As filed with the Securities and Exchange Commission on August 4, 2008June 13, 2019.
Registration Statement No. 333-152557
| UNITED STATES | ||||
| SECURITIES AND EXCHANGE COMMISSION | ||||
| Washington, D.C. 20549 | ||||
Amendment No. 2 to FORM S-1 | ||||
| REGISTRATION STATEMENT | ||||
| Under | ||||
| THE SECURITIES ACT OF 1933 | ||||
| SELLAS Life Sciences Group, Inc. | ||||
| (Exact name of registrant as specified in its charter) | ||||
| Delaware | 2834 | 20-8099512 | ||
| (State or other jurisdiction of incorporation or organization) | (Primary Standard Industrial Classification Code Number) | (I.R.S. Employer Identification |
60 Prescott Street15 West 38thSt., 10th Floor
New York, NY 10018
(917) 438-4353
| (Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices) |
Angelos M. Stergiou, M.D., Sc.D., h.c.
President and telephone number, including area code, of registrant’s principal executive offices)
SELLAS Life Sciences Group, Inc.
15 West 38thSt., 10th Floor
New York, NY 10018
(917) 438-4353
| (Name, address, including zip code, and telephone number, including area code, of agent for service) |
RXi Pharmaceuticals Corporation
Copies to:
Marc Rubenstein, Esq.
Joel I. Papernik Cliff M. Silverman | Barbara Wood Executive Vice President, General Counsel & Secretary SELLAS Life Sciences Group, Inc. 15 West 38th St., 10th Floor New York, NY 10018 Tel: (917) 438-4353 | Yvan-Claude J. Pierre Daniel I. Goldberg Marianne Sarrazin Cooley LLP 55 Hudson Yards New York, NY 10001 Tel: (212) 479-6000 |
Approximate date of commencement of proposed sale to public:As soon as practicable after this registration statement becomesRegistration Statement is declared effective.
If any of the securities being registered on this formForm are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box: box.þx
If this formForm is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering: offering.o¨
If this formForm is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering: offering.o¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering: offering.o¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company or emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer” andfiler,” “smaller reporting company,” and “emerging growth company” inRule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | ¨ | Accelerated filer | ¨ |
| Non-accelerated filer | x | Smaller reporting company | x |
| Emerging growth company | ¨ |
If an emerging growth company, indicate by check mark if the registrant has elected not check if a smaller reporting company)
CALCULATION OF REGISTRATION FEE
| Title of Each Class of Securities to be Registered | Proposed Maximum Aggregate Offering Price(1) | Amount of Registration Fee(2)(7) | ||||||
| Common Stock, $0.0001 par value per share(2)(3)(4) | $ | 23,000,000 | $ | 2,787.60 | ||||
| Pre-Funded Common Stock Purchase Warrants and Shares of Common Stock, $0.0001 par value per share, underlying Pre-Funded Common Stock Purchase Warrants(2)(3) | ||||||||
| Common Stock Purchase Warrants(2)(3)(5) | ||||||||
| Shares of Common Stock, $0.0001 par value per share, underlying Common Stock Purchase Warrants(2)(3)(4) | $ | 25,300,000(6) | 3,066.36 | |||||
| Total Registration Fee | $ | 48,300,000 | $ | 5,853.96 | ||||
| (1) | Estimated solely for the purpose of calculating the amount of registration fee pursuant to Rule 457(o) under the Securities Act of 1933, as amended (the “Securities Act”). |
| (2) | Pursuant to Rule 416, under the Securities Act the securities being registered hereunder include such indeterminate number of additional shares of common stock as may be issued after the date hereof as a result of stock splits, stock dividends or similar transactions. |
| (3) | The proposed maximum offering price of the common stock and accompanying warrants proposed to be sold in the offering will be reduced on a dollar-for-dollar basis based on the offering price of any pre-funded warrants offered and sold in the offering. |
| (4) | Includes the aggregate offering price of additional shares of common stock and/or warrants the underwriters have the option to purchase. |
| (5) | There will be issued a warrant to purchase one (1) share of common stock for every share or pre-funded warrant offered (at an exercise price of 110% of the public offering price). |
(6) | Relates to the shares of Common Stock, $0.0001 par value per share, underlying the Common Stock Purchase Warrants, if such Common Stock Purchase Warrants are exercised for cash. If such Common Stock Purchase Warrants are exercised cashlessly, then the underlying shares of Common Stock, $0.0001 par value per share, shall be covered by the Registration Fee in respect of the Common Stock, $0.0001 par value per share and accompanying Common Stock Purchase Warrants. |
| (7) | Filing fee of $5,575.20 previously paid. |
The registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine. |
The information contained in this preliminary prospectus is not complete and may be changed. We may not sell these securities until the Registration Statement shall become effective on such date asregistration statement filed with the Securities and Exchange Commission acting pursuantis effective. This preliminary prospectus is not an offer to said Section 8(a), may determine.
SUBJECT TO COMPLETION, DATED AUGUST 4, 2008JUNE 13, 2019
PRELIMINARY PROSPECTUS


RXi PHARMACEUTICALS CORPORATION
Pre-funded Warrants to the resalePurchase Shares of Common Stock
Common Warrants to Purchase up to 1,103,29947,619,048 Shares of Common Stock
We are offering 47,619,048 shares of our common stock and warrants to purchase up to an aggregate of 47,619,048 shares of our common stock (and the shares of common stock that are issuable from time to time upon exercise of RXi Pharmaceuticals Corporation (“RXi” or “the Company”)the common warrants). We are also offering to each purchaser whose purchase of shares of common stock in this offering would otherwise result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% of our outstanding common stock immediately following the consummation of this offering, the opportunity to purchase, if the purchaser so chooses, pre-funded warrants, in lieu of shares of common stock. Each pre-funded warrant will be exercisable for one share of our common stock. The purchase price of each pre-funded warrant will equal the price per share at which the shares of common stock are being sold to the public in this offering, minus $0.01, and the exercise price of each pre-funded warrant will be $0.01 per share. This prospectus also relates to the shares of common stock issuable upon exercise of any pre-funded warrants sold in this offering. For each pre-funded warrant we sell, the number of shares of common stock we are offering will be decreased on a one-for-one basis. Each share of common stock and pre-funded warrant is being sold together with a warrant to purchase one (1) share of our common stock, at an exercise price of $ per share (representing 110% of the public offering price).Because we will issue a common warrant to purchase one (1) share of our common stock for each share of our common stock and for each pre-funded warrant sold in this offering, the number of common warrants sold in this offering will not change as a result of a change in the mix of the shares of our common stock and pre-funded warrants sold. The common warrants will be exercisable immediately and will expire five years from the date of issuance. The shares of common stock or pre-funded warrants, and the accompanying common warrants, can only be purchased together in this offering but will be offered for resale by certain stockholders of the Company listed in this prospectus (the “Selling Stockholders”).
Our common stock is quotedlisted on the NASDAQThe Nasdaq Capital Market under the symbol “RXII”. On August 1, 2008, the“SLS.” The last reported closingsale price of the Company’sfor our common stock on the NASDAQThe Nasdaq Capital Market on June 11, 2019 was $7.40$0.42 per share.
Investing in our common stock involves a high degree of risk. See “Risk Factors” beginning on page 6.
Per Accompanying Common Warrant | Per Pre- Accompanying Common Warrant | Total(1) | ||||||||||
| Public offering price(2) | $ | $ | $ | |||||||||
| Underwriting discounts and commissions(3) | $ | $ | $ | |||||||||
| Proceeds, before expenses, to us | $ | $ | $ | |||||||||
| (1) | Assumes no sale of pre-funded warrants. |
| (2) | The public offering price is $ per share of common stock and $0.01 per accompanying common warrant and $ per pre-funded warrant and $0.01 per accompanying common warrant. |
| (3) | See the section entitled “Underwriting” beginning on page 59 of this prospectus for a description of the compensation payable to the underwriters. |
We have also granted an option to the underwriters to purchase up to 7,142,857 additional shares of common stock and/or common warrants on the same terms and conditions set forth above from us within 45 days after the date of this prospectus to cover over-allotments, if any.
Neither the Securities and Exchange Commission, or the SEC, nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The underwriters expect to deliver the shares of common stock and any pre-funded warrant and accompanying common warrant to purchasers on or about , 2019.
Sole Book-Running Manager
A.G.P.
Co-Manager
Maxim Group, LLC
The date of this prospectus is , 2008.
TABLE OF CONTENTS
i
You should not assume that the information contained in this prospectus is accurate on any date subsequent to the date set forth on the front of the document or that any prospectus supplement, as well as information contained in a document that we have previously filed or in the future will file with the SEC and incorporateincorporated by reference into this prospectus orherein is correct on any prospectus supplement, is accurate only as ofdate subsequent to the date of the document incorporated by reference, even though this prospectus is delivered, or securities are sold, on a later date.
No action is being taken in any jurisdiction outside the United States to permit a public offering of our common stock or possession or distribution of this prospectus in that jurisdiction. Person who come into possession of this prospectus in jurisdictions outside the United States are required to inform themselves about and to observe any restrictions as to this offering and the distribution of this prospectus applicable to that jurisdiction.
“SELLAS Life Sciences Group, Inc.,” “SELLAS,” the SELLAS logo, and other trademarks or service marks of SELLAS appearing in this prospectus are the property of our company. Other third-party logos and product/trade names are registered trademarks or trade names of their respective companies. Solely for convenience, trademarks and tradenames referred to in this prospectus appear (after the first usage) without the ® and ™ symbols, but those references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or that the applicable owner will not assert its rights, to these trademarks and tradenames.
This prospectus supplementcontains or incorporates by reference summaries of certain provisions contained in some of the document containing that information, asdocuments described herein, but reference is made to the case may be.
ii
| i |
This summary highlights information contained in other parts of this prospectus. In addition toBecause it is only a summary, it does not contain all the information you should consider before investing in our securities and it is qualified in its entirety by, and should be read in conjunction with, the more detailed information appearing elsewhere in this summary, we urge you toprospectus and the information incorporated by reference herein. You should read the entire prospectusall such documents carefully, especially the risks relating to our business and common stock discussed under the heading “Risk Factors”risk factors and our audited consolidated financial statements.statements and the related notes included herein, before deciding to buy our securities. Unless the context requires otherwise, references in this prospectus to “SELLAS,” “Company,” “we,” “us” and “our” refer to SELLAS Life Sciences Group, Inc. and our subsidiaries.
Company Overview
We are a discovery-stagelate-stage biopharmaceutical company pursuingfocused on the development and potential commercialization of proprietary therapeutics basednovel cancer immunotherapeutics for a broad range of indications.
Pipeline
Galinpepimut-S, or GPS
Our lead product candidate, galinpepimut-S, or GPS, is a cancer immunotherapeutic agent licensed from Memorial Sloan Kettering Cancer Center, or MSK, that targets the Wilms tumor 1, or WT1, protein, which is present in 20 or more cancer types. Based on RNA interference (RNAi)its mechanism of action as a directly immunizing agent, GPS has potential as a monotherapy or in combination with other immunotherapeutic agents to address a broad spectrum of hematologic, or blood, cancers and solid tumor indications.
In November 2018, following discussions with the U.S. Food and Drug Administration, or FDA, regarding a clinical trial design and biostatistical plan, we commenced preparations for a Phase 3 trial for GPS monotherapy in patients with acute myeloid leukemia, or AML, in the maintenance setting after achievement of their second complete remission, or CRem2, following successful completion of second-line antileukemic therapy. This trial is expected to serve as the basis for a Biologics License Application, or BLA, submission, subject to positive results. We are currently ready to start this Phase 3 trial, pending receipt of funding. The study is expected to enroll approximately 116 patients at approximately 50 clinical sites in the United States and Europe and is contemplated to have a planned interim safety and futility analysis after 80 events (deaths).
In December 2018, we initiated a Phase 1/2 multi-arm ("basket" type) clinical study of GPS in combination with Merck & Co., Inc.’s anti-PD-1 therapy, Keytruda® (pembrolizumab). We plan to enroll approximately 90 patients at up to 20 centers in the United States. The initial tumor types to be treated will be AML (in patients having achieved partial response as their best hematological response after four cycles of therapy with hypomethylating agents), and ovarian cancer (second or third line), to be followed by triple negative breast cancer, or TNBC, (second line), small cell lung cancer, or SCLC, (second line), and colorectal cancer (third or fourth line).
GPS was granted Orphan Drug Product Designations from the FDA as well as Orphan Medicinal Product Designations from the European Medicines Agency, or EMA, for GPS in AML, malignant pleural mesothelioma, or MPM, and multiple myeloma, or MM, as well as Fast Track Designation for AML, MPM, and MM from the FDA.
Nelipepimut-S or NPS
Nelipepimut-S, or NPS, is a cancer immunotherapy targeting the human epidermal growth factor receptor, or HER2, expressing cancers. Data presented in 2018 from our Phase 2b clinical trial of the combination of trastuzumab (Herceptin®) plus NPS in HER1/2+ breast cancer patients in the adjuvant setting to prevent recurrences showed a clinically and statistically significant improvement in the disease-free survival, or DFS, rate for the treatmentTNBC cohort at 24 months for patients treated with NPS plus trastuzumab of human diseases.92.6% compared to 70.2% for those treated with trastuzumab alone. In October 2018, the Data Safety Monitoring Board, or DSMB, unanimously concluded that the final analysis of the Phase 2b study data, with a median follow-up of 26 months, confirmed that TNBC patients should be the key target population for the development of trastuzumab plus NPS in the adjuvant setting in early-stage HER2 1+/2+ breast cancer patients. We believe RNAi-based therapeuticsare having ongoing discussions with the FDA since January 2019 to define an optimal path for further development of the combination of NPS plus trastuzumab in TNBC.
FBP-targeting bivalent vaccine (GALE-301/-302)
GALE-301 and GALE 302 are cancer immunotherapies that target the E39 peptide derived from the folate binding protein, or FBP. In a Phase 1/2a investigator sponsored trial, or IST, assessing GALE-301 in ovarian and endometrial cancers, we observed improvement in the 24-month DFS rate, in a small number of patients treated with the optimal dose. We are evaluating GALE-301/302 for potential internal development in a Phase 2 setting for ovarian cancer, strategic partnership, or other type of candidate rationalization.
| 1 |
The chart below summarizes the current status of our clinical development pipeline:

Our Strategy
We seek to use our expertise and understanding of cancer immunotherapy and general cancer therapeutic product development to advance novel products that have the potential to effectively treat a broad arraytransform the current standard of diseases by interfering with (sometimes referred to as silencing) the expression of targeted disease-associated genes. Our initial focus is on the treatment of neurological diseases, metabolic diseases and cancer.
1
| · | Continue to rapidly advance our pipeline of product candidates through clinical development, specifically our potential first-in-class, lead immunotherapy product candidate, GPS, which we are currently developing in both monotherapy and combination therapy settings. |
| · | Evaluate the potential for collaboration and license agreements with | |
Cancer Immunotherapy Market Overview
According to the 2018 “Global Oncology Trends” report by the IQVIA Institute, the global market for cancer drugs (including immunotherapy drugs) is expected to reach $200 billion by the end of 2022, growing at a compound annual growth rate, or CAGR, of 10-13% between 2017 and 2022. According to a 2018 report by Data Bridge Market Research (Pune, India), MarketsandMarkets, the global cancer immunotherapy market is expected to reach $202.89 billion by 2025, growing at a CAGR of 14.1% during the forecast period of 2018 to 2025. We expect that the first category of FDA-approved immunotherapies, immune synapse modulators (which includes checkpoint inhibitors and immune synapse co-stimulators), is likely to reach and exceed 90% of the immunotherapy market in the coming years, leaving approximately 10% for the other three major categories, which include peptide cancer active immunizers such as our product candidates, including GPS, NPS and the earlier stage peptide immunizers in our pipeline.
2
GPS targets malignancies and tumors characterized by an overexpression of the WT1 protein. The WT1 protein is one of the most widely expressed cancer proteins in multiple malignancies. A 2009 pilot project regarding the prioritization of cancer antigens conducted by the National Cancer Institute, or NCI, a division of the National Institutes of Health, or NIH, ranked the WT1 protein as a top priority for immunotherapy. WT1 is a protein that resides in the cell’s nucleus and participates in the process of cancer formation and progression. As such, it is classified as an “oncogene.” WT1 plays a key role in the development of the kidneys in fetal life, but then almost disappears from normal organs and tissues. In a wide variety of cancers (20 or more cancer types), WT1 becomes detectable again in the cells of these cancers. WT1 appears in large amounts (i.e., becomes “overexpressed”) in numerous hematological malignancies, including AML, MM and CML, as well as in many solid tumors such as MPM, gastrointestinal cancers (such as colorectal cancer), glioblastoma multiforme, TNBC, ovarian cancer and small-cell lung cancer. Overall, WT1 is expressed in at least 50% of tumor pathology specimens in 20 or more cancer types.
Our business is subject to RXi
| · | We have incurred substantial losses since our inception and anticipate that we will continue to incur substantial and increasing losses for the foreseeable future. |
| We will require substantial additional financing to achieve our goals, and a failure to obtain this necessary capital when needed could force us to delay, limit, reduce or terminate our product development or commercialization efforts. |
| · | We currently have no source of revenues. We may never generate revenues or achieve profitability. |
| · | We expect to continue to incur significant operating and non-operating expenses, which may make it difficult for us to secure sufficient financing and may lead to uncertainty about our ability to continue as a going concern. |
| · | We have announced that we are considering strategic alternatives in order to maximize stockholder value. We may not be able to |
| · | We have been involved in multiple legal and governmental proceedings, and may | |
| We are dependent on technologies we license, and if we lose the right to license such technologies or we fail to license new technologies in the future, our ability to develop new products would be harmed, and if we fail to meet our obligations under our license agreements, we may lose the ability to develop our product candidates. |
| · | We are currently a clinical-stage biopharmaceutical company with product candidates in clinical development. If we are unable to successfully develop and commercialize product candidates or experiences significant delays in doing so, our business may be materially harmed. |
| · | Our future success is dependent on the regulatory approval of our product candidates. |
Additional Information
For additional information related to our business and operations, please refer to the reports incorporated herein by reference, including our Annual Report on Form 10-K for the year ended December 31, 2018 as filed with the SEC on March 22, 2019, as amended on April 30, 2019 or the 2018 Form 10-K, our Quarterly Report on Form 10-Q for the quarterly period ended March 31, 2019 as filed with the SEC on May 15, 2019, or the 2019 Form 10-Q, and our Current Reports on Form 8-K as filed with the SEC, as described in the section entitled “Incorporation of Documents by Reference” beginning on page 63 of this prospectus.
Our Corporate Information
We were incorporated on April 3, 2006 in Delaware as Argonaut Pharmaceuticals, Inc. in Delaware on April 3,On November 28, 2006, and we changed our name to RXi Pharmaceuticals Corporation and began operations in January 2007. On September 26, 2011, we changed our name to Galena Biopharma, Inc. In December, 2017, we completed the business combination with the privately held Bermuda exempted company, Sellas Life Sciences Group Ltd., or Private SELLAS, which we refer to throughout the registration statement of which this prospectus forms as part of the “Merger.” As a result of the Merger, our business is now substantially comprised of the business of Private SELLAS. Upon completion of the Merger, we changed our name from “Galena Biopharma, Inc.” to “SELLAS Life Sciences Group, Inc.,” our common stock began trading on November 28, 2006. The Nasdaq Capital Market under a new ticker symbol “SLS” on January 2, 2018 and our financial statements became those of Private SELLAS.
Our principal executive office isoffices are located at 60 Prescott15 West 38th Street, Worcester, Massachusetts 0160510th Floor, New York, NY 10018, and our telephonephone number is(508) 767-3861. (917) 438-4353. Our Internetwebsite address is www.rxipharma.com. Our website and thewww.sellaslife.com. The information contained on, or that site, or connected to that site,can be accessed through, our website is not a part of or incorporated by reference into this prospectus.
| 3 |
The Offering
| Common stock offered by us | 47,619,048shares (54,761,905 shares if the underwriters exercise their over-allotment option in full), assuming the sale of our shares of common stock at an assumed public offering price of $0.42 per share, which is the last reported sale price of our common stock on Nasdaq on June 11, 2019, and no sale of any pre-funded warrants. | |
| We | ||
| Common warrants offered by us | We are also offering common warrants to purchase up to an aggregate of 47,619,048 shares of our common stock (or common warrants to purchase 54,761,905 shares of our common stock if the underwriters exercise their over-allotment option in full). Each share of our common stock and each pre-funded warrant is being sold together with a common warrant to purchase one share of our common stock. Each common warrant will have an exercise price of $ per share (representing 110% of the public offering price), will be immediately exercisable and will expire on the fifth anniversary of the original issuance date. This prospectus also relates to the offering of the shares of common stock issuable upon exercise of the common warrants. | |
| Option to purchase additional securities | The underwriter has a 45-day option to purchase up to an additional 7,142,857 shares of common stock and/or common warrants to purchase up to an additional 7,142,857 shares of our common stock from us at the public offering price, less underwriting discounts and commissions. | |
| Common stock to be outstanding after this offering | 72,795,523shares (assuming the sale of $20,000,000of our shares of common stock at an assumed public offering price of $0.42 per share, which is the last reported sale price of our common stock on Nasdaq on June 11, 2019, no sale of any pre-funded warrants, no exercise of any of the common warrants issued in this offering, and no exercise of the underwriters’ over-allotment option). | |
| Use of Proceeds | We estimate the net proceeds to us from this offering, after deducting underwriting discounts and commissions and estimated offering expenses payable by us, will be approximately $18.1 million, assuming a combined public offering price of $0.42 per share and accompanying common warrant, which is the last reported sale price of our common stock on Nasdaq on June11, 2019. The actual offering price for the offered securities will be as determined between us and the underwriters at the time of pricing, and may be at a discount to the current market price. We intend to use the net proceeds from this offering to commence a Phase 3 study for GPS monotherapy in AML CRem2 patients and to continue our Phase 1/2 basket type study of GPS in combination with pembrolizumab, and for general corporate purposes. See the section entitled “Use of Proceeds” beginning on page 46 of this prospectus. | |
| Risk Factors | An investment in our securities involves a high degree of risk. See the section entitled “Risk Factors” beginning on page 7 of this prospectus and the similarly entitled sections in the documents incorporated by reference into this prospectus. | |
Nasdaq Capital Market symbol | “SLS.” We do not intend to list the pre-funded warrants or the common warrants on any securities exchange or nationally recognized trading system. |
3
Upon completion of this offering, we intend to take such steps as are necessary to reduce the exercise price of all of the warrants that were issued on July 16, 2018 and that remain outstanding, to the public offering priceper share of common stock and accompanying common warrant.
Outstanding Shares
Except as otherwise indicated herein, the number of shares of our common stock to be outstanding after this offering is based on 24,176,475 shares of common stock outstanding as of March 31, 2019 and excludes:
| · | 17,698,061 shares of common stock issuable as of the date hereof upon the exercise of common stock warrants outstanding as of March 31, 2019 at a weighted average exercise price of $5.31 per share (including an aggregate of 1,000,000 shares of common stock issued between April 1, 2019 and May 31, 2019 upon the exercise of warrants pursuant to that certain warrant exercise agreement dated March 6, 2019) and 1,000,000 additional shares of common stock issuable as of the date hereof upon exercise of warrants issued subsequent to March 31, 2019, which have an exercise price of $1.40 per share; |
| · | 1,334,321 shares of common stock issuable upon the exercise of stock options outstanding as of March 31, 2019 at a weighted-average exercise price of $11.94 per share; |
| · | 12,758 shares of common stock issuable upon settlement of outstanding restricted stock unit, or RSU, awards as of March 31, 2019; |
| · | 362,208 shares of common stock available for future issuance under the 2017 Equity Incentive Plan, or the 2017 Plan, as of March 31, 2019; |
| · | 265,131 shares of common stock available for future issuance under the Employee Stock Purchase Plan, or the ESPP, as of March 31, 2019; and |
| · | The exercise by the underwriters of their over-allotment option to purchase additional shares of common stock and/or common warrants. |
| 4 |
Summary HistoricalCondensed Consolidated Financial Information
The following table sets forth our summary condensed consolidated financial data for the periods indicated. We derived the following condensed consolidated statements of operations data for the years ended December 31, 2018 and 2017 from our audited consolidated financial statements and the related notes appearing in the 2018 Form 10-K, which is incorporated by reference into this prospectus. We derived the following condensed consolidated statements of operations data for the three months ended March 31, 2019 and 2018 and the following condensed consolidated balance sheet data as of March 31, 2019 from our unaudited interim condensed consolidated financial statements and the related notes appearing in the 2019 Form 10-Q, which is incorporated by reference into this prospectus. The unaudited interim condensed consolidated financial statements have been prepared on a basis consistent with our audited consolidated financial statements incorporated by reference in this prospectus and include, in our opinion, all adjustments, consisting only of normal recurring adjustments, necessary for the fair presentation of the financial information in those statements.
The following summary historical financial informationdata should be read together with our consolidated financial statements and related notes appearing in conjunction with “Management’sthe 2018 Form 10-K and in the 2019 Form 10-Q, as well as in the sections entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the financial statements and corresponding notes to financial statements included elsewhereOperations” in this prospectus.
| Year Ended December 31, | Three Months Ended March 31, | |||||||||||||||
| 2018 | 2017 | 2019 | 2018 | |||||||||||||
| (in thousands) | ||||||||||||||||
| (unaudited) | ||||||||||||||||
| Statement of Operations Data: | ||||||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 8,767 | $ | 6,067 | $ | 1,859 | $ | 1,804 | ||||||||
| General and administrative | 12,772 | 15,089 | 2,500 | 3,880 | ||||||||||||
| In-process research and development impairment charge | 9,550 | — | — | — | ||||||||||||
| Severance costs | — | 1,883 | — | — | ||||||||||||
| Total operating expenses and loss from operations | (31,089 | ) | (23,039 | ) | (4,359 | ) | (5,684 | ) | ||||||||
| Change in fair value of warrant liability | 5,300 | — | 195 | 1,881 | ||||||||||||
| Change in fair value of the contingent consideration | (3,032 | ) | — | (387 | ) | (3,411 | ) | |||||||||
| Loss on settlement of liability-classified warrants | (727 | ) | — | — | (685 | ) | ||||||||||
| Gain on extinguishment of debt | 766 | — | — | — | ||||||||||||
| Interest expense, net | (266 | ) | (462 | ) | 14 | (96 | ) | |||||||||
| Income tax (benefit) expense | (1,378 | ) | 253 | — | — | |||||||||||
| Net loss | (27,670 | ) | (23,754 | ) | (4,537 | ) | (7,995 | ) | ||||||||
| Deemed dividend on conversion of 2015 Sely Note | — | (675 | ) | — | — | |||||||||||
| Deemed dividend arising from the issuance of common stock to Series A convertible preferred stockholders under most favored nation provision | (8,654 | ) | ||||||||||||||
| Deemed dividend arising from beneficial conversion feature of convertible preferred stock | (4,436 | ) | — | — | (1,968 | ) | ||||||||||
| Impact of anti-dilution protection on liability-classified warrants | (491 | ) | (439 | ) | ||||||||||||
| Net loss attributable to common stock stockholders | $ | (41,251 | ) | $ | (24,429 | ) | $ | (4,976 | ) | $ | (9,963 | ) | ||||
| Basic and diluted loss per share to common stock stockholders | $ | (3.15 | ) | $ | (10.44 | ) | $ | (0.22 | ) | $ | (1.67 | ) | ||||
| 5 |
| As of March 31, 2019 | ||||||||||||
(in thousands) Unaudited | Actual | Pro forma(1) | Pro forma as adjusted(2) | |||||||||
| Balance Sheet Data: | ||||||||||||
| Cash and cash equivalents | $ | 2,576 | $ | 3,621 | $ | 21,741 | ||||||
| Restricted cash and cash equivalents | 114 | 114 | 114 | |||||||||
| Working capital (deficit) | (2,388 | ) | (1,343 | ) | 16,777 | |||||||
| Total assets | 14,927 | 15,972 | 34,092 | |||||||||
| Total liabilities | 11,977 | 11,977 | 11,977 | |||||||||
| Accumulated deficit | (86,392 | ) | (86,392 | ) | (86,392 | ) | ||||||
| Total stockholders' equity | 2,950 | 3,995 | 22,115 | |||||||||
| (1) | The pro forma amounts give effect of the exercise of 1,000,000 warrants between April 1, 2019 and May 31, 2019 to purchase shares of common stock at an exercise price of $1.10 per share, resulting in net cash proceeds received of approximately $1.0 million after deducting commissions and offering expenses. |
| (2) | Pro forma as adjusted amounts reflect the pro forma adjustment described in footnote (1) as well as the sale of47,619,048 shares of our common stock and accompanying common warrants in this offering at an assumed public offering price of $0.42 per share, after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us, and assuming no pre-funded warrants are issued. |
| 6 |
Risks Relating to Our Financial Position and Capital Needs
We have incurred substantial losses since our inception and anticipate that we will continue to incur substantial and increasing losses for the foreseeable future.
We are a clinical stage biopharmaceutical company focused on development of novel cancer immunotherapies for a broad range of cancer indications. Investment in biopharmaceutical product development is highly speculative because it entails substantial upfront capital expenditures and significant risk that a product candidate will fail to prove effective, gain regulatory approval or become commercially viable. We do not have any products approved by regulatory authorities and have not generated any revenues from collaboration and licensing agreements or product sales to date, and have incurred significant research, development and other expenses related to our ongoing operations and expect to continue to incur such expenses. As a result, we have not been profitable and have incurred significant operating losses in every reporting period since our inception. For the three months ended March 31, 2019 and the years ended December 31, 2007, 2006, 20052018 and 2004.2017, we reported a net loss of $4.5 million, $27.7 million and $23.8 million respectively. As of March 31, 2019 and December 31, 2018, we had an accumulated deficit of $86.4 million and $81.9 million respectively.
We do not expect to generate revenues for many years, if at all. We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate these losses to increase as we continue to research, develop and seek regulatory approvals for our product candidates and any additional product candidates we may acquire, and potentially begin to commercialize product candidates that may achieve regulatory approval. We may also encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business. The information presentedsize of our future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues. Our expenses will further increase as we:
• | conduct additional clinical trials of our lead product, GPS, including the Phase 3 clinical trials evaluating GPS for AML and other cancers; | |
• | continue to develop immunotherapy programs for NPS; | |
• | pursue research and development of our other product candidates, including GALE-301 (a vaccine against the E39 peptide derived from the folate binding protein, or FBP) and GALE-302 (a vaccine against the J65 peptide derived from FBP); | |
• | in-license or acquire the rights to, and pursue development of, other products, product candidates or technologies; | |
• | hire additional clinical, manufacturing, quality control, quality assurance and scientific personnel; | |
• | seek marketing approval for any product candidates that successfully complete clinical trials; | |
• | develop our outsourced manufacturing and commercial activities and establish sales, marketing and distribution capabilities, if we receive, or expect to receive, marketing approval for any product candidates; | |
• | maintain, expand and protect our intellectual property portfolio; and | |
• | add operational, financial and management information systems and personnel. |
We need significant additional financing to fund our operations and complete the development and, if approved, the commercialization of our product candidates. If we are unable to raise capital when needed, we could be forced to delay, reduce or eliminate our product development programs or commercialization efforts.
We expect our existing cash as of March 31, 2019 and the proceeds received in May 2019 upon the exercise of common stock warrants will enable us to fund our operating expenses through the first half of 2019. Our existing cash will not be sufficient to complete development and obtain regulatory approval for any of our lead product candidates, and we will need to raise significant additional capital to help us do so. In addition, our operating plan may change as a result of many factors currently unknown to us, and we may need additional funds sooner than planned.
| 7 |
We expect to expend substantial resources for the three-month periods ended March 31, 2008foreseeable future to continue the clinical development and 2007manufacturing of our product candidates and the advancement and expansion of our preclinical research pipeline, in particular the Phase 1/2 basket study of GPS in combination with pembrolizumab and our planned Phase 3 study of GPS in AML. These expenditures will include costs associated with research and development, potentially acquiring new product candidates or technologies, conducting preclinical studies and clinical trials and potentially obtaining regulatory approvals and manufacturing products, as well as marketing and selling products approved for sale, if any.
Our future capital requirements depend on many factors, including:
4
| Period from | ||||||||||||||||||||||||||||
| January 1, | For the | For the | ||||||||||||||||||||||||||
| 2003 (Date of | Three | Three | For the | For the | For the | For the | ||||||||||||||||||||||
| Inception) | Months | Months | Year | Year | Year | Year | ||||||||||||||||||||||
| through | Ended | Ended | Ended | Ended | Ended | Ended | ||||||||||||||||||||||
| March 31, | March 31, | March 31, | December 31, | December 31, | December 31, | December 31, | ||||||||||||||||||||||
| 2008 | 2008 | 2007 | 2007 | 2006 | 2005 | 2004 | ||||||||||||||||||||||
| (Successor) | (Successor) | (Successor) | (Successor) | (Predecessor) | (Predecessor) | (Predecessor) | ||||||||||||||||||||||
| (In thousands, except share and per share amounts) | ||||||||||||||||||||||||||||
Statement Expenses Data: | ||||||||||||||||||||||||||||
| Expenses: | ||||||||||||||||||||||||||||
| Research and development | $ | 16,376 | $ | 1,088 | $ | 824 | $ | 6,747 | $ | 1,772 | $ | 2,080 | $ | 2,814 | ||||||||||||||
| General and administrative | 7,816 | 1,625 | 588 | 4,666 | 633 | 129 | 458 | |||||||||||||||||||||
| Operating loss | (24,192 | ) | (2,713 | ) | (1,412 | ) | (11,413 | ) | (2,405 | ) | (2,209 | ) | (3,272 | ) | ||||||||||||||
| Interest income | 523 | 75 | — | 448 | — | — | — | |||||||||||||||||||||
| Other expense | (8 | ) | (8 | ) | — | — | — | — | — | |||||||||||||||||||
| Loss before income taxes | (23,677 | ) | (2,646 | ) | (1,412 | ) | (10,965 | ) | (2,405 | ) | (2,209 | ) | (3,272 | ) | ||||||||||||||
| Income taxes | — | — | — | (25 | ) | — | — | — | ||||||||||||||||||||
| Net loss | $ | (23,677 | ) | $ | (2,646 | ) | $ | (1,412 | ) | $ | (10,990 | ) | $ | (2,405 | ) | $ | (2,209 | ) | $ | (3,272 | ) | |||||||
| Basic and diluted net loss per share | N/A | $ | (0.21 | ) | $ | (0.17 | ) | $ | (0.99 | ) | N/A | N/A | N/A | |||||||||||||||
| Weighted average shares outstanding, basic and diluted | N/A | 12,684,432 | 8,117,016 | 11,113,137 | N/A | N/A | N/A | |||||||||||||||||||||
| As of | As of | As of | As of | As of | As of | |||||||||||||||||||
| March 31, | December 31, | December 31, | December 31, | December 31, | December 31, | |||||||||||||||||||
| 2008 | 2007 | 2006 | 2006 | 2005 | 2004 | |||||||||||||||||||
| (Successor) | (Successor) | (Successor) | (Predecessor) | (Predecessor) | (Predecessor) | |||||||||||||||||||
Balance Sheet Data: | ||||||||||||||||||||||||
| Cash and cash equivalents | $ | 9,857 | $ | 1,763 | $ | 2 | $ | — | $ | — | $ | — | ||||||||||||
| Short term investments | — | 9,952 | — | — | — | — | ||||||||||||||||||
| Total current assets | 10,179 | 11,737 | 2 | — | — | — | ||||||||||||||||||
| Working capital | 8,405 | 10,413 | 2 | (318 | ) | (500 | ) | (968 | ) | |||||||||||||||
| Total assets | 10,580 | 12,147 | 2 | 57 | 50 | — | ||||||||||||||||||
| Due to parent | (67 | ) | (207 | ) | — | — | — | — | ||||||||||||||||
| Total stockholders’ equity | 8,806 | 10,823 | 2 | — | — | — | ||||||||||||||||||
| Parent company’s net deficit | — | — | — | (268 | ) | (450 | ) | (968 | ) | |||||||||||||||
5
Additional funds may not demonstrate in patients the chemical and pharmacological properties ascribedbe available when we need them on terms that are acceptable to them in laboratory studies, and they may interact with human biological systems in unforeseen, ineffectiveus, or even harmful ways.
We have announced that we are considering strategic alternatives in order to compete successfully we may need to be first to marketmaximize stockholder value, including financings, strategic alliances, acquisitions or to demonstrate that our RNAi based products are superior to therapies based on different technologies. If we are not first to market or are unable to demonstrate such superiority, any products for which we are able to obtain approval may not be successful. For example, Isis Pharmaceuticals, Inc. has begun pre-clinical toxicology studies for an antisense-based therapeutic product candidate, for which the FDA has granted orphan drug status, that targets the same gene for ALS that we intend to target. If Isis is able to successfully bring this treatment to market before we are able to complete the development of an RNAi therapeutic in this area, even if our development efforts are successful, we may not receive any market advantages that we would have benefited from if ours were the first such therapeutic product available on the market. Furthermore, under U.S. law, if a competitor has orphan drug status for a product and if our product candidate is determined to be contained within the competitor’s product for the same indication or disease, then that competitor would have market exclusivity and approval of our product for that indication or disease could potentially be blocked for seven years. Note that Isis’ product candidate for ALS should not present this challenge to any of our potential
6
We have announced that we are considering all strategic alternatives that may be available to us to maximize stockholder value, including financings, strategic alliances, acquisitions or the third party relationshipspossible sale of the company. We currently have no agreements or commitments to engage in any specific strategic transactions, and our exploration of various strategic alternatives may not result in any specific action or transaction. To the extent that this engagement results in a transaction, our business objectives may change depending upon the nature of the transaction. There can be no assurance that we will enter into any transaction as a result of the engagement.
| 8 |
Furthermore, if we determine to engage in a strategic transaction, we cannot predict the impact that such strategic transaction might have on our operations or stock price. We also cannot predict the impact on our stock price if we fail to enter into a transaction.
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our product candidates on unfavorable terms to us.
We may seek additional capital through a variety of means, including through private and public equity offerings and debt financings, collaborations, strategic alliances and marketing, distribution or licensing arrangements. To the extent that we raise additional capital (including this offering) through the sale of equity or convertible debt securities, or through the issuance of shares under management or other types of contracts, or upon the exercise or conversion of outstanding derivative securities, the ownership interests of our stockholders will be diluted, and the terms of such financings may include liquidation or other preferences, anti-dilution rights, conversion and exercise price adjustments and other provisions that adversely affect the rights of our stockholders, including rights, preferences and privileges that are necessarysenior to those of our holders of common stock in the event of a liquidation. For example, in March and May 2018, we issued convertible preferred stock which contained rights, preferences and privileges which were senior to those of holders of our common stock. Such preferred stock is no longer outstanding. In such event, there is a possibility that once all senior claims are settled, there may be no assets remaining to pay out to the holders of our common stock. Debt financing, if available, could include covenants limiting or restricting our ability to take certain actions, such as incurring additional debt, making capital expenditures, entering into licensing arrangements, or declaring dividends and may require us to grant security interests in our assets, including our intellectual property, and for our subsidiaries to guarantee our obligations. If we raise additional funds through collaborations, strategic alliances, or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, products or product candidates or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to grant rights to develop and market products or potentially commercialize some or allproduct candidates that we would otherwise prefer to develop and market ourselves.
There is substantial doubt about our ability to continue as a going concern.
As of our product candidates.
We currently have no source of revenues. We may never generate revenues or achieve profitability.
Currently, we do not generate any revenues from product candidatessales or otherwise. Even if we obtainare able to successfully achieve regulatory approval for our product candidates, we do not know when we will generate revenues or become profitable, if at all. Our ability to generate revenues from product sales and achieve profitability will depend on our ability to successfully commercialize them.
| 9 |
protect our rights in our intellectual property portfolio.
Our revenues for any product candidate for which regulatory approval is obtained will be dependent, in part, upon the size of the markets in the territories for which it gains regulatory approval, the accepted price for the product, the ability to get reimbursement at any price, and whether we own the commercial rights for that territory. If the number of our addressable disease patients is not as significant as our estimates, the indication approved by regulatory authorities is narrower than we expect, or the reasonably accepted population for treatment is narrowed by competition, physician choice or treatment guidelines, we may not generate significant revenues from UMMS.sales of such products, even if approved. In addition, we anticipate incurring significant costs associated with commercializing any approved product candidate. As a result, even if we generate revenues, we may not become profitable and may need to obtain additional funding to continue operations. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and may be forced to reduce our operations.
The Tax Cuts and Jobs Act could adversely affect our business and financial condition.
H.R. 1, “An Act to provide for reconciliation pursuant to title II and V of the concurrent resolution on the budget for fiscal year 2018,” informally entitled the Tax Cuts and Jobs Act, or the Tax Act, enacted on December 22, 2017, among other things, contains significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a single rate of 21%, limitation of the tax deduction for interest expense to 30% of adjusted taxable income (except for certain small businesses), limitation of the deduction for net operating losses carried forward from taxable years beginning after December 31, 2017 to 80% of current year taxable income and elimination of net operating loss carrybacks, one time taxation of offshore earnings at reduced rates regardless of whether they are repatriated, elimination of U.S. tax on foreign earnings (subject to certain important exceptions), providing immediate deductions for certain new investments instead of deductions for depreciation expense over time, and modifying or repealing many business deductions and credits (including reduction of tax credits under the Orphan Drug Act). Notwithstanding the reduction in the corporate income tax rate, the overall impact of the Tax Act is uncertain and our business and financial condition could be adversely affected. In addition, it is uncertain if and to what extent various states will conform to the Tax Act.
Our ability to use net operating losses to offset future taxable income may be subject to limitations.
As of December 31, 2018, we had federal and state net operating loss carryforwards of approximately $19.7 million and $3.7 million, respectively. Our NOLs generated in tax years ending on or prior to December 31, 2017 are only permitted to be carried forward for 20 years under applicable U.S. tax laws, and will begin to expire, if not utilized, beginning in 2027. These NOL carryforwards could expire unused and be unavailable to offset future income tax liabilities. Under the Tax Act, federal NOLs incurred in tax years ending after December 31, 2017 may be carried forward indefinitely, but the deductibility of such federal NOLs is limited. It is anticipated that refinementuncertain if andscale-up to what extent various states will conform to the Tax Act, or whether any further regulatory changes may be adopted in the synthesis methods willfuture that could minimize its applicability. In addition, under Section 382 of the Internal Revenue Code of 1986, as amended, and certain corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50% change, by value, in the ownership of its equity over a three-year period, the corporation’s ability to use its pre-change NOL carryforwards and other pre-change tax attributes to offset its post-change income may be performed under contractlimited. The Merger constituted an ownership change and as such, our ability to use our NOL carryforwards is materially limited, which may harm our future operating results by effectively increasing our future tax obligations.
| 10 |
Risks Related to the Development and Regulatory Approval of Our Product Candidates
Clinical-stage biopharmaceutical companies with this manufacturer. However, as the nanotransportersproduct candidates in clinical development face a wide range of challenging activities which may entail substantial risk.
We are unique chemicals, the costs of synthesis are not currently known and there is potential for technical challengesa clinical-stage biopharmaceutical company with respect toscale-up.
If we do not achieve one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully develop and commercialize our product candidates, which could materially harm our business.
Our lead product candidate, GPS, represents a new therapeutic approach that presents significant challenges.
Our future success is substantially dependent on the successful development of WT1 peptide immunotherapies in general and GPS in particular. Because this program represents a new approach to cancer immunotherapy for clinical use. The chemistry, manufacturingthe treatment of cancer and controlsother diseases, developing and commercializing GPS subjects us to a number of challenges, including:
| 11 |
Moreover, public perception of safety issues, including adoption of new therapeutics or novel approaches to treatment, may adversely influence the willingness of subjects to participate in increasedclinical trials, or if approved, of physicians to subscribe to the novel treatment mechanics. Physicians, hospitals and third-party payors often are slow to adopt new products, technologies and treatment practices that require additional educational upfront costs and delays in manufacturing. Additionally, although we are not currently aware of any such litigation or threatened litigation or challenge, if we have litigation or threatened litigation for or challenge to the composition of our products candidates in the future, manufacturers may refuse to manufacture such compounds.
We may find it difficult to enroll patients in our clinical trials given the limited number of patients who have the diseases for which our product candidates are being studied which could delay or prevent the start of clinical trials for our product candidates.
Identifying and qualifying patients to participate in clinical trials of our productscurrent and future product candidates is essential to our success. The timing of our clinical trials depends in development mustpart on the rate at which we can recruit patients to participate in clinical trials of our product candidates, and we may experience delays in our clinical trials if we encounter difficulties in enrollment. If we experience delays in our clinical trials, the timeline for obtaining regulatory approval of our product candidates will most likely be delayed.
Many factors may affect our ability to identify, enroll and maintain qualified patients, including the following:
| 12 |
For example, in our planned AML, Phase 3 clinical trial for GPS, only patients who meet specific inclusion criteria will enter the study. Primary entry restrictions include being greater than or equal to 60 years of age, having received upfront treatment with chemotherapy agents only, having achieved complete remission or CRem, as well as demonstrating adequate hematologic recovery. The estimated prevalence of AML is 12,000 to 20,000 cases in the United States (across all ages) and only a subset of this group satisfies the enrollment criteria for our AML Phase 3 clinical trial.
We may not be able to initiate or continue to support clinical trials of our product candidates for one or more indications, or any future product candidates if we are unable to locate and enroll a sufficient number of eligible participants in these trials as required by the FDA or similar foreign governmental agencies before they can be marketed. The process for obtaining FDA approvalother regulatory authorities. Even if we are able to enroll a sufficient number of patients in our clinical trials, if the pace of enrollment is both time-consuming and costly, with no certainty of a successful outcome. This process typically includes the conduct of extensive preclinical and clinical testing, which may take longer or cost moreslower than we anticipate,expect, the development costs for our product candidates may increase and the completion of our trials may prove
7
If we experience delays in the completion of, or termination of, any clinical trials of our current or future product candidates, the commercial prospects of our product candidates could be harmed, and our ability to generate product revenue from any of these product candidates could be delayed or prevented. In addition, any delays in completing our clinical trials would likely increase our overall costs, impair product candidate development and jeopardize our ability to obtain regulatory approval relative to our current plans. Any of these occurrences may harm our business, financial condition, and prospects significantly.
The results of preclinical studies or earlier clinical trials are not necessarily predictive of future results. Our existing product candidates in clinical trials, and any other product candidates that may appear to be promising at early stages of developmentadvance into clinical trials, may not successfully reachhave favorable results in later clinical trials or receive regulatory approval.
Success in preclinical studies and early clinical trials does not ensure that later clinical trials will generate adequate data to demonstrate the market for aefficacy and safety of an investigational drug. A number of reasons. The results of pre-clinical and initial clinical testing of these products may not necessarily indicate the results that will be obtained from later or more extensive testing. Companiescompanies in the pharmaceutical and biotechnology industries, including those with greater resources and experience than us, have suffered significant setbacks in advanced clinical trials, even after obtainingseeing promising results in earlier trials.
Despite the results reported in earlier preclinical studies or clinical trials for our product candidates, we do not know whether the clinical trials we may conduct will demonstrate adequate efficacy and safety to result in regulatory approval to market GPS or any of our product candidates for a drug candidate atparticular indication, either as a monotherapy or in combination, in any timeparticular jurisdiction. Efficacy data from prospectively designed trials may differ significantly from those obtained from retrospective subgroup analyses. If later-stage clinical trials do not produce favorable results, our ability to achieve regulatory approval for various reasons, includingGPS may be adversely impacted. Even if we believe that we have adequate data to support an application for regulatory approval to market any of our current or they believe the subjects or patients participating in such trials are being exposed to unacceptable health risks. Among other reasons, adverse side effects of a drug candidate on subjects or patients in a clinical trial could result infuture product candidates, the FDA or other regulatory authorities suspendingmay not agree and may require that we conduct additional clinical trials.
Clinical drug development involves a lengthy and expensive process with an uncertain outcome.
Clinical testing is expensive and can take many years to complete, with the outcome inherently uncertain. Failure can occur at any time during the clinical trial process. Before obtaining approval from regulatory authorities for the sale of any product candidate, we must conduct extensive clinical trials to demonstrate the safety and efficacy of our product candidates in humans. Prior to initiating clinical trials, a sponsor must complete extensive preclinical testing of a product candidate, including, in most cases, preclinical efficacy experiments as well IND-enabling toxicology studies. These experiments and studies may be time-consuming and expensive to complete. The necessary preclinical testing may not be completed successfully for a preclinical product candidate and a potentially promising product candidate may therefore never be tested in humans. Once it commences, clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. A failure of one or terminatingmore clinical trials can occur at any stage of testing. The outcome of preclinical testing and early clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Moreover, preclinical and refusingclinical data are often susceptible to approve avarying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval of their products. We may experience numerous unforeseen events during drug development that could delay or prevent our ability to receive marketing approval or commercialize our product candidates. In particular, drug candidate for any or all indications of use.
| 13 |
We may experience delays in our ongoing or future clinical trials, and we do not know whether planned clinical trials will begin or enroll subjects on time, will need to be redesigned or will be completed on schedule, if at all. There can be no assurance that the timing, cost or outcomeFDA will not put clinical trials of any of our drug development efforts, includingproduct candidates on clinical hold in the following:
8
| 14 |
If we experience delays in the completion or termination of any clinical trial of our product candidates, the approval and commercial prospects of such product candidates will be harmed, delaying our ability to generate product revenues from such product candidate and our costs will most likely increase. The required regulatory approvals may also be delayed, thereby jeopardizing our ability to commence product sales and generate revenues and the period of commercial exclusivity for our products may be decreased. Regulatory approval of our product candidates may be denied for the same reasons that caused the delay.
Risks associated with operating in foreign countries could materially adversely affect our product development.
We may conduct future studies in countries outside of the United States. Consequently, we may be subject to risks related to operating in foreign countries. Risks associated with conducting operations in foreign countries include:
| • | differing regulatory requirements for drug approvals and regulation of approved drugs in foreign countries; more stringent privacy requirements for data to be supplied to our operations in the United States,e.g., General Data Protection Regulation in the European Union; |
Our current and future product candidates, the methods used to deliver them or their dosage levels may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label or result in significant negative consequences following any regulatory approval.
Undesirable side effects caused by our current or future product candidates, their delivery methods or dosage levels could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval or termination of clinical trials by the FDA or other comparable foreign regulatory authority; an independent DSMB that is governing our clinical trials; or an IRB, that approves and, monitors biomedical research to protect the rights and welfare of human subjects. For example, although no high-grade delayed type hypersensitivity in the skin or systemic anaphylaxis events have been noted after GPS administration in patients treated in our clinical studies to date, it is theoretically possible that such toxicities, or other type of adverse events, may occur in future clinical studies. As a result of safety or toxicity issues that we may experience in our clinical trials, or negative or inconclusive results from the clinical trials of others for drug candidates similar to our own, we may not receive approval to market any product candidates, which could prevent us from ever generating revenues or achieving profitability. Results of our trials could reveal an unacceptably high severity and incidence of side effects. In such an event, our trials could be suspended or terminated, and the FDA or comparable foreign regulatory authorities could order us to cease further development of or deny approval of our product candidates for any or all targeted indications. The drug-related side effects could also affect patient recruitment or the ability of enrolled subjects to complete the trial or result in potential product liability claims. Any of these occurrences may have a material adverse effect on our business, results of operations, financial condition, cash flows and future prospects.
| 15 |
Additionally, if any of our product candidates receives regulatory approval, and we or others later identify undesirable side effects caused by such product, a number of potentially significant negative consequences could result, including that:
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved.
Our product development program may not uncover all possible adverse events that patients who take our product candidates may experience. The number of subjects exposed to product candidates and the average exposure time in the clinical development program may be inadequate to detect rare adverse events or chance findings that may only be detected once the product is administered to more patients and for greater periods of time.
Clinical trials by their nature utilize a sample of the potential patient population. However, with a limited number of subjects and limited duration of exposure, we cannot be fully assured that rare and severe side effects of our product candidates will be uncovered. Such rare and severe side effects may only be uncovered with a significantly larger number of patients exposed to our product candidates. If such safety problems occur or are identified after our product candidates reaches the market, the FDA may require that we amend the labeling of the product or recall the product, or may even withdraw approval for the product.
Our future success is dependent on the regulatory approval of our product candidates.
Our business is dependent on our ability to generate revenueobtain regulatory approval for our product candidates in a timely manner. We cannot commercialize product candidates in the United States without first obtaining regulatory approval for the product from the particular drugFDA. Similarly, we cannot commercialize product candidates outside of the United States without obtaining regulatory approval from comparable foreign regulatory authorities. Before obtaining regulatory approvals for the commercial sale of any product candidate for a target indication, we must demonstrate with substantial evidence gathered in preclinical studies and clinical trials, generally including two well-controlled Phase 3 trials, that the product candidate is safe and effective for use for that target indication and that the manufacturing facilities, processes and controls are adequate with respect to such product candidate.
The time required to obtain approval by the FDA and comparable foreign regulatory authorities is unpredictable but typically takes many years following the commencement of preclinical studies and clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions.
Even if a product candidate were to successfully obtain approval from the FDA and comparable foreign regulatory authorities, any approval might contain significant limitations related to use restrictions for specified age groups, warnings, precautions or contraindications, or may be subject to numerous foreignburdensome post-approval study or risk management requirements. Also, any regulatory requirements governingapproval of our current or future product candidates, once obtained, may be withdrawn.
| 16 |
Our current and future product candidates could fail to receive regulatory approval from the conductFDA.
We have not obtained regulatory approval for any product candidate and it is possible that our existing product candidates or any future product candidates will not obtain regulatory approval, for many reasons, including:
• | failure to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; | |
• | disagreement with our interpretation of data from preclinical studies or clinical trials; | |
• | the insufficiency of data collected from clinical trials of our product candidates to support the submission and filing of a BLA, NDA or other submission or to obtain regulatory approval; | |
• | the insufficiency of a single Phase 3 clinical trial of GPS in AML for regulatory approval in that indication; | |
• | failure to obtain approval of our manufacturing processes or facilities of third-party manufacturers with whom we contract for clinical and commercial supplies or our own manufacturing facility; or | |
• | changes in the approval policies or regulations that render our preclinical and clinical data insufficient for approval. |
The FDA or a comparable foreign regulatory authority may require more information, including additional preclinical or clinical data to support approval or additional studies, which may delay or prevent approval and our commercialization plans, or we may decide to abandon the development program. If we were to obtain approval, regulatory authorities may approve any of our product candidates for fewer or more limited indications than we request (including failing to approve the most commercially promising indications), may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve a product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate.
If we are unable to obtain regulatory approval for one of our product candidates in one or more jurisdictions, or any approval contains significant limitations, we may not be able to obtain sufficient funding to continue the development of that product or generate revenues attributable to that product candidate.
We currently have Orphan Drug designation for certain product candidates, and may seek Orphan Drug Product designation for additional product candidates or indications, which might not be received or provide the intended benefit thereof.
Regulatory authorities in some jurisdictions, including the United States and Europe, may designate drugs for relatively small patient populations as Orphan Drugs. Under the Orphan Drug Act, the FDA may designate a product as an Orphan Drug if it is a drug intended to treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals annually in the United States. We have received Orphan Drug Product designations from the FDA for GPS in AML, MPM and MM as well as Orphan Medicinal Product designations from the EMA for GPS in AML, MPM and MM. We also have received Orphan Drug Product designation for GALE-301 and GALE-302 from the FDA. Although we have received Orphan Drug Product designation for GPS, GALE-301 and GALE-302, there is no guarantee that these products will be successfully approved by the FDA, that they will be commercially successful in the marketplace, or that another product will not be approved for the same indication ahead of our product candidate.
Even if we obtain Orphan Drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different drugs can be approved for the same condition. Even after an Orphan Drug is approved, the FDA can subsequently approve another drug for the same condition if the FDA concludes that the later drug is clinically superior in that it is shown to be safer, more effective or makes a major contribution to patient care. In addition, Orphan Drug exclusivity may be lost if the FDA or EMA determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the drug to meet the needs of patients with the rare disease or condition.
| 17 |
We currently have Fast Track designation for certain product candidates and may seek Fast Track designation for additional product candidates or indications, which might not be received or provide the intended benefits thereof.
If a product candidate is intended for the treatment of a serious condition and nonclinical or clinical data demonstrate the potential to address unmet medical need for this condition, a product sponsor may apply to the FDA for Fast Track designation, which may or may not be granted by the FDA. The FDA has given us Fast Track designation for GPS in AML and MPM and for NPS for the adjuvant treatment of patients with early state breast cancer with low to intermediate HER2 expression following standard of care upfront therapy (surgery plus chemotherapy +/- radiotherapy).
However, Fast Track designation does not ensure that we will receive marketing approval or that approval will be granted within any particular timeframe. We may not experience a faster development or regulatory review or approval process with Fast Track designation compared to conventional FDA procedures. In addition, the FDA may withdraw Fast Track designation if it believes that the designation is no longer supported by data from our clinical development program. Fast Track designation alone does not guarantee qualification for the FDA’s priority review procedures.
Failure to obtain regulatory approval in international jurisdictions would prevent our product candidates from being marketed abroad.
In addition to regulations in the United States, to market and sell our product candidates in the European Union, United Kingdom, many Asian countries and other jurisdictions, we must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one regulatory authority outside the United States does not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA. The regulatory approval process outside the United States generally includes all of the risks associated with obtaining FDA approval described above as well as risks attributable to the satisfaction of local regulations in foreign jurisdictions. ApprovalThe approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ substantially from that required to obtain FDA approval. We may not be able to obtain approvals from regulatory authorities outside the United States on a timely basis, if at all. Clinical trials accepted in one country may not be accepted by regulatory authorities in other countries. In addition, many countries outside the United States require that a product be approved for reimbursement before it can be approved for sale in that country. A product candidate that has been approved for sale in a particular country may not receive reimbursement approval in that country.
We may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market. If we are unable to obtain approval of any of our current or future product candidates by regulatory authorities in the European Union, United Kingdom, Asia or elsewhere, the commercial prospects of that product candidate may be significantly diminished, our business prospects could decline and this could materially adversely affect our business, results of operations and financial condition.
Even if our current and future product candidates receive regulatory approval, they may still face future development and regulatory difficulties.
Even if we obtain regulatory approval for a product candidate, that approval would be subject to ongoing requirements by the FDA does not assureand comparable foreign regulatory authorities governing the manufacture, quality control, further development, labeling, packaging, storage, distribution, adverse event reporting, safety surveillance, import, export, advertising, promotion, recordkeeping and reporting of safety and other post-marketing information. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance by us and/or our contract manufacturing organizations, or CMOs, and CROs for any post-approval clinical trials that we may conduct. The safety profile of any product will continue to be closely monitored by the FDA and comparable foreign regulatory authorities after approval. If the FDA or comparable foreign regulatory authorities become aware of new safety information after approval of any of our product candidates, they may require labeling changes or establishment of a risk evaluation and mitigation strategy, impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies or post-market surveillance.
In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with Good Manufacturing Practices, or cGMP, Good Clinical Practices, or GCP, and other regulations. If we or a regulatory agency discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product, the manufacturing facility or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing. If we, our product candidates or the manufacturing facilities for our product candidates fail to comply with applicable regulatory requirements, a regulatory agency may:
| 18 |
The occurrence of any event or penalty described above may inhibit our ability to successfully commercialize our products and generate revenues.
Advertising and promotion of any product candidate that obtains approval in the United States will be heavily scrutinized by the FDA, the DOJ, the Office of Inspector General of HHS, state attorneys general, members of Congress and the public. A company can make only those claims relating to safety and efficacy, purity and potency that are approved by the FDA and in accordance with the provisions of the approved label. Additionally, advertising and promotion of any product candidate that obtains approval outside of the United States.
Even if we obtain regulatory approval for a product, we will remain subject to ongoing regulatory requirements.
Even if our product candidates are approved, we will be subject to ongoing regulatory requirements with respect to manufacturing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping, conduct of post-marketing clinical trials and submission of safety, efficacy and other post-approval information, including both federal and state requirements in the United States and requirements of comparable foreign regulatory agenciesauthorities.
Manufacturers and manufacturers’ facilities are required to delivercontinuously comply with FDA and comparable foreign regulatory authority requirements, including ensuring quality control and manufacturing procedures conform to cGMP, regulations and corresponding foreign regulatory manufacturing requirements. Accordingly, we and our contract manufacturers will be subject to continual review and inspections to assess compliance with cGMP and adherence to commitments made in any NDA submission to the FDA or any other type of domestic or foreign marketing authorization application.
Any regulatory approvals we receive for any of our product candidates may be subject to limitations on the approved indicated uses for which the product candidate may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the product candidate. We will be required to report adverse reactions and production problems, if any, to the FDA and comparable foreign regulatory authorities. Any new legislation addressing drug candidates,safety issues could result in delays in product development or commercialization, or increased costs to assure compliance. If our original marketing approval for a product candidate was obtained through an accelerated approval pathway, we may needcould be required to modifyconduct a successful post-marketing clinical trial to confirm the designclinical benefit for our products. An unsuccessful post-marketing clinical trial or failure to complete such a trial could result in the withdrawal of marketing approval.
If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, or it disagrees with the promotion, marketing or labeling of these delivery vehicles for some products wea product, the regulatory agency may develop. In such an event, the FDA may regulateimpose restrictions on that product or us, including requiring withdrawal of the product asfrom the market. If we fail to comply with applicable regulatory requirements, a combinationregulatory agency or enforcement authority may, among other things:
| 19 |
• | issue warning letters; | |
• | impose civil or criminal penalties; | |
• | suspend or withdraw regulatory approval; | |
• | suspend any of our ongoing clinical trials; | |
• | refuse to approve pending applications or supplements to approved applications submitted by us; | |
• | impose restrictions on our operations, including closing our contract manufacturers’ facilities; or | |
• | require a product recall. |
Any government investigation of a drugalleged violations of law would require us to expend significant time and a device or require additional approvals or clearances for the modified delivery. Additionally, it has been observedresources in at least one previous clinical trial, conducted by another company, that delivery vehicles similarresponse and could generate adverse publicity. Any failure to the delivery vehiclein-licensed from UMMScomply with ongoing regulatory requirements may cause toxicity, which could delay or prevent approval of this delivery vehicle.
Risks Related to Our Manufacturing
If we are not successful in developing pre-clinicalWe have limited to no manufacturing, sales, marketing or distribution capability and must rely upon third parties for such.
We currently have agreements with various third-party manufacturing facilities for production of our product candidates for research and development and testing purposes. We depend on these manufacturers to meet our deadlines, quality standards and specifications. Our reliance on third parties for the manufacture of our active pharmaceutical ingredient and drug product and, in the future, any approved products, creates a dependency that could severely disrupt our research and development, our clinical testing, and ultimately our sales and marketing efforts if the source of such supply proves to be unreliable or unavailable. If the contracted manufacturing source is unreliable or unavailable, we willmay not be able to commencemanufacture clinical trials in humans or obtain approvaldrug supplies of our product candidates, and our preclinical and clinical testing programs may not be able to move forward and our entire business plan could fail.
Both the active pharmaceutical ingredient and drug product for our product candidates.
We are subject to a multitude of manufacturing risks, any of which could substantially increase our costs and limit supply of our product candidates.
We and our CMOs will need to conduct significant development work for each product candidate for each target indication for studies, trials and commercial launch readiness. For example, the processes by which GPS is manufactured were initially developed by MSK for clinical purposes. Concurrent with the license of GPS, we acquired certain supplies intended for clinical use from MSK. These MSK clinical supplies may not be adequate for future clinical studies. We intend to improve the existing processes for GPS in connection with more advanced clinical trials or commercialization efforts we may undertake in the future. Developing commercially viable manufacturing processes is a difficult, expensive and uncertain task, and there are risks associated with scaling to the level required for advanced clinical trials or commercialization, including cost overruns, potential problems with process scale-up, process reproducibility, stability issues, consistency and timely availability of reagents or raw materials. The manufacturing facilities in which our product candidates will be made could be adversely affected by earthquakes and other natural disasters, equipment failures, labor shortages, power failures, and numerous other factors.
Additionally, the process of manufacturing our product candidates is complex, highly regulated and subject to several risks, including but not limited to:
| 20 |
• | reduced production yields, product defects, and other supply disruptions due to deviations, even minor, from normal manufacturing and distribution processes; | |
• | unexpected product defects; | |
• | microbial, viral, or other contaminations in our product candidates or in the manufacturing facilities in which our product candidates are made, which may result in the closure of such manufacturing facilities for an extended period of time to allow for the investigation and remediation of the contamination; | |
• | adverse impact on the active ingredient of GPS as a result of potential contamination from the presence of heavy metals which can lead to higher than acceptable rates of impurities resulting in the active ingredient being unacceptable for use; and | |
• | adverse impact on the manufacturing of GPS as a result of potential contamination from excess water and oxygen which can lead to higher than acceptable levels of impurities resulting in the drug product being unacceptable for use. |
Any adverse developments affecting manufacturing operations for our product candidates may result in shipment delays, inventory shortages, lot failures, withdrawals or recalls or other interruptions in the supply of our drug substance and drug product, which could delay the development of our product candidates. We may also have to write off inventory, incur other charges and expenses for supply of drug product that fails to meet specifications, undertake costly remediation efforts, or seek more costly manufacturing alternatives. Inability to meet the demand for our product candidates could damage our reputation and the reputation of our products among physicians, healthcare payors, patients or the medical community, and cancer treatment centers, which could adversely affect our ability to operate our business and our results of operations.
In the clinical trials using GPS and NPS, GM-CSF is also administered and its availability is dependent upon a third-party manufacturer, which may or may not reliably provide GM-CSF, thus jeopardizing the completion of the trials.
Some of our product candidates are administered in combination with GM-CSF, which is available in both liquid and lyophilized forms exclusively from one manufacturer. We will continue to be dependent on that manufacturer for our supply of GM-CSF in connection with the ongoing GPS and NPS trials and the potential commercial manufacture of these programs. We have not entered into a dedicated supply agreement with the manufacturer for GM-CSF, and instead rely on purchase orders to meet our supply needs. Any temporary interruptions or discontinuation of the availability of GM-CSF, or any determination by us to change the GM-CSF used with GPS or NPS, could have a material adverse effect on our clinical trials and any commercialization of the assets.
If any of our CMOs’ clinical manufacturing facilities are damaged or destroyed or production at such facilities is otherwise interrupted, our business and prospects would be negatively affected.
If our CMOs’ manufacturing facilities or the equipment in them is damaged or destroyed, we may not be able to quickly or inexpensively replace our manufacturing capacity or replace it at all. In the event of a temporary or protracted loss of this facility or equipment, we might not be able to transfer manufacturing to another CMO. Even if we could transfer manufacturing to another CMO, the shift would likely be expensive and time-consuming, particularly because the new drug discovery phasefacility would need to comply with the necessary regulatory requirements and we would need FDA approval before selling any products manufactured at that facility. Such an event could delay our clinical trials or reduce our product sales.
Although we currently maintain insurance coverage against damage to our property and to cover business interruption and research and development restoration expenses, our insurance coverage may not reimburse us, or may not be sufficient to reimburse us, for any expenses or losses we may suffer. We may be unable to meet our requirements for our product candidates if there were a catastrophic event or failure of our current manufacturing facility or processes.
Risks Related to Our Dependence on Third Parties and Our License Agreements
We rely on third parties to conduct our preclinical studies and clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, or if we lose any of our CROs or other key third-party vendors, we may not be able to obtain regulatory approval for or commercialize our current or future product candidates on a timely basis, if at all.
Our internal capacity for clinical trial execution and management is limited and therefore we rely heavily on third parties. We have relied upon and plan to continue to rely upon third-party CROs, vendors and contractors to monitor and manage data for our ongoing preclinical and clinical programs. For example, our collaborating investigators at MSK, along with their clinical and clinical operations teams, manage the conduct of the ongoing clinical trials for GPS as well as perform the analysis, publication and presentation of data and results related to this program. We also rely on collaborating investigators, along with their clinical and clinical operations teams, at MSK for the collection and transfer of various types of follow-up data regarding studies previously conducted by MSK.
| 21 |
We plan to rely on CROs and other third-party vendors for all currently contemplated clinical studies, with services to be rendered by such CROs ranging from, in the case of assorted Phase 2 trials, specific and need-tailored (e.g., data management and biostatistics) only to, in the case of our immune combination (PD1 blocker) Phase 2 trials and our planned Phase 3 trial for GPS in AML, all-encompassing. We rely on these parties for the execution of our preclinical studies and clinical trials, including the proper and timely conduct of our clinical trials, and we control only some aspects of their activities. Outsourcing these functions involves risk that third parties may not yet identifiedperform to our standards, may not produce results or data in a timely manner or may fail to perform at all.
While we have agreements governing the commitments of our third-party vendor services, we have limited influence over their actual performance. Nevertheless, we are responsible for ensuring that each of our trials is conducted in accordance with the applicable protocol and legal, regulatory and scientific standards, and our reliance on the CROs does not relieve us of our regulatory responsibilities.
If our company, or any lead compounds for therapeutic developmentof our partners or CROs, fail to comply with applicable regulations and good clinical practices, the clinical data generated in our initial areas of focus. RNA interference is a relatively new scientific field,clinical trials may be deemed unreliable and the technologiesFDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our regulatory applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with applicable requirements. In addition, our clinical trials must be conducted with product produced under cGMP and other requirements. We are stillalso required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database, clinicaltrials.gov, within a specified timeframe. Failure to comply also would violate federal requirements in the early stageUnited States and could result in other penalties, which would delay the regulatory approval process and result in adverse publicity.
Our CROs, third-party vendors and contractors are not our employees, and except for remedies available to us under our agreements with such CROs, third-party vendors and contractors, we cannot control whether or not they devote sufficient time and resources, including experienced staff, to our ongoing clinical, nonclinical and preclinical programs. They may also have relationships with other entities, some of development. We have no compounds in pre-clinical toxicology studies,which may be our competitors. If CROs, third-party vendors and contractors do not successfully carry out their contractual duties or obligations or meet expected deadlines or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated and we may not be able to advance anyobtain regulatory approval for or successfully commercialize our current or future product candidate throughcandidates. CRO, vendor or contractor errors could cause our results of operations and the pre-clinical stage into clinical trials. Additionally,commercial prospects for our development efforts may never resultcurrent or future product candidates to be harmed, our costs to increase and our ability to generate revenues to be delayed.
In addition, the use of third-party service providers requires us to disclose our proprietary information to these parties, which could increase the risk that this information will be misappropriated. We currently have a small number of employees, which limits the internal resources we have available to identify and monitor our third-party providers. To the extent we are unable to identify and successfully manage the performance of third-party service providers in the identificationfuture, our business may be adversely affected. Though we carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
If any of a pre-clinical candidate which we are able to successfully develop into a drug. Even if we are able to designate a lead candidate,our relationships with our third-party CROs, third-party vendors or contractors terminate, we may not be able to identify data that would support entering such a candidate into clinical trials. Furthermore, even if we successfully enter into clinical studies,arrangements with alternative CROs, third-party vendors or contractors on a timely basis, on commercially reasonable terms or at all.
Our CROs, third-party vendors and contractors have the results from pre-clinical testingright to terminate their agreements with us in the event of a drug candidate may not predictan uncured material breach. In addition, some of our CROs, third-party vendors and contractors have an ability to terminate their respective agreements with us if it can be reasonably demonstrated that the results that will be obtained on human clinical trials.
9
10
11
| 22 |
Other companies or organizations may assert patent rights that prevent us from developing our products.
12
We currently are dependent on licenses from third parties for our key technologies including licenses from UMMS and from Cold Spring Harbor Laboratory, relating to fundamental RNAi technologies.our product candidates. Our current licenses impose, and any future licenses we enter into are likely to impose, various development, funding, royalty, diligence, sublicensing, insurance and other obligations on us. If our license with respect to any of these technologies is terminated for any reason, the development of the products contemplated by the licenses would be delayed, or suspended altogether, while we seek to license similar technology or develop new non-infringing technology. The costs of obtaining new licenses are high,high. For example, we are entirely dependent on our license from MSK to allow us to develop and many patents in the RNAi fieldcommercialize our lead product candidate, GPS, and any loss of or challenge to our license agreement with MSK could have a material and adverse effect on our business and result of operations.
Under certain license agreements that we have already been exclusively licensedentered into, we have minimum dollar amounts per year that we are obligated to third parties, including our competitors. If any of our existing licenses is terminated,spend on the development of the technology we have licensed from our contract partners and other obligations to maintain certain licenses. If we fail to meet such requirements under any of our licenses or if we fail to comply with any other obligations under these licenses, we may be in breach of our obligations under such agreements, which may result in the loss of the technology licensed.
In addition, our business depends on our ability to license therapeutic compounds from third parties. If we fail to meet our obligations under our license agreements, we may lose the ability to develop our product candidates, which would adversely affect our business.
We have in-licensed a significant portion of our intellectual property from MSK. If we breach our license agreement with MSK, we could lose the ability to continue the development and potential commercialization of GPS.
We do not currently own any patents or patent applications related to our lead product candidate, GPS. GPS is licensed-in from MSK and includes an exclusive license to United States and foreign patent applications. Under the MSK license agreement, we are subject to various obligations, including diligence obligations with respect to funding, development and commercialization activities, payment obligations upon achievement of certain milestones and royalties on product sales, as well as other material obligations. If there is any conflict, dispute, disagreement or issue of nonperformance between us and MSK regarding our rights or obligations under the license agreements, including any such conflict, dispute or disagreement arising from our failure to satisfy diligence or payment obligations under any such agreement, we may be liable to pay damages and MSK may have a right to terminate the affected license. In 2018, we did not make certain required payments to MSK, which entitles MSK to terminate the license agreement if we are unable to make such payments after notice. To date, we have not received such a notice from MSK. The loss of our license agreement with MSK could materially adversely affect our ability to proceed to utilize the affected intellectual property in our development efforts, our ability to enter into future collaboration, licensing and/or marketing agreements for GPS and our ability to commercialize GPS. The risks described elsewhere pertaining to our patents and other intellectual property rights also apply to the intellectual property rights that we license, and any failure by us or our licensors to obtain, maintain and enforce these rights could have a material adverse effect on our business.
We may not realize the benefits of our strategic alliances that we may form in the future.
We may form strategic alliances, create joint ventures or collaborations or enter into licensing arrangements with third parties that we believe will complement or augment our existing business. These relationships, or those like them, may require us to incur nonrecurring and other charges, increase our near- and long-term expenditures, issue securities that dilute our existing stockholders or disrupt our management and business. In addition, we face significant competition in seeking appropriate strategic alliances and the negotiation process is time-consuming and complex. Moreover, we may not be successful in our efforts to establish a strategic alliance or other alternative arrangements for any future product candidates and programs because our research and development pipeline may be insufficient, our product candidates and programs may be deemed to be at too early a stage of development for collaborative effort and third parties may not view our product candidates and programs as having the requisite potential to demonstrate safety and efficacy. If we license products contemplatedor acquire businesses, we may not be able to realize the benefit of such transactions if we are unable to successfully integrate them with our existing operations and company culture. We cannot be certain that, following a strategic transaction or license, we will achieve the revenues or specific net income that justifies such transaction. Any delays in entering into new strategic alliances agreements related to our product candidates could also delay the development and commercialization of our product candidates and reduce their competitiveness even if they reach the market.
Our business involves the use of hazardous materials and we and our third-party manufacturers and suppliers must comply with environmental, health and safety laws and regulations, which can be expensive and restrict how we do business.
Our third-party manufacturers’ and suppliers’ activities involve the controlled storage, use and disposal of hazardous materials. We and our manufacturers and suppliers are subject to laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials even after we sell or otherwise dispose of the products. In some cases, these hazardous materials and various wastes resulting from their use will be stored at our contractors or manufacturers’ facilities pending use and disposal. We cannot completely eliminate the risk of contamination, which could cause injury to our employees and others, environmental damage resulting in costly cleanup and liabilities under applicable laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. Although we expect that the safety procedures utilized by our third-party contractors and manufacturers for handling and disposing of these materials will generally comply with the licensesstandards prescribed by these laws and regulations, we cannot guarantee that this will be the case or eliminate the risk of accidental contamination or injury from these materials. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources. We do not currently carry biological or hazardous waste insurance coverage and our property and casualty, and general liability insurance policies specifically exclude coverage for damages and fines arising from biological or hazardous waste exposure or contamination.
| 23 |
We may not be delayedable to establish or terminatedmaintain the third-party relationships that are necessary to develop or potentially commercialize some or all of our product candidates.
We expect to depend on collaborators, partners, licensees, clinical research organizations and other third parties to support our discovery efforts, to formulate product candidates, to manufacture our product candidates, and to conduct clinical trials for some or all of our product candidates. We cannot guarantee that we will be able to successfully negotiate agreements for or maintain relationships with collaborators, partners, licensees, clinical investigators, vendors and other third parties on favorable terms, if at all. Our ability to successfully negotiate such agreements will depend on, among other things, potential partners’ evaluation of the superiority of our technology over competing technologies and the quality of the preclinical and clinical data that we have generated, and the perceived risks specific to developing our product candidates. If we are unable to obtain or maintain these agreements, we may not be able to clinically develop, formulate, manufacture, obtain regulatory approvals for or commercialize our product candidates. We cannot necessarily control the amount or timing of resources that our contract partners will devote to our research and development programs, product candidates or potential product candidates, and we cannot guarantee that these parties will fulfill their obligations to us under these arrangements in a timely fashion. We may not be able to readily terminate any such agreements with contract partners even if such contract partners do not fulfill their obligations to us.
In addition, we may receive notices from third parties from time to time alleging that our technology or product candidates infringe upon the intellectual property rights of those third parties. Any assertion by third parties that our activities or product candidates infringe upon the intellectual property rights of third parties may adversely affect our ability to secure strategic partners or licensees for our technology or product candidates or our ability to secure or maintain manufacturers for our compounds.
Risks Related to Our Intellectual Property
We may not be able to obtain and enforce patent rights or other intellectual property rights that cover our product candidates and that are of sufficient breadth to prevent third parties from competing against us.
Our success with respect to our product candidates will depend in part on our ability to obtain and maintain patent protection in the United States and abroad, to preserve our trade secrets, and to prevent third parties from infringing upon our proprietary rights. We seek to protect our proprietary position by filing in the United States and in certain foreign jurisdictions patent applications related to our novel technologies and product candidates that are important to our business. The patent prosecution process is expensive and time-consuming, and we may not be able to negotiate additional licenses on acceptable terms, iffile and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. In addition, we may not pursue or obtain patent protection in all which wouldmajor markets. Our competitors may be able to design around our owned or licensed patents by developing similar or alternative peptides or technologies without infringing our intellectual property rights. Moreover, in some circumstances, we do not have the right to control the preparation, filing or prosecution of patent applications, or to maintain the patents, covering technology that we license from third parties or covering technology that a material adverse effect oncollaboration or commercialization partner may develop. In some circumstances, our licensors have the right to enforce the licensed patents without our involvement or consent, or to decide not to enforce or to allow us to enforce the licensed patents. Therefore, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions and has in recent years been the subject of much litigation. In addition, the laws of foreign jurisdictions may not protect our rights to the same extent as the laws of the United States. For example, European patent law restricts the patentability of methods of treatment of the human body more than U.S. law does. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot be defective, they may expose uscertain that we or our licensors were the first to claimsmake the inventions claimed in our owned or licensed patents or pending patent applications, or that we or our licensors were the first to file for personal injury by patients in clinical trialspatent protection of such inventions. Moreover, the U.S. Patent and Trademark Office, or USPTO, might require that the term of a patent issuing from a pending patent application be disclaimed and limited to the term of another patent that is commonly owned or names a common inventor. As a result, the issuance, scope, validity, term, enforceability and commercial value of our patent rights are highly uncertain.
| 24 |
Our pending and future patent applications, and any collaboration or commercialization partner’s pending and future patent applications, may not result in patents being issued which protect our technology or products, in whole or by patients usingin part, or which effectively prevent others from commercializing competitive technologies and products.
During prosecution of any patent application, the issuance of any patents based on the application may depend upon our commercially marketed products. Even ifor their ability to generate additional preclinical or clinical data that support the marketing of one or morepatentability of our products is approved by the FDA, usersproposed claims. We or any collaboration or commercialization partner may claim that such products caused unintended adverse effects. We will seek to obtain clinical trial insurance for clinical trials that we conduct, as well as liability insurance for any products that we market. There can be no assurance that we willnot be able to obtain insurance in the amounts we seek,generate sufficient additional data on a timely basis, or at all. We anticipate that licensees who develop our products will carry liability insurance coveringMoreover, changes in either the clinical testing and marketingpatent laws or interpretation of those products. There is no assurance, however, that any insurance maintained by us or our licensees will prove adequatethe patent laws in the eventUnited States or other countries may diminish the value of our or a claim against us. Evencollaboration or commercialization partner’s patents or narrow the scope of our or their patent protection.
Changes in either the patent laws or in the interpretations of patent laws in the United States or abroad may diminish the value of our intellectual property.
On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to the U.S. patent law. These include provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. Accordingly, it is not clear what, if claims asserted against us are unsuccessful, they may divert management’s attention fromany, impact the Leahy-Smith Act will have on the operation of our operationsbusiness. However, the Leahy-Smith Act, in particular the first-to-file provision and we may have to incur substantialour implementation, could increase the uncertainties and costs to defend such claims.
In addition, U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances in certain situations. From time to time, the U.S. Supreme Court, other federal courts, the U.S. Congress, or interpretation by the USPTO may change the standards of patentability and any such changes could have a negative impact on our business.
Some cases decided by the U.S. Supreme Court have involved questions of when claims reciting abstract ideas, laws of nature, natural phenomena and/or natural products are eligible for a patent, regardless of whether the claimed subject matter is otherwise novel and inventive. These cases include Association for Molecular Pathology v. Myriad Genetics, Inc., 569 U.S. 576 (2013), also known as the Myriad decision; Alice Corp. v. CLS Bank International, 573 U.S. 13-298 (2014), also known as the Alice decision; and Mayo Collaborative Services v. Prometheus Laboratories, Inc., also known as the Prometheus decision, 566 U.S. 66 (2012). The full impact of these decisions is not yet known. In view of these and subsequent court decisions, the USPTO has issued materials to patent examiners providing guidance for determining the patent eligibility of claims reciting laws of nature, natural phenomena, or natural products.
Our current product candidates include products, or components, derived to various extents from nature; therefore, these decisions and their interpretation by the courts and the USPTO may impact prosecution, defense, and enforcement of certain types of patent claims in our patent portfolio. In addition to increasing uncertainty with regard to our ability to obtain future patents, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on these and other decisions by U.S. Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change or be interpreted in unpredictable ways that would weaken our ability to obtain some patent claims or to enforce patents that may issue to us in the future. In addition, these events may adversely affect our ability to defend patents that may issue in procedures in the USPTO or in U.S. courts.
While we intend to take actions reasonably necessary to enforce our patent rights, we may not be able to detect infringement of our own or in-licensed patents, which may be especially difficult for methods of manufacturing or formulation products.
We depend, in part, on our licensors and collaborators to protect a substantial portion of our proprietary rights. In addition, third parties may challenge our in-licensed patents and any of our own patents that we may obtain, which could result in the invalidation or unenforceability of some or all of the relevant patent claims. Litigation or other proceedings to enforce or defend intellectual property rights is very complex, expensive, and may divert our management’s attention from our core business and may result in unfavorable results that could adversely affect our ability to prevent third parties from competing with us.
| 25 |
If another party has reason to assert a substantial new question of patentability against any of our claims in our own and in-licensed patents, the third party can request that the patent claims be reexamined, which may result in a loss of scope of some claims or a loss of the entire patent. In addition to potential infringement suits, and interference and reexamination proceedings, we may become a party to patent opposition proceedings where either the patentability of the inventions subject of our patents are challenged, or we are challenging the patents of others. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful. As the medical device, biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that others may assert our commercial product and/or product candidates infringe their patent rights. If a third-party’s patents were found to cover our commercial product and product candidates, proprietary technologies or our uses, we or our collaborators could be enjoined by a court and required to pay damages and could be unable to continue to commercialize our products or use our proprietary technologies unless we or it obtained a license to the patent. A license may not be available to us or our collaborators on acceptable terms, if at all. In addition, during litigation, the patent holder could obtain a preliminary injunction or other equitable relief, which could prohibit us from making, using or selling our commercial product and product candidates pending a trial on the merits, which could be years away.
Our product candidates may face competition sooner than expected after the expiration of our composition of matter patent protection for such products.
Our composition of matter patents for certain of our product candidates have expired or will expire prior to any product approval. We intend to sellseek data exclusivity or market exclusivity for our GPS as well as our NPS, GALE-301 and GALE-302 product candidates provided under the Federal Food, Drug and Cosmetic Act, or FDCA, and similar laws in other countries. We believe that these product candidates will qualify for 12 years of data exclusivity under the Biologics Price Competition and Innovation Act of 2009, or BPCIA. Under the BPCIA, an application for a biosimilar product or BLA cannot be submitted to the FDA until four years, or if approved by the FDA, until 12 years, after the original brand product identified as the reference product is approved under a BLA. The BPCIA provides an abbreviated pathway for the approval of biosimilar and interchangeable biological products. The abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on our similarity to an existing brand product. The law is complex and continues to be interpreted and implemented by the FDA. There is also a risk that the U.S. Congress could amend the BPCIA to shorten this exclusivity period, potentially creating the opportunity for biosimilar competition sooner than anticipated after the expiration of our patent protection. Moreover, the extent to which a biosimilar, once approved, will be substituted for any reference product in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
Even if, as we expect, GPS, NPS, GALE-301 and GALE-302 are considered to be reference products eligible for 12 years of exclusivity under the BPCIA or qualify for five years of exclusivity as drugs under the FDCA, another company could market competing products if the FDA approves a full BLA or full NDA for such product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of the products.
In some countries outside of the United States, peptide vaccines, such as GPS, NPS, GALE-301 and GALE-302, are regulated as chemical drugs rather than as biologics and may or may not be eligible for non-patent exclusivity.
If we are sued for infringing the intellectual property rights of third parties, such litigation could be costly and time-consuming and could prevent or delay our development and commercialization efforts.
Our commercial success depends, in part, on us and our collaborators not infringing the patents and proprietary rights of third parties. There is a substantial amount of litigation and other adversarial proceedings, both within and outside the United States, involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries, including patent infringement lawsuits, interference or derivation proceedings, oppositions, andinter partes and post-grant review proceedings before the USPTO and non-U.S. patent offices. Numerous U.S. and non-U.S. issued patents and pending patent applications owned by third parties exist in the fields in which we are developing and may develop our current and future product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, and as our product pipeline grows, the risk increases that our product candidates may be subject to claims of infringement of third parties’ patent rights as it may not always be clear to industry participants, including us, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform or predictable.
If we are sued for patent infringement, we would need to demonstrate that our product candidates, products and methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid, and we may not be able to do this. Proving that a patent is invalid is difficult. If any issued third-party patents were held by a court of competent jurisdiction to cover aspects of our materials, formulations, methods of manufacture or methods for treatment, we could be forced, including by court order, to cease developing, manufacturing or commercializing the relevant product candidate until such patent expired. Alternatively, we may be required to obtain a license from such third party in order to use the infringing technology and to continue developing, manufacturing or marketing the infringing product candidate. We could be prevented from commercializing a product candidate or be forced to cease some aspect of our business operations, if, as a result of actual or threatened patent infringement claims, we are unable to enter into licenses on acceptable terms. In addition, parties making claims against us may also obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize one or more of our product candidates.
| 26 |
Defending against claims of patent infringement or misappropriation of trade secrets could be costly and time consuming, regardless of the outcome. Thus, even if we were to ultimately prevail, or to settle at an early stage, such litigation could burden us with substantial unanticipated costs. In addition, litigation or threatened litigation could result in significant demands on the time and attention of our management team, distracting them from the pursuit of other company business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees if we are found to have willfully infringed a patent, or to redesign our infringing product candidates, which may be impossible or require substantial time and monetary expenditure. we may also elect to enter into license agreements in order to settle patent infringement claims prior to litigation, and any such license agreement may require us to pay royalties and other fees that could be significant. During the course of any patent or other intellectual property litigation, there could be public announcements of the results of hearings, rulings on motions, and other interim proceedings in the litigation. If securities analysts or investors regard these announcements as negative, the perceived value of our product candidates, programs or intellectual property could be diminished. Accordingly, the market price of our shares of common stock may decline.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting, enforcing and defending patents on our current and future product candidates in all countries throughout the world would be prohibitively expensive. We or our licensors’ intellectual property rights in certain countries outside the United States may be less extensive than those in the United States. In addition, the laws of certain foreign countries do not protect intellectual property rights to the same extent as laws in the United States. Consequently, we and our licensors may not be able to prevent third parties from practicing our and our licensors’ inventions in countries outside the United States, or from selling or importing infringing products made using our and our licensors’ inventions in and into the United States or other jurisdictions. Competitors may use our and our licensors’ technologies in jurisdictions where we have not obtained patent protection or where we do not have exclusive rights under the relevant patent(s) to develop their own products and, further, may export otherwise infringing products to territories where we and our licensors have patent protection but where enforcement is not as strong as that in the United States. These infringing products may compete with our product candidates in jurisdictions where we or our licensors have no issued patents or where we do not have exclusive rights under the relevant patent(s), or our patent claims and other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us and our licensors to stop the infringement of our and our licensors’ patents or marketing of competing products in violation of our and our licensors’ proprietary rights generally. Proceedings to enforce our and our licensors’ patent rights in foreign jurisdictions could result in substantial costs and divert our attention from other aspects of our business, could put our and our licensors’ patents at risk of being invalidated or interpreted narrowly, could put our and our licensors’ patent applications at risk of not issuing, and could provoke third parties to assert claims against us or our licensors. We or our licensors may not prevail in any lawsuit that we or our licensors initiate, and even if we or our licensors are successful the damages or other remedies awarded, if any, may not be commercially meaningful.
We may become involved in lawsuits to protect or enforce our intellectual property, which could be expensive, time-consuming and unsuccessful and have a material adverse effect on the success of our business and on our stock price.
Third parties may infringe our patents, the patents of our licensors, or misappropriate or otherwise violate our or our licensors’ intellectual property rights. We and our licensors’ patent applications cannot be enforced against third parties practicing the technology claimed in such applications unless and until a patent issues from such applications, and then only to the extent the issued claims cover the technology. In the future, we or our licensors may elect to initiate legal proceedings to enforce or defend our or our licensors’ intellectual property rights, to protect our or our licensors’ trade secrets or to determine the validity or scope of intellectual property rights we own or control. Any claims that we assert against perceived infringers could also provoke these parties to assert counterclaims against us alleging that we infringe their intellectual property rights or that our intellectual property rights are invalid. In addition, third parties may initiate legal proceedings against us or our licensors to challenge the validity or scope of intellectual property rights we own or control. The proceedings can be expensive and time-consuming. Many of our or our licensors’ adversaries in these proceedings may have the ability to dedicate substantially greater resources to prosecuting these legal actions than we or our licensors can. Accordingly, despite our or our licensors’ efforts, we or our licensors may not be able to prevent third parties from infringing upon or misappropriating intellectual property rights we own or control, particularly in countries where the laws may not protect our rights as fully as in the United States. Litigation could result in substantial costs and diversion of management resources, which could harm our business and financial results. In addition, in an infringement proceeding, a court may decide that a patent owned by or licensed to us is invalid or unenforceable, in whole or in part, or may refuse to stop the other party from using the technology at issue on the grounds that our or our licensors’ patents do not cover the technology in question. An adverse result in any litigation proceeding could put one or more of our or our licensors’ patents at risk of being invalidated, held unenforceable or interpreted narrowly.
| 27 |
Interference or derivation proceedings provoked by third parties, brought by us or our licensors or collaborators, or brought by the USPTO or any non-U.S. patent authority may be necessary to determine the priority of inventions or matters of inventorship with respect to our or our licensors’ patents or patent applications. We may also become involved in other proceedings, such as reexamination or opposition proceedings,inter partesreview, post-grant review or other pre-issuance or post-grant proceedings in the USPTO or its foreign counterparts relating to our intellectual property or the intellectual property of others. An unfavorable outcome in any such proceeding could require us or our licensors to cease using the related technology and commercializing the affected product candidate, or to attempt to license rights to it from the prevailing party.
Our business could be harmed if the prevailing party does not offer us or our licensors a license on commercially reasonable terms if any license is offered at all. Even if we or our licensors obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us or our licensors. In addition, if the breadth or strength of protection provided by our or our licensor’s patents and patent applications is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current and future product candidates. Even if we successfully defend such litigation or proceeding, we may incur substantial costs and it may distract our management and other employees. We could be found liable for monetary damages, including treble damages and attorneys’ fees, if we are found to have willfully infringed a patent.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of shares of our common stock.
Although we have taken steps to protect our trade secrets and unpatented know-how, by entering into confidentiality agreements with third parties, and proprietary information and invention agreements with certain employees, consultants and advisors, third parties may still obtain this information or we may be unable to protect our rights.
Proprietary trade secrets and unpatented know-how are also very important to our business. We also have limited control over the protection of trade secrets used by our licensors, collaborators and suppliers. There can be no assurance that binding agreements will not be breached, that we would have adequate remedies for any breach, or that our trade secrets and unpatented know-how will not otherwise become known or be independently discovered by our competitors. If trade secrets are independently discovered, we would not be able to prevent their use. Enforcing a claim that a third party illegally obtained and is using our trade secrets or unpatented know-how is expensive and time consuming, and the outcome is unpredictable.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed to us alleged trade secrets of their other clients or former employers. As is common in the biotechnology and pharmaceutical industry, certain of our employees were formerly employed by other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Moreover, we engage the services of consultants to assist us in the development of our commercial product and product candidates, many of whom were previously employed at or may have previously been or are currently providing consulting services to, other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We may be subject to claims that these employees and consultants or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers or their former or current customers. Litigation may be necessary to defend against these types of claims. Even if we are successful in defending against any such claims, any such litigation would likely be protracted, expensive, a distraction to our management team, not viewed favorably by investors and other third parties, and may potentially result in an unfavorable outcome.
If we are unable to protect the confidentiality of our trade secrets and other proprietary information, the value of our technology could be materially adversely affected, and our business could be harmed.
In addition to seeking the protection afforded by patents, we rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable or that we elect not to patent, processes for which patents are difficult to enforce, and other elements of our technology, discovery and development processes that involve proprietary know-how, information or technology that is not covered by patents. Any disclosure to or misappropriation by third parties of our confidential proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, including by enabling them to develop and commercialize products substantially similar to or competitive with our current or future product candidates, thus eroding our competitive position in the market. Trade secrets can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements and invention assignment agreements with our employees, consultants, and outside scientific advisors, contractors and collaborators. These agreements are designed to protect our proprietary information. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, or outside scientific advisors might intentionally or inadvertently disclose our trade secrets or confidential, proprietary information to competitors. In addition, competitors may otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. If any of our confidential proprietary information were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent such competitor from using that technology or information to compete with us, which could harm our competitive position.
| 28 |
Enforcing a claim that a third party illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, the laws of certain foreign countries do not protect proprietary rights such as trade secrets to the same extent or in the same manner as the laws of the United States. Misappropriation or unauthorized disclosure of our trade secrets to third parties could impair our competitive advantage in the market and could materially adversely affect our business, results of operations and financial condition.
Some intellectual property that we have in-licensed, if created as a result of government funded programs, may be subject to certain federal regulations.
Some of theagreements covering the intellectual property rights we have licensed provide that to the extent that such rights are derived from the use of U.S. government funding, those rights may therefore be subject to certain federal regulations. As a result, the U.S. government may have certain rights to intellectual property embodied in our current or future product candidates pursuant to the Bayh-Dole Act of 1980, or Bayh-Dole Act. These U.S. government rights in certain inventions developed under a government-funded program include a non-exclusive, non-transferable, irrevocable worldwide license to use inventions for any governmental purpose. In addition, the U.S. government has the right to require us to grant exclusive, partially exclusive or non-exclusive licenses to any of these inventions to a third party if it determines that: (i) adequate steps have not been taken to commercialize the invention, (ii) government action is necessary to meet public health or safety needs or (iii) government action is necessary to meet requirements for public use under federal regulations (also referred to as "march-in rights"). The U.S. government also has the right to take title to these inventions if we, or the applicable licensor, fail to disclose the invention to the government and fail to file an application to register the intellectual property within specified time limits. Intellectual property generated under a government funded program is also subject to certain reporting requirements, compliance with which may require us or the applicable licensor to expend substantial resources. In addition, the U.S. government requires that any products embodying the subject invention or produced through the use of the subject invention be manufactured substantially in the United States. The manufacturing preference requirement can be waived if the owner of the intellectual property can show that reasonable but unsuccessful efforts have been made to grant licenses on similar terms to potential licensees that would be likely to manufacture substantially in the United States or that under the circumstances domestic manufacture is not commercially feasible. This preference for U.S. manufacturers may limit our ability to contract with non-U.S. product manufacturers for products covered by such intellectual property. To the extent any of our current or future intellectual property is generated through the use of U.S. government funding, the provisions of the Bayh-Dole Act may similarly apply.
Risks Related to Commercialization of Our Current and Future Product Candidates
Our commercial success depends upon attaining significant market acceptance of our current and future product candidates, if approved, among physicians, patients, healthcare payors and cancer treatment centers.
Even if we obtain regulatory approval for any of our current or future product candidates, the products may not gain market acceptance among physicians, healthcare payors, patients or the medical community, including cancer treatment centers. Market acceptance of any product candidates for which we receive approval depends on a number of factors, including:
If any of our current and future product candidates are approved but fail to achieve market acceptance among physicians, patients, healthcare payors or cancer treatment centers, we will not be able to generate significant revenues, which would compromise our ability to become profitable.
| 29 |
Even if we are able to commercialize our current or future product candidates, the products may not receive coverage and adequate reimbursement from third-party payors in the United States and in other countries in which we seek to commercialize our products, which could harm our business.
Our ability to commercialize any product successfully will depend, in part, on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from third-party payors, including government health administration authorities, private health insurers and other domestic and international government programs, private insurance plans and managed care programs. Most third-party payors may deny reimbursement if they determine that a medical product was not used in accordance with cost-effective treatment methods, as determined by the third-party payor, or was used for an unapproved indication. organizations.
Third-party payors also may refuse to reimburse for experimental proceduresdetermine which medications they will cover and devices. Furthermore, because our programs areestablish reimbursement levels. A primary trend in the early stages of development, we are unable at this timehealthcare industry is cost containment. Third-party payors have attempted to determine their cost-effectivenesscontrol costs by limiting coverage and the level or methodamount of reimbursement.reimbursement for particular medications. Increasingly, the third-party payors who reimburse patients are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. IfThird-party payors may also seek additional clinical evidence, beyond the data required to obtain regulatory approval, demonstrating clinical benefit and value in specific patient populations before covering our products for those patients. We cannot be sure that coverage and adequate reimbursement will be available for any product that we commercialize and, if coverage is available, what the level of reimbursement will be. Coverage and reimbursement may impact the demand for, or the price of, any product candidate for which we areobtain regulatory approval. If reimbursement is not available or is available only at limited levels, we may not be able to chargesuccessfully commercialize any product candidate for any productswhich we develop is inadequate in light of our development and other costs, our profitability could be adversely effected.
13
Recently enacted and future legislation, including potentially unfavorable pricing regulations, may increase the difficulty and cost for us to obtain regulatory approval of and commercialize our current or future product candidates and affect the prices we may obtain.
The regulations that govern, among other things, regulatory approvals, coverage, pricing and reimbursement for new drug products vary widely from country to country. In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay regulatory approval of our current or future product candidates, restrict or regulate post-approval activities and affect our ability to successfully sell any product candidates for which we obtain regulatory approval. In the United States, the European Union, United Kingdom and other potentially significant markets for our current and future product candidates, government authorities and third-party payors are increasingly attempting to contain health care costs by limiting both coverage andlimit or regulate the levelprice of reimbursement for medical products and services. Aservices, particularly for new and innovative products and therapies, which has resulted in lower average selling prices. Legislative and regulatory proposals have also been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. For example, the Centers for Medicare & Medicaid Services, or CMS, issued a final rule, effective on July 9, 2019, that requires direct-to-consumer television advertisements of prescription drugs and biological products, for which payment is available through or under Medicare or Medicaid, to include in the advertisements the Wholesale Acquisition Cost, or list price, of that drug or biological product if it is equal to or greater than $35 for a monthly supply or usual course of treatment. Prescription drugs and biological products that are in violation of these requirements will be included on a public list. Congress and the Trump administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs. At the state level, legislatures are increasingly passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our product candidates or additional pricing pressures. We cannot be sure whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the regulatory approvals of our product candidates, if any, may be.
| 30 |
Healthcare legislative measures aimed at reducing healthcare costs may have a material adverse effect on our business and results of operations.
Third-party payors, whether domestic or foreign, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. In both the United States and certain international jurisdictions, there have been a number of legislative and regulatory changes to the health care system that could impact our ability to sell our products profitably. In particular, in 2010, the Affordable Care Act, or ACA, was enacted, which, among other things, subjected biologic products to potential competition by lower-cost biosimilars, addressed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program, extended the Medicaid Drug Rebate Program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations, subjected manufacturers to new annual fees and taxes for certain branded prescription drugs, and provided incentives to programs that increase the federal government’s comparative effectiveness research. Since its enactment, there have been judicial and Congressional challenges to certain aspects of the ACA, as well as recent efforts by the current U.S. administration to repeal or repeal and replace certain aspects of the ACA. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas, or the Texas District Court Judge, ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as a part of the Tax Act, the remaining provisions of the ACA are invalid as well. While the Texas District Court Judge, as well as the Trump Administration and CMS, have stated that the ruling will have no immediate effect, it is unclear how this decision, subsequent appeals and other efforts to repeal and replace the ACA will impact the ACA. Until there is more certainty concerning the future of the ACA, it will be difficult to predict its full impact and influence on our business.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. In August 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect in 2013, and will remain in effect through 2027 unless additional Congressional action is taken. The American Taxpayer Relief Act of 2012 further reduced Medicare payments to several providers, including hospitals and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels directed at containing or lowering the cost of healthcare. We cannot predict the initiatives that may be adopted in the future. The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare services to change contain or reduce costs of healthcare and/or impose price controls may adversely affect:
We expect that the ACA, as well as other healthcare reform measures that may be adopted in the future, may result in additional reductions in Medicare and other healthcare funding, more rigorous coverage criteria, lower reimbursement and new payment methodologies. This could lower the price that we receive for any approved product. Any denial in coverage or reduction in reimbursement from Medicare or other government-funded programs may result in a similar denial or reduction in payments from private payors, which may prevent us from being able to generate sufficient revenue, attain profitability or commercialize our product candidates, if approved.
| 31 |
Price controls may be imposed in foreign markets, which may adversely affect our future profitability.
In some countries, particularly member states of the European Union, the pricing of prescription drugs is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after receipt of regulatory approval for a product. In addition, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various European Union member states and parallel distribution, or arbitrage between low-priced and high-priced member states, can further reduce prices. In some countries, we or our collaborators may be required to conduct a clinical trial or other studies that compare the cost-effectiveness of our product candidates to other available therapies in order to obtain or maintain reimbursement or pricing approval. Publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be adversely affected.
Risks Related to Healthcare Compliance Regulations
Our relationships with customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings. If we or they are unable to comply with these provisions, we may become subject to civil and criminal investigations and proceedings that could have a material adverse effect on our business, financial condition and prospects.
Healthcare providers, physicians and third-party payors will play a primary role in the recommendation and prescription of any product candidates for which we obtain regulatory approval. Our current and future arrangements with healthcare providers, healthcare entities, third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we research, develop and will market, sell and distribute our products. As a pharmaceutical company, even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are applicable to our business. Restrictions under applicable federal and state healthcare laws and regulations that may affect our ability to operate include the following:
| 32 |
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, disgorgement, exclusion from government funded healthcare programs, such as Medicare and Medicaid, integrity oversight and reporting obligations, and the curtailment or restructuring of our operations. If any physicians or other healthcare providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could cause significant liability for us and harm our reputation.
We are exposed to the risk of employee fraud or other misconduct, including intentional failures to comply with FDA regulations or similar regulations of comparable foreign regulatory authorities, provide accurate information to the FDA or comparable foreign regulatory authorities, comply with manufacturing standards we have established, comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable foreign regulatory authorities, report financial information or data accurately or disclose unauthorized activities to us. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and integrity oversight and reporting obligations.
We have been involved in multiple legal and governmental proceedings, and may in the future be involved in proceedings, relating to the commercial activities of our predecessor that could adversely affect our financial condition and our business.
Our predecessor company, Galena, was involved in multiple legal and governmental proceedings, including stockholder class actions, both state and federal, some of which are ongoing. These legal and governmental actions (the “Galena Legacy Matters”), included allegations relating to federal securities law violations, claims under the False Claims Act and Anti-Kickback Statute, claims regarding breaches of contract, and other stockholder allegations, including claims of breaches of fiduciary duty by our former directors, and fentanyl related litigation.
In December 2015, we announced we had received a subpoena from the U.S. Attorney’s Office for the District of New Jersey, or the USAO NJ, requesting the production of a broad range of documents pertaining to marketing and promotional practices related to Abstral, a fentanyl or synthetic opioid product, that we sold to Sentynl Therapeutics Inc., or Sentynl, in November 2015. In January 2016, we announced that the U.S. Attorney’s Office of New Jersey, of USAO NJ and the Department of Justice, or DOJ, were conducting a criminal and civil investigation of us, which came to involve criminal investigations with respect to possibly one or more then-current and/or former employees. On September 8, 2017, the DOJ announced a civil settlement agreement with our company regarding certain of the marketing and promotional practices at issue in the USAO NJ and DOJ’s investigation. The settlement involved a civil resolution agreement and a civil payment of approximately $7.551 million, plus interest accrued since the date of reaching an agreement in principle in return for a release of federal government claims against our company in connection with the covered conduct in investigation. The civil payment was fully paid by us on or about December 29, 2017. The settlement did not include releases of criminal claims by the USAO NJ and DOJ or claims by state agencies or administrative claims by the Department of Health and Human Services, or HHS, but each of these government authorities indicated that they had no present intention to pursue claims in connection with the investigation. A qui tam action had been filed against us and others as described in our settlement agreement with DOJ and USAO NJ. As set forth in that settlement agreement, for a release of all claims against us and our former officers and directors and dismissal with prejudice of the qui tam lawsuit, the relator received a portion of the $7.551 million payment to the federal government. As a result of the payment of the settlement amount, the federal government and the relator filed a stipulation of dismissal with prejudice as to their claims against us in the qui tam lawsuit. In a separate settlement agreement, we paid $0.3 million in cash to the relator’s counsel for the statutorily mandated attorney’s fees.
| 33 |
We also received a subpoena from the U.S. Attorney’s Office for the Southern District of New York, or USAO SDNY, in February 2018, seeking documents related to specific prescribing physicians for Abstral who have been subsequently indicted, to which we responded. To our knowledge, we are not a target or subject of that investigation and have had no further interaction with the USAO SDNY with regard to the matter after responding to the subpoena.
A federal investigation led by the U.S. Attorney’s Office for the Southern District of Alabama, or the SDAL, of two of the high-prescribing physicians for Abstral (fentanyl) sublingual tablets resulted in the criminal prosecution of the two physicians for alleged violations of the federal False Claims Act and other federal statutes. On April 28, 2016, a second superseding indictment was filed in the criminal case, which added additional information about the defendant physicians and provided information regarding the facts and circumstances involving a rebate agreement between us and the defendant physicians’ pharmacy as well as their ownership of our common stock. The criminal trial, which began on January 4, 2017, concluded with a jury verdict on February 23, 2017 finding these physicians guilty on 19 of 20 counts. In May 2017, one physician was sentenced to 20 years in prison, and the other physician was sentenced to 21 years in prison. At the end of the SDAL case, SDAL dismissed count 18 of the indictment charging that the physicians conspired, through the C&R Pharmacy, to receive illegal kickbacks in exchange for prescribing Abstral. To our knowledge, we were not a target or subject of that investigation.
There continues to be significant litigation and governmental activity generally in the fentanyl and opioid area, and this activity is expected to continue and may increase in the future. We cannot assure you we will not become subject to additional significant legal or governmental proceedings relating to Galena’s former Abstral business in the future. Moreover, in addition to these ongoing and prior matters, we may be exposed to claims, or other legal or governmental actions in the future relating to violations of the False Claims Act, Anti-Kickback Statute, the Affordable Care Act, or any other applicable state or federal statutes or regulations, and thereby be subject to penalties, such as civil and criminal penalties, damages, fines, or an administrative action of exclusion from government health care reimbursement programs.
There can be no assurance that we will not be exposed to other liabilities or risks, including potential liabilities and risks not currently known to us, resulting from the prior operations of Galena. We can make no assurances as to the time or resources that will need to be devoted to the Galena Legacy Matters, or any new or future matters resulting from the prior operations of Galena or their outcome, or the impact, if any, that these matters or any resulting legal or governmental proceedings may have on our business or financial condition but any further action in respect of any such matter by a governmental agency could have a material adverse effect on our results of operation and our business and prospects.
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of any products that we may develop.
We face an inherent risk of product liability exposure related to the testing of our current or future product candidates in human clinical trials and will face an even greater risk if we commercially sell any products that we may develop. Product liability claims may be brought against us by subjects enrolled in our clinical trials, patients, healthcare providers or others using, administering or selling our products. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| 34 |
We currently hold product liability insurance coverage at a level that we believe is customary for similarly situated companies and adequate to provide us with insurance coverage for foreseeable risks, but which may not be adequate to cover all liabilities that we may incur. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise. We intend to expand our insurance coverage for products to include the sale of commercial products if we obtain regulatory approval for our product candidates in development, but we may be unable to obtain commercially reasonable product liability insurance for any products that receive regulatory approval. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us, particularly if judgments exceed our insurance coverage, could decrease our cash and adversely affect our business.
We face product liability exposure from prior sales of Abstral and Zuplenz (ondansetron) and, if successful claims are brought against us, we may incur substantial liability if our insurance coverage for those claims is inadequate.
Because we previously sold Abstral and Zuplenz (ondansetron), an anti-emetic, oral soluble film for chemotherapy-induced nausea we are exposed to possible product liability claims. In November 2015, Galena sold the rights to Abstral to Sentynl, and in December 2015, Galena sold the rights to Zuplenz to Midatech Pharma, PLC, or Midatech. Under the respective asset purchase agreements with Sentynl and Midatech, our future obligations under our former agreements with Orexo AB and MonoSol Rx have been assumed by Sentynl and Midatech, respectively, except that we will continue to be responsible for chargebacks, rebates, patient assistance and certain other product distribution channel liabilities related to Abstral and Zuplenz for a specified period of time post-closing. We are also required to indemnify Sentynl and Midatech for contractual or product liability claims arising from actions occurring prior to the sale date. With respect to Zuplenz, we will continue to be responsible for any downstream returns from end user customers or returns from wholesalers from inventory existing as of December 24, 2015 that was sold by us prior to December 24, 2015.
We do not consider our responsibilities with regard to Sentynl and Midatech to be material, but if substantial unknown liabilities were to arise, it could have a material adverse effect on our financial condition. If we cannot successfully defend ourselves against product liability claims we could incur substantial liabilities, regardless of merit or eventual outcome. A successful product liability claim or series of claims brought against us could cause our stock price to decline and, if judgments exceed our insurance coverage, could decrease our cash and have a material adverse effect on our business, results of operations, financial condition and prospects.
Risks Related to our Business Operations
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. As a result, stockholders could lose confidence in our financial and other public reports, which would harm our business, the trading price of our common stock and our ability to raise additional capital in the future.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation, could cause us to fail to meet our reporting obligations. Ineffective internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of Common Stock, and which could impact our ability to raise capital in the future. In addition, any future testing by us conducted in connection with Section 404 of the Sarbanes-Oxley Act of 2002, as amended (“SOX”), or any required subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses or that may require prospective or retroactive changes to our consolidated financial statements or identify other areas for further attention or improvement.
We are required, pursuant to Section 404 of SOX, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting as of December 31, 2018. However, our independent registered public accounting firm is not required to attest to the effectiveness of our internal control over financial reporting pursuant to Section 404. Under the supervision and with the participation of our Chief Executive Officer and Vice President Finance and Interim Principal Accounting Officer, our management conducted an evaluation of the effectiveness of our internal control over financial reporting based on the guidelines in the Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on that evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2018. An independent assessment of the effectiveness of our internal controls could detect problems that our management’s assessment might not. Undetected material weaknesses in our internal controls could lead to financial statement restatements and require us to incur the expense of remediation.
| 35 |
We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do.
We face competition from numerous pharmaceutical and biotechnology enterprises, as well as from academic institutions, government agencies and private and public research institutions for our current product candidates. Our commercial opportunities will be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer side effects or are less expensive than any products that we may develop. Competition could result in reduced sales and pricing pressure on our current or future product candidates, if approved, which in turn would reduce our ability to generate meaningful revenues and have a negative impact on our results of operations. In addition, significant delays in the development of our product candidates could allow our competitors to bring products to market before we do and impair our ability to commercialize our product candidates. The biotechnology industry, including the cancer immunotherapy market, is intensely competitive and involves a high degree of risk. We compete with other companies that have far greater experience and financial, research and technical resources than us. Potential competitors in the United States and worldwide are numerous and include pharmaceutical and biotechnology companies, educational institutions and research foundations, many of which have substantially greater capital resources, marketing experience, research and development staffs and facilities than ours. Some of our competitors may develop and commercialize products that compete directly with those incorporating our technology or may introduce products to market earlier than our products or on a more cost-effective basis. In addition, our technology may be subject to competition from other major healthcare marketstechnology or methods developed using techniques other than those developed by traditional biotechnology methods. Our competitors compete with us in recruiting and retaining qualified scientific and management personnel as well as in acquiring technologies complementary to our technology. Our company and our collaborators may face competition with respect to product efficacy and safety, ease of use and adaptability to various modes of administration, acceptance by physicians, the timing and scope of regulatory approvals, availability of resources, reimbursement coverage, price and patent position, including the potentially dominant patent positions of others. An inability to successfully complete our product development or commercializing those product candidates could result in our having limited prospects for establishing market share or generating revenue from our technology.
There are several agents in clinical development in similar settings to our planned Phase 3 AML clinical development program for GPS. The most advanced of these products is oral Vidaza (azacytidine) (also known as CC-486), under development by Celgene Corporation, which is anticipated to report results from a registration-enabling Phase 3 study (named the QUAZAR or CC-486-AML-001 study) by the end of 2019. There are several of other investigational immunotherapies advancing through Phase 2 and Phase 3 trials for target indications that we believe are also potential target indications for GPS. If these or other therapies are successful in their development, it could negatively impact our ability to enroll our clinical trials and could negatively impact the commercial potential of GPS.
We are also planning a clinical development program in combination with cancer checkpoint inhibitors. This is a highly competitive field, with hundreds of such combination trials with various checkpoint inhibitors ongoing. If one or more of these combinations produce positive results in indications that we believe are targets for GPS (either in combination or in stand-alone administration) this could increase the difficulty for us to conduct our trials and could negatively impact our path to regulatory approval and our ability to successfully commercialize our products.
Many of our competitors or potential competitors have been proposed in recent years. These proposals have included prescription drug benefit legislation recently enactedsignificantly greater established presence in the United Statesmarket, financial resources and healthcare reform legislation enacted by certain states. Levels of reimbursementexpertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do, and as a result may decreasehave a competitive advantage over us. Mergers and acquisitions in the future,pharmaceutical and future legislation, regulation or reimbursement policies of third-party payorsbiotechnology industries may adversely affect the demand for and price levelsresult in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies and technology licenses complementary to our programs or potentially advantageous to our business.
As a result of these factors, these competitors may obtain regulatory approval of their products before we are able to obtain patent protection or other intellectual property rights, which will limit our ability to develop or commercialize our current or future product candidates. Our competitors may also develop drugs that are safer, more effective, more widely used and cheaper than ours, and may also be more successful than us in manufacturing and marketing their products. IfThese appreciable advantages could render our customers are not reimbursed for our products, they may reduceproduct candidates obsolete or discontinue purchasesnoncompetitive before we can recover the expenses of development and commercialization.
| 36 |
We enter into various contracts in the normal course of our products,business in which we may be required to indemnify the other party to the contract under certain specific scenarios. In the event we have to perform under these indemnification provisions, it could have a material adverse effect on our business, financial condition and results of operations.
In the normal course of business, we periodically enter into academic, commercial, service, collaboration, licensing, consulting and localitiesother agreements that contain indemnification provisions. With respect to our academic and other research agreements, we typically agree to indemnify the institution and related parties from losses arising from claims relating to the products, processes or services made, used, sold or performed pursuant to the agreements for which we have established drug importation programssecured licenses, and from claims arising from our or our sublicensees’ exercise of rights under the agreement. With respect to our collaboration agreements, we indemnify our collaborators from any third-party product liability claims that could result from the production, use or consumption of the product, as well as for alleged infringements of any patent or other intellectual property right by a third party. With respect to consultants, we indemnify them from claims arising from the good faith performance of their citizens, and federal drug import legislation has been introduced in Congress. The Medicare Prescription Drug Plan legislation, which became law in December 2003, required the Secretary of Health and Human Services to promulgate regulations for drug reimportation from Canada into the United Statesservices.
Should our obligations under some circumstances, including when the drugs are sold at a lower price than in the United States. The Secretary, however, retained the discretion not to implement a drug reimportation planan indemnification provision exceed applicable insurance coverage or if he finds that the benefits do not outweigh the costs, and has so far declined to approve a reimportation plan. Proponents of drug reimportation may attempt to pass legislation that would directly allow reimportation under certain circumstances. Legislation or regulations allowing the reimportation of drugs, if enacted, could decrease the price we receivewere denied insurance coverage for any products thatclaim, our business, financial condition and results of operations could be adversely affected. Similarly, if we may developare relying on a collaborator to indemnify us and the collaborator is denied insurance coverage for the claim or the indemnification obligation exceeds the applicable insurance coverage, and if the collaborator does not have other assets available to indemnify us, our business, financial condition and results of operations could be adversely affected.
Significant disruptions of information technology systems, computer system failures or breaches of information security could adversely affect our business.
We rely to a large extent upon sophisticated information technology systems to operate our business. In the ordinary course of business, we collect, store and transmit large amounts of confidential information (including, but not limited to, personal information and intellectual property). We also have outsourced significant elements of our operations to third parties, including significant elements of our information technology infrastructure and, as a result, we are managing many independent vendor relationships with third parties who may or could have access to our confidential information. The size and complexity of our information technology and information security systems, and those of our third-party vendors with whom we contract (and the large amounts of confidential information that is present on them), make such systems potentially vulnerable to service interruptions or to security breaches from inadvertent or intentional actions by our employees or vendors, or from malicious attacks by third parties. Such attacks are of ever-increasing levels of sophistication and are made by groups and individuals with a wide range of motives (including, but not limited to, industrial espionage and market manipulation) and expertise. While we have invested significantly in the protection of data and information technology, there can be no assurance that our efforts will prevent service interruptions or security breaches.
Our internal computer systems, and those of MSK, our CROs, our CMOs, and other business vendors on which we rely, are vulnerable to damage from computer viruses, unauthorized access, natural disasters, fire, terrorism, war and telecommunication and electrical failures. We exercise little or no control over these third parties, which increases our vulnerability to problems with their systems. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our drug development programs. Any interruption or breach in our systems could adversely affect our business operations and/or result in the loss of critical or sensitive confidential information or intellectual property, and could result in financial, legal, business and reputational harm to us or allow third parties to gain material, inside information that they use to trade in our securities. For example, the loss of clinical trial data from completed or ongoing clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability, the further development of our current and future revenuesproduct candidates could be delayed and prospectsour business could be otherwise adversely affected.
We will likely need to grow the size of our organization in the future, and we may experience difficulties in managing this growth.
As of June 6, 2019, we had 6 full-time employees. Depending on the outcome of our review of our strategic alternatives, we may need to grow the size of our organization in order to support our continued development and potential commercialization of our product candidates. As our development and commercialization plans and strategies continue to develop, our need for profitability.additional managerial, operational, manufacturing, sales, marketing, financial and other resources may increase. Our management, personnel and systems currently in place may not be adequate to support this future growth. Future growth would impose significant added responsibilities on members of management, including:
| 37 |
If our operations expand, we will also need to manage additional relationships with various strategic partners, suppliers and other third parties. Our future financial performance and our ability to commercialize our product candidates and to compete effectively will depend, in part, on our ability to manage any future growth effectively, as well as our ability to develop a sales and marketing force when appropriate for our company. To that end, we must be able to manage our development efforts and preclinical studies and clinical trials effectively and hire, train and integrate additional management, research and development, manufacturing, administrative and sales and marketing personnel. The failure to accomplish any of these tasks could prevent us from successfully growing our company.
The requirements of being a public company may strain our resources, divert management’s attention and affect our ability to attract and retain qualified board members.
As a public company, we are subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act, the Dodd-Frank Act, the listing requirements of Nasdaq and other applicable securities rules and regulations. Compliance with these rules and regulations has increased, and will likely continue to increase, our legal and financial compliance costs, make some activities more difficult, time-consuming or costly, and place significant strain on our personnel, systems and resources. In addition, changing laws, regulations and standards relating to corporate governance and public disclosure are creating uncertainty for public companies, increasing legal and financial compliance costs and making some activities more time consuming. These laws, regulations and standards are subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time. This could result in continuing uncertainty regarding compliance matters, higher administrative expenses and a diversion of management’s time and attention. Further, if our compliance efforts differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us and our business may be harmed. Being a public company that is subject to these rules and regulations also makes it more expensive for us to obtain and retain director and officer liability insurance, and we may in the future be required to accept reduced coverage or incur substantially higher costs to obtain or retain adequate coverage. These factors could also make it more difficult for us to attract and retain qualified members of our board of directors and qualified executive officers.
Our common stock may be delisted from the Nasdaq Capital Market which could negatively impact the price of our common stock, liquidity and our ability to access the capital markets.
The listing standards of the Nasdaq Capital Market provide that a company, in order to qualify for continued listing, must maintain a minimum stock price of $1.00 and satisfy standards relative to minimum stockholders’ equity, minimum market value of publicly held shares and various additional requirements. If we fail to comply with all listing standards applicable to issuers listed on the Nasdaq Capital Market, our common stock may be delisted. If our common stock is delisted, it could reduce the price of our common stock and the levels of liquidity available to our stockholders. In addition, the delisting of our common stock could materially adversely affect our access to the capital markets and any limitation on liquidity or reduction in the price of our common stock could materially adversely affect our ability to raise capital. Delisting from the Nasdaq Capital Market could also result in other negative consequences, including the potential loss of confidence by suppliers, customers and employees, the loss of institutional investor interest and fewer business development opportunities.
As previously reported, on May 31, 2019, we received a letter from Nasdaq indicating that, based upon the closing bid price of our common stock for the last 30 consecutive business days, we did not meet the minimum bid price of $1.00 per share required for continued listing on the Nasdaq Capital Market pursuant to Minimum Bid Price Rule. We have been provided an initial period of 180 calendar days, or until November 27, 2019, to regain compliance with the Minimum Bid Price Rule. The letter also indicated that if at any time before November 27, 2019 the closing bid price for our common stock is at least $1.00 for a minimum of ten consecutive business days, Nasdaq will provide written notification to the company that it complies with the Minimum Bid Price Rule. If we do not regain compliance with the Minimum Bid Price Rule by November 27, 2019, we may be eligible for a second compliance period of 180 calendar days, provided that we meet the continued listing requirement for market value of publicly held shares and all other initial listing standards for the Nasdaq Capital Market, with the exception of the bid price requirement, and notify Nasdaq of our intention to cure the deficiency during such second compliance period, including by effecting a reverse stock split, if necessary. If we do not regain compliance with the Minimum Bid Price Rule by November 27, 2019 and are not eligible for a second compliance period at that time, Nasdaq will provide written notification to us that our common stock may be delisted. At that time, we may appeal Nasdaq’s delisting determination to a Nasdaq hearings panel. If we timely appeal, our common stock would remain listed pending the panel’s decision. There can be no assurance that, if we do appeal the delisting determination by Nasdaq to the panel, such appeal would be successful.
We have in the past, and may in the future, become involved in securities class action litigation that could divert management’s attention and harm our business, and insurance coverage may not be sufficient to cover all costs and damages.
In the past, securities class action or stockholder derivative litigation often follows certain significant business transactions, such as the sale of a business division or announcement of a merger. Additionally, securities class action or stockholder derivative litigation has become common in our industry following the announcement of negative data or adverse events. We have in the past, and may in the future, become involved in this type of litigation. Litigation often is expensive and diverts management’s attention and resources, which could adversely affect the continuing company’s business.
Our future success depends on our ability to retain our executive officers and to attract, retain and motivate qualified personnel.
We are highly dependent upon our personnel, including Dr. Angelos M. Stergiou (M.D., Sc.D. h.c.), our President and Chief Executive Officer, and member of our board of directors. Our employment agreement with Dr. Stergiou does not prevent him from terminating his employment with us at any time. The loss of Dr. Stergiou’s services could impede the achievement of our research, development and commercialization objectives. We have not obtained, do not own, nor are we the beneficiary of, key-person life insurance.
Governance changes, becoming subject to enhanced regulatory requirements and increased responsibilities associated with becoming a public company may influence our management personnel and our employees to terminate their employment with us. To enhance our ability to retain our executive management personnel, we have entered into retention agreements with certain executive officers and may find it beneficial to enter into additional retention agreements with other key personnel in the future, potentially increasing payroll and operating expenses.
| 38 |
Our future growth and success depend on our ability to recruit, retain, manage and motivate our employees. The loss of any member of our senior management team or the inability to hire or retain experienced management personnel could compromise our ability to execute our business plan and harm our operating results. Because of the specialized scientific and managerial nature of our business, we rely heavily on our ability to attract and retain qualified scientific, technical and managerial personnel. The competition for qualified personnel in the pharmaceutical field is intense and as a result, we may be unable to continue to attract and retain qualified personnel we may not be able to design, develop, market or sell our products or successfully managenecessary for the development of our business.
If we and our named executive officers and SAB members. The continued service of our named executive officers and SAB members is criticalthird-party manufacturers fail to our success. We have entered into employment agreements with our named executive officers, all of which can be terminated by such persons on short or no notice. The loss of any of our named executive officers or SAB members, or our inability to identify, attract, retain and integrate additional qualified key personnel, could make it difficult for us to manage our business successfully and achieve our business objectives.
14
We and disposal of these materials and specific waste products. Weour third-party manufacturers are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures exposure to blood-borne pathogens and the handling, use, storage, treatment and disposal of biohazardoushazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. The costOur operations also produce hazardous waste products. We generally contract with third parties for the disposal of compliance with these lawsmaterials and regulationswastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from us or our third-party manufacturers’ use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and may adversely affect capital expenditures to the extentpenalties.
Although we are required to procure expensive capital equipment to meet regulatory requirements.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or penalties if we violate any of these laws or regulations.
Risks Relating to Ownership of Our Common Stock
We need to secure additional capital which may cause dilution to you and our existing stockholders, provide subsequent investors with rights and preference that are senior to yours, restrict our operations or cash flows that we would have achieved as a separate company during the periods presented or those that we will achieve in the future. Priorrequire us to the contribution of our RNAi assets from CytRx, our RNAi research and development activities were conducted by CytRx as part of its broader operations, rather than as an independent division or subsidiary, and were primarily conducted through sponsored research arrangements rather than through internal activities. CytRx also performed various corporate functions relatingrelinquish rights to our business, as discussed above. Our historical financial information reflects allocations of indirect expenses from CytRx for these and similar functions. We believe that these allocations are comparable to the expenses we would have incurred had we operated as a separate company, although we may incur higher expenses as a separate company.
15
16
The market price and trading volume of shares of our common stock may be volatilevolatile.
The market forprice of shares of our common stock.stock has exhibited substantial volatility recently. Between January 2, 2018 and June 11, 2019, the trading price of shares of our common stock as reported on Nasdaq ranged from a low of $0.40 to a high of $11.09. The market price of shares of our common stock could continue to fluctuate significantly for many reasons, including the following factors:
17
Factors beyond our control may also have an impact on the market price of shares of our common stock. For example, to the extent that other large companies within our industry experience declines in their stock price, our stock price may decline as well. In addition, whenprices, the market price of a company’s common stock drops significantly, stockholders often institute securities class action lawsuits against the company. A lawsuit against us could cause us to incur substantial costs and could divert the time and attention of our management and other resources.
Inadequate funding for the FDA, the SEC and other government agencies could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or approximately 46%commercialized in a timely manner or otherwise prevent those agencies from performing normal business functions on which the operation of our outstanding shares. Webusiness may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have agreedfluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, including beginning on December 22, 2018, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA and the SEC, have had to furlough critical FDA, SEC and other government employees and stop critical activities. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, upon request by CytRx,completion of this offering and in our operations as a public company, future government shutdowns could impact our ability to access the public markets and obtain necessary capital in order to properly capitalize and continue our operations.
Risks Relating to this Offering and Ownership of Our Common Stock
Management will have broad discretion as to the use of the proceeds from this offering and we may not use the proceeds effectively.
Our management will have broad discretion with respect to the use our best efforts to cause allof proceeds of this offering, including for any of the purposes described in the section entitled “Use of Proceeds” beginning on page 46 of this prospectus. You will be relying on the judgment of our shares issuedmanagement regarding the application of the proceeds of this offering. The results and effectiveness of the use of proceeds are uncertain, and we could spend the proceeds in ways that you do not agree with or that do not improve our results of operations or enhance the value of our common stock. Our failure to CytRx pursuantapply these funds effectively could harm our business, delay the development of our product candidates and cause the price of our common stock to the two contribution agreements we entered into in relation to our initial capitalization to be registered under the Securities Act, with certain exceptions, with all expenses incurred in connection with any such registrationdecline.
| 40 |
You will be borne by us.
You will incur immediate and substantial dilution as a dividendresult of this offering. After giving effect to the sale by us of securities offered in this offering at an assumed public offering price of $0.42 per share of common stock and accompanying common warrant, and after deducting underwriting discounts and commissions and estimated offering expenses payable by us, investors in this offering can expect an immediate dilution of approximately $0.26 per share. See the section entitled “Dilution” beginning on page 50 of this prospectus for a more detailed discussion of the dilution you will incur if you purchase shares in this offering. The discussion above assumes (i) no sale of pre-funded warrants, which, if sold, would reduce the number of shares of common stock that we are offering on a one-for-one basis, and (ii) no exercise of the common warrants being offered in this offering.
There is no public market for the pre-funded warrants or distributionthe common warrants being offered in this offering.
There is no established public trading market for the pre-funded warrants or the common warrants being offered in this offering, and we do not expect a market to develop. In addition, we do not intend to apply to list the pre-funded warrants or the common warrants on any securities exchange or nationally recognized trading system, including The Nasdaq Capital Market. Without an active market, the liquidity of the pre-funded warrants and the common warrants will be limited.
Holders of pre-funded warrants or common warrants purchased in this offering will have no rights as common stockholders until such holders exercise their warrants and acquire our common stock.
Until holders of pre-funded warrants acquire shares of our common stock upon exercise thereof, holders of warrants will have no rights with respect to shares currently owned by CytRx, in other registration statements that we may file with the SEC on behalfshares of our companycommon stock underlying such warrants. Upon exercise of the pre-funded warrants or our security holders. The availabilitycommon warrants, the holders will be entitled to exercise the rights of a common stockholder only as to matters for which the record date occurs after the exercise date.
Future sales of substantial amounts of our shares held by CytRx and UMMS for resale publicly, as well as any actualcommon stock, or the possibility that such sales of these shares,could occur, could adversely affect the market price of our shares.
Future sales in the value of our shares owned by CytRx from time to time were to exceed 40% of the value of CytRx’s total assets, CytRx may be deemed an “investment company” within the meaning of the Investment Company Act of 1940 and become subject to the stringent regulations applicable to investment companies. In this event, CytRx would likely seek to promptly sell or otherwise disposepublic market of shares of our common stock, including shares referred to in order to avoid becoming an inadvertent investment company. Any such salesthe foregoing risk factors or other disposition by CytRxshares issued upon exercise of our shares,outstanding stock options or warrants, or the possibility of suchperception by the market that these sales or disposition, could adversely affectoccur, could lower the market price of our shares.
| 41 |
As of any new securities sold or issued by us so as to maintain its percentage ownership of us at the time of any such sale andMarch 31, 2019, we had reserved for issuance which is currently approximately 46% of our outstanding shares. The exercise by CytRx of its preemptive rights may impair our ability to raise funds, or adversely affect the terms on which we are able to raise funds, as we may not be able to offer to new investors the quantity of our stock that they may desire to purchase.
Certain of our securityholders have registration rights and they can require us, subject to certain limitations, to register their securities for resale, or require us to include their securities for resale in any offering of our common stock we may propose. Any such resales into the public market could place downward pressure on the price of our common stock.
We have issued and may issue additional preferred stock in the future, and the terms of the memberspreferred stock may reduce the value of our common stock.
We are authorized to issue up to five million shares of preferred stock in one or more series. Our board of directors are not affiliated (as defined)may determine the terms of future preferred stock offerings without further action by our stockholders. If we issue shares of preferred stock, it could affect stockholder rights or reduce the market value of our outstanding common stock. In particular, specific rights granted to future holders of preferred stock may include voting rights, preferences as to dividends and liquidation, conversion and redemption rights, sinking fund provisions, and restrictions on our ability to merge with CytRx. However, by virtue of its stock ownership, CytRx may be able to significantly influence the outcome of matters required to be submittedor sell our assets to a votethird party.
We have in the past and expect in the future to settle legal claims through the issuance of freely tradable shares of our stockholders, including any proposed amendmentscommon stock, which will result in dilution to holders of our certificate of incorporationcommon stock and approval of mergers and other significant corporate transactions. This concentration of ownership may adversely affect the market price of our common stock by:
18
Anti-takeover provisions of our certificateAmended and Restated Certificate of incorporationIncorporation and by-lawsour Amended and Restated Bylaws and provisions of Delaware law could delay or prevent a change of control that you may favor.control.
Anti-takeover provisions of our certificateAmended and Restated Certificate of incorporationIncorporation and by-lawsour Amended and provisions of Delaware lawRestated Bylaws may discourage, delay or prevent a merger or other change of control that stockholders may consider favorable or may impede the ability of the holders of our common stock to change our management.management and may be constrained by other contractual agreements with third parties. These provisions of our certificateAmended and Restated Certificate of incorporationIncorporation and by-laws,our Amended and Restated Bylaws, among other things:
| divide our |
| limit the right of |
| · | prohibit stockholders from acting by written consent; |
| regulate how stockholders may present proposals or nominate directors for election at annual meetings of |
| authorize our |
In addition, Section 203 of the Delaware General Corporation Law provides that, subject to limited exceptions, persons that acquire, or are affiliated with a person that acquires, more than 15% of the outstanding voting stock of a Delaware corporation such as our company shall not engage in any business combination with that corporation, including by merger, consolidation or acquisitions of additional shares for a three-year period following the date on which that person or itsour affiliate crosses the 15% stock ownership threshold. Section 203 could operate to delay or prevent a change of control of us.
We are, and in the future may be, subject to legal or governmental proceedings that could adversely affect our company.financial condition and our business.
Our predecessor company, Galena, was involved in multiple legal and governmental proceedings, including stockholder class actions, both state and federal, some of which are ongoing and to which the combined company continues to be subject. These legal and governmental actions, which we refer to as the Galena Legacy Matters, included allegations relating to federal securities law violations, claims under the False Claims Act and Anti-Kickback Statute, claims regarding breaches of contract, and other stockholder allegations, including claims of breaches of fiduciary duty by our former directors, and fentanyl related litigation. As a result of a cease and desist order issued by the SEC on April 10, 2017 and our related settlement with the SEC, or the SEC Settlement, we are currently an “ineligible issuer” as the term is defined under Rule 405 promulgated under the Securities Act. This could make it more difficult for us to raise necessary financing in the future. If we fail to comply with the terms of the SEC Settlement in the future, it could have significant additional adverse consequences to us. A number of the Galena Legacy Matters relate to Galena’s former commercial activities associated with Abstral, a fentanyl, or synthetic opioid, product. There continues to be significant litigation and governmental activity generally in the fentanyl and opioid area, and this activity is expected to continue and may increase in the future. We cannot assure you we will not become subject to additional significant legal or governmental proceedings relating to Galena’s former Abstral business in the future.
| 42 |
These Galena Legacy Matters have required and continue to require our management and board of directors to devote a significant amount of time and resources to defending such claims and addressing such allegations, rather than focusing on executing on our business plans and operations. We may, acquirein the future, become subject to additional legal and governmental actions that will also require us to expend time and resources. The settlement of the Galena Legacy Matters has resulted in substantial payments, some of which have not been covered by our insurance policies. We may continue to incur substantial unreimbursed legal fees and other businessesexpenses in connection with the Galena Legacy Matters. These ongoing and other future legal and governmental proceedings may not qualify for coverage under, or form joint ventures thatmay exceed the limit of, our applicable directors and officers liability insurance policies and could have a material adverse effect on our financial condition, liquidity, and results of operations. An unfavorable outcome in any of these matters could damage our business and reputation or result in additional claims or proceedings against us. Moreover, in addition to these ongoing and prior matters, we may be unsuccessfulexposed to claims as a result of the Merger, or other legal or governmental actions in the future, which could result in the payment of additional amounts and could adversely dilute your ownershiphave a material adverse effect on our financial condition and results of our company.
If our common stock becomes subject to the penny stock rules, it may pursue future acquisitionsbe more difficult to sell our common stock.
The SEC has adopted rules that regulate broker-dealer practices in connection with transactions in penny stocks. Penny stocks are generally equity securities with a price of other complementary businessesless than $5.00 (other than securities registered on certain national securities exchanges or authorized for quotation on certain automated quotation systems, provided that current price and technology licensing arrangements. We also may pursue strategic alliances. We have no experiencevolume information with respect to acquiring other companies and limited experience with respect to the formation of collaborations, strategic alliances and joint ventures. If we were to make any acquisitions, we may not be able to integrate these acquisitions successfully into our existing business and we could assume unknown or contingent liabilities. We also could experience adverse effects on our reported results of operations from acquisition related charges, amortization of acquired technology and other intangibles and impairment charges relating to write-offs of goodwill and other intangible assets from time to time following the acquisition. Integration of an acquired company also may require management resources that otherwise would be available for ongoing development of our existing business. We may not identify or complete these transactions in a timely manner, on a cost-effective basis,such securities is provided by the exchange or at all,system). The OTC Bulletin Board does not meet such requirements and we may not realizeif the anticipated benefits of any acquisition, technology license or strategic alliance.
19
We have never declared or paid cash dividends on our common stock and we do not anticipate paying cash dividends on our common stock in the foreseeable future.
Our business requires significant funding. We currently plan to invest all available funds and future managementearnings in the development and organizational structuregrowth of our business and do not anticipate paying any cash dividends on our common stock in the foreseeable future and are allprohibited by the terms of our outstanding indebtedness from paying dividends on any common stock, except with the prior consent of our lenders. As a result, capital appreciation, if any, of our common stock will be our stockholders’ sole source of potential gain for the foreseeable future.
| 43 |
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the documents incorporated by reference into this prospectus include forward-looking statements. Forward-looking statements are not guaranteeswithin the meaning of performance. TheySection 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934, as amended that relate to future events or our future financial performance and involve known and unknown risks, uncertainties and assumptionsother factors that may cause our actual results, levels of activity, performance or achievements to differ materially from any future results, levels of activity, performance or achievements expressed or implied by anythese forward-looking statement.
| · | our projected financial position and estimated cash burn rate; |
| · | our estimates regarding expenses, future revenues and capital requirements; |
| · | our ability to continue as a going concern; |
| · | our need to raise substantial additional capital to fund our operations; |
| · | the success, cost and timing of our clinical trials; |
| · | our dependence on third parties in the conduct of our clinical trials; |
| · | our ability to obtain the necessary regulatory approvals to market and commercialize our product candidates; |
| · | the potential that results of preclinical and clinical trials indicate our current product candidates or any future product candidates we may seek to develop are unsafe or ineffective; |
| · | the results of market research conducted by us or others; |
| · | our ability to obtain and maintain intellectual property protection for our current product candidates; |
| · | our ability to protect our intellectual property rights and the potential for us to incur substantial costs from lawsuits to enforce or protect our intellectual property rights; |
| · | the possibility that a third party may claim we have infringed, misappropriated or otherwise violated their intellectual property rights and that we may incur substantial costs and be required to devote substantial time defending against these claims; |
| · | our reliance on third-party suppliers and manufacturers; |
| · | the success of competing therapies and products that are or become available; |
| · | our ability to expand our organization to accommodate potential growth and our ability to retain and attract key personnel; |
| · | the potential for us to incur substantial costs resulting from product liability lawsuits against us and the potential for these product liability lawsuits to cause us to limit our commercialization of our product candidates; |
| · | market acceptance of our product candidates, the size and growth of the potential markets for our current product candidates and any future product candidates we may seek to develop, and our ability to serve those markets; and |
| · | the successful development of our commercialization capabilities, including sales and marketing capabilities. |
| 44 |
Our current product candidates are undergoing clinical development and have not been approved by the FDA or the European Commission. These product candidates have not been, nor may they ever be, approved by any regulatory agency or competent authorities nor marketed anywhere in the world.
We may not actually achieve the plans, intentions or expectations disclosed in our ability to achieve results described in any forward-looking statements. Stockholders are cautionedstatements, and you should not to place undue reliance on suchour forward-looking statements. Forward-looking statements which speak onlyshould be regarded solely as ofour current plans, estimates and beliefs. We have included important factors in the date ofcautionary statements included in this prospectus. We assume no obligation and expressly disclaim any duty to update any forward-looking statement to reflect events or circumstances afterdocument, particularly in the datesection entitled “Risk Factors” beginning on page 7 of this prospectus that we believe could cause actual results or events to reflectdiffer materially from the occurrence of unanticipated events. In addition,forward-looking statements that we cannotmake. Moreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of each factorall factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements contained in this prospectus.
| 45 |
20
We will not receive anyestimate that the net proceeds from theour issuance and sale of our common stock byand pre-funded warrants and accompanying common warrants in this offering will be approximately $18.1 million, after deducting the Selling Stockholders. The Selling Stockholders will pay any underwriting discounts and commissions and estimated offering expenses incurredpayable by themus and assuming a public offering price of $0.42 per share and accompanying common warrant, which is the last reported sale price of our common stock on Nasdaq on June 11, 2019, and excluding the proceeds, if any, from the exercise of any common warrants issued in disposingthis offering.
As of March 31, 2019, we had cash and cash equivalents of approximately $2.6 million. We intend to use the net proceeds from this offering to commence a Phase 3 study for GPS monotherapy in AML CRem2 patients and to continue our Phase 1/2 basket type study of GPS in combination with pembrolizumab, and for general corporate purposes.
This expected use of net proceeds from this offering and our existing cash and cash equivalents represents our intentions based upon our current plans and business conditions, which could change in the future as our plans and business conditions evolve. The amounts and timing of our actual expenditures may vary significantly depending on numerous factors, including the progress of our development, the status of and results from clinical trials, as well as any collaborations that we may enter with third parties for GPS or any other product candidates we may seek to develop, and any unforeseen cash needs. As a result, our management will retain broad discretion over the allocation of the shares.net proceeds from this offering. We have no current agreements, commitments or understandings for any material acquisitions or licenses of any products, businesses or technologies.
We anticipate existing cash and cash equivalents and the net proceeds from this offering will bear all other costs, fees and expenses incurredbe sufficient to fund our planned operations through December 31, 2020. We plan to raise additional capital in effecting the issuance and registrationfuture to fund the completion of the shares covered byclinical development of our current product candidates and our ongoing working capital requirements.
As of the date of this prospectus, we cannot predict with certainty all the uses for the net proceeds to be received upon the completion of this offering or the amounts we will spend on the uses set forth above. Pending our use of the net proceeds from this offering, we intend to invest a portion of the net proceeds in a variety of capital preservation investments, including without limitation, all registrationshort-term, interest-bearing instruments and filing fees, NASDAQ Capital Market feesU.S. government securities.
| 46 |
Dividend Policy
We have never declared or paid any cash dividends on our common stock and fees and expenses of our counsel and our accountants.
| 47 |
The following table sets forth for the periods indicated the highour cash and low sales prices of our common stock on the Nasdaq Capital Market.
2008 | High | Low | ||||||
| First Quarter (commencing on March 12, 2008) | $ | 23.95 | $ | 6.01 | ||||
| Second Quarter | $ | 10.12 | $ | 5.22 | ||||
| Third Quarter (through July 23, 2008) | $ | 8.11 | $ | 6.42 | ||||
21
| Number of | ||||||||||||||||||||
| Beneficial Ownership of Selling Stockholders Before this Offering | Shares | Beneficial Ownership Upon Completion of this Offering (Assuming all Shares Offered hereby are Sold) | ||||||||||||||||||
| Number of | Being | Number of | ||||||||||||||||||
Name | Shares | Percent | Offered | Shares | Percent | |||||||||||||||
| Investment Funds Affiliated with Fidelity Investments(1) | 2,049,622 | 14.9 | % | 1,023,299 | 1,026,323 | 7.5 | % | |||||||||||||
| RHP Master Fund, Ltd | 50,000 | * | 50,000 | — | — | |||||||||||||||
| Stephen S. Galliker(1) | 85,000 | * | 10,000 | 75,000 | * | |||||||||||||||
| Sandford J. Hillsberg(1) | 85,000 | * | 10,000 | 75,000 | * | |||||||||||||||
| Mark J. Ahn, Ph.D.(1) | 85,000 | * | 10,000 | 75,000 | * | |||||||||||||||
| · | on an actual basis as of March 31, 2019; |
| on a pro forma basis to reflect the exercise of 1,000,000 warrants between April 1, 2019 and May 31, 2019 to purchase shares of common stock at an exercise price of $1.10 per share, resulting in net cash proceeds received of approximately $1.0 million after deducting commissions and offering expenses; and |
| · | on an as adjusted basis to give further effect to the | |
22
23
24
| Actual | Pro Forma | Pro Forma As Adjusted(1) | ||||||||||
| (in thousands) | ||||||||||||
| Cash and cash equivalents | $ | 2,576 | $ | 3,621 | $ | 21,741 | ||||||
| Stockholders’ equity: | ||||||||||||
| Preferred stock, $0.0001 par value per share: 5,000,000 shares authorized; Series A convertible preferred stock, 17,500 shares designated; No shares issued and outstanding actual and no shares issued and outstanding pro forma | — | — | — | |||||||||
| Common stock, $0.0001 par value per share: 350,000,000 shares authorized, 24,176,475 shares issued and outstanding; 25,176,475 shares issued and outstanding pro forma | 2 | 2 | 7 | |||||||||
| Additional paid-in capital | 89,340 | 90,385 | 108,500 | |||||||||
| Accumulated deficit | (86,392 | ) | (86,392 | ) | (86,392 | ) | ||||||
| Total stockholders’ equity | 2,950 | 3,995 | 22,115 | |||||||||
| Total capitalization | $ | 2,950 | $ | 3,995 | $ | 22,115 | ||||||
| Period from | ||||||||||||||||||||||||||||
| January 1, | For the | For the | ||||||||||||||||||||||||||
| 2003 (Date of | Three | Three | For the | For the | For the | For the | ||||||||||||||||||||||
| Inception) | Months | Months | Year | Year | Year | Year | ||||||||||||||||||||||
| through | Ended | Ended | Ended | Ended | Ended | Ended | ||||||||||||||||||||||
| March 31, | March 31, | March 31, | December 31, | December 31, | December 31, | December 31, | ||||||||||||||||||||||
| 2008 | 2008 | 2007 | 2007 | 2006 | 2005 | 2004 | ||||||||||||||||||||||
| (Successor) | (Successor) | (Successor) | (Successor) | (Predecessor) | (Predecessor) | (Predecessor) | ||||||||||||||||||||||
Statement Expenses Data: | ||||||||||||||||||||||||||||
| Expenses: | ||||||||||||||||||||||||||||
| Research and development | $ | 16,376 | $ | 1,088 | $ | 824 | $ | 6,747 | $ | 1,772 | $ | 2,080 | $ | 2,814 | ||||||||||||||
| General and administrative | 7,816 | 1,625 | 588 | 4,666 | 633 | 129 | 458 | |||||||||||||||||||||
| Operating loss | (24,192 | ) | (2,713 | ) | (1,412 | ) | (11,413 | ) | (2,405 | ) | (2,209 | ) | (3,272 | ) | ||||||||||||||
| Interest income | 523 | 75 | — | 448 | — | — | — | |||||||||||||||||||||
| Other expense | (8 | ) | (8 | ) | — | — | — | — | — | |||||||||||||||||||
| Loss before income taxes | (23,677 | ) | (2,646 | ) | (1,412 | ) | (10,965 | ) | (2,405 | ) | (2,209 | ) | (3,272 | ) | ||||||||||||||
| Income taxes | — | — | — | (25 | ) | — | — | — | ||||||||||||||||||||
| Net loss | $ | (23,677 | ) | $ | (2,646 | ) | $ | (1,412 | ) | $ | (10,990 | ) | $ | (2,405 | ) | $ | (2,209 | ) | $ | (3,272 | ) | |||||||
| Basic and diluted net loss per share | N/A | $ | (0.21 | ) | $ | (0.17 | ) | $ | (0.99 | ) | N/A | N/A | N/A | |||||||||||||||
| Weighted average shares outstanding, basic and diluted | N/A | 12,684,432 | 8,117,016 | 11,113,137 | N/A | N/A | N/A | |||||||||||||||||||||
Balance Sheet Data: | ||||||||||||||||||||||||||||
| As of | As of | As of | As of | As of | As of | |||||||||||||||||||
| March 31, | December 31, | December 31, | December 31, | December 31, | December 31, | |||||||||||||||||||
| 2008 | 2007 | 2006 | 2006 | 2005 | 2004 | |||||||||||||||||||
| (Successor) | (Successor) | (Successor) | (Predecessor) | (Predecessor) | (Predecessor) | |||||||||||||||||||
| Cash and cash equivalents | $ | 9,857 | $ | 1,763 | $ | 2 | $ | — | $ | — | $ | — | ||||||||||||
| Short term investments | — | 9,952 | — | — | — | — | ||||||||||||||||||
| Total current assets | 10,179 | 11,737 | 2 | — | — | — | ||||||||||||||||||
| Working capital | 8,405 | 10,413 | 2 | (318 | ) | (500 | ) | (968 | ) | |||||||||||||||
| Total assets | 10,580 | 12,147 | 2 | 57 | 50 | — | ||||||||||||||||||
| Due to parent | (67 | ) | (207 | ) | — | — | — | — | ||||||||||||||||
| Total stockholders’ equity | 8,806 | 10,823 | 2 | — | — | — | ||||||||||||||||||
| Parent company’s net deficit | — | — | — | (268 | ) | (450 | ) | (968 | ) | |||||||||||||||
25
| (1) | Each $0.10 increase (decrease) in the assumed public offering price per share and accompanying common warrant would increase (decrease) the amount of cash and cash equivalents, working capital, total assets, and total stockholders’ equity by approximately $4.4 million, assuming the number of securities offered by us, as set forth on the cover page of this prospectus, remains the same, no pre-funded warrants are issued, and after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
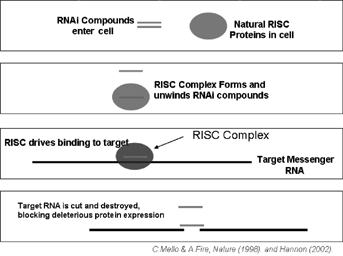
 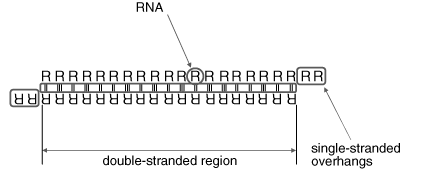
Except as
If you invest in our common stock in this offering, your ownership interest will be diluted immediately to the extent of the difference between the public offering price per share of our common stock and the as adjusted net tangible book value per share of our common stock after this offering. Our historical net tangible book deficit as of March 31, 2019 was $7.5 million, or $(0.31) per share of our common stock. Historical net tangible book deficit per share represents the amount of our total tangible assets less total liabilities, divided by Our pro forma net tangible book deficit as of March 31, 2019 was $6.4 million, or $(0.25) per share of our common stock. Pro forma net tangible book deficit per share represents total tangible assets less total liabilities, divided by the number of shares of our common stock outstanding as of March 31, 2019, after giving effect to After giving effect to the issuance and sale of 47,619,048 shares of our common stock in this offering at an assumed public offering price of $0.42 per share and accompanying common warrant, which is the last reported sale price of our common stock on Nasdaq on June 11, 2019, and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us, and assuming no issuance of pre-funded warrants,
Each $0.10 increase (decrease) in the assumed public offering price of $0.42 per share and accompanying common warrant, which is the last reported sale price of our common stock on Nasdaq on June 11, 2019, would increase (decrease) our as adjusted net tangible book value per share after this offering by approximately $0.06 and the dilution per share to new investors purchasing shares in this offering by $0.04 assuming the number of securities offered by us, as set forth on the cover page of this prospectus, remains the same, no pre-funded warrants are issued, and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. We may also increase or decrease the number of securities to be issued in this offering. Each increase (decrease) of 1.0 million shares and accompanying common warrants offered by us would increase (decrease) our as adjusted net tangible book value per share and the dilution per share and accompanying common warrant to new investors purchasing securities in this offering by $0.004 and ($0.002), respectively assuming that the assumed public offering price remains the same, no pre-funded warrants are Except as otherwise indicated
DESCRIPTION OF CAPITAL STOCK The following description of our capital stock General Our amended and restated certificate of incorporation authorizes us to issue up to 350,000,000 shares of common stock, $0.0001 par value As of
Common Stock Voting Each holder of our common stock Dividends Subject to preferences that may be applicable to any Liquidation In the event of our dissolution or liquidation, Rights and Preferences Holders of common stock Fully-paid All the outstanding shares of our common stock Our common stock is listed on The Nasdaq Capital Market under the symbol “SLS.” Preferred Stock Our board of directors has the authority, without further action by the stockholders, to issue up to 5,000,000 shares of preferred stock
Our board of directors Warrants Our outstanding warrants contain customary net exercise provisions and contain provisions for the adjustment of the exercise price and the number of shares issuable upon the exercise of the warrant in the event of certain stock dividends, stock splits, recapitalizations, reclassifications, consolidations and other fundamental transactions. In addition, from the original issue date until July 16, 2019 the initial exercise price and number of shares subject to purchase in the warrants issued in connection with our Series A Preferred are accompanying common warrant. Possible Anti-Takeover Effects of Provisions of Classified Board Our amended and restated certificate of incorporation and our amended and restated bylaws provide that our board of directors is divided into three classes. The directors designated as Class I directors have terms expiring at the annual meeting of stockholders in 2020. The directors designated as Class II directors will
our company.
Removal of Directors Our amended and Amendment Our amended and restated certificate of incorporation and Size of Board and Vacancies Our amended and restated bylaws provide that the number of directors on our board of directors is fixed exclusively by our board of directors. Newly created directorships resulting from any increase in our authorized number of directors will be filled by a majority of our board of directors then in office, provided that a majority of the entire board of directors, or a quorum, is present and any vacancies in our board of directors resulting from death, resignation, retirement, disqualification, removal from office or other cause will be filled generally by the majority vote of our remaining directors in office, even if less than a quorum is present.
Special Stockholder Meetings Our amended and restated certificate of incorporation provides that only the Chairman of our board of directors, our Chief Executive Officer or our board of directors pursuant to a resolution adopted by a majority of the total number of directors we would have if there were no vacancies may call special meetings of our stockholders. Stockholder Action by Unanimous Written Consent Our amended and restated certificate of incorporation expressly eliminates the right of our stockholders to act by written consent. Requirements for Advance Notification of Stockholder Nominations and Proposals Our amended and restated bylaws provide advance notice procedures with respect to stockholder proposals and nomination of candidates for election as directors other than nominations made by or at the direction of board of directors or a committee of our board of directors. No Cumulative Voting The DGCL provides that stockholders are denied the right to cumulate votes in the election of directors unless our certificate of incorporation provides otherwise. Our amended and restated certificate of incorporation does not provide for cumulative voting. Undesignated Preferred Stock The authority that is possessed by our board of directors to issue preferred stock could potentially be Authorized but Unissued Shares Our authorized but unissued shares of common stock and The above provisions may deter a hostile takeover or delay a change in control or management of our company. Transfer Agent and Registrar The transfer agent and registrar for our capital stock is
DESCRIPTION OF SECURITIES WE ARE OFFERING We are offering (i) 47,619,048 shares of our common stock or pre-funded warrants and (ii) common warrants to purchase up to 47,619,048 shares of our common stock. Each share of common stock and pre-funded warrant is being sold together with a common warrant to purchase one (1) share of common stock. The shares of common stock, pre-funded warrants and accompanying common warrants will be issued separately. We are also registering the Common Stock The material terms and provisions of our Pre-Funded Warrants The following summary of certain terms and provisions of pre-funded warrants that are being offered hereby is not Duration and Exercise Price Each pre-funded warrant offered hereby will have an initial exercise price per share equal to $0.01. The pre-funded warrants will be immediately exercisable and may be Exercisability The pre-funded warrants will be exercisable, at the option of each holder, in whole or in part, by delivering to us a Cashless Exercise In lieu of making the cash payment otherwise contemplated to be made to us upon such exercise in payment of the aggregate exercise price, the holder may elect instead to receive upon such exercise (either in whole or Fundamental Transaction In the event of a fundamental transaction, as described in the pre-funded warrants and generally including any reorganization, recapitalization or reclassification of our common stock, the sale, transfer or other disposition of all or substantially all of our properties or assets, our consolidation or merger with or into another person, the acquisition of more than 50% of our outstanding common stock, or any person or group becoming the beneficial owner of 50% of the Transferability Subject to applicable laws, a pre-funded warrant may be transferred at the option of the holder upon surrender of the pre-funded warrant to us together with the appropriate instruments of transfer. Exchange Listing We do not intend to list the pre-funded warrants on any securities exchange or nationally recognized trading system. Rights as a Stockholder Except as otherwise provided in the pre-funded warrants or by Common Warrants The following summary of certain terms and provisions of the Duration and Exercise Price Each common warrant offered hereby will have an initial exercise price per share equal to $ per share (representing 110% of the public offering price). The common warrants will be immediately exercisable and will expire on the fifth anniversary of the original issuance date. The exercise price and number of shares of common stock issuable upon exercise is subject to appropriate adjustment in the event of stock dividends, stock splits, reorganizations or similar events affecting our common stock and the exercise price. See also “Antidilution” below. The common warrants will be issued separately from the common stock and pre-funded warrants, and may be transferred separately immediately thereafter. A common warrant to purchase one share of our common stock will be issued for every one share of common stock (or pre-funded warrant, as applicable) purchased in this offering. Exercisability The common warrants will be exercisable, at the option of each holder, in whole or in part, by delivering a duly executed exercise notice accompanied by payment in full for the number of shares of our common stock purchased upon such exercise (except in the case of a cashless exercise as discussed below). A holder (together with its affiliates) may not exercise any portion of the common warrant to the extent that the holder would own more than [4.99][9.99]% of the outstanding common stock immediately after exercise, except that upon at least 61 days’ prior notice from the holder to us, the holder may increase the amount of ownership of outstanding stock after exercising the holder’s warrants. No fractional shares of common stock will be issued in connection with the exercise of a common warrant. In lieu of fractional shares, we will pay the holder an amount in cash equal to the fractional amount multiplied by the exercise price. Cashless Exercise If, at the time a holder exercises its common warrants, a registration statement registering the issuance of the shares of common stock underlying the common warrants under the Securities Act is not then effective or available, then in lieu of making the cash payment otherwise contemplated to be made to us upon such exercise in payment of the aggregate exercise price, the holder may elect instead to receive upon such exercise (either in whole or in part) the net number of shares of common stock determined according to a formula set forth in the common warrants.In addition, the common warrants also provide that, beginning 45 days after issuance, each warrant may be exercised at the option of the holder on a cashless basis, in whole or in part for a whole number of shares, for three (3) shares of common stock, if during the period of time between the date that is 45 days after issuance and the date that is 15 months after issuance, the five-day volume weighted average price of our common stock during any five (5) consecutive trading days is lower than the then-applicable exercise price per share. Anti-Dilution In addition to the adjustments noted above under “Duration and Exercise Price,” common warrants also contain anti-dilution protection upon the issuance of any Fundamental Transaction In the event of a fundamental transaction, as described in the common warrants and generally including any reorganization, recapitalization or reclassification of our common stock, the sale, transfer or other disposition of all or substantially all of our properties or assets, our consolidation or merger with or into another person, the acquisition of more than Transferability Subject to applicable laws, a common warrant may be transferred at the option of the holder upon surrender of the common warrant together with the appropriate instruments of transfer. Exchange Listing We do not intend to list the common warrants on any securities exchange or nationally recognized trading system. Right as a Stockholder Except as otherwise provided in the common warrants or by virtue of such holder’s ownership of shares of
MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES TO HOLDERS OF COMMON STOCK AND WARRANTS The following is a summary of the material U.S. federal income tax consequences of the acquisition, ownership and disposition of our common stock and the pre-funded warrants, and the acquisition, ownership, exercise, expiration or disposition of the common warrants, but does not purport to This summary also does not address the tax considerations arising under the laws of
In addition, if a partnership, including any entity or arrangement classified as a You are urged to consult your tax advisor with respect to the application of the U.S. federal income tax laws to your particular situation, as well as any tax consequences of the purchase, ownership and disposition of our common stock or pre-funded warrants or common warrants arising under the U.S. federal estate or gift tax rules or under the laws of any U.S. state or local or any non-U.S. or other taxing jurisdiction or under any applicable tax treaty. Definition of a U.S. Holder For purposes of this summary, a “U.S. Holder” is any beneficial owner of our common stock or pre-funded warrants or common warrants that is a “U.S. person,” and is not a partnership, or an entity treated as a partnership or disregarded from its owner, each for U.S. federal income tax purposes. A U.S. person is any person that, for U.S. federal income tax purposes, is or is treated as any of the following:
For purposes of this summary, a “Non-U.S. Holder” is any beneficial owner of our common stock or pre-funded warrants or common warrants that is not a U.S. Holder or a partnership, or other entity treated as a partnership or disregarded from its owner, each for U.S. federal income tax purposes. Treatment of Pre-funded Warrants Although it is not entirely free from doubt, a pre-funded warrant should be treated as a share of our common stock for U.S. federal income tax purposes and a holder of pre-funded warrants should generally be taxed in the same manner as a holder of common stock, as described below. Accordingly, no gain or loss should be recognized upon the exercise of a pre-funded warrant and, upon exercise, the holding period of a pre-funded warrant should carry over to the share of common stock received. Similarly, the tax basis of the pre-funded warrant should carry over to the share of common stock received upon exercise, increased by the exercise price of $0.01. Each holder should consult his, her or its own tax advisor regarding the risks associated with the acquisition of pre-funded warrants pursuant to this offering (including potential alternative characterizations). The balance of this discussion generally assumes that the characterization described above is respected for U.S. federal income tax purposes. Tax Consequences to U.S. Holders Distributions on Common Stock As discussed above under “Dividend Information – Dividend Policy,” we do not currently expect to make distributions on our common stock. In the event that we do make distributions of cash or other property, distributions paid on common stock, other than certain pro rata distributions of common stock, will be treated as a dividend to the extent paid out of our current or accumulated earnings and profits and will be includible in income by the U.S. Holder and taxable as ordinary income when received. If a distribution exceeds our current and accumulated earnings and profits, the excess will be first treated as a tax-free return of the U.S. Holder’s investment, up to the U.S. Holder’s tax basis in the common stock. Any remaining excess will be treated as a capital gain. Subject to applicable limitations, dividends paid to certain non-corporate U.S. Holders may be eligible for taxation as “qualified dividend income” and therefore may be taxable at rates applicable to long-term capital gains. U.S. Holders should consult their tax advisers regarding the availability of the reduced tax rate on dividends in their particular circumstances. Dividends received by a corporate U.S. Holder will be eligible for the dividends-received deduction if the U.S. Holder meets certain holding period and other applicable requirements. Constructive Dividends on Common Warrants Under Section 305 of the Code, an adjustment to the number of shares of common stock that will be issued on the exercise of the common warrants, or an adjustment to the exercise price of the common warrants, may be treated as a constructive distribution to a U.S. Holder of the common warrants if, and to the extent that, such adjustment has the effect of increasing such U.S. Holder’s proportionate interest in our “earnings and profits” or assets, depending on the circumstances of such adjustment (for example, if such adjustment is to compensate for a distribution of cash or other property to our stockholders). Adjustments to the exercise price of a common warrant made pursuant to a bona fide reasonable adjustment formula that has the effect of preventing dilution of the interest of the holders of the warrants should generally not result in a constructive distribution. Any constructive distributions would generally be subject to the tax treatment described above under “Dividends on Common Stock”. Sale or Other Disposition of Common Stock For U.S. federal income tax purposes, gain or loss realized on the sale or other disposition of common stock will be capital gain or loss, and will be long-term capital gain or loss if the U.S. Holder held the common stock for more than one year. The amount of the gain or loss will equal the difference between the U.S. Holder’s tax basis in the common stock disposed of and the amount realized on the disposition. Long-term capital gains recognized by non-corporate U.S. Holders will be subject to reduced tax rates. The deductibility of capital losses is subject to limitations. Sale or Other Disposition, Exercise or Expiration of Common Warrants For U.S. federal income tax purposes, gain or loss realized on the sale or other disposition of a common warrant (other than by exercise) will be capital gain or loss and will be long-term capital gain or loss if the U.S. Holder held the warrant for more than one year at the time of the sale or other disposition. The amount of the gain or loss will equal the difference between the U.S. Holder’s tax basis in the common warrant disposed of and the amount realized on the disposition. In general, a U.S. Holder will not be required to recognize income, gain or loss upon the exercise of a common warrant by payment of the exercise price, except to the extent of cash paid in lieu of a fractional share. A U.S. Holder’s tax basis in a share of common stock received upon exercise will be equal to the sum of (1) the U.S. Holder’s tax basis in the common warrant and (2) the exercise price of the common warrant. A U.S. Holder’s holding period in the stock received upon exercise will commence on the day or the day after such U.S. Holder exercises the common warrant. No discussion is provided herein regarding the U.S. federal income tax treatment on the exercise of a common warrant on a cashless basis, and U.S. Holders are urged to consult their tax advisors as to the exercise of a common warrant on a cashless basis. If a common warrant expires without being exercised, a U.S. Holder will recognize a capital loss in an amount equal to such U.S. Holder’s tax basis in the common warrant. This loss will be long-term capital loss if, at the time of the expiration, the U.S. Holder’s holding period in the common warrant is more than one year. The deductibility of capital losses is subject to limitations. Tax Consequences to Non-U.S. Holders Distributions As discussed in the section entitled “Dividend Information—Dividend Policy,” we do not anticipate paying any dividends on our common stock in the foreseeable future. If we make distributions on our common stock or on our common warrants (as described above under “Constructive Dividends on Common Warrants”), those payments will constitute dividends for U.S. federal income tax purposes to the extent we have current or accumulated earnings and profits, as determined under U.S. federal income tax principles. To the extent those distributions exceed both our current and our accumulated earnings and profits, they will constitute a return of capital and will first reduce a Non-U.S. Holder’s basis in our common stock or common warrants, as applicable, but not below zero. Any excess will be treated as capital gain and will be treated as described below under the “—Gain on Sale or Other Disposition of Common Stock or Common Warrants” section. Any such distributions would be subject to the discussions below regarding back-up withholding and Foreign Account Tax Compliance Act, or FATCA. Subject to the discussion below on effectively connected income, any dividend paid to a Non-U.S. Holder generally will be subject to U.S. withholding tax either at a rate of 30% of the gross amount of the dividend or such lower rate as may be specified by an applicable income tax treaty. To receive a reduced treaty rate, a Non-U.S. Holder must provide us or our agent with an IRS Form W-8BEN, IRS Form W-8 BEN-E or another appropriate version of IRS Form W-8 (or a successor form), which must be updated periodically, and which, in each case, must certify qualification for the reduced rate. Non-U.S. Holders should consult their tax advisors regarding their entitlement to benefits under any applicable income tax treaty.
Dividends paid to a Non-U.S. Holder that are effectively connected with the Non-U.S. Holder’s conduct of a trade or business within the United States and that are not eligible for relief from U.S. (net basis) income tax under an applicable income tax treaty, generally are exempt from the (gross basis) withholding tax described above. To obtain this exemption from withholding tax, the Non-U.S. Holder must provide the applicable withholding agent with an IRS Form W-8ECI or successor form or other applicable IRS Form W-8 certifying that the dividends are effectively connected with the Non-U.S. Holder’s conduct of a trade or business within the United States. Such effectively connected dividends, if not eligible for relief under a tax treaty, would not be subject to a withholding tax, but would be taxed at the same graduated rates applicable to U.S. persons, net of certain deductions and credits and if, in addition, the Non-U.S. Holder is a corporation, may also be subject to a branch profits tax at a rate of 30% (or such lower rate as may be specified by an applicable income tax treaty). If you are eligible for a reduced rate of withholding tax pursuant to a tax treaty, you may be able to obtain a refund of any excess amounts withheld if you timely file an appropriate claim for refund with the IRS. Exercise or Expiration of Common Warrants In general, a Non-U.S. Holder will not be required to recognize income, gain or loss upon the exercise of a common warrant by payment of the exercise price, except to the extent of cash paid in lieu of a fractional share. However, no discussion is provided herein regarding the U.S. federal income tax treatment on the exercise of a common warrant on a cashless basis, and Non-U.S. Holders are urged to consult their tax advisors as to the exercise of a common warrant on a cashless basis. If a common warrant expires without being exercised, a Non-U.S. Holder that is engaged in a U.S. trade or business to which any income from the common warrant would be effectively connected or who is present in the United States for a period or periods aggregating 183 days or more during the calendar year in which the expiration occurs (and certain other conditions are met) will recognize a capital loss in an amount equal to such Non-U.S. Holder’s tax basis in the common warrant. Gain on Sale or Other Disposition of Common Stock or Common Warrants Subject to the discussion below regarding backup withholding and FATCA, a Non-U.S. Holder generally will not be required to pay U.S. federal income tax on any gain realized upon the sale or other disposition of our common stock or common warrants unless:
Information Reporting and Backup Withholding Information returns may be filed with the IRS in connection with distributions on common stock or constructive dividends on common warrants, and the proceeds of a sale or other disposition of common stock or common warrants. A non-exempt U.S. Holder may be subject to U.S. backup withholding on these payments if it fails to provide its taxpayer identification number to the withholding agent and comply with certification procedures or otherwise establish an exemption from backup withholding. A Non-U.S. Holder may be subject to U.S. information reporting and backup withholding on these payments unless the Non-U.S. Holder complies with certification procedures to establish that it is not a U.S. person (within the meaning of the Code). The certification requirements generally will be satisfied if the Non-U.S. Holder provides the applicable withholding agent with a statement on the applicable IRS Form W-8BEN or IRS Form W-8BEN-E (or suitable substitute or successor form), together with all appropriate attachments, signed under penalties of perjury, stating, among other things, that such Non-U.S. Holder is not a U.S. Person. Applicable Treasury Regulations provide alternative methods for satisfying this requirement. In addition, the amount of distributions on common stock or constructive dividends on common stock paid to a Non-U.S. Holder, and the amount of any U.S. federal tax withheld therefrom, must be reported annually to the IRS and the holder. This information may be made available by the IRS under the provisions of an applicable tax treaty or agreement to the tax authorities of the country in which the Non-U.S. Holder resides.
Payment of the proceeds of the sale or other disposition of common stock or common warrants to or through a non-U.S. office of a U.S. broker or of a non-U.S. broker with certain specified U.S. connections generally will be subject to information reporting requirements, but not backup withholding, unless the Non-U.S. Holder certifies under penalties of perjury that it is not a U.S. person or an exemption otherwise applies. Payments of the proceeds of a sale or other disposition of common stock or common warrants to or through a U.S. office of a broker generally will be subject to information reporting and backup withholding, unless the Non-U.S. Holder certifies under penalties of perjury that it is not a U.S. person or otherwise establishes an exemption. Backup withholding is not an additional tax. The amount of any backup withholding from a payment generally will be allowed as a credit against the holder’s U.S. federal income tax liability and may entitle the holder to a refund, provided that the required information is timely furnished to the IRS. Foreign Account Tax Compliance Act FATCA imposes withholding tax on certain types of payments made to foreign financial institutions and certain other non-U.S. entities. The legislation imposes a 30% withholding tax on dividends on, or, subject to the discussion of certain proposed Treasury Regulations below, gross proceeds from the sale or other disposition of, our common stock or common warrants paid to a “foreign financial institution” or to certain “non-financial foreign entities” (each as defined in the Code), unless (i) the foreign financial institution undertakes certain diligence and reporting obligations, (ii) the non-financial foreign entity either certifies it does not have any “substantial United States owners” (as defined in the Code) or furnishes identifying information regarding each substantial United States owner, or (iii) the foreign financial institution or non-financial foreign entity otherwise qualifies for an exemption from these rules. If the payee is a foreign financial institution and is subject to the diligence and reporting requirements in (i) above, it must enter into an agreement with the U.S. Treasury requiring, among other things, that it undertake to identify accounts held by “specified United States persons” or “United States-owned foreign entities” (each as defined in the Code), annually report certain information about such accounts, and withhold 30% on payments to account holders whose actions prevent it from complying with these reporting and other requirements. If the country in which a payee is resident has entered into an “intergovernmental agreement” with the United States regarding FATCA, that agreement may permit the payee to report to that country rather than to the U.S. Department of the Treasury. The U.S. Treasury recently released proposed Treasury Regulations which, if finalized in their present form, would eliminate the federal withholding tax of 30% applicable to the gross proceeds of a sale or other disposition of our common stock. In its preamble to such proposed Treasury Regulations, the U.S. Treasury stated that taxpayers may generally rely on the proposed regulations until final regulations are issued. Prospective investors should consult their own tax advisors regarding the possible impact of these rules on their investment in our common stock or common warrants, and the possible impact of these rules on the entities through which they hold our common stock or common warrants, including, without limitation, the process and deadlines for meeting the applicable requirements to prevent the imposition of this 30% withholding tax under FATCA. The preceding discussion of U.S. federal tax considerations is for general information only. It is not tax advice. Each prospective investor should consult its tax advisor regarding the particular U.S. federal, state and local and non-U.S. tax consequences of purchasing, holding and disposing of our common stock or common warrants, including the consequences of any proposed change in applicable laws.
We and the underwriters named below have entered into an underwriting agreement with respect to the shares of common stock or pre-funded warrants and accompanying common warrants being offered. A.G.P./Alliance Global Partners, LLC is acting as the sole book-running manager and representative of the underwriters in this offering. In connection with this offering and subject to certain terms and conditions, the underwriters have agreed to purchase, and we have agreed to sell, all of the securities in this offering to the underwriters.
The underwriters have agreed to purchase all the securities offered by us other than those covered by the over-allotment option to purchase additional securities described below, if it purchases any such securities, and the underwriters’ obligations are several, which means that each underwriter is required to
The underwriters are offering the securities, subject to prior sale, when, as and if issued to and accepted by them, subject to approval of legal matters by the representative’s counsel and other conditions specified in the underwriting agreement. The underwriters reserve the right to withdraw, cancel or modify offers to the public and to reject orders in whole or in part. Over-allotment Option We have also granted the underwriters an option, exercisable for up to 45 days from the date of this prospectus, to purchase up to an additional 7,142,857 shares of common stock and/or common warrants, at the public offering price, less underwriting discounts and commissions. The underwriters may exercise the option solely to cover over-allotments. If the over-allotment option is exercised in full, the total public offering price, underwriting compensation (including discounts, but not including any other compensation described hereunder) and proceeds to us before offering expenses will be approximately $ million, $ million and $ million, respectively, and excluding the proceeds, if any, from the exercise of the pre-funded warrants and common warrants offered hereby. Indemnification We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act and liabilities arising from breaches of representations and warranties contained in the underwriting agreement, or to contribute to payments that the underwriters may be required to make in respect of those liabilities. Underwriter Compensation We have agreed to sell the securities to the underwriters at the combined offering price of $ per share of common stock (or per pre-funded warrant, as applicable) and accompanying common warrants, which represents the offering price of such securities set forth on the cover page of this prospectus, less the applicable 7% underwriting discount. We have also agreed to pay a non-accountable expense allowance to the underwriter equal to $25,000. In addition, we have agreed to reimburse the underwriters for accountable legal expenses incurred by it in connection with this transaction in the amount of $90,000. The total expenses of the underwriter which are subject to payment or reimbursement by the company hereunder shall not exceed $115,000. We estimate that the total expenses of the offering payable by us, excluding the total underwriting discount, will be approximately $480,000. Discount, Commissions and Expenses The underwriters have advised us that they propose to offer the shares of common stock and accompanying warrants at the public offering price set forth on the cover page of this prospectus and to certain dealers at that price less a concession not in excess of $ per share of common stock and accompanying warrant. After this offering, the public offering price and concession to dealers may be changed by the representative. No such change shall change the amount of proceeds to be received by us as set forth on the cover page of this prospectus. The shares of common stock and accompanying warrants are offered by the underwriters as stated herein, subject to receipt and acceptance by them and subject to their right to reject any order in whole or in part. The underwriters have informed us that they do not intended to confirm sales to any accounts over which they exercise discretionary authority.
The following table summarizes the underwriting discount we will pay to the underwriters. These amounts are shown assuming both no exercise and full exercise of the over-allotment option.
(1) Excluding the proceeds, if any, from the exercise of the pre-funded warrants and common warrants. Lock-Up Agreements and Trading Restrictions Our executive officers and directors have agreed to a 90-day “lock-up” from the effective date of this prospectus of shares of common stock that they beneficially own, including the issuance of common stock upon the exercise of currently outstanding convertible securities and options and options which may be issued. This means that, for a period of 90 days following the effective date of this prospectus, such persons may not offer, sell, pledge or otherwise dispose of these securities without the prior written consent of the representative. The representative has no present intention to waive or shorten the lock-up period; however, the terms of the lock-up agreements may be waived at its discretion. In determining whether to waive the terms of the lockup agreements, the representative may base its decision on its assessment of the relative strengths of the securities markets and companies similar to ours in general, and the trading pattern of, and demand for, our securities in general. In addition, the underwriting agreement provides that we will not, for a period of 90 days following the effective date of this prospectus, offer, sell or distribute any of our securities, without the prior written consent of the representative, subject to Beginning on the closing date of Stabilization The rules of the SEC generally prohibit the underwriters from trading in our securities on the open market during this offering. However, the underwriters are allowed to engage in
If the underwriters commence these activities, they may
Listing Our common stock is listed on
Electronic Distribution A prospectus in electronic format may be made available on websites or through other online services maintained by the underwriters of this offering, or by their affiliates. Other than the prospectus in electronic format, the information on any underwriters’ website and any information contained in any other website maintained by an underwriter is not part of this prospectus or the registration statement of which this prospectus forms a part, has not been approved and/or endorsed by us or the representative in its capacity as an underwriter. Other Relationships The representative and its affiliates have Except for services provided in connection with this offering, and except as set forth in this section, the representative has not provided any investment banking or other financial services during the 180-day period preceding Notice to In relation to each Member State of the
For the purposes of
The representative has represented, warranted and agreed that:
European Economic Area In particular, this document does not constitute an approved prospectus in accordance with
For the purposes of this provision, the expression an “offer of securities to the public” in relation to any of the securities in any Relevant Member State means the communication in any form and by any means of sufficient information on
LEGAL MATTERS The validity of the EXPERTS Our consolidated financial statements WHERE YOU CAN FIND MORE INFORMATION We are a reporting company and file annual, quarterly and current reports, proxy statements and other information with the SEC. We have filed with the SEC a registration statement onForm S-1 under the Securities Act with respect to the www.sec.gov. We maintain a website at
INCORPORATION OF DOCUMENTS BY REFERENCE The SEC allows us to
We also incorporate by reference any future filings (other than current reports furnished under Item 2.02 or Item 7.01 of Form 8-K and exhibits filed on such form that are related to such items unless such Form 8-K expressly provides to the contrary) made We will furnish without charge to each person, including any beneficial owner, to whom a prospectus is delivered, upon written or oral request, a copy of any or all of 10th Floor, New York, New York 10018. Our phone number is (917) 438-4353. You should rely only on
47,619,048 Shares of Common Stock Pre-Funded Warrants to Purchase Shares of Common Stock Common Warrants to Purchase up to 47,619,048 Shares of Common Stock PROSPECTUS Sole Book-Running Manager A.G.P. Co-Manager Maxim Group, LLC , 2019
PART II
The following table sets forth
As permitted by Section
These limitations of liability do not affect the As permitted by Section 145 of
Our amended and restated certificate of incorporation and bylaws, each as amended, which are filed as Exhibits 3.1 and 3.3, provide for the indemnification provisions described above and elsewhere herein. We have entered into separate indemnification agreements with our directors and officers that may be broader than the specific indemnification provisions contained in II-1 We have entered into indemnification agreements with our directors and executive officers, in addition to the indemnification provided for in our amended and restated certificate of incorporation and amended and restated bylaws, and intend to enter into indemnification agreements with any new directors and executive officers in the future. We have purchased and currently intend to maintain insurance on behalf of each and every person who is or was a director or officer of our company against any loss arising from any claim asserted against him or her and incurred by him or her in any such capacity, subject to certain exclusions.
Since March 31, 2016, the registrant has issued unregistered securities to the persons, as described below. None of these transactions involved any underwriters, underwriting discounts or commissions, except as specified below, or any
II-2
II-3 II-4 II-5 II-6
II-7
The undersigned registrant hereby undertakes:
provided, however, that paragraphs (a)(1)(i), (ii), and (iii) of this section do not
The undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
The undersigned registrant hereby II-8 Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers, and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the The undersigned registrant hereby undertakes that:
II-9
SIGNATURES Pursuant to the requirements of the Securities Act of 1933, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized in the
Pursuant to the requirements of the Securities Act of 1933, as amended, this registration statement on Form S-1 has been signed
II-10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
