDA of an IND application for human clinical testing, which must become effective before human clinical trials commence;
•approval by anone or more independent institutional review board,boards, or IRB, representingIRBs, or ethics committees covering each clinical site before each clinical trial may be initiated;
•performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug for each indication and conducted in accordance with good clinical practices, or GCP;GCPs;
•preparation and submission to the FDA of an NDA;
•FDA acceptance, review and approval of the NDA, which might include an Advisory Committee review;
•satisfactory completion of an FDA inspection of the manufacturing facilities at which the drug, or components thereof, are made to assess compliance with current good manufacturing practices, or cGMPs;
•satisfactory completion of FDA audits of clinical trial sites to assure compliance with GCPs and the integrity of the clinical data; and
•agreement for compliance with any post-approval requirements, including Risk Evaluation and Mitigation Strategies, or REMS, and post-approval studies required by the FDA.
The testing and approval process requires substantial time, effort and financial resources, and the receipt and timing of any approval is uncertain. The FDA may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
Preclinical and Human Clinical Trials in Support of an NDA
Preclinical studies include laboratory evaluations of the drug candidate, as well as in vitro and animal studies to assess the potential safety and efficacy of the drug candidate. The conduct of preclinical studies is subject to federal regulations and requirements including GLP regulations. The results of the preclinical studies, together with manufacturing information and analytical data, among other things, are submitted to the FDA as part of the IND, which must become effective before human clinical trials may commence. The IND will become effective automatically 30 days after receipt by the FDA, unless the FDA raises concerns or questions about the conduct of the trials as outlined in the IND prior to that time and places a clinical hold on the IND. In this case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can proceed. The FDA may nevertheless initiate a clinical hold after the 30 days if, for example, significant public health risks arise.
Clinical trials involve the administration of the drug candidate to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent in writing for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the
IND. Each clinical trial must be reviewed and approved by an IRB at each ofone or more IRBs or Ethics Committees covering the sites at which the trial will be conducted. The IRB or Ethics Committee will consider, among other things, ethical factors, the safety of human subjects and the possible liability of the institution.
Clinical trials are typically conducted in three sequential phases prior to approval, but the phases may overlap or be combined. These phases generally include the following:
•Phase 1. Phase 1 clinical trials represent the initial introduction of a drug candidate into human subjects, frequently healthy volunteers. In Phase 1, the drug candidate is usually tested for safety, including adverse effects, dosage tolerance, absorption, distribution, metabolism, excretion and pharmacodynamics.
•Phase 2. Phase 2 clinical trials usually involve studies in a limited patient population to (1) evaluate the efficacy of the drug candidate for specific indications, (2) determine dosage tolerance and optimal dosage and (3) identify possible adverse effects and safety risks.
•Phase 3. If a drug candidate is found to be potentially effective and to have an acceptable safety profile in Phase 2 clinical trials, the clinical trial program will be expanded to Phase 3 clinical trials to further demonstrate clinical efficacy, optimal dosage and safety within an expanded patient population at geographically dispersed clinical trial sites.
Post-approval trials, sometimes referred to as Phase 4 clinical trials, may be conducted after approval to gain additional experience from the treatment of patients in the intended therapeutic indication and to document a clinical benefit in the case of drugs approved under accelerated approval regulations, or when otherwise requested or required by the FDA in the form of post-market requirements or commitments. Failure to promptly conduct any required Phase 4 clinical trials could result in enforcement action or withdrawal of approval. Companies that conduct certain clinical trials are also are required to register them and post the results of completed clinical trials on a government-sponsored database, such as ClinicalTrials.gov in the United States, within certain timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
Submission and Review of an NDA
The results of preclinical studies and clinical trials, together with detailed information on the drug’s manufacture, composition, quality, controls and proposed labeling, among other things, are submitted to the FDA in the form of an NDA, requesting approval to market the drug for one or more indications. The application must be accompanied by a significant user fee payment, which typically increases annually, although waivers may be granted in limited cases. The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. The FDA has substantial discretion in the approval process and may refuse to file or approve any application or decide that the data is insufficient for approval and require additional preclinical, clinical or other studies.
Once an NDA has been accepted for filing, the FDA sets a user fee goal date that informs the applicant of the specific date by which the FDA intends to complete its review. This goal date is typically 10 months from the date that the FDA accepts the filing. The review process can be extended by FDA requests for additional information or clarification. The FDA reviews NDAs to determine, among other things, whether the proposed drug is safe and effective for its intended use, and whether the drug is being manufactured in accordance with cGMPs to assure and preserve the drug’s identity, strength, quality and purity. Before approving an NDA, the FDA typically will inspect the facilities at which the drug is manufactured and will not approve the drug unless the manufacturing facilities comply with cGMPs. Additionally, the FDA will typically inspect one or more clinical trial sites for compliance with GCP and integrity of the data supporting safety and efficacy.
During the approval process, the FDA also will determine whether a REMS is necessary to assure the safe use of the drug. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring and the use of patient registries. If the FDA concludes a REMS is needed, the sponsor of the application must submit a proposed REMS, and the FDA will not approve the application without an approved REMS, if required. A REMS can substantially increase the costs of obtaining approval. The FDA may also convene an advisory committee of external experts to provide input on certain review issues relating to risk, benefit and interpretation of clinical trial data. The FDA may delay approval of an NDA if applicable regulatory criteria are not satisfied and/or the FDA requires additional testing or information.
On the basis of the FDA’s evaluation of the NDA and accompanying information, including the results of the inspection of the manufacturing facilities and clinical trial sites, the FDA will issue either an approval of the NDA or a Complete Response Letter, detailing the deficiencies in the submission and the additional testing or information required for reconsideration of the application. Even with submission of this additional information, the FDA may ultimately decide that the application does not satisfy the regulatory criteria for approval.
If the FDA approves a new drug, it may limit the approved indications for use of the drug. It may also require that contraindications, warnings or precautions be included in the drug labeling, such as a special warning, known as a boxed warning, to highlight a particular safety risk. In addition, the FDA may call for post-approval studies, including Phase 4 clinical trials, to further assess the drug’s safety after approval. The agency may also require testing and surveillance programs to monitor the drug after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms, including REMS, to help ensure that the benefits of the drug outweigh the potential risks. The FDA may prevent or limit further marketing of a drug based on the results of post-market studies or surveillance programs.
After approval, many types of changes to the approved drug, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Fast Track, Breakthrough Therapy and BreakthroughPriority Review Designations
The FDA is authorized to designate certain drugs for expedited programs intended to facilitate and expedite development and review if they are intended to address an unmet medical need in the treatment of a serious or life-threatening disease or condition. These programs are fast track designation, breakthrough therapy designation and priority review designation.
TheFast track designation may be granted by the FDA may designateto a drug for fast track designation if it is intended, whether alone or in combination with one or more other drugs, for the treatment of a serious or life-threatening disease or condition, and it demonstratesnonclinical or clinical date demonstrate the potential to address unmet medical needsneed for such a disease or condition. For fast track designated drugs, sponsors may have a higher number of interactions with the FDA. In addition, the FDA may review sections of the NDA for a fast track designated drug on a rolling basis before the complete application is submitted. Fast track designation may be rescinded if the qualifying criteria are no longer met.
TheBreakthrough therapy designation may be granted by the FDA may designateto a drug for breakthrough designation if the drug is intended to treat a serious or life-threatening disease or condition and that preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on a clinically significant endpoint(s) over available therapies. The featurefeatures of this program allowsprovide the same advantages of the fast track designation, but also intensive FDA guidance to promote efficient development and FDA organizational commitment. Breakthrough therapy designation may be rescinded if the qualifying criteria are no longer met.
Priority review designation may be granted by the FDA to an application (original or efficacy supplement) for a drug that treats a serious condition and, if approved, would provide a significant improvement in safety or effectiveness, among other qualifying criteria. Priority review provides a shorter clock for review of marketing application (i.e. six months compared with the 10-month standard review) following acceptance of the NDA.
Accelerated Approval Pathway
The FDA may grant accelerated approval, under Subpart H of 21 CFR Part 314, to a drug for a serious or life-threatening condition that provides meaningful therapeutic advantage to patients over existing treatmentsavailable therapies based upon a determination that the drug hasdemonstrates an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit. The FDA may also grant accelerated approval for such a disease or condition when the drug hasdemonstrates an effect on an intermediate clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality, or IMM, and that is reasonably likely to predict an effect on IMM or other clinical benefit, taking into account the severity, rarity or prevalence of the condition and the availability or lack of alternative treatments. Drugs granted accelerated approval must meet the same statutory standards for safety and effectiveness as those granted traditional approval.
For the purposes of accelerated approval, a surrogate endpoint is a marker, such as a laboratory measurement, radiographic image, physical sign or other measure that is thought to predict clinical benefit, but is not itself a measure of clinical benefit. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. An intermediate clinical endpoint is a measurement of a therapeutic effect that is considered reasonably likely to predict the clinical benefit of a drug, such as andrug's effect on IMM.IMM or other clinical benefit. The FDA has limited experience with accelerated approvals based on intermediate clinical endpoints, but has indicatedstated its belief that such endpoints generally may support accelerated approval where the therapeutic effect measured by the endpoint is not itself a clinical benefit and basis for traditional approval, if there is a basis for concluding that the therapeutic effect is reasonably likely to predict the ultimate clinical benefit of a drug.
The accelerated approval pathway is most oftenhas been primarily used in settings in whichwhere the course of a disease is long and an extended period of time is required to measure the intended clinical benefit of a drug, even if the effect on the surrogate or intermediate clinical endpoint occurs rapidly. Thus, accelerated approval has been used extensively in the development and approval of drugs for treatment of a variety of cancers in which the goal of therapy is generally to improve survival or decrease morbidity and the duration of the typical disease course requires lengthy and sometimes large trials to demonstrate a clinical or survival benefit. The benefit of accelerated approval derives from the potential to receive approval based on surrogate endpoints sooner than possible for trials with clinical or survival endpoints, rather than deriving from any explicit shortening of the FDA approval timeline, as is the case with priority review.
The accelerated approval pathway is usually contingent on a sponsor’s agreement to conduct, in a diligent manner, confirmatory studies to verify and describe the drug’s clinical benefit. Asbenefit, the design of which must be agreed upon with the FDA prior to approval. No later than the date of the accelerated approval, the FDA must specify the conditions for a result, apost-approval trial or trials required to be conducted with respect to the drug, candidate approved on this basis is subject to rigorous post-marketing compliance requirements,which may include enrollment targets, the trial protocol and milestones, including the completiontarget date of Phase 4 ortrial completion. Sponsors must submit progress reports on required post-approval clinical trials to confirm the effect on the clinical endpoint.every six months. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, would allow the FDA to initiate expedited proceedings to withdraw approval of the drug. All promotional materials for drug candidates approved under accelerated regulations are subject to prior review by the FDA.
Post-Approval Requirements
In additionaladdition to the post-approval requirements specific to an accelerated approval pathway, there are other post-approval requirements whateverapply regardless of the registration pathway.
Approved drugs that are manufactured or distributed in the United States pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, drug sampling and distribution, advertising and promotion and reporting of adverse experiences with the drug. After approval, most changes to the approved drug, such as adding new indications or other labeling claims and some manufacturing and supplier changes are subject to prior FDA review and approval. There also are continuing, annual program user fee requirements for marketed drugs, as well as new application fees for certain supplemental applications.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-marketing testing, including Phase 4 clinical trials, and surveillance programs to further assess and monitor the drug’s safety and effectiveness after commercialization. The FDA may also require a REMS, which could involve requirements for, among other things, medication guides, special trainings for prescribers and dispensers, patient registries, and elements to assure safe use.
In addition, entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. The FDA has promulgated specific requirements for drug cGMPs. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP requirements and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, theThe FDA may issue enforcement letters or withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the drug reaches the market. Corrective action could delay drug distribution and require significant time and financial expenditures. Later discovery of previously unknown problems with a drug, including adverse events or AEs of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
•restrictions on the marketing or manufacturing of the drug, suspension of the approval, complete withdrawal of the drug from the market or product recalls;
•fines, warning letters or holds on post-approval clinical trials;
•refusal of the FDA to approve applications or supplements to approved applications, or suspension or revocation of drug approvals;
•drug seizure or detention, or refusal to permit the import or export of drugs; or
•injunctions or the imposition of civil or criminal penalties.
The FDA strictly regulates marketing, labeling, advertising and promotion of drugs that are placed on the market. Drugs may be promoted only for the approved indications and in accordancea manner consistent with the provisions of thefinal approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including investigation by federal and state authorities. However, physicians may, in their independent medical judgment, prescribe legally available products for off-label uses. The FDA does not regulate the practice of medicine and the behavior of physicians in their choice of treatments but the FDA does restrict manufacturer’s promotional communications on the subject of off-label use of their products.
Section 505(b)(2) NDAs
As an alternative path to FDA approval for modifications to formulations or uses of drugs previously approved by the FDA, an applicant may submit an NDA as described under Section 505(b)(2) of the FDCA. SectionA 505(b)(2) was enacted as partapplication is submitted under section 505(b)(1) of the Hatch-
Waxman Amendments.FDCA and approved under section 505(c). The 505(b)(2) pathway allows for flexibility in the characteristics of the proposed product without having to conduct studies on what is already known about the product. A Section 505(b)(2) NDA is an application that contains full reports of investigations of safety and effectiveness, but where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by orreference. A 505(b)(2) application may rely on, for whom the investigations were conducted. This type of application permits reliance for such approvals onexample, published literature or on an FDAFDA's finding of safety, effectiveness or both for an approved drug product. As such, under Section 505(b)(2), the FDA may rely, for approval of an NDA, on data not developed by the applicant. The FDA may also require companies505(b)(2) applicants to perform additional studies or measurements, including clinical trials, to support the changechanges from the approved branded reference drug.
US Pediatric Studies and Exclusivity
The Pediatric Research Equity Act of 2003 (“PREA”) requires all applications (or supplements thereto) submitted under section 505 of the FDCA (i.e., NDA, 505(b)(2), or supplement to the same) for a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration to contain a pediatric assessment unless the applicant has obtained a waiver or deferral. It authorizes the FDA to require holders of approved NDAs for marketed drugs to conduct pediatric studies under certain circumstances. The required clinical assessment must evaluate the safety and effectiveness of the product for the claimed indications in all relevant pediatric subpopulations and support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The sponsor or FDA may request a deferral of required pediatric clinical trials for some or all of the pediatric subpopulations. A deferral may be granted for several reasons, including a finding that the drug is ready for approval for use in adults before pediatric clinical trials are complete or that additional safety or effectiveness data needs to be collected before the pediatric clinical trials begin. The FDA must send a non-compliance letter to any sponsor that fails to submit the required assessment, keep a deferral current or fails to submit a request for approval of a pediatric formulation. The PREA pediatric studies requirement do not apply to certain drugs with orphan drug designation.
In addition, the Best Pharmaceuticals for Children Act (“BPCA”), provides NDA holders a six-month extension of any eligible exclusivity—patent or nonpatent—for a drug, if a sponsor conducts clinical trials in children in response to a written request from the FDA. The issuance of a written request does not require the sponsor to undertake the described clinical trials. If the sponsor does undertake the clinical trials and submits pediatric data that fairly respond to the written request, the FDA may then approvegrant six-months exclusivity. The data do not need to show the new product candidate forto be effective in the new indication sought by the 505(b)(2) applicant.pediatric population studied. The six-month exclusivity attaches to all existing, eligible exclusivity and patents.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug intended to treat a rare disease or condition, which is a disease or condition that affects fewer than 200,000 individuals in the United States or, if it affects more than 200,000 individuals in the United States, there is no reasonable expectation that the cost of developing and making a drug product available in the United States for this type of disease or condition will be recovered from sales of the product. Orphan drug designation must be requested before submitting an NDA.a marketing application for the drug for the orphan use. After the FDA grants orphan designation, the FDA publicly discloses the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA.use. The designation of such drug entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has orphan drug designation subsequently receives the first FDA approval of the drug for the disease or condition for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug or biological product for the same use or indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity or inability to manufacture the product in sufficient quantities. The designation of such drug also entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers. Competitors, however, may receive approval of different products for the indication for which the orphan product has exclusivity or obtain approval for the same product but for a different indication for which the orphan product has exclusivity.
Orphan exclusivity also could block the approval of one of our products for seven years if a competitor obtains approval of the same drug as defined by the FDA for an indication we intend to pursue or are pursuing or if our product candidate is determined to be contained within the competitor’s product for the same indication or disease. If an orphan designated product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan exclusivity.
–FDA Regulation of In Vitro Diagnostics
Under the FDCA, in vitro diagnostics are regulated as medical devices. In the United States, the FDCA and its implementing regulations, and other federal and state statutes and regulations govern, among other things, medical device design and development, preclinical and clinical testing, premarket clearance or approval, registration and listing, manufacturing, labeling, storage, advertising and promotion, sales and distribution, export and import, and post-market surveillance. Unless an exemption applies, diagnostic tests require marketing clearance or approval from the FDA prior to commercial distribution. The two primary types of FDA marketing authorization applicable to a medical device are premarket notification, also called 510(k) clearance, and premarket approval, or PMA; however, other devices may be commercialized after the FDA grants a de novo request.
Device Classification
Under the FDCA, medical devices are classified into one of three classes—Class I, Class II or Class III—depending on the degree of risk associated with each medical device and the extent of control needed to provide reasonable assurances with respect to safety and effectiveness.
Class I devices are those for which safety and effectiveness can be reasonably assured by adherence to a set of regulations, referred to as General Controls, which often require compliance with the applicable portions of the FDA’s Quality System Regulation, or QSR, facility registration and product listing, reporting of adverse events and malfunctions, and appropriate, truthful and non-misleading labeling and promotional materials. Most Class I products are exempt from the premarket notification requirements.
Class II devices are those that are subject to the General Controls, as well as Special Controls, which can include performance standards, guidelines and post market surveillance. Most Class II devices are subject to premarket review and clearance by the FDA. Premarket review and clearance by the FDA for Class II devices is accomplished through the 510(k) premarket notification process. Under the 510(k) process, the manufacturer must submit to the FDA a premarket notification, demonstrating that the device is “substantially equivalent,” as defined in the statute, to either:
•a device that was legally marketed prior to May 28, 1976, the date upon which the Medical Device Amendments of 1976 were enacted, or
•another commercially available, similar device that was cleared through the 510(k) process.
To be “substantially equivalent,” the proposed device must have the same intended use as the predicate device, and either have the same technological characteristics as the predicate device or have different technological characteristics and not raise different questions of safety or effectiveness than the predicate device. Clinical data are sometimes required to support substantial equivalence.
After a 510(k) notice is submitted, the FDA determines whether to accept it for substantive review. If it lacks necessary information for substantive review, the FDA will refuse to accept the 510(k) notification.. If it is accepted for filing, the FDA begins a substantive review. If the FDA agrees that the device is substantially equivalent, it will grant clearance to commercially market the device.
The PMA Process
If the FDA determines that the device is not “substantially equivalent” to a predicate device, or if the device is classified into Class III by operation of law, the device sponsor must then fulfill the much more rigorous premarketing requirements of the PMA process, or seek classification of the device through the de novo process by submitting a de novo request. A manufacturer can also submit a direct de novo request if the manufacturer is unable to identify an appropriate predicate device and the new device or new use of the device presents a moderate or low risk. In response to a de novo request, FDA may classify the device into class I or II. When FDA grants a de novo request, the device is granted marketing authorization and further can serve as a predicate for future devices of that type, including for 510(k)s.
Class III devices include devices deemed by the FDA to pose the greatest risk such as life-supporting or life-sustaining devices, or implantable devices, in addition to those deemed not substantially equivalent following the 510(k) process. The safety and effectiveness of Class III devices cannot be reasonably assured solely by the General Controls and Special Controls described above. Therefore, these devices are subject to the PMA application process, which is generally more costly and time consuming than the 510(k) process. Through the PMA application process, the applicant must submit data and information demonstratingto demonstrate reasonable assurance of the safety and effectiveness of the device for its intended use to the FDA’s satisfaction. Accordingly, a PMA application typically includes, but is not limited to, extensive technical information regarding device design and development, preclinical and clinical study data, manufacturing information, labeling and financial disclosure information for the clinical investigators in device studies. The PMA application must provide valid scientific evidence that demonstrates to the FDA’s satisfaction reasonable assurance of the safety and effectiveness of the device for its intended use. Overall, the FDA review of a PMA application generally takes between one and three years, but may take significantly longer.
Laboratory-developedIf the FDA determines that a device is not “substantially equivalent” to a predicate device pursuant to a 510(k) submission, or if the device is classified into Class III by operation of law, the device sponsor must then fulfill the much more rigorous premarketing requirements of the PMA process, described above, or seek classification of the device through the de novo process by submitting a de novo request. This process allows a manufacturer whose novel device is automatically classified into Class III to request down-classification of its medical device into Class I or Class II on the basis that the device presents low or moderate risk. If the manufacturer seeks reclassification into Class II, the manufacturer must include a draft proposal for special controls that are necessary to provide a reasonable assurance of the safety and effectiveness of the medical device. The FDA may reject the reclassification petition if it identifies a legally marketed predicate device that would be appropriate for a 510(k) or that general controls would be inadequate to control the risks and special controls cannot be developed. In response to a de novo request, FDA may classify the device into class I or II. When FDA grants a de novo request, the device is granted marketing authorization and further can serve as a predicate for future devices of that type, including for 510(k)s.
Laboratory Developed Tests (LDTs)
LDTs haveThe FDA has generally been considered LDTs to be testsin vitro diagnostic products that are intended for clinical use and that are designed, manufactured and used within a single laboratory.clinical laboratory certified under the Clinical Laboratory Improvement Amendments of 1988 (“CLIA”) and meeting the regulator requirements under CLIA to perform high complexity testing. The FDA takes the position that it has the authority to regulate such tests as devices under the FDCA. TheHistorically, the FDA has historically exercised enforcement discretion, meaning FDA has not enforced premarket review or other applicable FDA requirements with respect to most LDTs. In addition, the New York State Department of Health, or NYSDOH, separately approves certain LDTs offered to New York State patients. The laboratory partner to whom we license our technology will be responsible for obtaining the requisite approvals for our LDT in New York, and maintaining CLIA-certification and state clinical laboratory licenses, where applicable.
On October 3, 2014,In September 2023, the FDA issued two draft guidance documents regarding oversightproposed a rule to amend its regulations to make explicit that in vitro diagnostic products are devices under the FDCA, including when the manufacturer of LDTs. These draft guidance documents proposed more active oversight over LDTs. The draft guidance documents have been the subject of considerable controversy, and in November 2016,vitro diagnostic is a laboratory. At the same time, FDA announced that it would notits coordinate intent to phase out its general enforcement discretion approach over a period of years should the proposed rule be finalizing the 2014 draft guidance documents. On January 13, 2017, thefinalized and at which time FDA issuedwill publish a discussion paper which laid out elements of a possible revised future LDT regulatory framework, but did not establish any regulatory requirements. The FDA’s efforts to regulate LDTs have prompted the drafting of legislation governing diagnostic products and services, including LDTs. Congress or FDA may still act to provide further direction on the regulation of LDTs.final phaseout policy.
–European Union Regulation for Drug Development and Registration
Privacy and Security
We may be subject to diverse laws and regulations relating to data privacy and security as a result of our employee data or other personal information that we may collect. In addition, if we do collect personal data as part of any clinical trials or other testing, we would be subject to regulatory obligations. This includes, (i) in the European Union, or EU, and the European Economic Area, or EEA, the EU General Data Protection Regulation, or EU GDPR, (ii) in the United Kingdom, or UK, the UK GDPR. EU member states are also able to legislate separately on health and genetic information, and we must comply with these local laws where we operate. For example, in France, the conduct of clinical trials is subject to compliance with reference methodologies (such as MR-001) imposing stringent rules to process health-related information.
Preclinical and Clinical Development
In the European Economic Area or EEA (which is comprised of the 27 Member States of the European Union plus Norway, Iceland and Liechtenstein), our drug candidates are also subject to extensive regulatory requirements. As in the United States, medicinal products can only be marketed if a marketing authorization from the competent regulatory agenciesauthorities has been obtained.
Similar to the United States, the various phases of preclinical and clinical research in the European UnionEEA are subject to significant regulatory controls. Although
In the EU Clinical Trials Directive 2001/20/EC has sought to harmonize the European UnionEEA, clinical trials regulatory framework, setting out common rules forare governed by the control and authorization of clinical trials in the European Union, the EU Member States have transposed and applied the provisions of the Directive differently. This has led to significant variations in the Member State regimes. To improve the current system,Clinical Trials Regulation (EU) No 536/2014, on clinical trials on medicinal products for human use,or CTR, which repealed Directive 2001/20/EC, was adopted on April 16, 2014 and published in the European Official Journal on May 27, 2014. The Regulation entered into application on January 31, 2022.2022 repealing and replacing the former Clinical Trials Directive 2001/20, or CTD, and related national implementing legislation of EEA countries.
The Regulation aims at harmonizingCTR is intended to harmonize and streamlining thestreamline clinical trials authorization process, simplifying adverse eventtrial authorizations, simplify adverse-event reporting procedures, improvingimprove the supervision of clinical trials and increasing their transparency. The main characteristics ofSpecifically, the Regulation, include:which is directly applicable in all EEA countries, introduces a streamlined application procedure viathrough a single entrysingle-entry point, the “EU portal”;"EU portal", the Clinical Trials Information System, or CTIS; a single set of documents to be prepared and submitted for the applicationapplication; as well as simplified reporting procedures for clinical trial sponsors; and asponsors. A harmonized procedure for the assessment of applications for clinical trials whichhas been introduced and is divided ininto two parts. Part I assessment is assessed first by a single “reference” Member State whose conclusions are then assessedled by the competent authorities of a reporting Member State selected by the trial sponsor and relates to clinical trial aspects that are considered to be scientifically harmonized across EEA countries. This assessment is then submitted to the competent authorities of all EUconcerned Member States in which an applicationthe trial is to be conducted for authorization of a clinical trial has been submitted, the “concerned” Member States.their review. Part II is assessed separately by the competent authorities and Ethics Committees in each concerned Member State. Strict deadlines have been established forIndividual EEA countries retain the assessmentpower to authorize the conduct of clinical trial applications. trials on their territory.
The role of the relevant ethics committees in the assessment procedureextent to which on-going clinical trials will continue to be governed by the national lawCTR will depend on the duration of the concerned Member State. However, overall related timelinesindividual clinical trial. Sponsors could choose to submit a clinical trial application under either the CTD or the CTR until January 31, 2023. For clinical trials in relation to which application for approval was made on the basis of the CTD before January 31, 2022, the CTD will continue to apply on a transitional basis for three years. If authorized, those clinical trials will be definedgoverned by the Clinical Trials Regulation.CTD until January 31, 2025. By that date, all ongoing trials will become subject to the provisions of the CTR. The CTR will apply to clinical trials from an earlier date if the clinical trial has already transitioned to the CTR framework.
European Union Drug Review and Approval
In the EEA, medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA.
To obtain an MA for a product in the EEA, an applicant must submit a Marketing AuthorizationsAuthorization Application, or MAA, either under a centralized procedure administered by the EMA or one of the procedures administered by the Competent Authorities of EEA countries (decentralized procedure, national procedure or mutual recognition procedure). An MA may be granted either centrally (EU MA) or nationally (National MA).only to an applicant established in the EEA.
The EUcentralized procedure provides for the grant of a single MA is issued centrally by the European Commission through the Centralized Procedure, based on the opinion of the CHMP of the EMA andthat is valid throughoutfor all EEA countries. Pursuant to Regulation (EC) No 726/2004, the entire territory of the EEA. The Centralized Procedurecentralized procedure is mandatorycompulsory for certain types ofspecific products, such as biotechnologyincluding for (i) medicinal products derived from biotechnological processes, (ii) products designated as orphan medicinal products, and(iii) advanced therapy medicinal products, containingor ATMPs, and (iv) products with a new active substance indicated for the treatment of HIV/AIDS, cancer, neurodegenerative disorders,diseases, diabetes, auto-immune and other immune dysfunctions and viral diseases. The Centralized Procedure is optional forFor products containingwith a new active substance not yet authorized inindicated for the EEA,treatment of other diseases and products that are highly innovative or for products that constitutewhich a significant therapeutic, scientific or technical innovation or which arecentralized process is in the interest of patients, authorization through the centralized procedure is optional on related approval. Under the centralized procedure, the EMA’s Committee for Medicinal Products for Human Use, or CHMP, conducts the initial assessment of a product. The CHMP is also responsible for several post-authorization and maintenance activities, such as the assessment of modifications or extensions to an existing MA.
Under the centralized procedure in the EU, the maximum timeframe for the evaluation of an MAA is 210 days, excluding clock stops when additional information or written or oral explanation is to be provided by the applicant in response to questions of the CHMP. Accelerated assessment may be granted by the CHMP in exceptional cases, when a medicinal product targeting an unmet medical need is expected to be of major interest from the point of view of public health and, in particular, from the European Union.viewpoint of therapeutic innovation. If the CHMP accepts a request for accelerated assessment, the time limit of 210 days will be reduced to 150 days (excluding clock stops). The CHMP can, however, revert to the standard time limit for the centralized procedure if it considers that it is no longer appropriate to conduct an accelerated assessment.
National MAs are issued nationally
Unlike the centralized authorization procedure, the decentralized MA procedure requires a separate application to, and leads to separate approval by, the competent authorities of each EEA country in which the product is to be marketed. This application is identical to the application that would be submitted to the EMA for authorization through the centralized procedure. The reference Member State prepares a draft assessment and drafts of the related materials within 120 days after receipt of a valid application. The resulting assessment report is submitted to the concerned Member States who, within 90 days of receipt, must decide whether to approve the assessment report and related materials. If a concerned Member State cannot approve the assessment report and related materials due to concerns relating to a potential serious risk to public health, disputed elements may be referred to the Heads of Medicines Agencies’ Coordination Group for Mutual Recognition and Decentralised Procedures – Human, or CMDh, for review. The subsequent decision of the European Commission is binding on all EEA countries.
The mutual recognition procedure allows companies that have a medicinal product already authorized in one EEA country to apply for this authorization to be recognized by the competent authorities in other EEA countries. Like the decentralized procedure, the mutual recognition procedure is based on the acceptance by the competent authorities of EEA countries of the MA of a medicinal product by the competent authorities of other EEA countries. The holder of a national MA may submit an application to the competent authority of an EEA country requesting that this authority recognize the MA delivered by the competent authority of another EEA country.
An MA has, in principle, an initial validity of five years. The MA may be renewed after five years on the basis of a re-evaluation of the risk-benefit balance by the EMA or by the competent authority of the EEA and only cover their respective territory. National MAs are available for products not falling withincountry in which the mandatory scope oforiginal MA was granted. To support the Centralized Procedure. We do not foresee that any of our current drug candidates will be suitable for a Nationalapplication, the MA as they fall within the mandatory criteria for the Centralized Procedure. Therefore, our drug candidates should be approved through EU MAs.
Under the above-described procedures, before granting the MA,holder must provide the EMA or the competent authoritiesauthority with a consolidated version of the Member States ofeCTD (Common Technical Document) providing up-to-date data concerning the EEA make an assessment of the risk-benefit balancequality, safety and efficacy of the product, including all variations introduced since the MA was granted, at least nine months before the MA ceases to be valid. The European Commission or the competent authorities of EEA countries may decide on justified grounds relating to pharmacovigilance, to proceed with one further five year renewal period for the MA. Once subsequently definitively renewed, the MA shall be valid for an unlimited period. Any authorization which is not followed by the actual placing of the medicinal product on the basisEEA market (for a centralized MA) or on the market of scientific criteria concerning its quality,the authorizing EEA country within three years after authorization ceases to be valid (the so-called sunset clause).
Innovative products that target an unmet medical need and are expected to be of major public health interest may be eligible for a number of expedited development and review programs, such as the Priority Medicines, or PRIME, scheme, which provides incentives similar to the breakthrough therapy designation in the U.S. PRIME is a voluntary scheme aimed at enhancing the EMA’s support for the development of medicinal products that target unmet medical needs. Eligible products must target conditions for which there is an unmet medical need (there is no satisfactory method of diagnosis, prevention or treatment in the EU or, if there is, the new medicinal product will bring a major therapeutic advantage) and they must demonstrate the potential to address the unmet medical need by introducing new methods of therapy or improving existing ones. Benefits accrue to sponsors of product candidates with PRIME designation, including but not limited to, early and proactive regulatory dialogue with the EMA, frequent discussions on clinical trial designs and other development program elements, and potentially accelerated MAA assessment once a dossier has been submitted.
In the EEA, a “conditional” MA may be granted in cases where all the required safety and efficacy.efficacy data are not yet available. The EMAEuropean Commission may givegrant a positive opinion for conditional marketing authorization based on interim clinical dataMA for a medicinal product for human use if (1)it is demonstrated that all of the risk-benefitfollowing criteria are met: (i) the benefit-risk balance of the medicinal product is positive, (2)positive; (ii) it is likely that the applicant will be in a positionable to provide comprehensive data post-authorization; (iii) the required comprehensive clinical trial data, (3)medicinal product fulfills an unmet medical needs will be fulfilledneed; and (4)(iv) the benefit to public health of the immediate availability on the marketto patients of the medicinal product outweighsis greater than the risk inherent in the fact that additional data are still required. Specific obligations, including with respectThe conditional MA is subject to conditions to be fulfilled for generating the completion of ongoingmissing data or new studies, and with respect to the collection of pharmacovigilance data, may be specified in the conditional marketing authorization. Conditional marketing authorizations areensuring increased safety measures. It is valid for one year and maymust be renewed annually until all related conditions have been fulfilled. Once any pending studies are provided, the conditional MA can be converted into a traditional MA. However, if the conditions are not fulfilled within the timeframe set by the EMA and approved by the European Commission, the MA will cease to be renewed.
An MA may also be granted “under exceptional circumstances” where the applicant can show that it is unable to provide comprehensive data on efficacy and safety under normal conditions of use even after the product has been authorized and subject to specific procedures being introduced. These circumstances may arise in particular when the intended indications are very rare and, in the state of scientific knowledge at that time, it is not possible to provide comprehensive information, or when generating data may be contrary to generally accepted ethical principles. Like a conditional MA, an MA granted in exceptional circumstances is reserved to medicinal products intended to be authorized for treatment of rare diseases or unmet medical needs for which the applicant does not hold a complete data set that is required for the grant of a standard MA. However, unlike the conditional MA, an applicant for authorization in exceptional circumstances is not subsequently required to provide the missing data. Although the MA “under exceptional circumstances” is granted definitively, the risk-benefit balance of the medicinal product is reviewed annually, and the MA will be withdrawn if the risk-benefit balance remains positive,ratio is no longer favorable.
EU Pediatric Development
In the EEA, Regulation (EC) No 1901/2006 provides that all marketing authorization applications for new medicinal products must include the results of trials conducted in the pediatric population, in compliance with a pediatric investigation plan, or PIP, agreed with the EMA’s Pediatric Committee, or PDCO. The PIP sets out the timing and after an assessmentmeasures proposed to generate data to support a pediatric indication of the needmedicinal product for additionalwhich marketing authorization is being sought. The PDCO may grant a deferral of the obligation to implement some or modified conditions.
In additionall of the measures provided in the PIP until there are sufficient data to an MA, various other requirements applydemonstrate the efficacy and safety of the product in adults. Furthermore, the obligation to provide pediatric clinical trial data can be waived by the PDCO when these data are not needed or appropriate because the product is likely to be ineffective or unsafe in children, the disease or condition for which the product is intended occurs only in adult populations, or when the product does not represent a significant therapeutic benefit over existing treatments for pediatric patients. Once the marketing authorization is obtained in all EEA countries and study results are included in the product information, even when negative, the product is eligible for a six-month extension to the manufacturing and placing onSupplementary Protection Certificate, or SPC, if any is in effect at the EU markettime of medicinal products. Manufactureauthorization or, in the case of orphan medicinal products, a two-year extension of orphan market exclusivity.
Orphan Drugs in the EU requires a manufacturing authorization, and import of medicinal products into the EU requires a manufacturing authorization allowing for import. The manufacturing authorization holder must comply with various requirements set out in the applicable EU laws, regulations and guidance. These requirements include compliance with EU GMP standards when manufacturing medicinal products and APIs, including the manufacture of APIs outside of the EU with the intention to import the APIs into the EU. Similarly, the distribution of medicinal products within the EU is subject to compliance with the applicable EU laws, regulations and guidelines, including the requirement to hold appropriate authorizations for distribution granted by the competent authorities of the EU Member States. MA holders and/or manufacturing and import authorization, or MIA holders and/or distribution authorization holders may be subject to civil, criminal or administrative sanctions, including suspension of manufacturing authorization, in case of non-compliance with the EU or EU Member States’ requirements applicable to the manufacturing of medicinal products.
Orphan Drugs
In the EEA, Regulation (EC) No 141/2000, as amended,implemented by Regulation (EC) No. 847/2000, provides that a drug will be designated as an orphan drug if its sponsor can establish:
•that it is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting not more than five in ten thousand persons in the European Union when the application is made, or that it is intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition in the European Union and that without incentives it is unlikely that the marketing of the drug in the European Union would generate sufficient return to justify the necessary investment; and
•that there exists no satisfactory method of diagnosis, prevention or treatment of the condition in question that has been authorized in the European Union or, if such method exists, that the drug will be of significant benefit to those affected by that condition.
Regulation (EC) No 847/2000 sets out further provisions for implementation of the criteria for designation of a drug as an orphan drug. An application for the designation of a drug as an orphan drug must be submitted at any stage of development of the drug but before filing of a MA application.MAA. A MA for an orphan drug may only include indications designated as orphan. For non-orphan indications treated with the same active pharmaceutical ingredient, as a separate MA has to be sought.
Orphan medicinal product designation entitles an applicant to incentives such fee reductions or fee waivers, protocol assistance, and access to the centralized marketing authorization procedure. If an EU MA in respect of an orphan drug is granted pursuant to Regulation (EC) No 726/2004, regulatory authorities will not,the EMA cannot, for a period of usually 10 years, accept another application for a MA, or grant a MA or accept an application to extend an existing MA, for the same therapeutic indication, in respect of a similar drug. This period may however be reduced to six years if, at the end of the fifth year, it is established, in respect of the drug concerned, that the criteria for orphan drug designation are no longer met, in other words,including, when it is shown on the basis of available evidence that the product is sufficiently profitable not to justify maintenance of market exclusivity.exclusivity or where the prevalence of the condition has increased above the threshold. The exclusivity period may increase to 12 years if, among other things, the MAA includes the results of studies from an agreed pediatric investigation plan. Notwithstanding the foregoing, a MA may be granted, for the same therapeutic indication, to a similar drug if:
•the holder of the MA for the original orphan drug has given its consent to the second applicant;
•the holder of the MAmanufacturer for the original orphan drug is unable to supply sufficient quantities of the drug; or
•the second applicant can establish in the application that the second drug, although similar to the orphan drug already authorized, is safer, more effective or otherwise clinically superior.
Regulation (EC) No 847/2000 lays down definitions of the concepts ‘similar drug’medicinal product’ and ‘clinical superiority’.
Other incentives available to orphan drugs in the European Union include financial incentives such as a reduction of fees or fee waivers and protocol assistance. Orphan drug designation does not shorten the duration of the regulatory review and approval process.
EU Data and Market Exclusivity
The EU provides opportunities for data and market exclusivity related to MAs. Upon receiving marketing authorization, innovative, medicinal products are generally entitled to receive eight years of data exclusivity and 10 years of market exclusivity. Data exclusivity, if granted, prevents regulatory authorities in the EUgeneric or biosimilar applicants from referencing the innovator’s data to assesspre-clinical and clinical trial contained in the dossier of the reference product when submitting a generic application or biosimilar applicationMAA for eight years from the date of authorization of the innovative product, after whichreference product. During the additional two-year period of market exclusivity, a generic or biosimilar MAA can be submitted, and the innovator’s data may be referenced. The market exclusivity period prevents a successfulreferenced, but no generic or biosimilar applicant from commercializing its product can be marketed in the EU until ten years have elapsed from the initial MA of the reference product in the EU. The overall ten-year period may, occasionally,will be extended for a further year to a maximum of 11 years if, during the first eight years of those ten years, the MA holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. However, there is no guarantee that a product will be considered by the EU’s regulatory authorities to be a new chemical/biological entity, and products may not qualify for data exclusivity. In the EEA, there is a special regime for biosimilars, or biological medicinal products that are similar to a reference medicinal product but that do not meet the definition of a generic medicinal product. For such products, the results of appropriate preclinical or clinical trials must be provided in support of an MAA. Guidelines from the EMA detail the type of quantity of supplementary data to be provided for different types of biological product.
EU Regulatory Requirements after Marketing Authorization
Where an MA is granted in relation to a medicinal product in the EU, the holder of the MA is required to comply with a range of regulatory requirements applicable to the manufacturing, marketing, promotion and sale of medicinal products.
Similar to the United States, both MA holders and manufacturers of medicinal products are subject to comprehensive regulatory oversight by the EMA, the European Commission and/or the competent regulatory authorities of the individual EU Member States.EEA countries. The holder of an MA must establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions and submission of periodic safety update reports, or PSURs.
All new MAAs must include a risk management plan, or RMP, describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the MA. Such risk-minimization measures or post-authorization obligations may include additional safety monitoring, more frequent submission of PSURs, or the conduct of additional clinical trials or post-authorization safety studies.
In the EEA, the advertising and promotion of medicinal products are subject to both EU and EEA countries’ laws governing promotion of medicinal products, interactions with physicians and other healthcare professionals, misleading and comparative advertising and unfair commercial practices. Although general requirements for advertising and promotion of medicinal products are established under EU legislation, the details are governed by regulations in individual EEA countries and can differ from one country to another. For example, applicable laws require that promotional materials and advertising in relation to medicinal products comply with the product’s Summary of Product Characteristics, or SmPC, as approved by the competent authorities in connection with an MA. The SmPC is the document that provides information to physicians concerning the safe and effective use of the product. Promotional activity that does not comply with the SmPC is considered off-label and is prohibited in the EEA. Direct-to-consumer advertising of prescription medicinal products is also prohibited in the EEA.
In Vitro Diagnostics
The regulations on IVDs are currently harmonized through the Directive 98/79/EC on in vitro diagnostic medical devices (the IVD Directive), which will be replaced byOn 26 May 2022, Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR) from May 26, 2022, with certain exceptions for earlier(IVDs), or the IVDR, entered into application, repealing and transitional periods for later application.replacing Directive 98/79/EC concerning IVDs, or IVDD. The IVDR and its associated guidance documents and harmonized standards govern, among other things, device design and development, preclinical and clinical or performance testing, premarket conformity assessment, registration and listing, manufacturing, labeling, storage, claims, sales and distribution, export and import and post-market surveillance, vigilance, and market surveillance of IVDs.surveillance. IVDs must comply with the General Safety and Performance Requirements, or GSPRs, set out in Annex I of the IVDR. Compliance with these requirements is a prerequisite to be able to affix the CE mark to devices, without which they cannot be marketed or sold in the EEA. To demonstrate compliance with the GSPRs provided in the IVDR and obtain the right to affix the CE mark, medical devices manufacturers must undergo a conformity assessment procedure, which varies according to the type of IVD and its classification. Apart from low risk IVDs (Class A which are not sterile), in relation to which the manufacturer may issue an EU Declaration of Conformity based on a self-assessment of the conformity of its products with the GSPRs, a conformity assessment procedure requires the intervention of a Notified Body, which is an organization designated by a competent authority of an EEA country to conduct conformity assessments. Depending on the relevant conformity assessment procedure, the Notified Body audits and examines the technical documentation and the quality system for the manufacture, design and final inspection of the medical devices. The Notified Body issues a CE Certificate of Conformity following successful completion of a conformity assessment procedure conducted in relation to the IVDmedical device and its manufacturer and their conformity with the GSPRs. This Certificate and the related conformity assessment process entitles the manufacturer to affix the CE mark to its IVDsmedical devices after having prepared and signed a related EC Declaration of Conformity.
As a general rule, demonstration of conformity of medical devices and their manufacturers with the GSPRs must be based, among other things, on the evaluation of clinical data supporting the safety and performance of the products during normal conditions of use. Specifically, a manufacturer must demonstrate that the device achieves its intended performance during normal conditions of use and that the known and foreseeable risks, and any adverse events, are minimized and acceptable when weighed against the benefits of its intended performance, and that any claims made about the performance and safety of the device (e.g., product labeling and instructions for use) are supported by suitable evidence. This assessment must be based on clinical data, which can be obtained from (1) clinical studies conducted on the devices being assessed, (2) scientific literature from similar devices whose equivalence with the assessed device can be demonstrated or (3) both clinical studies and scientific literature. The conduct of clinical studies in the EEA is governed by detailed regulatory obligations. These may include the requirement of prior authorization by the Competent Authorities of the country in which the study takes place and the requirement to obtain a positive opinion from a competent Ethics Committee. This process can be expensive and time-consuming. After a device is placed on the market, it remains subject to significant regulatory requirements.
French Regulatory Framework on Transfer of Values to Health Care Professionals
The French Public Health Code provides for two sets of requirements regarding the transfer of values by health care companies to health care professionals:
•The Anti-Benefit regime prohibits companies that produce or market healthcare products or provide services related to healthcare products, or healthcare companies, from offering or promising benefits in cash or kind to healthcare professionals admitted to practice in France (Article L.1453-3 of the French Public Health Code). In certain limited circumstances, benefits may be excluded from this general prohibition. Exceptions include benefits of negligible value (Article L.1453-6 of the French Public Health Code). Additional exceptions apply to benefits such as remuneration, compensation or disbursements to healthcare professionals in relation to scientific research, speaker fees or hospitality provided in the course of scientific event. This includes benefits provided on the basis of a prior written agreement concluded between the parties where, depending on the amount of the benefit, the benefit is either notified to or authorized by the French competent authority prior to granting the benefit (Article L.1453-7 of the French Public Health Code).
•The Transparency or Sunshine regime, set out by Article L.1453-1 of the Public Health Code, requires healthcare companies manufacturing or marketing health care products (medicinal products, medical devices, etc.) in France to publicly disclose the advantagesbenefits and fees paid to healthcare professionals amountingadmitted to practice in France where the related amount is 10 euros or above, as well as theabove. The related agreements concluded withbetween the latter,parties, along with detailed information about each agreement (the precise subject matter of the agreement, the date of signature of the agreement, its end date, the total amount paid to the healthcare professional, etc.). must also be disclosed. Information must be submitted to the website https://www.entreprisestransparence.sante.gouv.fr and will be disclosed twice a year through this website.
•–The Anti-Gift regime, regarding the general prohibition of payments from pharmaceutical and device manufacturers to healthcare professionals (Article L.1453-3 of the French Public Health Code), except in certain circumstances in particular scientific research, speaker fees and hospitality provided in the course of scientific event. The Anti-Gift regime was modified by the implementation of the provisions of Ordinance n° 2017-49 of January 19, 2017. The new regime includes a prior declaration or prior authorization procedure for the transfers of values which do not fall under the above-mentioned prohibition. The related Decree concerning the advantages granted by persons manufacturing or marketing health products or services was finally published on June 17, 2020 and entered into force on October 1, 2020.Reimbursement
Reimbursement
Significant uncertainty exists in the United States as to the coverage and reimbursement status of any drug candidates for which we obtain regulatory approval. Sales of our products will depend, in part, on the extent to which our products, once approved, will be covered and reimbursed by third-party payors, such as government health programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly reducing reimbursement levels for medical products and services. The process for determining whether a third-party payor will provide coverage for a drug product typically is separate from the process for setting the price of a drug product or for establishing the reimbursement rate that a payor will pay for the drug product once coverage is approved. Third-party payors may limit coverage to specific drug products on an approved list, also known as a formulary, which might not include all of the approved drugs for a particular indication.
To secure coverage and reimbursement for any product candidate that might be approved for sale, we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the product candidate.
These costs are in addition to the costs required to obtain FDA or other comparable regulatory approvals. Whether or not we conduct such studies, our drug candidates may not be considered medically necessary or cost-effective. A third-party payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Further, no uniform policy for coverage and reimbursement exists in the United States, and coverage and reimbursement can differ significantly from payor to payor. One payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage, and adequate reimbursement, for the product. Third-party reimbursement may not be sufficient to enable us to realize an appropriate return on our investment in product development.
In January 2019, we entered into a license agreement with Labcorp to enable them to further develop and deploy NIS4 in the context of clinical research. Initially, we will enable Labcorp through its subsidiary Covance to market and sell an LDT powered by NIS4t in the context of clinical research studies. Covance will not seek or receive third-party insurance reimbursement because clinical trial sponsors will directly cover testing costs. In September 2020, we signed a five-year exclusive license agreement with Labcorp to allow them to develop and commercialize an LDT powered by NIS4 technology for use in routine clinical diagnostic testing in the United States and Canada.
As anWith respect to NASHNext®, the LDT powered by NIS4® technology, Labcorp, as the laboratory partner, will beis responsible for marketing the product to healthcare providers and is responsible for seeking coverage and reimbursement from third party payors, including Medicare and Medicaid. We will separatelySeparately, our strategy is to seek FDA marketing authorization for a kit-based IVD powered by NIS4NIS4® or its improvements to allow us to commercialize the test within the United States as a medical device. In parallel, we intend to progress towards submitting an application for a data packageCE Certificate of Conformity to a European Notified Body in the EEA to enable CE marking, and associated marketing approval in key European markets in 2021.alone or with a potential future partner. In Europe, we are still finalizing our plans but are considering, if approved,the appropriate approvals or certifications are obtained, selling the IVD powered by NIS4NIS4® through a distributor or commercial partner to independent, smaller laboratories, as there are fewer large central laboratories in these regions. We, or our collaborators, will be required to obtain coverage and reimbursement for this test separate and apart from the coverage and reimbursement we plan to seek for our product candidates, onceif approved. There is significant uncertainty regarding our ability to obtain coverage and adequate reimbursement in some or all commercial territories for this test for the same reasons applicable to our product candidates.
The containment of healthcare costs has become a priority of federal, state and foreign governments, and the prices of drugs have been a focus in this effort. The United States federal government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement, utilization management and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Decreases in third-party reimbursement for our drug candidates or a decision by a third-party payor to not cover our drug candidates could reduce physician usage of the drug candidates and could have a material adverse effect on our sales, results of operations and financial condition.
In addition, in some foreign countries, the proposed pricing and reimbursement for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing and reimbursement vary widely from country to country.
The complexity of this process explains why, there can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our drug candidates. Historically, products launched in the EEA do not follow price structures of the United States and generally prices tend to be significantly lower.
Many EEA countries periodically review their reimbursement of medicinal products, which could have an adverse impact on reimbursement status. In addition, we expect that legislators, policymakers and healthcare insurance funds in the EEA, pricing and reimbursement schemes vary widely from country to country. Some countries will continueprovide that products may be marketed only after a reimbursement price has been agreed. Other countries may require the completion of additional studies that compare the cost-effectiveness of a particular product candidate to propose and implement cost-containing measures, such as lower maximum prices, lower or lack of reimbursement coverage and incentives to use cheaper, usually generic, products as an alternative to branded products, and/or branded productscurrently available through parallel import to keep healthcare costs down. Moreover,therapies (so called health technology assessments) in order to obtain reimbursement for our products inor pricing approval. For example, some EEA countries we may be requiredapprove a specific price for a product, or they may instead adopt a system of direct or indirect controls on the profitability of the company placing the product on the market. Other EEA countries allow companies to compilefix their own prices for products but monitor and control prescription volumes and issue guidance to physicians to limit prescriptions. Recently, many EEA countries have increased the amount of discounts that pharmaceutical companies are requirement to offer. These efforts could continue as countries attempt to manage healthcare expenditures. The downward pressure on healthcare costs in general, particularly prescription products, has become intense. As a result, increasingly high barriers are being erected to the entry of new products onto national markets. Political, economic, and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various EEA countries, and parallel trade (arbitrage between low-priced and high-priced member states), can further reduce prices.
In addition, some EEA countries may require the completion of additional data comparingstudies that compare the cost-effectiveness of our productsa particular medicinal product candidate to othercurrently available therapies. Health Technology Assessment, or HTA, of medicinal products is becoming an increasingly common part of the pricing and reimbursement procedures in some EU member states,EEA countries, including those representing the larger markets. The HTA process, which is currently governed by national laws in each EU member state,EEA country, is the procedure to assess therapeutic, economic and societal impact of a given medicinal product in the national healthcare systems of the individual country. The outcome of an HTA will often influence the pricing and reimbursement status granted to these medicinal products by the competent authorities of individual EU member states.EEA countries. The extent to which pricing and reimbursement decisions are influenced by the HTA of the specific medicinal product currently varies between EEA countries. In December June 2021, the EU Parliament adopted the HTA Regulation which aims to harmonize the clinical benefit assessment of HTA across the EU,EEA, the consequences of which remain unknown at this time. The anticipated revenue from and growth prospects for products in the EEA could be negatively affected by the HTA Regulation.
Legislators, policymakers and healthcare insurance funds in the EEA may continue to propose and implement cost-containing measures to keep healthcare costs down; particularly due to the financial strain that the COVID-19 pandemic has placed on national healthcare systems of the EEA countries. These measures could include limitations on the prices of medicinal products or the level of reimbursement available for these products from governmental authorities or third party payors. Further, an increasing number of EEA and other foreign countries use prices for medicinal products established in other countries as “reference prices” to help determine the price of the product in their own territory. Consequently, a downward trend in prices of medicinal products in some countries could contribute to similar downward trends elsewhere.
–Healthcare Reform
In the United States and some foreign jurisdictions, there have been, and continue to be, several legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of product candidates, restrict or regulate post-approval activities, and affect the ability to profitably sell product candidates for which marketing approval is obtained. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, ACA, enacted in the United States in March 2010, has already had and is expected to continue to have, a significant impact on the healthcare industry. The ACA, has expanded coverage for the uninsured while at the same time containing overall healthcare costs. With regard to pharmaceutical products, among other things, the ACA expanded and increased industry rebates for drugs covered under Medicaid programs and made changes to the coverage requirements under the Medicare Part D program.
There have been judicial, executive and Congressional challenges, as well as challenges by the executive branch toa number of proposed and enacted health reform measures that have impacted certain aspects of the ACA. For example, President Trump signed Executive Orders and other directives designed to delay the implementation of certain provisions of the ACA or otherwise circumvent some of the requirements for health insurance mandated by the ACA. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the ACA. While Congress has not passed comprehensive repeal legislation, it has enacted laws that modify certain provisions of the ACA such as removing penalties, starting January 1, 2019, for not complying with the ACA’s individual mandate to carry health insurance, eliminating the implementation of certain ACA-mandated fees, and increasing the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D. For example, on June 17, 2021, the U.S. Supreme Court dismissed a challenge on procedural grounds that argued that the ACA is unconstitutional in its entirety because the individual mandate was repealed by Congress. Thus, the ACA will remain in effect in its current form. Moreover prior to the U.S. Supreme Court ruling,August 16, 2022, President Biden signed an Executive Order on January 28, 2021 to establish a Special Enrollment Period to seekthe Inflation Reduction Act of 2022, or IRA, into law, which among other things, extends enhanced subsidies for individuals purchasing health insurance coverage in ACA marketplaces through plan year 2025. The IRA also eliminates the ACA marketplace. The Executive Order also requires that some government agencies review"donut hole" under the Medicare Part D program beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost and reconsider their current policies and practices that may undermine access to health care, including, among others, the review of Medicaid demonstration and waiver policies including work requirements, and policies that may present unnecessary barriers to accessing health care, Medicaid or ACA coverage.creating a new manufacturer discount program. It is possible that the ACA will be subject to additional judicial or Congressional challenges in the future. It is unclear how any such challenges or the health reform measures of the Biden Administration will affect the ACA.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. For example, on August 2, 2011, the Budget Control Act of 2011 among other things, created measures for spending reductions by Congress. Specifically, the Joint Select Committee on Deficit Reduction was created to recommend to Congress proposals in spending reductions. The Joint Select Committee on Deficit Reduction did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2012 through 2021, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, started in April 2013 and which, due to subsequent legislative amendments, including the BBA, will stay in effect through 2031 unless additional Congressional action is taken. These Medicare sequester reductions will be suspended from May 1, 2020 through March 31, 2022 due to the COVID-19 pandemic. Under current legislation, the actual reduction in Medicare payments will vary from 1% in 2022 to up to 3% in the final fiscal year of the sequester. Additionally, on January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, or the ATRA. The ATRA, among other things, reduced Medicare payments to several providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Congress is considering additional health reform measures.
Recently, there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products. Such scrutiny has resulted in several recent U.S Presidential Orders, U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the cost of drugs under Medicare and reform government program reimbursement methodologies for drug products. At the federal level, for example, the Trump administration used several means to propose or implement a reform of drug pricing, notably through federal budget proposals, executive orders and political initiatives. For example, on July 24, 2020 and September 13, 2020, the Trump administration announced several executive orders related to prescription drug pricing, which attempted to implement several of the administration’s proposals. The FDA concurrently issued a rule and guidance in September 2020, implementing part of the Executive Order on Prescription Drug Importation, providing guidance to the States to prepare and submit importation plans for drugs from Canada. Further, on November 20, 2020,IRA, among other things (i) directs the Department of Health and Human Services, or HHS, issued a final rule forto negotiate the removalprice of safe harbor protection for rebates involving prescription pharmaceuticals to plan sponsorscertain high-expenditure, single-source drugs and biologics covered under Medicare part D, either directly or through the certain pharmacy benefit managers, unless the rebate is required by law. The implementation of the rule has been delayed by the Infrastructure Investment and Jobs Act to January 1, 2026. T The rule also created a new safe harbor protection for(ii) imposes rebates reflected at the point of sale, as well as a new safe harbor protection for certain fixed fees that manufacturers pay to pharmacy benefit managers for services rendered, of which the implementation has also been postponed until January 1, 2026. On November 20, 2020, the Center for Medicare & Medicaid Services issued an interim final rule implementing the Most Favored Nation Executive Order signed by President Trump, which would closely align payments for someunder Medicare Part B drugs administered by physiciansand Medicare Part D to the lowest cost paidpenalize price increases that outpace inflation. These provisions will take effect progressively starting in other economically advanced countries as of January 1, 2021 As a result of litigation challenging the Most Favored Nation model, on December 27, 2021, CMS published a final rule that rescinds the Most Favored Nation model interim final rule. In July 2021,fiscal year 2023, although they may be subject to legal challenges. Additionally, the Biden administration released an executive order "Promoting Competition in the American Economy", with multiple provisions aimed at prescription drugs. In response to Biden'sadditional executive order on September 9, 2021,October 14, 2022, directing HHS released a Comprehensive Planto report on how the Center for Addressing High Drug Prices that outlines principlesMedicare and Medicaid Innovation can be further leveraged to test new models for lowering drug pricing reformcosts for Medicare and sets out a variety of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can take to advance these principles. No legislation or administrative actions have been finalized to implement these principles. In addition, Congress is considering drug pricing as part of other reform initiatives. It is unclear whether these or similar policy initiatives will be implemented in the future.Medicaid beneficiaries. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Moreover, it is possible that additional governmental measures will be taken in response to the Covid-19 pandemic.
–Other U.S. Healthcare Laws and Compliance Requirements
Our business operations in the United States and our arrangements with clinical investigators, healthcare providers, consultants, third-party payors and patients expose us to broadly applicable federal and state fraud and abuse and other healthcare laws. These laws may impact, among other things, our research, and if approved, proposed sales, marketing and education programs of our drug candidates. The laws that may affect our ability to operate include, among others:
•the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering or paying remuneration (including any kickback, bribe or rebate), directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, lease, order, or recommendation of, an item, good, facility or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs;
•federal civil and criminal false claims laws, including the federal civil False Claims Act, which can be enforced by private individuals acting on behalf of the federal government, through civil whistleblower or qui tam actions, and civil monetary penalty laws, which prohibits individuals and entities from, among other things, knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent, or making a false statement or record material to payment of a false claim or avoiding, decreasing, or concealing an obligation to pay money to the federal government, including for example, providing inaccurate billing or coding information to customers or promoting a product off-label;
•the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created additional federal criminal statutes that prohibit, among other things, knowingly and willfully executing or attempting to execute a scheme to defraud any healthcare benefit program, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willingly falsifying, concealing or covering up a material fact or making materially false statements, fictitious, or fraudulent statements in connection with the delivery of or payment for healthcare benefits, items, or services;
•the federal Physician Payments Sunshine Act, enacted as part of the ACA, which requires applicable manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, with specific exceptions, to track and annually report to CMS payments and other transfers of value provided to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain other healthcare professionals (such as physician assistants and nurse practitioners), and teaching hospitals and certain ownership and investment interests held by physicians and their immediate family members. ;members;
•HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their implementing regulations, which imposes certain requirements on certain healthcare providers, health plans, and healthcare clearinghouses, known as covered entities, and their business associates, which are individuals and entities that perform functions or activities on behalf of covered entities that involve protected health information, relating to the privacy, security and transmission of protected health information; and
•State and foreign equivalents of each of the above federal laws and regulations, such as: state anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers; state marketing and/or transparency laws applicable to manufacturers that may be broader in scope than the federal requirements; state laws that require biopharmaceutical companies to comply with the biopharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government; state and local laws that require the registration of pharmaceutical sales representatives; and state and/or foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect as HIPAA, thus complicating compliance efforts.
The ACA broadened the reach of the federal fraud and abuse laws by, among other things, amending the intent requirement of the U.S. federal Anti-Kickback Statute and certain federal criminal healthcare fraud statutes. Pursuant to the statutory amendment, a person or entity no longer needsdoes not need to have actual knowledge of these statutes or specific intent to violate them in order to have committed a violation. In addition, the ACA provides that the government may assert that a claim including items or services resulting from a violation of the U.S. federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act or the civil monetary penalties laws.Act.
Efforts to ensure that our business arrangements with third parties comply with applicable healthcare laws involves substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to, for example, significant administrative, civil, and/or criminal penalties, damages, fines, disgorgement, contractual damages, reputational harm, diminished profits and future earnings, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws and the curtailment or restructuring of our operations. If the physicians or other healthcare providers or entities with whom we expect to do business are found to be not in compliance with applicable laws, they may be subject to significant administrative, civil, and/or criminal sanctions, including exclusions from government funded healthcare programs.
| | | | | |
| C. | Organizational Structure |
In 2021,September 2022, we announced that as partfinalized the acquisition of our new corporate strategy,Versantis AG, and its U.S.-based wholly-owned subsidiary, Versantis, Inc. Versantis, Inc. does not currently have any operational activities. For more information about the acquisition, see Note 30 - "Acquisitions”. In June 2023, we expected to distinguish our pharmaceutical and diagnostic technologies activities, in order to ensure more independent management and growth. In light of the COVID-19 pandemic, the lack of approved therapeutic options in NASH and the current lack of reimbursement for NASHnext, our partner Labcorp has experiencedliquidated Versantis Inc., a slow commercial uptake. As a result, wewholly-owned subsidiary which did not have decided to postpone the creation of a dedicated diagnostic entity in the near term.any operational activities.
| | | | | |
| D. | Property, Plants and Equipment |
Our corporate headquarters are located in Loos, France. To date, the total surface occupied is approximately 5,500 square meters of office space. The lease for our Loos headquarters continues through March 2029. We also lease office space in Paris, France, and,in Cambridge, Massachusetts for our U.S. subsidiary, GenfitGENFIT Corp., and in Cambridge, Massachusetts.Zurich, Switzerland, for our Swiss subsidiary, Versantis AG.
| | | | | |
| Item 4A. | Unresolved Staff Comments. |
Not applicable.
| | | | | |
| Item 5. | Operating and Financial Review and Prospects. |
Overview
We are a late-stage clinical biopharmaceutical company dedicated to the discovery and development of innovative drug candidates and diagnostic solutions targeting metabolic and liver-related diseases where there is considerable unmet medical need. We are a leaderpioneer in the fielddiscovery and development of nuclear receptor-based drug discoverydrugs for liver diseases with a rich history and strong scientific heritage spanning almost two decades. Since 2016, we had been evaluating our most advanced drug candidate, elafibranor, in a pivotal Phase 3 clinical trial (the RESOLVE-IT trial) as a potential treatment for nonalcoholic steatohepatitis, or NASH. , On May 11, 2020, we publishedOur pipeline now covers the topline data from the interim analysis of the RESOLVE-IT trial. In these interim results, elafibranor did not demonstrate a statistically significant effect on the primary surrogate endpoint which was NASH resolution without worsening of fibrosis nor did it achieve the key secondary endpoints. These results led us, after a detailed review of the whole dataset, to initiate the trial termination process for RESOLVE-IT at the end of July 2020 for lack of efficacy, but not due to safety reasons. In May 2021, we announced, following the termination of all development of elafibranor in NASH and the redefinition of our clinical products portfolio, that we had redeployed our R&D effort across three franchises covering therapeutic areas where patients have littlewith the following drugs at different development stages (preclinical, Phase 1, Phase 2 and Phase 3), with different mechanisms of action:
Primary Bilary Cholangitis, or no treatment and/or diagnostics options: cholestatic diseases, PBC:
•Elafibranor
Acute on Chronic Liver Failure, (ACLF),or ACLF:
•VS-01-ACLF,
•Nitazoxanide, or NTZ,
•SRT-015,
•CML-022,
Hepatic Encephalopathy, or HE:
•VS-02-HE
Cholangiocarcinoma, or CCA:
•GNS561
Urea Cycle Disorder, or UCD, and diagnostics.Organic Acidemia, or OA:
Elafibranor is currently being evaluated•VS-01-HAC
In addition, our pipeline includes a diagnostic franchise including NIS2+® in Metabolic dysfunction-Associated Steatohepatitis (MASH, previously known as NASH, for nonalcoholic steatohepatitis) and TS-01 focusing on blood ammonia levels.
In September 2022, we acquired Versantis AG, a potential treatment for primary biliary cholangitis, or PBC. PBC is an autoimmune disease unrelatedprivate Swiss-based clinical stage biotechnology company, focused on addressing the growing unmet medical needs in liver diseases. With the acquisition, we added the following Versantis' assets to the metabolic origins of NASHour pipeline: VS-01, VS-02 and is independent from our evaluation of elafibranor in NASH. Our phase 3 trial of elafibranor in PBC, ELATIVE, began in 2020. The first patient first visit in the ELATIVE trial took place on September 24, 2020. Enrollment is expected to be completed in the second quarter 2022 for the double-blind part of ELATIVE, which is the part to be used to support accelerated approval and we remain committed to delivering ELATIVE topline data in the second quarter 2023.TS-01.
The worldwide development and commercialization rights in elafibranor for the treatment of PBC and other indications were licensed to Ipsen Pharma SAS, or Ipsen, through a partnership agreement signed in December 2021, the Ipsen Agreement, with the exception of Greater China, which is licensed to Terns Pharmaceuticals, Inc., or Terns, in NASHMASH and PBC since June 2019. In June 2023, we and Ipsen announced positive 52-week interim topline data from the pivotal ELATIVE® Phase 3 trial.
In additionVS-01-ACLF, our first ACLF program, is currently in a Phase 2a proof of concept study (UNVEIL-IT®) and interim data from the trial are targeted for the second half of 2024.
Our second ACLF program is aimed at developing the repurposed drug nitazoxanide (NTZ) either as a standalone treatment or in combination with another treatment. A new formulation of NTZ is currently under development to PBC, our cholestatic diseases programpermit greater dosing flexibility and to optimize dose-response in patients with ACLF, which are known to have varying degrees of organ impairment or failure. The new formulation is also includes cholangiocarcinoma orintended to optimize the benefit/risk profile in this patient population. A Phase 2a proof of concept study in patients with ACLF grades 1 and 2 is targeted to initiate in the first half of 2025.
We are also developing GNS561 in CCA following the execution in December 2021 of an exclusive license to develop and commercialize GNS561 in the United States, Canada and Europe (including United Kingdom and Switzerland) from Genoscience Pharma,Pharma. Enrollment for CCA. Ourthe Phase 21b/2a clinical trial for GNS561 began in CCA2023 and is expected to start inprovide preliminary data by the second halfend of 2022.2024.
TheIn 2023, we in-licensed two additional investigational drugs in ACLF. SRT-015 is an ASK1 inhibitor in-licensed from Seal Rock Therapeutics in acute liver diseases. It targets the inhibition of cellular apoptosis, inflammation and fibrosis. A First-in-Human study with an intravenous formulation of SRT-015 is planned in the first program in our ACLF franchise aims at developingquarter of 2025. CLM-022 is a small molecule inhibitor targeting the repurposed drug nitazoxanide (NTZ). A Phase 1 open-label, non-randomized, 2-center, repeated-dose, parallel-group studyNLRP3 inflammasome in-licensed from Celloram. Preclinical Proof of Concept is expected to provide preliminary insight into NTZ pharmacokinetics,be obtained or PK, and safety in the setting of hepatic impairment in the second half of 2022. A second Phase 1 study to evaluate NTZ PK and safety is planned to initiate in the first half of 2022. The results of these studies will inform the potential need for dose adjustment in future studies to be conducted in patients with cirrhosis and hepatic impairment.attained by end-2024.
A key differentiator of our development strategy is our NASHMASH biomarker-based diagnostic program, called NIS4,NIS4® and its improvement, NIS2+®, a technology which we are developing to power a new in vitro diagnostic, or IVD, test to identify patients with NASHMASH who may be appropriate candidates for drug therapy. In January 2019, we entered into a first license agreement with Labcorp to allow Labcorp to develop and commercialize NIS4NIS4® in the clinical research space through their drug development subsidiary, Covance. Since then, Covance has made significant progress in the deployment of NIS4 in several clinical trials conducted by leading players in the pharmaceutical industry. A second exclusive license agreement with Labcorp to allow them to develop and commercialize an LDT powered by NIS4NIS4® technology for use in routine clinical diagnostic testing in the United States and Canada was signed in September 2020 and in April 2021, Labcorp launched commercialization of NASHnext,NASHNext®, an LDT powered by our NIS4NIS4® technology. The technology is also licensed to Q Squared Solutions LLC (Q2) in the clinical research field.
AlthoughIn 2021, we recorded revenue in 2019 from the receipt of an upfront payment under our collaborationthe Ipsen Agreement (part of which was recognized in 2022 and license agreement with Terns Pharmaceuticals2023). In 2023, we recorded revenue from a first milestone payment from Ipsen, which was triggered by the acceptance of the NDA filing by the FDA and MAA by the EMA for accelerated approval of elafibranor in 2021 from the receipt of an upfront payment under our agreement with Ipsen,PBC in December 2023. However, we have never generated significant revenues from product sales. WeWhile we expect to receive additional milestone payments and revenues from the Ipsen agreement in 2024 if elafibranor receives marketing authorization in PBC and is commercialized, we do not expect to generate material revenue from other product sales unless and until we successfully complete clinical development of, obtain marketing approval for and commercialize our drug candidates and LDT and IVD test.tests. Clinical development, regulatory approval and commercial launch of a product candidate or diagnostic can take several years and are subject to significant uncertainty.
Historically, we have financed our operations and growth through issuances of share capital and convertible bonds, through conditional advances and subsidies from Banque Publique d'Investissement (BPI France) and, from research tax credits.credits and through the upfront milestone of €120 million from the Ipsen agreement. In 2006, we completed the initial public offering of our ordinary shares on the Alternext market of Euronext in Paris and transferred to the Euronext Paris in April 2014. Between 2010 and 2016, we raised a total of over €220 million in gross proceeds from the issuance of ordinary shares. In October 2017, we issued €180 million in convertible bonds. In March 2019, we completed a global offering consisting of an initial public offering of our American Depositary Shares, or ADSs, in the United States, and a private placement of our ordinary shares in Europe and other countries outside the United States, including France. Aggregate gross proceeds from the global offering, before deducting underwriting discounts and commissions and offering expenses payable by us, were approximately $155.4 million. Additionally, in 2021, Ipsen also became a shareholder of GENFIT through the purchase of 3,985,239 newly issued shares representing 8% of GENFIT after issuance, via a €28 million investment. There have been no subsequent equity raises.
Since our inception, we have incurred significant operating losses. Our net loss was €65.1 million and €101.2 million for the years ended December 31, 2019 and 2020, respectively. For the year ended December 31, 2021, we had aour net profit ofgain was €67.3 million, owingprimarily due to the upfront payment received from Ipsen. FollowingFor the results ofyear ended December 31, 2022, our net loss was €23.7 million. For the Phase 3 RESOLVE-IT trial in the first half of 2020, we implemented a cost savings plan to reduce operational expenses, including a workforce reduction plan and eliminate non-essential expenses, which contributed to reducingyear ended December 31, 2023, our cash flows used in operating activities from €96.4 million in 2020 to €44.0 million in 2021 (excluding the upfront payment received from Ipsen in 2021). Nevertheless, as we continue our efforts to identify product candidates with the highest potential, conduct preclinical studies and clinical trials and advance the development of our diagnostic test, we expect that our cash used in operational activities will increase to €65 million in 2022 (excluding exceptional items of €30.0 million payable in 2022 in relation to the upfront payment received from Ipsen in late 2021, i.e. primarily VAT collected and corporate income tax, as well as potential costs and expenses in future business development activities such as in-licensing). This estimate takes into account our projected cash flows from operating activities and government funding of research programs. We have based this estimate on assumptions that may prove to be wrong. Our net losses may fluctuate significantly from quarter to quarter and year to year, notably depending on the timing of our clinical trials and our expenditures on other research and development activities. Also, we could use our available capital resources sooner than we currently expect.loss was €28.9 million.
Financial Operations Overview
Revenue and Other Income
For the year ended December 31, 2020, our revenue was €0.8 million, mainly due to a one-time transaction as well as the income recognized within the scope of our license agreements with Labcorp. For the year ended December 31, 2021, our revenue was €80.1 million, mainly from the receipt of the €120 million upfront payment from Ipsen, out of which €80 million is recognized as 2021 revenue, after deduction of €40 million deferred revenue, which will gradually be recognized as revenue following the completion of the ELATIVE double-blind study, pursuant to IFRS 15. Other income recognized in 2021 is related to licensing agreements with Labcorp for the deployment of NIS4 technology in NASH. For the year ended December 31, 2020, our revenue was €0.8 million, mainly due to a one-time transaction as well as the income recognized within the scope of our license agreements with Labcorp.
In 2019, we entered into two licensing agreements, one with Terns with respect to the development and commercialization of elafibranor in Greater China, or the Terns Agreement, and one with Covance, Labcorp’s drug development business, with respect to the development and deployment of a test powered by NIS4NIS4® technology in the clinical research space. Pursuant to our agreement withthe Terns Agreement, we received an upfront payment of $35 million in 2019, and are eligible for up to $193 million in clinical, regulatory and commercial milestone payments, as well as mid-teen percentage royalties (For more information see Note 4.4.1 29 - "Commitments and contingent liabilities"to our consolidated financial statements)statements included in this annual report). In September 2020, we entered into a second agreement with Labcorp, for a five-year exclusive licensing agreement with Labcorp to develop and commercialize an LDT powered by NIS4by NIS4® technology and its improvements in the clinical diagnostic market.Inmarket. In May 2021, we signed a worldwide, non-exclusive license agreement with Q Squared Solutions LLC, to broaden the availability of NIS4® technology and its improvements in the clinical research field. In December 2021, we entered into a long-term strategic partnership for global collaboration withthe Ipsen Agreement granting Ipsen an exclusive worldwide (excluding Greater China which is licensed to Terns) license to develop, manufacture and commercialize elafibranor, for people living with PBC, and in other indications. Under the agreement,Ipsen Agreement, Ipsen will pay GENFITus up to €480 million, comprisingwhich is comprised of an upfront cash payment of €120 million, as well as regulatory, commercial, and sales-based milestone payments of up to €360 million, plus tiered double-digit royalties of up to 20%. (For more information see Note 29 - "Commitments and contingent liabilities" to our consolidated financial statements included in this annual report). Other than pursuant to these three agreements, we do not expect to receive any revenue from any of our product candidates until we obtain regulatory approval and commercialize such products, or until we potentially enter into collaborative agreements with third parties for the development and commercialization of such candidates.
In 2022, we entered into the Inventory Purchase Agreement signed with Ipsen, pursuant to which Ipsen purchased inventory of elafibranor active pharmaceutical ingredient and drug product during the second half of 2022 with the prospect of transferring the conduct of the ELATIVE® study to Ipsen. In addition, we entered into the Transition Services Agreement with Ipsen, which essentially outlines the scope of services to facilitate the transition of some activities related to the Phase 3 clinical trial evaluating elafibranor in Primary Biliary Cholangitis.
In 2023, we entered into the Part B Transition Services Agreement with Ipsen, in order to facilitate the transition of certain services related to the Phase 3 ELATIVE® clinical trial until the complete transfer of the responsibility of the trial to Ipsen.
Our other income results principally from the research tax credits. We expect to continue to be eligible for these tax credits and subsidies for so long as we incur eligible expenses.
CIR Research Tax Credit
We benefit from a tax credit known as Crédit d’Impôt Recherche, or CIR, which is granted by French tax authorities to encourage companies to conduct technical and scientific research. Companies demonstrating that they have expenses that meet the required criteria, including research expenses located in France or within the European Union or in another state that is a party to the agreement in the European Economic Area that has concluded a tax treaty with France that contains an administrative assistance clause, receive a tax credit that can be used against the payment of French corporate income tax due for the fiscal year in which the expenses were incurred and the three fiscal years thereafter, or, as applicable, can be reimbursed for the excess portion. The expenses taken into account for the calculation of the CIR only involve certain eligible research and development expenses. The subcontracting expenses are limited to an amount equal to €10 million.
The main characteristics of the CIR are the following:
•the CIR results in a cash inflow from the tax authorities paid to us as we are not subject to corporate income tax;
•a company’s corporate income tax liability does not limit the amount of the CIR—a company which meets certain criteria in terms of sales, headcount or assets to be considered a small/mid sizemid-size company and that does not pay any corporate income tax can request cash payment of the research tax credit; and
•the CIR is not included in the determination of the corporate income tax.
We have concluded that the CIR meets the definition of a government grant as defined in IAS 20, Accounting for Government Grants and Disclosure of Government Assistance, and, as a result, it has been classified as other income within operating income in our statement of operations.
Exchange Gain on trade receivables and liabilities
We also recognize in other operating income within “other income” the exchange gains on trade receivables because we determined that they are attributable to the related revenue and other income initialinitially recognized.
Operating Expenses
Research and Development Expenses
We engage in substantial research and development (R&D) efforts to develop our drug and diagnostic candidates. Research and development expenses include:
•raw materials and consumables, such as lab supplies, used in research and development activities;
•fees and costs paid to third parties, such as clinical research organizations and scientific advisors, for clinical trial and other research and development activities, including services subcontracted to research partners for technical or regulatory reasons;
•employee-related costs and costs related to external employees seconded to us for clinical development, biometrics and information technology; and
•intellectual property fees related to the filing of patents.
•The provision recognized in 2019 due to the research tax credit dispute with the French revenue services and reversed in 2020
Research and development activities are central to our business model. Drug candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials, such as the RESOLVE-IT and ELATIVEELATIVE® trials. We expect that our research and development expenses will increase compared to 20212023 for the foreseeable future, asfuture. As we continue to advance our efforts to identify potentialcurrent product candidates, conduct preclinical studies and conduct clinical trials, we expect that our cash used in operational activities will amount to €75 million in 2024. This estimate takes into account our projected cash flows from operating activities and government funding of research programs. We have based this estimate on assumptions that may prove to be wrong. Our net losses may fluctuate significantly from quarter to quarter and year to year, notably depending on the timing of our clinical trials and advance theour expenditures on other research and development ofactivities. Also, we could use our diagnostic test.available capital resources sooner than we currently expect. They may also fluctuate depending on the next steps initiated in the clinical development of our drug candidates, new development programs, which we may decide to start, and progress in the development of our diagnostic test. The impact of the RESOLVE-IT study in NASH on our 2022 results will be limited as the residual amount for termination costs is estimated to be €0.5 million.tests.
We generally do not track our research and development expenses by product candidate. However, the substantial majority of our direct expenses incurred, such as for contract research organizations, or CROs, and other contracted research and development activities, as well as raw materials, relate to elafibranor, our lead drug candidate.
General and Administrative Expenses
General and administrative expenses include:
•employee-related costs for executive, intellectual property, finance, legal and human resources and communications functions;
•facility-related costs;
•grants to The NASH Epidemiology Institute (formerly The NASH Education Program) for 2018 and 2019, primarily to finance International NASH Day;
•fees for third-party providers of administrative services, including legal, audit and accounting, press relations and communication services, security and reception and recruiting; and
•intellectual property fees for the registration and maintenance of our patents.
The cost-saving plan initiated in September 2020 has led to a decrease in generalGeneral and administrative expenses in 2021, compared to 2020. However, expenses will remain significant over the next several years due to expenses associated with being a public company in the United States, including costs related to audit, legal, regulatory and tax-related services associated with maintaining compliance with U.S. exchange listing and SEC requirements, director and officer insurance premiums, investor relations and litigation costs. In particular, we will continue to incur additional expenses associated with accounting and internal control over financial reporting to comply with the Sarbanes-Oxley Act of 2002 in the United States.
Marketing and Market Access Expenses
Marketing and market access expenses include:
•employee-related costs for marketing, and business development functions;
•facility-related costs; and
•fees for third-party providers of marketing and pre-commercialization services including market surveys, brand strategy, medical communication and market access services.
The cost-saving plan initiated in the summer of 2020 allowed us to decrease our marketingReorganization and market access expenses in 2021. We expect that they will not increase significantly in 2022.Restructuring Expenses
Reorganization and restructuring expenses include:
•thethe accruals and provisions recognized within the scope of the reduction in force plan;
•the extraordinary amortization, loss of value and impairment of fixed assets recognized within the scope of the reorganization of GENFIT;reorganization;
•the impairment of the right of use of the leased equipment and premises,premises;
•the portion of the OCEANEOCEANEs renegotiation expenses recognized in 2021; and
•the provision recognized for some of the costs of the closing process for the RESOLVE-ITRESOLVE-IT® study, which, after detailed analysis, we concluded do not have any future economic advantage for the PBC program.
Financial Income (Expense)
Financial income relates primarily to interest income received from cash and cash equivalents deposits. Our cash and cash equivalents have been deposited primarily in cash accounts and term deposit accounts with short maturities, as well as medium term notes or UCITSUndertaking for Collective Investment in Transferable Securities and therefore generate only a modest amount of interest income.
Financial expense relates primarily to interest expense on our outstanding convertible bonds as well as interest expense for bank loans and for leases. We also incur foreign exchange losses related to our purchases of services in U.S. dollars and Swiss Francs, which amounts are recorded as financial expense and interest expenses due to leases in application of IFRS16.IFRS 16.
FinancialIn 2021, financial income includes notablyincluded the one-time buyback bonus of €35.6 million issued from the renegotiation of the OCEANEs completed in January 2021 (See note 22 "Financial Income").2021.
Our results of operations for the years ended December 31, 2019, 20202021, 2022 and 20212023 are summarized in the table below.
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | Notes | | Year ended |
| (in € thousands, except earnings per share data) | | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| | | | | | | | |
| Revenues and other income | | | | | | | |
| Revenue | 19 | | 30,839 | | 765 | | 80,069 |
| Other income | 19 | | 10,122 | | 6,993 | | 5,510 |
| Revenues and other income | | | 40,961 | | 7,758 | | 85,579 |
| | | | | | | | |
| Operating expenses and other operating income (expenses) | | | | | | | |
| Research and development expenses | 20 | | (66,170) | | (59,097) | | (35,166) |
| General and administrative expenses | 20 | | (17,265) | | (14,270) | | (16,153) |
| Marketing and market access expenses | 20 | | (13,708) | | (11,216) | | (1,539) |
| Reorganization and restructuring expenses | 20 | | — | | (5,308) | | (142) |
| Other operating income (expenses) | 20 | | (1,649) | | (764) | | (763) |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| Operating income (loss) | | | (57,832) | | (82,897) | | 31,816 |
| | | | | | | | |
| Financial income (1) | 22 | | 5,221 | | 6,544 | | 44,780 |
| Financial expenses | 22 | | (13,110) | | (25,296) | | (7,122) |
| Financial profit (loss) | | | (7,889) | | (18,752) | | 37,658 |
| | | | | | | | |
| Net profit (loss) before tax | | | (65,721) | | (101,649) | | 69,474 |
| | | | | | | | |
| Income tax benefit (expense) | 23 | | 576 | | 428 | | (2,215) |
| | | | | | | | |
| Net profit (loss) | | | (65,144) | | (101,221) | | 67,259 |
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | Notes | | Year ended |
| (in € thousands, except earnings per share data) | | | 2021/12/31 | | 2022/12/31 | | 2023/12/31 |
| | | | | | | | |
| Revenues and other income | | | | | | | |
| Revenue | 7 | | 80,069 | | 20,195 | | 28,565 |
| Other income | 7 | | 5,510 | | 6,371 | | 9,610 |
| Revenues and other income | | | 85,579 | | 26,566 | | 38,176 |
| | | | | | | | |
| Operating expenses and other operating income (expenses) | | | | | | | |
| Research and development expenses | 8 | | (35,166) | | (35,818) | | (46,503) |
| General and administrative expenses | 8 | | (16,153) | | (16,405) | | (17,741) |
| Marketing and market access expenses | 8 | | (1,539) | | (992) | | (876) |
| Reorganization and restructuring income (expenses) | 8 | | (142) | | 11 | | 505 |
| Other operating expenses | 8 | | (763) | | (652) | | (141) |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| Operating income (loss) | | | 31,816 | | (27,289) | | (26,580) |
| | | | | | | | |
| Financial income | 10 | | 44,780 | | 8,212 | | 3,680 |
| Financial expenses | 10 | | (7,122) | | (4,758) | | (5,614) |
| Financial profit (loss) | | | 37,658 | | 3,453 | | (1,934) |
| | | | | | | | |
| Net profit (loss) before tax | | | 69,474 | | (23,836) | | (28,514) |
| | | | | | | | |
| Income tax benefit (expense) | 11 | | (2,215) | | 116 | | (380) |
| | | | | | | | |
| Net profit (loss) | | | 67,259 | | (23,719) | | (28,894) |
Comparisons for the Years Ended December 31, 20202022 and 2023
A discussion and analysis of our financial condition and operating results for the year ended December 31, 2022 as compared to the year ended December 31, 2021 is included in Item 5 of our Annual Report on Form 20-F for the year ended December 31, 2022, filed with the SEC on April 18, 2023 and is incorporated herein by reference.
Revenue
Revenue of €0.8amounted to €20.2 million during the year ended December 31, 2020 was2022, which includes €15.9 million attributable to the result of a one-time transaction and thepartial recognition of the €40.0 million deferred income from 2021 related to the Ipsen Agreement, €1.0 million in revenue undergenerated from the Labcorp licensing agreements.services we rendered to Ipsen in accordance with the Transition Services Agreement signed in April 2022, which outlines the scope of services to facilitate the transition of certain activities related to the Phase 3 ELATIVE® clinical trial evaluating elafibranor in PBC and €3.3 million that was recognized as revenue in accordance with the Inventory Purchase Agreement signed with Ipsen in July 2022, pursuant to which Ipsen purchased inventory of the elafibranor active pharmaceutical ingredient and drug product during the second half of 2022 with the prospect of transferring the conduct of the ELATIVE® study to Ipsen.
Revenue of €80.1amounted to €28.6 million during the year ended December 31, 20212023. €13.3 million was attributable to a milestone invoiced to Ipsen in December 2023 in accordance with the resultIpsen Licence and Collaboration Agreement. This milestone was earned following the NDA filing acceptance by the FDA and MAA filing acceptance by the EMA for accelerated approval of elafibranor. €8.7 million in revenue was attributable to the partial recognition of the upfront payment for a licensing agreement with Ipsen recognized in 2021. Regarding the Application of IFRS15€40.0 million deferred income from 2021 related to the IPSEN LicenseIpsen Licence and Collaboration Agreement. €6.5 million in revenue was generated from the services rendered under the Transition Services Agreement and Part B Transition Services Agreement, signed in 2021, see Note 19 "Operating Income".April 2022 and September 2023 respectively by GENFIT and Ipsen, in order to facilitate the transition of certain services related to the Phase 3 ELATIVE® clinical trial until the complete transfer of the responsibility of the trial to Ipsen. €0.1 million was attributable to other ancillary activities. Other Income
Other income for the years ended December 31, 20202022 and 20212023 consisted of the following:
| | | | | | | | | | | | | | | |
| | | | | |
| | | | | |
| | | | | |
| Other income | | Year ended |
| (in € thousands) | | 2020/12/31 | | 2021/12/31 |
| CIR tax credit | | 6,020 | | 5,282 |
| Other operating income | | 968 | | 223 |
| Government grants and subsidies | | 5 | | 5 |
| TOTAL | | 6,993 | | 5,510 |
| | | | | | | | | | | | | | | |
| | | | | |
| | | | | |
| | | | | |
| Other income | | Year ended |
| (in € thousands) | | 2022/12/31 | | 2023/12/31 |
| CIR tax credit | | 6,017 | | 5,807 |
| Other operating income | | 320 | | 464 |
| Government grants and subsidies | | 34 | | 3,340 |
| TOTAL | | 6,371 | | 9,610 |
During the year ended December 31, 2020,Changes in other income amounted to €7.0 million.
During the year ended December 31, 2021 other income amounted to €5.5 million.
The decrease in Other Income compared to the previous year is mainly due to:
CIR tax credit
–Decreasing CIR tax credit (research tax credit granted by the French tax authorities) from €7.9€6.0 million for 2020, and2022 to €5.3€5.8 million for 20212023 due to lowerless eligible research and development activitiesexpenditures in 2021 compared to 2020; as2023. As a reminder, CIR tax credit for 2020 was partially balanced with an expense of €1.9 million related toresult, the settlement of a dispute regardingslight drop in the CIR in 2023 is mainly linked to a reduction in eligible personnel costs.
Note: There is currently a tax credit forinspection taking place by the years 2010, 2011, 2012 and 2014; andFrench tax authorities. For further information, see Note 11 - "Income tax". Other operating income
–DecreasingIncreasing foreign exchange gains related to trade receivables, which is included in the Operatingother operating income, whichand amounted to €0.2€0.5 million in 2021,2023, compared to €1.0€0.3 million in 2020.2022.
Government grants and subsidies
Operating Expenses
The tables below summarize our operating expenses for the years ended December 31, 20202022 and 2021.2023.
Operating Expenses for the Year Ended December 31, 20212023
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| Operating expenses and other operating income (expenses) | Year ended | | Of which : |
| | 2021/12/31 | | Raw | | Contracted | | Employee | | Other | Depreciation, | Gain / |
| | | | materials | | research and | | expenses | | expenses | amortization | (loss) on |
| | | | and | | development | | | | (maintenance, | and | disposal of |
| | | | consumables | | activities | | | | fees, travel, | impairment | property, |
| | | | used | | conducted by | | | | taxes…) | charges | plant and |
| (in € thousands) | | | | | third parties | | | | | | equipment |
| Research and development expenses | (35,166) | | (1,305) | | (18,808) | | (8,192) | | (4,593) | (2,247) | (19) |
| General and administrative expenses | (16,153) | | (161) | | (85) | | (7,379) | | (8,003) | (541) | 15 |
| Marketing and market access expenses | (1,539) | | (1) | | (1) | | (783) | | (741) | (13) | — |
| Reorganization and restructuring expenses | (142) | | (5) | | — | | — | | (2,343) | 2,206 | — |
| Other operating income (expenses) | (763) | | — | | — | | — | | (338) | 4 | (429) |
| TOTAL | (53,763) | | (1,472) | | (18,895) | | (16,354) | | (16,019) | (591) | (433) |
| (*) : including reversals | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| Operating expenses and other operating income (expenses) | Year ended | | Of which : |
| | 2023/12/31 | | Raw | | Contracted | | Employee | | Other | Depreciation, | Gain / |
| | | | materials | | research and | | expenses | | expenses | amortization | (loss) on |
| | | | and | | development | | | | (maintenance, | and | disposal of |
| | | | consumables | | activities | | | | fees, travel, | impairment | property, |
| | | | used | | conducted by | | | | taxes…) | charges | plant and |
| (in € thousands) | | | | | third parties | | | | | | equipment |
| Research and development expenses | (46,503) | | (1,831) | | (23,455) | | (12,475) | | (7,452) | (1,291) | — |
| General and administrative expenses | (17,741) | | (337) | | (205) | | (7,486) | | (9,396) | (317) | — |
| Marketing and market access expenses | (876) | | (4) | | (1) | | (556) | | (300) | (14) | — |
| Reorganization and restructuring expenses | 505 | | — | | — | | — | | — | 505 | — |
| Other operating income (expenses) | (141) | | — | | — | | — | | (222) | — | 81 |
| TOTAL | (64,756) | | (2,172) | | (23,661) | | (20,517) | | (17,370) | (1,117) | 81 |
| | | | | | | | | | | | |
Operating Expenses for the Year Ended December 31, 20202022
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| Operating expenses and other operating income (expenses) | Year ended | | Of which : |
| | 2020/12/31 | | Raw | | Contracted | | Employee | | Other | Depreciation, | Gain / |
| | | | materials | | research and | | expenses | | expenses | amortization | (loss) on |
| | | | and | | development | | | | (maintenance, | and | disposal of |
| | | | consumables | | activities | | | | fees, travel, | impairment | property, |
| | | | used | | conducted by | | | | taxes…) | charges | plant and |
| (in € thousands) | | | | | third parties | | | | | | equipment |
| Research and development expenses | (59,097) | | (1,876) | | (39,216) | | (11,554) | | (5,465) | (985) | — |
| General and administrative expenses | (14,270) | | (202) | | (92) | | (6,936) | | (6,545) | (495) | — |
| Marketing and market access expenses | (11,216) | | (7) | | (2) | | (1,298) | | (9,818) | (90) | — |
| Reorganization and restructuring expenses | (5,308) | | — | | — | | 8 | | (2,141) | (3,175) | — |
| Other operating income (expenses) | (764) | | — | | — | | — | | (684) | — | (80) |
| TOTAL | (90,655) | | (2,085) | | (39,310) | | (19,779) | | (24,655) | (4,746) | (80) |
| (*) : including reversals | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| Operating expenses and other operating income (expenses) | Year ended | | Of which : |
| | 2022/12/31 | | Raw | | Contracted | | Employee | | Other | Depreciation, | Gain / |
| | | | materials | | research and | | expenses | | expenses | amortization | (loss) on |
| | | | and | | development | | | | (maintenance, | and | disposal of |
| | | | consumables | | activities | | | | fees, travel, | impairment | property, |
| | | | used | | conducted by | | | | taxes…) | charges | plant and |
| (in € thousands) | | | | | third parties | | | | | | equipment |
| Research and development expenses | (35,818) | | (1,876) | | (17,407) | | (10,029) | | (5,177) | (1,328) | — |
| General and administrative income (expenses) | (16,405) | | (248) | | (71) | | (6,772) | | (9,168) | (146) | — |
| Marketing and market access expenses | (992) | | (3) | | (1) | | (565) | | (416) | (6) | — |
| Reorganization and restructuring income (expenses) | 11 | | — | | — | | — | | — | 11 | — |
| Other operating income (expenses) | (652) | | — | | — | | — | | (667) | — | 16 |
| TOTAL | (53,855) | | (2,128) | | (17,479) | | (17,366) | | (15,429) | (1,469) | 16 |
| | | | | | | | | | | | |
Research and Development Expenses
Research and development expenses totaled €59.1 million, or 65.2% of our total operating expenses, forFor the year ended December 31, 2020.2022, research and development expenses totaled €35.8 million, or 66.5% of our total operating expenses. These expenses consisted notablywere comprised of €39.2€17.4 million in contracted research and development conducted by third parties, €11.6€10.0 million in employee expenses, and €1.9€5.2 million in consumables. The clinical development costs related to the RESOLVE-IT Phase 3 trial evaluating elafibranorother expenses, €1.3 million in NASH remained high notwithstanding the decision made in July 2020 to terminate the study. The suspension or termination of some Phase 1 and Phase 2 trials related to NASH contributed to the decrease in contracting costs for 2020 compared to the previous year. To a lesser extent, the development costs related to the PBC and NTZ programs also generated contracting costs in 2020, but those were much less significant than the aforementioned trials.
We also incurred €11.6 million of employee-related expenses, consisting of wages, salaries, social security and pension costs and share-based compensation paid to employees in research and development functions. The changes in employee expenses for employees in research and development were mainly due to the reduction in force (82 vs. 127 employees) balanced with the change in employee profile, noting that bonuses granted to these employees for their involvement in the development of the Group affected these expenses in 2019 and such bonuses were not granted in 2020.
The depreciation, amortization and impairment charges amounted to €2.9 million reduced by the reversal of a provision ofand €1.9 million which was recognized in 2019 relating to the research tax credit dispute with the French revenue services.raw materials and consumables.
Research and development expenses totaled €35.2 million, or 65.4% of our total operating expenses, forFor the year ended December 31, 2021.2023, research and development expenses totaled €46.5 million, or 71.3% of our total operating expenses. These expenses were comprised €18.8of €23.5 million in contracted research and development conducted by third parties, and€12.5 million in employee expenses, €7.5 million in other expenses, €1.3 million in depreciation, amortization and impairment charges and €1.8 million in raw materials and consumables.
The clinicalincrease of €6.1 million in contracted research and development conducted by third parties is mainly due to:
–Increasing costs related to the RESOLVE-IT Phase 3 trial evaluating elafibranor in NASH continued to decrease over 2021 following the decision made in July 2020 to terminate the study. The suspension or terminationGNS 561 product candidate of some Phase 1 and Phase 2 trials related to NASH contributed to the decrease in contracting costs for 2021. The decrease in the development costs related to these trials in NASH was partly balanced with increasing development€4.8 million,
–Increasing costs related to the PBCVS-01 product candidate of €4.0 million,
–Decreasing costs related to the NTZ product candidate of €3.7 million,
–Decreasing costs related to the elafibranor program in Primary Sclerosing Cholangitis, or PSC, of €0.2 million,
–Increasing costs related to NIS4® and NTZ programs.its improvements of €0.1 million, and
We also incurred €8.2–Accrual reversals of €1.1 million related to the RESOLVE-IT® study in MASH in 2022 which did not repeat in 2023.
The increase of employee-related€2.5 million in employee expenses, consisting of wages, salaries, social security, and pension costs and share-based compensation paid to employees in the research and development functions. The changes in employee expenses for employees in research and development werefunction is mainly due to the reductionan increase in force (73workforce (91 employees in 2022 vs. 82 employees)96 employees in 2023).
The depreciation, amortization and impairment charges impacted the results byincrease of €2.2 million i.e. a €0.7in other expenses is mainly due to strategic consulting R&D services amounting to €1.4 million, decrease compared to 2020 (after adjustment for the reversal, in 2020,increased intellectual property costs of the €1.9€0.2 million, provisionincreased costs related to ELATIVE® of €0.2 million, and increased costs related to VS-01 and the settlementVersantis subsidiary of the research tax credit dispute with the French revenue services).€0.4 million.
We expect our research and development expenses to increase in the foreseeable future2024 compared to 2021,2023, as we continue our efforts to identify potential product candidates, conduct preclinical studies and clinical trials and advance the development of our diagnostic test.tests. They may fluctuate depending on the next steps initiated in the clinical development of our drug candidates, new development programs, which we may decide to start, and progress in the development of our diagnostic test. The impact of the RESOLVE-IT study in NASH on our 2022 results will be limited as the residual amount for termination costs is estimated to be lower than €0.5 million.tests.
General and Administrative Expenses
General and administrative expenses totaled €14.3 million, or 15.7% of our total operating expenses, forFor the year ended December 31, 2020.2022, general and administrative expenses totaled €16.4 million, or 30.5% of our total operating expenses. These expenses consistedwere composed primarily inof employee-related expenses, consisting of wages, salaries, social security and pension costs and share-based compensation paid to employees in general and administrative function for €6.9of €6.8 million, as well as €6.5€9.2 million in other expenses. The reduction in non-R&D and marketing employee expenses was mainly due to the reduction in force (— vs. — employees), and the reduction in compensation as bonuses granted to these employees for their involvement in the development of the Group which affected these expenses in 2019 and such bonuses were not granted in 2020, with the impact being partly balanced with the change in profile of these employees.
The impact of the insurance specific to the year of the IPO and costs following the listing on NASDAQ in 2019 mainly explain the relative decrease of general and administrative expenses in operational expenses for 2020.
General and administrative expenses totaled €16.2 million, or 30.0% of our total operating expenses, forFor the year ended December 31, 2021.2023, general and administrative expenses totaled €17.7 million, or 27.2% of our total operating expenses. These expenses consistedwere composed primarily inof employee-related expenses, consisting of wages, salaries, social security and pension costs and share-based compensation paid to employees in general and administrative function for €7.4of €7.5 million, as well as €8.0€9.4 million in other expenses.
The increase of €0.7 million in non-R&D and marketing employee expenses wasconsisting of wages, salaries, social security, pension costs and share-based compensation paid to employees in the general and administrative function is mainly due to thean increase in force (—workforce (57 employees in 2022 vs. — employees)63 employees in 2023).
The increase of €0.2 million in other expenses in the general and administrative expenses wasfunction is mainly due to increasing directors and officers insurance premiums.increased consulting fees related to the Versantis subsidiary.
The general and administrative expenses will remain significant over the next several years due to expenses associated with being a public company in the United States, including costs related to audit, legal, regulatory and tax-related services associated with maintaining compliance with U.S. exchange listing and SEC requirements, director and officer insurance premiums, investor relations and litigation costs. In particular, we will continue to incur additional expenses associated with accounting and internal control over financial reporting to comply with the Sarbanes-Oxley Act of 2002 in the United States.
In addition, we will also incur future expenses associated with the Corporate Sustainability Reporting Directive (CSRD).Marketing and Market Access Expenses
MarketingFor the year ended December 31, 2022, marketing and market access expenses totaled €11.2€1.0 million, or 12.4%1.8% of our total operating expenses, for the year ended December 31, 2020.expenses. These expenses consisted primarily of €10 million of other expenses, in particular, related to preparation for the potential marketing of elafibranor and NIS4 in NASH. The services performed include market surveys, brand strategy, medical communication and market access services. We also incurred €1.3 million in employee-related expenses, consisting of wages, salaries, social security and pension costs paid to employees in marketing and business development functions. This decrease of €1.5 million was primarily due to the cost saving plan linked to the termination of the development program of elafibranor in NASH.
Marketing and market access expenses totaled €1.5 million, or 2.9% of our total operating expenses, for the year ended December 31, 2021. These expenses consisted primarily of €0.7€0.4 million of other expenses, including market surveys, brand strategy, medical communication and market access services. We also incurred €0.8€0.6 million in employee-related expenses, consisting of wages, salaries, social security and pension costs paid to employees in marketing and business development functions.
This decrease of €9.7 million was primarily due to the cost saving plan linked to the termination of the development program of elafibranor in NASH, which allowed us to effectively terminate the other expenses related to preparation for the potential marketing of elafibranor in NASH. The reduction in marketing and market access employee expenses was mainly due to the reduction in force (5 vs. 2 employees).
We anticipate that our marketing and market access costs will not increase significantly in 2022.
Reorganization and Restructuration Expenses
Reorganization and restructuration expenses totaled €5.3 million, or 5.9% of our total operating expenses, forFor the year ended December 31, 2020.2023, marketing and market access expenses totaled €0.9 million, or 1.3% of our total operating expenses. These expenses consisted primarily of expenses and accruals related to employees within the scope of the reduction in force (Plan de Sauvegarde de l’Emploi or PSE) of €1.9 million, the part of renegotiation fees for the OCEANE recognized in 2020 for €0.8 million, the amortization and impairment loss of fixed assets recognized following the reorganization, the impairment of rights of use of leased premises and leased equipment notably for €2.2 million, and the provision of €0.4 million recognized for some termination costs of the RESOLVE-IT study.
Reorganization and restructuration expenses totaled €0.1 million, or 0.3% of our total operating expenses, for the year ended December 31, 2021. These expenses consisted primarily of the part of renegotiation fees for the OCEANE recognized in 2021 for €2.3 million, the reversal of the impairment of rights related to the use of leased premises for €0.7 million, following the relocation of our Paris office, and to leased equipment for €0.4 million, following the sale of certain equipment, the reversal of amortization and impairment loss related to fixed assets for €0.4 million, the reversal of accruals related to employees within the scope of the reduction in force (Plan de Sauvegarde de l’Emploi or PSE) for €0.4 million and the reversal of the provision of €0.4 million previously recognized for some termination costs of the RESOLVE-IT study.
Financial Income (Expense)
Our net financial expense for the year ended December 31, 2020 was €18.8 million, consisting primarily of €11.6 million of interest expense on our convertible bonds, and €13.5 million of foreign exchange losses, offset partially by €5.0 million in foreign exchange gain on cash and cash equivalents and €1.4 million in interest income. The exchange result was a loss of €8.5 million and is notably related to the exchange rate fluctuations on the cash held in US dollars, as the Company made the decision to keep part of its cash in US dollars. These cash holdings in US dollars are to be used to pay directly expenses in US dollars (natural currency hedge).
Our net financial income for the year ended December 31, 2021 was €37.7 million, consisting primarily of the financial income of €35.6 million corresponding to a repurchase bonus following the renegotiation of the OCEANEs in January 2021, €4.8 million of interest expense on our convertible bonds, €8.9 million in foreign exchange gain on cash and cash equivalents, offset partially by €2.2 million of foreign exchange losses, and €0.3 million in interest income.
The exchange result was a gain of €6.7 million and is notably related to the exchange rate fluctuations on the cash held in US dollars, as the Company made the decision to keep part of its cash in US dollars. These cash holdings in US dollars are to be used to pay directly expenses in US dollars (natural currency hedge).
Comparisons for the Years Ended December 31, 2019 and 2020
Revenue
Revenue of €30.8 million during the year ended December 31, 2019 related to the recognition of the revenue related to the license transferred to Terns under the Terns licensing agreement, after deduction of deferred revenue. Deferred revenue amounted to €0.1 million, which corresponds to our expected revenue in relation with the costs to be incurred to assist Terns under the Terns licensing agreement.
Revenue of €0.8 million during the year ended December 31, 2020 was the result of a one-time transaction and the recognition of the revenue under the Labcorp licensing agreements. The decrease in revenue compared to 2019 is due to the one-time nature of the payment for a licensing agreement with Terns recognized in 2019.
Other Income
Other income for the years ended December 31, 2019 and 2020 consisted of the following:
| | | | | | | | | | | | | | | |
| | | | | |
| | | | | |
| | | | | |
| Other income | | Year ended |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 |
| CIR tax credit | | 8,125 | | 6,020 |
| Other operating income | | 1,992 | | 968 |
| Government grants and subsidies | | 5 | | 5 |
| TOTAL | | 10,122 | | 6,993 |
During the year ended December 31, 2019 we had foreign exchange gains linked to trade receivables linked to services denominated in U.S. dollars, which amounted to €2.0 million and are recorded as Other operating income (see table above ).
During the year ended December 31, 2020, other income amounted to €7.0 million. The decrease compared to the previous year is mainly due to:
–the effect of the expense recorded in 2020 related to the end of the research tax credit dispute for 2010, 2011, 2012 and 2014. This expense was balanced with the reversal of the provision recognized in 2019. The research tax credit for the year 2020 amounted to €7.9 million; and
–the decrease in foreign exchange gains related to trade receivables, which amounted to €1.0 million and are recorded as Other operating income.
Operating Expenses
The tables below summarize our operating expenses for the years ended December 31, 2019 and 2020.
Operating Expenses for the Year Ended December 31, 2019
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| Operating expenses and other operating income (expenses) | Year ended | | Of which : |
| | 2019/12/31 | | Raw | | Contracted | | Employee | | Other | | Depreciation, | | Gain / |
| | | | materials | | research and | | expenses | | expenses | | amortization | | (loss) on |
| | | | and | | development | | | | (maintenance, | | and | | disposal of |
| | | | consumables | | activities | | | | fees, travel, | | impairment | | property, |
| | | | used | | conducted by | | | | taxes…) | | charges | | plant and |
| (in € thousands) | | | | | third parties | | | | | | | | equipment |
| Research and development expenses | (66,170) | | (2,017) | | (41,509) | | (11,740) | | (6,188) | | (4,716) | | 0 |
| General and administrative expenses | (17,265) | | (177) | | (59) | | (7,598) | | (8,972) | | (458) | | 0 |
| Marketing and market access expenses | (13,708) | | (8) | | 0 | | (1,645) | | (11,979) | | (76) | | 0 |
| Reorganization and restructuring expenses | 0 | | 0 | | 0 | | 0 | | 0 | | 0 | | 0 |
| Other operating income (expenses) | (1,649) | | 0 | | 0 | | 0 | | (1,668) | | 0 | | 19 |
| TOTAL | (98,793) | | (2,202) | | (41,568) | | (20,984) | | (28,807) | | (5,251) | | 19 |
| (*) : including reversals | | | | | | | | | | | | | |
Operating Expenses for the Year Ended December 31, 2020
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| Operating expenses and other operating income (expenses) | Year ended | | Of which : |
| | 2020/12/31 | | Raw | | Contracted | | Employee | | Other | Depreciation, | Gain / |
| | | | materials | | research and | | expenses | | expenses | amortization | (loss) on |
| | | | and | | development | | | | (maintenance, | and | disposal of |
| | | | consumables | | activities | | | | fees, travel, | impairment | property, |
| | | | used | | conducted by | | | | taxes…) | charges | plant and |
| (in € thousands) | | | | | third parties | | | | | | equipment |
| Research and development expenses | (59,097) | | (1,876) | | (39,216) | | (11,554) | | (5,465) | (985) | — |
| General and administrative expenses | (14,270) | | (202) | | (92) | | (6,936) | | (6,545) | (495) | — |
| Marketing and market access expenses | (11,216) | | (7) | | (2) | | (1,298) | | (9,818) | (90) | — |
| Reorganization and restructuring expenses | (5,308) | | — | | — | | 8 | | (2,141) | (3,175) | — |
| Other operating income (expenses) | (764) | | — | | — | | — | | (684) | — | (80) |
| TOTAL | (90,655) | | (2,085) | | (39,310) | | (19,779) | | (24,655) | (4,746) | (80) |
| (*) : including reversals | | | | | | | | | | | |
Research and Development Expenses
Research and development expenses totaled €66.2 million, or 67% of our total operating expenses, for the year ended December 31, 2019. These expenses consisted primarily of €41.5 million in contracted research and development conducted by third parties, the substantial majority of which were incurred in connection with the progression of our RESOLVE-IT Phase 3 trial. The clinical development costs related to the RESOLVE-IT Phase 3 trial, were lower in 2019 than in 2018 due in particular to a revised estimate of expensed yet unbilled investigator costs which led a decrease in costs of €7.0 million. To a lesser extent, the development costs related to the PBC and NTZ programs also generated subcontracting costs in 2019.
We also incurred €11.7 million of employee-related expenses, consisting of wages, salaries, social security and pension costs and share-based compensation paid to employees in research and development functions and patents. This increase of €2.3 million of employee-related expenses over the prior year was primarily due to changes in seniority, increase in headcount, wage increases and bonuses for our workforce in the research and development functions. Other expenses of €6.2 million consisted primarily of maintenance and other facility costs, as well as employee travel expenses and third-party fees incurred for seconded employees in research and development functions.
The depreciation, amortization and impairment charges totaled €4.7 million, consisting of a provision of €1.9 million with respect to the research tax credit (more information is provided in Note 20 of our consolidated financial statements for the year ended December 31, 2020), and due to additional depreciation due to the adoption of IFRS 16.Research and development expenses totaled €59.1 million, or 65% of our total operating expenses, for the year ended December 31, 2020. These expenses consisted primarily of €39.2 million in contracted research and development conducted by third parties and €1.9 million of consumables. The clinical development costs related to the RESOLVE-IT Phase 3 trial evaluating elafibranor in NASH remained high notwithstanding the decision made in July 2020 to terminate the study. The suspension or termination of some Phase 1 and Phase 2 trials related to NASH contributed to the decrease in contracting costs for 2020. To a lesser extent, the development costs related to the PBC and NTZ programs also generated contracting costs in 2020, but those were much less significant than the aforementioned trials.
We also incurred €11.6 million of employee-related expenses, consisting of wages, salaries, social security and pension costs and share-based compensation paid to employees in research and development functions. The changes in employee expenses for employees in research and development are mainly due to the reduction in force (82 vs. 127 employees) balanced with the change in employee profile, noting that bonuses granted to these employees for their involvement in the development of the Group affected these expenses in 2019 and such bonuses were not granted in 2020.
The changes in depreciation, amortization and impairment charges is mainly related to the provision of €1.9 million recognized in 2019 due to the research tax credit dispute with the French revenue services and reversed in 2020, and to the consequence of the application of IFRS16 in 2019, while in 2020, the amortization impacted the results for up to €2.9 million.
We expect our research and development expenses to decrease in the foreseeable future compared to 2020 . They will nonetheless remain significant and may increase depending on the next steps initiated in the clinical development of our drug candidates and progress in the development of our diagnostic test. Moreover, the RESOLVE-IT study in NASH will continue to impact our 2021 results insofar as the residual amount for termination costs is estimated to be between €8 and 10 million in 2021.
General and Administrative Expenses
General and administrative expenses totaled €17.3 million, or 17% of our total operating expenses, for the year ended December 31, 2019. These expenses consisted primarily of €9.0 million of other expenses, in particular, related to the insurance costs and audit and communication costs and third-party fees incurred for seconded employees in general and administrative functions. This increase compared to 2018 of €3.5 million was primarily due to an increase related to the insurance costs and costs following the listing on NASDAQ .We also incurred €7.6 million in employee-related expenses, consisting of wages, salaries, social security and pension costs and share-based compensation paid to employees in general and administrative functions. This increase compared to 2018 of €3.8 million was primarily due to an increase in headcount, changes in seniority, wage increases and to the bonuses paid to employees in those functions.
General and administrative expenses totaled €14.3 million, or 15.7% of our total operating expenses, for the year ended December 31, 2020. These expenses consisted primarily in employee-related expenses, consisting of wages, salaries, social security and pension costs and share-based compensation paid to employees in general and administrative function for €6.9 million, as well as €6.5 million in other expenses. The reduction in non-R&D and marketing employee expenses was mainly due to the reduction in force (— vs. — employees), and the reduction in compensation as bonuses granted to these employees for their involvement in the development of the Group which affected these expenses in 2019 and such bonuses were not granted in 2020, with the impact being partly balanced with the change in profile of these employees.
The impact of the insurance specific to the year of the IPO and costs following the listing on NASDAQ in 2019 mainly explain the relative decrease of general and administrative expenses in operational expenses for 2020.
We anticipate that the cost-saving plan initiated in September 2020 will lead to a decrease in general and administrative expenses over 2021. However, they will remain significant due to expenses associated with being a public company in the United States, including costs related to audit, legal, regulatory and tax-related services associated with maintaining compliance with U.S. exchange listing and SEC requirements, director and officer insurance premiums, investor relations and litigation costs. In particular, we will continue to incur additional accounting expenses to comply with the Sarbanes-Oxley Act of 2002 in the United States.
Marketing and Market Access Expenses
Marketing and market access expenses totaled €13.7 million, or 14% of our total operating expenses, for the year ended December 31, 2019. These expenses consisted primarily of €12.0 million of other expenses, in particular, related to preparation for the potential marketing of elafibranor and NIS4 in NASH. The services performed include market surveys, brand strategy, medical communication and market access services. We also incurred €1.6€0.6 million in employee-related expenses, consisting of wages, salaries, social security and pension costs paid to employees in marketing and business development functions. This increase of €1.2 million was primarily due to an increase in headcount, changes in seniority, wage increases.
Marketing and market access expenses totaled €11.2 million, or 12% of our total operating expenses, for the year ended December 31, 2020. These expenses consisted primarily of €10 million of other expenses, in particular, related to preparation for the potential marketing of elafibranor and NIS4 in NASH. The services performed include market surveys, brand strategy, medical communication and market access services. We also incurred €1.3 million in employee-related expenses, consisting of wages, salaries, social security and pension costs paid to employees in marketing and business development functions. This decrease of €1.5 million was primarily due to the cost saving plan linked to the termination of the development program of elafibranor in NASH.
We expectdo not anticipate that the decrease in our marketing and market access costs initiated in the summer of 2020 with our cost-saving plan will continue in 2021.increase significantly.
Reorganization and Restructuration Expenses
ReorganizationFor the year ended December 31, 2022, reorganization and restructuration expenses included mainly expenses and accruals related to employees withinwere not significant.
For the scopeyear ended December 31, 2023, the €0.5 million amount consisted of unused office space provision reversals as the RESOLVE-IT® study is complete. As a reminder, a provision was recorded in 2020 as part of the reduction in force (Plan de Sauvegarde de l’Emploi or PSE) of €1.9 million, the part of renegotiation fees for the OCEANE recognized in 2020 for €0.8 million, the amortization and impairment loss of fixed assets recognizedreorganization following the reorganization, the impairment of rights of use of leased premises and leased equipment notably for €2.2 million, and the provision of €0.4 million recognized for some termination costs of the RESOLVE-IT study.
Financial Income (Expense)
Our net financial income (expense) for the year ended December 31, 20192022 was €(7.9)€3.5 million, consisting primarily of €11.3 million of interest expense on our convertible bonds, and €1.7 million of foreign exchange losses, offset partially by € 2.3€7.5 million in foreign exchange gain on cash and cash equivalents and €2.6€0.7 million in interest income. The increaseincome, partially offset by €4.3 million of interest expense, €0.3 million in foreign exchange losses, and €0.1 million in other financial income is due to the increase in interest on term accounts as we keep some of our cash in US dollars. Interest rates received on investments in US dollars were higher than for investments in euros.
Our net financial expenseloss for the year ended December 31, 20202023 was €(18.8)€1.9 million, consisting primarily of €11.6 million of interest expense on our convertible bonds, and €13.5 million of foreign exchange losses, offset partially by €5€0.5 million in foreign exchange gain on cash and cash equivalents, and €1.4€3.2 million in interest income. income, offset by €4.6 million of interest expense, and €1.0 million in foreign exchange losses.
The foreign exchange result was a net loss of €8.5€0.5 million and is notably related to the exchange rate fluctuations on the cash held in USU.S. dollars, as the Companywe made the decision to keep part of itsour cash in USU.S. dollars. These cash holdings in USU.S. dollars are to be used to pay directly expenses in USU.S. dollars directly (natural currency hedge).
| | | | | |
| B. | Liquidity and Capital Resources |
Overview
As of December 31, 2019, 20202022 and 2021,2023, we had €276.7 million, €171.0€136.0 million and €258.8€77.8 million respectively, in cash and cash equivalents. In addition, as of December 31, 2022, we had €4.6 million in other current financial assets which consisted of a single short-term instrument whose term was 180 days. Cash, cash equivalents, and other current financial assets are used to finance key business activities, notably research and developments expenses.
Since our inception, we have financed our operations primarily through the issuance of new ordinary shares and bonds convertible into new ordinary shares in public offerings and private financing transactions.transactions, as well as an upfront payment pursuant to our collaboration with Ipsen. In 2006, we completed the initial public offering of our ordinary shares on the Alternext market of Euronext in Paris. The listing of our ordinary shares was transferred to the regulated market of Euronext Paris in 2014. Between 2010 and 2016, we raised a total of over €220.0 million in gross proceeds from the issuance of additional ordinary shares for cash. In October 2017, we issued €180.0 million in bonds convertible into new ordinary shares or exchangeable for existing ordinary shares. In March 2019, we completed a global offering consisting of an initial public offering of our American Depositary Shares, or ADSs in the United States, and a private placement of our ordinary shares in Europe and other countries outside the United States, including France. Aggregate gross proceeds from the global offering, before deducting underwriting discounts and commissions and offering expenses paid by us, were approximately $155.4 million. Additionally, in 2021, Ipsen also became a shareholder of GENFIT through the purchase of 3,985,239 newly issued shares representing 8% of GENFIT S.A after issuance, via a €28 million investment. There have been no subsequent equity raises.
We have also financed our operations through historical collaborative research alliances, as well as research tax credits and subsidies granted by various public institutions, such as BPI France Institutions. We also entered into conditionalthe Terns Agreement and repayable advances agreements with governmental entities and had a liability of €3.2 million, €3.2 million and €3.2 million associated with these types of arrangements as of December 31, 2019, 2020 and 2021, respectively. Additional information is provided in the note 12Ipsen Agreement. Pursuant to our consolidated financial statements under the captions “Subsidies and Refundable and Conditional Advances” and “Loans and Borrowings”. We also entered into loans with commercial banks and BPI France. In 2021,Terns Agreement, we entered into three bank loans for a total nominal amount of €15.2 million, granted in the context of the COVID-19 pandemic, including:
•A €11.0 million loan in June 2021 by a pool of four French commercial banks,
•A €2.0 million loan in July 2021 by BPI France,
both of which are 90% guaranteed by the French government (State-Guaranteed Loans or Prêts Garantis par l’Etat "PGE") and carry an initial term of one year with repayment options up to six years, as well as a €2.2 million subsidized loan in November 2021 by BPI France, with an initial term of six years.
Our loans with commercial banks and BPI France had an outstanding balance of €2.6 million, €1.5 million and €15.8 million as of December 31, 2019, 2020 and 2021, respectively.
In 2019, our cash and cash equivalents were increased byreceived an upfront payment of $35 million of which $34.9in 2019. Pursuant to the Ipsen Agreement, we received a €120 million was recognized as revenue in 2019, pursuant to a licensing and collaboration agreement with Terns.
In 2021, our cash and cash equivalents were also increased significantly by an upfront payment of €120 million,in 2021, out of which €80 million was recognized as revenue in 2021, pursuant to a licensing and collaboration agreement with Ipsen, after deduction of €40 million was deducted as deferred revenue, which will be recognized as revenue following the completion of the ELATIVE double-blind study, in accordance with IFRS 15.revenue. We also received €28 million from Ipsen as a result of their purchase of an 8% equity stake in GENFIT,
Followingus in 2021. During the resultsyears ended December 31, 2022 and 2023, we recognized €15.9 million and €8.7 million respectively, out of the Phase 3 RESOLVE-IT trial,€40.0 million deferred income from Ipsen. In addition, during the year ended December 31, 2023, we implementedrecognized a cost savings plan to reduce operational expenses, including a workforce reduction plan and eliminate non-essential expenses, which contributed to reducing our cash flows used in operating activities from €96.4€13.3 million in 2020 to €44.0 million in 2021 (excluding the upfront payment received from Ipsen in 2021). We will incur higher expenses and substantial operating losses over the next several years, as we continue our efforts to identify potential product candidates, conduct preclinical studies and clinical trials and advance the development of diagnostic tests based on our NIS4 technology. We expect that our cash flows used in operating activities will increase to €65 million in 2022 (excluding VAT, corporate tax and employee profit-sharing to be paid in 2022 relatedmilestone pursuant to the initial upfront received from Ipsen inAgreement following the previous year). This estimate takes into accountNDA filing acceptance by the FDA and MAA filing acceptance by the EMA for accelerated approval of elafibranor.
Additionally, we have financed our projected cash flow from operations and government fundingthrough the receipt of research programs. We have based this estimate on assumptions that may prove to be wrongtax credits and we could use our available capital resources sooner than we currently expect.
In light of this, we will likely continue relying on some or all of these sources of financing, as well as potential milestone payments and royalties that may result from licensing agreements for our drug candidates, diagnostic solutions and results of our research programs,subsidies granted by various public institutions, such as ourBPI France, conditional and repayable advances agreements with Ipsen, Labcorpgovernmental entities, loans with commercial banks and Terns.BPI France and the issuance of convertible bonds.
Cash Flows
The table below summarizes our cash flows for the years ended December 31, 2019, 20202021, 2022 and 2021:2023:
| | | | | | | | | | | | | | | | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | Year ended |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Cash flows provided by (used in) operating activities | | (47,680) | | (96,371) | | 99,915 |
| Cash flows provided by (used in) investment activities | | 327 | | (966) | | (3,377) |
| Cash flows provided by (used in) financing activities | | 116,860 | | (8,256) | | (8,916) |
| | | 69,507 | | (105,593) | | 87,622 |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | Year ended |
| (in € thousands) | | | | 2021/12/31 | 2022/12/31 | | 2023/12/31 |
| Cash flows provided by (used in) operating activities | | | | 99,915 | (72,638) | | (55,429) |
| Cash flows provided by (used in) investment activities | | | | (3,377) | (46,266) | | 2,234 |
| Cash flows provided by (used in) financing activities | | | | (8,916) | (3,786) | | (5,098) |
| | | | | 87,622 | (122,690) | | (58,293) |
Operating Activities
Cash used inprovided by (used in) operating activities was €47.7€99.9 million, €96.4€(72.6) million and €(55.4) million for the years ended December 31, 20192021, 2022 and 2020 respectively and cash provided by operating activities was €99.9 million for2023, respectively.
For the year ended December 31,2021.
With respect to the 20192022 period, this amount primarily resulted from our net loss of €65.1€23.7 million, largely the result of ourin addition to significant research and development efforts as we progressed our Phase 3 clinical trial of elafibranor in NASHincluding ELATIVE®, GNS561, and prepared for the potential commercialization of elafibranor in NASH,VS-01, adjusted by €17.8€6. million in non-cash and financial expenses, a decrease in payables and other adjustmentsliabilities of €-0.4€46.2 million, and an increase in receivables of €8.6 million.
With respect Specifically regarding the decrease in payables and other liabilities, this is primarily attributable to a one-time payment in early 2022 amounting to €24.0 million of VAT related to the 2020Ipsen upfront initial payment of €120 million from 2021.
For the 2023 period, this amount primarily resulted from our net loss of €101.2€28.9 million, largely the result of ourin addition to significant research and development efforts as we incurred significant costs for RESOLVE-IT, our Phase 3 clinical trialincluding ELATIVE®, GNS561, and VS-01, an increase in receivables of elafibranor€17.4 million, a decrease in NASH,payables and before implementing the cost saving plan during the summer 2020, for the preparation for the potential commercializationother liabilities of elafibranor in NASH,€10.4 million, taxes paid of €0.5 million, and adjusted by €16€1.8 million in non-cash and financial expenses and other adjustments of €-11.1 million.
With respect toexpenses. Specifically regarding the 2021 period,increase in receivables, this amountis primarily resulted from our net profit of €67.3 million largely the result of :
•The receipt of a €120 million initial upfront payment pursuantattributable to a licensing and collaboration agreement with Ipsen,
•Our significant research and development efforts as we incurred significant costs for ELATIVE, our Phase 3 clinical trial of elafibranormilestone payment recognized in PBC,
•Adjusted by (€27,0)2023 amounting to €13.3 million in non-cash and financial expenses, including the recognition of a €35.6 million repurchase bonus related to Ipsen as discussed in the partial buyback of our convertible bonds,revenue related paragraphs in the above section entitled "Comparisons for the Years Ended December 31, 2022 and
•Other adjustments of €59.7 million, including the recognition of a €40.0 million deferred income and €24.0 million of VAT collected, both of which are related to the initial upfront payment from Ipsen. 2023."
Investing Activities
Cash provided by investing(used in) investment activities was €0.3€(3.4) million, €(46.3) million and €2.2 million for the year ended December 31, 2019, due to the reimbursement by the landlord of the costs associated with the expansion of our corporate headquarters when construction was completed in April 2019 . Cash used in investing activities was €1 million for the year ended December 31, 2020 and consisted primarily of equipment. Cash used in investing activities was €3.4 million for the yearyears ended December 31, 2021, 2022 and 2023, respectively.
For the 2022 period, this consisted primarily of the
€3.0Versantis acquisition of €41.5 million
subscription(net of
new ordinary sharescash acquired) and short term investments of
Genoscience Pharma (more information is provided in Note 8€5.0 million. For the 2023 period, this consisted primarily of our consolidated financial statements, included in this report).a liquidation of a short term investment of €4.6 million offset by acquisitions of intangible assets and property, plant and equipment (net of disposals) of €2.3 million.
Financing Activities
For the 2019 period, cashCash provided by (used in) financing activities was €116.9€(8.9) million, €(3.8) million and primarily consisted of €125.3€(5.1) million in net proceeds fromfor the March 2019 global offering, partially offset by the repayment of loansyears ended December 31, 2021, 2022 and borrowings and including the impact of lease payments due to the implementation of IFRS16 on January 1, 2019.2023, respectively.
For the 20202022 period, cash used in financing activities was €8.3 million, whichthis amount consisted primarily of (€7.8)€2.2 million in interest paid on our convertible bondsdebt and (€2.1)€1.7 million in repayments of loans and lease repayments.repayments, offset by €0.1 million in financial interest payments received.
For the 20212023 period, cash used in financing activities was €8.9 million, whichthis amount consisted primarily of (€47.5) million used for the partial buy-back of our OCEANEs, €28.0 million of equity investment received from Ipsen, €15,2 million provided by new bank loans, and (€4.8)€2.2 million in interest paid on our convertible bonds.debt and €4.6 million in repayments of loans and lease repayments, offset by €1.7 million in financial interest payments received.
Restriction on use of capital
With the exception of deposits and guarantees (€0.3 million) recognized in non-current and current financial assets as of December 31, 2023, the Company is not faced with any restrictions as to the availability of its capital.
Currencies
Operating and Capital Expenditure Requirements
Since our inception, we have incurred significant operating losses. Our net loss was €65.1€23.7 million and €101.2€28.9 million for the years ended December 31,201931, 2022 and 2020,2023, respectively. For the year ended December 31, 2021 we had net profit of €67.3 million owing to the upfront payment received from Ipsen. Following the results of the Phase 3 RESOLVE-IT trial in May 2020, we implemented a cost savings plan to reduce operational expenses, including a workforce reduction plan and eliminate non-essential expenses. Nevertheless, weWe expect to incur higher expenses and substantial operating losses over the next several years, as we:
•initiate and conduct our planned preclinical studies and clinical trials of our drug candidates, including ELATIVE,in particular, our Phase 32 clinical trial of elafibranorVS-01 for the treatment of PBC,ACLF and our Phase 1 clinical1b/2 trial of NTZ for the treatmentGNS561 in CCA, as well as pre-clinical development of ACLF;SRT-15 and CLM-022 ;
•continue and complete the validation and development of NIS4NIS4® and its improvements for NASH;diagnosis of at-risk MASH;
•continue the research and development of our other drug candidates, including planned and future preclinical studies and clinical trials, notably a Phase 2 clinical program of GNS561 in CCA expected to start in the first half of 2022;trials;
•seek to discover and develop additional drug candidates and explore combination therapies for our existing drug candidates;
•continue our efforts to identify potential product candidates,.candidates;
•seek regulatory approval for an IVD powered by NIS4NIS4® or its variations and any drug candidates that successfully complete clinical trials;
•assist with the scale-up of our subcontractors’ manufacturing capabilities in order to support the launch of additional clinical trials and the commercialization of our drug candidates, if approved;
•establish a sales and marketing infrastructure for the commercialization of our drug candidates and diagnostic candidates, if approved, in certain geographies, either on our own or in partnership with a third party;
•maintain, expand and protect our intellectual property portfolio;
•hire additional clinical, quality control and scientific personnel; and
•add operational, financial and management information systems and personnel, including personnel to support our product development and commercialization efforts and our operations as a public company listed in the United States.
Our present and future funding requirements will depend on many factors, including, among other things:
•the size, progress, timing and completion of our clinical trials of elafibranor and our other current or future product candidates;
•the number of potential new product candidates we identify and decide to develop;
•the costs involved in filing patent applications and maintaining and enforcing patents or defending against claims of infringement raised by third parties;
•the time and costs involved in obtaining regulatory approval for our product candidates and any delays we may encounter as a result of evolving regulatory requirements or adverse results with respect to any of these product candidates;
•selling and marketing activities undertaken in connection with the anticipated commercialization of elafibranor and our other current or future product candidates, including other product candidates in preclinical development, together with the costs involved in the creation of an effective sales and marketing organization; and
•the amount of revenues, if any, we may derive either directly, or in the form of royalty payments from any future potential collaboration agreements.
Until such time, if ever, that we can generate substantial revenue from product sales, we expect to finance these expenses and our operating activities through a combination of our existing liquidity, equity offerings, debt financings, collaborations, strategic alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a holder of ordinary shares or ADSs. Debt financing, if available, may involve agreements that include covenants that would further limit or restrict our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends.
If we raise funds through additional collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us.
If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves, which could materially adversely affect our business, financial condition and results of operations.
We believe that our existing cash and cash equivalents as of December 31, 2021,2023, will enable us to fund our operating expenses and capital expenditure requirements for at least the next 12 months at least. We have based this estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect.
For more information as to the risks associated with our future funding needs, see the section of this annual report titled “Risk Factors.”
Consistent with customary practice in the French securities market, we entered into a liquidity agreement (contrat de liquidité) with Crédit Industriel et Commercial S.A. ("CIC") in August 2013. The liquidity agreement was entered into in accordance with applicable laws and regulations in France. The liquidity agreement authorizes CIC to carry out market purchases and sales of our shares on Euronext Paris. The amount is classified in other non-current financial assets in our statement of financial position. At December 31, 2023, 147,812 shares and $0.5 million were in the liquidity account. The liquidity agreement has a term of one year and will renew automatically unless otherwise terminated by either party. On a half-yearly basis, we provide a report on the use of the liquidity contract through a press release.
Disclosure of Contractual Obligations
We enter into contracts in the normal course of business with CROs and contract manufacturing organizations, or CMOs, for clinical trials, preclinical studies and clinical manufacturing, and with vendors for pre-commercial activities, research and development activities, research supplies and other services and products for operating purposes. These contracts generally provide for termination upon notice. Such agreements may be terminated at will.
We have entered into a licensing agreement with Genoscience Pharma whereby we are obligated to pay royalties and milestone payments based on future events that are uncertain and therefore they constitute contingent liabilities not recognized in our consolidated financial statements for the period ending December 31, 2021.2023.
We have entered into a share purchase agreement with the former shareholders of Versantis AG whereby we are obligated to pay milestone payments based on future events that are uncertain and therefore they constitute contingent liabilities not recognized in our consolidated financial statements for the period ending December 31, 2023.
We have entered into a licensing agreement with Seal Rock Therapeutics pursuant to which we may be required to pay royalties and milestone payments based on future events that are uncertain and therefore they constitute contingent liabilities not recognized in our consolidated financial statements for the period ending December 31, 2023.
We have entered into a licensing agreement with Celloram, Inc. pursuant to which we may be required to pay royalties and milestone payments based on future events that are uncertain and therefore they constitute contingent liabilities not recognized in our consolidated financial statements for the period ending December 31, 2023.
Subsidies and Refundable and Conditional Advances
We have received financial assistance from BPI France, and other governmental organizations in connection with the development of our product candidates. BPI France’s mission is to provide assistance and support to emerging French enterprises to facilitate the development and commercialization of innovative technologies. Such funding, in the form of refundable and conditional advances, is intended to finance our research and development efforts and the recruitment of specific personnel.
We account for non-refundable subsidies as other income ratably over the duration of the funded project. Funds received in the form of refundable advances are recognized as financial liabilities, as we are obligated to reimburse BPI France for such refundable advances in cash based on a repayment schedule if specified conditions are met.
As of December 31, 2019, December 31, 2020 and December 31, 2021 respectively,2022, we had one outstanding repayable advance from BPI France with an aggregate remaining balance of €3.2 million. This advance, in an amount of €3.2 million, is a conditional advance we received in our capacity as leader of a research consortium initiated in 2008 called IT-DIAB to follow patients at risk for type 2 diabetes. The program ended on December 31, 2014. The conditional advance is not refundable except in the event of technical or commercial success of the consortium’s activities, defined as the sale of related drugs or diagnostic devices developed using research results. We would then be required to repay the advance, plus an additional specified amount, based on a percentage of any revenues generated from the licensing of such products over a 10-year period. The maximum amount that we would behave been required to pay under this arrangement iswas €14.8 million, inclusive of the €3.2 million advance to be repaid.
As provided in the contract, we sent a letter to BPI France in December 2019 in order to notify it of our Labcorp and Terns contracts while indicating that elafibranor was now aimed at treating hepatic diseases and no longer type 2 diabetes as provided for in the agreement. We proposed to BPI France to acknowledge the failure of the IT DIABIT-DIAB project. Following this letter, the parties met in March 2020 for the presentation of our arguments, and were in contact again in June 2020 following the results of the RESOLVE ITRESOLVE-IT® trial. We sent another letter in November 2020. We are awaiting a proposal from
On October 20, 2023, BPI France on newagreed to formally recognize the failure of the project and therefore write off their outstanding receivable as previously mentioned. As of December 31, 2023, GENFIT had no remaining obligation associated with this, and thus the liability was reversed with the amount recognized in "Other income" in the 2023 consolidated statement of operations. Refer to Note 20.2.1 - "Breakdown of other loans and borrowings - Refundable and conditional advances" to our consolidated financial terms related tostatements included in this situation and a draft amendment to the repayable advance agreement.
Convertible Bonds
In October 2017, we issued convertible bonds (OCEANEs) for gross proceeds of €180.0 million. The convertible bonds carrymillion, with a fixed interest ratematurity date initially of 3.5%, with an effective interest rate of 7.2%, payable semi-annually in arrears in April and October.October 16, 2022.
On November 23, 2020, we presented to all OCEANEOCEANEs bondholders a two-prong renegotiation offer:
•A partial buyback of the outstanding OCEANEs for a maximum amount of 3,048,780 OCEANEs at €16.40 per bond; and
•An amendment of the terms of the remaining OCEANEs to extend their maturity (by 3three years) and increase the conversion ratio (to 5.5 shares per bond).
At the Shareholders’ and Bondholders’ Meetings on January 25, 2021, the shareholders and bondholders approved this renegotiation offer.
Following the shareholders’offer and bondholders’ decisions, GENFITwe completed the partial buyback of 2,895,260 OCEANEs at a price of €16.40 (including accrued interest of €0.30) per bond for a total buyback cost of €47.48 million. The settlement operations occurredmillion on January 29, 2021. The repurchased OCEANEs wereWe then cancelled by GENFIT.the repurchase of OCEANEs. Following the renegotiation, the OCEANEs bear interest at an annual nominal rate of 3.50% payable semi-annually in arrears on April 16 and October 16 of each year (or the following business day if this date is not a business day). The OCEANEs will be redeemed at par on October 16, 2025 (or the following business day if this date is not a business day). The effective interest rate is 8.8%.
We incurred and recognized fees related to this renegotiation (including financial advising, counsel fees and meeting costs) in the amount of €0.7 million and €2.3 million in 2020 and 2021 respectively.
Following conversionThe nominal unit value of the OCEANEs intowas set at €29.60. The OCEANEs conversion ratio is 5.5 shares up until April 1, 2022, which ledfor one OCEANE, subject to any subsequent adjustments.
The OCEANEs may be redeemed early at the creationoption of 6,941,875 newthe Company, under certain conditions. Specifically, the OCEANEs may be redeemed early at the option of the Company from November 3, 2023 onward if i) the mathematical average of the volume-weighted average price of GENFIT shares on the residualregulated market of Euronext in Paris and ii) the conversion ratio of the shares in force (over a period of 20 trading days) exceeds 150% of the nominal convertible debt, initially reducedvalue of the OCEANEs bonds.
As of December 31, 2023, there were 1,923,662 OCEANEs outstanding, and the maximum dilution to a nominal amountGENFIT's current share capital in the event of €94.3 million through the partial buyback transaction, was further reduced by a nominal amount of €37.4 million,full conversion would be 21.29%, with approximately €56.9 million nominal amount outstanding asoutstanding.
The OCEANEs are admitted to trading on Euronext Access (the free market of April 1, 2022.Euronext in Paris).
Bank Loans
At December 31, 2019, 2020 and 2021, we hadWe have borrowed under multiple bank loans primarily intended to finance the acquisition of scientific and information technology equipment forequipment. As of December 31, 2022 and 2023, the total principal amount outstanding of €2.6 million, €1.5was €15.2 million and €15.9€11.6 million, , respectively. These bank loans carry fixed interest rates of between 0.36%0.40% and 2.0%2.25% and are generally payable over periods ranging from three to five years from the original date of the loan.
In 2021, we entered into three new bank loans for a total nominal amount of €15.2 million, granted in the context of the COVID-19 pandemic, including:
•AnA €11.0 million loan in June 2021 by a pool of four French commercial banks,
•A €2.0 million loan in July 2021 by BPI France,
both of which•A €2.2 million subsidized loan in November 2021 by BPI France,
The June 2021 and July 2021 bank loans are 90% guaranteed by the French government (State-Guaranteed Loans or Prêts Garantis par l’Etat "PGE") and carry an initial term of one year with repayment options up to six years, as well as a€2.2 million subsidized loan inand the November 2021 by BPI France, withbank loan carries an initial term of six years.
In 2022 and 2023, we did not enter into any additional loan agreements.
Operating Leases
As of December 31, 2021, operating2023, leases subject to IFRS 16 consist of real estate leases for our offices located in Loos, France and Zurich, Switzerland, and lease agreements for scientific equipment.
In the second half of 2021, GENFIT SA and GENFIT CORP terminated the lease agreements for their offices, respectively located Additionally, we rent coworking spaces in Paris, France and Cambridge, MA which they both relocated to a coworking space. The rental of these office spaces, as a service contract, is no longerare not considered a leaseleases pursuant to IFRS 16.
Pension and Employee Benefits
French law requires payment of a lump sum retirement indemnity to employees based on years of service and annual compensation at retirement. Benefits do not vest prior to retirement. The amount presented in the table included in Note 12.425 – “Maturities of Financial Liabilities”- “Employee Benefits" in the Notes to theour consolidated financial statements included in this annual report, represents the present value of the estimated future benefits to be paid, applying a number of assumptions, including dates of expected retirement, life expectancies, salary growth rates and a discount rate.
| | | | | |
| C. | Research and Development, Patents and Licenses, etc. |
| | | | | |
| E. | Critical Accounting Estimates |
| | | | | |
| Item 6. | Directors, Senior Management and Employees. |
| | | | | |
| A. | Directors and Senior Management |
In January 2021, we announced the appointment to the Executive Committee of Pascal Caisey, Chief Commercial Officer, as well as Philippe Motté, as Chief Regulatory and Quality Officer. On February 26, 2021, the Board of Directors appointed Jean-François Tiné to replace Philippe Moons who resigned from his position as member of the Board. This appointment was ratified by the shareholders at the Shareholders Meeting held on June 30, 2021.
On March 11, 2021, the Board of Directors, in accordance with Article 24 of our by-laws, appointed Philippe Moons as board observer until the Shareholders’ Meeting called to approve the financial statements for the year ended December 31, 2021. Until that date, his role will be to provide an advisory opinion on questions that may arise with respect to the application of our by-laws, the Charter of the Board of Directors, with a view to supporting good corporate governance; in a particular context where three new directors have joined the Board of Directors since June 2020.
Although the board observer is not a director and is prohibited from interfering in any way in the management of the Company, the observer is invited to participate in all meetings of the Board of Directors and is subject to the Board charter in particular with respect to confidentiality and duties of loyalty.
Finally, on April 21, 2021, we announced the appointment of Thomas Baetz as Chief Financial Officer, as well as Stefanie Magner as Chief Compliance Officer, and their appointment to the Executive Committee.
Following the signature of the Collaboration and License Agreement with Ipsen, and as part of Ipsen's purchase of an 8% equity stake in Genfit, our Board of Directors will propose to the shareholders at the May 25, 2022 annual shareholders' meeting that Ipsen Pharma SAS become a member of the Board of Directors.
In March 2022,August 2023, we appointed two new members to the Executive Committee, Emilie Desodt,Sakina Sayah Jeanne, Executive Vice-President Human Resources,Research & Translational Science, and John Brozek,Tom Huijbers, Executive Vice-President Data & Information Technology.Regulatory.
In June 2023, Sandra Silvestri, M.D., Ph.D., replaced Steven Hildemann M.D., Ph.D., on the Board of Directors of the Company as representative of Ipsen, the legal entity that holds the Board seat.
The following table sets forth information concerning our senior management and directorsDirectors as of April 1, 2022.2024. Unless otherwise stated, the address for our senior management and directors is c/o GENFIT S.A., Parc Eurasanté, 885 avenue Eugène Avinée, 59120 Loos, France.
| | | | | | | | | | | | | | |
| Name | | Age | | Position(s) |
| Senior Management | | | | |
| Pascal Prigent | | 5456 | | Chief Executive Officer |
| Carol Addy, M.D. | | 6264 | | Chief Medical Officer |
| Thomas Baetz | | 4850 | | Chief Financial Officer |
| John Brozek | | 4547 | | VP,EVP, Data & Information Technology |
| Pascal Caisey | | 5456 | | Chief CommercialOperating Officer |
| Emilie Desodt | | 3941 | | VP,EVP, Human Resources |
| Dean Hum, Ph.D | | 5961 | | Chief OperatingScientific Officer |
| Laurent Lannoo | | 5254 | | Corporate Secretary, Director of Legal Affairs |
| Stefanie Magner, J.D. | | 4143 | | Chief Compliance Officer, VPEVP International Legal Affairs |
| Jean-Christophe Marcoux | | 4446 | | Chief StrategyCorporate Affairs Officer, Head of Investor Relations, Head of ESG |
| Meriam Kabbaj, Ph.D. | | 50 | | Chief Technology Officer |
Philippe MottéSakina Sayah-Jeanne | | 6252 | | EVP Research & Translational Science |
| Tom Huijbers | | Chief52 | | EVP Regulatory and Quality Officer |
| Non-Employee Directors | | | | |
Jean-François Mouney (1)(6)(7)(9) | | 6668 | | Chairman of the Board of Directors |
Xavier Guille des Buttes (2)(3)(7)(8)(9) | | 8082 | | Vice-Chairman of the Board of Directors |
| Eric Baclet (1)(2) | | 6264 | | Director |
Frédéric Desdouits-(7) | | 54 | | Director |
Katherine Kalin (7)(8) | | 5961 | | Director |
| Catherine Larue, Ph.D (1)(10) | | 6668 | | Director |
| Anne-Hélène Monsellato (4) | | 5456 | | Director |
Philippe Moons (8) | | 7072 | | Board observer (censeur) |
| Florence Séjourné (5) | | 52 | | Director |
| Sandra Silvestri, M.D., Ph.D. (6) | | 50 | | Director |
Jean-François Tiné (7)(8) | | 6567 | | Director |
(1)Member of the Nomination and Compensation Committee.
(2)Member of the Audit Committee.
(3)ChairmanChair of the Nomination and Compensation Committee.
(4)Chair of the Audit Committee.
(5)As representative of Biotech Avenir SAS, the legal entity that holds this boardBoard seat.
(6)ChairmanAs representative of IPSEN, the legal entity that holds this Board seat, since June 2023.
(7)Chair of the Strategy and Alliances Committee
(7)(8)Member of the Strategy and Alliances Committee
(8)(9)Resigned as DirectorMember of the BoardEnvironmental, Social, Governance Committee
(10) Chair of Directors on February 26, 2021, now serves as board observer.the Environmental, Social, Governance Committee
Senior Management
Pascal Prigenthas served as our Chief Executive Officer since September 2019. He served as our Executive Vice President, Marketing and Development from May 2018 to September 2019. Prior to that, he served as Vice President of Marketing—U.S. Vaccines for GlaxoSmithKline USA from April 2014 to November 2017. Prior to this, he was Vice President and General Manager of GlaxoSmithKline Romania from January 2011 to March 2014. He also served in various roles at Eli Lilly and its affiliates from 1996 through January 2011. Mr. Prigent is a graduate of Reims Management School, now known as NEOMA Business School, in Reims, France and earned his MBA from INSEAD in Fontainebleau, France.
Carol Addy has served as our Chief Medical Officer since September 2019. Prior to this, Dr. Addy held various leadership roles, including most recently, Chief Medical Officer at Health Management Resources, a subsidiary of Merck & Co., from November 2013 to August 2019, and as Associate Director, Director and Senior Principal Scientist at Merck Research Laboratories from June 2003 to November 2013. In addition to an M.D. degree, she holds a Masters of Medical Science from Harvard Medical School, and has also been an endocrinology consultant for MIT Medical.
Thomas Baetz has served as our Chief Financial Officer since April 1, 2021. He has extensive global finance experience across the investment banking and biotech industries. Prior to joining our company, Mr. Baetz was a Healthcare Director at Dragon Financial Partners, where he specialized in licensing agreements and fundraising consultancy for European biotechs. Before that, he was Group Chief Financial Officer and Head of Asia-Pacific for four years at Impeto Medical, a medtech company based in Hong-Kong and Paris, where he oversaw the corporate and business development in China until 2017. Prior to moving to Asia, he held key senior management positions, specializing in mergers and acquisitions, financial control, and consultancy among other areas. M.Mr. Baetz earned his MSc. in Finance and Actuarial Science from ENSAE and his BA from ESCP Europe.
John Brozek was appointed to the Executive Committee in March 2022.2022 as Executive Vice-President, Data & Information Technology. He holds three master’s degrees respectively in Cell and Molecular Biology from Lille University, Bioinformatics from Paris 7 University and Information Technology from Amiens University. He started his career in 2001 as Bioinformatician with IT-omics, a startup specializing in Information Systems design and data mining for biotech companies. In 2005, he joined GENFIT where he progressively took the lead of In Silico activities providing support in bioinformatics, biostatistics and Information Systems design. Since 2016, in addition to managing the In Silico activities, he leads the IT Department as Vice-President Data & Information Technology where he has been focusing on a global Information System renewal project while continuing to develop data related projects (data science and business intelligence).
Pascal Caisey joined GENFIT in September 2019 as Executive Vice President of Commercial Development, becoming Chief Commercial Officer in January 2021 and was appointed Chief Operating Officer in March 2022. He has vast pharmaceutical business experience, holding roles with GSK, BMS, Pfizer, Schering Plough and most recently Boehringer Ingelheim, where he oversaw, as the European Business Manager, the commercial launch of empagliflozin in Europe. Mr. Caisey is a registered nurse and holds an MBA from École des Hautes Études Commerciales (HEC) in Paris.
Emilie Desodt joined GENFIT in January 2018 as Human Resources Director and was appointed to the Executive Committee in March 2022.2022 as the Executive Vice-President, Human Resources. Ms. Desodt has been working in Human Resources for the past 18 years in various operational and strategic positions. Prior to joining GENFIT, she was in charge of HR activities, first at regional level (Americas & Middle East) then at global level at the Lesaffre Group. She has also held various HR roles of increasing responsibilities within General Electric. Ms. Desodt holds a bachelor’s degree in computer sciences (MIAGE) and a master’s degree in HR Development.
Dean Hum, Ph.D has served as our Chief Operating Officer since September 2018 and prior to that served as our Chief Scientific Officer since 2000 and2000. He also served as a member of our former Executive Board from May 2014 until the change in management and administration in June 2017. He earned a Ph.D in Biochemistry from McGill University in Montreal in 1990. He is an expert in the regulation of gene expression and nuclear receptors associated with endocrine and cardio metabolic diseases. Prior to becoming a Professor at Laval University in Quebec from 1994 to 2000, Dr. Hum held a research position at the University of California in San Francisco from 1990 to 1994. Dr. Hum coordinates our research and development activities with our Chief Executive Officer and in close collaboration with our other scientific officers and project managers. He is also a president and member of the board of directors of our wholly owned subsidiary, Genfit Corp., and a member of the Management Committee of our wholly owned subsidiary Genfit Pharmaceuticals SAS.
Laurent Lannoo has served as our Corporate Secretary and Director of Legal Affairs since 2008. From 2005 to 2008, he served in various roles at the Coeur et Artères foundation, including as chairman of its executive board from 2007 to 2008 and as corporate secretary from 2005 to 2006. Prior to that, from 1996 to 2005, he was in charge of finance and administration for Eurasanté, the public agency for the economic development of healthcare activities in the Nord-Pas de Calais region of France. He began his professional career at M&M, a consulting firm, in 1994, becoming partner in 1996. Mr. Lannoo graduated from Lille Law School with a degree in Business Law.
Stefanie Magner has served as our Chief Compliance Officer and VPExecutive Vice-President International Legal Affairs since March 2021, after joining our company in 2016 as Deputy Director of Legal Affairs. Prior to joining Genfit,GENFIT, she spent nearly 10 years at the Paris offices of the global U.S. law firm Jones Day, advising issuers, many in the biotech space, and banks on a variety of corporate, cross-border securities and M&A transactions, including several U.S. IPOs. She is admitted to practice law in New York and is a former member of the Paris Bar. She graduated from the University of Pennsylvania with a Bachelor of Arts in International Relations and French, as well as an international diploma from Sciences-Po Paris. She received her U.S. law degree from Washington College of Law at the American University in Washington D.C. and holds a Masters of Business Litigation from the Université de Paris X – Nanterre.
Jean-Christophe Marcoux has served as our Chief Corporate Affairs Officer, Head of Investor Relations, Head of ESG (previously titled Chief Strategy OfficerOfficer) since 2016, after joining2016. He joined our company in 2015 to play a cross-disciplinary role regarding tactical, strategic and operational matters. He is an engineer and graduated from INSA Lyon in France, having spent part of his time at the University of Leeds in England. In addition, he also holds a degree in Strategic Management and Economic Intelligence from EGE in France. From 2000 to 2015, he led international projects and programs in a variety of industrial sectors, in particular in Europe and Asia, and with clients and colleagues in the United States. In 2012, he joined IQVIA (formerly known as IMS Health, and later Quintiles IMS)Health), a global information and technology services company for clients in the healthcare industry, where he led projects in healthcare systems, such as patient longitudinal studies, forecasting, targeting, profiling, prospective analyses, digital healthcare and innovation. Since 2021, he is responsible for the Company's extra-financial reporting and activities, covering the challenges of corporate social and environmental responsibility.
Philippe MottéMeriam Kabbaj, Ph.D has served as our Chief Regulatory and QualityTechnology Officer since January 2021 after joiningDecember 2022. She is pharmacist by training (University of Geneva) and received her Master and PhD in Pharmaceutical Sciences from the University of Montreal. She acquired clinical drug development experience and was exposed to quality assurance and regulatory affairs in a leading Contract Research Organization (Celerion, formerly MDS Pharma Sciences) specialized in applied translational medicine, where she held many key operational and leadership positions. After a 10 year deep dive in the pharmaceutical industry, she co-founded Versantis where she successfully led the development of VS-01 from an academic prototype to a clinical lead compound and supported the fundraising activities. As a result of the acquisition of Versantis by GENFIT in June 2020September 2022, Meriam joined the Executive Committee as Chief Technology Officer in charge of CMC, Analytical Chemistry and Non-Clinical Development activities.
Sakina Sayah-Jeanne joined GENFIT on April 3, 2023 as Executive Vice-President Research & Translational Science, member of the Executive Committee. Sakina has more than 20 years pharmaceutical industry experience, including 7 years at GENFIT. Sakina obtained her PhD in Molecular and Cellular Biology in 1998, specializing in Neuro-Immunology, at the University of Rouen (Role of the complement in central nervous system pathologies with an inflammatory component). In 1999, Sakina joined Innothera, a French pharmaceutical group, as Project Leader, Pharmacology (Neurogenic pain). In 2002, Sakina joined GABA Laboratoire, as Scientific Attaché (oral and dental care). Sakina joined GENFIT in 2003 as a Project Leader, Preclinical. She then became Director of Therapeutic Target Research in 2005 (Cardiometabolic diseases, Alzheimer’s disease). In 2011, Sakina joined DaVolterra as the Manager of the Preclinical Research, to define and manage the nonclinical strategy for mechanistic and proof-of-concept studies for the product under development (Gut microbiome protection for the prevention of infectious diseases). In 2015, she was appointed Senior Vice PresidentDirector, Translational and Transversal R&D, where she was in charge of Globalproducing decision support for clinical development of the product, addressing questions around dose regimen and safe use for the different indications and populations, developing argumentations and defending Sponsor’s position statements with regulatory authorities (Gut microbiome protection for (i) prevention of infectious diseases, (ii) efficacy of anti-cancer treatments).
Tom Huijbers joined GENFIT on March 13, 2023 as Executive Vice-President Regulatory, member of the Executive Committee. Tom has more than 20 years of experience in the pharmaceutical industry. In 1999, he graduated with a Master of Science in Medicinal Chemistry and Molecular Pharmacology, from the University of Groningen (Netherlands). The same year, he joined the Janssen Research Foundation (Belgium) as Associate Manager Regulatory Affairs. Mr. Motté’s previous commercial and regulatory roles includeIn 2004, he joined Grünenthal GmbH based in Germany, as Regulatory Affairs Manager. As of 2006, he became Senior Regulatory Affairs Manager before holding the positions with Sanofi, GSK, Roche, Ipsen, and AbbVie. Prior to joining GENFIT, he was Vice President of Associate Director Global Regulatory Affairs, then Senior Director Global Regulatory Affairs between 2009 and Chief Access Officer (safety, quality, regulatory,2018. In 2018, Tom became Vice President, Head Development Strategy & Intelligence in Grünenthal’s Innovation Unit Devices and market access)Technologies. Before joining GENFIT, Tom had worked since 2020 at MedDay Pharmaceuticals. Mr. Motté holds a PharmD from the Paris-Descartes UniversityPinney Associates (USA) and a PhD in Human Biology (major Experimental Oncology) from the Paris-Sud University, completed Postdoctoral Research at Harvard Medical School and the Massachusetts General Hospital Cancer Center, earnedHarm Reduction Therapeutics (USA), as an MBA from the ESCP-EAP European School of Management (Paris), and is certified as a Pharmacien Responsable.independent Regulatory Affairs Consultant.
Non-Employee Directors
Jean-François Mouney has served as Chairman of our boardBoard of directorsDirectors since June 2017. Mr. Mouney also served as our Chief Executive Officer from September 1999 to September 2019. Mr. Mouney served as Chairman of our Executive Board from September 1999 to June 2017, when we changed our management structure. He co-founded GenfitGENFIT in 1999 after having been actively involved in the incubation of the companyCompany since 1997. Prior to this, he founded, managed and developed several companies specializing in high-performance materials, particularly in the aeronautical industry. In 1992, he founded M&M, a consultancy firm specializing in health economics. He was responsible for carrying out a feasibility study for the economic development agency, Eurasanté, within the field of health and biology in Nord-Pas-de-Calais region of France and was appointed Chief Executive Officer of this agency. He has continued to serve in this role since its launchagency when the agency was launched in 1995. Mr. Mouney has also served as Deputy Chairman of the “Nutrition, Health and Longevity” research hub between 2008 and 2016 and as an Advisor to the Banque de France since 2008.from 2008 to 2023. Mr. Mouney is a graduate of ESCP-Europe Business School, and holds a masters degree in Economics from the University of Lille.
Xavier Guille des Buttes served as member of our former Supervisory Board since 2006 and has served as a member of our boardBoard of directorsDirectors since June 2017. Mr. Guille des Buttes was educated at the Ecole Supérieure des Sciences Commerciales d’Angers, the Institut de gestion prévisionnelle et de contrôle de gestion, and has spent his entire career in the pharmaceutical industry. He has held a number of executive positions for more than 30 years, particularly in the French subsidiary of the German Group Schering AG, where, from 1974 to 2006, he successively held the positions of Marketing Director, General Manager of the Pharmaceutical Division and Chairman of the boardBoard of directors.Directors. As a member of our former Supervisory Board from October 2006, he chaired the Supervisory Board from April 2008 to June 2017, when he became Vice-Chairman of our Board of Directors following the change in administration and management. In addition to his responsibilities at Genfit,GENFIT, he also serves as director of several private companies.
Eric Baclet joined our boardBoard of directorsDirectors in 2020. In 1987, he began his extensive experience in the pharmaceutical industry with Eli Lilly and since the late 1990s until 2017, held executive or corporate officer positions in various countries where Eli Lilly and Company has a presence (North Africa, Belgium, the United States, China and Italy). From 2009 to 2013, Mr. Baclet was President and General Manager of Lilly China and most recently from 2014 to 2017, President of Lilly Italy and General Manager of Lilly Italian Hub. He is a seasoned executive with extensive experience gleaned from senior executive positions, having built and managed diverse and multicultural teams involved in the biopharmaceutical value chain throughout the world. From this background Mr. Eric Baclet has acquired extensive experience in international management from initial clinical development to final commercialization. Mr. Baclet has been responsible for portfolio strategies, international brand development, global marketing projects, global sales operations and the management of various geographic areas and countries. He currently serves as a board member of AIF Pharma Lux (Amanys Pharma) and AIF Pharma NA Board member (Future Pharmaceutical Industries); Mr. Baclet holds a Pharmacy degree from the University René Descartes.
Frédéric Desdouits served as member of our former Supervisory Board since 2014 and has served as a member of our board of directors since our change in management and administration in June 2017. Mr. Desdouits is the Chief Executive Officer of Treefrog Therapeutics, a cell therapy company based in Pessac, France. Until June 30, 2020, he was Managing Director of Seqens CDMO Business Unit and before that served as CEO for PCAS SA until March 23, 2020,a publicly listed affiliate of Seqens. Prior to joining Seqens (former Novacap) in 2017, he was head of Business Development, Acquisition and Market Intelligence at Pierre Fabre Group since 2011, and North American Pharma Director from January 2016. He was also a member of the pharmaceuticals executive board and of the development products board. Prior to joining Pierre Fabre, from 2004 to 2011, Mr. Desdouits was Managing Partner at Bionest Partners, a consulting and transaction firm based in Paris and New York specializing in healthcare and biotechnology. From 2007 to 2011, he was the founding Managing Partner of Bionest Partners Finance, a boutique specialized in value strategy and fund raising for emerging bio-companies. Between 1997 and 2004, Mr. Desdouits was a partner in charge of Pharmaceutical and Biotechnology sectors at Exane BNP-Paribas, an investment company. Prior to that, Mr. Desdouits worked in research from 1996 to 1997 at GlaxoWellcome in France (now GSK), as a consultant for Hoechst in the USA from 1995 to 1997 and was a Ph.D student from 1992 to 1995 with a grant from Rhône-Poulenc in France (now Sanofi). Between 2010 and 2011, he was a member of the Pre-Phase III DPU Blood & Vessels board at Sanofi Aventis (now Sanofi) in Chilly-Mazarin, France. Mr. Desdouits was a member of the supervisory board of CiToxLab (now Charles River). Between 2008 and 2011, Mr. Desdouits was a board member at Exonhit Therapeutics (now Eurobio Scientific) and member of the Mergers and Acquisitions subcommittee, and from 2015 to 2017, was an observer on the Orphelia Pharma Board of Directors. Mr. Desdouits graduated from Ecole Polytechnique (Palaiseau, France), obtained a M.S. in pharmacology and a Ph.D in Neurosciences at University Paris VI and Collège de France and studied from 1994 to 1996 at the Rockefeller University in New York. He is a CEFA (Certified European Financial Analyst) and Certified in Global Management from INSEAD.
Katherine Kalin joined our boardBoard of directorsDirectors in 2020. She brings more than 25 years of experience as a senior executive in healthcare and professional services, most recently at Celgene Corporation,services. Her healthcare industry experience spans pharmaceuticals, medical devices, diagnostics and digital health. From 1990 to 2002, Katherine was a partner in the global healthcare practice of McKinsey & Company, where she led corporate strategy from 2012 to 2017 andserved clients across a range of healthcare disciplines. In 2002, Katherine joined Johnson & Johnson where she held leadership roles in marketing, sales and new business development, from 2002until 2011. From 2012 to 2011. Prior to that, she was a partner in the global healthcare practice of McKinsey & Company from 1990 to 2002. Her healthcare industry experience spans diagnostics, medical devices, pharmaceuticals and digital health.2017, Katherine led corporate strategy at Celgene Corporation. She began her career as an investment banker in Corporate Finance at Nomura in Tokyo, Japan and London, UK. Ms. Kalin currently serves as a non-executive director on the board of Athersys,Sellas Life Sciences, a publicly-traded, USlate-stage clinical biopharmaceutical company, and as a member of the boardBoard of directorsDirectors of Brown Advisory LLC, an independent investment and strategic advisory firm and of FemHealth Ventures, a women’s health venture capital firm. She is also a member of the Advisory Board of Stardog, an enterprise data company, and a board member of PRIMARI Analytics, an artificial intelligence startup. She has a B.A. from Durham University, U.K., and an M.B.A. from Harvard Business School.
Catherine Larue, Ph.D has served as a member of our boardBoard of directorsDirectors since 2017. Since September 2020, she runs a consulting business in the biotechnology and diagnostic fields.fields (CoDx). From 2012 to 2020, Dr. Larue was CEO of the Integrated Biobank of Luxembourg (IBBL), where she led the development of the biobanking strategy and new initiatives in the field of personalized medicine. During this period, she also served as interim CEO of the Luxembourg Institute of Health (LIH), a biomedical research institute, between 2016 and 2017. Prior to joining the IBBL, Dr. Larue piloted GENFIT’s biomarker program until 2012. Dr. Larue began her career as team leader at Sanofi at the Montpellier, France based research and development center in the cardiovascular research department. She later joined Sanofi Diagnostics Pasteur, as Director of Research and Development for France and U.S. and then spent 11 years at the Bio-Rad group, holding different management positions. She participated in the discovery of several innovative biomarkers and the commercialization of dozens of diagnostic products. Dr. Larue holds a doctorate in experimental biology and an accreditation to direct research (Habilitation à Diriger la Recherche, or HDR) from the University of Rouen, a university degree in clinical oncology from the University of Paris VI and an executive MBA from St. John’s University (New York).
Anne-Hélène Monsellato has served as a member of our boardBoard of directorsDirectors and the chair of our Audit Committee since 2017. SinceFrom May 2015 to March 2023, she has beenwas an independent member of the Supervisory Committee and the Chairman of the Audit and Risk Committee of Euronav, a Belgian crude oil tanker company listed on the New York Stock Exchange and Euronext Brussels. In addition, she serves as the Vice President and Treasurer of the Board of Trustees of the American Center for Art and Culture, a U.S. private foundation based in New York, which operates the American cultural center in Paris, France. From 2005 until 2013, Ms. Monsellato served as a Partner with Ernst & Young (now EY), Paris, after having served as Auditor and, Manager for the firm starting in 1990. During her time at EY, she gained extensive experience in financial communication, IFRS, cross border listing transactions in(in particular with the United States,States), internal control over financial reporting and risk management, as well as financial statements audits and audits of internal control over financial reporting. She was involved with several companies in the pharmaceutical and biotechnology sector. Ms. Monsellato is an active member of the French association of Directors (IFA) since 2013 in particular with the Club of Audit Committee' Chairs, and the ESG Committee, where she regularly contributes to publications (November 2023 : Durabilité : les nouveaux engagements du conseil; February 2024 : Le conseil et la cybersécurité) and the European Confederation of Directors’ Association.Association (ecoDa). She was a member of the Consultative Working Group for the ESMA Corporate Reporting Standing Committee for 2019-2020.2019-2020, and she is a member of the EFRAG community for the development of the listed SMEs standards (LSME ESRS). Ms. Monsellato has been a Certified Public Accountant in France since 2008 and received a board member certification from IFA-Sciences Po in 2014. She graduated from EM Lyon in 1990 with a degree in Business Management. Ms Monsellato attended the Executive Education program "Governance & Climate" of Université Paris Dauphine-PSL in November 2023, a dedicated training for executives and board members providing a framework for reflection and action at corporate governance level in the face of complex and urgent climate-related issues. Ms Monsellato attended the Executive Education program "Governance & Climate" of Université Paris Dauphine-PSL in November 2023, a dedicated training for executives and board members providing a framework for reflection and action at corporate governance level in the face of complex and urgent climate-related issues.
Philippe Moonsserved as member of our former supervisory boardSupervisory Board since 2015 and has served as a member of our boardBoard of directorsDirectors since June 2017. In February 2021, he resigned from his position as director on the Board of Directors, but will remain as a boardBoard observer. Mr. Moons graduated from the Institut Catholique des Arts et Métiers de Lille and received an MBA from the Ecole des Hautes Etudes Commerciales du Nord (EDHEC), and began his career as a business engineer at Delattre Leviver, part of the Creusot-Loire Group, a French industrial Group. In 1989, he joined Finorpa, a venture capital and growth capital company, operating under the aegis of the Group “Charbonnage de France” in the Nord-Pas-de-Calais region of France. Between 2006 and 2015, he was in charge at Finorpa of supporting and financing several companies in their early-stage activities or development phases, in particular in the fields of biology and health. Mr. Moons was a member of the executive board of Finovam, a regional venture capital company, established in 2014 to strengthen the emergence and provide seed capital to innovative businesses, primarily technological projects in the Nord-Pas-de-Calais region, until 2015.
Florence Séjourné has served as a member of our boardBoard of directorsDirectors since June 2017 as representative of SAS Biotech Avenir. She was a member of our former Supervisory Board from 1999 until the change in our management and administration in June 2017. Ms. Séjourné co-founded our company and served as our chief operating officer,Chief Operating Officer, business development director, industrial alliances coordinator and member of our former Executive Board from 1999 to 2008. SinceFrom 2008 to 2022, she has beenwas the Chairwoman and CEO of Da Volterra, a clinical-stage biotechnology company developing novel Microbiota Protective therapies for protection against antibiotics residues, in particular in cancer and blood disorders. From 1997Since September 2022, Ms. Séjourné has been appointed CEO of a newly formed biopharmaceutical company founded as a joint venture by Boehringer Ingelheim, Evotec SE and bioMérieux, named AUROBAC THERAPEUTICS, to 1999, she was in chargecreate the next generation of the biopharmaceutical sector for Eurasanté, the economic development agency.antimicrobials along with actionable diagnostics to fight AntiMicrobial Resistance. Ms. Séjourné graduated from the Ecole des Mines of Paris with a degree in Biotechnology and holds a master’s degree in Pharmacy from the University of Illinois in Chicago.
Sandra Silvestri, MD., Ph.D, has served as a member of our Board of Directors since June 2023 as representative of Ipsen. Sandra Silvestri, M.D., Ph.D., joined Ipsen in 2023 as Executive Vice President, Chief Medical Officer and Head of Global Medical Affairs, Patient Safety and Patient Affairs. Prior to joining Ipsen, Ms. Silvestri held multiple leadership roles at Sanofi, where she was most recently SVP Chief Medical Officer for General Medicine GBU, leading a team of 1,600 medical employees worldwide. She also held several leadership positions at Eli Lilly in multiple disease areas including diabetology, endocrinology, neuroscience, immunology, dermatology, and oncology. Sandra Silvestri is a medical doctor specialized in endocrinology and metabolic diseases. She has been an investigator in several clinical studies, published numerous book chapters as well as scientific articles in international journals, been a speaker in several national and international congresses, and from 2017 to 2023 she led the Gender Balance Board and Global Network at Sanofi. She stays active as a professor at the medical schools of the University of Florence, Italy, and Descartes University in Paris. She has lived in Italy, Denmark, the U.S., France, and speaks Italian, English and French.
Jean-François Tiné joined the Board of Directors in 2021. He iswas a seasoned senior investment banking executive until 2022, having most recently served since 2017 as Chairman of Equity Capital Markets at Natixis Corporate & Investment Banking after joining Natixis in 2005 as Global Head of Equity Capital Markets. He began his career in various sales, trading and syndication positions in the London and Paris capital markets at Union Bancaire Privée, Crédit Suisse, First Boston and Bank of America. In 1993, he became an associate at MC Securities in London, before being appointed three years later as Global Head of Equity Syndicate at Société Générale in Paris.
Family Arrangements and Selection Arrangements
There are no family relationships between any of the members of our senior management or boardBoard of directors.Directors. Except as described below, there are no arrangements or understandings with major shareholders, customers, suppliers or others, pursuant to which any member of our senior management or boardBoard of directorsDirectors was selected as such.
Pursuant to an investment agreement entered into with Ipsen on December 16, 2021 pursuant to which Ipsen became a shareholder of GENFIT through the purchase of 3,985,239 newly issued shares representing 8% of GENFIT S.A after issuance, our Board of Directors will proposeshareholders, at the next shareholders’annual shareholders meeting currently expected to occurheld on May 25, 2022, thatappointed Ipsen becomes a board member, to be represented by Steven Hildemann.as Director.
Director Compensation
At our general meeting of shareholders held on June 30, 2021,May 24, 2023, shareholders renewed the total annual attendance fees (jetons de présence) to be distributed among non-employee directorsDirectors at €600,000 for the period beginning with the shareholders’ general meeting of June 30, 2021May 24, 2023 until the next shareholders’ general meeting, currently expected to occur on May 25, 2022.22, 2024. The following table sets forth information regarding the compensation earned by our non-employee directorsDirectors for service on our boardBoard of directorsDirectors during the year ended December 31, 2021,2023, which consisted solely of attendance fees, with the exception of our Chairman, Jean-François Mouney.
| | | | | | | | |
| NAME | | (€) |
| Jean-François Mouney(1) | | 332,416359,014 | |
| Eric Baclet | | 62,88147,960 | |
| Xavier Guille des Buttes | | 89,82280,660 | |
Frédéric Desdouits | | 43,600 | |
| Katherine Kalin | | 43,60039,240 | |
| Catherine Larue, Ph.D. | | 47,65143,600 | |
| Anne-Hélène Monsellato | | 52,32039,240 | |
Philippe MoonsMoons(2) | | 16,9867,630 | |
Florence Séjourné, as representative of SAS Biotech Avenir SAS | | — | |
| Sandra Silvestri, MD., Ph.D., as representative of IPSEN | | — | |
| Jean-François Tiné | | 39,72443,600 | |
(1)Mr. Mouney’s compensation includes his fixed compensation, directors’Directors’ fees and social security charges. See below “Chairman of the Board Compensation” for more details.
(2)Philippe Moons is an observer on the Board of Directors
We compensate all the members of the Board of Directors, with the exception of the permanent representativerepresentatives of Biotech Avenir a shareholderSAS and Ipsen, both shareholders of the Company and a non-independent member of the Board of Directors. If Ipsen Pharma SAS' nomination by the shareholders is approved at the next annual general meeting, Ipsen Pharma SAS would also not be entitled to receive directors fees.Company. Director compensation includes a fixed part for each directorDirector and a variable part depending on their attendance. The fixed part varies according to:
•the role played by each directorDirector on the Board of Directors and the Committees;committees; and
•the function of Vice-Chairman of the Board of Directors or Chairman of a specialized committee, which is better compensated.committee.
Given the frequency of meetings observed in recent years, the variable portion linked to attendance is greater than the fixed portion.
Directors fees are allocated as follows:
| | | | | | | | | | | | | | |
| (in euros) | | Annual fixed amount (1) | | Variable amount (per director and per meeting) |
| Board member | | 10,000 | | | 2,500 | |
| Board committee member | | 2,500 | | | 2,500 | |
| Vice-Chairman of the Board of Directors | | 10,000 | | | — | |
| Chairman of a Board committee | | 5,000 | | | — | |
| (1) For Board members joining during the course of the fiscal year, calculated pro-rata to number of months spent on the Board of Directors. Amounts may be cumulative. |
The Board of Directors may also compensate members on an exceptional basis for special assignments, within the meaning of article L.225-84L.225-46 of the French Commercial Code. To date, no special assignments have been given to any of the board members.
The Board of Directors, in accordance with the Articles of Association, decided on March 11, 2021 to appoint Philippe Moons as an observer. Mr. Moons’ compensation is deducted from the overall budget of €600,000 allocated by the Shareholders Meeting to Directors, at the rate of €1,250 per meeting of the Board of Directors and the ESG Committee in which he attends.
Chairman of the Board Compensation – Jean-François Mouney
The various components of the overall annual compensation of Mr. Mouney for his duties within the GenfitGENFIT group during the fiscal year ended December 31, 20212023 are summarized below:
•gross fixed compensation under article L.225-47L.22-10-16 of the French Commercial Code;
•attendance fees for participation in the work of the committees of the Board of Directors (as a member and/or chairman)Chairman), according to the distribution decided by the Board of DirectorsDirectors; and
•other benefits related to his position including use of a company vehicle and eligibility for the Group’s life insurance and health insurance benefits.
Mr. Mouney does not have an employment contract with the Company.
Fixed Compensation
Mr. Mouney received a gross fixed compensation of €192,996.€220,500.
Attendance Fees
Mr. Mouney also received gross compensation of €32,382€42,500 as Chairman of the Board of Directors, which amount includes directors’Directors’ fees for his participation in certain Board committees (Compensation(Nomination and NominationsCompensation Committee, Strategy and Alliances Committee and ESG Committee).
Other Compensation
The benefits in kind granted to Mr. Mouney for the year ended December 31, 20212023 consisted of the use of a company car valued at €7,200 and eligibility for the Group’s life insurance and health insurance benefits.
Chief Executive Officer Compensation – Pascal Prigent
Our only executive officer under French law is our chief executive officer. Chief Executive Officer.
Mr. Prigent's compensation for the fiscal year ended December 31, 2023 is composed of:
•fixed compensation;
•variable compensation (annual assessment);
•equity awards subject to presence and performance conditions;
•other benefits:
◦change of control and severance benefits, and
◦use of a company vehicle and eligibility for the Group’s life insurance and health insurance benefits.
Mr. Prigent does not have an employment contract with the Company.
The following table sets forth information regarding compensation earned during the year ended December 31, 20212023 by Mr. Prigent.
| | NAME AND PRINCIPAL POSITION | NAME AND PRINCIPAL POSITION | | FIXED
COMPENSATION | | VARIABLE
COMPENSATION (1) | | EQUITY
AWARDS | | ALL OTHER
COMPENSATION | | TOTAL | NAME AND PRINCIPAL POSITION | | FIXED
COMPENSATION | | VARIABLE
COMPENSATION (1) | | EQUITY
AWARDS (1) | | ALL OTHER
COMPENSATION | | TOTAL |
| (€) | | (€) | | (€) | | (€) | | (€) | (€) | | (€) |
| Pascal Prigent, Chief Executive Officer | Pascal Prigent, Chief Executive Officer | | 325,008 | | 162,504 | | 58,400 | | 16,229 | | 562,141 | Pascal Prigent, Chief Executive Officer | | 393,750 | | 147,656 | | 106,568 | | 16,250 | | 664,224 |
(1)Variable compensation and equity awards subject to “Say-on-Pay” approval of the Shareholders’ Meeting to be called to approve the financial statements for the year ended December 31, 2021.2023.
The various components of the overall annual compensation of Mr. Prigent for his duties as Chief Executive Officer of the GenfitGENFIT group during the fiscal year ended December 31, 20212023 are summarized below:
Fixed Compensation
Through his executive officer contract (contrat de mandat social), Mr. Prigent received a gross fixed compensation of €325,008.€393,750.
Variable Compensation
After evaluating the performance conditions relating to the variable compensation of the Chief Executive Officer, the Board of Directors has determined that the Chief Executive Officer’s variable compensation will be €147,656. This amount represents 37% of the Chief Executive Officer's fixed compensation.
The Board of Directors has determined that 75% of the CEO’sChief Executive Officer's objectives were achieved in 2023.
The 2023 objectives of the Chief Executive Officer and their weighting in the annual assessment of his performance were defined at the beginning of the financial year by the Board of Directors around the following four pillars/assessment criteria:
–Reinforcement of the Company's R&D pipeline through the acquisition of rights to new innovative compounds, partnership agreements or progress in internal research programs (representing a relative weight in the evaluation of the performance of 30%);
–Execution of the Company's main R&D programs with reference to the progress of the various clinical studies assessing elafibranor in PBC, NTZ and VS-01 in ACLF and GNS561 in CCA, and the deployment of the NIS4® and NIS2+® diagnostic technologies (representing a relative weight in the evaluation of the performance of 30%);
–The Company's financial performance with reference to the evolution of the Company's stock market valuation and the execution of the forward-looking cash management plan (representing a relative weight in the evaluation of the performance of 20%); and
–Implementation of the Company's CSR policy with reference to the execution of the 2023 roadmap, as defined on the recommendation of the ESG Committee and described in the extra-financial performance report, with reference to the overall extra-financial performance, as measured according to a panel of reference indices, and with reference, finally, to measurable indicators, in particular the diversity and satisfaction of the Company's employees (representing a relative weight in the evaluation of the performance of 20%).
The Board of Directors evaluated the performance of the Chief Executive Officer as follows:
–Reinforcement of the Company's R&D pipeline: 83.33% of the objective achieved, considering the addition to the portfolio of the new programs (SRT-015 et CLM-022) strengthening the Company's ACLF franchise;
–Execution of the Company's main R&D programs: 66.66% of the objective achieved considering (i) the positive interim results of the Phase 3 ELATIVE® trial evaluating elafibranor in PBC, which led to the filing of regulatory submissions for marketing authorization with the FDA and EMA, and to a milestone payment from Ipsen, (ii) the Phase 1 clinical data from the development of NTZ in ACLF, confirming a favorable safety profile, (iii) the rate of patient recruitment in the Phase 2 UNVEIL-IT® trial evaluating the VS-01 drug candidate in ACLF and in the Phase 1b/2a trial evaluating the GNS561 drug candidate in CCA and (iv) the several publications in well-known scientific journals on the performance of NIS2+® as a screening tool in MASH clinical trials and its use in clinical trials evaluating drug candidates in MASH;
–The Company's financial performance: 50% of the objective achieved considering the evolution of the Company's stock market valuation and the execution of the forward-looking cash management plan; and
–Implementation of the Company's CSR policy: 100% of the objective achieved considering the execution of the 2023 roadmap, the Company's progress on a panel of reference indices and on indicators measuring, in particular, the diversity and satisfaction of the Company's employees.
The Chief Executive Officer was not present during the Board of Directors discussion of his performance.
All variable compensation will be €162,504. Variable compensationis subject to approval at the upcoming Shareholders’ Meeting scheduled on May 22, 2024 called to approve the financial statements for the year ended December 31, 2023.
Equity Awards
During the year ended December 31, 2023, Mr. Prigent received a grant of 35,000 stock options (SO D 2023) and 10,000 free shares (AGA D 2023) with vesting subject to presence and performance conditions. The performance conditions attached to the stock options and free shares granted in 2023 are linked to internal and external conditions, in particular, the acquisition of new programs in accordance with the Group's strategy, clinical and regulatory advances in R&D programs and stock price. The performance conditions are detailed hereafter. The grant of these instruments is subject to approval at the upcoming Shareholders’ Meeting called to approve the financial statements for the year ended December 31, 2021.2023.
Equity Awards
DuringIn January 2024, after the year endedrecognition of the fulfillment of the presence condition and the assessment of the performance conditions (as of December 31, 2021, Mr. Prigent received a grant2023) of the stock option plan SO D 2020 of which the Chief Executive Officer is the beneficiary, 35,000 SO D 2020 stock options (SO 2021)have vested in favor of the Chief Executive Officer, i.e. the maximum amount provided for by the plan regulations in respect of the achievement of the performance criteria, considering:
–the announcement on June 30, 2023 of positive interim results from the Phase 3 ELATIVE® clinical trial, leading to the filing of regulatory submissions for marketing authorization with vesting subjectthe FDA and EMA by Ipsen in December 2023 (criteria a) ii) and iii) of the SO D 2020 plan, representing 50% of the stock options granted);
–the use of NIS4® technology in more than twenty clinical trials under the partnership with Labcorp and Q2, and recognition of NIS4® performance in several leading scientific publications during the measurement period (criteria b) ii) of the SO D 2020 plan, representing 25% of the stock options granted); and
–the completion in 2022 and 2023 of the Phase 1 clinical trials evaluating the safety, tolerability and pharmacokinetics of NTZ as part of its development program in ACLF, the acquisition of rights to new molecules under licensing agreements signed in 2021 and 2023 with Genoscience, Seal Rock Therapeutics and Celloram respectively, and finally, the acquisition, in 2022, of Versantis AG (criteria c) i) and ii) of the SO D 2020 plan, representing 25% of the stock options granted).
The performance conditions and 15,000 free shares (AGAof the SO D 2021).2020 plan which was adopted by the Board of Directors in 2020 are detailed below.
Other Compensation
Mr. Prigent received use of a company car valued at €6,609,€6,687, and was eligible for the Group’s life insurance and healthcare plans and the payment of premiums for unemployment insurance Social Security for Business Managers (GSC) whose purpose is to guarantee, which guarantees the payment of compensation in the event of unemployment (up to 55% of net professional tax income for the uncapped share for 12 months following the loss of the position) given that corporate officers are not eligible for standard French unemployment benefits, valued at €9,621.€9,563.
Change of Control and Severance Benefits
Mr. Prigent also benefits from a severance payment falling within the scope of Article L.225-42-1 of the French Commercial Code equal to 1218 months’ gross compensation, calculated on the basis of the last 12 months, increased, where applicable, by the amount of annual variable compensation due for the previous fiscal year and it would be paid if, and only if, one of the following three performance conditions is achieved at the time that his post is terminated:
•elafibranor has been granted marketing authorization by the FDA or EMA in NASH or PBC or that NIS4 has been granted FDA approval or obtained CE marking in Europe;PBC;
•a license agreement for elafibranorNTZ, GNS561, VS-01 or NTZVS-02 has been signed for the USU.S. market and / or for at least two of the five major European markets (Germany, France, Italy, United Kingdom Spainand Spain) and / or for Japan);Japan; or
•we have merged with or intothere is a biopharmaceutical group with a transaction value at least equal to our market capitalization.takeover of the Company.
Mr. Prigent also benefits from a non-compete indemnity equal to 12 months of gross fixed compensation, calculated on the basis of the gross amounts due for the past twelve months end, and where applicable, by the amount of the annual variable compensation due for the previous year. The amounts which he may receive under a non-compete indemnity are not cumulative with his severance payment and vice-versa. The non-compete covenant would not apply to the Chief Executive Officer if he leaves the Company, for whatever reason, either by decision of the Board of Directors or at his initiative, following a takeover of the Company.
Compensation recovery policy
In October 2022, the SEC adopted rules, pursuant to Rule 10D-1 under the Securities Exchange Act of 1934, as amended, or the Exchange Act, requiring national securities exchanges and national securities associations, such as Nasdaq, to amend their relevant listing standards no later than November 28, 2023 to require listed companies to adopt a written compensation recovery (clawback) policy providing for the recovery, in the event of a required accounting restatement, of incentive-based compensation received by the Chief Executive Officer and certain other “executive officers” as defined in Rule 10D-1(d) under the Exchange Act that is wholly or partially contingent on the attainment of financial performance criteria based on reported financial information that has been determined to be erroneous and has required restatement of the financial statements for accounting purposes. Our Board of Directors adopted at its meeting on March 28, 2023 a written compensation recovery policy, or the Recovery Policy, which, in accordance with French law, was presented to our shareholders and approved at our Annual General Meeting held on May 24, 2023. That policy is now in force with respect to the Chief Executive Officer and other executive officers, subject to compliance with applicable local laws and is included as an exhibit to this annual report for the year ended December 31, 2023.
Limitations on Liability and Indemnification Matters
Under French law, provisions of bylaws that limit the liability of directorsDirectors are ineffective. However, French law allows sociétés anonymes to contract for and maintain liability insurance against civil liabilities incurred by any of their directorsDirectors and officers involved in a third-party action, provided that they acted in good faith and within their capacities as directorsDirectors or officers of the company. Criminal liability cannot be indemnified under French law, whether directly by the company or through liability insurance.
We have liability insurance for our directorsDirectors and officers and insurance coverage for liability under the Securities Act. We have also entered into agreements with our directorsDirectors and senior management to provide contractual indemnification. With certain exceptions and subject to limitations on indemnification under French law, these agreements will provide for indemnification for damages and expenses including, among other things, attorneys’ fees, judgments and settlement amounts incurred by any of these individuals in any action or proceeding arising out of his or her actions in that capacity. We believe that this insurance and these agreements are necessary to attract qualified directorsDirectors and members of senior management.
Certain of our non-employee directorsDirectors may, through their relationships with their employers or partnerships, be insured against certain liabilities in their capacity as members of our boardBoard of directors.Directors.
These agreements may discourage shareholders from bringing a lawsuit against our directorsDirectors and senior management for breach of their fiduciary duty. These provisions also may have the effect of reducing the likelihood of derivative litigation against directorsDirectors and senior management, even though such an action, if successful, might otherwise benefit us and our shareholders. Furthermore, a shareholder’s investment may be adversely affected to the extent we pay the costs of settlement and damage awards against directorsDirectors and officers pursuant to these insurance agreements.
Equity Incentives
We believe our ability to grant equity incentives is a valuable and necessary compensation tool that allows us to attract and retain the best available personnel for positions of substantial responsibility, provides additional incentives to our employees, senior management and directorsDirectors and promotes the success of our business. Due to French corporate law and tax considerations, we have historically granted several different equity incentive instruments to our directors,Directors, senior management, employees and other service providers, including:
•redeemable share warrants (otherwise known as bons de souscription et/ou d’acquisition d’actions remboursables, or BSAAR);
•share warrants (otherwise known as bons de souscription d’actions, or BSA), which have historically only been granted to non-employee directors;Directors;
•restricted, or free, shares (otherwise known as actions gratuites, or AGA); and
•stock options (otherwise known as options de souscription et/ou d’achat d’actions, or SO).
Our boardBoard of directorsDirectors has authority to grant these equity incentive instruments and the aggregate amount authorized to be granted under these instruments must be approved by a two-thirds majority of the votes held by our shareholders present, represented or voting by authorized means, at the relevant extraordinary shareholders’ meeting. Once approved by our shareholders, our boardBoard of directorsDirectors can grant share warrants (BSA) for up to 18 months, and restricted (free) shares (AGA) and stock options (SO) for up to 38 months from the date of the applicable shareholders’ approval. The authority of our boardBoard of directorsDirectors to grant equity incentives may be extended or increased only by extraordinary shareholders’ meetings. As a result, we typically request that our shareholders authorize new pools of equity incentive instruments at every annual shareholders’ meetings.
We have three outstandingtwo types of share-based compensation plans for our senior management, directorscertain Directors and employees, the BSA plan, the AGA plan and the SO plan. In general, share warrants no longer continuevesting of our stock options and free shares is subject to vest following termination of thecontinued employment office or service of the holder and all vested sharesstock options must be exercised within post-termination exercise periods set forth in the grant documents. In the event of certain changes in our share capital structure, such as a consolidation or share split or dividend, French law and applicable grant documentation provides for appropriate adjustments of the numbers of shares issuable and/or the exercise price of the outstanding warrants.
As of April 1, 20222024, share warrants, stock options and free shares were outstanding allowing for the potential purchase and/or free allocation of an aggregate of 769,7781,400,700 ordinary shares.
Share Warrants (BSA)
ShareIn the past, share warrants have beenwere granted to the independent members of the former supervisory boardSupervisory Board and of the boardBoard of directorsDirectors and scientific consultants. Similar to options, share warrants entitle a holder to exercise the warrant for the underlying vested shares at an exercise price per share determined by our boardBoard of directorsDirectors and at least equal to the fair market value of an ordinary share on the date of grant. However, unlike options, the exercise price per share is fixed as of the date of implementation of the plans pursuant to which the warrants may be granted, rather than as of the date of grant of the individual warrants.
Pursuant to delegations granted by our shareholders, our boardBoard of directors,Directors, determines the recipients of the warrants, the dates of grant, the number and exercise price of the share warrants to be granted, the number of shares issuable upon exercise and certain other terms and conditions of the share warrants, including the period of their exercisability and their vesting schedule.
As of December 31, 2021, we have two outstandingApril 1, 2024, only the BSA 2019 share warrants plan is outstanding, whose beneficiaries are exclusively outside consultants. The two BSA 2017 plans as follows:expired without any warrants having been exercised.
| | Plan title | Plan title | | BSA 2017-A | | BSA 2017-B | | BSA 2019 | Plan title | | BSA 2017-A | | BSA 2017-B | | BSA 2019 |
| Meeting date | Meeting date | | June 16, 2017 | | June 16, 2017 | | June 15, 2018 | Meeting date | | June 16, 2017 | | June 15, 2018 |
| Dates of allocation | Dates of allocation | | November 21, 2017 | | November 21, 2017 | | October 31, 2019 | Dates of allocation | | November 21, 2017 | | October 31, 2019 |
| Exercise conditions(1) | Exercise conditions(1) | | 1 warrant / 1 share | | 1 warrant / 1 share | Exercise conditions(1) | | 1 warrant / 1 share | | 1 warrant / 1 share |
| Subscription periods | Subscription periods | | From December 11, 2017 to December 26, 2017 | | From July 1, 2018 to July 15, 2018 | | From October 31, 2019 to November 30, 2019 | Subscription periods | | From December 11, 2017 to December 26, 2017 | | From July 1, 2018 to July 15, 2018 | | From October 31, 2019 to November 30, 2019 |
| Total number of BSAs granted | Total number of BSAs granted | | 18,345 | | 18,345 | | 35,070 | Total number of BSAs granted | | 18,345 | | 35,070 |
Start date for the exercise of
the BSAs | Start date for the exercise of
the BSAs | | July 1, 2018 | | July 16, 2018 | | July 1, 2019 | Start date for the exercise of
the BSAs | | July 1, 2018 | | July 16, 2018 | | July 1, 2019 |
| BSA expiry date | BSA expiry date | | June 30, 2022 | | July 15, 2022 | | May 31, 2024 | BSA expiry date | | June 30, 2022 | | July 15, 2022 | | May 31, 2024 |
| BSA issuance price | BSA issuance price | | €2.00 | | €2.00 | | €1.23 | BSA issuance price | | €2.00 | | €1.23 |
| BSA exercise price per share | BSA exercise price per share | | €19.97 | | €19.97 | | €12.32 | BSA exercise price per share | | €19.97 | | €12.32 |
Number of shares subscribed as of
December 31, 2021 | | 0 | | 0 | | 0 |
| Warrants cancelled or lapsed | | 0 | | 0 | | 0 |
Warrants remaining
as of December 31, 2021 | | 18,345 | | 18,345 | | 35,070 |
Number of shares subscribed as of
December 31, 2023 | | Number of shares subscribed as of
December 31, 2023 | | 0 |
| BSA cancelled or lapsed | | BSA cancelled or lapsed | | 18,345 | | 0 |
BSA remaining
as of December 31, 2023 | | BSA remaining
as of December 31, 2023 | | 0 | | 35,070 |
(1)Exercisable by tranches of a minimum of 2,000 BSA, or a multiple thereof, except for outstanding balance under 2,000.
Free Shares (AGA)
Free shares may be granted to any individual employed by us or by any affiliated company. Free shares may also be granted to our chairmanChairman of the boardBoard of directors, chief executive officer (directeurDirectors (président du conseil d'administration), Chief Executive Officer (directeur général)ral) and deputy executive officers (directeurs(directeurs généralraux délégués). No free shares may be granted to the Directors. During the year ended December 31, 2021,2023, Mr. Prigent, our chief executive officer, received a free share grant. Mr. Mouney our chairman of our board, did not receive any free shares. We currently do not have any deputy executive officers. However, under French law, the maximum number of shares that may be granted shall not exceed 10% of the share capital as at the date of grant of the free shares (30%(40% if the allocation benefits all employees).
Our boardBoard of directorsDirectors has the authority to administer the free shares plans. Our boardBoard of directorsDirectors determines the recipients, the dates of grant, the number of free shares to be granted and the terms and conditions of the free shares, including the length of their vesting period (starting on the grant date, during which the beneficiary holds a right to acquire shares for free but has not yet acquired any shares) and holding period (starting when the shares are issued and definitively acquired but may not be transferred by the recipient) within the limits determined by the shareholders. Our shareholders have determined that the vesting period should be set by the boardBoard of directorsDirectors and should not be less than two yearsone year from the date of grant and that the optimal holding period should be set by the boardBoard of directors.Directors. From the beginning of the vesting period, the cumulated vesting and holding period should not be less than three years.
The boardBoard of directorsDirectors has the authority to modify awards outstanding under our AGA plans, subject to the consent of the beneficiary for any modification adverse to such beneficiary. For example, the board has the authority to release a beneficiary from the continued service condition during the vesting period after the termination of the employment.
The free shares granted under our AGA plans will be definitively acquired at the end of the vesting period as set by our boardBoard of directorsDirectors subject to performance conditions and continued service during the vesting period, except if the board releases a given beneficiary from this condition upon termination of his or her employment contract. At the end of the vesting period, the beneficiary will be the owner of the shares. However, the shares may not be sold, transferred or pledged during the holding period. In the event of disability before the end of the vesting period, the free shares shall be definitively acquired by the beneficiary on the date of disability. In the event the beneficiary dies during the vesting period, the free shares shall be definitively acquired at the date of the request of allocation made by his or her beneficiaries in the framework of the inheritance provided that such request is made within six months from the date of death.
As of April 1, 2022,2024, our free shares plans will vest, subject to performance conditions and continued employment, as follows:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | MEETING DATE | | DATE OF
ALLOCATION | | NUMBER OF FREE
SHARES GRANTED | | VESTING DATE
(SUBJECT TO
CONDITIONS)(1) | | STOCK PRICE ON
ALLOCATION
DATE | | FREE SHARES
VESTED |
| AGA D and S 2016-1 | | June 21, 2016 | | December 15, 2016 | | 20,520 | | December 16, 2019 | | €20.79 | | 17,484 |
| AGA D and S 2016-2 | | June 21, 2016 | | December 15, 2016 | | 10,189 | | December 16, 2019 | | €20.79 | | 7,796 |
| AGA D and S 2017-1 | | June 16, 2017 | | December 21, 2017 | | 27,472 | | January 1, 2021 | | €21.95 | | 19,400 |
| AGA D and S 2017-2 | | June 16, 2017 | | December 21, 2017 | | 13,730 | | January 1, 2021 | | €21.95 | | 8,021 |
| AGA D and S 2018 | | June 15, 2018 | | November 22, 2018 | | 35,800 | | January 1, 2021 | | €20.02 | | 21,741 |
| AGA D and S 2019 | | June 15, 2018 | | July 18, 2019 | | 36,782 | | September 17, 2022 | | €17.06 | | 0 |
| AGA D and S 2021 | | November 27, 2019 | | March 30, 2021 | | 51,600 | | March 31, 2024 | | €4.00 | | 0 |
| | | | | | | | | | | | | | | | | |
| Plan title | AGA D and S 2021 | AGA D and S 2022 | AGA D and S 2023 | AGA D and S 2024 |
| Date of Shareholders' Meeting | November 27, 2019 | May 25, 2022 | May 25, 2022 | March 24, 2023 |
| Date of allocation | March 30, 2021 | (S) | October 14, 2022 | March 10, 2023 | March 5, 2024 |
| March 17, 2021 | (D) |
| Vesting conditions | (1) | (1) | (1) | (1) |
| Number of free shares granted to employees | 32,400 | 38,900 | 30,100 | 47,900 |
| Number of free shares granted to the Chief Executive Officer: | 15,000 | 20,000 | 10,000 | 20,000 |
–Pascal Prigent | 15,000 | 20,000 | 10,000 | 20,000 |
| Vesting date (subject to vesting conditions) | April 1, 2024 | October 17, 2025 | March 14, 2026 | March 16, 2027 |
| Stock price on allocation date | 4,00 € (S) | 4,08 € | 4,05 € | 3,19 € |
| 4,15 € (D) |
| Number of lapsed or voided shares | 21,400 | 2400 | 1500 | 0 |
Number of free shares vested(2) | 26000 | 0 | 0 | 0 |
| Number of outstanding free shares | 0 | 56,500 | 38,600 | 67,900 |
(1)Subject to meeting performance conditions (detailed below) and continued employment with us.
Stock Options (SO)
Stock options may be granted to any individual employed by us or by any affiliated company. Stock options may also be granted to our chairmanChairman of the boardBoard of directors, chief executive officer (directeurDirectors (président du conseil d'administration), Chief Executive Officer (directeur général)ral) and deputy executive officers (directeurs(directeurs généralraux délégués). No stock options may be granted to the Directors. In addition, incentive stock options may not be granted to owners of shares possessing 10% or more of the share capital of our company.
Since 2016, the boardBoard of directors,Directors, using the authorizations granted to them by the extraordinary shareholders’ meeting, has granted stock options to the CEOChief Executive Officer and certain senior managers. These stock options were put in place as motivation and retention instruments for the current teams, to recruit new talents interested in participating in our future development and include them in obtaining operational and financial objectives.
These stock options allow us to continue to offer to new employees competitive packages compared to other companies in our sector, in particular U.S. companies; substantiate in shares a portion of the total profit-sharing of our employees, this contributing to the alignment of their interests with those of shareholders; and motivate the employees to achieve long-term objectives, and particularly to retain some of them by establishing a direct link between their level of profit sharing and the evolution of the stock price.
Stock options issued pursuant to these plans provide the holder with the right to purchase a specified number of ordinary shares from us at a fixed exercise price payable at the time the stock option is exercised, as determined by our boardBoard of directors.Directors. The plans generally provide that the exercise price for any stock option will be no less than 80% of the volume weighted average price of the 20 market trading days prior to the day of the boardBoard of directors’Directors’ decision to grant the options. Starting from 2020, stock options granted to the Chief Executive Officer are granted without discount. The vesting of the stock options is subject to performance conditions and the continued presence in our Company.company. These conditions are evaluated over a period of three years and reflect our mid-term objectives. Incentive stock options and non-statutory stock options may be granted under the SO plans.
Our boardBoard of directors,Directors, and in certain cases our CEO,Chief Executive Officer, has the authority to administer and interpret the SO plans. Subject to the terms and conditions of the stock option plan, our boardBoard of directorsDirectors determines the recipients, dates of grant, exercise price, number of stock options to be granted and the terms and conditions of the stock options, including the length of their vesting schedules. Our boardBoard of directorsDirectors is not required to grant stock options with vesting and exercise terms that are the same for every participant. The term of each stock option granted under the SO plans will generally be 10 years from the date of grant. Further, stock options will generally terminate on the earlier of when the beneficiary ceases to be an employee of our Companycompany or upon certain transactions involving our Company.company.
Our boardBoard of directorsDirectors has the authority to modify awards outstanding under our SO plans, subject to the written consent of the beneficiary for any modification adverse to such beneficiary. For example, our boardBoard of directorsDirectors has the authority to extend a post-termination exercise period.
Stock options granted under the SO plans generally may not be sold, transferred or pledged in any manner other than by will or by the laws of descent or distribution. In the event of disability, unless otherwise resolved by our boardBoard of directors,Directors, the beneficiary’s right to exercise the vested portion of his or her stock option generally terminates six months after the last day of such beneficiary’s service, but in any event no later than the expiration of the maximum term of the applicable stock options. In the event the beneficiary dies during the vesting period, then, unless otherwise resolved by our boardBoard of directors,Directors, the beneficiary’s estate or any recipient by inheritance or bequest may exercise any portion of the stock option vested at the time of the beneficiary’s death within the six months following the date of death, but in any event no later than the expiration of the maximum term of the applicable stock options.
The main terms of the SO plans are as follows:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Plan title | | SO 2016-1 | | SO 2016-2 | | SO 2017-1 | | SO 2017-2 | | SO 2018 | | SO 2019 | | SO US 2019-2 | | SO 2020 | | SO 2021 |
| Meeting date | | June 21, 2016 | | June 21, 2016 | | June 16, 2017 | | June 16, 2017 | | June 15, 2018 | | June 15, 2018 | | November 27, 2019 | | November 27, 2019 | | June 30, 2021 |
| Dates of allocation | | December 15, 2016 | | December 15, 2016 | | November 21, 2017 | | November 21, 2017 | | November 7, 2018 | | July 18, 2019 | | November 27, 2019 | | December 11, 2020 | | October 18, 2021 / October 19, 2021 |
| Exercise conditions(1) | | 1 option / 1 share
| | | | |
| Total number of SOs granted | | 48,917 | | 24,458 | | 72,830 | | 36,420 | | 139,500 | | 138,500 | | 13,350 | | 187,500 | | 201,875 |
| Start date for the exercise of the SOs | | December 16, 2019 | | December 16, 2019 | | January 1, 2021 | | January 1, 2021 | | January 1, 2022 | | September 17, 2022 | | January 17, 2023 | | January 1, 2024 | | October 19, 2024 / October 20, 2024 |
| SO expiry date | | December 16, 2026 | | December 16, 2026 | | December 31, 2027 | | December 31, 2027 | | December 31, 2028 | | September 17, 2029 | | January 17, 2030 | | January 1, 2031 | | October 19, 2031 / October 20, 2031 |
| SO exercise price per share | | €15.79/€21.12(2) | | €15.79/€21.12 | | €17.91/€22.54(3) | | €17.91/€22.54 | | €16.00/€21.65(4) | | €13.99/€16.90(5) | | €14.31 | | €4.52(6) | | €2.61/€3.26/€3.22(7) |
Number of SO exercised as of December 31,
2021 | | — | | — | | — | | — | | — | | — | | — | | — | | — |
| SO voided or lapsed | | 14,519 | | 9,150 | | 29,619 | | 18,655 | | 61,458 | | 53,815 | | 13,350 | | 21,250 | | 7,500 |
| SO vested as of December 31, 2021 | | 34,398 | | 15,308 | | 43,212 | | 17,765 | | 78,042 | | — | | — | | | | — |
| SO remaining to vest as of December 31, 2021 | | — | | — | | — | | — | | | | 84,685 | | — | | 166,250 | | 194,375 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Plan title | SO 2016-1 | SO
2016-2 | SO
2017-1 | SO
2017-2 | SO
2018 | SO 2018 US | SO 2019 | SO 2019 US | SO
2019 US-2 | SO
2020 | SO 2020 US |
| Date of Shareholders' Meeting | June 21, 2016 | June 21, 2016 | June 16, 2017 | June 16, 2017 | June 15, 2018 | June 15, 2018 | June 15, 2018 | June 15, 2018 | November 27, 2019 | November 27, 2019 | November 27, 2019 |
| Date of allocation | December 15, 2016 | December 15, 2016 | November 21, 2017 | November 21, 2017 | November 7, 2018 | November 7, 2018 | July 18, 2019 | July 18, 2019 | November 27, 2019 | December 11, 2020 | December 11, 2020 |
| Exercise conditions | (1) (2) | (1) (2) | (1) (2) | (1) (2) | (1) (2) | (1) (2) | (1) (2) | (1) (2) | (1) (2) | (1) (2) | (1) (2) |
| Number of stock options granted to employees | 42 250 | 21 125 | 61 497 | 30 753 | 95 000 | 17 500 | 82 750 | 30 620 | 13 350 | 103 750 | 56,250 |
| Number of stock options granted to the Chief Executive Officer | 6,667 | 3,333 | 11,333 | 5,667 | 27,000 | 0 | 25,130 | 0 | 0 | 35,000 | 0 |
–Jean-François Mouney | 6,667 | 3,333 | 11,333 | 5,667 | 17,000 | 0 | 15,130 | 0 | 0 | 0 | 0 |
–Pascal Prigent | 0 | 0 | 0 | 0 | 10,000 | 0 | 10,000 | 0 | 0 | 35,000 | 0 |
Start date for the exercise of the stock options(3) | December 16, 2019 | December 16, 2019 | January 1, 2021 | January 1, 2021 | January 1, 2022 | January 1, 2022 | September 17, 2022 | September 17, 2022 | January 17, 2023 | December 31, 2023 | December 31, 2023 |
| Stock options expiry date | December 16, 2026 | December 16, 2026 | January 1, 2027 | January 1, 2027 | January 1, 2028 | September 30, 2028 | September 17, 2029 | September 17, 2029 | January 17, 2030 | December 31, 2027 | December 31, 2027 |
Stock options exercise price per share(4) | €15.79 | €15.79 | €17.91 | €17.91 | €16.00 | €21.65 | €13.99 | €16.90 | €14.31 | €3.50 (C) | €4.52 |
| €4.38 (D) |
| Number of stock options exercised as of April 1, 2024 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Number of lapsed or voided stock options | 14,519 | 9,151 | 29,618 | 18,655 | 53,671 | 7 787 | 56,537 | 25,507 | 13,350 | 22,500 | 28,750 |
| Number of stock options vested | 34,398 | 15,307 | 43,212 | 17,765 | 68,329 | 9,713 | 51,343 | 5,113 | 0 | 116,250 | 27,500 |
| Number of stock options remaining to vest as of April 1, 2024 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Plan title | SO
2021 | SO 2021 US | SO 2022 | SO US 2022 | SO SU 2022 | SO 2023 | SO US 2023 | SO SU 2023 | SO 2024 | SO US 2024 | SO SU 2024 |
| Date of Shareholders' Meeting | June 30, 2021 | June 30, 2021 | May 25, 2022 | May 25, 2022 | May 25, 2022 | May 25, 2022 | May 25, 2022 | May 25, 2022 | May 24, 2023 | May 24, 2023 | May 24, 2023 |
| Date of allocation | October 18, 2021 | October 18, 2021 | October 14, 2022 | October 14, 2022 | October 14, 2022 | March 10, 2023 | March 10, 2023 | March 10, 2023 | March 5, 2024 | March 5, 2024 | March 5, 2024 |
| Exercise conditions | (1) (2) | (1) (2) | (1) (2) | (1) (2) | (1) (2) | (1) (2) | (1) (2) | (1) (2) | (1) (2) | (1) (2) | (1) (2) |
| Number of stock options granted to employees | 134,375 | 32,500 | 131,000 | 34,625 | 8,750 | 108,700 | 30,200 | 16,300 | 156,875 | 20,625 | 21,250 |
| Number of stock options granted to the Chief Executive Officer | 35,000 | 0 | 35,000 | 0 | 0 | 35,000 | 0 | 0 | 35,000 | 0 | 0 |
–Jean-François Mouney | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | | 0 | 0 |
–Pascal Prigent | 35,000 | 0 | 35,000 | 0 | 0 | 35,000 | 0 | 0 | 35,000 | 0 | 0 |
Start date for the exercise of the stock options(3) | October 20, 2024 | October 20, 2024 | October 18, 2025 | October 18, 2025 | December 3, 2025 | March 14, 2026 | March 14, 2026 | March 14, 2026 | March 16, 2027 | March 16, 2027 | March 16, 2027 |
| Stock options expiry date | October 20, 2031 | October 20, 2031 | October 17, 2032 | October 17, 2032 | December 3, 2032 | March 13, 2033 | March 13, 2033 | March 13, 2033 | March 15, 2034 | March 15, 2034 | March 15, 2034 |
Stock options exercise price per share(4) | €2.61 (C) | €3.22 | €3.12 (C) | €3.94 | €2.95 | €3.26 (C) | €4.05 | €3.26 | €2.74 (C) | €3.30 | €2.74 |
| €3.26 (D) | €3.91 (D) | $4.07 (D) | €3.42 (D) |
| Number of stock options exercised as of April 1, 2024 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Number of lapsed or voided stock options | 14,000 | 7,500 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Number of stock options vested | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Number of stock options remaining to vest as of April 1, 2023 | 155,375 | 25,000 | 166,000 | 34,625 | 8,750 | 143,700 | 30,200 | 16,300 | 191,875 | 20,625 | 21,250 |
(1)ExercisableOne share per stock option exercised; exercisable by 1/3one-third of the number of stock options held by each beneficiary.
(2)ExercisePerformance conditions (detailed below).
(3)Subject to meeting performance and presence conditions.
(4)The exercise price of the stock options was set at €15.7980% of the arithmetic average of Innate's volume-weighted average share prices for SO 2016-1 and SO 2016-2 and €21.12the twenty trading days preceding the grant date, except for SO US 2016-1 and SO US 2016-2.D, for which the exercise price of the stock options was set at 100% of the arithmetic average of Innate's volume-weighted average share prices for the twenty trading days preceding the grant date. (C): SO exercise price for French employees (SO C) ; (D): SO exercise price for the Chief Executive Officer (SO D).
(3)Exercise price at €17.91 for SO 2016-1 and SO 2016-2 and €22.54 for SO US 2016-1 and SO US 2016-2.
(4)Exercise price at €16.00 for SO 2018 and €21.65 for SO US 2018.
(5)Exercise price at €13.99 for the SO 2019 and €16.90 for the SO US 2019.
(6) Exercise price at €3.50 for the SO 2020, €4.52 for the SO US 2020, and €4.38 for the SO 2020 granted to Pascal Prigent.
(7) Exercise price at €2.61 for the SO 2021, €3.22 for the SO US 2021 and €3.26 for the SO 2021 granted to Pascal Prigent.
Until 2020, all of our stock option plans (SO and SO US) and our AGA D free share plans were subject to internal performance conditions related to our R&D programs, and to external performance conditions related to our stock price. The other free share plans (AGA S) are subject only to internal performance conditions, as further described below.
InSince then, and starting with the 2020 stock option plans, the Board of Directors decided that the 2020 stock option and AGA plans would only be subject to internal performance conditions, with the exception of the planAGA D plans dedicated to the CEO,Chief Executive Officer, which would have both internal and external performance conditions.
| | | | | |
| Plan | | | | | | | | Nature of performance conditions |
SO US 2019-2 Plans
| Evaluation date for performance conditionsconditions: 1/9/2023 | Nature of internalInternal conditions -
|
SO 2017-2
SO US 2017-2
AGA D 2017-2 | 12/31/2020 | 66 2/3%3 % of the instrumentsStock Options will be exercisable or definitively allocated, regardless of the evolution of the stock market priceif at least if at least one of the three following conditions is met:
fulfilled: (i) if an application forelafibranor has been granted marketing authorization for a product (elafibranor for NASH) is examined by the European Medicines Agency (EMA) or the U.S. Food and Drug Administration (FDA); in MASH or
ifa licensing agreement pertaining to elafibranor or NTZ has been signed for the launch ofU.S. market and/or for at least one clinical trial amongtwo of the following is authorized by the EMA five major European markets (Germany, France, Italy, United Kingdom, Spain) and/or the FDA, either:
• Phase IIIJapan; or (iii) at least two clinical trials of or which aim to record a new product (NTZ program) or a new indication for Elafibranor (PBC);
• Clinical trials with a product in Phase II (Elafibranor) within a NASH subpopulation; or
(iii) if we enter into at least one licensing agreement for our product candidates in one or several territories.
drug registration are underway. |
Nature of externalExternal conditions
|
33 1/3% of the instruments will be exercisable or definitively allocated in proportion to the evolution of the stock market price, as follows:
(i) if the Final Price is strictly lower than the Initial Price, the number exercisable or definitively allocated is equal to 0;
(ii) if the Final Price is between (i) a value equal to or higher than the Initial Price and (ii) a value lower than the Ceiling Price, the number exercisable or definitively allocated is equal to: [(Final Price / Initial Price)-1]/2 x 1/3 of number of instruments; or
(iii) if the Final Price is equal to or higher than the Ceiling Price, the number exercisable or definitively allocated is equal to the entire one-third of the instruments granted.
|
| | | | | | | | |
| | |
Plans | Evaluation date for performance conditions | Nature of internal conditions |
AGA S 2017-2 | 12/31/2020 | The free shares will be definitively allocated upon the same internal performance conditions as the SO 2017-2, SO US 2017-2 and AGA D 2017-2 plans.
|
| | | | | | | | |
| | |
Plans | Evaluation date for performance conditions | Nature of internal conditions |
SO 2018
SO US 2018
AGA D 2018 | 12/31/2021 | 66 2/3 % of the instruments will be exercisable or definitively vested, and 100% of the free shares for the AGA S 2018 will be vested, regardless of the variation of the stock market price, if one of the three following conditions is met:
(i) if an application for marketing authorization for elafibranor for the treatment of NASH is submitted to the European Medicines Agency (EMA) or the U.S. Food and Drug Administration (FDA); or
(ii) if authorization to launch at least one new clinical trial among the following trials is obtained:
• Phase III or Phase II/III clinical trial evaluating a new product (NTZ);
• Phase III or Phase II/III clinical trial evaluating elafibranor in PBC
• ��Phase III clinical trial evaluating elafibranor in a NASH subpopulation; or
(iii) if we enter into at least one licensing agreement for our product candidates in one or several territories.
|
Nature of external conditions
|
33 1/3% of the Stock Options will be exercisable, in proportion to the variation of our stock market price as per the following breakdown:
(i)
(iii)
(iii)
|
| | | | | | | | |
| | |
| Plans | Nature of performance conditions |
SO D 2020 SO C 2020 SO US 2020
Evaluation date for performance conditionsconditions: 12/31/2023 | Nature of internal conditions |
AGA S 2018 | 12/31/2021 | The free shares will be definitively allocated upon the same internal performance conditions as the SO 2018, SO US 2018 and AGA D 2018 plans.
|
| | | | | | | | |
| | |
Plans | Evaluation date for performance conditions | Nature of internal conditions |
SO 2019
SO US 2019
AGA D 2019 | 7/31/2022 | 66 2/3% of the instruments will be exercisable or definitively vested, and 100% of the free shares for the AGA S 2019 will be vested, regardless of the variation of the stock market price of our shares, if at least one of the three following conditions is fulfilled:
(i) if a marketing authorization is granted or an application for marketing authorization is examined:
• by the European Medicines Agency (EMA) or the U.S. Food and Drug Administration (FDA) for elafibranor for NASH; or:
• by the U.S. Food and Drug Administration (FDA)/the competent European authorities in the field of IVD for NIS4 for NASH; or:
(ii) if at least two of the four clinical trial among the following trials have delivered their principal results or are ongoing:
• Phase III clinical trials for elafibranor for PBC; or
• clinical trial evaluating elafibranor’s efficacy in NASH pediatric patients; or
• Phase IIb clinical trial or clinical trial aimed at registration for NTZ in fibrosis; or
• Clinical trial evaluating elafibranor or NTZ in combination therapy for NASH or for hepatic fibrosis; or:
(iii) if we enter into at least one new licensing agreement for our product candidates in one or several territories.
|
Nature of external conditions
|
33 1/3 % of the instruments will be exercisable or definitively vested, in proportion to the variation of our stock market price as per the following breakdown:
(i) if the Final Price is strictly lower than the Initial Price, the number of the Stock Options exercisable is equal to 0;
(ii) if the Final Price is between (i) a value equal to or higher than the Initial Price and (ii) a value lower than the Ceiling Price, the number of Stock Options exercisable is equal to: [(Final Price / Initial Price)-1]/2 x 1/3 of number of Stock Options; or
(iii) if the Final Price is equal to or higher than the Ceiling Price, the number of Stock Options exercisable is equal to the entire one-third of the Stock Options allocated. |
| | | | | | | | |
| | |
Plans | Evaluation date for performance conditions | Nature of internal conditions |
SO US 2019-2
| 1/9/2023 | 66 2/3 % of the instruments will be exercisable if at least if at least one of the three following conditions is fulfilled:
(i) if elafibranor has been granted marketing authorization by the European Medicines Agency (EMA) or the U.S. Food and Drug Administration (FDA) in NASH or PBC or NIS4 has been authorized by FDA or received CE marking from the EMA;
(ii) a licensing agreement pertaining to elafibranor or NTZ has been signed for the U.S. market and/or for at least two of the five major European markets (Germany, France, Italy, United Kingdom, Spain) and/or Japan; or
(iii) at least two clinical trials for drug registration are underway.
|
Nature of external conditions
|
33 1/3 % of the instruments will be exercisable, in proportion to the variation of our stock market price as per the following breakdown:
(i) if the Final Price is strictly lower than the Initial Price, the number of the Stock Options exercisable is equal to 0;
(ii) if the Final Price is between (i) a value equal to or higher than the Initial Price and (ii) a value lower than the Ceiling Price, the number of Stock Options exercisable is equal to: [(Final Price / Initial Price)-1]/2 x 1/3 of number of Stock Options; or
(iii) if the Final Price is equal to or higher than the Ceiling Price, the number of Stock Options exercisable is equal to the entire one-third of the Stock Options allocated. |
| | | | | | | | |
| | |
Plans | Evaluation date for performance conditions | Nature of internal conditions |
AGA S 2019 | 7/31/2022 | The free shares will definitively vest upon the same internal performance conditions as the SO 2019, SO US 2019 and AGA D 2019 plans. |
| | | | | | | | |
Plans | Evaluation date for performance conditions | Nature of internal conditions |
SO 2020
SO US 2020 | 12/21/2023 | a) 50% of the Stock Options will be exercisable if at least one of the following three conditions relating to PBC and ELATIVEELATIVE® is fulfilled:
i.“LastELATIVEELATIVE® in the fourth quarter of 2022 or earlier;
ii.IfELATIVEELATIVE® are released to the market before or during the first half of 2023;
iii.if
NIS 4NIS4® diagnostic is fulfilled:
i.ifNASHMASH player (“big pharma”, biotech company, institution, etc.) is entered into by the Company;
ii.the NIS 4
i.initiation
ii.if |
| | | | | | | | |
| Plans | Nature of performance conditions |
SO D 2021 SO C 2021 SO US 2021
Evaluation date for performance conditionsconditions: 10/20/2024 | Nature of internal conditions |
SO C 2021
SO D 2021
SO US 2021 | 12/21/2023 | a.a) 50% of the Stock Options will be exercisable if at least one of the following three conditions relating to the development of elafibranor in PBC and to the ELATIVEELATIVE® clinical trial is fulfilled:
i.ELATIVE (i) ELATIVE® topline results are released to the market before or during the second quarter of 2023;
ii. (ii) a new drug application is filed for elafibranor in PBC with the Food and Drug Administration (FDA) or the European Medicines Agency (EMA) in the second half of 2023 or before;
iii. (iii) elafibranor is approved by a regulatory authority in 2024.
b. b) 15% of the Stock Options will be exercisable if at least one of the following two conditions relating to the development of NTZ and the ACLF franchise is fulfilled:
i. (i) a phasePhase 2 clinical study or a more advanced clinical study evaluating NTZ is in ongoing or was carried out;
ii. (ii) the Company develops or acquires the rights to a new molecule (including through repositioning) for development in ACLF.
a. c) 15% of the Stock Options will be exercisable if at least one of the following two conditions relating to the NIS4NIS4® diagnostic technology is fulfilled:
i. (i) if a research and development partnership agreement relating to the implementation of the NIS4NIS4® diagnostic technology into an IVD test with at least one major NASHMASH player (“big pharma”, biotech company, institution, etc.) is entered into by the Company;
ii. (ii) Labcorp’s NASHnextNASHNext® LDT is reimbursed by at least three payers in the United States (insurance, integrated system, etc).
a. d) 20% of the Stock Options will be exercisable if at least one of the following two conditions relating to the development of the product pipeline of the Company is fulfilled:
i. (i) At least one new molecule (excluding elafibranor and NTZ) is developed by the Company or the Company has acquired development rights to a new molecule outside of the ACLF franchise (performance already covered by b(ii) above);
ii. (ii) At least two phasePhase 2 clinical studies or more advanced clinical studies are ongoing or have been completed ;completed; not including a phasePhase 2 clinical study or more advanced clinical study in NTZ (performance already covered by b(i) above).
|
| | | | | |
| Plans | Nature of performance conditions |
AGA S 2021 AGA D 2021
Evaluation date for performance conditions: 3/31/2024 |
External conditions - Each applicable portion of all 15,000 Free Shares under the AGA D 2022 plan, as each Internal Conditions above is met, is then subject to the External Condition according to the methods described below. The degree of fulfillment of the External Condition relating to the Company's stock market price will be determined according to the relative performance of GENFIT shares. Each applicable portion of all 15,000 Free Shares under the AGA D 2021 plan, as each Internal Conditions above is met, will be definitively acquired per the following conditions: (a) No AGA D 2021 shall vest if the Final Price is strictly lower than the Initial Price; (b) If the Final Price is between (i) a value equal to or greater than the Initial Price and (ii) a value lower than the Ceiling Price, the number of AGA D 2021 definitively allocated will be equal to:[(Final Price / Initial Price) -1] x 1/2 of the number of AGA D 2021 instruments (c) All AGA D 2021 if the Final Price is equal to or higher than the Ceiling Price. The notions of “Final Price”, “Initial Price” and “Ceiling Price” are defined in the plan regulations. |
| | | | | | | | |
| | |
| Plans | Nature of performance conditions |
SO D 2022 SO C 2022 SO US 2022 SO SU 2022 AGA S 2022 AGA D 2022
Evaluation date for performance conditions: - 10/17/2025 for SO D 2022/SO C 2022/SO US 2022/AGA S 2022/ AGA D 2022 - 12/3/2025 for SO SU 2022 | Internal conditions - a) 50% of the instruments SO D 2022/SO C 2022/SO US 2022/ SO SU 2022/AGA S 2022 will be exercisable or definitively vest, and 10,000 of the Free Shares for the AGA D 2022 will vest, if during the 2022 financial year and then at any time during the Vesting Period, 3 new R&D programs (at the rate of one third of these 2022 instruments per new program) complete the Company's R&D program portfolio (as it was at 12/31/2021); that these programs are at the so-called clinical development stage when this addition is made or that they reach this stage afterwards and that this addition originates: (i) a business-development operation (licensing-in, M&A, etc.), or (ii) the identification of new opportunities resulting from internal research (repositioning). b) 25% of the instruments SO D 2022/SO C 2022/SO US 2022/ SO SU 2022/AGA S 2022 will be exercisable or definitively vest, and 5,000 of the Free Shares for the AGA D 2022 will vest, if at least one of the following three conditions relating to the development of the elafibranor development program is fulfilled: (i) obtaining the main results of the first part of the ELATIVE® trial in the second quarter of 2023; (ii) filing of a Marketing Authorization Application for elafibranor in the second half of 2023; (iii) marketing authorization for elafibranor in 2024. c) 15% of the instruments SO D 2022/SO C 2022/SO US 2022/ SO SU 2022/AGA S 2022 will be exercisable or definitively vest, and 3,000 of the Free Shares for the AGA D 2022 will vest, if at least one of the following two conditions relating to the development of the NTZ program in the ACLF is fulfilled: (i) First clinical results in 2022; (ii) start of a Phase 2 clinical trial in the first half of 2023. d) 10% of instruments SO D 2022/SO C 2022/SO US 2022/ SO SU 2022/AGA S 2022 will be exercisable or definitively vest, and 2,000 of the Free Shares for the AGA D 2022 will vest, if as part of the development of the GNS561 program, a Phase 2b trial starts in the first half of 2023. External conditions - Each applicable portion of all 20,000 Free Shares under the AGA D 2022 plan, as each Internal Conditions above is met, is then subject to the External Condition according to the methods described below. The degree of fulfillment of the External Condition relating to the Company's stock market price will be determined according to the relative performance of GENFIT shares. Each applicable portion of all 20,000 Free Shares under the AGA D 2022 plan, as each Internal Conditions above is met, will be definitively acquired per the following conditions: (a) No AGA D 2022 shall vest if the Final Price is strictly lower than the Initial Price; (b) If the Final Price is between (i) a value equal to or greater than the Initial Price and (ii) a value lower than the Ceiling Price, the number of AGA D 2022 definitively allocated will be equal to:[(Final Price / Initial Price) -1] x 1/2 of the number of AGA D 2022 instruments (c) All AGA D 2022 if the Final Price is equal to or higher than the Ceiling Price. The notions of “Final Price”, “Initial Price” and “Ceiling Price” are defined in the plan regulations. |
| | | | | |
| Plans | Nature of internalperformance conditions |
AGA D2021SO D 2023
SO C 2023 SO US 2023 SO SU 2023 AGA S 20212023 AGA D 2023
Evaluation date for performance conditions: 3/13/2026 | 3/31/2024Internal conditions
| The free shares - a) 50% of the instruments SO D 2023/SO C 2023/SO US 2023/ SO SU 2023/AGA S 2023 will be exercisable or definitively vest, uponand 5,000 of the same internal performance conditions asFree Shares for the AGA D 2023 will be vest, if during 2023 and then at any time during the Vesting Period, 2 new R&D programs (at the rate of one-half of these 2023 instruments per new program), join the Company’s R&D pipeline (as evaluated at December 31, 2022) ; and that these programs are at the clinical development stage at the time they join the pipeline or that they later enter this stage, following: (i) A business development transaction (in-licensing, M&A, etc.) or, (ii) Identification of new opportunities resulting from in-house research (program going from preclinical development stage to clinical development stage). b) 25% of the instruments SO 2020 and D 2023/SO C 2023/SO US 2020 Plans..2023/ SO SU 2023/AGA S 2023 will be exercisable or definitively vest, and 2,500 of the Free Shares for the AGA D 2023 will vest, if at least one of the two following conditions related to development of elafibranor in PBC is met: (i) Filing of the Marketing Authorization Application in the fourth quarter of 2023 (in Europe or the United States); (ii) Marketing Authorization obtained in 2024 (in Europe or the United States). c) 15% of the instruments SO D 2023/SO C 2023/SO US 2023/ SO SU 2023/AGA S 2023 will be exercisable or definitively vest, and 1,500 of the Free Shares for the AGA D 2023 will vest, if at least one of the two following conditions related to the development of the ACLF program is met: (i) VS-01 in ACLF: top-line results from the Phase 2 study obtained in 2024 or communication of final results on the Phase 2 study in 2025; (ii) NTZ : start of a Phase 2 clinical trial in the second half of 2023. d) 10% of the instruments SO D 2023/SO C 2023/SO US 2023/ SO SU 2023/AGA S 2023 will be exercisable or definitively vest, and 1,000 of the Free Shares for the AGA D 2023 will vest, if intermediate results in the Phase 1b/2 of GNS561 are obtained in the fourth quarter 2024 or final results obtained in 2025.External conditions - Each applicable portion of all 10,000 Free Shares under the AGA D 2023 plan, as each Internal Conditions above is met, is then subject to the External Condition according to the methods described below. The degree of fulfillment of the External Condition relating to the Company's stock market price will be determined according to the relative performance of GENFIT shares. Each applicable portion of all 10,000 Free Shares under the AGA D 2023 plan, as each Internal Conditions above is met, will be definitively acquired per the following conditions: (a) No AGA D 2023 shall vest if the Final Price is strictly lower than the Initial Price; (b) If the Final Price is between (i) a value equal to or greater than the Initial Price and (ii) a value lower than the Ceiling Price, the number of AGA D 2023 definitively allocated will be equal to:[(Final Price / Initial Price) -1] x the number of AGA D 2023 instruments (c) All AGA D 2023 if the Final Price is equal to or higher than the Ceiling Price. The notions of “Final Price”, “Initial Price” and “Ceiling Price” are defined in the plan regulations.
|
| | | | | |
| Plans | Nature of performance conditions |
SO D 2024 SO C 2024 SO US 2024 SO SU 2024 AGA S 2024 AGA D 2024
Evaluation date for performance conditions: 3/15/2027 | Internal conditions - a) 10% of the instruments SO D 2024/SO C 2024/SO US 2024/ SO SU 2024/AGA S 2024 will be exercisable or definitively vest, and 1,000 of the Free Shares for the AGA D 2024 will be vest, if elafibranor obtains marketing authorization from the FDA or EMA in accordance with the road map. b) 30% of the instruments SO D 2024/SO C 2024/SO US 2024/ SO SU 2024/AGA S 2024 will be exercisable or definitively vest, and 3,000 of the Free Shares for the AGA D 2024 will vest, if at least one of the three following conditions relating to the development of VS-01 is met: (i) Interim results of the UNVEIL-IT® study are obtained in accordance with the road map; (ii) Final results of the UNVEIL-IT® study are obtained in accordance with the road map; (iii) Positive clinical results obtained and communicated in at least one ACLF sub-population or ACLF-related indication in accordance with the road map. c) 10% of the instruments SO D 2024/SO C 2024/SO US 2024/ SO SU 2024/AGA S 2024 will be exercisable or definitively vest, and 1,000 of the Free Shares for the AGA D 2024 will vest, if at least one of the two following conditions relating to the development of GNS561 is met: (i) Interim biomarker data from the ongoing phase 1b/2 is obtained in accordance with the road map; (ii) Final results for the Phase 1b part of the Phase 1b/2 study are obtained in accordance with the road map. d) 15% of the instruments SO D 2024/SO C 2024/SO US 2024/ SO SU 2024/AGA S 2024 will be exercisable or definitively vest, and 1,500 of the Free Shares for the AGA D 2024 will vest, if at least one of the three following conditions relating to the development of NTZ and SRT-015 is met: (i) Start of a phase 1b/2 study of NTZ in ACLF in accordance with the road map; (ii) Final results of a phase 1b/2 study of NTZ in ACLF in accordance with the road map and finalization of preclinical development of SRT-015 in 2024 which would allow, as necessary, the start of a first in human study of SRT-015 in accordance with the road map; (iii) Results of the first in human study of SRT-015 in accordance with the road map. e) 10% of the instruments SO D 2024/SO C 2024/SO US 2024/ SO SU 2024/AGA S 2024 will be exercisable or definitively vest, and 1,000 of the Free Shares for the AGA D 2024 will vest, if, with respect to one of the other programs in the Company’s pipeline in preclinical development at the time of this allocation decision (VS01 UCD/OA, VS02, CLM-022, …), at least one clinical trial is ongoing in accordance with the road map. f) 25% of the instruments SO D 2024/SO C 2024/SO US 2024/ SO SU 2024/AGA S 2024 will be exercisable or definitively vest, and 2,500 of the Free Shares for the AGA D 2024 will vest, if, at any time during the Vesting Period, two of the programs in the Company's pipeline at the date of the Grant Decision have delivered clinical results in humans enabling them to be considered for further development, resulting in the initiation of a phase 2b clinical trial or a phase 3 clinical trial, or the granting of accelerated approval. External conditions - Each applicable portion of all 10,000 Free Shares under the AGA D 2024 plan, as each Internal Conditions above is met, is then subject to the External Condition according to the methods described below. The degree of fulfillment of the External Condition relating to the Company's stock market price will be determined according to the relative performance of GENFIT shares. Each applicable portion of all 10,000 Free Shares under the AGA D 2024 plan, as each Internal Conditions above is met, will be definitively acquired per the following conditions: (a) No AGA D 2024 shall vest if the Final Price is strictly lower than the Initial Price; (b) If the Final Price is between (i) a value equal to or greater than the Initial Price and (ii) a value lower than the Ceiling Price, the number of AGA D 2024 definitively allocated will be equal to:[(Final Price / Initial Price) -1] x 1/2 of the number of AGA D 2024 instruments (c) All AGA D 2024 if the Final Price is equal to or higher than the Ceiling Price. The notions of “Final Price”, “Initial Price” and “Ceiling Price” are defined in the plan regulations. |
Board Composition
Until June 2017, our company had a two-tier corporate governance system: an executive board (directoire) was responsible for managing the company and a supervisory board (conseil de surveillance) oversaw and advised the executive board. We have now established a board of directors. Under French law and our bylaws, our boardBoard of directorsDirectors must be comprised of between three and 18 members. Their term of office, inIn accordance with our bylaws, isdirectors are appointed for a term of five years. Directors are appointed, reappointed to their position, or removed by the company’s ordinary general meeting. Directors chosen or appointed to fill a vacancy must be elected by our boardBoard of directorsDirectors for the remaining duration of the current term of the vacant director.Director. The appointment must then be ratified at the next shareholders’ general meeting. In the event the boardBoard of directorsDirectors would be comprised of less than three directorsDirectors as a result of a vacancy or removal, the remaining directorsDirectors shall immediately convene a shareholders’ general meeting to elect one or several new directorsDirectors so there are at least three directorsDirectors serving on the boardBoard of directors,Directors, in accordance with French law.
The annual meeting called to approve our financial statement for the year ended on December 31, 2022 and held on May 24, 2023 resolved to amend our articles of association to reduce the terms of our Directors from five to three years. The annual meeting further decided that this amendment of the articles of association will only take effect at the initial expiry of the terms of office of the Directors in office as of the date of the annual meeting and that in the event of the death, resignation or dismissal of the Directors in office on the date of the annual meeting: (i) in the case of co-optation following a death or resignation, the Director appointed in replacement of the deceased or resigning Director will be appointed for the remaining term of office of the replaced Director, (ii) in any other case, the new Director shall be appointed for a term of office of three years.Our boardBoard of directorsDirectors currently consists of nine members,Directors, one of which is a citizen or resident of the United States, and one boardBoard observer. As permitted by French law, onetwo of our directors,Directors, SAS Biotech Avenir, is aand Ipsen, are legal entity. This entity hasentities. These entities have designated, an individual,respectively, individuals, Florence Séjourné, and Dr. Sandra Silvestri, to represent itthem and to act on itstheir behalf at meetings of our boardBoard of directors.Directors. Ms. Séjourné hasand Dr. Silvestri have the same responsibilities to us and to our shareholders as shethey would have if shethey had been elected to our boardBoard of directorsDirectors in hertheir individual capacity. None of our directorsDirectors serve pursuant to a service contract providing benefits upon termination of service as a director.Director.
The following table sets forth the names of our directors,Directors, the years of their initial appointment as directorsDirectors of our boardBoard or our former supervisory boardSupervisory Board or our former executive boardExecutive Board and the expiration dates of their current term.
| | CURRENT
POSITION | | YEAR OF
INITIAL
APPOINTMENT | | TERM
EXPIRATION
YEAR |
| | CURRENT
POSITION | | | | CURRENT
POSITION | | YEAR OF
INITIAL
APPOINTMENT | | | TERM
EXPIRATION
YEAR |
| Jean-François Mouney | Jean-François Mouney | | Chairman | | 1999 | (1) | | 2022 | Jean-François Mouney | | Chairman | | 1999 | (1) | | 2027 |
| Xavier Guille des Buttes | Xavier Guille des Buttes | | Vice Chairman | | 2006 | (2) | | 2022 | Xavier Guille des Buttes | | Vice Chairman | | 2006 | (2) | | 2027 |
| Eric Baclet | Eric Baclet | | Director | | 2020 | | 2025 | Eric Baclet | | Director | | 2020 | | | 2025 |
| Frédéric Desdouits | | Director | | 2014 | (3) | | 2022 |
| IPSEN, represented by Dr. Sandra Silvestri | | IPSEN, represented by Dr. Sandra Silvestri | | Director | | 2022 | | | 2027 |
| Katherine Kalin | Katherine Kalin | | Director | | 2020 | | 2025 | Katherine Kalin | | Director | | 2020 | | | 2025 |
| Catherine Larue | Catherine Larue | | Director | | 2017 | | 2022 | Catherine Larue | | Director | | 2017 | | | 2027 |
| Anne-Hélène Monsellato | Anne-Hélène Monsellato | | Director | | 2017 | | 2022 | Anne-Hélène Monsellato | | Director | | 2017 | | | 2027 |
| Philippe Moons | Philippe Moons | | Observer | | 2015 | (4) | | 2022 | Philippe Moons | | Observer | | 2015 | (3) | | 2027 |
| SAS Biotech Avenir represented by Florence Séjourné | | Director | | 2010 | (5) | | 2022 |
| Biotech Avenir SAS represented by Florence Séjourné | | Biotech Avenir SAS represented by Florence Séjourné | | Director | | 2010 | (4) | | 2027 |
| Jean-François Tiné | Jean-François Tiné | | Director | | 2020 | (6) | | 2022 | Jean-François Tiné | | Director | | 2020 | (5) | | 2027 |
(1)As member of the former executive boardExecutive Board of our company and was subsequently appointed as a member of our board of directorsDirector at our combined general meeting in June 2017 and elected as chairmanChairman and chief executive officerChief Executive Officer of our company. Mr. Mouney resigned as chief executive officerChief Executive Officer of our company in September 2019 but continues to serve as chairmanChairman of our boardBoard of directors.Directors.
(2)As member of the former supervisory boardSupervisory Board and was subsequently appointed as a member of our board of directorsDirector at our combined general meeting in June 2017 and elected as vice chairman.Vice Chairman.
(3)As member of the former supervisory boardSupervisory Board and was subsequently appointed as a member of our board of directors at our combined general meeting in June 2017. For personal reasons and in accordance with the Board, Mr Desdouits has decided not to seek renewal of his board position, which expires at our next annual shareholder's meeting to be held on May 25, 2022.
(4)As member of the former supervisory board and was subsequently appointed as a member of our board of directorsDirector at our combined general meeting in June 2017. He resigned as a directorDirector on February 26, 2021 but will remain as an observer on the Board of Directors until the 2022 Shareholders Meeting.Directors.
(5)(4)Biotech Avenir SAS was appointed to the former supervisory boardSupervisory Board for the first time on incorporation of the companyCompany on September 15, 1999. Ms. Séjourné has been its permanent representative since 2010, first to the former supervisory boardSupervisory Board and later to the boardBoard of directorsDirectors of our company.
(6)(5)Appointed by the Board of Directors on February 26, 2021 to replace Philippe Moons on the Board of Directors. His appointment was approved by the Shareholders' Meeting on June 30, 2021 and he willto serve out the remainder of the term of Philippe Moons which will endended at the shareholders meeting called to approve the financial statements for the year ended December 31, 2021 scheduled to be held on May 25, 2022.
At His appointment was renewed by the upcoming annual shareholders' meeting, scheduled to be held on May 25, 2022 the Board of Directors has proposed to the shareholders to re-appoint Catherine Larue, Anne-Hélène Monsellato, Florence Séjourné (as permanent representative of BIOTECH AVENIR SA), Xavier Guille des Buttes, Jean-François Mouney, and Jean-François Tiné. The Board of Directors also proposes a resolution to appoint Ipsen Pharma SAS to the Board, with Steven Hildemann as its permanent representative.shareholders' meeting.
In 2021,2023, the Board of Directors met 15nine times, with an average participation rate of 93 % of Board members.Directors.
The average participation rates for each Board memberDirector at Board of Directors’ meetings was:
Mr. Jean-François Mouney : 100 % ;
Mr. Eric Baclet : 100%
Mr. Xavier Guille des Buttes: 100 % ;
IPSEN (represented by Mr. Frédéric DesdouitsSteven Hildemann) (until June, 2023) : 87%75%;
IPSEN (represented by Ms. Sandra Silvestri) (since June 2023): 100%
Ms. Katherine Kalin: 87%78%;
Ms. Catherine Larue : 87 %100% ;
Ms. Anne-Hélène Monsellato : 100 %100% ;
Mr. Philippe Moons : 100 %;
SAS Biotech Avenir SAS (represented by Ms. Florence Séjourné) : 73 %.78%.
Mr. Jean-François Tiné: 100%.
Board Diversity
Since January 1, 2017, under French law, the number of directorsDirectors of each gender may not be less than 40% of the total number of directors.Directors. Any appointment made in violation of this limit that is not remedied within six months of this appointment will be null and void. Any appointment which remedies a violation of the 40% gender limit must be ratified by our shareholders at the next ordinary general meeting.meeting pursuant to French Law.
The Nominations and Compensation Committee endeavors to seek nominees representing diverse experience in the drug development and diagnostics business, finance and other areas that are relevant to our activities. Furthermore, our boardBoard of Directors is committed to actively seeking out highly qualified women and individuals from minority groups to include in the pool from which Board nominees are chosen.
Pursuant to Nasdaq Listing Rule 5605(f) the table below provides certain highlights of the composition of our boardBoard members to the extent we are permitted to disclose such information under French law.
Board Diversity Matrix as of December 31, 20212023
| | Country of Principal Executive Offices: | Country of Principal Executive Offices: | France | Country of Principal Executive Offices: | France |
| Foreign Private Issuer: | Foreign Private Issuer: | Yes | Foreign Private Issuer: | Yes |
| Disclosure Prohibited under Home Country Law: | Disclosure Prohibited under Home Country Law: | No | Disclosure Prohibited under Home Country Law: | No |
| Total Number of Directors: | Total Number of Directors: | 9 | Total Number of Directors: | 9 |
| Part I: Gender Identity | | Female | Male | Non-Binary | Did Not Disclose Gender |
| Part I: Gender Identity | |
| Part I: Gender Identity | |
| Female | | | Female | Male | Non-Binary | Did Not Disclose Gender |
| Directors | Directors | 4 | 5 | 0 | 0 | Directors | 5 | 4 | 0 | 0 |
Part II: Demographic Background | | Underrepresented Individual in Home Country Jurisdiction | Underrepresented Individual in Home Country Jurisdiction | - | Underrepresented Individual in Home Country Jurisdiction | - |
| LGBTQ+ | LGBTQ+ | - | LGBTQ+ | - |
| Did Not Disclose Demographic Background | Did Not Disclose Demographic Background | 9 | Did Not Disclose Demographic Background | 9 |
Director Independence
As a foreign private issuer, under the listing requirements and rules of the Nasdaq Global Select Market, we are not required to have independent directorsDirectors on our boardBoard of directors,Directors, except to the extent that our audit committeeAudit Committee is required to consist exclusively of independent directors.Directors. Nevertheless, our boardBoard of directorsDirectors has undertaken a review of the independence of the directorsDirectors and considered whether any directorDirector has a material relationship with us that could compromise his or her ability to exercise independent judgment in carrying out his or her responsibilities. Based upon information requested from, and provided by, each directorDirector concerning such director’sDirector’s background, employment and affiliations, including family relationships, our boardBoard of directorsDirectors determined that all of our directors,Directors, except for Jean-François Mouney who previously served as our CEO, anddue to his ownership through Biotech Avenir, Florence Séjourné, as representative of Biotech Avenir, and Dr. Sandra Silvestri, as representative of IPSEN, qualify as “independent directors” as defined under applicable rules of the Nasdaq Global Select Market and the independence requirements contemplated by Rule 10A-3 under the Exchange Act. In making these determinations, our boardBoard of directorsDirectors considered the current and prior relationships that each non-employee directorDirector has with our company and all other facts and circumstances that our boardBoard of directorsDirectors deemed relevant in determining their independence, including the beneficial ownership of our ordinary shares by each non-employee directorDirector and his or her affiliated entities (if any).
Role of the Board in Risk Oversight
Our boardBoard of directorsDirectors is primarily responsible for the oversight of our risk management activities and has delegated to the audit committeeAudit Committee the responsibility to assist our boardBoard in this task. The audit committeeAudit Committee also monitors our system of disclosure controls and procedures and internal control over financial reporting and reviews contingent financial liabilities. The audit committee,Audit Committee, among other things, examines our balance sheet commitments and risks and the relevance of risk monitoring procedures. While our boardBoard oversees our risk management, our management is responsible for day-to-day risk management processes. Our boardBoard of directorsDirectors expects our management to consider risk and risk management in each business decision, to proactively develop and monitor risk management strategies and processes for day-to-day activities and to effectively implement risk management strategies adopted by the boardBoard of directors.Directors. We believe this division of responsibilities is the most effective approach for addressing the risks we face.
Corporate Governance Practices
As a French Ssociéociété anonymeAnonyme, we are subject to various corporate governance requirements under French law. When we listed our shares on Euronext Paris in 2014, we elected to refer to the Middlenext Governance Code providing guidance to mid and small cap companies. In addition, as a foreign private issuer listed on the Nasdaq Global Select Market, we are subject to Nasdaq corporate governance listing standards. However, the corporate governance standards provide that foreign private issuers, as defined in the rules promulgated under the US Securities Exchange Act of 1934, as amended (the “Exchange Act”), are permitted, pursuant to Nasdaq Listing Rule 5615(a)(3), to follow home country corporate governance practices in lieu of Nasdaq rules,Listing Rules, with certain exceptions. We rely on these exemptions for foreign private issuers and follow French corporate governance practices in lieu of the Nasdaq corporate governance rules,Listing Rules, which would otherwise require that (1) a majority of our boardBoard of directorsDirectors consist of independent directors; (2) we establish a nominating and corporate governance committee; and (3) our remunerationnomination committee be composed entirely of independent directors.directors; (3) our compensation committee be composed entirely of independent directors; and (4) our independent directors hold regularly scheduled meetings at which only independent directors are present.
The following is a summary of how certain of our corporate governance practices differ from U.S. companies listed on Nasdaq:
•Audit Committee. As a foreign private issuer, we are required to comply with Rule 10A-3 ofunder the Exchange Act, relating to audit committee composition and responsibilities. Rule 10A-3 provides that the audit committee must have direct responsibility for the nomination, compensation and choice of our auditors, as well as control over the performance of their duties, management of complaints made, and selection of consultants. However, under Rule 10A-3, if the laws of a foreign private issuer’s home country require that any such matter be approved by the boardBoard of directorsDirectors or the shareholders, the audit committee’s responsibilities or powers with respect to such matter may instead be advisory. Under French law, the audit committee may only have an advisory role and appointment of our statutory auditors, in particular, must be decided by the shareholders at our annual meeting.
In addition,•Quorum Requirements. Nasdaq rulesListing Rules require that a listed company specify that the quorum for any meeting of the holders of common stock be at least 33 1/3% of the outstanding shares of the company’s voting stock. We follow our French home country practice, rather than complying with these Nasdaq Listing Rules. Consistent with French law, our bylaws provide that awhen first convened, the quorum at the shareholders meeting requires the presence of shareholders having at least (1) 20% of the shares entitled to vote in the case of an ordinary shareholders’ general meeting or at an extraordinary shareholders’ general meeting where shareholders are voting on a capital increase by capitalization of reserves, profits or share premium, or (2) 25% of the shares entitled to vote in the case of any other extraordinary shareholders’ general meeting. If a quorum is not present, the meeting is adjourned. There is no quorum requirement when an ordinary general meeting is reconvened, but the reconvened meeting may consider only questions which were on the agenda of the adjourned meeting. When an extraordinary general meeting is reconvened, the quorum required is 20% of the shares entitled to vote, except where the reconvened meeting is considering capital increases through capitalization of reserves, profits or share premium. For these matters, no quorum is required at the reconvened meeting. The reconvened meeting may consider only questions that were on the agenda of the adjourned meeting. If a quorum is not present at a reconvened meeting requiring a quorum, then the meeting may be adjourned for a maximum of two months.
Board Committees
The boardBoard of directorsDirectors has established an audit committee,Audit Committee, a nominationNomination and compensation committee,Compensation Committee, a strategyStrategy and alliances committee,Alliances Committee, and most recently, we created an ESG committee,Committee, in accordance with the Middlenext Code requirements. Subject to available exemptions, the composition and functioning of all of our committees complies with all applicable requirements of the French Commercial Code, the Exchange Act, the Nasdaq Global Select Market and SEC rules and regulations.
In accordance with French law, committees of our boardBoard of directorsDirectors have only an advisory role and can only make recommendations to our boardBoard of directors.Directors. As a result, decisions will be made by our boardBoard of directorsDirectors taking into account non-binding recommendations of the relevant boardBoard committee.
In addition of the Board committees, in 2023, the Board of Directors appointed Mr. Eric Baclet, Director, to chair and coordinate a cybersecurity working group including the Executive Vice-President, Data & Information Technology and other key GENFIT employees. Eric Baclet reports regularly to the Board of Directors on cybersecurity matters, allowing the Board of Directors to provide effective oversight of management’s assessment and management of the cybersecurity risks.
In 2023, the Board of Directors also appointed Mr. Jean-François Mouney and Mr. Jean-François Tiné to chair and coordinate a financial strategy working group including the Chief Financial Officer and other key GENFIT employees. Jean-François Mouney and Jean-François Tiné report regularly to the Board of Directors on financial strategy, allowing the Board of Directors oversight of key financial issues of the Company.
Audit Committee.
Our audit committeeAudit Committee assists our boardBoard of directorsDirectors in its oversight of our corporate accounting and financial reporting and submits the selection of our statutory auditors, their remunerationcompensation and independence for approval. Ms. Anne-Hélène Monsellato, Mr. Xavier Guille dedes Buttes and Mr. Eric Baclet (who replaced Mr. Philippe Moons starting February 26, 2021) currently serve on our audit committee.Audit Committee. Ms. Monsellato is the chairperson of our audit committee.Audit Committee. Our boardBoard has determined that each member is independent within the meaning of the applicable listing rules and the independence requirements contemplated by Rule 10A-3 under the Exchange Act. Our boardBoard of directorsDirectors has further determined that Ms. Monsellato is an “audit committee financial expert” as defined by SEC rules and regulations and that each of the members qualifies as financially sophisticated under the applicable Nasdaq listing rules. The principal responsibility of our audit committeeAudit Committee is to monitor the existence and efficacy of the company’sCompany’s financial audit and risk control procedures on an ongoing basis.
Our boardBoard of directorsDirectors has specifically assigned the following duties to the audit committee:Audit Committee:
•monitoring the financial reporting process provided by the company.Company. In this respect, it examines in particular the consistency and the relevance of the accounting standards and methods used by the company,Company, and the advisability of any modification of the accounting methods. Special attention is paid by the audit committeeAudit Committee to reviewing the accounting policies used for the valuation of significant or unusual transactions. The audit committeeAudit Committee may make recommendations, in particular to ensure the integrity of the financial reporting process provided by the company,Company, control the integrity of the financial information provided by the companyCompany and, in particular, review the consistency and relevance of the accounting standards and methods retained by the company;Company;
•monitoring of the effectiveness of the internal control and risk management systems, as well as of the internal audit, as regards the procedures relating to the preparation and processing of accounting and financial information, without it undermining its independence. If necessary, it alerts the boardBoard of directorsDirectors in the event of an irregularity or anomaly identified in the company’sCompany’s financial statements or control procedures. The audit committeeAudit Committee assists the boardBoard of directorsDirectors in drafting the report on internal control;
•monitoring the appointment and renewal process of the statutory auditors. For this purpose, and in accordance with the regulations, the audit committeeAudit Committee issues a recommendation to the boardBoard of directorsDirectors on the statutory auditors proposed for appointment and / or renewal by the shareholders’ general meeting;
•monitoring of the performance by the Statutory Auditors of their mission, taking into account, where appropriate, the findings and conclusions of the Haut conseil du commissariat aux comptes(replaced by the Haute Autorité de l'Audit in 2024) following the audits carried out, in accordance with the regulations;
•monitoring by the statutory auditors of the conditions of independence under the conditions and in the manner provided for by the regulations, and in particular those mentioned in Article 6 of Regulation (EU) No. 537/2014. The audit committeeAudit Committee takes the necessary measures to implement paragraph 3 of Article 4 of this Regulation;
•pre-approval of the provision of services of the statutory auditors in compliance with the applicable regulations; and
•the regular report to the boardBoard of directorsDirectors on the performance of its duties. The audit committeeAudit Committee also reports on the results of the certification of the financial statements, how this mission has contributed to the integrity of financial reporting and the role it has played in this process. It informs the boardBoard of directorsDirectors without delay of any difficulty encountered.
In 2021,2023, the audit committeeAudit Committee met four4 times, with an average participation rate of 100% of committee members.
Nomination and Compensation Committee.
Mr. Xavier Guille des Buttes, Dr. Catherine Larue, Mr. Eric Baclet and Mr. Jean-François Mouney currently serve on our nominationNomination and compensation committee.Compensation Committee. Mr. Guille des Buttes is the chairperson of our nominationNomination and compensation committee.Compensation Committee.
Our boardBoard of directorsDirectors has specifically assigned the following duties to the nominationNomination and compensation committee:Compensation Committee:
•ensure the professionalism and objectivity of the appointment procedure for senior executives and corporate officers and senior management of the company.Company. In particular, it is in charge of making any proposal regarding the size and the desirable balance of the composition of the boardBoard of directorsDirectors in view of the structure and evolution of the shareholding of our company, as well as the requirements for good corporate governance, including the proportion of independent directorsDirectors at our boardBoard of directors,Directors, examine boardBoard committee membership, including in relation to the new ESG committee.Committee. Its mission is to research and assess potential candidates as well as the opportunity to renew mandates; and reviews the future succession of our company’s chairmanChairman and chief executive officer;Chief Executive Officer;
•assess the status of each of its board membersDirectors relative to other relations they might have with our company, which may compromise his or her free judgment or trigger potential conflicts of interest with us; the nominationNomination and compensation committeeCompensation Committee must also organize a procedure to select future independent members of the Board of Directors; and
•make proposals to the boardBoard of directorsDirectors concerning the elements of compensation or benefits granted to senior executives, corporate officers and senior management, including directors’Directors’ attendance fees and salaries, allowances or remuneration of any kind that such persons may receive under an employment contract or company contract with our company, the indemnities and benefits due upon termination of their employment, function or subsequent to this, the allocation of warrants, stock options or free shares, or any form of long-term incentive in the capital of the company.Company. In this respect, the nominationNomination and compensation committeeCompensation Committee assesses the scale of the compensation offered by the companyCompany in comparison with those practiced on the market and gives its recommendations to the boardBoard of directorsDirectors on the remuneration levels and the breakdown between the various elements of the compensation, as well as the changes in compensation that may be proposed by the companyCompany to its senior management and corporate officers.
In 2021,2023, the Nomination and Compensation Committee met six3 times, with an average participation rate of 96%100% of committee members.
Strategy and Alliances Committee.
Mr. Jean-François Mouney, Mr. Xavier Guille des Buttes, Mr. Frédéric Desdouits, Ms. Katherine Kalin and Mr. Jean-François Tiné (since his appointment to the Board of Directors on February 26, 2021) currently serve on our strategyStrategy and alliances committee.Alliances Committee. Mr. Jean-François Mouney is chairmanChairman of our strategyStrategy and alliances committee.Alliances Committee.
Our boardBoard of directorsDirectors has specifically assigned the following duties to the strategyStrategy and alliances committee:Alliances Committee:
•analyze business and corporate development opportunities, including strategic opportunities for acquisition or licensing of product rights or mergers and acquisitions with other companies;
•evaluate potential target products and companies;
•review the feasibility of any potential transactions.
In 2021,2023, the strategyStrategy and alliances committeeAlliances Committee met three7 times, with an average participation rate of 100% of committee members.
ESG Committee
Ms. Catherine Larue, Mr. Xavier Guille des Buttes and Mr. Jean-François Mouney currently serve on our ESG committee. Ms.Committee. Dr. Catherine Larue is the chairwoman of our ESG committee.
The ESG Committee was created in October 2021, in accordance with the new R8 recommendation of the Middlenext Code, with the mission of ensuring that the Company adequately addresses the economic and societal challenges related to its corporate purpose of proposing therapeutic and diagnostic solutions intended adressto address unmet medical needs of patients around the world.
Our boardBoard of directorsDirectors has specifically assigned the following duties to the ESG committee:Committee:
•review the Company's strategy, ambitions, policies and commitments in terms of social responsibility (Ethics and compliance, Human Rights, Hygiene / Health / Safety of people, Environment);
•ensure the Company's level of commitment to non-financial performance, ethics and social and environmental responsibility in relation to stakeholders’ expectations;
•ensure implementation of actions in these areas; and
•make recommendations in this regard to the Board of Directors.
The ESG Committee works in conjunctionmay be called upon to work with the Board's other specialized committees, notably the Nomination and Compensation Committee to define the components of social responsibility to be integrated into compensation policies and the development of diversity criteria within the Company as well as with the Audit Committee to manage the risks specific to the social responsibility of the Company.on issues that also concern them.
In 2021,2023, the ESG committeeCommittee met once,2 times, with a participation rate of 100% of committee members.
As of December 31, 2021,2023, we had 122159 employees. Of these employees, 7396 were engaged in research and development and services related to research and development activities, 4761 were engaged in administration and management, which includes finance, investor relations, information systems, human resources and legal, and 2 were engaged in marketing and commercial activities.
Of these 122159 employees, 112138 were employed by GenfitGENFIT S.A. and 10, 12 were employed by our U.S. subsidiary, Genfit Corp.GENFIT Corp, and 9 were employed by our Swiss subsidiary, Versantis AG. Employees employed by GenfitGENFIT S.A. are mainly based in France, and employees employed by GenfitGENFIT Corp. are mainly based in our Cambridge, Massachusetts office.office and employees employed by Versantis AG are mainly based in Zurich, Switzerland.
Pursuant to French law, employees employed by GenfitGENFIT S.A. are subject to the pharmaceutical industry collective bargaining agreement. We consider our relationship with our employees to be good.
| | | | | |
| F. | Disclosure of a registrant’s action to recover erroneously awarded compensation |
Not applicable.
| | | | | |
| Item 7. | Major Shareholders and Related Party Transactions. |
The following table sets forth, as of April 1, 2022,2024, information regarding beneficial ownership of our ordinary shares by:
•each person, or group of affiliated persons, known by us to beneficially own more than 5% of our ordinary shares;
•each member of our senior management;
•each of our directors; and
•all of our senior management and directors as a group.
Beneficial ownership is determined according to the rules of the SEC and generally means that a person has beneficial ownership of a security if he, she or it possesses sole or shared voting or investment power of that security, including free shares that vest by June 1, 2022,2024 the date that is 60 days after April 1, 2022,2024, and stock options and warrants that are currently exercisable or exercisable by June 1, 2022.2024. Shares subject to options and warrants currently exercisable or exercisable by June 1, 20222024 are deemed to be outstanding for computing the percentage ownership of the person holding these options or warrants and the percentage ownership of any group of which the holder is a member, but are not deemed outstanding for computing the percentage of any other person. Shares subject to free shares and stock options are not included, as no free shares nor stock options are currently vested because the requisite performance conditions have not been met.
Except as indicated by the footnotes below, we believe, based on the information furnished to us, that the persons named in the table below have sole voting and investment power with respect to all shares shown that they beneficially own, subject to community property laws where applicable. The information does not necessarily indicate beneficial ownership for any other purpose, including for purposes of Sections 13(d) and 13(g) of the Securities Act.
Our calculation of the percentage of beneficial ownership is based on 49,815,489.0049,860,983 of our ordinary shares outstanding as of April 1, 2022.2024.
Unless otherwise indicated, the address of each beneficial owner listed in the table below is c/o GenfitGENFIT S.A., Parc Eurasanté, 885, avenue Eugène Avinée, 59120 Loos, France.
| | | | | | | | | | | | | | |
| Name of Beneficial Owner | | Number of
Ordinary Shares | | Percentage |
|
| Significant Shareholders: | | | | |
| Biotech Avenir SAS(1) | | 1,888,618 | 1 | 3.79% |
| Ipsen Pharma SAS | | 3,985,239 | | |
| Directors and Senior Management: | | | | |
| Jean-François Mouney(2) | | 1,945,453 | | 3.91% |
| Pascal Prigent(3) | | 27,403 | | * |
| Dean Hum, Ph.D(4) | | 33,303 | | * |
| Carol Addy | | 0 | | — |
| Jean-Christophe Marcoux(5) | | 14,822 | | * |
| Laurent Lannoo(6) | | 23,637 | | * |
| Thomas Baetz | | 0 | | — |
| Pascal Caisey | | 0 | | — |
| Philippe Motté | | 0 | | — |
| Stefanie Magner(7) | | 12,758 | | * |
| Emilie Desodt (8) | | 3,305 | | * |
| John Brozek (9) | | 1,701 | | * |
| Xavier Guille Des Buttes(10) | | 6,842 | | * |
| | | | | | | | | | | | | | |
| Name of Beneficial Owner | | Number of
Ordinary Shares | | Percentage |
|
| Catherine Larue, Ph.D(11) | | 5,000 | | * |
| Anne-Hélène Monsellato(12) | | 5,000 | | * |
| Frédéric Desdouits(13) | | 5,111 | | * |
| Florence Séjourné | | 0 | | — |
| Philippe Moons(14) | | 5,310 | | * |
| Katherine Kalin (15) | | 5,000 | | * |
| Eric Baclet (16) | | 1,200 | | * |
| Jean-François Tiné | | — | | | — |
| All directors and senior management as a group (21 people)(17) | | 2,090,839 | | 4.2% |
| | | | | | | | | | | | | | |
| Name of Beneficial Owner | | Number of
Ordinary Shares | | Percentage |
|
| Significant Shareholders: | | | | |
| Biotech Avenir SAS(1) | | 1,888,618 | | 3.79% |
| Ipsen Pharma SAS(2) | | 3,985,239 | | 7.99% |
| Members of the board of directors and senior management: | | | | |
| Jean-François Mouney(3) | | 1,976,539 | | 3.96% |
| Pascal Prigent(4) | | 79,074 | | * |
| Dean Hum, Ph.D(5) | | 53,275 | | * |
| Carol Addy, M.D. | | 10,000 | | * |
| Jean-Christophe Marcoux(6) | | 31,625 | | * |
| Laurent Lannoo(7) | | 43,084 | | * |
| Thomas Baetz | | — | | — |
| Pascal Caisey(8) | | 15,969 | | * |
| Meriam Kabbaj | | — | | — |
| Stefanie Magner(9) | | 28,491 | | * |
| Emilie Desodt (10) | | 12,616 | | * |
| John Brozek (11) | | 20,349 | | * |
| Sakina Sayah-Jeanne | | — | | — |
| Tom Huijbers | | — | | — |
| Xavier Guille Des Buttes(12) | | 1,842 | | * |
| Catherine Larue, Ph.D | | — | | — |
| Anne-Hélène Monsellato | | — | | — |
| Sandra Silvestri, M.D., Ph.D.(2) | | — | | — |
| Florence Séjourné(1) | | — | | — |
| Philippe Moons(13) | | 1,040 | | * |
| Katherine Kalin (14) | | 5,000 | | * |
| Eric Baclet (15) | | 1,200 | | * |
| Jean-François Tiné (16) | | 10,600 | | * |
| All members of the board of directors and senior management as a group (23 people)(17) | | 2,290,704 | | 4.59% |
*Represents beneficial ownership of less than 1%
(1)Biotech Avenir SAS is our holding company. Mr. Mouney, the Chairman of our board of directors, and our Chief Executive Officer, is also the Chief Executive Officer and Chairman of the Management Committee of Biotech Avenir and holds 17.1% of its share capital. Florence Séjourné, who represents Biotech Avenir on our board of directors, is also a member of the Management Committee of Biotech Avenir and holds 9.9% of its share capital. Dean Hum holds 6.2% of its share capital, Laurent Lannoo, who is a member of the Management Committee of Biotech Avenir, holds less than 0.03% of its share capital and John Brozek holds 0.13% of its share capital.
(2)Sandra Silvestri represents Ipsen Pharma SAS (through Ipsen) on our board of directors. The Ipsen shares are subject to a lock-up period ending, on the earlier of the date on which the EMA makes a formal recommendation to the European Commission for the marketing authorization of elafibranor in PBC, the date on which the U.S. FDA grants approval of elafibranor in PBC or in the event the ELATIVE® trial does not meet its primary endpoint.
(3)Consists of 1,945,4531,935,212 ordinary shares, of which 1,888,618 shares are held directly by Biotech Avenir, 31,240and 41,327 stock options that are exercisable within 60 days of April 1, 2022.2024.
(3)(4)Consists of 20,70430,708 ordinary shares and 6,69948,366 stock options that are exercisable within 60 days of April 1, 2022.2024.
(4)(5)Consists of 6,72410,293 ordinary shares and 27,17942,982 stock options that are exercisable within 60 days of April 1, 2022.2024.
(5)(6)Consists of 2,2004,670 ordinary shares and 12,62226,955 stock options that are exercisable within 60 days of April 1, 2022.2024.
(6)(7)Consists of 9,03611,236 ordinary shares and 14,60131,848 stock options that are exercisable within 60 days of April 1, 2022.2024.
(7)(8)Consists of 1,5404,719 ordinary shares and 11,21811,250 stock options that are exercisable within 60 days of April 1, 2022.2024.
(8)(9)Consists of 14102,760 ordinary shares and 1,67525,731 stock options that are exercisable within 60 days of April 1, 2022, and 220 ordinary shares underlying OCEANE convertible bonds.2024.
(9)(10)Consists of 17011,738 ordinary shares and 4,81610,658 stock options that are exercisable within 60 days of April 1, 2022.2024, and 220 ordinary shares underlying OCEANEs convertible bonds.
(10)(11)Consists of 1,8426,910 ordinary shares and 5,000 BSA share warrants13,439 stock options that are exercisable within 60 days of April 1, 2022.
(11)Consists of 5,000 BSA share warrants that are exercisable within 60 days of April 1, 20222024.
(12)Consists of 5,000 BSA share warrants that are exercisable within 60 days of April 1, 2022.1,842 ordinary shares.
(13)Consists of 1111,040 ordinary shares and 5,000 BSA share warrants that are exercisable within 60 daysshares. Philippe Moons is an observer on the Board of April 1, 2022.Directors.
(14)Consists of 310 ordinary shares and 5,000 BSA share warrants that are exercisable within 60 days of April 1, 2022
(15)Consists of 5,000 ADS.
(15)Consists of 1,200 ordinary shares.
(16)Consists of 1,20010,600 ordinary shares.
(17)Includes 1,888,618 shares held directly by Biotech AvenirAvenir.
As of April 1, 2024, to the best of our knowledge, we believe that we are not directly or indirectly owned or controlled by another corporation, by any foreign government or by any other natural or legal persons.
Significant Changes in Percentage Ownership
In 2021, following the exercise by certain bondholders of their OCEANE convertible bonds, the percentage of share capital held by Biotech Avenir SAS decreased from 4.13% at December 31, 2020 to 3.79% at December 31, 2021.
On December 16, 2021, Ipsen Pharma SAS purchased 3,985,239 newly issued shares representing 8% of GENFIT S.A shares after issuance, in conjunction with the signature of the global licensing and collaboration agreement for elafibranor.
Other than described above, thereThere were no significant changes in the percentage ownership held by our principal shareholders during the year ended December 31, 2021.2023.
Voting Rights
A double voting right is attached to each registered share whichthat is held in the name of the same shareholder for at least two years. Any of our principal shareholders who have held our ordinary shares in registered form for at least two years have this double voting right.
Shareholders in the United States
As of April 27, 2022,1, 2024, to the best of our knowledge, 2,854,793approximately 1,440,000 of our outstanding ordinary shares (including ordinary shares in the form of ADSs) or approximately 5.7%3.31% were held by 1117 shareholders of record in the United States, including The Bank of New York Mellon, the depositary of our ADR program. The actual number of holders is greater than these numbers of record holders, and includes beneficial owners whose ordinary shares or ADSs are held in street name by brokers and other nominees. This number of holders of record also does not include holders whose shares may be held in trust by other entities.
| | | | | |
| B. | Related Party Transactions |
Since January 1, 2021,2023, we have engaged in the following transactions with our directors, senior management and holders of more than 5% of our outstanding voting securities and their affiliates, which we refer to as our related parties.
Directors
Biotech AvenirChief Executive Officer
In accordance with the decision of the Shareholders’ Meeting of November 27, 2019, the Chief Executive Officer benefits from a non-compete indemnity equal to (i) twelve months of fixed compensation, calculated on the basis of the gross amounts due to for the past twelve months and (ii) increased, where applicable, by the amount of the annual variable compensation due for the previous year.
This compensation is intended to compensate the prohibition made to the Chief Executive Officer, for a period of 12 months following the termination of his functions, for whatever reason, to collaborate in any way whatsoever with certain companies carrying out an activity directly competing with the Company. By decision of March 28, 2023, the Board of Directors has specified that this non-competition covenant will not apply to the Chief Executive Officer if he leaves the Company, for whatever reason, either by decision of the Board of Directors or at his initiative, following a takeover of the Company.
By decision of March 28, 2023, the Board of Directors has updated the severance pay of the Chief Executive Officer.
As a result, the Chief Executive Officer is eligible to receive, except in the case he is terminated on the basis of serious misconduct within the meaning of labor law, severance pay equal to (i) eighteen months of fixed compensation, calculated on the basis of the gross amounts due for the past twelve months and (ii) increased, where applicable, by the amount of the annual variable compensation due for the previous year. This compensation would be paid one month after his effective termination, provided that at least one of the following criteria or events has occurred (updated by the Board of Directors):
•elafibranor has been granted marketing authorization by the FDA or EMA in PBC;
•a license agreement for NTZ, GNS561, VS-01 or VS-02 has been signed for the U.S. market and / or for at least two of the five major European markets (Germany, France, Italy, United Kingdom, Spain and / or for Japan); or
•there is a takeover of the Company.
Compliance with these performance conditions will be assessed by the Board of Directors, taking into account the best interests of the Company, before any payment is made and after receiving the formal input from the Nomination and Compensation Committee.
The compensation will not be paid if, on his own initiative, the Chief Executive Officer leaves the Company to exercise new functions or changes functions within the Group, or even if he has the possibility of exercising in the short term his retirement rights.
Any amount paid under the non-compete clause will count as money owed for severance pay and vice versa.
Biotech Avenir SAS our
Biotech Avenir SAS, a management holding company, holds 3.79% of our share capital and 7.17%6.65% of our voting rights, as of April 1, 2022.2024. Mr. Mouney, the Chairman of our board of directors and, until September 2019, our Chief Executive Officer, is also Chairman of the Management Committee of Biotech Avenir SAS and holds 17.1% of its share capital. Florence Séjourné, who represents Biotech Avenir SAS on our board of directors, is also member of the Management Committee of Biotech Avenir SAS and holds 9.9% of its share capital. Dean Hum holds 6,2%6.2% of its share capital, Laurent Lannoo, who is a member of the Management Committee of Biotech Avenir SAS, holds less than 0,03%0.03% of its share capital and John Brozek holds 0,13%0.13% of its share capital. The registered office of Biotech Avenir SAS is located at the same address as our principal executive offices, without charge to Biotech Avenir.Avenir SAS.
Shareholders’ Agreement
A Shareholders’ Agreement binds all shareholders who held equity in our company prior to the private placement we carried out before the admission of our ordinary shares, on December 19, 2006, to trading on the Alternext stock exchange managed by Euronext Paris. In particular, this Shareholders’ Agreement grants a right of first refusal to Biotech Avenir or to any shareholder it designates, provided said shareholder is a signatory of the Shareholders’ Agreement, in the event that a shareholder who is a party to the Shareholders’ Agreement plans an off-market sale of its shares, insofar as the projected sale, plus any other sales carried out in a given year, represents at least 2% of our total share capital.
The parties to the Shareholders’ Agreement that hold our shares include the Université de Lille, Fondation partenariale de l’Université de Lille, Finorpa SCR, Biotech Avenir SAS, and two of our directors Messrs. Mouney and Guille de Buttes.des Buttes and Charles Woler.
This Shareholders’ Agreement became effective on December 19, 2006, and remained effective for an initial 10-year period, after which the Shareholders’ Agreement was, and may continue to be, automatically renewed for successive one-year periods.
The Shareholders’ Agreement was amended on January 30, 2018 as part of the restructuring of the University of Lille, whereby on January 1, 2018, the three universities of Lille (the universities of Lille I, Lille II and Lille III) merged into a single university (the Université de Lille). In this context, the Université de Lille II Droit et Santé (now Université de Lille) made a donation of 200,000 ordinary shares at the end of 2017 to the foundation, Fondation partenariale de l’Université de Lille, which is now one of our shareholders and a party to the Shareholders’ Agreement.
Ipsen Pharma SAS
Collaboration and license agreement
On December 16, 2021, we entered into a license and collaboration agreement withan exclusive collaboration and license agreement with Ipsen for the development and commercialization of elafibranor in PBC and other indications.indications (the Ipsen Collaboration and License Agreement). On the same date, we also entered into an investment agreement pursuant to which Ipsen became a shareholder of GENFIT through the purchase of 3,985,239 newly issued shares representing 8% of GENFIT S.A after issuance and, following approval by our shareholders at the shareholders' meeting on May 25, 2022, Ipsen became a member of our Board of Directors, undertook to propose at the next shareholders’ meeting scheduled for May 25, 2022, that Ipsen becomes a board member.currently represented by Dr. Sandra Silvestri. Ipsen therefore qualifies as a related person.
Pursuant to the license and collaboration agreement, we and Ipsen must enter into additional service and supply agreements in 2022, the value of which is expected to exceed $120,000, and therefore, will be related party transactions under our Related Person Transaction Policy.
Transition Services Agreement
The Transition Services Agreement (the "TSA") signed between the Company and Ipsen on April 6, 2022, pursuant to the Ipsen Collaboration and License Agreement, was approved by the Board of Directors on April 6, 2022 in accordance with the Company's Related Party Transactions policy.
The TSA governs the performance of a number of transition services by the Company in relation to the ongoing ELATIVE® trial, the Phase 3 clinical trial evaluating elafibranor in PBC and the financial conditions thereof. These services are mainly related to preparing the second phase of the ELATIVE® trial as well as certain regulatory tasks such as preparation of the conditional marketing authorization application for elafibranor in PBC. The services are being performed on an arms-length basis.
Part B Transition Services Agreement
The TSA was supplemented by a “Part B Transition Services Agreement” (the “Part B Agreement”) signed between the parties following approval by the Company's Board of Directors on September 19, 2023 in accordance with the policy relating to transactions between related parties and the Company.
The Part B Agreement governs the conditions under which a certain number of transition services have been and continue to be carried out by the Company until the total transfer of responsibility for the trial is turned over to Ipsen, and in particular the terms of compensation for these services during the specific period when some patients had completed the treatment corresponding to the first part of the clinical trial and initiated the treatment of the second part while others had not. These services are independent of those provided for by the TSA and the Ipsen Collaboration and License Agreement.
In 2023, €6.5 million in revenue was attributable to the services rendered under the TSA and the Part B Agreement.
Related Person Transaction Policy
We comply with French law regarding approval of transactions with related parties. We have adopted a related person transaction policy that sets forth our procedures for the identification, review, consideration and approval or ratification of related person transactions. For purposes of our policy only, a related person transaction is defined as (1) any transaction, arrangement or relationship (or any series of similar transactions, arrangements or relationships) in which we and any related person are, were or will be participants in and the amount involved exceeds $120,000, or (2) any agreement or similar transaction under French law which falls within the scope of Article L. 225-38 of the French Commercial Code. A related person is any director, member of senior management or beneficial owner of more than 5% of any class of our voting securities, including any of their immediate family members and any entity owned or controlled by such persons.
Under the policy, if a transaction has been identified as a related person transaction, including any transaction that was not a related person transaction when originally consummated or any transaction that was not initially identified as a related person transaction prior to consummation, our management must present information regarding the related person transaction to our board of directors for review, consideration and approval or ratification. The presentation must include a description of, among other things, the material facts, the interests, direct and indirect, of the related persons, the benefits to us of the transaction and whether the transaction is on terms that are comparable to the terms available to or from, as the case may be, an unrelated third party or to or from employees generally. Under the policy, we will collect information that we deem reasonably necessary from each director, member of senior management and, to the extent feasible, significant shareholder to enable us to identify any existing or potential related-person transactions and to effectuate the terms of the policy.
In addition, under our Code of Business Conduct, our employees and directors have an affirmative responsibility to disclose any transaction or relationship that reasonably could be expected to give rise to a conflict of interest.
In considering related person transactions, our board of directors will take into account the relevant available facts and circumstances including, but not limited to:
•the risks, costs and benefits to us;
•the impact on a director’s independence in the event that the related person is a director, immediate family member of a director or an entity with which a director is affiliated;
•the availability of other sources for comparable services or products; and
•the terms available to or from, as the case may be, unrelated third parties or to or from employees generally.
The policy requires that, in determining whether to approve, ratify or reject a related person transaction, our board of directors must consider, in light of known circumstances, whether the transaction is in, or is not inconsistent with, our best interests and those of our shareholders, as our board of directors determines in the good faith exercise of its discretion.
With the exception of the agreements with Ipsen, all of the transactions described above were entered into prior to the adoption of the written policy, but all were approved by our board of directors to the extent required by, and in compliance with, French law.
| | | | | |
| C. | Interests of Experts and Counsel |
Not applicable.
| | | | | |
| Item 8. | Financial Information. |
| | | | | |
| A. | Consolidated Statements and Other Financial Information |
Consolidated Financial Statements
Our consolidated financial statements are appended at the end of this annual report, starting at page F-1, and are incorporated by reference herein.
Dividend Distribution Policy
We have never declared or paid any dividends on our ordinary shares. We do not anticipate paying cash dividends on our equity securities in the foreseeable future and intend to retain all available funds and any future earnings for use in the operation and expansion of our business, given our state of development.
Subject to the requirements of French law and our bylaws, dividends may only be distributed from our distributable profits, plus any amounts held in our available reserves which are reserves other than legal and statutory and revaluation surplus. See “Item 10.B—10.B - "Additional Information - Memorandum and Articles of Association” for further details on the limitations on our ability to declare and pay dividends. Dividend distributions, if any in the future, will be made in euros and converted into U.S. dollars with respect to the ADSs, as provided in the deposit agreement. Legal Proceedings
From time to time, we may be involved in various claims and legal proceedings relating to claims arising out of our operations, including those described in Note 2275 - "Litigation" of our consolidated financial statements for the year ended December 31, 20212023 appended to this annual report. On May 14, 2020, following our announcement that elafibranor had not achieved the primary or key secondary endpoints of the RESOLVE-ITRESOLVE-IT® trial, a purported shareholder class action complaint, captioned Schwartz v. GenfitGENFIT S.A. et al., was filed in state court in the Commonwealth of Massachusetts, naming us, our board of directors and certain members of our senior management as defendants. The complaint alleged that we made materially misleading statements about the development of elafibranor in connection with our U.S. initial public offering in violation of U.S. federal securities laws. The complaint sought unspecified compensatory damages. In October 2020, the plaintiff voluntarily withdrew its action filed in state court in the Commonwealth of Massachusetts.
However, in December 2020, the same plaintiff filed a purported shareholder class action complaint in state court in the State of New York, alleging claims substantially similar to those in the previous complaint against the same defendants, as well as the underwriters of our U.S. initial public offering. In March 2021, we and the other defendants filed a motion to dismiss the claims before state court in the States of New York. In August 2021, the court grantedSupreme Court of the motion andState of New York, New York County, dismissed the complaint with prejudice. In September 2021, theThe plaintiff filed a notice of appeal toappealed, and in December 2022, the Supreme Court, Appellate Division, First Department and perfectedaffirmed the dismissal of the complaint, except that it deleted the phrase “with prejudice” from the Supreme Court’s judgment. The time to appeal on March 9, 2022. We intend to vigorously defend the lower court’s decision inof the appellate court.Appellate Division has expired.
Other than the legal proceeding described above, we are not currently a party to any legal proceedings that, in the opinion of our management, are likely to have a material adverse effect on our business. Regardless of outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Not applicable
| | | | | |
| Item 9. | The Offer and Listing. |
| | | | | |
| A. | Offer and Listing Details |
Our ADS have been listed on the Nasdaq Global Select Market under the symbol “GNFT” since March 27, 2019. Prior to that date, there was no public trading market for ADSs. Our ordinary shares have been trading on Euronext Paris under the symbol “GNFT” since 2006. Prior to that date, there was no public trading market for our ordinary shares. Our convertible bonds (OCEANE)(OCEANEs) have been traded on Euronext Access in Paris under the symbol “GNFAA” since October 16, 2017.
Not applicable.
Our ADSs have been listed on the Nasdaq Global Select Market under the symbol “GNFT” since March 27, 2019 and our ordinary shares have been trading on Euronext Paris under the symbol “GNFT” since 2006. Our convertible bonds (OCEANE) are(OCEANEs) have been traded on Euronext Access in Paris under GNFAA since October 16, 2017.
Not applicable.
Not applicable.
Not applicable.
| | | | | |
| Item 10. | Additional Information. |
Not applicable.
| | | | | |
| B. | Memorandum and Articles of Association |
The information set forth in the final prospectus dated March 27, 2019 as part of our Registration Statement on Form F-1 (File No. 333-229907), declared effective by the SEC on March 26, 2019, under the heading “Limitations Affecting Shareholders of a French Company” and the information in Exhibit 2.42.3 "Description of Securities" hereto is incorporated herein by reference.
Collaboration and License Agreement with Ipsen Pharma SAS
On December 16, 2021, we entered into an exclusive collaboration and license agreement with Ipsen Pharma SAS or Ipsen, a global, mid-sized biopharmaceutical company focused on transformative medicines in Oncology, Rare Disease and Neuroscience, as well as Consumer Healthcare products. Under the agreement, Ipsen has an exclusive worldwide (excluding Greater China which is licensed to Terns) license to develop, manufacture and commercialize elafibranor, our proprietary investigational compound, for people living with PBC, and in any other indications.
Under the terms of the agreement, we received an upfront cash payment of €120m, and are eligible for regulatory, commercial, and sales-based milestone payments up to €360m, plus tiered double-digit royalties of up to 20%.
In December 2023, a first milestone payment from Ipsen was due in an amount of €13.3 million and was received in February 2024.We remain responsible for the Phase III ELATIVE3 ELATIVE® trial until the completion of the double-blind period. Ipsen will assume responsibility for all additional clinical development, including completion of the long-term extension period of the ELATIVEELATIVE® trial, and global commercialization (excluding Greater China which is licensed to Terns). At the date of this report, the responsibility of the pursuit of the trial is almost entirely transferred to Ipsen (only one clinical investigation site remains to be transferred).
This newly established strategic partnership will also provide Ipsen with access to our research capabilities and other clinical programs through rights to first negotiation.
In addition, pursuant to an investment agreement entered into on the same date as the collaboration and licensing agreement, Ipsen also became a shareholder of GENFIT through the purchase of 3,985,239 newly issued shares representing 8% of GENFIT S.A after issuance, via a €28m investment. The new shares are subject to a lock-up period ending, on the earlier of the date on which the EMA makes a formal recommendation to the European Commission for the marketing authorization of elafibranor in PBC, the date on which the U.S. FDA grants approval of elafibranor in PBC (PDUFA date June 10, 2024) or in the event the ELATIVEELATIVE® trial does not meet its primary endpoint. In addition,Following approval by our shareholders at the shareholders' meeting on May 25, 2022, Ipsen became a member of our Board of Directors undertook to propose a resolution at the next shareholders’ meeting that Ipsen becomes a board member.Directors.
The summary provided above does not purport to be complete and is qualified in its entirety by reference to the complete agreement, which is attached as an exhibit to this annual report.
Collaboration and License Agreement with Terns Pharmaceuticals, Inc.
On June 24, 2019, we entered into a collaboration and license agreement with Terns Pharmaceuticals, Inc., or Terns, a global biopharmaceutical company based in the United States and China with a focus on developing novel and combination therapies to treat liver disease. Under the agreement, Terns will have the rights to develop and commercialize elafibranor, our proprietary investigational compound, in mainland China, Hong Kong, Macau and Taiwan, which we refer to as Greater China, for the treatment of NASHMASH and PBC.
Under the terms of the licensing agreement, we received an upfront payment from Terns of $35 million and will be eligible to receive up to $193 million in potential clinical, regulatory and commercial milestone payments. Terns obtains the exclusive rights to develop, register and market elafibranor in Greater China for both NASHMASH and PBC. Upon commercial launch of elafibranor for the treatment of NASHMASH in Greater China, we will be entitled to receive mid-teen percentage royalties from Terns based on sales in the territory.
As part of the deal, we and Terns will also undertake joint research and development projects in liver disease, including the development of elafibranor in combination with Terns’ proprietary compounds.
The summary provided above does not purport to be complete and is qualified in its entirety by reference to the complete agreement, which is an exhibit to this annual report.
Share Purchase Agreement for the Acquisition of Versantis AG
On September 19, 2022, we announced we had signed an exclusive agreement to acquire all the shares and voting rights of Versantis AG, or Versantis, a private Swiss-based clinical stage biotechnology company, and its U.S. subsidiary, Versantis, Inc., focused on addressing the growing unmet medical needs in liver diseases.
With this acquisition, we acquired Versantis' pipeline, which includes Versantis' main asset VS-01, a liposomal-based therapeutic product candidate currently in clinical development as a potential therapy for ACLF and HAC. In addition, it's second asset, VS-02 is a pre-clinical oral and colon-active, drug candidate being developed for the chronic management of HE. Finally, TS-01, a point-of-care diagnostic device in prototype development for at-home measurement of ammonia in the blood, is in-licensed by Versantis from ETH Zurich.
The deal included an initial consideration of CHF40.0 million due at closing plus a CHF2.8 million cash adjustment, with contingent consideration of up to CHF65 million upon positive Phase 2 results for VS-01 and VS-02 and regulatory approval of VS-01. In addition, the former owners of Versantis are eligible to receive 1/3 of the net proceeds resulting from the sale of VS-01’s pediatric review voucher to a third party, or 1/3 of the fair market value of this pediatric review voucher if we opt to apply it to one of our own programs.
The summary provided above does not purport to be complete and is qualified in its entirety by reference to the complete agreement, which is attached as an exhibit to this annual report.
Convertible Bonds (OCEANEs)
In October 2017, we issued convertible bonds (OCEANEs) for gross proceeds of €180.0 million, with a maturity date initially of October 16, 2022.
On November 23, 2020, we presented to all OCEANEs bondholders a two-prong renegotiation offer:
•A partial buyback of the outstanding OCEANEs for a maximum amount of 3,048,780 OCEANEs at €16.40 per bond; and
•An amendment of the terms of the remaining OCEANEs to extend their maturity (by 3 years) and increase the conversion ratio (to 5.5 shares per bond).
At the Shareholders’ and Bondholders’ Meetings on January 25, 2021, the shareholders and bondholders approved this renegotiation offer and we completed the partial buyback of 2,895,260 OCEANEs at a price of €16.40 (including accrued interest of €0.30) per bond for a total buyback cost of €47.48 million on January 29, 2021. We then cancelled the repurchase of OCEANEs. Following the renegotiation, the OCEANEs bear interest at an annual nominal rate of 3.50% payable semi-annually in arrears on April 16 and October 16 of each year (or the following business day if this date is not a business day). The OCEANEs will be redeemed at par on October 16, 2025 (or the following business day if this date is not a business day). The effective interest rate is 8.8%.
The nominal unit value of the OCEANEs was set at €29.60. The OCEANEs conversion ratio is 5.5 shares for one OCEANE, subject to any subsequent adjustments.
The OCEANEs may be redeemed early at the option of the Company, under certain conditions. Specifically, the OCEANEs may be redeemed early at the option of the Company from November 6, 2020 onward if i) the mathematical average of the volume-weighted average price of GENFIT shares on the regulated market of Euronext in Paris and ii) the conversion ratio of the shares in force (over a period of 20 trading days) exceeds 150% of the nominal value of the OCEANEs bonds.
As of April 1, 2024, there were 1,923,662 OCEANEs outstanding, and represent 21.2% of the share capital of the Company. The maximum dilution to GENFIT’s share capital in the event of full conversion would be 17.5%, with approximately €56.9 million nominal amount outstanding.
The OCEANEs are admitted to trading on Euronext Access (the free market of Euronext in Paris).
Under current French foreign exchange control regulations there are no limitations on the amount of cash payments that we may remit to residents of foreign countries. Laws and regulations concerning foreign exchange controls do, however, require that all payments or transfers of funds made by a French resident to a non-resident such as dividend payments be handled by an accredited intermediary. All registered banks and substantially all credit institutions in France are accredited intermediaries.
The following describes material U.S. federal income tax and French tax considerations relating to the acquisition, ownership and disposition of ADSs by a U.S. holder (as defined below). This summary addresses these tax considerations only for U.S. holders that will hold such ADSs as capital assets (generally, property held for investment). This summary does not address all U.S. federal income tax and French tax matters that may be relevant to a particular U.S. holder.holder, such as the effects of Section 451(b) of the U.S. Internal Revenue Code of 1986, as amended, or the Code. This summary does not address tax considerations applicable to a holder of ADSs that may be subject to special tax rules including, without limitation, the following:
•banks, financial institutions or insurance companies;
•brokers, dealers or traders in securities, currencies, commodities, or notional principal contracts;
•tax-exempt entities or organizations, including an “individual retirement account” or “Roth IRA” as defined in Section 408 or 408A of the Code (as defined below), respectively;
•real estate investment trusts, regulated investment companies or grantor trusts;
•persons that hold the ADSs as part of a “hedging,” “integrated,”“integrated”, “wash sale” or “conversion” transaction or as a position in a “straddle” for U.S. federal income tax purposes;
•S corporations, partnerships, or other entities or arrangements classified as partnerships for U.S. federal income tax purposes;
•certain former citizens or long term residents of the United States;
•persons that received ADSs as compensation for the performance of services;
•persons acquiring ADSs in connection with a trade or business conducted outside of the United States, including a permanent establishment or a fixed base in France;
•holders that elect to apply the provisions of Section 1400Z-2 of the Code to any gain realized upon a disposition of our ADSs;
•holders that own directly, indirectly, or through attribution 10% or more of the voting power or value of our ADSs and shares or, in the case of the discussion of French tax consequences, 5% or more of the voting stock or our share capital; and
•holders that have a “functional currency” other than the U.S. dollar.
Holders of ADSs who fall within one of the categories above are advised to consult their usual tax advisor regarding the specific tax consequences which may apply to their particular situation.
For the purposes of this description, a “U.S. holder” is a beneficial owner of ADSs that is (or is treated as), for U.S. federal income tax purposes:
•an individual who is a citizen or resident of the United States;
•a corporation, or other entity taxable as a corporation, created or organized in or under the laws of the United States, any state therein or the District of Columbia;
•an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or
•a trust, if a court within the United States is able to exercise primary supervision over its administration and one or more U.S. persons have the authority to control all of the substantial decisions of such trust, or if such trust has a valid election in effect under applicable U.S. Treasury Regulations to be treated as a U.S. person.
If a partnership (or any other entity treated as a partnership for U.S. federal income tax purposes) holds ADSs, the tax consequences relating to an investment in the ADSs will depend in part upon the status of the partner and the activities of the partnership. Such a partner or partnership should consult his, her or its tax advisor regarding the specific tax considerations of acquiring, owning and disposing of the ADSs in its particular circumstances.
The discussion in this section is based in part upon the representations of the depositary and the assumption that each obligation in the deposit agreement and any related agreement will be performed in accordance with its terms.
Persons considering an investment in the ADSs should consult their own tax advisors as to the particular tax consequences applicable to them relating to the acquisition, ownership and disposition of the ADSs, including the applicability of U.S. federal, state and local tax laws, French tax laws and other non-U.S. tax laws.
Material French Tax Considerations
The following describes the material French income tax consequences to U.S. holdersHolders of purchasing, owning and disposing of our ordinary shares or ADSs.
This discussion does not purport to be a complete analysis or listing of all potential tax effects of the acquisition, ownership or disposition of our ordinary shares or ADSs to any particular investor, and does not discuss tax considerations that arise from rules of general application or that are generally assumed to be known by investors. All of the following is subject to change. Such changes could apply retroactively and could affect the consequences described below.
In 2011, France introduced a comprehensive set ofFrench tax rules applicable to French assets that are held by or in foreign trusts. These rulestrusts generally provide inter alia for the inclusion of trust assets in the settlor’s net assets for the purpose of applying the former French wealth tax (replaced by the French real estate wealth tax, as from January 1, 2018), for the application of French gift and death dutiesestate tax to French assets held in trust, for a specific tax on capital on the French assets of foreign trusts not already subject to the former French wealth tax (replaced by the French real estate wealth tax as from January 1, 2018) and for a number of French tax reporting and disclosure obligations. The following discussion does not address the French tax consequences applicable to securities (including ordinary shares or ADSs) held in trusts. If our ordinary shares or ADSs are held in trust, the grantor, trustee and beneficiary are advised to consult their own tax advisor regarding the specific tax consequences of acquiring, owning and disposing of such securities.
The description of the French income tax and real estate wealth tax consequences set forth below is based on the Convention Between the Government of the United States of America and the Government of the French Republic for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion with Respect to Taxes on Income and Capital of August 31, 1994, or the U.S.-France Tax Treaty, which came into force on December 30, 1995 (as amended by any subsequent protocols, including the protocol of January 13, 2009), and the tax guidelines issued by the French tax authorities in force as of the date of this annual report.
For the purposes of this discussion, the term “U.S. Holder” means a beneficial owner of securities that is (1) an individual who is a U.S. citizen or resident for U.S. federal income tax purposes, (2) a U.S. domestic corporation or certain other entities created or organized in or under the laws of the United States or any state thereof, or (3) otherwise subject to U.S. federal income taxation on a net income basis in respect of securities.
If a partnership (or any other entity treated as partnership for U.S. federal income tax purposes) holds ordinary shares or ADSs, the tax treatment of the partnership and a partner in such partnership generally will depend upon the status of the partner and the activities of the partnership. If a U.S. Holder is a partner in a partnership that holds securities, such holder is urged to consult its own tax advisor regarding the specific tax consequences of acquiring, owning and disposing of securities.
This discussion applies only to investors that hold ordinary shares or ADSs as capital assets that have the U.S. dollar as their functional currency, that are entitled to U.S.-France Tax Treaty benefits under the “Limitation on Benefits” provision contained in the Treaty.
U.S.-France Tax Treaty, and whose ownership of the ordinary shares or ADSs is not effectively connected to a permanent establishment or a fixed base in France. Certain U.S. Holders (including, but not limited to, U.S. expatriates, partnerships or other entities classified as partnerships for U.S. federal income tax purposes, banks, insurance companies, regulated investment companies, tax-exempt organizations, financial institutions, persons subject to the alternative minimum tax, persons who acquired the securities pursuant to the exercise of employee share options or otherwise as compensation, persons that own (directly, indirectly or by attribution) 5% or more of our voting stock or 5% or more of our outstanding share capital, dealers in securities or currencies, brokers, mutual funds, individual retirement or other tax-deferred accounts persons that elect to mark their securities to market for U.S. federal income tax purposes and persons holding securities as a position in a synthetic security, straddle or conversion transaction) may be subject to special rules not discussed below.U.S. holdersHolders are urged to consult their own tax advisors regarding the tax consequences of the purchase, ownership and disposition of securities in light of their particular circumstances, especially with regard to the “Limitations on Benefits” provision contained in the U.S.-France Tax Treaty.
Estate and Gift Taxes
In general, a transfer of securities by gift or by reason of death of a U.S. holderHolder that would otherwise be subject to French gift or inheritance tax, respectively, will not be subject to such French tax by reason of the Convention Betweenbetween the Government of the United States of America and the Government of the French Republic for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion with Respect to Taxes on Estates, Inheritances and Gifts, dated November 24, 1978 (as amended from time to time), unless (1) the donor or the transferor is domiciled in France at the time of making the gift or at the time of his or her death, or (2) the securities were used in, or held for use in, the conduct of a business through a permanent establishment or a fixed base in France.
Financial Transactions Tax and Registration Dutieson Financial Transactions
Pursuant to Article 235 ter ZD of the French tax code (Code général des impôts, the “FTC”), purchases of shares or ADSs of a French company listed on a regulated market of the European Union or on a foreign regulated market formally acknowledged by the French Financial Market Authority (AMF) are subject to a 0.3% French tax on financial transactions provided that, broadly, the issuer’s market capitalization exceeds 1 billion euros as of December 1 of the taxation year. A list of companies whose market capitalization exceeds 1 billion euros as of December 1 of the taxation year within the meaning of Article 235 ter ZD of the FTC is published by the French tax authorities on an annual basis in their official guidelines. Pursuant to the official guidelines BOI-ANNX-000467BOI-ANNX-000467-20/12/2023 issued on December 29, 2021,20, 2023, we are currently not included in such list.
Moreover, Nasdaq Global Select Market, on which ADSs are listed, is not currently acknowledged by the AMF but this may change in the future.
As a consequence, neither the ADSs nor the ordinary shares are currently within the scope of the French tax on financial transactions.
Purchases of our securities may be subject to such tax in the future provided that our market capitalization exceeds 1 billion euros as of December 1 of the taxation year and that the Nasdaq Global Select Market is acknowledged by the AMF.
Registration Duties
In the case where Article 235 ter ZD of the FTC is not applicable, transfers of shares issued by a French company which are listed on a regulated or organized market within the meaning of the French Monetary and Financial Code are subject to uncapped registration duties at the rate of 0.1% if the transfer is evidenced by a written statement (“acte”) executed either in France or outside France. As ordinary shares of our company are listed on Euronext Paris, which is an organized market within the meaning of the French Monetary and Financial Code, their transfer should be subject to uncapped registration duties at the rate of 0.1% in case of the existence of a written statement (“acte”) and provided that Article 235 ter ZD of the FTC is not applicable. Although there is no case law or official guidelines published by the French tax authorities on this point, transfer of ADSs should remain outside of the scope of the aforementioned 0.1% registration duties. U.S. Holders are urged to consult their own tax advisor about the possible application of the registration duty upon the transfer of ADSs.
Tax on Sale or Other Disposals
As a matter of principle, under French tax law, and to the extent GENFIT is not a real estate company for the purpose of Article 244 bis A of the FTC,a U.S. holderHolder should not be subject to any French tax on any capital gain from the sale, exchange, repurchase or redemption by us of ordinary shares or ADSs, provided such U.S. holderHolder is not a French tax resident for French tax purposes and has not held more than 25% of our dividend rights, known as “droits aux bénéfices sociaux,” at any time during the preceding five years, either directly or indirectly, and, as relates to individuals, alone or with relatives (asand, it has not transferred ordinary shares or ADSs as part of redemption by GENFIT, in which case the proceeds may under certain circumstances be partially or fully characterized as dividends under French domestic law and, as result, be subject to French dividend withholding tax.
As an exception, a U.S holderHolder domiciled, established or incorporated in certain non-cooperative States or territories as defined in Article 238-0 A of the FTC, except for those mentioned in paragraph 2 bis-2° of the same Article, should be subject to a 75% withholding tax in France on any such capital gain, regardless of the fraction of the dividend rights it holds, subject to safe-harbor provisions and the more favorable provisions of the U.S.-France Tax Treaty. The list of non-cooperative states or territories is published by decree and is in principle updated annually. This list was last updated on February 3, 2023, and currently includes American Samoa, Anguilla, the Bahamas, the British Virgin Islands, Fiji, Guam, Palaos, Panama, Samoa, Seychelles, Trinidad and Tobago, Turk and Caicos, the United States Virgin Islands and Vanuatu. States referred to in Article 238-0 A, 2 bis-2° of the FTC, and thus outside of the scope of Article 244 bis B of the FTC, are currently American Samoa, Fiji, Guam, Palaos, Samoa, Trinidad and Tobago and the United States Virgin Islands.
Under application of the U.S.-France Tax Treaty, a U.S. holderHolder who is a U.S. resident for purposes of the U.S.-France Tax Treaty and entitled to Treaty benefit will not be subject to French tax on any such capital gain unless the ordinary shares or the ADSs form part of the business property of a permanent establishment or fixed base that the U.S. holderHolder has in France. U.S. holdersHolders who own ordinary shares or ADSs through U.S. partnerships that are not resident for U.S.-France Tax Treaty purposes are advised to consult their own tax advisors regarding their French tax treatment and their eligibility for Treaty benefits in light of their own particular circumstances.
A U.S. holderHolder that is not a U.S. resident for U.S.-France Tax Treaty purposes or is not entitled to Treaty benefit (and in both cases is not domiciled, established or incorporated in certain non-cooperative States or territories as defined in Article 238-0 A of the FTC, except for those mentioned in paragraph 2-bis-2°) and has held more than 25% of our dividend rights, known as “droits aux bénéfices sociaux,” at any time during the preceding five years, either directly or indirectly, and, as relates to individuals, alone or with relatives will be subject to a levy in France at the rate (1) of 12.8% for individuals and (2) 25% for fiscal years beginning on or after January 1st, 2022, for legal persons. However, eligible non-French tax resident legal entities may claim a refund of the 25% French levy to the extent such tax exceeds the amount that would have been due under French corporate income tax if they had been French tax residents. This refund mechanism is only available to certain legal entities. Non-French tax resident legal entities are advised to consult their own tax adviser regarding their French tax treatment and their eligibility to this refund mechanism.
The above French provisions expressly apply to sale, repurchase or redemption by us of ordinary shares. U.S. Holders who own ordinary shares or ADSs through U.S. partnerships that are not resident for U.S.-France Tax Treaty purposes are advised to consult their own tax adviser regarding their French tax treatment and their eligibility for Treaty benefits in light of their own particular circumstances.
Special rules apply to U.S. holdersHolders who are residents of more than one country.
Taxation of Dividends
Dividends paid by a French corporation to non-residents of France are generally subject to French withholding tax at a rate of (i) 25% for fiscal years beginning on or after January 1st, 2022, for payments benefiting legal persons which are not French tax residents, and (ii) 12.8% for payments benefiting individuals who are not French tax residents. Dividends paid by a French corporation in certain non-cooperative States or territories, as defined in Article 238-0 A of the FTC (except for those mentioned in paragraph 2-bis-2°), will generally be subject to French withholding tax at a rate of 75%, save for the safe-harbor provisions to apply. However, eligible U.S. holdersHolders which are legal entities and entitled to U.S.-France Tax Treaty benefits under the “Limitation on Benefits” provision contained in the U.S.-France Tax Treaty who are U.S. residents, as defined pursuant to the provisions of the U.S.-France Tax Treaty, will not be subject to this 25% or 75% withholding tax rate, but may be subject to the withholding tax at a reduced rate (as described below).
Under the U.S.-France Tax Treaty, the rate of French withholding tax on dividends paid to an eligible U.S. holderHolder who is a U.S. resident as defined pursuant to the provisions of the U.S.-France Tax Treaty and whose ownership of the ordinary shares or ADSs is not effectively connected with a permanent establishment or fixed base that such U.S. holderHolder has in France, is generally reduced to 15%, or to 5% if such U.S. holderHolder is a corporation and owns directly or indirectly at least 10% of the share capital of the issuer; such U.S. holderHolder may claim a refund from the French tax authorities of the amount withheld in excess of the U.S.-France Tax Treaty rates of 15% or 5%, if any.
For U.S. holdersHolders that are not individuals but are U.S. residents, as defined pursuant to the provisions of the U.S.-France Tax Treaty, the requirements for eligibility for Treaty benefits, including the reduced 5% or 15% withholding tax rates contained in the “Limitation on Benefits” provision of the U.S.-France Tax Treaty, are complex, and certain technical changes were made to these requirements by the protocol of January 13, 2009. U.S. holdersHolders are advised to consult their own tax advisors regarding their eligibility for Treaty benefits in light of their own particular circumstances.
Dividends paid to an eligible U.S. holderHolder may immediately be subject to the reduced rates of 5% or 15% provided that:
•such holder establishes before the date of payment that it is a U.S. resident under the U.S.-France Tax Treaty by completing and providing the depositary with a treaty form (Form 5000) in accordance with French guidelines (BOI-INT-DG-20-20-20-20 dated September 12, 2012;2012); or
•the depositary or other financial institution managing the securities account in the U.S. of such holder provides the French paying agent with a document listing certain information about the U.S. holderHolder and its ordinary shares or ADSs and a certificate whereby the financial institution managing the U.S. holder’sHolder’s securities account in the United States takes full responsibility for the accuracy of the information provided in the document.
Otherwise, dividends paid to a U.S. holder,Holder, if such U.S. holderHolder is a legal person, will be subject to French withholding tax at the rate of 25%, or 75% if paid in certain non-cooperative States or territories (as defined in Article 238-0 A of the FTC - except for those mentioned in paragraph 2 bis-2°), and then reduced at a later date to 5% or 15%, provided that such holder duly completes and provides through the French paying agent, the French tax authorities with the treaty forms Form 5000 and Form 5001 before December 31(due to recent case law regarding status of the second calendar year following the year during which the dividend is paid.limitation for filing a withholding tax claim; U.S. Holders are advised to consult their own tax advisors in this respect).
Certain qualifying pension funds and certain other tax-exempt entities areand certain US residents may be subject to the same generalspecific filing requirements as other U.S. holders except that they may haverequirements. They are advised to supply additional documentation evidencingconsult their entitlement to these benefits.own tax advisors on this point.
Form 5000 and Form 5001, together with instructions, will be provided by the depositary to all U.S. holdersHolders registered with the depositary. The depositary will arrange for the filing with the French tax authorities of all such forms properly completed and executed by U.S. holdersHolders of ordinary shares or ADSs and returned to the depositary in sufficient time so that they may be filed with the French tax authorities before the distribution in order to immediately obtain a reduced withholding tax rate. Otherwise, the depositary must withhold tax at the full rate of 25% or 75% as applicable. In that case, the U.S. holdersHolders may claim a refund from the French tax authorities of the excess withholding tax.
In any case, individual taxpayers who are not fiscally domiciled in France should not have to comply with these procedures if the French withholding tax applying to them is lower than 15%. In particular, since the withholding tax rate applicable under French domestic law to U.S. Holders who are individuals does not exceed the cap provided in the U.S.-France Tax Treaty (i.e., 15%), the 12.8% rate shall apply, without any reduction provided under the U.S.-France Tax Treaty (except in the particular situation when the dividends are paid to such U.S. Holders out of France in a non-cooperative State or territory as defined in Article 238-0 A of the FTC other than those mentioned in 2° of 2 bis of the same Article 238-0 A of the FTC and are subject to the 75% withholding tax in France).
In addition, please note that pursuant to Article 235 quater of the FTC (introduced by the French finance bill No. 2019-1479 for 2020) and under certain conditions (in particular, in addition to certain reporting obligations, the interest held in the distributing company must not enable the beneficiary to participate effectively in the management or control of that company and the beneficiary company is located in a country that has signed an administrative assistance agreement with France to combat tax evasion and avoidance, as well as an administrative assistance agreement on tax collection, and that is not a non-cooperative country), a corporate U.S. Holder which is in a tax loss position or which tax result is nil due to offset of tax losses (French Administrative Supreme Court, October 18, 2022, n° 466329) for the fiscal year during which the dividend is received may be entitled to a deferral regime, and obtain a withholding tax refund. The tax deferral ends in respect of the first financial year during which this U.S. Holder is in a profit making position, as well as in the cases set out in Article 235 quater of the FTC. The refund must be claimed within the same period applicable to claim related to taxes other than local taxes. Also, pursuant to Article 235 quinquies of the FTC and under certain conditions, a corporate U.S. Holder may be entitled to a refund of a fraction of the withholding tax, up to the difference between the withholding tax paid (on a gross basis) and the withholding tax based on the dividend net of the expenses incurred for the acquisition and conservation directly related to the income, provided (i) that these expenses would have been tax deductible had the U.S. Holder been established in France, and (ii) that the tax rules in the United States do not allow the U.S. Holder to offset the withholding tax.
Real Estate Wealth Tax
As from January 1, 2018, the French wealth tax (impôt de solidarité sur la fortune) is repealed and replaced by the French real estate wealth tax (impôt sur la fortune immobilière)re, or IFI). The scope of such new tax is narrowed to real estate assets (and certain assets deemed to be real estate assets) or rights, held directly or indirectly through one or more legal entities and whose net taxable assets amount to at least €1,300,000.
Broadly, subject to provisions of double tax treaties and to certain exceptions, individuals who are not residents of France for tax purposes within the meaning of Article 4 B of the FTC, are subject to real estate wealth tax (impôt sur la fortune immobilière) in France in respect of the portion of the value of their shares of our company representing French real estate assets (Article 965, 2° of the FTC). Some exceptions are provided by the FTC. For instance, any participations representing less than 10% of the share capital of an operational company andIn particular, GENFIT’s ordinary shares representing real estate for the professional use of the company considered shallor ADSs owned by a U.S. Holder should not fall within the scope of the French real estate wealth tax (impôt sur la fortune immobilière).IFI provided that such U.S. Holder does not own (together with the members of his/her household) directly or indirectly a shareholding exceeding 10% of the financial rights and voting rights of GENFIT. U.S. Holders holding directly or indirectly a shareholding exceeding 10% of the financial rights and voting rights of GENFIT should seek additional advice.
Under the U.S.-France Tax Treaty (the provisions of which should be applicable to this new real estate wealth tax (impôt sur la fortune immobilière) in France)IFI), the French real estate wealth tax (impôt sur la fortune immobilière)IFI will however generally not apply to shares that are held by U.S. Holders who (1) own, alone or with related persons, directly or indirectly, shares in our company which give rise to less than 25% of the rights in the company’s earnings, and (2) do not own their shares in connection with a permanent establishment or a fixed base through which the U.S. Holder carries on business or performs personal services in France.
U.S. Holders are advised to consult their usual tax advisor regarding the specific tax consequences which may apply to their particular situation with respect to such French real estate wealth tax (impôt sur la fortune immobilière).IFI.
Material U.S. Federal Income Tax Considerations
This section discusses the material U.S. federal income tax considerations relating to the acquisition, ownership and disposition of ADSs by a U.S. holder. This description does not address the U.S. federal estate, gift, or alternative minimum tax considerations, or any U.S. state, local, or non-U.S. tax considerations, of the acquisition, ownership and disposition of the ADSs.
This description is based on the U.S. Internal Revenue Code of 1986, as amended, or the Code, existing, proposed and temporary U.S. Treasury Regulations promulgated thereunder and administrative and judicial interpretations thereof, in each case as in effect and available on the date hereof. All the foregoing is subject to change, which change could apply retroactively, and to differing interpretations, all of which could affect the tax considerations described below. There can be no assurances that the U.S. Internal Revenue Service, or the IRS, will not take a position concerning the tax consequences of the acquisition, ownership and disposition of the ADSs or that such a position would not be sustained by a court. We have not obtained, nor do we intend to obtain, a ruling with respect to the U.S. federal income tax considerations in the purchase, ownership or disposition of our ADSs. Accordingly, holders should consult their own tax advisers concerning the U.S. federal, state, local and non-U.S. tax consequences of acquiring, owning and disposing of the ADSs in their particular circumstances.
The discussion below assumes that the representations contained in the deposit agreement are true and that the obligations in the deposit agreement and any related agreements will be complied with in accordance with their terms.
In general, and taking into account the earlier assumptions, for U.S. federal income and French tax purposes, a U.S. holder holding ADSs will be treated as the owner of the shares represented by the ADSs. Exchanges of shares for ADSs, and ADSs for shares, generally will not be subject to U.S. federal income or to French tax.
In addition, this discussion in this section is limited to holders who are not resident in France for purposes of the U.S.-France Tax Treaty.
Passive Foreign Investment Company Considerations.Considerations
If we are classified as a “passivepassive foreign investment company,” or a PFIC, in any taxable year, a U.S. holder will be subject to special rules generally intended to reduce or eliminate any benefits from the deferral of U.S. federal income tax that a U.S. holder could derive from investing in a non-U.S. company that does not distribute all of its earnings on a current basis.
We will be classified as a PFIC for U.S. federal income tax purposes in any taxable year in which, after applying certain look-through rules with respect to the income and assets of our subsidiaries, either: (1) at least 75% of theour gross income is “passive income” or (2) at least 50% of the quarterly weighted-average value of our total gross assets (which would generally be measured by fair market value of our assets, and for which purpose the total value of our assets may be determined in part by the market value of the ADSs and our ordinary shares, which are subject to change) is attributable to assets that produce “passive income” or are held for the production of “passive income.”
Passive income for this purpose generally includes dividends, interest, royalties, rents, gains from commodities and securities transactions, the excess of gains over losses from the disposition of assets which produce passive income, and includes amounts derived by reason of the temporary investment of funds raised in offerings of the ADSs. If a non-U.S. corporation owns directly or indirectly at least 25% by value of the stock of another corporation or partnership, the non-U.S. corporation is treated for purposes of the PFIC tests as owning its proportionate share of the assets of thesuch other corporation or partnership and as receiving directly its proportionate share of thesuch other corporation’s or partnership's income. The determination of whether we are a PFIC is a fact-intensive determination made on an annual basis and the applicable law is subject to varying interpretation. If we are classified as a PFIC in any taxable year during which a U.S. holder owns our ordinary shares or ADSs, such U.S. holder will be subject to special tax rules discussed below and could suffer adverse tax consequences.
The fair market value of our assets may be determined in large part by reference to the market price of the ADSs and our ordinary shares, which is likely to continue to fluctuate. Therefore, fluctuationsFluctuations in the market price of our ordinary shares or ADSs may result in our being a PFIC for any taxable year. In addition, the composition of our income and assets will be affected by how, and how quickly, we use the cash proceeds from our offerings. BasedAlthough the matter is not free from doubt, based on our analysis of our income, assets, activities and market capitalization for our taxable year ended December 31, 2021,2023, we do not believe that we were classified as a PFIC for the taxable year ended December 31, 2021.2023. Whether we are a PFIC for any taxable year will depend on our assets and income (including whether we receive certain non-refundable grants or subsidies, and whether such amounts along with reimbursements of certain refundable research tax credits and certain intercompany service payments will constitute gross income for purposes of the PFIC income test) in each year, and because this is a factual determination made annually after the end of each taxable year, there can be no assurance that we will not be considered a PFIC in any taxable year. In addition, we hold a substantial amount of cash and cash equivalents. Because the calculationequivalents, which are generally treated as a passive asset for purposes of the value of our assets may be based in part on the value of our ordinary shares or ADSs, the value of which may fluctuate considerably, ourdetermining PFIC status. Our PFIC status may change from year to year and it is difficult to predict whether we will be a PFIC for the current year or any future year. Therefore, we have not yet made any determination as to our expected PFIC status for the current taxable year. However, we could continue to be considered a PFIC for the current taxable year or a future taxable year if the current percentage of our passive assets compared to our total assets remains the same or increases. Even if we determine that we are not a PFIC after the close of a taxable year, thereThere can be no assurance that the IRS will agree with our conclusion.conclusion with respect to any taxable year that we were not a PFIC for such taxable year. Our U.S. counsel expresses no opinion regarding our conclusions or our expectations regarding our PFIC status.
If we are or become classified as a PFIC in any year with respect to which a U.S. holder owns our ordinary shares or ADSs, we will continue to be treated as a PFIC with respect to such U.S. holder in all succeeding years during which the U.S. holder owns the ordinary shares or ADSs, regardless of whether we continue to meet the tests described above, unless we cease to be a PFIC and the U.S. holder has made a “deemed sale” election under the PFIC rules or is eligible to make and makes a mark-to-market election (as described below), with respect to all taxable years during such U.S. holder’s holding period in which we are a PFIC. If the “deemed sale” election is made, a U.S. holder will be deemed to have sold the ordinary shares or ADSs the U.S. holder holds at their fair market value as of the date of such deemed sale, and any gain from such deemed sale would be subject to the rules described below. After the deemed sale election, so long as we do not become a PFIC in a subsequent taxable year, the U.S. holder’s ordinary shares or ADSs with respect to which such election was made will not be treated as shares in a PFIC and the U.S. holder will not be subject to the rules described below with respect to any “excess distribution” the U.S. holder receives from us or any gain from an actual sale or other disposition of the ordinary shares or ADSs. U.S. holders should consult their tax advisors as to the possibility and consequences of making a deemed sale election if such election becomes available.
If we are or become a PFIC, and you are a U.S. holder that does not make one of the elections described above (and below in further detail), a special tax regime will apply to both (a) any “excess distribution” by us to you (generally, your ratable portion of distributions in any year which are greater than 125% of the average annual distribution received by you in the shorter of the three preceding years or your holding period for the ADSs) and (b) any gain realized on the sale or other disposition of the ADSs. Under this regime, any excess distribution andor realized gain will be treated as ordinary income and will be subject to tax as if (a) the excess distribution or gain had been realized ratably over your holding period in the ADSs, (b) the amount deemed realized in each year had been subject to tax in each year of that holding period at the highest marginal rate for such year (other than income allocated to the current period or any taxable period before we became a PFIC, which would be subject to tax at the U.S. holder’s regular ordinary income rate for the current year and would not be subject to the interest charge discussed below), and (c) the interest charge generally applicable to underpayments of tax had been imposed on the taxes deemed to have been payable in those years. In addition, dividend distributions made to you will not qualify for the lower rates of taxation applicable to qualified dividends as discussed below under the heading “Distributions.”
Certain elections may alleviate some of the adverse consequences of PFIC status and would result in an alternative treatment of the ADSs. If a U.S. holder makes a mark-to-market election, the U.S. holder generally will recognize as ordinary income any excess of the fair market value of the ADSs at the end of each taxable year over theirthe U.S. holder's adjusted tax basis in such ADSs, and will recognize an ordinary loss in respect of any excess of the adjusted tax basis of the ADSs over their fair market value at the end of the taxable year (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). If a U.S. holder makes the election, the U.S. holder’s tax basis in the ADSs will be adjusted to reflect these income or loss amounts. Any gain recognized on the sale or other disposition of ADSs in a year when we are a PFIC will be treated as ordinary income and any loss will be treated as an ordinary loss (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). The mark-to-market election is available only if we are a PFIC and the ADSs are “regularly traded” on a “qualified exchange.” The ADSs will be treated as “regularly traded” in any calendar year in which more than a de minimis quantity of the ADSs are traded on a qualified exchange on at least 15 days during each calendar quarter (subject to the rule that trades that have as one of their principal purposes the meeting of the trading requirement as disregarded). The Nasdaq Global Select Market is a qualified exchange for this purpose and, consequently, if the ADSs are regularly traded, the mark-to-market election will be available to a U.S. holder.
However, a mark-to-market election generally cannot be made for equity interests in any lower-tier PFICs that we own, unless shares of such lower-tier PFIC are themselves “marketable.” As a result, even if a U.S. holder validly makes a mark-to-market election with respect to our ordinary shares or ADSs, the U.S. holder may continue to be subject to the PFIC rules (described above) with respect to its indirect interest in any of our investments that are treated as an equity interest in a PFIC for U.S. federal income tax purposes. U.S. holders should consult their tax advisors as to the availability and desirability of a mark-to-market election, as well as the impact of such election on interests in any lower-tier PFICs.
Notwithstanding our belief that we were classified as a PFIC for the taxable year ended December 31, 2021, weWe do not currently intend to provide the information necessary for U.S. holders to make qualified electing fund elections for such taxable year or any other taxable year for which we are treated as a PFIC. U.S. holders should consult their tax advisors to determine whether any of these elections would be available and if so, what the consequences of the alternative treatments would be in their particular circumstances.
If we are determined to be a PFIC, the general tax treatment for U.S. holders described in this section would apply to indirect distributions and gains deemed to be realized by U.S. holders in respect of any of our subsidiaries that also may be determined to be PFICs. U.S. holders should consult their tax advisors regarding the application of the PFIC rules to our subsidiaries.
If a U.S. holder owns ADSs during any taxable year in which we are a PFIC, the U.S. holder generally will be required to file an IRS Form 8621 (Information Return by a Shareholder of a Passive Foreign Investment Company or Qualified Electing Fund) with respect to the company, generally with the U.S. holder’s federal income tax return for that year. If we are a PFIC for a given taxable year, then youU.S. holders should consult yourtheir tax advisor concerning yoursuch annual filing requirements.
The U.S. federal income tax rules relating to PFICs are complex. U.S. holders and (and prospective U.S. holders) are urged to consult their own tax advisers with respect to the acquisition, ownership and disposition of the ADSs, the consequences to them of an investment in a PFIC, any elections available with respect to the ADSs and the IRS information reporting obligations with respect to the acquisition, ownership and disposition of the ADSs.
Distributions.Distributions
Subject to the discussion under “— Passive Foreign Investment Company Considerations,” above, the gross amount of any distribution (including any amounts withheld in respect of foreign tax) actually or constructively received by a U.S. holder with respect to ADSs will generally be taxable to the U.S. holder as a dividend to the extent of the U.S. holder’s pro rata share of our current and accumulated earnings and profits as determined under U.S. federal income tax principles. Distributions in excess of earnings and profits will generally be non-taxable to the U.S. holder to the extent of, and will be applied against and reduce, the U.S. holder’s adjusted tax basis in the ADSs. Distributions in excess of earnings and profits and such adjusted tax basis will generally be taxable to the U.S. holder as either long-term or short-term capital gain depending upon whether the U.S. holder has held the ADSs for more than one year as of the time such distribution is received. However, since we may not calculate our earnings and profits under U.S. federal income tax principles, it is expected that any distribution will be reported as a dividend, even if that distribution would otherwise be treated as a non-taxable return of capital or as capital gain under the rules described above. Non-corporate U.S. holders may qualify for the preferential rates of taxation with respect to dividends on ADSs applicable to long-term capital gains (i.e., gains from the sale of capital assets held for more than one year) and qualified dividend income (as discussed below) if we are a “qualified foreign corporation” and certain other requirements (discussed below) are met. A non-U.S. corporation (other than a corporation that is classified as a PFIC for the taxable year in which the dividend is paid or the preceding taxable year) generally will be considered to be a qualified foreign corporation (a) if it is eligible for the benefits of a comprehensive tax treaty with the United States which the Secretary of Treasury of the United States determines is satisfactory for such purposes of this provision and which includes an exchange of information provision, or (b) with respect to any dividend it pays on ADSs which are readily tradable on an established securities market in the United States. Our ADSs are currently listed on the Nasdaq Global Select Market, which is an established securities market in the United States, and we expect the ADSs to be readily tradable on the Nasdaq Global Select Market. However, there can be no assurance in this regard. The Company, which is incorporated under the laws of France, believes that it qualifies as a resident of France for purposes of, and is eligible for the benefits of, the Convention between the Government of the United States of America and the Government of the French Republic for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion with Respect to Taxes on Income and Capital, signed on August 31, 1994, as amended and currently in force, or the U.S.-France Tax Treaty, although there can be no assurance in this regard. Further, the IRS has determined that the U.S.-France Tax Treaty is satisfactory for purposes of the qualified dividend rules and that it includes an exchange-of-information program. Therefore, subject to the discussion under “— Passive Foreign Investment Company Considerations,” above, such dividends will generally be “qualified dividend income” in the hands of individual U.S. holders if such dividends are paid in a taxable year in which we were not a PFIC and were not a PFIC in the preceding taxable year, provided that a holding period requirement (more than 60 days of ownership, without protection from the risk of loss, during the 121-day period beginning 60 days before the ex-dividend date) and certain other requirements are met. The dividends will not be eligible for the dividends-received deduction generally allowed to corporate U.S. holders.
ASubject to applicable limitations and the Final FTC Treasury Regulations (as defined below), a U.S. holder generally may claim the amount of any French withholding tax on a distribution not exceeding the rate provided by the U.S.-France Tax Treaty as either a deduction from gross income or a credit against its U.S. federal income tax liability. French taxes withheld in excess of the rate applicable with respect to such U.S. holder under the U.S.-France Tax Treaty will not be eligible for a credit against a U.S. holder’s federal income tax liability. Treasury Regulations issued on December 28, 2021, which apply to foreign taxes paid or accrued in taxable years beginning on or after December 28, 2021, or the Final FTC Treasury Regulations, impose additional requirements for foreign taxes to be eligible for credit. However, the IRS has indicated that taxpayers may defer the application of many of the additional requirements until further notice. U.S. holders should consult their tax advisors regarding the availability of foreign tax credits for any amounts withheld with respect to dividends on ADSs or ordinary shares, including under the Final FTC Treasury Regulations.
The foreign tax credit is subject to numerous complex limitations that must be determined and applied on an individual basis. Generally, the credit cannot exceed the proportionate share of a U.S. holder’s U.S. federal income tax liability that such U.S. holder’s taxable income bears to such U.S. holder’s worldwide taxable income. In applying this limitation, a U.S. holder’s various items of income and deduction must be classified, under complex rules, as either “foreign source” or “U.S. source.” This limitation is calculated separately with respect to specific categories of income. The amount of a distribution with respect to the ADSs that is treated as a “dividend” may be lower for U.S. federal income tax purposes than it is for French income tax purposes, potentially resulting in a reduced foreign tax credit for the U.S. holder. In addition, the creditability of foreign taxes could be affected by actions taken by intermediaries in the chain of ownership between the holders of ADSs and our company if, as a result of such actions, the holders of ADSs are not properly treated as beneficial owners of the underlying ordinary shares. Each U.S. holder should consult its own tax advisors regarding the foreign tax credit rules.
In general, the amount of a distribution paid to a U.S. holder in a foreign currency will be the U.S. dollar value of the foreign currency calculated by reference to the spot exchange rate on the day the Depositary receives the distribution, regardless of whether the foreign currency is converted into U.S. dollars at that time. Any foreign currency gain or loss a U.S. holder realizes on a subsequent conversion of foreign currency into U.S. dollars will be U.S. source ordinary income or loss. If dividends received in a foreign currency are converted into U.S. dollars on the day they are received, a U.S. holder should not be required to recognize foreign currency gain or loss in respect of the dividend.
Sale, Exchange or Other Taxable Disposition of the ADSs.
A U.S. holder will generally recognize gain or loss for U.S. federal income tax purposes upon the sale, exchange or other taxable disposition of ADSs in an amount equal to the difference between the U.S. dollar value of the amount realized from such sale or exchange and the U.S. holder’s adjusted tax basis in those ADSs, determined in U.S. dollars. Subject to the discussion under “— Passive Foreign Investment Company Considerations” above, this gain or loss will generally be a capital gain or loss. The adjusted tax basis in the ADSs generally will be equal to the cost of such ADSs. Capital gain from the sale, exchange or other taxable disposition of ADSs by a non-corporate U.S. holder is generally eligible for a preferential rate of taxation applicable to capital gains, if the non-corporate U.S. holder’s holding period determined at the time of such sale, exchange or other taxable disposition for such ADSs exceeds one year (i.e., such gain is long-term taxable gain). The deductibility of capital losses for U.S. federal income tax purposes is subject to limitations. Any such gain or loss that a U.S. holder recognizes generally will be treated as U.S. source gain or loss for foreign tax credit limitation purposes.
For a cash basis taxpayer, units of foreign currency paid or received are translated into U.S. dollars at the spot rate on the settlement date of the purchase or sale. In that case, no foreign currency exchange gain or loss will result from currency fluctuations between the trade date and the settlement date of such a purchase or sale. An accrual basis taxpayer, however, may elect the same treatment required of cash basis taxpayers with respect to purchases and sales of the ADSs that are traded on an established securities market, provided the election is applied consistently from year to year. Such election may not be changed without the consent of the IRS. For an accrual basis taxpayer who does not make such election, units of foreign currency paid or received are translated into U.S. dollars at the spot rate on the trade date of the purchase or sale. Such an accrual basis taxpayer may recognize exchange gain or loss based on currency fluctuations between the trade date and the settlement date. Any foreign currency gain or loss a U.S. holder realizes will be U.S. source ordinary income or loss.
Medicare Tax.
Certain U.S. holders that are individuals, estates or trusts are subject to a 3.8% tax on all or a portion of their “net investment income,” which may include all or a portion of their dividend income and net gains from the disposition of ADSs. Each U.S. holder that is an individual, estate or trust is urged to consult its tax advisors regarding the applicability of the Medicare tax to its income and gains in respect of its investment in the ADSs.
Backup Withholding and Information Reporting.Reporting
U.S. holders generally will be subject to information reporting requirements with respect to dividends on ADSs and on the proceeds from the sale, exchange or disposition of ADSs that are paid within the United States or through U.S.-related financial intermediaries, unless the U.S. holder is an “exempt recipient.”recipient". In addition, U.S. holders may be subject to backup withholding on such payments, unless the U.S. holder provides a taxpayer identification number and a duly executed IRS Form W-9 or otherwise establishes an exemption. Backup withholding is not an additional tax, and the amount of any backup withholding will be allowed as a credit against a U.S. holder’s U.S. federal income tax liability and may entitle such holder to a refund, provided that the required information is timely furnished to the IRS.
Foreign Asset Reporting.Reporting
Certain individual U.S. holders are required to report information relating to an interest in the ADSs, subject to certain exceptions (including an exception for shares held in accounts maintained by U.S. financial institutions) by filing IRS Form 8938 (Statement of Specified Foreign Financial Assets) with their federal income tax return. U.S. holders are urged to consult their tax advisors regarding their information reporting obligations, if any, with respect to their ownership and disposition of the ADSs.
THE DISCUSSION ABOVE IS A SUMMARY OF THE MATERIAL FRENCH AND U.S. FEDERAL INCOME TAX CONSEQUENCES OF AN INVESTMENT IN OUR ADSs OR ORDINARY SHARES AND IS BASED UPON LAWS AND RELEVANT INTERPRETATIONS THEREOF IN EFFECT AS OF THE DATE OF THIS ANNUAL REPORT, ALL OF WHICH ARE SUBJECT TO CHANGE, POSSIBLY WITH RETROACTIVE EFFECT. EACH PROSPECTIVE INVESTOR IS URGED TO CONSULT ITS OWN TAX ADVISOR ABOUT THE TAX CONSEQUENCES TO IT OF AN INVESTMENT IN ADSs OR ORDINARY SHARES IN LIGHT OF THE INVESTOR’S OWN CIRCUMSTANCES.
| | | | | |
| F. | Dividends and Paying Agents |
Not applicable.
Not applicable.
We are subject to the information reporting requirements of the Exchange Act applicable to foreign private issuers and under those requirements will file reports with the SEC. Those reports may be inspected without charge at the locations described below. As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file periodic reports and financial statements with the SEC as frequently or as promptly as United States companies whose securities are registered under the Exchange Act. Nevertheless, we will file with the SEC an Annual Report on Form 20-F containing financial statements that have been examined and reported on, with and opinion expressed by an independent registered public accounting firm.
We maintain a corporate website at www.genfit.com. We intend to post our annual report on our website promptly following it being filed with the SEC. Information contained on, or that can be accessed through, our website does not constitute a part of this annual report. We have included our website address in this annual report solely as an inactive textual reference.
The Securities and Exchange Commission maintains a website (www.sec.gov) that contains reports, proxy and information statements and other information regarding registrants, such as GENFIT S.A., that file electronically with the SEC.
With respect to references made in this annual report to any contract or other document of our company, such references are not necessarily complete and you should refer to the exhibits attached or incorporated by reference to this annual report for copies of the actual contract or document.
Not required.
J. Annual Report to Security Holders
If we are required to provide an annual report to security holders in response to the requirements of Form 6-K, we will submit the annual report to security holders in electronic format in accordance with the EDGAR Filer Manual.
| | | | | |
| Item 11. | Quantitative and Qualitative Disclosures About Market Risk. |
Foreign Currency Exchange Risk
We use the euro as our functional currency and the majority of our operations are denominated in euros. However, a portion of our operating expenses is denominated in U.S. dollars and Swiss Francs (notably due to the acquisition of Versantis in 2022), as well as a significant portion of our cash and cash equivalent.equivalents. As result, we may be exposed to foreign currency risk.
Our overall exposure to the foreign exchange risk depends, in particular, on:
•the currencies in which we receivesreceive our revenues;
•the currencies chosen when agreements are entered into, such as licensing agreements, or co-marketing or co-development agreements;
•the location of clinical trials on drug or biomarker candidates;
•the ability for our co-contracting parties to indirectly transfer foreign exchange risk to us;
•our foreign exchange risk policy; and
•the fluctuation of foreign currencies against the euro.
For the years ended December 31, 20202022 and December 31, 2021,2023, expenses in U.S. dollars totaled $47.3$14.9 million and $12.6$15.3 million respectively, based on the exchange rate in effect at December 31, 20202022 and December 31, 2021.2023. As a result, an adverse 10% change in the exchange rate for the U.S. dollar against the euro would have resulted in a foreign exchange rate loss of approximately €4.3€1.6 million and €1.2€1.5 million for the years 20202022 and 20212023 respectively.
As atFor the years ended December 31, 20202022 and December 31, 2021, cash and cash equivalents2023, expenses in U.S. dollarsSwiss Francs totaled $111.2CHF2.0 million and $81.7CHF4.7 million respectively, based on the exchange rate in effect at December 31, 20202022 and December 31, 2021. 2023. As a result, an adverse 10% change in the exchange rate for the Swiss Franc against the euro would have resulted in a foreign exchange rate loss of approximately €0.2 million and €0.6 million for the years 2022 and 2023 respectively.
As of December 31, 2022 and December 31, 2023, cash and cash equivalents in U.S. dollars totaled $34.2 and $22.0 million respectively, based on the exchange rate in effect at December 31, 2022 and December 31, 2023. As a result, an adverse 10% change in the exchange rate for the U.S. dollar against the euro would have resulted in a foreign exchange rate loss of approximately €8.2€2.9 million and €6.6€1.8 million for the years 20202022 and 20212023 respectively.
As of December 31, 2022 and December 31, 2023, cash and cash equivalents in Swiss Francs totaled CHF2.3 million and CHF1.1 million respectively, based on the exchange rate in effect at December 31, 2022. As a result, an adverse 10% change in the exchange rate for the Swiss Franc against the euro would have resulted in a foreign exchange rate loss of approximately €0.2 million and €0.1 million for the years 2022 and 2023 respectively.
For the year ended December 31, 2020,2022, we recorded a total net foreign exchange lossexchange gain of €8.2€7.1 million (cumulating operating and financial exposure), including a realized lossgain of €2.7€7.5 million. For thethe year ended December 31, 2021,2023, we recorded a total net foreign exchange gain loss of €6.6€0.5 million (operating(operating and financial), including a realized gain of €0.8€0.4 million. Any such historical gains or losses do not predict the future impact of foreign exchange rate risks.
We maintain a balance between euros, U.S. dollars and US dollarsSwiss Francs in line with the projected outflows of expected resources in order to naturally cover the risk and therefore hold a significant portion of our cash in US dollars.U.S. dollars and to a lesser extent Swiss Francs. Given the significant portion of our operations denominated in USU.S. dollars and Swiss Francs, we decided to limit the conversions into euros of our USU.S. dollar denominated cash obtained notably fromand the conversions into euros of our March 2019 Nasdaq IPO in US dollars, andSwiss Franc denominated cash. We do not to use any specific hedging arrangements. However, as the majority of our expenses are denominated in euros,euros, we could be required to convert U.S. Dollarsdollars into euros or Swiss Francs into euros, and are therefore exposed to a foreign exchange risk. As of December 31, 2021,2023, we did not have foreign exchange rate hedging tools or contracts in place.
In the future, and in particular with respect to our clinical trials and the funding of our USU.S. subsidiary and our Swiss subsidiary, we will continue to have a significant portion of transactions denominated in currencies other than the euro or indirectly exposed to currency risk, and as a result, we will continue to have exposure to this risk.
Interest Rate Risk
We believe we have low exposure to interest rate risk.
Our financial liabilities, which consist primarily of convertible bonds, bank loans and government refundable or conditional advances, that carry no interest or fixed interest rates, and therefore are not subject to interest rate risk, with the exception of the state-guaranteed loans (PGE), the interest rates of which willmay be revised in case of their extension beyond their initialcurrent maturity, which in turn could lead to an increase in interest in the future.
With respect to our financial assets, which consist primarily of cash and cash equivalents, our exposure is also limited, as these assets are held on euro and USU.S. dollar denominated demand deposits, term deposits with progressive rates, or invested in euro and USU.S. dollar denominated medium-term negotiable notes or in euro denominated UCITs (Undertakings for the Collective Investment of Transferable Securities). While these interest-earning instruments carry a degree of interest rate risk, historical fluctuations in interest income in comparison to the average balance have not been significant, notably in the context of low market rates.
Credit Risk
We believe that the credit risk related to our cash and cash equivalents is not significant in light of the quality of the financial institutions at which such funds are held.
Liquidity Risk
We had €263.2€81.9 million in cash and cash equivalentsequivalents and other financial assets, including €258.8€77.8 million in cash and cash equivalents, as of December 31, 20212023 and as a result, do not believe that we are exposed to short-term liquidity risk. In addition, our loans and borrowings mainly consist of bonds convertible or exchangeable into new or existing shares (OCEANE)(OCEANEs), repayable for a nominal amount of €56.9 million on October 16, 2025 (seeNote 20.1 Note 12.1 “Breakdown- “Breakdown of convertibleconvertible loan”to our consolidated financial statements included in this annual report). We estimate that we will be able to fund our operating expenses and capital expenditure requirements for the next 12 months at least based on our existing cash and cash equivalents, and the reimbursement of research tax credits. Thiscredits and expected future milestones. Furthermore, this is based on current assumptions and programs, and does not include exceptional events. More specifically, this estimate includes our expectations to receive future milestone revenue in 2024, subject to approval by applicable regulatory authorities and US and European commercial launches of elafibranor in PBC by Ipsen. Lastly, this estimate is based on our current business plan and does not include any other potential milestones payable to or from us, nor any additional expenditures resulting from the potential in-licensing or acquisition of additional product candidates or technologies, or any associated development we may pursue. We have based this estimate on assumptions that may be incorrect and we may use our capital resources sooner than anticipated.
We may need to seek additional funds, through public or private equity or debt financings, government or other third-party funding, marketing and distribution arrangements and other partnerships, strategic alliances and licensing arrangements or a combination of these approaches. However, no assurance can be given at this time as to whether we will be able to achieve these financing objectives.
| | Detail of calculation of net cash | |
| Detail of calculation of net cash | |
| Detail of calculation of net cash | Detail of calculation of net cash | | As of | | | As of |
| (in € thousands) | (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 | (in € thousands) | | | | 2021/12/31 | 2022/12/31 | | 2023/12/31 |
| Cash and cash equivalents | Cash and cash equivalents | | 276,748 | | 171,029 | | 258,756 | Cash and cash equivalents | | | | 258,756 | 136,001 | | 77,789 |
| Current convertible loans | Current convertible loans | | 1,312 | | 1,312 | | 415 | Current convertible loans | | | | 415 | 415 | | 415 |
| Other current loans and borrowings | Other current loans and borrowings | | 3,226 | | 3,035 | | 1,773 | Other current loans and borrowings | | | | 1,773 | 4,665 | | 7,510 |
| Non-current convertible loans | Non-current convertible loans | | 164,142 | | 169,470 | | 47,682 | Non-current convertible loans | | | | 47,682 | 49,861 | | 52,206 |
| Other non-current loans and borrowings | Other non-current loans and borrowings | | 14,939 | | 11,873 | | 24,365 | Other non-current loans and borrowings | | | | 24,365 | 20,334 | | 10,047 |
| Net cash | Net cash | | 93,129 | | (14,662) | | 184,521 | Net cash | | | | 184,521 | 60,726 | | 7,610 |
Inflation Risk
We do not believe that inflation has had a material effect on our business, financial condition or results of operations in 2021. 2023. If our costs were to become subject to significant inflationary pressures, we may not be able to fully offset such higher costs, as we do not generate significant revenue from product sales. Our inability or failure to do so could harm our business, financial condition and results of operations.
| | | | | |
| Item 11C. | Interim Periods. |
Not applicable.
| | | | | |
| Item 12. | Description of Securities Other than Equity Securities. |
Not applicable.
Not applicable.
Not applicable.
| | | | | |
| D. | American Depositary Shares |
The Bank of New York Mellon, as depositary, registers and delivers American Depositary Shares, or ADSs. Each ADS represents one ordinary share (or a right to receive one ordinary share) deposited with BNP Paribas Securities Services, as custodian for the depositary in France. Each ADS will also represent any other securities, cash or other property that may be held by the depositary. The depositary’s office at which the ADSs are administered and its principal executive office are located at 240 Greenwich Street, New York, New York 10286.
A deposit agreement among us, the depositary and the ADS holders sets out the ADS holder rights as well as the rights and obligations of the depositary. New York law governs the deposit agreement and the ADSs. A copy of the deposit agreement is incorporated by reference as an exhibit to this annual report.
Fees and Charges
Pursuant to the terms of the deposit agreement, the holders of ADSs will be required to pay the following fees:
| | | | | | | | |
| Persons depositing or withdrawing ordinary shares or ADS holders must pay: | | For: |
| $5.00 (or less) per 100 ADSs (or portion of 100 ADSs) | | • Issuance of ADSs, including issuances resulting from a distribution of ordinary shares or rights or other property
• Cancellation of ADSs for the purpose of withdrawal, including if the deposit agreement terminates |
| $.05 (or less) per ADS | | • Any cash distribution to ADS holders |
| A fee equivalent to the fee that would be payable if securities distributed to you had been ordinary shares and the ordinary shares had been deposited for issuance of ADSs | | • Distribution of securities distributed to holders of deposited securities (including rights) that are distributed by the depositary to ADS holders |
| $.05 (or less) per ADS per calendar year | | • Depositary services |
| Registration or transfer fees | | • Transfer and registration of ordinary shares on our share register to or from the name of the depositary or its agent when you deposit or withdraw ordinary shares |
| Expenses of the depositary | | • Cable (including SWIFT) and facsimile transmissions (when expressly provided in the deposit agreement)
• Converting foreign currency to U.S. dollars |
| Taxes and other governmental charges the depositary or the custodian has to pay on any ADSs or ordinary shares underlying ADSs, such as stock transfer taxes, stamp duty or withholding taxes | | • As necessary |
| Any charges incurred by the depositary or its agents for servicing the deposited securities | | • As necessary |
The depositary collects its fees for delivery and surrender of ADSs directly from investors depositing ordinary shares or surrendering ADSs for the purpose of withdrawal or from intermediaries acting for them. The depositary collects fees for making distributions to investors by deducting those fees from the amounts distributed or by selling a portion of distributable property to pay the fees. The depositary may collect its annual fee for depositary services by deduction from cash distributions or by directly billing investors or by charging the book-entry system accounts of participants acting for them. The depositary may collect any of its fees by deduction from any cash distribution payable (or by selling a portion of securities or other property distributable) to ADS holders that are obligated to pay those fees. The depositary may generally refuse to provide fee-attracting services until its fees for those services are paid.
From time to time, the depositary may make payments to us to reimburse us for costs and expenses generally arising out of establishment and maintenance of the ADS program, waive fees and expenses for services provided to us by the depositary or share revenue from the fees collected from ADS holders. In performing its duties under the deposit agreement, the depositary may use brokers, dealers, foreign currency dealers or other service providers that are owned by or affiliated with the depositary and that may earn or share fees, spreads or commissions.
The depositary may convert currency itself or through any of its affiliates and, in those cases, acts as principal for its own account and not as agent, advisor, broker or fiduciary on behalf of any other person and earns revenue, including, without limitation, transaction spreads, that it will retain for its own account. The revenue is based on, among other things, the difference between the exchange rate assigned to the currency conversion made under the deposit agreement and the rate that the depositary or its affiliate receives when buying or selling foreign currency for its own account. The depositary makes no representation that the exchange rate used or obtained in any currency conversion under the deposit agreement will be the most favorable rate that could be obtained at the time or that the method by which that rate will be determined will be the most favorable to ADS holders, subject to the depositary’s obligations under the deposit agreement. The methodology used to determine exchange rates used in currency conversions is available upon request.
Payment of Taxes
ADS holders are responsible for any taxes or other governmental charges payable on their ADSs or on the deposited securities represented by any of their ADSs. The depositary may refuse to register any transfer of ADSs or allow an ADS holder to withdraw the deposited securities represented by his or her ADSs until those taxes or other charges are paid. It may apply payments owed to the ADS holder or sell deposited securities represented by the ADS holder’s American Depositary SharesADSs to pay any taxes owed and such ADS holder will remain liable for any deficiency. If the depositary sells deposited securities, it will, if appropriate, reduce the number of ADSs to reflect the sale and pay to ADS holders any proceeds, or send to ADS holders any property, remaining after it has paid the taxes. An ADS holder’s obligation to pay taxes and indemnify us and the depositorydepositary against any tax claims will survive the transfer or surrender of his or her ADSs, the withdrawal of the deposited ordinary shares as well as the termination of the deposit agreement.
PART II
| | | | | |
| Item 13. | Defaults, Dividend Arrearages and Delinquencies. |
Not applicable.
| | | | | |
| Item 14. | Material Modifications to the Rights of Security Holders and Use of Proceeds. |
In October 2017, we issued convertible bonds for gross proceeds of €180.0 million. The convertible bonds carrycarried a fixed interest rate of 3.5%, with an effective interest rate of 7.2%, payable semi-annually in arrears in April and October.
On November 23, 2020, we presented to all OCEANEOCEANEs bondholders a two-prong renegotiation offer :offer:
•A partial buyback of the outstanding OCEANEs for a maximum amount of 3,048,780 OCEANEs at €16.40 per bond; and
•An amendment of the terms of the remaining OCEANEs to extend their maturity (by 3 years) and increase the conversion ratio (to 5.5 shares per bond).
At the Shareholders’ and Bondholders’ Meetings on January 25, 2021, the shareholders and bondholders approved this renegotiation offer.
Following the shareholders’ and bondholders’ decisions, GENFIT completed the partial buyback of 2,895,260 OCEANEs at a price of €16.40 (including accrued interest of €0.30) for a total buyback cost of €47.48 million. The settlement operations occurred on January 29, 2021. The repurchased OCEANEs were then cancelled by GENFIT. The convertible bonds carry a fixed interest rate of 3.5%, with an effective interest rate of 8.8%, payable semi-annually in arrears in April and October.
Following conversion of the OCEANEs into shares up until April 1, 2022,2024, which led to the creation of 6,941,875 new shares, the residual nominal convertible debt, initially reduced to a nominal amount of €94.3 million through the partial buyback transaction, was further reduced by a nominal amount of €37.4 million, with approximately €56.9 million nominal amount outstanding as of April 1, 2022. 2024.
| | | | | |
| Item 15. | Disclosure Controls and Procedures. |
| | | | | |
| A. | Disclosure Controls and Procedures |
We maintain “disclosure controls and procedures,” as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, that are designed to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the Securities and Exchange Commission. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed in our reports filed or submitted under the Exchange Act is accumulated and communicated to management, including our chief executive officer (principal executive officer) and chief financial officer (principal financial officer), as appropriate, to allow timely decisions regarding required disclosure.
Our principal executive officer and principal financial officer, after evaluating the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) of the Exchange Act) as of December 31, 2021,2023, have concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
| | | | | |
| B. | Management’s Annual Report on Internal Control Over Financial Reporting |
Management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Management assessed the effectiveness of internal control over financial reporting as of December 31, 20212023 based on the framework in “Internal Control - Integrated Framework” (2013 framework) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on that assessment, management has concluded that, as December 31, 2021,2023, the Company’s internal control over financial reporting was effective to provide reasonable assurance regarding the reliability of its financial reporting and the preparation of its financial statements for external purposes, in accordance with generally accepted accounting principles. Due to its inherent limitations, internal control over financial reporting may not prevent or detect misstatements and can only provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
| | | | | |
| C. | Attestation Report of the Registered Public Accounting Firm |
This annual report does not include an attestation report of the Company’s registered public accounting firm due to a transition period established by rules of the Securities and Exchange Commission for emerging growth companies.
| | | | | |
| D. | Changes in Internal Control Over Financial Reporting |
There were no changes to our internal control over financial reporting that occurred during the period covered by this Form 20-F that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| | | | | |
| Item 16A. | Audit Committee Financial Expert. |
Our boardBoard of directorsDirectors has determined that Ms. Anne-Hélène Monsellato is an “audit committee financial expert” as defined by SEC rules and regulations and has the requisite financial sophistication under the applicable rules and regulations of the Nasdaq Stock Market. Ms. Monsellato is independent as such term is defined in Rule 10A-3 under the Exchange Act and under the listing standards of the Nasdaq Stock Market.
| | | | | |
| Item 16B. | Code of Business Conduct and Ethics. |
We have adopted a Code of Business Conduct and Ethics, or the Code of Conduct, applicable to all of our employees, senior management and directors. The Code of Conduct is available on our website at www.genfit.com. We expect that any amendments to the Code of Conduct, or any waivers of its requirements, will be disclosed on our website.
www.genfit.com.
| | | | | |
| Item 16C. | Principal Accountant Fees and Services. |
Ernst & Young et Autres, or E&Y, served as our independent registered public accounting firm for 20202022 and 2021.2023. Our accountants billed the following fees to us for professional services in each of those fiscal years :
| | | | | | | | | | | | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | As of |
| (in € thousands) | | | 2020/12/31 | | 2021/12/31 |
| Audit fees | | | 462 | | 337 |
| Audit-related fees | | | 30 | | 6 |
| Tax fees | | | — | | — |
| Other fees | | | — | | — |
| TOTAL | | | 492 | | 343 |
| | | | | | | | | | | | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | As of |
| (in € thousands) | | | 2022/12/31 | | 2023/12/31 |
| Audit fees | | | 420 | | 635 |
| Audit-related fees | | | 5 | | 34 |
| Tax fees | | | — | | — |
| Other fees | | | — | | — |
| TOTAL | | | 425 | | 669 |
“Audit Fees” are the aggregate fees billed for the audit of our annual financial statements. This category also includes services that E&Y provides, such as consents and assistance with and review of documents filed with the SEC.
“Audit-Related Fees” are the aggregate fees billed for assurance and related services that are reasonably related to the performance of the audit and are not reported under Audit Fees.
“Tax Fees” are the aggregate fees billed for professional services rendered by E&Y for tax compliance, tax advice and tax planning related services.
“Other Fees” are any additional amounts billed for products and services provided by E&Y.
There were no “Tax Fees” or “Other Fees” billed or paid during 20202022 or 2021.2023.
| | | | | | | | |
| Auditor Name | Auditor Location | Auditor Firm ID |
| Ernst & Young et Autres | Paris, France | 1704 |
Audit and Non-Audit Services Pre-Approval Policy
The audit committeeAudit Committee has responsibility for appointing, setting compensation of and overseeing the work of the independent registered public accounting firm. In recognition of this responsibility, the audit committeeAudit Committee has adopted a policy governing the pre-approval of all audit and permitted non-audit services performed by our independent registered public accounting firm to ensure that the provision of such services does not impair the independent registered public accounting firm’s independence from us and our management. Unless a type of service to be provided by our independent registered public accounting firm has received general pre-approval from the audit committee,Audit Committee, it requires specific pre-approval by the audit committee.Audit Committee. The payment for any proposed services in excess of pre-approved cost levels requires specific pre-approval by the Audit Committee. All audit committee.and non-audit services rendered by our independent registered public accounting firm in 2023 were pre-approved by the Audit Committee.
Pursuant to its pre-approval policy, the audit committeeAudit Committee may delegate its authority to pre-approve services to the chairperson of the audit committee.Audit Committee. The decisions of the chairperson to grant pre-approvals must be presented to the full audit committeeAudit Committee at its next scheduled meeting. The audit committeeAudit Committee may not delegate its responsibilities to pre-approve services to the management.
| | | | | |
| Item 16D. | Exemptions from the Listing Standards for Audit Committees. |
Not applicable.
| | | | | |
| Item 16E. | Purchases of Equity Securities by the Issuer and Affiliated Purchasers. |
Not applicable.
| | | | | |
| Item 16F. | Change in Registrant’s Certifying Accountant. |
Not applicable.Ernst & Young et Autres and Grant Thornton, joint statutory auditors of the Company, appointments will expire at the end of the Annual Shareholders’ Meeting called to approve the financial statements for the year ended December 31, 2023.
Our Audit Committee oversaw the selection of the auditors to be appointed by the Annual Shareholders’ Meeting in 2024 following a call for tenders. As part of the tender process, the Audit Committee paid particular attention to the quality of the information made available to the candidate firms, and to the fairness of the process. At the end of this process, the Committee's recommendation was to propose the renewal of Ernst & Young et Autres (EY) and Grant Thornton term of office as statutory auditors for an additional six-year period, pursuant to the French Law. The main factors taken into account in the renewal of the auditors, and in particular of EY (the only auditor of the Company that issues a Report of Independent Registered Public Accounting Firm on the Consolidated Financial Statements on Form 20-F), were the sectoral and PCAOB standards experience of the EY partner proposed to manage our account (rotation), enabling a renewed approach (including with regard to the potential use of technology in auditing), and the desire to capitalize on efforts to improve auditing conditions, in particular the implementation in 2023 of test run with a view to the auditors' assessment of internal control over financial reporting in 2024 (regarding section 404b of the Sarbanes-Oxley Act).
The Board of Directors, at its meeting of January 24, 2024 approved the Audit Committee’s recommendation. Consequently, the Board of Directors will propose to the Annual Shareholders’ Meeting to be held in May 22, 2024 to renew Ernst & Young et Autres and Grant Thornton as joint statutory auditors for a six-year term, i.e. until the Annual Shareholders’ Meeting to be held in 2030, which will approve the financial statements for the year 2029.
| | | | | |
| Item 16G. | Corporate Governance. |
As a French société anonyme,, we are subject to various corporate governance requirements under French law. In addition, as a foreign private issuer listed on the Nasdaq Global Select Market, we are subject to Nasdaq corporate governance listing standards. However, the corporate governance standards provide that foreign private issuers, as defined in the rules promulgated under the US Securities Exchange Act of 1934, as amended, (the “Exchange Act”), are permitted, pursuant to Nasdaq Listing Rule 5615(a)(3), to follow home country corporate governance practices in lieu of Nasdaq rules,Listing Rules, with certain exceptions. Currently, we rely on these exemptions for foreign private issuers and follow French corporate governance practices in lieu of the Nasdaq corporate governance rules,Listing Rules, which would otherwise require that (1) a majority of our boardBoard of directorsDirectors consist of independent directors; (2) we establish a nominating and corporate governance committee; and (3) our remunerationnomination committee be composed entirely of independent directors.directors; (3) our compensation committee be composed entirely of independent directors; and (4) our independent directors hold regularly scheduled meetings at which only independent directors are present.
The following is a summary of the significant ways in which our corporate governance practices differ from those followed by U.S. companies listed on Nasdaq:
•Audit Committee. As a foreign private issuer, we are required to comply with Rule 10A-3 ofunder the Exchange Act, relating to audit committee composition and responsibilities. Rule 10A-3 provides that the audit committee must have direct responsibility for the nomination, compensation and choice of our auditors, as well as control over the performance of their duties, management of complaints made, and selection of consultants. However, under Rule 10A-3, if the laws of a foreign private issuer’s home country require that any such matter be approved by the boardBoard of directorsDirectors or the shareholders, the audit committee’s responsibilities or powers with respect to such matter may instead be advisory. Under French law, the audit committee may only have an advisory role and appointment of our statutory auditors, in particular, must be decided by the shareholders at our annual meeting.
•Quorum Requirements. Nasdaq rulesListing Rules require that a listed company specify that the quorum for any meeting of the holders of common stock be at least 33 1/3% of the outstanding shares of the company’s voting stock. We follow our French home country practice, rather than complying with these Nasdaq Listing Rules. Consistent with French law, our bylaws provide that awhen first convened, the quorum at the shareholders meeting requires the presence of shareholders having at least (1) 20% of the shares entitled to vote in the case of an ordinary shareholders’ general meeting or at an extraordinary shareholders’ general meeting where shareholders are voting on a capital increase by capitalization of reserves, profits or share premium, or (2) 25% of the shares entitled to vote in the case of any other extraordinary shareholders’ general meeting. If a quorum is not present, the meeting is adjourned. There is no quorum requirement when an ordinary general meeting is reconvened, but the reconvened meeting may consider only questions which were on the agenda of the adjourned meeting. When an extraordinary general meeting is reconvened, the quorum required is 20% of the shares entitled to vote, except where the reconvened meeting is considering capital increases through capitalization of reserves, profits or share premium. For these matters, no quorum is required at the reconvened meeting. The reconvened meeting may consider only questions that were on the agenda of the adjourned meeting. If a quorum is not present at a reconvened meeting requiring a quorum, then the meeting may be adjourned for a maximum of two months.
•Authorization for issuances of securities. The Company follows French law with respect to shareholder approval requirements in lieu of the various shareholder approval requirements of Nasdaq Listing Rule 5635, which requires a Nasdaq listed company to obtain shareholder approval prior to certain issuances of securities, including: (a) issuances in connection with the acquisition of the stock or assets of another company if upon issuance the issued shares will equal 20% or more of the number of shares or voting power outstanding prior to the issuance, or if certain specified persons have a 5% or greater interest in the assets or company to be acquired (Nasdaq Listing Rule 5635(a)); (b) issuances or potential issuances that will result in a change of control of us (Nasdaq Listing Rule 5635(b)); (c) issuances in connection with equity compensation arrangements (Nasdaq Listing Rule 5635(c)); and (d) 20% or greater issuances in transactions other than public offerings, as defined in the Nasdaq Listing Rules (Nasdaq Listing Rule 5635(d)). Under French law, the Company’s shareholders may approve issuances of equity, as a general matter, through the adoption of delegation of authority resolutions at the Company’s shareholders’ meeting pursuant to which shareholders may delegate their authority to the Board of Directors to increase the Company’s share capital within specified parameters set by the shareholders, which may include a time limitation to carry out the share capital increase, the cancellation of their preferential subscription rights to the benefit of named persons or a category of persons, specified price limitations and/or specific or aggregate limitations on the size of the share capital increase. Due to differences between French law and corporate governance practices and Nasdaq Listing Rule 5635, the Company follows French home country practice, rather than complying with this Nasdaq Listing Rule.
Other than as set forth above, we currently intend to comply with the corporate governance listing standards of Nasdaq to the extent possible under French law. However, we may choose to change such practices to follow home country practice in the future.
| | | | | |
| Item 16H. | Mine Safety Disclosure. |
Not applicable.
| | | | | |
| Item 16I. | Disclosure Regarding Foreign Jurisdictions that Prevent Inspections. |
Not applicable.
| | | | | |
| Item 16J. | Insider trading policies |
Not applicable.
Risk management and strategy
GENFIT’s business is heavily dependent on our computer network and the use of information technology, or IT, systems, whether maintained directly by GENFIT or through external IT providers, including cloud-based applications. As a result, damage from computer viruses, unauthorized access, telecommunication and electrical failures can cause significant disruption to our operations.
We have implemented and maintain various information security processes to assess and manage the security, integrity, and availability of our IT systems, and safeguards to protect our data and that of patients participating in our clinical trials, our employees, and partners. To identify and mitigate cybersecurity risks, counteract threats, and limit and/or prevent disruptions to our IT systems, we have implemented detailed cybersecurity policies and procedures.
These processes are prioritized across all organizational levels, with cybersecurity acknowledged as a critical risk within the core enterprise risks that we regularly evaluate and address as an integral part of our risk management plan. As part of this plan, we also conduct periodic assessments of our assets, including IT assets, to evaluate the effectiveness of applicable security controls. In the past we have also commissioned third-party audits of our security controls.
Additionally, as part of our approach to third-party risk management, we generally assess our external partners to determine whether their cybersecurity standards meet our specifications prior to engagement. In addition, we have migrated some tools to cloud-based applications, which can offer increased assurances as to security upgrades and swiftness of remediation in the event of disruptions, to which we would not normally have access to in a closed environment.
Employees across all levels and departments receive training on cybersecurity policies through an extensive "read and understood" process and are informed about cybersecurity risks via digital ongoing and annual awareness training programs conducted through the IT department. Employees are required to report IT security incidents to the cybersecurity team through a dedicated communication channel, and if necessary, by contacting a member of the IT team.
In partnership with our internal cybersecurity team, a specialized third-party service provider responsible for managing our Cyber Security Operations Center investigates security incidents and alerts such as virus detection, abnormal traffic or unauthorized software installation. This includes identifying the type of threat, determining the scope of the incident, and assessing the severity of each situation.
Governance
Our cybersecurity initiatives are subject to ongoing monitoring and regular reporting to senior management and the GENFIT Board of Directors.
The IT Security Manager, or ITSM, in collaboration with the Executive Vice-President, Data & Information Technology who is also known as the Chief Information Officer, or CIO, leads our cybersecurity risk management efforts, aligning these initiatives with the strategic objectives established by our executive leaders. With nearly a decade of expertise in information security and technology, our ITSM plays a pivotal role in safeguarding our digital assets. Our CIO has more than twenty years of experience in information technology management and strategic planning and reports directly to the Chief Operating Officer or COO. The CIO is responsible for guiding our technology strategy, overseeing technology deployment, and managing operations. Our CIO regularly updates a working group established specifically by the Board of Directors in 2023 in order to oversee our cybersecurity status. This includes briefings on any recent incidents and our responses, testing of cybersecurity systems and third-party activities.
This cybersecurity working group is chaired by a member of the Board of Directors and includes the CIO, the ITSM and other key GENFIT employees. The chair of the cybersecurity working group meets and reports regularly to the Board of Directors on cybersecurity matters, allowing the Board of Directors to provide effective oversight of management’s assessment and management of the cybersecurity risks, in particular to reinforce transparency and accountability in our cyber strategies.
In addition, we have developed a procedure that details how we classify incidents, management of any incidents, and internal and external communication thereof. In accordance with that procedure, major or critical incidents are escalated for review to our Cyber Crisis Committee, which is comprised of various members of the Executive Committee, including our CEO. This committee is responsible for identifying and evaluating cybersecurity incidents. Our CEO reports directly to our Board of Directors regarding incidents identified as material by the Cyber Crisis Committee. This committee meets on an ad hoc basis as required to manage cybersecurity incidents.
As of the filing of this Form 20-F, we are not aware of any cyber-attacks that have occurred over the last three years that have materially affected, or are reasonably likely to materially affect us, including our business strategy, results of operations or financial condition. Although we have put in place the cybersecurity processes described above, we remain exposed to cybersecurity attacks and incidents and misuse or manipulation of any of our IT systems, which could have a material adverse effect on our business strategy, results of operations or financial condition. You should refer to the section of this annual report titled Item 3.D - "Key Information - Risk Factors” for additional information about these risks.
PART III
| | | | | |
| Item 17. | Financial Statements. |
See pages F-1 through F-62F-56 of this annual report.
| | | | | |
| Item 18. | Financial Statements. |
Not applicable.
| | Incorporation by Reference |
| | | | Incorporation by Reference | | | | | | Incorporation by Reference |
| Exhibit | Exhibit | | Description | | Schedule/
Form | | File
Number | | Exhibit | | File Date | Exhibit | | Description | | Schedule/
Form | | File
Number | | Exhibit | | File Date |
| 1.2* | | | | | | | | | | |
| 1.1* | |
| 2.1 | |
| 2.1 | |
| 2.1 | | | | | F-6 | | 333-230265 | | 4.1 | | 3/14/2019 |
| 2.2 | 2.2 | | | | F-6 | | 333-230265 | | 4.2 | | 3/14/2019 | 2.2 | | | | F-6 | | 333-230265 | | 4.1 | | 3/14/2019 |
| 2.3 | | | | F-6 | | 333-230265 | | 4.3 | | 3/14/2019 |
| 2.4* | | | |
| 2.3* | |
| 4.1† | |
| 4.1† | |
| 4.1† | 4.1† | | | | F-1 | | 333-229907 | | 10.1 | | 2/27/2019 | | | | 20-F | | 001-38844 | | 4.3 | | 5/27/2020 |
| 4.2† | 4.2† | | | | 20-F | | 001-38844 | | 4.3 | | 5/27/2020 | 4.2† | | | | 20-F | | 001-38844 | | 4.5 | | 5/27/2020 |
| 4.3† | 4.3† | | | | F-1 | | 333-229907 | | 10.4 | | 2/27/2019 | 4.3† | | | | 20-F | | 001-38844 | | 4.5 | | 4/29/2022 |
| 4.4† | 4.4† | | | | 20-F | | 001-38844 | | 4.5 | | 5/27/2020 | 4.4† | | | | 20-F | | 001-38844 | | 4.5 | | 4/18/2023 |
| 4.5* | | | |
| 4.6† | | | | F-1 | | 333-229907 | | 10.3 | | 2/27/2019 |
| 4.5† | | 4.5† | | | | 20-F | | 001-38844 | | 4.6 | | 4/18/2023 |
| 4.6†* | |
| 4.7† | |
| 4.7† | |
| 4.7† | 4.7† | | | | 20-F | | 001-38844 | | 4.7 | | 5/27/2020 | | | | F-1 | | 333-229907 | | 10.3 | | 2/27/2019 |
| 4.8† | 4.8† | | | | 20-F | | 001-38844 | | 4.8† | | | | 20-F | | 001-38844 | | 4.7 | | 5/27/2020 |
| 4.9* | | | |
| 4.9† | | 4.9† | | | | 20-F | | 001-38844 | | 4.8 | | 4/23/2021 |
| 4.10† | 4.10† | | | | F-1 | | 333-229907 | | 10.5 | | 2/27/2019 | 4.10† | | | | 20-F | | 001-38844 | | 4.9 | | 4/29/2022 |
| 4.11#† | | | | 20-F | | 001-38844 | | 4.9 | | 5/27/2020 |
| 4.12*# | | | |
| 8.1 | | | | F-1 | | 333-229907 | | 21.1 | | 2/27/2019 |
| 4.11† | | 4.11† | | | | 20-F | | 001-38844 | | 4.11 | | 4/18/2023 |
| 4.12† | | 4.12† | | | | 20-F | | 001-38844 | | 4.12 | | 4/18/2023 |
| 4.13†* | |
| 4.14 | |
| 4.14 | |
| 4.14 | | | | | F-1 | | 333-229907 | | 10.5 | | 2/27/2019 |
| 4.15# | | 4.15# | | | | 20-F | | 001-38844 | | 4.9 | | 5/27/2020 |
| 4.16# | | 4.16# | | | | 20-F | | 001-38844 | | 4.12 | | 4/29/2022 |
| 4.17# | | 4.17# | | | | 20-F | | 001-38844 | | 4.16 | | 4/18/2023 |
| 4.18 | | 4.18 | | | | 20-F | | 001-38844 | | 4.16 | | 4/18/2023 |
| 8.1* | |
| 12.1* | |
| 12.1* | |
| 12.1* | 12.1* | | | |
| 12.2* | 12.2* | | | |
| 12.2* | |
| 12.2* | |
| 13.1** | |
| 13.1** | |
| 13.1** | 13.1** | | | |
| 13.2** | 13.2** | | | |
| 13.2** | |
| 13.2** | |
| 15.1* | |
| 15.1* | |
| 15.1* | |
| 97.1 * | |
| 97.1 * | |
| 97.1 * | |
| | | | | | | | | | | | | | |
| 101.INS* | | XBRL Instance Document | | |
| 101.SCH* | | XBRL Taxonomy Extension Schema Document | | |
| 101.CAL* | | XBRL Taxonomy Extension Calculation Linkbase Document | | |
| 101.DEF* | | XBRL Taxonomy Extension Definition Linkbase Document | | |
| 101.LAB* | | XBRL Taxonomy Extension Label Linkbase Document | | |
| 101.PRE* | | XBRL Taxonomy Extension Presentation Linkbase Document | | |
| 104 | | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101) | | |
| | | | | |
| † | Indicates a management contract or any compensatory plan, contract or arrangement. |
| | | | | |
| # | Certain portions of this exhibit have been omitted because they are not material and would likely cause competitive harm to the registrant if disclosed. |
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
| | | | | | | | |
| | | GENFIT S.A. |
| | | |
| | By: | /s/ Pascal Prigent |
| | | Pascal Prigent |
| | | Chief Executive Officer |
Date: April 29, 20225, 2024
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| | | | | |
| Page |
Consolidated Financial Statements as of and for the Years Ended December 31, 2019, 2020, 20212023 | |
| |
| |
| |
| |
| |
| |
| |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and the Board of Directors
Genfit of GENFIT S.A.
Opinion on the Financial Statements
We have audited the accompanying consolidated statements of financial position of GenfitGENFIT S.A. (“the Group”)(the Group) as of December 31, 2019, 20202023 and 2021,2022, the related consolidated statements of operations, other comprehensive loss,income (loss), cash flows and changes in equity for each of the three years in the period ended December 31, 2021,2023, and the related notes(collectively (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Group at December 31, 2019, 20202023 and 2021,2022, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2021,2023, in conformity with International Financial Reporting Standards (“IFRS”) as issued by the International Accounting Standards Board.
Adoption of New Accounting Standard
As discussed in Note 4.7 to the consolidated financial statements, starting January 1, 2019, the Group changed its method of accounting for leases due to the adoption of IFRS 16 ”Leases”.
Basis for Opinion
These financial statements are the responsibility of the Group's management. Our responsibility is to express an opinion on the Group’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Group in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Group is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Group's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ ERNSTErnst & YOUNGYoung et Autres
We haveErnst & Young et Autres has served as the Group’s auditor since 1999
Paris,Paris-La Défense, France
April 29, 20225, 2024
CONSOLIDATED STATEMENTS OF FINANCIAL POSITION
(amounts in thousands of euros)
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| ASSETS | | Notes | | As of |
| (in € thousands) | | | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Current assets | | | | | | | | |
| Cash and cash equivalents | | 6 | | 276,748 | | 171,029 | | 258,756 |
| Current trade and others receivables | | 9 | | 12,033 | | 11,919 | | 7,236 |
| | | | | | | | | |
| Other current assets | | 11 | | 1,968 | | 1,765 | | 2,101 |
| Inventories | | — | | 4 | | 4 | | 4 |
| | | | | | | | | |
| Total - Current assets | | | | 290,753 | | 184,717 | | 268,097 |
| Non-current assets | | | | | | | | |
| | | | | | | | | |
| Intangible assets | | 7 | | 920 | | 297 | | 174 |
| Property, plant and equipment | | 8 | | 16,453 | | 11,648 | | 9,015 |
| Non-current trade and other receivables | | 9 | | — | | — | | 3 |
| Other non-current financial assets | | 10 | | 1,727 | | 1,458 | | 4,431 |
| | | | | | | | | |
| Deferred tax assets | | 22 | | — | | — | | — |
| Total - Non-current assets | | | | 19,099 | | 13,403 | | 13,623 |
| Total - Assets | | | | 309,853 | | 198,119 | | 281,720 |
| | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| ASSETS | | Notes | | | As of |
| (in € thousands) | | | | | | 2022/12/31 | | 2023/12/31 |
| Current assets | | | | | | | | |
| Cash and cash equivalents | | 13 | | | | 136,001 | | 77,789 |
| Current trade and others receivables | | 16 | | | | 15,906 | | 32,707 |
| Other current financial assets | | 18 | | | | 4,550 | | — |
| Other current assets | | 19 | | | | 1,998 | | 2,615 |
| Inventories | | — | | | | 4 | | 4 |
| | | | | | | | | |
| Total - Current assets | | | | | | 158,459 | | 113,115 |
| Non-current assets | | | | | | | | |
| | | | | | | | | |
| Intangible assets | | 14 | | | | 43,957 | | 48,761 |
| Property, plant and equipment | | 15 | | | | 8,210 | | 7,872 |
| | | | | | | | | |
| Other non-current financial assets | | 18 | | | | 4,914 | | 4,125 |
| | | | | | | | | |
| Deferred tax assets | | 11 | | | | — | | — |
| Total - Non-current assets | | | | | | 57,081 | | 60,758 |
| Total - Assets | | | | | | 215,540 | | 173,872 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| SHAREHOLDERS' EQUITY AND LIABILITIES | | Notes | | As of |
| (in € thousands) | | | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Current liabilities | | | | | | | | |
| Current convertible loans | | 12 | | 1,312 | | 1,312 | | 415 |
| Other current loans and borrowings | | 12 | | 3,226 | | 3,035 | | 1,773 |
| Current trade and other payables | | 14 | | 36,917 | | 25,564 | | 40,988 |
| Current deferred income and revenue | | 15 | | 139 | | 124 | | 14,298 |
| Current provisions | | 16 | | 2,061 | | 1,031 | | 313 |
| | | | | | | | | |
| Other current tax liabilities | | 22 | | — | | — | | 5,051 |
| Total - Current liabilities | | | | 43,657 | | 31,067 | | 62,837 |
| Non-current liabilities | | | | | | | | |
| Non-current convertible loans | | 12 | | 164,142 | | 169,470 | | 47,682 |
| Other non-current loans and borrowings | | 12 | | 14,939 | | 11,873 | | 24,365 |
| Non-current trade and other payables | | 14 | | 450 | | 450 | | 450 |
| Non-current deferred income and revenue | | 15 | | — | | — | | 25,821 |
| | | | | | | | | |
| Non-current employee benefits | | 17 | | 1,408 | | 922 | | 864 |
| Deferred tax liabilities | | 22 | | 1,193 | | 767 | | 602 |
| Total - Non-current liabilities | | | | 182,132 | | 183,482 | | 99,786 |
| Shareholders' equity | | | | | | | | |
| Share capital | | 18 | | 9,715 | | 9,722 | | 12,454 |
| Share premium | | — | | 377,821 | | 379,057 | | 444,438 |
| Retained earnings (accumulated deficit) | | 18 | | (238,340) | | (303,897) | | (405,076) |
| Currency translation adjustment | | — | | 14 | | (92) | | 22 |
| Net profit (loss) | | — | | (65,144) | | (101,221) | | 67,259 |
| Total shareholders' equity - Group share | | | | 84,065 | | (16,430) | | 119,097 |
| Non-controlling interests | | — | | — | | — | | — |
| Total - Shareholders' equity | | | | 84,065 | | (16,430) | | 119,097 |
| Total - Shareholders' equity & liabilities | | | | 309,853 | | 198,119 | | 281,720 |
| | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| SHAREHOLDERS' EQUITY AND LIABILITIES | | Notes | | | As of |
| (in € thousands) | | | | | | 2022/12/31 | | 2023/12/31 |
| Current liabilities | | | | | | | | |
| Current convertible loans | | 20 | | | | 415 | | 415 |
| Other current loans and borrowings | | 20 | | | | 4,665 | | 7,510 |
| Current trade and other payables | | 22 | | | | 14,845 | | 18,799 |
| Current deferred income and revenue | | 23 | | | | 14,479 | | 11,692 |
| Current provisions | | 24 | | | | 61 | | 40 |
| | | | | | | | | |
| Other current tax liabilities | | 11 | | | | 4,906 | | 23 |
| Total - Current liabilities | | | | | | 39,370 | | 38,480 |
| Non-current liabilities | | | | | | | | |
| Non-current convertible loans | | 20 | | | | 49,861 | | 52,206 |
| Other non-current loans and borrowings | | 20 | | | | 20,334 | | 10,047 |
| Non-current trade and other payables | | 22 | | | | 448 | | — |
| Non-current deferred income and revenue | | 23 | | | | 9,706 | | 3,755 |
| | | | | | | | | |
| Non-current employee benefits | | 25 | | | | 782 | | 978 |
| Deferred tax liabilities | | 11 | | | | 510 | | 455 |
| Total - Non-current liabilities | | | | | | 81,641 | | 67,441 |
| Shareholders' equity | | | | | | | | |
| Share capital | | 26 | | | | 12,459 | | 12,459 |
| Share premium | | — | | | | 444,683 | | 445,261 |
| Retained earnings (accumulated deficit) | | — | | | | (337,550) | | (361,870) |
| Currency translation adjustment | | — | | | | (1,344) | | 996 |
| Net profit (loss) | | — | | | | (23,719) | | (28,894) |
| | | | | | | | | |
| | | | | | | | | |
| Total - Shareholders' equity | | | | | | 94,528 | | 67,951 |
| Total - Shareholders' equity & liabilities | | | | | | 215,540 | | 173,872 |
The accompanying notes form an integral part of these consolidated financial statements.
CONSOLIDATED STATEMENTS OF OPERATIONS
(amounts in thousands of euros, except per share data)
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | Notes | | Year ended |
| (in € thousands, except earnings per share data) | | | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Revenues and other income | | | | | | | | |
| Revenue | | 19 | | 30,839 | | 765 | | 80,069 |
| Other income | | 19 | | 10,122 | | 6,993 | | 5,510 |
| Revenues and other income | | | | 40,961 | | 7,758 | | 85,579 |
| | | | | | | | | |
| Operating expenses and other operating income (expenses) | | | | | | | | |
| Research and development expenses | | 20 | | (66,170) | | (59,097) | | (35,166) |
| General and administrative expenses | | 20 | | (17,265) | | (14,270) | | (16,153) |
| Marketing and market access expenses | | 20 | | (13,708) | | (11,216) | | (1,539) |
| Reorganization and restructuring expenses | | 20 | | — | | (5,308) | | (142) |
| Other operating income (expenses) | | 20 | | (1,649) | | (764) | | (763) |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| Operating income (loss) | | | | (57,832) | | (82,897) | | 31,816 |
| | | | | | | | | |
| Financial income (1) | | 22 | | 5,221 | | 6,544 | | 44,780 |
| Financial expenses | | 22 | | (13,110) | | (25,296) | | (7,122) |
| Financial profit (loss) | | | | (7,889) | | (18,752) | | 37,658 |
| | | | | | | | | |
| Net profit (loss) before tax | | | | (65,721) | | (101,649) | | 69,474 |
| | | | | | | | | |
| Income tax benefit (expense) | | 23 | | 576 | | 428 | | (2,215) |
| | | | | | | | | |
| Net profit (loss) | | | | (65,144) | | (101,221) | | 67,259 |
| Attributable to owners of the Company | | | | (65,144) | | (101,221) | | 67,259 |
| Attributable to non-controlling interests | | | | — | | — | | — |
| | | | | | | | | |
| Basic and diluted earnings (loss) per share | | | | | | | | |
| Basic earnings (loss) per share (€/share) | | 24 | | (1.76) | | (2.60) | | 1.51 |
| Diluted earnings (loss) per share (€/share) | | 24 | | (1.76) | | (2.60) | | 1.23 |
| (1): Of which Financial income incurred by renegotiating the convertible bond debt OCEANE | | | | — | | — | | 35,578 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | Notes | | Year ended |
| (in € thousands, except earnings per share data) | | | | 2021/12/31 | | 2022/12/31 | | 2023/12/31 |
| Revenues and other income | | | | | | | | |
| Revenue | | 7 | | 80,069 | | 20,195 | | 28,565 |
| Other income | | 7 | | 5,510 | | 6,371 | | 9,610 |
| Revenues and other income | | | | 85,579 | | 26,566 | | 38,176 |
| | | | | | | | | |
| Operating expenses and other operating income (expenses) | | | | | | | | |
| Research and development expenses | | 8 | | (35,166) | | (35,818) | | (46,503) |
| General and administrative expenses | | 8 | | (16,153) | | (16,405) | | (17,741) |
| Marketing and market access expenses | | 8 | | (1,539) | | (992) | | (876) |
| Reorganization and restructuring income (expenses) | | 8 | | (142) | | 11 | | 505 |
| Other operating expenses | | 8 | | (763) | | (652) | | (141) |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| Operating income (loss) | | | | 31,816 | | (27,289) | | (26,580) |
| | | | | | | | | |
| Financial income | | 10 | | 44,780 | | 8,212 | | 3,680 |
| Financial expenses | | 10 | | (7,122) | | (4,758) | | (5,614) |
| Financial profit (loss) | | | | 37,658 | | 3,453 | | (1,934) |
| | | | | | | | | |
| Net profit (loss) before tax | | | | 69,474 | | (23,836) | | (28,514) |
| | | | | | | | | |
| Income tax benefit (expense) | | 11 | | (2,215) | | 116 | | (380) |
| | | | | | | | | |
| Net profit (loss) | | | | 67,259 | | (23,719) | | (28,894) |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| Basic and diluted earnings (loss) per share | | | | | | | | |
| Basic earnings (loss) per share (€/share) | | 12 | | 1.51 | | (0.48) | | (0.58) |
| Diluted earnings (loss) per share (€/share) | | 12 | | 1.23 | | (0.48) | | (0.58) |
| | | | | | | | | |
The accompanying notes form an integral part of these consolidated financial statements.
CONSOLIDATED STATEMENTS OF OTHER COMPREHENSIVE LOSSINCOME (LOSS)
(amounts in thousands of euros)
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | Notes | | Year ended |
| (in € thousands) | | | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| | | | | | | | | |
| Net profit (loss) | | | | (65,144) | | (101,221) | | 67,259 |
| | | | | | | | | |
| Actuarial gains and losses net of tax | | | | (168) | | 196 | | 216 |
| Other comprehensive income (loss) | | | | | | | | |
| that will never be reclassified to profit or loss | | | | (168) | | 196 | | 216 |
| | | | | | | | | |
| Exchange differences on translation of foreign operations | | | | 8 | | (106) | | 113 |
| Other comprehensive income (loss) | | | | | | | | |
| that are or may be reclassified to profit or loss | | | | 8 | | (106) | | 113 |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| Total comprehensive income (loss) | | | | (65,304) | | (101,131) | | 67,589 |
| Attributable to owners of the Company | | | | (65,304) | | (101,131) | | 67,589 |
| Attributable to non-controlling interests | | | | — | | — | | — |
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | Notes | | Year ended |
| (in € thousands) | | | | 2021/12/31 | | 2022/12/31 | | 2023/12/31 |
| | | | | | | | | |
| Net profit (loss) | | | | 67,259 | | (23,719) | | (28,894) |
| | | | | | | | | |
| Actuarial gains and losses net of tax | | 25 | | 216 | | 258 | | (51) |
| Change in fair value of equity instruments included in financial assets and financial liabilities | | 18 | | — | | — | | (785) |
| Other comprehensive income (loss) | | | | | | | | |
| that will never be reclassified to profit or loss | | | | 216 | | 258 | | (836) |
| | | | | | | | | |
| Exchange differences on translation of foreign operations | | | | 113 | | (1,366) | | 2,340 |
| Other comprehensive income (loss) | | | | | | | | |
| that are or may be reclassified to profit or loss | | | | 113 | | (1,366) | | 2,340 |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | |
| Total comprehensive income (loss) | | | | 67,589 | | (24,827) | | (27,390) |
| | | | | | | | | |
| | | | | | | | | |
The accompanying notes form an integral part of these consolidated financial statements.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(amounts in thousands of euros)
| | Year ended | | Year ended | | Year ended |
| | | Notes | |
| | | Notes | |
| | | Notes | | | Year ended |
| (in € thousands) | (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 | | (in € thousands) | | | | 2021/12/31 | | 2022/12/31 | | 2023/12/31 |
| | Cash flows from operating activities | Cash flows from operating activities | |
| Cash flows from operating activities | |
| Cash flows from operating activities | |
| + Net profit (loss) | + Net profit (loss) | | (65,144) | | | (101,221) | | | 67,259 | |
| + Non-controlling interests | | — | | | — | | | — | |
| + Net profit (loss) | |
| + Net profit (loss) | |
| | Reconciliation of net loss to net cash used in operating activities | |
| | Reconciliation of net loss to net cash used in operating activities | |
| | Reconciliation of net loss to net cash used in operating activities | Reconciliation of net loss to net cash used in operating activities | |
| Adjustments for: | Adjustments for: | |
| Adjustments for: | |
| Adjustments for: | |
| | + Depreciation and amortization on tangible and intangible assets | + Depreciation and amortization on tangible and intangible assets | | 3,263 | | | 3,559 | | | 2,742 | |
| + Impairment and provision for litigation | | 357 | | | 3,015 | | | (1,996) | |
| | + Depreciation and amortization on tangible and intangible assets | |
| | + Depreciation and amortization on tangible and intangible assets | |
| | + Impairment and provisions | |
| | + Impairment and provisions | |
| | + Impairment and provisions | |
| | + Expenses related to share-based compensation | + Expenses related to share-based compensation | | 1,657 | | | 1,236 | | | 470 | |
| - Gain on disposal of property, plant and equipment | | (19) | | | 80 | | | 420 | |
| | + Expenses related to share-based compensation | |
| | + Expenses related to share-based compensation | |
| | - Loss (gain) on disposal of property, plant and equipment | |
| - Loss (gain) on disposal of property, plant and equipment | |
| - Loss (gain) on disposal of property, plant and equipment | |
| | + Net finance expenses (revenue) | + Net finance expenses (revenue) | | 11,437 | | | 10,335 | | | 4,663 | |
| | + Net finance expenses (revenue) | |
| | + Net finance expenses (revenue) | |
| | + Income tax expense (benefit) | + Income tax expense (benefit) | | (576) | | | (428) | | | 2,215 | |
| | + Income tax expense (benefit) | |
| | + Income tax expense (benefit) | |
| | + Other non-cash items | + Other non-cash items | | 1,702 | | | (1,818) | | | (35,538) | |
| including Research Tax Credit litigation and Income incurred by renegotiating the convertible bond debt OCEANE | |
| + Other non-cash items | |
| + Other non-cash items | |
| | Operating cash flows before change in working capital | Operating cash flows before change in working capital | | (47,324) | | | (85,242) | | | 40,235 | |
| Change in: | | | | | | |
| | Operating cash flows before change in working capital | |
| | Operating cash flows before change in working capital | |
| | Decrease (increase) in trade receivables and other assets | Decrease (increase) in trade receivables and other assets | | (1,640) | | | 318 | | | 4,344 | |
| | Decrease (increase) in trade receivables and other assets | |
| | Decrease (increase) in trade receivables and other assets | |
| | (Decrease) increase in trade payables and other liabilities | (Decrease) increase in trade payables and other liabilities | | 1,284 | | | (11,447) | | | 55,335 | |
| | (Decrease) increase in trade payables and other liabilities | |
| | (Decrease) increase in trade payables and other liabilities | |
| | Change in working capital | Change in working capital | | (356) | | | (11,129) | | | 59,680 | |
| | Change in working capital | |
| | Change in working capital | |
| | Income tax paid | Income tax paid | | — | | | — | | | — | |
| Income tax paid | |
| Income tax paid | |
| | Net cash flows provided by (used in) in operating activities | Net cash flows provided by (used in) in operating activities | | (47,680) | | | (96,371) | | | 99,915 | |
| Net cash flows provided by (used in) in operating activities | |
| Net cash flows provided by (used in) in operating activities | |
| | Cash flows from investment activities | Cash flows from investment activities | | | | | | |
| Cash flows from investment activities | |
| Cash flows from investment activities | |
| | - Acquisition net of cash acquired (Versantis intangible) | |
| | - Acquisition net of cash acquired (Versantis intangible) | |
| | - Acquisition net of cash acquired (Versantis intangible) | |
| | - Acquisition of other intangible assets | |
| | - Acquisition of other intangible assets | |
| | - Acquisition of other intangible assets | |
| | - Acquisition of property, plant and equipment | - Acquisition of property, plant and equipment | | (2,030) | | | (900) | | | (537) | |
| | - Acquisition of property, plant and equipment | |
| | - Acquisition of property, plant and equipment | |
| | + Proceeds from disposal of / reimbursement of property, plant and equipment | + Proceeds from disposal of / reimbursement of property, plant and equipment | | 2,517 | | | — | | | 309 | |
| | + Proceeds from disposal of / reimbursement of property, plant and equipment | |
| | + Proceeds from disposal of / reimbursement of property, plant and equipment | |
| | - Acquisition of financial instruments | - Acquisition of financial instruments | | (160) | | | (66) | | | (3,148) | |
| | - Acquisition of financial instruments | |
| | - Acquisition of financial instruments | |
| | + Proceeds from disposal of financial instruments | |
| | + Proceeds from disposal of financial instruments | |
| | + Proceeds from disposal of financial instruments | |
| | Net cash flows provided by (used in ) investment activities | Net cash flows provided by (used in ) investment activities | | 327 | | | (966) | | | (3,377) | |
| | Net cash flows provided by (used in ) investment activities | |
| | Net cash flows provided by (used in ) investment activities | |
| | Cash flows from financing activities | Cash flows from financing activities | | | | | | |
| Cash flows from financing activities | |
| Cash flows from financing activities | |
| | + Proceeds from issue of share capital (net) | + Proceeds from issue of share capital (net) | | 126,486 | | | 7 | | | 27,972 | |
| + Proceeds from subscription / exercise of share warrants | | 43 | | | — | | | — | |
| | + Proceeds from issue of share capital (net) | |
| | + Proceeds from issue of share capital (net) | |
| | + Proceeds from new loans and borrowings net of issue costs | + Proceeds from new loans and borrowings net of issue costs | | — | | | — | | | 15,270 | |
| | + Proceeds from new loans and borrowings net of issue costs | |
| | + Proceeds from new loans and borrowings net of issue costs | |
| | - Repayments of loans and borrowings | - Repayments of loans and borrowings | | (6) | | | 207 | | | (48,436) | |
| | - Repayments of loans and borrowings | |
| | - Repayments of loans and borrowings | |
| | - Payments on lease debts | - Payments on lease debts | | (1,877) | | | (2,150) | | | (1,887) | |
| | - Payments on lease debts | |
| | - Payments on lease debts | |
| | - Financial interests paid (including finance lease) | - Financial interests paid (including finance lease) | | (7,785) | | | (7,762) | | | (2,109) | |
| | - Financial interests paid (including finance lease) | |
| | - Financial interests paid (including finance lease) | |
| | + Financial interests received | + Financial interests received | | — | | | 1,442 | | | 274 | |
| | + Financial interests received | |
| | + Financial interests received | |
| | Net cash flows provided by (used in ) financing activities | Net cash flows provided by (used in ) financing activities | | 116,860 | | | (8,256) | | | (8,916) | |
| | Net cash flows provided by (used in ) financing activities | |
| | Net cash flows provided by (used in ) financing activities | |
| | Increase (decrease) in cash and cash equivalents | Increase (decrease) in cash and cash equivalents | | 69,508 | | | (105,593) | | | 87,622 | |
| Increase (decrease) in cash and cash equivalents | |
| Increase (decrease) in cash and cash equivalents | |
| | Cash and cash equivalents at the beginning of the period | Cash and cash equivalents at the beginning of the period | | 207,240 | | | 276,748 | | | 171,029 | |
| Cash and cash equivalents at the beginning of the period | |
| Cash and cash equivalents at the beginning of the period | |
| | Effects of exchange rate changes on cash | Effects of exchange rate changes on cash | | — | | | (126) | | | 105 | |
| Effects of exchange rate changes on cash | |
| Effects of exchange rate changes on cash | |
| | Cash and cash equivalents at the end of the period | Cash and cash equivalents at the end of the period | | 276,748 | | | 171,029 | | | 258,756 | |
| | | Cash and cash equivalents at the end of the period | |
| | Cash and cash equivalents at the end of the period | |
The accompanying notes form an integral part of these consolidated financial statements.
In the above table, the amount of "Other non-cash items" includes the bonus generated by the partial OCEANE buyback following the renegotiation completed in January 2021 for the amount of €35,578 in 2021.
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY
(Amounts in thousands of euros, except for number of shares)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
| | | Share capital | | Share | | Treasury | | Retained | | Currency | | Net | | Total | | Non- | | Total |
| | | Number | | Share | | premium | | shares | | earnings | | translation | | profit | | shareholders' | | controlling | | shareholders' |
| | | of shares | | capital | | | | | | (accumulated | | adjustment | | (loss) | | equity | | interests | | equity |
| (in € thousands) | | | | | | | | | | deficit) | | | | | | Group share | | | | |
| | | | | | | | | | | | | | | | | | | | | |
| As of January 01, 2019 | | 31,183,921 | | 7,796 | | 251,554 | | (730) | | (158,167) | | 6 | | (79,521) | | 20,939 | | — | | 20,939 |
| Net profit (loss) | | | | | | | | | | | | | | (65,144) | | (65,144) | | | | (65,144) |
| Other comprehensive income (loss) | | | | | | | | | | (168) | | 8 | | | | (160) | | | | (160) |
| Total comprehensive income (loss) | | 0 | | — | | — | | — | | (168) | | 8 | | (65,144) | | (65,304) | | — | | (65,304) |
| Allocation of prior period profit (loss) | | | | | | | | | | (79,521) | | | | 79,521 | | — | | | | — |
| Capital increase | | 7,674,696 | | 1,919 | | 124,567 | | | | (7) | | | | | | 126,479 | | | | 126,479 |
| | | | | | | | | | | | | | | | | | | | | |
| Share-based compensation | | | | | | 1,657 | | | | | | | | | | 1,657 | | | | 1,657 |
| Treasury shares | | | | | | | | 252 | | | | | | | | 252 | | | | 252 |
| Other movements | | | | | | 43 | | | | | | | | | | 43 | | | | 43 |
| As of December 31, 2019 | | 38,858,617 | | 9,715 | | 377,821 | | (478) | | (237,862) | | 14 | | (65,144) | | 84,065 | | — | | 84,065 |
| Net profit (loss) | | | | | | | | | | | | | | (101,221) | | (101,221) | | | | (101,221) |
| Other comprehensive income (loss) | | | | | | | | | | 196 | | (106) | | | | 90 | | | | 90 |
| Total comprehensive income (loss) | | — | | — | | — | | — | | 196 | | (106) | | (101,221) | | (101,131) | | — | | (101,131) |
| Allocation of prior period profit (loss) | | | | | | | | | | (65,144) | | | | 65,144 | | — | | | | — |
| Capital increase | | 29,762 | | 7 | | — | | | | (7) | | | | | | — | | | | — |
| | | | | | | | | | | | | | | | | | | | | |
| Share-based compensation | | | | | | 1,236 | | | | | | | | | | 1,236 | | | | 1,236 |
| Treasury shares | | | | | | | | (333) | | | | | | | | (333) | | | | (333) |
| Other movements | | | | | | — | | | | (268) | | | | | | (268) | | | | (268) |
| As of December 31, 2020 | | 38,888,379 | | 9,722 | | 379,057 | | (811) | | (303,086) | | (92) | | (101,221) | | (16,430) | | — | | (16,430) |
| Net profit (loss) | | | | | | | | | | | | | | 67,259 | | 67,259 | | | | 67,259 |
| Other comprehensive income (loss) | | | | | | | | | | 216 | | 113 | | | | 330 | | | | 330 |
| Total comprehensive income (loss) | | — | | — | | — | | — | | 216 | | 113 | | 67,259 | | 67,589 | | — | | 67,589 |
| Allocation of prior period profit (loss) | | | | | | | | | | (101,221) | | | | 101,221 | | — | | | | — |
| Capital increase | | 10,927,110 | | 2,732 | | 62,600 | | | | — | | | | | | 65,332 | | | | 65,332 |
| Equity component of OCEANE net of deferred taxes | | | | | | 2,311 | | | | | | | | | | 2,311 | | | | 2,311 |
| Share-based compensation | | | | | | 470 | | | | | | | | | | 470 | | | | 470 |
| Treasury shares | | | | | | | | (174) | | | | | | | | (174) | | | | (174) |
| Other movements | | | | | | — | | | | | | | | | | — | | | | — |
| As of December 31, 2021 | | 49,815,489 | | 12,454 | | 444,438 | | (986) | | (404,090) | | 22 | | 67,259 | | 119,097 | | — | | 119,097 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
| | | Share capital | | Share | | Treasury | | Retained | | Currency | | Net | | | | | | Total |
| | | Number | | Share | | premium | | shares | | earnings | | translation | | profit | | | | | | shareholders' |
| | | of shares | | capital | | | | | | (accumulated | | adjustment | | (loss) | | | | | | equity |
| (in € thousands) | | | | | | | | | | deficit) | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
| As of January 01, 2021 | | 38,888,379 | | 9,722 | | 379,057 | | (811) | | (303,086) | | (92) | | (101,221) | | | | | | (16,430) |
| Net profit (loss) | | | | | | | | | | | | | | 67,259 | | | | | | 67,259 |
| Other comprehensive income (loss) | | | | | | | | | | 216 | | 113 | | | | | | | | 330 |
| Total comprehensive income (loss) | | — | | — | | — | | — | | 216 | | 113 | | 67,259 | | | | | | 67,589 |
| Allocation of prior period profit (loss) | | | | | | | | | | (101,221) | | | | 101,221 | | | | | | — |
| Capital increase | | 10,927,110 | | 2,732 | | 62,600 | | | | — | | | | | | | | | | 65,332 |
| Equity component of OCEANE net of deferred taxes | | | | | | 2,311 | | | | | | | | | | | | | | 2,311 |
| Share-based compensation | | | | | | 470 | | | | | | | | | | | | | | 470 |
| Treasury shares | | | | | | | | (174) | | | | | | | | | | | | (174) |
| | | | | | | | | | | | | | | | | | | | | |
| As of December 31, 2021 | | 49,815,489 | | 12,454 | | 444,438 | | (986) | | (404,090) | | 22 | | 67,259 | | | | | | 119,097 |
| Net profit (loss) | | | | | | | | | | | | | | (23,719) | | | | | | (23,719) |
| Other comprehensive income (loss) | | | | | | | | | | 258 | | (1,366) | | | | | | | | (1,108) |
| Total comprehensive income (loss) | | — | | — | | — | | — | | 258 | | (1,366) | | (23,719) | | | | | | (24,827) |
| Allocation of prior period profit (loss) | | | | | | | | | | 67,259 | | | | (67,259) | | | | | | — |
| Capital increase | | 19,494 | | 5 | | — | | | | (5) | | | | | | | | | | — |
| | | | | | | | | | | | | | | | | | | | | |
| Share-based compensation | | | | | | 245 | | | | | | | | | | | | | | 245 |
| Treasury shares | | | | | | | | 8 | | | | | | | | | | | | 8 |
| Other movements | | | | | | — | | | | 5 | | | | | | | | | | 5 |
| As of December 31, 2022 | | 49,834,983 | | 12,459 | | 444,683 | | (978) | | (336,573) | | (1,344) | | (23,719) | | | | | | 94,528 |
| Net profit (loss) | | | | | | | | | | | | | | (28,894) | | | | | | (28,894) |
| Other comprehensive income (loss) | | | | | | | | | | (836) | | 2,340 | | | | | | | | 1,504 |
| Total comprehensive income (loss) | | — | | — | | — | | — | | (836) | | 2,340 | | (28,894) | | | | | | (27,390) |
| Allocation of prior period profit (loss) | | | | | | | | | | (23,719) | | | | 23,719 | | | | | | — |
| | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
| Share-based compensation | | | | | | 578 | | | | | | | | | | | | | | 578 |
| Treasury shares | | | | | | | | 8 | | | | | | | | | | | | 8 |
| Other movements | | | | | | — | | | | 227 | | | | | | | | | | 227 |
| As of December 31, 2023 | | 49,834,983 | | 12,459 | | 445,261 | | (970) | | (360,901) | | 996 | | (28,894) | | | | | | 67,951 |
The accompanying notes form an integral part of these consolidated financial statements.
*As a reminder, the expenses incurred in 2019 in relation to the Initial Public Offering are deducted from the share issue premium.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(amounts in thousands of euros, except for numbers of shares and per share amounts, and unless stated otherwise)
1.THE COMPANY
Founded in 1999 under the laws of France, GENFIT S.A. (the "Company") is a late-stage biopharmaceutical company dedicated to the discovery and development of innovative drugs and diagnostic tools in therapeutic areas of high unmet need due in particular to the lack of effective treatments or diagnostic solutions and/or the increase in patients worldwide.
The Company focuses its research and development (R&D) efforts on the potential marketing of therapeutic and diagnostic solutions to combat certain metabolic, inflammatory, autoimmune and fibrotic diseases affecting in particular the liver (such as Primary Biliary Cholangitis or PBC) and more generally gastroenterological diseases. The head office address is : 885 Avenue Eugène Avinée – 59120 Loos, FRANCE France.
The consolidated financial statements of the Company include the financial statements of GENFIT S.A. and those of its wholly-owned subsidiaries: GENFIT CORPCORP. (U.S. subsidiary), Versantis AG (Swiss subsidiary), Versantis, Inc. (U.S. Subsidiary, liquidated prior to December 31, 2023), and GENFIT PHARMACEUTICALS SAS (French subsidiary)subsidiary, liquidated prior to December 31, 2022) (together referred to in these notes to the consolidated financial statements as "GENFIT" or the "Group" or “we “ or “us”).
2.MAJOR EVENTS IN THE PERIOD AND EVENTS AFTER THE PERIOD
2.1Termination of RESOLVE IT and the development program of elafibranor in NASH
Following the decision by the Company in July 2020 to terminate its Phase 3 RESOLVE-IT trial (see 2020 Form 20-F), the impacts of the RESOLVE-IT termination process, and more broadly the discontinuation of the elafibranor development program in NASH continued to have a significant impact in 2021.
Impact on subcontracting costs
The termination of the development program of elafibranor in NASH and the closing of the RESOLVE-IT study in particular involve external costs notably due to regulatory activities, expenses related to final patient visits, site closures, clinical data recording, finalizing the Clinical Study Report, updating the Trial Master File, and invoking a supply contract termination clause, etc.
Overall, the amount of contracting costs recognized in 2020 for the RESOLVE-IT trial was €25.4 million, including €9.7 million after the termination of the trial. In 2021, those costs amounted to €3.3 million. The Company estimates that further costs to be recognized in 2022 will not exceed €500.
As a reminder, the analysis completed in 2020 under IAS 37 led the Company to consider that :
•The costs incurred in relation with the termination of the RESOLVE-IT trial benefit the ELATIVE trial evaluating elafibranor in PBC (notably the preparation of a drug safety file for elafibranor) with the exception of some administrative fees and fees for the destruction of drug tablets;
•If the RESOLVE-IT study had not been initiated, Genfit would have needed to complete similar work (preparation of a drug safety file for elafibranor) to the work to be completed in 2021 as part of the closing of the RESOLVE-IT study and bear the related costs for the needs of elafibranorMajor events in the PBC project (ELATIVE).
•Therefore, these costs benefiting the ELATIVE trial were not provisioned insofar as Genfit intends to complete this trial (which may not be designated as onerous contract under IAS 37).
In keeping with this analysis, the fees that cannot be tied to the ELATIVE trial (administrative fees and fees for the destruction of drug tablets) were accrued in 2020 for €378. Considering the €366 provision reversal recognized in 2021 (of which €265 was used), this provision has been reduced to €12 at December 31, 2021.
Impact on scientific equipment leased and owned
The Group has analyzed the impact of the closing of RESOLVE-IT and its decision to reorganize its activities on its scientific equipment. An inventory of the equipment that could be sold, kept as a spare, or disposed of, was completed in the second half of 2020.
Leased equipment
The impairment loss for this equipment of €503 recognized in 2020 in order to account for the estimated loss in comparison to the net book value of the rights of use of the asset has been reduced to €62 at December 31, 2021 following the purchase and effective sale of some of this equipment in 2021.
Owned equipment
The impairment loss for this equipment of €363 recognized in 2020 in order to account for the estimated loss in comparison to the net book value, has been reduced to €25 at December 31, 2021, following the effective sale of some of this equipment in 2021.
Premises
At December 31, 2020, the Company recognized an impairment loss of the right of use of its rented premises in Lille and Paris (France) for €1,275 had been recognized (including fittings and fixtures), as part of the premises were no longer in use.
As the Company signed an agreement for a negotiated early termination of its lease agreement for its Paris premises, allowing for a relocation of the Paris office during the second half of 2021. The corresponding impairment has been reduced to €596 at December 31, 2021.
Reorganization and reduction in force
Following the reorganization and reduction in force plan (plan de sauvegarde de l’emploi or “PSE”) implemented by the company during the second half of 2020, the consolidated headcount was reduced to 130 employees at December 31, 2020, compared to 203 at June 30, 2020.
Accrued liabilities for notice periods, voluntary and involuntary separation severances related to the PSE, recognized for a total amount of €1,326 in 2020 were disbursed in 2021.
Apart from the aforementioned severances, a provision for support measures such as return-to-work bonuses, trainings, business start-up assistance granted within the scope of the PSE, which was recognized for €523 in 2020, was reduced to €171 at December 31, 2021, following a reversal of €352 (of which €189 was used).
2.22.1.1Renegotiation of the convertible bond debt (OCEANEs)Positive Results from Phase 3 ELATIVE® trial
OCEANEs partial buybackPositive Results
On June 30, 2023, GENFIT announced positive topline data from the pivotal ELATIVE® Phase 3 trial. In the trial, the efficacy and amendmentssafety of terms
In January 2021, the Company announced the success of the partial buyback operation and amendment of terms of the 6,081,081 Convertible and/or Exchangeable Bonds for New or Existing Shares ("OCEANEs") withelafibranor, an October 2022 maturity that it had issued within the scope of the €180 million convertible loan subscribed in October 2017. This renegotiation offer of the bond debt -approved by more than 98% of voting shareholders- allowed the Group to extend the maturity of the debt until October 16, 2025 and reduce its nominal amount in half. The conversion ratio changed from 1 OCEANE for one share to one OCEANE for 5.5 shares.
Reduction of the residual bond debt due to the share conversion of renegotiated OCEANEs
Following the implementation of the partial buyback operation and amendment of terms of the OCEANEs, some of the new OCEANEs were subject to a request for share conversion : respectively, 552,238, 483,330, 216,591 and 10,000 in January, February, March and August 2021.
Thus, the share capital of the Company reached €11,457,562.50, divided in 45,830,250 fully paid shares (amount before the capital increase subscribed by Ipsen in December 2021).
At the date of the approval of these consolidated Financial Statements :
•The Company has not received any new request for share conversion,
•The number of outstanding OCEANEsinvestigational dual α,δ PPAR agonist, is 1,923,662,
•The residual bond debt amounts for €56,940, representing less than one third of the initial bond debt of €180,000.
Considering the aforementioned conversions and taking into consideration the capital increase subscribed by Ipsen in December 2021, the share capital of the Company reached a nominal amount of €12,453,872.25, divided in 49,815,489 fully paid shares, and the maximum dilution in case of conversion of all convertible bonds would be 18% (in % of share capital at December 31, 2021).
The fees related to this renegotiation (i.e. mainly, financial advising, counsel fees, shareholders and bondholders meeting costs) have been recognized as Reorganization and Restructuring Expenses in the consolidated Financial Statements for an amount of €745 in 2020 and €2,303 in 2021.
2.3COVID-19 and Financial Assistance
COVID-19
The unprecedented spread of COVID-19 – characterized as a pandemic by the World Health Organization on March 11, 2020 – is impacting the global health and business ecosystem, Genfit included.
We have been working with our contract research organizations (CRO), clinical trial sites and clinical investigators to review regularly our estimatesbeing assessed for the execution of our programs taking into account the evolution of the pandemic and its impact on our activities.
Appropriate measures have been implemented, including virtual appointments, biological evaluations performed by local laboratories and delivery of the drug candidate to the patients’ homes, to ensure, when necessary, the safety of participants in our clinical trials under the current circumstances.
Following the implementation of the measures taken in collaboration with our CRO, we have been able to minimize the disruption on our Phase 3 ELATIVE clinical trial evaluating elafibranor in PBC, which recruited its first patient in September 2020. At the beginning of the trial, and taking into account the pandemic situation, we estimated that the recruitment period would span over 18 months, and we currently remain generally in line with that estimate. However, the rapid progression of the highly contagious Omicron variant has caused additional difficulties in the recruitmenttreatment of patients and throughout our clinical trial activities. The infection rate andwith the measures taken to slow down its spread have caused some patients to delay their visits or to be screened twice as they had exceeded the screening window. The recent worsening of the pandemic situation has also caused significant additional administrative delays for clinical sites and regulatory agencies due to staffing shortages, at a time when the number of clinical trials remains high. This has particularly affected areas which were already significantly delayed, such as Latin America. Even though we do not expect that these recent difficulties will affect our estimates for the availability of ELATIVE topline data, we are currently working with our CRO to assess the magnitude of the impact on patient recruitment timelines.
Financial Assistance
In 2021, we also secured 3 new bank loans for a total nominal amount of roughly €15.3 million, granted in the context of the COVID-19 pandemic, including (in rounded figures):
•An €11.0 million loan in June 2021 by a pool of 4 French commercial banks,
•A €2.0 million loan in July 2021 by BPI France,
Both of which are 90% guaranteed by the French government (State-Guaranteed Loans or Prêts Garantis par l’Etat "PGE") and carry an initial term of one year with repayment options up to six years, as well as
•A €2.3 million subsidized loan in November 2021 by BPI France, with an initial term of 6 years.
2.4. Signature of a Licensing and Partnership Agreement with Ipsen
In December 2021, GENFIT and Ipsen have entered into an exclusive licensing agreement for elafibranor, a Phase III asset evaluated inrare cholestatic liver disease, Primary Biliary Cholangitis (PBC), as partwho have an inadequate response or intolerance to the current standard of a long-term global partnership ("Collaboration and License Agreement")care therapy, ursodeoxycholic acid (UDCA).
The agreement gives Ipsen exclusive worldwide license (withtrial met its primary composite endpoint, with 51% of patients on elafibranor 80mg achieving a cholestasis response compared with 4% on placebo (p<0.0001). Cholestasis response is defined in the exceptiontrial as alkaline phosphatase (ALP) <1.67 x upper limit of China, Hong Kong, Taiwan,normal (ULN), an ALP decrease ≥ 15 percent and Macau where Terns Pharmaceuticals holds the exclusive license to developtotal bilirubin (TB) ≤ ULN at 52 weeks. ALP and commercialize elafibranor) to developbilirubin are important predictors of disease progression. Reductions in levels of both can indicate reduced cholestatic injury and commercialize elafibranor, GENFIT's first-in-class drug candidate, a PPAR alpha and PPAR delta agonist, for people living with PBC, a rare chronic inflammatoryimproved liver disease.function.
The Collaborationfirst secondary endpoint, normalization of ALP at Week 52, was also met with statistically significant improvements for investigational elafibranor compared with placebo. For the other secondary endpoint, a trend for pruritus improvement was observed with a greater decrease from baseline in the PBC Worst Itch NRS score for patients on elafibranor compared to placebo, which did not reach statistical significance. In the study, elafibranor was generally well tolerated with a safety profile consistent with that observed in previously reported studies.
December Filing
On December 7, 2023, IPSEN announced that the U.S. Food and License Agreement qualifiesDrug Administration (FDA) accepted the New Drug Application (NDA) for investigational elafibranor. The target FDA PDUFA date under priority review is June 10, 2024. The European Medicines Agency (EMA) also validated Ipsen’s Marketing Authorization Application (MAA) for elafibranor, and the review of the submission to the EMA’s Committee for Medicinal Products for Human Use (CHMP) began on 26 October 2023. Furthermore, a third simultaneous regulatory filing of elafibranor has been validated for review by the UK Medicines and Healthcare products Regulatory Agency (MHRA). Acceptance of filings in the US and Europe have triggered the first milestone payment for GENFIT totaling €13.3 million which has been recorded as a contract under IFRS 15,revenue in 2023. For more information on forthcoming milestones, see Note 7 - "Revenues and meets the criteria under IFRS 15.9.other income" to our consolidated financial statements included in this annual report. Under this agreement:
•2.1.2Seal Rock licence agreement
Summary
On May 31, 2023, GENFIT remains responsibleannounced the signing of a licensing agreement for the Phase III ELATIVE trial untilexclusive worldwide rights to the completionASK1 inhibitor SRT-015 with Seal Rock Therapeutics, a clinical-stage company based in Seattle, Washington developing kinase inhibitors.
SRT-015 is an injectable therapy intended for use in acute liver conditions, and GENFIT has acquired the rights to SRT-015 for use in liver conditions for which an injectable therapy is intended to be administered over a period of 21 consecutive days or less, including Acute on Chronic Liver Failure (ACLF) support during this period. Preclinical and clinical evidence support ASK1 inhibition as a relevant therapeutic strategy in multi-system disorders such as ACLF.
Total purchase price and contingent milestone payments
Under the terms of the double-blind period. Ipsen will assume responsibility for all additional clinical development, including completion of the long-term extension period of the ELATIVE trial, and commercialization.
•GENFIT received from Ipsen an upfront cash payment of €120 million in December 2021 (with an additional €24 million in collected VAT), of which €80 million was recognized as revenue in 2021. The remainder of this upfront payment (€40 million) has been recognized in 2021 as deferred revenue and will be recognized as revenue throughout the execution of the double-blind period of the ELATIVE study, in accordance with IFRS 15.
•GENFITagreement, Seal Rock is also eligible for milestone payments up to €360 million. These milestone payments constitute future variable income, dependent on the completion of key steps related to the development and sales of the licensed products. As such, in accordance with IFRS 15, this income will be recognized as revenue depending on the completion of these milestones.
•GENFIT is also eligible for tiered double-digit royalties of up to 20%€100 million (of which €2 million have been paid in 2023 as detailed below), appliedincluding regulatory, clinical and commercial milestone payments, as well as tiered royalties. SeeNote 29 - "Commitments and contingent liabilities" for further details.Accounting treatment of milestones paid and to be paid
Under the annual salesterms of licensed products realized by Ipsen. As such,the agreement, GENFIT made an upfront payment in the amount of €2 million to Seal Rock in exchange for acquiring the know-how and rights of use to SRT-015 as described above. In accordance with IFRS 15,IAS 38 - Intangible assets, this income will be recognized as revenue depending onamount was capitalized and allocated to Intangible assets. Further, given the realization of these sales.
Beyond the collaboration between GENFIT and Ipsen in PBC, this agreement also constitutes a strategic partnership, allowing Ipsen to access the research skills of GENFIT and other clinical programs, including some rights to first negotiation (while not being constitutive of a service obligation under IFRS 15).
At the same time, Ipsen also became a shareholder of GENFIT through the purchase of 3,985,239 newly issued shares representing 8% of GENFIT S.A after issuance, via a €28 million investment. The new shares were issued pursuant to the twentieth resolution of GENFIT’s 30 June 2021 shareholders’ meeting and are subject, to a lock-up period ending, in the event of positive ELATIVE results, on the earliernature of the date on which the EMA makesintangible asset, it was determined to have a formal recommendation to the European Commission for the marketing authorisationdefinite useful life of elafibranor in PBC or the date on which the U.S. FDA grants approval of elafibranor in PBC. In addition, the Board of Directors of GENFIT will propose at the next shareholders’ meeting that Ipsen becomes a board member.
2.5.Signature of a Licensing Agreement20 years, consistent with Genoscience Pharma
On December 16, 2021, GENFIT completed the acquisition of exclusive rights from Genoscience Pharma to develop and commercialize the investigational treatment GNS561 in cholangiocarcinoma (CCA)patent lifetimes in the United States Canada and Europe, including the United KingdomEuropean Union. Amortization will start based on the remaining patent term upon EMA/FDA regulatory approval and Switzerland.
GNS561 is a novel clinical-stage autophagy/PPT1 inhibitor developed by Genoscience Pharma and cholangiocarcinoma is an orphan disease. It has completed preclinical studies and a Phase 1b trial confirming the rationale for targeting cholangiocarcinoma, a rare biliary tract malignancy with high mortality and with limited treatment options.
Under the agreement, Genoscience Pharma is eligible for clinical and regulatory milestone payments for up to €50 million and tiered royalties.
The first payable milestones are contingent on positive Phase 2 clinical trial results in CCA, and may total up to €20 million, if applicable.
As such, under IAS 37, these payments constitute contingent liabilities not recognized in our consolidated financial statements for the period ending December 31, 2021.
The following payable milestones are contingent on positive Phase 3 results. These payments, when due,until then will be subject to a review to determine if theyan annual impairment test in accordance with IAS 38 - Intangible Assets and IAS 36 - Impairment of assets. As future milestones for this agreement are eligible for activation pursuant to IAS 38. If so,paid, they will be recorded as capital upon disbursement. Otherwise, they also constitute contingent liabilities which willanalyzed and be recognized when due.
In addition, we also have a right of first negotiation with respect to any license or assignment, or option for a license or an assignment, with any third party to develop or commercialize other Genoscience assets in the field of CCA,either i) capitalized and subject to the
extent Genosciencesame annual impairment test or ii) expensed as incurred. For further information, refer to Note 14 - "Goodwill and Intangible Assets".2.1.3Celloram licence agreement
On July 28, 2023, GENFIT licensed the exclusive worldwide rights to CLM-022, a first-in-class inflammasome inhibitor, from Celloram Inc., a Cleveland-based biotechnology company. GENFIT will leverage Celloram’s acquired scientific insights on this molecule, to finalize IND enabling studies of this preclinical stage asset and secure an IND for future clinical trials. A preclinical proof-of-concept is looking to partnertargeted for 2024.
Total purchase price and contingent milestone payments
Under the asset with a third party or receives a spontaneous offer for collaboration.
For the period commencing on the dateterms of the agreement,
untilCelloram is eligible for payments of up to €160 million, including regulatory, clinical and commercial milestones, as well as tiered royalties. See Note 29 - "Commitments and contingent liabilities" for further details.2.2Major events after the first regulatory approval of GNS561 for commercialization, Genoscience Pharma has the right to repurchase the license to GNS561 in CCA at a pre-determined price in the event that Genoscience Pharma receives an offer from a third party to acquire or obtain a license to GNS561 in all indications, provided that Genfit shall first have the opportunity to negotiate the acquisition or license to GNS561 in all indications or match the offer from the third party.
GENFIT also purchased a 10% equity stake in Genoscience Pharma through the subscription of new ordinary shares for an amount of €3.1 million fully paid up in December 2021.
Not applicable.
3.BASIS OF PRESENTATION
The Consolidated Financial Statements of GENFIT have been prepared in accordance with International Financial Reporting Standards (“IFRS”) as issued by the International Accounting Standards Board (“IASB”), and in accordance with IFRS as adopted by the European Union at December 31, 2021.2023. The Comparative information is presented as of and for the years ended December 31, 20192022 and December 31, 2020.2021.
The consolidated financial statements have been prepared using the historical cost measurement basis except for certain assets and liabilities that are measured at fair value in accordance with the IFRS general principles of fair presentation, going concern, accrual basis of accounting, consistency of presentation, materiality and aggregationaggregation.
These consolidated financial statements for the year ended December 31, 20212023 were prepared under the responsibility of the Board of Directors that approved such statements on April 6, 2022.3, 2024.
The term IFRS includes International Financial Reporting Standards ("IFRS"), International Accounting Standards (the "IAS"), as well as the Interpretations issued by the Standards Interpretation Committee (the "SIC"), and the International Financial Reporting Interpretations Committee ("IFRIC").
The principal accounting methods used to prepare the Consolidated Financial Statements are described below.
All financial information (unless indicated otherwise) is presented in thousands of euros (€).
3.1.Changes in accounting policies and new standards or amendments
With the exception of those mentioned below, theThe accounting policies applicable for these consolidated annual financial statements are the same as those applied to the previous consolidated annual financial statements.
The following new standards are applicable from January 1, 2021,2023, but do not have any material effect on the Group’s financial statements as of and for the year ended December 31, 2021.2023.
•IFRS 17 Insurance Contracts (including Amendments to IFRS 17 issued in June 2020 and Amendment to IFRS 4: Extension17 - Initial Application of the Temporary Exemption from Applying IFRS 917 and IFRS 9—Comparative Information issued in December 2021)
•AmendmentAmendments to IFRS 9, IAS 39 and IFRS 7: Interest Rate Benchmark reform– Phase 28 – Definition of Accounting Estimates
•AmendmentAmendments to IAS 1 and IFRS 16: COVID-19-Related Rent Concessions beyond 30 June 2021Practice Statement 2 – Disclosure of Accounting Policies
Some Agenda Decisions by the IFRS IC had an effect on the Group's consolidated Financial Statements, as of and for the year ended December 31, 2021.
The impact of the May 2021 IFRS IC decision•Amendments to IAS 12 – Deferred Tax related to Attributing Benefits to Periods of Service (IAS 19) has been recorded in the 2021 financial statements. The IFRS IC decision has resulted inAssets and Liabilities arising from a review by the Group of the calculation method for the retirement benefit plans under the terms of which:
a.employees are entitled to a retirement benefit provided they are employed by the Company upon retirement (with loss of entitlement in case of early retirement),
b.the retirement benefit depends on length of service, but is capped at a number of years of service.
This change in method resulted in the reduction of the provision for retirement benefits by €226 recognized retrospectively in the Group's equity on the opening balance sheet for the 2020 period.
The April 2021 IFRS IC decision related to accounting for configuration or customization costs in a Software as a Service (SaaS) arrangement was also impactful. The review of these configuration or customization costs by the Group has been completed and resulted in a reduction in the Group's equity of €495 recognized retrospectively on the opening balance sheet for the 2020 period.
3.2.Standards, interpretations and amendments issued but not yet effective
The GENFIT Group has not identified anyamendments and modifications to the standards or amendments issued and in force and anticipated as ofbelow are applicable for financial years beginning after January 1, 2021 or applicable2024, as specified below. GENFIT is in the process of assessing these amendments and modifications to the periods starting as of January 1, 2022 that maystandards, however they are not expected to have a significantmaterial impact on the Group's consolidated financial statements, notably:statements.
–IFRS 17 Insurance Contracts, effective in 2023
–Amendments to IAS 37 Onerous contracts – Cost of fulfilling a contract, effective in 2022,
–•Amendments to IFRS 3 Reference to the Conceptual Framework,16 – Lease Liability in a Sale and Leaseback, effective in 20222024,
–Amendments to IAS 16 Property, Plant and Equipment: Proceeds before Intended Use , effective in 2023,
–•Amendments to IAS 1 and Practice Statement 2 Disclosure of Accounting Policies, effective in 2023,
–Amendments to IAS 8 Definition of Accounting Estimates, effective in 2023,
–Amendments to IAS 12 Deferred Tax related to Assets and Liabilities arising from a Single Transaction, effective in 2023,
–Amendments to IAS 1 Classification of Liabilities as Current or Non-current (including Amendment to IAS 1 – Classification of Liabilities as Current or Non-current – Deferral of Effective Date issued in July 2020), effective in 2024,
–•Annual IFRS improvements, 2018-2020 cycle,Amendments to IAS 1 – Non-current Liabilities with Covenants, effective in 2022.2024,
•Amendments to IAS 21 – Lack of Exchangeability, effective in 2025.
4.SUMMARY OF SIGNIFICANTMATERIAL ACCOUNTING POLICIESINFORMATION
4.1.Use of estimates and judgments
In preparing these consolidated financial statements, management makes judgments, estimates and assumptions that affect the application of accounting policies and the reported amounts of assets and liabilities, incomes and expenses. Actual amounts may differ from these estimates.
The estimates and underlying assumptions are reviewed on an ongoing basis. Revisions to accounting estimates are recognized in the period in which the estimates are revised and in any future periods affected.
TheSignificant estimates and underlying assumptions mainly relate to research tax credits (see the following:
4.2.Consolidation
Going concern
The consolidated financial statements were prepared on a going concern basis. The Group believes it has sufficient resources to continue operating for at least twelve months following the consolidated financial statements’ publication.
When assessing going concern, the Group’s Board of Directors considers mainly the following factors :
The liquidity available at the statement of financial position date, milestones whose collection is considered highly probable (subject to approval by applicable regulatory authorities and US and European commercial launches of elafibranor in PBC), the cash spend projections for the next 12-month period as from the date of the financial statements are issued, and the availability of other funding.
4.2.ConsolidationConsolidated entities
The Group controls an entity when it is exposed to variable returns from its involvement with the entity, and it has the ability to affect those returns through its power over the entity.
The Group controls all the entities included in the scope of consolidation.
Versantis Inc was dissolved on June 2, 2023. All assets and liabilities of the company were transferred to Versantis AG. The impact to the financial statements was not material.
4.3.Foreign currency
4.3.1.Foreign currency transactions
Transactions in foreign currencies are translated into the respective functional currencies of the entities of the Group at the exchange rates applicable at the transaction dates. Monetary assets and liabilities denominated in foreign currencies are translated into the functional currency at the reporting date.
The resulting exchange gains or losses are recognized in the statements of operations.
4.3.2.Translation of foreign subsidiary financial statements
The assets and liabilities of foreign operations having a functional currency different from the euro are translated into euros at the closing exchange rate. The income and expenses of foreign operations are translated into euros at the exchange rates effective at the transaction dates or using the average exchange rate for the reporting period unless this method cannot be applied due to significant exchange rate fluctuations.
Gains and losses arising from foreign operations are recognized in the statement of other comprehensive loss. When a foreign operation is partly or fully divested, the associated share of gains and losses recognized in the currency translation reserve is transferred to the statements of operations.
The Group’s presentation currency is the euro, which is also the functional currency of GENFIT S.A.
The functional currency of GENFIT CORP and Versantis, Inc. is the U.S. dollar. The applicable exchange rates used to translate the financial statements of this entity for each of the periods are as follows:
| | Year ended |
| | Year ended | | | | Year ended |
| Ratio : 1 US dollars (USD) = x euros (EUR) | Ratio : 1 US dollars (USD) = x euros (EUR) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 | Ratio : 1 US dollars (USD) = x euros (EUR) | | 2021/12/31 | | 2022/12/31 | | 2023/12/31 |
| Exchange rate at period end | Exchange rate at period end | | 0.89015 | | 0.81493 | | 0.88292 | Exchange rate at period end | | 0.88292 | | 0.93756 | | 0.90498 |
| Average exchange rate for the period | Average exchange rate for the period | | 0.89341 | | 0.87755 | | 0.84542 | Average exchange rate for the period | | 0.84542 | | 0.95105 | | 0.92471 |
|
The functional currency of Versantis AG is the Swiss Franc. The applicable exchange rates used to translate the financial statements of this entity for each of the periods are as follows:
| | | | | | | | | | | | | | | |
| Ratio : 1 CH franc (CHF) = x euros (EUR) | | Year ended |
| | | 2021/12/31 | 2022/12/31 | 2023/12/31 |
| Exchange rate at period end | | N/A | 1.01554 | 1.07991 |
| Average exchange rate for the period | | N/A | 1.01710 | 1.02936 |
Note that the average rate immediately above for the year ended December 31, 2022 is based on the period between September 29, 2022 (the date of the acquisition of Versantis AG) and December 31, 2022.
4.4.5.SEGMENT INFORMATION
The Board of Directors and Chief Executive Officer are the chief operating decision makers.
The Board of Directors and the Chief Executive Officer oversee the operations and manage the business as one segment with a single activity; namely, the research and development of innovative medicines and diagnostic solutions, the marketing of which depends on the success of the clinical development phase.
The assets, liabilities and operating income (loss) are mainly located in France and in Switzerland (the latter as a result of the acquisition of Versantis in September 2022).
Revenue breakdown by geographical area
| | | | | | | | | | | | | | | |
| Revenue by destination | | Year ended |
| (in € thousands) | | 12/31/2021 | 12/31/2022 | 12/31/2023 |
| Revenue from France | | 100 | % | 100 | % | 100 | % |
| Revenue from other countries | | — | % | — | % | — | % |
| TOTAL | | 100 | % | 100 | % | 100 | % |
In 2023, 2022, and 2021 revenue was generated entirely in France. Substantially all revenue was generated from Ipsen in 2023.
Non-current assets by geographical area
Non-current assets break down by geographical area as follows:
| | | | | | | | | | | | | | | | | | | | | | | |
| NON-CURRENT ASSETS | As of December 31, 2022 | | As of December 31, 2023 |
| (thousands of euros) | France | Switzerland | Total | | France | Switzerland | Total |
| TOTAL | 12,923 | 44,158 | 57,081 | | 13,869 | 46,889 | 60,758 |
6.FINANCIAL RISKS MANAGEMENT
The Group may be exposed to the following risks arising from financial instruments: foreign exchange risk, interest rate risk, liquidity risk and credit risk.
6.1.Foreign exchange risk
The Group's overall exposure to the foreign exchange risk depends, in particular, on:
•the currencies in which it receives its revenues;
•the currencies chosen when agreements are entered into, such as licensing agreements, or co-marketing or co-development agreements;
•the location of clinical trials on drug or biomarker candidates;
•the ability, for its co-contracting parties to indirectly transfer foreign exchange risk to the Company;
•the Group’s foreign exchange risk policy; and
•the fluctuation of foreign currencies against the euro.
Given the significant portion of its operations denominated in US dollars, the Company has chosen to limit conversions into euros from its US dollars reserves, which resulted from funds received from the listing of its securities on the Nasdaq in March 2019 in US dollars. The Company has not entered into hedging agreements, opting instead to use its cash in US dollars to meet expenses denominated in said currency in subsequent years.
The following table shows the sensitivity of the Group's cash and cash equivalent and expenses in U.S. dollars to a variation of 10% of the U.S. dollar against the euro in 2021, 2022 and 2023.
| | | | | | | | | | | | | | | | | | | |
| Sensitivity of the Group's cash and cash equivalents to a variation of +/- 10% | | | As of |
| of the US dollar against the euro | | | | | | |
| (in € thousands or in US dollar thousands, as applicable) | | | | 2022/12/31 | | 2023/12/31 |
| Cash and cash equivalents denominated in US dollars | | | | 34,192 | | 22,023 |
| Equivalent in euros, on the basis of the exchange rate described below | | | | 32,057 | | 19,930 |
| Equivalent in euros, in the event of an increase of 10% of US dollar vs euro | | | | 35,619 | | 22,145 |
| Equivalent in euros, in the event of a decrease of 10% of US dollar vs euro | | | | 29,143 | | 18,119 |
| | | | | | | | | | | | | | | | | | | | | |
| Sensitivity of the Group's expenses to a variation of +/- 10% | | Year ended |
| of the US dollar against the euro | | | | | | |
| (in € thousands or in US dollar thousands, as applicable) | | 2021/12/31 | | 2022/12/31 | | 2023/12/31 |
| Expenses denominated in US dollars | | 12,566 | | 14,884 | | 15,326 |
| Equivalent in euros, on the basis of the exchange rate described below | | 11,095 | | 13,955 | | 13,870 |
| Equivalent in euros, in the event of an increase of 10% of US dollar vs euro | | 12,328 | | 15,506 | | 15,411 |
| Equivalent in euros, in the event of a decrease of 10% of US dollar vs euro | | 10,086 | | 12,686 | | 12,609 |
2023/12/31: Equivalent in euros, on the basis of 1 euro = 1.1050 dollars US.
2022/12/31: Equivalent in euros, on the basis of 1 euro = 1.0666 dollars US.
2021/12/31: Equivalent in euros, on the basis of 1 euro = 1.1326 dollars US.
The following table shows the sensitivity of the Group's cash and cash equivalent and expenses in Swiss Francs to a variation of 10% of the Swiss Franc against the euro in 2023.
| | | | | | | | | | | | | | | |
| Sensitivity of the Group's cash and cash equivalents to a variation of +/- 10% | | As of |
| of the CH franc against the euro | | | | |
| (in € thousands or in CH franc thousands, as applicable) | | 2021/12/31 | 2022/12/31 | 2023/12/31 |
| Cash and cash equivalents denominated in CH franc | | N/A | 2,321 | 1,111 |
| Equivalent in euros, on the basis of the exchange rate described below | | N/A | 2,357 | 1,200 |
| Equivalent in euros, in the event of an increase of 10% of CH franc vs euro | | N/A | 2,618 | 1,333 |
| Equivalent in euros, in the event of a decrease of 10% of CH franc vs euro | | N/A | 2,142 | 1,091 |
| | | | | |
| Sensitivity of the Group's expenses to a variation of +/- 10% | | Year ended |
| of the CH franc against the euro | | | | |
| (in € thousands or in CH franc thousands, as applicable) | | 2021/12/31 | 2022/12/31 | 2023/12/31 |
| Expenses denominated in CH franc | | N/A | 2,016 | 4,678 |
| Equivalent in euros, on the basis of the exchange rate described below | | N/A | 2,048 | 5,052 |
| Equivalent in euros, in the event of an increase of 10% of CH franc vs euro | | N/A | 2,275 | 5,614 |
| Equivalent in euros, in the event of a decrease of 10% of CH franc vs euro | | N/A | 1,862 | 4,593 |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
2023/12/31: Equivalent in euros, on the basis of a 1 euro = 0.9260 CHF
2022/12/31: Equivalent in euros on the basis of a 1 euro = 0.9847 CHF
The following table shows the Group's cash and cash equivalent and financial assets by currency (EUR, USD, CHF).
| | | | | | | | | | | | | | | | | |
| Cash, cash equivalents and financial assets | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| At origin, denominated in EUR | | | | | | |
| Cash and cash equivalents | | | | 101,536 | | | 56,593 | |
| Current and non current financial assets | | | | 9,456 | | | 4,095 | |
| Total | | | | 110,993 | | | 60,689 | |
| At origin, denominated in USD | | | | | | |
| Cash and cash equivalents | | | | 32,057 | | | 19,931 | |
| Current and non current financial assets | | | | 7 | | | 15 | |
| Total | | | | 32,064 | | | 19,946 | |
| At origin, denominated in CHF | | | | | | |
| Cash and cash equivalents | | | | 2,358 | | | 1,200 | |
| Current and non current financial assets | | | | — | | | 14 | |
| Total | | | | 2,358 | | | 1,214 | |
| Total, in EUR | | | | | | |
| Cash and cash equivalents | | | | 136,001 | | | 77,789 | |
| Current and non current financial assets | | | | 9,464 | | | 4,125 | |
| Total | | | | 145,464 | | | 81,913 | |
6.2.Interest rate risk
As of December 31, 2023, the Group was only liable for governmental advances or conditional advances and bank loans with no interest or interest at a fixed rate, generally below market rate.
As of December 31, 2022 and 2023, the Group's financial liabilities totaled €75.3 million and €70.2 million respectively (net of the equity component of the convertible loan and debt issue costs). Current borrowings are at a fixed rate. The Group's exposure to interest rate risk through its financial assets is also insignificant since these assets are mainly euro-denominated Undertakings for the Collective Investment of Transferable Securities (UCITs), medium-term negotiable notes or term deposits with progressive rates denominated in euros or US dollars.
6.3.Liquidity risk
The Group's loans and borrowings mainly consist of bonds convertible or exchangeable into new or existing shares (OCEANE), repayable for an nominal amount of €57 million on October 16, 2025, and bank loans. See Note20 -"Loans and borrowings" for additional information. The Group has conducted a specific review of its liquidity risk and considers that it is able to meet its future maturities. On December 31, 2022 and 2023, we had €136,001 and €77,789 respectively, in cash and cash equivalents. (In addition, as of December 31, 2022, the Group had €4,550 in other current financial assets which consisted of a single short-term instrument whose term was 180 days.)
The Group does not believe it is exposed to short-term liquidity risk. The Company believes that its cash and cash equivalents and current financial instruments are sufficient to ensure its financing for the next 12 months, in light of its current projects and obligations.
If the Group's funds are insufficient to cover any additional financing needs, the Group would require additional financing. The conditions and arrangements for any such new financing would depend, among other factors, on economic and market conditions that are beyond the Group's control.
6.4.Credit risk
Credit risk is the risk of financial loss if a customer or counterparty to a financial asset defaults on their contractual commitments. The Group is exposed to credit risk due to trade receivables and other financial assets.
The Group's policy is to manage this risk by transacting with third parties with good credit standards.
7.REVENUES AND OTHER INCOME
7.1.Revenues from ongoing activities under client agreementscontracts with customers
The Group’s accounting policies associated with revenue are as follows:
4.4.1.IFRS 15Accounting policy overview
Under IFRS 15, revenue is recognized when the Company fulfills a performance obligation by providing separate goods or services to a customer, i.e., when the customer obtains control of those goods or services. An asset is transferred when the customer obtains control of that asset or service.
Under this standard, each contract must be analyzed, on a case-by-case basis, in order to verify whether it contains performance obligations towards third parties, and, if applicable, to identify their nature in order to determine the appropriate accounting of amounts that the Company has received or is entitled to receive from third parties, for example:
•The transfer of control over the intellectual property, via a license granted by the Company, as it exists at the time of the sale, the date of which will determine that of the revenue recognition;
•If the license is considered as a right of access to the intellectual property of the Company over the life of the license, the revenue would be recognized over this lifetime;
•The supply of products whose revenues would be recognized at the time of transfer of control of the delivered products,products; and
•Potential revenue from milestones, or from royalties or royalties based on sales, would not be recognized until the achievement of the milestone or completion of the sale.
Financial statement line item detail
In 2023, the total revenues and other income amounted to €38,176 (€26,566 in 2022, and €85,579 in 2021).
Revenue amounted to €28,565 in 2023 (€20,195 in 2022, and €80,069 in 2021).
Revenue is primarily composed of:
1.Licensing Agreement (Ipsen). In December 2021, GENFIT and Ipsen entered into an exclusive licensing agreement for elafibranor, a Phase 3 asset evaluated in Primary Biliary Cholangitis (PBC), as part of a long-term global partnership ("Collaboration and License Agreement").
◦In 2023, €13.3 million was attributable to a milestone invoiced to Ipsen in December 2023 in accordance with the Collaboration and Licensing agreement signed in December 2021. This milestone was earned following the NDA filing acceptance by the FDA and MAA filing acceptance by the EMA for accelerated approval of elafibranor. €8.7 million was attributable to previously deferred revenue of €40 million from 2021 as noted immediately below, in line with the progress in the ELATIVE® clinical study and related expenses incurred during the period.
◦In 2022, €15.9 million was attributable to previously deferred revenue of €40 million from 2021 as noted immediately below, in line with the progress in the ELATIVE® clinical study and expenses incurred during the period.
◦In 2021, €80 million was attributable to the recognition of the initial payment received from Ipsen pursuant to the license agreement entered into in December 2021 (of the total amount of €120 million) for the granting of the licence. The remaining balance of the initial payment, i.e. 40 million euros, was recorded as deferred income.
2.Transition Services Agreements (Ipsen). GENFIT and Ipsen entered into the Transition Services Agreement and Part B Transition Services Agreement, signed in April 2022 and September 2023 respectively, in order to facilitate the transition of certain services related to the Phase 3 ELATIVE® clinical trial until the complete transfer of the responsibility of the trial to Ipsen.
◦In 2023, the services provided under this contract generated €6.5 million in revenue.
◦In 2022, the services provided under this contract generated €1 million in revenue.
3.Inventory Purchase Agreement (Ipsen). GENFIT and Ipsen also entered into an Inventory Purchase Agreement in 2022, which provided for the purchase by Ipsen of batches of active ingredients and elafibranor products during the second half of 2022.
◦In 2023, no revenue was generated under this contract.
◦In 2022, inventory sold to Ipsen under this contract generated €3.3 million in revenue.
4.Other revenue
◦In 2023 and 2022 other revenue was not significant.
◦In 2021, other revenue recognized relates to license agreements with Labcorp for the deployment of NIS4® diagnostic technology in the field of MASH, amounting to €69.
Application of IFRS 15 to the Ipsen License Agreement signed in 2021
Performance obligations
We have identified that the agreement provides for four distinct performance obligations:
•The license for elafibranor,
•The completion of the ELATIVE® Phase 3 trial until the end of the double-blind period,
•The knowledge transfer related to elafibranor, as well as support for Ipsen in future undertakings and processes, and
•The provision of drug tablets that may be needed by Ipsen to conduct their clinical trials.
The compensation under this agreement consists of an upfront payment, milestone payments, and royalties on future sales of elafibranor by Ipsen. Besides, it must be noted that, with respect to (i) support services other than the knowledge transfer and (ii) the provision of drug tablets, the agreement provides for separate prices covering all costs borne by the Company to provide those goods and services, therefore constituting in each case an individual and distinct sale price for the relevant goods or service, which is not included in the aforementioned price elements.
We estimate the individual sale price of the clinical trial phase to be €40 million, including forecasted external costs, personnel expenses for the relevant staff, indirect costs pertaining to the work environment of such staff, augmented of a customary margin rate for CRO (Clinical Research Organization) contracting. This calculation of the individual sale price for the clinical trial phase reflects observable price conditions as recommended under IFRS 15.79.c. We used the same method to calculate the individual sale price of the knowledge transfer.
Revenue accounting treatment for periods presented
Regarding the recognition of revenue related to the license, we have applied the following methods:
•The upfront payment, minus the portion of prices allocated to knowledge transfer services and clinical phase execution, has been recognized at the date of transfer of control, i.e. December 16, 2021 according to the above, as it is a static license (without implication or associated service provision);
•Milestone payments constitute variable and uncertain income, which would be, if applicable, recognized in revenue at the time they become highly probable, which means, in this case, due by Ipsen. In 2023, the €13.3 million milestone was triggered by the acceptance of the New Drug Application (NDA) filing by the US Food and Drug Administration (FDA) and Marketing Authorization Application (MAA) by the European Medicines Agency (EMA) for accelerated approval of elafibranor in Primary Biliary Cholangitis (PBC).
Regarding the recognition of revenue related to the Phase 3 ELATIVE® trial until the end of the double-blind period, we have applied the following method:
•The part of the upfront payment allocated to this service will be recognized progressively as completion progresses.
Accounting treatment for future milestones and royalties
•Milestone payments constitute variable and uncertain income, which would be, if applicable, recognized in revenue at the time they become highly probable, which means, in this case, due by Ipsen. In addition, the future milestones that we may receive in 2024, subject to approval by applicable regulatory authorities and US and European commercial launches of elafibranor in PBC by Ipsen, represents a total of up to approximately €75.2 million.
•Royalties will be progressively recognized in revenue as sales are completed by Ipsen, in accordance with the IFRS 15 exception for royalties constituting variable income.
Application of IFRS 15 to the Ipsen Transition Services Agreement and Inventory Purchase Agreement signed in 2022
In 2022, GENFIT and Ipsen entered into a Transition Services Agreement, which outlines the scope of services to facilitate the transition of some activities related to the Phase 3 clinical trial evaluating elafibranor in Primary Biliary Cholangitis (PBC). This agreement is a supplementary follow-on to the Collaboration and License Agreement mentioned above. We evaluated the agreement under IFRS 15 and we concluded that the services constitute a single performance obligation for which revenue is recognized as services are performed.
In 2022, GENFIT and Ipsen entered into an Inventory Purchase Agreement, pursuant to which Ipsen purchased inventory from GENFIT, namely the elafibranor active pharmaceutical ingredient and related drug product, during the second half of 2022 with the prospect of transferring the conduct of the ELATIVE® study to Ipsen. We evaluated the agreement under IFRS 15 and we concluded that the services constitute a single performance obligation for which revenue is recognized when inventory is provided to Ipsen.
Application of IFRS 15 to the Ipsen Part B Transition Services Agreement signed in 2023
In 2023, GENFIT and Ipsen entered into a Part B Transition Services Agreement, which outlines the scope of services to facilitate the transition of some activities related to the Phase 3 clinical trial evaluating elafibranor in Primary Biliary Cholangitis (PBC). This agreement is a supplementary follow-on to the Collaboration and License Agreement mentioned above. We evaluated the agreement under IFRS 15 and we concluded that the services constitute a single performance obligation for which revenue is recognized as services are performed.
4.5.7.2.Other income
7.2.1.Research tax credit
The Research Tax Credit ("Crédit d'Impôt Recherche," or "CIR") is granted to entities by the French tax authorities in order to encourage them to conduct technical and scientific research. Entities that demonstrate that their research expenditures meet the required CIR criteria receive a tax credit that may be used for the payment of their income tax due for the fiscal year in which the expenditures were incurred, as well as in the next three years. If taxes due are not sufficient to cover the full amount of tax credit at the end of the three-year period, the difference is paid in cash to the entity by the tax authorities. If a company meets certain criteria in terms of sales, headcount or assets to be considered a small/mid-size company, immediate payment of the Research Tax Credit can be requested. The Group meets such criteria.
The Group applies for CIR for research expenditures incurred in each fiscal year and recognizes the amount claimed in the line item "Other income" in the statements of operations in the same fiscal year. In the notes to the financial statements, the amount claimed is recognized under the heading "Research tax credit" (see Note 16- "Trade and other receivables" and the table below). The breakdown of Other income is as follows:
| | | | | | | | | | | | | | | | | | | | | |
| Other income | | Year ended |
| (in € thousands) | | 2021/12/31 | | 2022/12/31 | | 2023/12/31 |
| CIR tax credit | | 5,282 | | | 6,017 | | | 5,807 | |
| Other operating income | | 223 | | | 320 | | | 464 | |
| Government grants and subsidies | | 5 | | | 34 | | | 3,340 | |
| TOTAL | | 5,510 | | | 6,371 | | | 9,610 | |
The research tax credit (CIR) amounted to €5,807 in 2023 (€6,017 in 2022 and €5,282 in 2021), due to a slight reduction in eligible research and development expenses. Note that there is also a tax inspection currently taking place as explained in Note 11 - "Income tax". Other operating income amounted to €464 in 2023 (€320 in 2022 and €223 in 2021), mainly comprised of exchange gains on trade receivables.
Government grants and subsidies amounted to €3,340 in 2023 (€34 in 2022 and €5 in 2021). The increase is from one-time cancellation of €3,229 thousand refundable government grant from Bpifrance (the BPI France IT-DIAB) as part of a framework innovation aid agreement involving several scientific partners and for which the Group was the lead partner. For further information refer to Note 20.2.1 - "Breakdown and other Loans and Borrowings - Refundable and conditional advances". 8.OPERATING EXPENSES
Accounting policies - Research and development expenses
Research expenses are recorded in the financial statements as expenses.
In accordance with IAS 38, Intangible Assets, development expenses are recognized as intangible assets only if all the following criteria are met:
•Technical feasibility necessary for the completion of the development project;
•Intention on the Group's part to complete the project and to utilize it;
•Capacity to utilize the intangible asset;
•Proof of the probability of future economic benefits associated with the asset;
•Availability of the technical, financial, and other resources for completing the project; and
•Reliable evaluation of the expenses attributed to the intangible asset during its development.
As of the date of these financial statements, these criteria have not all been met for expenses incurred in the periods presented.
Accounting policies - Classification of operating expenses
Research and development expenses include:
•employee-related costs;
•costs related to external employees seconded to the Company (such as clinical development, biometrics and IT…);
•lab supplies and facility costs;
•fees paid to scientific advisers and contracted research and development activities conducted by third parties;
•intellectual property fees corresponding to the filing of the Group's patents, and
•provision and reversals of provisions in relation to the Research Tax Credit dispute.
Contracted research and development activities conducted by third parties include services subcontracted to research partners for technical and/or regulatory reasons. In particular, this includes the production of active ingredients and therapeutic units, all or a part of clinical trials and preclinical trials that are necessary to the development of GENFIT's drug candidates and biomarker candidates.
Research and development expenses at each reporting date take into account estimates for ongoing activities subcontracted as part of the clinical trials and not yet invoiced, on the basis of detailed information provided by subcontractors and reviewed by the Group’s internal departments. The accuracy of these estimates for some types of expenses improves with the progression of the trials and the review of their determination methods. For regulatory reasons, research services for clinical trials and the production of active ingredients and therapeutic units are contracted out to third parties.
General and administrative expenses include:
•employee-related costs for executive, business development, intellectual property, finance, legal and human resources and communications functions;
•facility-related costs;
•marketing, legal, audit and accounting fees;
•press relations and communications firm fees;
•the cost of external employees seconded to the Company (such as security, reception, and accounting...);
•other service costs (recruitment, etc.); and
•intellectual property fees corresponding to the maintenance of the Group's patents.
Marketing and market access expenses include:
•employee-related costs for marketing and business development functions; and
•marketing, and market access firm fees.
Reorganization and restructuring expenses include:
•(2022 and prior) the accruals and provisions recognized within the scope of the reduction in force plan;
•(2022 and prior) the extraordinary amortization, loss of value and impairment of fixed assets recognized within the scope of the reorganization of GENFIT;
•(2022 and prior) the impairment of the right of use of the leased equipment and premises;
•(2022 and prior) the portion of the OCEANEs renegotiation expenses;
•(2023 and prior) the provision (and subsequent reversals) recognized for some of the costs of the closing process for the RESOLVE-IT® study, which, after detailed analysis, do not have any future economic advantage for the PBC program.
Other operating expenses include:
•Legal fees, audit and accounting fees;
•Advisor fees (banking, press relations, communication, IT, market access, marketing, scientific advising);
•Intellectual property expenses, including in particular the charges and fees incurred by the Company for patent applications and maintenance;
•Expenses related to insurance, notably those triggered by the Company listing on the Nasdaq since 2019;
•Expenses related to the rental, use, and maintenance of the Group's premises;
•Expenses related to external personnel contracted out to the company (safety and security, front desk, clinical and IT services); and
•Expenses related to travel and conferences, including mainly employee travel costs as well as scientific, medical, financial and business development conference registration fees.
Financial statement line item detail
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Operating expenses and other operating income (expenses) | | Year ended | | Of which : |
| | | 2021/12/31 | | Raw | | Contracted | | Employee | | Other | | Depreciation, | | Gain / |
| | | | | materials | | research and | | expenses | | expenses | | amortization | | (loss) on |
| | | | | and | | development | | | | (maintenance, | | and | | disposal of |
| | | | | consumables | | activities | | | | fees, travel, | | impairment | | property, |
| | | | | used | | conducted by | | | | taxes…) | | charges | | plant and |
| (in € thousands) | | | | | | third parties | | | | | | | | equipment |
| Research and development expenses | | (35,166) | | | (1,305) | | | (18,808) | | | (8,192) | | | (4,593) | | | (2,247) | | | (19) | |
| General and administrative income (expenses) | | (16,153) | | | (161) | | | (85) | | | (7,379) | | | (8,003) | | | (541) | | | 15 | |
| Marketing and market access expenses | | (1,539) | | | (1) | | | (1) | | | (783) | | | (741) | | | (13) | | | — | |
| Reorganization and restructuring income (expenses) | | (142) | | | (5) | | | — | | | — | | | (2,343) | | | 2,206 | | | — | |
| Other operating income (expenses) | | (763) | | | — | | | — | | | — | | | (338) | | | 4 | | | (429) | |
| TOTAL | | (53,763) | | | (1,472) | | | (18,895) | | | (16,354) | | | (16,019) | | | (591) | | | (433) | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Operating expenses and other operating income (expenses) | | Year ended | | Of which : |
| | | 2022/12/31 | | Raw | | Contracted | | Employee | | Other | | Depreciation, | | Gain / |
| | | | | materials | | research and | | expenses | | expenses | | amortization | | (loss) on |
| | | | | and | | development | | | | (maintenance, | | and | | disposal of |
| | | | | consumables | | activities | | | | fees, travel, | | impairment | | property, |
| | | | | used | | conducted by | | | | taxes…) | | charges | | plant and |
| (in € thousands) | | | | | | third parties | | | | | | | | equipment |
| Research and development expenses | | (35,818) | | | (1,876) | | | (17,407) | | | (10,029) | | | (5,177) | | | (1,328) | | | — | |
| General and administrative expenses | | (16,405) | | | (248) | | | (71) | | | (6,772) | | | (9,168) | | | (146) | | | — | |
| Marketing and market access expenses | | (992) | | | (3) | | | (1) | | | (565) | | | (416) | | | (6) | | | — | |
| Reorganization and restructuring income (expenses) | | 11 | | | — | | | — | | | — | | | — | | | 11 | | | — | |
| Other operating income (expenses) | | (652) | | | — | | | — | | | — | | | (667) | | | — | | | 16 | |
| TOTAL | | (53,855) | | | (2,128) | | | (17,479) | | | (17,366) | | | (15,429) | | | (1,469) | | | 16 | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Operating expenses and other operating income (expenses) | | Year ended | | Of which : |
| | | 2023/12/31 | | Raw | | Contracted | | Employee | | Other | | Depreciation, | | Gain / |
| | | | | materials | | research and | | expenses | | expenses | | amortization | | (loss) on |
| | | | | and | | development | | | | (maintenance, | | and | | disposal of |
| | | | | consumables | | activities | | | | fees, travel, | | impairment | | property, |
| | | | | used | | conducted by | | | | taxes…) | | charges | | plant and |
| (in € thousands) | | | | | | third parties | | | | | | | | equipment |
| Research and development expenses | | (46,503) | | | (1,831) | | | (23,455) | | | (12,475) | | | (7,452) | | | (1,291) | | | — | |
| General and administrative expenses | | (17,741) | | | (337) | | | (205) | | | (7,486) | | | (9,396) | | | (317) | | | — | |
| Marketing and market access expenses | | (876) | | | (4) | | | (1) | | | (556) | | | (300) | | | (14) | | | — | |
| Reorganization and restructuring income (expenses) | | 505 | | | — | | | — | | | — | | | — | | | 505 | | | — | |
| Other operating income (expenses) | | (141) | | | — | | | — | | | — | | | (222) | | | — | | | 81 | |
| TOTAL | | (64,756) | | | (2,172) | | | (23,661) | | | (20,517) | | | (17,370) | | | (1,117) | | | 81 | |
| | | | | | | | | | | | | | | |
2022 Activity
Research and Development Expenses
The increase in research and development costs is generally explained by the increase in costs related to new programs and product candidates, in particular NTZ, VS-01 and GNS561, offset by the sharp reduction in study costs related to RESOLVE-IT®.
General and Administrative Expenses
The increase in general and administrative expenses is broadly explained by the increase in costs related to liability insurance, the increase in costs related to consulting fees, and other charges in the normal course of business.
Marketing and Market Access Expenses
This decrease is mainly explained by the decrease in marketing activity in the United States and France.
Reorganization and Restructuration Expenses
Reorganization and restructuration expenses were not significant.
2023 Activity
Research and Development Expenses
The increase in research and development costs is generally explained by the increase in costs related to new programs and product candidates, in particular VS-01 and GNS561, offset by a reduction in costs related to NTZ.
General and Administrative Expenses
The increase in general and administrative expenses is broadly explained by an increase headcount in the normal course of business.
Marketing and Market Access Expenses
This decrease is mainly explained by the decrease in marketing activity in the United States and France.
Reorganization and Restructuration Expenses
Reorganization and restructuration expenses were not significant. The income in 2023 relates solely to the reversal of a prior year provision related to unused office space.
Employee expenses
Employee expenses and number of employees were as follows:
| | | | | | | | | | | | | | | | | | | | | |
| Employee expenses | | Year ended |
| (in € thousands) | | 2021/12/31 | | 2022/12/31 | | 2023/12/31 |
| Wages and salaries | | (10,328) | | | (12,188) | | | (14,524) | |
| Social security costs | | (4,775) | | | (4,765) | | | (5,296) | |
| Changes in pension provision | | (154) | | | (169) | | | (119) | |
| Employee profit-sharing | | (628) | | | — | | | — | |
| Share-based compensation | | (470) | | | (245) | | | (578) | |
| TOTAL | | (16,354) | | | (17,366) | | | (20,517) | |
| | | | | | | | | | | | | | | | | | | | | |
| Number of employees at year-end | | Year ended |
| | | 2021/12/31 | | 2022/12/31 | | 2023/12/31 |
| Average number of employees | | 122 | | 133 | | 154 |
| Number of employees | | | | | | |
| Research and development | | 55 | | 73 | | 78 |
| Services related to research and development | | 18 | | 18 | | 18 |
| Administration and management | | 47 | | 55 | | 61 |
| Marketing and commercial | | 2 | | 2 | | 2 |
| TOTAL | | 122 | | | 148 | | | 159 | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
The increase in employee expenses resulted mainly from an increase in workforce of the average headcount from 133 in 2022 to 154 in 2023.
9.Share-based compensation
Accounting policies
The fair value of equity-settled share-based compensation granted to employees, officers, board members and consultants as determined on the grant date is recognized as a compensation expense with a corresponding increase in equity, over the vesting period. The amount recognized as an expense is adjusted to reflect the actual number of awards for which the related service and non-market performance conditions are expected to be met.
Evaluation models
The fair value of equity-settled share-based compensation granted to employees are measured using i) the Black-Scholes model for share warrants ("Bons de Souscriptions d'Actions" or "BSA") and stock options ("SO") and ii) the Monte Carlo model for free shares ("actions gratuites" or "AGA").
Data and key assumptions used in evaluations
For evaluating BSAs, the following data and key assumptions are utilized in accordance with IFRS 2 - Share based payment: issue price, exercise price, expected volatility, exercise period, expected dividends, risk free interest rate (based on government bonds), and conversion ratio.
For evaluating AGAs, the following data and key assumptions are utilized in accordance with IFRS 2 - Share based payment: grant date, share price at grant date, expected volatility, vesting period, expected dividends, risk free interest rate (based on government bonds), conversion ratio, and expected employee turnover.
For evaluating SOs, the following data and key assumptions are utilized in accordance with IFRS 2 - Share based payment: grant date, share price at grant date, exercise price, expected volatility, vesting period, exercise period, expected dividends, risk free interest rate (based on government bonds), conversion ratio, and expected employee turnover.
Regarding SOs and AGAs, market conditions are taken into account in the determination of the fair value of the plans award. For share-based compensation awards with non-vesting conditions, the grant date fair value of the share-based compensation is measured to reflect such conditions and there is no adjustment for differences between expected and actual outcomes.
Volatility assumptions in the above tables are determined by reference to the Company's historical share price observed on the grant date over a two- and three-year period prior to the grant date, adjusted for extreme variations, if any.
Consultants
GENFIT may also grant equity-settled share-based compensation in exchange for services to consultants who are not considered employees. In such cases, the value of the services is measured when they are rendered by the consultants and the share-based compensation exchanged for the services is measured at an equal amount. If the value of the services cannot be measured reliably, then such value is measured with reference to the fair value of the equity instruments granted.
Financial detail
Share-based compensation granted to employees and executive officers corresponds to stock options and free shares.
Share-based compensation granted to board members and consultants corresponds to share warrants. For the measurement of this share-based compensation, the Group determined that under IFRS its consultants were not equivalent to employees.
Under these programs, holders of vested instruments are entitled to subscribe to shares of the Company at a predetermined exercise price. All of the plans are equity settled.
In 2023 and 2022, only SO and AGA plans were granted as share-based compensation.
The expense recognized during 2023 pursuant to IFRS 2 was €578 (compared to €245 at December 31, 2022 and €470 at December 31, 2021).
The table below shows the share-based compensation by plan:
| | | | | | | | | | | | | | | | | | | | | |
| Share-based compensation - expense | | Year ended |
| (in € thousands) | | 2021/12/31 | | 2022/12/31 | | 2023/12/31 |
| SO 2018 | | 186 | | | — | | | — | |
| SO US 2018 | | 24 | | | — | | | — | |
| AGA S 2019 | | 39 | | | 50 | | | — | |
| AGA D 2019 | | 16 | | | 6 | | | — | |
| SO 2019 | | 105 | | | (21) | | | — | |
| SO 2019 - US | | (11) | | | (16) | | | — | |
| BSA 2019 | | — | | | — | | | — | |
| SO US 2019 | | (7) | | | — | | | — | |
| SO D 2020 | | 14 | | | 14 | | | 14 | |
| SO C 2020 | | 40 | | | 40 | | | 40 | |
| SO US 2020 | | 19 | | | 19 | | | (7) | |
| AGA S 2021 | | 29 | | | 32 | | | 34 | |
| AGA D 2021 | | 5 | | | 7 | | | 7 | |
| SO D 2021 | | 2 | | | 13 | | | 12 | |
| SO C2021 | | 9 | | | 55 | | | 50 | |
| SO US 2021 | | 2 | | | 9 | | | 9 | |
| AGA S 2022 | | — | | | 11 | | | 49 | |
| AGA D 2022 | | — | | | 2 | | | 11 | |
| SO D 2022 | | — | | | 4 | | | 18 | |
| SO C 2022 | | — | | | 17 | | | 83 | |
| SO US 2022 | | — | | | 4 | | | 18 | |
| SO SU 2022 | | — | | | — | | | 4 | |
| AGA S 2023 | | — | | | — | | | 47 | |
| AGA D 2023 | | — | | | — | | | 12 | |
| SO D 2023 | | — | | | — | | | 31 | |
| SO C 2023 | | — | | | — | | | 104 | |
| SO US 2023 | | — | | | — | | | 27 | |
| SO SU 2023 | | — | | | — | | | 16 | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| TOTAL | | 470 | | | 245 | | | 578 | |
9.1. Share warrants
The following table summarizes the data relating to share warrants and the assumptions used for the measurement thereof, in accordance with IFRS 2—Share-based Payment:
| | | | | | | | | | | |
| Share warrants (BSA) | 2019 | 2017 |
| BSA 2019 | BSA 2017-A | BSA 2017-B |
| Option pricing model | Black Scholes |
| Fair value per IFRS 2 | €0.75 | | €3.78 | | €3.81 | |
| Issue price | €1.23 | | €2.00 | | €2.00 | |
| Exercise price | €12.32 | | €19.97 | | €19.97 | |
| Expected volatility | 40.0 | % | 36.4 | % | 35.7 | % |
| End of exercise period | 2024/05/31 | 2022/06/30 | 2022/07/15 |
| Expected dividends | 0 | % | 0 | % | 0 | % |
| Risk free interest rate | 0 | % | 0 | % | 0 | % |
| Conversion ratio | 1:1 | 1:1 | 1:1 |
The services performed by the consultants are mainly:
•to evaluate product development plans and propose, if necessary, changes to strategic or technical approaches;
•to advise the Company's management and the Scientific Board in identifying strategies and selecting drug candidates, based in particular on the scientific results obtained by the Group (new therapeutic targets, new compounds); and
•to assist and advise the Group in its alliance strategies, such as external growth-supporting synergies (acquisition of new competencies and the purchase of operating rights, drug candidates and innovative technologies, etc.)
Information on share warrants activity is as follows for 2023:
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Grant Date | Type | BSAs issued | BSAs outstanding as of January 1, 2023 | BSAs awarded | BSAs exercised | BSAs cancelled/forfeited | BSAs outstanding as of December 31, 2023 | BSAs exercisable as of December 31, 2023 |
| 31/10/2019 | BSA 2019 | 35,070 | 35,070 | 0 | 0 | 0 | 35,070 | 35,070 |
| TOTAL | | | 35,070 | 0 | 0 | 0 | 35,070 | 35,070 |
Information on share warrants activity is as follows for 2022:
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Grant Date | Type | BSAs issued | BSAs outstanding as of January 1, 2022 | BSAs awarded | BSAs exercised | BSAs cancelled/forfeited | BSAs outstanding
as of December 31, 2022 | BSAs exercisable as of December 31, 2022 |
| 31/10/2019 | BSA 2019 | 35,070 | 35,070 | 0 | 0 | 0 | 35,070 | 35,070 |
| 06/12/2017 | BSA 2017-A | 18,345 | 18,345 | 0 | 0 | 18,345 | 0 | 0 |
| 06/12/2017 | BSA 2017-B | 18,345 | 18,345 | 0 | 0 | 18,345 | 0 | 0 |
| TOTAL | | | 71,760 | 0 | 0 | 36,690 | 35,070 | 35,070 |
9.2. Free shares (actions gratuites attribuées or AGA)
The following table summarizes the data relating to free shares and the assumptions used for the measurement thereof, in accordance with IFRS 2—Share-based Payment:
| | | | | | | | | | | | | | | | | |
| Free Shares (AGA) | 2023 | 2022 | 2021 | 2019 |
AGA D & S 2023 | AGA D & S
2022 | AGA S
2021 | AGA D
2021 | AGA D & S
2019 |
| Option pricing model | Monte Carlo |
| Fair value per IFRS 2 | €4.05 | €4.08 | | €4.00 | | €4.15 | | €17.06 | |
| Grant date | 03/10/2023 | 10/14/2022 | 03/30/2021 | 03/17/2021 | 07/18/2019 |
| Share price at grant date | €4.05 | €4.08 | | €4.00 | | €4.15 | | €17.06 | |
| Expected volatility | 84.3 | % | 50 | % | 51 | % | 51 | % | 40.2 | % |
| Vesting period | From 03/10/2023 to 03/13/2026 | From 10/14/2022 to 10/16/2025 | From 03/30/2021 to 03/31/2024 | From 03/17/2021 to 03/31/2024 | From 07/18/2019 to 09/16/2022 |
| Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % |
| Risk free interest rate | 3.06 | % | 2.24 | % | -0.59 | % | -0.59 | % | 0 | % |
| Conversion ratio | 1:1 | 1:1 | 1:1 | 1:1 | 1:1 |
| Expected employee turnover | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % |
The final allocation of free shares is subject to continued employment with the Group and performance conditions.
Information on free shares activity is as follows for 2023:
| | | | | | | | | | | | | | | | | | | | | | | |
| Grant Date | Type | AGAs
issued | AGAs outstanding as of January 1, 2023 | AGAs awarded | AGAs vested | AGAs cancelled/ forfeited | AGAs outstanding as of December 31, 2023 |
| 03/13/2023 | AGA S 2023 | 30,900 | | 30,900 | | 2,300 | 28,600 |
| 03/13/2023 | AGA D 2023 | 10,000 | | 10,000 | | | 10,000 |
| 10/14/2022 | AGA S 2022 | 39,200 | 38,900 | | | 2,400 | 36,500 |
| 10/14/2022 | AGA D 2022 | 20,000 | 20,000 | | | | 20,000 |
| 03/30/2021 | AGA S 2021 | 32,400 | 26,300 | | | 300 | 26,000 |
| 03/17/2021 | AGA D 2021 | 15,000 | 15,000 | | | | 15,000 |
| TOTAL | | | 100,200 | 40,900 | 0 | 5,000 | 136,100 |
Information on free shares activity is as follows for 2022:
| | | | | | | | | | | | | | | | | | | | | | | |
| Grant Date | Type | AGAs
issued | AGAs
outstanding as of January 1, 2022 | AGAs awarded | AGAs vested | AGAs cancelled/ forfeited | AGAs
outstanding as of December 31, 2022 |
| 10/14/2022 | AGA S 2022 | 39,200 | | 39,200 | | 300 | 38,900 |
| 10/14/2022 | AGA D 2022 | 20,000 | | 20,000 | | | 20,000 |
| 03/30/2021 | AGA S 2021 | 32,400 | 29,000 | | | 2,700 | 26,300 |
| 03/17/2021 | AGA D 2021 | 15,000 | 15,000 | | | | 15,000 |
| 07/18/2019 | AGA S 2019 | 17,556 | 10,782 | | 10,782 | | 0 |
| 07/18/2019 | AGA D 2019 | 19,070 | 13,068 | | 8,712 | 4,356 | 0 |
| TOTAL | | | 67,850 | 59,200 | 19,494 | 7,356 | 100,200 |
9.3. Stock options (options de souscription d'actions or SO)
The following tables summarize the data relating to stock options and the assumptions used for the measurement thereof, in accordance with IFRS 2—Share-based Payment:
| | | | | | | | | | | | | | | | | |
| Stock options (SO) | 2023 | |
| SO SU 2023 | SO D 2023 | SO C 2023 | SO US 2023 | | | |
| Option pricing model | Black Scholes |
| Fair value per IFRS 2 | €2.39 | €2.19 | €2.39 | €2.19 | | | |
| Grant date | 3/13/2023 | 3/13/2023 | 3/13/2023 | 3/13/2023 | | | |
| Share price at grant date | €4.00 | €4.00 | €4.00 | €4.00 | | | |
| Exercise price | €3.26 | €4.07 | €3.26 | €4.05 | | | |
| Expected volatility | 83.74 | % | 83.74 | % | 83.74 | % | 83.74 | % | | | |
| Vesting period | From 03/13/2023 to 03/13/2026 | |
| Exercise period | From 03/14/2023 to 03/13/2033 | |
| Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | | | |
| Risk free interest rate | 2.75 | % | 2.75 | % | 2.75 | % | 2.75 | % | | | |
| Conversion ratio | 1:1 | 1:1 | 1:1 | 1:1 | | | |
| Expected employee turnover | 0 | % | 0 | % | 0 | % | 0 | % | | | |
| | | | | | | | | | | | | | | | | | | | | | | |
| Stock options (SO) | 2022 | 2021 |
| SO SU 2022 | SO D 2022 | SO C 2022 | SO US 2022 | SO D 2021 | SO C 2021 | SO US 2021 |
| Option pricing model | Black Scholes |
| Fair value per IFRS 2 | €1.40 | | €1.57 | | €1.90 | | €1.56 | | €1.06 | | €1.30 | | €1.07 | |
| Grant date | 12/2/2022 | 10/17/2022 | 10/17/2022 | 10/17/2022 | 10/20/2021 | 10/20/2021 | 10/20/2021 |
| Share price at grant date | €3.46 | | €4.16 | | €4.16 | | €4.16 | | €3.24 | | €3.24 | | €3.24 | |
| Exercise price | €2.95 | | €3.91 | | €3.12 | | €3.94 | | €3.26 | | €2.61 | | €3.22 | |
| Expected volatility | 49.0 | % | 50.0 | % | 50.0 | % | 50.0 | % | 50.0 | % | 50.0 | % | 50.0 | % |
| Vesting period | From 3/12/2022 to 3/12/2025 | From 17/10/2022 to 17/10/2025 | From 20/10/2021 to 20/10/2024 |
| Exercise period | From 3/12/2022 to 3/12/2032 | From 18/10/2025 to 17/10/2032 | From 21/10/2024 to 21/10/2031 |
| Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % |
| Risk free interest rate | 2.1 | % | 2.24 | % | 2.24 | % | 2.24 | % | -0.6 | % | -0.6 | % | -0.6 | % |
| Conversion ratio | 1:1 | 1:1 | 1:1 | 1:1 | 1:1 | 1:1 | 1:1 |
| Expected employee turnover | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % |
| | | | | | | | | | | | | | | | | | | | |
| Stock options (SO) | 2020 | 2019 |
| SO D 2020 | SO C 2020 | SO US 2020 | SO 2019 | SO US 1 2019 | SO US 2 2019 |
| Option pricing model | Black Scholes |
| Fair value per IFRS 2 | €1.16 | | €1.46 | | €1.12 | | €4.59 | | €3.67 | | €3.23 | |
| Grant date | 12/31/2020 | 12/31/2020 | 12/31/2020 | 7/18/2019 | 7/18/2019 | 11/27/2019 |
| Share price at grant date | €3.99 | | €3.99 | | €3.99 | | €17.06 | | €17.06 | | €14.50 | |
| Exercise price | €4.38 | | €3.50 | | €4.52 | | €13.99 | | €16.90 | | €14.31 | |
| Expected volatility | 49.0 | % | 49.0 | % | 49.0 | % | 40.0 | % | 40.0 | % | 40.0 | % |
| Vesting period | From 31/12/2020 to 31/12/2023 | From 18/07/2019 to 16/09/2022 | From 27/11/2019 to 16/01/2023 |
| Exercise period | From 01/01/2024 to 31/12/2027 | From 17/09/2022 to 17/09/2029 | From 17/01/2023 to 17/01/2030 |
| Expected dividends | 0.0 | % | 0.0 | % | 0.0 | % | 0.0 | % | 0.0 | % | 0.0 | % |
| Risk free interest rate | -0.7 | % | -0.7 | % | -0.7 | % | 0.0 | % | 0.0 | % | 0.0 | % |
| Conversion ratio | 1:1 | 1:1 | 1:1 | 1:1 | 1:1 | 1:1 |
| Expected employee turnover | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % |
| | | | | | | | | | | | | | |
| Stock options (SO) | 2018 | 2017 | 2016 |
| SO 2018 | SO US 2018 | SO 2017 | SO 2016 |
| Option pricing model | Black Scholes |
| Fair value per IFRS 2 | €9.32 | | €6.90 | | €9.32 | | €10.30 | |
| Grant date | 11/7/2018 | 11/7/2018 | 12/6/2017 | 12/15/2016 |
| Share price at grant date | €22.10 | | €22.10 | | €21.95 | | €20.79 | |
| Exercise price | €16.00 | | €21.65 | | €17.91 | | €15.79 | |
| Expected volatility | 44.1 | % | 44.1 | % | 53.7 | % | 63.0 | % |
| Vesting period | From 07/11/2018 to 31/12/2021 | From 06/12/2017 to 31/12/2020 | From 15/12/2016 to 15/12/2019 |
| Exercise period | From 01/01/2022 to 31/12/2028 | From 01/01/2021 to 31/12/2027 | From 16/12/2019 to 16/12/2026 |
| Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % |
| Risk free interest rate | 0.0 | % | 0.0 | % | 0.0 | % | 0.0 | % |
| Conversion ratio | 1:1 | 1:1 | 1:1 | 1:1 |
| Expected employee turnover | 15 | % | 15 | % | 15 | % | 15 | % |
In 2019, the Group revised its estimate of the number of equity instruments expected to be vested taking into account the number of lapsed instruments noted after 4 years of successive plans. As a result, GENFIT revised the turnover rate assumption, which was estimated at 15%, to a rate of 0%, taking into account recent observations and the actual number of lapsed instruments at each closing.
Definitive vesting is subject to continued employment with the Group and performance conditions.
Information on stock options activity is as follows for 2023:
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Grant Date | Type | SO
issued | SO outstanding as of January 1, 2023 | SO awarded | SO cancelled/ forfeited | SO exercised | SO outstanding as of December 31, 2023 | SO exercisable as of December 31, 2023 |
| 03/13/2023 | SO SU 2023 | 16,300 | | 16,300 | | | 16,300 | |
| 03/13/2023 | SO D 2023 | 35,000 | | 35,000 | | | 35,000 | |
| 03/13/2023 | SO C 2023 | 108,700 | | 108,700 | | | 108,700 | |
| 03/13/2023 | SO US 2023 | 30,200 | | 30,200 | | | 30,200 | |
| 02/12/2022 | SO SU 2022 | 8,750 | 8,750 | | | | 8,750 | 0 |
| 10/17/2022 | SO D 2022 | 35,000 | 35,000 | | | | 35,000 | 0 |
| 10/17/2022 | SO C 2022 | 131,000 | 131,000 | | | | 131,000 | 0 |
| 10/17/2022 | SO US 2022 | 34,625 | 34,625 | | | | 34,625 | 0 |
| 10/20/2021 | SO D 2021 | 35,000 | 35,000 | | | | 35,000 | 0 |
| 10/20/2021 | SO C 2021 | 134,375 | 124,375 | | 4,000 | | 120,375 | 0 |
| 10/20/2021 | SO US 2021 | 32,500 | 25,000 | | | | 25,000 | 0 |
| 12/31/2020 | SO D 2020 | 35,000 | 35,000 | | | | 35,000 | 35,000 |
| 12/31/2020 | SO C 2020 | 103,750 | 81,250 | | | | 81,250 | 81,250 |
| 12/31/2020 | SO US 2020 | 56,250 | 50,000 | | 22,500 | | 27,500 | 27,500 |
| 07/18/2019 | SO 2019 | 107,880 | 51,343 | | | | 51,343 | 51,343 |
| 07/18/2019 | SO US 1 2019 | 30,620 | 5,113 | | | | 5,113 | 5,113 |
| 11/07/2018 | SO 2018 | 122,000 | 68,329 | | | | 68,329 | 68,329 |
| 11/07/2018 | SO US 2018 | 17,500 | 9,713 | | | | 9,713 | 9,713 |
| 12/06/2017 | SO 2017-1 | 64,164 | 43,212 | | | | 43,212 | 43,212 |
| 12/06/2017 | SO 2017-2 | 32,086 | 17,765 | | | | 17,765 | 17,765 |
| 12/15/2016 | SO 2016-1 | 41,917 | 34,398 | | | | 34,398 | 34,398 |
| 12/15/2016 | SO 2016-2 | 20,958 | 15,308 | | | | 15,308 | 15,308 |
| TOTAL | | | 805,181 | 190,200 | 26,500 | 0 | 968,881 | 388,931 |
Information on stock options activity is as follows for 2022:
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Grant Date | Type | SO
issued | SO outstanding as of January 1, 2022 | SO awarded | SO cancelled/ forfeited | SO exercised | SO outstanding
as of December 31, 2022 | SO exercisable
as of December 31, 2022 |
| 02/12/2022 | SO SU 2022 | 8,750 | | 8,750 | | | 8,750 | 0 |
| 10/17/2022 | SO D 2022 | 35,000 | | 35,000 | | | 35,000 | 0 |
| 10/17/2022 | SO C 2022 | 131,000 | | 131,000 | | | 131,000 | 0 |
| 10/17/2022 | SO US 2022 | 34,625 | | 34,625 | | | 34,625 | 0 |
| 10/20/2021 | SO D 2021 | 35,000 | 35,000 | | | | 35,000 | 0 |
| 10/20/2021 | SO C 2021 | 134,375 | 134,375 | | 10,000 | | 124,375 | 0 |
| 10/20/2021 | SO US 2021 | 32,500 | 25,000 | | | | 25,000 | 0 |
| 12/31/2020 | SO D 2020 | 35,000 | 35,000 | | | | 35,000 | 0 |
| 12/31/2020 | SO C 2020 | 103,750 | 81,250 | | | | 81,250 | 0 |
| 12/31/2020 | SO US 2020 | 56,250 | 50,000 | | | | 50,000 | 0 |
| 07/18/2019 | SO 2019 | 107,880 | 77,015 | | 25,672 | | 51,343 | 51,343 |
| 07/18/2019 | SO US 1 2019 | 30,620 | 7,670 | | 2,557 | | 5,113 | 5,113 |
| 11/07/2018 | SO 2018 | 122,000 | 68,329 | | | | 68,329 | 68,329 |
| 11/07/2018 | SO US 2018 | 17,500 | 9,713 | | | | 9,713 | 9,713 |
| 12/06/2017 | SO 2017-1 | 64,164 | 43,212 | | | | 43,212 | 43,212 |
| 12/06/2017 | SO 2017-2 | 32,086 | 17,765 | | | | 17,765 | 17,765 |
| 12/15/2016 | SO 2016-1 | 41,917 | 34,398 | | | | 34,398 | 34,398 |
| 12/15/2016 | SO 2016-2 | 20,958 | 15,308 | | | | 15,308 | 15,308 |
| TOTAL | | | 634,035 | 209,375 | 38,229 | 0 | 805,181 | 245,181 |
9.5.Performance conditions
The SO and SO US stock option plans as well as certain free share plans (AGA "D") implemented in 2016, 2017, 2018 and 2019 are subject to internal performance conditions related to the progress of the Group's research and development programs, and to external performance conditions related to the evolution of the Company's stock price.
The other free share plans (AGA "S") and SO plans implemented starting in 2020 are subject only to internal performance conditions.
Performance conditions of 2023 plans
| | | | | |
| Plans | Nature of performance conditions |
SO D 2023 SO C 2023 SO US 2023 SO SU 2023 AGA S 2023 AGA D 2023
Evaluation date for performance conditions: 3/13/2026 | Internal conditions - a) 50% of the instruments SO D 2023/SO C 2023/SO US 2023/ SO SU 2023/AGA S 2023 will be exercisable or definitively vest, and 5,000 of the Free Shares for the AGA D 2023 will be vest, if during 2023 and then at any time during the Vesting Period, 2 new R&D programs (at the rate of one-half of these 2023 instruments per new program), join the Company’s R&D pipeline (as evaluated at December 31, 2022) ; and that these programs are at the clinical development stage at the time they join the pipeline or that they later enter this stage, following: (i) A business development transaction (in-licensing, M&A, etc.) or, (ii) Identification of new opportunities resulting from in-house research (program going from preclinical development stage to clinical development stage). b) 25% of the instruments SO D 2023/SO C 2023/SO US 2023/ SO SU 2023/AGA S 2023 will be exercisable or definitively vest, and 2,500 of the Free Shares for the AGA D 2023 will vest, if at least one of the two following conditions related to development of elafibranor in PBC is met: (i) Filing of the Marketing Authorization Application in the fourth quarter of 2023 (in Europe or the United States); (ii) Marketing Authorization obtained in 2024 (in Europe or the United States). c) 15% of the instruments SO D 2023/SO C 2023/SO US 2023/ SO SU 2023/AGA S 2023 will be exercisable or definitively vest, and 1,500 of the Free Shares for the AGA D 2023 will vest, if at least one of the two following conditions related to the development of the ACLF program is met: (i) VS-01 in ACLF: top-line results from the Phase 2 study obtained in 2024 or communication of final results on the Phase 2 study in 2025; (ii) NTZ : start of a Phase 2 clinical trial in the second half of 2023. d) 10% of the instruments SO D 2023/SO C 2023/SO US 2023/ SO SU 2023/AGA S 2023 will be exercisable or definitively vest, and 1,000 of the Free Shares for the AGA D 2023 will vest, if intermediate results in the Phase 1b/2 of GNS561 are obtained in the fourth quarter 2024 or final results obtained in 2025. External conditions - Each applicable portion of all 10,000 Free Shares under the AGA D 2023 plan, as each Internal Conditions above is met, is then subject to the External Condition according to the methods described below. The degree of fulfillment of the External Condition relating to the Company's stock market price will be determined according to the relative performance of GENFIT shares. Each applicable portion of all 10,000 Free Shares under the AGA D 2023 plan, as each Internal Conditions above is met, will be definitively acquired per the following conditions: (a) No AGA D 2023 shall vest if the Final Price is strictly lower than the Initial Price; (b) If the Final Price is between (i) a value equal to or greater than the Initial Price and (ii) a value lower than the Ceiling Price, the number of AGA D 2023 definitively allocated will be equal to: [(Final Price / Initial Price) -1] x 1/2 of the number of AGA D 2023 instruments (c) All AGA D 2023 if the Final Price is equal to or higher than the Ceiling Price. The notions of “Final Price”, “Initial Price” and “Ceiling Price” are defined in the plan regulations.
|
Performance conditions of 2022 plans
| | | | | |
| Plans | Nature of performance conditions |
SO D 2022 SO C 2022 SO US 2022 SO SU 2022 AGA S 2022 AGA D 2022
Evaluation date for performance conditions: - 10/17/2025 for SO D 2022/SO C 2022/SO US 2022/AGA S 2022/AGA D 2022 - 12/3/2025 for SO SU 2022 | Internal conditions - a) 50% of the instruments SO D 2022/SO C 2022/SO US 2022/ SO SU 2022/AGA S 2022 will be exercisable or definitively vest, and 10,000 of the Free Shares for the AGA D 2022 will vest, if during the 2022 financial year and then at any time during the Vesting Period, 3 new R&D programs (at the rate of one third of these 2022 instruments per new program) complete the Company's R&D program portfolio (as it was at 12/31/2021); that these programs are at the so-called clinical development stage when this addition is made or that they reach this stage afterwards and that this addition originates: (i) a business-development operation (licensing-in, M&A, etc.), or (ii) the identification of new opportunities resulting from internal research (repositioning). b) 25% of the instruments SO D 2022/SO C 2022/SO US 2022/ SO SU 2022/AGA S 2022 will be exercisable or definitively vest, and 5,000 of the Free Shares for the AGA D 2022 will vest, if at least one of the following three conditions relating to the development of the elafibranor development program is fulfilled: (i) obtaining the main results of the first part of the ELATIVE® trial in the second quarter of 2023; (ii) filing of a Marketing Authorization Application for elafibranor in the second half of 2023; (iii) marketing authorization for elafibranor in 2024. c) 15% of the instruments SO D 2022/SO C 2022/SO US 2022/ SO SU 2022/AGA S 2022 will be exercisable or definitively vest, and 3,000 of the Free Shares for the AGA D 2022 will vest, if at least one of the following two conditions relating to the development of the NTZ program in the ACLF is fulfilled: (i) First clinical results in 2022; (ii) Start of a Phase 2 clinical trial in the first half of 2023. d) 10% of instruments SO D 2022/SO C 2022/SO US 2022/ SO SU 2022/AGA S 2022 will be exercisable or definitively vest, and 2,000 of the Free Shares for the AGA D 2022 will vest, if as part of the development of the GNS561 program, a Phase 2b trial starts in the first half of 2023. External conditions - Each applicable portion of all 20,000 Free Shares under the AGA D 2022 plan, as each Internal Conditions above is met, is then subject to the External Condition according to the methods described below. The degree of fulfillment of the External Condition relating to the Company's stock market price will be determined according to the relative performance of GENFIT shares. Each applicable portion of all 20,000 Free Shares under the AGA D 2022 plan, as each Internal Conditions above is met, will be definitively acquired per the following conditions: (a) No AGA D 2022 shall vest if the Final Price is strictly lower than the Initial Price; (b) If the Final Price is between (i) a value equal to or greater than the Initial Price and (ii) a value lower than the Ceiling Price, the number of AGA D 2022 definitively allocated will be equal to:[(Final Price / Initial Price) -1] x 1/2 of the number of AGA D 2022 instruments (c) All AGA D 2022 if the Final Price is equal to or higher than the Ceiling Price. The notions of “Final Price”, “Initial Price” and “Ceiling Price” are defined in the plan regulations. |
10.FINANCIAL INCOME AND EXPENSES
| | | | | | | | | | | | | | | | | | | | | |
| Financial income and expenses | | Year ended |
| (in € thousands) | | 2021/12/31 | | 2022/12/31 | | 2023/12/31 |
| | | | | | | |
| Financial income | | | | | | |
| Interest income | | 274 | | 137 | | 1,709 |
| Foreign exchange gain | | 8,876 | | 7,470 | | 452 |
| Financial income from renegotiating the convertible bond debt OCEANE | | 35,578 | | 0 | | 0 |
| Other financial income | | 52 | | 605 | | 1,519 |
| TOTAL - Financial income | | 44,780 | | 8,212 | | 3,680 |
| Financial expenses | | | | | | |
| Interest expenses | | (4,846) | | (4,341) | | (4,553) |
| Interest expenses for leases | | (109) | | (69) | | (71) |
| Foreign exchange losses | | (2,163) | | (340) | | (966) |
| Other financial expenses | | (5) | | (8) | | (23) |
| TOTAL - Financial expenses | | (7,122) | | (4,758) | | (5,614) |
| FINANCIAL GAIN (LOSS) | | 37,658 | | 3,453 | | (1,934) |
Interest income recognized is almost exclusively related to current financial assets. Other financial income similarly is almost exclusively related to accrued interest income for ongoing current financial assets at the end of the year.
The financial expenses are related to the interest of the OCEANEs and they mainly relate to the payment of coupons at the rate of 3.5% and the amortization of the discount of the bond debt at the effective interest rate of 8.8% to accrete the bond debt up to the amount that will be repaid (or converted) at maturity, recognizing a theoretical annual interest accrual as a result of the accretion on the period of an amount equivalent to the equity component at an effective interest rate.
The portion of financial gain related to currency exchange is a net loss of €515 in 2023 notably due to the difference in currency exchange recognized on the cash equivalents and other current financial assets in US dollars, as GENFIT has decided to keep some of its cash in US dollars. See Note 13 - “Cash and cash equivalents”. These cash investments in US dollars are to be used to pay directly expenses in US dollars (natural currency hedge). Financial income in 2021 included notably the one-time buyback bonus of €35.6 million issued from the renegotiation of the OCEANEs completed in said year.
11.INCOME TAX
Accounting policies
Income tax expense (or benefit) comprises current tax expense (or benefit) and deferred tax expense (or benefit), as applicable.
Deferred taxes are recognized for all the temporary differences arising from the difference between the tax basis and the accounting basis of assets and liabilities.
Deferred tax assets are recognized for unused tax losses, unused tax credits and temporary deductible differences to the extent that:
•it is probable that future taxable profit will be available against which they can be used; or
•if there are deferred tax liabilities for the same entity in the same tax jurisdiction on which they can be applied.
Financial detail
As of December 31, 2023 and 2022, corporate income tax payable by the Parent company GENFIT SA amounted to €23 and €4,906, respectively, which is recognized as "Other current tax liabilities" in the consolidated financial statements. It should be noted that in 2022 the Company recorded an income tax receivable of €5,282, related to previous years, in "Trade and other receivables" in the consolidated statement of financial position. In 2023, the Company presented the amounts net on the financial statements to reflect the net income tax filing position.
We benefited from a reduced tax rate on part of the income from the licensing agreement signed with Ipsen pursuant to Article 238 of the French Tax Code.
The determination of the income tax expense recognized in the consolidated financial statements, which amounted to (an expense) of €380 for 2023, is summarized in the table "Effective tax rate" hereunder.
Change in legislation
In 2017, the United States Congress passed the Tax Cuts and Jobs Act of 2017, which included a tax law change on Section 174 of the Internal Revenue Code. Research and development costs specified under Section 174 of the Code must be capitalized and amortized pro rata over 5 years for domestic expenditures and 15 years for foreign expenditures. Said provision came into effect for tax years commencing after December 31, 2021.
Effective tax rate
| | | | | | | | | | | | | | | |
| | | Year ended |
| (in € thousands) | | 2021/12/31 | 2022/12/31 | 2023/12/31 |
| Net profit (loss) | | 67,259 | (23,719) | (28,894) |
| Income tax benefit (expense) | | (2,215) | 116 | (380) |
| Net profit (loss) before tax | | 69,474 | (23,836) | (28,514) |
| Tax rate in France | | 27.37 | % | 25.00 | % | 25.00 | % |
| Theoretical tax benefit (expense) | | (19,018) | 5,959 | 7,129 |
| Increase / decrease in tax benefit arising from : | | | | |
| Tax credits | | 1,512 | 1,504 | 1,452 |
| Permanent differences | | 833 | (31) | (153) |
| Differences between rates | | 7,323 | (67) | (840) |
| Tax losses for the period, unrecognised as deferred tax assets | | 0 | (7,037) | (7,832) |
| Utilisation of previously unrecognised tax losses | | 5,590 | 0 | 0 |
| IFRS adjustments without tax incidence | | (129) | (61) | (145) |
| Non recognition of deffered tax assets related to temporary differences | | (24) | 331 | 454 |
| Recognition of deferred tax assets against deferred tax liabilities | | 430 | (453) | (418) |
| Tax effects related to the renegociation of the convertible debt | | 1,370 | 0 | 0 |
| Other | | (102) | (29) | (28) |
| | | | | |
| Income tax benefit (expense) recognised in profit or loss | | (2,215) | 116 | (380) |
| Effective income tax rate | | (3.19) | % | (0.49) | % | 1.33 | % |
Tax Inspection
We are subject to a tax audit by the French revenue service on our tax returns or operations subject to review on the 2019 and 2020 periods (including the Research Tax Credit claimed for these periods), which started on December 10, 2021 and is still ongoing at the date of this document.
As of December 2023, on the consolidated statement of financial position, the total amount corresponding to line item "Trade and other receivables" is €32,707. Of this amount, per Note 16 - "Trade and other receivables", the amount corresponding to subcategory "Research tax credit" is €12,200. The breakdown of this amount is as follows: •€5,807 relating to 2023,
•€6,017 relating to 2022, and
•€372 relating to 2021 (€5,282 relating to 2021, netted against the related tax payable balance of €4,906).
This balance has not yet been reimbursed as there is currently a tax inspection taking place by the French tax authorities.
11.1.Losses available for offsetting against future taxable income
At December 31, 2023, 2022 and 2021, the tax loss carry forwards for the Company amounted to €523,392, €477,149 and €449,679, respectively.
Such carry forwards can be offset against future taxable profit within a limit of €1.0 million per year plus 50% of the profit exceeding this limit. Remaining unused losses will continue to be carried forward indefinitely.
In 2021, the amount of tax loss carry forwards used to offset taxable profit were €33.7 million.
11.2.Deferred tax assets and liabilities
The Group's main sources of deferred tax assets and liabilities as of December 31, 2022 and 2023 related to:
•Temporary differences, recognized:
◦OCEANEs: a deferred tax liability for €1,770 and €1,183 as of December 31, 2022 and 2023, respectively, and a deferred tax asset for €1,260 and €842 as of December 31, 2022 and 2023,
◦Linked to other sources (tax-driven amortization): a deferred tax liability of €113.
•Temporary differences, unrecognized
◦Post-employment benefits: a deferred tax asset for €195 and €244, as of December 31, 2022 and 2023, respectively,
◦GENFIT Corp: a deferred tax asset for €1,275 as of December 31, 2023.
•Tax loss carry forwards
Deferred taxes detail
The Company offsets its deferred tax assets and liabilities as permitted by IAS 12, resulting in a net deferred tax liability of €455 as of December 31, 2023 (€510 as of December 31, 2022).
| | | | | | | | | | | | | | | | | | |
| | | As of | Impact on | Impact on the | As of |
| (in € thousands) | | 2021/12/31 | equity | profit/loss | 2022/12/31 |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| Deferred tax liabilities / convertible loan OCEANE | | (2,315) | — | 545 | (1,770) |
| Deferred tax assets / convertible loan OCEANE | | 1,712 | — | (453) | 1,260 |
| Deferred tax liabilities / exceptional depreciation on acquisition costs | | — | — | — | — |
| TOTAL | | (602) | — | 93 | (510) |
| | | | | | | | | | | | | | | | | | |
| | | As of | Impact on | Impact on the | As of |
| (in € thousands) | | 2022/12/31 | equity | profit/loss | 2023/12/31 |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| Deferred tax liabilities / convertible loan OCEANE | | (1,770) | — | 586 | (1,183) |
| Deferred tax assets / convertible loan OCEANE | | 1,260 | — | (418) | 842 |
| Deferred tax liabilities / exceptional depreciation on acquisition costs | | — | — | (113) | (113) |
| TOTAL | | (510) | — | 55 | (455) |
Other than as it relates to deferred tax assets recognized based on the available deferred tax liabilities, no other deferred tax asset has been recognized as it is not probable that taxable profit will be available to offset deductible temporary differences and tax loss carry forwards.
12.Earnings (loss) per share
Basic earnings (loss) per share are calculated by dividing profit or loss attributable to the Company's ordinary shareholders by the weighted average number of ordinary shares outstanding during the period, excluding shares held by GENFIT.
Diluted earnings (loss) per share are calculated by adjusting profit attributable to ordinary shareholders and the average number of ordinary shares outstanding weighted for the effects of all potentially dilutive instruments (share warrants, redeemable share warrants, free shares, stock options and bonds convertible into new and/or existing shares).
The components of the earnings (loss) per share computation are as follows:
| | | | | | | | | | | | | | | | | | | | | |
| Earnings per share | | Year ended |
| | | 2021/12/31 | | 2022/12/31 | | 2023/12/31 |
| | | | | | | |
| Profit (loss) for the period (in € thousands) | | 67,259 | | | (23,719) | | | (28,894) | |
| Weighted average number of ordinary shares used to calculate basic earnings (loss) per share | | 44,739,756 | | | 49,673,936 | | | 49,700,223 | |
| Basic earnings (loss) per share (€/share) | | 1.51 | | | (0.48) | | | (0.58) | |
| | | | | | | |
| | | | | | | |
| Weighted average number of ordinary shares used to calculate diluted earnings (loss) per share | | 55,613,634 | | | 49,673,936 | | | 49,700,223 | |
| Diluted earnings (loss) per share (€/share) | | 1.23 | | | (0.48) | | | (0.58) | |
The following table summarizes the potential common shares not included in the computation of diluted earnings per share because their impact would have been antidilutive:
| | | | | | | | | | | | | | | |
| Potential common shares not included in the computation of diluted earnings per share | | | | Year ended |
| | | | | 2023/12/31 |
| BSA | | | | 35,070 |
| STOCK OPTIONS | | | | 968,881 |
| AGA | | | | 136,100 |
| OCEANES | | | | 10,580,141 |
13.CASH AND CASH EQUIVALENTS
Cash and cash equivalents comprise cash on hand, bank accounts and term deposits, together with short-term deposits and highly liquid investments. They are readily convertible to a known amount of cash and thus present a negligible risk of a change in value. They also include Undertakings for Collective Investments in Transferable Securities (UCITs) whose characteristics allow them to be classified as cash and cash equivalents.
Initially recognized at their purchase cost at the transaction date, investments are subsequently measured at fair value. Changes in fair value are recognized in net financial income (expenses).
The main components of cash equivalents were:
•UCITS and interest-bearing current accounts, available immediately;
•Term accounts, available within the contractual maturities or by the way of early exit with no penalty; and
•Negotiable medium-term notes, available with a quarterly maturity or by the way of early exit with no penalty.
These investments, summarized in the tables below, are short-term, highly liquid and subject to insignificant risk of changes in value.
| | | | | | | | | | | | | | | | | |
| Cash and cash equivalents | | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| Short-term deposits | | | | 119,090 | | | 67,530 | |
| Cash on hand and bank accounts | | | | 16,910 | | | 10,258 | |
| TOTAL | | | | 136,001 | | | 77,789 | |
| | | | | | | | | | | | | | | | | |
| Short-term deposits | | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| | | | | | | |
| TERM ACCOUNTS | | | | 119,090 | | | 67,530 | |
| | | | | | | |
| | | | | | | |
| TOTAL | | | | 119,090 | | | 67,530 | |
| | | | | | | |
14.GOODWILL AND INTANGIBLE ASSETS
Goodwill
The company does not have any goodwill.
Intangible assets
Intangible assets mainly consist of software and operating licenses acquired by the Group. They are recognized at cost less accumulated amortization and impairment. Amortization expense is recorded on a straight-line basis over the estimated useful lives of the intangible assets. The estimated useful lives of both software and license agreements are between 1 and 8 years.
In the event of an acquisition not qualifying as a business combination under IFRS 3, GENFIT initially records the acquired asset at cost of the consideration transferred, excluding variable payments that are dependent on future events. No liability is recognized initially for these contingent payments. A liability will be recorded when the condition that triggers the obligation occurs.
The variable payments that would be due if the asset acquired complies with agreed-upon specifications at specific dates in the future are recognized as an adjustment to the cost of the related asset.
Seal Rock licence agreement (2023)
As previously noted in Note 2 - "Major events in the period and events after the period", on May 31, 2023, GENFIT announced the signing of a licensing agreement for the exclusive worldwide rights to the injectable formulation of ASK1 inhibitor SRT-015 in acute liver disease with Seal Rock Therapeutics, a clinical-stage company based in Seattle, USA. Under the terms of the agreement, GENFIT made an upfront payment in the amount of €2 million to Seal Rock in exchange for acquiring the know-how and rights of use to SRT-015 as described above. The addition is recorded in the table below under line item "Other intangibles."
In accordance with IAS 38 - Intangible assets, this amount was capitalized and allocated to Intangible assets. Further, given the nature of the intangible asset, it was determined to have a definite useful life of 20 years, consistent with patent lifetimes in the United States and the European Union. Amortization will start based on the remaining patent term upon EMA/FDA regulatory approval and until then will be subject to an annual impairment test in accordance with IAS 38 - Intangible Assets. As future milestones for this agreement are paid, they will be analyzed and be either i) capitalized and subject to the same annual impairment test or ii) expensed as incurred. The annual impairment test will be based on a valuation methodology including an income approach using discounted cash flow techniques for the injectable formulation of ASK1 inhibitor SRT-015 in acute liver disease.
In 2023, no indications of impairment was identified.
Versantis (2022)
As noted in Note 30 - "Acquisitions", on September 29, 2022, GENFIT acquired Versantis AG, a private Swiss-based clinical stage biotechnology company focused on addressing the growing unmet medical needs in liver diseases. The Phase 2 ready program, VS-01-ACLF, a program in scavenging liposomes technology, was deemed to be the asset with substantially all attributable value in accordance with the optional concentration test of fair value under paragraph B7A of IFRS 3. Of the total acquisition price paid of €46.6 million, €43.9 million was allocated to Intangible assets in accordance with IAS 38 - Intangible Assets. The difference between that amount and the acquisition price corresponds to the other assets acquired and liabilities assumed as part of the transaction. Further, given the nature of the intangible asset, it was determined to have a definite useful life of 20 years, consistent with patents lifetimes in the United States and the European Union. Amortization will start upon EMA/FDA regulatory approval and until then will be subject to an annual impairment test in accordance with IAS 38 - Intangible Assets.
In accordance with IAS 36, we performed an annual impairment test in 2023 related to the Versantis intangible asset (and in general whenever there is a triggering event), which was based on the excess earnings method using discounted cash flow techniques for the scientific research program VS-01. The aforementioned income method utilizes management’s estimates of future operating revenue, cash flows discounted using a weighted-average cost of capital that reflects market participant assumptions, and the expected success rate of the program based on similar external programs. Based on our analysis performed as of December 31, 2023, the initial valuation of €43.9 million is still appropriate and no impairment loss has been recognized.
The period over which management has projected its cash flows spans through 2036. The drug price growth rate used to extrapolate cash flow projections is 2%. Furthermore, we have performed the following sensitivity analyses in order to determine if a reasonably possible change in a key assumption on which we have based our determination of the recoverable amount would cause the carrying amount of the intangible asset to exceed its recoverable amount.
Values assigned to each key assumption
Discount rate: 12%
The amount by which the value assigned to the weighted average cost of capital must change in order for the recoverable amount to be equal to the carrying amount: 5.4%
Overall expected success rate of the program: 15.1%
The amount by which the value assigned to the expected rate of success of the program must change in order for the recoverable amount to be equal to the carrying amount: 6.2%
Indicators of impairment considered by the Group as part of the implementation of the impairment test above are as follows:
•Failure of or unfavorable data from our clinical trials
•Competition from other clinical trial programs covering the same indications as our drug candidates
•Availability of necessary financing
The following tables show the variations in intangible assets for the years ended December 31, 2022 and 2023:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | As of | | Increase | | Decrease | | Translation | | Reclassification | | As of |
| (in € thousands) | | 12/31/2021 | | | | | | adjustments | | | | 12/31/2022 |
| Gross | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Software | | 1,294 | | | 81 | | | (398) | | | — | | | — | | | 977 | |
| Patents | | 70 | | | 281 | | | — | | | — | | | — | | | 351 | |
| | | | | | | | | | | | | |
| Other intangibles | | — | | | 43,569 | | | — | | | — | | | — | | | 43,569 | |
| TOTAL—Gross | | 1,364 | | | 43,931 | | | (398) | | | — | | | — | | | 44,897 | |
| Accumulated depreciation and impairment | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Software | | (1,190) | | | (79) | | | 329 | | | — | | | — | | | (940) | |
| Patents | | — | | | — | | | — | | | — | | | — | | | — | |
| | | | | | | | | | | | | |
| Other intangibles | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated depreciation and impairment | | (1,190) | | | (79) | | | 329 | | | — | | | — | | | (940) | |
| TOTAL - Net | | 174 | | | 43,852 | | | (69) | | | — | | | — | | | 43,957 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | As of | | Increase | | Decrease | | Translation | | Reclassification | | As of |
| (in € thousands) | | 12/31/2022 | | | | | | adjustments | | | | 2023/12/31 |
| Gross | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Software | | 977 | | | 24 | | | (45) | | | — | | | — | | | 955 | |
| Patents | | 351 | | | — | | | — | | | — | | | 18 | | | 369 | |
| | | | | | | | | | | | | |
| Other intangibles | | 43,569 | | | 2,050 | | | — | | | 2,746 | | | — | | | 48,366 | |
| TOTAL—Gross | | 44,897 | | | 2,074 | | | (45) | | | 2,746 | | | 18 | | | 49,690 | |
| Accumulated depreciation and impairment | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Software | | (940) | | | (63) | | | 75 | | | — | | | — | | | (928) | |
| Patents | | — | | | — | | | — | | | — | | | — | | | — | |
| | | | | | | | | | | | | |
| Other intangibles | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated depreciation and impairment | | (940) | | | (63) | | | 75 | | | — | | | — | | | (928) | |
| TOTAL - Net | | 43,957 | | | 2,010 | | | 29 | | | 2,746 | | | 18 | | | 48,761 | |
4.6.15.PROPERTY, PLANT AND EQUIPMENT
Property, plantPlant and equipmentEquipment
Property, plant and equipment are initially recognized at cost. Cost includes expenditures that are directly attributable to the acquisition of the asset. Routine maintenance costs are expensed as incurred.
Subsequently, depreciation expense is recognized on a straight-line basis over the estimated useful lives of the assets. If components of property, plant and equipment have different useful lives, they are accounted for separately. Depreciation methods, useful lives and residual values are reviewed at each reporting date and adjusted, if appropriate.
Estimated useful lives are as follows:
| | | | | |
| Building on non-freehold land | 10 years |
| Fittings and fixtures | Between 9 and 25 years |
| Scientific equipment | Between 2 and 12 years |
| Computer equipment | Between 2 and 5 years |
| Furniture | Between 4 and 10 years |
| Vehicles | Between 4 and 6 years |
Any gain or loss on disposal of an item of property, plant and equipment is determined by comparing the proceeds from disposal with the carrying amount of the item. The net amount is recognized in the consolidated statements of operations under the line item "Other operating income (expenses)."
4.7.Leases
IFRS 16 introduces for the lessee a single model of accounting on the balance sheet for leases. The lessee recognizes a "right of use" asset which represents its right to use the underlying asset, and a lease liability for its obligation to pay the rent.
The Group recognizes a "right of use" asset and a lease liability at the start of the lease term. The "right of use" asset is initially measured at cost and then at cost less any amortization and accumulated impairment losses. The amount can be adjusted based on certain revaluations of the lease liability.
The lease liability is initially measured at the discounted value of the rents owed and not yet paid at the start date of the contract. The discount rate used is the implicit interest rate of the contract or, if it cannot be easily determined, the Company’s incremental borrowing rate of the lessee. The Group generally uses the latter as the discount rate.
The lease liability is then adjusted by the interest expense minus the amounts of rent paid. It is revalued in the event of a change in future rents following a change in the index or rate, a new estimate of the amount to be paid under a residual value guarantee or, where applicable, a revaluation of the exercise of an option to purchase or to extend, or the non-exercise of an option to terminate (which then becomes reasonably certain).
The Group has exercised its judgment in determining the term of the lease agreements that provide for extension options. The fact that the Group has determined that it is reasonably certain to exercise such options has an impact on the lease term used and has a significant impact on the amount of lease debt and the "right of use" asset in the accounts. The amount of short term or low value leases which are not included in the IFRS 16model16 model is not material.
The following tables show the variations in tangible assets for the years ended December 31, 2022 and 2023:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Property, plant and equipment - Variations | | As of As of | | Increase | | Decrease | | Translation | | Reclassification | | As of As of |
| (in € thousands) | | As of 2021/12/31 | | | | | | adjustments | | | | As of 2022/12/31 |
| Gross | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | 10,311 | | | 610 | | | — | | | — | | | — | | | 10,921 | |
| Scientific equipment | | 6,320 | | | 228 | | | (82) | | | — | | | — | | | 6,467 | |
| Fittings | | 1,474 | | | 61 | | | — | | | — | | | 2 | | | 1,537 | |
| Vehicles | | 91 | | | — | | | — | | | — | | | — | | | 91 | |
| Computer equipment | | 1,542 | | | 98 | | | (149) | | | — | | | 8 | | | 1,500 | |
| Furniture | | 279 | | | — | | | — | | | — | | | — | | | 279 | |
| In progress | | — | | | 16 | | | — | | | — | | | (16) | | | — | |
| TOTAL - Gross | | 20,017 | | | 1,014 | | | (230) | | | — | | | (7) | | | 20,794 | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Accumulated depreciation | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | (2,900) | | | (1,033) | | | — | | | — | | | — | | | (3,934) | |
| Scientific equipment | | (4,868) | | | (697) | | | 79 | | | 4 | | | — | | | (5,481) | |
| Fittings | | (888) | | | (95) | | | — | | | (2) | | | — | | | (985) | |
| Vehicles | | (31) | | | (12) | | | — | | | — | | | — | | | (43) | |
| Computer equipment | | (1,403) | | | (105) | | | 148 | | | (5) | | | — | | | (1,365) | |
| Furniture | | (213) | | | (10) | | | — | | | — | | | — | | | (223) | |
| In progress | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated depreciation | | (10,304) | | | (1,953) | | | 227 | | | (3) | | | — | | | (12,032) | |
| Accumulated impairment | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | (503) | | | — | | | 48 | | | — | | | — | | | (455) | |
| Scientific equipment | | (87) | | | — | | | 28 | | | — | | | — | | | (59) | |
| Fittings | | (93) | | | — | | | 69 | | | — | | | — | | | (24) | |
| Vehicles | | — | | | — | | | — | | | — | | | — | | | — | |
| Computer equipment | | (12) | | | — | | | 2 | | | — | | | — | | | (10) | |
| Furniture | | (3) | | | — | | | — | | | — | | | — | | | (3) | |
| In progress | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated impairment | | (699) | | | — | | | 147 | | | — | | | — | | | (552) | |
| TOTAL - Net | | 9,015 | | | (939) | | | 144 | | | (3) | | | (7) | | | 8,210 | |
4.8.Impairment of tangible assets, intangible assets and goodwill
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Property, plant and equipment - Variations | | As of | | Increase | | Decrease | | Translation | | Reclassification | | As of |
| (in € thousands) | | 2022/12/31 | | | | | | adjustments | | | | 2023/12/31 |
| Gross | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | 10,921 | | | 427 | | | — | | | — | | | 19 | | | 11,367 | |
| Scientific equipment | | 6,467 | | | 207 | | | (1,378) | | | — | | | — | | | 5,295 | |
| Fittings | | 1,537 | | | 33 | | | (7) | | | — | | | — | | | 1,563 | |
| Vehicles | | 91 | | | — | | | — | | | — | | | — | | | 91 | |
| Computer equipment | | 1,500 | | | 150 | | | (32) | | | — | | | (4) | | | 1,613 | |
| Furniture | | 279 | | | 5 | | | (9) | | | — | | | — | | | 274 | |
| In progress | | — | | | 16 | | | — | | | — | | | (16) | | | — | |
| TOTAL - Gross | | 20,794 | | | 839 | | | (1,426) | | | — | | | (3) | | | 20,204 | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Accumulated depreciation | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | (3,934) | | | (1,127) | | | — | | | (3) | | | — | | | (5,064) | |
| Scientific equipment | | (5,481) | | | (296) | | | 1,307 | | | (1) | | | — | | | (4,471) | |
| Fittings | | (985) | | | (100) | | | 2 | | | — | | | — | | | (1,083) | |
| Vehicles | | (43) | | | (12) | | | — | | | — | | | — | | | (55) | |
| Computer equipment | | (1,365) | | | (91) | | | 24 | | | 11 | | | — | | | (1,421) | |
| Furniture | | (223) | | | (11) | | | 5 | | | 1 | | | — | | | (228) | |
| In progress | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated depreciation | | (12,032) | | | (1,637) | | | 1,338 | | | 8 | | | — | | | (12,323) | |
| Accumulated impairment | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | (455) | | | — | | | 455 | | | — | | | — | | | — | |
| Scientific equipment | | (59) | | | — | | | 51 | | | — | | | — | | | (9) | |
| Fittings | | (24) | | | — | | | 24 | | | — | | | — | | | — | |
| Vehicles | | — | | | — | | | — | | | — | | | — | | | — | |
| Computer equipment | | (10) | | | — | | | 10 | | | — | | | — | | | — | |
| Furniture | | (3) | | | — | | | 3 | | | — | | | — | | | — | |
| In progress | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated impairment | | (552) | | | — | | | 543 | | | — | | | — | | | (9) | |
| TOTAL - Net | | 8,210 | | | (798) | | | 455 | | | 8 | | | (3) | | | 7,872 | |
Assets related to contracts that were originally classified as legacy finance leases are scientific equipment and are accounted for under IFRS 16. Their net carrying value as of December 31, 2022 and 2023 amounted to €27 and €0 respectively.
Amortization
Amortization of an asset starts when it becomes available for use. The Company does not have any goodwill.asset should be in the location and condition that is required for it to be operating in the manner intended by management, which – in the case of in process research and development (IPR&D) acquired from Versantis, will happen once it receives regulatory and marketing approval. Until that point, it is tested for impairment annually in accordance with the requirements of IAS 36. The asset is tested for impairment by comparing its recoverable amount with its carrying amount once a year, at a minimum. An additional impairment test is required whenever there is an indication that an intangible asset may be impaired.
Impairment
If indicators of impairment are identified, amortizable intangible assets and depreciable tangible assets are subject to an impairment test under the provisions of IAS 36, Impairment of Assets.
The Group has considered that the discontinued use of some equipment following the termination of RESOLVE-ITRESOLVE-IT® as well as the decision to no longer use part of the leased premises were indicative of an impairment loss requiring the completion of an impairment test of property, plant and equipment or of the rights of use recognized in the statement of financial position for this equipment and lease agreements.
The recovery value of an asset is the higher value between the value in use and the fair value less costs of divestment. The value in use is evaluated in relation to the future forecasted cash flows, discounted at current interest rates, before tax, which reflects the current market appreciation of the time value of money and the risks specific to the asset. In the present case, the recovery value of the tested assets corresponds to their fair value less costs of divestments.divestment.
The impacts related to the impairment (and any reversals thereof) of tangible assets and rights of use related to equipment and premises that are no longer in use due to the discontinuation of the RESOLVE-ITRESOLVE-IT® study are recognized in the consolidated statement of operations under “Reorganization and restructuring costs”.
Some equipment belonging to the Group and others under a leasing agreement were no longer in use following the reorganization of the group’s activities and the termination of the RESOLVE-IT® trial decided in mid-2020.
4.9.Financial instrumentsThis indication of loss of value led the Group to conduct an impairment test over owned and leased equipment, based on the value at which this equipment may be divested (on the basis of agreements with the lessors on the early purchase of the equipment and near-term purchase offers) in order to determine the recovery value.
IFRS 9 “Financial Instruments” takes into accountIn 2022, part of these elements, mainly scientific equipment, were sold. As a result the following three aspects of booking financial instruments :
accumulated impairment for these equipments was reduced to €97, including:•Classification€59 for scientific equipment (of which €31 related to owned equipment and measurement;€28 of leased equipment),
•Impairment and;€24 for fittings, and
•Hedge accounting.€13 for computer equipment and furniture,
Loans and borrowings are initially measured at fair value and subsequently recorded at amortized cost.including associated liabilities.
The IFRS 13 "Fair value measurement" amendment determines three categoriesIn 2023, part of financial instruments accordingthese elements, mainly scientific equipment, were sold. As a result the related accumulated impairment was reduced to a fair value hierarchy :€9, including:
•Level 1 : Fair value measured€9 for scientific equipment (of which €9 related to owned equipment and €0 of leased equipment).
It should be noted that the reversal of provisions related to unused premises in the amount of €455 corresponds to the reorganization of the RESOLVE-IT® study, now substantially complete.
Supplemental IFRS 16 Disclosures
Right of use assets and accumulated amortization
In accordance with IFRS 16, the Group has chosen not to present the right of use separately from other assets and has added them to the fixed assets of the same nature as the underlying leased assets.
Therefore, the right of use assets and related accumulated amortization as of December 31, 2022 included in the table above affect:
•The line item “Building on the basis of quoted prices in active markets for identical assets or liabilities;non-freehold land" amounting to €10,665 and €3,839, respectively;
•Level 2 : Fair value measuredThe line item "Scientific equipment", amounting to €1,502 and €1,475 respectively.
Therefore, the right of use assets and related accumulated amortization as of December 31, 2023 included in the table above affect:
•The line item “Building on the basis of valuation methods relying on quoted prices for similar assets, liabilities or observable inputs in active markets;non-freehold land" amounting to €11,067 and €4,940, respectively;
•Level 3 : Fair value measured on the basisThe line item "Scientific equipment", amounting to €741 and €741 respectively.
Right of valuation methods relying entirely oruse additions
There were no Right of use asset additions in part on unobservable inputs such as quoted prices in inactive markets or the valuation based on multiples for non-listed securities.2023.
4.10.16.InventoriesTRADE AND OTHER RECEIVABLES
The Company recognizes inventories of laboratory consumables in connection with its former co-research agreements.
These inventories are measured at the lower of cost and net realizable value. Cost is determined using the weighted average cost method.
4.11.Trade and other receivablesAccounting policies
Trade and other receivables are recognized at fair value, which is the nominal value of invoices unless payment terms require a material adjustment for the time value discounting effect at market interest rates. Trade receivables are subsequently measured at amortized cost. Impairment losses on trade accounts receivable are estimated using the expected loss method, in order to take account of the risk of payment default throughout the lifetime of the receivables .receivables.
Receivables are classified as current assets, except for those with a maturity exceeding 12 months after the reporting date, according to IFRS 9 standards ("expected credit loss").
Trade and other receivables consisted of the following:
| | | | | | | | | | | | | | | | | |
| Trade and other receivables - Total | | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| Trade receivables, net | | | | 3,188 | | | 18,526 | |
| Research tax credit | | | | 11,299 | | | 12,200 | |
| Social security costs receivables | | | | 1 | | | — | |
| VAT receivables | | | | 1,288 | | | 1,476 | |
| Grants receivables | | | | 4 | | | 7 | |
| Other receivables | | | | 126 | | | 498 | |
| TOTAL | | | | 15,906 | | | 32,707 | |
| Of which : Current | | | | 15,906 | | | 32,707 | |
| Of which : Non-current | | | | — | | | — | |
Trade receivables, net
Trade receivables amounted to €18,526 as of December 31, 2023. The balance mainly corresponds to revenue related to the our milestone receivable with Ipsen. Per IFRS 7.35(h), we have concluded that the expected credit loss on this amount is €0.
Trade receivables amounted to €3,188 as of December 31, 2022. The balance mainly corresponds to revenue related to the inventory purchase agreement with Ipsen.
Research tax credit
The research tax credit receivable for the year 2022 amounted to €11,299.
The research tax credit receivable for the year 2023 amounts to €12,200.
VAT receivables
The VAT receivable amounted to €1,476 at December 31, 2023.
The VAT receivable amounted to €1,288 at December 31, 2022.
Other receivables
The line item “other receivables” primarily consists of credit notes from suppliers for €498 and €126, respectively, as of December 31, 2023 and December 31, 2022.
4.12.17.Other financial assetsINVENTORIES
The Company recognizes inventories of laboratory consumables.
These inventories are measured at the lower of cost and net realizable value. Cost is determined using the weighted average cost method.
18.OTHER FINANCIAL ASSETS
Accounting policies
A financial asset is initially recognized as measured at amortized cost, at fair value through other comprehensive income - debt instrument, at fair value through other comprehensive income - equity instrument, or at fair value through profit or loss.
Financial assets will not be reclassified after initial recognition, unless we change our economic model of financial asset management. If so, all affected financial assets would be reclassified as of the first day of the first reporting period following the change in economic model. No such reclasses have taken place in any period presented herein.
A financial asset is measured at amortized cost if both of the following conditions are met, and if it is not measure at fair value through profit or loss:
•Its ownership is part of an economic model of which the objective is to hold assets in order to receive its contractual cash flows;
•Its contractual conditions provide for cash flows at defined dates, which correspond only to principal payments and interest on the remaining principal amount.
A debt instrument is measured at fair value through other comprehensive income if both of the following conditions are met, and if it is not measure at fair value through profit or loss:
•Its ownership is part of an economic model of which the goal is met through both the receipt of contractual cash flows and the sale of financial assets;
•Its contractual conditions provide for cash flows at defined dates, which correspond only to principal payments and interest on the remaining principal amount.
At the time of initial recognition of an equity instrument that is not held for trading, we may irrevocably choose to present future changes in fair value in other comprehensive income. This choice is made for each investment.
All financial assets that are not categorized as measured at amortized cost or at fair value through other comprehensive income as previously described are measured at fair value through profit or loss.
Financial detail
Other financial assets consisted of the following:
| | | | | | | | | | | | | | | | | |
| Financial assets - Total | | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| Non consolidated equity investments | | | | 3,133 | | | 2,348 | |
| Other investments | | | | 483 | | | 471 | |
| Financial investments | | | | 4,550 | | | — | |
| Loans | | | | 428 | | | 472 | |
| | | | | | | |
| Deposits and guarantees | | | | 335 | | | 303 | |
| Liquidity contract | | | | 534 | | | 531 | |
| TOTAL | | | | 9,464 | | | 4,125 | |
| Of which : Current | | | | 4,550 | | | — | |
| Of which : Non-current | | | | 4,914 | | | 4,125 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Financial assets - Variations | | | As of | | Increase | | Decrease | | As of |
| (in € thousands) | | | 31/12/2022 | | | | | | 31/12/2023 |
| Non consolidated equity investments | | | 3,133 | | | — | | | (785) | | | 2,348 | |
| Other investments | | | 483 | | | 0 | | (12) | | | 471 |
| Financial investments | | | 4,550 | | | — | | | (4,550) | | | — | |
| Loans | | | 428 | | | 44 | | | — | | | 472 | |
| | | | | | | | | | |
| Deposits and guarantees | | | 335 | | | 35 | | | (68) | | | 303 | |
| Liquidity contract | | | 534 | | 0 | | (3) | | 531 |
| TOTAL | | | 9,464 | | | 80 | | | (5,419) | | | 4,125 | |
4.13.Cash and cash equivalents
Cash and cash equivalents comprise cash on hand, bank accounts and term deposits, together with short-term deposits and highly liquid investments. They are readily convertible to a knownThe total amount of cash and thus presentfinancial assets of the Company was €9,464 at December 31, 2022, as is €4,125 at December 31, 2023. This change is mainly due to the short term financial asset with a negligible riskterm of 180 days as well as a changerecorded impairment related to the Company's shares in value. They also include UndertakingsGenoscience Pharma.
Non-consolidated equity investments
As of December 31, 2023, the value of "Non-consolidated equity investments" totaled €2,348, which relates solely to our equity purchase in Genoscience Pharma.
The gross value of the investment (and the initial transaction amount from 2021) totals €3,133.
The net value of the investment (including an impairment of €785) totals 2,348.
Since the transaction occurred, no shares have been sold.
We did not complete the equity purchase in Genoscience Pharma for Collective Investmentstrading purposes. Therefore, pursuant to IFRS 9, we elected to classify the equity in Transferable Securities (UCITs) whose characteristics allow them to be classifiedGenoscience Pharma we acquired in December 2021 as cash and cash equivalents.
Initiallyequity instruments recognized at their purchase cost atfair value through other comprehensive income (OCI). At the transaction date, investments are subsequentlytime of initial recognition in 2021, this investment in equity instruments has been measured at fair value. Changesvalue, inclusive of acquisition costs related to the purchase. The amount recognized on the balance sheet at December 31, 2021 corresponds to the subscription price agreed upon between the parties as representative of the value of Genoscience Pharma a few days before closing of the period. For future closings, changes in fair value on these equity instruments are recognized as OCI. This OCI may not be reused as profit or loss, including in the case of a sale. If applicable, only dividends related to the investment in equity instruments will be recognized as profit provided that all conditions are met.
For 2023, and in accordance with IFRS 13, we updated our estimated of the fair value of our equity stake in Genoscience Pharma, which was based on a valuation methodology including a royalty based income approach using discounted cash flow techniques for the company's main scientific research programs. The aforementioned income method utilizes management’s estimates of future operating results, cash flows discounted using a weighted-average cost of capital that reflects market participant assumptions, and the expected success rate of each program. Based on our analysis performed as of December 31, 2023, an impairment loss of €785 was recognized in OCI.
The period over which management has projected its cash flows spans through 2039. The growth rate used to extrapolate cash flow projections is 1%. Furthermore, we have performed the following sensitivity analyses in order to determine the change in value of the asset by modifying certain key assumptions.
Values assigned to each key assumption
Discount rate: 12.4%
The amount by which the asset would decrease if the weighted average cost of capital increased by 1%: €159
Overall expected success rate: 12.9%
The amount by which the asset would decrease if the estimated overall success rate decreased by 1%: €212
Indicators of impairment considered by the Group as part of the implementation of the impairment test above are as follows:
•Failure of or unfavorable data from our clinical trials
•Competition from other clinical trial programs covering the same indications as our drug candidates
•Availability of necessary financing
Other investments
As of December 31, 2023, the value of "Other investments" totaled €471. The balance relates solely to our investment in CAPTECH SANTE.
On May 24, 2022, GENFIT undertook to subscribe for 50 units of the CAPTECH SANTE Professional Equity Fund (Fonds Professionnel de Capital Investissement – FPCI) in the amount of €500. On June 25, 2022, the management company made an initial call for funds from GENFIT in an amount equal to 35% of the subscription amount, i.e. €175, which GENFIT paid. The remaining subscription amount of €325 must be paid upon successive calls from the fund management company.
GENFIT’s investment in CAPTECH SANTE constitutes a debt instrument that does not meet the SPPI (solely payments of principal and interest) criterion test. It is therefore classified as a financial asset recognized at fair value through profit or loss. This investment is also consistent with a regular way purchase of a financial asset. GENFIT has opted to use the trade date as date of initial recognition. An amount of €500 was therefore recognized in the Group’s balance sheet on May 24, 2022.
As of December 31, 2023, a loss of €12 was recognized based on the net financial income (expenses).asset value of the units as of said date.
Financial investments
As of December 31, 2023, and 2022, the value of "Financial investments" totaled €0 and €4,550 respectively. This change related solely to a short term investment whose term was 180 days, liquidated in 2023.
Liquidity contract
Consistent with customary practice in the French securities market, we entered into a liquidity agreement (contrat de liquidité) with Crédit Industriel et Commercial S.A. ("CIC") in August 2013. The liquidity agreement was entered into in accordance with applicable laws and regulations in France. The liquidity agreement authorizes CIC to carry out market purchases and sales of our shares on Euronext Paris.
As of December 31, 2023, the liquidity account had a cash balance of €531, and as of December 31, 2022 a cash balance of €534.
CIC holds the following number of GENFIT shares on behalf of the Company, recorded as a deduction in equity:
| | | | | | | | | | | | | | | | |
| Financial assets - Current | | | | As of |
| | | | 2022/12/31 | | 2023/12/31 |
| Number of shares (recorded as a deduction from equity) | | | | 138,691 | | | 147,812 | |
4.14.19.EquityOTHER ASSETS
Share capital comprises ordinary sharesOther current assets of €2,615 at December 31, 2023 and ordinary shares with double voting rights classified in equity. Costs directly attributable€1,998 at December 31, 2022, consisting of prepaid expenses related to the issue of ordinary shares or share options are recognized as a reduction in the share premium.
The liquidity agreement consists of a share buyback program contracted to an investment service provider. Purchases and sales of the Company's shares carried out under the contract are recognized directly in shareholders’ equity under treasury shares. See note 10 “Other financial income”.current operating expenses.
4.15.20.Loans and borrowingsLOANS AND BORROWINGS
Accounting policies
Financial liabilities are initially recognized at fair value, net of directly attributable transaction costs, and are subsequently measured at amortized cost using the effective interest rate method.
The Group derecognizes financial liabilities when the contractual obligations are discharged, cancelled or expire.
The bonds convertible or exchangeable into new or existing shares (OCEANE—(OCEANEs—see Section 12.1, "Note 20.1 - "Breakdown of convertible loanloan"") are recognized as follows: in accordance with IAS 32, Financial Instruments—Presentation, , if a financial instrument has different components and the characteristics indicate that some should be classified as liabilities and others as equity, the issuer must recognize the different components separately. The liability component is measured, at the date of issuance, at its fair value on the basis of future contractual cash flows discounted at market rates (taking into consideration the issuer's credit risk) of a debt having similar characteristics but without the conversion option.
The value of the conversion option is measured by the difference between the bond's issue price and the fair value of the liability component. After deduction of the pro rata portion of expenses related to the transaction, this amount is recognized in the line item "Share premium" under shareholders' equity and is subject to a calculation of deferred tax according to IAS 12.28.
The liability component (after deduction of the pro rata portion of the transaction expenses attributed to the liability and the conversion option) is measured at amortized cost. A non-monetary interest expense, recorded in net loss is calculated using an effective interest rate to progressively bring the debt component up to the amount which will be repaid (or converted) at maturity. A deferred tax liability is calculated on the basis of this amount. The shareholders' equity component is not remeasured. See
Introduction
On October 16, 2017, the Company issued 6,081,081 OCEANEs at par with a nominal unit value of €29.60 per bond for an aggregate nominal amount of €180 million and the debt was renegotiated in 2020 and 2021.
As of December 31, 2022 and 2023, key terms and conditions and balances are as follows:
Updated balances
| | | | | | | | | |
| As of 31/12/2022 : | | |
| Number of bonds | | 1,923,662 |
| Nominal amount of the loan | | 56,940,395.20€ |
| Nominal unit value of the bonds | | 29.60€ |
| Effective interest rate | | 8.8% |
| As of 31/12/2023 : | | |
| Number of bonds | | 1,923,662 |
| Nominal amount of the loan | | 56,940,395.20€ |
| Nominal unit value of the bonds | | 29.60€ |
| Effective interest rate | | 8.8% |
Nominal annual interest rate
The nominal annual interest rate is 3.5%, payable semi-annually in arrears.
Repayment Terms
Final reimbursement is scheduled for October 16, 2025.
Redemption prior to maturity at the option of the convertible bondCompany is possible if the arithmetic volume-weighted average price of GENFIT's listed share price and the then prevailing conversion ratio over a 20 day trading period exceeds 1.5 times the nominal value of the OCEANEs.
Conversion ratio and terms
The conversion ratio is 5.5 ordinary shares per bond.
There are no specific terms that need to be met for a holder of OCEANEs to convert their debt (OCEANEs)”into GENFIT shares.
There were no conversions in 2022 or 2023.
Conversion / exchange premium
The conversion / exchange premium is 30% relative to GENFIT's reference share price (22.77€).
Maximum Dilution
The potential issuance of new shares upon conversion requests of the outstanding OCEANEs would represent 21.2% of the share capital of the Company at December 31, 2022 (representing a 17.5% dilution if all OCEANEs were converted).
The potential issuance of new shares upon conversion requests of the outstanding OCEANEs would represent 21.2% of the share capital of the Company at December 31, 2023 (representing a 17.5% dilution if all OCEANEs were converted).
Deferred taxes
Current and non current balances
| | | | | | | | | | | | | | | | | |
| Convertible loans - Total | | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| Convertible loans | | | | 50,276 | | | 52,622 | |
| TOTAL | | | | 50,276 | | 52,622 |
| | | | | | | | | | | | | | | | | |
| Convertible loans - Current | | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| Convertible loans | | | | 415 | | | 415 | |
| TOTAL | | | | 415 | | 415 |
| | | | | | | | | | | | | | | | | |
| Convertible loans - Non current | | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| Convertible loans | | | | 49,861 | | | 52,206 | |
| TOTAL | | | | 49,861 | | 52,206 |
20.2.Breakdown of other loans and borrowings
Other loans and borrowings consisted of the following:
| | | | | | | | | | | | | | | | | |
| Other loans and borrowings - Total | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| Refundable and conditional advances | | | | 3,229 | | | — | |
| Bank loans | | | | 15,196 | | | 11,578 | |
| | | | | | | |
| | | | | | | |
| Obligations under leases | | | | 6,559 | | | 5,884 | |
| Accrued interests | | | | 14 | | | 7 | |
| | | | | | | |
| Bank overdrafts | | | | — | | | 89 | |
| TOTAL | | | | 24,999 | | 17,557 |
| | | | | | | | | | | | | | | | | |
| Other loans and borrowings - Current | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| Refundable and conditional advances | | | | — | | | — | |
| Bank loans | | | | 3,619 | | | 6,339 | |
| | | | | | | |
| | | | | | | |
| Obligations under leases | | | | 1,032 | | | 1,076 | |
| Accrued interests | | | | 14 | | | 7 | |
| | | | | | | |
| Bank overdrafts | | | | — | | | 89 | |
| TOTAL | | | | 4,665 | | | 7,510 | |
| | | | | | | |
| | | | | | | | | | | | | | | | | |
| Other loans and borrowings - Non current | | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| Refundable and conditional advances | | | | 3,229 | | | — | |
| Bank loans | | | | 11,578 | | | 5,239 | |
| | | | | | | |
| | | | | | | |
| Obligations under leases | | | | 5,527 | | | 4,808 | |
| Accrued interests | | | | — | | | — | |
| | | | | | | |
| Bank overdrafts | | | | — | | | — | |
| TOTAL | | | | 20,334 | | | 10,047 | |
20.2.1.Refundable and conditional advances
The following table summarizes advances outstanding at December 31, 2023 and 2022.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Refundable and conditional advances—general overview | | Grant date | | Total amount allocated | | Receipts | | Cancellations | | Effects of discounting | | Net book value As of 2023/12/31 |
| (in € thousands) | | | | | | | | | | | | |
| BPI FRANCE - IT-DIAB | | 12/23/2008 | | 3,229 | | | 3,229 | | | (3,229) | | | — | | | — | |
| Development of a global strategy for the prevention and management of type 2 diabetes | | | | | | | | | | | | |
| TOTAL | | | | 3,229 | | | 3,229 | | | (3,229) | | | — | | | — | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Refundable and conditional advances—general overview | | Grant date | | Total amount allocated | | Receipts | | Repayments | | Effects of discounting | | Net book value As of 2022/12/31 |
| (in € thousands) | | | | | | | | | | | | |
| BPI FRANCE - IT-DIAB | | 12/23/2008 | | 3,229 | | | 3,229 | | | — | | | — | | | 3,229 | |
| Development of a global strategy for the prevention and management of type 2 diabetes | | | | | | | | | | | | |
| TOTAL | | | | 3,229 | | | 3,229 | | | — | | | — | | | 3,229 | |
BPI France - IT-DIAB
On December 23, 2008, the Group received an advance from Bpifrance (the BPI France IT-DIAB) as part of a framework innovation aid agreement involving several scientific partners and for which the Group was the lead partner. The contribution expected at each stage by each of the partners in respect of work carried out and results achieved is defined in the framework agreement. With respect to the Group, the aid consisted of a €3,229 conditional advance and a €3,947 non-repayable government grant.
The conditional advance is not refundable except in the event of success. The program ended on December 31, 2014. In the event of success, defined as the commercial spin-offs of the IT-Diab program which involves products for the accounting appliedtreatment or diagnosis of type 2 diabetes, in that case, the financial returns generated will be used initially to repay the €3,229 conditional advance and the agreement stipulates that the conditional advance will be regarded as repaid in full when the total payments made in this regards by the recipient, discounted at the rate of 5.19%, equal the total amount, discounted at the same rate, of the aid paid. Any further amounts will be classified as additional payments, up to a maximum amount of €14,800.
As provided in the project assistance contract, we sent a letter to Bpifrance in December 2019 in order to notify it of our Labcorp and Terns Pharmaceuticals contracts while indicating that elafibranor was now aimed at treating hepatic diseases and no longer type 2 diabetes as provided for in the aid agreement. We proposed to Bpifrance to establish a statement of abandonment of the IT-DIAB project on which the above advance is based. Following this letter, the parties met in March 2020 for the presentation of our arguments, and in June 2020 following the publication of the results of the RESOLVE-IT® study, and a new letter was sent in November 2020.
On October 20, 2023, BPI France agreed to formally recognize the failure of the project and therefore write off their outstanding receivable as previously mentioned. As of December 31, 2023, GENFIT had no remaining obligation associated with this, and thus the liability was reversed with the related income recorded in "Other income" on the consolidated statement of operations.
20.2.2.Bank loans
In the context of the COVID-19 pandemic, in 2021 the Company secured several State-Guaranteed Loans (or "Prêt Garanti par l'Etat (PGE) Bancaire") and Subsidized Loans (or "BPI Prêt Taux Bonifié").
Bank loans consisted of the following as of December 31, 2022 and 2023 with the renegotiation.following interest rates and repayment terms:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Bank loans | | Loan | | Facility | | Interest | | Available As of 2023/12/31 | | Installments | | | | Outstanding As of 2022/12/31 | | Outstanding As of 2023/12/31 |
| (in € thousands) | | date | | size | | rate | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| BNP 4 | | April 2017 | | 800 | | | 0.87 | % | | — | | | 60 monthly | | | | 54 | | | — | |
| AUTRES | | - | | — | | | — | % | | — | | | 0 | | | | 17 | | | 13 | |
| CDN PGE | | June 2021 | | 900 | | | 1.36 | % | | — | | | 8 quarterly | | | | 900 | | | 675 | |
| CIC PGE | | June 2021 | | 2,200 | | | 0.75 | % | | — | | | 8 quarterly | | | | 2,200 | | | 1,650 | |
| BNP PGE | | June 2021 | | 4,900 | | | 0.45 | % | | — | | | 8 quarterly | | | | 4,900 | | | 3,675 | |
| NATIXIS PGE | | June 2021 | | 3,000 | | | 0.40 | % | | — | | | 8 quarterly | | | | 3,000 | | | 2,250 | |
| BPI PGE | | July 2021 | | 2,000 | | | 2.25 | % | | — | | | 16 quarterly | | | | 1,900 | | | 1,500 | |
| BPI PRÊT TAUX BONIFIE | | November 2021 | | 2,250 | | | 2.25 | % | | — | | | 20 quarterly | | | | 2,250 | | | 1,820 | |
| TOTAL | | | | | | | | | | | | | | 15,221 | | | 11,583 | |
The effective interest rates are follows for the PGE loans:
•CDN PGE (loan of €900): 2.08% per annum
•CIC PGE (loan of €2,200): 1.46% per annum
•BNP PGE (loan of €4,900): 1.16% per annum
•NATIXIS PGE (loan of €3,000): 1.11% per annum
•BPI PGE (loan of €2,000): 1.65% per annum
20.2.3.Maturities of financial liabilities
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Maturity of financial liabilities | | As of | | Less than | | Less than | | Less than | | Less than | | Less than | | More than |
| (in € thousands) | | 2023/12/31 | | 1 year | | 2 years | | 3 years | | 4 years | | 5 years | | 5 years |
| | | | | | | | | | | | | | | |
| TOTAL - Refundable and conditional advances | | — | | | — | | | — | | | — | | | — | | | — | | | — | |
| Convertible loans | | 57,356 | | | 415 | | | 56,940 | | | — | | | — | | | — | | | — | |
| Bank loans | | 11,578 | | | 6,339 | | | 3,601 | | | 867 | | | 771 | | | — | | | — | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| Leases | | 5,884 | | | 1,076 | | | 1,088 | | | 1,101 | | | 1,114 | | | 1,127 | | | 378 | |
| Accrued interests | | 7 | | | 7 | | | — | | | — | | | — | | | — | | | — | |
| | | | | | | | | | | | | | | |
| Bank overdrafts | | 89 | | | 89 | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Other loans and borrowings | | 74,913 | | | 7,926 | | | 61,630 | | | 1,968 | | | 1,884 | | | 1,127 | | | 378 | |
| TOTAL | | 74,913 | | | 7,926 | | | 61,630 | | | 1,968 | | | 1,884 | | | 1,127 | | | 378 | |
The values in the table above are contractual, undiscounted values.
21.FAIR VALUE OF FINANCIAL INSTRUMENTS
Accounting policies
IFRS 9 “Financial Instruments” takes into account the following three aspects of booking financial instruments :
•Classification and measurement;
•Impairment and;
•Hedge accounting.
Loans and borrowings are initially measured at fair value and subsequently recorded at amortized cost.
Pursuant to IFRS 7 – Financial Instruments: Disclosures, the financial instruments are presented into three categories according to a hierarchical method used to establish their fair value.
If financial instruments are measured at fair value, they are measured according to a hierarchy comprising three levels of valuation inputs:
•Level 1: Fair value measured on the basis of quoted prices in active markets for identical assets or liabilities;
•Level 2: Fair value measured on the basis of valuation methods relying on quoted prices for similar assets, liabilities or observable inputs in active markets;
•Level 3: Fair value measured on the basis of valuation methods relying entirely or in part on unobservable inputs such as quoted prices in inactive markets or the valuation based on multiples for non-listed securities.
Financial detail
The following tables provide the financial assets and liabilities carrying values by category and fair values as of December 31, 2023 and December 31, 2022:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | As of 31/12/2022 |
| | | Carrying value | | Fair value |
| | | As per | | Assets at | | Assets at | | Assets at | | Debt at | | Level 1 | | Level 2 | Level 3 |
| | | statement of | | fair value | | fair value | | amortized | | amortized | | | | | |
| | | financial | | through | | through OCI | | cost | | cost | | | | | |
| (in € thousands) | | position | | profit & loss | | | | | | | | | | | |
| Assets | | | | | | | | | | | | | | | |
| Equity investments | | 3,133 | | | | | 3,133 | | | | | | | | | | 3,133 | |
| Other investments | | 483 | | | 483 | | | | | | | | | | | | 483 | |
| Financial investments | | 4,550 | | | 4,550 | | | | | | | | | 4,550 | | | | |
| Loans | | 428 | | | | | | | 428 | | | | | | | 428 | | |
| | | | | | | | | | | | | | | | |
| Deposits and guarantees | | 335 | | | | | | | 335 | | | | | | | 335 | | |
| Liquidity contracts | | 534 | | | 534 | | | | | | | | | 534 | | | | |
| Trade receivables | | 3,188 | | | | | | | 3,188 | | | | | | | 3,188 | | |
| Cash and cash equivalents | | 136,001 | | | 136,001 | | | | | | | | | 136,001 | | | | |
| TOTAL - Assets | | 148,653 | | | 141,568 | | | 3,133 | | | 3,951 | | | — | | | 141,085 | | | 3,951 | | 3,617 | |
| Liabilities | | | | | | | | | | | | | | | |
| Conditional advances | | 3,229 | | | | | | | | | 3,229 | | | | | | 3,229 | |
| Convertible loans | | 50,276 | | | | | | | | | 50,276 | | | | | 52,708 | | |
| Bank loans | | 15,196 | | | | | | | | | 15,196 | | | | | 15,196 | | |
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | |
| Obligations under finance leases | | 6,559 | | | | | | | | | 6,559 | | | | | 6,559 | | |
| Accrued interests | | 14 | | | | | | | | | 14 | | | | | 14 | | |
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | |
| Trade payables | | 8,613 | | | | | | | | | 8,613 | | | | | 8,613 | | |
| Other payables | | 1,325 | | | | | | | | | 1,325 | | | | | 1,325 | | |
| TOTAL - Liabilities | | 85,214 | | | — | | | — | | | — | | | 85,214 | | | — | | | 84,416 | | 3,229 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | As of 31/12/2023 |
| | | Carrying value | | Fair value |
| | | As per | | Assets at | | Assets at | | Assets at | | Debt at | | Level 1 | | Level 2 | Level 3 |
| | | statement of | | fair value | | fair value | | amortized | | amortized | | | | | |
| | | financial | | through | | through OCI | | cost | | cost | | | | | |
| (in € thousands) | | position | | profit & loss | | | | | | | | | | | |
| Assets | | | | | | | | | | | | | | | |
| Equity investments | | 2,348 | | | | | 2,348 | | | | | | | | | | 2,348 | |
| Other investments | | 471 | | | 471 | | | | | | | | | | | | 471 | |
| | | | | | | | | | | | | | | | |
| Loans | | 472 | | | | | | | 472 | | | | | | | 472 | | |
| | | | | | | | | | | | | | | | |
| Deposits and guarantees | | 303 | | | | | | | 303 | | | | | | | 303 | | |
| Liquidity contracts | | 531 | | | 531 | | | | | | | | | 531 | | | | |
| Trade receivables | | 18,526 | | | | | | | 18,526 | | | | | | | 18,526 | | |
| Cash and cash equivalents | | 77,789 | | | 77,789 | | | | | | | | | 77,789 | | | | |
| TOTAL - Assets | | 100,439 | | | 78,790 | | | 2,348 | | | 19,300 | | | — | | | 78,319 | | | 19,300 | | 2,819 | |
| Liabilities | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | |
| Convertible loans | | 52,622 | | | | | | | | | 52,622 | | | | | 51,939 | | |
| Bank loans | | 11,578 | | | | | | | | | 11,578 | | | | | 11,578 | | |
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | |
| Obligations under finance leases | | 5,884 | | | | | | | | | 5,884 | | | | | 5,884 | | |
| Accrued interests | | 7 | | | | | | | | | 7 | | | | | 7 | | |
| | | | | | | | | | | | | | | | |
| Bank overdrafts | | 89 | | | | | | | | | 89 | | | | | 89 | | |
| Trade payables | | 10,448 | | | | | | | | | 10,448 | | | | | 10,448 | | |
| Other payables | | 914 | | | | | | | | | 914 | | | | | 914 | | |
| TOTAL - Liabilities | | 81,541 | | | — | | | — | | | — | | | 81,541 | | | — | | | 80,858 | | — | |
It should be noted that the section above “Equity investments” concerns the nonconsolidated equity investments in Genoscience. The gross value of the asset is €3,133 and was partially impaired in 2023. See Note 18 - "Other financial assets".
4.16.22.Trade and other payablesTRADE AND OTHER PAYABLES
Trade and other payables are initially recognized at the fair value of the amount due. This value is usually the nominal value, due to the relatively short period of time between the recognition of the instrument and its repayment.
Financial detail
Trade and other payables consisted of the following:
| | | | | | | | | | | | | | | | | |
| Trade and other payables - Total | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| Trade payables | | | | 8,613 | | | 10,448 | |
| Social security costs payables | | | | 4,838 | | | 4,188 | |
| VAT payables | | | | 200 | | | 3,139 | |
| Taxes payables | | | | 316 | | | 110 | |
| Other payables | | | | 1,325 | | | 914 | |
| TOTAL | | | | 15,293 | | | 18,799 | |
| | | | | | | | | | | | | | | | | |
| Trade and other payables - Current | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| Trade payables | | | | 8,613 | | | 10,448 | |
| Social security costs payables | | | | 4,838 | | | 4,188 | |
| VAT payables | | | | 200 | | | 3,139 | |
| Taxes payables | | | | 316 | | | 110 | |
| Other payables | | | | 877 | | | 914 | |
| TOTAL | | | | 14,845 | | | 18,799 | |
| | | | | | | | | | | | | | | | | |
| | | | | | | |
| Trade and other payables - Non current | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| Other payables | | | | 448 | | | — | |
| TOTAL | | | | 448 | | | — | |
| | | | | | | |
| | | | | | | |
At December 31, 2023, trade payables amounted to €10,448 (€8,613 at December 31, 2022). This change is primarily due to an increase in accrued expenses relating to yet unbilled amounts from the clinical trial sites via the Clinical Research Organizations (CROs) in charge of the Company's clinical trials. (€4,765 and €3,924 at December 31, 2023 and 2022 respectively). The timeframe in which those invoices will be received by the Company is unknown and may be spread out over a long period after the services have been performed.
23.DEFERRED INCOME AND REVENUE
Out of the €120 million upfront payment received from Ipsen in application of the licensing agreement signed in December 2021, an amount of €40 million was recognized as Deferred income in 2021. The Deferred income is recognized as revenue as GENFIT carries out its part of the double-blind ELATIVE® study, based on the progress made relative to the originally developed budget. As of December 31, 2022, the Company considers that this initial budget is still appropriate based on progress performed.
In 2022, €15.9 million of said balance was recognized as revenue. As of December 31, 2022, €24.1 million of Deferred income remains, of which €14.4 million relates to Current deferred income and of which €9.7 million relates to Non-current deferred income, which was determined based on the original budget.
In 2023, €8.7 million of said balance was recognized as revenue. As of December 31, 2023, €15.3 million of Deferred income remains, of which €11.6 million relates to Current deferred income and of which €3.8 million relates to Non-current deferred income, which was determined based on the original budget.
4.17.24.Provisions
Accounting policies
In accordance with IAS 37, Provisions Contingent Liabilities and Contingent Assets, provisions are recognized when the Group has a present obligation (legal, regulatory, contractual or constructive) as a result of a past event, for which it is probable that an outflow of resources will be required to settle the obligation, and of which the amount can be estimated reliably.
The amount recognized as a provision is the best estimate at the reporting date of the expenditure required to settle the present obligation.
Provisions are discounted when the time value effect is material.
A provision for reorganization is recognized when the Group has approved a formal and detailed plan for its reorganization and has either started to implement it or publicly disclosed it.
A provision for onerous contract is estimated at the actual value of the lowest expected cost of either the cancellation or the execution of the contract, the latter being established on the basis of the additional costs required to fulfill the obligations stipulated by the contract. Before a provision is established, the Group recognizes any impairment loss that occurred on the assets dedicated to this contract (see Note 2.1 “Termination of RESOLVE ITcontract. Financial detail
At December 31, 2023 and the development program of elafibranor in NASH”).
It is of note that, pursuant to IAS 37, our obligations under the terms of the agreement we entered into with Genoscience Pharma constitute contingent liabilities not recognized in the Company's consolidated financial statements at December 31, 2021 (see2022, this line item amounted to €40 and €61, respectively.
| | | | | | | | | | | | | | | | | | | | | |
| Change in provisions | | As of | Increase | Decrease | Decrease | As of |
| (in € thousands) | | 2022/12/31 | | (used) | (unused) | 2023/12/31 |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| Provision for charges | | 61 | 0 | (21) | 0 | 40 |
| TOTAL | | 61 | 0 | (21) | 0 | 40 |
Future milestone and revenue based royalty payments may be recorded pursuant to Contingent liability under IAS 37 or intangible asset under IAS 38. Under IAS 38, we record a provision when we have a present obligation, whether legal or constructive, as a result of a past event; it is probable that an outflow of resources embodying economic benefits will be required to settle the obligation; and a reliable estimate can be made of the amount of the outflow of resources. Under IAS 38, we record intangible asset when it is probable that the expected future economic benefits that are attributes to the assets will flow to us and the cost of asset can be measured reliably.
4.18.25.Employee benefitsEMPLOYEE BENEFITS
Accounting policies
The Group's pension schemes and other post-employment benefits consist of defined benefit plans and defined contribution plans.
4.18.1.25.1.Defined benefit plans
Defined benefit plans relate to French retirement benefit plans under which the Group is committed to guaranteeing a specific amount or level of contractually defined benefits. The obligation arising from these plans is measured on an actuarial basis using the projected unit credit method. The method consists of measuring the obligation based on a projected end-of-career salary and vested rights at the measurement date, according to the provisions of the collective bargaining agreement, corporate agreements and applicable law.
Actuarial assumptions are used to determine the benefit obligations. The amount of future payments is determined on the basis of demographic and financial assumptions such as mortality, staff turnover, pay increases and age at retirement, and then discounted to their present value. The discount rate used is the yield at the reporting date on AA credit-rated bonds with maturity dates that approximate the expected payments for the Group's obligations.
Re-measurements of the net defined benefit liability which comprise actuarial gains and losses are recognized in the statements of other comprehensive loss.
The Group determines the net interest expense on the net defined benefit liability for the period by applying the discount rate used to measure the defined benefit obligation at the beginning of the annual period to the then-net defined benefit liability, taking into account any changes in the net defined benefit liability during the period as a result of contributions and benefit payments.
4.18.2.25.2.Defined contribution plans
Under defined contribution plans, the management of plans is performed by an external organization, to which the Group pays regular contributions. Payments made by the Group in respect of these plans are recognized as an expense for the period in the statements of operations.
4.18.3.25.3.Short-term employee benefits
A liability is recognized for the amount expected to be paid under short-term cash bonus or profit-sharing plans if the Group has a present legal or constructive obligation to pay the amount as a result of past service provided by the employee, and the obligation can be estimated reliably.
4.19.Other income
4.19.1.Government grants
The Group received until 2016 various forms of government grants. This government aid is provided for and managed by French state-owned entities, and specifically "BPI France" (" Banque Publique d'Investissement "), formerly named "OSEO Innovation".
Subsidies received are non-refundable. Conditional advances received are interest-free or are subject to low interest rates depending on contractual provisions.
Conditional advances related to research programs
Conditional advances that are interest-free or subject to low interest rates are intended to finance research program’s needs.
In accordance with IAS 20, Accounting for Government Grants and Disclosure of Government Assistance, the advantage resulting from interest-free or low interest rates as compared to a market interest rate is considered and accounted for as a government grant. A financial liability is recognized for proceeds received from the advance less the grant, and interest expense is subsequently recorded under the effective interest rate method using a market interest rate.
The grant portion of conditional advances is treated as a grant related to income.
For advances granted by BPI France, repayment is required in the event of commercial success. In addition, if the Group decides to stop the research program, the conditional advance may be required to be repaid. If a program is unsuccessful, a pre-determined amount may be repayable. The remaining amount, if any, is then considered as a grant and written off in the line item "Other income" in the statements of operations.
4.19.2.Research tax credit
The Research Tax Credit (" Crédit d'Impôt Recherche ", or "CIR") is granted to entities by the French tax authorities in order to encourage them to conduct technical and scientific research. Entities that demonstrate that their research expenditures meet the required CIR criteria receive a tax credit that may be used for the payment of their income tax due for the fiscal year in which the expenditures were incurred, as well as in the next three years. If taxes due are not sufficient to cover the full amount of tax credit at the end of the three-year period, the difference is paid in cash to the entity by the tax authorities. If a company meets certain criteria in terms of sales, headcount or assets to be considered a small/mid-size company, immediate payment of the Research Tax Credit can be requested. The Group meets such criteria.
The Group applies for CIR for research expenditures incurred in each fiscal year and recognizes the amount claimed in the line item "Other income" in the statements of operations in the same fiscal year. In the notes to the financial statements, the amount claimed is recognized under the heading "Research tax credit" (see Note 9, "Trade and other receivables" and Note 19 "Operating Income").
4.20.Research and development expenses
In accordance with IAS 38, Intangible Assets, development expenses are recognized as intangible assets only if all the following criteria are met:
•Technical feasibility necessary for the completion of the development project;
•Intention on the Group's part to complete the project and to utilize it;
•Capacity to utilize the intangible asset;
•Proof of the probability of future economic benefits associated with the asset;
•Availability of the technical, financial, and other resources for completing the project; and
•Reliable evaluation of the expenses attributed to the intangible asset during its development.
As of the date of these financial statements none of these criteria have been met.
4.21.Classification of operating expenses
Research and development expenses include:
•employee-related costs;
•costs related to external employees seconded to the Company (such as clinical development, biometrics and IT…);
•lab supplies and facility costs;
•fees paid to scientific advisers and contracted research and development activities conducted by third parties;
•intellectual property fees corresponding to the filing of the Group's patents and,
•provision and reversals of provisions in relation to the Research Tax Credit dispute.
Contracted research and development activities conducted by third parties include services subcontracted to research partners for technical and/or regulatory reasons. In particular, this includes the production of active ingredients and therapeutic units, all or a part of clinical trials and preclinical trials that are necessary to the development of GENFIT's drug candidates and biomarker candidates.
General and administrative expenses include:
•employee-related costs for executive, business development, intellectual property, finance, legal and human resources and communications functions;
•facility-related costs;
•marketing, legal, audit and accounting fees;
•press relations and communications firm fees;
•the cost of external employees seconded to the Company (such as security, reception, and accounting..);
•other service costs (recruitment, etc.);
•intellectual property fees corresponding to the maintenance of the Group's patents.
Marketing and market access expenses include:
•employee-related costs for marketing and business development functions;
•marketing, and market access firm fees;
Extraordinary reorganization and restructuring expenses include:
•the accruals and provisions recognized within the scope of the reduction in force plan;
•the extraordinary amortization, loss of value and impairment of fixed assets recognized within the scope of the reorganization of GENFIT;
•the impairment of the right of use of the leased equipment and premises;
•the portion of the OCEANE renegotiation expenses;
•the provision recognized for some of the costs of the closing process for the RESOLVE-IT study, which, after detailed analysis, do not have any future economic advantage for the PBC program.
•
4.22.Share-based compensation
The fair value of equity-settled share-based compensation granted to employees, officers, board members and consultants as determined on the grant date is recognized as a compensation expense with a corresponding increase in equity, over the vesting period. The amount recognized as an expense is adjusted to reflect the actual number of awards for which the related service and non-market performance conditions are expected to be met.
The fair values of equity-settled share-based compensation granted to employees are measured using the Black-Scholes model with respect to the share warrants (BSA) and redeemable share warrants (BSAAR) and using the Monte Carlo model for the stock options (SO) and free shares (AGA). Measurement inputs include share price on the measurement date, the exercise price of the instrument, expected volatility, expected maturity of the instruments, expected dividends, and the risk-free interest rate (based on government bonds). With respect to the redeemable share warrants, service and non-market performance conditions attached to the transactions are not taken into consideration in determining fair value but are taken into consideration related to recognition of expense. Regarding the stock options and free shares, market conditions are taken into account in the determination of the fair value of the plans award. For share-based compensation awards with non-vesting conditions, the grant date fair value of the share-based compensation is measured to reflect such conditions and there is no adjustment for differences between expected and actual outcomes.
GENFIT may also grant equity-settled share-based compensation in exchange for services to consultants who are not considered employees. In such cases, the value of the services is measured when they are rendered by the consultants and the share-based compensation exchanged for the services is measured at an equal amount. If the value of the services cannot be measured reliably, then such value is measured with reference to the fair value of the equity instruments granted.
Share-based compensation granted to consultants consists of share warrants, some of which may be redeemed at GENFIT's discretion.
Share-based compensation granted to employees consists of redeemable share warrants, stock options and free shares.
4.23.Income tax
Income tax expense (or benefit) comprises current tax expense (or benefit) and deferred tax expense (or benefit), as applicable.
Deferred taxes are recognized for all the temporary differences arising from the difference between the tax basis and the accounting basis of assets and liabilities.
Deferred tax assets are recognized for unused tax losses, unused tax credits and temporary deductible differences to the extent that :
•it is probable that future taxable profit will be available against which they can be used; or
•if there are deferred tax liabilities for the same entity in the same tax jurisdiction on which they can be applied.
•
4.24.Earnings (loss) per share
Basic earnings (loss) per share are calculated by dividing profit or loss attributable to the Company's ordinary shareholders by the weighted average number of ordinary shares outstanding during the period.
Diluted earnings (loss) per share are calculated by adjusting profit attributable to ordinary shareholders and the average number of ordinary shares outstanding weighted for the effects of all potentially dilutive instruments (share warrants, redeemable share warrants, free shares, stock options and bonds convertible into new and/or existing shares).
4.25.Operating segments
The Board of Directors and Chief Executive Officer are the chief operating decision makers.
The Board of Directors and the Chief Executive Officer oversee the operations and manage the business as 1 segment with a single activity; namely, the research and development of innovative medicines and diagnostic solutions, the marketing of which depends on the success of the clinical development phase.
5.FINANCIAL RISKS MANAGEMENT
The Group may be exposed to the following risks arising from financial instruments : foreign exchange risk, interest rate risk, liquidity risk and credit risk.
5.1.Foreign exchange risk
The Group's overall exposure to the foreign exchange risk depends, in particular, on :
•the currencies in which it receives its revenues;
•the currencies chosen when agreements are entered into, such as licensing agreements, or co-marketing or co-development agreements;
•the location of clinical trials on drug or biomarker candidates;
•the ability, for its co-contracting parties to indirectly transfer foreign exchange risk to the Company;
•the Group’s foreign exchange risk policy; and
•the fluctuation of foreign currencies against the euro.
Given the significant portion of its operations denominated in US dollars, the Group decided to limit the conversions into euros of its US dollar denominated cash, issued notably from its March 2019 Nasdaq IPO in US dollars, and not to use any specific hedging arrangements, in order to cover expenses denominated in US dollars over the coming years.
The following table shows the sensitivity of the Group's cash and cash equivalent and expenses in U.S. dollars to a variation of 10% of the U.S. dollar against the euro in 2019, 2020 and 2021. It includes, in particular, the upfront payment in December 2021 by Ipsen for the licensing agreement entered into with GENFIT and its equity purchase in the Company :
| | | | | | | | | | | | | | | | | | | | | |
| Sensitivity of the Group's cash and cash equivalents to a variation of +/- 10% | | As of |
| of the US dollar against the euro | | | | | | |
| (in € thousands or in US dollar thousands, as applicable) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Cash and cash equivalents denominated in US dollars | | 153,438 | | 111,221 | | 81,713 |
| Equivalent in euros, on the basis of the exchange rate described below | | 136,582 | | 90,637 | | 72,146 |
| Equivalent in euros, in the event of an increase of 10% of US dollar vs euro | | 151,758 | | 100,708 | | 80,163 |
| Equivalent in euros, in the event of a decrease of 10% of US dollar vs euro | | 124,166 | | 82,398 | | 65,588 |
| | | | | | | | | | | | | | | | | | | | | |
| Sensitivity of the Group's expenses to a variation of +/- 10% | | Year ended |
| of the US dollar against the euro | | | | | | |
| (in € thousands or in US dollar thousands, as applicable) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Expenses denominated in US dollars | | 40,355 | | 47,277 | | 12,566 |
| Equivalent in euros, on the basis of the exchange rate described below | | 35,922 | | 38,528 | | 11,095 |
| Equivalent in euros, in the event of an increase of 10% of US dollar vs euro | | 39,914 | | 42,808 | | 12,328 |
| Equivalent in euros, in the event of a decrease of 10% of US dollar vs euro | | 32,657 | | 35,025 | | 10,086 |
2021/12/31 : Equivalent in euros, on the basis of 1 euro = 1.13261 dollars US
2020/12/31 : Equivalent in euros, on the basis of 1 euro =1.2271 dollars US
2019/12/31 : Equivalent in euros, on the basis of 1 euro = 1.1234 dollars US
| | | | | | | | | | | | | | | | | | | | | |
| Cash, cash equivalents and financial assets | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| At origin, denominated in EUR | | | | | | |
| Cash and cash equivalents | | 139,863 | | | 80,391 | | | 186,609 | |
| Current and non current financial assets | | 1,614 | | | 1,391 | | | 4,355 | |
| Total | | 141,477 | | | 81,782 | | | 190,964 | |
| At origin, denominated in USD | | | | | | |
| Cash and cash equivalents | | 136,884 | | | 90,637 | | | 72,147 | |
| Current and non current financial assets | | 113 | | | 67 | | | 76 | |
| Total | | 136,997 | | | 90,704 | | | 72,223 | |
| Total, in EUR | | | | | | |
| Cash and cash equivalents | | 276,748 | | | 171,029 | | | 258,756 | |
| Current and non current financial assets | | 1,727 | | | 1,458 | | | 4,431 | |
| Total | | 278,474 | | | 172,486 | | | 263,187 | |
5.2.Interest rate risk
As of December 31, 2021, the Group was only liable for governmental advances or conditional advances and bank loans with no interest or interest at a fixed rate, generally below market rate, with the exemption of the state-guaranteed loans (PGE), the variable rate of which could lead to an increase in interest in the future.
As of December 31, 2019, 2020 and 2021, the Group's financial liabilities totaled €183.6 million, €185.7 million and €74.2 million respectively (net of the equity component of the convertible loan and debt issue costs). Current borrowings are at a fixed rate. The Group's exposure to interest rate risk through its financial assets is also insignificant due to low market rates and since these assets are mainly euro-denominated Undertakings for the Collective Investment of Transferable Securities (UCITs), medium-term negotiable notes or term deposits with progressive rates denominated in euros or US dollars.
5.3.Liquidity risk
The Group's loans and borrowings mainly consist of bonds convertible or exchangeable into new or existing shares (OCEANE), initially repayable for an nominal amount of €57 million on October 16, 2025 (see Note 12.1 “Breakdown of convertible loan”), government advances for research projects and bank loans. For conditional advances, reimbursement of the principal is subject to the commercial success of the related research project (see Note 12.2.1 “Refundable and conditional advances”).The Company has conducted a specific review of its liquidity risk and considers that it is able to meet its future maturities. On December 31, 2019, 2020 and 2021, the Group had €278,474, €172,486 , and €263,187 respectively in cash and cash equivalents and other financial assets. The Company does not believe it is exposed to short-term liquidity risk. The Company believes that the Group's cash and cash equivalents and current financial instruments are sufficient to ensure its financing, in light of its current projects and obligations and of the renegotiation, effective in 2021, of its obligations pertaining to the OCEANE debt, including the extension of the maturity date, for at least the next twelve months.
If the Group's funds are insufficient to cover any additional financing needs, the Group would require additional financing. The conditions and arrangements for any such new financing would depend, among other factors, on economic and market conditions that are beyond the Group's control.
5.4.Credit risk
Credit risk is the risk of financial loss if a customer or counterparty to a financial asset defaults on their contractual commitments. The Group is exposed to credit risk due to trade receivables and other financial assets.
The Group's policy is to manage this risk by transacting with third parties with good credit standards.
6.CASH AND CASH EQUIVALENTS
The main components of cash equivalents were:
•UCITS and interest-bearing current accounts, available immediately;
•Term accounts, available within the contractual maturities or by the way of early exit with no penalty; and
•Negotiable medium-term notes, available with a quarterly maturity or by the way of early exit with no penalty.
These investments, summarized in the tables below, are short-term, highly liquid and subject to insignificant risk of changes in value.
| | | | | | | | | | | | | | | | | | | | | |
| Cash and cash equivalents | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Short-term deposits | | 263,147 | | | 166,034 | | | 69,045 | |
| Cash on hand and bank accounts | | 13,601 | | | 4,995 | | | 189,711 | |
| TOTAL | | 276,748 | | | 171,029 | | | 258,756 | |
| | | | | | | | | | | | | | | | | | | | | |
| Short-term deposits | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| UCITS | | 3,096 | | | 2,060 | | | — | |
| TERM ACCOUNTS | | 215,018 | | | 143,827 | | | 69,045 | |
| | | | | | | |
| INTEREST-BEARING CURRENT ACCOUNT | | 45,033 | | | 20,147 | | | — | |
| TOTAL | | 263,147 | | | 166,034 | | | 69,045 | |
| | | | | | | |
7.INTANGIBLE ASSETS
Intangible assets consist mainly of office and administrative software as well as scientific software purchased by the Group.
The following tables show the variations in intangible assets for the years ended December 31, 2019, 2020 and 2021:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | As of | | Increase | | Decrease | | Translation | | Reclassification | | As of |
| (in € thousands) | | 2018/12/31 | | | | | | adjustments | | | | 2019/12/31 |
| Gross | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Software | | 2,049 | | | 653 | | | (29) | | | — | | | 378 | | | 2,739 | |
| Patents | | 21 | | | 70 | | | — | | | — | | | — | | | 91 | |
| | | | | | | | | | | | | |
| Other intangibles | | 313 | | | 65 | | | (313) | | | — | | | (378) | | | — | |
| TOTAL—Gross | | 2,384 | | | 788 | | | (342) | | | — | | | — | | | 2,830 | |
| Accumulated depreciation and impairment | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Software | | (1,567) | | | (350) | | | 29 | | | — | | | — | | | (2) | |
| Patents | | (21) | | | — | | | — | | | — | | | — | | | (21) | |
| | | | | | | | | | | | | |
| Other intangibles | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated depreciation and impairment | | (1,588) | | | (350) | | | 29 | | | — | | | — | | | (1,910) | |
| TOTAL - Net | | 796 | | | 438 | | | (313) | | | — | | | — | | | 920 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | As of | | Increase | | Decrease | | Translation | | Reclassification | | As of |
| (in € thousands) | | 12/31/2019 | | | | | | adjustments | | | | 12/31/2020 |
| Gross | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Software | | 1,948 | | | 231 | | | (691) | | | — | | | (48) | | | 1,440 | |
| Patents | | 91 | | | — | | | — | | | — | | | — | | | 91 | |
| | | | | | | | | | | | | |
| Other intangibles | | — | | | (24) | | | (25) | | | — | | | 48 | | | — | |
| TOTAL—Gross | | 2,039 | | | 207 | | | (715) | | | — | | | — | | | 1,531 | |
| Accumulated depreciation and impairment | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Software | | (1,592) | | | (309) | | | 688 | | | — | | | — | | | (1,213) | |
| Patents | | (21) | | | — | | | — | | | — | | | — | | | (21) | |
| | | | | | | | | | | | | |
| Other intangibles | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated depreciation and impairment | | (1,613) | | | (310) | | | 688 | | | — | | | — | | | (1,234) | |
| TOTAL - Net | | 426 | | | (102) | | | (27) | | | — | | | — | | | 297 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | As of | | Increase | | Decrease | | Translation | | Reclassification | | As of |
| (in € thousands) | | 12/31/2020 | | | | | | adjustments | | | | 2021/12/31 |
| Gross | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Software | | 1,440 | | | 126 | | | (255) | | | — | | | (17) | | | 1,294 | |
| Patents | | 91 | | | — | | | (21) | | | — | | | — | | | 70 | |
| | | | | | | | | | | | | |
| Other intangibles | | — | | | — | | | (17) | | | — | | | 17 | | | — | |
| TOTAL—Gross | | 1,531 | | | 126 | | | (293) | | | — | | | — | | | 1,364 | |
| Accumulated depreciation and impairment | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Software | | (1,213) | | | (152) | | | 176 | | | — | | | — | | | (1,190) | |
| Patents | | (21) | | | — | | | 21 | | | — | | | — | | | — | |
| | | | | | | | | | | | | |
| Other intangibles | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated depreciation and impairment | | (1,234) | | | (152) | | | 197 | | | — | | | — | | | (1,190) | |
| TOTAL - Net | | 297 | | | (26) | | | (96) | | | — | | | — | | | 174 | |
The implementation for the April 2021 IFRIC decision on IAS 38 had a negative impact of €495 on intangible assets on the opening balance sheet for 2020.
8.PROPERTY, PLANT AND EQUIPMENT
The following tables show the variations in tangible assets for the years ended December 31, 2019, 2020 and 2021:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Property, plant and equipment - Variations | | As of As of | | Increase | | Decrease | | Translation | | Reclassification | | As of As of |
| (in € thousands) | | As of 2018/12/31 | | | | | | adjustments | | | | As of 2019/12/31 |
| Gross | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | 1,458 | | | 12,218 | | | — | | | — | | | (1,447) | | | 12,229 | |
| Scientific equipment | | 10,879 | | | 556 | | | (120) | | | — | | | (54) | | | 11,260 | |
| Fittings | | 1,531 | | | 66 | | | — | | | — | | | (5) | | | 1,592 | |
| Vehicles | | 99 | | | — | | | — | | | — | | | — | | | 99 | |
| Computer equipment | | 1,446 | | | 227 | | | (15) | | | — | | | 11 | | | 1,669 | |
| Furniture | | 361 | | | 31 | | | (3) | | | — | | | — | | | 389 | |
| In progress | | — | | | 241 | | | (1,737) | | | — | | | 1,496 | | | — | |
| TOTAL - Gross | | 15,774 | | | 13,339 | | | (1,875) | | | — | | | — | | | 27,238 | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Accumulated depreciation | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | (1) | | | (1,215) | | | — | | | — | | | — | | | (1,216) | |
| Scientific equipment | | (5,988) | | | (1,303) | | | 119 | | | — | | | — | | | (7,172) | |
| Fittings | | (769) | | | (105) | | | — | | | — | | | — | | | (875) | |
| Vehicles | | (45) | | | (21) | | | — | | | — | | | — | | | (66) | |
| Computer equipment | | (915) | | | (252) | | | 12 | | | — | | | — | | | (1,155) | |
| Furniture | | (292) | | | (13) | | | 3 | | | — | | | — | | | (303) | |
| In progress | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated depreciation | | (8,010) | | | (2,909) | | | 133 | | | — | | | — | | | (10,785) | |
| Accumulated impairment | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | — | | | — | | | — | | | — | | | — | | | — | |
| Scientific equipment | | — | | | — | | | — | | | — | | | — | | | — | |
| Fittings | | — | | | — | | | — | | | — | | | — | | | — | |
| Vehicles | | — | | | — | | | — | | | — | | | — | | | — | |
| Computer equipment | | — | | | — | | | — | | | — | | | — | | | — | |
| Furniture | | — | | | — | | | — | | | — | | | — | | | — | |
| In progress | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated impairment | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Net | | 7,765 | | | 10,429 | | | (1,741) | | | — | | | — | | | 16,453 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Property, plant and equipment - Variations | | As of As of | | Increase | | Decrease | | Translation | | Reclassification | | As of As of |
| (in € thousands) | | As of 2019/12/31 | | | | | | adjustments | | | | As of 2020/12/31 |
| Gross | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | 12,229 | | | — | | | — | | | — | | | (62) | | | 12,167 | |
| Scientific equipment | | 11,260 | | | 450 | | | (2,630) | | | — | | | — | | | 9,080 | |
| Fittings | | 1,592 | | | 233 | | | (113) | | | — | | | (9) | | | 1,703 | |
| Vehicles | | 99 | | | — | | | — | | | — | | | — | | | 99 | |
| Computer equipment | | 1,669 | | | 69 | | | (194) | | | — | | | (11) | | | 1,534 | |
| Furniture | | 389 | | | 8 | | | (68) | | | — | | | — | | | 329 | |
| In progress | | — | | | 15 | | | (17) | | | — | | | 2 | | | — | |
| TOTAL - Gross | | 27,238 | | | 775 | | | (3,022) | | | — | | | (80) | | | 24,911 | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Accumulated depreciation | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | (1,216) | | | (1,398) | | | 10 | | | — | | | — | | | (2,603) | |
| Scientific equipment | | (7,172) | | | (1,368) | | | 2,588 | | | — | | | — | | | (5,952) | |
| Fittings | | (875) | | | (218) | | | 107 | | | 4 | | | — | | | (982) | |
| Vehicles | | (66) | | | (20) | | | — | | | — | | | — | | | (85) | |
| Computer equipment | | (1,155) | | | (260) | | | 193 | | | 4 | | | — | | | (1,217) | |
| Furniture | | (303) | | | (15) | | | 68 | | | (1) | | | — | | | (251) | |
| In progress | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated depreciation | | (10,785) | | | (3,279) | | | 2,967 | | | 7 | | | — | | | (11,090) | |
| Accumulated impairment | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | — | | | (1,182) | | | — | | | — | | | — | | | (1,182) | |
| Scientific equipment | | — | | | (866) | | | — | | | — | | | — | | | (866) | |
| Fittings | | — | | | (93) | | | — | | | — | | | — | | | (93) | |
| Vehicles | | — | | | — | | | — | | | — | | | — | | | — | |
| Computer equipment | | — | | | (27) | | | — | | | — | | | — | | | (27) | |
| Furniture | | — | | | (3) | | | — | | | — | | | — | | | (3) | |
| In progress | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated impairment | | — | | | (2,172) | | | — | | | — | | | — | | | (2,172) | |
| TOTAL - Net | | 16,453 | | | (4,676) | | | (56) | | | 7 | | | (80) | | | 11,648 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Property, plant and equipment - Variations | | As of | | Increase | | Decrease | | Translation | | Reclassification | | As of |
| (in € thousands) | | 2020/12/31 | | | | | | adjustments | | | | 2021/12/31 |
| Gross | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | 12,167 | | | — | | | (1,912) | | | — | | | 56 | | | 10,311 | |
| Scientific equipment | | 9,080 | | | 71 | | | (2,831) | | | — | | | — | | | 6,320 | |
| Fittings | | 1,703 | | | (4) | | | (234) | | | — | | | 9 | | | 1,474 | |
| Vehicles | | 99 | | | 60 | | | (67) | | | — | | | — | | | 91 | |
| Computer equipment | | 1,534 | | | 30 | | | (18) | | | — | | | (4) | | | 1,542 | |
| Furniture | | 329 | | | — | | | (50) | | | — | | | — | | | 279 | |
| In progress | | — | | | 330 | | | (342) | | | — | | | 12 | | | — | |
| TOTAL - Gross | | 24,911 | | | 487 | | | (5,454) | | | — | | | 74 | | | 20,017 | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Accumulated depreciation | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | (2,603) | | | (1,417) | | | 1,120 | | | — | | | — | | | (2,900) | |
| Scientific equipment | | (5,952) | | | (1,061) | | | 2,145 | | | — | | | — | | | (4,868) | |
| Fittings | | (982) | | | (91) | | | 190 | | | (5) | | | — | | | (888) | |
| Vehicles | | (85) | | | (13) | | | 67 | | | — | | | — | | | (31) | |
| Computer equipment | | (1,217) | | | (195) | | | 14 | | | (6) | | | — | | | (1,403) | |
| Furniture | | (251) | | | (12) | | | 50 | | | — | | | — | | | (213) | |
| In progress | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated depreciation | | (11,090) | | | (2,789) | | | 3,587 | | | (11) | | | — | | | (10,304) | |
| Accumulated impairment | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Buildings on non-freehold land | | (1,182) | | | — | | | 679 | | | — | | | — | | | (503) | |
| Scientific equipment | | (866) | | | — | | | 779 | | | — | | | — | | | (87) | |
| Fittings | | (93) | | | — | | | — | | | — | | | — | | | (93) | |
| Vehicles | | — | | | — | | | — | | | — | | | — | | | — | |
| Computer equipment | | (27) | | | — | | | 15 | | | — | | | — | | | (12) | |
| Furniture | | (3) | | | — | | | — | | | — | | | — | | | (3) | |
| In progress | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL - Accumulated impairment | | (2,172) | | | — | | | 1,473 | | | — | | | — | | | (699) | |
| TOTAL - Net | | 11,648 | | | (2,302) | | | (394) | | | (11) | | | 74 | | | 9,015 | |
Assets related to contracts that were classified as finance leases under IAS 17 are scientific equipment. These contracts are accounted for in the same manner under IFRS 16. Their net carrying value as of December 31, 2019, 2020 and 2021 amounted to €1,413, €873 and €154 respectively.
Impairment test of assets under IAS 36
Some equipment belonging to the Group and others under a leasing agreement were no longer in use following the reorganization of the group’s activities and the termination of the RESOLVE-IT trial decided in mid-2020.
This indication of loss of value led the Group to conduct an impairment test over owned and leased equipment, based on the the value at which this equipment may be divested (on the basis of agreements with the lessors on the early purchase of the equipment and near-term purchase offers) in order to determine the recovery value.
The tests had resulted in 2020 in the recognition of an impairment of €990 relating to scientific equipment, fittings, computer equipment and furniture, including:
•€866 for scientific equipment (of which €363 related to owned equipment and €503 of leased equipment),
•€93 for fittings, and
•€30 for computer equipment and furniture,
including associated liabilities.
In 2021, part of these elements, mainly scientific equipment, were sold. As a result the accumulated impairment for these equipments was reduced to €196 as at December 31, 2021, including:
•€87 for scientific equipment (of which €25 related to owned equipment and €62 of leased equipment),
•€93 for fittings, and
•€15 for computer equipment and furniture,
including associated liabilities.
Similarly, parts of the leased premises (a portion of the office space in Paris and of the former laboratories at headquarters) were no longer in use. The vacant space is segmented and separate from the premises that will continue to be occupied. An impairment test of the rights of use of this space has also been performed.
In the financial statements for the period ending December 31, 2020, the recovery value had been estimated to be null considering that subleasing is prohibited for the office space in Paris and that the COVID-19 related health situation creates significant uncertainty on the potential to sublease the space in Loos. Subleasing was therefore not under consideration for the foreseeable future.
The test of the rights of use pertaining to these premises in 2020 had resulted in the recognition of an impairment of €1,182.
In 2021, the Company signed an agreement for a negotiated early termination of its lease agreement for its Paris premises, allowing for a relocation of the Paris office, thus the corresponding impairment was reduced to €503 as at December 31, 2021.
Reminders
In accordance with IFRS 16, the Group has chosen not to present the right of use separately from other assets and has added them to the fixed assets of the same nature as the underlying leased assets.
Therefore, the rights of use and related amortization as of December 31, 2021 included in the table affect:
•The line item “Building on non freehold land”, of €10,056 and €2,831, respectively;
•The line item "Scientific equipment", at €1,369 and €1,255 respectively.
In 2021 GENFIT SA and GENFIT CORP terminated the respective lease agreements for their offices, respectively located in Paris, France and Cambridge, MA, which they both relocated to a coworking space. The rental of these office spaces, as a service contract, no longer falls under IFRS 16. The impact of this change in 2021 is limited as both relocations happened during the second half of the year.
9.TRADE AND OTHER RECEIVABLES
Trade and other receivables consisted of the following:
| | | | | | | | | | | | | | | | | | | | | |
| Trade and other receivables - Total | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Trade receivables, net | | 207 | | | 793 | | | 57 | |
| Research tax credit | | 9,585 | | | 7,911 | | | 5,282 | |
| Social security costs receivables | | 5 | | | 24 | | | 4 | |
| VAT receivables | | 1,814 | | | 2,766 | | | 1,038 | |
| Grants receivables | | 3 | | | 3 | | | 5 | |
| Other receivables | | 420 | | | 422 | | | 852 | |
| TOTAL | | 12,033 | | | 11,919 | | | 7,239 | |
| | | | | | | | | | | | | | | | | | | | | |
| Trade and other receivables - Current | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Trade receivables, net | | 207 | | | 793 | | | 57 | |
| Research tax credit | | 9,585 | | | 7,911 | | | 5,282 | |
| Social security costs receivables | | 5 | | | 24 | | | 4 | |
| VAT receivables | | 1,814 | | | 2,766 | | | 1,038 | |
| Grants receivables | | 3 | | | 3 | | | 3 | |
| Other receivables | | 420 | | | 422 | | | 852 | |
| TOTAL | | 12,033 | | | 11,919 | | | 7,236 | |
| | | | | | | | | | | | | | | | | | | | | |
| Trade and other receivables - Non-current | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Trade receivables, net | | — | | | — | | | — | |
| Research tax credit | | — | | | — | | | — | |
| Social security costs receivables | | — | | | — | | | — | |
| VAT receivables | | — | | | — | | | — | |
| Grants receivables | | — | | | — | | | 3 | |
| Other receivables | | — | | | — | | | — | |
| TOTAL | | — | | | — | | | 3 | |
Research tax credit
The research tax credit due for 2019, amounting to €9,585, was received in June 2020.
The research tax credit due for 2020, amounting to €7,911, was received in October 2021.
The research tax credit receivable for the year 2021 amounts to €5,282.
VAT receivables
The VAT receivable amounted to €1,038 at December 31, 2021.
The last VAT receivable due for 2021 was received in January 2022.
In comparison, the VAT receivable amount of €2,766 recognized at December 31, 2020 was notably due to an audit on the basis of documents, by the French revenue services, of the VAT refund requests started in August 2020, which as a consequence, increased the time for a refund from the French revenue services. This audit was completed in 2021 and the Company received €2,766 in 2021.
Other receivables
The line item “other receivables” primarily consists of credit notes from suppliers for €€752, €406, and €408 respectively as of December 31, 2021, December 31, 2020 and December 31, 2019.
10.OTHER FINANCIAL ASSETS
Other financial assets consisted of the following:
| | | | | | | | | | | | | | | | | | | | | |
| Financial assets - Total | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Equity investments | | — | | | — | | | 3,133 | |
| | | | | | | |
| Loans | | 307 | | | 352 | | | 388 | |
| | | | | | | |
| Deposits and guarantees | | 396 | | | 418 | | | 397 | |
| Liquidity contract | | 1,023 | | | 688 | | | 513 | |
| TOTAL | | 1,727 | | | 1,458 | | | 4,431 | |
| | | | | | | | | | | | | | | | | | | | | |
| Financial assets - Current | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| | | | | | | |
| | | | | | | |
| Loans | | — | | | — | | | — | |
| | | | | | | |
| Deposits and guarantees | | — | | | — | | | — | |
| Liquidity contract | | — | | | — | | | — | |
| TOTAL | | — | | | — | | | — | |
| | | | | | | | | | | | | | | | | | | | | |
| Financial assets - Non current | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Equity investments | | — | | | — | | | 3,133 | |
| | | | | | | |
| Loans | | 307 | | | 352 | | | 388 | |
| | | | | | | |
| Deposits and guarantees | | 396 | | | 418 | | | 397 | |
| Liquidity contract | | 1,023 | | | 688 | | | 513 | |
| TOTAL | | 1,727 | | | 1,458 | | | 4,431 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Financial assets - Variations | | | As of | | Increase | | Decrease | | As of | |
| (in € thousands) | | | 31/12/2020 | | | | | | 31/12/2021 | |
| Equity investments | | | — | | | 3,133 | | | — | | | 3,133 | | |
| | | | | | | | | | | |
| Loans | | | 352 | | | 36 | | | — | | | 388 | | |
| | | | | | | | | | | |
| Deposits and guarantees | | | 418 | | | 107 | | | (128) | | | 397 | | |
| Liquidity contract | | | 688 | | 0 | | (175) | | 513 | |
| TOTAL | | | 1,458 | | | 3,276 | | | (303) | | | 4,431 | | |
The total amount of financial assets of the Company was €4,431 at December 31, 2021, up from €1,458 at December 31, 2020. This change is mainly due to the €3,000 investment representing a 10% equity stake taken by the Company in Genoscience Pharma in December 2021 through the subscription of new ordinary shares, plus directly related costs.
The liquidity contract consists of a share buyback program contracted to an investment service provider in order to facilitate the listing of the Group's shares (the liquidity cash balance hereunder represents funds held by investment service provider, which are not available for general use to the Company):
•As of December 31, 2021, the liquidity account had a cash balance of €513, and CMC-CIC Market Solutions holds on behalf of Genfit 137,012 shares, recorded as a deduction from equity.
•As of December 31, 2020, the liquidity account had a cash balance of €688, and CMC-CIC Market Solutions holds on behalf of Genfit 88,929 shares, recorded as a deduction from equity.
•As of December 31, 2019, the liquidity account had a cash balance of €1,023, and CMC-CIC Market Solutions holds on behalf of Genfit 18,132 shares, recorded as a deduction from equity.
About the recognition of equity investment in Genoscience Pharma subscribed in December 2021:
We have not completed the equity purchase in Genoscience Pharma for trading purposes. Therefore, pursuant to IFRS 9, we have elected to classify the equity in Genoscience Pharma we acquired in December 2021 as equity instruments recognized at fair value through other comprehensive income (OCI).
Pursuant to IFRS 9.5.1.1, at the time of initial recognition in 2021, this investment in equity instruments has been measured at fair value, augmented of the transaction costs related to the purchase.
The amount recognized in the balance sheet at December 31, 2021 corresponds to the subscription price agreed upon between the parties as representative of the value of Genoscience Pharma a few days before closing of the period.
In accordance with IFRS 9.5.7.1b, for future closings, changes in fair value on these equity instruments will be recognized as OCI. This OCI may not be reused as profit or loss, including in the case of a sale.
If applicable, only dividends related to the investment in equity instruments will be recognized as profit provided that all conditions are met (IFRS 9.5.7.6 and 5.7.1A).
At this stage, and in accordance with the amendment to IFRS 13, as equity in Genoscience Pharma is not publicly traded on active markets, the fair value of this equity is measured on the basis of valuation methods relying entirely or in part on unobservable inputs such as quoted prices in inactive markets or the valuation based on multiples for non-listed securities.
11.OTHER ASSETS
Other assets of €2,101 at December 31, 2021, €1,765 at December 31, 2020, €1,968 at December 31, 2019 and respectively, consisted of prepaid expenses related to current operating expenses.
12.LOANS AND BORROWINGS
12.1.Breakdown of convertible loan
On October 16, 2017, the Company issued 6,081,081 OCEANEs at par with a nominal unit value of €29.60 per bond for an aggregate nominal amount of €180 million. This debt was renegotiated in January 2021, and share conversions were executed during the period.
| | | | | | | |
| At origin (10/16/2017) : | | |
| Number of bonds | | 6,081,081 |
| Nominal amount of the loan | | 179,999,997.60€ |
| Nominal unit value of the bonds | | 29.60€ |
| Conversion / exchange premium | | 30% |
| | | To GENFIT's reference share price : |
| | | 22.77€ |
| Annual nominal interest rate | | 3.5% |
| | | Payable semi-annually in arrears |
| Annual nominal interest rate | | 7.2% |
| Offering | | 10/16/2017 |
| | | At par |
| Redemption | | 10/16/2022 |
| | | Redemption prior to maturity at the option of the Company from |
| | | 11/6/2020 |
| | | if the arithmetic volume-weighted average price of |
| | | GENFIT's listed share price and the then prevailing conversion ratio over a |
| | | 20 |
| | | trading period exceeds |
| | | 150% |
| | | of the nominal value of the OCEANEs. |
| After OCEANEs buyback : | | |
| Number of bonds | | 3,185,821 |
| Nominal amount of the loan | | 94,300,301.60€ |
| Nominal unit value of the bonds | | 29.60€ |
| Effective interest rate | | 8.8% |
| As of 06/30/2021 : | | |
| Number of bonds | | 1,923,662 |
| Nominal amount of the loan | | 56,940,395.20€ |
| Nominal unit value of the bonds | | 29.60€ |
| Effective interest rate | | 8.8% |
OCEANEs Buyback and Amendment of Terms
On November 23, 2020, GENFIT proposed to all OCEANE bondholders a renegotiation offer involving two interdependent components:
•A partial buyback of the outstanding OCEANEs for a maximum amount of 3,048,780 OCEANEs at €16.40 per bond; and
•An amendment of the terms of the remaining OCEANEs allowing to extend their maturity (by 3 years) and increase the conversion ratio (to 5.5 ordinary shares per bond).
The completion of these commitments for partial repurchase, made in late 2020, remained entirely subject to approval of the new terms of the OCEANEs, by both the Shareholders’ and Bondholders’ Meetings, which on January 25, 2021, approved this renegotiation offer. Following the shareholders’ and bondholders’ decisions, GENFIT completed the partial buyback of 2,895,260 OCEANEs at a price of €16.40 (including accrued interest of €0.30) for a total buyback cost of €47.48 million. The settlement operations occurred on January 29, 2021. The repurchased OCEANEs were then cancelled by GENFIT.
For the non-cancelled, renegotiated OCEANEs (“OCEANEs 2022”) (i.e. 3,185,821 remaining OCEANEs), the maturity is extended to October 16, 2025 and the conversion ratio changed from 1 OCEANE for 1 share to 1 OCEANE for 5.5 shares. The nominal amount and the payout value of the remaining OCEANEs remains unchanged at €29.60 per bond.
This renegotiation operation of the OCEANE has been recognized in the consolidated accounts for the half-year ended June 30, 2021, as:
•the derecognition of the full initial OCEANE as of January 25, 2021 against a payment of €47.48 million, and
•the issuance of 3,185,821 new amended OCEANEs.
As the conversion option for the new OCEANEs (2025 maturity) fits the definition of an equity instrument under IAS 32 (Financial Instruments: Presentation), the components of this new OCEANE (debt vs. equity) has been recognized separately on January 25, 2021, in accordance with the accounting rules and methods presented in this note.
The obligation and option components have been valued separately. The option component has been valued using a traditional binomial model.
The hypotheses considered to calculate the fair value of these new OCEANEs are the following:
•credit spread in the 874/976 bps interval
•volatility: first level: 30% second level: 35%
•no-risk rate: 5-year Euros swap equals -0.45%
On this basis, at January 25, 2021, the fair value of a new amended OCEANEs has been estimated at €27.80, of which a debt component of €24.12 and a €3.68 component that has been recognized in equity.
Accounting impacts of the debt renegotiation
On January 25, 2021 an amount of €94.8 million was derecognized and an amount of €76.8 million was recognized for the amended obligations, in exchange of:
•An increase in equity of €11.7 million before deferred taxes (corresponding to the recognition of the value of the conversion option of the amended OCEANE);
•The payment of €47.5 million for the OCEANEs partial buyback; and
•The recognition of a financial gain (buyback bonus) of €35.6 million before tax.
Accounting impacts of the conversions completed following the debt renegotiation
Following the implementation of the partial buyback operation and the approval of the amendment of the terms of the OCEANEs:
•552,238 of the new OCEANEs were subject to a request for share conversion in January 2021. On February 4, 2021, as a result of these conversion requests, a capital increase of €759,327.25 has been recognized, corresponding to the creation of 3,037,309 new shares. This conversion of 552,238 new OCEANEs resulted in a reduction in financial debt for the Group of €13.32 million.
•483,330 of the new OCEANEs were subject to a request for share conversion in February 2021. On March 2, 2021, as a result of these conversion requests, a capital increase of €664,578.75 has been recognized, corresponding to the creation of 2,658,312 new shares. This conversion of 483,330 new OCEANEs resulted in a reduction in financial debt for the Group of €11.66 million.
•216,591 of the new OCEANEs were subject to a request for share conversion in March 2021. On April 6, 2021, as a result of these conversion requests, a capital increase of €297,812.50 has been recognized, corresponding to the creation of 1,191,250 new shares. This conversion of 216,591 new OCEANEs resulted in a reduction in financial debt for the Group of €5.2 million.
•10,000 of the new OCEANEs were subject to a request for share conversion in August 2021. On September 1, 2021, as a result of these conversion requests, a capital increase of €13,750 has been recognized, corresponding to the creation of 55,000 new shares. This conversion of 10,000 new OCEANEs resulted in a reduction in financial debt for the Group of €0.2 million.
The potential issuance of new shares upon conversion requests of the outstanding OCEANEs would represent 21.24% of the share capital of the Company at December 31, 2021.
All fees and commission paid in relation to this operation have been directly recognized as operating expenses. The fees disbursed have been recognized in the financial statements for a total of €745 in 2020 and €2,303 in 2021.
Deferred tax assets and deferred tax liabilities recognized in the balance sheet at December 31, 2020 related to the 2022 OCEANEs for respectively €1.3 million and €2.0 million have been recognized in the profit and loss account in 2021.
A deferred tax liability related to the new OCEANEs has been recognized on January 25, 2021 with an impact on share capital for an amount of €4.4 million. A deferred tax asset was recognized on January 25, 2021 with an impact on the profit and loss statement under the allocation of tax loss carry forwards on the deferred tax liability reversal for €2.8 million.
| | | | | | | | | | | | | | | | | | | | | |
| Convertible loans - Total | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Convertible loans | | 165,454 | | | 170,782 | | | 48,097 | |
| TOTAL | | 165,454 | | 170,782 | | 48,097 |
| | | | | | | | | | | | | | | | | | | | | |
| Convertible loans - Current | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Convertible loans | | 1,312 | | | 1,312 | | | 415 | |
| TOTAL | | 1,312 | | 1,312 | | 415 |
| | | | | | | | | | | | | | | | | | | | | |
| Convertible loans - Non current | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Convertible loans | | 164,142 | | | 169,470 | | | 47,682 | |
| TOTAL | | 164,142 | | 169,470 | | 47,682 |
12.2.Breakdown of other loans and borrowings
Other loans and borrowings consisted of the following:
| | | | | | | | | | | | | | | | | | | | | |
| Other loans and borrowings - Total | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Refundable and conditional advances | | 3,229 | | | 3,229 | | | 3,229 | |
| Bank loans | | 2,645 | | 1,540 | | | 15,824 | |
| | | | | | | |
| | | | | | | |
| Obligations under leases | | 12,281 | | | 10,131 | | | 7,069 | |
| Accrued interests | | 1 | | 1 | | | 16 | |
| Other financial loans and borrowings | | 7 | | | 7 | | | — | |
| | | | | | | |
| TOTAL | | 18,165 | | 14,908 | | 26,138 |
| | | | | | | | | | | | | | | | | | | | | |
| Other loans and borrowings - Current | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Refundable and conditional advances | | — | | | — | | | — | |
| Bank loans | | 1,105 | | 942 | | | 667 | |
| | | | | | | |
| | | | | | | |
| Obligations under leases | | 2,112 | | | 2,085 | | | 1,089 | |
| Accrued interests | | 1 | | 1 | | | 16 | |
| Other financial loans and borrowings | | 7 | | | 7 | | | — | |
| | | | | | | |
| TOTAL | | 3,226 | | | 3,035 | | | 1,773 | |
| | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
| Other loans and borrowings - Non current | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Refundable and conditional advances | | 3,229 | | | 3,229 | | | 3,229 | |
| Bank loans | | 1,540 | | 598 | | | 15,156 | |
| | | | | | | |
| | | | | | | |
| Obligations under leases | | 10,169 | | | 8,046 | | | 5,980 | |
| Accrued interests | | — | | | — | | | — | |
| Other financial loans and borrowings | | — | | | — | | | — | |
| | | | | | | |
| TOTAL | | 14,939 | | | 11,873 | | | 24,365 | |
12.2.1.Refundable and conditional advances
The following table summarizes advances outstanding at December 31, 2021, 2020 and 2019.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Refundable and conditional advances—general overview | | Grant date | | Total amount allocated | | Receipts | | Repayments | | Effects of discounting | | Net book value As of 2021/12/31 |
| (in € thousands) | | | | | | | | | | | | |
| BPI FRANCE - IT-DIAB | | 12/23/2008 | | 3,229 | | | 3,229 | | | — | | | — | | | 3,229 | |
| Development of a global strategy for the prevention and management of type 2 diabetes | | | | | | | | | | | | |
| TOTAL | | | | 3,229 | | | 3,229 | | | — | | | — | | | 3,229 | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Refundable and conditional advances—general overview | | Grant date | | Total amount allocated | | Receipts | | Repayments | | Effects of discounting | | Net book value As of 2020/12/31 |
| (in € thousands) | | | | | | | | | | | | |
| BPI FRANCE - IT-DIAB | | 12/23/2008 | | 3,229 | | | 3,229 | | | — | | | — | | | 3,229 | |
| Development of a global strategy for the prevention and management of type 2 diabetes | | | | | | | | | | | | |
| TOTAL | | | | 3,229 | | | 3,229 | | | — | | | — | | | 3,229 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Refundable and conditional advances—general overview | | Grant date | | Total amount allocated | | Receipts | | Repayments | | Effects of discounting | | Net book value As of 2019/12/31 |
| | | | | | |
| (in € thousands) | | | | | | | | | | | | Net book value As of |
| BPI FRANCE - IT-DIAB | | 12/23/2008 | | 3,229 | | | 3,229 | | | — | | | — | | | 3,229 | |
| Development of a global strategy for the prevention and management of type 2 diabetes | | | | | | | | | | | | |
| TOTAL | | | | 3,229 | | | 3,229 | | | — | | | — | | | 3,229 | |
| | | | | |
BPI FRANCE IT-DIAB | On December 23, 2008, the Group received an advance from BPI France (the BPI France IT-DIAB) as part of a framework innovation aid agreement involving several scientific partners and for which the Group was the lead partner. The contribution expected at each stage by each of the partners in respect of work carried out and results achieved is defined in the framework agreement. With respect to the Group, the aid consisted of a €3,229 conditional advance and a €3,947 non-repayable government grant. |
| |
| The conditional advance is not refundable except in the event of success. The program ended on December 31, 2014. In the event of success, defined as the commercial spin-offs of the IT-Diab program which involves products for the treatment or diagnosis of type 2 diabetes, in that case, the financial returns generated will be used initially to repay the €3,229 conditional advance and the agreement stipulates that the conditional advance will be regarded as repaid in full when the total payments made in this regards by the recipient, discounted at the rate of 5.19%, equal the total amount, discounted at the same rate, of the aid paid. Any further amounts will be classified as additional payments, up to a maximum amount of €14,800. |
As provided in the project assistance contract, we sent a letter to BPI in December 2019 in order to notify it of our Labcorp and Terns contracts while indicating that elafibranor was now aimed at treating hepatic diseases and no longer type 2 diabetes as provided for in the aid agreement. We proposed to BPI to establish a statement of abandonment of the IT DIAB project on which the above advance is based. Following this letter, the parties met in March 2020 for the presentation of our arguments, and in June 2020 following the publication of the results of the RESOLVE-IT study, and a new letter was sent in November 2020. In this context, we are awaiting a proposal from BPI on new financial terms related to this situation and a draft amendment to the repayable advance agreement. Until we receive a response from BPI France, we consider that the fair value of this liability corresponds to the amount paid by BPI FRANCE.
12.2.2.Bank loans
In the context of the COVID-19 pandemic, the Company secured:
•A State-Guaranteed Loan (or "Prêt Garanti par l'Etat (PGE) Bancaire") for an amount of €11,000 (€10,919 net of fees), granted on June 24, 2021 by a syndicate of 4 French banks and paid out on June 29, 2021, 90% guaranteed by the French government with an initial term of one year with repayment options up to six years;
•A State-Guaranteed Loan (or "Prêt Garanti par l'Etat (PGE) Bpifrance") for an amount of €2,000 (€1,985 net of fees) granted on July 20, 2021 by Bpifrance and paid out on July 23, 2021, 90% guaranteed by the French government with an initial term of one year with repayment options up to six years;
•A Subsidized Loan for an amount of €2,250 (€2,250 net of fees) granted on November 23, 2021 by Bpifrance and paid out on November 26, 2021, with an initial term of six years.
The Company already intends to use the options to delay the repayment of both state-guaranteed loans above. Besides, the Prêt Garanti par l'Etat Bancaire above provides for a mandatory early repayment in full of the loans in case of a cash repayment of the existing bond debt (share conversions are not included in this provision).
The repayment hypothesis used in these consolidated financial statements includes:
•8 linear quarterly payments between September 29, 2023 and June 29, 2025 for the PGE Bancaire, and
•16 linear quarterly payments between October 23, 2022 and July 23, 2026 for the PGE Bpifrance.
The subsidized loan provides for a 4-quarter deferment of capital amortization, followed by 20 equal quarterly payments (amortization and interest) between February 28, 2023 and November 30, 2027.
Regarding the PGE Bancaire, the first-year interest rate is null (—%) and that of the following years will be communicated by the banks at the time an extension is requested. Moreover, the guarantee provided by the French State is compensated through a commission named "guarantee premium" (which increases progressively from 0.25% in the first year to 1% in the third year and beyond).
Regarding the PGE Bpifrance, the first-year interest rate is 1.85% (including 0.28% for the state guarantee) and that of the following years will be communicated by Bpifrance at the time an extension is requested. Moreover, this loans includes a one-year deferment on interest.
Regarding the subsidized loan, it bears a fixed interest rate at 2.25%.
Based on the above:
•The effective interest rate determined for the PGE Bancaire is 0.75% per year;
•The effective interest rate determined for the PGE Bpifrance is 1.95% per year;
•The effective interest rate determined for the Subsidized Loan is 2.27%.
The company has determined after analysis under IFRS that the subsidized loan should be treated in the same manner as the PGEs and that the review pursuant to IAS20 should not apply, in light of the facts, notably, that this subsidized loan:
•Constitutes Government Assistance under the "Umbrella" Scheme Notified by the French Government to the European Commission under the following references: State Aid SA.56985 (2020/N) - France - COVID-19: Temporary Framework to support companies;
•Has therefore not been granted to the Company in connection with research expenses on a particular project or investment;
•Supports the Company's cash position, similarly to the aforementioned PGEs, negotiated with and granted by Bpifrance in addition to these PGEs.
Thus, these loans are recognized using the effective interest rate method (with the aforementioned rates) and their respective IFRS values at December 31, 2021 are:
•€10,958 (of which zero at less than a year) for the PGE Bancaire;
•€2,002 (of which €125 at less than a year) for the PGE Bpifrance; and
•€2,250 (of which zero at less than a year) for the Subsidized Loan.
Bank loans consisted of the following as of December 31, 2019:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Bank loans | | Loan | | Facility | | Interest | | Available As of 2019/12/31 | | Installments | | Outstanding As of 2019/12/31 |
| (in € thousands) | | date | | size | | rate | | | | |
| CDN 3 | | April 2016 | | 500 | | | 0.72 | % | | — | | | 60 monthly | | 135 | |
| CDN 4 | | June 2017 | | 600 | | | 0.36 | % | | — | | | 48 monthly | | 226 | |
| CDN 5 | | November 2018 | | 500 | | | 0.46 | % | | — | | | 48 monthly | | 365 | |
| CIC 4 | | December 2016 | | 265 | | | 0.69 | % | | — | | | 60 monthly | | 111 | |
| CIC 5 | | July 2017 | | 1,000 | | | 0.69 | % | | — | | | 60 monthly | | 554 | |
| BNP 2 | | June 2016 | | 500 | | | 0.80 | % | | — | | | 20 quarterly | | 177 | |
| BNP 3 | | October 2016 | | 1,050 | | | 0.80 | % | | — | | | 20 quarterly | | 525 | |
| BNP 4 | | April 2017 | | 800 | | | 0.87 | % | | — | | | 60 monthly | | 537 | |
| AUTRES | | - | | — | | | — | % | | — | | | 0 | | 14 | |
| CDN PGE | | June 2021 | | — | | | (*) | | — | | | 8 quaterly | | — | |
| CIC PGE | | June 2021 | | — | | | (*) | | — | | | 8 quaterly | | — | |
| BNP PGE | | June 2021 | | — | | | (*) | | — | | | 8 quaterly | | — | |
| NATIXIS PGE | | June 2021 | | — | | | (*) | | — | | | 8 quaterly | | — | |
| BPI PGE | | July 2021 | | — | | | 1.85 | % | | — | | | 16 quaterly | | — | |
| BPI PRÊT TAUX BONIFIE | | November 2021 | | — | | | 2.25 | % | | — | | | 20 quaterly | | — | |
| TOTAL | | | | 5,215 | | | 0 | | — | | | 0 | | 2,645 | |
Bank loans consisted of the following as of December 31,2020:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Bank loans | | Loan | | Facility | | Interest | | Outstanding As of 2020/12/31 | | Installments | | Outstanding As of 2020/12/31 |
| (in € thousands) | | date | | size | | rate | | | | |
| CDN 3 | | April 2016 | | 500 | | | 0.72 | % | | — | | | 60 monthly | | 34 | |
| CDN 4 | | June 2017 | | 600 | | | 0.36 | % | | — | | | 48 monthly | | 75 | |
| CDN 5 | | November 2018 | | 500 | | | 0.46 | % | | — | | | 48 monthly | | 241 | |
| CIC 4 | | December 2016 | | 265 | | | 0.69 | % | | — | | | 60 monthly | | 58 | |
| CIC 5 | | July 2017 | | 1,000 | | | 0.69 | % | | — | | | 60 monthly | | 354 | |
| BNP 2 | | June 2016 | | 500 | | | 0.80 | % | | — | | | 20 quarterly | | 76 | |
| BNP 3 | | October 2016 | | 1,050 | | | 0.80 | % | | — | | | 20 quarterly | | 315 | |
| BNP 4 | | April 2017 | | 800 | | | 0.87 | % | | — | | | 60 monthly | | 377 | |
| AUTRES | | - | | — | | | 0.00 | % | | — | | | 0 | | 9 | |
| CDN PGE | | June 2021 | | — | | | (*) | | — | | | 8 quaterly | | — | |
| CIC PGE | | June 2021 | | — | | | (*) | | — | | | 8 quaterly | | — | |
| BNP PGE | | June 2021 | | — | | | (*) | | — | | | 8 quaterly | | — | |
| NATIXIS PGE | | June 2021 | | — | | | (*) | | — | | | 8 quaterly | | — | |
| BPI PGE | | July 2021 | | — | | | 1.85 | % | | — | | | 16 quaterly | | — | |
| BPI PRÊT TAUX BONIFIE | | November 2021 | | — | | | 2.25 | % | | — | | | 20 quaterly | | — | |
| TOTAL | | | | 5,215 | | | 0 | | — | | | 0 | | 1,540 | |
Bank loans consisted of the following as of December 31, 2021:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Bank loans | | Loan | | Facility | | Interest | | Available | | Installments | | Outstanding |
| | | date | | size | | rate | | as of | | | | as of |
| (in € thousands) | | | | | | | | 2021/12/31 | | | | 2021/12/31 |
| CDN 3 | | April 2016 | | 500 | | | 0.72 | % | | — | | | 60 monthly | | — | |
| CDN 4 | | June 2017 | | 600 | | | 0.36 | % | | — | | | 48 monthly | | — | |
| CDN 5 | | November 2018 | | 500 | | | 0.46 | % | | — | | | 48 monthly | | 115 | |
| CIC 4 | | December 2016 | | 265 | | | 0.69 | % | | — | | | 60 monthly | | 4 | |
| CIC 5 | | July 2017 | | 1,000 | | | 0.69 | % | | — | | | 60 monthly | | 152 | |
| BNP 2 | | June 2016 | | 500 | | | 0.80 | % | | — | | | 20 quarterly | | — | |
| BNP 3 | | October 2016 | | 1,050 | | | 0.80 | % | | — | | | 20 quarterly | | 105 | |
| BNP 4 | | April 2017 | | 800 | | | 0.87 | % | | — | | | 60 monthly | | 217 | |
| AUTRES | | - | | — | | | 0.00 | % | | — | | | 0 | | 20 | |
| CDN PGE | | June 2021 | | 900 | | | (*) | | — | | | 8 quaterly | | 900 | |
| CIC PGE | | June 2021 | | 2,200 | | | (*) | | — | | | 8 quaterly | | 2,200 | |
| BNP PGE | | June 2021 | | 4,900 | | | (*) | | — | | | 8 quaterly | | 4,900 | |
| NATIXIS PGE | | June 2021 | | 3,000 | | | (*) | | — | | | 8 quaterly | | 3,000 | |
| BPI PGE | | July 2021 | | 2,000 | | | 1.85 | % | | — | | | 16 quaterly | | 2,000 | |
| BPI PRÊT TAUX BONIFIE | | November 2021 | | 2,250 | | | 2.25 | % | | — | | | 20 quaterly | | 2,250 | |
| TOTAL | | | | 20,465 | | | 0 | | — | | | 0 | | 15,864 | |
(*) Will be defined at the end of the prorogation date.
12.4 Maturities of financial liabilities
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Maturity of financial liabilities | | As of | | Less than | | Less than | | Less than | | Less than | | Less than | | More than |
| (in € thousands) | | 2021-12-31 | | 1 year | | 2 years | | 3 years | | 4 years | | 5 years | | 5 years |
| BPI FRANCE - IT-DIAB | | 3,229 | | | — | | | — | | | — | | | — | | | — | | | 3,229 | |
| TOTAL - Refundable and conditional advances | | 3,229 | | | — | | | — | | | — | | | — | | | — | | | 3,229 | |
| Convertible loans | | 48,097 | | | 415 | | | — | | | — | | | 47,682 | | | — | | | — | |
| Bank loans | | 15,824 | | | 667 | | | 3,728 | | | 6,424 | | | 3,694 | | | 840 | | | 470 | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| Leases | | 7,069 | | | 1,089 | | | 939 | | | 925 | | | 932 | | | 942 | | | 2,242 | |
| Accrued interests | | 16 | | | 16 | | | — | | | — | | | — | | | — | | | — | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| TOTAL - Other loans and borrowings | | 71,006 | | | 2,188 | | | 4,667 | | | 7,349 | | | 52,308 | | | 1,782 | | | 2,712 | |
| TOTAL | | 74,235 | | | 2,188 | | | 4,667 | | | 7,349 | | | 52,308 | | | 1,782 | | | 5,941 | |
Based on the nominal amount of €56,940 at December 31, 2021, the convertible bond results in the payment of yearly interest of €1,993 (payable in 2 biannual installments). Its repayment is due on October 16, 2025.
13. FAIR VALUE OF FINANCIAL INSTRUMENTS
The following tables provide the financial assets and liabilities carrying values by category and fair values as of December 31, 2021, December 31, 2020 and December 31, 2019:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | As of 31/12/2019 |
| | | Carrying value | | Fair value |
| | | As per | | Assets at | | Loans & | | Debt at | | Level 1 | | Level 2 | | Level 3 |
| | | statement of | | fair value | | receivables | | amortized | | | | | | |
| | | financial | | through | | | | cost | | | | | | |
| (in € thousands) | | position | | profit & loss | | | | | | | | | | |
| Assets | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| Loans | | 307 | | | | | 307 | | | | | | | 307 | | | |
| | | | | | | | | | | | | | | |
| Deposits and guarantees | | 396 | | | | | 396 | | | | | | | 396 | | | |
| Trade receivables | | 207 | | | | | 207 | | | | | | | 207 | | | |
| Cash and cash equivalents | | 276,748 | | | 276,748 | | | | | | | 276,748 | | | | | |
| TOTAL - Assets | | 277,658 | | | 276,748 | | | 911 | | | — | | | 276,748 | | | 911 | | | — | |
| Liabilities | | | | | | | | | | | | | | |
| Conditional advances | | 3,229 | | | | | | | 3,229 | | | | | | | 3,229 | |
| Convertible loans | | 165,454 | | | | | | | 165,454 | | | | | 165,454 | | | |
| Bank loans | | 2,645 | | | | | | | 2,645 | | | | | 2,645 | | | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| Obligations under finance leases | | 12,281 | | | | | | | 12,281 | | | | | 12,281 | | | |
| Accrued interests | | 1 | | | | | | | 1 | | | | | 1 | | | |
| Other financial loans and borrowings | | 7 | | | | | | | 7 | | | | | 7 | | | |
| | | | | | | | | | | | | | | |
| Trade payables | | 32,753 | | | | | | | 32,753 | | | | | 32,753 | | | |
| Other payables | | 527 | | | | | | | 527 | | | | | 527 | | | |
| TOTAL - Liabilities | | 216,898 | | | — | | | — | | | 216,898 | | | — | | | 213,669 | | | 3,229 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | As of 31/12/2020 |
| | | Carrying value | | Fair value |
| | | As per | | Assets at | | Loans & | | Debt at | | Level 1 | | Level 2 | | Level 3 |
| | | statement of | | fair value | | receivables | | amortized | | | | | | |
| | | financial | | through | | | | cost | | | | | | |
| (in € thousands) | | position | | profit & loss | | | | | | | | | | |
| Assets | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| Loans | | 352 | | | | | 352 | | | | | | | 352 | | | |
| | | | | | | | | | | | | | | |
| Deposits and guarantees | | 418 | | | | | 418 | | | | | | | 418 | | | |
| Trade receivables | | 793 | | | | | 793 | | | | | | | 793 | | | |
| Cash and cash equivalents | | 171,029 | | | 171,029 | | | | | | | 171,029 | | | | | |
| TOTAL - Assets | | 172,592 | | | 171,029 | | | 1,563 | | | 0 | | 171,029 | | | 1,563 | | | 0 |
| Liabilities | | | | | | | | | | | | | | |
| Conditional advances | | 3,229 | | | | | | | 3,229 | | | | | | | 3,229 | |
| Convertible loans | | 170,782 | | | | | | | 170,782 | | | | | 170,782 | | | |
| Bank loans | | 1,540 | | | | | | | 1,540 | | | | | 1,540 | | | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| Obligations under finance leases | | 10,131 | | | | | | | 10,131 | | | | | 10,131 | | | |
| | | | | | | | | | | | | | | |
| Other financial loans and borrowings | | 7 | | | | | | | 7 | | | | | 7 | | | |
| | | | | | | | | | | | | | | |
| Trade payables | | 20,337 | | | | | | | 20,337 | | | | | 20,337 | | | |
| Other payables | | 569 | | | | | | | 569 | | | | | 569 | | | |
| TOTAL - Liabilities | | 206,596 | | | 0 | | 0 | | 206,596 | | | 0 | | 203,367 | | | 3,229 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | As of 31/12/2021 |
| | | Carrying value | | Fair value |
| | | As per | | Assets at | | Loans & | | Debt at | | Level 1 | | Level 2 | | Level 3 |
| | | statement of | | fair value | | receivables | | amortized | | | | | | |
| | | financial | | through | | | | cost | | | | | | |
| (in € thousands) | | position | | profit & loss | | | | | | | | | | |
| Assets | | | | | | | | | | | | | | |
| Equity investments | | 3,133 | | | 3,133 | | | | | | | | | | | 3,133 | |
| | | | | | | | | | | | | | | |
| Loans | | 388 | | | | | 388 | | | | | | | 388 | | | |
| | | | | | | | | | | | | | | |
| Deposits and guarantees | | 397 | | | | | 397 | | | | | | | 397 | | | |
| Trade receivables | | 57 | | | | | 57 | | | | | | | 57 | | | |
| Cash and cash equivalents | | 258,756 | | | 258,756 | | | | | | | 258,756 | | | | | |
| TOTAL - Assets | | 262,731 | | | 261,889 | | | 842 | | | — | | | 258,756 | | | 842 | | | 3,133 | |
| Liabilities | | | | | | | | | | | | | | |
| Conditional advances | | 3,229 | | | | | | | 3,229 | | | | | | | 3,229 | |
| Convertible loans | | 48,097 | | | | | | | 48,097 | | | | | 48,097 | | | |
| Bank loans | | 15,824 | | | | | | | 15,824 | | | | | 15,824 | | | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| Obligations under finance leases | | 7,069 | | | | | | | 7,069 | | | | | 7,069 | | | |
| Accrued interests | | 16 | | | | | | | 16 | | | | | 16 | | | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| Trade payables | | 12,304 | | | | | | | 12,304 | | | | | 12,304 | | | |
| Other payables | | 579 | | | | | | | 579 | | | | | 579 | | | |
| TOTAL - Liabilities | | 87,118 | | | — | | | — | | | 87,118 | | | — | | | 83,889 | | | 3,229 | |
About the equity investment in the table above: As the equity investment was made on December 16, 2021, we believe that fair market value as of December 31, 2021 is consistent with the cost of the investment.
14.TRADE AND OTHER PAYABLES
Trade and other payables consisted of the following:
| | | | | | | | | | | | | | | | | | | | | |
| Trade and other payables - Total | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Trade payables (*) | | 32,753 | | | 20,337 | | | 12,304 | |
| Social security costs payables | | 3,598 | | | 4,477 | | | 4,087 | |
| VAT payables | | 2 | | | 314 | | | 23,725 | |
| Taxes payables | | 487 | | | 319 | | | 744 | |
| Other payables | | 527 | | | 569 | | | 579 | |
| TOTAL | | 37,368 | | | 26,015 | | | 41,438 | |
| | | | | | | | | | | | | | | | | | | | | |
| Trade and other payables - Current | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Trade payables | | 32,753 | | | 20,337 | | | 12,304 | |
| Social security costs payables | | 3,598 | | | 4,477 | | | 4,087 | |
| VAT payables | | 2 | | | 314 | | | 23,725 | |
| Taxes payables | | 487 | | | 319 | | | 744 | |
| Other payables | | 76 | | | 118 | | | 128 | |
| TOTAL | | 36,917 | | | 25,564 | | | 40,988 | |
| | | | | | | | | | | | | | | | | | | | | |
| Trade and other payables - Non current | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Trade payables | | — | | | — | | | — | |
| Social security costs payables | | — | | | — | | | — | |
| VAT payables | | — | | | — | | | — | |
| Taxes payables | | — | | | — | | | — | |
| Other payables | | 450 | | | 450 | | | 450 | |
| TOTAL | | 450 | | | 450 | | | 450 | |
| | | | | | | |
| (*) Of which : Accrued expenses | | 18,682 | | | 13,809 | | | 6,201 | |
At December 31, 2021, trade payables amounted to €12,304 (€20,337 at December 31, 2020). This change is due to the reduction in operating expenses.
Trade payables include a significant portion of accrued expenses (€6,201 and €13,809 at December 31, 2021 and 2020 respectively), relating to yet unbilled amounts from the clinical trial sites via the Clinical Research Organizations (CROs) in charge of the Company's clinical trials.
The timeframe in which those invoices will be received by the Company is unknown and may be spread out over a long period after the services have been performed.
While at December 31, 2020, accrued expenses related to RESOLVE-IT, amounted to $9.6 million (€7.8 million) and €2.3 million, i.e. a total of €10.1 million, at December 31, 2021, they amounted to $0.9 million (€0.8 million) and €0.4 million, i.e. a total of €1.2 million). This reduction reflects the increase in RESOLVE-IT clinical trial sites closures following the decision taken mid-2020 to discontinue this study.
The VAT debt amounted to €23,725 at December 31, 2021 (€314 at December 31, 2020). This increase is related to the VAT amount collected on the upfront payment received from Ipsen in December 2021.
15.DEFERRED INCOME AND REVENUE
Out of the €120 million upfront payment received from Ipsen in application of the licensing agreement signed in December 2021, an amount of €40 million was recognized as Deferred income in 2021 (see: Note 19 "Operating Income"), of which €14,179 was recognized as Current deferred income, and €25,821 was recognized as Non-current deferred income.
16.PROVISIONS
At December 31, 2021, 2020 and 2019, this line item amounted to €313, €1,031, and €2,061, respectively.
This change mainly reflects provision reversals recorded in 2021 related to:
•Some administrative costs and costs related to the destruction of drug tablets following the decision taken mid-2020 to discontinue the RESOLVE-IT study : reversal of €366 (of which €265 was used), with the corresponding provision amounting to €12 at December 31, 2021;
•The estimated support costs related to the reduction in force plan (PSE) implemented starting in late 2020 (return-to-work bonuses, trainings, business start-up assistance and various other benefits) : reversal of €352 (of which €189 was used), with the corresponding provision amounting to €171 at December 31, 2021.
17.EMPLOYEE BENEFITSDetailed breakdown
In France, pension funds are generally financed by employer and employee contributions and are accounted for as a defined contribution plan with the employer contributions recognized as expense as incurred. The Group has no actuarial liabilities in connection with these plans. Related expenses recorded for the years ended December 31, 2021, 20202023 and 2019December 31, 2022 amounted to €774, €923 and €927€948, €876, respectively.
French law also requires payment of a lump sum retirement indemnity to employees based on years of service and annual compensation at retirement, which are accounted for as a defined benefit plan. Benefits do not vest prior to retirement. The liability is calculated as the present value of estimated future benefits to be paid, applying the projected unit credit method whereby each period of service is seen as giving rise to an additional unit of benefit entitlement, each unit being measured separately to build up the final liability. At December 31, 2021, 20202023 and 2019December 31, 2022 pension provisions recorded were €864 , €922€978 and €1,408 ,€782, respectively.
As part of the measurement of the retirement indemnity to employees, the following assumptions were used for all categories of employees in 2019, 20202022 and 2021:2023:
| | | | | | |
| Population | Permanent staff |
| Retirement age | 65 |
| Terms of retirement | Initiated by the employee |
| Life expectancy | On the basis of the INSEE table (1) |
| Probability of continued presence in the company at retirement age | On the basis of the DARES table |
(1)INSEE is the French National Institute of Statistics; DARES is the French Bureau of Studies and Statistics
| | | | | | | | | | | | | | | | | | | | | |
| Rate | | As of |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Salary growth rate - in 2022 | | 5.80 | % | | 3.00 | % | | 3.00 | % |
| Salary growth rate - beyond | | 3.00 | % | | 3.00 | % | | 3.00 | % |
| Discount rate (iboxx) | | 0.75 | % | | 0.50 | % | | 0.87 | % |
| | | | | | | | | | | | | | | | | |
| Rate | | | | As of |
| (in € thousands) | | | | 2022/12/31 | | 2023/12/31 |
| Salary growth rate - in 2023 | | | | 3.00 | % | | 3.00 | % |
| Salary growth rate - beyond | | | | 3.00 | % | | 3.00 | % |
| Discount rate (iboxx) | | | | 3.25 | % | | 3.59 | % |
The discount rates are based on the market yield at December 31, 2019, 20202022 and 20212023 on high-quality corporate bonds.
The following table presents the changes in the present value of the defined benefit obligation:
| | | | | | | | | |
| Changes in the present value of the defined benefit obligation | | | As of
| (in € thousands) | | | 2021/12/31
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| Defined benefit obligation as of January 01, 20192022 | | 1864 | |
| Current service cost | | —169 | |
| Interest cost on benefit obligation | | —8 | |
| Actuarial losses / (gains) on obligation | | — | |
| Past service costs | | — (258) | |
| Service paid to employees | | — | |
| Defined benefit obligation as of December 31, 20192022 | | 1782 | |
| Defined benefit obligation as of January 01, 2020 - as adjusted | | 1,181 | |
| Current service cost | | 181137 | |
| Interest cost on benefit obligation | | 1125 | |
| Actuarial losses / (gains) on obligation | | (255)— | |
| Past service costs | | (196)51 | |
| Service paid to employees | | — (18) | |
| Defined benefit obligation as of December 31, 20202023 | | 922978 | |
| Current service cost | | 154 | |
| Interest cost on benefit obligation | | 5 | |
| Actuarial losses / (gains) on obligation | | — | |
| Past service costs | | (216) | |
| Service paid to employees | | — | |
| Defined benefit obligation as of December 31, 2021 | | 864 | |
The amounts in the table above reflect the impact of the April 2021 IFRIC decision on IAS 19 related to Attributing Benefits to Periods of Service. This decision resulted in a reversal for €226 of the provision at January 1, 2020 mainly due to the retirement indemnities in France, which, before this decision, were spread out as of the arrival of the employee in the Company and now only start from the date from which each service year grants benefit entitlements.
The reduction of €255 related to the cost of past services/change in settlement terms recognized between January 1, 2020 and December 31, 2020 in connection with the settlement of the rights of those employees who left the Group, notably within the scope of the PSE.
Sensitivity of the Group’s retirement and post-employment benefits to a variation of the discount rate :rate:
| | | | | | | | | | | | | | | |
| | | Retirement and post-employment benefits |
| Sensitivity of the Group's retirement and post-employment benefits | | Changes in | | Impact / |
| to a variation of the discount rate | | assumptions / | | present value of |
| (in € thousands) | | discount rate | | the undertaking |
| + | | 0.25 | % | | (30) | |
| - | | 0.25 | % | | 31 | |
| | | | | | | | | | | | | | | |
| | | Retirement and post-employment benefits |
| Sensitivity of the Group's retirement and post-employment benefits | | Changes in | | Impact / |
| to a variation of the discount rate | | assumptions / | | present value of |
| (in € thousands) | | discount rate | | the undertaking |
| + | | 0.25 | % | | (28) | |
| - | | 0.25 | % | | 29 | |
The following assumed (undiscounted) benefit payments under the Company's French retirement indemnity are expected to be paid as follows:
| | | | | |
| 2024 | 0 |
| 2025 | 36 |
| 2026 | 0 |
| 2027 | 68 |
| 2028 | 62 |
| Years 2029 and thereafter | 3,035 |
18.26.EQUITY
Accounting policies
Share capital comprises ordinary shares and ordinary shares with double voting rights classified in equity. Costs directly attributable to the issue of ordinary shares or share options are recognized as a reduction in the share premium.
The Group has a liquidity agreement, contracted to an investment service provider. Purchases and sales of the Company's shares carried out under the contract are recognized directly in shareholders’ equity under treasury shares. See Note 18 - “Other financial assets”. Detailed breakdown
Share capital
| | | | | | | | | | | | | | | | | |
| Number of shares | | | | As of |
| | | | | 2022/12/31 | | 2023/12/31 |
| Ordinary shares issued (€0.25 par value per share) | | | | 49,834,983 | | 49,834,983 |
| Convertible preferred shares registered | | | | 0 | | 0 |
| Total shares issued | | | | 49,834,983 | | 49,834,983 |
| Less treasury shares | | | | 0 | | 0 |
| Outstanding shares | | | | 49,834,983 | | 49,834,983 |
Ordinary shares are classified under shareholders' equity. Any shareholder, regardless of nationality, whose shares are fully paid-in and registered for at least two years, is entitled to double voting rights under the conditions prescribed by law (Article 32 of the Company's bylaws).
At December 31, 2021, 2,347,639 shares have been held for more than two years and entitle their holders to double voting rights (4.71% of the issued share capital).
Changes in share capital in 20212023
On February 4, 2021, as a result of share conversion requests in January 2021, a capital increase of €759,327.25 has been recognized, corresponding to the creation of 3,037,309 new shares.
On March 2, 2021, as a result of share conversion requests in February 2021, a capital increase of €664,578.75 has been recognized, corresponding to the creation of 2,658,312 new shares.
On April 6, 2021, as a result of share conversion requests in March 2021, a capital increase of €297,812.50 has been recognized, corresponding to the creation of 1,191,250 new shares.
On September 1, 2021, as a result of share conversion requests in August 2021, a capital increase of €13,750 has been recognized, corresponding to the creation of 55,000 new shares.
The Chief Executive Officer, acting on a decision and delegation from the Board of Directors on December 16, 2021, recognized on December 22, 2021 the execution of a capital increase for the benefit of Ipsen Pharma SAS. 3,985,239 new shares were created (and €28 million was collected from Ipsen Pharma SAS) on this occasion. The share capital was increased accordingly.None.
At December 31, 2021, the total number of shares comprising the share capital, taking into account the above, was 49,815,489 shares.
At December 31, 2021, the share capital amounts to €12,453,872.25 represented by 49,815,489 fully authorized, subscribed and paid-up shares with a nominal value of €0.25 per share. This number does not include instruments granting access to share capital which have been issued by the Company and granted to certain directors, employees and consultants of the Group including stock options, free shares (AGA) that have not fully vested and share warrants (BSA) or the shares underlying our OCEANE convertible bonds.
At December 31, 2021,2023, the remaining unused authorizations to issue additional share-based compensation or other share-based instruments (stock options, free shares and share warrants) represent a total of 323,125525,000 shares.
Changes in share capital in 2020
On January 26, 2021, the Chief Executive Officer, acting on a decision and delegation from the Board of Directors on November 27, 2019, determined that some of the performance and attendance conditions of the AGA D 2017-2 and AGA D 2018 and all of the AGA S 2017-2 and AGA S 2018 free shares had been satisfied as of December 31, 2020. 29,762 free shares were thus definitively vested and the same number of new shares were created. The share capital was increased accordingly.
At December 31, 2020, the total number of shares comprising the share capital, taking into account the above, was 38,888,379 shares.
Changes in share capital in 2019
The Chairman and CEO, acting on a decision and delegation from the Board of Directors on March 13, 2019, decided on March 26, 2019, in accordance with the 17th and 18th resolutions of the Shareholders Meeting of June 15, 2018, to proceed with a capital increase by offering ordinary shares in the form of American Depositary Shares in the United States and a private placement of ordinary shares in Europe and other countries outside the United States. This transaction led to the issuance of 7,647,500 new shares representing a subscription of a gross amount of €137.6 million. Settlement-delivery took place on March 29, 2019 and the share capital has been increased accordingly. See note 2.1 “Initial Public Offering on the Nasdaq Global Select Market”.In addition, the Chief Executive Officer, acting on a decision and delegation from the Board of Directors on November 27, 2019, determined on December 16, 2019, with retroactive effect to December 15, 2019, that some of the performance and attendance conditions of the AGA D 2016-1 and AGA D 2016-2 and all of the AGA S 2016-2 free shares had been satisfied as of December 15, 2019. 7,796 free shares were thus definitively vested and the same number of new shares were created. The share capital was increased accordingly.
Finally, the Chief Executive Officer, acting on a decision and delegation from the Board of Directors on November 21, 2017, determined on January 2, 2020, with retroactive effect to December 31, 2019, that some of the performance and attendance conditions of the AGA D 2017-1 and all of the AGA S 2017-1 free shares has been satisfied. As a result, 19,400 free shares definitively vested and the same number of new shares were created and the share capital was increased accordingly.
At December 31, 2019, the total number of shares comprising the share capital, taking into account the above, was 38,858,617 shares.
Retained earnings (accumulated deficit)
In the present consolidated financial statements, the amount of "Retained earnings (accumulated deficit)" for 2020 reflect the April 2021 IFRIC decision on IAS 19 and IAS 38 (see Note 7 "Intangible Assets" and Note 17 "Employee Benefits"), resulting in an increase of €268 in the opening balance sheet for 2020.
19.OPERATING INCOME
In 2021, the total operating income amounted to €85,579 (€7,758 in 2020, and €40,961 in 2019).
The revenue amounted to €80,069 in 2021 (€765 in 2020, and €30,839 in 2019). It includes mainly the recognition of an upfront payment received from Ipsen in application of the licensing agreement signed in December 2021 for an amount of €80 million in 2021. The remainder of this upfront payment (€40 million) has been recognized in 2021 as deferred revenue and will be recognized as revenue throughout the execution of the double-blind period of the ELATIVE study, in accordance with IFRS 15. Other revenue recognized in 2021 relates to the license agreement with Labcorp for the deployment of NIS4 diagnostic technology in NASH.
In comparison, the revenue in 2020 mainly originated from the income generated by the license agreements with Labcorp and one-off revenue resulting from the sale of goods and services notably within the scope of the license and collaboration agreement with Terns Pharmaceuticals. The revenue in 2019 related to the the license transferred to Terns under the Terns licensing agreement.
Application of IFRS15 to the IPSEN License Agreement signed in 2021
Pursuant to IFRS 15, 27 ,28 and 29, we have identified that the agreement provides for 4 distinct performance obligations:
•The license for elafibranor,
•The completion of the ELATIVE Phase 3 trial until the end of the double-blind period,
•The knowledge transfer related to elafibranor, as well as support for Ipsen in future undertakings and processes, and
•The provision of drug tablets that may be needed by Ipsen to conduct their clinical trials.
The compensation under this agreement consists of an upfront payment, milestone payments, and royalties on future sales of elafibranor by Ipsen. Besides, it must be noted that, with respect to (i) support services other than the knowledge transfer and (ii) the provision of drug tablets, the agreement provides for separate prices covering all costs born by the Company to provide those goods and services, therefore constituting in each case an individual and distinct sale price for the relevant goods or service, which is not included in the aforementioned price elements.
We estimate the individual sale price of the clinical trial phase to be €40 million, including forecasted external costs, personnel expenses for the relevant staff, indirect costs pertaining to the work environment of such staff, augmented of a customary margin rate for CRO (Clinical Research Organization) contracting. This calculation of the individual sale price for the clinical trial phase reflects observable price conditions as recommended under IFRS 15.79.c. We used the same method to calculate the individual sale price of the knowledge transfer.
Regarding the calculation of the individual sale price of the license, we have analyzed recommended methods under IFRS 15.79 and determined that method (c) is the most relevant, considering in particular that the amount of this individual sale price is variable and partly uncertain. Thus, we applied the "residual" method, which stipulates that the individual sale price of the license corresponds to the difference between the total amount of the price and the individual sale prices of the knowledge transfer and the clinical trial phase. Moreover, referring to IFRS 15.B61, we determined that the date of transfer of control over the license corresponds to the date of the knowledge transfer, i.e. December 16, 2021, when key elements of the know-how were made available to Ipsen.
Regarding the recognition of revenue related to the license, we have chosen the following methods:
•The upfront payment, minus the portion of prices allocated to knowledge transfer services and clinical phase execution, has been recognized at the date of transfer of control, i.e. December 16, 2021 according to the above, as it is a static license (without implication or associated service provision);
•Milestone payments constitute variable and uncertain income, which would be, if applicable, recognized in revenue at the time they become highly probable, which means, in this case, due by Ipsen;
•Royalties would be progressively recognized in revenue as sales are completed by Ipsen, in accordance with the IFRS 15 exception for royalties constituting variable income.
Regarding the recognition of revenue related to the Phase 3 ELATIVE trial until the end of the double-blind period, we have chosen the following method:
• The part of the upfront payment allocated to this service will be recognized progressively as completion progresses.
Regarding the recognition of revenue related to the knowledge transfer, we have chosen the following method:
•The part of the upfront payment allocated to this service has been recognized on December 16, 2021 in accordance with the above.
It must be noted that the 8% equity purchase by Ipsen in the Company mentioned in note 2.4, under the terms of which Ipsen is represented in the Company's Board of Directors, has been completed on the basis of a subscription price agreed upon by the parties as representative of the value of GENFIT at the time, as we had secured future financing and created favorable conditions for the completion of the development and commercial launch of our main program. Therefore, the amount paid by Ipsen for its equity purchase does not interfere in the determination of the price of the licensing and collaboration agreement signed in December 2021 (including the Upfront Payment and other payments due for milestones identified above) and has been entirely recognized in the Group's equity.Application of IFRS15 to the TERNS PHARMACEUTICALS License Agreement signed in 2019
The Company identified 3 performance obligations under the license agreement with Terns:
•An exclusive license, with the right to sub-license, to develop, manufacture, distribute and promote elafibranor in NASH and PBC in Greater China;
•A transfer to Terns Pharmaceuticals of the Company’s Licensed Know-How and data regarding elafibranor and related support until the Marketing Authorization Application by Terns Pharmaceuticals; and
•Supply by the Company to Terns Pharmaceuticals of drug product to carry out its clinical trials in Greater China. The supply of drug product following the market authorization would be subject to a separate agreement if applicable.
Under the terms of the licensing agreement, the Company has received or could potentially receive:
•A $35 million non-refundable Upfront Payment payable within 10business days from June 24, 2019 upon the transfer of the existing Company’s Licensed Know-How. This Upfront Payment was received on July 3, 2019;
•Development Milestone Payments upon the achievement of the development milestones for the licensed product;
•Commercial Milestone Payments upon the achievement of commercial milestones depending on reaching certain aggregate thresholds;
•Mid-teen percentage Royalties based on sales by Terns Pharmaceuticals in Greater China; and
•Compensation for the supply of drug product for the clinical trials on a cost-plus basis.
The potential Development and Commercial Milestone payments may represent up to $193 million.
Under IFRS 15, the allocation and recognition of revenue was determined as follows based on the fair value of each of the performance obligations:
•The $35 million upfront payment was allocated to the license and the transfer of the existing know-how and related support to Terns Pharmaceuticals based on an estimate of the latter measured as the maximum estimated value to be incurred by the Company’s employees and management for the support given to Terns Pharmaceuticals. On this basis, $34.9 million was recognized as revenue in 2019 and $0.1 million was deferred to future periods. No such revenue was recognized in 2020 and 2021.
•Development and Commercial Milestones Payments whose payment depends on the achievement of certain scientific, regulatory or commercial objectives, as provided in the contract, are variable compensation that will be recognized as revenue when the milestones are met. No amounts were recognized in 2019, 2020 and 2021.
•Royalties on commercial sales by Terns Pharmaceuticals will be recognized as revenue pursuant to information given to the Company by Terns Pharmaceuticals, under the terms and timeframes set out in the agreement.No amounts were recognized in 2019, 2020 and 2021.
•Revenue on Supply for drug product will be recognized based on the delivery of drug product to Terns Pharmaceuticals. No amounts were recognized in 2019 and 2021. Revenue in 2020 was immaterial.
As part of this agreement, Genfit and Terns Pharmaceuticals will also undertake joint research and development projects in liver disease, including the development of elafibranor in combination with Terns Pharmaceuticals’ proprietary compounds. This collaboration agreement is only potential at the date of signing the license agreement and does not yet constitute a reciprocal commitment at December 31, 2020. It therefore has no accounting impact at this time.
This contract contains several delivery obligations. As a result, the Company has ensured, as required by IFRS 15, that the revenue allocation of the transaction corresponds to the fair value of each obligation.
Other Income
Other income consisted of the following:
| | | | | | | | | | | | | | | | | | | | | |
| Other income | | Year ended |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| CIR tax credit | | 8,125 | | | 6,020 | | | 5,282 | |
| Other operating income | | 1,992 | | | 968 | | | 223 | |
| Government grants and subsidies | | 5 | | | 5 | | | 5 | |
| TOTAL | | 10,122 | | | 6,993 | | | 5,510 | |
The research tax credit (CIR) amounted to €5,282 in 2021 (€6,020 in 2020), due to the reduction in research and development expenses.
In comparison, the 2020 Research Tax Credit amounted to €7,911, partially balanced with the expense amounting to €1,892corresponding to the resolution of the dispute on the 2010, 2011, 2012 and 2014 Research Tax Credit.
During 2021, the Group recognized €223 in “Other operating income” (€968 in 2020 and €1,992 in 2019), mainly comprised of exchange gains on trade receivables.
20.OPERATING EXPENSES
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Operating expenses and other operating income (expenses) | | Year ended | | Of which : |
| | | 2019/12/31 | | Raw | | Contracted | | Employee | | Other | | Depreciation, | | Gain / |
| | | | | materials | | research and | | expenses | | expenses | | amortization | | (loss) on |
| | | | | and | | development | | | | (maintenance, | | and | | disposal of |
| | | | | consumables | | activities | | | | fees, travel, | | impairment | | property, |
| | | | | used | | conducted by | | | | taxes…) | | charges | | plant and |
| (in € thousands) | | | | | | third parties | | | | | | | | equipment |
| Research and development expenses | | (66,170) | | | (2,017) | | | (41,509) | | | (11,740) | | | (6,188) | | | (4,716) | | | — | |
| General and administrative expenses | | (17,265) | | | (177) | | | (59) | | | (7,598) | | | (8,972) | | | (458) | | | — | |
| Marketing and market access expenses | | (13,708) | | | (8) | | | — | | | (1,645) | | | (11,979) | | | (76) | | | — | |
| Reorganization and restructuring expenses | | — | | | — | | | — | | | — | | | — | | | — | | | — | |
| Other operating income (expenses) | | (1,649) | | | — | | | — | | | — | | | (1,668) | | | — | | | 19 | |
| TOTAL | | (98,793) | | | (2,202) | | | (41,568) | | | (20,984) | | | (28,807) | | | (5,251) | | | 19 | |
| (*) : including reversals | | | | | | | | | | | | | | |
The depreciation, amortization and impairment charges totaled €4.7 million, consists of a provision of €1.8 million with respect to the research tax credit litigation and due to additional depreciation due to the adoption of IFRS 16. The reversal of this provision of €1.8 million with respect to the research tax credit litigation was booked in 2020 (see above ).
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Operating expenses and other operating income (expenses) | | Year ended | | Of which : |
| | | 2020/12/31 | | Raw | | Contracted | | Employee | | Other | | Depreciation, | | Gain / |
| | | | | materials | | research and | | expenses | | expenses | | amortization | | (loss) on |
| | | | | and | | development | | | | (maintenance, | | and | | disposal of |
| | | | | consumables | | activities | | | | fees, travel, | | impairment | | property, |
| | | | | used | | conducted by | | | | taxes…) | | charges | | plant and |
| (in € thousands) | | | | | | third parties | | | | | | | | equipment |
| Research and development expenses | | (59,097) | | | (1,876) | | | (39,216) | | | (11,554) | | | (5,465) | | | (985) | | | — | |
| General and administrative expenses | | (14,270) | | | (202) | | | (92) | | | (6,936) | | | (6,545) | | | (495) | | | — | |
| Marketing and market access expenses | | (11,216) | | | (7) | | | (2) | | | (1,298) | | | (9,818) | | | (90) | | | — | |
| Reorganization and restructuring expenses | | (5,308) | | | — | | | — | | | 8 | | | (2,141) | | | (3,175) | | | — | |
| Other operating income (expenses) | | (764) | | | — | | | — | | | — | | | (684) | | | — | | | (80) | |
| TOTAL | | (90,655) | | | (2,085) | | | (39,310) | | | (19,779) | | | (24,655) | | | (4,746) | | | (80) | |
| (*) : including reversals | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Operating expenses and other operating income (expenses) | | Year ended | | Of which : |
| | | 2021/12/31 | | Raw | | Contracted | | Employee | | Other | | Depreciation, | | Gain / |
| | | | | materials | | research and | | expenses | | expenses | | amortization | | (loss) on |
| | | | | and | | development | | | | (maintenance, | | and | | disposal of |
| | | | | consumables | | activities | | | | fees, travel, | | impairment | | property, |
| | | | | used | | conducted by | | | | taxes…) | | charges | | plant and |
| (in € thousands) | | | | | | third parties | | | | | | | | equipment |
| Research and development expenses | | (35,166) | | | (1,305) | | | (18,808) | | | (8,192) | | | (4,593) | | | (2,247) | | | (19) | |
| General and administrative expenses | | (16,153) | | | (161) | | | (85) | | | (7,379) | | | (8,003) | | | (541) | | | 15 | |
| Marketing and market access expenses | | (1,539) | | | (1) | | | (1) | | | (783) | | | (741) | | | (13) | | | — | |
| Reorganization and restructuring expenses | | (142) | | | (5) | | | — | | | — | | | (2,343) | | | 2,206 | | | — | |
| Other operating income (expenses) | | (763) | | | — | | | — | | | — | | | (338) | | | 4 | | | (429) | |
| TOTAL | | (53,763) | | | (1,472) | | | (18,895) | | | (16,354) | | | (16,019) | | | (591) | | | (433) | |
| (*) : including reversals | | | | | | | | | | | | | | |
Research and development expenses at each reporting date take into account estimates for ongoing activities subcontracted as part of the clinical trials and not yet invoiced, on the basis of detailed information provided by subcontractors and reviewed by the Group’s internal departments. The accuracy of these estimates for some types of expenses improves with the progression of the trials and the review of their determination methods. As a reminder: for regulatory reasons, research services for clinical trials and the production of active ingredients and therapeutic units are contracted out to third parties.
The decrease in "Employee expenses" mainly reflects:
•The reduced full-year headcount between 2020 and 2021, with an average headcounts decrease from 193 in 2020 to 122 in 2021 and the reduction in share-based compensation (BSA, BSAAR, SO and AGA) with no impact on the cash flow at €1,236 in 2020 and €470 in 2021,
• Partially compensated by the evolution of employee profiles and the profit sharing granted to employees in 2021 for a total amount of €628.
The "Other expenses mainly include:
•Legal fees, audit and accounting fees;
•Advisor fees (banking, press relations, communication, IT, market access, marketing, scientific advising);
•Intellectual property expenses, including in particular the charges and fees incurred by the Company for patent applications and maintenance;
•Expenses related to insurance, notably those triggered by the Company listing on the Nasdaq since 2019;
•Expenses related to the rental, use, and maintenance of the Group's premises;
•Expenses related to external personnel contracted out to the company (safety and security, front desk, clinical and IT services);
•Expenses related to travel and conferences, including mainly employee travel costs as well as scientific, medical, financial and business development conference registration fees.
The change in “Other Expenses” was mainly due to the reduction in marketing and market access expenses following the cessation of the pre-marketing of elafibranor in NASH.
The change in “Net amortization, depreciation and provisions” resulted notably from:
•Reversals in 2021 of impairments previously recognized in 2020 following the termination of RESOLVE-IT: These reversals are notably related to (i) the reorganization of vacant premises (reversal of €679 following the termination of the lease agreement and the relocation from the Company offices in Paris in 2021), (ii) some leased equipment (reversal of €441 following their sale in 2021), and (iii) some scientific and IT equipment (reversal of €374); and
•Reversals in 2021 of provisions previously recognized in 2020 related to (i) training and support commitments by the Company within the scope of the reduction in force plan (PSE) signed in October 2020 (reversal of €352 following the partial expiration of these commitments in 2021), and (iii) some administrative costs and costs related to the destruction of drug tablets following the termination of the RESOLVE-IT study (reversal of €366).
The above reversals are categorized as "Reorganization and restructuring costs".
As a reminder, in 2020, the "Net amortization, depreciation and provisions" included the reversal of the provision, which was previously recognized in 2019 related to the 2010, 2011, 2012 and 2014 research tax credit dispute, following the settlement of this dispute in 2020.
Employee expenses
Employee expenses and number of employees were as follows:
| | | | | | | | | | | | | | | | | | | | | |
| Employee expenses | | Year ended |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Wages and salaries | | (14,018) | | | (13,570) | | | (10,328) | |
| Social security costs | | (5,171) | | | (5,047) | | | (4,775) | |
| Changes in pension provision | | (138) | | | 74 | | | (154) | |
| Employee profit-sharing | | — | | | — | | | (628) | |
| Share-based compensation | | (1,657) | | | (1,236) | | | (470) | |
| TOTAL | | (20,984) | | | (19,779) | | | (16,354) | |
| | | | | | | | | | | | | | | | | | | | | |
| Number of employees at year-end - detail | | Year ended |
| | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Average number of employees | | 175 | | 193 | | 122 |
| Number of employees | | | | | | |
| Research and development | | 108 | | 66 | | 55 |
| Services related to research and development | | 19 | | 16 | | 18 |
| Administration and management | | 60 | | 43 | | 47 |
| Marketing and commercial | | 7 | | 5 | | 2 |
| TOTAL | | 194 | | | 130 | | | 122 | |
| Number of employees | | | | | | |
| Senior staff | | 144 | | 105 | | 97 |
| Staff | | 45 | | 23 | | 21 |
| Others (apprentices) | | 5 | | 2 | | 4 |
| TOTAL | | 194 | | 130 | | 122 |
| Number of employees | | | | | | |
| Male | | 78 | | 52 | | 45 |
| Female | | 116 | | 78 | | 77 |
| TOTAL | | 194 | | 130 | | 122 |
The decrease in employee expenses resulted mainly from lower salaries and social security costs due to the reduction of the average headcount from 193 in 2020 down to 122 in 2021, which was partially compensated by the evolution of employee profiles.
The decrease in share-based compensation (BSA, BSAAR, SO and AGA with no impact on the cash flow) reflected the reduction of the workforce, coupled with the decreasing expenses recognized for the 2016-2019 AGA and SO plans (see Note 21 "Share-Based Compensation").As the Company recorded a net profit in 2021, it granted a profit-sharing plan to its employees in accordance with the French Law, totaling €628 (payable in the first helf of 2022).
21.SHARE-BASED COMPENSATION
Share-based compensation is granted by the Group to employees, executive officers, board members and consultants.
Share-based compensation granted to employees and executive officers in 2014 through 2020 corresponds to redeemable share warrants ("Bons de Souscriptions et/ou d'Acquisition d'Actions" or "BSAAR"), stock options ("SO") and free shares ("actions gratuites" or "AGA")
Share-based compensation granted to board members and consultants in 2014, 2015, 2017 and 2019 corresponds to share warrants ("Bons de Souscriptions d'Actions" or "BSA").
For the measurement of this share-based compensation, the Group has determined that under IFRS its consultants were not equivalent to employees.
Under these programs, holders of vested instruments are entitled to subscribe to shares of the Company at a pre-determined exercise price. All of the plans are equity settled.
No instruments were exercised during 2021, 2020, and 2019.
In 2021, only SO and AGA plans were granted as share-based compensation, the terms and conditions of which are discussed below.
The expense recognized during 2021 pursuant to IFRS 2 was €470 (compared to €1,236 at December 31, 2020 and €1,656 at December 31, 2019).
In 2019, the Group revised its estimate of the number of equity instruments expected to be vested taking into account the number of lapsed instruments noted after 4 years of successive plans. As a result, Genfit revised the turnover rate assumption, which was estimated at 15%, to a rate of 0%, taking into account recent observations and the actual number of lapsed instruments at each closing.
The table below shows the share-based compensation under each plan according to the change in estimate mentioned above .
| | | | | | | | | | | | | | | | | | | | | |
| Share-based compensation - expense | | Year ended |
| | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| AGA S 2016-1 | | — | | | — | | | — | |
| AGA S 2016-2 | | 44 | | | — | | | — | |
| AGA D 2016-1 | | 21 | | | 21 | | | — | |
| AGA D 2016-2 | | 39 | | | 6 | | | — | |
| SO 2016-1 | | 213 | | | 49 | | | — | |
| SO 2016-2 | | 93 | | | 13 | | | — | |
| SO US 2016-1 | | (24) | | | — | | | — | |
| SO US 2016-2 | | (11) | | | — | | | — | |
| AGA S 2017-1 | | 209 | | | — | | | — | |
| AGA S 2017-2 | | 45 | | | 13 | | | — | |
| AGA D 2017-1 | | 190 | | | — | | | — | |
| AGA D 2017-2 | | 56 | | | 4 | | | — | |
| SO 2017-1 | | 27 | | | 335 | | | — | |
| SO 2017-2 | | 2 | | | 110 | | | — | |
| SO US 2017-1 | | (4) | | | — | | | — | |
| SO US 2017-2 | | (6) | | | — | | | — | |
| BSA-2017-A | | — | | | — | | | — | |
| BSA-2017-B | | — | | | — | | | — | |
| AGA S 2018 | | 148 | | | 62 | | | — | |
| AGA D 2018 | | 135 | | | 65 | | | — | |
| SO 2018 | | 285 | | | 225 | | | 186 | |
| SO US 2018 | | 25 | | | 24 | | | 24 | |
| AGA S 2019 | | 41 | | | 55 | | | 39 | |
| AGA D 2019 | | 35 | | | 63 | | | 16 | |
| SO 2019 | | 70 | | | 123 | | | 105 | |
| SO 2019 - US | | 16 | | | 35 | | | (11) | |
| BSA 2019 | | 7 | | | 20 | | | — | |
| SO US 2019 | | 1 | | | 14 | | | (7) | |
| SO D 2020 | | — | | | — | | | 14 | |
| SO C 2020 | | — | | | — | | | 40 | |
| SO US 2020 | | — | | | — | | | 19 | |
| AGA S 2021 | | — | | | — | | | 29 | |
| AGA D 2021 | | — | | | — | | | 5 | |
| SO D 2021 | | — | | | — | | | 2 | |
| SO C2021 | | — | | | — | | | 9 | |
| SO US 2021 | | — | | | — | | | 2 | |
| TOTAL | | 1,656 | | | 1,236 | | | 470 | |
21.1. Share warrants ( bons de souscription d'actions or BSA)
The key terms and conditions related to each program are detailed in the following tables:
| | | | | | | | | | | | | | | | | | | | | | | |
| Share-based compensation | 2019 | 2017 | 2015 | 2014 |
| Share warrants ( BSA ) |
| | | BSA 2017-A | BSA 2017-B | BSA 2015-A | BSA 2015-B | BSA 2014-A | BSA 2014-B |
| Date of the Shareholders meeting | 06/15/2018 | 06/16/2017 | 04/02/2014 | 04/02/2014 |
| Date of the Management Board meeting | | | 01/09/2015 | 07/24/2014 |
| Date of the decision and delegation of the Board of Directors to the CEO | 10/18/2019 | 11/21/2017 | | |
| Date of the CEO decision | 10/31/2019 | 12/6/2017 | | |
| Beneficiaries | Consultants | Consultants and officers | Consultants and officers | Consultants and officers |
| Total number of BSA subscribed | 35,070 | 18,345 | 18,345 | 12,860 | 12,860 | 46,765 | 46,765 |
| Total number of BSA voided | 0 | 0 | 0 | 12,860 | 12,860 | 46,765 | 46,765 |
| Total number of BSA remaining | 35,070 | 18,345 | 18,345 | 0 | 0 | 0 | 0 |
| Issue Price | €1.23 | €2.00 | €0.01 | €0.01 |
| Exercise price | €12.32 | €19.97 | €35.95 | €23.50 |
| Estimated fair value - according to IFRS 2 | €0.75 | €3.78 | €3.81 | €25.33 /€26.89 | €25.33 /€26.31 | €15.61/€24.84 | €15.61/€24.85 |
| End of exercise period | 05/31/2024 | 06/30/2022 | 07/15/2022 | 05/31/2019 | 11/30/2019 | 09/30/2018 | 02/28/2019 |
| Valuation method used | Black & Scholes |
| Expected dividends | 0% | 0% | 0% | 0% |
| Expected volatility | 40.0% | 36.4% | 35.7% | 74.9% | 74.9% |
| Risk-free interest rate | 0% | 0.0% | 0.4% | 0.4% |
| Expected life | 0.7 years | 6 years | 4 years | 4 years |
The services performed by the consultants are mainly:
•to evaluate product development plans and propose, if necessary, changes to strategic or technical approaches;
•to advise the Company's management and the Scientific Board in identifying strategies and selecting drug candidates, based in particular on the scientific results obtained by the Group (new therapeutic targets, new compounds); and
•to assist and advise the Group in its alliance strategies, such as external growth-supporting synergies (acquisition of new competencies and the purchase of operating rights, drug candidates and innovative technologies, etc.).
•
21.2. Redeemable warrants ( bons de souscription et/ou d'acquisition d'actions remboursables or BSAAR)
At December 31, 2021,, all of the BSAAR issued by the Company in 2016 became void without being exercised.
21.3. Free shares ( actions gratuites attribuées or AGA)
The key terms and conditions related to each program are detailed in the following tables:
| | | | | | | | | | | | | | | | | | | | | | | |
| Share-based compensation | 2019 | 2019 | 2018 | 2017 | 2016 |
| Free shares (AGA) | AGA D | AGA S | | | |
| | Officers(1) | Employees | Officers(1) | Employees | AGA D and S | AGA D and S 2017-1 and 2017-2 | AGA D and S
2016-1 and 2016-2 |
| | | | | |
| Date of the Shareholders meeting | 06/15/2018 | 06/15/2018 | 06/16/2017 | 06/21/2016 |
Date of the Management Board
meeting | | | | 12/15/2016 |
| Date of the decision and delegation of the Board of Directors to the CEO | 07/18/2019 | 11/07/2018 | 11/22/2017 | |
Date of the Executive
Board Meeting/CEO decision | 07/18/2019 | 11/22/2018 | 12/06/2017 | |
| Total number of AGA subscribed | 3,000 | 16,070 | 0 | 17,556 | 35,800 | 41,196 | 30,709 |
| Total number of AGA voided | 0 | 3,360 | 0 | 5,226 | 14,059 | 13,775 | 5,429 |
Total number of AGA definitively
vested | 0 | 0 | 0 | 0 | 21,741 | 27,421 | 25,280 |
| Total number of AGA remaining | 3,000 | 12,710 | 0 | 12,330 | — | — | 0 |
| Vesting period | From 07/18/2019 to 09/16/2022 | From 07/18/2019
to 09/16/2022 | From 12/06/2017
to 12/31/2020 | From 12/15/2016
to 12/15/2019 |
| Valuation method used | Monte Carlo |
Price of the share at the time
of allocation | €17.06 | €20.02 | €21.95 | €20.78 |
| Expected dividends | 0% | 0% | 0% | 0% |
| Expected volatility | 40.2% | 38.0% | 53.7% | 63.0% |
| Risk-free interest rate | 0.0% | 0.0% | 0.0% | 0.0% |
| Turnover rate | 0.00% | 15.00% | 15.00% | 15.00% |
| (1) : Chairman and CEO | | | | | | | |
| | | | | | | | |
Share-based compensation
Free shares(AGA) | 2021 |
| AGA S | AGA D |
| Employees | Officers (1) |
| Date of the Shareholders' Meeting | 11/27/2019 | 11/27/2019 |
| Date of the decision and delegation of the Board of Directors to the CEO | 02//26/2021 | 03/17/2021 |
| Date of the CEO decision | 03/30/2021 | 0 |
| Total number of AGA subscribed | 32,400 | 15,000 |
| Total number of AGA voided | 3,400 | 0 |
Total number of AGA definitively
vested | 0 | 0 |
| Total number of AGA remaining | 29,000 | 15,000 |
| Vesting period | From 03/30/2021 to 03/31/2024 | From 03/17/2021 to 03/31/2024 |
| Valuation method used | Monte Carlo | Monte Carlo |
Price of the share at the time
of allocation | 4.00 € | 4.15 € |
| Expected dividends | 0 % | 0% |
| Expected volatility | 51 | % | 51 | % |
| Risk-free interest rate | -0.59% | -0.59% |
| Turnover rate | 0.0 % | 0.0% |
| (1) Chief Executive Officer. |
The final allocation of free shares is subject to continued employment with the Group and performance conditions.
21.4. Stock options (options de souscription d'actions or SO)
The key terms and conditions related to each program are detailed in the following tables:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | |
| Share-based compensation | 2020 | 2019 | 2018 | 2017 | 2016 |
| Stock option SO | SO | SO US | SO 2019 | SO US 1 | SO US 2 | SO 2018 | SO US 2018 | SO 1 and 2 2017 | SO US 2017 | SO 1 and 2 2016 | SO US 2016 |
| | Officers (1) | Employees | Employees | Employees and Officers | Employees | Employees | Employees and Officers | Employees | Employees and Offices | Employees | Employees and Offices | Employees |
| |
| Date of the Shareholders meeting | 11/27/2019 | 06/15/18 | 11/27/2019 | 06/15/18 | 06/16/17 | 06/21/16 |
| Date of the Management Board meeting | | | | | | | 12/15/2016 |
| Date of the decision and delegation of the Board of Directors to the CEO | 12/11/2020 | 07/18/19 | | 11/27/2019 | 11/7/2018 | 11/21/2017 | |
| Date of the CEO decision | 12/11/2020 | 07/18/19 | | 11/27/2019 | 11/7/2018 | 12/6/2017 | |
| Total number of SO subscribed | 35,000 | 103,750 | 56,250 | 107,880 | 30,620 | 13,350 | 122,000 | 17,500 | 96,250 | 13,000 | 62,875 | 10,500 |
| Total number of SO voided | 0 | 0 | 0 | 13,350 | 7,000 | 4,450 | 50,322 | 7,787 | 35,273 | 13,000 | 13,169 | 10,500 |
| Total number of SO definitively vested | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 60,977 | 0 | 49,706 | 0 |
| Total number of SO remaining | 35,000 | 103,750 | 48,750 | 94,530 | 23,620 | 8,900 | 71,678 | 9,713 | 0 | 0 | 0 | 0 |
| Exercise price | €4.38 | €3.50 | €4.52 | €13.99 | €16.90 | €14.31 | €16.00 | €21.65 | €17.91 | €22.54 | €15.79 | €21.12 |
| Vesting period | From 12/31/2020 to 12/31/2023 | From 07/18/2019 to 09/16/2022 and From 11/27/2019 to 01/16/2023 | From 11/07/2018 to 12/31/2021 | From 12/06/2017 to 12/31/2020 | From 12/15/2016 to 12/15/2019 |
| Exercice period | From 01/01/2024 to 12/31/2027 | From 09/17/2022 to 09/17/2029 and From 01/17/2023 to 01/17/2030 | From 01/01/2022 to 12/31/2028 | From 01/01/2021 to 12/31/2027 | From 12/16/2019 to 12/16/2026 |
| Fair value | €1.16 | €1.46 | 1.12 | €4.59 | €3.67 | €3.23 | €9.32 | €6.90 | €9.32 | €10.30 | €8.52 |
| Valuation method used | Black-Scholes |
| Price of the share at the time of allocation | €3.99 | €3.99 | €17.06 | | €14.50 | €22.12 | €21.95 | €20.79 |
| Expected dividends | 0% | 0% | 0% | | 0% | 0% | 0% | 0% |
| Expected volatility | 49.0% | 49.0% | | | 40.0% | 44.1% | 53.7% | 63.0% |
| Risk-free interest rate | -0.7% | -0.7% | 0.0% | | 0.0% | 0.0% | 0.0% | 0.0% |
| Turnover rate | 0.00% | 0.00% | 0.00% | | 0.00% | 15.00% | 15.00% | 15.00% |
| | | | | | | | | | | |
| 2021 |
| SO | SO US |
Share-based compensation
| | | |
| Stock Options (SO) | Officers (1) | Employees | Employees |
| Date of the Shareholders' Meeting | 06/30/2021 |
| Date of the decision and delegation of the Board of Directors to the CEO | 10/18/2021 |
| Date of the CEO Decision | 10/19/2021 |
| Total number of SO subscribed | 35000 | 134375 | 32500 |
| Total number of SO voided | 0 | 0 | 7500 |
| Total number of SO definitely vested | 0 | 0 | 0 |
| Total number of SO remaining | 35000 | 134375 | 25000 |
| Exercise price | €3.26 | €2.61 | €3.22 |
| Vesting period | From 10/20/2021 to 10/20/2024 |
| Exercise period | From 10/21/2024 to 10/21/2031 |
| Fair value | 1.06 | 1.30 | 1.07 |
| Valuation method used | Black-Scholes |
| Price of the share (€) at the time of allocation | 3.24 | 3.24 | 3.24 |
| Expected dividends | 0.00 | 0.00 |
| Expected volatility | 50 | % | 50 | % |
| Risk-free interest rate | (0.59) | % | (0.59) | % |
| Turnover rate | 0.00 | 0.00 |
(1)Chief Executive Officer
Volatility assumptions in the above tables are determined by reference to the Company's historical share price observed on the grant date over a two- and three-year period prior to the grant date, adjusted for extreme variations, if any.
Definitive vesting is subject to continued employment with the Group and performance conditions.
21.5.Performance conditions
The SO and SO US stock option plans as well as certain free share plans (AGA "D") implemented in 2016, 2017, 2018 and 2019 are subject to internal performance conditions related to the progress of the Group's research and development programs, and to external performance conditions related to the evolution of the Company's stock price.
The other free share plans (AGA "S") and SO and AGA plans implemented starting in 2020 are subject only to internal performance conditions.
21.5.1. Performance conditions of the 2021 plans
| | | | | | | | |
Plans | Evaluation date for performance conditions | Nature of internal conditions |
SO C 2021
SO D 2021
SO US 2021 | 10/20/2024 | a.50% of the Stock Options will be exercisable if at least one of the following three conditions relating to the development of elafibranor in PBC and to the ELATIVE clinical trial is fulfilled:
i.ELATIVE top-line results are released to the market before or during the second quarter of 2023;
ii.a new drug application is filed for elafibranor in PBC with the Food and Drug Administration (FDA) or the European Medicines Agency (EMA) in the second half of 2023 or before;
iii.elafibranor is approved by a regulatory authority in 2024.
b.15% of the Stock Options will be exercisable if at least one of the following two conditions relating to the development of NTZ and the ACLF franchise is fulfilled:
i.a phase 2 clinical study or a more advanced clinical study evaluating NTZ is in ongoing or was carried out;
ii.the Company develops or acquires the rights to a new molecule (including through repositioning) for development in ACLF.
a.15% of the Stock Options will be exercisable if at least one of the following two conditions relating to the NIS4 diagnostic technology is fulfilled:
i.if a research and development partnership agreement relating to the implementation of the NIS4 diagnostic technology into an IVD test with at least one major NASH player (“big pharma”, biotech company, institution, etc.) is entered into by the Company;
ii.Labcorp’s NASHnext LDT is reimbursed by at least three payers in the United States (insurance, integrated system, etc).
a.20% of the Stock Options will be exercisable if at least one of the following two conditions relating to the development of the product pipeline of the Company is fulfilled:
i.At least one new molecule (excluding elafibranor and NTZ) is developed by the Company or the Company has acquired development rights to a new molecule outside of the ACLF franchise (performance already covered by b(ii) above);
ii.At least two phase 2 clinical studies or more advanced clinical studies are ongoing or have been completed ; not including a phase 2 clinical study or more advanced clinical study in NTZ (performance already covered by b(i) above).
|
| | | | | | | | |
| | |
Plans | Evaluation date for performance conditions | Nature of internal conditions |
AGA D 2021
AGA S 2021
| 3/31/2024 | 50% of the 2021 free shares will vest if at least one of the three following conditions regarding development of elafibranor in PBC and the ELATIVE trial is met:
(i) « Last Patient Visit »of ELATIVE in the 4th quarter 2022 or before;
(ii) results of ELATIVE are communicated to the market before or during the first half 2023 ;
(iii) the filing of an marketing authorization application for elafibranor in PBC with the FDA or EMA in 2023.
|
| 25% of the free shares will vest if at least one of the two following conditions related to NIS4 diagnostic technology are met :
(i) an R&D agreement for an IVD test using NIS4 technology is signed with at least one major NASH player (big pharma, biotech, institution, etc) ;
(ii) use of NIS4 technology in at least 20 clinical studies.
|
25% of the free shares will vest if at least one of the two following conditions related to the Company's product pipeline is met: :
(i) a clinical study in a new indication with elafibranor or NTZ is underway or was completed ;
(ii) development or in-licensing or acquisition of rights to a new compound by the Company.
|
| | |
22.FINANCIAL INCOME AND EXPENSES
| | | | | | | | | | | | | | | | | | | | | |
| Financial income and expenses | | Year ended |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| | | | | | | |
| | | | | | | |
| Interest income | | 2,626 | | 1,442 | | 274 |
| Foreign exchange gain | | 2,272 | | 4,983 | | 8,876 |
| Financial income occured by renegotiating the convertible bond debt OCEANE | | 0 | | 0 | | 35,578 |
| Other financial income | | 324 | | 119 | | 52 |
| TOTAL - Financial income | | 5,221 | | 6,544 | | 44,780 |
| Financial expenses | | | | | | |
| Interest expenses | | (11,289) | | (11,643) | | (4,846) |
| Interest expenses for leases | | (148) | | (134) | | (109) |
| Foreign exchange losses | | (1,657) | | (13,508) | | (2,163) |
| Other financial expenses | | (17) | | (11) | | (5) |
| TOTAL - Financial expenses | | (13,110) | | (25,296) | | (7,122) |
| FINANCIAL GAIN (LOSS) | | (7,889) | | (18,752) | | 37,658 |
Financial income includes notably the one-time buyback bonus of €35.6 million issued from the renegotiation of the OCEANEs completed in January 2021.
Interest income recognized is almost exclusively related to investments in US dollars, of which the return has decreased in 2021.
The financial expenses are related to the interest of the OCEANE and they mainly relate to the payment of coupons at the rate of 3.5% and the amortization of the discount of the bond debt at the effective interest rate of 8.8% to accrete the bond debt up to the amount that will be repaid (or converted) at maturity, recognizing a theoretical annual interest accrual as a result of the accretion on the period of an amount equivalent to the equity component at an effective interest rate.
The portion of financial gain related to currency exchange is a gain of €6,713 in 2021 notably due to the difference in currency exchange recognized on the cash investments in US dollars, as GENFIT has decided to keep some of its cash in US dollars. See Note 6 “Cash and cash equivalents”. These cash investments in US dollars are to be used to pay directly expenses in US dollars (natural currency hedge).
23.INCOME TAX
For 2021, the corporate income tax payable of the Parent company GENFIT SA amounted to €5,051, which is recognized as "Other current tax liabilities" in the consolidated financial statements.
It is of note that we benefited from a reduced tax rate on part of the income from the licensing agreement signed with Ipsen pursuant to Article 238 of the French Tax Code.
The determination of the income tax expense recognized in the consolidated financial statements, which amounted to €2,215 for 2021, is summarized in the table "Effective tax rate" hereunder.
Breakdown of deferred taxes by nature
| | | | | | | | | | | | | | | | | | |
| | | As of | Impact on | Impact on the | As of |
| (in € thousands) | | 12/31/2019 | equity | profit/loss | 12/31/2020 |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| Deferred tax liabilities | | (3,182) | — | 1,132 | (2,050) |
| Deferred tax assets | | 1,988 | — | (706) | 1,282 |
| TOTAL | | (1,193) | — | 426 | (768) |
| | | | | | | | | | | | | | | | | | |
| | | As of | Impact on | Impact on the | As of |
| (in € thousands) | | 12/31/2020 | equity | profit/loss | 12/31/2021 |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| Deferred tax liabilities | | (2,049) | (2,721) | 2,455 | (2,314) |
| Deferred tax assets | | 1,282 | — | 430 | 1,712 |
| TOTAL | | (767) | (2,721) | 2,885 | (602) |
Effective tax rate
| | | | | | | | | | | | | | | |
| | | Year ended |
| (in € thousands) | | 2019/12/31 | 2020/12/31 | 2021/12/31 |
| Profit (loss) for the period | | (65,144) | (101,221) | 67,259 |
| Tax gain (expense) | | 576 | 428 | (2,215) |
| Profit (loss) for the period before taxes | | (65,721) | (101,649) | 69,474 |
| Tax rate in France | | 32.02 | % | 28.92 | % | 27.37 | % |
| Theoretical group tax rate in France | | 21,046 | 29,401 | (19,018) |
| Increase / decrease in tax benefit arising from : | | | | |
| Tax credits | | 2,588 | 1,739 | 1,512 |
| Permanent differences | | 3,341 | (404) | 833 |
| Differences between rates | | 125 | 172 | 7,323 |
| Tax losses for the period, unrecognised as deferred tax assets | | (25,274) | (28,603) | 0 |
| Utilisation of previously unrecognised tax losses | | 0 | 0 | 5,590 |
| IFRS adjustments without tax incidence | | (530) | (358) | (129) |
| Non recognition of deffered tax assets related to temporary differences | | (114) | (775) | (24) |
| Recognition of deferred tax assets against deferred tax liabilities | | (889) | (706) | 430 |
| Tax effects related to the renegociation of the convertible debt | | 0 | 0 | 1,370 |
| Others | | 284 | (39) | (102) |
| | | | | |
| Income tax expense recognised in profit or loss | | 576 | 428 | (2,215) |
| Effective income rate | | (0.88) | % | (0.42) | % | (3.19) | % |
We are subject to a tax audit by the French revenue service on our tax returns or operations subject to review on the 2019 and 2020 periods (including the Research Tax Credit claimed for these periods), which started on December 10, 2021 and is still ongoing at the date of this document.
23.1.Losses available for offsetting against future taxable income
At December 31, 2019, 2020 and 2021, the tax loss carry forwards for the Company amounted to €384,471, €483,356 and €450,679, respectively.
Such carry forwards can be offset against future taxable profit within a limit of €1.0 million per year plus 50% of the profit exceeding this limit. Remaining unused losses will continue to be carried forward indefinitely.
In 2021, the amount of tax loss carry forwards used to offset taxable profit were €33.7 million.
23.2.Deferred tax assets and liabilities
The Group's main sources of deferred tax assets and liabilities as of December 31, 2019 and 2020 related to:
Tax loss carry forwards: €384,471 and €483,356 respectively;
•Temporary differences:
◦related to the OCEANEs: a net deferred tax liability for €1,193 and €2,049 as of December 31, 2019 and 2020, respectively and
◦related to post-employment benefits: €352 and €287, respectively, each offset by a deferred tax asset of the same amount.
The Group's main sources of deferred tax assets and liabilities as of December 31, 2021 related to:
•Tax loss carry forwards: €450,679 (compared to €483,356 at December 31, 2020);
•Temporary differences related to:
•the OCEANEs: a deferred tax liability of €2,314 and an asset of 1,712, i.e., a net deferred tax liability of €602; and
•post-employment benefits: a deferred tax liability of €216 offset by an asset of the same amount;
The Company offsets its deferred tax assets and liabilities (€1,712 and €2,314, respectively), as permitted by IAS 12, resulting in a net deferred tax liability of €602 as of December 31, 2021.
The deferred income tax benefit for the period is mainly due to the decrease in the net deferred tax liability over the period.
Other than as it relates to deferred tax assets recognized based on the available deferred tax liabilities, no other deferred tax asset has been recognized as it is not probable that taxable profit will be available to offset deductible temporary differences and tax loss carry forwards.
24.EARNINGS (LOSS) PER SHARE
Basic earnings per share are calculated by dividing profit attributable to our ordinary shareholders by the weighted average number of ordinary shares outstanding during the period.
Diluted earnings per share is calculated by adjusting profit attributable to ordinary shareholders and the weighted average number of ordinary shares outstanding, for the effects of all potentially dilutive ordinary shares (share warrants, free shares, stock options, OCEANE convertible bonds).
The components of the earnings (loss) per share computation are as follows:
| | | | | | | | | | | | | | | | | | | | | |
| Earnings per share | | Year ended |
| | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| | | | | | | |
| Profit (loss) for the period (in € thousands) | | (65,144) | | | (101,221) | | | 67,259 | |
| Weighted average number of ordinary shares used to calculate basic earnings (loss) per share | | 36,987,982 | | | 38,858,617 | | | 44,739,756 | |
| Basic earnings (loss) per share (€/share) | | (1.76) | | | (2.60) | | | 1.51 | |
| | | | | | | |
| | | | | | | |
| Weighted average number of ordinary shares used to calculate diluted earnings (loss) per share | | 36,987,982 | | | 38,858,617 | | | 55,613,634 | |
| Diluted earnings (loss) per share (€/share) | | (1.76) | | | (2.60) | | | 1.23 | |
25.27.LITIGATION AND CONTINGENT LIABILITIES
Class Action
In May 2020, following the Group announcement on the interim results of our RESOLVE-ITRESOLVE-IT® Phase 3 clinical trial in which elafibranor had not achieved the primary or key secondary endpoints, a purported shareholder class action complaint was filed in state court in the Commonwealth of Massachusetts, naming the Group, the board of directors and certain members of the senior management as defendants, alleging that defendants made materially misleading statements about the development of elafibranor in connection with our U.S. initial public offering in violation of U.S. federal securities laws.
In October 2020, the plaintiff voluntarily dismissed the Commonwealth of Massachusetts action, but in December 2020, the same plaintiff filed a purported shareholder class action complaint in state court in the State of New York, alleging claims substantially similar to those in the previous complaint against the same defendants, as well as the underwriters of our U.S. initial public offering.
In March 2021, the Company and the other defendants filed a motion to dismiss. In August 2021, the courtSupreme Court of the State of New York, New York County, granted the motion and dismissed the complaint with prejudice. In September 2021, theThe plaintiff filed a notice of appeal toappealed and in December 2022, the Supreme Court, Appellate Division, First Department and perfectedaffirmed the dismissal of the complaint, except that it deleted the phrase “with prejudice” from the Supreme Court’s judgment. The time to appeal on March 9, 2022. We intend to vigorously defend the lower court’s decision of the Appellate Division expired in the appellate court.January 2023.
26.28.RELATED PARTIES
Biotech Avenir SAS and The NASH Epidemiology Institute™ , an endowment fund set up by the Company, are related parties within the meaningCompensation of IAS 24.9.key management personnel
The registered officeaggregate compensation of Biotech Avenir SASthe members of the Company’s Board of Directors (including the Chairman of the Board) and that of The NASH Epidemiology Institute™ are located atto the same address asChief Executive Officer includes the Company. These domiciliations are provided without charge.following:
| | | | | | | | | | | | | | | | | | | | | |
| | | Year ended |
| (in € thousands) | | 2021/12/31 | | 2022/12/31 | | 2023/12/31 |
| Fixed compensation owed | | 518 | | 585 | | 614 |
| Variable compensation owed | | 163 | | 169 | | 170 |
| Attendance fees - board of Directors | | 488 | | 421 | | 381 |
| Contributions in-kind | | 23 | | 21 | | 23 |
| Share-based payments | | 58 | | 74 | | 72 |
| Employer contributions | | 443 | | 410 | | 396 |
| Consulting fees | | 0 | | 0 | | 0 |
| TOTAL | | 1,693 | | 1,680 | | 1,656 |
Biotech Avenir
Biotech Avenir SAS is a holding company incorporated in 2001 by the Company's founders. Most of its share capital is currently held by individuals, i.e. the 4four co-founders of the Company and 12twelve Company employees.
Jean-François Mouney, the Chairman of the Company, is also the Chairman of Biotech Avenir SAS.
At December 31, 2021,2023, Biotech Avenir SAS held 3.79% of the share capital of the Company.
The Company did not carry out any transactions with Biotech Avenir in 2021, 2020,2023, 2022, or 2019 ,2021, with the exception of the domiciliation without charge.
The NASH Epidemiology Institute™
The NASH Education Program™ (which became The NASH Epidemiology Institute™ ) endowment fund was created in November 2016 at the initiative of the Group to develop and finance disease awareness activities targeting medical professionals and the general public.
It was dissolved on December 31, 2020, and in that context, the endowment fund had a positive balance of €17. These funds were transferred to the benefit of the Fondation de France in 2021.
PCAS Group
Mr. Frédéric Desdouits, member of the Genfit Board of Directors since June 2014 , was appointed CEO of PCAS Group in March 2019. Elafibranor’s principal active ingredient has been made by a PCAS Group production unit since 2013, and as Mr. Frédéric Desdouits became PCAS Group’s CEO, he temporarily became a related party as defined by IAS 24.9 until his resignation from this position in March 2020.
In January 2020, the Company signed a Memorandum of Understanding with PCAS Group setting out the conditions under which the PCAS Group would set up a second manufacturing source for the active ingredient used in the composition of elafibranor, as part of initiatives to secure the supply chain, and undertake the necessary investments to carry out this goal and to increase the production capacity of the active ingredient in view of a potential future marketing authorization. The cost to carry out technology transfers between the current manufacturing unit and the second source, €255, was to be borne by PCAS, except in case of termination of the RESOLVE-IT program. Due to the termination decision enacted on July 22, 2020, these costs were included in the closing costs of the study that were recognized in 2020. This Memorandum of Understanding was terminated in 2020 following the RESOLVE-IT topline data readout.
Ipsen Pharma SAS
The licensing agreement signed with Ipsen Pharma SAS in December 2021 provides for a certain number of service agreements to bethat were signed with the Company in 2022.2022 and 2023, notably the Inventory Purchase Agreement, the Transition Services Agreement and the Part B Transition Services Agreement.
These agreements cover support for Ipsen in future proceedings and processes (other than knowledge transfer) and the provision of drug tablets which Ipsen may require to execute its clinical trial. As per the agreement signed with Ipsen in December 2021, the prices under these agreements will cover all costs bornborne by the Company to provide the relevant goods and services, without economic benefit for Ipsen.
27.29.COMPENSATION OF CORPORATE OFFICERSCOMMITMENTS AND CONTINGENT LIABILITIES
On September 2, 2019,Accounting policies
In accordance with IAS 37, Provisions Contingent Liabilities and Contingent Assets, provisions are recognized when the BoardGroup has a present obligation (legal, regulatory, contractual or constructive) as a result of Directors accepteda past event, for which it is probable that an outflow of resources will be required to settle the resignationobligation, and of which the amount can be estimated reliably.
Future milestone and revenue based royalty payments may be recorded pursuant to Contingent liability under IAS 37 or intangible asset under IAS 38. We record a provision when we have a present obligation, whether legal or constructive, as a result of a past event; it is probable that an outflow of resources embodying economic benefits will be required to settle the obligation; and a reliable estimate can be made of the Chairman and Chief Executive Officeramount of the Companyoutflow of resources. Under IAS 38, we record intangible asset when it is probable that the expected future economic benefits that are attributes to the assets will flow to us and decided to separate the rolescost of Chairman of the Board of Directors and Chief Executive Officer of Genfit SA with effect from September 16, 2019.
At the same meeting, the Board of Directors appointed the Chief Executive Officer of the Company and confirmed the former Chairman and Chief Executive Officer in his functions as Chairman of the Board of Directors and member of certain committees of the Company's Board of Directors.asset can be measured reliably.
Under these conditions, the following table details the compensation paid to the Chairman and Chief Executive Officer during the period from January 1, 2019 to September 16, 2019, and the compensation paid to the Chief Executive Officer in 2021, 2020 and during the period from September 16 to December 31, 2019 (after the change in governance) and the years in which the amounts were recognized in the statement of operations.
| | | | | | | | | | | | | | | | | | | | | |
| Compensation paid to the Chairman | | Year ended |
| during the period from January 1, 2019 to September 16, 2019 | | | | | | |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Short-term employee benefits (gross + employer's social contributions, paid) | | 1,338 | | | — | | | — | |
| Post-employment pension & medical benefits | | — | | | — | | | — | |
| Share-based payment transactions | | 109 | | | — | | | — | |
| Director fees Genfit Corp (net) | | 22 | | | — | | | — | |
| TOTAL | | 1,469 | | | — | | | — | |
| | | | | | | |
| Compensation paid to the Chief Executive Officer | | Year ended |
| | | | | | | |
| (in € thousands) | | 2019/12/31 | | 2020/12/31 | | 2021/12/31 |
| Short-term employee benefits (gross + employer's social contributions, paid) | | 140 | | | 534 | | | 628 | |
| Post-employment pension & medical benefits | | — | | | — | | | — | |
| Attendance fees | | — | | | — | | | — | |
| Share-based payment transactions | | — | | | 41 | | | 58 | |
| Director fees Genfit Corp (net) | | — | | | — | | | — | |
| TOTAL | | 140 | | | 575 | | | 686 | |
(1)Within this total, only the portion of the amounts paid in 2019 to the Chairman and Chief Executive Officer pursuant to the 13th resolution of the Annual General Meeting of June 13, 2019, for the part of the Incentive Plan corresponding to that portion of work on the initial public offering of the Company on the Nasdaq Global Select Market carried out in 2018, i.e., ¾ of the amount due, i.e. a gross amount of €562,893. However, the Chairman of the Board of Directors decided to forgo the balance amount of €187,631, which had been recorded in 2019.
The Chief Executive Officer’s corporate contract contains a clause whereby, in the event of termination, he would receive a non-compete indemnity equal to:
(i)twelve (12) months of fixed compensation, calculated on the basis of the gross amounts due for the past twelve months ended and
(ii)increased, where applicable, by the amount of the annual variable compensation due for the previous year. This compensation is intended to compensate the prohibition made to the Chief Executive Officer, for a period of 12 months following the termination of his functions, for whatever reason, to work in any way whatsoever with certain companies carrying out a directly competitive activity of the Company.
In addition, the Chief Executive Officer, except in the event of gross negligence within the meaning of labor law, shall receive severance pay equal to:
(i)twelve (12) months of fixed compensation, calculated on the basis of the gross amounts due for the twelve past completed months and
(ii)increased, where applicable, by the amount of annual variable compensation due for the previous year.
This compensation will be paid one month after his effective termination of activity within the Group. The compensation will not be paid if, on his initiative, the Chief Executive Officer leaves the Company to exercise new functions or changes functions within the Group, or even if he has the possibility of asserting in the short term his retirement rights. It is also specified that any sum paid under the non-competition clause will be deducted from the sums due under the severance pay and vice versa. The total and maximum commitment represented by this indemnity (gross, employer charges and payroll tax) as of December 31, 2021 would amount to €523.
The Chairman of the Board of Directors, Jean-François Mouney, receives a fixed compensation. He also has use of a company vehicle and the Group’s insurance and disability plan. These benefits are totaled in the table below in the "Other remuneration" line. The Chairman of the Board of Directors also receives attendance fees granted for his participation in the work of some of the committees of the Board of Directors.
The directors' fees and other compensation due and paid to the non executive directors are as follows:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Amounts
due* | | Amounts
paid* | | Amounts
due* | | Amounts
paid* | | Amounts
due* | | Amounts
paid* |
| Attendance fees and other forms of remuneration payable to each of the non executive officer | | During the year ended December 31, |
| (in euros) | | 2019 | | 2020 | | 2021 |
| Jean-François MOUNEY (1) | | | | | | | | | | | | |
| Attendance fees | | 14,791 | | | 633 | | | 49,937 | | | 49,939 | | | 45,863 | | | 39,232 | |
| Other remuneration | | 88,874 | | | 88,874 | | | 286,469 | | | 286,469 | | | 286,553 | | | 286,553 | |
| TOTAL | | 103,665 | | | 89,507 | | | 336,406 | | | 336,408 | | | 332,416 | | | 325,785 | |
| Xavier GUILLE DES BUTTES | | | | | | | | | | | | |
| Attendance fees | | 68,016 | | | 67,580 | | | 85,020 | | | 80,660 | | | 89,822 | | | 85,020 | |
| Other remuneration | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL | | 68,016 | | | 67,580 | | | 85,020 | | | 80,660 | | | 89,822 | | | 85,020 | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Frédéric DESDOUITS | | | | | | | | | | | | |
| Attendance fees | | 33,136 | | | 30,302 | | | 53,360 | | | 44,640 | | | 43,600 | | | 47,960 | |
| Other remuneration | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL | | 33,136 | | | 30,302 | | | 53,360 | | | 44,640 | | | 43,600 | | | 47,960 | |
| BIOTECH AVENIR | | | | | | | | | | | | |
| Représenté par Florence Séjourné | | | | | | | | | | | | |
| Attendance fees | | — | | | — | | | — | | | — | | | — | | | — | |
| Other remuneration | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL | | — | | | — | | | — | | | — | | | — | | | — | |
| Philippe MOONS | | | | | | | | | | | | |
| Attendance fees | | 36,188 | | | 41,202 | | | 45,780 | | | 41,420 | | | 16,986 | | | 25,161 | |
| Other remuneration | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL | | 36,188 | | | 41,202 | | | 45,780 | | | 41,420 | | | 16,986 | | | 25,161 | |
| Anne-Hélène MONSELLATO | | | | | | | | | | | | |
| Attendance fees | | 44,472 | | | 53,410 | | | 50,140 | | | 45,780 | | | 52,320 | | | 52,320 | |
| Other remuneration | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL | | 44,472 | | | 53,410 | | | 50,140 | | | 45,780 | | | 52,320 | | | 52,320 | |
| Catherine LARUE | | | | | | | | | | | | |
| Attendance fees | | 33,136 | | | 28,122 | | | 43,600 | | | 43,600 | | | 47,651 | | | 45,780 | |
| Other remuneration | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL | | 33,136 | | | 28,122 | | | 43,600 | | | 43,600 | | | 47,651 | | | 45,780 | |
| Katherine KALIN | | | | | | | | | | | | |
| Attendance fees | | — | | | — | | | 15,805 | | | 4,360 | | | 43,600 | | | 39,240 | |
| Other remuneration | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL | | — | | | — | | | 15,805 | | | 4,360 | | | 43,600 | | | 39,240 | |
| Eric BACLET | | | | | | | | | | | | |
| Attendance fees | | — | | | — | | | 20,165 | | | 6,540 | | | 62,881 | | | 62,336 | |
| Other remuneration | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL | | — | | | — | | | 20,165 | | | 6,540 | | | 62,881 | | | 62,336 | |
| Jean-François TINE | | | | | | | | | | | | |
| Attendance fees | | — | | | — | | | — | | | — | | | 39,724 | | | 23,919 | |
| Other remuneration | | — | | | — | | | — | | | — | | | — | | | — | |
| TOTAL | | — | | | — | | | — | | | — | | | 39,724 | | | 23,919 | |
| TOTAL | | 318,613 | | | 310,123 | | | 650,276 | | | 603,408 | | | 729,000 | | | 707,521 | |
(1)
The attendance fees and other remuneration for Jean-François Mouney above correspond to amounts due for the period from September 16, 2019 to December 31, 2019 and the full year 2020 and 2021.
In addition, the Company has provided corporate officers, directors and members of the Executive Committee a “directors and officers” insurance against claims relating to certain actions they may take in the performance of their duties. For the 12 months of the calendar year 2021, the insurance premium for the implementation of this insurance coverage amounted to €2,052 (€1,316 in 2020 and €3,146 in 2019)
28.COMMITMENTS
Obligations under the terms of subcontracting agreements
The Group enters into contracts for its business needs with clinical research organizations (CROs) for clinical trials, as well as with Contract Manufacturing Organizations (CMOs) for clinical and commercial supply manufacturing, commercial and pre-commercial activities, research and development activities and other services and products for operating purposes. The Group’s agreements generally provide for termination with specified periods of advance notice.
Such agreements are generally cancellable contracts and not included in the description of the Group’s contractual obligations and commitments.
Obligations under the terms of lease agreements
The Company has guaranteed its rental payment obligation under the lease agreement for the headquarters in Loos, France in the amount of €600 at December 31, 2023 (€600 at December 31, 2022).
Contingent liabilities
•Obligations under the terms of license and collaboration agreementsagreement with Genoscience
The Company has entered into a licensing agreement with Genoscience Pharma whereby we are obligated to pay royalties and milestone payments based on future events that are uncertain and therefore they constitute contingent liabilities not recognized in the Company's consolidated financial statements for the period ending December 31, 2021.2023.
Under the agreement, Genoscience Pharma is eligible for clinical and regulatory milestone payments for up to €50 million and tiered royalties. The first payable milestones are contingent on positive Phase 2 clinical trial results in CCA, and may total up to €20 million, if applicable.
Additional milestones are contingent on positive Phase 3 results. These payments, when due, will be subject to a review to determine if they are eligible for activation pursuant to IAS 38. If so, they will be recorded as capital upon disbursement. Otherwise, they also constitute contingent liabilities which will be recognized when due.
In addition, we also have a right of first negotiation with respect to any license or assignment, or option for a license or an assignment, with any third party to develop or commercialize other Genoscience assets in the field of CCA, to the extent Genoscience is looking to partner the asset with a third party or receives a spontaneous offer for collaboration.
For the period commencing on the date of the agreement until the first regulatory approval of GNS561 for commercialization, Genoscience Pharma has the right to repurchase the license to GNS561 in CCA at a pre-determined price in the event that Genoscience Pharma receives an offer from a third party to acquire or obtain a license to GNS561 in all indications, provided that GENFIT shall first have the opportunity to negotiate the acquisition or license to GNS561 in all indications or match the offer from the third party.
These obligations constitute contingent liabilities not recognized in the Company's consolidated financial statements at December 31, 2022 or December 31, 2023.
•Obligations underrelated to the Versantis acquisition
The company entered into an agreement with the former shareholders of Versantis whereby we are obligated to pay milestone payments based on future events that are uncertain and there therefore they constitute contingent liabilities not recognized in the Company's consolidated financial statements for the period ending December 31, 2023.
Milestone payments total up to 65 million CHF, contingent on the following outcomes:
•positive Phase 2 results related to VS-01-ACLF,
•regulatory approval of VS-01-ACLF, and
•positive Phase 2 results related to VS-02.
Furthermore, the former shareholders of Versantis are eligible to receive 1/3 of the net proceeds resulting from the potential sale of the Priority Review Voucher of VS-01’s pediatric application by GENFIT to a third party, or 1/3 of the fair market value of this Voucher if GENFIT opts to apply it to one of its own programs.
•Obligations related to the licensing agreement with Seal Rock Therapeutics
On May 31, 2023, GENFIT announced the signing of a licensing agreement for the exclusive worldwide rights to the ASK1 inhibitor SRT-015 with Seal Rock Therapeutics, a clinical-stage company based in Seattle, Washington.
Under the terms of lease agreementsthe agreement, Seal Rock is eligible for payments of up to €100 million (of which €2 million have been paid in 2023 ), subject to certain regulatory, clinical and commercial outcomes.
Seal Rock is likewise eligible for tiered royalties, applied to the annual sales of licensed products realized by GENFIT.
These obligations constitute contingent liabilities not recognized in the Company's consolidated financial statements at December 31, 2023.
•Obligations related to the licensing agreement with Celloram
On July 28, 2023, GENFIT licensed the exclusive worldwide rights to CLM-022, a first-in-class inflammasome inhibitor, from Celloram Inc., a Cleveland-based biotechnology company.
Under the terms of the agreement:
1.Celloram is eligible for payments of up to €160 million (of which €50 have been paid in 2023), subject to certain regulatory, clinical and commercial outcomes.
2.Celloram is likewise eligible for tiered royalties, applied to the annual sales of licensed products realized by GENFIT.
The conditional payments will be subject to analysis when they are incurred to determine if they are eligible for capitalization in accordance with IAS 38. If so, they will be capitalized. Otherwise, they will be expensed as incurred. In addition, a first milestone of €50 was paid in 2023. This milestone was capitalized in the “Intangible assets” line item on the statement of financial position.
These obligations constitute contingent liabilities not recognized in the Company's consolidated financial statements at December 31, 2023.
Contingent Assets
•Contingent assets related to the licensing agreement with IPSEN
In December 2021, GENFIT and Ipsen Pharma SAS ("Ipsen") entered into an exclusive worldwide licensing agreement (except for China, Hong Kong, Taiwan and Macao, which apply to Terns as noted below) for elafibranor, a Phase 3 asset evaluated in Primary Biliary Cholangitis (PBC), as part of a long-term global partnership ("Collaboration and License Agreement"). Under this agreement we could receive milestone payments based on future events that are uncertain and therefore they constitute contingent assets not recognized in the Company's consolidated financial statements for the period ending December 31, 2023.
•GENFIT is also eligible for total milestone payments up to €360 million (of which €13.3 million has been paid in 2024). These milestone payments constitute future variable income, dependent on the completion of key steps related to the development and sales of the licensed products. As such, in accordance with IFRS 15, this income will be recognized as revenue depending on the completion of these milestones. No such milestone payments were made in 2021 or 2022. In 2023, a milestone of €13.3 million was recognized as revenue and is recognized on the balance sheet in Accounts receivable as of December 31, 2023. Furthermore, we expect to receive future milestone revenue in 2024, subject to approval by applicable regulatory authorities and US and European commercial launches of elafibranor in PBC by Ipsen, representing a total of approximately €75.2 million.
•GENFIT is eligible for tiered double-digit royalties of up to 20%, applied to the annual sales of licensed products realized by Ipsen. As such, in accordance with IFRS 15, this income will be recognized as revenue depending on the realization of these sales. No such royalties were earned in 2021, 2022 or 2023.
•Contingent assets related to the licencing agreement with Terns Pharma
The Company has guaranteed its rental payment obligation underentered into a licensing agreement with Terns Pharma whereby we could receive milestone payments based on future events that are uncertain and therefore they constitute contingent assets not recognized in the lease agreementCompany's consolidated financial statements for the headquartersperiod ending December 31, 2023. The licensing agreement with Terns concerns China, Hong Kong, Taiwan and Macao.
Milestones include Development Milestone Payments upon the achievement of the development milestones for the licensed product and Commercial Milestone Payments upon the achievement of commercial milestones depending on reaching certain aggregate thresholds. There are also potential mid-teen royalties based on sales by Terns Pharmaceuticals in Loos, FranceGreater China. The potential Development and Commercial Milestone payments may represent up to $193 million.
30.ACQUISITIONS
Acquisition of the Clinical-stage Biopharmaceutical Company Versantis
On September 19, 2022, the Company announced it had signed an exclusive agreement with Versantis AG ("Versantis") to acquire all the shares and voting rights of Versantis, a private Swiss-based clinical stage biotechnology company focused on addressing the growing unmet medical needs in liver diseases. This acquisition aims at:
1.Consolidating GENFIT’s position as a leader in acute-on-chronic liver failure (ACLF)
2.Significantly expanding GENFIT’s pipeline with VS-01-ACLF, a Phase 2 ready program based on first-in-class scavenging liposomes technology, VS-01-HAC, a pediatric program focused on Urea Cycle Disorder (UCD), and VS-02-HE, an early-stage program focused on hepatic encephalopathy (HE), and
3.Combining Versantis’ expertise with GENFIT’s know-how in conducting complex development programs in liver diseases, to strengthen and accelerate research and development
The deal closed effective September 29, 2022.
Total purchase price and contingent milestone payments
This transaction includes:
•an initial payment of 40 million CHF (€41.9 million) due and paid at the date of closing,
•a net cash adjustment payment of 2.8 million CHF (€2.9 million) at the end of the year in accordance with the terms of the acquisition agreement
•additional milestone payments of up to 65 million CHF contingent on the following outcomes:
◦positive Phase 2 results related to VS-01-ACLF,
◦regulatory approval of VS-01-ACLF, and
◦positive Phase 2 results related to VS-02.
Furthermore, the former shareholders of Versantis are eligible to receive 1/3 of the net proceeds resulting from the potential sale of the Priority Review Voucher of VS-01’s pediatric application by GENFIT to a third party, or 1/3 of the fair market value of this Voucher if GENFIT opts to apply it to one of its own programs.
Acquisition costs totaled €1.8 million.
The impact of this acquisition as reflected within the line item "Acquisition net of cash acquired" in the consolidated statement of cash flows is a net cash outflow of €41.5 million.
Accounting treatment - IFRS 3
Paragraph B7B sets out an optional test (the concentration test) to permit a simplified assessment of whether an acquired set of activities and assets is not a business. An entity may elect to apply, or not apply, the test. The concentration test is met if substantially all of the fair value of the gross assets acquired is concentrated in a single identifiable asset or group of similar identifiable assets.
GENFIT chose to use the available option stated as per IFRS 3 and perform a concentration test to determine if the transaction qualifies as a business combination or asset acquisition. In accordance with the concentration test methodology as defined in paragraph B7B of IFRS 3, the acquisition of Versantis by GENFIT was determined to be an asset acquisition based on the VS-01-ACLF program because its fair value represents more than 90% of the value of all assets acquired. Therefore, the acquisition costs of €1.8 million were included and capitalized in the total cost of the operation to determine the net book value of the VS-01-ACLF program on the date of the acquisition. Conditional milestone payments were not included in this analysis.
Accounting treatment - IAS 21
Consistent with paragraph 8 of IAS 21, Versantis AG is considered as a foreign operation as its activities are conducted primarily in Swiss Francs. The Swiss Franc is thus also considered to be Versantis AG's functional currency. Versantis AG's subsidiary, Versantis Inc., is considered as a foreign operation as its activities are conducted primarily in US Dollars. The US Dollar is thus considered to be Versantis Inc.'s functional currency. For further information on converting and presenting Versantis' activity in euros, refer to Note 4.3.2 - "Foreign currency - Translation of foreign subsidiary financial statements". Note that on the Consolidated Statements of Other Comprehensive Income and Loss, for the period ending December 31, 2022, on line item "Other comprehensive income (loss) that are or may be reclassified to profit or loss," substantially all of the loss amount of €600 at€1.4 million is due to the application of IAS 21 for Versantis.
Supplementary information
The consolidated value of net assets acquired of Versantis AG and Versantis, Inc. on September 29, 2022 is as follows :
| | | | | |
| Total acquired assets and liabilities, in thousands of euros | 29/9/2022 |
| Cash and cash equivalents | 5,076 |
| Current trade and others receivables | 209 |
| Other current assets | 78 |
| Intangible assets | 45,323 |
| Property, plant and equipment | 326 |
| Other non-current financial assets | 14 |
| Total acquired assets | 51,026 |
| |
| Current trade and other payables | 3,202 |
| Current provisions | 858 |
| Other current tax liabilities | 63 |
| Lease liabilities | 302 |
| Total acquired liabilities | 4,425 |
| |
| Total purchase price | 46,601 |
The exchange rate used above to convert the assets and liabilities of Versantis AG into euros was 1.04843 (1 CHF = 1.04843) on September 29, 2022. The exchange rate used above to convert the assets and liabilities of Versantis, Inc. into euros was 0.9706 (1 USD = 0.9706) on September 29, 2022.
The net book value of assets and liabilities as of December 31, 2021, €600 at2022 is as follows, per the application of IAS 21:
| | | | | |
| Net assets, in thousands of euros | 31/12/2022 |
| Cash and cash equivalents | 2,168 |
| Current trade and others receivables | 17 |
| Other current assets | 197 |
| Intangible assets | 43,850 |
| Property, plant and equipment | 295 |
| Other non-current financial assets | 13 |
| Total assets | 46,540 |
| |
| Current trade and other payables | 1,614 |
| Current provisions | 672 |
| Other current tax liabilities | 33 |
| Lease liabilities | 282 |
| Total liabilities | 2,601 |
| |
| Net assets | 43,939 |
The exchange rate used above to convert the assets and liabilities of Versantis AG into euros was 1.01554 (1 CHF = 1.01554) on December 31, 2020,2022. The exchange rate used above to convert the assets and €542 atliabilities of Versantis, Inc. into euros was 0.93756 (1 USD = 0.93756) on December 31, 2019.2022.
Research and development expenses for the period between September 29, 2022 and December 31, 2022 attributable to Versantis total €1,187 thousand. If the acquisition had taken place on January 1, 2022, research and development expenses would have been €5,833 thousand.
General and administrative expenses for the period between September 29, 2022 and December 31, 2022 attributable to Versantis total €228 thousand.
31.SUPPLEMENTAL CASH FLOW INFORMATION
Supplemental cash flow information
Disclosure of non-cash financing and investing activities
Accrued property, plant and equipment, 2023: €42
Accrued property, plant and equipment, 2022: €142
Accrued property, plant and equipment, 2021: €76