may be liable for damages.
Our research and development activities involve the controlled use of potentially hazardous substances, including chemical and biological materials by our third-party manufacturers. Our manufacturers are subject to federal, state and local laws and regulations in the United States governing the use, manufacture, storage, handling and disposal of medical, radioactive and hazardous materials. Although we believe that our manufacturers’ procedures for using, handling, storing and disposing of these materials comply with legally prescribed standards, we cannot completely eliminate the risk of contamination or injury resulting from medical, radioactive or hazardous materials. As a result of any such contamination or injury we may incur liability or local, city, state or federal authorities may curtail the use of these materials and interrupt our business operations. In the event of an accident, we could be held liable for damages or penalized with fines, and the liability could
exceed our resources. We do not have any insurance for liabilities arising from medical radioactive or hazardous materials. Compliance with applicable environmental laws and regulations is expensive, and current or future environmental regulations may impair our research, development and production efforts, which could harm our business, prospects, financial condition or results of operations.
Risks Related to Our Financial Position and Capital Requirements
We have a limited operating history, have incurred significant operating losses since our inception and anticipate that we will continue to incur losses for the foreseeable future.
Our operations began in 2005 and we have only a limited operating history upon which you can evaluate our business and prospects. Our operations to date have been limited to conducting product development activities for emricasan and performing research and development with respect to our clinical and preclinical programs. In addition, as an early stage company, we have limited experience and have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the pharmaceutical area. Nor have we demonstrated an ability to obtain regulatory approval for or to commercialize a drug candidate. Consequently, any predictions about our future performance may not be as accurate as they would be if we had a history of successfully developing and commercializing pharmaceutical products.
To date, we have financed our operations primarily through private placements of convertible debt and preferred stock, and we have incurred significant operating losses since our inception, including consolidated net losses of $12.0 million and $8.7 million for the years ended December 31, 2011 and 2012, respectively, and $2.3 million for the three months ended March 31, 2013. As of March 31, 2013, we had an accumulated deficit of $61.1 million. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our stockholders’ equity and working capital. Our losses have resulted principally from costs incurred in our research and development activities. We anticipate that our operating losses will substantially increase over the next several years as we execute our plan to expand our research, development and commercialization activities, including the clinical development and planned commercialization of our drug candidate, emricasan, and incur the additional costs of operating as a public company. In addition, if we obtain regulatory approval of emricasan, we may incur significant sales and marketing expenses. Because of the numerous risks and uncertainties associated with developing pharmaceutical products, we are unable to predict the extent of any future losses or whether or when we will become profitable, if ever.
Our independent registered public accounting firm has included an explanatory paragraph relating to our ability to continue as a going concern in its report on our audited financial statements included in this prospectus.
Our report from our independent registered public accounting firm for the year ended December 31, 2012 includes an explanatory paragraph stating that our recurring losses from operations and negative cash flows raise substantial doubt about our ability to continue as a going concern. If we are unable to obtain sufficient funding, our business, prospects, financial condition and results of operations will be materially and adversely affected and we may be unable to continue as a going concern. If we are unable to continue as a going concern, we may have to liquidate our assets and may receive less than the value at which those assets are carried on our audited consolidated financial statements, and it is likely that investors will lose all or a part of their investment. After this offering, future reports from our independent registered public accounting firm may also contain statements expressing doubt about our ability to continue as a going concern. If we seek additional financing to fund our business activities in the future and there remains doubt about our ability to continue as a going concern, investors or other financing sources may be unwilling to provide additional funding on commercially reasonable terms or at all.
We have not generated any revenues to date from product sales. We may never achieve or sustain profitability, which could depressnegatively affect the market price of our common stock, and could cause you to lose all orregardless of our actual operating performance. Further, a part of your investment.
Our ability to become profitable depends on our ability to develop and commercialize emricasan. To date, we have no products approved for commercial sale and have not generated any revenues from sales of any drug
candidate, and we do not know when, or if, we will generate revenuesdecline in the future. We do not anticipate generating revenues, if any, from sales of emricasan for at least the next several yearsfinancial markets and we will never generate revenues from emricasan if we do not obtain regulatory approval for emricasan. Our ability to generate future revenues depends heavily onrelated factors beyond our success in:
developing and securing U.S. and/or foreign regulatory approvals for emricasan;
manufacturing commercial quantities of emricasan at acceptable cost;
achieving broad market acceptance of emricasan in the medical community and with third-party payors and patients;
commercializing emricasan, assuming we receive regulatory approval; and
pursuing clinical development of emricasan in additional indications.
Even if we do generate product sales, we may never achieve or sustain profitability. Our failure to become and remain profitable would depress the market price of our common stock and could impair our ability to raise capital, expand our business, diversify our product offerings or continue our operations.
If we fail to obtain additional financing, we may be unable to complete the development and commercialization of emricasan.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to spend substantial amounts to continue the clinical development of emricasan, including our planned Phase 2 and Phase 3 clinical trials. If approved, we will require significant additional amounts in order to launch and commercialize emricasan, including building our own commercial capabilities to sell, market, and distribute emricasan in the United States and the EU.
We estimate that our net proceeds from this offering will be approximately $ million, based upon an assumed initial public offering price of $ per share (the midpoint of the price range set forth on the cover page of this prospectus), after deducting the estimated underwriting discounts and commissions and offering expenses payable by us. We believe that such proceeds together with our existing cash and cash equivalents will be sufficient to fund our operations for at least the next 18 months. In particular, we expect that the net proceeds from this offering will allow us to complete our planned Phase 2b ACLF trial, Phase 2b/3 HCV-POLT trial and Phase 2b CLF trial. However, changing circumstancescontrol may cause usour stock price to consume capital significantly faster than we currently anticipate,decline rapidly and we may need to spend more money than currently expected because of circumstances beyond our control. We will require additional capital for the further development and commercialization of emricasan and may need to raise additional funds sooner if we choose to expand more rapidly than we presently anticipate.
We cannot be certain that additional funding will be available on acceptable terms, or at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back or discontinue the development or commercialization of emricasan or other research and development initiatives. We also could be required to seek collaborators for emricasan at an earlier stage than otherwise would be desirable or on terms that are less favorable than might otherwise be available or relinquish or license on unfavorable terms our rights to emricasan in markets where we otherwise would seek to pursue development or commercialization ourselves.
Any of the above events could significantly harm our business, prospects, financial condition and results of operations and cause the price of our common stock to decline.unexpectedly.
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies or drugproduct candidate.
We may seek additional capital through a combination of public and private equity offerings, debt financings, strategic partnerships and alliances and licensing arrangements. ToWe have a purchase agreement in place with Lincoln Park to sell up to $10.0 million worth of shares of our common stock, from time to time, to Lincoln Park, under which $8.5 million remains available for future sale as of September 30, 2020. Any sales under the Lincoln Park arrangement, and to the extent that we raise additional capital through the sale of equity or convertible debt securities, yourthe ownership interestinterests of our stockholders will be diluted, and the terms may include liquidation or other preferences that adversely affect yourthe rights as a stockholder.of our stockholders. The incurrence of
indebtedness would result in increased fixed payment obligations and could involve certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or drugproduct candidate, or grant licenses on terms unfavorable to us.
Our ability to utilize our net operating loss carryforwards and certain other tax attributes may be limited.
Under Section 382 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an “ownership change” (generally defined as a greater than 50% change (by value) in its equity ownership over a three year period), the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes to offset its post-change income may be limited. As a result of our most recent private placements and other transactions that have occurred over the past three years, we may have experienced, and, upon completion of this offering, may experience, an “ownership change.” We may also experience ownership changes in the future as a result of subsequent shifts in our stock ownership. As of December 31, 2012, we had federal and state net operating loss carryforwards of approximately $52.3 million and $51.2 million, respectively, and federal and state research and development credits of $1.3 million and $745,000, respectively, which could be limited if we experience an “ownership change.”
Unstable market and economic conditions may have serious adverse consequences on our business, financial condition and stock price.
As widely reported, global credit and financial markets have experienced extreme disruptions in the past several years, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in unemployment rates, and uncertainty about economic stability. There can be no assurance that further deterioration in credit and financial markets and confidence in economic conditions will not occur. Our general business strategy may be adversely affected by any such economic downturn, volatile business environment or continued unpredictable and unstable market conditions. If the current equity and credit markets deteriorate, or do not improve, it may make any necessary debt or equity financing more difficult, more costly, and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance and stock price and could require us to delay or abandon clinical development plans. In addition, there is a risk that one or more of our current service providers, manufacturers and other partners may not survive these difficult economic times, which could directly affect our ability to attain our operating goals on schedule and on budget.
At March 31, 2013, we had approximately $5.1 million of cash, cash equivalents and short-term investments. While we are not aware of any downgrades, material losses, or other significant deterioration in the fair value of our cash equivalents since March 31, 2013, no assurance can be given that further deterioration of the global credit and financial markets would not negatively impact our current portfolio of cash equivalents or marketable securities or our ability to meet our financing objectives. Furthermore, our stock price may decline due in part to the volatility of the stock market and the general economic downturn.
Risks Related to Our Intellectual Property
If our efforts to protect the proprietary nature of the intellectual property related to our technologies are not adequate, we may not be able to compete effectively in our market.
We rely upon a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to our technologies. Any disclosure to or misappropriation by third parties of our confidential proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, thus eroding our competitive position in our market.
Composition-of-matter patents on the active pharmaceutical ingredient and crystalline forms are generally considered to be the strongest form of intellectual property protection for pharmaceutical products, as such
patents provide protection without regard to any method of use. We cannot be certain that the claims in our patent applications covering composition-of-matter and crystalline forms of emricasan will be considered patentable by the United States Patent and Trademark Office, or the U.S. PTO, courts in the United States, or by the patent offices and courts in foreign countries. Method-of-use patents protect the use of a product for the specified method. This type of patent does not prevent a competitor from making and marketing a product that is identical to our product for an indication that is outside the scope of the patented method. Moreover, even if competitors do not actively promote their product for our targeted indications, physicians may prescribe these products “off-label.” Although off-label prescriptions may infringe or contribute to the infringement of method-of-use patents, the practice is common and such infringement is difficult to prevent or prosecute.
The strength of patents in the biotechnology and pharmaceutical field involves complex legal and scientific questions and can be uncertain. Some of our patents related to emricasan were acquired from a predecessor owner and were therefore not written by us or our attorneys, and we did not have control over the drafting and prosecution of these patents. Further, the former patent owners might not have given the same attention to the drafting and early prosecution of these patents and applications as we would have if we had been the owners of the patents and applications and had control over the drafting and prosecution. In addition, the former patent owners may not have been completely familiar with U.S. patent law, possibly resulting in inadequate disclosure and/or claims. This could result in findings of invalidity or unenforceability of the patents we own or patents issuing with reduced claim scope.
In addition, the patent applications that we own or that we may license may fail to result in issued patents in the United States or in other foreign countries. Even if the patents do successfully issue, third parties may challenge the validity, enforceability or scope thereof, which may result in such patents being narrowed, invalidated or held unenforceable. Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property or prevent others from designing around our claims. If the breadth or strength of protection provided by the patent applications we hold with respect to emricasan is threatened, it could dissuade companies from collaborating with us to develop, and threaten our ability to commercialize, emricasan. Further, if we encounter delays in our clinical trials, the period of time during which we could market emricasan under patent protection would be reduced. Since patent applications in the United States and most other countries are confidential for a period of time after filing, we cannot be certain that we were the first to file any patent application related to emricasan. Furthermore, for applications in which all claims are entitled to a priority date before March 16, 2013, an interference proceeding can be provoked by a third-party or instituted by the U.S. PTO, to determine who was the first to invent any of the subject matter covered by the patent claims of our applications. For applications containing a claim not entitled to priority before March 16, 2013, there is greater level of uncertainty in the patent law with the passage of the America Invents Act (2012) which brings into effect significant changes to the U.S. patent laws that are yet untried and untested, and which introduces new procedures for challenging pending patent applications and issued patents. A primary change under this reform is creating a “first to file” system in the U.S. This will require us to be cognizant going forward of the time from invention to filing of a patent application.
In addition to the protection afforded by patents, we seek to rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable, processes for which patents are difficult to enforce and any other elements of our drug discovery and development processes that involve proprietary know-how, information or technology that is not covered by patents. Although we require all of our employees to assign their inventions to us, and require all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology to enter into confidentiality agreements, we cannot be certain that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. Furthermore, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad. If we are unable to prevent unauthorized material disclosure of our intellectual property to third parties, we will not be able to establish or maintain a competitive advantage in our market, which could materially adversely affect our business, operating results and financial condition.
Third-party claims of intellectual property infringement may prevent or delay our drug discovery and development efforts.
Our commercial success depends in part on our avoiding infringement of the patents and proprietary rights of third parties. There is a substantial amount of litigation involving patents and other intellectual property rights in the biotechnology and pharmaceutical industries, as well as administrative proceedings for challenging patents, including interference and reexamination proceedings before the U.S. PTO or oppositions and other comparable proceedings in foreign jurisdictions. Recently, under U.S. patent reform, new procedures includinginter partes review and post grant review have been implemented. As stated above, this reform is untried and untested and will bring uncertainty to the possibility of challenge to our patents in the future. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are developing emricasan. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that emricasan may give rise to claims of infringement of the patent rights of others.
Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents of which we are currently unaware with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of emricasan. Because patent applications can take many years to issue, there may be currently pending patent applications which may later result in issued patents that emricasan may infringe. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. If any third-party patents were held by a court of competent jurisdiction to cover the manufacturing process of emricasan, any molecules formed during the manufacturing process or any final product itself, the holders of any such patents may be able to block our ability to commercialize the drug candidate unless we obtained a license under the applicable patents, or until such patents expire or they are finally determined to be held invalid or unenforceable. Similarly, if any third-party patent were held by a court of competent jurisdiction to cover aspects of our formulations, processes for manufacture or methods of use, including combination therapy or patient selection methods, the holders of any such patent may be able to block our ability to develop and commercialize the drug candidate unless we obtained a license or until such patent expires or is finally determined to be held invalid or unenforceable. In either case, such a license may not be available on commercially reasonable terms or at all. If we are unable to obtain a necessary license to a third-party patent on commercially reasonable terms, or at all, our ability to commercialize emricasan may be impaired or delayed, which could in turn significantly harm our business.
Parties making claims against us may seek and obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize emricasan. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, obtain one or more licenses from third parties, pay royalties or redesign our infringing products, which may be impossible or require substantial time and monetary expenditure. We cannot predict whether any such license would be available at all or whether it would be available on commercially reasonable terms. Furthermore, even in the absence of litigation, we may need to obtain licenses from third parties to advance our research or allow commercialization of emricasan. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In that event, we would be unable to further develop and commercialize emricasan, which could harm our business significantly.
We may be involved in lawsuits to protect or enforce our patents, which could be expensive, time-consuming and unsuccessful.
Competitors may infringe our patents. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that one or more of our patents is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated, held unenforceable, or interpreted narrowly and could put our patent applications at risk of not
issuing. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, obtain one or more licenses from third parties, pay royalties or redesign our infringing products, which may be impossible or require substantial time and monetary expenditure.
Interference proceedings provoked by third parties or brought by the U.S. PTO may be necessary to determine the priority of inventions with respect to our patents or patent applications. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent misappropriation of our trade secrets or confidential information, particularly in countries where the laws may not protect those rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the U.S. PTO and foreign patent agencies in several stages over the lifetime of the patent. The U.S. PTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. In such an event, our competitors might be able to enter the market, which would have a material adverse effect on our business.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties.
We have received confidential and proprietary information from third parties. In addition, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies. We may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed confidential information of these third parties or our employees’ former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial cost and be a distraction to our management and employees.
Risks Related to This Offering and Ownership of our Common Stock
We do not know whether an active, liquid and orderly trading market will develop for our common stock or what the market price of our common stock will be and as a result it may be difficult for you to sell your shares of our common stock.
Prior to this offering there has been no public market for shares of our common stock. Although we expect that our common stock will be approved for listing on The NASDAQ Global Market, an active trading market for our shares may never develop or be sustained following this offering. You may not be able to sell your shares quickly or at the market price if trading in shares of our common stock is not active. The initial public offering price for our common stock will be determined through negotiations with the underwriters, and the negotiated price may
not be indicative of the market price of the common stock after the offering. As a result of these and other factors, you may be unable to resell your shares of our common stock at or above the initial public offering price. Further, an inactive market may also impair our ability to raise capital by selling shares of our common stock and may impair our ability to enter into strategic partnerships or acquire companies or products by using our shares of common stock as consideration.
The price of our stock may be volatile, and you could lose all or part of your investment.
The trading price of our common stock following this offering is likely to be highly volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control, including limited trading volume. In addition to the factors discussed in this “Risk Factors” section and elsewhere in this prospectus, these factors include:
the commencement, enrollment or results of our planned Phase 2 and Phase 3 clinical trials of emricasan or any future clinical trials we may conduct, or changes in the development status of emricasan;
any delay in our regulatory filings for emricasan and any adverse development or perceived adverse development with respect to the applicable regulatory authority’s review of such filings, including without limitation the FDA’s issuance of a “refusal to file” letter or a request for additional information;
adverse results or delays in clinical trials;
our decision to initiate a clinical trial, not to initiate a clinical trial or to terminate an existing clinical trial;
adverse regulatory decisions, including failure to receive regulatory approval for emricasan;
changes in laws or regulations applicable to our products, including but not limited to clinical trial requirements for approvals;
adverse developments concerning our manufacturers;
our inability to obtain adequate product supply for any approved drug product or inability to do so at acceptable prices;
our inability to establish collaborations if needed;
our failure to commercialize emricasan;
additions or departures of key scientific or management personnel;
unanticipated serious safety concerns related to the use of emricasan;
introduction of new products or services offered by us or our competitors;
announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our competitors;
our ability to effectively manage our growth;
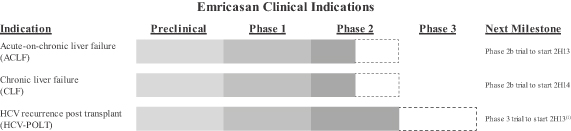
the size and growth, if any, of the ACLF, CLF and HCV-POLT markets and other targeted markets;
our ability to successfully enter new markets;
actual or anticipated variations in quarterly operating results;
our failure to meet the estimates and projections of the investment community or that we may otherwise provide to the public;
publication of research reports about us or our industry or positive or negative recommendations or withdrawal of research coverage by securities analysts;
changes in the market valuations of similar companies;
overall performance of the equity markets;
sales of our common stock by us or our stockholders in the future;
trading volume of our common stock;
changes in accounting practices;
ineffectiveness of our internal controls;
disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies;
significant lawsuits, including patent or stockholder litigation;
general political and economic conditions; and
other events or factors, many of which are beyond our control.
In addition, the stock market in general, and The NASDAQ Global Market and biotechnology companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance. If the market price of our common stock after this offering does not exceed the initial public offering price, you may not realize any return on your investment in us and may lose some or all of your investment. In the past, securities class action litigation has often been instituted against companies following periods of volatility in the market price of a company’s securities. This type of litigation, if instituted, could result in substantial costs and a diversion of management’s attention and resources, which would harm our business, operating results or financial condition.
We do not intend to pay dividends on our common stock so any returns will be limited to the value of our stock.
We have never declared or paid any cash dividend on our common stock. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. In addition, our ability to pay cash dividends is currently prohibited by the terms of a promissory note in the principal amount of $1.0 million issued by us to Pfizer Inc. in July 2010. Any return to stockholders will therefore be limited to the appreciation of their stock.
Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
Prior to this offering, our executive officers, directors, 5% stockholders and their affiliates owned approximately % of our voting stock and, upon the closing of this offering, that same group will hold approximately % of our outstanding voting stock (assuming no exercise of the underwriters’ over-allotment option) in each case assuming an initial public offering price of $ per share (the midpoint of the price range set forth on the cover page of this prospectus). Therefore, even after this offering, these stockholders will have the ability to influence us through this ownership position. These stockholders may be able to determine all matters requiring stockholder approval. For example, these stockholders may be able to control elections of directors, amendments of our organizational documents, or approval of any merger, sale of assets, or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that you may feel are in your best interest as one of our stockholders. In addition, entities affiliated with Aberdare Ventures, Advent Private Equity, Coöperative Gilde Healthcare II U.A., MPM Capital, Roche Finance Ltd, AgeChem Venture Fund L.P., Hale BioPharma Ventures LLC and our chief executive officer, each of which is a current stockholder, have indicated an interest in purchasing an aggregate of approximately $10.0 million of shares of our common stock in this offering. The above discussed ownership percentage upon completion of this offering does not reflect the potential purchase of any shares in this offering by such entities. If these entities purchase shares of our common stock in this offering, assuming an initial public offering price of $ per share (the midpoint of the price range set forth on the cover page of this prospectus), upon completion of this offering, our executive officers, directors, 5% stockholders and their affiliates will hold approximately % of our voting stock (assuming no exercise of the underwriters’ over-allotment option).
If you purchase our common stock in this offering, you will incur immediate and substantial dilution in the book value of your shares.
The initial public offering price is substantially higher than the net tangible book value per share of our common stock. Investors purchasing common stock in this offering will pay a price per share that substantially exceeds the book value of our tangible assets after subtracting our liabilities. As a result, investors purchasing common stock in this offering will incur immediate dilution of $ per share, based upon an assumed initial public offering price of $ per share (the midpoint of the price range set forth on the cover page of this prospectus). Further, investors purchasing common stock in this offering will contribute approximately % of the total amount invested by stockholders since our inception, but will own only approximately % of the shares of common stock outstanding after giving effect to this offering.
This dilution is due to our investors who purchased shares prior to this offering having paid substantially less when they purchased their shares than the price offered to the public in this offering and the exercise of stock options granted to our employees. To the extent outstanding options or warrants are exercised, there will be further dilution to new investors. As a result of the dilution to investors purchasing shares in this offering, investors may receive significantly less than the purchase price paid in this offering, if anything, in the event of our liquidation. For a further description of the dilution that you will experience immediately after this offering, see “Dilution.”
We are an emerging growth company, and we cannot be certain if the reduced reporting requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are an emerging growth company, as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in this prospectus and our periodic reports and proxy statements and exemptions from the requirements of holding nonbinding advisory votes on executive compensation and stockholder approval of any golden parachute payments not previously approved. We could be an emerging growth company for up to five years following the year in which we complete this offering, although circumstances could cause us to lose that status earlier, including if the market value of our common stock held by non-affiliates exceeds $700.0 million as of any June 30 before that time or if we have total annual gross revenue of $1.0 billion or more during any fiscal year before that time, in which cases we would no longer be an emerging growth company as of the following December 31 or, if we issue more than $1.0 billion in non-convertible debt during any three year period before that time, we would cease to be an emerging growth company immediately. Even after we no longer qualify as an emerging growth company, we may still qualify as a “smaller reporting company” which would allow us to take advantage of many of the same exemptions from disclosure requirements including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act and reduced disclosure obligations regarding executive compensation in this prospectus and our periodic reports and proxy statements. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
Under the JOBS Act, emerging growth companies can also delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies. As a result, changes in rules of U.S. generally accepted accounting principles or their interpretation, the adoption of new guidance or the application of existing guidance to changes in our business could significantly affect our financial position and results of operations.
We will incur significant increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives.
As a public company, we will incur significant legal, accounting and other expenses that we did not incur as a private company. We will be subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, which will require, among other things, that we file with the Securities and Exchange Commission, or the SEC, annual, quarterly and current reports with respect to our business and financial condition. In addition, the Sarbanes-Oxley Act, as well as rules subsequently adopted by the SEC and The NASDAQ Global Market to implement provisions of the Sarbanes-Oxley Act, impose significant requirements on public companies, including requiring establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. Further, in July 2010, the Dodd-Frank Wall Street Reform and Consumer Protection Act, or the Dodd-Frank Act, was enacted. There are significant corporate governance and executive compensation relatedAnti-takeover provisions in the Dodd-Frank Act that require the SEC to adopt additional rules and regulations in these areas such as “say on pay” and proxy access. Recent legislation permits emerging growth
companies to implement many of these requirements over a longer period and up to five years from the pricing of this offering. We intend to take advantage of this new legislation but cannot guarantee that we will not be required to implement these requirements sooner than budgeted or planned and thereby incur unexpected expenses. Stockholder activism, the current political environment and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact the manner in which we operate our business in ways we cannot currently anticipate.
We expect the rules and regulations applicable to public companies to substantially increase our legal and financial compliance costs and to make some activities more time-consuming and costly. If these requirements divert the attention of our management and personnel from other business concerns, they could have a material adverse effect on our business, financial condition and results of operations. The increased costs will decrease our net income or increase our consolidated net loss, and may require us to reduce costs in other areas of our business or increase the prices of our products or services. For example, we expect these rules and regulations to make it more difficult and more expensive for us to obtain director and officer liability insurance and we may be required to incur substantial costs to maintain the same or similar coverage. We cannot predict or estimate the amount or timing of additional costs we may incur to respond to these requirements. The impact of these requirements could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees or as executive officers.
Sales of a substantial number of shares of our common stock by our existing stockholders in the public market could cause our stock price to fall.
If our existing stockholders sell, or indicate an intention to sell, substantial amounts of our common stock in the public market after the lock-up and other legal restrictions on resale discussed in this prospectus lapse, the trading price of our common stock could decline. Based on shares of common stock outstanding as of March 31, 2013, upon the closing of this offering we will have outstanding a total of shares of common stock, assuming net exercises of all outstanding warrants, no exercise of the underwriters’ over-allotment option and no exercise of outstanding options. Of these shares, only the shares of common stock sold in this offering by us, plus any shares sold upon exercise of the underwriters’ over-allotment option, will be freely tradable without restriction in the public market immediately following this offering. Stifel, Nicolaus & Company, Incorporated and Piper Jaffray & Co., however, may, in their sole discretion, permit our officers, directors and other stockholders who are subject to these lock-up agreements to sell shares prior to the expiration of the lock-up agreements.
We expect that the lock-up agreements pertaining to this offering will expire 180 days from the date of this prospectus. After the lock-up agreements expire, up to an additional shares of common stock will be eligible for sale in the public market, of which shares are held by directors, executive officers and other affiliates and will be subject to volume limitations under Rule 144 under the Securities Act, assuming an initial public offering price of $ per share (the midpoint of the price range set forth on the cover page of this prospectus). In addition, shares of common stock that are either subject to outstanding options or reserved for future issuance under our employee benefit plans will become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules, the lock-up agreements and Rule 144 and Rule 701 under the Securities Act. If these additional shares of common stock are sold, or if it is perceived that they will be sold, in the public market, the trading price of our common stock could decline.
After this offering, the holders of shares of our common stock, or % of our total outstanding common stock as of March 31, 2013, will be entitled to rights with respect to the registration of their shares under the Securities Act, subject to the 180-day lock-up agreements described above and assuming an initial public offering price of $ per share (the midpoint of the price range set forth on the cover page of this prospectus). See “Description of Capital Stock—Registration Rights.” Registration of these shares under the Securities Act would result in the shares becoming freely tradable without restriction under the Securities Act, except for shares held by affiliates, as defined in Rule 144 under the Securities Act. Any sales of securities by these stockholders could have a material adverse effect on the trading price of our common stock.
Future sales and issuances of our common stock or rights to purchase common stock, including pursuant to our equity incentive plans, could result in additional dilution of the percentage ownership of our stockholders and could cause our stock price to fall.
We expect that significant additional capital may be needed in the future to continue our planned operations, including conducting clinical trials, commercialization efforts, expanded research and development activities and costs associated with operating a public company. To raise capital, we may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell common stock, convertible securities or other equity securities, investors may be materially diluted by subsequent sales. Such sales may also result in material dilution to our existing stockholders, and new investors could gain rights, preferences and privileges senior to the holders of our common stock, including shares of common stock sold in this offering.
Pursuant to our 2013 equity incentive plan, or the 2013 plan, which will become effective immediately prior to the closing of this offering, our management is authorized to grant stock options to our employees, directors and consultants. The number of shares available for future grant under the 2013 plan will automatically increase each year by an amount equal to 4% of all shares of our capital stock outstanding as of January 1stof each year, subject to the ability of our board of directors to take action to reduce the size of such increase in any given year. Unless our board of directors elects not to increase the number of shares available for future grant each year, our stockholders may experience additional dilution, which could cause our stock price to fall.
We could be subject to securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because pharmaceutical companies have experienced significant stock price volatility in recent years. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
We have broad discretion in the use of the net proceeds from this offering and may not use them effectively.
Our management will have broad discretion in the application of the net proceeds from this offering, including for any of the purposes described in the section entitled “Use of Proceeds,” and you will not have the opportunity as part of your investment decision to assess whether the net proceeds are being used appropriately. Because of the number and variability of factors that will determine our use of the net proceeds from this offering, their ultimate use may vary substantially from their currently intended use. Our management might not apply our net proceeds in ways that ultimately increase the value of your investment. We expect to use the net proceeds from this offering to fund the clinical development of emricasan and to fund working capital and for general corporate purposes. The failure by our management to apply these funds effectively could harm our business. Pending their use, we may invest the net proceeds from this offering in short-term, investment-grade, interest-bearing securities. These investments may not yield a favorable return to our stockholders. If we do not invest or apply the net proceeds from this offering in ways that enhance stockholder value, we may fail to achieve expected financial results, which could cause our stock price to decline.
Anti-takeover provisions under our charter documents and under Delaware law could delay or prevent a changemake an acquisition of control which could limit the market price of our common stockus more difficult and may prevent or frustrate attempts by our stockholders to replace or remove our current management.
Our amended and restatedProvisions in our certificate of incorporation and amended and restated bylaws which are to become effective at or prior to the closing of this offering, contain provisions that couldmay delay or prevent an acquisition or a change of control of our company or changes in ourmanagement. These provisions include a classified board of directors, that our stockholders might consider favorable. Some of these provisions include:
a board of directors divided into three classes serving staggered three-year terms, such that not all members of the board will be elected at one time;
a prohibition on stockholder action throughactions by written consent which requires that all stockholder actions be taken at a meeting of our stockholders;
a requirement that special meetings of stockholders be called only by the chairman of the board of directors,combined organization’s stockholders and the chief executive officer, the president or by a majority of the total number of authorized directors;
advance notice requirements for stockholder proposals and nominations for election to our board of directors;
| • | | a requirement that no member of our board of directors may be removed from office by our stockholders except for cause and, in addition to any other vote required by law, upon the approval of not less than 66 2⁄3% of all outstanding shares of our voting stock then entitled to vote in the election of directors;
|
| • | | a requirement of approval of not less than 66 2⁄3% of all outstanding shares of our voting stock to amend any bylaws by stockholder action or to amend specific provisions of our certificate of incorporation; and
|
the authorityability of the board of directors to issue preferred stock on terms determined by the board of directors without stockholder approval and which preferred stock may include rights superior to the rights of the holders of common stock.
approval. In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporate Law,DGCL, which may prohibit certain business combinations withprohibits stockholders owning in excess of 15% or more of our outstanding voting stock from merging or combining with us. Although we believe these provisions collectively will provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our board of directors, they would apply even if the offer may be considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove then current management by making it more difficult for stockholders to replace members of the board of directors, which is responsible for appointing the members of management.
Our pre-Merger net operating loss carryforwards and certain other tax attributes may be subject to limitations. The pre-Merger net operating loss carryforwards and certain other tax attributes of us may also be subject to limitations as a result of ownership changes resulting from the Merger.
In general, a corporation that undergoes an “ownership change” as defined in Section 382 of the Code, is subject to limitations on our ability to utilize our pre-change net operating loss carryforwards to offset future taxable income. In general, an ownership change occurs if the aggregate stock ownership of certain stockholders, generally stockholders beneficially owning five percent or more of a corporation’s common stock, applying certain look-through and aggregation rules, increases by more than 50 percentage points over such stockholders’ lowest percentage ownership during the testing period, generally three years. We may have experienced ownership changes in the past and we may experience ownership changes in the future. In addition, the closing of the merger may result in an ownership change for us, which could result in limitations on the use of our federal and state net
The PPACA established the Center for Medicare and its proper function is indispensable for many critical metabolic functions,Medicaid Innovation within CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending. Funding has been allocated to support the regulation of lipid and sugar metabolism, the production of important proteins, including those involved in blood clotting, and purification of blood. There are over 100 described diseasesmission of the liver,Center for Medicare and becauseMedicaid Innovation from 2011 to 2019.
Many of its many functions, thesethe details regarding the implementation of the PPACA are yet to be determined, and, at this time, the full effect of the PPACA on our business remains unclear. Further, there have been recent public announcements by members of the U.S. Congress, President Trump and his administration regarding their plans to repeal and replace the PPACA. For example, on December 22, 2017, President Trump signed into law the Tax Cuts and Jobs Act of 2017, which, among other things, eliminated the individual mandate requiring most Americans (other than those who qualify for a hardship exemption) to carry a minimum level of health coverage, effective January 1, 2019. We cannot predict the ultimate form or timing of any repeal or replacement of the PPACA or the effect such a repeal or replacement would have on our business.
Chemistry, Manufacturing, and Controls
We have successfully developed production processes that are scalable and economically viable. All of the derivatives of the manufacturing process can be highly debilitating and life-threatening unless effectively treated. Common causesused, creating a spectrum of liver disease include viral infections, such as HCV and HBV, obesity, chronic excessive alcohol use or autoimmune diseases. Many people with active liver disease remain undiagnosed largely because liver disease patients are often asymptomaticproducts for many years. The NIH estimates that 5.5 million Americans have chronic liver disease or cirrhosis, and liver disease is the twelfth leading cause of death in the United States. According to the EASL, 29 million Europeans have chronic liver disease and liver disease represents approximately two percent of deaths annually.
Liver disease is often first detected as active hepatitis, which is defined as inflammation of the liver. Hepatitis is easily detected by a routine laboratory test to measure blood levels of the liver enzyme ALT. ALT is elevated in almost all liver diseases and represents an overall measure of liver inflammation and liver cell death. As liver disease progresses, fibrotic scar tissue will begin to replace healthy liver tissue and over time will reduce the liver’s ability to function properly. A liver biopsy is used to diagnose fibrosis and determine how much liver scarring has developed. If fibrosis is allowed to progress, it will lead to cirrhosis. As liver cirrhosis becomes progressively worse, all aspects of liver function will dramatically decline.
Some patients with liver cirrhosis have a partially functioning liver, which is referred to as compensated liver disease, and may appear asymptomatic for long periods of time. When the liver is unable to perform its normal functions this is referred to as decompensated liver disease. ACLF occurs in patients who are in relatively stable condition until an acute event sets off a rapid deterioration of liver function. The morbidity and mortality of the patient population with ACLF we plan to study is high, with approximately 45% of patients dying or progressing to multi-organ failure or requiring transplant within 28 days of hospitalization as a result of the acute decompensating episode. If the patient survives the acute decompensating event, they may return to a stable state. Patients with CLF suffer from continual disease progression which may eventually lead them to require liver transplantation. Despite advances in liver transplantation, morbidity and mortality in the CLF patient population remains high with some patients ineligible for a liver transplant and others unable to be matched with a suitable donor liver.
Patients who receive liver transplants as a result of HCV infection are at risk of accelerated fibrosis of the transplanted liver. This occurs because residual HCV is still present in the patient’s blood and can immediately infect the new liver, thus increasing the risk of accelerated inflammation and fibrosis. This patient population is referred to as HCV-POLT. Liver fibrosis can be scored using the standard Ishak Fibrosis Score, which stages the
severity of fibrosis and/or cirrhosis on a 0-6 scale. Approximately 55% of the patient population we plan to study will progress more than one stage in fibrosis score within two years of transplant and 50% of these HCV-POLT patients will have stage 3 fibrosis within five years after their transplants. If emricasan demonstrates the ability to halt the progression of fibrosis in the HCV-POLT population, we believe that this could serve as a basis to evaluate emricasan for additional indications in patients at earlier stages of liver fibrosis resulting from HCV, HBV, obesity, chronic excessive alcohol use or autoimmune diseases.
The Role of Apoptosis, Necrosis and Inflammation in Liver Disease
The death of cells and resulting inflammation play an important role in the progression of many liver diseases. In general, cells can die by either of two major mechanisms, apoptosis, a form of programmed cell death, or necrosis. Both of these mechanisms can produce a state of acute and/or chronic inflammation as shown in Figure 1.
Figure 1. Apoptosis and Necrosis: The Two Main Pathways of Liver Cell Death
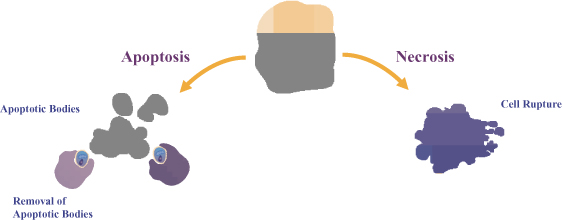
High levels of noxious stimuli can rapidly overwhelm the cell’s natural protective mechanisms, leading to a rupture of the cell and subsequent release of its contents into the surrounding tissue. This process is known as necrosis and results in a highly pro-inflammatory response, further damaging the surrounding tissue. In contrast, the programmed cell death mechanism, termed apoptosis, is a highly controlled and tightly regulated process that involves the orderly condensation and dismantling of the cell leading to its subsequent rapid and specific removal from the surrounding tissue by specialized cells. However, under conditions of excessive stress as often observed in disease, the production of apoptotic cells outpaces the body’s ability to effectively remove them from the surrounding tissue. This results in an accumulation of shed cell fragments known as apoptotic bodies which are taken up by surrounding cells and can stimulate additional cell death. Disease-driven excessive apoptosis results in the development of scar tissue or fibrosis which can lead to tissue destruction and eventually reduce the capacity of an organ to function normally.
Markers of Liver Cell Death
ALT is an enzyme that is produced in liver cells and is naturally found in the blood of healthy individuals. In liver disease, liver cells are damaged and as a consequence, ALT is released into the blood increasing ALT levels above the normal range. Physicians routinely test blood levels of ALT to monitor the health of a patient’s liver. ALT level is a clinically important biochemical marker of the severity of liver inflammation and ongoing liver disease. Elevated levels of ALT represent general markers of liver cell death and inflammation without regard to any specific mechanism. Aspartate aminotransferase, or AST, is a second enzyme found in the blood that is produced in the liver and routinely measured by physicians along with ALT. As with ALT, AST is often elevated in liver disease and, like ALT, is considered an overall marker of liver inflammation. We have measured both ALT and AST levels in our clinical studies, and have observed similar effects of emricasan on both enzymes. However, because ALT is considered more liver specific and the pattern of changes we have observed in AST levels has been similar to those seen in ALT levels, our discussion will focus primarily on ALT.
Another important marker of liver cell death is a protein fragment called cCK18. During apoptosis, a key structural protein within the cell called Cytokeratin 18, or CK18, is specifically cleaved by caspases which results in the release of cCK18 into the blood stream. cCK18 is easily detected in the blood with a commercially-available test and is a mechanism-specific biomarker of apoptosis and caspase activity. Importantly, cCK18 is also present in healthy subjects and multiple studies have demonstrated an approximate basal level in healthy subjects.
Numerous independent clinical trials and published studies have demonstrated the utility of cCK18 for detecting and gauging the severity of ongoing liver disease across a variety of disease etiologies. These studiesmarkets from one core technology.
We have demonstrated correlations between diseaseestablished in-house research, development and cCK18 levelsmanufacturing capabilities in patientsour corporate headquarters, however, we do not intend to continue manufacturing commercial or clinical material in-house moving forward. We intend to secure manufacturing agreements with ACLF, CLF, HCV, NASHa contract research organization for our own clinical and various other liver disease indications. For example, it has been shown that in HCV patients,commercial supply. Currently, we are not a party to any manufacturing agreements.
We currently manufacture commercial quantities of our CCM skin care ingredient for Allergan, who formulates the severity of liver disease was correlated with cCK18 levels and apoptosis, such that the more severe the disease, the higher the serum level of cCK18. In ACLF patients, studies have shown that blood levels of cCK18 were higher in non-surviving patients than in patients that survived. In CLF patients, studies have shown that cCK18 levelsingredient into their skin care product lines. However, we are elevated and correlate with liver inflammation and cholestasis. In patients with recurrent HCV-POLT, it has been shown that cCK18 levels and apoptosis were significantly elevated in liver biopsies as determined by immunohistochemical analysis. We believe these studies demonstrate the relationship between elevated cCK18 levels and severity of liver disease and that cCK18 is a valid and important biomarker of excessive apoptosis in liver disease.
The Model for End-Stage Liver Disease, or MELD, is a scoring system for assessing the severity of chronic liver disease. MELD is a composite score that is calculated using bilirubin levels, creatinine levels and International Normalized Ratio, or INR, which is a tool for assessing blood clotting times. MELD scores, which range from 6-40, are considered to be a valuable predictor of three-month survival. MELD score is also generally used to prioritize patients on the liver transplant list, with the average MELD score of 20 in patients undergoing liver transplant. In our planned clinical trials, we will study a subset of patients with ACLF and CLF who have MELD scores ranging from 20 to 30.
Emricasan
Emricasan is a first-in-class, proprietary and orally active caspase protease inhibitor designed to slow or halt the progression of chronic liver disease caused by fibrosis and cirrhosis. To date, emricasan has been administered to over 500 subjects in six Phase 1 and four Phase 2 clinical trials, and has been generally well-tolerated in both healthy volunteers and patients with liver disease. Emricasan has also been extensively profiled inin vitro tests and studied in many preclinical models of human disease.
Mechanism of Action
Emricasan works by inhibiting caspases, which are a family of related enzymes that play an important role as modulators of critical cellular functions, including functions that result in apoptosis and inflammation. Caspase activation and regulation is tightly controlled through a number of mechanisms. All caspases are expressed as enzymatically inactive forms known as pro-caspases which can be activated following a variety of cellular insults or stimuli. Seven caspases are specifically involved in the process of apoptosis while three caspases specifically activate pro-inflammatory cytokines and are not directly involvedconducting a technology transfer to Allergan, which will enable our commercial partner to utilize their own or third-party facilities. We anticipate exiting the commercial manufacturing business in apoptosis as shown in Figure 2.
Figure 2. Emricasan is a Potent Inhibitor of Apoptotic and Inflammatory Caspases
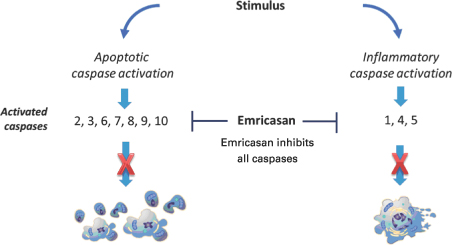
Caspase mediated apoptosis is driven primarily by the activity of caspases 3 and 7 which, by virtue of their enzymatic activity, cleave a wide variety of cellular proteins and result in dismantling of the cell. Other apoptotic caspase family members are principally involved in sensing and transmitting signals from either outside or inside the cell. These signals converge to activate pro-caspases 3 and 7, enabling them to carry out the process of apoptosis.
CK18 is one key structural protein that is cleaved by caspases 3 and 7 in a highly specific manner. The product of this cleavage is a small protein fragment, cCK18. This fragment is contained within the apoptotic cell fragments and is easily detected in serum using a commercially available monoclonal antibody assay. This monoclonal antibody, M30, is used routinely in clinical studies as a measure of apoptosis.
While healthy individuals have normal levels of apoptosis, excessive levels of apoptosis associated with disease can overwhelm the body’s normal clearance mechanisms. Reducing excessive levels of apoptosis reestablishes balance between apoptotic activity and normal clearance mechanisms and brings inflammation and other drivers of disease progression under control. As a result, we believe targeting caspases that drive both apoptosis and inflammation in disease offers a unique and potentially powerful therapeutic approach for the treatment of both acute and chronic liver disease.
Testing inin vitroenzyme assays demonstrated that emricasan efficiently inhibits all human caspases at low nanomolar concentrations. Preclinical studies have demonstrated that emricasan is highly selective for the caspase family of enzymes with little to no activity against other enzyme systems. These studies have also shown that emricasan potently inhibits the apoptosis of cells regardless of the apoptotic stimuli and that it is a potent inhibitor of caspase-mediated pro-inflammatory cytokines. Emricasan has been examined in various preclinical models of liver disease. In these models, caspase activity was demonstrated to be inhibited, as determined by histological examination, in liver tissue. Based on our evaluation of emricasan inin vitro systems, cellular assays and disease models, we believe emricasan’s mechanism of action has been well characterized.
Clinical Data
2021. To date, emricasan has been studied in over 500 subjects in six Phase 1 clinical trialswe have successfully transferred our process with two third-party contract manufacturing organizations (CMOs) and four Phase 2 clinical trials. This includes a total of 153 healthy volunteers, 306 subjects with elevated ALT levels and 53 liver transplant subjects receiving single or multiple doses of emricasan ranging from 1 to 500 mg per day orally or 0.1 to 10 mg/kg per day intravenously for up to 12 weeks. Emricasan has demonstrated evidence of a beneficial effect on serological biomarkers in patients with chronic liver disease independent of the cause of disease. Favorable changes have been observed in functional biomarkers of liver damage and inflammation, such as ALT and AST, and mechanistic biomarkers, such as cCK18 and caspase activity, indicating that emricasan works by the presumed mechanism of action of inhibiting apoptosis of liver cells. Importantly, clinical trials have also
demonstrated that emricasan does not inhibit normal levels of caspase activity in healthy individuals. Emricasan has been generally well-tolerated in clinical trials completed to date.
Phase 2b Dose Response Study in HCV Patients (Study A8491003)
Study A8491003, or the 003 trial, was a Phase 2b, randomized, multicenter, placebo-controlled, double-blind, parallel group, dose response trial. The trial was designed to evaluate the safety and efficacy of emricasan in patients with chronic HCV infection who were unresponsive to antiviral therapy and who had compensated liver disease with or without fibrosis. Patients with cirrhosis or hepatocellular carcinoma were excluded from the trial. The trial enrolled 204 HCV patients across three oral emricasan dose arms of twice-daily, or BID, 5 mg, 25 mg and 50 mg and one placebo arm. The primary endpoint in the study was changes from Baseline in ALT and AST levels over a period of 12 weeks. This study also measured cCK18 levels, and caspase 3 and 7 activity as exploratory biomarkers. In this trial, emricasan treatment resulted in statistically significant reductions in the primary endpoints of ALT and AST levels as well as statistically significant reductions in cCK18 levels and caspase 3 and 7 activity.
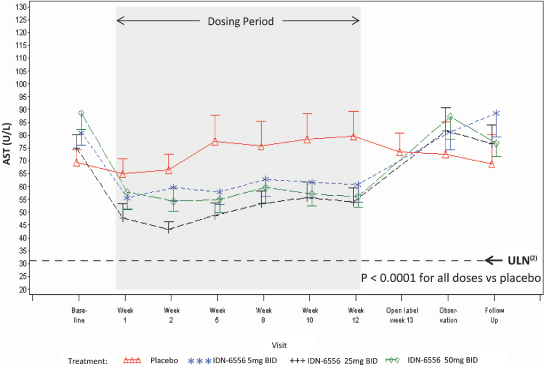
As shown in Figure 3 below, the changes in ALT demonstrated in the 003 trial were statistically significant in each of the emricasan treatment groups compared with the placebo group. The decreases in ALT were seen by day 7, the first time post-dosing that ALT was measured, and the decreases were maintained throughout the treatment period (up to 12 weeks) in all emricasan treatment groups. Discontinuation of emricasan at the end of the treatment period was followed by a gradual return of ALT towards baseline levels.
Figure 3. Change in ALT (Mean ± SEM(1)) from Baseline Following BID Dosing in Subjects with HCV
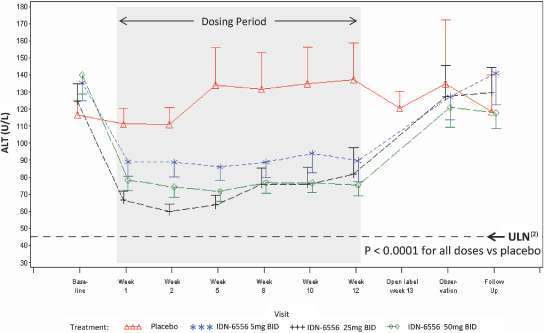
(1) | Standard Error of the Mean.
|
(2) | Upper limit of normal for males.
|
In addition to ALT levels, the 003 trial also examined changes in AST levels. As shown in Figure 4 below, the reductions in AST levels demonstrated in the 003 trial were also statistically significant in each of the emricasan treatment groups compared with the placebo group. Consistent with the ALT results, reductions in AST levels were seen as early as seven days and were maintained throughout the treatment period. At the end of treatment
AST levels gradually returned to baseline levels. During the 003 trial, biochemical flare, which is defined as ALT or AST values twice as high as the baseline value while on emricasan treatment, or overshoot, which is defined as ALT or AST values twice as high as the baseline value after stopping emricasan treatment, occurred in patients randomized to both placebo and emricasan. Twenty-one patients in the trial experienced flare and/or overshoot; six of these patients had both flare and overshoot; six of these patients had flare only; and nine of these patients had overshoot only. Of the six patients with flare and overshoot, four were in the placebo group, one was in the 5 mg group, and one was in the 25 mg group. Of the six patients with flare only, two were in the placebo group, one was in the 5 mg group, and three were in the 50 mg group. Of the nine patients with overshoot only, one was in the placebo group, five were in the 5 mg group, two were in the 25 mg group, and one was in the 50 mg group. These data suggest that the occurrence may be part of the natural variability of ALT or AST levels in the patient population under study. All subjects were followed up until levels had returned to baseline levels and there were no reports by the investigator of any clinical concern.
Figure 4. Change in AST (Mean ± SEM(1)) from Baseline Following BID Dosing in Subjects with HCV
(1) | Standard Error of the Mean.
|
(2) | Upper limit of normal for males.
|
The 003 trial data also provide evidence that emricasan reduces cCK18 levels from baseline in patients with elevated cCK18 levels, as shown in Figure 5 below. Statistically significant reductions in cCK18 levels were reported as early as three hours after dosing and were still evident following ten weeks of treatment, within each of the 5 mg, 25 mg and 50 mg dose arms compared to baseline values in the relevant dose group. Importantly, in the 003 trial, after ten weeks of dosing, cCK18 levels in all emricasan treatment groups were similar to the baseline level of cCK18 in healthy volunteers as established in our Phase 1 trial (see the description of study IDN-6556-03 below) and as generally reported from independent trials. We believe this observation suggests that normal levels of caspase activity remain intact. We also believe that by returning apoptosis to normalized levels, emricasan may enable the balance between apoptosis and the body’s normal clearance mechanism for apoptosis to be restored.
Figure 5. Change in cCK18 from Baseline Following BID Dosing in Subjects with HCV
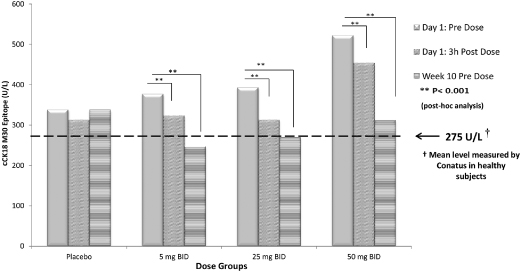
The 003 trial also included measurements of caspase 3 and 7 enzymatic activity. As shown in Figure 6 below, emricasan significantly reduced caspase 3 and 7 activity in a pattern similar to its effect on cCK18. We believe these data demonstrate that emricasan rapidly reduces elevated levels of caspase enzymatic activity and, as a consequence, excessive apoptosis in these patients. Since caspase 3 and 7 are known to be involved in the cleavage of CK18 which produces cCK18, we also believe these data suggest that the effect of emricasan on cCK18 is a result of inhibiting caspase activity. In addition, consistent with the cCK18 data, emricasan did not eliminate all caspase 3 and 7 activity in these patients. We believe this suggests that emricasan does not interfere with normal base levels of caspase activity or apoptosis, which is important in establishing the overall safety profile of emricasan.
Figure 6. Change in Caspase 3 and 7 Enzymatic Activity from Baseline Following
BID Dosing in Subjects with HCV
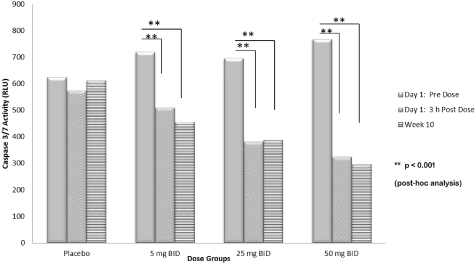
In the 003 trial, emricasan was generally observed to be well-tolerated. The most commonly reported adverse events in emricasan-treated subjects were headache, fatigue, nausea and diarrhea, most of which were mild to moderate in severity. Thirteen subjects withdrew from the study including seven in the placebo group, three from
the 5 mg, two from the 50 mg and one from the 25 mg emricasan groups. Nineteen adverse events reported by 14 subjects were considered severe with the greatest incidence in the placebo and 5 mg emricasan-treated groups (seven events each) and the lowest incidence in the 25 mg treatment group (one event). Severe adverse events were varied and showed no pattern across the treatment groups. The majority of adverse events had been resolved by the end of the study and the numbers of continuing events were similar for each of the patient cohorts. In addition, no concerning changes in any of the laboratory parameters and no clinically relevant changes in vital signs, electrocardiograms, physical examinations, or liver ultrasound scans could be attributed to emricasan.
Phase 2 Ascending Dose Study in Patients with Hepatic Impairment (Study A8491004)
Study A8491004, or the 004 trial, was a Phase 2, randomized, multicenter, placebo-controlled, double-blind, ascending dose trial in 105 patients with mild to moderate hepatic impairment. The trial was designed to evaluate the safety, tolerability, and pharmacokinetics of several dosing regimens of orally administered emricasan in these patients. The secondary objective of the trial was to evaluate the effects of emricasan on ALT and AST, as markers of efficacy. The trial was conducted at seven study sites and emricasan was administered orally for up to three times daily for 14 days. The study predominantly included patients with HCV liver disease, and also included limited numbers of patients with liver disease attributed to other causes, including HBV, NASH and primary biliary cirrhosis/primary sclerosing cholangitis. While once-daily, or QD, and BID dosing in the HCV patients demonstrated significant reductions in ALT from baseline, the BID dosing cohorts demonstrated greater percentage decreases of ALT levels than QD dosing.
In the 004 trial, 25 HCV patients were administered emricasan once per day for 14 days. As set forth in Figure 7 below, ALT reductions were rapid and sustained during the 14-day dosing period with a 30% to 40% reduction from baseline. While ALT decreases were statistically significantly different than placebo for QD dosing at 25 mg, 100 mg and 200 mg (p-values ranging from 0.0041 to <0.0001), those seen at a QD dose of 5 mg were less dramatic. After treatment with emricasan was completed, ALT levels returned to pre-treatment levels.
Figure 7. Percentage Change in ALT from Baseline Following QD Dosing in Subjects with HCV
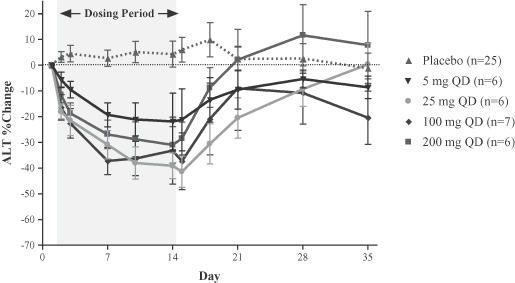
The 004 trial also examined BID, and three times daily, or TID, dosing of emricasan. In the trial, 30 patients received BID dosing at different dose levels and six patients were treated TID. As set forth in Figure 8 below, ALT reductions were rapid and sustained during the 14-day dosing period. In general, decreases in ALT were more pronounced than with QD dosing, with ALT reductions from baseline ranging from 39% to 56%. One cohort of patients was treated with 5 mg TID dosing. The results from this dosing group were similar to the BID
dosing groups. Patients with liver disease from causes other than HCV were dosed at 100 mg BID. Most of these patients had reductions in ALT similar to those observed in the HCV patients. All dosing groups were statistically significantly different than placebo (p-values ranging from 0.0041 to <0.0001).
Figure 8. Percentage Change in ALT from Baseline Following BID and TID Dosing in Subjects with HCV
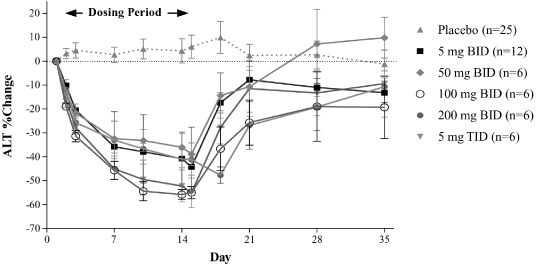
In all of the patient populations in this study, emricasan was generally well-tolerated. The most commonly reported adverse events related to emricasan were upper abdominal pain, dyspepsia, fatigue, dizziness, and headache. No subject was discontinued due to an adverse event. Importantly, in both the HCV and HBV infected patients studied, no increases in viral load parameters were observed.
Phase 2 Ascending Dose Crossover Study in Patients with HCV and Liver Fibrosis (Study A8491010)
Study A8491010 was a Phase 2, randomized double-blind, placebo-controlled crossover dose response study of emricasan in 24 patients with chronic HCV infection and liver fibrosis conducted by Pfizer. This study assessed the effects of BID dosing of emricasan on ALT and AST levels in these patients. For each patient, the study consisted of a screening visit, a two-week baseline period, three 14-day study periods separated by a minimum washout period of two weeks, and a two-week follow-up period after the last treatment period. Each patient was to receive three of five possible treatments (emricasan 0.5, 1, 2.5, 5 mg or placebo) BID for 13 days and QD on the final day of each study period. This trial was voluntarily discontinued early due to an unanticipated finding of inflammatory infiltrates in mice in a preclinical study that was ongoing concurrently with the clinical study and was unrelated to this Phase 2 trial. Pfizer notified the FDA of the findings in mice and the discontinuation of the trial, which resulted in the agency placing a clinical hold on the study of emricasan in 2007. At the time the clinical hold was imposed, 18 of 24 subjects had completed study A8491010. Because the study was discontinued prematurely, formal statistical tests were not performed. However, reductions in ALT in the 5 mg BID dose group were similar to the results of the 5 mg BID dose groups in the 003 and 004 trials. As described in the Emricasan History section below, the clinical hold was lifted in January 2013.
Clinical Studies in Organ Preservation in Transplant Patients
Preclinical studies demonstrated that emricasan is effective in protecting organs from damage that can occur during transplantation due to ischemia or reduced oxygen during isolation of donor organs, and reperfusion injury resulting from rapid exposure to oxygen following transplantation.
Study A8491002 was a Phase 2 randomized, placebo-controlled, double-blind, parallel group study to evaluate the effects of emricasan when administered in liver transplantation storage and flush solutions used in the preparation of the donated liver, and when administered to the recipient via IV during the first 24 hours after liver transplantation. Ninety-nine patients were randomized into one of four groups. In the first group, the liver was treated with placebo in the storage and flush solution and the patient was given placebo following
transplantation. In the second group the liver was treated with 15 µg/mL emricasan in the storage and flush solution, but the patient received placebo following transplantation. The third group was treated with a lower concentration of emricasan, 5 µg/mL, in the storage and flush solution and the patient received 0.5 mg/kg emricasan for 24 hours by IV administration following transplantation. The fourth group was treated with 15 µg/mL emricasan in the storage and flush solution and the patient received 0.5 mg/kg emricasan for 24 hours by IV administration following transplantation.
The co-primary endpoints were peak absolute change from baseline in ALT and AST measured up to three days post-transplantation. Large increases in both ALT and AST occurred in all groups reflecting liver injury typically occurring after liver transplantation. The outcome on the co-primary endpoints was not different between the placebo and the treatment groups possibly due to the short duration of drug treatment (24 hours) following transplantation. Serum markers of liver cell apoptosis, cCK18, were reduced in all groups receiving drug as compared to placebo. In addition, the level of liver cell apoptosis in liver tissue as determined by quantitation of cells with caspase 3 and 7 activity was reduced in all groups receiving emricasan, suggesting that the drug has activity through the anticipated mechanism of action.
Generally, the adverse events reported in the study were reflective of the severity of disease in the patient population. There were 1,240 adverse events reported in the 99 subjects, with similar numbers reported across the four study groups, as discussed above. There were 79 serious adverse events (6.4%) reported by 32 patients in this study. Of all the adverse events, 15 events were reported as possibly treatment-related but none were reported as probably or definitely treatment-related. The type and frequency of adverse events was similar across all groups, including placebo. There were deaths reported in all treatment groups (two in the placebo treatment group, and one in each of the emricasan treatment groups). These data in total support the conclusion that treatment with emricasan in this study has been generally well-tolerated.
Health Canada study NCT01653899, an investigator sponsored clinical trial, has been initiated at the University of Alberta to evaluate the safety and efficacy of islet cell culture and short-term (14 days) oral administration of emricasan in subjects with established Type 1 diabetes mellitus with unstable glycemic control. Health Canada approved the application in April 2012 and the trial is underway. This trial is an open label pilot study funded by a grant from the Juvenile Diabetes Research Foundation. Patients enrolled in this trial are not able to adequately regulate their blood glucose levels with insulin. The patients undergo pancreatic islet cell transplantation, a procedure referred to as the Edmonton procedure, with the goal of eliminating their need for insulin. One objective of this trial is to determine whether emricasan can improve upon the success rate of this so-called Edmonton procedure by reducing the amount of islet cell death after transplantation. Patients are dosed orally for 14 days following islet transplantation and are then monitored for their ability to control blood glucose without the need for insulin.
Phase 1 Studies
We have conducted six Phase 1 trials in subjects with both single and multiple-dose administration of emricasan. The objective of these trials was to examine the safety, tolerability and pharmacokinetics of emricasan. As shown in Figure 9 below, emricasan was generally well-tolerated in all six Phase 1 trials.
Figure 9. Emricasan Phase 1 Clinical Trial Summary
To understand the activity of emricasan on caspase activity in healthy subjects, a Phase 1 trial (study IDN-6556-03) in 15 subjects was conducted. In the trial, the levels of cCK18 in healthy human subjects was measured pre-dosing and then after dosing at different time intervals up to 12 hours post dosing. In this study, patients were administered 25 mg of emricasan BID as part of a drug-drug interaction study for 24 days with blood levels of cCK18 measured serially on days one, 17 and 24. As shown in Figure 10 below, dosing with emricasan did not cause meaningful decreases in cCK18 from predose levels in healthy subjects. We believe this demonstrates that emricasan does not interfere with the normal level of caspase activity and apoptosis in humans.
Figure 10. Mean Serum Levels of cCK18 in Healthy Subjects Following 25 mg BID of Emricasan in 15 Subjects
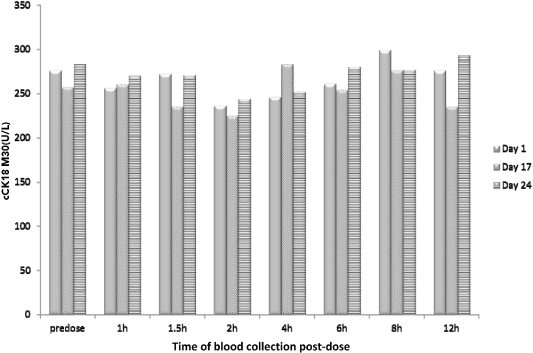
Emricasan History
Emricasan was initially discovered and developed by researchers at Idun, where the company was developing a new class of drugs that modulate caspases involved in the apoptosis and inflammation pathways. Idun, co-founded by Nobel Prize winner H. Robert Horvitz, Ph.D. for his work in the apoptosis field, was uniquely
positioned as a leading expert in translating apoptosis research into drug development candidates. Emricasan was Idun’s lead program when Pfizer acquired the company for approximately $298 million in 2005.
When we acquired emricasan from Pfizer in 2010, emricasan was on clinical hold in the United States due to an observation of inflammatory infiltrates in mice that Pfizer saw in a preclinical study and reported to the FDA in 2007. Pfizer performed additional preclinical studies attempting to characterize the nature of the inflammatory infiltrates, but did not carry out a formal carcinogenicity study to evaluate whether or not the infiltrates progressed to cancer. These infiltrates observed in mice were not observed in any other species. In 2008, Pfizer stopped work on the program. After acquiring emricasan, we conducted a thorough internal review of these studies and commissioned several independent experts to review all of the available data. The analysis provided by these experts unanimously concluded that these inflammatory infiltrates did not represent pre-cancerous lesions, nor were these infiltrates likely to progress to cancer. Additionally, a comprehensive analysis of available apoptosis literature supported the conclusion that the infiltrates were not likely to be precursors to cancer.
In April 2011, we met with the FDA to discuss plans for reinitiating clinical development of emricasan. We proposed conducting a carcinogenicity study designed to reproduce the previously observed findings of inflammatory infiltrates and determine whether they progress to cancer. We proposed using the Tg.rasH2 transgenic mouse model, which is known to be predisposed toward tumor development. The FDA agreed with the study design, and agreed that if the study reproduced the previously observed inflammatory infiltrates, but did not produce cancer, the issue which generated the clinical hold would be resolved.
This study was completed successfully in 2012. The inflammatory infiltrates were reproduced, and there was no evidence of tumor formation. In summary, treatment with emricasan for 26-weeks did not result in an increase in the incidence of tumors in Tg.rasH2 mice. The results were submitted to the FDA in preparation for a meeting in October 2012. The FDA reviewed the data and agreed with the study conclusion. We subsequently filed a new investigational new drug application, or IND, for emricasan for HCV-POLT, which was formally cleared in January 2013, effectively removing the clinical hold. In addition, the FDA has accepted this Tg.rasH2 carcinogenicity study as one of two carcinogenicity studies required for registration. We plan to perform a two-year rat carcinogenicity study, concurrent with the Phase 2b/3 HCV-POLT study, as the second carcinogenicity study.
Emricasan in ACLF
Medical Need and Market Opportunity
ACLF occurs in patients who have compensated or decompensated cirrhosis but are usually relatively stable. In these patients, some acute event sets off a rapid deterioration of liver function. The cause of this acute episode of decompensation may include toxins, such as alcohol, metabolic abnormalities and infections. The morbidity and mortality of patients with ACLF is high, and approximately 45% of the patient population that we intend to treat will die or develop multi-organ failure as a result of the decompensation episode within 28 days of hospitalization. We estimate that there are 150,000 patients in the United States and the EU diagnosed with symptoms consistent with our target population in ACLF. According to a 2011 publication in the Journal of Hepatology, the in-hospital mortality rateengage an existing partner for these acute deteriorating patients is greater than 50% and the total annual charges associated with ICU admissions alone are $3 billion, equating to a mean charge of $116,000 per admission.
Liver function in ACLF patients deteriorates rapidly. At the time of hospitalization, patients may present with MELD scores such as those seen on the transplant waiting list. Although the exact mechanisms remain unclear, massive liver cell loss involving excessive apoptosis and necrosis are known contributing factors leading to progressive dysfunction. There is evidence that serum markers of caspase-driven apoptosis such as cCK18 are elevated in these patients. In addition, independent studies have shown that increased cCK18 levels in this patient population are associated with worse prognosis.
The rapid deterioration in liver function, which may be exacerbated by an altered immune response, leads to life-threatening complications such as renal failure, increased susceptibility to infection, hepatic coma and systemic hemodynamic dysfunction. Current goals of the medical treatment for ACLF are to reverse precipitating factors, support failing organs and prevent further deterioration in liver function in order to give the liver time to
repair. Medical intervention for ACLF involves treatment of the underlying cause of the acute event. Patients who progress to multi-organ failure may require specific therapies for this such as medications for cardiac failure, mechanical ventilation for respiratory failure or dialysis for renal failure. Liver transplantation is required in some subjects to improve survival and quality of life. There are currently no approved therapies with a specific indication for the treatment of ACLF, and to our knowledge, there are only a limited number of clinical studies currently being conducted in patients with either liver cirrhosis or liver failure. We believe the ACLF population is in high medical need of an efficacious and well-tolerated therapy to prevent progression to multi-organ failure and, ultimately, premature death.
Development Plans
The ACLF studies will assess the safety and efficacy of emricasan in reducing cell death and inflammation in order to improve time to clinical worsening, or TTCW, which is defined as the first occurrence of liver transplant, progression to next organ failure or death in patients with acute liver failure in the presence of cirrhosis. Two studies are being planned in this patient population as follows.
Phase 2b Dose Ranging Study
In this Phase 2b study, we plan to enroll 60 patients with a history of liver cirrhosis that have been admitted to the hospital for an acute deterioration of liver function for more than 24 hours. Patients will be required to have a MELD score between 20 and 30 as well as certain other inclusion criteria related to creatinine levels, bilirubin levels and INR. Patients with uncontrolled infections will be excluded from the study. Patients will be equally randomized to receive either placebo, 5 mg, 25 mg or 50 mg emricasan BID orally. The primary objective in this 28-day dosing study will be to explore the pharmacokinetics and pharmacodynamics together with the safety of emricasan in this patient population. We also plan to measure TTCW, which is anticipated to be the primary efficacy endpoint for the planned Phase 3 studies in ACLF and the Phase 2b study in CLF. Changes in ALT, AST, cCK18 and the components of the MELD score will also be measured in the study.
We plan to initiate this Phase 2b clinical trial in the United Kingdom in the second half of 2013 and anticipate results from the trial will be available during the first half of 2014. The data from this trial will be used in supportcontract manufacturing of our planned end of Phase 2b meetings with both U.S. and EU regulatory authorities to finalize the protocol for our planned Phase 3 trial in ACLF.
Phase 3 Registration Study
The Phase 3 registration study for ACLF is expected to be an international study that will include approximately 400 patients with the same inclusion and exclusion criteria as the Phase 2b study. We expect that the primary endpoint will be TTCW assessed after 28 days of dosing. Changes in ALT, AST and cCK18 and components of MELD score will also be measured in this study. In order to be able to provide important prescribing information to physicians, at the end of the 28-day dosing period, patients randomized to receive emricasan during the initial 28 days of the study will be re-randomized to receive either emricasan or placebo for an additional five months. Therefore, we expect emricasan to be dosed up to six months in this study. Patients randomized to placebo will remain on placebo throughout the study.
Based on our discussions with regulatory authorities in the United Kingdom and Germany, we believe that the ACLF Phase 3 clinical trial could suffice as a single registration study for these geographies if the results are compelling. We will discuss a similar strategy for emricasan approval in the United States with the FDA at our end of Phase 2 meeting to be held after the completion of our Phase 2b trial. We plan to submit applications for orphan drug designations for ACLF in the United States and the EU in the second half of 2013.
Subject to the results of our planned clinical trials and any regulatory-approved product labeling, we currently anticipate that emricasan, if approved for this indication, would be prescribed by physicians to be dosed for 28 days, but potentially as long as six months, in the ACLF patient population.
Emricasan in CLF
Medical Need and Market Opportunity
CLF involves the progressive destruction of liver cells over time resulting in fibrosis and cirrhosis. The cause of the chronic decompensation or liver failure may vary and, similar to ACLF, includes infections, such as subacute bacterial peritonitis, HCV or HBV, metabolic causes, such as NASH, autoimmune diseases and alcohol. Eventually, these patients will progress to the point where, if eligible, they may require transplantation. The difference between this population and ACLF is that in ACLF, patients have an acute event that triggers a decompensatory event that may be reversed whereas in CLF, the event is of a chronic nature and it is unlikely that the liver function will improve. Objectives for the management of patients with CLF will be either to ensure that they stabilize to the point that they are eligible for transplant or that they survive until the time of transplant. The same therapies as those employed for the management of ACLF and mentioned above are generally employed. In CLF, apoptosis is elevated as determined by serological biomarkers and histology, which correlates with clinical worsening. Independent published studies have shown that cCK18 levels are elevated in CLF patients and correlate with extent of liver inflammation and cholestasis.
Although we are not planning to exclusively study patients with CLF on the liver transplant waitlist, many of these patients may also be included in the study. According to the U.S. Department of Health and Human Services, there were over 5,800 adult liver transplants performed in the United States in 2011 and approximately 2,500 died while waiting for transplant and another 500 subsequently became ineligible for a liver transplant. We estimate that there are approximately 5,000 CLF patients in our target population on the transplant waitlist in the United States and the EU. The median wait time for a liver transplant for the subset of patients with MELD scores of 19-24 is 106 days and the mortality rate is approximately 25% during that time frame, according to the United Network for Organ Sharing database. We estimate that there will be at least the same number of patients with CLF who are not candidates for liver transplantation but who might also benefit from emricasan. Given its mechanism of action, emricasan has the potential to improve patients’ ability to survive longer while waiting for a liver transplant or potentially make them eligible for transplant.
Development Plans
A Phase 2b study in patients with CLF is being planned. This study will be designed to include approximately 100 patients with MELD scores at entry of 20-30. It is expected that emricasan will be dosed for one to three months and the endpoints in this study will include TTCW as well as ALT, AST and cCK18 levels. The data from the ACLF dose ranging study is also expected to serve as the basis for dose selection in this trial. Depending on our interactions with regulatory authorities, this study might also be designed as a second registration study in support of the ACLF indication. We anticipate commencing this trial in the second half of 2014 after completion of the ACLF Phase 2b study.
Subject to the results of our planned clinical trials and any regulatory-approved product labeling, we currently anticipate that emricasan, if approved for this indication, would be prescribed by physicians to be dosed for one to three months in the CLF patient population.
Emricasan in HCV-POLT
Medical Need and Market Opportunity
According to the Organ Procurement and Transplantation Network and EASL, of the approximately 12,000 patients in the United States and EU who receive liver transplants each year, it is estimated that 30% are HCV-infected. Since immune system suppression is required following liver transplantation, 100% of these patients experience HCV recurrence that can lead to fibrosis and cirrhosis. It is estimated that 50% of these patients develop cirrhosis within two years of transplantation. We refer to this population as the HCV-POLT population, which we estimate to be 50,000 patients in the United States and EU.
In HCV-POLT patients, due to the lack of easily-administered and effective options to address the underlying HCV, patients are monitored closely for the presence of fibrosis and its rate of progression using ALT measurements and biopsies to determine need for treatment. Physicians currently treat these patients when they
have an acute flare of HCV together with some sign of fibrosis evident on liver histology. Current treatment options are largely limited to interferon-based regimens that are not well-tolerated or particularly efficacious in this population. Given the lack of effective treatment options, physicians have begun to implement newer treatments such as boceprevir and telaprevir in these patients despite limited data for those treatments in this post-transplant population.
The HCV landscape is expected to evolve dramatically over the next five to ten years with the introduction of interferon-free regimens, which are expected to reach the market as soon as 2015, and next-generation interferon-free regimens, which may reach the market by as early as 2016, with greater efficacy and tolerability over the current antiviral therapies. While these novel treatments are expected to drive high cure rates in treatment-naïve patients, even with 100% treatment rates and 90% cure rates, we believe the number of annual transplants needed will significantly exceed the available supply of transplants. Based on market research we commissioned, we estimate that treatment rates would need to be 100% and cure rates would have to exceed 97% before the supply of liver transplants would outstrip the need for transplant on an annual basis, without considering the current backlog. The immuno-compromised status of post-transplant patients and characteristics of viral infection in patients that have previously failed HCV treatments are likely to limit cure rates in the HCV-POLT population, even with the new therapies. We believe emricasan could play an important role in extending life for patients that still require a transplant as a result of not achieving a cure by next-generation agents.
Development Plans
We are planning a randomized, double-blind, placebo-controlled trial in the HCV-POLT population that we expect may serve as a single Phase 3 registration study to support a marketing authorization application, or MAA, filing in the EU. While the study is designated a Phase 3 study in the EU, the study is designated a Phase 2b study in the United States. We plan to seek further discussions with the FDA to determine if the study may be used to support the filing of an NDA with the FDA upon completion. This 260-patient study will enroll patients who have undergone a liver transplant and show demonstrable fibrosis on liver histology. Patients eligible for this study will include those who have had unsuccessful antiviral treatment prior to or following liver transplant; those who in the opinion of the treating physician are not suitable candidates for antiviral treatment; and/or those who are unable to tolerate antiviral treatment for HCV. Patients will be randomized to receive either placebo or 25 mg of emricasan BID. The study will be a two-year dosing study with a three-year follow up. The primary endpoint will be liver histology, specifically the presence or absence of disease progression as measured by the standard Ishak Fibrosis Score, which stages the severity of fibrosis and/or cirrhosis on a 0-6 scale. Changes in ALT, AST and cCK18 will also be measured in this study. We expect to initiate this trial in the second half of 2013.
We have filed for orphan drug designation for the HCV-POLT indication in the EU and the United States, but have not yet received responses from the regulatory bodies in these jurisdictions. A European Scientific Advice meeting with the Committee for Medicinal Products for Human Use (CHMP) for HCV-POLT is being planned for mid-2013. This Scientific Advice meeting will define the specific requirements and deliverables that will be required in order to qualify for single registration study status throughout Europe. It will also define the patient safety databases that will be required as well as the additional supportive clinical studies such as drug-drug interaction studies (if any) that will be required for the MAA in Europe.
Subject to the results of our planned clinical trials and any regulatory-approved product labeling, we currently anticipate that emricasan, if approved for this indication, would be prescribed by physicians to be dosed for up to two years in the HCV-POLT patient population.
Future Indications
Due to its mechanism of action and the presence of apoptosis and inflammation in many liver diseases, we believe there may be several patient populations that could potentially benefit from emricasan, including those that have previously failed HCV treatment and those with NASH, alcoholic liver disease, non-alcoholic fatty liver disease, viral hepatitis and other chronic liver diseases. At this time, we do not plan to explore these indications; however we may seek partners to pursue the evaluation of these potential indications. We are
currently supporting a pilot study funded by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) in patients with alcoholic hepatitis. This exploratory study is a placebo-controlled study which will be conducted at three centers in the United States over four years and will assess patient survival as the primary endpoint.
Commercialization Strategy
We expect that the majority of ACLF, CLF and HCV-POLT patients will be treated at tertiary care centers and transplant centers and therefore can be addressed with a targeted sales force. We intend to build our own commercial infrastructure in North America and the EU to target these centers. We believe we are well-positioned to retain commercialization rights for emricasan for ACLF, CLF and HCV-POLT in the United States and the EU while considering opportunities to partner in other territories or for other indications.
Manufacturing
Pfizer completed a significant portion of the manufacturing process optimization needed to provide an efficient synthesis of active pharmaceutical ingredient, or API, and scale-up for registration trials. API was successfully produced under current Good Manufacturing Practices, or cGMP, conditions, and a strategy to scale up the API for commercialization is in development. We have adequate cGMP API to support Phase 2 and Phase 3 clinical trials. We have secured a contractor that is capable of making sufficient quantities of the drug product. Both API and the drug product have demonstrated sufficient stability characteristics in our studies conducted to date. A number of dosage forms are being developed including capsule, powder-in-capsule, tablet, and alternate dosage forms suitable for pediatric administration.
Competition
The biopharmaceutical industry is characterized by intense competition and rapid innovation. Although we believe that we hold a leading position in our understanding of caspase inhibition related to liver disease, our competitors may be able to develop other compounds or drugs that are able to achieve similar or better results. Our potential competitors include major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical companies and universities and other research institutions. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. We believe the key competitive factors that will affect the development and commercial success of our product candidates are efficacy, safety and tolerability profile, reliability, convenience of dosing, price and reimbursement.going forward.
There are currently no therapeutic products approved for the treatment of ACLF, CLF or HCV-POLT. There are a number of marketed therapeutics used in each of these diseases to try to remove the underlying cause of the disease and prevent further liver injury. For example, if the liver damage is a result of HBV or HCV, marketed antiviral medications may be used to treat the virus that led to liver damage. If the liver damage is a result of alcoholic hepatitis, marketed alcohol addiction drugs may be used. If the liver damage is a result of obesity, diet and exercise may be prescribed along with marketed therapeutics. If the liver damage is a result of NASH, marketed drugs such as insulin sensitizers (e.g., metformin), antihyperlipidemic agents (e.g., gemfibrozil), pentoxifylline and ursodiol are generally used, although none of these are approved for NASH. In addition to the marketed drugs for those indications, there are drugs in development for each of these indications. Although these marketed therapies and those in development may be efficacious, all of them take time to show an effect and as long as the underlying conditions persist there will continue to be damage to the liver. Emricasan is the only therapeutic we are aware of that is being developed specifically to reduce the level of apoptosis in the liver and as a result it may be used with these other therapies.
In addition, the HCV landscape is expected to evolve dramatically over the next five to ten years with the introduction of new interferon-free regimens, which are expected to reach the market as soon as 2015, and next generation interferon-free regimens, which may reach the market by as early as 2016, with greater efficacy and tolerability over the current antiviral therapies. Based on market research we commissioned, we estimate that treatment rates pre-transplantation would need to be 100% and cure rates would have to exceed 97% before the supply of liver transplants would outstrip the need for transplant on an annual basis. Therefore, we expect that there will continue to be a significant unmet need in the HCV-POLT population.
Material Contracts
Pfizer Inc.
In July 2010, we entered into a Stock Purchase Agreement with Pfizer pursuant to which we acquired all of the outstanding capital stock of Idun, a wholly-owned subsidiary of Pfizer at the time, in consideration for an upfront payment of $250,000 and a promissory note in the principal amount of $1.0 million. The promissory note matures in July 2020, subject to acceleration upon specified events of default, including a change of control transaction, our failure to timely pay any principal or interest when due, our failure to timely provide certain financial information to Pfizer, the creation of any lien on our property other than permitted liens, any disposition of our business or property other than permitted transfers, our payment of dividends or other distributions on our equity securities, our incurrence of any indebtedness other than permitted indebtedness, our involvement in liquidation, dissolution, bankruptcy or similar proceedings, our failure to notify Pfizer of certain material adverse events, our failure to repay any indebtedness that causes an aggregate of $2.0 million or more in such indebtedness to accelerate in maturity, and the rendering of certain judgments against us. The note bears interest at 7% per year, compounded quarterly. Interest is payable on a quarterly basis during the term of the note. We have the right to prepay the promissory note at any time. We will also be required to make additional payments to Pfizer totaling $18.0 million upon the achievement of specified regulatory milestones relating to emricasan.
Idun Pharmaceuticals, Inc.
In January 2013, we conducted a spin-off of our subsidiary Idun Pharmaceuticals, Inc., or Idun Pharma (which we had acquired from Pfizer in the transaction described above), to our stockholders at that time. Immediately prior to the spin-off, all rights relating to emricasan were distributed to us pursuant to a distribution agreement. The assets remaining in Idun Pharma at the time of the spin-off consisted solely of intellectual property rights and license and collaboration agreements unrelated to emricasan. The spin-off was conducted as a dividend of all of the outstanding capital stock of Idun Pharma to our stockholders and, as a result, we no longer own any capital stock of Idun Pharma. The aggregate value of Idun Pharma at the time of the spin-off was deemed to be $9.6 million based on the valuation of an independent appraisal firm.
Also in connection with the spin-off, we contributed $500,000 to Idun Pharma to provide for its initial working capital requirements and entered into a transition services agreement to provide operating services to Idun Pharma, generally consisting of accounting support, technology license administration and intellectual property maintenance. Idun Pharma must pay us for all direct costs as well as overhead and general and administrative expenses incurred in performing these services. As of March 31, 2013, Idun Pharma had not made any payments to us for any services provided under the transition services agreement. The initial term of the transition services agreement ends on December 31, 2013, and will renew automatically for successive one year periods unless terminated by either Idun Pharma or us upon 90 days’ prior notice to the other party.
Idun Pharma Sublicense Agreement
In March 2013, we entered into a sublicense agreement with Idun Pharma in which we were granted the right to use the patent rights and know-how related to the screening and identification of emricasan. These rights were previously granted to Idun Pharma under license agreements with Thomas Jefferson University, or TJU. Under the sublicense, we are required to pay directly to TJU a royalty of less than one percent on net sales of emricasan. We also have the right to grant further sublicenses to third parties and are required to pay TJU a portion of any such sublicense revenue we receive. The sublicense agreement will expire upon the date which there are no longer any valid claims in any patents or patent applications sublicensed to us, unless earlier terminated. Idun Pharma may terminate the agreement if we substantially fail to perform or otherwise materially breach any of the material terms, covenants or provisions of the sublicense agreement, and we do not cure any such breach within 60 days of receipt of written notice from Idun Pharma specifying the breach. The agreement may also be terminated if the underlying license agreements between Idun Pharma and TJU are terminated. The underlying license agreements may be terminated by either Idun Pharma or TJU if the other party substantially fails to perform or otherwise materially breaches any of the material terms, covenants or provisions of the underlying license agreements, and the breaching party does not cure any such breach within 90 days of receipt
of written notice from the non-breaching party specifying the breach. Idun Pharma may also elect upon 30 days’ prior written notice to terminate its rights and obligations under one of the underlying license agreements with respect to any patent applications or patents licensed to it, or to terminate such underlying license agreement in its entirety. In the event that either of the underlying license agreements are terminated, Idun Pharma is obligated to assign and transfer to TJU all rights under sublicenses granted by Idun Pharma.
Intellectual Property
The proprietary natureAs of December 28, 2020, we hold or control eleven issued U.S. patents, five pending U.S. patent applications, and protection forforty patents in various jurisdictions outside the United States related to our product candidates and discovery programs and know-howcore technology. Additionally, we are importantpursuing twenty-three corresponding patent applications that are pending in various foreign jurisdictions, including two applications that are pending in accordance with the Patent Cooperation Treaty (“PCT”). Further advancement of our intellectual property portfolio will require the filing of patent applications related to our business.proprietary manufacturing process and product candidates. We have soughtpatents extending into the late 2020s, and 2030 as well as trade secrets protecting our intellectual property. Our patent prosecution strategy includes exploration of opportunities to expand our patent life in order to broaden our existing patent portfolio.
Below is a further description of certain of our key issued patents, including the method of protection, expiration date, number of related patents issued in foreign jurisdictions and the product candidates to which each patent relates. We currently hold or control:
three patents issued in the United States (U.S. Patent Nos. 10,538,736, 8,257,947 and internationally for emricascan, crystalline forms of emricasan8,524,494) and certain methods of treatment with emricasan. In addition, we have patent protection covering certain other preclinical stage compounds. Our policy is to pursue, maintain and defend patent rights whether developed internally or licensed from third parties and to protect the technology, inventions and improvements that are commercially important to the development of our business.
Our commercial success will dependthirty-one patents issued in part on obtaining and maintaining patent protection and trade secret protection of our current and future product candidates and the methods used to develop and manufacture them, as well as successfully defending these patents against third-party challenges. Our ability to stop third parties from making, using, selling, offering to sell or importing our products depends on the extent to which we have rights under valid and enforceable patents that cover these activities. We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed by us in the future, nor can we be sure that any of our existing patents or any patents that may be granted to us in the future will be commercially useful in protecting our product candidates, discovery programs and processes. For this and more comprehensive risks related to our intellectual property, please see “Risk Factors—Risks Related to Our Intellectual Property.”
Our patent portfolio for emricasan contains patentsforeign jurisdictions directed to the composition of matter, crystalline forms and methods of use. As of April 25, 2013 we have received three U.S. patents and corresponding foreign patents production
% of the outstanding shares of our common stock as of the last day of our immediately preceding fiscal year; or
such other amount as our board of directors may determine.
The 2013 Plan will also provide for an aggregate limit of shares of common stock that may be issued under the 2013 Plan over the course of its ten-year term.
Shares issued pursuant to awards under the 2013 Plan that we repurchase or that are forfeited, as well as shares used to pay the exercise price of an award or to satisfy the tax withholding obligations related to an award, will become available for future grant under the 2013 Plan. In addition, to the extent that an award is paid out in cash rather than shares, such cash payment will not reduce the number of shares available for issuance under the 2013 Plan.
Plan Administration. The compensation committee of our board of directors will administer the 2013 Plan (except with respect to any award granted to “independent directors” (as defined in the 2013 Plan), which must be administered by our full board of directors). Following the completion of this offering, to administer the 2013 Plan, our compensation committee must consist solely of at least two members of our board of directors, each of whom is a “non-employee director” for purposes of Rule 16b-3 under the Securities Exchange Act of 1934, as amended, or the Exchange Act, and, with respect to awards that are intended to constitute performance-based compensation under Section 162(m) of the Internal Revenue Code of 1986, as amended, or the Code, an “outside director” for purposes of Section 162(m). Subject to the terms and conditions of the 2013 Plan, our compensation committee has the authority to select the persons to whom awards are to be made, to determine the type or types of awards to be granted to each person, the number of awards to grant, the number of shares to be subject to such awards, and the terms and conditions of such awards, and to make all other determinations and decisions and to take all other actions necessary or advisable for the administration of the 2013 Plan. Our compensation committee is also authorized to establish, adopt, amend or revise rules relating to administration of the 2013 Plan. Our board of directors may at any time revest in itself the authority to administer the 2013 Plan.
Eligibility. Options, stock appreciation rights, or SARs, restricted stock and other awards under the 2013 Plan may be granted to individuals who are then our officers or employees or are the officers or employees of any of our subsidiaries or parent entities. Such awards may also be granted to our non-employee directors and consultants but only employees may be granted incentive stock options, or ISOs. As of December 31, 2012, there were eight non-employee directors and approximately 14 employees who would have been eligible for awards under the 2013 Plan had it been in effect on such date. At such time after the completion of this offering when we are subject to the requirements of Section 162(m) of the Code, the maximum number of shares that may be subject to awards granted under the 2013 Plan to any individual (other than non-employee directors) in any calendar year cannot exceed and the maximum amount that may be paid to a participant in cash during any
calendar year with respect to one or more cash based awards under the 2013 Plan is $ . In addition, the maximum number of shares that may be subject to awards granted under the 2013 Plan to any non-empolyee director in any calendar year cannot exceed .
Awards. The 2013 Plan provides that our compensation committee (or the board of directors, in the case of awards to non-employee directors) may grant or issue stock options, SARs, restricted stock, restricted stock units, dividend equivalents, stock payments and performance awards, or any combination thereof. Our compensation committee (or the board of directors, in the case of awards to non-employee directors) will consider each award grant subjectively, considering factors such as the individual performance of the recipient and the anticipated contribution of the recipient to the attainment of our long-term goals. Each award will be set forth in a separate agreement with the person receiving the award and will indicate the type, terms and conditions of the award.
Nonqualified stock options, or NQSOs, will provide for the right to purchase shares of our common stock at a specified price which may not be less than the fair market value of a share of common stock on the date of grant, and usually will become exercisable (at the discretion of our compensation committee or our board of directors, in the case of awards to non-employee directors) in one or more installments after the grant date, subject to the participant’s continued employment or service with us and/or subject to the satisfaction of performance targets established by our compensation committee (or our board of directors, in the case of awards to non-employee directors). NQSOs may be granted for any term specified by our compensation committee (or our board of directors, in the case of awards to non-employee directors).
ISOs will be designed to comply with the provisions of the Code and will be subject to specified restrictions contained in the Code. Among such restrictions, ISOs must have an exercise price of not less than the fair market value of a share of common stock on the date of grant, may only be granted to employees, must expire within a specified period of time following the optionee’s termination of employment, and must be exercised within ten years after the date of grant. In the case of an ISO granted to an individual who owns (or is deemed to own) more than 10% of the total combined voting power of all classes of our capital stock, the 2013 Plan provides that the exercise price must be at least 110% of the fair market value of a share of common stock on the date of grant and the ISO must expire upon the fifth anniversary of the date of grant.
Restricted stock may be granted to participants and made subject to such restrictions as may be determined by our compensation committee (or our board of directors, in the case of awards to non-employee directors). Typically, restricted stock may be forfeited for no consideration if the conditions or restrictions are not met, and it may not be sold or otherwise transferred to third parties until the restrictions are removed or expire. Recipients of restricted stock, unlike recipients of options, may have voting rights and may receive dividends, if any, prior to the time when the restrictions lapse.
Restricted stock units may be awarded to participants, typically without payment of consideration or for a nominal purchase price, but subject to vesting conditions including continued employment or performance criteria established by our compensation committee (or our board of directors, in the case of awards to non-employee directors). Like restricted stock, restricted stock units may not be sold or otherwise transferred or hypothecated until vesting conditions are removed or expire. Unlike restricted stock, stock underlying restricted stock units will not be issued until the restricted stock units have vested, and recipients of restricted stock units generally will have no voting or dividend rights prior to the time when vesting conditions are satisfied.
SARs granted under the 2013 Plan typically will provide for payments to the holder based upon increases in the price of our common stock over the exercise price of the SAR. Except as required by Section 162(m) of the Code with respect to SARs intended to qualify as performance-based compensation as described in Section 162(m) of the Code, there are no restrictions specified in the 2013 Plan on the exercise of SARs or the amount of gain realizable therefrom. Our compensation committee (or the board of directors, in the case of awards to non-employee directors) may elect to pay SARs in cash or in common stock or in a combination of both.
Dividend equivalents represent the value of the dividends, if any, per share paid by us, calculated with reference to the number of shares covered by the stock options, SARs or other awards held by the participant.
Performance awards may be granted by our compensation committee on an individual or group basis. Generally, these awards will be based upon the attainment of specific performance goals that are established by our compensation committee and relate to one or more performance criteria on a specified date or dates determined by our compensation committee. Any such cash bonus paid to a “covered employee” within the meaning of Section 162(m) of the Code may be, but need not be, qualified performance-based compensation as described below and will be paid in cash.
Stock payments may be authorized by our compensation committee (or our board of directors, in the case of awards to non-employee directors) in the form of common stock or an option or other right to purchase common stock as part of any bonus, deferred compensation or other arrangement, made in lieu of all or any part of compensation, that would otherwise be payable to employees, consultants or members of our board of directors.
Transferability of Awards. Unless the administrator provides otherwise, our 2013 Plan generally does not allow for the transfer of awards and only the recipient of an option or SAR may exercise such an award during his or her lifetime.
Qualified Performance-Based Compensation. The compensation committee may designate employees as “covered employees” whose compensation for a given fiscal year may be subject to the limit on deductible compensation imposed by Section 162(m) of the Code. The compensation committee may grant to such covered employees restricted stock, dividend equivalents, stock payments, restricted stock units, cash bonuses and other stock-based awards that are paid, vest or become exercisable upon the attainment of company performance criteria which are related to one or more of the following performance criteria as applicable to our performance or the performance of a division, business unit or an individual: operating or other costs and expenses, improvements in expense levels, cash flow (including, but not limited to, operating cash flow and free cash flow), return on assets, return on capital, stockholders’ equity, return on stockholders’ equity, total stockholder return, return on sales, gross or net profit or operating margin, working capital, net earnings (either before or after interest, taxes, depreciation and amortization), gross or net sales or revenue, net income (either before or after taxes), adjusted net income, operating earnings, earnings per share of stock, adjusted earnings per share of stock, price per share of stock, regulatory body approval for commercialization of a product, capital raised in financing transactions or other financing milestones, market recognition (including but not limited to awards and analyst ratings), financial ratios, implementation or completion of critical projects, market share, economic value, comparisons with various stock market indices, and implementation, completion or attainment of objectively determinable objectives relating to research, development, regulatory, commercial or strategic milestones or development. These performance criteria may be measured in absolute terms or as compared to performance in an earlier period or as compared to any incremental increase or decrease or as compared to results of a peer group or to market performance indicators or indices.
The compensation committee may provide that one or more objectively determinable adjustments will be made to one or more of the performance goals established for any performance period. Such adjustments may include one or more of the following: items related to a change in accounting principle, items relating to financing activities, expenses for restructuring or productivity initiatives, other non-operating items, items related to acquisitions, items attributable to the business operations of any entity acquired by us during the performance period, items related to the disposal of a business or segment of a business, items related to discontinued operations that do not qualify as a segment of a business under applicable accounting standards, items attributable to any stock dividend, stock split, combination or exchange of shares occurring during the performance period, any other items of significant income or expense which are determined to be appropriate adjustments, items relating to unusual or extraordinary corporate transactions, events or developments, items related to amortization of acquired intangible assets, items that are outside the scope of our core, on-going business activities, items related to acquired in-process research and development, items relating to changes in tax laws, items relating to major licensing or partnership arrangements, items relating to asset impairment charges, items relating to gains and losses for litigation, arbitration or contractual settlements, or items relating to any other unusual or nonrecurring events or changes in applicable laws, accounting principles or business conditions.
Forfeiture, Recoupment and Clawback Provisions. Pursuant to its general authority to determine the terms and conditions applicable to awards under the 2013 Plan, the compensation committee has the right to provide, in an award agreement or otherwise, that an award shall be subject to the provisions of any recoupment or clawback policies implemented by us, including, without limitation, any recoupment or clawback policies adopted to comply with the requirements of the Dodd-Frank Wall Street Reform and Consumer Protection Act and any rules or regulations promulgated thereunder.
Adjustments. If there is any stock dividend, stock split, combination or exchange of shares, merger, consolidation or other distribution (other than normal cash dividends) of our assets to stockholders, or any other change affecting the shares of our common stock or the share price of our common stock other than an equity restructuring (as defined in the 2013 Plan), the plan administrator may make such equitable adjustments, if any, as the plan administrator in its discretion may deem appropriate to reflect such change with respect to (1) the aggregate number and type of shares that may be issued under the 2013 Plan (including, but not limited to, adjustments of the number of shares available under the 2013 Plan and the maximum number of shares which may be subject to one or more awards to a participant pursuant to the 2013 Plan during any calendar year), (2) the number and kind of shares, or other securities or property, subject to outstanding awards, (3) the number and kind of shares, or other securities or property, for which automatic grants are to be subsequently made to new and continuing non-employee directors, (4) the terms and conditions of any outstanding awards (including, without limitation, any applicable performance targets or criteria with respect thereto), and (5) the grant or exercise price per share for any outstanding awards under the 2013 Plan. If there is any equity restructuring, (1) the number and type of securities subject to each outstanding award and the grant or exercise price per share for each outstanding award, if applicable, will be proportionately adjusted, and (2) the plan administrator will make proportionate adjustments to reflect such equity restructuring with respect to the aggregate number and type of shares that may be issued under the 2013 Plan (including, but not limited to, adjustments of the number of shares available under the 2013 Plan and the maximum number of shares which may be subject to one or more awards to a participant pursuant to the 2013 Plan during any calendar year). Adjustments in the event of an equity restructuring will not be discretionary. Any adjustment affecting an award intended as “qualified performance-based compensation” will be made consistent with the requirements of Section 162(m) of the Code. The plan administrator also has the authority under the 2013 Plan to take certain other actions with respect to outstanding awards in the event of a corporate transaction, including provision for the cash-out, termination, assumption or substitution of such awards.
Corporate Transactions. In the event of a change in control where the acquirer does not assume awards granted under the 2013 Plan, awards issued under the 2013 Plan will be subject to accelerated vesting such that 100% of the awards will become vested and exercisable or payable, as applicable. Under the 2013 Plan, a change in control is generally defined as:
a transaction or series of related transactions (other than an offering of our stock to the general public through a registration statement filed with the Securities and Exchange Commission, or SEC) whereby any person or entity or related group of persons or entities (other than us, our subsidiaries, an employee benefit plan maintained by us or any of our subsidiaries or a person or entity that, prior to such transaction, directly or indirectly controls, is controlled by, or is under common control with, us) directly or indirectly acquires beneficial ownership (within the meaning of Rule 13d-3 under the Exchange Act) of 50% or more of the total combined voting power of our securities outstanding immediately after such acquisition;
during any two-year period, individuals who, at the beginning of such period, constitute our board of directors together with any new director(s) whose election by our board of directors or nomination for election by our stockholders was approved by a vote of at least two-thirds of the directors then still in office who either were directors at the beginning of the two-year period or whose election or nomination for election was previously so approved, cease for any reason to constitute a majority of our board of directors;
our consummation (whether we are directly or indirectly involved through one or more intermediaries) of (x) a merger, consolidation, reorganization, or business combination or (y) the sale or other disposition of all or substantially all of our assets in any single transaction or series of transactions or (z) the acquisition of assets or stock of another entity, in each case other than a transaction:
which results in our voting securities outstanding immediately before the transaction continuing to represent (either by remaining outstanding or by being converted into our voting securities or the voting securities of the person that, as a result of the transaction, controls us, directly or indirectly, or owns, directly or indirectly, all or substantially all of our assets or otherwise succeeds to our business (we or such person being referred to as a successor entity)) directly or indirectly, at least 50% of the combined voting power of the successor entity’s outstanding voting securities immediately after the transaction; and
after which no person or group beneficially owns voting securities representing 50% or more of the combined voting power of the successor entity; provided, however, that no person or group is treated as beneficially owning 50% or more of combined voting power of the successor entity solely as a result of the voting power held in us prior to the consummation of the transaction; or
our stockholders approve a liquidation or dissolution of the company.
Amendment, Termination. Our board of directors has the authority to amend, suspend or terminate the 2013 Plan at any time. However, stockholder approval of any amendment to the 2013 Plan will be obtained to the extent necessary to comply with any applicable law, regulation or stock exchange rule. Additionally, stockholder approval is required within 12 months of an increase in the maximum number of shares issuable under the 2013 Plan or that may be issued to an individual in any calendar year. Except as necessary to comply with Section 409A of the Code, no amendment, suspension or termination of the 2013 Plan will impair the rights or obligations of a holder under an award theretofore granted, unless such award expressly so provides or such holder consents. If not terminated earlier by our board of directors, the 2013 Plan will terminate on the tenth anniversary of the date of its initial approval by our board of directors.
Repricing Permitted. Our compensation committee (or the board of directors, in the case of awards to non-employee directors) shall have the authority, without the approval of our stockholders, to authorize the amendment of any outstanding award to reduce its price per share and to provide that an award will be canceled and replaced with the grant of an award having a lesser price per share.
Securities Laws and Federal Income Taxes. The 2013 Plan is designed to comply with various securities and federal tax laws as follows:
Securities Laws. The 2013 Plan is intended to conform to all provisions of the Securities Act of 1933, as amended, and the Exchange Act and any and all regulations and rules promulgated by the SEC thereunder, including, without limitation, Rule 16b-3. The 2013 Plan will be administered, and awards will be granted and may be exercised, only in such a manner as to conform to such laws, rules and regulations.
Federal Income Tax Consequences. The material federal income tax consequences of the 2013 Plan under current federal income tax law are summarized in the following discussion, which deals with the general tax principles applicable to the 2013 Plan. The following discussion is based upon laws, regulations, rulings and decisions now in effect, all of which are subject to change. Foreign, state and local tax laws, and employment, estate and gift tax considerations are not discussed due to the fact that they may vary depending on individual circumstances and from locality to locality.
Stock Options and Stock Appreciation Rights. A 2013 Plan participant generally will not recognize taxable income and we generally will not be entitled to a tax deduction upon the grant of a stock option or stock appreciation right. The tax consequences of exercising a stock option and the subsequent disposition of the shares received upon exercise will depend upon whether the option qualifies as an ISO as defined in Section 422 of the Code. The 2013 Plan permits the grant of options that are intended to qualify as ISOs as well as options that are not intended to so qualify; however, ISOs generally may be granted only to our employees and employees of our parent or subsidiary corporations, if any. Upon exercising an option that does not qualify as an ISO when the fair market value of our stock is higher than the exercise price of the option, a 2013 Plan participant generally will recognize taxable income at ordinary income tax rates equal to the excess of the fair market value of the stock on the date of exercise over the purchase price, and we (or our subsidiaries, if any) generally will be entitled to a corresponding tax deduction for compensation expense, in the amount equal to the amount by which the fair market value of the shares purchased exceeds the purchase price for the shares. Upon a subsequent sale or other disposition of the option shares, the
| participant will recognize a short-term or long-term capital gain or loss in the amount of the difference between the sales price of the shares and the participant’s tax basis in the shares.
|
Upon exercising an ISO, a 2013 Plan participant generally will not recognize taxable income, and we will not be entitled to a tax deduction for compensation expense. However, upon exercise, the amount by which the fair market value of the shares purchased exceeds the purchase price will be an item of adjustment for alternative minimum tax purposes. The participant will recognize taxable income upon a sale or other taxable disposition of the option shares. For federal income tax purposes, dispositions are divided into two categories: qualifying and disqualifying. A qualifying disposition generally occurs if the sale or other disposition is made more than two years after the date the option was granted and more than one year after the date the shares are transferred upon exercise. If the sale or disposition occurs before these two periods are satisfied, then a disqualifying disposition generally will result.
Upon a qualifying disposition of ISO shares, the participant will recognize long-term capital gain in an amount equal to the excess of the amount realized upon the sale or other disposition of the shares over their purchase price. If there is a disqualifying disposition of the shares, then the excess of the fair market value of the shares on the exercise date (or, if less, the price at which the shares are sold) over their purchase price will be taxable as ordinary income to the participant. If there is a disqualifying disposition in the same year of exercise, it eliminates the item of adjustment for alternative minimum tax purposes. Any additional gain or loss recognized upon the disposition will be recognized as a capital gain or loss by the participant.
We will not be entitled to any tax deduction if the participant makes a qualifying disposition of ISO shares. If the participant makes a disqualifying disposition of the shares, we should be entitled to a tax deduction for compensation expense in the amount of the ordinary income recognized by the participant.
Upon exercising or settling a SAR, a 2013 Plan participant will recognize taxable income at ordinary income tax rates, and we should be entitled to a corresponding tax deduction for compensation expense, in the amount paid or value of the shares issued upon exercise or settlement. Payments in shares will be valued at the fair market value of the shares at the time of the payment, and upon the subsequent disposition of the shares the participant will recognize a short-term or long-term capital gain or loss in the amount of the difference between the sales price of the shares and the participant’s tax basis in the shares.
Restricted Stock and Restricted Stock Units. A 2013 Plan participant generally will not recognize taxable income at ordinary income tax rates and we generally will not be entitled to a tax deduction upon the grant of restricted stock or restricted stock units. Upon the termination of restrictions on restricted stock or the payment of restricted stock units, the participant will recognize taxable income at ordinary income tax rates, and we should be entitled to a corresponding tax deduction for compensation expense, in the amount paid to the participant or the amount by which the then fair market value of the shares received by the participant exceeds the amount, if any, paid for them. Upon the subsequent disposition of any shares, the participant will recognize a short-term or long-term capital gain or loss in the amount of the difference between the sales price of the shares and the participant’s tax basis in the shares. However, a 2013 Plan participant granted restricted stock that is subject to forfeiture or repurchase through a vesting schedule such that it is subject to a “risk of forfeiture” (as defined in Section 83 of the Code) may make an election under Section 83(b) of the Code to recognize taxable income at ordinary income tax rates, at the time of the grant, in an amount equal to the fair market value of the shares of common stock on the date of grant, less the amount paid, if any, for such shares. We will be entitled to a corresponding tax deduction for compensation, in the amount recognized as taxable income by the participant. If a timely Section 83(b) election is made, the participant will not recognize any additional ordinary income on the termination of restrictions on restricted stock, and we will not be entitled to any additional tax deduction.
Dividend Equivalents, Stock Payment Awards and Cash-Based Awards. A 2013 Plan participant will not recognize taxable income and we will not be entitled to a tax deduction upon the grant of dividend equivalents, stock payment awards or cash-based awards until cash or shares are paid or distributed to the participant. At that time, any cash payments or the fair market value of shares that the participant receives will be taxable to the participant at ordinary income tax rates and we should be entitled to a corresponding
| tax deduction for compensation expense. Payments in shares will be valued at the fair market value of the shares at the time of the payment, and upon the subsequent disposition of the shares, the participant will recognize a short-term or long-term capital gain or loss in the amount of the difference between the sales price of the shares and the participant’s tax basis in the shares.
|
Section 409A of the Code. Certain types of awards under the 2013 Plan may constitute, or provide for, a deferral of compensation under Section 409A. Unless certain requirements set forth in Section 409A are complied with, holders of such awards may be taxed earlier than would otherwise be the case (e.g., at the time of vesting instead of the time of payment) and may be subject to an additional 20% federal income tax (and, potentially, certain interest penalties). To the extent applicable, the 2013 Plan and awards granted under the 2013 Plan will be structured and interpreted to comply with Section 409A and the Department of Treasury regulations and other interpretive guidance that may be issued pursuant to Section 409A.
Section 162(m) Limitation. In general, under Section 162(m) of the Code, income tax deductions of publicly held corporations may be limited to the extent total compensation (including base salary, annual bonus, stock option exercises and non-qualified benefits paid) for certain executive officers exceeds $1 million (less the amount of any “excess parachute payments” as defined in Section 280G of the Code) in any one year. However, under Section 162(m), the deduction limit does not apply to certain “performance-based compensation” if an independent compensation committee determines performance goals and if the material terms of the performance-based compensation are disclosed to and approved by our stockholders. In particular, stock options and SARs will satisfy the “performance-based compensation” exception if the awards are made by a qualifying compensation committee, the plan sets the maximum number of shares that can be granted to any person within a specified period and the compensation is based solely on an increase in the stock price after the grant date. Specifically, the option exercise price must be equal to or greater than the fair market value of the stock subject to the award on the grant date. Under a Section 162(m) transition rule for compensation plans of corporations which are privately held and which become publicly held in an initial public offering, certain awards under the 2013 Plan will not be subject to Section 162(m) until a specified transition date, which is the earlier of (1) the material modification of the 2013 Plan, (2) the issuance of all employer stock and other compensation that has been allocated under the 2013 Plan, or (3) the first annual meeting of stockholders at which directors are to be elected that occurs after the close of the third calendar year following the calendar year in which the initial public offering occurs. After the transition date, rights or awards granted under the 2013 Plan, other than options and SARs, will not qualify as “performance-based compensation” for purposes of Section 162(m) unless such rights or awards are granted or vest upon pre-established objective performance goals, the material terms of which are disclosed to and approved by our stockholders.
We have attempted to structure the 2013 Plan in such a manner that, after the transition date, the compensation attributable to stock options and SARs which meet the other requirements of Section 162(m) will not be subject to the $1 million limitation. We have not, however, requested a ruling from the Internal Revenue Service or an opinion of counsel regarding this issue.
2006 Equity Incentive Plan
On March 27, 2006, our board of directors and our stockholders approved the Conatus Pharmaceuticals, Inc. 2006 Equity Incentive Plan, or the 2006 Plan.
As of March 31, 2013, a total of 8,500,000 shares of our common stock are reserved for issuance under the 2006 Plan. As of March 31, 2013, 4,899,000 shares of our common stock were subject to outstanding option awards and 69,500 shares of our common stock remained available for future issuance. After the effective date of the 2013 Plan, no additional awards will be granted under the 2006 Plan.
Administration. The compensation committee of our board of directors administers the 2006 Plan, except with respect to any award granted to non-employee directors (as defined in the 2006 Plan), which must be administered by our full board of directors. Subject to the terms and conditions of the 2006 Plan, the administrator has the authority to select the persons to whom awards are to be made, to determine the type or types of awards to be granted to each person, determine the number of awards to grant, determine the number of
shares to be subject to such awards, and the terms and conditions of such awards, and make all other determinations and decisions and to take all other actions necessary or advisable for the administration of the 2006 Plan. The plan administrator is also authorized to establish, adopt, amend or revise rules relating to administration of the 2006 Plan, subject to certain restrictions.
Eligibility. Options, SARs, restricted stock and other awards under the 2006 Plan may be granted to individuals who are then our employees, consultants and members of our board of directors or are employees, consultants or directors of our subsidiaries or parent entities. Only employees may be granted ISOs.
Awards. The 2006 Plan provides that our administrator may grant or issue stock options, restricted stock, restricted stock units, SARs, dividend equivalents, stock payments, or any combination thereof. The administrator considers each award grant subjectively, considering factors such as the individual performance of the recipient and the anticipated contribution of the recipient to the attainment of our long-term goals. Each award is set forth in a separate agreement with the person receiving the award and indicates the type, terms and conditions of the award.
NQSOs provide for the right to purchase shares of our common stock at a specified price which may not be less than the fair market value of a share of stock on the date of grant, and usually will become exercisable (at the discretion of our compensation committee or the board of directors, in the case of awards to non-employee directors) in one or more installments after the grant date, subject to the participant’s continued employment or service with us and/or subject to the satisfaction of performance targets established by our compensation committee (or the board of directors, in the case of awards to non-employee directors). NQSOs may be granted for any term specified by our compensation committee (or the board of directors, in the case of awards to non-employee directors), but the term may not exceed ten years.
ISOs are designed to comply with the provisions of the Internal Revenue Code and are subject to specified restrictions contained in the Internal Revenue Code applicable to ISOs. Among such restrictions, ISOs must have an exercise price of not less than the fair market value of a share of common stock on the date of grant, may only be granted to employees, must expire within a specified period of time following the optionee’s termination of employment, and must be exercised within the ten years after the date of grant. In the case of an ISO granted to an individual who owns (or is deemed to own) more than 10% of the total combined voting power of all classes of our capital stock on the date of grant, the 2006 Plan provides that the exercise price must be at least 110% of the fair market value of a share of common stock on the date of grant and the ISO must expire on the fifth anniversary of the date of its grant.
Restricted stock may be granted to participants and made subject to such restrictions as may be determined by the administrator. Typically, restricted stock may be repurchased by us at the original purchase price or, if no cash consideration was paid for such stock, forfeited for no consideration if the conditions or restrictions are not met, and the restricted stock may not be sold or otherwise transferred to third parties until restrictions are removed or expire. Recipients of restricted stock, unlike recipients of options, may have voting rights and may receive dividends, if any, prior to when the restrictions lapse.
Restricted stock units may be awarded to participants, typically without payment of consideration or for a nominal purchase price, but subject to vesting conditions including continued employment or performance criteria established by the administrator. Like restricted stock, restricted stock units may not be sold or otherwise transferred or hypothecated until vesting conditions are removed or expire. Unlike restricted stock, stock underlying restricted stock units will not be issued until some time after the restricted stock units have vested, and recipients of restricted stock units generally will have no voting or dividend rights prior to the time when vesting conditions are satisfied and the shares have been issued.
SARs typically will provide for payments to the holder based upon increases in the price of our common stock over the exercise price of the SAR. There are no restrictions specified in the 2006 Plan on the exercise of SARs or the amount of gain realizable therefrom. Subject to certain terms and conditions of the 2006 Plan, the administrator may elect to pay SARs in cash or in common stock or in a combination of both.
Dividend equivalents may be awarded to participants and represent the value of the dividends, if any, per share paid by us, calculated with reference to the number of shares covered by the stock options, SARs or other awards held by the participant.
Stock payments may be authorized by the administrator in the form of common stock or an option or other right to purchase common stock as part of any bonus, deferred compensation or other arrangement, made in lieu of all or any part of compensation, that would otherwise be payable to employees, consultants or members of our board of directors.
Corporate Transactions. In the event of a change of control where the acquiror does not assume awards granted under the 2006 Plan, awards issued under the 2006 Plan will be subject to accelerated vesting such that 100% of the awards will become vested and exercisable or payable, as applicable, immediately prior to the change in control. Under the 2006 Plan, a change of control is generally defined as:
a transaction or series of related transactions whereby any person or entity or related group of persons or entities (other than us, our subsidiaries, an employee benefit plan maintained by us or any of our subsidiaries or a person or entity that, prior to such transaction, directly or indirectly controls, is controlled by, or is under common control with, us) directly or indirectly acquires beneficial ownership (within the meaning of Rule 13d-3 under the Exchange Act) of 50% or more of the total combined voting power of our securities outstanding immediately after such acquisition;
during any two-year period, individuals who, at the beginning of such period, constitute our board of directors together with any new director(s) whose election by our board of directors or nomination for election by our stockholders was approved by a vote of at least two-thirds of the directors then still in office who either were directors at the beginning of the two-year period or whose election or nomination for election was previously so approved, cease for any reason to constitute a majority of our board of directors;
our consummation (whether we are directly or indirectly involved through one or more intermediaries) of (1) a merger, consolidation, reorganization, or business combination, (2) the sale or other disposition of all or substantially all of our assets or (3) the acquisition of assets or stock of another entity, in each case other than a transaction that results in our voting securities outstanding immediately before the transaction continuing to represent, directly or indirectly, at least 50% of the combined voting power of the successor entity’s outstanding voting securities immediately after the transaction, and after which no person or entity beneficially owns voting securities representing 50% or more of the combined voting power of the acquiring company that is not attributable to voting power held in the company prior to such transaction; or
the approval by our stockholders of a liquidation or dissolution of our company.
Amendment and Termination of the 2006 Plan. Our board of directors may terminate, amend or modify the 2006 Plan. However, stockholder approval of any amendment to the 2006 Plan must be obtained to the extent necessary and desirable to comply with any applicable law, regulation or stock exchange rule, or for any amendment to the 2006 Plan that increases the number of shares available under the 2006 Plan. The administrator may, with the consent of the affected option holders, cancel any or all outstanding options under the 2006 Plan and grant new options in substitution. If not terminated earlier by the compensation committee or the board of directors, the 2006 Plan will terminate on March 26, 2016.
Securities Laws and Federal Income Taxes. The 2006 Plan is designed to comply with applicable securities laws in the same manner as described above in the description of the 2013 Plan under the heading “—2013 Equity Incentive Award Plan—Securities Laws and Federal Income Taxes—Securities Laws.” The general federal tax consequences of awards under the 2006 Plan are the same as those described above in the description of the 2013 Plan under the heading “—2013 Equity Incentive Award Plan—Securities Laws and Federal Income Taxes—Federal Income Taxes.”
2013 Employee Stock Purchase Plan
Concurrently with this offering, we intend to establish the Conatus Pharmaceuticals, Inc. 2013 Employee Stock Purchase Plan, or the ESPP. We expect our board of directors to adopt, and our stockholders to approve, the ESPP prior to the completion of this offering. The ESPP will become effective on the business day prior to the public trading date of our common stock. Our executive officers and all of our other employees will be allowed to participate in our ESPP, subject to the eligibility requirements described below. The material terms of the ESPP, as it is currently contemplated, are summarized below. Our board of directors is still in the process of developing, approving and implementing the ESPP and, accordingly, this summary is subject to change.
A total of shares of our common stock will initially be reserved for issuance under our ESPP. In addition, the number of shares available for issuance under the ESPP will be annually increased on the first day of each fiscal year during the term of the ESPP, beginning with the 2014 fiscal year, by an amount equal to the least of:
% of the outstanding shares of our common stock as of the last day of our immediately preceding fiscal year; or
such other amount as may be determined by our board of directors.
The ESPP will also provide for an aggregate limit of shares of common stock that may be issued under the ESPP during the term of the ESPP.
Our board of directors or its committee has full and exclusive authority to interpret the terms of the ESPP and determine eligibility. We expect that our compensation committee will be the initial administrator of the ESPP.
Our employees are eligible to participate in the ESPP if they are customarily employed by us or any participating subsidiary for at least 20 hours per week and more than five months in any calendar year. However, an employee may not be granted rights to purchase stock under our ESPP if such employee, immediately after the grant, would own (directly or through attribution) stock possessing 5% or more of the total combined voting power or value of all classes of our common or other class of stock.
Our ESPP is intended to qualify under Code Section 423 and stock will be offered under the ESPP during offering periods. The length of the offering periods under the ESPP will be determined by our compensation committee and may be up to 27 months long. Employee payroll deductions will be used to purchase shares on each purchase date during an offering period. The purchase dates will be determined by the compensation committee for each offering period, but will generally be the last day in each offering period. Offering periods under the ESPP will commence when determined by our compensation committee. The compensation committee may, in its discretion, modify the terms of future offering periods.
Our ESPP permits participants to purchase common stock through payroll deductions of up to % of their eligible compensation, which includes a participant’s gross base compensation for services to the company, excluding overtime payments, sales commissions, incentive compensation, bonuses, expense reimbursements, fringe benefits and other special payments. A participant may purchase a maximum of shares of common stock during each offering period. In addition, no employee will be permitted to accrue the right to purchase stock under the ESPP at a rate in excess of $25,000 worth of shares during any calendar year during which such a purchase right is outstanding (based on the fair market value per share of our common stock as of the first day of the offering period).
On the first trading day of each offering period, each participant automatically is granted an option to purchase shares of our common stock. The option expires at the end of the offering period or upon termination of employment, whichever is earlier, but is exercised at the end of each purchase period to the extent of the payroll deductions accumulated during such purchase period. The purchase price of the shares will be 85% of the lower of the fair market value of our common stock on the first trading day of the offering period or on the applicable purchase date. Participants may end their participation at any time during an offering period, and will be paid their accrued payroll deductions that have not yet been used to purchase shares of common stock. Participation ends automatically upon termination of employment with us.
A participant may not transfer rights granted under the ESPP other than by will, the laws of descent and distribution or as otherwise provided under the ESPP.
In the event of certain significant transactions or a change in control (as defined in the ESPP), the compensation committee may provide for: (1) either the replacement or termination of outstanding rights in exchange for cash; (2) the assumption or substitution of outstanding rights by the successor or survivor corporation or parent or subsidiary thereof, if any; (3) the adjustment in the number and type of shares of stock subject to outstanding rights; (4) the use of participants’ accumulated payroll deductions to purchase stock on a new purchase date prior to the next purchase date and termination of any rights under ongoing offering periods;
or (5) the termination of all outstanding rights. Under the ESPP, a change in control has the same definition as given to such term in the 2013 Plan.
The compensation committee may amend, suspend or terminate the ESPP. However, stockholder approval of any amendment to the ESPP will be obtained for any amendment which changes the aggregate number or type of shares that may be sold pursuant to rights under the ESPP, changes the corporations or classes of corporations whose employees are eligible to participate in the ESPP or changes the ESPP in any manner that would cause the ESPP to no longer be an employee stock purchase plan within the meaning of Section 423(b) of the Code. The ESPP will terminate no later than the tenth anniversary of the ESPP’s initial adoption by our board of directors.
Securities Laws. The ESPP has been designed to comply with various securities laws in the same manner as described above in the description of the 2013 Plan.
Federal Income Taxes. The material federal income tax consequences of the ESPP under current federal income tax law are summarized in the following discussion, which deals with the general tax principles applicable to the ESPP. The following discussion is based upon laws, regulations, rulings and decisions now in effect, all of which are subject to change. Foreign, state and local tax laws, and employment, estate and gift tax considerations are not discussed due to the fact that they may vary depending on individual circumstances and from locality to locality.
The ESPP, and the right of participants to make purchases thereunder, is intended to qualify under the provisions of Section 423 of the Code. Under the applicable Code provisions, no income will be taxable to a participant until the sale or other disposition of the shares purchased under the ESPP. This means that an eligible employee will not recognize taxable income on the date the employee is granted an option under the ESPP (i.e., the first day of the offering period). In addition, the employee will not recognize taxable income upon the purchase of shares. Upon such sale or disposition, the participant will generally be subject to tax in an amount that depends upon the length of time such shares are held by the participant prior to disposing of them. If the shares are sold or disposed of more than two years from the first day of the offering period during which the shares were purchased and more than one year from the date of purchase, or if the participant dies while holding the shares, the participant (or his or her estate) will recognize ordinary income measured as the lesser of: (1) the excess of the fair market value of the shares at the time of such sale or disposition over the purchase price; or (2) an amount equal to 15% of the fair market value of the shares as of the first day of the offering period. Any additional gain will be treated as long-term capital gain. If the shares are held for the holding periods described above but are sold for a price that is less than the purchase price, there is no ordinary income and the participating employee has a long-term capital loss for the difference between the sale price and the purchase price.
If the shares are sold or otherwise disposed of before the expiration of the holding periods described above, the participant will recognize ordinary income generally measured as the excess of the fair market value of the shares on the date the shares are purchased over the purchase price and we will be entitled to a tax deduction for compensation expense in the amount of ordinary income recognized by the employee. Any additional gain or loss on such sale or disposition will be long-term or short-term capital gain or loss, depending on how long the shares were held following the date they were purchased by the participant prior to disposing of them. If the shares are sold or otherwise disposed of before the expiration of the holding periods described above but are sold for a price that is less than the purchase price, the participant will recognize ordinary income equal to the excess of the fair market value of the shares on the date of purchase over the purchase price (and we will be entitled to a corresponding deduction), but the participant generally will be able to report a capital loss equal to the difference between the sales price of the shares and the fair market value of the shares on the date of purchase.
Limitations of Liability and Indemnification Matters
Our amended and restated certificate of incorporation and our amended and restated bylaws provide that we will indemnify our directors and officers to the fullest extent permitted by the Delaware General Corporation Law, which prohibits our amended and restated certificate of incorporation from limiting the liability of our directors for the following:
any breach of the director’s duty of loyalty to us or our stockholders;
acts or omissions not in good faith or that involve intentional misconduct or a knowing violation of law;
unlawful payment of dividends or unlawful stock repurchases or redemptions; or
any transaction from which the director derived an improper personal benefit.
Our amended and restated certificate of incorporation and our amended and restated bylaws also provide that if Delaware law is amended to authorize corporate action further eliminating or limiting the personal liability of a director, then the liability of our directors will be eliminated or limited to the fullest extent permitted by Delaware law, as so amended. This limitation of liability does not apply to liabilities arising under the federal securities laws and does not affect the availability of equitable remedies such as injunctive relief or rescission.
Our amended and restated certificate of incorporation and our amended and restated bylaws also provide that we shall have the power to indemnify our employees and agents to the fullest extent permitted by law. Our amended and restated bylaws also permit us to secure insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in this capacity, regardless of whether our amended and restated bylaws would permit indemnification. We have obtained directors’ and officers’ liability insurance.
We have entered into separate indemnification agreements with our directors and executive officers, in addition to indemnification provided for in our amended and restated certificate of incorporation and amended and restated bylaws. These agreements, among other things, provide for indemnification of our directors and executive officers for expenses, judgments, fines and settlement amounts incurred by this person in any action or proceeding arising out of this person’s services as a director or executive officer or at our request. We believe that these provisions in our amended and restated certificate of incorporation and amended and restated bylaws and indemnification agreements are necessary to attract and retain qualified persons as directors and executive officers.
The above description of the indemnification provisions of our amended and restated certificate of incorporation, our amended and restated bylaws and our indemnification agreements is not complete and is qualified in its entirety by reference to these documents, each of which is incorporated by reference as an exhibit to this registration statement to which this prospectus forms a part.
The limitation of liability and indemnification provisions in our amended and restated certificate of incorporation and amended and restated bylaws may discourage stockholders from bringing a lawsuit against directors for breach of their fiduciary duties. They may also reduce the likelihood of derivative litigation against directors and officers, even though an action, if successful, might benefit us and our stockholders. A stockholder’s investment may be harmed to the extent we pay the costs of settlement and damage awards against directors and officers pursuant to these indemnification provisions. Insofar as indemnification for liabilities under the Securities Act may be permitted to directors, officers or persons controlling us pursuant to the foregoing provisions, we have been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable. There is no pending litigation or proceeding naming any of our directors or officers as to which indemnification is being sought, nor are we aware of any pending or threatened litigation that may result in claims for indemnification by any director or officer.
CERTAIN RELATIONSHIPS AND RELATED PERSON TRANSACTIONS
The following includes a summary of transactions since January 1, 2010 to which we have been a party in which the amount involved exceeded or will exceed $120,000, and in which any of our directors, executive officers or, to our knowledge, beneficial owners of more than 5% of our capital stock or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest, other than equity and other compensation, termination, change in control and other arrangements, which are described under “Executive and Director Compensation.” We also describe below certain other transactions with our directors, executive officers and stockholders.
Preferred Stock Financings and Convertible Notes and Warrant Financing
Series A Convertible Preferred Stock Financing. From October 2006 through December 2006 and in May 2007, we issued and sold to investors in private placements an aggregate of 42,494,218 shares of our Series A convertible preferred stock at a purchase price of $0.75 per share, for aggregate consideration of approximately $31.9 million.
Series B Convertible Preferred Stock Financing. In February and March 2011, we issued and sold to investors in private placements an aggregate of 36,417,224 shares of our Series B convertible preferred stock at a purchase price of $0.90 per share, for aggregate consideration of approximately $32.8 million.
Convertible Note and Warrant Financings. In March 2010, we entered into a note and warrant purchase agreement with certain existing investors pursuant to which we sold, in private placements in March 2010 and October 2010, an aggregate of $5.0 million of convertible promissory notes, or the 2010 Notes, and issued warrants exercisable to purchase shares of our Series A convertible preferred stock, or the 2010 Warrants. The 2010 notes accrued interest at a rate of 8% per annum and were due and payable on the earlier of (1) any date after July 31, 2011 upon which holders of 66 2/3% of the outstanding principal amount and accrued interest on all such 2010 Notes demanded repayment, or (2) the occurrence of a change of control of the company, subject in each case to their earlier conversion in the event we completed a qualified equity financing. In connection with the Series B financing, the principal amount of the 2010 Notes and accrued interest thereon was automatically converted into an aggregate of 5,861,667 shares of our Series B convertible preferred stock in February 2011. The 2010 Warrants are exercisable for an aggregate of 2,333,320 shares of Series A convertible preferred stock at an exercise price of $0.01 per share. We expect these warrants to be net exercised in connection with the completion of this offering, resulting in the issuance of an aggregate of shares of common stock, assuming an initial public offering price of $ per share (the midpoint of the price range set forth on the cover page of this prospectus). If these warrants are not exercised prior to the completion of this offering, they will terminate.
In May 2013, we entered into a note and warrant purchase agreement with certain existing investors pursuant to which we sold, in a private placement in May 2013, an aggregate of $1.0 million of convertible promissory notes, or the 2013 Notes, and issued warrants exercisable to purchase shares of our Series B convertible preferred stock, or the 2013 Warrants. The 2013 Notes accrue interest at a rate of 6% per annum and are due and payable on the earlier of (1) any date after November 30, 2013 upon which holders of 75% of the outstanding principal amount of all such 2013 Notes demand repayment, or (2) the occurrence of a change of control of the company, subject in each case to their earlier conversion in the event we complete a qualified initial public offering or private placement of debt and/or equity. In connection with the completion of this offering, the 2013 Notes (including accrued interest thereon) will automatically convert into shares of common stock, assuming an initial public offering price of $ per share (the midpoint of the price range listed on the cover page of this prospectus) and assuming the conversion occurs on , 2013 (the expected closing date of this offering). The 2013 Warrants are exercisable for an aggregate of 1,124,026 shares of Series B convertible preferred stock at an exercise price of $0.90 per share. In connection with the completion of this offering, the 2013 Warrants will become exercisable for an aggregate of shares of common stock, at an exercise price of $ per share. The 2013 Warrants will expire on May 30, 2018.
The following table sets forth the aggregate number of these securities acquired by the listed directors, executive officers or holders of more than 5% of our capital stock, or their affiliates. Each share of, or warrants exercisable for shares of, convertible preferred stock identified in the following table will convert into one share of, or warrants exercisable for, common stock upon completion of this offering, except as described below.
| | | | | | | | | | | | | | | | | | | | | | | | |
Participants | | Series A
Convertible
Preferred Stock | | | Series B
Convertible
Preferred Stock | | | Series A
Warrants | | | Principal
Amount of
2010 Notes | | | Series B
Warrants | | | Principal
Amount of
2013 Notes | |
5% or Greater Stockholders(1) | | | | | | | | | | | | | | | | | | | | | | | | |
Entities affiliated with Aberdare Ventures(2) | | | 10,729,941 | | | | 5,983,862 | | | | 619,766 | | | $ | 1,328,075 | | | | 43,333 | | | $ | 118,336 | |
Entities affiliated with Advent Private Equity(3) | | | 10,000,000 | | | | 5,576,786 | | | | 577,599 | | | $ | 1,237,728 | | | | 76,667 | | | $ | 209,364 | |
Entities affiliated with Bay City Capital(4) | | | 10,000,000 | | | | 5,576,789 | | | | 577,604 | | | $ | 1,237,728 | | | | — | | | | — | |
Coöperative Gilde Healthcare II U.A. | | | 6,666,668 | | | | 3,717,861 | | | | 385,070 | | | $ | 825,152 | | | | 222,569 | | | $ | 182,340 | |
Entities affiliated with MPM Capital(5) | | | — | | | | 8,333,334 | | | | — | | | | — | | | | 265,837 | | | $ | 182,056 | |
AgeChem Venture Fund L.P. | | | — | | | | 5,555,556 | | | | — | | | | — | | | | 214,262 | | | $ | 136,542 | |
Entities affiliated with Roche Finance Ltd(6) | | | 2,999,999 | | | | 1,673,036 | | | | 173,281 | | | $ | 371,318 | | | | 277,622 | | | $ | 154,748 | |
| | | | | | |
Executive Officers and Directors(1) | | | | | | | | | | | | | | | | | | | | | | | | |
Steven J. Mento, Ph.D.(7) | | | 786,417 | | | | — | | | | — | | | | — | | | | 20,256 | | | $ | 15,202 | |
Charles J. Cashion(8) | | | 355,352 | | | | — | | | | — | | | | — | | | | — | | | | — | |
Alfred P. Spada, Ph.D. (9) | | | 363,927 | | | | — | | | | — | | | | — | | | | — | | | | — | |
David F. Hale(10) | | | 162,356 | | | | — | | | | — | | | | — | | | | 3,480 | | | $ | 2,851 | |
(1) | Additional details regarding these stockholders and their equity holdings are provided in “Principal Stockholders.”
|
(2) | Represents securities acquired by Aberdare Ventures III, L.P. and Aberdare Partners III, L.P.
|
(3) | Represents securities acquired by Advent Private Equity Fund III ‘A’, Advent Private Equity Fund III ‘B’, Advent Private Equity Fund III ‘C’, Advent Private Equity Fund III ‘D’, Advent Private Equity Fund III GmbH & Co KG, Advent Private Equity Fund III Affiliates, Advent Management III Limited Partnership, Advent Private Equity Fund IV and Advent Management IV Limited Partnership.
|
(4) | Represents securities acquired by Bay City Capital Fund IV Co-Investment Fund, L.P. and Bay City Capital Fund IV, L.P. The convertible preferred stock purchased by such entities was converted into 1,557,678 shares of common stock in May 2013, reducing such entities’ aggregate ownership of our capital stock to less than 5%.
|
(5) | Represents securities acquired by MPM BioVentures IV-QP, L.P., MPM BioVentures IV GmbH & Co. Beteiligungs KG, MPM Asset Management Investors BV4 LLC, MPM BioVentures V, L.P. and MPM Asset Management Investors BV5 LLC.
|
(6) | Represents securities acquired by Roche Finance Ltd and Roche Holdings, Inc.
|
(7) | Represents securities acquired by a family trust.
|
(8) | Represents securities acquired by a family trust.
|
(9) | Represents securities acquired by a family trust.
|
(10) | Represents securities acquired by Hale BioPharma Ventures LLC.
|
Some of our directors are associated with our principal stockholders as indicated in the table below:
| | |
Director
| | Principal Stockholder
|
| |
Paul H. Klingenstein
| | Entities affiliated with Aberdare Ventures
|
| |
Louis Lacasse
| | AgeChem Venture Fund L.P.
|
| |
Shahzad Malik, M.D.
| | Entities affiliated with Advent Private Equity
|
| |
Marc Perret
| | Coöperative Gilde Healthcare II U.A.
|
| |
James Scopa
| | Entities affiliated with MPM Capital
|
Idun Pharmaceuticals, Inc.
In January 2013, we conducted a spin-off of our former subsidiary, Idun Pharmaceuticals, Inc., or Idun, to our stockholders at that time. Immediately prior to the spin-off, all rights relating to emricasan were distributed from Idun to us pursuant to a distribution agreement. The assets remaining in Idun at the time of the spin-off consisted solely of intellectual property rights and license and collaboration agreements unrelated to emricasan.
The spin-off was conducted as a dividend of all of the outstanding capital stock of Idun to our stockholders and, as a result, we no longer own any capital stock of Idun. The aggregate value of Idun at the time of the spin-off was deemed to be $9.6 million based on the valuation of an independent appraisal firm. Because the dividend was taxable to our stockholders and required us to withhold amounts with respect to certain of our non-U.S. stockholders, we agreed to advance the withholding amounts for these non-U.S. stockholders in exchange for promissory notes of equal amount.
Also in connection with the spin-off, we contributed $500,000 to Idun to provide for its initial working capital requirements and entered into a transition services agreement to provide operating services to Idun, generally consisting of accounting support, technology license administration and intellectual property maintenance. Idun must pay us for all direct costs as well as overhead and general and administrative expenses incurred in performing these services. As of March 31, 2013, Idun has not made any payment to us for services provided under the transition services agreement. The initial term of the transition services agreement ends on December 31, 2013, and will renew automatically for successive one year periods until terminated by either Idun or us upon 90 days’ prior notice to the other party.
In March 2013, we entered into a sublicense agreement with Idun in which we were granted the right to use the patent rights and know-how related to the screening and identification of emricasan. These rights were previously granted to Idun under license agreements with Thomas Jefferson University, or TJU. Under the sublicense, we are required to pay directly to TJU a royalty of less than one percent on net sales of emricasan. We also have the right to grant further sublicenses to third parties and are required to pay TJU a portion of any such sublicense revenue we receive. The sublicense agreement will expire upon the date which there are no longer any valid claims in any patents or patent applications sublicensed to us, unless earlier terminated. Idun may terminate the agreement upon a material uncured default. Each of Drs. Mento and Spada and Mr. Cashion continue to serve as officers of Idun in the same capacity as their officer positions with us, and all of our directors currently serve as directors of Idun.
Investor Rights Agreement
We entered into a first amended and restated investor rights agreement in February 2011 with the holders of our convertible preferred stock, including entities with which certain of our directors are affiliated. This agreement provides for certain rights relating to the registration of their shares of common stock and common stock issuable upon conversion of their convertible preferred stock, a right of first refusal to purchase future securities sold by us and certain additional covenants made by us. Except for the registration rights (including the related provisions pursuant to which we have agreed to indemnify the parties to the investor rights agreement), all rights under this agreement will terminate upon completion of this offering. The registration rights will continue following this offering and will terminate seven years following the completion of this offering, or for any particular holder with registration rights, at such time following this offering when all securities held by that stockholder subject to registration rights may be sold pursuant to Rule 144 under the Securities Act in a three-month period. See “Description of Capital Stock—Registration Rights” for additional information.
Voting Agreement
We entered into a first amended and restated voting agreement in February 2011 by and among us and certain of our stockholders, pursuant to which the following directors were each elected to serve as members on our board of directors and, as of the date of this prospectus, continue to so serve: Drs. Malik, Mento and Van Wart and Messrs. Hale, Klingenstein, Lacasse, Perret and Scopa. Pursuant to the voting agreement, Dr. Mento, as our Chief Executive Officer, was initially selected to serve on our board of directors as a representative of holders of our common stock, as designated by a majority of our common stockholders. Dr. Malik and Messrs.
Klingenstein, Lacasse, Perret and Scopa were initially selected to serve on our board of directors as representatives of holders of our convertible preferred stock, as designated by Advent Private Equity Fund IV, Aberdare Ventures III, L.P., AgeChem Venture Fund L.P., Coöperative Gilde Healthcare II U.A. and MPM BioVentures V, L.P., respectively. Mr. Hale and Dr. Van Wart were initially selected to serve on our board of directors as designated by a majority of our common and convertible preferred stock holders, voting together as a single class.
The voting agreement will terminate upon the closing of this offering, and members previously elected to our board of directors pursuant to this agreement will continue to serve as directors until they resign, are removed or their successors are duly elected by holders of our common stock. The composition of our board of directors after this offering is described in more detail under “Management—Board Composition and Election of Directors.”
Participation in this Offering
Entities affiliated with Aberdare Ventures, Advent Private Equity, Coöperative Gilde Healthcare II U.A., MPM Capital, Roche Finance Ltd, AgeChem Venture Fund L.P., Hale BioPharma Ventures LLC and our chief executive officer, each of which is a current stockholder, have indicated an interest in purchasing an aggregate of approximately $10.0 million of shares of our common stock in this offering. However, because indications of interest are not binding agreements or commitments to purchase, the underwriters may determine to sell more, less or no shares in this offering to any of these entities, or any of these entities may determine to purchase more, less or no shares in this offering.
Employment Agreements
We have entered into employment agreements with our named executive officers. For more information regarding these agreements, see the section in this prospectus entitled “Executive and Director Compensation—Narrative Disclosure to Compensation Tables.”
Indemnification Agreements
We have entered into indemnification agreements with each of our directors and executive officers prior to the closing of this offering. These agreements, among other things, require us or will require us to indemnify each director (and in certain cases their related venture capital funds) and executive officer to the fullest extent permitted by Delaware law, including indemnification of expenses such as attorneys’ fees, judgments, fines and settlement amounts incurred by the director or executive officer in any action or proceeding, including any action or proceeding by or in right of us, arising out of the person’s services as a director or executive officer.
Our amended and restated certificate of incorporation and our amended and restated bylaws provide that we will indemnify each of our directors and officers to the fullest extent permitted by the Delaware General Corporation Law. Further, we have entered into indemnification agreements with each of our directors and officers, and we have purchased a policy of directors’ and officers’ liability insurance that insures our directors and officers against the cost of defense, settlement or payment of a judgment under certain circumstances. For further information, see “Executive and Director Compensation—Limitations of Liability and Indemnification Matters.”
Stock Option Grants to Executive Officers and Directors
We have granted stock options to our executive officers and certain of our directors as more fully described in the section entitled “Executive and Director Compensation.”
Policies and Procedures for Related Person Transactions
Our board of directors has adopted a written related person transaction policy, to be effective upon the consummation of this offering, setting forth the policies and procedures for the review and approval or ratification of related-person transactions. This policy will cover, with certain exceptions set forth in Item 404 of Regulation S-K under the Securities Act, any transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships in which we were or are to be a participant, where the amount involved exceeds $120,000 and a related person had or will have a direct or indirect material interest, including,
without limitation, purchases of goods or services by or from the related person or entities in which the related person has a material interest, indebtedness, guarantees of indebtedness and employment by us of a related person. In reviewing and approving any such transactions, our audit committee is tasked to consider all relevant facts and circumstances, including, but not limited to, whether the transaction is on terms comparable to those that could be obtained in an arm’s length transaction and the extent of the related person’s interest in the transaction. All of the transactions described in this section occurred prior to the adoption of this policy.
PRINCIPAL STOCKHOLDERS
The following table sets forth certain information with respect to the beneficial ownership of our commoncapital stock as of June 12, 2013, and as adjusted to reflect the saleSeptember 30, 2020 by:
each of shares of common stock in this offering, by:our directors;
each of our named executive officers;
all of our current directors and executive officers and directors as a group; and
each person, or group of affiliated persons, known by us to beneficially own more than 5% of our common stock.
The numberBeneficial ownership has been determined in accordance with the rules of shares beneficially owned by each stockholderthe SEC and the information is determined under rules issued by the SEC. Under these rules, beneficial ownership includes any shares as to which a person has sole or shared voting power or investment power. Applicable percentage ownership is based on 76,190,081 shares of common stock outstanding on June 12, 2013, which gives effect to the conversion of all outstanding shares of convertible preferred stock into shares of common stockand the conversion of all outstanding warrants to purchase shares of our convertible preferred stock into warrants to purchase shares of our common stock. Our calculationnot necessarily indicative of beneficial ownership after the offering gives additional effect to (1) the issuance of shares of our common stock in connection with the completion of this offering as a result of the automatic conversion of the $1.0 million in aggregate principal amount of convertible promissory notes we issued in May 2013, or the 2013 Notes (including accrued interest thereon), assuming an initial public offering price of $ per share (the midpoint of the price range listed on the cover page of this prospectus) and assuming the conversion occurs on , 2013 (the expected closing date of this offering), and (2) the issuance of shares of common stock as a result of the expected net exercise of outstanding warrants, or the 2010 Warrants, in connection with the completion of this offering, assuming an initial public offering price of $ �� per share (the midpoint of the price range set forth on the cover page of this prospectus), which 2010 Warrants will terminate if unexercised prior to the completion of this offering. In computing the number of shares beneficially owned by a person and the percentage ownership of that person, shares of common stock subject to options, warrants or other rights held by such person that are currently exercisable or will become exercisable within 60 days of June 12, 2013 are considered outstanding, although these shares are not considered outstanding for purposes of computing the percentage ownership of any other person.
Entities affiliated with Aberdare Ventures, Advent Private Equity, Coöperative Gilde Healthcare II U.A., MPM Capital, Roche Finance Ltd, AgeChem Venture Fund L.P., Hale BioPharma Ventures LLC andpurpose. Unless otherwise indicated below, to our chief executive officer, each of which is a current stockholder, have indicated an interest in purchasing an aggregate of approximately $10.0 million of shares of our common stock in this offering. The information set forthknowledge, the persons or entities identified in the table below does not reflect the potential purchase of any shares in this offering by these stockholders.
Unless otherwise indicated, the address of each beneficial owner listed below is c/o Conatus Pharmaceuticals Inc., 4365 Executive Drive, Suite 200, San Diego, CA 92121. We believe, based on information provided to us, that each of the stockholders listed below hashave sole voting and sole investment power with respect to theall shares known as beneficially owned, by the stockholder unless noted otherwise, subject to community property laws where applicable.
| | | | | | | | | | | | | | |
| | | Shares Beneficially Owned
Prior to Offering | | | Shares Beneficially Owned
After Offering | |
Name of Beneficial Owner | | Number | | | Percentage | | | Number | | Percentage | |
5% or Greater Stockholders | | | | | | | | | | | | | | |
Entities affiliated with Aberdare Ventures(1) | | | 17,876,902 | | | | 23.3 | % | | | | | % | |
One Embarcadero Center, Suite 4000 | | | | | | | | | | | | | | |
San Francisco, CA 94111 | | | | | | | | | | | | | | |
| | | | |
Entities affiliated with Advent Private Equity(2) | | | 16,231,052 | | | | 21.1 | % | | | | | % | |
25 Buckingham Gate | | | | | | | | | | | | | | |
London, United Kingdom | | | | | | | | | | | | | | |
SW1E 6LD | | | | | | | | | | | | | | |
| | | | |
Coöperative Gilde Healthcare II U.A. (3) | | | 10,922,168 | | | | 14.3 | % | | | | | % | |
Newtonlaan 91 | | | | | | | | | | | | | | |
P.O. Box 85067 | | | | | | | | | | | | | | |
3508 AB Utrecht | | | | | | | | | | | | | | |
The Netherlands | | | | | | | | | | | | | | |
| | | | |
Entities affiliated with MPM Capital(4) | | | 8,599,171 | | | | 11.2 | % | | | | | % | |
c/o MPM Asset Management | | | | | | | | | | | | | | |
200 Clarendon Street, 54th Floor | | | | | | | | | | | | | | |
Boston, MA 02116 | | | | | | | | | | | | | | |
| | | | |
AgeChem Venture Fund L.P.(5) | | | 5,769,818 | | | | 7.6 | % | | | | | % | |
1 Westmount Square, Suite 800 | | | | | | | | | | | | | | |
Montreal, Quebec H3Z 2P9 | | | | | | | | | | | | | | |
Canada | | | | | | | | | | | | | | |
| | | | |
Entities affiliated with Roche Finance Ltd(6) | | | 5,123,938 | | | | 6.7 | % | | | | | % | |
Roche Finance Ltd Grenzacherstrasse 122 4070 Basel Switzerland | | | | | | | | | | | | | | |
| | | | |
Named Executive Officers and Directors | | | | | | | | | | | | | | |
Steven J. Mento, Ph.D.(7) | | | 5,126,673 | | | | 6.6 | % | | | | | % | |
Alfred P. Spada, Ph.D.(8) | | | 2,388,927 | | | | 3.1 | % | | | | | | |
Gary C. Burgess, M.B., Ch.B. M.Med.(9) | | | 1,000,000 | | | | 1.3 | % | | | | | % | |
David F. Hale(10) | | | 1,365,836 | | | | 1.8 | % | | | | | % | |
Paul H. Klingenstein(1) | | | 17,876,902 | | | | 23.3 | % | | | | | % | |
Louis Lacasse(5) | | | 5,769,818 | | | | 7.6 | % | | | | | | |
Shahzad Malik, M.D.(2) | | | 16,231,052 | | | | 21.1 | % | | | | | % | |
Marc Perret(3) | | | 10,922,168 | | | | 14.3 | % | | | | | % | |
James Scopa(4) | | | 8,599,171 | | | | 11.2 | % | | | | | % | |
Harold E. Van Wart, Ph.D.(11) | | | 250,000 | | | | * | | | | | | | |
All executive officers and directors as a group (11 persons)(12) | | | 71,285,899 | | | | 87.2 | % | | | | | % | |
| | | | | | | | |
Name and Address of Beneficial Owner | | Number of
Shares
Beneficially
Owned | | | Percentage
of Shares
Beneficially
Owned(1) | |
5% Stockholders | | | | | | | | |
Lordship Ventures(2) | | | 2,356,597 | | | | 18.8 | % |
John Paul Dejoria(3) | | | 1,260,938 | | | | 10.1 | % |
Directors and Executive Officers(4) | | | | | | | | |
Jonathan Jackson(5) | | | 2,356,597 | | | | 18.8 | % |
Gail K Naughton, Ph.D.(6) | | | 806,589 | | | | 6.3 | % |
Hayden Yizhuo Zhang(7) | | | 600,983 | | | | 4.8 | % |
Richard W. Pascoe(8) | | | 191,943 | | | | 1.5 | % |
Steven J. Mento(9) | | | 117,866 | | | | * | |
Martin Latterich(10) | | | 54,977 | | | | * | |
David H. Crean, Ph.D.(11) | | | 54,383 | | | | * | |
Stephen Chang, Ph.D.(12) | | | 52,733 | | | | * | |
Brian Satz(13) | | | 32,810 | | | | * | |
Daniel L. Kisner, M.D.(14) | | | 13,446 | | | | | |
All current directors and executive officers as a group (12 person)(15) | | | 4,282,327 | | | | 34.0 | % |
| * | Less than 1%.one percent. |
| (1) | Includes (a) 16,817,922Percentage ownership is calculated based on 12,509,355 shares of our common stock 605,513 shares of common stock issuable upon the exercise of warrants and 42,336 shares of common stock issuable upon the exercise of 2013 Warrants held by Aberdare Ventures III, L.P., and (b) 395,881 shares of common stock, 14,253 shares of common stock issuable upon the exercise of 2010 Warrants and 997 shares of common stock issuable upon the exercise of 2013 Warrants held by Aberdare Partners III, L.P. In addition, beneficial ownership after the offering gives additional effect to the issuance of and shares of common stock as a result of the
|
| expected net exercise of the 2010 Warrants held by Aberdare Ventures III, L.P. and Aberdare Partners III, L.P., respectively, in connection with the completion of this offering, assuming an initial public offering price of $ per share (the midpoint of the price range set forthoutstanding on the cover page of this prospectus). Mr. Klingenstein serves as Manager of Aberdare GP III, L.L.C., the general partner of Aberdare Ventures III, L.P. and Aberdare Partners III, L.P. Aberdare GP III, L.L.C. owns no shares directly. Mr. Klingenstein shares voting and investment control over the shares owned by Aberdare Ventures III, L.P. and Aberdare Partners III, L.P., and may be deemed to beneficially own such shares. Mr. Klingenstein disclaims beneficial ownership of the shares held by Aberdare Ventures III, L.P. and Aberdare Partners III, L.P., except to the extent of his pecuniary interest therein.September 30, 2020.
|
| (2) | Includes (a) 5,551,195Represents shares of our common stock beneficially owned by Lordship Ventures. Consists of shares of common stock.
|
| (3) | Represents shares of our common stock beneficially owned by John Paul Dejoria. Consists of shares of common stock. Mr. Dejoria holds 119,516 shares of his common stock in the John Paul DeJoria Family Trust. Mr. Dejoria is the Trustee of John Paul DeJoria Family Trust. |
| (4) | Unless otherwise indicated, the address for each of our executive officers and directors is c/o 10655 Sorrento Valley Road, Ste 200, San Diego, California, 92121. |
| (5) | Mr. Jackson is Chairman of Lordship Ventures and has beneficial ownership over our securities owned by Lordship. Consists of shares of common stock. |
| (6) | Consists of 426,526 shares of common stock, 205,845which are owned by the Gail K. Naughton Revocable Trust, dated January 19, 2018, and 380,063 shares of common stock issuable upon the exercise of 2010 Warrants and 27,322 sharesstock options exercisable within 60 days of common stock issuable uponSeptember 30, 2020. Dr. Naughton is the exercise of 2013 Warrants held by Advent Private Equity Fund III ‘A’, (b) 2,720,121 shares of common stock, 100,865 shares of common stock issuable upon the exercise of 2010 Warrants and 13,388 shares of common stock issuable upon the exercise of 2013 Warrants held by Advent Private Equity Fund III ‘B’, (c) 758,770 shares of common stock, 28,135 shares of common stock issuable upon the exercise of 2010 Warrants and 3,735 shares of common stock issuable upon the exercise of 2013 Warrants held by Advent Private Equity Fund III ‘C’, (d) 1,492,488 shares of common stock, 55,342 shares of common stock issuable upon the exercise of 2010 Warrants and 7,346 shares of common stock issuable upon the exercise of 2013 Warrants held by Advent Private Equity Fund III ‘D’, (e) 214,746 shares of common stock, 7,962 shares of common stock issuable upon the exercise of 2010 Warrants and 1,057 shares of common stock issuable upon the exercise of 2013 Warrants held by Advent Private Equity Fund III GmbH & Co KG, (f) 178,955 shares of common stock, 6,635 shares of common stock issuable upon the exercise of 2010 Warrants and 881 shares of common stock issuable upon the exercise of 2013 Warrants held by Advent Private Equity Fund III Affiliates, (g) 53,687 shares of common stock, 1,990 shares of common stock issuable upon the exercise of 2010 Warrants and 264 shares of common stock issuable upon the exercise of 2013 Warrants held by Advent Management III Limited Partnership, (h) 4,561,213 shares of common stock, 169,135 shares of common stock issuable upon the exercise of 2010 Warrants and 22,450 shares of common stock issuable upon the exercise of 2013 Warrants held by Advent Private Equity Fund IV, and (i) 45,611 shares of common stock, 1,690 shares of common stock issuable upon the exercise of 2010 Warrants and 224 shares of common stock issuable upon the exercise of 2013 Warrants held by Advent Management IV Limited Partnership. In addition, beneficial ownership after the offering gives additional effect to the issuance of , , , , , , , and shares of common stock as a resultTrustee of the expected net exerciseGail K. Naughton Revocable Trust, dated January 19, 2018. |
| (7) | Hayden Yizhuo Zhang is the Investment Director of the 2010 Warrants held by Advent Private Equity Fund III ‘A’, Advent Private Equity Fund III ‘B’, Advent Private Equity Fund III ‘C’, Advent Private Equity Fund III ‘D’, Advent Private Equity Fund III GmbH & Co KG, Advent Private Equity Fund III Affiliates, Advent Management III Limited Partnership, Advent Private Equity Fund IV and Advent Management IV Limited Partnership, respectively, in connection with the completion of this offering, assuming an initial public offering price of $ per share (the midpoint of the price range set forth on the cover page of this prospectus). Advent Venture Partners LLP owns 100% of Advent Management III Limited, which is General Partner of Advent Management III Limited Partnership, which is General Partner of each of Advent Private Equity Fund III “A”, Advent Private Equity Fund III “B”, Advent Private Equity Fund III “C”, Advent Private Equity Fund III “D” and Advent Private Equity Fund III Affiliates. Advent Venture Partners LLP also owns 100% of AdventPineworld Capital Limited and Advent Private Equity Fund IV. Advent Limited owns 100% of Advent Private Equity GmbH, which is General Partner of Advent Private Equity Fund III GmbH & Co. KG. Voting and investmenthas voting power over the shares held by each named fund may be deemed to be shared with Advent Venture Partners LLP due to the affiliate relationship described above. Each named fund disclaims beneficial ownershipPineworld. Consists of the others’ shares. Dr. Malik is a general partner of Advent Venture Partners LLP and shares voting and investment power over the shares, and disclaims beneficial ownership of such shares except to the extent of his pecuniary interest therein. |
(3) | Includes 10,384,529 shares of common stock, 385,070 shares of common stock issuable upon the exercise of 2010 Warrants and 222,569 shares of common stock issuable upon the exercise of 2013 Warrants. The manager of Coöperative Gilde Healthcare II U.A. is Gilde Healthcare II Management B.V., which isstock.
|
footnotes continued on following page
the gain is effectively connected with the non-U.S. holder’s conduct of a trade or business in the United States, and if required by an applicable tax treaty, attributable to a permanent establishment maintained by the non-U.S. holder in the United States;
the non-U.S. holder is a nonresident alien individual present in the United States for 183 days or more during the taxable year of the sale or disposition, and certain other requirements are met; or
our common stock constitutes a United States real property interest by reason of our status as a United States real property holding corporation (“USRPHC”) for United States federal income tax purposes at any time within the shorter of (i) the five-year period ending on the date of the sale or disposition of our common stock or (ii) the non-U.S. holder’s holding period for our common stock.
Unless an applicable treaty provides otherwise, the gain described in the first bullet point above generally will be subject to United States federal income tax on a net income basis in the same manner as if such non-U.S. holder were a U.S. person. A non-U.S. holder that is a corporation also may be subject to a branch profits tax equal to 30% (or such lower rate specified by an applicable tax treaty) of its effectively connected earnings and profits for the taxable year, as adjusted for certain items. Non-U.S. holders should consult their tax advisors regarding any applicable tax treaties that may provide for different rules.
Gain described in the second bullet point above generally will be subject to United States federal income tax at a flat 30% rate (or such lower rate specified by an applicable income tax treaty), but may be offset by United States source capital losses of the non-U.S. holder (even though the individual is not considered a resident of the United States), provided that the non-U.S. holder has timely filed U.S. federal income tax returns with respect to such losses.
With respect to the third bullet point above, we believe that we are not currently, and we do not anticipate becoming, a USRPHC. However, because the determination of whether we are a USRPHC depends on the fair market value of our United States real property interests relative to the fair market value of our other business assets, there can be no assurance that we will not become a USRPHC in the future. In the event we do become a USRPHC, as long as our common stock is regularly traded on an established securities market, our common stock will be treated as a U.S. real property interest only with respect to a non-U.S. holder that actually or constructively held more than 5% of our common stock at any time during the shorter of (i) the five-year period ending on the date of the sale or disposition of our common stock or (ii) the non-U.S. holder’s holding period for our common stock. If gain on the sale or other taxable disposition of our common stock were subject to taxation under the third bullet point above, the non-U.S. holder would be subject to regular United States federal income tax with respect to such gain in generally the same manner as a U.S. person.
Information Reporting and Backup Withholding
Generally, we must report annually to the IRS and to each non-U.S. holder the amount of dividends paid to such non-U.S. holder and the amount, if any, of tax withheld with respect to those dividends. This information also may be made available under a specific treaty or agreement with the tax authorities in the country in which the non-U.S. holder resides or is established. Under certain circumstances, the Code imposes backup withholding on certain reportable payments. Backup withholding, however, generally will not apply to payments of dividends to a non-U.S. holder of our common stock provided the non-U.S. holder furnishes to us or our paying agent the required certification as to its non-U.S. status, such as by providing a valid IRS Form W-8BEN or IRS Form W-8ECI, or otherwise establishes an exemption. Notwithstanding the foregoing, backup withholding may apply if either we or our paying agent has actual knowledge, or reason to know, that the holder is a U.S. person that is not an exempt recipient.
Unless a non-U.S. holder complies with certification procedures to establish that it is not a U.S. person, information returns may be filed with the IRS in connection with, and the non-U.S. holder may be subject to
backup withholding on the proceeds from, a sale or other disposition of our common stock. The certification procedures described in the above paragraph will satisfy these certification requirements as well.
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules may be allowed as a refund or a credit against a non-U.S. holder’s United States federal income tax liability, provided the required information is timely furnished to the IRS.
Foreign Accounts
Withholding taxes may apply to certain types of payments made to “foreign financial institutions” (as specially defined under those rules) and certain other non-U.S. entities. The failure to comply with additional certification, information reporting and other specified requirements could result in a withholding tax being imposed on payments of dividends and sales proceeds to foreign intermediaries and certain non-U.S. holders. A 30% withholding tax is imposed on dividends on, or gross proceeds from the sale or other disposition of, our common stock paid to a foreign financial institution or to a foreign non-financial entity, unless (i) the foreign financial institution undertakes certain diligence and reporting obligations, (ii) the foreign non-financial entity either certifies it does not have any substantial United States owners or furnishes identifying information regarding each substantial United States owner, or (iii) the foreign financial institution or foreign non-financial entity otherwise qualifies for an exemption from these rules. If the payee is a foreign financial institution and is subject to the diligence and reporting requirements in clause (i) above, it must enter into an agreement with the United States Treasury requiring, among other things, that it undertake to identify accounts held by certain United States persons or United States-owned foreign entities, annually report certain information about such accounts, and withhold 30% on payments to non-compliant foreign financial institutions and certain other account holders.
Recently issued final Treasury Regulations provide that such rules will generally apply to payments of dividends made on or after January 1, 2014 and to payments of gross proceeds from a sale or other disposition of common stock on or after January 1, 2017. Prospective investors should consult their tax advisors regarding these withholding provisions.
UNDERWRITING
Subject to the terms and conditions set forth in an underwriting agreement, each of the underwriters named below has severally agreed to purchase from us the aggregate number of shares of common stock set forth opposite their respective names below:
| | |
Underwriters
| | Number of Shares |
Stifel, Nicolaus & Company, Incorporated
| | |
Piper Jaffray & Co.
| | |
JMP Securities LLC
| | |
SunTrust Robinson Humphrey, Inc.
| | |
| | |
Total
| | |
| | |
The underwriting agreement provides that the obligations of the several underwriters are subject to various conditions, including approval of legal matters by counsel. The nature of the underwriters’ obligations commits them to purchase and pay for all of the shares of common stock listed above if any are purchased. The underwriters reserve the right to withdraw, cancel or modify offers to the public and to reject orders in whole or in part.
The underwriters expect to deliver the shares of common stock to purchasers on or about , 2013.
Over-Allotment Option
We have granted a 30-day over-allotment option to the underwriters to purchase up to a total of additional shares of our common stock from us, at the initial public offering price, less the underwriting discounts and commissions payable by us, as set forth on the cover page of this prospectus. If the underwriters exercise this option in whole or in part, then each of the underwriters will be separately committed, subject to the conditions described in the underwriting agreement, to purchase the additional shares of our common stock in proportion to their respective commitments set forth in the table above. We will pay the expenses associated with the exercise of the over-allotment option.
Lock-Up Agreements
We and the holders (including all of our directors and executive officers) of substantially all of the shares of our common stock outstanding prior to this offering have agreed that, without the prior written consent of each of Stifel, Nicolaus & Company, Incorporated and Piper Jaffray & Co., we and they will not directly or indirectly:
offer, sell, contract to sell (including any short sale), pledge, hypothecate, establish an open “put equivalent position” within the meaning of Rule 16a-1(h) under the Exchange Act, grant any option, right or warrant for the sale of, purchase any option or contract to sell, sell any option or contract to purchase, or otherwise encumber, dispose of or transfer, or grant any rights with respect to, directly or indirectly, any shares of common stock or securities convertible into or exchangeable or exercisable for any shares of common stock;
enter into a transaction which would have the same effect, or enter into any swap, hedge or other arrangement that transfers, in whole or in part, any of the economic consequences of ownership of the common stock, whether any such transaction is to be settled by delivery of the common stock or other securities, in cash or otherwise; or
publicly disclose the intention to do any of the foregoing,
for a period of 180 days after the date of this prospectus. However, in the case of our officers, directors and stockholders, these lock-up restrictions will not apply to:
bona fide gifts made by the holder;
the surrender or forfeiture of shares of common stock to us to satisfy tax withholding obligations upon exercise or vesting of stock options or equity awards;
transfers of common stock or any security convertible into or exercisable for common stock to an immediate family member, an immediate family member of a domestic partner or a trust for the benefit of the undersigned, a domestic partner or an immediate family member;
transfers of shares of common stock or any security convertible into or exercisable for common stock to any corporation, partnership, limited liability company or other entity all of the beneficial ownership interests of which are held exclusively by the holder, a domestic partner and/or one or more family members of the holder or the holder’s domestic partner in a transaction not involving a disposition for value;
transfers of shares of common stock or any security convertible into or exercisable for common stock upon death by will or intestate succession;
distributions of shares of common stock or securities convertible into or exercisable for common stock to members, partners or stockholders of the holder;
the exercise of any option, warrant or other right to acquire shares of common stock, the settlement of any stock-settled stock appreciation rights, restricted stock or restricted stock units, or the conversion of any convertible security into our securities;
securities transferred to one or more affiliates of the holder and distributions of securities to partners, members or stockholders of the holder;
transactions relating to securities acquired in open market transactions after the date of this prospectus;
the entry into a trading plan established pursuant to Rule 10b5-1 under the Exchange Act, provided that such plan does not provide for any sales or other dispositions of shares of common stock during the 180-day restricted period; or
any shares purchased by the holder in this offering.
Except for transfers related to securities acquired on the open market or in this offering or to the surrender or forfeiture of shares of common stock to us to satisfy tax withholding obligations upon exercise or vesting of stock options or equity awards, as described above, any transferee under the excepted transfers above must agree in writing, prior to the transfer, to be bound by the lock-up agreements.
Additionally, in our case, the lock-up restrictions will not apply to:
shares sold in this offering;
equity based awards granted pursuant to our equity incentive plans referred to in this prospectus, including any amendments to those plans, and shares of common stock issued upon the exercise of any equity based awards;
shares of common stock issued upon the conversion of outstanding securities described in this prospectus;
the filing of a registration statement on Form S-8 relating to the registration of shares issuable pursuant to our equity incentive plans;
shares of common stock issued in satisfaction of the accumulated dividend on our outstanding convertible preferred stock; or
shares of common stock or any securities convertible into, or exercisable, or exchangeable for, shares of common stock, issued to lenders or financial institutions in connection with bona fide debt or credit arrangements as long as (x) the aggregate number of shares of common stock issued or issuable does not exceed 5% of the number of shares of common stock outstanding immediately after this offering, and (y) each recipient of any such shares or other securities agrees to restrictions on the resale of such securities that are consistent with the lock-up agreements described above.
Stifel, Nicolaus & Company, Incorporated and Piper Jaffray & Co., in their sole discretion, may release the common stock and other securities subject to the lock-up agreements described above in whole or in part at any time with or without notice. When determining whether or not to release common stock and other securities from lock-up agreements, Stifel, Nicolaus & Company, Incorporated and Piper Jaffray & Co. will consider, among other factors, the holder’s reasons for requesting the release, the number of shares of common stock and other securities for which the release is being requested and market conditions at the time.
Determination of Offering Price
Prior to this offering, there has been no public market for our common stock. The initial public offering price will be determined through negotiations between us and Stifel, Nicolaus & Company, Incorporated and
Piper Jaffray & Co., as representatives of the several underwriters. In addition to prevailing market conditions, the factors to be considered in determining the initial public offering price will include:
the information set forth in this prospectus and otherwise available to the representatives;
our history and prospects, including our past and present financial performance and our prospects for future earnings;
the history and prospects of companies in our industry;
prior offerings of those companies;
an assessment of our management and their experience;
general conditions of the securities markets at the time of the offering; and
other factors as we deem relevant.
We cannot assure you that an active or orderly trading market will develop for our common stock or that our common stock will trade in the public markets subsequent to this offering at or above the initial public offering price.
Commissions and Expenses
The underwriters propose to offer the shares of common stock directly to the public at the initial public offering price set forth on the cover page of this prospectus, and at this price less a concession not in excess of $ per share of common stock to other dealers specified in a master agreement among underwriters who are members of the Financial Industry Regulatory Authority, Inc. The underwriters may allow, and the other dealers specified may reallow, concessions not in excess of $ per share of common stock to these other dealers. After this offering, the offering price, concessions, and other selling terms may be changed by the underwriters. Our common stock is offered subject to receipt and acceptance by the underwriters and to certain other conditions, including the right to reject orders in whole or in part.
The following table summarizes the compensation to be paid to the underwriters by us and the proceeds, before expenses, payable to us:
| | | | | | |
| | | | Total |
| | Per Share | | Without
Over-Allotment | | With
Over-Allotment |
Public offering price
| | | | | | |
Underwriting discounts and commissions
| | | | | | |
Proceeds, before expenses, to us
| | | | | | |
We estimate that the total expenses of the offering payable by us, including registration, filing and listing fees, printing fees and legal and accounting expenses, but excluding underwriting discounts and commissions, will be approximately $ million.
Indemnification of Underwriters
We will indemnify the underwriters against some civil liabilities, including liabilities under the Securities Act. If we are unable to provide this indemnification, we will contribute to payments the underwriters may be required to make in respect of those liabilities.
NASDAQ Market Listing
We have applied to have our common stock listed on The NASDAQ Global Market under the symbol “CNAT.”
Short Sales, Stabilizing Transactions and Penalty Bids
In order to facilitate this offering, persons participating in this offering may engage in transactions that stabilize, maintain, or otherwise affect the price of our common stock during and after this offering. Specifically,
the underwriters may engage in the following activities in accordance with the rules of the Securities and Exchange Commission.
Short sales.Short sales involve the sales by the underwriters of a greater number of shares than they are required to purchase in the offering. Covered short sales are short sales made in an amount not greater than the underwriters’ over-allotment option to purchase additional shares from us in this offering. The underwriters may close out any covered short position by either exercising their over-allotment option to purchase shares or purchasing shares in the open market. In determining the source of shares to close out the covered short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase shares through the over-allotment option. Naked short sales are any short sales in excess of such over-allotment option. The underwriters must close out any naked short position by purchasing shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that there may be downward pressure on the price of the common stock in the open market after pricing that could adversely affect investors who purchase in this offering.
Stabilizing transactions.The underwriters may make bids for or purchases of the shares for the purpose of pegging, fixing, or maintaining the price of the shares, so long as stabilizing bids do not exceed a specified maximum.
Penalty bids.If the underwriters purchase shares in the open market in a stabilizing transaction or syndicate covering transaction, they may reclaim a selling concession from the underwriters and selling group members who sold those shares as part of this offering. Stabilization and syndicate covering transactions may cause the price of the shares to be higher than it would be in the absence of these transactions. The imposition of a penalty bid might also have an effect on the price of the shares if it discourages resales of the shares.
The transactions above may occur on The NASDAQ Global Market or otherwise. Neither we nor the underwriters make any representation or prediction as to the effect that the transactions described above may have on the price of the shares. If these transactions are commenced, they may be discontinued without notice at any time.
Discretionary Sales
The underwriters have informed us that they do not expect to confirm sales of common stock offered by this prospectus to accounts over which they exercise discretionary authority without obtaining the specific approval of the account holder.
Electronic Distribution
A prospectus in electronic format may be made available on the internet sites or through other online services maintained by one or more of the underwriters participating in this offering, or by their affiliates. Other than the prospectus in electronic format, the information on any underwriter’s web site and any information contained in any other web site maintained by an underwriter is not part of the prospectus or the registration statement of which this prospectus forms a part, has not been approved and/or endorsed by us or any underwriter in its capacity as underwriter and should not be relied upon by investors.
Relationships
The underwriters and their respective affiliates are full service financial institutions engaged in various activities, which may include securities trading, commercial and investment banking, financial advisory, investment management, principal investment, hedging, financing and brokerage activities. Certain of the underwriters and their affiliates have in the past provided, and may in the future from time to time provide, investment banking and other financing and banking services to us, for which they have in the past received, and may in the future receive, customary fees and reimbursement for their expenses. In the ordinary course of their various business activities, the underwriters and their respective affiliates may make or hold a broad array of investments and actively trade debt and equity securities (or related derivative securities) and financial instruments (including bank loans) for their own account and for the accounts of their customers and may at any
time hold long and short positions in such securities and instruments. Such investment and securities activities may involve our securities and instruments.
European Economic Area
In relation to each member state of the European Economic Area that has implemented the Prospectus Directive (each, a relevant member state), with effect from and including the date on which the Prospectus Directive is implemented in that relevant member state (the relevant implementation date), an offer of securities described in this prospectus may not be made to the public in that relevant member state other than:
to any legal entity that is authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities;
to any legal entity that has two or more of (1) an average of at least 250 employees during the last financial year; (2) a total balance sheet of more than €43,000,000 and (3) an annual net turnover of more than €50,000,000, as shown in its last annual or consolidated accounts;
to fewer than 100 natural or legal persons (other than qualified investors as defined in the Prospectus Directive) subject to obtaining the prior consent of the representatives; or
in any other circumstances that do not require the publication of a prospectus pursuant to Article 3 of the Prospectus Directive,
provided that no such offer of securities shall require us or any underwriter to publish a prospectus pursuant to Article 3 of the Prospectus Directive. For purposes of this provision, the expression an “offer of securities to the public” in any relevant member state means the communication in any form and by any means of sufficient information on the terms of the offer and the securities to be offered so as to enable an investor to decide to purchase or subscribe the securities, as the expression may be varied in that member state by any measure implementing the Prospectus Directive in that member state, and the expression “Prospectus Directive” means Directive 2003/71/EC and includes any relevant implementing measure in each relevant member state.
We have not authorized and do not authorize the making of any offer of securities through any financial intermediary on their behalf, other than offers made by the underwriters with a view to the final placement of the securities as contemplated in this prospectus. Accordingly, no purchaser of the securities, other than the underwriters, is authorized to make any further offer of the securities on behalf of us or the underwriters.
United Kingdom
This prospectus is only being distributed to, and is only directed at, persons in the United Kingdom that are qualified investors within the meaning of Article 2(1)(e) of the Prospectus Directive (Qualified Investors) that are also (i) investment professionals falling within Article 19(5) of the Financial Services and Markets Act 2000 (Financial Promotion) Order 2005 (the Order) or (ii) high net worth entities, and other persons to whom it may lawfully be communicated, falling within Article 49(2)(a) to (d) of the Order (all such persons together being referred to as relevant persons). This prospectus and its contents are confidential and should not be distributed, published or reproduced (in whole or in part) or disclosed by recipients to any other persons in the United Kingdom. Any person in the United Kingdom that is not a relevant person should not act or rely on this document or any of its contents.
France
This prospectus has not been prepared in the context of a public offering of financial securities in France within the meaning of Article L.411-1 of the French Code Monétaire et Financier and Title I of Book II of the Reglement Général of the Autorité des marchés financiers (the “AMF”) and therefore has not been and will not be filed with the AMF for prior approval or submitted for clearance to the AMF. Consequently, the shares of our common stock may not be, directly or indirectly, offered or sold to the public in France and offers and sales of the shares of our common stock may only be made in France to qualified investors (investisseurs qualifiés) acting for their own, as defined in and in accordance with Articles L.411-2 and D.411-1 to D.411-4, D.734-1, D.744-1, D.754-1 and D.764-1 of the French Code Monétaire et Financier. Neither this prospectus nor any other offering material may be released, issued or distributed to the public in France or used in connection with any offer for
subscription on sale of the shares of our common stock to the public in France. The subsequent direct or indirect retransfer of the shares of our common stock to the public in France may only be made in compliance with Articles L.411-1, L.411-2, L.412-1 and L.621-8 through L.621-8-3 of the French Code Monétaire et Financier.
Notice to Residents of Germany
Each person who is in possession of this prospectus is aware of the fact that no German securities prospectus (wertpapierprospekt) within the meaning of the securities prospectus act (wertpapier-prospektgesetz, the “act”) of the federal republic of Germany has been or will be published with respect to the shares of our common stock. In particular, each underwriter has represented that it has not engaged and has agreed that it will not engage in a public offering in the federal republic of Germany (ôffertliches angebot) within the meaning of the act with respect to any of the shares of our common stock otherwise than in accordance with the act and all other applicable legal and regulatory requirements.
Notice to Residents of Switzerland
The securities which are the subject of the offering contemplated by this prospectus may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange, or SIX, or on any other stock exchange or regulated trading facility in Switzerland. This prospectus has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. None of this prospectus or any other offering or marketing material relating to the securities or the offering may be publicly distributed or otherwise made publicly available in Switzerland.
None of this prospectus or any other offering or marketing material relating to the offering, us or the securities have been or will be filed with or approved by any Swiss regulatory authority. In particular, this prospectus will not be filed with, and the offer of securities will not be supervised by, the Swiss Financial Market Supervisory Authority FINMA and the offer of securities has not been and will not be authorized under the Swiss Federal Act on Collective Investment Schemes, or CISA. The investor protection afforded to acquirers of interests in collective investment schemes under the CISA does not extend to acquirers of the securities.
Notice to Residents of the Netherlands
The offering of the shares of our common stock is not a public offering in The Netherlands. The shares of our common stock may not be offered or sold to individuals or legal entities in The Netherlands unless (i) a prospectus relating to the offer is available to the public, which has been approved by the Dutch Authority for the Financial Markets (Autoriteit Financiële Markten) or by the competent supervisory authority of another state that is a member of the European Union or party to the Agreement on the European Economic Area, as amended or (ii) an exception or exemption applies to the offer pursuant to Article 5:3 of The Netherlands Financial Supervision Act (Wet op het financieel toezicht) or Article 53 paragraph 2 or 3 of the Exemption Regulation of the Financial Supervision Act, for instance due to the offer targeting exclusively “qualified investors” (gekwalificeerde beleggers) within the meaning of Article 1:1 of The Netherlands Financial Supervision Act.
Notice to Residents of Japan
The underwriters will not offer or sell any of the shares of our common stock directly or indirectly in Japan or to, or for the benefit of, any Japanese person or to others, for re-offering or re-sale directly or indirectly in Japan or to any Japanese person, except in each case pursuant to an exemption from the registration requirements of, and otherwise in compliance with, the Financial Instruments and Exchange Law of Japan and any other applicable laws and regulations of Japan. For purposes of this paragraph, “Japanese person” means any person resident in Japan, including any corporation or other entity organized under the laws of Japan.
Notice to Residents of Hong Kong
The underwriters and each of their affiliates have not (1) offered or sold, and will not offer or sell, in Hong Kong, by means of any document, any shares of our common stock other than (a) to “professional investors”
within the meaning of the Securities and Futures Ordinance (Cap. 571) of Hong Kong and any rules made under that Ordinance or (b) in other circumstances which do not result in the document being a “prospectus” as defined in the Companies Ordinance (Cap. 32) of Hong Kong or which do not constitute an offer to the public within the meaning of that Ordinance; and (2) issued or had in its possession for the purposes of issue, and will not issue or have in its possession for the purposes of issue, whether in Hong Kong or elsewhere any advertisement, invitation or document relating to the shares of our common stock which is directed at, or the contents of which are likely to be accessed or read by, the public in Hong Kong (except if permitted to do so under the securities laws of Hong Kong) other than with respect to the shares of our common stock which are or are intended to be disposed of only to persons outside Hong Kong or only to “professional investors” within the meaning of the Securities and Futures Ordinance and any rules made under that Ordinance. The contents of this document have not been reviewed by any regulatory authority in Hong Kong. You are advised to exercise caution in relation to the offer. If you are in any doubt about any of the contents of this document, you should obtain independent professional advice.
Notice to Residents of Singapore
This document has not been registered as a prospectus with the Monetary Authority of Singapore. Accordingly, this document and any other document or material in connection with the offer or sale, or invitation for subscription or purchase, of shares of our common stock may not be circulated or distributed, nor may shares of our common stock be offered or sold, or be made the subject of an invitation for subscription or purchase, whether directly or indirectly, to persons in Singapore other than (i) to an institutional investor under Section 274 of the Securities and Futures Act, Chapter 289 of Singapore (the “Securities and Futures Act”), (ii) to a relevant person, or any person pursuant to Section 275(1A), and in accordance with the conditions, specified in Section 275 of the Securities and Futures Act or (iii) otherwise pursuant to, and in accordance with the conditions of, any other applicable provision of the Securities and Futures Act.
Where shares of our common stock are subscribed or purchased under Section 275 by a relevant person, which is:
(a) | a corporation (which is not an accredited investor) the sole business of which is to hold investments and the entire share capital of which is owned by one or more individuals, each of whom is an accredited investor; or
|
(b) | a trust (where the trustee is not an accredited investor) whose sole purpose is to hold investments and each beneficiary is an accredited investor,
|
shares, debentures and units of shares and debentures of that corporation or the beneficiaries’ rights and interest in that trust shall not be transferable for six months after that corporation or that trust has acquired the shares of our common stock under Section 275 except:
(1) | to an institutional investor or to a relevant person, or to any person pursuant to an offer that is made on terms that such rights or interest are acquired at a consideration of not less than $200,000 (or its equivalent in a foreign currency) for each transaction, whether such amount is to be paid for in cash or by exchange of securities or other assets;
|
(2) | where no consideration is given for the transfer; or
|
LEGAL MATTERS
The validity of the sharesissuance of common stocksecurities offered hereby will beby this prospectus has been passed upon for us by Latham & WatkinsDLA Piper LLP (US), San Diego, California. Goodwin ProcterDiego. Thompson Hine LLP, New York, New York has actedis acting as counsel forto the underwriters in connection with certain legal matters related to this offering.Placement Agent.
EXPERTS
Ernst & Young LLP, independent registered public accounting firm, has audited our consolidated financial statements at December 31, 20112019 and 2012,2018, and for each of the twothree years in the period ended December 31, 2012, and for the period from July 13, 2005 (inception) to December 31, 2012,2019, as set forth in their report (which contains an explanatory paragraph describing conditions that raise substantial doubt about our ability to continue as a going concern as described in Note 1 to the consolidated financial statements). We havereport. We’ve included our financial statements in the prospectus and elsewhere in the registration statement in reliance on Ernst & Young LLP’s report, given on their authority as experts in accounting and auditing.
The consolidated financial statements of Private Histogen (Histogen, Inc. now Histogen Therapeutics, Inc.) as of and for the years ended December 31, 2019 and 2018 included in this registration statement and related prospectus, have been audited by Mayer Hoffman McCann P.C., independent registered public accounting firm, as set forth in their report (which report includes an explanatory paragraph regarding the existence of substantial doubt about the Company’s ability to continue as a going concern) included in this prospectus in reliance on the report of Mayer Hoffman McCann P.C., given on the authority of such firm as experts in auditing and accounting in giving said reports.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the Securities and Exchange Commission a registration statement on Form S-1 under the Securities Act with respect to the shares of common stock offered hereby. This prospectus, which constitutes a part of the registration statement, does not contain all of the information set forth in the registration statement or the exhibits and schedules filed therewith. For further information about us and the common stock offered hereby, we refer you to the registration statement and the exhibits and schedules filed thereto. Statements contained in this prospectus regarding the contents of any contract or any other document that is filed as an exhibit to the registration statement are not necessarily complete, and each such statement is qualified in all respects by reference to the full text of such contract or other document filed as an exhibit to the registration statement. Upon completion of this offering, we will be required to file periodic reports, proxy statements and other information with the SEC. The SEC maintains an internet site that contains reports, proxy and information statements and other information regarding Histogen Inc. and other issuers that file electronically with the SEC. The address of the SEC internet site is www.sec.gov. In addition, we make available on or through our Internet site copies of these reports as soon as reasonably practicable after we electronically file or furnish them to the SEC. Our Internet site can be found at www.histogen.com