
As filed with the Securities and Exchange Commission on May 14, 2004March 31, 2017
Registration No. 333- 333-216663
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form S-1
Amendment No. 1 to
FORMS-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
Targacept, Inc.
CATALYST BIOSCIENCES, INC.
(Exact name of registrantRegistrant as specified in its charter)
| Delaware | 2834 | 56-2020050 | ||
(State or other jurisdiction of
| (Primary Standard Industrial Classification Code Number) | (I.R.S. Employer Identification Number) |
200 East First Street, Suite 300260 Littlefield Avenue
Winston-Salem, North Carolina 27101South San Francisco, California 94080
(650)(336) 480-2100266-8674
(Address, including zip code, and telephone number, including area code, of registrant’sRegistrant’s principal executive offices)
J. Donald deBethizy
Nassim Usman, Ph.D.
Chief Executive Officer
Targacept,Catalyst Biosciences, Inc.
200 East First Street, Suite 300South San Francisco, California 94080
(650)Winston-Salem, North Carolina 27101266-8674
(336) 480-2100
(Name, address, including zip code, and telephone number,
including area code, of agent for service)
Copies to:
Stephen B. Thau
Alfredo B. D. Silva
Morrison & Foerster LLP
755 Page Mill Road
Palo Alto, CA 94304
(650)813-5600
|
|
|
Approximate date of commencement of proposed sale of the securities to the public: public:
As soon as practicable after the effective date of this Registration Statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, as amended (the “Securities Act”), check the following box. ¨☒
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, anon-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule¨12b-2 of the Exchange Act.
If delivery of the Prospectus is expected to be made pursuant to Rule 434, please check the following box. ¨
| Large accelerated filer | ☐ | Accelerated filer | ☐ | |||
| Non-accelerated filer | ☐ (Do not check if a smaller reporting company) | Smaller reporting company | ☒ |
CALCULATION OF REGISTRATION FEE
Title of Each Class of Securities to be Registered | Proposed Maximum Aggregate Offering Price(1) | Amount of Registration Fee(2) | ||
Common Stock, par value $0.001 per share | $86,250,000 | $10,927.88 | ||
| ||||
Title of each class of securities to be registered(1) | Proposed maximum aggregate offering price(1)(2) | Amount of registration fee | ||
Class A Units consisting of: | ||||
(i) Shares of common stock, par value $0.001 per share | 5,250,000 | 608.48 | ||
(ii) Warrants to purchase common stock | ||||
Class B Units consisting of: | ||||
(i) Shares of Series A Preferred Stock, par value $0.001 per share | 12,000,000 | 1,390.88 | ||
(ii) Shares of common stock issuable on conversion of Series A Preferred Stock(3) | ||||
(iii) Warrants to purchase common stock | ||||
Common stock issuable upon exercise of warrants | $9,918,750 | 1,149.58 | ||
Total | $27,168,750 | $3,148.86 | ||
| ||||
| (1) | Estimated solely for the purpose of |
| (2) | Includes the price of additional shares of common stock and warrants to purchase shares of common stock that the underwriters have the option to purchase to cover over-allotments, if any. |
| (3) |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. WeThese securities may not sell these securitiesbe sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities, and we areit is not soliciting offersan offer to buy these securities in any statejurisdiction where the offer or sale is not permitted.
PROSPECTUS (Subject to Completion)
Issued May 14, 2004
SharesSubject to completion, dated March 31, 2017
285,714 Class A Units consisting of common stock and warrants and
12,000 Class B Units consisting of shares of Series A Preferred Stock and warrants
(and 1,857,143 shares of common stock underlying shares of
Series A Preferred Stock and warrants)

COMMON STOCK
Targacept, Inc. isWe are offering 285,714 Class A Units, with each Class A Unit consisting of one share of common stock, par value $0.001 per share (the “common stock”) and a warrant to purchase half of one share of our common stock (together with the shares of its common stock. This is our initial public offering and no public market currently exists for our shares. We anticipate thatstock underlying such warrants, the initial“Class A Units”) at a public offering price will be between $ andof $ per share.Class A Unit. Each warrant included in the Class A Units entitles its holder to purchase half of one share of common stock at an exercise price per share of $ .
We have appliedare also offering to havethose purchasers whose purchase of Class A Units in this offering would result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock following the consummation of this offering, the opportunity to purchase, if they so choose, in lieu of the number of Class A Units that would result in ownership in excess of 4.99% (or, at the election of the purchaser, 9.99%), 12,000 Class B Units. Each Class B Unit will consist of one share of Series A Preferred Stock, par value $0.001 per share (the “Series A Preferred Stock”), convertible into 95.24 shares of common stock and warrants to purchase 47.62 shares of our common stock approved(together with the shares of common stock underlying such shares of Series A Preferred Stock and such warrants, the “Class B Units” and, together with the Class A Units, the “units”) at a public offering price of $ per Class B Unit. Each warrant included in the Class B Units entitles its holder to purchase 47.62 shares of common stock at an exercise price per share of $ .
The Class A Units and Class B Units have no stand-alone rights and will not be certificated or issued as stand-alone securities. The shares of common stock, Series A Preferred Stock and warrants comprising such units are immediately separable and will be issued separately in this offering. The underwriters have the option to purchase additional shares of common stock and/or warrants to purchase shares of common stock solely to cover over-allotments, if any, at the price to the public less the underwriting discounts and commissions. The over-allotment option may be used to purchase shares of common stock, or warrants, or any combination thereof, as determined by the underwriters, but such purchases cannot exceed an aggregate of 15% of the number of shares of common stock (including the number of shares of common stock issuable upon conversion of shares of Series A Preferred Stock) and warrants sold in the primary offering. The over-allotment option is exercisable for listing45 days from the date of this prospectus.
Our common stock is listed on the NASDAQ NationalThe Nasdaq Capital Market under the symbol “TRGT.”
Investing in“CBIO”. The closing price of our common stock on March 29, 2017, as reported by The Nasdaq Capital Market, was $10.51 per share. All share and warrant numbers of the securities being offered included in this prospectus are based on an assumed public offering price per share of $10.50. We do not intend to apply for listing of the warrants offered hereby or the shares of Series A Preferred Stock on any securities exchange or trading system.
Investing in the units involves risks. Seea high degree of risk. Before making any investment in these securities, you should consider carefully the risks and uncertainties in the section entitled “Risk Factors” beginning on page 7.13 of this prospectus.
PRICE$ A SHARE
|
|
| Total | |||||||||
| $ | $ | $ | |||||||||
| $ | $ | $ | |||||||||
Proceeds, before expenses, to Catalyst Biosciences, Inc. | $ | $ | $ | |||||||||
| (1) | The public offering price and underwriting discount corresponds to (x) in respect of the Class A Units (i) a public offering price per share of common stock of $ and (ii) a public offering price per share underlying the warrants of $ and (y) in respect of the Class B Units (i) a public offering price per share of Series A Preferred Stock of $ and (ii) a public offering price per share underlying the warrants of $ . |
| (2) | We have also agreed to reimburse for certain expenses. See “Underwriting.” |
| (3) | We have granted a45-day day option to the underwriter to purchase additional shares of common stock and/or warrants to purchase shares of common stock (up to 15% of the number of shares of common stock (including the number of shares of common stock issuable upon conversion of shares of Series A Preferred Stock) and warrants sold in the primary offering) solely to cover over-allotments, if any. |
We have grantedNeither the underwriters the right to purchase up to an additional shares of our common stock to cover over-allotments.
The Securities and Exchange Commission andnor any state securities regulators have notcommission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense. The securities are not being offered in any jurisdiction where the offer is not permitted.
Morgan Stanley & Co. Incorporated expects to deliver the shares to purchasers on
Ladenburg Thalmann
The date of this prospectus is , 2004.
MORGAN STANLEY
DEUTSCHE BANK SECURITIES
CIBC WORLD MARKETS
PACIFIC GROWTH EQUITIES, LLC
, 2004
1 | ||||
| 1 | ||||
| 7 | ||||
| 7 | |||
| 8 | ||||
| 9 | ||||
| 13 | ||||
| 43 | |||
| 45 | ||||
| 46 | ||||
| 47 | ||||
| 48 | ||||
| 49 | ||||
| 53 | ||||
| 61 | ||||
| 65 | ||||
| 65 | ||||
| 66 | ||||
| 67 | ||||
You should rely only on the information contained in this prospectus.prospectus or in any related free writing prospectus filed by us with the Securities and Exchange Commission, or the SEC. We have not, and the underwriters and their affiliates have not, authorized anyone to provide you with any information different from the informationor to make any representation not contained in this prospectus. We are offeringdo not, and the underwriters and their affiliates do not, take any responsibility for, and can provide no assurance as to the reliability of, any information that others may provide to you. This prospectus is not an offer to sell shares of common stock, and seeking offersor an offer to buy shares of common stock, onlyunits in jurisdictionsany jurisdiction where offers and sales are not permitted. The information contained in this prospectus is accurate only as of its date, regardless of the datetime of delivery of this prospectus regardless of when this prospectus is delivered or when any sale of our common stock occurs.units. You should also read and consider the information in the documents to which we have referred you under the caption “Where You Can Find More Information” in the prospectus.
Until , 2004, 25 days after the commencement of this offering, all dealers that buy, sell or trade shares of our common stock, whether or not participating in this offering, may be required to deliver a prospectus. This delivery requirement is in addition to the dealers’ obligation to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
For investors outside the United States: Neither we nor any of the underwriters have done anything that would permit thisa public offering of the units or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. You are required toPersons outside the United States who come into possession of this prospectus must inform yourselvesthemselves about, and to observe any restrictions relating to, thisthe offering of the units and the distribution of this prospectus.prospectus outside of the United States.
The following summary highlightsis qualified in its entirety by, and should be read together with, the more detailed information and financial statements and related notes thereto appearing elsewhere in this prospectus. It may not contain all of the information that may be important toBefore you in deciding whetherdecide to invest in our common stock. Youstock, you should read the entire prospectus carefully, including the “Risk Factors” sectionrisk factors and the financial statements and related notes appearing at the end ofincluded in this prospectus before making an investment decision.
TARGACEPT, INC.
We are a biopharmaceutical company engagedUnless the context requires otherwise, in this prospectus the design, discoveryterms “Catalyst,” the “Company,” “we,” “us” and development of a new class of drugs“our” refer to treat multiple diseases and disorders of the nervous system by selectively targeting neuronal nicotinic acetylcholine receptors, or NNRs. NNRs are found on nerve cells throughout the nervous system and serve as key regulators of nervous system activity. Our product candidates are designed to selectively target specific NNR subtypes to promote therapeutic effects while limiting or eliminating adverse side effects. In addition to a marketed product, Inversine, we have four product candidates in clinical development and multiple ongoing preclinical programs. Our pipeline includes:
We trace our scientific lineage to a research program initiated by R.J. Reynolds Tobacco Company in 1982 to study the activity and effects of nicotine, a compound that interacts non-selectively with all nicotinic acetylcholine receptors. There is a significant amount of published clinical data relating to nicotine, including studies in which individuals with ADHD, cognitive impairment, and ulcerative colitis showed therapeutic improvement when treated with the nicotine patch. We have used this clinical data,Catalyst Biosciences, Inc., together with our deep understanding of the biological characteristics and functions of NNRs that we have built over more than 20 years, to validate NNRs as potential therapeutic targets. We have also developed an expertise in designing small molecules that can selectively interact with specific NNR subtypes, with the objective of eliciting a desired therapeutic effect while limiting or potentially eliminating side effects such as those typically seen with nicotine. We have built an extensive patent estate covering the structure or therapeutic use of small molecules designed to regulate the nervous system by selectively modulating specific NNR subtypes.
We develop product candidates using our proprietary databases and computer-based molecular design technologies, which we refer to collectively as Pentad. Together with our proprietary target validation assays and novel screening methods, Pentad enables us to efficiently identify, prioritize, characterize and optimize novel compounds.
We have entered into two collaboration agreements with Aventis Pharma SA relating to the development of treatments for Alzheimer’s disease and other diseases of the central nervous system. In addition, we have entered into a collaboration agreement with Dr. Falk Pharma GmbH relating to the development of a treatment for ulcerative colitis.
Our Product Pipeline
TC-1734. TC-1734 is a novel small molecule that we are developing as an oral treatment for a range of cognitive impairment in the elderly thatits subsidiary, Catalyst Bio, Inc. This prospectus includes age associated memory impairment, commonly referred to as AAMI, and mild cognitive impairment, commonly referred to as MCI. We are currently conducting a Phase II clinical trial of TC-1734 in 56 elderly persons classified with AAMI and have completed two arms of the trial. We are also currently conducting a Phase II clinical trial in 40 elderly persons classified with MCI. We have previously evaluated TC-1734 in 84 healthy volunteers in four Phase I clinical trials. In our clinical trials to date, TC-1734 was well tolerated at doses at which trial subjects showed signs of improvement in cognitive function. Subject to the results of discussions with the FDA, we anticipate commencing a separate Phase II clinical trial designed to evaluate the efficacy of TC-1734 in the fourth quarter of 2004. We are also evaluating TC-1734 for potential additional clinical development for indications marked by cognitive impairment that are not specific to the elderly, such as ADHD, schizophrenia and various forms of dementia.
TC-5231. TC-5231 is a small molecule that we are developing as an oral treatment for ADHD. TC-5231 is a low-dose reformulation of mecamylamine hydrochloride, the active ingredient in our FDA-approved product, Inversine. Inversine is approved in the United States for the management of moderately severe to severe essential hypertension at average daily doses of 25mg. However, our market research suggests that Inversine is prescribed predominantly for the treatment of Tourette’s syndrome and other neuropsychiatric disorders at doses ranging from 2.5mg to 7.5mg. We are evaluating TC-5231 in doses between 0.2mg and 1.0mg in two Phase II clinical trials, one in ADHD in children and adolescents and the other in ADHD in young adults.
TC-2403. TC-2403 is a small molecule that we are developing for the treatment of ulcerative colitis in collaboration with Dr. Falk Pharma GmbH. We are currently conducting a Phase II clinical trial of an enema formulation of the compound designed to induce remission of acute episodes of a form of ulcerative colitis known as left-sided colitis. In addition, we are developing a delayed release oral formulation of the compound designed to deliver the drug to the entire colon to induce and maintain remission of all forms of ulcerative colitis. We expect to complete the oral formulation of this product candidate in the fourth quarter of 2004.
TC-2696. TC-2696 is a novel small molecule that we are developing as an oral treatment for acute post-operative pain. We are currently conducting a Phase I clinical trial of TC-2696. Depending on clinical trial results, available resources and other considerations, we may pursue development of TC-2696 for other classes of pain as well.
Business Strategy
Our goal is to become a leader in the discovery, development and commercialization of novel drugs that selectively target NNRs in order to treat diseases and disorders where there is significant medical need and commercial potential. To achieve this goal, we are pursuing the following strategies:
Risks Associated with Our Business
Our business is subject to numerous risks, as more fully described in the section entitled “Risk Factors” immediately following this prospectus summary. We have a limited operating history and have incurred substantial net losses since our incorporation in 1997. We expect to continue to incur substantial losses for the foreseeable future. Inversine is the only product that we have available for commercial sale, and it generates limited revenues. All of our other product candidates are undergoing clinical trials or are in early stages of development, and failure is common and can occur at any stage of development. Our ability to generate product revenue in the future will depend heavily on the successful development and commercialization of these product candidates. Even if we succeed in developing and commercializing one or more of our product candidates, we may never generate sufficient sales revenue to achieve and then sustain profitability.
Company History
Our history traces back to 1982 when R.J. Reynolds Tobacco Company initiated a program to study the activity and effects of nicotine in the body. We were incorporated in Delaware as a wholly owned subsidiary of RJR and became an independent company in August 2000. Our executive offices are located at 200 East First Street, Suite 300, Winston-Salem, North Carolina 27101, and our telephone number is (336) 480-2100. Our web site is located at www.targacept.com. Information contained on our web site is not incorporated by reference into, and does not form any part of, this prospectus. We have included our website address in this document as an inactive textual reference only. Our trademarks, include Targacept® and Inversine®. Other service marks trademarks and trade names appearingowned by us or other companies. All trademarks, service marks and trade names included in this prospectus are the property of their respective owners. Unless
We are a clinical-stage biopharmaceutical company focused on developing novel medicines to address serious medical conditions for patients who need new or better treatment options. We have used a scientific approach to engineer several novel protease-based therapeutic candidates. We are focusing our product development efforts in the context requires otherwise, referencesfield of hemostasis (the process that regulates bleeding) and have a mission to develop valuable therapies for individuals with hemophilia.
With drug candidates in this prospectusclinical and advanced preclinical development, we are a leader in the field of prophylactic subcutaneously (SQ) dosed coagulation factor therapies for individuals with hemophilia. We have assembled an experienced management team, scientists and advisors with subject matter expertise, a strategic collaborator, an enabling technology platform, and a leading intellectual property position in the fields of protease therapeutics to advance our clinical and preclinical pipeline.
Our Focus—Hemophilia
Hemophilia is a rare but serious bleeding disorder that results from a genetic or an acquired deficiency of a protein required for normal blood coagulation. There are two major types of hemophilia, A and B, that are caused by alterations in Factor VIII or Factor IX genes, respectively, with a corresponding deficiency in the affected proteins. The disease is X chromosome-linked, meaning that most people who inherit the disorder and suffer from symptoms are male. However, female carriers of mutations in Factor VIII or Factor IX can also have reduced clotting factor levels.
Individuals with hemophilia suffer from spontaneous bleeding episodes and substantially prolonged bleeding times that can become limb- or life-threatening following injury or trauma. In cases of severe hemophilia, spontaneous bleeding into muscles or joints is frequent and often results in permanent, disabling joint damage. Individuals with hemophilia are currently treated with replacement therapy of key coagulation proteins, Factor VIII for hemophilia A or Factor IX for hemophilia B.
Our Pipeline of Product Candidates
We are currently focused on the clinical development of improved, next-generation subcutaneous prophylaxis using enhanced potency Factor VIIa and Factor IX variants.
The following table summarizes our development programs.
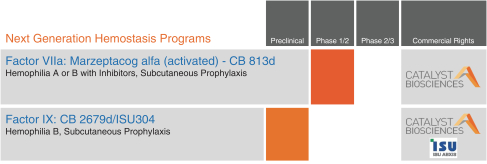
Our Technology
We are applying our substantial expertise in protease engineering and our proprietary product discovery platform to create, engineer and characterize protease drug candidates. Proteases regulate several complex biological cascades, or sequenced biochemical reactions, including the coagulation cascade (a mechanism of blood clotting) in hemophilia andnon-hemophilia settings and the complement cascade that causes inflammation and tissue damage in certain diseases. Our protease expertise allowed us to improve the biochemical and pharmacological properties of currently marketed hemophilia protease drugs, specifically Factors VIIa, IX and Xa and to create completely novel proteases that cleave disease-causing proteins, specifically complement Factor 3 (C3) for the potential treatment of dryage-related macular degeneration (Dry AMD) and renal delayed graft function (DGF).
We estimate the total market for our product candidates is $3.4 billion. Based on industry reports, annual worldwide sales in 2016 for Factor VIIa recombinant products for individuals with hemophilia A or B with an inhibitor were approximately $1.4 billion, and prothrombin complex concentrate products used to treat individuals with hemophilia A or B with an inhibitor were $0.8 billion. Worldwide sales in 2016 for Factor IX products for individuals with hemophilia B were approximately $1.2 billion.
We believe that the shortcomings of currently approved therapies, including a requirement for intravenous infusion, are barriers to prophylactic treatment strategies that, if surmounted, could provide meaningfully improved long-term clinical outcomes for individuals with hemophilia.
The substantially enhanced potency of marzeptacog alfa (activated) and CB 2679d/ISU304 compared with existing treatment options may allow for effective subcutaneous prophylactic treatment of individuals with hemophilia A or B with an inhibitor or individuals with hemophilia B, respectively. Our engineered hemostasis proteases are designed to overcome current treatment limitations by allowing delivery via subcutaneous injection which we believe will facilitate effective prophylactic treatment, especially in children, which represent approximately 40% of individuals with hemophilia, and may ultimately deliver substantially better outcomes for individuals with hemophilia.
Subcutaneous dosing results in progressive increases in the levels of our protease factors until they reach a stable blood level therapeutic target range (ideally mild hemophilia to normal). Conversely, dosing by intravenous (IV) infusions results in very high factor levels in the blood initially, but the factor level then falls rapidly to a trough level at a range that is measured as moderate or severe hemophilia, triggering the next dose. These results are illustrated in the diagram below.
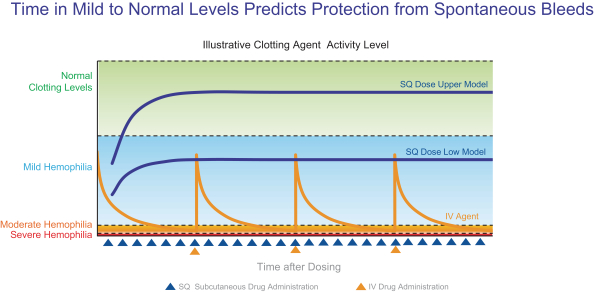
Stable factor levels could potentially yield a significant improvement in outcomes and have the added benefit of convenience over competing intravenous therapeutics, particularly when administered to children where venous access is challenging.
Factor VIIa
Our most advanced product candidate is marzeptacog alfa (activated) (formerly CB 813d), a next-generation Factor VIIa variant, was tested in an intravenous Phase 1 clinical trial that was completed in February 2015 to evaluate the safety, tolerability, pharmacokinetics, pharmacodynamics and coagulation activity of marzeptacog alfa (activated) in severe hemophilia A and B with and without an inhibitor.
Marzeptacog alfa (activated) is initially being developed for the prophylactic treatment of individuals with severe hemophilia A or B with inhibitors. Pfizer Inc. (“Pfizer”) filed the Investigational New Drug Application (IND) with the FDA for the Phase 1 trial in August 2011 for adult males with hemophilia A or B, with or without an inhibitor to Factor VIII or Factor IX. We have received the IND application filed with the FDA from Pfizer and plan to initiate the Phase 2 portion of a Phase 2/3 clinical subcutaneous prophylaxis efficacy trial in the fourth quarter of 2017. Marzeptacog alfa (activated) has received orphan drug designation in the United States from the FDA.
On June 29, 2009, we entered into a Research and License Agreement with Wyeth Pharmaceuticals, Inc., subsequently acquired by and referred to herein as Pfizer, whereby we and Pfizer collaborated on the development of novel human Factor VIIa products and we granted Pfizer the exclusive rights to develop and commercialize the licensed products on a worldwide basis. On April 2, 2015, Pfizer notified us that it was exercising its right to terminate the research and license agreement, and on December 8, 2016, we signed a definitive agreement related to the “company,” “we,” “us,”termination of the Pfizer agreement. Pursuant to this termination agreement, Pfizer granted us an exclusive license to Pfizer’s proprietary rights for manufacturing materials and “our” referprocesses that apply to Targacept, Inc.
IND application and documentation related to the development, manufacturing and testing of the Factor VIIa products as well as the orphan drug designation. Pursuant to this agreement, we agreed to make contingent cash payments to Pfizer in an aggregate amount equal to up to $17.5 million, payable upon the achievement of clinical, regulatory and commercial milestones. Following commercialization of any of Factor VIIa products, Pfizer would also receive a single-digit royalty on net product sales on aTHE OFFERINGcountry-by-country basis for a predefined royalty term.
In the Phase 1 clinical trial of intravenous marzeptacog alfa (activated) conducted by Pfizer, 25 individuals with severe hemophilia A or B with and without an inhibitor were enrolled and treated. Clinical endpoints included safety, tolerability, pharmacokinetics and clot-forming activity, such as prothrombin time, or PT, activated partial thromboplastin time, or aPTT, thrombin-antithrombin activity and others. Results showed that single doses of marzeptacog alfa (activated) were well tolerated when administered to individuals with hemophilia A and B, and there were no instances of bleeding or thrombosis. As shown in the graph below, marzeptacog alfa (activated) demonstrated pharmacological efficacy as measured by significant shortening of aPTT (activated partial thromboplastin time) and PT (prothrombin time) for up to24-hours post dosing. The results were presented in a poster session at the International Society on Thrombosis and Haemostasis (ISTH) Meeting held in Toronto, Canada from June 20 to 25, 2015.
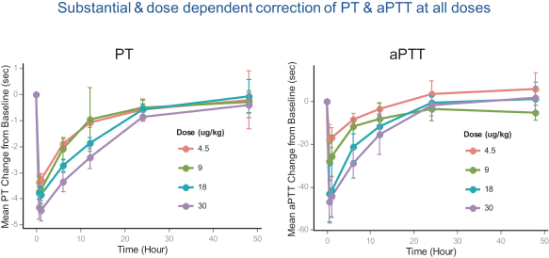
We designed marzeptacog alfa (activated) to combine higher clot-generating activity, or potency, at the site of bleeding and improved duration of action in vivo to allow for the effective, long-term, prophylaxis in individuals with hemophilia with an inhibitor. We anticipate that this product candidate, if approved, could be used prophylactically to prevent bleeding episodes with subcutaneous administration that may be superior to intravenous infusions. We have previously demonstrated in several bleeding models that marzeptacog alfa (activated) can treat or prevent bleeding when dosed intravenously. The next step required to develop marzeptacog alfa (activated) for subcutaneous use was to test its ability to correct bleeding times in hemophilia models and to achieve sufficient plasma (blood) levels of activity when dosed subcutaneously.
During the past nine months, we have presented data at scientific conferences demonstrating that daily subcutaneous administration in hemophilia B mice and hemophilia A dogs resulted in steady-state blood levels of marzeptacog alfa (activated) that correct the hemophilia coagulation impairment present at baseline as measured by whole blood clotting time and aPTT.
Factor IX
Our next most advanced product candidate is CB 2679d/ISU304, is a highly potent, next generation coagulation, Factor IX variant that is under development by our collaborator ISU Abxis and has been approved for human clinical trials by the Korean Food and Drug Administration. The National Hemophilia Foundation has recommended chronic, prophylactic treatment as the optimal therapy for individuals with severe hemophilia B. We intend to enter Phase 1/2 clinical development with our collaborator ISU Abxis in the second quarter of 2017, and clinical data is expected in the second half of 2017. We entered into aco-development agreement with ISU Abxis in 2013. Under the ISU Abxis agreement we licensed our proprietary human Factor IX products to ISU Abxis for initial development in South Korea. ISU Abxis is responsible for manufacturing, preclinical development activities and clinical development through aproof-of-concept Phase 1/2 study in individuals with hemophilia B. We have the sole rights and responsibility for worldwide development, manufacture, and commercialization of Factor IX products after Phase 1/2 development. ISU Abxis may exercise its right of first refusal to acquire commercialization rights in South Korea, in which case they would be entitled to profit sharing on worldwide sales.
CB 2679d/ISU304 has demonstrated in a hemophilia B mouse animal study higher potency than BeneFIX®, Pfizer’s currently marketed Factor IX therapeutic, and Alprolix®, Bioverativ’s approved FactorIX-Fc fusion protein, and may allow for subcutaneous prophylactic treatment of individuals with hemophilia B.
Factor Xa
We also have several Factor Xa variants that have demonstrated efficacy in several preclinical models and have the potential to be used as a universalpro-coagulant. We have delayed initiating further work on our Factor Xa therapeutic program at this time to focus our efforts on the Factor VIIa and Factor IX clinical programs.
Our Strategy
Our goal is to build a clinical-stage biopharmaceutical company whose mission is to develop valuable therapies for individuals with hemophilia who need new or better treatment options. Key elements of our strategy to achieve this goal are to:
|
| Advance the Clinical Development of our Lead Product Candidates:Our most advanced drug candidate, marzeptacog alfa (activated), for the treatment of hemophilia and to facilitate surgery in hemophilia, has completed a Phase 1 clinical trial evaluating safety and tolerability as well as pharmacokinetics, pharmacodynamics and coagulation activity. We expect that we will advance marzeptacog alfa (activated) into the Phase 2 portion of a Phase 2/3 subcutaneous dosing clinical efficacy trial in individuals with hemophilia A or B with an inhibitor in the fourth quarter of 2017. In addition, we expect that our collaborator ISU Abxis will initiate a Phase 1/2 subcutaneous dosing clinical trial of CB 2679d/ISU304, our next-generation Factor IX drug candidate in individuals with hemophilia B, in the second quarter of 2017, and clinical data is expected in the second half of 2017. |
| • | Leverage Existing Strategic Factor IX Collaboration:We have established a strategic collaboration with ISU Abxis for its CB 2679d/ISU304 program. We are entitled to up front and milestone payments and have retained worldwide commercialization rights, except for ISU Abxis’ right of first refusal for commercialization rights in South Korea, and subject to a future profit sharing arrangement. We believe our Factor IX collaboration contributes to our ability to advance our Factor IX product candidate through clinical development. |
| • | Build a Hemostasis Franchise:We intend to build on our recent clinical and preclinical success in Factor VIIa and Factor IX by advancing our Factor VIIa program into the Phase 2 portion of a Phase 2/3 subcutaneous dosing clinical efficacy trial in the fourth quarter of 2017. The combination of the wholly owned Factor VIIa product candidate entering a Phase 2/3 clinical efficacy trial and the Factor IX product candidate entering a Phase 1/2 clinical trial may allow us to build a strong hemostasis franchise. |
We continue to explore licensing opportunities for our anti-complement programs in DGF and Dry AMD so that we can focus our efforts and resources on advancing marzeptacog alfa (activated) and CB 2679d/ISU304 through Phase 2/3 and Phase 1/2 clinical trials, respectively.
Market Opportunity
Hemophilia A occurs in approximately 1 in 5,000 male births, and hemophilia B in 1 in 30,000 male births. The prevalence of hemophilia A and B in the United States is approximately 20,000 individuals out of an estimated 400,000 individuals worldwide.
Currently there is no cure for hemophilia. Treatment usually involves management of acute bleeding episodes or prophylactic treatment through factor replacement therapy by infusion of individuals’ missing Factor VIII or IX.
A complication for individuals with hemophilia who are receiving factor replacement therapy is the production of antibodies, also called inhibitors, that inactivate the replacement factor. The overall prevalence of inhibitor formation is up to 30% in individuals with hemophilia A and up to 5% in individuals with hemophilia B. Individuals with an inhibitor are treated with what are known as bypassing agents that initiate coagulation by a pathway that is independent of Factor VIII or Factor IX, the proteins that are deficient or inactivated in individuals with hemophilia A and B respectively. Currently available bypassing agents include recombinant Factor VIIa, NovoSeven® RT produced by Novo Nordisk and activated prothrombin complex concentrates, marketed as FEIBA by Shire. NovoSeven® was first approved in 1999 and is indicated for treatment of bleeding episodes, prevention of bleeding during surgeries in individuals with hemophilia A or B with an inhibitor, and individuals with congenital Factor VII deficiency. In 2006, it was approved for the treatment of acquired hemophilia. NovoSeven® RT was approved in 2014 and is also indicated for treatment of Glanzmann’s thrombasthenia. Sales of NovoSeven® RT in 2016, which we estimate based on our research, were $1.4 billion. FEIBA is approved for use in individuals with hemophilia A or B with an inhibitor, which we estimate, based on our research, had 2016 sales of $0.8 billion. Based on our market research, the treatable Factor VIIa patient population in the world’s seven major markets is approximately 3,000 patients.
Based on our research, we estimate worldwide sales of all FactorIX-containing products for the treatment of hemophilia B in 2016 were approximately $1.2 billion, including approximately $0.7 billion as reported by Pfizer, Inc. for its BeneFIX® product and $0.3 billion as reported by Bioverativ and Swedish Orphan Biovitrum for their Alprolix® product, and we estimate that the worldwide Factor IX patient population is approximately 9,700 patients.
Intellectual Property
We have established a broad intellectual property portfolio including patents and patent applications covering the identification, selection, optimization, and manufacture of human proteases, the composition of matter and methods of use of our product candidates and related technology, and other inventions that are important to our business.
As more fully described below, as of January 8, 2017, our patent portfolio included approximately 112 patents; including 13 issued and allowed U.S. patents and 99 foreign granted and accepted patents, and 4 U.S. patent applications, plus an additional 64 pending foreign patent applications. We also rely on trade secrets and careful monitoring of our proprietary information to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection.
All of our patents and applications were internally developed and assigned to us, except for one pending South Korean patent application that isco-owned. Our current patents and patent applications include:
59 patents, including 1 issued U.S. patent, and 16 patent applications, including 1 U.S. patent application, covering modified Factor VII polypeptides, such as our lead product candidate, marzeptacog alfa (activated),
|
We commenced operations in 2002 and are a Delaware corporation. On August 20, 2015, we completed our business combination between Targacept, Inc. and Catalyst Bio, Inc. (“Catalyst Bio”), which was incorporated in Delaware in 1997. Following the completion of the merger, the business conducted by the Company became primarily the business conducted by Catalyst Bio prior to the merger. In this prospectus, we refer to the business combination as the “merger” and to the Company prior to the merger as “Targacept.” Discussions of historical results reflect the results of Catalyst Bio prior to the completion of the merger and do not include the historical results of Targacept prior to the completion of the merger.
Our corporate headquarters are in South San Francisco, California, 94080. Our telephone number is (650)871-0761, and our website address iswww.catalystbiosciences.com. The information on or accessible through our website does not constitute part of this prospectus or any accompanying prospectus supplement and should not be relied upon in connection with making any investment in our securities.
On February 10, 2017, we effected a reverse stock split of our shares of common stock at a ratio ofone-for-fifteen (“2017 Reverse Stock Split”). The 2017 Reverse Stock Split was approved by our stockholders at our special meeting of stockholders held on February 2, 2017. As a result of the 2017 Reverse Stock Split, every fifteen (15) shares of our common stock outstanding was automatically changed and reclassified into one (1) new share of common stock. Holders of common stock that would have otherwise received a fractional share of common stock pursuant to the 2017 Reverse Stock Split received cash in lieu of the fractional share. Unless indicated otherwise, the numbers set forth in this prospectus have been adjusted to reflect the 2017 Reverse Stock Split.
After the completion of this offering, we expect that our Board of Directors will approve and recommend to our stockholders an increase to the number of shares of common stock reserved for issuance under our 2015 Stock Incentive Plan, or the creation of a new stock incentive plan with additional shares. Shares reserved for issuance under any such plans, if approved by stockholders to the extent required by applicable laws and regulations, may be issued by the Board of Directors, or a committee of the Board of Directors, to employees, consultants and directors of the Company, including our current officers and directors. The amount of such increase has not been determined, but could equal up to 20% or more of our total number of shares outstanding or issuable after this offering, including shares issuable upon the exercise of options and warrants. The final determination of the amount of such increase will be made by the Board of Directors or a committee thereof and will be subject to stockholder approval to the extent required by applicable laws or regulations. Any issuance of such shares could dilute the ownership of our other stockholders.
During the first quarter of 2017, under our at the market offering program with JonesTrading Institutional Services LLC (“JonesTrading”), we sold an aggregate of 439,880 shares of our common stock for approximately $5.34 million of net proceeds, including the sale of 241,600 shares of common stock on March 29, 2017, to be
issued at settlement on April 3, 2017, for approximately $3.5 million of net proceeds. Without giving effect to the receipt of proceeds from the foregoing March 29, 2017 sale or any other subsequent events, as of March 28, 2017, we had cash and cash equivalents of approximately $15.5 million. The foregoing financial information should not be viewed as a substitute for full interim financial statements as of and four the three months ended March 31, 2017 prepared in accordance with generally accepted accounting principles and reviewed by our auditors, which we will subsequently provide.
Investing in our securities involves substantial risk, and our business is subject to numerous risks and uncertainties. You should carefully consider all of the information set forth in this prospectus and, in particular, the information under the heading “Risk Factors,” prior to making an investment in our securities. Some of these risks include:
We file electronically with the Securities and Exchange Commission, or SEC, our annual reports on Form10-K, quarterly reports on Form10-Q and current reports on Form8-K pursuant to Section 13(a) or 15(d) of the Exchange Act. We make available on our website at www.catalystbiosciences.com, free of charge, copies of these reports, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC.
The public may read or copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street NE, Washington, D.C. 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at1-800-SEC-0330. The SEC maintains a website that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of that website iswww.sec.gov.
The information in or accessible through the websites referred to above are not incorporated into, and are not considered part of, this filing. Further, our references to the URLs for these websites are intended to be inactive textual references only.
The Offering
Class A Units offered by us | We are offering 285,714 Class A Units. Each Class A Unit consists of one share of common stock and a warrant to purchase half of one shares of our common stock (together with the shares of common stock underlying such warrants). | |
Offering price per Class A Unit | $ | |
Class B Units offered by us | We are also offering to those purchasers whose purchase of Class A Units in this offering would result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock following the consummation of this offering, the opportunity to purchase, in lieu of the number of Class A Units that would result in ownership in excess of 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock, 12,000 Class B Units. Each Class B Unit will consist of one share of Series A Preferred Stock, par value $0.001 per share, convertible into 95.24 shares of common stock and a warrant to purchase 47.62 shares of our common stock (together with the shares of our common stock underlying such shares of Series A Preferred Stock and warrants). | |
Offering price per Class B Unit | $ | |
Overallotment option | The underwriters have the option to purchase up to additional shares of common stock, and/or warrants to purchase shares of common stock solely to cover over-allotments, if any, at the price to the public less the underwriting discounts and commissions. The over-allotment option may be used to purchase shares of common stock, or warrants, or any combination thereof, as determined by the underwriters, but such purchases cannot exceed an aggregate of 15% of the number of shares of common stock (including the number of shares of common stock issuable upon conversion of shares of Series A Preferred Stock) and warrants sold in the primary offering. The over-allotment option is exercisable for 45 days from the date of this prospectus. | |
Description of warrants | The warrants will be exercisable beginning on the date of issuance and expire on the five (5) year anniversary of the date of issuance at an initial exercise price per share equal to , subject to appropriate adjustment in the event of recapitalization events, stock dividends, stock splits, stock combinations, reclassifications, reorganizations or similar events affecting our common stock. | |
Description of Series A Preferred Stock | Each share of Series A Preferred Stock is convertible at any time at the holder’s option into 95.24 shares of common stock. | |
| Notwithstanding the foregoing, we shall not effect any conversion of Series A Preferred Stock, with certain exceptions, to the extent that, after giving effect to an attempted conversion, the holder of shares of Series A Preferred Stock (together with such holder’s affiliates, and any persons acting as a group together with such holder or any of such holder’s affiliates) would beneficially own a number of shares of our common stock in excess of 4.99% (or, at the election of the purchaser, 9.99%) of the shares of our common stock then outstanding after giving effect to such exercise. For additional information, see “Description of Securities—Preferred Stock” on page 53 of this prospectus. | ||
Shares of common stock outstanding before this offering | 1,000,036 shares | |
Shares of Series A Preferred Stock outstanding before this offering | None | |
Shares of common stock outstanding after this offering | 1,285,750 shares | |
| 12,000 shares | |
| Use of proceeds |
| We estimate that the net proceeds to us from this offering will be approximately $13,470,000 million, based on an assumed offering price of $10.50 per Class A Unit and |
| Risk factors | You should carefully read and consider the information set forth under “Risk Factors” | |
Nasdaq Capital Market common stock symbol | CBIO | |
No listing of Series A Preferred Stock or | We do not intend to apply for listing of the shares of | |
|
|
The number of shares of common stock to be outstanding before this offering and to be outstanding after this offering in the table above is based on 1,000,036 shares of common stock outstanding as of March 28, 2017 and excludes:
The number of shares of our common stock that will be outstanding immediately after this offering excludes:
Unless otherwise indicated, all information contained in this prospectus assumes:
In addition, unless otherwise noted, all information in this prospectus gives effect to the one-for- reverse stock split of our common stock that will be effective prior to the completion of this offering.
SUMMARY FINANCIAL DATAits overallotment option.
The following tables summarize our financial data. You should read the following summary financial data together with our financial statements and the related notes appearing at the end of this prospectus and the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section and other financial information included in this prospectus.
The pro forma net loss attributable to common stockholders per share information is computed using the weighted average number of common shares outstanding, after giving pro forma effect to the conversion of all outstanding shares of our convertible preferred stock into 73,739,905 shares of common stock concurrently with the completion of this offering, as if the conversion had occurred at the date of the original issuance. This pro forma information does not give effect to the exercise of an outstanding warrant.
| Year ended December 31, | Three months ended March 31, | |||||||||||||||||||
| 2001 | 2002 | 2003 | 2003 | 2004 | ||||||||||||||||
| (unaudited) | ||||||||||||||||||||
| (in thousands, except share and per share data) | ||||||||||||||||||||
Statement of Operations Data: | ||||||||||||||||||||
Net revenue | $ | 1,703 | $ | 2,286 | $ | 2,458 | $ | 691 | $ | 497 | ||||||||||
Operating expenses: | ||||||||||||||||||||
Research and development | 8,152 | 16,244 | 18,179 | 4,069 | 6,050 | |||||||||||||||
General and administrative | 2,302 | 4,135 | 3,600 | 697 | 1,109 | |||||||||||||||
Cost of product sales | — | 244 | 743 | 200 | 182 | |||||||||||||||
Purchased in-process research and development | — | 2,666 | — | — | — | |||||||||||||||
Total operating expenses | 10,454 | 23,289 | 22,522 | 4,966 | 7,341 | |||||||||||||||
Loss from operations | (8,751 | ) | (21,003 | ) | (20,064 | ) | (4,275 | ) | (6,844 | ) | ||||||||||
Interest and dividend income | 1,449 | 88 | 791 | 124 | 231 | |||||||||||||||
Interest expense | — | (103 | ) | (122 | ) | (34 | ) | (25 | ) | |||||||||||
Loss on disposal of fixed assets | — | (54 | ) | — | — | — | ||||||||||||||
Net loss | (7,302 | ) | (21,072 | ) | (19,395 | ) | (4,185 | ) | (6,638 | ) | ||||||||||
Preferred stock accretion | (3,808 | ) | (4,173 | ) | (8,341 | ) | (1,915 | ) | (2,142 | ) | ||||||||||
Net loss attributable to common stockholders | $ | (11,110 | ) | $ | (25,245 | ) | $ | (27,736 | ) | $ | (6,100 | ) | $ | (8,780 | ) | |||||
Basic and diluted net loss per share applicable to common stockholders | $ | (26.80 | ) | $ | (45.28 | ) | $ | (33.91 | ) | $ | (9.48 | ) | $ | (7.89 | ) | |||||
Shares used to compute basic and diluted net loss per share | $ | 414,624 | $ | 557,492 | $ | 817,894 | $ | 643,571 | $ | 1,112,591 | ||||||||||
Pro forma basic and diluted net loss per share applicable to common stockholders (unaudited) | $ | (0.27 | ) | $ | (0.09 | ) | ||||||||||||||
Shares used to compute pro forma basic and diluted net loss per share (unaudited) | 71,118,629 | 74,852,496 | ||||||||||||||||||
The pro forma balance sheet information gives effect to the conversion of all outstanding shares of our convertible preferred stock into 73,739,905 shares of common stock concurrently with the completion of this offering. The pro forma as adjusted balance sheet information gives further effect to:
| As of March 31, 2004 | ||||||||||
| Actual | Pro Forma | Pro Forma As Adjusted | ||||||||
(unaudited) (in thousands) | ||||||||||
Balance Sheet Data: | ||||||||||
Cash, cash equivalents and short-term investments | $ | 36,847 | $ | 36,847 | ||||||
Working capital | 33,663 | 33,663 | ||||||||
Total assets | 40,806 | 40,806 | ||||||||
Long-term debt, net of current portion | 1,298 | 1,298 | ||||||||
Redeemable convertible preferred stock | 132,276 | — | ||||||||
Accumulated deficit | (82,818 | ) | (82,818 | ) | ||||||
Total stockholders’ equity (deficit) | (99,510 | ) | 32,766 | |||||||
InvestingAn investment in our common stocksecurities involves a high degree of risk. You should carefully consider the risks described in our Annual Report on Form10-K for the year ended December 31, 2016, as updated by any other document that we subsequently file with the Securities and uncertaintiesExchange Commission and that is incorporated by reference into this prospectus, the risks described below together withand all of the other information contained in this prospectus and incorporated by reference into this prospectus, including our financial statements and related notes, before deciding to investinvesting in our common stock.securities. If any of these risksthe possible events described in those sections or below actually occurs,occur, our business, business prospects, financial condition,cash flow, results of operations or cash flows would likely suffer, maybe materially. Thisfinancial condition could causebe harmed. In this case, the trading price of our common stock tocould decline, and you couldmight lose partall or allpart of your investment.
The following is a discussion of the risk factors that we believe are material to us at this time. These risks and uncertainties are not the only ones facing us and there may be additional matters that we are unaware of or that we currently consider immaterial. All of these could adversely affect our business, results of operations, financial condition and cash flows.
Risks Relatedrelated to Our Financial Resultsour financial condition and Need for Additional Financingcapital requirements
We have incurred significant losses since our inception, and anticipate that we willare expected to continue to incur substantialsignificant losses for the foreseeable future. We may never achieve or sustain profitability.
We were incorporated in 1997are a clinical-stage biotechnology company, and operated as a wholly owned subsidiary of R.J. Reynolds Tobacco Company until August 2000.we have not yet generated significant revenues. We have a limited operating history and have incurred substantial net losses in each year since our inception.inception in August 2002, including net losses of $16.9 million and $14.8 million for the years ended December 31, 2016 and 2015, respectively. As of MarchDecember 31, 2004,2016, we had an accumulated deficit of $82.8$148.0 million. Our net loss was $6.6 million
We are still in the early stages of development of our product candidates, and have no products approved for the three months ended March 31, 2004, $19.4 million for the fiscal year ended December 31, 2003commercial sale. To date, we have financed our operations primarily from private placements of convertible preferred stock, payments under collaboration agreements, and $21.1 million for the fiscal year ended December 31, 2002. Our lossesto a lesser extent through issuances of shares of common stock.
We have resulted principally from costs incurred in connection withdevoted most of our financial resources to research and development, activities, including clinical trials, and from general and administrative expenses associated with our operations.preclinical development activities. We expect to continue to incur substantialsignificant expenses and operating losses over the next several years. Our operating losses may fluctuate significantly from quarter to quarter and year to year. We are expected to continue to incur significant expenses and increasing operating losses for at least the foreseeablenext several years, and our expenses will increase substantially if and as we:
In addition, in connection with the license granted to us by Pfizer, we agreed to make contingent cash payments to Pfizer in an aggregate amount equal to up to $17.5 million, payable upon the achievement of clinical, regulatory and commercial milestones, the timing of which is uncertain. Following commercialization of any of Factor VIIa products, Pfizer would also receive a single-digit royalty on net product sales on acountry-by-country basis for a predefined royalty term. See “Business Overview—Factor VIIa” in this prospectus.
Further, in connection with an initial statement of work under the Development and Manufacturing Agreement that we have entered into with CMC ICOS Biologics, Inc. (“CMC”), we have agreed to a total of $3.8 million in payments to CMC, subject to the completion of work relating to the manufacturing development of marzeptacog alfa (activated). See “Item 1—Business—Collaborations” in our Annual Report on Form10-K for the year ended December 31, 2016.
To become and remain profitable, we must succeed in developing and eventually commercializing products that generate significant revenue. This will require us to be successful in a range of challenging activities, including completing preclinical testing and clinical trials of product candidates, obtaining regulatory approval for these product candidates and manufacturing, marketing and selling any products for which regulatory approval is obtained. We are only in the preliminary stages of most of these activities. We may never succeed in these activities and, even if we do, we may never generate revenues that are significant enough to achieve profitability.
Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Failure to become and remain profitable would depress the value of the company and could impair our ability to raise capital, expand our business, maintain research and development efforts, diversify product offerings or even continue operations. A decline in the value of the Company could also cause you to lose all or part of your investment.
We will continue to need additional capital in the future. If we are unable to raise sufficient capital in the future, we will be forced to delay, reduce or eliminate product development programs.
Developing pharmaceutical products, including conducting preclinical studies and clinical trials, is expensive. We expect our research and development expenses to increase substantially following completionwith our ongoing activities, particularly activities related to the continued clinical development of this offering asmarzeptacog alfa (activated), including a clinical efficacy trial and, if Phase 1 clinical trials of CB 2679d/ISU304 are successful, an efficacy trial for that compound. Until we expand our clinical trial activity and ascan generate a sufficient revenue from our product candidates, advanceif ever, we expect to finance future cash needs through public or private equity offerings, debt financings, corporate collaborations and/or licensing arrangements. Additional funds may not be available when we need them on terms that are acceptable, or at all. If adequate funds are not available, we may be required to delay, reduce the scope of or eliminate one or more of our research or development cycle. programs.
In August 2015, we issued $37.0 million in aggregate principal amount of redeemable convertible notes to former Targacept stockholders as part of a dividend immediately prior to the completion of the merger (the“Pre-Closing Dividend”), with an amount equal to the total principal deposited in an escrow account for the benefit of the noteholders. The notes may be redeemed for cash or repaid upon maturity, holders may also elect to convert any principal amount of the notes into shares of common stock at a price of $137.85 per share on or before February 19, 2018. As of December 31, 2016, $17.3 million in aggregate principal has been redeemed and $0.3 million had been converted to common stock. Except for this arrangement, we have no commitments or
arrangements for any additional financing to fund our research and development programs. There can be no assurance regarding the amount of the notes that will be redeemed or the portion of the remaining $19.4 million in capital that will become available to us.
We also expectbelieve that our general and administrative costscurrent available cash, together with the proceeds from this offering will be sufficient to increase substantially as we expandfund our infrastructure. As a result,operations for at least 18 months. However, we will need to generate significant revenuesraise substantial additional capital to pay these costscomplete the development and achieve profitability.commercialization of marzeptacog alfa (activated) and CB 2679d/ISU304, and depending on the availability of capital, may need to delay development of some of our product candidates.
InversineBecause successful development of our product candidates is our only current source of product revenue. We acquired the rights to Inversine in August 2002. Sales of Inversine generated revenues of only $188,000 for the three months ended March 31, 2004 and $815,000 for the year ended December 31, 2003. Inversine is approved in the United States for the treatment of moderately severe to severe essential hypertension. However, we believe that the substantial majority of Inversine sales are derived from prescriptions written by a very limited number of physicians for the treatment of Tourette’s syndrome and other neuropsychiatric disorders. If any of these physicians were to change their prescribing habits, Inversine sales would suffer. We do not expect that sales of Inversine will increase substantially in the future.
Ifuncertain, we are unable to develop and commercialize any of our product candidates, if development is delayed or if sales revenue from any of our product candidates that receives marketing approval is insufficient, we may never become profitable. Even if we do become profitable, we may not be ableestimate the actual funds required to sustain or increase our profitability on a quarterly or annual basis.
We will require substantial additional financing and our failure to obtain additional funding when needed could force us to delay, reduce or eliminate our product development programs or commercialization efforts.
We will require substantial future capital in order to continue to conduct thecomplete research and development clinical and regulatory activities necessary to bringcommercialize our product candidates to market and to establish marketing and sales capabilities.products under development. Our future capitalfunding requirements, both near and long-term, will depend on many factors, including:including, but not limited to:
Raising additional funds by issuing securities or through licensing arrangements may cause dilution to stockholders, restrict our general and administrative expenses.
operations or require us to relinquish proprietary rights.
In addition, we may seek additional capital due to market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership intereststockholders will be diluted, and the terms of these new securities may include liquidation or other preferences that adversely affect yourthe rights as a stockholder. of common stockholders.
Debt financing, if available at all, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through collaboration andcollaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, or product candidates or future revenue streams or grant licenses on terms that are not favorable to us.
Based on our current operating plan, We may also seek to access the public or private capital markets whenever conditions are favorable, even if we expectdo not have an immediate need for additional capital at that our existing capital resourcestime. There can be no assurance that we will be able to obtain additional funding if, and the net proceeds from this offering will enable uswhen necessary. If we are unable to maintain currently planned operations through the first half of 2006. However, our operating plan may change as a result of many factors including those described above, and we may need additional funds sooner than planned to meet operational needs and capital requirements for product development and commercialization. Other than a modest amount of committed equipmentobtain adequate financing we currently have no credit facility or committed sources of capital. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available on a timely basis, we may:could be required to delay, curtail or eliminate one or more, or all, of our development programs or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Demand™ Sales Agreement with JonesTrading. In accordance with the terms of the sales agreement, as of March 29, 2017, we have sold 479,681 shares of our common stock having an aggregate offering price of $6.5 million through JonesTrading. Any additional sales in the public market of our common stock under the shelf registration statement could adversely affect prevailing market prices for our common stock.
We have no history of clinical development or commercialization of pharmaceutical products, which may make it difficult to evaluate the prospects for the company’s future viability.
We began operations in August 2002. Our operations to date have been limited to financing and staffing the company, developing our technology and product candidates and establishing collaborations. We have not yet demonstrated an ability to successfully conduct a clinical trial, obtain marketing approvals, manufacture a product for clinical trials or at commercial scale, or arrange for a third-party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization. Consequently, predictions about the company’s future success or viability may not be as accurate as they could be if we had a longer operating history or a history of successfully developing and commercializing pharmaceutical products.
Risks related to the discovery, development and commercialization of our product candidates
We are substantially dependent upon the success of marzeptacog alfa (activated) and CB 2679d/ISU304.
The failure of marzeptacog alfa (activated) or CB 2679d/ISU304 to commence anticipated clinical trials or achieve successful clinical trial endpoints, delays in clinical development generally, unanticipated adverse side effects or any other adverse developments or information related to marzeptacog alfa (activated) or CB 2679d/ISU304 would significantly harm our business, its prospects and the value of the company’s common stock. We expect to advance marzeptacog alfa (activated) into a Phase 2 clinical efficacy trial in individuals with hemophilia A or B with an inhibitor and to advance CB 2679d/ISU304 into a Phase 1/2 clinical trial in individuals with hemophilia B. There is no guarantee that the results of these clinical trials, if they occur, will be positive or will not generate unanticipated safety concerns. The Phase 1 clinical trial of marzeptacog alfa (activated) was a single-dose escalation trial that would not, compared to multi-dose trials, be expected to exclude the possibility of an immunological response to marzeptacog alfa (activated) in individuals who received the product candidate. After completion of the dosing portion of the Phase 1 clinical trial, Pfizer observed a positive result in an assay for a potentialnon-neutralizing anti-drug antibody in a single individual at a time point 60 days post-dosing that was not confirmed by testing of a subsequent,follow-up blood draw. Additional confirmatory testing indicated that this was due to a false positive assay result; however, there can be no assurance thatanti-marzeptacog alfa (activated) antibodies will not be observed in subsequent trials. If subsequent multi-dose trials of marzeptacog alfa (activated) or of CB 2679d/ISU304 demonstrate a treatment-related neutralizing immunological response in individuals, development of such product could be halted. Even if the next trials of marzeptacog alfa (activated) are positive, marzeptacog alfa (activated) may require substantial additional trials and other testing before approving marzeptacog alfa (activated) for marketing, and CB 2679d/ISU304 will require additional trials and other testing before receiving approval for marketing.
Marzeptacog alfa (activated) and CB 2679d/ISU304 are not expected to be commercially available in the near term, if at all. Further, the commercial success of each product candidate will depend upon its acceptance by physicians, patients, third-party payors and other key decision-makers as a therapeutic and cost effective alternative to currently available products. If we are unable to successfully develop, obtain regulatory approval for and commercialize marzeptacog alfa (activated) and CB 2679d/ISU304, our ability to generate revenue from product sales will be significantly delayed and our business will be materially and adversely affected, and we may not be able to earn sufficient revenues to continue as a going concern.
Even if the FDA or other regulatory agency approves marzeptacog alfa (activated) or CB 2679d/ISU304, the approval may impose significant restrictions on the indicated uses, conditions for use, labeling, advertising, promotion, marketing and/or production of such product and may impose ongoing commitments or requirements
for post-approval studies, including additional research and development and clinical trials. The FDA and other agencies also may impose various civil or criminal sanctions for failure to comply with regulatory requirements, including withdrawal of product approval. Regulatory approval from authorities in other foreign countries will be needed to market marzeptacog alfa (activated) or CB 2679d/ISU304 in those countries. Approval by one regulatory authority does not ensure approval by regulatory authorities in other jurisdictions. If we fail to obtain approvals from foreign jurisdictions, the geographic market for marzeptacog alfa (activated) or CB 2679d/ISU304 would be limited.
We plan to conduct clinical trials for onesubcutaneous dosing trials of marzeptacog alfa (activated) and CB 2679d/ISU304, which is an untested route of administration for these product candidates in humans.
We expect to commence a subcutaneous prophylaxis clinical trial of marzeptacog alfa (activated) in 2017 and for ISU Abxis to commence a subcutaneous clinical trial of CB 2679d/ISU304 in the second quarter of 2017 and clinical data is expected in the second half of 2017. Neither product candidate has previously been studied in human clinical trials using subcutaneous dosing. There can be no assurance that either product will achieve efficacious levels of biological activity when administered subcutaneously. There can also be no assurance that the clinical trial results will be positive or morethat the clinical trials will not generate unanticipated safety concerns. The failure of either product to achieve successful clinical trial endpoints, delays in clinical trial commencement or in clinical development generally, unanticipated adverse side effects, adverse immunological responses, or any other adverse developments or information related to our product candidates would significantly harm our business, its prospects and the value of our common stock.
Marzeptacog alfa (activated) and CB 2679d/ISU304 may cause the generation of antibodies, which could prevent their further development.
Both marzeptacog alfa (activated) and CB 2679d/ISU304 are protein molecules which may cause the generation of antibodies in individuals who receive them. The Phase 1 clinical trial of marzeptacog alfa (activated) was a single-dose intravenous escalation trial that would not, compared to multi-dose trials or higher dose administered subcutaneously, be expected to exclude the possibility of an immunological response to marzeptacog alfa (activated) in individuals who received the product candidates;
If subsequent multi-dose trials demonstrate a treatment-related neutralizing immunological response in individuals, development of marzeptacog alfa (activated) or of CB 2679d/ISU304 could be halted.
We are transitioning manufacturing and clinical activities related to marzeptacog alfa (activated) from Pfizer to CMC and continuing to optimize the manufacturing process. This process will be lengthy and its outcome uncertain.
Pfizer, through its wholly-owned subsidiary Wyeth, conducted the Phase 1 clinical trial of marzeptacog alfa (activated) pursuant to a research and license agreement. Pfizer terminated this agreement effective June 1, 2015.
In March 2016, we engaged CMC to conduct manufacturing development and, upon successful development of the manufacturing process, manufacture the marzeptacog alfa (activated) that we intend to use in our establishment of salesclinical trials on afee-for-services basis. During 2016, we also worked with Pfizer to transition manufacturing capabilities from Pfizer to CMC, and marketing capabilities or other activitiesin December 2016, Pfizer granted us an exclusive license to its proprietary rights for manufacturing materials and processes that may be necessaryapply to commercialize our product candidates; or
Factor VIIa variants, CB 813a and marzeptacog
alfa (activated). Pfizer also transferred the IND and documentation related to the Risks RelatedDevelopmentdevelopment, manufacturing and Regulatory Approvaltesting of Our Product Candidates
Our success depends substantially onWe are very early in our most advanceddevelopment efforts and have only one product candidate that has completed a Phase 1 clinical trial. All our other product candidates which are still underin preclinical development. If we are unable to bring any or all of theseobtain regulatory clearance and commercialize our product candidates to market, or experience significant delays in doing so, our ability to generate product revenue and our likelihood of successbusiness will be materially harmed.
Inversine isWe are very early in our development efforts and have only marketedone product and generates limited revenues. Our most advancedcandidate that has completed a Phase 1 clinical trial, marzeptacog alfa (activated). All our other product candidates are still in preclinical development. Following the Korean Ministry of Food and Drug Safety’s approval in March 2017 of the IND application for CB 2679d/ISU304, we expect to advance CB 2679d/ISU304 into Phase I1/2 clinical trial in individuals with hemophilia B. We also expect to advance marzeptacog alfa (activated) into a Phase 2 clinical efficacy trial in individuals with hemophilia A or Phase II clinical trials.B with an inhibitor; however, the FDA may require additionalpre-clinical testing before we are permitted to commence subcutaneous dosing trials of marzeptacog alpha (activated). Moreover, engineered protease biopharmaceuticals are a relatively new class of therapeutics. There can be no assurance as to the length of the trial period, the number of individuals the FDA will require to be enrolled in the trials to establish the safety, efficacy, purity and potency of the engineered protease products, or that the data generated in these trials will be acceptable to the FDA or foreign regulatory agencies to support marketing approval. Our ability to generate product revenue in the futurerevenues, which we do not expect will occur for many years, if ever, will depend heavily on the successful development and eventual commercialization of theseour product candidates. Our other product candidates are in various stages of preclinical development. AnyThe success of our product candidates could be unsuccessful if it:will depend on several factors, including the following:
We do not expect any
If we do not obtain the regulatory approvals requiredachieve one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to market and sellsuccessfully commercialize our product candidates, which would materially harm our ability to generate product revenue will be materially impairedbusiness.
Clinical drug development involves a lengthy and expensive process with an uncertain outcome. Results from our business willsuccessful Phase 1 trials may not be successful.
The preclinical laboratory testing,confirmed in later trials, and if serious adverse or unacceptable side effects are identified during the development manufacturing and clinical trials of product candidates that we develop, whether independently or in collaboration with a third party, as well as their distribution, sale and marketing, are regulated by the FDA and other federal, state and local governmental authorities in the United States and by similar agencies in other countries. We must receive regulatory approval of each product candidate before we can market and sell it. We have only limited experience in pursuing regulatory approvals. Securing FDA approval requires the submission of extensive preclinical and clinical data and information about the chemistry and manufacture of, and control procedures for, each potential product. In addition, the supporting information submitted to the FDA for each therapeutic indication must establish the safety and efficacy of the product candidate. The marketing approval process takes many years, requires the expenditure of substantial resources, is subject to delays and can vary substantially based upon the type, complexity and novelty of the product candidates involved. In addition to the time and expense involved, the process is uncertain and we may never receive the required regulatory approvals. In addition, the FDA, the U.S. Congress and foreign regulatory authorities may from time to time change approval policies or adopt new laws or regulations, either of which could prevent or delay our receipt of required approvals. Even if we receive regulatory approval to market a particular product candidate, the approval will be subject to limitations on the indicated uses for which it may be marketed and may not permit labeling claims that are necessary or desirable for its promotion.
According to the FDA, a Phase I clinical trial program typically takes several months to complete, a Phase II clinical trial program typically takes several months to two years to complete and a Phase III clinical trial program typically takes one to four years to complete. Industry sources report that the preparation and submission of a new drug application, or NDA, which is required for regulatory approval in the United States, generally takes six months to one year to complete after completion of a pivotal clinical trial. Industry sources also report that approximately 10% to 15% of all NDAs accepted for filing by the FDA are not approved and that FDA approval, if granted, usually takes approximately one year after submission, although it may take longer if additional information is required by the FDA. In addition, the Pharmaceutical Research and Manufacturers of America reports that only one out of five product candidates that enter clinical trials will ultimately be approved by the FDA for commercial sale.
The FDA may delay, limit or deny approval of any of our product candidates for many reasons. For example:
In particular, because drugs that target NNRs are a new class of drugs, the FDA and other applicable regulatory authorities may require more preclinical or clinical data for our product candidates or more time to evaluate that data than we currently anticipate. If we obtain the requisite regulatory approval for a particular product candidate, the approval may not extend to all indications for which we have sought approval, which could limit the use of the product and adversely impact our potential revenues.
In addition, we currently intend to pursue marketing approval for TC-1734 for cognitive impairment in the elderly, including AAMI and MCI. Neither the FDA nor, to our knowledge, any foreign regulatory authority has approved a drug indicated for use either for cognitive impairment in the elderly generally or for specific conditions such as AAMI or MCI. Furthermore, neither AAMI nor MCI is listed in the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, or DSM-IV, the manual published by the American Psychiatric Association to establish diagnostic criteria. We do not know if the FDA or any other such regulatory authority will be willing to recognize AAMI or MCI as a defined condition or disease and grant approval of our product candidate for these indications.
Even if the FDA approves a product candidate for marketing and sale in the United States, applicable regulatory authorities in other countries may not approve the product candidate or may subject their approval to conditions such as additional product testing or otherwise cause delays. The regulatory approval process varies among countries, but generally includes all of the risks associated with obtaining FDA approval. In addition, many countries require a separate review process prior to marketing to determine whether their respective national health insurance schemes will pay for newly approved products, as well as the price that may be charged for a product. This process will cause delays in the marketing of any of our product candidates that receives marketing approval and could adversely impact our revenues and results of operations.
If clinical trials for our product candidates are not successful, we will not be able to obtain regulatory approval for and commercialize them.
To obtain the requisite regulatory approvals to market and sell any of our product candidates, we must demonstrate, through extensivemay need to abandon or limit our development of some of our product candidates.
Clinical testing is expensive, time consuming and uncertain as to outcome. We cannot guarantee that any preclinical studies and clinical trials that the product candidate is safe and effective in humans.will be conducted as planned or completed on schedule, if at all. The number of clinical trials required to obtain approval varies depending on the particular product candidate, the disease or condition for which it is in development and the regulations applicable to it. Preclinical studies and clinical trials are lengthy and expensive, difficult to design and implement and subject to a historically high rate of failure. The development of each of our product candidates involves significant risks at each stageis susceptible to the risk of testing. A failure of one or more of our clinical trials could occur at any stage of testing. If we experience difficulties or failuresdrug development, including failure to demonstrate efficacy in our clinical trials, or if we are not able to design our clinical trials with clear criteria to determine the efficacy of our product candidates, our product candidates may never be approved for sale or become commercially available.
Success in preclinical studies of a product candidate may not be predictive of similar results in humans during clinical trials. In addition, successful results from early clinical trials of a product candidate may not be replicated in later clinical trials. In particular, in our Phase I and Phase II clinical trials of TC-1734, our product candidate in development for the treatment of cognitive impairment in the elderly, we have used a battery of tests developed by CDR Ltd. to assess each subject’s cognitive function. The CDR test battery is different from the test battery that is most often used to assess the efficacy of drugs for the treatment of Alzheimer’s disease, the most common form of dementia. We plan to meet with the FDA to discuss the use of the CDR test battery in our future clinical trials of TC-1734. If, based on the discussions with the FDA, we use an additional or a different test battery for our future clinical trials of TC-1734, there would be a greater risk that the results of our Phase I and Phase II clinical trials of TC-1734 will not be predictive of similar results of those future clinical trials.
We may not be able to obtain authority from the FDA, other applicable regulatory authorities or the institutional review boards at our intended investigational sites to commence or complete our clinical trials.
Before a clinical trial may commence inor across a suitable population of patients, the United States, we must submit an investigational new drug application,occurrence of severe or IND, containing preclinical studies, chemistry, manufacturing, controlmedically or commercially unacceptable adverse events, failure to comply with protocols or applicable regulatory requirements and other information and a study protocol to the FDA. Ifdetermination by the FDA doesor any comparable foreign regulatory authority that a drug product is not object within 30 days after submission of the IND, then the trial may commence. If commenced, we, the FDA, other applicable regulatory authorities or institutional review boards may delay, suspend or terminate clinical trials of a product candidate at any timeapprovable. It is possible that even if among other reasons, we or they believe the subjects or patients participating in the clinical trials are being exposed to unacceptable health risks or for other reasons.
If we do not prove in clinical trials that our product candidates are safe and effective, we will not obtain marketing approvals from the FDA and other applicable regulatory authorities. In particular, one or more of our product candidates mayhas a beneficial effect, that effect will not exhibitbe detected during clinical evaluation as a result of one or more of a variety of factors, including the expected therapeutic results in humans, may cause harmful side effectssize, duration, design, measurements, conduct or may have other unexpected characteristics that preclude regulatory approval for any or all indicationsanalysis of use or limit commercial use if approved. As of April 30, 2004, fourour clinical trials. Conversely, as a result of the 78 patients that had participated in the ongoing Phase IIsame factors, our clinical trialtrials may indicate an apparent positive effect of TC-2403, oura product candidate forthat is greater than the treatment of ulcerative colitis, had experienced an elevationactual positive effect, if any. Similarly, in liver enzymes in excess of three times the upper limit of normal. These patients were withdrawn from the trial and their liver enzymes returned to within normal limits. Because the trial is double blind, we do not know if these patients were administered TC-2403 or a placebo. If some or all of these patients were administered TC-2403 and if futureour clinical trials we may fail to detect toxicity of TC-2403 show similar or more prevalent elevations of liver enzymes, the FDA or other regulatory authorities may not grant us approval to market TC-2403.Also, our product candidate TC-5231, in development for ADHD, is a low-dose reformulation of mecamylamine hydrochloride. Mecamylamine hydrochloride is approved at a high dose as Inversine for the treatment of moderately severe to severe essential hypertension. If clinical trials show that TC-5231 has a similar effect on blood pressure as Inversine, the FDA or other regulatory authorities may not grant us approval to market TC-5231 for the treatment of ADHD in children or at all.
Our research and preclinical programs and product candidates target diseases that are not well understood. For example, there is only limited scientific understanding of the causes of cognitive impairment, including AAMI, MCI and Alzheimer’s disease, ADHD, schizophrenia and depression and anxiety. In addition, there are no approved NNR-targeted drugs that treat these diseases, and there is only limited scientific understanding of the relationships between these diseases and the neurological pathways targetedintolerability caused by our product candidates, or mistakenly believe that our product candidates are toxic or not well tolerated when that is not in fact the case.
In addition, the outcome of preclinical studies and research and preclinical programs. These uncertainties increase the risk that one or more of ourearly clinical trials willmay not be successful.predictive of the success of later clinical trials. For example, the Phase 1 clinical trial of marzeptacog alfa (activated) was a single dose trial, and adverse immunological reactions such as the development of a neutralizing anti-drug antibody would not be likely to appear until patients received multiple doses in later trials.
IfMany companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials after achieving positive results in earlier development, and we may face similar setbacks. The design of a clinical trial can determine whether our results will support approval of a product and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. We have limited experience in designing clinical trials and may be unable to design and execute a clinical trial to support marketing approval. In addition, preclinical and clinical data are often susceptible to varying interpretations and analyses. Many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval for the product candidates. Even if we believe that the results of clinical trials for our product candidates are prolongedwarrant marketing approval, the FDA or delayed, we would be unable to commercializecomparable foreign regulatory authorities may disagree and may not grant marketing approval of our product candidates on a timely basis, which would require uscandidates.
In some instances, there can be significant variability in safety or efficacy results between different clinical trials of the same product candidate due to incur additional costsnumerous factors, including changes in trial procedures set forth in protocols, differences in the size and delay our receipttype of any revenues from potential product sales.
We cannot predict whether we will encounter problems with anythe patient populations, changes in and adherence to the dosing regimen and other clinical trial protocols and the rate of our completed, ongoingdropout among clinical trial participants. Any Phase 2, Phase 3 or plannedother clinical trials that will cause us or any regulatory authority to delay or suspend those clinical trials or delaywe may conduct may not demonstrate the analysis of data derived from them. A number of events, including any of the following, could delay the completion of our ongoingefficacy and planned clinical trials and negatively impact our abilitysafety necessary to obtain regulatory approval for, and to market and sell, a particularour product candidate:candidates.
trials as required by the FDA or resultssimilar regulatory authorities outside the United States. In particular, there is a relatively small number of individuals with hemophilia, which may cause delays in enrollment of clinical trials of marzeptacog alfa (activated) in individuals with hemophilia A or B with an inhibitor or CB 2679d/ISU304 in individuals with hemophilia B. In addition, some of our competitors have ongoing clinical trials for product candidates that are inconsistent with earlier results, that necessitate additionaltreat the same indications as our product candidates, and patients who would otherwise be eligible for our clinical study;trials may instead enroll in clinical trials of our competitors’ product candidates.
Patient enrollment is affected by other factors including:
Clinical trials require sufficient patient enrollment, which is a function of many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites, the availability of effective treatments for the relevant disease and the eligibility criteria for the study in question;
Our inability to enroll a sufficient number of patients in a timely or cost-effective manner. Furthermore, enrolled patients may drop out of our clinical trials, and thereby impair the validity or statistical significance of the clinical trials.
Prior to commencing clinical trials in the United States, we must submit an IND to the FDA. We are currently conducting clinical trials outside the United States for our product candidate TC-2696 and have not submitted an IND to enable us to conduct clinical trials of that product candidate in the United States.
Our product candidate TC-5231 is a low-dose reformulation of the active ingredient in Inversine, which was approved in the 1950s. If the FDA determines that the safety and tolerability data that were used to support regulatory approval of Inversine at that time are incomplete under current standards, outdated or otherwise not in compliance with current guidelines, the FDA may not accept the data as support for a potential regulatory submission by us for approval of TC-5231. The FDA has indicated to us that we may be required to conduct lengthy non-clinical carcinogenicity studies before we could submit an NDA for the use of TC-5231 to treat ADHD in children. These carcinogenicity studies are routinely conducted prior to submission of an NDA today but were not performed on Inversine prior to its approval. If we are required to conduct these carcinogenicity studies, our development costs for TC-5231 will increase and regulatory approval and receipt of any revenues from potential sales of TC-5231 may be delayed.
We do not know whether our clinical trials will begin as planned, will needresult in significant delays and could require us to be restructuredabandon one or will be completed on schedule, if at all. Delays in ourmore clinical trials willaltogether. Enrollment delays in clinical trials conducted by us may also result in increased development costs for our product candidates. In addition, if our clinical trials are delayed, our competitors may be ablecandidates, which would cause the value of the company to bring products to market before we dodecline and the commercial viability of our product candidates could be limited.
Our product candidates will remain subject to ongoing regulatory review even if they receive marketing approval. If we fail to comply with continuing regulations, we could lose these approvals and the sale of our products could be suspended.
Even if we receive regulatory approval to market a particular product candidate, the approval could be conditioned on us conducting additional costly post-approval studies or could limit the indications for use included in our labeling. Moreover, the product may later cause adverse effects that limit or prevent its widespread use, force us to withdraw it from the market or impede or delay our ability to obtain regulatory approvalsadditional financing.
Risks related to our reliance on third parties
We depend on our collaborative relationship with ISU Abxis for the Phase 1 development of CB 2679d/ISU304.
We have a collaboration agreement with ISU Abxis for preclinical and Phase 1/2 development of an improved, next-generation Factor IX product, CB 2679d/ISU304. Under our agreement with ISU Abxis, ISU Abxis is responsible for manufacturing and Phase 1/2 clinical trials of this product candidate, and we depend on ISU Abxis to complete these activities.
Our ability to generate revenues from this arrangement will depend on the ability of ISU Abxis to successfully perform the functions assigned to it in additional countries. In addition,this arrangement, and accordingly, any failure by ISU Abxis to develop this product candidate could adversely affect our cash flows. Further, this collaboration agreement may not lead to development or commercialization of this product candidate in the manufacturermost efficient manner or at all, and ISU Abxis has the right to abandon research or development projects and terminate applicable agreements, including funding obligations, prior to or upon the expiration of the product and its facilities will continue to beagreed upon terms. We are subject to FDA review and periodic inspections to ensure adherence to applicable regulations. After receiving marketing approval, the manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion and record keeping related to the product will remain subject to extensive regulatory requirements.
We may be slow to adapt, or we may never adapt, to changes in existing regulatory requirements or adoption of new regulatory requirements.
If we fail to comply with the regulatory requirements of the FDA and other applicable U.S. and foreign regulatory authorities or previously unknown problems with our products, manufacturers or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions, including:
Because we have a number of compounds and are considering a variety of target indications, we may expendrisks associated with our limited resources to pursue a particular candidate or indication and fail to capitalizedependence on candidates or indications that are more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we must focus on research programs and product candidates for the specific indications that we believe are the most promising. As a result, we may forego or delay pursuit of opportunities with other product candidates or other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. In addition, we may spend valuable time and managerial and financial resources on research programs and product candidates for specific indications that ultimately do not yield any commercially viable products. Furthermore, if we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been advantageous for us to retain sole development and commercialization rights.
We may not be successful in our efforts to identify or discover additional product candidates.
A key element of our strategy is to develop and commercialize selective NNR-targeted drugs. We seek to do so through our understanding of the role of specific NNRs in the nervous system, our scientific expertise and the use of Pentad.
Other than our four clinical stage product candidates, all of our research and development programs are at a preclinical stage. A significant portion of the research that we are conducting involves new and unproven compounds. Research programs to identify new product candidates require substantial technical, financial and human resources. These research programs may initially show promise in identifying potential product candidates, yet fail to yield product candidates for clinical development for a number of reasons, including:ISU Abxis:
Additional product candidates resulting from these research programs will require the commitment of substantial time and financial resources for further preclinical research and clinical development.
If we are unable to develop suitable product candidates through internal research programs, we will not be able to increase our revenues in future periods, which could result in significant harm to our financial position and adversely impact our stock price.
Risks Related to Our Dependence on Third Parties
We depend on collaborations with third parties for the development and commercialization of some of our product candidates. If these collaborations are not successful, we may not be able to capitalize on the market potential of these product candidates.
We have granted worldwide exclusive commercialization rights to Aventis Pharma SA with respect to TC-4959 for the treatment or prevention of Alzheimer’s disease. TC-4959 is the sole compound remaining under consideration for continued development and potential commercialization under our collaboration with Aventis relating to our compounds. We also have granted exclusive commercialization rights to Dr. Falk Pharma GmbH with respect to TC-2403 for the treatment or prevention of ulcerative colitis and other gastrointestinal and liver diseases in specified European countries, Russia, the Commonwealth of Independent States countries, Egypt and Israel. We have limited control overany decisions by ISU Abxis regarding the amount and timing of resources that our collaborators dedicate toresource expenditures for the development or commercialization of our licensed product candidates. OurCB 2679d/ISU304, and may have limited or no ability to generate royalties from our collaborators depends on our collaborators’ abilitiescontrol such decisions with respect to establish the safety and efficacy of ourother product candidates subject to obtain regulatory approvals and to achieve market acceptance. If either Aventis or Dr. Falk Pharma does not perform as contemplated under our agreements with them, our potential for revenue from the related product candidates will be adversely impacted. In addition, Aventis and Dr. Falk Pharma may terminate our collaborative agreements under certain conditions on short notice and at their sole discretion. Furthermore, our collaboration agreements with Aventis currently prohibit us from developing or commercializing product candidates for the treatment or prevention of Alzheimer’s disease. As a result, we cannot currently seek to develop our product candidate TC-1734 for Alzheimer’s disease.
In addition to our current collaboration agreements, we also intend to selectively enter into collaborative and other strategic alliances with leading pharmaceutical and biotechnology companies where our collaborator has particular therapeutic expertise in a target indication or where the target indication represents a large, primary care market.
In general, strategic collaborations involving our product candidates pose the following risks to us:
We expect to seek to establish additional collaborations, and, if we are not able to establish them on commercially reasonable terms, we may have to alter our development and commercialization plans.
Our drug development programs and the potential commercialization of our product candidates will require substantial additional cash to fund expenses. Accordingly, we may seek one or more additional collaborators for the development and commercialization of one or more of our product candidates. For example, we may seek a new collaborator to develop marzeptacog alfa (activated) and might also seek collaborators for CB 2689d/ISU304 or our earlier stage programs. In addition, full development efforts on the use of our novel proteases for the treatment of DGF or dry AMD will likely involve significant cost, and we do not expect to conduct any such efforts except in collaboration with one or more partners who are willing to pay for such costs.
We face significant competition in seeking appropriate collaborators. Whether we can reach a definitive agreement with a collaborator will depend, among other things, upon our assessment of the applicablecollaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of preclinical trials, the likelihood of approval by the FDA or comparable foreign regulatory authorities and the regulatory pathway for any such approval, the potential market for the product candidates.
candidate, the costs and complexities of manufacturing and delivering the product to patients and the potential of competing products. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available for collaboration and whether such a collaboration could be more attractive than the one with us. There can also be no assurance that any collaboration agreements will be on favorable terms.
Collaborations are complex and time consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators. If a present or future collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development program could be delayed, diminished or terminated. For example, Aventis recently announced an agreement to merge with Sanofi-Synthelabo. We do not know what effect, if any, this will have on our collaborations with Aventis.
If we do not establish additional collaborations, we may have to alter our development plans.
Our drug development programs and potential commercialization of our product candidates will require substantial additional cash to fund expenses. Our strategy includes selectively collaborating with leading pharmaceutical and biotechnology companies to assist us in furthering development and potential commercialization of some of our product candidates. We intend to do so especially for target indications in which our potential collaborator has particular therapeutic expertise or that involve a large, primary care market that must be served by large sales and marketing organizations. We face significant competition in seeking appropriate collaborators and these collaborations are complex and time-consuming to negotiate and document. We may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. If that werewe are unable to occur,do so, we may have to curtail the development of a particularthe product candidate for which we are seeking to collaborate, reduce or delay itsour development program or one or more of our other development programs, delay itsour potential commercialization or reduce the scope of ourany sales or marketing activities, orand increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we willmay not be able to bringfurther develop our product candidates or bring them to market and generate product revenue.
We contract If ourmanufacturers fail to devote sufficient resources to our concerns, or if their performance is substandard, our clinical trials and product introductions may be delayed or there may be a shortagewith third parties for the manufacture of commercial supply.
Our product candidates require precise, high quality manufacturing. We have limited internal manufacturing capability. We have historically manufactured our product candidates only in small quantities for preclinical testing and have contracted withexpect to continue to do so for clinical testing and commercialization. This reliance on third parties increases the risk that we will not have sufficient quantities of our product candidates or products or such quantities at an acceptable cost, which could delay, prevent or impair our development or commercialization efforts.
We currently have no internal capabilities to manufacture in collaboration with us, our product candidates for clinical use or for preclinical trials and, in the case of Inversine, for commercial sale. If any of our product candidates is approved by the FDAfollowing good manufacturing practices, or by foreign regulatory authorities for marketing and sale, it will need to be manufactured in substantially larger, commercial quantities. Our experience in the manufacture of drugs in commercial quantities is limited to our contractual arrangements with third parties to manufacture Inversine and its active ingredient.
GMP, or good laboratory practices, or GLP. We intend to continueexpect to rely on one or more third-party manufacturerscontractors to supply, storemanufacture, package, label and distribute our product candidates for our clinical trialssupplies and to manufacture commercial suppliesquantities of any product candidate that is approvedwe commercialize following approval for sale. Our reliancemarketing by applicable regulatory authorities. We also expect to rely on one or more third-party manufacturers will expose uscontractors to risks that could delay or prevent the initiation or completion ofmanufacture our product candidates for use in our clinical trials, the submission of applications for regulatory approvals, the
approval of our products by the FDA or the commercialization of our products or result in higher costs or lost product revenues. In particular, contract manufacturers:trials. Reliance on such third-party contractors entails risks, including:
We may incur delays in product development resulting from the need to identify or qualify manufacturers for our product candidates. Our current and anticipated future dependence upon others for the manufacture of our product candidates may adversely affect our future profit margins and our ability to commercialize any products that receive marketing approval on a timely and competitive basis.
We and our contract manufacturers will be subject to significant regulation with respect to manufacturing our products. The manufacturing facilities on which we will rely may not continue to meet regulatory requirements and have limited capacity.
All entities involved in the preparation of therapeutics for clinical studies or commercial sale, including any contract manufacturers for our product candidates, are subject to extensive regulation. Components of a finished therapeutic product approved for commercial sale or used in late-stage clinical studies must be manufactured in accordance with GMP. These regulations govern manufacturing processes and procedures (including record keeping) and the implementation and operation of quality systems to control and assure the quality of investigational products and products approved for sale. Poor control of production processes can lead to the introduction of adventitious agents or other contaminants, or to inadvertent changes in the properties or stability of our product candidates that may not be detectable in final product testing. We or our contract manufacturers must supply all necessary documentation in support of a BLA on a timely basis and must adhere to the FDA’s
good manufacturinglaboratory practices, or cGMPs, requiredGLP, and GMP regulations enforced by the FDA through its facilities inspection program. Our facilities and quality systems and the facilities and quality systems of some or all our third-party contractors must pass apre-approval inspection for FDAcompliance with the applicable regulations as a condition of regulatory approval of our product candidates. In addition, the regulatory authorities may, at any time, audit or inspect a manufacturing facility involved with the preparation of our product candidates or the associated quality systems for compliance with the regulations applicable to the activities being conducted. If these facilities do not pass apre-approval plant inspection or do not have a GMP compliance status acceptable for the FDA, FDA approval of the products will not be granted.
The regulatory authorities also may, at any time following approval of a product for sale, audit our manufacturing facilities or those of our third- party contractors. If any such inspection or audit identifies a failure to comply with applicable regulations or if a violation of our product specifications or applicable regulations occurs independent of such an inspection or audit, we or the relevant regulatory authority may require remedial measures that may be costly and/or time-consuming for us or a third-party to implement and that may include the temporary or permanent suspension of a clinical study or commercial sales or the temporary or permanent closure of a facility. Any such remedial measures imposed upon us or third parties with whom we contract could materially harm our business.
If we or any of our third-party manufacturers fail to document their adherencemaintain regulatory compliance, the FDA can impose regulatory sanctions including, among other things, refusal to cGMPs, eitherapprove a pending application for a new drug product or biologic product, or revocation of apre-existing approval. As a result, our business, financial condition and results of operations may be materially harmed.
Additionally, if supply from one approved manufacturer is interrupted, there could be a significant disruption in commercial supply. An alternative manufacturer would need to be qualified through a BLA supplement which could leadresult in further delay. The regulatory agencies may also require additional studies if a new manufacturer is relied upon for commercial production. Switching manufacturers may involve substantial costs and is likely to significant delaysresult in a delay in our desired clinical and commercial timelines.
These factors could cause the availabilitydelay of material for clinical study and delaystudies, regulatory submissions, required approvals or prevent filing or approvalcommercialization of marketing applications for our product candidates;candidates, cause us to incur higher costs and
meet contractual requirements, and we are unable to secure one or more replacement suppliers capable of production at a substantially equivalent cost, our clinical studies may be delayed or we could lose potential revenue.
We expect to rely initially on a single contract manufacturer for each ofthird parties to conduct our product candidates. Currently, we have separate arrangements with third-party manufacturers, each of which is a sole supplierclinical trials, and those third parties may not perform satisfactorily, including failing to us,meet deadlines for the active ingredientcompletion of Inversinesuch trials.
We expect to rely on third parties such as contract research organizations, or CROs, medical institutions and the finished tablets of Inversine. Changingclinical investigators to enroll qualified patients and conduct, supervise and monitor clinical trials. Our reliance on these or any manufacturer that we subsequently engage for a particular product or product candidate may be difficult, as the number of potential manufacturers is limited and we will have to compete with third parties for access to those manufacturing facilities. cGMP manufacturing processes and procedures typically must be reviewed and approved by the FDA and changing manufacturers may require re-validation of any new facility for cGMP compliance, which would likely be costly and time-consuming. We mayclinical development activities will reduce our control over these activities. Our reliance on these third parties, however, will not be able to engage replacement manufacturers on acceptable terms quickly or at all. In addition, our contract manufacturers located in foreign countries may be subject to import limitations or bans. As a result, if anyrelieve us of our contract manufacturers is unable, for whatever reason, to supply the contracted amounts of Inversine or any other productregulatory responsibilities, including ensuring that we successfully bring to market, a shortage would result which would have a negative impact on our revenues.
Drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA, the United States Drug Enforcement Agency and corresponding state and foreign agencies to ensure strict compliance with cGMPs, other government regulations and corresponding foreign standards. While we are obligated to audit the performance of third-party contractors, we do not have control over our third-party manufacturers’ compliance with these regulations and standards. Failure by our third-party manufacturers or us to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure of the government to grant pre-market approval of drugs, delays, suspension or withdrawal of approvals, seizures or recalls of product, operating restrictions and criminal prosecutions.
If third parties on which we rely for clinical trials do not perform as contractually required or as we expect, we may not be able to obtain regulatory approval for or commercialize our product candidates.
We do not have the ability to independently conduct the clinical trials required to obtain regulatory approval for our product candidates. We depend on independent clinical investigators and, in some cases, contract research organizations and other third-party service providers to conduct the clinical trials of our product candidates and expect to continue to do so. We rely heavily on these parties for successful execution of our clinical trials, but we do not control many aspects of their activities. Nonetheless, westudies are responsible for confirming that each of our clinical trials is conducted in accordance with its generalgood clinical practices, or GCP, and the investigational plan and protocol. Moreover,protocols contained in the FDA requires us to complyrelevant regulatory application, such as an investigational new drug application, or IND. In addition, the CROs with regulations and standards, commonly referred to as Good Clinical Practices, for conducting and recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the trial participants are adequately protected. Our reliance on third parties thatwhom we do not control does not relieve us of these responsibilities and requirements. Third partiescontract may not complete activities on schedule, or may not conduct our preclinical studies or clinical trialsstudies in accordance with regulatory requirements or the respective trial plans and protocols. The failure ofour clinical study design. If these third parties todo not successfully carry out their obligations could delaycontractual duties or prevent the development, approvalmeet expected deadlines, our efforts to obtain regulatory approvals for, and commercialization ofto commercialize, our product candidates may be delayed or prevented.
Risks related to employee matters, managing growth and our business operations
Our future success depends on our ability to retain key executives and to attract, retain and motivate qualified personnel.
Our ability to compete in the highly competitive biotechnology and pharmaceutical industries depends upon our ability to attract and retain highly qualified managerial, scientific and medical personnel. We are highly dependent on our management and scientific personnel, including our President and Chief Executive Officer, Dr. Usman, our Chief Medical Officer, Dr. Levy, our Chief Financial Officer, Fletcher Payne, and our Senior Vice President of Technical Operations, Andrew Hetherington. We do not maintain “key man” insurance policies on the lives of these individuals or the lives of any of our other employees. In addition, we will need to add personnel to achieve our business objectives. The loss of the services of any of our executive officers, other key employees, and our inability to find suitable replacements, or our inability to hire new clinical development and manufacturing personnel, could result in enforcement actiondelays in product development and harm our business.
We conduct operations at our facility in the San Francisco Bay Area. This region is headquarters to many other biopharmaceutical companies and many academic and research institutions. Competition for skilled personnel in our market is intense and may limit our ability to hire and retain highly qualified personnel on acceptable terms or at all.
To induce valuable employees to remain at Catalyst, in addition to salary and cash incentives, we have provided stock options that vest over time. The value to employees of stock options that vest over time may be significantly affected by movements in the company’s stock price that are beyond our control, and may at any time be insufficient to counteract more lucrative offers from other companies. Despite our efforts to retain valuable employees, members of management and scientific and development teams may terminate their employment with the company on short notice. Our employees are underat-will employment arrangements, which means that any of our employees can leave employment with Catalyst at any time, with or without notice. Failure to retain, replace or recruit personnel could harm our business.
Our employees, principal investigators, consultants and commercial partners may engage in misconduct or other improper activities, includingnon-compliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of fraud or other misconduct by our employees, principal investigators, consultants and collaborators. Misconduct by these parties could include intentional failures to comply with the regulations of the FDA andnon-U.S. regulators, to provide accurate information to the FDA andnon-U.S. regulators, to comply with healthcare fraud and abuse laws and regulations in the United States and abroad, to report financial information or data accurately or to disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Such misconduct could also involve the improper use of information obtained in the course of clinical studies that could result in regulatory sanctions and cause serious harm to our reputation. It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us.us and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
We will continue to incur significant increased costs as a result of operating as a public company, and our new management is required to devote substantial time to compliance initiatives, particularly after the completion of aone-year transition period to full compliance.
Upon the completion of the merger between Targacept and Catalyst Bio, the employment of the teams that historically operated the business of Targacept and its financial reporting was terminated, and substantially all of our current employees, including our finance staff, were the employees of Catalyst Bio from before the merger or are new hires. Accordingly, prior to the merger, we had never operated our current business as a public company. As a public company, we have and will continue to incur significant legal, accounting and other expenses that we did not incur as a private company, including costs associated with public company reporting and corporate governance requirements, in order to comply with the rules and regulations imposed by the Sarbanes-Oxley Act and the Dodd-Frank Wall Street Reform and Consumer Protection, or the Dodd-Frank Act, as well as rules implemented by the SEC and Nasdaq. Stockholder activism, the political environment and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact the manner in which we operate our business in ways that are not currently anticipated. Our management and other personnel need to devote a substantial amount of time to these compliance initiatives. In addition, these rules and regulations make it difficult and expensive for us to obtain director and officer liability insurance, and we may be required to incur substantial costs to maintain our current levels of such coverage. We expect that we will annually incur significant additional expenses to comply with the requirements imposed on us as a public company.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal controls over financial reporting and disclosure controls and procedures. In particular, as a public company, we are required to perform system and process evaluations and testing of our internal control over financial reporting to allow management and our independent registered public accounting firm to report on the effectiveness of our internal controls over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act. However, our independent registered public accounting firm was not required to report on the effectiveness of our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act for the year ended December 31, 2016, based on the SEC’s guidance for reporting over smaller reporting companies. In addition, our testing, or the subsequent testing in the future by our independent registered public accounting firm, may reveal deficiencies in our internal control over financial reporting that may be deemed to be material weaknesses. Our compliance with Section 404 will require that we incur substantial accounting expense and management time on compliance-related issues. Moreover, if we are not able to comply with the requirements of Section 404 in a timely manner, or if we or our independent registered public accounting firm identify deficiencies in our internal control over financial reporting that are deemed to be material weaknesses, we could lose investor confidence in the accuracy and completeness of our financial reports, which could cause our stock price to decline.
We or the third parties upon whom we depend may be adversely affected by earthquakes or other natural disasters and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
Our offices are located in the San Francisco Bay Area, which is prone to earthquakes. Earthquakes or other natural disasters could severely disrupt our operations, and have a material adverse effect on our business, results of operations, financial condition and prospects. If a natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as the manufacturing facilities of our third-party contract manufacturers, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. The disaster recovery and business continuity plans we have in place currently are limited and are unlikely to prove adequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans that, particularly when taken together with our lack of earthquake insurance, could have a material adverse effect on our business.
Risks Relatedrelated to Our Intellectual Propertyour intellectual property
If we are unable to obtain, protect ouror enforce intellectual property effectively, our competitors may develop and market similar products and the value of our technology and our ability to compete would be damaged.
Our continued success depends significantly on our ability to obtain and maintain meaningful intellectual property protection for our product candidates, technology and know-how. We generally seek to protect our compounds and technologies by, among other methods, filing United States and foreign patent applicationsrights related to our proprietary technology that is important to the development of our business. We file patent applications directed to our product candidates, we may not be able to compete effectively in an effortour markets.
We rely upon a combination of patents, trade secret protection and confidentiality agreements to establishprotect the intellectual property positions regarding new chemical entities and usesrelated to our product candidates. The strength of patents in the treatment of disease.
The patent positions of companies like ours are generally uncertainbiotechnology and involvepharmaceutical field involves complex legal and factual questions. Our ability to maintainscientific questions and solidify our proprietary position for our technology will depend on our success in obtaining effective patent claims and enforcing claims that are granted. We do not know whether any of our patent applications or those patent applications that we license will result in the issuance of any patents. Moreover, our issued patents and those that may issue in the future, or those licensed to us, maycan be challenged, invalidated, rendered unenforceable or circumvented, any of which could limit our ability to stop competitors from marketing related products for the term of our patent protection. In addition, the rights granted under any issued patents may not provide us with competitive advantages against competitors with similar compounds or technologies. Furthermore, our competitors may independently develop similar technologies in a manner that does not infringe our patents or other intellectual property.
Although we own or otherwise have rights to a number of patents, these patents may not effectively exclude competitors from engaging in activities that compete with us. Furthermore, the issuance of a patent is not conclusive as to its validity or enforceability and thirduncertain. Third parties may challenge the validity, enforceability or enforceabilityscope of our patents. Becausepatents that, may result in those patents being narrowed or invalidated. The patent applications that we own may fail to result in issued patents with claims that cover our product candidates in the United States and manyor in other foreign countriescountries. Furthermore, even if they are confidential for a period of time after filing,unchallenged, our patents and because publications of discoveries in the scientific literature often lag behind actual discoveries, we cannot be certain that we were the first to make the inventions claimed in our issued U.S. patents or pending patent applications or that we were the first to file for protection of the inventions set forth in the foreign patents or patent applications. It is possible that a competitor may successfully challenge our patents or that challenges will result in the elimination or narrowing of patent claims and, therefore, reduce our patent protection.
Because of the extensive time required for development, testing and regulatory review of a new drug, it is possible that any related patent may expire before any of our product candidates can be commercialized or remain in force for only a short period following commercialization. In either case, this would reduce any advantages of the patent. The patent laws of various foreign countries in which we intend to compete may not adequately protect our intellectual property, provide exclusivity for our product candidates or prevent others from designing around our claims. Certain of our patents also cover processes, for which enforcement can be difficult. Any of these outcomes could impair our ability to prevent competition from third parties that, may have an adverse impact on our business.
If the patents or patent applications we hold or havein-licensed for our programs or product candidates are invalidated or fail to issue, if their breadth or strength of protection is threatened, or if they fail to provide meaningful exclusivity for our product candidates, it could threaten our ability to commercialize future products. Further, if we encounter delays in regulatory approvals, the period of time during which we could market a product candidate under patent protection could be reduced. In addition, patents have a limited lifespan. In the United States, the natural expiration of a patent is generally 20 years after it is filed. Various extensions may be available; however the life of a patent, and the protection it affords, is limited. Once the patent life has expired for a product, we may be open to competition from generic medications.
In addition to the protection afforded by patents, we rely on trade secret protection and confidentiality agreements to protect proprietaryknow-how that is not patentable or that we elect not to patent and other elements of our product candidate discovery and development processes that involve proprietaryknow-how, information or technology that is not covered by patents. However, trade secrets can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with our employees, consultants, scientific advisors and contractors. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors.
Although we expect all of our employees and consultants to assign their inventions to us, and all of our employees, consultants, advisors and any third parties who have access to our proprietaryknow-how, information or technology to enter into confidentiality agreements, we cannot provide any assurances that all such agreements have been duly executed or that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. Misappropriation or unauthorized disclosure of our trade secrets could impair our competitive position and may have a material adverse effect on our business. Additionally, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating the trade secret. In addition, others may independently discover our trade secrets and proprietary information.
Further, filing, prosecuting and defending patents on product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. ChangesAs a result, we may encounter significant problems in either patent laws or in interpretations of patent lawsprotecting and defending our intellectual property both in the United
States and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection.
abroad. If we are unable to protect the confidentiality of our proprietary information and know-how, the commercial value of our technology and product candidates could be reduced.
In addition to patents, we rely on protection of trade secrets, know-how and confidential and proprietary information to maintain our competitive position. To maintain the confidentiality of trade secrets and proprietary information, we generally enter into confidentiality agreements with our employees, consultants, contractors and collaborative partners upon the commencement of our relationship with them. These agreements typically require that all confidential information developed by the individual or made known to the individual by us during the courseprevent material disclosure of the individual’s relationship with us be kept confidential and not disclosednon-patented intellectual property related to our technologies to third parties. However,parties, and there is no guarantee that we may not obtain these agreements in all circumstances, and individuals with whom wewill have these agreements may not comply with their terms. Even if obtained, these agreements may not provide meaningfulany such enforceable trade secret protection, for
our trade secrets or other proprietary information or an adequate remedy in the event of their unauthorized use or disclosure. The loss or exposure of our trade secrets or other proprietary information could impair our competitive position.
We also typically enter into agreements with employees that provide inventions conceived by them in the course of rendering services to us are our exclusive property and, where appropriate, we enter into similar agreements with consultants and contractors. To the extent that our employees, consultants or contractors use technology or know-how owned by others in their work for us, disputes may arise as to the rights in related inventions.
If we fail to comply with our obligations in our intellectual property licenses with third parties, we could lose license rights that are important to our business.
We are a party to various license agreements. In particular, we license patent rights for the method of use of TC-5231, our product candidate in development for attention deficit hyperactivity disorder, and TC-2696, our product candidate in development for pain. We may enter into additional licenses in the future. Our existing licenses impose, and we expect future licenses will impose, various diligence, milestone payment, royalty, insurance and other obligations on us. If we fail to comply with these obligations, the licensor may have the right to terminate the license, in which event we might not be able to market any product that is covered by the licensed patents.
Our patent protection for any particular compound may be limited to a particular method of use or indication such that, if a third party were to obtain approval of the compound for use in another indication, we could be subject to competition arising from off-label use.
Although we generally seek the broadest patent protection available for our proprietary compounds, we may not be able to obtainestablish or maintain a competitive advantage in our market, which could materially adversely affect our business, results of operations and financial condition.
Third-party claims of intellectual property infringement or challenging the inventorship or ownership of our patents may prevent or delay our development and commercialization efforts.
Our commercial success depends in part on avoiding infringement of the patents and proprietary rights of third parties. There is a substantial amount of litigation, both within and outside the United States, involving patent protection forand other intellectual property rights in the actual composition of any particular compoundbiotechnology and pharmaceutical industries, including patent infringement lawsuits, interferences, oppositions andinter partesreexamination proceedings before the U.S. Patent and Trademark Office, or U.S. PTO, and corresponding foreign patent offices. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are pursuing development candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our product candidates may be limitedsubject to protectingclaims of infringement of the patent rights of third parties.
Third parties may assert that the manufacture, use or sale of our product candidates infringes patents held by such third parties, or that we are employing their proprietary technology without authorization. For example, we are aware of a new methodpatent that has been issued in Europe (with counterparts in Australia, China, Japan, Poland, and South Korea) and includes a claim that may read on marzeptacog alfa (activated). An opposition proceeding with respect to this patent sustained the patent, and we filed an appeal on November 11, 2016. There can also be no assurance whether or not the claims of such patent would be found to read on marzeptacog alfa (activated) even if a claim survives the opposition. There may be third-party patents or patent applications with claims to compositions of matter, materials, formulations, methods of manufacture or methods for treatment related to the use for the compoundor manufacture of our product candidates. Because patent applications can take many years to issue, there may be currently pending patent applications that may later result in issued patents that our product candidates may infringe.
In addition, we have received confidential and proprietary information from third parties, and we employ individuals who were previously employed at other biotechnology or pharmaceutical companies. We may be subject to claims that our employees, consultants or independent contractors have inadvertently or otherwise restricted inused or disclosed confidential information of these third parties or our employees’ former employers. Litigation may be necessary to defend against these claims.
Parties making claims against us may obtain injunctive or other equitable relief that could effectively block our ability to prevent others from exploiting the compound. For example, we have composition of matter patent coverage in the United States on only twofurther develop and commercialize one or more of our four clinical stage compounds, TC-1734 and TC-2696. We relyproduct candidates unless we redesigned infringing products (which may be impossible) or obtained a license under the applicable patents (which may not be available on method of use patent coverage in the United States on our two other clinical stage compounds, TC-5231, for neuropsychiatric disorders, and TC-2403, for inflammatory bowel disease, including ulcerative colitis. Accordingly, we would likely be unable to prevent others from manufacturing TC-5231commercially reasonable terms or TC-2403at all), or from marketing either of them for any use that is not protected by our patent rights. If a third party were to receive marketing approval for either compound for another use, physicians could nevertheless prescribe it for indications that are not described in the product’s labeling or approved by the FDA or other regulatory authorities. Even if we have patent protection for the prescribed indication, as a practical matter, we would have little recourse as a result of this off-label use. In that event, our revenues from the commercialization of the compound would likely be adversely affected.until such patents expire.
We may be involved in lawsuits to protect or enforce our patentspatents.
Competitors may infringe our patents. To counter infringement or unauthorized use, we or our collaborators may be required to file infringement claims that couldcan be expensive and time-consuming.
We In addition, in an infringement proceeding, a court may initiate patent litigation against third partiesdecide that one of our patents is not valid, is unenforceable and/or is not infringed, or may refuse to protect or enforcestop the other party from using the technology at issue on the grounds that our patent rights and we may be similarly sued by third parties. We may also become subject to interference or opposition proceedings conductedpatents do not cover the technology in the patent and trademark offices of various countries to determine our entitlement to patents. The defense and prosecution of intellectual property suits, interference proceedings and related legal and administrative proceedings, if necessary, would be costly and divert our technical and management personnel from conducting our business. Moreover, we may not prevail in any of these suits.question. An adverse determination ofresult in any litigation or proceedingdefense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly interpreted and could put our patent applications at risk of not being issued and issuing.
Interference proceedings provoked by third parties or brought by us may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our licensors. An unfavorable outcome
could require us to cease using the related technology or to attempt to license rights from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. We may not be able to prevent, us from protectingalone or with our licensors, misappropriation of our intellectual property rights, particularly in countries where the laws may not protect suchthose rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that disclosure of some of our confidential information could be compelledcompromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our common stock.
Intellectual property litigation could cause us to spend substantial resources and distract our personnel from their normal responsibilities.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims, regardless of their merit, would cause us to incur significant expenses, and could distract our technical and management personnel from their normal responsibilities. In the information compromised.event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, in addition to paying royalties, redesign infringing products or obtain one or more licenses from third parties that, may be impossible or require substantial time and monetary expenditure. In addition, during the course of this kind of litigation, there could be public announcements of the results of hearings, motions or other interim proceedings or developments that,and if perceived as negative by securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the trading price of our common stock. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. We may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could compromise our ability to compete in the marketplace.
Claims byWe may need to license certain intellectual property from third parties, that we infringe their proprietary rightsand such licenses may result in liability for damagesnot be available or prevent or delay our development and commercialization efforts.
Our success depends in part on avoiding the infringement of other parties’ patents and proprietary rights. Patents may issue from patent applications of which we are unaware, and avoiding patent infringement may be difficult. We may infringe or it may be alleged that we infringe third-party patents. If a third party were to file a patent infringement suit against us, we could be forced to stop or delay research, development, manufacturing or sales of any infringing product in the country or countries covered by the patent infringed, unless we can obtain a license from the patent holder. Any necessary license may not be available on acceptable termscommercially reasonable terms.
A third-party may hold intellectual property, including patent rights, that is important or at all, particularly ifnecessary to the development of our products. It may be necessary for us to use the patented or proprietary technology of third party is developing or marketing a product competitive with the infringing product. Even ifparties to commercialize our products, in which case we are ablewould be required to obtain a license the rights may be nonexclusive, which would give our competitors access to the same intellectual property.
We also may be required to pay substantial damages to the patent holder in the event of an infringement. These damages could in some circumstances be triple the actual damages the patent holder incurs. If we have supplied infringing products to third parties for marketing or have licensed third parties to manufacture, use or market infringing products, we may be obligated to indemnifyfrom these third parties for any damages they mayon commercially reasonable terms, or our business could be requiredharmed, possibly materially.
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, and changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to payprotect our products.
On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law, including provisions that affect the way patent holderapplications will be prosecuted and for any losses they may sustain themselves as a result.
Any successful infringement action brought against us may also adversely affect marketingpatent litigation. The U.S. PTO is currently developing regulations and procedures to govern administration of the infringing product in other markets not covered byLeahy-Smith Act, and many of the infringement action, as well as our marketing of other products based on similar technology. Furthermore, we may suffer adverse consequences from a successful infringement action against us even if the action is subsequently reversed on appeal, nullified through another action or resolved by settlementsubstantive changes to patent law associated with the patent holder. The damages or other remedies awarded,Leahy-Smith Act, and in particular, the first to file provisions, were enacted March 16, 2013. However, it is not clear what, if any, may be significant. As a result, any infringement action against us would likely delayimpact the Leahy-Smith Act will have on the operation of our business.
Risks related to regulatory approval process, harm our competitive position, be very costly and require significant time and attention of our key management and technical personnel.
Risks Related to Commercialization
Even if approved for sale, our product candidates and other legal compliance matters
If we are not able to obtain, or if there are delays in obtaining, required regulatory approvals, we will not be able to commercialize our product candidates, and our ability to generate revenue will be materially impaired.
While we have multiple drug candidates in clinical and advanced preclinical development for a range of diseases, we have not yet submitted biologics license applications, or BLAs, for our engineered human proteases to the FDA, or similar approval filings to comparable foreign authorities. Submission of a BLA requires extensive preclinical and clinical data and supporting information that demonstrates the product candidate’s safety, purity, and potency, also known as safety and effectiveness, for each desired indication. A BLA must also include significant information regarding the chemistry, manufacturing and controls for the product. One of our product candidates, marzeptacog alfa (activated), has completed a Phase 1 clinical trial. However, failure of one or more clinical trials can occur at any stage in the clinical trial process. Accordingly, the regulatory pathway for our product candidates is still uncertain, complex, and lengthy, and ultimately approval may not gain market acceptancebe obtained.
We may experience delays in completing planned clinical trials for a variety of reasons, including delays related to:
The commercial success
We could also experience delays in obtaining approval if physicians encounter unresolved ethical issues associated with enrolling patients in clinical trials of our productsproduct candidates in lieu of prescribing existing treatments that have established safety and efficacy profiles given the serious nature of the diseases for the core indications for our product candidates. Additionally, a clinical trial may be suspended or terminated by us, the IRBs for the institutions in which we may obtain marketing approval fromthe trials are being conducted, the Data Monitoring Committee for the trial, or by the FDA or other regulatory authorities for a number of reasons, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues, or adverse side effects, failure to demonstrate a benefit from using a product candidate, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. If we experience termination of, or delays in the completion of, any clinical trial of our product candidates, our ability to commercialize our product candidates will depend uponbe harmed and our ability to generate revenue will be materially impaired. Additionally, delays in completing trials will increase costs, slow down our product development and approval process, and impair our ability to commence product sales and generate revenue. Many of the factors that could create or lead to a delay in the commencement or completion of clinical trials may ultimately lead to the denial of regulatory approval for our product candidates.
The FDA may disagree with our regulatory plan and we may fail to obtain regulatory approval of our product candidates.
The results of clinical trials we conduct may not support regulatory approval of our product candidates. Our product candidates could ultimately fail to receive regulatory approval for many reasons, including the following:
Our relationships with customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, physicians and third-party payors will play a primary role in the recommendation and prescription of any product candidates for which we obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we would market, sell and distribute our products. As a pharmaceutical company, even though we do not and may not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. These regulations include:
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any physicians or other healthcare providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Recently enacted and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates and affect the prices for our product candidates.
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of
our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any product candidates for which we obtain marketing approval.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or the MMA, changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for physician-administered drugs. In addition, this legislation provided authority for limiting the number of drugs that will be covered in any therapeutic class. Cost reduction initiatives and other provisions of this legislation could decrease the coverage and price that we receive for any approved products. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. Therefore, any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private payors.
More recently, in March 2010, then-President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively the PPACA, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms.
Among the provisions of the PPACA of importance to our potential product candidates are the following:
In addition, other legislative changes have been proposed and adopted since the PPACA was enacted. These changes included aggregate reductions in Medicare payments to providers of up to 2% per fiscal year, starting in 2013. In January 2013, then-President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several providers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding.
We expect that the PPACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we or our
collaborators may receive for any approved product. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our products.
Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We cannot be sure whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our product candidates, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could harm our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. From time to time and in the future, our operations may involve the use of hazardous and flammable materials, including chemicals and biological materials, and may also produce hazardous waste products. Even if we contract with third parties for the disposal of these materials and waste products, we cannot completely eliminate the risk of contamination or injury resulting from these materials. In the event of contamination or injury resulting from the use or disposal of our hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
We maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, but this insurance may not provide adequate coverage against potential liabilities. However, we do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us.
In addition, we may incur substantial costs to comply with current or future environmental, health and safety laws and regulations. Current or future environmental laws and regulations may impair our research, development or production efforts that, could adversely affect our business, financial condition, results of operations or prospects. In addition, failure to comply with these laws and regulations may result in substantial fines, penalties or other sanctions.
We face potential product liability, and, if successful claims are brought against us, we may incur substantial liability and costs. If the use of our product candidates harms patients, or is perceived to harm patients even when such harm is unrelated to our product candidates, regulatory approvals could be revoked or otherwise negatively impacted and we could be subject to costly and damaging product liability claims.
The use of our product candidates in clinical trials and the sale of any products for which we obtain marketing approval exposes us to the risk of product liability claims. Product liability claims might be brought against us by consumers, healthcare providers, pharmaceutical companies or others selling or otherwise coming into contact with our products. There is a risk that our product candidates may induce adverse events. If we cannot successfully defend against product liability claims, we could incur substantial liability and costs. In addition, regardless of merit or eventual outcome, product liability claims may result in:
We carry product liability insurance of $10,000,000 per occurrence and $10,000,000 aggregate limit. We believe our product liability insurance coverage is sufficient for our current clinical programs; however, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. If we obtain marketing approval for product candidates, we intend to expand our insurance coverage to include the sale of commercial products; however, we may be unable to obtain product liability insurance on commercially reasonable terms or in adequate amounts. On occasion, large judgments have been awarded in class action lawsuits based on drugs or medical treatments that had unanticipated adverse effects. A successful product liability claim or series of claims brought against us could cause our stock price to decline and, if judgments exceed insurance coverage, could adversely affect our results of operations and business.
Patients with the diseases targeted by our product candidates are often already in severe and advanced stages of disease and have both known and unknown significantpre-existing and potentially life-threatening health risks. During treatment, patients may suffer adverse events, including death, for reasons that may be related to our product candidates. Such events could subject us to costly litigation, require us to pay substantial amounts of money to injured patients, delay, negatively impact or end our opportunity to receive or maintain regulatory approval to market our products, or require us to suspend or abandon our commercialization efforts. Even in a circumstance in which we do not believe that an adverse event is related to our products, the investigation into the circumstance may be time-consuming or inconclusive. These investigations may interrupt our sales efforts, delay our regulatory approval process in other countries, or impact and limit the type of regulatory approvals our product candidates receive or maintain. As a result of these factors, a product liability claim, even if successfully defended, could have a material adverse effect on our business, financial condition, results of operations, or cash flows.
Risks related to commercialization of our product candidates
Even if any of our product candidates receives marketing approval, we may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success.
If any of our product candidates receives marketing approval, we may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors and others in the medical community. For example, current hemophilia treatments like NovoSeven® are well established in the medical community, and third-party payors as clinically useful, cost-effectivedoctors may continue to rely on these treatments. If our product candidates do not achieve an adequate level of acceptance, we may not generate significant product revenues and safe. Many of the products that we are developing are based upon technologies or therapeutic approaches that are relatively new and unproven. As a result, it may be more difficult for us to achieve market acceptance of our products.
not become profitable. The degree of market acceptance of any drug dependsour product candidates, if approved for commercial sale, will depend on a number ofseveral factors, such as:including:
Our product labelingcandidates are years away from regulatory approval.
Marzeptacog alfa (activated) and CB 2679d/ISU304 are not expected to be commercially available for several years, if at all. Further, the commercial success of either product candidate will depend upon its acceptance by physicians, individuals, third-party payors and other key decision-makers as a therapeutic and cost effective alternative to products available at the time, which may include competing products currently under development by others. See “We face substantial competition that may result in others discovering, developing or commercializing products before or more successfully than we do.” If we are unable to successfully develop, obtain regulatory approval for and commercialize marzeptacog alfa (activated) or CB 2679d/ISU304, our ability to generate revenue from product insert required bysales will be significantly delayed and our business will be materially and adversely affected, and we may not be able to earn sufficient revenues to continue as a going concern.
Even if the FDA or other regulatory agency approves marzeptacog alfa (activated) or CB 2679d/ISU304, the approval may impose significant restrictions on the indicated uses, conditions for use, labeling, advertising, promotion, marketing and/or production of such product and may impose ongoing commitments or requirements for post-approval studies, including additional research and development and clinical trials. The FDA and other agencies also may impose various civil or criminal sanctions for failure to comply with regulatory requirements, including withdrawal of product approval. Regulatory approval from authorities in foreign countries will be needed to market marzeptacog alfa (activated) or CB 2679d/ISU304 in those countries. Approval by one regulatory authority does not ensure approval by regulatory authorities in other countries.
jurisdictions. If we fail to obtain approvals from foreign jurisdictions, the geographic market for marzeptacog alfa (activated) or CB 2679d/ISU304 would be limited.
In addition, perceptions about the relationship or similarity between our product candidates and nicotine could limit their market potential. Our product candidates derive their therapeutic effects by interacting with NNRs. Nicotine, which can have significantly negative health effects, also interacts with NNRs. Accordingly, our product candidates may be perceived by some to be nicotine or to be closely related to nicotine, particularly in light of the shared derivative names, “nicotine” and neuronal “nicotinic” receptors, and the fact that our company was launched originally as a research group within, and then as a subsidiary of, R.J. Reynolds Tobacco Company. This potential perception could result in a reluctance by patients to take, or by physicians to prescribe, any of our product candidates that receives marketing approval and affect our revenues.
We currently have limited marketing and sales staff, and ifIf we are unable to enter into collaborations or other arrangements with third parties to marketestablish sales, marketing and sell our product candidates or to develop our own internal marketing capability,distribution capabilities, we may not be successful in commercializing our products.
product candidates if they are approved.
We currently have limitednot yet established a sales, marketing andor product distribution experience. Our experience is limitedinfrastructure for our other product candidates, which are still in preclinical or early clinical development. Except for ISU Abxis’ potential rights to commercialize CB 2679d/ISU304 in South Korea, we generally expect to retain commercial rights for the company’s hemophilia product candidates. We believe that it will be possible to access the United States hemophilia market through a focused, specialized sales force. However, we have not yet developed a commercial strategy for hemophilia products outside of the United States, or for any other of our contractual arrangements with third parties to market and sell Inversine,product candidates. To achieve commercial success for any product for which we acquired in 2002 and which generates only limited sales. We currently have no internal sales or distribution capabilities. Althoughobtain marketing approval, we intendwill need to build an internal sales force and expand our marketing capabilities in areas where specialists heavily influence our target markets, such as neurology and psychiatry, we also intend to seek to further augment our sales, marketing and distribution capabilities through arrangements with third parties. In particular, our strategy includes selectively entering into collaborations and other strategic alliances with respect to product candidates for disease indications withestablish a sales and distribution characteristics requiringmarketing organization within the United States, and develop a largestrategy for sales force. outside of the United States.
There are risks involved with establishing our owninternal sales, marketing and distribution capabilities. For example, recruiting and training a sales force is expensive and time consuming and could delay any product launch. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel. If we are unable to establish sales, marketing and distribution capabilities as well as in enteringand enter into additional arrangements with third parties to perform these services. Developingservices, then our own sales forceproduct revenues and profitability, if any, are likely to be lower than if we were to market, sell and distribute any products that we develop ourselves.
We face substantial competition that may result in others discovering, developing or commercializing products before or more successfully than we do.
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition, and a strong emphasis on proprietary products. We face potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions and governmental agencies, and public and private research institutions. Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future.
Specifically, there are a large number of companies developing or marketing treatments for hemophilia, including many major pharmaceutical and biotechnology companies, including Novo Nordisk, which has developed NovoSeven®, a human recombinant coagulation Factor VIIa indicated for treatment of bleeding episodes that has been approved for use in treatment of hemophilia A or B individuals with an inhibitor to Factor VIII or Factor IX and in individuals with Factor VII deficiency and Glanzmann’s thrombasthenia, Shire (formerly Baxter), which has developed BAX 817, a biosimilar of NovoSeven® that recently completed an intravenous Phase 3 clinical trial and has filed for marketing approval, Roche, which is developing ACE910/Emicizumab, a recombinant humanized bispecific antibody that binds to activated Factor IX and Factor X to mimic the cofactor function of Factor VIII and has been granted breakthrough therapy designation by the FDA to potentially treat hemophilia A, Alnylam, which is developing an investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia, OPKO Biologics, whose recombinant Factor VIIa product that may also be administered subcutaneously is in a Phase 1/2 clinical trial, and CSL Behring, which is developing an albumin-linked Factor VIIa that has an extended half-life. We are also aware of many companies focused on developing gene therapies that may compete with our planned hemophilia B indication, as well as several companies addressing other methods for modifying genes and regulating gene expression.
Our commercial opportunity in different indications could be reduced or eliminated if competitors develop and market products or therapies that are more convenient to use, more effective, less expensive, and time-consumingsafer to use than our products. Furthermore, if competitors gain FDA approval faster than we do, we may be unable to establish a strong market presence or to gain market share. The key competitive factors affecting the success of all our product candidates, if approved, are likely to be their efficacy, safety, convenience, price, the level of generic competition, and could delaythe availability of reimbursement from government and other third-party payors.
Many of the companies against which we are competing or against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical, biotechnology and diagnostic industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and individual registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Even if we commercialize any product launch. Wecandidates, the products may not be successful in entering into arrangements with third parties on terms that are favorablebecome subject to us or at all. Also, we would have little control over such third parties, and any of them may fail to devote the necessary resources and attention to sell, market or distribute our products effectively. If we do not establish sales and distribution capabilities successfully, either on our own or in collaboration with third parties, we may not successfully commercialize our products.
Unfavorableunfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives applicable tothat, would harm our products could limit our potential product revenue.
business.
The regulations governingthat govern marketing approvals, pricing, coverage and reimbursement for new drug pricing and reimbursementproducts vary widely from country to country. Current and future legislation may significantly change the approval requirements in ways that could involve additional costs and cause delays in obtaining approvals. Some countries require approval of the sale price of a drug before it can be marketed and, inmarketed. In many of these countries, the pricing review period begins only after marketing or product licensing approval is granted. In some countries,foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. Although we monitor these regulations, our product candidates are currently in the development stage and we will not be able to assess the impact of price regulations for at least several years.
As a result, we may obtain regulatorymarketing approval for a product in a particular country, but then be subject to price regulations that delay theour commercial launch of the product, possibly for lengthy time periods, and may negatively impact the revenues we are able to derivegenerate from salesthe sale of the product in that country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates, even if our product candidates obtain marketing approval.
Successful commercialization of our productsOur ability to commercialize any product candidates successfully also will also depend in part on the extent to which coverage and adequate paymentreimbursement for ourthese products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and third-party payors. If we succeed in bringing a product candidate to the market, it may not be considered cost-effectivepayors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement to the patient may not be available or sufficient to allow us to sell it at a satisfactory price. Because our product candidates arelevels. A primary trend in the development stage, we are unable at this time to determine their cost-effectiveness. We may need to conduct expensive studies in order to demonstrate
cost-effectiveness. Moreover,U.S. healthcare industry and elsewhere is cost containment. Government authorities and third-party payors frequently requirehave attempted to control costs by limiting coverage and the amount of reimbursement for certain medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are increasingly challenging the prices charged for medical products. BecauseCoverage and reimbursement may not be available for any product that we or our product candidatescollaborators commercialize and, even if these are in the development stage, we do not knowavailable, the level of reimbursement if any, we will receive for any products that we are able to successfully develop. If the reimbursement for any of our product candidates is inadequate in light of our development and other costs, our ability to achieve profitability couldmay not be affected.
We believe that the efforts of governments and third-party payors to contain or reduce the cost of healthcare will continue tosatisfactory. Reimbursement may affect the business and financial condition of pharmaceutical and biopharmaceutical companies. A number of legislative and regulatory proposals to change the healthcare system in the United States and other major healthcare markets have been proposed and adopted in recent years. For example, the U.S. Congress recently enacted a limited prescription drug benefit for Medicare recipients. While the program established by this statute may increase demand for, any products that we are able to successfully develop, if we participate in this program, our prices will be negotiated with drug procurement organizations for Medicare beneficiaries and are likely to be lower than prices we might otherwise obtain. If successfully developed, our product candidate for cognitive impairment in the elderly, TC-1734, could be particularly affected by this law because of its elderly target patient population. Non-Medicare third-party drug procurement organizations may also baseor the price they are willing to pay on the rate paid by drug procurement organizations for Medicare beneficiaries. In addition, ongoing initiatives in the United States have and will continue to increase pressure on drug pricing. The announcement or adoption of, any such initiative could have an adverse effect on potential revenues from any product candidate that receives marketing approval. Obtaining and maintaining adequate reimbursement for our products may be difficult. We may be required to conduct expensive pharmacoeconomic studies to justify coverage and reimbursement or the level of reimbursement relative to other therapies. If coverage and adequate reimbursement are not available or reimbursement is available only to limited levels, we may successfully develop.
If our competitors develop and market drugs that are less expensive, more effective or safer than ours, if they develop and market products faster than we do, or if they have better sales and marketing capabilities than we do, any products we are able to commercialize may not generate initial or ongoing revenues.
The development and commercialization of new drugs is highly competitive. Our business is characterized by extensive research efforts and rapid developments. We expect intense competition in our target markets as new products and advanced technologies become available. Our competitors include large pharmaceutical, biotechnology and other companies and research institutions, many of which have greater financial, technical and other resources and personnel and more experience in research, clinical development, regulatory and drug commercialization than we have. Our competitors may:
Competitive products may render our product candidates obsolete or noncompetitive before we can recover our development or commercialization expenses.
We also face substantial competition within the area of NNR-targeted therapeutics. We believe that several prominent pharmaceutical companies have product candidates that target NNRs in development, including Pfizer, with a compound in Phase III clinical trials for smoking cessation, and Abbott Laboratories, with one
compound in Phase I for pain and another in Phase II for Alzheimer’s disease, ADHD and schizophrenia. We expect that we will face increased competition in the future if NNR-targeted therapeutics are further validated and companies initiate or expand programs focused on NNRs, whether independently or by collaboration or acquisition.
Any products that we arebe able to successfully develop and commercialize in the future could be subject to competition from lower priced generic drugs. In particular, if mecamylamine hydrochloride is effective and receives regulatory approval for the treatment of ADHD at the low doses that we are currently developing as TC-5231, physicians could prescribe partial tablets of a generic version of Inversine for the off-label treatment of ADHD. We are currently evaluating mecamylamine hydrochloride at doses ranging from 0.2mg and 1.0mg. If we determine that the higher dose is more effective or otherwise more desirable for use in treating ADHD, physicians may view the higher dose as more similar to Inversine and be even more likely to prescribe partial tablets of Inversine for the off-label treatment of ADHD. In addition, the manufacturer of a generic product could challenge our patents as invalid or not infringed and subject us to expensive litigation. We do not know if we would prevail in litigation and succeed in keeping the generic product out of the market until our patent protection expires.
If we successfully develop and obtain approval for our product candidates, we will face competition based on the safety and effectiveness of our products, the timing and scope of regulatory approvals, the availability and cost of supply, marketing and sales capabilities, reimbursement coverage, price, patent position and other factors. Our competitors may develop or commercialize more effective or more affordable products, or obtain more effective patent protection, than we do. Accordingly, our competitors may commercialize products more rapidly or effectively than we do.
If approved, our product candidates will compete for a share of the existing market with numerous approved products. There are currently no approved products for AAMI or MCI. We believe that the primary competitive products for use in the other indications that we are currently targeting include:
We may have substantial exposure to product liability claims and may not have adequate insurance to pay them.
We face an inherent business risk of exposure to product liability claims if the use of our products is alleged to have resulted in harm to others. This risk exists for product candidates in clinical trials, whether or not theany product candidate is subsequently approved for commercial sale, as well as for products in commercial
distribution. Any product liability claims arising in the future against us or any third party that we have agreed to indemnify, regardless of its merit or eventual adjudication, could be costly and significantly divert management’s attention from conducting our business or adversely affect our reputation and the demand for our products.
We have secured product liability insurance coverage with limits of $8 million per occurrence and $8 million in the aggregate. Our current insurance coverage may not reimburse us or may not be sufficient to reimburse us for any expenses or losses we may incur. We intend to expand our coverage with respect to any products for which we obtain marketing approval. However, additional insurance may not be available to cover our potential liabilities fully or
There may be prohibitively expensive. In addition, some potential product liability claimssignificant delays in obtaining reimbursement for newly approved drugs, and coverage may be excluded from coverage undermore limited than the terms ofpurposes for which the policy. Our inability to obtain adequate insurance coverage at an acceptable cost could prevent or impede the commercialization of our products.
Our business activities involve hazardous materials, which could subject us to significant liability.
Our research and development activities involve, and any future manufacturing processes that we conduct may involve, the use of hazardous materials, including hazardous chemicals and radioactive materials. Accordingly, we are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of these materials. We incur significant costs to comply with these laws and regulations. Moreover, despite precautionary procedures that we implement, we cannot eliminate the risk of accidental contamination or injury from these materials. In the event of an accident or environmental discharge, we may be held liable for any resulting damages, which could exceed our financial resources.
If our promotional activities fail to comply with the regulations and guidelines of the FDA and other applicable regulatory authorities, we may be subject to warnings or enforcement actions that could harm our business.
Physicians may prescribe drugs for uses that are not described in the product’s labeling for uses that differ from those tested in clinical studies anddrug is approved by the FDA or similar regulatory authorities outside the United States. Moreover, eligibility for reimbursement does not imply that a drug will be paid for in other countries. Regulatory authorities generally doall cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim reimbursement levels for new drugs, if applicable, may also not regulatebe sufficient to cover our costs and may not be made permanent.
Reimbursement rates may vary according to the behavior of physicians in their choice of treatments. Regulatory authorities do, however, restrict communications on the subject of off-label use. Companies cannot actively promote approved drugs for off-label uses but, in some countries outsideuse of the European Union,drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may disseminate articles publishedbe sold at lower prices than in peer-reviewed journals that discuss off-label uses ofthe United States. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies. Our inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for any approved products to physicians. To the extent allowed,that we may in the future disseminate peer-reviewed articlesdevelop could have a material adverse effect on our operating results, ability to raise capital needed to commercialize products and overall financial condition.
The insurance coverage and reimbursement status of newly-approved products is uncertain. Failure to physicians. We do not currently promote Inversineobtain or maintain adequate coverage and reimbursement for off-label use in the treatmentnew or current products could limit our ability to market those products and decrease our ability to generate revenue.
The availability and extent of Tourette’s syndrome or any other neuropsychiatric disorder. However, if we undertake any promotional activities in the futurereimbursement by governmental and private payors is essential for Inversine or any other product candidate that we aremost patients to be able to commercialize and our activities fail to comply with these regulations or guidelines, we may be subject to warnings from, or enforcement action by, these authorities.
Risks Related to Employees and Managing Growth
If we lose our key personnel or are unable to attract and retain additional skilled personnel, we may be unable to successfully develop and commercializeafford expensive treatments. Sales of our product candidates or effectively compete in our industry.
Our performance dependswill depend substantially, both domestically and abroad, on the performanceextent to which the costs of our seniorproduct candidates will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management and key scientific, technical and managerial personnel, including our Chief Executive Officer and President, J. Donald deBethizy, and our Vice President, Clinical Development and Regulatory Affairs, Geoffrey C. Dunbar. Our executive officers, including these individuals, can terminate their employment agreements with us at any time. The loss of the services of any of them may significantly delayorganizations, or prevent the achievement of product research and developmentreimbursed by government health administration authorities, private health coverage insurers and other business objectives. We also rely on consultants and advisorsthird-party payors. If reimbursement is not available, or is available only to assist us in formulating our research and development strategy. All of our consultants and advisors are either self-employed or employed by other organizations, and they may have other commitments, including consulting or advisory contracts with other organizations, that may affect their ability to contribute to us.
Our ability to operate successfully and manage our potential future growth will depend on our ability to identify, recruit and retain additional qualified scientific, technical, financial and managerial personnel. There is currently a shortage of skilled executives in our industry, andlimited levels, we face intense competition for such personnel. We may not be able to continuesuccessfully commercialize our product candidates. Even if coverage is provided, the approved reimbursement amount may not be high enough to attractallow us to establish or maintain pricing sufficient to realize a sufficient return on our investment.
There is significant uncertainty related to the insurance coverage and retain personnelreimbursement of newly approved products. Moreover, increasing efforts by governmental and third-party payors, in the United States and abroad, to cap or reduce healthcare costs may cause such organizations to limit both coverage and level of reimbursement for new products approved and, as a result, they may not cover or provide adequate payment for our product candidates. We expect to experience pricing pressures in connection with the advanced qualifications necessary for the growthsale of any of our business.product candidates, due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
WeIf the market opportunities for our product candidates are smaller than expected, our revenues may encounter difficulties in managingbe adversely affected and our growth, which could increase our losses.
business may suffer.
We expectfocus our research and product development on hemostasis and inflammation treatment. Our projections of both the number of people who suffer from related conditions, as well as the subset of people with these conditions who have the potential to benefit from treatment with our employeesproduct candidates, are based on estimates. These estimates may prove to be incorrect and new studies may change the scopeestimated incidence or prevalence of our operationsthese diseases. The number of patients in the United States, Europe and elsewhere may turn out to grow following completion of this offering. Continued growth may place a significant strain on our managerial, operational and financial resources, in particular as we expand our focus beyond drug discovery and development to commercialization. To manage our anticipated growth, we must continue to implement and improve our managerial, operational and financial systems and controls and reporting processes and procedures, to expand our facilities and to continue to recruit and train additional qualified personnel. Webe lower than expected or may not be ableotherwise amenable to managetreatment with our growth effectively. Moreover, weproducts, or new patients may discover deficiencies in existing systemsbecome increasingly difficult to identify or gain access to, all of which would adversely affect our results of operations and controls thatour business.
Risks related to our common stock
Our common stock ownership is concentrated with our executive officers and directors, and their respective affiliates, which limits your ability to influence corporate matters.
Our significant stockholders, acting together, have the ability to affect matters submitted to our stockholders for approval, including the approval of significant transactions. This concentration of ownership may have the effect of delaying, deferring or preventing a strategic transaction, even if such a transaction would benefit other stockholders. As a result, the market price of our common stock could expose us to an increased risk of incurring financial or accounting irregularities or fraud.be adversely affected.
Risks Related to Our Common Stock and this Offering
The market price of our common stock may behas historically been highly volatile. You may not be able to resell your shares at or above the initial public offering price.
There has been no public market for our common stock prior to this offering, and it is possible that no active trading market for our common stock will develop or continue following this offering. You may not be able to sell your shares quickly or at the market price if trading in our common stock is not active. The initial public offering price for the shares will be determined by negotiation with representatives of the underwriters and may not be indicative of prices that will prevail in the trading market. Please see “Underwriters” for more information regarding our arrangement with the underwriters and the factors to be considered in setting the initial public offering price.
We expect that the trading price of our common stock is likely to behas historically been highly volatile in response to factors that are beyond our control. Theand the volume of common shares traded has been relatively low. Additionally, the stock market in general has recently experienced extreme price and volume fluctuations. The market prices of securities of pharmaceutical, biopharmaceutical and biotechnology companies in particular have been extremely volatile and have experienced fluctuations that have often been unrelated or disproportionate to operating performance. Factors giving rise to this volatility may include:
Fluctuations in operating performance of those companies. These broad market fluctuationsresults could result in extreme fluctuations inadversely affect the price of our common stock, which could cause a decline in the value of your shares.
If you purchase shares of our common stock in this offering, you will experience immediate and substantial dilution of your investment.
The offering price of our common stock will be substantially higher than the net tangible book value per share of our existing capital stock. As a result, purchasers of our common stock in this offering will incur immediate and substantial dilution of $ in net tangible book value per share of common stock, based on an assumed public offering price of $ per share, and will incur additional dilution if outstanding stock options and warrants with exercise prices below the public offering price are exercised. See “Dilution” for a more detailed discussion of the dilution new investors will incur in this offering.
If our operating results fluctuate significantly, our stock price may decline and result in losses to you.
Our operating results are likely to fluctuate significantly from quarter to quarter and year to year. These fluctuations could cause our stock price to decline. Some of the factors that couldmay cause our operating results to
fluctuate include:
Due to fluctuations in our operating results, a period-to-period comparisonUnited States.Period-to-period comparisons of our historical and future financial results of operations may not be a goodmeaningful, and investors should not rely on them as an indication of our future performance. In any particular quarter or quarters, our operating results could be belowFluctuating losses may fail to meet the expectations of securities analysts or investors andinvestors. Failure to meet these expectations may cause the price of our common stock price couldto decline.
If our existing stockholders sellSales of a substantialsignificant number of shares of our common stock in the public markets, or the perception that such sales could occur, could depress the market price of our stock price may decline.common stock.
SalesOur current trading volumes are modest, and sales of a substantial number of shares of our common stock in the public market, following this offeringor the perception that these sales could occur, could cause the market price to decline. Such sales also might make it more difficult for us to sell equity securities in the future at a time and at a price that we deem appropriate. After this offering,As part of thePre-Closing Dividend, we issued $37.0 million in aggregate principal amount of redeemable convertible notes. At the option of the note holders, those notes will be redeemable at any time on or before February 19, 2018 or convertible into shares of the Company at a conversion rate of $137.85 per share. As of December 31, 2016, the balance of these redeemable convertible notes was $19.4 million, convertible into approximately 141 thousand shares of our common stock. In addition, we have approximatelyalso registered all of the shares of common stock outstanding. If there are morethat we may issue under our outstanding stock options and employee stock incentive plans, and as of December 31, 2016, approximately 141 thousand shares of common stock were issuable upon the exercise of outstanding stock options at a weighted average exercise price of $128.25 per share, approximately 86 thousand additional shares of common stock were reserved for issuance under our stock option plan, and approximately 12 thousand shares of common stock were issuable upon the exercise of outstanding warrants at a weighted exercise price of $145.50 per share. Conversion or exercise of these securities into shares of our common stock offered for sale than buyers are willingwill cause dilution to purchase,the other holders of our common stock, and all such stock may be sold in the public market after conversion or exercise, subject to restrictions under the securities laws, which may lead to a decline in the market price of our common stock.
Stockholders may experience dilution of their ownership interests because of future issuances of common stock may declineunder our equity incentive plans
After the completion of this offering, we expect that our Board of Directors will approve and recommend to a market price at which buyers are willingour stockholders an increase to purchase the offered shares and sellers remain willing to sell the shares. The number of shares of our common stock availablereserved for sale inissuance under our 2015 Stock Incentive Plan, or the public market is limited by restrictionscreation of a new stock incentive plan with additional shares. Shares reserved for issuance under federal securities laws and under lock-up agreements that substantially all of our stockholders have entered into with the underwriters. Except in limited circumstances, these lock-up agreements restrict our stockholders from selling or otherwise disposing of their shares for a period of 180 days after the date of this prospectus without the prior written consent of Morgan Stanley & Co. Incorporated on behalf of the underwriters. However, Morgan Stanley may, in its sole discretion, release all or any portion of the common stock from the restrictions of the lock-up agreements.
Additionally, of the shares of our common stock thatsuch plans may be issued by the Board of Directors, or a committee of the Board of Directors, to employees, consultants and directors of the Company, including our current officers and directors. The amount of such increase has not been determined but could equal up to 20% or more of our total number of shares outstanding or issuable after this offering, including shares issuable upon the exercise of options outstanding asand warrants. The final determination of , 2004, approximately sharesthe amount of such increase will be vested and eligible for sale 180 days aftermade by the dateBoard of this prospectus. ForDirectors or a further descriptioncommittee thereof. Any issuance of such shares would dilute the eligibilityownership of shares for sale into the public market following the offering, see “Shares Eligible for Future Sale.” In the future, we may issue additional shares to our employees, directors or consultants, in connection with corporate alliances or acquisitions or to raise capital. Accordingly, sales of a substantial number of shares of our common stock in the public market could occur at any time.
Management may invest or spend the proceeds of this offering in ways in which you may not agree or in ways that may not yield a favorable return to our stockholders.
Management will retain broad discretion over the use of proceeds from this offering. Stockholders may not agree with such uses, and our use of the proceeds may not yield a significant return or any return at all for our stockholders. We intend to use the proceeds from this offering for research and development and other general corporate purposes. Because of the number and variability of factors that will determine our use of the proceeds from this offering, their ultimate use may vary substantially from their currently intended use.
Concentration of ownership among our existing executive officers, directors and principal stockholders may prevent new investors from influencing significant corporate decisions.
Following the completion of this offering, our executive officers, directors and their affiliates will beneficially own or control approximately % of the outstanding shares of our common stock. Accordingly, our current executive officers, directors and their affiliates will have substantial control over the
outcome of corporate actions requiring stockholder approval, including the election of directors, any merger, consolidation or sale of all or substantially all of our assets or any other significant corporate transactions, as well as our management and affairs. The concentration of ownership may also delay or prevent a change of control of us at a premium price if these stockholders oppose it, even if it would benefit our other stockholders.
Provisions ofAnti-takeover provisions in our charter bylawsdocuments and provisions of Delaware law may make an acquisition more difficult and could result in the entrenchment of us ormanagement.
We are incorporated in Delaware. Anti-takeover provisions of Delaware law and our charter documents may make a change in ourcontrol or efforts to remove management more difficult.
Certain Also, under Delaware law, our board of directors may adopt additional anti-takeover measures. The existence of the following provisions of Delaware
law and our restated certificate of incorporation and amended and restated bylaws that will be in effect upon the completion of this offering could discourage, delay or prevent a merger, acquisition or other change in control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. These provisions also could limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market pricestock.
Our restated certificate of our common stock. Stockholders who wish to participate in these transactions may not have the opportunity to do so. Furthermore, these provisions could prevent or frustrate attempts by our stockholders to replace or remove our management. These provisions:
In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which may, unless certain criteria are met, prohibit large stockholders, in particular those owning 15% or moreacquire a majority of our outstanding voting stock and vote the stock they acquire to remove management or directors.
Our restated certificate also provides staggered terms for the members of our board of directors, and that directors may be removed by stockholders only by vote of the holders of 66 2/3% of voting shares then outstanding. In addition, our amended and restated bylaws do not permit stockholders to call special or annual meetings of stockholders, or to act by written consent without a meeting. These provisions may prevent our voting stock unless the holder has held the stock for three years or, among other things, the board of directors has approved the transaction. Our board of directors could rely on Delaware law to prevent or delay an acquisition.
We are a smaller reporting company and we cannot be certain if the reduced disclosure requirements applicable to smaller reporting companies will make our common stock less attractive to investors.
We are currently a “smaller reporting company” as defined in the Securities Exchange Act of 1934, and are thus allowed to provide simplified executive compensation disclosures in our filings, are exempt from mergingthe provisions of Section 404(b) of the Sarbanes-Oxley Act requiring that an independent registered public accounting firm provide an attestation report on the effectiveness of internal control over financial reporting and have certain other decreased disclosure obligations in our SEC filings. We cannot predict whether investors will find our common stock less attractive because of our reliance on any of these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
We have in the past and may in the future fail to meet the continued listing requirements of The Nasdaq Capital Market. Our ability to publicly or combiningprivately sell equity securities and the liquidity of our common stock could be adversely affected if we are delisted from The Nasdaq Capital Market.
As previously disclosed, on November 29, 2016, we received a notification letter from the Listing Qualifications Department of The Nasdaq Capital Market indicating that we were not in compliance with usthe $1.00 minimum bid requirement. We were given a period of 180 days from the notification to regain compliance, by having the closing bid price of our common stock must exceed $1.00 for a prescribedminimum of ten (10) consecutive trading days. In connection with our effecting a1-for-15 reverse stock split of our common stock, we regained compliance with the minimum bid price requirement for continued listing on Nasdaq within the applicable time period as of time.February 28, 2017.
There is no assurance, however, that we will be able to maintain compliance with Nasdaq’s listing requirements in the future. If our common stock were delisted from Nasdaq, among other things, it would likely lead to a number of negative implications, including an adverse effect on the price of our common stock, reduced liquidity in our common stock, the loss of federal preemption of state securities laws and greater difficulty in obtaining financing. In the event of a delisting, we would take actions to restore our compliance with Nasdaq’s listing requirements, but we can provide no assurance that any such action taken by us would allow our common stock to become listed again, stabilize the market price or improve the liquidity of our common stock, prevent our common stock from dropping below the Nasdaq minimum bid price requirement or prevent futurenon-compliance with Nasdaq’s listing requirements.
Risks Related to this Offering
Management will have broad discretion as to the use of proceeds from this offering and we may use the net proceeds in ways with which you may disagree.
We intend to use the net proceeds of this offering for net working capital and general corporate purposes, which may include development of our clinical and preclinical product candidates, intellectual property protection and enforcement, capital expenditures, investments,in-licenses and acquisitions. Our management will have broad discretion in the application of the net proceeds from this offering and could spend the proceeds in ways that do not improve our results of operations or enhance the value of our common stock. Accordingly, you will be relying on the judgment of our management on the use of net proceeds, and you will not have the opportunity, as part of your investment decision, to assess whether the proceeds are being used appropriately. Our failure to apply these funds effectively could have a material adverse effect on our business, delay the development of our product candidates and cause the price of our common stock to decline.
The offering price will be set by our Board of Directors and does not necessarily indicate the actual or market value of our common stock.
Our Board of Directors will approve the offering price and other terms of this offering after considering, among other things: the number of shares authorized in our certificate of incorporation; the current market price of our common stock; trading prices of our common stock over time; the volatility of our common stock; our current financial condition and the prospects for our future cash flows; the availability of and likely cost of capital of other potential sources of capital; and market and economic conditions at the time of the offering. The offering price is not intended to bear any relationship to the book value of our assets or our past operations, cash flows, losses, financial condition, net worth or any other established criteria used to value securities. The offering price may not be indicative of the fair value of the common stock.
The Series A Preferred Stock is an unlisted security and there is no public market for it.
There is no established public trading market for the Series A Preferred Stock, and we do not expect a market to develop. In addition, the Series A Preferred Stock is not listed, and we do not intend to apply for listing of the Series A Preferred Stock on any securities exchange or trading system. Without an active market, the liquidity of the Series A Preferred Stock is limited, and investors may be unable to liquidate their investments in the Series A Preferred Stock.
The warrants may not have any value.
The warrants will be exercisable for five years from the closing date at an initial exercise price per share of $ . In the event that the price of a share of our common stock does not exceed the exercise price of the warrants during the period when the warrants are exercisable, the warrants may not have any value.
The warrants are subject to an issuer call.
If, after the date that is 180 days after the closing date, (i) the volume weighted average price for each of 30 consecutive trading days (the “Measurement Period”), which Measurement Period commences after the date that is 180 days after the closing date, exceeds 300% of the exercise price (subject to adjustment for forward and reverse stock splits, recapitalizations, stock dividends and the like after the initial exercise date), (ii) the average daily volume for such Measurement Period exceeds $500,000 per trading day and, (iii) the warrant holder is not in possession of any materialnon-public information which was provided by the Company, then the Company may, within 1 trading day of the end of such Measurement Period, call for cancellation of all or any portion of the warrants for which an exercise notice has not yet been delivered for consideration equal to $0.001 per warrant share. The Company’s right to call the warrants shall be exercised ratably among the holders based on the then outstanding warrants. You may be unable to reinvest your proceeds from the call in an investment with a return that is as high as the return on the warrants would have been if they had not been called.
A warrant does not entitle the holder to any rights as common stockholders until the holder exercises the warrant for shares of our common stock.
Until you acquire shares of our common stock upon exercise of your warrants, the warrants will not provide you any rights as a common stockholder. Upon exercise of your warrants, you will be entitled to exercise the rights of a common stockholder only as to matters for which the record date occurs on or after the exercise date.
SPECIALCAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
SomeThis prospectus contains or incorporates by reference “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as “may,” “should,” “anticipate,” “estimate,” “expect,” “projects,” “intends,” “plans,” “believes” and words and terms of similar substance used in connection with any discussion of future operating or financial performance, identify forward-looking statements. Forward-looking statements under “Prospectus Summary,” “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” “Business” and elsewhereincluded or incorporated by reference in this prospectus constitute forward-looking statements. Theseinclude, for example, statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements. While we believe that we have a reasonable basis for each forward-looking statement contained in this prospectus, we caution you that these statements are based on a combination of facts and factors currently known by us and projections of the future, about which we cannot be certain. Many factors affect our ability to achieve our objectives, including the following:about:
Forward-looking statements represent management’s present judgment regarding future events. Because forward-looking statements relate to the future, they are subject to inherent uncertainties, risks and changes in circumstances that are difficult to predict and many of third-party manufacturers with which we contract to provide a supplyare outside of our product candidates.
In addition,control. Our actual results and financial condition may differ materially from those indicated in the forward-looking statements. Therefore, you should refer to the “Risk Factors” sectionnot rely on any of this prospectus for a discussion of otherthese forward-looking statements. Important factors that maycould cause our actual results and financial condition to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you thatindicated in the forward-looking statements ininclude, among others, the risk factors identified under the caption “Risk Factors”, beginning on page 13 of this prospectus, and in the other the documents we have filed, or will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all. The Private Securities Litigation Reform Act of 1995 and Section 27A offile, with the Securities Act of 1933 do not protect any forward-looking statements that we make in connection with this offering.
You should read this prospectus completely. In some cases, you can identify forward-looking statements by the following words: “may,” “will,” “could,” “would,” “should,” “expect,” “intend,” “plan,” “anticipate,” “believe,” “estimate,” “predict,” “project,” “potential,” “continue,” “ongoing” or the negative of these terms or other comparable terminology, although not all forward-looking statements contain these words. We may not update these forward-looking statements even though our situation may change in the future. We qualify all the forward-lookingand Exchange Commission. Forward-looking statements contained in this prospectus speak as of the date hereof and the Company does not undertake to update any of these forward-looking statements to reflect a change in its views or events or circumstances that occur after such date.
In evaluating our business, prospective investors should carefully consider these factors in addition to the other information set forth in this prospectus, including under the caption “Risk Factors.” All forward-looking statements included in this document are based on information available to us on the date hereof. We disclaim any intent to update any forward-looking statements.
In this prospectus, we refer to information regarding potential markets for our drug candidates and other industry data. Unless otherwise expressly stated, we obtained this industry, business, market and other data from reports, research surveys, studies and similar data prepared by third parties, industry, medical and general publications, government data and similar sources. In some cases, we do not expressly refer to the foregoing cautionary statements.sources from which this data is derived. In that regard, when we refer to one or more sources of this type of data in any paragraph, you should assume that other data of this type appearing in the same paragraph is derived from the same sources, unless otherwise expressly stated or the context otherwise requires. We believe that all such information has been obtained from reliable sources that are customarily relied upon by companies in our industry. However, we have not independently verified any such information.
We estimate that ourthe net proceeds from the sale of shares of our common stock in this offering will be approximately $$13.47 million, or approximately $ million ifbased on an assumed offering price of $10.50 per Class A Unit and $1,000 per Class B Unit, after deducting the underwriting discounts and commissions and estimated offering expenses payable by us. If the underwriters exercise their over-allotment option in full, assuming an initial public offering price of $ per share andwe estimate that our net proceeds will be approximately $15.72 million, after deducting estimatedthe underwriting discounts and commissions and estimated offering expenses payable by us.
We will not receive any additional proceeds from any future conversions of the Series A Preferred Stock. We will only receive additional proceeds from the exercise of the warrants issuable in connection with this offering if the warrants are exercised and the holders of such warrants pay the exercise price in cash upon such exercise and do not utilize the cashless exercise provision of the warrants.
We expectintend to use thenet proceeds of thefrom this offering to fund:
acquisitions. We may also use a portion of the proceeds for the potential acquisition of, or investment in, technologies, products or companies that complement our business, although we have no current understandings, commitments or agreements to do so.
The amounts and timing of our actual expenditures will depend upon numerous factors, including the status of our development and commercialization efforts,not yet determined the amount of net proceeds actually raised in this offering, the amount of cash generated through our existing strategic collaborations,to be used specifically for any additional strategic collaborations into which we may enter and sales of Inversine. We have not determined the amounts we plan to spend in any of the areas listed aboveparticular purpose or the timing of these expenditures. Accordingly, our management will have significant discretion and flexibility in applying the net proceeds from the sale of these securities.
Our common stock trades on The Nasdaq Capital Market under the symbol “CBIO.” The last reported sale price for our common stock on March 29, 2017 was $10.51 per share. As of March 29, 2017, we had approximately 123 holders of record of our common stock. The number of record holders was determined from the records of our transfer agent and does not include beneficial owners of common stock whose shares are held in the names of various security brokers, dealers, and registered clearing agencies. A description of the common stock that we are issuing in this offering is set forth under the heading “Description of Securities” beginning on page 53 of this offering.prospectus.
The following table sets forth for the periods indicated the high and low sale prices per share of our common stock as reported on The Nasdaq Capital Market, but as adjusted to reflect the 2017 Reverse Stock Split:
Fiscal Year ended December 31, 2015 First Quarter Second Quarter Third Quarter Fourth Quarter Fiscal Year ending December 31, 2016 First Quarter Second Quarter Third Quarter Fourth Quarter Fiscal Year ending December 31, 2017 First Quarter (through March 29, 2017)Until the funds are used as described above, we intend to invest the net proceeds from this offering in short-term interest-bearing, investment grade securities. High Low $ 45.75 $ 37.50 $ 43.65 $ 33.90 $ 177.00 $ 33.15 $ 85.35 $ 30.00 $ 47.25 $ 24.30 $ 28.20 $ 18.15 $ 23.40 $ 17.25 $ 18.30 $ 8.10 $ 15.01 $ 4.73
We have never declared or paid cash dividends on any of our shares of capital stock. We currently intend to retain all available funds and any future earnings, if any, to financefund the expansiondevelopment and growthexpansion of our business and we do not anticipate paying any cash dividends in the foreseeable future. Any future determination related to dividend policy will be made at the discretion of our board of directors.
We will not pay any dividends on shares of common stock (other than dividends in the form of common stock) unless and until such time as we pay dividends on each share of Series A Preferred Stock on anas-converted basis. Other than as set forth in the previous sentence, no other dividends will be paid on the Series A Preferred Stock and we will pay no dividends (other than dividends in the form of common stock) on shares of common Stock unless we simultaneously comply with the previous sentence.
The following table sets forth our cash and cash equivalents and our capitalization as of December 31, 2016:
You should read this table together with “Item 5—Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our Annual Report onForm 10-K filed on March 8, 2017 and with our consolidated financial statements and the accompanying notes contained in this prospectus.
| As of December 31, 2016 | ||||||||
| Actual | As Adjusted | |||||||
| (In thousands, except share data) | ||||||||
Cash and cash equivalents | $ | 17,064 | $ | 30,534 | ||||
|
|
|
| |||||
Long term liabilities, less current portion | 54 | 54 | ||||||
Stockholders’ equity (deficit) | ||||||||
Preferred Stock, par value $0.001 per share; 5,000,000 shares authorized; no shares issued and outstanding as of December 31, 2016, actual; and 12,000 shares issued and outstanding, as adjusted | — | — | ||||||
Common Stock, par value $0.001 per share; 100,000,000 shares authorized; 801,756 shares issued and outstanding as of December 31, 2016, actual; and 1,087,470 shares issued and outstanding, as adjusted | 1 | 1 | ||||||
Additionalpaid-in capital | 164,053 | 179,053 | ||||||
Accumulated deficit | (147,982 | ) | (147,982 | ) | ||||
|
|
|
| |||||
Total stockholders’ equity | 16,071 | 31,071 | ||||||
|
|
|
| |||||
Total capitalization | 16,125 | 31,125 | ||||||
|
|
|
| |||||
The number of shares of common stock to be outstanding before this offering and to be outstanding after this offering in the table above is based on 801,756 shares of common stock outstanding as of December 31, 2016 and excludes:
SECURITY OWNERSHIP OF BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth certain information with respect to the beneficial ownership of our common stock as of March 28, 2017, for:
| (1) | each person or group of affiliated persons known by us to be the beneficial owner of more than 5% of our common stock; |
| (2) | each of our named executive officers; |
| (3) | each of our directors; and |
| (4) | all current executive officers and directors as a group. |
Applicable percentage ownership is based on 1,000,036 shares of common stock outstanding at March 28, 2017. We have determined beneficial ownership in accordance with SEC rules. The information does not necessarily indicate beneficial ownership for any other purpose. Under these rules, the number of shares of common stock deemed outstanding includes shares issuable upon exercise of options or warrants, or the conversion of convertible notes, held by the respective person or group that may be exercised or converted within 60 days after March 28, 2017. For purposes of calculating each person’s or group’s percentage ownership, stock options and warrants exercisable, and notes convertible, within 60 days after March 28, 2017 are included for that person or group, but not the stock options of any other person or group.
Unless otherwise indicated and subject to applicable community property laws, to our knowledge, each stockholder named in the following table possesses sole voting and investment power over the shares listed. Unless otherwise noted below, the address of each person listed in the table is c/o Catalyst Biosciences, Inc., 260 Littlefield Ave, South San Francisco, CA 94080.
Name 5% or Greater Stockholders Funds affiliated with Essex Woodlands Health Ventures 335 Bryant Street, 3rd Floor Palo Alto, CA 94301 New Enterprise 10, Limited Partnership and affiliates 1954 Greenspring Drive, Suite 600 Timonium, Maryland 21093 HealthCare Ventures VIII, L.P. 47 Thorndike Street, SuiteB1-11954 Cambridge, MA 02141 Johnson & Johnson Innovation—JJDC, Inc. 410 George Street New Brunswick, NJ 08901 Morgenthaler Partners VIII, L.P. 2710 Sand Hill Road, Suite 100 Menlo Park, CA 94025 Rosetta Capital V L.P. c/o The Accounts Bureau Limited 83 Victoria Street London, SW1H OHW United Kingdom Directors and Named Executive Officers Nassim Usman, Ph.D. Fletcher Payne Howard Levy, M.B.B.Ch., Ph.D., M.M.M. Edwin L. Madison, Ph.D. Harold E. Selick, Ph.D. Stephen A. Hill, M.D. Augustine Lawlor Jeff Himawan, Ph.D. John P. Richard Errol B. De Souza All Directors and Executive Officers as a Group (9 persons) Number of Shares
Owned and Nature of
Beneficial Ownership Percent
of Class 81,767 (1) 8.15 % 79,216 (2) 7.65 % 71,247 (3) 7.11 % 68,609 (4) 6.85 % 60,369 (5) 6.03 % 52,405 (6) 5.24 % 17,650 (7) 1.74 % 5,528 (8) * 1,806 (9) * 9,895 (10) 0.98 % 2,350 (11) * 23,072 (12) 2.26 % 72,274 (3)(13) 7.21 % 81,767 (1) 8.15 % 4,496 (14) * 4,533 (15) * 213,476 (16) 21.22 %
| * | Indicates less than 1% of class. |
| (1) | The information reported is based on a Schedule 13D filed with the SEC on August 31, 2015 which reports that, as of August 20, 2015, (i) Essex Woodlands Health Ventures Fund VIII, L.P. (“Essex VIII”) directly holds 74,103 shares, which include 2,850 shares issuable upon the exercise of warrants within 60 days, (ii) Essex Woodlands Health Ventures FundVIII-A, L.P. (“EssexVIII-A”) directly holds 5,342 shares which include 255 shares issuable upon the exercise of warrants within 60 days), and (iii) Essex Woodlands Health Ventures FundVIII-A, L.P. (“EssexVIII-B”) directly holds 2,322 shares, which include 89 shares issuable upon the exercise of warrants within 60 days. Essex Woodlands Health Ventures VIII, L.P. (the “GP Partnership”) is the general partner of Essex VIII, EssexVIII-A, and EssexVIII-B. Essex Woodlands Health Ventures VIII, LLC (“Essex VIII LLC”) is the general partner of the GP Partnership. Essex VIII LLC, as the general partner of the GP Partnership, may be deemed to have sole voting investment power with respect to 81,767 shares comprising of (i) 78,623 shares and (ii) 3,144 shares that may be purchased upon the exercise of warrants within 60 days. Essex VIII LLC disclaims beneficial ownership to 81,767 shares comprising of (i) 78,623 shares and (ii) 3,144 shares that may be purchased upon the exercise of warrants within 60 days, except to the extent of its pecuniary interest. Dr. Jeff |
| Himawan, James Currie, Marty Sutter, Immanuel Thangaraj, Petri Vainio, Ron Eastman, Steve Wiggins and Guido Neels (the “Managers”) may also be deemed to have shared dispositive power and voting power with respect to 81,767 shares comprising of (i) 78,623 shares and (ii) 3,144 shares that may be purchased upon the exercise of warrants within 60 days. The GP Partnership disclaims beneficial ownership of the shares except to the extent of its pecuniary interest therein. |
| (2) | The information reported is based on a Schedule 13D/A filed with the SEC on March 29, 2017, which reports that, as of February 10, 2017, New Enterprise Associates 10, Limited Partnership (“NEA 10”), is the record owner of 43,462 shares of our common stock (the “NEA 10 Shares”) and is deemed to beneficially own 35,754 shares of our common stock (together with the NEA 10 Shares, the “Securities”) issuable upon the conversion of $4,928,707.28 in aggregate principal amount of redeemable convertible notes issued by the Company to NEA 10 on August 19, 2015, convertible within 60 days of issuance. As the sole general partner of NEA 10, NEA Partners 10, Limited Partnership (“NEA Partners 10”) may be deemed to own beneficially the Securities. As the individual general partners of NEA Partners 10, each of Michael James Barrett, Peter J. Barris and Scott D. Sandell also may be deemed to own beneficially the Securities. |
| (3) | The information reported is based on a Schedule 13D filed with the SEC on August 31, 2015 which reports that, as of August 20, 2015, Healthcare Ventures VIII, L.P. (“HCVVIII”) directly beneficially owns 71,247 shares which include 1,846 shares that may be purchased upon the exercise of warrants within 60 days. Each of James H. Cavanaugh, Ph.D., Harold R. Werner, John W. Littlechild, Christopher Mirabelli, Ph.D., and Augustine Lawlor are the managing directors of HealthCare Ventures VIII, LLC (“HCPVIIILLC”), the general partner of HealthCare Partners VIII, L.P. (“HCPVIII”), which is the general partner of HCVVIII. HCPVIIILLC and HCPVIII may be deemed to indirectly beneficially own 71,247 shares, which include 1,846 shares that may be purchased upon the exercise of warrants within 60 days. Each of Drs. Cavanaugh and Mirabelli and Messrs. Werner, Littlechild and Lawlor may be deemed to indirectly beneficially own 71,247 shares, which include 1,846 shares that may be purchased upon the exercise of warrants within 60 days. HCVVIII, HCPVIII, HCPVIIILLC. Drs. Cavanaugh and Mirabelli and Messrs. Werner, Littlechild and Lawlor share the power to vote and direct the vote and to dispose of and direct the disposition of the shares beneficially owned by HCVVIII. |
| (4) | The information reported is based on a Schedule 13G filed with the SEC on August 28, 2015 which reports that as of August 20, 2015, Johnson & Johnson Innovation-JJDC, Inc. (“JJDC”) directly beneficially owns 68,609 shares, which includes 1,658 shares issuable upon exercise of warrants within 60 days. JJDC is a wholly-owned subsidiary of Johnson & Johnson, a New Jersey corporation (“J&J”). J&J may be deemed to indirectly beneficially own the securities that are directly beneficially owned by JJDC. |
| (5) | The information reported is based on a Schedule 13D filed with the SEC on August 31, 2015 which reports that as of August 20, 2015, Morgenthaler Partners VIII, L.P. (“MP LP”) is the record holder of 59,125 shares and 1,244 shares that may be purchased upon the exercise of warrants that are exercisable within 60 days. Morgenthaler Management Partners VIII, LLC (“MMP LLC”) is the general partner of MP LP and may be deemed to beneficially own the 59,125 shares and 1,244 shares that may be purchased upon the exercise of warrants that are exercisable within 60 days. MMP LLC shares voting control and investment power over the 59,125 shares and 1,244 shares that may be purchased upon the exercise of warrants that are exercisable within 60 days with Ralph Christoffersen, Ph.D., Robert Bellas, John Lutsi, Gary Morgenthaler, Robery Pavey and Gary Little (the “Members”), each of whom disclaim beneficial ownership over the 59,125 shares and 1,244 shares that may be purchased upon the exercise of warrants that are exercisable within 60 days. The Members are the members of MMP LLC. |
| (6) | The information reported is based on a Schedule 13D filed with the SEC on August 28, 2015 which reports that as of August 20, 2015, Rosetta Capital V GP Limited (the “GPCo”) is the record holder of 51,455 shares and 950 shares that may be purchased upon the exercise of warrants that are exercisable within 60 days on behalf of Rosetta Capital V LP (“Rosetta V”). Rosetta V, Rosetta Capital V GP LP (the “GP”), and Rosetta Capital Limited (“Rosetta Capital”) are management vehicles within the Rosetta Capital group. Rosetta Capital has management control over all of the shares directly held by Rosetta V, and Rosetta Capital has management control over Rosetta V. The GP, the GPCo and Rosetta Capital control Rosetta V through their respective direct and indirect interests in the Rosetta V partnership and pursuant to a |
| management agreement, and may be deemed to share beneficial ownership of the 51,455 shares and 950 shares that may be purchased upon the exercise of warrants that are exercisable within 60 days by virtue of their ability to collectively direct decision of Rosetta V. Rosetta Capital is the general partner of the GPCo and was appointed the manager of Rosetta V and therefore, it may be deemed to beneficially own the 51,455 shares and 950 shares that may be purchased upon the exercise of warrants that are exercisable within 60 days. |
| (7) | Consists of (i) 4,056 shares and 1 share issuable upon the exercise of warrants within 60 days held by the Usman Family Trust, of which Dr. Usman is aco-trustee with Susan L. Usman, (ii) 1,169 shares and (iii) 12,424 shares issuable upon the exercise of options within 60 days. |
| (8) | Consists of (i) 1,668 shares held by Charles and Nancy Payne 2000 Trust, of which Mr. Payne is a trustee and (ii) 3,860 shares issuable upon the exercise of options within 60 days. |
| (9) | Consists of 1,806 shares issuable upon the exercise of options within 60 days. |
| (10) | Consists of (i) 2,067 shares and (ii) 7,827 shares issuable upon the exercise of options within 60 days. |
| (11) | Consists of (i) 1,082 shares, (ii) 1,224 shares issuable upon the exercise of options within 60 days and (iii) 19 shares issuable upon the exercise of warrants within 60 days. Also includes 25 shares held directly by Dr. Selick’s wife. |
| (12) | Consists of (i) 1,380 shares, (ii) 20,166 shares issuable upon the exercise of options within 60 days, and (iii) 1,526 shares issuable upon the conversion of $210,397 in aggregate principal amount of redeemable convertible notes issued by the Company on August 19, 2015, as reported on a Form 4 filed on August 18, 2015, in respect of the shares, held by Dr. Hill and convertible within 60 days. |
| (13) | Consists of 1,027 shares issuable upon the exercise of options within 60 days. |
| (14) | Consists of (i) 217 shares, (ii) 4,101 shares issuable upon the exercise of options within 60 days and (iii) 178 shares issuable upon the conversion of $24,635 in aggregate principal amount of redeemable convertible notes convertible within 60 days. |
| (15) | Consists of (i) 146 shares, (ii) 4,267 shares issuable upon the exercise of options within 60 days and (iii) 120 shares of common stock issuable upon the conversion of $16,543.00 in aggregate principal amount of redeemable convertible notes convertible within 60 days. |
| (16) | Includes (i) 157,767 shares, (ii) 48,875 shares of subject to options exercisable within 60 days, 5,010 shares subject to warrants exercisable within 60 days, and 1,824 shares issuable upon the conversion of redeemable convertible notes convertible within 60 days. |
Units
We are offering 285,714 Class A Units, with each Class A Unit consisting of one share of common stock and a warrant to purchase half of one share of our common stock at an exercise price per share of $ , together with the shares of common stock underlying such warrants, at a public offering price of $ per Class A Unit of $ . The Class A Units will not be certificated and the shares of common stock and warrants part of such units are immediately separable and will be issued separately in this offering.
We are also offering to those purchasers whose purchase of Class A Units in this offering would result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock following the consummation of this offering, the opportunity to purchase, in lieu of the number of Class A Units that would result in ownership in excess of 4.99% (or, at the election of the purchaser, 9.99%), 12,000 Class B Units. Each Class B Unit will consist of one share of Series A Preferred Stock, par value $0.001 per share, convertible into 95.24 shares of common stock and a warrant to purchase 47.62 shares of our common stock at an exercise price per share of $ , together with the shares of common stock underlying such shares of Series A Preferred Stock and warrants, at a public offering price of $ per Class B Unit. The Class B Units will not be certificated and the shares of Series A Preferred Stock and the warrants part of such units are immediately separable and will be issued separately in this offering.
Description of Capital Stock
The following description of our common stock and preferred stock summarizes the material terms and provisions of the common stock and preferred stock that we may issue in connection with this offering. It may not contain all the information that is important to you. For the complete terms of our common stock and preferred stock, please refer to our Fourth Amended and Restated Certificate of Incorporation, as amended (the “restated certificate of incorporation”) and our amended and restated bylaws, which are filed as exhibits to the registration statement which includes this prospectus. The Delaware General Corporation Law (“DGCL”) may also affect the terms of these securities.
Common Stock
Under our restated certificate of incorporation, we have authority to issue 100,000,000 shares of our common stock, par value $0.001 per share. As of March 28, 2017, 1,000,036 shares of our common stock were issued and outstanding. All shares of our common stock will, when issued, be duly authorized, fully paid and nonassessable.
Dividends. Subject to preferential dividend rights of any other class or series of stock, the holders of shares of our common stock are entitled to receive dividends, including dividends of our stock, as and when declared by our board of directors, subject to any limitations imposed by law and to the rights of the holders, if any, of our preferred stock. We have never paid cash dividends on our common stock, except with respect to a cash dividend paid in connection with the closing of the merger between Targacept and Catalyst Bio. We do not anticipate paying periodic cash dividends on our common stock for the foreseeable future. Any future determination about the payment of dividends will be made at the discretion of our board of directors and will depend onupon our financial condition, results of operations,earnings, if any, capital requirements, operating and financial conditions and on such other factors that ouras the board of directors deems relevant.
Liquidation. In addition, the terms of any future debtevent we are liquidated, dissolved or credit facility may preclude us from paying dividends.
The following table sets forth our capitalization at March 31, 2004:
If the liabilities, each holder of the warrant instead exercises the warrant in full for cash, we would issue 1,612,903 shares of common stock for cash proceeds of approximately $3.1 million.
You should read this table together with our financial statements and the related notes appearing at the end of this prospectus and the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section and other financial information included in this prospectus.
| As of March 31, 2004 | |||||||||||
| Actual | Pro Forma | Pro Forma As Adjusted | |||||||||
(unaudited) (in thousands, except | |||||||||||
Total long-term obligations | $ | 1,935 | $ | 1,935 | $ | ||||||
Redeemable convertible preferred stock, $0.001 par value, 60,736,705 shares authorized, issued and outstanding actual; no shares authorized, issued or outstanding pro forma and pro forma as adjusted | 132,276 | — | |||||||||
Stockholders’ equity (deficit): | |||||||||||
Common stock, $0.001 par value, 85,000,000 shares authorized actual, pro forma and pro forma as adjusted; 1,164,524 issued and outstanding actual; shares issued and outstanding pro forma; shares issued and outstanding pro forma as adjusted | 1 | 1 | |||||||||
Preferred stock, $0.001 par value, no shares authorized, issued or outstanding actual and pro forma; shares authorized and no shares issued and outstanding pro forma as adjusted | — | — | |||||||||
Capital in excess of par value | 6,378 | 115,330 | |||||||||
Excess of fair value of Series A preferred stock over cash received | (23,250 | ) | — | ||||||||
Common stock warrants | 214 | 214 | |||||||||
Accumulated deficit | (82,818 | ) | (82,818 | ) | |||||||
Accumulated other comprehensive loss | (35 | ) | (35 | ) | |||||||
Total stockholders’ equity (deficit) | (99,510 | ) | (32,766 | ) | |||||||
Total capitalization | $ | 34,701 | $ | 34,701 | $ | ||||||
The table above does not include:
If you invest in our common stock, your interest will be diluted immediately to the extent of the difference between the initial public offering price per share of our common stock and the pro forma net tangible book value of our common stock immediately after completion of this offering.
The historical net tangible book value of our common stock as of March 31, 2004 was approximately $(100,089), or approximately $(85.95) per share, based on 1,164,524 shares of common stock outstanding as of March 31, 2004. Historical net tangible book value per share represents our total tangible assets less total liabilities divided by the actual number of our common stock outstanding.
As of March 31, 2004, the pro forma net tangible book value of our common stock was approximately $ million, or approximately $ per share. Pro forma net tangible book value per share represents our total tangible assets less total liabilities divided by the pro forma number of shares of our common stock outstanding, after giving effect to a one-for- reverse stock split of our common stock that will be effective prior to the completion of this offering, the conversion of all outstanding shares of our convertible preferred stock into 73,739,905 shares of common stock concurrently with the completion of this offering, and the issuance of 1,612,903 shares of common stock upon the exercise of an outstanding warrant in full for cash concurrently with the completion of this offering.
Assuming the sale of the shares of our common stock offered by this prospectus at an assumed initial public offering price of $ per share, after deducting estimated underwriting discounts and commissions and offering expenses payable by us, our pro forma net tangible book value as of March 31, 2004 would have been $ , or $ per share. This represents an immediate increase in pro forma net tangible book value of $ per share to our existing stockholders and an immediate dilution in pro forma net tangible book value of $ per share to new investors purchasing in this offering at the initial public offering price. The following table illustrates this dilution on a per share basis:
Assumed initial public offering price per share | $ | ||||||
Historical net tangible book value per share | $ | (89.95 | ) | ||||
Increase attributable to the conversion of the convertible preferred stock | |||||||
Pro forma net tangible book value per share before this offering | ( | ) | |||||
Increase per share attributable to new investors | |||||||
Pro forma net tangible book value per share after this offering | |||||||
Dilution per share to new investors | $ | ||||||
The following table summarizes, on a pro forma basis as of March 31, 2004, the total number of shares of common stock purchased from us, the total consideration paid and the average price per share paid by existing stockholders and by new investors purchasing shares in this offering at an assumed initial public offering price of $ per share, before deducting estimated underwriting discounts and commissions and offering expenses payable by us.
| Shares Purchased | Total Consideration | Average Price per Share | ||||||||||||
| Number | Percent | Amount | Percent | |||||||||||
Existing stockholders | % | $ | % | $ | ||||||||||
New investors | ||||||||||||||
Total | 100 | % | $ | 100 | % | |||||||||
The share data in the table above is based on shares outstanding as of March 31, 2004, counting as outstanding the 73,739,905 shares of common stock underlying all outstanding convertible preferred stock, and excludes:
If the underwriters exercise their over-allotment in full, the following will occur:
You should read the following selected financial data together with our financial statements and the related notes appearing at the end of this prospectus and the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section and other financial data included in this prospectus.
We have derived the statement of operations data for the years ended December 31, 2001, 2002 and 2003 and the balance sheet data as of December 31, 2002 and 2003 from our audited financial statements included in this prospectus. We have derived the statement of operations data for the year ended December 31, 2000 and the balance sheet data as of December 31, 2000 and 2001 from our audited financial statements not included in this prospectus. We became an independent company in August 2000, prior to which we were a wholly owned subsidiary of R.J. Reynolds Tobacco Company. We have derived the statement of operations data for the year ended December 31, 1999 and the balance sheet data as of December 31, 1999 from our unaudited financial statements not included in this prospectus. We have derived the statement of operations data for the three months ended March 31, 2003 and 2004 and the balance sheet data as of March 31, 2003 and 2004 from our unaudited financial statements included in this prospectus. In the opinion of our management, these unaudited financial statements reflect all adjustments, consisting only of normal recurring adjustments, necessary for a fair presentation of these financial statements. Our historical results for any prior or interim period are not necessarily indicative of the results to be expected for the fiscal year ending December 31, 2004 or for any other period.
The pro forma net loss attributable to common stockholders per share information is computed using the weighted average number of common shares outstanding, after giving pro forma effect to the conversion of all outstanding shares of our convertible preferred stock into 73,739,905 shares of common stock concurrently with the completion of this offering, as if the conversion had occurred at the date of the original issuance.
| Year ended December 31, | Three months ended March 31, | |||||||||||||||||||||||||||
| 1999 | 2000 | 2001 | 2002 | 2003 | 2003 | 2004 | ||||||||||||||||||||||
| (unaudited) | (unaudited) | |||||||||||||||||||||||||||
| (in thousands, except share and per share data) | ||||||||||||||||||||||||||||
Statement of Operations Data: | ||||||||||||||||||||||||||||
Net revenue | $ | 2,350 | $ | 2,351 | $ | 1,703 | $ | 2,286 | $ | 2,458 | $ | 691 | $ | 497 | ||||||||||||||
Operating expenses: | ||||||||||||||||||||||||||||
Research and development | 2,104 | 3,675 | 8,152 | 16,244 | 18,179 | 4,069 | 6,050 | |||||||||||||||||||||
General and administrative | 1,436 | 1,653 | 2,302 | 4,135 | 3,600 | 697 | 1,109 | |||||||||||||||||||||
Cost of product sales | — | — | — | 244 | 743 | 200 | 182 | |||||||||||||||||||||
Purchased in-process research and development | — | — | — | 2,666 | — | — | — | |||||||||||||||||||||
Total operating expenses | 3,540 | 5,328 | 10,454 | 23,289 | 22,522 | 4,966 | 7,341 | |||||||||||||||||||||
Loss from operations | (1,190 | ) | (2,977 | ) | (8,751 | ) | (21,003 | ) | (20,064 | ) | (4,275 | ) | (6,844 | ) | ||||||||||||||
Interest and dividend income | — | 536 | 1,449 | 88 | 791 | 124 | 231 | |||||||||||||||||||||
Interest expense | — | — | — | (103 | ) | (122 | ) | (34 | ) | (25 | ) | |||||||||||||||||
Loss on disposal of fixed assets | — | — | — | (54 | ) | — | — | — | ||||||||||||||||||||
Net loss | (1,190 | ) | (2,441 | ) | (7,302 | ) | (21,072 | ) | (19,395 | ) | (4,185 | ) | (6,638 | ) | ||||||||||||||
Preferred stock accretion | — | (981 | ) | (3,808 | ) | (4,173 | ) | (8,341 | ) | (1,915 | ) | (2,142 | ) | |||||||||||||||
Net loss attributable to common stockholders | $ | (1,190 | ) | $ | (3,422 | ) | $ | (11,110 | ) | $ | (25,245 | ) | $ | (27,736 | ) | $ | (6,100 | ) | $ | (8,780 | ) | |||||||
Basic and diluted net loss per share applicable to common stockholders | $ | (2,380.00 | ) | $ | (30.58 | ) | $ | (26.80 | ) | $ | (45.28 | ) | $ | (33.91 | ) | $ | (9.48 | ) | $ | (7.89 | ) | |||||||
Shares used to compute basic and diluted net loss per share | 500 | 111,915 | 414,624 | 557,492 | 817,894 | 643,571 | 1,112,591 | |||||||||||||||||||||
Pro forma basic and diluted net loss per share applicable to common stockholders (unaudited) | $ | (0.27 | ) | $ | (0.09 | ) | ||||||||||||||||||||||
Shares used to compute pro forma basic and diluted net loss per share (unaudited) | 71,118,629 | 74,852,496 | ||||||||||||||||||||||||||
Three months ended March 31, Balance Sheet Data: Cash, cash equivalents and short-term investments Working capital Total assets Long-term debt, net of current portion Redeemable convertible preferred stock Accumulated deficit Total stockholders’ equity (deficit) Year ended December 31, 1999 2000 2001 2002 2003 2003 2004 (unaudited) (unaudited) (in thousands, except share and per share data) $ — $ 28,053 $ 21,180 $ 49,361 $ 42,977 $ 58,207 $ 36,847 1,305 27,654 20,371 46,685 40,526 56,534 33,663 1,804 29,338 24,396 54,379 47,390 62,274 40,806 — — — 2,088 1,462 2,037 1,298 — 54,418 58,365 108,026 130,134 123,741 132,276 (6,335 ) (9,946 ) (21,057 ) (46,302 ) (74,038 ) (52,402 ) (82,818 ) 36 (27,314 ) (38,268 ) (63,335 ) (90,796 ) (69,409 ) (99,510 )
MANAGEMENT’S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and the related notes appearing at the end of this prospectus. Some of the information contained in this discussion and analysis or set forth elsewhere in this prospectus, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. You should read the “Risk Factors” section of this prospectus for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are a biopharmaceutical company engaged in the design, discovery and development of a new class of drugs to treat multiple diseases and disorders by selectively targeting a class of receptors known as neuronal nicotinic acetylcholine receptors, or NNRs. We are developing small molecules designed to selectively target NNRs to treat diseases and disorders of the nervous system. Our product development pipeline consists of four product candidates in clinical development and multiple ongoing preclinical programs for target indications in which we believe that NNRs can be exploited for therapeutic benefit. In addition, we market Inversine, which we believe is the only FDA-approved product that is designed to target an NNR.
We trace our scientific lineage to a research program initiated by R.J. Reynolds Tobacco Company in 1982 to study the activity and effects of nicotine in the body and the function of nicotinic acetylcholine receptors. We were incorporated in Delaware in 1997 as a wholly owned subsidiary of RJR. In August 2000, we became an independent company when we issued shares of our series B convertible preferred stock to outside investors.
We have devoted substantially all of our resources to the discovery and development of our product candidates and technologies, including the design, conduct and management of preclinical and clinical studies and related manufacturing, regulatory and clinical affairs, and intellectual property prosecution. Through 1998, we received all of our funding from RJR. At the end of 1998, we entered into a collaboration agreement with the predecessor company to Aventis Pharma SA. We received an upfront license fee and research support payments under this agreement which, together with a modest amount of additional financial support from RJR, funded our activities through August 2000. Since August 2000, we have funded our operations primarily through the private placement of equity securities and, to a much lesser extent, through payments we received from our collaborators, Aventis and Dr. Falk Pharma GmbH, equipment and building lease incentive financing, sales of our product Inversine and government grants.
We have never been profitable. As of March 31, 2004, we had an accumulated deficit of $82.8 million. We expect to continue to incur substantial losses for the foreseeable future. We expect our research and development expenses to increase substantially following completion of this offering as we expand our clinical trial activity, as our product candidates advance through the development cycle and as we invest in additional product opportunities and research programs. We also expect our general and administrative expenses to increase substantially as we expand our infrastructure. Clinical trials and preclinical studies are time-consuming, expensive and may never yield a product that will generate revenue. A substantial portion of our revenue for the next several years will depend on our entering into new collaborations. Our revenue may vary substantially from quarter to quarter and year to year. We believe that period-to-period comparisons of our results of operations are not meaningful and should not be relied upon as indicative of our future performance.
We currently have one product available in the market, Inversine. We acquired rights to Inversine in August 2002. Inversine is approved for the management of moderately severe to severe essential hypertension. However,
our market research suggests that it is prescribed predominantly for the treatment of Tourette’s syndrome and other neuropsychiatric disorders. Sales of Inversine generated revenue of $815,000 for the year ended December 31, 2003 and $188,000 for the three months ended March 31, 2004.
We have entered into two collaboration agreements with Aventis. Our first collaboration agreement with Aventis covers the research, development and commercialization of a specified group of our compounds for the treatment of Alzheimer’s disease. There is only one compound remaining in the collaboration, TC-4959, which is in the late preclinical evaluation stage. Our second collaboration agreement with Aventis covers the research, development and commercialization of Aventis compounds for the treatment of Alzheimer’s disease and other central nervous system diseases. As of March 31, 2004, we had received a total of $8.0 million in upfront license fees and payments for research and development services under the two agreements. In addition to royalties on potential product sales, we could receive up to $30.0 million under the first agreement upon the achievement of pre-commercialization development and regulatory milestones, up to $8.0 million under the second agreement upon the achievement of pre-commercialization development and regulatory milestones related to Alzheimer’s disease and up to $8.0 million under the second agreement for each other central nervous system disease upon the achievement of pre-commercialization development and regulatory milestones related to that disease.
We have also entered into a collaboration agreement with Dr. Falk Pharma covering the development and commercialization of our product candidate TC-2403 for the treatment of ulcerative colitis. Upon effectiveness of the collaboration agreement in January 2001, Dr. Falk Pharma paid us a $1.0 million upfront license fee and purchased $1.0 million of our common stock. Pursuant to the terms of the agreement, we share TC-2403 development costs equally with Dr. Falk Pharma.
Critical Accounting Policies and Estimates
Our management’s discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses. On an ongoing basis, we evaluate these estimates and judgments, including those described below. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. These estimates and assumptions form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results and experiences may differ materially from these estimates.
While our significant accounting policies are more fully described in Note 2 to our financial statements appearing at the end of this prospectus, we believe that the following accounting policies are the most critical to aid you in fully understanding and evaluating our reported financial results and affect the more significant judgments and estimates that we use in the preparation of our financial statements.
Revenue Recognition
Our collaboration agreements contain multiple elements, including non-refundable upfront license fees, research payments for ongoing research and development, payments associated with achieving development and regulatory milestones and royalties to be paid based on specified percentages of net product sales or net profits, if any. We consider a variety of factors in determining the appropriate method of revenue recognition under these arrangements such as whether the elements are separable, whether there are determinable fair values and whether there is a unique earnings process associated with a particular element of an agreement.
We recognize research fee revenue from research services performed under our collaboration agreements as work is performed. We defer upfront payments and amortize them over the estimated research and development period. All revenue that we have recognized to date under these collaborations, or under government grants, is non-refundable. We recognize revenue from milestones with substantive performance risk upon achievement of
the milestone. We have not yet received payment of any such milestone-based revenues. We record product sales revenues when goods are shipped, at which point title has passed, and we establish an allowance for estimated returns at that time.
Accrued Expenses
As part of the process of preparing financial statements, we are required to estimate accrued expenses. This process involves identifying services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual cost. We make these estimates as of each balance sheet date in our financial statements. Examples of estimated accrued expenses include:
We base our expenses related to clinical trials on our estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and clinical research organizations that conduct and manage clinical trials on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Payments under the contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we will adjust the accrual accordingly. If we do not identify costs that have begun to be incurred or we underestimate or overestimate the level of services performed or the costs of these services, our actual expenses could differ from our estimates.
Purchased In-process Research and Development Expense
We determine the amount of any acquired in-process research and development expense based on an analysis of the cash flows that we expect to be generated by products that may arise from in-process technologies that we have acquired. As part of this analysis, we review the project rights that we acquire to determine the stage of their development, the probability of demonstrating sufficient safety and efficacy in clinical trials to obtain regulatory approval and product specific risk factors inherent in the drug development process. The product specific risk factors that we review include the type of drug under development, the likelihood of regulatory approval, manufacturing process capability, scientific rationale, preclinical and clinical safety and efficacy data, target product profile and development plans. Different estimates and assumptions for any of these factors would, if changed, result in a different estimate of in-process research and development expense.
In August 2002, we acquired from Layton Bioscience, Inc. marketing and trademark rights to Inversine and patent rights related to its active ingredient for cash consideration of $3.5 million. In allocating the purchase price, including the amount of in-process research and development, we considered an appraisal prepared by an independent appraiser using established valuation techniques for the pharmaceutical industry. We allocated approximately $2.7 million of the purchase price to in-process research and development, which we expensed in connection with the acquisition.
Stock-Based Compensation
We account for our employee stock-based compensation in accordance with Accounting Principles Board Opinion No. 25 and related interpretations, or APB 25. Under APB 25, we do not recognize compensation expense when we issue stock options to employees and non-employee directors, unless the exercise price is below the fair market value of the underlying common stock on the date of grant. We recognize this compensation expense over the vesting periods of the shares purchasable upon exercise of options. We recorded deferred stock-based compensation related to stock options granted to employees and directors of $92,600 during 2001, $129,700 during 2002 and $65,300 during 2003. We amortize our deferred stock-based compensation on a straight-line basis over the related option vesting periods, which range from immediate vesting to four years.
As required by Statement of Financial Accounting Standards, or SFAS, No. 123,Accounting for Stock-Based Compensation, as amended by SFAS No. 148,Accounting for Stock-Based Compensation—Transition and Disclosure, our financial statement footnotes disclose on a pro forma basis the amount of compensation expense that we would have recorded had we applied the fair value option methodology described in SFAS 123. Had we recorded all of our stock-based compensation using the SFAS 123 fair value methodology, our compensation expense would have been approximately $450,000 greater and our diluted net loss per share attributable to common stockholders would have been approximately $0.55 greater in 2003. For more information, you should refer to Note 2 to our financial statements included at the end of this prospectus.
Financial Operations Overview
Revenue
Inversine is our only commercial product generating revenue. Sales of Inversine generated revenue of $815,000 for the year ended December 31, 2003 and $188,000 for the three months ended March 31, 2004. We have entered into an exclusive distribution agreement with a third party for the distribution of Inversine. We do not have or use a sales force or actively promote Inversine. Accordingly, we do not anticipate any significant increase in Inversine sales. If any of the very limited number of physicians that most often prescribe Inversine were to cease to do so, revenue generated by Inversine sales would likely be substantially less. We have no other commercial products for sale and do not anticipate that we will have any other commercial products for sale for at least the next several years.
Other revenue has consisted primarily of amounts earned for providing research and development services under our two collaboration agreements with Aventis and non-refundable upfront license fees that we received in connection with our first agreement with Aventis and our collaboration agreement with Dr. Falk Pharma. We recognize these non-refundable upfront license fees over the estimated research period of each of these agreements. We received research support payments from Aventis of $1.4 million for the year ended December 31, 2002, $1.3 million for the year ended December 31, 2003 and $100,000 for the three months ended March 31, 2004. We are currently receiving research support payments from Aventis only under our collaboration agreement with Aventis that relates to Aventis compounds. The research term of that agreement ends in December 2004.
In 2003, we were awarded a grant from the National Institute of Standards and Technology through its Advanced Technology Program. The terms of the grant provide for us to receive up to $1.9 million over a three-year period to help fund the development of sophisticated new computer simulation software designed to more accurately predict biological and toxicological effects of drugs. The grant provides for reimbursement of costs that we incur to perform specified work that is designed to meet the objectives of the grant. We recognize grant revenues as we perform the work and incur reimbursable costs.
Research and Development Expense
Since our inception, we have focused our activities on our drug discovery and development programs. We expense research and development expenses as they are incurred. Research and development expenses represented approximately 78% of our total expenses for the year ended December 31, 2001, 81% for the year ended December 31, 2002, 81% for the year ended December 31, 2003 and 82% for the three months ended March 31, 2004.
Research and development expenses include expenses associated with:
We use our employee and infrastructure resources across several projects. Consistent with our focus on the development of a class of drugs with potential uses in multiple indications, many of our costs are not attributable to a specifically identified project. Instead, these costs are directed to broadly applicable research efforts. Accordingly, we do not account for internal research and development costs on a project-by-project basis. As a result, we cannot state precisely the total costs incurred for each of our clinical and preclinical projects on a project-by-project basis.
The following table shows, for the periods presented, total payments that we made to third parties for preclinical study support, clinical supplies and clinical trial services for each of our four clinical-stage product candidates:
| Year ended December 31, | Three months ended March 31, | |||||||||||
Product Candidate | 2001 | 2002 | 2003 | 2004 | ||||||||
| (in thousands) | ||||||||||||
TC-1734 | $ | — | $ | 976 | $ | 3,557 | $ | 1,045 | ||||
TC-5231 | — | 61 | 852 | 781 | ||||||||
TC-2403 | 922 | 2,656 | 1,290 | 419 | ||||||||
TC-2696 | — | — | 893 | 533 | ||||||||
Total: | $ | 922 | $ | 3,693 | $ | 6,592 | $ | 2,778 | ||||
We expect to continue to incur substantial research and development expenses for the foreseeable future. We anticipate that these expenses will increase substantially in 2004 and in subsequent years as we continue to advance our clinical stage product candidates through the development process, to advance additional product candidates into clinical trials and to invest in promising product opportunities in our research programs.
Our expenditures on current and future preclinical and clinical development programs are subject to numerous uncertainties in timing and cost to completion. We test compounds in numerous preclinical studies for safety, toxicology and efficacy. We then conduct clinical trials for those product candidates that we determine to be the most promising. If we do not establish a collaboration covering the development of a particular product candidate, we fund these trials ourselves. As we obtain results from clinical trials, we may elect to discontinue or delay trials for some product candidates in order to focus our resources on more promising product candidates. Completion of clinical trials by us or our collaborators may take several years or more, but the length of time generally varies substantially according to the type, complexity, novelty and intended use of a product candidate. The cost of clinical trials may vary significantly over the life of a project as a result of a variety of factors, including:
We have not received FDA or foreign regulatory marketing approval for any of our product candidates that are in development. In order to achieve marketing approval, the FDA or foreign regulatory agencies must conclude that our or our collaborators’ clinical data establishes the safety and efficacy of the product candidates. Furthermore, our strategy includes entering into collaborations with third parties to participate in the development and commercialization of some of our product candidates. In situations in which third parties have control over the preclinical development or clinical trial process for a product candidate, the estimated completion date is largely under control of that third party and not under our control. We cannot forecast with any degree of certainty which of our product candidates will be subject to future collaborations or how such arrangements would affect our development plans or capital requirements.
As a result of the uncertainties discussed above, we are unable to determine the duration and completion costs of our research and development projects or when and to what extent we will generate revenues from the commercialization and sale of any of our development stage product candidates.
General and Administrative Expense
General and administrative expense consists principally of salaries and other related costs for personnel in executive, finance, accounting, business development, information technology and human resource functions. Other general and administrative expenses include facility costs not otherwise included in research and development expense, patent related costs, and professional fees for consulting, legal and accounting services. We expect that general and administrative expense will increase during 2004 and subsequent years due to increased payroll, expanded infrastructure, increased consulting, legal, accounting and investor relations expenses associated with being a public company and costs incurred to secure collaborations with respect to any of our product candidates.
Cost of Product Sales
Cost of product sales are those costs related directly to the sale of Inversine and are principally comprised of cost of goods sold, FDA product license fees, distribution expenses, product royalty obligations and product liability insurance.
Purchased In-process Research and Development Expense
Purchased in-process research and development expense consists of an allocated portion of the purchase price for the marketing rights to Inversine and related assets that we acquired in August 2002. We expensed the entire allocated portion as of the date of acquisition. We have not recorded purchased in-process research and development expense in any period other than 2002.
Interest and Dividend Income
Interest and dividend income consists of interest and dividends earned on our cash, cash equivalents and short-term investments.
Interest Expense
Interest expense consists of interest incurred to finance equipment, office furniture and fixtures.
Income Taxes
We have incurred net operating losses since our incorporation in 1997 and consequently have not paid federal, state or foreign income taxes in any period. As of December 31, 2003, we had net operating loss carryforwards of approximately $46.1 million. Pursuant to Section 382 of the Internal Revenue Code of 1986, the annual utilization of our net operating loss carryforwards may be limited if we experience a change in ownership of more than 50% within a three-year period. As a result of this offering, we may experience such an ownership change. Accordingly, our net operating loss carryforwards available to offset future federal taxable income arising before such ownership changes may be limited. For financial reporting purposes, we have recorded a valuation allowance to fully offset the deferred tax asset related to these carryforwards because realization of the benefit was uncertain.
Results of Operations
Three Months ended March 31, 2004 and 2003
Revenue. We recognized revenue of $497,000 for the three months ended March 31, 2004 compared to $691,000 for the corresponding period in 2003. The decrease resulted from a decrease of $178,000 in research fee revenue generated from our collaboration agreement with Aventis relating to Aventis compounds, resulting from less research activity under that agreement. The decrease also resulted from a reduction of $123,000 in Inversine sales in the first quarter of 2004 compared to the first quarter of 2003. These decreases in revenue were partially offset by $107,000 of grant revenue recognized in the first quarter of 2004. We did not recognize any grant revenue in the first quarter of 2003. The reduced level of Inversine sales in the first quarter of 2004 reflects a decrease from higher than expected Inversine sales in the first quarter of 2003. We believe that the higher sales in the first quarter of 2003 resulted from a build-up of demand for Inversine in the latter part of 2002 due primarily to a limited commercial supply of the product during the transition period following our acquisition of the rights to Inversine in August 2002. We began selling Inversine in December 2002.
Research and Development Expense. Research and development expense increased by $1.9 million, or 46%, to $6.0 million for the three months ended March 31, 2004, from $4.1 million for the corresponding period in 2003. The increase was primarily attributable to the costs associated with our four product candidates in clinical trials in the first quarter of 2004, compared to only two product candidates in clinical trials in the first
quarter of 2003. During the three months ended March 31, 2004, we estimate that approximately 17% of our total research and development expenses were payments made to third parties in connection with our TC-1734 program for cognitive impairment in the elderly, 13% were payments made to third parties in connection with our TC-5231 program for ADHD, 7% were payments made to third parties in connection with our portion of the costs of our TC-2403 program for ulcerative colitis and 9% were payments made to third parties in connection with our TC-2696 program for pain. We spent the remaining 54% of our total research and development expenses on salaries, benefits, and infrastructure costs for our internal research and development capabilities and on other earlier stage programs and research efforts.
General and Administrative Expense. General and administrative expense increased by $412,000, or 59%, to $1.1 million for the three-month period ended March 31, 2004, from $697,000 for the corresponding period in 2003. This increase resulted from our investment in development of the administrative infrastructure necessary to enable us to expand our operations, to support our development efforts and to fulfill the additional reporting and regulatory requirements applicable to a public company. The increase was principally attributable to increased expenses of $229,000 related to expansion of our business development staff and an increase in spending on business development pursuits, additional patent related expenses and increases in our legal and other professional fees.
Cost of Product Sales. Cost of product sales for the three months ended March 31, 2004 was $181,000 compared to $199,000 for the corresponding period in 2003. All of these costs related to sales of Inversine.
Interest and Dividend Income. Interest and dividend income increased by $106,000 to $230,000 for the three months ended March 31, 2004, from $124,000 for the corresponding period in 2003. The increase was primarily attributable to receiving higher rates of return on our short-term investments.
Interest Expense. Interest expense was $25,000 for the three months ended March 31, 2004 compared to $34,000 for the corresponding period in 2003. The decrease was attributable to a lower average outstanding principal balance on our equipment financing facility. In April 2004, we financed an additional $1.0 million of eligible fixed assets under this facility. We expect our interest expense to increase as a result of this additional indebtedness.
Years ended December 31, 2003 and 2002
Revenue. Revenue increased to $2.5 million for the year ended December 31, 2003 from $2.3 million for 2002. The increase resulted primarily from the inclusion of a full year of Inversine sales in 2003 of $815,000, compared to a partial year of Inversine sales in 2002 of $243,000. We began selling Inversine in December 2002. License fee revenues decreased to $270,000 in 2003 from $635,000 in 2003 primarily as a result of revisions to the estimated research terms used as the basis for revenue recognition of the non-refundable upfront license fees that we received in our collaborations with Aventis and Dr. Falk Pharma.
Research and Development Expense. Research and development expense increased by $2.0 million, or 12%, to $18.2 million for the year ended December 31, 2003, from $16.2 million for 2002. The increase resulted principally from increased spending of $2.9 million on our later stage clinical programs and increased personnel and infrastructure costs of $539,000 associated with the expansion of our internal clinical development and regulatory affairs capabilities. This was offset in part by a decrease of $1.8 million resulting from the conclusion in 2002 of a program to screen several of our preclinical candidates to select those to advance into clinical trials. During the year ended December 31, 2003, we estimate that approximately 20% of our total research and development expenses were payments made to third parties in connection with our TC-1734 program for cognitive impairment in the elderly, 5% were payments made to third parties in connection with our TC-5231 program for ADHD, 7% were payments made to third parties in connection with our portion of the costs of our TC-2403 program for ulcerative colitis and 5% were payments made to third parties in connection with our TC-2696 program for pain. We spent the remaining 63% of our total research and development expense on salaries, benefits, and infrastructure costs associated with our internal research and development capabilities and on other earlier stage programs and research efforts.
General and Administrative Expense. General and administrative expense decreased by $536,000, or 13%, to $3.6 million for the year ended December 31, 2003, from $4.1 million for 2002. This decrease resulted principally from a decrease of $330,000 from the costs incurred in 2002 associated with our relocation to a new leased facility, severance costs of $257,000 and a reduction in patent related costs in 2003 compared to 2002. In 2003 we increased our spending on the development of the administrative infrastructure necessary to enable us to expand our operations, support our development efforts and facilitate the additional reporting and regulatory requirements related to becoming a public company.
Cost of Product Sales. Cost of product sales increased to $743,000 for the year ended December 31, 2003, from $244,000 for 2002. The increase in cost of product sales for 2003 resulted primarily from increased sales of Inversine.
Interest and Dividend Income. Interest and dividend income increased by $703,000 to $791,000 for the year ended December 31, 2003, from $88,000 for 2002. The increase resulted from substantially higher average cash balances during 2003 as a result of the funds we received from the sale of shares of our series C convertible preferred stock. We raised $45.5 million in this financing in November 2002 and $13.8 million in March 2003.
Interest Expense. Interest expense was $123,000 for the year ended December 31, 2003 compared to $103,000 for 2002.
Years ended December 31, 2002 and December 31, 2001
Revenue. Revenue increased to $2.3 million for the year ended December 31, 2002 from $1.7 million for 2001. The increase resulted principally from an increase of $289,000 in research fee revenue generated from our Aventis collaboration as a result of higher levels of research activity and Inversine sales of $243,000 in December 2002 when we began selling the product.
Research and Development Expense. Research and development expense increased by $8.0 million, or 98%, to $16.2 million for the year ended December 31, 2002, from $8.2 million for 2001. The increase resulted principally from $1.8 million in costs attributable to a program to screen several of our preclinical candidates to ascertain those to advance into clinical trials, increased spending of $1.7 million to fund our portion of the costs of third-party services in connection with the TC-2403 program for ulcerative colitis, and $1.0 million for preclinical studies, toxicology and regulatory expenses directed towards advancing TC-1734 into clinical trials. We also incurred increased salary and benefits costs as we expanded our product development capabilities with the hiring of experienced key personnel. In addition, we incurred increased infrastructure costs in connection with our relocation in March 2002 to a new leased facility which includes expanded lab and research space. This also resulted in higher depreciation charges associated with the new equipment and furnishings that we acquired.
General and Administrative Expense. General and administrative expense increased by $1.8 million, or 80%, to $4.1 million for the year ended December 31, 2002, from $2.3 million for 2001. This increase resulted from our investment in the administrative infrastructure necessary to enable us to expand our operations, to support our development efforts and to attract and hire members of executive management. In the first quarter of 2002, we relocated our operations to a new leased facility. The costs of the relocation were approximately $330,000, and the new facility increased our occupancy related costs in 2002 by $661,000 compared to 2001. Salary and benefits expenses increased by $498,000, which included the costs associated with the addition of a chief financial officer in the first quarter of 2002 and severance costs. In 2002 we also increased our spending on professional fees related to business development, legal and public relations services.
Cost of Product Sales. Cost of product sales for the year ended December 31, 2002 was $244,000. All of these sales related to Inversine, which we began selling in December 2002. We had no cost of product sales expenses for the year ended December 31, 2001.
Interest and Dividend Income. Interest and dividend income decreased by $1.4 million to $88,000 for the year ended December 31, 2002. The decrease resulted from substantially lower average cash balances during 2002 compared to 2001 and lower interest rates during the year.
Interest Expense. Interest expense was $103,000 for the year ended December 31, 2002. The interest expense related to outstanding indebtedness on a loan facility established to finance equipment and other fixed assets. We had no interest expense for the year ended December 31, 2001.
Liquidity and Capital Resources
Sources of Liquidity
Since we became an independent company in 2000, we have financed our operations and internal growth primarily through private placements of convertible preferred stock. We have derived aggregate net proceeds of $88.4 million from these private placements. We have received additional funding from upfront license fees and payments for research and development services under collaboration agreements, equipment and building lease incentive financing, government grants and interest income. As of March 31, 2004, we have received $9.9 million under our collaboration agreements. We are currently receiving research support payments under only one of these agreements, and the research term of that agreement ends in December 2004. We began generating revenues from product sales of Inversine in December 2002. To date, Inversine sales have not been a significant source of cash and we do not expect them to be a significant source in the future.
Our cash, cash equivalents and short-term investments were $36.8 million as of March 31, 2004, $43.0 million as of December 31, 2003 and $49.4 million as of December 31, 2002.
Cash Flows
Net cash used for operating activities was $5.8 million for the three months ended March 31, 2004, reflecting a net loss of $6.6 million offset primarily by research support payments. Net cash used for operating activities was $19.3 million for the year ended December 31, 2003, primarily reflecting a net loss occurring for this period of $19.4 million. Net cash used for operating activities was $17.1 million for the year ended December 31, 2002, reflecting a net loss of $21.1 million partially offset by non-cash charges for acquired in-process research and development of $2.7 million and depreciation and amortization of $1.0 million. Accounts payable and accrued expenses increased by $1.5 million as of December 31, 2002, compared to December 31, 2001 primarily as a result of an increase in outsourced development activities with contract research organizations.
Net cash used in investing activities was $137,000 for the three months ended March 31, 2004, and $545,000 for the year ended December 31, 2003. These amounts exclude cash flows from the purchase and sale of investments and were primarily to purchase equipment for use in expanding our internal research and development activities. Investing activities for the year ended December 31, 2002, exclusive of cash flows from the purchase and sale of investments, included the use of $1.3 million for the purchase of equipment and furniture, the use of $3.5 million for the purchase of the marketing rights to Inversine and related assets from Layton Bioscience, Inc. and the receipt of a $2.0 million rent incentive from the owner of our facilities in connection with our entering into a lease with a minimum five-year term.
Net cash used in financing activities was $49,000 for the three months ended March 31, 2004. As of March 31, 2004, we had borrowing capacity of $2.0 million available under our equipment financing loan facility. We borrowed $1.0 million under the facility in April 2004 to finance equipment that we had previously purchased. Net cash provided by financing activities for the year ended December 31, 2003 was $13.4 million and consisted principally of net proceeds of $13.8 million from the issuance of shares of our series C convertible preferred stock, and proceeds of $239,000 received in connection with the purchase of our common stock upon the exercise of stock options, partially offset by $637,000 of principal repayments on equipment financing. Net cash provided by financing activities for the year ended December 31, 2002 was $48.2 million and consisted principally of net proceeds of $45.5 million from the issuance of shares of our series C convertible preferred
stock and proceeds of $3.0 million from long-term debt, comprised of a $2.5 million equipment financing loan repayable over 48 months and a $500,000 incentive loan from the City of Winston-Salem which requires no repayments and carries no interest charges for the initial five years, partially offset by $325,000 of equipment financing principal repayments.
In May 2002, we borrowed $2.5 million from R.J. Reynolds Tobacco Holdings, Inc. to finance equipment and other fixed assets that we had previously purchased. The borrowing bears a fixed interest rate of 6.6%, is payable in 48 equal monthly installments and matures in May 2006. In January 2004, we amended the terms of our loan facility to permit us to borrow up to an additional $2.0 million in 2004 in up to three separate borrowings. Each borrowing would bear a fixed interest rate equal to a theoretical four-year U.S. Treasury Rate on the disbursement date plus 3.5%, be payable in 48 equal monthly installments and be secured by specified tangible fixed assets determined sufficient by the lender at the time of disbursement. In April 2004, we borrowed $1.0 million under the amended loan facility to finance equipment. This borrowing bears a fixed interest rate of 5.9%, is payable in 48 equal monthly installments and matures in April 2008. All borrowings under the loan facility are secured by specified tangible fixed assets. As of May 1, 2004, the outstanding principal balance under the loan facility was $2.3 million.
Funding Requirements
We have incurred significant losses since our inception. As of March 31, 2004, we had an accumulated deficit of $82.8 million. We expect to continue to incur substantial operating losses for the foreseeable future. Our future capital requirements are difficult to forecast and will depend on many factors, including:
Although we currently have not specifically identified any material commitments for capital expenditures, we anticipate that implementing our strategy will require substantial increases in our capital expenditures and other capital commitments. Considering our current spending levels on our existing programs and a likely future increase in activity on these programs, estimates of future staff growth and related spending and the estimated capital requirements of potential future acquisitions, we expect that our existing capital resources, together with the net proceeds of this offering, will be sufficient to fund our operations through June 2006.
We do not expect to generate sufficient cash from our operations to sustain our business for the foreseeable future. Unless we are able to do so, we expect our continuing operating losses to result in increases in our cash used in operations over the next several quarters and years. To the extent our capital resources are insufficient to meet future capital requirements, we will need to finance future cash needs through public or private equity offerings, debt financings or corporate collaboration and licensing arrangements. Additional equity or debt financing, or corporate collaboration and licensing arrangements, may not be available on acceptable terms, if at all. If adequate funds are not available, we may be required to delay, reduce the scope of or eliminate our research and development programs, reduce our planned commercialization efforts, or obtain funds through arrangements with collaborators or others that may require us to relinquish rights to certain drug candidates that we might otherwise seek to develop or commercialize independently. Additionally, any future equity funding may dilute the ownership of our equity investors.
We cannot estimate the completion dates and costs of our current internal research and development programs due to inherent uncertainties in outcomes of clinical trials and regulatory approvals of our product candidates. We cannot be certain that we will be able to successfully complete our research and development projects or successfully find collaboration or distribution partners for our product candidates. Our failure to complete our research and development projects could have a material adverse effect on our financial position or results of operations.
To date, inflation has not had a material effect on our business.
Contractual Obligations
The following table summarizes our significant contractual obligations and commercial commitments as of December 31, 2003:
Payments due by period (in thousands) | |||||||||||||||
Contractual Obligations | Total | 2004 | 2005-2007 | 2008-2009 | After 2009 | ||||||||||
Long-term debt | $ | 2,038 | $ | 576 | $ | 962 | $ | 163 | $ | 337 | |||||
Operating leases | 5,216 | 1,456 | 2,911 | 849 | — | ||||||||||
Other contractual obligations | 4,220 | 3,984 | 230 | 6 | — | ||||||||||
Total | $ | 11,474 | $ | 6,016 | $ | 4,103 | $ | 1,018 | $ | 337 | |||||
The amounts reflected in the above table do not include contingent payments for milestones and royalties on potential product sales that we may become obligated to make under technology license agreements to which we are a party.
Quantitative and Qualitative Disclosures about Market Risk
The primary objective of our investment activities is to preserve our capital to fund operations. We also seek to maximize income from our investments without assuming significant risk. To achieve our objectives, we maintain a portfolio of cash equivalents and short-term investments in a variety of securities of high credit quality. As of March 31, 2004, we had cash, cash equivalents and short-term investments of $36.8 million consisting of cash and highly liquid investments deposited in, and invested through, highly rated financial institutions in the United States. A portion of our investments may be subject to interest rate risk and could fall in value if market interest rates increase. However, because our investments are short-term in duration, we believe that our exposure to interest rate risk is not significant and a 1% movement in market interest rates would not have a significant impact on the total value of our portfolio. We actively monitor changes in interest rates. We adjusted our portfolio in April 2004 to eliminate positions in investments that were subject to potentially significant interest rate risks.
We contract for the conduct of certain of our clinical trials and other research and development and manufacturing activities with contract research organizations, investigational sites and manufacturers in Europe. We may be subject to exposure to fluctuations in foreign exchange rates in connection with these agreements. We do not hedge our foreign currency exposures. We have not used derivative financial instruments for speculation or trading purposes.
Recent Accounting Pronouncements
During 2003, the Financial Accounting Standards Board, or the FASB, issued Interpretation No. 46,Consolidation of Variable Interest Entities, or FIN 46, which is an interpretation of Accounting Research Bulletin No. 51,Consolidated Financial Statements. FIN 46 requires that, if an entity has a controlling interest in a variable interest entity, the assets, liabilities and results of activities of the variable interest entity should be included in the consolidated financial statements of the entity. FIN 46 is effective immediately for all new variable interest entities created or acquired after January 31, 2003. For variable interest entities created or acquired prior to February 1, 2003, the provisions of FIN 46 must be applied in the first interim or annual period beginning after June 15, 2003. We implemented the provisions of FIN 46 for our financial statements for the year ended December 31, 2003. We have no investment in or contractual relationship or other business relationship with a variable interest entity. Therefore, the adoption of FIN 46 did not affect our financial position or results of operations.
In May 2003, the FASB issued SFAS No. 150,Accounting for Certain Financial Instruments with Characteristics of both Liabilities and Equity. SFAS 150 establishes how a company classifies and measures certain financial instruments with characteristics of both liabilities and equity, including redeemable convertible preferred stock. SFAS 150 is effective for financial instruments entered into or modified after May 31, 2003 and otherwise effective at the beginning of the interim period commencing July 1, 2003, except for mandatory redeemable financial instruments of nonpublic companies. The adoption of SFAS 150 did not affect our financial position or results of operations.
Overview
We are a biopharmaceutical company engaged in the design, discovery and development of a new class of drugs to treat multiple diseases and disorders by selectively targeting neuronal nicotinic acetylcholine receptors, or NNRs. NNRs are found on nerve cells throughout the human nervous system and serve as key regulators of nervous system activity. We trace our scientific lineage to a research program initiated by R.J. Reynolds Tobacco Company in 1982 to study the activity and effects of nicotine, a compound that interacts non-selectively with all nicotinic acetylcholine receptors. Since that time, we have developed a deep understanding of the biological characteristics and functions of NNRs and have learned that compounds that interact with NNRs have the potential to achieve therapeutic benefits by modulating their activity. We have built an extensive patent estate covering the structure or therapeutic use of small molecules designed to regulate the nervous system by selectively modulating specific NNR subtypes. We are developing drugs that target NNRs to treat diseases and disorders of the nervous system.
Our product development pipeline consists of four product candidates in clinical development and multiple ongoing preclinical programs for target indications in which we believe that NNRs can be exploited for therapeutic benefit. Our pipeline includes:
Our product candidate for ADHD, TC-5231, is a low-dose reformulation of mecamylamine hydrochloride. Mecamylamine hydrochloride is the active ingredient in our commercial product Inversine.
We believe that Inversine is the only FDA-approved product designed to target an NNR. Inversine is approved for the management of moderately severe to severe essential hypertension, a high blood pressure disorder. Our market research suggests, however, that Inversine is prescribed predominantly for the treatment of Tourette’s syndrome and other neuropsychiatric disorders at lower doses than indicated for the treatment of hypertension. Because we recognized the potential for mecamylamine hydrochloride as a treatment for ADHD, we acquired patent rights covering its use for neuropsychiatric disorders, as well as the marketing rights to Inversine, in 2002.
We develop product candidates using our proprietary databases and computer-based molecular design technologies, which we refer to collectively as Pentad. Critical to the power of Pentad are extensive biological data for a library of diverse compounds that we have developed and gathered over the last 20 years. Together with our proprietary target validation assays and novel screening methods, Pentad enables us to efficiently identify, prioritize, characterize and optimize novel compounds designed to selectively target specific NNR subtypes in an effort to achieve desired therapeutic results while limiting or potentially eliminating side effects.
We have entered into two collaboration agreements with Aventis Pharma SA. Our first collaboration agreement with Aventis covers the research, development and commercialization of specified Targacept compounds for the treatment or prevention of Alzheimer’s disease. Our second collaboration with Aventis covers the research, development and commercialization of Aventis compounds for the treatment or prevention of Alzheimer’s disease and other diseases of the central nervous system. In addition, we have entered into a
collaboration agreement with Dr. Falk Pharma GmbH, a German company with a focus on treatments for gastrointestinal diseases, which covers the research, development and commercialization of our product candidate TC-2403 for the treatment or prevention of ulcerative colitis.
Role of NNRs in the Nervous System
The human nervous system is a massive communications network that sends and receives information throughout the body via billions of specialized nerve cells known as neurons. Neurons continually gather information about the body’s internal and external environment and send signals to the brain. These signals pass from one neuron to another when electrical impulses of a communicating neuron are converted into chemicals called neurotransmitters. This occurs at the gap between the communicating neuron and the receiving neuron known as the synapse. When released by a communicating neuron, neurotransmitters bind to specialized proteins known as receptors located across the synapse on the receiving neuron to enable the signal to continue. The major neurotransmitters in the brain are dopamine, serotonin, norepinephrine, glutamate, gamma-aminobutyric acid, or GABA, and acetylcholine.
NNRs are a class of receptors found in the nervous system that play a critical role in modulating the release of neurotransmitters to regulate nervous system activity. When the neurotransmitter acetylcholine is released from a nearby neuron, called an interneuron, and binds to an NNR on the communicating neuron, the flow of neurotransmitters from the communicating neuron to the receiving neuron is adjusted by the NNR. This action, known as neuromodulation, results in a greater release of neurotransmitters across the synapse when the nervous system is understimulated and a lesser release of neurotransmitters across the synapse when the nervous system is overstimulated. As neuromodulators, NNRs serve as the nervous system’s self-adjusting “volume knob.”
The nervous system will not operate properly if the relative levels of key neurotransmitters in the brain are not maintained in a normal balance. A disruption in this balance can cause many common nervous system diseases and disorders. We believe that compounds that target NNRs to trigger their activity can be used to treat these diseases and disorders.
The following diagrams illustrate the role of NNRs in neuromodulation. In the illustration on the left, the release of a limited amount of acetylcholine from the interneuron causes the NNRs to release a limited amount of neurotransmitters across the synapse. In the illustration on the right, the release of more acetylcholine from the interneuron causes the NNRs to release a greater amount of neurotransmitters.
NNRs are comprised of five protein subunits that are arranged like staves of a barrel around a central pore. Each different combination of five subunits represents an NNR subtype. There are several subtypes, each of which is identified by Greek letters. Scientific evidence has established that individual NNR subtypes have
particular functions in the body that are relevant to a number of debilitating diseases and disorders, as set forth below.
|
|
| ||
|
| |||
|
| |||
| ||||
|
| |||
|
| |||
Our scientists and their former colleagues at R.J. Reynolds Tobacco Company have played a prominent role in the growth of knowledge about NNRs, as well as the effects of compounds that mimic the action of acetylcholine and interact with different NNR subtypes. For example, we believe that nicotine’s well-documented abilities to enhance attention, learning and memory result primarily from its interaction with thea4ß2 NNR and with thea7 NNR in the brain. Many published studies evaluating the effects of nicotine in humans and animals, as well as published studies showing the prevalence of diseases such as Alzheimer’s disease and Parkinson’s disease in non-smokers as compared to smokers, suggest the therapeutic effects of compounds such as nicotine that interact with NNRs. However, despite their positive effects, these compounds have historically not been desirable as therapeutic products because they have not been sufficiently selective. This means that these compounds interact not only with NNRs, but also with nicotinic acetylcholine receptors in the muscles and ganglia that are associated with adverse effects such as increased heart rate, high blood pressure, irregular heartbeat, nausea, vomiting and a dangerous slowing of breathing known as respiratory depression.
Based on our years of focus on NNRs and the expertise we have built over that time, we are developing product candidates that are designed to interact selectively with specific NNR subtypes to promote therapeutic effects while limiting or potentially eliminating side effects.
Our Business Strategy
Our goal is to become a leader in the discovery, development and commercialization of novel drugs that selectively target NNRs in order to treat diseases and disorders where there is significant medical need and commercial potential. To achieve this goal, we are pursuing the following strategies:
Opportunities in Our Target Indications
Because NNRs are so widespread in the body, we believe that there are a number of areas in which compounds that target NNRs could provide a therapeutic benefit, including:
Our primary product development focus is on diseases and disorders of the CNS, which represent a major segment of the global healthcare environment. The Reuters Business Insight Healthcare Report estimated the global CNS pharmaceutical market to be $58 billion in 2002. Four of the top ten selling drugs in the world in 2002, Pfizer’s Celebrex and Zoloft, Eli Lilly’s Zyprexa and GlaxoSmithKline’s Paxil/Seroxat, treat diseases and disorders of the CNS.However, despite their commercial success, many current CNS drugs are only moderately effective or are accompanied by significant side effects or other drawbacks. Accordingly, we believe that substantial opportunities exist for new therapies that address CNS disorders.
We are currently conducting clinical trials for the use of our product candidates in the treatment of cognitive impairment in the elderly, ADHD, ulcerative colitis and pain.
Cognitive Impairment in the Elderly
Cognition refers to a collection of mental processes that enable the acquisition, storage, retrieval and use of information. Cognitive functions such as attention, learning and memory underlie fundamental day-to-day activities. Impairment of these functions can impact an individual’s ability to function effectively and, in severe cases, to maintain quality of life. Cognitive impairment is particularly prominent in the growing elderly population. The most severe form of cognitive impairment is dementia, which can render a person unable to care for himself or herself. The most common form of dementia in the elderly is Alzheimer’s disease. Cognitive impairment without dementia ranges in severity from age associated memory impairment, commonly known as AAMI, to mild cognitive impairment, commonly known as MCI.
The term AAMI describes a common condition characterized by gradual memory loss or other cognitive impairment that generally occurs with normal aging. A person who is at least 50 years of age and scores at least one standard deviation below the mean established for young adults on a standardized memory test without evidence of dementia, neurological illness or other medical cause may be classified with AAMI. The term AAMI is not currently listed inDiagnostic and Statistical Manual of Mental Disorders, Fourth Edition, or DSM-IV, the manual published by the American Psychiatric Association to establish diagnostic criteria. However, DSM-IV does list the term “age related cognitive decline,” which is often used by the medical community interchangeably with AAMI, as an “objectively identified decline in cognitive functioning consequent to the aging process that is within normal limits given the person’s age.” A number of published clinical trials have been conducted in AAMI or similarly characterized conditions. Although estimates of the prevalence of AAMI in the elderly vary greatly because of varying methodologies and definitions of AAMI, one published study indicates that AAMI may affect as many as 38% of people over age 65. Based on a 2000 report of the Federal Interagency Forum on Aging-Related Statistics, this represents over 13 million people in the United States alone. The Federal Interagency Forum report projects that the number of people in the United States age 65 or older will double by 2030.
MCI is typically marked by memory problems that are more severe than in AAMI, but without other characteristics that would result in a diagnosis of dementia. A person who scores at least one and one-half standard deviations below the mean established for his or her age-matched peers on a standardized memory test may be classified with MCI. Datamonitor, a provider of business information for the pharmaceutical and other industries, estimates that MCI affects nearly 15 million people in the world’s seven major pharmaceutical markets, which are the United States, France, Germany, Italy, Spain, United Kingdom and Japan, including approximately 5 million in the United States. Researchers have estimated that 10% to 15% of persons with MCI are diagnosed with Alzheimer’s disease each year. Like AAMI, MCI is not currently listed in DSM-IV. However, there is ongoing discussion in the medical community as to its status as a distinct clinical classification. The FDA has acknowledged reviewing clinical trial protocols for MCI, and we are aware of clinical trials that have been conducted for MCI.
There are currently no products approved by the FDA for the treatment of AAMI or MCI. We believe that, when AAMI or MCI is treated at all, physicians typically prescribe the same drugs that are used to treat Alzheimer’s disease. These consist primarily of a class of drugs known as acetylcholinesterase inhibitors such as Aricept, Reminyl and Exelon. We believe that these drugs have limitations in that only about half of Alzheimer’s disease patients who take them show symptomatic improvement and they do not substantially delay the progressive deterioration and death of cells in the brain that can lead to more severe impairment and debilitation.
Attention Deficit Hyperactivity Disorder
ADHD is the most commonly diagnosed childhood behavioral disorder. ADHD is characterized by varying degrees of developmentally inappropriate inattention, hyperactivity and impulsivity. Children with ADHD may have difficulty functioning at home, at school and in peer relationships. The disorder has been linked to long-term adverse effects on academic performance, success in the workplace and social and emotional development. According to a published study, two-thirds of children diagnosed with ADHD will continue to show attention deficit symptoms into adult life, although the hyperactivity typically seen in children is diminished.
Datamonitor estimates that ADHD affects approximately 8 million youths in the world’s seven major pharmaceutical markets, including 4.8 million in the United States. According to Datamonitor, the worldwide market for ADHD drugs was approximately $1.3 billion in 2002.
We believe that the currently available treatments for ADHD have significant drawbacks or limitations. In particular, many current treatments such as Ritalin and Adderall are stimulants. Parents and physicians are often reluctant to administer stimulants to children because of concerns over addiction, abuse and side effects such as weight loss. One non-stimulant, Strattera, has been approved by the FDA to treat ADHD. However, it can cause side effects such as constipation, nausea and vomiting and may not be as effective in treating ADHD as stimulants.
Ulcerative Colitis
Ulcerative colitis is a chronic form of inflammatory bowel disease that is characterized by inflammation of the lining of the colon. The various types of ulcerative colitis are classified according to the location and extent of the inflammation, including left-sided colitis, which involves inflammation that extends up the left or descending colon, and pancolitis, which affects the entire colon. We believe that left-sided colitis represents about 40% to 60% of all cases of ulcerative colitis. The majority of ulcerative colitis patients suffer from repeated acute episodes.
Datamonitor estimates that ulcerative colitis affects approximately 870,000 people in the world’s seven major pharmaceutical markets, including approximately 320,000 people in the United States.
Ulcerative colitis is typically treated with a variety of forms of the drug commonly known as 5-ASA, such as Asacol. More severe cases of ulcerative colitis are treated with steroids. We believe that there are limitations to these treatments, as the National Institute of Diabetes and Digestive and Kidney Diseases estimates that 25% to 40% of all ulcerative colitis patients ultimately require surgery to remove all or a portion of the colon.
Pain
There are two general categories of pain, nociceptive and neuropathic. Pain occurs when base nerve endings known as pain receptors are activated and a pain signal is transmitted through the nervous system to the brain. With nociceptive pain, the pain signal starts with damage to tissue and is typically accompanied by inflammation. Nociceptive pain can be either acute, such as that experienced following surgery, or chronic. With neuropathic pain, the pain signal results from inflammation of the peripheral nerves or other injury to the nervous system itself. A common form of neuropathic pain is sciatica, which is characterized by compression of the sciatic nerve resulting in leg and back pain. Neuropathic pain also arises from diabetes, cancer and exposure to chemotherapy or radiation.
Datamonitor estimates that approximately 75 million people in the world’s seven major pharmaceutical markets suffer annually from acute nociceptive pain following a surgical procedure. Pharmaprojects, a healthcare publication, estimates that 26 million people worldwide suffer annually from some form of neuropathic pain. According to Datamonitor, the worldwide market for pain therapies was approximately $17 billion in 2001.
There is no single product available to treat all types of pain, and we believe that there are limitations to the existing treatments for each individual type of pain. Acute pain is typically treated with a class of drugs known as opioids such as morphine. Prolonged use of opioids, however, may result in a tolerance to the drug, ultimately making it ineffective. In addition, the use of opioids can result in addiction and abuse. As a result, physicians are often reluctant to prescribe opioids for an extended period of time or at all. Chronic pain is most often treated with a class of drugs known as non-steroidal anti-inflammatory drugs. These drugs are often not sufficiently effective. In a nationwide survey of over 1,000 adults conducted in the United States in August 2003, only 58% of chronic pain sufferers rated their prescription medications as very or somewhat effective. No class of drugs, including opioids and non-steroidal anti-inflammatory drugs, has demonstrated consistent effectiveness in treating neuropathic pain.
Our Product Development Pipeline
Our product development pipeline consists of four product candidates in clinical development and multiple ongoing preclinical programs for target indications in which we believe that NNRs can be exploited for therapeutic benefit. We also have one marketed product, Inversine, that is approved in the United States for the management of moderately severe to severe essential hypertension and that we believe is prescribed predominantly for the treatment of Tourette’s syndrome and other neuropsychiatric disorders. Except for Inversine, neither the FDA nor any foreign regulatory authority has approved any of our product candidates for marketing.
Our product candidates in clinical development are summarized in the table below.
|
|
|
|
| ||||
| ||||||||
|
| |||||||
|
| |||||||
| ||||||||
|
|
| ||||||
TC-1734
TC-1734 is a novel small molecule that we are developing as an oral treatment for a range of cognitive impairment in the elderly that includes AAMI and MCI. We are currently conducting a Phase II clinical trial of TC-1734 in 56 elderly persons classified with AAMI and have completed two arms of the trial. We are also currently conducting a Phase II clinical trial in 40 elderly persons classified with MCI. We have previously evaluated TC-1734 in 84 healthy volunteers in four Phase I clinical trials.
While the exact causes of AAMI and MCI are unknown, the aging process is generally accompanied by a decline of cognitive function linked to a progressive deterioration and death of cells in the brain. This is known as neurodegeneration. If neurodegeneration reaches a more advanced stage, a person becomes debilitated and unable to care for himself or herself. In addition, published studies have shown, among other things, that patients with Alzheimer’s disease have deficient levels of acetylcholine and other key neurotransmitters in the brain. We believe that these neurotransmitter levels are also deficient, perhaps to a lesser degree, in persons with AAMI and MCI.
Published studies with humans have shown a reduced number ofa4ß2 NNRs in persons with dementia, suggesting the involvement ofa4ß2 in cognition. In our preclinical animal studies, TC-1734 triggered activity ofa4ß2, enhanced the release of acetylcholine, enhanced memory and showed meaningful separation between the doses at which positive effects on memory and side effects were first seen. In two preclinical in vitro studies that we conducted, TC-1734 protected neuronal cells from deterioration and death, a process known as neuroprotection. Based on these results and published studies that link neuroprotection to exposure to nicotine, a non-selective activator of all NNRs with particularly strong activity ata4ß2, we believe that TC-1734 has the potential to prevent or delay neurodegeneration.
We are also evaluating TC-1734 for potential additional clinical development for indications marked by cognitive impairment that are not specific to the elderly, such as ADHD, schizophrenia and various forms of dementia. We are restricted from pursuing development of TC-1734 for the treatment or prevention of Alzheimer’s disease while our collaborative agreement with Aventis Pharma SA relating to the development of our compound TC-4959 remains in effect.
Clinical Development of TC-1734
Phase I Clinical Trials. During 2003, we completed four Phase I clinical trials of TC-1734 in 84 healthy volunteers in which the compound was well tolerated. The results of these trials are summarized below.
Phase II Clinical Trials. We are currently conducting a double blind, placebo-controlled Phase II clinical trial of TC-1734 in 56 elderly volunteers classified with AAMI. We are conducting the trial at multiple sites in the United Kingdom under a clinical trial exemption, the United Kingdom equivalent to an IND. The primary objective of this trial is to assess the safety and tolerability of TC-1734 in elderly subjects compared to placebo. The secondary objectives of this trial are to assess the efficacy of TC-1734 in improving cognitive function and changes in mood state.
We are also currently conducting a second double blind, placebo-controlled clinical trial of TC-1734 in 40 elderly volunteers classified with MCI. This trial has similar objectives to the AAMI trial and is also being conducted in the United Kingdom.
In the AAMI trial, the subjects have been divided into three dose groups – 20 subjects are in the 50mg group, 20 subjects are in the 100mg group and 16 subjects are in the 150mg group. In the MCI trial, the subjects have been divided into 50mg and 100mg dose groups of 20 subjects each. In both trials, each subject is initially dosed either with the applicable dose of TC-1734 or a placebo daily over a three-week period. Then, after a two-week period without being dosed, each subject is changed to be dosed with either a placebo or TC-1734, as the case may be, daily for another three-week period. Each subject takes TC-1734 or a placebo before eating on the day of dosing. During the trials, routine safety measures are recorded and pharmacokinetic assessments are made for each subject. In addition, subjects are assessed for changes in cognitive function and mood state before and after dosing on the first day of the three-week dosing period and then again on the last day of the dosing period. The trials are double blind, meaning that neither the subjects nor the clinical investigators know during the trials which subjects are receiving TC-1734 and which are receiving the placebo.
In the trials, we are testing subjects for changes in cognitive function using a computer-based test battery developed by CDR Ltd. This test battery includes measures of attention, speed of cognitive processes and memory that assess the ability to react to stimuli, recognize words and pictures and recall words. CDR has indicated that its battery has been used to assess cognitive performance in over 500 clinical trials worldwide.
We also used the CDR test battery in our Phase I clinical trials of TC-1734. We selected it because of its comprehensive measures and CDR’s extensive database of test results in unimpaired persons that enable assessment of statistical significance.
As of April 30, 2004, we have received data from the 40 subjects in the 50mg and 100mg arms of the AAMI trial. At each of these doses, TC-1734 was well tolerated, with no serious adverse events reported. In addition, subjects in the 50mg dose group that received TC-1734 showed improvements in attention, speed of cognitive processes and memory as compared to subjects receiving placebo. These findings were consistent with the effects seen in our Phase I trials. The positive effects that we observed in the 50mg dose group were less pronounced than in the 100mg dose group, which is consistent with the dose-related effects on cognition that we observed in our preclinical animal studies. In the 150mg dose group, three out of eight subjects treated with TC-1734 experienced side effects such as headache, lightheadedness, dizziness and vomiting and dropped out of the trial. In light of these effects, we ceased dosing new subjects at 150mg. The results of the AAMI trial suggest that TC-1734 is well tolerated at a dose range of up to 100mg, that 150mg is the maximum tolerated dose of TC-1734 for this trial design and that the compound had positive effects on cognition at a dose within the tolerated range.
To generate additional data related to the tolerability of TC-1734, we are currently enrolling eight additional elderly volunteers classified with AAMI to be tested at a dose of 150mg, after having eaten, using the same trial design. This will enable us to assess the impact of food on the tolerability of TC-1734 by comparing it in volunteers dosed at 150mg who have eaten and in volunteers dosed at 150mg who have not eaten. To further assess the tolerability of TC-1734, we are also enrolling additional elderly volunteers classified with AAMI to be tested at a dose of 125mg without having eaten using the same trial design.
Plans for Future Development. We plan to meet with the FDA regarding whether cognitive impairment in the elderly is an indication for which the FDA would approve a drug and, if so, the additional trials that we would need to perform to support an application for approval of TC-1734 for the treatment of this indication. Following the discussions with the FDA, we expect to evaluate the specific target indication or indications for continued clinical development of TC-1734. Subject to the results of our discussions with the FDA, we anticipate commencing a separate Phase II clinical trial designed to evaluate the efficacy of TC-1734 in the fourth quarter of 2004.
TC-5231
TC-5231 is a small molecule that we are developing as an oral treatment for ADHD. TC-5231 is a low-dose reformulation of mecamylamine hydrochloride, the active ingredient in our FDA-approved product, Inversine. Inversine is approved in the United States for the management of moderately severe to severe essential hypertension at average daily doses of 25mg. Our market research suggests, however, that Inversine is prescribed predominantly for the treatment of Tourette’s syndrome and other neuropsychiatric disorders at doses ranging from 2.5mg to 7.5mg. We have reformulated mecamylamine hydrochloride as TC-5231 in a liquid gel cap. We are evaluating this product candidate in doses between 0.2mg and 1.0mg in two Phase II clinical trials, one in ADHD in children and adolescents and one in ADHD in young adults.
In published studies conducted by third parties, mecamylamine hydrochloride at low doses:
Although the means by which mecamylamine hydrochloride acts to cause these effects is not known, scientists have suggested that it may prevent interference with the activity of thea4ß2 NNR, which regulates neurotransmitters involved in attentional processes such as dopamine, norepinephrine and serotonin, or it may blocka4ß2 from releasing the neurotransmitter GABA, which inhibits attentional processes.
The results of human and animal studies in which nicotine was shown to improve cognition also suggest the potential for drugs that target NNRs to treat ADHD. For example, in a placebo-controlled study with 34 adult non-smokers conducted by researchers at Duke University, nicotine was as effective in improving symptoms of ADHD as the active ingredient in Ritalin, a stimulant that is commonly prescribed for the treatment of ADHD. At low doses, mecamylamine hydrochloride shows pharmacological effects that are similar to those of nicotine.
Based on these study results, we believe that TC-5231 may have a therapeutic effect in the treatment of ADHD. Moreover, decades of adult use of Inversine at substantially higher doses than TC-5231 suggest that the compound may not exhibit the side effects characteristic of existing treatments for ADHD.
Clinical Development of TC-5231
Phase II Trial in Children and Adolescents. We are conducting a flexible dose, double blind, placebo-controlled, Phase II clinical trial of TC-5231 in children and adolescents with ADHD. We plan to enroll 150 to 180 patients between the ages of 6 and 17 in the trial at up to 17 sites in the United States. As of April 30, 2004, we had enrolled 128 patients. The primary objective of this trial is to determine whether TC-5231 is effective in the treatment of symptoms of ADHD in subjects within this age group. The trial will also assess the safety and tolerability and the pharmacokinetics of TC-5231. In addition, because mecamylamine hydrochloride at the Inversine dose is used to treat forms of hypertension, an independent safety board is monitoring the effects of TC-5231 on subjects’ blood pressure.
In the trial, each patient is randomly selected to receive either drug or a placebo each day for six weeks. Each patient selected to receive the drug is administered a 0.2mg dose of TC-5231 for the first two weeks of the trial. Based on the investigating physician’s assessment of tolerability of the 0.2mg dose, the physician can elect to increase the dose to 0.5mg for the next two weeks, or can elect to maintain the dose for the next two weeks at 0.2mg. Following that second two-week period, the investigating physician can again elect to increase the dose either to 0.7mg or 1.0mg, or maintain the existing dose for a third two-week period. The maximum dosage for subjects under the age of 13 is 0.7mg and the maximum dosage for subjects aged 13 to 17 is 1.0mg. The primary efficacy endpoint in this trial is the change after six weeks of therapy in a subject’s score on a standard rating system for ADHD patients known as the ADHD rating scale. This is an 18-item rating scale in which the investigating physician measures attention, hyperactivity and impulsivity of the subject. Secondary endpoints include changes in ratings on other standard rating scales. We expect the results of this trial to be available in the fourth quarter of 2004.
Phase II Trial in Young Adults. We have contracted with a physician-investigator to conduct a placebo-controlled, Phase II clinical trial of TC-5231 on our behalf. This trial is being conducted in the United States with 12 ADHD patients between the ages of 17 and 24. In the trial, the investigator doses patients once per week over a five-week period. Each week patients receive one of a 0.2mg, 0.5mg or 1.0mg dose of TC-5231, nicotine administered via patch or a placebo. The primary objective of the trial is to measure effects of TC-5231 on sustained attention. In particular, the trial measures the time needed for a patient to cancel a planned movement, referred to as “stop signal reaction time,” as well as other aspects of attention. In addition, the investigator is measuring performance on a word recognition test. The investigator has advised us that the results will be available in the second half of 2004.
Plans for Future Development. If the results of our Phase II clinical trials are favorable, we plan to conduct Phase III clinical trials of TC-5231 for the treatment of ADHD in children and possibly adults.
TC-2403
TC-2403 is a small molecule that we are developing for the treatment of ulcerative colitis in collaboration with Dr. Falk Pharma GmbH. We are currently conducting a Phase II clinical trial of an enema formulation of the compound designed to induce remission of acute episodes of left-sided colitis. In addition, we are developing a delayed release oral formulation of the compound designed to deliver the drug to the entire colon to induce and
maintain remission of all forms of ulcerative colitis. We expect to complete the oral formulation of this product candidate in the fourth quarter of 2004.
Ulcerative colitis is characterized by inflammation of the lining of the colon. Our preclinical studies of TC-2403 suggest that the compound interacts with thea3b2 NNR that is present in nerve endings in the gastrointestinal tract. We believe thata3b2 may play an important role in preventing inflammation and maintaining the lining of the colon. In other preclinical in vitro studies that we conducted, TC-2403 inhibited the release of proteins called cytokines that cause inflammation in humans and did not interact significantly with nicotinic acetylcholine receptors in the peripheral tissues associated with side effects like nausea and cardiovascular effects.
TC-2403 is related structurally to nicotine, and studies have shown that nicotine has positive effects for limited periods in treating the symptoms of ulcerative colitis. Specifically, in published third-party studies involving humans and animals, nicotine modulated the release of acetylcholine from nerves, increased secretion of mucus in the colon, inhibited the production of cytokines and reduced chemicals called prostaglandins that are involved in the process of inflammation. Collectively, these effects would help to maintain the normal structure of the colon. Also, in published studies conducted at the Mayo Clinic, nicotine was administered to non-smoking ulcerative colitis patients either via a nicotine patch or an enema. In these studies, remission of ulterative colitis symptoms and tissue healing was observed in approximately 40% of patients treated with the patch, as compared to 9% of patients treated with placebo, and approximately 70% of patients treated with an enema. Unlike the patch study, the enema study was not placebo controlled. However, serious side effects like nausea were observed in patients treated with nicotine, particularly when administered via patch. Because TC-2403 is designed to selectively target thea3b2 NNR, we believe that it may have positive effects on ulcerative colitis similar to those of nicotine, but without the side effects.
Clinical Development of TC-2403
Phase I Clinical Trials. We conducted two Phase I, placebo-controlled clinical trials of the enema formulation of TC-2403, including:
In each trial, TC-2403 was well tolerated and we observed no clinically significant adverse events.
Phase II Clinical Trial. We are sponsoring a double blind, placebo-controlled, dose range finding Phase II clinical trial to determine whether TC-2403 in the enema formulation is effective in treating mild to moderate left-sided colitis. The trial is being conducted at 48 sites in North America and 27 sites in three Eastern European countries. We plan to enroll between 176 and 240 ulcerative colitis patients for the trial. Because patient recruitment rates for ulcerative colitis are relatively low, we are using a large number of clinical sites in an effort to complete the trial by the end of 2004. As of April 30, 2004, approximately 78 patients had been enrolled. Two contract research organizations are monitoring and managing the trial for us.
In the trial, patients are administered an enema with 100mg, 200mg or 400mg of TC-2403 or a placebo each day over six weeks. Endoscopic examinations of the colon are made at the beginning and end of the six-week period and routine safety assessments are made throughout the trial. The primary efficacy endpoint of the trial is the change in patients’ U.S. Disease Activity Index for ulcerative colitis from baseline, as compared to placebo. This index measures stool frequency, rectal bleeding, appearance of the lining of the colon and a physician’s global rating of disease severity. A secondary endpoint is the change in patients’ European Union’s Clinical Activity Index for ulcerative colitis, which measures items such as number of stools, blood in stools, abdominal
pain and cramps, general well-being, presence or absence of fever and clinical manifestations such as arthritis outside of the intestines, as a secondary measure.
As of April 30, 2004, four of the 78 patients that had participated in the trial experienced an elevation in liver enzymes in excess of three times the upper limit of normal. These patients were withdrawn from the trial and their liver enzymes returned to within normal limits. Because the trial is double blind, we do not know if these patients were administered TC-2403 or a placebo.
The design of this trial is adaptive, meaning that we can adjust the dosages and the number of participating patients. We plan to have an analysis of the available data from the trial conducted by an independent third party in the third quarter of 2004 to enable us to select the most relevant dosages for the remainder of the trial and to determine the total number of patients to be studied. We expect the complete results of this trial to be available in the fourth quarter of 2004.
Plans for Future Development. We plan to file an IND or a foreign equivalent to conduct a Phase I clinical trial of the oral formulation of TC-2403 in the first half of 2005. If the results of the planned Phase I trial of the oral formulation and the ongoing Phase II trial of the enema formulation are favorable, we expect that we and Dr. Falk Pharma will further assess the development plans for both the oral and enema formulations.
TC-2696
TC-2696 is a novel small molecule that we are developing as an oral treatment for acute post-operative pain. We are currently conducting a Phase I clinical trial of TC-2696. Depending on clinical trial results, available resources and other considerations, we may pursue development of TC-2696 for other classes of pain as well.
In our preclinical in vitro studies of TC-2696, we found the compound to be a potent activator of thea4b2 NNR and to avoid interaction with nicotinic acetylcholine receptors in the muscles and ganglia that are associated with side effects. Published studies conducted by third parties have shown that compounds that activatea4b2 have pain-relieving effects in animals. These effects may be caused in part by the activation of NNRs that are abundant in CNS pathways to block the transmission of pain signals to the brain. In our preclinical animal studies, TC-2696:
Clinical Development of TC-2696
Phase I Clinical Trial. We are currently conducting a placebo-controlled Phase I single rising dose clinical trial of TC-2696 conducted to determine its safety and tolerability profile in healthy volunteers. The trial is being conducted in France in 38 healthy volunteers divided into dose groups of 2mg, 5mg, 10mg, 20mg, 50mg, 100mg and 200mg. In addition to evaluating safety and tolerability, we designed the trial to include a number of surrogate measures, including inflammation and pain relief. We expect the results of this trial to be available in the second half of 2004.
Plans for Future Development. To further assess the safety and tolerability profile of TC-2696, we expect to commence a Phase I multiple rising dose clinical trial upon completion of our Phase I single rising dose trial. If the results of the Phase I trials are favorable, we expect to commence a Phase II clinical trial of TC-2696 in the treatment of acute post-operative pain in the first half of 2005.
Inversine
Inversine is currently our only marketed product. Inversine is approved in the United States for the management of moderately severe to severe essential hypertension. Our market research suggests, however, that Inversine is prescribed predominantly for the treatment of Tourette’s syndrome and other neuropsychiatric disorders. Inversine has been approved for marketing since the 1950s. We acquired marketing rights to the product in August 2002.
We have reformulated mecamylamine hydrochloride and are developing it in a lower dose as TC-5231 for the treatment of ADHD. We are also exploring the potential use of mecamylamine hydrochloride in the same dose as Inversine for additional indications such as depression.
Our Preclinical Research Programs
We focus our preclinical research efforts on indications for which we believe that selective NNR-targeted drugs have the potential for use in the treatment of disease and for which we believe we can efficiently develop marketable product candidates. In selecting our target indications, we have considered a number of factors, including:
Based on our consideration of these factors, we currently have ongoing preclinical research programs for Alzheimer’s disease, schizophrenia, depression and anxiety, smoking cessation and obesity. Each of these indications represents a substantial market opportunity. Our current research objective is to file at least one IND or foreign equivalent each year beginning in 2005.
Alzheimer’s disease
Alzheimer’s disease progressively impairs memory, reason, judgment, language and eventually the ability to carry out simple tasks. While the exact cause of Alzheimer’s disease is unknown, the decline of cognitive function is linked to a progressive deterioration and death of cells in the brain. In addition, published studies have shown, among other things, that Alzheimer’s patients have deficient levels of acetylcholine.
TC-4959 is our compound that we are developing in collaboration with Aventis for the treatment or prevention of Alzheimer’s disease. TC-4959 induced significant increases in acetylcholine levels in preclinical in vitro studies by interacting with thea4b2 NNR and improved memory-related performance in rodent models.
An executive committee formed under our collaboration agreement with Aventis that covers the development of TC-4959 is responsible for determining whether to advance TC-4959 into clinical trials. We expect Aventis to complete the necessary preclinical studies to enable the committee to make that determination in the fourth quarter of 2004. If the compound is advanced into clinical trials, Aventis would be responsible for all clinical development and potential commercialization. If the compound is not advanced, this collaboration agreement with Aventis will terminate.
Schizophrenia
Schizophrenia is a chronic, severe and disabling form of psychosis. Although the precise cause of schizophrenia is unknown, the disease is thought to be associated with an imbalance of neurotransmitter levels in the brain, particularly dopamine levels. Because NNRs act to regulate levels of neurotransmitters in the brain, we believe that NNRs may be useful targets for schizophrenia therapies. A number of published studies have indicated an association between thea7 NNR and schizophrenia. In a survey of experts in connection with a National Institute of Mental Health schizophrenia initiative,a7 was selected more often than any other target as the target of most interest in the development of treatments for psychosis. In addition, because schizophrenic patients are frequently cognitively impaired, we believe that thea4ß2 NNR, which plays a role in cognition, also may be associated with schizophrenia. These studies further suggest the potential relevance of NNRs as targets for the treatment of schizophrenia. In addition, published studies have linked nicotine to improvements in the ability to filter or disregard unremarkable stimuli, a common symptom of schizophrenia, and cognitive impairment in schizophrenic patients.
We are evaluating a number of compounds for the treatment of schizophrenia in preclinical studies. Some of these compounds are designed to interact selectively witha7 and others are designed to interact selectively with botha7 anda4ß2.
Depression and Anxiety
Depression is thought to be associated with the disruption and imbalance in the brain of the neurotransmitters dopamine, norepinephrine and serotonin. As noted above, because NNRs act to regulate levels of these key neurotransmitters in the brain, we believe that they may be useful targets for depression therapies. Because patients are often diagnosed with both depression and anxiety, we believe that NNRs may also be useful targets for anxiety therapies. A number of reported studies in humans and animals have linked nicotine to improvements in symptoms of depression. In particular, depressed patients who were administered nicotine via patch had short-term improvements in symptoms after only the second day of treatment based on a reduction in scores on the Hamilton Rating Scale, an accepted rating scale for depression. Because many current anti-depressant therapies do not take effect for an extended period, the rapid onset of action of nicotine in these studies suggests a potentially significant advantage for NNR-targeted therapeutics. In addition, in animal studies conducted by third parties, nicotine and other compounds that act on NNRs have shown greater potency than, and similar anti-depressant effects as, common anti-depressant therapies such as selective serotonin reuptake inhibitors and tricyclics.
We are currently evaluating two compounds for the treatment of depression and anxiety. In preclinical evaluation, each of these compounds showed activity in various rodent models of depression and general anxiety disorder and panic disorder. In depression models, the compounds showed greater potency than, and comparable anti-depressant effects as, selective serotonin reuptake inhibitors and tricyclics. We are currently undertaking the additional preclinical toxicology studies necessary to support an IND filing to initiate human clinical trials of these compounds.
Smoking Cessation
Due primarily to nicotine’s addictive effects, it is very difficult to quit smoking. Published animal studies have linked nicotine’s addictive effects to the release of dopamine in regions in the brain involved in feelings of reward and pleasure. Although the specific NNR implicated in the regulation of dopamine is not fully characterized, several reported studies suggest that the ß2 NNR may be involved. These studies have shown that selectively blocking ß2 reduced the rewarding effects of nicotine in mice. Other studies have shown that mice deficient in ß2 failed to respond to nicotine and had reduced activity in the brain regions associated with reward and pleasure. We are evaluating a number of compounds in a variety of animal models of smoking cessation and nicotine dependence for advancement in our smoking cessation program.
Obesity
A number of published studies have demonstrated that non-smokers generally weigh significantly more than smokers, and nicotine is believed to be responsible. These studies have also shown that smokers gain weight when they stop smoking. Moreover, reported studies with animals have shown that food intake and body weight gain are reduced following repeated administration of nicotine and that the effects are reversed when the nicotine administration is stopped.
As part of our evaluation of our compounds for other indications, we also assess each compound for a preliminary signal of its ability to induce weight loss. We are collecting this data and currently plan to conduct additional preclinical evaluation of the most promising compounds for obesity in 2005.
Our Drug Discovery Technologies—Pentad
We use proprietary databases and computer-based molecular design technologies to identify promising product candidates. We refer to these technologies collectively as Pentad.
We designed Pentad to predict the likelihood that novel compounds will interact with various NNRs, the degree of the interaction and the potential of these compounds to be developed as drugs based on projected pharmacokinetic profiles. Pentad consists of sophisticated computer-based simulation methodologies and extensive biological data from a library of diverse compounds that we have developed and gathered over 20 years. To date, we have applied Pentad specifically in the discovery and optimization of NNR-targeted therapeutics, but we believe it has application to a wide range of targets.
Pentad’s virtual screening enables us to more rapidly identify clinically-viable compounds than we believe could be achieved using traditional laboratory synthesis and screening methods. This allows us to reduce drug development time by focusing our resources on compounds that we believe have a greater likelihood of clinical success. Our use of Pentad to design new classes of compounds selective for thea7 NNR is an example of its capabilities. We conducted virtual screening of nearly 11,000 compounds and, based on the results, synthesized 115 of them. In preclinical tests, 43 of the synthesized compounds were highly selective to thea7 NNR, showed low binding affinity for NNRs involved in side effects, were bioavailable and passed the blood-brain barrier. We identified the 43 compounds in only six months and are currently evaluating many of these compounds as part of our schizophrenia program.
Strategic Collaborations
We have entered into two collaboration agreements with Aventis Pharma SA. One agreement relates to the development and potential commercialization of specified Targacept compounds and the other relates to the development and potential commercialization of Aventis compounds. We have also entered into a collaboration agreement with Dr. Falk Pharma GmbH.
Aventis Pharma SA
Targacept Compounds. In January 2002, we entered into an amended and restated collaborative research and license agreement with Aventis that replaced our original agreement entered into in December 1998. Under the agreement, we granted Aventis an exclusive, worldwide, sublicensable license under our patent rights and know-how, excluding Pentad, to develop and commercialize specified Targacept compounds for the treatment or prevention of Alzheimer’s disease. The agreement restricts both us and Aventis from pursuing the development or commercialization of compounds with specified activity at thea4b2 ora7 NNRs for Alzheimer’s disease during the term of the agreement, except under the agreement or our other collaboration agreement with Aventis, which is described below.
Aventis paid us a non-refundable upfront license fee of $2 million at the time of execution of the original agreement in 1998. Aventis also made research support payments to us under the agreement until December 2002. In addition, we could receive up to an aggregate of $30 million from Aventis upon the achievement of specified pre-commercialization development and regulatory milestones. We are also entitled to receive royalties based on net sales by Aventis, its affiliates and its sublicensees of products developed under the agreement. Our right to receive royalties would continue on a country-by-country and product-by-product basis until the later of ten years from the first commercial sale of a product in that country or the expiration of the patent rights covering the product in that country.
An executive committee comprised of an equal number of representatives from Aventis and us is responsible for determining whether to advance our compounds into clinical development. Our compound TC-4959, which is in late preclinical evaluation, is the only compound that remains in consideration for continued development and potential commercialization under the agreement. If the members of the executive committee representing either Aventis or us desire to advance TC-4959 into clinical development and the members representing the other party object, TC-4959 will not advance into clinical development and neither party may then develop or commercialize the compound for any use. If the executive committee selects TC-4959 for advancement, Aventis will control clinical development and be responsible for conducting all preclinical research and clinical development activities and for obtaining all regulatory approvals. Aventis would be required to use commercially reasonable efforts to develop TC-4959 and, if Aventis receives regulatory approval, commercialize the compound for the licensed indication in the United States, the European Union and Japan.
All of our compounds that were not advanced into clinical development have been removed from the collaboration, with all rights reverting to us. For example, TC-1734, our product candidate for cognitive impairment in the elderly, was removed from the collaboration in this manner. The terms of the collaboration agreement restrict us from pursuing the development of TC-1734 for the treatment or prevention of Alzheimer’s disease while the agreement remains in effect.
Unless otherwise agreed, the agreement terminates six months after the end of the research term if a compound has not advanced into clinical development. The agreement provides for the research term to end on December 31, 2002. Although neither we nor Aventis has taken action to formally extend the research term, Aventis is continuing to evaluate TC-4959 for possible advancement under the agreement and to report its progress to our project team representatives at regular meetings. If TC-4959 is not advanced into clinical development, the collaboration agreement would terminate. Either party may terminate the agreement in the event of an uncured material breach by the other party. However, if a breach by Aventis is limited to a particular key market, we can terminate the agreement only as applied to that market. Upon termination of the agreement, the licenses that we granted to Aventis terminate, except that, in the event of a partial termination, only those licenses applicable to the terminated aspects of the agreement terminate.
Aventis Compounds. In January 2002, we also entered into a second collaborative research, development and commercialization agreement with Aventis. Under the agreement, we granted Aventis a worldwide, sublicensable license under our patent rights and know-how, excluding Pentad, to develop and commercialize designated Aventis compounds for Alzheimer’s disease and other CNS disorders. Aventis retains worldwide commercialization rights for all compounds that it develops under the agreement.
The agreement restricts us, during the research term, from developing and commercializing compounds with specified activity at thea4b2 ora7 NNRs for the treatment or prevention of Alzheimer’s disease. This restriction applies only if both our collaboration agreement with Aventis relating to the development and potential commercialization of our compounds remains in effect and Aventis is using commercially reasonable efforts to develop and commercialize a compound under that agreement for the treatment or prevention of Alzheimer’s disease in the United States, the European Union and Japan. Except with respect to any compounds that we in-license from Aventis under the agreement as described below, we are not restricted in any way under the agreement from developing or commercializing compounds for other CNS disorders.
We could receive up to $8 million upon the achievement of specified pre-commercialization development and regulatory milestones related to Alzheimer’s disease and up to $8 million for each other CNS indication upon the achievement of specified pre-commercialization development and regulatory milestones. We are also entitled to receive royalties based on net sales by Aventis, its affiliates and sublicensees of each product under the agreement. Our right to receive royalties would continue on a country-by-country and product-by-product basis until the later of ten years from the first commercial sale of a product in that country or the expiration of the patent rights covering the product in that country.
An executive committee comprised of an equal number of representatives from us and Aventis is responsible for initially recommending further development of any compound under the agreement. Aventis also has the independent right to recommend further development of any compound. A scientific review board from Aventis determines whether to advance a compound into clinical development. Under the agreement, Aventis is responsible for conducting all preclinical and clinical development activities and obtaining all required regulatory approvals for compounds selected for advancement. Any compound that Aventis initially selects for advancement but ultimately rejects is terminated from the collaboration and becomes available to us for in-licensing. We would be permitted to in-license the terminated compound for use in indications other than the treatment or prevention of CNS disorders or for use in specified CNS indications if Aventis is not developing its own product for those indications, subject to our making milestone and royalty payments to Aventis.
If not terminated earlier, this agreement terminates upon expiration of all royalty and other payment obligations. In addition, either party may terminate the agreement in the event of an uncured material breach by the other party. However, if Aventis is developing or commercializing more than one compound or product, or if we are developing or commercializing more than one compound or product that has been terminated from the collaboration and in-licensed by us, and the breach relates to a particular compound or product, then the non-breaching party can terminate the agreement only as applied to that compound or product. If Aventis terminates the agreement other than as a result of an uncured breach by us and our collaboration agreement with Aventis relating to the development and commercialization of our compounds is still in effect, we would continue to be entitled to receive milestone and royalty payments for specified compounds. In that case, we would be entitled to milestone and royalty payments for any Aventis compounds subject to the agreement that Aventis later develops for use in a CNS indication or for any other compound that targets thea4ß2 ora7 NNR that Aventis later develops for the treatment or prevention of Alzheimer’s disease.
Dr. Falk Pharma GmbH
In January 2001, we entered into a collaborative research, development and license agreement with Dr. Falk Pharma. The agreement provides for the research, development, and commercialization of our compound TC-2403 and, if selected by Dr. Falk Pharma and us, at least one additional compound for the treatment or prevention of ulcerative colitis and other gastrointestinal or liver diseases. Phase II clinical trials of TC-2403 are currently ongoing.
Under the agreement, we granted Dr. Falk Pharma a license under our patent rights and know-how relating to TC-2403 and additional compounds that may become part of the agreement, excluding Pentad, in a territory in Europe consisting of all of the major European pharmaceutical markets, Russia and the Commonwealth of Independent States countries, Egypt and Israel. The license is exclusive in the licensed territory with respect to the sale of compounds for the treatment or prevention of ulcerative colitis and other gastrointestinal or liver diseases and joint, or co-exclusive, with us with respect to all other purposes. We retained all commercialization rights in the rest of the world. Dr. Falk Pharma granted us a license under its patent rights and know-how relating to TC-2403 and additional compounds that may become part of the agreement to develop and commercialize pharmaceutical products for the treatment or prevention of ulcerative colitis and other gastrointestinal or liver diseases in the rest of the world.
Upon effectiveness of this agreement, Dr. Falk Pharma paid us a non-refundable upfront license fee of $1.0 million and purchased $1.0 million of our common stock. We are also entitled to receive a percentage of net profits from the sale of licensed products by Dr. Falk, its affiliates and its sublicensees in its licensed territory. Dr. Falk Pharma is entitled to receive royalties from us based on net sales by us, our affiliates and sublicensees of products developed under the agreement in the United States and Japan. Our right to receive a percentage of Dr. Falk Pharma’s net profits and Dr. Falk Pharma’s right to receive royalties would continue on a country-by-country and product-by-product basis until the earlier of 12 years from the first commercial sale of a product in that country or the expiration of the patent rights covering the product in that country.
We are jointly responsible with Dr. Falk Pharma for all clinical development in the territories licensed to Dr. Falk Pharma, and we share all related development expenses evenly. To the extent that particular clinical development is required as a condition to commercialization in the rest of the world, we would be responsible for that clinical development and related expenses. Dr. Falk Pharma may terminate a compound’s development under the agreement in its sole discretion after completion of Phase II clinical evaluation. If the development of a compound is terminated and the compound is not designated as a back-up compound for possible future development, then the compound is removed from the agreement. Dr. Falk Pharma is required to use commercially reasonable efforts to commercialize products under the agreement in each country in its licensed territory, and we are required to use commercially reasonable efforts to commercialize products under the agreement in the United States and Japan. If we fail to use commercially reasonable efforts to commercialize licensed products in the United States or Japan, the percentage of net profits that Dr. Falk Pharma would otherwise pay to us for sales in its licensed territory would be equitably adjusted to reflect Dr. Falk Pharma’s loss of anticipated royalty revenue from sales by us in the rest of the world.
We may terminate Dr. Falk Pharma’s commercialization rights for a compound in a country in its licensed territory if Dr. Falk Pharma does not use commercially reasonable efforts to commercialize the product in that country without sound business or commercial reasons. If we terminate these rights for a product in all of France, Germany and the United Kingdom, the agreement would no longer apply to that product and we could commercialize that product ourselves without obligation to Dr. Falk Pharma. We may also terminate the agreement with thirty days notice to Dr. Falk Pharma if required regulatory approvals have not been obtained for at least one compound in at least one of France, Germany or the United Kingdom by a specified date. Also, either party may terminate the agreement in the event of an uncured material breach by the other party, including a failure to use commercially reasonable efforts to commercialize products. However, in the case of a breach by Dr. Falk Pharma with respect to a particular compound, product or market, our right to terminate would be limited to the applicable compound, product or market. Upon termination of the agreement, the licenses that we granted to Dr. Falk Pharma terminate.
Patents and Proprietary Rights
We actively seek to protect the proprietary technology that we consider important to our business, including chemical species, compositions and forms, their methods of use and processes for their manufacture, as well as modified constructs of naturally-expressed receptors, in the United States and other key pharmaceutical markets. We also rely upon trade secrets and contracts to protect our proprietary information.
As of April 30, 2004, our patent estate includes 59 patents issued in the United States, four patent applications allowed in the United States and not yet issued, 20 patent applications pending in the United States, and numerous issued patents and patent applications pending in countries outside the United States. Our issued patents and pending patent applications in the United States include composition of matter coverage on a number of different structural families of compounds. Individual patents extend for varying periods depending on the date of filing of the patent application or the date of patent issuance and the legal term of patents in the countries in which they are obtained. The actual protection afforded by a patent varies from country to country and depends upon many factors, including the type of patent, the scope of its coverage and the availability of legal remedies in a particular country.
We consider the following United States patents that we own or license to be most important to the protection of our clinical stage product candidates.
|
|
| ||
|
| |||
| ||||
|
| |||
|
| |||
|
|
| ||
In addition, we have later-expiring patents relating to some of these product candidates that cover a particular form or composition, use as part of combination therapy or method of preparation or use. These patents could provide additional or a longer period of protection. We also have patent applications pending that seek equivalent or substantially comparable protection for our product candidates in key international markets.
License Agreements
We are parties to five license agreements that are important to our business.
University of South Florida Research Foundation
Pursuant to a license agreement with the University of South Florida Research Foundation, or USFRF, we hold an exclusive worldwide license to patent and patent applications owned by USFRF for use in the development and commercialization of mecamylamine hydrochloride, which we refer to as TC-5231, and other specified compounds. The licensed patents and patent applications include an issued patent covering methods of use for the treatment of ADHD, Tourette’s syndrome and nicotine-responsive neuropsychiatric disorders and pending patent applications covering the pharmaceutical composition of the components of mecamylamine hydrochloride. Under the agreement, we are obligated to pay to USFRF:
The aggregate annual license fees are creditable, up to a specified amount per year, against future royalties.
We are required to use commercially reasonable efforts to develop or to market and sell a product covered by the agreement. In particular, we are required to spend a specified minimum amount on research and development of products covered by the agreement each year until we receive marketing approval for a covered product. If USFRF believes that we are not meeting our diligence obligation, it is entitled to terminate the agreement following a cure period. If we do not agree with USFRF’s determination, we can submit the matter to binding arbitration. In addition, if we have not received marketing approval of a product covered by the agreement on or before December 31, 2012, USFRF can make our license nonexclusive.
We may terminate the agreement at any time. If not earlier terminated, the agreement will terminate upon expiration of the last to expire of the licensed patent rights.
Virginia Commonwealth University Intellectual Property Foundation
Pursuant to a license agreement with Virginia Commonwealth University Intellectual Property Foundation, or VCUIPF, we hold a non-exclusive worldwide license to patents covering a method of use of a family of compounds that includes TC-2696 for eliciting an analgesic effect. Under the agreement, we are obligated to pay to VCUIPF:
We are required to use reasonable efforts to bring one or more products covered by the agreement to market. We may terminate the agreement at any time with 90 days notice. If the agreement is not earlier terminated, our obligation to pay royalties under the agreement will terminate upon expiration of the licensed patent rights.
Wake Forest University Health Sciences
Pursuant to a license agreement with Wake Forest University Health Sciences, or WFUHS, we hold an exclusive worldwide license to patents covering a method of use of a family of compounds that includes TC-2696 for the treatment of chronic or female-specific pain. Under the agreement, we paid WFUHS a non-refundable upfront license fee of $25,000 and are obligated to pay to WFUHS:
We are required to use commercially reasonable efforts to pursue the development of at least one product covered by the agreement and to bring at least one such product to market. We may terminate the agreement at any time with 60 days notice. If not earlier terminated, the agreement will terminate upon expiration of the last to expire of the licensed patent rights.
University of Kentucky Research Foundation
Pursuant to a sponsored research agreement, the University of Kentucky Research Foundation, or UKRF, agreed to assign to R.J. Reynolds Tobacco Company rights to inventions that resulted in patents related to TC-1734, TC-2696 and other discovery-stage compounds in our portfolio. These patents were subsequently assigned by RJR to us in August 2000. Under the sponsored research agreement and a subsequent license agreement with UKRF, we are obligated to pay royalties to UKRF with respect to products covered by these patents. In addition, under the license agreement, RJR paid UKRF an upfront license fee of $20,000.
Medical College of Georgia Research Institute
Pursuant to a license agreement with Medical College of Georgia Research Institute, or MCGRI, we hold an exclusive worldwide license to a patent covering a method of use of a substance that stimulates the activity of nicotinic acetylcholine receptors by inhibiting the activity of another class of receptors, a method of use of increasing the presence of a therapeutic substance to treat neurodegeneration and a screening method. Under the agreement, we paid MCGRI an upfront license fee of $25,000 and are obligated to pay to MCGRI:
If not earlier terminated, the agreement will terminate upon the earlier of expiration of the licensed patent rights or July 2027.
Trade Secrets
In addition to patents, we rely upon unpatented trade secrets and know-how and continuing technological innovation to develop and maintain our competitive position. For example, we maintain Pentad as an unpatented trade secret. We seek to protect our proprietary information, in part, using confidentiality agreements with our commercial partners, collaborators, employees and consultants and invention assignment agreements with our employees. We also have confidentiality agreements or invention assignment agreements with some of our commercial partners and consultants. These agreements are designed to protect our proprietary information and, in the case of the invention assignment agreements, to grant us ownership of technologies that are developed through a relationship with a third party. These agreements may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our commercial partners, collaborators, employees and consultants use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
Sales and Marketing
We currently have limited sales and distribution capabilities and limited experience in marketing and selling pharmaceutical products. Our current strategy is to selectively enter into collaboration agreements with third parties for target indications in which our potential collaborator has particular expertise or that involve a large, primary care market that must be served by large sales and marketing organizations. In entering into these collaboration agreements, our goal will be to maintain co-promotion or co-commercialization rights in the United States and, in some cases, other markets. In order to implement our strategy successfully, we must develop a specialized sales and marketing organization with sufficient technical expertise. Our product currently available in the market, Inversine, is distributed by a third party pursuant to an exclusive distribution agreement.
Manufacturing
All of our drug candidates are organic compounds of low molecular weight, commonly referred to as small molecules. We have selected these compounds in part for their ease of synthesis and the low cost of their starting materials. All of our current product candidates are manufactured in a simple synthetic process from readily available starting materials. We expect to continue to develop drug candidates that can be produced cost-effectively by third-party contract manufacturers.
We are able to manufacture the quantities of our product candidates necessary for relatively short preclinical toxicology studies ourselves. We believe that this allows us to accelerate the drug development process by not having to rely on a third party for all of our manufacturing needs. However, we do rely and expect to continue to rely on a number of contract manufacturers to produce enough of our product candidates for use in more lengthy preclinical research. We also depend on these contract manufacturers to manufacture our product candidates in accordance with current good manufacturing practices, or cGMP, for use in clinical trials. We will ultimately depend on contract manufacturers for the manufacture of our products for commercial sale, as well as for process development.
Third parties currently manufacture Inversine and its active ingredient for us. Also, we have entered into a development and production agreement with Siegfried Ltd. Under this agreement, Siegfried has agreed to provide us with process development services and clinical trial material at specified rates for product candidates that we elect to introduce into the agreement. We have also agreed, following marketing approval or anticipated marketing approval of any product candidate for which Siegfried performs services under the agreement, to negotiate for a separate multi-year commercial supply agreement with Siegfried for a substantial percentage of our contracted supply needs for that product candidate, except in limited circumstances. Beginning in February 2006, either we or Siegfried can terminate the agreement at any time on 12 months notice or immediately in the event of an uncured material breach by the other party. Contract manufacturers are subject to extensive governmental regulation.
Competition
Our industry is subject to rapid and intense technological change. We face, and will continue to face, worldwide competition from biotechnology, biopharmaceutical and pharmaceutical companies, research institutions, government agencies and academic institutions. Many of these competitors are established in the CNS field and are developing and commercializing pharmaceutical products that would compete with our product candidates that are approved for marketing. Many of our competitors and potential competitors have more resources than we do and have already successfully developed and marketed drugs. Mergers and acquisitions in the pharmaceutical industry may result in even greater resources being concentrated in our competitors.
We also face substantial competition within the area of NNR-targeted therapeutics. We are aware of several prominent pharmaceutical companies with NNR-targeted product candidates in development, including Pfizer, with an NNR-targeted compound in Phase III for smoking cessation, and Abbott Laboratories, with an NNR-targeted compound in Phase II for Alzheimer’s disease, ADHD and schizophrenia and a second NNR-targeted compound in Phase I for pain. In addition, we believe that other companies have active NNR-based research programs, including, Merck & Co., AstraZeneca, Eli Lilly, Sanofi-Synthélabo, Memory Pharmaceuticals, Critical Therapeutics and NeuroSearch A/S. We expect to face increased competition in the future if NNR-targeted therapeutics are further validated and if companies initiate or grow NNR-based programs or otherwise enter the CNS market.
In addition, there are several pharmaceutical companies in the United States and globally that currently market and sell drugs for indications that we are targeting. There are currently no approved products for AAMI or MCI. We believe that the primary competitive products for use in the other indications that we currently target include:
Furthermore, pharmaceutical and biotechnology companies are currently developing additional treatments for the indications that we are targeting that may be approved for marketing and sale prior to any approval of our product candidates.
We expect to compete based upon, among other things, the efficacy of our products and favorable side effect profiles. Our ability to compete successfully will depend on our continued ability to attract and retain skilled and experienced scientific, clinical development and executive personnel, to identify and develop viable product candidates and to exploit these products and compounds commercially before others are able to develop competitive products. In addition, our ability to compete may be affected by insurers and other third-party payors encouraging the use of generic products. This may have the effect of making branded products less attractive from a cost perspective to buyers.
Government Regulation
Drug Regulation in the United States
The research, testing, manufacture and marketing of drug products are extensively regulated by the FDA and other governmental authorities in the United States. The Federal Food, Drug and Cosmetic Act and other federal and state statutes and regulations regulate the research, development, testing, manufacture, storage, record keeping, labeling, promotion and marketing and distribution of drug products.
The steps ordinarily required before a new drug may be marketed in the United States include:
Preclinical tests typically include laboratory evaluation of product chemistry, formulation and stability, as well as animal studies to evaluate toxicity and metabolism. The results of preclinical tests are submitted to the FDA as
part of an IND. The FDA requires a 30-day waiting period after the filing of an IND before clinical testing in humans may begin. If the FDA has not advised otherwise within this 30-day period, the proposed trial may begin. If the FDA has comments or questions, they must be resolved to the satisfaction of the FDA before the trial can begin. In addition, the FDA may halt proposed or ongoing clinical trials at any time, in which event the trial cannot commence or recommence without FDA authorization and then only under terms authorized by the FDA. The IND application process may be extremely costly and substantially delay development of product candidates. Moreover, positive results in preclinical tests do not ensure positive results in clinical trials.
Clinical trials involve the administration of the drug to healthy volunteers or patients under the supervision of a qualified principal investigator. Clinical trials must be conducted in compliance with federal regulations and requirements and under established protocols. These protocols detail the objectives of the clinical trial, the parameters to be used in monitoring safety and the efficacy criteria to be evaluated. The study protocol and informed consent information for patients in clinical trials must also be approved by an institutional review board at each institution where the clinical trials are conducted.
Clinical evaluation involves a time-consuming and costly process, typically involving the following three phases:
The FDA, the study sponsor and the institutional review boards reviewing each clinical trial site closely monitor the progress of each of the three phases of clinical trials that are conducted in the United States. They may change or terminate the testing based upon the data accumulated to that point and their assessment of the relative risks and benefits to the patient.
Upon successful completion of Phase III trials, a company may submit an NDA including the results of preclinical studies and clinical trials and data relating to the product candidate’s chemistry, pharmacology, manufacture, safety and effectiveness to the FDA in order to obtain approval to market the product in the United States. This submission is expensive, both in terms of studies required to generate and compile the requisite data and the significant user fees required for NDA submission.
The FDA has 45 days from its receipt of an NDA to determine if it will accept the filing for a substantive review. The FDA may refuse the filing, which would result in the loss of 50% of the application user fee. If the FDA accepts the filing, it begins an in-depth review. Under current performance goals, the FDA has either 180 or 365 days to respond, depending upon whether the review is classified by the FDA as priority or standard. The FDA often extends the review timeline by requesting additional information or clarification. The FDA may refer issues to an advisory committee for review, evaluation and recommendation as to whether the application should be approved, but the FDA is not bound by any recommendation of an advisory committee.
If the FDA’s evaluation of the NDA and the manufacturing facilities are favorable, the FDA may issue an approval letter, or, in many cases, an approvable letter followed by an approval letter. An approvable letter usually contains a number of conditions that must be met in order to secure final approval. If the FDA decides that the conditions have been met, it will issue an approval letter. An approval letter makes a drug available for physicians to prescribe in the United States, but authorizes commercial marketing of the drug only for specific
indications. After a drug has been approved for a particular indication, other trials and studies may be conducted to explore its use for treatment of new indications.
The FDA may also refuse to approve an NDA, or may issue a not approvable letter. A not approvable letter outlines the deficiencies in the submission and often requires additional testing or information. Even if the applicant completes the additional testing and submits additional information, the FDA may ultimately decide that the application does not satisfy the regulatory criteria for approval.
Satisfaction of FDA pre-market approval requirements for new drugs typically takes several years. The actual time required may vary substantially based upon the type, complexity and novelty of the product or target disease. Government regulation may delay or prevent marketing of potential products for a considerable period of time and require costly procedures. Success in early stage clinical trials does not assure success in later stage clinical trials. Data obtained from clinical activities is not always conclusive and may be susceptible to varying interpretations that could delay, limit or prevent regulatory approval.
Even if a drug receives regulatory approval, the FDA may require post-marketing studies, sometimes referred to as Phase IV studies, to monitor the effects of approved drugs and may limit further marketing based on the results of these post-marketing studies. Moreover, the FDA may impose restrictions on the drug or withdraw its approval if a company does not stay in compliance with pre- and post-market regulatory standards or if problems relating to safety or effectiveness of the drug occur after it reaches the marketplace. The FDA has broad post-market regulatory and enforcement powers, including the ability to levy fines and civil penalties, suspend or delay issuance of approvals, seize or recall products and withdraw approvals.
Once an NDA is approved, the product it covers becomes a listed drug that can, in turn, be cited by potential competitors in support of approval of an abbreviated new drug application, or ANDA. An ANDA provides for marketing of a drug product that is therapeutically equivalent to a marketed drug. This means, among other things, that it has the same active ingredients in the same strengths and dosage form as the listed drug, and has been demonstrated to be bioequivalent to the listed drug. There is generally no requirement, other than the requirement for evidence of bioequivalence, for an ANDA applicant to conduct or submit results of preclinical tests or clinical trials to establish the safety or efficacy of its drug product. Drugs approved in this way are commonly referred to as generic equivalents to the listed drug, are listed as such by the FDA and can generally be substituted by pharmacists under prescriptions written for the original listed drug.
Federal law provides for a period of three years of exclusivity following approval of a drug that contains previously approved active ingredients but is approved in a new dosage, dosage form or route of administration, or for a new use if new clinical trials were required to support the approval. During this three-year exclusivity period, the FDA cannot grant approval of an ANDA for a generic version of the listed drug. However, the FDA can approve generic equivalents of that listed drug based on other listed drugs with the same active ingredient, such as a generic that is the same in every way but its indication for use, and thus the value of this exclusivity may be limited. Federal law also provides a period of five years of exclusivity following approval of a drug that does not contain any previously approved active ingredients. During the five-year exclusivity period, no ANDA for a generic version of the listed drug can be submitted unless the submission accompanies a challenge to a listed patent, in which case the submission may be made four years following the original product approval.
In addition, applicants submitting an ANDA for a drug that has listed patents are required to make one of four certifications regarding each listed patent, which may include certifying that one or more listed patents are invalid or not infringed. If an applicant certifies invalidity or non-infringement, it is required to provide notice of its filing to the new drug application sponsor and the patent holder. If the patent holder then initiates a suit for patent infringement against the ANDA applicant within 45 days of receipt of the notice, the FDA cannot grant effective approval of the ANDA until either 30 months has passed or there has been a court decision holding that the patents in question are invalid or not infringed. The first of the ANDA applicants submitting substantially complete applications certifying that listed patents for a particular product are invalid or not infringed may qualify for an exclusivity period of 180 days, which runs from the date the generic product is first marketed.
Until any effective 180-day exclusivity expires, the FDA cannot grant effective approval of subsequently submitted ANDAs.
The manufacturers of approved drugs and their manufacturing facilities are subject to continuous review and periodic inspections by the FDA and must comply with the FDA’s current good manufacturing process, or cGMP, regulations. A manufacturer will be subject to possible legal or regulatory action, such as suspension of manufacturing, seizure of product or recall of a product, if it does not comply with the FDA’s rules. We intend to contract with third parties to manufacture our products, and our ability to control their compliance with FDA requirements will be limited.
We must also notify the FDA of any change in an approved product beyond variations already allowed in the approval. Changes to the product, its labeling or its manufacturing could require prior FDA approval and may require further clinical investigations to support the change. Such approvals may be expensive and time-consuming, and if not approved, the product will not be allowed to be marketed as modified.
The FDA also imposes a number of complex regulations on entities that advertise and promote marketed pharmaceuticals. These regulations include requirements for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet. The FDA enforces the regulations under the Federal Food Drug and Cosmetic Act. Failure to abide by these regulations can result in penalties, including the issuance of a warning letter mandating the correction of deviations from FDA standards or the publication of corrective advertising. They may also include a requirement that future advertising and promotional materials be pre-cleared by the FDA, as well as civil and criminal investigations and prosecutions.
Holders of an NDA are also subject to laws and regulations regarding non-clinical laboratory practices that support human safety and product distribution, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances. In each of these areas, the FDA has broad regulatory and enforcement powers, including the ability to levy fines and civil penalties, suspend or delay issuance of approvals, seize or recall products, and withdraw approvals.
From time to time, legislation is drafted and introduced in the U.S. Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of drug products. In addition, the FDA regulations and guidance are often revised or reinterpreted in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted, or FDA regulations, guidance or interpretations changed, or what the impact of these changes, if any, may be.
Fast Track Designation
Congress enacted the Food and Drug Administration Modernization Act of 1997, or FDAMA, in part, to ensure the timely availability of safe and effective drugs, biologics and medical devices by expediting the FDA review process for some new products. FDAMA establishes a statutory program for the approval of a so-called fast track product, defined as a new drug or biologic intended for the treatment of a serious or life-threatening condition that demonstrates the potential to address unmet medical needs for that condition. Under the fast track program, the sponsor of a new drug or biologic may request the FDA to designate the drug or biologic as a fast track product at any time during clinical development of the product. Fast track designation provides for an expedited review of a product, which is intended to accelerate FDA approval. Although we have not yet requested fast track designation for any of our product candidates, we may seek fast track designation in the future. We will never be sure that we will obtain fast track designation. We cannot predict the ultimate impact, if any, of the fast track process on the timing or likelihood of FDA approval of any of our potential products.
Drug Regulation Outside the United States
In addition to U.S. regulations, we are subject to a variety of foreign regulations governing clinical trials and potential commercial sales and distribution of our products and product candidates. Even if we obtain FDA
approval for a product, we must obtain approval of a product by the regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
Under European Union regulatory systems, we may submit marketing authorizations either under a centralized or decentralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. The decentralized procedure provides for mutual recognition of national approval decisions. Under this latter procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the applications and assessment report, each member state must decide whether to recognize approval.
Third-Party Reimbursement
In the United States, European Union and elsewhere, sales of pharmaceutical products depend in part on the availability of reimbursement to the patient from third-party payors, such as government health administrative authorities, managed care providers and private insurance plans. Third-party payors are increasingly challenging the prices charged for medical products and services and examining their cost-effectiveness. For example, the European Union generally provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement. It is possible that none of our product candidates that receive marketing approval will be considered cost-effective or that reimbursement to patients will not be sufficient to allow us to maintain price levels that enable us to realize a satisfactory return on our investment in product development.
Price Controls
In the United States there have been, and we expect that there will continue to be, a number of federal and state proposals to implement governmental pricing control on pharmaceutical products. In some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union generally provides options for its member states to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. We do not know whether any country that has price controls will allow favorable pricing arrangements for any of our product candidates.
Employees
As of April 30, 2004, we had 72 full-time employees, 30 of whom are Ph.D.s, M.D.s or both. Our management believes that relations with our employees are good. None of our employees is represented under a collective bargaining agreement.
Property and Facilities
We lease approximately 40,000 square feet of laboratory and office space located in the Piedmont Triad Research Park in Winston-Salem, North Carolina. We have rights generally exercisable until August 2005 to lease additional space in this facility as and when it becomes available. The term of our lease expires August 1, 2007, and we have renewal options for an additional five-year term. We believe that these facilities are adequate to satisfy our current needs.
Legal Proceedings
We are not currently a party to any material legal proceedings.
Executive Officers and Directors
The name, age and position of our executive officers and directors as of April 30, 2004 are as follows:
|
|
| ||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
Mark Skaletsky has been a member of our board of directors since February 2001 and has been our Chairman since January 2002. Since March 2001, he has been the chairman and chief executive officer of Trine Pharmaceuticals, Inc., formerly Essential Therapeutics, Inc., a privately held drug discovery and development company. From May 1993 to January 2001, Mr. Skaletsky was the president and chief executive officer of GelTex Pharmaceuticals, Inc., a publicly traded pharmaceutical company. Mr. Skaletsky is a member of the boards of directors of Paradigm Genetics, Inc., Isis Pharmaceuticals, Inc., ImmunoGen, Inc. and Advanced Magnetics, Inc., each of which is a publicly traded company. Essential Therapeutics and its wholly owned subsidiaries filed for protection under Chapter 11 of the United States Bankruptcy Code in May 2003. The plan of reorganization for Essential Therapeutics became effective in October 2003 by order of the United States Bankruptcy Court for the District of Delaware, and Essential Therapeutics was renamed Trine Pharmaceuticals, Inc. in November 2003.
J. Donald deBethizy, Ph.D. has been our Chief Executive Officer and a member of our board of directors since August 2000. Dr. deBethizy has been our President since March 1997. From March 1985 to March 1997, Dr. deBethizy worked for R.J. Reynolds Tobacco Company in various capacities, most recently as vice president of product evaluation, research and development. Dr. deBethizy has been an adjunct professor in the Department of Physiology and Pharmacology at Wake Forest University School of Medicine since October 1991 and has been an adjunct professor of toxicology in the Integrated Toxicology Program at Duke University since May 1988.
Merouane Bencherif, M.D., Ph.D.has been our Vice President, Preclinical Research since August 2002. He was our Vice President, Biological Sciences from August 2000 to August 2002 and our Senior Manager and Director of Pharmacology and Clinical Sciences from February 1999 to August 2000. From July 1993 to February 1999, Dr. Bencherif worked for R.J. Reynolds Tobacco Company’s Research and Development (Pharmacology) Department in various capacities as a scientist, most recently as a master scientist from March 1998 to February 1999. Dr. Bencherif was an adjunct assistant professor from March 1996 to March 2002 and, since March 2002, has been an associate professor in the Department of Physiology and Pharmacology at Wake Forest University School of Medicine.
Jeffrey P. Brennan has been our Vice President, Business and Commercial Development since September 2003. From September 2000 to May 2003, Mr. Brennan was vice president, commercial development at Sanofi-Synthélabo Inc., a publicly traded global pharmaceutical company based in Paris, France. From November 1996 to September 2000, Mr. Brennan served as vice president, business development at Sanofi-Synthélabo.
William S. Caldwell, Ph.D. has been our Vice President, Drug Discovery and Development since August 2000. From January 1999 to August 2000, Dr. Caldwell was our Director, Chemistry and Operations.
Geoffrey C. Dunbar, M.D. has been our Vice President, Clinical Development and Regulatory Affairs since June 2001. From January 1997 to June 2001, Dr. Dunbar was vice president, clinical development – neurosciences at Bristol-Myers Squibb Company, a publicly traded global pharmaceutical company.
Alan A. Musso has been our Vice President, Chief Financial Officer, Treasurer and Secretary since February 2002. From February 2001 to February 2002, Mr. Musso was vice president and chief financial officer of Osiris Therapeutics, Inc., a privately held biotechnology company. From April 1997 to February 2001, Mr. Musso was the chief financial officer for Cato Research & Cato Holding Company, a privately held global contract research organization. Mr. Musso also was the chief financial officer of Vascular Genetics, Inc., a privately held gene therapy company, from October 1997 to February 2000. In addition, Mr. Musso was employed by Pfizer Inc., a publicly traded global pharmaceutical company, from April 1989 to December 1994, first as a senior auditor and then as a general accounting manager for one of Pfizer’s manufacturing facilities. Mr. Musso is a certified public accountant and a certified management accountant.
M. James Barrett, Ph.D. has been a member of our board of directors since December 2002. Since September 2001, Dr. Barrett has been a general partner of New Enterprise Associates, a venture capital firm that focuses on the medical and life sciences and information technology industries. From 1997 to 2001, he was chairman and chief executive officer of Sensors for Medicine and Science, Inc., a privately held company that he founded and which develops optical chemical sensing technologies. He continues to serve as its chairman and is a member of the boards of directors of the publicly traded companies MedImmune, Inc. and Pharmion Corporation.
Charles A. Blixt has been a member of our board of directors since August 2000. Since January 1998, he has been executive vice president and general counsel of R.J. Reynolds Tobacco Company. Since June 1999, he has been executive vice president, general counsel and assistant secretary of R.J. Reynolds Tobacco Holdings, Inc., the parent company of R.J. Reynolds Tobacco Company.
G. Steven Burrill has been a member of our board of directors since August 2000. Since January 1994, he has been chief executive officer of Burrill & Company LLC, a merchant bank that he founded. Prior to founding Burrill & Company LLC, Mr. Burrill spent 27 years with Ernst & Young LLP, including the last 17 as a partner of the firm. He is chairman of the board of Paradigm Genetics, Inc. and a member of the boards of directors of DepoMed, Inc. and Third Wave Technologies, Inc., each of which is a publicly traded company.
Errol B. De Souza, Ph.D.has been a member of our board of directors since January 2004. Since March 2003, he has been president, chief executive officer and a director of Archemix Corporation, a privately held biotechnology company. From September 2002 to March 2003, he was president, chief executive officer and a director of Synaptic Pharmaceutical Corporation, a publicly traded biopharmaceutical company that was acquired by H. Lundbeck A/S in March 2003. From December 1999 to September 2002, he was senior vice president and site head of U.S. drug innovation & approval (research and development) of Aventis Pharma SA, a pharmaceutical company formed by the merger of Hoechst Marion Roussel and Rhone-Poulenc Rorer Inc. From September 1998 until December 1999, Dr. De Souza was senior vice president and global head, lead generation of Hoechst Marion Roussel. In 1992, Dr. De Souza co-founded Neurocrine Biosciences, Inc., a publicly traded biopharmaceutical company. Dr. De Souza is a member of the boards of directors of IDEXX Laboratories, Inc. and Palatin Technologies, Inc., each of which is a publicly traded company.
Elaine V. Jones, Ph.D. has been a member of our board of directors since August 2000. Since August 2003, she has been a general partner of EuclidSR Associates, L.P., which is the general partner of EuclidSR
Partners, L.P., a venture capital fund that focuses on life sciences and information technology companies. Dr. Jones was an investment manager from June 1999 to September 2001, and was a vice president from September 2001 to August 2003, for S.R. One, Limited, a venture capital subsidiary of SmithKline Beecham.
John P. Richardhas been a member of our board of directors since November 2002. Since April 1999, he has been an independent biotechnology consultant. He also has been a business advisor to GPC Biotech AG, a drug discovery and development company based in Munich, Germany and traded on the Frankfurt Stock Exchange, since April 1999.Prior to April 1999, Mr. Richard served as executive vice president, business development of SEQUUS Pharmaceuticals, Inc., a publicly traded biotechnology company that became a wholly owned subsidiary of ALZA Corporation in March 1999.
Alan G. Walton, Ph.D.has been a member of our board of directors since March 2003. He joined Oxford Partners, a venture capital firm, as a general partner in March 1987. In July 1992, Dr. Walton founded Oxford Bioscience Partners, a life science venture capital firm where he is a general partner. He is chairman of the board of directors of the management company Oxford Bioscience IV Corporation and serves as a member of the board of directors of Alexandria Real Estate Equities, Inc., a publicly traded company. Dr. Walton is also a founder of Human Genome Sciences, Inc. and Gene Logic Inc., both of which are publicly traded companies.
Board Composition
Our board of directors consists of nine members, each of whom was elected in accordance with the terms of a stockholders agreement that will terminate upon the completion of this offering. With the exception of Dr. deBethizy, all of our directors are “independent directors” within the meaning of NASDAQ regulations. There are no family relationships among any of our directors or executive officers.
Following the completion of this offering, our board of directors will consist of nine members divided into three classes:
We will determine the classification of each director after the offering has been completed. At each annual meeting of stockholders after the initial classification, or at a special meeting in lieu of an annual meeting, a class of directors will be elected to serve for a three-year term to succeed the directors of the same class whose terms are then expiring.
Board Committees
Audit Committee. The members of our audit committee are Messrs. Burrill, Blixt and Richard. Mr. Burrill chairs the committee. The audit committee assists the board of directors in its oversight of our accounting, financial reporting and internal control functions. Specific responsibilities of our audit committee include:
Compensation Committee. The members of our compensation committee are Mr. Skaletsky, Dr. Barrett and Dr. Jones. Mr. Skaletsky chairs the committee. The purpose of our compensation committee is to discharge
the responsibilities of our board of directors relating to compensation of our executive officers. Specific responsibilities of our compensation committee include:
Governance and Nominating Committee. Upon the completion of this offering, the members of our governance and nominating committee will be Messrs. Skaletsky and Blixt and Dr. De Souza. Mr. Skaletsky will chair the committee. Specific responsibilities of our governance and nominating committee will include:
Compensation Committee Interlocks and Insider Participation
None of our executive officers serves as a member of the board of directors or compensation committee, or other committee serving an equivalent function, of any other entity that has one or more of its executive officers serving as a member of our board of directors or our compensation committee. None of the members of our compensation committee has ever been our employee.
Director Compensation
In the past, each of our directors who is not our employee has received a nonqualified stock option to purchase 25,000 shares of our common stock upon his or her initial election to our board of directors. Additionally, upon each non-employee director’s annual reelection, he or she has been granted a nonqualified stock option to purchase 7,500 shares of common stock. However, our chairman received a nonqualified stock option to purchase 35,000 shares upon his or her initial election and a nonqualified stock option to purchase 12,500 shares upon his or her annual reelection. Each of these options:
In lieu of any such nonqualified stock option, each non-employee director could elect to receive a restricted stock award for the same number of shares of stock at a purchase price of $0.01 per share. Each non-employee director that is not a designee of one of our investors or a group of our investors has received, in addition to the equity compensation described above, cash compensation in the amount of $10,000 per year as an annual
retainer. Each director is reimbursed for expenses incurred in connection with his or her attendance at meetings of the board of directors and its committees. We have not historically paid any additional compensation for service on any committees of the board of directors.
We have adopted a new director compensation program that will become effective concurrently with the completion of this offering. Each non-employee director will receive an annual cash retainer of $20,000 payable in quarterly installments. Each member of a committee of the board will receive an additional annual cash retainer of $2,500, the chairman of our audit committee will receive an additional annual cash retainer of $7,500 and the chairman of each of our compensation and governance and nominating committees will each receive an additional annual cash retainer of $2,500. Each non-employee director also will receive an option for 25,000 shares upon initial election as a director and an option for 7,500 shares upon annual reelection. The chairman of the board will receive an option for 10,000 shares upon initial election as chairman, in addition to the option for 25,000 shares upon initial election as a director, and an option for 12,500 shares upon annual reelection. For more information, please see “Executive Compensation—Stock-Based Plans—2004 Stock Incentive Plan.”
Executive Compensation
The following table sets forth other information regarding compensation awarded to, earned by or paid to our chief executive officer and our five other most highly compensated executive officers during the year ended December 31, 2003 whose annual salary and bonus exceeded $100,000 during the year ended December 31, 2003. We refer to these officers in this prospectus as our named executive officers.
Summary Compensation Table
| Annual Compensation | Long-Term Compensation | ||||||||||
Name and Principal Position | Salary | Bonus | Shares Underlying Options (#) (1) | All Other Compensation (2) | |||||||
J. Donald deBethizy, Ph.D. President and Chief Executive Officer | $ | 275,000 | $ | 66,000 | 1,969,332 | $ | 12,000 | ||||
Merouane Bencherif, M.D., Ph.D. Vice President, Preclinical Research | 161,000 | 38,640 | 585,623 | 11,485 | |||||||
Jeffrey P. Brennan (3) Vice President, Business and Commercial Development | 75,000 | 13,500 | 169,000 | 4,500 | |||||||
William S. Caldwell, Ph.D. Vice President, Drug Discovery and Development | 161,750 | 29,115 | 534,385 | 11,314 | |||||||
Geoffrey C. Dunbar, Ph.D. Vice President, Clinical Development and Regulatory Affairs | 246,750 | 44,415 | 668,396 | 12,000 | |||||||
Alan A. Musso Vice President, Chief Financial Officer, Treasurer and Secretary | 181,731(4) | 32,400 | 533,671 | 12,000 | |||||||
Stock Options
The following table sets forth information regarding grants of stock options to purchase shares of our common stock to our named executive officers during the year ended December 31, 2003.
The potential realizable values set forth in the following table are calculated based on the term of the option at the time of grant and reflect gains that could be achieved for the options if exercised at the end of the option term. The 5% and 10% assumed annual rates of compounded stock price appreciation are required by the Securities and Exchange Commission and do not represent our estimate or projection of our future stock price performance. Actual gains, if any, on stock option exercises depend on the future performance of the common stock and the date on which the options are exercised.
Option Grants in Last Fiscal Year
|
|
|
|
| ||||||||||||
| ||||||||||||||||
| ||||||||||||||||
| ||||||||||||||||
| ||||||||||||||||
| ||||||||||||||||
| ||||||||||||||||
Option Exercises and Year-End Option Values
The following table sets forth information regarding the number of shares of our common stock issued upon option exercises by our named executive officers during the year ended December 31, 2003 and the value realized by our named executive officers. The table also sets forth information regarding the number and value of unexercised stock options held by our named executive officers as of December 31, 2003. There was no public trading market for our common stock as of December 31, 2003. Accordingly, as permitted by the rules of the Securities and Exchange Commission, we have calculated the value of the unexercised in-the-money options at fiscal year end by determining the difference between the exercise price per share and an assumed fair market value of our common stock as of December 31, 2003 equal to an assumed initial offering price of $ per share, the mid-point of the estimated price range shown on the cover page of this prospectus.
Aggregated Option Exercises in Last Fiscal Year and Fiscal Year-End Option Values
Number of Shares Acquired on Exercise | Value Realized | Number of Securities Underlying Unexercised Options Held at December 31, 2003 | Value of Unexercised In-the-Money Options at December 31, 2003 | ||||||||||||
Name | Exercisable | Unexercisable | Exercisable | Unexercisable | |||||||||||
J. Donald deBethizy, Ph.D. | 101,530 | $ | 21,321 | 898,338 (1) | 1,390,772 | $ | $ | ||||||||
Merouane Bencherif, M.D., Ph.D. | — | — | 376,270 | 413,048 | |||||||||||
Jeffrey P. Brennan | — | — | 52,334 (1) | 116,666 | |||||||||||
William S. Caldwell, Ph.D. | — | — | 378,306 (1) | 359,893 | |||||||||||
Geoffrey C. Dunbar, Ph.D. | — | — | 399,503 (1) | 458,120 | |||||||||||
Alan A. Musso | 6,000 | 70 | 267,929 (1) | 424,742 | |||||||||||
Employment Agreements
We have entered into employment agreements with each of our named executive officers. Each employment agreement continues until terminated by either party to the agreement, with the exception of Mr. Brennan’s employment agreement, which is set to expire on December 31, 2007.
Under the terms of these employment agreements, Dr. deBethizy is employed as our Chief Executive Officer and President at a minimum annual base salary of $225,000; Dr. Dunbar is employed as our Vice President, Clinical Development and Regulatory Affairs at a minimum annual base salary of $246,750; Mr. Brennan is employed as our Vice President, Business and Commercial Development at a minimum annual base salary of $225,000; Mr. Musso is employed as our Vice President and Chief Financial Officer at a minimum annual base salary of $180,000; Dr. Bencherif is employed as our Vice President, Preclinical Research at a minimum annual base salary of $135,000; and Dr. Caldwell is employed as our Vice President, Drug Discovery and Development at a minimum annual base salary of $135,000. For 2004, the base salary of Dr. deBethizy is $283,250; the base salary of Dr. Dunbar is $254,150; the base salary of Mr. Brennan is $225,000; the base salary of Mr. Musso is $190,000; the base salary of Dr. Bencherif is $170,000; and the base salary of Dr. Caldwell is $170,000.
The employment agreements provide that the annual base salaries of each of the named executive officers will be reviewed and are subject to increase in accordance with our policies and procedures, and in addition, will be increased annually as necessary to be consistent with the median base salaries of employees in similar positions at comparable companies as described in the then current Radford Biotechnology Compensation Report.
In addition to annual base salary, each named executive officer is eligible to receive awards under our 2000 equity incentive plan and earn an annual bonus equal to a percentage of his annual base salary. The employment agreements provide that Dr. deBethizy is eligible to earn an annual bonus of up to 35% of his annual base salary; each of Dr. Dunbar and Mr. Brennan is eligible to earn an annual bonus of up to 30% of his annual base salary; and each of Mr. Musso and Drs. Bencherif and Caldwell is eligible to earn an annual bonus of up to 25% of his annual base salary. In 2001, our board of directors increased the annual bonus for Dr. deBethizy to up to 40% of his annual base salary. In 2002, our board of directors increased the annual bonus for each of Drs. Bencherif and Caldwell to up to 30% of his annual base salary and in 2003 increased the annual bonus for Mr. Musso to up to 30% of his annual base salary. Our board of directors or compensation committee, in their discretion, may increase the annual bonus for each named executive officer beyond these percentages.
Under the terms of the employment agreements, the named executive officers cannot disclose any of our proprietary information during the periods of their employment. In addition, the employment agreements prohibit the named executive officers from soliciting, on behalf of themselves or any entity other than us, any of our customers or clients for the period of employment and nine months following termination of employment, and in the case of Dr. deBethizy, one year following termination. Furthermore, any inventions, discoveries, improvements and developments made by the named executive officers during their employment with us become and remain our property.
If a named executive officer’s employment terminates for any reason, the named executive officer is entitled to receive a lump sum equal to any base salary, bonus and other compensation earned and due but not paid through the effective date of termination. In addition, if we terminate a named executive officer’s employment other than for just cause or a named executive officer terminates his employment for good reason, in each case as that term is defined in his agreement, he is entitled to receive:
Stock Option and Other Compensation Plans
2000 Equity Incentive Plan
We maintain a 2000 equity incentive plan, which we refer to as our 2000 plan, that our board of directors and stockholders have approved. As of March 31, 2004, an aggregate of 9,216,657 shares of common stock had been authorized for issuance under our 2000 plan, of which options to purchase an aggregate of 8,024,394 shares of common stock were outstanding at a weighted average exercise price of $0.64 per share, 85,000 shares of common stock were issued and outstanding in the form of restricted stock and 448,274 shares of common stock were available for future grant. Upon completion of this offering, 1,113,760 shares of common stock subject to unvested options outstanding as of March 31, 2004 will immediately vest.
Our 2000 plan provides for the grant of a variety of stock-based awards, including incentive stock options, nonqualified stock options, stock appreciation rights, performance awards and restricted stock, to our employees, directors, independent contractors, consultants and advisors.
Administration of the Plan. Our 2000 plan is administered by the compensation committee of our board of directors, which, among other things, determines the terms and recipients of grants under the 2000 plan.
Options. Recipients of stock options under our 2000 plan have the right to purchase a stated number of shares of common stock at a stated exercise price, subject to any other terms and conditions that may be stated in connection with the option grant. We may grant options at an exercise price equal to, less than or greater than the fair market value of our common stock on the date of grant, except that we may not grant incentive stock options and options intended to qualify as performance-based compensation under Section 422 of the Internal Revenue Code to optionees holding more than 10% of the voting power of all shares of our capital stock at an exercise price less than 110% of the fair market value of our common stock on the date of grant. Grant recipients may pay the exercise price of stock options by various methods permitted under our 2000 plan. Unless modified with respect to any particular grant:
Stock Awards. We may grant stock awards to participants subject to certain restrictions or no restrictions. Until they are vested and earned, unless an individual award agreement provides otherwise, grantees will not have the right to vote shares of restricted stock or the right to receive dividends or other distributions paid on such shares. If a grantee’s employment or other service terminates during the restriction period or if any other conditions are not met, the restricted stock still subject to restrictions will terminate, unless an individual award agreement provides otherwise, and the shares must be immediately returned to us.
Significant Transactions. If:
all awards outstanding under our 2000 plan would become immediately vested and exercisable unless, in the case of a merger, consolidation, share exchange or asset sale or disposition, the board of directors or compensation committee determines that outstanding awards will not become immediately vested and exercisable because steps have been taken, such as the assumption of the awards or substitution of substantially equivalent awards by the other party, as it deems equitable to protect the rights of participants in our 2000 plan. Upon completion of this offering, all awards outstanding under our 2000 plan granted prior to August 20, 2003 would become immediately vested and exercisable.
Termination and Amendment. We may grant awards under our 2000 plan until August 21, 2010, unless our 2000 plan is terminated prior to that date. The board of directors may amend or terminate our 2000 plan at any time, subject to the rights of holders of outstanding awards. Our 2004 stock incentive plan, which we refer to as our 2004 plan, is intended to serve as the successor equity incentive program to our 2000 plan. However, our board of directors may not amend our 2000 plan without stockholder approval if stockholder approval is required in order for grants of incentive stock options to meet the requirements of Section 422 of the Internal Revenue Code, or if stockholder approval is required in order to exempt compensation under our 2000 plan from the deduction limit under Section 162(m) of the Internal Revenue Code.
2004 Stock Incentive Plan
Introduction. Our 2004 plan is intended to serve as the successor equity incentive program to our 2000 plan.Our 2004 plan will become effective on the date that the underwriting agreement for this offering is signed. At that time, all of the shares reserved for grant under our 2000 plan will be transferred to our 2004 plan and no further options will be granted under our 2000 plan.
Subject to adjustments as provided in our 2004 plan, the maximum number of shares that we may issue pursuant to awards granted under our 2004 plan may not exceed the sum of (i)shares and (ii) up toshares of common stock (a) remaining available for issuance as of the effective date under our 2000 plan or any other employee stock incentive plan that we maintain prior to the effective date and/or (b) subject to an award granted under our 2000 plan or any other prior plan, which award is forfeited, cancelled, terminated, expires or lapses for any reason, without the issuance of shares pursuant to the award. The maximum number of shares of common stock that we may issue under our 2004 plan pursuant to the grant of (i) incentive stock options isand (ii) restricted awards is. In any calendar year, (i) we may not grant to any participant options and stock appreciation rights, or SARs, that are not related to an option for more thanshares of common stock; (ii) we may not grant to any participant awards for more thanshares of common stock; and (iii) participants may not receive awards paid in cash having an aggregate dollar value in excess of $, subject to adjustments as provided in our 2004 plan. For purposes of these restrictions, we will treat an option and related SAR as a single award. The following will not be included in calculating the share limitations set forth above: (i) dividends, including dividends paid in shares of common stock, or dividend equivalents paid in cash in connection with outstanding awards; (ii) awards which by their terms are settled in cash rather than the issuance of shares; (iii) any shares subject to an award under our 2004 plan that is forfeited, cancelled, terminated, expires or lapses for any reason without the issuance of shares underlying the award; and (iv) any shares a participant surrenders or we withhold to pay the option or purchase price for an award or use to satisfy any tax withholding requirement in connection with the exercise, vesting or earning of an award if, in accordance with the terms of our 2004 plan, a participant pays such option or purchase price or satisfies such tax withholding by either tendering previously owned shares or having us withhold shares.
We may adjust the number of shares reserved for issuance under our 2004 plan and the terms of awards in the event of an adjustment in our capital stock structure or one of our affiliates due to a merger, stock split, stock dividend or similar event.
Purpose and Eligibility. The purpose of our 2004 plan is to encourage and enable selected employees and our directors and independent contractors to acquire or increase their holdings of common stock and other proprietary interests in us in order to promote a closer identification of their interests with those of us and our stockholders, thereby further stimulating their efforts to enhance our efficiency, soundness, profitability, growth and stockholder value. The purpose will be carried out by the granting of awards to selected participants. We may grant awards under our 2004 plan which include incentive stock options and nonqualified stock options; SARs; restricted awards in the form of restricted stock awards and restricted stock units; performance awards in the form of performance shares and performance units; phantom stock awards; director options in the form of
initial options and annual options; and dividend equivalent awards. We discuss the material terms of each type of award below.
Administration; Amendment and Termination. Our board of directors, or upon its delegation, the compensation committee of our board of directors, will administer our 2004 plan. In this discussion, we refer to our board of directors and the compensation committee collectively as the administrator. Under the terms of our 2004 plan, the administrator has full and final authority to take any action with respect to our 2004 plan, including, without limitation, the authority to: (i) determine all matters relating to awards, including selection of individuals to be granted awards, the types of awards, the number of shares, if any, of common stock subject to an award, and the terms, conditions, restrictions and limitations of an award; (ii) prescribe the form or forms of agreements evidencing awards granted under our 2004 plan; (iii) establish, amend and rescind rules and regulations for the administration of our 2004 plan; and (iv) construe and interpret our 2004 plan, awards and award agreements made under the plan, interpret rules and regulations for administering the plan and make all other determinations deemed necessary or advisable for administering the plan.
In certain circumstances and subject to certain terms and conditions, the administrator may delegate to one or more of our officers the authority to grant awards, and to make any or all of the determinations reserved for the administrator in our 2004 plan with respect to such awards.
Our board of directors may amend, alter or terminate our 2004 plan at any time, subject to the following: (i) stockholder approval is required of any amendment if such approval is required by applicable law, rule or regulation; and (ii) except for anti-dilution adjustments made under our 2004 plan, the option price for any outstanding option or base price of any outstanding SAR may not be decreased after the date of grant, nor may any participant surrender any outstanding option or SAR to us as consideration for the grant of a new option or SAR with a lower option or base price than the original option or SAR, as the case may be, without stockholder approval of any such action.
The administrator has the authority to make adjustments to awards upon the occurrence of certain unusual or nonrecurring events, if the administrator determines that such adjustments are appropriate in order to prevent dilution or enlargement of the benefits or potential benefits intended to be made available under our 2004 plan or necessary or appropriate to comply with applicable laws, rules or regulations. The administrator may cause any award granted under our 2004 plan to be cancelled in consideration of an alternative award or cash payment of an equivalent cash value, as determined by the administrator, made to the holder of the cancelled award. The administrator also may determine, in its discretion, that a participant’s rights, payments and/or benefits with respect to an award, including but not limited to any shares issued or issuable and/or cash paid or payable with respect to an award, will be subject to reduction, cancellation, forfeiture or recoupment upon the occurrence of certain specified events, in addition to any otherwise applicable vesting or performance conditions of an award.
Options. Our 2004 plan authorizes the grant of both incentive stock options and nonqualified stock options, both of which are exercisable for shares of common stock, although incentive stock options may only be granted to our employees. The administrator will determine the option price at which a participant may exercise an option and the option price must be:
Unless an individual award agreement provides otherwise, a participant may pay the option price in the form of cash or cash equivalent; in addition, where the administrator and applicable laws, rules and regulations permit, a participant may also make payment:
At the time of option grant, the administrator will determine the term and conditions of an option and the period or periods during which a participant may exercise an option and, in the case of incentive stock options, the option term may not exceed 10 years, or five years with respect to an employee who owns stock and who possesses more than 10% of the total combined voting power of all classes of our stock or stock of our parent or subsidiary corporation, if any. Options are also subject to certain restrictions on exercise if the participant terminates employment. The administrator has authority to establish other terms and conditions related to options.
Director Options. Each non-employee director who is first elected or appointed to our board of directors after the public offering date will receive an initial option to purchase 25,000 shares of common stock on the fifth business day after such director is first elected or appointed to our board of directors. A non-employee director who is first elected or appointed as chairman of the board also will receive an initial option for 12,500 shares. In addition, we will grant to each non-employee director, on an annual basis commencing with the 2005 annual meeting of stockholders, a director option to purchase 7,500 shares of common stock, or, in the case of the chairman of the board, an annual option for 12,500 shares, on the fifth business day after the annual or other stockholders meeting, provided that such director continues to serve as a member of our board of directors as of such date. Director options will be designated as nonqualified options. The option price at which a director may exercise a director option will be 100% of the fair market value per share of the common stock on the date the option is granted. Each initial option will vest and become exercisable on the first anniversary of the date of grant with respect to one-third of the shares subject to the option. Each initial option will vest with respect to the remaining two-thirds of the shares subject to the option on a pro rata quarterly basis over the next two years, so that the option will be vested in full as of the third anniversary of the date of grant if the director continues in service during such period. Each annual option will vest in full on the first anniversary of the date of grant. The term of a director option may not exceed 10 years from the date of grant. Director options are also subject to certain restrictions on exercise if the director’s service on our board of directors terminates. The administrator also has authority to establish other terms and conditions related to director options.
Stock Appreciation Rights. Under the terms of our 2004 plan, we may grant SARs to the holder of an option with respect to all or a portion of the shares of common stock subject to the option or we may grant SARs separately. The holder of an SAR may receive consideration paid either (i) in cash; (ii) shares of common stock valued at fair market value on the date of the SAR exercise; or (iii) a combination of cash and shares of common stock, as the administrator determines. Upon exercise of an SAR, a participant is entitled to receive from us consideration in an amount determined by multiplying:
Notwithstanding the foregoing, the administrator may limit the amount payable in its discretion. The base price may be no less than 100% of the fair market value per share of the common stock on the date the SAR is granted. We may pay consideration upon exercise of an SAR currently or on a deferred basis.
SARs are exercisable according to the terms established by the administrator and stated in the applicable award agreement. Upon the exercise of an SAR granted to the holder of an option, the option is deemed to be cancelled to the extent of the number of shares as to which the holder of an option exercises the SAR. No participant may exercise an SAR more than 10 years after it was granted, or such shorter period as may apply to related options. Each award agreement will set forth the extent to which the holder of an SAR will have the right to exercise an SAR following termination of the holder’s employment or service with us.
Restricted Awards. Subject to the limitations of our 2004 plan, the administrator may in its sole discretion grant restricted awards to such individuals in such numbers, upon such terms and at such times as the administrator shall determine. Restricted awards may be in the form of restricted stock awards and/or restricted stock units that are subject to certain conditions, which conditions must be met in order for the restricted award to vest and be earned, in whole or in part, and no longer subject to forfeiture. Restricted stock awards may be payable in shares of common stock. Restricted stock units may be payable in cash or whole shares of common stock, or partly in cash and partly in whole shares of common stock, in accordance with the terms of our 2004 plan and the discretion of the administrator.
The administrator has authority to determine the nature, length and starting date of the period during which a participant may earn a restricted award and will determine the conditions that must be met in order for a restricted award to be granted or to vest or be earned. These conditions may include:
However, restricted awards that vest based solely on continued service or the passage of time will be subject to a minimum restriction period of one year, except in the case of restricted awards assumed or substituted in connection with mergers or other business transactions, restricted awards granted in connection with hiring a participant and/or restricted awards granted under an incentive compensation or bonus program.
In the case of restricted awards based upon performance criteria, or a combination of performance criteria and continued service, the administrator will determine the performance measures applicable to such restricted awards, which performance measures may be based upon such corporate, business unit or division and/or individual performance factors and criteria as the administrator in its discretion may deem appropriate; provided, however, that such performance factors will be limited to the specific performance measures listed below.
The administrator has authority to determine whether and to what degree restricted awards have vested and been earned and are payable. If a participant’s employment or service is terminated for any reason and all or any part of a restricted award has not vested or been earned pursuant to the terms of our 2004 plan and the individual award, the participant will forfeit the award unless the administrator determines otherwise.
Performance Awards. Subject to the limitations of our 2004 plan, the administrator may in its discretion grant performance awards to such eligible individuals upon such terms and conditions and at such times as the administrator shall determine. Performance awards may be in the form of performance shares and/or performance units. An award of a performance share is a grant of a right to receive shares of our common stock, the cash value thereof or a combination thereof in the administrator’s discretion, which is contingent upon the achievement of performance or other objectives during a specified period and which has a value on the date of grant equal to the fair market value of a share of our common stock. An award of a performance unit is a grant of a right to receive shares of our common stock or a designated dollar value amount of common stock that is contingent upon the achievement of performance or other objectives during a specified period, and that has an initial value determined in a dollar amount established by the administrator at the time of grant.
The administrator has the authority to determine the nature, length and starting date of the period during which a participant may earn a performance award and will determine the conditions that must be met in order for a performance award to be granted or to vest or be earned. These conditions may include specific performance objectives, continued service or employment for a certain period of time, or a combination of such conditions. In the case of performance awards based on performance criteria, the administrator will determine the performance measures applicable to such awards, which performance measures may be based upon such corporate, business unit or division and/or individual performance factors and criteria as the administrator in its discretion may deem appropriate; provided, however, that such performance factors will be limited to the specific performance measures listed below.
The administrator has authority to determine whether and to what degree performance awards have been earned and are payable. If a participant’s employment or service is terminated for any reason and all or part of a performance award has not been earned pursuant to the terms of our 2004 plan and the individual award agreement, the participant will forfeit the award unless the administrator determines otherwise.
Phantom Stock Awards. Subject to the limitations of our 2004 plan, the administrator may in its discretion grant phantom stock awards to such eligible individuals in such numbers, upon such terms and at such times as the administrator shall determine. An award of phantom stock is an award of a number of hypothetical share units with respect to shares of our common stock, with a value based on the fair market value of a share of common stock.
The administrator has the authority to determine whether and to what degree phantom stock awards have vested and are payable. Upon vesting of all or part of a phantom stock award and satisfaction of other terms and conditions that the administrator determines, the holder of a phantom stock award will be entitled to a payment of an amount equal to the fair market value of one share of our common stock with respect to each such phantom stock unitratably in all assets that has vested. We may make payment in cash, shares of common stock, or a combination of cash and stock, as determined by the administrator. The administrator may determine the forms and terms of payment of phantom stock awards in accordance with our 2004 plan. If a participant’s employment or service is terminated for any reason and all or any part of a phantom stock award has not vested and become payable pursuant to the terms of our 2004 plan and the individual award, the participant will forfeit the award unless the administrator determines otherwise.
Dividend and Dividend Equivalents. The administrator may, in its sole discretion, provide that awards granted under our 2004 plan earn dividends or dividend equivalents. We may pay such dividends or dividend equivalents currently or credit such dividends or dividend equivalents to a participant’s account,remain, subject to such restrictions and conditions as the administrator may establish with respect to the crediting of an account, including reinvestment in additional shares of common stock or share equivalents.
Change in Control. Upon a change in control as defined in our 2004 plan, and unless an award agreement provides otherwise, our 2004 plan provides that: (i) all options and SARs outstanding as of the date of the change in control will become fully exercisable, whether or not then otherwise exercisable; and (ii) any restrictions applicable to any restricted award, performance award and/or phantom stock award will be deemed to have been
met, and such awards will become fully vested, earned and payable to the fullest extent of the original grant of the applicable award. However, our 2004 plan authorizes the administrator, in the event of a merger, share exchange, reorganization or other business combination affecting us or one of our affiliates, to determine that any or all awards will not vest or become exercisable on an accelerated basis, if we or the surviving or acquiring corporation takes action, including but not limited to the assumption of awards or the grant of substitute awards, that, in the opinion of the administrator, is equitable or appropriate to protect the rights and interest of participants under our 2004 plan.
Transferability. Incentive stock options are not transferable other than by will or the laws of intestate succession or, in the administrator’s discretion, as may otherwise be permitted in accordance with Section 422 of the Internal Revenue Code and related regulations. Nonqualified stock options, director options and SARs are not transferable other than by will or the laws of intestate succession, except as permitted by the administrator in a manner consistent with the registration provisions of the Securities Act. Restricted awards, performance awards and phantom stock awards that have not vested and been earned are not transferable, including by sale, assignment, pledge or hypothecation, other than by will or the laws of intestate succession, and participants may not sell, transfer, assign, pledge or otherwise encumber shares subject to such awards until the restriction period and/or performance period has expired and until all conditions to vesting and/or earning the award have been met.
General Federal Income Tax Consequences. Under current federal laws, in general, recipients of awards and grants of nonqualified stock options, SARs, restricted stock, dividend equivalents, performance awards and stock payments under our 2004 plan are taxable under Section 83 of the Internal Revenue Code upon their receipt of common stock or cash with respect to such awards or grants and, subject to Section 162(m) of the Internal Revenue Code and certain reporting requirements, we will be entitled to an income tax deduction with respect to the amounts taxable as ordinary income to such recipients. Under Sections 421 and 422 of the Internal Revenue Code, recipients of incentive stock options are generally not taxable on their receipt of common stock upon their exercises of incentive stock options if the option stock is held for specified minimum holding periods and, in such event, we would not be entitled to income tax deductions with respect to such exercises.
Performance-Based Compensation—Section 162(m) Requirements. Our 2004 plan is structured to comply with the requirements imposed by Section 162(m) of the Internal Revenue Code and related regulations in order to preserve, to the extent practicable, our tax deduction for awards made under our 2004 plan to covered employees. Section 162(m) of the Internal Revenue Code generally denies an employer a deduction for compensation paid to covered employees, which are generally the named executive officers, of a publicly held corporation in excess of $1,000,000 unless the compensation is exempt from the $1,000,000 limitation because it is performance-based compensation.
In order to qualify as performance-based compensation, we must pay the compensation under our 2004 plan to covered employees under pre-established objective performance goals that a committee comprised of outside directors determines and certifies. In addition to other requirements for the performance-based exception, we must advise stockholders, and stockholders must approve, the material terms or changes in material terms of the performance goals under which compensation is to be paid. Material terms include the individuals eligible to receive compensation, a description of the business criteria on which the performance goals are based, and either the maximum amount of the compensation to be paid or the formula used to calculate the amount of compensation if the performance goals are met.
As proposed, our 2004 plan limits the maximum amount of awards that we may grant to any employee. In particular, in any calendar year, (i) we may not grant to any participant options and SARs that are not relatedgranted to an option for more thanshares of common stock; (ii) we may not grant to any participant awards for more thanshares of common stock; and (iii) no participant may receive awards paid in cash having an aggregate dollar value in excess of $. Further, with respect to performance-based restricted awards and performance awards, and in some cases, certain other types of awards, payable to covered employees that are intended to be eligible for the compensation limitation exception available under Section 162(m) and related regulations, our 2004 plan limits performance measures to one or more of the following: cash flow, return on
equity, return on assets, earnings per share, achievement of clinical development or regulatory milestones, operations expense efficiency milestones, consolidated earnings before or after taxes and including earnings before interest, taxes, depreciation and amortization, net income, operating income, book value per share, return on investment, return on capital, improvements in capital structure, expense management, profitability of an identifiable business unit or product, maintenance or improvement of profit margins, stock price or total stockholder return, market share, revenues or sales, costs, working capital, economic wealth created, strategic business criteria, efficiency ratios, achievement of division, group, function or corporate financial, strategic or operational goals and comparisons with stock market indices or performances of metrics of peer companies.
To the extent that Section 162(m) of the Internal Revenue Code is applicable, the administrator will, within the time and in the manner prescribed by Section 162(m) of the Internal Revenue Code and related regulations, define in an objective fashion the manner of calculating the performance measures it selects to use for participants during any specific performance period. We may adjust or modify such performance factors due to extraordinary items, transactions, events or developments, or in recognition of, or in anticipation of, any other unusual or nonrecurring events affecting us or our financial statements, or in response to, or in anticipation of, changes in applicable laws, regulations, accounting principles or business conditions, in each case as the administrator may determine.
Targacept Retirement Savings Plan—401(k) Plan
Our employees are eligible to participate in our 401(k) plan. Under our 401(k) plan, eligible employees may elect to make a salary reduction contribution up to the statutorily prescribed annual limit. The 401(k) plan is intended to qualify under Section 401 of the Internal Revenue Code, so that the contributions by our employees will be deductible when made and income earned on 401(k) plan contributions will not be taxable to our employees until withdrawals are made. We match the contributions of our eligible employees at up to a maximum of 6% of an eligible employee’s salary.
Limitation of Liability and Indemnification
Our certificate of incorporation includes a provision that eliminates the personal liability of our directors for monetary damages to the fullest extent permitted by Section 102(b)(7) of the Delaware General Corporation Law. Under that statute, a director’s liability for monetary damages to us or our stockholders may not be limited with respect to:
Our bylaws provide that we will indemnify and hold harmless any person who is made or threatened to be made a party to any matter because he or she is or was our director or officer or was serving as a director, officer or trustee of another entity, employee benefit plan or enterprise at our request to the fullest extent permitted by the Delaware General Corporation Law. Prior to the completion of this offering, we plan to enter into agreements to indemnify our directors and officers. These agreements, among other things, will indemnify our directors and officers for certain expenses, including attorneys’ fees, judgments, fines and settlement amounts incurred by any such person in any action or proceeding, including any action by us arising out of such person’s services as our director or officer, any of our subsidiaries from time to time or any other company or enterprise to which the person provides services at our request. We believe that these provisions and agreements are necessary to attract and retain qualified persons as directors and officers. Currently, there is no pending litigation or proceeding involving any of our directors, officers or employees for which indemnification is sought, nor are we aware of any threatened litigation that may result in claims for indemnification. We currently maintain directors’ and officers’ liability insurance for each of our directors and officers.
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
Since August 22, 2000, we have engaged in the following transactions with our directors and executive officers and holders of more than 5% of our voting securities and affiliates of our directors, executive officers and 5% stockholders.
Stock Issuances
Issuance of Common Stock
On August 22, 2000, at the time that we became an independent company, we issued and sold an aggregate of 309,424 shares of our common stock at a purchase price per share of $0.001 for an aggregate purchase price of approximately $309. The following table sets forth the number of shares of common stock sold to our founders.
Name | Number of Shares of Common Stock | Aggregate Purchase Price | |||
J. Donald deBethizy, Ph.D. | 135,373 | $ | 135 | ||
Merouane Bencherif, M.D., Ph.D. | 58,017 | 58 | |||
William S. Caldwell, Ph.D. | 58,017 | 58 | |||
Patrick M. Lippiello, Ph.D. | 58,017 | 58 | |||
On August 8, 2002, we issued 47,500 shares of restricted stock to Mr. Skaletsky for an aggregate purchase price of $475. On June 11, 2003, we issued 12,500 shares of restricted stock to Mr. Skaletsky for an aggregate purchase price of $125.
On April 18, 2003, we issued 25,000 shares of restricted stock to Mr. Richard for an aggregate purchase price of $250.
Issuance of Series A Convertible Preferred Stock
On August 22, 2000, we recapitalized our 500 outstanding shares of common stock held by R.J. Reynolds Tobacco Company, our then parent corporation, into 5,000,000 shares of series A convertible preferred stock and a warrant to purchase 1,612,903 shares of common stock at an exercise price of $1.95 per share. All of the shares of series A convertible preferred stock and the warrant were subsequently assigned to R.J. Reynolds Tobacco Holdings, Inc. Each share of series A convertible preferred stock will convert into one share of common stock concurrently with the completion of this offering. The warrant will be cancelled if it is not exercised prior to the completion of this offering. If R.J. Reynolds exercises the warrant in full for cash, we would issue 1,612,903 shares of common stock and receive cash proceeds of approximately $3.1 million. If R.J. Reynolds exercises the warrant on a cashless basis, we would issue shares of common stock, based on an assumed initial public offering price of $ per share.
Mr. Blixt, one of our directors, is the executive vice president and general counsel of R.J. Reynolds Tobacco Company and is executive vice president, general counsel and assistant secretary of its parent company, R.J. Reynolds Tobacco Holdings, Inc.
Issuance of Series B Convertible Preferred Stock
On August 22, November 30, December 5 and December 19, 2000, we issued and sold an aggregate of 6,537,634 shares of our series B convertible preferred stock at a purchase price per share of $4.65 for an aggregate purchase price of approximately $30.4 million. On January 26, 2001, we issued an additional 29,933 shares of our series B convertible preferred stock in partial satisfaction of an outstanding payment obligation. The following table sets forth the number of shares of series B convertible preferred stock sold to our 5% stockholders and their affiliates.
Name | Number of Shares of Series B Preferred Stock | Aggregate Purchase Price | |||
EuclidSR Partners, L.P. | 2,021,505 | $ | 9,399,998 | ||
Burrill & Company LLC | 1,075,269 | 5,000,001 | |||
Advent Private Equity Fund II | 1,075,269 | 5,000,001 | |||
These shares of our series B convertible preferred stock will convert into an aggregate of 9,948,718 shares of our common stock concurrently with the completion of this offering.
Dr. Jones, one of our directors, is a general partner of EuclidSR Associates, L.P., the general partner of EuclidSR Partners, L.P. Mr. Burrill, one of our directors, is the chief executive officer of Burrill & Company LLC.
Issuance of Series C Convertible Preferred Stock
On November 26, 2002 and March 14, 2003, we issued and sold an aggregate of 49,169,138 shares of our series C convertible preferred stock at a purchase price per share of $1.21 for an aggregate purchase price of approximately $59.5 million. The following table sets forth the number of shares of series C convertible preferred stock sold to our 5% stockholders and their affiliates.
Name | Number of Shares of Series C Preferred Stock | Aggregate Purchase Price | |||
New Enterprise Associates 10, Limited Partnership | 12,396,694 | $ | 15,000,000 | ||
Nomura International plc | 8,264,462 | 9,999,999 | |||
Oxford Bioscience Partners IV L.P. | 6,198,347 | 7,500,000 | |||
EuclidSR Partners, L.P. | 5,123,966 | 6,199,999 | |||
Burrill & Company LLC | 1,540,440 | 1,863,932 | |||
Advent Private Equity Fund II | 1,540,440 | 1,863,932 | |||
These shares of our series C convertible preferred stock will convert into an aggregate of 37,882,020 shares of our common stock concurrently with the completion of this offering.
Dr. Barrett, one of our directors, is a general partner of NEA Partners 10, Limited Partnership, the general partner of New Enterprise Associates 10, Limited Partnership. Dr. Walton, one of our directors, is a general partner of OBP Management IV L.P., the general partner of Oxford Bioscience Partners IV L.P.
Registration Rights
Pursuant to the terms of an investor rights agreement that we entered into with the holders of ourany class or series A, series B and series C convertibleof preferred stock on November 26, 2002, we granted registration rights to these holders. For a more detailed description of these registration rights, see “Description of Capital Stock—Registration Rights.”stock.
Loan Agreement
In May 2002, we borrowed $2.5 million from R.J. Reynolds Tobacco Holdings, Inc. to finance equipment and other fixed assets. The borrowing bears a fixed interest rate of 6.6%, is payable in 48 equal monthly installments and matures in May 2006. In January 2004, we amended the terms of our loan facility to permit us to borrow up to an additional $2.0 million in 2004 in up to three separate borrowings. Each borrowing would bear a fixed interest rate equalVoting Rights. For all matters submitted to a theoretical four-year U.S. Treasury Rate on the disbursement date plus 3.5%, be payable in 48 equal monthly installments and be secured by specified tangible fixed assets that R.J. Reynolds determined to be sufficient at the timevote of disbursement. In April 2004, we borrowed $1.0 million under the amended loan facility to finance equipment. The borrowing bears a fixed interest ratestockholders, each holder of 5.9%, is payable in 48 equal monthly installments and matures in April 2008. All borrowings under the loan facility are secured by specified tangible fixed assets. We believe that the terms of the loan facility are no less favorable than those that we could have obtained from an unaffiliated third party. As of May 1, 2004, the outstanding principal balance under the loan facility was $2.3 million.
Director Compensation
For information regarding stock options granted to our non-employee directors, see “Management—Director Compensation.”
Executive Compensation and Employment Agreements
For information regarding the compensation of our executive officers, see “Management—Executive Compensation” and “—Stock Options.” For information regarding employment agreements with our executive officers, see “Management—Employment Agreements.”
The following table sets forth information regarding the beneficial ownership of our common stock as of April 30, 2004 and on an as adjusted basis to reflect the sale of the common stock offered in this offering by:
The number of shares of common stock beneficially owned by each stockholder is determined under rules issued by the Securities and Exchange Commission and includes voting or investment power with respect to securities. Under these rules, beneficial ownership includes any shares as to which the individual or entity has sole or shared voting power or investment power and includes any shares that an individual or entity has the right to acquire beneficial ownership of within 60 days of April 30, 2004 through the exercise of any warrant, stock option or other right. Unless otherwise indicated, the address of all listed stockholders is c/o Targacept, Inc., 200 East First Street, Suite 300, Winston-Salem, North Carolina 27101. Each of the stockholders listed has sole voting and investment power with respect to the shares beneficially owned by the stockholder unless noted otherwise, subject to community property laws where applicable.
| Percentage of Shares Beneficially Owned (1) | ||||||
Name and Address of Beneficial Owner | Number of Shares Beneficially Owned | Before Offering | After Offering | |||
5% Stockholders Entities affiliated with New Enterprise Associates (2) 1119 St. Paul Street Baltimore, Maryland 21202 | 13,425,357 | 17.8% | ||||
Entities affiliated with EuclidSR Partners, L.P. (3) 45 Rockefeller Plaza, Suite 3240 New York, New York 10111 | 10,396,226 | 13.8% | ||||
Nomura International plc (4) Nomura House 1 St. Martins le Grand London EC1A 4NP England | 8,928,571 | 11.8% | ||||
Entities affiliated with Oxford Bioscience Partners (5) 222 Berkeley Street, Suite 1650 Boston, Massachusetts 02116 | 6,728,928 | 8.9% | ||||
R.J. Reynolds Tobacco Holdings, Inc. (6) 401 North Main Street Winston-Salem, North Carolina 27102 | 6,669,628 | 8.7% | ||||
Entities affiliated with Burrill & Company LLC (7) One Embarcadero Center, Suite 2700 San Francisco, California 94111 | 4,268,328 | 5.7% | ||||
Entities affiliated with Advent Private Equity Fund II (8) 25 Buckingham Gate London SW1E 6LD England | 4,228,328 | 5.6% | ||||
Name and Address of Beneficial Owner Executive Officers and Directors J. Donald deBethizy, Ph.D. (9) Merouane Bencherif, M.D., Ph.D. (10) Jeffrey P. Brennan (11) William S. Caldwell, Ph.D. (12) Geoffrey C. Dunbar, M.D. (13) Alan A. Musso (14) Mark Skaletsky M. James Barrett, Ph.D. (15) Charles A. Blixt (16) G. Steven Burrill (17) Errol B. De Souza, Ph.D. Elaine V. Jones, Ph.D. (18) John P. Richard (19) Alan G. Walton, Ph.D. (20) All executive officers and directors as a group (14 persons) (21) Percentage of Shares
Beneficially Owned (1) Number of Shares
Beneficially Owned Before
Offering After
Offering 1,433,663 1.9 % 509,014 * 65,667 * 503,963 * 474,273 * 341,155 * 60,000 * 13,425,357 17.8 % 6,669,628 8.7 % 4,268,328 5.7 % — — 10,396,226 13.8 % 32,500 * 6,728,928 8.9 % 44,908,702 56.3 %
The following description of our capital stock and provisions of our certificate of incorporation and bylaws are summaries and are qualified by reference to the certificate of incorporation and the bylaws that will be in effect upon completion of this offering. Copies of these documents have been filed with the Securities and Exchange Commission as exhibits to our registration statement of which this prospectus forms a part. The descriptions of the common stock and preferred stock reflect changes to our capital structure that will occur concurrently with the completion of this offering.
Upon completion of this offering, our authorized capital stock will consist of shares of common stock, par value $0.001 per share, and shares of undesignated preferred stock, par value $0.001 per share.
As of March 31, 2004, we had outstanding:
As of March 31, 2004, we also had outstanding a warrant to purchase 1,612,903 shares of common stock at an exercise price of $1.95 per share.
All of our outstanding shares of preferred stock will convert into 73,739,905 shares of common stock concurrently with the completion of this offering. In addition, the warrant will be cancelled if it is not exercised prior to the completion of this offering. If R.J. Reynolds exercises the warrant in full for cash, we would issue 1,612,903 shares of common stock and receive cash proceeds of approximately $3.1 million. If R.J. Reynolds exercises the warrant on a cashless basis, we would issue shares of common stock, based on an assumed initial public offering price of $ per share.
Common Stock
Each holder of common stock is entitled to one vote for each share on all matters submittedregistered in his or her name. Except as may be required by law and in connection with some significant actions, such as mergers, consolidations, or amendments to our restated certificate of incorporation that affect the rights of stockholders, holders of our common stock vote together as a vote of the stockholders, includingsingle class. There is no cumulative voting in the election of our directors, and there are no cumulative voting rights. Subject to preferenceswhich means that, may be applicablesubject to any shares of preferred stockrights to elect directors that may become outstanding from time to time, holders of common stock are entitled to receive, ratably, dividends declared from time to time by our board of directors, if any, out of funds legally available for that purpose. In the event of our liquidation, dissolution or winding up, holders of common stock will be entitled to share ratably in the net assets legally available for distribution to stockholders after the payment of all of our debts and other liabilities and the satisfaction of any liquidation preference granted to the holders of any sharesclass or series of preferred stock, then outstanding. Holdersa plurality of the votes cast at a meeting of stockholders at which a quorum is present is sufficient to elect a director.
Other Rights and Restrictions. Subject to the preferential rights of any other class or series of stock, all shares of our common stock have equal dividend, distribution, liquidation and other rights, and have no preference, appraisal or exchange rights, except for any appraisal rights provided by Delaware law. Furthermore, holders of our common stock have no conversion, preemptive or other subscription rights, and there are no redemption or sinking fund provisions applicableor redemption rights, or preemptive rights to subscribe for any of our securities. Our restated certificate of incorporation and our bylaws do not restrict the ability of a holder of our common stock to transfer his or her shares of our common stock.
The rights, powers, preferences and privileges of the holders of our common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of preferred stock thatwhich we may designate and issue in the future.
Listing. Our common stock is listed on The Nasdaq Capital Market under the symbol “CBIO.”
Transfer Agent and Registrar. The transfer agent for our common stock is American Stock Transfer & Trust Company, LLC. Its address is 6201 15th Avenue, Brooklyn, NY 11219.
Preferred Stock
Under our restated certificate of incorporation, we have authority, subject to any limitations prescribed by law and without further stockholder approval, to issue from time to time up to 5,000,000 shares of preferred stock, par value $0.001 per share, in one or more series. As of March 28, 2017, no shares of preferred stock were issued and outstanding.
Upon completionPursuant to our restated certificate of incorporation, we are authorized to issue “blank check” preferred stock, which may be issued from time to time in one or more series upon authorization by our board of directors. Our board of directors, without further approval of the stockholders, is authorized to fix the designation, powers, preferences, relative, participating optional or other special rights, and any qualifications, limitations and restrictions applicable to each series of the preferred stock. The issuance of preferred stock, while providing flexibility in connection with possible acquisitions and other corporate purposes could, among other things, adversely affect the voting power or rights of the holders of our common stock and, under certain circumstances, make it more difficult for a third party to gain control of us, discourage bids for our common stock at a premium or otherwise adversely affect the market price of the common stock.
In connection with this offering, our board of directors will designate shares of our preferred stock as Series A Preferred Stock. The preferences and rights of the Series A Preferred Stock will be as set forth in a Certificate of Designation (the “Series A Certificate of Designation”) filed as an exhibit to the registration statement of which this prospectus is a part.
In the event of a liquidation, the holders of Series A Preferred Stock will be entitled to participate on anas-converted-to-common-stock basis with holders of the common stock in any distribution of assets of the Company to the holders of the common stock. The Series A Certificate of Designation will provide, among other things, that we shall not pay any dividends on shares of common stock (other than dividends in the form of common stock) unless and until such time as we pay dividends on each share of Series A Preferred Stock on anas-converted basis. Other than as set forth in the previous sentence, the Series A Certificate of Designation will provide that no other dividends shall be paid on shares of Series A Preferred Stock and that we shall pay no
dividends (other than dividends in the form of common stock) on shares of common stock unless we simultaneously comply with the previous sentence. The Series A Certificate of Designation will not provide for any restriction on the repurchase of Series A Preferred Stock by us while there is any arrearage in the payment of dividends on the Series A Preferred Stock. There will be no sinking fund provisions applicable to the Series A Preferred Stock.
With certain exceptions, as described in the Series A Certificate of Designation, the Series A Preferred Stock will have no voting rights. However, as long as any shares of Series A Preferred Stock remain outstanding, the Series A Certificate of Designation will provide that we shall not, without the affirmative vote of holders of a majority of the then-outstanding shares of Series A Preferred Stock, (a) alter or change adversely the powers, preferences or rights given to the Series A Preferred Stock or alter or amend the Series A Certificate of Designation, (b) increase the number of authorized without stockholder approval,shares of Series A Preferred Stock or (c) effect a stock split or reverse stock split of the Series A Preferred Stock or any like event.
Each share of Series A Preferred Stock will be convertible at any time at the holder’s option into 95.24 shares of common stock, which conversion ratio will be subject to issue upadjustment for stock splits, stock dividends, distributions, subdivisions and combinations. Notwithstanding the foregoing, the Series A Certificate of Designation will further provide that we shall not effect any conversion of the Series A Preferred Stock, with certain exceptions, to the extent that, after giving effect to an aggregateattempted conversion, the holder of Series A Preferred Stock (together with such holder’s affiliates, and any persons acting as a group together with such holder or any of such holder’s affiliates) would beneficially own a number of shares of preferredCommon Stock in excess of 4.99% (or, at the election of the purchaser, 9.99%) of the shares of our common stock in one or more series andthen outstanding after giving effect to fixsuch exercise (the “Preferred Stock Beneficial Ownership Limitation”).
Additionally, subject to certain exceptions, at any time prior to the rights, preferences, and powers granted to or imposed upon the preferred stock, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences. We cannot state with certainty the actual
effectsthree year anniversary of the issuance of the Series A Preferred Stock, subject to the Preferred Stock Beneficial Ownership Limitation, we will have the right to cause each holder of the Series A Preferred Stock to convert all or part of such holder’s Series A Preferred Stock in the event that (i) the volume weighted average price of our common stock for 30 consecutive trading days (the “Measurement Period”) exceeds 300% of the conversion price of the preferred stock issued in this offering (subject to adjustment for forward and reverse stock splits, recapitalizations, stock dividends and similar transactions), (ii) the average daily trading volume for such Measurement Period exceeds $500,000 per trading day and (iii) the holder is not in possession of any information that constitutes or might constitute, materialnon-public information which was provided by the Company and subject to the Preferred Beneficial Ownership Limitation. Our right to cause each holder of the Series A Preferred Stock to convert all or part of such holder’s Series A Preferred Stock shall be exercised ratably among the holders of the then outstanding Series A Preferred Stock.
We do not intend to apply for listing of the Series A Preferred Stock on any securities exchange or other trading system.
The transfer agent for our Series A Preferred Stock will be American Stock Transfer & Trust Company, LLC.
Description of Warrants Included in the Units
The material terms and provisions of the warrants being offered pursuant to this prospectus are summarized below. This summary of some provisions of the warrants is not complete. For the complete terms of the warrants, you should refer to the form of warrant filed as an exhibit to the registration statement of which this prospectus is a part. Pursuant to a warrant agency agreement between us and American Stock Transfer & Trust Company, LLC, as warrant agent, the warrants will be issued in book-entry form and shall initially be represented only by one or more global warrants deposited with the warrant agent, as custodian on behalf of The Depository Trust Company, or DTC, and registered in the name of Cede & Co., a nominee of DTC, or as otherwise directed by DTC.
Each Class A Unit includes a warrant to purchase half of one share of our common stock and each Class B Unit issued in this offering includes a warrants to purchase 47.62 shares of preferredour common stock at a price equal to $ per share at any time for up to five (5) years after the date of the closing of this offering. The warrants issued in this offering will be governed by the terms of a global warrant held in book-entry form. The holder of a warrant will not be deemed a holder of our underlying common stock until the warrant is exercised.
Subject to certain limitations as described below the warrants are immediately exercisable upon issuance on the closing date and expire on the five (5) year anniversary of the closing date. Subject to limited exceptions, a holder of warrants will not have the right to exercise any portion of its warrants if the holder (together with such holder’s affiliates, and any persons acting as a group together with such holder or any of such holder’s affiliates) would beneficially own a number of shares of common stock in excess of 4.99% (or, at the election of the purchaser, 9.99%) of the shares of our Common Stock then outstanding after giving effect to such exercise.
The exercise price and the number of shares issuable upon exercise of the warrants is subject to appropriate adjustment in the event of recapitalization events, stock dividends, stock splits, stock combinations, reclassifications, reorganizations or similar events affecting our common stock. The warrant holders must pay the exercise price in cash upon exercise of the warrants, unless such warrant holders are utilizing the cashless exercise provision of the warrants. On the expiration date, unexercised warrants will automatically be exercised via the “cashless” exercise provision.
In addition, in the event we consummate a merger or consolidation with or into another person or other reorganization event in which our common shares are converted or exchanged for securities, cash or other property, or we sell, lease, license, assign, transfer, convey or otherwise dispose of all or substantially all of our assets or we or another person acquire 50% or more of our outstanding shares of common stock, then following such event, the holders of the warrants will be entitled to receive upon exercise of such warrants the same kind and amount of securities, cash or property which the holders would have received had they exercised their warrants immediately prior to such fundamental transaction. Any successor to us or surviving entity shall assume the obligations under the warrants.
Upon the holder’s exercise of a warrant, we will issue the shares of common stock issuable upon exercise of the warrant within three trading days following our receipt of a notice of exercise, provided that payment of the exercise price has been made (unless exercised via the “cashless” exercise provision). In addition, in the event we consummate a merger or consolidation with or into another person or other reorganization event in which our common shares are converted or exchanged for securities, cash or other property, or we sell, lease, license, assign, transfer, convey or otherwise dispose of all or substantially all of our assets or we or another person acquire 50% or more of our outstanding shares of common stock, then following such event, the holders of the warrants will be entitled to receive upon exercise of the warrants the same kind and amount of securities, cash or property which the holders would have received had they exercised the warrants immediately prior to such fundamental transaction. Any successor to us or surviving entity shall assume the obligations under the warrants.
Upon the holder’s exercise of a warrant, we will issue the shares of common stock issuable upon exercise of the warrant within three trading days following our receipt of a notice of exercise, provided that payment of the exercise price has been made (unless exercised via the “cashless” exercise provision). Prior to the exercise of any warrants to purchase common stock, holders of the warrants will not have any of the rights of holders of the common stock untilpurchasable upon exercise, including the boardright to vote, except as set forth therein.
Warrant holders may exercise warrants only if the issuance of directors determines the specific rightsshares of common stock upon exercise of the warrants is covered by an effective registration statement, or an exemption from registration is available under the Securities Act and the securities laws of the state in which the holder resides. We intend to use commercially reasonable efforts to have the registration statement, of which this prospectus forms a part, effective when the warrants are exercised. The warrant holders must pay the exercise price in cash upon exercise of the warrants unless there is not an effective registration statement or, if required, there is not an effective state law registration
or exemption covering the issuance of the shares underlying the warrants (in which case, the warrants may only be exercised via a “cashless” exercise provision).
The warrants are callable by us in certain circumstances. Subject to certain exceptions, in the event that the warrants are outstanding, if, after the closing date, (i) the volume weighted average price of our common stock for each of 30 consecutive trading days (the “Measurement Period”), which Measurement Period commences on the closing date, exceeds 300% of the exercise price (subject to adjustment for forward and reverse stock splits, recapitalizations, stock dividends and similar transactions after the initial exercise date), (ii) the average daily trading volume for such Measurement Period exceeds $500,000 per trading day and (iii) the warrant holder is not in possession of any information that constitutes or might constitute, materialnon-public information which was provided by the Company, and subject to the Beneficial Ownership Limitation, then we may, within one trading day of the end of such Measurement Period, upon notice (a “Call Notice”), call for cancellation of all or any portion of the warrants for which a notice of exercise has not yet been delivered (a “Call”) for consideration equal to $0.001 per warrant share. Any portion of a warrant subject to such Call Notice for which a notice of exercise shall not have been received by the Call Date (as hereinafter defined) will be canceled at 6:30 p.m. (New York City time) on the tenth trading day after the date the Call Notice is sent by the Company (such date and time, the “Call Date”). Our right to call the warrants shall be exercised ratably among the holders based on the then outstanding warrants.
We do not intend to apply for listing of the warrants on any securities exchange or other trading system.
Outstanding Debt Securities
On August 19, 2015, we issued to our stockholdersnon-interest bearing redeemable convertible notes (the “Convertible Notes”) in the aggregate principal amount of $37.0 million. The Convertible Notes do not bear interest. The principal amounts under the Convertible Notes are convertible, at the option of each noteholder, into cash or into shares of our common stock at a conversion rate of $137.85 per share, and are payable in cash, if not previously redeemed or converted, at maturity on February 19, 2018, the30-month anniversary of the closing of the issuance of the Convertible Notes.
On August 19, 2015, we also entered into an indenture (the “Convertible Notes Indenture”) with American Stock Transfer & Trust Company, LLC, as trustee, and an escrow agreement with American Stock Transfer & Trust Company, LLC and Delaware Trust Company, LLC, as escrow agent, under which $37.0 million, which represents the initial principal amount of the Convertible Notes, was deposited in a segregated escrow account for the benefit of the holders of the preferred stock. Some of these effects might potentially include:
Holders of the Convertible Notes may submit conversion notices, which are irrevocable, instructing the trustee to convert such the Convertible Notes into shares of the common stock;
We do not currently have any plans to issue anywhole shares of preferredcommon stock following this offering.
Options
issuable upon conversion as promptly as practicable (and in any event within 10 business days). The trustee will in turn release to us the respective amount of restricted cash to cover the stock issuance.
As of March 31, 2004, options28, 2017, Convertible Notes in an aggregate principal amount of approximately $12.8 million remained outstanding.
Outstanding Warrants
Prior to this offering, as of March 28, 2017, we have outstanding warrants to purchase 8,024,394 shares of common stock as follows: (i) at a weighted average exercise priceany time until the5-year anniversary of $0.64 per share were outstanding.
Registration Rights
After this offering, holdersthe original date of approximatelyissuance in 2014, warrants to purchase an aggregate of 2,487 shares of our common stock will haveat an exercise price of $499.05 per share, (ii) at any time until the right to require us to register the sales of their shares under the Securities Act, under the terms of an agreement between us and the holders of these securities. Subject to limitations specified in this agreement, these registration rights include the following:
Demand Registration Rights. Beginning six months after the completion of this offering, subject to specified limitations, two separate constituencies5-year anniversary of the holdersoriginal date of registrable securities may require that we register partissuance in 2015, warrants to purchase an aggregate of these securities for sale under the Securities Act. Each constituency may make one such demand.
Incidental Registration Rights. If we register any9,467 shares of our common stock underat an exercise price of $49.91 per share and (iii) at any time until seven years from the Securities Act, solely for cash, either for our own account or fordate of the accountcompletion of other security holders, the holders ofmerger between Targacept and Catalyst Bio, a warrant to purchase 85 shares of registrable securities are entitled to noticeour common stock at an exercise price of $392.70. As of December 31, 2016, the registration and to include theirfair value of these warrants was immaterial.
Certain Effects of Authorized but Unissued Stock
We have shares of common stock inand preferred stock available for future issuance without stockholder approval. We may issue these additional shares for a variety of corporate purposes, including future public or private offerings to raise additional capital or to facilitate corporate acquisitions or for payment as a dividend on our capital stock. The existence of unissued and unreserved preferred stock may enable our board of directors to issue shares of preferred stock with terms that could render more difficult or discourage a third-party attempt to obtain control of us by means of a merger, tender offer, proxy contest or otherwise, thereby protecting the registration. These rights have been waived for this offering.
Form S-3 Registration Rights. Ifcontinuity of our management. In addition, if we become eligible to file registration statements on Form S-3,issue additional preferred stock, the issuance could adversely affect the voting power of holders of registrable securities can require us to register their registrable securities on Form S-3 ifcommon stock and the total gross proceedslikelihood that holders of common stock will receive dividend payments or payments upon liquidation.
Anti-Takeover Effects of Provisions of Our Charter Documents
Our restated certificate of incorporation provides for our board of directors to be received by them together would be at least $1.0 million.divided into three classes serving staggered terms.
ApproximatelyLimitations and Expenses. With specified exceptions, a holder’s right to include shares in a registration statement is subject to the right of the underwriters to limit the number of shares included in the offering. We are generally required to pay all expenses of registration, including the fees and expenses of one legal counsel to the registering security holders up to a prescribed maximum amount, but excluding underwriters’ discounts and commissions.one-third
Anti-Takeover Provisions
We are subject to Section 203 of the Delaware General Corporation Law, an anti-takeover statute. Subject to certain exceptions, Section 203 prohibits a publicly held Delaware corporation from engaging in a business combination with an interested stockholder for three years following the date that the person became an interested stockholder, unless the interested stockholder attained such status with the approval of our board of directors or unlesswill be elected each year. The provision for a classified board could prevent a party who acquires control of a majority of the business combination is approved in a prescribed manner. A business combination includes, among other things, a merger or consolidation involving us and the interested stockholder and the sale of more than 10% of our assets. In general, an interested stockholder is any entity or person beneficially owning 15% or more of our outstanding voting stock and any entity or person affiliated with or controlling or controlled by that entity or person.
Certain provisions of our certificate of incorporation and bylaws that will be in effect upon completion of this offering could make the acquisition of us through a tender offer, proxy contest or other means, or the removal of incumbent officers and directors, more difficult. These provisions may discourage certain types of coercive takeover practices and takeover bids and encourage persons seeking to acquirefrom obtaining control of us to first negotiate with our board of directors. We believe that the benefits of retaining the ability to negotiate with a proponent of an unfriendly or unsolicited proposal outweigh the potential disadvantages of discouraging such a proposal. These provisions may make it more difficult for stockholders to take specific corporate actions and could have the effect of delaying or preventing a change in our control.
In particular, our certificate of incorporation or bylaws provide for the following:
Staggered Board of Directors and Number of Directors. Our board of directors is divided into three classes of the same or nearly the same number of directors serving staggered three-year terms, which means that only one class of directors may be elected at a particular stockholders meeting. Also, the authorized number of directors comprising our board of directors may only be changed by resolution of our board of directors. As a result, the replacement of incumbent directors may be more difficult and third parties may be discouraged from seeking to circumvent the anti-takeover provisions of our certificate of incorporation and bylaws by replacing our incumbent directors.
Limitations on Calling Special Meetings of Stockholders. Under Delaware law, a special meeting of stockholders may be called by the board of directors or by any other person authorized to do so in the certificate of incorporation or the bylaws. Our certificate of incorporation and bylaws do not permit our stockholders to call a special meeting. As a result, a stockholder could not force stockholder consideration of a proposal over the opposition of the board of directors by calling a special meeting. The restriction on the ability of stockholders to call a special meeting means that a proposal to replace the board of directors also could be delayed until the nextsecond annual meeting.stockholders meeting following the date the acquirer obtains the controlling stock interest. The classified board provision could discourage a potential acquirer from making a tender offer or otherwise attempting to obtain control of Catalyst and could increase the likelihood that incumbent directors will retain their positions. Our restated certificate of incorporation provides that directors may be removed with or without cause by the affirmative vote of the holders of at least 66 2/3% of the voting power of all outstanding stock.
Our restated certificate of incorporation requires that certain amendments to the restated certificate of incorporation and amendments by the stockholders of our bylaws require the affirmative vote of at least 66 2/3% of the voting power of all outstanding stock. These provisions could discourage a potential acquirer from making a tender offer or otherwise attempting to obtain control of the Company and could delay changes in management.
Advance Notice Procedures.Our amended and restated bylaws establish an advance notice procedure for stockholder proposals to be brought before an annual stockholders meeting, of our stockholders, including proposed nominations of persons for election to theour board of directors. At an annual stockholders meeting, stockholders may only consider proposals or nominations specified in the notice of meeting or brought before the meeting by or at the direction of theour board of directors. Stockholders may also consider a proposal or nomination by a person who was a stockholder of record on the record date for the meeting, and on the date that notice of the proposal or nomination was given, who is entitled to vote at the meeting and who has given to our secretarythe Secretary of the Company timely written notice, in proper form, of his or her intention to bring that business before the annual stockholders meeting. The amended and restated bylaws do not give our board of directors the power to approve or disapprove stockholder nominations of candidates or proposals regarding other business to be conducted at a special or annual meeting of the stockholders. However our bylaws may have the effect of precluding the conduct of certain business at a meeting if the proper procedures are not followed. These provisions may also discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain control of us.the Company.
Our amended and restated bylaws provide that only our board of directors, the chairperson of the board, the President or the Chief Executive Officer may call a special meeting of stockholders. Because our stockholders do not have the right to call a special meeting, a stockholder could not force stockholder consideration of a proposal over the opposition of our board of directors by calling a special meeting of stockholders prior to such time as a majority of our board of directors, the chairperson of the board, the President or the Chief Executive Officer believed the matter should be considered or until the next annual meeting provided that the requestor met the notice requirements. The restriction on the ability of stockholders to call a special meeting means that a proposal to replace the board also could be delayed until the next annual stockholders meeting.
Prohibition of Stockholder Action by Written Consent. Delaware law provides that, unless prohibited by theOur restated certificate of incorporation does not allow stockholders may execute an actionto act by written consent in lieuwithout a meeting. Without the availability of a stockholder meeting. Our certificate of incorporation prohibits stockholder action by written consent, which may lengthen the amount of time required to take stockholder actions becausestockholder’s actions by written consent, are not subject to the minimum notice requirement of a stockholders’ meeting. The prohibition of stockholder action by written consent may deter hostile takeover attempts because a holder that controlledcontrolling a majority of our capital stock would not be able to amend our bylaws or remove directors without holding a stockholders meeting andstockholders’ meeting.
Anti-Takeover Effects of Provisions of Delaware Law
We are subject to the provisions of Section 203 of the DGCL, or Section 203. Under Section 203, we would havegenerally be prohibited from engaging in any business combination with any interested stockholder for a period of three years following the time that this stockholder became an interested stockholder unless:
Under Section 203, a “business combination” includes: In general, Section 203 defines an interested stockholder as an entity or person beneficially owning 15% or more of the outstanding voting stock of the corporation and any entity or person affiliated with or controlling or controlled by such entity or person.Undesignated Preferred Stock. Ourat or subsequent to such time, the business combination is approved by our board of directors isand authorized to issue up to shares of our preferred stock in oneat a special or more seriesannual stockholders meeting, and to fix the rights, preferences, designation and powers granted to or imposed upon the preferred stock, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences. The existence of this ability could discourage an attempt to take control of us through a merger, tender offer, proxy contest or other means.With the exception of the provision relating to the issuance of preferred stock, which can be amended with the approval of a majority of the outstanding shares of stock entitled to vote, none of these provisions can be amended without the approval of at least two-thirds of our outstanding shares of stock entitled to vote. In addition,not by written consent, by the affirmative vote of two-thirdsat least 66 2/3% of ourthe outstanding sharesvoting stock that is not owned by the interested stockholder.any merger or consolidation involving the corporation and the interested stockholder;any sale, transfer, pledge or other disposition of 10% or more of the assets of the corporation involving the interested stockholder;any transaction that results in the issuance or transfer by the corporation of any stock entitledof the corporation to vote is requiredthe interested stockholder, subject to amend provisionslimited exceptions;any transaction involving the corporation that has the effect of ourincreasing the proportionate share of the stock of any class or series of the corporation beneficially owned by the interested stockholder; orthe receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation.
Limitation of Liability and Indemnification
Our restated certificate of incorporation or bylaws relating to exculpation and indemnification of directors and officers, the number, election, qualification, term of office, resignation or removal of directors and the filling of director vacancies.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is .
NASDAQ National Market
We have applied to have our common stock listed on the NASDAQ National Market under the symbol “TRGT.”
SHARES ELIGIBLE FOR FUTURE SALE
Prior to this offering, there has been no public market for our common stock and we cannot assure you that a liquid trading market for our common stock will develop or be sustained after this offering. Future sales of substantial amounts of our common stock in the public market following this offering, or the anticipation of those sales, could adversely affect market prices prevailing from time to time and could impair our ability to raise capital by the sale of our equity securities.
Upon completion of this offering, we will have outstanding shares of common stock, after giving effect to the conversion of all outstanding shares of our convertible preferred stock into 73,739,905 shares of common stock concurrently with the completion of this offering and our issuance of shares of common stock upon the exercise of an outstanding warrant that will be cancelled if not exercised concurrently with the completion of this offering, assuming that the warrant is exercised on a cashless basis based on an assumed initial public offering price of $ per share. If the warrant is exercised in full for cash, we would issue 1,612,903 shares of our common stock.
All of the shares sold in this offering will be freely tradable without restriction unless purchased by our “affiliates,” as that term is defined in Rule 144 under the Securities Act. The remaining shares of common stock to be outstanding after this offering are “restricted securities” under Rule 144. All of these restricted securities will be subject to the 180-day lock-up period described below. Immediately after the 180-day period, shares will be freely tradable under Rule 144(k) and shares will be eligible for resale under Rule 144, subject to volume limitations.
Restricted securities may be sold in the public market only if registered or if they qualify for an exemption from registration under Rules 144 or 701 under the Securities Act. These rules are summarized below.
Rule 144
In general, under Rule 144, beginning 90 days after the date of this prospectus, a person who has beneficially owned shares of our common stock for at least one year would be entitled to sell within any three-month period a number of shares that does not exceed the greater of:
Sales under Rule 144 are also subject to manner of sale provisions and notice requirements and to the availability of current public information about us.
Rule 144(k)
Subject to the lock-up agreements described below, shares of our common stock eligible for sale under Rule 144(k) may be sold immediately after the completion of this offering. In general, under Rule 144(k), a person may sell shares of common stock acquired from us immediately after the completion of this offering, without regard to manner of sale, the availability of public information or volume, if:
Rule 701
In general, under Rule 701, any of our employees, consultants or advisors who purchased shares from us in connection with a qualified compensatory stock plan or other written agreement is eligible to resell those shares 90 days after the effective date of this offering in reliance on Rule 144, but without compliance with various restrictions, including the holding period, contained in Rule 144.
Lock-up Agreements
The holders of substantially all of our currently outstanding stock have agreed that, without the prior written consent of Morgan Stanley & Co. Incorporated on behalf of the underwriters and subject to the exceptions described in the section entitled “Underwriters” in this prospectus they will not, during the period ending 180 days after the date of this prospectus:
whether any transaction described above is to be settled by delivery of shares of our common stock or such other securities, in cash or otherwise.
Stock Options
After the completion of this offering, we intend to file a registration statement on Form S-8 under the Securities Act to register all of the shares of common stock subject to issuance upon exercise of outstanding options granted under, or reserved for future issuance under, our 2000 plan and our 2004 plan. Shares of common stock issued under the Form S-8 upon exercise of options will be available for sale in the public market, subject to Rule 144 volume limitations applicable to affiliates and subject to the contractual restrictions described above. As of March 31, 2004, options to purchase 8,024,394 shares of common stock were outstanding under our 2000 plan with a weighted average exercise price of $0.64, of which approximately 4,078,011 were vested and exercisable with a weighted average exercise price of $0.62 and an additional 448,274 shares were reserved for issuance under our 2000 plan. Upon completion of this offering, an additional shares of common stock will be reserved for issuance under our 2004 plan.
Registration Rights
Upon completion of this offering, the holders of shares of our common stock will be entitled to registration rights. Registration of the sale of these shares upon exercise of these rights would make them freely tradable without restriction under the Securities Act. For more information regarding these registration rights, see “Description of Capital Stock—Registration Rights.”
Under the terms and subject to the conditions contained in an underwriting agreement dated the date of this prospectus, the underwriters named below, for whom Morgan Stanley & Co. Incorporated, Deutsche Bank Securities Inc., CIBC World Markets Corp. and Pacific Growth Equities, LLC, are acting as representatives, have severally agreed to purchase, and we have agreed to sell to them, severally, the number of shares indicated below:
| ||
| ||
| ||
| ||
| ||
| ||
The underwriters are offering the shares of common stock subject to their acceptance of the shares from us and subject to prior sale. The underwriting agreement provides that the obligations of the several underwriters to pay for and accept delivery of the shares of common stock offered by this prospectus are subject to the approval of specified legal matters by their counsel and to other conditions. The underwriters are obligated to take and pay for all of the shares of common stock offered by this prospectus if any such shares are taken. However, the underwriters are not required to take or pay for the shares covered by the underwriters’ over-allotment option described below.
The underwriters initially propose to offer part of the shares of common stock directly to the public at the public offering price listed on the cover page of this prospectus and part to certain dealers at a price that represents a concession not in excess of $ per share under the public offering price. No underwriter may allow, and no dealer may reallow, any concession to other underwriters or to certain dealers. After the initial offering of the shares of common stock, the offering price and other selling terms may from time to time be varied by the representatives.
We have granted to the underwriters an option, exercisable for 30 days from the date of this prospectus, to purchase up to an aggregate of additional shares of common stock at the public offering price listed on the cover page of this prospectus, less underwriting discounts and commissions. The underwriters may exercise this option solely for the purpose of covering over-allotments, if any, made in connection with the offering of the shares of common stock offered by this prospectus. To the extent the option is exercised, each underwriter will become obligated, subject to certain conditions, to purchase approximately the same percentage of the additional shares of common stock as the number listed next to the underwriter’s name in the preceding table bears to the total number of shares of common stock listed next to the names of all underwriters in the preceding table. If the underwriters’ option is exercised in full, the total price to the public would be $ , the total underwriters’ discounts and commissions would be $ and the total proceeds to us would be $ .
The underwriters have informed us that they do not intend sales to discretionary accounts to exceed five percent of the total number of shares of common stock offered by them.
We and all of our directors and officers and holders of substantially all of our outstanding stock have agreed that, without the prior written consent of Morgan Stanley & Co. Incorporated on behalf of the underwriters, we and they will not, during the period ending 180 days after the date of this prospectus:
whether any transaction described above is to be settled by delivery of shares of our common stock or such other securities, in cash or otherwise.
The 180-day restricted period described in the preceding paragraph will be extended if:
in which case the restrictions described in the preceding paragraph will continue to apply until the expiration of the 18-day period beginning on the issuance of the earnings release or the occurrence of the material news or material event.
These restrictions do not apply to:
provided that in the case of each of the last three transactions, each donee, distributee, transferee and recipient agrees to be subject to the restrictions described in the immediately preceding paragraph, no filing under Section 16 of the Securities Exchange Act of 1934, as amended, is required in connection with these transactions, other than a filing on a Form 5 made after the expiration of the 180-day period, and no transaction includes a disposition for value.
The following table shows the underwriting discounts and commissions that we are to pay to the underwriters in connection with this offering. These amounts are shown assuming both no exercise and full exercise of the underwriters’ option to purchase additional shares of our common stock.
| ||||||
| ||||||
In addition, we estimate that the expenses of this offering payable by us, other than underwriting discounts and commissions, will be $ .
In order to facilitate the offering of the common stock, the underwriters may engage in transactions that stabilize, maintain or otherwise affect the price of the common stock. Specifically, the underwriters may sell
more shares than they are obligated to purchase under the underwriting agreement, creating a short position. A short sale is covered if the short position is no greater than the number of shares available for purchase by the underwriters under the over-allotment option. The underwriters can close out a covered short sale by exercising the over-allotment option or purchasing shares in the open market. In determining the source of shares to close out a covered short sale, the underwriters will consider, among other things, the open market price of shares compared to the price available under the over-allotment option. The underwriters may also sell shares in excess of the over-allotment option, creating a naked short position. The underwriters must close out any naked short position by purchasing shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that there may be downward pressure on the price of the common stock in the open market after pricing that could adversely affect investors who purchase in this offering. In addition, to stabilize the price of the common stock, the underwriters may bid for, and purchase, shares of common stock in the open market. Finally, the underwriting syndicate may reclaim selling concessions allowed to an underwriter or a dealer for distributing the common stock in this offering, if the syndicate repurchases previously distributed common stock in transactions to cover syndicate short positions or to stabilize the price of the common stock. Any of these activities may stabilize or maintain the market price of the common stock above independent market levels. The underwriters are not required to engage in these activities, and may end any of these activities at any time.
We have applied for quotation of our common stock on the NASDAQ National Market under the symbol “TRGT.”
We and the underwriters have agreed to indemnify each other against certain liabilities, including liabilities under the Securities Act.
Directed Share Program
At our request, the underwriters have reserved for sale, at the initial public offering price, up to shares offered by this prospectus to directors, officers, employees and other individuals associated with us and members of their respective families and friends through a directed share program. The number of shares of our common stock available for sale to the general public in the offering will be reduced to the extent these persons purchase these reserved shares. Any reserved shares not purchased by these persons will be offered by the underwriters to the general public on the same basis as the other shares offered by this prospectus. Recipients of reserved shares will be required to agree with the underwriters not to sell, transfer, assign, pledge or hypothecate these shares for a period of days after purchasing the shares.
Pricing of the Offering
Prior to the offering, there has been no public market for our common stock. The initial public offering price will be determined by negotiations between us and the representatives. Among the factors to be considered in determining the initial public offering price of the shares will be our future prospects and those of our industry in general, our sales, earnings and other financial operating information in recent periods, and the price-earnings ratios, price-sales ratios, market prices of securities and financial and operating information of companies engaged in activities similar to ours. The estimated initial public offering price range set forth on the cover page of this preliminary prospectus is subject to change as a result of market conditions and other factors.
Certain legal matters with respect to the validity of the shares of common stock offered hereby will be passed upon for us by Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C., Boston, Massachusetts and by Womble Carlyle Sandridge & Rice, PLLC, Winston-Salem, North Carolina. Hale and Dorr LLP, New York, New York, has acted as counsel for the underwriters in connection with certain legal matters related to this offering.
Ernst & Young LLP, independent auditors, have audited our financial statements as of December 31, 2003 and 2002 and for each of the three years in the period ended December 31, 2003, as set forth in their report. We have included our financial statements in the prospectus and elsewhere in the registration statement in reliance on Ernst & Young LLP’s report, given on their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the Securities and Exchange Commission a registration statement on Form S-1 under the Securities Act with respect to the shares of common stock to be sold in this offering. This prospectus does not contain all of the information set forth in the registration statement. You should refer to the registration statement for additional information regarding us and the shares of our common stock to be sold in this offering. Whenever we reference any contract, agreement or other document in this prospectus, the reference is not necessarily complete and you should refer to the exhibits to the registration statement for the actual contract, agreement or other document. In each instance, reference is made to such exhibits and each such statement is qualified in all respects by such reference. In addition, when this offering is completed, we will become subject to the information and reporting requirements of the Exchange Act and, in accordance with the Exchange Act, will file periodic reports, proxy statements and other information with the Securities and Exchange Commission.
You can read the registration statement and our future filings with the Securities and Exchange Commission over the Internet at the Securities and Exchange Commission’s website at http://www.sec.gov. You may also read and copy any document that we file with the Securities and Exchange Commission at its public reference room at 450 Fifth Street, N.W., Washington, D.C. 20549.
You may obtain copies of the documents at prescribed rates by writing to the Public Reference Section of the Securities and Exchange Commission at 450 Fifth Street, N.W., Washington, D.C. 20549. Please call the Securities and Exchange Commission at 1-800-SEC-0330 for further information on the operation of the public reference room. Upon approval of our common stock for listing on the NASDAQ National Market, such reports, proxy and information statements and other information may also be inspected at the offices of NASDAQ Operations, 1735 K Street, N.W., Washington, D.C. 20006.
INDEX TO THE FINANCIAL STATEMENTS
| ||
Report of Independent Auditors
The Board of Directors
Targacept, Inc.
We have audited the accompanying balance sheets of Targacept, Inc. as of December 31, 2002 and 2003, and the related statements of operations, redeemable convertible preferred stock and stockholders’ equity (deficit) and cash flows for each of the three years in the period ended December 31, 2003. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with auditing standards generally accepted in the United States. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Targacept, Inc. at December 31, 2002 and 2003, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2003, in conformity with accounting principles generally accepted in the United States.
ERNST & YOUNG LLP
January 22, 2004, except for Note 17,
as to which the date is May 13, 2004
Greensboro, North Carolina
The foregoing report is in the form that will be signed upon the completion of the restatement of capital accounts described in Note 17 to the financial statements.
/S/ ERNST & YOUNG LLP
May 13, 2004
Greensboro, North Carolina
Balance Sheets
| December 31, | March 31, | Pro forma Stockholders’ equity as of March 31, 2004 | |||||||||||||
| 2002 | 2003 | 2004 | |||||||||||||
| (unaudited) | |||||||||||||||
Assets | |||||||||||||||
Current assets: | |||||||||||||||
Cash and cash equivalents | $ | 44,353,320 | $ | 11,609,157 | $ | 5,482,910 | $ | ||||||||
Short-term investments | 5,008,139 | 31,367,500 | 31,364,260 | ||||||||||||
Research fees and accounts receivable | 1,335,648 | 818,618 | 224,674 | ||||||||||||
Inventories | 169,536 | 118,520 | 114,812 | ||||||||||||
Prepaid expenses | 423,016 | 514,552 | 698,488 | ||||||||||||
Total current assets | 51,289,659 | 44,428,347 | 37,885,144 | ||||||||||||
Property and equipment, net | 2,462,944 | 2,373,035 | 2,342,108 | ||||||||||||
Intangible assets, net of accumulated amortization of $15,735, $53,499 and $62,940 at December 31, 2002, 2003 and March 31, 2004, respectively | 626,265 | 588,501 | 579,060 | ||||||||||||
Total assets | $ | 54,378,868 | $ | 47,389,883 | $ | 40,806,312 | $ | ||||||||
Liabilities, redeemable convertible preferred stock and stockholders’ equity (deficit) | |||||||||||||||
Current liabilities: | |||||||||||||||
Accounts payable | $ | 1,503,170 | $ | 1,246,975 | $ | 1,224,603 | $ | ||||||||
Accrued expenses | 1,477,034 | 1,406,778 | 1,687,928 | ||||||||||||
Current portion of long-term debt | 586,816 | 576,072 | 637,137 | ||||||||||||
Current portion of deferred rent incentive | 402,647 | 402,647 | 402,647 | ||||||||||||
Current portion of deferred license fee revenue | 634,881 | 269,537 | 269,537 | ||||||||||||
Total current liabilities | 4,604,548 | 3,902,009 | 4,221,852 | ||||||||||||
Long-term debt, net of current portion | 2,088,293 | 1,461,554 | 1,298,317 | ||||||||||||
Deferred rent incentive, net of current portion | 1,442,818 | 1,040,171 | 939,509 | ||||||||||||
Deferred license fee revenue, net of current portion | 1,551,875 | 1,647,687 | 1,580,303 | ||||||||||||
Total liabilities | 9,687,534 | 8,051,421 | 8,039,981 | ||||||||||||
Commitments | |||||||||||||||
Redeemable convertible preferred stock: | |||||||||||||||
Series A, $0.001 par value, 5,000,000 shares authorized, issued and outstanding, aggregate liquidation preference of $26,826,253, $28,496,497 and $28,914,058 at December 31, 2002, 2003, and March 31, 2004, respectively, or $4.65 per share plus accreted redemption value | 26,826,253 | 28,496,497 | 28,914,058 | — | |||||||||||
Series B, $0.001 par value, 6,567,567 shares authorized, issued and outstanding, aggregate liquidation preference of $35,346,675, $37,484,419 and $38,018,854 at December 31, 2002, 2003, and March 31, 2004, respectively, or $4.65 per share, plus accreted redemption value | 35,346,675 | 37,484,419 | 38,018,854 | — | |||||||||||
Series C, $0.001 par value, 37,764,180, 49,169,138 and 49,169,138 shares authorized, issued and outstanding at December 31, 2002, 2003, and March 31, 2004, respectively, aggregate liquidation preference of $45,853,329, $64,153,421 and $65,343,314 at December 31, 2002, 2003, and March 31, 2004, or $1.21 per share, plus accreted redemption value | 45,853,329 | 64,153,421 | 65,343,314 | — | |||||||||||
Total redeemable convertible preferred stock | 108,026,257 | 130,134,337 | 132,276,226 | — | |||||||||||
Stockholders’ equity (deficit): | |||||||||||||||
Common stock, $0.001 par value, 75,000,000, 85,000,000 and 85,000,000 shares authorized at December 31, 2002, 2003, and March 31, 2004, respectively, 624,584, 1,082,771 and 1,164,524 shares issued and outstanding at December 31, 2002, 2003, and March 31, 2004, respectively. | 625 | 1,083 | 1,165 | ||||||||||||
Capital in excess of par value | 6,002,692 | 6,306,513 | 6,377,745 | ||||||||||||
Excess of fair value of Series A preferred stock over cash received | (23,250,000 | ) | (23,250,000 | ) | (23,250,000 | ) | |||||||||
Common stock warrants | 213,710 | 213,710 | 213,710 | ||||||||||||
Accumulated deficit | (46,301,950 | ) | (74,037,899 | ) | (82,817,723 | ) | |||||||||
Accumulated other comprehensive loss | — | (29,282 | ) | (34,792 | ) | ||||||||||
Total stockholders’ equity (deficit) | (63,334,923 | ) | (90,795,875 | ) | (99,509,895 | ) | |||||||||
Total liabilities, redeemable convertible preferred stock and stockholders’ equity (deficit) | $ | 54,378,868 | $ | 47,389,883 | $ | 40,806,312 | $ | ||||||||
See accompanying notes.
Statements of Operations
| Year ended December 31, | Three months ended March 31, | |||||||||||||||||||
| 2001 | 2002 | 2003 | 2003 | 2004 | ||||||||||||||||
| (unaudited) | ||||||||||||||||||||
Revenue: | ||||||||||||||||||||
Research fee revenue | $ | 1,100,000 | $ | 1,388,824 | $ | 1,302,500 | $ | 312,500 | $ | 134,674 | ||||||||||
License fee revenue | 602,807 | 634,881 | 269,532 | 67,383 | 67,384 | |||||||||||||||
Product sales | — | 242,861 | 814,724 | 310,913 | 187,511 | |||||||||||||||
Other | 687 | 19,792 | 71,529 | — | 107,370 | |||||||||||||||
Net revenue | 1,703,494 | 2,286,358 | 2,458,285 | 690,796 | 496,939 | |||||||||||||||
Operating expenses: | ||||||||||||||||||||
Research and development | 8,151,785 | 16,243,888 | 18,179,542 | 4,069,188 | 6,049,828 | |||||||||||||||
General and administrative | 2,302,161 | 4,135,262 | 3,599,673 | 697,269 | 1,109,254 | |||||||||||||||
Cost of product sales | — | 243,718 | 742,941 | 199,404 | 181,450 | |||||||||||||||
Purchased in-process research and development | — | 2,666,000 | — | — | — | |||||||||||||||
Total operating expenses | 10,453,946 | 23,288,868 | 22,522,156 | 4,965,861 | 7,340,532 | |||||||||||||||
Loss from operations | (8,750,452 | ) | (21,002,510 | ) | (20,063,871 | ) | (4,275,065 | ) | (6,843,593 | ) | ||||||||||
Other income (expense): | ||||||||||||||||||||
Interest and dividend income | 1,448,182 | 87,691 | 791,339 | 124,341 | 230,187 | |||||||||||||||
Interest expense | — | (102,891 | ) | (122,789 | ) | (34,318 | ) | (24,529 | ) | |||||||||||
Loss on disposal of fixed assets | — | (53,996 | ) | — | — | — | ||||||||||||||
Total other income (expense) | 1,448,182 | (69,196 | ) | 668,550 | 90,023 | 205,658 | ||||||||||||||
Net loss | (7,302,270 | ) | (21,071,706 | ) | (19,395,321 | ) | (4,185,042 | ) | (6,637,935 | ) | ||||||||||
Preferred stock accretion | (3,807,988 | ) | (4,173,545 | ) | (8,340,628 | ) | (1,914,958 | ) | (2,141,889 | ) | ||||||||||
Net loss attributable to common stockholders | $ | (11,110,258 | ) | $ | (25,245,251 | ) | $ | (27,735,949 | ) | $ | (6,100,000 | ) | $ | (8,779,824 | ) | |||||
Basic and diluted net loss attributable to common stockholders per share | $ | (26.80 | ) | $ | (45.28 | ) | $ | (33.91 | ) | $ | (9.48 | ) | $ | (7.89 | ) | |||||
Weighted average common shares outstanding—basic and diluted | 414,624 | 557,492 | 817,894 | 643,571 | 1,112,591 | |||||||||||||||
Pro forma basic and diluted net loss per share attributable to common stockholders assuming conversion of preferred stock (unaudited) | $ | (0.27 | ) | $ | (0.09 | ) | ||||||||||||||
Pro forma weighted average shares outstanding—basic and diluted (unaudited) | 71,118,629 | 74,852,496 | ||||||||||||||||||
See accompanying notes.
Statements of Redeemable Convertible Preferred Stock and Stockholders’ Equity (Deficit)
| Redeemable Convertible Preferred Stock | Common Stock | Capital in Excess of Par Value | Excess of Fair Value of Series A Preferred over Cash Received | Common Stock Warrants | Deferred Compensation | Accumulated Deficit | Accumulated Other Comprehensive Loss | Total Stockholders’ Deficit | |||||||||||||||||||||||||||||||||
| Series A | Series B | Series C | Shares | Amount | |||||||||||||||||||||||||||||||||||||
Balances at December 31, 2000 | $ | 23,485,765 | $ | 30,932,000 | $ | — | 309,424 | $ | 309 | $ | 5,760,958 | $ | (23,250,000 | ) | $ | 213,710 | $ | (92,638 | ) | $ | (9,946,441 | ) | $ | — | $ | (27,314,102 | ) | ||||||||||||||
Stock issuance costs | — | — | — | — | — | (42,496 | ) | — | — | — | — | — | (42,496 | ) | |||||||||||||||||||||||||||
Issuance of 29,933 shares of Series B redeemable convertible preferred stock at $4.65 per share | — | 139,188 | — | — | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||
Issuance of 111,111 shares of common stock valued at $0.68 per share, related to collaborative research and development agreement | — | — | — | 111,111 | 111 | 75,445 | — | — | — | — | — | 75,556 | |||||||||||||||||||||||||||||
Issuance of 65,565 shares of common stock at $0.001 per share par value, related to exercise of stock options | — | — | — | 65,565 | 66 | 30,749 | — | — | — | — | — | 30,815 | |||||||||||||||||||||||||||||
Accreted redemption value for common stock warrants attached to Series A redeemable convertible preferred stock | 42,744 | — | — | — | — | — | — | — | — | (42,744 | ) | — | (42,744 | ) | |||||||||||||||||||||||||||
Accreted redemption value for Series A and Series B redeemable convertible preferred stock | 1,627,500 | 2,137,744 | — | — | — | — | — | — | — | (3,765,244 | ) | — | (3,765,244 | ) | |||||||||||||||||||||||||||
Amortization of deferred compensation | — | — | — | — | — | — | — | — | 92,638 | — | — | 92,638 | |||||||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | — | — | — | (7,302,270 | ) | — | (7,302,270 | ) | |||||||||||||||||||||||||||
Balances at December 31, 2001 | 25,156,009 | 33,208,932 | — | 486,100 | 486 | 5,824,656 | (23,250,000 | ) | 213,710 | — | (21,056,699 | ) | — | (38,267,847 | ) | ||||||||||||||||||||||||||
Stock issuance costs | — | — | (206,887 | ) | — | — | — | — | — | — | — | — | — | ||||||||||||||||||||||||||||
Issuance of 37,764,180 shares of Series C redeemable convertible preferred stock at $1.21 per share | — | — | 45,694,658 | — | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||
Issuance of 138,484 shares of common stock at $0.001 per share par value, related to exercise of stock options | — | — | — | 138,484 | 139 | 178,036 | — | — | — | — | — | 178,175 | |||||||||||||||||||||||||||||
Accreted redemption value for common stock warrants attached to Series A redeemable convertible preferred stock | 42,744 | — | — | — | — | — | — | — | — | (42,744 | ) | — | (42,744 | ) | |||||||||||||||||||||||||||
Accreted redemption value for Series A, Series B, and Series C redeemable convertible preferred stock | 1,627,500 | 2,137,743 | 365,558 | — | — | — | — | — | — | (4,130,801 | ) | — | (4,130,801 | ) | |||||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | — | — | — | (21,071,706 | ) | — | (21,071,706 | ) | |||||||||||||||||||||||||||
Balances at December 31, 2002 | $ | 26,826,253 | $ | 35,346,675 | $ | 45,853,329 | 624,584 | $ | 625 | $ | 6,002,692 | $ | (23,250,000 | ) | $ | 213,710 | $ | — | $ | (46,301,950 | ) | $ | — | $ | (63,334,923 | ) | |||||||||||||||
Targacept, Inc.
Statements of Redeemable Convertible Preferred Stock and Stockholders’ Equity (Deficit)—(continued)
| Redeemable Convertible Preferred Stock | Common Stock | Capital in Excess of Par Value | Excess of Fair Value of Series A Preferred over Cash Received | Common Stock Warrants | Deferred Compensation | Accumulated Deficit | Accumulated Other Comprehensive Loss | Total Stockholders’ Deficit | ||||||||||||||||||||||||||||||||
| Series A | Series B | Series C | Shares | Amount | ||||||||||||||||||||||||||||||||||||
Balances at December 31, 2002 (carried forward) | $ | 26,826,253 | $ | 35,346,675 | $ | 45,853,329 | 624,584 | $ | 625 | $ | 6,002,692 | $ | (23,250,000 | ) | $ | 213,710 | $ | — | $ | (46,301,950 | ) | $ | — | $ | (63,334,923 | ) | ||||||||||||||
Stock issuance costs | — | — | (32,548 | ) | — | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
Issuance of 11,404,958 shares of Series C redeemable convertible preferred stock at $1.21 per share | — | — | 13,800,000 | — | — | — | — | — | — | — | — | — | ||||||||||||||||||||||||||||
Issuance of 458,187 shares of common stock at $0.001 per share par value, related to exercise of stock options | — | — | — | 458,187 | 458 | 303,821 | — | — | — | — | — | 304,279 | ||||||||||||||||||||||||||||
Accreted redemption value for common stock warrants attached to Series A redeemable convertible preferred stock | 42,744 | — | — | — | — | — | — | — | — | (42,744 | ) | — | (42,744 | ) | ||||||||||||||||||||||||||
Accreted redemption value for Series A, Series B, and Series C redeemable convertible preferred stock | 1,627,500 | 2,137,744 | 4,532,640 | — | — | — | — | — | — | (8,297,884 | ) | — | (8,297,884 | ) | ||||||||||||||||||||||||||
Net change in unrealized holding loss on available-for-sale securities | — | — | — | — | — | — | — | — | — | — | (29,282 | ) | (29,282 | ) | ||||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | — | — | — | (19,395,321 | ) | — | (19,395,321 | ) | ||||||||||||||||||||||||||
Comprehensive loss | (19,424,603 | ) | ||||||||||||||||||||||||||||||||||||||
Balances at December 31, 2003 | 28,496,497 | 37,484,419 | 64,153,421 | 1,082,771 | 1,083 | 6,306,513 | (23,250,000 | ) | 213,710 | — | (74,037,899 | ) | (29,282 | ) | (90,795,875 | ) | ||||||||||||||||||||||||
Issuance of 81,753 shares of common stock at $0.001 per share par value, related to exercise of stock options (unaudited) | — | — | — | 81,753 | 82 | 71,232 | — | — | — | — | — | 71,314 | ||||||||||||||||||||||||||||
Accreted redemption value for common stock warrants attached to Series A redeemable convertible preferred stock (unaudited) | 10,686 | — | — | — | — | — | — | — | — | (10,686 | ) | — | (10,686 | ) | ||||||||||||||||||||||||||
Accreted redemption value for Series A Series B and Series C redeemable convertible preferred stock (unaudited) | 406,875 | 534,435 | 1,189,893 | — | — | — | — | — | — | (2,131,203 | ) | — | (2,131,203 | ) | ||||||||||||||||||||||||||
Net change in unrealized holding loss on available-for-sale securities (unaudited) | — | — | — | — | — | — | — | — | — | — | (5,510 | ) | (5,510 | ) | ||||||||||||||||||||||||||
Net loss (unaudited) | — | — | — | — | — | — | — | — | — | (6,637,935 | ) | (6,637,935 | ) | |||||||||||||||||||||||||||
Comprehensive loss (unaudited) | (6,643,445 | ) | ||||||||||||||||||||||||||||||||||||||
Balances at March 31, 2004 (unaudited) | $ | 28,914,058 | $ | 38,018,854 | $ | 65,343,314 | 1,164,524 | $ | 1,165 | $ | 6,637,745 | $ | (23,250,000 | ) | $ | 213,710 | $ | — | $ | (82,817,723 | ) | $ | (34,792 | ) | $ | (99,509,895 | ) | |||||||||||||
See accompanying notes.
Statements of Cash Flows
| Year ended December 31, | Three months ended March 31, | |||||||||||||||||||
| 2001 | 2002 | 2003 | 2003 | 2004 | ||||||||||||||||
| (unaudited) | ||||||||||||||||||||
Operating activities | ||||||||||||||||||||
Net loss | $ | (7,302,270 | ) | $ | (21,071,706 | ) | $ | (19,395,321 | ) | $ | (4,185,042 | ) | $ | (6,637,935 | ) | |||||
Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||||||||||
Depreciation and amortization | 308,594 | 958,105 | 672,927 | 162,616 | 177,423 | |||||||||||||||
Loss on disposal of equipment | — | 53,996 | — | — | — | |||||||||||||||
Write-off of in-process research and development | — | 2,666,000 | — | — | — | |||||||||||||||
Non-cash compensation expense | 92,638 | 129,710 | 65,325 | 4,188 | 18,444 | |||||||||||||||
Recognition of deferred rent incentive | — | (167,769 | ) | (402,647 | ) | (100,662 | ) | (100,662 | ) | |||||||||||
Realized loss on sale of investments | — | — | 20,978 | — | 10,482 | |||||||||||||||
Amortization of discount on held-to-maturity investments | (460,701 | ) | (48,446 | ) | — | — | — | |||||||||||||
Changes in operating assets and liabilities, excluding the effects from acquired assets and liabilities: | ||||||||||||||||||||
Research fees and accounts receivable | (210,448 | ) | (713,109 | ) | 517,030 | 731,483 | 593,944 | |||||||||||||
Inventories | — | 22,464 | 51,016 | 6,340 | 3,708 | |||||||||||||||
Prepaid expenses and accrued interest receivable | (555,929 | ) | 158,605 | (205,054 | ) | 125,276 | (102,852 | ) | ||||||||||||
Accounts payable and accrued expenses | 742,837 | 1,503,132 | (326,451 | ) | (1,535,622 | ) | 258,778 | |||||||||||||
Deferred license fee revenue | 1,321,637 | (634,881 | ) | (269,532 | ) | (67,383 | ) | (67,384 | ) | |||||||||||
Net cash used in operating activities | (6,063,642 | ) | (17,143,899 | ) | (19,271,729 | ) | (4,858,806 | ) | (5,846,054 | ) | ||||||||||
Investment activities | ||||||||||||||||||||
Purchase of investments | (29,284,891 | ) | (11,500,000 | ) | (84,796,103 | ) | (43,500,000 | ) | (22,144,148 | ) | ||||||||||
Proceeds from sale of investments | 32,700,000 | 26,400,000 | 58,500,000 | — | 22,050,312 | |||||||||||||||
Purchase of property and equipment | (1,522,064 | ) | (1,281,840 | ) | (545,254 | ) | (99,715 | ) | (137,055 | ) | ||||||||||
Proceeds from rent incentive | — | 2,013,234 | — | — | — | |||||||||||||||
Purchase of Inversine product | — | (3,500,000 | ) | — | — | — | ||||||||||||||
Net cash provided by (used in) investing activities | 1,893,045 | 12,131,394 | (26,841,357 | ) | (43,599,715 | ) | (230,891 | ) | ||||||||||||
Financing activities | ||||||||||||||||||||
Proceeds from borrowing of long-term debt | — | 3,000,000 | — | 101,289 | — | |||||||||||||||
Principal payments on long-term debt | — | (324,891 | ) | (637,483 | ) | (143,102 | ) | (102,172 | ) | |||||||||||
Proceeds from issuance of redeemable convertible preferred stock, net of transaction costs | 96,692 | 45,487,771 | 13,767,452 | 13,767,452 | — | |||||||||||||||
Proceeds from issuance of common stock | 106,371 | 48,465 | 238,954 | 54,014 | 52,870 | |||||||||||||||
Net cash provided by (used in) financing activities | 203,063 | 48,211,345 | 13,368,923 | 13,779,653 | (49,302 | ) | ||||||||||||||
Net (decrease) increase in cash and cash equivalents | (3,967,534 | ) | 43,198,840 | (32,744,163 | ) | (34,678,868 | ) | (6,126,247 | ) | |||||||||||
Cash and cash equivalents at beginning of period | 5,122,014 | 1,154,480 | 44,353,320 | 44,353,320 | 11,609,157 | |||||||||||||||
Cash and cash equivalents at end of period | $ | 1,154,480 | $ | 44,353,320 | $ | 11,609,157 | $ | 9,674,452 | $ | 5,482,910 | ||||||||||
See accompanying notes.
Notes to Financial Statements
December 31, 2003
1. The Company and Nature of Operations
Targacept, Inc., a Delaware corporation (the “Company”), was formed on March 7, 1997. The Company is a biopharmaceutical company engaged in the design, discovery and development of a new class of drugs to treat multiple diseases and disorders by selectively targeting a class of receptors known as neuronal nicotinic acetylcholine receptors, or NNRs. Its facilities are located in Winston-Salem, North Carolina.
The accompanying financial statements have been prepared on a going concern basis. The Company has incurred operating losses since its inception and expects to incur substantial additional losses for the forseeable future. As a result, the Company will require substantial additional funds, and plans to seek collaborative agreements, research funding, and private or public equity or debt financing to meet such needs. If such funds are not available, management may need to reassess its plans. Even if the Company does not have an immediate need for additional cash, it may seek access to the private or public equity markets if and when conditions are favorable. There is no assurance that such additional funds will be available for the Company to finance its operations on acceptable terms, if at all.
The accompanying balance sheet as of March 31, 2004, the statements of operations and statements of cash flows for the three months ended March 31, 2003 and 2004 and the statement of redeemable convertible preferred stock and stockholders’ equity (deficit) for the three months ended March 31, 2004 are unaudited. The unaudited interim financial statements have been prepared on the same basis as the annual financial statements and, in the opinion of the Company’s management, reflect all adjustments, which include only normal recurring adjustments necessary to present fairly the Company’s financial position and results of operations and cash flows for the three months ended March 31, 2003 and 2004. The financial data and other information disclosed in these notes to financial statements related to the three-month periods are unaudited. The results for the three months ended March 31, 2004 are not necessarily indicative of the results to be expected for the year ending December 31, 2004 or for any other interim period or for any other future year.
2. Summary of Significant Accounting Policies
Cash and Cash Equivalents
The Company considers cash equivalents to be those investments, which are highly liquid, readily convertible to cash, and which mature within three months from the date of purchase.
Investments
In accordance with the Company’s investment policy, surplus cash is invested with high quality financial institutions in money market accounts, certificates of deposit, and a mutual fund that invests in Government National Mortgage Association and other mortgage-backed securities, United States Government debt and other asset-backed securities with AAA credit ratings. The Company determines the appropriate classification of marketable securities at the time of purchase and reevaluates such designation as of each balance sheet date. All marketable securities have been classified as available-for-sale and are stated at market value with the unrealized holding gains and losses reported as a component of stockholders’ equity in comprehensive loss. Interest and dividend income on investments, as well as realized gains and losses, are included in “Interest and dividend income.” The cost of securities sold is based on the specific identification method.
Research Fees and Accounts Receivable
Substantially all of the Company’s research fees and accounts receivable are related to the collaborative research and license agreements discussed in Note 15, and trade sales of Inversine. All of the Company’s trade
Targacept, Inc.
Notes to Financial Statements—(continued)
2. Summary of Significant Accounting Policies (continued)
accounts receivable are due from customers located within the United States. The Company makes judgments with respect to the collectibility of trade accounts receivable based on historical experience and current economic trends. Actual losses could differ from those estimates.
During 2001, 2002, 2003 and the three months ended March 31, 2004, the Company recognized revenues of $1,703,000, $2,024,000, $1,572,000 and $202,000, respectively, or 99%, 89%, 64% and 40%, respectively, of total revenues, from two collaborative research and license agreements discussed in Note 15.
Inventories
Inventories are stated at the lower of cost or market. Cost is determined by the weighted-average method.
Property and Equipment and Intangible Assets
Property and equipment consists primarily of lab equipment, office furniture and fixtures and leasehold improvements and is recorded at cost. Depreciation is computed using the straight-line method over the estimated useful lives of the assets ranging from three to ten years. Lab equipment is typically depreciated over 3-5 years, office furniture and fixtures are typically depreciated over 7-10 years, and leasehold improvements are amortized over the life of the applicable lease.
Intangible assets consist of patents acquired (See Note 16). The intangible assets are being amortized to research and development expense on a straight-line basis over the remaining useful life of the patents, or a period of 17 years from the date of acquisition.
The Company assesses the net realizable value of its long-lived assets and evaluates such assets for impairment whenever events or changes in business circumstances indicate that the carrying amount of an asset may not be recoverable. An impairment loss would be recognized when estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition are less than its carrying amount. An impairment loss, if recognized, would be based on the excess of the carrying value of the impaired asset over its respective fair value. Through March 31, 2004, there has been no such impairment.
Patents and research and development costs
The Company capitalizes the costs of patents purchased from external sources. Expenditures that are related to the development of new products and processes, including filing and maintenance costs of patents created internally, are expensed as incurred until such time as technological feasibility has been established and the products are approved by regulatory agencies for introduction to the market.
Research and development costs are expensed when incurred and include related salaries, contractor fees, administrative expenses and allocations of research related overhead costs.
The Company directly reduces expenses for amounts reimbursed pursuant to cost sharing agreements. During 2001, 2002, 2003 and the three months ended March 31, 2004, research and development expenses were reduced by $412,000, $514,000, $131,000 and $69,000, respectively, for costs reimbursed primarily by Dr. Falk Pharma, GmbH and under the terms of the collaborations described in Note 15.
Clinical Trials Accounting
The Company records accruals based on estimates of the services received and efforts expended pursuant to contracts with numerous clinical trial centers and contract research organizations. In the normal course of
Targacept, Inc.
Notes to Financial Statements—(continued)
2. Summary of Significant Accounting Policies (continued)
business, the Company contracts with third parties to perform various clinical trial activities in the ongoing development of potential products. The financial terms of these agreements are subject to negotiation and variation from contract to contract and may result in uneven payment flows. Payments under the contracts depend on factors such as the achievement of certain events, the successful recruitment of patients, the completion of portions of the clinical trial or similar conditions. The objective of the Company’s accrual policy is to match the recording of expenses in its financial statements to the actual services received and efforts expended. As such, expense accruals related to clinical trials are recognized based on the Company’s estimate of the degree of completion of the event or events specified in the specific clinical study or trial contract.
Deferred Rent Incentive
In August 2002, the Company received $2,013,000 as an incentive to lease its current office space. The incentive is being recognized monthly over the life of the lease on a straight-line basis as a reduction to the lease expense in general and administrative expenses. The Company recognized $168,000, $403,000 and $101,000 of the incentive during 2002, 2003 and the three months ended March 31, 2004, respectively.
Redeemable Convertible Preferred Stock
The carrying value of redeemable convertible preferred stock is increased by periodic accretions so that the carrying amount will equal the redemption amount at the earliest redemption date. These increases are affected through charges to accumulated deficit.
Fair Value of Financial Instruments
The carrying amounts of cash and cash equivalents, short-term investments, accounts receivable, accounts payable, accrued expenses and redeemable convertible preferred stock are considered to be representative of their respective fair values. The fair value of long-term debt is $2,590,000, $1,961,000 and $1,867,000 at December 31, 2002, 2003 and March 31, 2004, respectively. The Company estimates that the fair value of long-term debt using discounted cash flows based on its incremental borrowing rates for similar debt.
Credit Risk
Financial instruments that potentially subject the Company to credit risk consist principally of cash and short-term investments. The Company places its cash and cash equivalents with high-credit quality financial institutions. The Company has established guidelines for investment of its excess cash designed to emphasize safety, liquidity and preservation of capital.
Revenue Recognition
The Company uses revenue recognition criteria in Staff Accounting Bulletin No. 101,Revenue Recognition in Financial Statements, as amended by Staff Accounting Bulletin No. 104,Revision of Topic 13, and Emerging Issues Task Force (“EITF”) Issue 00-21,Revenue Arrangements with Multiple Deliverables. Revenues are recorded under collaboration agreements at the time of performance of services. Research fee revenues are earned and recognized in accordance with contract provisions. License fees for access to the Company’s intellectual properties are recognized ratably over the contracted period in accordance with the provisions of the contract. Amounts received in advance of performance are recorded as deferred revenue and amortized in the statements of operations into revenue over the estimated life of the research and development period. Revenues from milestones are only recognized upon achievement of the milestone criteria. Product sales revenues are recorded when goods are shipped, at which point title has passed.
Targacept, Inc.
Notes to Financial Statements—(continued)
2. Summary of Significant Accounting Policies (continued)
Shipping and Handling Costs
During 2002, 2003 and the three months ended March 31, 2004, $22,000, $173,000 and $41,000, respectively, of shipping and handling costs were included in cost of product sales.
Income Taxes
The liability method is used in accounting for income taxes as required by Statement of Financial Accounting Standards (“SFAS”) No. 109,Accounting for Income Taxes. Under this method, deferred tax assets and liabilities are recognized for operating loss and tax credit carryforwards and for the future tax consequences attributable to the differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in the results of operations in the period that includes the enactment date. A valuation allowance is recorded to reduce the carrying amounts of deferred tax assets unless it is more likely than not that such assets will be realized. Currently there is no provision for income taxes as the Company has incurred net losses to date.
Comprehensive Loss
SFAS No. 130,Reporting Comprehensive Income, requires components of other comprehensive loss, including unrealized gains and losses on available-for-sale securities, to be included as part of total comprehensive loss. The components of comprehensive loss are included in the statements of redeemable convertible preferred stock and stockholders’ equity (deficit).
Net Loss Per Share Attributable to Common Stockholders
The Company computes net loss per share attributable to common stockholders in accordance with SFAS No. 128,Earnings Per Share (“SFAS 128”). Under the provisions of SFAS 128, basic net loss per share attributable to common stockholders (“Basic EPS”) is computed by dividing net loss attributable to common stockholders by the weighted average number of common shares outstanding. Diluted net loss per share attributable to common stockholders (“Diluted EPS”) is computed by dividing net loss attributable to common stockholders by the weighted average number of common shares and dilutive common share equivalents then outstanding. Common share equivalents consist of the incremental common shares issuable upon the conversion of preferred stock, shares issuable upon the exercise of stock options and shares issuable upon the exercise of warrants. For the periods presented, Diluted EPS is identical to Basic EPS because common share equivalents are excluded from the calculation, as their effect is antidilutive.
Pro Forma Stockholders’ Equity and Pro Forma Net Loss Per Share
The Company’s Board of Directors has authorized management of the Company to file a registration statement with the Securities and Exchange Commission permitting the Company to sell shares of its common stock to the public in an initial public offering (the “IPO”). If the IPO is closed under the terms presently anticipated, all of the redeemable convertible preferred stock outstanding at the time of the IPO will automatically convert into 73,739,905 shares of common stock. Unaudited pro forma stockholders’ equity, as adjusted for the assumed conversion of the preferred stock, is set forth on the accompanying balance sheets. Unaudited pro forma basic and diluted net loss per share is computed using the weighted average number of common shares outstanding, including the pro forma effects of the conversion of outstanding redeemable
Targacept, Inc.
Notes to Financial Statements—(continued)
2. Summary of Significant Accounting Policies (continued)
convertible preferred stock into shares of the Company’s common stock effective upon the completion of the Company’s planned IPO as if such conversion had occurred at January 1, 2003, or the date of issuance, if later.
The following table sets forth the computation of basic and diluted, and unaudited pro forma basic and diluted, net loss per share attributable to common stockholders:
| Year ended December 31, | Three months ended March 31, | |||||||||||||||||||
| 2001 | 2002 | 2003 | 2003 | 2004 | ||||||||||||||||
| (unaudited) | ||||||||||||||||||||
Historical | ||||||||||||||||||||
Numerator: | ||||||||||||||||||||
Net loss attributable to common stockholders | $ | (11,110,258 | ) | $ | (25,245,251 | ) | $ | (27,735,949 | ) | $ | (6,100,000 | ) | $ | (8,779,824 | ) | |||||
Denominator: | ||||||||||||||||||||
Weighted-average common shares outstanding | 414,624 | 557,492 | 817,894 | 643,571 | 1,112,591 | |||||||||||||||
Basic and diluted net loss per share attributable to common stockholders | $ | (26.80 | ) | $ | (45.28 | ) | $ | (33.91 | ) | $ | (9.48 | ) | $ | (7.89 | ) | |||||
Pro forma | ||||||||||||||||||||
Numerator: | ||||||||||||||||||||
Net loss attributable to common stockholders | $ | (19,395,321 | ) | $ | (6,637,935 | ) | ||||||||||||||
Denominator: | ||||||||||||||||||||
Shares used above | 817,894 | 1,112,591 | ||||||||||||||||||
Pro forma adjustments to reflect assumed conversion of preferred stock, on a weighted average basis (unaudited) | 70,300,735 | 73,739,905 | ||||||||||||||||||
Shares used to compute pro forma basic and diluted net loss per share attributable to common stockholders (unaudited) | 71,118,629 | 74,852,496 | ||||||||||||||||||
Pro forma basic and diluted net loss per share attributable to common stockholders (unaudited) | $ | (0.27 | ) | $ | (0.09 | ) | ||||||||||||||
Targacept, Inc.
Notes to Financial Statements—(continued)
2. Summary of Significant Accounting Policies (continued)
The Company has excluded all outstanding stock options and warrants from the calculation of net loss per share attributable to common stockholders because such securities are antidilutive for all periods presented. Had the Company been in a net income position, these securities may have been included in the calculation. These potentially dilutive securities consist of the following on a weighted average basis:
| Year Ended December 31, | Three Months ended March 31, | |||||||||
| 2001 | 2002 | 2003 | 2003 | 2004 | ||||||
Outstanding common stock options | 1,496,707 | 2,150,650 | 4,601,270 | 3,604,941 | 8,000,521 | |||||
Redeemable convertible preferred stock | 11,567,567 | 15,980,048 | 70,300,735 | 59,792,159 | 73,739,905 | |||||
Outstanding warrants | 1,612,903 | 1,612,903 | 1,612,903 | 1,612,903 | 1,612,903 | |||||
Total | 14,677,177 | 19,743,601 | 76,514,908 | 65,010,003 | 83,353,329 | |||||
Stock-Based Compensation
In December 2002, the Financial Accounting Standards Board (“FASB”) issued SFAS No. 148,Accounting for Stock-Based Compensation—Transition and Disclosure. SFAS No. 148 amends SFAS No. 123 to provide alternative methods of transition for a voluntary change to the fair value based method of accounting for stock-based employee compensation. In addition, SFAS No. 148 amends the disclosure provisions of SFAS No. 123 and Accounting Principles Board (“APB”) Opinion No. 28,Interim Financial Reporting, to require more prominent disclosure in the summary of significant accounting policies about the method of accounting for the effects of an entity’s accounting policy with respect to stock-based employee stock compensation and the effect of the method used on reported net loss results.
The Company has elected to continue to account for stock options granted to employees using the intrinsic-value method as prescribed by APB Opinion No. 25,Accounting for Stock Issued to Employees, and, thus recognizes no compensation expense for options granted with exercise prices equal to the fair market value of the Company’s common stock on the date of grant. The information regarding net loss as required by SFAS No. 123 has been determined as if the Company had accounted for its employee stock options under the fair value method of that Statement.
The following table illustrates the weighted-average assumptions for the Black-Scholes model used in determining the fair value of options granted to employees:
Year ended December 31, | Three months ended March 31, | ||||||||||||||
| 2001 | 2002 | 2003 | 2003 | 2004 | |||||||||||
Dividend yield | — | — | — | — | — | ||||||||||
Risk-free interest rate | 4.0 | % | 3.5 | % | 2.8 | % | 2.7 | % | 2.8 | % | |||||
Volatility | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 | ||||||||||
Expected life | 4 years | 4 years | 4 years | 4 years | 4 years | ||||||||||
Targacept, Inc.
Notes to Financial Statements—(continued)
2. Summary of Significant Accounting Policies (continued)
For purposes of disclosures pursuant to SFAS No. 123, as amended by SFAS No. 148, the estimated fair value of the options is amortized to expense over the options’ vesting period. The following table illustrates the effect on net loss if the Company had applied the fair value recognition provisions of SFAS No. 123:
| Year Ended December 31, | Three months ended March 31, | |||||||||||||||||||
| 2001 | 2002 | 2003 | 2003 | 2004 | ||||||||||||||||
Net loss attributable to common stockholders, as reported | $ | (11,110,258 | ) | $ | (25,245,251 | ) | $ | (27,735,949 | ) | $ | (6,100,000 | ) | $ | (8,779,824 | ) | |||||
Add: Stock-based employee compensation expense included in reported net income, net of related tax effects | 92,638 | 129,710 | 65,325 | 4,188 | 18,444 | |||||||||||||||
Deduct: Stock-based employee compensation expense determined under fair value based method for all awards, net of related tax effects | (205,710 | ) | (312,830 | ) | (515,405 | ) | (87,714 | ) | (233,319 | ) | ||||||||||
Pro forma net loss | $ | (11,223,330 | ) | $ | (25,428,371 | ) | $ | (28,186,029 | ) | $ | (6,183,526 | ) | $ | (8,994,699 | ) | |||||
Net loss per share: | ||||||||||||||||||||
Basic and diluted, as reported | $ | (26.80 | ) | $ | (45.28 | ) | $ | (33.91 | ) | $ | (9.48 | ) | $ | (7.89 | ) | |||||
Basic and diluted, pro forma | $ | (27.06 | ) | $ | (45.61 | ) | $ | (34.46 | ) | $ | (9.61 | ) | $ | (8.08 | ) | |||||
Stock compensation arrangements to non-employees are accounted for in accordance with SFAS No. 123, as amended by SFAS No. 148, and EITF No. 96-18,Accounting for Equity Instruments That Are Issued to Other than Employees for Acquiring, or in Conjunction with Selling, Goods or Services,using a fair value approach.
Recent Accounting Pronouncements
In 2003, FASB Statement No. 150,Accounting for Certain Financial Instruments with Characteristics of both Liabilities and Equity (“SFAS 150”), was issued. This statement establishes how a company classifies and measures certain financial instruments with characteristics of both liabilities and equity, including redeemable convertible preferred stock. This statement is effective for financial instruments entered into or modified after May 31, 2003, and otherwise effective at the beginning of the interim period commencing July 1, 2003, except for mandatory redeemable financial instruments of nonpublic companies. The FASB has indefinitely deferred implementation of certain provisions of SFAS 150. The adoption of SFAS 150 did not affect the financial position or results of operations of the Company.
In 2003, the FASB issued Interpretation No. 46,Consolidation of Variable Interest Entities (“FIN 46”), which is an interpretation of Accounting Research Bulletin No. 51,Consolidated Financial Statements. FIN 46 requires that if an entity has a controlling interest in a variable interest entity, the assets, liabilities, and results of activities of the variable interest entity should be included in the consolidated financial statements of the entity. FIN 46 is effective immediately for all new variable interest entities created or acquired after January 31, 2003. The Company implemented the provisions of FIN 46 for its financial statements for the year ending December 31, 2003 for any variable interest entities created before February 1, 2003. The adoption of FIN 46 did not affect the financial position or results of operations of the Company.
Targacept, Inc.
Notes to Financial Statements—(continued)
2. Summary of Significant Accounting Policies (continued)
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
Reclassifications
Certain reclassifications have been made to the prior year financial statements to conform to the current year presentation. These reclassifications had no impact on net loss or previously recorded amounts.
3. Short-term investments
The following is a summary of available-for-sale securities as of December 31, 2002, 2003 and March 31, 2004:
December 31, 2002 | Amortized Cost | Unrealized Gains | Unrealized Losses | Fair Value | ||||||||
Certificates of deposit | $ | 5,000,000 | $ | — | $ | — | $ | 5,000,000 | ||||
Interest receivable | 8,139 | — | — | 8,139 | ||||||||
Total | $ | 5,008,139 | $ | — | $ | — | $ | 5,008,139 | ||||
December 31, 2003 | Amortized Cost | Unrealized Gains | Unrealized Losses | Fair Value | |||||||||
Adjustable rate mortgage fund | $ | 20,177,033 | $ | — | $ | (29,282 | ) | $ | 20,147,751 | ||||
Certificates of deposit | 11,098,092 | — | — | 11,098,092 | |||||||||
Interest receivable | 121,657 | — | — | 121,657 | |||||||||
Total | $ | 31,396,782 | $ | — | $ | (29,282 | ) | $ | 31,367,500 | ||||
March 31, 2004 | Amortized Cost | Unrealized Gains | Unrealized Losses | Fair Value | |||||||||
Adjustable rate mortgage fund | $ | 15,377,586 | $ | — | $ | (34,793 | ) | $ | 15,302,793 | ||||
Certificates of deposit | 16,020,895 | — | — | 16,020,895 | |||||||||
Interest receivable | 40,572 | — | — | 40,572 | |||||||||
Total | $ | 31,439,053 | $ | — | $ | (34,793 | ) | $ | 31,364,260 | ||||
The adjustable rate mortgage fund investment has remained in an unrealized loss position for less than 6 months. Accordingly the Company believes this is a temporary decline. The Company recognized $1,448,000, $88,000, $591,000 and $59,000 of interest income during 2001, 2002, 2003 and the three months ended March 31, 2004, respectively. The Company recognized $200,000 and $171,000 of dividend income during 2003 and the three months ended March 31, 2004, respectively.
Targacept, Inc.
Notes to Financial Statements—(continued)
4. Inventories
Inventories consisted of the following:
| December 31, | March 31, | ||||||||
| 2002 | 2003 | 2004 | |||||||
Raw materials | $ | 137,706 | $ | 46,988 | $ | 46,988 | |||
Work-in-process | 9,042 | 6,892 | 6,892 | ||||||
Finished goods | 22,788 | 64,640 | 60,932 | ||||||
| $ | 169,536 | $ | 118,520 | $ | 114,812 | ||||
5. Property and equipment
Property and equipment consists of the following:
| December 31, | March 31, | ||||||||
| 2002 | 2003 | 2004 | |||||||
Lab equipment | $ | 4,674,369 | $ | 5,057,345 | $ | 5,056,768 | |||
Office furniture and fixtures | 1,030,406 | 1,192,685 | 1,295,933 | ||||||
Leasehold improvements | 87,875 | 87,875 | 87,875 | ||||||
| 5,792,650 | 6,337,905 | 6,440,576 | |||||||
Less: accumulated depreciation | 3,329,706 | 3,964,870 | 4,098,468 | ||||||
Property and equipment, net | $ | 2,462,944 | $ | 2,373,035 | $ | 2,342,108 | |||
The Company recorded $309,000, $577,000, $635,000 and $168,000 of depreciation expense during 2001, 2002, 2003 and the three months ended March 31, 2004, respectively.
6. Intangible Assets
Intangible assets consist of the following:
| December 31, | March 31, | |||||||||||
| 2002 | 2003 | 2004 | ||||||||||
Patents (See Note 16) | $ | 642,000 | $ | 642,000 | $ | 642,000 | ||||||
Less: accumulated amortization | (15,735 | ) | (53,499 | ) | (62,940 | ) | ||||||
Total | $ | 626,265 | $ | 588,501 | $ | 579,060 | ||||||
The Company recognized amortization expense of $0, $16,000, $38,000 and $9,000 in 2001, 2002, 2003 and the three months ended March 31, 2004, respectively. The Company expects to recognize $38,000 of amortization expense in each of the next five years.
7. Accrued Expenses
Accrued expenses consists of the following:
| December 31, | March 31, | ||||||||
| 2002 | 2003 | 2004 | |||||||
Clinical trials costs | $ | 361,119 | $ | 623,158 | $ | 1,265,409 | |||
Employee compensation | 860,368 | 676,900 | 311,273 | ||||||
Other | 255,547 | 106,720 | 111,246 | ||||||
| $ | 1,477,034 | $ | 1,406,778 | $ | 1,687,928 | ||||
Targacept, Inc.
Notes to Financial Statements—(continued)
8. Long-term debt
During 2002, the Company entered into agreements to borrow $500,000 from the City of Winston-Salem and $2,500,000 from R.J. Reynolds Tobacco Company (“RJRT”). The note payable to the City of Winston-Salem matures on April 19, 2012, is non-interest bearing until April 2007 and, thereafter, bears interest between 5% and 7% depending on the gross revenues of the Company until maturity. No payments are due on the City of Winston-Salem note until the 5-year anniversary of the loan. The note payable to RJRT accrues interest at 6.6%, is repayable in monthly payments of $59,403 through the maturity date of May 1, 2006, and is secured by equipment owned by the Company with a book value of approximately $2,373,000, net of accumulated depreciation, at December 31, 2003. The Company paid $91,000, $135,000 and $25,000 for interest under the RJRT note during 2002, 2003 and the three months ended March 31, 2004, respectively.
Maturities of long-term debt are as follows at December 31, 2003:
2004 | $ | 576,072 | |
2005 | 669,379 | ||
2006 | 292,175 | ||
2007 | 69,438 | ||
2008 | 93,830 | ||
Thereafter | 336,732 | ||
| $ | 2,037,626 | ||
9. Redeemable Preferred Stock
In August 2000, the Company issued 5,000,000 shares of its Series A redeemable convertible preferred stock (the “Series A”) to RJRT, and completed a private placement of 6,537,634 of its Series B redeemable convertible preferred stock (the “Series B”) generating cash of $29,073,000, net of offering costs.
In January 2001, the Company issued 29,933 shares of Series B to three consultants in partial payment of consulting fees owed by the Company.
In November 2002, the Company completed a private placement of 37,764,180 shares of its Series C redeemable convertible preferred stock (the “Series C”) and received cash of $45,488,000, net of offering costs.
In March 2003, the Company completed a private placement of an additional 11,404,958 shares of Series C and received cash of $13,767,452, net of offering costs.
The following is a summary of the rights, preferences and terms of the Company’s outstanding series of redeemable convertible preferred stock:
Conversion
Each share of Series A, Series B and Series C is convertible, at the option of the holder thereof, at any time and from time to time, and without the payment of additional consideration by the holder thereof, into fully paid and nonassessable shares of the Company’s common stock. As of December 31, 2003 and March 31, 2004, conversion of the Series A, Series B and Series C would result in the issuance of 5,000,000, 15,619,675, and 53,120,230 shares of common stock, respectively. Future sales of equity at prices below the respective conversion prices could result in adjustments to the number of shares of common stock into which each series of preferred stock is convertible.
Targacept, Inc.
Notes to Financial Statements—(continued)
9. Redeemable Preferred Stock (continued)
Automatic conversion of the Series A, Series B and Series C into fully paid and nonassessable shares of common stock, without the payment of additional consideration by the holders thereof, would occur immediately upon the closing of the sale of the Company’s common stock in a firm commitment, underwritten public offering registered under the Securities Act of 1933 in which (i) the price per share equals or exceeds $3.60 (subject to certain adjustments) and (ii) the gross proceeds to the Company are not less than $50,000,000. The accrued but unpaid cumulative dividend on the Series C shall, if not yet declared, be forfeited upon conversion of the Series C.
Dividends
Dividends accrue daily on each share of Series C on a cumulative basis at the rate of 8% per annum and are recorded as an increase to Series C and an increase to accumulated deficit. Cumulative dividends may be declared and paid at any time and shall be payable upon liquidation or redemption. At December 31, 2003 and March 31, 2004, cumulative accrued dividends on the Series C stock totaled $4,898,000 and $6,088,000, respectively.
Dividends on the Series A, Series B and Series C are payable when and if declared by the Board of Directors. No dividend may be paid on the common stock without the approval of the holders of a majority of the then outstanding shares of Series A and Series B, considered together on an as-converted basis, and the holders of 65% of the Series C. No dividend may be declared or paid on either the Series A or the Series B unless, simultaneously with such declaration or payment, the same dividend per share is declared or paid on both the Series A and the Series B, as well as the Series C, and any unpaid cumulative dividends on the Series C are declared and paid in full.
Voting
Each holder of the Series A, Series B and Series C is entitled to the number of votes equal to the number of shares of common stock into which such holder’s shares are convertible on the applicable record date. In addition, certain actions by the Company require the approval of one or more of (i) the holders of a majority of the outstanding shares of Series A, (ii) the holders of at least two-thirds of the outstanding shares of Series B, (iii) the holders of a majority of the outstanding shares of Series A and Series B, considered together on an as-converted basis, and/or (iv) the holders of at least 65% of the outstanding shares of Series C.
Liquidation Preference
In the event of any liquidation, dissolution or winding up of the Company, the holders of the Series C shares have preference and are entitled to receive an amount per share equal to the greater of (i) their initial purchase price per share plus any accrued or declared and unpaid dividends on such share or (ii) the amount per share of Series C that such holders would receive if all of the Series A, Series B and Series C were converted to common stock immediately prior to such liquidation, dissolution or winding up.
Next, the holders of Series A and Series B are entitled to receive, on apari passubasis, an amount equal to their initial purchase price per share plus any declared and unpaid dividends on such shares. Any assets of the Company remaining after the payments specified above shall be distributed on apari passubasis among the holders of common stock and, on an as-converted to common stock basis, Series A, Series B and Series C. Unless the holders of a prescribed number of shares of Series A, Series B and/or Series C otherwise elect, certain fundamental transactions involving the Company shall be treated as a liquidation for the Series A, Series B and/or Series C, as the case may be.
Targacept, Inc.
Notes to Financial Statements—(continued)
9. Redeemable Preferred Stock (continued)
Mandatory Redemption
At any time after November 26, 2008, upon demand by the holders of at least 65% of the outstanding shares of Series C, all of the outstanding shares of Series C shall be redeemed in cash in an amount per share equal to the initial purchase price per share (subject to certain adjustments) plus any accrued or declared and unpaid dividends on such shares.
At any time after the later of August 22, 2005 or the date on which no shares of Series C are outstanding, a number of outstanding shares of Series A or Series B elected upon demand by the holders of a majority of the outstanding shares of Series A (in the case of Series A) or a majority of the outstanding shares of Series B (in the case of Series B) shall be redeemed in an amount per share equal to $4.65 (subject to certain adjustments) plus (i) any previously declared but unpaid dividends on such share and (ii) an amount equal to $0.081375 per share (subject to certain adjustments) multiplied by the number of complete three-month periods that have elapsed from the date such share was originally issued to the redemption date. The Company may satisfy its redemption obligation with respect to the Series A and/or the Series B in cash or by paying a portion in cash and issuing a promissory note that meets certain prescribed conditions for the remaining amount.
10. Stockholders’ Equity (Deficit)
Prior to August 22, 2000, the Company was a wholly owned subsidiary of RJRT. On August 22, 2000, the Company reclassified its 500 shares of common stock as 5,000,000 shares of Series A and a warrant to purchase 1,612,903 shares of the Company’s common stock at $4.65 per share. On the same date, the Company sold 5,892,473 shares of the Company’s Series B to an investor group. The Company then issued 309,424 shares of the common stock to management, at par value, for proceeds of $309, which was less than fair value. As a result, the Company recorded compensation expense of $145,120. An aggregate of 645,161 shares of Series B redeemable convertible preferred stock were subsequently sold in a second offering to certain of the Company’s investors.
On January 2, 2001, the Company amended its Certificate of Incorporation to increase the number of authorized shares of preferred stock to 11,567,567 shares and issued 29,933 shares of Series B to consultants in exchange for the partial satisfaction of a cash liability on January 26, 2001.
On November 26, 2002, the Company amended its Certificate of Incorporation to increase the number of authorized shares of common stock to 75,000,000 and preferred stock to 49,331,747 and issued 37,764,180 shares of Series C.
On March 14, 2003, the Company amended its Certificate of Incorporation to increase the number of authorized shares of common stock to 85,000,000 and preferred stock to 60,736,705 and issued 11,404,958 shares of Series C.
In conjunction with the issuance of Series A, the Company issued a warrant to purchase 1,612,903 shares of the Company’s common stock at an original exercise price of $4.65 per share (subject to certain adjustments). In connection with the Company’s issuance of Series C and price adjustment provisions of the warrant, the conversion price of the warrant was adjusted to $1.95. The warrant is exercisable only upon the earlier of an initial public offering or a change in control. The fair value of the warrant is a direct cost of obtaining capital. As such, the fair value has been recorded in stockholders’ equity, with the offset recorded as a decrease in the redemption value of the Series A. The Company will accrete to the redemption value of the Series A at the earliest date of redemption, or until August 2005 through an increase in redemption value to Series A and an increase to retained deficit. The fair value of the warrant to purchase 1,612,903 shares of the Company’s
Targacept, Inc.
Notes to Financial Statements—(continued)
10. Stockholders’ Equity (Deficit) (continued)
common stock was estimated at the grant date to be $213,710 or $0.53 per share. The Company considered the anti-dilution features, the contingencies surrounding the limited opportunities for exercise, and the warrant’s priorities over common stock options in relation to the fair value of the Company’s common stock at the date of issuance when estimating the fair value of the warrant.
At December 31, 2003 and March 31, 2004, the Company had reserved shares of common stock for future issuance as follows:
December 31, 2003 | March 31, 2004 | |||
Convertible preferred stock | 73,739,905 | 73,739,905 | ||
Warrant | 1,612,903 | 1,612,903 | ||
Options | 8,554,421 | 8,472,668 | ||
| 83,907,229 | 83,825,476 | |||
11. Income Taxes
There is no income tax provision (benefit) for federal or state income taxes as the Company has incurred operating losses since inception.
The Company’s effective tax rate differs from the federal income tax rate for the following reasons:
| Year ended December 31, | Three months ended March 31, | |||||||||||
| 2001 | 2002 | 2003 | 2004 | |||||||||
Expected federal income tax benefit at statutory rate | (34 | )% | (34 | )% | (34 | )% | (34 | )% | ||||
Increase (decrease) resulting from: | ||||||||||||
Research and development credits | — | (1 | ) | (6 | ) | (2 | ) | |||||
Purchased in-process research and development | — | 4 | — | — | ||||||||
State income tax expense, net of federal benefit | (5 | ) | (4 | ) | (5 | ) | (5 | ) | ||||
Change in valuation allowance | 38 | 35 | 44 | 41 | ||||||||
Other | 1 | — | 1 | — | ||||||||
| — | % | — | % | — | % | — | % | |||||
At December 31, 2002 and 2003 and March 31, 2004, the Company had net operating loss carryforwards for income tax purposes of approximately $25,317,000, $46,090,000 and $53,007,000, respectively, and research and development tax credits of approximately $299,000, $1,456,000 and $1,594,000, respectively. The federal net operating loss carryforwards begin to expire in 2020. State net operating loss carryforwards begin to expire in 2015. The research and development tax credits will begin to expire in 2021.
Utilization of the net operating loss carryforwards and credits may be subject to a substantial annual limitation due to ownership change limitations provided by the Internal Revenue Code of 1986, as amended, and similar state provisions. The Company has not performed a detail analysis to determine whether an ownership change under Section 382 of the Internal Revenue Code occurred. The effect of an ownership change would be the imposition of an annual limitation on the use of net operating loss carryforwards attributable to periods before the change.
Targacept, Inc.
Notes to Financial Statements—(continued)
11. Income Taxes (continued)
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The Company’s net deferred tax assets relate primarily to its net operating loss carryforwards. A valuation allowance has been recognized to offset the deferred tax assets related to those carryforwards. If and when recognized, the tax benefit for those items will be reflected in current operations of the period in which the benefit is recorded as a reduction of income tax expense. The utilization of the loss carryforwards to reduce future income taxes will depend on the Company’s ability to generate sufficient taxable income prior to the expiration of the net operating loss carryforwards. For the years ended December 31, 2002, 2003 and the three months ended March 31, 2004, the valuation allowance increased approximately $7,380,000, $8,617,000 and $2,694,000, respectively.
Significant components of the Company’s deferred tax assets (liabilities) are as follows:
| December 31, | March 31, | |||||||||||
| 2002 | 2003 | 2004 | ||||||||||
Deferred tax assets: | ||||||||||||
Net operating loss carryforward | $ | 9,760,723 | $ | 17,769,377 | $ | 20,436,139 | ||||||
Research and development tax credit | 299,266 | 1,456,438 | 1,594,235 | |||||||||
Equipment and other | 226,338 | — | — | |||||||||
Patents | 682,413 | 641,008 | 641,008 | |||||||||
Unearned revenue | 843,084 | 739,167 | 635,251 | |||||||||
Total gross deferred tax assets | 11,811,824 | 20,605,990 | 23,306,633 | |||||||||
Valuation allowance | (11,811,824 | ) | (20,428,878 | ) | (23,122,851 | ) | ||||||
Net deferred tax asset | — | 177,112 | 183,782 | |||||||||
Deferred tax liabilities: | ||||||||||||
Equipment and other | — | (177,112 | ) | (183,782 | ) | |||||||
Net deferred tax asset | $ | — | $ | — | $ | — | ||||||
Targacept, Inc.
Notes to Financial Statements—(continued)
12. Equity Incentive Plan
On August 22, 2000, the Company established an Equity Incentive Plan (the “Plan”) and authorized the issuance of up to 2,011,259 shares under the Plan to attract and retain employees, directors and certain independent contractors, consultants and advisors and to allow them to participate in the growth of the Company. During 2001, the number of shares authorized for issuance under the Plan was increased to 2,611,259. In conjunction with the Series C financing, the Company authorized the issuance of an additional 3,000,000 shares, increasing the number of authorized shares to 5,611,259. Upon the issuance of the additional Series C shares in March 2003, the number of authorized shares was increased to 9,216,657. Awards may be made to participants under the Plan in the form of incentive and nonqualified stock options, restricted stock, stock appreciation rights, stock awards, and performance awards. Eligible participants under the Plan include employees, directors and certain independent contractors, consultants or advisors of the Company or a related corporation. The vesting periods for awards made under the Plan are determined at the discretion of the Plan administrator. The exercise price of incentive options granted under the Plan may not be less than 100% of the fair market value of the common stock on the date of grant, as determined by the Plan Administrator. The following summarizes stock option activity and balances:
| Options Granted | Price | Weighted Average Exercise Price Per Share | |||||||
Outstanding at January 1, 2001 | 1,234,999 | 0.47 | $ | 0.47 | |||||
Granted | 440,000 | 0.47-4.65 | 0.90 | ||||||
Forfeited | (31,717 | ) | 0.47 | 0.47 | |||||
Exercised | (65,565 | ) | 0.47 | 0.47 | |||||
Outstanding at December 31, 2001 | 1,577,717 | 0.47-4.65 | 0.59 | ||||||
Granted | 838,550 | 0.01-0.68 | 0.58 | ||||||
Forfeited | (18,672 | ) | 0.56 | 0.56 | |||||
Exercised | (90,984 | ) | 0.47-0.68 | 0.53 | |||||
Outstanding at December 31, 2002 | 2,306,611 | 0.47-4.65 | 0.59 | ||||||
Granted | 5,996,095 | 0.01-0.75 | 0.67 | ||||||
Forfeited | (35,980 | ) | 0.01-0.68 | 0.26 | |||||
Exercised | (458,187 | ) | 0.01-0.68 | 0.52 | |||||
Outstanding at December 31, 2003 | 7,808,539 | 0.01-0.75 | 0.65 | ||||||
Granted | 322,608 | 0.01-0.75 | 0.69 | ||||||
Forfeited | (25,000 | ) | 4.65 | 4.65 | |||||
Exercised | (81,753 | ) | 0.47-0.75 | 0.65 | |||||
Outstanding at March 31, 2004 | 8,024,394 | $ | 0.01-0.75 | $ | 0.64 | ||||
Targacept, Inc.
Notes to Financial Statements—(continued)
12. Equity Incentive Plan (continued)
The weighted average fair value of options granted during 2001, 2002, 2003 and the three months ended March 31, 2004 was $0.38, $0.46, $0.41 and $0.47, respectively. At December 31, 2001, 2002, 2003 and March 31, 2004, 83,305, 958,059, 3,538,218 and 4,078,011 options, respectively, were exercisable at a weighted-average price of $0.47, $0.64, $0.63 and $0.62, respectively.
A summary of options outstanding as of December 31, 2003 is as follows:
| Options Outstanding | Options Exercisable | |||||||||||
Exercise | Number Outstanding | Weighted Average Remaining Contractual Exercise Life | Weighted Average Exercise Price | Number Exercisable | Weighted Average Price | |||||||
0.01 | 184,225 | 9.0 | $ | 0.01 | 184,225 | $ | 0.01 | |||||
0.47 | 919,437 | 6.7 | 0.47 | 692,331 | 0.47 | |||||||
0.68 | 6,629,877 | 9.4 | 0.68 | 2,636,662 | 0.68 | |||||||
0.75 | 50,000 | 9.9 | 0.75 | — | — | |||||||
4.65 | 25,000 | 7.1 | 4.65 | 25,000 | 4.65 | |||||||
| 7,808,539 | 9.1 | $ | 0.65 | 3,538,218 | $ | 0.63 | ||||||
A summary of options outstanding as of March 31, 2004 (unaudited) is as follows:
| Options Outstanding | Options Exercisable | |||||||||||
Exercise | Number Outstanding | Weighted Average Remaining Contractual Exercise Life | Weighted Average Exercise Price | Number Exercisable | Weighted Average Price | |||||||
0.01 | 209,225 | 8.9 | $ | 0.01 | 184,225 | $ | 0.01 | |||||
0.47 | 901,142 | 6.5 | 0.47 | 748,519 | 0.47 | |||||||
0.68 | 6,583,523 | 9.1 | 0.68 | 2,884,713 | 0.68 | |||||||
0.75 | 330,504 | 9.8 | 0.75 | 260,554 | 0.75 | |||||||
| 8,024,394 | 8.9 | $ | 0.64 | 4,078,011 | $ | 0.62 | ||||||
During 2001, the Company granted 25,000 options above fair value at a weighted-average exercise price of $4.65. The options had a weighted-average fair value of $0. During 2002 and 2003, respectively, the Company granted 130,000 and 97,500 options below fair value at an exercise price of $0.01 and fair value of $0.67 per share. The fair value of these shares was recorded as compensation expense in the amounts $129,710 and $65,325, during 2002 and 2003, respectively. During the three months ended March 31, 2004, the Company granted 25,000 options below fair value at an exercise price of $0.01 and a fair value of $0.74 per share. The fair value of these shares was recorded as compensation expense in the amount of approximately $18,000.
13. Leases
Prior to March 1, 2002, the Company leased its office space and certain equipment under a non-cancelable, one-year operating lease agreement with RJRT. Rent expense incurred by the Company under the RJRT lease was approximately $609,000 and $106,000 for the years ended December 31, 2001 and 2002, respectively. The Company has no future minimum lease payments to RJRT as of December 31, 2003.
Targacept, Inc.
Notes to Financial Statements—(continued)
13. Leases (continued)
On March 1, 2002, the Company entered into an agreement with Wake Forest University to lease an office and research facility in Winston-Salem, North Carolina with an initial term that extends through July 31, 2007. The lease contains a renewal option for up to one additional five-year term, with a lease rate similar to the original agreement. Rent expense incurred by the Company under this lease was approximately $904,000, $1,456,000 and $364,000 for the years ended December 31, 2002, 2003 and the three months ended March 31, 2004, respectively. Rent expense is offset by the monthly recognition of the deferred rent incentive discussed in Note 2. At December 31, 2003, the Company has the following future minimum lease payments in relation to this lease:
2004 | $ | 1,455,552 | |
2005 | 1,455,552 | ||
2006 | 1,455,552 | ||
2007 | 849,072 | ||
2008 and thereafter | — | ||
| $ | 5,215,728 | ||
14. Retirement Savings Plan
The Company has a 401(k) retirement plan that covers substantially all of its employees. This plan provides for the Company to make 100% matching contributions up to a maximum of 6% of employees’ eligible compensation. The Company contributed $171,000, $290,000, $298,000 and $114,000 to the plan for the years ended December 31, 2001, 2002, 2003 and the three months ended March 31, 2004, respectively.
15. Collaborative Research and License Agreements
Aventis Pharma
In December 1998, the Company entered into a collaborative research and license agreement with Aventis Pharma (“Aventis”) whereby the Company and Aventis agreed to collaborate on the discovery, development and commercialization of nicotinic agonists for use in prevention of certain human diseases. Under the agreement, Aventis was granted a license under certain patent rights and knowledge to develop, manufacture and commercialize certain compounds. The agreement provides for the payment of research fees on a “fee for service” basis for development work that the Company agreed to perform. For the years ended December 31, 2001, 2002, and 2003 and the three months ended March 31, 2004, these fees were $1,100,000, $1,389,000, $1,303,000 and $135,000, respectively. The Company is entitled to receive milestone payments under the contract at specified dates during the development period. The Company did not receive milestone payments under the agreement during 2001, 2002, 2003 or the three months ended March 31, 2004. In addition, Aventis agreed to make royalty payments based on net sales of products developed and sold. In general, either party may terminate the agreement in the event of a material breach by the other party, including a material breach of research obligations or the issuance of third-party patent rights to a competitor. Additionally, Aventis may terminate the agreement without cause by providing the Company with 30 days, written notice at any time after the research term, in which case all rights to the product candidate would revert to the Company. All royalty and other payment obligations of the parties survive any termination of the agreement.
During 1999, the Company received a one-time non-refundable license fee payment of $2,000,000 to enter into this agreement. The product candidate subject to the agreement has not completed the research and clinical development process. Accordingly, the Company has deferred recognition of the license fee and is amortizing it
Targacept, Inc.
Notes to Financial Statements—(continued)
15. Collaborative Research and License Agreements (continued)
over the expected term of the research and development period. The Company recognized $250,000, $250,000, $100,000 and $25,000 of the license fee payment during 2001, 2002, 2003 and the three months ended March 31, 2004, respectively.
On January 21, 2002, the Company entered into a second collaborative research and license agreement with Aventis to discover and develop drugs, derived from the Aventis library of compounds for the treatment of Alzheimer’s disease and other disorders of the central nervous system. The second agreement was structured similarly to the first agreement. The research terms of the agreement will extend for two years.
Dr. Falk Pharma
On January 26, 2001, the Company entered into a collaborative research development and license agreement with Dr. Falk Pharma GmbH (“Dr. Falk Pharma”), a German corporation, pursuant to which the parties agreed to collaborate to research, develop and commercialize nicotinic therapeutics for use in the prevention or treatment of ulcerative colitis and other gastrointestinal and liver diseases. Upon execution of the agreement, Dr. Falk Pharma paid the Company a $1,000,000 license fee and purchased 111,111 shares of the Company’s common stock for $1,000,000. The Company is continuing to advance the compound subject to the agreement through the research and clinical development process. Therefore, the Company has deferred recognition of the license fee payment and is amortizing it over the expected term of the research and development period. To account for the $1,000,000 in proceeds for the common stock, the Company used the estimated fair value of the common stock to value the shares issued to arrive at a total equity value of $75,556, with the remaining proceeds of $924,494 allocated to deferred revenue. This deferred revenue is also being amortized over the expected term of the research and development period. The Company recognized $353,000, $385,000, $170,000 and $41,000 of deferred revenue under this agreement during 2001, 2002, 2003 and the three months ended March 31, 2004, respectively. As of March 31, 2004, deferred revenue under this agreement was approximately $975,000 and was included in deferred license fee revenue in the accompanying balance sheet.
Dr. Falk Pharma agreed to make royalty payments based on net profits from products containing compounds developed under the agreement. For the years ended December 31, 2001, 2002, 2003 and the three months ended March 31, 2004, the Company did not pay or receive any royalties related to this agreement.
16. Acquisition of Inversine product
On August 5, 2002, the Company purchased from Layton Bioscience the Inversine product line, inventory and related patent rights for cash consideration of $3,500,000. The purchase was made in order to further the Company’s science and portfolio of compounds, and to further the Company’s development in certain neuropsychiatric indications.
This transaction was accounted for as an acquisition of assets. The aggregate purchase price was allocated to the assets acquired based on their fair values as follows:
| Amount | |||
Inventories | $ | 192,000 | |
Intangible assets acquired: | |||
Core technology | 296,000 | ||
Developed product technology | 346,000 | ||
In-process research and development | 2,666,000 | ||
Aggregate purchase price | $ | 3,500,000 | |
Targacept, Inc.
Notes to Financial Statements—(continued)
16. Acquisition of Inversine product (continued)
In determining the total consideration as well as the allocation of the purchase price including the amount of in-process research and development, the Company considered as part of its analysis an appraisal prepared by an independent appraiser that used established valuation techniques appropriate for the pharmaceutical industry. The amount allocated to in-process research and development was expensed upon acquisition. A one-time charge of $2,666,000 for purchased in-process research and development arising from the acquisition has been reflected in the Statement of Operations for the year ended December 31, 2002.
17. Subsequent Event
On , 2004 the Company’s Board of Directors adopted, and on , 2004 the stockholders approved, a to reverse stock split of the Company’s common stock effective as of . All common stock and per common share amounts for all periods presented in the accompanying financial statements have been restated to reflect the effect of this common stock split.
18. Selected Quarterly Financial Data (Unaudited)
| 2002 Quarter | ||||||||||||||||
| First | Second | Third | Fourth | |||||||||||||
Net revenue | $ | 489,891 | $ | 412,339 | $ | 471,219 | $ | 912,909 | ||||||||
Gross profit (loss) on product sales | — | — | — | (857 | ) | |||||||||||
Operating loss | (4,335,806 | ) | (3,537,219 | ) | (8,250,014 | ) | (4,879,471 | ) | ||||||||
Net loss | (4,421,557 | ) | (3,510,492 | ) | (8,250,530 | ) | (4,889,127 | ) | ||||||||
Net loss attributable to common stockholders | (5,373,554 | ) | (4,462,489 | ) | (9,202,527 | ) | (6,206,681 | ) | ||||||||
Basic and diluted net loss per share attributable to common stockholders(1) | $ | (10.83 | ) | $ | (8.38 | ) | $ | (15.81 | ) | $ | (10.05 | ) | ||||
Weighted average common shares outstanding—basic and diluted | 496,382 | 532,673 | 581,894 | 617,423 | ||||||||||||
| 2003 Quarter | ||||||||||||||||
| First | Second | Third | Fourth | |||||||||||||
Net revenue | $ | 690,796 | $ | 526,517 | $ | 598,840 | $ | 642,132 | ||||||||
Gross profit (loss) on product sales | 111,509 | (67,923 | ) | (957 | ) | 29,154 | ||||||||||
Operating loss | (4,275,065 | ) | (5,997,799 | ) | (4,525,572 | ) | (5,265,435 | ) | ||||||||
Net loss | (4,185,042 | ) | (5,775,002 | ) | (4,387,882 | ) | (5,047,395 | ) | ||||||||
Net loss attributable to common stockholders | (6,100,000 | ) | (7,916,891 | ) | (6,529,771 | ) | (7,189,287 | ) | ||||||||
Basic and diluted net loss per share attributable to common stockholders(1) | $ | (9.48 | ) | $ | (10.99 | ) | $ | (7.15 | ) | $ | (7.26 | ) | ||||
Weighted average common shares outstanding—basic and diluted | 643,571 | 720,093 | 913,185 | 989,874 | ||||||||||||
Diluted EPS is identical to Basic EPS since common stock equivalent shares are excluded from the calculation, as their effect is anti-dilutive.
Part II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
The following table sets forth the expenses in connection with the offering, all of which will be borne by us. All amounts shown are estimates except for the Securities and Exchange Commission registration fee, the NASDAQ National Market listing fee and the NASD filing fee.
Securities and Exchange Commission registration fee | $ | 10,928 | |
NASDAQ National Market listing fee | 100,000 | ||
NASD filing fee | 9,125 | ||
Blue sky fees and expenses | * | ||
Accounting fees and expenses | * | ||
Legal fees and expenses | * | ||
Transfer agent and registrar fees and expenses | * | ||
Printing and engraving expenses | * | ||
Miscellaneous | * | ||
Total | $ | * | |
Item 14. Indemnification of Directors and Officers.
Our Second Amended and Restated Certificate of Incorporation, as amended and in effect as of the date of this registration statement, and our Third Amended and Restated Certificate of Incorporation to be in effect upon completion of this offering (as may be in effect from time to time, the “Certificate”) provide that, except to the extent prohibited by the Delaware General Corporation Law, as amended (the “DGCL”), our directors shall not be personally liable to us or our stockholders for monetary damages for any breach of fiduciary duty while serving as a director, except for liability for breach of the director’s duty of loyalty to us or our directors. stockholders, for acts or omissions not in good faith or involving intentional misconduct or a knowing violation of law, for payment of dividends or approval of stock purchases or redemptions that are prohibited by the DGCL, or for any transaction from which the director derived an improper personal benefit.
Under the DGCL, our directors have a fiduciary duty to us that is not eliminated by this provision of the Certificaterestated certificate of incorporation and, in appropriate circumstances, equitable remedies such as injunctive or other forms ofnon-monetary relief will remain available. In addition, each director will continue to be subject to liability under the DGCL for breach of the director’s duty of loyalty to us or our stockholders, for acts or omissions that are found by a court of competent jurisdiction to be not in good faith or involving intentional misconduct, for knowing violations of law, for actions leading to improper personal benefit to the director and for payment of dividends or approval of stock repurchases or redemptions that are prohibited by the DGCL. This provision also does not affect our directors’ responsibilities under any other laws, such as federal securities laws or state or federal environmental laws.
Section 145 of the DGCL empowers a corporation to indemnify its directors and officers against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlements actually and reasonably incurred by them in connection with any action, suit or proceeding brought by third parties by reason of the fact that they were or are directors or officers of the corporation, if they acted in good faith, in a manner they reasonably believed to be in or not opposed to the best interests of the corporation and, with respect to any criminal action or proceeding, had no reasonable cause to believe that their conduct was unlawful. The DGCL provides further that the indemnification permitted thereunder shall not be deemed exclusive of any other rights to which the directors and officers may be entitled under the corporation’s bylaws, any agreement, a vote of stockholders or otherwise. Our restated certificate of incorporation provides that, to the fullest extent permitted by Section 145 of the DGCL, we shall indemnify any person who is or was a director or officer of us, or is or was serving at our request as a director, officer or trustee of another corporation, partnership, joint venture, trust, employee benefit plan or other enterprise, against the expenses, liabilities or other matters referred to in or covered by Section 145 of the DGCL. Our amended and restated bylaws provide that we will indemnify any person who was or is a party or threatened to be made a party to any proceeding by reason of the fact that such person is or was a director or officer of us or is or was serving at our request as a director, officer or trustee of another corporation, partnership, joint venture, trust, employee benefit plan or other enterprise to the fullest extent permitted by the DGCL.
In addition, we have entered into indemnification agreements with each of our directors and with certain of our executive officers. Pursuant to the indemnification agreements, we has agreed to indemnify and hold harmless these directors and officers to the fullest extent permitted by the DGCL. The agreements generally cover expenses that a director or officer incurs or amounts that a director or officer becomes obligated to pay because of any proceeding to which he or she is made or threatened to be made a party or participant by reason of his or her service as a current or former director, officer, employee or agent of the Company. The agreements also provide for the advancement of expenses to the directors and officers subject to specified conditions. There are certain exceptions to our obligation to indemnify the directors and officers, including any intentional malfeasance or act where the director or officer did not in good faith believe he or she was acting in our best interests, with respect to “short-swing” profit claims under Section 16(b) of the 1934 Act and, with certain exceptions, with respect to proceedings that he or she initiates.
Section 145 of the DGCL also empowers a corporation to purchase insurance for its officers and directors for such liabilities. We maintain liability insurance for our officers and directors.
We have entered into an underwriting agreement dated , 2017 with Ladenburg Thalmann & Co. Inc., as the representative of the underwriters (the “representative”) named below and the sole book-running manager of this offering. Subject to the terms and conditions of the underwriting agreement, the underwriters have agreed to purchase the number of our securities set forth opposite its name below.
Underwriters | Class A Units | Class B Units | ||||||
Ladenburg Thalmann & Co. Inc. | ||||||||
Total | ||||||||
A copy of the underwriting agreement will be filed as an exhibit to the registration statement of which this prospectus is part.
We have been advised by the underwriters that they propose to offer the units directly to the public at the public offering price set forth on the cover page of this prospectus. The underwriters may sell Class A Units or Class B Units separately to purchasers or may sell a combination of Class A Units and Class B Units to purchasers in any proportion. Any securities sold by the underwriters to securities dealers will be sold at the public offering price less a selling concession not in excess of $ per share. The underwriters may allow and these selected dealers mayre-allow a concession of not more than $ per share to other brokers and dealers.
The underwriting agreement provides that subject to the satisfaction or waiver by the representative of the conditions contained in the underwriting agreement, the underwriters are obligated to purchase and pay for all of the units offered by this prospectus.
No action has been taken by us or the underwriters that would permit a public offering of the units, or the shares of common stock, shares of preferred stock, shares of common stock underlying the preferred stock and warrants to purchase common stock included in the units, in any jurisdiction outside the United States where action for that purpose is required. None of our securities included in this offering may be offered or sold, directly or indirectly, nor may this prospectus or any other offering material or advertisements in connection with the offer and sales of any of the securities offered hereby be distributed or published in any jurisdiction except under circumstances that will result in compliance with the applicable rules and regulations of that jurisdiction. Persons who receive this prospectus are advised to inform themselves about and to observe any restrictions relating to this offering of securities and the distribution of this prospectus. This prospectus is neither an offer to sell nor a solicitation of any offer to buy the securities in any jurisdiction where that would not be permitted or legal.
The underwriters have advised us that they do not intend to confirm sales to any account over which they exercise discretionary authority.
Underwriting Discount and Expenses
The following table summarizes the underwriting discount and commission to be paid to the underwriters by us.
| Per Class A Unit(1) | Per Class B Unit(1) | Total | ||||||||||
Public offering price | ||||||||||||
Underwriting discount to be paid to the underwriters by us(2) | ||||||||||||
Proceeds to us (before expenses) | ||||||||||||
| (1) | The public offering price and underwriting discount corresponds to (x) in respect of the Class A Units (i) a public offering price per share of common stock of $ and (ii) a public offering price per warrant of $ and (y) in respect of the Class B Units (i) a public offering price per share of Series A Preferred Stock of $ and (ii) a public offering price per warrant of $ . |
| (2) | We have granted a 45 day option to the underwriter to purchase additional shares of common stock and/or warrants to purchase shares of common stock (up to 15% of the number of shares of common stock (including the number of shares of common stock issuable upon conversion of shares of Series A Preferred Stock) and the number of shares of common stock underlying the warrants sold in the primary offering) at the public offering price per share of common stock and the public offering price per warrant set forth above less the underwriting discounts and commissions, solely to cover over-allotments, if any. |
We estimate the total expenses payable by us for this offering to be approximately $1,530,000 which amount includes (i) the underwriting discount of $1,200,000 ($1,380,000 if the underwriters’ over-allotment option is exercised in full) and (ii) reimbursement of the accountable expenses of the representative equal to $120,000 including the legal fees of the representative being paid by us and (iii) other estimated company expenses of approximately $210,000 which includes legal, accounting, printing costs and various fees associated with the registration and listing of our shares.
The securities we are offering are being offered by the underwriters subject to certain conditions specified in the underwriting agreement.
Over-allotment Option
We have granted to the underwriters an option exercisable not later than 45 days after the date of this prospectus to purchase up to a number of additional shares of common stock and/or warrants to purchase shares of common stock not to exceed 15% of the number of shares of common stock sold in the primary offering (including the number of shares of common stock issuable upon conversion of shares of Series A Preferred Stock, but excluding shares of common stock underlying the warrants issued in this offering and any shares of common stock issued upon any exercise of the underwriter’s over-allotment option) and/or 15% of the warrants sold in the primary offering at the public offering price per share of common stock and the public offering price per warrant set forth on the cover page hereto less the underwriting discounts and commissions. The underwriters may exercise the option solely to cover overallotments, if any, made in connection with this offering. If any additional shares of common stock and/or warrants are purchased pursuant to the over-allotment option, the underwriters will offer these shares of common stock and/or warrants on the same terms as those on which the other securities are being offered.
Determination of Offering Price
Our common stock is currently traded on The Nasdaq Capital Market under the symbol “CBIO.” On March 29, 2017 the closing price of our common stock was $10.51 per share. We do not intend to apply for listing of the Series A Preferred Stock or the warrants on any securities exchange or other trading system.
The public offering price of the securities offered by this prospectus will be determined by negotiation between us and the underwriters. Among the factors that will be considered in determining the public offering price of the shares:
The offering price stated on the cover page of this prospectus should not be considered an indication of the actual value of the shares of common stock or shares of preferred stock sold in this offering. That price is subject to change as a result of market conditions and other factors and we cannot assure you that the shares of common stock sold in this offering can be resold at or above the public offering price.
Lock-up Agreements
Our officers and directors and certain affiliated funds, representing 29.4% of our outstanding shares, are expected to agree with the representative to be subject to alock-up period of 90 days following the date of this prospectus. This means that, during the applicablelock-up period, such persons may not offer for sale, contract to sell, sell, distribute, grant any option, right or warrant to purchase, pledge, hypothecate or otherwise dispose of, directly or indirectly, any shares of our common stock or any securities convertible into, or exercisable or exchangeable for, shares of our common stock. Certain limited transfers are permitted during thelock-up period if the transferee agrees to theselock-up restrictions. We have also agreed, in the underwriting agreement, to similarlock-up restrictions on the issuance and sale of our securities for 90 days following the effectiveness of the underwriting agreement, although we will be permitted to issue stock options or stock awards to directors, officers and employees under our existing plans. Thelock-up period is subject to an additional extension to accommodate for our reports of financial results or material news releases. The representative may, in its sole discretion and without notice, waive the terms of any of theselock-up agreements.
Other Relationships
Upon completion of this offering, we have granted the representative a right of first refusal to act as lead orco-lead bookrunner or lead orco-lead placement agent in connection with any subsequent public or private offering of equity securities or other capital markets financing by us. This right of first refusal extends for 12 months from the closing date of this offering. The terms of any such engagement of the representative will be determined by separate agreement.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is American Stock Transfer & Trust Company, LLC.
Stabilization, Short Positions and Penalty Bids
The underwriters may engage in syndicate covering transactions stabilizing transactions and penalty bids or purchases for the purpose of pegging, fixing or maintaining the price of our common stock:
These syndicate covering transactions, stabilizing transactions, and penalty bids may have the effect of raising or maintaining the market prices of our securities or preventing or retarding a decline in the market prices of our securities. As a result the price of our common stock may be higher than the price that might otherwise exist in the open market. Neither we nor the underwriters make any representation or prediction as to the effect that the transactions described above may have on the price of our common stock. These transactions may be effected on The Nasdaq Capital Market, in theover-the-counter market or on any other trading market and, if commenced, may be discontinued at any time.
In connection with this offering, the underwriters also may engage in passive market making transactions in our common stock in accordance with Regulation M during a period before the commencement of offers or sales of shares of our common stock in this offering and extending through the completion of the distribution. In general, a passive market maker must display its bid at a price not in excess of the highest independent bid for that security. However, if all independent bids are lowered below the passive market maker’s bid that bid must then be lowered when specific purchase limits are exceeded. Passive market making may stabilize the market price of the securities at a level above that which might otherwise prevail in the open market and, if commenced, may be discontinued at any time.
Neither we, nor the underwriters make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the prices of our securities. In addition, neither we nor the underwriters make any representation that the underwriters will engage in these transactions or that any transactions, once commenced will not be discontinued without notice.
Indemnification
We have agreed to indemnify the underwriters against certain liabilities, including certain liabilities arising under the Securities Act or to contribute to payments that the underwriters may be required to make for these liabilities.
Certain legal matters relating to the issuance of the securities offered by this prospectus will be passed upon for us by Morrison & Foerster LLP, Palo Alto, California. Certain legal matters in connection with this offering will be passed upon for the underwriter by Ellenoff Grossman & Schole LLP.
The consolidated balance sheets of Catalyst Biosciences, lnc. as of December 31, 2016 and 2015, and the related consolidated statements of operations, comprehensive loss, convertible preferred stock and stockholders’ equity (deficit) and cash flows for each of the years in the two-year period ended December 31, 2016, have been audited by EisnerAmper LLP, independent registered public accounting firm, as stated in their report which is incorporated herein by reference. Such financial statements have been incorporated herein by reference in reliance on the report of such firm given upon their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed a registration statement on FormS-1 with the SEC covering the units we are offering by this prospectus. This prospectus does not include all of the information contained in the registration statement. You should refer to the registration statement and its exhibits for additional information. Whenever we make reference in this prospectus to any of our contracts, agreements or other documents, the references are not necessarily complete and you should refer to the exhibits filed as part of the registration statement for copies of the actual contract, agreement or other document.
We file annual, quarterly and other periodic reports, proxy statements and other information with the Securities and Exchange Commission. You can read our Securities and Exchange Commission filings, including this registration statement, over the Internet at the Securities and Exchange Commission’s website at www.sec.gov. You may also read and copy any document we file with the Securities and Exchange Commission at its public reference facilities at 100 F Street NE, Washington, D.C. 20549. You may also obtain copies of these documents at prescribed rates by writing to the Public Reference Section of the Securities and Exchange Commission at 100 F Street NE, Washington, D.C. 20549. Please call the Securities and Exchange Commission at1-800-SEC-0330 for further information on the operation of the public reference facilities.
Our Internet address iswww.catalystbiosciences.com. There we make available free of charge, on or through the investor relations section of our website, annual reports on Form10-K, quarterly reports on Form10-Q, current reports on Form8-K and amendments to those reports filed pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after we electronically file such material with the Securities and Exchange Commission. The information found on our website is not part of this prospectus and investors should not rely on any such information in deciding whether to invest.
INCORPORATION OF CERTAIN INFORMATION BY REFERENCE
We have elected to incorporate the following documents into this prospectus, together with all exhibits filed therewith or incorporated therein by reference, to the extent not otherwise amended or superseded by the contents of this prospectus:
In addition, we incorporate by reference in this prospectus any future filings we make with the SEC under Sections 13(a), 13(c), 14, or 15(d) of the Exchange Act (excluding any information furnished and not filed with the SEC) after the date on which the registration statement that includes this prospectus was initially filed with the SEC (including all such documents we may file with the SEC after the date of the initial registration statement and prior to the effectiveness of the registration statement) and until all offerings under this prospectus are terminated.
Any statement contained in a document incorporated by reference herein shall be deemed to be modified or superseded for all purposes to the extent that a statement contained in this prospectus or in any other subsequently filed document which is also incorporated or deemed to be incorporated by reference, modifies or supersedes such statement. Any statement so modified or superseded shall not be deemed, except as so modified or superseded, to constitute a part of this prospectus. You may request a copy of these filings (other than an exhibit to a filing unless that exhibit is specifically incorporated by reference into that filing) at no cost by writing, telephoning ore-mailing us at the following address, telephone number ore-mail address:
Catalyst Biosciences, Inc.
260 Littlefield Avenue
South San Francisco, CA 94080
Tel: (650)871-0761
Attn: Fletcher Payne
fpayne@catbio.com
Copies of these filings are also available through the “Investors” section of our website at www.catalystbiosciences.com. For other ways to obtain a copy of these filings, please refer to “Prospectus Summary—Available Information.”

285,714 Class A Units consisting of common stock and warrants and
12,000 Class B Units consisting of shares of Series A Preferred Stock and warrants
(and 1,857,143 shares of common stock underlying shares of
Series A Preferred Stock and warrants)
PROSPECTUS
Ladenburg Thalmann
, 2017
PART II
Information Not Required in Prospectus
ITEM 13. OTHER EXPENSES OF ISSUANCE AND DISTRIBUTION
The following table sets forth the fees and expenses, other than placement agent fees and expenses, payable in connection with the registration of the common stock hereunder. All amounts are estimates except the SEC registration fee and the FINRA filing fee.
Item | Amount to be paid | |||
SEC registration fee | $ | 3,149 | ||
FINRA filing fee | 4,575 | |||
Printing expenses | 40,000 | |||
Legal fees and expenses | 125,000 | |||
Accounting fees and expenses | 20,000 | |||
Transfer Agent fees and expenses | 15,000 | |||
Miscellaneous expenses | 2,276 | |||
|
| |||
Total | $ | 210,000 | ||
ITEM 14. INDEMNIFICATION OF DIRECTORS AND OFFICERS
Our Fourth Amended and Restated Certificate of Incorporation, as amended and as may be further amended and in effect from time to time, which we refer to as the restated certificate of incorporation, provides that our directors shall not be personally liable to us or our stockholders for monetary damages for any breach of fiduciary duty as a director, except for liability for breach of the director’s duty of loyalty to us or our stockholders, for acts or omissions not in good faith or involving intentional misconduct or a knowing violation of law, for payment of dividends or approval of stock purchases or redemptions that are prohibited by the General Corporation Law of the State of Delaware, as amended, which we refer to as the DGCL, or for any transaction from which the director derived an improper personal benefit. Under the DGCL, our directors have a fiduciary duty to us that is not eliminated by this provision of the restated certificate of incorporation and, in appropriate circumstances, equitable remedies such as injunctive or other forms ofnon-monetary relief will remain available. This provision also does not affect our directors’ responsibilities under any other laws, such as federal securities laws or state or federal environmental laws.
Section 145 of the DGCL empowers a corporation to indemnify its directors and officers against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlements actually and reasonably incurred by them in connection with any action, suit or proceeding brought by third parties by reason of the fact that they were or are directors or officers of the corporation, if they acted in good faith, in a manner they reasonably believed to be in or not opposed to the best interests of the corporation and, with respect to any criminal action or proceeding, had no reasonable cause to believe that their conduct was unlawful. The DGCL provides further that the indemnification permitted thereunder shall not be deemed exclusive of any other rights to which the directors and officers may be entitled under the corporation’s bylaws, any agreement, a vote of stockholders or otherwise. The Certificaterestated certificate of incorporation provides that, to the fullest extent permitted by Section 145 of the DGCL, we shall indemnify any person who is or was a director or officer of us, or is or was serving at our request as a director, officer or trustee of another corporation, partnership, joint venture, trust, employee benefit plan or other enterprise, against the expenses, liabilities or other matters referred to in or covered by Section 145 of the DGCL. Our amended and restated bylaws provide that we will indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding (whether civil, criminal, administrative or investigative) by reason of the fact that such person is or was a director or officer of us or is or was serving at our request as a director, officer or trustee of another corporation, partnership, joint venture, trust, employee benefit plan or other enterprise against expensesto the fullest extent permitted by the DGCL. In
II-1
(addition, we have entered into agreements with each of our directors and officers under which, among other things, we have agreed to indemnify the director or officer against expenses incurred in any proceeding, including attorneys’ fees), judgments, fines and amounts paidany action by us, in settlement actually and reasonably incurredwhich the director or officer was, is or is threatened to be made a party or a participant by such person in connection with such action, suitreason of his or proceeding.her status as a present or former director, officer, employee or agent of us or, at our request, any other corporation, partnership, joint venture, trust, employee benefit plan or other enterprise. At present, there is no pending litigation or proceeding involving any director or officer as to which indemnification will be required or permitted, under the Certificate and we are not aware of any threatened litigation or proceeding that may result in a claim for such indemnification.
Section 145 of the DGCL also empowers a corporation to purchase insurance for its officers and directors for such liabilities. We maintain liability insurance for our officers and directors.
ITEM 15. RECENT SALES OF UNREGISTERED SECURITIES
Item 15. Recent Sales of Unregistered Securities.
Set forth below is information regarding shares of common stock and preferred stock issued, and options and warrants granted, by us since August 22, 2000. Also included is the consideration, if any, received by us for such shares, options and warrants and information relating to the section of the Securities Act, or rule of the Securities and Exchange Commission, under which exemption from registration was claimed.
(a) Issuance of Securities
1. On August 22, 2000, at the time that we became an independent company, we issued an aggregate of 309,424 shares of our common stock at a purchase price per share of $0.001 to each of Dr. deBethizy, Dr. Bencherif, Dr. Caldwell and Dr. Patrick M. Lippiello for an aggregate purchase price of approximately $309.
2. On August 22, 2000, we recapitalized our 500 outstanding shares of common stock held by R.J. Reynolds Tobacco Company, our then parent corporation, into 5,000,000 shares of series A convertible preferred stock and a warrant to purchase 1,612,903 shares of common stock at an exercise price of $1.95 per share. All of the shares of series A convertible preferred stock and the warrant were issued to R.J. Reynolds Tobacco Company, which subsequently assigned them to R.J. Reynolds Tobacco Holdings, Inc. Each share of series A convertible preferred stock will convert into one share of common stock concurrently with the completion of this offering.
3. On August 22, 2000, we issued and sold an aggregate of 5,892,473 shares of our series B convertible preferred stock at a purchase price per share of $4.65 to investors affiliated with EuclidSR Partners, L.P., Burrill & Company LLC, Societe Generale Asset Management Finance (which subsequently assigned its shares to FCPR SGAM Biotechnology Fund), Genavent Venture Fund, Auriga Ventures, Advent Private Equity Fund II, FCPR CDC-Innovation 2000 and Longleaf Venture Fund, LLC (now known as Academy Venture Fund, LLC) for an aggregate purchase price of approximately $27.4 million. These shares will convert into common stock at the rate of approximately 2.38 shares of common stock for each share of series B convertible preferred stock concurrently with the completion of this offering.
4. On November 30, December 5 and December 19, 2000, we issued and sold an aggregate of 645,161 shares of our series B convertible preferred stock at a purchase price per share of $4.65 to investors affiliated with EuclidSR Partners, L.P. and Longleaf Venture Fund, LLC (now known as Academy Venture Fund, LLC) for an aggregate purchase price of approximately $3.0 million. These shares will convert into common stock at the rate of approximately 2.38 shares of common stock for each share of series B convertible preferred stock concurrently with the completion of this offering.
5. On January 26, 2001, we issued and sold an aggregate of 29,933 shares of our series B convertible preferred stock, valued at $4.65 per share, to Andre L. Lamotte, Joseph F. Lovett and Jeffrey D. Wager in partial satisfaction of amounts payable by us for consulting services rendered. The aggregate amount of consideration was approximately $139,000. These shares will convert into common stock at the rate of one share of common stock for each share of series B convertible preferred stock concurrently with the completion of this offering.
II-2
6. On January 30, 2001, we issued and sold an aggregate of 111,111 shares of our common stock at a purchase price of $9.00 per share to Dr. Falk Pharma GmbH, our collaborative partner, for an aggregate purchase price of approximately $1.0 million.
7. On August 8, 2002 and June 11, 2003, we issued and sold an aggregate of 60,000 shares of restricted stock to Mr. Skaletsky for an aggregate purchase price of $600.
8. On November 26, 2002, we issued and sold an aggregate of 37,764,180 shares of our series C convertible preferred stock at a purchase price per share of $1.21 to investors affiliated with Nomura International plc, New Enterprise Associates 10, Limited Partnership, CDIB Bioscience Ventures I, Inc., Easton Hunt Capital Partners, L.P., EuclidSR Partners, L.P., Burrill & Company LLC, Genavent Venture Fund, FCPR SGAM Biotechnology Fund, Auriga Ventures, FCPR CDC-Innovation 2000, Advent Private Equity Fund II and Academy Venture Fund, LLC for an aggregate purchase price of approximately $45.7 million. These shares will convert into common stock at the rate of approximately 1.08 shares of common stock for each share of series C convertible preferred stock concurrently with the completion of this offering.
9. On March 14, 2003, we issued and sold an aggregate of 11,404,958 shares of our series C convertible preferred stock at a purchase price per share of $1.21 to investors affiliated with JAFCO G-9(A) Venture Capital, Rock Castle Ventures, Cogene Biotech Ventures Five, Bison Capital, LLC and Oxford Bioscience Partners IV L.P. for an aggregate purchase price of approximately $13.8 million. These shares will convert into common stock at the rate of approximately 1.08 shares of common stock for each share of series C convertible preferred stock concurrently with the completion of this offering.
10. On April 18, 2003,2016, we issued and sold an aggregate of 25,000 shares of restricted stock to Mr. Richard for an aggregate purchase price of $250.
11. Since inception to April 30, 2004, we have granted:
The weighted average exercise price of all options to purchase shares of common stock granted since inception to April 30, 2004 is $0.64 per share. As of April 30, 2004, there were 53 holders of record of shares of our common stock.
(b) No underwriters were involved in the foregoing sales of securities. The securities described in paragraphs (a)(1)—(10) of this Item 15 were issued to a combination of foreign and U.S. investors in reliance upon the exemption from the registration requirements of the Securities Act, as set forth in Sections 3(a)(9) or 4(2) under the Securities Act and Rule 506 of Regulation D promulgated thereunder relative to sales by an issuer not involving any public offering, to the extent an exemption from such registration was required. All purchasers of shares of our convertible preferred stock described in paragraph (a)(1)—(10) of this Item 15 represented to us in connection with their purchase that they were accredited investors and were acquiring the shares for investment and not distribution, that they could bear the risks of the investment and could hold the securities for an indefinite period of time. The purchasers received written disclosures that the securities had not been registered under the Securities Act and that any resale must be madenew hire inducement grant pursuant to a registration or an available exemption from such registration.
II-3
All of the foregoing securities are deemed restricted securities for purposes of the Securities Act. All certificates representing the issued shares of common stock described in this Item 15 included appropriate legends setting forth that the securities had not been registeredNasdaq Listing Rule 5635(c)(4) and the applicable restrictions on transfer.
The issuance of stock options and the common stock issuable upon the exercise of such options as described in paragraph (a)(11) of this Item 15 were issued pursuant to written compensatory plans or arrangements with our employees, directors and consultants, in reliance on the exemption provided by Rule 701 promulgated under the Securities Act. All recipients either received adequate information about us or had access, through employment or other relationships, to such information.
Item 16. Exhibits and Financial Statement Schedules.
(a) Exhibits.
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
II-4
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
II-5
(b) Financial Statement Schedules.
All informationDr. Levy’s option grant is for which provision is made in the applicable accounting regulationspurchase of an aggregate of 6,666 shares of our common stock at $22.80 per share, the closing trading price on the date of the Securities and Exchange Commission is either included ingrant, as adjusted to reflect the financial statements or is not required under2017 Reverse Stock Split. The grant was approved by the related instructions or is inapplicable, and therefore has been omitted.
Item 17. Undertakings.
compensation committee of our board of directors. The undersigned registrant hereby undertakes to providestock options were granted pursuant to the underwriters atCompany’s 2016 Inducement Stock Incentive Plan, which was approved by our board of directors on April 15, 2016.
ITEM 16. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
See the closing specified inExhibit Index included following the underwriting agreement, certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.signature page of this registration statement.
ITEM 17. UNDERTAKINGS
Insofar as indemnification for liabilities arising under the Securities Act of 1933, as amended, or the Act, may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, described herein, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
The undersigned registrant hereby undertakes that:undertakes:
| (1) | To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement: |
(1) For purposes of determining(i) To include any liability underprospectus required by section 10(a)(3) of the Securities Act of 1933,1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information omittedset forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed as a part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed bywith the registrantCommission pursuant to Rule 424(b)(1) or (4) or 497(h) under if, in the Securities Act shall be deemedaggregate, the changes in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement.
II-2
(iii) To include any material information with respect to be partthe plan of thisdistribution not previously disclosed in the registration statement as ofor any material change to such information in the time it was declared effective.registration statement;
(2) For
| (2) | That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment |
| (3) | To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering. |
| (4) | That, for the purpose of determining liability of the registrant under the Securities Act of 1933 to any purchaser in the initial distribution of the securities: The undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser: |
(i) Any preliminary prospectus or prospectus of the undersigned registrant relating to the securities offered therein, andoffering required to be filed pursuant to Rule 424;
(ii) Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
(iii) The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and
(iv) Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
| (5) | For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective. |
| (6) | For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
II-6
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrantRegistrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized in the City of Winston-Salem,South San Francisco, State of North Carolina,California, on May 14, 2004.March 30, 2017.
By: |
| |
| Nassim Usman, Ph.D. | ||
| President and Chief Executive Officer | |
POWER OF ATTORNEY
Each person whose signature appears below hereby constitutes and appoints J. Donald deBethizy and Alan A. Musso, or either of them, as his or her attorney-in-fact, each with full power of substitution, for him or her in any and all capacities, to sign any and all amendments to this registration statement on Form S-1, including post-effective amendments and any and all new registration statements filed pursuant to Rule 462 under the Securities Act, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each said attorney-in-fact or his or her substitute or substitutes may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities indicated on May 14, 2004.and as of the dates indicated.
Signature | Title | Date | ||||||
/s/ Nassim Usman, Ph.D. Nassim Usman, Ph.D. |
and Director (Principal Executive Officer) | March 31, 2017 | ||||||
/s/ Fletcher Payne Fletcher Payne |
| |||||||
|
(Principal Financial Officer and Principal Accounting Officer) | March 31, 2017 | ||||||
* Harold E. Selick, Ph.D. |
Director |
| ||||||
* Errol B. De Souza, Ph.D. |
Director |
| ||||||
* Jeff Himawan, Ph.D. |
Director |
| ||||||
* Augustine Lawlor | Director | March 31, 2017 | ||||||
* John P. Richard | Director | March 31, 2017 | ||||||
* Stephen M. Hill, M.D. | Director | March 31, 2017 | ||||||
| * By: | /S/ NASSIM USMAN, PH.D. | |||||
|
| |||||
II-7
EXHIBIT INDEX
| Exhibit Number | Description | |
| 1.1* | Form of Underwriting | |
| |
| |
| 2.1(b) | Amendment No. 1 to Agreement and Plan of Merger by and among Targacept, Talos Merger Sub, Inc., and Catalyst dated May 6, 2015 (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form8-K, as filed with the SEC on May 12, 2015) | |
| 2.1(c) | Amendment No. 2 to Agreement and Plan of Merger by and among Targacept, Talos Merger Sub, Inc., and Catalyst dated May 13, 2015 (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form8-K, as filed with the SEC on May 14, 2015) | |
| 3.1 | Fourth Amended and Restated Certificate of Incorporation of the Company (incorporated by reference to Exhibit 4.1 to the Company’s FormS-8 (Reg.No. 333-133881),as | |
| | Certificate of Amendment to | |
| ||
| | Second Certificate of Amendment to the Fourth Amended and Restated | |
| |
| |
| |
| |
| |
| |
| 4.2 | Form of Global Security (incorporated by reference to Annex G, Exhibit A to the | |
| 4.3 | Form of Warrant to Purchase | |
| 4.4 | Form of Warrant to Purchase Stock of Catalyst Biosciences, Inc., issued to purchasers of convertible promissory notes (incorporated by reference to Exhibit 4.5 to the Company’s Form10-K, filed with the SEC on March 9, 2016) | |
| 4.5* | Form of Warrant to be Issued in Offering | |
| 4.6* | Form of Series A Preferred Stock Certificate of the Company | |
| 5.1* | Opinion of | |
| ||
| 10.1** | Catalyst Biosciences, Inc. (formerly Targacept, Inc.) 2015 Stock Incentive Plan (As Amended and Restated Effective June 9, 2016) (incorporated by reference to Appendix A to the Company’s Definitive Proxy Statement (FileNo. 000-51173), filed with the SEC on April 25, 2016 | |
| 10.2** | Catalyst Biosciences, Inc. 2016 Inducement Stock Incentive Plan (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form8-K, as filed with the SEC on April 20, 2016) | |
| Exhibit Number | Description | |
| 10.3** | Offer Letter, executed February 21, 2006, by and between Catalyst and Dr. Nassim Usman (incorporated by reference to Exhibit 10.35 to the Company’s FormS-4 (Reg.No. 333-204423), filed with the SEC on May 22, 2015) | |
| 10.4** | Offer Letter, dated March 30, 2015, by and between Catalyst and Fletcher Payne (incorporated by reference to Exhibit 10.39 to the Company’s FormS-4 (Reg.No. 333-204423), filed with the SEC on May 22, 2015) | |
| 10.5** | Offer Letter, executed April 27, 2012, by and between Catalyst and Dr. Harold E. Selick (incorporated by reference to Exhibit 10.34 to the Company’s FormS-4 (Reg.No. 333-204423), filed with the SEC on May 22, 2015) | |
| 10.6** | Nonqualified Stock Option Agreement, dated December 3, 2012, by and between the Company and Stephen A. Hill (incorporated by reference to Exhibit 99.1 to the Company’s Registration Statement on FormS-8, as filed with the SEC on January 4, 2013 (RegistrationNo. 333-185888)) | |
| 10.7 | Sublease Agreement, dated February 23, 2015, by and between Catalyst Biosciences, Inc. and Reset Therapeutics, Inc. (incorporated by reference to Exhibit 10.29 to the Company’s FormS-4 (Reg.No. 333-204423), filed with the SEC on May 22, 2015) | |
| 10.8(a)+ | License and Collaboration Agreement, dated September 16, 2013, by and between Catalyst and ISU Abxis (incorporated by reference to Exhibit 10.30(a) to the Company’s FormS-4 (Reg.No. 333-204423), filed with the SEC on May 22, 2015) | |
| 10.8(b)+ | Amendment No. 1 to License and Collaboration Agreement, dated October 31, 2014, by and between Catalyst and ISU Abxis (incorporated by reference to Exhibit 10.30(b) to the Company’s FormS-4 (Reg.No. 333-204423), filed with the SEC on May 22, 2015) | |
| 10.9** | Catalyst’s 2004 Stock Plan (incorporated by reference to Exhibit 10.31(a) to the Company’sForm S-4 (Reg.No. 333-204423), filed with the SEC on May 22, 2015) | |
| 10.10** | Consulting Agreement, dated January 14, 2015, by and between the Catalyst and Fletcher Payne (incorporated by reference to Exhibit 10.38 to the Company’s FormS-4 (Reg.No. 333-204423), filed with the SEC on May 22, 2015) | |
| 10.11(a)** | Stock Option Agreement—Early Exercise, No. 427, dated January 22, 2015, by and between Catalyst and Fletcher Payne (incorporated by reference to Exhibit 10.40(a) to the Company’s FormS-4 (Reg.No. 333-204423), filed with the SEC on May 22, 2015) | |
| 10.11(b)** | Stock Option Agreement—Early Exercise, No. 428, dated January 22, 2015, by and between Catalyst and Fletcher Payne (incorporated by reference to Exhibit 10.40(b) to the Company’s FormS-4 (Reg.No. 333-204423), filed with the SEC on May 22, 2015) | |
| 10.11(c)** | Stock Option Agreement—Early Exercise, No. 429, dated May 8, 2015, by and between Catalyst and Fletcher Payne (incorporated by reference to Exhibit 10.40(c) to the Company’s FormS-4 (Reg.No. 333-204423), filed with the SEC on May 22, 2015) | |
| 10.12+ | Development and Manufacturing Services Agreement, by and between CMC ICOS Biologics, Inc. and the Company, dated as of May 20, 2016 (incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form10-Q, filed with the SEC on August 4, 2016) | |
| 10.13** | Separation Agreement, dated September 14, 2016, between the Company and Edwin Madison (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form8-K, as filed with the SEC on September 16, 2016) | |
| 10.14** | Form of Indemnification Agreement between the Company and each of its directors and | |
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| ||
| Exhibit Number | Description | |
| ||
| ||
| ||
| 10.16+ |
| |
| ||
| ||
| ||
| ||
| ||
| ||
| ||
Consent of | ||
| ||
| ||
| 24.1 | Power of Attorney (included | |
| * | To be filed by amendment. |
| ** | Indicates a management contract or compensatory plan or arrangement. |
| *** | Filed herewith. |
| + |