As filed with the Securities and Exchange Commission on May 17, 2010
RegistrationNo. 333-166146333-
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 2to
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
REGENERX BIOPHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 2834 | 52-1253406 | ||
| (State or other jurisdiction of incorporation or organization) | (Primary Standard Industrial Classification Code Number) | (I.R.S. Employer Identification Number) |
15245 Shady Grove Road, Suite 470
Rockville, MD 20850
(301) 208-9191
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
J.J. Finkelstein
President and Chief Executive Officer
15245 Shady Grove Road, Suite 470
Rockville, MD 20850
(301) 208-9191
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Todd A. Taylor, Esq. Minneapolis, Minnesota 55402
|
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. þ
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration number of the earlier effective registration statement for the same offering. o¨
| Proposed Maximum | Proposed Maximum | Amount of | ||||||||||||||||
| Title of Each Class of | Amount to be | Offering | Aggregate | Registration | ||||||||||||||
| Securities Being Registered | Registered(1) | Price per Security(2) | Offering Price(2) | Fee | ||||||||||||||
| Units, each consisting of one share of common stock, $0.001 par value per share, and 0.4 warrants to purchase common stock(1) | 13,225,000 units | $ | 0.56 | $ | 7,406,000 | $ | 528.05 | |||||||||||
| Common Stock, $0.001 par value per share, included in the Units(1) | 13,225,000 shares | — | — | —(3 | ) | |||||||||||||
| Warrants included in the Units(1) | 5,290,000 warrants | — | — | —(3 | ) | |||||||||||||
| Shares of common stock underlying the warrants included in the Units | 5,290,000 shares | $ | 0.62 | $ | 3,279,800 | $ | 233.85 | |||||||||||
| Representative’s Warrant | 1 warrant | $ | 100.00 | $ | 100 | $ | 0.01 | |||||||||||
| Shares of common stock underlying the Representative’s Warrant | 805,000 shares | $ | 0.62 | $ | 499,100 | $ | 35.59 | |||||||||||
| Total | $ | $ | 11,185,000 | $ | 797.50(5 | ) | ||||||||||||
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule12b-2 of the Exchange Act. (Check one):
Large accelerated filer | Accelerated filer | Non-accelerated filer ¨ (Do not check if a smaller reporting company) | Smaller reporting company þ |
CALCULATION OF REGISTRATION FEE (1)
| Title of Each Class of Securities to be Registered | Amount to be Registered(1) | Proposed Maximum Offering Price Per Share (2) | Proposed Maximum Aggregate Offering Price | Amount of Registration Fee | ||||||||||||
| Common stock, $0.001 par value per share | 10,551,471 | $ | 0.40 | $ | 4,220,588 | $ | 425.01 | |||||||||
| Total | $ | 4,220,588 | $ | 425.01 | ||||||||||||
| (1) | Pursuant to Rule 416 under the Securities Act, the shares being registered hereunder include such indeterminate number of shares as may be issuable with respect to the shares being registered hereunder as a result of stock splits, stock dividends or similar transactions. | |
| (2) | Estimated solely for purposes of calculating the amount of the registration fee pursuant to Rule 457. The offering price per share and the aggregate offering price are based upon the average of the high and low prices of the registrant’s common stock as reported on the OTCQB on July 19, 2016. |
The registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment that specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
A registration statement relating to these securities has been filed with the Securities and Exchange Commission. These securities may not be sold nor may offers to buy be accepted prior to the time the registration statement becomes effective. This prospectus shall not constitute an offer to sell or the solicitation of an offer to buy nor shall there be any sale of these securities in any state in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state.
PROSPECTUS SUBJECT TO COMPLETION, DATED MAY 17, 2010July 20, 2016
PROSPECTUS
11,500,000 Units

5,404,412 Shares Issuable upon the Exercise of Warrants

The selling stockholders identified beginning on page 27 of this prospectus are offering 11,500,000 units, each unit consistingon a resale basis a total of one share10,551,471 shares of our common stock, and 0.4 warrants to purchase common stock at anof which 5,404,412 are issuable upon the exercise price per whole share equal to 110% of outstanding warrants. We will not receive any proceeds from the closing bid pricesale of our common stock onthese shares by the date of this prospectus. No fractional warrants will be issued. The units will separate immediately and the common stock and warrants will be issued separately. There will be no market for the units. Each unit will be sold at a purchase price of $ .
Our common stock is currently listedquoted on the NYSE Amex stock exchangeOTCQB under the symbol “RGN.“RGRX.” On May 14, 2010,July , 2016, the last reported sale price of our common stock on the NYSE AmexOTCQB was $0.56$0. per share. Currently, no public market exists for the warrants offered by this prospectus. It is anticipated that the warrants will be quoted on the OTC Bulletin Board under the symbol “ ” promptly after the date of this prospectus. We cannot assure you that our common stock will continue to be listed on the NYSE Amex or that our warrants will continue to be quoted on the OTC Bulletin Board.
Investing in our securitiescommon stock involves a high degree of risk. See “Risk Factors”
beginning on page 710 of this prospectus for a discussion of information that should be
considered in connection with an investment in our securities.
45-day option to purchase up to an additional 1,725,000 units from us on the same terms and conditions as set forth above.
| Prospectus Summary | 1 | |||
| 10 | ||||
| 24 | ||||
| Use of Proceeds | 26 | |||
| 26 | ||||
| Selling Stockholders | 27 | |||
| Plan of Distribution | 28 | |||
| Capitalization | 31 | |||
| Price Range of Common Stock and Dividend Policy | 32 | |||
| 34 | ||||
| 43 | ||||
| Management | 58 | |||
| 63 | ||||
| Certain Relationships and Related Party Transactions | ||||
| 65 | ||||
| 71 | ||||
| Description of Securities | 72 | |||
| 75 | ||||
| Experts | 75 | |||
| Where You Can Find Additional Information | ||||
| F-1 | ||||
You should rely only on the information contained in this prospectus and any related free writing prospectus we may authorize to be delivered to you. We have not, and the underwriters have not authorized any person to provide you with different information. If anyone provides you with different or inconsistent information, you should not rely on it. Neither this prospectus nor any related free writing prospectus is an offer to sell, nor are they seeking an offer to buy, these securities in any state where the offer or solicitation is not permitted. The information contained in this prospectus is complete and accurate as of the date on the front cover of this prospectus, but information may have changed since that date.
This prospectus includes statistical and other industry and market data that we obtained from industry publications and research, surveys and studies conducted by third parties. Industry publications and third-party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. While we believe that these industry publications and third-party research, surveys and studies are reliable, we have not independently verified such data and we do not make any representation as to the accuracy of the information.
The items in the following summary are described in more detail later in this prospectus. This summary does not contain all of the information you should consider. Before investing in our securities, you should read the entire prospectus carefully, including the “Risk Factors” beginning on page 710 and the financial statements and related notes beginning onpage F-1. Unless the context indicates otherwise, (i) as used in this prospectus, the terms “RegeneRx,” “our company,” “we,” “us” and “our” refer to RegeneRx Biopharmaceuticals, Inc. and (ii)
Overview
RegeneRx Biopharmaceuticals, Inc. (“RegeneRx” or the information in this prospectus assumes no exercise of the underwriters’ over-allotment option.
| · | RGN-259, a preservative-free topical eye drop for regeneration of corneal tissues damaged by injury, disease or other pathology; |
| RGN-352, an injectable | ||
| RGN-137, a |
We have a fourthare continuing strategic partnership discussions with biotechnology and pharmaceutical companies regarding the further clinical development of all of our product candidate, RGN-457, in preclinical development. RGN-457 is an inhaled formulation of Tß4 targeting cystic fibrosis and other pulmonary diseases.
In addition to our fourthree pharmaceutical product candidates, we are also pursuingevaluating the commercial development of peptide fragments and derivatives of Tß4 for potential cosmeceutical use. Cosmeceuticalsand other personal care uses. These fragments are cosmetic products with biologically active ingredients.select amino acid sequences, and variations thereof, within the Tß4 molecule that have demonstrated activity in severalin vitro preclinical research studies that we have sponsored. We believe the biological activities of these fragments may be useful, for example, in developing novel cosmeceutical products for the anti-aging market.
Overview of our product candidates and their development status:
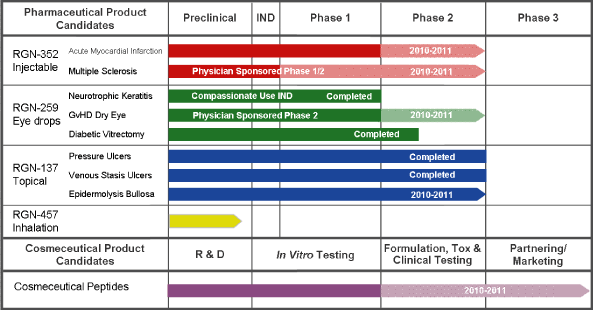
1
Although it is recognized that wound healing and tissue regeneration are complex processes, most companies working to develop new drugs in this area have focused primarily on the development of growth factors to stimulate healing only and have, to date, failed to demonstrate dramatic improvements in the healing process. Unlike growth factors, numerous preclinical animal research hasstudies, published by independent researchers, have identified several important biological activities ofinvolving Tß4 that we believe make it potentially useful as a wound healing, repair and tissue regenerating agent, including:
| · | ||
| Progenitor (Stem) Cell Recruitment and Differentiation. Independent research published in the journal Nature in November 2006 featured the discovery that Tß4 is the key signaling molecule that recruits and triggers adult epicardial progenitor cells, or EPCs, to differentiate into coronary blood |
| 1 |
| · | Actin Regulation. Tß4 regulates actin, which comprises up to 10% of the protein of non-muscle cells in the body and plays a central role in cell structure and in the movement of cells. Independent research studies have indicated that Tß4 stimulates the migration of human keratinocytes, or skin cells, as well as corneal epithelial cells that protect the eye, human endothelial cells and progenitor cells of the heart and brain. Endothelial cells are the major cell type responsible for the formation of new blood vessels, a process known as angiogenesis. Certain of these studies conducted at the National Institutes of Health, or NIH, were the first to suggest the role of Tß4 in wound healing. The data from these studies encouraged us to license the rights to Tß4 from the NIH in 2001 and to launch an initial clinical development program that targeted the use Tß4 for chronic dermal wounds. | |
| · | Reduction of Inflammation and scar tissue formation. Uncontrolled inflammation is the underlying basis of many pathologies and injuries. Independent research has shown that Tß4 is a potent anti-inflammatory agent in skin cells and in corneal epithelial cells in the eye. Tß4 has also been shown to decrease the levels of inflammatory mediators and to significantly reduce the influx of inflammatory cells in the reperfused heart of animals. More recent preclinical research suggests that Tß4 blocks activation of the NFκB pathway, which is involved in DNA activation of inflammatory mediators, thereby modulating inflammation in the body. This anti-inflammatory activity may explain, in part, the mechanism by which Tß4 appeared to improve functional outcome in the mouse multiple sclerosis model described above, as well as promoting repair in the heart and skin. In the skin, it has been shown to reduce scar formation by reduction of infiltration of myofibroblasts. Identifying a factor such as Tß4 that reduces scarring and blocks activation of NFκB suggests that Tß4 could have additional important therapeutic applications for inflammation-related diseases, such as cancer, osteoarthritis, rheumatic diseases, autoimmune diseases, inflammatory pulmonary disease and pancreatitis. | |
| · | ||
| the prevention of disease.Laminin-5 promotes cell migration and maintains cell-cell and cell-matrix contacts for intact tissues which are important for preventing fluid loss and bacterial infection. | ||
| Anti-Apoptosis. Tß4 has been shown to prevent apoptosis, or programmed cell |
Tß4 has shown efficacy in heart repair and regeneration in numerous animal models. A 2004 paper inNature showed that it could reduce the lesion size, improve cardiac function and promote survival. The 2006 Nature publication mentioned above further concluded that Tß4’s interaction with EPCs resulted in the formation of Tß4 and have formulated it for various routes of administration, targeting medical indications with significant unmet needs and market potential, as well as orphan indicationscardiomyocytes that we believe could also provide substantial commercial value. Our product candidates are intended to provide solutions to these medical indications and to offer improvements to current standards of care.
| 2 |
Similar research in the area of brain and central nervous system tissues also showed efficacy of repair and regeneration was published in the journal Neuroscience in 2009. This publication concluded that Tß4 triggered the differentiation of oligodendrocyte progenitor cells to form myelin-producing oligodendrocytes, which led to the remyelination of axons in the brain of mice with experimental autoimmune encephalomyelitis, or EAE. This mouse model is an accepted small animal model for the study of multiple sclerosis. Research published in the Journal of Neurosurgery in 2010 and also in the Journal of Neurological Science in 2014 showed that Tß4 could improve functional neurological outcome in an animal stroke model. A second study was published in the Journal of Neurosurgery in 2011 demonstrating that administration of Tß4 can guide specific typessignificantly improve histological and functional outcomes in rats with traumatic brain injury, or TBI, indicating that Tß4 has considerable therapeutic potential for patients with TBI. More recently, researchers studying Tß4 under a material transfer agreement (MTA) found that Tß4 had beneficial effects in animal models of stem cellsperipheral neuropathy, one of the major complications of diabetes. This research was published in the Journal of Neurobiology of Disease in December 2012 and appears to corroborate previous findings using Tß4 for repair of central nervous system disorders. A paper in Neuropharmacology in 2014 found many benefits of Tß4 administration in a rat model of spinal cord injury, including decreased lesion size at 7 days, increased neural and oligodendrocyte survival, increase levels of myelin basic protein (a marker of mature oligodendrocytes), decreased ED1 (a marker of activated microglia/macrophages), and decreased proinflammatory cytokines. Thus, Tß4 has efficacy for repair and regeneration in several nervous system injury models including MS, TBI, stroke, peripheral neuropathy, and spinal cord injury and there will likely be additional applications in this area. We believe that these various biological activities work in concert to play a vital role in the healing and repair of injured or damaged tissue and suggest that Tß4 is an essential component of the tissue protection and regeneration process that may lead to many potential medical applications. All of our product candidates utilize Tß4 as the active pharmaceutical ingredient (API), which is manufactured by solid-phase peptide synthesis and is an exact copy of the naturally occurring peptide. We have created three distinct formulations for various routes of administration and medical indications.
Our Product Candidates
RGN-259
RGN-259 is our proprietary preservative-free eye drop formulation of Thymosin beta 4. In September 2011, we completed a Phase 2a exploratory clinical trial evaluating the safety and efficacy of RGN-259 in 72 patients with moderate dry eye syndrome. Patients were randomly assigned to receive either RGN-259 or placebo in this double-masked, placebo-controlled trial. All patients received either RGN-259 (0.1% concentration) or placebo, twice daily for 30 days. Various signs and symptoms of dry eye, such as the degree of ocular surface damage, ocular itching, burning and grittiness, among others, were graded periodically during and following the treatment period. The trial was conducted by Ora Inc., an ophthalmic contract research organization that specializes in dry eye research and clinical trials, and utilized Ora’s Controlled Adverse Environment (CAESM) chamber, which is a model that exacerbates and standardizes signs and symptoms in the dry eye patient.
In November 2011, we reported preliminary safety and efficacy results from the outer layertrial. RGN-259 was deemed safe and well-tolerated, with no observed drug-related adverse events.
The co-primary outcome measures evaluated in the trial were inferior corneal fluorescein staining and decreased ocular discomfort on day 29, 24 hours after CAESMchallenge. Various secondary outcome efficacy measures were also evaluated in the trial. These outcome measures were based on the best available animal data at the time but without the benefit of any actual human clinical experience in dry eye. While the study did not meet statistical significance for reducing inferior corneal fluorescein staining, it did show a positive trend in this exploratory trial. RGN-259 did, however, show a statistically significant efficacy result in the other co-primary endpoint of decreased ocular discomfort and also demonstrated statistical significance in several secondary endpoints such as reduction of central corneal and superior corneal staining, important signs in dry eye patients and approvable endpoints by the FDA.
Key outcome measures were as follows:
| · | Patients receiving RGN-259 experienced a 325% greater reduction from baseline in central corneal fluorescein staining compared to placebo at the 24 hour recovery period (p = 0.0075). Reduction of fluorescein staining is indicative of a reduction in ocular surface damage of the central cornea; |
| 3 |
| · | Patients receiving RGN-259 experienced a 257% greater reduction from baseline in exacerbation of superior corneal fluorescein staining in the CAESM chamber as compared to the placebo (p = 0.0210); and | |
| · | Patients receiving RGN-259 experienced a 27.3% greater reduction in exacerbation of ocular discomfort at day 28 during a 75-minute challenge in the CAESM chamber compared to the placebo group (p = 0.0244). Reduction indicates that RGN-259 can slow progression of ocular symptoms in patients with dry eye syndrome. | |
| · | Other CAESM-related findings, such as peripheral (combination of the average of superior and inferior) corneal staining reduction, were observed having statistical significance, while others had positive trends after treatment with RGN-259. These observations are in line with the known biological properties and mechanisms of action of RGN-259 reported in various nonclinical studies. |
With respect to inferior corneal fluorescein staining, we did see a positive trend toward improvement, at day 28 during exposure to adverse conditions in the CAESMchamber in patients receiving RGN-259 compared to placebo, although this improvement was not deemed to be statistically significant (p = 0.0968).
Statistical significance (p value) of ≤ 0.05 is the generally accepted threshold for showing an outcome did not happen merely by chance.
The co-primary outcome measures, selected at the outset of this initial Phase 2a exploratory trial, were based on the best available animal data at the time but without the benefit of any actual human clinical experience in dry eye. Therefore, we believe that not having met one of the hearttwo co-primary outcome measures at this stage is not as important as identifying statistically significant outcomes that could potentially serve as approvable endpoints in later stage or in pivotal Phase 3 clinical trials. We believe that the statistically significant observation of reduction in central and/or superior corneal staining, as well as symptom improvements observed in the trial and described above, reflect actual patient benefits and would represent acceptable outcome measures to generate new myocardial blood vesselsthe FDA for use in follow-up Phase 2b or confirmatory pivotal Phase 3 trials. We prepared a clinical study report for submission to the FDA that describes the results of the exploratory Phase 2a clinical trial and tissuethe results were published in an appropriate medical journal.
In June 2012, we reported preliminary results from a double-masked, vehicle-controlled, physician-sponsored Phase 2 clinical trial evaluating RGN-259 for the treatment of nine patients (18 eyes) with severe dry eye. RGN-259 was observed to be safe and well-tolerated and met key efficacy objectives with statistically significant sign and symptom improvements, compared to vehicle control, at injured sites. various time intervals, including 28 days post-treatment.
In the trial, nine patients with severe dry eye (18 eyes) were treated with RGN-259 or vehicle control six times daily over a period of 28 days. They were evaluated upon entering the study after a two-week washout period, at weekly intervals during the treatment phase, at the end of the 28-day treatment period, and at a follow-up visit 28 days after treatment. Statistically significant differences in sign and symptom assessments, such as ocular discomfort and corneal fluorescein staining, were seen at various time points throughout the study. Of particular note were the differences between RGN-259 and vehicle control 28 days post-treatment, or the follow-up period. The RGN-259-treated group had a 35.1% reduction of ocular discomfort (symptom) compared to vehicle control (p = 0.0141), and a 59.1% reduction of total corneal fluorescein staining (sign) compared to vehicle control (p = 0.0108) at 28 days after treatment showing that the repair was sustained long after treatment cessation.
Consistent with the reduction of ocular discomfort and fluorescein staining at the 28-day follow-up visit, other improvements seen in the RGN-259-treated patients included tear film breakup time and increased tear volume production. Likewise, these improvements were seen at other time points in the study. These results were recently published in an appropriate medical journal.
In May 2016, we reported the results of a 317-patient Phase 2/3 trial conducted by our U.S. joint venture ReGenTree (see the discussion below under “Strategic Patnerships – U.S. Joint Venture (ReGenTree, LLC)”). In the trial, RGN-259 demonstrated statistically significant improvements in both signs and symptoms of dry eye with 0.05% and 0.1% RGN-259 compared to placebo in a dose dependent manner during a 28-day dosing period. While the primary outcome measures were not met, several key related pre-specified endpoints and subgroups of patients with more severe dry eye showed statistically significant treatment effects. These results confirm the findings from the previous Phase 2 trial providing clear direction for the clinical regulatory pathway and remaining registration trials for RGN-259.
| 4 |
RGN-259 was evaluated using the Controlled Adverse Environment (CAESM) Model developed by Ora, Inc., the product development firm managing the program. The CAESM was utilized to screen and enroll an enriched patient population and measure a patient's ability to withstand an acute adverse environmental challenge of the ocular surface.
On the final day of dosing (Day 28), patients receiving 0.1% RGN-259 had a statistically significant reduction in ocular discomfort during CAESM exposure when compared to placebo (Intent-to-Treat Population (ITT), p=0.043). Importantly, this result was also observed in the previous Phase 2 trial in patients treated with 0.1% RGN-259 (ITT, p=0.024), thereby demonstrating a symptom endpoint in two independent trials. A statistically significant ocular discomfort improvement after CAESMexposure on Day 28 was also observed in the 0.05% and 0.1% RGN-259 treatment arms when compared to placebo (ITT, p=0.0366 and p=0.0072, respectively) indicating a dose dependent response.
Efficacy in an environmental setting was also demonstrated in more symptomatic patients at baseline, with statistically significant improvements in ocular discomfort observed at day 28 prior to CAESM in patients receiving 0.05% and 0.1% RGN-259 compared to placebo (p=0.022 and p=0.006, respectively). These data suggest that RGN-259 has a fast-acting treatment effect on a dry eye symptom during exposure to an adverse environment as well as in the natural environment after 28 days of dosing.
RGN-259 also improved a common objective endpoint – ocular surface staining after 28 days of dosing in patients with compromised tear film break-up time at baseline. In this population, patients receiving 0.1% RGN-259 had a statistically significant reduction in corneal fluorescein staining prior to entering the CAESM on Day 28 when compared to placebo (p=0.034). The same result was observed in the previous Phase 2 trial for patients treated with 0.1% RGN-259, although it was not statistically significant in the smaller sample size of this previous Phase 2 trial. Additionally, a change from baseline analysis (Day 28 minus Day 0) demonstrated a statistically significant improvement in inferior corneal staining for the 0.1% RGN-259 treatment arm when compared to placebo (p=0.003). This finding was also observed at Day 14 compared to placebo (p=0.035). These data suggest that RGN-259 has a fast-acting treatment effect on a dry eye sign after 14 and 28 days of dosing.
There were no significant drug-related adverse or serious adverse events and RGN-259 was well-tolerated and comfortable for the patients with no irritation upon instillation.
ReGenTree intends to meet with the FDA this summer to provide full results of the trial and its plan to conduct a confirmatory Phase 3 study to start by the fourth quarter of 2016.
RGN-352
During 2009, we completed a Phase 11a and Phase 1b clinical trial evaluating the safety, tolerability and pharmacokinetics of the intravenous administration of RGN-352 in 60 healthy subjects. The product candidate was well-tolerated, and there were no reported drug-related adverse events.
| 5 |
In addition to the potential application of this offering to initiate and conduct this Phase 2 clinical trial, although depending on the amount of proceeds we may not be able to complete the trial without additional capital. Depending on our capital resources, we may conduct the trial while also continuing strategic partnership discussions with biotechnology and pharmaceutical companiesRGN-352 for the further developmenttreatment of RGN-352. In May 2010, we were awarded a $3 million grant from the National Heart, Lung and Blood Institute, one of the institutes of the NIH, to support the further development of RGN-352.
2
RGN-137
Clinical Development — Epidermolysis Bullosa (EB). In 2005, we initially sought for RGN-259. Based on these data and slower than expected patient accrualbegan enrolling patients in a Phase 2 ophthalmic wound healing trial that we had initiated with RGN-259, in 2009, we closed the Phase 2 trial after enrolling the initial low-dose cohort. The results from evaluating this initial cohort indicated increased corneal epithelial thickening and reduced cell and flare inflammation in the Tß4-treated patients, as compared to patients who were administered placebo. We believe these results are indicative of Tß4’s activities in corneal re-epithelialization and healing.
3
Clinical Development — Pressure Ulcers. In late 2005, we began enrolling patients in a Phase 2 clinical trial designed to assess the safety and Polandeffectiveness of RGN-137 for the treatment of patients with chronic pressure ulcers, commonly known as bedsores. In this randomized, double-blind, placebo-controlled, dose-response trial, 15 clinical sites in the United States enrolled a total of 72 patients to evaluate the safety, tolerability, and wound healing effectiveness of three different concentrations of RGN-137 compared to placebo. RGN-137 was applied topically to patients’ ulcers, once daily for up to 84 consecutive days. Patients in the trial were between 19 and 85 years old and had at least one stable Stage III or IV pressure ulcer with a surface area between 5 and 70 cm2. Stage III and IV pressure ulcers are full thickness wounds that penetrate through the skin and muscle, sometimes completely to the bone.
In January 2009, we reported final data from this trial. RGN-137 was well-tolerated at all three dose levels studied, with no dose-limiting adverse events, which achieved the primary objective of the study. As for efficacy, all Tß4 doses performed similarly compared to placebo, with no statistically significant efficacy results. However, patients treated with the middle dose showed a 17% improvement of wound healing, which was the highest rate among the three active doses evaluated. The improvement in ulcer healing in this middle dose group following nine weeks of treatment was equal to the improvement in patients treated with placebo after 12 weeks of treatment. A follow-on evaluation, reported at the 3rd International Symposium on the Thymosins in Health and Disease in March 2012, showed that for those pressure ulcer patients’ wounds that healed, RGN-137 mid dose (0.02% Tß4 gel product) accelerated wound closure with a median time to healing of 22 days as compared to 57 days for the placebo. Although those results are clinically significant, they were not statistically significant.
| 6 |
Clinical Development — Venous Stasis Ulcers. In mid-2006 we began enrolling patients in a Phase 2 clinical trial designed to assess the safety and effectiveness of RGN-137 for the treatment of patients with venous stasis ulcers.
In 2009, we reported final data from that trial. All doses of RGN-137 were well tolerated. More patients achieved healing in the RGN-137 mid dose (0.03% Tß4 gel product) than in any other dose group. The mid dose showed both an increased incidence of wound healing and a faster healing time compared to placebo. The mid dose decreased the median time to healing by 45% among those wounds that completely healed. A follow-on evaluation, reported at the 3rd International Symposium on the Thymosins in Health and Disease in March 2012, showed that for those venous stasis ulcer patients’ wounds greater than 3 cm2 that healed, the RGN-137 mid dose (0.03% Tß4 gel product) accelerated wound closure with a median time to healing of 49 days as compared to 78 days for the placebo. Those results were both clinically and statistically significant.
GtreeBNT.In March 2014, we entered into a License Agreement with GtreeBNT to license certain development and commercialization rights for eachRGN-137 in the U.S.
Strategic Partnerships
Lee’s Pharmaceuticals.In July 2012, we entered into a License Agreement with Lee’s Pharmaceutical (HK) Limited (“Lee’s”), headquartered in Hong Kong, for the license of Thymosin Beta 4 in any pharmaceutical form, including our RGN-259, RGN-352 and RGN-137 product candidates, in China, Hong Kong, Macau and Taiwan. Lee’s has filed an investigational new drug candidateapplication IND with the Chinese FDA to conduct a Phase 2, randomized, double-masked, dose-response clinical trial with RGN-259 in China for dry-eye syndrome. Lee's recently informed us that it received notice from China's FDA (CFDA) declining its investigational new drug (IND) application for a Phase 2b dry eye clinical trial because the API (active pharmaceutical ingredient or Tß4) was manufactured outside of China. The API was manufactured in the U.S. and provided to Lee's by RegeneRx pursuant to a license agreement to develop RGN-259 ophthalmic eye drops in the licensed territory. Due to this unexpected regulatory hurdle, Lee's is modifying its clinical program to conduct the Phase 2b dry eye trial in Hong Kong and Taiwan using the Tß4 supplied by RegeneRx while awaiting the manufacturing of Tß4 in China for a subsequent Phase 3 registration trial. We do not know when the Hong Kong/Taiwan trial will begin enrollment of patients. Under this revised strategy, Lee's believes it should be able to begin a Phase 3 registration trial in China sooner rather than waiting on the production of Tß4 in China before initiating a Phase 2b trial.
GtreeBNT.In March 2014, we entered into a License Agreement with GtreeBNT Co., Ltd., a Korean pharmaceutical company (“Gtree”) and shareholder of the Company, for the license of RGN-259. GtreeBNT licensed certain development and commercialization rights for RGN-259, in Asia (excluding China, Hong Kong, Macau and Taiwan). GtreeBNT is currently our peptide fragments,second largest shareholder. Gtree filed an IND with certain exceptions. For example,the Korean Ministry of Food and Drug Safety to conduct a Phase 2b/3 study with RGN-259 in patients with dry eye syndrome and in July 2015 received approval to conduct the trial. GtreeBNT has informed us that given its immediate focus on the two U.S. trials, it is considering the best timing for the Korean trial.
U.S. Joint Venture (ReGenTree, LLC).On January 28, 2015, we believeannounced that we can commercially developentered into a Joint Venture Agreement (the “Joint Venture Agreement”) with GtreeBNT. The Joint Venture Agreement provides for the creation of an entity (the “Joint Venture” or “ReGenTree”), owned by us and marketRGN-137GtreeBNT, that will commercialize RGN-259 for EB, since the patient population is smalltreatment of dry eye and well-defined, as is the population of pediatric dermatologists who specialize in treating this disease. We continue to hold strategic discussions with pharmaceutical and biotechnology companies at each development milestone for each product candidate.
4
Our ownership interest in ReGenTree is 49% and will be reduced to 42% following completion of the Phase 2b trial for Dry Eye Syndrome. Based on when, and if, ReGenTree achieves certain additional development milestones in the U.S. with RGN-259, our equity ownership may be incrementally reduced to between 42% and 25%, with 25% being the final equity ownership upon FDA approval of an NDA for Dry Eye Syndrome in the U.S. In addition to our equity ownership, RegeneRx retains a royalty on net sales that varies between single and low double digits, depending on whether commercial sales are made by ReGenTree or a licensee. In the event the ReGenTree entity is acquired or there is a change of control that occurs following achievement of an NDA, RegeneRx shall be entitled to 40% of all change of control proceeds paid or payable and will forgo any future royalties.
The numberNK trial, a smaller study in an orphan population, has enrolled seven patients thus far with a goal of 46. Of the eight original clinical sites for the study, six are enrolling patients, one has yet to receive IRB approval to begin enrolling patients, and one was unable to consummate a clinical contract with ReGenTree. ReGenTree is considering adding additional sites to accelerate patient enrollment.
GtreeBNT has developed the CMC (chemistry, manufacturing and controls) dossier required for Phase 3 clinical trials and commercialization in the U.S. and in Korea. This comprehensive and critical effort ensures that final drug product manufacturing, packaging, stability, purity, reproducibility, etc., meets regulatory guidelines and product specifications. The product of this activity is the current product format being utilized in the U.S. trials being conducted by ReGenTree and will also be utilized in the planned clinical activity to be conducted by GtreeBNT under the RGN-259 license agreement for Pan Asia.
In May 2015, ReGenTree completed a Phase 2/3 study with regard to RGN-259. See the discussion above under “Our Product Candidates – RGN-259.”
Risk Factors
As with most biopharmaceutical product candidates, the development of our product candidates is subject to numerous risks, including the risk of delays in or discontinuation of development from lack of financing, inability to obtain necessary regulatory approvals to market the products, unforeseen safety issues relating to the products and dependence on third party collaborators to conduct research and development of the products. Because we are an early stage company with a very limited history of operations, we are also subject to many risks associated with early-stage companies. For a more detailed discussion of some of the risks you should consider before purchasing shares of our common stock that will be outstanding immediately afteror other securities issued by us, you are urged to carefully review and consider the section entitled “Risk Factors” beginning on page10 of this prospectus.
The Offering
The selling stockholders identified beginning on page 27 of this prospectus are offering is based on 60,406,828 a resale basis a total of10,551,471shares of our common stock, of which 5,404,412 are issuable upon the exercise of outstanding warrants. The total value of all the common stock offered pursuant to this prospectus is approximately $4.5 million, based upon a per share price of $0.43, which represents the average of the high and low prices of our common stock as of March 31, 2010, and excludes:
| Common stock offered | 10,551,471 shares | |
| Common stock outstanding before the offering (1) | 101,640,092 shares | |
| Common stock outstanding after the offering (2) | 112,191,563 shares |
| 8 |
| Use of Proceeds | We will receive none of the proceeds from the sale of the shares by the selling stockholders, except for the warrant exercise price upon exercise of the warrants, which would be used for working capital and other general corporate purposes. | |
| OTCQB Symbol | RGRX |
| (1) | Based on the number of shares outstanding as of | |
| convertible promissory notes. | ||
| Assumes the issuance of all shares offered hereby that are issuable upon exercise of |
5
| Year Ended | Three Months Ended | |||||||||||||||
| December 31, | March 31, | |||||||||||||||
| 2009 | 2008 | 2010 | 2009 | |||||||||||||
Statement of Operations Data: | ||||||||||||||||
| Sponsored research revenue | $ | — | $ | 168,412 | $ | — | $ | — | ||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | 3,724,514 | 7,149,808 | 470,434 | 1,661,600 | ||||||||||||
| General and administrative | 2,781,790 | 3,805,346 | 678,068 | 859,568 | ||||||||||||
| Total operating expenses | 6,506,304 | 10,955,154 | 1,148,502 | 2,521,168 | ||||||||||||
| Loss from operations | (6,506,304 | ) | (10,786,742 | ) | (1,148,502 | ) | (2,521,168 | ) | ||||||||
| Interest income | 12,444 | 149,777 | 2,793 | 6,518 | ||||||||||||
| Net loss | $ | (6,493,860 | ) | $ | (10,636,965 | ) | $ | (1,145,709 | ) | $ | (2,514,650 | ) | ||||
| Basic and diluted net loss per share | $ | (0.12 | ) | $ | (0.21 | ) | $ | (0.02 | ) | $ | (0.05 | ) | ||||
| Shares used to compute basic and diluted net loss per share | 55,680,525 | 50,967,617 | 60,406,828 | 53,622,491 | ||||||||||||
| As of March 31, 2010 | ||||||||
| Actual | As Adjusted | |||||||
Balance Sheet Data: | ||||||||
| Cash and cash equivalents | $ | 3,189,990 | $ | 8,637,974 | ||||
| Working capital | 2,689,485 | 8,096,771 | ||||||
| Total assets | 3,455,854 | 8,903,838 | ||||||
| Common stock | 60,407 | 71,907 | ||||||
Additionalpaid-in capital | 88,276,191 | 93,712,675 | ||||||
| Accumulated deficit | (85,647,113 | ) | (85,647,113 | ) | ||||
| Total stockholders’ equity | 2,689,485 | 8,137,469 | ||||||
6
Investing in our securities involves a high degree of risk. You should carefully consider the risks described below, as well as the other information included in this prospectus, before you decide to purchase our securities. If any of the following risks actually occurs, they may harm our business, prospects, financial condition and operating results. As a result, the trading price of our securities could decline and you could lose part or all of your investment.
Risks Related to Our Liquidity and Need for Financing
Even after giving effect to the proceeds of this offering, we estimate that our capital resources will only be sufficient to fund our operations into the second quarter of 2011.
In addition to our current development objectives, weWe will need substantial additional capital for the continued development of product candidates through marketing approval and for our longer-term future operations.
We anticipate that substantial new capital resources will be required to continue our longer-term independent product development efforts, including any and all follow-on trials that will result from our current clinical programs beyond those currently contemplated, and to scale up manufacturing processes for our product candidates. TheHowever, the actual amount of funds that we will need will be determined by many factors, some of which are beyond our control. These factors include, without limitation:
| the scope of our clinical trials, which is significantly influenced by the quality of clinical data achieved as trials are completed and the requirements established by regulatory authorities; | ||
| the speed with which we complete our clinical trials, which depends on our ability to attract and enroll qualifying patients and the quality of the work performed by our clinical | ||
| investigators and contract research organizations chosen to conduct the studies; | ||
| · | the time required to prosecute, enforce and defend our intellectual property rights, which depends on evolving legal regimes and infringement claims that may arise between us and third parties; | |
| the ability to manufacture at scales sufficient to supply commercial quantities of any of our product candidates that receive regulatory approval, which may require levels of effort not currently anticipated; and |
7
| · | the successful commercialization of our product candidates, which will depend on our ability to either create or partner with an effective commercialization organization and which could be delayed or prevented by the emergence of equal or more effective therapies. |
Emerging biotechnology companies like us may raise capital through corporate collaborations and by licensing intellectual property rights to other biotechnology or pharmaceutical enterprises. We intend to pursue this strategy, but there can be no assurance that we will be able to enter into additional license agreements with respect to our intellectual property or product development programs on commercially reasonable terms, if at all. There are substantial challenges and risks that will make it difficult to successfully implement any of these alternatives. If we are successful in raising additional capital through such a license or collaboration, we may have to give up valuable rights to our intellectual property. In addition, the business priorities of a strategic partner may change over time, which creates the possibility that the interests of the strategic partner in developing our technology may diminish whichand could have a potentially material negative impact on the value of our interest in the licensed intellectual property or product candidates.
Further, if we raise additional funds by selling shares of our common stock or securities convertible into our common stock the ownership interest of our existing stockholders may be significantly diluted. If additional funds are raised through the issuance of preferred stock or debt securities, these securities are likely to have rights, preferences and privileges senior to our common stock and may involve significant fees, interest expense, restrictive covenants or the granting of security interests in our assets.
Our failure to successfully address our long-term liquidity requirements would have a material negative impact on our business, including the possibility of surrendering our rights to some technologies or product opportunities, delaying our clinical trials or ceasing our operations.
| 10 |
We have incurred losses since inception and expect to incur significant losses in the foreseeable future and may never become profitable.
We have not commercialized any product candidates to date and incurred net operating losses every year since our inception in 1982. We believe these losses will continue for the foreseeable future, and may increase, as we pursue our product development efforts related to Tß4. As of March 31, 2010,2016, our accumulated deficit totaled approximately $85.6$108 million.
As we expand our research and development efforts and seek to obtain regulatory approval of our product candidates to make them commercially viable, we anticipate substantial and increasing operating losses. Our ability to generate additional revenues and to become profitable will depend largely on our ability, alone or through the efforts of third-party licensees and collaborators, to efficiently and successfully complete the development of our product candidates, obtain necessary regulatory approvals for commercialization,scale-up commercial quantity manufacturing capabilities either internally or through third-party suppliers, and market our product candidates. There can be no assurance that we will achieve any of these objectives or that we will ever become profitable or be able to maintain profitability. Even if we do achieve profitability, we cannot predict the level of such profitability. If we sustain losses over an extended period of time and are not otherwise able to raise necessary funds to continue our development efforts and maintain our operations, we may be forced to cease operations.
We are currently not in compliance with NYSE Amex rules regarding
Our common stock is quoted on the minimum shareholders’ equity requirement and are at risk of being delisted from the NYSE Amex stock exchange,over-the-counter market, which may subjectsubjects us to the SEC’s penny stock rules and may decrease the liquidity of our common stock.
8
In addition, if our common stock is delisted, our ability to raise additional capital may be impaired because of the less liquid nature of the OTC Bulletin Board and the pink sheets.over-the-counter markets. While we cannot guarantee that we would be able to complete an equity financing on acceptable terms, or at all, we believe that dilution from any equity financing while our shares are quoted on the OTC Bulletin Board or the pink sheetsan over-the-counter market would likely be substantially greater than if we were to complete thea financing while our common stock is traded on a national securities exchange. Further, we are unable to use short-form registration statements on Form S-3 for the NYSE Amex exchange.
Our common stock is delisted, it may also become subject to penny stock rules.rules, which impose additional sales practice requirements on broker-dealers who sell our common stock. The SEC Commissiongenerally defines “penny stock” as an equity security that has a market price of less than $5.00 per share, subject to certain exceptions. We are not currently subject to the penny stock rules because our common stock qualifies for an exception to the SEC’s penny stock rules for companies that have an equity security that is quoted on an exchange. However, if we were delisted, our common stock would become subject to the penny stock rules, which impose additional sales practice requirements on broker-dealers who sell our common stock. If our common stock were considered penny stock, theThe ability of broker-dealers to sell our common stock and the ability of our stockholders to sell their shares in the secondary market wouldwill be limited and, as a result, the market liquidity for our common stock wouldwill likely be adversely affected. We cannot assure you that trading in our securities will not be subject to these or other regulations in the future.
The report of our independent registered public accounting firm containsas of December 31, 2015 contained explanatory language that substantial doubt exists about our ability to continue as a going concern.
The report of our independent registered public accounting firm on our financial statements for the year ended December 31, 20092015 contains explanatory language that substantial doubt exists about our ability to continue as a going concern, without raising additional capital. Althoughconcern. However, since December 31, 2015, we expect thathave raised an aggregate of approximately $1.9 million through an agreement to expand the proceedsterritory of a license agreement and a private offering of stock and warrants. We believe these funds will fund operations for at least the 12 months following the date of this offeringProspectus. However, we will provide us the abilityneed to seek additional sources of capital to fund our long term operations, into the second quarter of 2011,and if we will continue to have a need for additional financing after this offering, which we may not be able to complete either on favorable terms or at all. As a result, we cannot assure you that, after taking into account the proceeds of this offering and our anticipated expenditures for the remainder of 2010, the report of our independent registered public accounting firm for the year ending December 31, 2010 will not express substantial doubt about our ability to continue as a going concern. If this were to occur, we would once again face the needare unable to obtain sufficient financing in the short term,to support and the failure to do socomplete these activities, then we would, in all likelihood, createexperience severe liquidity problems and cause us tomay have to curtail our operations. If we were to curtail our operations, we couldmay be placed into bankruptcy or undergo liquidation, the result of which wouldwill adversely affect the value of our common stock.shares.
| 11 |
9
Our planned Phase 2 clinical trial of RGN-352 was placed on clinical hold by the FDA in March 2011 due to non-compliance of cGMP regulations by a contract manufacturer and we are unsure when, if ever, we will be able to resume this trial.
In the second half of 2010, we implemented the development plans for our phase 2 clinical trial to evaluate RGN-352 in patients who have suffered an acute myocardial infarction, or AMI. We had planned to begin enrolling patients near the end of the first quarter of 2011. However, in March 2011, we were notified by the FDA that the trial was placed on clinical hold as a result of our contract manufacturer’s alleged failure to comply with current Good Manufacturing Practice (“cGMP”) regulations. The FDA has prohibited us from using any of the active drug or placebo manufactured by this manufacturer in human trials, which will require us to identify a cGMP-compliant manufacturer and to have new material produced in the event that we seek to resume this trial. We have also learned that the contract manufacturer has closed its manufacturing facility and has filed for bankruptcy protection. Significant preparatory time and procedures will be required before any new suitable manufacturer would be able to manufacture RGN-352 for the AMI trial. Since we are unable to estimate the length of time that the trial will be on clinical hold, we have elected to cease activities on this trial until the FDA clinical hold is resolved and the requisite funding might be secured. Consequently, there can be no assurance that we will be able to timely initiate trial activities or complete this trial, if at all.
All of our drug candidates are based on a single compound that has yet to be proven effective in human subjects.compound.
Our current primary business focus is the development of Tß4, and its analogues, derivatives and fragments, for the improvementregeneration and accelerated repair of damaged tissue from non-healing dermal and corneal wounds, cardiac function, the acceleration of corneal healing, the treatment of non-healing woundsinjury, central/peripheral nervous system diseases and other conditions.conditions, as well as an improvement in various functions, such as, but not limited to, cardiac and neurological. Unlike many pharmaceutical companies that have a number of unique chemical entities in development, we are dependent on a single molecule, formulated for different routes of administration and different clinical indications, for our potential commercial success. As a result, any common safety or efficacy concerns for Tß4-based products that cross formulations would have a much greater impact on our business prospects than if our product pipeline were more diversified.
We may never be able to commercialize our product candidates.
Although Tß4 has shown biological activity inin vitro studies and in vivo animal models and while we observed clinical activity and efficacious outcomes in our recent RGN-259 Phase 2a trial and earlier Phase 2 dermal trials, we cannot assure you that our product candidates will exhibit activity or importance in humans.humans in large-scale trials. Our drug candidates are still in research and development, and we do not expect them to be commercially available for the foreseeable future, if at all. Only a small number of research and development programs ultimately result in commercially successful drugs. Potential products that appear to be promising at early stages of development may not reach the market for a number of reasons. These include the possibility that the potential products may:
| be found ineffective or cause harmful side effects during preclinical studies or clinical trials; | ||
| fail to receive necessary regulatory approvals; | ||
| be precluded from commercialization by proprietary rights of third parties; | ||
| be difficult to manufacture on a large scale; or | ||
| be uneconomical or otherwise fail to achieve market acceptance. |
If any of these potential problems occurs, we may never successfully market Tß4-based products.
| 12 |
We are subject to intense government regulation, and we may not receive regulatory approvals for our drug candidates.
Our product candidates will require regulatory approvals prior to sale. In particular, therapeutic agents are subject to stringent approval processes, prior to commercial marketing, by the FDA and by comparable agencies in most foreign countries. The process of obtaining FDA and corresponding foreign approvals is costly and time-consuming, and we cannot assure you that such approvals will be granted. Also, the regulations we are subject to change frequently and such changes could cause delays in the development of our product candidates.
Three of our drug candidates are currently in the clinical development stage, and we cannot be certain that we or our collaborators will successfully complete the clinical trials necessary to receive regulatory product approvals. The regulatory approval process is lengthy, unpredictable and expensive. To obtain regulatory approvals in the United States, we or a collaborator must ultimately demonstrate to the satisfaction of the U.S. Food and Drug Administration, or FDA that our product candidates are sufficiently safe and effective for their proposed administration to humans. Many factors, known and unknown, can adversely impact clinical trials and the ability to evaluate a product candidate’s safety and efficacy, including:
| the FDA or other health regulatory authorities, or institutional review boards, or IRBs, do not approve a clinical trial protocol or place a clinical trial on hold; | ||
| suitable patients do not enroll in a clinical trial in sufficient numbers or at the expected rate, for reasons such as the size of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the perceptions of investigators and patients regarding safety, and the availability of other treatment options; |
10
| clinical trial data is adversely affected by trial conduct or patient withdrawal prior to completion of the trial; | ||
| there may be competition with ongoing clinical trials and scheduling conflicts with participating clinicians; | ||
| patients experience serious adverse events, including adverse side effects of our drug candidates, for a variety of reasons that may or may not be related to our product candidates, including the advanced stage of their disease and other medical problems; | ||
| patients in the placebo or untreated control group exhibit greater than expected improvements or fewer than expected adverse events; | ||
| third-party clinical investigators do not perform the clinical trials on the anticipated schedule or consistent with the clinical trial protocol and good clinical practices, or other third-party organizations do not perform data collection and analysis in a timely or accurate manner; | ||
| service providers, collaborators or co-sponsors do not adequately perform their obligations in relation to the clinical trial or cause the trial to be delayed or terminated; | ||
| we are unable to obtain a sufficient supply of manufactured clinical trial materials; | ||
| regulatory inspections of manufacturing facilities, which may, among other things, require us or aco-sponsor to undertake corrective action or suspend the clinical | ||
| the interim results of the clinical trial are inconclusive or negative; | ||
| the clinical trial, although approved and completed, generates data that is not considered by the FDA or others to be clinically relevant or sufficient to demonstrate safety and efficacy; and | ||
| · | changes in governmental regulations or administrative actions affect the conduct of the clinical trial or the interpretation of its results. |
There can be no assurance that our clinical trials will in fact demonstrate, to the satisfaction of the FDA and others, that our product candidates are sufficiently safe or effective. The FDA or we may also restrict or suspend our clinical trials at any time if either believesit is believed that we are exposing the subjects participating in the trials are being exposed to unacceptable health risks.
Clinical trials for product candidates such as ours are often conducted with patients who have more advanced forms of a particular condition or other unrelated conditions. For example, in clinical trials for our product candidate RGN-137, we have studied patients who are not only suffering from chronic epidermal wounds but who are also older and much more likely to have other serious adverse conditions. During the course of treatment with our product candidates, patients could die or suffer other adverse events for reasons that may or may not be related to the drug candidate being tested. Further, and as a consequence ofthat all of our drug candidates beingare based on Tß4, crossover risk exists such that a patient in one trial may be adversely impacted by one drug candidate, and that adverse event may have implications for our other trials and other drug candidates. However, even if unrelated to our product candidates, such adverse events can nevertheless negatively impact our clinical trials, and our business prospects would suffer.
These factors, many of which may be outside of our control, may have a negative impact on our business by making it difficult to advance product candidates or by reducing or eliminating their potential or perceived value. As a consequence, we may need to perform more or larger clinical trials than planned. Further, if we are forced to contribute greater financial and clinical resources to a study, valuable resources will be diverted from other areas of our business. If we fail to complete or if we experience material delays in completing our clinical trials as currently planned, or we otherwise fail to commence or complete, or experience delays in, any of our other present or planned clinical trials, including as a result of the actions of third parties upon which we rely for these functions, our ability to conduct our business as currently planned could materially suffer.
11
We have only limited resources, experience with and capacity to conduct requisite testing and clinical trials of our drug candidates. As a result, we rely and expect to continue to rely on third-party service providers and collaborators, including corporate partners, licensors and contract research organizations, or CROs, to perform a number of activities relating to the development of our drug candidates, including the design and conduct of clinical trials, and potentially the obtaining of regulatory approvals. For example, we currently rely on several third-party contractors to manufacture and formulate Tß4 into the product candidates used in our clinical trials, develop assays to assess Tß4’s effectiveness in complex biological systems, recruit clinical investigators and sites to participate in our trials, manage the clinical trial process and collect, evaluate and report clinical results.
We may not be able to maintain or expand our current arrangements with these third parties or maintain such relationships on favorable terms. Our agreements with these third parties may also contain provisions that restrict our ability to develop and test our product candidates or that give third parties rights to control aspects of our product development and clinical programs. In addition, conflicts may arise with our collaborators, such as conflicts concerning the interpretation of clinical data, the achievement of milestones, the interpretation of financial provisions or the ownership of intellectual property developed during the collaboration. If any conflicts arise with our existing or future collaborators, they may act in their self-interest, which may be adverse to our best interests. Any failure to maintain our collaborative agreements and any conflicts with our collaborators could delay or prevent us from developing our product candidates. We and our collaborators may fail to develop products covered by our present and future collaborations if, among other things:
| we do not achieve our objectives under our collaboration agreements; | ||
| · | we or our collaborators are unable to obtain patent protection for the products or proprietary technologies we develop in our collaborations; | |
| we are unable to manage multiple simultaneous product development collaborations; | ||
| our collaborators become competitors of ours or enter into agreements with our competitors; | ||
| we or our collaborators encounter regulatory hurdles that prevent commercialization of our product candidates; or | ||
| we develop products and processes or enter into additional collaborations that conflict with the business objectives of our other collaborators. |
We also have less control over the timing and other aspects of our clinical trials than if we conducted the monitoring and supervision entirely on our own. Third parties may not perform their responsibilities for our clinical trials on our anticipated schedule or consistent with a clinical trial protocol or applicable regulations. We also rely on clinical research organizations to perform much of our data management and analysis. They may not provide these services as required or in a timely manner. If any of these parties do not meet deadlines or follow proper procedures, including procedures required by law, the preclinical studies and clinical trials may take longer than expected, may be delayed or may be terminated, which would have a materially negative impact on our product development efforts. If we were forced to find a replacement entity to perform any of our preclinical studies or clinical trials, we may not be able to find a suitable entity on favorable terms or at all. Even if we were able to find a replacement, resulting delays in the tests or trials may result in significant additional expenditures and delays in obtaining regulatory approval for drug candidates, which could have a material adverse impact on our results of operations and business prospects.
GtreeBNT Co., Ltd. has limited drug development experience.
In March 2014 we completed two licensing agreements for the development and commercialization of RGN-259 and RGN-137 in certain territories, with GtreeBNT, headquartered outside of Seoul, Korea. In January 2015 we entered into a Joint Venture Agreement with GtreeBNT and entered into a license agreement with the Joint Venture, pursuant to which we granted to the Joint Venture the right to develop and exclusively commercialize RGN-259 in the United States. Although we will share control of the Joint Venture with GtreeBNT, GtreeBNT will have greater control over the Joint Venture than we will meaning that GtreeBNT will have significant control over the commercialization of RGN-259.
Historically, GtreeBNT’s business focus has been in the IT software industry in Korea with strong IP positions addressing specific software tools and apps such as optimized multimedia software for smart phones. GtreeBNT made a strategic decision in November 2013 to expand into the biopharmaceutical business through selected strategic alliances with biopharmaceutical companies in the U.S. and EU. The collaboration with RegeneRx is the first strategic investment in this initiative. While GtreeBNT has hired executives and staff with significant pharmaceutical experience, the company has no internal drug development experience. As a result, GtreeBNT may face more and different challenges in the development of these product candidates than would more established pharmaceutical companies.
We are subject to intense competition from companies with greater resources and more mature products, which may result in our competitors developing or commercializing products before or more successfully than we do.
We are engaged in a business that is highly competitive. Research and development activities for the development of drugs to treat indications within our focus are being sponsored or conducted by private and
12
| 15 |
With respect to our product candidate RGN-259, there are also numerous ophthalmic companies developing drugs for corneal wound healing and other front-of-the-eye diseases and injuries, including dry eye syndrome. Amniotic membranes have been successfully used to treat corneal wounds in certain cases, as have topical steroids and antibacterial agents. Most specialty ophthalmic companies have a number of products on the market that could compete with RGN-259. There are numerous antibiotics to treat eye infections to promote corneal wound healing and many eye lubrication products that are soothing to the eye and help eye healing, many of which are sold without prescriptions. Companies also market steroids to treat certain conditions within our area of interest. Allergan, Inc. markets Restasis™, Ophthalmic Emulsion, the only commercially available and FDA-approved eye drop to treat dry eye. Restasis, and other products, have been approved for marketing in certain other countries where we have licensed RGN-259. Shire PLC is developing is product candidate, Lifitegrast, and is in pivotal Phase 3 clinical trials in the U.S. Shire has said it plans to resubmit an NDA for Lifitegrast in 2016.
We have initially targeted our product candidate RGN-352 for cardiovascular indications. Most large pharmaceutical companies and many smaller biomedical companies are vigorously pursuing the development of therapeutics to treat patients after heart attacks and for other cardiovascular indications. With respect to our product candidate RGN-259 for corneal defects, there are also numerous ophthalmic companies developing drugs for corneal wound healing and otheroutside-of-the-eye diseases and injuries. Amniotic membranes have been successfully used to treat corneal wounds in certain cases, as have topical steroids and antibacterial agents.
With respect to our product candidate RGN-137 for wound healing, Johnson & Johnson has previously marketed RegranextmRegranex™ for this purpose in patients with diabetic foot ulcers. Other companies, such as Novartis, are developing and marketing artificial skins, which we believe could also compete with RGN-137. Moreover, wound healing is a large and highly fragmented marketplace attracting many companies, large and small, to develop products for treating acute and chronic wounds, including, for example, honey-based ointments, hyperbaric oxygen therapy, and low frequency cavitational ultrasound.
Even if approved for marketing, our technologies and product candidates are unproven and they may fail to gain market acceptance.
Our product candidates, all of which are based on the molecule Tß4, are new and unproven and there is no guarantee that health care providers or patients will be interested in our product candidates, even if they are approved for use. If any of our product candidates are approved by the FDA, our success will depend in part on our ability to demonstrate sufficient clinical benefits, reliability, safety, and cost effectiveness of our product candidates relative to other approaches, as well as on our ability to continue to develop our product candidates to respond to competitive and technological changes. If the market does not accept our product candidates, when and if we are able to commercialize them, then we may never become profitable. Factors that could delay, inhibit or prevent market acceptance of our product candidates may include:
| the timing and receipt of marketing approvals; | ||
| the safety and efficacy of the products; | ||
| the emergence of equivalent or superior products; | ||
| the cost-effectiveness of the products; and | ||
| ineffective marketing. |
It is difficult to predict the future growth of our business, if any, and the size of the market for our product candidates because the markets are continually evolving. There can be no assurance that our product candidates will prove superior to products that may currently be available or may become available in the future or that our research and development activities will result in any commercially profitable products.
| 16 |
13
Although we do not currently have any marketable products, our ability to produce revenues ultimately depends on our ability to sell our product candidates if and when they are approved by the FDA and other regulatory authorities. We currently have no experience in marketing or selling pharmaceutical products, and we do not have a marketing and sales staff or distribution capabilities. Developing a marketing and sales force is also time-consuming and could delay the launch of new products or expansion of existing product sales. In addition, we will compete with many companies that currently have extensive and well-funded marketing and sales operations. If we fail to establish successful marketing and sales capabilities or fail to enter into successful marketing arrangements with third parties, our ability to generate revenues will suffer.
If we enter markets outside the United States our business will be subject to political, economic, legal and social risks in those markets, which could adversely affect our business.
There are significant regulatory and legal barriers to entering markets outside the United States that we must overcome if we seek regulatory approval to market our product candidates in countries other than the United States. We would be subject to the burden of complying with a wide variety of national and local laws, including multiple and possibly overlapping and conflicting laws. We also may experience difficulties adapting to new cultures, business customs and legal systems. Any sales and operations outside the United States would be subject to political, economic and social uncertainties including, among others:
| changes and limits in import and export controls; | ||
| increases in custom duties and tariffs; | ||
| changes in currency exchange rates; | ||
| economic and political instability; | ||
| changes in government regulations and laws; | ||
| absence in some jurisdictions of effective laws to protect our intellectual property rights; and | ||
| currency transfer and other restrictions and regulations that may limit our ability to sell certain product candidates or repatriate profits to the United States. |
Any changes related to these and other factors could adversely affect our business if and to the extent we enter markets outside the United States.
Governmental and third-party payors may subject any product candidates we develop to sales and pharmaceutical pricing controls that could limit our product revenues and delay profitability.
The successful commercialization of our product candidates, if they are approved by the FDA, will likely depend on our ability to obtain reimbursement for the cost of the product and treatment. Government authorities, private health insurers and other organizations, such as health maintenance organizations, are increasingly seeking to lower the prices charged for medical products and services. Also, the trend toward managed health care in the United States, the growth of healthcare maintenance organizations, and recently enacted legislation reforming healthcare and proposals to reform government insurance programs could have a significant influence on the purchase of healthcare services and products, resulting in lower prices and reducing demand for our product candidates. The cost containment measures that healthcare providers are instituting and any healthcare reform could reduce our ability to sell our product candidates and may have a material adverse effect on our operations. We cannot assure you that reimbursement in the United States or foreign countries will be available for any of our product candidates, and that any reimbursement granted will be maintained, or that limits on reimbursement available from third-party payors will not reduce the demand for, or the price of, our product candidates. The lack or inadequacy of third-party reimbursements for our product candidates would decrease the potential profitability of our operations. We cannot forecast what
14
| 17 |
We have no manufacturing or formulation capabilities and are dependent upon third-party suppliers to provide us with our product candidates. If these suppliers do not manufacture our product candidates in sufficient quantities, at acceptable quality levels and at acceptable cost, or if we are unable to identify suitable replacement suppliers if needed, our clinical development efforts could be delayed, prevented or impaired.
We do not own or operate manufacturing facilities and have little experience in manufacturing pharmaceutical products. We currently rely, and expect to continue to rely, primarily on peptide manufacturers to supply us with Tß4 for further formulation into our product candidates. We have historically engaged three separate smaller drug formulation contractors for the formulation of clinical grade product candidates, one for each of our three product candidates in clinical development.development, although, as described in this report, the contractor we engaged to formulate and vial RGN-352 filed for bankruptcy and closed its manufacturing facility, and our clinical trial involving RGN-352 has been placed on clinical hold. We currently do not have an alternative source of supply for either Tß4 or the individual drug candidates. If these suppliers, together or individually, are not able to supply us with either Tß4 or individual product candidates on a timely basis, in sufficient quantities, at acceptable levels of quality and at a competitive price, or if we are unable to identify a replacement manufacturer to perform these functions on acceptable terms as needed, our development programs could be seriously jeopardized.
The clinical hold on our RGN-352 trial will require us to have new material manufactured by a cGMP-compliant manufacturer in the event that we seek to resume this trial. Significant preparatory time and procedures will be required before any new manufacturer would be able to manufacture RGN-352 for the AMI trial, due to the time required for revalidation of processes and assays related to such production that were already in place with the original manufacturer. Since we are unable to estimate the length of time that the trial will be on clinical hold, we have elected to cease activities on this trial until the FDA clinical hold is resolved and the requisite funding might be secured.
Other risks of relying solely on single suppliers for each of our product candidates include:
| · | ||
| · | ||
| · | ||
| · | ||
| · | ||
| · | ||
| · | ||
| · | an individual supplier may not be able to obtain the raw materials or validated drug containers in sufficient quantities, at acceptable costs or in sufficient time to complete the manufacture, formulation and delivery of our product candidates. |
Our suppliers may use hazardous and biological materials in their businesses. Any claims relating to improper handling, storage or disposal of these materials could be time-consuming and costly to us, and we are not insured against such claims.
Our product candidates and processes involve the controlled storage, use and disposal by our suppliers of certain hazardous and biological materials and waste products. We and our suppliers and other collaborators are subject to federal, state and local regulations governing the use, manufacture, storage, handling and disposal of materials and waste products. Even if we and these suppliers and collaborators comply with the standards prescribed by law and regulation, the risk of accidental contamination or injury from hazardous materials cannot be completely eliminated. In the event of an accident, we could be held liable for any damages that result, and we do not carry insurance for this type of claim. We may also incur significant costs to comply with current or future environmental laws and regulations.
15
We may be subject to product liability claims as a result of our testing, manufacturing, and marketing of drugs. In addition, the use of our product candidates, when and if developed and sold, will expose us to the risk of product liability claims. Product liability may result from harm to patients using our product candidates, such as a complication that was either not communicated as a potential side effect or was more extreme than anticipated. We require all patients enrolled in our clinical trials to sign consents, which explain various risks involved with participating in the trial. However, patient consents provide only a limited level of protection, and it may be alleged that the consent did not address or did not adequately address a risk that the patient suffered. Additionally, we will generally be required to indemnify our clinical product manufacturers, clinical trial centers, medical professionals and other parties conducting related activities in connection with losses they may incur through their involvement in the clinical trials.
Our ability to reduce our liability exposure for human clinical trials and commercial sales, if any, of Tß4 is dependent in part on our ability to obtain sufficient product liability insurance or to collaborate with third parties that have adequate insurance. Although we intend to obtain and maintain product liability insurance coverage if we gain approval to market any of our product candidates, we cannot guarantee that product liability insurance will continue to be available to us on acceptable terms, or at all, or that its coverage will be sufficient to cover all claims against us. A product liability claim, even one without merit or for which we have substantial coverage, could result in significant legal defense costs, thereby potentially exposing us to expenses significantly in excess of our revenues, as well as harm to our reputation and distraction of our management.
If any of our key employees discontinue their services with us, our efforts to develop our business may be delayed.
We are highly dependent on the principal members of our management team. The loss of our chairmanChairman and chief scientific advisor,Chief Scientific Officer, Allan Goldstein, or our chief executive officer,Chief Executive Officer, J.J. Finkelstein could prevent or significantly delay the achievement of our goals. We have employment agreements with Dr. Goldstein and Mr. Finkelstein. For part of 2009, we effected salary reductions for certain of our employees, including Dr. Goldstein and Mr. Finkelstein. Although their salaries were restored effective as of October 1, 2009, we cannot assure you that they,Dr. Goldstein or Mr. Finkelstein, or any other key employees mayor consultants, will not elect to terminate their employment as a result of the salary reductions or for other reasons.consulting arrangements. In addition, we do not maintain a key man life insurance policy with respect to Dr. Goldstein or Mr. Finkelstein.any of our management personnel. In the future, we anticipate that we maywill also need to add additional management and other personnel. Competition for qualified personnel in our industry is intense, and our success will depend in part on our ability to attract and retain highly skilled personnel. We cannot assure you that our efforts to attract or retain such personnel will be successful.
| 19 |
Mauro Bove, a member of our Board, iswas also a director and officer of entities affiliated with Sigma-Tau and is a relationshipdirector of Lee’s Pharmaceuticals, relationships which could give rise to a conflict of interest involvingfor Mr. Bove.
Mauro Bove is a member of our Board of Directors, is alsoand, until March 31, 2014, was a director and officer of entities affiliated with Sigma-Tau, which collectively make up our largest stockholder group. At this time Mr. Bove remains engaged with Sigma-Tau as a consultant. Sigma-Tau has subsequently merged into Alfa Wassermann, S.p.A., an Italian pharmaceutical company. Sigma-Tau/Alfa Wassermann, previously provided us with significant funding may continue doing so in the future, and is also our strategic partner in Europe with respect to the development of certain of our drug candidates. During 2008 and 2009, weWe have issued shares of common stock, convertible promissory notes and common stock warrants to Sigma-Tau and its affiliates in four separateseveral private placement financing transactions, but we retained the right to repurchase some of these shares under certain circumstances.
16
Additionally, Mr. Bove is a non-executive director of Lee’s Pharmaceuticals, in which affiliates of Sigma-Tau/Alfa Wassermann have a significant equity interest. In July 2012, we entered into a license agreement for TB4 in any pharmaceutical form, including our RGN-259, RGN-352 and RGN-137 product candidates for development in China, Hong Kong, Macau and Taiwan. There can be no assurance that we will ever receive any further payments from Lee’s under the agreement. As a result of Mr. Bove’s relationship with Lee’s and Sigma-Tau, Mr. Bove may have interests that are different from our other stockholders in connection with these and other agreements and circumstances that may require the exercise of the Board’s discretion with respect to Lee’s or Sigma-Tau. These conflicts could result in decisions that are not in the best interest of our other stockholders.
Risks Related To Our Intellectual Property
We are heavilypartially reliant on our license from the National Institutes of Health for the rights to Tß4, and any loss of these rights wouldcould adversely affect our business.
We have received an exclusive worldwide license to intellectual property discovered at the National Institutes of Health, or NIH, pertaining to the use of Tß4 in wound healing and tissue repair. The intellectual property rights from this license, along with independent patent applications we have filed, as well as patents and patent applications under licenses we acquired, form the basis for our current commercial development focus with Tß4. ThisThe NIH license terminates upon the last to expire of the patent applications that are filed, or any patents that may issue from such applications, in connection with the license. This license requires us to pay a minimum annual royalty to the NIH, regardless of the success of our product development efforts, plus certain other royalties upon the sale of products created by the intellectual property granted under the license. This license may be terminated for a number of reasons, including our non-paymentIn 2013 we amended certain provisions of the exclusive license; we were permitted to credit amounts paid to prosecute or maintain the licensed patent rights during the 2013 calendar year against the 2013 minimum annual royalty or lack of continued product development, among others.$25,000. Beginning in 2014, the minimum annual royalty is $2,000. While to date we believe that we have complied with all requirements to maintain the license, the loss of this license would have a materialan adverse effect on our business and business prospectsprospects.
We may not be able to maintain broad patent protection for our product candidates, which could limit the commercial potential of our product candidates.
Our success will depend in part on our ability to obtain, defend and may require usenforce patents, both in the United States and abroad. We have attempted to cease developmentcreate a substantial intellectual property portfolio, submitting patent applications for various compositions of matter, methods of use and fragments and derivatives of Tß4. As described elsewhere in this report, we currently do not have adequate financial resources to fund our ongoing business activities substantially beyond 12 months without additional funding. As a result of our current linefinancial condition, we continuously evaluate our issued patents and patent applications and may decide to limit their therapeutic and/or geographic coverage in an effort to enhance our ability to focus on certain medical conditions and countries within our financial constraints. As a result, we may not be able to protect our intellectual property rights in indications and/or territories that we otherwise would, and, therefore, our ability to commercialize Tß4, if at all, could be substantially limited, which could have a material adverse impact on our future results of Tß4-based product candidates.operations.
| 20 |
If we are not able to maintain adequate patent protection for our product candidates, we may be unable to prevent our competitors from using our technology or technology that we license.
Our success will depend in substantial part on our ability to obtain, defend and enforce patents, maintain trade secrets and operate without infringing upon the proprietary rights of others, both in the United States and abroad. Pursuant to an exclusive worldwide license from the NIH, we have exclusive rights to use Tß4 in the treatment of non-healing wounds. While patents covering our use of Tß4 have issued in some countries, we cannot guarantee whether or when corresponding patents will be issued, or the scope of any patents that may be issued, in other countries. We have attempted to create a substantial intellectual property portfolio, submitting patent applications for various compositions of matter, methods of use and fragments and derivatives of Tß4. We have also in-licensed other intellectual property rights from third parties that could be subject to the same risks as our own patents. If any of these patent applications do not issue, or do not issue in certain countries, or are not enforceable, the ability to commercialize Tß4 in various medical indications could be substantially limited or eliminated.
In addition, the patent positions of the products being developed by us and our collaborators involve complex legal and factual uncertainties. As a result, we cannot assure you that any patent applications filed by us, or by others under which we have rights, will result in patents being issued in the United States or foreign countries. In addition, there can be no assurance that any patents will be issued from any pending or future patent applications of ours or our collaborators, that the scope of any patent protection will be sufficient to provide us with competitive advantages, that any patents obtained by us or our collaborators will be held valid if subsequently challenged or that others will not claim rights in or ownership of the patents and other proprietary rights we or our collaborators may hold. Unauthorized parties may try to copy aspects of our product candidates and technologies or obtain and use information we consider proprietary. Policing the unauthorized use of our proprietary rights is difficult. We cannot guarantee that no harm or threat will be made to our or our collaborators’ intellectual property. In addition, changes in, or different interpretations of, patent laws in the United States and other countries may also adversely affect the scope of our patent protection and our competitive situation.
17
Additionally, there is certain subject matter that is patentable in the United States but not generally patentable outside of the United States. Differences in what constitutes patentable subject matter in various countries may limit the protection we can obtain outside of the United States. For example, methods of treating humans are not patentable in many countries outside of the United States. These and other issues may prevent us from obtaining patent protection outside of the United States, which would have a material adverse effect on our business, financial condition and results of operations.
Changes to U.S. patent laws could materially reduce any value our patent portfolio may have.
The value of our patents depends in part on their duration. A shorter period of patent protection could lessen the value of our rights under any patents that may be obtained and may decrease revenues derived from its patents. For example, the U.S. patent laws were previously amended to change the term of patent protection from 17 years following patent issuance to 20 years from the earliest effective filing date of the application. Because the time from filing to issuance of biotechnology applications may be more than three years depending on the subject matter, a20-year patent term from the filing date may result in substantially shorter patent protection. Future changes to patent laws could shorten our period of patent exclusivity and may decrease the revenues that we might derive from the patents and the value of our patent portfolio.portfolio.
| 21 |
We may not have adequate protection for our unpatented proprietary information, which could adversely affect our competitive position.
In addition to our patents, we also rely on trade secrets, know-how, continuing technological innovations and licensing opportunities to develop and maintain our competitive position. However, others may independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets or disclose our technology. To protect our trade secrets, we may enter into confidentiality agreements with employees, consultants and potential collaborators. However, we may not have such agreements in place with all such parties and, where we do, these agreements may not provide meaningful protection of our trade secrets or adequate remedies in the event of unauthorized use or disclosure of such information. Also, our trade secrets or know-how may become known through other means or be independently discovered by our competitors. Any of these events could prevent us from developing or commercializing our product candidates.
We may be subject to claims that we or our employees have wrongfully used or disclosed alleged trade secrets of former employers.
As is commonplace in the biotechnology industry, we employ now, and may hire in the future, individuals who were previously employed at other biotechnology or pharmaceutical companies, including competitors or potential competitors. Although there are no claims currently pending against us, we may be subject to claims that we or certain employees have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and would be a significant distraction to management.
18
Our common stock price is volatile, and has had limited trading volume,our stock is highly illiquid, and any investment in our securities could decline substantially in value.
For the period from January 1, 20092015 through July 11, 2016, the dateclosing price of this prospectus, our closingcommon stock price has fluctuated between prices of $0.42ranged from $0.0.28 to $1.75 per share,$0.75, with an average daily trading volume of approximately 80,00060,000 shares. In light of our small size and limited resources, as well as the uncertainties and risks that can affect our business and industry, our stock price is expected to continue to be highly volatile and can be subject to substantial drops, with or even in the absence of news affecting our business. The following factors, in addition to the other risk factors described in this prospectus,report, and the potentially low volume of trades in our common stock since it is not listed on a national securities exchange, may have a significant impact on the market price of our common stock, some of which are beyond our control:
| results of | ||
| commercial success of approved products; | ||
| corporate partnerships; | ||
| technological innovations by us or competitors; | ||
| changes in laws and government regulations both in the U.S. and overseas; | ||
| changes in key personnel at our company; | ||
| developments concerning proprietary rights, including patents and litigation matters; | ||
| · | public perception relating to the commercial value or safety of any of our product candidates; | |
| · | ||
| anticipated or unanticipated changes in our financial performance; | ||
| general trends related to the biopharmaceutical and biotechnological industries; and | ||
| general conditions in the stock market. |
The stock market in general has recently experienced relatively large price and volume fluctuations. In particular, the market prices of securities of smaller biotechnology companies have experienced dramatic fluctuations that often have been unrelated or disproportionate to the operating results of these companies. Continued market fluctuations could result in extreme volatility in the price of our common stock, which could cause a decline in its value. You should also be aware that price volatility may be worse if the trading volume of the common stock remains limited or declines.
Our principal stockholders have significant voting power and may take actions that may not be in the best interests of our other stockholders.
Our officers, directors and principal stockholders together will control approximately 40%62.7% of our outstanding common stock. Included in this group is Essetifin (f/k/a Sigma-Tau) and its affiliates, which together hold outstanding shares representing approximately 33.2% of our outstanding common stock not including the exercise of options or warrants. Included in this group is Sigma-Tau,and GtreeBNT which will holdowns approximately 33%18.3% of our outstanding common stock. These stockholders also hold options, warrants, convertible promissory notes and stock followingpurchase rights that provide them with the offering, excluding the effect of any options and warrants, and not giving effectright to any potential participation by Sigma-Tau in this offering. Sigma-Tau has expressed an interest in purchasing units in this offering. A portion of theacquire significantly more shares of common stock currently held by Sigma-Tau, representing approximately 18% of our outstanding common stock, is subject to voting agreements under which westock. Accordingly, if these stockholders acted together they could control the voting poweroutcome of these shares. We cannot assure you that these voting agreements would prevent Sigma-Tau from taking actions not in your best interests and effectively exercising control over us. These voting agreements are currently scheduled to expire between June 2010 and September 2012. After their expiration, we will have no control over the voting of these shares controlled by Sigma-Tau, including with respect to the
19
An active trading market for the warrants being sold in this offering may not develop.
If securities or industry analysts do not publish research or reports or publish unfavorable research about our business, the price of our common stock and other securities and their trading volume could decline.
The trading market for our common stock and other securities will depend in part on the research and reports that securities or industry analysts publish about us or our business. We do not currentlyUntil recently, we have and may never obtainhad no research coverage by securities and industry analysts.analysts, and there is no guarantee that we will have research coverage in the future. If securities or industry analysts do not commence or maintain coverage of us, the trading price for our common stock and other securities would be negatively affected. In the event we obtain securities or industry analyst coverage, if one or more of the analysts who covers us downgrades our securities, the price of our securities would likely decline. If one or more of these analysts ceases to cover us or fails to publish regular reports on us, interest in the purchase of our securities could decrease, which could cause the price of our common stock and other securities and their trading volume to decline.
Our rights to repurchase certain shares of stock held by Sigma-Tau expire over time, and we may never be able or elect to exercise these rights.
A significant portion of our total outstanding shares are restricted from immediate resale but may be sold into the market in the near future. This could cause the market price of our common stock to drop significantly, even if our business is doing well.
20
As of March 31, 2010,2016, there were outstanding options to purchase an aggregate of 4,914,1127,621,211 shares of our common stock at exercise prices ranging from $0.28 per share to $3.82 per share, of which optionsunder our 2000 and 2010 incentive equity plans and outstanding warrants to purchase 3,613,069 shares were exercisable as of such date. As of March 31, 2010, there were warrants outstanding to purchase 7,933,8515,804,412 shares of our common stock, at a weighted average exercise pricestock. In addition to the outstanding options and warrants we have also issued five series of $2.01 per share. We will issue additional warrants as partconvertible promissory notes, which are presently convertible into an aggregate of the units being sold in this offering, with an exercise price equal to $ per share.13,683,334 shares of our common stock. The exercise of options and warrants or the conversion of notes at prices below the market price of our common stock could adversely affect the price of shares of our common stock. Additional dilution may result from the issuance of shares of our capital stock in connection with collaborations or manufacturing arrangements or in connection with other financing efforts.
| 23 |
Any issuance of our common stock that is not made solely to then-existing stockholders proportionate to their interests, such as in the case of a stock dividend or stock split, will result in dilution to each stockholder by reducing his, her or its percentage ownership of the total outstanding shares. Moreover, if we issue options or warrants to purchase our common stock in the future and those options or warrants are exercised or we issue restricted stock, stockholders may experience further dilution. Holders of shares of our common stock have no preemptive rights that entitle them to purchase their pro rata share of any offering of shares of any class or series.
In addition, certainmost of the outstanding warrants to purchase shares of our common stock currently containhave an exercise price above the current market price for theour common stock, or above-market warrants, as will the warrants issued in connection with this offering.stock. As a result, these warrants may not be exercised prior to their expiration, andin which case we maywould not realize any proceeds from their exercise.
Our certificate of incorporation our stockholder rights plan and Delaware law contain provisions that could discourage or prevent a takeover or other change in control, even if such a transaction would be beneficial to our stockholders, which could affect our stock price adversely and prevent attempts by our stockholders to replace or remove our current management.
Our certificate of incorporation provides our Board with the power to issue shares of preferred stock without stockholder approval. In addition, under our stockholder rights plan, our Board has the discretion to issue certain rights to purchase our capital stock to our stockholders when a person acquires in excess of 25% of our outstanding common shares. These provisions may make it more difficult for stockholders to take corporate actions and may have the effect of delaying or preventing a change in control, even if such actions or change in control would be in your best interests. In addition, we are subject to the anti-takeover provisions of Section 203 of the Delaware General Corporation Law. Subject to specified exceptions, this section provides that a corporation may not engage in any business combination with any interested stockholder, as defined in that statute, during the three-year period following the time that such stockholder becomes an interested stockholder. This provision could also have the effect of delaying or preventing a change of control of our company. The foregoing factors could reduce the price that investors or an acquirer might be willing to pay in the future for shares of our common stock.
21
The stock market has from time to time experienced significant price and volume fluctuations that have affected the market prices for the common stock of pharmaceutical and biotechnology companies. These broad market fluctuations may cause the market price of our common stock to decline. In the past, following periods of volatility in the market price of a particular company’s securities, securities class action litigation has often been brought against that company. We may become involved in this type of litigation in the future. Litigation often is expensive and diverts management’s attention and resources, which could hurt our business, operating results and financial condition.
We have broad discretion in the use of proceeds from this offering and may invest or spend the proceeds in ways with which you do not agree and in ways that may not yield a return.
This prospectus contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act, that involve substantial risks and uncertainties. The forward-looking statements are contained principally in the sections entitled “Prospectus Summary,” “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Business,” but are also contained elsewhere in this prospectus. In some cases, you can identify forward-looking statements by the words “may,” “might,” “will,” “could,” “would,” “should,” “expect,” “intend,” “plan,” “objective,” “anticipate,” “believe,” “estimate,” “predict,” “project,” “potential,” “continue” and “ongoing,” or the negative of these terms, or other comparable terminology intended to identify statements about the future. These statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements. Although we believe that we have a reasonable basis for each forward-looking statement contained in this prospectus, we caution you that these statements are based on a
22
| • | the progress, outcome, timing or success of preclinical studies and clinical trials; | |
| • | the expected timing of clinical trials and availability of data from those trials; | |
| • | our ability to obtain and maintain regulatory approval for our product candidates from the FDA or foreign regulatory authorities; | |
| • | future demand for our product candidates and our ability to sustain such demand; | |
| • | the size of the potential market for our product candidates; | |
| • | our plans to seek collaborative relationships and the success of those relationships; | |
| • | the success of competing therapies that are or become available; | |
| • | our compliance with federal, state and foreign regulatory requirements, and regulatory developments that impact those requirements; | |
| • | our estimates and assumptions with respect to disease incidence; | |
| • | our intellectual property and our strategies regarding filing additional patent applications to attempt to strengthen our intellectual property rights; | |
| • | our ability to retain key | |
| • | estimates of our future financial performance; | |
| • | our ability to | |
| • | anticipated trends and challenges in our business. |
In addition, you should refer to the “Risk Factors” section of this prospectus for a discussion of other important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this prospectus will prove to be accurate or that we will achieve the plans, intentions or expectations expressed or implied in our forward-looking statements. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all. Any forward-looking statements we make in this prospectus speak only as of its date, and we undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
You should read this prospectus and the documents that we reference in this prospectus and have filed as exhibits to the registration statement, of which this prospectus is a part, completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
| 25 |
23
We estimatewill receive none of the proceeds from the sale of the shares by the selling stockholders, except for the warrant exercise price upon exercise of the warrants, which would be used for working capital and other general corporate purposes.
General
On June 27, 2016, we entered into a Securities Purchase Agreement with certain purchasers identified therein pursuant to which we agreed to sell, and the purchasers agreed to purchase, an aggregate of 5,147,059 shares of common stock and warrants to purchase 5,147,059 shares of common stock, which we refer to as the 2016 Offering, and in conjunction with the closing of such transaction we issued warrants to purchase 257,353 shares of common stock to our placement agent.
The purchase agreement contains customary representations, warranties and covenants by each of us and the purchasers. In addition, the purchase agreement provides that each purchaser has a right, subject to certain exceptions described in the agreement, to participate in future issuances of equity and debt securities by us for a period of 12 months following the effective date of this registration statement, and certain price protections that provide for the grant of additional shares of common stock if we sell shares for less than $0.34 per share (the purchase price in the 2016 Offering) during such 12-month period. Moreover, we agreed, subject to certain exceptions, not to sell or announce the sale of our securities for five months from the effective date of this registration statement.
Description of the Warrants
The following description is qualified in its entirety by the terms and conditions of the warrants sold to the purchasers in the 2016 Offering, which we refer to as the Warrants, the form of which is incorporated by reference into the registration statement of which this prospectus forms a part. The following description may not contain all the information with respect to the Warrants that is important to you. We encourage you to read the form of Warrant in its entirety.
The Warrants have a term that extends for the longer of five and one half years from the date of grant or five years from the effective date of this registration statement and an exercise price of $0.51 per share. The applicable exercise price of the Warrants is subject to a full-ratchet anti-dilution adjustment in the event we make future issuances of common stock or rights to acquire common stock (subject to certain exceptions) at a per share price less than the applicable warrant exercise price, meaning that the net proceeds from our issuance and saleexercise price of 11,500,000 units in this offeringthe Warrants will be approximately $5.4 million,reduced to such lesser sale price.
The Warrants are required to be exercised for cash, provided that if during the term of the Warrants there is not an effective registration statement under the Securities Act covering the resale of the shares issuable upon exercise of the Warrants, then the Warrants may be exercised on a cashless (net exercise) basis.
Registration Rights Agreement
The following description is qualified in its entirety by the terms and conditions set forth in the registration rights agreement with respect to the 2016 Offering incorporated by reference to the registration statement that contains this prospectus hereto, which we refer to as the Registration Rights Agreement. The following description may not contain all the information with respect to such registration rights important to you. We encourage you to read the Registration Rights Agreement.
| 26 |
In connection with the entry into the purchase agreement, and as contemplated thereby, on June 27, 2016, we entered into a Registration Rights Agreement with the purchasers. Pursuant to the terms of the Registration Rights Agreement, we agreed to file, within 30 days of the date of the Registration Rights Agreement, a registration statement under the Securities Act covering the resale of the shares of common stock sold pursuant to the purchase agreement and the shares of common stock issuable upon exercise of the Warrants, and to cause such registration statement to be declared effective by the Securities and Exchange Commission (the “Commission”) as soon as practicable thereafter, but not later than 60 days following the date of the Registration Rights Agreement or approximately $6.3 million90 days if the underwriters exercise their over-allotment optionregistration statement is subject to full review by theCommission. If we do not meet such deadlines, we will be required to pay liquidated damages to the purchasers in full, based upon an assumed public offeringamount equal to 1.5% of the aggregate purchase price paid by such purchaser for the Units per month until the registration statement is filed or declared effective.
This prospectus covers the resale by the selling stockholders identified below of $0.56 per unit (based on the last reported sale price10,551,471 shares of our common stock, on May 14, 2010), after deducting estimated underwriting discounts and commissions,of which 5,404,4121 shares are issuable upon the 1% corporate finance fee and estimated offering expenses payable by us.
| Number of | ||||||||||||||||||||
| shares | ||||||||||||||||||||
| Number of | offered by | Beneficial | ||||||||||||||||||
| Shares | outstanding | selling | ownership | |||||||||||||||||
| beneficially | shares | stockholder | after | |||||||||||||||||
| owned | offered by | upon | offering (1) | |||||||||||||||||
| before | selling | exercise of | Number | |||||||||||||||||
| Selling Stockholder | offering (1) | stockholder | warrants | of shares | Percent | |||||||||||||||
| Sabby Healthcare Master Fund, Ltd. (2)(3) | 0 | 3,676,471 | 3,676,471 | 10,294,118 | 9.2 | % | ||||||||||||||
| Sabby Volatility Warrant Master Fund, Ltd.(2)(3) | 0 | 1,470,588 | 1,470,588 | 10,294,118 | 9.2 | % | ||||||||||||||
| Maxim Partners LLC | 0 | 0 | 257,353 | 257,353 | * | |||||||||||||||
| TOTALS | 5,147,059 | 5,404,412 | 10,551,471 | 9.4 | % | |||||||||||||||
* denotes less than 1%
| (1) | Beneficial ownership is determined in accordance with Rule 13d-3 under the Exchange Act, and includes any shares as to which the security or stockholder has sole or shared voting power or investment power, and also any shares which the security or stockholder has the right to acquire within 60 days of the date hereof, whether through the exercise or conversion of any stock option, convertible security, warrant or other right. The indication herein that shares are beneficially owned is not an admission on the part of the security or stockholder that he, she or it is a direct or indirect beneficial owner of those shares. Percentage of shares beneficially owned after the resale of all the shares offered by this prospectus assumes there are outstanding 112,191,563 shares of common stock, including all shares offered hereby that are issuable upon exercise of warrants. | |
| (2) | Notwithstanding the number of shares of our common stock shown as beneficially owned by the selling stockholder in the table above, the held by the selling stockholder provide that the selling stockholder may not exercise such Warrants to the extent that the selling stockholder would beneficially own in excess of 4.99% of our outstanding common stock immediately after giving effect to such exercise. | |
| (3) | Each of Sabby Healthcare Master Fund, Ltd. and Sabby Volatility Warrant Master Fund, Ltd. (collectively, the “Sabby Funds”) has indicated to us that Hal Mintz has voting and investment power over the shares held by it. Each of the Sabby Funds has also indicated to us that Sabby Management, LLC serves as its investment manager, that Hal Mintz is the manager of Sabby Management, LLC and that each of Sabby Management, LLC and Hal Mintz disclaim beneficial ownership over these shares except to the extent of any pecuniary interest therein. In addition to the shares offered hereby, beneficial ownership also includes: (i) with respect to Sabby Healthcare Master Fund, Ltd., 3,676,471 shares of our common stock issuable upon the exercise of Warrants; and (ii) with respect to Sabby Volatility Warrant Master Fund, Ltd., 1,470,588 shares of our common stock issuable upon the exercise of Warrants. |
| 27 |
ADDITIONAL DISCLOSURE REGARDING TRANSACTIONS
BETWEEN THE COMPANY AND THE SELLING STOCKHOLDERS
Payments in connection with 2016 Offering
The following table summarizes the total payments and potential Phase 1/2 clinical trialpayments to the selling stockholders and their affiliates in connection with our 2016 Offering.
| Amounts paid as of June 27, 2016 | Potential Future Payment Obligations | Total | ||||||||||
| Placement Fees (1) | $ | 140,000 | $ | - | $ | 140,000 | ||||||
| Placement Warrants (2) | 83,799 | - | 83,799 | |||||||||
| Potential Liquidated Damages for Delayed Registration (3) | - | 26,250 | 26,250 | |||||||||
| Legal Fees (4) | 25,000 | - | 25,000 | |||||||||
| Total payments and potential payments | $ | 248,799 | $ | 26,250 | $ | 275,049 | ||||||
| (1) | We engaged Maxim Group LLC as placement agent in connection with the 2016 Offering pursuant to the terms of an engagement letter, which provided for a fee of 8% of the proceeds of an offering completed under the terms of such engagement letter. | |
| (2) | In addition to the cash fee described in footnote (1), we also issued five-year warrants to purchase 257,353 shares of common stock at an initial exercise price of $0.374 per share (the “Maxim Warrants”) to Maxim Partners LLC (“Maxim”). On the date of the grant of the maxim Warrants, they had an estimated fair value of $83,799. | |
| (3) | Under the Registration Rights Agreement, we agreed to file, a registration statement under the Securities Act covering the resale of the shares of common stock issued as part of the 2016 Offering and the shares of common stock issuable upon exercise of the 2016 Warrants and to cause such registration statement to be declared effective by the Commission as soon as practicable thereafter, but not later than 90th day following the date of the Registration Rights Agreement. If we do not meet such timing requirement, then we are required to pay liquidated damages to the purchasers in an amount equal to 1.5% of the aggregate purchase price paid by such purchaser in the 2016 Offering per month until the registration statement is filed or declared effective, as applicable. The amount in the table assumes payment of such liquidated damages for a one month period, although there is no cap on the amount we could be required to pay under the Registration Rights Agreement. We are required to maintain the effectiveness of the registration statement until all of the shares covered thereby are sold. | |
| (4) | We paid Maxim the amount of $25,000 as reimbursement of legal fees incurred in connection with negotiating the 2016 Offering. |
Each Selling Stockholder (the “Selling Stockholders”) of RGN-352the securities and any of their pledgees, assignees and successors-in-interest may, from time to time, sell any or all of their securities covered hereby on the OTCQB or any other stock exchange, market or trading facility on which the securities are traded or in patients with multiple sclerosis. We also expectprivate transactions. These sales may be at fixed or negotiated prices.
A Selling Stockholder may use any one or more of the following methods when selling securities:
| 28 |
The Selling Stockholders may also sell securities under Rule 144 under the Securities Act of 1933, as amended (the “Securities Act”), if available, rather than under this prospectus.
Broker-dealers engaged by the Selling Stockholders may arrange for other brokers-dealers to participate in sales. Broker-dealers may receive commissions or discounts from the Selling Stockholders (or, if any broker-dealer acts as agent for the purchaser of securities, from the purchaser) in amounts to be negotiated, but, except as set forth in a supplement to this Prospectus, in the case of an agency transaction not in excess of a customary brokerage commission in compliance with FINRA Rule 2440; and in the case of a principal transaction a markup or markdown in compliance with FINRA IM-2440.
In connection with the sale of the securities or interests therein, the Selling Stockholders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the securities in the course of hedging the positions they assume. The Selling Stockholders may also sell securities short and deliver these securities to close out their short positions, or loan or pledge the securities to broker-dealers that in turn may sell these securities. The Selling Stockholders may also enter into option or other transactions with broker-dealers or other financial institutions or create one or more derivative securities which require the delivery to such broker-dealer or other financial institution of securities offered by this prospectus, which securities such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).
The Selling Stockholders and any broker-dealers or agents that are involved in selling the securities may be deemed to be “underwriters” within the meaning of the Securities Act in connection with such sales. In such event, any commissions received by such broker-dealers or agents and any profit on the resale of the securities purchased by them may be deemed to be underwriting commissions or discounts under the Securities Act. Each Selling Stockholder has informed the Company that it does not have any written or oral agreement or understanding, directly or indirectly, with any person to distribute the securities.
We are required to pay certain fees and expenses incurred by us incident to the registration of the securities.
We agreed to indemnify the Selling Stockholders against certain losses, claims, damages and liabilities, including working capital.liabilities under the Securities Act.
We agreed to keep this prospectus effective until the earlier of (i) the date on which the securities may be resold by the Selling Stockholders without registration and without regard to any volume or manner-of-sale limitations by reason of Rule 144, without the requirement for the Company to be in compliance with the current public information under Rule 144 under the Securities Act or any other rule of similar effect or (ii) all of the securities have been sold pursuant to this prospectus or Rule 144 under the Securities Act or any other rule of similar effect. The resale securities will be sold only through registered or licensed brokers or dealers if required under applicable state securities laws. In addition, in certain states, the resale securities covered hereby may not be sold unless they have been registered or qualified for sale in the applicable state or an exemption from the registration or qualification requirement is available and is complied with.
| 29 |
Under applicable rules and regulations under the Exchange Act, any person engaged in the distribution of net proceeds from this offering represents our intentions based upon our present plansthe resale securities may not simultaneously engage in market making activities with respect to the common stock for the applicable restricted period, as defined in Regulation M, prior to the commencement of the distribution. In addition, the Selling Stockholders will be subject to applicable provisions of the Exchange Act and business conditions.the rules and regulations thereunder, including Regulation M, which may limit the timing of purchases and sales of the common stock by the Selling Stockholders or any other person. We will not be able to complete the contemplated Phase 2 clinical trial of RGN-352 without additional capital. Further, we expect that the net proceeds will not be sufficient to complete clinical trials to obtain regulatory approval for the marketing of any of our current product candidates. As described elsewhere in this prospectus, the completion of these trials may be delayed for a number of reasons. As of the datemake copies of this prospectus we cannot predict with certainty allavailable to the Selling Stockholders and have informed them of the particular uses for the proceedsneed to deliver a copy of this offeringprospectus to each purchaser at or prior to the amounts that we will actually spend ontime of the uses set forth above. The amount and timing of actual expenditures may vary significantly depending upon a number of factors, such assale (including by compliance with Rule 172 under the progress of our development efforts, regulatory requirements, commercialization efforts in the event that we obtain regulatory approval, the amount of cash, if any, we generate from strategic collaborations that we may enter into or from other sources, and the amount of cash used by operations. Accordingly, we will have significant flexibility in applying the net proceeds of this offering.Securities Act).
| 30 |
24
CAPITALIZATION
The following table sets forth our cash and cash equivalents and our capitalization as of March 31, 2010:
| • | on an actual basis; and |
| • | on an as adjusted basis to give effect to |
| Actual | As Adjusted | |||||||
| (In thousands) | ||||||||
| Cash and cash equivalents | $ | 3,190 | $ | 8,638 | ||||
| Stockholders’ equity: | ||||||||
| Preferred stock, $0.001 per share, 1,000,000 shares authorized, no shares issued or outstanding, actual or as adjusted | — | — | ||||||
| Common stock, $0.001 par value, 100,000,000 shares authorized, 60,406,828 shares issued and outstanding, actual; 100,000,000 shares authorized, 79,406,828 shares issued and outstanding, as adjusted; | 60 | 72 | ||||||
Additionalpaid-in-capital | 88,276 | 93,713 | ||||||
| Accumulated deficit | (85,647 | ) | (85,647 | ) | ||||
| Total stockholders’ equity | 2,689 | 8,137 | ||||||
| Total capitalization | $ | 2,689 | $ | 8,137 | ||||
| Actual | As Adjusted | |||||||
| (In thousands) | ||||||||
| Cash and cash equivalents | $ | 80 | $ | 1,690 | ||||
| Convertible long-term debt | $ | 1,001 | $ | 1,001 | ||||
| Stockholders’ equity: | ||||||||
| Preferred stock, $.001 par value per share, 1,000,000 shares authorized; no shares issued | - | - | ||||||
| Common stock, par value $.001 per share, 200,000,000 shares authorized, 101,640,092 issued and outstanding | 102 | 107 | ||||||
| Additional paid-in-capital | 98,402 | 99,942 | ||||||
| Accumulated deficit | (108,454 | ) | (108,454 | ) | ||||
| Total stockholders’ equity (deficit) | (9,950 | ) | (8,405 | ) | ||||
| Total capitalization | $ | (8,949 | ) | $ | (7,404 | ) | ||
The number of shares of common stock outstanding in the table above does not include:
| • |
| • |
| • | ||
| • | 400,000 shares of our common stock issuable upon the exercise of outstanding warrants |
25
Our common stock tradesis quoted on the NYSE Amex, previously known as the American Stock Exchange,OTC Bulletin Board under the symbol “RGN.“RGRX.” On July 11, 2016 the trading of our common stock closed at a price $0.45 per share. The prices, as presented below, representfollowing table provides the highesthigh and lowest bid or salelow sales prices for our common stock for each quarterly period within the two most recent fiscal years as reported by quarterthe OTC Bulletin Board, as quoted onappropriate. The quotations reported by the NYSE Amex. On May 14, 2010,OTC Bulletin Board reflect inter-dealer prices, without retail mark-up, mark-down or commission and may not represent actual transactions.
The following table sets forth the last sale price ofhigh and low bid prices for our common stock as reported onfor the NYSE Amex was $0.56 per share.
2008 | High | Low | ||||||
| First Quarter | $ | 1.10 | $ | 0.80 | ||||
| Second Quarter | $ | 1.92 | $ | 0.83 | ||||
| Third Quarter | $ | 1.43 | $ | 1.02 | ||||
| Fourth Quarter | $ | 1.66 | $ | 0.85 | ||||
2009 | High | Low | ||||||
| First Quarter | $ | 1.75 | $ | 0.42 | ||||
| Second Quarter | $ | 0.85 | $ | 0.45 | ||||
| Third Quarter | $ | 1.12 | $ | 0.52 | ||||
| Fourth Quarter | $ | 0.83 | $ | 0.55 | ||||
2010 | High | Low | ||||||
| First Quarter | $ | 0.65 | $ | 0.53 | ||||
| Second Quarter (through May 14, 2010) | $ | 0.68 | $ | 0.46 | ||||
| 2016 (through July 11) | 2015 | 2014 | ||||||||||||||||||||||
| High | Low | High | Low | High | Low | |||||||||||||||||||
| First Quarter | $ | 0.75 | $ | 0.37 | $ | 0.38 | $ | 0.13 | 0.28 | 0.05 | ||||||||||||||
| Second Quarter | $ | 0.75 | $ | 0.28 | $ | 0.60 | $ | 0.25 | 0.27 | 0.15 | ||||||||||||||
| Third Quarter | $ | 0.47 | $ | 0.35 | $ | 0.50 | $ | 0.32 | 0.20 | 0.10 | ||||||||||||||
| Fourth Quarter | $ | - | $ | - | $ | 0.45 | $ | 0.35 | 0.18 | 0.12 | ||||||||||||||
As of April 14, 2010,July 11, 2016, we had 851739 holders of record of our common stock.
We have never declared or paid anya cash dividend on our common stock. We are anearly-stage company and have not historically generated, and do not anticipate generating in the forseeable future, revenues sufficient to enable us to declare or pay cash dividends on our common stock or any other securities. We anticipate that we will retain allstock. All of our future earnings, if any, for use in the operation of our businessfunds are committed to clinical research and we do not anticipate payingthat any cash dividends in the foreseeable future.
26
| Assumed public offering price per share of common stock underlying each unit | $ | 0.56 | ||||||
| Actual net tangible book value per share as of March 31, 2010 | $ | 0.04 | ||||||
| Increase in net tangible book value per share attributable to new investors participating in this offering | 0.07 | |||||||
| Net tangible book value per share after this offering | 0.11 | |||||||
| Dilution per share to investors participating in this offering | $ | 0.45 | ||||||
| 32 |
27
You should read the following selected financial data together with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and accompanying notes included later in this prospectus. The selected financial data in this section is not intended to replace our financial statements and the accompanying notes.
We have derived the selected balance sheet data as of December 31, 20092015 and 20082014 and the selected statement of operations data for the years ended December 31, 20092015 and 20082014 from our audited financial statements that are included in this prospectus. We have derived the selected balance sheet data as of December 31, 2007, 20062013, 2012 and 20052011 and the selected statement of operations data for the years ended December 31, 2007, 20062013, 2012 and 20052011 from our audited financial statements that are not included in this prospectus. We have derived the selected statement of operations data for three months ended March 31, 2010 and 2009 and the selected balance sheet data as of March 31, 20102016 and 2015 and the selected statement of operations data for the three-month periods ended March 31, 2016 and 2015 from our unaudited financial statements that are included in this prospectus.
Our historical results are not necessarily indicative of the results to be expected in any future period.
| Year Ended December 31, | Three Months Ended | |||||||||||||||||||||||||||
| 2015 | 2014 | 2013 | 2012 | 2011 | 2016 | 2015 | ||||||||||||||||||||||
| Statement of Operations Data: | ||||||||||||||||||||||||||||
| Revenue | $ | 60,612 | $ | - | $ | 29,484 | $ | 1,342,844 | $ | 1,511,572 | $ | 55,190 | $ | 40,000 | ||||||||||||||
| Operating expenses: | ||||||||||||||||||||||||||||
| Research and development | 203,185 | 367,275 | 188,643 | 1,161,967 | 5,067,085 | 102,901 | 36,472 | |||||||||||||||||||||
| General and administrative | 1,566,962 | 1,188,211 | 755,092 | 1,022,207 | 2,456,028 | 404,710 | 394,500 | |||||||||||||||||||||
| Total operating expenses | 1,770,147 | 1,555,486 | 943,735 | 2,184,174 | 7,523,113 | 507,611 | 430,972 | |||||||||||||||||||||
| Loss from operations | (1,709,535 | ) | (1,555,486 | ) | (914,251 | ) | (841,330 | ) | (6,011,541 | ) | (452,421 | ) | (390,972 | ) | ||||||||||||||
| Interest and other income | 101 | 153 | 16 | 26 | 4,489 | - | 77 | |||||||||||||||||||||
| Interest expense | 172,883 | 183,473 | 100,941 | 5,698 | - | 43,101 | 42,629 | |||||||||||||||||||||
| Change in fair value of derivative | (3,388,166 | ) | (1,014,836 | ) | 343,833 | - | - | (2,883,334 | ) | (1,285,166 | ) | |||||||||||||||||
| Net loss | $ | (5,270,483 | ) | $ | (2,753,642 | ) | $ | (671,343 | ) | $ | (847,002 | ) | $ | (6,007,052 | ) | $ | (3,378,856 | ) | $ | (1,718,690 | ) | |||||||
| Basic and diluted net loss per share | $ | (0.05 | ) | $ | (0.03 | ) | $ | (0.01 | ) | $ | (0.01 | ) | $ | (0.07 | ) | $ | (0.03 | ) | $ | (0.02 | ) | |||||||
| Shares used to compute basic and diluted net loss per share | 101,527,676 | 93,186,443 | 81,733,247 | 81,627,638 | 80,135,099 | 101,640,092 | 101,316,580 | |||||||||||||||||||||
| As of December 31, | As of March 31, | |||||||||||||||||||||||||||
| 2015 | 2014 | 2013 | 2012 | 2011 | 2016 | 2015 | ||||||||||||||||||||||
| Balance Sheet Data: | ||||||||||||||||||||||||||||
| Cash and cash equivalents | $ | 317,627 | $ | 844,043 | $ | 6,306 | $ | 141,905 | $ | 116,042 | $ | 79,834 | $ | 1,062,499 | ||||||||||||||
| Working capital (deficiency) | (17,114 | ) | 520,949 | (583,655 | ) | (362,815 | ) | (455,514 | ) | (360,123 | ) | 646,767 | ||||||||||||||||
| Total assets | 358,223 | 949,191 | 42,730 | 356,621 | 383,594 | 113,750 | 1,131,967 | |||||||||||||||||||||
| Total liabilities | 7,100,619 | 2,660,810 | 1,664,139 | 1,379,910 | 810,659 | 10,063,433 | 4,534,137 | |||||||||||||||||||||
| Accumulated deficit | (105,074,838 | ) | (99,804,355 | ) | (97,050,713 | ) | (96,379,370 | ) | (95,532,368 | ) | (108,453,694 | ) | (101,523,045 | ) | ||||||||||||||
| Stockholders’ deficiency | (6,742,396 | ) | (1,711,619 | ) | (1,621,409 | ) | (1,023,289 | ) | (427,065 | ) | (9,949,683 | ) | (3,402,170 | ) | ||||||||||||||
| 33 |
| Three Months Ended | ||||||||||||||||||||||||||||
| Year Ended December 31, | March 31, | |||||||||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | 2010 | 2009 | ||||||||||||||||||||||
Statement of Operations Data: | ||||||||||||||||||||||||||||
| Sponsored research revenue | $ | — | $ | 168,412 | $ | 240,324 | $ | 272,491 | $ | — | $ | — | $ | — | ||||||||||||||
| Operating expenses: | ||||||||||||||||||||||||||||
| Research and development | 3,724,514 | 7,149,808 | 8,887,255 | 6,396,524 | 3,155,735 | 470,434 | 1,661,600 | |||||||||||||||||||||
| General and administrative | 2,781,790 | 3,805,346 | 3,197,685 | 2,665,652 | 2,513,792 | 678,068 | 859,568 | |||||||||||||||||||||
| Total operating expenses | 6,506,304 | 10,955,154 | 12,084,940 | 9,062,176 | 5,669,527 | 1,148,502 | 2,521,168 | |||||||||||||||||||||
| Loss from operations | (6,506,304 | ) | (10,786,742 | ) | (11,844,616 | ) | (8,789,685 | ) | (5,669,527 | ) | (1,148,502 | ) | (2,521,168 | ) | ||||||||||||||
| Interest income | 12,444 | 149,777 | 666,458 | 522,704 | 214,676 | 2,793 | 6,518 | |||||||||||||||||||||
| Net loss | $ | (6,493,860 | ) | $ | (10,636,965 | ) | $ | (11,178,158 | ) | $ | (8,266,981 | ) | $ | (5,454,851 | ) | $ | (1,145,709 | ) | $ | (2,514,650 | ) | |||||||
| Basic and diluted net loss per share | $ | (0.12 | ) | $ | (0.21 | ) | $ | (0.24 | ) | $ | (0.21 | ) | $ | (0.15 | ) | $ | (0.02 | ) | $ | (0.05 | ) | |||||||
| Shares used to compute basic and diluted net loss per share | 55,680,525 | 50,967,617 | 46,465,982 | 40,116,367 | 36,843,609 | 60,406,828 | 53,622,491 | |||||||||||||||||||||
| As of | ||||||||||||||||||||||||||||
| As of December 31, | March 31, | |||||||||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | 2010 | |||||||||||||||||||||||
Balance Sheet Data: | ||||||||||||||||||||||||||||
| Cash and cash equivalents | $ | 4,355,768 | $ | 5,655,367 | $ | 3,696,878 | $ | 13,052,308 | $ | 4,896,143 | $ | 3,189,990 | ||||||||||||||||
| Short-term investments | — | — | 4,579,592 | 4,000,000 | 2,679,693 | — | ||||||||||||||||||||||
| Working capital | 3,671,910 | 4,565,932 | 6,102,596 | 16,187,188 | 6,939,195 | 2,648,787 | ||||||||||||||||||||||
| Total assets | 4,583,754 | 5,922,576 | 8,621,793 | 17,501,625 | 7,724,634 | 3,455,854 | ||||||||||||||||||||||
| Total liabilities | 880,404 | 1,325,912 | 2,469,069 | 1,249,290 | 714,127 | 766,369 | ||||||||||||||||||||||
| Accumulated deficit | (84,501,404 | ) | (78,007,544 | ) | (67,405,579 | ) | (56,227,421 | ) | (47,960,440 | ) | (85,647,113 | ) | ||||||||||||||||
| Stockholders’ equity | 3,703,350 | 4,596,664 | 6,152,724 | 16,252,335 | 7,010,507 | 2,689,485 | ||||||||||||||||||||||
28
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of our operations together with our financial statements and the related notes to those statements included later in this prospectus. In addition to historical financial information, this discussion contains forward-looking statements reflecting our current plans, estimates, beliefs and expectations that involve risks and uncertainties. As a result of many important factors, particularly those set forth under “Special Note Regarding Forward-Looking Statements” and “Risk Factors,” our actual results and the timing of events may differ materially from those anticipated in these forward-looking statements.
You should read the following discussion and analysis together with our consolidated financial statements and the related notes included elsewhere in this registration statement.
Business Overview
We are a biopharmaceutical company focused on the development of a novel therapeutic peptide, Thymosin beta 4, or Tß4, for tissue and organ protection, repair, and regeneration. We have formulated Tß4 into three distinct product candidates currently in clinical development:
• RGN-259, a preservative-free topical eye drop for regeneration of corneal tissues damaged by injury, disease or other pathology • RGN-352, an injectable formulation to treat cardiovascular diseases, central and peripheral nervous system diseases, | ||
• RGN-137, a topical gel for dermal wounds and reduction of scar tissue.
We are continuing strategic partnership discussions with biotechnology and pharmaceutical companies regarding the further clinical development of all of our product candidates.
In addition to our fourthree pharmaceutical product candidates, we are also pursuingevaluating the commercial developmentpotential use of peptide fragments and derivatives of Tß4 for cosmeceutical use. We believe the biological activities of these fragments may be useful, for example, in developing novel cosmeceutical products for the anti-aging market.
29
Current Clinical Status
On January 28, 2015, we announced that we had entered into a Joint Venture Agreement with GtreeBNT Co., Ltd., a Korean pharma company and shareholder of the dateCompany. The Joint Venture Agreement provides for the creation of this prospectus, we believe we have sufficient liquidityan entity, ReGenTree, LLC, jointly owned by us and capital resourcesGtreeBNT that will commercialize RGN-259 for treatment of dry eye and neurotrophic keratopathy, an orphan indication in the United States. GtreeBNT is be responsible for funding all product development and commercialization efforts, and holds a majority interest in ReGenTree. In March 2015, GtreeBNT reported that it received $7.28 million to fund our operations, including our ongoing clinical trials and other research initiatives, intoexpand international development of the third quarter of 2010, without considering any proceeds from this offering or any other sources of funding. With the proceeds of this offering, we believe that weproduct candidate, RGN-259 (designated GBT-201 in Korea). The $7.28 million will be ableused for development of RGN-259/GBT-201 for dry eye syndrome and neurotrophic keratopathy in the U.S. through the U.S. joint venture, ReGenTree, LLC.
| 34 |
In September 2015, ReGenTree began a Phase 2b/3 clinical trial in patients with dry eye syndrome (“DES”) and a Phase 3 clinical trial in patients with neurotrophic keratopathy (“NK”), both in the U.S. In May 2016, we reported the results of a 317-patient Phase 2/3 trial conducted by our U.S. joint venture ReGenTree. In the trial, RGN-259 demonstrated statistically significant improvements in both signs and symptoms of dry eye with 0.05% and 0.1% RGN-259 compared to initiateplacebo in a dose dependent manner during a 28-day dosing period. While the primary outcome measures were not met, several key related pre-specified endpoints and conduct at least a portionsubgroups of our contemplated Phase 2 AMI trial of RGN-352 as well as to complete our ongoingpatients with more severe dry eye showed statistically significant treatment effects. These results confirm the findings from the previous Phase 2 trial providing clear direction for the clinical regulatory pathway and remaining registration trials for RGN-259. The NK trial, a smaller study in an orphan population, has enrolled seven patients thus far with a goal of RGN-137 in EB46. Of the eight original clinical sites for the study, six are enrolling patients, one has yet to receive IRB approval to begin enrolling patients, and one was unable to supportconsummate a clinical contract with ReGenTree. ReGenTree is considering adding additional sites to accelerate patient enrollment.
Currently, we have active partnerships in three major territories: the compassionate use studiesU.S., China and Pan Asia. Our partners have been moving forward and making progress in each territory. In each case, the cost of development is being borne by our partners with no financial obligation for RegeneRx. Patient accrual, treatment, and follow-up for these ophthalmic trials are, in general, relatively fast, as opposed to most other clinical efforts.
We still have significant clinical assets to develop, primarily RGN-352 (injectable formulation of Tß4 for cardiac and CNS disorders) in the U.S., Pan Asia, and Europe, and RGN-259 andin the proposed Phase 1/2 trial of RGN-352 in multiple sclerosis patients.EU. Our goal is to wait until the results are obtained from the current ophthalmic clinical trials before moving into the EU with RGN-259. If successful, this should allow us to obtain a higher value for the asset at that time. However, we will need substantial additional funds beyond the proceeds of this offering in order to initiate and complete further clinical trials beyond those currently contemplated andintend to continue to develop RGN-352, either by obtaining grants to fund our operations.
Financial Operations Overview
We have never generated product revenues, and we do not expect to generate product revenues until the FDA approves one of our product candidates, if ever, and we begin marketing and selling it. Subject to the availability of financing, we expect to invest increasingly significant amounts in the furtherance of our current clinical programs and may add additional preclinicalnonclinical studies and new clinical trials as we explore the potential of our current product candidates in other indications and explore new formulations of Tß4-based product candidates. As we expand our clinical development initiatives, we expect to incur substantial and increasing losses. Accordingly, we will need to generate significant product revenues in order to ultimately achieve and then maintain profitability. Also, we expect that we will need to raise substantial additional capital in addition to the proceeds of this offering in order to meet product development requirements. We cannot assure investors that such capital will be available when needed, on acceptable terms, or at all.
Most of our expenditures to date have been for research and development, or R&D, activities and general and administrative, or G&A, activities. R&D costs include all of the wholly-allocable costs associated with our various clinical programs passed through to us by our outsourced vendors. Those costs include manufacturing Tß4 and peptide fragments, formulation of Tß4 into our product candidates, stability studies for both Tß4, and the various formulations, preclinical toxicology, safety and pharmacokinetic studies, clinical trial management, medical oversight, laboratory evaluations, statistical data analysis, regulatory compliance, quality assurance
30
R&D expenditures are subject to the risks and uncertainties associated with clinical trials and the FDA review and approval process. As a result, these expenses could exceed our expectations, possibly materially. We are uncertain as to what we will incur in future research and development costs for our clinical studies, as these amounts are subject to the outcome of current studies, management’smanagement's continuing assessment of the economics of each individual research and development project and the internal competition for project funding.
| 35 |
G&A costs include outside professional fees for legal, business development, audit and accounting services, including the costs to maintain our intellectual property portfolio.services. G&A also includes cash and non-cash compensation, employee benefits, travel and other miscellaneous costs of our internal G&A personnel, threetwo in total, who are wholly dedicated to G&A efforts. G&A also includes a proration of our common infrastructure costs for office space, and communications.
Critical Accounting Policies
We prepare our financial statements in conformity with accounting principles generally accepted in the United States. Such accounting principles require that our management make estimates and assumptions that affect the amounts reported in our financial statements and accompanying notes. Our actual results could differ materially from those estimates. The items in our financial statements that have required us to make significant estimates and judgments are as follows:
Revenue Recognition
We recognize revenue in accordance with the authoritative guidance for revenue recognition. We recognize revenue when all of the following criteria are met: (i) persuasive evidence of an arrangement exists, (ii) delivery (or passage of title) has occurred or services have been rendered, (iii) the seller's price to the buyer is fixed or determinable, and (iv) collectability is reasonably assured. We also comply with the authoritative guidance for revenue recognition regarding arrangements with multiple deliverables. Multiple-element arrangements are analyzed to determine whether the deliverables, which may include a license together with performance obligations such as providing a clinical supply of product and steering committee services, can be separated or whether they must be accounted for as a single unit of accounting. Revenue associated with licensing agreements consists of non-refundable upfront license fees and milestone payments. Non-refundable upfront license fees received under license agreements, whereby continued performance or future obligations are considered inconsequential to the relevant license technology, are recognized as revenue upon delivery of the technology.
Whenever we determine that an arrangement should be accounted for as a single unit of accounting, we must determine the period over which the performance obligations will be performed and revenue will be recognized. Revenue will be recognized using either a relative performance or straight-line method. We recognize revenue using the relative performance method provided that the we can reasonably estimate the level of effort required to complete our performance obligations under an arrangement and such performance obligations are provided on a best-efforts basis. Revenue recognized is limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the relative performance method, as of each reporting period.
If we cannot reasonably estimate the level of effort required to complete our performance obligations under an arrangement, the performance obligations are provided on a best-efforts basis and we can reasonably estimate when the performance obligation ceases or the remaining obligations become inconsequential and perfunctory, then the total payments under the arrangement, excluding royalties and payments contingent upon achievement of substantive milestones, would be recognized as revenue on a straight-line basis over the period we expect to complete our performance obligations. Revenue is limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the straight-line basis, as of the period ending date.
If we cannot reasonably estimate when our performance obligation either ceases or becomes inconsequential and perfunctory, then revenue is deferred until we can reasonably estimate when the performance obligation ceases or becomes inconsequential. Revenue is then recognized over the remaining estimated period of performance.
We recognize consideration that is contingent upon the achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone is substantive in its entirety. A milestone is considered substantive when it meets all of the following criteria:
| · | The consideration is commensurate with either the entity's performance to achieve the milestone or the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity's performance to achieve the milestone; |
| 36 |
| · | The consideration relates solely to past performance; and | |
| · | The consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. |
A milestone is defined as an event (i) that can only be achieved based in whole or in part on either the entity's performance or on the occurrence of a specific outcome resulting from the entity's performance, (ii) for which there is substantive uncertainty at the date the arrangement is entered into that the event will be achieved and (iii) that would result in additional payments being due to us.
Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue in our accompanying balance sheets.
Variable Interest Entities
The Company has determined that the Joint Venture is a “variable interest entity”, since the total equity investment at risk is not sufficient to permit the Joint Venture to finance its activities without additional subordinated financial support. Further, because of GtreeBNT’s majority equity stake in the Joint Venture, voting control, control of the board of directors, and substantive management rights, and given that the Company does not have the power to direct the Joint Venture’s activities that most significantly impact its economic performance, the Company determined that it is not the primary beneficiary of the Joint Venture and therefore is not required to consolidate the Joint Venture. The Company reports its equity stake in the Joint Venture using the equity method of accounting because, while it does not control the Joint Venture, the Company can exert significant influence over the Joint Ventures activities by virtue of its board representation.
Because the Company is not obligated to fund the Joint Venture, and has not provided any financial support and has no commitment to provide financial support in the future to the Joint Venture, the carrying value of its investment in the Joint Venture is zero. As a result, the Company is not recognizing its share (currently 49%) of the Joint Venture’s operating losses and will not recognize any such losses until the Joint Venture produces net income (as opposed to net losses) and at that point the Company will reduce its share of the Joint Venture’s net income by its share of previously suspended net losses. As of December 31, 2015, because it has not provided any financial support, the Company has no financial exposure as a result of its variable interest in the Joint Venture.
Convertible Notes with Detachable Warrants.
In accordance with Accounting Standards Codification (“ASC”) 470-20,Debt with Conversion and Other Options, the proceeds received from convertible notes are allocated between the convertible notes and the detachable warrants based on the relative fair value of the convertible notes without the warrants and the relative fair value of the warrants. The portion of the proceeds allocated to the warrants is recognized as additional paid-in capital and a debt discount. The debt discount related to warrants is accreted into interest expense through maturity of the notes.
Derivative Financial Instruments.
Derivative financial instruments consist of financial instruments or other contracts that contain a notional amount and one or more underlying variables (e.g. interest rate, security price or other variable), which require no initial net investment and permit net settlement. Derivative financial instruments may be free-standing or embedded in other financial instruments. Further, derivative financial instruments are initially, and subsequently, measured at fair value and recorded as liabilities or, in rare instances, assets.
The Company does not use derivative financial instruments to hedge exposures to cash-flow, market or foreign-currency risks. However, the Company has issued financial instruments including warrants that are either (i) not afforded equity classification, (ii) embody risks not clearly and closely related to host contracts, or (iii) may be net-cash settled by the counterparty. In certain instances, these instruments are required to be carried as derivative liabilities, at fair value, in the Company’s financial statements. In other instances these instruments are classified as equity instruments in the Company’s financial statements.
| 37 |
The Company estimates the fair values of its derivative financial instrument using the Black-Scholes option pricing model because it embodies all of the requisite assumptions (including trading volatility, estimated terms and risk free rates) necessary to fair value these instruments. Estimating fair values of derivative financial instruments requires the development of significant and subjective estimates that may, and are likely to, change over the duration of the instrument with related changes in internal and external market factors. In addition, option-based techniques are highly volatile and sensitive to changes in the trading market price of the Company’s common stock, which has a high-historical volatility. Since derivative financial instruments are initially and subsequently carried at fair values, the Company’s operating results reflect the volatility in these estimate and assumption changes in each reporting period.
Share-Based Payment
Share-based payment
We account for share-based compensation based on the estimated grant date fair value of the award using the Black-Scholes option-pricing model. The estimated grant date fair value is recognized over the requisite service period.
Determining the appropriate fair value model and calculating the fair value of share-based payment awards require the input of highly subjective assumptions, including the expected life of the share-based payment awards and stock price volatility. Since our historical data is limited, the expected life was determined in accordance with SECCommission Staff Accounting Bulletin No. 107 guidance for “plain vanilla” options. Since our historical trading volume is relatively low, we estimated the expected volatility based on monthly closing prices for a period consistent with the expected life of the option.
The assumptions used in calculating the fair value of share-based payment awards represent management’s best estimates, but these estimates involve inherent uncertainties and the application of management judgment. As a result, if factors change and we use different assumptions, our stock-based compensation expense could be materially different in the future. In addition, we are required to estimate the expected forfeiture rate and only recognize expense for those shares expected to vest. If our actual forfeiture rate is materially different from our estimate, the stock-based compensation expense could be significantly different from what we have recorded in the current period. See NoteNotes 2 and 9 to our financial statements included in this prospectusthe Financial Statements for the year ended December 31, 2015 for a further discussion on stock-based compensation and the relative ranges of our historical, underlying assumptions.
Costs of Preclinical Studiespre-clinical studies and Clinical Trialsclinical trials
We accrue estimated costs for preclinicalpre-clinical studies and clinical trials conducted by contract research organizations and participating hospitals. These costs are a significant component of research and development expenses. We accrue costs for preclinicalpre-clinical studies and clinical trials performed by contract research organizations based on estimates of work performed under the contracts. Costs of setting up hospital sites for participation in trials are accrued immediately. Hospital costs related to patient enrollment are accrued as patients are entered in the trial.
Results of Operations
Comparison of years ended December 31, 2015 and 2014
Revenues. For the year ended December 31, 2015, we recorded revenue in the amount of $60,612, $40,000 of this revenue related to the sale of unformulated Tß4 to GtreeBNT for use in their product development work in Korea. There were no associated costs with this transaction as the cost of Tß4 had been expensed in a prior period. We also recorded revenue in the amount of $20,612 which reflects the amortization of the upfront license fee over the life of the ReGenTree license agreement of 25 years. We did not record any revenue for the year ended December 31, 2014.
Expenses — Research and development. For the year ended December 31, 2015, our R&D expenditures decreased by $164,000, or 45%, to $203,000, from approximately $367,000 in 2014. The decrease from 2014 reflects the execution of the our strategy to out license or partner our development programs in addition to our entry into the ReGenTree joint venture agreement in 2015 under which we are not responsible for the development activity or costs. The 2014 R&D expenditures were primarily related to the engagement of part-time personnel, outside consultants and CROs to evaluate our readiness to proceed directly into a Phase 3 clinical trial for neurotrophic keratopathy (NK) under the orphan designation received in late 2013. In 2015 our R&D expense includes $73,000 of non-cash stock based compensation expense versus $48,000 in 2014.
| 38 |
31
Comparison of the three months ended March 31, 20102016 and 20092015
Research and Development Expense.Revenues.For the three months ended March 31, 2010, our R&D expenses decreased by approximately $1.2 million, or 72%,2016, we recorded revenue in the amount of $55,190. $45,088 related to $470,000,the sale of unformulated Tß4 to G-treeBNT for use in their product development work in Korea and the balance of $10,102 related to the amortization of the license fees received from approximately $1.7 million for the same period in 2009. The decrease was primarily the result of reduced clinical activity in 2010 as compared to 2009. During the three months ended March 31, 2009, we concluded our Phase 2 clinical trials evaluating RGN-137 in patients with pressure ulcers and venous stasis ulcers, as well as the clinical portion of our Phase 1 trial evaluating the safety of RGN-352 in healthy subjects. In January 2009, we also terminated the clinical portion of our Phase 2 clinical trial evaluating the safety and efficacy of RGN-259 to treat diabetic patients whose corneal epithelium had been scraped during vitrectomy surgery.
32
G&A Expenses. For the three months ended March 31, 2010 as compared2016, our G&A expenses increased by approximately $10,000, or 3%, to $405,000, from $395,000 for the same period in 2009.
Net Loss.
33
Liquidity and Capital Resources
We have not commercialized any of our product candidates to date and have incurred significant losses since inception. We have primarily financed our operations through the issuance of common stock and common stock warrants in private and public financings, althoughissuance of convertible debt, and, as discussed below, we have recently been awarded a government grant and intend to apply for additional federal cash grants and tax credits.will continue to pursue other governmental funding sources. The report of our independent registered public accounting firm regarding our financial statements as of and for the year ended December 31, 20092015 contains an explanatory paragraph regarding our ability to continue as a going concern based upon our history of net losses and dependence on future financing in order to meet our planned operating activities.
We had cash and cash equivalents totaling $3.2 million, $4.4 million and $5.7 millionof $79,834 at March 31, 2010, December 31, 20092016. This amount coupled with a $250,000 payment that was received on April 6, 2016 pursuant to an amendment of the RGN-259 license agreement with ReGenTree and 2008, respectively. The decrease during the threeapproximately $1.6 million proceed from the 2016 offering should fund our planned operations for at least the 12 months ended March 31, 2010 was largely the result of our net loss during this period. The $1.3 million decrease during 2009 was the result of $6.2 million used in operating activities, offset by $4.9 million in cash raised through the private placement of common stock and warrants. As offollowing the date of this prospectus,Prospectus. This estimate does not include receipt of any funds from grants, new partnerships or the raising of additional capital if the market climate warrants. Additionally, we have approximately $2.7 million of cash and cash equivalents. Based on our current operations, we believe our existing cash resources, along with the expected net proceeds of this offering, will be adequateintend to fund our operations into the second quarter of 2011.
Net Cash Used in Operating Activities.
Net cash used in operating activities was approximately $1.2 million and $1.8 million$238,000 for the three months ended March 31, 2010 and 2009, respectively. In both periods, the net cash used in2016 compared to approximately $218,000 provided by operating activities was primarily the result of our net losses during the periods. Included in these net losses were non-cash expenses related to employee stock compensation and depreciation
34
Net cash used in operating activities was approximately $6.2 million$525,000 and $10.6 million$1,704,000 for the years ended December 31, 20092015 and 2008,2014, respectively. While ourOur reported net loss for the year ended December 31, 20092015 was approximately $6.5 million, it$5,270,000, which included approximately $0.8 million$366,000 in non-cash expenses primarily non-cash share-based compensation, which was offset by approximately $0.5 millionas well as an unrealized loss of $3,388,000 as a result of the increase in valuation of our convertible debt derivative component. In 2015 our statement of cash usedflows reflects an additional $979,000 related to retire current liabilitiespayments received under the license agreement with the Joint Venture as compared to the liabilitieswell as a net decrease in accounts payable and accrued expenses of $80,000 and a decrease in prepaid expenses of $62,000. For 2014 we reported as of December 31, 2008. Oura net loss of $2,754,000 which included $302,000 of non-cash expenses as well as an unrealized loss of $1,015,000 as a result of the increase in 2008valuation of $10.6 million approximated theour convertible debt derivative component. In 2014 we also experienced a net decrease in accounts payable and accrued expenses of $207,000 and well as an increase in prepaid expenses of $60,000.
| 39 |
Net Cash Used in Investing Activities.
Net cash used in operatinginvesting activities in the same period as the non-cash share based compensation expenses of approximately $1.1 million were fully offset by a similar reduction in liabilities as compared to those reported as of December 31, 2007.
There were no investing activities duringfor the three monthsmonth periods ended March 31, 2009. In 2008, we sold all of our short-term, highly-liquid, investment-grade financial instruments that had more than a90-day maturity from the date of purchase2016 and invested the proceeds in cash equivalents.
Net Cash Provided by Financing Activities. There were no financing activities during either of the three months ended March 31, 2010 or 2009.
Net cash provided by financing activities totaled approximately $4.9 million$0 and $7.9 million$2,555,000 for the years ended December 31, 20092015 and 2008,2014, respectively. In both periods, these net2014 the cash provided by financing activities consisted of the proceeds result from the issuancesale of common stock to GtreeBNT in March ($1,500,000) and warrantsAugust ($1,000,000) and $55,000 from the sale of convertible debt offerings as discussed in Note 7 to purchase common stock.
There were no financing activities for the three month periods ended March 31, 2016 and 2015.
Future Funding Requirements
The expenditures that will be necessary to execute our business plan are subject to numerous uncertainties that may adversely affect our liquidity and capital resources. As described elsewhereCurrently, RegeneRx has active partnerships in this prospectus, during 2009 we completed two Phase 2three major territories: the U.S., China and Pan Asia. Our partners have been moving forward and making progress in each territory. In each case, the cost of development is being borne by our partners with no financial obligation for RegeneRx. Patient accrual, treatment, and follow-up for ophthalmic trials are, in general, relatively fast, as opposed to most other clinical efforts, top line data from the U.S. dry eye trial was released in early May and data from the NK study toward the end of 2016 or possibly later.
We still have significant clinical assets to develop, primarily RGN-352 (injectable formulation of Tß4 for cardiac and CNS disorders) in the U.S., Pan Asia, and Europe, and RGN-259 in the EU. Our goal is to wait until the results are obtained from the current ophthalmic clinical trials closed one additional Phase 2 clinical trial and completedbefore moving into the EU with RGN-259. If successful, this should allow us to obtain a Phase 1 clinical trial. Currently, we are actively enrolling patients in one Phase 2 trial,higher value for RGN-137 in EB patients, while we are supporting small compassionate use studies of RGN-259. Subject to available funding,the asset at that time. However, we intend to commencecontinue to develop RGN-352, either by obtaining grants to fund a Phase 22a clinical trial of RGN-352 for AMI patients in the second half of 2010cardiovascular or central nervous system fields or finding a suitable partner with the resources and support a Phase 1/2 clinical trial of RGN-352 for MS patients,capabilities to develop it as well as a physician-sponsored Phase 2 clinical trial of RGN-259 in patientswe have with dry eye secondary to graft versus host disease. We currently do not have sufficient capital resources to continue clinical development beyond the third quarter of 2010, without giving effect to this offering. As described elsewhere in this prospectus, we were recently awarded a grant from the NIH, which will allow us to continue the development of RGN-352 without exhausting our current capital resources.
In addition, the length of time required for clinical trials varies substantially according to the type, complexity, novelty and intended use of a product candidate. Some of the factors that could impact our liquidity and capital needs include, but are not limited to:
| the progress of our clinical trials; | ||
| the progress of our research activities; | ||
| the number and scope of our research programs; | ||
| the progress of our preclinical development activities; | ||
| the costs involved in preparing, filing, prosecuting, maintaining, enforcing and defending patent and other intellectual property claims; |
35
| · | the costs related to development and manufacture of preclinical, clinical and validation lots for regulatory purposes and commercialization of drug supply associated with our product candidates; | |
| our ability to enter into corporate collaborations and the terms and success of these collaborations; | ||
| the costs and timing of regulatory approvals; and | ||
| the costs of establishing manufacturing, sales and distribution capabilities. |
In addition, the duration and the cost of clinical trials may vary significantly over the life of a project as a result of differences arising during the clinical trial protocol, including, among others, the following:
| the number of patients that ultimately participate in the trial; | ||
| · | the duration of patientfollow-up that seems appropriate in view of the results; | |
| the number of clinical sites included in the trials; and | ||
| the length of time required to enroll suitable patient subjects. |
Also, we test our potential product candidates in numerous preclinical studies to identify indications for which they may be product candidates.efficacious. We may conduct multiple clinical trials to cover a variety of indications for each product candidate. As we obtain results from trials, we may elect to discontinue clinical trials for certain product candidates or for certain indications in order to focus our resources on more promising product candidates or indications.
Our proprietary product candidates also have not yet achieved FDA regulatory approval, which is required before we can market them as therapeutic products. In order to proceed to subsequent clinical trial stages and to ultimately achieve regulatory approval, the FDA must conclude that our clinical data establish safety and efficacy. Historically, the results from preclinical studies and early clinical trials have often not been predictive of results obtained in later clinical trials. A number of new drugs and biologics have shown promising results in clinical trials, but subsequently failed to establish sufficient safety and efficacy data to obtain necessary regulatory approvals.
We were previously committed under an office space lease through January 2013 that requires average baseand continued to occupy the space on a month to month basis through May 2014. Beginning in June 2014 we consolidated our office space and amended our lease agreement for the reduced space. The new lease commitment is for 36 months and our rental payments offor this period will be approximately $7,300$4,500 per month.
Sources of Liquidity
We have not commercialized any of our product candidates to date and have primarily financed our operations through the issuance of common stock and common stock warrants in private and public financings. Sigma-Tau has historically provided significant equity capitalfinancings in addition to us. a series of five convertible debt placements from October 2012 to January 2014.
In 2009, Sigma-Tau providedMarch 2014 we entered into a strategic transaction with GtreeBNT which includes the purchase of approximately 19.6 million shares of common stock by G-GtreeBNT in two tranches, GtreeBNT is now our second largest stockholder.
In June 2016, we completed the 2016 Offering, through which we raised net proceeds of approximately $1.6 million. Following completion of the 2016 Offering, we believe we have sufficient capital resources to continue operations for at least the 12 months following the date of this Prospectus.
On January 28, 2015, we announced that we had entered into a Joint Venture Agreement with GtreeBNT, a shareholder of the Company. The Joint Venture Agreement provides for the creation of an entity, ReGenTree, LLC, jointly owned by us and GtreeBNT, which will commercialize RGN-259 for treatment of dry eye and neurotrophic keratopathy, an orphan indication in the United States. GtreeBNT is responsible for funding all product development and commercialization efforts.
RegeneRx’s ownership interest in ReGenTree is 49% and will be reduced to 42% following completion of the Phase 2b dry eye clinical trial. Based on when, and if, certain additional development milestones are achieved in the U.S. with RGN-259, our equity ownership may be incrementally reduced to between 42% and 25%, with 25% being the final equity ownership upon approval of an NDA for Dry Eye Syndrome in the U.S. In conjunction with the Joint Venture Agreement, we also entered into a royalty-bearing license agreement (the “License Agreement”) with ReGenTree pursuant to which we granted to ReGenTree the right to develop and exclusively commercialize RGN-259 in the United States. We received a total of $1 million in gross proceeds outtwo tranches under the terms of the approximately $5.3 millionLicense Agreement. The first tranche of $500,000 was received in total grossMarch 2015 and a second in the amount of $500,000, was received in September 2015.
We are also entitled to royalties as a percentage of net sales ranging from the single digits to the low-double digits based on the medical indications approved and whether the Joint Venture commercializes products directly or through a third party. In the event the ReGenTree entity is acquired or there is a change of control that occurs following achievement of an NDA, RegeneRx shall be entitled to 40% of all change of control proceeds raised duringpaid or payable and will forgo any future royalties. RegeneRx possesses one of three board seats and certain major decisions and transactions within ReGenTree, such as commercialization strategy, mergers, and acquisitions, require RegeneRx’s board designee’s consent. Additionally, we intend to continue to pursue additional partnering activities, particularly for RGN-352, our injectable systemic product candidate for cardiac and central nervous system indications.
| 41 |
Licensing Agreements
As noted above, we have entered into two strategic agreements with GtreeBNT. GtreeBNT licensed the year,development and commercialization rights for RGN-259, in Asia (excluding China, Hong Kong, Macau and Taiwan) while also licensing the development and commercialization rights for RGN-137 in the U.S. In January 2015 we entered into a joint venture and licensing agreement with new investors providingGtreeBNT that will commercialize RGN-259 for treatment of dry eye and neurotrophic keratitis in the remaining $3.7 million. Sigma-Tau provided allUnited States, as well as any other indications within the field of ophthalmology. The license agreements provide for the opportunity for us to receive milestone payments upon specified commercial events and royalty payments in connection with any commercial sales of the $8.0 millionlicensed products in gross proceeds raised during 2008.
We have a license agreement with a subsidiary of Sigma-TauSigma-Tau/Alfa Wassermann that provides the opportunity for us to receive milestone payments upon specified events and royalty payments uponin connection with commercial sales of Tß4 in Europe. However, we have not received any milestone payments to date, and there can be no assurance that we will be able to attain such milestones and generate any such payments under the agreement.
We also have entered into a result of recent government initiatives, we have engaged a consulting firm to assist us in identifying sources of Federal government funding. We are pursuing federally-sourced funding and were recently awarded a $3 million grant from the NIH, as described in this prospectus. Recently enacted healthcare reform legislation alsolicense agreement with Lee’s Pharmaceuticals that provides for a qualifying therapeutic discovery project credit, or Therapeutic Credit, as an incentivethe opportunity for small biotechnology companies like us. The Therapeutic Credit will allow small businessesus to apply for a federal grantreceive milestone payments upon specified events and royalty payments in an amount equal to 50%connection with any commercial sales of their investmentTß4-based products in qualifying therapeutic discovery projects for 2009China, Hong Kong, Macau and 2010. Qualifying therapeutic discovery projects include those designed to treat or
36
Government Grants
We have previously received significant government funding under this program. We are also collaborating with the Federal government in evaluatingand continue to pursue such funding for both RGN-259 our sterile eye drop product candidate, in animals exposedas well as RGN-352, although there can be no guarantee that we will be able to caustic agents, and we believe that our other formulations may also be of interest in healing damaged tissues for indications that result from battlefield or homeland security situations.
Other Financing Sources
Other potential sources of outside capital include entering into additional strategic business relationships, publicadditional issuances of equity securities or private sales of shares of our capital stock, or debt financing or other similar financial instruments. While we sold common stock and warrants to purchase common stock to Sigma-Tau and new investors in the fourth quarter of 2009, we do not have any committed sources of outside capital at this time. Consequently, there can be no assurance that we will be able to obtain additional capital in sufficient amounts, on acceptable terms, or at all.
Our failure to successfully address ongoing liquidity requirements wouldcould have a materially negative impact on our business, including the possibility of surrendering our rights to some technologies or product opportunities, delaying our clinical trials, or ceasing operations.
Off Balance Sheet Arrangements
We do not have any off-balance sheet arrangements, as such term is defined in Item 303(a)(4) ofRegulation S-K.
| 42 |
General
RegeneRx Biopharmaceuticals, Inc. (“RegeneRx” or the short-term nature of these investments, if market interest rates differed by 10% from their levels as of March 31, 2010, the change in fair value of our financial instruments would not have been material.
37
• RGN-259, a preservative-free topical eye drop for regeneration of corneal tissues damaged by injury, disease or other pathology; • RGN-352, an injectable formulation to treat cardiovascular diseases, central and peripheral nervous system diseases, |
• RGN-137, a topical gel for dermal wounds and reduction of scar tissue.
We are continuing strategic partnership discussions with biotechnology and pharmaceutical companies regarding the further clinical development of all of our product candidates.
In addition to our fourthree pharmaceutical product candidates, we are also pursuingevaluating the commercial development of peptide fragments and derivatives of Tß4 for potential cosmeceutical use. Cosmeceuticalsand other personal care uses. These fragments are cosmetic products with biologically active ingredients.select amino acid sequences, and variations thereof, within the Tß4 molecule that have demonstrated activity in severalin vitro preclinical research studies that we have sponsored. We believe the biological activities of these fragments may be useful, for example, in developing novel cosmeceutical products for the anti-aging market. Our strategy is to collaborate with another company to develop cosmeceutical formulations based on these peptides.
Current Clinical Status
On January 28, 2015, we announced that we had entered into a Joint Venture Agreement (the “Joint Venture Agreement”) with GtreeBNT Co., Ltd., a Korean pharma company (“GtreeBNT”) and shareholder of the Company. The Joint Venture Agreement provides for the creation of an entity, ReGenTree, LLC (the “Joint Venture” or “ReGenTree”), jointly owned by us and GtreeBNT, that will commercialize RGN-259 for treatment of dry eye and neurotrophic keratopathy, an orphan indication in the United States. GtreeBNT will be responsible for funding all product development and commercialization efforts, and holds a majority interest of ReGenTree that varies depending on development milestones achieved and eventual commercialization path, if successful. In conjunction with the Joint Venture Agreement, we also entered into a royalty-bearing license agreement (the “License Agreement”) with ReGenTree pursuant to which we granted to ReGenTree the right to develop and exclusively commercialize RGN-259 in the United States. We received a total of $1 million in two tranches under the terms of the License Agreement. The first tranche of $500,000 was received in March 2015 and a second in the amount of $500,000, was received in September 2015. We are also entitled to royalties as a percentage of net sales ranging from the single digits to the low-double digits based on the medical indications approved and whether the Joint Venture commercializes products directly or through a third party. RegeneRx possesses one of three board seats and certain major decisions and transactions within ReGenTree, such as commercialization strategy, mergers, and acquisitions, require RegeneRx’s board designee’s consent. In March 2015, GtreeBNT received $7.28 million to expand international development of the product candidate, RGN-259 (designated GBT-201 in Korea).
Our ownership interest in ReGenTree is 49% and will be reduced to 42% following completion of the Phase 2b trial for Dry Eye Syndrome. Based on when, and if, ReGenTree achieves certain additional development milestones in the U.S. with RGN-259, our equity ownership may be incrementally reduced to between 42% and 25%, with 25% being the final equity ownership upon FDA approval of an NDA for Dry Eye Syndrome in the U.S. In addition to our equity ownership, RegeneRx retains a royalty on net sales that varies between single and low double digits, depending on whether commercial sales are made by ReGenTree or a licensee. In the event the ReGenTree entity is acquired, or there is a change of control that occurs following achievement of an NDA RegeneRx shall be entitled to 40% of all change of control proceeds paid or payable and will forgo any future royalties.
| 43 |
In September 2015, ReGenTree began a Phase 2b/3 clinical trial in patients with dry eye syndrome (“DES”) and a Phase 3 clinical trial in patients with neurotrophic keratopathy (“NK”), both in the U.S. In January 2016, the DES had completed enrollment of all patients in the trial. The last patient received the last treatment in February. We expect to report top line data from the DES trial at the end of April 2016. The NK trial, a smaller study in an orphan population, has enrolled seven patients thus far with a goal of 46. Of the eight original clinical sites for the study, six are enrolling patients, one has yet to receive IRB approval to begin enrolling patients, and one was unable to consummate a clinical contract with ReGenTree. ReGenTree is considering adding additional sites to accelerate patient enrollment.
Currently, we have active partnerships in three major territories: the U.S., China and Pan Asia. Our partners have been moving forward and making progress in each territory. In each case, the cost of development is being borne by our partners with no financial obligation for RegeneRx. Patient accrual, treatment, and follow-up for ophthalmic trials are, in general, relatively fast, as opposed to most other clinical efforts, so preliminary data from the U.S. dry eye trial should be forthcoming in the second quarter of 2016 and data from the NK study toward the end of 2016 or possibly later.
We still have significant clinical assets to develop, primarily RGN-352 (injectable formulation of Tß4 for cardiac and CNS disorders) in the U.S., Pan Asia, and Europe, and RGN-259 in the EU. Our goal is to wait until the results are obtained from the current ophthalmic clinical trials before moving into the EU with RGN-259. If successful, this should allow us to obtain a higher value for the asset at that time. However, we intend to continue to develop RGN-352, either by obtaining grants to fund a Phase 2a clinical trial in the cardiovascular or central nervous system fields or finding a suitable partner with the resources and capabilities to develop it as we have with RGN-259.
In addition to these RGN-259 development activities, we intend to continue to pursue additional partnering activities, particularly for RGN-352, our injectable systemic product candidate for cardiac and central nervous system indications.
We anticipate incurring additional operating losses in the future as we continue to explore the potential clinical benefits of Tß4-based product candidates over multiple indications. To fund further development and clinical trials we have entered into a series of strategic partnerships under licensing and joint venture agreements (see Note 4) where our partners are responsible for advancing development of our product candidates with multiple clinical trials.
Overview of Tß4
Tß4 is a synthetic copy of a naturally occurring43-amino acid peptide that was originally isolated from bovine thymus glands. It plays a vital role in cell structure and motility and in the protection, regeneration, remodeling and healing of tissues.
Although it is recognized that wound healing is aand tissue regeneration are complex process,processes, most companies working to develop new drugs in this area have focused primarily on the development of growth factors to stimulate healing only and have, to date, failed to demonstrate dramatic improvements in the healing process. Unlike growth factors, numerous preclinical animal studies, published by independent researchers, have identified several important biological activities involving Tß4 that we believe make it potentially useful as a wound healing, repair and tissue regenerating agent. These activities include:
| · | Progenitor (Stem) Cell Recruitment and Differentiation. |
38
| models of multiple sclerosis, stroke, and traumatic brain injury. | ||
| · | Actin Regulation. Tß4 regulates actin, which comprises up to 10% of the protein of non-muscle cells in the body and plays a central role in cell structure and in the movement of cells. |
| · | Reduction of |
| · | Collagen and Laminin-5 Stimulation. Tß4 has a number of additional biological activities shown to reduce inflammation, stimulate the formation of collagen, and up-regulate the expression of laminin-5, a subepithelial basement membrane protein. Both collagen and laminin-5 are central to healthy tissue, wound repair and the prevention of | |
| disease.Laminin-5 promotes cell migration and maintains cell-cell and cell-matrix contacts for intact tissues which are important for preventing fluid loss and bacterial infection. | ||
| · |
Tß4 has shown efficacy in heart repair and regeneration in numerous animal models. A 2004 paper inNature showed that it could reduce the lesion size, improve cardiac function and promote survival. The 2006 Nature publication mentioned above further concluded that Tß4’s interaction with EPCs resulted in the formation of cardiomyocytes that repaired damaged myocardium, or heart tissue, in mice after an induced acute myocardial infarction, or AMI, commonly known as a heart attack. Research published in the journal Circulation showed Tß4’s cardioprotective effects in a pig ischemic-reperfusion model. This pig model is accepted as an important model upon which to base human clinical research, as pigs are larger mammals, the anatomy of the pig heart is similar to that of the human heart, and vascular response processes are completed five to six times faster in pigs than in humans, so that long-term results can be obtained in a relatively short period of time. This research also identified Tß4’s interaction with EPCs as the underlying basis of cardioprotection through the differentiation of EPCs into cardiomyocytes, yielding statistically significant cardiac functional recovery results when compared to the administration of placebo.
| 45 |
Similar research in the area of brain and central nervous system tissues also showed efficacy of repair and regeneration was published in the journal Neuroscience in 2009. This publication concluded that Tß4 triggered the differentiation of oligodendrocyte progenitor cells to form myelin-producing oligodendrocytes, which led to the remyelination of axons in the brain of mice with experimental autoimmune encephalomyelitis, or EAE. This mouse model is an accepted small animal model for the study of multiple sclerosis. Research published in the Journal of Neurosurgery in 2010 and also in the Journal of Neurological Science in 2014 showed that Tß4 could improve functional neurological outcome in an animal stroke model. A second study was published in the Journal of Neurosurgery in 2011 demonstrating that administration of Tß4 can significantly improve histological and functional outcomes in rats with traumatic brain injury, or TBI, indicating that Tß4 has considerable therapeutic potential for patients with TBI. More recently, researchers studying Tß4 under a material transfer agreement (MTA) found that Tß4 had beneficial effects in animal models of peripheral neuropathy, one of the major complications of diabetes. This research was published in the Journal of Neurobiology of Disease in December 2012 and appears to corroborate previous findings using Tß4 for repair of central nervous system disorders. A paper in Neuropharmacology in 2014 found many benefits of Tß4 administration in a rat model of spinal cord injury, including decreased lesion size at 7 days, increased neural and oligodendrocyte survival, increase levels of myelin basic protein (a marker of mature oligodendrocytes), decreased ED1 (a marker of activated microglia/macrophages), and decreased proinflammatory cytokines. Thus, Tß4 has efficacy for repair and regeneration in several nervous system injury models including MS, TBI, stroke, peripheral neuropathy, and spinal cord injury and there will likely be additional applications in this area. We believe that these various biological activities work togetherin concert to play a vital role in the healing and repair of injured or damaged tissue and suggest that Tß4 is an essential component of the tissue protection and regeneration process that may lead to many potential medical applications. All of our product candidates are based onutilize Tß4 as the active pharmaceutical ingredient (API), which is manufactured as a syntheticby solid-phase peptide synthesis and is an exact copy of the naturally occurring peptide and formulatedpeptide. We have created three distinct formulations for various routes of administration and applications.
Our Product Candidates
RGN-352
RGN-259
GN-259
RGN-259 is an injectableour proprietary preservative-free eye drop formulation of Thymosin beta 4. In September 2011, we completed a Phase 2a exploratory clinical trial evaluating the safety and efficacy of RGN-259 in 72 patients with moderate dry eye syndrome. Patients were randomly assigned to receive either RGN-259 or placebo in this double-masked, placebo-controlled trial. All patients received either RGN-259 (0.1% concentration) or placebo, twice daily for 30 days. Various signs and symptoms of dry eye, such as the degree of ocular surface damage, ocular itching, burning and grittiness, among others, were graded periodically during and following the treatment period. The trial was conducted by Ora Inc., an ophthalmic contract research organization that specializes in dry eye research and clinical trials, and utilized Ora’s Controlled Adverse Environment (CAESM) chamber, which is a model that exacerbates and standardizes signs and symptoms in the dry eye patient.
In November 2011, we reported preliminary safety and efficacy results from the trial. RGN-259 was deemed safe and well-tolerated, with no observed drug-related adverse events.
The co-primary outcome measures evaluated in the trial were inferior corneal fluorescein staining and decreased ocular discomfort on day 29, 24 hours after CAESMchallenge. Various secondary outcome efficacy measures were also evaluated in the trial. These outcome measures were based on the best available animal data at the time but without the benefit of any actual human clinical experience in dry eye. While the study did not meet statistical significance for reducing inferior corneal fluorescein staining, it did show a positive trend in this exploratory trial. RGN-259 did, however, show a statistically significant efficacy result in the other co-primary endpoint of decreased ocular discomfort and also demonstrated statistical significance in several secondary endpoints such as reduction of central corneal and superior corneal staining, important signs in dry eye patients and approvable endpoints by the FDA.
| 46 |
Key outcome measures were as follows:
| · | Patients receiving RGN-259 experienced a 325% greater reduction from baseline in central corneal fluorescein staining compared to placebo at the 24 hour recovery period (p = 0.0075). Reduction of fluorescein staining is indicative of a reduction in ocular surface damage of the central cornea; | |
| · | Patients receiving RGN-259 experienced a 257% greater reduction from baseline in exacerbation of superior corneal fluorescein staining in the CAESM chamber as compared to the placebo (p = 0.0210); and | |
| · | Patients receiving RGN-259 experienced a 27.3% greater reduction in exacerbation of ocular discomfort at day 28 during a 75-minute challenge in the CAESM chamber compared to the placebo group (p = 0.0244). Reduction indicates that RGN-259 can slow progression of ocular symptoms in patients with dry eye syndrome. | |
| · | Other CAESM-related findings, such as peripheral (combination of the average of superior and inferior) corneal staining reduction, were observed having statistical significance, while others had positive trends after treatment with RGN-259. These observations are in line with the known biological properties and mechanisms of action of RGN-259 reported in various nonclinical studies. |
With respect to inferior corneal fluorescein staining, we did see a positive trend toward improvement, at day 28 during exposure to adverse conditions in the CAESM chamber in patients receiving RGN-259 compared to placebo, although this improvement was not deemed to be statistically significant (p = 0.0968).
Statistical significance (p value) of ≤ 0.05 is the generally accepted threshold for showing an outcome did not happen merely by chance.
The co-primary outcome measures, selected at the outset of this initial Phase 2a exploratory trial, were based on the best available animal data at the time but without the benefit of any actual human clinical experience in dry eye. Therefore, we believe that not having met one of the two co-primary outcome measures at this stage is not as important as identifying statistically significant outcomes that could potentially serve as approvable endpoints in later stage or in pivotal Phase 3 clinical trials. We believe that the statistically significant observation of reduction in central and/or superior corneal staining, as well as symptom improvements observed in the trial and described above, reflect actual patient benefits and would represent acceptable outcome measures to the FDA for use in follow-up Phase 2b or confirmatory pivotal Phase 3 trials. We prepared a clinical study report for submission to the FDA that describes the results of the exploratory Phase 2a clinical trial and the results were published in an appropriate medical journal.
In June 2012, we reported preliminary results from a double-masked, vehicle-controlled, physician-sponsored Phase 2 clinical trial evaluating RGN-259 for the treatment of nine patients (18 eyes) with severe dry eye. RGN-259 was observed to be safe and well-tolerated and met key efficacy objectives with statistically significant sign and symptom improvements, compared to vehicle control, at various time intervals, including 28 days post-treatment.
��
In the trial, nine patients with severe dry eye (18 eyes) were treated with RGN-259 or vehicle control six times daily over a period of 28 days. They were evaluated upon entering the study after a two-week washout period, at weekly intervals during the treatment phase, at the end of the 28-day treatment period, and at a follow-up visit 28 days after treatment. Statistically significant differences in sign and symptom assessments, such as ocular discomfort and corneal fluorescein staining, were seen at various time points throughout the study. Of particular note were the differences between RGN-259 and vehicle control 28 days post-treatment, or the follow-up period. The RGN-259-treated group had a 35.1% reduction of ocular discomfort (symptom) compared to vehicle control (p = 0.0141), and a 59.1% reduction of total corneal fluorescein staining (sign) compared to vehicle control (p = 0.0108) at 28 days after treatment showing that the repair was sustained long after treatment cessation.
Consistent with the reduction of ocular discomfort and fluorescein staining at the 28-day follow-up visit, other improvements seen in the RGN-259-treated patients included tear film breakup time and increased tear volume production. Likewise, these improvements were seen at other time points in the study. These results were recently published in an appropriate medical journal.
| 47 |
Strategic Partnerships
Lee’s Pharmaceuticals.In July 2012, we entered into a License Agreement with Lee’s Pharmaceutical (HK) Limited (“Lee’s”), headquartered in Hong Kong, for the license of Thymosin Beta 4 in any pharmaceutical form, including our RGN-259, RGN-352 and RGN-137 product candidates, in China, Hong Kong, Macau and Taiwan. Lee’s has filed an investigational new drug application IND with the Chinese FDA to conduct a Phase 2, randomized, double-masked, dose-response clinical trial with RGN-259 in China for dry-eye syndrome. Lee's recently informed us that it received notice from China's FDA (CFDA) declining its investigational new drug (IND) application for a Phase 2b dry eye clinical trial because the API (active pharmaceutical ingredient or Tß4) was manufactured outside of China. The API was manufactured in the U.S. and provided to Lee's by RegeneRx pursuant to a license agreement to develop RGN-259 ophthalmic eye drops in the licensed territory. Due to this unexpected regulatory hurdle, Lee's is modifying its clinical program to conduct the Phase 2b dry eye trial in Hong Kong and Taiwan using the Tß4 supplied by RegeneRx while awaiting the manufacturing of Tß4 in China for systemic administration.a subsequent Phase 3 registration trial. We have initially targeted RGN-352do not know when the Hong Kong/Taiwan trial will begin enrollment of patients. Under this revised strategy, Lee's believes it should be able to begin a Phase 3 registration trial in China sooner rather than waiting on the production of Tß4 in China before initiating a Phase 2b trial.
GtreeBNT.In March 2014, we entered into a License Agreement with GtreeBNT for the license of RGN-259. GtreeBNT licensed certain development and commercialization rights for RGN-259, in Asia (excluding China, Hong Kong, Macau and Taiwan). GtreeBNT is currently our second largest shareholder. Gtree filed an IND with the Korean Ministry of Food and Drug Safety to conduct a Phase 2b/3 study with RGN-259 in patients with dry eye syndrome and in July 2015 received approval to conduct the trial. GtreeBNT has informed us that given its immediate focus on the two U.S. trials, it is considering the best timing for the Korean trial.
U.S. Joint Venture (ReGenTree, LLC).On January 28, 2015, we announced that we entered into a Joint Venture Agreement (the “Joint Venture Agreement”) with GtreeBNT. The Joint Venture Agreement provides for the creation of an entity (the “Joint Venture” or “ReGenTree”), owned by us and GtreeBNT, that will commercialize RGN-259 for treatment of dry eye and neurotrophic keratopathy in the United States. GtreeBNT will be responsible for funding product development and commercialization efforts and holds a majority interest of ReGenTree. RegeneRx possesses one of three board seats and certain major decisions and transactions within ReGenTree, such as commercialization strategy, mergers, and acquisitions, require RegeneRx’s board designee’s consent. In conjunction with the Joint Venture Agreement, we also entered into a royalty-bearing license agreement (the “License Agreement”) with ReGenTree pursuant to which we granted to ReGenTree the right to develop and exclusively commercialize RGN-259 in the United States.
Our ownership interest in ReGenTree is 49% and will be reduced to 42% following completion of the Phase 2b trial for Dry Eye Syndrome. Based on when, and if, ReGenTree achieves certain additional development milestones in the U.S. with RGN-259, our equity ownership may be incrementally reduced to between 42% and 25%, with 25% being the final equity ownership upon FDA approval of an NDA for Dry Eye Syndrome in the U.S. In addition to our equity ownership, RegeneRx retains a royalty on net sales that varies between single and low double digits, depending on whether commercial sales are made by ReGenTree or a licensee. In the event the ReGenTree entity is acquired or there is a change of control that occurs following achievement of an NDA, RegeneRx shall be entitled to 40% of all change of control proceeds paid or payable and will forgo any future royalties.
The NK trial, a smaller study in an orphan population, has enrolled seven patients thus far with a goal of 46. Of the eight original clinical sites for the study, six are enrolling patients, one has yet to receive IRB approval to begin enrolling patients, and one was unable to consummate a clinical contract with ReGenTree. ReGenTree is considering adding additional sites to accelerate patient enrollment.
| 48 |
GtreeBNT has developed the CMC (chemistry, manufacturing and controls) dossier required for Phase 3 clinical trials and commercialization in the U.S. and in Korea. This comprehensive and critical effort ensures that final drug product manufacturing, packaging, stability, purity, reproducibility, etc., meets regulatory guidelines and product specifications. The product of this activity is the current product format being utilized in the U.S. trials being conducted by ReGenTree and will also be utilized in the planned clinical activity to be conducted by GtreeBNT under the RGN-259 license agreement for Pan Asia.
In May 2016, we reported the results of a 317-patient Phase 2/3 trial conducted by our U.S. joint venture ReGenTree (see the discussion below under “Strategic Patnerships – U.S. Joint Venture (ReGenTree, LLC)”). In the trial, RGN-259 demonstrated statistically significant improvements in both signs and symptoms of dry eye with 0.05% and 0.1% RGN-259 compared to placebo in a dose dependent manner during a 28-day dosing period. While the primary outcome measures were not met, several key related pre-specified endpoints and subgroups of patients with more severe dry eye showed statistically significant treatment effects. These results confirm the findings from the previous Phase 2 trial providing clear direction for the clinical regulatory pathway and remaining registration trials for RGN-259.
RGN-259 was evaluated using the Controlled Adverse Environment (CAESM) Model developed by Ora, Inc., the product development firm managing the program. The CAESM was utilized to screen and enroll an enriched patient population and measure a patient's ability to withstand an acute adverse environmental challenge of the ocular surface.
On the final day of dosing (Day 28), patients receiving 0.1% RGN-259 had a statistically significant reduction in ocular discomfort during CAESMexposure when compared to placebo (Intent-to-Treat Population (ITT), p=0.043). Importantly, this result was also observed in the previous Phase 2 trial in patients treated with 0.1% RGN-259 (ITT, p=0.024), thereby demonstrating a symptom endpoint in two independent trials. A statistically significant ocular discomfort improvement after CAESM exposure on Day 28 was also observed in the 0.05% and 0.1% RGN-259 treatment arms when compared to placebo (ITT, p=0.0366 and p=0.0072, respectively) indicating a dose dependent response.
Efficacy in an environmental setting was also demonstrated in more symptomatic patients at baseline, with statistically significant improvements in ocular discomfort observed at day 28 prior to CAESM in patients receiving 0.05% and 0.1% RGN-259 compared to placebo (p=0.022 and p=0.006, respectively). These data suggest that RGN-259 has a fast-acting treatment effect on a dry eye symptom during exposure to an adverse environment as well as in the natural environment after 28 days of dosing.
RGN-259 also improved a common objective endpoint – ocular surface staining after 28 days of dosing in patients with compromised tear film break-up time at baseline. In this population, patients receiving 0.1% RGN-259 had a statistically significant reduction in corneal fluorescein staining prior to entering the CAESM on Day 28 when compared to placebo (p=0.034). The same result was observed in the previous Phase 2 trial for patients who have suffered an acute myocardial infarction, or AMI, commonly known as a heart attack. Preclinical research publishedtreated with 0.1% RGN-259, although it was not statistically significant in the scientific journalNaturesmaller sample size of this previous Phase 2 trial. Additionally, a change from baseline analysis (Day 28 minus Day 0) demonstrated a statistically significant improvement in inferior corneal staining for the 0.1% RGN-259 treatment arm when compared to placebo (p=0.003). This finding was also observed at Day 14 compared to placebo (p=0.035). These data suggest that RGN-259 has indicated that Tß4 can guide specific typesa fast-acting treatment effect on a dry eye sign after 14 and 28 days of stem cells fromdosing.
There were no significant drug-related adverse or serious adverse events and RGN-259 was well-tolerated and comfortable for the outer layerpatients with no irritation upon instillation.
ReGenTree intends to meet with the FDA this summer to provide full results of the hearttrial and its plan to generate new myocardial blood vessels and tissue at injured sites.conduct a confirmatory Phase 3 study to start by the fourth quarter of 2016.
| 49 |
Clinical Development. In
RGN-352
During 2009, we completed a Phase 11a and Phase 1b clinical trial evaluating the safety, tolerability and the pharmacokinetics of the intravenous administration of RGN-352. We also designed this trial to explore the use of RGN-352 in other indications in which acute administration of Tß4 may be warranted. We conducted the Phase
39
In addition to observe RGN-352’s cardioprotective effects and its ability to salvage and regenerate damaged cardiac tissue and improve cardiac function after a heart attack. We intend to use a portionthe potential application of the proceeds of this offering to initiate and conduct at least a portion of this Phase 2 clinical trial, although we will not be able to complete the trial without additional capital. In May 2010, we were awarded a $3 million grant from the NIH’s Blood, Heart and Lung Institute to support the further development of RGN-352. We expect that the Phase 2 trial design will allow for an interim review of patient data from an initial group of evaluated patients, and we currently expect that the proceeds of this offering will be sufficient to reach this point in the trial. Depending on our capital resources, we may conduct the trial while continuing strategic partnership discussions with biotechnology and pharmaceutical companiesRGN-352 for the further clinical developmenttreatment of RGN-352.
Based on thisour Phase 1 data and the preclinical research discussed above, we intend to supportare evaluating various opportunities for government funding for a proposed Phase 1/22a clinical trial to be conducted at a major U.S. medical center under a physician-sponsored INDshow proof-of-concept in ordereach case while also talking with prospective strategic partners with the interest, capabilities and resources to evaluate the therapeutic potential of RGN-352 in patients with MS. We are planning to supply RGN-352 and provide clinical and regulatory guidance for the trial. We believe that we can support this trial from our existing capital resources, although we intend to use a portion of the proceeds from this offering to provide additional support.
Clinical Development. Emerging human clinical data from two compassionate use studies have demonstrated the ability of RGN-259 to repair and regenerate corneal tissue. A corneal specialist has received approval from the FDA to treat up to ten patients with neurotrophic keratitis, or NK, with RGN-259. NK is a rare degenerative corneal disease induced by a nerve impairment. The most common causes of NK include the herpes zoster virus. To date, nine patients have been treated in an open label protocol for periods of 28 or 49 days. The NK patients being evaluated have non-healing defects that have lasted at least six weeks and up to greater than ten years.
40
RGN-137
41
| 50 |
In January 2009, we reported final data from this trial. RGN-137 was well-tolerated at all three dose levels studied, with no dose-limiting adverse events, which achieved the primary objective of the study. As for efficacy, all Tß4 doses performed similarly compared to placebo, with no statistically significant efficacy results. PatientsHowever, patients treated with the middle dose showed a 17% rateimprovement of wound healing, which was the highest rate among the three active doses evaluated. The improvement in ulcer healing in this middle dose group following nine weeks of treatment was equal to the improvement in patients treated with placebo after 12 weeks of treatment.
Clinical Development — Venous Stasis Ulcers.In 2006,mid-2006 we began enrolling patients in a Phase 2 clinical trial designed to assess the safety and effectiveness of RGN-137 for the treatment of patients with venous stasis ulcers. Venous stasis ulcers are a common type of chronic wound that develops on the ankle or lower leg in patients with chronic vascular disease. In these patients blood flow in the lower extremities is impaired leading to venous hypertension, edema (swelling) and mild redness and scaling of the skin that gradually progresses to ulceration. In this randomized, double-blind, placebo-controlled, dose-response trial, eight clinicalstudy, 8 European sites in Italy (N=5) and Poland enrolled a total of 73(N=3) make up the 72 patients randomized to evaluate the safety, tolerability, and wound healing effectiveness ofreceive three different concentrations of RGN-137 compared toor placebo. RGN-137 or placebo was applied topically to thepatients’ ulcers once daily for up to 84 consecutive days. Patients in the trialA patient’s ulcer size and ulcer stability for enrollment were between 18 to 79 years old and had at least one venous stasis ulcer with a surface area between 3 and 30 cm2. We were the sponsor of the trial, and it was conducted and funded by Sigma-Tau.
In March 2009, we reported final data from thethat trial. RGN-137 was well-tolerated at all three dose levels, with no dose-limiting adverse events, which achieved the primary objective of the study. Thirty-three percent (33%) of the patients who received the middle doseAll doses of RGN-137 had their ulcers heal completely afterwere well tolerated. More patients achieved healing in the 12 weeksRGN-137 mid dose (0.03% Tß4 gel product) than in any other dose group. The mid dose showed both an increased incidence of treatment,wound healing and a faster healing time compared to 24% of patients receiving the placebo, 16% of the patients receiving the lowest drugplacebo. The mid dose and 17% of patients receiving the highest drug dose. Of the patients receiving the middle dose whose ulcers healed completely,decreased the median time to complete healing decreased by approximately 45%, among those wounds that completely healed. A follow-on evaluation, reported at the 3rd International Symposium on the Thymosins in Health and Disease in March 2012, showed that for those venous stasis ulcer patients’ wounds greater than 3 cm2 that healed, the RGN-137 mid dose (0.03% Tß4 gel product) accelerated wound closure with a median time to healing of 49 days as compared to a 37% decrease in78 days for the time to healing for patients in the placebo-treated group. None of the differences observed between RGN-137placebo. Those results were both clinically and placebo were statistically significant.
Future Plans. Once
GtreeBNT.In March 2014, we complete our Phase 2 EB trial and evaluate the results, we will evaluate its potential value for acceleration of dermal wound healing and whetherentered into a License Agreement with GtreeBNT to continue clinical development of this product candidate. We believe that preclinical research indicating the ability of Tß4 to reduce scarring in rats, complimented by reduced scarring in the hearts of mice and pigs after an induced heart attack, may also be relevant in suggesting that Tß4 may be effective in reducing dermal scar tissue. Subject to available funding, we plan to continue research and development of RGN-137 for this potential application.
42
Our Strategy
We seek to maximize the value of our product candidates by advancing their clinical development and then identifying suitable partners for further development, regulatory approval, and marketing. We intend to engage in strategic partnerships with companies with clinical development and commercialization strengths in desired pharmaceutical therapeutic fields. We are actively seeking partners with suitable infrastructure, expertise and a long-term initiative in our medical fields of interest. To that end, we have entered several important licensing and joint venture agreements with pharmaceutical companies to develop our product candidates.
Our ownership interest in ReGenTree is 49% and will be reduced to 42% following completion of the Phase 2b trial for Dry Eye Syndrome. Based on when, and if, ReGenTree achieves certain additional development milestones in the U.S. with RGN-259, our equity ownership may be incrementally reduced to between 42% and 25%, with 25% being the final equity ownership upon FDA approval of an NDA for Dry Eye Syndrome in the U.S. In addition to our equity ownership, RegeneRx retains a royalty on net sales that varies between single and low double digits, depending on whether commercial sales are made by ReGenTree or a licensee. In the event the ReGenTree entity is acquired or there is a change of control that occurs following achievement of an NDA, RegeneRx shall be entitled to 40% of all change of control proceeds paid or payable and will forgo any future royalties.
The Joint Venture with GtreeBNT follows two previous transactions with GtreeBNT signed in March 2014 when we had entered into License Agreements for the license of our RGN-259 and RGN-137 product candidates. GtreeBNT licensed the development and commercialization rights for RGN-259 in Asia (excluding China, Hong Kong, Macau and Taiwan) while also licensing the development and commercialization rights for RGN-137 in the U.S.
| 51 |
We have entered into a License Agreement with Lee’s Pharmaceutical (HK) Limited, headquartered in 2004, weHong Kong, for the license of Thymosin Beta 4 in any pharmaceutical form, including our RGN-259, RGN-352 and RGN-137 product candidates, in China, Hong Kong, Macau and Taiwan. Lee’s is an affiliate of Sigma-Tau, which collectively with its affiliates is our largest stockholder.
We previously entered into a strategic partnership with Defiante Farmaceutica S.A., (“Defiante”), formerly a wholly-owned subsidiary of Sigma-Tau Group, a leading international pharmaceutical company, which collectively comprise our largest shareholder, or Sigma-Tau, for development and marketing of RGN-137 and RGN-352 for specified indications in Europe and other contiguous countries. Defiante merged with Sigma-Tau also fundedIndustrie Farmaceutiche Riunite S.p.A. in 2013 and co-managedSigma-Tau recently merged with Alfa Wasserman S.p.A., an Italian pharmaceutical company. Currently, there is no ongoing development of our Phase 2 clinical trial of RGN-137 in Europe for the treatment of venous stasis ulcers.
Manufacturing
We use a major contract manufacturer to produce bulk Tß4, which is the active pharmaceutical ingredient, or API, in our product candidates by an established and proven manufacturing process known as solid-phase peptide synthesis, and we are in the early stages of qualifying backup manufacturers.synthesis. While we do not currently have long-term supply agreements in place, we and ReGenTree intend to establish a long-term supply arrangement with at least one manufacturer once practicable. No assurance can be given, however, that such agreements will be negotiated on favorable terms, or at all. Contractors are selected on the basis of their supply capability, ability to produce a drug substanceproduct in accordance with currentCurrent Good Manufacturing Practice, or cGMP, requirements of the FDA and ability to meet our established specifications.
We also use a number of outside contract manufacturers to formulate bulk Tß4 into our product candidates.candidates, RGN-137, RGN-259 and RGN-352. We use separate manufacturers for each formulation of Tß4. All of these formulations may require modifications, along with additional studies, as we move throughadvance our clinical development programs.programs through commercialization.
One of the compelling reasons to create a joint venture with GtreeBNT to develop RGN-259 in the U.S. for ophthalmology products was their recent manufacturing experience gained from their development of RGN-259 in Korea. This experience has allowed ReGenTree to move rapidly from Phase 2 to Phase 3 clinical trials in the U.S. without duplication of required Chemistry, Manufacturing, and Control (CMC) efforts, which are quite substantial when moving into Phase 3 and in anticipation of commercialization. GtreeBNT has been working with companies to manufacture RGN-259 in blow-filled sealed containers, which are currently being utilized for Phase 3 clinical trials and will be used for commercial marketing upon FDA approval.
As described elsewhere in this report, in 2011 our formulation and vialing contractor for RGN-352 underwent a manufacturing inspection by the FDA and was found not to be in compliance with cGMP, resulting in a clinical hold of our Phase 2 AMI clinical trial. This company has since closed its manufacturing facility and filed for bankruptcy protection. If we are to continue clinical development of RGN-352, we will need to secure a cGMP-compliant formulation and filling manufacturer of RGN-352. We have identified several cGMP-compliant companies able to perform this service.
| 52 |
Competition
We are engaged in a business that is highly competitive, and our target medical indications are ones with significant unmet needs. Moreover, the cosmetic and cosmeceutical industries are rapidly developing new products based on new scientific research. Consequently, there are many enterprises, both domestic and foreign, pursuing therapies and products that could compete with ours. Most of these entities have financial and human resources that are substantially greater than ours, specifically with regard to the conduct of clinical research and development activities, clinical testing and in obtaining the regulatory approvals necessary to market pharmaceutical products. Brief descriptions of some of these competitive products follow:
43
RGN-259. Most specialty ophthalmic companies have a number of products on the market that could compete with RGN-259. There are numerous antibiotics to treat eye infections to promote corneal wound healing and many eye lubrication products that are soothing to the eye and help eye healing, many of which are sold without prescriptions. Companies also market steroids to treat certain conditions within our area of interest. Allergan, Inc. markets Restasis™, Ophthalmic Emulsion, the only commercially available and FDA-approved eye drop to treat dry eye.Restasis, and other products, have been approved for marketing in certain other countries where we have licensed RGN-259. Shire PLC is developing its product candidate, Lifitegrast, and is in pivotal Phase 3 clinical trials in the U.S. Shire has said it plans to resubmit an NDA for Lifitegrast in 2016. We believe RGN-259 is different than any other product candidate for dry eye in that it actively promotes repair using a multi-faceted approach of increasing cell migration and laminin-5 production, and decreasing inflammation and apoptosis.
RGN-352. Currently, there are no approved pharmaceutical products for regenerating cardiac tissue following a heart attack, nor are there approved pharmaceutical products for regeneration of nervous tissue or for the remyelination of axons of patients with multiple sclerosis or patients suffering from traumatic brain injury. However, many pharmaceutical companies and research organizations are developing products, pharmacologic and stem cell therapies and technologies that are intended to prevent cardiac damage, improve cardiac function, and regenerate cardiac muscle after a heart attack. There are also companies developing products that are purported to remyelinate neurons and provide functional improvement for patients suffering from multiple sclerosis, stroke, traumatic brain injury, and peripheral neuropathy. If we, or a partner, were to successfully develop RGN-352 for cardiovascular or central nervous system indications, such products would have to compete with other drugs or therapies currently being developed or marketed by large pharmaceutical companies for similar indications.
RGN-137. There are numerous companies developing new pharmaceutical products for wound healing. Products and therapies such as antibiotics, honey-based ointments, silver-based compounds and low frequency cavitational ultrasound are also used to treat certain types of dermal wounds. Moreover, dermal wound healing is a large and highly fragmented marketplace that includes numerous therapeutic products and medical devices for treating acute and chronic dermal wounds.
Government Regulation
In the United States, the Federal Food, Drug, and Cosmetic Act, as amended, or FFDCA, and the regulations promulgated thereunder, and other federal and state statutes and regulations govern, among other things, the testing, manufacturing, labeling, storing, recordkeeping, distribution, advertising and promotion of our product candidates. Regulation by governmental authorities in the United States and foreign countries will be a significant factor in the manufacturing and potential marketing of our product candidates and in our ongoing research and product development activities. Any product candidate we develop will require regulatory approval by governmental agencies prior to commercialization. In particular, human therapeutic products are subject to rigorous preclinical studies, clinical trials and other approval procedures by the FDA and similar health authorities in foreign countries. The process of obtaining these approvals and subsequent compliance with appropriate federal and state statutes and regulations requires the expenditure of substantial resources.
Preclinical studies must ordinarily be conducted to evaluate an investigational new drug’s potential safety by toxicology studies and potential efficacy by pharmacology studies. The results of these studies, among other things, are submitted to the FDA as part of an Investigational New Drug Application, or IND, which must be reviewed by the FDA before clinical trials can begin. Typically, clinical evaluation involves a three-stage process. Phase 1 clinical trials are conducted with a small number of healthy volunteers to determine the safety profile and the pattern of drug absorption, distribution, metabolism and excretion, and to assess the drug’s effect on the patient. Phase 2, or therapeutic exploratory, trials are conducted with somewhat larger groups of patients, who are selected by relatively narrow criteria yielding a more homogenous population that is afflicted with the target disease, in order to determine preliminary efficacy, optimal dosages and expanded evidence of safety. Phase 2 trials should allow for the determination of the dose to be used in Phase 3 clinical trials. Phase 3, or therapeutic confirmatory, large scale, multi-center, comparative trials are conducted with patients afflicted with a target disease in order to provide enough data for the statistical proof of safety and efficacy required by the FDA and other regulatory authorities. The primary objective of Phase 3 clinical trials
44
| 53 |
The results of all of these preclinical studies and clinical trials, along with detailed information on manufacturing, are submitted to the FDA in the form of a New Drug Application, or NDA, for approval to commence commercial sales. The FDA’s review of an NDA requires the payment of a user fee currently in excess of $1$1.8 million, which may be waived for the first NDA submitted by a qualifying small business. In responding to an NDA, the FDA may refuse to file the application if the FDA determines that the application does not satisfy its regulatory approval criteria, request additional information or grant marketing approval. Therefore, even if we complete Phase 3 clinical trials for our product candidates and submit an NDA to the FDA, there can be no assurance that the FDA will grant marketing approval, or if granted, that it will be granted on a timely basis. If the FDA does approve a product candidate, it may require, among other things, post-marketing testing, including potentially expensive Phase 4 trials, which monitor the safety of the drug. In addition, the FDA may in some circumstances impose risk evaluation and mitigation strategies that may be difficult and expensive to administer. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems occur after the product reaches the market.
Among the conditions for NDA approval is the requirement that the applicable clinical, pharmacovigilance, quality control and manufacturing procedures conform on an ongoing basis with current Good Clinical Practices, Good Laboratory Practices, current Good Manufacturing Practices, and computer information system validation standards. During the review of an NDA, the FDA will perform a pre-licensing inspection of select clinical sites, manufacturing facilities and the related quality control records to determine the applicant’s compliance with these requirements. To assure compliance, applicants must continue to expend time, money and effort in the area of training, production and quality control. After approval of any product, manufacturers are subject to periodic inspections by the FDA. If a company fails to comply with FDA regulatory requirements, FDA may pursue a wide range of remedial actions, including seizure of products, corrective actions, warning letters and fines.
We have received orphan drug designation from the FDA for Tß4RGN-137 for the treatment of EB.EB and RGN-259 for the treatment of neurotrophic keratopathy or NK, (now to be developed by ReGenTree). The FDA may designate a product or products as having orphan drug status to treat a disease or condition that affects less than 200,000 individuals in the United States, or, if patients of a disease number more than 200,000, the sponsor can establish that it does not realistically anticipate its product sales will be sufficient to recover its costs. If a product candidate is designated as an orphan drug, then the sponsor may receive incentives to undertake the development and marketing of the product, including grants for clinical trials, as well as a waiver of the user fees for submission of an NDA application. For example, as described above, we received a grant of approximately $681,000 infrom the aggregateFDA for our ongoing Phase 2 clinical trial of RGN-137 to treat patients with EB.
Generally, if a product with an orphan drug designation subsequently receives the first marketing approval for the indication for which it has such designation, the product is entitled to marketing exclusivity for a period of seven years in the United States.States and ten years in the EU. There may be multiple designations of orphan drug status for a given drug and for different indications. Orphan drug designation does not guarantee that a product candidate will be approved by the FDA for marketing for the designation, and even if a sponsor of a product candidate for an indication for use with an orphan drug designation is the first to obtain FDA approval of an NDA for that designation and obtains marketing exclusivity, another sponsor’s application for the same drug product may be approved by the FDA during the period of exclusivity if the FDA concludes that the competing product is clinically superior. In this instance, the orphan designation and marketing exclusivity originally granted would be lost in favor of the clinically superior product.
| 54 |
Intellectual Property
We have applied for or hold over 60 worldwide patents onand patent applications covering peptide compositions, uses and formulations related to dermal and ophthalmic indications and other organ and tissue repair activities, as well as for cosmetic and consumer product applications. In 2001, we entered into a license agreement with the NIH under
45
We hold a U.S. patent relating to the use of Tß4 for treatment of alopecia, an autoimmune skin disease that results in hair loss, which expires in 2017, with corresponding patents in Europe and Singapore that expire in 2018. In 2006, we were issued a patent in China for the use of Tß4 to treat EB, which expires in 2022.
We hold a research agreement with The George Washington University, or GWU, we funded Tß4 research at GWU and received a sole and exclusive worldwide licenseU.S. patent relating to any resulting patents. While we no longer fund any research under this agreement, we remain obligated to pay GWU a royalty of 4% of the net sales, if any, of specified products covered by patents issued in connection with the agreement. Pursuant to the research agreement, we have exclusive rights to patent applications filed in the United States and in Europe disclosing the use of Tß4 for the treatment of septic shockcongestive heart failure. This patent issued in January 2012, and associated syndromes, including Adult Respiratory Distress Syndrome. Two U.S. patents covered by this agreement have been issued, whichwill expire in 20132027. Other patent applications for our various product candidates, if issued, will offer protection in the U.S. and 2014.
We have also filed numerous additional U.S. and international patent applications covering various compositions, uses, formulations and other components of Tß4, as well as for novel peptides resulting from our research efforts.efforts, the latest of which were filed during 2013. There can be no assurance that these, or any other future patent applications under which we have rights, will result in the issuance of a patent or that any patent issued will not be subject to challenge or opposition. In the case of a claim of patent infringement by or against us, there can be no assurance that we will be able to afford the expense of any litigation that may be necessary to enforce our proprietary rights.
We have also evaluated a number of our patents and patent applications in certain territories to determine whether it is cost-effective to continue to maintain or prosecute them. In some cases, we have determined that the value or potential value of such patents and/or applications is not worth the continued effort or expense and have either ceased efforts to pursue specific patents or abandoned any that have short expiries or cover countries of minimal strategic interest to us or our partners. We will continue to evaluate our portfolio and take such actions from time to time as appropriate.
Material Agreements
National Institutes of Health
We have entered into a license agreement with NIH under which we are obligated to pay an annual minimum royalty of $25,000.$2,000. In 2013 we amended certain provisions of the exclusive license; we were permitted to credit amounts paid to prosecute or maintain the licensed patent rights during 2013 calendar year against the 2013 minimum annual royalty. Beginning in 2014 the minimum annual royalty is $2,000. Additionally, we are obligated to pay the NIH a percentage of sales of qualifying product candidates, if any. There have been no such sales to date. Through the date of this Prospectus, we have complied with all minimum royalty requirements, and no milestone payments have been required under the agreement.
| 55 |
Defiante/Sigma-Tau
Defiante/Sigma-Tau/Alfa Wassermann
We have exclusively licensed certain internal and external wound healing European rights to Tß4 to Defiante, Farmaceutica, S.A., or Defiante, a Portuguese company that is a wholly owned subsidiary of Sigma-Tau.which merged with Sigma-Tau Industrie Farmaceutiche Riunite S. P. A. in 2013. In 2015, Sigma-Tau merged into Alfa Wassermann, an Italian pharma company. These licensed rights to Tß4 include its use to treat indications that are the subject of all of our current dermal clinical trials of RGN-137, as well as the treatment of heart attacks. The license excludes the use of Tß4 in any ophthalmic indications and other indications that are disease-based and not the result of a wound. Under the agreement, Sigma-Tau willAlfa Wassermann may develop Tß4 for the treatment of internal and external wounds in Europe and certain other contiguous and geographically relevant countries. The license agreement expires on acountry-by-country basis upon the later of the expiration of the last to expire of any granted patent in the territory having at least one valid claim covering the products then on the market, the expiration of any other exclusive or proprietary marketing rights, or January 2016.
Under the license agreement, Sigma-TauAlfa Wassermann is obligated to pay us a royalty on commercial sales, if any, and we will supply all required Tß4 for development. Upon the completion of a Phase 2 clinical trial for the covered indications that yields positive results in terms of efficacy and safety, Sigma-TauAlfa Wassermann must either pay us a $5 million milestone payment or initiate and fund a pivotal Phase 3 clinical trial for the applicable product candidate in order to maintain the license. As described elsewhere in this prospectus, in 2009, weWe have completed
46
The license agreement with Defiante also contains future clinical and regulatory milestones in the licensed territory. If those milestones are attained, certain performance criteria regarding commercial registration and minimum annual royalties will be payable to us in each licensed country. The agreement does not prevent us from sublicensing the technology in countries outside the licensed territory and has no impact on any U.S. rights. RegeneRx may seek to reacquire the licensed rights back from Alfa Wassermann at some point in the future
Lee’s Pharmaceuticals
On July 15, 2012, we entered into a License Agreement with Lee’s Pharmaceutical for the license of Tß4 in any pharmaceutical formulation, including our RGN-259, RGN-352 and RGN-137 product candidates, in China, Hong Kong, Macau and Taiwan. Lee’s paid us $200,000 upon signing of a term sheet with respect to the transaction on March 27, 2012, and Lee’s paid us an additional $200,000 upon signing of the definitive license agreement.
The terms of the license agreement include aggregate potential milestone payments of up to $3.6 million and royalties ranging from low double digit to high single digit royalties on commercial sales, if any.
Under the agreement, Lee’s is responsible for all developmental costs associated with each product candidate. We provided Tß4 to Lee’s at no charge for a Phase 2 ophthalmic clinical trial and will provide Tß4 to Lee’s for all other developmental and clinical work at a price equal to our cost.
The two companies have discussed Lee’s development plans and we have continued to provide information as requested. Lee’s previously filed an investigational new drug application IND with the Chinese FDA (CFDA) to conduct begin phase 2, randomized, double-masked, dose-response clinical trial with RGN-259 in China for dry-eye syndrome. Recently, Lee’s received notice from the CFDA that it would not grant an IND to begin Phase 2 studies because the API (active pharmaceutical ingredient or Tß4) was not manufactured in China. Therefore, Lee’s has informed us that they intend to initiate Phase 2 in Hong Kong (HK) and will proceed under their regulatory process while establishing API manufacturing capabilities in mainland China for future Phase 3 studies. It is believed that this strategy will facilitate the initiation of Phase 2 clinical trials in Lee’s licensed territory. We have not been informed of a projected starting date for Phase 2 in HK.
| 56 |
GtreeBNT
On March 7, 2014, we entered into license agreements with GtreeBNT Co., Ltd. The two Licensing Agreements are for the license of territorial rights to two of our Thymosin Beta 4-based products candidates, RGN-259 and RGN-137.
Under the License Agreement for RGN-259, our preservative-free eye drop product candidate, GtreeBNT will have the right to develop and commercialize RGN-259 in Asia (excluding China, Hong Kong, Taiwan, and Macau). The rights will be exclusive in Korea, Japan, Australia, New Zealand, Brunei, Cambodia, East Timor, Indonesia, Laos, Malaysia, Mongolia, Myanmar (Burma), Philippines, Singapore, Thailand, Vietnam, and Kazakhstan, and semi-exclusive in India, Pakistan, Bangladesh, Bhutan, Maldives, Nepal, Sri Lanka, Kyrgyzstan, Tajikistan, Turkmenistan and Uzbekistan, collectively, the Territory (the “259 Territory”). Under the 259 License Agreement we are eligible to receive aggregate potential milestone payments of up to $3.5 million. In addition, we are eligible to receive royalties of a low double digit percentage of any commercial sales of the licensed product sold by GtreeBNT in the 259 Territory.
Under the License Agreement for RGN-137, our topical dermal gel product candidate, GtreeBNT will have the exclusive right to develop and commercialize RGN-137 in the U.S. (the”137 Territory”). Under the 137 License Agreement we are eligible to receive aggregate potential milestone payments of up to $3.5 million. In addition, we are eligible to receive royalties of a low double digit percentage of any commercial sales of the Company’s licensed product sold by GtreeBNT in the 137 Territory.
Each license agreement contains diligence provisions which require the initiation of certain clinical trials within certain time periods that, if not met, would result in the loss of rights or exclusivity in certain countries. GtreeBNT will pay for all developmental costs associated with each product candidate. We will provide a certain limited amount of Tß4 to GtreeBNT at no charge for initial clinical trials in Korea, Japan and Australia for RGN-259 and in the U.S. for RGN-137 and will provide Tß4 to GtreeBNT for all other developmental and clinical work on a cost plus basis. We retain the manufacturing and supply rights for Tß4 in the respective Territories and the parties will negotiate in good faith an exclusive supply agreement for Tß4 as soon as practicable. We will also have the right to exclusively license any improvements made by GtreeBNT to our products outside of the licensed territory on a royalty-free basis.
The two firms have created a joint development committee and continue to discuss and the development of the licensed products and share information relating thereto. Both companies will also share all non-clinical and clinical data and other information related to development of the licensed product candidates.
U.S. Joint Venture
On January 28, 2015, the Company entered into the Joint Venture Agreement with GtreeBNT, a shareholder in the Company and licensee in certain Pan Asian countries. The Joint Venture Agreement provides for the creation of the Joint Venture, ReGenTree, LLC (“ReGenTree”), jointly owned by the Company and GtreeBNT that will commercialize RGN-259 for treatment of dry eye and neurotrophic keratopathy in the United States, as well as any other relevant ophthalmic indications.
GtreeBNT is solely responsible for funding all of the product development and commercialization efforts of ReGenTree. GtreeBNT made an initial contribution of $3 million in cash and received an initial equity stake of 51%. Our ownership interest in ReGenTree is 49% and will be reduced to 42% upon completion of the Phase 2b trial for Dry Eye Syndrome. Based on when, and if, ReGenTree achieves certain additional development milestones in the U.S. with RGN-259, our equity ownership may be incrementally reduced to between 42% and 25%, with 25% being the final equity ownership upon FDA approval of an NDA for Dry Eye Syndrome in the U.S. In addition to our equity ownership, RegeneRx retains a royalty on net sales that varies between single and low double digits, depending on whether commercial sales are made by ReGenTree or a licensee. In the event the ReGenTree entity is acquired or there is a change of control that occurs following achievement of an NDA, RegeneRx shall be entitled to 40% of all change of control proceeds paid or payable and will forgo any future royalties.
| 57 |
The Company is not required or otherwise obligated to provide financial support to ReGenTree.
ReGenTree is responsible for executing all development and commercialization activities under the License Agreement, which activities will be directed by a joint development committee comprised of representatives of the Company and GtreeBNT. The License Agreement has a term that extends to the later of the expiration of the last patent covered by the License Agreement or 25 years from the first commercial sale under the License Agreement. The License Agreement may be earlier terminated if the Joint Venture fails to meet certain commercialization milestones, or if either party breaches the License Agreement and fails to cure such breach, or as a result of government action that limits the ability of the Joint Venture to commercialize the product, as a result of a challenge to a licensed patent, following termination of the license between the Company and certain agencies of the United States federal government, or upon the bankruptcy of either party.
Development Agreements
We have entered into agreements with outside service providers for the manufacture and development of Tß4, the formulation of Tß4 into our product candidates, the conduct of nonclinical safety, toxicology and efficacy studies in animal models, and the management and execution of clinical trials in humans. Terms of these agreements vary in that they can last from a few months to more than a year in duration. Certain of these agreements require initial up front payments ranging from 25% to 50% of the total estimated cost. For additional information regarding our research and development expenses over the past two years, see “Management’s Discussion and Analysis of Financial Condition and Results of Operations — Results of Operations” in this prospectus.
Employees
We currently have nine full-timethree full time employees including our President and one part-time employee,CEO and wealso employ two part time employees. We also retain severalseven independent contractors on an as-needed basis.contractors. We believe that we have good relations with our employees.
Corporate Information
We were incorporated in Delaware in 1982 under the name Alpha 1 Biomedicals, Inc. In 2000, we changed our corporate name to RegeneRx Biopharmaceuticals, Inc. Our principal executive office is located at 15245 Shady Grove Road, Suite 470, Rockville, Maryland 20850. Our telephone number is(301) 208-9191.
Available Information
Our corporate website iswww.regenerx.com. Our electronic filings with the U.S. Securities and Exchange Commission, including our annual report on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K, and any amendments to these reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, are not currently a partyavailable free of charge through our website as soon as reasonably practicable after we have electronically filed such information with, or furnished such information to, or engaged in any material legal proceedings. However, we may be subject to various claims and legal actions arising in the ordinary course of business from time to time.
47
MANAGEMENT
Executive Officers and Directors
The following table sets forth as of the date of this prospectusJuly 11, 2016 the name, age and position of each person who serves as an executive officer or director of our company. There are no family relationships among any of our executive officers or directors, with the exception that Mr. Finkelstein is the first cousin of Dr. Goldstein’s wife.
| 58 |
We seek to assemble a board that, as a whole, possesses the appropriate balance of professional and industry knowledge.knowledge, financial expertise and high-level management experience necessary to oversee and direct our business. To that end, our board intends to maintain membership of directors who complement and strengthen the skills of other members and who also exhibit integrity, collegiality, sound business judgment and other qualities that we view as critical to effective functioning of the board. The brief biographies below include information, as of the date of this prospectus, regarding the specific and particular experience, qualifications, attributes or skills of each director or nominee that led the board to believe that the director should serve on the board.
| Name | Age | Position | ||||
| Executive Officers: | ||||||
| Mr. J.J. Finkelstein | 64 | President, Chief Executive Officer and Director | ||||
| Directors: | ||||||
| 63 | Director | |||||
| Mr. Joseph C. McNay | 82 | Director | ||||
| Mr. Mauro Bove | ||||||
| 61 | Director | |||||
Mr. Finkelstein has served as our President and Chief Executive Officer and a member of our Board of Directors since 2002. Mr. Finkelstein also served as our Chief Executive Officer from 1984 to 1989 and as the Vice Chairman of our Board of Directors from 1989 to 1991. Mr. Finkelstein has worked as an executive officer and consultant in the bioscience industry for the past 2834 years, including serving from 1989 to 1996 as chief executive officer of Cryomedical Sciences, Inc., a publicly-traded medical device company. Mr. Finkelstein has significant experience in developing early-stage companies. He has been responsible for the regulatory approval and marketing of several medical devices in the U.S. and abroad. Mr. Finkelstein has previously served on the executive committee of the Board of Directors of the Technology Council of Maryland since 2006,and MdBio, Inc. since 1998 and currently chairs the MdBio Foundation, all of which are non-profit entities that support bioscience development and education in the State of Maryland. Mr. Finkelstein received a business degree in finance from the University of Texas. The Board believes that Mr. Finkelstein’s history and long tenure as our Chief Executive Officer positions him to contribute to the Board his extensive knowledge of our company and to provide Board continuity. In addition, the Board believes that his experience at prior companies has provided him with operational and industry expertise, as well as leadership skills that are important to the Board.
48
| 59 |
Mr. HindinElseyhas served as a member of our Board of Directors since 2002.September 2010. Currently Mr. Hindin has been an entrepreneur during his more than 40 year careerElsey serves as CFO of Senseonics, Inc. a medical device company focused on continuous glucose monitoring. From May 2014 until February 2015 Mr. Elsey served as chief financial officer of Regado Biosciences, a public, late-stage clinical development biopharmaceutical company. From December 2012 to February 2014 Mr. Elsey served as chief financial officer of LifeCell, Inc., a privately held regenerative medicine company. From June 2005 to December 2012, he served in numerous finance capacities, most recently as senior vice president and is currently the principal stockholder of Chicken Out Rotisserie,chief financial officer, at Emergent BioSolutions Inc., which operates 15 restaurants in three states and the District of Columbia. Mr. Hindin hasa publicly held biopharmaceutical company. He served since 1987 as a member and since 1989 as the chairmandirector of the board of directors of The Institute for Advanced Studies in Aging & Geriatric Medicine, or IASIA, a non-profit corporation that disseminates medical information to the public as well as providing the pharmaceutical industry with an independent source for testing vaccinesfinance and drugs for the elderly. Mr. Hindin’s entrepreneurial background includes several companies and commenced with Britches of Georgetown,administration at IGEN International, Inc., a clothing retailer specializing in the sale of upscale men’spublicly held biotechnology company, and women’s apparel and accessories which he co-founded.its successor BioVeris Corporation, from April 2000 to June 2005. Prior to joining IGEN, Mr. Hindin has alsoElsey served as Chairmandirector of the Board of Hinsilblon Laboratories Ltd.,finance at Applera, a genomics and sequencing company, basedand in Fort Myers, Florida which sells odor neutralization productsseveral finance positions at International Business Machines, Inc. He received an M.B.A. in finance and delivery systems, since 1990. Finally,a B.A. in economics from Michigan State University. Mr. Hindin has served as President of Adworks Inc,Elsey is a Washington D.C.-based advertising and marketing consulting agency, since 1987. During 2009, Mr. Hindin filed for personal bankruptcy under the U.S. Bankruptcy Code and is currently involved in proceedings related to the matter.certified management accountant. The Board believes that Mr. Hindin’s extensiveElsey’s experience as an entrepreneur will be increasinglychief financial officer of a public company is particularly valuable to the Board as we seekour business in that it positions him to expandcontribute to our board’s and finance our operations.
Mr. McNay has served as a member of our Board of Directors since 2002. He is currently Chairman, Chief Investment Officer and Managing Principal of Essex Investment Management Company, LLC, positions he has held since 1976 when he founded Essex. He has direct portfolio management responsibilities for a variety of funds and on behalf of private clients. He is also a member of the firm’s Management Board. Prior to founding Essex, Mr. McNay was Executive Vice President and Director of Endowment Management & Research Corp. from 1967. Prior to that, Mr. McNay was Vice President and Senior Portfolio Manager at the
49
Mr. Bovehas served as a member of our Board of Directors since 2004 and has more than 2530 years of business and management experience within the pharmaceutical industry. Mr. Bove is currently the Head ofserving as a Business Development consultant to emerging pharmaceutical companies in Asia, including Lee’s Pharmaceuticals after leading for more than 20 years Corporate & Business Development and serves on the board of Sigma-Tau Finanziaria S.p.A., the holding company of Sigma-Tau Group, a leading international pharmaceutical company and certain Sigma-Tau affiliates, positions he has held since 1993. Sigma-Tau(Sigma-Tau Finanziaria S.p.A. and its affiliates are collectively our largest stockholder.stockholder). Mr. Bove, who resigned this role with Sigma-Tau on March 31, 2014, has also held a number of senior positions in business, licensing and corporate development within Sigma-Tau Group, which has subsidiaries in most European countries and the United States.Group. Mr. Bove obtained his law degree at the University of Parma, Italy, in 1980. In 1985, he attended the Academy of American and International Laws at the International and Comparative Law Center, Dallas, Texas. The Board believes that Mr. Bove’s extensive business and management experience within the pharmaceutical industry allows him to recognize and advise the Board with respect to recent industry developments.
Independence of the Board of Directors
Under NYSE Amex listing standards, a majority of the members of a listed company’s board of directors must qualify as “independent,” as affirmatively determined by the Board. The Boardboard. Although our common stock is no longer listed on the NYSE Amex exchange, we have determined the independence of our directors using the NYSE Amex definitions of independence. Our board consults with counsel to ensure that the Board’sits determinations are consistent with relevant securities and other laws and regulations regarding the definition of “independent,” including those set forth in pertinent listing standards of the NYSE Amex, as in effect from time to time.
Consistent with these considerations, after review of all relevant identified transactions or relationships between each director, or any of his family members, and us,our company, our senior management and our independent auditors, the Boardour board has affirmatively determined that the following fourthree directors are independent directors within the meaning of the applicable NYSE Amex listing standards: Mr. Hindin,Elsey, Mr. Bove and Mr. McNay and Dr. Bowles.McNay. In making this determination, the Boardboard found that none of the these directors had a material or
50
| 60 |
In determining the independence of Mr. Bove, the board of directors took into account the significant ownership of our common stock by Sigma-Tau and its affiliates.affiliates and our License Agreement with Lee’s Pharmaceuticals. The board of directors does not believe that any of the transactions with Lee’s or Sigma-Tau and its affiliates described in this prospectus has interfered or would reasonably be expected to interfere with Mr. Bove’s exercise of independent judgment in carrying out his responsibilities as a director of our company.
Information Regarding Committees of the Board of Directors
The Board has two standing committees: an Audit Committee and a Compensation Committee. The Board does not have a separate nominating and corporate governance committee. Rather, the independent members of the full Board perform the functions of a nominating and corporate governance committee.
Below is a description of each committee of the Board. Each of the committees has authority to engage legal counsel or other experts or consultants, as it deems appropriate to carry out its responsibilities. The Board has determined that each member of each committee meets the applicable NYSE Amex rules and regulations regarding “independence” and that each member is free of any relationship that would impair his individual exercise of independent judgment with regard to our company.
Audit Committee
The Audit Committeemembers of the Board consists ofaudit committee are Messrs. Hindin and McNay and Dr. Bowles, withElsey. Mr. Hindin actingMcNay serves as the Chairmanchairman of the audit committee. The Audit Committee meets no less than quarterly with management and our independent registered public accounting firm, both jointly and separately, has sole authority to engage and terminate the engagement of our independent registered public accounting firm, and reviews our financial reporting process on behalf of the Board. The Audit Committee operates under a formal written charter available on our website at www.regenerx.com.
Each member of the Audit Committee is an independent director determined in accordance with both Section 121A of the NYSE Amex listing standards andRule 10A-3 of the Exchange Act. Furthermore, the BoardOur board of directors has also determined that Messrs. Hindineach of Mr. McNay and McNay qualifyMr. Elsey qualifies as “auditan audit committee financial experts”expert, as defined under SECin applicable Commissio rules.
The Audit Committee pre-approves all audit and non-audit engagement fees, and terms and services. On an ongoing basis, management communicates specific projects and categories of services for which advance approval of the Audit Committee is required. The Audit Committee reviews these requests and advises management and the independent auditors if the Audit Committee pre-approves the engagement of the independent auditors for such projects and services. On a periodic basis, the independent auditors report to the Audit Committee the actual spending for such projects and services compared to the approved amounts.
Compensation Committee
The Compensation Committee is composedmembers of four directors:the compensation committee are Messrs. Hindin, McNay, Elsey and Bove and Dr. Bowles. All members of our Compensation Committee are independent,with Mr. Elsey acting as independence is currently defined in Section 803Athe Chairman of the NYSE Amex listing standards.committee. The Compensation Committee has adopted a written charter that is available to stockholders on our website at www.regenerx.com.
The Compensation Committee of the Board acts on behalf of the Board to review, adopt and oversee our compensation strategy, policies, plans and programs, including:
| • | establishment of corporate and individual performance objectives relevant to the compensation of our chief executive officer, other executive officers and Board members; | |
| • | evaluation of performance in light of these stated objectives; |
51
| • | review and approval of the compensation and other terms of employment or service, including severance andchange-in-control arrangements, of our Chief Executive Officer and the other executive officers; and | |
| 61 |
| • | administration of our equity compensation plans and other similar plan and programs. |
Nominating and Corporate Governance
The Board does not have a standing nominating and corporate governance committee. Instead, pursuant to Section 804 of the NYSE Amex listing standards, the independent members of the Board, currently consisting of Messrs. Hindin, McNay andElsey, Bove and Dr. Bowles,McNay, are responsible for performing key nominating and corporate governance activities on behalf of the Board, including identifying, reviewing and evaluating candidates to serve as our directors of the Company, reviewing and evaluating incumbent directors, selecting candidates for election to the Board, making recommendations to the Board regarding the membership of the committees of the Board, assessing the performance of management and developing and maintaining a set of corporate governance principles for us. All members of the Board performing the role of a nominating and corporate governance committee are “independent” as defined in Section 803A of the NYSE Amex listing standards.
Director Compensation
The following table setsets forth certain information for the fiscal year ended December 31, 20092015 with respect to the compensation of our directors. Mr. Finkelstein’s compensation is disclosed in the Summary Compensation Table below,above, and he does not receive any additional compensation for his service as a director. Dr. Goldstein is an employee of our company and his compensation as an employee is set forth in the table below. He does not receive any additional compensation for his service as a director.
The Company had in effect a non-employee director is eligiblecompensation policy which was suspended in November 2011 by our Board of Directors elected to receive an annualhelp the company preserve capital and consistent with this certain fees accrued in 2011 were forfeited and no retainer or meeting fees were paid to non-employee directors in 2014 or 2015.
In March 2014, in view of the Board’s November 2011 decision to temporarily cease paying cash retainercompensation to non-employee directors for Board and committee service, the Board elected to make director option grants to all directors to compensate them for serving during 2014 and 2015. In 2015 each independent director was granted 100,000 options in each February and June with exercise prices per share of $13,905. The chairman of each of our audit committee$0.19 and compensation committee is eligible to receive a supplemental annual cash retainer of $10,300. Mr. Hindin currently serves as the chairman of each$0.36, respectively. Each of these committees.
We also reimburse directors for expenses incurred in attending meetings of the board and other events attended on our behalf and at our request.
52
| Fees Earned | ||||||||||||||||
| or Paid | Option | All Other | ||||||||||||||
| in Cash | Awards | Compensation | Total | |||||||||||||
| Name | ($)(1) | ($) | ($) | ($) | ||||||||||||
| Allan Goldstein, Ph.D. | — | 63,552 | 92,712 | (2) | 156,264 | |||||||||||
| R. Don Elsey | — | 38,623 | — | 38,623 | ||||||||||||
| Joseph McNay | — | 38,623 | — | 38,623 | ||||||||||||
| Mauro Bove | — | 38,623 | — | 38,623 | ||||||||||||
| Fees Earned | ||||||||||||||||
| or Paid | Option | All Other | ||||||||||||||
| in Cash | Awards | Compensation | Total | |||||||||||||
Name | ($) | ($)(1) | ($) | ($) | ||||||||||||
| Allan Goldstein, Ph.D. | — | 71,348 | 162,466 | (2) | 233,814 | |||||||||||
| Richard Hindin | 37,877 | 16,039 | — | 53,916 | ||||||||||||
| L. Thompson Bowles M.D., Ph.D. | 21,105 | 11,875 | — | 32,980 | ||||||||||||
| Joseph McNay | 18,028 | 10,917 | — | 28,945 | ||||||||||||
| Mauro Bove | 16,292 | 10,459 | — | 26,751 | ||||||||||||
| (1) |
Options held by each Board member as of December 31, 2015, are as follows:
| Allan Goldstein, Ph.D. | ||||
| Joseph McNay | ||||
| Mauro Bove |
| 62 |
| (2) | In addition to being Chairman of our Board of Directors, Dr. Goldstein also serves as our Chief |
53
We entered into an employment agreement with Dr. Goldstein on April 16, 2014 for him to serve as our Chief Science Officer. Dr. Goldstein’s employment agreement had an initial one-year term, which has been and will be automatically renewed for additional one-year periods unless either we or Mr. Goldstein elect not to renew it. Dr. Goldstein’s annual base salary was $75,000 and was increased to $90,000 on January 1, 2015. Dr. Goldstein’s salary may not be adjusted downward without his written consent, except in a circumstance which is part of a general reduction or other concessionary arrangement affecting all employees or affecting senior executive officers. Dr. Goldstein is also eligible to receive an annual bonus in an amount established by the Board and is entitled to participate in and receive all standard employee benefits and to participate in all of our applicable incentive plans, including stock option, stock, bonus, savings and retirement plans.
Summary Compensation Table The following table shows, for the fiscal years ended December 31, Of note, our annual rates of compensation for our named executive 20092015 and 2008,2014, compensation awarded to or paid to, or earned by, our chief executive officer andwho was our two other most highly compensatedonly named executive officers during 2009 who were serving as executive officers at December 31, 2009.officer for fiscal 2015. For purposes of this prospectus,report, we sometimes refer to these officersour chief executive officer as theour named executive officers.officersofficer and all employees were reduced effective December 1, 2011. Beginning in effect at December 31, 2008January 2012, all employees became part-time hourly employees with reduced work schedules. Additionally, in January 2012, we discontinued providing employee health benefits and 2009 remain the same. However, given our limited cash resources during 2009, the named executive officers other than Mr. Crockford had their annual base salaries reduced by 35% for the period fromcompany-sponsored 401(k) matching contributions. On April 1 to September 30, 2009. Consequently, the salary amounts set forth in the following table may differ from the disclosed annual base salaries currently in effect.In return for the 35% salary reduction,16, 2014 we entered into a new employment agreement with Mr. Finkelstein under which Mr. Finkelstein’s base salary was set at $125,000 annually, and on January 1, 2015 Mr. Lyons received optionsFinkelstein’s base salary was increased to purchase 172,122 and 116,592 shares, respectively, of our common stock at an exercise price of $0.57 per share. Effective October 1, 2009, their salaries were restored to the levels in effect at December 31, 2008 and, therefore, the options ceased vesting as of September 30, 2009 but remain exercisable in accordance with the terms of our stock option plan. The number of shares vested and outstanding from these option grants are set forth in the table within the “Outstanding Equity Awards at December 31, 2009” section below.
| Option | All Other | |||||||||||||||||||||||
| Salary | Bonus | Awards(1) | Compensation(2) | Total | ||||||||||||||||||||
| Name and Principal Position | Year | ($) | ($) | ($) | ($) | ($) | ||||||||||||||||||
| J.J. Finkelstein, President and | 2015 | 154,519 | — | 127,104 | 3,360 | 284,983 | ||||||||||||||||||
| Chief Executive Officer | 2014 | 110,813 | — | 71,630 | 3,360 | 185,803 | ||||||||||||||||||
| Non-Equity | ||||||||||||||||||||||||||||
| Incentive | ||||||||||||||||||||||||||||
| Option | Plan | All Other | ||||||||||||||||||||||||||
| Salary(1) | Bonus(2) | Awards(3) | Compensation(4) | Compensation(5) | Total | |||||||||||||||||||||||
Name and Principal Position | Year | ($) | ($) | ($) | ($) | ($) | ($) | |||||||||||||||||||||
| J.J. Finkelstein, President and | 2009 | 244,608 | 18,720 | 116,198 | — | 13,005 | 392,531 | |||||||||||||||||||||
| Chief Executive Officer | 2008 | 299,520 | 22,464 | 86,137 | 14,976 | 17,690 | 440,787 | |||||||||||||||||||||
| C. Neil Lyons, | 2009 | 167,093 | 11,140 | 74,395 | — | 4,999 | 257,627 | |||||||||||||||||||||
| Chief Financial Officer | 2008 | 200,817 | 12,152 | 68,848 | 10,127 | 8,508 | 300,452 | |||||||||||||||||||||
| David R. Crockford, | 2009 | 210,223 | 5,781 | — | — | 6,818 | 222,822 | |||||||||||||||||||||
| Vice President, Clinical and Regulatory Affairs | 2008 | 209,203 | 12,613 | 51,682 | 10,511 | 11,681 | 295,690 | |||||||||||||||||||||
| (1) | ||
| (2) | The 2014 & 2015 amount reflects payment of life insurance premiums for Mr. Finkelstein in the |
Employment Agreements; Potential Payments Upon Termination or Change in Control
Employment Agreement with our named executive officers. These employment agreements contain severance and other provisions that may provide for payments to the named executive officers following termination of employment with us in specified circumstances. The following is a summary of the material terms of these employment agreements with our named executive officers.
We entered into an employment agreement with Mr. Finkelstein in January 2002on April 16, 2014 for him to serve as our president and chief executive officer. Mr. Finkelstein’s employment agreement hadhas an initial three-year term, which is automatically renewed for additional one-year periods unless either we or Mr. Finkelstein elect not to renew it. This agreement was amended and restated during 2008 and again in 2009. Mr. Finkelstein’s annual base salary is $299,520.was $125,000, which was increased to $150,000 on January 1, 2015. Mr. Finkelstein’s salary may not be adjusted downward
54
Mr. Finkelstein is eligible to receive options to purchase common stock under our Amended and Restated 2000 Stock Option and Incentive Plan, which we refer to in this prospectus as the 2000 Plan.equity incentive plans. The decision to grant any such options and the terms of such options are within the discretion of our board of directorsBoard or the compensation committee thereof. All vested options are exercisable for a period of time following any termination of Mr. Finkelstein’s employment as may be set forth in the 2000 Planapplicable benefit plan or in any option agreement between Mr. Finkelstein and us.
In the event that Mr. Finkelstein’s employment is terminated by us without “cause” or by Mr. Finkelstein for “good reason,” each as defined in his employment agreement, or if Mr. Finkelstein voluntarily terminates his employment within 12 months following a “change in control,” as defined in his employment agreement, then in each case, subject to Mr. Finkelstein’s entering into and not revoking a release of claims in a form acceptable to us, Mr. Finkelstein will be entitled to receive (i) a lump sum severance payment in an amount equal to one-half of his then annual base salary if within the first anniversary date of this Agreement; or (ii) a lump sum payment in an amount equal to three-fourths of his then annual base salary if within the first anniversary date and second anniversary date of this Agreement; or (iii) a lump sum payment in an amount equal to his then annual base salary if any time after the second anniversary date of this Agreement, less all federal and state withholdings. In the event of a “change in control,” as defined in his employment agreement and Mr. Finkelstein is involuntarily terminated within 12 months after a change in control event or within 12 months after a change in control event he resigns his employment for “good reason”, then the Company shall (i) pay Mr. Finkelstein, in a lump sum cash payment, an amount equal to his annual base salary then in effect (or ifon the date of his base salary istermination from employment, less than the amountany applicable federal and state taxes and withholdings. In addition, in effect as of March 31, 2009, the base salary in effect as of March 31, 2009), plus (ii)each instance Mr. Finkelstein would also be eligible to receive (i) any earned bonus and (iii) ifaccrued vacation pay, and (ii) to the extent that he timely elects and remainsis eligible for continuationand participates in a Company sponsored health insurance plan the Company shall pay or reimburse Executive for the amount of healthcare benefits, that portionany insurance premiums for a twelve-month period, but these payments shall be limited to the amount of the continued healthcare premiums that we were payingbeing paid by the Company for Executive’s coverage or the amount being reimbursed for insurance premiums immediately prior to the date of his termination for a period of 12 months, in each case less applicable taxes and withholdings. If Mr. Finkelstein’s employment had been terminated for any of the reasons described in this paragraph as of December 31, 2009, he would have been entitled to receive a lump sum payment of $299,520, less taxes and withholdings, plus continuation of healthcare benefits with a value of $8,772.
In addition, if Mr. Finkelstein’s employment is terminated without “cause,” or if there is a “change in control” event, in each case as defined in either the 2000 Planapplicable benefit plan or in Mr. Finkelstein’s employment agreement, then the unvested portion of Mr. Finkelstein’s options outstanding as of December 31, 2009options would accelerate in full.
55
56
The following table shows certain information regarding outstanding equity awards at December 31, 20092015 for the named executive officers,officer, all of which were stock options.
| Number of Shares Underlying Unexercised Options (#) | Number of Shares Underlying Unexercised Options (#) | Option Exercise Price | Option | |||||||||||||||
| Name | Exercisable | Unexercisable | ($) | Expiration Date | Note | |||||||||||||
| Mr. Finkelstein | 114,748 | — | 0.57 | 4/10/2019 | ||||||||||||||
| 125,000 | — | 0.76 | 10/11/2016 | |||||||||||||||
| 125,000 | — | 0.27 | 07/14/2017 | |||||||||||||||
| 125,000 | — | 0.22 | 8/3/2018 | |||||||||||||||
| 80,135 | — | 0.16 | 12/12/2018 | |||||||||||||||
| 375,000 | 125,000 | 0.14 | 1/24/2019 | (1 | ) | |||||||||||||
| 35,000 | — | 0.16 | 4/4/2019 | |||||||||||||||
| 250,000 | 250,000 | 0.21 | 3/25/2021 | (2 | ) | |||||||||||||
| 125,000 | 375,000 | 0.36 | 6/30/2022 | (2 | ) | |||||||||||||
| Number of | ||||||||||||||||||||
| Shares | Number of Shares | |||||||||||||||||||
| Underlying | Underlying | |||||||||||||||||||
| Unexercised | Unexercised | Option | ||||||||||||||||||
| Options (#) | Options (#) | Exercise Price | Option | |||||||||||||||||
Name | Exercisable | Unexercisable | ($) | Expiration Date | Note | |||||||||||||||
| Mr. Finkelstein | 500,000 | — | 0.33 | 1/1/2012 | ||||||||||||||||
| 100,000 | — | 3.21 | 4/1/2015 | |||||||||||||||||
| 62,500 | 62,500 | 2.34 | 3/15/2014 | (1 | ) | |||||||||||||||
| 31,250 | 93,750 | 1.15 | 4/15/2015 | (1 | ) | |||||||||||||||
| 114,748 | — | 0.57 | 4/10/2019 | |||||||||||||||||
| — | 125,000 | 0.76 | 10/11/2016 | (1 | ) | |||||||||||||||
| Mr. Lyons | 133,332 | 66,668 | 3.10 | 4/7/2015 | (2 | ) | ||||||||||||||
| 37,500 | 37,500 | 2.34 | 3/15/2014 | (1 | ) | |||||||||||||||
| 18,750 | 56,250 | 1.50 | 6/15/2015 | (1 | ) | |||||||||||||||
| 77,728 | — | 0.57 | 4/10/2019 | |||||||||||||||||
| — | 75,000 | 0.76 | 10/11/2016 | (1 | ) | |||||||||||||||
| Mr. Crockford | 15,000 | — | 1.07 | 7/1/2013 | ||||||||||||||||
| 125,000 | — | 0.86 | 1/1/2014 | |||||||||||||||||
| 70,000 | 30,000 | 3.21 | 4/1/2015 | (3 | ) | |||||||||||||||
| 17,500 | 7,500 | 3.82 | 5/25/2015 | (3 | ) | |||||||||||||||
| 25,000 | 25,000 | 2.15 | 1/16/2014 | (1 | ) | |||||||||||||||
| 37,500 | 37,500 | 2.34 | 3/15/2014 | (1 | ) | |||||||||||||||
| 18,750 | 56,250 | 1.15 | 4/15/2015 | (1 | ) | |||||||||||||||
| (1) | This option vests in equal installments on the first four anniversaries of the grant date. In each case these options were granted seven years prior to the listed expiration dates. | |
| (2) | This option vests in equal installments upon grant and on the first | |
| 64 |
Post-Employment Compensation
We do not maintain any plans providing for payment or other benefits at, following, or in connection with retirement other than a 401(k) plan madewhich was available to all employees.employees through 2011. The Company did not make any plan contributions in 2014 or 2015. In addition, we do not maintain any non-qualified deferred compensation plans.
57
The following table provides information as of December 31, 20092015 about the securities authorized for issuance to our employees, directors and other eligible participants under our equity compensation plans, consisting solely of the Amended and Restated 2000 Plan.
| Number of Securities | ||||||||||||
| Remaining Available for | ||||||||||||
| Number of Securities to | Future Issuance Under | |||||||||||
| be Issued Upon Exercise | Weighted-Average Exercise | Equity Compensation Plans | ||||||||||
| of Outstanding Options, | Price of Outstanding Options, | (Excluding Securities | ||||||||||
| Warrants and Rights | Warrants and Rights | Reflected in Column (a)) | ||||||||||
Plan Category | (a) | (b) | (c) | |||||||||
| Equity compensation plans approved by security holders | 4,914,112 | $ | 1.53 | 1,550,888 | ||||||||
| Equity compensation plans not approved by security holders | — | — | — | |||||||||
| Total | 4,914,112 | $ | 1.53 | 1,550,888 | ||||||||
58
| Number of securities | ||||||||||||
| remaining available for | ||||||||||||
| Number of securities to | future issuance under | |||||||||||
| be issued upon exercise | Weighted-average exercise | equity compensation plans | ||||||||||
| of outstanding options, | price of outstanding options, | (excluding securities | ||||||||||
| warrants and rights | warrants and rights | reflected in column (a)) | ||||||||||
| Plan Category | (a) | (b) | (c) | |||||||||
| Equity compensation plans approved by security holders | 7,131,211 | $ | 0.30 | 1,548,029 | ||||||||
| Equity compensation plans not approved by security holders | — | — | — | |||||||||
| Total | 7,131,211 | $ | 0.30 | 1,548,029 | ||||||||
59
60
Related Party Transactions
Described below are transactions and series of relatedsimilar transactions since January 1, 2007that occurred during fiscal 2013, 2014 or 2015 to which we have beenwere a party or are a party in which:
| · | the amounts involved exceeded or will exceed $120,000; and |
| · | a director, executive officer, beneficial owner of more than five percent of any class of our voting securities or any member of their immediate family had or will have a direct or indirect material interest. |
| 65 |
U.S. Joint Venture
On January 28, 2015, we announced that we entered into a Joint Venture Agreement with GtreeBNT. The Joint Venture Agreement provides for the creation of ReGenTree, owned by us and GtreeBNT, that will commercialize RGN-259 for treatment of dry eye and neurotrophic keratopathy in the United States, or other ophthalmic indications in the U.S. GtreeBNT will be responsible for funding product development and commercialization efforts, and hold a participant,majority interest, of ReGenTree. In conjunction with the Joint Venture Agreement, we also entered into a royalty-bearing license agreement (the “License Agreement”) with ReGenTree pursuant to which we granted to ReGenTree the right to develop and exclusively commercialize RGN-259 in the United States.
In September 2015, ReGenTree began a multi-centered, randomized, double-masked Phase 2b/3 clinical trial in patients with dry eye syndrome (“DES”) and a multi-centered, randomized, double-masked Phase 3 clinical trial in patients with neurotrophic keratopathy (“NK”), both in the U.S. The DES trial has completed full enrollment, treatment and follow-up of all patients. Top line data from the DES trial was released in early May and additional clinical work is being planned for late 2016.
The study, which the amount involved exceeded or will exceed one percentenrolled 317 subjects, tested two doses of RGN-259 eye drops containing 0.05% and 0.1% concentrations of the averageTβ4 four times daily for 28 days vs. placebo. On the 28th and final day of ourdosing, patients were subject to the Controlled Adverse Environment (CAE) challenge. Co-primary endpoints were total assets at yearcorneal fluorescein staining score change on the 28th day pre-CAE and post-CAE (sign) and total ocular discomfort score change on the 28th day pre-CAE and post-CAE (symptom). Although the co-primary endpoints were not met, we demonstrated RGN-259’s protective efficacy in improving a sign and symptom of dry eye syndrome, which is in line with the results observed in the previous Phase 2a trial. Importantly, we identified approvable sign and symptom endpoints, which were met with statistical significance. ReGenTree expects to meet with the FDA this summer and initiate a confirmatory Phase 3 trial before the end of 2016.
RGN-259 was safe and well-tolerated and comfortable for the last two completed fiscal yearspatients with no irritation upon instillation. There were no significant drug-related adverse events for both concentrations. The safety profile is consistent with that observed in the previous Phase 2a trial, which had a twice daily instillation regimen.
The NK trial, a smaller study in an orphan population, has enrolled eight patients thus far with a goal of 46. Of the eight original clinical sites for the study, six are enrolling patients, one has yet to receive IRB approval to begin enrolling patients, and one was unable to consummate a clinical contract with ReGenTree. ReGenTree is considering adding additional sites to accelerate patient enrollment.
GtreeBNT has developed the CMC (chemistry, manufacturing and controls) dossier required for Phase 3 clinical trials and commercialization in the U.S. and in which anyKorea. This comprehensive and critical effort ensures that final drug product manufacturing, packaging, stability, purity, reproducibility, etc., meets regulatory guidelines and product specifications. The product of our executive officers, directors or beneficial holders of more than five percent of our capital stock had or will have a direct or indirect material interest, or any immediate family member of, or person sharingthis activity is the household with, any of these individuals, had or will have a direct or indirect material interest, other than executive and director compensation arrangements, including the employment, termination of employment and change of control arrangements, which are describedcurrent product format being utilized in the section of this prospectus entitled “Executive Compensation.”
Other Licensing Agreements with GtreeBNT
In addition to the Joint Venture Agreement, we have entered into four financing transactionstwo license agreements on March 7, 2014 with Sigma-TauGtreeBNT. The two Licensing Agreements are for the license of territorial rights to RGN-259 and its affiliatesRGN-137.
Under the License Agreement for RGN-259, GtreeBNT has the right to develop and commercialize RGN-259 in Asia (excluding China, Hong Kong, Taiwan, and Macau). The rights are exclusive in Korea, Japan, Australia, New Zealand, Brunei, Cambodia, East Timor, Indonesia, Laos, Malaysia, Mongolia, Myanmar (Burma), Philippines, Singapore, Thailand, Vietnam, and Kazakhstan, and semi-exclusive in India, Pakistan, Bangladesh, Bhutan, Maldives, Nepal, Sri Lanka, Kyrgyzstan, Tajikistan, Turkmenistan and Uzbekistan, collectively, the Territory (the “259 Territory”). Under the RGN-259 License Agreement we are eligible to receive aggregate potential milestone payments of up to $3.5 million. In addition, we are eligible to receive royalties of a low double-digit percentage of any commercial sales of the licensed product sold by GtreeBNT in the 259 Territory.
Under the License Agreement for RGN-137, GtreeBNT has the exclusive right to develop and commercialize RGN-137 in the U.S. (the “137 Territory”). Under the 137 License Agreement we are eligible to receive aggregate potential milestone payments of up to $3.5 million. In addition, we are eligible to receive royalties of a low double-digit percentage of any commercial sales of the Company’s licensed product sold by GtreeBNT in the 137 Territory.
Each license agreement contains diligence provisions which require the initiation of certain clinical trials within certain time periods that, if not met, would result in the loss of rights or exclusivity in certain countries. GtreeBNT will pay for all developmental costs associated with each product candidate. We will provide a certain limited amount of Tβ4 to GtreeBNT at no charge for initial clinical trials in Korea, Japan and Australia for RGN-259 and in the U.S. for RGN-137 and will provide Tβ4 to GtreeBNT for all other developmental and clinical work on a cost plus basis. We retain the manufacturing and supply rights for Tβ4 in the respective Territories and the parties will negotiate in good faith an exclusive supply agreement for Tβ4 as described below. As described above, Mauro Bove, onesoon as practicable. We also have the right to exclusively license any improvements made by GtreeBNT to our products outside of our directors, is an officerthe licensed territory on a royalty free basis.
The two firms will create a joint development committee to discuss and agree on the development of Sigma-Tau. Eachthe licensed products and share information relating thereto. Both companies will also share all non-clinical and clinical data and other information related to development of these transactions was approved by our Boardthe licensed product candidates.
| 66 |
Private Placement of Directors and our audit committee, following disclosure of Mr. Bove’s potential interests in these transactions.
On February 29, 2008,October 19, 2012, we issued 2,500,000Sinaf a convertible promissory note for $200,000 and a warrant to purchase 266,667 shares at a purchase price of $0.15 per share. Additionally, the convertible promissory note is convertible into 1,333,333 shares of common stock at $0.15 cents per share. The notes issued in October 2012 were originally to mature after twenty-four (24) months from issuance. In order to conserve the Company’s capital, in October 2014 the holders agreed to extend the maturity date to October 19, 2017, all other terms were unchanged.
On October 19, 2012, we completed a private placement of convertible notes (the “October 2012 Notes”) with four accredited investors raising an aggregate of $300,000 in gross proceeds.
Warrants. In connection with the issuance of the October 2012 Notes, we also issued warrants to each investor. The warrants are exercisable for an aggregate of Chaumiere-Consultadoria e Servicos SDC Unipessoal LDA, or Chaumiere, which400,000 shares of common stock with an exercise price of $0.15 per share for a period of five years.
Investors. The investors in the October 2012 Notes, and the principal amount of their respective October 2012 Notes and number of shares of common stock issuable upon exercise of their respective warrants, are as set forth below:
| Investor | October 2012 Note Principal | Warrants | ||||||
| SINAF S.A. | $ | 200,000 | 266,667 | |||||
| Joseph C. McNay | $ | 50,000 | 66,667 | |||||
| Allan L. Goldstein | $ | 35,000 | 46,666 | |||||
| J.J. Finkelstein | $ | 15,000 | 20,000 | |||||
Sinaf S. A. is an indirecta direct wholly-owned subsidiary of an entityAptafin S.p.A., or Aptafin. Aptafin is owned directly by Paolo Cavazza and members of his family, who directly and indirectly own 38% of Sigma-Tau,Sigma-Tau. The other investors are members of our Board of Directors, including Mr. Finkelstein who serves as our chief executive officer and Inverlochy-Consultadoria e Servicos (S.U.) LDA, or Inverlochy,member of our Board of Directors, Mr. McNay who serves as a member of Board of Directors, and Dr. Goldstein who serves as our Chief Scientific Officer and as Chairman of our Board of Directors.
2014 Convertible Notes
On January 7, 2014, we completed a private placement of convertible notes raising an entity wholly owned by Claudio Cavazza, who directly and indirectly owns 57%aggregate of Sigma-Tau,$55,000 in gross proceeds (the “January 2014 Notes”). The January 2014 Notes pay interest at a purchase pricerate of $1.005% per share in a private placement. The purchase agreements provide that the purchasers may not transfer the shares through December 31, 2010, except for transfers to affiliates, that we, rather than the purchasers, have all voting rights in respectannum, mature 60 months after their date of the shares until December 31, 2010,issuance and that we had the right to repurchase the shares at a price of $2.00 per share until December 31, 2009, which right has expired, and that we have the right to repurchase the shares at a price of $2.50 per share until December 31, 2010. We also issued warrants to each of Chaumiere and Inverlochy to purchase 500,000are convertible into shares of our common stock at an exercisea conversion price of $1.60$0.06 per share. share (subject to adjustment as described in the January 2014 Notes) at any time prior to repayment, at the election of the Investor. In the aggregate, the Notes were initially convertible into up to 916,667 shares of our common stock.
At any time prior to maturity of the January 2014 Notes, with the consent of the holders of a majority in interest of the January 2014 Notes, we may prepay the outstanding principal amount of the January 2014 Notes plus unpaid accrued interest without penalty. Upon the commission of any act of bankruptcy by the Company, the execution by the Company of a general assignment for the benefit of creditors, the filing by or against the Company of a petition in bankruptcy or any petition for relief under the federal bankruptcy act or the continuation of such petition without dismissal for a period of 90 days or more, or the appointment of a receiver or trustee to take possession of the property or assets of the Company, the outstanding principal and all accrued interest on the January 2014 Notes will accelerate and automatically become immediately due and payable.
The warrants have vestedInvestors in fullthe offering included three directors of the Company, Dr. Goldstein who serves as the Company’s Chief Scientific Officer and Chairman of December 31, 2009our Board of Directors, Mr. McNay who serves as a member of Board of Directors, and L. Thompson Bowles who previously served as a member of our Board of Directors. The original principal amounts of their respective Notes are as set forth below:
| Investor | Note Principal | |||
| Joseph C. McNay | $ | 25,000 | ||
| Allan L. Goldstein | $ | 10,000 | ||
| L. Thompson Bowles | $ | 5,000 | ||
| 67 |
2013 Convertible Notes
On March 29, 2013, we completed a private placement of convertible notes (the “March 2013 Notes”) raising an aggregate of $225,000 in gross proceeds. The March 2013 Notes bear interest at a rate of five percent (5%) per annum, mature sixty (60) months after their date of issuance and are exercisable until December 31, 2010.
At any time prior to maturity of the March 2013 Notes, with the consent of the holders of a majority in interest of the March 2013 Notes, we may prepay the outstanding principal amount of the March 2013 Notes plus unpaid accrued interest without penalty. Upon the commission of any act of bankruptcy by the Company, the execution by the Company of a general assignment for the benefit of creditors, the filing by or against the Company of a petition in bankruptcy or any petition for relief under the Federal bankruptcy act or the continuation of such petition without dismissal for a period of ninety (90) days or more, or the appointment of a receiver or trustee to take possession of the property or assets of the Company, the outstanding principal and all accrued interest on the March 2013 Notes will accelerate and automatically become immediately due and payable.
The warrants were vestedinvestors in full uponthe offering included two directors of the Company, Dr. Goldstein, the Company’s Chief Scientific Officer and Chairman of our Board of Directors, and Mr. McNay, an independent director. The original principal amounts of their respective March 2013 Notes are as set forth below:
| Investor | Note Principal | |||
| Joseph C. McNay | $ | 50,000 | ||
| Allan L. Goldstein | $ | 25,000 | ||
On July 5, 2013, we completed a private placement of convertible notes (the “July 2013 Notes”) raising an aggregate of $100,000 in gross proceeds. The July 2013 Notes bear interest at a rate of five percent (5%) per annum, mature sixty (60) months after their date of issuance and are exercisable until December 31, 2011.
At any time prior to maturity of the July 2013 Notes, with the consent of the holders of a majority in interest of the July 2013 Notes, we may prepay the outstanding principal amount of the July 2013 Notes plus unpaid accrued interest without penalty. Upon the commission of any act of bankruptcy by the Company, the execution by the Company of a general assignment for the benefit of creditors, the filing by or against the Company of a petition in bankruptcy or any petition for relief under the Federal bankruptcy act or the continuation of such petition without dismissal for a period of ninety (90) days or more, or the appointment of a receiver or trustee to take possession of the property or assets of the Company, the outstanding principal and all accrued interest on the July 2013 Notes will accelerate and automatically become immediately due and payable.
The warrant was fully vested uponinvestors in the offering included four directors of the Company, Mr. Finkelstein who serves as the Company’s Chief Executive Officer and member of our Board of Directors, Dr. Goldstein who serves as the Company’s Chief Scientific Officer and Chairman of our Board of Directors, Mr. McNay who serves as a member of Board of Directors, and L. Thompson Bowles who previously served as a member of our Board of Directors. The original principal amounts of their respective July 2013 Notes are as set forth below:
| Investor | Note Principal | |||
| Joseph C. McNay | $ | 50,000 | ||
| Allan L. Goldstein | $ | 10,000 | ||
| J.J. Finkelstein | $ | 5,000 | ||
| L. Thompson Bowles | $ | 5,000 | ||
| 68 |
On September 11, 2013, we completed a private placement of convertible notes raising an aggregate of $321,000 in gross proceeds (the “September 2013 Notes”). The September 2013 Notes bear interest at a rate of five percent (5%) per annum, mature sixty (60) months after their date of issuance and is exercisable until April 30, 2012.
At any time prior to maturity of the September 2013 Notes, with the consent of the holders of a majority in interest of the September 2013 Notes, we may prepay the outstanding principal amount of the September 2013 Notes plus unpaid accrued interest without penalty. Upon the commission of any act of bankruptcy by the Company, the execution by the Company of a general assignment for the benefit of creditors, the filing by or against the Company of a petition in bankruptcy or any petition for relief under the federal bankruptcy act or the continuation of such petition without dismissal for a period of ninety (90) days or more, or the appointment of a receiver or trustee to take possession of the property or assets of the Company, the outstanding principal and all accrued interest on the September 2013 Notes will accelerate and automatically become immediately due and payable.
The warrant was fully vested upon issuanceinvestors in the offering included an affiliate, four directors of the Company, and is exercisable untiltwo unaffiliated accredited investors. The principal amounts of the affiliate and directors respective September 30, 2014.
| Investor | Note Principal | |||
| SINAF S.A. | $ | 150,000 | ||
| Joseph C. McNay | $ | 100,000 | ||
| Allan L. Goldstein | $ | 11,000 | ||
| L. Thompson Bowles | $ | 5,000 | ||
| R. Don Elsey | $ | 5,000 | ||
Indemnification of Officers and Directors
Our restated certificate of incorporation limits the personal liability of directors for breach of fiduciary duty to the maximum extent permitted by the General Corporation Law of the State of Delaware, referred to herein as the DGCL. Our restated certificate of incorporation provides that no director will have personal
61
| • | any breach of their duty of loyalty to us or our stockholders; | |
| • | acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law; | |
| • | voting or assenting to unlawful payments of dividends or other distributions; or | |
| • | any transaction from which the director derived an improper personal benefit. |
Any amendment to or repeal of these provisions will not eliminate or reduce the effect of these provisions in respect of any act or failure to act, or any cause of action, suit or claim that would accrue or arise prior to any amendment or repeal or adoption of an inconsistent provision. If the DGCL is amended to provide for further limitations on the personal liability of directors of corporations, then the personal liability of our directors will be further limited in accordance with the DGCL.
Section 145 of the DGCL provides that a Delaware corporation may indemnify any persons who are, or are threatened to be made, parties to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of such corporation), by reason of the fact that such person was an officer, director, employee or agent of such corporation, or is or was serving at the request of such person as an officer, director, employee or agent of another corporation or enterprise. Our amended and restated bylaws include such a provision. The indemnity may include expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with such action, suit or proceeding, provided that such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation’s best interests and, with respect to any criminal action or proceeding, had no reasonable cause to believe that his or her conduct was unlawful.
| 69 |
Section 145 of the DGCL also provides that a Delaware corporation may indemnify any persons who are, or are threatened to be made, a party to any threatened, pending or completed action or suit by or in the right of the corporation by reason of the fact that such person was a director, officer, employee or agent of such corporation, or is or was serving at the request of such corporation as a director, officer, employee or agent of another corporation or enterprise. Our amended and restated bylaws contain such a provision. The indemnity may include expenses (including attorneys’ fees) actually and reasonably incurred by such person in connection with the defense or settlement of such action or suit provided such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation’s best interests except that no indemnification is permitted without judicial approval if the officer or director is adjudged to be liable to the corporation. Where an officer or director is successful on the merits or otherwise in the defense of any action referred to above, the corporation must indemnify him or her against the expenses that such officer or director has actually and reasonably incurred.
Expenses incurred by any indemnitee in defending or investigating a threatened or pending action, suit or proceeding in advance of its final disposition shall be paid by us upon delivery to us of an undertaking, by or on behalf of such indemnitee, to repay all amounts so advanced if it shall ultimately be determined that such indemnitee is not entitled to be indemnified by us. No advance will be made by us if a determination is reasonably and promptly made by our board of directors by a majority vote of a quorum of disinterested directors, or if such a quorum is not obtainable or, even if obtainable, a quorum of disinterested directors so directs, by independent legal counsel in a written opinion, that, based upon the facts known to the board or counsel at the time such determination is made, such person did not meet the applicable standard of conduct in order to be indemnified.
At present, there is no pending litigation or proceeding involving any of our directors or officers as to which indemnification is required or permitted, and we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
We have an insurance policy covering our officers and directors with respect to certain liabilities, including liabilities arising under the Securities Act or otherwise.
| 70 |
62
The following table sets forth certain information regarding the beneficial ownership of our common stock as of April 28, 2010June 26, 2016 by (i) each director; (ii) each named executive officer; (iii) all currently serving executive officers and directors as adjusted to reflect the sale of shares in this offering by:
| Beneficial Ownership(1) | ||||||||
| Beneficial Owner | Number of Shares | Percent of Total | ||||||
| 5% Stockholders: | ||||||||
| Entities affiliated previously affiliated with Essetifin S.p.A., Via Sudafrica, 20, Rome, Italy 00144 | 35,489,418 | (2) | 33.2 | % | ||||
| GtreeBNT Co., Ltd. 22nd FL, Parkview Tower, 248 Jungjail-ro, Bundang-gu, Seongnam-si, Gyeonggi-do 463-863, Republic of Korea | 19,583,333 | (3) | 18.3 | % | ||||
| Named Executive Officers and Directors: | ||||||||
| J.J. Finkelstein | 3,235,854 | (4) | 3.0 | % | ||||
| Allan L. Goldstein | 3,291,653 | (5) | 3.0 | % | ||||
| Joseph C. McNay | 5,792,135 | (6) | 5.2 | % | ||||
| Mauro Bove | 339,655 | (7) | * | |||||
| R. Don Elsey | 383,333 | (8) | * | |||||
| All directors and executive officers as a group (5 persons) | 13,042,630 | (9) | 11.2 | % | ||||
| Number of | Percentage of Shares | |||||||||||
| Shares | Beneficially Owned | |||||||||||
| Beneficially | Before | After | ||||||||||
Name of Beneficial Owner | Owned | Offering | Offering | |||||||||
5% Stockholders: | ||||||||||||
| Entities affiliated with Sigma-Tau Finanziaria, S.p.A. Via Sudafrica, 20, Rome, Italy 00144 | 30,142,859 | (1) | 46.9 | % | 41.9 | % | ||||||
Named Executive Officers and Directors: | ||||||||||||
| J.J. Finkelstein | 2,299,636 | (2) | 3.8 | % | 3.2 | % | ||||||
| Allan L. Goldstein | 2,152,538 | (3) | 3.5 | % | 3.0 | % | ||||||
| Richard J. Hindin | 1,175,459 | (4) | 1.9 | % | 1.6 | % | ||||||
| Joseph C. McNay | 1,537,135 | (5) | 2.5 | % | 2.1 | % | ||||||
| Mauro Bove | 197,155 | (6) | * | * | ||||||||
| L. Thompson Bowles | 124,843 | (7) | * | * | ||||||||
| C. Neil Lyons | 348,143 | (8) | * | * | ||||||||
| David R. Crockford | 396,250 | (9) | * | * | ||||||||
| All current directors and executive officers as a group (8 persons) | 8,231,159 | (10) | 13.0 | % | 11.0 | % | ||||||
| * | Less than one percent. |
| (1) |
| (2) | ||
| Consists of 984,615 shares of common stock held of record held by Essetifin S.p.A. (f/k/a Sigma-Tau Finanziaria, S.p.A.) (“ |
63
| 71 |
| (3) | Consists of 19,583,333 shares of common stock held of record by GtreeBNT which were acquired in two equity purchases in March 2014 and August 2014. The beneficial ownership of GtreeBNT is derived from its Schedule 13D/A filed on February 1, 2015. |
| (4) | Consists of 1,377,638 shares of common stock held of record by Mr. Finkelstein, 1,654,883 shares of common stock issuable upon exercise of options, 20,000 shares of common stock issuable upon exercise of warrants and |
| (5) | Consists of 806,743 shares of common stock held of record by Dr. Goldstein, 1,166,667 shares of common stock issuable upon conversion of a convertible promissory note, 1,159,077 shares of common stock issuable upon exercise of options and 46,666 shares of common stock issuable upon exercise of warrants, in each case exercisable within 60 days of June 26, 2016. |
| (6) | Consists of 1,339,111 shares of common stock held of record by Mr. |
| (7) | Consists of 324,655 shares of common stock issuable upon exercise of options exercisable within 60 days of |
| (8) | Consists of 300,000 shares of common stock issuable upon exercise of options and 83,333 shares of common stock issuable upon conversion of a convertible promissory note, in each case exercisable within 60 days of |
| (9) | Consists of |
64
As of the date of this prospectus, our certificate of incorporation authorizes us to issue 100,000,000200,000,000 shares of common stock, par value $0.001 per share, and 1,000,000 shares of preferred stock, par value $0.001 per share. As of March 31, 2010, 60,406,828June 30, 2016, 107,787,151 shares of common stock were outstanding and no shares of preferred stock were outstanding. Our board of directors and stockholders have approved an amendment to our restated certificate of incorporation to increase the authorized number of shares of common stock from 100,000,000 shares to 200,000,000 shares. We expect to effect this amendment to our restated certificate of incorporation following the completion of this offering.
As of March 31, 2010,June 30, 2016, we also had outstanding:
| · | ||
| · | 5,804,412 shares of our common stock issuable upon the exercise of outstanding warrants at a |
| · | 13,683,334 shares of our common stock issuable upon conversion of convertible promissory notes. |
| 72 |
The following summary description of our capital stock is based on the provisions of our certificate of incorporation, including the certificate of designation for our Series A Participating Cumulative Preferred Stock described below, as well as our bylaws our stockholder rights plan and the applicable provisions of the Delaware General Corporation Law. This information is qualified entirely by reference to the applicable provisions of our restated certificate of incorporation, as amended to date, our bylaws, as amended to date, our stockholder rights plan and the Delaware General Corporation Law. For information on how to obtain copies of our certificate of incorporation bylaws and stockholder rights plan,bylaws, which are exhibits to the registration statement of which this prospectus is a part, see “Where You Can Find Additional Information.”
Common Stock
Voting Rights. Each holder of our common stock is entitled to one vote for each share on all matters submitted to a vote of the stockholders, including the election of directors. Our certificate of incorporation and bylaws do not provide for cumulative voting rights. Because of this, the holders of a majority of the shares of common stock entitled to vote in any election of directors can elect all of the directors standing for election, if they should so choose.
Dividends. Subject to preferences that may be applicable to any then outstanding preferred stock, holders of common stock are entitled to receive dividends, if any, as may be declared from time to time by our board of directors out of legally available funds.
Liquidation. In the event of our liquidation, dissolution or winding up, holders of common stock will be entitled to share ratably in the net assets legally available for distribution to stockholders after the payment of all of our debts and other liabilities and the satisfaction of any liquidation preference granted to the holders of any then outstanding shares of preferred stock.
Rights and Preferences. Holders of common stock have no preemptive, conversion, subscription or other rights, and there are no redemption or sinking fund provisions applicable to the common stock. The rights,
65
Fully Paid and Nonassessable. All of our outstanding shares of common stock are, and the shares of common stock to be issued in this offering will be, fully paid and nonassessable.
Warrants to Be Issued as Part of the Units
The material terms and provisions of the warrants being offered pursuantsold in the 2016 Offering, which we refer to this prospectusas the Warrants, are summarizedset forth below. This summary is subject to, and qualified in its entirety by, the form of warrant agreement and warrant certificate included as exhibitsan exhibit to the registration statement filed with the SECCommission of which this prospectus is a part. You should review copies of these items for a complete description of the terms and conditions applicable to the warrants.
Each whole warrantWarrant entitles the registered holder to purchase one share of our common stock at a price equal to $ , which represents 110% of the closing bid price per share of our common stock on the date of this prospectus.$0.51. The warrants may only be exercised for cash. The warrantscash, provided that if during the term of the Warrants there is not an effective registration statement under the Securities Act covering the resale of the shares issuable upon exercise of the Warrants, then the Warrants may be exercised beginning 30 days after original issuanceon a cashless (net exercise) basis.. The Warrants expire upon the later of five and will expireone half years from the date they were issued or five years from the date of issuance at 5:00 p.m., New York City time.
66
The exercise price and number of shares of common stock issuable on exercise of the warrants may be adjusted in certain circumstances, including but not limited to in the event of a stock split, stock dividend, recapitalization, reorganization, merger or consolidation. However,The exercise price of Warrants is subject to a “full-ratchet” anti-dilution provision for a period of one year from the warrants will not be adjusted foreffective date of this Registration Statement, such that in the issuancesevent the Company makes an issuance of common stock or securities convertible or exercisable into common stock(subject to customary exceptions) at a price below their respectiveper share less than the applicable exercise prices.
Preferred Stock
Under our certificate of incorporation, our board of directors has the authority, without further action by the stockholders, to issue up to 1,000,000 shares of preferred stock in one or more series, to establish from time to time the number of shares to be included in each such series, to fix the rights, preferences and privileges of the shares of each wholly unissued series and any qualifications, limitations or restrictions thereon and to increase or decrease the number of shares of any such series, but not below the number of shares of such series then outstanding. Our board of directors has designated 200,000 of the 1,000,000 authorized shares of preferred stock as Series A Participating Cumulative Preferred Stock, none of which shares are outstanding but which could be issued under the terms of the stockholder rights plan.outstanding.
| 73 |
Our board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of the common stock. The issuance of preferred stock, while providing flexibility in connection with possible acquisitions and other corporate purposes, could, among other things, have the effect of delaying, deferring or preventing a change in control
67
Delaware Anti-takeover Law and Certain Provisions of Our Amended and Restated Certificate of Incorporation and Bylaws
Delaware law. We are subject to Section 203 of the Delaware General Corporation Law. Section 203 generally prohibits a public Delaware corporation from engaging in a business combination with an interested stockholder for a period of three years after the date of the transaction in which the person became an interested stockholder, unless:
| prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder; | ||
| the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for this purpose shares owned by persons who are directors and also officers and shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or | ||
| on or subsequent to the date of the transaction, the business combination is approved by the board and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least |
Section 203 defines a business combination to include:
| any merger or consolidation involving the corporation and the interested stockholder; | ||
| any sale, transfer, pledge or other disposition involving the interested stockholder of 10% or more of the assets of the corporation; | ||
| subject to exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; | ||
| any transaction involving the corporation that has the effect of increasing the proportionate share of the stock or any class or series of the corporation beneficially owned by the interested stockholder; and | ||
| the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation. |
In general, Section 203 defines an interested stockholder as any entity or person beneficially owning 15% or more of the outstanding voting stock of the corporation and any entity or person affiliated with or controlling or controlled by the entity or person.
Certificate of Incorporation and Bylaws. Provisions of our restated certificate of incorporation and amended and restated bylaws may delay or discourage transactions involving an actual or potential change in our control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares, or transactions that our stockholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our common stock.
| 74 |
68
| permit our board of directors to issue up to 1,000,000 shares of preferred stock, with any rights, preferences and privileges as they may designate, including the right to approve an acquisition or other change in our control; | ||
| provide that the authorized number of directors, which may not be less than three nor more than seven, may be changed only by resolution of the board of directors; | ||
| provide that stockholders seeking to nominate candidates for election as directors at a meeting of stockholders must provide notice in writing in a timely manner, and also specify requirements as to the form and content of a stockholder’s notice; | ||
| do not provide for cumulative voting rights, therefore allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election, if they should so choose; and | ||
| provide that special meetings of our stockholders may be called only by the chairman of the board, our president or by the board of directors. |
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is American Stock Transfer & Trust Company. The transfer agent and registrar’s address is 6201 15th Street, Brooklyn, NY 11219.
NYSE Amex ListingOTC Bulletin Board Quotation of our Common Stock
Our common stock is currently listedquoted on the NYSE AmexOTC Bulletin Board under the trading symbol “RGN.“RGRX.”
69
70
71
72
73
74
75
The validity of the securities being offered by this prospectus will behas been passed upon for us by CooleyFredrikson & Byron, P.A., Minneapolis, Minnesota.
Our financial statements as of and for the years ended December 31, 2015 and December 31, 2014 included in this prospectus have been audited by CohnReznick LLP, Reston, Virginia. The underwriters are being represented by Lowenstein Sandler PC, Roseland, New Jersey.
We have filed with the We are subject to the information reporting requirements of the Exchange Act, and we file reports, proxy statements and other information with the SECCommission a registration statement onForm S-1 under the Securities Act, with respect to the securities being offered by this prospectus. This prospectus does not contain all of the information in the registration statement and its exhibits. For further information with respect to RegeneRx and the securities offered by this prospectus, we refer you to the registration statement and its exhibits. Statements contained in this prospectus as to the contents of any contract or any other document referred to are not necessarily complete, and in each instance, we refer you to the copy of the contract or other document filed as an exhibit to the registration statement. Each of these statements is qualified in all respects by this reference.SEC.Commission. You can read our SECCommission filings, including the registration statement, over the Internet at the SEC’sCommission’s website atwww.sec.gov. You may also read and copy any document we file with the SECCommission at its public reference facilities at 100 F Street, N.E., Washington, D.C. 20549. You may also obtain copies of these documents at prescribed rates by writing to the Public Reference Section of the SECCommission at 100 F Street, N.E., Washington, D.C. 20549. Please call the SECCommission at1-800-SEC-0330 for further information on the operation of the public reference facilities.76
| 75 |
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling the registrant pursuant to the foregoing provisions, the registrant has been informed that in the opinion of the
SECURITIES ACT LIABILITYSECCommission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable.76 77
F-1
RegeneRx Biopharmaceuticals, Inc.
Condensed Balance Sheets
| March 31, | December 31, | |||||||
| 2016 | 2015 | |||||||
| (Unaudited) | (See Note 1) | |||||||
| ASSETS | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | $ | 79,834 | $ | 317,627 | ||||
| Prepaid expenses and other current assets | 18,451 | 24,300 | ||||||
| Total current assets | 98,285 | 341,927 | ||||||
| Property and equipment, net of accumulated depreciation of $89,626 and $88,794 | 9,713 | 10,544 | ||||||
| Other assets | 5,752 | 5,752 | ||||||
| Total assets | $ | 113,750 | $ | 358,223 | ||||
| LIABILITIES AND STOCKHOLDERS' DEFICIT | ||||||||
| Current liabilities | ||||||||
| Accounts payable | $ | 174,642 | $ | 141,130 | ||||
| Unearned revenue | 40,408 | - | ||||||
| Accrued expenses | 243,358 | 217,911 | ||||||
| Total current liabilities | 458,408 | 359,041 | ||||||
| Long-Term liabilities | ||||||||
| Unearned revenue | 1,328,878 | 1,379,388 | ||||||
| Convertible promisory note | 300,000 | 300,000 | ||||||
| Convertible promisory notes, net of derivative liability | 419,477 | 388,854 | ||||||
| Fair value of derivative liability | 7,556,670 | 4,673,336 | ||||||
| Total liabilities | 10,063,433 | 7,100,619 | ||||||
| Commitments and contingencies | ||||||||
| Stockholders' deficit | ||||||||
| Preferred stock, $.001 par value per share, 1,000,000 shares authorized; no shares issued | - | - | ||||||
| Common stock, par value $.001 per share, 200,000,000 shares authorized, 101,640,092 issued and outstanding | 101,640 | 101,640 | ||||||
| Additional paid-in capital | 98,402,371 | 98,230,802 | ||||||
| Accumulated deficit | (108,453,694 | ) | (105,074,838 | ) | ||||
| Total stockholders' deficit | (9,949,683 | ) | (6,742,396 | ) | ||||
| Total liabilities and stockholders' deficit | $ | 113,750 | $ | 358,223 | ||||
The accompanying notes are an integral part of these condensed financial statements.
| F-2 |
RegeneRx Biopharmaceuticals, Inc.Condensed Statements of Operations
| Three Months ended March 31, | ||||||||
| 2016 | 2015 | |||||||
| (Unaudited) | (Unaudited) | |||||||
| Revenues | $ | 55,190 | $ | 40,000 | ||||
| Operating expenses | ||||||||
| Research and development | 102,901 | 36,472 | ||||||
| General and administrative | 404,710 | 394,500 | ||||||
| Total operating expenses | 507,611 | 430,972 | ||||||
| Loss from operations | (452,421 | ) | (390,972 | ) | ||||
| Interest and other income | - | 77 | ||||||
| Interest expense | (43,101 | ) | (42,629 | ) | ||||
| Change in fair value of derivative | (2,883,334 | ) | (1,285,166 | ) | ||||
| Net loss | $ | (3,378,856 | ) | $ | (1,718,690 | ) | ||
| Basic and diluted net loss per common share | $ | (0.03 | ) | $ | (0.02 | ) | ||
| Weighted average number of common shares outstanding | 101,640,092 | 101,316,580 | ||||||
The accompanying notes are an integral part of these condensed financial statements.
| F-3 |
RegeneRx Biopharmaceuticals, Inc.
Condensed Statements of Cash Flows
| For the Three months ended March 31, | ||||||||
| 2016 | 2015 | |||||||
| (Unaudited) | (Unaudited) | |||||||
| Operating activities: | ||||||||
| Net loss | $ | (3,378,856 | ) | $ | (1,718,690 | ) | ||
| Adjustments to reconcile net loss to net cash provided by (used in) operating activities: | ||||||||
| Depreciation and amortization | 831 | 909 | ||||||
| Share-based compensation | 171,569 | 28,139 | ||||||
| Non-cash interest expense | 30,623 | 30,288 | ||||||
| Change in fair value of derivative | 2,883,334 | 1,285,166 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | 5,849 | 34,771 | ||||||
| Accounts payable | 33,512 | 28,535 | ||||||
| Accrued expenses | 25,447 | 29,338 | ||||||
| Unearned revenue | (10,102 | ) | 500,000 | |||||
| Net cash provided by (used in) operating activities | (237,793 | ) | 218,456 | |||||
| Net (decrease) increase in cash and cash equivalents | (237,793 | ) | 218,456 | |||||
| Cash and cash equivalents at beginning of period | 317,627 | 844,043 | ||||||
| Cash and cash equivalents at end of period | $ | 79,834 | $ | 1,062,499 | ||||
The accompanying notes are an integral part of these condensed financial statements.
| F-4 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRMRegeneRx Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
For the three months ended March 31, 2016 and 2015 (Unaudited)
| 1. | organization, business overview and basis of presentation |
Organization and Nature of Operations.
RegeneRx Biopharmaceuticals, Inc. (“RegeneRx”, the “Company”, “We”, “Us”, “Our”), a Delaware corporation, was incorporated in 1982. We are focused on the discovery and development of novel molecules to accelerate tissue and organ repair. Our operations are confined to one business segment: the development and marketing of product candidates based on Thymosin Beta 4 (“Tß4”), an amino acid peptide.
Management Plans to Address Operating Conditions.
On January 28, 2015, we announced that we had entered into a Joint Venture Agreement (the “Joint Venture Agreement”) with GtreeBNT Co., Ltd., a Korean pharma company (“GtreeBNT”) and shareholder of the Company. The Joint Venture Agreement provides for the creation of an entity, ReGenTree, LLC (the “Joint Venture” or “ReGenTree”), jointly owned by us and GtreeBNT, that will commercialize RGN-259 for treatment of dry eye and neurotrophic keratopathy, an orphan indication in the United States. GtreeBNT is responsible for funding all product development and commercialization efforts, and holds a majority interest of ReGenTree that varies depending on development milestones achieved and eventual commercialization path, if successful. In March 2015, GtreeBNT reported that it received $7.28 million to expand international development of the product candidate, RGN-259 (designated GBT-201 in Korea). The $7.28 million will be used for development of RGN-259/GBT-201 for dry eye syndrome and neurotrophic keratopathy in the U.S. through the U.S. joint venture, ReGenTree, LLC.
RegeneRx’s ownership interest in ReGenTree is 49% and will be reduced to 42% when the clinical study report is filed for the Phase 3 dry eye clinical trial. Based on when, and if, certain additional development milestones are achieved in the U.S. with RGN-259, our equity ownership may be incrementally reduced to between 42% and 25%, with 25% being the final equity ownership upon approval of an NDA for Dry Eye Syndrome in the U.S. In the event the ReGenTree entity is acquired or there is a change of control that occurs following achievement of an NDA, RegeneRx shall be entitled to 40% of all change of control proceeds paid or payable and will forgo any future royalties.
In conjunction with the Joint Venture Agreement, we also entered into a royalty-bearing license agreement (the “License Agreement”) with ReGenTree pursuant to which we granted to ReGenTree the right to develop and exclusively commercialize RGN-259 in the United States. We received a total of $1 million in two tranches under the terms of the License Agreement. The first tranche of $500,000 was received in March 2015 and a second in the amount of $500,000 was received in September 2015. On April 6, 2016 we received $250,000 from ReGenTree and executed an amendment to the license agreement on April 28, 2016. Under the amendment the territorial rights were expanded to include Canada. We are also entitled to royalties as a percentage of net sales ranging from the single digits to the low-double digits based on the medical indications approved and whether the Joint Venture commercializes products directly or through a third party. RegeneRx possesses one of three board seats and certain major decisions and transactions within ReGenTree, such as commercialization strategy, mergers, and acquisitions, require RegeneRx’s board designee’s consent.
In September 2015, ReGenTree began a Phase 2b/3 clinical trial in patients with dry eye syndrome (“DES”) and a Phase 3 clinical trial in patients with neurotrophic keratopathy (“NK”), both in the U.S. In January 2016, the DES had completed enrollment of all patients in the trial. The last patient received the last treatment in February. Top line data from the DES trial was released in early May and additional clinical work is being planned for late 2016.
| F-5 |
The study, which enrolled 317 subjects, tested two doses of RGN-259 eye drops containing 0.05% and 0.1% concentrations of the Tβ4 four times daily for 28 days vs. placebo. On the 28th and final day of dosing, patients were subject to the Controlled Adverse Environment (CAE) challenge. Co-primary endpoints were total corneal fluorescein staining score change on the 28th day pre-CAE and post-CAE (sign) and total ocular discomfort score change on the 28th day pre-CAE and post-CAE (symptom). Although the co-primary endpoints were not met, we demonstrated RGN-259’s protective efficacy in improving a sign and symptom of dry eye syndrome, which is in line with the results observed in the previous Phase 2a trial. Importantly, we identified approvable sign and symptom endpoints, which were met with statistical significance. ReGenTree expects to meet with the FDA this summer and initiate a confirmatory Phase 3 trial before the end of 2016.
RGN-259 was safe and well-tolerated and comfortable for the patients with no irritation upon instillation. There were no significant drug-related adverse events for both concentrations. The safety profile is consistent with that observed in the previous Phase 2a trial, which had a twice daily instillation regimen.
The NK trial, a smaller study in an orphan population, has enrolled eight patients thus far with a goal of 46. Of the eight original clinical sites for the study, six are enrolling patients, one has yet to receive IRB approval to begin enrolling patients, and one was unable to consummate a clinical contract with ReGenTree. ReGenTree is considering adding additional sites to accelerate patient enrollment.
Currently, we have active partnerships in three major territories: the U.S., China and Pan Asia. Our partners have been moving forward and making progress in each territory. In each case, the cost of development is being borne by our partners with no financial obligation for RegeneRx. We received $250,000 pursuant to the signing of a term sheet with ReGenTree in April 2016, with this amount coupled with our current cash, we believe we should be able to maintain our existing operations at the current level into the third quarter of 2016.
Lee's Pharmaceutical Ltd. (“Lee’s”), RegeneRx's licensee in China, Hong Kong, Macau and Taiwan, previously received notice from China's FDA (“CFDA”) declining its investigational new drug (“IND”) application for a Phase 2b dry eye clinical trial because the API (active pharmaceutical ingredient or Tß4) was manufactured outside of China. The API was manufactured in the U.S. and provided to Lee's by RegeneRx pursuant to a license agreement to develop RGN-259 ophthalmic eye drops in the licensed territory. Due to this unexpected regulatory hurdle, Lee's plans to modify its clinical program to conduct the Phase 2b dry eye trial in Hong Kong and Taiwan using the Tß4 supplied by RegeneRx while awaiting the manufacturing of Tß4 in China for a subsequent Phase 3 registration trial. We do not know when the Hong Kong/Taiwan trial will begin enrollment of patients although we have been providing documents and data recently for their clinical activities. Under this revised strategy, Lee's believes it should be able to begin a Phase 3 registration trial in China sooner, rather than waiting on the production of Tß4 in China before initiating a Phase 2b trial.
GtreeBNT, RegeneRx's licensee in Korea, Australia, Japan and a number of other countries in Asia, filed an IND with the Korean Ministry of Food and Drug Safety to conduct a Phase 2b/3 study with RGN-259 in patients with dry eye syndrome and in July 2015 received approval to conduct the trial. GtreeBNT has informed us that given its immediate focus on the two U.S. trials, it is considering the best timing for the Korean trial.
We still have significant clinical assets to develop, primarily RGN-352 (injectable formulation of Tß4 for cardiac and CNS disorders) in the U.S., Pan Asia, and Europe, and RGN-259 in the EU. Our goal is to wait until the results are obtained from the current ophthalmic clinical trials before moving into the EU with RGN-259. If successful, this should allow us to obtain a higher value for the asset at that time. However, we intend to continue to develop RGN-352, either by obtaining grants to fund a Phase 2a clinical trial in the cardiovascular or central nervous system fields or finding a suitable partner with the resources and capabilities to develop it as we have with RGN-259.
We anticipate incurring additional losses in the future as we continue to explore the potential clinical benefits of Tß4-based product candidates over multiple indications. We have entered into a series of strategic partnerships under licensing and joint venture agreements where our partners are responsible to advance development of our product candidates with multiple clinical trials started in 2015 and additional trials to begin in 2016. We will need additional funds to continue operations during the third quarter of 2016 as well as substantial additional funds in order to significantly advance development of our unlicensed programs. Accordingly, we will continue to evaluate opportunities to raise additional capital and are in the process of exploring various alternatives, including, without limitation, a public or private placement of our securities, debt financing, corporate collaboration and licensing arrangements, or the sale of our company or certain of our intellectual property rights.
| F-6 |
These factors raise substantial doubt about our ability to continue as a going concern. The accompanying financial statements have been prepared assuming that we will continue as a going concern. This basis of accounting contemplates the recovery of our assets and the satisfaction of our liabilities in the normal course of business.
Although we intend to continue to seek additional financing or additional strategic partners, we may not be able to complete a financing or corporate transaction, either on favorable terms or at all. If we are unable to complete a financing or strategic transaction, we may not be able to continue as a going concern after our funds have been exhausted, and we could be required to significantly curtail or cease operations, file for bankruptcy or liquidate and dissolve. There can be no assurance that we will be able to obtain any sources of funding. The financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts and classification of liabilities that might be necessary should we be forced to take any such actions.
In addition to our current operational requirements, we continually refine our operating strategy and evaluate alternative clinical uses of Tß4. However, substantial additional resources will be needed before we will be able to achieve sustained profitability. Consequently, we continually evaluate alternative sources of financing such as the sharing of development costs through strategic collaboration agreements. There can be no assurance that our financing efforts will be successful and, if we are not able to obtain sufficient levels of financing, we would delay certain clinical and/or research activities and our financial condition would be materially and adversely affected. Even if we are able to obtain sufficient funding, other factors including competition, dependence on third parties, uncertainty regarding patents, protection of proprietary rights, manufacturing of peptides, and technology obsolescence could have a significant impact on us and our operations.
To achieve profitability we, and/or a partner, must successfully conduct pre-clinical studies and clinical trials, obtain required regulatory approvals and successfully manufacture and market those pharmaceuticals we wish to commercialize. The time required to reach profitability is highly uncertain, and there can be no assurance that we will be able to achieve sustained profitability, if at all.
Basis of Presentation.
The accompanying unaudited interim financial statements reflect, in the opinion of management, all adjustments (consisting only of normal recurring adjustments) necessary for a fair presentation of our financial position, results of operations and cash flows for each period presented. These statements have been prepared in accordance with U.S. generally accepted accounting principles (“GAAP”) and with the rules and regulations of the SEC, for interim financial statements. Accordingly, they do not include all of the information and footnotes required by GAAP. The accounting policies underlying our unaudited interim financial statements are consistent with those underlying our audited annual financial statements. These unaudited interim financial statements should be read in conjunction with the audited annual financial statements as of and for the year ended December 31, 2015, and related notes thereto, included in our Annual Report on Form 10-K for the year ended December 31, 2015 (the “Annual Report”).
The accompanying December 31, 2015 financial information was derived from our audited financial statements included in the Annual Report. Operating results for the three-month period ended March 31, 2016 are not necessarily indicative of the results to be expected for the year ending December 31, 2016 or any other future period.
References in this Quarterly Report on Form 10-Q to “authoritative guidance” are to the Accounting Standards Codification issued by the Financial Accounting Standards Board (“FASB”).
| F-7 |
Use of Estimates.
The preparation of financial statements in conformity with accounting principles generally accepted in the United Stated of America (“U.S. GAAP”) requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Critical accounting policies involved in applying our accounting policies are those that require management to make assumptions about matters that are highly uncertain at the time the accounting estimate was made and those for which different estimates reasonably could have been used for the current period. Critical accounting estimates are also those which are reasonably likely to change from period to period, and would have a material impact on the presentation of our financial condition, changes in financial condition or results of operations. Our most critical accounting estimates relate to accounting policies for fair value measurements in connection with derivative liabilities, clinical trial accruals and share-based arrangements. Management bases its estimates on historical experience and on various other assumptions that it believes are reasonable under the circumstances. Actual results could differ from those estimates.
Convertible Notes with Detachable Warrants.
In accordance with Accounting Standards Codification (“ASC”) 470-20,Debt with Conversion and Other Options, the proceeds received from convertible notes are allocated between the convertible notes and the detachable warrants based on the relative fair value of the convertible notes without the warrants and the relative fair value of the warrants. The portion of the proceeds allocated to the warrants is recognized as additional paid-in capital and a debt discount. The debt discount related to warrants is accreted into interest expense through maturity of the notes.
Derivative Financial Instruments
Derivative financial instruments consist of financial instruments or other contracts that contain a notional amount and one or more underlying variables (e.g. interest rate, security price or other variable), which require no initial net investment and permit net settlement. Derivative financial instruments may be free-standing or embedded in other financial instruments. Further, derivative financial instruments are initially, and subsequently, measured at fair value and recorded as liabilities or, in rare instances, assets.
The Company does not use derivative financial instruments to hedge exposures to cash-flow, market or foreign-currency risks. However, the Company has issued financial instruments including warrants that are either (i) not afforded equity classification, (ii) embody risks not clearly and closely related to host contracts, or (iii) may be net-cash settled by the counterparty. In certain instances, these instruments are required to be carried as derivative liabilities, at fair value, in the Company’s financial statements. In other instances these instruments are classified as equity instruments in the Company’s financial statements.
The Company estimates the fair values of its derivative financial instrument using the Black-Scholes option pricing model because it embodies all of the requisite assumptions (including trading volatility, estimated terms and risk free rates) necessary to fair value these instruments. Estimating fair values of derivative financial instruments requires the development of significant and subjective estimates that may, and are likely to, change over the duration of the instrument with related changes in internal and external market factors. In addition, option-based techniques are highly volatile and sensitive to changes in the trading market price of the Company’s common stock, which has a high-historical volatility. Since derivative financial instruments are initially and subsequently carried at fair values, the Company’s operating results reflect the volatility in these estimate and assumption changes in each reporting period.
| F-8 |
Revenue Recognition.
We recognize revenue in accordance with the authoritative guidance for revenue recognition. We recognize revenue when all of the following criteria are met: (i) persuasive evidence of an arrangement exists, (ii) delivery (or passage of title) has occurred or services have been rendered, (iii) the seller's price to the buyer is fixed or determinable, and (iv) collectability is reasonably assured. We also comply with the authoritative guidance for revenue recognition regarding arrangements with multiple deliverables. Multiple-element arrangements are analyzed to determine whether the deliverables, which may include a license together with performance obligations such as providing a clinical supply of product and steering committee services, can be separated or whether they must be accounted for as a single unit of accounting. Revenue associated with licensing agreements consists of non-refundable upfront license fees and milestone payments. Non-refundable upfront license fees received under license agreements, whereby continued performance or future obligations are considered inconsequential to the relevant license technology, are recognized as revenue upon delivery of the technology.
Whenever we determine that an arrangement should be accounted for as a single unit of accounting, we must determine the period over which the performance obligations will be performed and revenue will be recognized. Revenue will be recognized using either a relative performance or straight-line method. We recognize revenue using the relative performance method provided that the we can reasonably estimate the level of effort required to complete our performance obligations under an arrangement and such performance obligations are provided on a best-efforts basis. Revenue recognized is limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the relative performance method, as of each reporting period.
If we cannot reasonably estimate the level of effort required to complete our performance obligations under an arrangement, the performance obligations are provided on a best-efforts basis and we can reasonably estimate when the performance obligation ceases or the remaining obligations become inconsequential and perfunctory, then the total payments under the arrangement, excluding royalties and payments contingent upon achievement of substantive milestones, would be recognized as revenue on a straight-line basis over the period we expect to complete our performance obligations. Revenue is limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the straight-line basis, as of the period ending date.
If we cannot reasonably estimate when our performance obligation either ceases or becomes inconsequential and perfunctory, then revenue is deferred until we can reasonably estimate when the performance obligation ceases or becomes inconsequential. Revenue is then recognized over the remaining estimated period of performance.
We recognize consideration that is contingent upon the achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone is substantive in its entirety. A milestone is considered substantive when it meets all of the following criteria:
| · | The consideration is commensurate with either the entity's performance to achieve the milestone or the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity's performance to achieve the milestone; |
| · | The consideration relates solely to past performance; and |
| · | The consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. |
A milestone is defined as an event (i) that can only be achieved based in whole or in part on either the entity's performance or on the occurrence of a specific outcome resulting from the entity's performance, (ii) for which there is substantive uncertainty at the date the arrangement is entered into that the event will be achieved and (iii) that would result in additional payments being due to us.
Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue in our accompanying balance sheets.
| F-9 |
Variable Interest Entities
The Company has determined that the Joint Venture is a “variable interest entity” and that its equity stake in the Joint Venture is a variable interest, since the total equity investment at risk is not sufficient to permit the Joint Venture to finance its activities without additional subordinated financial support. Further, because of GtreeBNT’s majority equity stake in the Joint Venture, voting control, control of the board of directors, and substantive management rights, and given that the Company does not have the power to direct the Joint Venture’s activities that most significantly impact its economic performance, the Company determined that it is not the primary beneficiary of the Joint Venture and therefore is not required to consolidate the Joint Venture. The Company reports its equity stake in the Joint Venture using the equity method of accounting because, while it does not control the Joint Venture, the Company can exert significant influence over the Joint Ventures activities by virtue of its large equity stake and its board representation.
Because the Company is not obligated to fund the Joint Venture, and has not provided any financial support to the Joint Venture, the carrying value of its investment in the Joint Venture is zero. As a result, the Company is not recognizing its share (49%) of the Joint Venture’s operating losses and will not recognize any such losses until the Joint Venture produces net income (as opposed to net losses) and at that point the Company will reduce its share of the Joint Venture’s net income by its share of previously suspended net losses. As of March 31, 2016, because it has not provided any financial support, the Company has no financial exposure as a result of its variable interest in the Joint Venture.
Research and Development.
Research and development (“R&D”) costs are expensed as incurred and include all of the wholly-allocable costs associated with our various clinical programs passed through to us by our outsourced vendors. Those costs include: manufacturing Tβ4; formulation of Tβ4 into the various product candidates; stability for both Tβ4 and the various formulations; pre-clinical toxicology; safety and pharmacokinetic studies; clinical trial management; medical oversight; laboratory evaluations; statistical data analysis; regulatory compliance; quality assurance; and other related activities. R&D includes cash and non-cash compensation, payroll taxes, travel and other miscellaneous costs of our internal R&D personnel, part-time hourly employees and external consultants dedicated to R&D efforts. R&D also includes a pro-ration of our common infrastructure costs for office space and communications.
Cost of Preclinical Studies and Clinical Trials.
We accrue estimated costs for preclinical studies based on estimates of work performed. We estimate expenses incurred for clinical trials that are in process based on patient enrollment and based on clinical data collection and management. Costs based on clinical data collection and management are recognized based on estimates of unbilled goods and services received in the reporting period. We monitor the progress of the trials and their related activities and adjust the accruals accordingly. Adjustments to accruals are charged to expense in the period in which the facts that give rise to the adjustment become known. In the event of early termination of a clinical trial, we would accrue an amount based on estimates of the remaining non-cancelable obligations associated with winding down the clinical trial.
Recent Accounting Pronouncements.
In August 2014, the FASB issued Accounting Standard Update (“ASU”) 2014-15,Presentation of Financial Statements – Going Concern. The new standard requires management to evaluate on a regular basis whether any conditions or events have arisen that could raise substantial doubt about the entity’s ability to continue as a going concern. The guidance 1) provides a definition for the term “substantial doubt,” 2) requires an evaluation every reporting period, interim periods included, 3) provides principles for considering the mitigating effect of management’s plans to alleviate the substantial doubt, 4) requires certain disclosures if the substantial doubt is alleviated as a result of management’s plans, 5) requires an express statement, as well as other disclosures, if the substantial doubt is not alleviated, and 6) requires an assessment period of one year from the date the financial statements are issued. The standard is effective for the Company’s reporting year beginning January 1, 2017 and early adoption is not permitted. The Company is currently evaluating the impact, if any, that this new accounting pronouncement will have on its financial statements.
| F-10 |
In May 2014, the FASB issued ASU 2014-09, Revenue from Contracts with Customers, which provides guidance for revenue recognition for contracts, superseding the previous revenue recognition requirements, along with most existing industry-specific guidance. The guidance requires an entity to review contracts in five steps: 1) identify the contract, 2) identify performance obligations, 3) determine the transaction price, 4) allocate the transaction price, and 5) recognize revenue. The new standard will result in enhanced disclosures regarding the nature, amount, timing and uncertainty of revenue arising from contracts with customers. The standard is effective for the Company’s reporting year beginning January 1, 2017 and early adoption is not permitted. The Company is currently evaluating the impact, if any, that this new accounting pronouncement will have on its financial statements.
In November 2015, the FASB issued new guidance on the balance sheet classification of deferred taxes. To simplify presentation, the new guidance requires that all deferred tax assets and liabilities, along with any related valuation allowance, be classified as noncurrent on the balance sheet. The accounting standard is effective for public business entities for annual reporting periods (including interim reporting periods within those periods) beginning after December 15, 2016. Early adoption is permitted. The adoption of this guidance did not have an impact on our financial statements.
In January 2016, the FASB issued ASU 2016-01,Financial Instruments-Recognition and Measurement of Financial Assets and Financial Liabilities. The accounting standard primarily affects the accounting for equity investments, financial liabilities under the fair value option, and the presentation and disclosure requirements for financial instruments. In addition, it includes a clarification related to the valuation allowance assessment when recognizing deferred tax assets resulting from unrealized losses on available-for-sale debt securities. The accounting guidance is effective for annual reporting periods (including interim periods within those periods) beginning after December 15, 2017. Early adoption is permitted for the provision to record fair value changes for financial liabilities under the fair value option resulting from instrument-specific credit risk in other comprehensive income. The Company is currently evaluating the impact, if any, that the pronouncement will have on the financial statements.
In February 2016, the FASB issued ASU 2016-02,Leases, which supersedes ASC Topic 840,Leases, and creates a new topic, ASC Topic 842,Leases. ASU 2016-02 requires lessees to recognize a lease liability and a lease asset for all leases, including operating leases, with a term greater than 12 months on its balance sheet. ASU 2016-02 also expands the required quantitative and qualitative disclosures surrounding leases. ASU 2016-02 is effective for the Company beginning January 1, 2019. Early adoption is permitted. The Company has determined that the adoption of ASU 2016-02 will currently have no impact on its financial statements.
In March 2016, the FASB issued ASU 2016-07,Equity Method and Joint Ventures which affects all entities that have an investment that becomes qualified for the equity method of accounting as a result of an increase in the level of ownership or degree of influence. ASU 2016-07 is effective for the Company beginning on January 1, 2017, early adoption is permitted. The Company is currently evaluating the effect this ASU will have on the financial statements.
| 2. | Net Loss per Common Share |
Net loss per common share for the three-month periods ended March 31, 2016 and 2015, is based on the weighted-average number of shares of common stock outstanding during the periods. Basic and diluted loss per share are identical for all periods presented as potentially dilutive securities have been excluded from the calculation of the diluted net loss per common share because the inclusion of such securities would be antidilutive. The potentially dilutive securities include 21,704,544 shares and 28,875,092 shares in 2016 and 2015, respectively, reserved for the conversion of convertible debt or exercise of outstanding options and warrants.
| F-11 |
| 3. | Stock-Based Compensation |
We measure stock-based compensation expense based on the grant date fair value of the awards, which is then recognized over the period which service is required to be provided. We estimate the value of our stock option awards on the date of grant using the Black-Scholes option pricing model and amortize that cost over the expected term of the grant. We recognized $171,569 and $28,139 in stock-based compensation expense for the three months ended March 31, 2016 and 2015, respectively. We expect to recognize the compensation cost related to non-vested options as of March 31, 2016 of $597,475 over the weighted average remaining recognition period of 1.1 years.
We used the following forward-looking range of assumptions to value the 940,000 stock options granted to employees, consultants and directors during the three months ended March 31, 2016 and the 325,000 stock options granted to employees, consultants and directors during the three months ended March 31, 2015:
| 2016 | 2015 | |||||||
| Dividend yield | 0.0 | % | 0.0 | % | ||||
| Risk-free rate of return | 1.41 | % | 1.53 | % | ||||
| Expected life in years | 4.5 - 7 | 4.75 | ||||||
| Volatility | 87-95% | 92 | % | |||||
| Forfeiture rate | 2.6 | % | 2.6 | % | ||||
| 4. | Income Taxes |
As of March 31, 2016, there have been no material changes to our uncertain tax positions disclosures as provided in Note 9 of the Annual Report. The tax returns for all years in the Company’s major tax jurisdictions are not settled as of January 1, 2016; no changes in settled tax years have occurred through March 31, 2016. Due to the existence of tax attribute carryforwards (which are currently offset by a full valuation allowance), the Company treats all years’ tax positions as unsettled due to the taxing authorities’ ability to modify these attributes.
| 5. | Fair Value Measurements |
The authoritative guidance for fair value measurements defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or the most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Market participants are buyers and sellers in the principal market that are (i) independent, (ii) knowledgeable, (iii) able to transact, and (iv) willing to transact. The guidance describes a fair value hierarchy based on the levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value which are the following:
Level 1 — Quoted prices in active markets for identical assets and liabilities.
Level 2 — Observable inputs other than quoted prices in active markets for identical assets and liabilities.
Level 3 — Unobservable inputs.
As of March 31, 2016 and December 31, 2015, our only qualifying assets that required measurement under the foregoing fair value hierarchy were money market funds included in Cash and Cash Equivalents valued at $79,834 and $317,627, respectively, using Level 1 inputs. Our March 31, 2016 and December 31, 2015 balance sheets reflect qualifying liabilities resulting from the price protection provision in the convertible promissory notes issued in March, July and September of 2013 and January 2014 (see Note 6). We evaluated the derivative liability embedded in the series of convertible notes to determine if an adjustment to the carrying value of the liability was required at March 31, 2016 using the following assumptions.
| F-12 |
| March 2013 | July 2013 | Sept 2013 | Jan 2014 | |||||||||||||
| Dividend yield | 0.00 | % | 0.00 | % | 0.00 | % | 0.00 | % | ||||||||
| Risk-free rate of return | 0.88 | % | 0.88 | % | 0.88 | % | 1.04 | % | ||||||||
| Expected life in years | 2 | 2.25 | 2.45 | 2.75 | ||||||||||||
| Volatility | 69.4 | % | 121.0 | % | 117.9 | % | 111.5 | % | ||||||||
Given the conditions surrounding the trading of the Company’s equity securities, the Company values its derivative instruments related to embedded conversion features from the issuance of convertible debentures in accordance with the Level 3 guidelines. For the three months ended March 31, 2016, the following table reconciles the beginning and ending balances for financial instruments that are recognized at fair value in these financial statements.
| Balance at | Balance at | |||||||||||||||
| December 31, | New | Change in | March 31, | |||||||||||||
| 2015 | Issuances | Fair Values | 2016 | |||||||||||||
| Level 3 - | ||||||||||||||||
| Derivative liabilities from: | ||||||||||||||||
| Conversion features | ||||||||||||||||
| March 2013 | $ | 1,500,000 | $ | - | $ | 900,000 | $ | 2,400,000 | ||||||||
| July 2013 | 666,667 | - | 416,667 | 1,083,334 | ||||||||||||
| September 2013 | 2,140,000 | - | 1,337,500 | 3,477,500 | ||||||||||||
| January 2014 | 366,669 | - | 229,167 | 595,836 | ||||||||||||
| Derivative instruments | $ | 4,673,336 | $ | - | $ | 2,883,334 | $ | 7,556,670 | ||||||||
| 6. | Convertible Notes |
2012 Convertible Note
On October 19, 2012 we completed a private placement of convertible notes (the “2012 Notes”) raising an aggregate of $300,000 in gross proceeds. The 2012 Notes were originally to mature after twenty-four (24) months from issuance. In order to conserve the Company’s capital, in October 2014 the Investors agreed to extend the maturity date to October 19, 2017, all other terms were unchanged. The 2012 Notes bear interest at a rate of five percent (5%) per annum and are convertible into shares of our common stock at a conversion price of fifteen cents ($0.15) per share (subject to adjustment as described in the 2012 Notes) at any time prior to repayment, at the election of the Investors. In the aggregate, the 2012 Notes are convertible into up to 2,000,000 shares of our common stock excluding interest.
| F-13 |
At any time prior to maturity of the 2012 Notes, with the consent of the holders of a majority in interest of the 2012 Notes, we may prepay the outstanding principal amount of the 2012 Notes plus unpaid accrued interest without penalty. Upon the commission of any act of bankruptcy by the Company, the execution by the Company of a general assignment for the benefit of creditors, the filing by or against the Company of a petition in bankruptcy or any petition for relief under the federal bankruptcy act or the continuation of such petition without dismissal for a period of ninety (90) days or more, or the appointment of a receiver or trustee to take possession of the property or assets of the Company, the outstanding principal and all accrued interest on the 2012 Notes will accelerate and automatically become immediately due and payable.
In connection with the issuance of the 2012 Notes we also issued warrants to each Investor. The warrants are exercisable for an aggregate of 400,000 shares of common stock with an exercise price of fifteen cents ($0.15) per share for a period of five years. The relative fair value of the warrants issued is $27,097, calculated using the Black-Scholes-Merton valuation model value of $0.07 with an expected and contractual life of 5 years, an assumed volatility of 74.36%, and a risk-free interest rate of 0.77%. The warrants were recorded as additional paid-in capital and a discount on the 2012 Notes of $27,097. Non-cash interest expense related to the debt discount during the three months ended March 31, 2016 and 2015 totaled $0 and $3,341, respectively.
The Investors, and the principal amount of their respective 2012 Notes and number of shares of common stock issuable upon exercise of their respective warrants, are as set forth below:
| Investor | Note Principal | Warrants | ||||||
| Sinaf S.A. | $ | 200,000 | 266,667 | |||||
| Joseph C. McNay | $ | 50,000 | 66,667 | |||||
| Allan L. Goldstein | $ | 35,000 | 46,666 | |||||
| J.J. Finkelstein | $ | 15,000 | 20,000 | |||||
Sinaf S. A. is a direct wholly-owned subsidiary of Aptafin S.p.A., or Aptafin. Aptafin is owned directly by Paolo Cavazza and members of his family, who directly and indirectly own 38% of Sigma-Tau, our largest stockholder. The other Investors are members of our Board of Directors including Mr. Finkelstein who serves as our CEO and also the Chairman of our Board of Directors and Dr. Goldstein who also serves as our Chief Scientific Advisor.
2013 Convertible Notes
On March 29, 2013, we completed a private placement of convertible notes (the “March 2013 Notes”) raising an aggregate of $225,000 in gross proceeds. The March 2013 Notes bear interest at a rate of five percent (5%) per annum, mature sixty (60) months after their date of issuance and are convertible into shares of our common stock at a conversion price of six cents ($0.06) per share (subject to adjustment as described in the March 2013 Notes) at any time prior to repayment, at the election of the investor. In the aggregate, the March 2013 Notes are initially convertible into up to 3,750,000 shares of our common stock.
At any time prior to maturity of the March 2013 Notes, with the consent of the holders of a majority in interest of the March 2013 Notes, we may prepay the outstanding principal amount of the March 2013 Notes plus unpaid accrued interest without penalty. Upon the commission of any act of bankruptcy by the Company, the execution by the Company of a general assignment for the benefit of creditors, the filing by or against the Company of a petition in bankruptcy or any petition for relief under the Federal bankruptcy act or the continuation of such petition without dismissal for a period of ninety (90) days or more, or the appointment of a receiver or trustee to take possession of the property or assets of the Company, the outstanding principal and all accrued interest on the March 2013 Notes will accelerate and automatically become immediately due and payable.
The investors in the offering included two directors of the Company, Dr. Goldstein and Joseph C. McNay, an outside director. The principal amounts of their respective March 2013 Notes are as set forth below:
| Investor | Note Principal | |||
| Joseph C. McNay | $ | 50,000 | ||
| Allan L. Goldstein | $ | 25,000 | ||
| F-14 |
The Company has evaluated the terms of the March 2013 Notes which contain a down round provision under which the conversion price could be decreased as a result of future equity offerings, as defined in the March 2013 Notes. The adjustment would reduce the conversion price of the March 2013 Notes to be equivalent to that of the newly issued stock or stock-related instruments. As a result, the Company concluded that the conversion feature represented an embedded conversion feature for accounting purposes and should be recognized as a derivative liability, requiring a mark-to-market adjustment at the end of each reporting period until the related March 2013 Notes have been settled. The bifurcated liability of $225,000 was recorded on the date of issuance which resulted in a residual debt value of $0. The discount related to the embedded feature will be accreted back to debt through the maturity of the notes.
On July 5, 2013, we completed a private placement of convertible notes (the “July 2013 Notes”) raising an aggregate of $100,000 in gross proceeds. The July 2013 Notes bear interest at a rate of five percent (5%) per annum, mature sixty (60) months after their date of issuance and are convertible into shares of our common stock at a conversion price of six cents ($0.06) per share (subject to adjustment as described in the July 2013 Notes) at any time prior to repayment, at the election of the investor. In the aggregate, the July 2013 Notes are initially convertible into up to 1,666,667 shares of our common stock.
At any time prior to maturity of the July 2013 Notes, with the consent of the holders of a majority in interest of the July 2013 Notes, we may prepay the outstanding principal amount of the July 2013 Notes plus unpaid accrued interest without penalty. Upon the commission of any act of bankruptcy by the Company, the execution by the Company of a general assignment for the benefit of creditors, the filing by or against the Company of a petition in bankruptcy or any petition for relief under the Federal bankruptcy act or the continuation of such petition without dismissal for a period of ninety (90) days or more, or the appointment of a receiver or trustee to take possession of the property or assets of the Company, the outstanding principal and all accrued interest on the July 2013 Notes will accelerate and automatically become immediately due and payable.
The investors in the offering included four directors of the Company, Mr. Finkelstein, Dr. Goldstein, Mr. McNay and L. Thompson Bowles, a former outside director. The principal amounts of their respective July 2013 Notes are as set forth below:
| Investor | Note Principal | |||
| Joseph C. McNay | $ | 50,000 | ||
| Allan L. Goldstein | $ | 10,000 | ||
| J.J. Finkelstein | $ | 5,000 | ||
| L. Thompson Bowles | $ | 5,000 | ||
The Company has evaluated the terms of the July 2013 Notes which contain a down round provision under which the conversion price could be decreased as a result of future equity offerings, as defined in the July 2013 Notes. The adjustment would reduce the conversion price of the July 2013 Notes to be equivalent to that of the newly issued stock or stock-related instruments. As a result, the Company concluded that the conversion feature represented an embedded conversion feature for accounting purposes and should be recognized as a derivative liability, requiring a mark-to-market adjustment at the end of each reporting period until the related July 2013 Notes have been settled. The bifurcated liability of $66,667 was recorded on the date of issuance which resulted in a residual debt value of $33,333. The discount related to the embedded feature will be accreted back to debt through the maturity of the notes.
On September 11, 2013, we completed a private placement of convertible notes raising an aggregate of $321,000 in gross proceeds (the “September 2013 Notes”). The September 2013 Notes bear interest at a rate of five percent (5%) per annum, mature sixty (60) months after their date of issuance and are convertible into shares of our common stock at a conversion price of six cents ($0.06) per share (subject to adjustment as described in the September 2013 Notes) at any time prior to repayment, at the election of the investor. In the aggregate, the September 2013 Notes are initially convertible into up to 5,350,000 shares of our common stock.
| F-15 |
At any time prior to maturity of the September 2013 Notes, with the consent of the holders of a majority in interest of the September 2013 Notes, we may prepay the outstanding principal amount of the September 2013 Notes plus unpaid accrued interest without penalty. Upon the commission of any act of bankruptcy by the Company, the execution by the Company of a general assignment for the benefit of creditors, the filing by or against the Company of a petition in bankruptcy or any petition for relief under the federal bankruptcy act or the continuation of such petition without dismissal for a period of ninety (90) days or more, or the appointment of a receiver or trustee to take possession of the property or assets of the Company, the outstanding principal and all accrued interest on the September 2013 Notes will accelerate and automatically become immediately due and payable.
The investors in the offering included an affiliate and four directors of the Company. The principal amounts of the affiliate and directors respective September 2013 Notes are as set forth below:
| Investor | Note Principal | |||
| SINAF S.A. | $ | 150,000 | ||
| Joseph C. McNay | $ | 100,000 | ||
| Allan L. Goldstein | $ | 11,000 | ||
| L. Thompson Bowles | $ | 5,000 | ||
| R. Don Elsey | $ | 5,000 | ||
The Company has evaluated the terms of the September 2013 Notes which contain a down round provision under which the conversion price could be decreased as a result of future equity offerings, as defined in the September 2013 Notes. The adjustment would reduce the conversion price of the September 2013 Notes to be equivalent to that of the newly issued stock or stock-related instruments. As a result, the Company concluded that the conversion feature represented an embedded conversion feature for accounting purposes and should be recognized as a derivative liability, requiring a mark-to-market adjustment at the end of each reporting period until the related September 2013 Notes have been settled. The bifurcated liability of $267,500 was recorded on the date of issuance which resulted in a residual debt value of $53,500. The discount related to the embedded feature will be accreted back to debt through the maturity of the notes.
2014 Convertible Notes
On January 7, 2014, we completed a private placement of convertible notes raising an aggregate of $55,000 in gross proceeds (the “January 2014 Notes”). The January 2014 Notes will pay interest at a rate of 5% per annum, mature 60 months after their date of issuance and are convertible into shares of our common stock at a conversion price of $0.06 per share (subject to adjustment as described in the January 2014 Notes) at any time prior to repayment, at the election of the Investor. In the aggregate, the Notes are initially convertible into up to 916,667 shares of our common stock.
At any time prior to maturity of the January 2014 Notes, with the consent of the holders of a majority in interest of the January 2014 Notes, we may prepay the outstanding principal amount of the January 2014 Notes plus unpaid accrued interest without penalty. Upon the commission of any act of bankruptcy by the Company, the execution by the Company of a general assignment for the benefit of creditors, the filing by or against the Company of a petition in bankruptcy or any petition for relief under the federal bankruptcy act or the continuation of such petition without dismissal for a period of 90 days or more, or the appointment of a receiver or trustee to take possession of the property or assets of the Company, the outstanding principal and all accrued interest on the January 2014 Notes will accelerate and automatically become immediately due and payable.
The Investors in the offering included three directors of the Company. The principal amounts of their respective Notes are as set forth below:
| Investor | Note Principal | |||
| Joseph C. McNay | $ | 25,000 | ||
| Allan L. Goldstein | $ | 10,000 | ||
| L. Thompson Bowles | $ | 5,000 | ||
| F-16 |
The Company has evaluated the terms of the January 2014 Notes which contain a down round provision under which the conversion price could be decreased as a result of future equity offerings, as defined in the January 2014 Notes. The adjustment would reduce the conversion price of the January 2014 Notes to be equivalent to that of the newly issued stock or stock-related instruments. As a result, the Company concluded that the conversion feature represented an embedded conversion feature for accounting purposes and should be recognized as a derivative liability, requiring a mark-to-market adjustment at the end of each reporting period until the related January 2014 Notes have been settled. The bifurcated liability of $55,000 was recorded on the date of issuance which resulted in a residual debt value of $0. The discount related to the embedded feature will be accreted back to debt through the maturity of the notes.
The Company recorded interest expense and discount accretion as set forth below:
| For the three months ended | ||||||||
| March 31, 2016 | March 31, 2015 | |||||||
| 2012 Notes | $ | 3,738 | $ | 3,699 | ||||
| March 2013 Notes | 14,024 | 13,870 | ||||||
| July 2013 Notes | 4,571 | 4,521 | ||||||
| September 2013 Notes | 17,340 | 17,149 | ||||||
| January 2014 Notes | 3,428 | 3,390 | ||||||
| Total interest expense | $ | 43,101 | $ | 42,629 | ||||
The fair value of the derivative liability is as follows:
| March 31, 2016 | December 31, 2015 | |||||||
| March 2013 Notes | $ | 2,400,000 | $ | 1,500,000 | ||||
| July 2013 Notes | 1,083,333 | 666,667 | ||||||
| September 2013 Notes | 3,477,500 | 2,140,000 | ||||||
| January 2014 Notes | 595,837 | 366,669 | ||||||
| Total Fair value of derivative liability | $ | 7,556,670 | $ | 4,673,336 | ||||
| F-17 |
The change in fair value of derivative liability is as follows:
| For the three months ended | ||||||||
| March 31, 2016 | March 31, 2015 | |||||||
| March 2013 Notes | $ | 900,000 | $ | 412,500 | ||||
| July 2013 Notes | 416,666 | 183,333 | ||||||
| September 2013 Notes | 1,337,500 | 588,500 | ||||||
| January 2014 Notes | 229,168 | 100,833 | ||||||
| Total change in fair value of derivative | $ | 2,883,334 | $ | 1,285,166 | ||||
| 7. | License agreement |
Joint Venture Agreement - ReGenTree
On January 28, 2015, the Company entered into the Joint Venture Agreement with GtreeBNT, a shareholder in the Company. The Joint Venture Agreement provides for the creation of the Joint Venture, jointly owned by the Company and GtreeBNT, which will commercialize RGN-259 for treatment of dry eye and neurotrophic keratopathy in the United States.
GtreeBNT is solely responsible for funding all the product development and commercialization efforts of the Joint Venture. GtreeBNT made an initial contribution of $3 million in cash and received an initial equity stake of 51%. RegeneRx’s ownership interest in ReGenTree is 49% and will be reduced to 42% when the clinical study report is filed for the Phase 3 dry eye clinical trial. Based on when, and if, certain additional development milestones are achieved in the U.S. with RGN-259, our equity ownership may be incrementally reduced to between 42% and 25%, with 25% being the final equity ownership upon approval of an NDA for Dry Eye Syndrome in the U.S. In addition to our equity ownership, RegeneRx retains a royalty on net sales that varies between single and low double digits, depending on whether commercial sales are made by ReGenTree or a licensee. In the event the ReGenTree entity is acquired or there is a change of control that occurs following achievement of an NDA, RegeneRx shall be entitled to 40% of all change of control proceeds paid or payable and will forgo any future royalties. The Company is not required or otherwise obligated to provide financial support to the Joint Venture.
The Joint Venture is responsible for executing all development and commercialization activities under the License Agreement, which activities will be directed by a joint development committee comprised of representatives of the Company and GtreeBNT. The License Agreement has a term that extends to the later of the expiration of the last patent covered by the License Agreement or 25 years from the first commercial sale under the License Agreement. The License Agreement may be earlier terminated if the Joint Venture fails to meet certain commercialization milestones, if either party breaches the License Agreement and fails to cure such breach, as a result of government action that limits the ability of the Joint Venture to commercialize the product, as a result of a challenge to a licensed patent, following termination of the license between the Company and certain agencies of the United States federal government, or upon the bankruptcy of either party.
| F-18 |
Under the License Agreement, the Company received $1.0 million in up-front payments and is entitled to receive royalties on the Joint Venture’s future sales of products. On April 6, 2016 we received $250,000 from ReGenTree and executed an amendment to the license agreement on April 28, 2016. Under the amendment the territorial rights were expanded to include Canada. The Company is accounting for the License Agreement with the Joint Venture as a revenue arrangement. The Company has determined that the deliverables within the License Agreement, including a delivered element (providing the license) and an undelivered element (participation on the joint development committee), do not have stand-alone value and, as such, are treated as a single unit of accounting. As a result, the Company is recognizing the up-front milestone payments as revenue ratably over the anticipated life of the joint development committee, or 25 years. The joint development committee commenced activities as of April 1, 2015 therefore the Company has begun recognizing the revenue for the license fee. Revenue will be recognized for future royalty payments as they are earned.
| 8. | Stockholders’ Equity |
The Company did not issue any additional shares of stock in the period ended March 31, 2016.
| 9. | Commitments |
The office lease commitment expires in June 2017 with rental payments of approximately $4,500 per month.
| F-19 |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders
of
We have audited the accompanying balance sheets of RegeneRx Biopharmaceuticals, Inc. (the “Company”) as of December 31, 20092015 and 2008,2014, and the related statements of operations, changes in stockholders’ equity,deficit and cash flows for each of the years in the two-year period ended December 31, 2009. The Company’sthen ended. RegeneRx Biopharmaceuticals, Inc.’s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of RegeneRx Biopharmaceuticals, Inc. as of December 31, 20092015 and 2008,2014, and the results of its operations and its cash flows for each of the years in the two-year periodthen ended, December 31, 2009 in conformity with accounting principles generally accepted in the United StatesStated of America.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As more fully described in Note 1 to the financial statements, the Company has experienced negative cash flows from operations since inception and is dependent upon future financing in order to meet its planned operating activities. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans regarding these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ REZNICK GROUP, P.C.CohnReznick LLP
Tysons, Virginia
April 8, 2016
| F-20 |
F-2
| December 31, | December 31, | |||||||
| 2009 | 2008 | |||||||
ASSETS | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | $ | 4,355,768 | $ | 5,655,367 | ||||
| Prepaid expenses and other current assets | 196,546 | 236,477 | ||||||
| Total current assets | 4,552,314 | 5,891,844 | ||||||
| Property and equipment, net of accumulated depreciation of $98,171 and $81,623 | 8,492 | 25,039 | ||||||
| Other assets | 22,948 | 5,693 | ||||||
| Total assets | $ | 4,583,754 | $ | 5,922,576 | ||||
LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities | ||||||||
| Accounts payable | $ | 140,206 | $ | 70,554 | ||||
| Accrued expenses | 740,198 | 1,255,358 | ||||||
| Total current liabilities | 880,404 | 1,325,912 | ||||||
| Commitments | — | — | ||||||
| Stockholders’ equity | ||||||||
| Preferred stock, $.001 par value per share, 1,000,000 shares authorized; no shares issued | — | — | ||||||
| Common stock, $.001 par value per share, 100,000,000 shares authorized; 60,406,828 and 53,622,491 issued and outstanding | 60,407 | 53,623 | ||||||
| Additional paid-in capital | 88,144,347 | 82,550,585 | ||||||
| Accumulated deficit | (84,501,404 | ) | (78,007,544 | ) | ||||
| Total stockholders’ equity | 3,703,350 | 4,596,664 | ||||||
| Total liabilities and stockholders’ equity | $ | 4,583,754 | $ | 5,922,576 | ||||
| December 31, | ||||||||
| 2015 | 2014 | |||||||
| ASSETS | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | $ | 317,627 | $ | 844,043 | ||||
| Prepaid expenses and other current assets | 24,300 | 86,525 | ||||||
| Total current assets | 341,927 | 930,568 | ||||||
| Property and equipment, net of accumulated depreciation of $88,794 and $85,392 | 10,544 | 12,871 | ||||||
| Other assets | 5,752 | 5,752 | ||||||
| Total assets | $ | 358,223 | $ | 949,191 | ||||
| LIABILITIES AND STOCKHOLDERS' DEFICIT | ||||||||
| Current liabilities | ||||||||
| Accounts payable | $ | 141,130 | $ | 232,610 | ||||
| Accrued expenses | 217,911 | 177,009 | ||||||
| Total current liabilities | 359,041 | 409,619 | ||||||
| Long-Term liabilities | ||||||||
| Unearned revenue | 1,379,388 | 400,000 | ||||||
| Convertible promisory note | 300,000 | 300,000 | ||||||
| Convertible promisory notes, net of derivative liability | 388,854 | 266,021 | ||||||
| Fair value of derivative liability | 4,673,336 | 1,285,170 | ||||||
| Total liabilities | 7,100,619 | 2,660,810 | ||||||
| Commitments and contingencies | - | - | ||||||
| Stockholders' deficit | ||||||||
| Preferred stock, $.001 par value per share, 1,000,000 shares authorized; no shares issued | - | - | ||||||
| Common stock, par value $.001 per share, 200,000,000 shares authorized, 101,640,092 and 101,316,580 issued and outstanding | 101,640 | 101,317 | ||||||
| Additional paid-in capital | 98,230,802 | 97,991,419 | ||||||
| Accumulated deficit | (105,074,838 | ) | (99,804,355 | ) | ||||
| Total stockholders' deficit | (6,742,396 | ) | (1,711,619 | ) | ||||
| Total liabilities and stockholders' deficit | $ | 358,223 | $ | 949,191 | ||||
The accompanying notes are an integral part of these financial statements.
| F-21 |
F-3
| Years Ended December 31, | ||||||||
| 2009 | 2008 | |||||||
| Sponsored research revenue | $ | — | $ | 168,412 | ||||
| Operating expenses | ||||||||
| Research and development | 3,724,514 | 7,149,808 | ||||||
| General and administrative | 2,781,790 | 3,805,346 | ||||||
| Total operating expenses | 6,506,304 | 10,955,154 | ||||||
| Loss from operations | (6,506,304 | ) | (10,786,742 | ) | ||||
| Interest income | 12,444 | 149,777 | ||||||
| Net loss | $ | (6,493,860 | ) | $ | (10,636,965 | ) | ||
| Basic and diluted net loss per common share | $ | (0.12 | ) | $ | (0.21 | ) | ||
| Weighted average number of common shares outstanding | 55,680,525 | 50,967,617 | ||||||
| Years ended December 31, | ||||||||
| 2015 | 2014 | |||||||
| Revenues | $ | 60,612 | $ | - | ||||
| Operating expenses | ||||||||
| Research and development | 203,185 | 367,275 | ||||||
| General and administrative | 1,566,962 | 1,188,211 | ||||||
| Total operating expenses | 1,770,147 | 1,555,486 | ||||||
| Loss from operations | (1,709,535 | ) | (1,555,486 | ) | ||||
| Interest and other income | 101 | 153 | ||||||
| Interest expense | (172,883 | ) | (183,473 | ) | ||||
| Change in fair value of derivative liability | (3,388,166 | ) | (1,014,836 | ) | ||||
| Net loss | $ | (5,270,483 | ) | $ | (2,753,642 | ) | ||
| Basic and diluted net loss per common share | $ | (0.05 | ) | $ | (0.03 | ) | ||
| Weighted average number of common shares outstanding | 101,527,676 | 93,186,443 | ||||||
The accompanying notes are an integral part of these financial statements.
| F-22 |
F-4
Years ended December 31, 20092015 and 2008
| Accumulated | ||||||||||||||||||||||||
| Other | Total | |||||||||||||||||||||||
| Common Stock | Additional | Accumulated | Comprehensive | Stockholders’ | ||||||||||||||||||||
| Shares | Amount | Paid-in Capital | Deficit | Income/(loss) | Equity | |||||||||||||||||||
| Balance, December 31, 2007 | 46,553,527 | $ | 46,554 | $ | 73,513,292 | $ | (67,405,579 | ) | $ | (1,543 | ) | $ | 6,152,724 | |||||||||||
| Cumulative effect of a change in accounting principle — ASC Topic 730 | — | — | — | 35,000 | — | 35,000 | ||||||||||||||||||
| Issuance of common stock, net of offering costs of $52,240 | 7,068,964 | 7,069 | 7,940,691 | — | — | 7,947,760 | ||||||||||||||||||
| Share-based compensation expense | — | — | 1,096,602 | — | — | 1,096,602 | ||||||||||||||||||
| Net loss | — | — | — | (10,636,965 | ) | — | (10,636,965 | ) | ||||||||||||||||
| Unrealized gain on available for sale securities | — | — | — | — | 1,543 | 1,543 | ||||||||||||||||||
| Total comprehensive loss | (10,635,422 | ) | ||||||||||||||||||||||
| Balance, December 31, 2008 | 53,622,491 | 53,623 | 82,550,585 | (78,007,544 | ) | $ | — | $ | 4,596,664 | |||||||||||||||
| Issuance of common stock, net of offering costs of $447,933 | 6,784,337 | 6,784 | 4,845,282 | — | — | 4,852,066 | ||||||||||||||||||
| Share-based compensation expense | — | — | 748,480 | — | — | 748,480 | ||||||||||||||||||
| Net loss | — | — | — | (6,493,860 | ) | — | (6,493,860 | ) | ||||||||||||||||
| Balance, December 31, 2009 | 60,406,828 | $ | 60,407 | $ | 88,144,347 | $ | (84,501,404 | ) | $ | — | $ | 3,703,350 | ||||||||||||
| Total | ||||||||||||||||||||
| Common stock | Additional | Accumulated | stockholders' | |||||||||||||||||
| Shares | Amount | paid-in capital | deficit | deficit | ||||||||||||||||
| Balance, December 31, 2013 | 81,733,247 | $ | 81,733 | $ | 95,347,571 | $ | (97,050,713 | ) | $ | (1,621,409 | ) | |||||||||
| Issuance of common stock to G-treeBNT | 19,583,333 | 19,584 | 2,480,416 | - | 2,500,000 | |||||||||||||||
| Share-based compensation expense | - | - | 163,432 | - | 163,432 | |||||||||||||||
| Net loss | - | - | - | (2,753,642 | ) | (2,753,642 | ) | |||||||||||||
| Balance, December 31, 2014 | 101,316,580 | $ | 101,317 | $ | 97,991,419 | $ | (99,804,355 | ) | $ | (1,711,619 | ) | |||||||||
| Issuance of common stock & warrants | 323,512 | 323 | 7,776 | - | 8,099 | |||||||||||||||
| Share-based compensation expense | - | - | 231,607 | - | 231,607 | |||||||||||||||
| Net loss | - | - | - | (5,270,483 | ) | (5,270,483 | ) | |||||||||||||
| Balance, December 31, 2015 | 101,640,092 | $ | 101,640 | $ | 98,230,802 | $ | (105,074,838 | ) | $ | (6,742,396 | ) | |||||||||
The accompanying notes are an integral part of these financial statements.
| F-23 |
F-5
| Years Ended December 31, | ||||||||
| 2009 | 2008 | |||||||
| Operating activities: | ||||||||
| Net loss | $ | (6,493,860 | ) | $ | (10,636,965 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | 16,547 | 19,396 | ||||||
| Non-cash share-based compensation | 748,480 | 1,096,602 | ||||||
| Gain on settlement of accrued expenses | (100,000 | ) | — | |||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable | — | 26,951 | ||||||
| Prepaid expenses and other current assets | 39,931 | 66,767 | ||||||
| Other assets | (17,255 | ) | — | |||||
| Accounts payable | 69,652 | (203,007 | ) | |||||
| Accrued expenses | (415,160 | ) | (940,150 | ) | ||||
| Net cash used in operating activities | (6,151,665 | ) | (10,570,406 | ) | ||||
| Investing activities: | ||||||||
| Sales/maturities of short-term investments | — | 4,581,135 | ||||||
| Net cash provided by investing activities | — | 4,581,135 | ||||||
| Financing activities: | ||||||||
| Net proceeds from issuance of common stock | 4,852,066 | 7,947,760 | ||||||
| Net cash provided by financing activities | 4,852,066 | 7,947,760 | ||||||
| Net (decrease) increase in cash and cash equivalents | (1,299,599 | ) | 1,958,489 | |||||
| Cash and cash equivalents at beginning of year | 5,655,367 | 3,696,878 | ||||||
| Cash and cash equivalents at end of year | $ | 4,355,768 | $ | 5,655,367 | ||||
| Years ended December 31, | ||||||||
| 2015 | 2014 | |||||||
| Operating activities: | ||||||||
| Net loss | $ | (5,270,483 | ) | $ | (2,753,642 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | 3,403 | 4,823 | ||||||
| Non-cash share-based compensation | 231,607 | 163,432 | ||||||
| Non-cash interest expense | 122,833 | 133,476 | ||||||
| Non-cash expense - issuance of stock for services | 8,100 | - | ||||||
| Change in fair value of derivative liability | 3,388,166 | 1,014,836 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | 62,225 | (60,226 | ) | |||||
| Accounts payable | (91,481 | ) | (344,602 | ) | ||||
| Accrued expenses | 40,902 | 137,960 | ||||||
| Unearned revenue | 979,388 | - | ||||||
| Net cash used in operating activities | (525,340 | ) | (1,703,943 | ) | ||||
| Investing activities: | ||||||||
| Purchase of property and equipment | (1,076 | ) | (13,320 | ) | ||||
| Net cash used in investing activities | (1,076 | ) | (13,320 | ) | ||||
| Financing activities: | ||||||||
| Proceeds from sale of common stock and issuance of warrants | - | 2,500,000 | ||||||
| Net proceeds from issuance of debt | - | 55,000 | ||||||
| Net cash provided by financing activities | - | 2,555,000 | ||||||
| Net increase (decrease) in cash and cash equivalents | (526,416 | ) | 837,737 | |||||
| Cash and cash equivalents at beginning of year | 844,043 | 6,306 | ||||||
| Cash and cash equivalents at end of year | $ | 317,627 | $ | 844,043 | ||||
The accompanying notes are an integral part of these financial statements.
| F-24 |
F-6
December 31, 2015
| 1. | ORGANIZATION AND BUSINESS |
Organization and Nature of Operations.
RegeneRx Biopharmaceuticals, Inc. (the(“RegeneRx”, the “Company”, “We”, “Us”, “Our”), a Delaware corporation, was incorporated in 1982. We are focused on the discovery and development of novel molecules to accelerate tissue and organ repair. Our operations are confined to one business segment: the development and marketing of product candidates based on Thymosin Beta 4 (“Tß4”), an amino acid peptide.
Management Plans to Address Operating Conditions.
On January 28, 2015, we announced that we had entered into a Joint Venture Agreement (the “Joint Venture Agreement”) with GtreeBNT Co., Ltd., a Korean pharma company (“GtreeBNT”) and shareholder of the Company. The Joint Venture Agreement provides for the creation of an entity, ReGenTree, LLC (the “Joint Venture” or “ReGenTree”), jointly owned by us and GtreeBNT, that will commercialize RGN-259 for treatment of dry eye and neurotrophic keratopathy, an orphan indication in the United States. GtreeBNT will be responsible for funding all product development and commercialization efforts, and holds a majority interest of ReGenTree that varies depending on development milestones achieved and eventual commercialization path, if successful. In March 2015, GtreeBNT reported that it received $7.28 million to expand international development of the product candidate, RGN-259 (designated GBT-201 in Korea). The $7.28 million will be used for development of RGN-259/GBT-201 for dry eye syndrome and neurotrophic keratopathy in the U.S. through the U.S. joint venture, ReGenTree, LLC.
RegeneRx’s ownership interest in ReGenTree is 49% and will be reduced to 42% when the clinical study report is filed for the Phase 3 dry eye clinical trial. Based on when, and if, certain additional development milestones are achieved in the U.S. with RGN-259, our equity ownership may be incrementally reduced to between 42% and 25%, with 25% being the final equity ownership upon approval of an NDA for Dry Eye Syndrome in the U.S. In the event the ReGenTree entity is acquired or there is a change of control that occurs following achievement of an NDA, RegeneRx shall be entitled to 40% of all change of control proceeds paid or payable and will forgo any future royalties.
In conjunction with the Joint Venture Agreement, we also entered into a royalty-bearing license agreement (the “License Agreement”) with ReGenTree pursuant to which we granted to ReGenTree the right to develop and exclusively commercialize RGN-259 in the United States. We received a total of $1 million in two tranches under the terms of the License Agreement. The first tranche of $500,000 was received in March 2015 and a second in the amount of $500,000 was received in September 2015. We are also entitled to royalties as a percentage of net sales ranging from the single digits to the low-double digits based on the medical indications approved and whether the Joint Venture commercializes products directly or through a third party. RegeneRx possesses one of three board seats and certain major decisions and transactions within ReGenTree, such as commercialization strategy, mergers, and acquisitions, require RegeneRx’s board designee’s consent.
In September 2015, ReGenTree began a Phase 2b/3 clinical trial in patients with dry eye syndrome (“DES”) and a Phase 3 clinical trial in patients with neurotrophic keratopathy (“NK”), both in the U.S. In January 2016, the DES had completed enrollment of all patients in the trial. The last patient received the last treatment in February. We expect to report top line data from the DES trial at the end of April 2016. The NK trial, a smaller study in an orphan population, has enrolled seven patients thus far with a goal of 46. Of the eight original clinical sites for the study, six are enrolling patients, one has yet to receive IRB approval to begin enrolling patients, and one was unable to consummate a clinical contract with ReGenTree. ReGenTree is considering adding additional sites to accelerate patient enrollment.
Currently, we have active partnerships in three major territories: the U.S., China and Pan Asia. Our partners have been moving forward and making progress in each territory. In each case, the cost of development is being borne by our partners with no financial obligation for RegeneRx. We received $250,000 pursuant to the signing of a term sheet with an affiliated entity in April 2016, with this amount coupled with our current cash, we believe we should be able to maintain our existing operations at the current level into the third quarter of 2016.
| F-25 |
Lee's Pharmaceutical Ltd. (“Lee’s”), RegeneRx's licensee in China, Hong Kong, Macau and Taiwan, recently received notice from China's FDA (“CFDA”) declining its investigational new drug (“IND”) application for a Phase 2b dry eye clinical trial because the API (active pharmaceutical ingredient or Tß4Tß4) was manufactured outside of China. The API was manufactured in the U.S. and provided to Lee's by RegeneRx pursuant to a license agreement to develop RGN-259 ophthalmic eye drops in the licensed territory. Due to this unexpected regulatory hurdle, Lee's plans to modify its clinical program to conduct the Phase 2b dry eye trial in Hong Kong and Taiwan using the Tß4 supplied by RegeneRx while awaiting the manufacturing of Tß4 in China for a subsequent Phase 3 registration trial. We do not know when the Hong Kong/Taiwan trial will begin enrollment of patients. Under this revised strategy, Lee's believes it should be able to begin a Phase 3 registration trial in China sooner, rather than waiting on the production of Tß4 in China before initiating a Phase 2b trial.
GtreeBNT, RegeneRx's licensee in Korea, Australia, Japan and a number of other countries in Asia. GtreeBNT filed an IND with the Korean Ministry of Food and Drug Safety to conduct a Phase 2b/3 study with RGN-259 in patients with dry eye syndrome and in July 2015 received approval to conduct the trial. GtreeBNT has informed us that given its immediate focus on the two U.S. trials, it is considering the best timing for the Korean trial.
We still have significant clinical assets to develop, primarily RGN-352 (injectable formulation of Tß4 for cardiac and CNS disorders) in the U.S., Pan Asia, and Europe, and RGN-259 in the EU. Our goal is to wait until the results are obtained from the current ophthalmic clinical trials before moving into the EU with RGN-259. If successful, this should allow us to obtain a higher value for the asset at that time. However, we intend to continue to develop RGN-352, either by obtaining grants to fund a Phase 2a clinical trial in the cardiovascular or central nervous system fields or finding a suitable partner with the resources and capabilities to develop it as we have with RGN-259.
We have incurred net losses of $6.5 million$5,270,000 and $10.6 million$2,754,000 for the years ended December 31, 20092015 and 2008,2014, respectively. Since inception, and through December 31, 2009,2015, we have an accumulated deficit of $84.5$105 million and we had cash and cash equivalents of $4.4 million$318,000 as of December 31, 2009. Based on our operating plan, we believe that our cash and cash equivalents will fund our operations into the third quarter of 2010.
These factors raise substantial doubt about our ability to continue as a going concern. The accompanying financial statements have been prepared assuming that we will continue as a going concern. This basis of accounting contemplates the recovery of our assets and the satisfaction of our liabilities in the normal course of business.
Although we intend to continue to seek additional financing or aadditional strategic partner,partners, we may not be able to complete a financing or corporate transaction, either on favorable terms or at all. If we are unable to complete a financing or strategic transaction, we may not be able to continue as a going concern after our funds have been exhausted, and we could be required to significantly curtail or cease operations, file for bankruptcy or liquidate and dissolve. There can be no assurance that we will be able to obtain any sources of funding. The financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts and classification of liabilities that might be necessary should we be forced to take any such actions.
In addition to our current operational requirements, we expect to continue to expend substantial funds to complete our planned product development efforts. Additionally, we continually refine our operating strategy and evaluate alternative clinical uses of Tß4. However, substantial additional resources will be needed before we will be able to achieve sustained profitability. Consequently, we continually evaluate alternative sources of financing such as the sharing of development costs through strategic collaboration agreements. There can be no assurance that our financing efforts will be successful and, if we are not able to obtain sufficient levels of financing, we would delay certain clinicaland/or research activities and our financial condition would be materially and adversely affected. Even if we are able to obtain sufficient funding, other factors including competition, dependence on third parties, uncertainty regarding patents, protection of proprietary rights, manufacturing of peptides, and technology obsolescence could have a significant impact on us and our operations.
To achieve profitability we, and/or a partner, must successfully conduct pre-clinical studies and clinical trials, obtain required regulatory approvals and successfully manufacture and market those pharmaceuticals we wish to commercialize. The time required to reach profitability is highly uncertain, and there can be no assurance that we will be able to achieve sustained profitability, if at all.
F-7
| 2. | SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
Use of Estimates. The preparation of financial statements in conformity with accounting principles generally accepted in the United Stated of America (“U.S. GAAP”) requires management to make certain estimates and assumptions that affect the reported earnings, financial position and various disclosures. Critical accounting policies involved in applying our accounting policies are those that require management to make assumptions about matters that are highly uncertain at the time the accounting estimate was made and those for which different estimates reasonably could have been used for the current period. Critical accounting estimates are also those which are reasonably likely to change from period to period, and would have a material impact on the presentation of our financial condition, changes in financial condition or results of operations. Our most critical accounting estimates relate to accounting policies for revenue recognition, clinical trial accruals and share-based arrangements. Management bases its estimates on historical experience and on various other assumptions that it believes are reasonable under the circumstances. Actual results could differ from these estimates.
| F-26 |
Cash and Cash Equivalents. Cash and cash equivalents consist of cash and highly-liquid investments with original maturities of three months or less when acquired and are stated at cost that approximates their fair market value.
Concentration of Credit Risk. Financial instruments, which potentially subject the Company to concentrations of credit risk, consist primarily of cash and cash equivalents. We limit our exposure to credit loss by placing our cash and cash equivalents with high quality financial institutions and, in accordance with our investment policy, in securities that are rated investment grade.
Property and Equipment. Property and equipment consists of office furniture and equipment, and is stated at cost and depreciated over the estimated useful lives of the assets (generally two to five years) using the straight-line method. Expenditures for maintenance and repairs which do not significantly prolong the useful lives of the assets are charged to expense as incurred. Depreciation expense was $16,547$3,403 and $19,396$4,823 for the years ended December 31, 20092015 and 2008,2014, respectively.
Impairment of Long-lived Assets. When we record long-lived assets our policy is to regularly perform reviews to determine if and when the carrying value of our long-lived assets becomes impaired. During the two years ended December 31, 2009 we did not report qualifying long-lived assets2015 and therefore2014 no impairment losses were recorded.
Sponsored Research Revenues.
Convertible Notes with Detachable Warrants. We account for non-refundable grantsIn accordance with Accounting Standards Codification (ASC) 470-20,Debt with Conversion and Other Options, the proceeds received from convertible notes are allocated between the convertible notes and the detachable warrants based on the relative fair value of the convertible notes without the warrants and the warrants. The portion of the proceeds allocated to the warrants is recognized as “Sponsored research revenues”additional paid-in capital and a debt discount. The debt discount related to warrants is accreted into interest expense through maturity of the notes.
Derivative Financial Instruments.Derivative financial instruments consist of financial instruments or other contracts that contain a notional amount and one or more underlying variables (e.g. interest rate, security price or other variable), which require no initial net investment and permit net settlement. Derivative financial instruments may be free-standing or embedded in other financial instruments. Further, derivative financial instruments are initially, and subsequently, measured at fair value and recorded as liabilities or, in rare instances, assets.
The Company does not use derivative financial instruments to hedge exposures to cash-flow, market or foreign-currency risks. However, the Company has issued financial instruments including warrants that are either (i) not afforded equity classification, (ii) embody risks not clearly and closely related to host contracts, or (iii) may be net-cash settled by the counterparty. In certain instances, these instruments are required to be carried as derivative liabilities, at fair value, in the accompanying statementsCompany’s financial statements.
The Company estimates the fair values of operations. Revenuesits derivative financial instrument using the Black-Scholes option pricing model because it embodies all of the requisite assumptions (including trading volatility, estimated terms and risk free rates) necessary to fair value these instruments. Estimating fair values of derivative financial instruments requires the development of significant and subjective estimates that may, and are likely to, change over the duration of the instrument with related changes in internal and external market factors. In addition, option-based techniques are highly volatile and sensitive to changes in the trading market price of the Company’s common stock, which has a high-historical volatility. Since derivative financial instruments are initially and subsequently carried at fair values, the Company’s operating results reflect the volatility in these estimate and assumption changes in each reporting period.
Revenue Recognition.We recognize revenue in accordance with the authoritative guidance for revenue recognition. We recognize revenue when all of the following criteria are met: (i) persuasive evidence of an arrangement exists, (ii) delivery (or passage of title) has occurred or services have been rendered, (iii) the seller's price to the buyer is fixed or determinable, and (iv) collectability is reasonably assured. We also comply with the authoritative guidance for revenue recognition regarding arrangements with multiple deliverables. Multiple-element arrangements are analyzed to determine whether the deliverables, which may include a license together with performance obligations such as providing a clinical supply of product and steering committee services, can be separated or whether they must be accounted for as a single unit of accounting. Revenue associated with licensing agreements consists of non-refundable upfront license fees and milestone payments. Non-refundable upfront license fees received under license agreements, whereby continued performance or future obligations are considered inconsequential to the relevant license technology, are recognized as revenue upon delivery of the technology.
| F-27 |
Whenever we determine that an arrangement should be accounted for as a single unit of accounting, we must determine the period over which the performance obligations will be performed and revenue will be recognized. Revenue will be recognized using either a relative performance or straight-line method. We recognize revenue using the relative performance method provided that the we can reasonably estimate the level of effort required to complete our performance obligations under an arrangement and such performance obligations are provided on a best-efforts basis. Revenue recognized is limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the relative performance method, as of each reporting period.
If we cannot reasonably estimate the level of effort required to complete our performance obligations under an arrangement, the performance obligations are provided on a best-efforts basis and we can reasonably estimate when the associated researchperformance obligation ceases or the remaining obligations become inconsequential and perfunctory, then the total payments under the arrangement, excluding royalties and payments contingent upon achievement of substantive milestones, would be recognized as revenue on a straight-line basis over the period we expect to complete our performance obligations. Revenue is limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the straight-line basis, as of the period ending date.
If we cannot reasonably estimate when our performance obligation either ceases or becomes inconsequential and perfunctory, then revenue is deferred until we can reasonably estimate when the performance obligation ceases or becomes inconsequential. Revenue is then recognized over the remaining estimated period of performance.
We recognize consideration that is contingent upon the achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone is substantive in its entirety. A milestone is considered substantive when it meets all of the following criteria:
| · | The consideration is commensurate with either the entity's performance to achieve the milestone or the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity's performance to achieve the milestone; |
| · | The consideration relates solely to past performance; and |
| · | The consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. |
A milestone is defined as an event (i) that can only be achieved based in whole or in part on either the entity's performance or on the occurrence of a specific outcome resulting from the entity's performance, (ii) for which there is substantive uncertainty at the date the arrangement is entered into that the event will be achieved and (iii) that would result in additional payments being due to us.
Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue in our accompanying condensed balance sheets.
Variable Interest Entities
The Company has been performeddetermined that the Joint Venture is a “variable interest entity”, since the total equity investment at risk is not sufficient to permit the Joint Venture to finance its activities without additional subordinated financial support. Further, because of GtreeBNT’s majority equity stake in the Joint Venture, voting control, control of the board of directors, and substantive management rights, and given that the related underlying costs are incurred.Company does not have the power to direct the Joint Venture’s activities that most significantly impact its economic performance, the Company determined that it is not the primary beneficiary of the Joint Venture and therefore is not required to consolidate the Joint Venture. The Company reports its equity stake in the Joint Venture using the equity method of accounting because, while it does not control the Joint Venture, the Company can exert significant influence over the Joint Ventures activities by virtue of its board representation.
Because the Company is not obligated to fund the Joint Venture, and has not provided any financial support and has no commitment to provide financial support in the future to the Joint Venture, the carrying value of its investment in the Joint Venture is zero. As a result, the Company is not recognizing its share (49%) of the Joint Venture’s operating losses and will not recognize any such losses until the Joint Venture produces net income (as opposed to net losses) and at that point the Company will reduce its share of the Joint Venture’s net income by its share of previously suspended net losses. As of December 31, 2015, because it has not provided any financial support, the Company has no financial exposure as a result of its variable interest in the Joint Venture.
| F-28 |
Research and Development.Development. Research and development (“R&D”) costs are expensed as incurred and include all of the wholly-allocable costs associated with our various clinical programs passed through to us by our outsourced vendors. Those costs include: manufacturing TßTb4; formulation of TßTb4 into the various product candidates; stability for both TßTb4 and the various formulations; pre-clinical toxicology; safety and pharmacokinetic studies; clinical trial management; medical oversight; laboratory evaluations; statistical data analysis; regulatory compliance; quality assurance; and other related activities. R&D includes cash and non-cash compensation, employee benefits, travel and other miscellaneous costs of our internal R&D personnel, sevenfour persons in total, who are wholly dedicated to R&D efforts. R&D also includes a pro-ration of our common infrastructure costs for office space and communications.
F-8
Patent Costs. Costs related to filing and pursuing patent applications are recognized as general and administrative expenses as incurred since recoverability of such expenditures is uncertain.
Income Taxes. Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. We recognize the effect of income tax positions only if those positions are more likely than not of being sustained. Recognized income tax positions are measured at the largest amount that is greater than 50% likely of being realized. Changes in recognition or measurement are reflected in the period in which the change in judgment occurs. The Company’sOur policy for recording interest and penalties associated with audits is that penalties and interest expense are recorded in “Income taxes” in the Company’sour statements of operations.
The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. Management considers the scheduled reversal of deferred tax liabilities, projected future taxable income, and tax planning strategies in making that assessment. We recorded a full valuation allowance against all estimated net deferred tax assets at December 31, 20092015 and 2008.2014. We have significant net operating loss carryforwards to potentially reduce future federal and state taxable income, and research and experimentation tax credit carryforwards available to potentially offset future federal and state income taxes. Use of our net operating loss and research and experimentation credit carryforwards may be limited due to changes in our ownership as defined within Section 382 of the Internal Revenue Code.
Net Loss Per Common Share. Net loss per common share for the years ended December 31, 20092015 and 2008,2014, respectively, is based on the weighted-average number of shares of common stock outstanding during the periods. Basic and diluted loss per share are identical for all periods presented as potentially dilutive securities have been excluded from the calculation of the diluted net loss per common share because the inclusion of such securities would be antidilutive. The potentially dilutive securities include 12,847,96322,621,951 shares and 9,366,59034,050,093 shares in 20092015 and 2008,2014, respectively, reserved for the exercise of outstanding options, warrants and warrants.
F-9
Fair Value of Financial Instruments. The carrying amounts of our financial instruments, as reflected in the accompanying balance sheets, approximate fair value. Financial instruments consist of cash and cash equivalents, accounts payable, and accounts payable.
Recent Accounting Pronouncements.
In February 2010,May 2014, the Financial Accounting Standards Board (“FASB”)FASB issued Accounting Standards Update (ASU 2010 — 09)ASU 2014-09, Revenue from Contracts with Customers, which provides guidance for revenue recognition for contracts, superseding the previous revenue recognition requirements, along with most existing industry-specific guidance. The guidance requires an entity to address potential practice issues associatedreview contracts in five steps: 1) identify the contract, 2) identify performance obligations, 3) determine the transaction price, 4) allocate the transaction price, and 5) recognize revenue. The new standard will result in enhanced disclosures regarding the nature, amount, timing and uncertainty of revenue arising from contracts with customers. The standard is effective for the Company’s reporting year beginning December 15, 2017 and early adoption is not permitted. The Company is currently evaluating the impact, if any, that this new accounting pronouncement will have on its financial statements.
| F-29 |
In April 2015, the FASB ASC 855 (formerly SFAS 165)issued ASU 2015-03,Interest – Imputation of Interest, “Subsequent Events.”which amends the presentation of debt issuance costs. These costs will now be presented as a direct reduction from the carrying amount of that debt liability. The ASU wasupdate is effective upon issuance and eliminated the requirement for entities that file or furnish financial statements issued for reporting periods beginning after December 15, 2015. This guidance should be applied on a retrospective basis with disclosures for a change in accounting principle applicable. The Company has not yet adopted this update and is currently evaluating the SEC to discloseimpact, if any, it may have on its financial condition and results of operations.
In November 2015, the date through which subsequent events have been evaluated in originallyFASB issued new guidance on the balance sheet classification of deferred taxes. To simplify presentation, the new guidance requires that all deferred tax assets and reissued financial statements. Otherliabilities, along with any related valuation allowance, be classified as noncurrent on the balance sheet. The accounting standard is effective for public business entities would continue to be required to disclose the date through which subsequent events have been evaluated; however, disclosures about the date would be required only in financial statements revised because of an error correction or retrospective application of U.S. GAAP. Ourfor annual reporting periods (including interim reporting periods within those periods) beginning after December 15, 2016. Early adoption is permitted. The adoption of this standard changed our presentation of subsequent events when preparing our financial statements.
In August 2009,January 2016, the FASB issued ASUNo. 2009-05, “Fair Value Measurementsa new accounting standard on recognition and Disclosures (ASC 820) — Measuring Liabilities at Fair Value” (ASU2009-05). ASU2009-05 providesmeasurement of financial assets and financial liabilities. The accounting standard primarily affects the accounting for equity investments, financial liabilities under the fair value option, and the presentation and disclosure requirements for financial instruments. In addition, it includes a clarification related to the valuation allowance assessment when recognizing deferred tax assets resulting from unrealized losses on available-for-sale debt securities. The accounting guidance is effective for annual reporting periods (including interim periods within those periods) beginning after December 15, 2017. Early adoption is permitted for the provision to record fair value changes for financial liabilities under the fair value option resulting from instrument-specific credit risk in other comprehensive income. The Company is currently evaluating the impact, if any, that the pronouncement will have on the financial statements.
In February 2016, the FASB issued ASU 2016-02,Leases, which supersedes ASC Topic 840,Leases, and creates a new topic, ASC Topic 842,Leases. ASU 2016-02 requires lessees to recognize a lease liability and a lease asset for all leases, including operating leases, with a term greater than 12 months on its balance sheet. ASU 2016-02 also expands the required quantitative and qualitative disclosures surrounding leases. ASU 2016-02 is effective for the Company beginningJanuary 1, 2019. Early adoption is permitted. The Company has determined that the adoption of ASU 2016-02 will currently have no impact on its financial statements.
In March 2016, the FASB issued ASU 2016-07,Equity Method and Joint Ventures affect all entities that have an investment that becomes qualified for the equity method of accounting as a result of an increase in circumstancesthe level of ownership or degree of influence. ASU 2016-07 is effective for the Company beginning onJanuary 1, 2017, early adoption is permitted. The Company is currently evaluating the effect this ASU will have on the consolidated financial statements.
| 3. | FAIR VALUE MEASUREMENTS |
The authoritative guidance for fair value measurements defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in which a quoted price in an activethe principal or the most advantageous market for the identicalasset or liability is not available,in an orderly transaction between market participants on the measurement date. Market participants are buyers and sellers in the principal market that are (i) independent, (ii) knowledgeable, (iii) able to transact, and (iv) willing to transact. The guidance describes a reporting entity is requiredfair value hierarchy based on the levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value using a valuation technique that useswhich are the quoted price of thefollowing:
F-10
At December 31, 20092015 and 2008, we held no qualifying liabilities, and2014, our only qualifying assets that required measurement under the foregoing fair value hierarchy were money market funds and U.S. Treasury Bills included in Cash and Cash Equivalents valued at $4.4 million$318,000 and $5.7 million,$844,000, respectively, which were valued using Level 1 inputs. Our December 31, 2015 balance sheet reflects qualifying liabilities resulting from the price protection provision in the convertible promissory notes issued in March, July and September of 2013 and January 2014 (see Note 7). We evaluated the derivative liability embedded in the series of convertible notes to determine if an adjustment to the carrying value of the liability was required at December 31, 2015 using the following assumptions.
| F-30 |
| March 2013 | July 2013 | Sept 2013 | Jan 2014 | |||||||||||||
| Dividend yield | 0.00 | % | 0.00 | % | 0.00 | % | 0.00 | % | ||||||||
| Risk-free rate of return | 0.92 | % | 0.92 | % | 0.92 | % | 0.92 | % | ||||||||
| Expected life in years | 2.25 | 2.5 | 2.7 | 3 | ||||||||||||
| Volatility | 119.5 | % | 114.0 | % | 110.7 | % | 104.9 | % | ||||||||
Given the conditions surrounding the trading of the Company’s equity securities, the Company values its derivative instruments related to embedded conversion features from the issuance of convertible debentures in accordance with the Level 3 guidelines. For the year ended December 31, 2015, the following table reconciles the beginning and ending balances for financial instruments that are recognized at fair value in these financial statements.
| Balance at | Balance at | |||||||||||||||
| December 31, | New | Change in | December 31, | |||||||||||||
| 2014 | Issuances | Fair Values | 2015 | |||||||||||||
| Level 3 - | ||||||||||||||||
| Derivative liabilities from: | ||||||||||||||||
| Conversion features | ||||||||||||||||
| March 2013 | $ | 412,500 | $ | - | $ | 1,087,500 | $ | 1,500,000 | ||||||||
| July 2013 | 183,334 | - | 483,333 | 666,667 | ||||||||||||
| September 2013 | 588,500 | - | 1,551,500 | 2,140,000 | ||||||||||||
| January 2014 | 100,836 | - | 265,833 | 366,669 | ||||||||||||
| Derivative instruments | $ | 1,285,170 | $ | - | $ | 3,388,166 | $ | 4,673,336 | ||||||||
| 4. | LICENSES, INTELLECTUAL PROPERTY, AND RELATED PARTY TRANSACTIONS |
We have an exclusive, worldwide licensing agreement with the National Institutes of Health (“NIH”) for all claims to TßTb4 within their broadly-defined patent application. In exchange for this exclusive worldwide license, we must make certain royalty and milestone payments to the NIH. Through December 31, 2009In 2013 we have complied with these requirements.amended certain provisions of the exclusive license; we were permitted to credit amounts paid to prosecute or maintain the licensed patent rights during 2013 calendar year against the 2013 minimum annual royalty of $25,000. Beginning in 2014 the minimum annual royalty is $2,000. No assurance can be given as to whether or when a patent will be issued, or as to any claims that may be included or excluded within the patent. We have also filed numerous additional patent applications covering various compositions, uses, formulations and other components of TßTb4, as well as to novel peptides resulting from our research efforts. Some of these patents have issued, while many patent applications are still pending. Minimum
We have also entered into an agreement with a university under the terms of which we have received an exclusive license to technology and intellectual property. The agreement, which is generally cancelable by us, provided for the payment of a license issue fee and/or minimum annual payments. The initial license fee of $25,000 was paid in 2010 and no minimum fees were due for the year ended December 31, 2011. Beginning in 2012, minimum annual maintenance fees are $5,000 annually which was paid in 2012 but has not been paid for each2013, 2014 or 2015 as of the years ended December 31, 2009date of this report. In addition, the agreements provide for payments upon the achievement of certain milestones in product development. The agreement also requires us to fund certain costs associated with the filing and 2008 were $25,000.prosecution of patent applications. In February 2013 this agreement was amended to include additional technology and intellectual property. The expanded license does not require payment of an initial license fee or additional annual maintenance fees but will be subject to payments upon the achievement of certain milestones for a product developed under the amended license of the additional technology and intellectual property.
All license fees are included in Research and Development in the accompanying statements of operations.
| F-31 |
We have entered into a License and Supply Agreement (the “Agreement”) with Defiante Farmaceutica S.A. (“Defiante”) a Portuguese company that is a wholly owned subsidiary of Sigma-Tau, S.p.A., an international pharmaceutical company and an affiliate of Sigma-Tau Finanziaria S.p.A., who together with its affiliates comprise our largest stockholder group (the “Sigma-Tau Group”). This Agreement grants to Defiante the exclusive right to use TßTb4 to conduct research and development activities in Europe. Under the Agreement, we will receive fees and royalty payments based on a percentage of specified sales of TßTb4-related products by Defiante. The term of the Agreement continues until the later of the expiration of any patents developed under the Agreement, the expiration of marketing rights, or December 31, 2016.
In furtherance2012, we entered into a License Agreement (the “Agreement”) with Lee’s Pharmaceutical (HK) Limited, headquartered in Hong Kong, for the license of Thymosin Beta 4 in any pharmaceutical form, including our RGN-259, RGN-352 and RGN-137 product candidates, in China, Hong Kong, Macau and Taiwan. Under the License Agreement, we are eligible to receive milestone payments and royalties, ranging from low double digit to high single digit percentages of any commercial sales of the licensed products. Lee’s will pay for all developmental costs associated with each product candidate. We will provide Tß4 to Lee’s at no charge for a Phase 2 ophthalmic clinical trial and will provide Tß4 to Lee’s for all other developmental and clinical work at a price equal to our cost. We will also have the right to exclusively license any improvements made by Lee’s to RegeneRx’s products outside of the licensed territory. Lee’s paid us $200,000 upon signing of a term sheet in March 2012, and Lee’s paid us an additional $200,000 upon signing of the definitive license agreement. Lee’s is an affiliate of Sigma-Tau, which collectively with its affiliates is our largest stockholder. As of December 31, 2015 and 2014, we have unearned revenue totaling $400,000 pursuant to this Agreement.
On March 7, 2014, we entered into license agreements with GtreeBNT Co., Ltd. The two Licensing Agreements are for the license of territorial rights Sigma-Tau Group fundedto two of our Thymosin Beta 4-based products candidates, RGN-259 and managedRGN-137.
Under the RegeneRx-sponsored Phase IILicense Agreement for RGN-259, our preservative-free eye drop product candidate, GtreeBNT will have the right to develop and commercialize RGN-259 in Asia (excluding China, Hong Kong, Taiwan, and Macau). The rights will be exclusive in Korea, Japan, Australia, New Zealand, Brunei, Cambodia, East Timor, Indonesia, Laos, Malaysia, Mongolia, Myanmar (Burma), Philippines, Singapore, Thailand, Vietnam, and Kazakhstan, and semi-exclusive in India, Pakistan, Bangladesh, Bhutan, Maldives, Nepal, Sri Lanka, Kyrgyzstan, Tajikistan, Turkmenistan and Uzbekistan, collectively, the Territory (the “259 Territory”). Under the 259 License Agreement we are eligible to receive aggregate potential milestone payments of up to $3.5 million. In addition, we are eligible to receive royalties of a low double digit percentage of any commercial sales of the licensed product sold by GtreeBNT in the 259 Territory.
Under the License Agreement for RGN-137, our topical dermal wound healinggel product candidate, GtreeBNT will have the exclusive right to develop and commercialize RGN-137 in the U.S. (the”137 Territory”). Under the 137 License Agreement we are eligible to receive aggregate potential milestone payments of up to $3.5 million. In addition, we are eligible to receive royalties of a low double digit percentage of any commercial sales of the Company’s licensed product sold by GtreeBNT in the 137 Territory.
Each license agreement contains diligence provisions which require the initiation of certain clinical trials within certain time periods that, if not met, would result in the loss of rights or exclusivity in certain countries. GtreeBNT will pay for all developmental costs associated with each product candidate. We will provide a certain limited amount of Tß4 to GtreeBNT at no charge for initial clinical trials in venous stasis ulcers conducted in ItalyKorea, Japan and Poland that concludedAustralia for RGN-259 and in the U.S. for RGN-137 and will provide Tß4 to GtreeBNT for all other developmental and clinical work on a cost plus basis. We have the right to exclusively license any improvements made by GtreeBNT to our products outside of the licensed territory on a royalty free basis. The two firms have created a joint development committee and continue to discuss the development of the licensed products and share information relating thereto. Both companies will also share all non-clinical and clinical data and other information related to development of the licensed product candidates.
On January 28, 2015, the Company entered into the Joint Venture Agreement with GtreeBNT, a shareholder in the Company. The Joint Venture Agreement provides for the creation of the Joint Venture, jointly owned by the Company and GtreeBNT, which will commercialize RGN-259 for treatment of dry eye and neurotrophic keratopathy in the United States.
GtreeBNT is solely responsible for funding all the product development and commercialization efforts of the Joint Venture. GtreeBNT made an initial contribution of $3 million in cash and received an initial equity stake of 51%. RegeneRx’s ownership interest in ReGenTree is 49% and will be reduced to 42% when the clinical study report is filed for the Phase 3 dry eye clinical trial. Based on when, and if, certain additional development milestones are achieved in the U.S. with RGN-259, our equity ownership may be incrementally reduced to between 42% and 25%, with 25% being the final equity ownership upon approval of an NDA for Dry Eye Syndrome in the U.S. In addition to our equity ownership, RegeneRx retains a royalty on net sales that varies between single and low double digits, depending on whether commercial sales are made by ReGenTree or a licensee. In the event the ReGenTree entity is acquired or there is a change of control that occurs following achievement of an NDA, RegeneRx shall be entitled to 40% of all change of control proceeds paid or payable and will forgo any future royalties. The Company is not required or otherwise obligated to provide financial support to the Joint Venture.
| F-32 |
The Joint Venture is responsible for executing all development and commercialization activities under the License Agreement, which activities will be directed by a joint development committee comprised of representatives of the Company and GtreeBNT. The License Agreement has a term that extends to the later of the expiration of the last patent covered by the License Agreement or 25 years from the first quartercommercial sale under the License Agreement. The License Agreement may be earlier terminated if the Joint Venture fails to meet certain commercialization milestones, if either party breaches the License Agreement and fails to cure such breach, as a result of 2009.
F-11
Under the License Agreement, the Company received $1.0 million in up-front payments and is entitled to receive royalties on the Joint Venture’s future sales of products. The Company is accounting for the License Agreement with the Joint Venture as a revenue arrangement. The Company has determined that the deliverables within the License Agreement, including a delivered element (providing the license) and an undelivered element (participation on the joint development committee), do not have stand-alone value and, as such, are treated as a single unit of accounting. As a result, the Company is recognizing the up-front milestone payments as revenue ratably over the anticipated life of the joint development committee, or 25 years. The joint development committee commenced activities as of April 1, 2015 therefore the Company has begun recognizing the revenue for the license fee. Revenue will be recognized for future royalty payments as they are earned.
| 5. | COMPOSITION OF CERTAIN FINANCIAL STATEMENT CAPTIONS |
Prepaid expenses and other current assets are comprised of the following:
| December 31, | ||||||||
| 2015 | 2014 | |||||||
| Prepaid insurance | $ | 10,552 | $ | 50,779 | ||||
| Prepaid and other | 13,748 | 35,746 | ||||||
| $ | 24,300 | $ | 86,525 | |||||
Accrued expenses are comprised of the following:
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Accrued clinical research | $ | 496,997 | $ | 944,283 | ||||
| Accrued professional fees | 122,590 | 155,000 | ||||||
| Accrued vacation | 35,300 | 61,714 | ||||||
| Accrued license fees | 30,000 | — | ||||||
| Accrued compensation | 28,995 | 84,361 | ||||||
| Other | 26,316 | 10,000 | ||||||
| $ | 740,198 | $ | 1,255,358 | |||||
| December 31, | ||||||||
| 2015 | 2014 | |||||||
| Accrued professional fees | $ | 31,788 | $ | 80,393 | ||||
| Accrued other | 30,000 | 2,954 | ||||||
| Accrued compensation | 22,249 | 9,838 | ||||||
| Accrued interest on convertible notes | 133,874 | 83,824 | ||||||
| $ | 217,911 | $ | 177,009 | |||||
| 6. | EMPLOYEE BENEFIT PLANS |
In 2014 we did not offer any Company sponsored health or retirement plans. In 2015 the Company provided health and dental insurance to one employee under a defined contributiongroup plan. No retirement plan that complies with Section 401(k)was in place for 2015.
| 7. | CONVERTIBLE NOTES |
2012 Convertible Note
On October 19, 2012 we completed a private placement of convertible notes (the “2012 Notes”) raising an aggregate of $300,000 in gross proceeds. The 2012 Notes were originally scheduled to mature after twenty-four (24) months from issuance. The 2012 Notes bear interest at a rate of five percent (5%) per annum and are convertible into shares of our common stock at a conversion price of fifteen cents ($0.15) per share (subject to adjustment as described in the 2012 Notes) at any time prior to repayment, at the election of the Internal Revenue Code (the “Code”). All employeesInvestors. In the aggregate, the 2012 Notes are convertible into up to 2,000,000 shares of our common stock excluding interest.
| F-33 |
At any time prior to maturity of the 2012 Notes, with the consent of the holders of a majority in interest of the 2012 Notes, we may prepay the outstanding principal amount of the 2012 Notes plus unpaid accrued interest without penalty. Upon the commission of any act of bankruptcy by the Company, the execution by the Company of a general assignment for the benefit of creditors, the filing by or against the Company of a petition in bankruptcy or any petition for relief under the federal bankruptcy act or the continuation of such petition without dismissal for a period of ninety (90) days or more, or the appointment of a receiver or trustee to take possession of the property or assets of the Company, are eligible to participate in the plan. The Company matches 100% of each participant’s voluntary contributions, subject to a maximum Company contribution of 4%outstanding principal and all accrued interest on the 2012 Notes will accelerate and automatically become immediately due and payable.
In connection with the issuance of the participant’s compensation.2012 Notes we also issued warrants to each Investor. The Company’s matching portion totaled $18,269warrants are exercisable for an aggregate of 400,000 shares of common stock with an exercise price of fifteen cents ($0.15) per share for a period of five years. The relative fair value of the warrants issued is $27,097, calculated using the Black-Scholes-Merton valuation model value of $0.07 with an expected and $51,494 forcontractual life of 5 years, an assumed volatility of 74.36%, and a risk-free interest rate of 0.77%. The warrants were recorded as additional paid-in-capital and a discount on the 2012 Notes of $27,097. Non-cash interest expense related to the debt discount during the years ended December 31, 20092014 totaled $10,854.
The Investors, and 2008, respectively. In order to conserve cash, the Company discontinued the matching contribution effective June 5, 2009principal amount of their respective 2012 Notes and reinstated it on March 1, 2010.
| Investor | Note Principal | Warrants | ||||||
| Sinaf S.A. | $ | 200,000 | 266,667 | |||||
| Joseph C. McNay | $ | 50,000 | 66,667 | |||||
| Allan L. Goldstein | $ | 35,000 | 46,666 | |||||
| J.J. Finkelstein | $ | 15,000 | 20,000 | |||||
Sinaf S. A. is a Rights Agreement, dated April 29, 1994, as amended, thatdirect wholly-owned subsidiary of Aptafin S.p.A., or Aptafin. Aptafin is intended to discourage an unsolicited change in controlowned directly by Paolo Cavazza and members of the Company. In general, if an entity acquires more than a 25% ownership interest in the Company without the endorsementhis family, who directly and indirectly own 38% of Sigma-Tau, our largest stockholder. The other Investors are members of our Board of Directors thenincluding Mr. Finkelstein who serves as our current stockholders (other thanCEO and also the acquiring entity)Chairman of our Board of Directors and Dr. Goldstein who also serves as our Chief Scientific Officer.
During 2014, the Company amended the existing October 2012 convertible debt agreement with the lenders, solely to extend the due date of the principal and accrued unpaid until interest October 19, 2017. No other terms of the original debt were amended or modified, and the lenders did not reduce the borrowed amount or change the interest rate of the debt. The Company considered the restructuring a troubled debt restructuring as a result of the Company’s financial condition (see Note 1 discussion of “going concern”). At the date of the amendment, all existing debt discounts and deferred financing fees were fully amortized and the amendment did not involve any additional fees paid to the lender or third parties; as such there was no gain recognized as a result of the amendment.
2013 Convertible Notes
On March 29, 2013, we completed a private placement of convertible notes (the “March 2013 Notes”) raising an aggregate of $225,000 in gross proceeds. The March 2013 Notes bear interest at a rate of five percent (5%) per annum, mature sixty (60) months after their date of issuance and are convertible into shares of our common stock at a conversion price of six cents ($0.06) per share (subject to adjustment as described in the March 2013 Notes) at any time prior to repayment, at the election of the investor. In the aggregate, the March 2013 Notes are initially convertible into up to 3,750,000 shares of our common stock.
At any time prior to maturity of the March 2013 Notes, with the consent of the holders of a majority in interest of the March 2013 Notes, we may prepay the outstanding principal amount of the March 2013 Notes plus unpaid accrued interest without penalty. Upon the commission of any act of bankruptcy by the Company, the execution by the Company of a general assignment for the benefit of creditors, the filing by or against the Company of a petition in bankruptcy or any petition for relief under the Federal bankruptcy act or the continuation of such petition without dismissal for a period of ninety (90) days or more, or the appointment of a receiver or trustee to take possession of the property or assets of the Company, the outstanding principal and all accrued interest on the March 2013 Notes will accelerate and automatically become immediately due and payable.
The investors in the offering included two directors of the Company, Dr. Goldstein and Joseph C. McNay, an outside director. The principal amounts of their respective March 2013 Notes are as set forth below:
| Investor | Note Principal | |||
| Joseph C. McNay | $ | 50,000 | ||
| Allan L. Goldstein | $ | 25,000 | ||
| F-34 |
The Company has evaluated the terms of the March 2013 Notes which contain a down round provision under which the conversion price could be decreased as a result of future equity offerings, as defined in the March 2013 Notes. The adjustment would reduce the conversion price of the March 2013 Notes to be equivalent to that of the newly issued stock or stock-related instruments. As a result, the Company concluded that the conversion feature represented an embedded conversion feature for accounting purposes and should be recognized as a derivative liability, requiring a mark-to-market adjustment at the end of each reporting period until the related March 2013 Notes have been settled. The bifurcated liability of $225,000 was recorded on the date of issuance which resulted in a residual debt value of $0. The discount related to the embedded feature will be issued a significant number of new shares,accreted as an addition to the effect of which would dilutedebt through the ownershipmaturity of the acquiring entitynotes.
On July 5, 2013, we completed a private placement of convertible notes (the “July 2013 Notes”) raising an aggregate of $100,000 in gross proceeds. The July 2013 Notes bear interest at a rate of five percent (5%) per annum, mature sixty (60) months after their date of issuance and are convertible into shares of our common stock at a conversion price of six cents ($0.06) per share (subject to adjustment as described in the July 2013 Notes) at any time prior to repayment, at the election of the investor. In the aggregate, the July 2013 Notes are initially convertible into up to 1,666,667 shares of our common stock.
At any time prior to maturity of the July 2013 Notes, with the consent of the holders of a majority in interest of the July 2013 Notes, we may prepay the outstanding principal amount of the July 2013 Notes plus unpaid accrued interest without penalty. Upon the commission of any act of bankruptcy by the Company, the execution by the Company of a general assignment for the benefit of creditors, the filing by or against the Company of a petition in bankruptcy or any petition for relief under the Federal bankruptcy act or the continuation of such petition without dismissal for a period of ninety (90) days or more, or the appointment of a receiver or trustee to take possession of the property or assets of the Company, the outstanding principal and all accrued interest on the July 2013 Notes will accelerate and automatically become immediately due and payable.
The investors in the offering included four directors of the Company, Mr. Finkelstein, Dr. Goldstein, Mr. McNay and L. Thompson Bowles, previously an outside director. The principal amounts of their respective July 2013 Notes are as set forth below:
| Investor | Note Principal | |||
| Joseph C. McNay | $ | 50,000 | ||
| Allan L. Goldstein | $ | 10,000 | ||
| J.J. Finkelstein | $ | 5,000 | ||
| L. Thompson Bowles | $ | 5,000 | ||
The Company has evaluated the terms of the July 2013 Notes which contain a down round provision under which the conversion price could delaybe decreased as a result of future equity offerings, as defined in the July 2013 Notes. The adjustment would reduce the conversion price of the July 2013 Notes to be equivalent to that of the newly issued stock or preventstock-related instruments. As a result, the Company concluded that the conversion feature represented an embedded conversion feature for accounting purposes and should be recognized as a derivative liability, requiring a mark-to-market adjustment at the end of each reporting period until the related July 2013 Notes have been settled. The bifurcated liability of $66,667 was recorded on the date of issuance which resulted in a residual debt value of $33,333. The discount related to the embedded feature will be accreted back to debt through the maturity of the notes.
On September 11, 2013, we completed a private placement of convertible notes raising an aggregate of $321,000 in gross proceeds (the “September 2013 Notes”). The September 2013 Notes bear interest at a rate of five percent (5%) per annum, mature sixty (60) months after their date of issuance and are convertible into shares of our common stock at a conversion price of six cents ($0.06) per share (subject to adjustment as described in the September 2013 Notes) at any time prior to repayment, at the election of the investor. In the aggregate, the September 2013 Notes are initially convertible into up to 5,350,000 shares of our common stock.
At any time prior to maturity of the September 2013 Notes, with the consent of the holders of a majority in interest of the September 2013 Notes, we may prepay the outstanding principal amount of the September 2013 Notes plus unpaid accrued interest without penalty. Upon the commission of any act of bankruptcy by the Company, the execution by the Company of a general assignment for the benefit of creditors, the filing by or against the Company of a petition in bankruptcy or any petition for relief under the federal bankruptcy act or the continuation of such petition without dismissal for a period of ninety (90) days or more, or the appointment of a receiver or trustee to take possession of the property or assets of the Company, the outstanding principal and all accrued interest on the September 2013 Notes will accelerate and automatically become immediately due and payable.
The investors in the offering included an affiliate and three current and one prior directors of the Company. The principal amounts of the affiliate and directors respective September 2013 Notes are as set forth below:
| F-35 |
| Investor | Note Principal | |||
| SINAF S.A. | $ | 150,000 | ||
| Joseph C. McNay | $ | 100,000 | ||
| Allan L. Goldstein | $ | 11,000 | ||
| L. Thompson Bowles | $ | 5,000 | ||
| R. Don Elsey | $ | 5,000 | ||
The Company has evaluated the terms of the September 2013 Notes which contain a down round provision under which the conversion price could be decreased as a result of future equity offerings, as defined in the September 2013 Notes. The adjustment would reduce the conversion price of the September 2013 Notes to be equivalent to that of the newly issued stock or stock-related instruments. As a result, the Company concluded that the conversion feature represented an embedded conversion feature for accounting purposes and should be recognized as a derivative liability, requiring a mark-to-market adjustment at the end of each reporting period until the related September 2013 Notes have been settled. The bifurcated liability of $267,500 was recorded on the date of issuance which resulted in a residual debt value of $53,500. The discount related to the embedded feature will be accreted back to debt through the maturity of the notes.
2014 Convertible Notes
On January 7, 2014, we completed a private placement of convertible notes raising an aggregate of $55,000 in gross proceeds (the “January 2014 Notes”). The January 2014 Notes bear interest at a rate of 5% per annum, mature 60 months after their date of issuance and are convertible into shares of our common stock at a conversion price of $0.06 per share (subject to adjustment as described in the January 2014 Notes) at any time prior to repayment, at the election of the Investor. In the aggregate, the Notes are initially convertible into up to 916,667 shares of our common stock.
At any time prior to maturity of the January 2014 Notes, with the consent of the holders of a majority in interest of the January 2014 Notes, we may prepay the outstanding principal amount of the January 2014 Notes plus unpaid accrued interest without penalty. Upon the commission of any act of bankruptcy by the Company, the execution by the Company of a general assignment for the benefit of creditors, the filing by or against the Company of a petition in bankruptcy or any petition for relief under the federal bankruptcy act or the continuation of such petition without dismissal for a period of 90 days or more, or the appointment of a receiver or trustee to take possession of the property or assets of the Company, the outstanding principal and all accrued interest on the January 2014 Notes will accelerate and automatically become immediately due and payable.
The Investors in the offering included two current and one prior directors of the Company. The principal amounts of their respective Notes are as set forth below:
| Investor | Note Principal | |||
| Joseph C. McNay | $ | 25,000 | ||
| Allan L. Goldstein | $ | 10,000 | ||
| L. Thompson Bowles | $ | 5,000 | ||
The Company has evaluated the terms of the January 2014 Notes which contain a down round provision under which the conversion price could be decreased as a result of future equity offerings, as defined in the January 2014 Notes. The adjustment would reduce the conversion price of the January 2014 Notes to be equivalent to that of the newly issued stock or stock-related instruments. As a result, the Company concluded that the conversion feature represented an embedded conversion feature for accounting purposes and should be recognized as a derivative liability, requiring a mark-to-market adjustment at the end of each reporting period until the related January 2014 Notes have been settled. The bifurcated liability of $55,000 was recorded on the date of issuance which resulted in a residual debt value of $0. The discount related to the embedded feature will be accreted back to debt through the maturity of the notes.
| F-36 |
The outstanding balance of the derivative liability is as follows:
| December 31, 2015 | December 31, 2014 | |||||||
| March 2013 Notes | $ | 1,500,000 | $ | 412,500 | ||||
| July 2013 Notes | 666,667 | 183,334 | ||||||
| September 2013 Notes | 2,140,000 | 588,500 | ||||||
| January 2014 notes | 366,669 | 100,836 | ||||||
| Total Fair value of derivative liability | $ | 4,673,336 | $ | 1,285,170 | ||||
The change in control.fair value of the derivative liability is as follows:
| For the twelve months ended | ||||||||
| December 31, 2015 | December 31, 2014 | |||||||
| March 2013 Notes | $ | 1,087,500 | $ | 337,500 | ||||
| July 2013 Notes | 483,333 | 150,000 | ||||||
| September 2013 Notes | 1,551,500 | 481,500 | ||||||
| January 2014 notes | 265,833 | 45,836 | ||||||
| Total change in fair value of derivative | $ | 3,388,166 | $ | 1,014,836 | ||||
The Company recorded interest expense and discount accretion as set forth below:
| For the twelve months ended | ||||||||
| December 31, 2015 | December 31, 2014 | |||||||
| 2012 Notes | $ | 15,000 | $ | 25,854 | ||||
| March 2013 Notes | 56,250 | 56,250 | ||||||
| July 2013 Notes | 18,333 | 18,333 | ||||||
| September 2013 Notes | 69,550 | 69,550 | ||||||
| January 2014 notes | 13,750 | 13,486 | ||||||
| Total interest expense | $ | 172,883 | $ | 183,473 | ||||
| F-37 |
| 8. | STOCKHOLDERS’ EQUITY |
Common Stock.In April 2015 we entered into a contract with an investor relations firm to provide services for six months. Under the agreement the Company paid $5,000 per month and issued 30,000 shares of common stock as compensation. In addition, in May 2015 the Company issued 293,512 shares of common stock pursuant to the “cashless” exercise of warrants issued in 2010.
On August 29, 2014, the Company received gross proceeds of $1,000,000 and pursuant to the warrant exercise issued 8,333,333 shares of common stock at $0.12 per share pursuant to the securities purchase and licensing agreements signed with GtreeBNT on March 7, 2014. Under the securities purchase agreement, GtreeBNT invested $1,350,000 for the issuance of 11,250,000 common shares at $0.12 per share and was required to invest an additional $1,000,000 at $0.12 per share on or before August 31, 2014. Under the terms of the security purchase agreement, GtreeBNT also has the right to make an optional investment to acquire an additional 5.5 million shares of common stock at $0.15 per share. Such optional investment right expired unexercised on January 31, 2015.
In addition, GtreeBNT agreed to pay the Company milestone payments upon the achievement of certain commercial sales milestones, as well as with royalties on commercial sales. As the security purchase and licensing agreements were signed in contemplation of each other and the execution of performance under the securities purchase agreement was stipulated as a condition for the retention of the rights granted under the licensing agreements, the three agreements were treated as a multiple-elements arrangement. Following the closing of the agreements, the Company determined that the total consideration received under the three agreements, totaling $1,500,000, should be allocated to identifiable elements within this multiple-elements arrangement (1) the equity investment in the Company’s common shares, including the purchase option and (2) the licensed development and commercialization rights under the two licensing agreements. The optional investment right was considered an equity instrument reduced from the Company’s equity, and its fair value of approximately $725,000 was calculated using the Black Scholes option pricing model at the issuance of this right. As the common shares were issued at a discount to the then market price of the Company’s common stock of $0.20 on the date of closing, all of the proceeds received were absorbed by the allocation to the common shares and the optional investment right leaving no allocation of proceeds to the licensed rights.
Registration Rights Agreements. In connection with the sale of certain equity instruments, we have entered into Registration Rights Agreements. Generally, these Agreements required us to file registration statements with the Securities and Exchange Commission to register common shares to permit re-sale of common shares previously sold under an exemption from registration or to register common shares that may be issued on exercise of outstanding warrants.
The Registration Rights Agreements usually require us to pay penalties for any failure or time delay in filing or maintaining the effectiveness of the required registration statements. These penalties are usually expressed as a fixed percentage, per month, of the original amount we received on issuance of the common shares, options or warrants. While to date we have not incurred any penalties under these agreements, if a penalty is determined to be probable we would recognize the amount as a contingent liability and not as a derivative instrument.
F-12
Stock Option and Incentive Plans. On July 14, 2010, at our Annual Meeting of Stockholders, our stockholders approved the 2010 Equity Incentive Plan (the “2010 Plan”). The terms of the 2010 Plan provide for the discretionary grant of incentive stock options, nonstatutory stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards, performance stock awards, other stock awards and performance cash awards to our employees, directors and consultants. At inception of the 2010 Plan, 5,000,000 shares of our common stock were reserved for future issuance. On September 10, 2014 at our Annual Meeting of Stockholders, our stockholders approved an increase in the number of shares available under the 2010 Equity Incentive Plan (the “2010 Plan”). The increase of 3,000,000 results in a total of 8,000,000 shares of common stock reserved for issuance.
We previously adopted an equity incentive plan, known as the Amended and Restated 2000 Stock Option and Incentive Plan as amended. Our Board(the “2000 Plan”). The 2000 Plan has a term of Directors (the “Board”) and stockholders have approvedten years that expired in December 2010. All outstanding option awards granted under the 2000 Stock OptionPlan will continue to be subject to the terms and Incentive Plan under whichconditions as set forth in the Board may grant options to purchase shares of our common stock. Options may only be granted to our directors, officers, employees, consultants or advisors,agreements evidencing such option awards and no single participant can receive more than 450,000 shares in any one year. The exercise price and term of any grant are determined by the Board at the time of grant but the exercise price may not be less than the fair market value of our common stock on the dateterms of the grant, and the term of the option shall not exceed ten years. As of December 31, 2009, there were 6,500,000 shares
F-13
| F-38 |
The following summarizes share-based compensation expense for the years ended December 31, 20092015 and 2008,2014, which was allocated as follows:
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Research and development | $ | 369,814 | $ | 440,850 | ||||
| General and administrative | 378,666 | 655,752 | ||||||
| $ | 748,480 | $ | 1,096,602 | |||||
| December 31, | ||||||||
| 2015 | 2014 | |||||||
| Research and development | $ | 72,766 | $ | 48,244 | ||||
| General and administrative | 158,841 | 115,188 | ||||||
| $ | 231,607 | $ | 163,432 | |||||
The following summarizes stock option activity for the years ended December 31, 20092015 and 2008:
| Options Outstanding | ||||||||||||||||
| Weighted | ||||||||||||||||
| Shares | Average | |||||||||||||||
| Available for | Number of | Exercise Price | Exercise | |||||||||||||
| Grant | Shares | Range | Price | |||||||||||||
| December 31, 2007 | 620,000 | 3,545,000 | $ | 0.28 - $3.82 | $ | 1.80 | ||||||||||
| Grants | (572,500 | ) | 572,500 | 1.14 - 1.50 | 1.23 | |||||||||||
| Exercises | — | — | — | — | ||||||||||||
| Cancellations | — | — | — | — | ||||||||||||
| Newly authorized | 2,300,000 | — | — | — | ||||||||||||
| December 31, 2008 | 2,347,500 | 4,117,500 | 0.28 - 3.82 | 1.72 | ||||||||||||
| Grants | (1,192,939 | ) | 1,192,939 | 0.57 - 0.76 | 0.64 | |||||||||||
| Exercises | — | — | — | — | ||||||||||||
| Cancellations | 396,327 | (396,327 | ) | 0.57 - 2.59 | 0.82 | |||||||||||
| December 31, 2009 | 1,550,888 | 4,914,112 | $ | 0.28 - $3.82 | $ | 1.53 | ||||||||||
| Options outstanding | ||||||||||||||||
| Weighted | ||||||||||||||||
| Shares | average | |||||||||||||||
| available for | Number of | Exercise price | exercise | |||||||||||||
| grant | shares | range | price | |||||||||||||
| December 31, 2013 | 1,959,036 | 5,999,599 | 0.14 – 3.82 | 1.02 | ||||||||||||
| Grants | — | 2,195,000 | 0.16 – 0.21 | 0.19 | ||||||||||||
| Exercises | — | — | — | — | ||||||||||||
| Cancellations* | (1,930,888 | ) | 0.16 – 3.82 | 1.54 | ||||||||||||
| December 31, 2014 | 3,273,029 | 6,263,711 | $ | 0.14 – 3.21 | $ | 0.58 | ||||||||||
| Grants | — | 1,725,000 | 0.19 – 0.40 | 0.33 | ||||||||||||
| Exercises | — | — | — | — | ||||||||||||
| Cancellations* | (857,500 | ) | 0.14 – 3.21 | 0.41 | ||||||||||||
| December 31, 2015 | 1,548,029 | 7,131,211 | $ | 0.14 – 3.00 | $ | 0.30 | ||||||||||
| Vested and expected to vest at December 31, 2015 | 7,020,866 | |||||||||||||||
| Exercisable at December 31, 2015 | 4,639,961 | |||||||||||||||
*Note: Cancellations in 2015 and a portion of the 2014 cancellations were for options issued out of the 2000 Equity Incentive Plan and therefore they are not available for reissuance.
The following summarizes information about stock options outstanding at December 31, 2009:2015:
| Outstanding options | Exercisable options | |||||||||||||||||||||||
| Weighted- | Weighted- | |||||||||||||||||||||||
| average | Weighted- | average | Weighted- | |||||||||||||||||||||
| Number of | remaining | average | Number of | remaining | average | |||||||||||||||||||
| shares | contractual | exercise | shares | contractual | exercise | |||||||||||||||||||
| Range of exercise prices | outstanding | life(in years) | price | exercisable | life(in years) | price | ||||||||||||||||||
| $0.14 – $0.36 | 6,361,971 | 4.6 | $ | 0.23 | 3,938,221 | 3.9 | $ | 0.20 | ||||||||||||||||
| $0.40 – $0.76 | 684,240 | 2.7 | 0.63 | 616,740 | 2.2 | 0.65 | ||||||||||||||||||
| $2.50 – $2.68 | 35,000 | 0.8 | 2.53 | 35,000 | 0.8 | 2.53 | ||||||||||||||||||
| $3.00 – $3.21 | 50,000 | 0.0 | 3.00 | 50,000 | 0.0 | 3.00 | ||||||||||||||||||
| 7,131,211 | 4.4 | 0.30 | 4,639,961 | 3.6 | 0.31 | |||||||||||||||||||
| Intrinsic value of in-the-money options, using the December 31, 2015 closing price of $0.44 | $ | 950,573 | $ | 3,960,721 | ||||||||||||||||||||
| F-39 |
| Outstanding Options | Exercisable Options | |||||||||||||||||||||||
| Weighted- | Weighted- | |||||||||||||||||||||||
| Average | Weighted- | Average | Weighted- | |||||||||||||||||||||
| Number of | Remaining | Average | Number of | Remaining | Average | |||||||||||||||||||
| Shares | Contractual | Exercise | Shares | Contractual | Exercise | |||||||||||||||||||
Range of Exercise Prices | Outstanding | Life (in Years) | Price | Exercisable | Life (in Years) | Price | ||||||||||||||||||
| $0.28-$0.86 | 2,151,612 | 4.5 | $ | 0.50 | 1,724,112 | 4.0 | $ | 0.43 | ||||||||||||||||
| $1.07-$1.93 | 827,500 | 5.0 | $ | 1.31 | 435,625 | 4.6 | $ | 1.40 | ||||||||||||||||
| $2.02-$2.68 | 860,000 | 4.3 | $ | 2.26 | 323,750 | 4.4 | $ | 2.31 | ||||||||||||||||
| $3.00-$3.82 | 1,075,000 | 5.4 | $ | 3.19 | 950,832 | 5.4 | $ | 3.19 | ||||||||||||||||
| 4,914,112 | 3,434,319 | |||||||||||||||||||||||
Intrinsic value ofin-the-money options, using the December 31, 2009 closing price of $0.55 | $ | 254,450 | $ | 254,450 | ||||||||||||||||||||
Determining the Fair Value of Options. We use the Black-Scholes valuation model to estimate the fair value of options granted. Black-Scholes considers a number of factors, including the market price and
F-14
| 2009 | 2008 | |||
| Dividend yield | 0.0% | 0.0% | ||
| Risk free rate of return | 1.9 - 2.3% | 0.8 - 3.7% | ||
| Expected life in years | 4.75 - 5.38 | 1.00 - 4.75 | ||
| Volatility | 71 - 72% | 68 - 82% | ||
| Forfeitures | 2.61% | — |
| 2015 | 2014 | |||||||
| Dividend yield | 0.0 | % | 0.0 | % | ||||
| Risk-free rate of return | 1.43-1.63 | % | 1.63-1.76 | % | ||||
| Expected life in years | 4.75 - 7 | 4 - 5 | ||||||
| Volatility | 90-94 | % | 91-98 | % | ||||
| Forfeiture rate | 2.6 | % | 2.6 | % | ||||
Our dividend yield assumption is based on the fact that we have never paid cash dividends and do not anticipate paying cash dividends in the foreseeable future. Our risk-free interest rate assumption is based on yields of U.S. Treasury notes in effect at the date of grant. Our expected life represents the period of time that options granted are expected to be outstanding and is calculated in accordance with the Securities and Exchange Commission (“SEC”) guidance provided in the SEC’s Staff Accounting Bulletin 107 (“SAB 107”), and SAB 110, using a “simplified” method. The Company has used the simplified method and will continue to use the simplified method as it does not have sufficient historical exercise data to provide a reasonable basis upon which to estimate an expected term. Our volatility assumption is based on reviews of the historical volatility of our common stock. We estimate forfeiture rates at the time of grant and adjust these estimates, if necessary, periodically based on the extent to which future actual forfeitures differ, or are expected to differ, from such estimates. Accordingly, we have estimated forfeiture percentages for the unvested portion of previously granted awards that remain outstanding at the date of adoption and for awards granted subsequent to the date of adoption. Forfeitures are estimated based on the demographics of current option holders and standard probabilities of employee turnover. Using Black-Scholes and these factors, the weighted average fair value of stock options granted to employees and directors was $0.39$0.33 for the year ended December 31, 2009 and $0.73 for the year ended December 31, 2008.
Warrants to Purchase Common Stock.Stock
The following table summarizes our warrant activity for 20092015 and 2008:
| Warrants Outstanding | ||||||||||||
| Weighted | ||||||||||||
| Average | ||||||||||||
| Number of | Exercise Price | Exercise | ||||||||||
| Shares | Range | Price | ||||||||||
| December 31, 2007 | 3,522,544 | $ | 2.75 - $4.06 | $ | 3.26 | |||||||
| Grants | 1,745,104 | 1.60 - 1.74 | 1.66 | |||||||||
| Exercises | — | — | — | |||||||||
| Cancellations | (18,558 | ) | 4.05 - 4.06 | 4.05 | ||||||||
| December 31, 2008 | 5,249,090 | 1.60 - 4.06 | 2.80 | |||||||||
| Grants | 3,129,011 | 0.91 - 1.12 | 1.10 | |||||||||
| Exercises | — | — | — | |||||||||
| Cancellations | (444,250 | ) | 4.06 | 4.06 | ||||||||
| December 31, 2009 | 7,933,851 | $ | 0.91 - $4.06 | $ | 2.01 | |||||||
F-15
| Warrants outstanding | ||||||||||||
| Weighted | ||||||||||||
| Number of | Exercise price | Average exercise | ||||||||||
| shares | range | price | ||||||||||
| December 31, 2013 | 11,468,901 | $ | 0.15 – 1.12 | $ | 0.64 | |||||||
| Grants | 13,833,333 | 0.12 – 0.15 | 0.18 | |||||||||
| Exercises | (8,333,333 | ) | 0.12 | 0.12 | ||||||||
| Cancellations | (2,865,853 | ) | 1.12 | 1.12 | ||||||||
| December 31, 2014 | 14,103,048 | $ | 0.15 – $0.56 | $ | 0.35 | |||||||
| Exercises | (957,641 | ) | 0.38 – 0.45 | 0.40 | ||||||||
| Cancellations | (11,338,000 | ) | 0.15 – 0.56 | 0.41 | ||||||||
| December 31, 2015 | 1,807,407 | $ | 0.15 – $0.38 | $ | 0.32 | |||||||
| INCOME TAXES |
As a result of its operating losses, the Company did not recognize a provision (benefit) for income taxes in its statements of operations for 2015 and 2014. The Company has provided a full valuation allowance against its net deferred tax assets, as it appears more likely than not that its net deferred tax assets will not be realized.
| F-40 |
Significant components of the Company’s deferred tax assets at December 31, 20092015 and 20082014 and related valuation reserves are presented below:
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryforwards | $ | 16,988,000 | $ | 18,370,000 | ||||
| Research and development tax credit carryforward | 1,710,000 | 1,628,000 | ||||||
| Charitable contribution carryforward | 37,000 | 39,000 | ||||||
| Accrued vacation | 8,000 | 12,000 | ||||||
| Accrued expenses | 163,000 | 150,000 | ||||||
| Amortization | 5,000 | 6,000 | ||||||
| Depreciation | 1,000 | — | ||||||
| Stock option expense | 975,000 | 919,000 | ||||||
| 19,887,000 | 21,124,000 | |||||||
| Less — valuation allowance | (19,887,000 | ) | (21,123,000 | ) | ||||
| Net deferred tax asset | — | 1,000 | ||||||
| Deferred tax liabilities: | ||||||||
| Depreciation | — | (1,000 | ) | |||||
| Net deferred tax amounts | $ | — | $ | — | ||||
| December 31, | ||||||||
| 2015 | 2014 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryforwards | $ | 17,856,000 | $ | 17,721,000 | ||||
| Research and development tax credit carryforward | 2,257,000 | 2,252,000 | ||||||
| Charitable contribution carryforward | 3,000 | 3,000 | ||||||
| Accrued expenses and deferred revenue | 565,000 | 164,000 | ||||||
| Amortization | 2,000 | 2,000 | ||||||
| Depreciation | 1,000 | 2,000 | ||||||
| Non-cash share based compensation | 1,083,000 | 1,044,000 | ||||||
| 21,767,000 | 21,188,000 | |||||||
| Less — valuation allowance | (21,767,000 | ) | (21,188,000 | ) | ||||
| Net deferred tax asset | $ | — | $ | — | ||||
At December 31, 2009,2015, we had net operating loss carryforwards for income tax purposes of approximately $43.1$45.3 million, which are available to offset future federal and state taxable income, if any, and, research and development tax credit carryforwards of approximately $1.7$2.3 million. The carryforwards, if not utilized, will expire in increments through 2029.
Section 382 of the Internal Revenue Code imposes substantial restrictions on the utilization of net operating losses and tax credits in the event of a corporation’s ownership change, as defined in Section 382 of the Code.change. During 2009, the Company completed a preliminary study to compute any limits on the net operating losses and credit carryforwards for purposes of Section 382. It was determined that the Company experienced a cumulative change in ownership, as defined by the regulations, in 2002. This change in ownership triggers an annual limitation on the Company’s ability to utilize certain U.S. federal and state net operating loss carryforwards and research tax credit carryforwards, resulting in the potential loss of approximately $9.8 million of net operating loss carryforwards and $0.2 million in research credit carryforwards. The Company has reduced the deferred tax assets associated with these carryforwards in its balance sheetsheets at December 31, 20092015 and 2008.
F-16
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Tax benefit at statutory rate | $ | (2,213,000 | ) | $ | (3,617,000 | ) | ||
| State taxes | (354,000 | ) | (579,000 | ) | ||||
| Permanent M-1s | 339,000 | 563,000 | ||||||
| Limited/expired net operating loss carryforwards | 3,546,000 | 6,150,000 | ||||||
| Limited/expired research and development tax credit carryforward | 120,000 | 284,000 | ||||||
| Research and development tax credit carryforward | (202,000 | ) | (504,000 | ) | ||||
| Change in effective tax rate | — | (455,000 | ) | |||||
| Change in valuation allowance | (1,236,000 | ) | (1,842,000 | |||||
| $ | — | $ | — | |||||
| December 31, | ||||||||
| 2015 | 2014 | |||||||
| Federal tax benefit at statutory rate | $ | (1,782,000 | ) | $ | (911,000 | ) | ||
| State taxes | (285,000 | ) | (146,000 | ) | ||||
| Change in fair value of derivative liabilities | 1,336,000 | 400,000 | ||||||
| Other permanent differences and other | 98,000 | 124,000 | ||||||
| Research and experimental tax credits | (8,000 | ) | (20,000 | ) | ||||
| Change in valuation allowance | 641,000 | 553,000 | ||||||
| $ | — | $ | — | |||||
As discussed in Note 2, we recognize the effect of income tax positions only if those positions more likely than not of being sustained. At December 31, 2009,2015 and December 31, 20082014, we had no gross unrecognized tax benefits. We do not expect any significant changes in unrecognized tax benefits over the next 12 months. In addition, we did not recognize any interest or penalties related to uncertain tax positions at December 31, 20092015 and 2008.2014.
The 2005 through 2015 tax years generally remain subject to examination by federal and most state tax authorities. In addition, we would remain open to examination for earlier years if we were to utilize net operating losses or tax credit carryforwards that originated prior to 2011.
| F-41 |
| COMMITMENTS |
Lease. Our rent expense, related solely to
Lease. Beginning in June 2014 we consolidated our office space for 2009 and 2008 was $91,183 and $100,196, respectively. We are committed under an office spaceamended our lease that expires on January 31, 2013 that requires the following approximate annual lease payments: $63,000, $94,000, $98,000 and $8,000agreement for the years ending December 31, 2010, through 2013, respectively.
Employment Continuity Agreements.Agreements. We have entered into employment contracts with our executive officers which provide for severance if the executive is dismissed without cause or under certain circumstances after a change of control in our ownership. At December 31, 20092015 these obligations, if triggered, could amount to a maximum of approximately $900,000$112,500 for termination without cause or $225,000 with a change of control in the aggregate.
F-17
| March 31, | December 31, | |||||||
| 2010 | 2009 | |||||||
| (Unaudited) | ||||||||
ASSETS | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | $ | 3,189,990 | $ | 4,355,768 | ||||
| Prepaid expenses and other current assets | 225,166 | 196,546 | ||||||
| Total current assets | 3,415,156 | 4,552,314 | ||||||
| Fixed assets, net of accumulated depreciation of $101,444 and $98,171 | 23,443 | 8,492 | ||||||
| Other assets | 17,255 | 22,948 | ||||||
| Total assets | $ | 3,455,854 | $ | 4,583,754 | ||||
LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities | ||||||||
| Accounts payable | $ | 177,718 | $ | 140,206 | ||||
| Accrued expenses | 588,651 | 740,198 | ||||||
| Total current liabilities | 766,369 | 880,404 | ||||||
| Commitments | — | — | ||||||
| Stockholders’ equity | ||||||||
| Preferred stock, $.001 par value per share, 1,000,000 authorized; no shares issued | — | — | ||||||
| Common stock, par value $.001 per share, 100,000,000 shares authorized; 60,406,828 issued and outstanding | 60,407 | 60,407 | ||||||
| Additional paid-in capital | 88,276,191 | 88,144,347 | ||||||
| Accumulated deficit | (85,647,113 | ) | (84,501,404 | ) | ||||
| Total stockholders’ equity | 2,689,485 | 3,703,350 | ||||||
| Total liabilities and stockholders’ equity | $ | 3,455,854 | $ | 4,583,754 | ||||
F-18
| Three Months Ended | ||||||||
| March 31, | ||||||||
| 2010 | 2009 | |||||||
| Revenues | $ | — | $ | — | ||||
| Operating expenses: | ||||||||
| Research and development | 470,434 | 1,661,600 | ||||||
| General and administrative | 678,068 | 859,568 | ||||||
| Total operating expenses | 1,148,502 | 2,521,168 | ||||||
| Loss from operations | (1,148,502 | ) | (2,521,168 | ) | ||||
| Interest income | 2,793 | 6,518 | ||||||
| Net loss | $ | (1,145,709 | ) | $ | (2,514,650 | ) | ||
| Basic and diluted net loss per common share | $ | (0.02 | ) | $ | (0.05 | ) | ||
| Weighted average number of common shares outstanding | 60,406,828 | 53,622,491 | ||||||
F-19
| For the Three Months Ended | ||||||||
| March 31, | ||||||||
| 2010 | 2009 | |||||||
| Operating activities: | ||||||||
| Net loss | $ | (1,145,709 | ) | $ | (2,514,650 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | 3,273 | 4,467 | ||||||
| Non-cash share-based compensation | 131,844 | 266,628 | ||||||
| Gain on settlement of accrued liabilities | (141,016 | ) | — | |||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | (28,620 | ) | 72,405 | |||||
| Other assets | 5,693 | — | ||||||
| Accounts payable | 37,512 | 301,075 | ||||||
| Accrued expenses | (10,531 | ) | 119,016 | |||||
| Net cash used in operating activities | (1,147,554 | ) | (1,751,059 | ) | ||||
| Investing activities: | ||||||||
| Purchase of fixed assets | (18,224 | ) | — | |||||
| Net cash used in investing activities | (18,224 | ) | — | |||||
| Net decrease in cash and cash equivalents | (1,165,778 | ) | (1,751,059 | ) | ||||
| Cash and cash equivalents at beginning of period | 4,355,768 | 5,655,367 | ||||||
| Cash and cash equivalents at end of period | $ | 3,189,990 | $ | 3,904,308 | ||||
F-20
On April 6, 2016, RegeneRx Biopharmaceuticals, Inc. (the “Company”, “We”, “Us”, “Our”"Company"), a Delaware corporation, was incorporated in 1982. We are focused on received $250,000 pursuant to the developmentsigning of a novel therapeutic peptide, Thymosin Beta 4 (“Tß4”), for tissueterm sheet with an affiliated entity. The parties are negotiating the expansion of the licensed rights, and organ protection, repair and regeneration. Our operations are confined to one business segment:full details of the development and marketingtransaction will be provided after execution of product candidates based on Tß4.
F-21
F-22
F-23
Issuable upon the Exercise of Warrants
, 2016
INFORMATION NOT REQUIRED IN PROSPECTUS
| Item 13. | Other Expenses of Issuance and Distribution. |
The following table sets forth all costs and expenses, other than underwriting discounts and commissions, payable by us in connection with the sale of the common stock being registered. All amounts shown are estimates except for the SECCommission registration fee, the FINRA filing fee and the NYSE Amex listing fee.
| Amount to | ||||
| be Paid | ||||
| SEC registration fee | $ | 798 | ||
| FINRA filing fee | 1,619 | |||
| NYSE Amex listing fee | 45,000 | |||
| Corporate finance fee payable to the representative of the underwriters | 64,400 | |||
| Printing and engraving expenses | 75,000 | |||
| Legal fees and expenses | 250,000 | |||
| Accounting fees and expenses | 25,000 | |||
| Transfer agent and registrar fees and expenses | 5,000 | |||
| Miscellaneous expenses | 10,000 | |||
| Total | $ | 476,816 | ||
| Amount to | ||||
| be Paid | ||||
| Commission registration fee | $ | 1,000 | ||
| Printing and engraving expenses | 5,000 | |||
| Legal fees and expenses | 40,000 | |||
| Accounting fees and expenses | 10,000 | |||
| Total | $ | 56,000 | ||
| Item 14. | Indemnification of Directors and Officers. |
We are incorporated under the laws of the State of Delaware. Section 102(b)(7) of the Delaware General Corporation Law, or DGCL, permits a corporation to provide in its certificate of incorporation that a director of the corporation shall not be personally liable to the corporation or its stockholders for monetary damages for breach of fiduciary duties as a director, except for liability for any:
| • | transaction from which the director derives an improper personal benefit; | |
| • | act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; | |
| • | unlawful payment of dividends or redemption of shares; or | |
| • | breach of a director’s duty of loyalty to the corporation or its stockholders. |
Our restated certificate of incorporation includes such a provision.
Section 145 of the DGCL provides that a Delaware corporation may indemnify any persons who are, or are threatened to be made, parties to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of such corporation), by reason of the fact that such person was an officer, director, employee or agent of such corporation, or is or was serving at the request of such person as an officer, director, employee or agent of another corporation or enterprise. Our amended and restated bylaws include such a provision. The indemnity may include expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with such action, suit or proceeding, provided that such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation’s best interests and, with respect to any criminal action or proceeding, had no reasonable cause to believe that his or her conduct was unlawful.
Section 145 of the DGCL also provides that a Delaware corporation may indemnify any persons who are, or are threatened to be made, a party to any threatened, pending or completed action or suit by or in the right of the corporation by reason of the fact that such person was a director, officer, employee or agent of such corporation, or is or was serving at the request of such corporation as a director, officer, employee or agent of another corporation or enterprise. Our amended and restated bylaws contain such a provision. The indemnity
II-1
Expenses incurred by any indemnitee in defending or investigating a threatened or pending action, suit or proceeding in advance of its final disposition shall be paid by us upon delivery to us of an undertaking, by or on behalf of such indemnitee, to repay all amounts so advanced if it shall ultimately be determined that such indemnitee is not entitled to be indemnified by us. No advance will be made by us if a determination is reasonably and promptly made by our board of directors by a majority vote of a quorum of disinterested directors, or if such a quorum is not obtainable or, even if obtainable, a quorum of disinterested directors so directs, by independent legal counsel in a written opinion, that, based upon the facts known to the board or counsel at the time such determination is made, such person did not meet the applicable standard of conduct in order to be indemnified.
At present, there is no pending litigation or proceeding involving any of our directors or officers as to which indemnification is required or permitted, and we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
We have an insurance policy covering our officers and directors with respect to certain liabilities, including liabilities arising under the Securities Act or otherwise.
We plan to enter into an underwriting agreement that provides that the underwriters are obligated, under some circumstances, to indemnify our directors, officers and controlling persons against specified liabilities, including liabilities under the Securities Act.
| Item 15. | Recent Sales of Unregistered Securities. |
The following list sets forth information regarding all unregistered securities sold by us since January 1, 20072013 through the date of this registration statement.
II-2
| · | On March 28, 2014,we issued 11,250,000 shares of common stock to GtreeBNT, Co. Ltd. for aggregate cash proceeds of $1,350,000. The offer, sale, and issuance of the shares to GtreeBNT were exempt from registration under the Securities Act under Section 4(2) of the Securities Act and Regulation D promulgated thereunder as transactions by an issuer not involving a public offering. GtreeBNT represented to the Company that it is an accredited investor as defined in Rule 501 promulgated under the Securities Act. |
| · | On August 29, 2014, we issued 8,333,333 shares of common stock to GtreeBNT, Co. Ltd. for aggregate cash proceeds of $1,000,000. The offer, sale, and issuance of the shares to GtreeBNT were exempt from registration under the Securities Act under Section 4(2) of the Securities Act and Regulation D promulgated thereunder as transactions by an issuer not involving a public offering. GtreeBNT represented to the Company that it is an accredited investor as defined in Rule 501 promulgated under the Securities Act. |
| · | On May 4, 2015, we issued 30,000 shares of our common stock, valued at approximately $16,500 to ProActive Capital Resources Group LLC in consideration for investor relations services. This issuance was exempt from registration under Section 4(2) of the Securities Act on the basis that the transactions did not involve a public offering. |
| · | On May 11, 2015, we issued 249,671 shares of our common stock to Lincoln Park Capital, LLC upon the cashless exercise of a warrant. This issuance was exempt from registration under Section 4(2) of the Securities Act on the basis that the transactions did not involve a public offering. |
| · | On June 29, 2016, we issued 5,147,047 shares of our common stock and warrants to purchase 5,147,047 shares of our common stock of our common stock to two purchasers in a private offering, and in connection with such private offering we issued warrants to purchase 257,353 shares of our common stock to the placement agent in such private offering. These issuances were exempt from registration under Rule 506 under Regulation D of the Securities Act and Section 4(2) of the Securities Act. |
| Item 16. | Exhibits and Financial Statement Schedules. |
| (a) | Exhibits |
| Exhibit No. | Description of Exhibit | Reference* | ||
| Exhibit | ||||
Number | Description of Document | |||
| 1 | .1 | Form of Underwriting Agreement. | ||
| 3 | .1# | Restated Certificate of Incorporation. | ||
| 3 | .2# | Certificate of Amendment of Restated Certificate of Incorporation. | ||
| 3 | .3# | Certificate of Amendment of Restated Certificate of Incorporation. | ||
| 3 | .4# | Certificate of Designation of Series A Participating Cumulative Preferred Stock. | ||
| 3 | .5(1) | Amended and Restated Bylaws adopted July 26, 2006. | ||
| 3 | .6(2) | Amendment to Amended and Restated Bylaws. | ||
| 4 | .1# | Specimen Common Stock Certificate. | ||
| 4 | .2# | Specimen Rights Certificate. | ||
| 4 | .3# | Rights Agreement, dated April 29, 1994, between the Company and American Stock Transfer & Trust Company, as Rights Agent. | ||
| 4 | .4# | Amendment No. 1 to Rights Agreement, dated March 4, 2004, between the Company and American Stock Transfer & Trust Company, as Rights Agent. | ||
| 4 | .5 | Form of Warrant Agreement. | ||
| 4 | .6 | Form of Warrant Certificate. | ||
| 4 | .7 | Form of Representative’s Warrant. | ||
| 5 | .1 | Opinion of Cooley LLP. | ||
| 10 | .1+(3) | Amended and Restated 2000 Stock Option and Incentive Plan, as amended. | ||
| 10 | .2*(4) | Patent License Agreement — Exclusive, dated January 24, 2001, between the Company and the U.S. Public Health Service. | ||
| 10 | .3*(5) | Thymosin Beta 4 License and Supply Agreement, dated January 21, 2004, between the Company and Defiante Farmaceutica S.A. | ||
| 10 | .4(6) | Lease by and between RegeneRx Biopharmaceuticals, Inc. and The Realty Associates Fund V, L.P., dated December 10, 2009. | ||
| 10 | .5+(8) | Second Amended and Restated Employment Agreement, dated March 11, 2009, between the Company and Allan L. Goldstein, as amended. | ||
| 10 | .6+(7) | Second Amended and Restated Employment Agreement, dated March 12, 2009, between the Company and J.J. Finkelstein, as amended. | ||
| 10 | .7+(7) | Second Amended and Restated Employment Agreement, dated March 31, 2009, between the Company and C. Neil Lyons, as amended. | ||
| 10 | .8+(7) | Second Amended and Restated Employment Agreement, dated March 31, 2009, between the Company and David Crockford. | ||
| 10 | .1(9) | Stock Purchase Agreement, dated as of June 23, 2005. | ||
| 10 | .2(10) | Form of Warrant to Purchase Common Stock, dated March 17, 2006. | ||
| 10 | .3(11) | Form of Warrant to Purchase Common Stock, dated December 18, 2006. | ||
| 10 | .4(11) | Registration Rights Agreement, dated as of December 15, 2006. | ||
| 10 | .5(12) | Securities Purchase Agreement, dated as of February 27, 2008. | ||
| 10 | .6(12) | Form of Warrant to Purchase Common Stock, dated February 29, 2008. | ||
| 10 | .7(13) | Securities Purchase Agreement, dated as of December 10, 2008. | ||
| 10 | .8(13) | Form of Warrant to Purchase Common Stock, dated December 10, 2008. | ||
II-3
| Exhibit | ||||
Number | Description of Document | |||
| 10 | .9(14) | Securities Purchase Agreement, dated as of April 13, 2009. | ||
| 10 | .10(14) | Form of Warrant to Purchase Common Stock, dated April 30, 2009. | ||
| 10 | .11(15) | Securities Purchase Agreement, dated as of September 30, 2009. | ||
| 10 | .12(15) | Form of Warrant to Purchase Common Stock, dated October 5, 2009. | ||
| 10 | .13(16) | Securities Purchase Agreement, dated as of September 30, 2009. | ||
| 10 | .14(16) | Form of Warrant to Purchase Common Stock, dated October 15, 2009. | ||
| 23 | .1 | Consent of Reznick Group, P.C., independent registered public accounting firm. | ||
| 23 | .2 | Consent of Cooley LLP (included in Exhibit 5.1). | ||
| 24 | .1# | Power of Attorney. Reference is made to Page II-6 of the Registration Statement onForm S-1 (FileNo. 333-166146) filed with the SEC on April 16, 2010. | ||
| Exhibit 3.1 to | ||||
| 3.2 | Certificate of Amendment to Restated Certificate of Incorporation | Exhibit 3.2 to Registration Statement on Form S-1 (File No. 333-166146) (filed April 16, 2010) | ||
| 3.3 | Certificate of Amendment to Restated Certificate of Incorporation | Exhibit 3.3 to Registration Statement on Form S-1 (File No. 333-166146) (filed April 16, 2010) | ||
| 3.4 | Certificate of Amendment of Restated Certificate of Incorporation | Exhibit 3.4 to Registration Statement on Form S-8 (File No. 333-168252) (filed July 21, 2010) | ||
| 3.5 | Certificate of Designation of Series A Participating Cumulative Preferred Stock | Exhibit 3.4 to Registration Statement on Form S-1 (File No. 333-166146) (filed April 16, 2010) | ||
| 3.6 | Amended and Restated Bylaws | Exhibit 3.4 to Quarterly Report onForm 10-Q (File No. 001-15070) for the quarter ended June 30, 2006 (filed August 14, 2006) | ||
| 3.7 | Amendment to Amended and Restated Bylaws | Exhibit 3.6 to Registration Statement on Form S-8 (File No. 333-152250) (filed July 10, 2008) | ||
| 4.1 | Specimen Common Stock Certificate | Exhibit 4.1 to Registration Statement on Form S-1 (File No. 333-166146) (filed April 16, 2010) | ||
| 4.2 | Specimen Rights Certificate | Exhibit 4.2 to Registration Statement on Form S-1 (File No. 333-166146) (filed April 16, 2010) | ||
| 4.3 | Rights Agreement, dated April 29, 1994, between the Company and American Stock Transfer & Trust Company, as Rights Agent | Exhibit 4.3 to Registration Statement on Form S-1 (File No. 333-166146) (filed April 16, 2010) | ||
| 4.4 | Amendment No. 1 to Rights Agreement, dated March 4, 2004, between the Company and American Stock Transfer & Trust Company, as Rights Agent | Exhibit 4.4 to Registration Statement on Form S-1 (File No. 333-166146) (filed April 16, 2010) | ||
| 4.5 | Warrant Agreement, dated May 21, 2010, between the Company and American Stock Transfer & Trust Company, as Warrant Agent | Exhibit 4.1 to Current Report on Form 8-K (File No. 001-15070) (filed May 21, 2010) | ||
| 4.6 | Form of Warrant Certificate | Exhibit 4.6 to Amendment No. 1 to Registration Statement on Form S-1 (File No. 333-166146) (filed May 17, 2010) | ||
| 4.7 | Form of Warrant Certificate | Exhibit 4.1 to Current Report on Form 8-K (File No. 001-15070) (filed July 1, 2016) |
| 4.8 | Placement Agent Warrant issued to Maxim Partners LLC | Exhibit 4.2 to Current Report on Form 8-K (File No. 001-15070) (filed July 1, 2016) | ||
| 5.1 | Opinion of Fredrikson & Byron, P.A. | Filed herewith | ||
| 10.1^ | Amended and Restated 2000 Stock Option and Incentive Plan, as amended | Annex A to the Company’s Proxy Statement on Schedule 14A (File No. 001-15070) (filed May 9, 2008) | ||
| 10.2^ | 2010 Equity Incentive Plan | Exhibit 10.1 to Current Report on Form 8-K (File No. 001-15070) (filed July 20, 2010) | ||
| 10.3 | Form of Stock Option Grant Notice and Stock Option Agreement under the 2010 Equity Incentive Plan | Exhibit 10.2 to Current Report on Form 8-K (File No. 001-15070) (filed July 20, 2010) | ||
| 10.4 | Patent License Agreement — Exclusive, dated January 24, 2001, between the Company and the U.S. Public Health Service | Exhibit B to Exhibit 10.1 to Amendment No. 1 to Quarterly Report on Form 10-Q for the quarter ended September 30, 2012 (File No. 001-15070) (filed January 16, 2013) | ||
| 10.5 | Thymosin Beta 4 License and Supply Agreement, dated January 21, 2004, between the Company and Defiante Farmaceutica S.A. | Exhibit 10.10 to Registration Statement on Form SB-2 (File No. 333-113417) (filed March 9, 2004)** | ||
| 10.6 | Lease, by and between the Company and The Realty Associates Fund V, L.P., dated December 10, 2009 | Exhibit 10.25 to Annual Report on Form 10-K for the year ended December 31, 2009 (File No. 001-15070) (filed March 31, 2010) | ||
| 10.7 | Form of Warrant to Purchase Common Stock dated April 30, 2009 | Exhibit 10.1 to Current Report on Form 8-K (File No. 001-15070) (filed April 16, 2009) | ||
| 10.8 | Form of Common Stock Purchase Warrant, dated October 5, 2009 | Exhibit 4.1 to Current Report on Form 8-K (File No. 001-15070) (filed September 30, 2009) | ||
| 10.9 | Form of Warrant, dated October 15, 2009 | Exhibit 4.1 to Current Report on Form 8-K (File No. 001-15070) (filed October 5, 2009) | ||
| 10.10 | Representative’s Warrant to Purchase Common Stock, dated May 21, 2010 | Exhibit 4.3 to Current Report on Form 8-K (File No. 001-15070) (filed May 21, 2010) | ||
| 10.11 | Registration Rights Agreement, dated January 4, 2011 | Exhibit 10.3 to Current Report on Form 8-K (File No. 001-15070) (filed January 7, 2011) | ||
| 10.12 | Warrant to Purchase Common Stock, dated January 7, 2011, issued to Lincoln Park Capital | Exhibit 4.1 to Current Report on Form 8-K (File No. 001-15070) (filed January 7, 2011) | ||
| 10.13 | Form of Warrant to Purchase Common Stock, dated January 7, 2011, issued to the Sigma-Tau Purchasers | Exhibit 4.2 to Current Report on Form 8-K (File No. 001-15070) (filed January 7, 2011) | ||
| 10.14^ | Amended and Restated Change in Control Agreement between the Company and J.J. Finkelstein, dated July 2, 2012 | Exhibit 10.8 to Current Report on Form 10-Q (File No. 001-15070) (filed August 14, 2012) | ||
| 10.15^ | Amended and Restated Change in Control Agreement between the Company and Allan L. Goldstein, dated July 2, 2012 | Exhibit 10.12 to Current Report on Form 10-Q (File No. 001-15070) (filed August 14, 2012) |
| 10.16 | Form of Convertible Promissory Note | Exhibit 4.1 to Current Report on Form 8-K (File No. 001-15070) (filed October 24, 2012) | ||
| 10.17 | Form of Warrant | Exhibit 4.2 to Current Report on Form 8-K (File No. 001-15070) (filed October 24, 2012) | ||
| 10.18 | Convertible Note and Warrant Purchase Agreement | Exhibit 10.1 to Current Report on Form 8-K (File No. 001-15070) (filed October 24, 2012) | ||
| 10.19 | License Agreement with Lee’s Pharmaceutical (HK) Limited | Exhibit 10.1 to Amendment No. 1 to Form 10-Q (File No. 001-15070) for the quarter ended September 30, 2012 (filed January 16, 2013)** | ||
| 10.20 | Form of Convertible Promissory Note | Exhibit 4.1 to Current Report on Form 8-K (File No. 001-15070) (filed April 2, 2013) | ||
| 10.21 | Convertible Note Purchase Agreement | Exhibit 10.1 to Current Report on Form 8-K (File No. 001-15070) (filed April 2, 2013) | ||
| 10.22 | Form of Convertible Promissory Note | Exhibit 4.1 to Current Report on Form 8-K (File No. 001-15070) (filed July 11, 2013) | ||
| 10.23 | Convertible Note Purchase Agreement | Exhibit 10.1 to Current Report on Form 8-K (File No. 001-15070) (filed July 11, 2013) | ||
| 10.24^ | Letter Agreement between the Company and J.J. Finkelstein, dated July 5, 2013 | Exhibit 10.2 to Current Report on Form 8-K (File No. 001-15070) (filed July 11, 2013) | ||
| 10.25^ | Letter Agreement between the Company and Allan L. Goldstein, dated July 5, 2013 | Exhibit 10.4 to Current Report on Form 8-K (File No. 001-15070) (filed July 11, 2013) | ||
| 10.26 | Form of Convertible Promissory Note | Exhibit 4.1 to Current Report on Form 8-K (File No. 001-15070) (filed September 19, 2013) | ||
| 10.27 | Convertible Note Purchase Agreement | Exhibit 10.1 to Current Report on Form 8-K (File No. 001-15070) (filed September 19, 2013) | ||
| 10.28 | Form of Convertible Promissory Note | Exhibit 4.1 to Current Report on Form 8-K (File No. 001-15070) (filed January 9, 2014) | ||
| 10.29 | Convertible Note Purchase Agreement | Exhibit 10.1 to Current Report on Form 8-K (File No. 001-15070) (filed January 9, 2014) | ||
| 10.30^ | Letter Agreement between the Company and J.J. Finkelstein, dated January 7, 2014 | Exhibit 10.2 to Current Report on Form 8-K (File No. 001-15070) (filed January 9, 2014) | ||
| 10.31 | Letter Agreement between the Company and Allan L. Goldstein, dated January 7, 2014 | Exhibit 10.3 to Quarterly Report on Form10-Q (File No. 001-15070) (filed January 9, 2014) | ||
| 10.32 | Securities Purchase Agreement | Exhibit 10.5 to Quarterly Report on Form10-Q (File No. 001-15070) (filed May 15, 2014) | ||
| 10.33 | License Agreement RGN-259 dated March 7, 2014 with GtreeBNT (formerly Digital Aria) | Exhibit 10.6 to Quarterly Report on Form10-Q (File No. 001-15070) (filed May 15, 2014)** |
| 10.34 | License Agreement RGN-137 dated March 7, 2014 with GtreeBNT (formerly Digital Aria) | Exhibit 10.7 to Quarterly Report on Form10-Q (File No. 001-15070) (filed May 15, 2014)** | ||
| 10.35^ | Executive Employment Agreement between the Company and J.J. Finkelstein dated April 16, 2014 | Exhibit 10.1 to Quarterly Report on Form10-Q (File No. 001-15070) (filed August 14, 2014) | ||
| 10.36^ | Executive Employment Agreement between the Company and Allan L. Goldstein dated April 16, 2014 | Exhibit 10.2 to Quarterly Report on Form10-Q (File No. 001-15070) (filed August 14, 2014) | ||
| 10.37^ | Executive Employment Agreement between the Company and Dane Saglio dated April 16, 2014 | Exhibit 10.3 to Quarterly Report on Form10-Q (File No. 001-15070) (filed August 14, 2014) | ||
| 10.38 | Form of First Amendment to Promissory Note dated October 3, 2014 | Exhibit 10.1 to Current Report on Form 8-K (File No. 001-15070) (filed October 9, 2014) | ||
| 10.39 | Joint Venture Agreement between the Company and GtreeBNT Co., Ltd. dated January 28, 2015 | Exhibit 10.1 to Quarterly Report on Form 10-Q (File No. 001-15070) (filed May 15, 2015) | ||
| 10.40 | License Agreement between the Company and ReGenTree, LLC dated January 28, 2015 | Exhibit 10.2 to Quarterly Report on Form 10-Q (File No. 001-15070) (filed May 15, 2015) | ||
| 10.41 | 2014 Amendment to Lease Agreement | Exhibit 10.41 to Annual Report on Form 10-K (File No. 001-15070) (filed April 11, 2016) | ||
| 10.42 | Securities Purchase Agreement, dated as of June 27, 2016, by and between the Company and the Purchasers identified therein | Exhibit 10.1 to Current Report on Form 8-K (File No. 001-15070) (filed July 1, 2016) | ||
| 10.43 | Registration Rights Agreement, dated as of June 27, 2016, by and between the Company and certain Purchasers identified therein | Exhibit 10.2 to Current Report on Form 8-K (File No. 001-15070) (filed July 1, 2016) | ||
| 23.1 | Consent of CohnReznick LLP | Filed herewith | ||
| 24.1 | Powers of Attorney | Included on signature page | ||
| 101 | The following materials from the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2015 and Quarterly Report on Form 10-Q for the quarter ended March 31, 2016, formatted in XBRL (eXtensible Business Reporting Language): (i) Balance Sheets at December 31, 2015 and 2014; (ii) Statements of Operations for the years ended December 31, 2015 and 2014; (iii) Statements of Changes in Stockholders’ Deficit for the years ended December 31, 2015 and 2014; (iv) Statements of Cash Flows for the years ended December 31, 2015 and 2014; (v) Notes to Financial Statements; (vi) Condensed Balance Sheets at March 31, 2016 and December 31, 2015; (vii) Condensed Statements of Operations for the three months ended March 31, 2016 and 2015; (viii) Condensed Statements of Cash Flows for the three months ended March 31, 2016 and 2015; and (ix) Notes to Condensed Financial Statements. | Filed herewith |
| * | Except where noted, the exhibits referred to in this column have heretofore been filed with the Securities and Exchange Commission | |
II-4
| ** | The registrant has been granted confidential treatment with respect to certain portions of this exhibit (indicated by asterisks), which have been filed separately with the Securities and Exchange Commission. |
| ^ | Compensatory plan, contract or arrangement. |
| Item 17. | Undertakings. |
The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement;
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability of the registrant under the Securities Act of 1933 to any purchaser in the initial distribution of the securities:
The undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424;
(ii) Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
II-5
(iv) Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the Registrant pursuant to the foregoing provisions, or otherwise, the Registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer or controlling person of the Registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
The undersigned Registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this Registration Statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this Registration Statement as of the time it was declared effective.
(2) For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-6
Pursuant to the requirements of the Securities Act, the Registrant has duly caused this Amendment No. 2 to Registration Statement on Form S-1 to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Rockville, State of Maryland, on the 17th20th day of May, 2010.
REGENERX BIOPHARMACEUTICALS, INC.
| By: | /s/ J.J. Finkelstein | |
J.J. Finkelstein President and Chief Executive Officer |
KNOW ALL BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints J.J. Finkelstein,
Pursuant to the requirements of the Securities Act, this Post-Effective Amendment No. 2 to Registration Statement has been signed by the following persons in the capacities and on the dates indicated.
Signature | Title | Date | ||||
/s/ J.J. Finkelstein | President and Chief Executive Officer and Director (Principal Executive Officer, Principal Financial Officer and Principal Accounting Officer) | July 20, 2016 | ||||
| J.J. Finkelstein | ||||||
/s/ | ||||||
| Allan L. Goldstein | ||||||
/s/ Joseph C. | ||||||
| Joseph C. McNay | ||||||
/s/ Mauro Bove | ||||||
Mauro Bove | ||||||
| Director | ||||||
II-7