PRELIMINARY PROSPECTUS SUBJECT TO COMPLETION DATED _____________.

PROSPECTUS18,333,450 SharesCARDO MEDICAL, INC.Common StockOTC Bulletin Board Trading Symbol: CDOM.OB
The selling stockholders may offer and sell from time to time up to an aggregate of 18,333,450 shares of Cardo Medical, Inc. (the "Company") common stock that they own or that they may acquire from us upon exercise of warrants. For information concerning the selling stockholders and the manner in which they may offer and sell shares of our common stock, see "Selling Stockholders" and "Plan of Distribution" in this prospectus.
We will not receive any proceeds from the sale by the selling stockholders of their shares of common stock other than the exercise price of the warrants if and when the warrants are exercised unless the warrants are exercised on a cashless basis.
On December 17, 2009, the last reported sale price for our common stock on the OTC Bulletin Board was $1.01 per share.
Investing in shares of our common stock involves a high degree of risk. You should purchase our common stock only if you can afford to lose your entire investment. See "Risk Factors," which begins on page 3.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The selling stockholders have not engaged any underwriter in connection with the sale of their shares of common stock. The selling stockholders may sell their shares of common stock in the public market based on the market price at the time of sale or at negotiated prices. The selling stockholders may also sell their shares in transactions that are not in the public market in the manner set forth under "Plan of Distribution."
You should rely only on the information contained in this prospectus. We have not authorized any dealer, salesperson or other person to provide you with information concerning us, except for the information contained in this prospectus. The information contained in this prospectus is complete and accurate only as of the date on the front cover page of this prospectus, regardless of the time of delivery of this prospectus or the sale of any common stock. This prospectus is not an offer to sell these securities and we are not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
The date of this prospectus is December [ ], 2009.
The information in this preliminary prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and it iswe are not soliciting an offeroffers to buy these securities in any statejurisdiction where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED JULY 15, 2019
BioCardia, Inc.

1,500,000 Units, Each Unit Consisting of
One Share of Common Stock and
a Warrant to Purchase One Share of Common Stock
We are offering 1,500,000 units, with each unit consisting of one share of common stock, $0.001 par value per share, and a warrant to purchase one share of common stock at an exercise price of $ per share (together with the shares of common stock underlying such warrants, the “Units”), of BioCardia, Inc., a Delaware corporation (the “Company”), in a firm commitment underwritten public offering at a public offering price of $ per Unit (this “Offering”).
The warrants offered hereby may be exercised from time to time beginning on the date of issuance and expire on 2022. Our Units have no stand-alone rights and will not be certificated or issued as stand-alone securities. The shares of our common stock and the warrants comprising our Units are immediately separable and will be issued separately in this Offering.
Our common stock is currently quoted on the OTCQB Marketplace (“OTCQB”) under the symbol “BCDA.” On July 12, 2019, the last reported price for our common stock as reported on the OTCQB was $10.00 per share, which gives effect to our one-for-nine reverse split of our issued and outstanding shares of common stock which began trading on a post-split basis on OTCQB on June 6, 2019. There is currently no public market for the offered warrants. We have applied to list our common stock and our warrants on the Nasdaq Capital Market under the symbols “BCDA” and “BCDAW.” There can be no assurance that we will be successful in listing our common stock or our warrants on the Nasdaq Capital Market.
Investing in our securities involves significant risks. You should review carefully the “Risk Factors” beginning on page 14 of this prospectus for a discussion of information that should be considered in connection with an investment in our securities.
The Securities and Exchange Commission and state securities regulators have not approved or disapproved of these securities, or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
Per Unit | Total | |||||||
Public offering price(1) | $ | |||||||
Underwriting discounts and commissions(2) | $ | |||||||
Proceeds, before expenses, to us(3) | $ | |||||||
(1) | The public offering price and underwriting discount corresponds to (i) a public offering price per share of common stock of $ and (ii) a public offering price per warrant of $ . |
(2) | We have also agreed to reimburse the underwriters for certain expenses. See “Underwriting” for additional information regarding total underwriter compensation. |
(3) | The amount of offering proceeds to us presented in this table does not give effect to any exercise of the warrants being issued in this Offering, if any. |
We have granted a 45-day option to the underwriters to purchase up to additional shares of common stock at a price of $ per share and/or additional warrants at a price of $ per warrant less, in each case, the underwriting discount solely to cover overallotments, if any. If the underwriters exercise the option in full, the total underwriting discounts and commissions payable to us will be $ and the total proceeds to us, before expenses, will be $ .
The underwriters expect to deliver the units against payment in New York, New York on , 2019.
Sole Book-Running Manager
Maxim Group LLC
Co-Managers
| Arcadia Securities | Dawson James Securities, Inc. |
The date of this prospectus is , 2019
TABLE OF CONTENTS
Page | |
About this Prospectus | i |
Prospectus Summary | 1 |
The Offering | 10 |
Risk Factors | 14 |
Special Note Regarding Forward-Looking Statements | 53 |
Use of Proceeds | 55 |
Dividend Policy | 56 |
Capitalization | 57 |
Dilution | 59 |
Business | 61 |
Properties | 89 |
Legal Proceedings | 89 |
Management’s Discussion | 90 |
Directors, Executive Officers and | 99 |
Executive | 108 |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 111 |
Certain Relationships and Related Transactions, | 114 |
Description of the Securities We Are Offering | 116 |
Material | 121 |
Shares Available for Future Sales | 127 |
Underwriting | 128 |
Legal Matters | 132 |
Experts | 132 |
Where You Can Find More Information | 132 |
Index to Consolidated Financial Statements | F-1 |
ABOUT THIS PROSPECTUS
The registration statement of which this prospectus forms a part that we have filed with the Securities and Exchange Commission, or SEC, includes exhibits that provide more detail of the matters discussed in this prospectus. You should read this prospectus and the related exhibits filed with the SEC, together with the additional information described under the heading “Where You Can Find More Information.”
You should rely only on the information contained in this prospectus and in any free writing prospectus prepared by or on behalf of us. We have not, and the underwriters have not, authorized anyone to provide you with information different from, or in addition to, that contained in this prospectus or any related free writing prospectus. This prospectus is an offer to sell only the securities offered hereby but only under circumstances and in jurisdictions where it is lawful to do so. The information contained in this prospectus is current only as of its date. Our business, financial condition, results of operations and prospects may have changed since that date.
We are not offering to sell or seeking offers to purchase these securities in any jurisdiction where the offer or sale is not permitted. We have not done anything that would permit this Offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States Persons outside the United States who come into possession of this prospectus and any free writing prospectus related to this Offering in jurisdictions outside the United States are required to inform themselves about and to observe any restrictions relating to this Offering and the distribution of this prospectus and any such free writing prospectus applicable to that jurisdiction.
Unless the context otherwise requires, the terms “BioCardia,” the “Company,” “we,” “us” and “our” refer to BioCardia, Inc. We have registered our name, logo and the trademarks “BioCardia,” “CardiAMP,” “CardiALLO,” and “Morph” in the United States. We have registered the trademarks “CardiAMP” and “CardiALLO” for use in connection with a biologic product, namely, a cell-based therapy product composed of bone marrow derived cells for medical use. We also have rights to use the “Helix” trademark in the United States. Other service marks, trademarks and trade names referred to in this prospectus are the property of their respective owners. Except as set forth above and solely for convenience, the trademarks and trade names in this prospectus are referred to without the ®, © and ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto.
PROSPECTUS SUMMARY
This summary highlights information contained in greater detail elsewhere in this prospectus. This summary does not contain all of the information that is important to you.you should consider before investing. You should read the entire prospectus carefully, including the Risk Factorssections titled “Risk Factors” and our consolidated financial statements“Management’s Discussion and related notes appearing elsewhereAnalysis of Financial Condition and Results of Operations.”
Unless otherwise indicated, all information in this prospectus before making an investment decision.
Our Business
Cardo Medical, Inc. ("Cardo" or the "Company"),reflects a Delaware corporation, is an orthopedic medical device company specializing in designing, developing and marketing reconstructive joint devices and spinal surgical devices. Reconstructive joint devices are used to replace knee, hip and other joints that have deteriorated through disease or injury. Spinal surgical devices involve products to stabilize the spine for fusion and reconstructive procedures. Within these areas, Cardo intends to focus on the higher-growth sectorsone-for-nine reverse stock split of the orthopedic industry, such as advanced minimally invasive instrumentation and bone-conserving high-performance implants. Cardo is focused on developing surgical devices that will enable surgeons to bridge the gap between soft tissue-driven sports medicine techniques and classical reconstructive surgical procedures. Cardo commercializes its reconstructive joint devices through its Cardo Orthopedics division and its spine devices through its Cardo Spine division.
GENERAL
On June 18, 2008, Cardo Medical, LLC, a California limited liability company, entered into a Merger Agreement and Plan of Reorganization with clickNsettle.com, Inc. ("CKST") and Cardo Acquisition, LLC, a California limited liability company and wholly-owned subsidiary of CKST. Upon the consummation of the transactions contemplated by the Merger Agreement, CKST acquired Cardo Medical, LLC through a merger of Cardo Medical, LLC with Cardo Acquisition, with Cardo Medical, LLC continuing as the surviving entity in the Merger and a wholly-owned subsidiary of CKST. Pursuant to the Merger Agreement, all of theour issued and outstanding units of Cardo Medical, LLC's membership interests were converted into the right to receive shares of the common stock of CKST. In connection with the consummation of the Merger, CKST approved through its stockholders an amendment to its Amended and Restated Certificate of Incorporation to change its name from "clickNsettle.com, Inc." to "Cardo Medical, Inc."
Our executive offices are located at 9701 Wilshire Blvd., Suite 1100, Beverly Hills, California 90212. Our telephone number is (310) 274-2036. Our website is www.cardomedical.com. Information on our website or any other website is not part of this prospectus. References to "we," "us," "our" and similar words in this prospectus refer to Cardo Medical, Inc. and its consolidated subsidiaries.
Sale of Securities to the Selling Stockholders
On October 27, 2009, we sold, in the first tranche of a private placement, 9,949,276 shares of common stock, options, restricted stock units and warrants, and the corresponding adjustment of all common stock and restricted stock unit prices per share and stock option and warrant exercise prices per share and conversion ratios. Our common stock began trading on a reverse stock split-adjusted basis on The OTC Market on the opening of trading on June 6, 2019.
Overview
We are a clinical-stage regenerative medicine company developing novel therapeutics for cardiovascular diseases with large unmet medical needs. Our lead therapeutic candidate is the investigational CardiAMP Cell Therapy System, which provides an autologous bone marrow derived cell therapy (using a patient’s own cells) for the treatment of two clinical indications: heart failure that develops after a heart attack and chronic myocardial ischemia. Chronic myocardial ischemia involves sustained poor blood perfusion to the heart muscle often at $0.35 per share. On November 13, 2009, we sold,the microvascular level. The CardiAMP Cell Therapy System is being developed to provide a comprehensive biotherapeutic solution, incorporating a proprietary molecular diagnostic to characterize the potency of a patient’s own bone marrow cells and determine if they are an optimal candidate for therapy, a proprietary point of care processing platform to prepare cells at the patient’s bedside, an optimized therapeutic formulation that builds on the total experience in the cardiac stem cell field to-date, and a proprietary interventional delivery system that easily navigates a patient’s vasculature to securely deliver cells in a routine cardiac catheterization procedure. Our second tranchetherapeutic candidate is the CardiALLO Cell Therapy System, an investigational culture expanded bone marrow derived allogenic “off the shelf” cell therapy from a donor that meets specified criteria, which has potential to be advanced for many clinical indications including heart failure.
We are committed to applying our expertise in the fields of autologous and allogeneic cell-based therapies to improve the lives of patients with cardiovascular conditions.
Market Overview
Adult bone marrow contains a large reservoir of stem and progenitor cells capable of differentiating into blood cells, blood vessel cells, and connective tissue cells. In addition, numerous pre-clinical cardiac studies have shown that cell-to-cell communication in which bone marrow-derived stem cells promotes microcirculatory adaptation, immune modulation and cell protection, facilitating cardiac recovery, in part via recruitment of other reparative cell types.
Bone marrow cell homing to the heart is believed to be part of the body’s natural repair process. After a heart attack or an acute injury to the heart, cells from bone marrow are known to home to the heart. For example, a population of bone marrow cells that express the surface marker CD34 has certain receptors, including CXC-4 and CXC-7 receptors, that home to the SDF-1 ligand in injured heart tissue. In heart failure, the heart may have fewer of these homing signals and a decreased ability to stimulate or recreate this signaling process, leading to a lower likelihood of heart tissue repair. A number of other bone marrow derived cells with unique cell surface markers have also been shown to have beneficial effects in animal models of heart failure and chronic myocardial ischemia disease when delivered directly to the heart.
Bone marrow derived cell-based therapy has been shown to have the potential to provide therapeutic benefit for patients with heart failure and chronic myocardial ischemia. In the past decade, intramyocardial delivery of bone marrow derived cell-based therapies in preclinical and clinical studies of heart failure and chronic myocardial ischemia has predominantly resulted in benefits, such as improvement in ventricular function, reduction in the area of dead heart tissue and increase in heart muscle blood flow, reduction in pain symptoms, reduced major adverse event rates, and reduced mortality.
Recent systematic review and meta-analysis of the scientific literature from 23 randomized controlled trials prior to 2013, covering more than 1,200 participants, was published by Fisher in Circulation Research in January 2015. The review found evidence that bone marrow cell treatment, including intramyocardial delivery of bone marrow cells, has improved left ventricle ejection fraction, or LVEF, and chronic ischemic heart disease. The authors of the review found evidence for a potential beneficial clinical effect in terms of mortality and performance status after at least one year post-treatment in people who suffer from chronic ischemic heart disease and heart failure. Results in heart failure trials indicate that bone marrow derived cell-based therapy leads to a reduction in deaths and readmission to hospital and improvements over standard treatment as measured by tests of heart function. This review concluded that further research is required to confirm the results. Published scientific papers provide clinical support for efficacy from randomized controlled clinical trials of intramyocardial delivery of bone marrow derived cells in closely related clinical conditions of chronic myocardial ischemia, diastolic heart failure, and subacute myocardial infarction.
Product Pipeline and Development Status
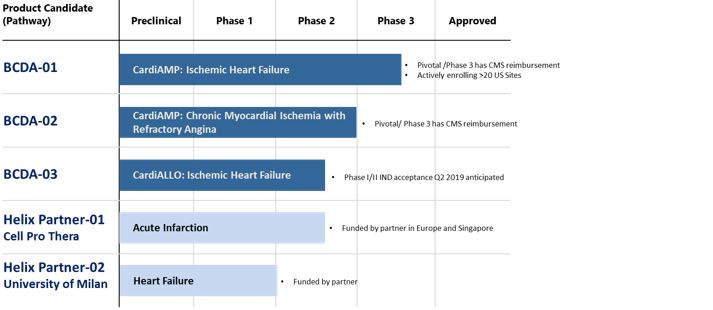
CardiAMP Cell Therapy System
The CardiAMP Cell Therapy System, or CardiAMP, is our lead therapeutic program being advanced for two clinical indications. This investigational cell therapy system is comprised of (i) a cell potency screening test, (ii) a point of care cell processing platform, and (iii) a biotherapeutic delivery system. In the screening process, the physician extracts a small sample of the patient’s bone marrow in an outpatient procedure performed under local anesthesia. The clinic sends the sample to a centralized diagnostic lab, which tests for identified biomarkers from which we generate a potency assay score for the patient. During the treatment for patients who are assessed as meeting the indication specific CardiAMP cell potency assay score, a doctor harvests and then prepares the patient’s own bone marrow mononuclear cells, or autologous cells, using our point of care cell processing platform, which a cardiologist then delivers into the heart using our proprietary biotherapeutic delivery system. We designed the entire procedure to be performed in approximately 60 to 90 minutes, which we believe is substantially faster than alternative cell-based therapies in development. The patient then leaves the hospital the same or next day.
CardiAMP Cells Phase I Heart Failure Study: Transendocardial Autologous Marrow Cells in Myocardial Infarction
The CardiAMP Phase I Transendocardial Autologous Marrow Cells in Myocardial Infarction or TABMMI trial enrolled 20 patients with ischemic systolic heart failure in an open label safety trial of bone marrow cells delivered with the Helix™ biotherapeutic delivery system at a dosage of 100 million cells. Results showed improvement in cardiac function as measured by left ventricular ejection fraction, improved exercise tolerance, and superior survival as compared to historical controls. The Phase I TABMMI study was submitted to the Argentine Administración Nacional de Medicamentos, Alimentos y Technología Médica.
In our TABMMI Phase I trial of CardiAMP cells, we enrolled 20 patients with previous evidence of having had a heart attack and who presented with a low ejection fraction of less than or equal to 40% and greater than or equal to 20%. Baseline evaluations included informed consent, history and physical examination, electrocardiogram, 24-hour Holter monitoring, echocardiography, routine blood tests and exercise tolerance testing. Reduced regional heart wall motion was coincident with the diseased coronary vessel in each patient. A total of 20 patients with heart failure (NYHA Class I, II and III) each received three to ten transendocardial infusions of cells using our Helix™ biotherapeutic delivery system in an open-label dose-escalation two cohort trial. Dosage administration ranged from 30 million to 130 million autologous bone marrow derived mononuclear cells, with an average of 96 million cells.
Bone marrow cells delivered in TABMMI demonstrated an excellent safety profile in this heart failure population, with no treatment related toxicities observed. The 20 patients who received CardiAMP cells, demonstrated improvements from baseline to both six-month and 12-month follow-up across a number of parameters important in heart failure, including statistically and clinically significant improvements in left ventricular, or LV, function (ejection fraction). The difference of average ejection fraction was statistically significant over baseline at all follow-up time points of 6 months, 12 months, and 24 months. Average exercise tolerance time showed an increase at all follow-up time points, but was only statistically significant at 12 months and 24 months.
A total of 12 adverse events were observed in six patients, although none were related to the investigational delivery or cell transplantation procedure. The complete results of the 20 patients at two-year follow-up have been published in the journal Eurointervention in 2011.
CardiAMP Cells Phase II Heart Failure Trial: Transendocardial Autologous Cells in Heart Failure Trial (TAC-HFT)
In our co-sponsored Phase II Transendocardial Autologous Cells in Heart Failure Trial, patients with ischemic systolic heart failure were randomized on a one to one basis into two double-blind, placebo-controlled trials: TACHFT-BMC and TACHFT-MSC. The IND for the TACHFT trial was filed with the U.S. Food and Drug Administration, or FDA Center for Biologics Evaluation and Research in 2008 by the University of Miami, the co-sponsor of the trial.
In the safety dose escalation roll-in cohort stage of the study, eight patients received treatment with either CardiAMP cells, or autologous bone marrow mesenchymal cells, or MSC, at dosages of 100 million or 200 million cells. In the randomized, placebo-controlled efficacy stage of the study, 29 patients received treatment with either CardiAMP cells or placebo and 30 patients received treatment with either MSCs or placebo. The mode of administration was 10 intramyocardial infusions per patient using our Helix™ biotherapeutic delivery system into the myocardium adjacent to and into the infarcted tissue. All subjects had ischemic systolic heart failure (NYHA Class I, II or III).
TACHFT-BMC met its primary safety endpoint at both dosages (100 million and 200 million cells) and treated patients had increased functional capacity, improved quality of life, symptoms and key markers of cardiac function predictive of survival, such as end systolic volume, or ESV. The TACHFT-BMC trial included a single dose of CardiAMP cells with a follow up observation period of 12 months. The Phase II, randomized, placebo-controlled study met its primary safety endpoint and demonstrated statistically significant and clinically meaningful improvements in secondary efficacy endpoints of functional capacity, as measured by the six minute walk distance (6MW), and in quality of life, as measured by the Minnesota Living with Heart Failure Questionnaire score. Phase II results were published in the journal of the American Medical Association in 2014 and were presented at the World Congress of Regenerative Medicine in 2015.
By way of comparison, previous investigational device exemption (IDE) pivotal clinical trials that led to FDA approval of Cardiac Resynchronization Therapy, in which a pacemaker stimulates both sides of a private placement, 7,808,561 sharespatient’s heart for the treatment of common stockheart failure followed the same IDE regulatory pathway and have had similar endpoints to the CardiAMP Heart Failure Trial. Pacemakers that pace both sides of the heart are intended for patients that are NYHA III and IV versus the CardiAMP Heart Failure Trial indication of NYHA II and III. Results from 5 out of 6 randomized pivotal trials for pacemakers that pace both sides of the heart have showed both smaller improvements in functional capacity as measured by the six minute walk test and smaller improvement in quality of life than the CardiAMP Phase II results. Although the benefits with pacemakers that pace both sides of the heart were less than observed in CardiAMP placebo controlled Phase II trial, these results for the permanently implantable pacemaker devices were sufficient to obtain FDA approval.
CardiAMP Cell Phase III Heart Failure Trial
The CardiAMP Heart Failure Trial is a Phase III, multi-center, randomized, double-blinded, sham-controlled study of up to 260 patients at $0.35 per share. 40 centers nationwide, which includes a 10-patient roll-in cohort.
The Phase III pivotal trial is designed to provide the primary support for the safety and efficacy of the CardiAMP Cell Therapy System. The primary endpoint is a clinical composite of six minute walk distance and major adverse cardiac and cerebrovascular events. Based on the results achieved in the Phase II trial, our Phase III pivotal trial is designed to have more than 95% probability of achieving a positive result with statistical significance. Statistical significance denotes the mathematical likelihood that the results observed are real and not due to chance.
Particularly novel aspects of this trial include a cell potency assay to screen subjects who are most likely to respond favorably to treatment, a point of care treatment method, use of a high target dose of 200 million cells and an efficient transcatheter delivery method that is associated with high cell retention. Success in the primary endpoint of the trial may lead to a new treatment for those suffering from heart failure in the aftermath of a heart attack. The trial design was published in the peer reviewed American Heart Journal in 2018.
The Department of Health & Human Services Centers for Medicare & Medicaid Services, or CMS, has designated the CardiAMP Heart Failure Trial as a qualifying trial for Medicare national coverage determination that routine costs of care will be covered for Medicare beneficiaries. Private insurance plans covering 50 million insured Americans follow this CMS reimbursement policy and are also anticipated to pay for these costs in the CardiAMP Heart Failure Trial. Covered costs today for both the treatment and control arms of the trial include patient screening, the CardiAMP Cell Therapy System and procedure, and clinical follow-up at one and two years after the procedure.
The Phase III CardiAMP Heart Failure Trial was initiated in the fourth quarter of 2016, and the first patient treated in Q1 2017. The Data Safety Monitoring Board (DSMB) safety review of the 10 patient roll in cohort treated at three clinical sites was completed successfully in the third quarter of 2017, and the trial is actively enrolling today at 22 clinical sites. Efficacy data from the primary endpoint in the open label roll in cohort were presented at the American Heart Association Scientific Sessions in 2018.
At the primary endpoint of exercise capacity at 12 months, the 10-patient roll-in cohort of the trial showed clinically meaningful improvement, walking an average of 46.4 meters more than baseline, although the improvement was not considered statistically significant (p=0.06). Eight of the 10 patients experienced improvement in their exercise capacity. This improvement is more than triple the average improvement over baseline reported in the CardiAMP-treated arm of the Phase II TAC-HFT-MNC trial, and greater than the average improvement seen in a number of pivotal trials for implantable pacemakers to treat heart failure.
In the private placement, we issued an aggregatesecondary efficacy endpoint of 17,757,837 sharesquality of common stocklife, patients showed a clinically meaningful improvement of 9.8 points relative to baseline, which was not statistically significant (p=0.33) in the small cohort. Seven of the 10 patients reported better quality of life after CardiAMP treatment. This was a greater improvement over baseline than was seen in the Phase II TAC-HFT-MNC trial of CardiAMP therapy.
The secondary efficacy endpoints of superiority relative to major adverse cardiac events (MACE) and survival were not possible to assess in this roll-in cohort as there is no control arm specific to this cohort. There were no treatment emergent major adverse cardiac events in this group at $0.35 per share,30 days, while there was one MACE event due to 74 accredited investors. In conjunction witha hospitalization at nine months. All MACE have not been adjudicated at this time. All patients from this cohort were alive and out of the private placement, Cardo Medical, Inc. issuedhospital at 12 months.
Echocardiographic core lab measures of cardiac function in the roll-in cohort at 12 months showed improvement in left ventricular ejection fraction of 4.1 percent (N=10, p=0.181), left ventricular end systolic volume (2.5 ml, p=0.502), neither of which were statistically significant in this small cohort relative to baseline. However, statistically significant improvement in total wall motion score (5.9 points, p=0.014) were observed.
We have enrolled 45 patients in the trial to date and enrollment is anticipated to begin accelerating due to additional compelling data presented from the roll in cohort, the addition of world class centers to the placement agent warrants to purchase 575,613 sharestrial, and the completion of competitive clinical programs. We anticipate a first interim readout from the trial in Q3 2019, a second interim readout in Q3 2020, that trial enrollment will be completed in Q3 2020 and that top line data will be available in Q3 2021.
Although clinical trial results show support for safety and efficacy in our Phase I TABMMI trial, Phase II TACHFT-BMC trial, and Phase III CardiAMP Heart Failure trial, the CardiAMP Cell Therapy System still remains investigational, and no claims regarding safety or efficacy can be made until the products are approved by the FDA.
We believe the remaining clinical efficacy risk is modest in light of the Company's common stock,Phase I, II, and III data available, and broader literature which supports CardiAMP Cell Therapy System as a number thattherapeutic candidate for heart failure secondary to having had a heart attack. The CardiAMP Cell Therapy System has the potential to significantly benefit patients who have limited options and provide a cost-effective therapy to help reduce the substantial heart failure hospitalization and care costs. Unlike other autologous cell therapies, the CardiAMP Cell Therapy System, is equivalentexpected to six percent (6%)have elegantly straightforward and low cost manufacturing and distribution, significantly enhancing its probability of commercial success.
CardiAMP Chronic Myocardial IschemiaPhase III PivotalTrial
In 2017, we submitted for approval of an investigational device exemption for the CardiAMP Cell Therapy System in a second related clinical indication of chronic myocardial ischemia based on the strength of our Phase I and II heart failure trial data, and the strength of the numberclinical data showing support for the efficacy of sharesone component of common stock soldour cell therapy (the CD34+ cells) in chronic myocardial ischemia. In January 2018, the FDA approved the IDE for the randomized controlled pivotal trial of autologous bone marrow mononuclear cells using the CardiAMP Cell Therapy System in patients with refractory chronic myocardial ischemia for up to 343 patients at up to 40 clinical sites in the private placement transaction to investors that were solicited byUnited States. This therapeutic approach uses the placement agent ("Approved Investors"), at an exercise price of $0.44 per share. The warrants issuedsame novel aspects as the CardiAMP Heart Failure Trial. An update to the placement agentstatistical analysis plan to enable an adaptive trial design is anticipated. Success in the private placement are sometimesprimary endpoint of the trial, which is exercise tolerance, may lead to a new treatment for those suffering from chronic myocardial ischemia and having sustained debilitating heart pain, referred to as refractory angina.
In 2018, CMS approved BioCardia’s request for the "Placement Agent Warrants"designation of the CardiAMP Chronic Myocardial Ischemia Trial as a qualifying trial for Medicare national coverage determination similar to the designation received for the CardiAMP Heart Failure Trial. It is anticipated that this second pivotal trial will build on and benefit from the experience and infrastructure from the CardiAMP Heart Failure Trial. With additional modest funding to support this program, there is potential for this trial to be activated in 2019.
CardiAMP Cell Therapy System - Other Indications
In the future, we may also explore the continued development of CardiAMP for use immediately after a heart attack and for treatment of heart failure with preserved ejection fraction, a form of heart failure wherein the amount of blood pumped from the heart’s left ventricle with each beat (ejection fraction) is greater than 50%.
CardiALLO Cell Therapy System
Our second therapeutic candidate is the CardiALLO Cell Therapy System, an investigational culture expanded bone marrow derived “off the shelf” mesenchymal stem cell therapy. CardiALLO cell therapy cells are expanded from Neurokinin-1 receptor positive bone marrow cells. These cells are being advanced to treat heart failure, but have potential for numerous therapeutic applications as these are anticipated to be the cells that respond to the release of Substance P. Substance P (“SP”) is a neuropeptide released from sensory nerves and is associated with the inflammatory processes and pain. The Placement Agent Warrants expireendogenous receptor for SP is the neurokinin-1 receptor (“NK1-receptor” or “NK1R”), which is distributed over cytoplasmic membranes of many cell types (for example neurons, glia, endothelia of capillaries and lymphatics, fibroblasts, stem cells, and white blood cells) in many tissues and organs. SP amplifies or excites most cellular processes. Elevation of serum, plasma, or tissue SP and/or its receptor NK1R has been associated with many diseases: sickle cell crisis, inflammatory bowel disease, major depression and related disorders, fibromyalgia rheumatological, and infections such as HIV/AIDS and respiratory syncytial virus, as well as in cancer. Our CardiALLO NK1R positive derived cells are believed to be an important subset of the cells that we have delivered in our previous preclinical and clinical mesenchymal stem cell studies. We believe this therapy presents the advantages of an "off the shelf" therapy that does not require tissue harvesting or cell processing. We have completed manufacturing validation runs of these cells at BioCardia to support future clinical studies. We are working to obtain FDA acceptance of an Investigational New Drug (“IND”) application for a Phase I/II trial for CardiALLO Cell Therapy System for the treatment of ischemic systolic heart failure in the fourth quarter of 2019.
The subset of patients we are targeting initially for the CardiALLO Heart Failure Trial are those that have been excluded from the CardiAMP Heart Failure Trial due to their lower cell potency assay scores. CardiALLO trial activation would likely enhance enrollment in the CardiAMP Heart Failure Trial. And if the CardiAMP trial is successful, as anticipated, there is the potential for the CardiALLO therapy indication to be designated as an orphan indication.
CardiALLO related Phase I /II Studies: POSEIDON, TAC-HFT-MSC, and TRIDENT
We have co-sponsored three clinical trials for MSCs for the treatment of ischemic systolic heart failure. In substantially similar trial designs, the POSEIDON Phase I/II trial compared autologous MSCs to allogeneic MSCs, the TACHFT-MSC Phase II trial compared autologous MSCs to placebo, and the TRIDENT Phase II compared allogenic MSCs at different doses. The first two trials shared common arms of autologous MSCs, enabling a bridge to placebo, leading us to conclude that allogeneic MSC therapy has potential to be superior to placebo. The IND for the TACHFT trial was filed with the FDA Center for Biologics Evaluation and Research in 2008 by the University of Miami, our co-sponsor for the trial. The POSEIDON trial and the TRIDENT trials were submitted by amendment under the same IND filed for the TACHFT study, and was co-sponsored by the University of Miami, the National Institutes of Health and us. The results from all three of these studies can be submitted to the FDA in support of an IND for the CardiALLO Cell Therapy System.
CardiALLO Development
CardiALLO is being advanced with an anticipated improved cell production strategy to be detailed in the Chemistry Manufacturing and Controls (CMC) of the IND in development. We believe the new CMC will reduce the likelihood of immune response to transplanted allogenic cells further, may enhance efficacy, and will enable commercial scale up and global distribution. CardiALLO will require more extensive clinical development than the CardiAMP Cell Therapy System, beginning with a Phase I/II trial that follows previous work, to confirm the results with the modified cell culture and dosage strategy.
We are performing our own CMC development work in BioCardia laboratories to accelerate the effort and secure additional intellectual property, and have taken steps to build the capacity at BioCardia to culture and supply the MSC cells for CardiALLO clinical development. Our development tissue culture laboratory is fully operational and our clinical manufacturing controlled environment room has been built. We expect to demonstrate support for the safety and efficacy of MSCs in our target patient population in a Phase I/II randomized controlled study. We expect the CardiALLO Phase II Heart Failure Trial will enroll patients with control, low dose and high dose groups using the Helix™ biotherapeutic delivery system and a similar inclusion criterion as the CardiAMP Heart Failure Trial. In the United States, CardiALLO Cell Therapy System is expected to be regulated by the FDA as a biologic product with a dedicated delivery system, our own Helix™ biotherapeutic delivery system.
The completed clinical studies show support for the safety and efficacy of both the CardiAMP Cell Therapy System and CardiALLO Cell Therapy System development programs; however, both product candidates remain investigational, and no claims regarding safety or efficacy can be made until the constituent products are approved by the FDA. As we engage in clinical trials of our therapeutic candidates, we have compensated and intend to compensate all parties performing the trials or studies (including all the parties identified in our Annual Report on November 13, 2014Form 10-K) only on terms that are standard and customary in clinical study arrangements.
These two therapeutic candidates provide compelling and synergistic approaches to replicating the natural response of bone marrow cells to cardiac injury. CardiAMP harnesses the potential of autologous minimally processed bone marrow cells, using an anticipated companion diagnostic to identify patients most likely to benefit from the therapy. CardiALLO utilizes mesenchymal stem cells from a donor that meets specified criteria and may be exercised on a cashless basis.
We paidappropriate for patients who are not optimal candidates for the placement agent for this offering a commission equal to eight percent (8%) of the gross proceeds from the offering that was received from Approved Investors. Additionally, the placement agent received (i) a cash non-accountable expense allowance equal to one percent (1%) of the gross proceeds of the offering received from Approved Investors; (ii) reimbursement of the placement agent's out-of-pocket expenses related to the offering, including its legal fees and expenses up to $40,000; and (iii) warrants to purchase 575,613 shares of common stock equal to six percent (6%) of the number of shares sold in the offering to Approved Investors at a exercise price of $0.44 per share.
1
The registration rights agreement entered into with subscribers in the offering requires us to use commercially reasonable efforts to have this registration statement declared effective by the Securities and Exchange Commission ("SEC") as soon as practicable, but in no event later than the one hundred twenty (120) calendar days after the final closing date or one hundred fifty (150) calendar days if the SEC reviews the registration statement.CardiAMP therapy.
Cell Processing and Cell Delivery Product Platforms
BioCardia has developed and secured exclusive rights to enabling cell processing and cell delivery products, which are used as part of our CardiAMP and CardiALLO therapies, and which we believe validate our approach and development expertise: (i) the CardiAMP cell processing platform, (ii) the CardiALLO cell processing platform, (iii) the Helix™ transendocardial biotherapeutic delivery system, and (iv) our Morph vascular access products.
• | CardiAMP cell processing platform - processes bone marrow aspirate at the point of care to concentrate mononuclear cells and prepare the dosage form. We expect the CardiAMP cell processing platform to be approved in the United States for ischemic systolic heart failure and/or chronic myocardial ischemia as part of the CardiAMP Cell Therapy System clinical development. The platform is currently cleared for use in the United States and in European Union for the preparation of a cell concentrate from bone marrow. The platform is approved in Japan with a broad regenerative medicine indication. The platform is under investigational use for the treatment of heart failure and chronic myocardial ischemia under BioCardia investigational device exemptions in the United States. | |
• | CardiALLO cell processing platform - processes young qualified donor marrow at BioCardia cell manufacturing facility to expand the cell subpopulation that is NK1R positive to produce a cryopreserved “off the shelf” cell dosage that may be shipped to hospitals for therapeutic delivery. This manufacturing facility is believed to be suitable and sufficient for the planned Phase I and Phase II clinical trials for a number of clinical indications including heart failure. |
2
| Helix™biotherapeutic delivery system - delivers therapeutics into the heart muscle with a penetrating helical needle from within the heart. This is a leading delivery platform in the field, which has increased safety and performance. We expect Helix™ to be approved in the United States as part of CardiAMP Cell Therapy System. The | |
|
| |
|
On April 1, 2019, CE Mark certification expired due to delays in certifying to the new ISO 13485:2016 standard, which defines new requirements in the quality management system required for medical devices. We anticipate being in compliance within calendar year 2019. Until we receive renewal of our EC certificate for CE marking, we will not be able to sell our delivery system in Europe. This is not expected to have a material impact on the Company because we have planned for such delay with our Biotherapeutic partners that use Helix™ in Europe. |
| • | Morph vascular access products - provides enhanced control for Helix™ in biotherapeutic delivery and for other common interventions. We have secured all necessary approvals in the United States and Europe. Currently there are seven Morph product model numbers approved for commercial sale in the United States via a 510(k) clearance. The Morph products are valued by physicians performing difficult vascular procedures worldwide and they have been used in more than 12,000 clinical procedures to date. We are working to receive FDA clearance for market release of new Morph product family members, which include the AVANCE bidirectional steerable introducers for use in transseptal procedures. These transseptal procedures may include atrial fibrillation ablation, patent foramen ovale and atrial septal defect repair, percutaneous mitral valve repair, and left atrial appendage closure, among others. FDA clearance of the AVANCE 510(k) submitted to the FDA during the first quarter of 2019 was received in May 2019. |
Business Strategy
We are committed to applying our expertise in the fields of autologous and allogeneic cell-based therapies to improve the lives of patients with cardiovascular conditions. We are pursuing the following business strategies:
● | Complete the ongoing 260 patient, 40 center Phase III pivotal IDE trial of CardiAMP Cell Therapy System for patients with ischemic systolic heart failure. |
● | Complete the FDA approved, CMS reimbursed, 343 patient, 40 center Phase III pivotal IDE trial of CardiAMP Cell Therapy System for patients with chronic myocardial ischemia. |
● | Obtain FDA approval and commercialize CardiAMP Cell Therapy System using a highly-targeted cardiology sales force in the United States. |
● | Advance our CardiALLO Cell Therapy System for the treatment of ischemic systolic heart failure. CardiALLO has the potential to benefit patients for whom the CardiAMP Cell Therapy System is not optimal due to the lower potency of their bone marrow cells. This therapy may present advantages for patients or physicians who wish to avoid bone marrow aspiration, and our development work builds on our clinical development capabilities established through our CardiAMP program. This program positions us to provide therapy to patients ineligible for CardiAMP. |
● | Expand CardiAMP and CardiALLO Cell Therapy Systems into additional cardiac indications. CardiAMP and CardiALLO Cell Therapy Systems have potential therapeutic benefits for multiple cardiovascular indications in addition to ischemic systolic heart failure and ischemic heart disease. |
● | Continue to develop and partner our Helix™biotherapeutic delivery system for use with other biotherapeutics. We plan to continue to make our Helix™ biotherapeutic delivery system available for use by qualified partners seeking to advance their own biotherapeutic candidates for similar indications. |
● | Continue to develop and commercialize Morph catheter products. We plan to continue to enhance the performance of our Morph catheter products to benefit the CardiAMP and CardiALLO Cell Therapy Systems, to enhance Helix™ partnering, and to grow revenues. |
Risks Associated With Our Business
Our business is subject to numerous risks, as more fully described in the “Risk Factors” section immediately following this prospectus summary. These risks include, among other:
We have a history of net losses and may not be able to achieve or sustain profitability, which raises substantial doubt about our ability to continue as a going concern.
Our success depends in large part on our ability to obtain FDA approval for, and successfully commercialize, CardiAMP. This therapeutic candidate is still in clinical trials, has been used in only a limited number of procedures and there is no long-term data on its safety and efficacy.
Our limited commercialization experience and number of approved products makes it difficult to evaluate our current business, predict our future prospects and forecast our financial performance and growth.
We may not be able to secure additional financing on favorable terms, or at all, to meet our future capital needs and our failure to obtain additional financing when needed could force us to delay, reduce or eliminate our therapeutic candidate development programs and commercialization efforts.
We may in the future be a party to intellectual property litigation or administrative proceedings that could be costly and could interfere with our ability to sell our products and therapeutic candidates, if approved.
Recent Developments
On July 5, 2019, we entered into a note purchase agreement pursuant to which we issued on such date $0.625 million in aggregate principal amount of convertible promissory notes, a portion of which was issued to certain of our officers and directors and a principal stockholder (or their respective affiliates). Interest on the convertible notes accrues at the rate of 14.0% per year. The unpaid principal amount of the convertible notes, together with all interest accrued but unpaid thereon, will be automatically converted into units (the “Private Placement Units”), each Private Placement Unit consisting of one share of common stock and a warrant to purchase one share of common stock (the “Private Placement Warrants”), upon the closing of this Offering at a conversion price equal to 50% of the price to the public in this Offering.
On July 11, 2019, we signed an extension to a 2017 development agreement with a global biotherapeutics leader for BioCardia’s Helix™ biotherapeutic delivery catheter system. The agreement is exclusive with respect to a class of biotherapeutic agents that BioCardia is not currently developing on its own or with any other party and is time limited. Under the terms of the initial pre-clinical phase of the relationship, BioCardia will receive an upfront payment of $1,000,000, a portion of which will be creditable to BioCardia biotherapeutic delivery systems, support and training.
Corporate Information
We were incorporated in Delaware in March 2002 as BioCardia DeviceCo, Inc. and changed our name to BioCardia, Inc. in August 2002. Our principal executive offices are located at 125 Shoreway Road, Suite B, San Carlos, CA 94070. Our telephone number is (650) 226-0120. Our website address is www.biocardia.com. Information contained in our website is not incorporated by reference into this prospectus, and should not be considered to be a part of this prospectus. You should not rely on our website or any such information in making your decision whether or not to purchase our common stock.
THE OFFERING
Issuer | BioCardia, Inc. |
|
|
| We |
___________
|
|
Offering price per Unit | $ |
Over-allotment option | We have granted |
|
|
Common stock to be outstanding immediately after this Offering | shares. |
Use of proceeds | We estimate the net proceeds to us from this Offering, after deducting underwriting discounts and commissions and estimated offering expenses payable by us, will be approximately $ million. We intend to use the |
Current Market for the common stock and warrants | Our common stock is currently quoted on the OTCQB under the symbol “BCDA.” There is currently no public market for the offered warrants. |
Proposed Nasdaq Capital Market Trading Symbols and Listings | We have applied to list our common stock and offered warrants on the Nasdaq Capital Market under the symbols “BCDA” and “BCDAW,” respectively. No assurance can be given that such listings will be approved or that a trading market will develop for our common stock or warrants. |
Underwriters’ warrants | We will issue to Maxim Group LLC, as the representative of the underwriters, upon closing of this Offering compensation warrants entitling the underwriters or their designees to purchase up to 3.5% of the aggregate number of shares of our Common Stock that we issue to retail investors in this Offering. The warrants will be exercisable for a five-year period, commencing six months following the effective date of this Offering at an exercise price per share equal to 110% of the public offering price of our Units offered hereby. See “Underwriting.” |
Lock-up | Our directors, executive officers, and stockholders who own 5% or more of the outstanding shares of our common stock have agreed with the underwriters not to offer for sale, issue, sell, contract to sell, pledge or otherwise dispose of any of our common stock or securities convertible into common stock for a period of 180 days commencing on the date of this prospectus. See “Underwriting.” |
Risk factors | You should read the “Risk Factors” section of this prospectus and the documents incorporated herein for a discussion of certain factors to consider carefully before deciding to purchase any of our securities. |
Outstanding Shares
The number of shares of common stock to be outstanding immediately following this Offering is based on 4,847,471 shares issued and outstanding at March 31, 2019. The number of shares of common stock to be outstanding immediately following this Offering does not take into account:
● | 606,249 shares of our common stock issuable upon the exercise of options outstanding as of March 31, 2019, with a weighted-average exercise price of $24.39 per share; |
● | 27,435 shares of our common stock issuable upon the vesting of restricted stock units outstanding as of March 31, 2019; |
● | 296,295 shares of our common stock issuable upon the exercise of the warrants outstanding as of March 31, 2019 with a weighted-average exercise price of $6.75 per share; |
● | 410,596 shares of our common stock reserved for future issuance under our equity compensation plans; |
● | the shares issuable upon the exercise of warrants to be issued with the Units in this Offering (or shares if the underwriters exercise their over-allotment option in full); |
● | the shares of our common stock that may be issued upon exercise of the underwriter warrants; and |
| ● | the shares of our common stock that may be issued upon exercise of the Private Placement Warrants. |
The number of shares of common stock to be outstanding immediately following this Offering includes:
● | the shares to be issued upon automatic conversion of our convertible notes into Private Placement Units upon completion of this Offering. |
Unless otherwise indicated, all information in this prospectus assumes no exercise of the outstanding options or the warrants being offered in this Offering or the underwriter warrants.
Unless otherwise indicated, all information in this prospectus reflects a one-for-nine reverse stock split of our issued and outstanding shares of common stock, options, restricted stock units and warrants, and the corresponding adjustment of all common stock and restricted stock unit prices per share and stock option and warrant exercise prices per share and conversion ratios. Our common stock began trading on a reverse stock split-adjusted basis on The OTC Market on the opening of trading on June 6, 2019.
RISK FACTORS
This registration statement includes "forward-looking statements" within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended ("Exchange Act"), including, in particular, certain statements about our plans, strategies and prospects. Although we believe that our plans, intentions and expectations reflected in or suggested by such forward-looking statements are reasonable, we cannot assure you that such plans, intentions or expectations will be achieved. Important factors that could cause our actual results to differ materially from our forward-looking statements include those set forth in this Risk Factors section.
An investment in theour securities is speculative and involves a high degree of risk. YouIn addition to the other information included in this prospectus, including the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” you should carefully consider the risk factorsrisks described below, togetherbefore making an investment decision with respect to the securities. We expect to update these Risk Factors from time to time in the periodic and current reports that we file with the otherSEC after the date of this prospectus. Please refer to these subsequent reports for additional information containedrelating to the risks associated with investing in this prospectus before making a decision to purchase our securities.common stock and the accompanying warrant. If any of thesuch risks described below occur, or if other risks not identified below occur,and uncertainties actually occurs, our business, business prospects, cash flow, financial condition, stock price and results of operations could be materially adversely affected. Under these circumstances,severely harmed. This could cause the trading price of our common stock couldand our warrants to decline, and you maycould lose all or part of your investment. Additional risksinvestment.
Risks Relating to Our Business
We will require additional financing in 2019 in order to continue the trial and uncertaintiesto continue operations at the current level.
Our current cash resources are not presently knownsufficient to us or thatfund operations at the expected level of activity through the third quarter of 2019. We will need additional capital to continue operations at the current level and to continue the Phase III trial. While we currently deem immaterial also may impair our financial condition and operations. Furthermore, referencesplan to "we," "us" and "our" are referencesraise additional capital to fund operations, including the trials, there can be no assurances as to the Company.availability of capital or the terms on which capital will be available.
We have a history of operating losses, and we may not be able to achieve or sustain profitability. In addition, we may be unable to the risk factors related to the offering set forth below, the risk factors set forth in the SEC filings, including the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2008,continue as a going concern.
We are a clinical-stage regenerative medicine company and future filings with the SEC are incorporated by reference into this prospectus.
You understand that, certain unique factors make an investment in the Company subject to a high degree of risk. You have been cautioned that an investment in the Company is speculative and involves significant risks, and that it is probably not possible to foresee and describe all of the business, economic and financial risk factors which may affect the Company.
3
This document contains "forward-looking statements," as that term is defined under the Private Securities Litigation Reform Act of 1995, or the PSLRA. Forward-looking statements include statements about our expectations, beliefs or intentions regarding our product development efforts, business, financial condition, results of operations, strategies or prospects. You can identify forward-looking statements by the fact that these statements do not relate strictly to historical or current matters. Rather, forward-looking statements relate to anticipated or expected events, activities, trends or results as of the date they are made. Because forward-looking statements relate to matters thatwe have not yet occurred, these statements are inherently subject to risks and uncertainties that could cause our actual results to differ materially from any future results expressed or implied by the forward-looking statements.
Any or allgenerated a profit. We have incurred net losses during each of our forward-looking statements in this document may turn out to be wrong. They can be affected by inaccurate assumptions we might make or by known or unknown risksfiscal years since our inception. Our net loss for the three months ended March 31, 2019 was $3.7 million and uncertainties. Many factors mentioned in our discussion in this document will be important in determining future results. Consequently, no forward-looking statement can be guaranteed. Actual future results may vary materially. While we may elect to update these forward-looking statements at some point in the future, we specifically disclaim any obligation to do so to reflect actual results, changes in assumptions or changes in other factors affecting forward-looking statements. We intend that all forward-looking statements be subject to the safe-harbor provisions of the PSLRA. These forward-looking statements are only predictions and reflect our viewsaccumulated deficit totaled approximately $90.0 million as of the date they are made with respectMarch 31, 2019. We do not know whether or when we will become profitable, if ever. We currently expect operating losses and negative cash flows to future events and financial performance.
Risks and uncertainties, the occurrence of which could adversely affect our business, include the following:
Risks Related to Our Business, Industry and Regulatory Matters
We expect to incur significant losses, either directly or indirectly through the companies in which we develop our products,continue for at least the next several years.
To date, our only approved or cleared products are our Morph universal deflectable guide catheters and Morph AccessPro sheaths, or Morph, in the United States and Europe and our HelixTM biotherapeutic delivery system, or Helix, in Europe. Our limited commercialization experience and number of approved products makes it difficult to evaluate our current business and predict our future prospects. Our short commercialization experience and limited number of approved products also makes it difficult for us to forecast our future financial performance and growth and such forecasts are limited and subject to a number of uncertainties, including our ability to successfully complete our Phase III pivotal trials in heart failure and chronic myocardial ischemia and obtain FDA approval for, and then successfully commercialize, the CardiAMP Cell Therapy System.
Our ability to generate sufficient revenue to achieve profitability depends on our ability, either alone or with strategic collaboration partners, to successfully complete the development of, and obtain the regulatory approvals necessary to commercialize our therapeutic candidates. We do not anticipate generating revenues from sales of the CardiAMP Cell Therapy System, the CardiALLO Cell Therapy System or any other biotherapeutic candidates within the next few years, and we cannot assure youmay never generate sales of these products.
Our unaudited consolidated financial statements as of and for the three months ended March 31, 2019 and the year ended December 31, 2018 have been prepared on the basis that we will ever be profitable.
continue as a going concern, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business. We expect to incurhave incurred significant losses over the next several years, either directly or indirectly through the companies in whichsince our inception and we develop our products, as we expand our research and development activities, apply for regulatory approvals, develop additional technology and expand our operations. We cannot assure youexpect that we will continue to incur losses as we aim to successfully execute our business plan and will be successful in sellingdependent on additional public or private financings, collaborations or licensing any of the products we might developarrangements with strategic partners, or predict the terms we may be ableadditional credit lines or other debt financing sources to obtain in any sales or licensing transaction.
We havefund continuing operations. Based on our cash balances, recurring losses since inception and our existing capital resources to fund our planned operations for a limited number of products currently available for sale andtwelve-month period, there is substantial doubt about our ability to continue as a high risk that our research and development efforts might not successfully generate any viable product candidates in the future.
We currently have nine products available for sale, all of which are in the early stages of distribution. Other than those nine products,going concern. As noted below, we are in the preliminary stages of product identification and development, and have identified only a few potential additional products. We have not yet conducted preclinical studies or clinical testing on these potential additional products. It is statistically unlikely that the few products that we have identified as potential candidates will actually lead to successful development efforts, and we do not expect any additional products resulting from our research to be commercially available for several years, if at all. Our leads for potential products will be subject to the risks and failures inherent in developing medical devices and products, including, but not limited to, the unanticipated problems relating to research and development, product testing, confirming intellectual property rights and non-infringement, regulatory compliance, manufacturing, marketing and competition. Additional expenses may exceed current estimates and, therefore, adversely affect our profitability.
We will need to raiseobtain additional funds in the future to fund our operations and research, and these funds may not be available on acceptable terms, if at all.
We anticipate spending significant amounts of cash on expanding our research and development, sales and marketing efforts, and product commercialization. We expect proceedsfunding from the recent private placement will be sufficient to provide working capital requirements for the next nine months. However, actual capital requirements may change as a result of various factors, including:
the success of our research and development efforts, and any changes in the breadth of our research and development programs;
4
results from preclinical studies and clinical trials conducted by us or our collaborative partners or licensees, if any;the number and timing of acquisitions and other strategic transactions;our ability to maintain and establish corporate relationships and research collaborations;our ability to manage growth and costs associated with this growth, and the costs associated with increased capital expenditures;the time and costs involved in filing, prosecuting, defending and enforcing patent and intellectual property claims;the cost and timing of obtaining and maintaining regulatory approval or clearance for our products and products in development;the expenses we incur in manufacturing and selling our products;the revenues generated by sales of our products;expenses that may be incurred in pursuing or defending any potential litigation; andthe costs associated with our employee retention programs and related benefits.
Our primary goal as it relates to liquidity and capital resources is to attain the appropriate level of debt and equity and the resultant cash to implement our business plan. We may need to raise additional funds, which may not be available to us on favorable terms, if at all. If we raise capital by issuing equity or debt securities,financings, which may require us to agree to burdensome covenants, grant security interests in our existing stockholders may experience dilutionassets, enter into collaboration and the new equity or debt securities may have rights, preferences and privileges senior to those of our existing stockholders. Further, if we raise additional funds through collaboration, licensing or other similar arrangements it may be necessarythat require us to relinquish commercial rights, to our existing or potential products or proprietary technologies, or to grant licenses on terms that are not favorablefavorable. No assurance can be given at this time as to us.whether we will be able to achieve our fundraising objectives, regardless of the terms. If adequate funds are not available, the Company may be required to reduce operating expenses, delay or reduce the scope of its product development programs, obtain funds through arrangements with others that may require the Company to relinquish rights to certain of its technologies or products that the Company would otherwise seek to develop or commercialize itself, or cease operations.
Our success depends in large part on our ability to obtain approval for, and successfully commercialize, the CardiAMP Cell Therapy System.
The long-term viability of our company is largely dependent on the successful development and commercialization of the CardiAMP Cell Therapy System. We are currently enrolling patients in a Phase III pivotal trial that will be used to support regulatory approval, and we do not have significant long-term data on the CardiAMP Cell Therapy System’s safety and efficacy in either heart failure or chronic myocardial ischemia. While we expect to successfully complete our ongoing Phase III pivotal trial of the CardiAMP Cell Therapy System in heart failure, there can be no guarantee that the study will be completed, that the primary endpoints will be achieved, or that we will receive regulatory approval for the sale and marketing in the United States. A number of companies in similar fields have suffered significant setbacks during clinical trials due to lack of efficacy or unacceptable safety issues, notwithstanding promising preliminary results. Because we are depending heavily on sales of the CardiAMP Cell Therapy System to achieve our revenue goals, failure to successfully complete the study and receive U.S. Food and Drug Administration, or FDA, approval, in a timely manner or at all, will harm our financial results and ability to become profitable. Even if we obtain regulatory approval, our ability to successfully market this product will be limited due to a number of factors, including regulatory restrictions in our labeling or requirements to obtain additional post-approval data, if any. In addition, there can be no guarantee that the CardiAMP Cell Therapy System will be accepted by the medical community as a valid alternative to currently available products. If we are unablecannot sell the CardiAMP Cell Therapy System as planned, our financial results will be harmed.
FDA acceptance of a Phase III pivotal trial is not a guarantee of an approval of a product candidate or any permissible claims about the product candidate. Failure to raise needed capital on terms acceptablesuccessfully complete our ongoing Phase III trial of CardiAMP in heart failure would significantly impair our financial results. Such a failure could (i) delay or prevent the CardiAMP Cell Therapy System from obtaining regulatory approval, (ii) require us to us, weperform another clinical trial, which will be expensive, may not be able to develop new products, enhance our existing products, execute our business plan, take advantage of future opportunities, respond to competitive pressures or unanticipated customer requirements or continue to operate our business. Any of these events could have a material adverse effect on our business, financial conditionsuccessful and results of operations.
Cost containment measures, pressure from our competitors and availability of medical reimbursement may impactwill significantly delay our ability to sellcommercialize the CardiAMP Cell Therapy System and (iii) impair our products at prices necessaryability to expandconvince hospitals and physicians of the benefits of our operationsCardiAMP Cell Therapy System product. Furthermore, even if we are granted regulatory clearances or approvals, they may include significant limitations on the indicated uses for CardiAMP, which may limit the market for this product.
Because the CardiAMP Cell Therapy System is, to our knowledge, the first cardiac cell-based therapy with an accepted pivotal trial that is to be regulated by the FDA via the premarket approval pathway, the approval process for the CardiAMP Cell Therapy System is uncertain.
Although we have obtained FDA acceptance of Phase III pivotal trials of the CardiAMP Cell Therapy System for the treatment of ischemic systolic heart failure and reach profitability.
Healthcare costs have risen significantly overchronic myocardial ischemia, this does not guarantee any particular outcome from regulatory review. To the past decadebest of our knowledge, the CardiAMP Cell Therapy System for the treatment of ischemic systolic heart failure is the first cardiac cell-based therapy with an accepted pivotal trial that is to be regulated by the FDA Center for Biologics Evaluation and numerous initiatives and reforms initiatedResearch, or CBER, via the premarket approval, or PMA, pathway requiring a single trial. The CardiAMP Cell Therapy System for the treatment of chronic myocardial ischemia is also to be regulated under the same IDE/PMA pathway. All other cardiac cell-based therapies in clinical trials are believed to be regulated by legislators, regulators and third-party payorsthe same agency, but as biologics which generally require two separate pivotal trials. There is no guarantee that the FDA will grant us regulatory clearance or approval to curb these costs have resulted inmarket the CardiAMP Cell Therapy System on the basis of a consolidation trend insingle pivotal trial, or that the healthcare industry, including hospitals. This has resulted in greater pricing and other competitive pressures and the exclusion of certain suppliers from important market segments as group purchasing organizations, independent delivery networks and large single accounts continue to consolidate purchasing decisions for some hospital customers. We expect that market demand, government regulation, third-party reimbursement policies and societal pressuresFDA will continue to changeallow us to develop the national and worldwide healthcare industry, resulting in further business consolidations and alliances among customers and competitors. This consolidation may reduce competition, exert downward pressure onCardiAMP Cell Therapy System via the pricesPMA pathway. Two well-controlled pivotal studies could be necessary to provide FDA assurance of our products and adversely impact our business, financial conditionsafety or results of operations.
Further, third-party payors ineffectiveness. If the United States and abroad continue to work to contain healthcare costs. The introduction of cost containment incentives, along with closer scrutiny of healthcare expenditures by both private health insurers and employers, has resulted in increased discounts and contractual adjustments to hospital charges for services performed and has shifted services between inpatient and outpatient settings. HospitalsFDA approval process does not occur as we anticipate or physicians may respond to these cost-containment pressures by substituting lower-cost products or other therapies for our products, the occurrence of which may adversely impact our business, financial condition and results of operation.
The market for orthopedic, knee and hip surgery devices is large and growing at a significant rate. Numerous new companies and technologies, as well as more established companies, have entered this market. New entrants to our markets include numerous niche companies with a singular product focus, as well as companies owned partially by surgeons, who may have greater access than we do to the surgeons who may use our products. As a result of this intensified competition, we believe there will be increasing pressure to reduce pricing of our medical devices. If we are unablerequired to price our products appropriately dueconduct more than one pivotal study to these competitive pressures or for other reasons, our profit margins will shrink and our ability to invest in and grow our business and achieve profitability will decrease.
5
Sales of our products will depend on the availability of adequate reimbursement from third-party payors (such as governmental programs, for example, Medicare and Medicaid, private insurance plans and managed care programs), both in terms of the sales volumes and prices of our products.
Healthcare providers, such as hospitals that purchase medical devices for treating their patients, generally rely on third-party payors to reimburse all or part of theobtain approval, we may incur substantial additional costs and fees associated with the procedures performed with these devices. These third-party payors may deny reimbursementdelays to obtain approval, if they feel that a device is not the most cost-effective treatment available, or was used for an unapproved indication. As such, surgeons are unlikely to use our products if they do not receive reimbursement adequate to cover the cost of their involvement in the surgical procedures. The failure of surgeons to use our products, or the diminished use by surgeons, mayat all, which would have a material adverse impact on our business, financial condition and resultsprospects.
Our CardiAMP and CardiALLO cell therapy system therapeutic candidates are based on novel technology, which makes it difficult to accurately and reliably predict the time and cost of operations.product development and subsequently obtaining regulatory approval. At the moment, no cell-based therapies have been approved in the United States for a cardiac indication.
The success of our business depends on our ability to develop and commercialize our therapeutic candidates, including CardiAMP. We have concentrated our product research and development efforts on our CardiAMP therapeutic candidate, a novel type of cell-based therapy. Our future success depends on the successful development of this therapeutic approach. There can be no assurance that any development problems we experience in the future related to our therapeutic candidates and products will not cause significant delays or unanticipated costs, or that such development problems can be solved. We may be unable to maintain and further develop sustainable, reproducible and scalable manufacturing processes, or transfer these processes to collaborators, which may prevent us from completing our clinical studies or commercializing our products on a timely or profitable basis, if at all.
In addition, the clinical study requirements of the FDA, the European Medicines Agency, or EMA, and other regulatory agencies and the criteria these regulators use to determine the safety and efficacy of a product candidate vary substantially according to the type, complexity, novelty, intended use and market of the potential product candidates. The regulatory approval process for novel product candidates such as our CardiAMP and CardiALLO Cell Therapy Systems may be more expensive and take longer than other, better known or extensively studied pharmaceutical or other product candidates to develop. In addition, adverse developments in clinical trials of cell-based products or therapies conducted by others may cause the FDA or other regulatory bodies to change the requirements for approval of any of our therapeutic candidates. At the moment, no other cell-based therapies have been approved in the United States for a cardiac indication, which makes it difficult to determine how long it will take or how much it will cost to obtain regulatory approvals for our therapeutic candidates in either the United States or elsewhere.
Regulatory requirements governing cell-based therapy products have changed frequently and may continue to change in the future. For example, the FDA established the Office of Cellular, Tissue and Gene Therapies within CBER to consolidate the review of gene therapy and related products, and the Cellular, Tissue and Gene Therapies Advisory Committee to advise CBER on its review. These regulatory authorities and advisory groups and the new requirements or guidelines they promulgate may lengthen the regulatory review process, require us to perform additional studies, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of our product candidates or lead to significant post-approval limitations or restrictions. As we advance our product candidates, we will be required to consult with the FDA and other regulatory authorities, and our products could be reviewed by the FDA’s advisory committee. We also believemust comply with applicable requirements, and if we fail to do so, we may be required to delay or discontinue development of our product candidates.
We will require substantial additional financing to achieve our goals, and our failure to obtain this necessary capital when needed could force us to delay, limit, reduce or terminate our product development or commercialization efforts.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to incur significant expenses and operating losses for the foreseeable future in connection with our planned research, development and product commercialization efforts, including our planned clinical trials for our CardiAMP and CardiALLO Cell Therapy System therapeutic candidates. In addition, we will require additional financing to achieve our goals and our failure to do so could adversely affect our commercialization efforts. We anticipate that futureour expenses will increase substantially if and as we:
continue the research and clinical development of our CardiAMP and CardiALLO Cell Therapy System therapeutic candidates;
initiate and advance our CardiAMP and CardiALLO Cell Therapy System therapeutic candidates into larger and more expensive clinical studies, including the ongoing Phase III pivotal trial for our CardiAMP Cell Therapy System therapeutic candidate in heart failure and our recently approved Phase III pivotal trial for our CardiAMP Cell Therapy System therapeutic candidate in chronic myocardial ischemia;
seek to identify, assess, acquire, and/or develop other product candidates and technologies;
seek regulatory and marketing approvals in multiple jurisdictions for our therapeutic candidates that successfully complete clinical studies;
build and maintain a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain marketing approval, or otherwise establish collaborations with third parties for the development and commercialization of our therapeutic candidates;
further develop and implement our manufacturing processes and expand our manufacturing capabilities and resources for commercial production;
seek coverage and reimbursement from third-party payors, including government and private payors for future products;
seek to maintain, protect and expand our intellectual property portfolio; and
seek to attract and retain skilled personnel.
If we were to experience any delays or encounter issues with any of the above, including clinical holds, failed studies, inconclusive or complex results, safety or efficacy issues, or other regulatory challenges that require longer follow-up of existing studies, additional major studies, or additional supportive studies in order to pursue marketing approval, it could further increase the costs associated with the above. Further, the net operating losses we incur may fluctuate significantly from quarter to quarter and year to year, such that a period-to-period comparison of our results of operations may not be a good indication of our future performance.
We have encountered, and may in the future encounter substantial delays in our clinical studies.
We have encountered, and may in the future encounter substantial delays in our clinical studies. We cannot guarantee that any preclinical testing or clinical trials will be conducted as planned or completed on schedule, if at all. As a result, we may not achieve our expected clinical milestones. A failure can occur at any stage of testing. Events that may prevent successful or timely commencement, enrollment or completion of clinical development include:
delays in raising, or inability to raise, sufficient capital to fund the planned trials;
delays in reaching a consensus with regulatory agencies on trial design;
changes in trial design;
inability to identify, recruit and train suitable clinical investigators;
inability to add new clinical trial sites;
delays in reaching agreement on acceptable terms for the performance of the trials with prospective clinical research organizations, or CROs, and clinical trial sites;
delays in obtaining required Institutional Review Board, or IRB, approval at each clinical trial site;
delays in recruiting suitable clinical sites and patients (i.e., subjects) to participate in clinical trials;
imposition of a clinical hold by regulatory agencies for any reason, including negative clinical results, safety concerns or as a result of an inspection of manufacturing or clinical operations or trial sites;
failure by us, CROs or other third parties to adhere to clinical trial requirements;
failure to perform in accordance with the FDA’s current Good Clinical Practices, or GCP, or applicable regulatory guidelines in other countries;
delays in the testing, validation, manufacturing and delivery to the clinical sites;
delays caused by patients not completing participation in a trial or not returning for post-treatment follow-up;
delays caused by clinical trial sites not completing a trial;
failure to demonstrate adequate efficacy;
occurrence of serious adverse events in clinical trials that are associated with the therapeutic candidates or products that are viewed to outweigh its potential benefits;
changes in regulatory requirements and guidance that require amending or submitting new clinical protocols; or
disagreements between us and the FDA or other regulatory agencies interpreting the data from our clinical trials.
Delays, including those caused by the above factors, can be costly and could negatively affect our ability to complete clinical trials for our therapeutic candidates. If we are not able to successfully complete clinical trials or are not able to do so in a timely and cost-effective manner, we will not be able to obtain regulatory approval and/or will not be able to commercialize our therapeutic candidates or products, which would have an adverse effect on our business. Clinical trial delays could also shorten any periods during which we may have the exclusive right to commercialize our therapeutic candidates or products or allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our therapeutic candidates or products and may harm our business and results of operations.
We may find it difficult to enroll patients in our clinical trials, which could delay or prevent development of our therapeutic candidates.
Identifying and qualifying patients to participate in clinical trials of our therapeutic candidates is critical to our success. The timing of our clinical trials depends on the speed at which we can recruit patients to participate in testing our therapeutic candidates as well as completion of required follow-up periods. In general, if patients are unwilling to participate in our cell-based therapy trials because of negative publicity from adverse events in the biotechnology or cell-based industries or for other reasons, including competitive clinical trials for similar patient populations, the timeline for recruiting patients, conducting trials and obtaining regulatory approval for our therapeutic candidates may be subjectdelayed. These delays could result in increased costs, delays in advancing our product development, delays in testing the effectiveness of our therapeutic candidates or termination of the clinical trials altogether.
Patient enrollment and completion of clinical trials are affected by factors including:
• | size of the patient population; | |
• | severity of the disease under investigation; | |
• | design of the trial protocol; | |
• | eligibility criteria for the particular trial; | |
• | perceived risks and benefits of the product candidate being tested; | |
• | proximity and availability of clinical trial sites for prospective patients; | |
• | availability of competing therapies and clinical trials; | |
• | efforts to facilitate timely enrollment in clinical trials; | |
• | patient referral practices of physicians; | |
• | ability to monitor patients adequately during and after treatment; and | |
• | the degree of treatment effect in event-driven trials. |
Once enrolled, patients may choose to increased restrictions bothdiscontinue their participation at any time during the trial, for any reason. Participants also may be terminated from the study at the initiative of the investigator, for example if they experience serious adverse clinical events or do not follow the study directions. If we are unable to maintain an adequate number of patients in our clinical trials, we may be required to delay or terminate an ongoing clinical trial, which would have an adverse effect on our business.
We depend on our license and distribution agreement with Biomet Biologics, LLC, and if we fail to comply with our obligations under this agreement, or if our rights under this agreement are otherwise reduced or terminated, we could lose intellectual property rights that are important to our business.
In October 2012, we entered into a license and distribution agreement with Biomet Biologics, LLC under which we obtained an exclusive, nontransferable, worldwide distribution right, patent license and trademark license to Biomet Biologic, LLC’s point of care cell processing platform. Under the terms of the agreement, we are obligated to pay Biomet Biologics, LLC a royalty based on the price of the disposables in the U.S. and international markets. If we sell our products internationally, market acceptance may depend, in part, upon the availabilityCardiAMP cell processing platform. A breach or termination of reimbursement within the prevailing healthcare payment systems. Reimbursement and healthcare payment systems in international markets vary significantly by country, and include both government sponsored healthcare and private insurance.
Future legislation, regulation or reimbursement policies of third-party payors maythis agreement would materially adversely affect the demandclinical development or commercialization strategy of our CardiAMP therapeutic candidate as currently planned. A reduction or elimination of our rights under this agreement may result in our having to negotiate new or reinstated arrangements on less favorable terms, or our not having sufficient intellectual property rights to operate our business as currently planned. The occurrence of such events could materially harm our business and financial condition.
We rely on third parties to conduct some or all aspects of our product manufacturing, diagnostic protocol development, research, and preclinical and clinical testing, and these third parties may not perform satisfactorily.
We do not currently, and do not expect to in the future, independently conduct all aspects of our product manufacturing, anticipated companion diagnostic testing, protocol development, research and monitoring and management of our ongoing preclinical and clinical programs. We currently rely, and expect to continue to rely, on third parties with respect to these items, and control only certain aspects of their activities.
Any of these third parties may terminate their engagements with us at any time. If we need to enter into alternative arrangements, our commercialization activities or our therapeutic candidate or companion diagnostic development activities may be delayed or suspended. Our reliance on these third parties for research and development activities, including the conduct of any IDE and IND-enabling studies, reduces our control over these activities but does not relieve us of our responsibility to ensure compliance with all required legal, regulatory and scientific standards and any applicable trial protocols. For example, for therapeutic candidates that we develop and commercialize on our own, we will remain responsible for ensuring that each of our IDE and IND-enabling studies and clinical trials are conducted in accordance with the trial plan and protocols.
If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our studies in accordance with regulatory requirements or our stated study plans and protocols, we may be delayed in completing, or unable to complete, the preclinical studies and clinical trials required to support future IDE and IND submissions and approval of our therapeutic candidates.
Reliance on third-party manufacturers entails exposure to risks to which we would not be subject if we manufactured the therapeutic candidates or companion diagnostic ourselves, including:
we may be unable to negotiate manufacturing agreements with third parties under commercially reasonable terms;
reduced control over the manufacturing process for our existing productstherapeutic candidates and companion diagnostic as a result of using third-party manufacturers for many aspects of manufacturing activities;
termination or nonrenewal of manufacturing agreements with third parties in a manner or at a time that may be costly or damaging to us or result in delays in the development or commercialization of our products currently undertherapeutic candidates or companion diagnostic; and
disruptions to the operations of our third-party manufacturers or suppliers caused by conditions unrelated to our business or operations, including the bankruptcy of the manufacturer or supplier.
Any of these events could lead to delays in the development and limitof our therapeutic candidates, including delays in our clinical trials, or failure to obtain regulatory approval for our therapeutic candidates, or it could impact our ability to sellsuccessfully commercialize our productscurrent therapeutic candidates, companion diagnostic or any future products. Some of these events could be the basis for FDA or other regulatory action, including injunction, recall, seizure or total or partial suspension of production.
We rely on a profitable basis. Also, third-party payorsthird parties to conduct, supervise and monitor our clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates and our business could be substantially harmed.
We rely on CROs and clinical trial sites to ensure our clinical trials are increasingly challengingconducted properly and on time. While we will have agreements governing their activities, we will have limited influence over their actual performance. We will control only certain aspects of our CROs’ activities. Nevertheless, we will be responsible for ensuring that each of our clinical trials is conducted in accordance with the prices chargedapplicable protocol, legal, regulatory and scientific standards, and our reliance on the CROs does not relieve us of our regulatory responsibilities.
We and our CROs are required to comply with the FDA’s GCPs for medical productsconducting, recording and services. Particularly inreporting the United States, third-party payors carefully review,results of clinical trials to assure that the data and increasingly challenge,reported results are credible and accurate and that the prices charged for proceduresrights, integrity and medical products. Also, greater numbersconfidentiality of insured individualsclinical trial participants are receiving (and will continue to receive overprotected. The FDA, the next decade) their medical care through managed care programs, which monitor and often require pre-approvalCompetent Authorities of the servicesMember States of the EEA, and comparable foreign regulatory authorities, enforce these GCPs through periodic inspections of trial sponsors, principal investigators and clinical trial sites. If we or our CROs fail to comply with applicable GCPs, the clinical data generated in our future clinical trials may be deemed unreliable and the FDA, the EMA, or other foreign regulatory authorities may require us to perform additional clinical trials before approving any marketing applications. Upon inspection, the FDA may determine that our clinical trials did not comply with GCPs. In addition, our future clinical trials will require a member will receive. Many managed care programssufficient number of test subjects to evaluate the safety and effectiveness of our therapeutic candidates. Accordingly, if our CROs fail to comply with these regulations or fail to recruit a sufficient number of patients, we may be required to repeat such clinical trials, which would delay the regulatory approval process.
Our CROs are payingnot our employees, and we are therefore unable to directly monitor whether or not they devote sufficient time and resources to our clinical and nonclinical programs. These CROs may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials or other product development activities that could harm our competitive position. If our CROs do not successfully carry out their providers on a capitated basis, which putscontractual duties or obligations, fail to meet expected deadlines, or if the providers at financial risk forquality or accuracy of the services provided to their patients by paying them a predetermined payment per member per month. Challenges by third-party payorsclinical data they obtain is compromised due to the prices charged for medical products and services, coupled with the increasing popularity of managed care programs, may result in hospitals and physicians seeking lower-cost alternativesfailure to adhere to our products,clinical protocols or regulatory requirements, or for any other reasons, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize, our therapeutic candidates. If any such event were to occur, our financial results and the occurrencecommercial prospects for our therapeutic candidates would be harmed, our costs could increase, and our ability to generate revenues could be delayed.
If any of our relationships with these third-party CROs terminate, we may not be able to enter into arrangements with alternative CROs or to do so on commercially reasonable terms. Further, switching or adding additional CROs involves additional costs and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which could materially adversely affectimpact our ability to meet our desired clinical development timelines. Though we carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
We may also rely on other third parties to store and distribute our products for the clinical trials that we conduct. Any performance failure on the part of our distributors could delay clinical development or marketing approval of our therapeutic candidates or commercialization of our products, if approved, producing additional losses and depriving us of potential product revenue.
We depend on third party vendors to manufacture some of our components and sub-assemblies, which could make us vulnerable to supply shortages and price fluctuations that could harm our business.
We currently manufacture some of our components and sub-assemblies internally and rely on third party vendors for other components and sub-assemblies used in our products and therapeutic candidates. Our reliance on third party vendors subjects us to a number of risks that could impact our ability to manufacture our products and therapeutic candidates and harm our business, including:
interruption of supply resulting from modifications to, or discontinuation of, a supplier’s operations;
delays in product shipments resulting from uncorrected defects, reliability issues or a supplier’s failure to consistently produce quality components;
price fluctuations due to a lack of long-term supply arrangements with our suppliers for key components;
inability to obtain adequate supply in a timely manner or on commercially reasonable terms;
difficulty identifying and qualifying alternative suppliers for components in a timely manner;
inability of the manufacturer or supplier to comply with Quality System Regulations, or QSRs, enforced by the FDA and state regulatory authorities;
inability to control the quality of products manufactured by third parties;
production delays related to the evaluation and testing of products from alternative suppliers and corresponding regulatory qualifications; and
delays in delivery by our suppliers due to changes in demand from us or their other customers.
Any significant delay or interruption in the supply of components or sub-assemblies, or our inability to obtain substitute components, sub-assemblies or materials from alternate sources at acceptable prices in a timely manner, could impair our ability to meet the demand of our customers and harm our business.
Our future commercial success depends upon attaining significant market acceptance of our therapeutic candidates, if approved, among physicians, patients and healthcare payors.
Even when product development is successful and regulatory approval has been obtained, our ability to generate significant revenue depends on the acceptance of our products by physicians, payors and patients. Many potential market participants have limited knowledge of, or experience with, cell-based products and therapies, so gaining market acceptance and overcoming any safety or efficacy concerns may be more challenging than for more traditional therapies. Our efforts to educate the medical community and third-party payors on the benefits of our therapeutic candidates may require significant resources and may never be successful. Such efforts to educate the marketplace may require more resources than are required by conventional therapies marketed by our competitors. We cannot assure you that our products will achieve the expected market acceptance and revenue if and when they obtain the requisite regulatory approvals. Alternatively, even if we obtain regulatory approval, that approval may be for indications or patient populations that are not as broad as intended or desired or may require labeling that includes significant use or distribution restrictions or safety warnings. The market acceptance of each of our therapeutic candidates will depend on a number of factors, including:
the efficacy and safety of the therapeutic candidate, as demonstrated in clinical trials;
the clinical indications for which the product is approved and the label approved by regulatory authorities for use with the product, including any warnings that may be required on the label;
acceptance by physicians and patients of the product as a safe and effective treatment;
the cost, safety and efficacy of treatment in relation to alternative treatments;
the continued projected growth of markets for our various indications;
relative convenience and ease of administration;
the prevalence and severity of adverse side effects; and
the effectiveness of our sales and marketing efforts.
Market acceptance is critical to our ability to generate significant revenue. Any therapeutic candidate, if approved and commercialized, may be accepted in only limited capacities or not at all. If any approved products are not accepted by the market to the extent that we expect, we may not be able to generate significant revenue and our business would suffer.
If we fail to attract and keep senior management and key scientific personnel, we may be unable to successfully develop our therapeutic candidates, conduct our clinical trials and commercialize our therapeutic candidates.
We are highly dependent on the members of our executive team, the loss of whose services may adversely impact the achievement of our objectives. Any of our executive officers could leave our employment at any time, as all of our employees are “at will” employees. Recruiting and retaining other qualified employees, consultants and advisors for our business, including scientific and technical personnel, will also be critical to our success.
Recruiting and retaining qualified scientific, clinical, manufacturing, sales and marketing personnel will also be critical to our success. We may not be able to attract and retain these personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us.
We will need to expand our organization and we may experience difficulties in managing this growth, which could disrupt our operations.
As we mature and expand our research and development and other pre-commercialization activities, we expect to expand our existing full-time employee base and to hire more consultants and contractors. In addition, we currently plan to commercialize the CardiAMP Cell Therapy System, if approved, using an internal sales force to selected cardiologists, interventional cardiologists and third-party payors in the United States. Our management may need to divert a disproportionate amount of its attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. We may not be able to effectively manage the expansion of our operations, which may result in weaknesses in our infrastructure, operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. Our expected growth could require significant capital expenditures and may divert financial resources from other projects, such as the development of additional product candidates. If our management is unable to effectively manage our growth, our expenses may increase more than expected, our ability to generate and/or grow revenues could be reduced, and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize product candidates and compete effectively will depend, in part, on our ability to effectively manage any future growth.
We face substantial competition, which may result in others discovering, developing or commercializing products before, or more successfully, than we do.
Our industry is highly competitive and subject to rapid change. The industry continues to expand and evolve as an increasing number of competitors and potential competitors enter the market. Astra Zeneca/Moderna, Blue Rock Therapeutics, Caladrius Biosciences, Celixr, Cesca Therapeutics, Celyad, Mesoblast, Tenaya Therapeutics, Vericel Corp, and Uniqure, among others. Many of our competitors, potentially including the aforementioned, have significantly greater development, financial, manufacturing, marketing, technical and human resources than we do. Large pharmaceutical and medical device companies, in particular, have extensive experience in clinical testing, obtaining regulatory approvals, recruiting patients and in manufacturing pharmaceutical and medical device products. Recent and potential future merger and acquisition activity in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. Established companies may also invest heavily to accelerate discovery and development of novel products that could make our therapeutic candidates obsolete. As a result of all of these factors, our competitors may succeed in obtaining patent protection and/or FDA approval or discovering, developing and commercializing our therapeutic candidates or competitors to our therapeutic candidates before we do. Specialized, smaller or early-stage companies may also prove to be significant competitors, particularly those with a focus and expertise in the stem cell industry and/or those with collaboration arrangements and other third-party payors. In addition, any new product that competes with an approved product must demonstrate compelling advantages in efficacy, convenience, tolerability and safety in order to overcome price competition and to be commercially successful. If we are not able to compete effectively against potential competitors, our business will not grow, and our financial condition and results of operations.operations will suffer.
Legislative
Even if we obtain regulatory approval for a product candidate, including our CardiAMP and CardiALLO Cell Therapy System therapeutic candidates, these products or administrative reformstherapies, along with our other regulated products, will be subject to ongoing regulatory scrutiny.
Even if we obtain regulatory approval or clearance in a jurisdiction, regulatory authorities may still impose significant restrictions on the indicated uses or marketing of our therapeutic candidates or impose ongoing requirements for potentially costly post-approval studies or post-market surveillance. For example, once a product receives regulatory approval or clearance for sale, we are obligated to monitor and report adverse events and any failure of a product to meet the specifications in the applicable regulatory approval or clearance. We must also submit new or supplemental applications and obtain FDA approval or clearance for certain changes to the approved or cleared product, product labeling or manufacturing process. Advertising and promotional materials must comply with FDA rules and are subject to FDA review, in addition to other potentially applicable federal and state laws.
In addition, product manufacturers and their facilities are subject to payment of user fees and continual review and periodic inspections by the FDA and other regulatory authorities for compliance with good manufacturing practices or QSRs and adherence to commitments made in the applicable regulatory approval. If we or a regulatory agency discovers previously unknown problems with a product such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions relative to that product or the manufacturing facility, including requiring recall or withdrawal of the product from the market or suspension of manufacturing.
If we fail to comply with applicable regulatory requirements following approval of any of our therapeutic candidates, a regulatory agency may impose the following:
restrictions on the marketing or manufacturing of our products, withdrawal of our products from the market, or voluntary or mandatory product recalls;
costly regulatory inspections;
fines, warning letters, or holds on clinical trials;
refusal by the FDA to approve pending applications or supplements to approved applications filed by us or our collaborators, or suspension or revocation of applicable regulatory approvals;
product seizure or detention, or refusal to permit the import or export of products; and
injunctions or the imposition of civil or criminal penalties by FDA or other regulatory bodies.
Any government investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. The occurrence of any event or penalty described above may inhibit our ability to commercialize our therapeutic candidates and generate revenues.
Our ability to compete is highly dependent on demonstrating the benefits of CardiAMP to physicians, hospitals and patients.
In order to generate sales, we must be able to clearly demonstrate that CardiAMP is both a more effective treatment system and less costly than alternative products and treatments offered by our competitors. If we are unable to convince physicians that CardiAMP leads to significant improvement in functional capacity, improved quality of life and reduced hospitalization, our business will suffer.
We may fail to demonstrate safety and efficacy to the satisfaction of applicable regulatory agencies.
We have not obtained regulatory approval for either our CardiAMP or CardiALLO Cell Therapy System therapeutic candidates. We must conduct extensive testing of our therapeutic candidates to demonstrate their safety and efficacy, including human clinical trials and, if applicable, preclinical animal testing, before we can obtain regulatory approval to market and sell them. Conducting such testing is a lengthy, time-consuming, and expensive process and there is a high rate of failure. Our current and completed preclinical and clinical results for our therapeutic candidates are not necessarily predictive of the results of our ongoing or future clinical trials. Promising results in preclinical studies of a therapeutic candidate may not be predictive of similar results in humans during clinical trials, and successful results from early human clinical trials of a therapeutic candidate may not be replicated in later and larger human clinical trials or in clinical trials for different indications. If the results of our ongoing or future clinical trials are negative or inconclusive with respect to the efficacy of our therapeutic candidates or if we or they do not meet the clinical endpoints with statistical significance or if there are safety concerns or adverse events associated with our therapeutic candidates, we may be prevented or delayed in obtaining marketing approval for our therapeutic candidates.
If we fail to obtain and maintain necessary regulatory clearances or approvals for our therapeutic candidates or products, or if clearances or approvals for our therapeutic candidates or products in additional indications are delayed or not issued, our commercial operations would be harmed.
We are required to timely file various reports with the FDA, require that we report to the regulatory authorities if our therapeutic candidates or products may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur. If these reports are not filed timely, regulators may impose sanctions and sales may suffer, and we may be subject to product liability or regulatory enforcement actions, all of which could harm our business.
If we initiate a correction or removal to reduce a risk to health posed, we would be required to submit a publicly available Correction and Removal report to the FDA and in many cases, similar reports to other regulatory agencies. This report could be classified by the FDA as a product recall which could lead to increased scrutiny by the FDA, other international regulatory agencies and our customers regarding the quality and safety of our therapeutic candidates or products. Furthermore, the submission of these reports has been and could be used by competitors against us in competitive situations and cause customers to delay purchase decisions or cancel orders and would harm our reputation.
The FDA and the Federal Trade Commission, or FTC, also regulate the advertising and promotion of our therapeutic candidates or products to ensure that the claims we make are consistent with our regulatory approvals, that there are adequate and reasonable data to substantiate the claims and that our promotional labeling and advertising is neither false nor misleading in any respect. If the FDA or FTC determines that any of our advertising or promotional claims are misleading, not substantiated or not permissible, we may be subject to enforcement actions, including warning letters, and we may be required to revise our promotional claims and make other corrections or restitutions.
FDA and state authorities have broad enforcement powers. Our failure to comply with applicable regulatory requirements could result in enforcement action by FDA or state agencies, which may include any of the following sanctions:
• | adverse publicity, warning letters, fines, injunctions, consent decrees and civil penalties; | |
• | repair, replacement, refunds, recall or seizure of our products; | |
• | operating restrictions, partial suspension or total shutdown of production; | |
• | refusing our requests for premarket approval of new products, new intended uses or modifications to existing products; | |
• | withdrawing premarket approvals that have already been granted; and | |
• | criminal prosecution. |
If any of these events were to occur, our business and financial condition would be harmed.
Serious adverse events or other safety risks could require us to abandon development and preclude, delay or limit approval of our therapeutic candidates or products or limit the scope of any approved indication or market acceptance.
Participants in clinical trials of our investigational cell-based therapies and products may experience adverse reactions or other undesirable side effects. While some of these can be anticipated, others may be unexpected. We cannot predict the frequency, duration, or severity of adverse reactions or undesirable side effects that may occur during clinical investigation. If any of our therapeutic candidates or products, prior to or after any approval for commercial sale, cause adverse events or are associated with other safety risks, a number of potentially significant negative consequences could result, including:
regulatory authorities may suspend (e.g., through a clinical hold) or terminate clinical trials;
regulatory authorities may deny regulatory approval of our therapeutic candidates or products;
regulatory authorities may restrict the indications or patient populations for which a therapeutic candidate or products is approved;
regulatory authorities may require certain labeling statements, such as warnings or contraindications or limitations on the indications for use, and/or impose restrictions on distribution in the form of a Risk Evaluation and Mitigation Strategy, or REMS, in connection with approval, if any;
regulatory authorities may withdraw their approval, require more onerous labeling statements or impose a more restrictive REMS than any therapeutic candidate or product that is approved;
we may be required to change the way the therapy or therapeutic candidate or product is administered or conduct additional clinical trials;
patient recruitment into our clinical trials may suffer;
we could be required to provide compensation to subjects for their injuries, e.g., if we are sued and found to be liable or if required by the laws of the relevant jurisdiction or by the policies of the clinical site; or
our reputation may suffer.
There can be no assurance that adverse events associated with our therapeutic candidates or products will not be observed, even where no prior adverse events have occurred. We may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to participants or if preliminary data demonstrate that our therapeutic candidates or products are unlikely to receive regulatory approval or are unlikely to be successfully commercialized. Regulatory agencies, IRBs or data safety monitoring boards may at any time recommend the temporary or permanent discontinuation of our clinical trials or request that we cease using investigators in the clinical trials if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements, or that they present an unacceptable safety risk to participants. If we elect or are forced to suspend or terminate a clinical trial for any reason this would have an adverse effect on our business.
Our therapeutic candidates are intended to treat patients who are extremely ill, and patient deaths that occur in our clinical trials could negatively impact our business even if they are not shown to be related to our therapeutic candidates.
Generally, patients remain at high risk following their treatment with our CardiAMP and CardiALLO therapeutic candidates. As a result, it is likely that we will observe severe adverse outcomes during our clinical trials for these therapeutic candidates, including patient death. If a significant number of study subject deaths were to occur, regardless of whether such deaths are attributable to our therapeutic candidates, our ability to obtain regulatory approval for the applicable therapeutic candidate may be adversely impacted and our business could be materially harmed.
If we or our suppliers fail to comply with the FDA’s QSRs, our manufacturing operations could be delayed or shut down and product sales could suffer.
Our manufacturing processes and those of our third-party suppliers are required to comply with the FDA’s QSRs, which covers the procedures and documentation of the design, testing, production, control, quality assurance, labeling, packaging, storage and shipping. We are also subject to similar state requirements and licenses. In addition, we must engage in extensive record keeping and reporting and must make available our manufacturing facilities and records for periodic unannounced inspections by governmental agencies, including the FDA, state authorities and comparable agencies in other countries. If we fail a Quality System inspection, our operations could be disrupted and our manufacturing interrupted. Failure to take adequate corrective action in response to an adverse Quality System inspection could result in, among other things, a shut-down of our manufacturing operations, significant fines, suspension of marketing clearances and approvals, seizures or recalls, operating restrictions and criminal prosecutions, any of which would cause our business to suffer. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with applicable regulatory requirements, which may result in manufacturing delays and cause our revenues to decline.
We have registered with the FDA as a medical device manufacturer and have obtained a manufacturing license from the California Department of Health Services, or CDHS. The FDA has broad post-market and regulatory enforcement powers. We are subject to unannounced inspections by the FDA and the Food and Drug Branch of CDHS to determine our compliance with the QSR and other regulations, and these inspections may include the manufacturing facilities of our suppliers. If the FDA or CDHS inspect our facility and discover compliance problems, we may have to shut down our facility and cease manufacturing until we can take the appropriate remedial steps to correct the audit findings. Taking corrective action may be expensive, time consuming and a distraction for management and if we experience a shutdown or delay at our manufacturing facility we may be unable to produce our products, which may have an adverse impact on our business.
The requirements to obtain regulatory approval of the FDA and regulators in other jurisdictions can be costly, time-consuming, and unpredictable. If we are unable to obtain timely regulatory approval for our therapeutic candidates, our business may be substantially harmed.
The regulatory approval process is expensive, and the time and resources required to obtain approval from the FDA or other regulatory authorities in other jurisdictions to sell any therapeutic candidate or product is uncertain and approval may take years. Whether regulatory approval will be granted is unpredictable and depends upon numerous factors, including the discretion of the regulatory authorities. For example, governing legislation, approval policies, regulations, regulatory policies, or the type and amount of preclinical and clinical data necessary to gain approval may change during the course of a therapeutic candidate’s clinical development and may vary among jurisdictions. It is possible that none of our existing or future therapeutic candidates will ever obtain regulatory approval, even if we expend substantial time and resources seeking such approval.
Further, regulatory requirements governing cell-based therapy products in particular have changed frequently and may continue to change in the future. For example, in November 2014, Japan’s parliament enacted new legislation to promote the safe and accelerated development of treatments using stem cells. The new Pharmaceuticals, Medical Devices and Other Therapeutic Products Act, or PMD Act, establishes a framework for expedited approval in Japan for regenerative medical products. As this is a new regulation, it is not clear yet what impact it will have on the operation of our business. Any regulatory review committees and advisory groups and any contemplated new guidelines may lengthen the regulatory review process, require us to perform additional studies, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of our therapeutic candidates or products or lead to significant post-approval limitations or restrictions. As we advance our therapeutic candidates or products, we will be required to consult with these regulatory and advisory groups and comply with applicable guidelines. If we fail to do so, we may be required to delay or discontinue development of our therapeutic candidates or products. Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a therapeutic candidate or product to market could decrease our ability to generate sufficient revenue to maintain our business.
Our therapeutic candidates could fail to receive regulatory approval for many reasons, including the following:
we may be unable to successfully complete our ongoing and future clinical trials of therapeutic candidates;
we may be unable to demonstrate to the satisfaction of the FDA or other regulatory authorities that a therapeutic candidate is safe, pure, and potent for any or all of a therapeutic candidate’s proposed indications;
we may be unable to demonstrate that a therapeutic candidate’s benefits outweigh the risk associated with the therapeutic candidate;
the FDA or other regulatory authorities may disagree with the design or implementation of our clinical trials;
the results of clinical trials may not meet the level of statistical significance required by the FDA or other regulatory authorities for approval;
the FDA or other regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials;
a decision by the FDA, other regulatory authorities or us to suspend or terminate a clinical trial at any time;
the data collected from clinical trials of our therapeutic candidates may be inconclusive or may not be sufficient to obtain regulatory approval in the United States or international reimbursement systemselsewhere;
the inability to obtain sufficient quantities of the therapeutic candidates for use in clinical trials;
our third-party manufacturers of supplies needed for manufacturing therapeutic candidates may fail to satisfy FDA or other regulatory requirements and may not pass inspections that may be required by FDA or other regulatory authorities;
the failure to comply with applicable regulatory requirements following approval of any of our therapeutic candidates may result in the refusal by the FDA or similar foreign regulatory agency to approve a pending PMA or a biologics license application, or BLA, or supplement to a PMA or BLA submitted by us for other indications or new therapeutic candidates or products; and
the approval policies or regulations of the FDA or other regulatory authorities outside of the United States may significantly change in a manner rendering our clinical data insufficient for approval.
We may gain regulatory approval for any of our therapeutic candidates in some but not all of the territories available and any future approvals may be for some but not all of the target indications, limiting their commercial potential. Regulatory requirements and timing of product approvals vary from country to country and some jurisdictions may require additional testing beyond what is required to obtain FDA approval. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other countries or by the FDA. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. In addition, regulatory approval does not specify pricing or reimbursement which may not match our expectations based on the results of our clinical data.
Even if we obtain and maintain approval for our therapeutic candidates or products from the FDA, we may never obtain approval for our therapeutic candidates or products outside of the United States, which would limit our market opportunities and adversely affect our business.
Approval in the United States by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. Sales of our therapeutic candidates or products, if approved, outside of the United States will be subject to foreign regulatory requirements governing clinical trials and marketing approval.
Even if the FDA grants marketing approval, comparable regulatory authorities of foreign countries must also approve the manufacturing and marketing in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from, and greater than, those in the United States, including additional preclinical studies or clinical trials. In many countries outside the United States, a therapeutic candidate or product must be approved for reimbursement before it can be approved for sale in that significantly reduces reimbursementcountry. In some cases, the price that we intend to charge, if approved, is also subject to approval. While we may decide to submit a request to the EMA for approval of our therapeutic candidates, including CardiAMP, as Advanced Therapeutic Medicinal Products, or ATMPs, in Europe, obtaining such approval is a lengthy and expensive process and the EMA has its own procedures for approval. Even if a therapeutic candidate or product is approved, the FDA or the EMA, as the case may be, may limit the indications for which it may be marketed, require extensive warnings on the product labeling or require expensive and time-consuming clinical trials or reporting as conditions of approval. Regulatory authorities in countries outside of the United States and Europe also have requirements for approval of therapeutic candidates or products with which we must comply prior to marketing in those countries. Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction in certain countries. Further, clinical trials conducted in one country may not be accepted by regulatory authorities in other countries and regulatory approval in one country does not ensure approval in any other country, while a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory approval process in others. Also, regulatory approval may be withdrawn. If we fail to comply with the regulatory requirements in international markets and/or receive applicable marketing approvals, our target market will be reduced and our ability to realize the full market potential of our therapeutic candidates or products will be harmed and our business will be adversely affected.
We may face competition from biosimilars due to changes in the regulatory environment.
We may face competition for the CardiALLO Cell Therapy System from biosimilars due to the changing regulatory environment. In the United States, the Biologics Price Competition and Innovation Act of 2009 created an abbreviated approval pathway for biological products that are demonstrated to be “highly similar,” or biosimilar to, or “interchangeable” with an FDA-approved innovator (original) biological product. This new pathway could allow competitors to reference data from innovator biological products already approved after 12 years from the time of approval. In Europe, a competitor may reference data from biological products already approved but will not be able to get on the market until 10 years after the time of approval. This 10-year period will be extended to 11 years if, during the first eight of those 10 years, the marketing authorization holder obtains an approval for one or more new therapeutic indications that bring significant clinical benefits compared with existing therapies. In addition, companies may be developing biosimilars in other countries that could compete with CardiALLO, if approved. Additionally, the FDA may approve our competitors’ products through a PMA pathway, similar to CardiAMP. If competitors are able to obtain marketing approval for biosimilars referencing CardiALLO, if approved, it may become subject to competition from such biosimilars with the attendant competitive pressure and consequences.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations may involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations may also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials, which could cause an interruption of our commercialization efforts, research and development efforts and business operations, environmental damage resulting in costly clean-up and liabilities under applicable laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. Although we believe that the safety procedures utilized by our third-party manufacturers for handling and disposing of these materials generally comply with the standards prescribed by these laws and regulations, we cannot guarantee that this is the case or eliminate the risk of accidental contamination or injury from these materials. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources and state or federal or other applicable authorities may curtail our use of certain materials and/or interrupt our business operations. Furthermore, environmental laws and regulations are complex, change frequently and have tended to become more stringent. We cannot predict the impact of such changes and cannot be certain of our future compliance. In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions. We do not currently carry biological or hazardous waste insurance coverage.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials or other work-related injuries, this insurance may not provide adequate coverage against potential liabilities.
We are subject to various federal and state fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute and the federal False Claims Act.
Even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, certain federal and state healthcare laws and regulations pertaining to fraud and abuse will be applicable to our business. Healthcare fraud and abuse regulations are complex and can be subject to varying interpretations as to whether or not a statute has been violated. The laws that may affect our ability to operate include:
the federal Anti-Kickback Statute which prohibits, among other things, the knowing and willful payment of remuneration to induce or reward patient referrals or the generation of business involving any item or service which may be payable by the federal health care programs (e.g., drugs, supplies, or health care services for Medicare or Medicaid patients);
the federal False Claims Act which prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment for government funds (e.g., payment from Medicare or Medicaid) or knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim for government funds;
the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and its implementing regulations, imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HIPAA imposes civil and criminal liability for the wrongful access or disclosure of protected health information;
the federal Physician Payments Sunshine Act, created under Section 6002 of the Patient Protection and Affordable Care Act, as amended, the ACA, requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report information related to certain payments or other transfers of value made or distributed to physicians and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, those physicians and teaching hospitals and to report annually certain ownership and investment interests held by physicians and their immediate family members;
the federal Food, Drug and Cosmetic Act which prohibits, among other things, the adulteration or misbranding of drugs and devices;
the U.S. Foreign Corrupt Practices Act which prohibits corrupt payments, gifts or transfers of value to non-U.S. officials; and
non-U.S. and U.S. state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers.
The federal fraud and abuse laws have been interpreted to apply to arrangements between medical device and pharmaceutical manufacturers and a variety of health care professional. Although the federal Anti-Kickback Statute has several statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, all elements of the potentially applicable exemption or safe harbor must be met in order for the arrangement to be protected, and prosecutors have interpreted the federal healthcare fraud statutes to attack a wide range of conduct by medical device and pharmaceutical companies. In addition, most states have statutes or regulations similar to the federal anti-kickback and federal false claims laws, which apply to items and services covered by Medicaid and other state programs, or, in several states, apply regardless of the payor. Administrative, civil and criminal sanctions may be imposed under these federal and state laws.
Further, the ACA, among other things, amended the intent standard under the Anti-Kickback Statute such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the ACA makes clear that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim under the federal False Claims Act. Any violations of these laws, or any action against us for violation of these laws, even if we successfully defend against it, could result in a material adverse effect on our medical devicesreputation, business, results of operations and financial condition.
Efforts to ensure that our business arrangements will comply with applicable healthcare laws may involve substantial costs. It is possible that governmental and enforcement authorities will conclude that our business practices do not comply with current or denies coverage forfuture statutes, regulations or case law interpreting applicable fraud and abuse or other healthcare laws and regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those proceduresactions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, disgorgement, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could harm our ability to operate our business and our results of operations. In addition, the clearance or approval and commercialization of any of our products outside the United States will also likely subject us to foreign equivalents of the healthcare laws mentioned above, among other foreign laws.
A failure to adequately protect private health information could result in severe harm to our reputation and subject us to significant liabilities, each of which could have a material adverse effect on our business, financial conditionbusiness.
Throughout the clinical trial process, we may obtain the private health information of our trial subjects. There are a number of state, federal and international laws protecting the privacy and security of health information and personal data. As part of the American Recovery and Reinvestment Act of 2009, or resultsARRA, Congress amended the privacy and security provisions of operations.
We believe that the overall escalating cost of medical products and services has led to, and will continue to lead to, increased pressuresHIPAA. HIPAA imposes limitations on the use and disclosure of an individual’s healthcare industryinformation by healthcare providers conducting certain electronic transactions, healthcare clearinghouses, and health insurance plans, collectively referred to reduceas covered entities. The HIPAA amendments also impose compliance obligations and corresponding penalties for non-compliance on certain individuals and entities that provide services to or perform certain functions on behalf of healthcare providers and other covered entities involving the costsuse or disclosure of productsindividually identifiable health information, collectively referred to as business associates. ARRA also made significant increases in the penalties for improper use or disclosure of an individual’s health information under HIPAA and services.extended enforcement authority to state attorneys general. The amendments also create notification requirements to federal regulators, and in some cases local and national media, for individuals whose health information has been inappropriately accessed or disclosed. Notification is not required under HIPAA if the health information that is improperly used or disclosed is deemed secured in accordance with certain encryption or other standards developed by the U.S. Department of Health and Human Services, or HHS. Most states have laws requiring notification of affected individuals and state regulators in the event of a breach of personal information, which is a broader class of information than the health information protected by HIPAA. Many state laws impose significant data security requirements, such as encryption or mandatory contractual terms to ensure ongoing protection of personal information. Activities outside of the United States implicate local and national data protection standards, impose additional compliance requirements and generate additional risks of enforcement for noncompliance. The European Union’s Data Protection Directive, Canada’s Personal Information Protection and Electronic Documents Act and other data protection, privacy and similar national, state/provincial and local laws may also restrict the access, use and disclosure of patient health information abroad. We cannot assure you that third-party reimbursementmay be required to expend significant capital and coverage will be availableother resources to ensure ongoing compliance with applicable privacy and data security laws, to protect against security breaches and hackers or adequate, or that future legislation, regulation, or reimbursement policiesto alleviate problems caused by such breaches.
A recall of third-party payors will not adversely affect the demand forany of our commercialized products, or our abilitythe discovery of serious safety issues, could have a significant negative impact on us.
The FDA and other relevant regulatory agencies have the authority to sell these products onrequire or request the recall in the event of material deficiencies or defects in design or manufacture or in the event an unacceptable risk to health. Manufacturers may, under their own initiative, also initiate a profitable basis. We also cannot assure you that our products will be considered cost-effective by third-party payors, that reimbursement will be availablerecall. A government-mandated or if available, that the third-party payors' reimbursement policies will not adversely affect our ability to sell our products profitably.
We must convince orthopedic and spine surgeons that our products are an attractive alternative to existing surgical treatments of orthopedic and spine disorders.
To be commercially successful, we believe that we will need surgeons to adopt our products as their preferred treatment option for their patients. Surgeons may be slow to adopt our products for the following reasons, among others:
lack of clinical evidence;the time that must be dedicated for training;lack of experience with our products;perceived risks generally associated with the use of new products and procedures;perceived risks associated with purchasing products from an early-stage medical device company;
6
costs associated with the purchase of new products and equipment; andlimited availability of reimbursement within healthcare payment systems.
We also believe that recommendations and support of our products by influential surgeons are essential for market acceptance and adoption. If we do not receive support from these surgeons, surgeons and hospitals may not use our products. As a result, we may not achieve expected revenues and may never become profitable.
Our business plan relies on certain assumptions about the market for our products, which, if incorrect, may adversely affect our business and profitability.
We believe that the aging of the general population and increasingly active lifestyles will continue and that these trends will increase the need for our medical products. However, the projected demand for our productsvoluntary recall could differ materially from actual demand if our assumptions regarding these trends and acceptance of our products by the medical community prove to be incorrect or do not materialize, or if non-surgical treatments gain more widespread acceptance as a viable alternative to our devices.
We expect to face significant competitionoccur as a result of the rapid technological changes in the medical devices industry, which couldan unacceptable risk to health, component failures, manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls would divert managerial and financial resources and have an adverse effect on our business,reputation, financial condition and operating results.
Further, under the FDA’s reporting regulations, we are required to report to the FDA any event that reasonably suggests that our products may have caused or results of operations.
The medical device market is highly competitive, subjectcontributed to rapid change, and significantly affected by new product introductions and other market activities of industry participants. We expect to encounter intense competition across our product lines and in each marketa death or serious injury or in which our products are sold from various medical device companies, manyproduct malfunctioned and, if the malfunction of which arethe same or similar product marketed by us were to recur, would likely cause or contribute to have greater financialdeath or serious injury. The FDA also requires reporting of serious, life-threatening, unexpected and marketing resources than us. Primary competitors are Zimmer, J&J/DePuy Orthopedics, Strykerother adverse experiences and Biomet in the hipssubmission of periodic safety reports and knees market, and Medtronic/Sofamor Danek, J&J/DePuy Spine and Synthes in the spine market. In addition, we will face competition from a wide range of companies that sell a singleother information. Malfunctions or a limited number of competitive products or which participate onlyother adverse event reports may result in a specific market segment, as well as from non-medical device companies, including pharmaceutical companies,voluntary or involuntary recall and other adverse actions, which may offer alternative therapies for disease states intended to be treated using our products.
Additionally, the medical device market is characterized by extensive research and development, and rapid technological change. Developments by other companies of new or improved products, processes or technologies may make our products or proposed products obsolete or less competitive and may negatively impact our revenues. We will be required to devote continued effortscould divert managerial and financial resources, to develop or acquire scientifically advanced technologies and products, apply our technologies cost-effectively across product lines and markets, attract and retain skilled development personnel, obtain patent and other protection for our technologies and products, obtain required regulatory and reimbursement approvals and successfully manufacture and market our products consistent with our quality standards. If we fail to develop new products or enhance existing products, it could have a material adverse effect on our business, financial condition or results of operations.
Many of our larger competitors are either publicly traded or divisions or subsidiaries of publicly traded companies, and enjoy several competitive advantages over us, including:
larger and more well-established distribution networks;established relationships with a greater number of surgeons, hospitals, other healthcare providers and third-party payors;products supported by long-term clinical data;greater experience in obtaining and maintaining regulatory approvals or clearances for products and product enhancements;greater name recognition;greater access to manufacturers, vendors and raw materials for manufacturing medical devices;more expansive portfolios of intellectual property rights; andgreater financial and other resources for product research and development, sales and marketing, intellectual property protection and litigation.
7
Hospitals, surgeons, distributors and agents may have existing relationships with other medical device companies that make it difficult for us to establish new relationships with them. As a result, we may not be able to sell and market our products effectively.
We believe that to sell and market our products effectively, we must establish relationships with key surgeons and hospitals in the field of orthopedic knee, hip and spinal surgery. Many of these key surgeons and hospitals already have long-standing relationships with large, better-known companies that dominate the medical devices industry through collaborative research programs and other relationships. Because of these existing relationships, some of which may be contractually enforced, surgeons and hospitals may be reluctant to adopt our products, particularly if our products compete with or have the potential to compete with products supported through their own collaborative research programs or by these existing relationships. Even if these surgeons and hospitals purchase our products, they may be unwilling to enter into collaborative relationships with us to promote joint marketing programs or to provide us with clinical and financial data.
We work primarily with a network of independent orthopedic product agents and distributors that generate sales leads for us, in addition to working with our own internal direct sales force. If these product agents and distributors believe that their relationship with us is less beneficial than other relationships they may have with more established or well-known medical device companies, they may be unwilling to continue their relationships with us, making it more difficult for us to sell and market our products effectively.
Our manufacturers may be unsuccessful in manufacturing products at the levels required to meet future market demand.
We are seeking to rapidly grow sales of our products, and, if we are successful, our growth may strain the ability of our manufacturers to manufacture an increasingly large supply of our products. Manufacturers regularly experience difficulties in scaling up production and our manufacturers may face difficulties in increasing their production levels.
Our manufacturers may not be able to manufacture our products with consistent and satisfactory quality or in sufficient quantities to meet demand, which could hurt our reputation, cause our customers to cancel orders or refrain from placing new orders for our products and reduce or slow growth of sales of our products. The increased production volume also could make it harder for us to maintain control over expenses, manage our relationships with our manufacturers, maintain good relations with our employees or otherwise manage our business.
We rely on single source manufacturers, which could impair our ability to meet demand for delivering our products in a timely manner or within our budget.
We rely on third-party manufacturers to manufacture our products. It is critical to our business that our contract manufacturers be able to provide us with products in substantial quantities, in accordance with agreed upon specifications, in compliance with regulatory requirements, at acceptable cost and on a timely basis. Our anticipated growth could strain the ability of manufacturers to deliver an increasingly large supply of products. If we are unable to obtain sufficient quantities of high quality products to meet customer demand on a timely and cost-effective basis, we could lose customers, our reputation could be harmed and our business could suffer.
We currently use up to seven manufacturers for each of our devices. Our dependence on these few manufacturers involves several risks, including limited control over pricing, availability, quality and delivery schedules. If any one or more of our manufacturers cease to provide us with sufficient quantities of our products in a timely manner or on terms acceptable to us, or cease to manufacture products of acceptable quality, we would have to seek alternative sources of manufacturing. We could experience delays while we locate and engage alternative qualified manufacturers, and we might be unable to engage alternative manufacturers on favorable terms, if at all. Any disruption or increased expenses relating to our supply source could harm our sales and marketing efforts and adversely affect our ability to generate revenue.
Our growth will depend on developing new products or product enhancements, requiring significant research and development, clinical trials and regulatory approvals, all of which are expensive and time-consuming and may not result in a commercially viable product.
8
We believe that it is important for us to continue to build a more complete product offering and to enhance the products we currently offer. Our success in this regard will depend in part on our ability to develop and introduce new products and product enhancements to keep pace with the rapidly changing medical device market. We cannot assure you that we will be able to successfully develop, obtain regulatory approval for or market new products or product enhancements, or that any of our future products or enhancements will be accepted by the surgeons who use our products or the payors who financially support many of the procedures performed with our products.
Factors affecting the success of any new product offering or enhancement to an existing product include our ability to:
properly identify and anticipate surgeon and patient needs;fund the development of and introduce new products or product enhancements in a timely manner;avoid infringing upon the intellectual property rights of third parties;obtain the necessary regulatory clearances or approvals for new products or product enhancements;demonstrate, if required, the safety and efficacy of new products with data from preclinical studies and clinical trials;provide adequate training to potential users of our products;receive adequate reimbursement; anddevelop an effective and dedicated marketing and distribution network.
If we do not develop new products or product enhancements in time to meet market demand or if there is insufficient demand for these products or enhancements, our results of operations may suffer.
If we choose to grow our business by acquiring new and complementary businesses, products or technologies, we may be unable to complete these acquisitions or successfully integrate them in a cost-effective and non-disruptive manner.
We believe that our success depends in part on our ability to continually enhancetimely manner and broaden our product offering in response to changing customer demands, competitive pressures and technologies and our ability to increase our market share. To achieve this growth, we have completed certain acquisitions, and intend to pursue other acquisitions of complementary businesses, products or technologies, in some cases instead of developing them ourselves. We may be unable to successfully complete any further acquisitions, or we may not be able to successfully integrate any acquired business, product or technology into our business or retain any key personnel, manufacturers or distributors. The success of any acquisition, investment or alliance undertaken will depend on a number of factors, including:
our ability to identify suitable opportunities;our ability to finance any acquisition, investment or alliance;whether we are able to establish an acquisition, investment or alliance on terms that are satisfactory to us, if at all;the strength of the other companies' underlying technology and ability to execute;intellectual property and litigation related to these technologies or businesses; andour ability to successfully integrate the acquired company or business with our existing business, including the ability to adequately fund acquired in-process research and development projects.
These efforts could be expensive and time-consuming, disrupt our ongoing business and distract management. If we are unable to integrate any acquired businesses, products or technologies effectively, our business, financial condition and results of operations will be materially adversely affected. For example, an acquisition could materially impair our operating results by causing us to incur debt or requiring us to amortize significant amounts of expenses, including non-cash acquisition costs, and acquired assets.
9
We rely on our independent sales distributors and sales representatives to market and sell our products.
We depend upon independent sales distributors and sales representatives to market and sell our products, in particular due to their sales and service expertise and relationships with customers in the marketplace. Independent distributors and sales representatives may terminate their relationships with us or devote insufficient sales efforts to our products for any number of reasons. We do not control our independent distributors and they may not be successful in implementing our marketing plans. If we fail to maintain our existing relationships with our independent distributors and sales representatives, our operations would suffer. Similarly, our failure to recruit and retain additional skilled, independent sales distributors and sales representatives could have an adverse effect on our operations. We may experience turnover with somereputation, financial condition and operating results. Similar reporting requirements exist in Europe and other jurisdictions.
Any adverse event involving our products could result in future voluntary corrective actions, such as recalls or customer notifications, or regulatory agency action, which could include inspection, mandatory recall or other enforcement action. Any corrective action, whether voluntary or involuntary, will require the dedication of our independent sales distributors, which could adversely affecttime and capital, distract management from operating our short-term financial results while we transition to new distributors. Our failure to manage these transitions effectively could negatively impact our operationsbusiness and profitability.
We are dependent on the services of Andrew A. Brooks, M.D. and Michael Kvitnitsky, and the loss of either of them couldmay harm our business.
Our success dependsreputation and financial results. For example, in part upon2014 we notified the continued serviceFDA that we were going to initiate a voluntary recall of Andrew A. Brooks, M.D., who serves as our Chairman of the Board and Chief Executive Officer, and Michael Kvitnitsky, who serves as our President and Chief Operating Officer. Dr. Brooks and Mr. Kvitnitsky are criticalMorph AccessPro product based on a manufacturing observation, which was completed to the overall management ofFDA’s satisfaction in the same year, and in 2017 we updated our Company as well asinstructions for use for the Helix™ and Morph catheter products to the development of our technology, our culture and our strategic direction. The loss of either Dr. Brooks or Mr. Kvitnitsky could have a material adverse effectprovide guidance on our business, results of operations and financial condition. We have not obtained and do not expect to obtain any key-person life insurance policies on Dr. Brooks or Mr. Kvitnitsky.
Failure to attract and retain skilled personnel and cultivate key academic collaborations will delay product development programs and business development efforts.
Our success will depend on our ability to continuously attract and retain highly qualified management and scientific personnel and on our ability to develop relationships with academic collaborators. The competition for qualified personnel and collaborators is intense. We cannot assure youknown potential risks. There can be no guarantee that we will be able to attractnot experience similar product recalls or retain personnel or cultivate academic collaborations. In addition, our collaborators may have arrangements with other companies to assist those companies in developing products that compete with ours. Our inability to hire or retain qualified personnel or cultivate academic collaborations would harm our business.
If we fail to properly manage our anticipated growth, our business could suffer.
We have experienced growth in, and will continue to pursue rapid growthchanges in the number of surgeons usingfuture with these products or our other products or therapeutic candidates, if approved.
Modifications to our products the types of products we offer and the number of states in which our products are sold. This growth has placed and may continue to place significant demands on our managerial, operational and financial resources and systems. We are currently focused on increasing the size and effectiveness of our sales force and distribution network, marketing activities, research and development efforts, inventory management systems, management team, accounting systems and corporate infrastructure. If we do not manage our growth effectively, the quality of our products, our relationships with surgeons, distributors and hospitals, and our reputation could suffer.
We must attract and retain qualified personnel and third-party distributors and manage and train them effectively. Personnel qualified in the design, development, production and marketing of our products are difficult to find and hire, and enhancements of information technology systems to support our growth are difficult to implement. In addition, we will need to carefully monitor and manage our surgeon services, and the quality assurance and efficiency of our manufacturers and distributors. This managing, training and monitoring will require allocation of valuable management resources and significant expense.
If we decide to market and sell our devices and products internationally, we would be subject to various risks relating to our international activities, which could negatively impact our business and financial results.
We currently do not marketreclassifications, new regulatory approvals or sell our products outside of the United States. However, we may actively pursue one or more international markets within the next few years, at which point we would be exposed to risks separate and distinct from those we face in our U.S. operations. Any international business we may engage in may be adversely affected by changing economic conditions in foreign countries, as well as U.S. laws that may affect the international
10
business operations of a U.S. company such as ours. In addition, increases or decreases in the value of the U.S. dollar relative to foreign currencies could affect our results of operations since international sales most likely would be denominated in the functional currency of the country in which the product is sold.
Certain additional or different risks inherent in engaging in international business include the following:
compliance with existing and changing foreign regulatory laws and requirements;export restrictions and controls and other government regulation relating to technology or medical devices;foreign laws and business practices favoring local companies;pricing pressures that we may experience internationally;the availability and level of reimbursement within prevailing foreign healthcare payment systems or insurance providers;shipping delays due to cross-border sales;longer payment cycles;difficulties and costs of establishing, staffing and managing foreign operations;potentially adverse tax consequences, tariffs and other trade barriers;difficulties in enforcing intellectual property rights;difficulties in enforcing agreements and collecting receivables through certain foreign legal systems;political and economic instability; andinternational terrorism and anti-American sentiment.
Our exposure to each of these risks may increase our costs, impair our ability to market and sell our products and require significant management attention, resulting in harm to our business and financial results.
We are subject to substantial governmental regulation that could change and thus force us to make modifications to how we develop, manufacture and price our products.
The medical device industry is regulated extensively by governmental authorities, principally the Food and Drug Administration, or the FDA, and corresponding state and foreign regulatory agencies. The FDA and other federal, state and foreign governmental agencies regulate, among other things, the development, manufacturing, clinical trials, marketing clearance and approval, promotion and sale of medical devices.
In particular, the FDA permits commercial distribution of a new medical device only after the device has received clearance under Section 510(k), or is the subject of an approved premarket approval application, or PMA. The FDA will approve marketing a medical device through the Section 510(k) process if it is demonstrated that the new product is substantially equivalent to other Section 510(k)-cleared products. The PMA process is more costly, lengthy and uncertain than the Section 510(k) clearance process. A PMA application must be supported by extensive data, including, but not limited to, technical, preclinical, clinical trial, manufacturing and labeling data, to demonstrate to the FDA's satisfaction the safety and efficacy of the device for its intended use. To date, all of our products, unless exempt, have been cleared through the Section 510(k) process. We have no experience in obtaining premarket approval.
Compliance with complex regulations is, and will continue to be, time-consuming, burdensome and expensive. Failure to comply with these regulations could jeopardize our ability to manufacture and sell our products and result in enforcement actions such as warning letters, fines, injunctions, civil penalties, termination of distribution, seizures of products, total or partial suspension of production, refusal of the FDA or other regulatory agencies to grant future clearances, or approvals, or withdrawals or suspensions of current clearances or approvals. These enforcement actions could result in higher than anticipated costs or lower than anticipated revenue and have a material adverse effect on our business, financial condition and results of operations. In the most egregious cases, we could face criminal sanctions, closure of the manufacturing facilities in which our products are manufactured, and prohibitions on the sales of our products.
Foreign governmental authorities that regulate the manufacture and sale of medical devices have become increasingly vigilant, and if we engage in sales of our products in foreign countries, these sales would be subject to rigorous foreign regulations. In these circumstances, we would rely heavily on our foreign independent sales
11
agencies to comply with the varying regulations, and any failures on their part could result in restrictions on the sale of our products in foreign countries. We currently do not sell any of our products internationally.
Federal regulatory reforms may adversely affect our ability to sell our products profitably.
Legislation may be drafted from time to time and introduced in Congress that could significantly change the statutory provisions governing the clearance or approval, manufacture and marketing of a medical device in the United States. In addition, FDA regulations and guidance often are revised or reinterpreted by the agency in ways that may significantly affect our business and our ability to commercialize our products. It is impossible to predict whether legislative changes will be enacted or FDA regulations, guidance or interpretations changed, and what the impact of these changes, if any, may be. For example, on September 27, 2007, Congress enacted, and the President signed into law, the Food and Drug Administration Amendments Act of 2007. This new legislation grants significant new powers to the FDA and imposes new obligations and requirements on both the FDA and FDA-regulated industries, including the medical device industry. In particular, this law requires, among other things, that the FDA propose, and ultimately implement, regulations that will require manufacturers to label medical devices with unique identifiers unless a waiver is received from the FDA. In addition, it reauthorizes the FDA to collect medical device user fees and amends the existing user fee program by, among other things, reducing device application fees and imposing new fees, including a new annual establishment registration fee. Also, the new law authorizes the FDA to establish a unique medical device identification system and expands the federal government's clinical trial registry and results databank to include, among other things, information on medical device clinical trials. While these new requirements undoubtedly will have a significant effect on the medical device industry, we cannot yet predict the extent of that effect on our Company. As regulations, guidance and interpretations are issued by the FDA relating to the new legislation, its impact on the industry, as well as our business, will become clearer. Compliance with those regulations could require us to take additional steps, and incur additional costs, in manufacturing and labeling products.
We have not yet collected long-term clinical data to support the safety of our products, and our products may, therefore, prove to be less safe and effective than initially thought.
We obtained clearance to offer all of our products that require FDA clearance or approval through the Section 510(k) clearance process, which is less rigorous than the PMA process and requires less supporting clinical data. As a result of using this expedited process, we currently lack the breadth of published long-term clinical data supporting the safety of our products and the benefits they offer that might have been generated using the PMA process. Because of the lack of this in-depth data, surgeons may be slow to adopt our products, we may not have comparative data that our competitors have or are generating, and we may be subject to greater regulatory and product liability risks. Further, future patient studies or clinical experience may indicate that treatment with our products does not improve outcomes. These results would reduce demand for our products, thereby preventing us from becoming profitable. If future results and experience indicate that our products cause unexpected or serious complications or other unforeseen negative effects, we could be subject to significant legal liability and harm to our business reputation. The medical device market has been particularly prone to costly product liability litigation. The time and costs of any product liability litigation we may face may materially adversely affect our business, financial condition or results of operations, even if we are ultimately victorious in any such litigation.
The FDA requires us to obtain new Section 510(k) clearances or premarket approvals for modifications to our approved products. Otherwise, we may have to cease marketing or to recall the modified products until clearancesnew CE marking is obtained.
Currently there are obtained.
Anysix Morph product family model numbers that have been approved for commercial use in the United States via a 510(k) clearance. A modification to these products could lead to a Section 510(k)-cleared device thatreclassification and could significantly affect its safety or efficacy, or that would constitute a major changeresult in its intended use, requires a new Section 510(k)further requirements (including additional clinical trials) to maintain each respective clearance or possibly, premarket approval. Under FDA regulations, every manufacturer must make this determination in the first instance, but the FDA may review any manufacturer's decision. The FDA may not agreeIf we fail to comply with any of our decisions regarding whether new clearances or approvals are necessary. If the FDA requires us to seek Section 510(k) clearance or premarket approval for any modification to a previously cleared product,such further requirements, we may be required to cease marketing or to recall the modified product until we obtain clearance or approval. Thisapproval, and we may expose usbe subject to significant regulatory fines or penalties.
12
In addition, CE Mark certification for three Morph product family model numbers expired in April 2019. While the Company is working to renew CE Mark certification for such products, there can be no assurance that renewal will be granted or that the business will not be materially adversely affected as a result of the loss of such certification or the failure to obtain such renewal.
The financial performance of our enabling and delivery products couldmay be subjectadversely affected by medical device tax provisions in the healthcare reform laws in the United States.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, the Affordable Care Act, imposes, among other things, an annual excise tax of 2.3% on any entity that manufactures or imports medical devices offered for sale in the United States beginning with tax year 2013. Under these provisions, the Congressional Research Service predicts that the total cost to recall if the FDA determines,medical device industry may be up to $20 billion over the next decade. On December 18, 2015, President Obama signed into law the Consolidated Appropriations Act, 2016 (H.R. 2029), which includes a two-year moratorium on the medical device excise tax. It amended section 4191 of the Internal Revenue Code to exempt medical device sales during the period of January 1, 2016 to December 31, 2017. On January 22, 2018, President Trump signed legislation that suspended the medical device excise tax through December 31, 2019. Absent further legislative action, the tax will be automatically reinstated for any reason,medical device sales starting on January 1, 2020. The financial impact this tax may have on our business is unclear and there can be no assurance that our productsbusiness will not be materially adversely affected by it.
We work with outside scientists and their institutions in developing therapeutic candidates and products. These scientists may have other commitments or conflicts of interest, which could limit our access to their expertise.
We work with scientific advisors and collaborators at academic research institutions in connection with our development programs. These scientific advisors serve as our link to the specific pools of trial participants we are targeting in that these advisors may:
identify individuals as potential candidates for study;
obtain their consent to participate in our research;
perform medical examinations and gather medical histories;
conduct the initial analysis of suitability of the individuals to participate in our research based on the foregoing; and
collect data and biological samples from trial participants periodically in accordance with our study protocols.
These scientists and collaborators are not safeour employees, rather they serve as either independent contractors or effective. Any recall or FDA requirementthe primary investigators under research collaboration agreements that we seek additional approvalshave with their sponsoring academic or clearances could result in delays, fines, costs associatedresearch institution. Such scientists and collaborators may have other commitments that would limit their availability to us. Although our scientific advisors generally agree not to do competing work, if an actual or potential conflict of interest between their work for us and their work for another entity arises, we may lose their services. It is also possible that some of our valuable proprietary knowledge may become publicly known through these scientific advisors if they breach their confidentiality agreements with modifying a product, loss of revenue,us, which would cause competitive harm to our reputation and lossbusiness.
The use, misuse or off-label use of customers and potential operating restrictions imposed by the FDA. Anyour products or therapies, if approved, may result in injuries that lead to product liability claimsuits, which could be costly to our business.
We are not permitted to make claims about the use of our marketed products and will not be permitted to make claims about the use of our therapeutic candidates, if approved, outside of their approved indications. Further, we are not and will not be able to proactively discuss or recall would divert managerialprovide information on off-label uses of such products, with very specific and financial resourceslimited exceptions. However, we cannot prevent a physician from using our products or therapeutic candidates, if approved, for off-label applications. Off-label use of our products or therapies, if approved, is more likely to result in complications that have serious consequences. Product liability claims are especially prevalent in our industry and could harm our reputation, divert management’s attention from our core business, be expensive to defend and may result in sizable damage awards against us. Although we maintain product liability insurance, the amount or breadth of our coverage may not be adequate for the claims that may be made against us. In addition, failure to follow FDA rules and guidelines relating to promotion and advertising can result in, among other things, the FDA’s refusal to approve a product or therapeutic candidate, the suspension or withdrawal of an approved product or therapy from the market, product recalls, fines, disgorgement of money, operating restrictions, injunctions or criminal prosecutions.
Our employees, principal investigators, consultants and collaboration partners may engage in misconduct or other improper activities, including noncompliance with customers. laws and regulatory standards and requirements and insider trading.
We cannot assure youare exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include failures to comply with FDA regulations, to provide accurate information to the FDA, to comply with manufacturing standards we have established, to comply with federal and state healthcare fraud and abuse laws and regulations, to report financial information or data accurately or to disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations restrict or prohibit a wide range of activity relating to pricing, discounting, marketing and promotion, sales commissions, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation, or a breach of insider trading laws. It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell our therapeutic candidates, if approved, we may be unable to generate any revenues.
We currently have a limited organization for the sales, marketing and distribution of products and the cost of establishing and maintaining such an organization may exceed the cost-effectiveness of doing so. In order to market any products that may be approved, including CardiAMP and CardiALLO Cell Therapy Systems, we must build our sales, distribution, marketing, managerial and other non-technical capabilities or make arrangements with third parties to perform these services. We have limited prior experience in the marketing, sale or distribution of approved products and there are significant risks involved in building and managing a sales organization, including our ability to hire, retain, and incentivize qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel, and effectively manage a geographically dispersed sales and marketing team. Any failure or delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of our therapeutic candidates.
Our strategy is to obtain FDA approval and market the CardiAMP Cell Therapy System for potential heart failure and chronic myocardial ischemia indications using a dedicated direct sales model focused on selected cardiologists and interventional cardiologists. We may in the future, choose to align ourselves with collaborators as part of our commercialization strategy, particularly outside of the United States, and our future collaboration partners, if any, may not dedicate sufficient resources to the commercialization of our therapeutic candidates or companion diagnostic or may otherwise fail in their commercialization due to factors beyond our control. If we are unable to establish effective collaborations to enable the sale of our therapeutic candidates and companion diagnostic to healthcare professionals and in geographical regions, including the United States, that will not be covered by our own marketing and sales force, or if our potential future collaboration partners do not successfully commercialize our therapeutic candidates or companion diagnostic, our ability to generate revenues from product sales, including sales of CardiAMP and CardiALLO Cell Therapy Systems, will be adversely affected.
Building an internal sales force involves many challenges, including:
• | recruiting and retaining talented people; | |
• | training employees that we recruit; | |
• | setting the appropriate system of incentives; | |
• | managing additional headcount; and | |
• | integrating a new business unit into an existing corporate architecture. |
If we are unable to build our own sales force or negotiate a strategic partnership for the commercialization of CardiAMP or CardiALLO Cell Therapy Systems in the United States, we may be forced to delay the potential commercialization of these therapies or reduce the scope of our sales and marketing activities for CardiAMP or CardiALLO Cell Therapy Systems. To fund commercialization activities, we will need to obtain additional capital, which may not be available to us on acceptable terms, or at all. If we do not have sufficient funds, we will not be able to bring CardiAMP or CardiALLO Cell Therapy Systems to market or generate product revenue.
If we are unable to establish adequate sales, marketing and distribution capabilities, whether independently or with third parties, we may not be able to generate sufficient product revenue and may not become profitable. We will be competing with many companies that currently have product liability claimsextensive and well-funded marketing and sales operations. Without an internal team or recallsthe support of a third party to perform marketing and sales functions, we may be unable to compete successfully against these more established companies.
In addition, there are risks involved with both establishing our own sales and marketing capabilities and entering into arrangements with third parties to perform these services. For example, recruiting and training a sales force is expensive and time-consuming and could delay any launch. If the commercial launch of a therapeutic candidate for which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
We have limited experience manufacturing our therapeutic candidates or products in commercial quantities, which could harm our business.
Because we have only limited experience in manufacturing therapeutic candidates or products in commercial quantities, we may encounter production delays or shortfalls. Such production delays or shortfalls may be caused by many factors, including the following:
we intend to significantly expand our manufacturing capacity, and our production processes may have to change to accommodate this growth;
key components and sub-assemblies of our products and therapeutic candidates are currently provided by a single supplier or limited number of suppliers, and we do not maintain large inventory levels of these components and sub-assemblies; if we experience a shortage in any of these components or sub-assemblies, we would need to identify and qualify new supply sources, which could increase our expenses and result in manufacturing delays;
we may experience a delay in completing validation and verification testing for new controlled-environment rooms at our manufacturing facilities;
we have limited experience in complying with FDA’s QSRs, which applies to the manufacture of our products and therapeutic candidates; and
to increase our manufacturing output significantly, we will have to attract and retain qualified employees, who are in short supply, for our manufacturing operations.
If we are unable to keep up with demand for our products, our revenues could be impaired, market acceptance for our products could be harmed and our customers might instead purchase our competitors’ products. Our inability to successfully manufacture our products would materially harm our business.
If we fail to obtain and sustain an adequate level of reimbursement for our products by third-party payors, sales and profitability would be adversely affected.
Our ability to commercialize any therapeutic candidates or products successfully will depend, in part, on the extent to which coverage and reimbursement for our therapeutic candidates or products and related treatments will be available from government healthcare programs, private health insurers, managed care plans, and other organizations. Additionally, even if there is a commercially viable market, if the level of third-party reimbursement is below our expectations, our revenue and profitability could be materially and adversely affected.
Third-party payors, such as government programs, including Medicare in the future,United States, or private healthcare insurers, carefully review and increasingly question the coverage of, and challenge the prices charged for medical products and services, and many third-party payors limit coverage of or reimbursement for newly approved therapies or products. Reimbursement rates and coverage from private health insurance companies vary depending on the company, the insurance plan and other factors. As a result, the coverage determination process will require us to provide scientific and clinical support for the use of our therapeutic candidates to each private health insurance company separately, with no assurance that these claimsadequate coverage and reimbursement will be obtained.
A current trend in the U.S. healthcare industry as well as in other countries around the world is toward cost containment, including a number of legislative and regulatory changes to the health care system that could impact our ability to sell our approved therapies or recalls wouldproducts profitably. In particular, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 revised the payment methodology for many products under Medicare in the United States, which has resulted in lower rates of reimbursement. In 2010, the Affordable Care Act was enacted. This expansion in the government’s role in the U.S. healthcare industry may further lower rates of reimbursement.
Other legislative changes have been proposed and adopted in the United States since the Affordable Care Act was enacted. President Donald Trump has made statements that suggest he plans to seek repeal of all or portions of the Affordable Care Act, and has stated that he will ask Congress to replace the current legislation with new legislation. There is uncertainty with respect to the impact President Trump’s Administration may have, if any, and any changes likely will take time to unfold, and could have an impact on coverage and reimbursement for healthcare items and services covered by plans that were authorized by the Affordable Care Act. However, we cannot predict the ultimate content, timing or effect of any healthcare reform legislation or the impact of potential legislation on us. In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. These changes included aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, effective April 1, 2013, which, due to subsequent legislative amendments, will stay in effect through 2027 unless additional Congressional action is taken. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several providers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on customers for our products, if approved, and accordingly, on our financial operations.
In 2017, the European Union released new regulations to ensure patient safety with the use of pharmaceuticals, medical devices and in-vitro diagnostics that will go into effect over a three-year period from 2020 to 2022. The new regulations replace predecessor directives and emphasize a global convergence of regulations. Marketing authorization timelines will become more protracted and the costs of operating in Europe will increase. A significantly more costly path to regulatory compliance is anticipated. Adjusting to the new Medical Device Regulation may prove to be costly and disruptive to our business.
Large public and private payors, managed care organizations, group purchasing organizations and similar organizations are exerting increasing influence on decisions regarding the use of, and reimbursement levels for, particular treatments. In particular, third-party payors may limit the covered indications. Cost-control initiatives could decrease the price we might establish, which could result in revenue and profitability being lower than anticipated.
There may be significant delays in obtaining coverage and reimbursement for newly approved therapies or products, and coverage may be more limited than the purposes for which the therapy or product is approved by the FDA or other regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that a therapy or product will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution expenses. Interim reimbursement levels, if applicable, may also be insufficient to cover our and any partner’s costs and may not be made permanent. Our inability to promptly obtain coverage and profitable payment rates from both government-funded and private payors for any approved therapies or products that we develop could have a material adverse effect on our business.operating results, our ability to raise capital needed to commercialize therapies or products and our overall financial condition.
Furthermore, reimbursement systems in international markets vary significantly by country and by region, and reimbursement approvals must be obtained on a country-by-country basis. In many countries, therapies or products cannot be commercially launched until reimbursement is approved and the negotiation process in some countries can exceed 12 months. In addition, pricing and reimbursement decisions in certain countries can be affected by decisions taken in other countries, which can lead to mandatory price reductions and/or additional reimbursement restrictions across a number of other countries, which may thereby adversely affect our sales and profitability. In the event that countries impose prices which are not sufficient to allow us to generate a profit, this would adversely affect sales and profitability.
IfPrice controls may be imposed in foreign markets, which may adversely affect our future profitability.
In some countries, particularly European Union member states, Japan, Australia and Canada, the pricing of therapies and products is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after receipt of marketing approval for a therapy or product. In addition, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various European Union member states and parallel distribution, or arbitrage between low-priced and high-priced member states, can further reduce prices. In some countries, we or our third-party manufacturers failpartners may be required to comply withconduct a clinical trial or other studies that compare the FDA's Quality System Regulations, the manufacturecost-effectiveness of our therapeutic candidates to other available therapies in order to obtain or maintain reimbursement or pricing approval. Publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. If reimbursement of our therapies or products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business, revenues or profitability could be interruptedadversely affected.
If the market opportunities for our therapeutic candidates or products are smaller than we believe they are, our revenues may be adversely affected and our business may suffer.
It is very difficult to estimate the future commercial potential of the CardiAMP Cell Therapy System, the CardiALLO Cell Therapy System, and our commercialized products due to factors such as safety and efficacy compared to other available treatments, changing standards of care, third-party payor reimbursement standards, patient and physician preferences, and the availability of competitive alternatives that may emerge. We believe that approximately 70% of the NYHA Class II and Class III ischemic systolic heart failure patients in the United States will be eligible for CardiAMP due to a sufficient CardiAMP potency assay score. However, if considerably less than approximately 70% of NYHA Class II and Class III ischemic heart failure patients are eligible for CardiAMP due to an insufficient CardiAMP potency assay score, it would significantly and negatively impact our business, financial condition and results of operations.
If product salesliability lawsuits are brought against us, we may incur substantial liabilities and operating results could suffer.may be required to limit commercialization of our therapeutic candidates or products.
We and someface an inherent risk of product liability as a result of the human clinical use of our third-party manufacturers aretherapeutic candidates and products and will face an even greater risk if we continue to commercialize our therapeutic candidates and products. For example, we may be sued if any therapy or product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of inherent dangers, negligence, strict liability, and a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to comply with the FDA's Quality System Regulation, or QSR, which covers the methodslimit commercialization. Even a successful defense would require significant financial and documentationmanagement resources. Regardless of the design, testing, production, control, quality assurance,merits or eventual outcome, liability claims may result in:
decreased demand, even if such products or therapies are approved;
injury to our reputation;
withdrawal of clinical trial participants;
costs to defend the related litigations;
a diversion of management’s time and our resources;
substantial monetary awards to trial participants or patients;
recalls, withdrawals, or labeling, packaging, sterilization, storagemarketing or promotional restrictions;
increased cost of liability insurance;
loss of revenue;
the inability to receive regulatory approvals or commercialize our approved products or therapies; and shipping
a decline in our share price.
Although we maintain product liability insurance with coverage that we believe is consistent with industry norms for companies at our stage of development, the amount or breadth of our products. In addition,coverage may not be adequate for the claims that may be made against us. Failure to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of products or therapies we develop. Additionally, our insurance policies have various exclusions, and our manufacturers willwe may be subject to a product liability claim for which we have no coverage or reduced coverage. Any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the regulations of foreign jurisdictions regarding the manufacturing process if we market our products overseas.
The FDA enforces the QSR through periodic and unannounced inspections of manufacturing facilities. If our facilities or thoselimits of our manufacturers failinsurance coverage. We will have to take satisfactory corrective actionpay any amounts awarded by a court or negotiated in response to an adverse QSR inspection, the FDA could take enforcement action, including any of the following sanctions:
customer notificationsa settlement that exceed our coverage limitations orrepair, replacement, refunds, recall, detentionthat are not covered by our insurance, and we may not have, orseizure of our products;operating restrictions or partial suspension or total shutdown of production;untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties;refusing or delaying requests for Section 510(k) clearance or PMA approvals of new products or modified products;withdrawing Section 510(k) clearances or PMA approvals;refusal to grant export approval for our products; orcriminal prosecution.
If we sell our products in the European Community, we will be required to maintain certain ISO certifications and must undergo periodic inspections by notified bodies to obtain and maintain these certifications. We cannot assure you that we or our manufacturers will be able to obtain, sufficient capital to pay such amounts.
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems and those of our current and any future CROs and other contractors, consultants and potential collaborators are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. For example, our systems have been impacted by computer viruses in the past, and while we have not experienced any material system failure, accident or maintain all required registrationssecurity breach that has resulted in lasting impacts to date, if such an event were to occur and certifications.cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Likewise, we rely on third parties for manufacturing our therapeutic candidates and conducting clinical trials, and similar events relating to their computer systems could also have a material adverse effect on our business. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development and commercialization of our therapeutic candidates could be delayed.
Any
Interruptions in supply or inventory loss may adversely affect our operating results and financial condition.
Our therapeutic candidates and products are manufactured and distributed using technically complex processes requiring specialized facilities, highly specific raw materials and other production constraints. The complexity of these factorsprocesses, as well as strict company and government standards for manufacture and storage, subjects us to production risks. While batches released for use in clinical trials or for commercialization undergo sample testing, some defects may only be identified following release. In addition, process deviations or unanticipated effects of approved process changes may result in these intermediate products not complying with stability requirements or specifications. The investigation and remediation of any identified problems can cause production delays, substantial expense, lost sales and delays of new product or therapy launches. Any supply interruption or the loss thereof could impairhinder our ability to producetimely distribute our approved products and satisfy demand. Any unforeseen storage failure or loss in supply could delay our clinical trials and, if our therapeutic candidates are approved, result in a cost-effectiveloss of our market share and timely manner in order to meetnegatively affect our customers' demands. revenues and operations.
We alsoor the third parties upon whom we depend may be required to bearadversely affected by earthquakes or other costs or take other actions that may have a negative impact on our future salesnatural disasters and our ability to generate profits
We are subject to various complex lawsbusiness continuity and regulations. Compliance with these laws and regulations is costly and time-consuming, and failure to comply with them can have adverse consequences ondisaster recovery plans may not adequately protect us from a serious disaster.
Earthquakes or other natural disasters could severely disrupt our business.
U.S. federal government entities, such as the Occupational Safety and Health Administration, the Environmental Protection Agency, the Internal Revenue Service, the Centers for Medicare and Medicaid Services and the U.S. Department of Veteran's Affairs, as well as the FDA and regulatory authorities in other states, have each been empowered to administer certain laws and regulations applicable to us. Many of the laws and regulations are complex, and compliance will require substantial time and effort by our officers, directors and employees and extensive consultations with our advisors. Because of this complexity, we cannot assure you that our efforts will be sufficient to ensure compliance with all of these laws and regulations at any given time.
We are subject to audit, investigation and litigation by each of these entities to ensure compliance, each of which also can be time-consuming, costly, divert the attention of senior managementoperations, and have a significantmaterial adverse effect on our business, evenresults of operations, financial condition and prospects. A majority of our management operates in our principal executive offices located in San Carlos, California and we currently manufacture our Helix™ and Morph products at this facility and use it for storage of our clinical trial materials. If our San Carlos offices were affected by a natural or man-made disaster, particularly those that are characteristic of the region, such as wildfires and earthquakes, or other business interruption, our ability to manage our domestic and foreign operations could be impaired, which could materially and adversely affect our results of operations and financial condition. If a natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as the manufacturing facilities of our third-party contract manufacturers, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. The disaster recovery and business continuity plans we have in place currently are limited and are unlikely to prove adequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which, particularly when taken together with our lack of earthquake insurance, could have a material adverse effect on our business. The ultimate impact of any such events on us, our significant suppliers and our general infrastructure is unknown.
Risks Relating to Our Intellectual Property
We may not be able to protect our proprietary technology in the marketplace.
Our success will depend, in part, on our ability to obtain patents, protect our trade secrets and operate without infringing on the proprietary rights of others. We rely upon a combination of patents, trade secret protection, and confidentiality agreements to protect the intellectual property of our therapeutic candidates and products. Patents might not be issued or granted with respect to our patent applications that are currently pending, and issued or granted patents might later be found to be invalid or unenforceable, be interpreted in a manner that does not adequately protect our current therapeutic candidates or products or any future therapeutic candidates or products, or fail to otherwise provide us with any competitive advantage. As such, we do not know the degree of future protection that we will have on our therapeutic candidates or products and technology, if any, and a failure to obtain adequate intellectual property protection with respect to our therapeutic candidates or products could have a material adverse impact on our business.
Filing, prosecuting and defending patents throughout the world would be prohibitively expensive, so our policy is to patent technology in jurisdictions with significant or otherwise relevant commercial opportunities or activities. However, patent protection may not be available for some of the therapeutic candidates or products we are found todeveloping. If we must spend significant time and money protecting or enforcing our patents, designing around patents held by others or licensing, potentially for large fees, patents or other proprietary rights held by others, our business, results of operations and financial condition may be harmed.
The patent protection of biotherapeutics is complex and uncertain.
The scope and extent of patent protection for our therapeutic candidates and products are particularly uncertain. To date, our principal therapeutic candidates have been based on specific subpopulations of known and naturally occurring adult stem cells. We anticipate that the therapeutic candidates or products we develop in compliancethe future will continue to include or be based on the extentsame or other naturally occurring stem cells or derivatives or products thereof. Although we have sought and expect to continue to seek patent protection for our therapeutic candidates and products, their methods of our non-compliance is deemed immaterial. If we are found touse, methods of manufacture, and methods of delivery, any or all of them may not be in compliance with any of these laws and regulations, we and, in some cases, our officers and directors may be subject to fines, penalties, criminal sanctionseffective patent protection. Publication of information related to our therapeutic candidates and products by us or others may prevent us from obtaining or enforcing patents relating to these products and therapeutic candidates. Furthermore, others may independently develop similar therapeutic candidates or products, may duplicate our therapeutic candidates or products, or may design around our patent rights. In addition, any of our issued patents may be declared invalid. If we fail to adequately protect our intellectual property, we may face competition from companies who attempt to create a generic therapeutic candidate or product to compete with our therapeutic candidates or products.
Filing, prosecuting and defending patents on therapeutic candidates or products in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own therapeutic candidates or products and further, may export otherwise infringing therapeutic candidates or products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These therapeutic candidates or products may compete with our current or future therapeutic candidates or products, if any, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries do not favor the enforcement of patents, trade secrets and other liability,intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for us to stop the infringement of our patents or marketing of competing therapeutic candidates or products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
We may be unable to adequately prevent disclosure of trade secrets and other proprietary information.
We maintain certain of our proprietary know-how and technological advances as trade secrets, especially where we do not believe patent protection is appropriate or obtainable, including, but not exclusively, with respect to certain aspects of the manufacturing of our therapeutic candidates or products. However, trade secrets are difficult to protect. We take a number of measures to protect our trade secrets including, limiting disclosure, physical security and confidentiality and non-disclosure agreements. We enter into confidentiality agreements with our employees, consultants, outside scientific collaborators, contract manufacturing partners, sponsored researchers and other advisors and third parties to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights. Failure to obtain or maintain trade secret protection, or failure to adequately protect our intellectual property could enable competitors to develop generic products or use our proprietary information to develop other therapeutic candidates or products that compete with our therapeutic candidates or products or cause additional, material adverse effects upon our business, results of operations and financial condition.
We may be forced to litigate to enforce or defend our intellectual property rights, and/or the intellectual property rights of our licensors.
We may be forced to litigate to enforce or defend our intellectual property rights against infringement by competitors, and to protect our trade secrets against unauthorized use. In so doing, we may place our intellectual property at risk of being invalidated, unenforceable, or limited or narrowed in scope and may no longer be used to prevent the manufacture and sale of competitive product. Further, an adverse result in any litigation or other proceedings before government agencies such as the United States Patent and Trademark Office, or the USPTO, may place pending applications at risk of non-issuance. Further, interference proceedings, derivation proceedings, entitlement proceedings, ex parte reexamination, inter partes reexamination, inter partes review, post-grant review, and opposition proceedings provoked by third parties or brought by the USPTO or any foreign patent authority may be used to challenge inventorship, ownership, claim scope, or validity of our patent applications. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential and proprietary information could be compromised by disclosure during this type of litigation.
Intellectual property disputes could cause us to spend substantial resources and distract our personnel from their normal responsibilities.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses and could distract our technical and/or management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the market price of our shares. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of litigation proceedings more effectively than we can because of their greater financial resources and personnel. In addition, the uncertainties associated with litigation could have a material adverse effect on our ability to raise the funds necessary to conduct our clinical trials, continue our internal research programs, in-license needed technology or enter into strategic collaborations that would help us bring our therapeutic candidates to market. As a result, uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
Patent reform legislation and recent court decisions could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law, including provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. The USPTO has and continues to develop and implement regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act. The full effect of these changes is currently unclear as the USPTO has not yet adopted all pertinent final rules and regulations, the courts have yet to address these provisions and the applicability of the Leahy-Smith Act and new regulations on specific patents, including our patents discussed herein, have not been determined and would need to be reviewed. Accordingly, it is not yet clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. As a result, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents, all of which could have a material adverse effect on our business.business and financial condition.
13
Risks Related to Our Financial Results
WeOn June 13, 2013, the U.S. Supreme Court decision in Association for Molecular Pathology v. Myriad Genetics, Inc., held that isolated DNA sequences are an orthopedic medical device company withnot patentable because they constitute a limited operating historyproduct of nature. The Supreme Court did not address stem cells in particular, and our business mayas a result, it is not become profitable.
We are an orthopedic medical device company with a limited operating history. We began commercial sales in 2007. We currentlyyet clear what, if any, impact this Supreme Court decision or future decisions will have on the following nine products with Section 510(k) marketing clearance from the FDA: (1) Cardo Align 360 Posterior-Stabilized Total Knee System; (2) Cardo Align 360 Cruciate Retaining Knee System; (3) Cardo Align 360 Unicompartmental Knee (used in partial knee replacement procedures); (4) Cardo Align 360 Patello-Femoral Replacement (used in partial knee replacement procedures); (5) Cardo Total Hip System (used in total hip replacement procedures); (6) Cardo Bipolar Hip System (two-piece product used in femoral head replacement procedures); (7) Cardo Monopolar Hip System (one-piece product used in femoral head replacement procedures); (8) Cardo Cervical Plate (used in neck fusion procedures); and (9) Cardo Pedicle Screw System (used in lumbar spine fusion procedures).
The successoperation of our business will depend, in part, onbusiness.
If third parties claim that our ability to develop and obtain regulatory clearancestherapeutic candidates or approvals for enhancements toother products infringe upon their intellectual property, commercialization of our productstherapeutic candidates or for planned products which we may be unable to do in a timely manner, or at all. Our success and ability to generate revenue or be profitable also depends on our ability to establish our sales and marketing force, generate product sales and control costs, all of which we may be unable to do. In addition, we may not be successful in our research and development efforts to develop enhancements of these products or to develop new products.
We have a limited history of operations upon which you can evaluate our business, and our operating expenses are increasing. We have yet to demonstrate that we can generate ongoing sufficient salesprofits could be adversely affected.
There is a substantial amount of our products to become profitable. The extent of our future operating losseslitigation, both within and outside the timing of profitability, if at all, are difficult to predict. Our lack of any significant operating history also limits your ability to make a comparative evaluation of us, our products and our prospects. Even if we do achieve profitability as planned, we may not be able to sustain or increase profitability on an ongoing basis.
Our quarterly financial results are likely to fluctuate significantly because our sales prospects are uncertain.
Our quarterly operating results are difficult to predict and may fluctuate significantly from period to period, particularly because our sales prospects are uncertain. These fluctuations also may affect our annual operating results and may cause those results to fluctuate unexpectedly from year to year. The level of our revenues and results of operations at any given time will be based primarily on the following factors:
our ability to increase sales of our products;our ability to develop, manufacture and market new products;results of clinical research and trials on our current or planned products;our ability to obtain regulatory approvals;legislative and reimbursement policy changes affecting the products we may offer or those of our competitors;the variability of the profit margins among the products we sell;our ability to expand and maintain an effective and dedicated sales force;pricing pressure from competitors applicable to our products;adverse third-party reimbursement outcomes;timing of new product launches, acquisitions, licenses or other significant events by us or our competitors;the ability of our manufacturers to timely provide us with an adequate supply of products and meet our quality requirements; andinterruption in the manufacturing or distribution of our products.
For all the foregoing reasons, it will be difficult for us to forecast demand for our products with any degree of certainty. In addition, we will be increasing our operating expenses as we build our commercial capabilities. Accordingly, we may experience significant, unanticipated quarterly losses. Because of these factors, our operating
14
results in one or more future quarters may fail to meet the expectations of securities analysts or investors and may adversely impact the trading price of our common stock.
Risks Related to Our Intellectual Property and Potential Litigation
If we cannot adequately protect our patentsUnited States, involving patent and other intellectual property rights in the biopharmaceutical industry. We may, from time to time, be notified of claims that we may lose market shareare infringing upon patents, trademarks, copyrights, or other intellectual property rights owned by third parties, and we cannot provide assurances that other companies will not, in the future, pursue such infringement claims against us or any third-party proprietary technologies we have licensed. Any such claims could also be expensive and time consuming to defend and divert management’s attention and resources and could delay or prevent us from commercializing our competitors and be unable to operate our business profitably.
therapeutic candidates or products. Our success depends on our ability to protect our proprietary rights to the technologies used in our products. We rely significantly on patent protection, as wellcompetitive position could suffer as a combination of trade secrets, know-how, continuing technological innovations, strategic alliances and licensing opportunities to develop, maintain and strengthen our competitive position. We also expect to pursue a policy of generally obtaining patent protection in both the United States and abroad for patentable subject matter in our proprietary devices and attempt to reviewresult. Although we have reviewed certain third-party patents and patent filings that we believe may be relevant to our therapeutic candidates or products, we have not conducted a freedom-to-operate search or analysis for our therapeutic candidates or products, and we may not be aware of patents or pending or future patent applications that, if issued, would block us from commercializing our therapeutic candidates or products. Thus, we cannot guarantee that our therapeutic candidates or products, or our commercialization thereof, do not and will not infringe any third party’s intellectual property.
From time to time, we have reviewed the extent they become known to develop an effective patent strategy, avoid infringing third-partyclaims of specific patents identify licensing opportunities and monitorowned by third parties. While we have concluded that no claims of any of these patents would be infringed by our products, that all relevant claims would expire before our products would be commercialized, or both, we cannot guarantee that the patent claims of others.owners would not disagree and conclude that our products would infringe these claims.
We have a number of U.S.If we do not obtain patent term extension in the United States under the Hatch-Waxman Act and in foreign patent applications pending in spine, hip and knee reconstructive surgery. Although we have filed these patent applications, we cannot assure you that any patents may issue or that, if they issue, these patents will adequately protect our rights or permit us to gain or keep any competitive advantage.
The U.S. Patent and Trademark Office, or USPTO, may deny or require significant narrowing of claims in our pending patent applications, and patents issued as a result ofcountries under similar legislation, thereby potentially extending the pending patent applications, if any, may not provide us with significant commercial protection or be issued in a form that is advantageous to us. We also could incur substantial costs in proceedings before the USPTO. These proceedings could result in adverse decisions as to the priorityterm of our inventions and the narrowingmarketing exclusivity of our therapeutic candidates or invalidation of claims in any patents that may issue. Any U.S. and foreign patents thatproducts, our business may be issued inmaterially harmed.
Depending on the future could subsequently be successfully challenged by otherstiming, duration and invalidated or rendered unenforceable, which could limit our ability to stop competitors fromspecifics of FDA marketing and selling related products.
Both the patent application process and the process of managing patent disputes can be time-consuming and expensive. Competitors may be able to design around our patents or develop products that provide outcomes that are comparable to our products. Although we have entered into confidentiality agreements and intellectual property assignment agreements with certainapproval of our employees, consultants and advisors astherapeutic candidates or products, if any, one of the waysU.S. patents covering each of such approved therapeutic candidate or product or the use thereof may be eligible for up to five years of patent term restoration under the Hatch-Waxman Act. The Hatch-Waxman Act allows a maximum of one patent to be extended per FDA approved product. Patent term extension also may be available in certain foreign countries upon regulatory approval of our therapeutic candidates, including by the EMA in the European Union or the Pharmaceutical and Medical Devices Agency in Japan. Nevertheless, we seek to protect our intellectual property and other proprietary technology, these agreements may not be enforceable or may not provide meaningful protection for our trade secrets or other proprietary informationgranted patent term extension either in the event of unauthorized use or disclosure or other breaches of the agreements. Furthermore, the laws of some foreign countries may not protect our intellectual property rights to the same extent as the laws of the United States if at all. Since mostor in any foreign country because of, our pending patent applications are for example, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. Moreover, the United States only, we lack a correspondingterm of extension, as well as the scope of patent protection in other countries. Thus,during any such extension, afforded by the governmental authority could be less than we request. In addition, if a patent we wish to extend is owned by another party and licensed to us, we may not be ableneed to stop a competitorobtain approval and cooperation from marketing products in other countries thatour licensor to request the extension.
If we are similarunable to someobtain patent term extension or restoration, or the term of our products.
Changes to intellectual property laws may negatively impact our ability to protect our intellectual property.
There are numerous proposed changes toany such extension is less than we request, the patent laws and rules of the USPTO,period during which if enacted, maywe will have a significant impact on our ability to protect our technology and enforce our intellectual property rights. For example, proposed changes to the patent rules of the USPTO were scheduled to take effect on November 1, 2007, which would have limited significantly the right to pursue continuation applications. On October 31, 2007, a temporary injunction was granted in a lawsuit against the USPTO which served to stay the applicationexclusively market our therapeutic candidates or products will be shortened and our competitors may obtain approval of the proposed rules. On April 1, 2008, the court issued its ruling that the proposedcompeting products following our patent rules were void, thus making the injunction permanent. If the ruling is successfully appealed, the proposed rules may take effectexpiration, and may adversely impact our ability to prevent others from designing around our existing patents.
Moreover, Congress is considering several significant changes to the U.S. patent laws, including changing from a "first to invent" to a "first inventor to file" system, requiring that patent lawsuits be brought in the forum of the defendant, requiring the apportionment of patent damages, and creating a post-grant opposition process to challenge patents after they have issued.
15
The medical device industry is characterized by patent and other intellectual property litigation, and we could become subject to litigation thatrevenue could be costly, result in diverting management's time and efforts, requirereduced, possibly materially.
Our reliance on third parties requires us to pay damages, and/or prevent us from marketingshare our existing or future products.
The medical device market in which we primarily participate is in large part technology-driven. Physician customers move quickly to new products and new technologies. As a result, intellectual property rights, particularly patents and trade secrets, playwhich increases the possibility that a significant role in product development and differentiation. However, intellectual property litigation to defendcompetitor will discover them or create market advantage is inherently complex, unpredictable, time-consuming and costly. Furthermore, appellate courts frequently overturn lower court patent decisions.
In addition, competing parties frequently file multiple suits to leverage patent portfolios across product lines, technologies and geographies and to balance risk and exposure between the parties. In some cases, several competitors are parties in the same proceeding, or in a series of related proceedings, or litigate multiple features of a single class of medical devices. These forces frequently drive settlement not only of individual cases, but also of a series of pending and potentially related and unrelated cases. In addition, although monetary and injunctive relief is typically sought, remedies and restitution generally are not determined until the conclusion of the proceedings and are frequently modified on appeal. Accordingly, the outcomes of individual cases are difficult to time, predict or quantify and are often dependent upon the outcomes of other cases in other geographies.
Certain product categories, including pedicle screws, have been subject to significant patent litigation in recent years. Since we sell orthopedic and spinal devices, such as pedicle screws, knee replacement devices, and cervical plates, and we recently introducedthat our Accin pedicle screw system, any related litigation could harm our business.
We also may have to take legal action in the future to protect our patents, trade secrets or know-how or to assert them against claimed infringement by others. Any legal action of that type could be costly and time-consuming, and we cannot assure you that any lawsuit will be successful. In addition,misappropriated or disclosed.
Because we may not have sufficient resources to enforcerely on third parties for manufacturing, and because we collaborate with various organizations and academic institutions on the advancement of our intellectual property rights or to defend our patents against a challenge.
Further,clinical trials, we intendmust, at times, share trade secrets with them. We seek to protect our proprietary technology in part through proprietary informationby entering into confidentiality agreements and, inventionsif applicable, material transfer agreements, consulting agreements or other similar agreements with our advisors, employees, third-party contractors and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, including our trade secrets. Despite the contractual provisions employed when working with third parties, the need to share trade secrets and other parties. These agreements with someconfidential information increases the risk that such trade secrets become known by our competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our employeesproprietary position is based, in part, on our know-how and consultants generally contain standard provisions requiring those individuals to assign to the employer, without additional consideration, inventions conceived or reduced to practice by them while employed or retained by the employer, subject to customary exceptions. If any of our employees, consultants or others breach these agreements, or if these agreements are found to be unenforceable, competitors may learntrade secrets, a competitor’s discovery of our trade secrets or other unauthorized use or disclosure would impair our competitive position and proprietary information.
For the reasons indicated above, enforcing our intellectual property rights may be costly, difficult and time-consuming. Even if successful, litigation to enforce our intellectual property rights or to defend our patents against challenge could be expensive and time-consuming and could divert our management's attention.
Patent infringement lawsuits brought against us could have a material adverse affecteffect on our commercialbusiness.
In addition, these agreements typically restrict the ability of our advisors, employees, third-party contractors and consultants to publish data potentially relating to our trade secrets, although our agreements may contain certain limited publication rights. For example, any academic institution that we may collaborate with in the future will usually expect to be granted rights to publish data arising out of such collaboration, provided that we are notified in advance and given the opportunity to delay publication for a limited time period in order for us to secure patent protection of intellectual property rights arising from the collaboration, in addition to the opportunity to remove confidential or trade secret information from any such publication. In the future we may also conduct joint research and development programs that may require us to share trade secrets under the terms of our research and development partnerships or similar agreements. Despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of our agreements with third parties, independent development or publication of information by any of our third-party collaborators. A competitor’s discovery of our trade secrets would impair our competitive position and have an adverse impact on our business.
Risks Related to Our Common Stock
An active trading market may not develop for our securities and you may not be able to sell your common stock or warrants at or above the offering price per share or the warrant exercise price per share.
Our common stock is currently quoted on the OTCQB Marketplace and there is not any significant trading activity in our common stock or market for shares of our common stock. We have applied to list our common stock and warrants on the Nasdaq Capital Market under the symbols “BCDA” and “BCDAW,” respectively. There can be no assurance that we will be successful in listing our common stock and/or our warrants on the Nasdaq Capital Market. Even if our common stock and warrants are listed on the Nasdaq Capital Market, we cannot predict the extent to which investor interest in our company will lead to the development of any active trading market in our common stock and/or warrants or how liquid the market for our common stock and/or warrants might become. If a market does not develop or is not sustained it may be difficult for you to sell your securities at the time you wish to sell them, at a price that is attractive to you, or at all. You may not be able to sell your common stock or warrants at or above the offering price or warrant exercise price per share.
The market price and trading volume of our securitiesmay be volatile and may be affected by economic conditions beyond our control.
The market price of our securities is likely to be volatile. Some specific factors that could negatively affect the price of our securities or result in fluctuations in its price and trading volume include:
results of clinical trials of our therapeutic candidates;
results of clinical trials of our competitors’ products;
regulatory actions with respect to our therapeutic candidates or products or our competitors’ products;
actual or anticipated fluctuations in our quarterly operating results or those of our competitors;
publication of research reports by securities analysts about us or our competitors in the industry;
our failure or the failure of our competitors to meet analysts’ projections or guidance that we or our competitors may give to the market;
issuances by us of debt or equity securities;
litigation involving our company, including stockholder litigation; investigations or audits by regulators into the operations of our company; or proceedings initiated by our competitors or clients;
strategic decisions by us or our competitors, such as acquisitions, divestitures, spin-offs, joint ventures, strategic investments or changes in business strategy;
the passage of legislation or other regulatory developments affecting us or our industry; fluctuations in the valuation of companies perceived by investors to be comparable to us;
trading volume of our common stock and warrants;
sales or perceived potential sales of our common stock and/or warrants by us, our directors, senior management or our stockholders in the future;
short selling or other market manipulation activities;
announcement or expectation of additional financing efforts;
terrorist acts, acts of war or periods of widespread civil unrest;
natural disasters and other calamities;
changes in market conditions for biopharmaceutical stocks; and
conditions in the U.S. financial markets or changes in general economic conditions.
If securities or industry analysts do not publish research or reports about our business, or if they issue an adverse or misleading opinion regarding our stock, the price and trading volume of our securities could decline.
The trading market for our securities will be influenced by the research and reports that industry or securities analysts publish about us or our business. We do not currently have and may never obtain research coverage by securities and industry analysts. If no or few securities or industry analysts commence coverage of us, the trading price for our securities would be negatively impacted. In the event we obtain securities or industry analyst coverage, if any of the analysts who cover us issue an adverse or misleading opinion regarding us, our business model, our intellectual property or our stock performance, or if our clinical trials and operating results fail to meet the expectations of analysts, the price of our securities would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, we could lose visibility in the financial markets, which in turn could cause the price or trading volume of our securities to decline.
Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
As of June 30, 2019, our executive officers, directors, 5% stockholders and their affiliates beneficially own approximately 54.9% of our voting stock. Therefore, these stockholders will have the ability to influence us through this ownership position. These stockholders may be able to determine all matters requiring stockholder approval. For example, these stockholders, acting together, may be able to control elections of directors, amendments of our organizational documents, or approval of any merger, sale of assets, or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our Common stock that you may believe are in your best interest as one of our stockholders.
Our financial controls and procedures may not be sufficient to accurately or timely report our financial condition or results of operations, which may adversely affect investor confidence in us and, as a result, the value of our common stock.
As a public company, we are required to maintain internal control over financial reporting and to report any material weaknesses in such internal controls. Section 404 of the Sarbanes-Oxley Act requires that we evaluate and determine the effectiveness of our internal control over financial reporting and provide a management report on internal control over financial reporting.
The effectiveness of our controls and procedures may in the future be limited by a variety of factors, including:
faulty human judgements and simple errors, omissions or mistakes;
fraudulent actions of an individual or collusion of two or more people;
inappropriate management override of procedures; and
the possibility that any enhancements to controls and procedures may still not be adequate to assure timely and accurate financial information.
If we identify material weaknesses in our internal control over financial reporting in the future, if we are unable to comply with the requirements of Section 404 in a timely manner, if we are unable to assert that our internal control over financial reporting is effective, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our Common Stock could be adversely affected, and we could become subject to investigations by the stock exchange on which our securities are listed, the SEC, or other regulatory authorities, which could require additional financial and management resources.
As a smaller reporting company, we are subject to scaled disclosure requirements that may make it more challenging for investors to analyze our results of operations and financial prospects.
Currently, we are a “smaller reporting company,” as defined by Rule 12b-2 of the Exchange Act. As a “smaller reporting company,” we are able to provide simplified executive compensation disclosures in our filings and have certain other decreased disclosure obligations in our filings with the SEC, including being required to provide only two years of audited financial statements in annual reports. Consequently, it may be more challenging for investors to analyze our results of operations and financial prospects.
Furthermore, we are a non-accelerated filer as defined by Rule 12b-2 of the Exchange Act, and, as such, are not required to provide an auditor attestation of management’s assessment of internal control over financial reporting, which is generally required for SEC reporting companies under Section 404(b) of the Sarbanes-Oxley Act. Because we are not required to, and have not, had our auditor’s provide an attestation of our management’s assessment of internal control over financial reporting, a material weakness in internal controls may remain undetected for a longer period.
We may be exposed to additional risks as a result of our reverse merger transaction.
We may be exposed to additional risks as a result of our “reverse merger” transaction and rules and regulations relating to shell companies or former shell companies. There has been increased focus in recent years by government agencies on transactions such as the reverse merger transaction, and we may be subject to increased scrutiny and/or restrictions by the SEC and other government agencies and holders of our securities as a result of the completion of that transaction. This may make it more difficult for us to obtain coverage from securities analysts of major brokerage firms. The occurrence of any such event could cause our business or stock price to suffer.
Our annual and quarterly operating results may fluctuate significantly or may fall below the expectations of investors or securities analysts, each of which may cause our stock price to fluctuate or decline.
We expect our operating results to be subject to annual and quarterly fluctuations. Our net loss and other operating results will be affected by numerous factors, including:
variations in the level of expenses related to our therapeutic candidates, products or future development programs;
if any of our therapeutic candidates receives regulatory approval, the level of underlying demand for these therapeutic candidates and wholesalers’ buying patterns;
addition or termination of clinical trials or funding support;
our execution of any collaborative, licensing or similar arrangements, and the timing of payments we may make or receive under these arrangements;
any intellectual property infringement lawsuit in which we may become involved;
regulatory developments affecting our therapeutic candidates or products or those of our competitors;
the timing and cost of, and level of investment in, research and development activities relating to our therapeutic candidates, which may change from time to time;
our ability to attract, hire and retain qualified personnel;
expenditures that we will or may incur to acquire or develop additional therapeutic candidates and technologies;
future accounting pronouncements or changes in our accounting policies;
the timing and success or failure of clinical studies for our therapeutic candidates or competing product candidates, or any other change in the competitive landscape of our industry, including consolidation among our competitors or partners;
the risk/benefit profile, cost and reimbursement policies with respect to our therapeutic candidates, if approved, and existing and potential future therapies or biologics that compete with our products or therapeutic candidates; and
the changing and volatile U.S., European and global economic environments.
If our annual or quarterly operating results fall below the expectations of investors or securities analysts, the price of our securities could decline substantially. Furthermore, any annual or quarterly fluctuations in our operating results may, in turn, cause the price of our stock to fluctuate substantially. We believe that annual and quarterly comparisons of our financial results are not necessarily meaningful and should not be relied upon as an indication of our future performance.
Raising additional funds through debt or equity financing could be dilutive and may cause the market price of our Common Stock to decline.
To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest may be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a stockholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take certain actions, such as incurring debt, making capital expenditures or declaring dividends. If we raise additional funds through collaborations, strategic collaborations or partnerships, or marketing, distribution or licensing arrangements with third parties, we may be required to limit valuable rights to our intellectual property, technologies, therapeutic candidates or future revenue streams, or grant licenses or other rights on terms that are not favorable to us. Furthermore, any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and sellcommercialize our products and to operate profitably.therapeutic candidates.
As theSales of a substantial number of entrants intoshares of our Common Stock in the public market increases, the possibilitycould cause our stock price to fall.
Sales of a patent infringement claim against us grows. While we make an effortsubstantial number of shares of our common stock in the public market or the perception that these sales might occur, could depress the market price of our common stock and could impair our ability to ensureraise capital through the sale of additional equity securities. We are unable to predict the effect that sales may have on the prevailing market price of our products do not infringe other parties' patentscommon stock.
Future sales and proprietary rights,issuances of our products and methods may be covered by patents held by our competitors. In addition, our competitors may assert that future products we may market infringe their patents.
A patent infringement suit brought against uscommon stock or any strategic partners or licensees may force us or any strategic partners or licensees to stop or delay developing, manufacturing or selling potential products that are claimed to infringe a third party's intellectual property, unless that party grants us or any strategic partners or licensees rights to use its intellectual property. In those cases, we may be requiredpurchase our common stock, including pursuant to obtain licenses to patents or proprietary rightsour equity incentive plans, could result in additional dilution of others in order to continue to commercialize our products. However, we may not be able to obtain any licenses required under any patents or proprietary rights of third parties on acceptable terms, or at all, and any licenses may require substantial royalties or other payments by us. Even if we, any strategic partners or licensees were able to
16
obtain rights to the third party's intellectual property, these rights may be non-exclusive, thereby giving our competitors access to the same intellectual property. Ultimately, we may be unable to commercialize somepercentage ownership of our potential products or may havestockholders and could cause our stock price to cease some of our business operations as a result of patent infringement claims, which could severely harm our business.fall.
We mayexpect that significant additional capital will be subject to damages resulting from claims that we or our employees or consultants have wrongfully used or disclosed alleged trade secrets of their former employers.
Some of our employees and consultants were previously employed or engaged at universities or other medical device companies, including our competitors or potential competitors. We couldneeded in the future be subject to claims that these employees and consultants, orcontinue our planned operations. To the extent we have inadvertently or otherwise used or disclosed trade secretsraise additional capital by issuing equity securities, our stockholders may experience substantial dilution. We may sell our common stock, convertible securities or other proprietary information of their former employers. Litigationequity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell our common stock, convertible securities or other equity securities in more than one transaction, investors may be necessarymaterially diluted by subsequent sales. These sales may also result in material dilution to defendour existing stockholders, and new investors could gain rights superior to our existing stockholders.
We are at risk of securities class action litigation.
In the past, securities class action litigation has often been brought against these claims.a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology companies have experienced significant stock price volatility in recent years. If we fail to defend against these claims, a court could order us to pay substantial damages and prohibit us from using technologies or features that are essential to our products and processes, if these technologies or features are found to incorporate or be derived from the trade secrets or other proprietary information of the former employers. In addition, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper or prevent our ability to commercialize certain potential products, which could severely harm our business. Even if we are successful in defending against these claims,face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be a distractionlimited.
We have incurred substantial losses during our history and do not expect to management.
Fluctuationsbecome profitable in the costnear future and availability of insurance could adversely affect our profitability or our risk management profile.
We hold a number of insurance policies, including product liability insurance, errors and omissions insurance, directors' and officers' liability insurance, property insurance, general liability insurance, employee benefits liability and workers' compensation insurance. If the costs of maintaining adequate insurance coverage increases significantly at any time, our operating results could be materially adversely impacted. Likewise, if anywe may never achieve profitability. None of our current insurance coverage should become unavailable to us or become economically impractical, we would be required to operate our business without indemnity from commercial insurance providers.
Potential future product liability claims and other litigation, including contract litigation, may adversely affect our business, reputation and ability to attract and retain customers.
Reconstructive and spine surgery involves a high risk of serious complications, including bleeding, nerve injury, paralysis and even death. As a result, we are exposed to potential product liability claims that are inherent inpre-Merger tax attributes remain available after the testing, manufacture and sale of medical devices for surgery procedures. Many of these medical devices are designed to be implanted in the human body for long periods of time or indefinitely. A number of factors could result in an unsafe condition or injury to, or death of, a patient with respect to these or other products that we manufacture or sell, including component failures, manufacturing flaws, design defects or inadequate disclosure of product-related risks or product-related information. These factors could result in product liability claims, a recall of one or more products or a safety alert relating to one or more products. Product liability claims may be brought by individuals or by groups seeking to represent a class.
In connection with our acquisition of the assets of Accin Corporation ("Accin") in May 2007 (through our ownership of Accelerated Innovation ("Accelerated Innovation"), one of our former subsidiaries) andMerger as a result of the reverse merger we completed in August 2008 (the "Merger"), we assumed the responsibility for any litigation or claims relatedlimitations Section 382 due to Accin'slack of business including product liability claims relating to products previously sold by Accin. The outcome of litigation, particularly class action lawsuits, is difficult to assess or quantify. Plaintiffs in these lawsuits often seek recovery of very large or indeterminate amounts, including not only actual damages, but also punitive damages. The magnitude of the potential loss relating to these lawsuits may remain unknown for substantial periods of time. In addition, the cost to defend against any future litigation may be significant.
Any product liability claim brought against us, with or without merit, could result in the increase of our insurance rates or the inability to secure coverage in the future. In addition, if our product liability insurance proves to be inadequate to pay a damage award, we may have to pay the excess out of our cash reserves, which may harm our financial condition. If longer-term patient results and experience indicate that our products or any component cause
17
tissue damage, motor impairment or other adverse effects, we could be subject to significant liability. Finally, even a meritless or unsuccessful product liability claim could harm our reputation in the industry, lead to significant legal fees and result in diverting management's attention from managing our business.
Even if any product liability loss is covered by an insurance policy, these policies have substantial retentions or deductibles that provide that insurance proceeds are not recoverable until the losses incurred exceed the amount of those retentions or deductibles.continuity. To the extent that we continue to generate taxable losses, unused losses will carry forward to offset future taxable income, if any, until such unused losses expire. Losses generated after 2017 do not have an expiration date. Under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an “ownership change,” generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period, the corporation’s ability to use its pre-change net operating loss carryforwards, or NOLs, and other pre-change tax attributes (such as research tax credits) to offset its post-change income or taxes may be limited. Our prior equity offerings and other changes in our stock ownership may have resulted in ownership changes. We have not performed an analysis to assess whether an ownership change has occurred. If we have experienced an ownership change at any time since our formation, utilization of our net operating loss carryforwards would be subject to an annual limitation under Section 382 of the Code. In addition, we may experience ownership changes in the future as a result of subsequent shifts in our stock ownership, some of which are below these retentionsoutside of our control. As a result, if we earn net taxable income, our ability to use our pre-change net operating loss carryforwards to offset U.S. federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us. In addition, at the state level, there may be periods during which the use of NOLs is suspended or deductibles, we will be responsible for paying these losses. Paying retentionsotherwise limited, which could accelerate or deductibles for a significant amount of claims couldpermanently increase state taxes owed.
Recent U.S. tax legislation and future changes to applicable U.S. or foreign tax laws and regulations may have a material adverse effect on our business, financial condition and results of operations.
Further, it is possible that we may in the future be substantially self-insured with respect to general and product liability claims. As a result of economic factors currently impacting the insurance industry, meaningful product liability insurance coverage also may become unavailable due to its economically prohibitive cost. The absence of significant third-party insurance coverage increases potential exposure to unanticipated claims and adverse decisions. As a result, product liability claims, product recalls and other litigation in the future, regardless of their outcome, could have a material adverse effect on our financial position, results of operations or liquidity.
Any claims relating to our making improper payments to physicians for consulting services, or other potential violations of regulations governing interactions between us and healthcare providers, could be time-consuming and costly.
Our relationship with surgeons, hospitals and the marketers of our productsWe are subject to scrutiny under various stateincome and federal anti-kickback, self-referral, false claims and similar laws, often referred to collectively as healthcare fraud and abuse laws. The federal anti-kickback laws prohibit unlawful inducements for the referral of business reimbursable under federally-funded health care programs, such as remuneration provided to physicians to induce them to use certain medical devices reimbursable by Medicare or Medicaid. Healthcare fraud and abuse laws are complex and subject to evolving interpretations, and even minor, inadvertent violations potentially can give rise to claims that the relevant law has been violated. Certain states in which we market our products have similar anti-kickback, anti-fee splitting and self-referral laws, imposing substantial penalties for violations. Any violations of these laws could result in a material adverse effect on the market price of our common stock, as well as our business, financial condition and results of operations. We cannot assure you that any of the healthcare fraud and abuse laws will not change or be interpretedother taxes in the futureU.S. Changes in a manner which restricts or adversely affects our business activities or relationships with surgeons, hospitals and marketers of our products. In addition, possible sanctions for violating these anti-kickback laws include monetary fines, civil and criminal penalties, exclusion from Medicare and Medicaid programs and forfeiture of amounts collected in violation of these prohibitions.
Federal anti-kickback laws and regulations prohibit any knowing and willful offer, payment, solicitationpolicy relating to taxes or receipt of any form of remuneration bytrade may have an individual or entity in return for, or to induce:
the referral of an individual for a service or product for which payment may be made by Medicare, Medicaid or other government-sponsored healthcare program; orpurchasing, leasing, ordering or arranging for any service or product for which payment may be made by a government-sponsored healthcare program.
We must comply with a variety of other laws, such as laws prohibiting false claims for reimbursement under Medicare and Medicaid, which also can be triggered by violations of federal anti-kickback laws; Healthcare Insurance Portability and Accountability Act of 1996, which protects the privacy of individually identifiable healthcare information; and the Federal Trade Commission Act and similar laws regulating advertisement and consumer protections. In certain cases, federal and state authorities pursue actions for false claims on the basis that manufacturers and distributors are promoting unapproved or off-label uses of their products.
Pursuant to FDA regulations, we can market our products only for cleared or approved uses. Although surgeons are permitted to use medical devices for indications other than those cleared or approved by the FDA based on their medical judgment, we are prohibited from promoting products for those off-label uses. We market our products and provide promotional materials and training programs to surgeons regarding the use of our products. Although we believe our marketing, promotional materials and training programs for surgeons do not constitute promotion of
18
unapproved uses of our products, if it is determined that our marketing, promotional materials or training programs constitute promotion of unapproved uses, we could be subject to significant fines in addition to regulatory enforcement actions, including the issuance of a warning letter, injunction, seizure and criminal penalty.
The scope and enforcement of these laws is uncertain and subject to rapid change, especially in light of the lack of applicable precedent and regulations. We cannot assure you that federal or state regulatory authorities will not challenge or investigate our current or future activities under these laws. Any challenge or investigation could have a material adverse effect on our business, financial condition and results of operations. Any state or federal regulatory review of us, regardlessFor example, the U.S. government recently enacted significant tax reform, and certain provisions of the outcome, wouldnew law may adversely affect us. Changes include, but are not limited to, a federal corporate tax rate decrease from 34% to 21% for tax years beginning after December 31, 2017, the transition of U.S. international taxation from a worldwide tax system to a more generally territorial system, and a one-time transition tax on the mandatory deemed repatriation of foreign earnings. The legislation is unclear in many respects and could be costlysubject to potential amendments and time-consuming. Additionally, we cannot predicttechnical corrections and will be subject to interpretations and implementing regulations by the impactTreasury and Internal Revenue Service, any of anywhich could mitigate or increase certain adverse effects of the legislation. In addition, it is unclear how these U.S. federal income tax changes will affect state and local taxation. Generally, future changes in theseapplicable U.S. or foreign tax laws and whetherregulations, or their interpretation and application could have an adverse effect on our business, financial conditions and results of operations. Changes with respect to the transition to a territorial tax system are generally expected to have little impact given our lack of foreign operations.
We do not theyintend to pay dividends on our common stock so any returns will be retroactive.limited to the value of our stock.
We have never declared or paid any cash dividends on our common stock. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. Any return to stockholders will therefore be limited to the appreciation of their stock.
Risks Related to Ownershipthis Offering
Resales of Our Common Stockour common stock, including the common stock issuable upon exercise of the warrants being offered in this Offering, in the public market by our stockholders as a result of this Offering may cause the market price of our common stock to fall.
Sales of a substantial number of shares of our common stock, including the shares of common stock issuable upon exercise of the warrants being offered in this Offering, in the public market could occur at any time. The issuance of new shares of our common stock, including the shares of common stock issuable upon exercise of the warrants being offered in this Offering, could result in resales of our common stock by our current stockholders concerned about the potential ownership dilution of their holdings. In turn, these resales could have the effect of depressing the market price for our common stock and consequently our warrants.
There may be future sales or other dilution of our equity, which may adversely affect the market price of our common stock.
Our
If our existing stockholders sell, or indicate an intention to sell, substantial amounts of our common stock in the public market after applicable legal restrictions on resale and the lock-up agreements, the trading price of our stock could decline. As of March 31, 2019, we had approximately 4,847,471 shares of common stock outstanding. Substantially all of such shares of common stock may be thinly traded.
Theresold in the public market. If substantial additional shares are sold, or if it is a very minimal public market for our common stock. We cannot predict how liquid the market for our common stock might become. Our common stock will likely be thinly traded compared to larger more widely known companies.
Trades of our common stock are conducted on the OTC Bulletin Board. We anticipate applying for listing of our common stock on NYSE AMEX LLC. We cannot ensureperceived that wethey will be able to satisfysold, in the listing standards of the NYSE AMEX LLC or that our common stock will be accepted for listing. Should we fail to satisfy the initial listing standards of the NYSE AMEX LLC, or our common stock is otherwise rejected for listing and remains listed on the OTC Bulletin Board or suspended from the OTC Bulletin Board,public market, the trading price of our common stock, and consequently our warrants, could suffer,decline.
You may experience future dilution as a result of future equity offerings.
In order to raise additional capital, we may in the trading marketfuture offer additional shares of our common stock or other securities convertible into or exchangeable for our common stock may be less liquid andthat could result in further dilution to investors purchasing our common stock price may be subject to increased volatility.
Furthermore, for companies whose securities are traded in the OTC Bulletin Board, it is more difficult to obtain accurate stock quotationsthis Offering or needed capital. Also, because major wire services generally do not publish press releases about these companies, it is also more difficult for them to obtain coverage for significant news and events.
In addition, the price at which our common stock may be sold is very unpredictable because there could be very few tradesresult in our common stock. We cannot predict the extent to which an active public market for our common stock will develop or be sustained at any time in the future. If our common stock is thinly traded, a large block of shares traded can lead to a dramatic fluctuation in the share price.
We expect thatdownward pressure on the price of our common stock and the warrants. We may sell shares of our common stock or other securities in any other offering at prices that are higher or lower than the prices paid by investors in this Offering, and investors purchasing shares or other securities in the future could have rights superior to existing stockholders.
Our management will fluctuate substantially, potentially adversely affectinghave broad discretion over the use of the net proceeds from this Offering, you may not agree with how we use the proceeds and the proceeds may not be invested successfully.
We have not designated any portion of the net proceeds from this Offering to be used for any particular purpose. Accordingly, our management will have broad discretion as to the use of the net proceeds from this Offering and could use them for purposes other than those contemplated at the time of commencement of this Offering. Accordingly, you will be relying on the judgment of our management with regard to the use of these net proceeds, and you will not have the opportunity, as part of your investment decision, to assess whether the proceeds are being used appropriately. It is possible that, pending their use, we may invest the net proceeds in a way that does not yield a favorable, or any, return for our company.
Even if this Offering is successful, we may need to raise additional funding to complete the development and commercialization of our product candidates. Additional financing may not be available on acceptable terms, or at all. Failure to obtain additional capital may force us to delay, limit, or terminate our product development efforts or other operations.
We estimate that our current cash and cash equivalents, along with the net proceeds from this Offering, will be sufficient for us to fund our operating expenses and capital expenditure requirements through the first half of 2020. Without giving effect to the anticipated net proceeds from this Offering, our existing capital resources are not sufficient to meet our projected operating requirements through the third quarter of 2019. This raises substantial doubt about our ability to continue as a going concern beyond one year from the date of investorsour unaudited consolidated financial statements for the three months ended March 31, 2019. The net proceeds from this Offering may remove such doubt regarding our ability to sell their shares.continue as a going concern. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect. In addition, the expected net proceeds of this Offering may not be sufficient for us to fund any of our product candidates through regulatory approval, and we may need to raise substantial additional capital to complete the development and commercialization of our product candidates. We may continue to seek funds through equity or debt financings, collaborative or other arrangements with corporate sources, or through other sources of financing. Additional funding may not be available to us on acceptable terms, or at all. Any failure to raise capital as and when needed, as a result of insufficient authorized shares or otherwise, could have a negative impact on our financial condition and on our ability to pursue our business plans and strategies.
You may experience immediate and substantial dilution in the book value per share of the common stock you purchase.
The public offering price per Unit is substantially higher than the net tangible book value per share of our common stock. Therefore, if you purchase securities in this Offering, you will pay an effective price per share of common stock you acquire that substantially exceeds our net tangible book value per share after this Offering. Assuming no exercise of the warrants being offered in this Offering, no value is attributed to such warrants and such warrants are classified as and accounted for as equity, you will experience immediate dilution of $ per share, representing the difference between our as adjusted net tangible book value per share after giving effect to this Offering and the public offering price per Unit. In addition, if previously issued options to acquire common stock are exercised at prices below the offering price or the accompanying warrants being offered in this Offering are accounted for as liabilities, you will experience further dilution. See “Dilution” for a more detailed discussion of the dilution you may incur in connection with this Offering.
This Offering may cause the trading price of our common stock to decrease.
The Unit price, together with the number of shares of common stock we propose to issue and ultimately will issue if this Offering is completed, may result in an immediate decrease in the market price of our common stock. This decrease may continue after the completion of this Offering. We cannot predict the effect, if any, that the availability of shares for future sale represented by the warrants issued in connection with this Offering will have on the market price of our common stock is likelyfrom time to be highly volatiletime. Further, if a substantial number of Units are purchased and subject to wide fluctuations in response to the following factors, manyholders of which are generally beyond our control. These factors may include:
volume and timingthe shares received upon exercise oforders for our products;- the
introduction of new products or product enhancements by us or our competitors; quarterly variations in our or our competitor's results of operations;announcements of technological or medical innovations for treating spine, knee and hip pathologies;our ability to develop, obtain regulatory clearance or approval for, and market new and enhanced products on a timely basis;changes in governmental regulations or in the status of our regulatory approvals, clearances or applications, including announcements of actions by the FDA or other regulatory agencies;changes in the availability of third-party reimbursement in the United States or other countries;the acquisition or divestiture of businesses, products, assets or technology;disputes, litigation or other developments with respect to intellectual property rights or other potential legal actions;sales of large blocks of our common stock, including sales by our executive officers and directors;
19
changes in earnings estimates or recommendations by securities analysts; andgeneral market conditions and other factors, including factors unrelated to our operating performance or the operating performance of our competitors.
Market price fluctuations may negatively affect the ability of investorsrelated warrants choose to sell oursome or all of the shares at consistent prices.
Securities analysts may elect not to report on our common stock or may issue negative reports that adversely affectunderlying the warrants, the resulting sales could depress the market price of our common stock.
At this time, no securities analyst provides research coverage of our common stock. Further, securities analysts may never provide this coverage in the future. Rules mandated by the Sarbanes Oxley Act of 2002 and other restrictions led to a number of fundamental changes in how analysts are reviewed and compensated. In particular, many investment banking firms are required to contract with independent financial analysts for their stock research. It may remain difficult for a company with a small market capitalization such as ours to attract independent financial analysts that will cover our common stock. If securities analysts do not cover our common stock, the lack of research coverage may adversely affect our actual and potential market price and trading volume.
The trading market for our common stock may be affected in part by the research and reports that industry or financial analysts publish about our business. If one or more analysts elect to cover our Company and then downgrade the stock, the stock price may decline rapidly. If one or more of these analysts cease coverage of our Company, we could lose visibility in the market, which in turn could cause our stock price to decline. This could have a negative effect on the market price of our shares.
Anti-takeover provisions in our charter documents and Delaware law may discourage or prevent a change in control, even if an acquisition would be beneficial to our stockholders, which could adversely affect our stock price and prevent attempts by our stockholders to replace or remove our current management.
Our Certificate of Incorporation and Bylaws contain provisions that could delay or prevent a change in control of our Company or changes in our Board of Directors that our stockholders might consider favorable. Some of these provisions:
impose limitations on our stockholders to call special stockholder meetings; andauthorize the issuance of preferred stock which can be created and issued by the Board of Directors without prior stockholder approval, with rights senior to those of the common stock.
In addition, we are subject to the provisions of Section 203 of the Delaware General Corporation Law, which may prohibit certain business combinations with stockholders owning 15% or more of our outstanding voting stock. These and other provisions in our Certificate of Incorporation, our Bylaws and Delaware law could make it more difficult for stockholders or potential acquirers to obtain control of our Board of Directors or initiate actions that are opposed by our then-current Board of Directors, including to delay or impede a merger, tender offer or proxy contest involving our Company. Any delay or prevention of a change in control transaction or changes in our Board of Directors could cause the market price of our common stock to decline.
● | impose limitations on our stockholders to call special stockholder meetings; and |
Because our common stock may be a "penny stock," it may be more difficult for investors to sell shares of our common stock, and the market price of our common stock may be adversely affected.
● | authorize the issuance of preferred stock which can be created and issued by the Board of Directors without prior stockholder approval, with rights senior to those of the common stock. |
Our common stock may be a "penny stock" if, among other things, the stock price is below $5.00 per share, we are not listed for trading on a national securities exchange or approved for quotation on the Nasdaq Stock Market or any other national stock exchange, or we have not met certain net tangible asset or average revenue requirements. Broker-dealers who sell penny stocks must provide purchasers of these stocks with a standardized risk-disclosure document prepared by the SEC. This document provides information about penny stocks and the nature and level of risks involved in investing in the penny-stock market. A broker also must give a purchaser, orally or in writing, bid and offer quotations and information regarding broker and salesperson compensation, make a written determination that the penny stock is a suitable investment for the purchaser, and obtain the purchaser's written agreement to the purchase. In addition, broker-dealers must provide customers that hold penny stock in their accounts with that broker-dealer a monthly statement containing price and market information relating to the penny stock. If a penny
20
stock is sold to an investor in violation of the penny stock rules, the investor may be able to cancel its purchase and get its money back.
If applicable, the penny stock rules may make it difficult for investors to sell their shares of our common stock. Because of the rules and restrictions applicable to a penny stock, there is less trading in penny stocks and the market price of our common stock may be adversely affected. Also, many brokers choose not to participate in penny stock transactions. Accordingly, investors may not always be able to resell their shares of our common stock publicly at times and prices that they feel are appropriate.
A significant number of shares are eligible for resale by our stockholders and the sale of those shares could adversely affect the stock price.
A number of our outstanding shares of common stock are eligible for resale by our stockholders. Furthermore, a significant number of additional shares will become eligible for resale within the next nine months, or sooner, if the Company elects to waive lock-up restrictions applicable to such shares. If our stockholders whose shares are, or hereafter become, eligible for resale sell, or indicate an intention to sell, substantial amounts of our common stock in the public market, the trading price of our common stock could decline.
Directors, executive officers, principal stockholders and affiliated entities own a significant percentage of our capital stock, and they may make decisions that you do not consider to be in the best interests of our stockholders.
As of December 14, 2009, our directors, executive officers, principal stockholders and affiliated entities beneficially owned, in the aggregate, approximately 58.1% of our outstanding voting securities. As a result, if some or all of them acted together, they would have the ability to exert substantial influence over the election of our Board of Directors and the outcome of issues requiring approval by our stockholders. This concentration of ownership also may have the effect of delaying or preventing a change in control of our Company that may be favored by other stockholders. This could prevent transactions in which stockholders might otherwise recover a premium for their shares over current market prices.
Our stock price could decline as a result of our failure to meet reporting and other regulatory requirements.
Our management team is responsible for our operations, reporting and compliance. Our failure to comply with the Sarbanes-Oxley Act, once our Company becomes subject thereto, and/or the reporting requirements and other provisions of securities laws could negatively affect our stock price and adversely affect our results of operations, cash flow and financial condition.
Operating as a small public company also requires us to make forward-looking statements about future operating results and to provide some guidance to the public markets. Our management team has limited experience serving in a managerial capacity in a public company and as a result projections may not be made timely or set at expected performance levels and could materially affect the price of our shares. Any failure to meet published forward-looking statements that adversely affect the stock price could result in losses to investors, stockholder lawsuits or other litigation, sanctions or restrictions issued by the SEC or any stock market upon which our stock is traded.
If we do not implement necessary improvements to our internal control over financial reporting in an efficient and timely manner, or if we discover additional deficiencies and weaknesses in existing systems and controls, we could be subject to regulatory enforcement and investors may lose confidence in our ability to operate in compliance with existing internal control rules and regulations, either of which could result in a decline in our share price.
Disclosure controls and procedures are controls and other procedures that are designed to ensure that information required to be disclosed in company reports filed or submitted under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC's rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed in Company reports filed or submitted under the Exchange Act is accumulated and communicated to management, including our chief executive officer and chief financial officer, as appropriate to allow timely decisions regarding required disclosure. As required by Rules 13a-15 and 15d-15 under the Exchange Act, our chief
21
executive officer and chief financial officer carried out an evaluation of the effectiveness of the design and operation of our disclosure controls and procedures as of September 30, 2009. Based upon their evaluation, and as a result of previously identified material weakness in internal control over financial reporting as discussed in our Annual Report on Form 10-K for the year ended December 31, 2008, they concluded that our disclosure controls and procedures were not effective as of September 30, 2009. The previously reported material weaknesses in internal control over financial reporting related to (1) adequate qualified staff necessary to effectively apply the process, and (2) methods and practices employed to report unusual transactions such as our reverse merger. Our management has discussed the material weakness in our internal control over financial reporting with our audit committee. In an effort to remediate the identified material weaknesses, we have documented our process and procedures governing our internal reporting. We also plan to implement further changes to our internal control over financial reporting, including (1) a re-evaluation of our staffing needs, and (2) analysis of unusual transactions as they are occurring to allow adequate time for multiple levels of review.
Through these steps, we believe we are addressing the deficiencies that affected our internal control over financial reporting. However, the effectiveness of any system of internal controls is subject to inherent limitations and we cannot assure you that our internal control over financial reporting will prevent or detect all errors. Also, management may not be able to implement necessary improvements to internal control over financial reporting in an efficient and timely manner and may discover additional deficiencies and weaknesses in existing systems and controls, especially if the systems and controls are tested by an increased rate of growth or the impact of acquisitions. In addition, upgrades or enhancements to computer systems could cause internal control weaknesses.
If we are unable to establish adequate internal controls over financial reporting systems, we may encounter difficulties in the audit or review of our financial statements by our independent public accountants, which in turn may have a material adverse effect on our ability to comply with the reporting obligations imposed upon us by the SEC.
If we fail to maintain an effective system of internal control, we may be unable to produce reliable financial reports or prevent fraud. If we are unable to assert that our internal control over financial reporting is effective at any time in the future, or if our independent registered public accounting firm is unable to attest to the effectiveness of internal controls, is unable to deliver a report at all or can deliver only a qualified report, we could be subject to regulatory enforcement and investors may lose confidence in our ability to operate in compliance with existing internal control rules and regulations, either of which could result in a decline in our share price.
Our status as a public company may make it more difficult to attract and retain officers and directors.
The Sarbanes-Oxley Act and new rules subsequently implemented by the SEC have required changes in corporate governance practices of public companies. As an operating public company, we expect these new rules and regulations to increase our compliance costs in 2009 and beyond and to make certain activities more time-consuming and costly than if we were not an operating public company. As an operating public company, we also expect that these new rules and regulations may make it more difficult and expensive for us to obtain director and officer liability insurance in the future, and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified persons to serve on our Board of Directors or as executive officers.
Compliance with changing regulations concerning corporate governance and public disclosure may result in additional expenses.
There have been changing laws, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act, new regulations promulgated by the SEC and rules promulgated by the NYSE AMEX LLC and other national securities exchanges. These new or changed laws, regulations and standards are subject to varying interpretations in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies, which could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. As a result, our efforts to comply with evolving laws, regulations and standards are likely to continue to result in increased general and administrative expenses and a diversion of management time and attention from revenue- generating activities to compliance activities. Our board members,
22
Chief Executive Officer and Chief Financial Officer could face an increased risk of personal liability in connection with the performance of their duties. As a result, we may have difficulty attracting and retaining qualified board members and executive officers, which could harm our business. If our efforts to comply with new or changed laws, regulations and standards differ from the activities intended by regulatory or governing bodies, we could be subject to liability under applicable laws or our reputation may be harmed.
We do not intend to pay cash dividends. Any return on investment may be limited to the value of our common stock, if any.
We have never declared or paid cash dividends on our capital stock (other than certain dividends that may have been paid by CKST in or before 2005). We currently expect to use available funds and any future earnings to develop, operate and expand our business and do not anticipate paying any cash dividends in the foreseeable future. In addition, the terms of any future debt or credit facility we may obtain may preclude us from paying any dividends. As a result, capital appreciation, if any, of our common stock will be an investor's only source of potential gain from our common stock for the foreseeable future.
Stockholders may experience significant dilution if future equity offerings are used to fund operations or acquire complementary businesses.
If future operations or acquisitions are financed through issuing equity securities, stockholders could experience significant dilution. In addition, securities issued in connection with future financing activities or potential acquisitions may have rights and preferences senior to the rights and preferences of our common stock. We expect to issue additional equity securities pursuant to employee benefit plans. The issuance of shares of our common stock upon the exercise of options may result in dilution to our stockholders.
Our Certificate of Incorporation grants our Board of Directors the power to designate and issue additional shares of common and/or preferred stock.
Our authorized capital consists of 750,000,000 shares of common stock and 50,000,000 shares of preferred stock. Our preferred stock may be designated into series pursuant to authority granted by our Certificate of Incorporation, and on approval from our Board of Directors. The Board of Directors, without any action by our stockholders, may designate and issue shares in classes or series as the Board of Directors deems appropriate and establish the rights, preferences and privileges of those shares, including dividends, liquidation and voting rights. The rights of holders of other classes or series of stock that may be issued could be superior to the rights of holders of our common shares. The designation and issuance of shares of capital stock having preferential rights could adversely affect other rights appurtenant to shares of our common stock. Furthermore, any issuances of additional stock (common or preferred) will dilute the percentage of ownership interest of then-current holders of our capital stock and may dilute our book value per share.
23
Some of the statements in this prospectus are "forward-looking statements," as that term is defined in the Private Securities Litigation Reform Act of 1995. Forward-looking statements may be identified by the use of words such as "may," "will," "should," "anticipate," "estimate," "expect," "plan," "believe," "predict," "potential," "project," "target," "forecast," "intend," "assume," "guide," "seek" and similar expressions. Forward-looking statements do not relate strictly to historical or current matters. Rather, forward-looking statements are predictive in nature and may depend upon or refer to future events, activities or conditions. Although we believe that these statements are based upon reasonable assumptions, we cannot provide any assurances regarding future results. We undertake no obligation to revise or update any forward-looking statements, or to make any other forward-looking statements, whether as a result of new information, future events or otherwise.
Because forward-looking statements relate to matters that have not yet occurred, these statements are inherently subject to risks and uncertainties. Many factors could cause our actual activities or results to differ materially from the activities and results anticipated in forward-looking statements. Information regarding our risk factors appears in "Risk Factors" beginning on page 3, which include, but are not limited to, the following:
We will need to raise additional funds in the future to fund our operations and research, and these funds may not be available on acceptable terms, if at all.We expect to incur significant losses, either directly or indirectly through the companies in which we develop our products, for at least the next several years, and we cannot assure you that we will ever be profitable.We have a limited number of products currently available for sale and there is a high risk that our research and development efforts might not successfully generate any viable product candidates in the future.Cost containment measures, pressure from our competitors and availability of medical reimbursement may impact our ability to sell our products at prices necessary to expand our operations and reach profitability.Sales of our products will depend on the availability of adequate reimbursement from third-party payors (such as governmental programs, for example, Medicare and Medicaid, private insurance plans and managed care programs) both in terms of the sales volumes and prices for our products.Legislative or administrative reforms to the United States or international reimbursement systems in a manner that significantly reduces reimbursement for procedures using our medical devices or denies coverage for those procedures could have a material adverse effect on our business, financial condition or results of operations.We must convince orthopedic and spine surgeons that our products are an attractive alternative to existing surgical treatments of orthopedic and spine disorders.Our business plan relies on certain assumptions about the market for our products, which, if incorrect, may adversely affect our business and profitability.We expect to face significant competition as a result of the rapid technological changes in the medical devices industry, which could have an adverse effect on our business, financial condition or results of operations.Hospitals, surgeons, distributors and agents may have existing relationships with other medical device companies that make it difficult for us to establish new relationships with them. As a result, we may not be able to sell and market our products effectively.Our manufacturers may be unsuccessful in manufacturing products at the levels required to meet future market demand.We rely on single source manufacturers, which could impair our ability to meet demand for delivering our products in a timely manner or within our budget.Our growth will depend on developing new products or product enhancements, requiring significant research and development, clinical trials and regulatory approvals, all of which are expensive and time-consuming and may not result in a commercially viable product.If we choose to grow our business by acquiring new and complementary businesses, products or technologies, we may be unable to complete these acquisitions or successfully integrate them in a cost-effective and non-disruptive manner.We rely on our independent sales distributors and sales representatives to market and sell our products.
24
We are dependent on the services of Andrew A. Brooks, M.D. and Michael Kvitnitsky, and the loss of either of them could harm our business.Failure to attract and retain skilled personnel and cultivate key academic collaborations will delay product development programs and business development efforts.If we fail to properly manage our anticipated growth, our business could suffer.If we decide to market and sell our devices and products internationally, we would be subject to various risks relating to our international activities, which could negatively impact our business and financial results.We are subject to substantial governmental regulation that could change and thus force us to make modifications to how we develop, manufacture and price our products.Federal regulatory reforms may adversely affect our ability to sell our products profitably.We have not yet collected long-term clinical data to support the safety of our products, and our products may, therefore, prove to be less safe and effective than initially thought.The FDA requires us to obtain new Section 510(k) clearances or premarket approvals for modifications to our approved products. Otherwise, we may have to cease marketing, or to recall, the modified products until clearances are obtained.If we or our third-party manufacturers fail to comply with the FDA's Quality System Regulations, the manufacture of our products could be interrupted and our product sales and operating results could suffer.We are subject to various complex laws and regulations. Compliance with these laws and regulations is costly and time- consuming, and failure to comply with them can have adverse consequences on our business.We are an orthopedic medical device company with a limited operating history and our business may not become profitable.Our quarterly financial results are likely to fluctuate significantly because our sales prospects are uncertain.If we cannot adequately protect our patents and other intellectual property rights, we may lose market share to our competitors and be unable to operate our business profitably.Changes to intellectual property laws may negatively impact our ability to protect our intellectual property.Patent infringement lawsuits brought against us could have a material adverse affect on our commercial success, and our ability to develop and sell our products and to operate profitably.The medical device industry is characterized by patent and other intellectual property litigation, and we could become subject to litigation that could be costly, result in diverting management's time and efforts, require us to pay damages, and/or prevent us from marketing our existing or future products.We may be subject to damages resulting from claims that we or our employees or consultants have wrongfully used or disclosed alleged trade secrets of their former employers.Fluctuations in the cost and availability of insurance could adversely affect our profitability or our risk management profile.Potential future product liability claims and other litigation, including contract litigation, may adversely affect our business, reputation and ability to attract and retain customers.Any claims relating to our making improper payments to physicians for consulting services, or other potential violations of regulations governing interactions between us and healthcare providers, could be time-consuming and costly.Our common stock may be thinly traded.We expect that the price of our common stock will fluctuate substantially, potentially adversely affecting the ability of investors to sell their shares.Securities analysts may elect not to report on our common stock or may issue negative reports that adversely affect the price of our common stock.Anti-takeover provisions in our charter documents and Delaware law may discourage or prevent a change in control, even if an acquisition would be beneficial to our stockholders, which could affect our stock price adversely and prevent attempts by our stockholders to replace or remove our current management.Because our common stock may be a "penny stock," it may be more difficult for investors to sell shares of our common stock, and the market price of our common stock may be adversely affected.A significant number of shares will become eligible for future sale by our stockholders and the sale of those shares could adversely affect the stock price.
25
Directors, executive officers, principal stockholders and affiliated entities own a significant percentage of our capital stock, and they may make decisions that you do not consider to be in the best interests of our stockholders.Our stock price could decline as a result of our failure to meet reporting and other regulatory requirements.If we do not implement necessary improvements to our internal control over financial reporting in an efficient and timely manner, or if we discover additional deficiencies and weaknesses in existing systems and controls, we could be subject to regulatory enforcement and investors may lose confidence in our ability to operate in compliance with existing internal control rules and regulations, either of which could result in a decline in our share price.Our status as a public company may make it more difficult to attract and retain officers and directors.Compliance with changing regulations concerning corporate governance and public disclosure may result in additional expenses.We do not intend to pay cash dividends. Any return on investment may be limited to the value of our common stock, if any.Stockholders may experience significant dilution if future equity offerings are used to fund operations or acquire complementary businesses.Our Certificate of Incorporation grants our Board of Directors the power to designate and issue additional shares of common and/or preferred stock.
Additional information concerning these risk factors can be found in "Risk Factors" beginning on page 3. Forward-looking statements in this prospectus should be evaluated in light of these important factors.
We are registering theses shares pursuant to registration rights granted to the selling stockholders. We are not selling any securities under this prospectus and we will not receive any proceeds from the sale by the selling stockholders of their common stock. If and when the placement agent exercises any placement agent warrants, we will receive the amount of the exercise price unless the warrants are exercised on a cashless basis. The Placement Agent Warrants are for the purchase of 575,613 shares of the Company's common stock, the number that is equivalent to six percent (6%) of the number of shares of common stock sold in the transaction to Approved Investors, at an exercise price of $0.44 per share. If all of the Placement Agent Warrants are exercised, the Company would receive approximately $253,270 in cash, unless any of the Placement Agent Warrants are exercised on a cashless basis. We expect that any proceeds which we receive from the exercise of the Placement Agent Warrants will be used for working capital and general corporate purposes.
The common stock to be sold by the selling stockholders is common stock that is currently issued and outstanding. Accordingly, there will be no dilution to our existing stockholders in connection with the offer and sale by the selling stockholders of common stock currently issued and outstanding.
However, 575,613 shares of common stock underlying the placement agent warrants are also being registered pursuant to this registration statement. Such shares are not currently issued and outstanding. If any of the placement agent warrants to purchase 575,613 shares of common stock are exercised, our stockholders may experience a reduction in their ownership interest in the Company, however such reduction would not be material.
Except as otherwise indicated, the following table sets forth certain information with respect to the beneficial ownership of our common stock including the names of the selling stockholders, the number of shares of common stock known by the Company to be owned beneficially by the selling stockholders as of December 14, 2009, the number of shares of our common stock that may be offered by the selling stockholders pursuant to this prospectus, the number of shares owned by the selling stockholders after completion of the offering and the percentage of shares to be owned by the selling stockholders after completion of the offering. Except for Frost Gamma Investments Trust, the Company knows of no selling stockholder that
26
will own more than 1% of our outstanding common stock after the sale of shares owned by such selling stockholder. After completion of the sale of the shares owned by Frost Gamma Investments Trust and offered by this prospectus, Frost Gamma Investments Trust would beneficially own 31,822,339 shares of common stock, representing 13.82% of our outstanding common stock, assuming no warrants are exercised by the placement agent, and 13.78% of the common stock, assuming all of the warrants held by the placement agent are exercised. The table has been prepared based upon a review of Exchange Act filings related to the Company and additional information furnished to us by or on behalf of the selling stockholders.
Name of Selling Stockholder | Shares of | Shares of | Shares of | Percent of |
Alan Mandel | 95,000 | 95,000 | 0 | 0.00% |
Alan Mandel | 285,714 | 285,714 | 0 | 0.00% |
Andrew Moon | 142,857 | 142,857 | 0 | 0.00% |
Barry Fine | 100,000 | 100,000 | 0 | 0.00% |
Bassan Investments LLC | 100,000 | 100,000 | 0 | 0.00% |
Benjamin Kaminash | 285,714 | 285,714 | 0 | 0.00% |
Bruce Pollack | 2,073,043 | 71,428 | 2,001,615(1) | * |
Canyon State Masonry Inc | 142,857 | 142,857 | 0 | 0.00% |
Chanel Gold Enterprise | 100,000 | 100,000 | 0 | 0.00% |
Charles David Stadterman | 100,000 | 100,000 | 0 | 0.00% |
Matthew Coffin TR UA 10/07/08 Coffin Family Trust | 1,428,571 | 1,428,571 | 0 | 0.00% |
Cyrus Hadidi | 750,000 | 750,000 | 0 | 0.00% |
David C Blatte | 2,073,043 | 71,428 | 2,001,615(1) | * |
David Mittler | 57,142 | 57,142 | 0 | 0.00% |
David H Shepard TR UA 01/02/02 David Haspel Shepard Rev Liv Trust | 285,714 | 285,714 | 0 | 0.00% |
David Thalheim | 100,000 | 100,000 | 0 | 0.00% |
Dominguez Investments | 142,857 | 142,857 | 0 | 0.00% |
Donald S Shepard | 142,857 | 142,857 | 0 | 0.00% |
Joseph S. Levy TR UA 06/17/98 Dr Joseph S Levy Rev Liv Trust | 71,428 | 71,428 | 0 | 0.00% |
Ed Baroody | 57,142 | 57,142 | 0 | 0.00% |
Edward C Gomez | 142,857 | 142,857 | 0 | 0.00% |
Frost Gamma Investments Trust | 33,250,911 | 1,428,572 | 31,822,339 | 13.82% |
George & Son Partners 2008-1 LLP | 94,285 | 94,285 | 0 | 0.00% |
Gilbert Hooper & Danielle Hooper JT TEN | 71,428 | 71,428 | 0 | 0.00% |
Glenn Marshak | 71,428 | 71,428 | 0 | 0.00% |
Gregory Schroeder & Silvia Schroeder TR UA 05/10/05 Gregory & Silvia Schroeder Fam Trust | 85,714 | 85,714 | 0 | 0.00% |
Hank Yunes & Marci Yunes JT TEN | 100,000 | 100,000 | 0 | 0.00% |
Hugo Reiter & Arlene Reiter JT TEN | 300,000 | 300,000 | 0 | 0.00% |
Ingrid K Pelerin TR UA 09/13/96 Ingrid K Pelerin Rev Liv Trust | 71,428 | 71,428 | 0 | 0.00% |
Ira Levy | 71,428 | 71,428 | 0 | 0.00% |
J & C Johnstone Family Limited Partnership | 100,000 | 100,000 | 0 | 0.00% |
27
Jack Parks 57,142 57,142 0 0.00% Jacqueline Simkin TTEE Amended & Restated DTD 12/16/03 Jacqueline Simkin Trust 2,545,510 285,714 2,259,796 * James J Bischoff & Lucinda G Bischoff TR UA 02/02/87 James & Lucinda Bischoff Fam Liv Trust 71,428 71,428 0 0.00% Jason Hirzel & Kelly Hirzel JT TEN 285,714 285,714 0 0.00% Jay R Flackoff 30,000 30,000 0 0.00% Jay G Goldman 142,857 142,857 0 0.00% Irv Goldman TR UA 04/24/95 Jay Goldman & Stephanie Goldman Irrev Trust 142,857 142,857 0 0.00% Jiansheng Zhao 100,000 100,000 0 0.00% Jim Miller 100,000 100,000 0 0.00% Joe Hadden 100,000 100,000 0 0.00% John Ritchie & Christine Ritchie JT TEN 285,714 285,714 0 0.00% John Radtke 100,000 100,000 0 0.00% Jorge Wolf 300,000 300,000 0 0.00% Josh Berman 142,857 142,857 0 0.00% Joshua Mandel 100,000 100,000 0 0.00% Lester Pollack 2,073,043 71,428 2,001,615(1) * Lonnie Ogulnick & Dara Ogulnick JT TEN 57,142 57,142 0 0.00% Louis Olivia and Stacy Olivia JT TEN 100,000 100,000 0 0.00% Michael Sinel 142,857 142,857 0 0.00% Mike Miller 71,428 71,428 0 0.00% Next View Capital LP 857,143 857,143 0 0.00% Paul Musschoot 100,000 100,000 0 0.00% Phillip George & Daughter Partners 2008-1 LLP 94,285 94,285 0 0.00% Phillip T George 458,707 97,142 361,565 * Portal Ventures LLC 2,000,000 2,000,000 0 0.00% Richard Paz & Dalit Paz JT TEN 57,142 57,142 0 0.00% Richard J Rosenstock 100,000 100,000 0 0.00% Richard Paul Yurich TR UA 06/11/08 The Richard Paul Yurich Rev Liv Trust 285,714 285,714 0 0.00% Rick Miner 100,000 100,000 0 0.00% Robert Margolis & Elizabeth Margolis JT TEN 285,714 285,714 0 0.00% Robert Bergmann 2,073,043 71,428 2,001,615(1) * Robert H Hartley 142,857 142,857 0 0.00% Robert Halm 57,142 57,142 0 0.00% Robert Sudack 40,000 40,000 0 0.00% Scoggin Capital Management LP II 1,428,571 1,428,571 0 0.00% Scoggin International Fund Ltd 1,428,571 1,428,571 0 0.00% Shari Notowitz 71,428 71,428 0 0.00% Tevis Margolis 71,428 71,428 0 0.00% VED Software Services 28,571 28,571 0 0.00% Virginia Myers & Ernie Myers JT TEN 285,714 285,714 0 0.00% Wendy F Lumish 100,000 100,000 0 0.00% W R Everett & Kathleen Everett JT TEN 100,000 100,000 0 0.00% Yukiyo Matsumura & Machi Liu JT TEN 200,000 200,000 0 0.00% Ladenburg Thalmann & Co. Inc. 575,613 575,613 0 0.00%
28
* Less than 1%.(1) The stockholder is a member of CP Cardio LLC which owns 2,001,615 shares of the Company.
__________
None of the selling stockholders has, or within the past three years has had, any position, office or material relationship with us or any of our predecessors or affiliates except as follows:
In consideration for investment banking services rendered to us by Ladenburg Thalmann & Co. Inc. ("Ladenburg"), as placement agent for the private placement offering, we issued to Ladenburg warrants to purchase 575,613 shares of the Company's common stock, a number of shares that is equivalent to six percent (6%) of the number of shares of common stock sold in the private placement to Approved Investors, at an exercise price of $0.44 per share. The services rendered by Ladenburg included serving as financial advisor to the Company in connection with raising equity and equity related capital for the Company. The warrants have an exercise price of $0.44 per share and a term of five years.
Frost Gamma Investments Trust is a 10% owner of the Company. Frost Gamma Investments Trust owned 33,250,911 shares of common stock of the Company prior to the offering. The Company sold 1,428,572 shares of common stock of the Company for the account of Frost Gamma Investments Trust. Frost Gamma Investments Trust owns 31,822,339 shares of common stock or 13.78% percent of the Company after completion of the offering.
Lonnie Ogulnick owns stock in the Company as Lonnie Ogulnick & Dara Ogulnick JT TEN. Lonnie Ogulnick is a registered financial advisor with Ladenburg and Managing Director of Ladenburg Thalmann Private Client Services.
Richard Rosenstock owns stock in the Company as Richard J Rosenstock. Richard Rosenstock is a registered broker with Ladenburg and a director of Ladenburg Thalmann Financial Services.
We are registering the shares of common stock to permit the resale of these shares of common stock by the holders of the common stock from time to time after the date of this prospectus. However, we will receive gross proceeds of up to approximately $253,270 from the issuance of shares of common stock being registered pursuant to the registration statement of which this prospectus forms a part in connection with the exercise of the placement agent warrants, if and when they are exercised, unless such warrants are exercised on a cashless basis. We will not receive any of the proceeds from the sale by the selling stockholders of the shares of common stock. We will bear all fees and expenses incident to our obligation to register the shares of common stock.
The selling stockholders may sell all or a portion of the shares of common stock beneficially owned by them and offered hereby from time to time directly or through one or more underwriters, broker-dealers or agents. If the shares of common stock are sold through underwriters or broker-dealers, the selling stockholders will be responsible for underwriting discounts or commissions or agent's commissions. The shares of common stock may be sold in one or more transactions at fixed prices, at prevailing market prices at the time of the sale, at varying prices determined at the time of sale, or at negotiated prices. These sales may be effected in transactions, which may involve crosses or block transactions,
on any national securities exchange or quotation service on which the securities may be listed or quoted at the time of sale;in the over-the-counter market;in transactions otherwise than on these exchanges or systems or in the over-the-counter market;through the writing of options, whether such options are listed on an options exchange or otherwise;ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers;
29
block trades in which the broker-dealer will attempt to sell the shares as agent but may position and resell a portion of the block as principal to facilitate the transaction;purchases by a broker-dealer as principal and resale by the broker-dealer for its account;an exchange distribution in accordance with the rules of the applicable exchange;privately negotiated transactions;sales pursuant to Rule 144;broker-dealers may agree with the selling securityholders to sell a specified number of such shares at a stipulated price per share;a combination of any such methods of sale; andany other method permitted pursuant to applicable law.
If the selling stockholders effect such transactions by selling shares of common stock to or through underwriters, broker-dealers or agents, such underwriters, broker-dealers or agents may receive commissions in the form of discounts, concessions or commissions from the selling stockholders or commissions from purchasers of the shares of common stock for whom they may act as agent or to whom they may sell as principal (which discounts, concessions or commissions as to particular underwriters, broker-dealers or agents may be in excess of those customary in the types of transactions involved). In connection with sales of the shares of common stock or otherwise, the selling stockholders may enter into hedging transactions with broker-dealers, which may in turn engage in short sales of the shares of common stock in the course of hedging in positions they assume. The selling stockholders may also sell shares of common stock short and deliver shares of common stock covered by this prospectus to close out short positions and to return borrowed shares in connection with such short sales. The selling stockholders may also loan or pledge shares of common stock to broker-dealers that in turn may sell such shares.
The selling stockholders may pledge or grant a security interest in some or all of the shares of common stock owned by them and, if they default in the performance of their secured obligations, the pledgees or secured parties may offer and sell the shares of common stock from time to time pursuant to this prospectus or any amendment to this prospectus under applicable provisions of the Securities Act of 1933, as amended, amending, if necessary, the list of selling stockholders to include the pledgee, transferee or other successors in interest as selling stockholders under this prospectus. The selling stockholders also may transfer and donate the shares of common stock in other circumstances in which case the transferees, donees, pledgees or other successors in interest will be the selling beneficial owners for purposes of this prospectus.
The selling stockholders and any broker-dealer participating in the distribution of the shares of common stock may be deemed to be "underwriters" within the meaning of the Securities Act, and any commission paid, or any discounts or concessions allowed to, any such broker-dealer may be deemed to be underwriting commissions or discounts under the Securities Act. At the time a particular offering of the shares of common stock is made, a prospectus supplement, if required, will be distributed which will set forth the aggregate amount of shares of common stock being offered and the terms of the offering, including the name or names of any broker-dealers or agents, any discounts, commissions and other terms constituting compensation from the selling stockholders and any discounts, commissions or concessions allowed or reallowed or paid to broker-dealers.
Under the securities laws of some states, the shares of common stock may be sold in such states only through registered or licensed brokers or dealers. In addition, in some states the shares of common stock may not be sold unless such shares have been registered or qualified for sale in such state or an exemption from registration or qualification is available and is complied with.
30
There can be no assurance that any selling stockholder will sell any or all of the shares of common stock registered pursuant to this registration statement, of which this prospectus forms a part.
The selling stockholders and any other person participating in such distribution will be subject to applicable provisions of the Securities Exchange Act of 1934, as amended, and the rules and regulations thereunder, including, without limitation, Regulation M of the Exchange Act, which may limit the timing of purchases and sales of any of the shares of common stock by the selling stockholders and any other participating person. Regulation M may also restrict the ability of any person engaged in the distribution of the shares of common stock to engage in market-making activities with respect to the shares of common stock. All of the foregoing may affect the marketability of the shares of common stock and the ability of any person or entity to engage in market-making activities with respect to the shares of common stock.
We will pay all expenses of the registration of the shares of common stock pursuant to the registration rights agreement, estimated to be $70,624 in total, including, without limitation, Securities and Exchange Commission filing fees and expenses of compliance with state securities or "blue sky" laws; provided, however, that a selling stockholder will pay all underwriting discounts and selling commissions, if any. We will indemnify the selling stockholders against liabilities, including some liabilities under the Securities Act, in accordance with the registration rights agreements, or the selling stockholders will be entitled to contribution. We may be indemnified by the selling stockholders against civil liabilities, including liabilities under the Securities Act, that may arise from any written information furnished to us by the selling stockholder specifically for use in this prospectus, in accordance with the related registration rights agreement, or we may be entitled to contribution.
Once sold under this registration statement, of which this prospectus forms a part, the shares of common stock will be freely tradable in the hands of persons other than our affiliates.
The Company appointed Ladenburg as the Company's exclusive placement agent for the private placement offering. There is a common principal stockholder of both the Company and of Ladenburg. Furthermore, certain senior managers of the placement agent are stockholders of the Company. After reviewing all information related to the transaction between the Company and Ladenburg, a potential related party transaction, the Company's Audit Committee approved the related party transaction. Ladenburg, as the placement agent, for acting in such capacity for the shares of common stock offered in the private placement offering, received: (i) a cash commission equal to eight percent (8%) of the gross proceeds from the offering that was received from Approved Investors; (ii) a cash non-accountable expense allowance equal to one percent (1%) of the gross proceeds of the offering to Approved Investors; (iii) reimbursement of Ladenburg's out-of-pocket expenses related to the offering, including its legal fees and expenses up to $40,000; and (iv) the issuance to Ladenburg of warrants to purchase 575,613 shares of common stock equal to six percent (6%) of the number of shares sold in the offering to Approved Investors, at an exercise price of $0.44 per share.
DESCRIPTION OF SECURITIES TO BE REGISTERED
Our authorized capital stock consists of 750,000,000 shares of common stock, with a par value of $0.001 per share and 50,000,000 shares of preferred stock.
Common Stock
As of December 14, 2009, there were 230,293,141 shares of our common stock issued and outstanding, held by 261 stockholders of record. Holders of common stock are entitled to (i) one vote for each share at all meetings of stockholders, (ii) receive, subject to the prior rights of holders, if any, of outstanding stock having prior rights as to dividends, dividends as may be declared by the Board of Director, and (iii) subject to the prior rights of holders, if any, of outstanding stock having prior rights as to asset distributions, our remaining assets upon liquidation, dissolution or winding up of our company. The holders of common stock have no preemptive or other subscription or conversion rights. There are no redemption or sinking fund provisions applicable to the common stock. All shares of common stock now outstanding are fully paid and nonassessable.
31
In conjunction with first tranche and the second tranche of the private placement that closed on October 27, 2009 and November 13, 2009 respectively, Cardo Medical, Inc. issued to the placement agent warrants to purchase 575,613 shares of the Company's common stock, at an exercise price of $0.44 per share. The Placement Agent Warrants expire on November 13, 2014. The shares of the Company's common stock underlying the Placement Agent Warrants are being registered under the registration statement of which this prospectus constitutes a part.
Preferred Stock
As of December 14, 2009, there were no shares of our preferred stock currently issued and outstanding.
Our Certificate of Incorporation and Bylaws contain provisions that could delay or prevent a change in control of our Company or changes in our Board of Directors that our stockholders might consider favorable. Some of these provisions:
impose limitations on our stockholders to call special stockholder meetings; andauthorize the issuance of preferred stock which can be created and issued by the Board of Directors without prior stockholder approval, with rights senior to those of the common stock.
In addition, we are subject to the provisions of Section 203 of the Delaware General Corporation Law, which may prohibit certain business combinations with stockholders owning 15% or more of our outstanding voting stock. These and other provisions in our Certificate of Incorporation, our Bylaws and Delaware law could make it more difficult for stockholders or potential acquirers to obtain control of our Board of Directors or initiate actions that are opposed by our then-current Board of Directors, including to delay or impede a merger, tender offer or proxy contest involving our Company. Any delay or prevention of a change in control transaction or changes in our Board of Directors could cause the market price of our common stock to decline.
Our
The unit price determined for this Offering is not an indication of the fair value of our common stock.
In determining the Unit price, our board of directors considered a number of factors, including, but not limited to, our need to raise capital in the near term to continue our operations, the current and historical trading prices of our common stock, is quoteda price that would increase the likelihood of participation in this Offering, the cost of capital from other sources, the value of the warrants being issued as components of the Unit and comparable precedent transactions. The Unit price does not necessarily bear any relationship to any established criteria for value. No valuation consultant or investment banker has opined upon the fairness or adequacy of the Unit price. You should not consider the Unit price as an indication of the value of our Company or our common stock.
Risks Related to the Warrants
The warrants may not have any value and if an active, liquid trading market for the warrants does not develop, you may not be able to sell your warrants quickly or at or above the price you paid for them.
The warrants issued in this Offering will be immediately exercisable and expire on , 2022. The warrants will have an initial exercise price equal to $ . In the event that our common stock price does not exceed the exercise price of the warrants during the period when the warrants are exercisable, the warrants may not have any value.
Prior to this Offering, there has been no public market for any of our warrants. We have applied to list the Warrants on the OTC Bulletin BoardNasdaq Capital Market. However, there can be no assurance that such listing will be approved. An active trading market may not develop for the warrants to be sold in this Offering or, if developed, may not be sustained, and the market for the warrants may be highly volatile or may decline regardless of our operating performance. The lack of an active market may impair your ability to sell your warrants at the time you wish to sell them or at a price that you consider reasonable.
Since the warrants are executory contracts, they may have no value in a bankruptcy or reorganization proceeding.
In the event a bankruptcy or reorganization proceeding is commenced by or against us, a bankruptcy court may hold that any unexercised warrants are executory contracts that are subject to rejection by us with the approval of the bankruptcy court. As a result, holders of the warrants may, even if we have sufficient funds, not be entitled to receive any consideration for their warrants or may receive an amount less than they would be entitled to if they had exercised their warrants prior to the commencement of any such bankruptcy or reorganization proceeding.
Holders of our warrants will have no rights as a common stockholder until they acquire our common stock.
Until investors acquire shares of our common stock upon exercise of the warrants being offered in this Offering, they will have no rights with respect to our common stock such as voting rights or the right to receive dividends. Upon exercise of such warrants, holders will be entitled to exercise the rights of a common stockholder only as to matters for which the record date occurs after the exercise date.
We may amend the terms of the warrants in a way that may be adverse to holders with the approval by the holders of a majority of the then outstanding warrants.
Our warrants will be issued in physical certificated form under a warrant agreement between Continental Stock Transfer and Trust Company, as warrant agent, and us. The warrant agreement provides that the terms of the warrants may be amended without the consent of any holder to cure any ambiguity or correct any defective provision. All other modifications or amendments, including any amendment to increase the exercise price of the warrants or shorten the exercise period of the warrants, shall require the written consent of the registered holders of a majority of the then outstanding warrants.
Our outstanding warrants may have an adverse effect on the market price of our common stock and make it more difficult to effect a business combination.
We will be issuing warrants to purchase shares of common stock as part of this Offering. To the extent we issue shares of common stock to effect a future business combination, the potential for the issuance of a substantial number of additional shares upon exercise of these warrants could make us a less attractive acquisition vehicle in the eyes of a target business. Such securities, when exercised, will increase the number of issued and outstanding ordinary shares and reduce the value of the shares issued to complete the business combination. Accordingly, our warrants may make it more difficult to effectuate a business combination or increase the cost of acquiring a target business. Additionally, the sale, or even the possibility of sale, of the shares of common stock underlying the warrants could have an adverse effect on the market price for our securities or on our ability to obtain future financing. If and to the extent these warrants are exercised, you may experience dilution to your holdings.
Certain of the possible adjustments to the warrants may result in a deemed distribution from us to a beneficial owner of a warrant that will be taxable, even though the beneficial owner does not receive a corresponding distribution of cash.
The exercise terms of the warrants being offered in this Offering may be adjusted in certain circumstances. An adjustment to the number of shares of common stock that will be issued on the exercise of the warrants or an adjustment to the exercise price of the warrants (or, in certain circumstances, a failure to make adjustments) may be treated as a taxable deemed distribution to a holder of the warrants, even if such holder does not receive any cash or other property in connection with the adjustment. Holders of the warrants should consult their tax advisors regarding the proper treatment of any adjustments to the warrants. For a more detailed discussion, see “—Material U.S. Federal Income Tax Considerations For Holders of Our Common Stock and Warrants.”
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains certain statements that constitute forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act. Any and all statements contained in this prospectus that are not statements of historical fact may be deemed forward-looking statements. Terms such as “may,” “might,” “would,” “should,” “could,” “project,” “estimate,” “pro-forma,” “predict,” “potential,” “strategy,” “anticipate,” “attempt,” “develop,” “plan,” “help,” “believe,” “continue,” “intend,” “expect,” “future” and terms of similar import (including the negative of any of the foregoing) may be intended to identify forward-looking statements. However, not all forward-looking statements may contain one or more of these identifying terms. Those statements appear in this prospectus, and include statements regarding the intent, belief or current expectations of the company and management that are subject to known and unknown risks, uncertainties and assumptions and other factors that could cause actual results and the timing of certain events to differ materially from future results expressed or implied by such forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to those discussed in the section titled “Risk Factors” set forth above.
Forward-looking statements in this prospectus may include, without limitation, statements regarding:
(i) the plans and objectives of management for future operations, including plans or objectives relating to the development of our cell therapy systems,
(ii) the timing and conduct of the clinical trials for our products, including statements regarding the timing, progress and results of current and future preclinical studies and clinical trials as well as our research and development programs;
(iii) the timing or likelihood of regulatory filing, approvals and required licenses for our cell therapy systems;
(iv) our ability to adequately protect our intellectual property rights and enforce such rights to avoid violation of the intellectual property rights of others;
(v) the timing, costs and other aspects of the commercial launch of our products;
(vi) our estimates regarding the market opportunity, clinical utility, potential advantages and market acceptance of our products;
(vii) the impact of government laws and regulations;
(viii) our ability to recruit and retain qualified clinical, regulatory and research and development personnel;
(ix) the availability of reimbursement or other forms of funding for our products from government and commercial payors;
(x) difficulties in maintaining commercial scale manufacturing capacity and capability and our ability to generate growth;
(xi) uncertainty in industry demand;
(xii) general economic conditions and market conditions in our industry;
(xiii) future sales of large blocks of our securities, which may adversely impact our share price;
(xiv) the depth of the trading symbol "CDOM.OB".market in our securities;
(xv) a projection of income (including income/loss), earnings (including earnings/loss) per share, capital expenditures, dividends, capital structure or other financial items,
(xvi) our future financial performance, including any such statement contained in a discussion and analysis of financial condition by management or in the results of operations included pursuant to the rules and regulations of the SEC, and
(xvii) the assumptions underlying or relating to any statement described in points (i), (ii) or (iii) above. These statements are not guarantees of future performance and are subject to numerous risks, uncertainties, and assumptions that are difficult to predict.
Because forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified, you should not rely upon forward-looking statements as predictions of future events. The events and circumstances reflected in the forward-looking statements may not be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. Except as required by applicable law, including the securities laws of the United States and the rules and regulations of the SEC, we do not assume any obligation to update any forward-looking statement. We disclaim any intention or obligation to update or revise any forward-looking statement contained herein, whether as a result of new information, future events or otherwise.
USE OF NAMED EXPERTS AND COUNSELPROCEEDS
None
INFORMATION WITH RESPECT TO THE REGISTRANTWe estimate that our net proceeds from this Offering will be approximately $ million, after deducting the underwriting discounts and commissions and estimated offering expenses payable by us. This estimate excludes the proceeds, if any, from exercise of the warrants sold in this Offering. If all of the warrants sold in this Offering were to be exercised in cash at the exercise price of $ per share, we would receive additional net proceeds of approximately $ million. We cannot predict when or if these warrants will be exercised. It is possible that these warrants may expire and may never be exercised.
Description
We intend to use the net proceeds for working capital and general corporate purposes, including, but not limited to, completing enrollment in the ongoing CardiAMP Cell Therapy pivotal trial for the treatment of Businessheart failure, the funding of clinical development and pursuing regulatory approval for our product candidates.
We have not determined the amount of net proceeds to be used specifically for such purposes. Pending the use of any net proceeds, we expect to invest the net proceeds in interest-bearing, marketable securities.
DIVIDEND POLICY
We have never declared or paid cash dividends on our common stock. We currently intend to retain all available funds and any future earnings for use in the operation of our business and do not anticipate paying any dividends on our common stock in the foreseeable future. Any future determination to declare dividends will be made at the discretion of our board of directors and will depend on our financial condition, operating results, capital requirements, general business conditions and other factors that our board of directors may deem relevant.
CAPITALIZATION
The following business descriptiontable presents a summary of our cash and cash equivalents and capitalization as of March 31, 2019:
on an actual basis;
You should be read the following table in conjunction with the consolidatedour historical financial statements and the related notes thereto incorporated by reference into this prospectus.
As of March 31, 2019(1) | ||||||||||||
(in thousands except for share and per share data)(unaudited) | ||||||||||||
|
| Pro Forma, | ||||||||||
| Actual | Pro Forma | As Adjusted | ||||||||||
Assets | ||||||||||||
Cash and cash equivalents | $ | 2,838 | $ | $ | ||||||||
Liabilities | ||||||||||||
Total liabilities | 4,339 | |||||||||||
Stockholders’ Equity | ||||||||||||
Preferred stock, $0.001 par value, 25,000,000 shares authorized; no shares issued and outstanding | ||||||||||||
Common stock, $0.001 par value, 100,000,000 shares authorized; 4,847,471 shares issued and outstanding | 44 | |||||||||||
Additional paid-in capital | 90,800 | |||||||||||
Accumulated deficit | (90,026 | ) | ||||||||||
Total Stockholders’ Equity | 818 | |||||||||||
Total Capitalization | 5,157 | |||||||||||
(1) Reflects a one-for-nine reverse stock split of our issued and outstanding shares of common stock, options, restricted stock units and warrants that became effective May 7, 2019 and the corresponding adjustment of all common stock and prices per share and stock option and warrant exercise prices per share and conversion ratios.
The foregoing as adjusted information is illustrative only, and our capitalization following the completion of this Offering will be adjusted based on the actual public offering price and other terms of this Offering determined at pricing. You should read this table together with our financial statements and the related notes appearing elsewhere in this registration statement.prospectus and the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section of this prospectus.
Organization
The table and discussion set forth in the table above excludes, as of March 31, 2019:
● | 606,249 shares of our common stock issuable upon the exercise of options outstanding as of March 31, 2019, with a weighted-average exercise price of $24.39 per share; |
● | 27,435 shares of our common stock issuable upon the vesting of restricted stock units outstanding as of March 31, 2019; |
● | 296,295 shares of our common stock issuable upon the exercise of the warrants outstanding as of March 31, 2019 with a weighted-average exercise price of $6.75 per share; |
● | 410,596 shares of our common stock reserved for future issuance under our equity compensation plans; |
● | the shares issuable upon the exercise of warrants to be issued in this Offering (or shares if the underwriters exercise their over-allotment option in full); |
● | the shares of our common stock that may be issued upon exercise of the underwriter warrants; and |
| ● | the shares of our common stock that may be issued upon exercise of the Private Placement Warrants. |
DILUTION
If you invest in the securities being offered by this prospectus, your interest will be diluted immediately to the extent of the difference between the public offering price per Unit and the adjusted net tangible book value per share of our common stock after this Offering.
The net tangible book value of our common stock as of March 31, 2019, was approximately ($0.6) million, or approximately ($0.12) per share. Net tangible book value per share represents the amount of our total tangible assets, excluding goodwill and intangible assets, less total liabilities, divided by the total number of shares of our common stock outstanding. Dilution per share to new investors represents the difference between the effective amount per share paid by purchasers for each share of common stock in this Offering and the net tangible book value per share of our common stock immediately following the completion of this Offering.
Dilution in net tangible book value per share represents the difference between the amount per share paid by purchasers in this Offering and the net tangible book value per share of our common stock immediately after this Offering. After giving effect to (i) the pro forma adjustments referenced under “Capitalization,” and (ii) the sale of Units in this Offering at a public offering price of $ per Unit (attributing no value to the warrants or proceeds from the sale of warrants being offered), and after deducting the underwriting discounts and commissions and the estimated offering expenses payable by us, our as adjusted net tangible book value as of March 31, 2019 would have been approximately $ per share of common stock. This represents an immediate increase in pro forma net tangible book value of $ per share to our existing stockholders and an immediate dilution of $ per share to investors purchasing shares of common stock in this Offering.
The following table illustrates this dilution on a per share basis assuming $ of value is attributable to each warrant issued as part of each unit:
Public offering price per Unit | $ | |||
Net tangible book value per share at March 31, 2019 | $ | ( 0.12 | ) | |
Increase to net tangible book value per share attributable to investors purchasing our common stock in this Offering | ||||
| Pro forma, net tangible book value per share after giving effect to the pro forma adjustments referenced under “Capitalization��� | ||||
Pro forma, as adjusted net tangible book value per share as of March 31, 2019, after giving effect to this Offering | ||||
Dilution of pro forma net tangible book value per share to investors purchasing our common stock in this Offering | $ |
Each $1.00 increase (decrease) in the assumed public offering price of $ per share, would increase (decrease) our pro forma as adjusted net tangible book deficit per share to new investors by $ , and would increase (decrease) dilution per share to new investors in this Offering by $ , assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting the estimated discounts and commissions and estimated offering expenses payable by us. In addition, to the extent any outstanding options to purchase common stock are exercised or any outstanding restricted stock units vest, new investors would experience further dilution. If the underwriters exercise their option to purchase additional shares in full, the pro forma as adjusted net tangible book value per share of our common stock after giving effect to this Offering would be approximately $ per share, and the dilution in pro forma as adjusted net tangible book value per share to investors in this Offering would be approximately $ per share of common stock.
The table and discussion set forth in the table above excludes, as of March 31, 2019:
● | 606,249 shares of our common stock issuable upon the exercise of options outstanding as of March 31, 2019, with a weighted-average exercise price of $24.39 per share; |
● | 27,435 shares of our common stock issuable upon the vesting of restricted stock units outstanding as of March 31, 2019; |
● | 296,295 shares of our common stock issuable upon the exercise of the warrants outstanding as of March 31, 2019 with a weighted-average exercise price of $6.75 per share; |
● | 410,596 shares of our common stock reserved for future issuance under our equity compensation plans; |
● | the shares issuable upon the exercise of warrants to be issued in this Offering. (or shares if the underwriters exercise their over-allotment option in full); |
● | the shares of our common stock that may be issued upon exercise of the underwriter warrants; and |
| ● | the shares of our common stock that may be issued upon exercise of the Private Placement Warrants. |
To the extent that any of these outstanding options are exercised or warrants are exercised or we issue additional shares under our equity incentive plans, there may be further dilution to new investors. In addition, we may choose to raise additional capital due to market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. To the extent that additional capital is raised through the sale of equity or convertible debt securities, the issuance of these securities could result in further dilution to our stockholders.
Unless otherwise indicated, information presented in this section reflects a one-for-nine reverse stock split of our issued and outstanding shares of common stock, options and warrants pursuant to an amendment to our certificate of incorporation effective May 7, 2019, and the corresponding adjustment of all common stock prices per share and stock option and warrant exercise prices per share and conversion ratios. Our common stock began trading on a reverse stock split-adjusted basis on The OTC Market on the opening of trading on June 6, 2019.
BUSINESS
Description of Business
We are a clinical-stage regenerative medicine company developing novel therapeutics for cardiovascular diseases with large unmet medical needs. Our lead therapeutic candidate is the investigational CardiAMP Cell Therapy System, which provides an autologous bone marrow derived cell therapy (using a patient’s own cells) for the treatment of two clinical indications: heart failure that develops after a heart attack and chronic myocardial ischemia. Chronic myocardial ischemia involves sustained poor blood perfusion to the heart muscle often at the microvascular level. The CardiAMP Cell Therapy System is being developed to provide a comprehensive biotherapeutic solution, incorporating a proprietary molecular diagnostic to characterize the potency of a patient’s own bone marrow cells and determine if they are an optimal candidate for therapy, a proprietary point of care processing platform to prepare cells at the patient’s bedside, an optimized therapeutic formulation that builds on the total experience in the cardiac stem cell field to-date, and a proprietary interventional delivery system that easily navigates a patient’s vasculature to securely deliver cells in a routine cardiac catheterization procedure. Our second therapeutic candidate is the CardiALLO Cell Therapy System, an investigational culture expanded bone marrow derived allogenic “off the shelf” cell therapy from a donor that meets specified criteria, which has potential to be advanced for many clinical indications including heart failure.
We are committed to applying our expertise in the fields of autologous and allogeneic cell-based therapies to improve the lives of patients with cardiovascular conditions.
Market Overview
Cardo
Adult bone marrow contains a large reservoir of stem and progenitor cells capable of differentiating into blood cells, blood vessel cells, and connective tissue cells. In addition, numerous pre-clinical cardiac studies have shown that cell-to-cell communication in which bone marrow-derived stem cells promotes microcirculatory adaptation, immune modulation and cell protection, facilitating cardiac recovery, in part via recruitment of other reparative cell types.
Bone marrow cell homing to the heart is believed to be part of the body’s natural repair process. After a heart attack or an acute injury to the heart, cells from bone marrow are known to home to the heart. For example, a population of bone marrow cells that express the surface marker CD34 has certain receptors, including CXC-4 and CXC-7 receptors, that home to the SDF-1 ligand in injured heart tissue. In heart failure, the heart may have fewer of these homing signals and a decreased ability to stimulate or recreate this signaling process, leading to a lower likelihood of heart tissue repair. A number of other bone marrow derived cells with unique cell surface markers have also been shown to have beneficial effects in animal models of heart failure and chronic myocardial ischemia disease when delivered directly to the heart.
Bone marrow derived cell-based therapy has been shown to have the potential to provide therapeutic benefit for patients with heart failure and chronic myocardial ischemia. In the past decade, intramyocardial delivery of bone marrow derived cell-based therapies in preclinical and clinical studies of heart failure and chronic myocardial ischemia has predominantly resulted in benefits, such as improvement in ventricular function, reduction in the area of dead heart tissue and increase in heart muscle blood flow, reduction in pain symptoms, reduced major adverse event rates, and reduced mortality.
Recent systematic review and meta-analysis of the scientific literature from 23 randomized controlled trials prior to 2013, covering more than 1,200 participants, was published by Fisher in Circulation Research in January 2015. The review found evidence that bone marrow cell treatment, including intramyocardial delivery of bone marrow cells, has improved left ventricle ejection fraction, or LVEF, and chronic ischemic heart disease. The authors of the review found evidence for a potential beneficial clinical effect in terms of mortality and performance status after at least one year post-treatment in people who suffer from chronic ischemic heart disease and heart failure. Results in heart failure trials indicate that bone marrow derived cell-based therapy leads to a reduction in deaths and readmission to hospital and improvements over standard treatment as measured by tests of heart function. This review concluded that further research is required to confirm the results. Published scientific papers provide clinical support for efficacy from randomized controlled clinical trials of intramyocardial delivery of bone marrow derived cells in closely related clinical conditions of chronic myocardial ischemia, diastolic heart failure, and subacute myocardial infarction.
Product Pipeline and Development Status
CardiAMP Cell Therapy System
The CardiAMP Cell Therapy System, or CardiAMP, is our lead therapeutic program being advanced for two clinical indications. This investigational cell therapy system is comprised of (i) a cell potency screening test, (ii) a point of care cell processing platform, and (iii) a biotherapeutic delivery system. In the screening process, the physician extracts a small sample of the patient's bone marrow in an outpatient procedure performed under local anesthesia. The clinic sends the sample to a centralized diagnostic lab, which tests for identified biomarkers from which we generate a potency assay score for the patient. During the treatment for patients who are assessed as meeting the indication specific CardiAMP cell potency assay score , a doctor harvests and then prepares the patient's own bone marrow mononuclear cells, or autologous cells, using our point of care cell processing platform, which a cardiologist then delivers into the heart using our proprietary biotherapeutic delivery system. We designed the entire procedure to be performed in approximately 60 to 90 minutes, which we believe is substantially faster than alternative cell-based therapies in development. The patient then leaves the hospital the same or next day.
CardiAMP Cells Phase I Heart Failure Study: Transendocardial Autologous Marrow Cells in Myocardial Infarction
The CardiAMP Phase I Transendocardial Autologous Marrow Cells in Myocardial Infarction or TABMMI trial enrolled 20 patients with ischemic systolic heart failure in an open label safety trial of bone marrow cells delivered with the Helix™ biotherapeutic delivery system at a dosage of 100 million cells. Results showed improvement in cardiac function as measured by left ventricular ejection fraction, improved exercise tolerance, and superior survival as compared to historical controls. The Phase I TABMMI study was submitted to the Argentine Administración Nacional de Medicamentos, Alimentos y Technología Médica.
In our TABMMI Phase I trial of CardiAMP cells, we enrolled 20 patients with previous evidence of having had a heart attack and who presented with a low ejection fraction of less than or equal to 40% and greater than or equal to 20%. Baseline evaluations included informed consent, history and physical examination, electrocardiogram, 24-hour Holter monitoring, echocardiography, routine blood tests and exercise tolerance testing. Reduced regional heart wall motion was coincident with the diseased coronary vessel in each patient. A total of 20 patients with heart failure (NYHA Class I, II and III) each received three to ten transendocardial infusions of cells using our Helix™ biotherapeutic delivery system in an open-label dose-escalation two cohort trial. Dosage administration ranged from 30 million to 130 million autologous bone marrow derived mononuclear cells, with an average of 96 million cells.
Bone marrow cells delivered in TABMMI demonstrated an excellent safety profile in this heart failure population, with no treatment related toxicities observed. The 20 patients who received CardiAMP cells, demonstrated improvements from baseline to both six-month and 12-month follow-up across a number of parameters important in heart failure, including statistically and clinically significant improvements in left ventricular, or LV, function (ejection fraction). The difference of average ejection fraction was statistically significant over baseline at all follow-up time points of 6 months, 12 months, and 24 months. Average exercise tolerance time showed an increase at all follow-up time points but was only statistically significant at 12 months and 24 months.
A total of 12 adverse events were observed in six patients, although none were related to the investigational delivery or cell transplantation procedure. The complete results of the 20 patients at two-year follow-up have been published in the journal Eurointervention in 2011.
CardiAMP Cells Phase II Heart Failure Trial: Transendocardial Autologous Cells in Heart Failure Trial (TAC-HFT)
In our co-sponsored Phase II Transendocardial Autologous Cells in Heart Failure Trial, patients with ischemic systolic heart failure were randomized on a one to one basis into two double-blind, placebo-controlled trials: TACHFT-BMC and TACHFT-MSC. The IND for the TACHFT trial was filed with the U.S. Food and Drug Administration, or FDA Center for Biologics Evaluation and Research in 2008 by the University of Miami, the co-sponsor of the trial.
In the safety dose escalation roll-in cohort stage of the study, eight patients received treatment with either CardiAMP cells, or autologous bone marrow mesenchymal cells, or MSC, at dosages of 100 million or 200 million cells. In the randomized, placebo-controlled efficacy stage of the study, 29 patients received treatment with either CardiAMP cells or placebo and 30 patients received treatment with either MSCs or placebo. The mode of administration was 10 intramyocardial infusions per patient using our Helix™ biotherapeutic delivery system into the myocardium adjacent to and into the infarcted tissue. All subjects had ischemic systolic heart failure (NYHA Class I, II or III).
TACHFT-BMC met its primary safety endpoint at both dosages (100 million and 200 million cells) and treated patients had increased functional capacity, improved quality of life, symptoms and key markers of cardiac function predictive of survival, such as end systolic volume, or ESV. The TACHFT-BMC trial included a single dose of CardiAMP cells with a follow up observation period of 12 months. The Phase II, randomized, placebo-controlled study met its primary safety endpoint and demonstrated statistically significant and clinically meaningful improvements in secondary efficacy endpoints of functional capacity, as measured by the six minute walk distance (6MW), and in quality of life, as measured by the Minnesota Living with Heart Failure Questionnaire score. Phase II results were published in the journal of the American Medical Inc. ("Cardo"Association in 2014 and were presented at the World Congress of Regenerative Medicine in 2015.
By way of comparison, previous investigational device exemption (IDE) pivotal clinical trials that led to FDA approval of Cardiac Resynchronization Therapy, in which a pacemaker stimulates both sides of a patient’s heart for the treatment of heart failure, followed the same IDE regulatory pathway and have had similar endpoints to the proposed CardiAMP Heart Failure Trial. Pacemakers that pace both sides of the heart are intended for patients that are NYHA III and IV versus the CardiAMP Heart Failure Trial indication of NYHA II and III. Results from 5 out of 6 randomized pivotal trials for pacemakers that pace both sides of the heart have showed both smaller improvements in functional capacity as measured by the six minute walk test and smaller improvement in quality of life than the CardiAMP Phase II results. Although the benefits with pacemakers that pace both sides of the heart were less than observed in CardiAMP placebo controlled Phase II trial, these results for the permanently implantable pacemaker devices were sufficient to obtain FDA approval.
CardiAMP Cell Phase III Heart Failure Trial
The CardiAMP Heart Failure Trial is a Phase III, multi-center, randomized, double-blinded, sham-controlled study of up to 260 patients at 40 centers nationwide, which includes a 10-patient roll-in cohort. The Phase III pivotal trial is designed to provide the primary support for the safety and efficacy of the CardiAMP Cell Therapy System. The primary endpoint is a clinical composite of six minute walk distance and major adverse cardiac and cerebrovascular events. Based on the results achieved in the Phase II trial, our Phase III pivotal trial is designed to have more than 95% probability of achieving a positive result with statistical significance. Statistical significance denotes the mathematical likelihood that the results observed are real and not due to chance.
Particularly novel aspects of this trial include a cell potency assay to screen subjects who are most likely to respond favorably to treatment, a point of care treatment method, use of a high target dose of 200 million cells and an efficient transcatheter delivery method that is associated with high cell retention. Success in the primary endpoint of the trial may lead to a new treatment for those suffering from heart failure in the aftermath of a heart attack. The trial design was published in the peer reviewed American Heart Journal in 2018.
The Department of Health & Human Services Centers for Medicare & Medicaid Services, or CMS, has designated the "Company"CardiAMP Heart Failure Trial as a qualifying trial for Medicare national coverage determination that routine costs of care will be covered for Medicare beneficiaries. Private insurance plans covering 50 million insured Americans follow this CMS reimbursement policy and are also anticipated to pay for these costs in the CardiAMP Heart Failure Trial. Covered costs today for both the treatment and control arms of the trial include patient screening, the CardiAMP Cell Therapy System and procedure, and clinical follow-up at one and two years after the procedure.
The Phase III CardiAMP Heart Failure Trial was initiated in the fourth quarter of 2016, and the first patient treated in Q1 2017. The Data Safety Monitoring Board (DSMB) safety review of the 10 patient roll in cohort treated at three clinical sites was completed successfully in the third quarter of 2017, and the trial is actively enrolling today at 22 clinical sites. Efficacy data from the primary endpoint in the open label roll in cohort were presented at the American Heart Association Scientific Sessions in 2018.
At the primary endpoint of exercise capacity at 12 months, the 10-patient roll-in cohort of the trial showed clinically meaningful improvement, walking an average of 46.4 meters more than baseline, although the improvement was not considered statistically significant (p=0.06). Eight of the 10 patients experienced improvement in their exercise capacity. This improvement is more than triple the average improvement over baseline reported in the CardiAMP-treated arm of the Phase II TAC-HFT-MNC trial, and greater than the average improvement seen in a number of pivotal trials for implantable pacemakers to treat heart failure.
In the secondary efficacy endpoint of quality of life, patients showed a clinically meaningful improvement of 9.8 points relative to baseline, which was not statistically significant (p=0.33) in the small cohort. Seven of the 10 patients reported better quality of life after CardiAMP treatment. This was a greater improvement over baseline than was seen in the Phase II TAC-HFT-MNC trial of CardiAMP therapy.
The secondary efficacy endpoints of superiority relative to major adverse cardiac events (MACE) and survival were not possible to assess in this roll-in cohort as there is no control arm specific to this cohort. There were no treatment emergent major adverse cardiac events in this group at 30 days, while there was one MACE event due to a hospitalization at nine months. All MACE have not been adjudicated at this time. All patients from this cohort were alive and out of the hospital at 12 months.
Echocardiographic core lab measures of cardiac function in the roll-in cohort at 12 months showed improvement in left ventricular ejection fraction of 4.1 percent (N=10, p=0.181), left ventricular end systolic volume (2.5 ml, p=0.502), neither of which were statistically significant in this small cohort relative to baseline. However, statistically significant improvement in total wall motion score (5.9 points, p=0.014) were observed.
We have enrolled 45 patients in the trial to date and enrollment is anticipated to begin accelerating due to additional compelling data presented from the roll in cohort, the addition of world class centers to the trial, and the completion of competitive clinical programs. We anticipate a first interim readout from the trial in Q3 2019, a second interim readout in Q3 2020, that trial enrollment will be completed in Q3 2020 and that top line data will be available in Q3 2021.
Although clinical trial results show support for safety and efficacy in our Phase I TABMMI trial, Phase II TACHFT-BMC trial, and Phase III CardiAMP Heart Failure trial, the CardiAMP Cell Therapy System still remains investigational, and no claims regarding safety or efficacy can be made until the products are approved by the FDA.
We believe the remaining clinical efficacy risk is modest in light of the Phase I, II, and III data available, and broader literature which supports CardiAMP Cell Therapy System as a therapeutic candidate for heart failure secondary to having had a heart attack. The CardiAMP Cell Therapy System has the potential to significantly benefit patients who have limited options and provide a cost-effective therapy to help reduce the substantial heart failure hospitalization and care costs. Unlike other autologous cell therapies, the CardiAMP Cell Therapy System, is expected to have elegantly straightforward and low cost manufacturing and distribution, significantly enhancing its probability of commercial success.
CardiAMP Chronic Myocardial IschemiaPhase III PivotalTrial
In 2017, we submitted for approval of an investigational device exemption for the CardiAMP Cell Therapy System in a second related clinical indication of chronic myocardial ischemia based on the strength of our Phase I and II heart failure trial data, and the strength of the clinical data showing support for the efficacy of one component of our cell therapy (the CD34+ cells) in chronic myocardial ischemia. In January 2018, the FDA approved the IDE for the randomized controlled pivotal trial of autologous bone marrow mononuclear cells using the CardiAMP Cell Therapy System in patients with refractory chronic myocardial ischemia for up to 343 patients at up to 40 clinical sites in the United States. This therapeutic approach uses the same novel aspects as the CardiAMP Heart Failure Trial. An update to the statistical analysis plan to enable an adaptive trial design is anticipated. Success in the primary endpoint of the trial, which is exercise tolerance, may lead to a new treatment for those suffering from chronic myocardial ischemia and having sustained debilitating heart pain, referred to as refractory angina.
In 2018, CMS approved BioCardia’s request for the designation of the CardiAMP Chronic Myocardial Ischemia Trial as a qualifying trial for Medicare national coverage determination similar to the designation received for the CardiAMP Heart Failure Trial. It is anticipated that this second pivotal trial will build on and benefit from the experience and infrastructure from the CardiAMP Heart Failure Trial. With additional modest funding to support this program, there is potential for this trial to be activated in 2019.
CardiAMP Cell Therapy System - Other Indications
In the future, we may also explore the continued development of CardiAMP for use immediately after a heart attack and for treatment of heart failure with preserved ejection fraction, a form of heart failure wherein the amount of blood pumped from the heart's left ventricle with each beat (ejection fraction) is greater than 50%.
CardiALLO Cell Therapy System
Our second therapeutic candidate is the CardiALLO Cell Therapy System, an investigational culture expanded bone marrow derived “off the shelf” mesenchymal stem cell therapy. CardiALLO cell therapy cells are expanded from Neurokinin-1 receptor positive bone marrow cells. These cells are being advanced to treat heart failure, but have potential for numerous therapeutic applications as these are anticipated to be the cells that respond to the release of Substance P. Substance P (“SP”) is a neuropeptide released from sensory nerves and is associated with the inflammatory processes and pain. The endogenous receptor for SP is the neurokinin-1 receptor (“NK1-receptor” or “NK1R”), which is distributed over cytoplasmic membranes of many cell types (for example neurons, glia, endothelia of capillaries and lymphatics, fibroblasts, stem cells, and white blood cells) in many tissues and organs. SP amplifies or excites most cellular processes. Elevation of serum, plasma, or tissue SP and/or its receptor NK1R has been associated with many diseases: sickle cell crisis, inflammatory bowel disease, major depression and related disorders, fibromyalgia rheumatological, and infections such as HIV/AIDS and respiratory syncytial virus, as well as in cancer. Our CardiALLO NK1R positive derived cells are believed to be an orthopedic medical device company specializingimportant subset of the cells that we have delivered in designing, developingour previous preclinical and marketingclinical mesenchymal stem cell studies. We believe this therapy presents the advantages of an "off the shelf" therapy that does not require tissue harvesting or cell processing. We have completed manufacturing validation runs of these cells at BioCardia to support future clinical studies. We are working to obtain FDA acceptance of an Investigational New Drug (“IND”) application for a Phase I/II trial for CardiALLO Cell Therapy System for the treatment of ischemic systolic heart failure in the second quarter of 2019.
The subset of patients we are targeting initially for the CardiALLO Heart Failure Trial are those that have been excluded from the CardiAMP Heart Failure Trial due to their lower cell potency assay scores. CardiALLO trial activation would likely enhance enrollment in the CardiAMP Heart Failure Trial. And if the CardiAMP trial is successful, as anticipated, there is the potential for the CardiALLO therapy indication to be designated as an orphan indication.
CardiALLO related Phase I /II Studies: POSEIDON, TAC-HFT-MSC, and TRIDENT
We have co-sponsored three clinical trials for MSCs for the treatment of ischemic systolic heart failure. In substantially similar trial designs, the POSEIDON Phase I/II trial compared autologous MSCs to allogeneic MSCs, the TACHFT-MSC Phase II trial compared autologous MSCs to placebo, and the TRIDENT Phase II compared allogenic MSCs at different doses. The first two trials shared common arms of autologous MSCs, enabling a bridge to placebo, leading us to conclude that allogeneic MSC therapy has potential to be superior to placebo. The IND for the TACHFT trial was filed with the FDA Center for Biologics Evaluation and Research in 2008 by the University of Miami, our co-sponsor for the trial. The POSEIDON trial and the TRIDENT trials were submitted by amendment under the same IND filed for the TACHFT study, and was co-sponsored by the University of Miami, the National Institutes of Health and us. The results from all three of these studies can be submitted to the FDA in support of an IND for the CardiALLO Cell Therapy System.
CardiALLO Development
CardiALLO is being advanced with an anticipated improved cell production strategy to be detailed in the Chemistry Manufacturing and Controls (CMC) of the IND in development. We believe the new CMC will reduce the likelihood of immune response to transplanted allogenic cells further, may enhance efficacy, and will enable commercial scale up and global distribution. CardiALLO will require more extensive clinical development than the CardiAMP Cell Therapy System, beginning with a Phase I/II trial that follows previous work, to confirm the results with the modified cell culture and dosage strategy.
We are performing our own CMC development work in BioCardia laboratories to accelerate the effort and secure additional intellectual property, and have taken steps to build the capacity at BioCardia to culture and supply the MSC cells for CardiALLO clinical development. Our development tissue culture laboratory is fully operational and our clinical manufacturing controlled environment room has been built. We expect to demonstrate support for the safety and efficacy of MSCs in our target patient population in a Phase I/II randomized controlled study. We expect the CardiALLO Phase II Heart Failure Trial will enroll patients with control, low dose and high performance reconstructive joint devicesdose groups using the Helix™ biotherapeutic delivery system and spinal surgical devices. Reconstructive joint devicesa similar inclusion criterion as the CardiAMP Heart Failure Trial. In the United States, CardiALLO Cell Therapy System is expected to be regulated by the FDA as a biologic product with a dedicated delivery system, our own Helix™ biotherapeutic delivery system.
The completed clinical studies show support for the safety and efficacy of both the CardiAMP Cell Therapy System and CardiALLO Cell Therapy System development programs; however, both product candidates remain investigational, and no claims regarding safety or efficacy can be made until the constituent products are approved by the FDA. As we engage in clinical trials of our therapeutic candidates, we have compensated and intend to compensate all parties performing the trials or studies (including all the parties identified in our Annual Report on Form 10-K) only on terms that are standard and customary in clinical study arrangements.
These two therapeutic candidates provide compelling and synergistic approaches to replicating the natural response of bone marrow cells to cardiac injury. CardiAMP harnesses the potential of autologous minimally processed bone marrow cells, using an anticipated companion diagnostic to identify patients most likely to benefit from the therapy. CardiALLO utilizes mesenchymal stem cells from a donor that meets specified criteria and may be appropriate for patients who are not optimal candidates for the CardiAMP therapy.
Cell Processing and Cell Delivery Product Platforms
BioCardia has developed and secured exclusive rights to enabling cell processing and cell delivery products, which are used as part of our CardiAMP and CardiALLO therapies, and which we believe validate our approach and development expertise: (i) the CardiAMP cell processing platform, (ii) the CardiALLO cell processing platform, (iii) the Helix™ transendocardial biotherapeutic delivery system, and (iv) our Morph vascular access products.
• | CardiAMP cell processing platform - processes bone marrow aspirate at the point of care to concentrate mononuclear cells and prepare the dosage form. We expect the CardiAMP cell processing platform to be approved in the United States for ischemic systolic heart failure and/or chronic myocardial ischemia as part of the CardiAMP Cell Therapy System clinical development. The platform is currently cleared for use in the United States and in European Union for the preparation of a cell concentrate from bone marrow. The platform is approved in Japan with a broad regenerative medicine indication. The platform is under investigational use for the treatment of heart failure and chronic myocardial ischemia under BioCardia investigational device exemptions in the United States. | |
• | CardiALLO cell processing platform - processes young qualified donor marrow at BioCardia cell manufacturing facility to expand the cell subpopulation that is NK1R positive to produce a cryopreserved “off the shelf” cell dosage that may be shipped to hospitals for therapeutic delivery. This manufacturing facility is believed to be suitable and sufficient for the planned Phase I and Phase II clinical trials for a number of clinical indications including heart failure. | |
• | Helix™biotherapeutic delivery system - delivers therapeutics into the heart muscle with a penetrating helical needle from within the heart. This is a leading delivery platform in the field, which has increased safety and performance. We expect Helix™ to be approved in the United States as part of CardiAMP Cell Therapy System. The system is under investigational use in the United States as part of our CardiAMP Cell Therapy System and CardiALLO Cell Therapy System development programs. We believe the Helix™ biotherapeutic delivery system is the world's safest and most efficient platform for cardiac therapeutic delivery. It has been used in more than 300 clinical procedures and is designed to be used in any catheterization laboratory in the world without the need for additional capital equipment. We supply our Helix™ biotherapeutic delivery system to selected partners developing other cell, gene, and protein therapeutic programs. These programs provide additional data, intellectual property rights, and opportunities to participate in the development of combination products for the treatment of cardiac diseases. On April 1, 2019, CE Mark certification expired due to delays in certifying to the new ISO 13485:2016 standard, which defines new requirements in the quality management system required for medical devices. We anticipate being in compliance within calendar year 2019. Until we receive renewal of our EC certificate for CE marking, we will not be able to sell our delivery system in Europe. This is not expected to have a material impact on the Company because we have planned for such delay with our Biotherapeutic partners that use Helix™ in Europe. | |
• | Morph vascular access products - provides enhanced control for Helix™ in biotherapeutic delivery and for other common interventions. We have secured all necessary approvals in the United States and Europe. Currently there are seven Morph product model numbers approved for commercial sale in the United States via a 510(k) clearance. The Morph products are valued by physicians performing difficult vascular procedures worldwide and they have been used in more than 12,000 clinical procedures to date. We are working to receive FDA clearance for market release of new Morph product family members, which include the AVANCE bidirectional steerable introducers for use in transseptal procedures. These transseptal procedures may include atrial fibrillation ablation, patent foramen ovale and atrial septal defect repair, percutaneous mitral valve repair, and left atrial appendage closure, among others. FDA clearance of the AVANCE 510(k) submitted to the FDA during the first quarter of 2019 was received in May 2019. |
Business Strategy
We are committed to replace knee, hipapplying our expertise in the fields of autologous and allogeneic cell-based therapies to improve the lives of patients with cardiovascular conditions. We are pursuing the following business strategies:
• | Complete the ongoing 260 patient, 40 center Phase III pivotal IDE trial of CardiAMP Cell Therapy System for patients with ischemic systolic heart failure. |
• | Complete the FDAapproved, CMS reimbursed,343 patient, 40 centerPhase III pivotal IDE trial of CardiAMP Cell Therapy System for patients with chronic myocardial ischemia. |
• | Obtain FDA approval and commercialize CardiAMPCell Therapy Systemusing a highly-targeted cardiology sales force in the United States. |
• | Advance our CardiALLOCell Therapy Systemfor the treatment of ischemic systolic heart failure. CardiALLO has the potential to benefit patients for whom the CardiAMP Cell Therapy System is not optimal due to the lower potency of their bone marrow cells. This therapy may present advantages for patients or physicians who wish to avoid bone marrow aspiration, and our development work builds on our clinical development capabilities established through our CardiAMP program. This program positions us to provide therapy to patients ineligible for CardiAMP. |
• | Expand CardiAMP and CardiALLOCell Therapy Systemsinto additional cardiac indications. CardiAMP and CardiALLO Cell Therapy Systems have potential therapeutic benefits for multiple cardiovascular indications in addition to ischemic systolic heart failure and ischemic heart disease. | |
• | Continue todevelop andpartner our Helix™biotherapeutic delivery system for use with other biotherapeutics. We plan to continue to make our Helix™ biotherapeutic delivery system available for use by qualified partners seeking to advance their own biotherapeutic candidates for similar indications. | |
• | Continue todevelop andcommercialize Morph catheter products. We plan to continue to enhance the performance of our Morph catheter products to benefit the CardiAMP and CardiALLO Cell Therapy Systems, to enhance Helix™ partnering, and to grow revenues. |
Intellectual Property
We strive to protect and enhance the proprietary technologies that we believe are important to our business and seek to obtain and maintain patents for any patentable aspects of our therapeutic candidates or products, including our anticipated companion diagnostic, their methods of use and any other inventions that are important to the development of our business. Our success will depend significantly on our ability to obtain and maintain patent and other joints thatproprietary protection for commercially important technology, inventions and know-how related to our business, defend and enforce our patents, maintain our licenses to use intellectual property owned by third parties, preserve the confidentiality of our trade secrets and operate without infringing the valid and enforceable patents and other proprietary rights of third parties. We also rely on know-how, continuing technological innovation and in-licensing opportunities to develop, strengthen, and maintain our proprietary position in the fields targeted by our therapeutic candidates.
We have deteriorated through diseasea large patent portfolio of issued and pending claims covering the CardiAMP Cell Therapy System, the CardiALLO Cell Therapy System, the Helix™ biotherapeutic delivery system product and the Morph vascular access catheter products. As of December 31, 2018, we had developed or injury. Spinal surgical devices involve productssecured rights to stabilize the spine for fusionover 70 issued or pending U.S. and reconstructive procedures. Within these areas, Cardo intends to focus on the higher-growth sectorsinternational patents or patent pending applications. We have sole ownership of the orthopedic industry, such as advanced minimally invasive instrumentationpatents that we consider to be material, other than the patents that we license exclusively from Biomet Biologics, LLC. Our issued U.S. patents expire between 2019 and bone-conserving high-performance implants. Cardo is focused on developing surgical devices that will enable surgeons2034, without taking into consideration patent term extension. Among these are five issued material US patents related to bridge the gapCardiAMP Cell Therapy System and the CardiALLO Cell Therapy System, which expire between soft tissue-driven sports medicine techniques2027 and classical reconstructive surgical procedures. Cardo commercializes its reconstructive joint devices through its Cardo Orthopedics division2029 without taking into consideration patent term extension. We maintain trade secrets covering a significant body of know-how and its spine devices through its Cardo Spine division.proprietary information related to our core therapeutic candidates, biotherapeutic delivery systems and technologies. As a result, we believe our intellectual property position provides us with substantial competitive advantages for the commercial development of novel therapeutics for cardiovascular diseases.
32
Our most recently issued United States Patents are listed below,
US Patent No. | Patent Title | Expiration on or after |
10,071,226 | Radial and trans-endocardial delivery catheter | 2034 |
10,035,982 | Method of preparing autologous cells and methods of use for therapy | 2029 |
9,945,854 | Methods of measuring therapeutic potency potential and defining dosages for autologous cell therapy | 2034 |
9,752,123 | Method of Preparing Autologous Cells and Methods of Use for Therapy | 2029 |
9,517,199 | Treatment for chronic myocardial infarct | 2027 |
9,504,642 | Treatment for chronic myocardial infarct | 2027 |
9,301,975 | Method of preparing autologous cells and method of use for therapy | 2029 |
8,496,926 | Treatment for chronic myocardial infarction | 2027 |
U.S. Regulatory Protection for CardiAMP and CardiALLO
In December 2006, Cardo initiatedaddition to patent and trade secret protection, we may receive a 12-year period of regulatory exclusivity from the FDA upon approval of CardiAMP Cell Therapy System and CardiALLO Cell Therapy System pursuant to the Biologics Price Competition and Innovation Act. The exclusivity period, if granted, will run from the time of FDA approval. This exclusivity period, if granted, will supplement the intellectual property protection discussed above, providing an additional barrier to entry for any competitor seeking approval for a bio-similar version of the CardiAMP or CardiALLO cell therapy systems.
In addition, it is possible to extend the patent term of at least one patent covering CardiAMP and CardiALLO Cell Therapy Systems following FDA approval. This patent term extension, or PTE, is intended to compensate a patent owner for the loss of patent term during the FDA approval process. If eligible, we may use a PTE to extend the term of one or more of the patents discussed above beyond the expected expiration date. Because CardiAMP and CardiALLO cell therapy systems may involve multiple simultaneous approvals under the IDE and IND applications, each pre-market approval, or PMA or biologics license application, or BLA, associated with the system approval is anticipated to have the ability to have an extended patent term.
Trademarks
We have registered or applied for registration of our name, logo and the trademarks "BioCardia," "CardiAMP," "CardiALLO," and "Morph" in the United States. We have registered or applied for registration of the trademarks "CardiAMP" and "CardiALLO" for use in connection with a biological product, namely, a cell-based therapy product composed of bone marrow derived cells for medical use. We also have rights to use the “Helix” trademark in the United States. We have registered Morph for use in connection with steerable vascular access technology. We intend to pursue additional registrations in markets outside the United States where we plan to sell our therapies and products.
Patent Term
The term of individual patents and patent applications will depend upon the legal term of the patents in the countries in which they are obtained. In most countries, the patent term is 20 years from the date of filing of the patent application (or parent application, if applicable). For example, if an international Patent Cooperation Treaty, or PCT, application is filed, any patent issuing from the PCT application in a specific country expires 20 years from the filing date of the PCT application. In the United States, however, if a patent was in force on June 8, 1995, or issued on an application that was filed before June 8, 1995, that patent will have a term that is the greater of 20 years from the filing date, or 17 years from the date of issue.
Under the Hatch-Waxman Act, the term of a patent that covers an FDA-approved drug, biological product may also be eligible for PTE. PTE permits restoration of a portion of the patent term of a U.S. patent as compensation for the patent term lost during product development and the FDA regulatory review process if approval of the application for the product is the first permitted commercial marketing of a drug or biological product containing the active ingredient. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of a BLA plus the time between the submission date of a BLA and the approval of that application. The Hatch-Waxman Act permits a PTE for only one patent applicable to an approved drug, and the maximum period of restoration is five years beyond the expiration of the patent. A PTE cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, and a patent can only be extended once, and thus, even if a single patent is applicable to multiple products, it can only be extended based on one product. Similar provisions may be available in Europe and certain other foreign jurisdictions to extend the term of a patent that covers an approved drug. When possible, depending upon the length of clinical trials and other factors involved in the filing of a BLA, we expect to apply for PTEs for patents covering our therapeutic candidates and products and their methods of use. For additional information on PTE, see "Government Regulation."
Proprietary Rights and Processes
We may rely, in some circumstances, on proprietary technology and processes (including trade secrets) to protect our technology. However, these can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with those who have access to our confidential information, including our employees, consultants, scientific advisors and contractors. We also seek to preserve the integrity and confidentiality of our proprietary technology and processes by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our proprietary technology and processes may otherwise become known or be independently discovered by competitors. To the extent that our employees, consultants, scientific advisors, contractors, or any future collaborators use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions. For this and more comprehensive risks related to our proprietary technology and processes, please see "Risk Factors-Risks Related to our Intellectual Property."
Recent Developments
In the fourth quarter of 2018, we entered into an agreement with CellProThera to expand the current collaboration to the SingXpand Clinical Trial in Singapore. The study will evaluate the safety and efficacy of in vitro expanded peripheral blood CD34+ stem cells output by the StemXpand® Automated Process and delivered via BioCardia’s Helix™ Biotherapeutic Delivery System for the treatment of patients soon after a heart attack. Under the terms of the agreement, CellProThera will fund completion of all regulatory and clinical activities undertaken by both firms for the clinical investigation. If the study results in regulatory approval of the product, CellProThera will have exclusive commercial rights in Singapore to the Helix™ Biotherapeutic Delivery System for the delivery of culture expanded CD34+ cells to treat patients who have suffered a recent heart attack. BioCardia will receive double-digit royalty payments on future sales of the Align 360 unicompartmental kneecombination product.
On December 24, 2018, the Company entered into a securities purchase agreement with entities affiliated with its existing investors, relating to an offering and sale of an aggregate of 592,592 shares of the Company’s common stock at a purchase price of $6.75 per share, and warrants to purchase up to one-half of the number of shares of common stock sold to an investor, up to an aggregate for all investors of 296,295 shares of Common Stock (the “Warrant Shares”) at an exercise price of $6.75 per share, for aggregate proceeds of $3.8 million, net of $200,000 in expenses. The warrants will expire on December 24, 2023. The warrants contain customary adjustments and are exercisable immediately for cash and after six months will also be exercisable on a cashless basis if there is no effective registration statement registering the resale of the Warrant Shares. The investors do not have registration rights in connection with any securities purchased. The closing of this Offering took place on December 24, 2018.
On July 5, 2019, the Company entered into a note purchase agreement pursuant to which we issued on such date $0.625 million in aggregate principal amount of convertible promissory notes, a portion of which was issued to certain of our officers and directors and a principal stockholder (or their respective affiliates). Interest on the convertible notes accrues at the rate of 14.0% per year. The unpaid principal amount of the convertible notes, together with all interest accrued but unpaid thereon, will be automatically converted into Private Placement Units upon the closing of this Offering at a conversion price equal to 50% of the price to the public in this Offering. Assuming a public offering price of $ per share, and the closing of this Offering occurs on , 2019, the $0.625 million principal amount of the outstanding convertible notes and interest thereon will convert into approximately Private Placement Units.
On July 11, 2019, we signed an extension to a 2017 development agreement with a global biotherapeutics leader for BioCardia’s Helix™ biotherapeutic delivery catheter system. The agreement is exclusive with respect to a class of biotherapeutic agents that BioCardia is not currently developing on its own or with any other party and is time limited. Under the terms of the initial pre-clinical phase of the relationship, BioCardia will receive an upfront payment of $1,000,000, a portion of which will be creditable to BioCardia biotherapeutic delivery systems, support and training.
Manufacturing
The CardiAMP cell processing platform is manufactured for us by our partner Biomet Biologics, LLC. We currently produce CardiALLO cells for preclinical development in our preclinical development tissue culture facility and anticipate manufacturing for clinical development in a newly installed clinical cell manufacturing facility, both in San Carlos, California. We currently manufacture our Helix™ biotherapeutic delivery system and Morph vascular access products in our San Carlos, California device manufacturing facility using components we source from third party suppliers.
Sales and Marketing
Our sales and marketing strategy is to market the CardiAMP and CardiALLO cell therapy systems, if approved by the FDA, for heart failure and chronic myocardial ischemia indications using a partial knee resurfacing devicededicated direct sales model focused on selected cardiologists. These physicians are typically affiliated with leading hospitals and medical centers and we believe that they tend to have well-established referral networks of interventional cardiologists and cardiac catheterization laboratories. We believe they represent a concentrated customer base suitable to a specialist care sales model. We believe that the CardiAMP and CardiALLO cell therapy systems will be adopted first by leading cardiologists at high-volume U.S. hospitals and medical centers, and progressively by a broader segment of the market. Cardiologists and interventional cardiologists have a history of early adoption of innovative products and technologies, in part because the rate of innovation in this sector has been sustained, and in part because of the large unmet medical needs of heart failure patients.
Competition
The biotechnology and pharmaceutical industries in which we operate are subject to rapid change and are characterized by intense competition to develop new technologies and proprietary products. We face potential competition from many different sources, including larger and better-funded companies. While we believe that the CardiAMP Cell Therapy System’s unique benefits provide us with competitive advantages, particularly given that CardiAMP is designed to be administered in a safe and short procedure, we have identified several companies which are active in the advancement of cell-based and gene-based therapeutic products in the heart failure and chronic myocardial ischemia indications. Not only must we compete with other companies that are focused on cell-based therapy treatments, any products that we may commercialize will have to compete with existing therapies and new therapies that may become available in the future.
Some of the companies that have historically been developing cell-based and gene-based therapies for cardiac indications include Abbott Laboratories, Athersys, Astra Zeneca/Moderna, Baxter/Baxalta, Blue Rock Therapeutics, Caladrius Biosciences, Celixr, Cesca Therapeutics, CapriCor Therapeutics, Celyad, CellProthera, Cytori Therapeutics, Juventas Therapeutics, Mesoblast, Osiris Therapeutics, Tenaya Therapeutics, Vericel Corp, Vestion, and Uniqure, some of which are in the clinical stages of development with their product candidates. There are also academic programs at University of Wisconsin, Stanford University, University of Milan and University of Washington that are also developing cell-based and gene-based therapies for cardiac indications.
However, these competitors may require delivery platforms for their own therapeutic programs. Because the clinical need is so large and our biotherapeutic delivery products have potential to enable multiple biotherapeutics, we view these companies also as potential collaborators and partners. To date, we have entered into agreements to provide our biotherapeutic delivery system to fourteen of these entities for various pre-clinical and clinical studies. Two are active in the clinic today. None of these relationships are believed to be material to our business at this time.
License Agreement with Biomet Biologics, LLC
In October 2012, we entered into a license and distribution agreement with Biomet Biologics, LLC under which we obtained an exclusive, nontransferable, worldwide distribution right, patent license and trademark license to a point of care cell processing platform. Under the terms of the agreement, we are obligated to pay a royalty based on the price of the disposables in the CardiAMP cell processing platform for the medial or lateral partduration of the knee. Cardoagreement. We expect the royalty payments to Biomet Biologics, LLC for the licensed product to amount to a low or mid-single digit percentage of the expected price that we will charge for the CardiAMP Cell Therapy System. The agreement has received approvala term of 10 years or the time the last patent pursuant to the agreement expires, whichever is later. The agreement may be terminated by Biomet Biologics, LLC for a failure by us to meet any milestone requirements, including minimum purchase requirements, as well as by either party upon 30 days prior written notice in the event of a breach of any material term by the other party. We have the right to terminate the agreement upon 90 days prior written notice in the event the safety, efficacy or comparative effectiveness of the product is insufficient to meet our commercial needs.
Technology Access Program for Biotherapeutic Delivery Systems
Our preclinical work with partners and collaborators generally takes place under Section 510(k)arrangements where we secure access to data, reports, and a non-exclusive license to delivery technology improvement inventions.
Clinical Research Agreements for Biotherapeutic Delivery Systems
Our clinical work with partners generally takes place under arrangements where we secure access to data, reports, and a non-exclusive license to technology improvement inventions. Financial terms of each agreement are anticipated to cover our costs and provide milestone payments. We hope to generate sales if any of our partners are successful with commercializing their products with our delivery platform.
Government Regulation
Biological products, including cell-based therapy products, and medical devices are subject to regulation under the Federal Food, Drug, and Cosmetic Act, ("Section 510(k)")or FD&C Act, and the Public Health Service Act, or PHS Act, and other federal, state, local and foreign statutes and regulations. Both the FD&C Act and the PHS Act and their corresponding regulations govern, among other things, the testing, manufacturing, safety, identity, potency and purity, efficacy, labeling, packaging, storage, record keeping, distribution, reporting, advertising and other promotional practices involving biological products. FDA acceptance must be obtained before clinical testing of an investigational biological and medical device begins, and each clinical trial protocol for its uniquely instrumented patello-femoral arthroplasty, a resurfacing devicecell-based therapy product is submitted to and reviewed by the FDA. FDA approval must be obtained before marketing of a biological product. Unless an exception applies, FDA approval or clearance is required for medical devices. The process of obtaining regulatory approvals and the backsubsequent compliance with applicable federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources and we may not be able to obtain the kneecaprequired regulatory approvals on a timely basis, or at all. To date, the FDA has never approved for commercial sale a cell-based therapy product intended to treat the heart.
Within the FDA, the Center for Biologics Evaluation and distal femur. Cardo hasResearch, or CBER, regulates cell-based therapy products. For products that use medical devices, including diagnostics, to deliver cell therapies, CBER works closely with the FDA’s Center for Devices and Radiological Health, or CDRH.
U.S. Biological Product Development and Regulatory Approval Process
Our CardiALLO therapeutic candidate will be regulated in the United States as a biological product. The process required by the FDA before a biological product may be tested and marketed in the United States generally involves the following:
• | completion of nonclinical laboratory tests and animal studies according to good laboratory practices, or GLP, regulations and applicable requirements for the humane use of laboratory animals or other applicable regulations; | |
• | submission to the FDA of an IND application, which must become effective before human clinical trials may begin and must be updated annually or when significant changes are made; | |
• | approval by an independent Institutional Review Board, or IRB, or ethics committee at each clinical site before the trial begins; | |
• | performance of adequate and well-controlled human clinical trials according to the FDA’s regulations, commonly referred to as good clinical practices, or GCPs, and any additional requirements for the protection of human research subjects and their health information, to establish the safety, purity and potency of the proposed biological product for its intended use; | |
• | preparation of and submission to the FDA of a BLA for marketing approval, after completion of all pivotal clinical trials; | |
• | satisfactory completion of an FDA Advisory Committee review, if applicable; | |
• | a determination by the FDA within 60 days of its receipt of a BLA to file the application for review; | |
• | satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the biological product is produced to assess compliance with GMP, to assure that the facilities, methods and controls are adequate to preserve the biological product’s identity, strength, quality and purity and, if applicable, the FDA’s current good tissue practices, or GTPs, for the use of human cellular and tissue products; | |
• | potential FDA audit of the nonclinical study and clinical trial sites that generated the data in support of the BLA; and | |
• | FDA review and approval, or licensure, of the BLA for particular indications for use in the United States, which must be updated annually when significant changes are made. |
The testing and approval process require substantial time, effort and financial resources, and we cannot be certain that any approvals for our therapeutic candidates or product candidates will be granted on a timely basis, if at all. Before testing any biological product candidate, including a cell-based therapy product, in humans, the product candidate enters the preclinical testing stage. Preclinical tests, also received FDA 510k approval for its Total Knee System which has both a posterior cruciate sacrificingreferred to as nonclinical studies, include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the product candidate. The conduct of the preclinical tests must comply with federal regulations and requirements including GLPs.
The clinical trial sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a posterior cruciate sparing component design. Cardo hasproposed clinical protocol, to the FDA as part of the IND. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also receivedimpose clinical holds on a biological product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA 510kimposes a clinical hold, trials may not recommence without FDA authorization and then only under terms authorized by the FDA. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that suspend or terminate such trials.
Clinical trials involve the administration of the biological product candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety, including stopping rules that assure a clinical trial will be stopped if certain adverse events should occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND. Clinical trials must be conducted and monitored in accordance with the FDA’s regulations comprising the GCP requirements, including the requirement that all research subjects provide informed consent. Further, each clinical trial must be reviewed and approved by an independent institutional review board, or IRB, at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent that must be signed by each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. Clinical trials also must be reviewed by an institutional biosafety committee, or IBC, a local institutional committee that reviews and oversees basic and clinical research conducted at that institution. The IBC assesses the safety of the research and identifies any potential risk to public health or the environment.
For purposes of BLA approval, for its total hip replacement system along with its monopolar and bipolar hip systems. Cardo received Section 510(k) approvals for its spinal lumbar fusion system and its cervical plate and screw systems. Cardo is actively engagedhuman clinical trials are typically conducted in three sequential phases that may overlap or be combined:
• | Phase I. The biological product is initially introduced into healthy human subjects and tested for safety. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients with the disease or condition. These studies are designed to test the safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans, the side effects associated with increasing doses and, if possible, to gain early evidence on effectiveness. | |
• | Phase II. The biological product is evaluated in a limited patient population with a specified disease or condition to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule. Multiple Phase II clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase III clinical trials. |
• | Phase III. Clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency, and safety in an expanded patient population at geographically dispersed clinical trial sites, to provide statistically significant evidence of clinical efficacy and to further test for safety. These clinical trials are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product approval and labeling. |
Post-approval clinical trials, sometimes referred to as Phase IV clinical trials, may be required by the FDA or voluntarily conducted after initial marketing approval to gain more information about the product, including long-term safety follow-up.
During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data, and clinical trial investigators. Annual progress reports detailing the results of the clinical trials must be submitted to the FDA. Written IND safety reports must be promptly submitted to the FDA, the NIH and the investigators for serious and unexpected adverse events, any findings from other studies, tests in laboratory animals or in vitro testing that suggest a significant risk for human subjects, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information qualifies for reporting. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days after the sponsor’s initial receipt of the information. Phase I, Phase II and Phase III clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor or its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk, including risks inferred from other unrelated trials. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the biological product has been associated with unexpected serious harm to patients.
Human cell-based therapy products administered directly into heart tissue are a relatively new category of therapeutics. Because this is a relatively new and expanding area of novel therapeutic interventions, there can be no assurance as to the length of the trial period, the number of highly innovative research and development projects for total knee arthroplasty, spinal motion preservation, fusion devices and minimally invasive approaches for treating an array of joint and spinal disorders.
As of December 14, 2009, Cardo employed 20 full-time employees.
Code of Business Conduct and Ethics
We have adopted a Code of Business Conduct and Ethics which is applicablepatients the FDA will require to all of our employees, officers and directors (including our principal executive officer, principal financial officer and principal accounting officer). A copy of the Code of Business Conduct and Ethics is available on the Company's website at www.cardomedical.com.
Recent Transactions
Cardo Medical, LLC ("Cardo LLC") was formed on April 6, 2007 as a California limited liability company for the purpose of acquiring an interestbe enrolled in the medical device business conducted by Accin Corporation directly and through Accin's interests in Cervical Xpand, LLC and Uni-Knee, LLC. Following Cardo LLC's organization:
Cardo LLC and Accin formed a Delaware limited liability company on April 20, 2007 under the name Accelerated Innovation, LLC;On May 21, 2007, Accin contributed substantially all of its business, properties and assets, including its majority interests in Cervical Xpand and Uni-Knee, to Accelerated Innovation in exchange for a 62.5% interest in Accelerated Innovation and the distribution referenced below in the amount of $3.75 million;Concurrently with the above, on May 21, 2007, Cardo LLC contributed $3.75 million to Accelerated Innovation in exchange for a 37.5% interest in Accelerated Innovation; andThe amount of $3.75 million was distributed by Accelerated Innovation to Accin.
Under the terms of Accelerated Innovation's Limited Liability Company Agreement, Cardo LLC was granted an option to purchase the 62.5% interest in Accelerated Innovation held by Accin for a purchase price of $6.25 million. Following the exercise of that option in June 2008, Cardo LLC acquired all of the interests in Accelerated Innovation held by Accin, and Accelerated Innovation became a wholly-owned subsidiary of Cardo LLC.
Prior to that, in February 2008, Cardo LLC entered into Membership Interest Purchase Agreements with the holders of the minority membership interests in Cervical Xpand and Uni-Knee. Cervical Xpand and Uni-Knee were formed as New Jersey limited liability companies on July 12, 2005 and May 10, 2006, respectively, for the purpose of conducting research and development activities. Prior to the closing of the transactions contemplated by the Membership Interest Purchase Agreements, Accelerated Innovation, as the assignee of Accin's assets, owned 52.083% of the membership interests in Cervical Xpand and 51.21% of the membership interests in Uni-Knee, and the minority holders held the remaining outstanding interests. Upon the closing of the transactions contemplated by the Membership Interest Purchase Agreements, in June 2008, Cardo LLC acquired the outstanding membership interests from the minority holders for an aggregate purchase price of $1,437,510 for the Cervical Xpand interests and $2,049,180 for the Uni-Knee interests. As a result, Cardo LLC owned all of the interests in Cervical Xpand and Uni-Knee directly and indirectly through its ownership of Accelerated Innovation.
33
On June 18, 2008, Cardo LLC entered into a Merger Agreement and Plan of Reorganization with clickNsettle.com, Inc. ("CKST") and Cardo Acquisition, LLC, a California limited liability company and wholly-owned subsidiary of CKST. Upon the consummation of the transactions contemplated by the Merger Agreement, CKST acquired Cardo LLC through a merger of Cardo LLC with Cardo Acquisition, with Cardo LLC continuing as the surviving entity in the Merger and a wholly-owned subsidiary of CKST. Pursuant to the Merger Agreement, all of the issued and outstanding units of Cardo LLC's membership interests were converted into the right to receive shares of the common stock of CKST.
On or about the signing of the Merger Agreement, Frost Gamma Investments Trust and other investors invested $12.975 million in Cardo LLC in exchange for units of Cardo LLC's membership interests. Dr. Phillip Frost, Chairman and Chief Executive Officer of Opko Health, Inc., is the trustee and beneficiary of Frost Gamma Investments Trust. Cardo LLC used approximately $9.7 million of the proceeds from these investments to close on the acquisition of the outstanding equity interests of three partially owned subsidiaries of Cardo LLC (Accelerated Innovation, LLC, Cervical Xpand, LLC and Uni-Knee, LLC), to repay an existing member loan (in the amount of $1.2 million) and for transaction expenses, and used the remaining funds to accelerate its research and product development.
Under the terms of the Merger Agreement, at the closing of the Merger, each Cardo LLC unit of membership interest issued and outstanding was converted into and exchanged for the right to receive 667,204.70995 shares of common stock of CKST. As a result of the Merger, CKST's stockholders and optionholders owned approximately 5.5% of the combined company on a fully diluted basis (or 11,298,979 shares of common stock outstanding and underlying options), the members of Cardo LLC, excluding the new investors, owned approximately 64.8% of the combined company on a fully diluted basis (or 133,440,942 shares of common stock), the new investors owned approximately 28.5% of the combined company on a fully diluted basis (or 58,641,701 shares of common stock), and optionholders of Cardo LLC owned approximately 1.2% of the combined company on a fully diluted basis (or 2,398,400 shares of common stock underlying those options).
Following the closing of the Merger, each of Cervical Xpand, Uni-Knee and Accelerated Innovation merged with and into Cardo Medical, LLC, which is now the sole subsidiary of the Company and Cardo LLC converted into a Delaware limited liability company.
On June 30, 2009, Cardo completed the first tranche of a private placement with investors to purchase 8,689,319 shares of Cardo's common stock, par value $0.001 per share, at a price of $0.35 per share for gross proceeds of $3,041,260. The common shares sold under this private placement have a 24-month lock up provision. The second tranche of this private placement closed on September 21, 2009 with the purchase of 485,714 shares of Cardo's common stock for gross proceeds of $170,000.
On October 27, 2009 and November 13, 2009, Cardo completed another private placement with investors to purchase an aggregate of 17,757,837 shares of Cardo's common stock, par value $0.001 per share, at a price of $0.35 per share for gross proceeds of $6,215,250.
Proceeds from these two private placements allowed us sufficient working capital to build inventory and instrumentationtrials in order to meet anticipated levels neededestablish the safety, efficacy, purity and potency of human cell-based therapy products, or that the data generated in these trials will be acceptable to expand our salesthe FDA to support marketing approval.
Concurrently with clinical trials, companies usually complete additional animal studies and to acquire substantially allmust also develop additional information about the physical characteristics of the assets of Vertebron, Inc. ("Vertebron").
Vertebron, a spinal implant device company located in Stratford, CT, designs, develops, manufactures and sells spinal implant products focused on fusion technology for the lumbar and cervical spinebiological product as well as motion preservation technologies. Cardo purchased allfinalize a process for manufacturing the product in commercial quantities in accordance with GMP requirements. To help reduce the risk of Vertebron's inventorythe introduction of adventitious agents with use of biological products, the PHS Act emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, fixed assetsamong other things, the sponsor must develop methods for testing the identity, strength, quality, potency and will retain 100% ownershippurity of all Vertebron's implant technologies for spinal surgery. We also acquired all intellectual property rights owned by Vertebron which includes 20 U.S. issued patentsthe final biological product. Additionally, appropriate packaging must be selected and patent applications. Through a previous licensing agreement, Cardo currently marketstested and distributesstability studies must be conducted to demonstrate that the PSS Pedicle Screw and SCP Cervical Plate systems. The Vertebron transaction will allow for Cardo to expandbiological product candidate does not undergo unacceptable deterioration over its existing domestic distribution. On April 21, 2009, Vertebron filed for Chapter 11 bankruptcy protection inshelf life.
After the Districtsuccessful completion of Connecticut. The acquisition is the resultclinical trials of a Chapter 11 auction process, approved by the United States Bankruptcy Court for the Districtbiological product, FDA approval of Connecticut.
34
We are headquartered in Beverly Hills, California. In connection with the consummationa BLA must be obtained before commercial marketing of the Merger, CKST approved through its stockholders an amendment to its Amendedbiological product. The BLA must include results of product development, laboratory and Restated Certificate of Incorporation to change its name from "clickNsettle.com, Inc." to "Cardo Medical, Inc." CKST's trading symbol was "CKST.OB," which has changed to "CDOM.OB" in connection with the name change. Cardo Medical's common stock is quotedanimal studies, human trials, information on the National Associationmanufacture and composition of Securities Dealers, Inc.'s, Over-the-Counter Bulletin Board,the product, proposed labeling and other relevant information. The FDA may grant deferrals for submission of data or the OTC Bulletin Board.
At December 14, 2009, we had approximately $4,956,000 in cash; however, in October 2009 we used $1,170,000 in order to complete the Vertebron, Inc. transactionfull or partial waivers. The testing and raised net proceeds of approximately $5,871,000 through a private placement. With this subsequent net cash infusion, the available funds are still not projected to meet all of our working capital needs for the next twelve months. The fact that we will sustain losses through the remainder of 2009approval processes require substantial time and the first two quarters of 2010,effort and may still require outside sources of additional capital to supplement operations continues to create an uncertainty about our ability to continue as a going concern.
Management intends to use borrowings and securities sales to mitigate the effects of our use of that cash. However, we cannot assure you that debt or equity financing, if and when required, will be available. Our ability to continue as a going concern is dependent upon receiving additional funds either through the issuance of debt or through common and/or preferred stock and the success of management's plan to expand sales. Although we may obtain external financing through the sales of our own securities, there can be no assurance that such financingthe FDA will accept the BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all.
Under the Prescription Drug User Fee Act, or PDUFA, as amended, each BLA must be accompanied by a significant user fee. The FDA adjusts the PDUFA user fees on an annual basis. PDUFA also imposes an annual product fee for biological products and an annual establishment fee on facilities used to manufacture prescription biological products. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on BLAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
Within 60 days following submission of the application, the FDA reviews a BLA submitted to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review of the BLA. The FDA reviews the BLA to determine, among other things, whether the proposed product is safe, pure and potent, for its intended use, and whether the product is being manufactured in accordance with GMP to assure and preserve the product’s identity, safety, strength, quality, potency and purity. The FDA may refer applications for novel biological products or biological products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. During the biological product approval process, the FDA also will determine whether a Risk Evaluation and Mitigation Strategy, or REMS, is necessary to assure the safe use of the biological product. If the FDA concludes a REMS is needed, the sponsor of the BLA must submit a proposed REMS. The FDA will not approve a BLA without a REMS, if required.
Before approving a BLA, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with GMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure that the clinical trials were conducted in compliance with IND trial requirements and GCP requirements. To assure GMP and GCP compliance, an applicant must incur significant expenditure of time, money and effort in the areas of training, record keeping, production, and quality control.
Notwithstanding the submission of relevant data and information, the FDA may ultimately decide that the BLA does not satisfy its regulatory criteria for approval and deny approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. If the agency decides not to approve the BLA in its present form, the FDA will issue a complete response letter that describes all of the specific deficiencies in the BLA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the BLA, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. The FDA may impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a risk management plan, or otherwise limit the scope of any approval. In addition, the FDA may require post-marketing clinical trials, sometimes referred to as Phase IV clinical trials, designed to further assess a biological product’s safety and effectiveness, and testing and surveillance programs to monitor the safety of approved therapies and products that have been commercialized.
The FDA has agreed to certain review goals under PDUFA and aims to complete its review of 90% of standard BLAs within ten months from filing and 90% of priority BLAs within six months from filing. The FDA does not always meet its PDUFA goal dates for standard and priority BLAs and its review goals are subject to change from time to time. The review process and the PDUFA goal date may be extended by three months if the FDA requests, or the BLA sponsor otherwise provides, additional information or clarification regarding information already provided in the submission within the last three months before the PDUFA goal date.
Fast Track Designation, Accelerated Approval, Priority Review and Breakthrough Therapy Programs
The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a new drug or biological product may request the FDA to designate the drug or biological product as a Fast Track product at any time during the clinical development of the product. Unique to a Fast Track product, the FDA may consider for review sections of the marketing application on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the application, the FDA agrees to accept sections of the application and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the application.
Other types of FDA programs intended to expedite development and review, such as priority review, accelerated approval and Breakthrough Therapy designation, also exist. A product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug or biological product designated for priority review in an effort to facilitate the review. Additionally, a product may be eligible for accelerated approval. Drug or biological products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical trials establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA may require that a sponsor of a drug or biological product receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product.
A product may also be eligible for receipt of a Breakthrough Therapy designation. The Breakthrough Therapy designation is intended to expedite the FDA’s review of a potential new drug for serious or life-threatening diseases where “preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.” The designation of a drug as a Breakthrough Therapy provides the same benefits as are available under the Fast Track program, as well as intensive FDA guidance on the product’s development program. Where appropriate, we intend to utilize regulatory programs that can help expedite our product development and commercialization efforts. However, Fast Track designation, priority review, accelerated approval and Breakthrough Therapy designation do not change the standards for approval, but may expedite the development or approval process.
Post-Approval Requirements
Maintaining substantial compliance with applicable federal, state and local statutes and regulations requires the expenditure of substantial time and financial resources. Rigorous and extensive FDA regulation of biological products continues after approval, particularly with respect to GMP. We will rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of any products that we may commercialize. Manufacturers of our products are required to comply with applicable requirements in the GMP regulations, including quality control and quality assurance and maintenance of records and documentation. Other post-approval requirements applicable to biological products include reporting of GMP deviations that may affect the identity, potency, purity and overall safety of a distributed product, record-keeping requirements, reporting of adverse effects, reporting updated safety and efficacy information, and complying with electronic record and signature requirements. After a BLA is approved, the product also may be subject to official lot release. As part of the manufacturing process, the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official release by the FDA, the manufacturer submits samples of each lot of product to the FDA together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot. In addition, the FDA conducts laboratory research related to the regulatory standards on the safety, purity and potency of biological products.
We also must comply with the FDA’s advertising and promotion requirements, such as those related to direct-to-consumer advertising, the prohibition on promoting products for uses or in-patient populations that are not described in the product’s approved labeling (known as “off-label use”), industry-sponsored scientific and educational activities, and promotional activities involving the internet. Discovery of previously unknown problems or the failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product or withdrawal of the product from the market as well as possible civil or criminal sanctions. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval may subject an applicant or manufacturer to administrative or judicial civil or criminal sanctions and adverse publicity. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval, clinical hold, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, mandated corrective advertising or communications with doctors, debarment, restitution, disgorgement of profits, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
Biological product manufacturers and other entities involved in the manufacture and distribution of approved biological products are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with GMPs and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain GMP compliance. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer or holder of an approved BLA, including withdrawal of the product from the market. In addition, changes to the manufacturing process or facility generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Abbreviated Licensure Pathway of Biological Products as Biosimilar or Interchangeable
The Affordable Care Act includes a subtitle called the Biologics Price Competition and Innovation Act of 2009 (“BPCIA”), which created an abbreviated approval pathway for biological products shown to be highly similar to an FDA-licensed reference biological product. The BPCIA attempts to minimize duplicative testing, and thereby lower development costs and increase patient access to affordable treatments. If our CardioAllo product is approved by the FDA, we could face competition from products regulated by the FDA as biosimilar products.
Biosimilarity means that the biological product is highly similar to the reference product notwithstanding minor differences in clinically inactive components; and that there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity and potency of the product. In addition, the law provides for a designation of “interchangeability” between the reference and biosimilar products, whereby the biosimilar may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product. The higher standard of interchangeability must be demonstrated by information sufficient to show that:
| ● | the proposed product is biosimilar to the reference product; | |
| ● | the proposed product is expected to produce the same clinical result as the reference product in any given patient; and |
| ● | for a product that is administered more than once to an individual, the risk to the patient in terms of safety or diminished efficacy of alternating or switching between the biosimilar and the reference product is no greater than the risk of using the reference product without such alternation or switch. |
FDA approval is required before a biosimilar may be marketed in the United States. However, complexities associated with the large and intricate structures of biological products and the process by which such products are manufactured pose significant hurdles to the FDA’s implementation of the law that are still being worked out by the FDA. For example, the FDA has discretion over the kind and amount of scientific evidence—laboratory, preclinical and/or clinical—required to demonstrate biosimilarity to a licensed biological product.
The FDA intends to consider the totality of the evidence, provided by a sponsor to support a demonstration of biosimilarity, and recommends that sponsors use a stepwise approach in the development of their biosimilar products. Biosimilar product applications thus may not be required to duplicate the entirety of preclinical and clinical testing used to establish the underlying safety and effectiveness of the reference product. However, the FDA may refuse to approve a biosimilar application if there is insufficient information to show that the active ingredients are the same or to demonstrate that any such financing wouldimpurities or differences in active ingredients do not affect the safety, purity or potency of the biosimilar product. In addition, as with BLAs, biosimilar product applications will not be on terms acceptableapproved unless the product is manufactured in facilities designed to us. If we are unable to fund our cash flow needs, we may have to reduce or stop planned growth or scale back operationsassure and reduce staff.preserve the biological product’s safety, purity and potency.
Nature of Business
The Company developssubmission of a biosimilar application does not guarantee that the FDA will accept the application for filing and distributes reconstructive orthopedicreview, as the FDA may refuse to accept applications that it finds are insufficiently complete. The FDA will treat a biosimilar application or supplement as incomplete if, among other reasons, any applicable user fees assessed under the Biosimilar User Fee Act of 2012 have not been paid. In addition, the FDA may accept an application for filing but deny approval on the basis that the sponsor has not demonstrated biosimilarity, in which case the sponsor may choose to conduct further analytical, preclinical or clinical studies and spinal surgerysubmit a BLA for licensure as a new biological product.
The timing of final FDA approval of a biosimilar for commercial distribution depends on a variety of factors, including whether the manufacturer of the branded product is entitled to one or more statutory exclusivity periods, during which time the FDA is prohibited from approving any products that are biosimilar to various medical organizations.the branded product. The Company worksFDA cannot approve a biosimilar application for twelve years from the date of first licensure of the reference product. Additionally, a biosimilar product sponsor may not submit an application for four years from the date of first licensure of the reference product. A reference product may also be entitled to exclusivity under other statutory provisions. For example, a reference product designated for a rare disease or condition (an “orphan drug”) may be entitled to seven years of exclusivity, in small, focused development teams in conjunction with physicianswhich case no product that is biosimilar to rapidly develop productsthe reference product may be approved until either the end of the twelve-year period provided under the biosimilarity statute or the end of the seven-year orphan drug exclusivity period, whichever occurs later. In certain circumstances, a regulatory exclusivity period can extend beyond the life of a patent, and thus block biosimilarity applications from conception to launch. The Company launchedbeing approved on or after the patent expiration date. In addition, the FDA may under certain circumstances extend the exclusivity period for the reference product by an additional six months if the FDA requests, and commenced salesthe manufacturer undertakes, studies on the effect of its first product in late 2006,children, a so-called pediatric extension.
The first biological product determined to be interchangeable with a branded product for any condition of use is also entitled to a period of exclusivity, during which was a high-performance, uni-compartmental knee replacement. The Company commenced salestime the FDA may not determine that another product is interchangeable with the reference product for any condition of its reconstructive products in 2007 and spine products in 2008.
Products
Following is a listing of our current products:
Knee Portfolio
Align 360 Unicompartmental Knee System - A uniquely instrumented high performance partial knee replacement that allows resurfacing of eitheruse. This exclusivity period extends until themedial or lateral compartmentsearlier of: (1) one year after the first commercial marketing of theknee. Thisfirst interchangeable product; (2) 18 months after resolution of a patent infringement against the applicant that submitted the application for the first interchangeable product,promotes the consistent balancingbased on a final court decision regarding all of theflexion and extension gaps for unicompartmental knee surgery.Align 360 Patellofemoral System - A uniquely instrumented and novel patellofemoral system that allows resurfacingpatents in the litigation or dismissal of thepatellofemoral joint. Thislitigation with or without prejudice; (3) 42 months after approval of the first interchangeable product, if a patent infringement suit against the applicant that submitted the application for the first interchangeable product isan anatomic system that addresses the diseasestill ongoing; or (4) 18 months after approval of thepatellofemoral joint.Align 360 Total Knee System - A uniquely instrumented high performance total knee system consisting of posterior-stabilizedfirst interchangeable product if the applicant that submitted the application for the first interchangeable product has not been sued.U.S. Premarket Clearance and
cruciate retaining femoral components
Hip Portfolio
Accin Total Hip System - A taperloc type of hip system that allows replacement of the ball and socket of the hip joint. This product offers a dual taper hip designApproval Requirements fortotal hip arthroplasty complemented by the Accin Bipolar and Monopolar Hip Systems for hip fracture applications.Accin Bipolar Hip System - A bipolar hip that allows replacement of the ball of the hip for either fracture, tumors or reconstruction from some other type of pathology.Accin Monopolar Hip System - A monopolar hip that allows replacement of the ball of the hip from either fracture, tumors or reconstruction from some other type of pathology.
35
Medical Devices
Spinal Product Line
Accin Lumbar Pedicle Screw/Rod System - A pedicle screw and rod system for instrumentation of lumbar spine fusion incorporatingUnless anevolutionary locking mechanism allowing for high screw angulation.Accin Cervical Plate/Screw System - An innovative low-profile system for cervical spine fusion incorporating an integrated, floating tapered-ring locking mechanismexemption applies, each medical device we wish tosimplify surgical procedure.
Our products listed above have received Section 510(k) approval. We have a number of earlier stage research and development projects underway, some of which have received Section 510(k) approval and others that may be submitted for regulatory approval in the future.
Orthopedic Industry
According to the 2007-2008 Orthopaedic Industry Annual Report published by Knowledge Enterprises, Inc., which we refer to herein as the Industry Annual Report, the worldwide market for orthopedic products in 2007 was estimated to be $32.5 billion, representing an 11.8% increase from the previous year. According to this report, bone and joint diseases account for half of all the chronic conditions in people over 50 years of age. With the predicted doubling of the aged population by the year 2020, the report suggests that demographics alone will drive growth in the global orthopedic industry. We also believe that the orthopedic industry will continue to grow due to an increasingly older population and extended life spansdistribute commercially in the United States and other developed countries worldwide.
According to the Industry Annual Report, the world's six largest replacement companies-Zimmer, Johnson & Johnson, Stryker, Smith & Nephew, Biomet and Wright Medical-generated 89% of joint product sales in 2007. We believe that the size of these companies often leads them to concentrate their marketing and research and development efforts on products that they believe will have a relatively high minimum threshold level of sales. As a result, there is an opportunity for a smaller orthopedic company, such as us, to focus on smaller, higher-growth sectors of the orthopedic market, while still offering a comprehensive product line to address the needs of its customers in a customized and interactive fashion.
Orthopedic devices are commonly divided into several primary sectors corresponding to the major subspecialties within the orthopedic field: reconstruction, trauma, arthroscopy, spine and biologics. Management's initial focus is on innovation related to reconstructive joint devices and spinal products, as discussed below.
Reconstructive Joint Device Market
Most reconstructive joint devices are used to replacerequire either prior premarket notification, or repair joints that have deteriorated as a result of disease or injury. Despite the availability of non-surgical treatment alternatives such as oral medications, injections and joint fluid supplementation of the knee, severe cases of disease or injury often require reconstructive joint surgery.
Reconstructive joint surgery involves modifying the bone area surrounding the affected joint and inserting one or more manufactured components, and also may involve using bone cement.
The reconstructive joint device market is generally divided into the areas of hips, knees and extremities. According to the Industry Annual Report, it is estimated that the worldwide reconstructive joint device market had sales of approximately $11.6 billion in 2007, with hip reconstruction and knee reconstruction representing the largest sectors.
Knee Reconstruction.The knee joint involves the surfaces of three distinct bones: the lower end of the femur, or thigh bone, the upper end of the tibia, or shin bone, and the patella, or kneecap. Cartilage on any of these surfaces can be damaged due to disease or injury, leading to pain and inflammation requiring knee reconstruction. According to the Industry Annual Report, knee reconstruction was the largest sector of the reconstructive joint device market in 2007, with estimated sales of approximately $5.9 billion worldwide.
36
One of the major trends in knee reconstruction includes the use of minimally invasive techniques to accomplish reconstructive goals with less damage to surrounding soft tissues. Our unicompartmental device has been designed to be inserted through small incision surgery with an innovative instrumentation approach. Our design approach was to develop an innovative instrumentation system to improve and simplify surgical technique for a clinically proven implant concept. We believe that our system allows the surgeon to simply and reproducibly balance both flexion and extension gaps. This is a general approach we plan to continue with our other products.
Hip Reconstruction. The hip joint is a ball-and-socket joint that enables the large range of motion that the hip performs in daily life. The hip joint is most commonly replaced due to degeneration of the cartilage between the head of the femur (the ball) and the acetabulum or hollow portion of the pelvis (the socket). This degeneration causes pain, stiffness and a reduction in hip mobility. According to the Industry Annual Report, it is estimated that the worldwide hip reconstruction market had sales of approximately $5.1 billion in 2007.
Similar to the knee reconstruction market, major trends in hip replacement procedures and implants are to extend implant life and to preserve bone stock for possible future procedures. New products have been developed that incorporate advances in bearing surfaces from the traditional polyethylene surface. These alternative bearing surfaces include metal-on-metal, cross-linked polyethylene and ceramic-on-ceramic combinations, which exhibit improved wear characteristics and lead to longer implant life. In addition to advances in bearing surfaces, implants that preserve more natural bone have been developed in order to minimize surgical trauma and recovery time for patients. These implants, known as bone- conserving implants, leave more of the hip bones intact, which may be beneficial given the likelihood of future revision replacement procedures as the average patient's lifetime increases. Bone-conserving procedures are intended to enable patients to delay their first total hip procedure and may significantly increase the time from the first procedure to the time when a revision replacement implant is required. Our hip product portfolio, currently consisting of three products, is focused on improving the surgical techniques for bone-conservative procedures. These products integrate implant designs that are based on predicate devices (i.e., a device with a similar design that has already received clearance) with successful long-term clinical histories. We are actively engaged in several research and development efforts to develop better instrumentation for less traumatic surgeries, improved component designs and bearing surfaces to increase longevity of our devices.
Spine Market
Back and neck pain is one of the leading causes of healthcare expenditures in the United States, with a direct cost of approximately $86 billion annually for diagnosis, treatment and rehabilitation, according to an article published in The Journal of the American Medical Association (published February 13, 2008). According to the Industry Annual Report, the U.S. market for lumbar and cervical spine fusion, which is the focus of our spinal business, was estimated to be over $3 billion in 2006 and over $3.6 billion in 2007, and is estimated to grow to more than $4.2 billion in 2008.
The spine consists of vertebrae, which are 29 separate bones connecting the skull to the pelvis. The vertebrae are joined together by soft tissue structures that provide the core of the human skeleton. Within the spinal column, the spinal cord, which is the body's central nerve pathway, is protected by the bony parts of the vertebrae. Nerves contained in the spinal column exit through the foramen openings to the rest of the body. Vertebrae are joined to each other in pairs which are often referred to as motion segments. These motion segments move by means of three joints: two facet joints and one spine disc. The facet joints provide stability and enable the spine to bend and twist while the discs absorb pressures and shocks to the vertebrae.
The four major categories of spine disorders are degenerative conditions, deformities, trauma and tumors. The largest market, and the focus of our spinal research and development business, is degenerative conditions of the facet joints and disc space. These conditions can result in instability and pressure on the nerve roots as they exit the spinal column, causing back pain or radiating pain in the arms or legs.
The recommended treatments for spine disorders depend on the severity and duration of the disorder. Initially, physicians will prescribe non-operative procedures, including bed rest, bracing, medication, lifestyle modification, exercise, physical therapy, chiropractic care and steroid injections. In most cases, non-surgical treatment options are effective; however, many patients do not respond to non-operative treatments and require spine surgery to alleviate their symptoms.
37
It is estimated that in excess of one million patients undergo spine surgery each year in the United States. The most common spine surgery procedures are: discectomy, which consists of the removal of all or part of a damaged disc; laminectomy, the removal of all or part of a lamina, or thin layer of bone, to relieve pinching of the nerve and narrowing of the spinal canal; and fusion, where two or more adjoining vertebrae are fused together to provide stability. All three of these procedures require access to the spine through either a traditional open approach or through smaller, less invasive methods using various types of retractors or other percutaneous techniques.
We believe that the implant market for spine surgery procedures will continue to grow because of the following market dynamics:
Demographics.The population cohort most likely to experience back pain is likely to grow as a result of our aging baby boomer population. The first baby boomers turned 62 in 2008, and over the next two decades we will see a substantial increase in our aging population. We believe that this generation of older people is less willing to compromise on reducing activity levels and is more interested in treatments that will allow a more rapid return to activities with shorter periods of disability.
Increased Acceptance of Implants.The implementation of implants for use in spine surgery has become the standard of care over the past decade. In the last five years, there has been a substantial and significant increase in the percentage of spinal fusion surgeries using implants. According to Millennium Research Group, an estimated 85% or more of all spinal fusion procedures involve an implant. The current generation of modern trained spine surgeons has accepted usage of implants as the gold standard for achieving optimal results.
Increased Demand for Newer Technologies. Because of the ubiquitous nature of back pain, the market is interested in newer technologies, such as motion preservation, and novel minimally invasive techniques which would potentially allow earlier intervention in the degenerative process of the spine for many patients.
Government Regulation
United States
Our products are regulated by the U.S. Food and Drug Administration, or the FDA, under the Federal Food, Drug, and Cosmetic Act. Some of our products also are regulated by state agencies. FDA regulations and the requirements of the Federal Food, Drug, and Cosmetic Act affect the pre-clinical and clinical testing, design, manufacture, safety, efficacy, labeling, storage, recordkeeping, advertising and promotion of our medical device products. FDA regulations govern, among other things, the following activities that we or our partners perform and will continue to perform:
product design and development;product testing;product manufacturing;product labeling;product storage;premarket510(k) clearance, orapproval;advertising and promotion; andproduct sales and distribution.
Generally, before we can market a new medical device, marketing clearance from the FDA must be obtained through either a pre- market notification under Section 510(k) or theprior approval of a pre-market approval, or PMA application.application from the FDA. The FDA typically grantsclassifies medical devices into one of three classes. Devices deemed to pose low to moderate risk are placed in either class I or II, which, absent an exemption, requires the manufacturer to file with the FDA a Section 510(k) clearance ifsubmission requesting permission for commercial distribution. This process is known as 510(k) clearance. Some low-risk devices are exempt from this requirement. Devices deemed by the applicant can establish thatFDA to pose the device isgreatest risk, such as life-sustaining, life-supporting or certain implantable devices, or devices deemed not substantially equivalent to a predicatepreviously cleared 510(k) device, (i.e., a device with a similar design that has already received clearance). It generally takes approximately three months from the dateare placed in class III, requiring approval of a Section 510(k) submission to obtain clearance, but it may take longer, particularly if a clinical trial is required. The FDA may find that a Section 510(k) clearance is not appropriate or that substantial equivalence has not been shown and, as a result, will require a PMA application.
38
Regulation of CardiAMP through the PMA Pathway
Combination products are therapeutic and diagnostic products that combine drugs, devices, and/or biological products. Because combination products involve components that would normally be regulated under different types of regulatory authorities, and frequently by different centers of the FDA, they raise regulatory, policy, and review management challenges. Differences in regulatory pathways for each component of the product can impact the regulatory processes for all aspects of product development and management, including preclinical testing, clinical investigation, marketing applications, manufacturing and quality control, adverse event reporting, promotion and advertising, and post-approval modifications.
A combination product is assigned to an FDA Agency Center or alternative organizational component that will have primary jurisdiction for its premarket review and regulation. For cell-based therapy and related products, the FDA established the Office of Cellular, Tissue and Gene Therapies within CBER to consolidate the review of such products, and the Cellular, Tissue and Gene Therapies Advisory Committee to advise CBER on its review. In our case, the CardiAMP Cell Therapy System involves minimal manipulation of cells within the procedure room, enabling it to be the first cardiac cell-based therapy we are aware of that CBER has indicated it will regulate through the PMA pathway.
PMA applications must be supported by valid scientific evidence, which typically requires extensive data, including technical, preclinical, clinical and manufacturing data, to demonstrate to the FDA’s satisfaction reasonable safety and effectiveness of the cell-based therapy. After a PMA application is deemed complete, the FDA will accept the application for filing and begin an in-depth review of the submitted information. During this review period, the FDA may request additional information or clarification of information already provided. Also during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. As part of its review of the PMA, the FDA will conduct a pre-approval inspection of the manufacturing facility or facilities to ensure compliance with the Quality System Regulation, or QSR, which requires manufacturers to follow design, testing, control, documentation and other quality assurance procedures. FDA review of an initial PMA application is required by statute to take between six to ten months, although the process typically takes longer, and may require several years to complete. If the FDA evaluations of both the PMA application and the manufacturing facilities are favorable, the FDA will either issue an approval letter or an approvable letter, which usually contains a number of conditions that must be met in order to secure the final approval of the PMA. If the FDA’s evaluation of the PMA or manufacturing facilities is not favorable, the FDA will deny approval of the PMA or issue a not approvable letter. A not approvable letter will outline the deficiencies in the application and, where practical, will identify what is necessary to make the PMA approvable. The FDA may also determine that additional clinical trials are necessary, in which case the PMA approval may be delayed for several months or years while the trials are conducted and then the data submitted in an amendment to the PMA. Once granted, PMA approval may be withdrawn by the FDA if compliance with post-approval requirements, conditions of approval or other regulatory standards is not maintained, or problems are identified following initial marketing.
The FDA may approve a PMA application with post-approval conditions intended to ensure the safety and effectiveness of the device typicallyincluding, among other things, restrictions on labeling, promotion, sale and distribution, collection of long-term follow-up data from patients in the clinical trial that supported approval, or new post-approval studies. Failure to comply with the conditions of approval can result in materially adverse enforcement action, including the resultsloss or withdrawal of the approval. PMA supplements are required for modifications that could affect device safety or effectiveness, including, for example, certain types of modifications to the device’s indication for use, manufacturing process, labeling and design. PMA supplements often require submission of the same type of information as an original PMA application, except that the supplement is limited to information needed to support any changes to the device covered by the original PMA application, and may not require as extensive clinical data or the convening of an advisory panel.
A clinical trial is almost always required to support a PMA application. We expect that the CardiAMP Cell Therapy System will require a single pivotal trial for PMA approval in the CardiAMP Heart Failure and CardiAMP Chronic Myocardial Ischemia trials. However, there is no guarantee that the FDA will grant us regulatory approval to market the CardiAMP Cell Therapy System on the basis of a single pivotal trial. Two well-controlled pivotal studies could be necessary to provide the FDA assurance of safety or effectiveness. In the United States, absent certain limited exceptions, human clinical trials bench tests and laboratory and animal studies. The PMAintended to support product clearance or approval require an IDE application, also must contain a complete description of the device and its components, and a detailed description of the methods, facilities and controls used to manufacture the device. In addition, the submission must include the proposed labeling and any training materials. The PMA application process can be expensive and generally takes significantly longer than the Section 510(k) process. Additionally,which the FDA may never approve the PMA application. As partreviews. Some types of the PMA application review process, the FDA generally will inspect the manufacturer's facilitiestrials deemed to ensure compliance with applicable quality system regulatorypresent “non-significant risk” are deemed to have an approved IDE once certain requirements which include quality control testing, control documentationare addressed, and other quality assurance procedures.
IRB approval is obtained. If human clinical trials of a medical device are required and the device presents a significant risk,“significant risk” to human health, as defined by FDA regulations, the sponsor of the trial must filesubmit an investigational device exemption, or IDE application to the FDA and obtain IDE approval prior to commencing the human clinical trials. The IDE application must be supported by appropriate data, typically includingsuch as animal and laboratory trial results, showing that it is safe to evaluate the resultsdevice in humans and that the trial protocol is scientifically sound. The IDE application must be approved in advance by the FDA for a specified number of animal and/or laboratory testing. Ifsubjects, unless the product is deemed a non-significant risk device and eligible for more abbreviated IDE requirements. Clinical trials for a significant risk device may begin once the IDE application is approved by the FDA and one or morethe responsible institutional review boards human clinical trials may begin at a specific number of investigational sites with a specific number of patients, as approved by the FDA. If the device presents a non-significant risk to the patient, a sponsor may begin the clinical trial after obtaining approval for the trial by one or more institutional review boards without separate approval from the FDA. Submissionsites. There can be no assurance that submission of an IDE will result in the ability to commence clinical trials. Additionally, after a trial begins, the FDA may place it on hold or terminate it if, among other reasons, it concludes that the clinical subjects are exposed to unacceptable health risks that outweigh the benefits of participation in the trial. During a trial, we are required to comply with the FDA’s IDE requirements for investigator selection, trial monitoring, reporting, record keeping and prohibitions on the promotion or commercialization of investigational devices or making safety or efficacy claims for them, among other things. We are also responsible for the appropriate labeling and distribution of investigational devices. Our clinical trials must be conducted in accordance with FDA regulations and federal and state regulations concerning human subject protection, including informed consent and healthcare privacy, and the clinical trials must be conducted pursuant to GCPs. The investigators must also obtain patient informed consent, rigorously follow the investigational plan and trial protocol, control the disposition of investigational devices and comply with all reporting and record keeping requirements, among other things. The FDA’s grant of permission to proceed with clinical trials does not give assuranceconstitute a binding commitment that the FDA will approveconsider the IDE and, if it is approved, we cannot assure you that the FDA will determinetrial design adequate to support marketing clearance or approval. In addition, there can be no assurance that the data derived fromgenerated during a clinical trial will meet the trials supportchosen study endpoints or otherwise produce results that will lead the safety and effectiveness ofFDA to grant marketing clearance or approval. Similarly, in Europe, the device or warrant the continuation of clinical trials. An IDE supplementtrial must be submitted to and approved by the FDA beforelocal ethics committee and in some cases, including trials of high-risk devices, by the Ministry of Health in the applicable country.
After a sponsordevice is placed on the market, it remains subject to significant regulatory requirements. Medical devices may be marketed only for the uses and indications for which they are cleared or investigator may make a changeapproved. Device manufacturers must also establish registration and device listings with the FDA. A medical device manufacturer’s manufacturing processes and those of its suppliers are required to the investigational plan that may affect its scientific soundness, study indication or the rights, safety or welfare of human subjects. The trial also must comply with the FDA's IDE regulationsapplicable portions of the QSR, which cover the methods and informeddocumentation of the design, testing, production, processes, controls, quality assurance, labeling, packaging and shipping of medical devices. Domestic facility records and manufacturing processes are subject to periodic unscheduled inspections by the FDA. The FDA also may inspect foreign facilities that export products to the United States.
Failure by us or our suppliers to comply with applicable regulatory requirements can result in enforcement action by the FDA or other regulatory authorities, which may result in sanctions and related consequences including, but not limited to:
adverse publicity, untitled letters or warning letters;
fines, injunctions, consent decrees and civil penalties;
recall, detention or seizure of our products;
operating restrictions, partial suspension or total shutdown of production;
refusal of or delay in granting our requests for 510(k) clearance or premarket approval of new products or modified products;
withdrawing 510(k) clearance or premarket approvals that are already granted;
refusal to grant export approval for our products;
criminal prosecution; and
unanticipated expenditures to address or defend such actions.
Because elements of the CardiAMP Cell Therapy System are already approved or cleared and manufactured for commercial use, we believe regulatory approval risks are primarily those of clinical safety and efficacy in each of the two indications being assessed under separate IDEs.
Regulation of Companion Diagnostics
Companion diagnostics are subject to regulation by the FDA, the EMA and other foreign regulatory authorities as medical devices and require separate regulatory clearance or approval prior to commercial use. We anticipate that the CardiAMP potency assay for each indication will require approval under a PMA submitted to the CDRH prior to commercialization. We and our third-party collaborators who may develop our companion diagnostics will work cooperatively to generate the data required for submission with the PMA application, and will remain in close contact with the CDRH to ensure that any changes in requirements are incorporated into the development plans. We further anticipate that regulatory approval of the CardiAMP potency assay for each indication will be a prerequisite to our ability to market the CardiAMP Cell Therapy System. Representatives of CDRH have participated in our meetings with CBER regarding CardiAMP Cell Therapy System to discuss the potential use of the CardiAMP potency assay, and we anticipate that future meetings will include representatives from both CBER and CDRH to ensure that the PMA submissions (for CardiAMP and the CardiAMP potency assay) are coordinated and subject to parallel review by these respective FDA centers. Accordingly, our objective is to align the development programs such that the CardiAMP potency assay will be developed and approved contemporaneously with CardiAMP.
On July 31, 2014 the FDA issued "Guidance for Industry: In Vitro Companion Diagnostic Devices," to help companies identify the need for companion diagnostics at an earlier stage in the drug development process and to plan for co-development of the drug and companion diagnostic test. The ultimate goal of the guidance is to stimulate early collaborations that will result in faster access to promising new treatments for patients living with serious and life-threatening diseases. According to the draft guidance, for novel products such as CardiAMP, the PMA for a companion diagnostic device should be developed and approved contemporaneously with the biological product. We believe our programs for the development of the CardiAMP potency assay are consistent with the draft guidance as proposed.
Coverage and Reimbursement
Sales of our products will depend, in part, on the extent to which our products will be covered by third-party payors, such as government healthcare programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly reducing reimbursements for medical products and services. In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Decreases in third-party reimbursement for our therapeutic candidates or a decision by a third-party payor to not cover our therapeutic candidates could reduce physician usage of our products once approved and have a material adverse effect on our sales, results of operations and financial condition.
Affordable Care Act
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, the Affordable Care Act, was enacted, which includes measures that have or will significantly change the way health care is financed by both governmental and private insurers. Among the provisions of the Affordable Care Act of greatest importance to the pharmaceutical industry are the following:
• | The Medicaid Drug Rebate Program requires pharmaceutical manufacturers to enter into and have in effect a national rebate agreement with the Secretary of the Department of Health and Human Services as a condition for states to receive federal matching funds for the manufacturer’s outpatient drugs furnished to Medicaid patients. Effective in 2010, the Affordable Care Act made several changes to the Medicaid Drug Rebate Program, including increasing pharmaceutical manufacturers’ rebate liability by raising the minimum basic Medicaid rebate on most branded prescription drugs and biologic agents from 15.1% of average manufacturer price (AMP) to 23.1% of AMP and adding a new rebate calculation for “line extensions” (i.e., new formulations, such as extended release formulations) of solid oral dosage forms of branded products, as well as potentially impacting their rebate liability by modifying the statutory definition of AMP. The Affordable Care Act also expanded the universe of Medicaid utilization subject to drug rebates by requiring pharmaceutical manufacturers to pay rebates on Medicaid managed care utilization as of 2010. Per a ruling by the U.S. Supreme Court in 2012, states have the option to expand their Medicaid programs which in turn expands the population eligible for Medicaid drug benefits. The Centers for Medicare & Medicaid Services, or CMS, has proposed to expand Medicaid rebate liability to the territories of the United States as well. In addition, the Affordable Care Act provides for the public availability of retail survey prices and certain weighted average AMPs under the Medicaid program. The implementation of this requirement by the CMS may also provide for the public availability of pharmacy acquisition of cost data, which could negatively impact our sales. |
In order for a pharmaceutical product to receive federal reimbursement under the Medicare Part B and Medicaid programs or to be sold directly to U.S. government agencies, the manufacturer must extend discounts to entities eligible to participate in the 340B drug pricing program. The required 340B discount on a given product is calculated based on the AMP and Medicaid rebate amounts reported by the manufacturer. Effective in 2010, the Affordable Care Act expanded the types of entities eligible to receive discounted 340B pricing, although, under the current state of the law, with the exception of children’s hospitals, these newly eligible entities will not be obtained from each subject.eligible to receive discounted 340B pricing on orphan drugs when used for the orphan indication. In July 2013, the Health Resources and Services Administration (HRSA) issued a final rule allowing the newly eligible entities to access discounted orphan drugs if used for non-orphan indications. While the final rule was vacated by a federal court ruling, HRSA has stated it will continue to allow discounts for orphan drugs when used for any indication other than for orphan indications. In addition, as 340B drug pricing is determined based on AMP and Medicaid rebate data, the revisions to the Medicaid rebate formula and AMP definition described above could cause the required 340B discount to increase.
• | Effective in 2011, the Affordable Care Act imposed a requirement on manufacturers of branded drugs and biologic agents to provide a 50% discount off the negotiated price of branded drugs dispensed to Medicare Part D patients in the coverage gap (i.e., “donut hole”). |
Effective in 2011, the Affordable Care Act imposed an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs, although this fee would not apply to sales of certain products approved exclusively for orphan indications.
The Affordable Care Act required pharmaceutical manufacturers to track certain financial arrangements with physicians and teaching hospitals, including any “transfer of value” made or distributed to such entities, as well as any ownership or investment interests held by physicians and their immediate family members. Manufacturers were required to begin tracking this information in 2013 and to report this information to CMS by March 2014.
As of 2010, a new Patient-Centered Outcomes Research Institute was established pursuant to the Affordable Care Act to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. The research conducted by the Patient-Centered Outcomes Research Institute may affect the market for certain pharmaceutical products.
There have been judicial and Congressional challenges and amendments to certain aspects of the Affordable Care Act, and with recent legislative activity we expect there could be additional challenges, amendments and attempts to repeal the Affordable Care Act. New state and federal healthcare reform measures could limit the amounts that federal and state governments will pay for our product candidates if we obtain regulatory approval for them and could have other impacts on consequences which cannot be reasonably predicted at this time.
Other Healthcare Laws and Compliance Requirements
If we obtain regulatory approval for any of our product candidates, we may be subject to various federal and state laws targeting fraud and abuse in the FDA believeshealthcare industry. These laws may impact, among other things, our proposed sale, marketing and education programs. In addition, we are notmay be subject to patient privacy regulations by both the federal government and the states in compliance with which we conduct our business. The laws may affect our ability to operate include:
the law, it can institute proceedingsfederal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to detaininduce, or seize products, issuein return for, the purchase or recommendation of an item or service reimbursable under a market withdrawal, enjoin future violationsfederal healthcare program, such as Medicare and seekMedicaid programs;
federal civil and criminal penalties against usfalse claims laws and our officerscivil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent;
the federal Health Insurance Portability and employees. If we failAccountability Act of 1996, or HIPAA, which created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters;
• | the federal transparency laws, including the federal Physician Payment Sunshine Act, that requires drug manufacturers to disclose payments and other transfers of value provided to physicians and teaching hospitals and ownership and investment interest held by such physicians and their immediate family members; |
HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH, and its implementing regulations, which imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; and
State law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require pharmaceutical companies to comply with these regulatory requirements, our business, financial conditionthe pharmaceutical industry’s voluntary compliance guidelines and results of operations could be harmed.
Thus far, all of our approved products have been clearedthe relevant compliance guidance promulgated by the FDA throughfederal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state laws governing the Section 510(k) pre-market notification process. Weprivacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have not needed to conduct any clinical trials to supportthe same effect, thus complicating compliance efforts.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our regulatory approvals. Regulations regarding the manufacture and sale of our products arefuture business activities could be subject to change. challenge under one or more of such laws. In addition, the Affordable Care Act broadened the reach of the fraud and abuse laws by, among other things, amending the intent requirement of the federal Anti-Kickback Statute and certain criminal healthcare fraud statutes. Pursuant to the statutory amendment, a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the Affordable Care Act provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the false claims laws or the civil monetary penalties statute.
We cannot predictare also subject to the effect, ifForeign Corrupt Practices Act, or FCPA, which prohibits improper payments or offers of payments to foreign governments and their officials for the purpose of obtaining or retaining business.
Safeguards we implement to discourage improper payments or offers of payments by our employees, consultants, and others may be ineffective, and violations of the FCPA and similar state laws may result in severe criminal or civil sanctions, or other liabilities or proceedings against us, any that these changes might have onof which would likely harm our reputation, business, financial condition and results of operations. In addition
If our operations are found to granting approvals for our products,be in violation of any of the FDA has the authority to randomly inspect us for compliance with regulatory requirementslaws described above or any other government regulations that apply to our operations. These requirements include labeling regulations, manufacturing regulations, quality system regulations, regulations governing unapproved or off-label uses and medical device regulations. Medical device regulations require a manufacturer to report to the FDA serious adverse events or certain types of malfunctions involving its products. The FDA inspects device and drug manufacturing facilities in the United States in order to assure compliance with applicable quality system regulations. As discussed in the section below titled "Manufacturing and Supply,"us, we currently outsource the manufacture of our products to third-party vendors.
Further, we aremay be subject to various federalpenalties, including civil and state laws concerning health care fraud and abuse, including false claims laws, anti- kickback laws and physician self-referral laws. Violations of these laws can result in criminal and/or civil punishment, including fines, imprisonment and, in the United States,penalties, exclusion from participation in government health care programs. The scopehealthcare programs, such as Medicare and Medicaid and imprisonment, damages, fines and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operation.
In addition to the foregoing, state and federal laws regarding environmental protection and hazardous substances, including the Occupational Safety and Health Act, the Resource Conservancy and Recovery Act and the Toxic Substances Control Act, affect our business. These and other laws govern our use, handling and disposal of various biological, chemical and radioactive substances used in, and wastes generated by, our operations. If our operations result in contamination of the environment or expose individuals to hazardous substances, we could be liable for damages and governmental fines. We believe that we are in material compliance with applicable environmental laws and that continued compliance therewith will not have a material adverse effect on our business. We cannot predict, however, how changes in these laws and relatedmay affect our future operations.
Government Regulation outside the United States
In addition to regulations is expanding and their interpretation is evolving. There is very little precedent related to these laws and regulations. Increased funding for enforcement of these laws and regulations has resulted in greater scrutiny of marketing practices in our industry and resulted in several investigations by various government authorities. If a governmental authority were to determine thatthe United States, we do not comply with these laws and regulations, then we and our officers and employees couldwill be subject to criminala variety of regulations in other jurisdictions governing, among other things, clinical trials and civil sanctions, including exclusionany commercial sales and distribution of our products. Because biologically sourced raw materials are subject to unique contamination risks, their use may be restricted in some countries.
Whether or not we obtain FDA approval or clearance for a product, we must obtain the requisite approvals or clearances from participationregulatory authorities in federalforeign countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application much like the PMA or IND prior to the commencement of human clinical trials. In Europe, for example, a Clinical Trial Authorization, or CTA, must be submitted to each country’s national health careauthority and an independent ethics committee, much like the FDA and the IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement programs.vary from country to country. In all cases, the clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
39
To obtain regulatory approval of an investigational biological product under European regulatory systems, we must submit a marketing authorization application. The application used to file the PMAs for CardiAMP Cell Therapy System and BLA for CardiALLO Cell Therapy System in the United States are similar to that required in Europe, with the exception of, among other things, country-specific document requirements. Europe also provides opportunities for market exclusivity. For example, in Europe, upon receiving marketing authorization, new chemical entities generally receive eight years of data exclusivity and an additional two years of market exclusivity. If granted, data exclusivity prevents regulatory authorities in Europe from referencing the innovator’s data to assess a generic application. During the additional two-year period of market exclusivity, a generic marketing authorization can be submitted, and the innovator’s data may be referenced, but no generic product can be marketed until the expiration of the market exclusivity. However, there is no guarantee that a product will be considered by Europe’s regulatory authorities to be a new chemical entity, and products may not qualify for data exclusivity.
International
The 10-year market exclusivity may be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria for orphan designation, for example, if the product is sufficiently profitable not to justify maintenance of market exclusivity. Additionally, marketing authorization may be granted to a similar product for the same indication at any time if:
the second applicant can establish that its product, although similar, is safer, more effective or otherwise clinically superior;
the applicant consents to a second orphan medicinal product application; or
the applicant cannot supply enough orphan medicinal product.
For other countries outside of Europe, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
In Europe, we expect both CardiAMP and CardiALLO Cell Therapy Systems to be regulated as advanced therapy medicinal products, or ATMPs. To provide for a common framework for the next few years,marketing of ATMPs, Regulation (EC) No 1394/2007 of the European Parliament and of the Council on advanced therapy medicinal products, or ATMP Regulation, was adopted in 2007. The ATMP Regulation was designed to ensure a high level of human health protection as well as the free movement of ATMPs in Europe. The cornerstone of the ATMP Regulation is that a marketing authorization must be obtained prior to the marketing of ATMPs. In turn, the marketing authorization can only be granted if, after a scientific assessment of the quality, efficacy and safety profile, it is demonstrated that the benefits outweigh the risks. The application for a marketing authorization must be submitted to the EMA and the final decision is taken by the European Commission. This procedure ensures that these products are assessed by a specialized body (the Committee for Advanced Therapies, or CAT) and that the marketing authorization is valid in all the European Union Member States.
The ATMP Regulation empowered the EMA to make scientific recommendations as to whether a given product should be considered an ATMP (hereinafter “classifications”). Additionally, it provided for a new instrument, the so-called certification procedure, designed as an incentive for small and medium sized enterprises, or SMEs, that were involved in the first stages of the development of ATMPs but lacked the resources to conduct clinical trials. Specifically, the certification that the quality and preclinical aspects of the development are in conformity with the relevant regulatory requirements was expected to help SMEs attract capital and to facilitate the transfer of research activities to entities with the capacity to market medicinal products.
The ATMP Regulation builds on the procedures, concepts, and requirements designed for chemical-based medicinal products. However, ATMPs present very different characteristics. Additionally, in contrast to chemical-based medicinal products, research in advanced therapies is –for the most part- conducted by academia, non-for-profit organizations, and SMEs, which only have limited financial resources and often lack exposure to the regulatory system that governs medicines.
If we planfail to seek requiredcomply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and complycriminal prosecution.
The advertising and promotion of our products in the EEA is subject to the provisions of the Medical Devices Directive, Directive 2006/114/EC concerning misleading and comparative advertising, and Directive 2005/29/EC on unfair commercial practices, as well as other national legislation in the EEA countries governing the advertising and promotion of medical devices. The European Commission has submitted a Proposal for a Regulation of the European Parliament and the Council on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009, to replace, inter alia, Directive 93/42/EEC and to amend regulations regarding medical devices in the European Union, which could result in changes in the regulatory requirements for medical devices in Europe. In Germany, the advertising and promotion of our products can also be subject to restrictions provided by the German Act Against Unfair Competition (Gesetzgegen den unlauteren Wettbewerb) and the law on the advertising of medicines (Heilmittelwerbegesetz), criminal law, and some codices of conduct with extensiveregard to medical products and medical devices among others. These laws may limit or restrict the advertising and promotion of our products to the general public and may impose limitations on our promotional activities with healthcare professionals.
Sales of medical devices are subject to foreign government regulations, governing product safety, quality, manufacturing and reimbursement processes inwhich vary substantially from country to country. In order to market our products in some major foreign markets, which may include countries in Latin America, Europeoutside the United States, we must obtain regulatory approvals or Asia. These regulations vary significantly from country to countryCE Certificates of Conformity and comply with respect to the nature of the particular medical device.extensive safety and quality regulations. The time required to obtain theseapproval by a foreign approvalscountry or to market our productsobtain a CE Certificate of Conformity may be longer or shorter than that required infor FDA clearance or approval, and the United States,requirements may differ. In the EEA, we are required to obtain Certificates of Conformity before drawing up an EC Declaration of Conformity and requirements foraffixing the CE Mark of conformity to our medical devices. Many other countries accept CE Certificates of Conformity or FDA clearance or approval may differ from FDA requirements.although others, such as Brazil, Canada and Japan require separate regulatory filings.
If
Employees
As of June 30, 2019, we sell anyhad 24 full-time employees, consisting of clinical development, product development, regulatory, manufacturing, quality, finance, administration, sales, and marketing. We also regularly use independent contractors across the organization to augment our regular staff. None of our products internationally, the products will be subject to certain foreign regulatory approvals. In order to market our product devices in the member countries of the European Union, we will be required to comply with the European Medical Devices Directives and obtain "CE" mark certification. CE mark certification is the European symbol of adherence to quality assurance standards and compliance with applicable European Medical Devices Directives. Under the European Medical Devices Directives, all medical devices including active implants must qualify for CE marking. We also would be required to comply with other foreign regulations, such as obtaining Ministry of Health Labor and Welfare approval in Japan, Health Protection Branch approval in Canada, and Therapeutic Goods Administration approval in Australia, if we market in those jurisdictions.
Research and Development
Our research and development engineering personnel have extensive experience in developing medical devices to treat joint and spine pathologies. Our engineers work closely with surgeons to design devices thatemployees are intended to improve patient care, simplify surgical techniques and reduce overall costs. In addition to constantly enhancing and improving our current product offerings, we are focusing our research and development efforts in novel approaches to total knee arthroplasty, spinal motion preservation devices and products that promote new fusion techniques and minimally invasive surgical techniques for reconstructive and spinal surgery. Our research and development efforts are part of our overall business plan to become a market leader in providing solutions for the reconstructive joint and spine markets. To further promote this strategy, we are focused on converting these research and development efforts into commercially viable products that incorporate minimally invasive techniques and quick recovery to improve patient outcomes across all of our products. Currently, our research and development staff is located in New Jersey,covered by collective bargaining agreements and we also engage the services of independent contractors in that state. However, we are considering expansion of this staff by hiring engineers in California as well. We expectconsider relations with our research and development costs to maintain the 2009 levels as we continue to expend significant resources to develop and commercialize our products and potential products.
At this time, we have one formal consulting arrangement with a surgeon. However, we work with other surgeons informally to obtain their feedback to enhance our products and to identify product candidates that we would like to develop. We plan to work closely with product opinion leaders to develop and enhance our product portfolio. In 2008, we spent approximately $1,332,000 on research and development and approximately $329,000 through the three quarters ended September 30, 2009.
Manufacturing and Supply
We do not have a manufacturing facility, and we currently do not intend to build manufacturing facilities of our own in the foreseeable future. We utilize third-party vendors to manufacture all of our implants and instruments, including components of our products, while internally performing product design and quality assurance. We currently use up to seven manufacturers for our devices.
Our outsourced manufacturing process typically involves machining semi-completed raw materials for both our metal and polyethylene components that make up our joint replacement systems. After being machined, the parts are inspected and processed in preparation for final polishing and finishing as needed. Prior to being packaged, our parts are inspected again to ensure that they are within approved specifications. We also use components in our devices that we acquire from other companies. We distribute both sterile and non-sterile implants and instruments.
Our outsourcing strategy is targeted at companies that meet FDA Quality Standards and our internal policies and procedures. Supplier performance is maintained and managed through a corrective action program intended to ensure that all product requirements are met or exceeded. We believe these manufacturing relationships minimize our capital investment, help control costs and allow us to compete with larger volume manufacturers of spine surgery and reconstructive surgical products.
40
We currently utilize a small number of manufacturers for our products and rely on a limited number of sources for our product components that are manufactured by third parties. In the future, we may consider manufacturing certain products or product components internally, if and when demand or quality requirements make it appropriate to do so.
Although we believe that alternative third-party manufacturers are available, we cannot assure you that we will be able to timely replace our third-party manufacturers immediately if one or more of them can no longer provide us with their manufacturing services. In addition, while we do not anticipate that we will encounter problems in obtaining adequate supplies of components, we cannot assure you that we will continueemployees to be able to obtain components under acceptable terms and in a timely manner.
Sales and Marketing
We mostly rely on third-party independent distributors to market and sell our products. In the future, we intend to increase the number of our internal sales and marketing personnel and further build our own sales and marketing infrastructure to market some of our products targeting surgeons in certain regions. We also intend to continue collaborating with third-party independent distributors, including large regional distributors.
Patents and Proprietary Technology; Trademarks
Patents
We have applied for U.S. and foreign patents covering several of our implant components, and some of our surgical instrumentation. As of December 14, 2009, we had 20 issued patents and 19 pending domestic and foreign patent applications covering seven devices.
Patents and intellectual property will continue to be an important aspect of the orthopedic and spine industry. In this regard, we intend to defend our intellectual property rights.good. We believe that our patents and products do not and will not infringe patents or violate proprietary rights of others, although it is possible that our existing patent rights may not be valid or that infringement of existing or future patents or proprietary rights may occur. If some of our intellectual property and agreements relating to our products are deemed invalid, that action may have a material adverse effect on our financial condition and results of operations.
The medical device industry is characterized by patent and other intellectual property litigation, and we could become subject to litigation that could be costly, result in diverting management's time and efforts, require us to pay damages and/or prevent us from marketing our existing or future products. Patent litigation typically involves complex factual and legal questions whose outcome is uncertain. Any claim relating to infringement of patents that is successfully asserted against us may require us to pay substantial damages. Even if successful, litigation to enforce our intellectual property rights or to defend our patents against challenge could be expensive and time-consuming and could divert our management's attention. Our success will depend in part on our not infringing patents issued to others, including our competitors and potential competitors. If our products are found to infringe the patents of others, the development, manufacture and sale of our products or potential products could be severely restricted or prohibited. Also, our competitors may independently develop similar technologies that are not restricted by other companies' patents, including ours. Due to the importance of our patents to our business, our market share can decline if we fail to protect our intellectual property rights.
A patent infringement suit brought against us or our partners may force us or our partners to halt the development, manufacture or sale of products or potential products that are claimed to be infringing, unless that party grants us or our partners rights to use its intellectual property. As a result, we may be required to obtain licenses to patents or proprietary rights of others in order to continue to commercialize our products, which we may not be able to do on acceptable terms, or at all. Even if we or any partner were able to obtain rights to the third party's intellectual property, these rights may be non-exclusive, thereby giving our competitors access to the same intellectual property. Ultimately, we may be unable to commercialize some of our products or potential products or may have to cease some of our business operations as a result of patent infringement claims, which could severely harm our business.
41
As more companies enter the orthopedic and spine market, the possibility of a patent infringement claim against us grows. While we try to ensure that our products do not infringe others' patents and proprietary rights, our products, potential products and methods may be covered by patents held by our competitors.
Trademarks
As of December 14, 2009, we had three registered trademarks with the U.S. Patent and Trademark Office, or USPTO, for the marks "Accin," "Align 360" and "Cardo Medical"; we have an application pending for the mark "A La Carte."
Competition
The orthopedic and spinal device industry is highly competitive and dominated by a number of large companies with substantially greater financial and other resources than we have. Our largest competitors in the orthopedic and spinal surgical device market are DePuy Orthopaedics, Inc. and DePuy Spine, Inc. (divisions of Johnson & Johnson Company), Zimmer, Inc. (a subsidiary of Zimmer Holdings, Inc.), Stryker Howmedica Osteonics (a subsidiary of Stryker Corporation), Smith & Nephew plc, Biomet Orthopedics, Inc. (a subsidiary of Biomet, Inc.), Medtronic Sofamor Danek, and Synthes Inc.
Companies in the industry compete on the basis of product features and design, innovation, service, thecontinued ability to maintain new product flow, relationships with key orthopedic surgeonsattract, hire and hospitals, the strength of their distribution networkretain qualified personnel.
Corporate Information
We were originally incorporated as NAM Corporation in Delaware on January 12, 1994 and price. While price is a key factor in the orthopedic market, other significant factors could negatively impactsubsequently changed our results of operations and financial condition, including: technological innovation, reimbursement rates, surgeon preference, ease of use, clinical results and service provided by us and our representatives.
Our products are, and any potential products we commercialize will be, subjectname to intense competition. Many of our current and potential competitors have substantially greater financial, technical and marketing resources than we do, and they may succeed in developing products that would render our products obsolete or noncompetitive. Many of these competitors also have significantly greater operating history and reputations than we do in our respective fields. We may not be able to compete successfully if we are unable to develop proprietary products that reach the market in a timely manner, receive adequate reimbursement and are safer, less invasive and less expensive than alternatives available for the same purpose. Because of the rapidly growing orthopedic market, we anticipate that companies will dedicate significant resources to developing competing products.
Regarding our spinal portfolio, we also face competition from a growing number of smaller companies with more limited product offerings and geographic reach than our larger competitors. These companies, who represent intense competition in specified markets, include Abbott Spine,clickNsettle.com, Inc. (a division of Abbott Laboratories, Inc.), Orthofix International N.V. (parent of Blackstonethen Cardo Medical, Inc.), Alphatec Spine Inc. (a subsidiary of Alphatec Holdings, Inc.), Globusthen Tiger X Medical, Inc., and Nuvasive,finally BioCardia, Inc. on October 26, 2016 in connection with a reverse merger transaction in which our wholly-owned subsidiary, Icicle Acquisition Corp., merged with and into BioCardia Lifesciences, Inc. (which was named BioCardia, Inc. prior to the merger), with BioCardia Lifesciences continuing as the surviving company. Following the completion of the reverse merger transaction, we assumed the business and operations of BioCardia Lifesciences and changed our name to BioCardia, Inc.
Product Liability and Insurance
We operate in only one business segment, which is a clinical-stage regenerative medicine company developing novel therapeutics for cardiovascular diseases with large unmet medical needs. See Note 1 to our consolidated financial statements for the year ended December 31, 2018 included in this prospectus. Our principal executive offices are subject to potential product liability risks that are inherent in the design, marketing and sale of orthopedic implants and surgical instrumentation.located at 125 Shoreway Road, Suite B, San Carlos, CA 94070. Our telephone number is (650) 226-0120.
Our website address is www.biocardia.com. We have implemented strict quality control measuresnot incorporated by reference into this prospectus the information on our website, and currently maintain product liability insuranceyou should not consider it to be a part of this prospectus.
PROPERTIES
Our principal executive office is located at 125 Shoreway Road, Suite B, San Carlos, CA 94070 in amountsa facility we lease encompassing 13,718 square feet of office, lab, and manufacturing space. The lease for this facility expires in 2021. We believe that our existing facilities are adequate for our current needs. If we believedetermine that additional or new facilities are typical in the industry for companies with a comparable size to ours. Our insurance premiums are paid as a percentage of sales. We evaluate our levels of product liability insurance annually, as well as the amount of retention carried compared to other comparable companies in the industry. Due to the volatility of the insurance marketplace, the value of the product liability insurance products delivered and the small number of providers of these products, there can be no guarantees as to whether we will be able to secure coverageneeded in the future, at a reasonable cost.
42
Third-Party Reimbursement
Sales of our products will depend on the availability of adequate reimbursement from third-party payors (such as governmental programs, for example, Medicare and Medicaid, private insurance plans and managed care programs), both in terms of the sales volumes and prices of our products. Healthcare providers, such as hospitals that purchase medical devices for treating their patients, generally rely on third-party payors to reimburse all or part of the costs and fees associated with the procedures performed with these devices. These third-party payors may deny reimbursement if they feel that a device is not the most cost-effective treatment available, or was used for an unapproved indication. As such, surgeons are unlikely to use our products if they do not receive reimbursement adequate to cover the cost of their involvement in the surgical procedures. We alsowe believe that future reimbursementsufficient options would be available to us on commercially reasonable terms.
LEGAL PROCEEDINGS
The Company may be subject to increased restrictions bothvarious claims, complaints, and legal actions that arise from time to time in the U.S. and internationally. If we sell our products internationally, market acceptance may depend, in part, uponnormal course of business. Management does not believe that the availabilityCompany is party to any currently pending legal proceedings as of reimbursement within the prevailing healthcare payment systems. Reimbursement and healthcare payment systems in international markets vary significantly by country, and include both government sponsored healthcare and private insurance.
Future legislation, regulationJune 30, 2019. There can be no assurance that existing or reimbursement policies of third-party payors may adversely affect the demand for our existing products or our products currently under development and limit our ability to sell our products on a profitable basis.
Also, third-party payors are increasingly challenging the prices charged for medical products and services. Particularlyfuture legal proceedings arising in the United States, third-party payors carefully review, and increasingly challenge, the prices charged for procedures and medical products. Also, greater numbersordinary course of insured individuals are receiving (andbusiness or otherwise will continue to receive over the next decade) their medical care through managed care programs, which monitor and often require pre-approval of the services that a member will receive. Many managed care programs are paying their providers on a capitated basis, which puts the providers at financial risk for the services provided to their patients by paying them a predetermined payment per member per month.
Legislative or administrative reforms to the U.S. or international reimbursement systems in a manner that significantly reduces reimbursement for procedures using our medical devices or denies coverage for those procedures couldnot have a material adverse effect on ourthe Company’s business, financial condition orposition, results of operations. We believe that the overall escalating costoperations, or cash flows.
Management’s Discussion and Analysis of medical productsFinancial Condition and services has led to, and will continue to lead to, increased pressures on the healthcare industry to reduce the costsResults of products and services. We cannot assure you that third-party reimbursement and coverage will be available or adequate, or that future legislation, regulation, or reimbursement policiesOperations
The following discussion of third-party payors will not adversely affect the demand for our products or our ability to sell these products on a profitable basis. We also cannot assure you that our products will be considered cost-effective by third-party payors, that reimbursement will be available or, if available, that the third-party payors' reimbursement policies will not adversely affect our ability to sell our products profitably.
Healthcare Fraud and Abuse
Our relationship with surgeons, hospitals and the marketers of our products are subject to scrutiny under various state and federal anti-kickback, self-referral, false claims and similar laws, often referred to collectively as healthcare fraud and abuse laws. The federal anti-kickback laws prohibit unlawful inducements for the referral of business reimbursable under federally-funded health care programs, such as remuneration provided to physicians to induce them to use certain medical devices reimbursable by Medicare or Medicaid. Healthcare fraud and abuse laws are complex and subject to evolving interpretations, and even minor, inadvertent violations potentially can give rise to claims that the relevant law has been violated. Certain states in which we market our products have similar anti-kickback, anti-fee splitting and self-referral laws, imposing substantial penalties for violations. Any violations of these laws could result in a material adverse effect on the market price of our common stock, as well as our business, financial condition and results of operations. We cannot assure you that any of the healthcare fraud and abuse laws will not change oroperations should be interpretedread in the future in a manner which restricts or adversely affects our business activities or relationshipsconjunction with surgeons, hospitals and marketers of our products. In addition, possible sanctions for violating these anti-kickback laws include monetary fines, civil and criminal penalties, exclusion from Medicare and Medicaid programs and forfeiture of amounts collected in violation of these prohibitions.
43
We must comply with a variety of other laws, such as laws prohibiting false claims for reimbursement under Medicare and Medicaid, which also can be triggered by violations of federal anti-kickback laws; Healthcare Insurance Portability and Accountability Act of 1996, which protects the privacy of individually identifiable healthcare information; and the Federal Trade Commission Act and similar laws regulating advertisement and consumer protections. In certain cases, federal and state authorities pursue actions for false claims on the basis that manufacturers and distributors are promoting unapproved or off-label uses of their products.
Management's Plan
To achieve our growth objectives, we are considering different strategies, including growth through acquisitions and raising additional capital. As a result, we are constantly and aggressively evaluating and we will continue to evaluate other companies and businesses for potential synergies that would add value to our existing operations.
Description of Property
As of September 30, 2009, the Company leases an office facility in Van Nuys, California (near Los Angeles) under a month-to-month operating lease. We also lease facilities in Beverly Hills and Clifton, New Jersey (near New York City) under operating leases that expire in July 2010 and August 2012, respectively.
Legal Proceedings
From time to time, we may be a party to legal proceedings incidental to our business. We do not believe that there are any proceedings threatened or pending against us, which, if determined adversely to us, would have a material effect on our financial position or results of operationsstatements and cash flows.
MARKET PRICE OF AND DIVIDENDS ON THE REGISTRANT'S COMMON EQUITY AND RELATED STOCKHOLDER MATTERS
Market for Common Stock
The Company's common stock currently trades on the OTC Bulletin Board under the symbol "CDOM.OB" The following table sets forth the quarterly high and low sales prices of our common stock for the fiscal years 2008 and 2007 and the first three quarters of 2009, as quoted on the OTC Bulletin Board. This information represents prices between dealers and does not include retail mark-ups, markdowns or commissions and may not represent actual transactions. All information related to stock price and numbers of common stock are post-split, which reflect a reverse split with clickNsettle.com which occurred in the month of March 2008.
| High | Low | |||||
| Fiscal Year 2007 | ||||||
| First Quarter | $2.50 | $0.52 | ||||
| Second Quarter | $1.20 | $0.70 | ||||
| Third Quarter | $5.00 | $0.75 | ||||
| Fourth Quarter | $6.20 | $3.00 | ||||
| Fiscal Year 2008 | ||||||
| First Quarter | $3.90 | $1.60 | ||||
| Second Quarter | $2.25 | $1.05 | ||||
| Third Quarter | $2.90 | $1.10 | ||||
| Fourth Quarter | $1.90 | $1.25 | ||||
| Fiscal Year 2009 | ||||||
| First Quarter | $2.25 | $0.52 | ||||
| Second Quarter | $1.50 | $0.65 | ||||
| Third Quarter | $1.25 | $0.60 | ||||
| Fourth Quarter (through December 17, 2009) | $1.01 | $0.51 |
44
As of December 14, 2009, there were approximately 261 registered holders of record of the common stock.
We have not paid any cash dividends on our common stock and do not plan to pay any such dividends in the foreseeable future. Our Board of Directors will determine our future dividend policy on the basis of many factors, including results of operations, capital requirements and general business conditions.
Equity Compensation Plan Information
The following table summarizes the number of outstanding options granted to employees, service providers and directors under the Company's compensation plans and arrangements as of the quarter ended September 30, 2009. For a description of equity compensation plans not approved by security holders see Note 12 to the Consolidated Financial Statements for the year ended December 31, 2008notes included elsewhere in this registration statement.
| Number of securities | Number of securities | ||||||
| to be issued upon | Weighted-average | remaining available | |||||
| exercise of | exercise price of | for future issuance | |||||
| outstanding options, | outstanding options, | under equity | |||||
| warrants and rights | warrants and rights | compensation plans | |||||
| Equity compensation plans | |||||||
| not approved by security holders | 2,358,400 | $ 0.23 | - | ||||
| Equity compensation plans | |||||||
| approved by security holders | - | - | - | ||||
| Total | 2,358,400 | $ 0.23 | - |
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITIONAND RESULTS OF OPERATIONSprospectus. This discussion contains certain forward-looking statements that involve risk and uncertainties. Our actual results may differ materially from those discussed below. Factors that could cause or contribute to such differences include, but are not limited to, those identified below and those set forth under the section titled “Risk Factors,” and other documents we file with the Securities and Exchange Commission. Historical results are not necessarily indicative of future results.
Special Note Regarding Smaller Reporting Company Status
As a result of having been a “smaller reporting company” (as defined in Rule 12b-2 of the Securities Exchange Act of 1934, as amended), we are allowed and have elected to omit certain information, including three years of year-to-year comparisons and tabular disclosure of contractual obligations, from this Management’s Discussion and Analysis of Financial Condition and Results of Operations; however, we have provided all information for the periods presented that we believe to be appropriate and necessary.
Overview
On August 22, 2016, the Company, Icicle Acquisition Corp., a Delaware corporation and our direct wholly-owned subsidiary, and BioCardia Lifesciences, Inc. (at the time, named BioCardia, Inc.) entered into an Agreement and Plan of Merger, or the Merger Agreement. On October 24, 2016, pursuant to the Merger Agreement, Icicle Acquisition Corp. merged with and into BioCardia Lifesciences, with BioCardia Lifesciences continuing as the surviving company. BioCardia Lifesciences was determined to be the accounting acquirer, and following the completion of the Merger, we assumed the business and operations of BioCardia Lifesciences and changed our name to BioCardia, Inc.
We are a clinical-stage regenerative medicine company developing novel therapeutics for cardiovascular diseases with large unmet medical needs. Our lead therapeutic candidate is the investigational CardiAMP Cell Therapy System, or CardiAMP, which provides an autologous bone marrow derived cell therapy (using a patient’s own cells) for the treatment of two clinical indications: heart failure that develops after a heart attack and chronic myocardial ischemia.
We initiated our U.S. Food and Drug Administration, or FDA, accepted Phase III pivotal trial for CardiAMP Cell Therapy in ischemic systolic heart failure, in December 2016. The CardiAMP Heart Failure Trial is a Phase III, multi-center, randomized, double-blinded, sham-controlled study of up to 260 patients at 40 centers nationwide, which includes a 10-patient roll-in cohort. The trial’s primary endpoint is a clinical composite of six-minute walk distance and major adverse cardiac and cerebrovascular events. In September 2017, the independent Data Safety Monitoring Board (DSMB) completed the pre-specified interim analysis of safety outcomes for the first 10 patients treated in the Phase III trial of its investigational CardiAMP cell therapy product. The DSMB indicated there were no significant safety concerns with the CardiAMP study results and recommended that the trial continue, as planned. Currently 22 world class medical centers are actively enrolling in the study. We anticipate that trial enrollment will be completed in Q3 2020.
In January 2018, the FDA approved a second IDE for the randomized controlled pivotal trial of autologous bone marrow mononuclear cells using the CardiAMP Cell Therapy System in patients with refractory chronic myocardial ischemia for up to 343 patients at up to 40 clinical sites in the United States. This therapeutic approach uses many of the same novel aspects as the CardiAMP Heart Failure Trial and leverages our experience and investment in the heart failure trial.
Our second therapeutic candidate is the CardiALLO Cell Therapy System, an investigational culture expanded bone marrow derived “off the shelf” mesenchymal cell therapy. We are actively working to secure FDA acceptance of an Investigational New Drug (“IND”) application for a Phase I/II trial for CardiALLO Cell Therapy System for the treatment of ischemic systolic heart failure. To date we have completed manufacturing validation runs of these cells at BioCardia to support future clinical studies and have received written input from the FDA on the protocol design and the chemistry manufacturing and controls. Our goal is to receive FDA acceptance of the IND in the fourth quarter of 2019.
We are committed to applying our expertise in the fields of autologous and allogeneic cell-based therapies to improve the lives of patients with cardiovascular conditions. As we engage in clinical trials of our therapeutic candidates, we have compensated and intend to compensate all parties performing the trials or studies (including all the parties identified in our Annual Report on Form 10-K) only on terms that are standard and customary in clinical study arrangements.
To date, we have devoted substantially all of our resources to research and development efforts relating to our therapeutic candidates and biotherapeutic delivery systems, including conducting clinical trials, developing manufacturing and sales capabilities, in-licensing related intellectual property, providing general and administrative support for these operations and protecting our intellectual property. We have also generated modest revenues from sales of our approved products. We have funded our operations primarily through the sales of equity and convertible debt securities, and certain government and private grants.
We have incurred net losses in each year since our inception. Our net losses were approximately $14.0 million, $12.3 million and $10.3 million for the years ended December 31, 2018, 2017 and 2016, respectively. As of March 31, 2019, we had an accumulated deficit of approximately $90.0 million. Substantially all of our net losses have resulted from costs incurred in connection with our research and development programs, clinical trials, intellectual property matters, building our manufacturing and sales capabilities, and from general and administrative costs associated with our operations. As discussed in more detail under “Liquidity and Capital Resources,” we have determined that there is substantial doubt about the Company's ability to continue as a going concern, and we plan to raise additional capital, potentially including debt and equity arrangements, to finance our future operations.
Financial Overview
Revenue
We currently have a portfolio of enabling and delivery products, from which we have generated modest revenue. These revenues include commercial sales of our Morph vascular access system in the US and EU and revenue from partnering agreements with corporate and academic institutions. Under these partnering agreements, we provide our Helix™ biotherapeutic delivery system and customer training and support for use in preclinical and clinical studies.
Cost of Goods Sold
Cost of goods sold includes the costs of raw materials and components, manufacturing personnel and facility costs and other indirect and overhead costs associated with manufacturing our enabling and delivery products.
Research and Development Expenses
Our research and development expenses consist primarily of:
salaries and related overhead expenses, which include share-based compensation and benefits for personnel in research and development functions;
fees paid to consultants and contract research organizations, or CROs, including in connection with our preclinical studies and clinical trials and other related clinical trial fees, such as for investigator grants, patient screening, laboratory work, clinical trial management and statistical compilation and analysis;
costs related to acquiring and manufacturing clinical trial materials;
costs related to compliance with regulatory requirements; and
payments related to licensed products and technologies.
We expense all research and development costs in the periods in which they are incurred. Costs for certain development activities are recognized based on an evaluation of the progress of completion of specific tasks using information and data provided to us by our vendors and clinical sites. Nonrefundable advance payments for goods or services to be received in future periods for use in research and development activities are deferred and capitalized. The capitalized amounts are then expensed as the related goods are delivered and the services are performed.
We plan to increase our research and development expenses for the foreseeable future as we continue the Pivotal CardiAMP Heart Failure Trial and the Pivotal CardiAMP Chronic Myocardial Ischemia Trial, further develop CardiAMP and CardiALLO Cell Therapy Systems, and subject to the availability of additional funding, further advance the development of other therapeutic candidates for additional indications. We typically use our employee and infrastructure resources across multiple research and development programs, and accordingly, we have not historically allocated resources specifically to our individual programs.
Selling, General and Administrative Expenses
Selling, general and administrative expenses consist primarily of salaries and related costs for employees in executive, finance and administration, sales, corporate development and administrative support functions, including share-based compensation expenses and benefits. Other selling, general and administrative expenses include sales commissions, rent, accounting and legal services, obtaining and maintaining patents, the cost of consultants, occupancy costs, insurance premiums and information systems costs.
Other Income (Expense)
Other income and expense consist primarily of interest income we earn on our cash, cash equivalents and investments, interest charges we incurred in periods when we have convertible debt outstanding, and changes in the fair value of our warrant and convertible shareholder note derivative liabilities in periods when we have warrants or convertible debt outstanding. Subsequent to the Merger, we have no interest charges related to the convertible debt and changes in the fair value of our warrant and convertible shareholder note derivative liabilities as such instruments were converted, cancelled or exchanged as part of the Merger.
Critical Accounting Policies and Estimates
Our management’s discussion and analysis of our financial condition and results of operations areis based on our financial statements, which we have prepared in accordance with generally accepted accounting principles generally accepted in the United States of America.States. The preparation of theseour financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenues and expenses during the reporting periods. On an ongoing basis, weliabilities. We evaluate these estimates and judgments including those described in greater detail below.on an ongoing basis. We base our estimates on historical experience and on various other factorsjudgements that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
The following discussion
We define our critical accounting policies as those that require us to make subjective estimates and analysis excludes thejudgments about matters that are uncertain and are likely to have a material impact of clickNsettle.com, Inc. ("CKST")'son our financial condition and results of operations prioras well as the specific manner in which we apply those principles. The following discussion addresses what we believe to be the critical accounting policies used in the preparation of our financial statements that require significant estimates and judgments.
Research and Development—Clinical Trial Accruals
As part of the process of preparing our financial statements, we are required to estimate our expenses resulting from our obligations under contracts with vendors and consultants and clinical site agreements in connection with conducting clinical trials. The financial terms of these contracts are subject to negotiations which vary from contract to contract and may result in payment flows that do not match the periods over which materials or services are provided to us under such contracts. Our clinical trial accrual is dependent upon the timely and accurate reporting of expenses of our CROs and other third-party vendors.
Our objective is to reflect the appropriate clinical trial expenses in our financial statements by matching those expenses with the period in which services and efforts are expended. We account for these expenses according to the Merger on August 29, 2008 because they were not material in relationprogress of the trial as measured by patient progression and the timing of various aspects of the trial. We determine accrual estimates through discussion with applicable personnel and outside service providers as to the progress or state of completion of clinical trials, or the services completed. During the course of a clinical trial, we adjust the rate of clinical trial expense recognition if actual results differ from the estimates. We make estimates of our accrued expenses as of each balance sheet date in our financial informationstatements based on facts and circumstances known at that time. Although we do not expect that our estimates will be materially different from amounts actually incurred, our understanding of status and timing of services performed relative to the actual status and timing of services performed may vary and may result in our reporting amounts that are too high or too low for any particular period. Through March 31, 2019, there had been no material adjustments to our prior period estimates of accrued expenses for clinical trials. However, due to the nature of estimates, we cannot provide assurance that we will not make changes to our estimates in the future as we become aware of additional information about the status or conduct of our clinical trials.
Share-Based Compensation
We measure and recognize share-based compensation expense for equity awards to employees, directors and consultants based on fair value at the grant date. We use the Black-Scholes-Merton option-pricing model, or BSM, to calculate the fair value of stock options, which includes subjective assumptions such as the risk free interest rate, the expected volatility in the value of the periods presented below.Company's common stock, and the expected term of the option. Restricted stock units (RSUs) are measured based on the fair market values of the underlying stock on the dates of grant. Share-based compensation expense recognized in the statements of operations is based on awards at the time of grant and is reduced for actual forfeitures at the time that the forfeitures occur. Compensation cost for employee share-based awards will be recognized over the vesting period of the applicable award on a straight-line basis.
All amounts, other than share amounts,
For options granted to nonemployees, we revalue the unearned portion of the share-based compensation and the resulting change in fair value is recognized in the statements of operations over the period the related services are stated in thousands.rendered.
As used in this "Management's Discussion and Analysis of Financial Condition and Results of Operations" except where the context otherwise requires, the term "we," "us," "our" or "Cardo" refers to the businessOperations
Comparison of Cardo Medical, Inc.Three Months Ended March 31, 2019 and 2018
45
The following discussion should be read together withtable summarizes our results of operations for the information containedthree months ended March 31, 2019 and 2018 (in thousands):
Three months ended March 31, | ||||||||
2019 | 2018 | |||||||
Revenue: | ||||||||
Net product revenue | $ | 76 | $ | 82 | ||||
Collaboration agreement revenue | 140 | 117 | ||||||
Total revenue | 216 | 199 | ||||||
Costs and expenses: | ||||||||
Cost of goods sold | 106 | 157 | ||||||
Research and development | 2,166 | 1,955 | ||||||
Selling, general and administrative | 1,631 | 1,707 | ||||||
Total costs and expenses | 3,903 | 3,819 | ||||||
Operating loss | (3,687 | ) | (3,620 | ) | ||||
Other income (expense): | ||||||||
Interest income | 23 | 36 | ||||||
Other income (expense) | (1 | ) | — | |||||
Total other income, net | 22 | 36 | ||||||
Net loss | $ | (3,665 | ) | $ | (3,584 | ) | ||
Revenue. Revenue increased by approximately $17,000 in the financial statementsthree months ended March 31, 2019 compared to the three months ended March 31, 2018 primarily due to increased revenue earned under programs with our collaborative partners. We expect collaboration agreement revenues to increase modestly in 2019. We expect net product revenue will be flat or increase modestly in the latter half of 2019 depending on demand for the new Morph AVANCE™ steerable introducer and related notes included elsewheresubject to our ability to secure funding for marketing and production.
Cost of Goods Sold. Cost of goods sold decreased by approximately $51,000 in this Registration Statement on Form S-1.
Overview
Cardo Medical, Inc. is an orthopedic medical device company specializingthe three months ended March 31, 2019 compared to the three months ended March 31, 2018 primarily due to a decrease in designing, developing and marketing high performance reconstructive joint devices and spinal surgical devices. Reconstructive joint devices are usedproduct sales volumes. We expect cost of goods sold to replace knee, hip and other joints that have deteriorated through disease or injury. Spinal surgical devices involve products to stabilize the spine for fusion and reconstructive procedures. Within these areas, Cardo intends to focusincrease moderately in 2019, depending on the higher-growth sectorssales volumes of Morph AVANCE™ in 2019.
Research and Development Expenses. Research and development expenses increased by approximately $211,000 in the three months ended March 31, 2019 compared to the three months ended March 31, 2018 primarily due to increased expenses incurred in conducting the ongoing pivotal CardiAMP Heart Failure Trial and in the development of the orthopedic industry, suchCardiALLO Cell Therapy System. These expenses include fees paid to consultants and contract research organizations (CRO) and additional personnel costs. We expect research and development expenses to continue to increase as advanced minimally invasive instrumentationenrollment accelerates in the CardiAMP Heart Failure Trial and bone-conserving high-performance implants. Cardo is focused on developing surgical devices that will enable surgeons to bridgewe further develop the gap between soft tissue-driven sports medicine techniquesCardiAMP and classical reconstructive surgical procedures. Cardo commercializes its reconstructive joint devices through its Cardo Orthopedics divisionCardiALLO platforms.
Selling, General and its spine devices through its Cardo Spine division. The Company launchedAdministrative Expenses. Selling, general and commenced sales of its first product in late 2006,administrative expenses for the three months ended March 31, 2019 totaled $1,631,000 which was a high-performance, uni-compartmental knee replacement. 4.5% decrease as compared to $1,707,000 for the three months ended March 31, 2018. We expect selling, general and administrative expenses for 2019 to remain relatively consistent in the latter half of 2019 compared to the same period in 2018.
Comparison of Years EndedDecember 31, 2018 and 2017
The Company commencedfollowing table summarizes our results of operations for the years ended December 31, 2018 and 2017 (in thousands):
Years ended December 31, | ||||||||
2018 | 2017 | |||||||
Revenue: | ||||||||
Net product revenue | $ | 282 | $ | 389 | ||||
Collaboration agreement revenue | 343 | 90 | ||||||
Total revenue | 625 | 479 | ||||||
Costs and expenses: | ||||||||
Cost of goods sold | 517 | 690 | ||||||
Research and development | 8,453 | 5,799 | ||||||
Selling, general and administrative | 5,757 | 6,395 | ||||||
Total costs and expenses | 14,727 | 12,884 | ||||||
Operating loss | (14,102 | ) | (12,405 | ) | ||||
Other income (expense): | ||||||||
Interest income | 118 | 95 | ||||||
Other income (expense) | (3 | ) | 2 | |||||
Total other income, net | 115 | 97 | ||||||
Net loss | $ | (13,987 | ) | $ | (12,308 | ) | ||
Revenue. Revenue increased by approximately $146,000 in 2018 compared to 2017 primarily due to an increase in collaboration agreement revenues, which offset decreases in net product revenues. We expect collaboration agreement revenues to continue to increase modestly. Net product revenue will be subject to customer demand for our new Morph product family members.
Cost of Goods Sold. Cost of goods sold decreased by approximately $173,000 in 2018 compared to 2017, primarily due to the decrease in net product revenue. We expect cost of goods sold to decrease in 2019 as manufacturing resources are focused on supporting clinical partners, development activities and the ongoing pivotal CardiAMP Heart Failure Trial.
Research and Development Expenses. Research and development expenses increased by approximately $2.7 million in 2018 compared to 2017 primarily due to expenses incurred in the execution of the pivotal CardiAMP Heart Failure Trial, development of the CardiALLO Cell Therapy System and our other therapeutic programs, including fees paid to consultants and contract research organizations (CRO), additional personnel costs and increased stock compensation expense. We expect research and development expenses to increase as we continue enrolling and treating patients in the CardiAMP Heart Failure Trial and further develop the CardiAMP and CardiALLO cell processing and delivery platforms.
Selling, General and Administrative Expenses. Selling, general and administrative expenses decreased by approximately $0.6 million in 2018 compared to 2017, primarily due to lower costs for consulting expense and personnel costs and other corporate expenses. We expect selling, general and administrative expenses for 2019 to remain relatively consistent with 2018.
Interest Income. Interest income for the year ended December 31, 2018 consisted primarily of interest income earned on cash equivalents and short-term investments.
Liquidity and Capital Resources
We have incurred net losses each year since our inception and as of March 31, 2019, we had an accumulated deficit of approximately $90.0 million. We anticipate that we will continue to incur net losses for at least the next several years.
We have funded our operations principally through the sales of its reconstructive products in 2007equity and spine products in 2008.
On June 18, 2008, Cardo Medical, LLC ("Cardo LLC") entered into a Merger Agreement and Plan of Reorganization with CKST and Cardo Acquisition, LLC, a California limited liability company and wholly-owned subsidiary of CKST. Upon the consummation of the transactions contemplated by the Merger Agreement, CKST acquired Cardo LLC through a merger of Cardo LLC with Cardo Acquisition, with Cardo LLC continuingconvertible debt securities as well as the surviving entity incash acquired through the Merger and a wholly-owned subsidiaryMerger. As of CKST. Pursuant to the Merger Agreement, all of the issued and outstanding units of Cardo LLC's membership interests were converted into the right to receive shares of the common stock of CKST.
On or about the signing of the Merger Agreement, Frost Gamma Investments Trust and other investors invested $12.975 million in Cardo LLC in exchange for units of Cardo LLC's membership interests. Dr. Phillip Frost, Chairman and Chief Executive Officer of Opko Health, Inc., is the trustee and beneficiary of Frost Gamma Investments Trust. Cardo LLC used approximately $9.7 million of the proceeds from these investments to close on the acquisition of the outstanding equity interests of three partially owned subsidiaries of Cardo LLC (Accelerated Innovation, LLC, Cervical Xpand, LLC and Uni-Knee, LLC), to repay an existing member loan (in the amount of $1.2 million) and for transaction expenses, and used the remaining funds to accelerate its research and product development. In addition, at the closing of the Merger, CKSTMarch 31, 2019, we had cash and cash equivalents of approximately $2.5$2.8 million.
On July 5, 2019, we entered into a note purchase agreement pursuant to which we issued on such date $0.625 million held in money market accountsaggregate principal amount of convertible promissory notes, a portion of which was issued to certain of our officers and certificatesdirectors and a principal stockholder (or their respective affiliates). Interest on the convertible notes accrues at the rate of deposit as well as accrued liabilities of approximately $354 thousand.
We are headquartered in Beverly Hills, California. In connection with the consummation14.0% per year. The unpaid principal amount of the Merger, CKST proposedconvertible notes, together with all interest accrued but unpaid thereon, will be automatically converted into Private Placement Units upon the closing of this Offering at a conversion price equal to its stockholders an amendment to its Amended and Restated Certificate50% of Incorporation to change its name from "clickNsettle.com, Inc." to "Cardo Medical, Inc." CKST's trading symbol was "CKST.OB," which has changed to "CDOM.OB" in connection with the name change. Cardo Medical's common stock is quoted on the National Association of Securities Dealers, Inc.'s, Over-the-Counter Bulletin Board, or the OTC Bulletin Board.
Critical Accounting Policies and Estimates
Our significant accounting policies are more fully described in the notes to our consolidated financial statements. Those material accounting estimates that we believe are the most critical to an investor's understanding of our financial results and condition are discussed immediately below and are particularly important to the portrayal of our financial position and results of operations and require the application of significant judgment by our management to determine the appropriate assumptions to be used in the determination of certain estimates.
46
Use of Estimates
Financial statements prepared in accordance with accounting principles generally accepted in the United States require management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Among other things, management makes estimates relating to allowances for doubtful accounts, excess and obsolete inventory items, the estimated depreciable lives of property and equipment, the impairment of goodwill and other intangible assets, share-based payment, deferred income tax assets and the allocation of the purchase price paid for the minority interests in Uni, Cervical and Accelerated Innovation. Given the short operating history of Cardo, actual results could differ from those estimates.
Revenue Recognition
The Company recognizes revenue when it's realizable and earned. The Company considers revenue to be realizable and earned when the following criteria are met: persuasive evidence of an arrangement exists, delivery has occurred or services have been rendered, the seller's price to the buyer is fixed or determinable, and collectability is reasonably assured. Persuasive evidence of the arrangements occurs when the Company receives a signed contract from the hospitalpublic in which the surgery will be performed. Within that contract is the price at which the hospital will buy the device. Delivery occurs on the day of surgery when the device is implanted by the surgeon. Collectability is reasonably assured as we have continuing relationships with the hospitals and we can pursue collections if necessary. As the Company does not accept returns and does not have any post-sale obligations, the date of revenue recognition is generally on the day of the surgery.this Offering.
Intangible and Long-Lived Assets
The Company evaluates long-lived assets for impairment whenever events or changes in circumstances indicate that their net book value may not be recoverable. When such factors and circumstances exist, the Company compares the projected undiscounted future cash flows associated with the related asset or group of assets over their estimated useful lives against their respective carrying amount. Impairment, if any, is based on the excess of the carrying amount over the fair value, based on market value when available, or discounted expected cash flows, of those assets and is recorded in the period in which the determination is made. The Company's management currently believes there is no impairment of its long-lived assets. There can be no assurance, however, that market conditions will not change or demand for the Company's products will continue. Either of these could result in future impairment of long-lived assets. The first step of the Company's goodwill impairment test compares the fair value of a reporting unit with its carrying amount, including goodwill. If the fair value of a reporting unit exceeds its carrying amount, goodwill of the reporting unit is considered not impaired, thus the second step of the impairment test is unnecessary. If the carrying amount of a reporting unit exceeds its fair value, the second step of the goodwill impairment test shall be performed to measure the amount of impairment loss, if any. The second step of the goodwill impairment test compares the implied fair value of reporting unit goodwill with the carrying amount of that goodwill. If the carrying amount of reporting unit goodwill exceeds the implied fair value of that goodwill, an impairment loss shall be recognized in an amount equal to that excess. The loss recognized cannot exceed the carrying amount of goodwill. After a goodwill impairment loss is recognized, the adjusted carrying amount of goodwill shall be its new accounting basis. Subsequent reversal of a previously recognized goodwill impairment loss is prohibited once the measurement of that loss is completed. The testing for impairment needs to be conducted at the reporting unit, or component level, which is one level below the operating unit. In Cardo's case, the operating units are the Reconstructive and Spine product lines. The reporting units are one level below that. In the case of the Reconstructive Division, the reporting units are the knee and hip products. For the Spine Division, the reporting units are the licensed and internally developed products.
Property and Equipment
Property and equipment are recorded at historical cost and depreciated on a straight-line basis over their estimated useful lives, which range from three to five years. This estimate is based on the useful life of the individual items. When items are retired or disposed of, income is charged or credited for the difference between the net book value of the asset and the proceeds realized thereon. Ordinary maintenance and repairs are charged to expense as incurred, and replacements and betterments are capitalized. This estimate is unlikely to experience any differences from what is reflected in the financial statements.
47
Share Based Payment
In order to determine compensation on options issued to consultants, and employees' options, the fair value of each option granted is estimated on the date of grant using the Black-Scholes option-pricing model. The Company estimates the requisite service period used in the Black-Scholes calculation based on an analysis of vesting and exercisability conditions, explicit, implicit, and/or derived service periods, and the probability of the satisfaction of any performance or service conditions. The Company also considers whether the requisite service has been rendered when recognizing compensation costs. Because the Black-Scholes option valuation model incorporate ranges of assumptions for inputs, those ranges are disclosed. Expected volatilities are based on the historical volatility of the components of the small cap sector of the Dow Jones medical equipment index for a period equal to the expected life of the Cardo options. It also measures the volatility of other public companies with similar size and industry characteristics to Cardo for the same period. These measurements are averaged and the result is used as expected volatility. As there is no history of option lives at Cardo, the expected term of options granted is the midpoint between the vesting periods and the contractual life of the options. The risk-free rate for periods within the contractual life of the option is based on the U.S. Treasury yield curve in effect at the time of grant. The forfeiture rate is based on an analysis of the nature of the recipients' jobs and relationships to the Company.
Income Taxes
On August 29, 2008, Cardo LLC consummated a reverse takeover of CKST thereby adopting CKST as the taxpaying entity.
The Company's deferred tax assets and liabilities are recognized to reflect the estimated future tax effects, calculated at currently effective tax rates, of future deductible or taxable amounts attributable to events that have been recognized on a cumulative basis in the financial statements. A valuation allowance related to a deferred tax asset is recorded when it is more likely than not that some portion of the deferred tax asset will not be realized. The estimated value of the deferred tax assets are subject to significant change based on the company's future profitability. Deferred tax assets and liabilities are adjusted for the effects of the changes in tax laws and rates of the date of enactment.
In June 2008, the Financial Accounting Standards Board ("FASB") sought to reduce the diversity in practice associated with certain aspects of measurement and recognition in accounting for income taxes. FASB prescribed a recognition threshold and measurement requirement for the financial statement recognition of a tax position that has been taken or is expected to be taken on a tax return and also provides guidance on de-recognition, classification, interest and penalties, accounting in interim periods, disclosure, and transition. As such, the Company may only recognize or continue to recognize tax positions that meet a "more likely than not" threshold. Based on this analysis, the Company's tax position is unlikely to change.
Inventory
Inventory is stated at the lower of cost or net realizable value as determined by assessing the gross profit less selling costs of each inventory item. Cost is determined on a first-in, first-out basis; and the inventory is comprised of work in process and finished goods. Work in process consists of fabrication costs paid relating to items not physically received. Finished goods are completed knee, spine and hip replacement products ready for resale to customers.
At each balance sheet date, the Company evaluates its ending inventories for excess quantities and obsolescence. This evaluation includes an analysis of sales levels by product type. Among other factors, the Company considers current product configurations, historical and forecasted demand, market conditions and product life cycles when determining the net realizable value of the inventory. Provisions are made to reduce excess or obsolete inventories to their estimated net realizable values. Once established, write-downs are considered permanent adjustments to the cost basis of the excess or obsolete inventory. The Company did not have any inventory considered by management to be excess or obsolete as of December 31, 2008 and September 30, 2009. Based on the forecasted sales amounts, we do not expect any changes in net realizable value in the near future.
48
Recent Accounting Pronouncements
In July 2009, the FASB issued ASC 105-10 (Prior authoritative literature: SFAS No. 168,The FASB Accounting Standards CodificationTM and the Hierarchy of Generally Accepted Accounting Principles, which will become the single source of authoritative GAAP recognized by the FASB. ASC 105-10 does not change current U.S. GAAP, but on the effective date, the FASB Accounting Standards Codification ("ASC")TM will supersede all then existing non-SEC accounting and reporting standards. The ASC is effective for interim and annual periods ending after September 15, 2009. Cardo adopted ASC 105-10 during the quarter ended September 30, 2009 and revised the referencing of GAAP accounting standards in our financial statements to reflect the new standards.
In May 2009, the FASB issued ASC 855-10 (Prior authoritative literature: SFAS No. 165, Subsequent Events) which provides guidance on management's assessment of subsequent events. It is not expected to significantly change practice because its guidance is similar to that in American Institute of Certified Public Accountants Professional Standards U.S. Auditing Standards Section 560, Subsequent Events, with some modifications. The Company adopted ASC 855-10 during the quarter ended June 30, 2009. The adoption of ASC 855-10 did not have a material impact on our results of operations, financial position or cash flows.
In April 2009, FASB issued ASC 820-10 (Prior authoritative literature: FSP No. FAS 157-4,Determining Fair Value When the Volume and Level of Activity for the Asset or Liability Have Significantly Decreased and Identifying Transactions That Are Not Orderly). It does not change the definition of fair value as previously detailed, but provides additional guidance for estimating fair value when the volume and level of activity for the asset or liability have significantly decreased. Cardo adopted ASC 820-10 during the quarter ended June 30, 2009. The adoption of ASC 820-10 did not have a material impact on our results of operations, financial position or cash flows.
In April 2009, the FASB issued ASC 320-10 (Prior authoritative literature: FSP No. FAS 115-2 and FAS 124-2,Recognition and Presentation of Other-Than-Temporary Impairments). ASC 320-10 amends the other-than-temporary impairment guidance in U.S. GAAP for debt securities and provides additional disclosure requirements for other-than-temporary impairments for debt and equity securities. ASC 320-10 addresses the determination as to when an investment is considered impaired, whether that impairment is other than temporary, and the measurement of an impairment loss. Cardo adopted ASC 320-10 during the quarter ended June 30, 2009. The adoption of ASC 320-10 did not have a material impact on our results of operations, financial position or cash flows.
In April 2009, the FASB issued ASC 825-10 (Prior authoritative literature: FSP No. FAS 107-1 and APB 28-1,Interim Disclosures about Fair Value of Financial Instruments). FASB ASC 825-10 requires that disclosures about the fair value of a company's financial instruments be made whenever summarized financial information for interim reporting periods is made. Cardo adopted ASC 825-10 during the quarter ended September 30, 2009. The adoption of ASC 825-10 did not have a material impact on our results of operations, financial position or cash flows.
In June 2009, the FASB issued the following new accounting standards, which remain authoritative until such time that each is integrated into the Codification:
SFAS No. 166,Accounting for Transfers of Financial Assets - an amendment of FASB Statement No. 140SFAS No. 167,Amendments to FASB Interpretation No. 46(R). SFAS No. 167 amends FASB Interpretation No. ("FIN") 46,Consolidation of Variable Interest Entities (revised December 2003) - an interpretation of ARB No. 51
SFAS No. 166 seeks to improve the relevance and comparability of the information that a reporting entity provides in its financial statements about transfers of financial assets; the effects of the transfer on its financial position, financial performance, and cash flows; and a transferor's continuing involvement, if any, in transferred financial assets. SFAS No. 166 eliminates the concept of a qualifying special-purpose entity, creates more stringent conditions for reporting a transfer of a portion of a financial asset as a sale, clarifies other sale-accounting criteria, and changes the initial measurement of a transferor's interest in transferred financial assets. SFAS No. 166 is effective for interim and annual reporting periods beginning after November 15, 2009. The Company has not completed its evaluation, but does not expect the adoption of SFAS No. 166 to have a material impact on our consolidated financial statements.
49
SFAS No. 167 requires an enterprise to determine whether its variable interest or interests give it a controlling financial interest in a variable interest entity. The primary beneficiary of a variable interest entity is the enterprise that has both (1) the power to direct the activities of a variable interest entity that most significantly impact the entity's economic performance, and (2) the obligation to absorb losses of the entity that could potentially be significant to the variable interest entity or the right to receive benefits from the entity that could potentially be significant to the variable interest entity. SFAS No. 167 also amends FIN 46(R) to require ongoing reassessments of whether an enterprise is the primary beneficiary of a variable interest entity. SFAS No. 167 is effective for interim and annual reporting periods beginning after November 15, 2009. The Company has not completed its evaluation, but does not expect the adoption of SFAS No. 167 to have a material impact on our consolidated financial statements.
Results of Operations for the Three Months Ended September 30, 2009 as Compared to the Three Months Ended September 30, 2008. (Amounts in thousands)
The following istable shows a comparisonsummary of our cash flows for the consolidated resultsperiods indicated (in thousands):
Three months ended March 31, | ||||||||
2019 | 2018 | |||||||
Net cash provided by (used in): | ||||||||
Operating activities | $ | (2,465 | ) | (3,036 | ) | |||
Investing activities | (55 | ) | (5 | ) | ||||
Financing activities | — | 5 | ||||||
Net decrease in cash and cash equivalents | $ | (2,520 | ) | $ | (3,036 | ) | ||
Cash Flows from Operating Activities. The decrease in overall spending for operating activities of operations for Cardo for$571,000 in the three months ended September 30, 2009 and 2008:
| Three Months Ended | ||||||||||
| September 30, | ||||||||||
| 2009 | 2008 | Variance | ||||||||
| (Restated) | ||||||||||
| Net sales | $ | 436 | $ | 411 | $ | 25 | ||||
| Cost of sales | 84 | 66 | 18 | |||||||
| Gross profit | 352 | 345 | 7 | |||||||
| Research and development expenses | 123 | 142 | (19) | |||||||
| Selling, general and administrative expenses | 1,440 | 1,089 | 351 | |||||||
| Loss from operations | (1,211) | (886) | (325) | |||||||
| Interest income (expense), net | 6 | 5 | 1 | |||||||
| Loss before income tax provision | (1,205) | (881) | (324) | |||||||
| Provision for income taxes | - | - | - | |||||||
| Net loss | (1,205) | (881) | (324) | |||||||
| Less: Net income attributable to non-controlling interest | - | - | - | |||||||
| Net loss attibutable to Cardo Medical, Inc. | $ | (1,205) | $ | (881) | $ | (324) | ||||
Revenues
During the quarter ended September 30, 2009, we generated revenues of $436March 31, 2019 compared to $411 for the same period in 2008. The wider acceptancethree months ended March 31, 2018 related primarily to payment of our Hip and Spine products by orthopedic and back surgeons has resulted in higher sales volumepersonnel related costs in the current year. Accordingly, sales of Hip and Spine products increased $122 in the current quarter as compared to 2008. We launched our Hip product in the first quarter of 2009 which resulted in $78 of sales during the quarterthree months ended September 30, 2009. Comparable sales for the same period in 2008March 31, 2018 that were nominal. The increases in Hip and Spine sales were offset by a drop in Knee sales in 2009 compared to 2008. Our Knee and Hip products accounted for 79% of total salesnot incurred during the three months ended September 30, 2009. During the quarter ended September 30, 2008, Knee products totaled 93% of sales.
Gross Profit
During the quarter ended September 30, 2009, we had cost of sales of $84 compared to $66March 31, 2019, board cash compensation that was deferred coupled with reduced professional fees during the quarter ended September 30, 2008. Our gross profit percentage was 80.7% for the three months ended September 30, 2009 comparedMarch 31, 2019. We expect spending to 83.9% for the corresponding period in 2008. This decrease is attributed to sales of Hip products in 2009, which generate lower margins than Knee products,increase as we continue enrolling and a slight decreasetreating patients in the profit marginCardiAMP Heart Failure Trial, further develop the CardiAMP and CardiALLO cell therapy systems and continue to strengthen and enhance the supporting organization.
Cash Flows from Knee products. During the quarter ended September 30, 2008, there were minimal sales of Hip products so the profit margin was indicative of our Knee product. As Cardo moves forward, we expect that our current product costs will be maintained and the gross profits shall remain mostly in-line with the current year percentages.
50
Research and Development Expenses
During the quarter ended September 30, 2009, we had research and development costs of $123 compared to $142 for the same period in 2008. The decrease in the current year is indicative of reduced research activity as most of our products are on the market. Research costs associated with our Total Knee product have slowed because it is nearing its launch in mid-2010; however, research is underway on a new Hip product so research and development expenses are likely to increase in the upcoming quarters.
Selling, General and Administrative Expenses
During the quarter ended September 30, 2009, we had selling general and administrative expenses of $1,440 compared to $1,089 in the same period of 2008. During 2009, labor and labor-related expenses increased $342 compared to 2008 as we expanded our operations in order to meet current and anticipated growth. Depreciation and amortization expense increased by $93 during the quarter ended September 30, 2009 because of the acquisition of additional instrumentation required to support base inventory levels and expected future sales increases, and the purchase of the non-controlling interest in Accelerated Innovation, LLC in June 2008 which resulted in $5,328 of amortizable intangible assets. Rent, travel and office expenses were slightly higher in 2009 as we added office space and our overall business activity was greater than it was in 2008. These cost increases were partially offset by a drop in professional fees during 2009 of $154 and commission expense which was higher in 2008 due to greater incentives to initiate sales. There were significant legal and accounting costs associated with the purchase of the non-controlling interest in Accelerated Innovation, LLC in June 2008 and the reverse merger transaction with clickNsettle.com, Inc. which was completed in August 2008.
Interest Income/(Expense)
During the quarter ended September 30, 2009, we had interest income of $6 compared to $5 during the quarter ended September 30, 2008. Interest income is earned on our excess cash balances, which were comparable in both periods.
Results of Operations for the Nine Months Ended September 30, 2009 as Compared to the Nine Months Ended September 30, 2008. (Amounts in thousands)
The following is a comparison of the consolidated results of operations for Cardo for the nine months ended September 30, 2009 and 2008:
| Nine Months Ended | ||||||||||
| September 30, | ||||||||||
| 2009 | 2008 | Variance | ||||||||
| (Restated) | ||||||||||
| Net sales | $ | 1,314 | $ | 932 | $ | 382 | ||||
| Cost of sales | 254 | 139 | 115 | |||||||
| Gross profit | 1,060 | 793 | 267 | |||||||
| Research and development expenses | 329 | 1,288 | (959) | |||||||
| Selling, general and administrative expenses | 4,521 | 2,488 | 2,033 | |||||||
| Loss from operations | (3,790) | (2,983) | (807) | |||||||
| Interest income (expense), net | 22 | (37) | 59 | |||||||
| Loss before income tax provision | (3,768) | (3,020) | (748) | |||||||
| Provision for income taxes | - | - | - | |||||||
| Net loss | (3,768) | (3,020) | (748) | |||||||
| Less: Net income attributable to non-controlling interest | - | (148) | 148 | |||||||
| Net loss attibutable to Cardo Medical, Inc. | $ | (3,768) | $ | (3,168) | $ | (600) | ||||
51
Revenues
During the nine months ended September 30, 2009, we generated revenues of $1,314 compared to $932 for the same period in 2008. The increase of $382 was attributed to the wider acceptance of our Knee, Hip and Spine products by orthopedic and back surgeons that resulted in higher sales volume in the current year. We launched our Hip product in the first quarter of 2009 which resulted in $278 of sales thus far in 2009 compared to $45 in 2008. Our Knee and Hip products accounted for 89% of total sales during the nine months ended September 30, 2009. Knee was the primary product with 90% of total sales during the nine months ended September 30, 2008.
Gross Profit
During the nine months ended September 30, 2009, we had cost of sales of $254 compared to $139 during 2008. Our gross profit percentage was 80.7% for the nine months ended September 30, 2009 compared to 85.1% for the corresponding period in 2008. This decrease was attributed to significant sales of Hip products in 2009, which generate lower margins than Knee products, and a slight decrease in the profit margin from Knee products. During the nine months ended September 30, 2008, sales of Hip products were minimal so the profit margin was mostly indicative of our Knee product. As Cardo moves forward, we expect that our current product costs will be maintained and the gross profits shall remain mostly in-line with the current year percentages.
Research and Development Expenses
During the nine months ended September 30, 2009, we had research and development costs of $329 compared to $1,288 for the same period in 2008. The decrease was primarily due to a one-time charge of $938 of in-process research and development acquired in connection with the purchase of the non- controlling interest in Accelerated Innovation, LLC in June 2008. The acquired in-process research and development related to development costs associated with our Uni Knee system. Aside from the in-process research and development, spending on research and development was fairly consistent from 2008 to 2009. During 2008, we incurred more costs associated with Hip products. Research costs associated with our Total Knee product have slowed because it is nearing its launch in mid-2010; however, research is underway on a new Hip product so research and development expenses are likely to increase some in the upcoming quarters.
Selling, General and Administrative Expenses
During the nine months ended September 30, 2009 our selling general and administrative expenses were $4,521 compared to $2,488 for the same period in 2008, a net increase of $2,033. During 2009, labor and labor-related expenses increased $1,626 compared to 2008 as we expanded our operations in order to meet current and anticipated growth. Depreciation and amortization expense increased by $558 during the quarter ended September 30, 2009 because of the acquisition of additional instrumentation required to support base inventory levels and expected future sales increases, and the purchase of the non-controlling interest in Accelerated Innovation, LLC in June 2008 which resulted in $5,328 of amortizable intangible assets. Commission expense increased accordingly with the increase in sales. Rent, travel, warehouse, insurance and office expenses were also higher during the nine months ended September 30, 2009 as we added office space and our overall business activity was greater than it was in 2008. These cost increases were partially offset by a drop in professional fees during 2009 of $442. There were significant legal and accounting costs associated with the purchase of the non-controlling interest in Accelerated Innovation, LLC in June 2008 and the reverse merger transaction (the "Merger") with clickNsettle.com, Inc. which was completed in August 2008.
Interest Income
During the nine months ended September 30, 2009, we had interest income of $22 for the nine months ended September 30, 2009 compared to interest expense of $37 in the corresponding period of 2008. In the first and second quarter of 2008, we had interest expense stemming from a short-term note payable which was fully repaid in July 2008. Interest income is earned on our excess cash balances, which were significantly higher throughout 2009 compared to 2008.
52
Segment Information
Our businesses are currently organized into the following two reportable segments; reconstructive products (the "Reconstructive Division") and spine products (the "Spine Division")Investing Activities. The Reconstructive Division segment is comprised of activity relating to Cardo's unicompartmental knee, patella-femoral products, and reconstructive knee products. The Spine Division segment is comprised of the spinal lumbar fusion system and cervical plate and screw systems.
These reportable segments are based on the nature of the products offered. Management evaluates performance and allocates resources based on several factors, of which the primary financial measure is segment operating results. Due to the distinct nature of the products in our Reconstructive Division, and the fact that it has a more developed market for its products, it is considered by management as a separate segment. Our Spine Division is still in the process of developing the market and obtaining instrumentation necessary to sell the products in greater quantities. As a result, the Spine Division is considered by management as a separate segment. The accounting policies of the reportable segments are the same as those described in Note 1.
As of September 30, 2009, the Reconstructive Division includes $1,233 of goodwill and $4,515 in other intangible assets relating to the Company's unicompartmental knee product. These amounts are expected to be deductible for income tax purposes.
The following table sets forth summarized financial results by reportable segment for the three and nine months ended September 30, 2009 and 2008:
| Reconstructive | Spine | ||||||||||
| Division | Division | Corporate | Total | ||||||||
| Nine Months Ended September 30, 2009 (unaudited) | |||||||||||
| Net sales | $ | 1,175 | $ | 139 | $ | - | $ | 1,314 | |||
| Total cost of sales and operating expenses | 226 | 28 | 3,972 | 4,226 | |||||||
| Depreciation and amortization | 848 | 4 | 26 | 878 | |||||||
| Interest income, net | - | - | 22 | 22 | |||||||
| Net income (loss) | $ | 101 | $ | 107 | $ | (3,976) | $ | (3,768) | |||
| Nine Months Ended September 30, 2008 (unaudited - restated) | |||||||||||
| Net sales | $ | 885 | $ | 47 | $ | - | $ | 932 | |||
| Total cost of sales and operating expenses | 125 | 14 | 3,590 | 3,729 | |||||||
| Depreciation and amortization | 319 | 3 | 12 | 334 | |||||||
| Interest expense, net | - | - | 37 | 37 | |||||||
| Net income (loss) | $ | 441 | $ | 30 | $ | (3,639) | $ | (3,168) | |||
| Three Months Ended September 30, 2009 (unaudited) | |||||||||||
| Net sales | $ | 351 | $ | 85 | $ | - | $ | 436 | |||
| Total cost of sales and operating expenses | 76 | 8 | 1,250 | 1,334 | |||||||
| Depreciation and amortization | 303 | 2 | 8 | 313 | |||||||
| Interest income, net | - | - | 6 | 6 | |||||||
| Net income (loss) | $ | (28) | $ | 75 | $ | (1,252) | $ | (1,205) | |||
| Three Months Ended September 30, 2008 (unaudited - restated) | |||||||||||
| Net sales | $ | 386 | $ | 25 | $ | - | $ | 411 | |||
| Total cost of sales and operating expenses | 62 | 8 | 1,011 | 1,081 | |||||||
| Depreciation and amortization | 211 | 1 | 4 | 216 | |||||||
| Interest expense, net | - | - | 5 | 5 | |||||||
| Net income (loss) | $ | 113 | $ | 16 | $ | (1,010) | $ | (881) | |||
53
All of the Company's net sales were attributable to activity in the United States. There were no long-lived assets held in foreign countries.
Liquidity and Capital Resources
Net cash used in operating activities was $3,630 for the nine months ended September 30, 2009 compared to $1,613 for the same period in 2008. The primary use of cash in 2009 beyond wages and other operating costs was the build-up of inventory which has increased $1,051 during the current year.
Net cash used in investing activities was $1,051 forof $55,000 during the ninethree months ended September 30, 2009 compared to $1,484 for the same period in 2008. The cash used by investment activities during the nine months ended September 30, 2009 was mostly attributed to the purchaseMarch 31, 2019 consisted primarily of equipment to accommodate our operational and corporate growth as well as additional instrumentation required in order to support current and anticipated future sales levels. The current year also includes $130purchases of deposits associated with the October 2009 purchase of certain Vertebron, Inc. assets.lab equipment.
Our net
Cash Flows from Financing Activities. Net cash provided by financing activities was $3,193 forof $5,000 during the ninethree months ended September 30, 2009 comparedMarch 31, 2018 consisted of the proceeds from the exercise of stock options.
Future Funding Requirements
To date, we have generated modest revenue from sales of our approved products. We do not know when, or if, we will generate any revenue from our development stage biotherapeutic programs. We do not expect to $6,775 forgenerate any revenue from sales of our CardiAMP or CardiALLO therapeutic candidates unless and until we obtain regulatory approval. At the same periodtime, we expect our expenses to increase in 2008. Duringconnection with our ongoing development activities, particularly as we continue the research, development and clinical trials of, and seek regulatory approval for, our therapeutic candidates. In addition, subject to obtaining regulatory approval for any of our therapeutic candidates and companion diagnostic, we expect to incur significant commercialization expenses for product sales, marketing, manufacturing and distribution. We anticipate that we will need additional funding in connection with our continuing operations.
Based upon our current operating plan, we believe that the cash and cash equivalents of $2.8 million as of March 31, 2019 are not sufficient to fund our operations through the third quarter of 2019. In order to continue to further the development of our lead therapeutic candidates, the CardiAMP Cell Therapy System, and our second therapeutic candidate, the CardiALLO Cell Therapy System, through and beyond the third quarter of 2019, we completed twowill be required to raise additional capital. We plan to raise additional capital, potentially including debt and equity investment transactionsarrangements, to provide us additional workingfinance our future operations. We have based our estimates on assumptions that may prove to be wrong, and we may use our available capital neededresources sooner than we currently expect. Because of the numerous risks and uncertainties associated with the development and commercialization of our therapeutic candidates, we are unable to maintainestimate the amounts of increased capital outlays and build certain inventory and instrumentation levels to meet expected increases in future sales and product development.
At September 30, 2009, we had $1,607 in cash; however, in October 2009 we used $1,170 in orderoperating expenditures necessary to complete the Vertebron, Inc. transaction and raised net proceeds of $3,189 through a private placement. With this subsequent net cash infusion, the available funds are still not projected to meet alldevelopment of our workingtherapeutic candidates.
Our future capital needsrequirements will depend on many factors, including:
the progress, costs, results and timing of our CardiAMP and CardiALLO clinical trials and related development programs;
FDA acceptance of our CardiAMP and CardiALLO therapies for heart failure and for other potential indications;
the outcome, costs and timing of seeking and obtaining FDA and any other regulatory approvals;
the costs associated with securing, establishing and maintaining commercialization and manufacturing capabilities;
the number and characteristics of product candidates that we pursue, including our product candidates in preclinical development;
the ability of our product candidates to progress through clinical development successfully;
our need to expand our research and development activities;
the costs of acquiring, licensing or investing in businesses, products, product candidates and technologies;
our ability to maintain, expand and defend the scope of our intellectual property portfolio, including the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights;
the general and administrative expenses related to be a public company;
our need and ability to hire additional management and scientific, medical and sales personnel;
the effect of competing technological and market developments; and
our need to implement additional internal systems and infrastructure, including financial and reporting systems.
Until such time that we can generate meaningful revenue from the sales of approved therapies and products, if ever, we expect to finance our operating activities through public or private equity or debt financings, government or other third-party funding, marketing and distribution arrangements, and other collaborations, strategic alliances and licensing arrangements or a combination of these approaches. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of our Common Stock holders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of our Common stock holders. Debt financing, if available, may involve agreements that include conversion discounts or covenants limiting or restricting our ability to take specific actions, such as incurring debt, making capital expenditures or declaring dividends. If we raise additional funds through government or other third-party funding, marketing and distribution arrangements or other collaborations, or strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs, products or therapeutic candidates or to grant licenses on terms that may not be favorable to us.
Our consolidated financial statements as of and for the next twelve months. The factthree months ended March 31, 2019 and the year ended December 31, 2018 have been prepared on the basis that Cardothe Company will sustain losses throughcontinue as a going concern, which contemplates the remainderrealization of 2009assets and still requires outside sourcessatisfaction of additional capitalliabilities in the ordinary course of business. Due to supplement operations continues to create an uncertaintythe factors described above, there is substantial doubt about ourthe Company’s ability to continue as a going concern.
Management intends to use borrowings and securities sales to mitigateconcern within one year after the effects of our use of that cash. However, we cannot assure you that debt or equity financing, if and when required, will be available.date these financial statements are issued. Our ability to continue as a going concern is dependent upon receivingwill depend in a large part, on our ability to raise additional capital. If adequate funds either through the issuance of debt or equity and the success of management's plan to expand sales. Althoughare not available, we may obtain external financing throughbe required to reduce operating expenses, delay or reduce the salesscope of our own securities,product development programs, obtain funds through arrangements with others that may require us to relinquish rights to certain of our technologies or products that we would otherwise seek to develop or commercialize ourselves, or cease operations. While we believe in the viability of our strategy to raise additional funds, there can be no assuranceassurances that such financingwe will be available, or if available,able to obtain additional capital on acceptable terms and in the amounts necessary to fully fund our operating needs.
The financial statements do not include any adjustments that any such financing would be on terms acceptable to us.might result from the outcome of this uncertainty. If we are unable to fund our cash flow needs,continue as a going concern, we may be forced to liquidate assets. In such a scenario, the values received for assets in liquidation or dissolution could be significantly lower than the values reflected in our financial statements.
Off-Balance Sheet Arrangements
During the periods presented, we did not have, nor do we currently have, any off-balance sheet arrangements as defined under the rules of the Securities and Exchange Commission.
Recent Accounting Pronouncements
See Note 2 of our notes to reduce or stop planned growth or scale back operationseach of our consolidated financial statements as of and reduce staff.
54
Results of Operations for the Year Ended Decemberthree months ended March 31, 2008 as Compared to the Year Ended December 31, 2007 (including the combined results of operations for Cardo2019 and Accin) (Amounts in thousands)
The following are the consolidated results of operations for Cardo for the year ended December 31, 2008 compared2018 for information regarding recent accounting pronouncements that are of significance or potential significance to an unaudited pro forma presentationus.
Directors, Executive Officers and Corporate Governance
Board of CardoDirectors
Our business affairs are managed under the direction of our board of directors, which is currently composed of eight members. All of our directors other than Peter Altman are independent within the meaning of the listing standards of the NASDAQ Stock Market LLC. Our board of directors is divided into three staggered classes of directors. At each annual meeting of stockholders, a class of directors will be elected for a three-year term to succeed the same class whose term is then expiring.
The following table sets forth the names, ages as of March 31, 2019, and Accin Corporation,certain other information for each of the directors with terms expiring at the 2019 Annual Meeting (who are also nominees for election as a director at the 2019 Annual Meeting) and for each of the continuing members of our board of directors:
Directors with Terms expiring at the Annual Meeting | Class | Age | Position | Director | Current | ||||||||||
Richard Krasno, Ph.D.(3) | III | 77 | Director | 2016 | 2019 | ||||||||||
Jay M. Moyes(1)(2) | III | 65 | Director | 2011 | 2019 | ||||||||||
Simon H. Stertzer, M.D.(2)(3) | III | 83 | Chairman of the Board of Directors | 2002 | 2019 | ||||||||||
Continuing Directors | |||||||||||||||
Peter Altman, Ph.D. | I | 52 | President, Chief Executive Officer and Director | 2002 | 2020 | ||||||||||
Fernando L. Fernandez(1) | I | 58 | Director | 2016 | 2020 | ||||||||||
Thomas Quertermous, M.D.(3) | II | 67 | Director | 2002 | 2021 | ||||||||||
Richard Pfenniger, Jr.(2) | II | 63 | Director | 2016 | 2021 | ||||||||||
Allan R. Tessler(1) | II | 82 | Director | 2012 | 2021 | ||||||||||
(1) | Member of the audit committee |
(2) | Member of the compensation committee |
(3) | Member of the nominating and corporate governance committee |
(4) | Service on our board of directors prior to 2016 noted in the narrative below includes service with BioCardia Lifesciences, Inc., the company we merged with in our reverse merger transaction in October 2016. |
Business Experience of Directors
Thomas Quertermous, M.D. has served on our board of directors since 2002. Dr. Quertermous is the William G. Irwin Professor of Medicine and Director of the Division of Cardiovascular Medicine at Stanford University since 1997. Dr. Quertermous came to Stanford from Vanderbilt University where he served as H.J. Morgan Professor of Medicine and Director of the Division of Cardiology. From 2006 to 2013, Dr. Quertermous served as a board member at Aviir, Inc., a company from which Cardo acquired its medical device business,providing metabolic tests and services for the year ended December 31, 2007, assuming they were combinedprevention and management of cardiovascular diseases. Dr. Quertermous received both a Master of Science degree in biophysics and theoretical biology and his Doctor of Medicine degree from the University of Chicago, where he also completed residency training in internal medicine. Subsequently, he served as clinical fellow in the Cardiac Unit at the beginningMassachusetts General Hospital and completed a research fellowship in the Department of the year in thousands.
| Cardo | |||||||||||||||||
| April 6, 2007, | Pro Forma | ||||||||||||||||
| Cardo | Inception, | Accin | Combined | ||||||||||||||
| Year Ended | Through | Five Months | Year Ended | ||||||||||||||
| December 31, | December 31, | Ended | December 31, | ||||||||||||||
| 2008 | 2007 | May 31, 2007 | 2007 | $ Change | % Change | ||||||||||||
| (unaudited) | |||||||||||||||||
| Net sales | $ | 1,268 | $ | 643 | $ | 157 | $ | 800 | $ | 468 | 58.5% | ||||||
| Cost of sales | 197 | 69 | 25 | 94 | 103 | 109.6% | |||||||||||
| Gross profit | 1,071 | 574 | 132 | 706 | 365 | 51.7% | |||||||||||
| Research and development expenses | 1,332 | 215 | 41 | 256 | 1,076 | 420.3% | |||||||||||
| Selling, general and administrative expenses | 3,914 | 671 | 250 | 921 | 2,993 | 325.0% | |||||||||||
| Impairment charges | 1,457 | - | - | - | 1,457 | 100.0% | |||||||||||
| Loss from operations | (5,632) | (312) | (159) | (471) | (5,161) | 1095.8% | |||||||||||
| Income (expense), net | (20) | 33 | 20 | 53 | (73) | - -137.7% | |||||||||||
| Loss before non-controlling interest | (5,652) | (279) | (139) | (418) | (5,234) | 1252.2% | |||||||||||
| Non-controlling interest in loss (earnings) of subsidiaries | (148) | (8) | 128 | 120 | (268) | - -223.3% | |||||||||||
| Net loss | $ | (5,800) | $ | (287) | $ | (11) | $ | (298) | $ | (5,502) | 1846.3% | ||||||
RevenuesGenetics at Harvard Medical School.
Net sales for the year ended December 31, 2008 increased by $468, or 58.5%,
We believe that Dr. Quertermous possesses specific attributes that qualify him to serve as compared to 2007. Accin, the company from which Cardo acquired its medical device business, launched and commenced sales of its first product in December 2006, a high performance, unicompartmental knee replacement product. As doctors became more familiar with our new product, they began using it more often. Total unicompartmental knee sales for 2008 amounted to $1,117, which represented an increase of $318 over 2007. In addition, during 2008, we began sales of our patellofemoral knee, hip and spine products, which contributed to an increase in sales of approximately $151 as compared to 2007.
Costs of Sales
Costs of sales for the year ended December 31, 2008 increased by $103, or 109.6%, as compared to the same period in 2007 primarily to due increased sales on the products mentioned above. Our gross profit percentage for 2008 was 84.5%, representing a decrease from the gross profit percentage of 88.3% in 2007. This decrease in gross profit percentage was primarily a result of a variation of sales mix during the year. During 2008, we began selling our patellofemoral knee, hip and spine products, which have lower gross margin percentages than do our knee products (75.5% for hip sales in and 72.2% for spine sales). This decreased our overall gross profit percentage, as virtually all sales during 2007 were knee products.
55
Research and Development Expenses
Research and development expenses for the year ended December 31, 2008 increased by $1,076, or 420.3%, from the same period in 2007. The increase was primarily due to $938 of in-process research and development expenses acquired in connection with the purchase of the non-controlling interest in Accelerated Innovation, LLC in June 2008. In addition, we increased prototype expenses in 2008 for production of our hip replacement prototypes. There were no expenditures for these items in 2007.
Selling, General and Administrative Expenses
Selling, general and administrative expenses for the year ended December 31, 2008 increased by $2,993, or 325.0%, as compared to the same period in 2007. During 2008, we incurred $978 in selling, general and administrative expenses relating primarily to legal and accounting fees associated with the reverse merger transaction with CKST which occurred on August 29, 2008, which we did not incur in 2007. We also incurred salary and payroll related expenses of $1,185 relating to additional employees hired in 2008. In addition, we incurred increased depreciation expense of $185 in 2008 as a result of increased capital expenditures for instrumentation and other equipment necessary to support our growth. We also incurred increased amortization expense of $376 in 2008 as a result of intangible assets acquired in connection with the purchase of the non-controlling interest in Accelerated Innovation, LLC in June 2008, as well as amortization of capitalized license fees.
Impairment Expenses
In 2008, we recognized $1,457 in goodwill impairment for the Cervical Xpand LLC purchase. There were no impairment charges in 2007.
Interest Income (Expense)
Net interest expense for the year ended December 31, 2008 amounted to $20, which consisted of interest expense of $48 relating to a note payable of $1,200 issued in February 2008 and repaid in July 2008, offset by interest income of $28. During 2007, our interest income amounted to $53. We had no interest expense in 2007, as we had no outstanding debt.
Liquidity and Capital Resources
Net cash used in operating activities was $2,696 for the year ended December 31, 2008 in contrast to $581 for April 6, 2007, inception, through December 31, 2007. The main uses of cash included expenses in connection with the reverse merger transaction, research and development costs and salaries.
Net cash used by investing activities was $1,836 for the year ended December 31, 2008 in contrast to an increase of cash from investing activities of $235 for April 6, 2007, inception, through December 31, 2007. The cash used by investment activities during the year ended December 31, 2008 primarily was attributable to the purchase of Cervical, Uni, Accelerated Innovation and the reverse merger with CKST, as well as purchases of instrumentation and other equipment of $515.
Our net cash provided by financing activities was $6,723 for the year ended December 31, 2008 in contrast to $1,250 for April 6, 2007, inception, through December 31, 2007. The cash provided by financing activities in 2008 consisted of the proceeds from a capital contribution in 2008, the proceeds of which were used to acquire the remaining minority interests of Cervical, Uni and Accelerated Innovation.
Over the next 12 months, we intend to use our capital to accelerate our research and product development, to add sales and financing personnel, to increase in-house vendor-related operations, to increase our inventory levels and for working capital.
In February 2008, Cardo borrowed $1,200 from the trustee of a member of Cardoour board of directors, including his expertise in the cardiovascular, biotechnology and therapeutic development industries.
Richard C. Pfenniger, Jr. was appointed to partially financeour board of directors in October 2016. From May 2014 to February 2015, Mr. Pfenniger served as Interim Chief Executive Officer of Vein Clinics of America, Inc., a medical group specializing in the acquisitiontreatment of vein disease. From January 2013 to May 2013, Mr. Pfenniger served as Interim Chief Executive Officer of IntegraMed America, Inc., an operator of the minority interestslargest U.S. network of Cervical Xpand and Uni-Knee for an aggregate purchase price of $3,487. This $1,200 note payablefertility centers. From October 2003 until October 2011, when it was repaid in July 2008.
56
On June 19, 2008, simultaneously with the signingacquired by Metropolitan Health, Inc., he served as Chairman of the Merger Agreement, Frost Gamma Investments Trustboard of directors and other investors invested $9,500 in Cardo in exchange for unitsPresident and Chief Executive Officer of Cardo's membership interests. Dr. Phillip Frost,Continucare Corporation, a provider of primary care physician and practice management services. Prior thereto, Mr. Pfenniger served as Chief Executive Officer of Whitman Education, Inc. from 1997 to 2003 and as Chief Operating Officer of IVAX Corporation from 1994 to 1997 after having served as the Senior Vice President – Legal Affairs from 1989 to 1994. Mr. Pfenniger currently serves as a director on the board of directors of OPKO Health, Inc., a pharmaceutical and medical diagnostic company, since 2008; on TransEnterix, Inc., a medical device company, since 2005; on GP Strategies, Inc., a corporate training and performance improvement company, since 2005; and on IntegraMed America, Inc. since 2012. Mr. Pfenniger holds a Juris Doctor degree from the University of Florida and a Bachelor of Business Administration degree from Florida Atlantic University.
We believe that Mr. Pfenniger possesses specific attributes that qualify him to serve as a member of our board of directors, including his expertise with public companies and the healthcare industry.
Allan R. Tessler has served on our board of directors since 2012. Mr. Tessler has served as Chairman and Chief Executive Officer of Opko Health,International Financial Group, Inc., is the trustee and beneficiary of Frost Gamma Investments Trust. Certain other investors invested an additional $3,475 in Cardo before the consummation of the Merger. Proceeds from these investments were used to close on the acquisition of the outstanding equity interests of Accelerated Innovation, Cervical Xpand and Uni-Knee, and to enable Cardo to accelerate its research and product development. Following the acquisitions, Cardo directly owns 100% of the equity interests of Accelerated Innovation, Cervical Xpand and Uni-Knee, as described above. Of these investment amounts, $1,600 remains available for use by us to accelerate our research and product development. To achieve our growth objectives, we are considering different strategies, including growth through acquisitions. As a result, we are evaluating and we will continue to evaluate other companies and businesses for potential synergies that would add value to our existing operations.
At March 27, 2009, we have $1,600 in cash which is not projected to meet all of our working capital needs for the next twelve months. The fact that the Company sustained losses in 2008 and still requires outside sources of additional capital to sustain operations has created an uncertainty about the Company's ability to continue as a going concern.
Management intends to use borrowings and securities sales to mitigate the effects of our use of that cash. However, we cannot assure you that debt or equity financing, if and when required, will be available. Our ability to continue as a going concern is dependent upon receiving additional funds either through the issuance of debt or through common and/or preferred stock and the success of management's plan to expand sales. Although we may obtain external financing through the sales of our own securities, there can be no assurance that such financing will be available, or if available, that any such financing would be on terms acceptable to us. If we are unable to fund our cash flow needs, we may have to reduce or stop planned growth or scale back operations and reduce staff.
Off-Balance Sheet Arrangements
We have no off-balance sheet financing arrangements.
Contractual Obligations
We had the following aggregate future minimum operating lease payments at September 30, 2009 (in thousands):
| Annual periods ending September 30 | ||
| 2010 | $ 182 | |
| 2011 | 81 | |
| 2012 | 74 | |
| $ 337 |
Forward Looking Statements-Safe Harbor
Our business, financial condition, results of operations, cash flows and prospects, and the prevailing market price and performance of our common stock, may be adversely affected by a number of factors, including the matters discussed in "Risk Factors". Certain statements and information set forth in this Form S-1, as well as other written or oral statements made from time to time by us or by our authorized executive officers on our behalf, constitute "forward-looking statements" within the meaning of the Federal Private Securities Litigation Reform Act of 1995. We intend for our forward-looking statements to be covered by the safe harbor provisions for forward-looking statements contained in the Private Securities Litigation Reform Act of 1995, and we set forth this statement and the risk factors set forth herein in order to comply with such safe harbor provisions. You should note that our forward-looking statements speak only as of the date of this Form S-1 or when made and we undertake no duty or obligation to update or revise our forward-looking statements, whether as a result of new information, future events or otherwise. Although we believe that the expectations, plans, intentions and projections reflected in our forward-looking statements are reasonable, such statements are subject to known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements. The risks, uncertainties and other factors that our stockholders and prospective investors should consider are included in "Risk Factors" beginning on page 3.
57
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
On October 24, 2008, we dismissed Pender Newkirk & Company LLP ("Pender") as our independent registered public accounting firm. Concurrent with this action on the same date, our audit committee appointed Stonefield Josephson, Inc. ("Stonefield") as our new independent registered public accounting firm. The decision to change accountants was approved by the audit committee and ratified by the Board of Directors.
The audit report of Pender on the financial statements of clickNsettle.com, Inc. as of and for the year ended June 30, 2008, did not contain any adverse opinion or disclaimer of opinion, nor were they qualified or modified as to uncertainty, audit scope, or accounting principles.
During the period from October 3, 2007, the date we hired Pender, to the end of the most recent fiscal year on June 30, 2008 and from July 1, 2008 to the date of our dismissal of Pender, there have been no disagreements with Pender on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedure, which if not resolved to Pender's satisfaction, would have caused it to make reference to the subject matter of the disagreement in connection with its reports. During the same period, there have been no reportable events, as that term is described in Item 304(a)(1)(v) of Regulation S-K.
We provided Pender with a copy of the foregoing disclosures. A copy of a letter from Pender to the Securities and Exchange Commission, dated October 24, 2008, was filed as Exhibit 16.1 to the Company's Current Report on Form 8-K filed on October 29, 2008. During the two most recent fiscal years, and the subsequent interim period prior to engaging Stonefield, neither the Company nor anyone on its behalf consulted Stonefield regarding the application of accounting principles to a specific transaction, either completed or proposed, or the type of audit opinion that might be rendered on the Company's financial statements, and no written or oral advice was provided by Stonefield that was a factor considered by the Company in reaching a decision as to the accounting, auditing or financial reporting issues as set forth in Item 304(a)(2)(i) and (ii) of Regulation S-K.
DIRECTORS AND EXECUTIVE OFFICERS
The following table sets forth information, as of December 14, 2009, concerning the individuals named below, including their ages and their positions as of December 14, 2009):
Note - our Directors serve a term of one year.
Business Experience of Directors and Executive Officers During the Past Five Years
Andrew A. Brooks, M.D.Dr. Brooks serves as our Chairman of the Board and Chief Executive Officer. since 1987. He founded Cardo Medical, LLC on April 6, 2007, and has served as the President and Chief Executive Officer and manager of Cardo and of Accelerated Innovation, LLC. Dr. Brooks has been in the private practice of orthopedic surgery since 1994, specializing in sports medicine, arthroscopy and joint reconstruction. He has previously served as a design consultant to major companies for joint reconstruction and sports medicine products. He currently maintains a part time surgical practice at the Southern California Orthopedic Institute in Van Nuys, California.
58
Dr. Brooks was a founder and managing partner of Specialty Surgical Centers, a group of multi-specialty outpatient surgical centers operating in Beverly Hills, Encino, Irvine, Arcadia and Westlake Village. These surgical centers were sold to Symbion Healthcare, Inc. in August 2005. Dr. Brooks currentlyalso serves as a managing partner of Specialty Surgical Center in Westlake Village. Dr. Brooks also co-founded the Ridgecrest Sports Rehabilitation Center in 1995, which was sold to a public company in February 1998.
Dr. Brooks is a graduate of the University of Southern California School of Medicine. He completed his residency in Orthopaedic Surgery at the University of Southern California, and subsequently completed a fellowship in arthroscopic reconstructive surgery and sports medicine at the Hughston Clinic in Columbus, Georgia. Dr. Brooks is board-certified by the American Board of Orthopaedic Surgery and is a Fellow of the American Academy of Orthopaedic Surgeons. He is also a Fellow of the American College of Surgeons and aboard member of the Arthroscopy Association of North America. He is an active member of the Los Angeles Chapter of the Young Presidents Organization.
Michael Kvitnitsky.Mr. Kvitnitsky serves as our President and Chief Operating Officeronline brokerage firm TD Ameritrade since November 2006, and as a directorboard member of our company. Since May 2007, Mr. Kvitnitsky has served as the Chief Operating Officer and manager of Cardo Medical, LLC and Accelerated Innovation, LLC. He also has served as the President and manager of Cervical Xpand, LLC, a developmental-stage spinal company,L Brands since July 2005, and of Uni-Knee, LLC, a developmental-stage orthopedic company, since May 2006. Mr. Kvitnitsky founded Accin Corporation, a medical device company, for which he has served as President, Chief Executive Officer and director since February 2005. Prior to that, Mr. Kvitnitsky was employed by Stryker Corporation (NYSE: SYK) from 1998 until January 2005; his last position with Stryker was Vice President, Innovation and Business Development, of Stryker International. His prior employment, from 1990 to 1998, included engineering and research positions with multinational medical device companies in the United States and, from 1986 to 1989, included research institutions in Ukraine.
Derrick Romine.Mr. Romine serves as our Chief Financial Officer and Secretary. Since February 2008, Mr. Romine has served as the Chief Financial Officer of Cardo Medical, LLC. Prior to joining Cardo, he worked for 18 years in all aspects of finance and strategy, including corporate restructuring, capital structure management and organizational development. Most recently, from 2004 to February 2008, Mr. Romine served as Controller for Specialty Surgical Centers, following its acquisition by Symbion Healthcare, Inc. From 2000 to 2004, Mr. Romine held a key financial position at Doane Pet Care, Inc. As Doane's Director of Financial Planning and Control, he orchestrated financial modeling for the largest private label pet food manufacturer globally. Prior to that, from 1997 to 2000, he worked in strategic projects as Director of Strategy & Analysis at Service Merchandise Corporation, a retail company,1987, where he focused specifically on corporate restructureis also Lead Director and capital management. Prior to 1997, Mr. Romine held various financial and operational positions in both the public and private sector.
Joseph Loggia.Mr. Loggia serves as a director of our company. Mr. Loggia has served as the Chief Executive Officer of Advanstar, Inc. and its wholly-owned subsidiary, Advanstar Communications, Inc., a leading worldwide media company providing integrated marketing solutions for the fashion, life sciences and powersports industries, since January 2004. As Chief Executive Officer, he led Advanstar's effort to develop and implement a new strategy, transforming the company from a traditional B2B publisher and trade show producer to a market-focused media company, culminating in a $1.14 billion saleChair of the company in 2007 to Veronis Suhler Stevenson, LLC, a private equity firm focusing on media, communications, information and education industries in North America and Europe. From 2001 through 2003,Finance Committee. Mr. LoggiaTessler has also served as President and Chief Operating Officer of Advanstar, leading the company's efforts to enhance its operating efficiencies and implementing state-of-the-art data systems, new business development procedures and rewards, and a new growth-based management compensation system. From 1995 through 1998, he served as President and Chief Executive Officer of MAGIC International, producer of the MAGIC Marketplace apparel trade show, leading the acquisition of MAGIC by Advanstar in 1998. Prior to joining MAGIC, Mr. Loggia, who is a certified public accountant, was a manager at the accounting firm of Coopers & Lybrand in its fraud and financial investigations division, after having spent 10 years in law enforcement.
59
Thomas H. Morgan.Mr. Morgan serves as a director of our company. He is the Managing Member of Morgan Exploration, LLC, Morgan Marathon, LLC and Morgan United, LLC. Since 1982, Mr. Morgan also has been the founder and President of Morgan Energy Corporation, an oil and gas exploration company. Prior to that, he worked for Conoco Oil Company and Gulf Oil Company. Mr. Morgan has drilled, developed and owned interests in thousands of oil and gas wells throughout the Rocky Mountain region, Texas and Oklahoma. Through other entities, since 1985, Mr. Morgan has owned and developed numerous shopping centers, apartment complexes, condo towers and luxury single-family residences throughout the United States.
Ronald N. Richards, Esq.Mr. Richards serves as a director of our company. Mr. Richards has represented Specialty Surgical Centers, as one of its litigation counsel, and other medical professionals and clinics throughout Southern California. Since 2000, he was the senior partner of Ronald Richards & Associates based in Beverly Hills, California. Since 2003, Mr. Richards has served as Secretary of Sierra Towers Homeowners Association. Mr. Richards was a professor of law at the San Fernando Valley College of Law from 2006 to 2007. He has had numerous published opinions in the state courts and federal courts of appeal. Mr. Richards lectures to other attorneys on various legal matters and has published works on various related medical topics. In 2008, he obtained a Certificate of Management from the Anderson School of Management at the University of California, Los Angeles . Mr. Richards received his law degree from University of La Verne in 1995 and his undergraduate degree from the University of California, Los Angeles, in 1991.
Steven D. Rubin. Mr. Rubin serves as a director of our company. Mr. Rubin has served as Executive Vice President-Administration and as a director of OPKO Health, Inc. since May 2007. Mr. Rubin served as the Senior Vice President, General Counsel and Secretary of IVAX from August 2001 until September 2006. Prior to joining IVAX, Mr. Rubin was Senior Vice President, General Counsel and Secretary with privately-held Telergy, Inc., a provider of business telecommunications and diverse optical network solutions, from early 2000 to August 2001. In addition, he was with the Miami law firm of Stearns Weaver Miller Weissler Alhadeff & Sitterson from 1986 to 2000, in the Corporate and Securities Department. Mr. Rubin had been a shareholder of that firm since 1991 and a director since 1998. Mr. Rubin currently serves on the board of directors of Dreams,Steel Partners Holding, since July 2009 and for Imperva Inc., since 2013. Mr. Tessler was Chief Executive Officer of Epoch Holding Corporation, an investment management company, from February 2000 to June 2004, and Chairman of its board of directors from May 1994 to December 2013; the Co-Chairman and Co-Chief Executive Officer of Interactive Data Corporation, a vertically integrated sports licensingsecurities market data supplier, from June 1992 to February 2000; and productsa co-founder and Chairman of the board of directors of Enhance Financial Services, a public insurance holding company, Safestitch Medical, Inc.,from 1986 to 2001. Mr. Tessler is a medical device company, Prolor Biotech, Inc.,member of the board of governors of the Boys & Girls Clubs of America. Mr. Tessler holds a Bachelor of Arts degree from Cornell University and a Bachelor of Laws degree from Cornell University Law School.
We believe that Mr. Tessler possesses specific attributes that qualify him to serve as a member of our board of directors, including an array of executive management and board positions he has served for publicly traded companies during his career.
Peter Altman, Ph.D. has served as our President and Chief Executive Officer since 2002, where he has global responsibility for the development, stage biopharmaceutical company, Kidville, Inc., which operates large, upscale facilities, cateringmanufacture and marketing of our therapeutic candidates and products. He was founding Chief Executive Officer from 1999 to newborns through five-year-old children2003 and their families and offers a wide rangeboard member of developmental classes for newborns-5 year olds, Non-Invasive Monitoring Systems, Inc., a medical device company, Castle Brands, Inc.,CareDx from 1999 to 2014, a developer of a gene based diagnostics to be used in chronic inflammatory diseases, including cardiac transplantation, coronary artery disease and marketersystemic lupus erythematosus. He was also founding Chief Executive Officer for Lumen Therapeutics from 2004 to 2005, an early-stage pharmaceutical company. He has 30 years of premium brand spirits, SearchMedia Holdings Limited, a leading nationwide multi-platform media company basedexperience in Shanghai, Chinalife science research and Neovasc, Inc., a company developingproduct development, is named inventor in 45 U.S. patents, and marketing medical specialty vascular devices.
Subbarao Uppaluri, Ph.D.Dr. Uppalurihas authored 40 scientific publications. Dr. Altman currently serves as a director on the board of directors of Oncocyclist Biotech, since 2018. He received his Ph.D. in Bioengineering/Pharmaceutical Chemistry from the University of California, San Francisco and University of California, Berkeley, his Management of Technology certificate from the Walter A. Haas School of Business at the University of California, Berkeley, and both his Master of Science and Bachelor of Science in Mechanical Engineering from the Columbia University School of Engineering and Applied Sciences. Dr. Altman has been elected Fellow of the American Heart Association.
We believe that Dr. Altman possesses specific attributes that qualify him to serve as a member of our company. Dr. Uppaluriboard of directors, including his extensive experience in the biotechnology, medical device and diagnostic industries and the operational insight and expertise he has accumulated as our President and Chief Executive Officer.
Fernando L. Fernandez was appointed to our board of directors in October 2016. Mr. Fernandez has served as Seniorthe Vice President of Finance and Chief Financial Officer of United Data Technologies, an information technology company, since November 2016. Mr. Fernandez served as the Market Vice President and Chief Financial Officer of the Care Delivery segment of Humana, Inc., a health and well-being company, from December 2012 to October 2016. From June 2004 to December 2012, Mr. Fernandez served as the Senior Vice President of Finance and Chief Financial Officer of Continucare Corporation, a medical care service company. He currently serves as a director for South Florida Business Forum since January 2018. Mr. Fernandez spent his early career in public accounting and finance functions at other companies, including Whitman Education Group, Inc., Frost-Nevada LP, and PriceWaterhouseCoopers LLP. Mr. Fernandez holds a Bachelor of Business Administration, Accounting from the University of Miami, and is a CPA.
We believe that Mr. Fernandez possesses specific attributes that qualify him to serve as a member of our board of directors, including his expertise in accounting and finance.
Richard Krasno, Ph.D. was appointed to our board of directors in October 2016. Dr. Krasno has served as a director of Ladenburg Thalmann since March 2015, Castle Brands, Inc. since 2014, and OPKO Health, Inc. since May 2007.2017. Dr. UppaluriKrasno served as the Vice President, Strategic Planningexecutive director of the William R. Kenan, Jr. Charitable Trust from 1999 to 2014 and, Treasurerfrom 1999 to 2010, as president of IVAXthe four affiliated funds. Prior to that, Dr. Krasno was the president of the Monterey Institute of International Studies in Monterey, California. From 2004 to 2012, Dr. Krasno also served as a director of the University of North Carolina Health Care System and served as chairman of the board of directors from 1997 until December 2006. Before joining IVAX, from 19872009 to August 1996, Dr. Uppaluri was Senior Vice President, Senior Financial Officer and Chief Investment Officer with Intercontinental Bank, a publicly traded commercial bank in Florida. In addition,2012. From 1981 to 1998, he served as president and chief executive officer of the Institute of International Education in various positions,New York. He also served as Deputy Assistant Secretary of Education in Washington, D.C. from 1979 to 1980. Mr. Krasno holds a Bachelor of Science from the University of Illinois and a Ph.D. from Stanford.
We believe that Mr. Krasno possesses specific attributes including Senior Vice President, Chief Investment Officerhis qualifications and Controller, at Peninsula Federal Savings & Loan Association, a publicly traded Florida S&L, from October 1983 to 1987. His prior employment, during 1974 to 1983, included engineering, marketingskills, including financial literacy and research positions with multinational companies and research institutes in Indiaexpertise, his managerial experience and the United States. Dr. Uppaluri currently servesknowledge and experience he has attained through his service as a director of publicly-traded corporations, which qualify him to serve as a member of our board of directors.
Jay M. Moyes has served on our board of directors since 2011. He has served on the board of directors of Kidville, Inc., which operates large, upscale facilities, cateringPuma Biotechnologies since April 2012, and on the board of directors of Achieve Life Sciences from 2018 to newborns through five-year-old childrenthe present and their familieson the board of directors and offersChairman of the Audit Committee of Osiris Therapeutics, a wide rangebiosurgical company, from May 2006 until December 2017. He also served as a member of developmental classes for newborns-5 year olds, Non-Invasive Monitoring Systems,the board of directors and Chairman of the Audit Committee of Integrated Diagnostics, a privately held molecular diagnostics company, from 2011 to 2016. From 2012 to 2014, Mr. Moyes served as a member of the board of directors of Amedica Corporation, a publicly traded orthopaedics company, and as Chief Financial Officer from 2013 to 2014. From 2008 to 2009, Mr. Moyes served as Chief Financial Officer of CareDx, a publicly traded molecular diagnostics company. Prior to that, he served as Chief Financial Officer of Myriad Genetics, Inc., a medical devicespublicly held healthcare diagnostics company, from June 1996 until his retirement in November 2007, and Winston Pharmaceuticals Inc.,as Vice President of Finance from July 1993 until July 2005. From 1991 to 1993, Mr. Moyes served as Vice President of Finance and Chief Financial Officer of Genmark, a specialty pharmaceutical company engaged inprivately held genetics company. Mr. Moyes held various positions with the discovery and developmentaccounting firm of products for pain management.
Relationship among Directors and Executive Officers
PursuantKPMG from 1979 to the terms1991. He also served as a member of the Merger Agreement, Dr. Brooks nominated Messrs. Kvitnitsky, Loggia, MorganBoard of Trustees of the Utah Life Science Association from 1999 to 2006. Mr. Moyes holds a Masters of Business Administration from the University of Utah, a Bachelor of Arts in economics from Weber State University, and Richards and himselfis formerly a Certified Public Accountant.
We believe that Mr. Moyes possesses specific attributes that qualify him to serve as a member of our board of directors, including his extensive background in finance and accounting in the life sciences industry.
Simon H. Stertzer, M.D. is Chairman of our board of directors and has served on our board of directors since 2002. Dr. Phillip Frost nominated Mr. RubinStertzer is a Professor of Medicine, Emeritus at the Stanford University School of Medicine, Division of Cardiovascular Medicine, and a Professor at the Stanford University Biodesign Program. He served as Assistant Resident in Medicine at New York University and later as Chief Medical Resident at New York University Division of Bellevue Hospital. Dr. UppaluriStertzer was a founder and board member of Arterial Vascular Engineering, an angioplasty balloon and stent company that went public in 1996 and was subsequently acquired by Medtronic. Dr. Stertzer served as Director of the Catheterization Laboratory at Lenox Hill Hospital from 1971 to 1983. He was the Director of Medical Research and Director of the Cardiac Catheterization Laboratory at the San Francisco Heart Institute from 1983 until 1993. He was appointed Professor of Medicine at Stanford University in 1998, and became Professor Emeritus at Stanford University in 2011. Dr. Stertzer received his Doctor of Medicine degree from New York University. He also earned a Certificat de Physiologie from University of Paris (Sorbonne) and had a fellowship at New York University Hospital in Cardiovascular Disease. Dr. Stertzer received a Bachelor of Arts degree in Humanities from Union College.
We believe that Dr. Stertzer possesses specific attributes that qualify him to serve as Chairman of our board of directors, including his historical association with our company and his expertise in interventional cardiology and the operational experience he has accumulated in the life sciences industry.
Director Independence
We are not currently subject to listing requirements of any national securities exchange that has requirements that a majority of the board of directors be “independent.” Nevertheless, we expect that our board of directors will determine that all of our directors, other than Dr. Altman, qualify as “independent” directors in accordance with listing requirements of The NASDAQ Stock Market, or NASDAQ. Dr. Altman is not considered independent because he is an employee of BioCardia. The NASDAQ independence definition includes a series of objective tests, such as that the director is not, and has not been for at least three years, one of our employees and that neither the director nor any of his family members has engaged in various types of business dealings with us.
Board Leadership Structure
Board Structure. Our board of directors has eight authorized seats divided into three classes (Class I, Class II and Class III) with staggered three-year terms. Three Class III directors are to be elected at the 2019 Annual Meeting to serve a three-year term expiring at the 2022 Annual Meeting of stockholders or until their respective successors have been elected and qualified. The Class I and Class II directors will continue to serve their respective terms until the respective 2020 and 2021 Annual Meetings of stockholders.
Board Leadership Structure. Our board of directors does not have a policy on whether or not the role of the Chief Executive Officer and Chairman should be separate or, if it is to be separate, whether the Chairman should be selected from the non-employee directors or be an employee. Currently, we operate with Dr. Altman serving as a director and our President and Chief Executive Officer and Dr. Stertzer serving as our Chairman. We believe that the separation of the Chairman and Chief Executive Officer positions suit the talents, expertise and experience that each of Drs. Altman and Stertzer bring to the Company.
Board Committees. Our board of directors has established an audit committee, a compensation committee and a nominating and corporate governance committee. The composition and responsibilities of each of the committees of our board of directors is described below. Members will serve on these committees until their resignation or until as otherwise determined by our board of directors. No
Family Relationships
There are no family relationships exist among any of the individuals who will serve as our directors or executive officers.
60
Board Committees
Summary Our board of directors has established an audit committee, a compensation committee and a nominating and corporate governance committee. The composition and responsibilities of each of the committees of our board of directors is described below. Members will serve on these committees until their resignation or until as otherwise determined by our board of directors.
Audit Committee
Our audit committee currently consists of Allan Tessler, who is the chair of the committee, Jay Moyes and Fernando Fernandez, each of whom are independent for Audit Committee purposes under the requirements of Financial Industry Regulatory Authority (“FINRA”) and the SEC. Mr. Tessler is an “audit committee financial expert” as the term is defined under SEC regulations. The audit committee operates under a written charter. The functions of the audit committee include:
overseeing the engagement of our independent registered accounting firm;
reviewing our audited financial statements and discussing them with the independent registered accounting firm and our management;
meeting with the independent registered accounting firm and our management to consider the adequacy of our internal controls; and
reviewing our financial plans, reporting recommendations to our full board of directors for approval and authorizing actions.
Both our independent registered accounting firm and internal financial personnel regularly meet with our audit committee and have unrestricted access to the audit committee.
Our audit committee operates under a written charter adopted by our board of directors, a current copy of which is available on the Corporate Governance portion of our website at investors.biocardia.com. During 2018, our audit committee held four meetings.
Compensation TableCommittee
Our compensation committee currently consists of Jay Moyes, who is the chair of the committee, Simon Stertzer and Richard Pfenniger, each of whom are independent in accordance with the NASDAQ Stock Market LLC standards. Each member of our compensation committee is also a non-employee director, as defined pursuant to Rule 16b-3 promulgated under the Securities and Exchange Act of 1934, as amended (the “Exchange Act”). The compensation committee operates under a written charter. The functions of the compensation committee include:
reviewing and, if deemed appropriate, recommending to our board of directors policies, practices and procedures relating to the compensation of our directors, officers and other managerial employees and the establishment and administration of our employee benefit plans;
determining or recommending to the board of directors the compensation of our executive officers; and
advising and consulting with our officers regarding managerial personnel and development.
Our compensation committee operates under a written charter adopted by our board of directors, a current copy of which is available on the Corporate Governance portion of our website at investors.biocardia.com.
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee consists of Simon Stertzer, who is the chair of the committee, Thomas Quertermous and Richard Krasno, each of whom are independent in accordance with the NASDAQ Stock Market LLC standards. The nomination committee operates under a written charter. The functions of the nominating and corporate governance include:
establishing standards for service on our board of directors;
identifying individuals qualified to become members of our board of directors and recommending director candidates for election or re-election to our board;
considering and making recommendations to our board of directors regarding the size and composition of the board of directors, committee composition and structure and procedures affecting directors;
reviewing compliance with relevant corporate government guidelines;
reviewing governance-related stockholder proposals and recommending Board responses; and
reviewing actual and potential conflicts of interest of Board members and corporate officer, other than related-party transactions reviewed by the Audit Committee, and approving or prohibiting any involvement of such persons in matters that may involve a conflict of interest or taking of a corporate opportunity.
Our nominating and corporate governance committee operates under a written charter adopted by our Board of Directors, a current copy of which is available on the Corporate Governance portion of our website at investors.biocardia.com.
Non-Employee Director Compensation
Cash and Equity Compensation
We compensate non-employee members of the board of directors. Directors who are also employees do not receive cash or equity compensation for service on the board of directors in addition to compensation payable for their service as our employees. The non-employee members of our board of directors are reimbursed for travel, lodging and other reasonable expenses incurred in attending board of directors or committee meetings. Our directors received equity grants annually at the fair market value of our common stock at the time of grant under our 2016 Plan.
In January 2017 our compensation policy for non-employee directors was established. The cash and equity components of our compensation policy for non-employee directors are set forth below:
Position | Annual Cash | Equity Grant | ||||||
Base Fee | $ | 40,000 | $ | 44,000 | ||||
Chairperson Fee | ||||||||
Chairman of the Board | 25,000 | |||||||
Audit Committee | 15,000 | |||||||
Compensation Committee | 10,000 | |||||||
Nominating and Corporate Governance Committee | 7,500 | |||||||
Committee Member Fee | ||||||||
Audit Committee | 7,500 | |||||||
Compensation Committee | 5,000 | |||||||
Nominating and Corporate governance | 3,750 | |||||||
Under our non-employee director compensation program, each non-employee director received an initial equity award in January of 2017 of either an option to purchase 2,472 shares of common stock or receive 1,703 restricted stock units which, in either case, vest over three years upon the anniversary of the grant date, subject to continued service through the vesting date. We expect additional annual equity grants may be made to our non-employee directors and that compensation for our non-employee directors will be competitive at the 50th percentile of our peer group. In July 2018, the board elected to defer quarterly payment of the cash portion of director compensation until the Company has raised sufficient financing.
Compensation for 2018
The following table sets forth a summary ofinformation concerning the compensation awarded to, paid to, or earned by or paid to the principal executive officers and principal financial officers of Cardo.
| Name and | ||||||
| Principal | All Other | |||||
| Position | Year | Salary | Bonus | Stock Options | Compensation | Total |
| Andrew A. Brooks (1) | 2008 | $ 250,000 | $ - | $ 29,250 | $ - | $ 279,250 |
| Mikhail Kvitnitsky (2) (3) | 2008 | 220,000 | - | 26,000 | - | 246,000 |
| 2007 | 52,000 | - | - | 30,276 | 82,276 | |
| Derrick Romine (4) | 2008 | 180,000 | - | 61,100 | - | 241,100 |
(1) Dr. Brooks currently serves as our Chairman of the Board and Chief Executive Officer. He has served as the President, Chief Executive Officer and manager of Cardo Medical ,LLC since May 2007 and Accelerated Innovation, LLC from May 2007 until its merger with Cardo Medical, LLC.
(2) Mr. Kvitnitsky currently serves as our President and Chief Operating Officer and as a directornon-employee members of our company. Mr. Kvitnitsky has served as the Chief Operating Officer and managerboard of Cardo Medical, LLC since May 2007 and Accelerated Innovation, LLC from May 2007 until its merger with Cardo Medical, LLC. He also has served as the President and manager of Cervical Xpand, LLC since July 2005, and of Uni-Knee, LLC since May 2006 until their mergers with Cardo Medial, LLC. Mr. Kvitnitsky founded Accin Corporation, for which he has served as President, Chief Executive Officer and director since February 2005. The information presented in this table reflects all compensation received by Mr. Kvitnitsky from Cardo, Accelerated Innovation, Cervical Xpand, Uni-Knee and Accin on a consolidated basis for the applicable periods.
(3) These amounts reflect 5% of the net receipts from the sale of the Align 360 unicompartmental knee product, which Mr. Kvitnitsky was entitled to receive per his compensation arrangement with us. See "Employment Agreements and Change in Control Arrangements-Compensation Arrangement with Michael Kvitnitsky" below.
(4) Mr. Romine currently serves as our Chief Financial Officer and Secretary. He has served as the Chief Financial Officer of Cardo Medical, LLC since February 2008.
Director Compensation
The following table sets forth a summary of compensation awarded to, earned by or paid to each directordirectors for the fiscal year ended December 31, 2008.2018:
Director | Fees Earned or Cash($)(1) | Stock Awards | Option Awards | Total ($) | ||||||||||||
Fernando L. Fernandez | $ | 47,500.00 | $ | 44,000.00 | — | $ | 91,500.00 | |||||||||
Richard Krasno, Ph.D. | $ | 43,750.00 | $ | 44,000.00 | — | $ | 87,750.00 | |||||||||
Jay M. Moyes | $ | 57,500.00 | $ | 44,000.00 | — | $ | 101,500.00 | |||||||||
Richard C. Pfenniger, Jr. | $ | 45,000.00 | $ | 44,000.00 | — | $ | 89,000.00 | |||||||||
Thomas Quertermous, M.D. | $ | 43,750.00 | $ | 44,000.00 | — | $ | 87,750.00 | |||||||||
Simon H. Stertzer, M.D. | $ | 77,500.00 | $ | 44,000.00 | — | $ | 121,500.00 | |||||||||
Allan R. Tessler | $ | 55,000.00 | $ | 44,000.00 | — | $ | 99,000.00 | |||||||||
(1) | In July 2018, the board elected to defer quarterly payment of the cash portion of director compensation until the Company has raised sufficient financing. |
(2) | This amount reflects the aggregate grant fair value computed in accordance with ASC Topic 718. The assumptions that we used to calculate these amounts are discussed in Notes 2 and 13 to our consolidated financial statements for the year ended December 31, 2018. |
The following table lists all outstanding equity awards held by our non-employee directors as of December 31, 2018.
Name | Aggregate Options | Aggregate Awards | ||||||
Fernando L. Fernandez | — | 4,729 | (1) | |||||
Richard Krasno, Ph.D. | — | 4,729 | (1) | |||||
Jay M. Moyes | 5,931 | (2) | 3,594 | (6) | ||||
Richard C. Pfenniger, Jr. | — | 4,729 | (1) | |||||
Thomas Quertermous, M.D. | 3,921 | (3) | 3,594 | (6) | ||||
Simon H. Stertzer, M.D. | 12,218 | (4) | 3,594 | (6) | ||||
Allan R. Tessler | 1,508 | (5) | 4,729 | (1) | ||||
(1) | Includes (i) 567 shares subject to a restricted stock award that vested on January 13, 2019, (ii) 3,594 shares subject to a restricted stock award that vests July 26, 2019, and (iii) 567 shares subject to a restricted stock award that vests January 13, 2020. |
(2) | Includes (i) 4,283 shares subject to an option, which are fully vested and immediately exercisable, (ii) 824 shares subject to an option that vested January 13, 2019, and (iii) 824 shares subject to an option that vest on January 13, 2020. |
(3) | Includes (i) 2,273 shares subject to an option, which are fully vested and immediately exercisable, (ii) 824 shares subject to an option that vested January 13, 2019, and (iii) 824 shares subject to an option that vest on January 13, 2020. |
(4) | Includes (i) 10,570 shares subject to an option which are fully vested and immediately exercisable, (ii) 824 shares subject to an option that vested January 13, 2019, and (iii) 824 shares subject to an option that vest on January 13, 2020. |
(5) | Includes 1,508 shares subject to an option, which are fully vested and immediately exercisable. |
(6) | Includes 3,594 shares subject to a restricted stock award that vests on July 26, 2020. |
Code of Ethics
We anticipate reimbursing each director for reasonable travel expenses relatedhave adopted a Code of Business Conduct and Ethics that applies to all of our (1) officers, (2) employees (including our principal executive officer, principal financial officer, principal accounting officer or controller and other employees who perform financial or accounting functions), and (3) agents and representatives, including our independent directors and consultants, who are not employees of ours, with regard to their BioCardia-related activities. Our code of business conduct and ethics is available on our website at www.biocardia.com under the heading “Corporate Governance” under the section titled “Investors”. We will post on this section of our website any amendment to our code of business conduct and ethics, as well as any waivers of our code of business conduct and ethics, that director's attendance at Boardare required to be disclosed by the rules of the SEC.
Executive Officers
The following table identifies certain information about our executive officers as of March 28, 2019. Officers are elected by our board of directors to hold office until their successors are elected and qualified.
Name | Age | Position | ||
Peter Altman, Ph.D. | 52 | President, Chief Executive Officer, and Director | ||
Henricus Duckers, M.D., Ph.D., FESC | 51 | Chief Medical Officer | ||
David McClung | 55 | Chief Financial Officer | ||
Phil Pesta | 53 | Vice President of Operations |
The service of our executive officers prior to 2016 noted in the narrative below includes service with BioCardia Lifesciences, Inc., the company we merged with in the reverse merger transaction in October 2016. For a brief biography of Dr. Altman, please see “Board of Directors and committee meetings.Corporate Governance−Nominees for Director.”
| Fees Earned or | All Other | ||
| Name | Paid In Cash | Compensation | Total |
| Joseph Loggia | $ - | $ 5,000 | $ 5,000 |
| Thomas H. Morgan | - | 5,000 | 5,000 |
| Ronald N. Richards, Esq. | - | 5,000 | 5,000 |
| Steven D. Rubin | - | 5,000 | 5,000 |
| Subbarao Uppaluri | - | 5,000 | 5,000 |
Compensation
Henricus Duckers has served as our Chief Medical Officer since 2016. From 2013 to 2016, Dr. Duckers was the Chair of NamedRegenerative Medicine and the Head of R&D in the Department of Cardiology and Pulmonology at the University Medical Center Utrecht. He has over 20 years of experience in cardiovascular research, is named inventor in ten U.S. patents, and has authored 140 scientific publications in cardiology, neurology and cell biology. Dr. Duckers studied Medicine and Pharmacy at the University of Utecht, as well as Management in Health Care (Univ. Rotterdam). From 1992 to 1993 he completed his Ph.D. at the Rudolf Magnus Institute for NeuroScience, Cum Laude, and obtained a registration as clinical pharmacologist. He was trained as an interventional cardiologist at the Thoraxcenter Rotterdam, where, he, among other notable achievements, also supervised the molecular cardiology program.
David McClung has served as our Chief Financial Officer since September 2017 and has been with the Company since September 2013, also serving as Vice President of Finance from March 2016 to August 2017 and as Senior Director of Finance & Controller from September 2013 to February 2016. Mr. McClung has more than 20 years of finance and accounting experience in publicly and privately financed organizations, including startup enterprises, large public companies and middle-market businesses. Before joining our company, Mr. McClung served as Director of Finance and Controller at Sonitus Medical, Inc., a privately-held manufacturer of an FDA cleared prosthetic hearing device for the treatment of single-sided deafness and conductive hearing loss, from June 2011 to August 2013. Prior to that, Mr. McClung served as Controller at NextWave Pharmaceuticals, Inc. a specialty pharmaceutical company acquired by Pfizer, Inc., from April 2010 to June 2011. Mr. McClung spent his early career in public accounting and finance functions at other companies, including Matson Navigation, Inc., The Clorox Company and KPMG LLP. Mr. McClung earned a Bachelor of Arts degree in Accounting from Georgia State University, graduating with honors. He is an actively licensed CPA and member of the AICPA and the California Society of CPAs.
Phil Pesta has served as our Vice President of Operations since July 2011. Mr. Pesta has more than 20 years of experience in the medical device industry, primarily in manufacturing and operations roles. Before joining our company, Mr. Pesta was with Boston Scientific. He was most recently responsible for developing the operations transfer plan for the divestiture of their neurovascular division to Stryker Corporation. Prior to that, Mr. Pesta held simultaneous roles as Director of Engineering at Boston Scientific’s electrophysiology division and Plant Manager at the embolic protection division. Earlier in his career, Mr. Pesta held positions in project management and manufacturing engineering at other companies, including Conceptus, Novare Surgical Systems, Medtronic Anneurx and Modified Polymer Components. He has facilitated the commercial launch of multiple products and is listed as an inventor on three U.S. patents. Mr. Pesta earned a Bachelor of Arts Degree in General Design Studies from San Jose State University.
EXECUTIVE COMPENSATION
Information relating to director compensation is provided under the section titled “Directors, Executive Officers and Corporate Governance.”
Commencing August 29, 2008,
Fiscal 2018 Summary Compensation Table
The following table sets forth total compensation paid to our named executive officers, who are comprised of (1) our principal executive officer and (2) our next two highest compensated executive officers other than the closing date ofprincipal executive officer.
Name and Principal Position | Year | Salary ($) | Bonus ($) | Stock ($)(1) | Option ($)(1) | All Other ($) | Total ($) | ||||||||||||||||||
Peter Altman, Ph.D. | 2018 | 360,000.00 | — | (2)(4) | 742,000.00 | (3) | 1,102,000.00 | ||||||||||||||||||
President, Chief Executive Officer, and Director | 2017 | 310,000.00 | 62,000.00 | — | — | — | 372,000.00 | ||||||||||||||||||
David McClung | 2018 | 300,000.00 | — | (2)(4) | 294,982.10 | (3) | 594,982.10 | ||||||||||||||||||
Chief Financial Officer | 2017 | 210,000.00 | 60,000.00 | — | — | — | 270,000.00 | ||||||||||||||||||
Henricus Duckers | 2018 | 350,000.00 | — | (2)(4) | 311,417.40 | (3) | 661,417.40 | ||||||||||||||||||
Chief Medical Officer | 2017 | 270,000.00 | 51,300.00 | — | — | — | 321,300.00 | ||||||||||||||||||
Phil Pesta | 2018 | 235,000.00 | — | (2)(4) | 129,850.00 | (3) | 364,850.00 | ||||||||||||||||||
Vice President of Operations | 2017 | 230,000.00 | 46,000.00 | — | — | — | 276,000.00 | ||||||||||||||||||
(1) | This amount reflects the aggregate grant fair value computed in accordance with ASC Topic 718. The assumptions that we used to calculate these amounts are discussed in Notes 2 and 13 to our consolidated financial statements for the year ended December 31, 2018. |
(2) | We use short-term cash incentive compensation to motivate our named executive officers to achieve our annual financial and operational objectives, while making progress towards our longer-term strategic and growth goals. |
Our executive 2018 bonus targets were set by our compensation committee in July 2018. Our executive bonus plan allows our compensation committee to provide cash incentive awards to employees selected by our compensation committee, including our named executive officers, which may but need not be based upon performance goals established by our compensation committee.
For 2018, the Merger, the annual compensationtarget and actual incentive amounts under our executive bonus plan for our named executive officers is as follows:were the following:
| Name and | |||
| Principal | (1) Bonus | ||
| Position | Salary | Potential | Total |
| Andrew A. Brooks, Chief Executive Officer | $ 250,000 | $ 250,000 | $ 500,000 |
| Mikhail Kvitnitsky, President and Chief Operating Officer | $ 220,000 | $ 220,000 | $ 440,000 |
| Derrick Romine, Chief Financial Officer and Secretary | $ 180,000 | $ 45,000 | $ 225,000 |
(1)
Named Executive Officer | Target Award Salary) | Target Award ($) | ||||||
Peter Altman | 40 | % | $ | 144,000.00 | ||||
Henricus Duckers | 25 | % | $ | 87,500.00 | ||||
David McClung | 25 | % | $ | 75,000.00 | ||||
Phil Pesta | 25 | % | $ | 58,750.00 | ||||
The amount reflected in this column reflects the maximum potential bonus thatamounts for our Board of Directors may grant to each of the abovenamed executive officers.
61
Equity Incentive Plan
Our Board of Directors will consider adopting and implementing an equity incentive plan, pursuant to which we may grant various types of equity and equity-based awards to our executives, employees and contractors, including awards of stock options and restricted stock. Awards made pursuant to the plan may be made subject to the attainment of performance goals relating to one or more business criteria. If adopted, this plan is intended to assist our company in attracting, retaining and motivating designated eligible employees and independent contractors of ours and our subsidiaries and to increase their interestofficers earned in the success of our company in order to promote our long-term interests. This plan will be designed to meet this intent by providing designated eligible persons with a proprietary interest in pursuingfiscal year ending December 31, 2018 are not calculable through the long-term growth, profitability and financial success of our company.latest practicable date.
The Compensation Committee of our Board of Directors, is expected to have complete authority, subject to the express provisions of the plan, to approve the employees or contractors to be granted awards, to determine the number of stock options or other awards to be granted, to set the terms and conditions of the awards, to remove or adjust any restrictions and conditions upon those awards, and to adopt rules and regulations, and to make all other determinations deemed necessary or desirable for the administration of this plan.
(3) | This option vests and becomes exercisable in substantially equal monthly installments over four years beginning on March 1, 2018. |
(4) | This amount was earned in the fiscal year ending December 31, 2017, but was not paid until 2018. |
Employment Agreements and Change in Control Arrangements
Michael Kvitnitsky Employment Agreement, as Terminated as of June 23, 2008
On January 31, 2005, Michael KvitnitskyPeter Altman
We have not entered into an employment agreement with Accin Corporation,Dr. Altman. Accordingly, he is employed on an at-will basis. Dr. Altman’s current annual base salary is $360,000.00 and he is eligible for an annual bonus equal to 40% of his base salary.
Dr. Altman is also eligible for equity compensation under which he servedour equity compensation plans, as Chief Executive Officerdetermined from time to time by the compensation committee of Accin. Accin assigned this agreement to Cardo's wholly-owned subsidiary, Accelerated Innovation, LLC, on May 21, 2007, along with substantially allour board of the other assets of Accin. On June 6, 2008, Mr. Kvitnitsky and Accelerated Innovationdirectors.
David McClung
We have not entered into an amendment to this employment agreement with Mr. McClung. Accordingly, he is employed on an at-will basis. Mr. McClung’s current annual base salary is $300,000.00 and he is eligible for an annual bonus equal to remove references25% of his base salary.
Mr. McClung is also eligible for equity compensation under our equity compensation plans, as determined from time to a shareholderstime by the compensation committee of our board of directors.
Henricus Duckers
We have not entered into an employment agreement with Dr. Duckers. Accordingly, he is employed on an at-will basis. Dr. Duckers’ current annual base salary is $350,000.00 and he is eligible for Accin. On June 23, 2008, Cardo acquired allan annual bonus equal to 25% of his base salary.
Dr. Duckers is also eligible for equity compensation under our equity compensation plans, as determined from time to time by the compensation committee of our board of directors.
Phil Pesta
We have not entered into an employment agreement with Phil Pesta. Accordingly, he is employed on an at-will basis. Mr. Pesta’s current annual base salary is $235,000.00 and he is eligible for an annual bonus equal to 25% of his base salary.
Mr. Pesta is also eligible for equity compensation under our equity compensation plans, as determined from time to time by the compensation committee of our board of directors.
Potential Payments on Termination or Change of Control
We entered into change of control and severance agreements with each of our named executive officers, effective as of the ownership interestscompletion of our Merger. Under each of these agreements, if, within the period three months prior to and 12 months following a “change of control” (such period, the “change in Accelerated Innovation held by Accin, and Accelerated Innovation became the wholly-owned subsidiary of Cardo. Upon the closing of this acquisition, Mr. Kvitnitsky and Accelerated Innovation terminated Mr. Kvitnitsky's employment agreement.
Prior to its termination, the term of the employment agreement was from June 1, 2005 through May 30, 2008, with automatic renewal thereafter for successive one-year periods ending on each May 30, unless (i) either party elected tocontrol period”), we terminate the employment agreement at the end of the then-current term by givingapplicable employee other than for “cause,” death or disability, or the other party four months' advance written notice, or unless the agreement was earlier terminated by Accelerated Innovationemployee resigns for "Cause" or by Mr. Kvitnitsky for "Good Reason," as“good reason” (as such terms are defined in the employment agreement. The employmentemployee’s change of control and severance agreement) and, within 60 days following the employee’s termination, the employee executes an irrevocable separation agreement was automatically renewed for one year on May 30, 2008 and terminated by mutual agreement effective June 23, 2008. Under this employment agreement, Mr. Kvitnitsky wasrelease of claims, the employee is entitled to receive (i) a lump sum payment equal to the following compensation and benefits:
Initialpercentage of the employee’s annual base salarysalary: 150% for Dr. Altman, 100% for Mr. McClung, 100% for Dr. Duckers and 100% for Mr. Pesta, (ii) a lump sum payment equal to the following percentage of $52,000; Eligibilitythe employee’s target annual bonus: 150% for Dr. Altman, 100% for Mr. McClung, 100% for Dr. Duckers and 100% for Mr. Pesta, (iii) reimbursement of premiums to receive stock options upon implementing a stock option plan; Eligibility to share in "milestone" payments; Five weeks of paid time off, including sick, vacation and personal days; Reimbursement for all reasonable and necessary business-related expenses; Participation in the life andmaintain group health insurance plans, 401(k) plancontinuation benefits pursuant to “COBRA” for employee and other employee benefit plansemployee’s dependents for 18 months for Dr. Altman, 12 months for Mr. McClung, 12 months for Dr. Duckers and programs generally made available12 months for Mr. Pesta, and (iv) accelerated vesting as to other employees; and certain severance benefits if his employment was terminated by Accelerated Innovation without Cause or by Mr. Kvitnitsky for Good Reason. If Mr. Kvitnitsky fails to perform his duties under this agreement or to otherwise comply with the terms of this agreement, or if Mr. Kvitnitsky does not cure that failure within 30 days after his receipt of written notice100% of the failure, hisemployee’s outstanding unvested equity awards.
Additionally, under each of these agreements, if, outside of the change in control period, we terminate the employment will be terminated. Mr. Kvitnitsky will be afforded a reasonable opportunity to be heard byof the Boardapplicable employee other than for cause, death or disability, or the employee resigns for good reason and, within 60 days following the employee’s termination, the employee executes an irrevocable separation agreement and release of Directors prior to termination.
Compensation Arrangement with Michael Kvitnitsky, as Terminated as of June 23, 2008
Michael Kvitnitsky wasclaims, the employee is entitled to receive 5% of net receipts from(i) a lump sum payment equal to the salefollowing percentage of the Align 360 unicompartmental knee product. Foremployee’s annual base salary: 100% for Dr. Altman, 50% for Mr. McClung, 50% for Dr. Duckers and 50% for Mr. Pesta, (ii) reimbursement of premiums to maintain group health insurance continuation benefits pursuant to “COBRA” for employee and employee’s dependents for 12 months for Dr. Altman, 6 months for Mr. McClung, 6 months for Dr. Duckers and 6 months for Mr. Pesta, and (iii) the year endedemployee’s outstanding unvested equity awards will vest as to an additional 24 months for Dr. Altman, 12 months for Mr. McClung, 12 months for Dr. Duckers and 12 months for Mr. Pesta.
Pursuant to the change of control and severance agreements, in the event any payment or benefit provided to our named executive officers would be subject to the excise tax imposed by Section 4999 of the Internal Revenue Code, as amended, or the Code (as a result of a payment being classified as a parachute payment under Section 280G of the Code), the applicable employee will receive such payment as would entitle him to receive the greatest after-tax benefit, even if it means that we pay him a lower aggregate payment so as to minimize or eliminate the potential excise tax imposed by Section 4999 of the Code.
Outstanding Equity Awards at 2018 Year-End
The following table sets forth summary information regarding the outstanding equity awards for each of the named executive officers as of December 31, 2006, Accin (the party from which Cardo acquired its medical device business in 2007) paid $100,0002018:
Option Awards(1)(2) | Stock Awards(2) | ||||||||||||||||||||||
Name | Grant Date | Number of | Number of | Option | Option Expiration | Number of Stock (#) | Market Value Stock | ||||||||||||||||
Peter Altman | 4/10/2010 | 754 | (4) | — | 16.20 | 4/10/2020 | — | — | |||||||||||||||
7/5/2014 | 35,290 | (4) | — | 16.20 | 7/5/2024 | — | — | ||||||||||||||||
8/19/2016 | 87,390 | (5) | 47,055 | 16.20 | 8/19/2026 | — | — | ||||||||||||||||
2/1/2018 | 11,111 | (5) | 33,333 | 23.40 | 2/1/2028 | — | — | ||||||||||||||||
David McClung | 6/23/2014 | 2,514 | (4) | — | 16.20 | 6/23/2024 | — | — | |||||||||||||||
8/9/2016 | 5,918 | (6) | 1,255 | 16.20 | 8/9/2026 | — | — | ||||||||||||||||
8/19/2016 | 4,811 | (7) | 2,590 | 16.20 | 8/19/2026 | — | — | ||||||||||||||||
2/1/2018 | 4,417 | (5) | 13,251 | 23.40 | 2/1/2028 | — | — | ||||||||||||||||
Henricus Duckers | 8/9/2016 | 12,586 | (8) | 5,393 | 16.20 | 8/9/2026 | — | — | |||||||||||||||
8/19/2016 | 9,168 | (7) | 4,937 | 16.20 | 8/19/2026 | — | — | ||||||||||||||||
2/1/2018 | 4,663 | (5) | 13,990 | 23.40 | 2/1/2028 | — | — | ||||||||||||||||
Phil Pesta | 7/28/2011 | 6,035 | (4) | — | 16.23 | 7/28/2021 | — | — | |||||||||||||||
7/9/2013 | 251 | (4) | — | 16.23 | 7/9/2023 | — | — | ||||||||||||||||
7/5/2014 | 4,705 | (4) | — | 16.20 | 7/5/2024 | — | — | ||||||||||||||||
8/19/2016 | 6,869 | (7) | 3,698 | 16.20 | 8/19/2026 | — | — | ||||||||||||||||
2/1/2018 | 1,944 | (5) | 5,833 | 23.40 | 2/1/2028 | — | — | ||||||||||||||||
(1) | Information for this table is depicted on an award-by-award basis unless the exercise price and expiration date are identical. |
(2) | Where applicable, share numbers have been adjusted to reflect each of the Company’s reverse stock splits, which became effective pursuant to amendments to our certificate of incorporation effective on November 2, 2017 and May 7, 2019, respectively. |
(3) | This column represents the fair value of a share of our common stock on the date of grant, as determined by our board of directors. |
(4) | This option is fully vested and immediately exercisable. |
(5) | This option vests and becomes exercisable in equal monthly installments over four years from the grant date. |
(6) | This option vests and becomes exercisable in equal monthly installments over four years beginning April 28, 2016. |
(7) | This option vests and becomes exercisable in equal monthly installments over four years beginning November 24, 2016. |
(8) | This option vests and becomes exercisable in equal monthly installments over four years from the grant date, subject to a one-year cliff. |
401(k) Savings Plan
We maintain a tax-qualified retirement plan, or our 401(k) plan, that provides eligible employees with an opportunity to Mr. Kvitnitsky under this arrangement, and paid him $6,863save for the period from January 1, 2007 through May 21, 2007 (the date of sale of Accin's business to Cardo). For the period from May 21, 2007 through December 31, 2007, Accelerated Innovation (which acquired Accin's medical device business) paid $22,918 to Mr. Kvitnitsky under this arrangement, and paid him $26,933 for the six months ended June 30, 2008.
62
Derrick Romine Employment Offer Letter
Derrick Romine serves as the Chief Financial Officer of Cardo, and will serve in that same capacity for Cardo,retirement on an at-will basis pursuant to an employment offer letter dated September 5, 2008. This offer letter provides that Mr. Romine will receive an annual base salary of $180,000, a discretionary bonus of up to a maximum amount of $45,000 based on specific performance objectives tied to Cardo meeting its financial targets, and reimbursement for normal business expenses. In addition, he is entitledtax-advantaged basis. Eligible employees are able to participate in all health insuranceour 401(k) plan as of the first day of the month following the date they meet our 401(k) plan’s eligibility requirements, and employee benefits adopted by Cardo and isparticipants are able to defer up to 100% of their eligible compensation subject to accrue three weeks of vacation during his first year of employment. The offer letter also confirmed the grant of options to Mr. Romine exercisable for units of membershipapplicable annual Internal Revenue Code limits. All participants’ interests in Cardo,their deferrals are 100% vested when contributed. Our 401(k) plan permits us to make matching contributions and discretionary contributions to eligible participants.
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to securities. In accordance with SEC rules, shares of our common stock which converted intomay be acquired upon exercise of stock options which are currently exercisable or which become exercisable within 60 days of June 30, 2019 are deemed beneficially owned by the holders of such options and are deemed outstanding for the purpose of computing the percentage of ownership of such person, but are not treated as outstanding for the purpose of computing the percentage of ownership of any other person.
As of June 30, 2019 there were 4,847,471 shares of common stock of Cardo upon completion of the Merger. See the section titled "Outstanding Option Grants" below for more information regarding the options granted to Mr. Romine.
If Cardo terminates Mr. Romine's employment without "Cause" (as defined below), or if he terminates his employment without "Good Reason" (as defined below), at any time on or prior to September 4, 2010, Mr. Romine will be entitled to the following severance benefits: Cardo will pay Mr. Romine the sum of six months of his then-current monthly salary as severance payment to be paid in bi-weekly installments so long as he does not work or otherwise provide services to a competitor of Cardo during that six-month period. Fifty percent of Mr. Romine's unvested options will become fully exercisable as of the date of termination of his employment and, together with any vested options at the termination date, may be exercised pursuant to the terms thereof within 90 days of the termination date (or one year after the termination date if Mr. Romine dies during that 90-day period). The remaining unvested options at the termination date, to the extent not then presently exercisable, shall terminate as of the termination date and shall not be exercisable thereafter.
If Mr. Romine is terminated for Cause, or if he voluntarily terminates his employment or resigns from his positions with the Company without Good Reason, he will not be entitled to the Severance Benefits. As used in the offer letter, the term "Cause" means an act or omission that constitutes fraud, deceit, intentional misconduct, a knowing violation of law, recklessness or gross negligence that materially and adversely has affected or affects the business of Cardo, a material breach of any of Mr. Romine's obligations under any written agreement with Cardo, or material nonperformance of his duties to Cardo which has not been cured after 15 days' written notice from Cardo setting forth in reasonable detail the nature of the nonperformance. As used in the offer letter, the term "Good Reason" means a material breach by Cardo of any of their obligations under any written agreement with Mr. Romine, a substantial and unusual reduction in his duties, responsibilities or authority, or receipt of instructions to take actions in violation of law that has not been cured after 15 days' written notice from Mr. Romine to Cardo and setting forth in reasonable detail the nature of the action giving rise to the claim of Good Reason.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
outstanding. The following tables settable sets forth information with respect to the beneficial ownership of our outstanding common stock, as of December 14, 2009, by (i) each nominee for directorstockholder known by us to be the beneficial owner of ours, (ii) each named executive officer identified in the Summary Compensation Table, (iii) all nominees for director and nominees for executive officers as a group, and (iv) each stockholder identified as beneficially owning greatermore than 5% of our common stock. Exceptstock (our only class of voting securities), (ii) each of our directors and executive officers, and (iii) all of our directors and executive officers as a group. To the best of our knowledge, except as otherwise indicated, below, each personof the persons named in the tablestable has sole voting and investment power with respect to allthe shares of our common stock beneficially owned by thatsuch person, except to the extent that authority issuch power may be shared by spouses under applicable law.with a spouse. To our knowledge, none of the shares reportedlisted below are pledgedheld under a voting trust or similar agreement, except as security.
For purposesnoted. Other than the Merger, to our knowledge, there is no arrangement, including any pledge by any person of our securities or any of our parents, the operation of which may at a subsequent date result in a change in control of the Company. The following tables,table excludes Private Placement Units issuable upon the conversion of convertible notes held by certain stockholders that will be automatically converted at the close of this Offering. See the section titled "Certain Relationships and Related Transactions, and Director Independence−Other Transactions" for additional information.
Unless otherwise noted below, the address of each person listed on the table is c/o BioCardia, Inc., 125 Shoreway Road, Suite B, San Carlos, CA 94070.
Name and Address of Beneficial Owner | Number of | Percentage | ||||||
5% Stockholders: | ||||||||
Entities affiliated with Stertzer Family Trust(2) | 965,511 | 17.8 | % | |||||
Frost Gamma Investments Trust(3) | 1,541,702 | 28.5 | % | |||||
Named Executive Officers and Directors: | ||||||||
Peter Altman, Ph.D.(4) | 233,170 | 4.3 | % | |||||
Henricus Duckers(5) | 30,470 | * | ||||||
Fernando L. Fernandez | 2,154 | * | ||||||
Richard Krasno | 2,154 | * | ||||||
David McClung(6) | 21,197 | * | ||||||
Jay M. Moyes(7) | 5,474 | * | ||||||
Phil Pesta | 21,392 | * | ||||||
Richard C. Pfenniger, Jr. | 7,709 | * | ||||||
Thomas Quertermous, M.D. | 15,184 | * | ||||||
Simon H. Stertzer, M.D.(2) | 965,511 | 17.8 | % | |||||
Allan R. Tessler(8) | 94,171 | 1.7 | % | |||||
All directors and executive officers as a group (13 people) | 2,968,747 | 54.9 | % | |||||
* | Represents beneficial ownership of less than 1%. |
(1) | Where applicable, share numbers have been adjusted to reflect each of the Company’s reverse stock splits, which became effective on November 2, 2017 and May 7, 2019, respectively. |
(2) | Consists of (i) 475,363 shares of common stock held by the Stertzer Family Trust, (ii) 230,704 shares of our common stock held by Windrock Enterprises L.L.C., (iii) 11,656 shares of our common stock held by the Stertzer Gamma Trust, (iv) 49,877 shares our common stock held by Stertzer Holdings LLC, (v) 1,333 shares of our common stock held by Dr. Stertzer and his spouse Kimberly Stertzer, (vi) 11,394 shares subject to options that are vested and exercisable within 60 days of June 30, 2019, held by Dr. Stertzer, (vii) 92,592 shares subject to warrants held by the Stertzer Family Trust, and (viii) 92,592 shares subject to warrants held by Windrock Enterprises L.L.C. Dr. Stertzer and his spouse are co-trustees of the Stertzer Family Trust, and sole members and managers of Windrock Enterprises L.L.C., and share voting and dispositive control over the shares held by the Stertzer Family Trust and Windrock Enterprises L.L.C. Dr. Stertzer is the grantor of the Stertzer Gamma Trust and may be deemed to have voting and dispositive control over the shares held by the Stertzer Gamma Trust. Dr. Stertzer may be deemed to have voting and dispositive control over the shares held by Stertzer Holdings LLC. |
(3) | Dr. Phillip Frost is the trustee and Frost Gamma Limited Partnership is the sole and exclusive beneficiary of Frost Gamma Investments Trust. Dr. Frost is one of two limited partners of Frost Gamma Limited Partnership. The general partner of Frost Gamma Limited Partnership is Frost Gamma, Inc. and the sole shareholder of Frost Gamma, Inc. is Frost-Nevada Corporation. Dr. Frost is also the sole shareholder of Frost-Nevada Corporation. The address for these entities is 4400 Biscayne Boulevard, Suite 1500, Miami, Florida 33137. |
(4) | Consists of (i) 84,227 shares of our common stock held by Dr. Altman, and (ii) 147,943 shares subject to options vested and exercisable within 60 days of June 30, 2019. |
(5) | Consists of 30,470 shares of our common stock held by Dr. Druckers. |
(6) | Consists of 21,197 shares of our common stock held by Mr. McClung. |
(7) | Consists of 367 shares of our common stock and 5,107 shares subject to options held by Mr. Moyes that are vested and exercisable within 60 days of June 30, 2019. |
(8) | Consists of (i) 2,154 shares of common stock held by Mr. Tessler, (ii) 1,508 shares subject to options held by Mr. Tessler that are exercisable within 60 days of June 30, 2019, (iii) 64,491 shares of our common stock held by ART/FGT Family Limited Partnership, and (iv) 13,009 shares of our common stock held by International Financial Group, (v) 13,009 shares of our common stock held by The Tessler Family Limited Partnership. Mr. Tessler and his spouse are limited partners of the ART/FGT Family Limited Partnership and share voting and dispositive control over the shares held by the ART/FGT Family Limited Partnership. The address for the ART/FGT Family Limited Partnership is 2500 Moose Wilson Road, Wilson, Wyoming 83014. Mr. Tessler may be deemed to have voting and dispositive control over the shares held by the Tessler Family Limited Partnership and International Financial Group. |
Equity Compensation Plan Information
The following table summarizes our equity compensation plan information as of December 31, 2018. Information is included for equity compensation plans approved by our stockholders and equity compensation plans not approved by our stockholders. We will not grant equity awards in the future under any of the equity compensation plans not approved by our stockholders included in the table below.
Plan Category | (a) Number of | (b) Weighted | (c) Number of | |||||||||
Equity compensation plans approved by stockholders(1) | 591,749 | $ | 25.92 | 215,345 | ||||||||
Equity compensation plans not approved by stockholders(3) | 46,553 | $ | 16.20 | — | ||||||||
Total | 638,302 | $ | 25.20 | 215,345 | ||||||||
(1) | Where applicable, share numbers have been adjusted to reflect each of the Company’s reverse stock splits, which became effective on November 2, 2017 and May 7, 2019, respectively. |
(2) | The weighted average exercise price is calculated based solely on outstanding stock options. It does not take into account the shares of our common stock underlying RSUs, which have no exercise price. |
(3) | In August 2016, the Company granted an option to purchase common stock outside of the Company’s stock option plans to a consultant. |
Certain Relationships and Related Transactions, and Director Independence
Policies and Procedures for Related Party Transactions
We have adopted a personformal policy that our executive officers, directors, holders of more than 5% of any class of our voting securities, and any member of the immediate family of and any entity affiliated with any of the foregoing persons, are not permitted to enter into a related party transaction with us without the prior consent of our audit committee, or other independent members of our board of directors if it is inappropriate for our audit committee to review such transaction due to a conflict of interest. Any request for us to enter into a transaction with an executive officer, director, principal stockholder, or any of their immediate family members or affiliates, in which the amount involved exceeds the lesser of $120,000 or one percent of the average of our total assets at year-end for the last two completed fiscal years must first be presented to our audit committee for review, consideration and approval. In approving or rejecting any such proposal, our audit committee is to consider the relevant facts and circumstances available and deemed relevant to the audit committee, including, but not limited to, whether the transaction is on terms no less favorable than terms generally available to an unaffiliated third party under the same or similar circumstances and the extent of the related party’s interest in the transaction. All of the transactions described above were entered into prior to the adoption of this policy.
Related Party Transactions
We describe below transactions and series of similar transactions, since January 1, 2018, to which we were a party or will be a party, in which:
• | the amounts involved exceeded or will exceed the lesser of $120,000 or one percent of the average of our total assets at year-end for the last two completed fiscal years; and | |
• | any of our directors, nominees for director, executive officers or holders of more than 5% of our outstanding capital stock, or any immediate family member of, or person sharing the household with, any of these individuals or entities, had or will have a direct or indirect material interest. |
Other than as described below, there has not been, nor is there any currently proposed, transactions or series of similar transactions to which we have been or will be a party.
Other Transactions
We have granted stock options to our named executive officers and certain of our directors. See the section titled “Executive Compensation−Outstanding Equity Awards at 2018 Year-End” for a description of these stock options.
We have entered into change of control and severance agreements with certain of our executive officers that provides for certain severance and change in control benefits. See the section titled “Executive Compensation−Potential Payments on Termination or Change of Control.”
On December 24, 2018, the Company entered into a securities purchase agreement with entities affiliated with Dr. Simon H. Stertzer, the Chairman of our Board of Directors and a beneficial owner of more than 5% of the outstanding shares of the Company’s common stock, and Frost Gamma Investments Trust, a beneficial owner of more than 5% of the outstanding shares of the Company’s common stock (the “Investors”), relating to an offering and sale (the “2018 Offering”) of an aggregate of 592,592 shares of the Company’s common stock at a purchase price of $6.75 per share, and warrants to purchase up to one-half of the number of shares of common stock sold to an Investor, up to an aggregate for all Investors of 296,295 shares of Common Stock (the “Warrant Shares”) at an exercise price of $6.75 per share, for aggregate net proceeds of $3.8 million. The warrants will expire on December 24, 2023. The warrants contain customary adjustments and are exercisable immediately for cash and after six months will also be exercisable on a cashless basis if there is no effective registration statement registering the resale of the Warrant Shares. The Investors do not have registration rights in connection with any securities that canpurchased in the 2018 Offering. The closing of the 2018 Offering took place on December 24, 2018.
On July 5, 2019, the Company entered into a note purchase agreement pursuant to which we issued on such date $0.625 million in aggregate principal amount of convertible promissory notes, a portion of which was issued to certain of our officers and directors and a principal stockholder (or their respective affiliates) in the following amounts:
Name of Related Party | Relationship | Principal Amount | ||||
Simon H. Stertzer, MD | Chairman of the Board and greater than 5% beneficial owner | $ | 200,000 | |||
Peter Altman, Ph.D. | President, Chief Executive Officer and Director | $ | 200,000 | |||
Allan R. Tessler | Director | $ | 50,000 | |||
Ian McNiece, Ph.D. | Chief Scientific Officer | $ | 50,000 | |||
David McClung | Chief Financial Officer | $ | 50,000 | |||
Henricus Duckers, MD | Chief Medical Officer | $ | 25,000 | |||
Interest on the convertible notes accrues at the rate of 14.0% per year. The unpaid principal amount of the convertible notes, together with all interest accrued but unpaid thereon, will be acquiredautomatically converted into Private Placement Units upon the closing of this Offering at a conversion price equal to 50% of the price to the public in this Offering. Assuming an initial public offering price of $ per share, and the closing of this Offering occurs on , 2019, the $0.625 million principal amount of the outstanding convertible notes and interest thereon will convert into approximately Private Placement Units.
Indemnification Agreements
We have entered into indemnification agreements with each of our directors and executive officers. The indemnification agreements, our amended and restated certificate of incorporation and our amended and restated bylaws require us to indemnify our directors to the fullest extent permitted by that person within 60 days from December 14, 2009Delaware law.
Information related to the independence of our directors is provided under the section titled “Directors, Executive Officers and Corporate Governance.”
DESCRIPTION OF THE SECURITIES WE ARE OFFERING
In this Offering, we are offering our Units, with each Unit consisting of one share of our common stock and a warrant to purchase one share of our common stock. Our Units will not be certificated and the shares of our common stock and the warrants part of such Units are immediately separable and will be issued separately in this Offering. We are also registering the shares of common stock issuable upon exercise of the warrants. These securities are being issued pursuant to an underwriting agreement between us and the underwriters. You should review the underwriting agreement and the form of warrant, each filed as exhibits to the registration statement of which this prospectus is a part, for a complete description of the terms and conditions applicable to the warrants. The following brief summary of the material terms and provisions of the warrants is subject to, and qualified in its entirety by, the form of warrant.
Unless otherwise indicated, information presented in this section reflects a one-for-nine reverse stock split of our issued and outstanding shares of common stock, options and warrants and/that became effective May 7, 2019, and the corresponding adjustment of all common stock prices per share and stock option and warrant exercise prices per share and conversion ratios. Our common stock began trading on a reverse stock split-adjusted basis on The OTC Market on the opening of trading on June 6, 2019.
Units
We are offering our Units, with each Unit consisting of one share of our common stock and a warrant to purchase one share of our common stock, together with the shares of our common stock underlying such warrants, at an assumed public offering price of $ per Unit. Our Units will not be certificated and the shares of our common stock and the warrants part of such Units are immediately separable and will be issued separately in this Offering.
Warrants for Common Stock
The material terms and provisions of the warrants being offered pursuant to this prospectus are summarized below. This summary of some provisions of the warrants is not complete. For the complete terms of the warrants, you should refer to the form of warrant filed as an exhibit to the registration statement of which this prospectus forms a part. The warrants that are purchased in this Offering as part of the units will be issued pursuant to a separate warrant agreement to be entered into between purchasers and the Company.
The warrants issued in this Offering entitle the registered holder to purchase share of our common stock at a price equal to $ per share, based on the assumed public offering price of $ per Unit, subject to adjustment as discussed below, immediately following the issuance of such warrant and terminating at 5:00 p.m., New York City time, three (3) years after the closing of this Offering. We have applied to list each warrant on the Nasdaq Capital Market under the symbol “BCDAW.”
The warrants will be issued pursuant to a Warrant Agent Agreement between us and Continental Stock Transfer & Trust, as Warrant Agent. Certain provisions of the warrants are set forth herein but are only a summary and are qualified in their entirety by the relevant provisions of such Warrant Agent Agreement.
The exercise price and number of shares of common stock issuable upon exercise of the warrants may be adjusted in certain circumstances, including in the event of a stock dividend or recapitalization, reorganization, merger or consolidation. However, the warrants will not be adjusted for issuances of common stock at prices below its exercise price.
The warrants may be exercised upon surrender of the warrant certificate on or prior to the expiration date at the offices of the Warrant Agent, with the exercise form on the reverse side of the warrant certificate completed and executed as indicated, accompanied by full payment of the exercise price, by certified or official bank check payable to us, for the number of warrants being exercised. Under the terms of the Warrant Agreement, we must use our best efforts to maintain the effectiveness of the registration statement and current prospectus relating to common stock issuable upon exercise of the warrants until the expiration of the warrants. If we fail to maintain the effectiveness of the registration statement and current prospectus relating to the common stock issuable upon exercise of the warrants, the holders of the warrants shall have the right to exercise the warrants solely via a cashless exercise feature provided for in the warrants, until such time as there is an effective registration statement and current prospectus. The warrant holders do not have the rights or privileges of holders of common stock or any voting rights until they exercise their warrants and receive shares of common stock. After the issuance of shares of common stock upon exercise of the warrants, each holder will be entitled to one vote for each share held of record on all matters to be voted on by stockholders.
A holder may not exercise any portion of a warrant to the extent that the holder, together with its affiliates and any other convertibleperson or exercisable securities. Each beneficial owner'sentity acting as a group, would own more than 4.99% of the outstanding common stock after exercise, as such percentage ownership is determined in accordance with the terms of the warrant, except that upon prior notice from the holder to us, the holder may waive such limitation up to a percentage not in excess of 9.99%.
No fractional shares of common stock will be issued upon exercise of the warrants. If, upon exercise of the warrant, a holder would be entitled to receive a fractional interest in a share, we will, upon exercise, pay a cash adjustment in respect of such fraction in an amount equal to such fraction multiplied by assuming that options,the exercise price. If multiple warrants are exercised by the holder at the same time, we shall pay a cash adjustment in respect of such final fraction in an amount equal to such fraction multiplied by the exercise price.
Capital Stock
The Company’s authorized capital stock consists of 125,000,000 shares of capital stock, par value $0.001 per share, of which 100,000,000 shares are common stock, par value $0.001 per share and other convertible or exercisable securities that are held by that person (but not those held by any other person) and that are convertible or exercisable within25,000,000 of preferred stock, par value $0.001 per share. As of March 31, 2019, the 60-day period have been exercised. The percentage of outstanding common sharesCompany has been calculated based upon 230,293,1414,847,471 shares of common stock outstanding held by approximately 243 stockholders of record, and no shares of preferred stock outstanding. Because many of our shares of common stock are held by brokers and other institutions on December 14, 2009. Nonebehalf of stockholders, we are unable to estimate the total number of stockholders represented by these record holders.
The following is a summary of the material provisions of the common stock and preferred stock provided for in our amended and restated certificate of incorporation and amended and restated bylaws. For additional detail about our capital stock, please refer to our certificate of incorporation and amended and restated bylaws, each as amended.
Common Stock
Our board of directors is authorized, without stockholder approval, to issue additional shares of our capital stock.
Voting
Each holder of our common stock is entitled to one vote for each share on all matters submitted to a vote of the stockholders, listedincluding the election of directors. Our amended and restated certificate of incorporation and amended and restated bylaws do not provide for cumulative voting rights. Because of this absence of cumulative voting, the holders of a majority of the shares of our common stock entitled to vote in any election of directors can elect all of the directors standing for election, if they should so choose. Subject to the rights of holders of any series of preferred stock with respect to the election of directors, a director may be removed from office by our stockholders only for cause and only by the affirmative vote of the holders of at least 66 2/3% in voting power of our stock entitled to vote thereon.
The Delaware General Corporation Law provides generally that the affirmative vote of a majority of the shares entitled to vote on any matter is required to amend a corporation’s certificate of incorporation or bylaws, unless the corporation’s certificate of incorporation or bylaws, as the case may be, requires a greater percentage. The affirmative vote of the holders of at least 66 2/3% in voting power of our stock entitled to vote thereon shall be required for our stockholders to amend, alter or repeal our amended and restated bylaws.
Dividend Rights
Subject to preferences that may be applicable to any then outstanding preferred stock, holders of our common stock are entitled to receive equally on a per share basis those dividends, if any, as may be declared from time to time by our board of directors out of legally available funds. We have never declared or paid cash dividends on any of our capital stock and currently do not anticipate paying any cash dividends after this Offering or in the foreseeable future.
Rights to Receive Liquidation Distributions
In the event of our liquidation, dissolution or winding up, holders of our common stock will be entitled to share ratably in the net assets legally available for distribution to stockholders after the payment of all of our debts and other liabilities and the satisfaction of any liquidation preferences that may be granted to the holders of any then outstanding shares of preferred stock.
Rights and Preferences
Holders of our common stock have no preemptive, conversion or subscription rights, and there are no redemption or sinking fund provisions applicable to our common stock. The rights, preferences and privileges of the holders of our common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of preferred stock, which we may designate and issue in the future.
Fully Paid and Nonassessable
All of our outstanding shares of common stock are, and the shares of common stock to be issued pursuant to this Offering, when paid for, will be fully paid and nonassessable.
Preferred Stock
Shares of preferred stock may be issued from time to time in one or more series, each of which will have such distinctive designation or title as shall be determined by our board of directors prior to the issuance of any shares thereof. Our board of directors may designate the rights, preferences, privileges and restrictions of the preferred stock, including dividend rights, conversion rights, voting rights, redemption rights, liquidation preference, sinking fund terms, and the number of shares constituting any series or the designation of any series. The issuance of preferred stock could have the effect of restricting dividends on our common stock, diluting the voting power of our common stock, impairing the liquidation rights of our common stock, or delaying, deterring, or preventing a change in control. Such issuance could have the effect of decreasing the market price of our common stock. No shares of preferred stock are outstanding, and we currently have no plans to issue any shares of preferred stock.
Stock Options
As of March 31, 2019, there were 606,249 shares of our common stock issuable upon exercise of outstanding stock options, at a weighted-average exercise price of $24.39 per share.
Effect of Certain Provisions of our Amended and Restated Certificate of Incorporation and Bylaws
Our amended and restated certificate of incorporation and amended and restated bylaws contain provisions that could have the effect of delaying, deferring, or discouraging another party from acquiring control of us. These provisions and certain provisions of Delaware law, which are summarized below, could discourage takeovers, coercive or otherwise. These provisions are also designed, in part, to encourage persons seeking to acquire control of us to negotiate first with our board of directors. We believe that the benefits of increased protection of our potential ability to negotiate with an unfriendly or unsolicited acquirer outweigh the disadvantages of discouraging a proposal to acquire us.
Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws
Our amended and restated certificate of incorporation and our amended and restated bylaws provide for the following:
● | Undesignated Preferred Stock. The ability to authorize undesignated preferred stock makes it possible for our board of directors to issue one or more series of preferred stock with voting or other rights or preferences that could impede the success of any attempt to change control of our company. These and other provisions may have the effect of deferring hostile takeovers or delaying changes in control or management of our company. |
● | Limits on Ability of Stockholders to Act by Written Consent or Call a Special Meeting. Our amended and restated certificate of incorporation provides that our stockholders may not act by written consent except in limited circumstances. In addition, our amended and restated certificate of incorporation requires that special meetings of the stockholders be called only by our board of directors, our chief executive officer or our president (in the absence of a chief executive officer). This limit on the ability of stockholders to act by written consent or call a special meeting may lengthen the amount of time required to take stockholder proposed actions. |
In addition, our amended and restated certificate of incorporation and amended and restated bylaws provide that special meetings of the stockholders may be called only by the chairperson of the board, the chief executive officer, the president (in the absence of a chief executive officer), or our board of directors. A stockholder may not call a special meeting, which may delay the ability of our stockholders to force consideration of a proposal or for holders controlling a majority of our capital stock to take any action, including the removal of directors.
● | Requirements for Advance Notification of Stockholder Nominations and Proposals. Our amended and restated bylaws establish advance notice procedures with respect to stockholder proposals and the nomination of candidates for election as directors, other than nominations made by or at the direction of our board of directors or a committee of the board of directors. |
● | Board Classification. Our board of directors is divided into three classes. The directors in each class are elected to serve for a three-year term, one class being elected each year by our stockholders. This system of electing and removing directors may tend to discourage a third party from making a tender offer or otherwise attempting to obtain control of us, because it generally makes it more difficult for stockholders to replace a majority of the directors. |
Anti-Takeover Effects of Delaware Law
Certain provisions of Delaware law contain provisions that could have the effect of delaying, deferring or discouraging another party from acquiring control of us. These provisions, which are summarized below, are expected to discourage certain types of coercive takeover practices and inadequate takeover bids. These provisions are also designed in part to encourage anyone seeking to acquire control of us to first negotiate with our board of directors. We believe that the advantages gained by protecting our ability to negotiate with any options,unsolicited and potentially unfriendly acquirer outweigh the disadvantages of discouraging such proposals, including those priced above the then-current market value of our common stock, because, among other reasons, the negotiation of such proposals could improve their terms.
Delaware Anti-Takeover Statute
We are subject to the provisions of Section 203 of the Delaware General Corporation Law regulating corporate takeovers. In general, Section 203 prohibits a publicly-held Delaware corporation from engaging, under certain circumstances, in a business combination with an interested stockholder for a period of three years following the date the person became an interested stockholder unless:
● | prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder; |
● | upon completion of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the voting stock outstanding, but not for determining the outstanding voting stock owned by the interested stockholder, (1) shares owned by persons who are directors and also officers and (2) shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or |
● | at or subsequent to the date of the transaction, the business combination is approved by the board of directors of the corporation and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock which is not owned by the interested stockholder. |
Generally, a business combination includes a merger, asset or stock sale, or other transaction resulting in a financial benefit to the interested stockholder. An interested stockholder is a person who, together with affiliates and associates, owns or, within three years prior to the determination of interested stockholder status, did own 15% or more of a corporation’s outstanding voting stock. We expect the existence of this provision to have an anti-takeover effect with respect to transactions our board of directors does not approve in advance. We also anticipate that Section 203 may discourage business combinations or other attempts that might result in a premium over the market price for the shares of our common stock held by our stockholders.
The provisions of Delaware law could have the effect of discouraging others from attempting hostile takeovers and, as a consequence, they may also inhibit temporary fluctuations in the market price of our common stock that often result from actual or rumored hostile takeover attempts. These provisions may also have the effect of preventing changes in our management. It is possible that these provisions could make it more difficult to accomplish transactions that our stockholders may otherwise deem to be in their best interests.
Transfer Agent, Registrar and Warrant Agent
The transfer agent and registrar for our common stock and the warrant agent for the warrants is Continental Stock Transfer & Trust. The address of Continental Stock Transfer & Trust is 1 State Street 30th Floor, New York, New York 10004-1561. Shares of our common stock and warrants will be issued in uncertificated form only, subject to limited circumstances.
Market Listing
Our common stock trades on the OTCQB tier of OTC Markets Group, Inc. under the symbol “BCDA.” We have applied to list our common stock and the offered warrants on the Nasdaq Capital Market under the symbols “BCDA” and “BCDAW,” respectively. No assurance can be given that such listings will be approved or that a trading market will develop for the common stock and/or the warrants.
MATERIAL U.S. FEDERAL INCOME TAX CONSIDERATIONS FOR HOLDERS OF OUR COMMON STOCK AND WARRANTS
The following discussion describes the material U.S. federal income tax consequences of the acquisition, ownership and disposition of our common stock and warrants acquired in this Offering. This discussion is based on the current provisions of the Internal Revenue Code of 1986, as amended, or the Code, existing and proposed U.S. Treasury regulations promulgated thereunder, and administrative rulings and court decisions in effect as of the date hereof, all of which are subject to change at any time, possibly with retroactive effect. No ruling has been or will be sought from the Internal Revenue Service, or IRS, with respect to the matters discussed below, and there can be no assurance the IRS will not take a contrary position regarding the tax consequences of the acquisition, ownership or disposition of our common stock and warrants, or that any such contrary position would not be sustained by a court.
We assume in this discussion that the shares of our common stock and warrants will be held as capital assets (generally, property held for investment). This discussion does not address all aspects of U.S. federal income taxes, does not discuss the potential application of the Medicare contribution tax on net investment income or the alternative minimum tax and does not deal with state or local taxes, U.S. federal gift and estate tax laws, except as specifically provided below with respect to non-U.S. holders, or any non-U.S. tax consequences that may be relevant to holders in light of their particular circumstances. This discussion also does not address the special tax rules applicable to particular holders, such as
● | financial institutions; | |
● | brokers or dealers in securities or currencies, or traders in securities that elect to use a mark-to-market method of accounting for their securities holdings; | |
● | tax-exempt organizations; | |
● | pension plans; | |
● | regulated investment companies, real estate investment trusts; | |
● | owners that hold our common stock or warrants as part of a straddle, hedge, conversion transaction, synthetic security or other integrated investment; | |
● | insurance companies; | |
● | persons that own, or are deemed to own, more than 5% of our capital stock (except to the extent specifically set forth below); | |
● | entities or arrangements treated as partnerships for U.S. federal income tax purposes and other pass-through entities (and partners or other investors therein); | |
● | controlled foreign corporations, passive foreign investment companies, or corporations that accumulate earnings to avoid U.S. federal income tax; | |
● | persons subject to special tax accounting rules as a result of any item of gross income with respect to our common stock or warrants being taken into account in an applicable financial statement; | |
● | persons deemed to sell our common stock or warrants under the constructive sale provisions of the Code; and | |
● | certain U.S. expatriates and certain former citizens or long-term residents of the United States. |
In addition, this discussion does not address the tax treatment of partnerships or other derivative securitiespass-through entities or persons who hold our common stock or warrants through partnerships or other entities that are pass-through entities for U.S. federal income tax purposes. A partner in a partnership or other pass-through entity that will hold our common stock or warrants should consult his, her or its own tax advisor regarding the tax consequences of the ownership and disposition of our common stock or warrants through a partnership or other pass-through entity, as applicable.
This discussion of U.S. federal income tax considerations is for general information purposes only and is not tax advice. Prospective investors should consult their own tax advisors regarding the U.S. federal, state, local and non-U.S. income and other tax considerations of acquiring, holding and disposing of our common stock and warrants.
For the purposes of this discussion, a “U.S. Holder” means a beneficial owner of our common stock or warrants that is, for U.S. federal income tax purposes: (a) an individual who is a citizen or resident of the United States, (b) a corporation (or other entity taxable as a corporation for U.S. federal income tax purposes), created or organized in or under the laws of the United States, any state thereof or the District of Columbia, (c) an estate the income of which is subject to U.S. federal income taxation regardless of its source, or (d) a trust if it (1) is subject to the primary supervision of a court within the United States and one or more U.S. persons (within the meaning of Section 7701(a)(30) of the Code) have the authority to control all substantial decisions of the trust or (2) has a valid election in effect under applicable U.S. Treasury regulations to be treated as a U.S. person. A “Non-U.S. Holder” is, for U.S. federal income tax purposes, a beneficial owner of common stock or warrants that is neither a U.S. Holder nor an entity or arrangement treated as a partnership for U.S. federal income tax purposes.
Allocation of Purchase Price
For U.S. federal income tax purposes, the shares of common stock and warrants acquired in this Offering will be treated as an “investment unit” consisting of one share of common stock and a warrant to acquire one share of our common stock. The purchase price for each investment unit will be allocated between these two components in proportion to their relative fair market values at the time the unit is purchased by the holder. This allocation of the purchase price for each unit will establish the holder’s initial tax basis for U.S. federal income tax purposes in the share of common stock and the warrant included in each unit. The separation of the share of common stock and the warrant included in each unit should not be a taxable event for U.S. federal income tax purposes. Each holder should consult his, her or its own tax advisor regarding the allocation of the purchase price for a unit.
Tax Considerations Applicable to U.S. Holders
Exercise and Expiration of Warrants
In general, a U.S. Holder will not recognize gain or loss for U.S. federal income tax purposes upon exercise of a warrant. The U.S. Holder will take a tax basis in the shares acquired on the exercise of a warrant equal to the exercise price of the warrant, increased by the U.S. Holder’s adjusted tax basis in the warrant exercised (as determined pursuant to the rules discussed above). The U.S. Holder’s holding period in the shares of our common stock acquired on exercise of the warrant will begin on the date of exercise of the warrant, and will not include any period for which the U.S. Holder held the warrant.
In certain limited circumstances, a U.S. Holder may be permitted to undertake a cashless exercise of warrants into our common stock. The U.S. federal income tax treatment of a cashless exercise of warrants into our common stock is unclear, and the tax consequences of a cashless exercise could differ from the consequences upon the exercise of a warrant described in the preceding paragraph. U.S. Holders should consult their own tax advisors regarding the U.S. federal income tax consequences of a cashless exercise of warrants.
The lapse or expiration of a warrant will be treated as if the U.S. Holder sold or exchanged the warrant and recognized a capital loss equal to the U.S. Holder's tax basis in the warrant. The deductibility of capital losses is subject to limitations.
Certain Adjustments to and Distributions on Warrants
Under Section 305 of the Code, an adjustment to the number of shares of common stock issued on the exercise of the warrants or an adjustment to the exercise price of the warrants may be treated as a constructive distribution to a U.S. Holder of the warrants if, and to the extent that, such adjustment has the effect of increasing such U.S. Holder’s proportionate interest in our “earnings and profits” or assets, depending on the circumstances of such adjustment (for example, if such adjustment is to compensate for a distribution of cash or other property to our shareholders). An adjustment made pursuant to a bona fide reasonable adjustment formula that has the effect of preventing dilution should generally not be considered to result in a constructive distribution. Any constructive distribution would be taxable whether or not there is an actual distribution of cash or other property to the holders of warrants. In certain circumstances, if we were to make a distribution in cash or other property with respect to our common stock after the issuance of the warrants, then we may make a corresponding distribution to the holders of the warrants. The taxation of a distribution received with respect to a warrant is unclear. It is possible such a distribution would be treated as a distribution (or constructive distribution), although other treatments are possible. For more information regarding the U.S. federal income tax considerations related to distributions, see the discussion below regarding “—Distributions.” U.S. Holders should consult their tax advisors regarding the proper treatment of any adjustments to the warrants and any distributions with respect to the warrants.
Distributions
As discussed above, we currently anticipate that we will retain future earnings, if any, to finance the growth and development of our business and do not intend to pay cash dividends in respect of our common stock in the foreseeable future. In the event that we do make distributions on our common stock to a U.S. Holder, those distributions generally will constitute dividends for U.S. tax purposes to the extent paid out of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles). Distributions in excess of our current and accumulated earnings and profits will constitute a return of capital that is applied against and reduces, but not below zero, a U.S. Holder’s adjusted tax basis in our common stock. Any remaining excess will be treated as gain realized on the sale or exchange of our common stock as described below under the section titled “– Disposition of Our Common Stock or Warrants.” Under current law, if certain requirements are met, a preferential U.S. federal income tax rate will apply to any dividends paid to the beneficial owner of our common stock who is an individual U.S. Holder and meets certain holding period requirements.
Distributions constituting dividends for U.S. federal income tax purposes that are convertiblemade to U.S. Holders that are corporate shareholders may qualify for the dividends received deduction, or exercisable within 60 daysDRD, which is generally available to corporate shareholders. No assurance can be given that we will have sufficient earnings and profits (as determined for U.S. federal income tax purposes) to cause any distributions to be eligible for a DRD. In addition, a DRD is available only if certain holding periods and other taxable income requirements are satisfied.
Disposition of Our Common Stock or Warrants
Upon a sale or other taxable disposition of our common stock or warrants, a U.S. Holder generally will recognize capital gain or loss in an amount equal to the difference between the amount realized and the U.S. Holder’s adjusted tax basis in the common stock or warrants. Capital gain or loss will constitute long-term capital gain or loss if the U.S. Holder’s holding period for the common stock or warrants exceeds one year. The deductibility of capital losses is subject to certain limitations. U.S. Holders who recognize losses with respect to a disposition of our common stock or warrants should consult their own tax advisors regarding the tax treatment of such losses.
Information Reporting and Backup Withholding
Information reporting requirements generally will apply to payments of dividends (including constructive dividends) on the common stock and warrants and to the proceeds of a sale or other disposition of common stock and warrants paid by us to a U.S. Holder unless such U.S. Holder is an exempt recipient, such as a corporation. Backup withholding will apply to those payments if the U.S. Holder fails to provide the holder's taxpayer identification number, or certification of exempt status, or if the holder otherwise fails to comply with applicable requirements to establish an exemption.
Backup withholding is not an additional tax. Rather, any amounts withheld under the backup withholding rules will be allowed as a refund or a credit against the U.S. Holder’s U.S. federal income tax liability provided the required information is timely furnished to the IRS. U.S. Holders should consult their own tax advisors regarding their qualification for exemption from December 14, 2009.information reporting and backup withholding and the procedure for obtaining such exemption.
63
Shares of Common Stock Beneficially Owned Amount and Nature of Directors and Officers Beneficial Ownership Percent of Class Andrew A. Brooks, M.D. 61,868,189 26.87 % Mikhail Kvitnitsky 28,956,653 12.57 % Joseph Loggia 8,000 * Thomas H. Morgan 7,863,615 3.42 % Ronald N. Richards, Esq. 675,205 * Derrick Romine 771,941 * Steven D. Rubin 53,197 * Subbarao Uppaluri, Ph.D. 346,967 * All directors and executive officers as a group (8 persons) 100,543,767 43.66 % *Indicates ownership of less than 1%.
Shares of Common Stock Beneficially Owned Number and Nature of 5% or More Shareholders (1) Beneficial Ownership Percent of Class Frost Gamma Investments Trust (2) 33,250,911 14.44 %
Exercise and Expiration of Warrants
(1) Based on information in separate Schedule 13D dated September 8, 2008, Andrew A. Brooks, M.D. and Michael Kvitnitsky also are 5%
In general, a Non-U.S. Holder will not recognize gain or more stockholders. The business addressloss for U.S. federal income tax purposes upon the exercise of Andrew A. Brooks and Michael Kvitnitsky is 9701 Wilshire Boulevard, Suite 1100, Beverly Hills, CA 90212.
(2) Based on information in Amendment No. 2 to Schedule 13D dated December 8, 2009, Frost Gamma Investments Trust holds 33,250,911warrants into shares of common stock. The U.S. federal income tax treatment of a cashless exercise of warrants into our common stock is unclear. A Non-U.S. Holder should consult his, her, or its own tax advisor regarding the U.S. federal income tax consequences of a cashless exercise of warrants.
The expiration of a warrant will be treated as if the Non-U.S. Holder sold or exchanged the warrant and recognized a capital loss equal to the Non-U.S. Holder’s tax basis in the warrant. However, a Non-U.S. Holder will not be able to utilize a loss recognized upon expiration of a warrant against the Non-U.S. Holder’s U.S. federal income tax liability unless the loss is effectively connected with the Non-U.S. Holder’s conduct of a trade or business addresswithin the United States (and, if an income tax treaty applies, is attributable to a permanent establishment or fixed base in the United States) or is treated as a U.S.-source loss and the Non-U.S. Holder is present 183 days or more in the taxable year of Frost Gamma Investments Trustdisposition and certain other conditions are met.
Certain Adjustments to and Distributions on Warrants
As described under “—U.S. Holders –Certain Adjustments to and Distributions on Warrants,” an adjustment to the warrants could result in a constructive distribution to a Non-U.S. Holder, which would be treated as described under “—Distributions” below, and the tax treatment of distributions on the warrants is 4400 Biscayne Boulevard, Suite 1500, Miami, Florida 33137. Phillip Frost, M.D.unclear. Any resulting withholding tax attributable to deemed dividends would be collected from other amounts payable or distributable to the Non-U.S. Holder. Non-U.S. Holders should consult their tax advisors regarding the proper treatment of any adjustments to and distributions on the warrants.
Distributions
As discussed above, we currently anticipate that we will retain future earnings, if any, to finance the growth and development of our business and do not intend to pay cash dividends in respect of our common stock in the foreseeable future. In the event that we do make distributions on our common stock to a Non-U.S. Holder, those distributions generally will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as described in “—U.S. Holders – Distributions.”
Any distribution (including constructive distributions) on our common stock that is the trustee and Frost Gamma Limited Partnership is the sole and exclusive beneficiary of Frost Gamma Investments Trust.
64
Outstanding Option Grants
In August 2008, Cardo LLC issued options exercisable for units of membership interests in Cardo LLC with an exercise price of $147,625 per unit (whichtreated as a dividend paid to a Non-U.S. Holder that is not less thaneffectively connected with the fair market valueholder’s conduct of a trade or business in the United States will generally be subject to withholding tax at a 30% rate or such lower rate as may be specified by an applicable income tax treaty between the United States and the Non-U.S. Holder’s country of residence. To obtain a reduced rate of withholding under a treaty, a Non-U.S. Holder generally will be required to provide the applicable withholding agent with a properly executed IRS Form W-8BEN, IRS Form W-8BEN-E or other appropriate form, certifying the Non-U.S. Holder’s entitlement to benefits under that treaty. Such form must be provided prior to the payment of dividends and must be updated periodically. If a Non-U.S. Holder holds stock through a financial institution or other agent acting on the dateholder's behalf, the holder will be required to provide appropriate documentation to such agent. The holder’s agent may then be required to provide certification to the applicable withholding agent, either directly or through other intermediaries. If you are eligible for a reduced rate of grant)U.S. withholding tax under an income tax treaty, you should consult with your own tax advisor to determine if you are able to obtain a refund or credit of any excess amounts withheld by timely filing an appropriate claim for a refund with the IRS.
We generally are not required to withhold tax on dividends paid (or constructive dividends deemed paid) to a Non-U.S. Holder that are effectively connected with the holder’s conduct of a trade or business within the United States (and, if required by an applicable income tax treaty, are attributable to a permanent establishment or fixed base that the holder maintains in the United States) if a properly executed IRS Form W-8ECI, stating that the dividends are so connected, is furnished to us (or, if stock is held through a financial institution or other agent, to the applicable withholding agent). Each option hasIn general, such effectively connected dividends will be subject to U.S. federal income tax on a termnet income basis at the regular graduated rates applicable to U.S. persons. A corporate Non-U.S. Holder receiving effectively connected dividends may also be subject to an additional “branch profits tax,” which is imposed, under certain circumstances, at a rate of 10 years and vests in equal installments over a five-year period commencing30% (or such lower rate as may be specified by an applicable treaty) on the first anniversarycorporate Non-U.S. Holder’s effectively connected earnings and profits, subject to certain adjustments.
See also the sections below titled “—Backup Withholding and Information Reporting” and “—Foreign Accounts” for additional withholding rules that may apply to dividends paid to certain foreign financial institutions or non-financial foreign entities.
Disposition of our Common Stock or Warrants
Subject to the discussions below under the sections titled “—Backup Withholding and Information Reporting” and “—Foreign Accounts” a Non-U.S. Holder generally will not be subject to U.S. federal income or withholding tax with respect to gain realized on a sale or other disposition of our common stock or warrants unless:
● | the gain is effectively connected with the Non-U.S. Holder’s conduct of a trade or business in the United States, and if an applicable income tax treaty so provides, the gain is attributable to a permanent establishment or fixed base maintained by the Non-U.S. Holder in the United States; in these cases, the Non-U.S. Holder will be taxed on a net income basis at the regular graduated rates and in the manner applicable to U.S. persons, and if the Non-U.S. Holder is a corporation, an additional branch profits tax at a rate of 30%, or a lower rate as may be specified by an applicable income tax treaty, may also apply; |
● | the Non-U.S. Holder is a nonresident alien present in the United States for 183 days or more in the taxable year of the disposition and certain other requirements are met, in which case the Non-U.S. Holder will be subject to a 30% tax (or such lower rate as may be specified by an applicable income tax treaty between the United States and such holder's country of residence) on the net gain derived from the disposition, which may be offset by certain U.S.-source capital losses of the Non-U.S. Holder, if any; or |
● | our common stock constitutes a U.S. real property interest because we are, or have been at any time during the five-year period preceding such disposition (or the Non-U.S. Holder’s holding period for the common stock or warrants, if shorter), a “U.S. real property holding corporation,” unless our common stock is regularly traded on an established securities market and the Non-U.S. Holder held no more than 5% of our outstanding common stock, directly or indirectly, during the shorter of the five-year period ending on the date of the disposition or the period that the Non-U.S. Holder held our common stock. Special rules may apply to the determination of the 5% threshold in the case of a holder of a warrant. Non-U.S. Holders are urged to consult their own tax advisors regarding the effect of holding our warrants on the calculation of such 5% threshold. Generally, a corporation is a “U.S. real property holding corporation” if the fair market value of its “U.S. real property interests” (as defined in the Code and applicable regulations) equals or exceeds 50% of the sum of the fair market value of its worldwide real property interests plus its other assets used or held for use in a trade or business. Although there can be no assurance, we believe that we are not currently, and we do not anticipate becoming, a “U.S. real property holding corporation” for U.S. federal income tax purposes. No assurance can be provided that our common stock will be regularly traded on an established securities market for purposes of the rules described above. Non-U.S. Holders are urged to consult their own tax advisors regarding the U.S. federal income tax considerations that could result if we are, or become, a “U.S. real property holding corporation.” |
Federal Estate Tax
Common stock owned or treated as owned by an individual who is not a citizen or resident of the dateUnited States (as specially defined for U.S. federal estate tax purposes) at the time of grant. In connection withdeath will be included in the Merger, these options converted into options exercisableindividual's gross estate for U.S. federal estate tax purposes and, therefore, may be subject to U.S. federal estate tax, unless an applicable estate tax or other treaty provides otherwise. The foregoing may also apply to warrants. A Non-U.S. Holder should consult his, her, or its own tax advisor regarding the U.S. federal estate tax consequences of the ownership or disposition of shares of our common stock with an exercise priceand warrants.
Backup Withholding and Information Reporting
We must report annually to the IRS and to each Non-U.S. Holder the gross amount of $0.22126 per share (which is not less than the fair market valuedistributions (including constructive distributions) on our common stock or warrants paid to such holder and the date of grant). The following table provides informationtax withheld, if any, with respect to (i)such distributions. Non-U.S. Holders may have to comply with specific certification procedures to establish that the nameholder is not a U.S. person (as defined in the Code) in order to avoid backup withholding at the applicable rate, currently 24%, with respect to dividends (or constructive dividends) on our common stock or warrants. Generally, a holder will comply with such procedures if it provides a properly executed IRS Form W-8BEN, IRS Form W-8BEN-E (or other applicable IRS Form W-8) or otherwise meets documentary evidence requirements for establishing that it is a Non-U.S. Holder, or otherwise establishes an exemption. Dividends paid to Non-U.S. Holders subject to withholding of U.S. federal income tax, as described above under the heading “—Dividends,” will generally be exempt from U.S. backup withholding.
Information reporting and relationbackup withholding generally will apply to the proceeds of a disposition of our common stock or warrants by a Non-U.S. Holder effected by or through the U.S. office of any broker, U.S. or foreign, unless the holder certifies its status as a Non-U.S. Holder and satisfies certain other requirements, or otherwise establishes an exemption. Generally, information reporting and backup withholding will not apply to a payment of disposition proceeds to a Non-U.S. Holder where the transaction is effected outside the United States through a non-U.S. office of a broker. However, for information reporting purposes, dispositions effected through a non-U.S. office of a broker with substantial U.S. ownership or operations generally will be treated in a manner similar to dispositions effected through a U.S. office of a broker. Non-U.S. Holders should consult their own tax advisors regarding the application of the optioneeinformation reporting and backup withholding rules to Cardo, (ii) the numberthem.
Copies of units of membership interests in Cardo thatinformation returns may be acquired pursuantmade available to an exercisethe tax authorities of the options,country in which the Non-U.S. Holder resides or is incorporated under the provisions of a specific treaty or agreement.
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules from a payment to a Non-U.S. Holder can be refunded or credited against the Non-U.S. Holder’s U.S. federal income tax liability, if any, provided that an appropriate claim is timely filed with the IRS.
Foreign Accounts
The Foreign Account Tax Compliance Act, or FATCA, generally imposes a 30% withholding tax on dividends (including constructive dividends) on, and (unless otherwise provided by the Treasury Secretary) gross proceeds from the sale or other disposition of, our common stock and warrants if paid to a non-U.S. entity unless (i) if the non-U.S. entity is a “foreign financial institution,” the non-U.S. entity undertakes certain due diligence, reporting, withholding, and certification obligations, (ii) if the non-U.S. entity is a “non-financial foreign entity,” the entity provides the withholding agent with either a certification that it does not have any substantial direct or indirect U.S. owners or provides information regarding direct and indirect U.S. owners of the entity, or (iii) the non-U.S. entity is otherwise exempt under FATCA.
Withholding under FATCA generally (1) applies to payments of dividends (including constructive dividends) on our common stock and warrants and (2) unless otherwise provided by the Treasury Secretary, will apply to payments of gross proceeds from a sale or other disposition of our common stock and warrants. The Treasury Secretary has issued proposed Treasury Regulations, which, if finalized in their present form, would eliminate withholding under FATCA with respect to payment of gross proceeds from a sale or other disposition of our common stock and warrants. In its preamble to such proposed Treasury Regulations, the U.S. Treasury stated that taxpayers may generally rely on the proposed Treasury Regulations until final regulations are issued. An intergovernmental agreement between the United States and an applicable foreign country may modify the requirements described in this section. Under certain circumstances, a holder may be eligible for refunds or credits of the tax. Holders should consult their own tax advisors regarding the possible implications of FATCA on their investment in our common stock or warrants.
The preceding discussion of material U.S. federal tax considerations is for information only. It is not tax advice. Prospective investors should consult their own tax advisors regarding the particular U.S. federal, state, local and non-U.S. tax consequences of purchasing, holding and disposing of our common stock or warrants, including the consequences of any proposed changes in applicable laws.
Shares Available for Future Sales
Future sales of our common stock in the public market, or the availability of such shares for sale in the public market, could adversely affect market prices prevailing from time to time. As described below, the sale of a portion of our shares will be limited after this Offering due to contractual and legal restrictions on resale. Nevertheless, sales of our common stock in the public market after such restrictions, lapse, or the perception that those sales may occur, could adversely affect the prevailing market price at such time and our ability to raise equity capital in the future.
Based on the number of shares of Cardo'sour common stock that mayoutstanding as of , 2019, upon the completion of this Offering, shares of our common stock will be acquired pursuant to anoutstanding, assuming Units are issued in this Offering and assuming no exercise of the options,underwriters’ overallotment option, or shares of our common stock will be outstanding, assuming Units are issued in each casethis Offering and the underwriters’ over-allotment option is exercised in full.
Except for shares subject to lock-up agreement, substantially all of our outstanding shares will be freely tradable except that any shares held by our affiliates, as that term is defined in Rule 144 under the Securities Act, may only be sold in compliance with the limitations described below.
Rule 144
In general, under Rule 144 of the Securities Act, as in effect on the date of this prospectus, any person who is not our affiliate at any time during the preceding three months, and who has beneficially owned the relevant shares of our common stock for at least six months, including the holding period of any prior owner other than one of our affiliates, would be entitled to sell an unlimited number of shares of our common stock into the public markets provided current public information about us is available, and, after owning such shares for at least one year, including the holding period of any prior owner other than one of our affiliates, would be entitled to sell an unlimited number of shares of our common stock into the public markets without restriction.
A person who is our affiliate or who was our affiliate at any time during the preceding three months, and who has beneficially owned restricted securities for at least six months, including the affiliates, is entitled to sell within any three-month period a number of shares that does not exceed the greater of:
1% of the number of shares of our common stock then outstanding, which will equal approximately shares, or approximately shares if the underwriters exercise their over-allotment option in full, immediately following this Offering, based on the number of shares of our common stock outstanding as of , 2019; or
the average weekly trading volume of our common stock during the four calendar weeks preceding the filing of a Form 144 notice by such person with respect to such sale, if our class of common stock is listed on Nasdaq, the New York Stock Exchange, or the NYSE American.
Sales under Rule 144 by our affiliates are also subject to certain manner of sale provisions and notice requirements and to the availability of current public information about us.
Lock-up Agreements
See the section titled “Underwriting” below for a detailed discussion.
UNDERWRITING
Subject to the terms and conditions of the underwriting agreement, the underwriters named below, through their representative, Maxim Group LLC, referred to herein as Maxim, have severally agreed to purchase from us on a firm commitment basis the following respective number of units at a public offering price less the underwriting discounts and commissions set forth on the cover page of this prospectus:
Name | Number of | |||
Maxim Group LLC | ||||
| Arcadia Securities, LLC | ||||
| Dawson James Securities, Inc. | ||||
Total: | ||||
The underwriting agreement provides that the obligation of the underwriters to purchase all of the units being offered to the public is subject to specific conditions, including the absence of any material adverse change in our business or in the financial markets and the receipt of certain legal opinions, certificates and letters from us, our counsel and the independent auditors. Subject to the terms of the options:underwriting agreement, the underwriters will purchase all of the units being offered to the public, other than those covered by the over-allotment option described below, if any of these units are purchased.
Over-Allotment Option
We have granted to the underwriters an option, exercisable not later than 45 days after the effective date of the registration statement, to purchase up to an additional shares of common stock and/or warrants to purchase up to shares of common stock solely to cover overallotments, if any, at the price to the public less the underwriting discounts and commissions set forth on the cover of this prospectus. To the extent that the underwriters exercise this option, each of the underwriters will become obligated, subject to conditions, to purchase approximately the same percentage of these additional shares of common stock and/or warrants as the number of units to be purchased by it in the above table bears to the total number of units offered by this prospectus. We will be obligated, pursuant to the option, to sell these additional shares of common stock and/or warrants to the underwriters to the extent the option is exercised. If any additional shares of common stock and/or warrants are purchased, the underwriters will offer the additional shares of common stock and/or warrants on the same terms as those on which the other units are being offered hereunder.
Commissions and Expenses
The underwriting discounts and commissions are 7% of the public offering price for investors directly introduced by Maxim (either in-person or via telephone) (the “Introduced Investors”) and 4.0% for all other investors that participate in this Offering other than the Introduced Investors. The 4.0% fee shall apply only up to a maximum amount of $3,000,000 independent of the quantum of funds raised by investors who are not Introduced Investors.
In addition, we have agreed to reimburse Maxim for out-of-pocket accountable expenses it incurs in connection with this Offering for up to $ , including, up to $10,000 for “road show” expenses, and the fees and disbursements of Maxim’s counsel up to a maximum of $95,000. We have paid $15,000 to Maxim as an advance to be applied towards out-of-pocket accountable expenses (the “Advance”). Any portion of the Advance shall be returned back to us to the extent not actually incurred.
We have been advised by the representative of the underwriters that the underwriters propose to offer the units to the public at the public offering price set forth on the cover of this prospectus and to dealers at a price that represents a concession not in excess of $ per unit under the public offering price of $ per unit. The underwriters may allow, and these dealers may re-allow, a concession of not more than $ per unit to other dealers. After the public offering, the representative of the underwriters may change the offering price and other selling terms.
The following table shows the per-unit and total public offering price, underwriting discounts and commissions, and proceeds before expenses to us. These amounts are shown assuming both no exercise and full exercise of the underwriters’ over-allotment option.
Total | ||||||||||||
Per unit | Without Over- Allotment | With Full Over- Allotment | ||||||||||
Public offering price | $ | |||||||||||
Underwriting discounts and | $ | |||||||||||
Proceeds, before expenses, to us | $ | |||||||||||
| $ | ||||||||||||
Equity Compensation Plan Information
The following table summarizesestimated offering expenses payable by us, exclusive of underwriting discounts and commissions, are approximately $ .
Representative Warrants
We have also agreed to issue to Maxim (or its permitted assignees) the warrants to purchase a number of outstanding options granted to employees, service providers and directors under the Company's compensation plans and arrangements as of the quarter ended September 30, 2009. For a description of equity compensation plans not approved by security holders see Note 12 to the Consolidated Financial Statements for the year ended December 31, 2008 included elsewhere in this registration statement.
| Number of securities | Number of securities | ||||||
| to be issued upon | Weighted-average | remaining available | |||||
| exercise of | exercise price of | for future issuance | |||||
| outstanding options, | outstanding options, | under equity | |||||
| warrants and rights | warrants and rights | compensation plans | |||||
| Equity compensation plans | |||||||
| not approved by security holders | 2,358,400 | $ 0.23 | - | ||||
| Equity compensation plans | |||||||
| approved by security holders | - | - | - | ||||
| Total | 2,358,400 | $ 0.23 | - |
65
TRANSACTIONS WITH RELATED PERSONS, PROMOTERS AND CERTAIN CONTROL PERSONS
The Company appointed Ladenburg as the Company's exclusive placement agent for the private placement offering that closed in October and November 2009. Dr. Phillip Frost, through Frost Gamma Investments Trust, is a common principal stockholder of both the Company and of Ladenburg. Furthermore, certain senior managers of the placement agent are stockholders of the Company. After reviewing all information related to the transaction between the Company and Ladenburg, a potential related party transaction, the Company's Audit Committee approved the related party transaction. Ladenburg, as the placement agent, for acting in such capacity for theour shares of common stock offeredup to an aggregate of 3.5% of the total number of shares sold to retail investors in the private placement offering, received: (i) a cash commissionthis Offering (“Representative’s Warrants”). The Representative’s Warrants will have an exercise price equal to eight percent (8%) of the gross proceeds from the offering that was received from Approved Investors; (ii) a cash non-accountable expense allowance equal to one percent (1%) of the gross proceeds110% of the offering to Approved Investors; (iii) reimbursementprice of Ladenburg's out-of-pocket expensesthe shares sold in this Offering and may be exercised on a cashless basis. The Representative’s Warrants are exercisable commencing 180 days after the effective date of the registration statement related to this Offering, and will expire five (5) years after the offering, including its legal feeseffective date. The Representative’s Warrants are not redeemable by us. The Representative’s Warrants also provide for unlimited “piggyback” registration rights at our expense with respect to the underlying shares for a period of seven (7) years commencing from the effective date of the registration statement related to this Offering. The Representative’s Warrants and expenses upthe shares underlying the Representative’s Warrants, have been deemed compensation by FINRA and are therefore subject to $40,000;a 180-day lock-up pursuant to Rule 5110(g)(1) of FINRA. The underwriters (or permitted assignees under the Rule) may not sell, transfer, assign, pledge or hypothecate the Representative’s Warrants or the securities underlying the Representative’s Warrants, nor will they engage in any hedging, short sale, derivative, put or call transaction that would result in the effective economic disposition of the Representative’s Warrants or the underlying securities for a period of 12 months from the effective date of this Offering, except to any FINRA member participating in this Offering and (iv)their bona fide officers or partners. The Representative’s Warrants will provide for adjustment in the issuancenumber and price of such Representative’s Warrants (and the shares underlying such Representative’s Warrants) in the event of recapitalization, merger or other structural transaction to Ladenburgprevent mechanical dilution or in the event of warrantsa future financing undertaken by us.
Lock-Up Agreements
Prior to purchase 575,613the completion of this Offering, we and each of our officers, directors, and stockholders who own 5% or more of our outstanding shares of common stock equalof the Company (and all holders of securities exercisable for or convertible into shares of common stock) as of the effective date of the registration statement will agree not to six percent (6%)offer, issue, sell, contract to sell, encumber, grant any option for the sale of or otherwise dispose of any securities of the Company without Maxim’s prior written consent, subject to customary exceptions, for a period of 180 days from the effective date of the effective date of the registration statement relating to this Offering.
Right of Participation
We have agreed to grant Maxim, upon successful completion of an offering of at least $13,000,000, for the twelve (12) month period following the effective date of the registration statement, a right of participation to act at minimum as a co-manager or co-placement agent, as the case may be, for the first future U.S. private or public offering of equity or equity linked securities of the Company with minimum economics in such offering of 20%.
Pricing of this Offering
Prior to this Offering, there has not been an active market for our common stock and there has been no public market for our warrants. The public offering price for our units will be determined through negotiations between us and the underwriter. Among the factors to be considered in these negotiations will be prevailing market conditions, our financial information, market valuations of other companies that we and the underwriter believe to be comparable to us, estimates of our business potential, the present state of our development and other factors deemed relevant.
We offer no assurances that the public offering price of our units will correspond to the price at which our common stock will trade in the public market subsequent to this Offering or that an active trading market for our common stock and warrants will develop and continue after this Offering.
Indemnification
The underwriting agreement provides that we will indemnify the underwriter against certain liabilities that may be incurred in connection with this Offering, including liabilities under the Securities Act, or to contribute payments that the underwriter may be required to make in respect thereof.
Listing
Our common stock trades on the OTCQB tier of OTC Markets Group, Inc. under the symbol “BCDA.” We have applied to list our common stock and warrants on the Nasdaq Capital Market under the symbols “BCDA” and “BCDAW,” respectively. No assurance can be given that such listings will be approved or that a trading market will develop for the common stock and/or the warrants.
Electronic Distribution
A prospectus in electronic format may be made available on websites or through other online services maintained by the underwriter of this Offering, or by its affiliates. Other than the prospectus in electronic format, the information on the underwriter’s website and any information contained in any other website maintained by an underwriter is not part of this prospectus or the registration statement of which this prospectus forms a part, has not been approved and/or endorsed by us or the underwriter in its capacity as underwriter, and should not be relied upon by investors.
Price Stabilization, Short Positions and Penalty Bids
In connection with this Offering, the underwriters may engage in stabilizing transactions, over-allotment transactions, syndicate covering transactions and penalty bids in accordance with Regulation M under the Exchange Act:
Stabilizing transactions permit bids to purchase securities so long as the stabilizing bids do not exceed a specified maximum.
Over-allotment involves sales by the underwriters of securities in excess of the number of shares soldsecurities the underwriters are obligated to purchase, which creates a syndicate short position. The short position may be either a covered short position or a naked short position. In a covered short position, the number of securities over-allotted by the underwriters is not greater than the number of securities that it may purchase in the offeringover-allotment option. In a naked short position, the number of securities involved is greater than the number of securities in the over-allotment option. The underwriters may close out any covered short position by either exercising the over-allotment option and/or purchasing securities in the open market.
Syndicate covering transactions involve purchases of securities in the open market after the distribution has been completed in order to Approved Investors, at an exercisecover syndicate short positions. In determining the source of securities to close out the short position, the underwriters will consider, among other things, the price of $0.44 per share. The approximate dollar valuesecurities available for purchase in the open market as compared to the price at which it may purchase securities through the over-allotment option. If the underwriters sell more securities than could be covered by the over-allotment option, a naked short position, the position can only be closed out by buying securities in the open market. A naked short position is more likely to be created if the underwriters are concerned that there could be downward pressure on the price of the related partysecurities in the open market after pricing that could adversely affect investors who purchase in this Offering.
Penalty bids permit the underwriters to reclaim a selling concession from a syndicate member when the securities originally sold by the syndicate member are purchased in a stabilizing or syndicate covering transaction is $317,198.to cover syndicate short positions.
CORPORATE GOVERNANCEThese stabilizing transactions, syndicate covering transactions and penalty bids may have the effect of raising or maintaining the market price of our securities or preventing or retarding a decline in the market price of our securities. As a result, the price of our securities may be higher than the price that might otherwise exist in the open market. Neither we nor the underwriters make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the price of our securities. In addition, neither we nor the underwriters make any representations that the underwriters will engage in these stabilizing transactions or that any transaction, once commenced, will not be discontinued without notice.
Director Independence
OfAffiliations
The underwriters and their respective affiliates may in the seven membersfuture provide, various investment banking and other financial services for us for which services they may in the future receive, customary fees. Except for services provided in connection with this Offering, none of the Boardunderwriters have provided any investment banking or other financial services to us and we do not expect to retain the underwriter to perform any investment banking or other financial services to us for at least 90 days after the date of Directors five are determined to be independent, they are Joseph Loggia, Thomas H. Morgan, Steven D. Rubin, Ronald N. Richards and Subbarao Uppaluri, all of whom are independent directors as determined by the rules of NYSE AMEX, LLC (formerly the American Stock Exchange).this prospectus.
Audit Committee
The members of the Audit Committee are Subbarao Uppaluri, Thomas H. Morgan and Joseph Loggia, all of whom are independent directors as determined by the rules of NYSE AMEX, LLC (formerly the American Stock Exchange). The responsibilities and duties of the Audit Committee consist of but are not limited to: (1) overseeing the financial reporting process; (2) meeting with our external auditors regarding audit results; (3) engaging and ensuring independence of our outside audit firm and (4) reviewing the effectiveness of the Company's internal controls. Our Board has determined that Mr. Loggia and Mr. Uppaluri qualify as Audit Committee financial experts within the meaning of applicable regulations of the SEC, promulgated pursuant to the Sarbanes-Oxley Act of 2002.
None
INCORPORATION OF CERTAIN INFORMATION BY REFERENCE
None
DISCLOSURE OF COMMISSION POSITION ON INDEMNIFICATION FOR SECURITIES ACT LIABILITIES
The Company's Amended and Restated Certificate of Incorporation provides that the Company will indemnify an officer, director, or former officer or director, to the fullest extent permitted under the General Corporation Law of the State of Delaware or any other applicable laws as presently or thereafter in effect.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers or persons controlling the registrant pursuant to the foregoing provisions, the registrant has been informed that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is therefore unenforceable.
The validity of the shares of common stocksecurities offered throughby this prospectus will be passed onupon by Akerman Senterfitt.Wilson Sonsini Goodrich & Rosati, P.C., Palo Alto, California. Certain members of, and investment partnerships comprised of members of, and persons associated with, Wilson Sonsini Goodrich & Rosati, P.C., own an aggregate of 15,191 shares of our common stock and convertible notes convertible into Private Placement Units at the closing of this Offering. Certain legal matters in connection with this Offering will be passed upon for the underwriters by Loeb & Loeb LLP, New York, New York.
66
EXPERTS
The consolidated financial statements of BioCardia, Inc. as of December 31, 2018 and 2017, and for each of the years in the three-year period ended December 31, 2007 and 2008, included in this prospectus to the extent and for the periods indicated in their reports,2018, have been audited by Stonefield Josephson, Inc., independent registered public accountants,included herein and are included hereinin the registration statement, in reliance upon the report of KPMG LLP, independent registered public accounting firm, appearing elsewhere herein, and upon the authority of suchsaid firm as experts in accounting and auditingauditing. The audit report covering the December 31, 2018 consolidated financial statements contains an explanatory paragraph that states that the Company’s recurring losses from operations and net capital deficiency raise substantial doubt about the entity’s ability to continue as a going concern. The consolidated financial statements do not include any adjustments that might result from the outcome of that uncertainty.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-1 under the Securities Act relating to the offering of these securities. The registration statement, including the attached exhibits and schedules, contains additional relevant information about us and the securities. This prospectus does not contain all of the information set forth in giving such reports.the registration statement and the exhibits and schedules thereto. For further information respecting our company and the shares offered by this prospectus, you should refer to the registration statement, including the exhibits and schedules thereto.
We file annual, quarterquarterly and periodicother reports, proxy statements and other information with the SecuritiesSEC. Our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, and Exchange Commission usingCurrent Reports on Form 8-K, including any amendments to those reports, and other information that we file with or furnish to the Commission's EDGAR system. You may inspect these documents and copy information from them at the Commission's public reference room at 100 F Street, NE, Washington, D.C. 20549. You may obtain information on the operationSEC pursuant to Section 13(a) or 15(d) of the public reference room by callingExchange Act can be accessed free of charge through the Internet. The SEC at 1-800-SEC-0330. The Commission maintains a weban Internet site that contains reports, proxy and information statements, and other information regarding registrantsissuers that file electronically with the Commission. The addressSEC at http://www.sec.gov. You may access the registration statement of suchwhich this prospectus is a part at the SEC’s Internet site.
We make available through our website, free of charge, copies of our SEC filings as soon as reasonably practicable after we electronically file or furnish them to the SEC on our Internet site, is http//www.sec.gov.
You should rely only on the information contained in this prospectus.www.biocardia.com. We have not authorized any person to provide you with any information that is different.
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
ITEM 13 - OTHER EXPENSES OF ISSUANCE AND DISTRIBUTION
| ||
|
| |
|
| |
|
| |
|
| |
|
|
All amounts are estimates, other than the SEC's registration fee.
We are paying all expenses of the offering listed above. No portion of these expenses will be borne by the selling stockholders. The selling stockholders; however, will pay any other expenses incurred in selling their common stock, including any brokerage commissions or costs of sale.
ITEM 14 - INDEMNIFICATION OF DIRECTORS AND OFFICERS
The Company's Amended and Restated Certificate of Incorporation provides that the Company will indemnify an officer, director, or former officer or director, to the fullest extent permitted under the General Corporation Law of the State of Delaware or any other applicable laws as presently or thereafter in effect. The Company is also party to an Indemnification Agreement with Directors and Officers of the Company filed as Exhibit 10.12. The Company must indemnify the Directors and Officers to the fullest extent permitted by law.
ITEM 15 - RECENT SALES OF UNREGISTERED SECURITIES
On June 30, 2009, Cardo Medical, Inc. closed on the sale of 8,689,319 shares of the Company common stock, par value $0.001 per share at a price of $0.35 per share for aggregate gross proceeds of $3,041,260. The offering of common stock was conducted by the Company in a non-brokered private placement to accredited investors pursuant to an exemption from registration provided by Section 4(2) of the Securities Act of 1933, as amended. The Company believes the exemption is available because the securities were not offered pursuant to a general solicitation and the status of the investors as "accredited investors" as defined in Regulation D of the Securities Act of 1933, as amended.
On September 21, 2009, Cardo Medical, Inc. closed on the sale of 485,714 shares of the Company common stock, par value $0.001 per share at a price of $0.35 per share for aggregate gross proceeds of $170,000. The offering of common stock was the second tranche of the June 30, 2009 offering referred to above. The Company conducted the offering of common stock in a non-brokered private placement to accredited investors pursuant to an exemption from registration provided by Section 4(2) of the Securities Act of 1933, as amended. The Company believes the exemption is available because the securities were not offered pursuant to a general solicitation and the status of the investors as "accredited investors" as defined in Regulation D of the Securities Act of 1933, as amended.
67
On October 27, 2009, Cardo Medical, Inc. sold 9,949,276 shares of common stock, par value $0.001 per share of the Company at a price of $0.35 per share for aggregate gross proceeds of $3,482,250 as part of a private placement for maximum gross proceeds of $6,500,000. The Company relied on the exemption provided by Section 4(2) of the Securities Act of 1933, as amended for the issuance of the shares of common stock in the Company, which exception the Company believed was available because the securities were not offered pursuant to a general solicitation and the status of the investors as "accredited investors" as defined in Regulation D of the Securities Act, as amended.
On November 13, 2009, Cardo Medical, inc. sold 7,808,561 shares of common stock, par value $0.001 per share of the Company at a price of $0.35 per share for aggregate gross proceeds of $2,733,000 in the second tranche of the prior private placement for maximum gross proceeds of $6,500,000. The November 13, 2009 closing completed the private placement. The Company relied on the exemption provided by Section 4(2) of the Securities Act of 1933, as amended for the issuance of the shares of common stock in the Company, which exception the Company believed was available because the securities were not offered pursuant to a general solicitation and the status of the investors as "accredited investors" as defined in Regulation D of the Securities Act, as amended.
ITEM 16 - EXHIBITS
EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
Incorporated By Reference | |||||
Exhibit Number | Exhibit Description | Form | File Number | Exhibit | Filing Date |
2.1 | Merger Agreement and Plan of Reorganization, dated as of June 18, 2008, by and among clickNsettle.com, Inc., Cardo Medical, LLC and Cardo Acquisition, LLC. | 8-K | 000-21419 | 2.1 | June 23, 2008 |
2.2 | First Amendment to Merger Agreement and Plan of Reorganization, dated as of August 29, 2008, by and among clickNsettle.com, Inc., Cardo Medical, LLC and Cardo Acquisition, LLC. | 8-K | 000-21419 | 2.2 | September 9, 2008 |
2.3 | Asset Purchase Agreement, dated September 30, 2009, by and among Cardo Medical, Inc. and Vertebron, Inc. | 8-K | 000-21419 | 2.1 | October 6, 2009 |
3.1 | Amended and Restated Certificate of Incorporation | 8-K | 000-21419 | 3.1 | March 18, 2008 |
3.2 | Amended and Restated Bylaws. | 8-K | 000-21419 | 3.1 | February 1, 2008 |
5.1# | Opinion of Akerman Senterfitt | ||||
10.1 | Escrow Agreement, dated as of August 29, 2008, by and among Chicago Title Company, clickNsettle.com, Inc. Andrew A. Brooks, M.D. and Mikhail Kvitnitsky. | 8-K | 000-21419 | 10.1 | September 9, 2008 |
10.2 | Form of Lockup Agreement. | 8-K | 000-21419 | 10.2 | September 9, 2008 |
10.3 | Lockup Agreement, dated August 29, 2008, for Derrick Romine. | 8-K | 000-21419 | 10.3 | September 9, 2008 |
10.4 | Amended and Restated 1996 Incentive and Nonqualified Stock Option Plan. | 10-KSB | September 28, 1998 | ||
68
10.5* Form of Cardo Medical, LLC Nonstatutory Option Agreement 8-K 000-21419 10.5 September 9, 2008 10.6 Stock Purchase Agreement, dated as of December 19, 2007, by and among clickNsettle.com, Inc., Frost Gamma Investments Trust, Dr. Jane Hsiao, Steven D. Rubin and Subbarao Uppaluri. 8-K 000-21419 10.1 December 21, 2007 10.7 First Amendment to Stock Purchase Agreement, dated as of January 31, 2008, by and among clickNsettle.com, Inc., Frost Gamma Investments Trust, Dr. Jane Hsiao, Steven D. Rubin and Subbarao Uppaluri. 8-K 000-21419 10.2 February 1, 2008 10.8* Employment Agreement, dated as of January 31, 2005, by and between Accelerated Innovation, LLC, as successor to Accin Corporation, and Mikhail Kvitnitsky. 8-K 000-21419 10.8 September 9, 2008 10.9* Amendment to Employment Agreement, dated as of June 6, 2008, by and between Accelerated Innovation, LLC and Mikhail Kvitnitsky. 8-K 000-21419 10.9 September 9, 2008 10.10* Termination Agreement, effective as of June 23, 2008, by and between Accelerated Innovation, LLC and Mikhail Kvitnitsky. 8-K 000-21419 10.10 September 9, 2008 10.11* Employment Offer Letter with Derrick Romine dated September 5, 2008. 8-K 000-21419 10.11 September 9, 2008 10.12 Form of Indemnification Agreement for officers and directors. 8-K 000-21419 10.12 September 9, 2008 10.13+ Agreement, dated as of August 22, 2006, by and between Accelerated Innovation, LLC, as successor to Accin Corporation, and Infinesse Corporation. 8-K 000-21419 10.13 September 9, 2008 10.14 Supplier Agreement, dated June 16, 2006, between Stelkast Company and Accelerated Innovation, LLC, as successor to Accin. 8-K 000-21419 10.14 September 9, 2008 10.15+ Contracted Services Agreement, dated September 1, 2007, by and between Accelerated Innovation, LLC and Summit Corporate Services, Inc. 8-K 000-21419 10.15 September 9, 2008 10.16 Agreement dated April 30, 2008, by and among Mikhail Kvitnitsky and Accelerated Innovation, LLC, Cervical Xpand, LLC and Uni-Knee, LLC. 8-K 000-21419 10.16 September 9, 2008 10.17 Agreement dated April 30, 2008, by and among John D. Kuczynski, Accelerated Innovation, LLC and Uni-Knee, LLC. 8-K 000-21419 10.17 September 9, 2008 10.18 Agreement dated April 30, 2008, by and among Richard H. Rothman, M.D., Ph.D., Accelerated Innovation, LLC and Cervical Xpand, LLC. 8-K 000-21419 10.18 September 9, 2008 10.19 Agreement dated April 30, 2008, by and among Todd J. Albert, M.D., Accelerated Innovation, LLC and Cervical Xpand, LLC. 8-K 000-21419 10.19 September 9, 2008
69
10.20 Agreement dated April 28, 2008, by and among, Rafail Zubok, Accelerated Innovation, LLC and Cervical Xpand, LLC. 8-K 000-21419 10.20 September 9, 2008 10.21* Nonstatutory Option Agreement, dated August 27, 2008, by and between Cardo Medical, LLC and Derrick Romine. 10-Q 000-21419 10.1 November 14, 2008 10.22 Form of Registration Rights Agreement, dated October 27, 2009, by and among Cardo Medical, Inc. and the Several Purchasers. 8-K 000-21419 10.1 October 29, 2009 21.1 Subsidiaries of clickNsettle.com, Inc. 8-K 000-21419 21.1 September 9, 2008 23.1# Consent of Stonefield Josephson, Inc. 23.2# Consent of Akerman Senterfitt (included in Exhibit 5.1)
|
| |
|
| |
|
|
ITEM 17 - UNDERTAKINGS
UNDERTAKINGS
(a) The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20 percent change in the maximum aggregate offering price set forth in the "Calculation of Registration Fee" table in the effective registration statement;
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement;
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initialbona fide offering thereof.
70
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) For determining liability under the Securities Act to any purchaser, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into this prospectus the registration statement or prospectus that isinformation on our website, and you should not consider it to be a part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.this prospectus.
(5) That, for the purpose of determining liability of the registrant under the Securities Act of 1933 to any purchaser in the initial distribution of the securities:
The undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424;
(ii) Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
(iii) The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and
(iv) Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
71
CARDO MEDICAL, INC.INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Documents for the fiscal year ended December 31, 2008, extracted from our annual report on Form 10-K filed with the Securities and Exchange Commission on March 31, 2009:
Report of Independent Registered Accounting Firm for the years ended December 31, 2008 and 2007
Financial Statements
Consolidated Balance Sheets at December 31, 2008 and 2007
Consolidated Statements of Operations and Comprehensive Loss for the years ended December 31, 2008 and 2007
Consolidated Statements of Shareholders' Equity (Deficiency) for the years ended December 31, 2008 and 2007
Consolidated Statements of Cash Flows for the years ended December 31, 2008 and 2007
NotesIndex to Consolidated Financial Statements
Unaudited Financial Statements for the Nine Months Ended September 30, 2009, extracted from our quarterly report on Form 10-Q filed with the Securities and Exchange Commission on November 12, 2009:
Financial Statements
Condensed Consolidated Balance Sheets at September 30, 2009 (Unaudited) and December 31, 2008
Condensed Consolidated Statement of Operations (Unaudited) - Three and Nine Months Ended September 30, 2009 and 2008
Condensed Consolidated Statements of Cash Flows (Unaudited) - Nine Months Ended September 30, 2009 and 2008
Notes to Condensed Consolidated Financial Statements (Unaudited)
F-1
Page | |
| Audited Consolidated Financial Statements for the Fiscal Years Ended December 31, 2018 and 2017 | |
Report of Independent Registered Public Accounting Firm |
|
Consolidated Balance Sheets | F-3 |
Consolidated Statements of | F-4 |
Consolidated Statements of Stockholders’ Equity (Deficit) | F-5 |
Consolidated Statements of Cash Flows | F-6 |
Notes to Consolidated Financial Statements | F-7 |
Unaudited Consolidated Financial Statements for the Three Months Ended March 31, 2019 | |
Consolidated Balance Sheets | F-23 |
Consolidated Statements of Operations | F-24 |
Consolidated Statements of Stockholders’ Equity (Deficit) | F-25 |
Consolidated Statements of Cash Flows | F-26 |
Notes to Consolidated Financial Statements | F-30 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors
BioCardia, Inc.:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of BioCardia, Inc. and its subsidiary (the Company) as of December 31, 2018 and 2017, the related consolidated statements of operations, stockholders’ equity (deficit), and cash flows for each of the years in the three-year period ended December 31, 2018, and the related notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2018 and 2017, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2018, in conformity with U.S. generally accepted accounting principles.
Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the consolidated financial statements, the Company has suffered recurring losses from operations and has a net capital deficiency that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
(signed) KPMG LLP
We have served as the Company’s auditor since 2012.
San Francisco, California
April 1, 2019
BIOCARDIA, INC.
Consolidated Balance Sheets
(In thousands, except share and per share data)
December 31, | ||||||||
| 2018 | 2017 | ||||||
| Assets | ||||||||
Current assets: | ||||||||
Cash and cash equivalents | $ | 5,358 | $ | 12,689 | ||||
Accounts receivable, net of allowance for doubtful accounts of $9 and $6 at December 31, 2018 and 2017, respectively | 274 | 95 | ||||||
Inventory | 141 | 191 | ||||||
Prepaid expenses | 445 | 340 | ||||||
Total current assets | 6,218 | 13,315 | ||||||
Property and equipment, net | 145 | 169 | ||||||
Other assets | 54 | 54 | ||||||
Total assets | $ | 6,417 | $ | 13,538 | ||||
Liabilities and Stockholders’ Equity | ||||||||
Current liabilities: | ||||||||
Accounts payable | $ | 1,020 | $ | 902 | ||||
Accrued expenses and other current liabilities | 1,528 | 1,263 | ||||||
Deferred revenue | — | 167 | ||||||
Total current liabilities | 2,548 | 2,332 | ||||||
Deferred rent | 77 | 81 | ||||||
Total liabilities | 2,625 | 2,413 | ||||||
Stockholders’ equity: | ||||||||
Preferred stock, $0.001 par value, 25,000,000 shares authorized as of December 31, 2018 and 2017; no shares issued and outstanding as of December 31, 2018 and 2017 | ||||||||
Common stock, $0.001 par value, 100,000,000 shares authorized as of December 31, 2018 and 2017; 43,611,240 and 38,218,660 shares issued and outstanding as of December 31, 2018 and 2017, respectively | 43 | 38 | ||||||
Additional paid-in capital | 90,110 | 83,537 | ||||||
Accumulated deficit | (86,361 | ) | (72,450 | ) | ||||
Total stockholders’ equity | 3,792 | 11,125 | ||||||
Total liabilities and stockholders’ equity | $ | 6,417 | $ | 13,538 | ||||
See accompanying notes to consolidated financial statements.
BIOCARDIA, INC.
Consolidated Statements of Operations
(In thousands, except share and per share amounts)
Years ended December 31, | ||||||||||||
2018 | 2017 | 2016 | ||||||||||
Revenue: | ||||||||||||
Net product revenue | $ | 282 | $ | 389 | $ | 517 | ||||||
Collaboration agreement revenue | 343 | 90 | 59 | |||||||||
Total revenue | 625 | 479 | 576 | |||||||||
Costs and expenses: | ||||||||||||
Cost of goods sold | 517 | 690 | 746 | |||||||||
Research and development | 8,453 | 5,799 | 3,330 | |||||||||
Selling, general and administrative | 5,757 | 6,395 | 4,108 | |||||||||
Total costs and expenses | 14,727 | 12,884 | 8,184 | |||||||||
Operating loss | (14,102 | ) | (12,405 | ) | (7,608 | ) | ||||||
Other income (expense): | ||||||||||||
Interest income | 118 | 95 | — | |||||||||
Interest expense | — | — | (1,736 | ) | ||||||||
Change in fair value of convertible preferred stock warrant liability | — | — | 250 | |||||||||
Change in fair value of maturity date preferred stock warrant liability | — | — | 10 | |||||||||
Change in fair value of convertible shareholder notes derivative liability | — | — | (1,224 | ) | ||||||||
Other expense | (3 | ) | 2 | (2 | ) | |||||||
| Other income (expense), net | 115 | 97 | (2,702 | ) | ||||||||
Net loss | $ | (13,987 | ) | $ | (12,308 | ) | $ | (10,310 | ) | |||
Net loss per share, basic and diluted | $ | (0.36 | ) | $ | (0.32 | ) | $ | (1.23 | ) | |||
Weighted-average shares used in computing net loss per share, basic and diluted | 38,377,606 | 38,160,543 | 8,368,284 | |||||||||
See accompanying notes to consolidated financial statements.
BIOCARDIA, INC.
Consolidated Statements of Stockholders’ Equity (Deficit)
(In thousands, except share data)
Convertible preferred stock | Common stock | Additional | Accumulated | |||||||||||||||||||||||||
Shares | Cost | Shares | Cost | paid in capital | deficit | Total | ||||||||||||||||||||||
Balance at December 31, 2015 | 110,500,514 | 46,030 | 1,578,962 | 2 | 358 | (49,832 | ) | (3,442 | ) | |||||||||||||||||||
Exchange of convertible preferred stock warrants to common stock | — | — | 10,807 | — | 25 | — | 25 | |||||||||||||||||||||
Reclassification of convertible shareholder notes derivative liability | — | — | — | — | 2,268 | — | 2,268 | |||||||||||||||||||||
Conversion of convertible notes into common stock | — | — | 8,091,103 | 8 | 12,148 | — | 12,156 | |||||||||||||||||||||
Conversion of preferred stock into common stock | (110,500,514 | ) | (46,030 | ) | 9,208,376 | 9 | 46,021 | — | — | |||||||||||||||||||
Issuance of common stock upon reverse merger | — | — | 19,228,595 | 19 | 18,902 | — | 18,921 | |||||||||||||||||||||
Exercise of stock options | — | — | 13,460 | — | 22 | — | 22 | |||||||||||||||||||||
Share-based compensation | — | — | — | — | 942 | — | 942 | |||||||||||||||||||||
Net loss | — | — | — | — | — | (10,310 | ) | (10,310 | ) | |||||||||||||||||||
Balance at December 31, 2016 | — | $ | — | 38,131,303 | $ | 38 | $ | 80,686 | $ | (60,142 | ) | $ | 20,582 | |||||||||||||||
Exercise of stock options | — | — | 87,357 | — | 144 | — | 144 | |||||||||||||||||||||
Share-based compensation | — | — | — | — | 2,707 | — | 2,707 | |||||||||||||||||||||
Net loss | — | — | — | — | — | (12,308 | ) | (12,308 | ) | |||||||||||||||||||
Balance at December 31, 2017 | — | $ | — | 38,218,660 | $ | 38 | $ | 83,537 | $ | (72,450 | ) | $ | 11,125 | |||||||||||||||
Adjustments to opening balance for change in accounting principle | — | — | — | — | — | 76 | 76 | |||||||||||||||||||||
Sale of common stock and warrants, net of issuance costs of $200 | — | — | 5,333,332 | 5 | 3,795 | — | 3,800 | |||||||||||||||||||||
Restricted stock units vested and issued | 57,108 | 0 | — | — | 0 | |||||||||||||||||||||||
Exercise of stock options | — | — | 2,140 | 0 | 4 | — | 4 | |||||||||||||||||||||
Share-based compensation | — | — | — | — | 2,774 | — | 2,774 | |||||||||||||||||||||
Net loss | — | — | — | — | — | (13,987 | ) | (13,987 | ) | |||||||||||||||||||
Balance at December 31, 2018 | — | $ | — | 43,611,240 | $ | 43 | $ | 90,110 | $ | (86,361 | ) | $ | 3,792 | |||||||||||||||
See accompanying notes to consolidated financial statements.
BIOCARDIA, INC.
Consolidated Statements of Cash Flows
(In thousands)
Years ended December 31, | ||||||||||||
2018 | 2017 | 2016 | ||||||||||
Operating activities: | ||||||||||||
Net loss | $ | (13,987 | ) | $ | (12,308 | ) | $ | (10,310 | ) | |||
Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
Write-off of inventory | — | — | 597 | |||||||||
Depreciation and amortization | 88 | 78 | 39 | |||||||||
Change in fair value of convertible preferred stock warrant liability | — | — | (250 | ) | ||||||||
Change in fair value of maturity date preferred stock warrant liability | — | — | (10 | ) | ||||||||
Change in fair value of convertible shareholder notes derivative liability | — | — | 1,224 | |||||||||
Share-based compensation | 2,774 | 2,707 | 942 | |||||||||
Non-cash interest expense on convertible shareholder notes | — | — | 1,736 | |||||||||
Changes in operating assets and liabilities: | ||||||||||||
Accounts receivable | (179 | ) | (21 | ) | 33 | |||||||
Inventory | 49 | (56 | ) | 27 | ||||||||
Prepaid expenses and other current assets | (104 | ) | 16 | (110 | ) | |||||||
Other assets | — | — | (11 | ) | ||||||||
Accounts payable | 119 | 377 | (17 | ) | ||||||||
Accrued liabilities excluding accrued interest on convertible note | 240 | 415 | 530 | |||||||||
Deferred revenue | (65 | ) | 96 | 32 | ||||||||
Deferred rent | (4 | ) | 25 | 26 | ||||||||
Net cash used in operating activities | (11,069 | ) | (8,671 | ) | (5,522 | ) | ||||||
Investing activities: | ||||||||||||
Purchase of property and equipment | (66 | ) | (136 | ) | — | |||||||
Cash acquired in reverse merger | — | — | 19,017 | |||||||||
Payment of transaction costs of reverse merger | — | — | (96 | ) | ||||||||
Purchase of short-term investments | — | (1,800 | ) | — | ||||||||
Maturity of short-term investments | — | 1,800 | — | |||||||||
| Net cash (used) provided by investing activities | (66 | ) | (136 | ) | 18,921 | |||||||
Financing activities: | ||||||||||||
Proceeds from sales of common stock and warrants, net of issuance costs of $200 | 3,800 | — | — | |||||||||
| Proceeds from issuance of convertible notes and warrants | — | — | 4,374 | |||||||||
Proceeds from the exercise of common stock options | 4 | 144 | 22 | |||||||||
Net cash provided by financing activities | 3,804 | 144 | 4,396 | |||||||||
| Net (decrease) increase in cash and cash equivalents | (7,331 | ) | (8,663 | ) | 17,795 | |||||||
Cash and cash equivalents at beginning of period | 12,689 | 21,352 | 3,557 | |||||||||
Cash and cash equivalents at end of period | $ | 5,358 | $ | 12,689 | $ | 21,352 | ||||||
Supplemental disclosure for noncash investing and financing activities: | ||||||||||||
Exchange of convertible preferred stock warrants for common stock | $ | — | $ | — | $ | 25 | ||||||
Conversion of convertible shareholder notes and related interest payable | $ | — | $ | — | $ | 12,156 | ||||||
Reclassification of convertible shareholder notes derivative liability | $ | — | $ | — | $ | 2,268 | ||||||
Conversion of preferred stock | $ | — | $ | — | $ | 46,030 | ||||||
See accompanying notes to consolidated financial statements.
(1) | Summary of Business |
(a) | Description of Business |
BioCardia, Inc., or the Company, is a clinical-stage regenerative medicine company developing novel therapeutics for cardiovascular diseases with large unmet medical needs. Its lead therapeutic candidate is the CardiAMP cell therapy system and its second therapeutic candidate is the CardiALLO cell therapy system. To date, the Company has devoted substantially all of its resources to research and development efforts relating to its therapeutic candidates and biotherapeutic delivery systems including conducting clinical trials, developing manufacturing and sales capabilities, in-licensing related intellectual property, providing general and administrative support for these operations and protecting its intellectual property.
The Company has three enabling device product lines: (1) the CardiAMP cell processing system; (2) the Helix biotherapeutic delivery system, or Helix; and (3) the Morph vascular access product line, or Morph. The Company manages its operations as a single segment for the purposes of assessing performance and making operating decisions.
(2) | Significant Accounting Policies |
(a) | Basis of Presentation and Consolidation |
These consolidated financial statements have been prepared in accordance with generally accepted accounting principles in the United States (GAAP) and include all adjustments necessary for the fair presentation of the Company’s financial position for the periods presented. The consolidated financial statements include the accounts of the Company and its wholly owned subsidiary. All material intercompany accounts and transactions have been eliminated during the consolidation process. The Company manages its operations as a single segment for the purposes of assessing performance and making operating decisions.
(b) | Liquidity - Going Concern |
The Company has incurred net losses and negative cash flows from operations since its inception and had an accumulated deficit of $86.4 million as of December 31, 2018. Management expects operating losses and negative cash flows to continue through at least the next several years. The Company expects to incur increasing costs as the pivotal CardiAMP Heart Failure trial is advanced and development of the CardiAMP and CardiALLO Cell Therapy Systems continue. Therefore, absent additional funding, management believes cash and cash equivalents of $5.4 million as of December 31, 2018 are not sufficient to fund the Company beyond the second quarter of 2019. These factors raise substantial doubt about the Company’s ability to continue as a going concern beyond one year from the date these financial statements are issued. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
The Company’s ability to continue as a going concern and to continue further development of its therapeutic candidates through and beyond the second quarter of 2019, will require the Company to raise additional capital. The Company plans to raise additional capital, potentially including debt and equity arrangements, to finance its future operations. While management believes this plan to raise additional funds will alleviate the conditions that raise substantial doubt, these plans are not entirely within its control and cannot be assessed as being probable of occurring. If adequate funds are not available, the Company may be required to reduce operating expenses, delay or reduce the scope of its product development programs, obtain funds through arrangements with others that may require the Company to relinquish rights to certain of its technologies or products that the Company would otherwise seek to develop or commercialize itself, or cease operations.
(c) | Use of Estimates |
The preparation of the financial statements in accordance with U.S. GAAP requires Company management to make certain estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ materially from those estimates. Significant items subject to such estimates and assumptions include the useful lives of property and equipment; allowances for doubtful accounts and sales returns; inventory valuation; and share-based compensation.
(d) | Cash Equivalents |
The Company classifies all highly liquid investments with an original maturity date of 90 days or less at the date of purchase as cash equivalents. The Company maintains its cash and cash equivalents with reputable financial institutions.
(e) | Concentration of Credit Risk |
Financial instruments that potentially subject the Company to a concentration of credit risk consist of cash and cash equivalents. The Company maintains its cash at financial institutions, which at times, exceed federally insured limits. At December 31, 2018, the Company’s cash was held by one financial institution and the amount on deposit was in excess of FDIC insurance limits. The Company has not recognized any losses from credit risks on such accounts since inception. The Company believes it is not exposed to significant credit risk on cash.
(f) | Accounts Receivable and Allowance for Doubtful Accounts |
Accounts receivable are recorded at the invoiced amount and do not bear interest. The Company considers the creditworthiness of its customers but does not require collateral in advance of a sale. The Company evaluates collectability and maintains an allowance for doubtful accounts for estimated losses inherent in its accounts receivable portfolio when necessary. The estimate is based on the Company’s historical write-off experience, customer creditworthiness, facts and circumstances specific to outstanding balances and payment terms. Account balances are charged off against the allowance after all means of collection have been exhausted and the potential for recovery is considered remote. The allowance for doubtful accounts was $9,000 and $6,000 as of December 31, 2018 and 2017, respectively.
(g) | Inventory |
Inventory is stated at the lower of cost or net realizable value. Cost is determined using the average-cost method. Net realizable value is the estimated selling price in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. The Company analyzes its inventory levels quarterly and writes down inventory that has become obsolete or has a cost basis in excess of its expected net realizable value or inventory quantities in excess of expected requirements. Excess requirements are determined based on comparison of existing inventories to forecasted sales, with consideration given to inventory shelf life. Expired inventory is disposed of and the related costs are recognized in cost of goods sold.
(h) | Property and Equipment, Net |
Property and equipment, net, are carried at cost less accumulated depreciation. Depreciation is calculated using the straight-line method over the estimated useful lives of the related assets, as described in the table below. Maintenance and repairs are expensed as incurred. When assets are retired or otherwise disposed of, the cost and the related accumulated depreciation and amortization are removed from the accounts and any resulting gain or loss is reflected in the accompanying consolidated statements of operations.
Asset | Estimated useful lives (in years) | |||
Computer equipment and software | 3 | |||
Laboratory and manufacturing equipment | 3 | |||
Furniture and fixtures | 3 | |||
Leasehold improvements | 5 years or lease term, if shorter | |||
(i) | Long-Lived Assets |
The carrying value of long-lived assets, including property and equipment, is reviewed for impairment whenever events or changes in circumstances indicate that the asset may not be recoverable. An impairment loss is recognized when the total of estimated future undiscounted cash flows, expected to result from the use of the asset and its eventual disposition, are less than its carrying amount. Impairment, if any, would be assessed using discounted cash flows or other appropriate measures of fair value. Through December 31, 2018, there have been no such impairment losses.
(j) | Clinical Trial Accruals |
As part of the process of preparing its consolidated financial statements, the Company is required to estimate its expenses resulting from its obligations under contracts with vendors and consultants and clinical site agreements in connection with conducting clinical trials. The financial terms of these contracts are subject to negotiation and may result in payment flows that do not match the periods over which materials or services are provided by the vendor under the contracts. The Company’s objective is to reflect the clinical trial expenses in its consolidated financial statements by matching those expenses with the period in which the services and efforts are expended. The Company accounts for these expenses according to the progress of the trial as measured by patient progression and the timing of various aspects of the trial. The Company makes estimates of its accrued expenses as of each balance sheet date in its consolidated financial statements based on the facts and circumstances known at that time. Although, the Company does not expect its estimates to be materially different from amounts actually incurred, its understanding of the status and timing of services relative to the actual status and timing of services performed may vary and may result in reported amounts that differ from the actual amounts incurred.
(k) | Derivatives |
The Company accounts for its derivative instruments as either assets or liabilities on the consolidated balance sheet and measures them at fair value. Derivatives are adjusted to fair value through other (expense) income, net in the consolidated statements of operations.
(l) | Deferred Rent |
The Company’s lease for its facility provides for fixed increases in minimum annual rental payments. The total amount of rental payments due over the lease term is charged to rent expense ratably over the life of the lease. Deferred rent consists of the difference between cash payments and the recognition of rent expense on a straight-line basis.
(m) | Revenue Recognition |
Net product revenue – We currently have a portfolio of enabling and delivery products. Revenue from product sales is recognized generally upon shipment to the end customer, which is when control of the product is deemed to be transferred. Product sale transactions are evidenced by customer purchase orders, customer contracts, invoices and/or related shipping documents.
Collaboration agreement revenue – Collaboration agreement revenue is income from agreements under partnering programs with corporate and academic institutions, wherein we provide biotherapeutic delivery systems and customer training and support for their use in clinical trials and studies. These programs provide additional clinical data, intellectual property rights and opportunities to participate in the development of combination products for the treatment of cardiac disease.
In determining the appropriate amount of revenue to be recognized as it fulfills its obligations under each of its agreements, the Company performs the following steps: (i) identification of the promised goods or services in the contract; (ii) determination of whether the promised goods or services are performance obligations, including whether they are distinct in the context of the contract; (iii) measurement of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition of revenue when (or as) the Company satisfies each performance obligation. As part of the accounting for these arrangements, the Company must develop assumptions that require judgment to determine the stand-alone selling price for each performance obligation identified in the contract. The Company uses key assumptions to determine the stand-alone selling price, which may include forecasted revenues, development timelines, reimbursement rates for personnel costs, discount rates and probabilities of technical and regulatory success.
Amounts received from customers in advance of revenue recognition are recorded as deferred revenue on the consolidated balance sheets.
(n) | Shipping Costs |
Costs incurred for the shipping of products to customers totaled approximately $6,000, $5,000 and $7,000 for the years ended December 31, 2018, 2017 and 2016, respectively, and are included in cost of goods sold in the accompanying consolidated statements of operations.
(o) | Product Warranties |
The Company provides a standard warranty of serviceability on all its products for the duration of the product’s shelf life, which is two years for Helix and Morph products currently. Estimated future warranty costs, if any, are accrued and charged to costs of goods sold in the period that the related revenue is recognized. Historical data and trends of product reliability and costs of repairing or replacing defective products are considered. Due to the low historical warranty claims experience, a general warranty accrual has not been required or recorded as of December 31, 2018 and 2017.
(p) | Research and Development |
The Company’s research and development costs are expensed as incurred. Research and development expense include the costs of basic research activities as well as other research, engineering, and technical effort required to develop new products or services or make significant improvement to an existing product or manufacturing process. Research and development costs also include pre-approval regulatory and clinical trial expenses and support costs for collaborative partnering programs wherein the Company provides biotherapeutic delivery systems and customer training and support for their use in clinical trials and studies. The Company’s research and development costs consist primarily of:
Salaries, benefits and other personnel-related expenses, including share-based compensation
Fees paid for services provided by clinical research organizations, research institutions, consultants and other outside service providers
Costs to acquire and manufacture materials used in research and development activities and clinical trials
Laboratory consumables and supplies
Facility-related expenses allocated to research and development activities
Fees to collaborators to license technology
Depreciation expense for equipment used for research and development and clinical purposes.
(q) | Share-Based Compensation |
The Company measures and recognizes share-based compensation expense for equity awards to employees, directors and consultants based on fair value at the grant date. The Company uses the Black-Scholes option pricing model to calculate fair value. Share-based compensation expense recognized in the consolidated statements of operations is based on the period the services are performed. The Company accounts for forfeitures as they occur. The compensation cost for restricted stock awards is based on the closing price of the Company’s common stock on the date of grant and recognized as compensation expense on a straight-line basis over the requisite service period.
For options granted to nonemployees, the Company revalues the unearned portion of the share-based compensation and the resulting change in fair value is recognized in the consolidated statements of operations over the period the related services are rendered.
The Black-Scholes option pricing model (BSM) requires the input of subjective assumptions, including the risk-free interest rate, the expected volatility in the value of the Company’s common stock, and the expected term of the option. These estimates involve inherent uncertainties and the application of management’s judgment. If factors change and different assumptions are used, our share-based compensation expense could be materially different in the future. These assumptions are estimated as follows:
Risk-free Interest Rate
The risk-free interest rate assumption is based on the zero-coupon U.S. Treasury instruments appropriate for the expected term of the stock option grants.
Expected Volatility
The Company has limited historical data of its own to utilize in determining expected volatility. As such we based our volatility assumption on a combined weighted average of our own historical data and that of a selected peer group. The peer group was developed based on companies in the biotechnology and medical device industries whose shares are publicly-traded.
Expected Term
The expected term represents the period of time that options are expected to be outstanding. As the Company does not have sufficient historical experience for determining the expected term of the stock options awards granted, the expected life is determined using the simplified method, which is an average of the contractual terms of the option and its ordinary vesting period.
(r) | Income Taxes |
The Company accounts for income taxes based on the asset and liability method whereby deferred tax asset and liability account balances are determined based on differences between the financial reporting and tax bases of assets, liabilities, operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. A valuation allowance is provided when it is more likely than not that some portion or all of the deferred tax assets will not be realized.
In evaluating the ability to recover its deferred income tax assets, the Company considers all available positive and negative evidence, including its operating results, forecasts of future taxable income, and ongoing tax planning. In the event the Company was to determine that it would be able to realize its deferred tax assets in the future in excess of their net recorded amount, it would make an adjustment to the valuation allowance, which would reduce the provision for income taxes. Conversely, in the event that all or part of the net deferred tax assets are determined not to be realizable in the future, an adjustment to the valuation allowance would be charged to earnings in the period such determination is made.
The Company recognizes and measures benefits for uncertain tax positions using a two-step approach. The first step is to evaluate the tax position taken or expected to be taken in a tax return by determining if the weight of available evidence indicates that it is more likely than not that the tax position will be sustained upon audit, including resolution of any related appeals or litigation processes. For tax positions that are more likely than not to be sustained upon audit, the second step is to measure the tax benefit as the largest amount that is more than 50% likely to be realized upon settlement. Significant judgment is required to evaluate uncertain tax positions. The Company evaluates its uncertain tax positions quarterly. Evaluations are based upon a number of factors, including the technical merits of the tax position, changes in facts or circumstances, changes in tax law, interactions with tax authorities during the course of audits, and effective settlement of audit issues. The Company’s policy is to recognize interest and penalties related to unrecognized tax benefits as a component of income tax expense in the consolidated statements of operations and accrued interest and penalties within accrued liabilities in the consolidated balance sheets. No such interest and penalties have been recorded to date.
(s) | Fair Value of Financial Instruments |
The Company applies fair value accounting for all financial assets and liabilities and nonfinancial assets and liabilities that are required to be recognized or disclosed at fair value in the consolidated financial statements. The Company defines fair value as the price that would be received from selling an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. Where observable prices or inputs are not available, valuation models are applied. These valuation techniques involve some level of management estimation and judgment, the degree of which is dependent on the price transparency for the instruments or market and the instruments complexity.
The Company’s financial assets and liabilities consist principally of cash and cash equivalents, accounts receivable, and accounts payable. The fair value of the Company’s cash equivalents is determined based on quoted prices in active markets for identical assets. The recorded values of the Company’s accounts receivable and accounts payable approximate their current fair values due to the relatively short-term nature of these accounts.
(t) | Net Loss per Share |
Basic net loss per share is calculated by dividing the net loss by the weighted average number of shares of common stock outstanding. Diluted net loss per share is computed by dividing the net loss by the weighted-average number of common share equivalents outstanding for the period determined using the treasury-stock method. Common stock equivalents are comprised of restricted stock units, warrants to purchase common stock and options outstanding under our stock option plans. For all periods presented, there is no difference in the number of shares used to calculate basic and diluted shares outstanding since the effects of potentially dilutive securities are antidilutive due to our net loss position.
(u) | Recently Adopted Accounting Pronouncements |
In May 2014, the Financial Accounting Standards Board, or FASB, issued Accounting Standards Update, or ASU, No. 2014-09, which amends the guidance for accounting for revenue from contracts with customers. This ASU supersedes the revenue recognition requirements in Topic 605, Revenue Recognition, and creates a new Topic 606, Revenue from Contracts with Customers. In 2015 and 2016, the FASB issued additional ASUs related to Topic 606 that delayed the effective date of the guidance and clarified various aspects of the new revenue guidance, including principal versus agent considerations, identifying performance obligations, and licensing, and they include other improvements and practical expedients. The Company adopted this new standard on January 1, 2018 using the cumulative-effect method. The impact of adoption was immaterial to the consolidated financial statements.
In May 2017, the FASB issued ASU No. 2017-09 Compensation – Stock Compensation (Topic 718) Scope of Modification Accounting. The amendments in ASU 2017-09 provide guidance about which changes to the terms or conditions of a share-based payment award require an entity to apply modification accounting in Topic 718. The Company adopted ASU 2016-01 on January 1, 2018, and the adoption did not have a material impact on its financial statements.
(v) | Recent Accounting Pronouncements |
In February 2016, the FASB issued ASU No. 2016-02—Leases, (Topic 842) (ASU 2016-02), as amended, which generally requires lessees to recognize operating and financing lease liabilities and corresponding right-of-use assets on the balance sheet and to provide enhanced disclosures surrounding the amount, timing and uncertainty of cash flows arising from leasing arrangements. In July 2018, the FASB issued ASU No. 2018-11, Leases (Topic 842): Targeted Improvements, or ASU No. 2018-11. In issuing ASU No. 2018-11, the FASB is permitting another transition method for ASU 2016-02, which allows the transition to the new lease standard by recognizing a cumulative-effect adjustment to the opening balance of accumulated deficit in the period of adoption. We will elect this transition method and package of practical expedients permitted under the transition guidance, which allows us to carryforward our historical lease classification, our assessment on whether a contract is or contains a lease, and our initial direct costs for any leases that exist prior to adoption of the new standard. We will also elect to combine lease and non-lease components and to keep leases with an initial term of 12 months or less off the balance sheet and recognize the associated lease payments in the statements of operations on a straight-line basis over the lease term. We will adopt the ASU on January 1, 2019 using the cumulative-effect method and are finalizing our assessment of the impact of the adoption of the ASU and expect to record a right-of-use asset of approximately $1.5 million and a corresponding lease liability of approximately $1.6 million to account for our facility lease.
In June 2018, the FASB issued ASU No. 2018-07, Stock Compensation (Topic 718): Improvements to Nonemployee Share-Based Payment Accounting. ASU 2018-07 is intended to reduce the cost and complexity and to improve financial reporting for nonemployee share-based payments. ASU 2018-07 expands the scope of Topic 718, Compensation-Stock Compensation (which currently only includes share-based payments to employees) to include share-based payments issued to nonemployees for goods or services. Consequently, the accounting for share-based payments to nonemployees and employees will be substantially aligned. ASU 2018-07 supersedes Subtopic 505-50, Equity-Based Payments to Non-Employees. ASU 2018-07 is effective for the Company for fiscal years beginning after December 15, 2018, including interim periods within that fiscal year and early adoption is permitted. The Company is currently assessing the impact of this standard on its consolidated financial statements.
In August 2018, the FASB issued ASU No. 2018-13, Fair Value Measurement (Topic 820): Disclosure Framework—Changes to the Disclosure Requirements for Fair Value Measurement. ASU 2018-13 considers cost and benefits, and removes, modifies and adds disclosure requirements in Topic 820. The amendments on changes in unrealized gains and losses, the range and weighted average of significant unobservable inputs used to develop Level 3 fair value measurements, and the narrative description of measurement uncertainty is to be applied prospectively for only the most recent interim or annual period presented in the initial fiscal year of adoption. All other amendments are to be applied retrospectively to all periods presented. ASU 2018-13 is effective for the Company for fiscal years beginning after December 15, 2019, including interim periods within that fiscal year and early adoption is permitted. The Company is currently assessing the impact of this standard on its consolidated financial statements.
In November 2018, the FASB issued ASU No. 2018-18, Collaborative Arrangements (Topic 808): Clarifying the Interaction Between Topic 808 and Topic 606 (“ASU 2018-18”). ASU 2018-18 clarifies when certain transactions between collaborative arrangement participants should be accounted for under Topic 606 and incorporates unit-of-account guidance consistent with Topic 606 to aid in this determination. ASU 2018-18 is effective for public companies for annual and interim periods beginning after December 15, 2019, with early adoption permitted. ASU 2018-18 should generally be applied retrospectively to the date of initial application of Topic 606. Management is currently assessing the impact ASU 2018-18 will have on the Company’s financial statements.
Other recent accounting pronouncements issued by the FASB, including its Emerging Issues Task Force, and the American Institute of Certified Public Accountants did not or are not believed by management to have a material impact on the Company’s financial statement presentation or disclosures.
(3) | Reverse Merger |
On October 24, 2016, we completed the Merger as discussed in Note 1. For financial reporting purposes, the Merger is accounted for as an asset acquisition by BioCardia Lifesciences rather than a business combination because we did not meet the definition of a business as defined by US GAAP as of immediately prior to the Merger. The fair value of the purchase consideration, consisting of Company common stock, was determined based on the fair value of the cash acquired by BioCardia Lifesciences of $19 million, which is considered the best indicator for the fair value of the purchase consideration. No other assets or liabilities were acquired by BioCardia Lifesciences. Transaction costs of $96,000 were recorded as a reduction to additional paid in capital. Additionally, pursuant to the Merger Agreement, upon consummation of the Merger, the Company assumed all of BioCardia Lifesciences, Inc. options outstanding immediately prior to the Merger at the same Exchange Ratio.
The Merger is considered a tax free reverse triangular merger for tax purposes pursuant to Internal Revenue Code Sections 368(a) and 368(a)(2)(E) in which the Company continues as the parent and BioCardia Lifesciences as the wholly owned subsidiary and is therefore treated as an acquisition of stock of BioCardia Lifesciences by the Company. Despite the stock acquisition treatment, none of our pre-Merger tax attributes remain available after the Merger as a result of limitations under Internal Revenue Code Section 382 due to lack of business continuity.
(4) | Fair Value Measurements |
The fair value of financial instruments reflects the amounts that the Company estimates to receive in connection with the sale of an asset or paid in connection with the transfer of a liability in an orderly transaction between market participants at the measurement date (exit price). The Company follows a fair value hierarchy that prioritizes the use of inputs used in valuation techniques into the following three levels:
Level 1 – quoted prices in active markets for identical assets and liabilities
Level 2 – observable inputs other than quoted prices in active markets for identical assets and liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities
Level 3 – unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
The following table sets forth the fair value of our financial assets measured on a recurring basis as of December 31, 2018 and 2017 and indicates the fair value hierarchy utilized to determine such fair value (in thousands).
As of December 31, 2018 | ||||||||||||||||
Level 1 | Level 2 | Level 3 | Total | |||||||||||||
Assets: | ||||||||||||||||
Money market funds | $ | 5,358 | $ | — | $ | — | $ | 5,358 | ||||||||
As of December 31, 2017 | ||||||||||||||||
Level 1 | Level 2 | Level 3 | Total | |||||||||||||
Assets: | ||||||||||||||||
Money market funds | $ | 12,689 | $ | — | $ | — | $ | 12,689 | ||||||||
(5) | Inventories |
Inventories are stated at the lower of cost or net realizable value using the average cost method. Inventories consist of the following (in thousands):
December 31, | ||||||||
2018 | 2017 | |||||||
Raw materials | $ | 79 | $ | 70 | ||||
Work in process | 39 | 92 | ||||||
Finished goods | 23 | 29 | ||||||
Total | $ | 141 | $ | 191 | ||||
Write downs for excess or expired inventory are based on management’s estimates of forecasted usage of inventories and are included in cost of goods sold. A significant change in the timing or level of demand for certain products as compared to forecasted amounts may result in recording additional write downs for excess or expired inventory in the future. Charges to cost of goods sold for inventory write-downs, reserve adjustments, scrap, shrinkage and expired inventories totaled approximately $12,000, $33,000 and $52,000 for the years ended December 31, 2018, 2017 and 2016, respectively.
(6) | Property and Equipment, Net |
Property and equipment, net consist of the following (in thousands):
December 31, | ||||||||
2018 | 2017 | |||||||
Computer equipment and software | $ | 119 | 106 | |||||
Laboratory and manufacturing equipment | 481 | 447 | ||||||
Furniture and fixtures | 55 | 48 | ||||||
Leasehold improvements | 332 | 326 | ||||||
Construction in progress | 3 | — | ||||||
Property and equipment, gross | 990 | 927 | ||||||
Less accumulated depreciation | (845 | ) | (758 | ) | ||||
Property and equipment, net | $ | 145 | 169 | |||||
Depreciation expense totaled approximately $88,000, $78,000 and $39,000 for the years ended December 31, 2018, 2017 and 2016, respectively. All of the Company’s property and equipment is located in the United States.
(7) | Commitments |
In November 2016, the Company entered into an amendment to its lease with respect to its office and laboratory space. The extended term of this lease is 60 months and commenced on January 1, 2017 and will expire on December 31, 2021. Rent expense is recognized on a straight-line basis over the life of the lease. Rental expense was approximately $601,000 for the years ended December 31, 2018 and 2017, and $321,000 for the year ended December 31, 2016. Future minimum lease payments under the lease as of December 31, 2018 are as follows (in thousands):
Years ending December 31: | ||||
2019 | $ | 612 | ||
2020 | 630 | |||
2021 | 649 | |||
Total | $ | 1,891 |
(8) | Collaborative Agreements |
The Company has entered into various collaborations related to clinical development. These agreements allow partners to utilize the Company’s enabling biotherapeutic delivery systems, including training and support during clinical and pre-clinical delivery of biotherapeutics. Under the terms of these agreements, the Company typically receives a use fee and payments for the systems and services provided. The Company gains access to certain data generated by its partners for use in its own product development efforts and also receives nonexclusive patent rights to any BioCardia technology improvement inventions.
(9) | Accrued Expenses and Other Current Liabilities |
Accrued expenses and other current liabilities consisted of the following (in thousands):
December 31, | ||||||||
2018 | 2017 | |||||||
Accrued expenses | $ | 495 | $ | 465 | ||||
Accrued clinical trial costs | 276 | 74 | ||||||
Grant liability | 645 | 663 | ||||||
Customer deposits | 112 | 61 | ||||||
Total | $ | 1,528 | $ | 1,263 | ||||
(10) | Sales of Unregistered Securities |
On December 24, 2018, the Company entered into a Securities Purchase Agreement (the “Securities Purchase Agreement”) with entities affiliated with its existing investors (the “Investors”), relating to an offering and sale (the “Offering”) of an aggregate of 5,333,332 shares of the Company’s common stock at a purchase price of $0.75 per share, and warrants to purchase up to one-half of the number of shares of common stock sold to an Investor, up to an aggregate for all Investors of 2,666,666 shares of Common Stock (the “Warrant Shares”) at an exercise price of $0.75 per share, for aggregate net proceeds of $3.8 million net of $200,000 expenses. Both the common stock and the common stock warrants are classified as equity and recorded on a relative fair value basis. The warrants will expire on December 24, 2023. The warrants contain customary adjustments and are exercisable immediately for cash and after six months will also be exercisable on a cashless basis if there is no effective registration statement registering the resale of the Warrant Shares. The Investors do not have registration rights in connection with any securities purchased in the Offering. The closing of the Offering took place on December 24, 2018.
(11) | Share-Based Compensation |
BioCardia Lifesciences adopted, and the BioCardia Lifesciences shareholders approved, the 2002 Stock Plan in 2002 (the “2002 Plan”), and the Company assumed the 2002 Plan in the Merger. We have not granted or do not intend to grant any additional awards under the 2002 Plan following the Merger. In 2016, BioCardia Lifesciences adopted, and the BioCardia Lifesciences shareholders approved, the 2016 Equity Incentive Plan (the “2016 Plan”), and the Company assumed the 2016 Plan in the Merger. We have granted awards, including incentive stock options and nonstatutory stock options, under the 2016 Plan following the Merger. Under the 2002 Plan and the 2016 Plan, the number of shares, terms, and vesting periods are determined by the Company’s board of directors or a committee thereof on an option-by-option basis. Options generally vest ratably over service periods of four years and expire ten years from the date of grant. The per share exercise price shall be no less than the fair market value on the date of grant. Compensation cost for employee share-based awards is based on the grant-date fair value and is recognized over the vesting period of the applicable award on a straight-line basis. As of December 31, 2018, 7,932,494 shares have been authorized for awards under the 2016 Plan.
The Company recognizes in the consolidated statements of operations the grant-date fair value of stock options and other equity-based compensation. Share-based compensation expense for the years ended December 31, 2018, 2017 and 2016 was recorded as follows (in thousands):
Years ended December 31, | ||||||||||||
2018 | 2017 | 2015 | ||||||||||
Cost of goods sold | $ | 143 | $ | 140 | $ | 14 | ||||||
Research and development | 953 | 678 | 127 | |||||||||
Selling, general and administrative | 1,678 | 1,889 | 801 | |||||||||
Share-based compensation expense | $ | 2,774 | $ | 2,707 | $ | 942 | ||||||
The following table summarizes activity under the Company’s stock option plans, including the 2002 Plan and the 2016 Plan and related information (in thousands, except share and per share amounts and term):
Options outstanding | ||||||||||||||||
Number of shares | Weighted average exercise price | Weighted average remaining contractual term (years) | Aggregate intrisinsic value | |||||||||||||
Balance, December 31, 2017 | 4,213,100 | $ | 2.96 | 8.1 | $ | 1,890 | ||||||||||
Stock options granted | 1,698,452 | 2.41 | ||||||||||||||
Stock options exercised | (2,488 | ) | 1.80 | |||||||||||||
Stock options canceled | (431,700 | ) | 2.92 | |||||||||||||
Balance, December 31, 2018 | 5,477,364 | $ | 2.80 | 7.8 | $ | - | ||||||||||
Exercisable and vested, December 31, 2018 | 2,346,990 | $ | 2.72 | 6.9 | $ | - | ||||||||||
The total intrinsic value of options exercised during the years ended December 31, 2018, 2017 and 2016 was approximately $3,000, $400,000 and $144,000, respectively. The weighted average grant-date fair value of options granted during the years ended December 31, 2018, 2017 and 2016 was $1.72, $5.80 and $1.33 per share, respectively.
Employee Share-Based Compensation
During the years ended December 31, 2018 and 2017, the Company granted stock options to certain non-employee directors and employees to purchase 1,698,452 and 796,399 shares of common stock, respectively. The fair value of each option grant was estimated on the date of the grant using the Black-Scholes option pricing model with the following assumptions:
Years ended December 31, | |||||||||||||||
2018 | 2017 | 2016 | |||||||||||||
Risk-free interest rate | 2.66 | - | 2.89% | 1.76 | - | 2.25% | 1.28 | - | 1.58% | ||||||
Volatility | 81 | - | 83% | 81 | - | 89% | 88% | ||||||||
Dividend yield | None | None | None | ||||||||||||
Expected term (in years) | 6.25 | 5.00 | - | 6.25 | 6.25 | ||||||||||
Unrecognized share-based compensation for non-employee directors and employee options granted through December 31, 2018 is approximately $5.0 million to be recognized over a remaining weighted average service period of 2.4 years.
Non-Employee Director Share-Based Compensation (RSUs)
During the year ended December 31, 2018, the Company granted to certain non-employee directors 226,471 restricted stock units, or RSUs. The fair value of each RSU is estimated on the closing market price on the grant date.
The following summarizes the activity of non-vested RSUs:
Weighted | ||||||||
average | ||||||||
grant date | ||||||||
Number of | fair value | |||||||
shares | per share | |||||||
Balance, December 31, 2017 | 97,996 | $ | 8.71 | |||||
RSUs granted | 226,471 | $ | 1.36 | |||||
RSUs vested | (57,108 | ) | $ | 7.03 | ||||
RSUs forfeited | - | — | ||||||
Balance, December 31, 2018 | 267,359 | $ | 2.84 | |||||
Unrecognized share-based compensation for employee RSUs granted through December 31, 2018 was approximately $235,000 to be recognized over a remaining weighted average service period of 1.0 years.
Nonemployee Share-Based Compensation
During the year ended December 31, 2018, 2017 and 2016, the Company granted options to purchase, zero, 46,254 and 490,849 shares of common stock to consultants. These options were granted in exchange for consulting services to be rendered and vest over the term specified in the grant, which correlates to the period the services are rendered. The Company recorded nonemployee share-based compensation expense of $176,000, $768,000 and $545,000 for the years ended December 31, 2018, 2017 and 2016 respectively. Unrecognized share-based compensation expense for nonemployee options granted through December 31, 2018 is approximately $204,000 to be recognized over a remaining weighted average service period of 1.8 years.
The Company accounts for share-based compensation arrangements with nonemployees, using the Black Scholes option pricing model, based on the fair value as these instruments vest. Accordingly, at each reporting date, the Company revalues the unearned portion of the share-based compensation and the resulting change in fair value is recognized in the consolidated statements of operations over the period the related services are rendered. The following assumptions were used to value the awards.
Years ended December 31, | |||||||||||||||
2018 | 2017 | 2016 | |||||||||||||
Risk-free interest rate | 2.80 | - | 2.95% | 2.25 | - | 2.40% | 1.60 | - | 2.42% | ||||||
Volatility | 78 | - | 85% | 81 | - | 87% | 89 | - | 91% | ||||||
Dividend yield | None | None | None | ||||||||||||
Expected term (in years) | 7.6 | - | 8.8 | 8.6 | - | 9.8 | 9.6 | - | 9.9 | ||||||
(12) | Concentrations |
Most of the Company’s customers are located in the United States. One customer accounted for approximately 29% of revenue in 2018 and no single customer accounted for more than 10% of revenue in 2017 or 2016. One customer accounted for 23% of accounts receivable at December 31, 2018, 20% of accounts receivable at December 31, 2017 and another customer accounted for 29% of accounts receivable at December 31, 2016.
(13) | Net Loss per Share |
The following table sets forth the computation of the basic and diluted net loss per share for the years ended December 31, 2018, 2017 and 2016 (in thousands, except share and per share data):
Years ended December 31, | ||||||||||||
2018 | 2017 | 2016 | ||||||||||
Numerator: | ||||||||||||
Net loss | $ | (13,987 | ) | $ | (12,308 | ) | $ | (10,310 | ) | |||
Denominator: | ||||||||||||
Weighted average shares used to compute net loss per share, basic and diluted | 38,377,606 | 38,160,543 | 8,368,284 | |||||||||
Net loss per share, basic and diluted | $ | (0.36 | ) | $ | (0.32 | ) | $ | (1.23 | ) | |||
The following weighted-average outstanding common stock equivalents were excluded from the computation of diluted net loss per share for the periods presented because including them would have been antidilutive:
December 31, | ||||||||||||
2018 | 2017 | 2016 | ||||||||||
Stock options to purchase common stock | 5,477,364 | 4,213,100 | 3,491,937 | |||||||||
Unvested restricted stock units | 267,359 | 97,996 | - | |||||||||
| Common stock warrants | 2,666,666 | - | - | |||||||||
Total | 8,411,389 | 4,311,096 | 3,491,937 | |||||||||
(14) | Income Taxes |
The Company’s provision for income taxes for the years ended December 31, 2018, 2017 and 2016 was $0 for all years.
The provision for income taxes differs from the amount which would result by applying the federal statutory income tax rate to pre-tax loss for the years ended December 31, 2018, 2017 and 2016. The reconciliation of the provision computed at the federal statutory rate to the Company’s provision (benefit) for income taxes was as follows (in thousands):
Years ended December 31, | ||||||||||||
2018 | 2017 | 2016 | ||||||||||
Tax at federal statutory rate | $ | (2,937 | ) | $ | (4,185 | ) | $ | (3,505 | ) | |||
State, net of federal benefit | (455 | ) | (1,238 | ) | (315 | ) | ||||||
Research and development credit | (236 | ) | (135 | ) | (89 | ) | ||||||
Stock-based compensation | 440 | 344 | 136 | |||||||||
Nondeductible interest | — | — | 590 | |||||||||
Warrant and derivative revaluation | — | — | 328 | |||||||||
Change in Federal tax rate | — | 8,172 | — | |||||||||
Other | 22 | 7 | 94 | |||||||||
Change in valuation allowance | 3,166 | (2,965 | ) | 2,761 | ||||||||
Total provision for income taxes | $ | — | $ | — | $ | — | ||||||
Deferred income taxes reflect the net tax effects of temporary differences between carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes as well as net operating loss and tax credit carryforwards, net of any adjustment for unrecognized tax benefits. The components of the net deferred income tax assets as of December 31, 2018 and 2017 were as follows (in thousands):
December 31, | ||||||||
2018 | 2017 | |||||||
Accrued compensation | $ | 84 | $ | 110 | ||||
Inventory adjustments | 276 | 449 | ||||||
Depreciation and amortization - noncurrent | 113 | 146 | ||||||
Share-based compensation | 648 | 558 | ||||||
Net operating loss and tax credit carryforwards - noncurrent | 20,698 | 17,368 | ||||||
Other | 12 | 34 | ||||||
Gross deferred tax asset | 21,831 | 18,665 | ||||||
Valuation allowance | (21,831 | ) | (18,665 | ) | ||||
Net deferred tax asset | $ | — | $ | — | ||||
The Company has approximately $69.2 million and $54.4 million of federal and state net operating loss carryforwards, respectively, as of December 31, 2018. For tax reporting purposes, operating loss carryforwards are available to offset future taxable income; such carryforwards expire in varying amounts beginning in 2022 and 2028 for federal and state purposes, respectively, with 2018 federal NOLs having no expiration date. Under current federal and California law, the amounts of and benefits from net operating losses carried forward may be impaired or limited in certain circumstances. Events which may cause limitations in the amount of net operating losses that the company may utilize in any one year include, but are not limited to, a cumulative ownership change of more than 50% over a three-year period.
Generally, utilization of the net operating loss carryforwards and credits may be subject to a substantial annual limitation due to the ownership change limitations provided by section 382, which discusses limitations on NOL carryforwards and certain built-in losses following ownership changes, and section 383, which discusses, special limitations on certain excess credits, etc., of the Internal Revenue Code (IRC) of 1986, as amended and similar state provisions. Accordingly, our ability to utilize net operating losses carryforwards may be limited, potentially significantly, as the result of such an “ownership change”. The Company has not yet performed a comprehensive study to determine if it has undergone any ownership changes. If the Company is able to potentially utilize its net operating loss carryforwards, it will perform a comprehensive section 382 study to determine what, if any, limitation on its ability to utilize its NOLs exists.
At December 31, 2018, the Company has federal and state research and development credits of approximately $2.0 million and $1.5 million available to offset future federal and state income taxes, respectively. The federal tax credit carryforward expires beginning in 2028. The state credit carryforwards have no expiration.
The Company does not believe that these assets are realizable on a more-likely than not basis; therefore, the net deferred tax assets have been fully offset by a valuation allowance. The Company did not have deferred tax liabilities as of December 31, 2018 or 2017. The net increase in the total valuation allowance for the year ending December 31, 2018 is $3.2 million, primarily from the net operating losses generated. The net decrease in the total valuation allowance for the year ending December 31, 2017 was $3.0 million, primarily from the decrease in Federal Tax Rate applied to deferred tax assets, with an offsetting increase from net operating losses generated.
No liability related to uncertain tax positions is reported in the financial statements.
The aggregate changes in the balance of gross unrecognized tax benefits were as follows (in thousands):
December 31, | ||||||||
2018 | 2017 | |||||||
Balance, beginning of year | $ | 725 | $ | 608 | ||||
Additions based on tax positions related to the current year | 166 | 117 | ||||||
Additions (reductions) for tax positions related to prior years | — | — | ||||||
Balance, end of year | $ | 891 | $ | 725 | ||||
Recognition of approximately $636,000 and $511,000 of unrecognized tax benefits would impact the effective rate at December 31, 2018 and 2017 respectively, if recognized. Contributing to the increase in amount impacting the rate in 2017 was the consideration of the federal tax rate change as a result of the Tax Act. Increases in 2018 relate to increased research and development activity.
The Company is subject to U.S. federal, California, Colorado, Florida and Minnesota income taxes. Tax regulations within each jurisdiction are subject to the interpretation of the related tax laws and regulations and require significant judgment to apply. The Company was incorporated in 2002 and is subject to U.S. federal, state, and local tax examinations by tax authorities for all prior years.
US Tax Reform - Impact of the Tax Cuts and Jobs Act
On December 22, 2017, the Tax Cuts and Jobs Act (H.R. 1) (the “Tax Act”) was signed into law. The Tax Act contains significant changes to corporate taxation, including; (i) the reduction of the corporate income tax rate from a maximum rate of 35% to 21%, (ii) the acceleration of expensing for certain business assets, (iii) the one-time transition tax related to the transition of U.S. international tax from a worldwide tax system to a territorial tax system, (iv) the repeal of the domestic production deduction, (v) additional limitations on the deductibility of interest expense, (vi) expanded limitations on executive compensation, (vii) acceleration of tax revenue recognition, (viii) capitalization of research and development expenditures and (ix) creation of new minimum taxes such as the base erosion anti-abuse tax (“BEAT”) and Global Intangible Low Taxed Income (“GILTI”) tax.
After the enactment of the Tax Act, the Securities and Exchange Commission ("SEC") staff issued Staff Accounting Bulletin No. 118 ("SAB 118") to address the application of U.S. GAAP in situations when an entity does not have the necessary information available, prepared or analyzed (including computations) in reasonable details to complete the accounting for certain income tax effects of the Act. The Company has made adjustments to reduce its deferred tax assets and liabilities as of December 31, 2017, based on the reduction of the U.S. federal corporate rate from 34% to 21% and assessed the reliability of its deferred tax assets based on its understanding of the provisions of the new law. As of December 31, 2018, the Company completed its assessment of the impact of the Tax Act and determined no additional adjustments are required.
The Company has considered required policy elections with respect to its treatment of potential base erosion anti-abuse tax (“BEAT”) and Global Intangible Low Taxed Income (“GILTI”). Companies can either account for taxes on BEAT and GILTI as incurred or recognize deferred taxes when basis differences exist that are expected to affect the amount of the BEAT and GILTI inclusion upon reversal. The Company has considered the provisions of the Act associated with BEAT and GILTI and noted that these are not applicable as of December 31, 2018. The Company expects to account for any taxes on BEAT and GILTI as incurred if applicable.
(15) | Contingencies |
The Company may be subject to various claims, complaints, and legal actions that arise from time to time in the normal course of business. Management is not aware of any current legal or administrative proceedings that are likely to have a material effect on the Company’s business, financial position, results of operations, or cash flows.
(16) | Grant Funding |
In June 2016, the Company entered into a grant agreement with Maryland Technology Development Corporation (“TEDCO”). TEDCO was created by the Maryland State Legislature in 1998 to facilitate the transfer and commercialization of technology from Maryland’s research universities and federal labs into the marketplace. TEDCO administers the Maryland Stem Cell Research Fund to promote State funded stem cell research and cures through financial assistance to public and private entities operating within the State. Under the agreement, TEDCO has agreed to provide the Company an amount not to exceed $750,000 to be used solely to finance the costs to conduct the research project entitled “Heart Failure Trial” over a period of three years.
As of December 31, 2018, the Company has received approximately $750,000 under the grant which is accounted for as a reduction to research and development expenses as the related qualifying costs are incurred. Approximately $105,000 of the qualifying costs had been incurred as of December 31, 2018. The remaining $645,000 was recorded as grant liability on the consolidated balance sheet at December 31, 2018. The amount is recorded as a liability as the amounts are refundable, should a default by the Company, as defined in the agreement, occur prior to incurring the qualifying costs.
(17) | Related Party Transactions |
In August 2016, the Company granted an option to purchase 418,977 shares of common stock, with 4-year vesting period and an exercise price of $1.80 per share, to OPKO Health, Inc. (“OPKO”) as consideration for consulting services to be provided by OPKO. The Company recorded $142,000 and $480,000 as share-based compensation expense related to the OPKO stock option during the years ended December 31, 2018 and 2017, respectively. The estimated grant-date fair value of the option was $5.3 million. The term of the consulting agreement is 4 years and will be automatically renewed for successive one year periods. The chairman and chief executive officer of OPKO is a beneficial owner of more than 5% of the outstanding shares of the Company’s common stock.
(18) | Employee Benefit Plans |
The Company’s U.S. employees are eligible to participate in a retirement and savings plan that qualifies under Section 401(k) of the Internal Revenue Code. Participating employees may contribute up to 75% of their pretax salary, but not more than statutory limits. The Company did not make any matching contributions during the years ended December 31, 2017 and 2016, but did make a $26,000 matching contribution in the year ending December 31, 2018.
PART I. FINANCIAL INFORMATION
ITEM 1. UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
BIOCARDIA, INC.
Condensed Consolidated Balance Sheets
(In thousands, except share and per share amounts)
March 31, | December 31, | |||||||
| 2019 | 2018 | ||||||
(unaudited) | ||||||||
| Assets | ||||||||
Current assets: | ||||||||
Cash and cash equivalents | $ | 2,838 | $ | 5,358 | ||||
Accounts receivable, net of allowance for doubtful accounts of $14 and $9 at March 31, 2019 and December 31, 2018 | 287 | 274 | ||||||
Inventory | 118 | 141 | ||||||
Prepaid expenses and other current assets | 285 | 445 | ||||||
Total current assets | 3,528 | 6,218 | ||||||
Property and equipment, net | 175 | 145 | ||||||
Operating lease right-of-use asset, net | 1,400 | — | ||||||
Other assets | 54 | 54 | ||||||
Total assets | $ | 5,157 | $ | 6,417 | ||||
Liabilities and Stockholders’ Equity | ||||||||
Current liabilities: | ||||||||
Accounts payable | $ | 1,139 | $ | 1,020 | ||||
Accrued expenses and other current liabilities | 1,715 | 1,528 | ||||||
Operating lease liability - current | 469 | — | ||||||
Total current liabilities | 3,323 | 2,548 | ||||||
Operating lease liability - noncurrent | 1,016 | — | ||||||
Deferred rent | — | 77 | ||||||
Total liabilities | 4,339 | 2,625 | ||||||
Stockholders’ equity: | ||||||||
Preferred stock, $0.001 par value, 25,000,000 shares authorized as of March 31, 2019 and December 31, 2018; no shares issued and outstanding as of March 31, 2019 and December 31, 2018 | — | — | ||||||
Common stock, $0.001 par value, 100,000,000 shares authorized as of March 31, 2019 and December 31, 2018; 43,631,684 and 43,611,240 shares issued and outstanding as of March 31, 2019 and December 31, 2018 | 44 | 43 | ||||||
Additional paid-in capital | 90,800 | 90,110 | ||||||
Accumulated deficit | (90,026 | ) | (86,361 | ) | ||||
Total stockholders’ equity | 818 | 3,792 | ||||||
Total liabilities and stockholders’ equity | $ | 5,157 | $ | 6,417 | ||||
See accompanying notes to condensed consolidated financial statements.
BIOCARDIA, INC.
Condensed Consolidated Statements of Operations
(In thousands, except share and per share amounts)
(unaudited)
Three months ended March 31, | ||||||||
2019 | 2018 | |||||||
Revenue: | ||||||||
Net product revenue | $ | 76 | $ | 82 | ||||
Collaboration agreement revenue | 140 | 117 | ||||||
Total revenue | 216 | 199 | ||||||
Costs and expenses: | ||||||||
Cost of goods sold | 106 | 157 | ||||||
Research and development | 2,166 | 1,955 | ||||||
Selling, general and administrative | 1,631 | 1,707 | ||||||
Total costs and expenses | 3,903 | 3,819 | ||||||
Operating loss | (3,687 | ) | (3,620 | ) | ||||
Other income (expense): | ||||||||
Interest income | 23 | 36 | ||||||
Other expense | (1 | ) | — | |||||
| Total other income, net | 22 | 36 | ||||||
Net loss | $ | (3,665 | ) | $ | (3,584 | ) | ||
Net loss per share, basic and diluted | $ | (0.08 | ) | $ | (0.09 | ) | ||
Weighted-average shares used in computing net loss per share, basic and diluted | 43,628,958 | 38,236,056 | ||||||
See accompanying notes to condensed consolidated financial statements.
BIOCARDIA, INC.
Condensed Consolidated Statements of Stockholders’ Equity (Deficit)
(In thousands, except share amounts)
(unaudited)
Common stock | Additional | Accumulated | ||||||||||||||||||
Shares | Cost | paid in capital | deficit | Total | ||||||||||||||||
Balance at December 31, 2018 | 43,611,240 | $ | 43 | $ | 90,110 | $ | (86,361 | ) | $ | 3,792 | ||||||||||
Restricted stock units vested and issued | 20,444 | 1 | — | — | 1 | |||||||||||||||
Share-based compensation | — | — | 690 | — | 690 | |||||||||||||||
Net loss | — | — | — | (3,665 | ) | (3,665 | ) | |||||||||||||
Balance at March 31, 2019 | 43,631,684 | $ | 44 | $ | 90,800 | $ | (90,026 | ) | $ | 818 | ||||||||||
Balance at December 31, 2017 | 38,218,660 | $ | 38 | $ | 83,537 | $ | (72,450 | ) | $ | 11,125 | ||||||||||
Adjustments to opening balance for change in accounting principle | — | — | — | 46 | 46 | |||||||||||||||
Restricted stock units vested and issued | 20,444 | — | — | — | — | |||||||||||||||
Exercise of stock options | 2,140 | — | 5 | — | 5 | |||||||||||||||
Share-based compensation | — | — | 615 | — | 615 | |||||||||||||||
Net loss | — | — | — | (3,584 | ) | (3,584 | ) | |||||||||||||
Balance at March 31, 2018 | 38,241,244 | $ | 38 | $ | 84,157 | $ | (75,988 | ) | $ | 8,207 | ||||||||||
See accompanying notes to condensed consolidated financial statements.
BIOCARDIA, INC.
Condensed Consolidated Statements of Cash Flows
(In thousands)
(unaudited)
Three months ended March 31, | ||||||||
2019 | 2018 | |||||||
Operating activities: | ||||||||
Net loss | $ | (3,665 | ) | $ | (3,584 | ) | ||
Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
Depreciation and amortization | 23 | 22 | ||||||
Amortization of right-of-use asset | 105 | — | ||||||
Share-based compensation | 690 | 615 | ||||||
Changes in operating assets and liabilities: | ||||||||
Accounts receivable | (13 | ) | (32 | ) | ||||
Inventory | 23 | 43 | ||||||
Prepaid expenses and other current assets | 160 | 63 | ||||||
Accounts payable | 122 | (164 | ) | |||||
Accrued expenses and other current liabilities | (36 | ) | 34 | |||||
Deferred revenue | — | (35 | ) | |||||
Deferred rent | — | 2 | ||||||
Operating lease liability - noncurrent | 126 | — | ||||||
Net cash used in operating activities | (2,465 | ) | (3,036 | ) | ||||
Investing activities: | ||||||||
Purchase of property and equipment | (55 | ) | (5 | ) | ||||
Net cash used in investing activities | (55 | ) | (5 | ) | ||||
Financing activities: | ||||||||
Proceeds from the exercise of common stock options | — | 5 | ||||||
Net cash provided by financing activities | — | 5 | ||||||
Net change in cash and cash equivalents | (2,520 | ) | (3,036 | ) | ||||
Cash and cash equivalents at beginning of period | 5,358 | 12,689 | ||||||
Cash and cash equivalents at end of period | $ | 2,838 | $ | 9,653 | ||||
See accompanying notes to condensed consolidated financial statements.
(1) | Summary of Business and Basis of Presentation |
(a) | Description of Business | |
BioCardia, Inc., (BioCardia or the Company), is a clinical-stage regenerative medicine company developing novel therapeutics for cardiovascular diseases with large unmet medical needs. The Company’s lead therapeutic candidate is the CardiAMP® cell therapy system and its second therapeutic candidate is the CardiALLO™ cell therapy system. To date, the Company has devoted substantially all its resources to research and development efforts relating to its therapeutic candidates and biotherapeutic delivery systems including conducting clinical trials, developing manufacturing and sales capabilities, in-licensing related intellectual property, providing general and administrative support for these operations and protecting its intellectual property. | ||
BioCardia also has three enabling device product lines: (1) the CardiAMP cell processing system; (2) the Helix™ biotherapeutic delivery system, or Helix; and (3) the Morph® vascular access product line, or Morph. The Company manages its operations as a single segment for the purposes of assessing performance and making operating decisions. |
(2) | Significant Accounting Policies |
(b) |
| |
| ||
The Company has incurred net losses
| ||
The Company’s ability to continue
|
| (c) | Use of Estimates
|
| The preparation of the financial statements |
(d) | Principles of Consolidation | |
| The condensed consolidated financial statements include the |
(e) | Changes to Significant Accounting Policies | |||||||||||||||||||||
The Company’s significant accounting policies are described in
Measurement of nonemployee awards - The measurement of equity-classified nonemployee awards is fixed at the grant date, and Operating lease right-of-use asset and liabilities - The Company will determine if an arrangement is a lease at the inception of the arrangement. All leases are assessed for The Company’s lease liabilities are recognized at the The Company's ROU assets are also recognized at
The
The Company’s lease contracts often include lease and non-lease components. The Company
The Company has
|
(f) | Recently Adopted Accounting Pronouncement
In February 2016, the FASB amended its guidance related to lease accounting. The amended guidance required lessees to recognize a majority of
|
The FASB made available several practical expedients in The most significant impact was the recognition of ROU assets and related lease liabilities for operating leases on the Condensed Consolidated Balance Sheet. The Company recognized ROU assets and related lease liabilities of $1,505,000 and $1,593,000 respectively, related to operating lease commitments, as of January 1, 2019. The operating lease ROU asset represents the lease liability, plus any lease payments made at or before the commencement date, less any lease incentives received. The amended guidance did not have In
|
|
| Recently Issued Accounting
|
| Recent accounting pronouncements issued by the FASB, |
(3) Fair Value Measurement
The fair value of financial instruments reflects the amounts that the Company estimates to receive in connection with the sale of an asset or paid in connection with the transfer of a liability in an orderly transaction between market participants at the measurement date (exit price). The Company follows a fair value hierarchy that prioritizes the use of inputs used in valuation techniques into the following three levels:
Level 1 – quoted prices in active markets for identical assets and liabilities
Level 2 – observable inputs other than quoted prices in active markets for identical assets and liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities
Level 3 – unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
The following table sets forth the fair value of its financial assets measured on a recurring basis as of March 31, 2019 and December 31, 2018 and indicates the fair value hierarchy utilized to determine such fair value (in thousands).
As of March 31, 2019 | ||||||||||||||||
Level 1 | Level 2 | Level 3 | Total | |||||||||||||
Assets: | ||||||||||||||||
Money market funds | $ | 2,838 | $ | — | $ | — | $ | 2,838 | ||||||||
As of December 31, 2018 | ||||||||||||||||
Level 1 | Level 2 | Level 3 | Total | |||||||||||||
Assets: | ||||||||||||||||
Money market funds | $ | 5,358 | $ | — | $ | — | $ | 5,358 | ||||||||
(4) Inventories
Inventories are stated at the lower of cost or net realizable value using the average cost method. Inventories consisted of the following (in thousands):
March 31, | December 31, | |||||||
2019 | 2018 | |||||||
Raw materials | $ | 82 | $ | 79 | ||||
Work in process | 27 | 39 | ||||||
Finished goods | 9 | 23 | ||||||
Total | $ | 118 | $ | 141 | ||||
Write downs for excess or expired inventory are based on management’s estimates of forecasted usage of inventories and are included in cost of goods sold. A significant change in the timing or level of demand for certain products as compared to forecasted amounts may result in recording additional write downs for excess or expired inventory in the future. Charges to cost of goods sold for inventory write-downs, reserve adjustments, scrap, shrinkage and expired inventories totaled approximately $2,000, and $2,000 for the three months ended March 31, 2019 and 2018, respectively.
(5) Property and Equipment, Net
Property and equipment, net consisted of the following (in thousands):
March 31, | December 31, | |||||||
2019 | 2018 | |||||||
Computer equipment and software | $ | 121 | 119 | |||||
Laboratory and manufacturing equipment | 532 | 481 | ||||||
Furniture and fixtures | 55 | 55 | ||||||
Leasehold improvements | 332 | 332 | ||||||
Construction in progress | 3 | 3 | ||||||
Property and equipment, gross | 1,043 | 990 | ||||||
Less accumulated depreciation | (868 | ) | (845 | ) | ||||
Property and equipment, net | $ | 175 | 145 | |||||
Depreciation expense totaled approximately $23,000 and $22,000 for the three months ended March 31, 2019 and 2018, respectively.
(6) Operating Lease Right-of-Use Asset, Net
The Company adopted the new lease standard on January 1, 2019 using the cumulative-effect method. Prior periods were not retrospectively adjusted and continue to be reported under the accounting standards in effect for those periods.
The Company determines if an arrangement is a lease at inception by assessing whether it conveys the right to control the use of an identified asset for a period of time in exchange for consideration. The Company’s operating lease is primarily related to a property lease for its laboratory and corporate offices. BioCardia’s lease agreement does not contain any material residual guarantees or material restrictive covenants, nor does it contain an additional lease extension.
ROU assets and lease liabilities are recognized at the lease commencement date based on the present value of lease payments over the lease term. The Company’s lease does not provide an implicit rate. The Company used an adjusted historical incremental borrowing rate, based on the information available at the approximate lease commencement date, to determine the present value of lease payments. The net lease asset was adjusted for deferred rent, lease incentives, and prepaid rent. Variable rent expense is made up of expenses for common area maintenance and shared utilities and were not included in the determination of the present value of lease payments. The Company has no finance leases. The new lease standard did not materially impact its condensed consolidated statements of operations.
The impact of the new lease standard on the March 31, 2019 was as follows (in thousands, except years and percentages):
March 31, 2019 | ||||
Straight-line rent expense recognized for operating lease | $ | 150 | ||
Variable rent expense recognized for operating lease | 75 | |||
| Total lease cost | $ | 225 | ||
Weighted average remaining lease term (in years) | 2.75 | |||
Weighted average discount rate | 12.05 | % | ||
Supplemental cash flow information related to the operating lease was as follows (in thousands):
March 31, 2019 | ||||
Cash paid for amounts included in the measurement of lease liabilities | $ | 153 | ||
Cash lease expense (imputed interest expense component of net income) | 45 | |||
Future minimum lease payments under the operating lease as of March 31, 2019 are as follows (in thousands):
Operating lease | ||||
March 31, 2019 | ||||
2019 (excluding the three months ended March 31, 2019) | $ | 459 | ||
2020 | 630 | |||
2021 | 649 | |||
Total undiscounted lease payments | $ | 1,738 | ||
Less: imputed interest | 253 | |||
Total operating lease liabilities | $ | 1,485 | ||
Rent expense under the operating lease was $150,000 and $153,000 for the three months ended March 31, 2019 and 2018, respectively. Prior to the Company’s adoption of the new leases standard, future minimum lease payments as of December 31, 2018, which were undiscounted, were as follows (in thousands):
Years ending December 31: | ||||
2019 | $ | 612 | ||
2020 | 630 | |||
2021 | 649 | |||
Total | $ | 1,891 |
(7) Accrued Expenses and Other Current Liabilities
Accrued expenses and other current liabilities consisted of the following (in thousands):
March 31, | December 31, | |||||||
2019 | 2018 | |||||||
Accrued expenses | $ | 596 | $ | 495 | ||||
Accrued clinical trial costs | 363 | 276 | ||||||
Grant liability | 638 | 645 | ||||||
Customer deposits | 118 | 112 | ||||||
Total | $ | 1,715 | $ | 1,528 | ||||
(8) Warrants for Common Stock
On December 24, 2018, the Company issued 2,666,666 warrants to purchase the Company’s common stock in connection with the sale of an aggregate of 5,333,332 shares of the Company’s common stock at a purchase price of $0.75 per share for aggregate proceeds of $3.8 million, net of $200,000 expenses. The warrants are exercisable immediately for cash and after six months will also be exercisable on a cashless basis if there is no effective registration statement registering the resale of the warrants. Warrants can be settled in unregistered shares. The warrants have an exercise price of $0.75 per share and will expire on December 24, 2023. The issued warrants are standalone financial instruments and were equity classified in accordance with US GAAP.
(9) Share-Based Compensation
The share-based compensation expense is recorded in cost of goods sold, research and development, and selling, general and administrative expenses based on the employee's respective function. No share-based compensation was capitalized during the periods presented. Share-based compensation expense for the three months ended March 31, 2019 and 2018 was recorded as follows (in thousands):
Three months ended March 31, | ||||||||
2019 | 2018 | |||||||
Cost of goods sold | $ | 46 | $ | 30 | ||||
Research and development | 242 | 197 | ||||||
Selling, general and administrative | 402 | 388 | ||||||
Share-based compensation expense | $ | 690 | $ | 615 | ||||
The following table summarizes the activity of stock options and related information:
Options outstanding | ||||||||
Number of shares | Weighted average exercise price | |||||||
Balance, December 31, 2018 | 5,477,364 | $ | 2.80 | |||||
Stock options granted | — | — | ||||||
Stock options exercised | — | — | ||||||
Stock options canceled | — | — | ||||||
Balance, March 31, 2019 | 5,477,364 | $ | 2.80 | |||||
Exercisable and vested, March 31, 2019 | 2,961,574 | $ | 2.71 | |||||
Unrecognized share-based compensation for employee and nonemployee options granted through March 31, 2019 is approximately $5.2 million to be recognized over a remaining weighted average service period of 2.2 years.
Non-Employee Director Share-Based Compensation (RSUs)
The following summarizes the activity of non-vested RSUs:
Weighted | ||||||||
average | ||||||||
grant date | ||||||||
Number of | fair value | |||||||
shares | per share | |||||||
Balance, December 31, 2018 | 267,359 | $ | 2.84 | |||||
RSUs granted | - | - | ||||||
RSUs vested | (20,444 | ) | $ | 11.04 | ||||
RSUs forfeited | - | - | ||||||
Balance, March 31, 2019 | 246,915 | $ | 2.16 | |||||
Unrecognized share-based compensation for employee RSUs granted through March 31, 2019 is approximately $235,000 to be recognized over a remaining weighted average service period of 0.8 years.
(10) Net Loss per Share
Basic net loss per share is computed by dividing net loss by the weighted-average number of common shares outstanding for the period. Diluted net loss per share is computed by dividing the net loss by the weighted-average number of common share equivalents outstanding for the period determined using the treasury-stock method. For all periods presented, there is no difference in the number of shares used to calculate basic and diluted shares outstanding since the effects of potentially dilutive securities are antidilutive due to its net loss position.
The following outstanding common stock equivalents were excluded from the computation of diluted net loss per share for the periods presented because including them would have been antidilutive:
March 31, | ||||||||
2019 | 2018 | |||||||
Stock options to purchase common stock | 5,477,364 | 3,816,927 | ||||||
Unvested restricted stock units | 246,915 | 61,333 | ||||||
Common stock warrants | 2,666,666 | - | ||||||
Total | 8,390,945 | 3,878,260 | ||||||
(11) Income Taxes
During the three months ended March 31, 2019 and 2018, there was no income tax expense or benefit for federal or state income taxes in the accompanying condensed consolidated statement of operations due to the Company’s net loss and a full valuation allowance on the resulting deferred tax assets.
As of March 31, 2019, the Company retains a full valuation allowance on its deferred tax assets in all jurisdictions. The realization of its deferred tax assets depends primarily on its ability to generate future taxable income which is uncertain. The Company does not believe that its deferred tax assets are realizable on a more-likely-than-not basis; therefore, the net deferred tax assets have been fully offset by a valuation allowance.
(12) Related Party Transactions
In August 2016, the Company granted an option to purchase 418,977 shares of common stock, with a 4-year vesting period and an exercise price of $1.80 per share, to OPKO Health, Inc. (“OPKO”) as consideration for consulting services to be provided by OPKO. BioCardia recorded approximately $22,000 (as adjusted for the adoption of ASU 2018-07) and $37,000 as share-based compensation expense related to the OPKO stock option during the three months ended March 31, 2019 and 2018, respectively. The estimated grant-date fair value of the option was $5.3 million. The term of the consulting agreement is 4 years and will be automatically renewed for successive one year periods. The chairman and chief executive officer of OPKO is a beneficial owner of more than 5% of the outstanding shares of the Company’s common stock.
(13) Subsequent Events
In April 2019, the Company submitted a Form S-1 Registration Statement (S-1) to the Securities and Exchange Commission in order to offer for sale units consisting of shares of common stock or some combination of common stock and warrants to purchase shares of common stock. Proposed maximum aggregate offering is approximately $18,000,000. The net cash realized by the Company will be less than the maximum aggregate offering due to offering expenses, underwriting discounts and commissions. The S-1 has not yet been declared effective by the Securities and Exchange Commission.
1,500,000 Units, Each Consisting of
One Share of Common Stock and
a Warrant to Purchase One Share of Common Stock
PROSPECTUS
|
Sole Book-Running Manager Maxim Group LLC Co-Managers
, 2019 Until , 2019 (the 25th day after the date of this prospectus), all dealers effecting transactions in these securities, whether or not participating in this Offering, may be required to deliver a prospectus. This delivery requirement is in addition to a dealer’s obligation to deliver a prospectus when acting as an underwriter and with respect to an unsold allotment or subscription. PART II INFORMATION NOT REQUIRED IN THE PROSPECTUS Item 13. Other Expenses of Issuance and Distribution. The following table sets forth all expenses to be paid by the Registrant, other than underwriting discounts and commissions, upon the completion of this Offering. All amounts shown are estimates except for the SEC registration fee and the FINRA filing fee.
Item 14. Indemnification of Directors and Officers. Section 102 of the General Corporation Law of the State of Delaware permits a corporation to eliminate the personal liability of directors of a corporation to the corporation or its stockholders for monetary damages for a breach of fiduciary duty as a director, except for breaches of the director’s duty of loyalty to the corporation or its stockholders, acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of a law, authorizations of the payments of a dividend or approval of a stock repurchase or redemption in violation of Delaware corporate law or for any transactions from which the director derived an improper personal benefit. Section 145 of the General Corporation Law of the State of Delaware provides that a corporation has the power to indemnify a director, officer, employee, or agent of the corporation and certain other persons serving at the request of the corporation in related capacities against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlements actually and reasonably incurred by the person in connection with a threatened, pending, or completed action, suit or proceeding to which he or she is or is threatened to be made a party by reason of such position, if such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the best interests of the corporation, and, in any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful, except that, in the case of actions brought by or in the right of the corporation, indemnification is limited to expenses (including attorneys’ fees) actually and reasonably incurred by the person in connection with defense or settlement of such action or suit and no indemnification shall be made with respect to any claim, issue, or matter as to which such person shall have been adjudged to be liable to the corporation unless and only to the extent that the Court of Chancery or other adjudicating court determines that, despite the adjudication of liability but in view of all of the circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses which the Court of Chancery or such other court shall deem proper. In addition, to the extent that a present or former director or officer of a corporation has been successful on the merits or otherwise in defense of any action, suit, or proceeding described above (or claim, issue, or matter therein), such person shall be indemnified against expenses (including attorneys’ fees) actually and reasonably incurred by such person in connection therewith. Expenses (including attorneys’ fees) incurred by an officer or director in defending any civil, criminal, administrative, or investigative action, suit, or proceeding may be advanced by the corporation upon receipt of an undertaking by such person to repay such amount if it is ultimately determined that such person is not entitled to indemnification by the corporation under Section 145 of the General Corporation Law of the State of Delaware. II-1 Our amended and restated certificate of incorporation provides for the indemnification of our directors to the fullest extent permissible under Delaware General Corporation Law. Our amended and restated bylaws provide for the indemnification of our directors and officers to the maximum extent permitted by the Delaware General Corporation Law. In addition, we have entered into indemnification agreements with our directors and officers, and we maintain insurance policies insuring our directors and officers against certain liabilities that they may incur in their capacity as officers and directors of our company. See also the undertakings set out in response to Item 17 herein. II-2 Item 15. Recent Sales of Unregistered Securities. The following is a summary of all securities that we have sold since January 1, 2016 without registration under the Securities Act of 1933, as amended (the “Securities Act”). On November 2, 2017, we executed a one-for-twelve reverse stock split of our common stock and pursuant to an amendment to our certificate of incorporation effective May 7, 2019, we executed a one-for-nine reverse stock split. Unless otherwise indicated, all per share amounts included in the summary below for transactions that occurred prior to such respective reverse stock splits are presented without giving effect to such respective reverse stock splits.
| On July 5, 2019, pursuant to the terms of
|
● | On December 24, 2018, pursuant to the
|
● | On October 24, 2016, our wholly-owned subsidiary, Icicle Acquisition Corp., a
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
● | In 2016, BioCardia issued convertible notes with an aggregate principal amount of $4.4 million to a group of accredited investors. |
● | In 2016, BioCardia granted OPKO Health, Inc. a stock option to purchase up to |
● | An aggregate of 647 shares of BioCardia common stock upon the exercise of options |
● | Stock
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
● | Stock options to purchase an aggregate of 1,191,004 shares of BioCardia common stock at an exercise price per share of $2.85 (approximately $16.20 giving effect to the November 2, 2017 and May 7, 2019 reverse stock splits) to certain of BioCardia’s directors, officers, employees, and consultants under BioCardia’s 2016 Equity Incentive Plan. |
None of the foregoing transactions involved any underwriters, underwriting discounts or commissions, or any public offering. We believe the offers, sales and issuances of the above securities were exempt from registration under the Securities Act by virtue of Section 4(a)(2) of the Securities Act because the issuance of securities to the recipients did not involve a public offering, or in reliance on Rule 701 because the transactions were pursuant to compensatory benefit plans or contracts relating to compensation as provided under such rule. The recipients of the securities in each of these transactions represented their intentions to acquire the securities for investment only and not with a view to or for sale in connection with any distribution thereof, and appropriate legends were placed upon the stock certificates issued in these transactions. All recipients had adequate access, through their relationships with us, to information about us. The sales of these securities were made without any general solicitation or advertising. The recipients of the securities in each of these transactions represented their intentions to acquire the securities for investment only and not with a view to or for sale in connection with any distribution thereof, and appropriate legends were placed upon the stock certificates issued in these transactions. All recipients had adequate access, through their relationships with us, to information about BioCardia.
Item 16. Exhibits and Financial Statement Schedules.
(a) See the Exhibit Index on the page immediately preceding the signature page hereto for a list of exhibits filed as part of this registration statement on Form S-1, which Exhibit Index is incorporated herein by reference.
(b) No financial statement schedules are provided because the information called for is not required or is shown either in the financial statements or the notes thereto.
Item 17. Undertakings.
The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by section 10(a)(3) of the Securities Act of 1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Securities and Exchange Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement.; provided, however, that paragraphs (1)(i), (1)(ii) and (1)(iii) above do not apply if the information required to be included in a post-effective amendment by those paragraphs is contained in reports filed with or furnished to the Securities and Exchange Commission by the registrant pursuant to Section 13 or Section 15(d) of the Securities Exchange Act of 1934 that are incorporated by reference in the registration statement, or is contained in a form of prospectus filed pursuant to Rule 424(b) that is part of the registration statement.
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser:
(A) Each prospectus filed by the registrant pursuant to Rule 424(b)(3) shall be deemed to be part of the registration statement as of the date the filed prospectus was deemed part of and included in the registration statement; and
(B) Each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(5) That for the purpose of determining liability of the registrant under the Securities Act of 1933 to any purchaser in the initial distribution of securities, the undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424;
(ii) Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
(iii) The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and
(iv) Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to any charter provision, by law or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act of 1933 and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
The undersigned registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(2) For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
EXHIBIT INDEX
Exhibit | Exhibit Title | |
1.1* | Form of Underwriting Agreement | |
2.1(1) | ||
2.2(2) | First Amendment to Agreement and Plan of Merger dated October 21, 2016 | |
3.1(3) | ||
3.2(4) | ||
3.3(5) | Certificate of Amendment of Amended and Restated Certificate of Incorporation effective May 7, 2019. | |
3.4(6) | ||
4.1(7) | ||
4.2(8)# | ||
4.3(9)# | Form of Stock Option Agreement under BioCardia 2002 Stock Plan | |
4.4(10)# | ||
4.5(11)# | Form of Stock Option Agreement under BioCardia 2016 Equity Incentive Plan | |
4.6(12)# | Form of Restricted Stock Unit Agreement under BioCardia 2016 Equity Inventive Plan | |
4.7(13) | ||
4.8(14) | Form of Convertible Promissory Note pursuant to a Note Purchase Agreement dated July 5, 2019 | |
4.9* | Form of Common Stock Purchase Warrant | |
4.10* | Form of Representative’s Warrant (included in Exhibit 1.1) | |
5.1* | Opinion of Wilson Sonsini Goodrich & Rosati, Professional Corporation | |
10.1(15)# | Form of Indemnification Agreement for | |
10.2(16)# | Form of | |
10.3(17) | ||
10.4(18) | First Amendment to
| |
10.5(19) | ||
10.6(20) | ||
10.7(21) † | ||
10.8(22) | Consulting Agreement, dated August 19, 2016, by and between the Company and OPKO Health, Inc. | |
| 10.9(14) | Note Purchase Agreement dated July 5, 2019 by and between the Company and the investors party thereto | |
23.1+ | Consent of KPMG LLP, Independent Registered Public Accounting Firm | |
23.2* | Consent of Wilson Sonsini Goodrich & Rosati, Professional Corporation (included in Exhibit 5.1) | |
24.1 | Power of Attorney (see signature page to this |
† | Confidential treatment has been granted with respect to certain portions of this Exhibit. |
# | Indicates management contract or compensatory plan or arrangement. |
* | To be filed by amendment. |
+ | Filed herewith. |
(1) | Previously filed as an exhibit to the Current Report on Form |
(2) | Previously filed as an exhibit to the |
(3) | Previously filed as an exhibit to the Current Report on Form 8-K filed by us on April 11, 2017. |
(4) | Previously filed as an exhibit to the Current Report on Form 8-K filed by us on November 7, 2017. |
(5) | Previously filed as an exhibit to the Current Report on Form 8-K filed by us on June 7, 2019. |
(6) | Previously filed as an exhibit to the Current Report on Form 8-K filed by us on April 11, 2017. |
(7) | Previously filed as an exhibit to the Current Report on Form 8-K filed by us on October 27, 2016. |
(8) | Previously filed as an exhibit to the Current Report on Form 8-K filed by us on October 27, 2016. |
(9) | Previously filed as an exhibit to the registration statement on Form S-8 filed by us on February 8, 2017. |
(10) | Previously filed as an exhibit to the registration statement on Form S-8 filed by us on February 8, 2017. |
(11) | Previously filed as an exhibit to the registration statement on Form S-8 filed by us on February 8, 2017. |
(12) | Previously filed as an exhibit to the registration statement on Form S-8 filed by us on February 8, 2017. |
(13) | Previously filed as an exhibit to the Current Report on Form 8-K filed by us on December 24, 2018. |
(14) | Previously filed as an exhibit to the Current Report on Form 8-K filed by us on July 5, 2019. |
(15) | Previously filed as an exhibit to the Current Report on Form 8-K filed by us on October 27, 2016. |
(16) | Previously filed as an exhibit to the Current Report on Form 10-K filed by us on March 30, 2017. |
(17) | Previously filed as an exhibit to the Current Report on Form 8-K filed by us on October 27, 2016. |
(18) | Previously filed as an exhibit to the Current Report on Form 8-K filed by us on October 27, 2016. |
(19) | Previously filed as an exhibit to the Current Report on Form 8-K filed by us on October 27, 2016. |
(20) | Previously filed as an exhibit to the Current Report on Form 10-K filed by us on March 30, 2017. |
(21) | Previously filed as an exhibit to the Current Report on Form 8-K filed by us on October 27, 2016. |
(22) | Previously filed as an exhibit to the Current Report on Form 10-K/A filed by us on July 20, 2017. |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of New York, State of New York, on July 15, 2019.
BIOCARDIA, INC. | |
By: | /s/ Peter Altman, Ph.D. |
Peter Altman, Ph.D. | |
President and Chief Executive Officer | |
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities indicated below:
Signature | Title |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| July 15, 2019 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Peter Altman, Ph.D. | President and Chief Executive | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Officer (Principal Executive Officer) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
* | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
David McClung | Chief Financial Officer (Principal | July 15, 2019 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Accounting and Financial Officer) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
* | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Simon H. Stertzer | Chairman of the
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
* Pursuant to power of attorney |
|
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||
|
|
| ||
| ||||
By: | /s/ Peter Altman, Ph.D. | |||
Peter Altman, Ph.D. | ||||
Attorney-in-fact |
EXHIBIT INDEX
The following exhibits have been filed as part of this Registration Statement on Form S-1.
Incorporated By Reference | ||||||
Exhibit Number | Exhibit Description | Form | File Number | Exhibit | Filing Date | |
2.1 | Merger Agreement and Plan of Reorganization, dated as of June 18, 2008, by and among clickNsettle.com, Inc., Cardo Medical, LLC and Cardo Acquisition, LLC. | 8-K | 000-21419 | 2.1 | June 23, 2008 | |
2.2 | First Amendment to Merger Agreement and Plan of Reorganization, dated as of August 29, 2008, by and among clickNsettle.com, Inc., Cardo Medical, LLC and Cardo Acquisition, LLC. | 8-K | 000-21419 | 2.2 | September 9, 2008 | |
2.3 | Asset Purchase Agreement, dated September 30, 2009, by and among Cardo Medical, Inc. and Vertebron, Inc. | 8-K | 000-21419 | 2.1 | October 6, 2009 | |
3.1 | Amended and Restated Certificate of Incorporation | 8-K | 000-21419 | 3.1 | March 18, 2008 | |
3.2 | Amended and Restated Bylaws. | 8-K | 000-21419 | 3.1 | February 1, 2008 | |
5.1# | Opinion of Akerman Senterfitt | |||||
10.1 | Escrow Agreement, dated as of August 29, 2008, by and among Chicago Title Company, clickNsettle.com, Inc. Andrew A. Brooks, M.D. and Mikhail Kvitnitsky. | 8-K | 000-21419 | 10.1 | September 9, 2008 | |
10.2 | Form of Lockup Agreement. | 8-K | 000-21419 | 10.2 | September 9, 2008 | |
10.3 | Lockup Agreement, dated August 29, 2008, for Derrick Romine. | 8-K | 000-21419 | 10.3 | September 9, 2008 | |
10.4 | Amended and Restated 1996 Incentive and Nonqualified Stock Option Plan. | 10-KSB | September 28, 1998 | |||
10.5* | Form of Cardo Medical, LLC Nonstatutory Option Agreement | 8-K | 000-21419 | 10.5 | September 9, 2008 | |
10.6 | Stock Purchase Agreement, dated as of December 19, 2007, by and among clickNsettle.com, Inc., Frost Gamma Investments Trust, Dr. Jane Hsiao, Steven D. Rubin and Subbarao Uppaluri. | 8-K | 000-21419 | 10.1 | December 21, 2007 | |
10.7 | First Amendment to Stock Purchase Agreement, dated as of January 31, 2008, by and among clickNsettle.com, Inc., Frost Gamma Investments Trust, Dr. Jane Hsiao, Steven D. Rubin and Subbarao Uppaluri. | 8-K | 000-21419 | 10.2 | February 1, 2008 | |
10.8* | Employment Agreement, dated as of January 31, 2005, by and between Accelerated Innovation, LLC, as successor to Accin Corporation, and Mikhail Kvitnitsky. | 8-K | 000-21419 | 10.8 | September 9, 2008 | |
10.9* | Amendment to Employment Agreement, dated as of June 6, 2008, by and between Accelerated Innovation, LLC and Mikhail Kvitnitsky. | 8-K | 000-21419 | 10.9 | September 9, 2008 | |
10.10* Termination Agreement, effective as of June 23, 2008, by and between Accelerated Innovation, LLC and Mikhail Kvitnitsky. 8-K 000-21419 10.10 September 9, 2008 10.11* Employment Offer Letter with Derrick Romine dated September 5, 2008. 8-K 000-21419 10.11 September 9, 2008 10.12 Form of Indemnification Agreement for officers and directors. 8-K 000-21419 10.12 September 9, 2008 10.13+ Agreement, dated as of August 22, 2006, by and between Accelerated Innovation, LLC, as successor to Accin Corporation, and Infinesse Corporation. 8-K 000-21419 10.13 September 9, 2008 10.14 Supplier Agreement, dated June 16, 2006, between Stelkast Company and Accelerated Innovation, LLC, as successor to Accin. 8-K 000-21419 10.14 September 9, 2008 10.15+ Contracted Services Agreement, dated September 1, 2007, by and between Accelerated Innovation, LLC and Summit Corporate Services, Inc. 8-K 000-21419 10.15 September 9, 2008 10.16 Agreement dated April 30, 2008, by and among Mikhail Kvitnitsky and Accelerated Innovation, LLC, Cervical Xpand, LLC and Uni-Knee, LLC. 8-K 000-21419 10.16 September 9, 2008 10.17 Agreement dated April 30, 2008, by and among John D. Kuczynski, Accelerated Innovation, LLC and Uni-Knee, LLC. 8-K 000-21419 10.17 September 9, 2008 10.18 Agreement dated April 30, 2008, by and among Richard H. Rothman, M.D., Ph.D., Accelerated Innovation, LLC and Cervical Xpand, LLC. 8-K 000-21419 10.18 September 9, 2008 10.19 Agreement dated April 30, 2008, by and among Todd J. Albert, M.D., Accelerated Innovation, LLC and Cervical Xpand, LLC. 8-K 000-21419 10.19 September 9, 2008 10.20 Agreement dated April 28, 2008, by and among, Rafail Zubok, Accelerated Innovation, LLC and Cervical Xpand, LLC. 8-K 000-21419 10.20 September 9, 2008 10.21* Nonstatutory Option Agreement, dated August 27, 2008, by and between Cardo Medical, LLC and Derrick Romine. 10-Q 000-21419 10.1 November 14, 2008 10.22 Form of Registration Rights Agreement, dated October 27, 2009, by and among Cardo Medical, Inc. and the Several Purchasers. 8-K 000-21419 10.1 October 29, 2009 21.1 Subsidiaries of clickNsettle.com, Inc. 8-K 000-21419 21.1 September 9, 2008 23.1# Consent of Stonefield Josephson, Inc. 23.2# Consent of Akerman Senterfitt (included in Exhibit 5.1)
|
| |||||
|
| |||||
|
| |||||