

Use these links to rapidly review the document
TABLE OF CONTENTS
TABLE OF CONTENTS 2Table of Contents
As filed with the Securities and Exchange Commission on June 14, 201329, 2016
Registration No. 333- 333-211769
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM S-1Form S-l
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
Onconova Therapeutics, Inc.
(Exact name of registrant as specified in its charter)
| Delaware (State or other jurisdiction of incorporation or organization) | 2834 (Primary Standard Industrial Classification Code Number) | 22-3627252 (I.R.S. Employer Identification |
Onconova Therapeutics, Inc.
375 Pheasant Run
Newtown, PA 18940
(267) 759-3680-759-3680
(Address, including zip code, and telephone number, including
area code, of registrant's principal executive offices)
Ramesh Kumar, Ph.D.
President and Chief Executive Officer
Onconova Therapeutics, Inc.
375 Pheasant Run
Newtown, PA 1894018954
(267) 759-3680
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Jeffery P. Libson
Donald R. Readlinger
Pepper Hamilton LLP
301 Carnegie Center, Suite 400
Princeton, NJ 08540-6227
609-951-4164
Approximate date of commencement of proposed sale to the public:
As soon as practicable after the effective date of this registration statement.
If any of the securities being registered on this formForm are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, as amended (the "Securities Act"), check the following box. oý
If this formForm is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this formForm is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this formForm is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check(check one):o
| Large | Accelerated filer o | Non-accelerated filer (Do not check if a smaller reporting company) | Smaller reporting company |
CALCULATION OF REGISTRATION FEE
| Title of each class of securities to be registered(1) | Proposed maximum aggregate offering price(2) | Amount of registration fee(3) | ||
|---|---|---|---|---|
Common Stock, $0.01 par value per share | $75,000,000 | $10,230 | ||
| ||||
| Title of securities to be registered | Proposed maximum aggregate offering price(1) | Amount of registration fee(5) | ||
|---|---|---|---|---|
Units, each consisting of one share of common stock, par value $0.01 per share ("Common Stock") and 0.75 of a warrant ("Warrant") to purchase one share of Common Stock ("Units") | $26,597,381 | $2,678 | ||
Non-transferable Rights to purchase Units(2) | — | — | ||
Common Stock, par value $0.01 per share included as part of the Units(3) | Included with Units above | — | ||
Warrants included as part of the Units(3) | Included with Units above | — | ||
Common Stock issuable upon exercise of the Warrants included in the Units(4) | $23,937,643 | $2,411 | ||
Pre-Funded Warrants in lieu of Common Stock included in Units(3) | Included with Units Above | — | ||
Common Stock issuable upon exercise of Pre-Funded Warrants(3)(4) | Included with Units Above | — | ||
Total | $50,535,024 | $5,089 | ||
| ||||
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall filefiles a further amendment thatwhich specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.determine.
The information in this preliminary prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission isdeclares our registration statement effective. This preliminary prospectus is not an offer to sell these securities and it iswe are not soliciting offersan offer to buy these securities in any state or other jurisdiction where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED JUNE 14, 2013Subject to completion, dated June 29, 2016
PRELIMINARY PROSPECTUS
Shares
Onconova Therapeutics, Inc.
Common Stock$ per share
This is the initial public offeringWe are distributing to holders of our common stock.stock and to holders of certain of our outstanding warrants who are entitled to participate in this offering, at no charge, non-transferable subscription rights to purchase units. Each unit, which we refer to as a Unit, consists of one share of common stock and 0.75 of a tradable warrant representing the right to purchase one share of common stock, which we refer to as the Warrants. We are sellingrefer to the offering that is the subject of this prospectus as the Rights Offering.
In the Rights Offering, you will receive 1.5 subscription rights for each share of common stock or each share of common stock underlying our participating warrants owned at 5:00 PM Eastern Time, on July 7, 2016, the record date of the Rights Offering, or the Record Date. The common stock and the Warrants comprising the Units will separate upon the effectiveness of the exercise of the rights and will be issued as separate securities, and the Units will not trade as a separate security. The subscription rights will not be tradable.
Each subscription right will entitle you to purchase one Unit, which we refer to as the Basic Subscription Right, at a subscription price per Unit to be determined, which we refer to as the Subscription Price. The Subscription Price per Unit is expected to be between $5.75 and $6.25. The exact Subscription Price per Unit will be determined by our board of directors or pricing committee prior to the effectiveness of the registration statement of which this prospectus is a part. The Warrants entitle the holder to purchase one share of common stock at an exercise price of $ per share, 120% of the per Unit price, from the date of issuance through its expiration on , 2021. If you exercise your Basic Subscription Rights in full, and other shareholders or participating warrant holders do not, you will be entitled to an over-subscription privilege to purchase a portion of the unsubscribed Units at the Subscription Price, subject to proration, which we refer to as the Over-Subscription Privilege. Each subscription right consists of a Basic Subscription Right and an Over-Subscription Privilege, which we refer to as the Subscription Right.
For certain investors whose subscriptions may result in the purchaser beneficially owning more than 4.99% of our outstanding common stock, such investors may elect to receive in the Rights Offering, in lieu of shares of common stock, certain pre-funded warrants (which we refer to as the Pre-Funded Warrants) to purchase the same amount of shares of common stock. If you do not wish to exceed the ownership threshold, you may elect to receive a Pre-Funded Warrant in this offering. We currently expect the initial public offering price to be between $ and $ perlieu of any share of common stock.
Westock underlying the Units for which you have grantedsubscribed. You will not be eligible to elect to receive Pre-Funded Warrants, except to the underwriters an option to purchase up to additionalextent that your beneficial ownership could exceed 4.99% of the shares of common stock outstanding following the consummation of the Rights Offering. Each Pre-Funded Warrant will have an exercise price of $0.01, and the subscription price per Unit for any such electing investors will be reduced to cover over-allotments.$ (which equals the Subscription Price for the other Units sold in the Rights Offering, less the $0.01 exercise price for each Pre-Funded Warrant). The Pre-Funded Warrants do not confer upon the holder any voting or any other rights of a shareholder of the Company.
This prospectus also relates to the offering of the shares of common stock issuable upon exercise of these Pre-Funded Warrants.
The Subscription Rights will expire if they are not exercised by 5:00 PM Eastern Time, on July 26, 2016. We may extend the Rights Offering for additional periods in our sole discretion. Once made, all exercises of Subscription Rights are irrevocable.
The Rights Offering is being conducted on a best-efforts basis. There is no minimum amount of proceeds necessary in order for us to close the Rights Offering. While none of our directors or executive officers has entered into any binding commitment or agreement to exercise Subscription Rights received in the Rights Offering, our directors and executive officers have indicated interests in subscribing for up to an aggregate of $3.0 million in the Rights Offering, subject to potential proration.
We have engaged Maxim Group LLC to act as dealer-manager in the Rights Offering.
Investing in our securities involves a high degree of risk. See the section entitled "Risk Factors" beginning on page 20 of this prospectus. You should carefully consider these risk factors, as well as the information contained in this prospectus, before you invest.
Wells Fargo Bank, N.A. will serve as the Subscription Agent for the Rights Offering. The Subscription Agent will hold the funds we receive from subscribers until we complete, abandon or terminate the Rights Offering. If you want to participate in this Rights Offering and you are the record holder of your shares or participating warrants, we recommend that you submit your subscription documents to the Subscription Agent well before the deadline. If you want to participate in this Rights Offering and you hold shares through your broker, dealer, bank, or other nominee, you should promptly contact your broker, dealer, bank, or other nominee and submit your subscription documents in accordance with the instructions and within the time period provided by your broker, dealer, bank, or other nominee. For a more detailed discussion, see "The Rights Offering—The Subscription Rights."
Our board of directors reserves the right to terminate the Rights Offering for any reason any time before the completion of the Rights Offering. If we terminate the Rights Offering, all subscription payments received will be returned as soon as practicable, without interest or penalty.
Our common stock is listed on the NASDAQ Capital Market, or NASDAQ, under the symbol "ONTX." On June 28, 2016, the last reported sale price of our common stock was $6.00. We have applied to list the Warrants on NASDAQ following their issuance under the symbol "ONTXW." The Subscription Rights are non-transferrable and will not be listed for trading on NASDAQ or any other stock exchange or market. You are urged to obtain a current price quote for our common stock onbefore exercising your Subscription Rights.
| Per Unit | Total(2) | |||
|---|---|---|---|---|
Subscription price | $ | $ | ||
Dealer-Manager fees and expenses(1) | $ | $ | ||
Proceeds to us, before expenses | $ | $ | ||
Investing indollar amount of the Units sold to any holders of Subscription Rights who were beneficial owners of shares of our common stock involves risks.prior to July 30, 2013, and (b) 8.0% of the dollar amount of the Units sold to any other holders of Subscription Rights. We will provide to the dealer-manager upon completion of the Rights Offering a non-accountable expense allowance equal to the lesser of $100,000 or 3% of the gross proceeds of the Rights Offering for expenses incurred in connection with the Rights Offering. We advanced $30,000 against out-of-pocket accountable expenses to Maxim Group LLC upon its engagement as a dealer-manager; provided that Maxim Group LLC will promptly reimburse to us (i) any portion of the advance not used for actual out-of-pocket expenses, if the Rights Offering is not completed, and (ii) the full amount of the advance, if the Rights Offering is completed. See "Risk Factors" beginning on page 10.
We are an "emerging growth company" under applicable Securities and Exchange Commission rules and will be eligible for reduced public company reporting requirements. See "Summary—ImplicationsOur board of Being an Emerging Growth Company."directors is making no recommendation regarding your exercise of the Subscription Rights. You should carefully consider whether to exercise your Subscription Rights before the expiration date. You may not revoke or revise any exercises of Subscription Rights once made unless we terminate the Rights Offering.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined ifpassed upon the adequacy or accuracy of this prospectus is truthful or complete.prospectus. Any representation to the contrary is a criminal offense.
Maxim Group LLC
| |||||||
| |||||||
| |||||||
The underwriters expect to deliver the shares to purchasers on or aboutdate of this Prospectus is , 2013 through the book-entry facilities of The Depository Trust Company.
, 20132016
We are responsible for the information contained in this prospectus. We have not authorized anyone to provide you with different information, and we take no responsibility for any other information others may give you. If anyone provides you with different or inconsistent information, you should not rely on it. We are not, and the underwriters are not, making an offer to sell these securities in any jurisdiction where the offer or sale is not permitted. You should not assume that the information contained in this prospectus is accurate as of any date other than the date on the front of this prospectus.
| | Page | |||
|---|---|---|---|---|
| 1 | |||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| 27 | |||
CAPITALIZATION | 28 | |||
DILUTION | 29 | |||
MARKET PRICE OF OUR COMMON STOCK AND RELATED STOCKHOLDER MATTERS | 30 | |||
DIVIDEND POLICY | 31 | |||
THE RIGHTS OFFERING | 31 | |||
MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES | 40 | |||
DESCRIPTION OF SECURITIES | 48 | |||
PLAN OF DISTRIBUTION | 53 | |||
EXPERTS | 54 | |||
| 54 | |||
WHERE YOU CAN FIND MORE INFORMATION | 54 | |||
INCORPORATION BY REFERENCE | 55 | |||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
Unless the context otherwise requires, references in this prospectus to "Onconova," "Onconova Therapeutics," "Company," "we," "us" and "our" refer to Onconova Therapeutics, Inc. and its consolidated subsidiaries. This prospectus is part of a registration statement that we have filed with the Securities and Exchange Commission, which we refer to as the SEC or the Commission, utilizing a registration process. It is important for you to read and consider all of the information contained in this prospectus and any applicable prospectus before making a decision whether to invest in the common stock. You should also read and consider the information contained in the exhibits filed with our registration statement, of which this prospectus is a part, as described in "Where You Can Find More Information" in this prospectus.
You should rely only on the information contained in this prospectus and any applicable prospectus supplement, including the information incorporated by reference. We have not authorized anyone to provide you with different information. We are not offering to sell or soliciting offers to buy, and will not sell, any securities in any jurisdiction where it is unlawful. You should assume that the information contained in this prospectus or any prospectus supplement, as well as information contained in a document that we have previously filed or in the future will file with the SEC is accurate only as of the date of this prospectus, the applicable prospectus supplement or the document containing that information, as the case may be.
i
QUESTIONS AND ANSWERS RELATING TO THE RIGHTS OFFERING
The following are examples of what we anticipate will be common questions about the Rights Offering. The answers are based on selected information included elsewhere in this prospectus. The following questions and answers do not contain all of the information that may be important to you and may not address all of the questions that you may have about the Rights Offering. This prospectus and the documents incorporated by reference into this prospectus contain more detailed descriptions of the terms and conditions of the Rights Offering and provide additional information about us and our business, including potential risks related to the Rights Offering, the Units offered hereby, and our business. We urge you to read this entire prospectus and the documents incorporated by reference into this prospectus.
Why are we conducting the Rights Offering?
We are conducting the Rights Offering to raise additional capital:
What is the Rights Offering?
We are distributing, at no charge, to record holders of our common stock and to holders of certain of our outstanding warrants who are entitled to participate in this Rights Offering pursuant to the terms of such warrants, non-transferable Subscription Rights to purchase Units at a price per Unit expected to be between $5.75 and $6.25. The exact Subscription Price per Unit will be determined by our board of directors or pricing committee prior to the effectiveness of the registration statement of which this prospectus is a part. The Subscription Rights will not be tradable. Each Unit consists of one share of common stock and 0.75 of a Warrant representing the right to purchase one share of common stock at an exercise price of $ per share, 120% of the per Unit price. Upon the effectiveness of the exercise of the Subscription Rights, the common stock and Warrants will immediately separate and will be issued as separate securities. We have applied to list the Warrants on NASDAQ under the symbol "ONTXW." You will receive 1.5 Subscription Rights for each share of common stock or each share of common stock underlying the participating warrants that you owned as of 5:00 PM Eastern Time, on the Record Date. Each Subscription Right entitles the record holder or holder of a participating warrant to a Basic Subscription Right and an Over-Subscription Privilege.
What are the Basic Subscription Rights?
For each whole share you owned or whole share underlying the participating warrants you owned as of the Record Date, you will receive 1.5 Basic Subscription Rights, each of which gives you the opportunity to purchase one share of our common stock and to receive 0.75 of a Warrant to purchase one additional share of our common stock for a price of $ per Unit. For example, if you owned 100 shares of common stock as of the Record Date, you will receive 150 Subscription Rights and will have the right to purchase 150 shares of our common stock and 112 Warrants to purchase one additional share of our common stock for $ per whole Unit (or a total payment of $ ). You may exercise all or a portion of your Basic Subscription Rights or you may choose not to exercise any Basic Subscription Rights at all.
If you are a record holder or a holder of participating warrants, the number of shares you may purchase pursuant to your Basic Subscription Rights is indicated on the enclosed Subscription Rights Statement. If you hold your shares in the name of a broker, dealer, bank, or other nominee who uses
the services of the Depository Trust Company, or DTC, you will not receive a Subscription Rights Statement. Instead, DTC will issue 1.5 Subscription Rights to your nominee record holder for each share of our common stock that you own as of the Record Date. If you are not contacted by your nominee, you should contact your nominee as soon as possible.
What is the Over-Subscription Privilege?
If you exercise your Basic Subscription Rights in full, you may also choose to exercise your Over-Subscription Privilege to purchase a portion of any Units that the other record holders and participating warrant holders do not purchase through the exercise of their Basic Subscription Rights. You should indicate on your Subscription Rights Statement, or the form provided by your nominee if your shares are held in the name of a nominee, how many additional Units you would like to purchase pursuant to your Over-Subscription Privilege.
If sufficient Units are available, we will seek to honor your Over-Subscription request in full. If Over-Subscription requests exceed the number of Units available, however, we will allocate the available Units pro-rata among the record holders and participating warrant holders exercising the Over-Subscription Privilege in proportion to the number of shares of our common stock each of those record holders owned and the number of shares of our common stock underlying the participating warrants held by each of those warrant holders on the Record Date, relative to the number of shares owned and the number of shares underlying participating warrants held on the Record Date by all record holders and warrant holders exercising the Over-Subscription Privilege. If this pro-rata allocation results in any record holders or warrant holders receiving a greater number of Units than the record holder or warrant holder subscribed for pursuant to the exercise of the Over-Subscription Privilege, then such record holder or warrant holder will be allocated only that number of Units for which the record holder or warrant holder oversubscribed, and the remaining Units will be allocated among all other record holders and warrant holders exercising the Over-Subscription Privilege on the same pro rata basis described above. The proration process will be repeated until all Units have been allocated. See "The Rights Offering—Limitation on the Purchase of Units" for a description of certain limitations on purchase.
To properly exercise your Over-Subscription Privilege, you must deliver to the Subscription Agent the subscription payment related to your Over-Subscription Privilege before the Rights Offering expires. See "The Rights Offering—The Subscription Rights—Over-Subscription Privilege." To the extent you properly exercise your Over-Subscription Privilege for an amount of Units that exceeds the number of unsubscribed Units available to you, any excess subscription payments will be returned to you as soon as practicable after the expiration of the Rights Offering, without interest or penalty.
Wells Fargo Bank, N.A., our Subscription Agent for the Rights Offering, will determine the Over-Subscription allocation based on the formula described above.
What are the terms of the Warrants?
Each Warrant entitles the holder to purchase one share of common stock at an exercise price of $ per share, 120% of the per Unit price, from the date of issuance through its expiration on , 2021. The Warrants will be exercisable by paying the exercise price in cash, or, solely during any period when a registration statement for the exercise of the Warrants is not in effect, exercisable on a cashless basis. After the one-year anniversary of issuance, we may redeem the Warrants for $0.001 per Warrant if the volume weighted average price of our common stock is above $ per share, 300% of the exercise price, for each of 10 consecutive trading days. Subject to certain exceptions, a holder may not exercise any portion of the Warrant to the extent that the holder would beneficially own more than 4.99% of the outstanding common stock after exercise.
What are the terms of the Pre-Funded Warrants?
The Pre-Funded Warrants will only be issued to certain investors whose subscriptions for Units in the Rights Offering may result in the purchaser beneficially owning more than 4.99% of our outstanding common stock following the consummation of the Rights Offering, and who elect to receive Pre-Funded Warrants in lieu of shares of common stock underlying the Units for which the investors have subscribed. You will not be eligible to elect to receive Pre-Funded Warrants, except to the extent that your beneficial ownership could exceed 4.99% of the shares of common stock outstanding following the consummation of the Rights Offering.
Each Pre-Funded Warrant entitles the holder to purchase one share of common stock at an exercise price of $0.01 per share, and the subscription price per Unit for any such electing investors will be reduced to $ (which equals the Subscription Price for the other Units sold in the Rights Offering, less the $0.01 exercise price for each Pre-Funded Warrant). Each Pre-Funded Warrant will be exercisable from the date of issuance through its expiration on , 2021. The Pre-Funded Warrants will be exercisable by paying the exercise price in cash or on a cashless basis in accordance with the terms of the Pre-Funded Warrants. The Pre-Funded Warrants will not be listed for trading on any stock exchange or market. The Pre-Funded Warrants do not confer upon the holder any voting or any other rights of a shareholder of the Company.
Will fractional shares be issued upon exercise of Subscription Rights or upon the exercise of Warrants?
No. We will not issue fractional shares of common stock in the Rights Offering. Rights holders will only be entitled to purchase a number of Units representing a whole number of shares of common stock, rounded down to the nearest whole number of Units a holder would otherwise be entitled to purchase. Any excess subscription payments received by the Subscription Agent will be returned as soon as practicable after expiration of the Rights Offering, without interest or penalty. Similarly, no fractional shares of common stock will be issued in connection with the exercise of a Warrant. If, upon exercise of a Warrant, the holder thereof would be entitled to receive a fractional share of common stock, upon exercise, the holder will only be entitled to receive a whole number of shares of common stock, rounded down to the nearest whole number.
What effect will the Rights Offering have on our outstanding common stock?
After our one-for-ten reverse stock split on May 31, 2016, 2,740,212 shares of our common stock were outstanding, along with participating warrants to purchase 96,842 shares of common stock. Based on the foregoing, and assuming no other transactions by us involving our common stock prior to the expiration of the Rights Offering, if the Rights Offering is fully subscribed, approximately 6,995,793 shares of our common stock will be issued and outstanding and Warrants to purchase approximately 3,191,686 additional shares of our common stock will be outstanding (excluding the currently outstanding warrants). The exact number of shares and Warrants that we will issue in this Rights Offering will depend on the number of Units that are subscribed for in the Rights Offering.
How was the Subscription Price determined?
In determining the Subscription Price, the pricing committee of our board of directors is expected to consider, among other things, the following factors:
In conjunction with the review of these factors, our pricing committee will also review our history and prospects, including our past and present earnings and cash requirements, our prospects for the future, the outlook for our industry and our current financial condition. Our pricing committee believes that the Subscription Price should be designed to provide an incentive to our current shareholders and holders of the participating warrants to participate in the Rights Offering and exercise their Basic Subscription Right and their Over-Subscription Privilege.
The Subscription Price does not necessarily bear any relationship to any established criteria for value. You should not consider the Subscription Price as an indication of actual value of our company or our common stock. We cannot assure you that the market price of our common stock will not decline during or after the Rights Offering. You should obtain a current price quote for our common stock before exercising your Subscription Rights and make your own assessment of our business and financial condition, our prospects for the future, and the terms of this Rights Offering. Once made, all exercises of Subscription Rights are irrevocable.
Am I required to exercise all of the Basic Subscription Rights I receive in the Rights Offering?
No. You may exercise any number of your Basic Subscription Rights, or you may choose not to exercise any Basic Subscription Rights. If you do not exercise any Basic Subscription Rights, the number of shares of our common stock or number of shares underlying our warrants you own will not change. However, if you choose to not exercise your Basic Subscription Rights in full, your proportionate ownership interest in our company will decrease. If you do not exercise your Basic Subscription Rights in full, you will not be entitled to exercise your Over-Subscription Privilege.
How soon must I act to exercise my Subscription Rights?
If you received a Subscription Rights Statement and elect to exercise any or all of your Subscription Rights, the Subscription Agent must receive your completed and signed Subscription Rights Statement and payment for both your Basic Subscription Rights and any Over-Subscription Privilege you elect to exercise, including final clearance of any uncertified check, before the Rights Offering expires on July 26, 2016, at 5:00 PM Eastern Time. If you hold your shares in the name of a broker, dealer, custodian bank, or other nominee, your nominee may establish a deadline before the expiration of the Rights Offering by which you must provide it with your instructions to exercise your Subscription Rights, along with the required subscription payment.
May I transfer my Subscription Rights?
No. The Subscription Rights may be exercised only by the shareholders or participating warrant holders to whom they are distributed, and they may not be sold, transferred, assigned or given away to anyone else, other than by operation of law. As a result, a Subscription Rights Statement may be completed only by the shareholder or participating warrant holder who receives the statement. The Subscription Rights will not be listed for trading on any stock exchange or market.
Will our directors and executive officers participate in the Rights Offering?
To the extent they hold common stock as of the Record Date, our directors and executive officers will be entitled to participate in the Rights Offering on the same terms and conditions applicable to
other Rights holders. While none of our directors or executive officers has entered into any binding commitment or agreement to exercise Subscription Rights received in the Rights Offering, our directors and executive officers have indicated interests in subscribing for up to an aggregate of $3.0 million in the Rights Offering, subject to potential proration.
Has the board of directors made a recommendation to shareholders and warrant holders regarding the Rights Offering?
No. Our board of directors is not making a recommendation regarding your exercise of the Subscription Rights. Stockholders and holders of the participating warrants who exercise Subscription Rights will incur investment risk on new money invested. We cannot predict the price at which our shares of common stock will trade after the Rights Offering. On June 28, 2016, the closing price of our common stock was $6.00 per share. The market price for our common stock may be above the Subscription Price or may be below the Subscription Price. If you exercise your Subscription Rights, you may not be able to sell the underlying shares of our common stock or Warrants in the future at the same price or a higher price. You should make your decision based on your assessment of our business and financial condition, our prospects for the future, the terms of the Rights Offering and the information contained in this prospectus. See "Risk Factors" for discussion of some of the risks involved in investing in our securities.
How do I exercise my Subscription Rights?
If you are a shareholder of record (meaning you hold your shares of our common stock in your name and not through a broker, dealer, bank, or other nominee) or a holder of the participating warrants and you wish to participate in the Rights Offering, you must deliver a properly completed and signed Subscription Rights Statement, together with payment of the Subscription Price for both your Basic Subscription Rights and any Over-Subscription Privilege you elect to exercise, to the Subscription Agent before 5:00 PM Eastern Time, on July 26, 2016. If you are exercising your Subscription Rights through your broker, dealer, bank, or other nominee, you should promptly contact your broker, dealer, bank, or other nominee and submit your subscription documents and payment for the Units subscribed for in accordance with the instructions and within the time period provided by your broker, dealer, bank or other nominee.
What if my shares are held in "street name"?
If you hold your shares of our common stock in the name of a broker, dealer, bank, or other nominee, then your broker, dealer, bank, or other nominee is the record holder of the shares you own. The record holder must exercise the Subscription Rights on your behalf. Therefore, you will need to have your record holder act for you.
If you wish to participate in this Rights Offering and purchase Units, please promptly contact the record holder of your shares. We will ask the record holder of your shares, who may be your broker, dealer, bank, or other nominee, to notify you of this Rights Offering.
What form of payment is required?
You must timely pay the full Subscription Price for the full number of Units you wish to acquire pursuant to the exercise of Subscription Rights by delivering to the Subscription Agent a:
If you send a payment that is insufficient to purchase the number of Units you requested, or if the number of Units you requested is not specified in the forms, the payment received will be applied to
exercise your Subscription Rights to the fullest extent possible based on the amount of the payment received.
When will I receive my new shares of common stock and Warrants?
The Subscription Agent will arrange for the issuance of the common stock and Warrants as soon as practicable after the expiration of the Rights Offering, payment for the Units subscribed for has cleared, and all prorating calculations and reductions contemplated by the terms of the Rights Offering have been effected. All shares and Warrants that you purchase in the Rights Offering will be issued in book-entry, or uncertificated, form meaning that you will receive a direct registration (DRS) account statement from our transfer agent reflecting ownership of these securities if you are a holder of record of shares or warrants. If you hold your shares in the name of a broker, dealer, bank, or other nominee, DTC will credit your account with your nominee with the securities you purchase in the Rights Offering.
When will I receive my Pre-Funded Warrants?
If you elect to receive any Pre-Funded Warrants, the Subscription Agent will arrange for the issuance of the Pre-Funded Warrants as soon as practicable after the expiration of the Rights Offering, payment for the Units subscribed for has cleared, and all prorating calculations and reductions contemplated by the terms of the Rights Offering have been effected. All Pre-Funded Warrants will be issued in physical form.
After I send in my payment and Subscription Rights Statement to the Subscription Agent, may I cancel my exercise of Subscription Rights?
No. Exercises of Subscription Rights are irrevocable unless the Rights Offering is terminated, even if you later learn information that you consider to be unfavorable to the exercise of your Subscription Rights. You should not exercise your Subscription Rights unless you are certain that you wish to purchase Units at the Subscription Price.
How much will our company receive from the Rights Offering?
Assuming that all 4,255,581 Units are sold in the Rights Offering, we estimate that the net proceeds from the Rights Offering will be approximately $23.0 million, based on the midpoint of the expected Subscription Price range of $6.00 per Unit, after deducting fees and expenses payable to the dealer-manager, and after deducting other expenses payable by us and excluding any proceeds received upon exercise of any Warrants issued in the Rights Offering.
Are there risks in exercising my Subscription Rights?
Yes. The exercise of your Subscription Rights involves risks. Exercising your Subscription Rights involves the purchase of additional shares of our common stock and Warrants to purchase common stock and you should consider this investment as carefully as you would consider any other investment. We cannot assure you that the market price of our common stock will exceed the Subscription Price, nor can we assure you that the market price of our common stock will not further decline during or after the Rights Offering. We also cannot assure you that you will be able to sell shares of our common stock or Warrants purchased in the Rights Offering at a price equal to or greater than the Subscription Price. In addition, you should carefully consider the risks described under the heading "Risk Factors" for discussion of some of the risks involved in investing in our securities.
Can the board of directors terminate or extend the Rights Offering?
Yes. Our board of directors may decide to terminate the Rights Offering at any time and for any reason before the expiration of the Rights Offering. We also have the right to extend the Rights Offering for additional periods in our sole discretion. We do not presently intend to extend the Rights Offering. We will notify shareholders and warrant holders if the Rights Offering is terminated or extended by issuing a press release.
If the Rights Offering is not completed or is terminated, will my subscription payment be refunded to me?
Yes. The Subscription Agent will hold all funds it receives in a segregated bank account until completion of the Rights Offering. If we do not complete the Rights Offering, all subscription payments received by the Subscription Agent will be returned as soon as practicable after the termination or expiration of the Rights Offering, without interest or penalty. If you own shares in "street name," it may take longer for you to receive your subscription payment because the Subscription Agent will return payments through the record holder of your shares.
How do I exercise my Rights if I live outside the United States?
The Subscription Agent will hold Subscription Rights Statements for shareholders having addresses outside the United States. To exercise Subscription Rights, foreign shareholders must notify the Subscription Agent and timely follow other procedures described in the section entitled "The Rights Offering—Foreign Shareholders."
What fees or charges apply if I purchase shares in the Rights Offering?
We are not charging any fee or sales commission to issue Subscription Rights to you or to issue shares or Warrants to you if you exercise your Subscription Rights. If you exercise your Subscription Rights through a broker, dealer, custodian bank, or other nominee, you are responsible for paying any fees your broker, dealer, bank, or other nominee may charge you.
What are the U.S. federal income tax consequences of exercising my Subscription Rights?
For U.S. federal income tax purposes, we do not believe you should recognize income or loss in connection with the receipt or exercise of Subscription Rights in the Rights Offering. You should consult your tax advisor as to the tax consequences of the Rights Offering in light of your particular circumstances. For a more detailed discussion, see "Material U.S. Federal Income Tax Consequences."
To whom should I send my forms and payment?
If your shares are held in the name of a broker, dealer, bank, or other nominee, then you should send your subscription documents and subscription payment to that broker, dealer, bank, or other nominee. If you are the record holder or a holder of participating warrants, then you should send your Subscription Rights Statement and payment of your subscription price to the Subscription Agent hand delivery, first class mail or courier service to:
By Mail:
Wells Fargo Bank, N.A.
Shareowner Services
Voluntary Corporate Actions
P.O. Box 64858
St. Paul, Minnesota 55164-0858
By Hand or Overnight Courier:
Wells Fargo Bank, N.A.
Shareowner Services
Voluntary Corporate Actions
1110 Centre Pointe Curve, Suite 101
Mendota Heights, Minnesota 55120
You or, if applicable, your nominee are solely responsible for completing delivery to the Subscription Agent of your subscription documents, Subscription Rights Statement and payment. You should allow sufficient time for delivery of your subscription materials to the Subscription Agent before the expiration of the Rights Offering at 5:00 PM Eastern Time on July 26, 2016.
Whom should I contact if I have other questions?
If you have other questions or need assistance, please contact the dealer-manager for the Rights Offering:
Maxim Group LLC
405 Lexington Avenue
New York, New York 10174
Attention Syndicate Department
Email: syndicate@maximgrp.com
Telephone: (212) 895-3745
Who is the dealer-manager?
Maxim Group LLC will act as dealer-manager for the Rights Offering. Under the terms and subject to the conditions contained in the dealer-manager agreement, the dealer-manager will use its best efforts to solicit the exercise of Subscription Rights. We have agreed to pay the dealer-manager certain fees for acting as dealer-manager and to reimburse the dealer-manager for certain out-of-pocket expenses incurred in connection with this offering. The dealer-manager is not underwriting or placing any of the Subscription Rights or the Units, shares of common stock or Warrants being issued in this offering, and does not make any recommendation with respect to such Subscription Rights (including with respect to the exercise or expiration of such Subscription Rights), Units, shares of common stock or Warrants. See "Plan of Distribution" for a discussion of the fees and expenses to be paid to the dealer-manager in connection with this Rights Offering.
The items in the following summary are described in more detail later in this prospectus and in the documents we incorporate by reference. This summary highlights certainprovides an overview of selected information about us and this offering contained elsewhere in this prospectus. Because it is only a summary, it does not contain all of the information that you should consider beforeconsider. Before investing in shares of our common stock and it is qualified in its entirety by, and should be read in conjunction with, the more detailed information appearing elsewhere in this prospectus. Before you decide to invest in our common stock, you should read the entire prospectus carefully, including the information set forth under the headings "Risk Factors" beginning on page 10 andFactors "and the consolidated financial statements and related notes and additional information included or incorporated by reference in this prospectus.
Unless the context indicates otherwise, as used in this prospectus, the terms "Onconova," "Onconova Therapeutics," "we," "us," "our," "our company" and "our business" refer to
OUR COMPANY
Our Business
Onconova Therapeutics, Inc.
Overview
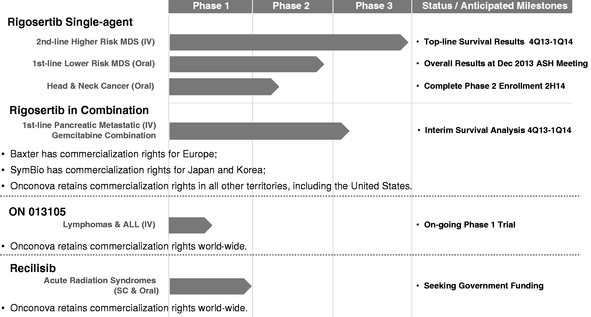
We are, sometimes referred to as "we" or the "Company," is a clinical-stage biopharmaceutical company focused on discovering and developing novel small molecule drug candidates to treat cancer. Using our proprietary chemistry platform, we have created an extensive library of targeted anti-cancer agents designed to work against specific cellular pathways that promote cancer.important to cancer cells. We believe that the drug candidates in our pipeline have the potential to be efficacious in a wide variety of cancers without causing harm to normal cells.cancers. We have threeone Phase 3 clinical-stage product candidate and two other clinical-stage product candidates (one of which is being developed for treatment of acute radiation syndromes) and sixseveral preclinical programs.
Rigosertib, Substantially all of our most advancedcurrent effort is focused on our lead product candidate, rigosertib. Rigosertib is being tested in both intravenous and oral formulations as a number of ongoing Phase 2single agent, and the oral formulation is also being tested in combination with azacitidine, in clinical trials for patients with myelodysplastic syndromes, or MDS, and related cancers.
In December 2015, we enrolled the first patient in a randomized controlled Phase 3 clinical trial of rigosertib IV in a population of patients with higher-risk MDS after failure of hypomethylating agent, or HMA, therapy. The trial, which we refer to as INSPIRE, is expected to enroll approximately 225 patients at more than 100 sites globally. The primary endpoint of INSPIRE is overall survival, and an interim analysis is anticipated. We anticipate reporting topline data from the INSPIRE trial in 2018.
In the first quarter of 2016, we took significant actions to conserve cash, including reduction in personnel and expenditures. While we will continue to take cash conservation actions where appropriate, our costs will increase in subsequent quarters as more INSPIRE sites open and more patients enroll in the INSPIRE trial. We believe that our cash and cash equivalents, prior to our receipt of any proceeds from this Rights Offering, together with anticipated contractual cost-sharing payments from Baxalta GmbH, or Baxalta, for a portion of the INSPIRE trial costs, will be sufficient to fund our ongoing trials and operations into the first quarter of 2017.
We are exploring various sources of funding for continued development of rigosertib in MDS and acute myelogenous leukemia, or AML, as well as our ongoing operations. Completion of the Rights Offering is not subject to us raising a minimum offering amount and therefore the net proceeds from the Rights Offering may be insufficient to meet our objectives. Even if we sell all of the Units subject to the Rights Offering, we will need to obtain additional financing in the future in order to fully fund rigosertib or any other product candidates through the regulatory approval process.
Due to our ongoing losses and our accumulated deficit in combination with other factors, the opinion of our independent registered public accounting firm on our audited consolidated financial statements for our fiscal year ended December 31, 2015 contains an explanatory paragraph regarding substantial doubt about our ability to continue as a going concern. Even if we sell all of the Units subject to the Rights Offering, if we are unable to obtain sufficient additional funding, through future financings or through strategic and collaborative transactions and arrangements, we may not have sufficient resources to complete our INSPIRE trial and we may continue to delay, scale-back or
eliminate certain of our planned research, drug discovery and development activities and certain other aspects of our operations, or we may not be able to continue as a going concern.
Rigosertib
Rigosertib is a small molecule that inhibits cellular signaling by acting as a Ras mimetic. This is believed to be mediated by the binding of rigosertib to the Ras-binding domain, or RBD, found in many Ras effector proteins, including the Raf and PI3K kinases. This mechanism of action provides a new approach to block the interactions between Ras and its targets containing RBD sites. Rigosertib is being tested as a single agent and in combination with azacitidine, in clinical trials of patients with MDS and related cancers. We have enrolled more than 1,200 patients in rigosertib clinical trials. We are conducting a pivotal Phase 3 trial ofparty to a license and development agreement with Baxalta, which granted Baxalta certain rights to commercialize rigosertib under a Special Protocol Assessment, or SPA, fromin Europe. The Baxalta agreement is scheduled to terminate August 30, 2016, at which time the U.S. Food and Drug Administration, or FDA, for higher risk myelodysplastic syndromes, or MDS. We expectEuropean rights will revert to report top-line overall survival results from this trial in the fourth quarter of 2013 or the first quarter of 2014.us at no cost. We are also evaluating rigosertib inparty to a Phase 3 trial for metastatic pancreatic cancer, in two Phase 2 trials for transfusion-dependent lower risk MDS, and in a Phase 2 trial for head and neck cancers. We have tested rigosertib in more than 850 patientscollaboration agreement with solid tumors and hematological diseases. Rigosertib has been granted orphan drug status for MDS in both the United States and Europe as well as orphan drug status for pancreatic cancer in the United States. Baxter Healthcare SA, or Baxter, a subsidiary of Baxter International Inc., has commercialization rights for rigosertib in Europe and SymBio Pharmaceuticals Limited, or SymBio, has commercializationwhich grants SymBio certain rights to commercialize rigosertib in Japan and Korea. We have retained development and commercialization rights to rigosertib in the rest of the world, including in the United States.States, although we will consider licensing commercialization rights to other territories as a source of additional funding.
Rigosertib IV for higher-risk MDS
Rigosertib is an inhibitorIn early 2014, we announced topline survival results from our "ONTIME" trial, a multi-center Phase 3 clinical trial of two important cellular signaling pathways: phosphoinositide 3-kinase, or PI3K,rigosertib IV as a single agent. The ONTIME trial did not meet its primary endpoint in the intent-to-treat population, although improvements in median overall survival were observed in various pre-specified and polo-like kinase, or PLK, bothexploratory subgroups of which are frequently over-active in cancer cells. PI3K signaling promotes the growth and survival of cells under stressful conditions, such as under low oxygen levels that are often found in tumors. By inhibiting the PI3K pathway in cancer cells, rigosertib promotes tumor cell apoptosis, or programmed cell death.higher-risk MDS patients.
During 2014 and 2015, we held meetings with the U.S. Food and Drug Administration, or FDA, European Medicines Agency, or EMA, and several European national regulatory authorities to discuss and seek guidance on a path for approval of rigosertib IV in higher-risk MDS patients whose disease had failed HMA therapy. After discussions with the FDA and EMA, we refined our patient eligibility criteria by defining a more homogenous patient population. After regulatory feedback, input from key opinion leaders in the U.S. and Europe and based on learnings from the ONTIME study, we designed a new randomized controlled Phase 3 trial, referred to as INSPIRE, with overall survival as a primary endpoint. The PLK pathwayINSPIRE trial is enrolling higher-risk MDS patients under 80 years of age who have progressed on, or failed to respond to, previous treatment with HMAs within the first nine months after initiation of HMA therapy, and had their last dose of HMA within six months prior to enrollment in the trial. The primary endpoint of this study is overall survival, and an interim analysis is anticipated. This randomized trial of approximately 225 patients is expected to be conducted at more than 100 sites globally. In August 2015, we submitted an updated investigational new drug application, or IND, to the FDA, and in August 2015 we submitted Clinical Trial Applications, or CTAs, with the United Kingdom, German and Austrian regulatory authorities for IV rigosertib as a treatment for higher-risk MDS after failure of HMA therapy. The first CTA has a critical rolebeen cleared by the Medicines and Healthcare products Regulatory Agency. The first patient in maintaining proper chromosome organizationthe INSPIRE trial was enrolled at the MD Anderson Cancer Center in December 2015 and, sorting during cell division. By modulatingas of May 18, 2016, 59 clinical sites are open (43 in the PLK pathway,U.S. and Europe and 16 in Japan) and can recruit patients. The first patient in Europe was enrolled on March 18, 2016.
Rigosertib oral in combination with azacitidine for MDS and AML
We have completed enrollment in the Phase 2 portion of an open label Phase 1/2 clinical trial testing rigosertib stops cancer cells at late stagesoral in combination with the approved dose of injectable azacitidine for patients with higher-risk MDS and AML. This study is based on our published preclinical data demonstrating synergistic activity of this combination. The Phase 2 portion of the cell division cycle, which leadstrial was designed to chromosome disorganization and death in these cells. In normal cells, rigosertib pauses progression of the cell cycle in the early stages, without causing harm or death to these cells.
Due to this dual effect of inhibiting both the PI3K and PLK pathways, and thereby effecting both tumor cell survival and division, we believe that rigosertib has potential to treat a variety of cancer types, including hematological diseases and solid tumors.
assess whether
We are testing both intravenous and oral formulationstreatment with rigosertib, in combination with the approved dose of rigosertib, referred to as rigosertib IV and rigosertib Oral, in clinical trials.
The Phase 2 trial included both front-line MDS patients in four early-stage Phase 1/2 trials of rigosertib IV involving 39 refractory(that is, patients not previously treated with HMAs) and MDS patients we observed objective responses in 12whose disease had failed prior HMA therapy (second-line patients). The presentation at ASH included results from a total of 37 MDS patients fivetreated with the recommended Phase 2 dose of whichoral rigosertib (560 mg AM/280 mg PM) plus the full standard dose of injectable azacitidine. The combination of oral rigosertib and azacitidine was generally well tolerated, with a median duration of treatment of four months (range 1 to 27 months).
At the time of the presentation, 30 MDS patients were complete bone marrow responses. To our knowledge, there are no other current Phase 3 trials in this patient population. If we achieve positive results in this trial, we intend to submit a New Drug Application, or NDA,evaluable for efficacy assessment per 2006 IWG, criteria. Twenty-three of 30 patients (77%) responded to the FDAcombination therapy, including six patients who had complete remissions. Hematologic improvement was observed in 13 of 26 patients that were evaluable for this part of the second halfanalysis. Notably, 16 of 2014, and19 (84%) HMA-naïve patients had a Marketing Authorization Application, or MAA,response to the European Medicines Agency, orcombination therapy and 7 of 11 (64%) patients whose disease had previously failed HMAs responded. As of December 2015, the EMA, by the fourth quartermedian duration of 2014 or the first quarter of 2015these responses had not yet been reached. Additional data collection for marketing approval of rigosertib IV.
A provider of marketing analyticsefficacy and data to the biopharmaceutical industry has estimated that, for 2011 in the United States, the diagnosed incidence of MDS was approximately 15,600 and the prevalence of MDS was approximately 52,000. According to the same marketing analytics firm, approximately 23% of MDS patients are estimated to fall into the categories of MDS characterized as higher risk.
Rigosertib Oraloral for lower-risk MDS
Higher-risk MDS patients suffer from a shortfall in lower risk MDS:normal circulating blood cells, or cytopenias, as well as elevated levels of cancer cells, or blasts in their bone marrow and peripheral blood, whereas lower-risk MDS patients suffer mainly from cytopenias, that is low levels of red blood cells, white blood cells and/or platelets. Thus, lower-risk MDS patients depend on transfusions and growth factors or other therapies to improve their low blood counts.
We are evaluatinghave explored single agent rigosertib Oraloral as a treatment for lower-risk MDS in two Phase 2 clinical trials, as a first-line treatment for09-05 and 09-07. In December 2013, we presented data at the Annual ASH Meeting from the 09-05 Phase 2 trial. To date, Phase 2 clinical data have shown encouraging signs of efficacy of single agent oral rigosertib in transfusion-dependent, lower risklower-risk MDS patients. The qualityRigosertib has been generally well tolerated, except for urinary side effects at higher dose levels. Additional clinical testing will be needed to evaluate dosing and schedule modifications and their impact on efficacy and toxicity of life of these patients could be significantly improved by lowering the number of required blood transfusions or eliminating the need for transfusions altogether. We reported initial response and safety dataoral rigosertib in lower-risk MDS patients.
Data presented from the first Phase 209-05 trial in June 2013 and expectalso suggested the potential of a genomic methylation assessment of bone marrow cells to complete enrollment and present overall results from this trial in December 2013. Upon completion of the first Phase 2 trial, we will meet with the FDA to discuss an approval pathway for rigosertib Oral as a first-line treatment in lower risk MDS patients. We expect to complete enrollment in the second Phase 2 trial in lower risk, transfusion-dependentprospectively identify lower-risk MDS patients who have failed erythroid stimulating agents inlikely to respond to oral rigosertib. We therefore expanded the second half09-05 trial by adding an additional cohort of 2014. Approximately 77% of MDS20 patients are estimated to fall into the categories of MDS characterized as lower risk.
To accelerate and broadenadvance the development of rigosertib, we entered into a development and licensing agreementthis genomic methylation test. Enrollment in this expansion cohort has been completed. We are working with Baxter in 2012academic collaborators to commercialize rigosertib in Europe. In 2011, we entered into a licensing agreement with SymBio to commercialize rigosertib in Japan and Korea. We have retained development and commercialization rights to rigosertib in the restrefine this genomic methylation test.
Other Programs
The vast majority of the world, includingCompany's efforts are now devoted to the
advanced stage development of rigosertib for unmet medical needs of MDS patients. Other programs are either paused, inactive or require only minimal internal resources and efforts.
United States.Briciclib
Briciclib, another of our product candidates, is a small molecule targeting an important intracellular regulatory protein, cyclin D1, which is often found at elevated levels in cancer cells. Cyclin D1 expression is regulated through a process termed cap-dependent translation, which requires the function of eukaryotic initiation factor 4E protein, or eIF4E. In vitro evidence indicates briciclib binds to eIF4E, blocking cap-dependent translation of cyclin D1 and other cancer proteins, such as c-MYC, leading to tumor cell death. We will explore a variety of alternatives for the commercialization of rigosertib in territories we currently retain, including direct commercialization, co-promotion or selective out-licensing of rights to a third party.
Our second clinical-stage product candidate,ON 013105, is inhave been conducting a Phase 1 multisite dose-escalation trial of briciclib in patients with relapsed oradvanced solid tumors refractory lymphoma, including an aggressive formto current therapies. Safety and efficacy assessments are complete in six of non-Hodgkin's lymphoma identified as mantle cell lymphoma, or MCL, and acute lymphoid leukemia, or ALL. A critical defect in many cancer cells is the uncontrolled expressionseven dose-escalation cohorts of cyclin D1, a protein essential for normal cell division. Cyclin D1 is over-expressed in several hematological diseases, including B-cell lymphomas, such as MCL. ON 013105 suppresses the accumulation of cyclin D1 in cancer cells. In 2011, we suspended enrollmentpatients in this Phase 1 trial because enrollmenttrial. As of patients was occurring slowly, and asDecember 2015, the briciclib IND is on full clinical hold following a result, our inventorydrug product lot testing failure. If we elect to continue development of ON 013105briciclib, we would be required to undertake appropriate remedial actions prior to re-initiating the clinical trial materials expired. We plan to restart enrollment in this trial with newly manufactured clinical trial materials atand completing the final dose-escalation cohort.
Recilisib
Recilisib is a new clinical trial site in the fourth quarter of 2013.
Our third clinical-stage product candidaterecilisib, is being developed in collaboration with the U.S. Department of Defense or DoD, for acute radiation syndromes, or ARS. We have conducted animal studies and clinical trials of recilisib under the FDA's Animal Efficacy Rule, which permits marketing approval for new medical countermeasures for which human efficacy studies are not feasible or ethical, by relying on evidence from animal studies in appropriate animal models to support efficacy in humans.syndromes. We have completed four Phase 1 trials to evaluate the safety and pharmacokinetics of recilisib in healthy human adult subjects using both subcutaneous and oral formulations. We have received orphan drug designationalso conducted animal studies and clinical trials of recilisib under the FDA's Animal Efficacy Rule, which permits marketing approval for new medical countermeasures for which conventional human efficacy studies are not feasible or ethical, by relying on evidence from studies in appropriate animal models to support efficacy in humans. Ongoing studies of recilisib, for ARS infocusing on animal models and biomarker development to assess the United States.
Theefficacy of recilisib are being conducted by third parties with government funding. We anticipate that any future development status of our three clinical-stage product candidates is summarized below:recilisib beyond these ongoing studies would be conducted solely with government funding or by collaboration.
Clinical-StagePreclinical Product Candidates
In addition to our three clinical-stage product candidates, we are advancing six preclinical programshave several product candidates that target kinases, cellular metabolism or division.
cell division in preclinical development. We have broad-based capabilities that span drug discovery and clinical development, from medicinal chemistry and evaluation in biochemical, cell-based and animal models, through Phase 3 trials and regulatory filings in the United States, Europe and India. Our discovery program is based on a proprietary chemistry platform comprising more than 150 novel core chemical structures. Our
chemistry and screening approaches aim to discover new drug candidates that increase efficacy and help overcome resistance to therapy in cancer cells, while minimizing their toxicity to normal cells. Our intellectual property portfolio includes more than 100 issued patents and over 90 patent applications, either owned by us or licensed exclusively to us, including patents covering our most advanced product candidate, rigosertib. These patents and licenses cover composition-of-matter, process, formulations and method-of-treatment claims for our clinical-stage product portfolio.
Our Strategy
We are committed to delivering novel treatments to cancer patients. We are focused on discovering and developing targeted small molecule anti-cancer product candidates. The key components of our strategy are to:
several lead molecules in our preclinical pipeline. We intend to explore additional collaborations to further the development of these product candidates.
Recent Developments
We effected a one-for-ten reverse stock split of our Programs
Risks Associated with Our Business
Our ability to implement our business strategy is subject to numerous risks and uncertainties. As a clinical-stage biopharmaceutical company, we face many risks inherent in our business and our industry generally. You should carefully consider all of the information set forth in this prospectus and, in particular, the information under the heading "Risk Factors," prior to making an investment in our common stock. These risks include, among others, the following:
Our Corporate Information
We were formed as Onconova Therapeutics, Inc., a corporation under the laws of the State ofincorporated in Delaware in December 1998 and commenced operations in January 1999. Our primaryprincipal executive offices are located at 375 Pheasant Run, Newtown, PAPennsylvania 18940, and our telephone number is (267) 759-3680. Our website address ishttp://www.onconova.com. www.onconova.com. The information contained in,on, or that can be accessed through, our website is not part of this prospectus.
We have registered several U.S. trademarks, including Onconova Therapeutics, Inc. All other trademarks, trade names or service marks referred to in this prospectus are the property of their respective owners.
Implications of Being an Emerging Growth Company
We arequalify as an "emerging growth company,"company" as defined in Section 2(a) of the SecuritiesJumpstart our Business Startups Act of 1933, as amended,2012, or the Securities Act, as modified by the Jumpstart Our Business Act, or JOBS Act. As such,
An emerging growth company may take advantage of specified reduced
we are eligible to take advantagereporting requirements and is relieved of exemptions from various reportingcertain other significant requirements that are otherwise generally applicable to other public companies that are not "emergingcompanies. As an emerging growth companies" including, but not limited to:company,
We may choosewe have elected to take advantage of some or all of the available exemptions. We have taken advantage of some of the reduced reporting burdens in this prospectus. Accordingly, the information contained herein may be different than the information you receive from other public companies in which you hold stock. We do not know if some investors will find our shares less attractive as a result of our utilization of these or other exemptions. The result may be a less active trading market for our shares and our share price may be more volatile.
In addition, Section 107 of the JOBS Act also provides thatuse an "emerging growth company" can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. In other words, an "emerging growth company" can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
We will remain an "emerging growth company"may take advantage of these provisions until the earliest of (a) the last day of the first fiscal year in whichJuly 24, 2018. However, if certain events occur prior to such date, including if we become a "large accelerated filer," our annual gross revenues exceed $1.0 billion (b) the date thator we become a "large accelerated filer" as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended, or the Exchange Act, which would occur if the market value of our shares that are held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter, (c) the date on which we have issuedissue more than $1.0 billion of non-convertible debt in nonconvertible debt during the precedingany three-year period, or (d) the last day of our fiscal year containing the fifth anniversary of the date on which shares of our common stock become publicly traded in the United States.
we will cease to be an emerging growth company.
SUMMARY CONSOLIDATED FINANCIAL DATA
The following summary consolidated statements of operations data for the years ended December 31, 2015 and 2014 and the consolidated balance sheet data as of December 31, 2015, have been derived from our audited consolidated financial statements that are included in the documents incorporated by reference into this prospectus. The summary historical financial information as of and for the three months ended March 31, 2016 has been derived from our unaudited consolidated financial statements that are included in the documents incorporated by reference into this prospectus. The unaudited financial information as of and for the three months ended March 31, 2016 has been prepared on the same basis as our audited financial statements and includes, in the opinion of management, all adjustments, consisting of only normal recurring adjustments, necessary to fairly present the data for such periods. All share and per share data has been restated to reflect our one-for-ten reverse stock split effective May 31, 2016. The historical financial data presented below is not necessarily indicative of our financial results in future periods. You should read the summary consolidated financial data in conjunction with those financial statements and the accompanying notes and "Management's Discussion and Analysis of Financial Condition and Results of Operations" in the documents incorporated by reference into this prospectus. Our consolidated financial statements are prepared and presented in accordance with United States generally accepted accounting principles, or U.S. GAAP.
Condensed Consolidated Statements of Operations Data:
| | For the Years ended December 31, | | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | For the three months ended March 31, 2016 | |||||||||
| | 2015 | 2014 | ||||||||
Revenue | $ | 11,456,000 | $ | 800,000 | $ | 1,474,000 | ||||
Operating expenses: | ||||||||||
General and administrative | 9,533,000 | 15,119,000 | 3,172,000 | |||||||
Research and development | 25,895,000 | 49,425,000 | 5,822,000 | |||||||
| | | | | | | | | | | |
Total operating expenses | 35,428,000 | 64,544,000 | 8,994,000 | |||||||
| | | | | | | | | | | |
Loss from operations | (23,972,000 | ) | (63,744,000 | ) | (7,520,000 | ) | ||||
Change in fair value of warrant liability | — | 20,000 | 271,000 | |||||||
Other income (expense), net | (35,000 | ) | (52,000 | ) | 9,000 | |||||
| | | | | | | | | | | |
Net loss before income taxes | (24,007,000 | ) | (63,776,000 | ) | (7,240,000 | ) | ||||
Income taxes | 16,000 | 19,000 | — | |||||||
| | | | | | | | | | | |
Net loss | (24,023,000 | ) | (63,795,000 | ) | (7,240,000 | ) | ||||
Net loss attributable to non-controlling interest | 44,000 | 113,000 | — | |||||||
| | | | | | | | | | | |
Net loss attributable to Onconova Therapeutics, Inc | (23,979,000 | ) | (63,682,000 | ) | $ | (7,240,000 | ) | |||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Net loss per share of common stock, basic and diluted | $ | (10.54 | ) | $ | (29.41 | ) | $ | (2.65 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Basic and diluted weighted average shares outstanding | 2,273,976 | 2,165,354 | 2,731,590 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Consolidated Balance Sheet Data:
(Stockholders' equity has been restated to reflect the one-for-ten
reverse stock split and decrease in authorized common shares from
75,000,000 to 25,000,000)
| | December 31, 2015 | March 31, 2016 | |||||
|---|---|---|---|---|---|---|---|
Assets | |||||||
Current assets: | |||||||
Cash and cash equivalents | $ | 19,799,000 | $ | 16,835,000 | |||
Receivables | 1,504,000 | 1,368,000 | |||||
Prepaid expenses and other current assets | 1,832,000 | 1,153,000 | |||||
Restricted cash | 50,000 | 50,000 | |||||
| | | | | | | | |
Total current assets | 23,185,000 | 19,406,000 | |||||
Property and equipment, net | 248,000 | 224,000 | |||||
Other non-current assets | 12,000 | 12,000 | |||||
| | | | | | | | |
Total assets | $ | 23,445,000 | $ | 19,642,000 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Liabilities and stockholders' equity | |||||||
Current liabilities: | |||||||
Accounts payable | $ | 3,421,000 | $ | 3,296,000 | |||
Accrued expenses and other current liabilities | 3,729,000 | 3,891,000 | |||||
Deferred revenue | 455,000 | 455,000 | |||||
| | | | | | | | |
Total current liabilities | 7,605,000 | 7,642,000 | |||||
Warrant liability | — | 295,000 | |||||
Deferred revenue, non-current | 5,000,000 | 4,886,000 | |||||
| | | | | | | | |
Total liabilities | 12,605,000 | 12,823,000 | |||||
| | | | | | | | |
Commitments and contingencies | |||||||
Stockholders' equity: | |||||||
Preferred stock, $0.01 par value, 5,000,000 authorized at December 31, 2015 and March 31, 2016, none issued and outstanding at March 31, 2016 and December 31, 2015 | — | — | |||||
Common stock, $0.01 par value, 25,000,000 authorized at December 31, 2015 and March 31, 2016, 2,546,419 and 2,740,212 shares issued and outstanding at December 31, 2015 and March 31, 2016 | 25,000 | 27,000 | |||||
Additional paid-in capital | 328,564,000 | 331,775,000 | |||||
Accumulated other comprehensive loss | (22,000 | ) | (16,000 | ) | |||
Accumulated deficit | (318,557,000 | ) | (325,797,000 | ) | |||
| | | | | | | | |
Total Onconova Therapeutics, Inc. stockholders' equity | 10,010,000 | 5,989,000 | |||||
Non-controlling interest | 830,000 | 830,000 | |||||
| | | | | | | | |
Total stockholders' equity | 10,840,000 | 6,819,000 | |||||
| | | | | | | | |
Total liabilities and stockholders' equity | $ | 23,445,000 | $ | 19,642,000 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
SUMMARY OF THE RIGHTS OFFERING
| We are distributing to you, at no charge, 1.5 non-transferable Subscription Rights to purchase one Unit for every share of our common stock that you owned on the Record Date, either as a holder of record or, in the case of shares | |
|
| |
|
| |
|
| |
| For certain investors whose subscriptions may result in the | ||
| Holders of our currently outstanding participating warrants will also be entitled, pursuant to the terms of such warrants, to receive 1.5 Subscription Rights to purchase one Unit for every share of common stock as if each of such participating warrants had been exercised immediately prior to the record date for the Rights Offering. | ||
|
| |
|
| |
Warrants | Each Warrant entitles the holder to purchase one share of | |
Unless otherwise noted, the information in this prospectus assumes:
The number of shares of common stock to be outstanding after this offering is based on 20,591,954 shares of common stock outstanding as of March 31, 2013, after giving effect to the conversion of our outstanding shares of preferred stock into 17,113,481 shares of common stock, and excludes as of that date:
SUMMARY CONSOLIDATED FINANCIAL DATA
The following table summarizes our historical financial data as of the dates indicated and for the periods then ended. We have derived the following statement of operations data for the years ended December 31, 2011 and 2012 from our audited consolidated financial statements included elsewhere in this prospectus. We have derived the following statement of operations data for the three months ended March 31, 2012 and 2013 and balance sheet data as of March 31, 2013 from our unaudited condensed consolidated financial statements included elsewhere in this prospectus. Our historical results are not necessarily indicative of the results that may be expected in the future, and our interim period results are not necessarily indicative of results to be expected for a full year or any other interim period. The summary of our consolidated financial data set forth below should be read together with our consolidated financial statements and the related notes to those statements, as well as "Management's Discussion and Analysis of Financial Condition and Results of Operations," appearing elsewhere in this prospectus.
| exercisable by paying the exercise price in cash, or, solely during any period when a registration statement for the exercise of the Warrants is not in effect, exercisable on a cashless basis. We have applied to list the Warrants on NASDAQ under the symbol "ONTXW," although there is no assurance that a sufficient number of Subscription Rights will be exercised so that the Warrants will meet the minimum listing criteria to be accepted for listing on NASDAQ. After the one-year anniversary of issuance, we may redeem the Warrants for $0.001 per Warrant if the volume weighted average price of our common stock is above $ per share, 300% of the exercise price, for each of 10 consecutive trading days. | ||
Pre-Funded Warrants | Each Pre-Funded Warrant entitles the holder to purchase one share of common stock at an exercise price of $0.01 per share, and the subscription price per Unit for any such electing investors will be reduced to $ (which equals the Subscription Price for the other Units sold in the Rights Offering, less the $0.01 exercise price for each Pre-Funded Warrant). Each Pre-Funded Warrant will be exercisable from the date of issuance through its expiration on , 2021. The Pre-Funded Warrants will be exercisable by paying the exercise price in cash or on a cashless basis. The Pre-Funded Warrants will not be listed for trading on any stock exchange or market. The Pre-Funded Warrants do not confer upon the holder any voting or any other rights of a shareholder of the Company. See "Description of Securities—Pre-Funded Warrants Issuable in the Rights Offering." | |
Record Date | 5:00 PM Eastern Time, July 7, 2016. | |
Subscription Right | Each Subscription Right consists of a Basic Subscription Right and an Over-Subscription Privilege. | |
Basic Subscription Rights | Basic Subscription Right will entitle you to purchase one Unit at the Subscription Price. | |
Over-Subscription Privilege | If you exercise your Basic Subscription Rights in full, you may also choose to purchase a portion of any Units that are not purchased by our other shareholders or holders of participating warrants through the exercise of their Basic Subscription Rights. You may subscribe for additional Units pursuant to this Over-Subscription Privilege, subject to proration described elsewhere. | |
Expiration date | The Subscription Rights will expire at 5:00 PM Eastern Time, on July 26, 2016. We reserve the right to extend the expiration date in our sole discretion. | |
Procedure for exercising Subscription Rights | To exercise your Subscription Rights, you must take the following steps: | |
| If you are a record holder of our common stock or a holder of participating warrants, you must deliver payment and a properly completed Subscription Rights Statement to the | ||
| | Year Ended December 31, | Three Months Ended March 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | 2012 | 2013 | |||||||||
Consolidated Statement of Operations Data: | |||||||||||||
Revenue | $ | 1,487,000 | $ | 46,190,000 | $ | 198,000 | $ | 1,116,000 | |||||
Operating expenses: | |||||||||||||
General and administrative | 6,436,000 | 15,707,000 | 2,460,000 | 3,346,000 | |||||||||
Research and development | 22,624,000 | 52,762,000 | 8,448,000 | 12,756,000 | |||||||||
Total operating expenses | 29,060,000 | 68,469,000 | 10,908,000 | 16,102,000 | |||||||||
Loss from operations | (27,573,000 | ) | (22,279,000 | ) | (10,710,000 | ) | (14,986,000 | ) | |||||
Change in fair value of warrant liability | 1,287,000 | 367,000 | (609,000 | ) | 14,000 | ||||||||
Interest expense | (19,000 | ) | (8,608,000 | ) | (21,000 | ) | — | ||||||
Other income, net | 11,000 | 608,000 | 541,000 | 127,000 | |||||||||
Net loss before income taxes expense | (26,294,000 | ) | (29,912,000 | ) | (10,799,000 | ) | (14,845,000 | ) | |||||
Income taxes | — | — | — | — | |||||||||
Net loss | (26,294,000 | ) | (29,912,000 | ) | (10,799,000 | ) | (14,845,000 | ) | |||||
Accretion of preferred stock | (4,020,000 | ) | (3,953,000 | ) | (1,231,000 | ) | (1,019,000 | ) | |||||
Net loss applicable to common stockholders | $ | (30,314,000 | ) | $ | (33,865,000 | ) | $ | (12,030,000 | ) | $ | (15,864,000 | ) | |
Per share information: | |||||||||||||
Net loss per share of common stock, basic and diluted(1) | $ | (10.64 | ) | $ | (11.51 | ) | $ | (4.15 | ) | $ | (4.56 | ) | |
Basic and diluted weighted average shares outstanding(1) | 2,849,159 | 2,941,782 | 2,897,347 | 3,475,673 | |||||||||
Pro forma net loss per share of common stock, basic and diluted(1) | $ | (2.01 | ) | $ | (0.77 | ) | |||||||
Basic and diluted pro forma weighted average shares outstanding(1) | 16,887,329 | 20,589,154 | |||||||||||
| | As of March 31, 2013 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | Actual | Pro Forma(2) | Pro Forma As Adjusted(3)(4) | |||||||
Consolidated Balance Sheet Data: | ||||||||||
Cash and cash equivalents | $ | 67,307,000 | $ | 67,307,000 | ||||||
Total assets | 70,759,000 | 70,759,000 | ||||||||
Total liabilities | 42,544,000 | 42,544,000 | ||||||||
Accumulated deficit | (183,205,000) | (183,205,000) | ||||||||
Total stockholders' (deficit) equity | (174,119,000) | 28,215,000 | ||||||||
| Subscription Agent to be received before 5:00 PM Eastern Time, on July 26, 2016. You may deliver the documents and payments by first class mail or courier service. If you use first class mail for this purpose, we recommend using registered mail, properly insured, with return receipt requested. | ||
| If you are a beneficial owner of shares that are registered in the name of a broker, dealer, custodian bank, or other nominee, you should instruct your broker, dealer, custodian bank, or other nominee to exercise your Subscription Rights on your behalf. Please follow the instructions of your nominee, who may require that you meet a deadline earlier than 5:00 PM Eastern Time, on July 26, 2016. | ||
Delivery of shares and Warrants | As soon as practicable after the expiration of the Rights Offering, the Subscription Agent will arrange for the issuance of the shares of common stock and Warrants purchased pursuant to the Rights Offering. All shares and Warrants that are purchased in the Rights Offering will be issued in book-entry, or uncertificated, form meaning that you will receive a direct registration (DRS) account statement from our transfer agent reflecting ownership of these securities if you are a holder of record of shares or warrants. If you hold your shares in the name of a custodian bank, broker, dealer, or other nominee, DTC will credit your account with your nominee with the securities you purchased in the Rights Offering. | |
Non-transferability of Subscription Rights | The Subscription Rights may not be sold, transferred, assigned or given away to anyone. The Subscription Rights will not be listed for trading on any stock exchange or market. | |
Transferability of Warrants | The Warrants will be separately transferable following their issuance and through their expiration five years from the issuance date. | |
No board recommendation | Our board of directors is not making a recommendation regarding your exercise of the Subscription Rights. You are urged to make your decision to invest based on your own assessment of our business and the Rights Offering. Please see "Risk Factors" for a discussion of some of the risks involved in investing in our securities. | |
Directors and Officers | While none of our directors or executive officers has entered into any binding commitment or agreement to exercise Subscription Rights received in the Rights Offering, our directors and executive officers have indicated interests in subscribing for up to an aggregate of $3.0 million in the Rights Offering, subject to potential proration. | |
No revocation | All exercises of Subscription Rights are irrevocable, even if you later learn of information that you consider to be unfavorable to the exercise of your Subscription Rights. | |
Use of proceeds | We intend to use the net proceeds from this Rights Offering to continue funding INSPIRE, our ongoing Phase 3 clinical trial of rigosertib IV in a population of patients with | |
| higher-risk MDS after failure of HMA therapy, and, to a lesser extent, for other development of our clinical and preclinical programs, other research and development activities, business development and general corporate purposes, which may include capital expenditures and funding our working capital needs. See "Use of Proceeds." | ||
Material U.S. federal income tax consequences | For U.S. federal income tax purposes, we do not believe you should recognize income or loss upon receipt or exercise of a Subscription Right. You should consult your own tax advisor as to the tax consequences of the Rights Offering in light of your particular circumstances. See "Material U.S. Federal Income Tax Consequences." | |
Extension and termination | Although we do not presently intend to do so, we may extend the Rights Offering for additional time in our sole discretion. Our board of directors may for any reason terminate the Rights Offering at any time before the completion of the Rights Offering. | |
Subscription Agent | Wells Fargo Bank, N.A. | |
Questions | If you have any questions about the Rights Offering, please contact the dealer-manager, Maxim Group LLC, at 405 Lexington Avenue, New York, New York 10174, Attention Syndicate Department, email: syndicate@maximgrp.com or telephone: (212) 895-3745. | |
Market for common stock | Our common stock is listed on NASDAQ under the symbol "ONTX." See "Market Price of our Common Stock and Related Stockholder Matters." | |
Risk factors | Before you exercise your Subscription Rights to purchase Units, you should be aware that there are risks associated with your investment, and you should carefully read and consider risks described in the section captioned "Risk Factors" together with all of the other information included in this prospectus. | |
Dealer-Manager | Maxim Group LLC. | |
Distribution arrangements | Under the terms and subject to the conditions contained in the dealer-manager agreement, the dealer-manager will use its best efforts to solicit the exercise of Subscription Rights. We have agreed to pay the dealer-manager certain fees for acting as dealer-manager and to reimburse the dealer-manager for certain out-of-pocket expenses incurred in connection with this offering. The dealer-manager is not underwriting or placing any of the Subscription Rights or the Units, shares of common stock or Warrants being issued in this offering, and does not make any recommendation with respect to such Subscription Rights (including with respect to the exercise or expiration of such Subscription Rights), Units, shares of common stock or Warrants. See "Plan of Distribution" for a discussion of the fees and expenses to be paid to the dealer-manager. |
Investing in our common stock involves a high degree of risk. YouOur business is influenced by many factors that are difficult to predict, and that involve uncertainties that may materially affect actual operating results, cash flows and financial condition. Before making an investment decision, you should carefully consider these risks, including those set forth below and those described in the "Risk Factors" section of our most recent Annual Report on Form 10-K, filed with the Commission on March 28, 2016, as revised or supplemented by our Quarterly Reports on Form 10-Q filed with the SEC since the filing of our most recent Annual Report on Form 10-K, each of which is incorporated by reference into this prospectus, and you should also carefully consider any other information we include or incorporate by reference in this prospectus.
Any of the risks describedwe describe below together withor in the other information containedincorporated herein by reference in this prospectus including our consolidated financial statements and the related notes appearing at the end of this prospectus, before making your decision to invest in shares of our common stock. We cannot assure you that any of the events discussed in the risk factors below will not occur. These risks could have a material and adverse impact oncause our business, results of operations, financial condition and cash flows and our future prospects would likely be materially and adversely affected. If that wereor operating results to happen, the tradingsuffer. The market price of our common stock could decline if one or more of these risks and youuncertainties develop into actual events. You could lose all or part of your investment.
Risks Related to Our Financial Position and Capital Needsthe Rights Offering
We have incurred significant losses since our inception and anticipate that we will continue to incur lossessubstantial expenses in connection with the future.Rights Offering, which may not return adequate value if the Rights Offering is ultimately not consummated or successful.
The estimated expenses for the Rights Offering are approximately $448,964, excluding fees and expenses of the dealer-manager that we have engaged to assist us with the Rights Offering. If the registration statement of which this prospectus is a part is not declared effective, the Rights Offering is not commenced or the Rights Offering is not ultimately consummated or successful, we will incur these expenses nonetheless. We arewill provide to the dealer-manager upon completion of the Rights Offering a clinical-stage biopharmaceutical company. Investmentnon-accountable expense allowance equal to the lesser of $100,000 or 3% of the gross proceeds of the Rights Offering for expenses incurred in biopharmaceutical product developmentconnection with the Rights Offering. We advanced $30,000 against out-of-pocket accountable expenses to Maxim Group LLC upon its engagement as a dealer-manager; provided that Maxim Group LLC will promptly reimburse to us (i) any portion of the advance not used for actual out-of-pocket expenses, if the Rights Offering is highly speculative because it entails substantial upfront capital expendituresnot completed, and significant risk that a product candidate will fail to gain regulatory approval or become commercially viable. We do not have any products approved by regulatory authorities for marketing and have not generated any revenue from product sales to date, and we continue to incur significant research, development and other expenses related to(ii) the full amount of the advance, if the Rights Offering is completed. See "Plan of Distribution."
Your interest in our ongoing operations. Ascompany may be diluted as a result we are not profitable and have incurred losses in every reporting period since our inception in 1998. For the year ended December 31, 2012 and the three months ended March 31, 2013, we reported a net loss of $29.9 million and $14.8 million, respectively, and we had an accumulated deficit of $183.2 million at March 31, 2013.
We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate these losses to increase as we continue the research and development of, and seek regulatory approvals for, our product candidates, and potentially begin to commercialize any products that may achieve regulatory approval. We may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business. The size of our future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues. If any of our product candidates fail in clinical trials or do not gain regulatory approval, or if approved, fail to achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
We have a limited operating history, which may make it difficult for you to evaluate the success of our business to date and to assess our future viability.this Rights Offering.
Our operations to date have been limited to organizing and staffingShareholders who do not fully exercise their Subscription Rights should expect that they will, at the completion of this offering, own a smaller proportional interest in our company acquiring productthan would otherwise be the case had they fully exercised their Basic Subscription Right and technology rights, discovering novel molecules and conducting product development activities for our product candidates. WeOver-Subscription Privilege. Further, the shares issuable upon the exercise of the Warrants to be issued pursuant to the Rights Offering will dilute the ownership interest of shareholders not participating in this offering or holders of Warrants issued pursuant to this offering who have not yet obtained regulatory approval for anyexercised them.
Further, because the price per Unit being offered may be substantially higher than the net tangible book value per share of our product candidates. Consequently, any predictions about our future success, performance or viabilitycommon stock, you may not be as accurate as they could be if we hadsuffer substantial dilution in the net tangible book value of the common stock you purchase in this offering. If you purchase Units in this offering at the Subscription Price, you may suffer immediate and substantial dilution in the net tangible book value of the common stock. See "Dilution" in this prospectus for a longer operating history or approved products onmore detailed discussion of the market.dilution which may incur in connection with this offering.
We currently have no sourceCompletion of product revenuethe Rights Offering is not subject to us raising a minimum offering amount and may never become profitable.we will still need additional funding to carry out our proposed operating activities, including our INSPIRE trial, after the Rights Offering.
To date, we haveCompletion of the Rights Offering is not generated any revenuessubject to us raising a minimum offering amount and therefore the net proceeds from commercial product sales. Our abilitythe Rights Offering may be insufficient to generate revenue from product sales and achieve profitability will depend uponmeet our ability to successfully commercialize products, including any of our current product candidates, or other product candidates that we may in-license or acquire in the future. Even if we are able to successfully achieve regulatory approval for these product candidates, we do not know when any of these products willobjectives, thereby
generate revenue fromincreasing the risk to investors in this offering, including investing in a company that continues to require capital. Even if we sell all of the Units subject to the Rights Offering, we will need to obtain additional financing in the future in order to fully fund rigosertib or any other product salescandidates through the regulatory approval process.
Due to our ongoing losses and our accumulated deficit in combination with other factors, the opinion of our independent registered public accounting firm on our audited consolidated financial statements for us, if at all. Ourour fiscal year ended December 31, 2015 contains an explanatory paragraph regarding substantial doubt about our ability to generate revenue from product sales from our current or future product candidates also depends oncontinue as a numbergoing concern. Even if we sell all of additional factors, including our ability to:
In addition, because of the numerous risks and uncertainties associated with product development, including that our product candidates may not advance through development or achieve the endpoints of applicable clinical trials,Rights Offering, if we are unable to predict the timingobtain sufficient additional funding, through future financings or amountthrough strategic and collaborative transactions and arrangements, we may not have sufficient resources to complete our INSPIRE trial and we may continue to delay, scale-back or eliminate certain of increased expenses,our planned research, drug discovery and development activities and certain other aspects of our operations, or when or if we willmay not be able to achieve or maintain profitability. Even if we are able to completecontinue as a going concern.
This Rights Offering may cause the development and regulatory process for any product candidates, we anticipate incurring significant costs associated with commercializing these products.
Even if we are able to generate revenues from the saletrading price of our products, we may not become profitable and may needcommon stock to obtain additional funding to continue operations. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce our operations.
We are likely to require additional capital to fund our operations and if we fail to obtain necessary financing, we may be unable to complete the development and potential commercialization of our product candidates.decrease.
Our operations have consumed substantial amountsThe Subscription Price, together with the number of cash since inception. We expectshares of common stock we propose to continue to spend substantial amounts to advanceissue and ultimately will issue if this Rights Offering is completed, may result in an immediate decrease in the clinical developmentmarket price of our product candidates and launch and commercializecommon stock. This decrease may continue after the completion of this Rights Offering. If that occurs, you may have committed to buy shares of common stock in the Rights Offering at a price greater than the prevailing market price. We cannot predict the effect, if any, product candidates for which we receive regulatory approval, including potentially building our own commercial organization. We believe that the net proceedsavailability of shares for future sale represented by the Warrants issued in connection with the Rights Offering, or the ability to trade the Warrants themselves, will have on the market price of our common stock from time to time. Further, if a substantial number of Subscription Rights are exercised and the holders of the shares received upon exercise of those Subscription Rights or the related Warrants choose to sell some or all of the shares underlying the Subscription Rights or the related Warrants, the resulting sales could depress the market price of our common stock. Following the exercise of your Subscription Rights you may not be able to sell your common stock at a price equal to or greater than the Subscription Price.
Because the exercise of your Subscription Rights is not revocable, you could be committed to buying shares of common stock above the prevailing market price.
Once you exercise your Subscription Rights, you may not revoke such exercise even if you later learn information that you consider to be unfavorable to the exercise of your Subscription Rights. The market price of our shares of common stock may decline prior to the expiration of this offering together with existing cashor a Subscribing Rights holder may not be able to sell shares of common stock purchased in this offering at a price equal to or greater than the Subscription Price. Until shares of our common stock are delivered upon expiration of the Rights Offering, you will not be able to sell or transfer the shares of our common stock that you purchase in the Rights Offering. Any such delivery will occur as soon as practicable after the Rights Offering has expired, payment for the shares of common stock and cash equivalentsattached Warrants subscribed for has cleared, and interest thereon, will be sufficient to fund our projected operating requirementsall prorating calculations and reductions contemplated by the terms of the Rights Offering have been effected.
If we terminate this offering for at least the next 12 months. However,any reason, we will likely require additional capitalhave no obligation other than to return subscription monies as soon as practicable.
We may decide, in our sole discretion and for any reason, to cancel or terminate the further development and potential commercialization of our product candidates and may also needRights Offering at any time prior to raise additional funds soonerthe expiration date. If this offering is cancelled or terminated, we will have no obligation with respect to pursue a more accelerated development of our product candidates.
Our forecast ofSubscription Rights that have been exercised except to return as soon as practicable, without interest, the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors, includingsubscription payments deposited with the factors discussed elsewhere in this "Risk Factors" section. We have based this estimate on assumptions that may prove to be wrong, andSubscription Agent.
we could utilize our available capital resources sooner than we currently expect. Our future funding requirements, both near and long-term, will depend on many factors, including, but not limited to:
If we are unable to expand our operations or otherwise capitalize on our business opportunities due to a lack of capital, our ability to become profitableterminate this offering and you have not exercised any Subscription Rights, such Subscription Rights will be compromised.
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
Until we can generate substantial revenue from product sales, if ever, we expect to seek additional capital through a combination of private and public equity offerings, debt financings, strategic collaborations and alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of existing stockholders will be diluted, and the terms may include liquidation or other preferences that adversely affect the rights of existing stockholders. Debt financing, if available, may involve agreements that include restrictive covenants limiting our ability to take important actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through strategic collaborations and alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us. If we are unable to raise additional funds through equity or debt financing when needed, we may be required to delay, limit, reduce or terminate our product development or commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Unstable market and economic conditions may have serious adverse consequences on our business, financial condition and stock price.
As has been widely reported, global credit and financial markets have been experiencing extreme disruptions over the past several years, including severely diminished liquidity and credit availability,
declines in consumer confidence, declines in economic growth, increases in unemployment rates and uncertainty about economic stability. There can be no assurance that further deterioration in credit and financial markets and confidence in economic conditions will not occur. Our general business strategy may be compromised by economic downturns and volatile business environment and unpredictable and unstable market conditions. If the current equity and credit markets deteriorate, or do not improve, it may make any necessary debt or equity financing more difficult to secure, more costly, or more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could harm our growth strategy, financial performance and stock price and could require us to delay or abandon our business and clinical development plans. In addition, there is a risk that one or more of our current service providers, manufacturers and other partners may not survive these difficult economic times, which could directly affect our ability to attain our operating goals on schedule and on budget.
Risks Related to Our Business and Industryexpire worthless.
Our future success is dependent primarily on the regulatory approval and commercialization of our product candidates, including rigosertib, which is currently undergoing Phase 3 clinical trials.
We do not have any products that have gained regulatory approval. Currently, our only clinical-stage product candidates are rigosertib, ON 013105 and recilisib, and rigosertib is our only late-stage product candidate.
As a result, our business is substantially dependent on our ability to obtain regulatory approval for, and, if approved, to successfully commercialize rigosertib and, to a lesser degree, ON 013105 and recilisib in a timely manner. We cannot commercialize product candidates in the United States without first obtaining regulatory approval for the product from the FDA; similarly, we cannot commercialize product candidates outside of the United States without obtaining regulatory approval from comparable foreign regulatory authorities. Before obtaining regulatory approvals for the commercial sale of any product candidate for a target indication, we must demonstrate with substantial evidence gathered in preclinical and well-controlled clinical studies, generally including two well-controlled Phase 3 trials, and, with respect to approval in the United States, to the satisfaction of the FDA, that the product candidate is safe and effective for use for that target indication and that the manufacturing facilities, processes and controls are adequate. Even if rigosertib or another product candidate were to successfully obtain approval from the FDA and comparable foreign regulatory authorities, any approval might contain significant limitations related to use restrictions for specified age groups, warnings, precautions or contraindications, orcommon stock price may be subject to burdensome post-approval study or risk management requirements. If we are unable to obtain regulatory approval for rigosertib in one or more jurisdictions, or any approval contains significant limitations, we may not be able to obtain sufficient funding or generate sufficient revenue to continue the development of ON 013105, recilisib, or any other product candidate that we may discover, in-license, develop or acquire in the future. Furthermore, even if we obtain regulatory approval for rigosertib, we will still need to develop a commercial organization, establish commercially viable pricing and obtain approval for adequate reimbursement from third-party and government payors. If we or our commercialization collaborators are unable to successfully commercialize rigosertib, we may not be able to earn sufficient revenues to continue our business.
Because the results of preclinical testing or earlier clinical studies are not necessarily predictive of future results, rigosertib, which is currently in Phase 3 clinical trials, or any other product candidate we advance into clinical trials may not have favorable results in later clinical trials or receive regulatory approval.
Success in preclinical testing and early clinical studies does not ensure that later clinical trials will generate adequate data to demonstrate the efficacy and safety of an investigational drug. A number of companies in the pharmaceutical and biotechnology industries, including those with greater resources and experience, have suffered significant setbacks in clinical trials, even after seeing promising results in
earlier clinical trials. Despite the results reported in earlier clinical trials for rigosertib and our other clinical-stage product candidates, we do not know whether the clinical trials we may conduct will demonstrate adequate efficacy and safety to result in regulatory approval to market any of our product candidates in any particular jurisdiction. If later-stage clinical trials do not produce favorable results, our ability to achieve regulatory approval for any of our product candidates may be adversely impacted.
Clinical drug development involves a lengthy and expensive process with an uncertain outcome.
Clinical testing is expensive, can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through preclinical studies and early clinical trials.
We may experience delays in our ongoing or future clinical trials and we do not know whether planned clinical trials will begin or enroll subjects on time, need to be redesigned or be completed on schedule, if at all. For example, we experienced a clinical hold with our initial IND submission for recilisib based on the need to conduct additional toxicology studies and to revise quality requirements for manufacture of the drug product. While we do not anticipate any future such delays, there can be no assurance that the FDA will not put clinical trials of recilisib or any other of our product candidates on clinical hold in the future. Clinical trials may be delayed, suspended or prematurely terminated for a variety of reasons, such as:
Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors including the size and nature of the patient population, the proximity of subjects to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, ability to obtain and maintain patient consents, risk that enrolled subjects will drop out before completion, competing clinical trials and clinicians' and patients' perceptions as to the potential advantages of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating. In 2011, we suspended enrollment in our Phase 1 trial of ON 013105 because enrollment of patients was occurring so slowly that our inventory of ON 013105 clinical trial material expired. We intend to restart enrollment in this trial with newly manufactured clinical trial materials at a new clinical trial site in the fourth quarter of 2013. Furthermore, we rely on CROs and clinical trial sites to ensure the proper and timely conduct of our clinical trials, and while we have agreements governing their committed activities, we have limited influence over their actual performance.
If we experience delays in the completion or termination of, any clinical trial of our product candidates, the commercial prospects of our product candidates will be harmed, and our ability to generate product revenues from any of these product candidates will be delayed. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues. In addition, many of the factors that could cause a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
The regulatory approval processes of the FDA and comparable foreign regulatory authorities are lengthy, time consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for our product candidates, our business will be substantially harmed.Rights Offering.
The time required to obtain approval by the FDA and comparable foreign regulatory authorities is unpredictable, but typically takes many years following the commencement of preclinical studies and clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate's clinical development and may vary among jurisdictions. We have not obtained regulatory approval for any product candidate, and it is possible that none of our existing product candidates or any product candidates we may discover, in-license or acquire and seek to develop in the future will ever obtain regulatory approval.
Our product candidates could fail to receive regulatory approval from the FDA or a comparable foreign regulatory authority for many reasons, including:
The FDA or a comparable foreign regulatory authority may require more information, including additional preclinical or clinical data to support approval, which may delay or prevent approval and our commercialization plans, or we may decide to abandon the development program altogether. Even if we do obtain regulatory approval, our product candidates may be approved for fewer or more limited indications than we request, approval contingent on the performance of costly post-marketing clinical trials, or approval with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. In addition, if our product candidate produces undesirable side effects or safety issues, the FDA may require the establishment of Risk Evaluation Mitigation Strategies, or REMS, or a comparable foreign regulatory authority may require the establishment of a similar strategy, that may, restrict distribution of our products and impose burdensome implementation requirements on us. Any of the foregoing scenarios could materially harm the commercial prospects for our product candidates.
We have had limited interactions with foreign regulatory authorities. Approval by the FDA does not ensure approval by foreign regulatory authorities and approval by one or more foreign regulatory authorities does not ensure approval by regulatory authorities in other countries or by the FDA. However, a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. We may not be able to file for regulatory approvals and even if we file we may not receive the necessary approvals to commercialize our products in any market.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following any marketing approval.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign regulatory authority. For example, even though rigosertib IV and rigosertib Oral have generally been well tolerated by patients in our earlier-stage clinical trials, in some cases there were side effects, some of which were severe. The most common drug-related adverse side effects reported by at least 10% of the 79 patients enrolled in the four Phase 1 and 2 trials of rigosertib IV with MDS or acute myeloid leukemia, or AML, were
gastrointestinal, such as nausea and diarrhea, constitutional, such as fatigue, urinary, such as dysuria and hematuria, or the presence of red blood cells in the urine, or hematologic, such as anemia. These side effects were generally mild or moderate in severity. Drug-related side effects that were Grade 3 or Grade 4, meaning they were more than mild or moderate in severity, that were reported in two or more patients in the four studies were decreased red blood cells, decreased platelets, neutropenia, or decreased neutrophils, leukopenia, or decreased white blood cells, frequent urination, dysuria, low blood sodium, increased blood clotting time, fever, fatigue and diarrhea. In patients enrolled in our rigosertib Oral studies in MDS, the most common side effects were urinary disorders. In our rigosertib Oral Phase 1 MDS study, hematuria was the most frequent dose-limiting toxicity, although some patients did experience decreased appetite, diarrhea or nausea. The most severe side effects, seen in two patients, were neutropenia, which occurred at Grade 3 in one patient and Grade 4 in one other patient, as well as urinary tract infection, fainting and shortness of breath. None of these side effects required interruption of the trial.
As a result of these side effects or further safety or toxicity issues that we may experience in our clinical trials in the future, we may not receive approval to market any product candidates, which could prevent us from ever generating revenues or achieving profitability. Results of our trials could reveal an unacceptably high severity and prevalence of side effects. In such an event, our trials could be suspended or terminated and the FDA or comparable foreign regulatory authorities could order us to cease further development of or deny approval of our product candidates for any or all targeted indications. The drug-related side effects could affect patient recruitment or the ability of enrolled subjects to complete the trial or result in potential product liability claims.
Additionally, if any of our product candidates receives marketing approval, and we or others later identify undesirable side effects caused by such product, a number of potentially significant negative consequences could result, including:
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved.
Even if our product candidates receive regulatory approval, they may still face future development and regulatory difficulties.
Even if we obtain regulatory approval for a product candidate, it would be subject to ongoing requirements by the FDA and comparable foreign regulatory authorities governing the manufacture, quality control, further development, labeling, packaging, storage, distribution, safety surveillance, import, export, advertising, promotion, recordkeeping and reporting of safety and other post-market information. The safety profile of any product will continue to be closely monitored by the FDA and comparable foreign regulatory authorities after approval. If the FDA or comparable foreign regulatory authorities become aware of new safety information after approval of any of our product candidates, they may require labeling changes or establishment of a REMS or similar strategy, impose significant restrictions on a product's indicated uses or marketing, or impose ongoing requirements for potentially
costly post-approval studies or post-market surveillance. For example, the label ultimately approved for rigosertib, if it achieves marketing approval, may include restrictions on use.
In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with current good manufacturing practices, or cGMP, and other regulations. If we or a regulatory agency discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product, the manufacturing facility or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing. If we, our product candidates or the manufacturing facilities for our product candidates fail to comply with applicable regulatory requirements, a regulatory agency may:
The occurrence of any event or penalty described above may inhibit our ability to commercialize our products and generate revenue.
Advertising and promotion of any product candidate that obtains approval in the United States will be heavily scrutinized by the FDA, the Department of Justice, or the DOJ, the Office of Inspector General of the Department of Health and Human Services, or HHS, state attorneys general, members of Congress and the public. Violations, including promotion of our products for unapproved or off-label uses, are subject to enforcement letters, inquiries and investigations, and civil and criminal sanctions by the FDA. Additionally, advertising and promotion of any product candidate that obtains approval outside of the United States will be heavily scrutinized by comparable foreign regulatory authorities.
In the United States, engaging in impermissible promotion of our products for off-label uses can also subject us to false claims litigation under federal and state statutes, which can lead to civil and criminal penalties and fines and agreements that materially restrict the manner in which we promote or distribute our drug products. These false claims statutes include the federal False Claims Act, which allows any individual to bring a lawsuit against a pharmaceutical company on behalf of the federal government alleging submission of false or fraudulent claims, or causing to present such false or fraudulent claims, for payment by a federal program such as Medicare or Medicaid. If the government prevails in the lawsuit, the individual will share in any fines or settlement funds. Since 2004, these False Claims Act lawsuits against pharmaceutical companies have increased significantly in volume and breadth, leading to several substantial civil and criminal settlements based on certain sales practices
promoting off-label drug uses. This growth in litigation has increased the risk that a pharmaceutical company will have to defend a false claim action, pay settlement fines or restitution, agree to comply with burdensome reporting and compliance obligations, and be excluded from the Medicare, Medicaid and other federal and state healthcare programs. If we do not lawfully promote our approved products, we may become subject to such litigation and, if we are not successful in defending against such actions, those actions could compromise our ability to become profitable.
Failure to obtain regulatory approval in international jurisdictions would prevent our product candidates from being marketed abroad.
In order to market and sell our products in the European Union and many other jurisdictions, including Japan and Korea, we must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ substantially from that required to obtain FDA approval. The regulatory approval process outside the United States generally includes all of the risks associated with obtaining FDA approval. In addition, in many countries outside the United States, it is required that the product be approved for reimbursement before the product can be approved for sale in that country. We may not obtain approvals from regulatory authorities outside the United States on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one regulatory authority outside the United States does not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA. We may not be able to file for marketing approvals and may not receive necessary approvals to commercialize our products in any market. If we are unable to obtain approval of any of our product candidates by regulatory authorities in the European Union, Japan, Korea or another country, the commercial prospects of that product candidate may be significantly diminished and our business prospects could decline.
Recently enacted and future legislation, including potentially unfavorable pricing regulations or other healthcare reform initiatives, may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates and affect the prices we may obtain.
The regulations that govern, among other things, marketing approvals, coverage, pricing and reimbursement for new drug products vary widely from country to country. In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to successfully sell any product candidates for which we obtain marketing approval.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or Medicare Modernization Act, changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for physician administered drugs. In recent years, Congress has considered further reductions in Medicare reimbursement for drugs administered by physicians. The Centers for Medicare and Medicaid Services, the agency that runs the Medicare program, also has the authority to revise reimbursement rates and to implement coverage restrictions for some drugs. Cost reduction initiatives and changes in coverage implemented through legislation or regulation could decrease utilization of and reimbursement for any approved products, which in turn would affect the price we can receive for those products. While the Medicare Modernization Act and Medicare regulations apply only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. Therefore, any reduction in reimbursement that results from federal legislation or regulation may result in a similar reduction in payments from private payors.
In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act and the Health Care and Education Affordability Reconciliation Act of 2010, or the Affordable Care Act, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on pharmaceutical and medical device manufacturers and impose additional health policy reforms. The Affordable Care Act expanded manufacturers' rebate liability to include covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations increased the minimum rebate due for innovator drugs from 15.1% of average manufacturer price, or AMP, to 23.1% of AMP and capped the total rebate amount for innovator drugs at 100% of AMP. The Affordable Care Act and subsequent legislation also changed the definition of AMP. Furthermore, the Affordable Care Act imposes a significant annual, nondeductible fee on companies that manufacture or import certain branded prescription drug products. Substantial new provisions affecting compliance have also been enacted, which may affect our business practices with healthcare practitioners, and a significant number of provisions are not yet, or have only recently become, effective. Although it is too early to determine the effect of the Affordable Care Act, it appears likely to continue the pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. More recently, on August 2, 2011, the President signed into law the Budget Control Act of 2011, which, among other things, creates the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation's automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on our customers and accordingly, our financial operations. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We cannot be sure whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our product candidates, if any, may be.
In the United States, the European Union and other potentially significant markets for our product candidates, government authorities and third-party payors are increasingly attempting to limit or regulate the price of medical products and services, particularly for new and innovative products and therapies, which has resulted in lower average selling prices. Furthermore, the increased emphasis on managed healthcare in the United States and on country and regional pricing and reimbursement controls in the European Union will put additional pressure on product pricing, reimbursement and usage, which may adversely affect our future product sales and results of operations. These pressures can arise from rules and practices of managed care groups, judicial decisions and governmental laws and regulations related to Medicare, Medicaid and healthcare reform, pharmaceutical reimbursement policies and pricing in general.
Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for a product candidate in a particular country, but then be subject to price regulations that delay our commercial launch of the product, possibly for lengthy time periods, which could negatively impact the
revenues we are able to generate from the sale of the product in that particular country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates even if our product candidates obtain marketing approval.
Laws and regulations governing international operations may preclude us from developing, manufacturing and selling certain product candidates outside of the United States and require us to develop and implement costly compliance programs.
As we expand our operations outside of the United States, we must comply with numerous laws and regulations in each jurisdiction in which we plan to operate. The creation and implementation of international business practices compliance programs is costly and such programs are difficult to enforce, particularly where reliance on third parties is required.
The Foreign Corrupt Practices Act, or FCPA, prohibits any U.S. individual or business from paying, offering, authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the United States to comply with certain accounting provisions requiring the company to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations. The anti-bribery provisions of the FCPA are enforced primarily by the DOJ. The Securities and Exchange Commission, or the SEC, is involved with enforcement of the books and records provisions of the FCPA.
Compliance with the FCPA is expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, the FCPA presents particular challenges in the pharmaceutical industry, because, in many countries, hospitals are operated by the government, and doctors and other hospital employees are considered foreign officials. Certain payments to hospitals in connection with clinical studies and other work have been deemed to be improper payments to government officials and have led to FCPA enforcement actions.
Various laws, regulations and executive orders also restrict the use and dissemination outside of the United States, or the sharing with certain non-U.S. nationals, of information classified for national security purposes, as well as certain products and technical data relating to those products. Our expanding presence outside of the United States will require us to dedicate additional resources to comply with these laws, and these laws may preclude us from developing, manufacturing, or selling certain products and product candidates outside of the United States, which could limit our growth potential and increase our development costs.
The failure to comply with laws governing international business practices may result in substantial penalties, including suspension or debarment from government contracting. Violation of the FCPA can result in significant civil and criminal penalties. Indictment alone under the FCPA can lead to suspension of the right to do business with the U.S. government until the pending claims are resolved. Conviction of a violation of the FCPA can result in long-term disqualification as a government contractor. The termination of a government contract or relationship as a result of our failure to satisfy any of our obligations under laws governing international business practices would have a negative impact on our operations and harm our reputation and ability to procure government contracts. The SEC also may suspend or bar issuers from trading securities on U.S. exchanges for violations of the FCPA's accounting provisions.
Even if we are able to commercialize our product candidates, the products may not receive coverage and adequate reimbursement from third-party payors, which could harm our business.
Our ability to commercialize any products successfully will depend, in part, on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, determine which medications they will cover and establish reimbursement levels. A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. Third-party payors may also seek additional clinical evidence, beyond the data required to obtain marketing approval, demonstrating clinical benefits and value in specific patient populations before covering our products for those patients. We cannot be sure that coverage and adequate reimbursement will be available for any product that we commercialize and, if reimbursement is available, what the level of reimbursement will be. Coverage and reimbursement may impact the demand for, or the price of, any product candidate for which we obtain marketing approval. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize any product candidate for which we obtain marketing approval.
There may be significant delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or comparable foreign regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may only be temporary. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies. Our inability to obtain coverage and profitable reimbursement rates from both government-funded and private payors for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell our product candidates, we may be unable to generate any revenue.
We do not currently have an organization for the sale, marketing and distribution of pharmaceutical products and the cost of establishing and maintaining such an organization may exceed the cost-effectiveness of doing so. In order to market any products that may be approved by the FDA and comparable foreign regulatory authorities, we must build our sales, marketing, managerial and other non-technical capabilities or make arrangements with third parties to perform these services. If we are unable to establish adequate sales, marketing and distribution capabilities, whether independently or with third parties, we may not be able to generate product revenue and may not become profitable. We will be competing with many companies that currently have extensive and well-funded sales and marketing operations. Without an internal commercial organization or the support of a third party to perform sales and marketing functions, we may be unable to compete successfully against these more established companies.
Our commercial success depends upon attaining significant market acceptance of our product candidates, if approved, among physicians, patients, healthcare payors and the major operators of cancer clinics.
Even if we obtain regulatory approval for any of our product candidates that we may develop or acquire in the future, the product may not gain market acceptance among physicians, healthcare payors, patients or the medical community. Market acceptance of any of our product candidates for which we receive approval depends on a number of factors, including:
If any of our product candidates are approved but fail to achieve market acceptance among physicians, patients, or healthcare payors, we will not be able to generate significant revenues, which would compromise our ability to become profitable.
Our relationships with customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, physicians and third-party payors will play a primary role in the recommendation and prescription of any product candidates for which we obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we would market, sell and distribute our products. As a pharmaceutical company, even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, federal and state healthcare laws and regulations pertaining to fraud and abuse and patients' rights are and will be applicable to our business. Restrictions under applicable federal and state healthcare laws and regulations that may affect our ability to operate include the following:
recommendation of, any good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid;
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any physicians or other healthcare providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could cause significant liability for us and harm our reputation.
We are exposed to the risk of employee fraud or other misconduct, including intentional failures to comply with FDA regulations or similar regulations of comparable foreign regulatory authorities, provide accurate information to the FDA or comparable foreign regulatory authorities, comply with manufacturing standards we have established, comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable foreign regulatory authorities, report financial information or data accurately or disclose unauthorized activities to us. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We will adopt a code of conduct for our directors, officers and employees, or the Code of Conduct, which will be effective as of consummation of this offering, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant fines or other sanctions.
We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do.
The development and commercialization of new drug products is highly competitive. We face competition with respect to our current product candidates, rigosertib, ON 013105 and recilisib, and will face competition with respect to any product candidates that we may seek to develop or commercialize in the future, from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. There are a number of large pharmaceutical and biotechnology companies that currently market and sell products or are pursuing the development of products for the treatment of the disease indications for which we are developing our product candidates. Some of these competitive products and therapies are based on scientific approaches that are the same as or similar to our approach, and others are based on entirely different approaches. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
Our product candidates are being developed for cancer therapeutics and radiation protection. There are a variety of available therapies and supportive care products marketed for cancer patients. In many cases, these drugs are administered in combination to enhance efficacy or to reduce side effects. Some of these drugs are branded and subject to patent protection, and others are available on a generic basis. Many of these approved drugs are well established therapies or products and are widely accepted by physicians, patients and third-party payors. Insurers and other third-party payors may also encourage the use of generic products. This may make it difficult for us to achieve market acceptance at desired levels in a timely manner to ensure viability of our business.
More established companies may have a competitive advantage over us due to their greater size, cash flows and institutional experience. Compared to us, many of our competitors may have significantly greater financial, technical and human resources.
As a result of these factors, our competitors may obtain regulatory approval of their products before we are able to obtain patent protection or other intellectual property rights which will limit our ability to develop or commercialize our product candidates. Our competitors may also develop drugs that are safer, more effective, more widely used and cheaper than ours, and may also be more
successful than us in manufacturing and marketing their products. These appreciable advantages could render our product candidates obsolete or non-competitive before we can recover the expenses of development and commercialization.
Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller and other early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific, management and commercial personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
If we breach the license agreement with Temple University pertaining to our clinical-stage product candidates, we could lose the ability to continue the development and potential commercialization of these product candidates.
In January 1999, we entered into an agreement with Temple, as subsequently amended, to obtain an exclusive, world-wide license to make, have made, use, sell, offer for sale and import several classes of novel compounds, including all three of our clinical-stage product candidates. If we fail to meet our obligations under this license agreement, Temple has the right to terminate our exclusive license, and upon the effective date of such termination, our right to use the licensed technology would terminate. While we would expect to exercise all rights and remedies available to us, including attempting to cure any breach by us, and otherwise seek to preserve our rights under the patents and other technology licensed to us, we may not be able to do so in a timely manner, at an acceptable cost or at all. Any uncured, material breach under the license agreement could result in our loss of exclusive rights and may lead to a complete termination of our product development and any commercialization efforts for the applicable product candidates.
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of any products that we may develop.
We face an inherent risk of product liability exposure related to the testing of our product candidates in human clinical trials and will face an even greater risk if we commercially sell any products that we may develop. Product liability claims may be brought against us by subjects enrolled in our clinical trials, patients, healthcare providers or others using, administering or selling our products. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
We currently hold $10.0 million in product liability insurance coverage in the aggregate, which may not be adequate to cover all liabilities that we may incur. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise. We intend to expand our insurance coverage for products to include the sale of commercial products if we obtain marketing approval for our product candidates in development, but we may be unable to obtain commercially reasonable product liability insurance for any products approved for marketing. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us, particularly if judgments exceed our insurance coverage, could decrease our cash and adversely affect our business.
We will need to grow the size of our organization, and we may experience difficulties in managing this growth.
As of June 1, 2013, we had 56 employees. As our development and commercialization plans and strategies develop, or as a result of any future acquisitions, we will need additional managerial, operational, sales, marketing, financial and other resources. Our management, personnel and systems currently in place may not be adequate to support this future growth. Future growth would impose significant added responsibilities on members of management, including:
As our operations expand, we will need to manage additional relationships with various strategic partners, suppliers and other third parties. Our future financial performance and our ability to commercialize our product candidates and to compete effectively will depend, in part, on our ability to manage any future growth effectively. To that end, we must be able to manage our development efforts and clinical trials effectively and hire, train and integrate additional management, administrative and sales and marketing personnel. Our failure to accomplish any of these tasks could prevent us from successfully growing our company.
Our future success depends on our ability to retain our executive officers and to attract, retain and motivate qualified personnel.
We are highly dependent upon Ramesh Kumar, Ph.D., President and Chief Executive Officer; Francois Wilhelm, M.D., Ph.D., Chief Medical Officer and Senior Vice President; Manoj Maniar, Ph.D., Senior Vice President, Product Development; Thomas McKearn, M.D., Ph.D., President, Research and Development; Ajay Bansal, Chief Financial Officer; Scott Megaffin, Senior Vice President, Commercial Development; David Stephon, Senior Vice President, Quality Management; and James Altland, Senior Vice President, Finance and Corporate Development. Although we have employment agreements with the persons named above, these agreements are at-will and do not prevent such persons from terminating their employment with us at any time. We do not maintain "key person" insurance for any of our executives or other employees, other than our President and Chief Executive Officer. The loss of the services of any of these persons could impede the achievement of our research, development and commercialization objectives.
If we are unable to attract and retain highly qualified employees, we may not be able to grow effectively.
Our future growth and success depend on our ability to recruit, retain, manage and motivate our employees. The loss of any member of our senior management team or the inability to hire or retain experienced management personnel could compromise our ability to execute our business plan and harm our operating results.
Because of the specialized scientific and managerial nature of our business, we rely heavily on our ability to attract and retain qualified scientific, technical and managerial personnel. The competition for qualified personnel in the pharmaceutical field is intense and as a result, we may be unable to continue to attract and retain qualified personnel necessary for the development of our business.
We may engage in future acquisitions that could disrupt our business, cause dilution to our stockholders and harm our financial condition and operating results.
While we currently have no specific plans to acquire any other businesses, we may, in the future, make acquisitions of, or investments in, companies that we believe have products or capabilities that are a strategic or commercial fit with our current product candidates and business or otherwise offer opportunities for our company. In connection with these acquisitions or investments, we may:
We may not be able to complete acquisitions on favorable terms, if at all. If we do complete an acquisition, we cannot assure you that it will ultimately strengthen our competitive position or that it will be viewed positively by customers, financial markets or investors. Furthermore, future acquisitions could pose numerous additional risks to our operations, including:
We may not be able to complete any acquisitions or effectively integrate the operations, products or personnel gained through any such acquisition.
Our business and operations would suffer in the event of computer system failures.
Despite the implementation of security measures, our internal computer systems, and those of our CROs and other third parties on which we rely, are vulnerable to damage from computer viruses, unauthorized access, natural disasters, fire, terrorism, war and telecommunication and electrical failures. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our drug development programs. For example, the loss of clinical trial data from completed, ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to our data or applications, or inappropriate disclosure
of confidential or proprietary information, we could incur liability and the further development of our product candidates could be delayed.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Although we maintain workers' compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological or hazardous materials.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
Business disruptions could seriously harm our future revenues and financial condition and increase our costs and expenses.
Our operations could be subject to earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or manmade disasters or business interruptions, for which we are predominantly self-insured. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses. We rely on third-party manufacturers to produce our product candidates. Our ability to obtain clinical supplies of product candidates could be disrupted, if the operations of these suppliers is affected by a man-made or natural disaster or other business interruption. The ultimate impact on us, our significant suppliers and our general infrastructure of being consolidated in certain geographical areas is unknown, but our operations and financial condition could suffer in the event of a major earthquake, fire or other natural disaster.
We are relying on the FDA's "Animal Efficacy Rule" to demonstrate efficacy of recilisib, which could result in delays or failure at any stage of recilisib's development process, increase our development costs and adversely affect the commercial prospects of recilisib.
Because humans are not normally exposed to radiation and it would be unethical to expose humans to such, effectiveness of recilisib cannot be demonstrated in humans, but instead, under the FDA's "Animal Efficacy Rule," can be demonstrated, in part, by utilizing animal models. This effect has to be demonstrated in more than one animal species expected to be predictive of a response in humans, but an effect in a single animal species may be acceptable if that animal model is sufficiently well-characterized for predicting a response in humans. The animal study endpoint must be clearly related to the desired benefit in humans and the information obtained from animal studies must allow selection of an effective dose in humans. Safety may be demonstrated in human studies.
We may not be able to sufficiently demonstrate the animal correlation to the satisfaction of the FDA, as these correlates are difficult to establish and are often unclear. The FDA may decide that our data are insufficient for approval and require additional preclinical, clinical or other studies, refuse to approve recilisib, or place restrictions on our ability to commercialize recilisib. Furthermore, other countries do not, at this time, have established criteria for review and approval of these types of products outside their normal review process. There is no "Animal Efficacy Rule" equivalent in countries other than the United States, and consequently there can be no assurance that we will be able to make a submission for marketing approval in foreign countries based on such animal data.
Risks Related to Our Dependence on Third Parties
We rely on third parties to conduct our preclinical and clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates.
We have relied upon and plan to continue to rely upon third-party CROs to monitor and manage data for our ongoing preclinical and clinical programs. We rely on these parties for execution of our preclinical and clinical trials, and we control only some aspects of their activities. Nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol and legal, regulatory and scientific standards, and our reliance on the CROs does not relieve us of our regulatory responsibilities. We also rely on third parties to assist in conducting our preclinical studies in accordance with Good Laboratory Practices, or GLP, and the Animal Welfare Act requirements. We and our CROs are required to comply with federal regulations and current Good Clinical Practices, or GCP, which are international standards meant to protect the rights and health of patients that are enforced by the FDA, the Competent Authorities of the Member States of the European Economic Area, or EEA, and comparable foreign regulatory authorities for all of our products in clinical development. Regulatory authorities enforce GCP through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of our CROs fail to comply with applicable GCP, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP requirements. In addition, our clinical trials must be conducted with product produced under cGMP requirements. Failure to comply with these regulations may require us to repeat preclinical and clinical trials, which would delay the regulatory approval process.
Our CROs are not our employees, and except for remedies available to us under our agreements with such CROs, we cannot control whether or not they devote sufficient time and resources to our ongoing clinical, nonclinical and preclinical programs. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. As a result, our results of operations and the commercial prospects for our product candidates would be harmed, our costs could increase and our ability to generate revenues could be delayed.
Because we have relied on third parties, our internal capacity to perform these functions is limited. Outsourcing these functions involves risk that third parties may not perform to our standards, may not produce results in a timely manner or may fail to perform at all. In addition, the use of third-party service providers requires us to disclose our proprietary information to these parties, which could increase the risk that this information will be misappropriated. We currently have a small number of employees, which limits the internal resources we have available to identify and monitor our third-party providers. To the extent we are unable to identify and successfully manage the performance of
third-party service providers in the future, our business may be adversely affected. Though we carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
If we lose our relationships with CROs, our drug development efforts could be delayed.
We rely on third-party vendors and CROs for preclinical studies and clinical trials related to our drug development efforts. Switching or adding additional CROs would involve additional cost and requires management time and focus. Our CROs have the right to terminate their agreements with us in the event of an uncured material breach. In addition, some of our CROs have an ability to terminate their respective agreements with us if it can be reasonably demonstrated that the safety of the subjects participating in our clinical trials warrants such termination, if we make a general assignment for the benefit of our creditors or if we are liquidated. Identifying, qualifying and managing performance of third-party service providers can be difficult, time consuming and cause delays in our development programs. In addition, there is a natural transition period when a new CRO commences work and the new CRO may not provide the same type or level of services as the original provider. If any of our relationships with our third-party CROs terminate, we may not be able to enter into arrangements with alternative CROs or to do so on commercially reasonable terms.
We have limited experience manufacturing our product candidates on a large clinical or commercial scale and have no manufacturing facility. We are dependent on third-party manufacturers for the manufacture of our most advanced product candidate as well as on third parties for our supply chain, and if we experience problems with any third parties, the manufacturing of our product candidates or products could be delayed.
We do not own or operate facilities for the manufacture of our product candidates. We currently have no plans to build our own clinical or commercial scale manufacturing capabilities. We currently rely on a single source contract manufacturing organization, or CMO, for the chemical manufacture of active pharmaceutical ingredient for rigosertib, another CMO for the production of the rigosertib intravenous formulation, and a third CMO for the production of the rigosertib oral formulation for Phase 3 clinical trials. To meet our projected needs for clinical supplies to support our activities through regulatory approval and commercial manufacturing, the CMOs with whom we currently work will need to increase the scale of production. We may need to identify additional CMOs for continued production of supply for our product candidates. We have not yet identified alternate suppliers in the event the current CMOs we utilize are unable to scale production, or if we otherwise experience any problems with them. Although alternative third-party suppliers with the necessary manufacturing and regulatory expertise and facilities exist, it could be expensive and take a significant amount of time to arrange for alternative suppliers. If we are unable to arrange for alternative third-party manufacturing sources, or to do so on commercially reasonable terms or in a timely manner, we may not be able to complete development of our product candidates, or market or distribute them.
Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured product candidates or products ourselves, including reliance on the third party for regulatory compliance and quality assurance, the possibility of breach of the manufacturing agreement by the third party because of factors beyond our control, including a failure to synthesize and manufacture our product candidates or any products we may eventually commercialize in accordance with our specifications, and the possibility of termination or nonrenewal of the agreement by the third party, based on its own business priorities, at a time that is costly or damaging to us. In addition, the FDA and other regulatory authorities require that our product candidates and any products that we may eventually commercialize be manufactured according to cGMP and similar foreign standards. Any failure by our third-party manufacturers to comply with cGMP or failure to scale up manufacturing processes, including any failure to deliver sufficient quantities of product candidates in a timely manner,
could lead to a delay in, or failure to obtain, regulatory approval of any of our product candidates. In addition, such failure could be the basis for the FDA to issue a warning letter, withdraw approvals for product candidates previously granted to us, or take other regulatory or legal action, including recall or seizure of outside supplies of the product candidate, total or partial suspension of production, suspension of ongoing clinical trials, refusal to approve pending applications or supplemental applications, detention or product, refusal to permit the import or export of products, injunction, or imposing civil and criminal penalties.
Any significant disruption in our supplier relationships could harm our business. Any significant delay in the supply of a product candidate or its key materials for an ongoing clinical study could considerably delay completion of our clinical studies, product testing and potential regulatory approval of our product candidates. If our manufacturers or we are unable to purchase these key materials after regulatory approval has been obtained for our product candidates, the commercial launch of our product candidates would be delayed or there would be a shortage in supply, which would impair our ability to generate revenues from the sale of our product candidates.
We have entered into collaboration agreements with SymBio Pharmaceuticals Limited and Baxter Healthcare SA for rigosertib development and commercialization in certain territories and we may elect to enter into additional licensing or collaboration agreements to partner rigosertib in territories currently retained by us. Our dependence on such relationships may adversely affect our business.
Because we have limited resources, we seek to enter into, and in the past we have entered into, collaboration agreements with other pharmaceutical companies. In July 2011, we entered into a license agreement with SymBio Pharmaceuticals Limited, or SymBio, as subsequently amended, granting an exclusive, royalty-bearing license for the development and commercialization of rigosertib in Japan and Korea. In September 2012, we entered into a development and license agreement with Baxter Healthcare SA, or Baxter, a subsidiary of Baxter International Inc., granting an exclusive, royalty-bearing license for the development and commercialization of rigosertib in Europe. We have also entered into a collaboration agreement for the further development of two of our preclinical oncology programs. Any failure by our partners to perform their obligations or any decision by our partners to terminate these agreements could negatively impact our ability to successfully develop, obtain regulatory approvals for and commercialize the applicable product candidate. In addition, any termination of our collaboration agreements will terminate the funding we may receive under the relevant collaboration agreement and may impair our ability to fund further development efforts and our progress in our development programs.
Our commercialization strategy for rigosertib in territories currently retained by us may depend on our ability to enter into agreements with collaborators to obtain assistance and funding for the development and potential commercialization of rigosertib in those territories. Despite our efforts, we may be unable to secure additional collaborative licensing or other arrangements that are necessary for us to further develop and commercialize rigosertib. Supporting diligence activities conducted by potential collaborators and negotiating the financial and other terms of a collaboration agreement are long and complex processes with uncertain results. Even if we are successful in entering into one or more collaboration agreements, collaborations may involve greater uncertainty for us, as we have less control over certain aspects of our collaborative programs than we do over our proprietary development and commercialization programs. We may determine that continuing a collaboration under the terms provided is not in our best interest, and we may terminate the collaboration. Our collaborators could delay or terminate their agreements, and as a result rigosertib may never be successfully commercialized.
Further, our future collaborators may develop alternative products or pursue alternative technologies either on their own or in collaboration with others, including our competitors, and the priorities or focus of our collaborators may shift such that rigosertib receives less attention or resources
than we would like, or they may be terminated altogether. Any such actions by our collaborators may adversely affect our business prospects and ability to earn revenues. In addition, we could have disputes with our current or future collaborators, such as the interpretation of terms in our agreements. Any such disagreements could lead to delays in the development or commercialization of rigosertib or could result in time-consuming and expensive litigation or arbitration, which may not be resolved in our favor.
With respect to our programs that are currently not the subject of collaborations, we may enter into agreements with collaborators to share in the burden of conducting clinical trials, manufacturing and marketing these product candidates. In addition, our ability to apply our proprietary technologies to develop proprietary compounds will depend on our ability to establish and maintain licensing arrangements or other collaborative arrangements with the holders of proprietary rights to such compounds. We may not be able to establish such arrangements on favorable terms or at all, and our future collaborative arrangements may not be successful.
Risks Related to Our Intellectual Property
If we are unable to protect our intellectual property rights, our competitive position could be harmed.
We depend on our ability to protect our proprietary technology. We rely on trade secret, patent, copyright and trademark laws, and confidentiality, licensing and other agreements with employees and third parties, all of which offer only limited protection. Our commercial success will depend in large part on our ability to obtain and maintain patent protection in the United States and other countries with respect to our proprietary technology and products. Where we have the right to do so under our license agreements, we seek to protect our proprietary position by filing patent applications in the United States and abroad related to our novel technologies and products that are important to our business. The patent positions of biotechnology and pharmaceutical companies generally are highly uncertain, involve complex legal and factual questions and have in recent years been the subject of much litigation. As a result, the issuance, scope, validity, enforceability and commercial value of our patents, including those patent rights licensed to us by third parties, are highly uncertain.
The steps we have taken to protect our proprietary rights may not be adequate to preclude misappropriation of our proprietary information or infringement of our intellectual property rights, both inside and outside the United States. The rights already granted under any of our currently issued patents and those that may be granted under future issued patents may not provide us with the proprietary protection or competitive advantages we are seeking. If we are unable to obtain and maintain patent protection for our technology and products, or if the scope of the patent protection obtained is not sufficient, our competitors could develop and commercialize technology and products similar or superior to ours, and our ability to successfully commercialize our technology and products may be adversely affected.
With respect to patent rights, we do not know whether any of the pending patent applications for any of our licensed compounds will result in the issuance of patents that protect our technology or products, or if any of our issued patents will effectively prevent others from commercializing competitive technologies and products. Our pending applications cannot be enforced against third parties practicing the technology claimed in such applications unless and until a patent issues from such applications. Further, the examination process may require us or our licensor to narrow the claims for our pending patent applications, which may limit the scope of patent protection that may be obtained if these applications issue. Because the issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, issued patents that we own or have licensed from third parties may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in the loss of patent protection, the narrowing of claims in such patents or the invalidity or unenforceability of such patents, which could limit our ability to stop others from using or
commercializing similar or identical technology and products, or limit the duration of the patent protection for our technology and products. Protecting against the unauthorized use of our patented technology, trademarks and other intellectual property rights is expensive, difficult and may in some cases not be possible. In some cases, it may be difficult or impossible to detect third-party infringement or misappropriation of our intellectual property rights, even in relation to issued patent claims, and proving any such infringement may be even more difficult.
We could be required to incur significant expenses to perfect our intellectual property rights, and our intellectual property rights may be inadequate to protect our competitive position.
The patent prosecution process is expensive and time-consuming, and we or our licensors may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we or our licensors will fail to identify patentable aspects of inventions made in the course of our development and commercialization activities before it is too late to obtain patent protection on them. Further, given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. We expect to seek extensions of patent terms in the United States and, if available, in other countries where we are prosecuting patents. In the United States, the Drug Price Competition and Patent Term Restoration Act of 1984 permits a patent term extension of up to five years beyond the expiration of the patent. However, the applicable authorities, including the FDA in the United States, and any equivalent regulatory authority in other countries, may not agree with our assessment of whether such extensions are available, and may refuse to grant extensions to our patents, or may grant more limited extensions than we request. If this occurs, our competitors may be able to take advantage of our investment in development and clinical trials by referencing our clinical and preclinical data and launch their product earlier than might otherwise be the case. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection. The laws of foreign countries may not protect our rights to the same extent as the laws of the United States, and these foreign laws may also be subject to change. For example, methods of treatment and manufacturing processes may not be patentable in certain jurisdictions. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing or in some cases not at all. Therefore we cannot be certain that we or our licensors were the first to make the inventions claimed in our owned or licensed patents or pending patent applications, or that we or our licensors were the first to file for patent protection of such inventions.
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. In particular, under the Leahy-Smith Act, the United States transitioned in March 2013 to a "first to file" system in which the first inventor to file a patent application will be entitled to the patent. Third parties are allowed to submit prior art before the issuance of a patent by the U.S. Patent and Trademark Office, or the USPTO, and may become involved in opposition, derivation, reexamination, inter-partes review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, which could adversely affect our competitive position.
The USPTO is currently developing regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, did not become effective until March 16, 2013. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submissions, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we or our licensors fail to maintain the patents and patent applications covering our product candidates, our competitive position would be adversely affected.
We may become involved in lawsuits to protect or enforce our intellectual property, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents or misappropriate or otherwise violate our intellectual property rights. To counter infringement or unauthorized use, litigation may be necessary in the future to enforce or defend our intellectual property rights, to protect our trade secrets or to determine the validity and scope of our own intellectual property rights or the proprietary rights of others. This can be expensive and time consuming. Many of our current and potential competitors have the ability to dedicate substantially greater resources to defend their intellectual property rights than we can. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing upon or misappropriating our intellectual property. Litigation could result in substantial costs and diversion of management resources. In addition, in an infringement proceeding, a court may decide that a patent owned by or licensed to us is invalid or unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation proceeding could put one or more of our patents at risk of being invalidated, held unenforceable or interpreted narrowly. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could harm our business.
Our commercial success depends upon our ability and the ability of our collaborators to develop, manufacture, market and sell our product candidates, and to use our proprietary technologies without infringing the proprietary rights of third parties. We may become party to, or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our products and technology, including interference or derivation proceedings before the USPTO. Third parties may
assert infringement claims against us based on existing patents or patents that may be granted in the future. If we are found to infringe a third party's intellectual property rights, we could be required to obtain a license from such third party to continue developing and commercializing our products and technology. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we are able to obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. We could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, in any such proceeding or litigation, we could be found liable for monetary damages. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could materially harm our business. Any claims by third parties that we have misappropriated their confidential information or trade secrets could have a similar negative impact on our business.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
Many of our employees, including our senior management, were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Some of these employees, including each member of our senior management, executed proprietary rights, non-disclosure and non-competition agreements in connection with such previous employment. Although we try to ensure that our employees do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these employees have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such employee's former employer. We are not aware of any threatened or pending claims related to these matters or concerning the agreements with our senior management, but in the future litigation may be necessary to defend against such claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
Intellectual property disputes could cause us to spend substantial resources and distract our personnel from their normal responsibilities.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses, and could distract our technical and management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the market price of our common stock. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could compromise our ability to compete in the marketplace.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for some of our technology and products, we also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position. We seek to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, CMOs, consultants, advisors and other third parties. We also generally enter into confidentiality and invention or patent assignment agreements with our employees and consultants. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts both within and outside the United States may be less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent such competitor from using that technology or information to compete with us, which could harm our competitive position.
Although we expect all of our employees to assign their inventions to us, and all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology to enter into confidentiality agreements, we cannot provide any assurances that all such agreements have been duly executed or that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. Additionally, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating the trade secret, In addition, others may independently discover our trade secrets and proprietary information. For example, the FDA, as part of its Transparency Initiative, is currently considering whether to make additional information publicly available on a routine basis, including information that we may consider to be trade secrets or other proprietary information, and it is not clear at the present time how the FDA's disclosure policies may change in the future, if at all.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on all of our product candidates throughout the world would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products, and may export otherwise infringing products to territories where we have patent protection, but where enforcement is not as strong as that in the United States. These products may compete with our products in jurisdictions where we do not have any issued patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
Risks Related to This Offering and Ownership of Our Common Stock
We do not know whether an active, liquid and orderly trading market will develop for our common stock or what the market price of our common stock will be and as a result it may be difficult for you to sell your shares of our common stock.
Prior to this offering there has been no market for shares of our common stock. An active trading market for our shares may never develop or be sustained following this offering. The initial public offering price for our common stock will be determined through negotiations with the underwriters, and the negotiated price may not be indicative of the market price of our common stock after this offering. The market value of our common stock may decrease from the initial public offering price. As a result of these and other factors, you may be unable to resell your shares of our common stock at or above the initial public offering price. The lack of an active market may impair your ability to sell your shares at the time you wish to sell them or at a price that you consider reasonable. The lack of an active market may also reduce the fair market value of your shares. Further, an inactive market may also impair our ability to raise capital by selling shares of our common stock and may impair our ability to enter into collaborations or acquire companies or products by using our shares of common stock as consideration. The market price of our stock may be volatile, and you could lose all or part of your investment.
The trading price of our common stock followingmay fluctuate substantially. The price of the common stock that will prevail in the market after this offering is likely tomay be highly volatile and could be subject to wide fluctuations in response to varioushigher or lower than the Subscription Price depending on many factors, some of which are beyond our control. In additioncontrol and may not be directly related to our operating performance. These factors include, but are not limited to, the factors discussed in this "Risk Factors" section and elsewhere in this prospectus, these factors include:following:
In addition,Additionally, the stock market in general, and pharmaceutical and biotechnology companies in particular, havehistorically has experienced extremesignificant price and volume fluctuations. These fluctuations that haveare often been unrelated or disproportionate to the operating performance of theseparticular companies. BroadThese broad market fluctuations may cause declines in the trading price and industry factors may negatively affectmarket value of our common stock.
We cannot assure you that the markettrading price of our common stock regardlesswill not decline after you elect to exercise your Subscription Rights. If that occurs, you may have committed to buy shares of our actual operating performance. The realization of any of these risks or any ofcommon stock in the Rights Offering at a broad range of other risks, including those described in these "Risk Factors,"price greater than the prevailing market price and could have a dramatic and material adverse impact onan immediate unrealized loss. Moreover, we cannot assure you that, following the market priceexercise of our common stock.
We mayyour Subscription Rights, you will be subjectable to securities litigation, which is expensive and could divert management attention.
The market price of oursell your common stock may be volatile, and inat a price equal to or greater than the past companies that have experienced volatility in the market price of their stock have been subject to securities class action litigation. We may be the target of this type of litigation in the future. Securities litigation against us
could resultSubscription Price, and you may lose all or part of your investment in substantial costs and divert our management's attention from other business concerns, which could seriously harm our business.
Our principal stockholders and management own a significant percentagecommon stock. Until shares are delivered upon expiration of our stock andthe Rights Offering, you will not be able to exert significant control over matters subject to stockholder approval.
Prior to this offering, our executive officers, directors, holderssell the shares of 5% or more of our capital stock together beneficially owned approximately 43.3% of our voting stock and, upon consummation of this offering, that same group will together hold approximately % of our outstanding voting stock, assuming no exercise of the underwriters' over-allotment option, no exercise of outstanding options and after giving effect to the issuance of shares in this offering. These stockholders may be able to determine the outcome of all matters requiring stockholder approval. For example, these stockholders may be able to control elections of directors, amendments of our organizational documents, or approval of any merger, sale of assets, or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that you may feel arepurchase in your best interest as onethe Rights Offering. Shares of our stockholders. The interestscommon stock purchased will be issued as soon as practicable after the Rights Offering has expired, payment for the shares of this group of stockholders may not always coincide with your interests or the interests of other stockholders and they may act in a manner that advances their best interests and not necessarily those of other stockholders, including seeking a premium value for their common stock and attached Warrants subscribed for has cleared, and all prorating calculations and reductions contemplated by the terms of the Rights Offering have been effected. We will not pay you interest on funds delivered to the Subscription Agent pursuant to your exercise of Subscription Rights.
Because we do not have any formal commitments from any of our shareholders to participate in the Rights Offering, the net proceeds we receive from the Rights Offering may be lower than we currently anticipate.
We do not have any formal commitments from any of our shareholders to participate in the Rights Offering, and we cannot assure you that any of our shareholders or warrant holders will exercise all or any part of their Basic Subscription Rights or their Over-Subscription Privilege. If our shareholders or participating warrant holders subscribe for fewer shares of our common stock than we currently anticipate, the net proceeds we receive from the Rights Offering could be significantly lower than we currently expect.
The Subscription Price determined for this offering is not an indication of the fair value of our common stock.
In determining the Subscription Price, the pricing committee of our board of directors considered a number of factors, including, but not limited to, the price at which our stockholders might affectbe willing to participate in the prevailing market priceRights Offering, the value of the Warrant being issued as a component of the Unit, historical and current trading prices for our common stock.
If you purchasestock, the amount of proceeds desired, the potential need for liquidity and capital, potential market conditions, and the desire to provide an opportunity to our common stock in this offering, you will incur immediate and substantial dilutionshareholders to participate in the bookRights Offering. In conjunction with its review of these factors, our pricing committee also reviewed a range of discounts to market value of your shares.
represented by the subscription prices in various prior Rights Offerings by other public companies. The initial public offering price is substantially higher than the net tangible book value per share of our common stock. Investors purchasing common stock in this offering will pay a price per share that substantially exceedsSubscription Price does not necessarily bear any relationship to the book value of our tangible assets, after subtractingresults of operations, cash flows, losses, financial condition or any other established criteria for value. You should not consider the Subscription Price as an indication of the fair value of our liabilities. As a result, investors purchasingcommon stock. After the date of this prospectus, our common stock may trade at prices above or below the Subscription Price.
If you do not act on a timely basis and follow subscription instructions, your exercise of Subscription Rights may be rejected.
Holders of Subscription Rights who desire to purchase shares of our common stock and attached Warrants in this offering will incur immediate dilution of $ per share, basedmust act on an assumed initial public offering price of $ per share, which isa timely basis to ensure that all required forms and payments are actually received by the mid-point of the price range set forthSubscription Agent prior to 5:00 PM Eastern Time, on the coverexpiration date, unless extended. If you are a beneficial owner of this prospectus. Further, investors purchasing common stock in this offering will contribute approximately % of the total amount invested by stockholders since our inception, but will own, as a result of such investment, only approximately % of the shares of common stock outstanding immediately following this offering.
Theand you wish to exercise of any of our outstanding options would resultyour Subscription Rights, you must act promptly to ensure that your broker, dealer, custodian bank, trustee or other nominee acts for you and that all required forms and payments are actually received by your broker, dealer, custodian bank, trustee or other nominee in additional dilution. As a result ofsufficient time to deliver such forms and payments to the dilutionSubscription Agent to investors purchasing sharesexercise the Subscription Rights granted in this offering investorsthat you beneficially own prior to 5:00 PM Eastern Time on the expiration date, as may receive significantly less thanbe extended. We will not be responsible if your broker, dealer, custodian bank, trustee or other nominee fails to ensure that all required forms and payments are actually received by the purchase price paidSubscription Agent prior to 5:00 PM Eastern Time, on the expiration date.
If you fail to complete and sign the required subscription forms, send an incorrect payment amount, or otherwise fail to follow the subscription procedures that apply to your exercise in this offering, if anything, inRights Offering, the event of our liquidation. Further, because weSubscription Agent may, need to raise additional capital to fund our clinical development programs, we may independing on the future sell substantial amounts of common stock or securities convertible into or exchangeable for common stock. These future issuances of equity or equity-linked securities, together with the exercise of outstanding options and any additional shares issued in connection with acquisitions, if any, may result in further dilution to investors.
We are an "emerging growth company" and we intend to take advantage of reduced disclosure and governance requirements applicable to emerging growth companies, which could result in our common stock being less attractive to investors.
We are an "emerging growth company," as defined in the JOBS Act, and we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. Wecircumstances, reject your subscription
or accept it only to the extent of the payment received. Neither we nor the Subscription Agent undertakes to contact you concerning an incomplete or incorrect subscription form or payment, nor are we under any obligation to correct such forms or payment. We have the sole discretion to determine whether a subscription exercise properly follows the subscription procedures.
You may not receive all of the Units for which you over-subscribe.
Holders who fully exercise their Basic Subscription Rights will be entitled to subscribe for an additional number of Units. Over-Subscription Privileges will be allocated pro rata among Rights holders who over-subscribed, based on the number of over-subscription Units to which they have subscribed. We cannot predict if investorsguarantee that you will find ourreceive any or the entire amount of Units for which you over-subscribed. If the prorated amount of Units allocated to you in connection with your Over-Subscription Privilege is less than your Over-Subscription Request, then the excess funds held by the Subscription Agent on your behalf will be returned to you, without interest, as soon as practicable after the Rights Offering has expired and all prorating calculations and reductions contemplated by the terms of the Rights Offering have been effected, and we will have no further obligations to you.
The receipt of Subscription Rights may be treated as a taxable distribution to you.
We believe the distribution of the Subscription Rights in this Rights Offering should be a non-taxable distribution to holders of shares of common stock less attractive because we will relyand the participating warrants under Section 305(a) of the Internal Revenue Code of 1986, as amended, or the "Code." Please see the discussion on these exemptions.the "Material U.S. Federal Income Tax Consequences" below. This position is not binding on the IRS, or the courts, however. If some investors findthis Rights Offering is deemed to be part of a "disproportionate distribution" under Section 305 of the Code, your receipt of Subscription Rights in this offering may be treated as the receipt of a taxable distribution to you equal to the fair market value of the Subscription Rights. Any such distribution would be treated as dividend income to the extent of our current and accumulated earnings and profits, if any, with any excess being treated as a return of capital to the extent thereof and then as capital gain. Each holder of shares of common stock less attractive asand each holder of a result,participating warrant is urged to consult his, her or its own tax advisor with respect to the particular tax consequences of this Rights Offering.
The Subscription Rights are not transferable, and there is no market for the Subscription Rights.
You may benot sell, transfer, assign or give away your Subscription Rights. Because the Subscription Rights are non-transferable, there is no market or other means for you to directly realize any value associated with the Subscription Rights. You must exercise the Subscription Rights to realize any potential value from your Subscription Rights.
Absence of a less activepublic trading market for our common stock and our stock pricethe Warrants may be more volatile. We may take advantage of these reporting exemptions until we are no longer an emerging growth company, which in certain circumstances could be for uplimit your ability to five years. See "Summary—Implications of Being an Emerging Growth Company."
Our status as an "emerging growth company" underresell the JOBS Act may make it more difficult to raise capital as and when we need it.Warrants.
Because ofThere is no established trading market for the exemptions from various reporting requirements providedWarrants to us as an "emerging growth company" we may be less attractiveissued pursuant to investorsthis offering, and it may be difficult for us to raise additional capital as and when we need it. Investors may be unable to compare our business with other companies in our industry if they believe that our financial accounting is not as transparent as other companies in our industry. If we are unable to raise additional capital as and when we need it, our financial condition and results of operations may be materially and adversely affected.
If we fail to maintain an effective system of internal control over financial reporting in the future, weWarrants may not be ablewidely distributed. We have applied to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us and, as a result,list the value of our common stock.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal controlsWarrants for financial reporting and disclosure controls and procedures. Commencing with our annual reporttrading on Form 10-K forNASDAQ under the year ending December 31, 2014, we will be required, under Section 404 of the Sarbanes-Oxley Act, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. This assessment will need to include disclosure of any material weaknesses identified by our management in our internal control over financial reporting. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting that results in more than a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. Section 404 of the Sarbanes-Oxley Act also generally requires an attestation from our independent registered public accounting firm on the effectiveness of our internal control over financial reporting. However, for as long as we remain an emerging growth company as defined in the JOBS Act, we intend to take advantage of the exemption permitting us not to comply with the independent registered public accounting firm attestation requirement.
Our compliance with Section 404 will require that we incur substantial accounting expense and expend significant management efforts. We currently do not have an internal audit group, and we will need to hire additional accounting and financial staff with appropriate public company experience and technical accounting knowledge, and compile the system and process documentation necessary to perform the evaluation needed to comply with Section 404. We may not be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. We cannot assure you thatsymbol "ONTXW," but there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. If we are unable to conclude that our internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness or significant deficiency in our internal control over financial reporting once that firm begin its Section 404 reviews, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of our common stock could decline, and we could be subject to
sanctions or investigations by the NASDAQ Stock Market, the SEC or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
If we are unable to successfully remediate the existing material weakness in our internal control over financial reporting, the accuracy and timing of our financial reporting may be adversely affected.
In connection with the audit of our consolidated financial statements for the year ended December 31, 2012, our management and independent registered public accounting firm identified control deficiencies in our internal control over financial reporting that together constitute a material weakness in our internal control over financial reporting. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our financial statements will not be prevented or detected on a timely basis. Our management and independent registered public accounting firm did not perform an evaluation of our internal control over financial reporting as of December 31, 2012 in accordance with the provisions of the Sarbanes-Oxley Act. Had we and our independent registered public accounting firm performed such an evaluation, additional control deficiencies may have been identified by management or our independent registered public accounting firm, and those control deficiencies could have also represented one or more material weaknesses.
Our management and independent registered public accounting firm identified a material weakness in our control over financial reporting attributable to the combination of our lack of sufficient financial reporting and accounting personnel with appropriate training in generally accepted accounting principles in the United States, or GAAP, and SEC rules and regulations with respect to financial reporting. As such, our controls over financial reporting were not designed or operating effectively, and as a result there were adjustments required in connection with closing our books and records and preparing our 2012 consolidated financial statements. The control deficiencies that we and our independent registered public accounting firm identified, and the adjustments recorded as a result, were as follows:
administrative expenses by $5.8 million and increase research and development expenses by $5.5 million.
These control deficiencies resulted in more than a remote likelihood that a material misstatement of our annual and interim financial statements would not be prevented or detected.
In an effort to remediate our material weakness, we have recently hired a Chief Financial Officer and a director of financial reporting. We intend to hire additional finance and accounting personnel with appropriate training, build our financial management and reporting infrastructure, and further develop and document our accounting policies and financial reporting procedures. There can be no assurance that wea sufficient number of Subscription Rights will be successful in pursuing these measuresexercised so that the Warrants will meet minimum listing criteria to be accepted for listing on NASDAQ or that these measuresa market will significantly improve or remediatedevelop for the material weakness described above. There is also no assurance that we have identified all of our material weaknesses or that we will not inWarrants. Even if a market for the future have additional material weaknesses. If we fail to remediateWarrants does develop, the material weakness or to meet the demands that will be placed upon us as a public company, including the requirementsprice of the Sarbanes-Oxley Act, weWarrants may fluctuate and liquidity may be limited. If the Warrants are not accepted for listing on NASDAQ or if a market for the Warrants does not develop, then purchasers of the Warrants may be unable to accurately report our financial results,resell the Warrants or reportsell them within the timeframes required by law or stock exchange regulations. Failure to comply with Section 404only at an unfavorable price for an extended period of time, if at all. Future trading prices of the Sarbanes-Oxley Act could also potentially subject us to sanctions or investigations by the SEC or other regulatory authorities. There is no assurance that weWarrants will be able to remediate the material weakness in a timely manner, or at all, or that in the future, additional material weaknesses will not exist or otherwise be discovered. If our efforts to remediate the material weakness identified are not successful, or if other material weaknesses or other deficiencies occur, our ability to accurately and timely report our financial position could be impaired, which could result in late filings of our annual and quarterly reports under the Exchange Act, restatements of our consolidated financial statements, a decline in our stock price, suspension or delisting of our common stock from the NASDAQ Global Market, and could adversely affect our reputation, results of operations and financial condition.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
Upon consummation of this offering, we will become subject to the periodic reporting requirements of the Exchange Act. Our disclosure controls and procedures are designed to reasonably assure that information required to be disclosed by us in reports we file or submit under the Exchange Act is accumulated and communicated to management, recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements or insufficient disclosures due to error or fraud may occur and not be detected.
We will incur increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives.
As a public company, we will incur significant legal, accounting and other expenses that we did not incur as a private company, and these expenses may increase even more after we are no longer an "emerging growth company." We will be subject to the reporting requirements of the Exchange Act,
the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Protection Act, as well as rules adopted, and to be adopted, by the SEC and NASDAQ Stock Market. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, we expect these rules and regulations to substantially increase our legal and financial compliance costs and to make some activities more time-consuming and costly. We estimate that we will incur approximately $2.0 to $3.0 million of incremental costs per year associated with being a publicly traded company, although it is possible that our actual incremental costs will be higher than we currently estimate. The increased costs will increase our consolidated net loss. For example, we expect these rules and regulations to make it more difficult and more expensive for us to obtain director and officer liability insurance and we may be required to incur substantial costs to maintain the sufficient coverage. We cannot predict or estimate the amount or timing of additional costs we may incur to respond to these requirements. The impact of these requirements could also make it more difficult for us to attract and retain qualified persons to servedepend on our board of directors, our board committees or as executive officers.
Because we do not anticipate paying any cash dividends on our capital stock in the foreseeable future, capital appreciation, if any, will be your sole source of gain.
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all of our future earnings, if any, to finance the growth and development of our business. In addition, the terms of any future debt agreements may preclude us from paying dividends. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future.
Sales of a substantial number of shares of our common stock in the public market could cause our stock price to fall.
Sales of a substantial number of shares of our common stock in the public market could occur at any time. These sales, or the perception in the market that the holders of a large number of shares intend to sell shares, could reduce the market price of our common stock. After this offering, we will have outstanding shares of common stock based on the number of shares outstanding as of March 31, 2013. This includes the shares that we are selling in this offering, which may be resold in the public market immediately without restriction, unless purchased by our affiliates. Of the remaining shares, shares of our common stock, will be restricted as a result of securities laws or lock-up agreements but will be able to be sold after the offering as described in the "Shares Eligible for Future Sale" section of this prospectus. Moreover, after this offering, holders of an aggregate of shares of our common stock will have rights, subject to certain conditions, to require us to file registration statements covering their shares or to include their shares in registration statements that we may file for ourselves or other stockholders. We also intend to register all shares of common stock that we may issue under our equity compensation plans. Once we register these shares, they can be freely sold in the public market upon issuance, subject to volume limitations applicable to affiliates and the lock-up agreements described in the "Underwriting" section of this prospectus.
Future sales and issuances of our common stock or rights to purchase common stock, including pursuant to our equity incentive plans, could result in additional dilution of the percentage ownership of our stockholders and could cause our stock price to fall.
We expect that significant additional capital will be needed in the future to continue our planned operations. To raise capital, we may sell substantial amounts of common stock or securities convertible into or exchangeable for common stock. These future issuances of common stock or common stock-related securities, together with the exercise of outstanding options and any additional shares issued in connection with acquisitions, if any, may result in material dilution to our investors. Such sales may also result in material dilution to our existing stockholders, and new investors could gain rights, preferences
and privileges senior to those of holders of our common stock, including shares of common stock sold in this offering.
Pursuant to our equity incentive plans, our compensation committee is authorized to grant equity-based incentive awards to our directors, executive officers and other employees and service providers, including officers, employees and service providers of our subsidiaries and affiliates. The number of shares of our common stock available for future grant under our 2007 Equity Compensation Plan, which became effective on December 10, 2007, was 385,643 as of March 31, 2013. Future option grants and issuances of common stock under our 2007 Equity Compensation Plan may have an adverse effect on the market price of our common stock.
We have broad discretion in the use of the net proceeds from this offering and may not use them effectively.
Although we currently intend to use the net proceeds from this offering in the manner described in "Use of Proceeds" elsewhere in this prospectus, our management will have broad discretion in the application of the net proceeds from this offering and could spend the proceeds in ways that do not improve our results of operations or enhance the value of our common stock. The failure by our management to apply these funds effectively could result in financial losses that could have a material adverse effect on our business, cause the market price of our common stock to decline and delay the development of our product candidates. Pending their use, we may invest the net proceeds from this offering in a manner that does not produce income or that loses value. If we do not invest the net proceeds from this offering in ways that enhance stockholder value, we may fail to achieve expected financial results, which could cause the price of our common stock to decline.
Some provisions of our charter documents and Delaware law may have anti-takeover effects that could discourage an acquisition of us by others, even if an acquisition would be beneficial to our stockholders and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our tenth amended and restated certificate of incorporation, or certificate of incorporation, and amended and restated bylaws, or bylaws, that will become effective in connection with consummation of this offering, as well as provisions of Delaware law, could make it more difficult for a third party to acquire us or increase the cost of acquiring us, even if doing so would benefit our stockholders, or remove our current management. These include provisions that will:many factors, including:
These provisions may frustrateThere is no market for the Pre-Funded Warrants.
There is no established trading market for the Pre-Funded Warrants to be issued pursuant to this offering, if any, and the Pre-Funded Warrants will not be listed for trading on any stock exchange or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, who are responsible for appointing the members of our management. Because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which may discourage, delay or prevent someone from acquiring us or merging with us whether or not it is desired by or beneficial to our stockholders. Under Delaware law, a corporation may not, in general, engage in a business combination with any holder of 15% or more of its capital stock unless the holder has held the stock for three years or, among other things, the board of directors has approved the transaction. Any provision of our amended and restated certificate of incorporation or amended and restated bylaws or Delaware law that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their sharesmarket.
The market price of our common stock and could also affectmay never exceed the exercise price that some investors are willing to pay for our common stock.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.of the Warrants issued in connection with this offering.
The tradingWarrants being issued in connection with this offering become exercisable upon issuance and will expire five years after issuance. We cannot provide you any assurance that the market forprice of our common stock will depend in partever exceed the exercise price of the Warrants prior to their date of expiration. Any Warrants not exercised by their date of expiration will expire worthless and we will be under no further obligation to the Warrant holder.
The Warrants may be redeemed on short notice. This may have an adverse impact on their price.
After the research and reports that securities or industry analysts publish about us or our business. Securities and industry analysts do not currently, andone-year anniversary of issuance, we may never, publish research on our company. If no securities or industry analysts commence coverageredeem the Warrants for $0.001 per Warrant once the closing price of our company,common stock has equaled or exceeded $ per share, 300% of the exercise price, subject to adjustment, for 10 consecutive trading pricedays. If we give notice of redemption, you will be forced to sell or exercise your Warrants or accept the redemption price. The notice of redemption could come at a time when it is not advisable or possible for ouryou to exercise the Warrants. As a result, you would be unable to benefit from owning the Warrants being redeemed.
The dealer-manager is not underwriting, nor acting as placement agent of, the Subscription Rights or the securities underlying the Subscription Rights.
Maxim Group LLC is acting as dealer-manager for this Rights Offering. Under the terms and subject to the conditions contained in the dealer-manager agreement, the dealer-manager will provide marketing assistance in connection with this offering. The dealer-manager is not underwriting or placing any of the Subscription Rights or the Units, shares of common stock would likelyor Warrants being issued in this offering, and does not make any recommendation with respect to such Subscription Rights (including with respect to the exercise or expiration of such Subscription Rights), Units, shares of common stock or Warrants. The dealer-manager will not be negatively impacted.subject to any liability to us in rendering the services contemplated by the dealer-manager agreement except for any act of bad faith or gross negligence by the dealer-manager. The services of the dealer-manager to us in connection with this offering cannot be construed as any assurance that this offering will be successful.
Since the Warrants and Pre-Funded Warrants are executory contracts, they may have no value in a bankruptcy or reorganization proceeding.
In the event securitiesa bankruptcy or industry analysts initiate coverage, if onereorganization proceeding is commenced by or moreagainst us, a bankruptcy court may hold that any unexercised Warrants and Pre-Funded Warrants are executory contracts that are subject to rejection by us with the approval of the analysts who cover us downgrade our stockbankruptcy court. As a result, holders of the Warrants and Pre-Funded Warrants may, even if we have sufficient funds, not be entitled to receive any consideration for their Warrants and Pre-Funded Warrants or publish inaccuratemay receive an amount less than they would be entitled to if they had exercised their Warrants and Pre-Funded Warrants prior to the commencement of any such bankruptcy or unfavorable research about our business, our stock price would likely decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our stock could decrease, which might cause our stock price and trading volume to decline.reorganization proceeding.
SPECIALCAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS AND INDUSTRY DATA
This prospectus includes forward-looking statements. We may, in some cases, use terms such as "believes," "estimates," "anticipates," "expects," "plans," "intends," "may," "could," "might," "will," "should," "approximately" or other words that convey uncertainty of future events or outcomes to identify these forward-looking statements. Forward-looking statements appear in a number of places throughout this prospectusreport and include statements regarding our intentions, beliefs, projections, outlook, analyses or current expectations concerning, among other things, our ongoing and planned preclinical development and clinical trials, the timing of and our ability to make regulatory filings and obtain and maintain regulatory approvals for our product candidates, protection of our intellectual property position,portfolio, the degree of clinical utility of our products, particularly in specific patient populations, our ability to develop commercial and manufacturing functions, expectations regarding clinical trial data, our results of operations, cash needs, spending of the proceeds from this offering, financial condition, liquidity, prospects, growth and strategies, the industry in which we operate and the trends that may affect the industry or us.
By their nature, forward-looking statements involve risks and uncertainties because they relate to events, competitive dynamics and industry change, and depend on the economic circumstances that may or may not occur in the future or may occur on longer or shorter timelines than anticipated. Although we believe that we have a reasonable basis for each forward-looking statement contained in this prospectus,report, we caution you that forward-looking statements are not guarantees of future performance and that our actual results of operations, financial condition and liquidity, and the development of the industry in which we operate may differ materially from the forward-looking statements contained in this prospectus.report. In addition, even if our results of operations, financial condition and liquidity, and events in the industry in which we operate are consistent with the forward-looking statements contained in this prospectus,report, they may not be predictive of results or developments in future periods.
Actual results could differ materially from our forward-looking statements due to a number of factors, including risks related to:
Any forward-looking statements that we make in this prospectus speak only as of the date of such statement, and we undertake no obligation to update such statements to reflect events or circumstances after the date of this prospectus or to reflect the occurrence of unanticipated events. Comparisons of results for current and any prior periods are not intended to express any future trends or indications of future performance, unless expressed as such, and should only be viewed as historical data.
You should also read carefully the factors described in the "Risk Factors" section of this prospectus and elsewherein our annual report on Form 10-K filed with the SEC on March 28, 2016, to better understand thesignificant risks and uncertainties inherent in our business and underlying any forward-looking statements. As a result of these factors, we cannot assure you thatactual results could differ materially and adversely from those anticipated or implied in the forward-looking statements in this prospectus will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements,report and you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified timeframe, or at all. The Private Securities Litigation Reform Act of 1995 and Section 27A of the Securities Act do not protectplace undue reliance on any forward-looking statements that we make in connection with this offering.
We obtained the industry, market and competitive position data in this prospectus from our own internal estimates and research as well as from industry and general publications and research surveys and studies conducted by third parties. Industry publications and surveys generally state that the information contained therein has been obtained from sources believed to be reliable. We believe this data is accurate in all material respects as of the date of this prospectus.
Table of Contentsstatements.
WeAssuming that all 4,255,581 Units are sold in the Rights Offering and no elections to receive Pre-Funded Warrants are made, we estimate that ourthe net proceeds from the sale of the shares of common stock in this offeringRights Offering will be approximately $$23.0 million, assuming an initial public offering price of $ per share, which isbased on the mid-pointmidpoint of the priceexpected Subscription Price range set forth on the cover page of this prospectus,$6.00 per Unit, after deducting fees and expenses payable to the estimated underwriting discountsdealer-manager, and commissions and estimated offering costsafter deducting other expenses payable by us. Ifus and excluding any proceeds received upon exercise of any Warrants issued in the underwriters exercise their over-allotment option in full, we estimate that our net proceeds from this offering will be approximately $ million.Rights Offering.
We intend to use our net proceeds from this offering for the overall development of our product candidates. We currently estimate that we will use approximately $ million of the net proceeds from this offeringRights Offering primarily to fund thecontinue funding our ongoing Phase 3 clinical developmenttrial of rigosertib IV in a population of patients with higher-risk MDS after failure of HMA therapy, which we refer to as "INSPIRE," and, approximately $ to fund thea lesser extent, for other development of our other clinical and preclinical programs. The balance will be usedprograms, for other research and development activities, business development and for general corporate purposes, which may include capital expenditures and funding our working capital and general corporate purposes. Pending the application of the net proceeds as described above, we intend to invest the net proceeds of the offering in short-term, investment-grade, interest-bearing securities.needs.
Our management will have broad discretion to allocatein the application of the net proceeds to us from this offering, and investors will be relying on the judgment of our management regarding the application of the proceeds from this offering. We reserve the rightwith regard to change the use of these proceeds as a resultnet proceeds. Pending the use of certain contingencies such as competitive developments, the results of our commercialization efforts and investment opportunities and other factors.
A $1.00 increase or decrease in the assumed initial public offering price of $ per share, which is the mid-point of the price range set forth on the cover page of this prospectus, would increase or decrease our net proceeds from this offering as described above, we intend to hold the net proceeds in cash or invest in short-term, investment-grade, interest-bearing instruments.
The following table presents our cash, cash equivalents and capitalization, as of March 31, 2016:
The table below does not reflect the exercise of Warrants issued in connection with the Rights Offering. The pro forma as adjusted information set forth below assumes equity accounting for the Warrants, is illustrative only and will be adjusted based on the Units sold. You should read this information in conjunction with our consolidated financial statements and notes thereto incorporated by reference into this prospectus.
| | March 31, 2016 (unaudited) | ||||||
|---|---|---|---|---|---|---|---|
| | Actual | Pro Forma as Adjusted | |||||
Cash and cash equivalents | $ | 16,835,000 | $ | 39,880,000 | |||
Long-term liabilities | 5,181,000 | 5,181,000 | |||||
Stockholders' equity*: | |||||||
Preferred stock, $0.01 par value, 5,000,000 authorized at March 31, 2016 and March 31, 2016 Pro Forma, none issued and outstanding at March 31, 2016 | — | — | |||||
Common stock, $0.01 par value, 25,000,000 authorized at March 31, 2016 and March 31, 2016 Pro Forma as Adjusted, 2,740,212 shares issued and outstanding at March 31, 2016 and 6,995,793 shares issued and outstanding at March 31, 2016 Pro Forma as Adjusted | 27,000 | 70,000 | |||||
Additional paid-in capital | 331,775,000 | 354,777,000 | |||||
Accumulated other comprehensive loss | (16,000 | ) | (16,000 | ) | |||
Accumulated deficit | (325,797,000 | ) | (325,797,000 | ) | |||
| | | | | | | | |
Total Onconova Therapeutics, Inc. stockholders' equity | 5,989,000 | 29,034,000 | |||||
Non-controlling interest | 830,000 | 830,000 | |||||
| | | | | | | | |
Total stockholders' equity | 6,819,000 | 29,864,000 | |||||
| | | | | | | | |
The information above is as of March 31, 2016 and excludes:
All share and per share data has been restated to reflect our one-for-ten reverse stock split effective May 31, 2016, subject to final adjustments for treatment of fractional share interests.
Purchasers of our common stock in the Rights Offering (and upon exercise of the Warrants issued pursuant to this Rights Offering) will experience an immediate dilution of the net tangible book value per share of our common stock. Our net tangible book value as of March 31, 2016 was approximately $ million, assuming that$6,819,000, or $2.49 per share of our common stock (based upon 2,740,212 shares of our common stock outstanding after giving effect to the one-for-ten reverse stock split on May 31, 2016). Net tangible book value per share is equal to our total net tangible book value, which is our total tangible assets less our total liabilities, divided by the number of shares offeredof our outstanding common stock. Dilution per share equals the difference between the amount per share paid by purchasers of shares of common stock in the Rights Offering and the net tangible book value per share of our common stock immediately after the Rights Offering.
Based on the sale by us as set forth onin this Rights Offering of a maximum of 4,255,581 Units (consisting of 4,255,581 shares of our common stock (assuming no Pre-Funded Warrants) and Warrants to purchase an aggregate of 3,191,686 shares of common stock upon exercise), at the cover pageassumed Subscription Price of this prospectus, remains$6.00 per Unit (the midpoint of the sameexpected range of $5.75 and $6.25 per Unit), and after deducting estimated offering expenses and dealer-manager fees and expenses payable by us of $2.5 million, and the application of the estimated underwriting discounts$23.0 million of net proceeds from the Rights Offering, assuming equity accounting for the Warrants, our pro forma net tangible book value as of March 31, 2016 would have been approximately $29.9 million, or $4.27 per share. This represents an immediate increase in pro forma net tangible book value to existing shareholders of $1.78 per share and commissions. Each increase or decrease of 1.0 million sharesan immediate dilution to purchasers in the numberRights Offering of shares offered by us$1.73 per share.
The following table illustrates this per-share dilution on a pro forma basis, assuming a fully subscribed for Rights Offering of 4,255,581 Units at the assumed initial public offeringSubscription Price of $6.00 per Unit (the midpoint of the expected range of $5.75 and $6.25 per Unit) (assuming no Pre-Funded Warrants and excluding any issuance of shares of common stock upon exercise of Warrants):
Subscription Price | $ | 6.00 | |||||
Net tangible book value per share as of March 31, 2016, before Rights Offering | $ | 2.49 | |||||
Increase in net tangible book value per share attributable to Rights Offering | $ | 1.78 | |||||
Pro forma net tangible book value per share as of March 31, 2016, after giving effect to Rights Offering | $ | 4.27 | |||||
Dilution in net tangible book value per share to purchasers | $ | 1.73 |
The information above is as of March 31, 2016 and excludes:
All share and per share data has been restated to reflect our one-for-ten reverse stock split effective May 31, 2016.
To the extent that outstanding options or warrants are exercised, the investor purchasing our common stock in this offering by approximately $ million. We do not expectwill experience further dilution. In addition, we may choose to raise additional capital due to market conditions or strategic considerations. To the extent that a changeadditional capital is raised through the sale of securities, the issuance of those securities could result in further dilution to our stockholders.
MARKET PRICE OF OUR COMMON STOCK AND RELATED STOCKHOLDER MATTERS
Our common stock began trading on the offeringNASDAQ Global Select Market on July 25, 2013 under the symbol "ONTX." Prior to that time, there was no public market for our common stock. On February 5, 2016, we transferred the listing of our shares to the NASDAQ Capital Market. The following table sets forth for the periods indicated the high and low intra-day sale prices per share of our common stock as reported on the NASDAQ Global Market or NASDAQ Capital Market, as adjusted to give effect to our one-for-ten reverse stock split effective May 31, 2016:
| | High | Low | |||||
|---|---|---|---|---|---|---|---|
Year Ending December 31, 2016 | |||||||
First Quarter | $ | 10.28 | $ | 3.23 | |||
Second Quarter (through June 28, 2016) | 8.17 | 3.80 | |||||
Year Ended December 31, 2015 | |||||||
First Quarter | $ | 44.30 | $ | 21.50 | |||
Second Quarter | 30.20 | 22.60 | |||||
Third Quarter | 40.00 | 13.20 | |||||
Fourth Quarter | 18.90 | 9.20 | |||||
Year Ended December 31, 2014 | |||||||
First Quarter | $ | 162.20 | 60.50 | ||||
Second Quarter | 64.90 | 41.00 | |||||
Third Quarter | 57.80 | 42.40 | |||||
Fourth Quarter | 50.00 | 32.40 | |||||
On June 28, 2016, the last reported sale price orof our common stock on the NASDAQ Capital Market was $6.00 per share. As of June 27, 2016, we had 138 holders of record of our common stock. The actual number of holders of common stock is greater than these numbers of record holders and includes stockholders who are beneficial owners, but whose shares are held in street name by these amounts would have a material effect on our usesbrokers and nominees. The number of the proceeds from this offering, although it might affect the amountholders of time prior to which we will need to seek additional capital.record also does not include stockholders whose shares may be held in trust by other entities.
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain all available funds and any future earnings to support our operationscorporation and finance the growth and development of our business. We do not intend to pay cash dividends on our common stock for the foreseeable future.
CAPITALIZATIONTHE RIGHTS OFFERING
The following table sets forthSubscription Rights
We are distributing to the record holders and to holders of certain outstanding warrants who are entitled to participate in this offering pursuant to the terms of such warrants, at no charge, non-transferable Subscription Rights to purchase one Unit at a subscription price per Unit to be determined. The Subscription Price per Unit is expected to be between $5.75 and $6.25. The exact Subscription Price per Unit will be determined by our cashboard of directors or pricing committee prior to the effectiveness of the registration statement of which this prospectus is a part. Each Basic Subscription Right will entitle you to purchase one share of our common stock and cash equivalents and capitalization0.75 of a Warrant for the purchase of one additional share of our common stock at an exercise price of $ per share, 120% of the per Unit price, from the date of issuance through its expiration on , 2021. Each record holder will receive 1.5 Subscription Rights for each whole share of our common stock owned by such record holder as of March 31, 2013:
Basic Subscription Rights
Your Basic Subscription Rights will entitle you to purchase one share of our common stock and 0.75 of a Warrant to purchase one share of our common stock at the exercise price described elsewhere in this prospectus. For example, if you owned 100 shares of our preferredcommon stock into 17,113,481as of the Record Date, you will receive 150 Subscription Rights and will have the right to purchase 150 shares of our common stock whichand Warrants to purchase 112 shares of our common stock for $ per whole Unit, or a total payment of $ . You may exercise all or a portion of your Basic Subscription Rights, or you may choose not to exercise any of your Basic Subscription Rights. If you do not exercise your Basic Subscription Rights in full, you will occur immediately priornot be entitled to consummationexercise your Over-Subscription Privilege.
Over-Subscription Privilege
If you exercise your Basic Subscription Rights in full, you may also choose to exercise your Over-Subscription Privilege. Subject to proration, if applicable, we will seek to honor the Over-Subscription Privilege requests in full. If Over-Subscription Privilege requests exceed the number of this offering;Units available, however, we will allocate the available Units pro rata among the record holders and
You should read the information in this table together with our consolidated financial statements and accompanying notes and "Management's Discussion and Analysis of Financial Condition and Results of Operations" appearing elsewhere in this prospectus.
| | As of March 31, 2013 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | Actual | Pro Forma | Pro Forma As Adjusted | |||||||
Cash and cash equivalents | $ | 67,307,000 | $ | 67,307,000 | $ | |||||
Preferred stock, $0.01 par value per share: | ||||||||||
Series A convertible preferred stock: 400,000 shares authorized, 107,000 shares issued and outstanding, actual; no shares authorized, issued or outstanding, pro forma and pro forma as adjusted | $ | 535,000 | $ | — | $ | — | ||||
Series B convertible preferred stock: 1,200,000 shares authorized, 1,107,189 shares issued and outstanding, actual; no shares authorized, issued or outstanding, pro forma and pro forma as adjusted | 12,733,000 | — | — | |||||||
Series C convertible preferred stock: 1,200,000 shares authorized, 1,069,946 shares issued and outstanding, actual; no shares authorized, issued or outstanding, pro forma and pro forma as adjusted | 7,618,000 | — | — | |||||||
Series D convertible preferred stock: 1,625,000 shares authorized, 1,583,568 shares issued and outstanding, actual; no shares authorized, issued or outstanding, pro forma and pro forma as adjusted | 18,211,000 | — | — | |||||||
Series E convertible preferred stock: 1,650,000 shares authorized, 1,633,082 shares issued and outstanding, actual; no shares authorized, issued or outstanding, pro forma and pro forma as adjusted | 18,780,000 | — | — | |||||||
Series F convertible preferred stock: 2,000,000 shares authorized, 2,000,000 shares issued and outstanding, actual; no shares authorized, issued or outstanding, pro forma and pro forma as adjusted | 23,000,000 | — | — | |||||||
| | As of March 31, 2013 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | Actual | Pro Forma | Pro Forma As Adjusted | |||||||
Series G convertible preferred stock: 2,700,000 shares authorized, 1,934,359 shares issued and outstanding, actual; no shares authorized, issued or outstanding, pro forma and pro forma as adjusted | 22,819,000 | — | — | |||||||
Series H convertible preferred stock: 2,042,950 shares authorized, 2,013,424 shares issued and outstanding, actual; no shares authorized, issued or outstanding, pro forma and pro forma as adjusted | 22,385,000 | — | — | |||||||
Series I convertible preferred stock: 2,700,000 shares authorized, 2,433,328 shares issued and outstanding, actual; no shares authorized, issued or outstanding, pro forma and pro forma as adjusted | 27,033,000 | — | — | |||||||
Series J convertible preferred stock: 3,030,303 shares authorized, 3,030,303 shares issued and outstanding, actual; no shares authorized, issued or outstanding, pro forma and pro forma as adjusted | 49,220,000 | — | — | |||||||
Total preferred stock | 202,334,000 | — | — | |||||||
Stockholders' (deficit) equity: | ||||||||||
Common stock, $0.01 par value per share: 30,145,155 shares authorized, 3,478,473 shares issued and outstanding, actual; 30,145,155 shares authorized, 20,591,954 shares issued and outstanding, pro forma; and shares authorized, shares issued and outstanding, pro forma as adjusted | 35,000 | 206,000 | ||||||||
Additional paid-in capital | 9,044,000 | 211,207,000 | ||||||||
Accumulated other comprehensive income | 7,000 | 7,000 | 7,000 | |||||||
Accumulated deficit | (183,205,000 | ) | (183,205,000 | ) | ||||||
Total stockholders' (deficit) equity | (174,119,000 | ) | 28,215,000 | |||||||
Total capitalization | $ | (174,119,000 | ) | $ | 28,215,000 | $ | ||||
The number of shares underlying participating warrants held by each of common stockthose warrant holders on the Record Date, relative to be outstanding afterthe number of shares owned or underlying participating warrants on the Record Date by all record holders and warrant holders exercising the Over-Subscription Privilege. If this offering is based on 20,591,954 sharespro rata allocation results in any record holder or warrant holder receiving a greater number of common stock outstanding as of March 31, 2013, and excludes as of that date:
Wells Fargo Bank, N.A., the Subscription Agent for the Rights Offering, will determine the over-subscription allocation based on the formula described above.
To the extent the aggregate subscription payment of the actual number of unsubscribed Units available to you pursuant to the Over-Subscription Privilege is less than the amount you actually paid in connection with the exercise of the Over-Subscription Privilege, you will be allocated only the number of unsubscribed Units available to you, and any excess subscription payments will be returned to you, without interest or penalty, as soon as practicable after expiration of the Rights Offering.
We can provide no assurances that you will actually be entitled to purchase the number of Units issuable upon the exercise of your Over-Subscription Privilege in full at the expiration of the Rights Offering. We will not be able to satisfy any requests for Units pursuant to the Over-Subscription Privilege if all of our shareholders and participating warrant holders exercise their Basic Subscription Rights in full, and we will only honor an Over-Subscription Privilege to the extent sufficient Units are available following the exercise of Basic Subscription Rights.
DILUTIONPre-Funded Warrants
If your subscription for Units in the Rights Offering may result in the your beneficial ownership of more than 4.99% of our outstanding common stock following the consummation of the Rights Offering, and you do not wish to exceed that ownership threshold, you may elect to receive a Pre-Funded Warrant to purchase one share of common stock in lieu of any share of common stock underlying the Units for which you have subscribed in excess of such threshold.
You may make an election to receive Pre-Funded Warrants in lieu of common stock, to the extent that your beneficial ownership would otherwise be above the ownership threshold. If you intend to do so, in addition to making your election on your Subscription Rights Statement, we ask that you contact the dealer-manager for the Rights Offering as follows:
Maxim Group LLC
405 Lexington Avenue
New York, New York 10174
Attention Syndicate Department
Email: syndicate@maximgrp.com
Telephone: (212) 895-3745
If you invest in our common stock in this offering, your ownership interest will be dilutedmake an election to receive Pre-Funded Warrants but fail to timely provide the extentrequired holder information for the issuance of any Pre-Funded Warrants, you may receive the difference between the initial public offering price per share of our common stock and the pro forma as adjusted net tangible book value per share of our common stock upon consummation of this offering. The historical net tangible book value (deficit) of our common stock as of March 31, 2013 was $(174.1) million, or $(50.06) per share. Historical net tangible book value (deficit) per share is determined by dividing the number of our outstanding shares of common stock into our total tangible assets (total assets less intangible assets) less total liabilities.underlying all of your subscribed Units.
Our Participating Warrant Holders
On a pro forma basis, after giving effectJanuary 11, 2016, we issued common stock purchase warrants to the conversion of all outstanding shares of our preferred stock into 17,113,481purchase up to 96,842 shares of our common stock at an exercise price equal to $11.50 per share, subject to customary adjustments and as adjusted for our one-for-ten reverse stock split effective May 31, 2016. These warrants, referred to as our "participating warrants," entitle the holders to participate in this Rights Offering as if each of such participating warrants had been exercised immediately prior to consummationthe record date for the Rights Offering. As a result, holders of our participating warrants are receiving Subscription Rights for an aggregate of approximately 96,842 Units in connection with this Rights Offering; provided, however, to the extent that any warrant holder's right to participate in this Rights Offering would result in the holder exceeding the beneficial ownership limitation set forth in the participating warrants, then the holder will not be entitled to participate in this Rights Offering to such extent and the portion of this offering, our net tangible book value at March 31, 2013 would have been $28.2 million, or $1.37 per share.
Investors purchasingRights Offering will be held in this offering will incur immediate and substantial dilution. After giving effect toabeyance for the sale of common stock offered in this offering assuming an initial public offering price of $ per share, which is the mid-pointbenefit of the price range set forth on the cover page of this prospectus, and after deducting the estimated underwriting discounts and commissions and estimated offering costs payable by us, our pro formaholder until such time, if ever, as adjusted net tangible book value as of December 31, 2012its right thereto would have been $ million, or $ per share. This represents an immediate increase in pro forma net tangible book value of $ share to existing stockholders, and an immediate dilutionnot result in the pro forma net tangible book value of $ per share to investors purchasing in this offering. The following table illustrates this per share dilution:
Assumed initial public offering price per share | $ | ||||||
Historical net tangible book value (deficit) per share as of March 31, 2013 | $ | (50.06 | ) | ||||
Pro forma increase in net tangible book value per share attributable to the conversion of all outstanding shares of our preferred stock into 17,113,481 shares of our common stock immediately prior to consummation of this offering | 51.43 | ||||||
Pro forma net tangible book value per share March 31, 2013 | 1.37 | ||||||
Increase in pro forma net tangible book value per share attributable to investors purchasing in this offering | |||||||
Pro forma as adjusted net tangible book value per share after this offering | |||||||
Dilution per share to investors purchasing in this offering | $ | ||||||
The following table summarizes, onholder exceeding the pro forma as adjusted basis described above as of March 31, 2013, the differences between the number of shares of common stock purchased from us, the total consideration paid and the average price per share paid by existing stockholders and by investors purchasing in this offering at an assumed initial public offering price of $ per share, before deducting estimated underwriting discounts and commissions and estimated offering costs payable by us.
| | Shares Purchased | Total Consideration | | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Average Price Per Share | |||||||||||||||
| | Number | Percent | Amount | Percent | ||||||||||||
Existing stockholders before this offering | % | $ | % | $ | ||||||||||||
Investors purchasing in this offering | ||||||||||||||||
Total | 100.0 | % | $ | 100.0 | % | |||||||||||
A $1.00 increase or decrease in the assumed initial public offering price of $ per share, which is the mid-point of the price range set forth on the cover page of this prospectus, would increase or decrease the pro forma as adjusted net tangible book value per share by $ , assuming the number of shares of common stock offered by us, as set forth on the cover page of this prospectus,applicable beneficial ownership limitation.
remains None of our other currently outstanding warrants is entitled to receive Subscription Rights in this offering.
Limitation on the samePurchase of Units
You may only purchase the number of whole Units purchasable upon exercise of the number of Basic Subscription Rights distributed to you in the Rights Offering, plus the Over-Subscription Privilege, if any. Accordingly, the number of Units that you may purchase in the Rights Offering is limited by the number of shares of our common stock you held on the Record Date or the number of shares underlying your outstanding participating warrants and by the extent to which other shareholders and participating warrant holders exercise their Basic Subscription Rights and Over-Subscription Privileges, which we cannot determine prior to completion of the Rights Offering.
Subscription Price
The Subscription Price per Unit is expected to be between $5.75 and $6.25. The exact Subscription Price per Unit will be determined by our board of directors or pricing committee prior to the effectiveness of the registration statement of which this prospectus is a part. The Subscription Price does not necessarily bear any relationship to our past or expected future results of operations, cash flows, current financial condition, or any other established criteria for value. No change will be made to the Subscription Price by reason of changes in the trading price of our common stock or other factor prior to the expiration of this Rights Offering.
Determination of Subscription Price
In the determining the Subscription Price, the board of directors or pricing committee may consider a variety of factors including those listed below:
The Subscription Price does not necessarily bear any relationship to any established criteria for value. No valuation consultant or investment banker has opined upon the fairness or adequacy of the Subscription Price. You should not consider the Subscription Price as an indication of actual value of our company or our common stock. You should not assume or expect that, after deducting estimated underwriting discountsthe Rights Offering, our shares of common stock will trade at or above the Subscription Price in any given time period. The market price of our common stock may decline during or after the Rights Offering. We cannot assure you that you will be able to sell the shares of our common stock purchased during the Rights Offering at a price equal to or greater than the Subscription Price. You should obtain a current price quote for our common stock before exercising your Subscription Rights and commissionsmake your own assessment of our
business and estimated offering expenses payablefinancial condition, our prospects for the future, and the terms of this Rights Offering. Once made, all exercises of Subscription Rights are irrevocable.
No Recombination
The common stock and Warrants comprising the Units will separate upon the effectiveness of the exercise of the Subscription Rights and will be issued as separate securities, and the Units will not trade as a separate security. Holders may not recombine shares of common stock and Warrants to receive a Unit.
Non-Transferability of Subscription Rights
The Subscription Rights are non-transferable (other than by us. Each increaseoperation of law) and, therefore, you may not sell, transfer, assign or decreasegive away your Subscription Rights to anyone. The Subscription Rights will not be listed for trading on any stock exchange or market.
Expiration Date; Extension
The subscription period, during which you may exercise your Subscription Rights, expires at 5:00 PM Eastern Time, on July 26, 2016, which is the expiration of 1.0 millionthe Rights Offering. If you do not exercise your Subscription Rights before that time, your Subscription Rights will expire and will no longer be exercisable. We will not be required to issue shares to you if the Subscription Agent receives your Subscription Rights Statement or your subscription payment after that time. We have the option to extend the Rights Offering in our sole discretion, although we do not presently intend to do so. We may extend the Rights Offering by giving oral or written notice to the Subscription Agent before the Rights Offering expires. If we elect to extend the Rights Offering, we will issue a press release announcing the extension no later than 9:00 AM Eastern Time, on the next business day after the most recently announced expiration date of the Rights Offering.
If you hold your shares of common stock in the name of a broker, dealer, custodian bank or other nominee, the nominee will exercise the Subscription Rights on your behalf in accordance with your instructions. Please note that the nominee may establish a deadline that may be before 5:00 PM Eastern Time, on July 26, 2016, which is the expiration date that we have established for the Rights Offering.
Termination
We may terminate the Rights Offering at any time and for any reason prior to the completion of the Rights Offering. If we terminate the Rights Offering, we will issue a press release notifying shareholders, warrant holders and the public of the termination.
Return of Funds upon Completion or Termination
The Subscription Agent will hold funds received in payment for shares in a segregated account pending completion of the Rights Offering. The Subscription Agent will hold this money until the Rights Offering is completed or is terminated. To the extent you properly exercise your Over-Subscription Privilege for an amount of Units that exceeds the number of unsubscribed Units available to you, any excess subscription payments will be returned to you as soon as practicable after the expiration of the Rights Offering, without interest or penalty. If the Rights Offering is terminated for any reason, all subscription payments received by the Subscription Agent will be returned as soon as practicable, without interest or penalty.
Shares of Our Common Stock Outstanding After the Rights Offering
After our one-for-ten reverse stock split on May 31, 2016, 2,740,212 shares of our common stock were outstanding, along with participating warrants to purchase 96,842 shares of common stock. Based on the foregoing, and assuming no other transactions by us involving our common stock prior to the expiration of the Rights Offering, and that no Pre-Funded Warrants are issued in lieu of common stock, if the Rights Offering is fully subscribed approximately 6,995,793 shares of our common stock will be issued and outstanding and Warrants to purchase approximately 3,191,686 additional shares of our common stock will be outstanding (excluding the currently outstanding warrants). The exact number of shares of common stock, offeredWarrants and Pre-Funded Warrants that we will issue in this Rights Offering will depend on the number of Units that are subscribed for in the Rights Offering and the elections of eligible investors to receive Pre-Funded Warrants.
Methods for Exercising Subscription Rights
The exercise of Subscription Rights is irrevocable and may not be cancelled or modified. You may exercise your Subscription Rights as follows:
Subscription by usRecord Holders
If you are a shareholder of record or a holder of participating warrants, the number of Units you may purchase pursuant to your Subscription Rights in indicated on the enclosed Subscription Rights Statement. You may exercise your Subscription Rights by properly completing and executing the Subscription Rights Statement and forwarding it, together with your full payment, to the Subscription Agent at the assumed public offering price would increaseaddress given below under "Subscription Agent," to be received before 5:00 PM Eastern Time, on July 26, 2016.
Subscription by Beneficial Owners
If you are a beneficial owner of shares of our common stock that are registered in the name of a broker, dealer, custodian bank, or decreaseother nominee, you will not receive a Subscription Rights Statement. Instead, we will issue one Subscription Right to such nominee record holder for all shares of our pro formacommon stock held by such nominee at the Record Date. If you are not contacted by your nominee, you should promptly contact your nominee in order to subscribe for shares in the Rights Offering and follow the instructions provided by your nominee.
To properly exercise your Over-Subscription Privilege, you must deliver the subscription payment related to your Over-Subscription Privilege before the Rights Offering expires. Because we will not know the total number of unsubscribed Units before the Rights Offering expires, if you wish to maximize the number of shares you purchase pursuant to your Over-Subscription Privilege, you will need to deliver payment in an amount equal to the aggregate subscription payment for the maximum number of Units that you wish to purchase.
Payment Method
Payments must be made in full in U.S. currency by cashier's check or by wire transfer, and payable to "Wells Fargo Shareowner Services, as adjusted net tangible book valueSubscription Agent for Onconova Therapeutics, Inc." You must timely pay the full subscription payment, including payment for the Over-Subscription Privilege, for the full number of Units of our common stock and Warrants you wish to acquire pursuant to the exercise of Subscription Rights by $ million, our pro formadelivering a:
You should read the instruction letter accompanying the Subscription Rights Statement carefully and strictly follow it.DO NOT SEND SUBSCRIPTION RIGHTS STATEMENTS OR PAYMENTS DIRECTLY TO US. We will not consider your subscription received until the Subscription Agent has received delivery of a properly completed and duly executed Subscription Rights Statement and payment of the full subscription payment.
The method of delivery of Subscription Rights Statements and payment of the subscription payment to the Subscription Agent will be at the risk of the holders of Subscription Rights. If sent by $ .mail, we recommend that you send those statements and payments by registered mail, properly insured, with return receipt requested, or by overnight courier, and that you allow a sufficient number of days to ensure delivery to the Subscription Agent before the Rights Offering expires.
Missing or Incomplete Subscription Forms or Payment
Except asIf you fail to complete and sign the Subscription Rights Statement or otherwise indicated,fail to follow the discussion and tables above assume nosubscription procedures that apply to the exercise of your Subscription Rights before the underwriters' over-allotment optionRights Offering expires, the Subscription Agent will reject your subscription or accept it to the extent of the payment received. Neither we nor our Subscription Agent undertakes any responsibility or action to contact you concerning an incomplete or incorrect subscription form, nor are we under any obligation to correct such forms. We have the sole discretion to determine whether a subscription exercise properly complies with the subscription procedures.
If you send a payment that is insufficient to purchase the number of shares you requested, or if the number of shares you requested is not specified in the forms, the payment received will be applied to exercise your Subscription Rights to the fullest extent possible based on the amount of the payment received. Any excess subscription payments received by the Subscription Agent will be returned, without interest or penalty, as soon as practicable following the expiration of the Rights Offering.
Issuance of common stock and no exerciseWarrants
The shares of any outstanding optionscommon stock and Warrants that are purchased in the Rights Offering as part of the Units will be issued in book-entry, or uncertificated, form meaning that you will receive a direct registration (DRS) account statement from our transfer agent reflecting ownership of these securities if you are a holder of record of shares or warrants. If you hold your shares of common stock in the underwriters' over-allotment optionname of a custodian bank, broker, dealer, or other nominee, DTC will credit your account with your nominee with the securities you purchased in the Rights Offering.
Any Pre-Funded Warrants that are purchased in the Rights Offering will be issued in physical form.
Subscription Agent
The Subscription Agent for the Rights Offering is exercisedWells Fargo Bank, N.A. The address to which Subscription Rights Statements and payments should be mailed or delivered by overnight courier is provided below. If sent by mail, we recommend that you send documents and payments by registered mail, properly insured, with return receipt requested, and that you allow a sufficient number of days to
ensure delivery to the Subscription Agent before the Rights Offering expires. Do not send or deliver these materials to us.
By Mail:
Wells Fargo Bank, N.A.
Shareowner Services
Voluntary Corporate Actions
P.O. Box 64858
St. Paul, Minnesota 55164-0858
By Hand or Overnight Courier:
Wells Fargo Bank, N.A.
Shareowner Services
Voluntary Corporate Actions
1110 Centre Pointe Curve, Suite 101
Mendota Heights, Minnesota 55120
If you deliver the Subscription Rights Statements in full,a manner different than that described in this prospectus, we may not honor the exercise of your Subscription Rights.
Dealer-Manager
You should direct any questions or requests for assistance concerning the method of subscribing for the shares of our common stock or for additional copies of this prospectus to the dealer-manager for the Rights Offering as follows:
Maxim Group LLC
405 Lexington Avenue
New York, New York 10174
Attention Syndicate Department
Email: syndicate@maximgrp.com
Telephone: (212) 895-3745
No Fractional Shares
We will not issue fractional shares of common stock in the Rights Offering. Rights holders will only be entitled to purchase a number of Units representing a whole number of shares of common stock, heldrounded down to the nearest whole number of Units a holder would otherwise be entitled to purchase. Any excess subscription payments received by existing stockholdersthe Subscription Agent will be reduced to %returned as soon as practicable after expiration of the totalRights Offering, without interest or penalty. Similarly, no fractional shares of common stock will be issued in connection with the exercise of a Warrant or Pre-Funded Warrant. If, upon exercise of a Warrant, the holder thereof would be entitled to receive a fractional share of common stock, upon exercise, the holder will only be entitled to receive a whole number of shares of common stock, rounded down to the nearest whole number.
Notice to Brokers and Nominees
If you are a broker, dealer, bank, or other nominee holder that holds shares of our common stock for the account of others on the Record Date, you should notify the beneficial owners of the shares for whom you are the nominee of the Rights Offering as soon as possible to learn their intentions with respect to exercising their Subscription Rights. If a beneficial owner of our common stock so instructs, you should complete the Subscription Rights Statement and submit it to the Subscription Agent with the proper subscription payment by the expiration date. You may exercise the number of Subscription
Rights to which all beneficial owners in the aggregate otherwise would have been entitled had they been direct holders of our common stock on the Record Date, provided that you, as a nominee record holder, make a proper showing to the Subscription Agent by submitting the form entitled "Nominee Holder Certification," which is provided with your Rights Offering materials. If you did not receive this form, you should contact our Subscription Agent to request a copy.
Validity of Subscriptions
We will resolve all questions regarding the validity and form of the exercise of your Subscription Rights, including time of receipt and eligibility to participate in the Rights Offering. Our determination will be outstanding upon consummationfinal and binding. Once made, subscriptions are irrevocable; we will not accept any alternative, conditional, or contingent subscriptions. We reserve the absolute right to reject any subscriptions not properly submitted or the acceptance of this offering,which would be unlawful. You must resolve any irregularities in connection with your subscriptions before the expiration date of the Rights Offering, unless we waive them in our sole discretion. Neither we nor the Subscription Agent is under any duty to notify you or your representative of defects in your subscriptions. A subscription will be considered accepted, subject to our right to withdraw or terminate the Rights Offering, only when the Subscription Agent receives a properly completed and duly executed Subscription Rights Statement and any other required documents and the numberfull subscription payment. Our interpretations of the terms and conditions of the Rights Offering will be final and binding.
Stockholder Rights
You will have no rights as a holder of the shares of our common stock you purchase in the Rights Offering until shares are issued in book-entry form or your account at your broker, dealer, bank, or other nominee is credited with the shares of our common stock purchased in the Rights Offering. Holders of Warrants issued in connection with the Rights Offering will not have rights as holders of our common stock until such Warrants are exercised and the shares of common stock held by investors purchasing inunderlying the Warrants are issued to the holder.
Foreign Shareholders
We will not mail this offeringprospectus or Subscription Rights Statements to shareholders with addresses that are outside the United States or that have an army post office or foreign post office address. The Subscription Agent will be increasedhold these Subscription Rights Statements for their account. To exercise Subscription Rights, our foreign shareholders must notify the Subscription Agent prior 5:00 PM Eastern Time, on July 26, 2016, the third business day prior to shares or %the expiration date, of your exercise of Subscription Rights and provide evidence satisfactory to us, such as a legal opinion from local counsel, that the exercise of such Subscription Rights does not violate the laws of the total numberjurisdiction in which such shareholder resides and payment by a U.S. bank in U.S. dollars before the expiration of the offer. If no notice is received by such time or the evidence presented is not satisfactory to us, the Subscription Rights represented thereby will expire.
No Revocation or Change
Once you submit the Subscription Rights Statement or have instructed your nominee of your subscription request, you are not allowed to revoke or change the exercise or request a refund of monies paid. All exercises of Subscription Rights are irrevocable, even if you learn information about us that you consider to be unfavorable. You should not exercise your Subscription Rights unless you are certain that you wish to purchase shares at the Subscription Price.
U.S. Federal Income Tax Treatment of Rights Distribution
For U.S. federal income tax purposes, we do not believe holders of shares of our common stock or warrants should recognize income or loss upon receipt or exercise of a Subscription Right. See "Material U.S. Federal Income Tax Consequences."
No Recommendation to Rights Holders
Our board of directors is not making a recommendation regarding your exercise of the Subscription Rights. Stockholders who exercise Subscription Rights risk investment loss on money invested. We cannot assure you that the market price of our common stock will reach or exceed the Subscription Price, and even if it does so, that it will not decline during or after the Rights Offering. We also cannot assure you that you will be able to sell shares of our common stock or Warrants purchased in the Rights Offering at a price equal to or greater than the Subscription Price. You should make your investment decision based on your assessment of our business and financial condition, our prospects for the future and the terms of this Rights Offering. Please see "Risk Factors" for a discussion of some of the risks involved in investing in our common stock.
Fees and Expenses
We will pay all fees charged by the Subscription Agent and by the dealer-manager. You are responsible for paying any other commissions, fees, taxes or other expenses incurred in connection with the exercise of your Subscription Rights.
Listing
The Subscription Rights may not be sold, transferred, assigned or given away to anyone, and will not be listed for trading on any stock exchange or market. We have applied to have the Warrants listed for trading on NASDAQ under the symbol "ONTXW," however, there is no assurance that a sufficient number of Subscription Rights will be exercised so that the Warrants will meet the minimum listing criteria to be outstanding upon consummation of this offering.
As of March 31, 2013, there were 3,722,188accepted for listing on NASDAQ. The shares of our common stock, issuable uponincluding the shares to be issued in the Rights Offering and the shares underlying the Warrants and any Pre-Funded Warrants to be issued in the Rights Offering, are traded on NASDAQ under the symbol "ONTX." The Pre-Funded Warrants will not be listed for trading on any stock exchange or market.
Important
Do not send Subscription Rights Statements directly to us. You are responsible for choosing the payment and delivery method for your Subscription Rights Statement and you bear the risks associated with such delivery. If you choose to deliver your Subscription Rights Statement and payment by mail, we recommend that you use registered mail, properly insured, with return receipt requested. We also recommend that you allow a sufficient number of days to ensure delivery to the Subscription Agent prior to the expiration time.
Distribution Arrangements
Maxim Group LLC is the dealer-manager for the Rights Offering. The dealer-manager will provide marketing assistance and advice to us in connection with the Rights Offering and will use its best efforts to solicit the exercise of options underSubscription Rights and participation in the 2007 Equity Compensation Plan, with a weighted average exercise price of $6.41 per share, and an aggregate of 385,643 shares of common stock were reserved for future issuance under our 2007 Equity Compensation Plan. As of March 31, 2013, there were 6,128 shares of common stock issuable upon exercise of warrants, with an exercise price of $9.79 per share. We may choose to raise additional capital through the sale of equityOver-Subscription Privilege. The dealer-manager is not underwriting or convertible debt securities due to market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. To the extent thatplacing any of these optionsthe Subscription Rights or warrants are exercised, new options are issued under our 2007 Equity Compensation Plan or we issue additionalthe Units, shares of common stock or other equity securitiesWarrants to be issued in the future, there willRights Offering, and does not make any recommendation with respect to such Subscription Rights (including with respect to the exercise or expiration of such Subscription Rights), Units, shares of common stock or Warrants. We have agreed to pay the dealer-manager certain fees and to reimburse the dealer-manager for certain out-of-pocket expenses incurred in connection with this offering. See "Plan of Distribution" for a discussion of the fees and expenses to be further dilutionpaid to investors purchasingthe dealer-manager in connection with this offering.Rights Offering.
SELECTED CONSOLIDATED FINANCIAL DATAMATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES
The following selected consolidated financial data should be read together with our consolidated financial statementsdiscussion is a summary of material U.S. federal income tax consequences relating to the receipt and accompanying notes and "Management's Discussion and Analysis of Financial Condition and Results of Operations" appearing elsewhere in this prospectus.
We have derived the following statement of operations data for the years ended December 31, 2011 and 2012 and balance sheet data as of December 31, 2012 from our audited consolidated financial statements included elsewhere in this prospectus. We have derived the following statement of operations data for the three months ended March 31, 2012 and 2013 and balance sheet data as of March 31, 2013 from our unaudited condensed consolidated financial statements included elsewhere in this prospectus. Our historical results are not necessarily indicativeexercise (or expiration) of the resultsSubscription Rights acquired through the Rights Offering and the ownership and disposition of shares of our common stock and Warrants received upon exercise of the Subscription Rights, Pre-Funded Warrants or Warrants.
This summary deals only with Subscription Rights acquired through the Rights Offering, shares of our common stock, Pre-Funded Warrants and Warrants acquired upon exercise of Subscription Rights and shares of our common stock acquired upon exercise of the Pre-Funded Warrants or Warrants, in each case, that are held as capital assets by a beneficial owner. This discussion does not address all aspects of U.S. federal income taxation that may be expectedrelevant to such a beneficial owner in light of their personal circumstances, including the future, and our interim period results are not necessarily indicative of results to be expected for a full year or any other interim period.
| | Year Ended December 31, | Three Months Ended March 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | 2012 | 2013 | |||||||||
Consolidated Statement of Operations Data: | |||||||||||||
Revenue | $ | 1,487,000 | $ | 46,190,000 | $ | 198,000 | $ | 1,116,000 | |||||
Operating expenses: | |||||||||||||
General and administrative | 6,436,000 | 15,707,000 | 2,460,000 | 3,346,000 | |||||||||
Research and development | 22,624,000 | 52,762,000 | 8,448,000 | 12,756,000 | |||||||||
Total operating expenses | 29,060,000 | 68,469,000 | 10,908,000 | 16,102,000 | |||||||||
Loss from operations | (27,573,000 | ) | (22,279,000 | ) | (10,710,000 | ) | (14,986,000 | ) | |||||
Change in fair value of warrant liability | 1,287,000 | 367,000 | (609,000 | ) | 14,000 | ||||||||
Interest expense | (19,000 | ) | (8,608,000 | ) | (21,000 | ) | — | ||||||
Other income, net | 11,000 | 608,000 | 541,000 | 127,000 | |||||||||
Net loss before income taxes expense | (26,294,000 | ) | (29,912,000 | ) | (10,799,000 | ) | (14,845,000 | ) | |||||
Income taxes | — | — | — | — | |||||||||
Net loss | (26,294,000 | ) | (29,912,000 | ) | (10,799,000 | ) | (14,845,000 | ) | |||||
Accretion of preferred stock | (4,020,000 | ) | (3,953,000 | ) | (1,231,000 | ) | (1,019,000 | ) | |||||
Net loss applicable to common stockholders | $ | (30,314,000 | ) | $ | (33,865,000 | ) | $ | (12,030,000 | ) | $ | (15,864,000 | ) | |
Per share information: | |||||||||||||
Net loss per share of common stock, basic and diluted(1) | $ | (10.64 | ) | $ | (11.51 | ) | $ | (4.15 | ) | $ | (4.56 | ) | |
Basic and diluted weighted average shares outstanding(1) | 2,849,159 | 2,941,782 | 2,897,347 | 3,475,673 | |||||||||
Pro forma net loss per share of common stock, basic and diluted(1) | $ | (2.01 | ) | $ | (0.77 | ) | |||||||
Basic and diluted pro forma weighted average shares outstanding(1) | 16,887,329 | 20,589,154 | |||||||||||
| | As of December 31, 2012 | As of March 31, 2013 | |||||
|---|---|---|---|---|---|---|---|
Consolidated Balance Sheet Data: | |||||||
Cash and cash equivalents | $ | 81,527,000 | $ | 67,307,000 | |||
Total assets | 83,852,000 | 70,759,000 | |||||
Total liabilities | 40,843,000 | 42,544,000 | |||||
Accumulated deficit | (168,360,000 | ) | (183,205,000 | ) | |||
Total stockholders' deficit | (158,306,000 | ) | (174,119,000 | ) | |||
MANAGEMENT'S DISCUSSION AND ANALYSIS OFFINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with our consolidated financial statements and related notes appearing in this prospectus. Some of the information contained in this discussion and analysis or set forth elsewhere in this prospectus, including information with respect to our plans and strategy for our business and related financing, includes forward looking statements that involve risks and uncertainties. As a result of many factors, including those factors set forth in the "Risk Factors" section of this prospectus, our actual results could differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are a clinical-stage biopharmaceutical company focused on discovering and developing novel small molecule drug candidates to treat cancer. Using our proprietary chemistry platform, we have created an extensive library of targeted anti-cancer agents designed to work against specific cellular pathways that promote cancer. We believe that the drug candidates in our pipeline have the potential to be efficacious in a wide variety of cancers without causing harm to normal cells. We have three clinical-stage product candidates and six preclinical programs.
Rigosertib, our most advanced product candidate, is being tested in a number of ongoing Phase 2 and Phase 3 clinical trials. We are conducting a pivotal Phase 3 trial of rigosertib under an SPA from the FDA for higher risk myelodysplastic syndromes, or MDS. We expect to report top-line overall survival results from this trial in the fourth quarter of 2013 or the first quarter of 2014. We are also evaluating rigosertib in a Phase 3 trial for metastatic pancreatic cancer, in two Phase 2 trials for transfusion-dependant lower risk MDS, and in a Phase 2 trial for head and neck cancers. Baxter has commercialization rights for rigosertib in Europe and SymBio has commercialization rights in Japan and Korea. We have retained development and commercialization rights to rigosertib in the rest of the world, including in the United States.
Our second clinical-stage product candidate, ON 013105, is in a Phase 1 trial in patients with relapsed or refractory lymphoma, including an aggressive form of non-Hodgkin's lymphoma known as mantle cell lymphoma, or MCL, and acute lymphoid leukemia, or ALL. We have suspended enrollment in this Phase 1 trial; however, we plan to restart its enrollment in the fourth quarter of 2013.
Our third clinical-stage product candidate, recilisib, is being developed in collaboration with the U.S. Department of Defense for acute radiation syndromes. We have conducted animal studies and clinical trials of recilisib under the FDA's Animal Efficacy Rule, which permits marketing approval for new medical countermeasures for which human efficacy studies are not feasible or ethical, by relying on evidence from animal studies in appropriate animal models to support efficacy in humans. We have completed four Phase 1 trials to evaluate the safety and pharmacokinetics of recilisib in healthy human adult subjects using both subcutaneous and oral formulations.
In addition to our three clinical-stage product candidates, we are advancing six preclinical programs that target kinases, cellular metabolism or division. We intend to explore additional collaborations to further the development of these product candidates as we focus internally on our more advanced programs.
We were incorporated in Delaware in December 1998 and commenced operations in January 1999. Our operations to date have included our organization and staffing, business planning, raising capital, in-licensing technology from research institutions, identifying potential product candidates, developing product candidates and building strategic alliances, as well as undertaking preclinical studies and clinical trials of our product candidates.
Since commencing operations we have dedicated a significant portion of our resources to our development efforts for our clinical-stage product candidates, particularly rigosertib. We incurred research and development expenses of $22.6 million and $52.8 million during the years ended December 31, 2011 and 2012, respectively, and $12.8 million during the three months ended March 31, 2013. We anticipate that a significant portion of our operating expenses will continue to be related to research and development as we continue to advance our preclinical programs and our clinical-stage product candidates. We have funded our operations primarily through the sale of preferred stock amounting to $144.7 million, including $50.0 million that Baxter invested in our preferred stock in 2012, as well as proceeds from the issuance of convertible debt and a stockholder loan amounting to $26.8 million in the aggregate, all of which was later converted intoother pass-through entities, persons holding Subscription Rights, shares of our preferredcommon stock, and upfront paymentsparticipating warrants, Pre-Funded Warrants or Warrants as part of $7.5 million from SymBio and $50.0 million from Baxtera hedging, integrated, conversion or constructive sale transaction or a straddle, financial institutions, brokers, dealers in securities or currencies, traders that elect to mark-to-market their securities, persons that acquired Subscription Rights, shares of our common stock, participating warrants, Pre-Funded Warrants or Warrants in connection with our collaboration agreements. We have also received an aggregate of $8.0 million from LLS under a funding agreement. Under our collaboration agreements with Baxter and SymBio, we are also eligible to receive an aggregate of up to $545.5 million upon the achievement of specified development and regulatory milestones and up to $280.0 million upon the achievement of specified commercialization milestones, as well as tiered royalties, at percentage rates ranging from the low-teens to low-twenties, on any future net sales of products resulting from these collaborations. As of December 31, 2012 and March 31, 2013, we had $81.5 million and $67.3 million in cash and cash equivalents, respectively.
Our net losses were $26.3 million and $29.9 million for the years ended December 31, 2011 and 2012, respectively, and $14.8 million for the three months ended March 31, 2013. We recognized revenues of $1.5 million and $46.2 million for the years ended December 31, 2011 and 2012, respectively, and $1.1 million for the three months ended March 31, 2013. As of March 31, 2013, we had an accumulated deficit of $183.2 million. We expect to incur significant expenses and operating losses for the foreseeable future as we continue the development and clinical trials of, and seek regulatory approval for, our product candidates, even as milestones under our license and collaboration agreements may be met. If we obtain regulatory approval for any of our product candidates, we expect to incur significant commercialization expenses. We do not currently have an organization for the sales, marketing and distribution of pharmaceutical products. We may rely on licensing and co-promotion agreements with strategic or collaborative partners for the commercialization of our products in the United States and other territories. If we choose to build a commercial infrastructure to support marketing in the United States for any of our product candidates that achieve regulatory approval, such commercial infrastructure could be expected to include a targeted, oncology sales force supported by sales management, internal sales support, an internal marketing group and distribution support. To develop the appropriate commercial infrastructure internally, we would have to invest financial and management resources, some of which would have to be deployed prior to having any certainty about marketing approval.
Furthermore, following consummation of this offering, we expect to incur additional costs associated with operating as a public company. Accordingly, we will seek to fund our operations primarily through public or private equity or debt financingsemployment or other sources. Other additional financing may not be available to us on acceptable terms, or at all. Our failure to raise capital as and when needed couldperformance of services, U.S. Holders (as defined below) that have a material adverse effect on our financial conditionfunctional currency other than the U.S. dollar, U.S. expatriates, and our ability to pursue our business strategy.
Collaboration Agreements
Baxter Healthcare SA
In September 2012, we entered into a development and license agreement with Baxter, granting Baxter an exclusive, royalty-bearing license for the research, development, commercialization and manufacture (in specified instances)certain former citizens or residents of rigosertib in all therapeutic indications in Europe. Under the Baxter agreement, we are obligated to use commercially reasonable efforts to, in accordance with a development plan agreed upon by the parties, direct, coordinate and manage the development of
rigosertib for MDS and pancreatic cancer. Under the agreement, if after a specified development event we elect not to move forward with the development of rigosertib for pancreatic cancer, Baxter may, at its own expense, develop rigosertib for pancreatic cancer on its own for the purposes of obtaining marketing approval. In addition, there is a specified mechanism set forth in the agreement to expand the scope of the collaboration for additional indications. Our agreement with Baxter is guided by a joint steering committee. If the joint steering committee is not able to make a decision by consensus, then any dispute would be resolved by specified executive officers of both parties.
Under the terms of the agreement, Baxter made an upfront payment of $50.0 million. We are eligible to receive pre-commercial milestone payments of up to an aggregate of $512.5 million if specified development and regulatory milestones are achieved. The potential pre-commercial development milestone payments to us include the following:
We may also receive up to $337.5 million in milestone payments for regulatory approvals of the three rigosertib indications specified in the arrangement with Baxter, each of which may be up to and in excess of $100.0 million. We are also potentially eligible to receive an additional $20.0 million pre-commercial milestone payment related to the timing of regulatory approval of rigosertib IV in higher risk MDS patients in Europe. In addition to these pre-commercial milestones, we are eligible to receive up to an aggregate of $250.0 million in milestone payments based on Baxter's achievement of pre-specified threshold levels of annual net sales of rigosertib. We are also entitled to receive royalties at percentage rates ranging from the low-teens to the low-twenties on net sales of rigosertib by Baxter in the licensed territory. In July 2012, Baxter also purchased $50.0 million of our Series J convertible preferred stock.
SymBio Pharmaceuticals Limited
In July 2011, we entered into a license agreement with SymBio, as subsequently amended, granting SymBio an exclusive, royalty-bearing license for the development and commercialization of rigosertib in Japan and Korea. Under the SymBio license agreement, SymBio is obligated to use commercially reasonable efforts to develop and obtain market approval for rigosertib inside the licensed territory and we have similar obligations outside of the licensed territory. We have also entered into an agreement with SymBio providing for us to supply them with development-stage product. Under the SymBio license agreement, we also agreed to supply commercial product to SymBio under specified terms that will be included in a commercial supply agreement to be negotiated prior to first commercial sale of rigosertib. We have also granted SymBio a right of first negotiation to license or obtain the rights to develop and commercialize compounds having a chemical structure similar to rigosertib in the licensed territory.
Under the terms of the SymBio license agreement, we received an upfront payment of $7.5 million. We are eligible to receive milestone payments of up to an aggregate of $33.0 million from SymBio upon the achievement of specified development and regulatory milestones for specified indications. Of the development milestones, $3.0 million is due after enrollment of the first patient in the event a decision is made, after our interim analysis, to start a Phase 3 clinical trial of rigosertib IV in combination with gemcitabine for pancreatic cancer patients in the United States. Of the regulatory milestones, $5.0 million is due upon receipt of marketing approval in the United States for rigosertib IV in higher risk MDS patients, $3.0 million is due upon receipt of marketing approval in Japan for
rigosertib IV in higher risk MDS patients, $5.0 million is due upon receipt of marketing approval in the United States for rigosertib Oral in lower risk MDS patients, $5.0 million is due upon receipt of marketing approval in Japan for rigosertib Oral in lower risk MDS patients, $5.0 million is due upon receipt of marketing approval in the United States for rigosertib IV in combination with gemcitabine in pancreatic cancer patients, and $3.0 million is due upon receipt of marketing approval in Japan for rigosertib IV in combination with gemcitabine in pancreatic cancer patients. Furthermore, upon receipt of marketing approval in the United States and Japan for an additional specified indication of rigosertib, which we are currently not pursuing, an aggregate of $4.0 million would be due. In addition to these pre-commercial milestones, we are eligible to receive tiered milestone payments of up to an aggregate of $30.0 million based upon annual net sales of rigosertib by SymBio in the licensed territory. Further, under the terms of the SymBio license agreement, SymBio is obligated to make royalty payments to us at percentage rates ranging from the mid-teens to 20% based on net sales of rigosertib by SymBio in the licensed territory.
The Leukemia and Lymphoma Society
In May 2010, we entered into a funding agreement with LLS to fund the development of rigosertib. Under the LLS funding agreement, we are obligated to use the funding received exclusively for the payment or reimbursement of the costs and expenses for clinical development activities for rigosertib. Under this agreement, we retain ownership and control of all intellectual property pertaining to works of authorship, inventions, know-how, information, data and proprietary material.
Under the LLS funding agreement, as amended, we received funding of $8.0 million from LLS through 2012. We have not received any funding in 2013 and we terminated the funding agreement effective as of March 2013. We are required to make specified payments to LLS, including payments payable upon execution of the first out-license; first approval for marketing by a regulatory body; completion of the first commercial sale of rigosertib; and achieving specified annual net sales levels of rigosertib. The extent of these payments and our obligations will depend on whether we out-license rights to develop or commercialize rigosertib to a third party, we commercialize rigosertib on our own or we combine with or are sold to another company. In addition, we will pay to LLS a single-digit percentage royalty of our net sales of rigosertib, if any. The sum of our payments to LLS is capped at three times the total funding received from LLS, or $24.0 million.
Financial Overview
Revenue
To date, we have derived revenue principally from activities pursuant to our collaboration arrangements with Baxter and SymBio as well as from grants and research agreements. The following table sets forth a summary of revenue recognized from our collaboration agreements and research agreements for the years ended December 31, 2011 and 2012 and for the three months ended March 31, 2012 and 2013:
| | Year Ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | |||||
Baxter license and collaboration revenue | $ | — | $ | 45,490,000 | |||
SymBio license and collaboration revenue | 227,000 | 503,000 | |||||
Research funding | 1,260,000 | 197,000 | |||||
| $ | 1,487,000 | $ | 46,190,000 | ||||
| | Three Months Ended March 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2012 | 2013 | |||||
Baxter license and collaboration revenue | $ | — | $ | 978,000 | |||
SymBio license and collaboration revenue | 113,000 | 138,000 | |||||
Research funding | 85,000 | — | |||||
| $ | 198,000 | $ | 1,116,000 | ||||
We have not generated any revenue from commercial product sales. In the future, if any of our product candidates currently under development are approved for commercial sale in the United States and Canada, we may generate revenue from product sales, or alternatively, we may choose to select a collaborator to commercialize our product candidates in these markets.
The Baxter collaboration agreement is considered to be a multiple-element arrangement for accounting purposes. We determined that there are two deliverables under the Baxter agreement; specifically, the license to rigosertib for Europe and the related research and development services that we are obligated to provide. We concluded that $42.4 million of the fixed and determinable $50.0 million upfront payment was associated with the license and $7.6 million was associated with the research and development services. We recognized the entire $42.4 million associated with the upfront license as revenue during the third quarter of 2012 upon the execution of the Baxter agreement, and we are recognizing the research and development services revenue of $7.6 million on the proportional performance method over the period of commitment and development, which we estimate to be through March 31, 2014, the period of our non-contingent obligations to perform research and development services sufficient to advance rigosertib. For the year ended December 31, 2012 and for the three months ended March 31, 2013, we recognized $3.1 million and $1.0 million, respectively, of research and development services revenue under the Baxter agreement.
The SymBio collaboration agreement is also considered to be a multiple-element arrangement for accounting purposes. We determined that there were three deliverables under the SymBio collaboration agreement; specifically, the license to rigosertib for Japan and Korea, our obligation to perform research and development services necessary for SymBio to seek approval in its territory and our obligation to participate on a joint steering committee. We concluded that these deliverables should be accounted for as a single unit of accounting. We determined that the $7.5 million upfront payment received in 2011 should be deferred and recognized as revenue on a straight-line basis through December 2027, reflecting our estimate of when we will complete our obligations under the agreement. For the years ended December 31, 2011 and 2012, we recognized revenues of $227,000 and $455,000, respectively, under the SymBio collaboration agreement. In addition, we recognized revenues of $48,000 for the year ended December 31, 2012 related to the supply agreement with SymBio. For the three months ended March 31, 2013, we recognized revenues of $113,000 under the SymBio collaboration agreement. In addition, we recognized revenues of $25,000 for the three months ended March 31, 2013 related to the supply agreement with SymBio.
The remaining revenue recognized during the years ended December 31, 2011 and 2012 of $1,260,000 and $197,000, respectively, pertained to research and development services provided under research grants.
Pursuant to our funding agreement with LLS, during the year ended December 31, 2012, we paid $1.0 million to LLS, which we recorded as research and development expenses. This payment reduced the maximum milestone and royalty payment obligation under this agreement to $23.0 million at December 31, 2012.
In addition, some of our obligations under the LLS funding agreement will remain in effect until the completion of specified milestones and payments to LLS. Assuming the successful outcome of the development activities covered by the LLS funding agreement and our receipt of necessary regulatory
approvals, we will be required to take commercially reasonable steps through March 2018 to advance the development of rigosertib in clinical trials and to bring rigosertib to practical application for MDS in a major market country, provided that we believe the product is safe and effective. We believe that we can satisfy our obligation by out-licensing rigosertib to, or partnering rigosertib with, a third party. We are required to report to LLS on our efforts and results with respect to continuing development of rigosertib. Our failure to perform these diligence obligations, even if we successfully achieve the specified development milestones, would require us to pay back to LLS the total amount of the funding we received from them, unless an exception applies. If LLS were to claim that such failure occurred and we disagreed with such claim, the dispute would be settled through binding arbitration.
As a result of the potential obligation to pay back to LLS the total amount of funding received under this arrangement, the $8.0 million of milestone payments we received through March 31, 2013 has been recorded as deferred revenue.
Preclinical Collaboration
We recently entered into a joint venture with GVK Biosciences Private Limited, or GVK, a CRO based in India, to collaborate on the development of two of our preclinical programs. GVK will initially make a capital contribution of $500,000 in exchange for a 10% interest in the joint venture and we will contribute a sub-license to the intellectual property related to the two programs in exchange for a 90% interest. GVK will be required to make additional capital contributions over time, subject to specified conditions, and its interest in the joint venture will increase to as much as 50%. At specified times, we will be entitled to buy back from GVK the rights to either of these two programs.
We currently anticipate that the joint venture will be consolidated in our financial statements, which means that we will include its assets and liabilities in our balance sheets and its expenses in our statements of operations. We do not expect the consolidation of the joint venture will initially have a material affect on our consolidated financial position or results of operations.
Operating Expenses
The following table summarizes our operating expenses for the years ended December 31, 2011 and 2012 and for the three months ended March 31, 2012 and 2013:
| | Year Ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | |||||
General and administrative | $ | 6,436,000 | $ | 15,707,000 | |||
Research and development | 22,624,000 | 52,762,000 | |||||
Total operating expenses | $ | 29,060,000 | $ | 68,469,000 | |||
| | Three Months Ended March 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2012 | 2013 | |||||
General and administrative | $ | 2,460,000 | $ | 3,346,000 | |||
Research and development | 8,448,000 | 12,756,000 | |||||
Total operating expenses | $ | 10,908,000 | $ | 16,102,000 | |||
General and Administrative Expenses
General and administrative expenses consist principally of salaries and related costs for executive and other administrative personnel, including stock-based compensation and travel expenses. Other general and administrative expenses include facility-related costs, communication expenses and professional fees for legal, patent review, consulting and accounting services.
For the years ended December 31, 2011 and 2012 and for the three months ended March 31, 2012 and 2013, our general and administrative expenses totaled approximately $6.4 million, $15.7 million, $2.5 million and $3.3 million, respectively. We anticipate that our general and administrative expenses will increase in the future with the continued research and development and potential commercialization of our product candidates and as we operate as a public company. These increases will likely include increased costs for insurance, costs related to the hiring of additional personnel and payments to outside consultants, investor relations, lawyers and accountants, among other expenses. Additionally, if and when we believe a regulatory approval of a product candidate appears likely, we anticipate an increase in payroll and expense as a result of our preparation for commercial operations, especially as it relates to the sales and marketing of our product candidates.
Research and Development Expenses
Our research and development expenses consist primarily of costs incurred for the development of our product candidates, which include:
Research and development costs are expensed as incurred. License fees and milestone payments we make related to in-licensed products and technology are expensed if it is determined that they have no alternative future use. We record costs for some development activities, such as clinical trials, based on an evaluation of the progress to completion of specific tasks using data such as patient enrollment, clinical site activations or information provided to us by our vendors.
Research and development activities are central to our business model. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We plan to increase our research and development expenses for the foreseeable future.
To date, our research and development expenses have related primarily to the development of rigosertib. In the years ended December 31, 2011 and 2012 and for the three months ended March 31, 2012 and 2013, we recorded approximately $22.6 million, $52.8 million, $8.4 million and $12.8 million, respectively, of research and development expenses. We do not currently utilize a formal time allocation system to capture expenses on a project-by-project basis because we are organized and record expense by functional department and our employees may allocate time to more than one development project. Accordingly, we do not allocate expenses to individual projects or product candidates, although we do allocate some portion of our research and development expenses by functional area and by compound, as shown below.
The following table summarizes our research and development expenses by functional area for the years ended December 31, 2011 and 2012:
| | Year Ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | |||||
Clinical development | $ | 10,926,000 | $ | 19,285,000 | |||
Personnel related | 4,020,000 | 4,876,000 | |||||
Consulting fees | 1,949,000 | 3,422,000 | |||||
Milestone payments | 1,875,000 | 13,500,000 | |||||
Manufacturing and formulation | 1,584,000 | 1,752,000 | |||||
Institutional research | 1,234,000 | 1,417,000 | |||||
Pre-clinical research | 667,000 | 1,672,000 | |||||
Laboratory costs | 366,000 | 193,000 | |||||
Stock-based compensation | 3,000 | 6,645,000 | |||||
| $ | 22,624,000 | $ | 52,762,000 | ||||
The following table summarizes our research and development expenses by functional area for the three months ended March 31, 2012 and 2013:
| | Three Months Ended March 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2012 | 2013 | |||||
Clinical development | $ | 4,194,000 | $ | 6,831,000 | |||
Personnel related | 1,193,000 | 1,882,000 | |||||
Consulting fees | 809,000 | 1,496,000 | |||||
Manufacturing and formulation | 624,000 | 152,000 | |||||
Institutional research | 255,000 | 359,000 | |||||
Pre-clinical research | 145,000 | 835,000 | |||||
Laboratory costs | 58,000 | 43,000 | |||||
Stock-based compensation | 1,170,000 | 1,158,000 | |||||
| $ | 8,448,000 | $ | 12,756,000 | ||||
The following table summarizes our research and development expenses by compound for the years ended December 31, 2011 and 2012:
| | Year Ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | |||||
Rigosertib | $ | 15,915,000 | $ | 38,683,000 | |||
Recilisib | 876,000 | 286,000 | |||||
ON 013105 | 202,000 | 274,000 | |||||
Other research and development | 1,608,000 | 1,998,000 | |||||
Personnel related and stock-based compensation | 4,023,000 | 11,521,000 | |||||
| $ | 22,624,000 | $ | 52,762,000 | ||||
The following table summarizes our research and development expenses by compound for the three months ended March 31, 2012 and 2013:
| | Three Months Ended March 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2012 | 2013 | |||||
Rigosertib | $ | 5,435,000 | $ | 9,183,000 | |||
Recilisib | 221,000 | 20,000 | |||||
ON 013105 | 68,000 | 58,000 | |||||
Other research and development | 361,000 | 455,000 | |||||
Personnel related and stock-based compensation | 2,363,000 | 3,040,000 | |||||
| $ | 8,448,000 | $ | 12,756,000 | ||||
It is difficult to determine with certainty the duration and completion costs of our current or future preclinical programs and clinical trials of our product candidates, or if, when or to what extent we will generate revenues from the commercialization and sale of any of our product candidates that obtain regulatory approval. We may never succeed in achieving regulatory approval for any of our product candidates. The duration, costs and timing of clinical trials and development of our product candidates will depend on a variety of factors, including the uncertainties of future clinical and preclinical studies, uncertainties in clinical trial enrollment rate and significant and changing government regulation. In addition, the probability of success for each product candidate will depend on numerous factors, including competition, manufacturing capability and commercial viability. We will determine which programs to pursue and how much to fund each program in response to the scientific and clinical success of each product candidate, as well as an assessment of each product candidate's commercial potential.
Change in Fair Value of Warrant Liability
We have issued warrants for the purchase of our Series G convertible preferred stock that we believe are financial instruments that may require a transfer of assets becausediscussion does not describe any tax consequences arising out of the redemption featurestax laws of the underlying preferred stock. Therefore, we have classified these warrantsany state, local or foreign jurisdiction, or any U.S. federal tax considerations other than income taxation (such as liabilities that we re-measure to fair value at each balance sheet date and we record the changes in the fair value of the warrant liability as either incomeestate, generation skipping or expense. Upon consummation of this offering, the underlying preferred stock will be converted to common stock, and the fair value of the warrant liability at that time will be reclassified to additional paid-in capital.
Interest Expense and Other Income, Net
Other income, net consists principally of interest income earned on cash and cash equivalent balances and income earned on our sale of New Jersey state net operating losses in 2012.gift taxation).
Interest expense for the years ended December 31, 2011 and 2012 consisted of cash paid and non-cash interest expense related to our prior loan from a stockholder and convertible promissory notes payable, as well as a charge for the unamortized contingent beneficial conversion feature upon conversion of those promissory notes into shares of Series I convertible preferred stock.
Accretion of Preferred Stock
We account for the redemption of premium and issuance costs on our preferred stock using the interest method, accreting such amounts to preferred stock from the date of issuance to the earliest date of redemption.
Critical Accounting Policies and Significant Judgments and Estimates
This management'sThe discussion and analysis of our financial condition and results of operationsbelow is based on our consolidated financial statements, which have been prepared in accordance with GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses and the disclosure of contingent assets and liabilities in our consolidated financial statements. On an ongoing basis, we evaluate our estimates and judgments, including those related to accrued expenses, revenue recognition, deferred revenue and stock-based compensation. We base our estimates on historical experience, known trends and events and various other factors that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are described in the notes to our consolidated financial statements appearing elsewhere in this prospectus, we believe the following accounting policies to be most critical to the judgments and estimates used in the preparation of our consolidated financial statements.
Revenue Recognition
We generate revenue primarily through collaborative research and license agreements. The terms of these agreements contain multiple deliverables, which may include licenses, research and development activities, participation in joint steering committees and product supply. The terms of these agreements may include nonrefundable upfront license fees, payments for research and development activities, payments based upon the achievementprovisions of specified milestones, royalty payments based on product sales derived from the collaboration, and payments for supplying product. In all instances, we recognize revenue only when the price is fixed or determinable, persuasive evidence of an arrangement exists, delivery has occurred or the services have been rendered, collectability of the resulting receivable is reasonably assured and we have fulfilled our performance obligations under the contract.
Effective January 1, 2011, we adopted the Financial Accounting Standards Board, or FASB, Accounting Standards Update, or ASU, No. 2009-13,Multiple-Deliverable Revenue Arrangements, or ASU 2009-13. This guidance, which applies to multiple-element arrangements entered into or materially modified on or after January 1, 2011, amends the criteria for separating and allocating consideration in a multiple-element arrangement by modifying the fair value requirements for revenue recognition and eliminating the use of the residual value method. The selling prices of deliverables under an arrangement may be derived using third-party evidence, or TPE, or a best estimate of selling price, or BESP, if vendor-specific objective evidence of fair value, or VSOE, is not available. The objective of BESP is to determine the price at which we would transact a sale if the element within the license agreement was sold on a standalone basis. Establishing BESP involves management's judgment and takes into account multiple factors, including market conditions and company-specific factors, such as those factors contemplated in negotiating the agreements as well as internally developed models that include assumptions related to market opportunity, discounted cash flows, estimated development costs, probability of success, and the time needed to commercialize a product candidate pursuant to the license. In validating the BESP, management considers whether changes in key assumptions used to determine the BESP will have a significant effect on the allocation of the arrangement consideration between the multiple deliverables. We may use third-party valuation specialists to assist us in determining BESP. Deliverables under the arrangement are separate units of accounting if (i) the delivered item has value to the customer on a standalone basis and (ii) if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item is considered probable and substantially within our control. The arrangement consideration that is fixed
or determinable at the inception of the arrangement is allocated to the separate units of accounting based on their relative selling prices. The appropriate revenue recognition model is applied to each element and revenue is accordingly recognized as each element is delivered. Management exercises significant judgment in determining whether a deliverable is a separate unit of accounting.
In determining the separate units of accounting, we evaluate whether the license has standalone value to the collaborator based on consideration of the relevant facts and circumstances for each arrangement. Factors considered in this determination include the research and development capabilities of the collaborator and the availability of relevant research expertise in the marketplace. In addition, we consider whether or not (i) the collaborator can use the license for its intended purpose without the receipt of the remaining deliverables, (ii) the value of the license is dependent on the undelivered items and (iii) the collaborator or other vendors can provide the undelivered items.
Under a collaborative research and license agreement, a steering committee is typically responsible for overseeing the general working relationships, determining the protocols to be followed in the research and development performed, and evaluating the results from the continued development of the product. We evaluate whether our participation in joint steering committees is a substantive obligation or whether the services are considered inconsequential or perfunctory. The factors we consider in determining if our participation in a joint steering committee is a substantive obligation include: (i) which party negotiated or requested the steering committee, (ii) how frequently the steering committee meets, (iii) whether or not there are any penalties or other recourse if we do not attend the steering committee meetings, (iv) which party has decision making authority on the steering committee and (v) whether or not the collaborator has the requisite experience and expertise associated with the research and development of the licensed intellectual property.
For all periods presented, whenever we determine that an element is delivered over a period of time, we recognize revenue using either a proportional performance model or a straight-line model over the period of performance, which is typically the research and development term. We typically use progress achieved under our various CRO contracts as the measure of performance. At each reporting period, we reassess our cumulative measure of performance and make appropriate adjustments, if necessary. We recognize revenue using the proportional performance model whenever we can make reasonably reliable estimates of the level of effort required to complete our performance obligations under an arrangement. We recognize revenue under the proportional performance model at each reporting period by multiplying the total expected payments under the contract, excluding royalties and payments contingent upon achievement of milestones, by the ratio of the level of effort incurred to date to the estimated total level of effort required to complete the performance obligations under the arrangement. Revenue is limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the proportional performance model as of each reporting period. Alternatively, if we cannot make reasonably reliable estimates of the level of effort required to complete our performance obligations under an arrangement, then we recognize revenue under the arrangement on a straight-line basis over the period expected to complete our performance obligations.
Incentive milestone payments may be triggered either by the results of our research efforts or by events external to us, such as regulatory approval to market a product. We recognize consideration that is contingent upon achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved, but only if the consideration earned from the achievement of a milestone meets all the criteria for the milestone to be considered substantive at the inception of the arrangement. For a milestone to be considered substantive, the consideration earned by achieving the milestone must be commensurate with either our performance to achieve the milestone or the enhancement of the value of the item delivered as a result of a specific outcome resulting from our performance to achieve the milestone, relate solely to our past performance and be reasonable relative to all deliverables and payment terms in the collaboration agreement.
For events for which the occurrences are contingent solely upon the passage of time or are the result of performance by a third party, we will recognize the contingent payments as revenue when payments are earned, the amounts are fixed and determinable and collectability is reasonably assured.
We will recognize royalty revenue, if any, as earned in accordance with the contract terms when third-party sales can be reliably measured and collectability is reasonably assured.
We recognized revenue of $45.5 million and $1.0 million during the year ended December 31, 2012 and during the three months ended March 31, 2013, respectively, under our license and collaboration agreement with Baxter. We recognized revenue of $227,000, $503,000 and $138,000 during the years ended December 31, 2011 and 2012 and for the three months ended March 31, 2013, respectively, under our license and collaboration agreement with SymBio. The remaining revenue recognized during the years ended December 31, 2011 and 2012 of $1,260,000 and $197,000, respectively, pertained to research and development services provided under research grants. The Baxter and SymBio agreements are the only agreements that are being accounted for under ASU 2009-13.
Research and Development Expenses
Research and development costs are charged to expense as incurred and include, but are not limited to, license fees related to the acquisition of in-licensed products, employee-related expenses, including salaries, benefits and travel, expenses incurred under agreements with CROs and investigative sites that conduct clinical trials and preclinical studies, the cost of acquiring, developing and manufacturing clinical trial materials, facilities, depreciation and other expenses, which include direct and allocated expenses for rent and maintenance of facilities, insurance and other supplies and costs associated with preclinical activities and regulatory operations.
We record costs for certain development activities, such as clinical trials, based on our evaluation of the progress to completion of specific tasks using data such as patient enrollment, clinical site activations, or information provided to us by our vendors on their actual costs incurred. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected in the consolidated financial statements as prepaid or accrued research and development expense, as the case may be.
Income Taxes
We recorded deferred tax assets of $55.3 million as of December 31, 2012, which have been fully offset by a valuation allowance due to uncertainties surrounding our ability to realize these tax benefits. The deferred tax assets are primarily composed of federal and state tax net operating loss, or NOL, carry forwards and research and development tax credit carry forwards. As of December 31, 2012, we had federal NOL carry forwards of $91.3 million, state NOL carry forwards of $75.1 million and research and development tax credit carry forwards of $13.9 million available to reduce future taxable income, if any. These federal NOL carry forwards will begin to expire at various dates starting in 2019. The state NOL carry forwards will begin to expire at various dates starting in 2016. In general, if we experience a greater than 50 percentage point aggregate change in ownership of specified significant stockholders over a three-year period, utilization of our pre-change NOL carry forwards will be subject to an annual limitation under Section 382 of the U.S. Internal Revenue Code of 1986, as amended, or the Code, the United States Treasury regulations promulgated thereunder, rulings and similar state laws. Such limitations may result in expiration of a portionjudicial decisions, as of the NOL carry forwards before utilizationdate hereof, and such authorities may be substantial.repealed, revoked or modified, perhaps retroactively. We have determinednot sought, and will not seek, any rulings from the Internal Revenue Service, or the IRS, regarding the matters discussed below. There can be no assurance that we have experienced ownership changes in the past and approximately $30.7 millionIRS or a court (if the matter were contested) will not take positions concerning the tax consequences of the receipt of Subscription Rights acquired through the Rights Offering by persons holding shares of our NOL carry forwards are subject to an annual limitation under Section 382common stock or participating warrants, the exercise (or expiration) of the Code. If we experience a Section 382Subscription Rights, the acquisition, ownership change in connection with this offering or as a resultand disposition of future changes in our stock ownership, some of which changes are outside our control, the tax benefits related to the NOL carry forwards may be further limited or lost.
Preferred Stock
We account for the redemption of premium and issuance costs on our preferred stock using the interest method, accreting such amounts to our preferred stock from the date of issuance to the earliest date of redemption.
Preferred Stock Warrants
Our outstanding warrants to purchase shares of preferred stock are classified as liabilities and recorded at fair value, regardless of the timing of the redemption feature or the redemption price or the likelihood of redemption of the underlying preferred stock. The warrants are subject to re-measurement at each balance sheet date and we recognize any change in fair value in our consolidated statements of operations as a change in fair value of warrant liability. Pursuant to the terms of these warrants, upon the conversion of the series of preferred stock underlying the warrants, the warrants automatically become exercisable for shares of common stock based upon the conversion ratio of the underlying series of preferred stock. The consummation of this offering will result in the conversion of all series of our preferred stock into common stock. Upon such conversion of the underlying series of preferred stock, the warrants will be classified as a component of equity and no longer be subject to re-measurement. We will continue to adjust the liability for changes in fair value until the earlier of the exercise or expiration of the warrants or the conversion of the underlying preferred stock. Our consolidated statements of operations for the period in which this offering occurs will be affected by any change in the fair value of the warrants from the end of the prior period through the time of conversion.
Stock-Based Compensation
Our stock option awards have been accounted for as liability awards as we, through our chairman of the board of directors, who is also a significant stockholder, have established a pattern of settling these awards for cash in the past. Accordingly, we have measured stock-based compensation expense at the end of each reporting period based on the intrinsic value of all outstanding vested stock options on each reporting date and recognize expense based on any increases in their intrinsic value since the last measurement date to the extent the stock options have vested. The intrinsic value represents the difference between the current fair value of our common stock and the contractual exercise pricesacquisition, ownership and disposition (or expiration) of the awards.
Stock-based compensation expense totaled $6,000, $13.8 million and $2.5 million for the years ended December 31, 2011 and 2012 and for the three months ended March 31, 2013, respectively. We record stock-based compensation expense as a component of research and development expensesPre-Funded Warrants or general and administrative expenses, depending on the function performed by the optionee. For the years ended December 31, 2011 and 2012 and the three months ended March 31, 2012 and 2013, we allocated stock-based compensation as follows:
| | Year Ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | |||||
General and administrative | $ | 3,000 | $ | 7,199,000 | |||
Research and development | 3,000 | 6,645,000 | |||||
Total | $ | 6,000 | $ | 13,844,000 | |||
| | Three Months Ended March 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2012 | 2013 | |||||
General and administrative | $ | 1,267,000 | $ | 1,306,000 | |||
Research and development | 1,170,000 | 1,158,000 | |||||
Total | $ | 2,437,000 | $ | 2,464,000 | |||
On April 23, 2013, we distributed a notification letter to all holders of stock options under our 2007 Equity Compensation Plan advising them that cash settlement transactions will no longer occur, unless, at the time of a cash settlement transaction, the option holder has held the common stock issuedWarrants acquired upon exercise of options forthe Subscription Rights that are different from those discussed below.
As used herein, a period"U.S. Holder" means a beneficial owner of greater than six months prior to such cash settlement transaction and that any such settlement would be at the fair value of the common stock on the date of such sale. Following this notification, we reclassified options outstanding under our 2007 Equity Compensation Plan from liabilities to stockholders' deficit within our consolidated balance sheets.
Upon issuing the notification, a modification to the liability awards occurred and the awards will be accounted for as equity awards from the date of modification with compensation expensed fixed at fair value at the modification date. As a result, we classified the amount previously recorded as a stock-based compensation liability to additional paid-in capital. In addition, we will recognize the remaining modified date fair value over the remaining service period, generally the vesting period, which we will recognize on a straight-line basis. The fair value of the modified awards will be estimated using the Black-Scholes valuation model. Awards granted to non-employees will also be valued using the Black-Scholes valuation model and will be subject to periodic adjustment until the underlying equity instruments vest.
Fair Value Estimates
We are required to estimate the fair value of the common stock underlying our stock-based awards when performing the fair value calculations using the intrinsic value method at each reporting date. We engaged an independent third-party valuation firm to assist our board of directors in determining the fair value of the common stock underlying our stock-based awards. All options to purchase shares of our common stock, have been granted with an exercise price per share no less than the fair value per shareparticipating warrants, Subscription Rights, shares of our common stock, underlying those options on the datePre-Funded Warrants and Warrants acquired upon exercise of grant, based on the information known to us on the date of grant. Accordingly, under the liability method of accounting, we have not recorded any stock-based compensation expense on the grant dates of our options. However, under the liability method, the liability for all outstanding vested stock-based awards is adjusted through our statement of operations, based on the current estimated fair valueSubscription Rights or shares of our common stock at each reporting date. Asacquired upon exercise of March 31, 2013,Pre-Funded Warrants or Warrants, as the liabilitycase may be, that is for all outstanding vested stock-based awards has been recorded based onU.S. federal income tax purposes: (1) an individual who is a citizen or resident of the fair valueUnited States; (2) a corporation (or other entity treated as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of our common stock on March 31, 2013 as determined by our boardthe United States or any state thereof or the District of directors withColumbia; (3) an estate the assistanceincome of an independent third-party valuation.
Inwhich is subject to U.S. federal income taxation regardless of its source; or (4) a trust (a) the absenceadministration of which is subject to the primary supervision of a public trading market for our common stock, on each grant date, we develop an estimatecourt within the United States and one or more United States persons as described in Section 7701(a)(30) of the fair valueCode have authority to control all substantial decisions of our common stockthe trust or (b) that has a valid election under the Treasury Regulations in ordereffect to determine an exercise price for the option grants based in part on input from the independent third-party valuation firm. We determined the fair value of our common stock using methodologies, approaches and assumptions consistent with the American Institute of Certified Public Accountants, or AICPA, Audit and Accounting Practice Aid Series:Valuation of Privately Held Company Equity Securities Issuedbe treated as Compensation, or the AICPA Practice Guide. In addition, our board of directors considered various objective and subjective factors, along with input from management and the independent third-party valuation firm, to determine the fair value of our common stock, including external market conditions affecting the biotechnology industry, trends within the biotechnology industry, the prices at which we sold shares of our different series of preferred stock, the superior rights and preferences of each series of preferred stock relativea United States person. A "Non-U.S. Holder" is such a beneficial
to our common stock atowner (other than an entity or arrangement that is treated as a partnership for U.S. federal income tax purposes) that is not a U.S. Holder.
If any entity or arrangement that is treated as a partnership for U.S. federal income tax purposes is the timerecord owner, the U.S. federal income tax treatment of each grant, our results of operations and financial position,a partner generally will depend upon the status of our research and development efforts, our stage of development and business strategy, the lack of an active public market for our common and our preferred stock,partner and the likelihood of achieving a liquidity event such as an initial public offering or sale of our company in light of prevailing market conditions.
The per share estimated fair value of common stock in the table below represents the determination by our board of directorsactivities of the fair valuepartnership. Holders that are partnerships (and partners in such partnerships) are urged to consult their own tax advisors.
HOLDERS OF SHARES OF OUR COMMON STOCK AND PARTICIPATING WARRANTS SHOULD CONSULT THEIR OWN TAX ADVISORS REGARDING THE APPLICATION OF THE U.S. FEDERAL INCOME TAX LAWS TO THEIR PARTICULAR SITUATIONS AND THE CONSEQUENCES UNDER FEDERAL ESTATE AND GIFT TAX LAWS, FOREIGN, STATE AND LOCAL LAWS AND TAX TREATIES OF THE RECEIPT, OWNERSHIP AND EXERCISE OF SUBSCRIPTION RIGHTS AND THE ACQUISITION, OWNERSHIP AND DISPOSITION OF SHARES OF OUR COMMON STOCK, PRE-FUNDED WARRANTS AND WARRANTS ACQUIRED UPON EXERCISE OF SUBSCRIPTION RIGHTS AND SHARES OF OUR COMMON STOCK ACQUIRED UPON EXERCISE OF PRE-FUNDED WARRANTS OR WARRANTS.
Tax Treatment of Pre-Funded Warrants
Any person that elects to receive Pre-Funded Warrants in lieu of our common stock asupon the exercise of Subscription Rights should consult their own tax advisor regarding the application of the dateU.S. federal income tax laws to their particular situation.
Tax Consequences to U.S. Holders
Taxation of grant, taking into considerationSubscription Rights
Receipt of Subscription Rights
Although the various objectiveauthorities governing transactions such as this Rights Offering are complex and subjective factors described above,do not speak directly to the consequences of certain aspects of this Rights Offering, including the conclusions, if applicable,inclusion of contemporaneous valuationsthe right to purchase Pre-Funded Warrants and Warrants in the Subscription Rights (rather than the right to purchase only shares of our common stock as discussed below. The following table presentsstock), the grant datesdistribution of Subscription Rights to participating warrant holders and related exercise prices of stock options granted to employees and non-employees from January 1, 2011 through March 20, 2013:
Date of issuance | Number of shares underlying option grants | Exercise price per option | Per share estimated fair value of common stock | Per share grant date intrinsic value of options | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
January 1, 2011 to February 1, 2012 | 554,049 | $ | 4.60 | $ | 4.60 | $ | — | ||||||
March 1, 2012 to July 11, 2012 | 49,270 | 5.65 | 5.65 | — | |||||||||
August 31, 2012 to March 20, 2013 | 1,573,360 | 9.96 | 9.96 | — | |||||||||
In determining the exercise priceseffects of the options set forthOver-Subscription Privilege, we do not believe your receipt of Subscription Rights pursuant to the Rights Offering should be treated as a taxable distribution with respect to your existing shares of common stock or participating warrants for U.S. federal income tax purposes. Pursuant to Section 305(a) of the Code, in general, the receipt by a shareholder or a participating warrant holder of a right to acquire stock or warrants should not be included in the table above granted from January 1, 2011 through March 20, 2013, our boardtaxable income of directors considered the most recent valuationsrecipient. The general rule of non-recognition in Section 305(a) is subject to exceptions in Section 305(b), which include "disproportionate distributions." A disproportionate distribution is a distribution or a series of distributions, including deemed distributions, that has the effect of the receipt of cash or other property by some shareholders and an increase in the proportionate interest of other shareholders in a corporation's assets or earnings and profits. During the last 36 months, we have not made any distributions of cash or non-stock property with respect to: (i) our common stock which were prepared asor (ii) our options or warrants to acquire common stock. Currently we do not intend to make any future distributions of September 30, 2010, February 29, 2012, and July 31, 2012, and based its determination in part on the analyses summarized below. On May 16, 2013, an independent third-party valuation was prepared as of March 31, 2013 to assist our board of directors in determining the exercise price of options to be issued after that date and to calculate the liability for our outstanding vested stock awards as of March 31, 2013.
Stock option grants from January 1, 2011 to February 1, 2012
Our board of directors granted stock options from January 1, 2011 through February 1, 2012,cash or non-stock property with each having an exercise price of $4.60 per share. The exercise price per share was supported by an independent third-party valuation as of September 30, 2010 that was performed in connection with our Series H convertible preferred stock financing. In conducting this valuation, we estimated the value ofrespect to: (i) our common stock usingor (ii) our options or warrants to acquire common stock; however, there is no guarantee that we will not make such distributions in the income approach to estimate our equity value. The income approach involves applying an appropriate risk-adjusted discount rate to projected cash flows based on forecasted revenue and costs. Forfuture.
Our position regarding the September 30, 2010 valuation, we used a risk-adjusted discount rate of 17% to discount our projected cash flows to the valuation date. To corroborate our equity value calculated under the income approach, we used the option-pricing back solve method, or OPM-BS, to estimate the equity value that corresponded to the pricing and termstax-free treatment of the Series H convertible preferred stock financing. The lead investor inSubscription Rights distribution is not binding on the financing was an unrelated investor, andIRS or the price forcourts. If this position is finally determined by the Series H convertible preferred stock of $9.79 per share was determined through negotiations withIRS or a court to be incorrect, whether on the investors. We then allocated the equity value among our preferred stock and common stock using the OPM-BS. For our OPM-BS analysis, we estimated the time to liquidity as three years and assumed an annual volatility rate of 64.0%. Our estimate of volatility was based on historical share price trading data for a group of 13 companies we considered comparable to ours. We applied a discount for lack of marketability of 34.0% to our common stock. The income approach and OPM-BS methodologies resulted in a similar equity value. Based on these factors, we concluded that our common stock had a fair value of $4.60 per share as of September 30, 2010.
We concludedbasis that the valueissuance of our company remained relatively unchanged from September 30, 2010 through February 1, 2012. This was primarily attributable to our continued efforts to obtainthe Subscription Rights is a "disproportionate distribution" or
financing to support our liquidity needs and funding of operating expenses. The specific facts and circumstances considered by our board of directors includedotherwise, the following:
Stock option grants from March 1, 2012 to July 11, 2012
Our board of directors granted stock options from March 1, 2012 through July 11, 2012, with each having an exercise price of $5.65 per share. The exercise price per share was supported by an independent third-party valuation as of February 29, 2012. The specific facts and circumstances considered by our board of directors for the February 29, 2012 valuation included the following:
the underlying stock, as well as the risk-free rate of 0.4%. In addition, the current restrictions on the marketability of our common stock were considered. For the February 29, 2012 valuation, we estimated a 31.7% discount for the lack of marketability.
Stock option grants from August 31, 2012 to March 20, 2013
Our board of directors granted stock options from August 31, 2012 through March 20, 2013, with each having an exercise price of $9.96 per share. The exercise price per share was supported by an independent third-party valuation as of July 31, 2012. The specific facts and circumstances considered by our board of directors for the July 31, 2012 valuation included the following:
Valuation as of March 31, 2013
We completed a valuation as of March 31, 2013 and determined the fair value of our common stock to be $11.06 per share. The specific facts and circumstances considered by our board of directors for this valuation included the following:
determination of equity value under various exit scenarios and an estimation of the return to common stock under each of them.
Determination of Estimated Offering Price
The midpoint of the price range for this offering as determined by us and the underwriters is $ per share. In comparison, our estimate of the fair value of our common stock was $11.06 per share as of the March 31, 2013 valuation, which was used for stock option grants subsequent to March 20, 2013. We note that, as is typical in initial public offerings, the price range was not derived using a formal determination of fair value, but was determined based upon discussions between us and the underwriters. Among the factors that were considered in setting this range were our prospects and the history of and prospects for our industry, the general condition of the securities markets and the recent market prices of, and the demand for, publicly traded common stock of generally comparable companies. We believe that the difference between the fair valueholders of our common stock as a dividend to the extent of March 31, 2013the holder's pro rata share of our current and accumulated earnings and profits, if any, with any excess being treated as a return of capital to the extent thereof and then as capital gain. Although no assurance can be given, it is anticipated that we will not have current and accumulated earnings and profits through the end of 2016. Further, if our position is incorrect, the treatment of holders of participating warrants may differ from the treatment of the Subscription Rights distribution to the holders of our common stock. The participating warrant holders may be treated in a manner similar to holders of our common stock but it is possible that they may be subject to different and adverse U.S. federal income tax consequences.
The following discussion is based upon the treatment of the Subscription Rights issuance as a non-taxable distribution with respect to your existing shares of common stock or participating warrants for U.S. federal income tax purposes.
Tax Basis in the Subscription Rights
If the fair market value of the Subscription Rights you receive is less than 15% of the fair market value of your existing shares of common stock or participating warrants (with respect to which the Subscription Rights are distributed) on the date you receive the Subscription Rights, the Subscription Rights will be allocated a zero dollar basis for U.S. federal income tax purposes, unless you elect to allocate your basis in your existing shares of common stock or participating warrants between your existing shares of common stock or participating warrants and the midpointSubscription Rights in proportion to the relative fair market values of the existing shares of common stock or participating warrants and the Subscription Rights, determined on the date of receipt of the Subscription Rights. If you choose to allocate basis between your existing common shares or participating warrants and the Subscription Rights, you must make this election on a statement included with your timely filed tax return (including extensions) for the taxable year in which you receive the Subscription Rights. Such an election is irrevocable.
However, if the fair market value of the Subscription Rights you receive is 15% or more of the fair market value of your existing shares of common stock or participating warrants on the date you receive the Subscription Rights, then you must allocate your basis in your existing shares of common stock or participating warrants between those shares or participating warrants and the Subscription Rights you receive in proportion to their fair market values determined on the date you receive the Subscription Rights.
The fair market value of the Subscription Rights on the date that the Subscription Rights are distributed is uncertain, and we have not obtained, and do not intend to obtain, an appraisal of the fair market value of the Subscription Rights on that date. In determining the fair market value of the Subscription Rights, you should consider all relevant facts and circumstances, including any difference between the Subscription Price of the Subscription Rights and the trading price range for this offering isof our shares of common stock on the resultdate that the Subscription Rights are distributed, the exercise price of these factors as well asthe Warrants, the length of the period during which the Subscription Rights may be exercised and the fact that the estimated initial public offering price range necessarily assumes thatSubscription Rights are non-transferable.
Exercise of Subscription Rights
Generally, you will not recognize gain or loss upon the initial public offering has occurred,effectiveness of the exercise of a public market for ourSubscription Right in the Rights Offering. Your adjusted tax basis, if any, in the Subscription Right plus the Subscription Price should be allocated between the new common stock has been created and our preferred stock converted into common stock in connection with this offering, and therefore excludes any discount for lack of marketability of our common stock, which was factored into the March 31, 2013 valuation.
There is inherent uncertainty in our forecasts and projections and, if we had made different assumptions and estimates than those described previously, the amount of our stock-based compensation expense, net loss, and net loss per share amounts could have been materially different.
Clinical Trial Expense
As partWarrant acquired upon exercise of the process of preparing our consolidated financial statements, we are required to estimate our accrued expenses. Our clinical trial accrual process seeks to account for expenses resulting from our obligations under contracts with vendors, consultants and CROs and clinical site agreementsSubscription Right. The basis in connection with conducting clinical trials. The financial terms of these contracts are subject to negotiations,the stock or participating warrants upon which vary from contract to contract and may result in payment flows that do not match the periods overSubscriptions Rights were issued which materials or services are provided to us under such contracts. Our objective is to reflect the appropriate clinical trial expenses in our consolidated financial statements by matching the appropriate expenses with the period in which services and efforts are expended. We account for these expenses accordingallocated to the progress ofSubscription Rights under the trial as measured by patient progression and the timing of various aspects of the trial. We determine accrual estimates through financial models that take into account discussion with applicable personnel and outside service providers as to the progress or state of completion of trials, or the services completed. During the course of a clinical trial, we adjust our clinical expense recognition if actual results differ from our estimates. We make estimates of our accrued expenses as of each balance sheet date in our consolidated financial statements based on the facts and circumstances known to us at that time. Our clinical trial accrual and prepaid assets are dependent, in part, upon the receipt of timely and accurate reporting from CROs and other third-party vendors. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in us reporting amounts that are too high or too low for any particular period.
Basic and Diluted Net Loss Per Share of Common Stock
We compute basic net loss per share of common stock by dividing net loss applicable to common stockholders by the weighted-average number of shares of common stock outstanding during the period, excluding the dilutive effects of preferred stock, warrants to purchase preferred stock and stock options. We compute diluted net loss per share of common stock by dividing the net loss applicable to common stockholders by the sum of the weighted-average number of shares of common stock outstanding during the period plus the potential dilutive effects of preferred stock and warrants to purchase preferred stock, and stock options outstanding during the period calculated in accordance with the treasury stock method, but such items are excluded if their effect is anti-dilutive. Because the impact of these items is anti-dilutive during periods of net loss, there was no difference between our basic and diluted net loss per share of common stock for the years ended December 31, 2011 and 2012 and for the three months ended March 31, 2012 and 2013.
JOBS Act
In April 2012, the JOBS Act was enacted. Section 107 of the JOBS Act provides that an "emerging growth company" can take advantage of an extended transition period for complying with new or revised accounting standards. Thus, an "emerging growth company" can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other companies.
Internal Control Over Financial Reporting
In preparing our consolidated financial statements as of and for the year ended December 31, 2012, we and our independent registered public accounting firm identified control deficienciesprior section entitled "Tax Basis in the design and operation of our internal control over financial reporting that together constituted a material weakness in our internal control over financial reporting. A material weakness is a deficiency,Subscription Rights" would be further allocated between the new common
stock and the Warrant acquired upon exercise of the Subscription Right in proportion to their relative fair market values on the date the Subscription Rights were distributed. The Subscription Price should be allocated between the new common stock and Warrant acquired upon exercise of the Subscription Right in proportion to their relative fair market values on the exercise date. These allocations will establish your initial tax basis for U.S. federal income tax purposes in your new common stock and Warrants. The holding period of shares of common stock or a combinationWarrant acquired upon exercise of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our financial statements will not be prevented or detected on a timely basis. The material weakness identified was that we did not have sufficient financial reporting and accounting staff with appropriate training in GAAP and SEC rules and regulations with respect to financial reporting. As such, our controls over financial reporting were not designed or operating effectively, and as a result there were adjustments required in connection with closing our books and records and preparing our 2012 consolidated financial statements. The control deficiencies that we and our independent registered public accounting firm identified, and the adjustments recorded as a result, were as follows:
If you exercise a Subscription Right received in the Rights Offering after disposing of the warrant liability by $367,000.
The material weakness in our internal control over financial reporting was attributable to our lack of sufficient financial reporting and accounting personnel with appropriate training in GAAP and SEC rules and regulations. In response to this material weakness, we have recently hired a Chief Financial Officer and a director of financial reporting, each with public company financial reporting experience. We intend to hire additional finance and accounting personnel with appropriate training, build our financial management and reporting infrastructure, and further develop and document our accounting policies and financial reporting procedures. However, we cannot assure you that we will be successful in pursuing these measures or that these measures will significantly improve or remediate the material weakness described above. We also cannot assure you that we have identified all of our existing material weaknesses, or that we will not in the future have additional material weaknesses.
We have not yet remediated the material weakness described above, and the remediation measures that we have implemented and intend to implement may be insufficient to address our existing material weakness or to identify or prevent additional material weaknesses. See "Risk Factors—Risks Relating to this Offering and Ownership of Our Common Stock—If we are unable to successfully remediate the existing material weakness in our internal control over financial reporting, the accuracy and timing of our financial reporting may be adversely affected."
Neither we nor our independent registered public accounting firm has performed an evaluation of our internal control over financial reporting during any period in accordance with the provisions of the Sarbanes-Oxley Act. In light of the control deficiencies and the resulting material weakness that were identified as a result of the limited procedures performed, we believe that it is possible that, had we and our independent registered public accounting firm performed an evaluation of our internal control over financial reporting in accordance with the provisions of the Sarbanes-Oxley Act, additional material weaknesses and significant control deficiencies may have been identified. However, for as long as we remain an "emerging growth company" as defined in the JOBS Act, we intend to take advantage of the exemption permitting us not to comply with the requirement that our independent registered public accounting firm provide an attestation on the effectiveness of our internal control over financial reporting.
Results of Operations
Comparison of the Three Months Ended March 31, 2012 and 2013
| | Three Months Ended March 31, | | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2012 | 2013 | Change | |||||||
Revenue | $ | 198,000 | $ | 1,116,000 | $ | 918,000 | ||||
Operating expenses: | ||||||||||
General and administrative | 2,460,000 | 3,346,000 | 886,000 | |||||||
Research and development | 8,448,000 | 12,756,000 | 4,308,000 | |||||||
Total operating expenses | 10,908,000 | 16,102,000 | 5,194,000 | |||||||
Loss from operations | (10,710,000 | ) | (14,986,000 | ) | (4,276,000 | ) | ||||
Change in fair value of warrant liability | (609,000 | ) | 14,000 | 623,000 | ||||||
Interest expense | (21,000 | ) | — | 21,000 | ||||||
Other income, net | 541,000 | 127,000 | (414,000 | ) | ||||||
Net loss before income taxes | (10,799,000 | ) | (14,845,000 | ) | (4,046,000 | ) | ||||
Income taxes | — | — | — | |||||||
Net loss | (10,799,000 | ) | (14,845,000 | ) | (4,046,000 | ) | ||||
Accretion of preferred stock | (1,231,000 | ) | (1,019,000 | ) | 212,000 | |||||
Net loss applicable to common stockholders | $ | (12,030,000 | ) | $ | (15,864,000 | ) | $ | (3,834,000 | ) | |
Revenues
Revenues increased by $0.9 million for the first quarter of 2013 when compared to the same period in 2012 primarily as a result of entering into the Baxter agreement in the third quarter of 2012.
General and administrative expenses
General and administrative expenses increased by $0.9 million, or 36.0%, from $2.5 million for the three months ended March 31, 2012 to $3.3 million for the three months ended March 31, 2013. The increase was primarily attributable to an increase of $0.1 million related to stock-based compensation due to the increase in the fair valueshares of our common stock an increase of $0.5 million in professional fees, an increase of $0.1 million in travel expenses and an increase of $0.1 million as a resultor participating warrants with respect to which such Subscription Right is received, then certain aspects of the increasetax treatment of the exercise of the Subscription Right are unclear, including (1) the allocation of the tax basis between the shares of common stock or participating warrants previously sold and the Subscription Right, (2) the impact of such allocation on the amount and timing of gain or loss recognized with respect to the shares of our common stock or participating warrants previously sold and (3) the impact of such allocation on the tax basis of the shares of our common stock and Warrants acquired upon exercise of the Subscription Right. If you exercise a Subscription Right received in the Rights Offering after disposing of shares of our common stock or participating warrants with respect to which the Subscription Right is received, you should consult with your own tax advisor.
Expiration of Subscription Rights
If you allow Subscription Rights received in the Rights Offering to expire, you should not recognize any gain or loss for U.S. federal income tax purposes, and you should re-allocate any portion of the tax basis in your existing common stock or participating warrants previously allocated to the Subscription Rights that have expired to the existing common stock or participating warrants.
Taxation of Warrants
Sale, Exchange, Redemption or other Taxable Disposition of Warrants
Upon the sale, exchange, redemption or other taxable disposition of a Warrant, in general, you will recognize taxable gain or loss measured by the difference, if any, between (i) the amount of cash and administrative headcount from ninethe fair market value of any property received upon such taxable disposition and (ii) your adjusted tax basis in the Warrant as determined pursuant to the rules discussed above. Your gain or loss generally will be capital gain or loss and generally will be long-term capital gain or loss if, at the endtime of March 31, 2012the sale or other disposition, your holding period for the Warrant is more than one year. The deductibility of capital losses is subject to 11 at March 31, 2013.limitations.
Research and development expensesExercise of Warrants
Research and development expensesUpon the exercise of a Warrant by paying the exercise price in cash, in general, you will not recognize gain or loss for U.S. federal income tax purposes, except to the extent you receive a cash payment for any such fractional share that would otherwise have been issuable upon exercise of the Warrant. Your initial tax basis in common stock received will equal your adjusted tax basis in the Warrant exercised (as determined pursuant to the rules discussed above), increased by $4.3 million, or 51.0%, from $8.5 millionthe amount of cash paid to exercise the Warrant and decreased by the adjusted tax basis allocable to any fractional share that would otherwise have been issuable upon exercise of the Warrant. Your holding period for the three months ended March 31, 2012 to $12.8 millionshares of our common stock received on exercise generally will commence on the day of exercise.
In certain circumstances, namely during any period when a registration statement for the three months ended March 31, 2013. Thisexercise of the Warrants is not in effect, the Warrants will be exercisable on a cashless basis. The tax consequences of a cashless exercise are not clear and could differ from the consequences described
increase was driven byabove, including the possibility that a cashless exercise could be a taxable event. You should consult your own tax advisor regarding the tax consequences of a cashless exercise of a Warrant.
Expiration of Warrants
If you allow a Warrant to expire, you will generally recognize a loss for U.S. federal income tax purposes equal to your adjusted tax basis in the Warrant. In general, such a loss will be a capital loss and will be a short-term or long-term capital loss depending on your holding period for the Warrant.
Certain Adjustments to the Warrants
Under Section 305 of the Code, an increase in clinical trial expensesadjustment to the number of $3.0 million for rigosertib,common shares that will be issued on the exercise of the Warrants, or an increaseadjustment to the exercise price of $0.7 million related to consulting services and an increase of $0.8 million related to an increase in research and development headcount from 25 at March 31, 2012 to 37 at March 31, 2013the Warrants, may be treated as a resultconstructive distribution to you if, and to the extent that, such adjustment has the effect of increasing your proportionate interest in our expanded researchearnings and development activities.
Change in fair valueprofits or assets, depending on the circumstances of warrant liability
The fair valuesuch adjustment (for example, if such adjustment is to compensate for a distribution of cash or other property to our shareholders). Adjustments to the exercise price of Warrants made pursuant to a bona fide reasonable adjustment formula that has the effect of preventing dilution of the warrant liability increased by $0.6 million duringinterest of the three months ended March 31, 2012 comparedholders of the Warrants should generally not be considered to a decrease of $14,000 during the three months ended March 31, 2013, which resultedresult in a commensurate increase inconstructive distribution. Any such constructive distribution would be taxable whether or not there is an actual distribution of cash or other expense and other income, respectively. The decrease inproperty. See the fair valuemore detailed discussion of the warrant liability in 2013 was primarily duerules applicable to a slight change indistributions made by us under the valueheading "Taxation of the liability related to the revaluation of the warrants outstanding. The increase in the fair value of the warrant liability in 2012 was related to the revaluation of the outstanding warrants.Common Stock—Distributions" below.
Interest expense
Interest expense decreased from $21,000 during the three months ended March 31, 2012 to zero for the three months ended March 31, 2013, as the promissory note outstanding in 2012 converted into shares of Series I convertible preferred stock in July 2012.
Other income, net
Other income, net, decreased by $0.4 million during the three months ended March 31, 2013 compared to the three months ended March 31, 2012. This decrease was largely the result of a $0.5 million gain recognized on our sale of New Jersey state NOL carry forwards in 2012.
ComparisonTaxation of Years Ended December 31, 2011 and 2012Common Stock
| | Year Ended December 31, | | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | Change | |||||||
Revenue | $ | 1,487,000 | $ | 46,190,000 | $ | 44,703,000 | ||||
Operating expenses: | ||||||||||
General and administrative | 6,436,000 | 15,707,000 | 9,271,000 | |||||||
Research and development | 22,624,000 | 52,762,000 | 30,138,000 | |||||||
Total operating expenses | 29,060,000 | 68,469,000 | 39,409,000 | |||||||
Loss from operations | (27,573,000 | ) | (22,279,000 | ) | 5,294,000 | |||||
Change in fair value of warrant liability | 1,287,000 | 367,000 | (920,000 | ) | ||||||
Interest expense | (19,000 | ) | (8,608,000 | ) | (8,589,000 | ) | ||||
Other income, net | 11,000 | 608,000 | 597,000 | |||||||
Net loss before income taxes | (26,294,000 | ) | (29,912,000 | ) | (3,618,000 | ) | ||||
Income taxes | — | — | — | |||||||
Net loss | (26,294,000 | ) | (29,912,000 | ) | (3,618,000 | ) | ||||
Accretion of preferred stock | (4,020,000 | ) | (3,953,000 | ) | 67,000 | |||||
Net loss applicable to common stockholders | $ | (30,314,000 | ) | $ | (33,865,000 | ) | $ | (3,551,000 | ) | |
RevenuesDistributions
Revenues increased by $44.7 million in 2012 when comparedDistributions with respect to 2011 primarily as a result of entering into the Baxter agreement in 2012. Of this increase, $42.4 million related to the value ascribed to the license, and was therefore recognized immediately upon signing of the agreement, and
$3.1 million related to the portion of development services revenue recognized in 2012 under the Baxter agreement. Those increases attributable to the Baxter agreement were partially offset by a $1.1 million reduction in research funding.
General and administrative expenses
General and administrative expenses increased by $9.3 million, or 144.0%, from $6.4 million for the year ended December 31, 2011 to $15.7 million for the year ended December 31, 2012. The increase was primarily attributable to an increase of $7.2 million related to stock-based compensation due to the increase in the fair valueshares of our common stock during the year, an increaseacquired upon exercise of $1.7 million in professional fees relatedSubscription Rights or upon exercise of Warrants will be taxable as dividend income when actually or constructively received to the negotiationextent of the Baxter agreement in 2012,our current or accumulated earnings and an increase of $0.4 millionprofits as a result of the increase in general and administrative headcount from six at the end of 2011determined for U.S. federal income tax purposes.
Dividend income received by certain non-corporate U.S. Holders with respect to nine at the end of 2012.
Research and development expenses
Research and development expenses increased by $30.1 million, or 133.0%, from $22.6 million for the year ended December 31, 2011 to $52.8 million for the year ended December 31, 2012. This increase was driven by a $12.5 million milestone due to Temple University, or Temple, in 2012 as a result of entering into the Baxter agreement in 2012 compared to a $1.9 million payment to Temple in 2011 as a result of entering into the SymBio agreement. The change was also due to an increase in clinical trial expenses of $8.4 million for rigosertib, $6.6 million in additional stock-based compensation due to the increase in the fair valueshares of our common stock duringgenerally will be "qualified dividends" subject to preferential rates of U.S. federal income tax, provided that the year, an increaseU.S. Holder meets applicable holding period and other requirements. Subject to similar exceptions for short-term and hedged positions, dividend income on our shares of $1.4 million relatedcommon stock paid to consulting services in connection withU.S. Holders that are domestic corporations generally will qualify for the rigosertib clinical trials, an increasedividends-received deduction. To the extent that the amount of $1.5 million in nonclinical trial-related costs for rigosertiba distribution exceeds our current and an increase of $0.9 million related to a change in researchaccumulated earnings and development headcount from 25 at the end of 2011 to 36 at the end of 2012profits, such distribution will be treated first as a resulttax-free return of our expanded research and development activities.
Change in fair value of warrant liability
The fair value of the warrant liability decreased by $1.3 million during the year ended December 31, 2011 compared to a decrease of $0.4 million during the year ended December 31, 2012, which in both cases resulted in a commensurate increase in other income. The decrease in the fair value of the warrant liability in 2012 was primarily duecapital to the expirationextent of Series G convertible preferred stock warrantsyour adjusted tax basis in 2012, which accounted for a decrease of $1.0 million in the value of the liability, which was partially offset by an increase of $0.6 million in the value of the liability related to the revaluation of the warrants outstanding. The decrease in the fair value of the warrant liability in 2011 was primarily due to the revaluation of the warrants outstanding.
Interest expense
Interest expense increased by $8.6 million during the year ended December 31, 2012 compared to the year ended December 31, 2011. In July 2012, the holders of our convertible notes elected to convert their outstanding principal and accrued interest intosuch shares of Series I convertible preferred stock. At the time of the conversion, there was $8.2 million in unamortized contingent beneficial conversion features that we immediately expensed.
Other income, net
Other income, net, increased by $0.6 million during the year ended December 31, 2012 compared to the year ended December 31, 2011. This increase was driven by a $0.5 million gain recognized on our sale of New Jersey state NOL carry forwards.
Liquidity and Capital Resources
Since our inception, we have incurred net losses and generally negative cash flows from our operations. We incurred net losses of $26.3 million and $29.9 million for the years ended December 31, 2011 and 2012, respectively, and $10.8 million and $14.8 million for the three months ended March 31, 2012 and 2013, respectively. Our operating activities used $6.1 million and $13.9 million of cash flows during the three months ended March 31, 2012 and 2013, respectively. At March 31, 2013, we had an accumulated deficit of $183.2 million, working capital of $41.7 million and cash and cash equivalents of $67.3 million. Historically, we have financed our operations principally through private placements of preferred stock and convertible debt. Through March 31, 2013, we have received gross proceeds of $171.5 million from the issuance of preferred stock and convertible debt. We have also financed our operations with the $57.5 million in upfront payments we received from SymBio and Baxter in 2011 and 2012.
Cash Flows
The following table summarizes our cash flows for the years ended December 31, 2011 and 2012 and the three months ended March 31, 2012 and 2013:
| | Year Ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | |||||
Net cash (used in) provided by: | |||||||
Operating activities | $ | (14,171,000 | ) | $ | 1,633,000 | ||
Investing activities | (241,000 | ) | (279,000 | ) | |||
Financing activities | 9,785,000 | 77,460,000 | |||||
Net (decrease) increase in cash and cash equivalents | $ | (4,627,000 | ) | $ | 78,814,000 | ||
| | Three Months Ended March 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2012 | 2013 | |||||
Net cash (used in) provided by: | |||||||
Operating activities | $ | (6,088,000 | ) | $ | (13,949,000 | ) | |
Investing activities | (4,000 | ) | (284,000 | ) | |||
Financing activities | 3,637,000 | 6,000 | |||||
Effect of foreign currency translation on cash | — | 7,000 | |||||
Net decrease in cash and cash equivalents | $ | (2,455,000 | ) | $ | (14,220,000 | ) | |
Net cash (used in) provided by operating activities
Net cash used in operating activities was $14.2 million for the year ended December 31, 2011 and consisted primarily of a net loss of $26.3 million and $0.9 million of noncash decreases primarily related to the change in fair value of our warrant liabilities. The cash used in operating activities was offset by the $7.7 million increase in deferred revenues primarily related to receipt of the upfront payment from SymBio under the research and collaboration agreement and the $5.4 million increase related to the change in operating assets and liabilities. The significant items in the change in operating assets and liabilities included a decrease in grant receivable of $1.7 million, a $2.2 million increase in accounts payable, and a $1.1 million increase in accrued expenses. The decrease in grant receivable was attributable to our receipt in 2011 of the grant payment from LLS. The increase in accounts payable and accrued expenses was primarily due to the timing of our payment of clinical trial costs related to the ongoing trials and development of our product candidates.
Net cash provided by operating activities was $1.6 million for the year ended December 31, 2012 and consisted primarily of noncash increases of $22.0 million and a $9.6 million increase related to the change in operating assets and liabilities that were offset by a net loss of $29.9 million. The noncash increases were primarily attributable to increases in stock-based compensation and recognition of debt discounts and beneficial conversion features of $8.2 million upon conversion of our convertible promissory notes into preferred stock. The significant items in the change in operating assets and liabilities included an increase in prepaid expenses and other current assets of $1.1 million, offset by increases in accrued expenses of $2.6 million and an increase in deferred revenues of $8.2 million. The increase in prepaid expenses and other current assets was primarily due to the prepayment of upfront costs for our Phase 3 clinical trials and continued development activities. The increase in accrued expenses was primarily due to the costs for our Phase 3 clinical trial activities. The increase in deferred revenues was primarily due to the receipt of payments from Baxter and LLS of $6.9 million and $4.1 million, respectively, pursuant to the terms of our agreements with such parties. These increases were partially offset by the recognition of the unamortized portions of upfront payments under our collaboration agreements with Baxter and SymBio for $3.1 million and $0.5 million, respectively.
Net cash used in operating activities was $6.1 million for the three months ended March 31, 2012 and consisted primarily of a net loss of $10.8 million, offset by $3.1 million of noncash items primarily related to the change in fair value of our warrant liabilities and stock-based compensation. The cash used in operating activities was further offset by the $1.0 million increase in deferred revenue primarily related to receipt of an upfront payment from LLS under the research and collaboration agreement and the $0.6 million increase related to the change in operating assets and liabilities. The significant items in the change in operating assets and liabilities included a decrease in prepaid expenses of $0.2 million, a $0.2 million increase in accounts payable and a $0.2 million increase in accrued expenses.
Net cash used in operating activities was $14.0 million for the three months ended March 31, 2013 and consisted primarily of a net loss of $14.8 million, partially offset by $2.5 million of stock-based compensation expense. A $1.6 million decrease related to the change in operating assets and liabilities drove the balance of cash used in operations. The significant items in the change in operating assets and liabilities included an increase in prepaid expenses and other current assets of $0.9 million and a decrease in deferred revenue of $1.1 million, offset by a net increase in accounts payable and accrued expenses of $0.4 million. The increase in prepaid expenses and other current assets was primarily due to the prepayment of upfront manufacturing costs and filing fees. The decrease in deferred revenue was due to the recognition of the unamortized portions of upfront payments under our collaboration agreements with Baxter and SymBio in the amounts of $1.0 million and $0.1 million, respectively.
Net cash used in investing activities
Net cash used in investing activities for the years ended December 31, 2011 and 2012 was $0.2 million and $0.3 million, respectively. Cash used in investing activities primarily consisted of purchases of fixed assets.
Net cash used in investing activities for the three months ended March 31, 2012 and 2013 was $4,000 and $0.3 million, respectively. Cash used in investing activities consisted of purchases of fixed assets.
Net cash provided by financing activities
Net cash provided by financing activities was $9.8 million for the year ended December 31, 2011, which was primarily due to $7.2 million in proceeds from the issuance of Series H convertible preferred stock, $1.9 million in proceeds from the issuance of Series G convertible preferred stock, $0.8 million in proceeds from our loan and security agreement with a bank that were restricted and to be used for
future debt repayment, $0.6 million in proceeds from a loan from our chairman of the board of directors, who is also a significant stockholder, and $0.2 million in proceeds upon the exercise of stock options. These proceeds were partially offset by $0.9 million in principal payments made under our loan and security agreement with a bank.
Net cash provided by financing activities was $77.5 million for the year ended December 31, 2012, which was primarily due to $47.8 million in proceeds from the issuance of Series J convertible preferred stock in connection with the Baxter equity investment, $25.8 million in proceeds upon the issuance of convertible debt that was subsequently converted into shares of Series I convertible preferred stock, $2.2 million in proceeds upon the exercise of warrants in exchange for shares of Series G convertible preferred stock, $0.4 million in proceeds from collection of a subscription receivable for our issuance of Series H convertible preferred stock and $1.3 million in proceeds upon the exercise of stock options.
Net cash provided by financing activities was $3.6 million for the three months ended March 31, 2012, which was primarily due to $0.4 million in proceeds from the issuance of Series H convertible preferred stock, $0.4 million in proceeds upon the exercise of warrants in exchange for shares of Series G convertible preferred stock and $2.8 million in proceeds from a loan from our chairman of the board of directors, who is also a significant stockholder.
Net cash provided by financing activities was $6,000 for the three months ended March 31, 2013, resulting from the exercise of stock options.
Operating and Capital Expenditure Requirements
We have not achieved profitability since our inception and we expect to continue to incur net losses for the foreseeable future. We expect our cash expenditures to increase in the near term as we fund our Phase 2 and Phase 3 clinical trials of rigosertib, as well as our clinical trials of our other earlier-stage product candidates and continuing preclinical activities. Following this offering, we will be a publicly traded company and will incur significant legal, accounting and other expenses that we were not required to incur as a private company. In addition, the Sarbanes-Oxley Act, as well as rules adopted by the SEC and the NASDAQ Stock Market, require public companies to implement specified corporate governance practices that are currently inapplicable to us as a private company. We expect these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly. We estimate that we will incur approximately $2.0 to $3.0 million of incremental costs per year associated with being a publicly traded company, although it is possible that our actual incremental costs will be higher than we currently estimate.
We believe that our existing capital resources, together with the net proceeds from this offering, will be sufficient to fund our operations for at least the next 12 months. However, we anticipate that we will need to raise substantial additional financing in the future to fund our operations. In order to meet these additional cash requirements, we may seek to sell additional equity or convertible debt securities that may result in dilution to our stockholders. If we raise additional funds through the issuance of convertible debt securities, these securities could have rights senior to those of our common stock and could contain covenants that restrict our operations. There can be no assurance that wethereafter as capital gain.
Dispositions
If you sell or otherwise dispose of shares of common stock acquired upon exercise of Subscription Rights or upon exercise of Warrants in a taxable transaction, you will generally recognize capital gain or loss equal to the difference between the amount realized and your adjusted tax basis in the shares. Such capital gain or loss will be able to obtain additional equitylong-term capital gain or debt financing on terms acceptable to us,loss if at all. Further, the achievement of milestones and receipt from Baxter and SymBio of milestone payments and royalties, even if rigosertibyour holding period for such shares is approved for commercial use in Baxter's and SymBio's licensed territories, are not assured. Our failure to obtain sufficient funds on acceptable terms when needed could have a negative impact on our business, results of operations, and financial condition. Our future capital requirements will depend on many factors, including:
Please see "Risk Factors" for additional risks associated with our substantial capital requirements.
Contractual Obligations and Commitments
The following is a summary of our long-term contractual cash obligations as of December 31, 2012:
| | Total | Less than one year | 1-3 Years | 3-5 Years | More than 5 Years | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Operating lease obligations | $ | 365,000 | $ | 256,000 | $ | 109,000 | $ | — | $ | — | ||||||
Total contractual obligations | $ | 365,000 | $ | 256,000 | $ | 109,000 | $ | — | $ | — | ||||||
Purchase Commitments
We have no material non-cancelable purchase commitments with contract manufacturers or service providers as we have generally contracted on a cancelable purchase order basis.
Milestone, Royalty-Based and Other Commitments
Under our license agreement with Temple to develop, manufacture, market and sell rigosertib related compounds and derivatives, we are obligated to pay annual license maintenance payments, as well as 25% of any sublicensing fees we receive. We are also required to pay a low-single digit percentage of our net sales of rigosertib as a royalty. During the year ended December 31, 2011, in connection with the execution of the SymBio agreement, we made a payment to Temple in the amount of $1.9 million. During the year ended December 31, 2012, in connection with the execution of the
Baxter agreement, we became obligated to make a payment to Temple in the amount of $12.5 million. Both of these payments were recorded as research and development expenses. There were no expenses related to this agreement during the three months ended March 31, 2013.
In May 2010, we entered into an agreement with LLS under which we were to conduct research in return for milestone payments, up to $10.0 million through 2013. This milestone payment amount was subsequently reduced to $8.0 million pursuant to an amendment signed in January 2013. In the event that the research is successful, we must proceed with commercialization of the product or repay the amount funded. In addition, we will owe to LLS regulatory and commercial milestone payments and royalties based on net sales of the product not to exceed three times the aggregate amount funded, or $24.0 million. During the year ended December 31, 2012, in connection with the execution of the Baxter agreement, we paid $1.0 million to LLS and we have recorded this amount in research and development expenses. This payment reduced the maximum contingent payment obligation under this agreement to $23.0 million at December 31, 2012, and there were no changes during the three months ended March 31, 2013.
Because the achievement and timing of these milestones and net sales is not fixed and determinable, our commitments under these agreements have not been included on our consolidated balance sheets or in the Contractual Obligations table above.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements, as defined by applicable SEC regulations.
Quantitative and Qualitative Disclosure About Market Risk
We are exposed to market risks in the ordinary course of our business. These market risks are principally limited to interest rate fluctuations.
We had cash and cash equivalents of $81.5 million and $67.3 million at December 31, 2012 and March 31, 2013, respectively, consisting primarily of funds in cash and money market accounts. The primary objective of our investment activities is to preserve principal and liquidity while maximizing income without significantly increasing risk. We do not enter into investments for trading or speculative purposes. Due to the short-term nature of our investment portfolio, we do not believe an immediate 10% increase in interest rates would have a material effect on the fair market value of our portfolio, and accordingly we do not expect our operating results or cash flows to be materially affected by a sudden change in market interest rates.
Segment Reporting
We view our operations and manage our business in one segment, which is the identification and development of oncology therapeutics.
Recent Accounting Pronouncements
Effective January 1, 2011, we prospectively adopted ASU 2009-13, which amended the guidance for revenue recognition related to multiple-deliverable revenue arrangements. The amendments in this guidance enabled vendors to account for products or services separately rather than as a combined unit upon meeting certain criteria and establish a hierarchy for determining the selling price of a deliverable. In addition, a vendor can determine a best estimate of selling price, in a manner that is consistent with that used to determine the price to sell the deliverable on a standalone basis, if a vendor does not have vendor-specific objective evidence or third party evidence of selling price. This guidance also eliminated the use of the residual method and requires a vendor to allocate revenue using the relative selling price method. The amendments were effective prospectively, with an option
for retrospective restatement of the financial statements, for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010 with early adoption permitted at the beginning of an entity's fiscal year. Our adoption of this new accounting standard did not have a significant impact on our consolidated financial position, results of operations or cash flows.
In June 2011, FASB issued ASU No. 2011-05, "Comprehensive Income (ASC Topic 220): Presentation of Comprehensive Income," or ASU 2011-05, which amended current comprehensive income guidance. This accounting update eliminated the option to present the components of other comprehensive income as part of the statement of stockholders' equity. Instead, comprehensive income must be presented in either a single continuous statement of comprehensive income, which contains two sections, net income and other comprehensive income, or in two separate but consecutive statements. ASU 2011-05 was effective for fiscal periods beginning after December 15, 2011 with early adoption permitted. Our retrospective adoption of ASU 2011-05 did not have a significant impact on our consolidated financial position, results of operations or cash flows.
In February 2013, FASB issued ASU No. 2013-02, "Comprehensive Income (Topic 220): Reporting of Amounts Reclassified Out of Accumulated Other Comprehensive Income," or ASU 2013-02. ASU 2013-02 requires companies to present either in a single note or parenthetically on the face of the financial statements, the effect of significant amounts reclassified from each component of accumulated other comprehensive income based on its source and the income statement line items affected by the reclassification. This guidance is effective for annual reporting periods beginning after December 15, 2012. We believe the adoption of this standard will not have a significant impact on our consolidated financial position, results of operations or cash flows.
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
In connection with this offering, Ernst & Young LLP became our independent registered public accounting firm effective as of February 27, 2013, and EisnerAmper LLP was dismissed as our independent registered public accounting firm effective as of December 17, 2012. The decision to appoint Ernst & Young LLP and dismiss EisnerAmper LLP was recommended by our audit committee and subsequently approved by our board of directors.
The report of EisnerAmper LLP on our financial statements for the year ended December 31, 2011 did not contain an adverse opinion or a disclaimer of opinion and was not qualified or modified as to uncertainty, audit scope or accounting principles.
In connection with the audit of our financial statements for the year ended December 31, 2011 and through EisnerAmper LLP's dismissal, there were no disagreements with EisnerAmper LLP on any matters of accounting principles or practices, financial statement disclosures or auditing scope or procedures, which if not resolved to EisnerAmper LLP's satisfaction would have caused EisnerAmper LLP to make reference to the matter in their report.
In connection with our audited financial statements for the year ended December 31, 2011 through EisnerAmper LLP's dismissal, there have been no reportable events with us as set forth in Item 304(a)(1)(v) of Regulation S-K.
We requested that EisnerAmper LLP furnish us with a letter addressed to the SEC stating whether it agrees with the above statements. A copy of the letter, dated June 14, 2013, is filed as an exhibit to the registration statement of which this prospectus forms a part.
Overview
We are a clinical-stage biopharmaceutical company focused on discovering and developing novel small molecule drug candidates to treat cancer. Using our proprietary chemistry platform, we have created an extensive library of targeted anti-cancer agents designed to work against specific cellular pathways that promote cancer. We believe that the drug candidates in our pipeline have the potential to be efficacious in a wide variety of cancers without causing harm to normal cells. We have three clinical-stage product candidates and six preclinical programs.
Rigosertib, our most advanced product candidate, is being tested in a number of ongoing Phase 2 and Phase 3 clinical trials. We are conducting a pivotal Phase 3 trial of rigosertib under a Special Protocol Assessment, or SPA, from the U.S. Food and Drug Administration, or FDA, for higher risk myelodysplastic syndromes, or MDS. We expect to report top-line overall survival results from this trial in the fourth quarter of 2013 or the first quarter of 2014. We are also evaluating rigosertib in a Phase 3 trial for metastatic pancreatic cancer, in two Phase 2 trials for transfusion-dependent lower risk MDS, and in a Phase 2 trial for head and neck cancers. We have tested rigosertib in more than 850 patients with solid tumors and hematological diseases. Rigosertib has been granted orphan drug status for MDS in both the United States and Europe as well as orphan drug status for pancreatic cancer in the United States. Baxter Healthcare SA, or Baxter, a subsidiary of Baxter International Inc., has commercialization rights for rigosertib in Europe and SymBio Pharmaceuticals Limited, or SymBio, has commercialization rights in Japan and Korea. We have retained development and commercialization rights to rigosertib in the rest of the world, including in the United States.
Rigosertib is an inhibitor of two important cellular signaling pathways: phosphoinositide 3-kinase, or PI3K, and polo-like kinase, or PLK, both of which are frequently over-active in cancer cells. PI3K signaling promotes the growth and survival of cells under stressful conditions, such as under low oxygen levels that are often found in tumors. By inhibiting the PI3K pathway in cancer cells, rigosertib promotes tumor cell apoptosis, or programmed cell death.
The PLK pathway has a critical role in maintaining proper chromosome organization and sorting during cell division. By modulating the PLK pathway, rigosertib stops cancer cells at late stages of the cell division cycle, which leads to chromosome disorganization and death in these cells. In normal cells, rigosertib pauses progression of the cell cycle in the early stages, without causing harm or death to these cells.
Due to this dual effect of inhibiting both the PI3K and PLK pathways, and thereby effecting both tumor cell survival and division, we believe that rigosertib has potential to treat a variety of cancer types, including hematological diseases and solid tumors.
We are testing both intravenous and oral formulations of rigosertib, referred to as rigosertib IV and rigosertib Oral, in clinical trials.
Drug Application, or NDA, to the FDA in the second half of 2014, and a Marketing Authorization Application, or MAA, to the European Medicines Agency, or the EMA, by the fourth quarter of 2014 or the first quarter of 2015, for marketing approval of rigosertib IV.
A provider of marketing analytics and data to the biopharmaceutical industry has estimated that, for 2011 in the United States, the diagnosed incidence of MDS was approximately 15,600 and the prevalence of MDS was approximately 52,000. According to the same marketing analytics firm, approximately 23% of MDS patients are estimated to fall into the categories of MDS characterized as higher risk.
To accelerate and broaden the development of rigosertib, we entered into a development and licensing agreement with Baxter in 2012 to commercialize rigosertib in Europe. In 2011, we entered into a licensing agreement with SymBio to commercialize rigosertib in Japan and Korea. We have retained development and commercialization rights to rigosertib in the rest of the world, including the United States. We will explore a variety of alternatives for the commercialization of rigosertib in territories we currently retain, including direct commercialization, co-promotion or selective out-licensing of rights to a third party.
Our second clinical-stage product candidate,ON 013105, is in a Phase 1 trial in patients with relapsed or refractory lymphoma, including an aggressive form of non-Hodgkin's lymphoma identified as mantle cell lymphoma, or MCL, and acute lymphoid leukemia, or ALL. A critical defect in many cancer cells is the uncontrolled expression of cyclin D1, a protein essential for normal cell division. Cyclin D1 is over-expressed in several hematological diseases, including B-cell lymphomas, such as MCL. ON 013105 suppresses the accumulation of cyclin D1 in cancer cells. In 2011, we suspended enrollment in this Phase 1 trial because enrollment of patients was occurring slowly, and as a result, our inventory of ON 013105 clinical trial materials expired. We plan to restart enrollment in this trial with newly manufactured clinical trial materials at a new clinical trial site in the fourth quarter of 2013.
Our third clinical-stage product candidate,recilisib, is being developed in collaboration with the U.S. Department of Defense, or DoD, for acute radiation syndromes, or ARS. We have conducted animal studies and clinical trials of recilisib under the FDA's Animal Efficacy Rule, which permits marketing approval for new medical countermeasures for which human efficacy studies are not feasible or ethical, by relying on evidence from animal studies in appropriate animal models to support efficacy in humans. We have completed four Phase 1 trials to evaluate the safety and pharmacokinetics of recilisib in healthy human adult subjects using both subcutaneous and oral formulations, referred to as recilisib SC and recilisib Oral. We have received orphan drug designation for recilisib for ARS in the United States.
In addition to our three clinical-stage product candidates, we are advancing six preclinical programs that target kinases, cellular metabolism or division.
We have broad-based capabilities that span drug discovery and clinical development, from medicinal chemistry and evaluation in biochemical, cell-based and animal models, through Phase 3 trials and regulatory filings in the United States, Europe and India. Our discovery program is based on a proprietary chemistry platform comprising more than 150 novel core chemical structures. Our chemistry and screening approaches aim to discover new drug candidates that increase efficacy and help overcome resistance to therapy in cancer cells, while minimizing their toxicity to normal cells. Our intellectual property portfolio includes more than 100 issued patents and over 90 patent applications, either owned by us or licensed exclusively to us, including patents covering our most advanced product candidate, rigosertib. These patents and licenses cover composition-of-matter, process, formulations and method-of-treatment claims for our clinical-stage product portfolio.
Our Strategy
We are committed to delivering novel treatments to cancer patients. We are focused on discovering and developing targeted small molecule anti-cancer product candidates. The key components of our strategy are to:
Our Product Candidates
Clinical Programs
The development status of our three clinical-stage product candidates is summarized below:
Clinical-Stage Product Candidates
Rigosertib—Inhibitor of Phosphoinositide 3-Kinase & Polo-Like Kinase Pathways
Overview
Rigosertib is our most advanced product candidate and has been extensively tested in clinical trials, which have collectively enrolled more than 850 patients. We believe that rigosertib has the potential to become the first approved second-line therapy for higher risk MDS patients. Also, we believe that rigosertib has the potential to be a first-line therapy for transfusion-dependent lower risk MDS patients and for metastatic pancreatic cancer patients.
Rigosertib IV is currently in several late-stage clinical trials, including a pivotal Phase 3 trial being conducted under an SPA from the FDA and Scientific Advice from the EMA for adult MDS patients whose disease has failed azacitidine or decitabine therapy. We completed enrollment in this trial in May 2013 and expect to report top-line overall survival results in the fourth quarter of 2013 or the first quarter of 2014.
Rigosertib IV is also being studied as a combination therapy with gemcitabine in a Phase 3 trial for treatment of patients with metastatic pancreatic cancer who have not previously received any chemotherapy. We have enrolled 150 patients in the first phase of this trial and expect results of the pre-planned interim analysis for overall survival in the fourth quarter of 2013 or the first quarter of 2014.
In addition, rigosertib Oral is being studied in Phase 2 trials in patients with transfusion-dependent lower risk MDS and in patients with head and neck cancers. We reported initial response and safety data from the first Phase 2 trial in lower risk MDS in June 2013 and expect to complete enrollment and present overall results from this trial in December 2013. We expect to complete enrollment for the head and neck cancers trial in the second half of 2014.
Rigosertib Inhibits Two Key Signaling Pathways Associated with Cancer Cell Growth and Survival
Rigosertib is an inhibitor of two important cellular signaling pathways, PI3K and PLK, both of which are frequently over-active in cancer cells. PI3K signaling promotes the growth and survival of cells under stressful conditions, such as under low oxygen levels that are often found in tumors. If the PI3K pathway is over-active, apoptosis of cancer cells is diminished, leading to excessive cellular growth. By inhibiting the PI3K pathway, rigosertib promotes tumor cell apoptosis. Rigosertib also influences signals along the PI3K pathway, such as those leading to the production of cyclin D1.
The PLK pathway plays a critical role in maintaining proper organization and sorting of chromosomes during cell division. Too much PLK activity in cancer cells results in uncontrolled proliferation. By modulating PLK pathway activity, rigosertib stops cancer cells at late stages of the cell division cycle, which leads to chromosome disorganization and death in these cells. In normal cells rigosertib pauses progression of the cell cycle in the early stages, without causing harm or death to these cells.
Due to this dual effect of inhibiting both the PI3K and PLK pathways, and thereby effecting both tumor cell survival and division, we believe that rigosertib has potential to treat a variety of cancer types, including hematological diseases and solid tumors.
Myelodysplastic Syndromes
MDS is a group of blood disorders that affect bone marrow function. MDS typically affects patients over the age of 65. In MDS, bone marrow becomes dysplastic, or defective. The blood cells produced do not develop normally, such that too few healthy blood cells are released into the blood stream, which leads to low blood cell counts, or cytopenias. Thus, many patients with MDS require frequent blood transfusions. In most cases, the disease worsens and the patient develops progressive bone marrow failure. In advanced stages of the disease, immature blood cells, or blasts, leave the bone
marrow and enter the blood stream, leading to acute myelogenous leukemia, or AML, which occurs in approximately one-third of patients with MDS.
Based on Surveillance Epidemiology and End Results (SEER) data from the National Cancer Institute, a marketing analytics firm has estimated the 2011 incidence of MDS at approximately 15,600 cases and the prevalence of MDS at approximately 52,000 cases in the United States. We believe that the actual incidence numbers may be higher, due to underdiagnosing and underreporting of new cases of MDS to centralized cancer registries, and that the incidence of MDS in the United States is likely to increase, due to an aging population, improved disease awareness and diagnostic precision, and an increase in the number of cases of secondary, often chemotherapy-induced, MDS.
MDS is typically diagnosed using routine blood tests or by observing symptoms, such as shortness of breath, weakness, easy bruising or bleeding, or fever with frequent infections. A diagnosis of MDS is confirmed by evaluating a bone marrow biopsy/aspirate showing dysplastic changes, and, in more advanced cases, the presence of excess blasts, meaning that blasts account for more than 5% of the total number of cells in the bone marrow. Because the bone marrow and blood cells in MDS patients can undergo different kinds of abnormal changes, several classification systems have been developed to gauge the severity of disease and help determine prognosis and treatment strategy. We use two standard classification systems, the French-American-British morphological classification system, or the FAB system, as modified by the World Health Organization, or WHO, and the International Prognostic Scoring System, or IPSS, to define patient inclusion criteria for our rigosertib trials in MDS:
Patients with RAEB-1, RAEB-2 or RAEB-t under the FAB/WHO criteria or patients with IPSS risk scores of Intermediate-2 or High are generally considered to have higher risk MDS, with a median survival of less than two years. According to a marketing analytics firm, approximately 23% of MDS patients are classified in these higher risk categories.
Patients with IPSS scores of Low and Intermediate-1 are generally considered to have lower risk MDS, with an overall survival of approximately three to six years. Approximately 77% of MDS patients are classified in these lower risk categories.
Treating Myelodysplastic Syndromes
Stem cell or bone marrow transplantation is a potentially curative therapy for MDS. However, since most patients with MDS are elderly and therefore ineligible for transplantation, this option is generally considered only for the small proportion of younger MDS patients.
We believe that most higher risk and some lower risk MDS patients are treated with hypomethylating drugs, azacitidine and decitabine, the hypomethylating drugs approved in the United States for treatment of MDS. A provider of information, services and technology for the healthcare industry estimates that in the year ended June 2012, approximately 12,500 MDS patients received treatment with hypomethylating agents. For 2012, revenues of azacitidine have been reported to be
approximately $327 million in the United States and revenues for decitabine have been reported to be approximately $225 million in North America.
A significant number of higher risk MDS patients fail or cannot tolerate treatment with azacitidine or decitabine, which represent the current standard of care for higher risk MDS patients, and almost all patients who initially respond to therapy eventually relapse. Median survival time of MDS patients who have failed hypomethylating drugs is less than six months. Accordingly, we believe that a new therapy that would extend survival in these refractory patients would represent a major contribution in the treatment of MDS.
Hypomethylating drugs work by inhibiting the methylation of DNA. Methylation is a biochemical process involving the addition of a methyl group to DNA and plays an important role in gene expression during cell division and differentiation. By inhibiting methylation, hypomethylating drugs cause the death of rapidly dividing cells, including cancer cells that are no longer responsive to normal growth control mechanisms. Hypomethylation may also restore normal function to genes that are critical for differentiation and proliferation. The mechanisms underlying resistance to hypomethylating agents in patients is not well understood. Studies performed with decitabine in cultured cell lines revealed lowered expression of enzymes required for drug transport and activation.
By contrast, rigosertib works by inhibiting the PI3K pathway, a cellular signaling pathway that promotes the growth and survival of cells under stressful conditions, such as under low oxygen levels that are often found in tumors, and by modulating PLK pathway activity, which leads to chromosome disorganization and death in cancer cells. We believe that, because rigosertib has a mechanism of action that is different from hypomethylating agents, it may be effective in patients who have failed treatment with those drugs. We have observed that rigosertib treatment resulted in bone marrow responses in some patients whose bone marrow blast cell counts had increased during prior treatment with hypomethylating agents.
In the case of lower risk MDS patients, those categorized as Low or Intermediate-1 risk with transfusion-dependent anemia and del(5q) cytogenetic abnormality are generally treated with lenalidomide (Revlimid®). For all other lower risk MDS patients, supportive care employing blood products, such as red blood cell and platelet transfusions, and erythroid stimulating agents, is the mainstay of therapy. Frequent transfusions are subject to many risks, including iron overload, blood borne infections and immune-related reactions. We believe that a therapeutic agent that could lower or eliminate the need for transfusions over an extended period of time would fulfill a significant unmet medical need for this patient population.
Clinical Development of Rigosertib IV in Higher Risk Myelodysplastic Syndromes
We filed an investigational new drug application, or IND, amendment with the FDA for rigosertib in MDS in July 2008. We are conducting a multi-center, pivotal Phase 3 trial with rigosertib IV as a single agent in patients with MDS who have failed prior azacitidine or decitabine therapy, which we refer to as our "ONTIME" trial. The protocol for this trial was reviewed and agreed upon with the FDA under an SPA. The EMA has also provided Scientific Advice, indicating that the study design should be adequate to meet the scientific and regulatory requirements to support efficacy claims for a marketing application.
The ONTIME trial is a randomized, controlled study, where eligible patients must have failed azacitidine or decitabine treatment, have excess blasts (5-30% blasts) and have at least one cytopenia. There is currently no approved drug for this group of patients and the current standard treatment consists of best supportive care, which is treatment intended to manage disease-related symptoms. In the ONTIME trial, both groups of patients receive best supportive care, with the treatment group of patients also receiving rigosertib. The study employs a 2:1 randomization in which two-thirds of the patients receive rigosertib plus best supportive care, and one-third of patients receive only best supportive care. The key assumption used to calculate the required size of the ONTIME trial was
based on hypothesized median survival of 30 weeks in the rigosertib treatment group and 17 weeks in the best supportive care group. A total sample size of 270 patients, with 180 patients in the rigosertib group and 90 patients in the best supportive care group, and a total number of 223 deaths yields greater than 95% statistical power to detect a significant difference in overall survival between the two groups.
The primary objective of the ONTIME trial is to ascertain whether treatment with rigosertib leads to an overall survival benefit. Secondary objectives include evaluation of other responses, such as improvements in bone marrow and cytogenetic and blood profiles, according to standard response criteria for MDS defined by the 2006 WHO International Working Group, and to measure patients' quality of life scores and times to transition to AML. In this study, rigosertib is administered as a continuous intravenous infusion over a period of three days with a bag change every 24 hours. Patients use an ambulatory pump, avoiding the need for hospital stays. We completed enrollment of 270 patients in the ONTIME trial in May 2013.
The ONTIME trial is being conducted at 42 sites in the United States and at 47 sites in Belgium, France, Germany, Italy and Spain. Among the 270 patients enrolled as of May 2013, 179 are enrolled at U.S. sites. The safety and conduct of the trial is being reviewed by a data safety monitoring committee at fixed intervals. Three such reviews have been held to date, each with the recommendation to continue the trial without change to the study protocol. We expect to report top-line overall survival results from this trial in the fourth quarter of 2013 or the first quarter of 2014.
Phase 1/2 Trial Results of Rigosertib IV in Patients with Myelodysplastic Syndromes
We conducted two Phase 1 studies, one Phase 2 study and one Phase 1/2 study at four sites, where we enrolled 79 patients with MDS or AML. Of these 79 patients, 39 were RAEB-1, RAEB-2, or RAEB-t MDS patients and who had previously failed treatment with hypomethylating agents. There were encouraging signs of activity, as determined by survival and bone marrow analyses, in this group of patients. The following graph is a probability-of-survival curve, known as a Kaplan-Meier curve, showing the length of survival of these 39 patients after initiation of rigosertib IV treatment. In this intent-to-treat analysis, the median survival was 36 weeks. Twenty-three patients survived at least six months, eight patients lived more than one year but less than two years, and three patients lived more than two years, including one alive at 142 weeks. According to several peer-reviewed published reports, median overall survival for MDS patients who have failed treatment with hypomethylating agents is less than six months.
Survival Data for 39 Refractory MDS Patients Receiving Rigosertib IV:Intent-to-Treat Analysis: Median Overall Survival = 36 Weeks
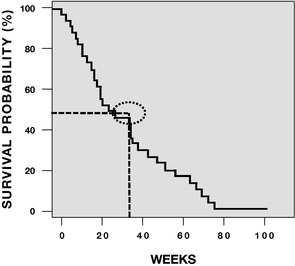
A follow-up bone marrow biopsy, to assess the effects of treatment on blast counts, was available for 30 of the 39 patients. The percentage change in bone marrow, or BM, blast count is shown in the graph below. Of the 30 evaluable patients, 21 had a reduction in BM blast count, and 12 patients in this subset demonstrated an objective response, meaning that they had a 50% or more reduction in BM blasts. Of the 12 patients with objective responses, five patients were determined to have a complete BM blast response, meaning that their blast count was reduced to 5% or less of BM cells present. Nine of the 30 evaluable patients had an increase in BM blast count, with three patients showing progression, meaning that they experienced an increase of 50% or more in BM blasts. During the period these patients received treatment, a subset of 37 patients had between one and four follow-up bone marrow biopsies. These biopsies showed that the cellularity, or distribution of normal cell types, was not decreased, suggesting that rigosertib was selectively reducing the BM blast counts in these patients, while being nontoxic to normal bone marrow cells.
Not all patients in a study may have follow-up bone marrow biopsies because these are invasive procedures and represent a risk of profuse bleeding, particularly in patients with severe thrombocytopenia, a condition in which the body does not produce enough platelets and a common occurrence in higher risk MDS patients. Such biopsies are also uncomfortable and sometimes painful procedures and patients may withhold consent for such procedures. Finally, early worsening of the overall clinical condition toward leukemia or death may cause follow-up bone marrow biopsies to be deferred. Since the primary endpoint of the ONTIME trial is overall survival, we anticipate that unavailable bone marrow biopsies would not have a material impact on the results of the trial.
Best Bone Marrow (BM) Responses After Rigosertib IV in30 RAEB-1, -2, -t MDS Patients Previously Treated withHypomethylating Agents
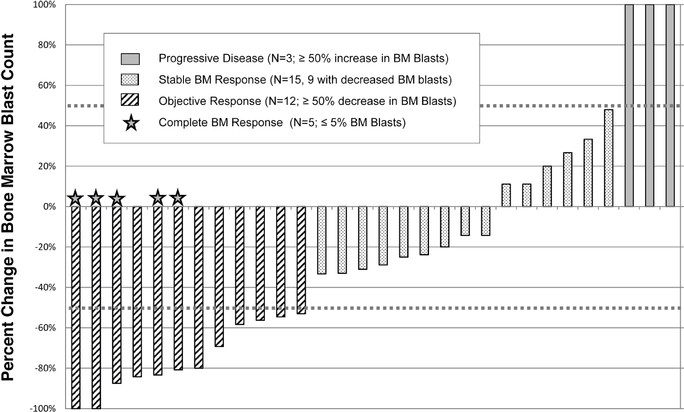
Further analysis, tabulated below, of BM results in comparison with the overall survival of these patients revealed a positive correlation between BM blast response and overall survival, with a statistically significant p-value of 0.003. The p-value is a widely used, conventional parameter for indicating statistical significance. A p-value of 0.05 or less represents statistical significance, meaning that there is a less than one-in-twenty likelihood that the observed results occurred by chance. Those patients with objective BM response and patients who had stable BM counts, meaning they had a less than 50% increase or decrease in BM blast count, exhibited a higher survival rate than patients who progressed during treatment or who were not evaluable for BM analysis. These initial results suggest that BM blast counts have predictive value for estimating overall survival in this group of patients.
Preliminary Analysis of BM Response and Overall Survival Data from39 MDS Patients in Four Single-Arm Phase 1 andPhase 1/2 Trials of Rigosertib IV in Patients with MDS
| | Objective BM Response (³ 50% blast decrease) | Stable BM Response (< 50% increase or decrease in blasts) | Progressive Disease (³ 50% blast increase) | Not Assessed (no biopsy) | P-value | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Number of patients | 12 | 15 | 3 | 9 | — | |||||||||||
Median overall survival (weeks) | 37 | 40 | 10 | 11 | 0.003 | |||||||||||
This observation is further elaborated in the following Kaplan-Meier graph, in which survival curves for groups of patients are shown based on their BM response category. Treated patients with a stable BM response or an objective BM response survived significantly longer than those patients who experienced progression or who could not be evaluated for BM response.
Overall Survival Data from 39 Refractory MDS Patients Receiving Rigosertib IV:Grouped by Bone Marrow Responses
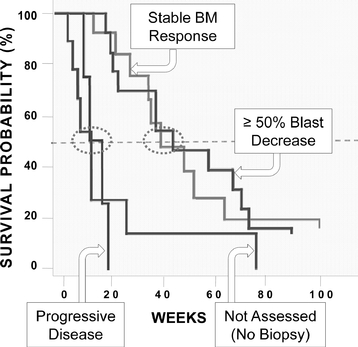
In these exploratory trials, we also compared the relationship between duration of rigosertib exposure and BM response. Overall, more patients who received three-day infusions of rigosertib every two weeks achieved improved BM blast responses compared to patients who received two-day infusions every week for three weeks of a four week cycle. Extending infusion duration beyond three days did not result in further improvements of BM blast responses. As a result, we chose a three-day dosing regimen for the Phase 3 trial.
Safety data for the 79 patients with MDS or AML in four Phase 1 and 2 trials are available. Rigosertib IV was generally well tolerated in these trials. The most frequent drug-related adverse events, occurring in at least 10% of patients, were nausea, diarrhea, fatigue, anemia, dysuria and hematuria. Drug-related adverse events that were Grade 3 or Grade 4 in severity, meaning that they were more than mild or moderate in toxicity, observed in at least two patients included anemia, thrombocytopenia, neutropenia, decreased white blood cells, urinary frequency, dysuria, decreased blood sodium, increased clotting time, fatigue, fever and diarrhea.
Clinical Development of Rigosertib Oral in Lower Risk Myelodysplastic Syndromes
We filed an IND amendment with the FDA for rigosertib Oral in MDS in February 2009. Based on Phase 1 trial results with rigosertib Oral, we believe that rigosertib has the potential to significantly reduce the transfusion needs and improve the quality of life for patients with lower risk MDS. Although median survival for these transfusion-dependent patients is five or more years, frequent transfusions are subject to risks and limitations, including iron overload, blood borne infections and immune related reactions. We believe that an oral therapeutic agent that could lower or eliminate the need for transfusions over an extended period of time would fulfill a significant unmet medical need for this patient population.
We are enrolling transfusion-dependent lower risk MDS patients in two Phase 2 trials. We reported an interim analysis of initial response and safety data from the first Phase 2 trial, which we refer to as our "ONTARGET" trial, at the June 2013 Annual Meeting of the American Society for Clinical Oncology, or ASCO. We expect to complete enrollment in this trial and to present overall results at the American Society of Hematology, or ASH, Annual Meeting in December 2013.
The ONTARGET trial is an open-label, multi-site trial testing the effect of rigosertib Oral on transfusion-dependent lower risk MDS patients. To be eligible for this study, patients must have received transfusions of at least four units of red blood cells during the eight weeks before randomization and patients can continue to receive transfusions and erythroid stimulating agents while in the trial. The primary objectives of this trial are to evaluate efficacy, as measured by transfusion independence, as well as safety. The study has a target enrollment of 60 patients, with enrollment open at four trial sites in the United States. Initially, patients were randomized 1:1 on an outpatient basis to one of two dosing regimens, receiving either 560 mg of rigosertib twice a day for 14 consecutive days of a 21-day cycle, which we refer to as the intermittent dosing arm, or 560 mg of rigosertib twice a day for 21 days of a 21-day circle, which we refer to as the continuous dosing arm. We were able to evaluate interim data from 26 evaluable patients in the intermittent dosing arm and eight evaluable patients in the continuous dosing arm.
Evaluation of the interim safety data indicated that rigosertib Oral was generally well tolerated, with the most frequently observed drug-related side effects being urologic in nature and believed to be related to the dosing regimen. In the continuous dosing arm of the study, five of the first nine patients experienced drug-related urinary side effects of Grade 2 or higher, meaning that they were more than mild in toxicity. Accordingly, the study protocol was amended to allow all patients to be treated with the intermittent dosing regimen. The most frequent urinary adverse events of Grade 2 or higher in the intermittent dosing arm were urinary urgency/frequency, which occurred in 38% of patients, dysuria, which occurred in 18%, and hematuria, or the presence of red blood cells in the urine, which occurred in 15%. Several of the patients who experienced dysuria reported improvements after they were administered oral hydration or sodium bicarbonate. Other adverse events of Grade 2 or higher included intermittent neutrophenia, or an abnormally low number of white blood cells that serve as the primary defense against infections, which occurred at Grade 3 in one patient and at Grade 4 in one other patient. Median onset of drug-related adverse events of Grade 2 or higher was 28 weeks in the intermittent dosing arm, compared with 12 weeks in the continuous dosing arm. Median duration of the treatment in the continuous dosing arm was 24 weeks. Because the trial is ongoing, median duration of the treatment in the intermittent dosing arm has not yet been determined. Renal function was unaffected and gastrointestinal adverse events and fatigue were infrequently observed. We are investigating ways to mitigate or eliminate urinary symptoms by employing oral hydration and sodium bicarbonate and adjusting the administered rigosertib dose and schedule, with the goal of reducing exposure of bladder mucosa to excreted rigosertib.
Initial response results showed that 13 of the 26 evaluable patients in the intermittent dosing arm and two of the eight evaluable patients in the continuous dosing arm achieved transfusion independence, defined as a period of at least eight consecutive weeks without any red blood cell transfusions. As shown in the graph below, in the 13 patients who achieved transfusion independence in the intermittent dosing arm, the duration of transfusion independence ranged from eight weeks to more than 48 weeks. Onset of independence ranged from one week to 24 weeks following the initiation of rigosertib dosing and two patients continued to benefit from therapy more than nine months after starting rigosertib.
Interim Analysis of Lower Risk Transfusion-Dependent MDS Patients Treated withRigosertib Oral: Duration of Transfusion Independence Achieved in 13 of 26 Evaluable Patients in the Intermittent Dosing Arm*
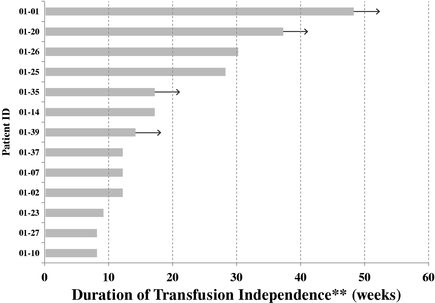
One of the aims of our ongoing trials is to correlate patient characteristics with transfusion response, thus potentially allowing for the selection of appropriate patients for future trials with rigosertib. Eleven of the 13 transfusion-independent patients in the intermittent dosing arm received one or more injections of erythroid stimulating agents during the time of rigosertib Oral administration, and the patterns of hemoglobin responses observed in some patients suggest a possible synergy between rigosertib Oral and erythroid stimulating agents.
We initiated a second multi-center Phase 2 trial in May 2013 in lower risk, transfusion-dependent MDS patients who have failed treatment with erythroid stimulating agents, in which all patients will receive the intermittent dosing schedule. We expect to complete enrollment for this trial in the second half of 2014.
Previously, we conducted a Phase 1 trial of rigosertib Oral to determine drug safety and tolerability and to monitor plasma levels of rigosertib Oral in Low, Intermediate-1, Intermediate-2 and High Risk MDS patients who had failed prior azacitidine, decitabine or lenalidomide therapy or injection of an erythroid stimulating agent. Pharmacokinetic analysis showed that plasma levels of rigosertib could be achieved that were at or above levels predicted to be pharmacodynamically active. Encouraging preliminary signs of activity were observed. Safety data are available in 33 patients from this trial. The most frequent drug-related adverse events, occurring in at least 10% of patients, were decreased appetite, diarrhea, nausea and dysuria. Drug-related adverse events that were Grade 3 or greater in severity occurred in two patients and included urinary tract infection, syncope, or fainting, and dyspnea, or shortness of breath. Hematuria was identified as the most frequent dose-limiting toxicity, occurring in two patients.
Metastatic Pancreatic Cancer
The American Cancer Society estimates that 45,220 new cases of pancreatic cancer will be diagnosed in the United States in 2013, and that 38,460 people are expected to die in the United States from the disease in 2013. Deaths from pancreatic cancer ranked fourth among cancer-related deaths in the United States in 2012 according to the American Cancer Society. At diagnosis, 50% of pancreatic cancer patients already have metastasis to the liver or peritoneal surface in the abdomen. Since 1975, the five-year survival rate of pancreatic cancer patients has only marginally improved, from 2% to 6%. The median survival for locally advanced and for metastatic disease, which collectively represent over 80% of cases, is about ten months and six months, respectively.
Currently, the best therapeutic option for pancreatic cancer patients is surgical resection to remove tumor-laden tissue. However, only 15% of newly diagnosed patients are candidates for this treatment option, and of these patients only about 20% survive to five years. For the remaining 85% of pancreatic cancer patients, gemcitabine is one of two currently prescribed, FDA approved therapies, but it provides only a marginal survival benefit over best supportive care, from five and a half to six months. The epidermal growth factor receptor inhibitor erlotinib (Tarceva®) has also received approval in the United States and Europe for metastatic pancreatic cancer, and has shown a modest increase of median survival and a 6% increase in one-year survival rates. Fluorouracil and mitomycin-C, older cytotoxic drugs, are also approved for the treatment of pancreatic cancer. Unapproved combination therapies like FOLFIRINOX, a chemotherapy regimen, are becoming a part of standard care, especially in patients with good performance status who can tolerate the toxic side effects associated with this regimen. We believe that metastatic pancreatic cancer remains an area of significant unmet medical need and presents a large market opportunity for the development of additional targeted therapies.
Clinical Development Rigosertib IV in Metastatic Pancreatic Cancer
We filed an original IND with the FDA for rigosertib in pancreatic cancer in December 2011. We are studying rigosertib as first-line therapy in a Phase 3 trial in advanced pancreatic cancer. In this trial, we are comparing a treatment combination of rigosertib IV and gemcitabine to a treatment with gemcitabine alone. After reviewing our Phase 1 results in 24 advanced pancreatic cancer patients and proposing a clinical plan to the FDA, the FDA authorized us to move directly from the Phase 1 trial to a two-stage Phase 3 trial, which we refer to as our "ONTRAC" trial.
ONTRAC is a multi-center, open-label, randomized controlled clinical trial in patients with histologically confirmed metastatic pancreatic cancer who received no prior chemotherapy, with overall survival as the primary endpoint. In March 2013, we completed enrollment of 150 patients in the first portion of this trial. Once 100 deaths have been reported in this patient population, a data safety monitoring committee will compare overall survival between the two arms of the trial and will also review the adequacy of the proposed sample size. This committee may recommend adjusting the target enrollment for the second portion of the study, beyond the initially planned 364 patients, or it may recommend early termination of the study. We believe that this study, if successful, could support an NDA submission for this indication. We expect results of the pre-planned interim analysis for overall survival of the 150 patients enrolled in the first portion of this trial in the fourth quarter of 2013 or the first quarter of 2014.
Rigosertib has been evaluated in combination with gemcitabine in 40 patients in a two-site Phase 1 trial. Of the patients we studied, 25 had advanced pancreatic cancer and 15 had other tumors. Efficacy was assessed according to a set of published international rules that define when cancer patients improve, or respond, stay the same, or stabilize, or worsen, or progress, during treatments, called Response Evaluation Criteria in Solid Tumors. Of the 37 patients with measurable disease, three achieved partial response, including one with metastatic pancreatic cancer previously treated with gemcitabine, and a fourth patient with gemcitabine-naive pancreatic cancer had an unconfirmed partial
response. Sixteen patients, including nine patients with metastatic pancreatic cancer, had an overall response of stable disease.
All 40 patients were evaluable for safety. A total of 37 patients in this study had drug-related adverse events. The most frequent drug-related adverse events, occurring in at least 10% of patients, were nausea, thrombocytopenia, fatigue, neutropenia, diarrhea, vomiting, anemia, leukopenia, pyrexia, constipation, abdominal pain, lymphopenia, aspartate transaminase increase and decreased appetite. Twenty-two patients had drug-related adverse events of Grade 3 or greater in severity, the most common of which were the hematological adverse events of neutropenia, thrombocytopenia, and lymphopenia. Hematological adverse events following administration of gemcitabine are well documented, including anemia, neutropenia, leukopenia and thrombocytopenia.
Clinical Development of Rigosertib Oral in Head and Neck Cancers
According to the National Cancer Institute, head and neck cancers accounts for approximately 3% of all new cancer cases in the United States, with approximately 52,000 cases diagnosed in 2012. Single-modality treatment with surgery or radiationnon-corporate U.S. Holder is generally recommended for the 30% to 40%taxed at preferential rates of patients with early-stage disease. Combined modality therapy with surgery, radiation or concurrent chemotherapy and radiation is utilized for patients with locally or regionally advanced disease. Patients with very advanced or recurrent disease are treated with platinum-based chemotherapy, cetuximab (Erbitux®) or both in combination. Expected overall survival in patients with head and neck cancers who have failed platinum-based therapy is about six months.
We filed an IND amendment with the FDA for rigosertib in head and neck cancer in November 2012. We conducted a Phase 1 trial with rigosertib Oral in 48 patients with various advanced solid tumors refractory to standard therapy, including six patients with head and neck cancers who had previously failed on platinum-based therapy. TwoU.S. federal income tax. The deductibility of these six head and neck cancers patients achieved durable responses to rigosertib therapy. One patient had a confirmed complete response, defined as the disappearance of chest and lung disease, and the other patient had a partial response, with a 53% decrease of liver metastasis. Both patients remained on therapy for an extended period of time, over 98 weeks for one and over 48 weeks for the other. We believe that these observations are encouraging for this patient population.
Rigosertib Oral is currently in a Phase 2 trial in relapsed or metastatic squamous cell carcinoma, with a focus on enrolling patients with head and neck cancers. The study will evaluate both human papilloma virus, or HPV, positive and HPV negative groups of patients. HPV infection presents a significant new therapeutic challenge in head and neck cancer patients. We expect to complete enrollment of 80 patients in this trial in the second half of 2014.
ON 013105—Targeted Anti-cancer Agent Modulating Cyclin D1
Overview
ON 013105 suppresses cyclin D1 accumulation in cancer cells. Cyclin D1 is a protein required for normal progression through the cell reproduction cycle, and it is over-expressed in several hematological diseases, including B-cell lymphomas, such as MCL. Based on the activity of ON 013105 in preclinical in vitro and in vivo models of MCL, we are currently evaluating ON 013105 in a Phase 1 trial in patients with relapsed or refractory lymphomas and ALL.
Cyclin D1 in Lymphoma
Lymphomas are a collection of blood cancers that develop in the lymphatic system, including the B and T lymphocytes. Lymphoma is the most common form of hematological disease in the developed world. Approximately 75,000 people in the United States were diagnosed with lymphoma in 2011. MCL is an aggressive subset of non-Hodgkin's lymphoma with a poor prognosis and high frequency of cyclin
D1 overexpression. Studies conducted in patients with MCL have shown that levels of cyclin D1 correlate with the proliferative rate of the malignant cells as well as decrease in overall survival.
Clinical Development in Lymphoma and Acute Lymphoid Leukemia
We filed an original IND with the FDA for ON 013105 in November 2008. We are conducting a Phase 1 dose-escalation study of ON 013105 in patients with relapsed or refractory lymphomas and ALL, in which we are evaluating the safety, pharmacokinetics and activity of a once-weekly intravenous infusion regimen. In the first two complete dose cohorts, ON 013105 was readily detectible in plasma and had a half-life of less than one hour. In 2011, we suspended enrollment in our Phase 1 trial of ON 013105 because enrollment of patients was occurring slowly and, as a result, our inventory of ON 013105 clinical trial materials expired. We plan to restart enrollment in this trial with newly manufactured clinical trial materials at a new clinical trial site in the fourth quarter of 2013.
Recilisib—Acute Radiation Syndromes Treatment
Overview
Recilisib is a small molecule with radiation protection properties. We are developing recilisib SC and recilisib Oral to address a need for medical countermeasures to treat the effects of ARS, specifically radiation-induced cytopenia. The DoD provided $10.2 million in government funding to us pursuant to a number of programs through 2011. Our strategy is to continue to seek support from government agencies and to develop recilisib under the FDA Animal Efficacy Rule.
Novel Mechanism of Action
Recilisib employs a novel mechanism of action that involves intracellular signaling and DNA damage repair pathways. In preclinical studies, cells treated with recilisib sustained less DNA damage upon exposure to ionizing radiation in comparison to untreated cells.
Clinical Development for Acute Radiation Syndromes
We filed original INDs with the FDA for recilisib SC and for recilisib Oral in April 2008 and May 2011, respectively. We have completed four Phase 1 trials with recilisib, three trials with recilisib SC in more than 50 healthy adults and one trial with recilisib Oral in nine healthy adults.
Preclinical Development for Acute Radiation Syndromes
We have conducted preclinical studies to evaluate the radioprotective effects of recilisib SC and recilisib Oral in collaboration with the Armed Forces Radiobiology Research Institute and Georgetown University. In these studies, protection from radiation injury by recilisib was observed when administered prophylactically prior to ionizing radiation exposure in cellular and animal models. In irradiated mice, the protective benefits included increased overall survival and an enhanced rate of recovery of the hematopoietic system and crypt cells lining the gut. We are also working with the Biomedical Advanced Research and Development Authority on this program.
Preclinical Programs
In addition to our three clinical-stage programs, we have developed a pipeline of preclinical programs. Our preclinical pipeline includes six programs that target kinases, cellular metabolism or division. We intend to explore additional collaborations to further the development of these product candidates as we focus internally on our more advanced programs.
ON 1231320—Inhibitor of Polo-like Kinase 2, or PLK2: PLK2 is critical for centriole duplication during cell division, or mitosis. ON 1231320 is a specific inhibitor of PLK2, and in preclinical studies, it induced mitotic arrest and reduced tumor burden in mice injected subcutaneously with colon tumor and triple-negative breast cancer cells.
ON 123300—Inhibitor of the Cell Cycle and Cancer Cell Metabolism: ON 123300 inhibits the activity of two kinases, cyclin-dependent kinase 4, or CDK4, and AMP-activated protein kinase 5, or ARK5. CDK4 is an essential component of the cell division machinery and is a well-established target for therapeutic development. ARK5 is believed to be involved in the regulation of cancer cell metabolic activity. We believe that ON 123300 may have promise as a brain tumor therapy. We observed that ON 123300 can kill glioblastoma tumor cells in vitro and we also observed that, in mouse brain tumor models, ON 0123300 can cross the blood-brain barrier.
ON 108600—Inhibitor of Cyclin-dependent Kinase 9 and Casein Kinase 2: ON 108600 is a dual inhibitor of two growth-regulatory kinases. Cyclin-dependent kinase 9 is over expressed in several cancers, including leukemias and lymphomas. Casein kinase 2 is overexpressed in a variety of tumor types and plays a role in oncogenic processes including DNA damage and repair. We believe that ON 108600 may invoke a novel mechanism of cancer cell lethality by inhibiting these two targets.
ON 044580—Non-ATP Dual Inhibitor of Janus Kinase 2, or JAK-2, and Bcr-Abl Kinase: ON 044580 inhibits mutant forms of the two target kinases, including JAK2V617F and imatinib-resistant Bcr-AblT315I. Three major myeloproliferative disorders, a group of diseases of the bone marrow in which excess cells are produced, are polythycemia vera, essential thrombocythemia and myeloid metaplasia with myelofibrosis. These disorders harbor mutations in JAK2, making this enzyme a potentially attractive therapeutic target for treating these disorders. Philadelphia chromosome-positive chronic myeloid leukemia cells make an abnormal protein called Bcr-Abl kinase, which is the clinical target of the approved inhibitor, imatinib (Gleevec®).
ON 24 Series of Compounds—Oral Anti-Tubulin Agents: Microtubules are organelles composed of a protein known as tubulin that help maintain cell shape, assist in cell movement and guide cell division. Interference with microtubule formation is an established anti-cancer strategy. ON 24 compounds cause tubulin to depolymerize, inducing mitotic arrest in cultured tumor cell lines.
ON 146 Series—Selective Inhibitors of PI3K alpha/delta Isoforms: Gain-of-function mutations in alpha and delta isoforms of PI3K are critical drivers of growth in several cancers. ON 146040 inhibits the growth of a variety of blood cancer cell lines, including Burkitt's lymphoma, MCL, multiple myeloma and chronic myeloid leukemia.
Our Proprietary Drug Discovery Platform
Our product candidates, designed for targeting cancer while protecting healthy cells, are derived from a novel chemistry platform and cell-based differential screening, which together define our discovery approach. Our chemical library contains more than 150 novel core chemical structures and thousands of unique compounds. Most are simple two-ring or three-ring structures that are built around a common core or signature element. Most kinase enzymes require the binding of adenosine triphosphate, or ATP, to function. Unlike most kinase inhibitors, our proprietary library includes many molecules that do not compete with the ATP binding site of kinases, which we believe may provide a more selective way to inhibit these enzymes.
Research and Development
Since commencing operations, we have dedicated a significant portion of our resources to the development of our clinical-stage product candidates, particularly rigosertib. We incurred research and development expenses of $22.6 million, $52.8 million and $12.8 million during the years ended December 31, 2011 and 2012 and the three months ended March 31, 2013, respectively. We anticipate that a significant portion of our operating expenses will continue to be related to research and development as we continue to advance our clinical-stage product candidates.
Collaborations
Baxter Healthcare SA
In September 2012, we entered into a development and license agreement with a subsidiary of Baxter International Inc., Baxter Healthcare SA, or Baxter, granting Baxter an exclusive, royalty-bearing license for the research, development, commercialization and manufacture (in specified instances) of rigosertib in all therapeutic indications in Europe. Under the Baxter agreement, we are obligated to use commercially reasonable efforts to, in accordance with a development plan agreed upon by the parties, direct, coordinate and manage the development of rigosertib for MDS and pancreatic cancer. Under the agreement, if after a specified development event we elect not to move forward with the development of rigosertib for pancreatic cancer, Baxter may, at its own expense, develop rigosertib for pancreatic cancer on its own for the purposes of obtaining marketing approval. In addition, there is a specified mechanism set forth in the agreement to expand the scope of the collaboration for additional indications. Our agreement with Baxter is guided by a joint steering committee. If the joint steering committee is not able to make a decision by consensus, then any dispute would be resolved by specified executive officers of both parties.
Under the terms of the agreement, Baxter made an upfront payment of $50.0 million. We are eligible to receive pre-commercial milestone payments of up to an aggregate of $512.5 million if specified development and regulatory milestones are achieved. The potential pre-commercial development milestone payments to us include the following:
We may also receive up to $337.5 million in milestone payments for regulatory approvals of the three rigosertib indications specified in the arrangement with Baxter, each of which may be up to and in excess of $100.0 million. We are also potentially eligible to receive an additional $20.0 million pre-commercial milestone payment related to the timing of regulatory approval of rigosertib IV in higher risk MDS patients in Europe. In addition to these pre-commercial milestones, we are eligible to receive up to an aggregate of $250.0 million in milestone payments based on Baxter's achievement of pre-specified threshold levels of annual net sales of rigosertib. We are also entitled to receive royalties at percentage rates ranging from the low-teens to the low-twenties on net sales of rigosertib by Baxter in the licensed territory.
Under the agreement, Baxter is obligated to pay us royalties, on a country-by-country basis in the licensed territory, until the later of the expiration of all valid claims of the patent rights licensed to Baxter that cover the manufacture, use, sale or importation of rigosertib in such country, and the expiration of regulatory-based exclusivity for rigosertib in such country. If the patent rights and
regulatory-based exclusivity expire in a particular country before a specified period of time after first commercial sale of rigosertib in that country, Baxter will pay us royalties at a reduced rate until the end of the specified period. In addition, unless we receive marketing approval for the use of rigosertib IV for MDS from the EMA or specified European Union countries without undertaking additional specified clinical-trials, the royalty rates may be reduced depending on when we receive marketing approval for the use of rigosertib IV for MDS from the EMA or specified European Union countries, and whether or not a competing product for refractory MDS has been approved within a specified period after our receipt of approval for rigosertib IV for MDS.
The agreement with Baxter will remain in effect until the expiration of all applicable royalty terms and satisfaction of all payment obligations in each licensed country, unless terminated earlier due to the uncured material breach or bankruptcy of a party, force majeure, or in the event of a specified commercial failure. We may terminate the agreement in the event that Baxter brings a challenge against us in relation to the licensed patents. Baxter may terminate the agreement without cause commencing after a specified period of time from the execution of the agreement.
In July 2012, Baxter also purchased $50.0 million of our Series J convertible preferred stock.
SymBio Pharmaceuticals Limited
In July 2011, we entered into a license agreement with SymBio Pharmaceuticals Limited, or SymBio, as subsequently amended, granting SymBio an exclusive, royalty-bearing license for the development and commercialization of rigosertib in Japan and Korea. Under the SymBio license agreement, SymBio is obligated to use commercially reasonable efforts to develop and obtain market approval for rigosertib inside the licensed territory and we have similar obligations outside of the licensed territory. We have also entered into an agreement with SymBio to supply them with development-stage product. Under the SymBio license agreement we also agreed to supply commercial product to SymBio under specified terms that will be included in a commercial supply agreement to be negotiated prior to the first commercial sale of rigosertib. We have also granted SymBio a right of first negotiation to license or obtain the rights to develop and commercialize compounds having a chemical structure similar to rigosertib in the licensed territory.
Under the terms of the SymBio license agreement, we received an upfront payment of $7.5 million. We are eligible to receive milestone payments of up to an aggregate of $33.0 million from SymBio upon the achievement of specified development and regulatory milestones for specified indications. Of the development milestones, $3.0 million is due after enrollment of the first patient in the event a decision is made, after our interim analysis, to start a Phase 3 clinical trial of rigosertib IV in combination with gemcitabine for pancreatic cancer patients in the United States. Of the regulatory milestones, $5.0 million is due upon receipt of marketing approval in the United States of rigosertib IV in higher risk MDS patients, $3.0 million is due upon receipt of marketing approval in Japan for rigosertib IV in higher risk MDS patients, $5.0 million is due upon receipt of marketing approval in the United States for rigosertib Oral in lower risk MDS patients, $5.0 million is due upon receipt of marketing approval in Japan for rigosertib Oral in lower risk MDS patients, $5.0 million is due upon receipt of marketing approval in the United States for rigosertib IV in combination with gemcitabine in pancreatic cancer patients, and $3.0 million is due upon receipt of marketing approval in Japan for rigosertib IV in combination with gemcitabine in pancreatic cancer patients. Furthermore, upon receipt of marketing approval in the United States and Japan for an additional specified indication of rigosertib, which we are currently not pursuing, an aggregate of $4.0 million would be due. In addition to these pre-commercial milestones, we are eligible to receive tiered milestone payments of up to an aggregate of $30.0 million based upon annual net sales of rigosertib by SymBio in the licensed territory. Further, under the terms of the SymBio license agreement, SymBio is obligated to make royalty payments to us at percentage rates ranging from the mid-teens to 20% based on net sales of rigosertib by SymBio in the licensed territory.
Royalties will be payable under the SymBio agreement on a country-by-country basis in the licensed territory, until the later of the expiration of marketing exclusivity in those countries, a specified period of time after first commercial sale of rigosertib in such country, or the expiration of all valid claims of the licensed patents covering rigosertib or the manufacture or use of rigosertib in such country. If no valid claim exists covering the composition of matter of rigosertib or the use of or treatment with rigosertib in a particular country before the expiration of the royalty term, and specified competing products achieve a specified market share percentage in such country, SymBio's obligation to pay us royalties will continue at a reduced royalty rate until the end of the royalty term. In addition, the applicable royalties payable to us may be reduced if SymBio is required to pay royalties to third parties for licenses to intellectual property rights necessary to develop, use, manufacture or commercialize rigosertib in the licensed territory.
The license agreement with SymBio will remain in effect until the expiration of the royalty term. However, the SymBio license agreement may be terminated earlier due to the uncured material breach or bankruptcy of a party, or force majeure. If SymBio terminates the license agreement in these circumstances, its licenses to rigosertib will survive, subject to SymBio's milestone and royalty obligations, which SymBio may elect to defer and offset against any damages that may be determined to be due from us. In addition, we may terminate the license agreement in the event that SymBio brings a challenge against us in relation to the licensed patents, and SymBio may terminate the license agreement without cause by providing us with written notice a specified period of time in advance of termination.
The Leukemia and Lymphoma Society
In May 2010, we entered into a funding agreement with The Leukemia and Lymphoma Society, or LLS, to fund the development of rigosertib. Under the LLS funding agreement, we are obligated to use the funding received exclusively for the payment or reimbursement of the costs and expenses for clinical development activities for rigosertib. Under this agreement, we retain ownership and control of all intellectual property pertaining to works of authorship, inventions, know-how, information, data and proprietary material.
Under the LLS funding agreement, as amended, we received funding of $8.0 million from LLS through 2012. We have not received any funding from LLS in 2013 and we terminated the funding agreement effective as of March 2013. We are required to make specified payments to LLS, including payments payable upon execution of the first out-license; first approval for marketing by a regulatory body; completion of the first commercial sale of rigosertib; and achieving specified annual net sales levels of rigosertib. The extent of these payments and our obligations will depend on whether we out-license rights to develop or commercialize rigosertib to a third party, we commercialize rigosertib on our own or we combine with or are sold to another company. In addition, we will pay to LLS a single-digit percentage royalty of our net sales of rigosertib, if any. The sum of our payments to LLS is capped at three times the total funding received from LLS, or $24.0 million.
In addition, some of our obligations under the funding agreement will remain in effect until the completion of specified milestones and payments to LLS. Assuming the successful outcome of the development activities covered by the LLS funding agreement and our receipt of necessary regulatory approvals, we will be required to take commercially reasonable steps through March 2018 to advance the development of rigosertib in clinical trials and to bring rigosertib to practical application for MDS in a major market country, provided that we reasonably believe the product is safe and effective. We believe that we can satisfy our obligation by out-licensing rigosertib to, or partnering rigosertib with, a third party. We are required to report to LLS on our efforts and results with respect to continuing development of rigosertib. Our failure to perform these diligence obligations, even if we successfully achieve the specified development milestones, would require us to pay back to LLS the total amount of
the funding we received from them, unless an exception applies. If LLS were to claim that such failure occurred and we disagreed with such claim, the dispute would be settled through binding arbitration.
Preclinical Collaboration
We recently formed a joint venture with GVK Biosciences Private Limited, or GVK, a contract research organization based in India, to collaborate on the development of our ON 1231320 and ON 108600 preclinical programs through the submission of an IND with the FDA or conducting proof of concept studies. GVK will initially make a monetary capital contribution in exchange for a 10% interest in the joint venture and we will contribute a sub-license to the intellectual property related to the two programs in exchange for a 90% interest. GVK will be required to make additional capital contributions over time, subject to specified conditions, and its interest in the joint venture will increase to as much as 50%. At specified times, we will be entitled to buy back from GVK the rights to either of these two programs.
Intellectual Property
Patents and Proprietary Rights
We have access to intellectual property through our internal research, a licensing agreement with Temple University, or Temple, and a research agreement with the Mount Sinai School of Medicine, or Mount Sinai.
License Agreement with Temple University
In January 1999, we entered into a license agreement with Temple as subsequently amended, to obtain an exclusive, world-wide license to certain Temple patents and technical information to make, have made, use, sell, offer for sale and import several classes of novel compounds, including our three clinical-stage product candidates, rigosertib, ON 013105 and recilisib.
Under the terms of the license agreement, we paid Temple a non-refundable up-front payment, and are required to pay annual license maintenance fees, as well as a low single-digit percentage of net sales as a royalty. In addition, we agreed to pay Temple 25% of any consideration received from any sublicensee of the licensed Temple patents and technical information, which does not include any royalties on sales, funds received for research and development or proceeds from any equity or debt investment.
The license agreement with Temple can be terminated by mutual agreement or due to the material breach or bankruptcy of either party. We may terminate the license agreement for any reason by giving Temple prior written notice.
Research Agreement with Mount Sinai School of Medicine
In May 2010, we entered into a research agreement with Mount Sinai. This agreement is described in more detail under the caption "Certain Relationships and Related Party Transactions—Research Agreement."
Rigosertib Patents
We own or exclusively license 64 issued patents and 18 pending patent applications covering composition-of-matter, process, formulation and various indications for method-of-use for rigosertib filed worldwide, including five patents and three patent applications in the United States. The U.S. composition-of-matter patent for rigosertib, which we in-license pursuant to the license agreement with Temple, expires in 2026. The U.S. method of treatment patent for rigosertib, which we also in-license from Temple, expires in 2025.
ON 013105 Patents
We own or exclusively license eight issued patents and five pending patent applications covering composition-of-matter, process, formulation and various indications for method-of-use for ON 013105 filed worldwide, including one patent in the United States. The U.S. composition-of-matter patent for ON 013105 expires in 2025.
Recilisib Patents
We own or exclusively license 43 issued patents and 38 pending patent applications covering composition of matter, formulation and various indications for method-of-use for recilisib filed worldwide, including four patents and five patent applications in the United States. The U.S. composition-of-matter patent for recilisib expires in 2020.
General Considerations
As with other biotechnology and pharmaceutical companies, our ability to maintain and solidify a proprietary position for our product candidates will depend upon our success in obtaining effective patent claims and enforcing those claims once granted.
Our commercial success will depend in part upon not infringing upon the proprietary rights of third parties. It is uncertain whether the issuance of any third-party patent would require us to alter our development or commercial strategies, or our product candidates or processes, obtain licenses or cease certain activities. The biotechnology and pharmaceutical industries are characterized by extensive litigation regarding patents and other intellectual property rights. If a third party commences a patent infringement action against us, or our collaborators, it could consume significant financial and management resources, regardless of the merit of the claims or the outcome of the litigation.
The term of a patent that covers an FDA-approved drug may be eligible for patent term extension, which provides patent term restoration as compensation for the patent term lost during the FDA regulatory review process. The Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the drug is under regulatory review. Patent extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved drug may be extended. Similar provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved drug. In the future, if and when our pharmaceutical products receive FDA approval, we expect to apply for patent term extensions on patents covering those products.
Furthermore, we may be able to obtain extension of patent term by adjustment of the said term under the provisions of 35 U.S.C.§154 if the issue of an original patent is delayed due to the failure of the U.S. Patent and Trademark Office. For example, we have received adjustments of 1,139 days extension to the patent term for the rigosertib composition of matter patent (US 7,598,232), 1,155 days extension for the patent covering the process for making rigosertib (US 8,143,453) and 751 days extension for rigosertib formulation patent (US 8,063,109) under the provisions of 35 U.S.C. §154.
Many pharmaceutical companies, biotechnology companies and academic institutions are competing with us in the field of oncology and filing patent applications potentially relevant to our business. Even when a third party patent is identified, we may conclude upon a thorough analysis, that we do not infringe upon the patent or that the patent is invalid. If the third-party patent owner disagrees with our conclusion and we continue with the business activity in question, we may be subject to patent litigation. Alternatively, we might decide to initiate litigation in an attempt to have a court
declare the third-party patent invalid or non-infringed by our activity. In either scenario, patent litigation typically is costly and time-consuming, and the outcome can be favorable or unfavorable.
In addition to patents, we rely upon unpatented trade secrets, know-how and continuing technological innovation to develop and maintain a competitive position. We seek to protect our proprietary information, in part, through confidentiality agreements with our employees, collaborators, contractors and consultants, and invention assignment agreements with our employees. We also have agreements requiring assignment of inventions with selected consultants and collaborators. The confidentiality agreements are designed to protect our proprietary information and, in the case of agreements or clauses requiring invention assignment, to grant us ownership of technologies that are developed through a relationship with a third party.
Competition
The pharmaceutical industry is highly competitive and subject to rapid and significant technological change. While we believe that our development experience and scientific knowledge provide us with competitive advantages, we face competition from both large and small pharmaceutical and biotechnology companies. There are a number of pharmaceutical companies, biotechnology companies, public and private universities and research organizations actively engaged in the research and development of products that may compete with our products. Many of these companies are multinational pharmaceutical or biotechnology organizations, which are pursuing the development of, or are currently marketing, pharmaceuticals that target the key oncology indications or cellular pathways on which we are focused.
It is probable that the increasing incidence and prevalence of cancer will lead to many more companies seeking to develop products and therapies for the treatment of unmet needs in oncology. Many of our competitors have significantly greater financial, technical and human resources than we have. Many of our competitors also have a significant advantage with respect to experience in the discovery and development of product candidates, as well as obtaining FDA and other regulatory approvals of products and the commercialization of those products. We anticipate intense and increasing competition as new drugs enter the market and as more advanced technologies become available. Our success will be based in part on our ability to identify, develop and manage a portfolio of drugs that are safer and more effective than competing products in the treatment of cancer patients.
Myelodysplastic Syndromes
There are several ongoing clinical trials aimed at expanding the use of approved chemotherapeutic and immunomodulatory agents in higher risk MDS. Companies competing in this space include Eisai Inc. (decitabine alone and in combination with clofarabine), Celgene Corporation (azacitidine in combination with lenalidomide or vorinostat (Zolinza®)), Genentech, Inc. (erlotinib), Cell Therapeutics, Inc. (tosedostat in combination with decitabine or cytarabine) and Cyclacel Pharmaceuticals, Inc. (sapacitabine). To our knowledge, there are no Phase 3 trials other than our trial for rigosertib being conducted for higher risk MDS patients who have failed treatment with hypomethylating agents. In the lower risk MDS market, we face competition from a number of companies in early-stage clinical trials, such as Celgene Corporation (lenalidomide), Telik, Inc. (ezatiostat (Telintra®)), Spectrum Pharmaceuticals, Inc. (belinostat as monotherapy and in combination with bortezomib (Velcade®)), Astex (SG-110) and Array BioPharma Inc (ARRY-614).
Pancreatic Cancer
There are a number of ongoing clinical programs for the treatment of pancreatic cancer. Companies competing in this space include Celgene Corporation, Astellas Pharma, Inc. and Threshold Pharmaceuticals, Inc. Recently, the FOLFIRINOX chemotherapy regimen, combining fluorouracil,
leucovorin, irinotecan and oxaliplatin, showed survival improvement over gemcitabine alone in metastatic pancreatic cancer. However, patients receiving FOLFIRINOX reported higher frequency of toxicities including febrile neutropenia that render its use restricted to selected patients with good performance status. In addition, recent studies have shown that albumin-bound paclitaxel (Abraxane®) in combination with gemcitabine helped patients with advanced pancreatic cancer live a median of 1.8 months longer than those given chemotherapy alone. Gemcitabine and erlotinib are two FDA-approved therapies for this disease. Fluorouracil and mitomycin-C are also approved for the treatment of pancreatic cancer, but these drugs are rarely used as single agents.
Refractory Lymphomas
Chemotherapy and radiation therapy are the two principal forms of treatment for non-Hodgkin's lymphoma. Stem cell transplantation is also used to treat some subtypes of non-Hodgkin's lymphoma. Other forms of treatment are emerging, and some are already approved for specific forms of non-Hodgkin's lymphoma. Most patients with refractory or relapsed disease receive second-line therapy, in some cases followed by stem cell transplantation. A number of targeted therapies have been approved for MCL, including bortezomib as a second-line treatment and lenalidomide for patients whose disease relapsed or progressed after two prior therapies, one of which included bortezomib. In addition, there are multiple programs currently in late-stage clinical development for this disease. Ibrutinib, which is a Bruton's tyrosine kinase inhibitor being developed by Pharmacyclics Inc., is being tested in Phase 2 and 3 trials for MCL.
Acute Radiation Syndromes
Competitors developing products to address ARS include Soligenix, Inc., Cellerant Therapeutics, Inc., and Cleveland BioLabs, Inc. Each of these companies is working with the U.S. government to develop its products through federal contracts and grants.
Manufacturing
Our product candidates are synthetic small molecules. Manufacturing activities must comply with FDA current good manufacturing practices, or cGMP, regulations. We conduct our manufacturing activities under individual purchase orders with third-party contract manufacturers, or CMOs. We have in place quality agreements with our key CMOs and are negotiating supply agreements with them. We have also established an internal quality management organization, which audits and qualifies CMOs in the United States and abroad.
One of our CMOs is currently validating its manufacturing process to synthesize the rigosertib active pharmaceutical ingredient, which we believe will enable us to launch and commercialize rigosertib IV if and when marketing approval is obtained. Another CMO produces rigosertib IV for use in our ongoing clinical trials. A third CMO produces rigosertib Oral for use in our ongoing clinical trials. We believe that the manufacturing processes for the active pharmaceutical ingredient and finished drug products for rigosertib have been developed to adequately support future development and commercial demands. We believe that our existing suppliers of the rigosertib active pharmaceutical ingredient and drug products would be capable of providing sufficient quantities of the rigosertib active pharmaceutical ingredient and drug products to meet anticipated full-scale commercial demands.
The FDA regulates and inspects equipment, facilities and processes used in manufacturing pharmaceutical products prior to approval. If we fail to comply with applicable cGMP requirements and conditions of product approval, the FDA may seek sanctions, including fines, civil penalties, injunctions, suspension of manufacturing operations, operating restrictions, withdrawal of FDA approval, seizure or recall of products and criminal prosecution. Although we periodically monitor the
FDA compliance of our third-party CMOs, we cannot be certain that our present or future third-party CMOs will consistently comply with cGMP and other applicable FDA regulatory requirements.
Commercial Operations
We do not currently have an organization for the sales, marketing and distribution of pharmaceutical products. We may rely on licensing and co-promotion agreements with strategic partners for the commercialization of our products in the United States and other territories. If we choose to build a commercial infrastructure to support marketing in the United States, such commercial infrastructure could be expected to include a targeted, oncology sales force supported by sales management, internal sales support, an internal marketing group and distribution support. To develop the appropriate commercial infrastructure internally, we would have to invest financial and management resources, some of which would have to be deployed prior to any confirmation that rigosertib will be approved.
Government Regulation
As a pharmaceutical company that operates in the United States, we are subject to extensive regulation by the FDA, and other federal, state, and local regulatory agencies. The Federal Food, Drug, and Cosmetic Act, or the FDC Act, and its implementing regulations set forth, among other things, requirements for the research, testing, development, manufacture, quality control, safety, effectiveness, approval, labeling, storage, record keeping, reporting, distribution, import, export, advertising and promotion of our products. Although the discussion below focuses on regulation in the United States, we anticipate seeking approval for, and marketing of, our products in other countries. Generally, our activities in other countries will be subject to regulation that is similar in nature and scope as that imposed in the United States, although there can be important differences. Additionally, some significant aspects of regulation in Europe are addressed in a centralized way through the EMA, but country-specific regulation remains essential in many respects. The process of obtaining regulatory marketing approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
United States Government Regulation
The FDA is the main regulatory body that controls pharmaceuticals in the United States, and its regulatory authority is based in the FDC Act. Pharmaceutical products are also subject to other federal, state and local statutes. A failure to comply explicitly with any requirements during the product development, approval, or post-approval periods, may lead to administrative or judicial sanctions. These sanctions could include the imposition by the FDA or an institutional review board, or IRB, of a hold on clinical trials, refusal to approve pending marketing applications or supplements, withdrawal of approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution.
The steps required before a new drug may be marketed in the United States generally include:
Clinical Trials
An IND is a request for authorization from the FDA to administer an investigational drug product to humans. This authorization is required before interstate shipping and administration of any new drug product to humans that is not the subject of an approved NDA. A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has neither commented on nor questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin. Clinical trials involve the administration of the investigational drug to patients under the supervision of qualified investigators following GCPs, an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators and monitors. Clinical trials are conducted under protocols that detail the parameters to be used in monitoring safety, and the efficacy criteria to be evaluated. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to the FDA as part of the IND. The informed written consent of each participating subject is required. The clinical investigation of an investigational drug is generally divided into three phases. Although the phases are usually conducted sequentially, they may overlap or be combined. The three phases of an investigation are as follows:
consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible.
The decision to terminate development of an investigational drug product may be made by either a health authority body, such as the FDA or IRB/ethics committees, or by a company for various reasons. The FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. In some cases, clinical trials are overseen by an independent group of qualified experts organized by the trial sponsor, or the clinical monitoring board. This group provides authorization for whether or not a trial may move forward at designated check points. These decisions are based on the limited access to data from the ongoing trial. The suspension or termination of development can occur during any phase of clinical trials if it is determined that the participants or patients are being exposed to an unacceptable health risk. In addition, there are requirements for the registration of ongoing clinical trials of drugs on public registries and the disclosure of certain information pertaining to the trials as well as clinical trial results after completion.
A sponsor may be able to request an SPA, the purpose of which is to reach agreement with the FDA on the Phase 3 clinical trial protocol design and analysis that will form the primary basis of an efficacy claim. A sponsor meeting the regulatory criteria may make a specific request for an SPA and provide information regarding the design and size of the proposed clinical trial. An SPA request must be made before the proposed trial begins, and all open issues must be resolved before the trial begins. If a written agreement is reached, it will be documented and made part of the record. The agreement will be binding on the FDA and may not be changed by the sponsor or the FDA after the trial begins except with the written agreement of the sponsor and the FDA or if the FDA determines that a substantial scientific issue essential to determining the safety or efficacy of the product candidate was identified after the testing began. An SPA is not binding if new circumstances arise, and there is no guarantee that a study will ultimately be adequate to support an approval even if the studylosses is subject to an SPA. Rigosertib is being tested in several advanced stage clinical trials, including a pivotal Phase 3 trial under an SPA. Having an SPA agreement does not guarantee that a product will receive FDA approval.
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, detailed investigational drug product information is submitted to the FDA in the form of a NDA to request market approval for the product in specified indications.
New Drug Applications
In order to obtain approval to market a drug in the United States, a marketing application must be submitted to the FDA that provides data establishing the safety and effectiveness of the drug product for the proposed indication. The application includes all relevant data available from pertinent preclinical and clinical trials, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product's chemistry, manufacturing, controls and proposed labeling, among other things. Data can come from company-sponsored clinical trials intended to test the safety and effectiveness of a product, or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and effectiveness of the investigational drug product to the satisfaction of the FDA.
In most cases, the NDA must be accompanied by a substantial user fee (currently exceeding $1,958,000); there may be some instances in which the user fee is waived. The FDA will initially review the NDA for completeness before it accepts the NDA for filing. The FDA has 60 days from its receipt
of an NDA to determine whether the application will be accepted for filing based on the agency's threshold determination that it is sufficiently complete to permit substantive review. After the NDA submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of NDAs. Most such applications for standard review drug products are reviewed within ten to twelve months. The FDA can extend this review by three months to consider certain late-submitted information or information intended to clarify information already provided in the submission. The FDA reviews the NDA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP. The FDA may refer applications for novel drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. After the FDA evaluates the NDA and the manufacturing facilities, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If, or when, those deficiencies have been addressed to the FDA's satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require a risk evaluation and mitigation strategy, or REMS, to help ensure that the benefits of the drug outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the drug. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug's safety or efficacy. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented. An NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing NDAs.
Advertising and Promotion
The FDA and other federal regulatory agencies closely regulate the marketing and promotion of drugs through, among other things, standards and regulations for direct-to-consumer advertising, communications regarding unapproved uses, industry-sponsored scientific and educational activities, and promotional activities involving the Internet. A product cannot be commercially promoted before it is approved. After approval, product promotion can include only those claims relating to safety and effectiveness that are consistent with the labeling approved by the FDA. Healthcare providers are permitted to prescribe drugs for "off-label" uses—that is, uses not approved by the FDA and therefore not described in the drug's labeling—because the FDA does not regulate the practice of medicine. However, FDA regulations impose stringent restrictions on manufacturers' communications regarding off-label uses. Broadly speaking, a manufacturer may not promote a drug for off-label use, but may engage in non-promotional, balanced communication regarding off-label use under specified conditions. Failure to comply with applicable FDA requirements and restrictions in this area may subject a company to adverse publicity and enforcement action by the FDA, the DOJ, or the Office of the Inspector General of HHS, as well as state authorities. This could subject a company to a range of penalties that could have a significant commercial impact, including civil and criminal fines and agreements that materially restrict the manner in which a company promotes or distributes drug products.
Post-Approval Regulations
After regulatory approval of a drug is obtained, a company is required to comply with a number of post-approval requirements. For example, as a condition of approval of an NDA, the FDA may require post-marketing testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product's safety and effectiveness after commercialization. Regulatory approval of oncology products often requires that patients in clinical trials be followed for long periods to determine the overall survival benefit of the drug. In addition, as a holder of an approved NDA, a company would be required to report adverse reactions and production problems to the FDA, to provide updated safety and efficacy information, and to comply with requirements concerning advertising and promotional labeling for any of its products. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval to assure and preserve the long term stability of the drug or biological product. The FDA periodically inspects manufacturing facilities to assess compliance with cGMP, which imposes extensive procedural and substantive record keeping requirements. In addition, changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon a company and any third-party manufacturers that a company may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our product candidates. Future FDA and state inspections may identify compliance issues at our facilities or at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of previously unknown problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved NDA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action that could delay or prohibit further marketing.
Newly discovered or developed safety or effectiveness data may require changes to a product's approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Also, new government requirements, including
those resulting from new legislation, may be established, or the FDA's policies may change, which could delay or prevent regulatory approval of our products under development.
FDA Animal Efficacy Rule for Approval of Medical Countermeasures
Marketing approval by the FDA for new medical countermeasures in situations for which human efficacy testing is not feasible or ethical, such as for ARS, is based on the so-called "Animal Efficacy Rule." Under this rule, FDA can rely on the evidence from animal studies to provide substantial prediction of effectiveness of an agent in humans, when coupled with:
The Hatch-Waxman Amendments to the FDC Act
Orange Book Listing
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent whose claims cover the applicant's product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA's Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential generic competitors in support of approval of an abbreviated new drug application, or ANDA. An ANDA provides for marketing of a drug product that has the same active ingredients in the same strengths and dosage form as the listed drug and has been shown through bioequivalence testing to be therapeutically equivalent to the listed drug. Other than the requirement for bioequivalence testing, ANDA applicants are not required to conduct, or submit results of, pre-clinical or clinical tests to prove the safety or effectiveness of their drug product. Drugs approved in this way are commonly referred to as "generic equivalents" to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA's Orange Book. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new product. The ANDA applicant may also elect to submit a statement certifying that its proposed ANDA label does not contain (or carves out) any language regarding the patented method-of-use rather than certify to a listed method-of-use patent. If the applicant does not challenge the listed patents, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired.
A certification that the new product will not infringe the already approved product's listed patents, or that such patents are invalid, is called a Paragraph IV certification. If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response
to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit, or a decision in the infringement case that is favorable to the ANDA applicant.
The ANDA application also will not be approved until any applicable non-patent exclusivity listed in the Orange Book for the referenced product has expired.
Exclusivity
Upon NDA approval of a new chemical entity, or NCE, which is a drug that contains no active moiety that has been approved by the FDA in any other NDA, that drug receives five years of marketing exclusivity during which the FDA cannot receive any ANDA seeking approval of a generic version of that drug. Certain changes to a drug, such as the addition of a new indication to the package insert, are associated with a three-year period of exclusivity during which the FDA cannot approve an ANDA for a generic drug that includes the change.
An ANDA may be submitted one year before NCE exclusivity expires if a Paragraph IV certification is filed. If there is no listed patent in the Orange Book, there may not be a Paragraph IV certification, and, thus, no ANDA may be filed before the expiration of the exclusivity period.
Patent Term Extension
After NDA approval, owners of relevant drug patents may apply for up to a five year patent extension. The allowable patent term extension is calculated as half of the drug's testing phase—the time between IND application and NDA submission—and all of the review phase—the time between NDA submission and approval up to a maximum of five years. The time can be shortened if the FDA determines that the applicant did not pursue approval with due diligence. The total patent term after the extension may not exceed 14 years.
The Foreign Corrupt Practices Act
The Foreign Corrupt Practices Act, or FCPA, prohibits any U.S. individual or business from paying, offering, or authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the United States to comply with accounting provisions requiring the company to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations.
Europe and Other International Government Regulation
In addition to regulations in the United States, we will be subject to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Some countries outside of the United States have a similar process that requires the submission of a clinical trial application, or CTA, much like the IND prior to the commencement of human clinical trials. In Europe, for example, a CTA must be submitted to each country's national health authority and an independent ethics committee, much like the FDA and IRB, respectively. Once the CTA is approved in accordance with a country's requirements, clinical trial development may proceed.
To obtain regulatory approval to commercialize a new drug under European Union regulatory systems, we must submit a marketing authorization application, or MAA. The MAA is similar to the NDA, with the exception of, among other things, country-specific document requirements.
For other countries outside of the European Union, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
Compliance
During all phases of development (pre- and post-marketing), failure to comply with applicable regulatory requirements may result in administrative or judicial sanctions. These sanctions could include the FDA's imposition of a clinical hold on trials, refusal to approve pending applications, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, product detention or refusal to permit the import or export of products, injunctions, fines, civil penalties or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us.
Other Special Regulatory Procedures
Orphan Drug Designation
The FDA may grant Orphan Drug Designation to drugs intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the United States, or, if the disease or condition affects more than 200,000 individuals in the United States, there is no reasonable expectation that the cost of developing and making the drug would be recovered from sales in the United States. In the European Union, the EMA's Committee for Orphan Medicinal Products, or COMP, grants Orphan Drug Designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than five in 10,000 persons in the European Union community. Additionally, designation is granted for products intended for the diagnosis, prevention or treatment of a life- threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in the European Union would be sufficient to justify the necessary investment in developing the drug.
In the United States, Orphan Drug Designation entitles a party to financial incentives, such as opportunities for grant funding towards clinical trial costs, tax credits for certain research and user fee waivers under certain circumstances. In addition, if a product receives the first FDA approval for the indication for which it has orphan designation, the product is entitled to seven years of market exclusivity, which means the FDA may not approve any other application for the same drug for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority over the product with orphan exclusivity. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition.
In the European Union, Orphan Drug Designation also entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity is granted following drug approval. This period may be reduced to six years if the Orphan Drug Designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Orphan drug designation must be requested before submission of an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of the regulatory review and approval process.
Priority Review (United States) and Accelerated Review (European Union)
Based on results of the Phase 3 clinical trial(s) submitted in an NDA, upon the request of an applicant, a priority review designation may be granted to a product by the FDA, which sets the target date for FDA action on the application at six months from FDA filing, or eight months from the sponsor's submission. Priority review is given where preliminary estimates indicate that a product, if approved, has the potential to provide a safe and effective therapy where no satisfactory alternative therapy exists, or a significant improvement compared to marketed products is possible. If criteria are not met for priority review, the standard FDA review period is ten months from FDA filing, or 12 months from sponsor submission. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
Under the Centralized Procedure in the European Union, the maximum timeframe for the evaluation of a marketing authorization application is 210 days (excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP). Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest, defined by three cumulative criteria: the seriousness of the disease (e.g., heavy disabling or life-threatening diseases) to be treated; the absence or insufficiency of an appropriate alternative therapeutic approach; and anticipation of high therapeutic benefit. In this circumstance, EMA ensures that the opinion of the CHMP is given within 150 days.
Pediatric Information
Under the Pediatric Research Equity Act, or PREA, NDAs or supplements to NDAs must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may grant full or partial waivers, or deferrals, for submission of data. Unless otherwise required by regulation, PREA does not apply to any drug for an indication for which orphan designation has been granted.
The Best Pharmaceuticals for Children Act, or BPCA, provides NDA holders a six-month extension of any exclusivity—patent or non-patent—for a drug if certain conditions are met. Conditions for exclusivity include the FDA's determination that information relating to the use of a new drug in the pediatric population may produce health benefits in that population, the FDA making a written request for pediatric studies, and the applicant agreeing to perform, and reporting on, the requested studies within the statutory timeframe. Applications under the BPCA are treated as priority applications, with all of the benefits that designation confers.
Healthcare Reform
In March 2010, the President of the United States signed one of the most significant healthcare reform measures in decades. The Affordable Care Act, substantially changes the way healthcare will be financed by both governmental and private insurers, and significantly impacts the pharmaceutical industry. The Affordable Care Act will impact existing government healthcare programs and will result in the development of new programs. For example, the Affordable Care Act provides for Medicare payment for performance initiatives and improvements to the physician quality reporting system and feedback program.
Among the Affordable Care Act's provisions of importance to the pharmaceutical industry are the following:
The Affordable Care Act also establishes an Independent Payment Advisory Board, or IPAB, to reduce the per capita rate of growth in Medicare spending. Beginning in 2014, IPAB is mandated to propose changes in Medicare payments if it determines that the rate of growth of Medicare expenditures exceeds target growth rates. The IPAB has broad discretion to propose policies to reduce expenditures, which may have a negative impact on payment rates for pharmaceutical products. A proposal made by the IPAB is required to be implemented by the U.S. government's Centers for Medicare & Medicaid Services unless Congress adopts a proposal with savings greater than those proposed by the IPAB. IPAB proposals may impact payments for physician and free-standing services beginning in 2015 and for hospital services beginning in 2020.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. On August 2, 2011, the President signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend proposals in spending reductions to Congress. The Joint Select Committee did not achieve its targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation's automatic reductions to several government programs. These reductions include aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on our customers and accordingly, our financial operations.
We anticipate that the Affordable Care Act will result in additional downward pressure on coverage and the price that we receive for any approved product, and could seriously harm our business. Any reduction in reimbursement from Medicare and other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our products. In addition, it is possible that there will be further legislation or regulation that could harm our business, financial condition, and results of operations.
Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any drug products for which we obtain regulatory approval. In the United States and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of reimbursement from third-party payors. Third-party payors include government health administrative authorities, managed care providers, private health insurers and other organizations. The process for determining whether a payor will provide coverage for a drug product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the drug product. Third-party payors may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA-approved drugs for a particular indication. Third-party payors are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain FDA approvals. Our product candidates may not be considered medically necessary or cost-effective. A payor's decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Adequate third party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
In 2003, the U.S. government enacted legislation providing a partial prescription drug benefit for Medicare recipients, which became effective at the beginning of 2006. Government payment for some of the costs of prescription drugs may increase demand for any products for which we receive marketing approval. However, to obtain payments under this program, we would be required to sell products to Medicare recipients through prescription drug plans operating pursuant to this legislation. These plans will likely negotiate discounted prices for our products. Federal, state and local governments in the U.S. continue to consider legislation to limit the growth of healthcare costs, including the cost of prescription drugs. Future legislation could limit payments for pharmaceuticals such as the drug candidates that we are developing.
Different pricing and reimbursement schemes exist in other countries. In the European Union, governments influence the price of pharmaceutical products through their pricing and reimbursement
rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed upon. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on healthcare costs in general, particularly prescription drugs, has become more intense. As a result, increasingly high barriers are being erected to the entry of new products. The European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. We may face competition for our product candidates from lower-priced products in foreign countries that have placed price controls on pharmaceutical products. In addition, in some countries, cross-border imports from low-priced markets exert a commercial pressure on pricing within a country.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, an increasing emphasis on managed care in the United States has increased and will continue to increase the pressure on pharmaceutical pricing. Coverage policies and third party reimbursement rates may change at any time.
Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Other Healthcare Laws and Compliance Requirements
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, and formulary managers on the other. Although there are a number of statutory exemptions and regulatory safe harbors protecting some business arrangements from prosecution, the exemptions and safe harbors are drawn narrowly and practices that involve remuneration intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Our practices may not in all cases meet all of the criteria for safe harbor protection from federal Anti-Kickback Statute liability. The reach of the Anti-Kickback Statute was broadened by the Affordable Care Act, which, among other things, amends the intent requirement of the federal Anti-Kickback Statute. Pursuant to the statutory amendment, a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. In addition, the Affordable Care Act provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act (discussed below) or the civil monetary penalties statute, which imposes penalties against any person who is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent.
The federal False Claims Act prohibits any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government or knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal
government. As a result of a modification made by the Fraud Enforcement and Recovery Act of 2009, a claim includes "any request or demand" for money or property presented to the U.S. government. Recently, several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies' marketing of the product for unapproved, and thus non-reimbursable, uses. HIPAA created new federal criminal statutes that prohibit knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private third-party payors and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Also, many states have similar fraud and abuse statutes or regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
In addition, we may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by HITECH, and its implementing regulations, imposes requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA's privacy and security standards directly applicable to "business associates"—independent contractors or agents of covered entities that receive or obtain protected health information in connection with providing a service on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney's fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in specified circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
In the United States, our activities are potentially subject to additional regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services, other divisions of HHS (e.g., the Office of Inspector General), the DOJ and individual U.S. Attorney offices within the DOJ, and state and local governments. If a drug product is reimbursed by Medicare or Medicaid, pricing and rebate programs must comply with, as applicable, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 as well as the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990, or the OBRA, and the Veterans Health Care Act of 1992, each as amended. Among other things, the OBRA requires drug manufacturers to pay rebates on prescription drugs to state Medicaid programs and empowers states to negotiate rebates on pharmaceutical prices, which may result in prices for our future products that will likely be lower than the prices we might otherwise obtain. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. Under the Veterans Health Care Act, or VHCA, drug companies are required to offer some drugs at a reduced price to a number of federal agencies including the U.S. Department of Veterans Affairs and DoD, the Public Health Service and some private Public Health Service designated entities in order to participate in other federal funding programs including Medicaid. Recent legislative changes require that discounted prices be offered for specified DoD purchases for its TRICARE program via a rebate system. Participation under the VHCA requires submission of pricing data and calculation of discounts and rebates pursuant to complex statutory formulas, as well as the entry into government procurement contracts governed by the Federal Acquisition Regulation.
Because of the breadth of these laws and the narrowness of available statutory and regulatory exemptions, it is possible that some of our business activities could be subject to challenge under one or more of such laws. If our operations are found to be in violation of any of the federal and state laws described above or any other governmental regulations that apply to us, we may be subject to penalties,
including criminal and significant civil monetary penalties, damages, fines, imprisonment, exclusion from participation in government programs, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre-marketing product approvals, private "qui tam" actions brought by individual whistleblowers in the name of the government or refusal to allow us to enter into supply contracts, including government contracts, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. To the extent that any of our products are sold in a foreign country, we may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws, and implementation of corporate compliance programs and reporting of payments or transfers of value to healthcare professionals.
In order to distribute products commercially, we must comply with state laws that require the registration of manufacturers and wholesale distributors of pharmaceutical products in a state, including, in some states, manufacturers and distributors who ship products into the state even if such manufacturers or distributors have no place of business within the state. Some states also impose requirements on manufacturers and distributors to establish the pedigree of product in the chain of distribution, including some states that require manufacturers and others to adopt new technology capable of tracking and tracing product as it moves through the distribution chain. Several states have enacted legislation requiring pharmaceutical companies to, among other things, establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities, and/ or register their sales representatives, as well as to prohibit pharmacies and other healthcare entities from providing specified physician prescribing data to pharmaceutical companies for use in sales and marketing, and to prohibit other specified sales and marketing practices. All of our activities are potentially subject to federal and state consumer protection and unfair competition laws.
Employees
As of June 1, 2013, we had 56 employees, of whom 17 hold Ph.D. degrees and five hold M.D. degrees. We have no collective bargaining agreements with our employees and have not experienced any work stoppages. We believe that relations with our employees are good.
Facilities
Our corporate headquarters and research facilities are located in Newtown, Pennsylvania, where we lease an aggregate of approximately 9,500 square feet of office and laboratory space, pursuant to lease agreements, the terms of which expire in March 2014 and September 2013, respectively. We have a second office located in Pennington, New Jersey, where we lease an aggregate of approximately 5,200 square feet of office space pursuant to lease agreements, the terms of which expire in February 2015 and October 2014, respectively. This facility houses our clinical development, clinical operations, regulatory and commercial personnel.
We believe that our existing facilities are adequate for our near-term needs. When our leases expire, we may exercise renewal options or look for additional or alternate space for our operations. We believe that suitable additional or alternative space would be available if required in the future on commercially reasonable terms.
Legal Proceedings
We are not a party to any legal proceedings.
Executive Officers, Directors and Other Significant Employees
The following table sets forth information regarding our executive officers, the directors to be serving following the listing of our common stock on the NASDAQ Global Market and other significant employees, including their respective ages as of June 1, 2013:
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
Ramesh Kumar, Ph.D. Dr. Kumar is one of our co-founders, and is currently our President and Chief Executive Officer, a position he has held since 1998, as well as a member of our board of directors. Prior to founding our company, Dr. Kumar held positions in research and development or management at Princeton University, Bristol-Myers Squibb Company, or Bristol-Myers Squibb, DNX Corp. (later Nextran Corp., a subsidiary of Baxter International Inc.) and Kimeragen, Inc. (later ValiGen Inc.), a genomics company, where he was President of the Genomics and Transgenics Division. Dr. Kumar received his Ph.D. in Molecular Biology from the University of Illinois, Chicago, and trained at the National Cancer Institute. Additionally, Dr. Kumar received his B.Sc. and M.Sc., both with honors, in Microbiology from Panjab University.
Our board of directors believes Dr. Kumar's perspective and experience as our co-founder, President and Chief Executive Officer, as well as his depth of operating and senior management experience in our industry, provide him with the qualifications and skills to serve as a director.
Michael B. Hoffman. Mr. Hoffman has served as Chairman of our board of directors since 2006 and as a member of our board of directors since December 2002. Since 2003, Mr. Hoffman has been a managing director of Riverstone Holdings LLC, or Riverstone, where he is principally responsible for investments in power and renewable energy. Before joining Riverstone, Mr. Hoffman was senior managing director and head of the mergers and acquisitions advisory business of The Blackstone Group L.P., or Blackstone, where he also served on the firm's principal group investment committee as well as its executive committee. Prior to joining Blackstone, Mr. Hoffman was managing director and
co-head of the mergers and acquisitions department at Smith Barney, Harris Upham & Co. Mr. Hoffman received his Bachelor's and Master's Degrees from Northwestern University and his M.B.A. from the Harvard Business School.
Our board of directors believes Mr. Hoffman's perspective and experience as an investor, as well as his educational background, provide him with the qualifications and skills to serve as a director.
Thomas McKearn, M.D., Ph.D. Dr. McKearn has served as our President, Research & Development since September 2012. Prior to joining us, Dr. McKearn served as Vice President, Medical Affairs and then as Vice President, Strategic Clinical Affairs at Agennix AG (formerly GPC Biotech GP), a biopharmaceutical company, from April 2002 to August 2012. Prior to joining Agennix AG, Dr. McKearn held several executive positions both in biotech and pharmaceutical organizations, including Executive Director of Strategic Science & Medicine at Bristol-Myers Squibb. Dr. McKearn was a founder of Cytogen Corporation in 1981 and later served as its Chief Executive Officer. He has served as a director of Advaxis, Inc., a publicly held biotechnology company focused on oncology, since 2004. Dr. McKearn has served on the faculty of the Medical School at the University of Pennsylvania. Dr. McKearn received his medical, graduate, and post-graduate training at the University of Chicago.
François E. Wilhelm, M.D., Ph.D. Dr. Wilhelm has served as our Chief Medical Officer and Senior Vice President since May 2008. Before joining us, Dr. Wilhelm held a variety of clinical development positions with several pharmaceutical and biotechnology companies, including Hoffmann-La Roche Ltd., Fujisawa Healthcare Inc., Pfizer Inc., The Procter & Gamble Company, Akros Pharma Inc. and Johnson and Johnson. Dr. Wilhelm is Board Certified in Rheumatology, receiving his medical degree from Paris University Medical School, his Ph.D. in Endocrinology and a Master's degree in Biostatistics, both from the Paris Sciences University.
Ajay Bansal. Mr. Bansal has served as our Chief Financial Officer and as a member of our board of directors since March 2013. He intends to resign from our board of directors prior to consummation of this offering. From May 2010 to March 2013, Mr. Bansal served as Chief Financial Officer of Complete Genomics Incorporated, a life sciences company. From June 2009 to January 2010, Mr. Bansal served as Chief Financial Officer of Lexicon Pharmaceuticals, Inc., a biopharmaceutical company. From October 2008 to June 2009 and from February 2010 to April 2010, Mr. Bansal was a consultant. From March 2006 to October 2008, Mr. Bansal served as Chief Financial Officer of Tercica, Inc., a biopharmaceutical company. From February 2003 to January 2006, Mr. Bansal served as Chief Financial Officer of Nektar Therapeutics, a biopharmaceutical company. Prior to joining Nektar Therapeutics, Mr. Bansal spent more than 15 years as a management consultant at Arthur D. Little, Inc., McKinsey & Company, Inc. and ZS Associates, Inc., in management roles at Novartis Corporation, a pharmaceuticals company, at Mehta Partners, a financial advisory firm, and at Capital One, a bank holding company. Mr. Bansal received a B.S. in Mechanical Engineering from the Indian Institute of Technology (Delhi) and an M.S. in Operations Research and an M.B.A. from Northwestern University.
Manoj Maniar, Ph.D. Dr. Maniar has served as our Senior Vice President, Product Development since August 2005. Prior to joining us, Dr. Maniar was with SRI International, Inc., a nonprofit research institute, where he served as Senior Director, Formulations and Drug Delivery. Dr. Maniar received his B.S. in Pharmacy from Bombay College of Pharmacy and his Ph.D. in Pharmaceutics from the University of Connecticut.
Henry S. Bienen, Ph.D. Dr. Bienen has served as a member of our board of directors since May 2009. He currently serves as the chairman of Rasmussen College, has served as the president emeritus of Northwestern University since 2009 and served as the president of Northwestern University from 1995 to 2009. Dr. Bienen was the James S. McDonnell Distinguished University Professor and Dean of the Woodrow Wilson School of Public and International Affairs at Princeton University prior to his
appointment at Northwestern. Dr. Bienen began his association with Princeton University in 1966, advancing from assistant professor to professor of politics and international affairs, and was then appointed the William Stewart Tod Professor of Politics and International Affairs in 1981 and the James S. McDonnell Distinguished University Professor in 1985. Dr. Bienen has served as a director of Gleacher & Company, a publicly held investment banking firm, since May 2010, and the Grosvenor Registered Multi Strategy Fund (TI 1), LLC, the Grosvenor Registered Multi Strategy Fund (TI 2), LLC, the Grosvenor Registered Multi Strategy Fund (TE), LLC and the Grosvenor Registered Multi Strategy Master Fund, LLC since April 2011. Dr. Bienen previously served on the boards of directors of The Bear Stearns Companies Inc. until its purchase by JP Morgan Chase & Co. in 2008, and SPSS Inc. from 2007 until 2009, when the company was sold to IBM Corporation. Dr. Bienen received his Bachelor's Degree with honors from Cornell University and both his Master's Degree and Ph.D., from the University of Chicago.
Our board of directors believes Dr. Bienen's perspective and experience as a director of a public company, as well as his educational background, provide him with the qualifications and skills to serve as a director.
Viren Mehta. Dr. Mehta has served as a member of our board of directors since February 2004. Dr. Mehta has been a managing member of Mehta Partners since 1997. Mehta Partners provides strategic advisory services to the biotechnology and pharmaceutical companies worldwide. Prior to founding Mehta Partners, Dr. Mehta co-founded Mehta and Isaly in 1989, and prior to that was a part of the strategic planning team of the International Division at Merck & Co., or Merck. Dr. Mehta earned a Doctor of Pharmacy at the University of Southern California, and an M.B.A. from the Anderson School of Business at the University of California, Los Angeles.
Our board of directors believes Dr. Mehta's perspective and experience in the life sciences industry as a biopharma fund manager, fund consultant and a strategic advisor to senior managers in the biopharma industry, as well as his educational background, provide him with the qualifications and skills to serve as a director.
E. Premkumar Reddy, Ph.D. Dr. Reddy is one of our scientific founders and has served as a member of our board of directors since February 1999. Since March 2010, Dr. Reddy has served as a Professor and Director of the Experimental Cancer Therapeutics Program at Mount Sinai School of Medicine, or Mount Sinai. From 1992 to February 2010, Dr. Reddy served as a Professor and Director of the Fels Institute for Cancer Research of Temple University. He was the founder and co-editor of the international journal of cancer research, Oncogene, published by Nature Publishing Group. Dr. Reddy received his B.Sc., M.Sc. and Ph.D. from Osmania University.
Our board of directors believes Dr. Reddy's perspective and experience as our co-founder, his educational background, as well as his experience in research and product development, provide him with the qualifications and skills to serve as a director.
James Altland. Mr. Altland has served as our Senior Vice President, Finance and Corporate Development since August 2007. Prior to joining us, Mr. Altland was a Partner of the Philadelphia office of Tatum LLC, a professional services firm, where he was Practice Leader for the life science segment. Mr. Altland received a B.S. in Accounting from the University of Akron and is a retired Certified Public Accountant.
Scott T. Megaffin. Mr. Megaffin has served as our Senior Vice President, Commercial Development since August 2010. Prior to that, he served as a commercial consultant for us from December 2009 to August 2010. From September 2009 to December 2009, Mr. Megaffin was a consultant. From July 2008 to September 2009, Mr. Megaffin was Vice President, Pain Marketing of Cephalon, Inc. From June 2006 to July 2008, Mr. Megaffin was the Vice President of Marketing of Adolor Corporation. Mr. Megaffin received his B.S. in Biology from Pittsburg State University.limitations.
David Stephon.Information Reporting and Backup Withholding Mr. Stephon has served as our Senior Vice President, Quality Management since February 2011. Prior to joining us, Mr. Stephon was the head of a pharmaceutical quality consulting business, David M. Stephon Consulting, LLC, from July 2010 to February 2011. Previously, Mr. Stephon was Vice President, Quality Management at Adolor Corporation from October 2002 to July 2010. Mr. Stephon received a M.S. in Chemistry from Lehigh University, and a B.S. in Chemistry from Muhlenberg College.
Clinical Advisory Board Members
We have established a clinical advisory board and we regularly seek advice and inputYou may be subject to information reporting and/or backup withholding with respect to the gross proceeds from these experienced clinical leaders on matters related to our research and development programs. The membersthe disposition of our clinical advisory board consist of experts across a range of key disciplines relevant to our programs. We intend to continue to leverage the broad expertise of our advisors by seeking their counsel on important topics relating to our drug discovery and development programs. Some members of our clinical advisory board have entered into consulting agreements with us covering their respective confidentiality, non-disclosure and proprietary rights matters and own or have ownedWarrants, shares of our common stock acquired through the exercise of Subscription Rights or optionsthrough the exercise of Warrants, or dividend payments. Backup withholding (currently at the rate of 28%) may apply under certain circumstances if you (1) fail to purchasefurnish your social security or other taxpayer identification number, or TIN, (2) furnish an incorrect TIN, (3) fail to report interest or dividends properly or (4) fail to provide a certified statement, signed under penalty of perjury, that the TIN provided is correct, that you are not subject to backup withholding and that you are a U.S. person for U.S. federal income tax purposes on IRS Form W-9. Any amount withheld from a payment under the backup withholding rules is allowable as a credit against (and may entitle you to a refund with respect to) your U.S. federal income tax liability, provided that the required information is timely furnished to the IRS. Certain persons are exempt from information reporting and backup withholding, including corporations and certain financial institutions, provided that they demonstrate this fact, if requested. You are urged to consult your own tax advisor as to your qualification for exemption from backup withholding and the procedure for obtaining such exemption.
Tax Consequences to Non-U.S. Holders
Taxation of the Subscription Rights
Receipt, Exercise and Expiration of the Subscription Rights
The discussion assumes that the receipt of Subscription Rights will be treated as a non-taxable distribution. See "Tax Consequences to U.S. Holders—Taxation of Subscription Rights—Receipt of Subscription Rights" above.
Exercise and Expiration of Warrants and Certain Adjustments to Warrants
Exercise of Warrants
In general, a Non-U.S. Holder will not recognize gain or loss for U.S. federal income tax purposes upon exercise of a Warrant, except to the extent the Non-U.S. Holder receives a cash payment for any such fractional share that would otherwise have been issuable upon exercise of the Warrant, which will be treated as a sale subject to the rules described under "Sale or Other Disposition of Common Stock or Warrants" below.
Expiration of Warrants
In general, a Non-U.S. Holder will not be able to utilize a loss recognized upon expiration of a Warrant against the Non-U.S. Holder's U.S. federal income tax liability unless the loss is effectively connected with the Non-U.S. Holder's conduct of a trade or business within the United States (and, if an income tax treaty so provides, is attributable to a permanent establishment in the United States) or is treated as a U.S.-source loss and the Non-U.S. Holder is present 183 days or more in the taxable year of disposition and certain other conditions are met.
Certain Adjustments to the Warrants
Under Section 305 of the Code, an adjustment to the number of common shares that will be issued on the exercise of the Warrants, or an adjustment to the exercise price of the Warrants, may be treated as a constructive distribution to a Non-U.S. Holder of the Warrants if, and to the extent that, such adjustment has the effect of increasing such Non-U.S. Holder's proportionate interest in our common stock."earnings and profits" or assets, depending on the circumstances of such adjustment (for example, if such adjustment is to compensate for a distribution of cash or other property to our shareholders). Adjustments to the exercise price of Warrants made pursuant to a bona fide reasonable adjustment
Allformula that has the effect of preventing dilution of the clinical advisors are employedinterest of the holders of the Warrants should generally not be considered to result in a constructive distribution. Any such constructive distribution would be taxable whether or not there is an actual distribution of cash or other property. See the more detailed discussion of the rules applicable to distributions made by or have consulting arrangements with other entities and devote only a small portionus under the heading "—Taxation of their time to us. Our current advisors are:
Table of ContentsDistributions on Common Stock" below.
Scientific Advisory BoardTaxation of Distributions on Common Stock
We have also established a scientific advisory board. We regularly seek advice and input from these experienced scientific leaders on matters relatedAny distributions of cash or property (including any adjustments to the Warrants described in the immediately preceding paragraph) made with respect to our research and development programs. The memberscommon stock generally will be subject to withholding tax to the extent paid out of our scientific advisory board consistcurrent or accumulated earnings and profits as determined for U.S. federal income tax purposes, if any, at a rate of experts across30% (or a rangelower rate prescribed by an applicable income tax treaty). In order to obtain a reduced withholding tax rate, if applicable, you will be required to provide a properly completed IRS Form W-8BEN or IRS Form W-8BEN-E, as applicable, certifying your entitlement to benefits under a treaty. In addition, you will not be subject to withholding tax if you provide an IRS Form W-8ECI certifying that the distributions are effectively connected with your conduct of key disciplines relevanta trade or business within the United States (and, if an applicable income tax treaty so provides, are attributable to our programsa permanent establishment within the United States); instead, you generally will be subject to U.S. federal income tax, net of certain deductions, with respect to such income at the same rates applicable to U.S. persons. If you are a corporation, a "branch profits tax" of 30% (or a lower rate prescribed by an applicable income tax treaty) also may apply to such effectively connected income.
Non-U.S. Holders may be required to periodically update their IRS Forms W-8.
Any distribution will also be subject to the discussion below under the heading "FATCA."
Sale or Other Disposition of Our Common Stock or Warrants
Subject to the discussion below regarding backup withholding and science. We intendFATCA, you generally will not be subject to continue to leverage the broad expertiseU.S. federal income tax on any gain realized on a sale or other disposition of our advisors by seeking their counsel on important topics relating to our drug discovery and development programs. Some members of our scientific advisory board have entered into consulting agreements with us covering their respective confidentiality, non-disclosure and proprietary rights matters and own or have owned shares of our common stock or optionsWarrants unless:
Gain that is effectively connected with your conduct of a trade or business within the United States (and, if an applicable income tax treaty so provides, is attributable to a permanent establishment within the United States) generally will be subject to U.S. federal income tax, net of certain deductions, at the same rates applicable to U.S. persons. If you are a corporation, a "branch profits tax" of 30% (or a lower rate prescribed in an applicable income tax treaty) also may apply to such effectively connected gain.
AllA domestic corporation is treated as a USRPHC if the fair market value of its United States real property interests equals or exceeds 50% of the scientific advisors are employed by or have consulting arrangements with other entities and devote only a small portionsum of their time to us. Our current advisors are:
Board Composition
Our board(1) the fair market value of directors currently consists of ten members, five of whom intend to resign prior to consummation of this offering. We expect that upon the listing of our common stock on the NASDAQ Global Market, our board of directors will consist of members. Prior to the listing of our common stock on the NASDAQ Global Market, we intend to appoint and to our board of directors and they have consented to so serve. Our ninth amended and restated certificate of incorporation currently in effect provides that our board of directors shall consist of ten directors. Our tenth amended and restated certificate of incorporation will provide that our board of directors will consist of not less than three nor more than 11 directors, as such number of directors may from time to time be fixed by our board of directors. Each director shall be elected to the board for a one-year term, to serve until the election and qualification of successor directors at the annual meeting of stockholders, or until the director's earlier removal, resignation or death.
Board Committees
Upon the listing of our common stock on the NASDAQ Global Market, our board of directors will have a standing audit committee, compensation committee and nominating and corporate governance committee. The members of our audit committee will consist of , and , with serving as chairman. The members of our compensation committee will consist of , and , with serving as chairman. The members of our nominating and corporate governance committee will consist of , and , with serving as chairman.its United States
Our boardreal property interests, (2) the fair market value of directors has undertakenits non-United States real property interests and (3) the fair market value of any other of its assets which are used or held for use in a review of the independence of our directorstrade or business. We believe that we are not currently, and has determined that all directors except are independenthave not been within the meaningrelevant testing period, a USRPHC. However, no assurance can be given that we will not become a USRPHC in the future. If we are a USRPHC or become a USRPHC in the future, a Non-U.S. Holder may still not be subject to U.S. federal income tax on a sale or other disposition if an exception for 5% or less shareholders applies. You are urged to consult your own tax advisor regarding the U.S. federal income tax considerations that could result if we are, or become, a USRPHC and with respect to the exception for 5% or less shareholders.
Information Reporting and Backup Withholding
Distributions on our common stock and the amount of Section 5605(a)(2)tax withheld, if any, with respect to such distributions will generally be subject to information reporting. If you comply with certification procedures to establish that you are not a United States person, additional information reporting and backup withholding should not generally apply to distributions on our common stock and information reporting and backup withholding should not generally apply to the proceeds from a sale or other disposition of the NASDAQ Stock Market listing rules and Rule 10A-3 under the Securities Act and that and meet the additional test for independence for audit committee members imposed by SEC regulations and Section 5605(c)(2)(A) of the NASDAQ Stock Market listing rules. The NASDAQ Stock Market listing rules require that each committee of our board of directors has at least one independent director on the listing dateWarrants or shares of our common stock. Generally, a Non-U.S. Holder will comply with such procedures if it provides a properly executed IRS Form W-8BEN or W-8BEN-E, as applicable, (or other applicable IRS Form W-8) or otherwise meets documentary evidence requirements for establishing that it is a Non-U.S. Holder, or otherwise establishes an exemption. The amount of any backup withholding will generally be allowed as a refund or credit against your U.S. federal income tax liability, provided that the required information is timely furnished to the IRS.
FATCA
Payments of dividends on our common stock hasto a majority of independent directors no later than 90 days after such date and be fully independent within one year after that date. The composition of our audit, compensation and nominating and corporate governance committees will satisfy these independence requirements in accordance with the phase-in schedule allowed by the NASDAQ Global Market.
Audit Committee
The primary purpose of our audit committeeNon-U.S. Holder will be subject to assista 30% withholding tax if the boardNon-U.S. Holder fails to provide the withholding agent with documentation sufficient to show that it is compliant with FATCA. Generally such documentation is provided on an executed and properly completed IRS Form W-8BEN or IRS Form W-8BEN-E, as applicable. If dividends are subject to the 30% withholding tax under FATCA, they will not be subject to the 30% withholding tax described above under "Tax Consequences to Non-U.S. Holders—Taxation of directorsDistributions on Common Stock." Starting in the oversight2019, payments of the integrity of our accounting and financial reporting process, the audits of our consolidated financial statements, and our compliance with legal and regulatory requirements. The functions of our audit committee will include, among other things:
With respect to reviewing and approving related-party transactions, our audit committee will review related-party transactions for potential conflicts of interests or other improprieties. Under SEC rules, related-party transactions are those transactions to which we are or may be a party in which the amount involved exceeds $120,000, and in which any of our directors or executive officers or any other related person had or will have a direct or indirect material interest, excluding, among other things, compensation arrangements with respect to employment and board membership. Our audit committee could approve a related-party transaction if it determines that the transaction is in our best interests. Our directors will be required to disclose to this committee or the full board of directors any potential conflict of interest, or personal interest in a transaction that our board is considering. Our executive
officers will be required to disclose any related-party transaction to the audit committee. We also plan to poll our directors on an annual basis with respect to related-party transactions and their service as an officer or director of other entities. Any director involved in a related-party transaction that is being reviewed or approved must recuse himself or herself from participation in any related deliberation or decision. Whenever possible, the transaction should be approved in advance and if not approved in advance, must be submitted for ratification as promptly as practical.
The financial literacy requirements of the SEC require that each member of our audit committee be able to read and understand fundamental financial statements. In addition, at least one member of our audit committee must qualify as an audit committee financial expert, as defined in Item 407(d)(5) of Regulation S-K promulgated under the Securities Act, and have financial sophistication in accordance with the NASDAQ Stock Market listing rules. Our board of directors has determined that qualifies as an audit committee financial expert.
Both our independent registered public accounting firm and management periodically will meet privately with our audit committee.
In connection with this offering, our board of directors intends to adopt a charter for the audit committee that complies with current federal and NASDAQ Stock Market rules relating to corporate governance. The charter will be available on our website athttp://www.onconova.com.
Compensation Committee
The primary purpose of our compensation committee will be to assist our board of directors in exercising its responsibilities relating to compensation of our executive officers and employees and to administer our equity compensation and other benefit plans. In carrying out these responsibilities, this committee will review all components of executive officer and employee compensation for consistency with its compensation philosophy, as in effect from time to time. The functions of our compensation committee will include, among other things:
In connection with this offering, our board of directors intends to adopt a charter for the compensation committee that complies with current federal and NASDAQ Stock Market rules relating to corporate governance. The charter will be available on our website athttp://www.onconova.com.
Nominating and Corporate Governance Committee
The primary purpose of our nominating and corporate governance committee will be to assist our board of directors in promoting the best interest of our company and our stockholders through the implementation of sound corporate governance principles and practices. The functions of our nominating and corporate governance committee will include, among other things:
In connection with this offering, our board of directors intends to adopt a charter for the nominating and corporate governance committee that complies with current federal and NASDAQ Stock Market rules relating to corporate governance. The charter will be available on our website athttp://www.onconova.com.
Code of Conduct for Employees, Executive Officers and Directors
In connection with the consummation of this offering, we plan to adopt a Code of Conduct applicable to all of our employees, executive officers and directors. Following the consummation of this offering, the Code of Conduct will be available on our website athttp://www.onconova.com. The audit committee of our board of directors will be responsible for overseeing the Code of Conduct and must approve any waivers of the Code of Conduct for employees, executive officers or directors. We expect that any amendments to the Code of Conduct, or any waivers of its requirements, will be disclosed on our website.
Compensation Committee Interlocks and Insider Participation
No member of our compensation committee has ever been an executive officer or employee of ours. None of our officers currently serves, or has served during the last completed year, on the board of directors, compensation committee or other committee serving an equivalent function, of any other entity that has one or more officers serving as a member of our board of directors or compensation committee.
EXECUTIVE AND DIRECTOR COMPENSATION
Summary Compensation Table
The following table sets forth information for the fiscal year ended December 31, 2012 concerning compensation of our principal executive officer and our three other executive officers who were serving as executive officers as of December 31, 2012. We refer to these four executives as our named executive officers.
Name and Principal Position | Year | Salary ($) | Bonus ($)(1) | Option Awards ($)(2) | All Other Compensation(3) ($) | Total ($) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Ramesh Kumar, Ph.D. | 2012 | 408,595 | 163,438 | — | 9,407 | 581,440 | |||||||||||||
President and Chief Executive Officer | |||||||||||||||||||
Francois Wilhelm, M.D., Ph.D. | 2012 | 374,587 | 112,376 | — | 10,167 | 497,130 | |||||||||||||
Chief Medical Officer and Senior Vice President | |||||||||||||||||||
Manoj Maniar, Ph.D. | 2012 | 326,795 | 98,039 | — | 9,422 | 434,256 | |||||||||||||
Senior Vice President of Development | |||||||||||||||||||
Thomas McKearn, M.D., Ph.D. | 2012 | 68,265 | 23,917 | — | 539 | 92,721 | |||||||||||||
President of Research and Development(5) | |||||||||||||||||||
| | | |||
|---|---|---|---|---|
December 31, 2012 Fair Value | ||||
Ramesh Kumar, Ph.D. | $ | 1,832,568 | ||
Francois Wilhelm, M.D., Ph.D. | 1,103,460 | |||
Manoj Maniar, Ph.D. | 634,528 | |||
Thomas McKearn, M.D., Ph.D. | 5,133 | |||
Employment Agreements
We have entered into employment agreements with our all of our named executive officers. The following is a summary of the material terms of each employment agreement. For complete terms, please see the respective employment agreements attached as exhibits to the registration statement of which this prospectus forms a part.
Ramesh Kumar, Ph.D.
We entered into an employment agreement with Dr. Kumar on April 1, 2007. The employment agreement provided for an initial term of four years, unless extended by mutual agreement of the parties. We and Dr. Kumar have mutually agreed to extend the term until March 31, 2015, unless sooner terminated in accordance with the terms of the agreement.
The employment agreement provided for an initial base salary of $299,076 and an annual bonus of up to 35% of such base salary, payable upon our achievement of revenue or profit objectives, specific business plan goals or other performance milestones mutually agreed to by Dr. Kumar and our board of directors, provided that Dr. Kumar remain employed by us throughout the performance year. The bonus may be paid in the form of cash, stock options, sharesexchange of our common stock or a combination thereof, at our compensation committee's discretion. Dr. Kumarother securities may also be entitledsubject to additional compensation in recognitionFATCA withholding absent proof of extraordinary contributions, at the sole discretion of our compensation committee.
Dr. Kumar is entitled to participate in all of our employee benefit plans and programs that are made generally available from time to time to our executive officers and is entitled to vacation benefits. Pursuant to his employment agreement, Dr. Kumar is entitled to term life insurance coverage in a face amount of $300,000, a reasonable transportation allowance if we relocate our research facility more than 40 miles from its present location, and up to $10,000 annually for educational programs related to the performance of his duties. If Dr. Kumar dies during his employment, we will be entitled to a $1 million death benefit under a "key man" life insurance policy. Dr. Kumar's employment agreement contains non-solicitation, non-competition, confidentiality and inventions assignment provisions that, among other things, prevent him from competing with us during the term of his employment and for a specified time thereafter.
If Dr. Kumar's employment is terminated due to his death, disability, by us for "cause" or by Dr. Kumar without "good reason" during the term of his employment agreement, we will pay to Dr. Kumar or his spouse or estate the balance of his accrued and unpaid salary, unreimbursed expenses, and unused accrued vacation time through the termination date.
If Dr. Kumar's employment is terminated by us without "cause" or by Dr. Kumar for "good reason," Dr. Kumar will be entitled to receive bi-weekly payments equal to his then-current bi-weekly base salary commencing on the date written notice of a termination without "cause" is delivered to Dr. Kumar or the date of termination for "good reason" and ending on the earlier of the date on which the term of the employment agreement would otherwise expire or the date Dr. Kumar accepts comparable employment, provided that if Dr. Kumar accepts such comparable employment within one year of the commencement of such payments, he shall continue to receive such payments until the earlier of the date on which the term of the employment agreement would otherwise expire or one year from such commencement. All incentive stock options that are unvested as of the date of such termination would fully vest as of the date of termination and would remain exercisable for three months from the date of termination.
Francois Wilhelm, M.D., Ph.D., Manoj Maniar, Ph.D. and Thomas McKearn, M.D., Ph.D.
We entered into an employment agreement with Dr. Wilhelm on April 17, 2008. The original employment agreement provided for a term of two years, unless extended by mutual agreement of the parties, with an automatic renewal for successive one-year periods unless written notification is provided by either party to the other at least ten business daysFATCA compliance prior to the expiration of the term. The current term has been extended through May 5, 2014, unless sooner terminated in accordance with the terms of the agreement.
We entered into an employment agreement with Dr. Maniar on January 1, 2007. The original employment agreement provided for a term of two years, unless extended by mutual agreement of the parties. The current term has been extended through May 31, 2014, unless sooner terminated in accordance with the terms of the agreement.2019.
We entered into an employment agreement with Dr. McKearn on September 1, 2012. The employment agreement provides for a term of two years, unless extended by mutual agreement of the parties, with an automatic renewal for successive one-year periods unless written notification is
provided by either party to the other at least ten business days prior to the expiration of the term, unless sooner terminated in accordance with the terms of the agreement.
Dr. Wilhelm's employment agreement provided for an initial base salary of $290,000 and an annual bonus of up to 30% of such base salary, based on our performance and the performance of Dr. Wilhelm. Dr. Maniar's employment agreement provided for an initial base salary of $236,500 and an annual bonus of up to 30% of such base salary, based on our performance and the performance of Dr. Maniar. Dr. McKearn's employment agreement provides for an initial base salary of $205,000 and an annual bonus of up to 35% of such base salary, based on our performance and the performance of Dr. McKearn. In each case, the bonus may be paid in the form of cash, stock options, shares of our common stock, or a combination thereof, at our compensation committee's discretion.
Drs. Wilhelm, Maniar and McKearn are entitled to participate in all of our employee benefit plans and programs that are made generally available from time to time to our executive officers and are entitled to vacation benefits and reimbursement of reasonable business expenses in conformance with our policies. Drs. Wilhelm, Maniar and McKearn's employment agreements contain non-solicitation, non-competition, confidentiality and inventions assignment provisions that, among other things, prevent the executive from competing with us during the term of his employment and for a specified time thereafter.
If Dr. Wilhelm, Dr. Maniar or Dr. McKearn's employment is terminated due to death, disability, by us for "cause," or by the executive without "good reason" (with 30 days' notice to us) during the term of his employment agreement, the agreement shall terminate and we shall pay to the executive or his spouse or estate the balance of his accrued and unpaid salary, unreimbursed expenses, and unused accrued vacation time through the termination date.
If Dr. Wilhelm's or Dr. McKearn's employment is terminated by us without "cause" or by the executive with "good reason" during the term of the employment agreement, the executive will continue to receive salary during a three-month period from the date of notice but shall not be required to perform his duties during such period. All stock options that are unvested as of the date of such termination shall fully vest and shall remain exercisable for three months from the date of termination.
We may terminate Dr. Maniar's employment during the term of the employment agreement without "cause" at any time upon the lesser of six months' or the remainder of the term of the employment agreement's written notice to Dr. Maniar. During the notice period, Dr. Maniar will continue to receive salary but shall not be required to perform his duties, provided that if Dr. Maniar accepts comparable employment during the notice period, all compensation and benefits shall cease as of the date of such acceptance. All stock options that are unvested as of the date of such termination shall fully vest upon written notice of such termination. If Dr. Maniar terminates his employment agreement with "good reason," we will pay him in accordance with the above terms and conditions as well.
If we, Dr. Wilhelm or Dr. McKearn provides written notice of intent to terminate their applicable employment agreements at the expiration of the term, the executive shall receive a lump sum payment equal to three months of his then-current base salary, subject to his execution of a general release of claims against us.
If any of the payments or benefits received by Drs. Kumar, Wilhelm, Maniar or McKearn shall be nondeductible to us by reason of Section 280G of the Code, such payments shall be reduced to the maximum amount which can be deducted by us, provided that we shall make all reasonable efforts to avoid rendering such payments or benefits nondeductible, including seeking stockholder approval if our board of directors determines that such seeking of approval shall have no adverse effect on us.
For purposes of the employment agreements, "cause" generally means (i) any gross failure of the executive (other than by reason of disability) to faithfully and professionally carry out his duties or to comply with any other material provision of his employment agreement, which continues after our written notice thereof or is not susceptible to remedy or relates to the same types of acts or omissions for which notice has previously been given, (ii) the executive's dishonesty or other willful misconduct, (iii) the executive's conviction for any felony or any other crime involving moral turpitude, whether or not relating to his employment, (iv) in accordance with applicable law, the executive's insobriety or use of illegal drugs either in the course of performing his duties or otherwise affecting his ability to perform such duties, (v) the executive's failure to comply with a lawful written direction of us or (vi) any wanton or willful dereliction of duties by the executive.
For purposes of the employment agreements, "good reason" generally means (i) a reduction in base salary by more than 20% in and for any twelve-month period, (ii) breach by us of any material provision of the employment agreement that continues without steps being taken to cure such breach for ten days after written notice thereof by the executive to us, or (iii) during the term of the employment agreement, the occurrence of (1) the sale or transfer of substantially all of our assets or (2) our merger or consolidation under circumstances where we are not the surviving entity or where persons having control of us immediately prior thereto are not in control of us immediately after.
Potential Payments Upon a Termination or Change in Control
As discussed under the caption "—Employment Agreements" above, we have agreements with our named executive officers pursuant to which they will receive severance payments upon certain termination events. The information below describes and quantifies certain compensation that would be available under our existing plans and arrangements if (i) the named executive officer was terminated as of December 31, 2012 or (ii) if a Change in Control, as defined herein, occurred on December 31, 2012 and the named executive officer had been subsequently terminated on the same date.
Acceleration of Equity Awards
Pursuant to the terms of each named executive officer's option agreements, in the event of a "Change in Control" that occurs during any time prior to such named executive officer's Termination of Service (as such terms are defined in our 2007 Equity Compensation Plan) with us, all stock options granted pursuant to such option agreement shall fully vest. See "—Equity Benefit Plans—2007 Equity Compensation Plan—Change in Control" for a summary of the definition of a Change in Control under the 2007 Equity Compensation Plan.
Termination Other than for Cause, Death or Disability; Resignation for Good Reason
Assuming a December 31, 2012 termination event, the aggregate value of the payment and benefits to which each named executive officer would be entitled in the event the named executive officer's employment is terminated for any reason other than for cause, death, or disability, or if the named
executive officer resigns for good reason, whether or not following a "change in control" as described above, would be as follows:
Name | Cash Severance ($)(1) | Benefits and Health Programs ($)(2) | Value of Accelerated Equity Awards ($)(3) | Total ($) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Ramesh Kumar, Ph.D. | 939,769 | — | 1,974,220 | 2,913,989 | |||||||||
Francois Wilhelm, M.D., Ph.D. | 93,647 | 5,115 | 1,254,565 | 1,353,327 | |||||||||
Manoj Maniar, Ph.D. | 163,398 | 7,644 | 719,514 | 890,556 | |||||||||
Thomas McKearn, M.D., Ph.D. | 51,250 | 2,977 | 5,133 | 59,360 | |||||||||
Risk Considerations in Our Compensation Program
Our board of directors is evaluating the philosophy and standards on which our compensation plans will be implemented. It is our belief that our compensation programs do not, and in the future will not, encourage inappropriate actions or risk taking by our executive officers. We do not believe that any risks arising from our employee compensation policies and practices are reasonably likely to have a material adverse effect on us. In addition, we do not believe that the mix and design of the components of our executive compensation program will encourage management to assume excessive risks. We believe that our current business process and planning cycle fosters the behaviors and controls that would mitigate the potential for adverse risk caused by the action of our executives.
Outstanding Equity Awards at Fiscal Year-End
The following table provides information regarding equity awards held by each of our named executive officers that were outstanding as of December 31, 2012.
Name | Number of Securities Underlying Unexercised Options (#) Exercisable | Number of Securities Underlying Unexercised Options (#) Unexercisable | Option Exercise Price ($) | Option Expiration Date | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Ramesh Kumar | 7,007 | 2.00 | 1/1/2016 | ||||||||||
| 140,000 | 4.50 | 4/1/2017 | |||||||||||
| 25,000 | 4.32 | 3/17/2020 | |||||||||||
| 69,863 | (1) | 30,137 | 4.32 | 3/17/2020 | |||||||||
| 70,000 | 4.60 | 12/10/2020 | |||||||||||
| 13,777 | 4.60 | 12/5/2021 | |||||||||||
| 125,000 | 9.96 | 12/19/2022 | |||||||||||
| — | (2) | 125,000 | 9.96 | 12/19/2022 | |||||||||
Francois E. Wilhelm | 100,000 | 4.50 | 5/6/2018 | ||||||||||
| 25,000 | 4.32 | 3/17/2020 | |||||||||||
| 34,932 | (1) | 15,068 | 4.32 | 3/17/2020 | |||||||||
| 25,000 | 4.60 | 12/10/2020 | |||||||||||
| 9,493 | (3) | 10,507 | 4.60 | 2/7/2021 | |||||||||
| 671 | (4) | 1,829 | 4.60 | 12/5/2021 | |||||||||
| 5,777 | 4.60 | 12/5/2021 | |||||||||||
| — | (2) | 40,000 | 9.96 | 12/19/2022 | |||||||||
| — | (2) | 20,000 | 9.96 | 12/19/2022 | |||||||||
Manoj Maniar | 15,000 | 4.50 | 8/1/2017 | ||||||||||
| 25,000 | 4.32 | 3/17/2020 | |||||||||||
| 34,932 | (1) | 15,068 | 4.32 | 3/17/2020 | |||||||||
| 10,000 | 4.60 | 12/10/2020 | |||||||||||
| 25,000 | 4.60 | 12/10/2020 | |||||||||||
| 5,040 | 4.60 | 12/5/2021 | |||||||||||
| — | (2) | 40,000 | 9.96 | 12/19/2022 | |||||||||
Thomas McKearn | 1,191 | 5.65 | 3/10/2022 | ||||||||||
| 550 | 9.96 | 8/31/2022 | |||||||||||
| — | (2) | 100,000 | 9.96 | 12/19/2022 | |||||||||
Equity Benefit Plans
2007 Equity Compensation Plan
Our board of directors adopted our 2007 Equity Compensation Plan in December 2007 for the purpose of attracting key employees, directors and consultants, inducing them to remain with us and encouraging them to increase their efforts to make our business more successful. Our stockholders approved the 2007 Equity Compensation Plan in January 2008. Our 2007 Equity Compensation Plan provides for the grant of stock options, stock appreciation rights, restricted stock, or other equity-based awards to our employees, directors and consultants. We have reserved 4,107,831 shares of common stock for issuance pursuant to our 2007 Equity Compensation Plan, subject to adjustment as set forth in the plan, of which 385,643 shares were available for future grant as of March 31, 2013. This summary is qualified in its entirety by the detailed provisions of our 2007 Equity Compensation Plan, which is filed as an exhibit to the registration statement of which this prospectus is a part.
Section 162(m) of the Code limits publicly held companies to an annual deduction forTHE PRECEDING DISCUSSION OF MATERIAL U.S. federal income tax purposes of $1,000,000 for compensation paid to each of their chief executive officer and their three highest compensated executive officers, other than the chief executive officer or the chief financial officer, determined at the end of each year, referred to as covered employees. However, performance-based compensation is excluded from this limitation. Our 2007 Equity Compensation Plan is designed to permit the compensation committee to grant awards that qualify as performance-based for purposes of satisfying the conditions of Section 162(m), but it is not required under our 2007 Equity Compensation Plan that awards qualify for this exception. To qualify as performance-based (i) the compensation must be paid solely on account of attainment of one or more pre-established objective performance goals; (ii) the performance goal under which compensation is paid must be established by a compensation committee comprised solely of two or more directors who qualify as outside directors; (iii) the material terms under which the compensation is to be paid must be disclosed to, and subsequently approved by, stockholders before payment is made; and (iv) the compensation committee must certify in writing before payment of the compensation that the performance goals and any other material terms were in fact satisfied.
Administration of our 2007 Equity Compensation Plan
Our 2007 Equity Compensation Plan will be administered by our compensation committee, as appointed by our board of directors, and such committee will determine all terms of awards under the plan. Each member of the compensation committee that administers the plan will be a "non-employee director" within the meaning of Rule 16b-3 of the Exchange Act and, at such times as we are subject to Section 162(m) of the Code, an "outside director" within the meaning of Section 162(m) of the Code. If no compensation committee is designated by our board of directors to act for these purposes, our board of directors will have the rights and responsibilities of our compensation committee under our 2007 Equity Compensation Plan. References below to the compensation committee include a reference to our board of directors or another committee appointed by our board of directors for those periods in which our board of directors or such other committee appointed by our board of directors is acting. Our compensation committee will, in its discretion, (i) authorize the granting of awards to key employees, directors and consultants, (ii) determine the eligibility of an employee, director or consultant to receive an award, (iii) determine the number of shares to be covered under any award agreement, (iv) make rules and regulations and establish procedures for the administration of the plan as it deems appropriate, (v) determine the extent, if any, to which awards shall be forfeited, (vi) interpret the plan and award agreements and (vii) take any other actions or make any other determinations or decisions that it deems necessary or appropriate in connection with the plan or the administration and interpretation thereof.
Eligibility
All of our key employees, directors and consultants and those of our related corporations designated by our compensation committee as eligible to participate in our 2007 Equity Compensation Plan are eligible to receive awards under the 2007 Equity Compensation Plan, provided that non-employee directors and consultants may not receive incentive stock options.
Share Authorization
We have reserved 4,107,831 shares of common stock for issuance under our 2007 Equity Compensation Plan. In connection with stock dividends, recapitalizations, forward or reverse stock splits, reorganizations, mergers, consolidations, spin-offs, combinations, repurchases or share exchanges, extraordinary or unusual cash distributions and other similar corporate transactions or events, our compensation committee will make appropriate adjustments that it deems equitable in the number and kind of shares subject to the plan and any outstanding awards, individual participant limits, incentive stock option limits, performance goals and the exercise or purchase price per share under any outstanding awards, as well as take any other such action as our compensation committee shall deemed necessary in its judgment to preserve the participants' rights in their respective awards substantially proportionate to the rights existing in such awards prior to such event. In addition, the number of shares available for issuance under our 2007 Equity Compensation Plan will be increased automatically on each anniversary of the effective date of the plan by (a) a number of shares that when added to the number of shares subject to outstanding awards not otherwise considered to be issued and outstanding shares plus the number of shares remaining available for future awards under our 2007 Equity Compensation Plan such new reserve equals 17% of our issued and outstanding shares on a fully diluted and converted basis or (b) such lesser number as may be determined by our board of directors, provided that in no case shall such increase result in the total number of shares available under our 2007 Equity Compensation Plan exceeding the number of our authorized shares minus the sum of (x) the number of our issued and outstanding shares, (y) the number of shares reserved by us for issuance upon exercise or conversion of other securities exercisable for or convertible into shares and (z) the number of shares subject to outstanding awards under our 2007 Equity Compensation Plan.
If any awards are forfeited or for any other reason are not payable under the plan, the shares of common stock subject to such awards will again be available for purposes of our 2007 Equity Compensation Plan.
The maximum aggregate number of shares that may be issued under our 2007 Equity Compensation Plan pursuant to incentive stock options was initially 2,069,292 shares, subject to annual increase by the lesser of the annual increase amount described above or 1,000,000 shares. No participant in our 2007 Equity Compensation Plan may receive awards for more than 300,000 shares in any calendar year.
Options
Our 2007 Equity Compensation Plan authorizes the compensation committee to grant incentive stock options (under Section 421 of the Code) and options that do not qualify as incentive stock options, or nonstatutory stock options. The exercise price of each option is determined by the compensation committee, provided that the price is equal to at least 100% of the fair market value of the shares of common stock on the date on which the option is granted. If we were to grant incentive stock options to any 10% stockholder, the exercise price may not be less than 110% of the fair market value of our shares of common stock on the date of grant.
The term of an option cannot exceed ten years from the date of grant. If we were to grant incentive stock options to any 10% stockholder, the term cannot exceed five years from the date of grant. Our compensation committee determines at what time or times each option may be exercised
and the period of time, if any, after termination of service during which options may be exercised, provided that no incentive stock option may be exercised later than three months following a termination of service other than as a result of death or disability or one year following death or disability. Options may be made exercisable in installments. The exercisability of options may be accelerated by our compensation committee.
The aggregate fair market value, determined at the time of grant, of our common stock with respect to incentive stock options that are exercisable for the first time by an optionee during any calendar year under all of our stock plans may not exceed $100,000. Options or portions thereof that exceed such limit will generally be treated as nonstatutory stock options.
The exercise price for any option is generally payable (1) in cash or a certified or banker's check, (2) if permitted by our compensation committee in its discretion, with the proceeds of a loan program by us or third-party sale program or a note acceptable to our compensation committee given as consideration under such a program, (3) if approved by our compensation committee in its discretion, by the surrender of shares of common stock with an aggregate fair market value on the date on which the option is exercised of the exercise price, (4) if approved by our compensation committee in its discretion, through the written election of the optionee to have shares withheld by us from the shares otherwise to be received, with such withheld shares having an aggregate fair market value on the date on which the option is exercised equal to the exercise price or (5) by any combination of such methods of payment or any other methods of payments acceptable to our compensation committee in its discretion.
Stock Appreciation Rights
Our 2007 Equity Compensation Plan authorizes our compensation committee to grant stock appreciation rights that provide the recipient with the right to receive, upon exercise of the stock appreciation right, cash, shares of common stock or a combination of the two. The amount that the recipient will receive upon exercise of the stock appreciation right will equal the excess of the fair market value of our common stock on the date of exercise over the base price of the stock appreciation right as determined by our compensation committee, which shall not be less than the shares' fair market value on the date of grant. Stock appreciation rights will become exercisable in accordance with terms determined by our compensation committee. Stock appreciation rights may be granted in tandem with an option grant or independently from an option grant. The term of a stock appreciation right cannot exceed ten years from the date of grant.
Restricted Stock
Our 2007 Equity Compensation Plan also provides for the grant of restricted stock. A restricted stock award is an award of shares of common stock that may be subject to restrictions on transferability and other restrictions as our compensation committee determines in its sole discretion on the date of grant. The restrictions, if any, may lapse over a specified period of time or through the satisfaction of conditions, in installments or otherwise, as our compensation committee may determine. A participant who receives restricted stock will have all of the rights of a stockholder as to those shares, including, without limitation, the right to vote and the right to receive dividends or distributions on the shares. During the period, if any, when restricted stock is non-transferable or forfeitable, a participant is prohibited from selling, transferring, pledging, anticipating, alienating, encumbering or assigning his or her restricted stock.
Performance Awards
Our 2007 Equity Compensation Plan permits the grant of performance-based stock awards that may qualify as performance-based compensation not subject to the $1,000,000 limitation on the income
tax deductibility of compensation paid to a covered executive officer imposed by Section 162(m) of the Code. Our compensation committee will determine the applicable performance period, the performance goals and such other conditions that apply to the performance-based award.
The performance goals that may be selected may be based upon: (i) the price of our common stock, (ii) our market share, (iii) our sales, (iv) earnings per share of our common stock, (v) return on our stockholder equity, (vi) our costs, (vii) our cash flow, (viii) return on our total assets, (ix) return on our invested capital, (x) return on our net assets, (xi) our operating income, (xii) our net income or (xiii) any other criteria specified by our compensation committee.
Other Equity-Based Awards
Our compensation committee may grant other types of equity-based awards under our 2007 Equity Compensation Plan. Other equity-based awards are payable in cash, shares of common stock or other equity, or a combination thereof, and may be restricted or unrestricted, as determined by our compensation committee. The terms and conditions that apply to other equity-based awards are determined by our compensation committee.
Change in Control
Upon a change in control, unless otherwise provided in the participant's award agreement, our compensation committee, in its discretion, may (i) accelerate the vesting and exercisability of all options and stock appreciation rights, (ii) cancel outstanding awards in exchange for a cash payment in an amount equal to the fair market value of restricted stock with respect to which the restricted period has expired or will expire in connection with such change in control or the excess, if any, of the fair market value of the common stock underlying the portion the option or stock appreciation right that is exercisability or become exercisable as of the date of such change in control over the exercise price or base price (or, if such exercise price or base price exceeds the fair market value, cancel such award without further obligation), (iii) terminate all such options and stock appreciation rights immediately prior to the change in control, provided that we provide participants an opportunity to exercise the options and stock appreciation rights within a specified period following the participants' receipt of written notice of such change in control or (iv) require the successor corporation to assume or substitute all outstanding awards on terms and conditions necessary to preserve the rights of participants with respect to such awards.
For purposes of this section only, a "change in control" shall mean (i) the acquisition in one or more transactions by any person other than us, its related corporations and its employee benefit plans, of beneficial ownership of 50% or more of the combined voting power of our then-outstanding voting securities, (ii) the individuals comprising our board of directors at the effective date of our 2007 Equity Compensation Plan ceasing to constitute a majority of our board of directors, provided that new members approved by a majority of the incumbent board shall be considered members of the incumbent board and reductions in size of the board shall not be considered to be a Change in Control, (iii) a merger or consolidation involving us if our stockholders immediately prior to the transaction do not own more than 65% of the voting power of our outstanding voting securities immediately following the transaction, as well as our complete liquidation or dissolution or a sale or disposition of all or substantially all of our assets, (iv) acceptance by our stockholders of shares in a share exchange if our stockholders immediately prior to the exchange do not own more than 65% of the voting power of our outstanding voting securities immediately following the exchange, or (v) in our compensation committee's discretion, a "change in control" as defined in any employment, consulting or similar agreement between us and a participant.
Amendment; Termination
Our board of directors may amend our 2007 Equity Compensation Plan or any outstanding awards at any time; provided that our board of directors may not make any amendment to our 2007 Equity Compensation Plan that would, if such amendment were not approved by the holders of a requisite percentage of our issued and outstanding voting capital stock, cause our 2007 Equity Compensation Plan to fail to comply with our stockholders agreement or any requirement of applicable law or regulation, without such approval. In addition, no amendment may, without the participant's consent, adversely affect the rights of such participation with respect to a previously granted award, except that such participant shall be deemed to consent if (i) there is an amendment to our stockholders agreement that affects our 2007 Equity Compensation Plan or the award agreement, (ii) the amendment is required to comply with applicable law or (iii) such amendment is necessary or desirable, in our compensation committee's discretion, in order to obtain desired tax or accounting treatment. Unless terminated sooner by our board of directors, our 2007 Equity Compensation Plan will terminate on December 10, 2017.
Retirement Benefits
In October 2007, we established a 401(k) Retirement Savings Plan. Employees are eligible to participate in the plan as soon as they join us if they are at least 21 years of age and work a minimum of 1,000 hours per year. We match $0.50 for every dollar of the first 6% of payroll that employees invest, up to the legal limit. Our contributions vest over four years at the rate of 25% per year. For the year ended December 31, 2012, we made approximately $159,000 in matching contributions.
Compensation of Directors
During 2012, we did not pay any cash compensation to our directors. Our board of directors has not adopted a formal non-employee director compensation policy. All of our directors are eligible to receive awards under the 2007 Equity Compensation Plan, provided that non-employee directors may not receive incentive stock options. The following table sets forth information concerning the compensation of our directors who were not also executive officers for the fiscal year ended December 31, 2012:
| |||||||
| |||||||
| |||||||
| |||||||
| |||||||
| |||||||
| |||||||
| |||||||
220,000 shares, 28,900 shares, 103,809 shares and 73,825 shares, respectively. None of our other directors who served during 2012 hold any options to purchase our common stock.
| | December 31, 2012 Fair Value | |||
|---|---|---|---|---|
Michael B. Hoffman | $ | 1,271,300 | ||
Henry S. Bienen | 152,286 | |||
Alan R. Williamson, Ph.D. | 515,760 | |||
E. Premkumar Reddy, Ph.D. | 847,129 | |||
CERTAIN RELATIONSHIPSFEDERAL INCOME TAX CONSEQUENCES IS NOT TAX ADVICE. HOLDERS OF SUBSCRIPTION RIGHTS, SHARES OF OUR COMMON STOCK AND RELATED PARTY TRANSACTIONS
PARTICIPATING WARRANTS SHOULD CONSULT THEIR OWN TAX ADVISORS REGARDING THE APPLICATION OF THE U.S. FEDERAL INCOME TAX LAWS TO THEIR PARTICULAR SITUATIONS AND THE CONSEQUENCES UNDER FEDERAL ESTATE AND GIFT TAX LAWS, FOREIGN, STATE AND LOCAL LAWS AND TAX TREATIES OF THE RECEIPT, OWNERSHIP AND EXERCISE OF SUBSCRIPTION RIGHTS AND THE ACQUISITION, OWNERSHIP AND DISPOSITION OF SHARES OF OUR COMMON STOCK, PRE-FUNDED WARRANTS AND WARRANTS ACQUIRED UPON EXERCISE OF SUBSCRIPTION RIGHTS AND SHARES OF OUR COMMON STOCK ACQUIRED UPON EXERCISE OF PRE-FUNDED WARRANTS OR WARRANTS.
The following is a description of transactions since January 1, 2010, to which we have been a party, in which the amount involved in the transaction exceeds $120,000, and in which any of our directors, executive officers or to our knowledge, beneficial owners of more than 5% of our capital stock or an affiliate or immediate family member thereof, had or will have a direct or indirect material interest, other than employment, compensation, termination and change in control arrangements with our named executive officers, which are described under "Executive and Director Compensation." We believe the terms obtained or consideration that we paid or received, as applicable, in connection with the transactions described below were comparable to terms available or the amounts that would be paid or received, as applicable, in arm's-length transactions with unrelated third parties.
After consummation of this offering, our audit committee will be responsible for the review, approval and ratification of related person transactions. The audit committee will review these transactions under our Code of Conduct, which will govern conflicts of interests, among other matters, and will be applicable to our employees, officers and directors.
Preferred Stock and Convertible Promissory Note Issuances
Issuance of Series H convertible preferred stock
In September 2010, November 2010, December 2010, February 2011, June 2011 and September 2011, we issued and sold an aggregate of 2,013,424 shares of our Series H convertible preferred stock at a price per share of $9.79, for aggregate consideration of approximately $19.7 million. In September 2010 and June 2011, we issued and sold an aggregate of 199,144 and 102,146 shares of our Series H convertible preferred stock, respectively, to an affiliate of Michael B. Hoffman, the Chairman of our board of directors and the beneficial owner of more than 5% of our capital stock, at a price per share of $9.79, for aggregate consideration of approximately $2.9 million. In September 2011, we issued and sold an aggregate of 510,725 shares of our Series H convertible preferred stock to an affiliate of Sarath Naru, a member of our current board of directors, at a price per share of $9.79, for aggregate consideration of approximately $5.0 million. For every share of Series H convertible preferred stock, the affiliates of Mr. Hoffman and Mr. Naru, respectively, will receive, upon conversion, one share of our common stock immediately prior to consummation of this offering.
Promissory Notes and Issuance of Series I convertible preferred stock
In April, 2012, June 2012, and July 2012, we issued and sold $26.4 million aggregate principal amount of our convertible promissory notes for an aggregate purchase price of $26.4 million. On July 25, 2012, $26.4 million aggregate principal amount of and $0.3 million of accrued interest on our convertible promissory notes were converted into 2,433,328 shares of our Series I convertible preferred stock at a price per share of $11.00.
The table below sets forth the aggregate principal amount of convertible promissory notes purchased by, and the number of shares of Series I convertible preferred stock subsequently issued to, our executive officers, directors and stockholders who held more than 5% of any class of our voting securities and their affiliates. For every share of Series I convertible preferred stock set forth in the
table below, the holder will receive, upon conversion, one share of our common stock immediately prior to consummation of this offering.
| | Aggregate Purchase Price of April 2012 Convertible Promissory Notes | Aggregate Purchase Price of June 2012 Convertible Promissory Notes | Aggregate Purchase Price of July 2012 Convertible Promissory Notes | Series I Convertible Preferred Stock Issued Upon Conversion of the Promissory Notes | Shares of Common Stock Issuable Upon Conversion of Series I Convertible Preferred Stock | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Directors and their affiliates | ||||||||||||||||
Affiliates of Michael B. Hoffman(1) | $ | 6,420,000 | $ | 1,297,000 | $ | 10,000,000 | 1,635,514 | 1,635,514 | ||||||||
Viren Mehta(2) | — | — | 250,000 | 22,771 | 22,771 | |||||||||||
Issuance of Series J convertible preferred stock
On July 27, 2012, we issued and sold an aggregate of 3,030,303 shares of our Series J convertible preferred stock to Baxter, the beneficial owner of more than 5% of our capital stock, at a price per share of $16.50, for aggregate consideration of $50.0 million. For every share of Series J convertible preferred stock, Baxter will receive, upon conversion, one share of our common stock immediately prior to consummation of this offering.
Indemnification Agreements
We intend to enter into indemnification agreements with our directors and executive officers. Under these agreements, we will agree to indemnify these persons against any and all expenses incurred by them resulting from their status as one of our directors or executive officers to the fullest extent permitted by Delaware law, our certificate of incorporation and our bylaws to be in effect upon the consummation of this offering, except in limited circumstances. In addition, these indemnification agreements will provide that, to the fullest extent permitted by Delaware law, we will pay for all expenses incurred by such persons in connection with a legal proceeding arising out of their service to us.
Agreements with Our Stockholders
We have entered into the eighth amended and restated stockholders' agreement, dated July 27, 2012, or the stockholders' agreement, with substantially all holders of our common and preferred stock that contains agreements with respect to the election of our board of directors, restrictions on transfer of shares, right of first offer, right of first refusal and co-sale and registration rights. For a description of the registration rights, see "Description of Capital Stock—Registration Rights." Certain of our current directors were elected pursuant to the terms of the stockholders' agreement or an antecedent version thereof. The provisions of the stockholders' agreement relating to the rights of first offer, rights of first refusal and co-sale rights shall terminate upon consummation of this offering.
Development and License Agreement
In September 2012, we entered into a development and license agreement with Baxter, the beneficial owner of more than 5% of our capital stock. For a description of the Baxter development and license agreement, see "Business—Collaborations—Baxter Healthcare SA."
Research Agreement
On May 3, 2010, as subsequently amended, we entered into a research agreement with Mount Sinai, with which E. Premkumar Reddy, Ph.D., a member of our board of directors and the beneficial owner of more than 5% of our capital stock, is affiliated. The research is undertaken by Mount Sinai on our behalf. Mount Sinai, in connection with us, will prepare applications for patents generated from the research. Results from all projects will belong exclusively to Mount Sinai, but we will have an exclusive option to license any inventions. The initial term of the research agreement was one year with options to extend by mutual agreement. The term of the agreement has been extended through July 2013. In 2010, 2011 and 2012 and for the three months ended March 31, 2013, we paid Mount Sinai an aggregate of $369,000, $554,000, $1.2 million and $56,000, respectively. As of March 31, 2013, $190,000 was due to Mount Sinai under the research agreement.
Loans
In December 2011, Michael B. Hoffman, the Chairman of our board of directors and the beneficial owner of more than 5% of our capital stock, advanced $620,000 to us to fund operations. Interest accrued at 10% per annum. In April 2012, we and Mr. Hoffman cancelled the loan in exchange for approximately $620,000 aggregate principal amount of our convertible promissory notes at the closing of our convertible promissory note offering described above.
Vendor Agreements
We outsource the synthesis of some of our chemical compounds to vendors in the United States and abroad. EPR Pharmaceuticals Pvt Ltd., an entity affiliated with E. Premkumar Reddy, Ph.D., a member of our board of directors and the beneficial owner of more than 5% of our capital stock, produced one of these compounds for us under purchase orders. The payments for these services for the years ended December 31, 2010, 2011 and 2012 and for the three months ended March 31, 2013 were approximately $230,000, $6,000, $157,000 and $28,000, respectively. There were no amounts due under purchase orders with EPR Pharmaceuticals Pvt Ltd. as of March 31, 2013.
We purchase chemical compounds from Zydus BSV Pharma Pvt. Limited, an entity affiliated with Pankaj R. Patel, a member of our current board of directors Mr. Patel will not continue as a director after this offering. We made purchases from Zydus during the years ended December 31, 2010, 2011 and 2012 and the three months ended March 31, 2013 in the amounts of $0, $451,000, $400,000 and $28,000, respectively. There were no amounts due under purchase orders with Zydus BSV Pharma Pvt. Limited as of March 31, 2013.
We purchase chemical compounds from Zyfine (A Division of Cadila Healthcare Limited), an entity affiliated with Pankaj R. Patel, a member of our current board of directors. Mr. Patel will not continue as a director after this offering. We made purchases from Zyfine during the years ended December 31, 2010, 2011 and 2012 and the three months ended March 31, 2013 in the amounts of $186,000, $519,000, $10,000 and $35,000, respectively. There were no amount due under purchase orders with Zyfine as of March 31, 2013.
Lease
We rent office space in Pennington, New Jersey from Zydus Healthcare, LLC, an entity affiliated with Pankaj R. Patel, a member of our current board of directors. Mr. Patel will not continue as a director after this offering. We made aggregate rent payments under the leases for the years ended December 31, 2010, 2011 and 2012 and for the three months ended March 31, 2013 of $0, $49,000, $55,000 and $33,000, respectively.
Consulting Agreements
We entered into a consulting agreement with E. Premkumar Reddy, Ph.D., a member of our board of directors and the beneficial owner of more than 5% of our capital stock, effective as of January 1, 2012 for consulting services rendered in addition to his membership on our board of directors. The consulting agreement provided for a term of one year, unless renewed by mutual agreement of the parties. The current term has been extended through December 31, 2013, unless sooner terminated in accordance with the terms of the agreement. We had previously entered in a consulting agreement with Dr. Reddy, effective as of March 1, 2010, which terminated effective as of December 31, 2011. The services provided by Dr. Reddy include guidance and opinions on Temple's intellectual property that has been licensed for commercialization through us, with certain restrictions relating to Dr. Reddy's other employment. Dr. Reddy is subject to certain confidentiality and use and intellectual property restrictions. The payments for these services for the years ended December 31, 2010, 2011 and 2012 and the three months ended March 31, 2013 were approximately $146,000, $158,000, $165,000 and $45,000, respectively. No amounts were due under the consulting agreement as of March 31, 2013.
Employment Agreement
We entered into an employment agreement with Ajay Bansal, our Chief Financial Officer as well as a member of our current board of directors, on March 20, 2013. The employment agreement provides for a term of employment of two years, with automatic renewals for one-year terms unless earlier terminated in accordance with its terms or on ten business days' written notice by either party.
The employment agreement provides for an initial base salary of $310,000 and an annual bonus of up to 30% of such base salary, based on our and Mr. Bansal's performance. The bonus may be paid in the form of cash, stock options, shares of our common stock, or a combination thereof, at our compensation committee's discretion.
Mr. Bansal is entitled to either participate in all of our employee benefit plans and programs that are made generally available from time to time to our executive officers or to receive $10,000 annually in lieu of health coverage under our medical plans, and is entitled to vacation benefits. Pursuant to his employment agreement, Mr. Bansal is eligible to be granted incentive stock options, with a vesting period of four years, in amount to be determined by our compensation committee. Mr. Bansal's employment agreement contains non-solicitation, non-competition, confidentiality and inventions assignment provisions that, among other things, prevent the executive from competing with us during the term of his employment and for a specified time thereafter.
If Mr. Bansal's employment is terminated due to his death, disability, by us for "cause" or by Mr. Bansal without "good reason" during the term provided under the employment agreement, we shall pay to Mr. Bansal or his spouse or estate the balance of his accrued and unpaid salary, unreimbursed expenses, and unused accrued vacation time through the termination date.
If Mr. Bansal's employment is terminated by us without "cause" or by Mr. Bansal with "good reason" during the term provided under the employment agreement, he will continue to receive salary during a three-month period from the date of notice but shall not be required to perform his duties
during such period. All stock options that are unvested as of the date of such termination shall fully vest and shall remain exercisable for three months from the date of termination.
If we or Mr. Bansal provides written notice of intent to terminate Mr. Bansal's employment agreements at the expiration of the term of his employment pursuant to the employment agreement, Mr. Bansal shall receive a lump sum payment equal to three months of his then-current base salary, subject to his execution of a general release of claims against us.
If any of the payments or benefits received by Mr. Bansal shall be nondeductible to us by reason of Section 280G of the Code such payments shall be reduced to the maximum amount which can be deducted by us, provided that we shall make all reasonable efforts to avoid rendering such payments or benefits nondeductible, including seeking stockholder approval if our board of directors determines that such seeking of approval shall have no adverse effect on us.
For purposes of Mr. Bansal's employment agreement, "cause" means (i) any gross failure of the executive (other than by reason of disability) to faithfully and professionally carry out his duties or to comply with any other material provision of his employment agreement, which continues after our written notice thereof or is not susceptible to remedy or relates to the same types of acts or omissions for which notice has previously been given, (ii) the executive's dishonesty or other willful misconduct, (iii) the executive's conviction for any felony or any other crime involving moral turpitude, whether or not relating to his employment, (iv) in accordance with applicable law, the executive's insobriety or use of illegal drugs either in the course of performing his duties or otherwise affecting his ability to perform such duties, (v) the executive's failure to comply with a lawful written direction of us or (vi) any wanton or willful dereliction of duties by the executive.
For purposes of Mr. Bansal's employment agreement, "good reason" means (i) a reduction in base salary by more than 20% in and for any twelve-month period, (ii) breach by us of any material provision of the employment agreement that continues without steps being taken to cure such breach for ten days after written notice thereof by the executive to us, or (iii) during the term of his employment pursuant to the employment agreement, the occurrence of (1) the sale or transfer of substantially all of our assets or (2) our merger or consolidation under circumstances where we are not the surviving entity or where persons having control of us immediately prior thereto are not in control of us immediately after.
The following table sets forth certain information regarding the beneficial ownership of our capital stock outstanding as of June 1, 2013 by:
The percentage ownership information shown in the table is based upon 20,591,954 shares of common stock outstanding as of June 1, 2013 after giving effect to the conversion of all outstanding shares of preferred stock into an aggregate of 17,113,481 shares of common stock immediately prior to consummation of this offering. The number of shares and percentage of shares beneficially owned after the offering gives effect to the issuance by us of shares of common stock in this offering. The percentage ownership information after this offering assumes no exercise of the underwriters' over-allotment option.
Each individual or entity shown in the table has furnished us with information with respect to beneficial ownership. We have determined beneficial ownership in accordance with the SEC's rules. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities. In addition, the rules include shares of common stock issuable pursuant to the exercise of stock options, warrants or other rights that are either immediately exercisable or exercisable on or before July 31, 2013, which is 60 days after June 1, 2013. These shares are deemed to be outstanding and beneficially owned by the person holding those rights for the purpose of computing the percentage ownership of that person, but they are not treated as outstanding for the purpose of computing the percentage ownership of any other person. Unless otherwise indicated, the persons or entities identified in this table have sole voting and investment power with respect to all shares shown as beneficially owned by them, subject to applicable community property laws.
Except as otherwise noted below, the address for each person or entity listed in the table is c/o Onconova Therapeutics, Inc., 375 Pheasant Run, Newtown, PA 18940.
| | | Percentage of shares beneficially owned | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Name and Address of Beneficial Owner | Number of shares beneficially owned | Before offering | After offering | |||||||
5% or greater stockholders: | ||||||||||
The Jane & Michael B. Hoffman 2013 Descendants Trust | 4,495,321 | 21.8 | % | % | ||||||
712 Fifth Avenue, 51st Fl. | ||||||||||
Michael B. Hoffman(1) | 4,765,321 | 22.8 | ||||||||
712 Fifth Avenue, 51st Fl. | ||||||||||
Baxter Healthcare SA | 3,030,303 | 14.7 | ||||||||
c/o Baxter Healthcare Corporation | ||||||||||
E. Premkumar Reddy(2) | 1,773,697 | 8.6 | ||||||||
Other Directors and Named Executive Officers: | ||||||||||
Ramesh Kumar, Ph.D.(3) | 823,400 | 3.9 | ||||||||
Thomas McKearn, M.D., Ph.D.(4) | 1,741 | * | ||||||||
François E. Wilhelm, M.D., Ph.D.(5) | 211,400 | 1.0 | ||||||||
Manoj Maniar, Ph.D.(6) | 122,232 | * | ||||||||
Ajay Bansal(7) | 58,451 | * | ||||||||
Henry S. Bienen, Ph.D.(8) | 54,329 | * | ||||||||
Viren Mehta(9) | 171,074 | * | ||||||||
Sarath Naru(10) | 510,725 | 2.5 | ||||||||
Pankaj R. Patel(11) | 867,925 | 4.2 | ||||||||
Alan R. Williamson, Ph.D.(12) | 116,316 | * | ||||||||
All current executive officers and directors as a group (12 persons)(13) | 9,476,611 | 43.3 | ||||||||
DESCRIPTION OF CAPITAL STOCKSECURITIES
Upon consummation of this offering, ourOur authorized capital stock will consistconsists of 25,000,000 shares of which will be designated as common stock, with a par value of $0.01 per share, and 5,000,000 shares of which will be designated as preferred stock, with a par value of $0.01 per share. As of MarchAfter our one-for-ten reverse stock split on May 31, 2013, there would have been 20,591,9542016, 2,740,212 shares of our common stock were outstanding, held by 250 stockholders of record, and no shares of our preferred stock, outstanding, in each case after giving effect to the conversion of all outstanding preferred stock immediately prior to consummation of this offering.were outstanding.
The following is a summary of our capital stock upon consummation of this offering. This summary does not purport to be complete and is qualified in its entirety by the provisions of our certificate of incorporation and bylaws to be in effect upon consummation of this offering, copies of which have been filed as exhibits to the registration statement of which this prospectus is a part.
Common Stock
Voting Rights
Each holder of common stock shall be entitled to one vote for each share on all matters submitted to a vote of the stockholders.
Dividends
Subject to the preferences that may be applicable to any outstanding preferred stock, holders of our common stock shall beare entitled to receive ratably any dividends that may be declared by theour board of directors out of funds legally available for that purpose.
Liquidation
Holders of our common stock are entitled to one vote for each share on all matters voted on by stockholders, including the election of directors. Holders of our common stock do not have any conversion, redemption, sinking fund or preemptive rights. In the event of our dissolution, liquidation dissolution or winding up, holders of our common stock shall beare entitled to share ratably in allany assets remaining after paymentthe satisfaction in full of liabilitiesthe prior rights of creditors and the aggregate liquidation preference of any outstanding preferred stock.
No Preemptive or Similar Rights
Ourstock then outstanding. The rights, preferences and privileges of the holders of our common stock shall not be entitled to preemptive rights and is notare subject to, conversion, redemption or sinking fund provisions.and may be adversely affected by, the rights of the holders of shares of any series of preferred stock that we may designate and issue in the future. All outstanding shares of our common stock are, and any shares of common stock that we may issue in the future will be, fully paid and non-assessable.
Preferred Stock
Immediately prior to consummationWe may issue any class of this offering, all outstanding shares of our preferred stock will be converted into an aggregate of 17,113,481 shares of common stock. Under our certificate of incorporation that will be in effect following consummation of this offering, ourany series. Our board of directors has the authority, subject to limitations prescribed byunder Delaware law, to issue preferred stock in one or more series, to establish from time to time the number of shares to be included in each series and to fix the designation, powers, preferences and rights of the shares of each series and any of its qualifications, limitations and restrictions. Our board of directors can also can increase or decrease the number of shares of any series, but not below the number of shares of that series then outstanding.
Our board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of the common stock. The issuance of preferred stock, while providing flexibility in connection with possible acquisitions and other corporate purposes, could, among other things, have the effect of delaying, deferring or preventing a change in control of our company and may adversely affect the market price of our
common stock and the voting and other rights of the holders of common stock. We have no current plan to issue any shares of preferred stock.
Warrants
InTradable Warrants Included in Units Issuable in the Rights Offering
The Warrants to be issued as a part of this Rights Offering will be separately transferable following their issuance and through their expiration five years from the date of issuance. The Warrants entitle the holder to purchase one share of common stock at an exercise price of $ per share, 120% of the per Unit price, from the date of issuance through its expiration on , 2021. We have applied to list the Warrants for trading on NASDAQ under the symbol "ONTXW," however, there is no assurance that a sufficient number of Subscription Rights will be exercised so that the Warrants will meet the minimum listing criteria to be accepted for listing on NASDAQ. The common stock underlying the Warrants, upon issuance, will also be traded on NASDAQ under the symbol "ONTX."
All Warrants that are purchased in the Rights Offering as part of the Units will be issued in book-entry, or uncertificated, form meaning that you will receive a direct registration (DRS) account statement from our transfer agent reflecting ownership of Warrants if you are a holder of record of shares or warrants. The Subscription Agent will arrange for the issuance of the Warrants as soon as
practicable after the expiration of the Rights Offering, payment for the Units subscribed for has cleared, and all prorating calculations and reductions contemplated by the terms of the Rights Offering have been effected. If you hold your shares of common stock in the name of a custodian bank, broker, dealer, or other nominee, DTC will credit your account with your nominee with the Warrants you purchased in the Rights Offering.
The Warrants will be exercisable by paying the exercise price in cash, or, solely during any period when a registration statement for the exercise of the Warrants is not in effect, exercisable on a cashless basis.
The exercise price of the Warrants and the number of shares of common stock issuable upon exercise of the Warrants are subject to adjustment in certain circumstances, including a stock split of, stock dividend on, or a subdivision, combination or recapitalization of the common stock.
Except as described below, a holder may not exercise any portion of the Warrant to the extent that the holder would beneficially own more than 4.99% of our outstanding common stock after exercise, except that upon at least 61 days' prior notice from the holder to us, the holder may increase the amount of ownership of outstanding stock after exercising the holder's Warrants up to 9.99% of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the Warrants. The foregoing limitation on exercise does not apply to any holder who beneficially owns in excess of 4.99% of our outstanding common stock immediately prior to the Rights Offering.
No public market currently exists for the Warrants. We have applied to list the Warrants on NASDAQ under the symbol "ONTXW," although there is no assurance that a sufficient number of Subscription Rights will be exercised so that the Warrants will meet the minimum listing criteria to be accepted for listing on NASDAQ. Even if the Warrants are listed on NASDAQ, an active public market for the Warrants may not be developed or sustained. Without an active trading market, the liquidity of the Warrants will be limited.
Subject to applicable laws and the restriction on transfer set forth in the warrant, the warrant may be transferred at the option of the holder upon surrender of the warrant to us together with the appropriate instruments of transfer.
After the one-year anniversary of issuance, we may redeem the Warrants for $0.001 per Warrant if our common stock is above $ per share, 300% of the exercise price, for each of 10 consecutive trading days.
The Warrants do not confer upon the holder any voting or any other rights of a shareholder of the Company. Upon notice to the Warrants holders, we have the right at any time and from time to time, to reduce the exercise price or to extend the Warrants termination date.
The Warrants will be issued pursuant to a warrant agreement by and between us and Wells Fargo Bank, N.A., as the warrant agent.
Pre-Funded Warrants Issuable in Lieu of Common Stock in the Rights Offering
Any Pre-Funded Warrants issued as a part of this Rights Offering will be separately transferable following their issuance and through their expiration five years from the date of issuance. The Pre-Funded Warrants entitle the holder to purchase one share of common stock at an exercise price of $0.01 per share, and the subscription price per Unit for any such electing investors will be reduced to $ (which equals the Subscription Price for the other Units sold in the Rights Offering, less the $0.01 exercise price for each Pre-Funded Warrant). Each Pre-Funded Warrant will be exercisable from the date of issuance through its expiration on , 2021. The Pre-Funded Warrants will not be listed for trading on any stock exchange or market.
Any Pre-Funded Warrants that are purchased in the Rights Offering as part of the Units will be issued in physical form. The Subscription Agent will arrange for the issuance of the Pre-Funded Warrants as soon as practicable after the expiration of the Rights Offering, payment for the Units subscribed for has cleared, and all prorating calculations and reductions contemplated by the terms of the Rights Offering have been effected.
The Pre-Funded Warrants will be exercisable by paying the exercise price in cash or on a cashless basis. The exercise price of the Pre-Funded Warrants and the number of shares of common stock issuable upon exercise of the Pre-Funded Warrants are subject to adjustment in certain circumstances, including a stock split of, stock dividend on, or a subdivision, combination or recapitalization of the common stock.
A holder may not exercise any portion of the Pre-Funded Warrant to the extent that the holder would beneficially own more than 4.99% of our outstanding common stock after exercise, except that upon at least 61 days' prior notice from the holder to us, the holder may increase the amount of ownership of outstanding stock after exercising the holder's Pre-Funded Warrant up to 9.99% of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the Pre-Funded Warrant.
Subject to applicable laws, the pre-funded warrants may be offered for sale, sold, transferred or assigned without our consent.
The Pre-Funded Warrants do not confer upon the holder any voting or any other rights of a shareholder of the Company.
Other Currently Outstanding Warrants
On January 11, 2016, we issued common stock purchase warrants, referred to as our "participating warrants," to purchase up to 96,842 shares of our common stock at an exercise price equal to $11.50 per share, subject to customary adjustments and as adjusted for our one-for-ten reverse stock split effective May 31, 2016. Upon the terms and subject to the limitations on exercise and the conditions set forth in the participating warrants, the participating warrants are exercisable at any time on or after July 11, 2016 and on or prior to July 11, 2021. The participating warrants expire on July 11, 2021. The participating warrants entitle the holder to participate in any dividend or distribution, including any distribution of rights to purchase common stock, to the holders of our common stock. Subject to limited exceptions, a holder of participating warrants will not have the right to exercise any portion of its participating warrants if the holder, together with its affiliates, would beneficially own in excess of 9.99% of the number of shares of our common stock outstanding immediately after giving effect to such exercise.
The participating warrants and the shares of our common stock issuable upon exercise of the participating warrants were offered and sold without registration under the Securities Act of 1933, as amended ("the Securities Act"), or state securities laws, in reliance on the exemptions provided by Section 4(a)(2) of the Act and/or Regulation D promulgated thereunder and in reliance on similar exemptions under applicable state laws. Accordingly, neither the participating warrants nor the shares of our common stock underlying the participating warrants may be offered or sold except pursuant to an effective registration statement under the Securities Act or pursuant to an available exemption from, or in a transaction not subject to, the registration requirements of the Securities Act and in accordance with applicable state securities laws.
Prior to our initial public offering, in connection with a credit facility, obtained in 2007, we issued a warrant to purchase 6,128 shares of Series G convertible preferred stock in June 2009. The warrant was immediately exercisable upon issuance. The warrantissuance and expires on May 12, 2015. Upon consummation of this offering, the expiration date of the warrant will be extended to the third anniversary ofJuly 30, 2016. Following the consummation of this offering. Furthermore, upon the consummation of thisour initial public offering and the conversion of theour Series G convertible preferred stock into shares of common stock, the warrant shall become exercisable for 6,128 shares of common stock. The exercise price per share is $9.79.
Registration Rights
We entered into the stockholders' agreement with substantially all holders of our common and preferred stock. Under the stockholders' agreement, holders of shares of our preferred stock have been granted registration rights with respect to the shares of common stock issuable upon conversion as further described below.
Demand Registration Rights
At any time after six months following consummation of this offering, the holders of 25% or more of the shares having demand registration rights may request that we register all or a portion of their shares of common stock. We will effect the registration as requested, unless, in the good faith judgment of our board of directors, such registration would be materially detrimental to us and our stockholders and should be delayed. We have the right to defer the filing of such registration statement once for up to 120 days during any 12-month period. We are not obligated to file a registration statement pursuant to this provision on more than two occasions. In addition, when we are eligible for the use of Form S-3, or any successor form, holders of a majority of the shares having demand registration rights may make unlimited requests that we register all or a portion of their common stock for sale under the Securities Act on Form S-3, or any successor form, so long as the aggregate price to the public in connection with any such offering is at least $500,000. However, we are not obligated to file a Form S-3 pursuant to this provision on more than two occasions in any 12-month period.
Piggyback/Incidental Registration Rights
In addition, if at any time we register any shares of our stock, the holders of all shares having registration rights are entitled to notice of the filing of the applicable registration statement and to include all or a portion of their common stock in the registration. Certain holders of these registration rights have waived this right to the extent it relates to this offering and we are seeking waivers from the remaining holders.
Other Provisions
In the event that any registration in which the holders of registrable shares participate pursuant to the stockholders' agreement is an underwritten public offering, the number of registrable shares to be included may, in specified circumstances, be limited due to market conditions. With the exception of this offering, from which all registrable shares may be excluded, the number of registrable shares to be excluded from registration pursuant to the above shall not be reduced below 20% of the shares to be offered.
We will pay all registration expenses, other than underwriting discounts and selling commissions, and the reasonable fees and expenses, other than underwriting discounts and selling commissions, and
the reasonable feescommon stock in July 2013, and expenses of a single special counsel for the selling stockholders, related to any demand or piggyback registration. The stockholders' agreement contains customary cross-indemnification provisions,subsequent adjustments pursuant to which we are obligated to indemnify the selling stockholdersadjustment provisions set forth in that warrant, after our one-for-ten reverse stock split on May 31, 2016, the eventwarrant was exercisable for approximately 582 shares of material misstatements or omissions in the registration statement attributable to us, and they are obligated to indemnify us for material misstatements or omissions in the registration statement attributable to them.common stock at an exercise price per share of approximately $103.08.
Delaware Anti-Takeover Law and Provisions ofin Our Certificate of Incorporation and Bylaws
Delaware Anti-Takeover Law
We are subject to Section 203 of the Delaware General Corporation Law. Section 203 generally prohibits a public Delaware corporation from engaging in a "business combination" with an "interested stockholder" for a period of three years after the date of the transaction in which the person became an interested stockholder, unless:
Section 203 defines a "business combination" to include:
In general, Section 203 defines an "interested stockholder" as any person that is:
Under specific circumstances, Section 203 makes it more difficult for an "interested stockholder" to effect various business combinations with a corporation for a three-year period, although the
stockholders may, by adopting an amendment to the corporation's certificate of incorporation or bylaws, elect not to be governed by this section, effective 12 months after adoption.
Our certificate of incorporation and bylaws do not exclude us from the restrictions of Section 203. We anticipate that the provisions of Section 203 might encourage companies interested in acquiring us to negotiate in advance with our board of directors since the stockholder approval requirement would be avoided if a majority of the directors then in office approve either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder.
Certificate of Incorporation and Bylaws
Provisions of our certificate of incorporation and bylaws to be in effect upon the consummation of this offering may delay or discourage transactions involving an actual or potential change of control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares, or transactions that our stockholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our common stock. Among other things, our certificate of incorporation and bylaws will:
Listing on the NASDAQ Global Market
We have applied to list our common stock on the NASDAQ Global Market under the symbol "ONTX."
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is . The transfer agent and registrar's addressWells Fargo Bank, N.A.
Listing
Our common stock is .listed on the Nasdaq Capital Market under the symbol "ONTX."
SHARES ELIGIBLE FOR FUTURE SALEPLAN OF DISTRIBUTION
Immediately priorOn or about July 6, 2016, we will distribute the Subscription Rights, Subscription Rights Statements and copies of this prospectus to this offering, there was no public market for our common stock. Future sales of substantial amountsthe holders of our common stock and participating warrants on the Record Date. Subscription Rights holders who wish to exercise their Subscription Rights and purchase Units must complete the Subscription Rights Statement and return it with payment for the shares to the Subscription Agent at the following address:
By Mail:
Wells Fargo Bank, N.A.
Shareowner Services
Voluntary Corporate Actions
P.O. Box 64858
St. Paul, Minnesota 55164-0858
By Hand or Overnight Courier:
Wells Fargo Bank, N.A.
Shareowner Services
Voluntary Corporate Actions
1110 Centre Pointe Curve, Suite 101
Mendota Heights, Minnesota 55120
See "The Rights Offering—Methods for Exercising Subscription Rights."
If you have any questions, you should contact the dealer-manager for the Rights Offering:
Maxim Group LLC
405 Lexington Avenue
New York, New York 10174
Attention Syndicate Department
Email: syndicate@maximgrp.com
Telephone: (212) 895-3745
Other than as described in this prospectus, we do not know of any existing agreements between any shareholder, broker, dealer, underwriter or agent relating to the sale or distribution of the underlying common stock.
Maxim Group LLC is the dealer-manager of this Rights Offering. We and Maxim may introduce one or more co-dealer-managers and one or more financial advisors to assist in the public market could adversely affect prevailing market prices. Furthermore, since only a limited number of sharesRights Offering. In any such event, Maxim Group LLC will be available for sale shortly afterthe lead dealer-manager. In such capacity, the dealer-manager will provide marketing assistance and advice to us in connection with this offering becauseand will solicit the exercise of contractualSubscription Rights and legal restrictions on resale described below, sales of substantial amounts of common stockparticipation in the public market afterOver-Subscription Privilege. The dealer-manager is not underwriting or placing any of the restrictions lapse could adversely affectSubscription Rights or the prevailing market price for our common stock as well as our ability to raise equity capital in the future.
After consummation of this offering,Units, shares of common stock, will be outstanding, assuming no exercise of the underwriters' over-allotment option. All of the shares soldWarrants or Pre-Funded Warrants being issued in this offering, will be freely tradable unless held by an affiliateand does not make any recommendation with respect to such Subscription Rights (including with respect to the exercise or expiration of ours. Except as set forth below, the remainingsuch Subscription Rights), Units, shares of common stock, outstanding after this offering will be restricted as a result of securities lawsWarrants or lock-up agreements. These remaining shares will generally become available for sale in the public market as follows:
Rule 144Pre-Funded Warrants.
In general, under Rule 144 underconnection with this Rights Offering, we have agreed to pay to the Securities Act as in effect on the date of this prospectus, beginning 90 days after the effective datedealer-manager a cash fee equal to (a) 4.5% of the registration statement of which this prospectus is a part, a person who is not an affiliate of ours at any time during the three months preceding a sale, and who has held their shares for at least six months, as measured by SEC rule, including the holding period of any prior owner other than one of our affiliates, may sell shares without restriction, provided current public information about us is available. In addition, under Rule 144, any person who is not an affiliate of ours at any time during the three months preceding a sale, and who has held their shares for at least one year, as measured by SEC rule, including the holding period of any prior owner other than one of our affiliates, would be entitled to sell an unlimited number of shares immediately upon consummation of this offering without regard to whether current public information about us is available.
Beginning 90 days after the effective datedollar amount of the registration statementUnits sold to any holders of which this prospectus is a part, a personSubscription Rights who is an affiliate of ours and who has beneficially owned restricted securities for at least six months, as measured by SEC rule, including the holding period of any prior owner other than one of our affiliates, is entitled to sell a number of restricted shares within any three-month period that does not exceed the greater of:
Sales of restricted shares under Rule 144 held by our affiliates are also subject to requirements regarding the manner of sale, notice and the availability of current public information about us. Rule 144 also requires that affiliates relying on Rule 144 to sell shares of our common stock that are not restricted shares must nonetheless comply with the same restrictions applicable to restricted shares, other than the holding period requirement.
Notwithstanding the availability of Rule 144, certain holders of our restricted shares have entered into lock-up agreements as described below and their restricted shares will become eligible for sale at the expirationdealer-manager upon completion of the restrictions set forth in those agreements.
Rule 701
Under Rule 701 underRights Offering a non-accountable expense allowance equal to the Securities Act, shareslesser of our common stock acquired upon the exercise of currently outstanding options$100,000 or pursuant to other rights granted under a written compensatory stock or option plan or other written agreement in compliance with Rule 701 may be resold, by:
As of March 31, 2013, options to purchase a total of 3,722,188 shares of common stock were outstanding, of which 2,334,784 were vested. Of the total number of shares of our common stock issuable under these options, are subject to contractual lock-up agreements with the underwriters described below under "Underwriting" and will become eligible for sale at the expiration of those agreements.
Lock-up Agreements
In connection with this offering, we, our officers and directors and certain of our stockholders have agreed with the underwriters, subject to certain exceptions, not to dispose of or hedge any shares of our common stock or securities convertible into or exchangeable for shares of common stock during the period from the date of the lock-up agreement continuing through the date 180 days after the date of this prospectus, except with the prior written consent of Citigroup Global Markets Inc. Citigroup Global Markets Inc. has advised us that they have no current intent or arrangement to release any of the shares subject to the lock-up agreements prior to the expiration of the lock-up period. The lock-up agreements permit stockholders to transfer common stock and other securities subject to the lock-up agreements in certain circumstances.
Following the lock-up periods set forth in the agreements described above, and assuming that the representatives of the underwriters do not release any parties from these agreements and that there is no extension of the lock-up period, all of the shares of our common stock that are restricted securities or are held by our affiliates as of the date of this prospectus will be eligible for sale in the public market in compliance with Rule 144 under the Securities Act.
Registrationconnection with the Rights
Upon consummation of this offering, the holders of shares of our common stock Offering. We advanced $30,000 against out-of-pocket accountable expenses to Maxim Group LLC upon its engagement as a dealer-manager; provided that Maxim Group LLC will be entitledpromptly reimburse to rights with respect to the registration of their shares under the Securities Act, subject to the lock-up arrangement described above. Registration of these shares under the Securities Act would result in the shares becoming freely tradable without restriction under the Securities Act, except for shares purchased by affiliates, immediately upon the effectivenessus (i) any portion of the registration statementadvance not used for actual out-of-pocket expenses, if the Rights Offering is not completed, and (ii) the full amount of which this prospectusthe advance, if the Rights Offering is a part. Any sales of securities by these stockholders could have a material adverse effect oncompleted.
From the trading price of our common stock. See "Description of Capital Stock—Registration Rights."
Equity Incentive Plans
We intend to file one or more registration statements on Form S-8 under the Securities Act after consummation of this offering to register the shares of our common stock that are issuable pursuant to our 2007 Equity Compensation Plan. The registration statements are expected to be filed and become effective as soon as practicableRecord Date until 90 days after the consummation of this offering. Accordingly, shares registered under the registration statements will be available for sale in the open market following their effective dates, subject to Rule 144 volume limitations and the lock-up arrangement described above, if applicable.
MATERIAL U.S. FEDERAL INCOME TAX CONSIDERATIONSFOR NON-U.S. HOLDERS OF OUR COMMON STOCK
The following discussion is a general summaryclosing of the material U.S. federal income tax consequences applicableRights Offering, we have agreed not to Non-U.S. Holdersissue, agree to issue or announce the issuance of acquiring, owning and disposing of our common stock as of the date hereof.
For the purposes of this discussion, a "Non-U.S. Holder" of our common stock means a holder that, for U.S. federal income tax purposes, is not a U.S. Holder. A "U.S. Holder" means a holder of our common stock that is for U.S. federal income tax purposes:
This summary does not consider specific facts and circumstances that may be relevant to a particular Non-U.S. Holder's tax particular circumstances and does not consider the state, local or non-U.S. tax consequences of an investment in our common stock. It also does not consider Non-U.S. Holders subject to special tax treatment under U.S. federal income tax laws (including partnerships or other pass-through entities, banks and insurance companies, regulated investment companies, real estate investment trusts, dealers in securities, holders of our common stock held as part of a "straddle," hedge," conversion transaction" or other risk-reduction transaction, controlled foreign corporations, passive foreign investment companies, companies that accumulate earnings to avoid U.S. federal income tax, foreign tax-exempt organizations, "expatriated entities," companies subject to the "stapled stock" rules, former U.S. citizens or residents and persons who hold or receive the shares of common stock as compensation). This summary is based on provisionsor common stock equivalents without the consent of the Code, applicable Treasury regulations, administrative pronouncements of the U.S. Internal Revenue Service, or IRS, and judicial decisions, all as in effect on the date hereof, and all of which aredealer-manager, subject to change, possibly oncertain exceptions including a retroactive basis,pre-existing agreement, equity awards, conversion of derivative securities and different interpretations.
This summary is general information only. It is not tax advice. We urge each prospective Non-U.S. Holder to consult their own tax advisor concerning the particular U.S. federal, state, local and non-U.S. income, estate and other tax consequences of the purchase, ownership and disposition of our common stock.
U.S. Tradein connection with any acquisitions, partnerships or Business Incomestrategic transactions.
For purposes ofWe have also agreed to indemnify the dealer-manager and its respective affiliates against certain liabilities arising under the Securities Act. The dealer-manager's participation in this discussion, dividend income and gain on the sale or other taxable disposition of shares of our common stock will be considered to be "U.S. trade or business income" if such dividend income or gain is (1) effectively connected with the conduct by a Non-U.S. Holder of a trade or business within the United States; and (2) in the case of a Non-U.S. Holder that is eligible for the benefits of an income tax treaty with the United States, attributable to a "permanent establishment" or "fixed base" maintained by the Non-U.S. Holder in the United States. Generally, U.S. trade or business income is not subject to U.S. federal withholding tax (provided the Non-U.S. Holder complies with applicable certification and disclosure requirements); instead, U.S. trade or business incomeoffering is subject to U.S. federal income tax on a net income basis at regular U.S. federal income tax ratescustomary conditions contained in the same manner as ifdealer-manager agreement, including the recipient were a U.S. person. Any U.S. trade or business income receivedreceipt by a
Tablethe dealer-manager of Contents
Non-U.S. Holder that is treated as a corporation also may be subject to a "branch profits tax" at a 30% rate, or such lower rate as provided under an applicable income tax treaty.
Distributions
Distributions of cash or property (other than certain stock distributions) that we pay with respect to our common stock (or certain redemptions that are treated as distributions with respect to our shares of common stock) will be taxable as dividends for U.S. federal income tax purposes to the extent paid outopinion of our current or accumulated earnings and profits as determined for U.S. federal income tax purposes. Subject to the discussion in "—Recently-Enacted Federal Tax Legislation" below, a Non-U.S. Holder generally will be subject to withholding of U.S. federal income tax at a rate of 30% of the gross amount of our distributions or such lower rate as may be specified by an applicable income tax treaty. In order to obtain a reduced rate of U.S. federal withholding tax under an applicable income tax treaty, a Non-U.S. Holder will be required to provide a properly executed IRS Form W-8BEN (or appropriate substitute or successor form) certifying its entitlement to benefits under the treaty. A Non-U.S. Holder of our common stock that is eligible for a reduced rate of U.S. federal withholding tax under an income tax treaty may obtain a refund or credit of any excess amounts withheld by timely filing an appropriate claim for refund with the IRS. A Non-U.S. Holder is encouraged to consult its own tax advisor regarding its possible entitlement to benefits under an income tax treaty. If the amount of a distribution exceeds our current and accumulated earnings and profits, such excess first will be treated as a tax-free return of capital to the extent of the Non-U.S. Holder's adjusted tax basis in our shares, and thereafter will be treated as capital gain. To the extent a distribution exceeds our current or accumulated earnings and profits, a Non-U.S. Holder of our common stock may obtain a refund or credit of any excess amounts withheld by timely filing an appropriate from for refund with the IRS. A Non-U.S. Holder's adjusted tax basis in our shares will generally be equal to the amount the Non-U.S. Holder paid for its shares, reduced by the amount of any distributions treated as a return of capital. See, "—Sale, Exchange or Other Disposition of Common Stock" below.
counsel. The U.S. federal withholding tax does not apply to dividends that are U.S. trade or business income, as described above, of a Non-U.S. Holder who provides a properly executed IRS Form W-8ECI (or appropriate substitute or successor form), certifying that the dividends are subject to tax as income effectively connected with the Non-U.S. Holder's conduct of a trade or business within the U.S.
Sale, Exchange or Other Disposition of Our Common Stock
Subject to the discussion in "—Recently-Enacted Federal Tax Legislation" below, a Non-U.S. Holder generally will not be subject to U.S. federal income tax or withholding tax in respect of any gain recognized on a sale, exchange or other disposition of shares of our common stock unless:
Gain described in the first bullet above will be subject to U.S. federal income tax in the manner described under "—U.S. Trade or Business Income." Gain described in the second bullet above will be subject to a flat 30% tax (or such lower rate specified by an applicable income tax treaty), but may be offset by certain U.S. source capital losses (even though the Non-U.S. Holder is not considered a
resident of the United States), provided that the Non-U.S. Holder has timely filed U.S. federal income tax returns with respect to such losses.
In general, a corporation is a "United States real property holding corporation" if the fair market value of its "U.S. real property interests" equals or exceeds 50% of the sum of the fair market value of its worldwide (domestic and foreign) real property interestsdealer-manager and its other assets used or held for use in a trade or business. For this purpose, real property interests generally include land, improvements and associated personal property. We believe that we have not been, and we are not and do not anticipate becoming, a "United States real property holding corporation" for U.S. federal income tax purposes. If we are or become a "United States real property holding corporation," a Non-U.S. Holder, nevertheless, will not be subjectaffiliates may provide to U.S. federal income or withholding tax in respect of any gain on a sale or other disposition of our common stock so long as shares of our common stock are "regularly traded on an established securities market" as defined under applicable Treasury regulations and a Non-U.S. Holder owns, actually and constructively, 5% or less of our shares at all times during the shorter of the five-year period ending on the date of disposition and such Non-U.S. Holder's holding period for our shares. Prospective investors should be aware that no assurance can be given that our shares will be so regularly traded when a Non-U.S. Holder sells its shares of our common stock.
U.S. Federal Estate Tax
Individual Non-U.S. Holders and entities, the property of which is potentially includible in an individual's gross estate for U.S. federal income tax purposes (for example, a trust funded by an individual and with respect to which the individual has retained certain interests or powers), should note that, unless an applicable tax treaty provides otherwise, shares of our common stock will be treated as U.S. situs property subject to U.S. federal estate tax.
Information Reporting Requirements and Backup Withholding
We must annually report to the IRS and to each Non-U.S. Holder any dividend income that is subject to U.S. federal withholding tax, or that is exempt from such withholding tax pursuant to an income tax treaty with the United States. Copies of these information returns also may be made available under the provisions of a specific treaty or agreement to the tax authorities of the country in which the Non-U.S. Holder resides. Under certain circumstances, the Code imposes a backup withholding obligation on certain reportable payments. Dividends paid to a Non-U.S. Holder of our common stock generally will be exempt from backup withholding if the Non-U.S. Holder provides a properly executed IRS Form W-8BEN (or appropriate substitute or successor form) or otherwise establishes an exemption.
The payment of the proceeds from the disposition of our common stock to or though the U.S. office of any broker, U.S. or foreign, will be subject to information reporting and possible backup withholding unless the owner certifies (usually on IRS Form W-8BEN) as to its non-U.S. status under penalties of perjury or otherwise establishes an exemption, provided that the broker does not have actual knowledge or reason to know that the holder is a U.S. person or that the conditions of any other exemption are not, in fact, satisfied. The payment of the proceeds from the disposition of our common stock to or through a non-U.S. office of a non-U.S. broker will not be subject to information reporting or backup withholding unless the non-U.S. broker has certain types of relationships with the United States (which we refer to as a United States related person). In the case of the payment of the proceeds from the disposition of our common stock to or through a non-U.S. office of a broker that is either a U.S. person or a United States related person, the Treasury Regulations require information reporting (but not the backup withholding) on the payment unless the broker has documentary evidence in its files that the owner is a non-U.S. holder and the broker has no knowledge to the contrary. Non-U.S. Holders should consult their own tax advisors on the application of information
reporting and backup withholding to them in their particular circumstances (including upon their disposition of our common stock).
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules from a payment to a Non-U.S. Holder will be credited against the Non-U.S. Holder's U.S. federal income tax liability, if any, with any excess withholding refunded to the Non-US. Holder, provided that the required information is furnished on a timely basis to the IRS.
Recently Enacted Legislation Affecting Taxation of Our Common Stock Held by or Through Non-U.S. Entities
Withholding taxes may apply to certain types of payments made to "foreign financial institutions" (as specifically defined in the Code) and certain other non-United States entities. Specifically, a 30% withholding tax may be imposed on distributions and gross proceeds from the sale, exchange or other disposition of our common stock paid to a foreign financial institution or to a non-financial foreign entity unless (1) the foreign financial institution undertakes certain diligence and reporting, (2) the non-financial foreign entity either certifies it does not have any substantial United States owners or furnishes identifying information regarding each substantial United States owner, or (3) the foreign financial institution or non-financial foreign entity otherwise qualifies for an exemption from these rules. If the payee is a foreign financial institution and is subject to the diligence and reporting requirements in clause (1) above, it must enter into an agreement with the IRS requiring, among other things, that it undertake to identify accounts held by certain United States persons or United States-owned foreign entities, annually report certain information about such accounts, and withhold 30% on payments to non-compliant foreign financial institutions and certain other account holders.
The withholding provisions above will generally apply to payments of dividends made on or after January 1, 2014 and to payments of gross proceeds from the sale or disposition of stock on or after January 1, 2017. Non-U.S. Holders are urged to consult their own tax advisors regarding the possible implications of this legislation on their investment in our common stock.
Citigroup Global Markets Inc. and Leerink Swann LLC are acting as joint book-running managers of the offering and as representatives of the underwriters named below. Subject to the terms and conditions stated in the underwriting agreement dated the date of this prospectus, each underwriter named below has severally agreed to purchase, and we have agreed to sell to that underwriter, the number of shares of common stock set forth opposite the underwriter's name.
| ||||
| ||||
| ||||
| ||||
| ||||
The underwriting agreement provides that the obligations of the underwriters to purchase the shares of common stock included in this offering are subject to approval of legal matters by counsel and to other conditions. The underwriters are obligated to purchase all the shares of common stock (other than those covered by the over-allotment option described below) if they purchase any of the shares of common stock.
Shares of common stock sold by the underwriters to the public will initially be offered at the initial public offering price set forth on the cover of this prospectus. Any shares of common stock sold by the underwriters to securities dealers may be sold at a discount from the initial public offering price not to exceed $ per share. If all the shares of common stock are not sold at the initial offering price, the underwriters may change the offering price and the other selling terms. The representatives have advised us that the underwriters do not intend to make sales to discretionary accounts.
If the underwriters sell more shares of common stock than the total number set forth in the table above, we have granted to the underwriters an option, exercisable for 30 days from the date of this prospectus, to purchase up to additional shares of common stock at the public offering price less the underwriting discount. The underwriters may exercise the option solely for the purpose of covering over-allotments, if any, in connection with this offering. To the extent the option is exercised, each underwriter must purchase a number of additional shares of common stock approximately proportionate to that underwriter's initial purchase commitment. Any shares of common stock issued or sold under the option will be issued and sold on the same terms and conditions as the other shares of common stock that are the subject of this offering.
We, our officers and directors and certain of our stockholders have agreed that, subject to specified limited exceptions, for a period of 180 days from the date of this prospectus, we and they will not, without the prior written consent of Citigroup Global Markets Inc., dispose of or hedge any shares or any securities convertible into or exchangeable for our common stock. Citigroup Global Markets Inc. in its sole discretion may release any of the securities subject to these lock-up agreements at any time, which, in the case of officers and directors, shall be with notice.
Prior to this offering, there has been no public market for our common stock. Consequently, the initial public offering price for the shares of our common stock will be determined by negotiations between us and the representatives. Among the factors considered in determining the initial public offering price will be our results of operations, our current financial condition, our future prospects, our markets, the economic conditions in and future prospects for the industry in which we compete, our management, and currently prevailing general conditions in the equity securities markets, including current market valuations of publicly traded companies considered comparable to our company. We
cannot assure you, however, that the price at which the shares of our common stock will sell in the public market after this offering will not be lower than the initial public offering price or that an active trading market in our common stock will develop and continue after this offering.
We have applied to list our common stock on the NASDAQ Global Market under the symbol "ONTX."
The following table shows the underwriting discounts and commissions that we are to pay to the underwriters in connection with this offering. These amounts are shown assuming both no exercise and full exercise of the underwriters' over-allotment option.
| |||||||
| |||||||
We estimate that the total expenses of this offering payable by us will be $ .
In connection with this offering, the underwriters may purchase and sell shares of common stock in the open market. Purchases and sales in the open market may include short sales, purchases to cover short positions, which may include purchases pursuant to the over-allotment option, and stabilizing purchases.
Purchases to cover short positions and stabilizing purchases, as well as other purchases by the underwriters for their own accounts, may have the effect of preventing or retarding a decline in the market price of the shares of common stock. They may also cause the price of the shares of common stock to be higher than the price that would otherwise exist in the open market in the absence of these transactions. The underwriters may conduct these transactions on the NASDAQ Global Market, in the
over-the-counter market or otherwise. If the underwriters commence any of these transactions, they may discontinue them at any time.
Conflicts of Interest
The underwriters are full service financial institutions engaged in various activities, which may include securities trading, commercial and investment banking, financial advisory, investment management, principal investment, hedging, financing and brokerage activities. The underwriters and their respective affiliates may, from time to time engage in transactions with and perform services for usthe future in the ordinary course of their business certain financial advisory, investment banking and other services for which they maywill be entitled to receive customary fees and reimbursement of expenses. In the ordinary course of their various business activities, the underwriters and their respective affiliates may make or hold a broad array of investments and actively trade debt and equity securities (or related derivative securities) and financial instruments (which may include bank loans and/or credit default swaps) for their own account and for the accounts of their customers and may at any time hold long and short positions in such securities and instruments. Such investments and securities activities may involve securities and/or instruments of ours or our affiliates. The underwriters and their affiliates may also make investment recommendations and/or publish or express independent research views in respect of such securities or financial instruments and may hold, or recommend to clients that they acquire, long and/or short positions in such securities and instruments.fees.
We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act, or to contribute to payments the underwriters may be required to make because of any of those liabilities.
Notice to Prospective Investors in the European Economic Area
In relation to each member state of the European Economic Area that has implemented the Prospectus Directive (each, a relevant member state), with effect from and including the date on which the Prospectus Directive is implemented in that relevant member state (the relevant implementation date), an offer of shares described in this prospectus may not be made to the public in that relevant member state other than:
provided that no such offer of shares shall require us or any underwriter to publish a prospectus pursuant to Article 3 of the Prospectus Directive.
For purposes of this provision, the expression an "offer of securities to the public" in any relevant member state means the communication in any form and by any means of sufficient information on the terms of the offer and the shares to be offered so as to enable an investor to decide to purchase or subscribe for the shares, as the expression may be varied in that member state by any measure implementing the Prospectus Directive in that member state, and the expression "Prospectus Directive" means Directive 2003/71/EC (and amendments thereto, including the 2010 PD Amending Directive, to the extent implemented in the relevant member state) and includes any relevant implementing measure in the relevant member state. The expression 2010 PD Amending Directive means Directive 2010/73/EU.
We have not authorized and do not authorize the making of any offer of shares through any financial intermediary on our behalf, other than offers made by the underwriters with a view to the final placement of the shares as contemplated in this prospectus. Accordingly, no purchaser of the shares, other than the underwriters, is authorized to make any further offer of the shares on behalf of us or the underwriters.
Notice to Prospective Investors in the United Kingdom
This prospectus is only being distributed to, and is only directed at, persons in the United Kingdom that are qualified investors within the meaning of Article 2(1)(e) of the Prospectus Directive that are also (i) investment professionals falling within Article 19(5) of the Financial Services and Markets Act 2000 (Financial Promotion) Order 2005 (the "Order") or (ii) high net worth entities, and other persons to whom it may lawfully be communicated, falling within Article 49(2)(a) to (d)Industry Regulatory Authority, Inc. The principal business address of the Order (each such person being referred to as a "relevant person"). This prospectus and its contents are confidential and should not be distributed, published or reproduced (in whole or in part) or disclosed by recipients to any other persons in the United Kingdom. Any person in the United Kingdom thatMaxim Group LLC is not a relevant person should not act or rely on this document or any of its contents.405 Lexington Avenue, New York, New York 10174.
Notice to Prospective Investors in Australia
No prospectus or other disclosure document (as defined in the Corporations Act 2001 (Cth) of Australia, or Corporations Act) in relation to the common stock has been or will be lodged with the Australian Securities & Investments Commission, or ASIC. This document has not been lodged with ASIC and is only directed to certain categories of exempt persons. Accordingly, if you receive this document in Australia:
Notice to Prospective Investors in France
Neither this prospectus nor any other offering material relating to the shares described in this prospectus has been submitted to the clearance procedures of theAutorité des Marchés Financiers or of the competent authority of another member state of the European Economic Area and notified to theAutorité des Marchés Financiers. The shares have not been offered or sold and will not be offered or
sold, directly or indirectly, to the public in France. Neither this prospectus nor any other offering material relating to the shares has been or will be:
Such offers, sales and distributions will be made in France only:
The shares may be resold directly or indirectly, only in compliance with articles L.411-1, L.411-2, L.412-1 and L.621-8 through L.621-8-3 of the FrenchCode monétaire et financier.
Notice to Prospective Investors in Hong Kong
The shares may not be offered or soldconsolidated financial statements of Onconova Therapeutics, Inc. appearing in Hong Kong by means of any document other than (i) in circumstances which do not constitute an offer to the public within the meaning of the Companies Ordinance (Cap. 32, Laws of Hong Kong), or (ii) to "professional investors" within the meaning of the Securities and Futures Ordinance (Cap. 571, Laws of Hong Kong) and any rules made thereunder, or (iii) in other circumstances which do not result in the document being a "prospectus" within the meaning of the Companies Ordinance (Cap. 32, Laws of Hong Kong) and no advertisement, invitation or document relating to the shares may be issued or may be in the possession of any personOnconova Therapeutics, Inc.'s Annual Report (Form 10-K) for the purpose of issue (in each case whether in Hong Kong or elsewhere), which is directed at, or the contents of which are likely to be accessed or readyear ended December 31, 2015 have been audited by theErnst & Young LLP, independent registered public in Hong Kong (except if permitted to do so under the laws of Hong Kong) other than with respect to shares which are or are intended to be disposed of only to persons outside Hong Kong or only to "professional investors" within the meaning of the Securities and Futures Ordinance (Cap. 571, Laws of Hong Kong) and any rules made thereunder.
Notice to Prospective Investors in Japan
The shares offered in this prospectus have not been and will not be registered under the Financial Instruments and Exchange Law of Japan. The shares have not been offered or sold and will not be offered or sold, directly or indirectly, in Japan or to or for the account of any resident of Japan (including any corporation or other entity organized under the laws of Japan), except (i) pursuant to an exemption from the registration requirements of the Financial Instruments and Exchange Law and (ii) in compliance with any other applicable requirements of Japanese law.
Notice to Prospective Investors in Singapore
This prospectus has not been registeredaccounting firm, as a prospectus with the Monetary Authority of Singapore. Accordingly, this prospectus and any other document or material in connection with the offer or sale, or invitation for subscription or purchase, of the shares may not be circulated or distributed, nor may the shares be offered or sold, or be made the subject of an invitation for subscription or purchase, whether directly or indirectly, to persons in Singapore other than (i) to an institutional investor under Section 274 of the Securities and Futures Act, Chapter 289 of Singapore, or the SFA, (ii) to a relevant person pursuant to Section 275(1), or any person pursuant to Section 275(1A), and in accordance with the conditions specified in Section 275 of the SFA or (iii) otherwise pursuant to, and in accordance with the conditions of, any other applicable provision of the SFA, in each case subject to compliance with the conditions set forth in their report thereon (which contains an explanatory paragraph describing conditions that raise substantial doubt about the SFA.
WhereCompany's ability to continue as a going concern as described in Note 1 to the sharesconsolidated financial statements), included therein and incorporated herein by reference. Such consolidated financial statements are subscribed or purchased under Section 275incorporated herein by reference in reliance upon such report given on the authority of the SFA by a relevant person which is:
shares, debentures and units of shares and debentures of that corporation or the beneficiaries' rights and interest (howsoever described) in that trust shall not be transferred within six months after that corporation or that trust has acquired the shares pursuant to an offer made under Section 275 of the SFA except:
Table of Contentsauditing.
The validity of the shares of common stock being offered by this prospectushereby and certain other legal matters will be passed upon for us by DechertPepper Hamilton LLP, New York, New York. Cooley LLP, Reston, Virginia, is acting as counsel for the underwriters in connection with this offering.
Our consolidated financial statements at December 31, 2012, and for the year then ended, appearing in this prospectus and registration statement have been audited by Ernst & Young LLP, independent registered public accounting firm, and at December 31, 2011, and for the year then ended, by EisnerAmper LLP, independent registered public accounting firm, as set forth in their respective reports thereon appearing elsewhere herein, and are included in reliance upon such reports given on the authority of such firms as experts in accounting and auditing.Philadelphia, Pennsylvania.
WHERE YOU CAN FIND MORE INFORMATION
We have filedfile annual, quarterly and current reports, proxy statements and other information with the Securities and Exchange Commission, or "SEC." You may read and copy any documents we file with the SEC at the SEC's Public Reference Room at 100 F Street, NE, Washington, D.C. 20549. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. In addition, our filings with the SEC are available to the public through the SEC's Internet site at http://www.sec.gov. Information about us is also available on our website at http://www.onconova.com. This URL and the SEC's URL above are intended to be inactive textual references only. The information on the SEC's website and our website is not part of, and is not incorporated into, this prospectus.
We have filed a registration statement on Form S-1 under the Securities Act of 1933, as amended, with respect to thecovering our shares of common stock being offered bysubject to this prospectus.offering, of which this prospectus forms a part. This prospectus, which constitutes a part of the registration statement,however, does not contain all of the information set forth in the registration statement and its exhibits. For further information with respect to us and the common stock offered by this prospectus, you should refer to the registration statement and the exhibits to the registration statement. For
further information concerning us and the securities we may offer and sell, you should read the entire registration statement and the exhibits to the registration statement. The registration statement has been filed as part of that document. Statementselectronically and may be obtained in any manner listed above. Any statements contained in this prospectus as toconcerning the contentsprovisions of any contract or any other document referred to are not necessarily complete, and, in each instance, we refer youreference is made to the copy of the contract or othersuch document filed as an exhibit to the registration statement.statement or otherwise filed with the SEC. Each of these statementssuch statement is qualified in all respectsits entirety by thissuch reference.
The SEC allows us to "incorporate by reference" the information we file with it, which means that we can readdisclose important information to you by referring you to those documents. The information incorporated by reference is considered to be part of this prospectus. We incorporate by reference the documents listed below:
We will provide without charge to each person, including any beneficial owner, to whom this prospectus is delivered, upon his or her written or oral request, a copy of these filings,any or all documents referred to above which have been or may be incorporated by reference into this prospectus but not delivered with this prospectus excluding exhibits to those documents unless they are specifically incorporated by reference into those documents. You can request those documents from us, at no cost, by writing or telephoning us at: Onconova Therapeutics, Inc., 375 Pheasant Run, Newtown, PAPennsylvania, 18940, (267) 759-3680.759-3036, Attention: Benjamin Hoffman.
Upon consummation of this offering, The most recent information that we will be subject to the information and periodic reporting requirements of the Exchange Act, and we will file periodic reports, proxy statements and other information with the SEC. These reports, proxy statementsSEC automatically updates and other information will be available for inspection and copying at the public reference room and website of the SEC referred to above. We also maintain a website athttp://www.onconova.com, at which you may access these materials free of charge as soon as reasonably practicable after they are electronically filed with, or furnished to, the SEC.supersedes older information. The information contained in or that canany such filing will be accessed through, our website is notdeemed to be a part of this prospectus.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
|
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Onconova Therapeutics, Inc.
We have audited the accompanying consolidated balance sheet of Onconova Therapeutics, Inc. (the Company) as of December 31, 2012, and the related consolidated statements of operations and comprehensive loss, redeemable convertible preferred stock and stockholders' deficit, and cash flows for the year ended December 31, 2012. These consolidated financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these consolidated financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. We were not engaged to perform an audit of the Company's internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall consolidated financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of Onconova Therapeutics, Inc. at December 31, 2012, and the consolidated results of its operations and its cash flows for the year ended December 31, 2012, in conformity with U.S. generally accepted accounting principles.
The consolidated financial statements referred to above have been prepared assuming that the Company will continue as a going concern. As more fully described in Note 1 to the consolidated financial statements, the Company has incurred losses from operations and will require additional capital to fund planned operations. In addition, certain series of the Company's Preferred Stock are currently redeemable. The Company does not have sufficient resources to fund planned operations and satisfy the redemption of Preferred Stock should the holders so elect. These conditions raise substantial doubt about the Company's ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ ERNST & YOUNG LLP
Philadelphia, PAMay 3, 2013
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders ofOnconova Therapeutics, Inc.
We have audited the accompanying balance sheet of Onconova Therapeutics, Inc. (the "Company") as of December 31, 2011 and the related statements of operations and comprehensive loss, redeemable convertible preferred stock and stockholders' deficit, and cash flows for the year ended December 31, 2011. The financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatements. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Onconova Therapeutics, Inc. as of December 31, 2011, and the results of its operations and its cash flows for the year ended December 31, 2011, in conformity with accounting principles generally accepted in the United States of America.
/s/ EISNERAMPER LLP
Edison, New JerseyMay 2, 2013
Onconova Therapeutics, Inc.Consolidated Balance Sheets
| | December 31, | Pro Forma | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | December 31, 2012 | |||||||
| | | | (unaudited) | |||||||
Assets | ||||||||||
Current assets: | ||||||||||
Cash and cash equivalents | $ | 2,713,000 | $ | 81,527,000 | ||||||
Grants receivable | 78,000 | — | ||||||||
Prepaid expenses and other current assets | 627,000 | 1,725,000 | ||||||||
Preferred stock subscription receivable | 400,000 | — | ||||||||
Total current assets | 3,818,000 | 83,252,000 | ||||||||
Property and equipment, net | 507,000 | 463,000 | ||||||||
Restricted cash | 125,000 | 125,000 | ||||||||
Other non-current assets | 12,000 | 12,000 | ||||||||
Total assets | $ | 4,462,000 | $ | 83,852,000 | ||||||
Liabilities, redeemable convertible preferred stock and stockholders' (deficit) equity | ||||||||||
Current liabilities: | ||||||||||
Accounts payable | $ | 5,614,000 | $ | 5,517,000 | ||||||
Accrued expenses and other current liabilities | 1,680,000 | 3,925,000 | ||||||||
Warrant liability | 1,064,000 | 62,000 | ||||||||
Stock option liability | 2,648,000 | 11,967,000 | ||||||||
Stockholder loan | 620,000 | — | ||||||||
Deferred revenue | 455,000 | 3,907,000 | ||||||||
Total current liabilities | 12,081,000 | 25,378,000 | ||||||||
Deferred revenue, non-current | 10,718,000 | 15,421,000 | ||||||||
Other | 85,000 | 44,000 | ||||||||
Total liabilities | 22,884,000 | 40,843,000 | ||||||||
Commitments and contingencies | ||||||||||
Redeemable convertible preferred stock, $0.01 par value per share, 12,817,950 and 18,548,253 shares authorized at December 31, 2011 and 2012, 11,227,169 and 16,912,199 shares issued and outstanding at December 31, 2011 and 2012, liquidation preference of $194,329,000 at December 31, 2012, and no shares issued and outstanding at December 31, 2012 (pro forma) | 119,997,000 | 201,315,000 | $ | — | ||||||
Stockholders' (deficit) equity: | ||||||||||
Common stock, $0.01 par value, 19,200,000 and 30,145,155 shares authorized at December 31, 2011 and 2012, 2,889,747 and 3,474,473 shares issued and outstanding at December 31, 2011 and 2012 and 20,587,954 shares issued and outstanding at December 31, 2012 (pro forma) | 29,000 | 35,000 | 206,000 | |||||||
Additional paid in capital | — | 10,019,000 | 211,163,000 | |||||||
Accumulated deficit | (138,448,000 | ) | (168,360,000 | ) | (168,360,000 | ) | ||||
Total stockholders' (deficit) equity | (138,419,000 | ) | (158,306,000 | ) | 43,009,000 | |||||
Total liabilities, redeemable convertible preferred stock and stockholders' (deficit) equity | $ | 4,462,000 | $ | 83,852,000 | $ | 83,852,000 | ||||
See accompanying notes to consolidated financial statements.
Onconova Therapeutics, Inc.Consolidated Statements of Operations and Comprehensive Loss
| | Year Ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | |||||
Revenue | $ | 1,487,000 | $ | 46,190,000 | |||
Operating expenses: | |||||||
General and administrative | 6,436,000 | 15,707,000 | |||||
Research and development | 22,624,000 | 52,762,000 | |||||
Total operating expenses | 29,060,000 | 68,469,000 | |||||
Loss from operations | (27,573,000 | ) | (22,279,000 | ) | |||
Change in fair value of warrant liability | 1,287,000 | 367,000 | |||||
Interest expense | (19,000 | ) | (8,608,000 | ) | |||
Other income, net | 11,000 | 608,000 | |||||
Net loss before income taxes | (26,294,000 | ) | (29,912,000 | ) | |||
Income taxes | — | — | |||||
Net loss | (26,294,000 | ) | (29,912,000 | ) | |||
Other comprehensive loss | — | — | |||||
Comprehensive loss | (26,294,000 | ) | (29,912,000 | ) | |||
Accretion of redeemable convertible preferred stock | (4,020,000 | ) | (3,953,000 | ) | |||
Net loss applicable to common stockholders | $ | (30,314,000 | ) | $ | (33,865,000 | ) | |
Per share information: | |||||||
Net loss per share of common stock, basic and diluted | $ | (10.64 | ) | $ | (11.51 | ) | |
Basic and diluted weighted average shares outstanding | 2,849,159 | 2,941,782 | |||||
Pro forma net loss per share of common stock, basic and diluted (unaudited) | $ | (2.01 | ) | ||||
Basic and diluted pro forma weighted average shares outstanding (unaudited) | 16,887,329 | ||||||
See accompanying notes to consolidated financial statements.
Onconova Therapeutics, Inc.Consolidated Statements of Redeemable Convertible Preferred Stock and Stockholders' Deficit
| | | | | Stockholders' Deficit | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Redeemable Convertible Preferred Stock | ||||||||||||||||||||||||
| | Common Stock | | | | |||||||||||||||||||||
| | | | Preferred Stock Subscribed | Additional Paid in Capital | Accumulated deficit | | |||||||||||||||||||
| | Shares | Amount | Shares | Amount | Total | ||||||||||||||||||||
Balance at January 1, 2011 | 10,211,835 | $ | 105,734,000 | $ | 300,000 | 2,756,585 | $ | 28,000 | $ | — | $ | (108,745,000 | ) | $ | (108,717,000 | ) | |||||||||
Issuance of preferred stock, net of issuance costs | 819,329 | 7,917,000 | (300,000 | ) | — | — | — | — | — | ||||||||||||||||
Exercise of stock options | — | — | — | 133,162 | 1,000 | 611,000 | — | 612,000 | |||||||||||||||||
Issuance of preferred stock upon exercise of warrants | 196,005 | 2,326,000 | — | — | — | — | — | — | |||||||||||||||||
Accretion of preferred stock to redemption value | — | 4,020,000 | — | — | — | (611,000 | ) | (3,409,000 | ) | (4,020,000 | ) | ||||||||||||||
Net loss | — | — | — | — | — | — | (26,294,000 | ) | (26,294,000 | ) | |||||||||||||||
Balance at December 31, 2011 | 11,227,169 | 119,997,000 | — | 2,889,747 | 29,000 | — | (138,448,000 | ) | (138,419,000 | ) | |||||||||||||||
Issuance of preferred stock, net of issuance costs | 3,030,303 | 47,796,000 | — | — | — | — | — | — | |||||||||||||||||
Exercise of stock options | — | — | — | 584,726 | 6,000 | 4,688,000 | — | 4,694,000 | |||||||||||||||||
Proceeds from stockholder in connection with settlement of stock option exercises | — | — | — | — | — | 3,943,000 | — | 3,943,000 | |||||||||||||||||
Settlement of stock option liabilities | — | — | — | — | — | (2,835,000 | ) | — | (2,835,000 | ) | |||||||||||||||
Issuance of preferred stock upon exercise of warrants | 221,399 | 2,802,000 | — | — | — | — | — | — | |||||||||||||||||
Exchange of convertible debt and preferred stock | 2,433,328 | 26,767,000 | — | — | — | — | — | — | |||||||||||||||||
Beneficial conversion feature on convertible debt | — | — | — | — | — | 8,176,000 | — | 8,176,000 | |||||||||||||||||
Accretion of preferred stock to redemption value | — | 3,953,000 | — | — | — | (3,953,000 | ) | — | (3,953,000 | ) | |||||||||||||||
Net loss | — | — | — | — | — | — | (29,912,000 | ) | (29,912,000 | ) | |||||||||||||||
Balance at December 31, 2012 | 16,912,199 | $ | 201,315,000 | $ | — | 3,474,473 | $ | 35,000 | $ | 10,019,000 | $ | (168,360,000 | ) | $ | (158,306,000 | ) | |||||||||
See accompanying notes to consolidated financial statements.
Onconova Therapeutics, Inc.Consolidated Statements of Cash Flows
| | Year Ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | |||||
Operating activities: | |||||||
Net loss | $ | (26,294,000 | ) | $ | (29,912,000 | ) | |
Adjustment to reconcile net loss to net cash (used in) provided by operating activities: | |||||||
Depreciation and amortization | 316,000 | 319,000 | |||||
Loss on asset disposal | — | 3,000 | |||||
Amortization of deferred financing fees | 21,000 | 15,000 | |||||
Amortization of debt discount | — | 8,176,000 | |||||
Change in fair value of warrant liabilities | (1,287,000 | ) | (367,000 | ) | |||
Stock compensation expense | 6,000 | 13,844,000 | |||||
Changes in assets and liabilities: | |||||||
Grants receivable | 1,730,000 | 78,000 | |||||
Prepaid expenses and other current assets | 253,000 | (1,098,000 | ) | ||||
Other assets | — | (15,000 | ) | ||||
Accounts payable | 2,230,000 | (97,000 | ) | ||||
Accrued expenses | 1,123,000 | 2,573,000 | |||||
Other liabilites | 58,000 | (41,000 | ) | ||||
Deferred revenue | 7,673,000 | 8,155,000 | |||||
Net cash (used in) provided by operating activities | (14,171,000 | ) | 1,633,000 | ||||
Investing activities: | |||||||
Payments for purchase of property and equipment | (256,000 | ) | (279,000 | ) | |||
Security deposits | 15,000 | — | |||||
Net cash used in investing activities | (241,000 | ) | (279,000 | ) | |||
Financing activities: | |||||||
Proceeds from the exercise of stock options | 154,000 | 165,000 | |||||
Proceeds from stockholder in connection with settlement of stock option exercises | — | 3,943,000 | |||||
Settlement of stock options | — | (2,835,000 | ) | ||||
Proceeds from the exercise of warrants | 1,918,000 | 2,167,000 | |||||
Proceeds from the sale of Series H preferred stock | 7,218,000 | 400,000 | |||||
Proceeds from the sale of Series J preferred stock | — | 47,796,000 | |||||
Repayments of long-term debt | (917,000 | ) | — | ||||
Release of cash restricted for debt repayment | 792,000 | — | |||||
Proceeds from stockholder loan and convertible debt | 620,000 | 25,824,000 | |||||
Net cash provided by financing activities | 9,785,000 | 77,460,000 | |||||
Net (decrease) increase in cash and cash equivalents | (4,627,000 | ) | 78,814,000 | ||||
Cash and cash equivalents at beginning of period | 7,340,000 | 2,713,000 | |||||
Cash and cash equivalents at end of period | $ | 2,713,000 | $ | 81,527,000 | |||
Supplemental disclosures of cash flow information: | |||||||
Interest paid | $ | 17,000 | $ | — | |||
See accompanying notes to consolidated financial statements.
Onconova Therapeutics, Inc.Notes to Consolidated Financial Statements
1. Nature of Business
The Company
Onconova Therapeutics, Inc. (the "Company") was incorporated in the State of Delaware on December 22, 1998 and commenced operations on January 1, 1999. The Company's headquarters are located in Newtown, Pennsylvania. The Company is a clinical-stage biopharmaceutical company focused on discovering and developing novel small molecule drug candidates to treat cancer. Using its proprietary chemistry platform, the Company has created an extensive library of targeted anti-cancer agents designed to work against specific cellular pathways that promote cancer. The Company believes that the drug candidates in its pipeline have the potential to be efficacious in a wide variety of cancers without causing harm to normal cells. The Company has three clinical-stage product candidates and six preclinical programs. To accelerate and broaden the development of rigosertib, the Company's most advanced product candidate, the Company entered into a collaboration and license agreement with Baxter Healthcare SA ("Baxter"), a subsidiary of Baxter International Inc., in 2012 to commercialize rigosertib in Europe. In 2011, the Company entered into a collaboration and license agreement with SymBio Pharmaceuticals Limited ("SymBio") to commercialize rigosertib in Japan and Korea. The Company has retained development and commercialization rights to rigosertib in the rest of the world, including the United States. During 2012, Onconova Europe GmbH was established as a wholly owned subsidiary of the Company for the purpose of further developing business in Europe.
During the year ended December 31, 2012, the Company began to generate significant revenue from its principal operations when it entered into the collaboration and licensing agreement with Baxter. Accordingly, it was determined that the Company emerged from the development stage.
Liquidity
The Company has incurred recurring operating losses since inception. For the year ended December 31, 2012, the Company incurred a net loss of $29,912,000 and as of December 31, 2012, the Company had generated an accumulated deficit of $168,360,000. The Company anticipates operating losses to continue for the foreseeable future due to, among other things, costs related to research funding, development of its product candidates and its preclinical programs, strategic alliances and the development of its administrative organization. The Company will require substantial additional financing to fund its operations and to continue to execute its strategy.
The Company has raised significant capital through the issuance of its redeemable convertible preferred stock, par value $0.01 per share, in ten series denominated as Series A through Series J ("Series A Preferred Stock" through "Series J Preferred Stock," respectively, and collectively the "Preferred Stock"). Upon written request of the holders of at least 66.67% of the then outstanding shares of Series A, Series B, and Series C Preferred Stock collectively, and upon written request of holders of at least a majority of the then outstanding shares of Series D, Series E, and Series F Preferred Stock collectively, as the case may be, the Company is required to redeem the requested number of outstanding shares of Series A Preferred Stock at $5.00 per share, Series B Preferred Stock at $11.50 per share, Series C Preferred Stock at $7.12 per share, and Series D, E and F Preferred Stock at $11.50 per share. Upon written request of holders of at least a majority of the then outstanding shares of Series G Preferred Stock, the Company is required to redeem the outstanding shares of Series G Preferred Stock at a price equal to $11.50 per share. At any time on or after September 21, 2013, upon written request of holders of at least a majority of the then outstanding shares of Series H Preferred Stock, the Company is required to redeem the outstanding shares of Series H Preferred
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
1. Nature of Business (Continued)
Stock at a price equal to $11.50 per share. At any time on or after July 25, 2015, upon written request of holders of at least a majority of the then outstanding shares of Series I Preferred Stock, the Company is required to redeem the outstanding shares of Series I Preferred Stock at a price equal to $11.50 per share. At any time on or after July 27, 2015, upon written request of holders of at least a majority of the then outstanding shares of Series J Preferred Stock, the Company is required to redeem the outstanding shares of Series J Preferred Stock at a price equal to $18.00 per share. At December 31, 2012, Preferred Stock with an aggregate redemption value of $103,122,000 was currently redeemable. During 2013, Preferred Stock with an aggregate redemption value of $23,154,000 will become redeemable at the option of the holder. The Company has not received any notice of redemption as of and through the date the financial statements were available for issuance.
The issued and outstanding shares of Preferred Stock contain conversion features which provide for automatic conversion into shares of common stock, par value $0.01 per share ("Common Stock"), of the Company upon the occurrence of a designated offering, which is defined as a publicly registered offering under the Securities Act of 1933, as amended, in which the gross proceeds after underwriting discount are not less than $25,000,000 at a per share price of at least $16.50 per share, or at a price of at least $11.50 per share with the consent of the holders of a majority of the outstanding Series J Preferred Stock. Due to this conversion provision, the Company expects the issued and outstanding shares of Preferred Stock to convert into shares of Common Stock upon the completion of a designated offering, at which time the redemption rights would terminate. However, there can be no assurances that the Company will complete a designated offering.
Management intends to fund future operations through additional equity offerings, licensing revenue, grants, government contracts and, if any of the Company's product candidates receive marketing approval, future sales of its products. There can be no assurance, however, that the Company will be successful in obtaining financing at the level needed to sustain operations or on terms acceptable to the Company, or that the Company will obtain approvals necessary to market its products or achieve profitability or sustainable, positive cash flow.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business. The Company's ability to continue as a going concern is dependent on its ability to raise additional capital to fund its research and development and commercial programs and meet its obligations, including the potential obligation related to the redemption of the Preferred Stock, which is outside of the Company's control, on a timely basis. If the Company is unable to successfully raise sufficient additional capital, through future debt or equity financings or through strategic and collaborative ventures with third parties, the Company will not have sufficient cash flows and liquidity to fund its planned business operations. In that event, the Company might be forced to limit many, if not all, of its programs and consider other means of creating value for its stockholders, such as licensing to others the development and commercialization of products that it considers valuable and would otherwise likely develop itself. If the Company is unable to raise the necessary capital, it may be forced to curtail all of its activities and, ultimately, potentially cease operations. Even if the Company is able to raise additional capital, such financings may only be available on unattractive terms, or could result in significant dilution of stockholders' interests. The consolidated financial statements do not include any adjustments relating to recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might be necessary should the Company be unable to continue in existence.
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
1. Nature of Business (Continued)
The Company faces many risks associated with companies in the early stages. It also faces risks inherent in its business and its industry generally. These risks include, among others, the following:
2. Summary of Significant Accounting Policies
Basis of Presentation
The consolidated financial statements are prepared in conformity with accounting principles generally accepted in the United States ("GAAP"). The information reported within the Company's financial statements through December 31, 2011 was based solely on the accounts of Onconova Therapeutics, Inc. In December 2012, Onconova Europe GmbH was established as a wholly owned subsidiary of the Company. The financial statements include the consolidated accounts of the Company and its wholly owned subsidiary. All significant intercompany transactions have been eliminated.
Unaudited Pro Forma Presentation
On May 1, 2013, the Company's board of directors authorized management of the Company to confidentially submit a registration statement to the Securities and Exchange Commission (the "SEC") for the Company to sell shares of Common Stock to the public. The unaudited pro forma balance sheet information as of December 31, 2012 assumes the conversion of all outstanding shares of Preferred Stock as of that date into 17,113,481 shares of Common Stock.
The unaudited pro forma net loss per share is computed using the weighted-average number of shares of Common Stock outstanding after giving pro forma effect to the conversion of all issued and outstanding shares of Preferred Stock during the year ended December 31, 2012 into shares of Common Stock as if such conversion had occurred at January 1, 2012, or the date of original issuance, if later.
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Segment Information
Operating segments are defined as components of an enterprise about which separate discrete information is available for evaluation by the chief operating decision maker, or decision-making group, in deciding how to allocate resources and in assessing performance. The Company views its operations and manages its business in one segment, which is the identification and development of oncology therapeutics.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, expenses, other comprehensive income and related disclosures. On an ongoing basis, management evaluates its estimates, including estimates related to clinical trial accruals, warrant liability, option liability and allocation of consideration to multiple element collaborative arrangements. The Company bases its estimates on historical experience and other market-specific or other relevant assumptions that it believes to be reasonable under the circumstances. Actual results may differ from those estimates or assumptions.
In addition, the Company utilizes estimates and assumptions in determining the fair value of its Common Stock. The Company granted stock options at exercise prices not less than the fair value of its Common Stock as determined by the board of directors, with input from management. Management uses the assistance of a third-party valuation firm in estimating the fair value of the Common Stock. The board of directors has determined the estimated fair value of the Common Stock based on a number of objective and subjective factors, including external market conditions affecting the biotechnology industry sector and the historic prices at which the Company sold shares of Preferred Stock.
Concentrations of Credit Risk and Off-Balance Sheet Risk
Financial instruments that potentially subject the Company to concentrations of credit risk are primarily cash, cash equivalents and restricted cash. The Company maintains a portion of its cash and cash equivalent balances in the form of money market accounts with financial institutions that management believes are creditworthy. The Company has no financial instruments with off-balance sheet risk of loss.
Cash and Cash Equivalents
The Company considers all highly liquid investments with original or remaining maturity from the date of purchase of three months or less to be cash equivalents. Cash and cash equivalents include bank demand deposits, marketable securities with maturities of three months or less at purchase, and money market funds that invest primarily in certificates of deposit, commercial paper and U.S. government and U.S. government agency obligations. Cash equivalents are reported at fair value.
Fair Value of Financial Instruments
The carrying amounts reported in the accompanying consolidated financial statements for cash and cash equivalents, accounts payable and accrued liabilities approximate their respective fair values
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
because of the short-term nature of these accounts. The fair value of the warrant liability is discussed in Note 4, "Fair Value Measurements."
Property and Equipment
Property and equipment are stated at cost, less accumulated depreciation. Property and equipment are depreciated using the straight-line method over the estimated useful lives of the assets. Leasehold improvements are amortized over the useful life of the asset or the lease term, whichever is shorter. Maintenance and repairs are expensed as incurred. The following estimated useful lives were used to depreciate the Company's assets:
| ||
| ||
| ||
|
Upon retirement or sale, the cost of the disposed asset and the related accumulated depreciation are removed from the accounts and any resulting gain or loss recognized.
The Company reviews long-lived assets for impairment when events or changes in circumstances indicate that the carrying value of the assets may not be recoverable. Recoverability is measured by comparison of the assets' book value to future net undiscounted cash flows that the assets are expected to generate. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount by which the book value of the assets exceeds their fair value, which is measured based on the projected discounted future net cash flows generated from the assets. No impairment losses have been recorded through December 31, 2012.
Restricted Cash
Under the Company's office lease, the Company is required to provide the landlord a $125,000 letter of credit, which is recorded as restricted cash on the consolidated balance sheets as of each of December 31, 2011 and 2012.
Revenue Recognition
Currently, the Company's revenue is generated primarily through collaborative research and license agreements. The terms of these agreements contain multiple deliverables which may include (i) licenses, (ii) research and development activities, (iii) participation in joint steering committees and (iv) product supply. The terms of these agreements may include nonrefundable upfront license fees, payments for research and development activities, payments based upon the achievement of certain milestones, royalty payments based on product sales derived from the collaboration, and payments for supplying product. In all instances, revenue is recognized only when the price is fixed or determinable, persuasive evidence of an arrangement exists, delivery has occurred or the services have been rendered, collectability of the resulting receivable is reasonably assured, and the Company has fulfilled its performance obligations under the contract.
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Effective January 1, 2011, the Company adopted the Financial Accounting Standards Board ("FASB") Accounting Standards Update ("ASU") No. 2009-13,Multiple-Deliverable Revenue Arrangements ("ASU 2009-13"). This guidance, which applies to multiple element arrangements entered into or materially modified on or after January 1, 2011, amends the criteria for separating and allocating consideration in a multiple element arrangement by modifying the fair value requirements for revenue recognition and eliminating the use of the residual value method. The selling prices of deliverables under an arrangement may be derived using third-party evidence ("TPE"), or a best estimate of selling price ("BESP"), if vendor-specific objective evidence of selling price ("VSOE") is not available. The objective of BESP is to determine the price at which the Company would transact a sale if the element within the license agreement was sold on a standalone basis. Establishing BESP involves management's judgment and considers multiple factors, including market conditions and company-specific factors, including those factors contemplated in negotiating the agreements, as well as internally developed models that include assumptions related to market opportunity, discounted cash flows, estimated development costs, probability of success and the time needed to commercialize a product candidate pursuant to the license. In validating the BESP, management considers whether changes in key assumptions used to determine the BESP will have a significant effect on the allocation of the arrangement consideration between the multiple deliverables. The Company may use third-party valuation specialists to assist it in determining BESP. Deliverables under the arrangement are separate units of accounting if (i) the delivered item has value to the customer on a standalone basis and (ii) if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item is considered probable and substantially within the Company's control. The arrangement consideration that is fixed or determinable at the inception of the arrangement is allocated to the separate units of accounting based on their relative selling prices. The appropriate revenue recognition model is applied to each element and revenue is accordingly recognized as each element is delivered. Management exercises significant judgment in determining whether a deliverable is a separate unit of accounting.
In determining the separate units of accounting, the Company evaluated whether the license had standalone value to the collaborator based on consideration of the relevant facts and circumstances for each arrangement. Factors considered in this determination included the research and development capabilities of the collaborator and the availability of relevant research expertise in the marketplace. In addition, the Company considered whether or not (i) the collaborator could use the license for its intended purpose without the receipt of the remaining deliverables, (ii) the value of the license was dependent on the undelivered items and (iii) the collaborator or other vendors could provide the undelivered items.
Under a collaborative research and license agreement, a steering committee is typically responsible for overseeing the general working relationships, determining the protocols to be followed in the research and development performed and evaluating the results from the continued development of the product. The Company evaluates whether its participation in joint steering committees is a substantive obligation or whether the services are considered inconsequential or perfunctory. The factors the Company considers in determining if its participation in a joint steering committee is a substantive obligation include: (i) which party negotiated or requested the steering committee, (ii) how frequently the steering committee meets, (iii) whether or not there are any penalties or other recourse if the Company does not attend the steering committee meetings, (iv) which party has decision making authority on the steering committee and (v) whether or not the collaborator has the requisite
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
experience and expertise associated with the research and development of the licensed intellectual property.
For all periods presented, whenever the Company determined that an element is delivered over a period of time, revenue was recognized using either a proportional performance model, if a pattern of performance can be determined or a straight-line model over the period of performance, which is typically the research and development term. Progress achieved under the Company's various clinical research organization contracts are typically used as the measure of performance when applying the proportional performance method. At the end of each reporting period, the Company reassessed its cumulative measure of performance and made appropriate adjustments, if necessary. The Company recognized revenue using the proportional performance model whenever the Company could make reasonably reliable estimates of the level of effort required to complete its performance obligations under an arrangement. Revenue recognized under the proportional performance model at each reporting period was determined by multiplying the total expected payments under the contract (excluding royalties and payments contingent upon achievement of milestones) by the ratio of the level of effort incurred to date to the estimated total level of effort required to complete the performance obligations under the arrangement. Revenue was limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the proportional performance model as of each reporting period. Alternatively, if the Company could not make reasonably reliable estimates of the level of effort required to complete its performance obligations under an arrangement, then revenue under the arrangement is recognized on a straight-line basis over the period expected to be required to complete the Company's performance obligations.
Incentive milestone payments may be triggered either by the results of the Company's research efforts or by events external to it, such as regulatory approval to market a product or attaining agreed-upon sales levels. Consideration that is contingent upon achievement of a milestone was recognized in its entirety as revenue in the period in which the milestone was achieved, but only if the consideration earned from the achievement of a milestone meets all the criteria for the milestone to be considered substantive at the inception of the arrangement. For a milestone to be considered substantive, the consideration earned by achieving the milestone must (i) be commensurate with either the Company's performance to achieve the milestone or the enhancement of the value of the item delivered as a result of a specific outcome resulting from the Company's performance to achieve the milestone, (ii) relate solely to past performance and (iii) be reasonable relative to all deliverables and payment terms in the collaboration agreement.
For events for which the occurrences are contingent solely upon the passage of time or are the result of performance by a third party, the contingent payments will be recognized as revenue when payments are earned, the amounts are fixed and determinable and collectability is reasonably assured.
Royalties are recorded as earned in accordance with the contract terms when third party sales can be reliably measured and collectability is reasonably assured.
The Company recognized revenue of $45,490,000 during the year ended December 31, 2012 as a result of its license and collaboration agreement with Baxter. The Company recognized revenue of $227,000 and $503,000 during the years ended December 31, 2011 and 2012, respectively, as a result of its license and collaboration agreement with SymBio. The remaining revenue recognized during the years ended December 31, 2011 and 2012 of $1,260,000 and $197,000, respectively, pertained to research and development services provided by the Company under certain research grants. The Baxter
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
and SymBio agreements are the only agreements that are being accounted for under ASU 2009-13. See Note 15, "License and Collaboration Agreements," for a further discussion of the agreements with Baxter and SymBio.
Research and Development Expenses
Research and development costs are charged to expense as incurred. These costs include, but are not limited to, license fees related to the acquisition of in-licensed products; employee-related expenses, including salaries, benefits and travel; expenses incurred under agreements with contract research organizations and investigative sites that conduct clinical trials and preclinical studies; the cost of acquiring, developing and manufacturing clinical trial materials; facilities, depreciation and other expenses, which include direct and allocated expenses for rent and maintenance of facilities, insurance and other supplies; and costs associated with preclinical activities and regulatory operations.
Costs for certain development activities, such as clinical trials, are recognized based on an evaluation of the progress to completion of specific tasks using data such as patient enrollment, clinical site activations, or information provided to the Company by its vendors with respect to their actual costs incurred. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected in the consolidated financial statements as prepaid or accrued research and development expense, as the case may be.
Comprehensive Loss
Comprehensive loss is defined as the change in equity of a business enterprise during a period from transactions and other events and circumstances from non-owner sources. Comprehensive loss was equal to net loss for all periods presented.
Income Taxes
The Company accounts for income taxes under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases using enacted tax rates in effect for the year in which the differences are expected to affect taxable income. The deferred tax asset primarily includes net operating loss and tax credit carry forwards, accrued expenses not currently deductible and the cumulative temporary differences related to certain research and patent costs, which have been charged to expense in the accompanying statements of operations but have been recorded as assets for income tax purposes. The portion of any deferred tax asset for which it is more likely than not that a tax benefit will not be realized must then be offset by recording a valuation allowance. A full valuation allowance has been established against all of the deferred tax assets (see Note 8, "Income Taxes"), as it is more likely than not that these assets will not be realized given the Company's history of operating losses. The Company recognizes the tax benefit from an uncertain tax position only if it is more likely than not to be sustained upon examination based on the technical merits of the position. The amount for which an exposure exists is measured as the largest amount of benefit determined on a cumulative probability basis that the Company believes is more likely than not to be realized upon ultimate settlement of the position.
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Preferred Stock
The Company accounts for the redemption premium and issuance costs on its Preferred Stock using the effective interest method, accreting such amounts to its Preferred Stock from the date of issuance to the earliest date of redemption.
Preferred Stock Warrants
Freestanding warrants that are related to the purchase of Preferred Stock are classified as liabilities and recorded at fair value regardless of the timing of the redemption feature or the redemption price or the likelihood of redemption. The warrants are subject to re-measurement at each balance sheet date and any change in fair value is recognized as a component of change in fair value of warrant liability in the consolidated statements of operations and comprehensive loss. Pursuant to the terms of these warrants, upon the conversion to Common Stock of the series of Preferred Stock underlying the warrant, the warrants automatically become exercisable for shares of Common Stock based upon the conversion ratio of the underlying Preferred Stock. The consummation of a designated offering (as described in Note 1) will result in the conversion of all outstanding shares of Preferred Stock into shares of Common Stock. Upon such conversion of the underlying series of Preferred Stock, the warrants will be classified as a component of equity and will no longer be subject to re-measurement. The Company will continue to adjust the liability for changes in fair value until the earlier of the exercise or expiration of the warrants or the conversion of the underlying Preferred Stock. The Preferred Stock warrants are classified as Level 3 liabilities (see Note 4 for a discussion of the fair value hierarchy).
Stock-Based Compensation Expense
The Company applies the provisions of FASB Accounting Standards Codification ("ASC") Topic 718, Compensation—Stock Compensation ("ASC 718"), which requires the measurement and recognition of compensation expense for all stock-based awards made to employees and non-employees, including employee stock options. Under certain circumstances, shares acquired upon the exercise of the Company's stock option awards have been purchased by the Company's chairman of the board, who is also a significant stockholder of the Company (See Note 11). As a result, the Company has established a pattern of providing cash settlement alternatives for option holders, and thus the Company's stock-based compensation awards have been accounted for as liability awards. The Company measures liability awards based on the award's intrinsic value on the grant date and then re-measures them at each reporting date until the date of settlement. Compensation expense is recognized on a straight-line basis over the requisite service period for each separately vesting portion of the award. Compensation expense for each period until settlement is based on the change in intrinsic value (or a portion of the change in intrinsic value, depending on the percentage of the requisite service that has been rendered at the reporting date). Changes in the intrinsic value of a liability that occur after the end of the requisite service period are considered compensation expense in the period in which the changes occur.
Clinical Trial Expense Accruals
As part of the process of preparing its financial statements, the Company is required to estimate its expenses resulting from its obligations under contracts with vendors, clinical research organizations
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
and consultants and under clinical site agreements in connection with conducting clinical trials. The financial terms of these contracts are subject to negotiations, which vary from contract to contract and may result in payment flows that do not match the periods over which materials or services are provided under such contracts. The Company's objective is to reflect the appropriate trial expenses in its financial statements by matching those expenses with the period in which services are performed and efforts are expended. The Company accounts for these expenses according to the progress of the trial as measured by patient progression and the timing of various aspects of the trial. The Company determines accrual estimates through financial models taking into account discussion with applicable personnel and outside service providers as to the progress or state of consummation of trials, or the services completed. During the course of a clinical trial, the Company adjusts its clinical expense recognition if actual results differ from its estimates. The Company makes estimates of its accrued expenses as of each balance sheet date based on the facts and circumstances known to it at that time. The Company's clinical trial accruals are dependent upon the timely and accurate reporting of contract research organizations and other third-party vendors. Although the Company does not expect its estimates to be materially different from amounts actually incurred, its understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in it reporting amounts that are too high or too low for any particular period. For the years ended December 31, 2011 and 2012, there were no material adjustments to the Company's prior period estimates of accrued expenses for clinical trials.
Collaboration Arrangements
A collaboration arrangement is defined as a contractual arrangement that has or may have significant financial milestones associated with success-based development, which include certain arrangements the Company has entered into regarding the research and development, manufacture and/or commercialization of products and product candidates. These collaborations generally provide for non-refundable, upfront license fees, research and development and commercial performance milestone payments, cost sharing and royalty payments. The collaboration agreements with third-parties are performed on a "best efforts" basis with no guarantee of either technological or commercial success. The Company evaluates whether an arrangement is a collaboration arrangement at its inception based on the facts and circumstances specific to the arrangement. The Company reevaluates whether an arrangement qualifies or continues to qualify as a collaboration arrangement whenever there is a change in the anticipated or actual ultimate commercial success of the endeavor. See Note 15, "License and Collaboration Agreements," for a discussion of the Company's current collaborations with Baxter and SymBio.
Basic and Diluted Net Loss Per Share of Common Stock
Basic net loss per share of common stock is computed by dividing net loss applicable to common stockholders by the weighted-average number of shares of Common Stock outstanding during the period, excluding the dilutive effects of Preferred Stock, warrants to purchase Preferred Stock and stock options. Diluted net loss per share of common stock is computed by dividing the net loss applicable to common stockholders by the sum of the weighted-average number of shares of Common Stock outstanding during the period plus the potential dilutive effects of Preferred Stock and warrants to purchase Preferred Stock, and stock options outstanding during the period calculated in accordance with the treasury stock method, but are excluded if their effect is anti-dilutive. Because the impact of
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
these items is anti-dilutive during periods of net loss, there was no difference between basic and diluted net loss per share of Common Stock for the years ended December 31, 2011 and 2012.
Recent Accounting Pronouncements
As described above, effective January 1, 2011, the Company prospectively adopted ASU 2009-13. The amendments in this guidance enable vendors to account for products or services separately rather than as a combined unit upon meeting certain criteria and to establish a hierarchy for determining the selling price of a deliverable. In addition, a vendor can determine a best estimate of selling price, in a manner that is consistent with that used to determine the price to sell the deliverable on a standalone basis, if the vendor does not have vendor-specific objective evidence or third-party evidence of selling price. This guidance also eliminates the use of the residual method and requires a vendor to allocate revenue using the relative selling price method. The Company's adoption of ASU 2009-13 did not have a significant impact on its consolidated financial position, results of operations or cash flows.
In June 2011, FASB issued ASU No. 2011-05, "Comprehensive Income (ASC Topic 220): Presentation of Comprehensive Income" ("ASU 2011-05"). This accounting update eliminates the option to present the components of other comprehensive income as part of the statement of stockholders' equity. Instead, comprehensive income must be presented in either a single continuous statement of comprehensive income, which contains two sections, net income and other comprehensive income, or in two separate but consecutive statements. ASU 2011-05 was effective for fiscal periods beginning after December 15, 2011 with early adoption permitted. The Company's retrospective adoption of ASU 2011-05 did not have a significant impact on its consolidated financial position, results of operations or cash flows.
In February 2013, FASB issued ASU No. 2013-02, "Comprehensive Income (Topic 220): Reporting of Amounts Reclassified Out of Accumulated Other Comprehensive Income" ("ASU 2013-02"). ASU 2013-02 requires companies to present either in a single note or parenthetically on the face of the financial statements, the effect of significant amounts reclassified from each component of accumulated other comprehensive income based on its source and the income statement line items affected by the reclassification. This guidance is effective for annual reporting periods beginning after December 15, 2012. The Company believes the adoption of this standard will not have a significant impact on its consolidated financial position, results of operations or cash flows.
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
3. Property and Equipment
Property and equipment and related accumulated depreciation are as follows:
| | December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | |||||
Laboratory equipment | $ | 729,000 | $ | 764,000 | |||
Software | 82,000 | 91,000 | |||||
Computer and office equipment | 359,000 | 323,000 | |||||
Leasehold improvements | 468,000 | 633,000 | |||||
| 1,638,000 | 1,811,000 | ||||||
Less accumulated depreciation | (1,131,000 | ) | (1,348,000 | ) | |||
Property and equipment, net | $ | 507,000 | $ | 463,000 | |||
Depreciation and amortization expense was $316,000 and $319,000 for the years ended December 31, 2011 and 2012, respectively.
4. Fair Value Measurements
The Company applies various valuation approaches in determining the fair value of its financial assets and liabilities within a hierarchy that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the asset or liability based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company's assumptions about the inputs that market participants would use in pricing the asset or liability and are developed based on the best information available under the circumstances. The fair value hierarchy is broken down into three levels based on the source of inputs as follows:
Level 1—Valuations based on unadjusted quoted prices in active markets for identical assets or liabilities that the Company has the ability to access.
Level 2—Valuations based on quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active and models for which all significant inputs are observable, either directly or indirectly.
Level 3—Valuations based on inputs that are unobservable and significant to the overall fair value measurement.
The availability of observable inputs can vary among the various types of financial assets and liabilities. To the extent that the valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. In certain cases, the inputs used to measure fair value may fall into different levels of the fair value hierarchy. In such cases, for financial statement disclosure purposes, the level in the fair value hierarchy within which the fair value measurement is classified is based on the lowest level input that is significant to the overall fair value measurement.
The Company had no assets or liabilities classified as Level 1 or Level 2. The Series G Preferred Stock warrants (see Note 10) are classified as Level 3. The fair values of these instruments are determined using models based on market observable inputs and management judgment. There were
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
4. Fair Value Measurements (Continued)
no material re-measurements of fair value during the years ended December 31, 2011 and 2012 with respect to financial assets and liabilities, other than those assets and liabilities that are measured at fair value on a recurring basis.
The Company has classified the Series G Preferred Stock warrants as a liability and has re-measured the liability to estimated fair value at December 31, 2011 and 2012, using the Black-Scholes option pricing model using the following assumptions: contractual life according to the remaining terms of the warrants, no dividend yield, weighted average risk-free interest rate of 0.01% and 0.31% at December 31, 2011 and 2012, respectively, and weighted average volatility of 62.42% and 64.87% at December 31, 2011 and 2012, respectively
The following fair value hierarchy table presents information about the Company's financial assets and liabilities measured at fair value on a recurring basis as of December 31, 2011 and 2012.
| | Fair Value Measurement As of December 31, 2011 | | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Balance As of December 31, 2011 | ||||||||||||
| | Level 1 | Level 2 | Level 3 | ||||||||||
Warrant liability | $ | — | $ | — | $ | 1,064,000 | $ | 1,064,000 | |||||
Total | $ | — | $ | — | $ | 1,064,000 | $ | 1,064,000 | |||||
| | Fair Value Measurement As of December 31, 2012 | | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Balance As of December 31, 2012 | ||||||||||||
| | Level 1 | Level 2 | Level 3 | ||||||||||
Warrant liability | $ | — | $ | — | $ | 62,000 | $ | 62,000 | |||||
Total | $ | — | $ | — | $ | 62,000 | $ | 62,000 | |||||
The following table presents a reconciliation of the Company's liabilities measured at fair value on a recurring basis using significant unobservable inputs (Level 3) for the years ended December 31, 2011 and 2012:
| | Warrant Liability | |||
|---|---|---|---|---|
Balance at December 31, 2010 | $ | 2,758,000 | ||
Settlements of warrant liability awards | (407,000 | ) | ||
Change in fair value upon re-measurement | (1,287,000 | ) | ||
Balance at December 31, 2011 | 1,064,000 | |||
Settlements of warrant liability awards | (635,000 | ) | ||
Expiration of warrant liability awards | (975,000 | ) | ||
Change in fair value upon re-measurement | 608,000 | |||
Balance at December 31, 2012 | $ | 62,000 | ||
The fair values of cash equivalents, accounts payable and accrued liabilities approximate their respective carrying values due to the short-term nature of these accounts. Based on the borrowing rates currently available to the Company for debt with similar terms and consideration of default and credit risk, the carrying value of the stockholder loan (see Note 7) approximated its fair value at
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
4. Fair Value Measurements (Continued)
December 31, 2011. There were no transfers between Level 1 and Level 2 in any of the periods reported.
5. Net Loss Per Share of Common Stock
The following table sets forth the computation of basic and diluted earnings per share for the years ended December 31, 2011 and 2012:
| | Year Ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | |||||
Basic and diluted net loss per share of common stock: | |||||||
Net loss | $ | (26,294,000 | ) | $ | (29,912,000 | ) | |
Accretion to redemption value of preferred stock | (4,020,000 | ) | (3,953,000 | ) | |||
Net loss applicable to common stockholders | $ | (30,314,000 | ) | $ | (33,865,000 | ) | |
Weighted average shares of common stock outstanding | 2,849,159 | 2,941,782 | |||||
Net loss per share of common stock—basic and diluted | $ | (10.64 | ) | $ | (11.51 | ) | |
The following potentially dilutive securities outstanding at December 31, 2011 and 2012 have been excluded from the computation of diluted weighted average shares outstanding, as they would be antidilutive:
| | December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | |||||
Preferred Stock | 11,431,700 | 17,113,481 | |||||
Warrants | 568,577 | 6,128 | |||||
Stock options | 2,702,468 | 3,418,228 | |||||
| 14,702,745 | 20,537,837 | ||||||
6. Accrued Expenses
Accrued expenses are as follows:
| | December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | |||||
Research and development | $ | 931,000 | $ | 3,521,000 | |||
Payroll | 431,000 | 247,000 | |||||
Other | 318,000 | 157,000 | |||||
| $ | 1,680,000 | $ | 3,925,000 | ||||
7. Debt
The Company had a Loan and Security Agreement with a bank, the outstanding principal of which was $917,000 at December 31, 2010. Interest on the loan was payable at a rate of London Inter Bank
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
7. Debt (Continued)
Offering Rate ("LIBOR") plus 2.75%. In connection with the Loan and Security Agreement, the Company issued 6,128 Series G Preferred Stock warrants (see Note 10). On August 11, 2011, the Company paid off the outstanding balance of the loan.
In December 2011, the chairman of the Company's board of directors, who is also a significant stockholder of the Company, advanced $620,000 to the Company to fund operations. Interest accrued at 10% per annum. The stockholder loan was exchanged for convertible promissory notes in April 2012 (see Note 9).
8. Income Taxes
The Company accounts for income taxes under FASB ASC 740 ("ASC 740"). Deferred income tax assets and liabilities are determined based upon differences between financial reporting and tax bases of assets and liabilities, which are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse.
As of December 31, 2012, the Company had federal net operating loss ("NOL") carry forwards of $91,261,000, state NOL carry forwards of $75,147,000 and research and development tax credit carry forwards of $13,923,000, which are available to reduce future taxable income. The federal NOL and tax credit carry forwards will begin to expire at various dates starting in 2019. The state NOL carry forwards will begin to expire at various dates starting in 2016. The NOL carry forwards are subject to review and possible adjustment by the Internal Revenue Service and state tax authorities. NOL and tax credit carry forwards may become subject to an annual limitation in the event of certain cumulative changes in the ownership interest of significant shareholders over a three-year period in excess of 50%, as defined under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, as well as similar state tax provisions. This could limit the amount of NOLs that the Company can utilize annually to offset future taxable income or tax liabilities. The amount of the annual limitation, if any, will be determined based on the value of the Company immediately prior to the ownership change. Subsequent ownership changes may further affect the limitation in future years.
The Company's reserves related to taxes are based on a determination of whether and how much of a tax benefit taken by the Company in its tax filings or positions is more likely than not to be realized. The Company recognized no material adjustment for unrecognized income tax benefits. Through December 31, 2012, the Company had no unrecognized tax benefits or related interest and penalties accrued.
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
8. Income Taxes (Continued)
The principal components of the Company's deferred tax assets are as follows:
| | December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | |||||
Deferred tax assets: | |||||||
Net operating loss carryovers | $ | 36,018,000 | $ | 35,921,000 | |||
R&D tax credits | 8,326,000 | 13,868,000 | |||||
Non-qualified stock options | 610,000 | 1,057,000 | |||||
Deferred revenue | 1,436,000 | 4,246,000 | |||||
Charitable contributions | 2,000 | 6,000 | |||||
Accrued expenses | 110,000 | 40,000 | |||||
Fixed assets | 77,000 | 159,000 | |||||
Deferred tax assets | 46,579,000 | 55,297,000 | |||||
Less valuation allowance | (46,579,000 | ) | (55,297,000 | ) | |||
Net deferred tax assets | $ | — | $ | — | |||
ASC 740 requires a valuation allowance to reduce the deferred tax assets reported if, based on the weight of available evidence, it is more likely than not that some portion or all of the deferred tax assets will not be realized. After consideration of all the evidence, both positive and negative, the Company has recorded a full valuation allowance against its deferred tax assets at December 31, 2011 and 2012, respectively, because the Company's management has determined that is it more likely than not that these assets will not be fully realized.
A reconciliation of income tax expense (benefit) at the statutory federal income tax rate and income taxes as reflected in the financial statements is as follows:
| | Year Ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2011 | 2012 | |||||
Federal income tax expense at statutory rate | 34.0 | % | 34.0 | % | |||
Permanent items | (5.6 | ) | (25.1 | ) | |||
State income tax, net of federal benefit | 6.2 | 1.8 | |||||
Tax credits | 22.1 | 18.5 | |||||
Deferred true-ups | (5.1 | ) | — | ||||
Change in valuation allowance | (51.6 | ) | (29.1 | ) | |||
Other | — | (0.1 | ) | ||||
Effective income tax rate | 0 | % | 0 | % | |||
9. Convertible Promissory Notes
In March 2012, the Company offered to its stockholders the opportunity to participate in a $30,000,000 private placement of convertible promissory notes (the "Convertible Debt Offering"). From April through July 2012, the Company had aggregate closings of the Convertible Debt Offering of $26,444,000, including $620,000 from the principal that remained outstanding on a stockholder loan at December 31, 2011.
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
9. Convertible Promissory Notes (Continued)
The convertible promissory notes issued in the Convertible Debt Offering carried a 10% interest rate calculated on the basis of a 360-day year and were scheduled to mature on June 29, 2013. The convertible promissory notes provided the holder the right to convert any portion of the outstanding principal and interest in exchange for equity instruments upon the completion of an initial public offering that generated aggregate proceeds in excess of $25,000,000 (the "IPO Scenario") or upon the completion of an equity offering that generated aggregate proceeds in excess of $15,000,000 (the "Equity Scenario"). In the event of a conversion under the IPO Scenario, the holder would have received shares of Common Stock equal to the offering price that was initially offered to the public. In the event of a conversion under the Equity Scenario, the holder would have received equity instruments equivalent to those issued in the Equity Scenario at a conversion price equal to the lower of $11.00 per share or the original issuance price of the underlying equity instrument. If the Company merged with another company while the convertible promissory notes were still outstanding, immediately after which the Company's stockholders owned less than 50% of the voting stock of the surviving company, then each convertible promissory note would have been required to be redeemed for an amount equal to two times the outstanding principal amount together with any unpaid and accrued interest.
In July 2012, the Company amended and restated its certificate of incorporation and designated Series I Preferred Stock. The Company and the holders of the convertible promissory notes amended the Equity Scenario provision of the notes to permit the holders to convert the convertible promissory notes into shares of Series I Preferred Stock at a conversion price of $11.00 per share.
In connection with the amendment of the convertible promissory notes, the notes were also analyzed to determine the existence of a beneficial conversion feature. The Company concluded that an $8,176,000 contingent beneficial conversion feature existed related to the Equity Scenario as of July 2012. The fair value of the Series I Preferred Stock used to calculate the value of the beneficial conversion feature was determined by management with the assistance of a third-party valuation firm.
On July 27, 2012, the holders of the convertible promissory notes exercised their right to convert the outstanding principal and interest into Series I Preferred Stock. Upon conversion, the holders received 2,443,328 shares of Series I Preferred Stock, and the Company recorded interest expense of $8,176,000, which was equal to the amount of the unamortized contingent beneficial conversion feature.
10. Preferred Stock and Stockholders' Deficit
Capitalization
As of December 31, 2012, the Company's ninth amended and restated certificate of incorporation reflected the following authorized shares: 48,693,408 shares of capital stock, consisting of (i) 400,000 shares of Series A Preferred Stock, (ii) 1,200,000 shares of Series B Preferred Stock, (iii) 1,200,000 shares of Series C Preferred Stock, (iv) 1,625,000 shares of Series D Preferred Stock, (v) 1,650,000 shares of Series E Preferred Stock, (vi) 2,000,000 shares of Series F Preferred Stock, (vii) 2,700,000 shares of Series G Preferred Stock, (viii) 2,042,950 of Series H Preferred Stock, (ix) 2,700,000 shares of Series I Preferred Stock, (x) 3,030,303 shares of Series J Preferred Stock and (xi) 30,145,155 shares of Common Stock.
The Company issued shares of Series H Preferred Stock in three closings in 2011: On February 17, 2011, the Company raised $700,000 in gross proceeds from the issuance of 71,488 shares of Series H Preferred Stock; On June 2, 2011, the Company raised $1,326,000 in gross proceeds from the issuance
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
10. Preferred Stock and Stockholders' Deficit (Continued)
of 135,391 shares of Series H Preferred Stock; and on September 19, 2011, the Company raised $5,996,000 in gross proceeds from the issuance of 612,450 shares of Series H Preferred Stock.
In July 2012, the Company issued 2,433,328 shares of Series I Preferred Stock in exchange for the conversion of the convertible promissory notes and accrued interest in the amount of $26,444,000 and $323,000, respectively (See Note 9, "Convertible Promissory Notes"). The effective conversion price was $11.00 per share. Additionally, in July 2012, the Company issued 3,030,303 shares of Series J Preferred Stock at $16.50 per share for gross proceeds of $50,000,000. Issuance costs associated with this offering were $2,204,000.
Series A Preferred Stock was originally issued at $5.00 per share; Series B Preferred Stock was issued at $5.75 per share; Series C Preferred Stock was issued at $3.56 per share; Series D Preferred Stock was issued at $4.67 per share; Series E Preferred Stock was issued at $9.76 per share; Series F Preferred Stock was issued at $11.00 per share; Series G and Series H Preferred Stock was issued at a price of $9.79 per share; Series I Preferred Stock was issued at a price of $11.00 per share; and Series J Preferred Stock was issued at a price of $16.50 per share.
The following is the composition of share capital as of the dates indicated:
| | Authorized | Issued and Outstanding | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | December 31, | December 31, | |||||||||||
| | 2011 | 2012 | 2011 | 2012 | |||||||||
Shares of $0.01 par value per share: | |||||||||||||
Common stock | 19,200,000 | 30,145,155 | 2,889,747 | 3,474,473 | |||||||||
Series A Preferred Stock | 400,000 | 400,000 | 107,000 | 107,000 | |||||||||
Series B Preferred Stock | 1,200,000 | 1,200,000 | 1,107,189 | 1,107,189 | |||||||||
Series C Preferred Stock | 1,200,000 | 1,200,000 | 1,069,946 | 1,069,946 | |||||||||
Series D Preferred Stock | 1,625,000 | 1,625,000 | 1,583,568 | 1,583,568 | |||||||||
Series E Preferred Stock | 1,650,000 | 1,650,000 | 1,633,082 | 1,633,082 | |||||||||
Series F Preferred Stock | 2,000,000 | 2,000,000 | 2,000,000 | 2,000,000 | |||||||||
Series G Preferred Stock | 2,700,000 | 2,700,000 | 1,712,960 | 1,934,359 | |||||||||
Series H Preferred Stock | 2,042,950 | 2,042,950 | 2,013,424 | 2,013,424 | |||||||||
Series I Preferred Stock | — | 2,700,000 | — | 2,433,328 | |||||||||
Series J Preferred Stock | — | 3,030,303 | — | 3,030,303 | |||||||||
Total Preferred Stock | 12,817,950 | 18,548,253 | 11,227,169 | 16,912,199 | |||||||||
Total | 32,017,950 | 48,693,408 | 14,116,916 | 20,386,672 | |||||||||
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
10. Preferred Stock and Stockholders' Deficit (Continued)
The following is the activity of the Preferred Stock for the years ended December 31, 2011 and 2012:
| | January 1, 2011 | Issuance of Preferred Stock | Exercise of warrants | Accretion of redemption premium and issuance costs on Preferred Stock | December 31, 2011 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Series A | ||||||||||||||||
Shares | 107,000 | — | — | — | 107,000 | |||||||||||
Amount | $ | 535,000 | $ | — | $ | — | $ | — | $ | 535,000 | ||||||
Series B | ||||||||||||||||
Shares | 1,107,189 | — | — | — | 1,107,189 | |||||||||||
Amount | $ | 12,733,000 | $ | — | $ | — | $ | — | $ | 12,733,000 | ||||||
Series C | ||||||||||||||||
Shares | 1,069,946 | — | — | — | 1,069,946 | |||||||||||
Amount | $ | 7,618,000 | $ | — | $ | — | $ | — | $ | 7,618,000 | ||||||
Series D | ||||||||||||||||
Shares | 1,583,568 | — | — | — | 1,583,568 | |||||||||||
Amount | $ | 18,211,000 | $ | — | $ | — | $ | — | $ | 18,211,000 | ||||||
Series E | ||||||||||||||||
Shares | 1,633,082 | — | — | — | 1,633,082 | |||||||||||
Amount | $ | 18,780,000 | $ | — | $ | — | $ | — | $ | 18,780,000 | ||||||
Series F | ||||||||||||||||
Shares | 2,000,000 | — | — | — | 2,000,000 | |||||||||||
Amount | $ | 23,000,000 | $ | — | $ | — | $ | — | $ | 23,000,000 | ||||||
Series G | ||||||||||||||||
Shares | 1,516,955 | — | 196,005 | — | 1,712,960 | |||||||||||
Amount | $ | 13,183,000 | $ | — | $ | 2,325,000 | $ | 3,066,000 | $ | 18,574,000 | ||||||
Series H | ||||||||||||||||
Shares | 1,194,095 | 819,329 | — | — | 2,013,424 | |||||||||||
Amount | $ | 11,674,000 | $ | 7,918,000 | $ | — | $ | 954,000 | $ | 20,546,000 | ||||||
Series I | ||||||||||||||||
Shares | — | — | — | — | — | |||||||||||
Amount | $ | — | $ | — | $ | — | $ | — | $ | — | ||||||
Series J | ||||||||||||||||
Shares | — | — | — | — | — | |||||||||||
Amount | $ | — | $ | — | $ | — | $ | — | $ | — | ||||||
Total | ||||||||||||||||
Shares | 10,211,835 | 819,329 | 196,005 | — | 11,227,169 | |||||||||||
Amount | $ | 105,734,000 | $ | 7,918,000 | $ | 2,325,000 | $ | 4,020,000 | $ | 119,997,000 | ||||||
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
10. Preferred Stock and Stockholders' Deficit (Continued)
| | January 1, 2012 | Issuance of Preferred Stock | Exercise of warrants | Accretion of redemption premium and issuance costs on Preferred Stock | December 31, 2012 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Series A | ||||||||||||||||
Shares | 107,000 | — | — | — | 107,000 | |||||||||||
Amount | $ | 535,000 | $ | — | $ | — | $ | — | $ | 535,000 | ||||||
Series B | ||||||||||||||||
Shares | 1,107,189 | — | — | — | 1,107,189 | |||||||||||
Amount | $ | 12,733,000 | $ | — | $ | — | $ | — | $ | 12,733,000 | ||||||
Series C | ||||||||||||||||
Shares | 1,069,946 | — | — | — | 1,069,946 | |||||||||||
Amount | $ | 7,618,000 | $ | — | $ | — | $ | — | $ | 7,618,000 | ||||||
Series D | ||||||||||||||||
Shares | 1,583,568 | — | — | — | 1,583,568 | |||||||||||
Amount | $ | 18,211,000 | $ | — | $ | — | $ | — | $ | 18,211,000 | ||||||
Series E | ||||||||||||||||
Shares | 1,633,082 | — | — | — | 1,633,082 | |||||||||||
Amount | $ | 18,780,000 | $ | — | $ | — | $ | — | $ | 18,780,000 | ||||||
Series F | ||||||||||||||||
Shares | 2,000,000 | — | — | — | 2,000,000 | |||||||||||
Amount | $ | 23,000,000 | $ | — | $ | — | $ | — | $ | 23,000,000 | ||||||
Series G | ||||||||||||||||
Shares | 1,712,960 | — | 221,399 | — | 1,934,359 | |||||||||||
Amount | $ | 18,574,000 | $ | — | $ | 2,802,000 | $ | 1,443,000 | $ | 22,819,000 | ||||||
Series H | ||||||||||||||||
Shares | 2,013,424 | — | — | — | 2,013,424 | |||||||||||
Amount | $ | 20,546,000 | $ | — | $ | — | $ | 1,459,000 | $ | 22,005,000 | ||||||
Series I | ||||||||||||||||
Shares | — | 2,433,328 | — | — | 2,433,328 | |||||||||||
Amount | $ | — | $ | 26,767,000 | $ | — | $ | 166,000 | $ | 26,933,000 | ||||||
Series J | ||||||||||||||||
Shares | — | 3,030,303 | — | — | 3,030,303 | |||||||||||
Amount | $ | — | $ | 47,796,000 | $ | — | $ | 885,000 | $ | 48,681,000 | ||||||
Total | ||||||||||||||||
Shares | 11,227,169 | 5,463,631 | 221,399 | — | 16,912,199 | |||||||||||
Amount | $ | 119,997,000 | $ | 74,563,000 | $ | 2,802,000 | $ | 3,953,000 | $ | 201,315,000 | ||||||
Voting
Each holder of outstanding shares of Preferred Stock has the right to one vote for each share of Common Stock into which such Preferred Stock could then be converted. The holders of shares of Preferred Stock have full voting rights and powers equal to the voting rights and powers of shares of Common Stock and are entitled to notice of any stockholders' meeting and vote together with the
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
10. Preferred Stock and Stockholders' Deficit (Continued)
holders of Common Stock, with respect to any question upon which holders of shares of Common Stock have the right to vote, as a single class, including without limitation, actions to increase or decrease the aggregate number of authorized shares of Common Stock and/or Preferred Stock.
Dividends
The holders of each share of Series A, Series B, Series C, Series D, Series E, Series F, Series G, Series H, Series I and Series J Preferred Stock are entitled to receive dividends when, as, and if declared by the Company's board of directors in the following order of preference: (i) the Series D, Series E, Series F Series G, Series H, Series I and Series J Preferred Stock, which rank pari passu; (ii) the Series B and Series C Preferred Stock, which rank pari passu; (iii) the Series A Preferred Stock; and then (iv) Common Stock.
Liquidation
The assets of the Company legally available for distribution to stockholders will be distributed in the following order of priority: (i) the holders of the shares of Series D, Series E, Series F, Series G, Series H, Series I and Series J Preferred Stock, which rank pari passu; (ii) the holders of the shares of Series B and Series C Preferred Stock, which rank pari passu; (iii) the holders of the shares of Series A Preferred Stock; and (iv) the holders of the shares of Common Stock. Each series of Preferred Stock is entitled to receive an amount per share equal to the greater of (1) the original issuance price for such series, plus all declared but unpaid dividends thereon, or (2) the amount that the holders of such series would receive per share of Common Stock if all shares of such series of Preferred Stock were converted to Common Stock immediately prior to such liquidation. If upon a deemed liquidation event, the assets of the Company are insufficient to make payment in full to all holders of a series of Preferred Stock, then such assets shall be distributed among the holders of such series of Preferred Stock at the time outstanding ratably in proportion to the full amount to which they would otherwise be respectively entitled. The holders of Common Stock are entitled to receive, after the payment of the liquidation preference of all Preferred Stock outstanding, the remaining assets of the Company on a pro rata basis.
Conversion
Each issued and outstanding share of Preferred Stock is convertible into Common Stock at the holder's option at any time after the date of issuance or automatically upon the occurrence of certain events as defined in the Company's ninth amended and restated certificate of incorporation, at a defined conversion rate. At December 31, 2012, the number of shares of Common Stock into which one share of each series of Preferred Stock was convertible was as follows: the Series A Preferred Stock, 1.07; the Series B Preferred Stock, 1.13; the Series C Preferred Stock, 1.00; the Series D Preferred Stock, 1.00; the Series E Preferred Stock, 1.00; the Series F Preferred Stock, 1.03; the Series G Preferred Stock, 1.00; the Series H Preferred Stock, 1.00; the Series I Preferred Stock, 1.00; and the Series J Preferred Stock, 1.00.
The conversion price for each share of Preferred Stock is subject to adjustment upon the occurrence of certain events. The conversion price of each share of a series of Preferred Stock is adjusted if the Company issues additional shares, subject to specified exceptions, at a price lower than
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
10. Preferred Stock and Stockholders' Deficit (Continued)
the current conversion price for such series, which is measured and recognized if the contingency occurs.
Redemption
To the extent it is then lawfully able to do so, the Company is required at any time, upon written request of the holders of at least 66.67% of the then outstanding Series A, Series B and Series C Preferred Stock collectively, or upon written request of at least a majority of the then outstanding shares of Series D, Series E and Series F Preferred Stock collectively, in each case as determined on an as-converted to common stock basis, to redeem the requested number of outstanding shares of Series A Preferred Stock at $5.00 per share, Series B Preferred Stock at $11.50 per share, and Series C Preferred Stock at $7.12 per share, and/or Series D, E and F Preferred Stock at $11.50 per share, as the case may be.
In addition, to the extent it is lawfully able to do so, the Company is required at any time, upon written request of the holders of at least a majority of the then outstanding shares of Series G Preferred Stock, to redeem, from the holders requesting such redemption, the requested number of outstanding shares of Series G Preferred Stock at $11.50 per share.
To the extent it is lawfully able to do so, the Company is required at any time on or after September 21, 2013, upon written request of the holders of at least a majority of the then outstanding shares of Series H Preferred Stock, to redeem, from the holders requesting such redemption, the requested number of outstanding shares of Series H Preferred Stock at $11.50 per share.
To the extent it is lawfully able to do so, the Company is required at any time on or after July 25, 2015, upon written request of the holders of at least a majority of the then outstanding shares of Series I Preferred Stock, to redeem, from the holders requesting such redemption, the requested number of outstanding shares of Series I Preferred Stock at $11.50 per share.
To the extent it is lawfully able to do so, the Company is required at any time on or after July 27, 2015, upon written request of the holders of at least a majority of the then outstanding shares of Series J Preferred Stock, to redeem, from the holders requesting such redemption, the requested number of outstanding shares of Series J Preferred Stock at $18.00 per share.
If, upon any applicable redemption date, defined as sixty days after the Company receives the written request for redemption, the funds of the Company legally available for redemption of Preferred Stock are insufficient to redeem the total number of shares to be redeemed on that date, those funds that are legally available shall be used to redeem the maximum possible number of shares, ratably among the holders of such shares to be redeemed. All remaining shares not redeemed shall remain outstanding until such time as additional funds become legally available for redemption.
If more than one series of Preferred Stock is contemporaneously subject to redemption, the redemption rights of the Preferred Stock shall follow the liquidation order of priority.
As of December 31, 2012, Preferred Stock with an aggregate redemption value of $103,122,000 was currently redeemable. During 2013 and 2015, additional shares of Preferred Stock with an aggregate redemption value of $23,154,000 and $82,529,000, respectively, will become redeemable at the option of the holders of Preferred Stock.
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
10. Preferred Stock and Stockholders' Deficit (Continued)
Series G Preferred Stock warrant transactions
The Company issued 6,128 Series G Preferred Stock warrants in connection with a Loan and Security Agreement (see Note 7). Additionally, the Company issued one Series G Preferred Stock warrant for every two shares of Series G Preferred Stock purchased in 2009 and 2010. The warrants were initially recorded at their fair value calculated using the Black-Scholes model, with the following weighted average assumptions: exercise price of $9.79, share price of $9.79, expected term of three years, risk-free rate of 1.52% and volatility of 85.46%. The warrants are classified as liabilities because they are exercisable for Preferred Stock, and the value of the warrants is adjusted to current fair value at each reporting period end. For the years ended December 31, 2011 and 2012, the Company recorded $1,287,000 and $367,000, respectively, in the consolidated statements of operations and comprehensive loss related to the change in the fair value of the outstanding warrants.
Series G Preferred Stock warrant transactions for the years ended December 31, 2011 and 2012 consisted of the following:
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
11. Stock-Based Compensation
In January 2008, the board of directors approved the 2007 Equity Compensation Plan (the "Plan"), which amended and restated the Company's 1999 Stock Based Compensation Plan, which provides for the granting of incentive and nonqualified stock options and restricted stock to its employees, directors and consultants at the discretion of the board of directors. Under the Plan, the Company increases the number of shares reserved for issuance under the Plan such that the number of reserved shares is equal to 17% of the fully diluted shares calculated annually on December 10th. At December 31, 2011 and 2012, 3,498,579 and 4,107,831 shares of Common Stock were reserved under the Plan, respectively. Stock options may be granted with exercise prices of not less than the estimated fair value of the Company's common stock on the date of grant and generally vest over a period of up to four years. Stock options granted under the Plan generally expire no later than ten years from the
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
11. Stock-Based Compensation (Continued)
date of grant. A summary of stock option activity for the years ended December 31, 2011 and 2012 is as follows:
| | Number of Options | Weighted- Average Exercise Price | Weighted- Average Remaining Contractual Term (in years) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Outstanding at December 31, 2010 | 2,430,659 | $ | 3.31 | 6.09 | ||||||
Granted | 459,233 | 4.60 | ||||||||
Exercised | (133,162 | ) | 1.15 | |||||||
Forfeited | (54,262 | ) | 4.33 | |||||||
Outstanding at December 31, 2011 | 2,702,468 | 3.61 | 6.39 | |||||||
Granted | 1,303,486 | 9.38 | ||||||||
Exercised | (584,726 | ) | 2.27 | |||||||
Forfeited | (3,000 | ) | 1.15 | |||||||
Outstanding at December 31, 2012 | 3,418,228 | $ | 6.04 | 7.52 | ||||||
Vested or expected to vest at December 31, 2012 | 3,418,228 | $ | 6.04 | 7.52 | ||||||
Exercisable at December 31, 2012 | 2,251,304 | $ | 4.63 | 6.47 | ||||||
At December 31, 2011 and 2012, the aggregate intrinsic value of the option liability recorded was $2,648,000 and $11,967,000, respectively. During 2011 and 2012, the Company granted 459,233 and 1,303,486 options, respectively, at an intrinsic value of $0 at the grant date. At certain times throughout the Company's history, the chairman of the Company's board of directors, who is also a significant stockholder of the Company (the "Significant Holder"), has afforded option holders the opportunity for liquidity in transactions in which options were exercised and the shares of Common Stock issued in connection therewith were simultaneously purchased by the Significant Holder (each, a "Purchase Transaction"). The Company issued an aggregate of 584,726 shares of Common Stock upon the exercise of options in 2012, of which 395,863 shares were then purchased by the Significant Holder in Purchase Transactions. The Company received proceeds of $3,943,000 from the Significant Holder of which $2,835,000 was paid to the option holders upon exercise and settlement of the option liabilities. Because the Company has established a pattern of providing cash settlement alternatives for option holders, the Company has accounted for its stock-based compensation awards as liability awards, the fair value of which is then re-measured at each balance sheet date. Upon the exercise of stock options in 2011 and 2012, stock option liabilities of $458,000 and $4,525,000, respectively, were reclassified to stockholders' deficit.
Stock-based compensation expense includes stock options granted to employees and non-employees and has been reported in the Company's statements of operations and comprehensive loss in either research and development expenses or general and administrative expenses depending on the function performed by the optionee. For the years ended December 31, 2011 and 2012, $3,000 and $6,645,000 was recorded in research and development expenses, respectively. For the years ended December 31, 2011 and 2012, $3,000 and $7,199,000 was recorded in general and administrative expenses, respectively. As of December 31, 2012, the Company had unrecognized stock-based compensation of $1,338,000 related to unvested stock options held by employees and non-employees which is expected to vest over
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
11. Stock-Based Compensation (Continued)
a weighted average period of 1.93 years. No net tax benefits related to the stock-based compensation costs have been recognized since the Company's inception.
A roll forward of the stock option liability balance for the years ended December 31, 2011 and 2012 is as follows:
Balance at December 31, 2010 | $ | 3,100,000 | ||
Change in intrinsic value upon re-measurement | 6,000 | |||
Settlements of option liability awards | (458,000 | ) | ||
Balance at December 31, 2011 | 2,648,000 | |||
Change in intrinsic value upon re-measurement | 13,844,000 | |||
Settlements of option liability awards | (4,525,000 | ) | ||
Balance at December 31, 2012 | $ | 11,967,000 | ||
Information with respect to stock options outstanding and exercisable at December 31, 2012 is as follows:
| Exercise Price | Shares | Weighted Average Remaining Contractual Life (years) | Exercisable | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| $ | 0.75 | 4,000 | 0.01 | 4,000 | |||||||
| 1.00 | 217,445 | 1.53 | 217,445 | ||||||||
| 1.15 | 2,000 | 0.01 | 2,000 | ||||||||
| 2.00 | 88,934 | 3.07 | 88,934 | ||||||||
| 4.32 | 529,150 | 7.19 | 462,849 | ||||||||
| 4.50 | 560,250 | 4.68 | 560,250 | ||||||||
| 4.60 | 790,414 | 8.33 | 622,121 | ||||||||
| 5.65 | 66,635 | 9.31 | 41,158 | ||||||||
| 9.96 | 1,159,400 | 9.72 | 252,547 | ||||||||
| Total | 3,418,228 | 7.52 | 2,251,304 | ||||||||
12. Employee Benefit Plan
In October 2007, the Company established a 401(k) Retirement Savings Plan. Employees are eligible to participate in the plan as soon as they join the Company if they are at least 21 years of age and work a minimum of 1,000 hours per year. The Company matches $0.50 for every dollar of the first 6% of payroll that employees invest, up to the legal limit. Employer contributions vest over four years at the rate of 25% per year. For the years ended December 31, 2011 and 2012, the Company contributed $131,000, and $159,000, respectively.
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
13. Commitments and Contingencies
Operating leases
In November 2010, the Company entered into a lease for 3,117 square feet of office space in Pennington, New Jersey. The lease had an original term of two years, with an option for two additional years. For the first two years of the lease, the Company was obligated to pay $4,400 per month, or $53,000 annually, beginning when possession of the facility was taken on February 1, 2011. The Company was required to provide the landlord a $125,000 letter of credit, the collateral for which is recorded as restricted cash on the consolidated balance sheets. This lease was renewed on February 1, 2012 with a 3.5% increase in the rent, to $5,000 per month. On October 2, 2012, the Company leased an additional 2,130 square feet of office space for $3,100 per month, or $38,000 annually.
In January 2007, the Company entered into a lease for 8,100 square feet of office and lab space in Newtown, Pennsylvania, and in October 2009, the Company and the landlord amended the lease to add three additional one-year options to extend the lease term. The Company exercised the first option for the period from April 1, 2012 to March 31, 2013 and the second option for the period from April 1, 2013 to March 31, 2014 for rent of $11,000 per month. In September 2012, the Company sub-leased an additional 1,356 square feet of office space for one year for $1,600 per month, or $19,000 annually.
Future minimum lease payments under these non-cancellable leases having terms in excess of one year as of December 31, 2012 are as follows:
| | December 31, 2012 | |||
|---|---|---|---|---|
2013 | $ | 256,000 | ||
2014 | 103,000 | |||
2015 | 6,000 | |||
Total minimum lease payments | $ | 365,000 | ||
Rent expense was $186,000 and $233,000 for the years ended December 31, 2011 and 2012, respectively.
Employment agreements
The Company has entered into employment agreements with certain of its executives. The agreements provide for, among other things, salary, bonus and severance payments.
14. Research Agreements
The Company has entered into various licensing and right-to-sublicense agreements with educational institutions for the exclusive use of patents and patent applications, as well as any patents that may develop from research being conducted by such educational institutions in the field of anticancer therapy, genes and proteins. Results from this research have been licensed to the Company pursuant to these agreements. Under one of these agreements with Temple University ("Temple"), the Company is required to make annual maintenance payments to Temple and royalty payments based upon a percentage of sales generated from any products covered by the licensed patents, with minimum specified royalty payments. As no sales had been generated through December 31, 2012 under the licensed patents, the Company did not incur any royalty expenses for the years ended December 31, 2011 and 2012. In addition, the Company is required to pay Temple 25% of any sublicensing fees received by the Company. In September 2011, the Company made a payment to Temple in the amount
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
14. Research Agreements (Continued)
of $1,875,000 in connection with the collaboration agreement the Company executed in July 2011 with SymBio. Such payment was recorded in the consolidated statement of operations as research and development expenses. In 2012, the Company became obligated to pay Temple $12,500,000 in connection with the collaboration agreement the Company executed in 2012 with Baxter. Such expense was recorded in the consolidated statement of operations as research and development expenses. As of December 31, 2012, $1,405,000 of this amount was not yet paid and was included in accrued expenses and other current liabilities in the consolidated balance sheets.
In May 2010, the Company signed a funding agreement with the Leukemia and Lymphoma Society ("LLS") to fund the development of rigosertib. Under this agreement, the Company was entitled to receive milestone payments of up to $10,000,000 through 2013 in connection with clinical trials to be conducted. The aggregate milestone payment amount was subsequently reduced to $8,000,000 pursuant to an amendment signed in January 2013, after which LLS was not obligated to fund any further amounts. In the event that the research is successful, the Company must proceed with commercialization of the licensed product or repay the amount funded. In addition, LLS is entitled to receive regulatory and commercial milestone payments and royalties from the Company based on the Company's net sales of the licensed product, with the amount of such royalties not to exceed three times the amount funded ($24,000,000). During the year ended December 31, 2012, in connection with the execution of the Baxter agreement (Note 15), the Company paid $1,000,000 to LLS and has recorded this amount in research and development expenses. This payment reduced the maximum milestone and royalty payment obligation under this agreement to $23,000,000 at December 31, 2012. During the years ended December 31, 2011 and 2012, the Company received milestone payments of $1,900,000 and $4,100,000, respectively. As a result of the potential obligation to repay the funds under this arrangement, the milestone payments received through December 31, 2012 amounting to $8,000,000 have been recorded as deferred revenue and will be recognized as revenue commencing with the resolution of the repayment contingency.
15. License and Collaboration Agreements
Baxter Agreement
In September 2012, the Company entered into a development and license agreement with Baxter granting Baxter an exclusive, royalty-bearing license for the research, development, commercialization and manufacture (in specified instances) of rigosertib in all therapeutic indications in Europe (the "Baxter Territory"). Baxter is a shareholder in the Company and invested in Series J Preferred Stock issued in July 2012.
Under the terms of the agreement, the Company is initially required to perform research and development to advance three initial rigosertib indications, rigosertib intravenous ("IV") in higher risk myelodysplastic syndrome ("MDS") patients, rigosertib IV in pancreatic cancer patients and rigosertib oral in lower risk MDS patients, through phase 3, phase 3 and phase 2 clinical trials, respectively.
If an additional phase 3 clinical trial beyond the current phase 3 clinical trial in process for rigosertib IV in higher risk MDS patients is required to obtain marketing approval in the Baxter Territory, the Company could require Baxter to fund a percentage of the costs of such additional trial up to a specified maximum. At the completion of the current phase 3 trial for rigosertib IV in pancreatic cancer and the current phase 2 trial for rigosertib oral in lower risk MDS patients and the review of the resulting data and findings, the Company and Baxter will decide whether or not to pursue
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
15. License and Collaboration Agreements (Continued)
further development of rigosertib for these indications. If the Company and Baxter mutually agree to progress the development of rigosertib IV in pancreatic cancer patients and rigosertib oral in lower risk MDS patients, then certain milestone payments will be payable to the Company, and the Company will be required to use its commercially reasonable efforts to progress the development of rigosertib for these indications to a drug approval application in the Baxter Territory. The Company and Baxter will work together for potential future rigosertib indications, beyond the initial indications noted above. Generally, if Baxter chooses to participate in the development of additional indications, Baxter will be responsible for a percentage of all research and development costs and expenses and the Company could earn additional milestone payments. Baxter has full responsibility for all commercialization activities for the product in the Baxter Territory, at Baxter's sole cost and expense.
The Company and Baxter have agreed to negotiate a supply agreement under terms satisfactory to both parties whereby the Company will supply Baxter with Baxter's required levels of product to support commercialization efforts in the Baxter Territory. Baxter also has the right to engage third parties for the manufacture and supply of its requirements for the licensed product.
Under the terms of the agreement, Baxter made an upfront payment of $50,000,000. The Company is eligible to receive pre-commercial milestone payments of up to an aggregate of $512,500,000 if specified development and regulatory milestones are achieved. The potential pre-commercial development milestone payments to us include the following:
The Company may also receive up to $337,500,000 in milestone payments for regulatory approvals of the three rigosertib indications specified in the arrangement with Baxter, each of which may be up to and in excess of $100,000,000. The Company is also potentially eligible to receive an additional $20,000,000 pre-commercial milestone payment related to the timing of regulatory approval of the MDS IV indication in Europe. In addition to these pre-commercial milestones, the Company is eligible to receive up to an aggregate of $250,000,000 in milestone payments based on Baxter's achievement of pre-specified threshold levels of annual net sales of rigosertib. The Company will also be entitled to receive royalties at percentage rates ranging from the low-teens to the low-twenties on net sales of rigosertib by Baxter in the Baxter Territory.
The agreement with Baxter will remain in effect until the expiration of all applicable royalty terms and satisfaction of all payment obligations in each licensed country, unless terminated earlier due to the uncured material breach or bankruptcy of a party, force majeure, or in the event of a specified commercial failure. The Company may terminate the agreement in the event that Baxter brings a challenge against it in relation to the licensed patents. Baxter may terminate the agreement without cause commencing after a specified period of time from the execution of the agreement.
The Company determined that the deliverables under the Baxter agreement include the exclusive, royalty-bearing, sublicensable license to rigosertib and the research and development services to be performed by the Company. The Company concluded that the license had standalone value to Baxter
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
15. License and Collaboration Agreements (Continued)
and was separable from the research and development services because the license is sublicensable, there are no restrictions as to Baxter's use of the license and Baxter has significant research capabilities in this field. The Company was not able to establish VSOE or TPE for either the license or the research and development services and instead allocated the arrangement consideration between the license and research and development services based on their relative selling prices using BESP. Management developed the BESP of the license using a discounted cash flow model, taking into consideration assumptions including the development and commercialization timeline, discount rate and probability of success. Management utilized a third party valuation specialist to assist with the determination of BESP of the license. Management estimated the selling price of the research and development services using third party costs and a discounted cash flow model. The estimated selling prices utilized assumptions including internal estimates of research and development personnel needed to perform the research and development services; and estimates of expected cash outflows to third parties for services and supplies over the expected period that the services will be performed.
The key assumptions in these models included the following market conditions and entity-specific factors: (a) the specific rights provided under the license, (b) the stage of development of rigosertib and estimated remaining development and commercialization timelines, (c) the probability of successfully developing and commercializing rigosertib, (d) the market size including the associated sales figures which generate royalty revenue, (e) cost of goods sold, which was assumed to be a specified percentage of revenues based on estimated cost of goods sold of a typical oncology product, (f) sales and marketing costs, which were based on the costs required to field an oncology sales force and marketing group, including external costs required to promote an oncology product, (g) the expected product life of rigosertib assuming commercialization and (h) the competitive environment. The Company utilized a discount rate of 16%, representing the cost of capital derived from returns on equity for comparable companies.
Based on management's analyses, it was determined that the BESP of the license was $120,000,000 and the BESP of the research and development services was $20,600,000. As noted above, the Company received an up-front payment of $50,000,000 under the Baxter agreement, which represents the allocable agreement consideration. Based on the respective BESPs, this payment was allocated $42,400,000 to the license and $7,600,000 to the research and development services. Since the delivery of the license occurred upon the execution of the Baxter agreement and there was no general right of return, $42,400,000 of the $50,000,000 upfront payment was recognized upon the execution of the Baxter agreement. The portion allocated to research and development services is being recognized over the period of performance on a proportional performance basis through March 31, 2014. Management estimated the period of performance to be the period necessary for completion of the non-contingent obligations to perform research and development services required to advance the three formulations of rigosertib described above. For the year ended December 31, 2012, the Company recognized $3,100,000 of research and development revenue under the Baxter agreement.
The Company and Baxter have agreed to establish a joint committee to facilitate the governance and oversight of the parties' activities under the agreements. Management considered whether participation on the joint committee may be a deliverable and determined that it was not a deliverable. Had management considered participation on the joint committee as a deliverable, it would not have had a material impact on the accounting for the arrangement based on the analysis of the estimated selling price of such participation.
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
15. License and Collaboration Agreements (Continued)
As noted above, in July 2012, Baxter purchased Series J Preferred Stock. Because the Series J Preferred Stock was acquired within several months of the Baxter development and license agreement, management considered whether the Preferred Stock was issued at fair value and if not, whether the consideration received for the Preferred Stock ($50,000,000) or for the collaboration and license agreement ($50,000,000) should be allocated in the financial statements in a manner differently than the prices stated in the agreements. Management, with the assistance of an outside valuation specialist, determined that the price paid by Baxter for the Series J Preferred Stock approximated its fair value, and therefore the consideration received under the agreements was allocated in accordance with terms of the individual agreements.
SymBio Agreement
In July 2011, the Company entered into a license agreement with SymBio, as subsequently amended, granting SymBio an exclusive, royalty-bearing license for the development and commercialization of rigosertib in Japan and Korea. Under the SymBio license agreement, SymBio is obligated to use commercially reasonable efforts to develop and obtain market approval for rigosertib inside the licensed territory and the Company has similar obligations outside of the licensed territory. The Company has also entered into an agreement with SymBio providing for it to supply SymBio with development-stage product. Under the SymBio license agreement, the Company also agreed to supply commercial product to SymBio under specified terms that will be included in a commercial supply agreement to be negotiated prior to the first commercial sale of rigosertib. The supply of development-stage product and the supply of commercial product will be at the Company's cost plus a defined profit margin. Sales of development-stage product have been de minimis. The Company has additionally granted SymBio a right of first negotiation to license or obtain the rights to develop and commercialize compounds having a chemical structure similar to rigosertib in the licensed territory.
Under the terms of the SymBio license agreement, the Company received an upfront payment of $7,500,000. The Company is eligible to receive milestone payments of up to an aggregate of $33,000,000 from SymBio upon the achievement of specified development and regulatory milestones for specified indications. Of the development milestones, $3,000,000 is due after enrollment of the first patient in the event a decision is made, after the Company's interim analysis, to start a phase 3 clinical trial of rigosertib IV in combination with gemcitabine for pancreatic cancer patients in the United States. Of the regulatory milestones, $5,000,000 is due upon receipt of marketing approval in the United States for rigosertib IV in higher risk MDS patients, $3,000,000 is due upon receipt of marketing approval in Japan for rigosertib IV in higher risk MDS patients, $5,000,000 is due upon receipt of marketing approval in the United States for rigosertib oral in lower risk MDS patients, $5,000,000 is due upon receipt of marketing approval in Japan for rigosertib oral in lower risk MDS patients, $5,000,000 is due upon receipt of marketing approval in the United States for rigosertib IV in combination with gemcitabine in pancreatic cancer patients, and $3,000,000 is due upon receipt of marketing approval in Japan for rigosertib IV in combination with gemcitabine in pancreatic cancer patients. Furthermore, upon receipt of marketing approval in the United States and Japan for an additional specified indication of rigosertib, which we are currently not pursuing, an aggregate of $4,000,000 would be due. In addition to these pre-commercial milestones, the Company is eligible to receive tiered milestone payments based upon annual net sales of rigosertib by SymBio of up to an aggregate of $30,000,000. Further, under the terms of the SymBio license agreement, SymBio will make royalty payments to the Company at percentage rates ranging from the mid-teens to 20% based on net sales of rigosertib by SymBio.
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
15. License and Collaboration Agreements (Continued)
Royalties will be payable under the SymBio agreement on a country-by-country basis in the licensed territory, until the later of the expiration of marketing exclusivity in those countries, a specified period of time after first commercial sale of rigosertib in such country, or the expiration of all valid claims of the licensed patents covering rigosertib or the manufacture or use of rigosertib in such country. If no valid claim exists covering the composition of matter of rigosertib or the use of or treatment with rigosertib in a particular country before the expiration of the royalty term, and specified competing products achieve a specified market share percentage in such country, SymBio's obligation to pay the Company royalties will continue at a reduced royalty rate until the end of the royalty term. In addition, the applicable royalties payable to the Company may be reduced if SymBio is required to pay royalties to third-parties for licenses to intellectual property rights necessary to develop, use, manufacture or commercialize rigosertib in the licensed territory. The license agreement with SymBio will remain in effect until the expiration of the royalty term. However, the SymBio license agreement may be terminated earlier due to the uncured material breach or bankruptcy of a party, or force majeure. If SymBio terminates the license agreement in these circumstances, its licenses to rigosertib will survive, subject to SymBio's milestone and royalty obligations, which SymBio may elect to defer and offset against any damages that may be determined to be due from the Company. In addition, the Company may terminate the license agreement in the event that SymBio brings a challenge against it in relation to the licensed patents, and SymBio may terminate the license agreement without cause by providing the Company with written notice within a specified period of time in advance of termination.
The Company determined that the deliverables under the SymBio agreement include the exclusive, royalty-bearing, sublicensable license to rigosertib, the research and development services to be provided by the Company and its obligation to serve on a joint committee. The Company concluded that the license did not have standalone value to SymBio and was not separable from the research and development services, because of the uncertainty of SymBio's ability to develop rigosertib in the SymBio territory on its own and the uncertainty of SymBio's ability to sublicense rigosertib and recover a substantial portion of the original upfront payment of $7,500,000 paid by SymBio to the Company.
The supply of rigosertib for SymBio's commercial requirements is contingent upon the receipt of regulatory approvals to commercialize rigosertib in Japan and Korea. Because the Company's commercial supply obligation was contingent upon the receipt of future regulatory approvals, and there were no binding commitments or firm purchase orders pending for commercial supply at or near the execution of the agreement, the commercial supply obligation is deemed to be contingent and is not valued as a deliverable under the SymBio agreement. If SymBio orders the supplies from the Company, the Company expects the pricing for this supply to equal its third-party manufacturing cost plus a pre-negotiated percentage, which will not result in a significant incremental discount to market rates.
Due to the lack of standalone value for the license, research and development services, and joint committee obligation, the upfront payment is being recognized ratably using the straight line method through December 2027, the expected term of the agreement. For the year ended December 31, 2011 and 2012, the Company recognized revenues of $227,000 and $455,000, respectively, under this agreement. In addition, the Company recognized revenues of $48,000 for the year ended December 31, 2012 related to the supply agreement with SymBio.
16. Joint Venture
In December 2012, the Company agreed to create a joint venture with GVK Biosciences Private Limited ("GVK BIO"), a private limited company located in India. The resulting joint venture,
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
16. Joint Venture (Continued)
GBO, LLC, a Delaware limited liability company ("GBO"), was formed on April 1, 2013. The purpose of GBO is to collaborate on and develop two new programs through filing of an investigational new drug application ("IND") and/or conducting proof of concept studies using the Company's technology platform.
GVK BIO will make an initial capital contribution of $500,000 in exchange for a 10% interest in GBO, and the Company will make an initial capital contribution of a sub-license to all the intellectual property controlled by the Company related to the two specified programs in exchange for a 90% interest. In addition, GVK BIO will make additional capital contributions (with a minimum of $500,000) necessary to complete the major studies plus the exercise of the proof on concept study option leading to deliverables under the programs including an IND approval for one product and completion of IND studies for the other product. The GVK BIO percentage interest in GBO accordingly may change from the initial 10% to up to 50%, depending on the amount of its total capital contributions.
For thirty days following the 15-month anniversary of the commencement of either of the two programs, the Company will have an option to (i) cancel the license and (ii) purchase all rights in and to that program. There are three of these buy-back scenarios depending on the stage of development of the underlying assets. GVK BIO will have operational control of GBO and the Company will have strategic and scientific controls. As of December 31, 2012, neither the Company nor GVK BIO had contributed any property to GBO.
17. Related-Party Transactions
In May 2010, the Company entered into a research agreement with the Mount Sinai School of Medicine ("Mount Sinai"), with which certain of its stockholders are affiliated. Mount Sinai is undertaking research on behalf of the Company on the terms set forth in the agreement. Mount Sinai, in connection with the Company, will prepare applications for patents generated from the research. Results from all projects will belong exclusively to Mount Sinai, but the Company will have an exclusive option to license any inventions. The initial term of this research agreement was one year, with options to extend by mutual agreement. The Company's initial payment to Mount Sinai under the agreement for the period of May 2010 to May 2011 was $738,000, to be paid in quarterly installments of $185,000. The agreement was subsequently amended to include the period of May 2011 through July 2012, for additional total consideration of $738,000 to be paid by the Company in quarterly installments of $185,000. The Company entered into a second amendment to the agreement during 2012, further extending the agreement period from July 2012 through July 2013 for additional total consideration of $758,000 to be paid in quarterly installments of $190,000. In 2011 and 2012, the Company paid Mount Sinai an aggregate of $554,000 and $1,230,000, respectively. At December 31, 2011 and 2012, the Company owed Mount Sinai $185,000 and $0, respectively, which is included in accounts payable on the consolidated balance sheets.
The Company outsources the synthesis of some of its chemical compounds to vendors in the United States and in foreign countries. A supplier, of which one of the Company's preferred stockholders is an owner, produces one of these compounds under contract. The Company's aggregate payments for these services for the years ended December 31, 2011 and 2012 were $6,000 and $157,000, respectively. The Company owed this supplier, as of December 31, 2011 and 2012, $0 and $107,000, respectively, which amounts are included in accounts payable in the accompanying consolidated balance sheets.
Onconova Therapeutics, Inc.
Notes to Consolidated Financial Statements (Continued)
17. Related-Party Transactions (Continued)
The Company purchases chemical compounds from a corporation owned by one of its stockholders. The Company's aggregate purchases from this supplier for the years ended December 31, 2011 and 2012 were $970,000 and $410,000, respectively. The Company owed this supplier, as of December 31, 2011 and 2012, $538,000 and $0, respectively, which amounts are included in accounts payable in the accompanying consolidated balance sheets. The Company also rents office space in Pennington, New Jersey from a related corporation.
18. Subsequent Events
The Company has completed an evaluation of all subsequent events through May 3, 2013, the date on which these financial statements were available to be issued, to ensurethe filing is made.
Information furnished under Items 2.02 or 7.01 (or corresponding information furnished under Item 9.01 or included as an exhibit) in any past or future Current Report on Form 8-K that these financial statements include appropriate disclosure of events both recognized in the consolidated financial statements as of December 31, 2012, and events which occurred subsequently but were not recognized in the financial statements.
As discussed in Note 11, at certain times throughout the Company's history, the Significant Holder has afforded option holders the opportunity for liquidity in Purchase Transactions. On April 23, 2013, the Company distributed a notification letter to all equity award holders under the Plan advising them that Purchase Transactions will no longer occur, unless, at the time of a Purchase Transaction, the option holder has held the Common Stock issued upon exercise of options for a period of greater than six months prior to selling such Common Stock to the Significant Holder and that any such sale to the Significant Holder would be at the fair value of the Common Stock on the date of such sale. Based on these new criteria for Purchase Transactions, the Company remeasured options outstanding under the Plan as of April 23, 2013 to their intrinsic value and reclassified such options from liabilities to stockholders' deficit within the Company's consolidated balance sheets.
Onconova Therapeutics, Inc.Condensed Consolidated Balance Sheets
| | | | Pro Forma | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | December 31, 2012 | March 31, 2013 | March 31, 2013 | |||||||
| | | (unaudited) | (unaudited) | |||||||
Assets | ||||||||||
Current assets: | ||||||||||
Cash and cash equivalents | $ | 81,527,000 | $ | 67,307,000 | ||||||
Prepaid expenses and other current assets | 1,725,000 | 2,666,000 | ||||||||
Total current assets | 83,252,000 | 69,973,000 | ||||||||
Property and equipment, net | 463,000 | 649,000 | ||||||||
Restricted cash | 125,000 | 125,000 | ||||||||
Other non-current assets | 12,000 | 12,000 | ||||||||
Total assets | $ | 83,852,000 | $ | 70,759,000 | ||||||
Liabilities, redeemable convertible preferred stock and stockholders' (deficit) equity | ||||||||||
Current liabilities: | ||||||||||
Accounts payable | $ | 5,517,000 | $ | 4,243,000 | ||||||
Accrued expenses and other current liabilities | 3,925,000 | 5,559,000 | ||||||||
Warrant liability | 62,000 | 48,000 | ||||||||
Stock option liability | 11,967,000 | 14,394,000 | ||||||||
Deferred revenue | 3,907,000 | 3,986,000 | ||||||||
Total current liabilities | 25,378,000 | 28,230,000 | ||||||||
Deferred revenue, non-current | 15,421,000 | 14,250,000 | ||||||||
Other | 44,000 | 64,000 | ||||||||
Total liabilities | 40,843,000 | 42,544,000 | ||||||||
Commitments and contingencies | ||||||||||
Redeemable convertible preferred stock, $0.01 par value per share, 18,548,253 shares authorized at December 31, 2012 and March 31, 2013, 16,912,199 shares issued and outstanding at December 31, 2012 and March 31, 2013, liquidation preference of $205,760,000 at March 31, 2013, and no shares issued and outstanding at March 31, 2013 (pro forma) | 201,315,000 | 202,334,000 | $ | — | ||||||
Stockholders' (deficit) equity: | ||||||||||
Common stock, $0.01 par value, 30,145,155 shares authorized at December 31, 2012 and March 31, 2013, 3,474,473 and 3,478,473 shares issued and outstanding at December 31, 2012 and March 31, 2013 and 20,591,954 shares issued and outstanding at March 31, 2013 (pro forma) | 35,000 | 35,000 | 206,000 | |||||||
Additional paid in capital | 10,019,000 | 9,044,000 | 211,207,000 | |||||||
Accumulated other comprehensive income | — | 7,000 | 7,000 | |||||||
Accumulated deficit | (168,360,000 | ) | (183,205,000 | ) | (183,205,000 | ) | ||||
Total stockholders' (deficit) equity | (158,306,000 | ) | (174,119,000 | ) | 28,215,000 | |||||
Total liabilities, redeemable convertible preferred stock and stockholders' (deficit) equity | $ | 83,852,000 | $ | 70,759,000 | $ | 70,759,000 | ||||
See accompanying notes to condensed consolidated financial statements.
Onconova Therapeutics, Inc.Condensed Consolidated Statements of Operations and Comprehensive Loss (unaudited)
| | Three Months Ended March 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2012 | 2013 | |||||
Revenue | $ | 198,000 | $ | 1,116,000 | |||
Operating expenses: | |||||||
General and administrative | 2,460,000 | 3,346,000 | |||||
Research and development | 8,448,000 | 12,756,000 | |||||
Total operating expenses | 10,908,000 | 16,102,000 | |||||
Loss from operations | (10,710,000 | ) | (14,986,000 | ) | |||
Change in fair value of warrant liability | (609,000 | ) | 14,000 | ||||
Interest expense | (21,000 | ) | — | ||||
Other income, net | 541,000 | 127,000 | |||||
Net loss before income taxes | (10,799,000 | ) | (14,845,000 | ) | |||
Income taxes | — | — | |||||
Net loss | (10,799,000 | ) | (14,845,000 | ) | |||
Other comprehensive income: | |||||||
Foreign currency translation | — | 7,000 | |||||
Comprehensive loss | $ | (10,799,000 | ) | $ | (14,838,000 | ) | |
Reconciliation of net loss to net loss applicable to common stockholders: | |||||||
Net loss | $ | (10,799,000 | ) | $ | (14,845,000 | ) | |
Accretion of redeemable convertible preferred stock | (1,231,000 | ) | (1,019,000 | ) | |||
Net loss applicable to common stockholders | $ | (12,030,000 | ) | $ | (15,864,000 | ) | |
Net loss per share of common stock, basic and diluted | $ | (4.15 | ) | $ | (4.56 | ) | |
Basic and diluted weighted average shares outstanding | 2,897,347 | 3,475,673 | |||||
Pro forma net loss per share of common stock, basic and diluted | $ | (0.77 | ) | ||||
Basic and diluted pro forma weighted average shares outstanding | 20,589,154 | ||||||
See accompanying notes to condensed consolidated financial statements.
Onconova Therapeutics, Inc.Condensed Consolidated Statements of Redeemable Convertible Preferred Stock and Stockholders' Deficit
| | | | Stockholders' Deficit | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Redeemable Convertible Preferred Stock | ||||||||||||||||||||||||
| | Common Stock | | Accumulated other comprehensive income | | | ||||||||||||||||||||
| | Additional Paid in Capital | Accumulated deficit | | ||||||||||||||||||||||
| | Shares | Amount | Shares | Amount | Total | ||||||||||||||||||||
Balance at December 31, 2011 | 11,227,169 | $ | 119,997,000 | 2,889,747 | $ | 29,000 | $ | — | $ | — | $ | (138,448,000 | ) | $ | (138,419,000 | ) | |||||||||
Issuance of preferred stock, net of issuance costs | 3,030,303 | 47,796,000 | — | — | — | — | — | — | |||||||||||||||||
Exercise of stock options | — | — | 584,726 | 6,000 | 4,688,000 | — | — | 4,694,000 | |||||||||||||||||
Proceeds from stockholder in connection with settlement of stock option exercises | — | — | — | — | 3,943,000 | — | — | 3,943,000 | |||||||||||||||||
Settlement of stock option liabilities | — | — | — | — | (2,835,000 | ) | — | — | (2,835,000 | ) | |||||||||||||||
Issuance of preferred stock upon exercise of warrants | 221,399 | 2,802,000 | — | — | — | — | — | — | |||||||||||||||||
Exchange of convertible debt and preferred stock | 2,433,328 | 26,767,000 | — | — | — | — | — | — | |||||||||||||||||
Beneficial conversion feature on convertible debt | — | — | — | — | 8,176,000 | — | — | 8,176,000 | |||||||||||||||||
Accretion of preferred stock to redemption value | — | 3,953,000 | — | — | (3,953,000 | ) | — | — | (3,953,000 | ) | |||||||||||||||
Net loss | — | — | — | — | — | — | (29,912,000 | ) | (29,912,000 | ) | |||||||||||||||
Balance at December 31, 2012 | 16,912,199 | 201,315,000 | 3,474,473 | 35,000 | 10,019,000 | — | (168,360,000 | ) | (158,306,000 | ) | |||||||||||||||
Exercise of stock options | — | — | 4,000 | — | 44,000 | — | — | 44,000 | |||||||||||||||||
Accretion of preferred stock to redemption value | — | 1,019,000 | — | — | (1,019,000 | ) | — | — | (1,019,000 | ) | |||||||||||||||
Other comprehensive income | — | — | — | — | — | 7,000 | — | 7,000 | |||||||||||||||||
Net loss | — | — | — | — | — | — | (14,845,000 | ) | (14,845,000 | ) | |||||||||||||||
Balance at March 31, 2013 (unaudited) | 16,912,199 | $ | 202,334,000 | 3,478,473 | $ | 35,000 | $ | 9,044,000 | $ | 7,000 | $ | (183,205,000 | ) | $ | (174,119,000 | ) | |||||||||
See accompanying notes to condensed consolidated financial statements.
Onconova Therapeutics, Inc.Condensed Consolidated Statements of Cash Flows (unaudited)
| | Three Months Ended March 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2012 | 2013 | |||||
Operating activities: | |||||||
Net loss | $ | (10,799,000 | ) | $ | (14,845,000 | ) | |
Adjustment to reconcile net loss to net cash used in operating activities: | |||||||
Depreciation and amortization | 79,000 | 98,000 | |||||
Change in fair value of warrant liabilities | 609,000 | (14,000 | ) | ||||
Stock compensation expense | 2,437,000 | 2,465,000 | |||||
Changes in assets and liabilities: | |||||||
Grants receivable | 60,000 | — | |||||
Prepaid expenses and other current assets | 198,000 | (941,000 | ) | ||||
Accounts payable | 221,000 | (1,274,000 | ) | ||||
Accrued expenses | 160,000 | 1,634,000 | |||||
Other liabilites | (40,000 | ) | 20,000 | ||||
Deferred revenue | 987,000 | (1,092,000 | ) | ||||
Net cash used in operating activities | (6,088,000 | ) | (13,949,000 | ) | |||
Investing activities: | |||||||
Payments for purchase of property and equipment | (4,000 | ) | (284,000 | ) | |||
Net cash used in investing activities | (4,000 | ) | (284,000 | ) | |||
Financing activities: | |||||||
Proceeds from the exercise of stock options | 9,000 | 6,000 | |||||
Proceeds from the exercise of warrants | 428,000 | — | |||||
Proceeds from the sale of Series H preferred stock | 400,000 | — | |||||
Proceeds from stockholder loan and convertible debt | 2,800,000 | — | |||||
Net cash provided by financing activities | 3,637,000 | 6,000 | |||||
Effect of foreign currency translation on cash | — | 7,000 | |||||
Net decrease in cash and cash equivalents | (2,455,000 | ) | (14,220,000 | ) | |||
Cash and cash equivalents at beginning of period | 2,713,000 | 81,527,000 | |||||
Cash and cash equivalents at end of period | $ | 258,000 | $ | 67,307,000 | |||
See accompanying notes to condensed consolidated financial statements.
Onconova Therapeutics, Inc.Notes to Condensed Consolidated Financial Statements(Unaudited)
1. Nature of Business
The Company
Onconova Therapeutics, Inc. (the "Company") was incorporated in the State of Delaware on December 22, 1998 and commenced operations on January 1, 1999. The Company's headquarters are located in Newtown, Pennsylvania. The Company is a clinical-stage biopharmaceutical company focused on discovering and developing novel small molecule drug candidates to treat cancer. Using its proprietary chemistry platform, the Company has created an extensive library of targeted anti-cancer agents designed to work against specific cellular pathways that promote cancer. The Company believes that the drug candidates in its pipeline have the potential to be efficacious in a wide variety of cancers without causing harm to normal cells. The Company has three clinical-stage product candidates and six preclinical programs. To accelerate and broaden the development of rigosertib, the Company's most advanced product candidate, the Company entered into a collaboration and license agreement with Baxter Healthcare SA ("Baxter"), a subsidiary of Baxter International Inc., in 2012 to commercialize rigosertib in Europe. In 2011, the Company entered into a collaboration and license agreement with SymBio Pharmaceuticals Limited ("SymBio") to commercialize rigosertib in Japan and Korea. The Company has retained development and commercialization rights to rigosertib in the rest of the world, including the United States. During 2012, Onconova Europe GmbH was established as a wholly owned subsidiary of the Company for the purpose of further developing business in Europe.
Liquidity
The Company has incurred recurring operating losses since inception. For the three months ended March 31, 2013, the Company incurred a net loss of $14,845,000 and as of March 31, 2013, the Company had generated an accumulated deficit of $183,205,000. The Company anticipates operating losses to continue for the foreseeable future due to, among other things, costs related to research funding, development of its product candidates and its preclinical programs, strategic alliances and the development of its administrative organization. The Company will require substantial additional financing to fund its operations and to continue to execute its strategy.
The Company has raised significant capital through the issuance of its redeemable convertible preferred stock, par value $0.01 per share, in ten series denominated as Series A through Series J ("Series A Preferred Stock" through "Series J Preferred Stock," respectively, and collectively the "Preferred Stock"). Upon written request of the holders of at least 66.67% of the then outstanding shares of Series A, Series B, and Series C Preferred Stock collectively, and upon written request of holders of at least a majority of the then outstanding shares of Series D, Series E, and Series F Preferred Stock collectively, as the case may be, the Company is required to redeem the requested number of outstanding shares of Series A Preferred Stock at $5.00 per share, Series B Preferred Stock at $11.50 per share, Series C Preferred Stock at $7.12 per share, and Series D, E and F Preferred Stock at $11.50 per share. Upon written request of holders of at least a majority of the then outstanding shares of Series G Preferred Stock, the Company is required to redeem the outstanding shares of Series G Preferred Stock at a price equal to $11.50 per share. At any time on or after September 21, 2013, upon written request of holders of at least a majority of the then outstanding shares of Series H Preferred Stock, the Company is required to redeem the outstanding shares of Series H Preferred Stock at a price equal to $11.50 per share. At any time on or after July 25, 2015, upon written request of holders of at least a majority of the then outstanding shares of Series I Preferred Stock, the Company is required to redeem the outstanding shares of Series I Preferred Stock at a price equal to
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
1. Nature of Business (Continued)
$11.50 per share. At any time on or after July 27, 2015, upon written request of holders of at least a majority of the then outstanding shares of Series J Preferred Stock, the Company is required to redeem the outstanding shares of Series J Preferred Stock at a price equal to $18.00 per share. At March 31, 2013, Preferred Stock with an aggregate redemption value of $103,122,000 was currently redeemable. During 2013, Preferred Stock with an aggregate redemption value of $23,154,000 will become redeemable at the option of the holder. The Company has not received any notice of redemption as of and through the date the financial statements were available for issuance.
The issued and outstanding shares of Preferred Stock contain conversion features which provide for automatic conversion into shares of common stock, par value $0.01 per share ("Common Stock"), of the Company upon the occurrence of a designated offering, which is defined as a publicly registered offering under the Securities Act of 1933, as amended, in which the gross proceeds after underwriting discount are not less than $25,000,000 at a per share price of at least $16.50 per share, or at a price of at least $11.50 per sharewe file with the consent of the holders of a majority of the outstanding Series J Preferred Stock. Due to this conversion provision, the Company expects the issued and outstanding shares of Preferred Stock to convert into shares of Common Stock upon the completion of a designated offering, at which time the redemption rights would terminate. However, there can be no assurances that the Company will complete a designated offering.
Management intends to fund future operations through additional equity offerings, licensing revenue, grants, government contracts and, if any of the Company's product candidates receive marketing approval, future sales of its products. There can be no assurance, however, that the Company will be successfulSEC, unless otherwise specified in obtaining financing at the level needed to sustain operations or on terms acceptable to the Company, or that the Company will obtain approvals necessary to market its products or achieve profitability or sustainable, positive cash flow.
The accompanying condensed consolidated financial statements have been prepared assuming that the Company will continue as a going concern, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business. The Company's ability to continue as a going concernsuch report, is dependent on its ability to raise additional capital to fund its research and development and commercial programs and meet its obligations, including the potential obligation related to the redemption of the Preferred Stock, which is outside of the Company's control, on a timely basis. If the Company is unable to successfully raise sufficient additional capital, through future debt or equity financings or through strategic and collaborative ventures with third parties, the Company will not have sufficient cash flows and liquidity to fund its planned business operations. In that event, the Company might be forced to limit many, if not all, of its programs and consider other means of creating value for its stockholders, such as licensing to others the development and commercialization of products that it considers valuable and would otherwise likely develop itself. If the Company is unable to raise the necessary capital, it may be forced to curtail all of its activities and, ultimately, potentially cease operations. Even if the Company is able to raise additional capital, such financings may only be available on unattractive terms, or could result in significant dilution of stockholders' interests. The consolidated financial statements do not include any adjustments relating to recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might be necessary should the Company be unable to continue in existence.
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
1. Nature of Business (Continued)
The Company faces many risks associated with companies in the early stages. It also faces risks inherent in its business and its industry generally. These risks include, among others, the following:
2. Summary of Significant Accounting Policies
Basis of Presentation
The condensed consolidated financial statements are prepared in conformity with accounting principles generally accepted in the United States ("GAAP") for interim financial information. Certain information and footnotes normally included in financial statements prepared in accordance with GAAP have been condensed or omitted pursuant to the rules and regulations of the Securities and Exchange Commission. In December 2012, Onconova Europe GmbH was established as a wholly owned subsidiary of the Company. The financial statements include the consolidated accounts of the Company and its wholly owned subsidiary. All significant intercompany transactions have been eliminated.
Unaudited Interim Financial Information
The accompanying condensed consolidated balance sheet as of March 31, 2013, condensed consolidated statements of operations and comprehensive loss and condensed consolidated statements of cash flows for the three months ended March 31, 2012 and 2013, and the condensed consolidated statement of redeemable convertible preferred stock and stockholders' deficit for the three months ended March 31, 2013 are unaudited. The interim unaudited condensed consolidated financial statements have been prepared on the same basis as the annual audited consolidated financial statements and, in the opinion of management, reflect all adjustments, which include only normal recurring adjustments, necessary for the fair statement of the Company's financial position as of March 31, 2013 and the results of its operations, its comprehensive loss and its cash flows for the three months ended March 31, 2012 and 2013. The financial data and other information disclosed in these
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
2. Summary of Significant Accounting Policies (Continued)
notes related to the three months ended March 31, 2012 and 2013 are unaudited. The results for the three months ended March 31, 2013 are not necessarily indicative of results to be expected for the year ending December 31, 2013, any other interim periods, or any future year or period.
Unaudited Pro Forma Presentation
On May 1, 2013, the Company's board of directors authorized management of the Company to confidentially submit a registration statement to the Securities and Exchange Commission (the "SEC") for the Company to sell shares of Common Stock to the public. The unaudited pro forma balance sheet information as of March 31, 2013 assumes the conversion of all outstanding shares of Preferred Stock as of that date into 17,113,481 shares of Common Stock.
The unaudited pro forma net loss per share is computed using the weighted-average number of shares of Common Stock outstanding after giving pro forma effect to the conversion of all issued and outstanding shares of Preferred Stock during the three months ended March 31, 2013 into shares of Common Stock as if such conversion had occurred at January 1, 2013, or the date of original issuance, if later.
Segment Information
Operating segments are defined as components of an enterprise about which separate discrete information is available for evaluationincorporated by the chief operating decision maker, or decision-making group, in deciding how to allocate resources and in assessing performance. The Company views its operations and manages its business in one segment, which is the identification and development of oncology therapeutics.
Significant Accounting Policies
The Company's significant accounting policies are disclosed in the audited consolidated financial statements for the year ended December 31, 2012 included elsewherereference in this prospectus. Since the date of those financial statements, there have been no changes to the Company's significant accounting policies.
Foreign Currency Translation
The reporting currency of the Company and its U.S. subsidiary is the U.S. dollar. The functional currency of the Company's non-U.S. subsidiary is the local currency. Assets and liabilities of the foreign subsidiary are translated into U.S. dollars based on exchange rates at the end of the period. Revenues and expenses are translated at average exchange rates during the reporting period. Gains and losses arising from the translation of assets and liabilities are included as a component of accumulated other comprehensive income. Gains and losses resulting from foreign currency transactions are reflected within the Company's results of operations. The Company has not utilized any foreign currency hedging strategies to mitigate the effect of its foreign currency exposure.
Recent Accounting Pronouncements
Effective January 1, 2011, the Company prospectively adopted the Financial Accounting Standards Board ("FASB") Accounting Standards Update ("ASU") No. 2009-13, Multiple-Deliverable Revenue
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
2. Summary of Significant Accounting Policies (Continued)
Arrangements ("ASU 2009-13"). The amendments in this guidance enable vendors to account for products or services separately rather than as a combined unit upon meeting certain criteria and to establish a hierarchy for determining the selling price of a deliverable. In addition, a vendor can determine a best estimate of selling price, in a manner that is consistent with that used to determine the price to sell the deliverable on a standalone basis, if the vendor does not have vendor-specific objective evidence or third-party evidence of selling price. This guidance also eliminates the use of the residual method and requires a vendor to allocate revenue using the relative selling price method. The Company's adoption of ASU 2009-13 did not have a significant impact on its consolidated financial position, results of operations or cash flows.
In June 2011, FASB issued ASU No. 2011-05, "Comprehensive Income (ASC Topic 220): Presentation of Comprehensive Income" ("ASU 2011-05"). This accounting update eliminates the option to present the components of other comprehensive income as part of the statement of stockholders' equity. Instead, comprehensive income must be presented in either a single continuous statement of comprehensive income, which contains two sections, net income and other comprehensive income, or in two separate but consecutive statements. ASU 2011-05 was effective for fiscal periods beginning after December 15, 2011 with early adoption permitted. The Company's retrospective adoption of ASU 2011-05 did not have a significant impact on its consolidated financial position, results of operations or cash flows.
In February 2013, FASB issued ASU No. 2013-02, "Comprehensive Income (Topic 220): Reporting of Amounts Reclassified Out of Accumulated Other Comprehensive Income" ("ASU 2013-02"). ASU 2013-02 requires companies to present either in a single note or parenthetically on the face of the financial statements, the effect of significant amounts reclassified from each component of accumulated other comprehensive income based on its source and the income statement line items affected by the reclassification. This guidance is effective for annual reporting periods beginning after December 15, 2012. The Company's adoption of ASU 2013-02 did not have a significant impact on its consolidated financial position, results of operations or cash flows.
3. Fair Value Measurements
The Company applies various valuation approaches in determining the fair value of its financial assets and liabilities within a hierarchy that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the asset or liability based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company's assumptions about the inputs that market participants would use in pricing the asset or liability and are developed based on the best information available under the circumstances. The fair value hierarchy is broken down into three levels based on the source of inputs as follows:
Level 1—Valuations based on unadjusted quoted prices in active markets for identical assets or liabilities that the Company has the ability to access.
Level 2—Valuations based on quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active and models for which all significant inputs are observable, either directly or indirectly.
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
3. Fair Value Measurements (Continued)
Level 3—Valuations based on inputs that are unobservable and significant to the overall fair value measurement.
The availability of observable inputs can vary among the various types of financial assets and liabilities. To the extent that the valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. In certain cases, the inputs used to measure fair value may fall into different levels of the fair value hierarchy. In such cases, for financial statement disclosure purposes, the level in the fair value hierarchy within which the fair value measurement is classified is based on the lowest level input that is significant to the overall fair value measurement.
The Company had no assets or liabilities classified as Level 1 or Level 2. The Series G Preferred Stock warrants (see Note 6) are classified as Level 3. The fair values of these instruments are determined using models based on market observable inputs and management judgment. There were no material re-measurements of fair value during the year ended December 31, 2012 and the three months ended March 31, 2013 with respect to financial assets and liabilities, other than those assets and liabilities that are measured at fair value on a recurring basis.
The Company has classified the Series G Preferred Stock warrants as a liability and has re-measured the liability to estimated fair value at December 31, 2012 and March 31, 2013, using the Black-Scholes option pricing model with the following assumptions: contractual life according to the remaining terms of the warrants, no dividend yield, weighted average risk-free interest rates of 0.31% and 0.26% at December 31, 2012 and March 31, 2013, respectively, and weighted average volatility of 64.87% and 63.70% at December 31, 2012 and March 31, 2013, respectively.
The following fair value hierarchy table presents information about the Company's financial assets and liabilities measured at fair value on a recurring basis as of December 31, 2012 and March 31, 2013:
| | Fair Value Measurement As of December 31, 2012 | | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Balance As of December 31, 2012 | ||||||||||||
| | Level 1 | Level 2 | Level 3 | ||||||||||
Warrant liability | $ | — | $ | — | $ | 62,000 | $ | 62,000 | |||||
Total | $ | — | $ | — | $ | 62,000 | $ | 62,000 | |||||
| | Fair Value Measurement As of March 31, 2013 | | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Balance As of March 31, 2013 | ||||||||||||
| | Level 1 | Level 2 | Level 3 | ||||||||||
Warrant liability | $ | — | $ | — | $ | 48,000 | $ | 48,000 | |||||
Total | $ | — | $ | — | $ | 48,000 | $ | 48,000 | |||||
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
3. Fair Value Measurements (Continued)
The following table presents a reconciliation of the Company's liabilities measured at fair value on a recurring basis using significant unobservable inputs (Level 3) for the three months ended March 31, 2013:
| | Warrant Liability | |||
|---|---|---|---|---|
Balance at December 31, 2012 | $ | 62,000 | ||
Change in fair value upon re-measurement | (14,000 | ) | ||
Balance at March 31, 2013 | $ | 48,000 | ||
The fair values of cash equivalents, accounts payable and accrued liabilities approximate their respective carrying values due to the short-term nature of these accounts. There were no transfers between Level 1 and Level 2 in any of the periods reported.
4. Net Loss Per Share of Common Stock
The following table sets forth the computation of basic and diluted earnings per share for the three months ended March 31, 2012 and 2013:
| | Three Months Ended March 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2012 | 2013 | |||||
Basic and diluted net loss per share of common stock: | |||||||
Net loss | $ | (10,799,000 | $ | (14,845,000 | ) | ||
Accretion to redemption value of preferred stock | (1,231,000 | ) | (1,019,000 | ) | |||
Net loss applicable to common stockholders | $ | (12,030,000 | ) | $ | (15,864,000 | ) | |
Weighted average shares of common stock outstanding | 2,897,347 | 3,475,673 | |||||
Net loss per share of common stock—basic and diluted | $ | (4.15 | ) | $ | (4.56 | ) | |
The following potentially dilutive securities outstanding at March 31, 2012 and 2013 have been excluded from the computation of diluted weighted average shares outstanding, as they would be antidilutive:
| | March 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2012 | 2013 | |||||
Preferred Stock | 11,475,875 | 17,113,481 | |||||
Warrants | 524,402 | 6,128 | |||||
Stock options | 2,788,004 | 3,722,188 | |||||
| 14,788,281 | 20,841,197 | ||||||
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
5. Accrued Expenses
Accrued expenses are as follows:
| | December 31, 2012 | March 31, 2013 | |||||
|---|---|---|---|---|---|---|---|
Research and development | $ | 3,521,000 | $ | 4,575,000 | |||
Payroll | 247,000 | 574,000 | |||||
Other | 157,000 | 410,000 | |||||
| $ | 3,925,000 | $ | 5,559,000 | ||||
6. Preferred Stock and Stockholders' Deficit
Capitalization
As of March 31, 2013, the Company's ninth amended and restated certificate of incorporation reflected the following authorized shares: 48,693,408 shares of capital stock, consisting of (i) 400,000 shares of Series A Preferred Stock, (ii) 1,200,000 shares of Series B Preferred Stock, (iii) 1,200,000 shares of Series C Preferred Stock, (iv) 1,625,000 shares of Series D Preferred Stock, (v) 1,650,000 shares of Series E Preferred Stock, (vi) 2,000,000 shares of Series F Preferred Stock, (vii) 2,700,000 shares of Series G Preferred Stock, (viii) 2,042,950 of Series H Preferred Stock, (ix) 2,700,000 shares of Series I Preferred Stock, (x) 3,030,303 shares of Series J Preferred Stock and (xi) 30,145,155 shares of Common Stock.
The Company issued shares of Series H Preferred Stock in three closings in 2011: On February 17, 2011, the Company raised $700,000 in gross proceeds from the issuance of 71,488 shares of Series H Preferred Stock; On June 2, 2011, the Company raised $1,326,000 in gross proceeds from the issuance of 135,391 shares of Series H Preferred Stock; and on September 19, 2011, the Company raised $5,996,000 in gross proceeds from the issuance of 612,450 shares of Series H Preferred Stock.
In July 2012, the Company issued 2,433,328 shares of Series I Preferred Stock in exchange for the conversion of the convertible promissory notes and accrued interest in the amount of $26,444,000 and $323,000, respectively. The effective conversion price was $11.00 per share. Additionally, in July 2012, the Company issued 3,030,303 shares of Series J Preferred Stock at $16.50 per share for gross proceeds of $50,000,000. Issuance costs associated with this offering were $2,204,000.
Series A Preferred Stock was originally issued at $5.00 per share; Series B Preferred Stock was issued at $5.75 per share; Series C Preferred Stock was issued at $3.56 per share; Series D Preferred Stock was issued at $4.67 per share; Series E Preferred Stock was issued at $9.76 per share; Series F Preferred Stock was issued at $11.00 per share; Series G and Series H Preferred Stock were issued at $9.79 per share; Series I Preferred Stock was issued at $11.00 per share; and Series J Preferred Stock was issued at $16.50 per share.
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
6. Preferred Stock and Stockholders' Deficit (Continued)
The following is the composition of share capital as of the dates indicated:
| | Authorized | Issued and Outstanding | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | December 31, 2012 | March 31, 2013 | December 31, 2012 | March 31, 2013 | |||||||||
Shares of $0.01 par value per share: | |||||||||||||
Common stock | 30,145,155 | 30,145,155 | 3,474,473 | 3,478,473 | |||||||||
Series A Preferred Stock | 400,000 | 400,000 | 107,000 | 107,000 | |||||||||
Series B Preferred Stock | 1,200,000 | 1,200,000 | 1,107,189 | 1,107,189 | |||||||||
Series C Preferred Stock | 1,200,000 | 1,200,000 | 1,069,946 | 1,069,946 | |||||||||
Series D Preferred Stock | 1,625,000 | 1,625,000 | 1,583,568 | 1,583,568 | |||||||||
Series E Preferred Stock | 1,650,000 | 1,650,000 | 1,633,082 | 1,633,082 | |||||||||
Series F Preferred Stock | 2,000,000 | 2,000,000 | 2,000,000 | 2,000,000 | |||||||||
Series G Preferred Stock | 2,700,000 | 2,700,000 | 1,934,359 | 1,934,359 | |||||||||
Series H Preferred Stock | 2,042,950 | 2,042,950 | 2,013,424 | 2,013,424 | |||||||||
Series I Preferred Stock | 2,700,000 | 2,700,000 | 2,433,328 | 2,433,328 | |||||||||
Series J Preferred Stock | 3,030,303 | 3,030,303 | 3,030,303 | 3,030,303 | |||||||||
Total Preferred Stock | 18,548,253 | 18,548,253 | 16,912,199 | 16,912,199 | |||||||||
Total | 48,693,408 | 48,693,408 | 20,386,672 | 20,390,672 | |||||||||
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
6. Preferred Stock and Stockholders' Deficit (Continued)
The following is the activity of the Preferred Stock for the three months ended March 31, 2013:
| | December 31, 2012 | Issuance of Preferred Stock | Exercise of warrants | Accretion of redemption premium and issuance costs on Preferred Stock | March 31, 2013 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Series A | ||||||||||||||||
Shares | 107,000 | — | — | — | 107,000 | |||||||||||
Amount | $ | 535,000 | $ | — | $ | — | $ | — | $ | 535,000 | ||||||
Series B | ||||||||||||||||
Shares | 1,107,189 | — | — | — | 1,107,189 | |||||||||||
Amount | $ | 12,733,000 | $ | — | $ | — | $ | — | $ | 12,733,000 | ||||||
Series C | ||||||||||||||||
Shares | 1,069,946 | — | — | — | 1,069,946 | |||||||||||
Amount | $ | 7,618,000 | $ | — | $ | — | $ | — | $ | 7,618,000 | ||||||
Series D | ||||||||||||||||
Shares | 1,583,568 | — | — | — | 1,583,568 | |||||||||||
Amount | $ | 18,211,000 | $ | — | $ | — | $ | — | $ | 18,211,000 | ||||||
Series E | ||||||||||||||||
Shares | 1,633,082 | — | — | — | 1,633,082 | |||||||||||
Amount | $ | 18,780,000 | $ | — | $ | — | $ | — | $ | 18,780,000 | ||||||
Series F | ||||||||||||||||
Shares | 2,000,000 | — | — | — | 2,000,000 | |||||||||||
Amount | $ | 23,000,000 | $ | — | $ | — | $ | — | $ | 23,000,000 | ||||||
Series G | ||||||||||||||||
Shares | 1,934,359 | — | — | — | 1,934,359 | |||||||||||
Amount | $ | 22,819,000 | $ | — | $ | — | $ | — | $ | 22,819,000 | ||||||
Series H | ||||||||||||||||
Shares | 2,013,424 | — | — | — | 2,013,424 | |||||||||||
Amount | $ | 22,005,000 | $ | — | $ | — | $ | 380,000 | $ | 22,385,000 | ||||||
Series I | ||||||||||||||||
Shares | 2,433,328 | — | — | — | 2,433,328 | |||||||||||
Amount | $ | 26,933,000 | $ | — | $ | — | $ | 100,000 | $ | 27,033,000 | ||||||
Series J | ||||||||||||||||
Shares | 3,030,303 | — | — | — | 3,030,303 | |||||||||||
Amount | $ | 48,681,000 | $ | — | $ | — | $ | 539,000 | $ | 49,220,000 | ||||||
Total | ||||||||||||||||
Shares | 16,912,199 | — | — | — | 16,912,199 | |||||||||||
Amount | $ | 201,315,000 | $ | — | $ | — | $ | 1,019,000 | $ | 202,334,000 | ||||||
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
6. Preferred Stock and Stockholders' Deficit (Continued)
Voting
Each holder of outstanding shares of Preferred Stock has the right to one vote for each share of Common Stock into which such Preferred Stock could then be converted. The holders of shares of Preferred Stock have full voting rights and powers equal to the voting rights and powers of shares of Common Stock and are entitled to notice of any stockholders' meeting and vote together with the holders of Common Stock, with respect to any question upon which holders of shares of Common Stock have the right to vote, as a single class, including without limitation, actions to increase or decrease the aggregate number of authorized shares of Common Stock and/or Preferred Stock.
Dividends
The holders of each share of Series A, Series B, Series C, Series D, Series E, Series F, Series G, Series H, Series I and Series J Preferred Stock are entitled to receive dividends when, as, and if declared by the Company's board of directors in the following order of preference: (i) the Series D, Series E, Series F Series G, Series H, Series I and Series J Preferred Stock, which rank pari passu; (ii) the Series B and Series C Preferred Stock, which rank pari passu; (iii) the Series A Preferred Stock; and then (iv) Common Stock.
Liquidation
The assets of the Company legally available for distribution to stockholders will be distributed in the following order of priority: (i) the holders of the shares of Series D, Series E, Series F, Series G, Series H, Series I and Series J Preferred Stock, which rank pari passu; (ii) the holders of the shares of Series B and Series C Preferred Stock, which rank pari passu; (iii) the holders of the shares of Series A Preferred Stock; and (iv) the holders of the shares of Common Stock. Each series of Preferred Stock is entitled to receive an amount per share equal to the greater of (1) the original issuance price for such series, plus all declared but unpaid dividends thereon, or (2) the amount that the holders of such series would receive per share of Common Stock if all shares of such series of Preferred Stock were converted to Common Stock immediately prior to such liquidation. If upon a deemed liquidation event, the assets of the Company are insufficient to make payment in full to all holders of a series of Preferred Stock, then such assets shall be distributed among the holders of such series of Preferred Stock at the time outstanding ratably in proportion to the full amount to which they would otherwise be respectively entitled. The holders of Common Stock are entitled to receive, after the payment of the liquidation preference of all Preferred Stock outstanding, the remaining assets of the Company on a pro rata basis.
Conversion
Each issued and outstanding share of Preferred Stock is convertible into Common Stock at the holder's option at any time after the date of issuance or automatically upon the occurrence of certain events as defined in the Company's ninth amended and restated certificate of incorporation, at a defined conversion rate. At March 31, 2013, the number of shares of Common Stock into which one share of each series of Preferred Stock was convertible was as follows: the Series A Preferred Stock, 1.07; the Series B Preferred Stock, 1.13; the Series C Preferred Stock, 1.00; the Series D Preferred Stock, 1.00; the Series E Preferred Stock, 1.00; the Series F Preferred Stock, 1.03; the Series G
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
6. Preferred Stock and Stockholders' Deficit (Continued)
Preferred Stock, 1.00; the Series H Preferred Stock, 1.00; the Series I Preferred Stock, 1.00; and the Series J Preferred Stock, 1.00.
The conversion price for each share of Preferred Stock is subject to adjustment upon the occurrence of certain events. The conversion price of each share of a series of Preferred Stock is adjusted if the Company issues additional shares, subject to specified exceptions, at a price lower than the current conversion price for such series, which is measured and recognized if the contingency occurs.
Redemption
To the extent it is then lawfully able to do so, the Company is required at any time, upon written request of the holders of at least 66.67% of the then outstanding Series A, Series B and Series C Preferred Stock collectively, or upon written request of the holders of at least a majority of the then outstanding shares of Series D, Series E and Series F Preferred Stock collectively, in each case as determined on an as-converted to Common Stock basis, to redeem the requested number of outstanding shares of Series A Preferred Stock at $5.00 per share, Series B Preferred Stock at $11.50 per share, and Series C Preferred Stock at $7.12 per share, and/or Series D, E and F Preferred Stock at $11.50 per share, as the case may be.
In addition, to the extent it is lawfully able to do so, the Company is required at any time, upon written request of the holders of at least a majority of the then outstanding shares of Series G Preferred Stock, to redeem, from the holders requesting such redemption, the requested number of outstanding shares of Series G Preferred Stock at $11.50 per share.
To the extent it is lawfully able to do so, the Company is required at any time on or after September 21, 2013, upon written request of the holders of at least a majority of the then outstanding shares of Series H Preferred Stock, to redeem, from the holders requesting such redemption, the requested number of outstanding shares of Series H Preferred Stock at $11.50 per share.
To the extent it is lawfully able to do so, the Company is required at any time on or after July 25, 2015, upon written request of the holders of at least a majority of the then outstanding shares of Series I Preferred Stock, to redeem, from the holders requesting such redemption, the requested number of outstanding shares of Series I Preferred Stock at $11.50 per share.
To the extent it is lawfully able to do so, the Company is required at any time on or after July 27, 2015, upon written request of the holders of at least a majority of the then outstanding shares of Series J Preferred Stock, to redeem, from the holders requesting such redemption, the requested number of outstanding shares of Series J Preferred Stock at $18.00 per share.
If, upon any applicable redemption date, defined as sixty days after the Company receives the written request for redemption, the funds of the Company legally available for redemption of Preferred Stock are insufficient to redeem the total number of shares to be redeemed on that date, those funds that are legally available shall be used to redeem the maximum possible number of shares, ratably among the holders of such shares to be redeemed. All remaining shares not redeemed shall remain outstanding until such time as additional funds become legally available for redemption.
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
6. Preferred Stock and Stockholders' Deficit (Continued)
If more than one series of Preferred Stock is contemporaneously subject to redemption, the redemption rights of the Preferred Stock shall follow the liquidation order of priority.
As of March 31, 2013, Preferred Stock with an aggregate redemption value of $103,122,000 was redeemable. During 2013 and 2015, additional shares of Preferred Stock with an aggregate redemption value of $23,154,000 and $82,529,000, respectively, will become redeemable at the option of the holders of Preferred Stock.
Series G Preferred Stock Warrant Transactions
The Company issued 6,128 Series G Preferred Stock warrants in connection with a Loan and Security Agreement. Additionally, the Company issued one Series G Preferred Stock warrant for every two shares of Series G Preferred Stock purchased in 2009 and 2010. The warrants were initially recorded at their fair value calculated using the Black-Scholes model, with the following weighted average assumptions: exercise price of $9.79, share price of $9.79, expected term of three years, risk-free rate of 1.52% and volatility of 85.46%. The warrants are classified as liabilities because they are exercisable for Preferred Stock, and the value of the warrants is adjusted to current fair value at each reporting period end. For the three months ended March 31, 2012 and 2013, the Company recorded $(609,000) and $14,000, respectively, in the consolidated statements of operations and comprehensive loss related to the change in the fair value of the outstanding warrants.
There were no Series G Preferred Stock warrant transactions during the three months ended March 31, 2013.
7. Stock-Based Compensation
In January 2008, the board of directors approved the 2007 Equity Compensation Plan (the "Plan"), which amended and restated the Company's 1999 Stock Based Compensation Plan, which provides for the granting of incentive and nonqualified stock options and restricted stock to its employees, directors and consultants at the discretion of the board of directors. Under the Plan, the Company increases the number of shares reserved for issuance under the Plan such that the number of reserved shares is equal to 17% of the fully diluted shares calculated annually on December 10th. At December 31, 2011 and 2012 and March 31, 2012 and 2013, 3,498,579, 4,107,831, 3,498,579 and 4,107,831 shares of Common Stock were reserved under the Plan, respectively. Stock options may be granted with exercise prices of not less than the estimated fair value of the Company's common stock on the date of grant and generally vest over a period of up to four years. Stock options granted under the Plan generally expire no later than ten years from the date of grant. A summary of stock option
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
7. Stock-Based Compensation (Continued)
activity for the year ended December 31, 2012 and the three months ended March 31, 2013 is as follows:
| | Number of Options | Weighted- Average Exercise Price | Weighted- Average Remaining Contractual Term (in years) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Outstanding at December 31, 2012 | 3,418,228 | $ | 6.04 | 7.52 | ||||||
Granted | 413,960 | 9.96 | ||||||||
Exercised | (4,000 | ) | 1.50 | |||||||
Forfeited | (106,000 | ) | 9.45 | |||||||
Outstanding at March 31, 2013 | 3,722,188 | $ | 6.41 | 7.32 | ||||||
Vested or expected to vest at March 31, 2013 | 3,722,188 | $ | 6.41 | 7.32 | ||||||
Exercisable at March 31, 2013 | 2,334,784 | $ | 4.83 | 6.37 | ||||||
At December 31, 2012 and March 31, 2013, the aggregate intrinsic value of the option liability recorded was $11,967,000 and $14,394,000, respectively. During the year ended December 31, 2012 and the three months ended March 31, 2013, the Company granted 1,303,486 and 413,960 options, respectively, at an intrinsic value of $0 at the grant date. At certain times throughout the Company's history, the chairman of the Company's board of directors, who is also a significant stockholder of the Company (the "Significant Holder"), has afforded option holders the opportunity for liquidity in transactions in which options were exercised and the shares of Common Stock issued in connection therewith were simultaneously purchased by the Significant Holder (each, a "Purchase Transaction"). The Company issued an aggregate of 584,726 shares of Common Stock upon the exercise of options in 2012, of which 395,863 shares were then purchased by the Significant Holder in Purchase Transactions. The Company received proceeds of $3,943,000 from the Significant Holder of which $2,835,000 was paid to the option holders upon exercise and settlement of the option liabilities. Because the Company has established a pattern of providing cash settlement alternatives for option holders, the Company has accounted for its stock-based compensation awards as liability awards, the fair value of which is then re-measured at each balance sheet date. Upon the exercise of stock options in 2012 and the three months ended March 31, 2013, stock option liabilities of $4,525,000 and $38,000, respectively, were reclassified to stockholders' deficit.
Stock-based compensation expense includes stock options granted to employees and non-employees and has been reported in the Company's statements of operations and comprehensive loss in either research and development expenses or general and administrative expenses depending on the function performed by the optionee. For the three months ended March 31, 2012 and 2013, $1,170,000 and $1,158,000 was recorded in research and development expenses, respectively. For the three months ended March 31, 2012 and 2013, $1,267,000 and $1,306,000 was recorded in general and administrative expenses, respectively. As of March 31, 2013, the Company had unrecognized stock-based compensation of $2,918,000 related to unvested stock options held by employees and non-employees which is expected to vest over a weighted average period of 2.20 years. No net tax benefits related to the stock-based compensation costs have been recognized since the Company's inception.
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
7. Stock-Based Compensation (Continued)
A roll forward of the stock option liability balance for the three months ended March 31, 2013 is as follows:
Balance at December 31, 2012 | $ | 11,967,000 | ||
Change in intrinsic value upon re-measurement | 2,465,000 | |||
Settlements of option liability awards | (38,000 | ) | ||
Balance at March 31, 2013 | $ | 14,394,000 | ||
Information with respect to stock options outstanding and exercisable at March 31, 2013 is as follows:
| Exercise Price | Shares | Weighted Average Remaining Contractual Life (years) | Exercisable | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| $ | 1.00 | 215,445 | 1.29 | 215,445 | |||||||
| 2.00 | 86,934 | 2.82 | 86,934 | ||||||||
| 4.32 | 529,150 | 6.94 | 476,410 | ||||||||
| 4.50 | 560,250 | 4.43 | 560,250 | ||||||||
| 4.60 | 810,072 | 6.95 | 645,433 | ||||||||
| 5.65 | 46,977 | 9.16 | 34,541 | ||||||||
| 9.96 | 1,473,360 | 9.83 | 315,771 | ||||||||
| 3,722,188 | 7.32 | 2,334,784 | |||||||||
On April 23, 2013, the Company distributed a notification letter to all equity award holders under the Plan advising them that Purchase Transactions will no longer occur, unless, at the time of a Purchase Transaction, the option holder has held the Common Stock issued upon exercise of options for a period of greater than six months prior to selling such Common Stock to the Significant Holder and that any such sale to the Significant Holder would be at the fair value of the Common Stock on the date of such sale. Based on these new criteria for Purchase Transactions, the Company remeasured options outstanding under the Plan as of April 23, 2013 to their intrinsic value and reclassified such options from liabilities to stockholders' deficit within the Company's consolidated balance sheets, which amounted to $14,480,000. The Company will recognize $2,831,000 of compensation expense over a weighted average vesting period of 2.16 years related to these modified awards.
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
8. Employee Benefit Plan
In October 2007, the Company established a 401(k) Retirement Savings Plan. Employees are eligible to participate in the plan as soon as they join the Company if they are at least 21 years of age and work a minimum of 1,000 hours per year. The Company matches $0.50 for every dollar of the first 6% of payroll that employees invest, up to the legal limit. Employer contributions vest over four years at the rate of 25% per year during the employees' first four years. Thereafter, contributions vest 100% immediately. For the three months ended March 31, 2012 and 2013, the Company contributed $24,000, and $36,000, respectively.
9. Commitments and Contingencies
Operating leases
In November 2010, the Company entered into a lease for 3,117 square feet of office space in Pennington, New Jersey. The lease had an original term of two years, with an option for two additional years. For the first two years of the lease, the Company was obligated to pay $4,400 per month, or $53,000 annually, beginning when possession of the facility was taken on February 1, 2011. The Company was required to provide the landlord a $125,000 letter of credit, the collateral for which is recorded as restricted cash on the consolidated balance sheets. This lease was renewed on February 1, 2012 with a 3.5% increase in the rent, to $5,000 per month. On October 2, 2012, the Company leased an additional 2,130 square feet of office space for $3,100 per month, or $38,000 annually.
In January 2007, the Company entered into a lease for 8,100 square feet of office and lab space in Newtown, Pennsylvania, and in October 2009, the Company and the landlord amended the lease to add three additional one-year options to extend the lease term. The Company exercised the first option for the period from April 1, 2012 to March 31, 2013 and the second option for the period from April 1, 2013 to March 31, 2014 for rent of $11,000 per month. In September 2012, the Company sub-leased an additional 1,356 square feet of office space for one year for $1,600 per month, or $19,000 annually.
Future minimum lease payments under these non-cancellable leases having terms in excess of one year as of December 31, 2012 are as follows:
| | December 31, 2012 | |||
|---|---|---|---|---|
2013 | $ | 256,000 | ||
2014 | 103,000 | |||
2015 | 6,000 | |||
Total minimum lease payments | $ | 365,000 | ||
Rent expense was $47,000 and $76,000 for the three months ended March 31, 2012 and 2013, respectively.
Employment agreements
The Company has entered into employment agreements with certain of its executives. The agreements provide for, among other things, salary, bonus and severance payments.
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
10. License and Collaboration Agreements
Baxter Agreement
In September 2012, the Company entered into a development and license agreement with Baxter granting Baxter an exclusive, royalty-bearing license for the research, development, commercialization and manufacture (in specified instances) of rigosertib in all therapeutic indications in Europe (the "Baxter Territory"). Baxter is a shareholder in the Company and invested in Series J Preferred Stock issued in July 2012.
Under the terms of the agreement, the Company is initially required to perform research and development to advance three initial rigosertib indications, rigosertib intravenous ("IV") in higher risk myelodysplastic syndrome ("MDS") patients, rigosertib IV in pancreatic cancer patients and rigosertib oral in lower risk MDS patients, through phase 3, phase 3 and phase 2 clinical trials, respectively.
If an additional phase 3 clinical trial beyond the current phase 3 clinical trial in process for rigosertib IV in higher risk MDS patients is required to obtain marketing approval in the Baxter Territory, the Company could require Baxter to fund a percentage of the costs of such additional trial up to a specified maximum. At the completion of the current phase 3 trial for rigosertib IV in pancreatic cancer and the current phase 2 trial for rigosertib oral in lower risk MDS patients and the review of the resulting data and findings, the Company and Baxter will decide whether or not to pursue further development of rigosertib for these indications. If the Company and Baxter mutually agree to progress the development of rigosertib IV in pancreatic cancer patients and rigosertib oral in lower risk MDS patients, then certain milestone payments will be payable to the Company, and the Company will be required to use its commercially reasonable efforts to progress the development of rigosertib for these indications to a drug approval application in the Baxter Territory. The Company and Baxter will work together for potential future rigosertib indications, beyond the initial indications noted above. Generally, if Baxter chooses to participate in the development of additional indications, Baxter will be responsible for a percentage of all research and development costs and expenses and the Company could earn additional milestone payments. Baxter has full responsibility for all commercialization activities for the product in the Baxter Territory, at Baxter's sole cost and expense.
The Company and Baxter have agreed to negotiate a supply agreement under terms satisfactory to both parties whereby the Company will supply Baxter with Baxter's required levels of product to support commercialization efforts in the Baxter Territory. Baxter also has the right to engage third parties for the manufacture and supply of its requirements for the licensed product.
Under the terms of the agreement, Baxter made an upfront payment of $50,000,000. The Company is eligible to receive pre-commercial milestone payments of up to an aggregate of $512,500,000 if specified development and regulatory milestones are achieved. The potential pre-commercial development milestone payments to us include the following:
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
10. License and Collaboration Agreements (Continued)
The Company may also receive up to $337,500,000 in milestone payments for regulatory approvals of the three rigosertib indications specified in the arrangement with Baxter, each of which may be up to and in excess of $100,000,000. The Company is also potentially eligible to receive an additional $20,000,000 pre-commercial milestone payment related to the timing of regulatory approval of the MDS IV indication in Europe. In addition to these pre-commercial milestones, the Company is eligible to receive up to an aggregate of $250,000,000 in milestone payments based on Baxter's achievement of pre-specified threshold levels of annual net sales of rigosertib. The Company will also be entitled to receive royalties at percentage rates ranging from the low-teens to the low-twenties on net sales of rigosertib by Baxter in the Baxter Territory.
The agreement with Baxter will remain in effect until the expiration of all applicable royalty terms and satisfaction of all payment obligations in each licensed country, unless terminated earlier due to the uncured material breach or bankruptcy of a party, force majeure, or in the event of a specified commercial failure. The Company may terminate the agreement in the event that Baxter brings a challenge against it in relation to the licensed patents. Baxter may terminate the agreement without cause commencing after a specified period of time from the execution of the agreement.
The Company determined that the deliverables under the Baxter agreement include the exclusive, royalty-bearing, sublicensable license to rigosertib and the research and development services to be performed by the Company. The Company concluded that the license had standalone value to Baxter and was separable from the research and development services because the license is sublicensable, there are no restrictions as to Baxter's use of the license and Baxter has significant research capabilities in this field. The Company was not able to establish vendor-specific objective evidence of selling price or third-party evidence for either the license or the research and development services and instead allocated the arrangement consideration between the license and research and development services based on their relative selling prices using best estimate of selling price ("BESP"). Management developed the BESP of the license using a discounted cash flow model, taking into consideration assumptions including the development and commercialization timeline, discount rate and probability of success. Management utilized a third party valuation specialist to assist with the determination of BESP of the license. Management estimated the selling price of the research and development services using third party costs and a discounted cash flow model. The estimated selling prices utilized assumptions including internal estimates of research and development personnel needed to perform the research and development services; and estimates of expected cash outflows to third parties for services and supplies over the expected period that the services will be performed.
The key assumptions in these models included the following market conditions and entity-specific factors: (a) the specific rights provided under the license, (b) the stage of development of rigosertib and estimated remaining development and commercialization timelines, (c) the probability of successfully developing and commercializing rigosertib, (d) the market size including the associated sales figures which generate royalty revenue, (e) cost of goods sold, which was assumed to be a specified percentage of revenues based on estimated cost of goods sold of a typical oncology product, (f) sales and marketing costs, which were based on the costs required to field an oncology sales force and marketing group, including external costs required to promote an oncology product, (g) the expected product life of rigosertib assuming commercialization and (h) the competitive environment.
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
10. License and Collaboration Agreements (Continued)
The Company utilized a discount rate of 16%, representing the cost of capital derived from returns on equity for comparable companies.
Based on management's analyses, it was determined that the BESP of the license was $120,000,000 and the BESP of the research and development services was $20,600,000. As noted above, the Company received an up-front payment of $50,000,000 under the Baxter agreement, which represents the allocable agreement consideration. Based on the respective BESPs, this payment was allocated $42,400,000 to the license and $7,600,000 to the research and development services. Since the delivery of the license occurred upon the execution of the Baxter agreement and there was no general right of return, $42,400,000 of the $50,000,000 upfront payment was recognized upon the execution of the Baxter agreement. The portion allocated to research and development services is being recognized over the period of performance on a proportional performance basis through March 31, 2014. Management estimated the period of performance to be the period necessary for completion of the non-contingent obligations to perform research and development services required to advance the three formulations of rigosertib described above. For the three months ended March 31, 2013, the Company recognized $978,000 of research and development revenue under the Baxter agreement.
The Company and Baxter have agreed to establish a joint committee to facilitate the governance and oversight of the parties' activities under the agreements. Management considered whether participation on the joint committee may be a deliverable and determined that it was not a deliverable. Had management considered participation on the joint committee as a deliverable, it would not have had a material impact on the accounting for the arrangement based on the analysis of the estimated selling price of such participation.
As noted above, in July 2012, Baxter purchased Series J Preferred Stock. Because the Series J Preferred Stock was acquired within several months of the Baxter development and license agreement, management considered whether the Preferred Stock was issued at fair value and if not, whether the consideration received for the Preferred Stock ($50,000,000) or for the collaboration and license agreement ($50,000,000) should be allocated in the financial statements in a manner differently than the prices stated in the agreements. Management, with the assistance of an outside valuation specialist, determined that the price paid by Baxter for the Series J Preferred Stock approximated its fair value, and therefore the consideration received under the agreements was allocated in accordance with terms of the individual agreements.
SymBio Agreement
In July 2011, the Company entered into a license agreement with SymBio, as subsequently amended, granting SymBio an exclusive, royalty-bearing license for the development and commercialization of rigosertib in Japan and Korea. Under the SymBio license agreement, SymBio is obligated to use commercially reasonable efforts to develop and obtain market approval for rigosertib inside the licensed territory and the Company has similar obligations outside of the licensed territory. The Company has also entered into an agreement with SymBio providing for it to supply SymBio with development-stage product. Under the SymBio license agreement, the Company also agreed to supply commercial product to SymBio under specified terms that will be included in a commercial supply agreement to be negotiated prior to the first commercial sale of rigosertib. The supply of development-stage product and the supply of commercial product will be at the Company's cost plus a defined profit
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
10. License and Collaboration Agreements (Continued)
margin. Sales of development-stage product have been de minimis. The Company has additionally granted SymBio a right of first negotiation to license or obtain the rights to develop and commercialize compounds having a chemical structure similar to rigosertib in the licensed territory.
Under the terms of the SymBio license agreement, the Company received an upfront payment of $7,500,000. The Company is eligible to receive milestone payments of up to an aggregate of $33,000,000 from SymBio upon the achievement of specified development and regulatory milestones for specified indications. Of the development milestones, $3,000,000 is due after enrollment of the first patient in the event a decision is made, after the Company's interim analysis, to start a phase 3 clinical trial of rigosertib IV in combination with gemcitabine for pancreatic cancer patients in the United States. Of the regulatory milestones, $5,000,000 is due upon receipt of marketing approval in the United States for rigosertib IV in higher risk MDS patients, $3,000,000 is due upon receipt of marketing approval in Japan for rigosertib IV in higher risk MDS patients, $5,000,000 is due upon receipt of marketing approval in the United States for rigosertib oral in lower risk MDS patients, $5,000,000 is due upon receipt of marketing approval in Japan for rigosertib oral in lower risk MDS patients, $5,000,000 is due upon receipt of marketing approval in the United States for rigosertib IV in combination with gemcitabine in pancreatic cancer patients, and $3,000,000 is due upon receipt of marketing approval in Japan for rigosertib IV in combination with gemcitabine for pancreatic cancer patients. Furthermore, upon receipt of marketing approval in the United States and Japan for an additional indication of rigosertib, which we are currently not pursuing, an aggregate of $4,000,000 would be due. In addition to these pre-commercial milestones, the Company is eligible to receive tiered milestone payments based upon annual net sales of rigosertib by SymBio of up to an aggregate of $30,000,000. Further, under the terms of the SymBio license agreement, SymBio will make royalty payments to the Company at percentage rates ranging from the mid-teens to 20% based on net sales of rigosertib by SymBio.
Royalties will be payable under the SymBio agreement on a country-by-country basis in the licensed territory, until the later of the expiration of marketing exclusivity in those countries, a specified period of time after first commercial sale of rigosertib in such country, or the expiration of all valid claims of the licensed patents covering rigosertib or the manufacture or use of rigosertib in such country. If no valid claim exists covering the composition of matter of rigosertib or the use of or treatment with rigosertib in a particular country before the expiration of the royalty term, and specified competing products achieve a specified market share percentage in such country, SymBio's obligation to pay the Company royalties will continue at a reduced royalty rate until the end of the royalty term. In addition, the applicable royalties payable to the Company may be reduced if SymBio is required to pay royalties to third-parties for licenses to intellectual property rights necessary to develop, use, manufacture or commercialize rigosertib in the licensed territory. The license agreement with SymBio will remain in effect until the expiration of the royalty term. However, the SymBio license agreement may be terminated earlier due to the uncured material breach or bankruptcy of a party, or force majeure. If SymBio terminates the license agreement in these circumstances, its licenses to rigosertib will survive, subject to SymBio's milestone and royalty obligations, which SymBio may elect to defer and offset against any damages that may be determined to be due from the Company. In addition, the Company may terminate the license agreement in the event that SymBio brings a challenge against it in relation to the licensed patents, and SymBio may terminate the license agreement without cause by providing the Company with written notice within a specified period of time in advance of termination.
Onconova Therapeutics, Inc.
Notes to Condensed Consolidated Financial Statements (Continued)
(Unaudited)
10. License and Collaboration Agreements (Continued)
The Company determined that the deliverables under the SymBio agreement include the exclusive, royalty-bearing, sublicensable license to rigosertib, the research and development services to be provided by the Company and its obligation to serve on a joint committee. The Company concluded that the license did not have standalone value to SymBio and was not separable from the research and development services, because of the uncertainty of SymBio's ability to develop rigosertib in the SymBio territory on its own and the uncertainty of SymBio's ability to sublicense rigosertib and recover a substantial portion of the original upfront payment of $7,500,000 paid by SymBio to the Company.
The supply of rigosertib for SymBio's commercial requirements is contingent upon the receipt of regulatory approvals to commercialize rigosertib in Japan and Korea. Because the Company's commercial supply obligation was contingent upon the receipt of future regulatory approvals, and there were no binding commitments or firm purchase orders pending for commercial supply at or near the execution of the agreement, the commercial supply obligation is deemed to be contingent and is not valued as a deliverable under the SymBio agreement. If SymBio orders the supplies from the Company, the Company expects the pricing for this supply to equal its third-party manufacturing cost plus a pre-negotiated percentage, which will not result in a significant incremental discount to market rates.
Due to the lack of standalone value for the license, research and development services, and joint committee obligation, the upfront payment is being recognized ratably using the straight line method through December 2027, the expected term of the agreement. For each of the three months ended March 31, 2012 and 2013, the Company recognized revenues of $113,000 under this agreement. In addition, the Company recognized revenues of $25,000 for the three months ended March 31, 2013 related to the supply agreement with SymBio.
Shares
PROSPECTUS
Consisting of an Aggregate of Up to Shares of Common Stock
and Warrants to Purchase Up to Shares of Common Stock
at a Subscription Price of $ Per Unit
Common StockDealer-Manager
PRELIMINARY PROSPECTUSMaxim Group LLC
, 20132016
Citigroup
Leerink Swann
Piper Jaffray
Janney Montgomery Scott
Through and including , 2013 (25 days after the commencement of this offering), all dealers that buy, sell or trade shares of our common stock, whether or not participating in this offering, may be required to deliver a prospectus. This delivery requirement is in addition to the dealers' obligation to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
PART IIINFORMATION NOT REQUIRED IN PROSPECTUSInformation Not Required In Prospectus
Item 13. Other Expenses of Issuance and Distribution.
The following table sets forth all costs andis a statement of estimated expenses other than underwriting discounts and commissions, paid or payable by us in connection with the saleissuance and distribution of the securities being registered, excluding dealer-manager fees. All expenses incurred with respect to the registration of the common stock being registered.will be borne by us. All amounts shown are estimates except for the SEC registration fee and the Financial Industry Regulatory Authority, or FINRA filing fee and the initial listing fee for the NASDAQ Global Market.fee.
| | Amount | |||
|---|---|---|---|---|
SEC registration fee | $ | 10,230 | ||
FINRA filing fee | 8,000 | |||
NASDAQ Global Market listing fee | * | |||
Blue sky qualification fees and expenses | * | |||
Printing expenses | * | |||
Legal fees and expenses | * | |||
Accounting fees and expenses | * | |||
Transfer agent and registrar fees and expenses | * | |||
Miscellaneous expenses | * | |||
Total | $ | * | ||
| | Amount to be Paid | |||
|---|---|---|---|---|
SEC Registration Fee | $ | 5,089 | ||
FINRA Filing Fee | 3,875 | |||
NASDAQ Fee | 5,000 | |||
Printing Expenses | 40,000 | |||
Legal Fees and Expenses | 200,000 | |||
Accounting Fees and Expenses | 75,000 | |||
Subscription Agent and Warrant Agent Fees and Expenses | 100,000 | |||
Miscellaneous Expenses | 20,000 | |||
| | | | | |
| $ | 448,964 | |||
| | | | | |
| | | | | |
| | | | | |
Item 14. Indemnification of Directors and Officers.
We are incorporated under the laws of the State of Delaware. Section 145 of the Delaware General Corporation Law provides that a Delaware corporation may indemnify any persons who are, or are threatened to be made, parties to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of such corporation), by reason of the fact that such person was an officer, director, employee or agent of such corporation, or is or was serving at the request of such person as an officer, director, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys' fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with such action, suit or proceeding, provided that such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation's best interests and, with respect to any criminal action or proceeding, had no reasonable cause to believe that his or her conduct was illegal.
A Delaware corporation may indemnify any persons who are, or are threatened to be made, a party to any threatened, pending or completed action or suit by or in the right of the corporation by reason of the fact that such person was a director, officer, employee or agent of such corporation, or is or was serving at the request of such corporation as a director, officer, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys' fees) actually and reasonably incurred by such person in connection with the defense or settlement of such action or suit provided that such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation's best interests except that no indemnification is permitted without judicial approval if the officer or director is adjudged to be liable to the corporation. Where an officer or director is successful on the merits or otherwise in the defense of any action referred to above, the corporation must indemnify him or her against the expenses which such officer or director has actually and reasonably incurred. Our certificate of incorporation and bylaws each of which will become effective upon consummation of this offering, provide for the indemnification of our directors and officers to the fullest extent permitted under the Delaware General Corporation Law.
II-1
Section 102(b)(7) of the Delaware General Corporation Law permits a corporation to provide in its certificate of incorporation that a director of the corporation shall not be personally liable to the
II-1
corporation or its stockholders for monetary damages for breach of fiduciary duties as a director, except for liability for any:
Our certificate of incorporation includes such a provision. Expenses incurred by any officer or director in defending any such action, suit or proceeding in advance of its final disposition shall be paid by us upon delivery to us of an undertaking, by or on behalf of such director or officer, to repay all amounts so advanced if it shall ultimately be determined that such director or officer is not entitled to be indemnified by us.
As permitted by the Delaware General Corporation Law, we intend to enterhave entered into indemnification agreements with our directors and executive officers. These agreements, among other things, will require us to indemnify each director and officer to the fullest extent permitted by law and advance expenses to each indemnitee in connection with any proceeding in which indemnification is available.
At present, there is no pending litigation or proceeding involving any of our directors or executive officers as to which indemnification is required or permitted, and we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
We have an insurance policy covering our officers and directors with respect to certain liabilities, including liabilities arising under the Securities Act of 1933, as amended, or the Securities Act, or otherwise.Act.
Item 15. Recent Sales of Unregistered Securities.
Set forth belowOn October 8, 2015, the Company entered into the Purchase Agreement with Lincoln Park, pursuant to which the Company has the right to sell to, and Lincoln Park is information regardingobligated to purchase from the Company, up to $16.5 million in shares of preferredthe Company's common stock, subject to certain limitations, from time to time, over the 36-month period commencing on the date that this registration statement is declared effective by the SEC and convertible promissory notesa final prospectus in connection therewith is filed. On October 8, 2015, Lincoln Park purchased 846,755 shares of the Company's common stock for a total purchase price of $1,500,000 as an initial purchase under the Purchase Agreement and the Company issued and options granted by us within200,000 shares of common stock pursuant to the past three years that wereterms of the Purchase Agreement as consideration for its commitment to purchase additional shares of common stock under the Purchase Agreement. The sale of such shares to Lincoln Park was not registered under the Securities Act. Also included is the consideration, if any, received by us for such shares, notes and options and information relating to the sectionAct because it was made in a transaction exempt from registration under Section 4(a)(2) of the Securities Act and/or ruleRule 506 promulgated thereunder.
On January 5, 2016, the Company entered into a Securities Purchase Agreement (the "Purchase Agreement") with an institutional investor (the "Investor") providing for the issuance and sale by the Company of 1,936,842 shares of the SEC, under which exemption from registration was claimed.
(a) Issuances of Capital Stock:
(1) On September 21, 2010, we issued an aggregate of 668,602 shares of our Series H convertible preferredCompany's common stock at a price per share of $9.79 for an aggregate purchase price of $6,545,613.58.
(2) On November 4, 2010, we issued an aggregate of 299,797$0.95 per share and warrants to purchase 968,421 shares of our Series H convertible preferredthe Company's common stock at a price per share(the "Private Placement Warrants") for aggregate gross proceeds of $9.79 for an aggregate purchase price of $2,935,012.63.
(3) On December 22, 2010, we issued an aggregate of 225,696$1,840,000. The shares of our Series H convertible preferredthe Company's common stock were offered pursuant to an effective shelf registration statement on Form S-3, declared effective by the SEC on November 20, 2014 (File No. 333-199219). The Private Placement Warrants were issued and sold without registration under the Securities Act in reliance on the exemptions provided by Section 4(a)(2) of the Securities Act and/or Regulation D promulgated thereunder and in reliance upon similar exemptions under applicable state laws. Each Private Placement Warrant shall be initially exercisable on the six (6) month anniversary of the issuance date at aan exercise price equal to $1.15 per share of $9.79 for an aggregate purchase price of $2,209,563.84.
(4) On February 17, 2011, we issued 71,488 shares of our shares of our Series H convertible preferred stock at a price per share of $9.79 for an aggregate purchase price of $699,867.52.
II-2
share of Common Stock, subject to customary adjustments, and have a term of exercise of five (5) On June 6, 2011, we issued an aggregateyears from the initial exercise date. H.C. Wainwright & Co., LLC acted as the Company's exclusive placement agent for the issuance and sale of 135,391the shares of our Series H convertible preferred stock at a price per share of $9.79 for an aggregate purchase price of $1,325,477.89.
(6) On September 19, 2011, we issued an aggregate of 612,450 shares of our Series H convertible preferred stock at a price per share of $9.79 for an aggregate purchase price of $5,995,885.50.
(7) On July 25, 2012, $26,444,316.00 aggregate principal amount and accrued interest thereon of our convertible promissory notes were converted into 2,433,328 shares of our Series I convertible preferred stock at a price per share of $11.00.
(8) On July 27, 2012, we issued an aggregate of 3,030,303 shares of our Series J convertible preferred stock at a price per share of $16.50 for an aggregate purchase price of $49,999,999.50.
(9) Since May 1, 2010, we issued an aggregate of 417,404 shares of our Series G convertible preferred stock at a price per share of $9.79 for an aggregate purchase price of $4,086,385 pursuant to the exercise of warrants.
(10) Since May 1, 2010, we issued an aggregate of 965,627 shares of our common stock at prices rangingand Private Placement Warrants, and was paid a cash fee equal to 7.5% of the gross proceeds received by the Company from $0.75 to $5.65 per share to certain of our employees, consultants and directors pursuant to the exercise of stock options under the Onconova Therapeutics, Inc. 2007 Equity Compensation Plan, for an aggregate purchase price of $1,703,824.
(b) Issuance of Convertible Notes
(1) On April 27, 2012, we sold $7,050,000.00 aggregate principal amount of our convertible promissory notes for an aggregate purchase price of $7,050,000.00.
(2) On June 29, 2012, we sold $4,619,000.00 aggregate principal amount of our convertible promissory notes for an aggregate purchase price of $4,619,000.00.
(3) On July 18, 2012, we sold $14,775,316.00 aggregate principal amount of our convertible promissory notes for an aggregate purchase price of $14,775,316.00.
(c) Grants of Stock Options:
(1) Since May 1, 2010, we have granted stock options to purchase an aggregate of 2,607,812 shares of our common stock with exercise prices ranging from $4.32 to $11.06 per share, to certain of our employees, consultants and directors in connection with services provided by such parties to us.
We deemed the offers, sales and issuancessale of the securities described in paragraphs (a)(1) through (a)(9) and (b)(1) through (b)(3) above to be exempt from registration under the Securities Act in reliance on Section 4(2) of the Securities Act, including in some cases, Regulation D and Rule 506 promulgated thereunder and/or Regulation S promulgated thereunder, relative to transactions by an issuer not involving a public offering, to the extent an exemption from such registration was required.
We deemed the issuance of the securities described in paragraph (a)(10) above and the grant of stock options described in paragraph (c)(1) above to be exempt from registration under the Securities Act in reliance on Rule 701 promulgated under the Securities Act as offers and sales of securities under written compensatory benefit plans and contracts relating to compensation in compliance with Rule 701 promulgated under the Securities Act and/or Section 4(2) of the Securities Act, relative to transactions by an issuer not involving a public offering, to the extent an exemption from such registration was required. Each of the recipients of securities in any transaction exempt from registration either received or had adequate access, through employment, business or other relationships, to information about us.
II-3
All purchasers of securities in transactions exempt from registration pursuant to Regulation D promulgated under the Securities Act described above represented to us in connection with their purchase that they were accredited investors and were acquiring the securities for investment purposes only and not with a view to, or for sale in connection with, any distribution thereof and that they could bear the risks of the investment and could hold the securities for an indefinite period of time. The purchasers received written disclosures that the securities had not been registered under the Securities Act and that any resale must be made pursuant to a registration statement or an available exemption from the registration requirements of the Securities Act.
All of the foregoing securities are deemed restricted securities for purposes of the Securities Act. The certificates representing the issued securities described in this Item 15 included appropriate legends setting forth that the applicable securities have not been registered and reciting the applicable restrictions on transfer. There were no underwriters employed in connection with any of the transactions set forthand was reimbursed by the Company for up to $50,000 in this Item 15.expenses.
Item 16. Exhibits and Financial Statement Schedules.
The exhibits to the registration statement are listed in the Exhibit Index to Exhibits attached to this registration statement which isand are incorporated herein by reference herein.
reference.
All schedules have been omitted because either they are provided becausenot required, are not applicable or the information called for is not required or is shown eitherotherwise set forth in the financial statements or notes.and related notes thereto.
The undersigned Registrantregistrant hereby undertakes:
Provided,however, that Paragraphs (1)(i), (1)(ii) and (1)(iii) of this section do not apply if the information required to be included in a post-effective amendment by those paragraphs is contained in reports filed with or furnished to the Commission by the registrant pursuant to section 13 or section 15(d) of the Exchange Act that are incorporated by reference in the registration statement, or is contained in a form of prospectus filed pursuant to Rule 424(b) that is part of the registration statement.
II-3
If the registrant is subject to Rule 430C (§230.430C of this chapter), each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A (§230.430A of this chapter), shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
The undersigned registrant hereby undertakes that, for purposes of determining any liability under the Securities Act of 1933, each filing of the registrant's annual report pursuant to providesection 13(a) or section 15(d) of the Securities Exchange Act of 1934 (and, where applicable, each filing of an employee benefit plan's annual report pursuant to section 15(d) of the Securities Exchange Act of 1934) that is incorporated by reference in the registration statement shall be deemed to be a new registration statement relating to the underwritersecurities offered therein, and the offering of such securities at that time shall be deemed to be the closing specified in the underwriting agreement certificates in such denominations and registered in such names as required by the underwriter to permit prompt delivery to each purchaser.initial bona fide offering thereof.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the Registrantregistrant has been advised that in the opinion of the SECSecurities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director,
II-4
officer or controlling person in connection with the securities being registered, the Registrantregistrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
The undersigned Registrantregistrant hereby undertakes that:
II-4II-5
Pursuant to the requirements of the Securities Act of 1933, as amended, the Registrant has duly caused this registration statementRegistration Statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the CityTownship of Newtown, Commonwealth of Pennsylvania, on the 14th29th day of June, 2013.2016.
| ONCONOVA THERAPEUTICS, INC. | ||||
By: | /s/ RAMESH KUMAR, PH.D. Ramesh Kumar, Ph.D. President and Chief Executive Officer | |||
Each of the undersigned directors and officers of Onconova Therapeutics, Inc. hereby constitutes and appoints each of Ramesh Kumar, Ph.D. and Ajay Bansal, his true and lawful attorneys-in-fact and agents with full power of substitution and resubstitution, for him and his name, place and stead, in any and all capacities, to execute any and all amendments (including post-effective amendments) to this registration statement, to sign any registration statement related to this registration statement filed pursuant to Rule 462(b) of the Securities Act of 1933, and to cause the same to be filed with all exhibits thereto, and all documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and desirable to be done in and about the premises as fully and to all intents and purposes as the undersigned might or could do in person, hereby ratifying and confirming all acts and things that said attorneys-in-fact and agents or any of them, or their or his substitute or substitutes may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, this registration statementRegistration Statement has been signed by the following persons in the capacities and on the dates indicated.
Signature | Title | Date | ||
|---|---|---|---|---|
| /s/ RAMESH KUMAR, PH.D. Ramesh Kumar, Ph.D. | Director, President and Chief Executive Officer (Principal Executive Officer and Principal Operating Officer) | June | ||
/s/ | ||||
June | ||||
Henry S. Bienen, Ph.D. | Director | June 29, 2016 | ||
* Jerome E. Groopman, M.D. | Director | June 29, 2016 | ||
* Michael B. Hoffman | Chairman, Board of Directors | June | ||
Director | June | |||
* Viren Mehta, Pharm.D | Director | June 29, 2016 |
II-5II-6
Signature | Title | Date | ||
|---|---|---|---|---|
| Director | June | |||
Director | June | |||
II-6 The undersigned by signing his name hereto signs and executes this Pre-Effective Amendment No. 1 to Registration Statement on Form S-1 pursuant to the Powers of Attorney executed by the above named signatories and previously filed with the Commission on June 1, 2016.
*By: | /s/ RAMESH KUMAR, PH.D. | |||||
| Ramesh Kumar, Ph.D., Attorney-in-Fact | ||||||
II-7
| Exhibit Number | Exhibit Description | ||
|---|---|---|---|
| 1.1 | Form of | ||
| 3.1 | |||
| 3.2 | |||
| 3.3 | |||
| 4.1 | Form of Certificate of Common | ||
| 4.2 | Eighth Amended and Restated Stockholders' Agreement, effective as of July 27, 2012, by and among Onconova Therapeutics, Inc. and certain stockholders named | ||
| 4.3 | |||
| 4.4 | Form of | ||
| 4.5 | Form of Subscription Rights Statement | ||
| 4.6 | † | Form of Warrant Certificate for Warrants underlying Units | |
| 4.7 | † | Form of Warrant Agreement | |
| 4.8 | † | Form of Pre-Funded Warrant | |
| 5.1 | Opinion of Pepper Hamilton LLP | ||
| 10.1 | * | Development and License Agreement, effective as of September 19, 2012, by and between Onconova Therapeutics, Inc. and Baxter Healthcare | |
| 10.2 | * | License Agreement, effective as of July 5, 2011, by and between Onconova Therapeutics, Inc. and SymBio Pharmaceuticals | |
| 10.3 | * | First Amendment to License Agreement, effective as of September 2, 2011, by and between Onconova Therapeutics, Inc. and SymBio Pharmaceuticals | |
| 10.4 | * | License Agreement, effective as of January 1, 1999, by and between Onconova Therapeutics, Inc. and Temple University—Of The Commonwealth System of Higher | |
| 10.5 | * | Amendment to License Agreement, effective as of September 1, 2000, by and between Temple University—Of The Commonwealth System of Higher Education and Onconova Therapeutics, Inc.(Incorporated by reference to Exhibit 10.5 to the Company's Registration Statement on Form S-1 filed on June 14, 2013). | |
II-8
| Exhibit Number | Exhibit Description | ||
|---|---|---|---|
10.6 | * | ||
| 10.7 | * | Definitive Agreement, effective as of May 12, 2010, by and between Onconova Therapeutics, Inc. and The Leukemia and Lymphoma | |
| 10.8 | * | First Amendment to Definitive Agreement, effective as of June 23, 2011, by and between Onconova Therapeutics, Inc. and The Leukemia and Lymphoma | |
| 10.9 | * | Second Amendment to Definitive Agreement, effective as of May 29, 2012, by and between Onconova Therapeutics, Inc. and The Leukemia and Lymphoma | |
| 10.10 | * | Third Amendment to Definitive Agreement, effective as of January 5, 2013, by and between Onconova Therapeutics, Inc. and The Leukemia and Lymphoma | |
| 10.11 | * | Termination of Agreement, effective as of February 5, 2013, by and between Onconova Therapeutics, Inc. and The Leukemia and Lymphoma | |
| 10.12 | * | Limited Liability Company Agreement of GBO, LLC, dated as of December 12, 2012, by and between Onconova Therapeutics, Inc. and GVK Biosciences Private | |
| 10.13 | |||
| 10.14 | Employment Agreement, effective as of July 1, 2015, by and between Onconova Therapeutics, Inc. and Ramesh Kumar, Ph.D.(Incorporated by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed on July 8, 2015). | ||
| 10.15 | Letter Agreement, effective as of January 1, 2016, by and between Onconova Therapeutics, Inc. and Ramesh Kumar, Ph.D.(Incorporated by reference to Exhibit 10.2 to the Company's Current Report on Form 8-K filed on February 17, 2016). | ||
| 10.16 | Amended and Restated Employment Agreement, effective as of July 1, 2015, by and between Onconova Therapeutics, Inc. and Thomas McKearn, M.D., Ph.D.(Incorporated by reference to Exhibit 10.2 to the Company's Current Report on Form 8-K filed on July 8, 2015). | ||
| 10.17 | Amended and Restated Employment Agreement, effective as of July 1, 2015, by and between Onconova Therapeutics, Inc. and Ajay Bansal.(Incorporated by reference to Exhibit 10.4 to the Company's Current Report on Form 8-K filed on July 8, 2015). | ||
| 10.18 | Consulting Agreement, effective as of January 1, 2012, by and between Onconova Therapeutics, Inc. and E. Premkumar Reddy, Ph.D., including Consultant Agreement Renewal, dated February 27, 2013(Incorporated by reference to Exhibit 10.23 to the Company's Registration Statement on Form S-1 filed on June 14, 2013) | ||
II-7II-9
| Exhibit Number | Exhibit Description | ||
|---|---|---|---|
| 10.20 | |||
| 10.21 | Onconova Therapeutics, Inc. 2013 Performance Bonus Plan(Incorporated by reference to Exhibit 10.26 to Pre-Effective Amendment | ||
| 10.22 | Employment Agreement, effective as of | ||
| 10.23 | Employment Agreement, effective as of | ||
| 10.24 | |||
| 10.25 | |||
| 10.26 | |||
| 10.27 | |||
Subsidiaries of Onconova Therapeutics, Inc.(Incorporated by reference to Exhibit 21.1 to the Company's Annual Report on Form 10-K filed on March 28,2016). | |||
| 23.1 | Consent of Ernst & Young, LLP. | ||
| 23.2 | † | Consent of | |
| 24.1 | D | Power of Attorney | |
| 99.1 | Form of Instructions as to Use of Subscription Rights Statements | ||
| 99.2 | Form of Letter to Shareholders who are Record Holders | ||
| 99.3 | Form For Use With Election to Receive Pre-Funded Warrants | ||
II-8II-10