
As filed with the U.S. Securities and Exchange Commission on August 25, 2022April 5, 2024
Registration No. 333-265429
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
AMENDMENT NO. 5
TO
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
SHUTTLE PHARMACEUTICALS HOLDINGS, INC.Shuttle Pharmaceuticals Holdings, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 2834 | 82-5089826 | ||
(State or other jurisdiction of | (Primary Standard Industrial Classification Code Number) | (I.R.S. Employer Identification |
One Research Court,401 Professional Drive, Suite 450260
Rockville, Maryland 20850Gaithersburg, MD 20879
Telephone: (240) 403-4212
(Address, including zip code, and telephone number, including
area code, of registrant’s principal executive office)offices)
Anatoly Dritschilo, M.D.
Chief Executive Officer
Shuttle Pharmaceuticals Holdings, Inc.
One Research Court, Suite 450
Rockville, Maryland 20850401 Professional Drive, Suite 260
Gaithersburg, MD 20879
(240) 403-4212
(Name, address, including zip code, and telephone number,
including area code, of agent for service)
Copies to:to:
Megan J. Penick
Dorsey & Whitney LLP
51 West 52nd Street
New York, New York 10019
(212) 415-9200
|
| |
|
| |
| ||
|
| |
|
| |
Approximate date of commencement of proposed sale to the public:public: As soon as practicable after the effective date of this Registration Statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box.box: ☐
If this Formform is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Formform is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Formform is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large“large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging“emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| Non-accelerated filer | ☒ | Smaller reporting company | ☒ |
| Emerging | ☒ |
If an emerging growth company indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ☐
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Sectionsection 8(a) of the Securities Act of 1933 as amended, or until the registration statement shall become effective on such date as the Securities and Exchange Commission acting pursuant to said Sectionsection 8(a), may determine.
EXPLANATORY NOTE
This Registration Statement contains two prospectuses, as set forth below.
The Resale Prospectus is substantively identical to the Public Offering Prospectus, except for the following principal points:
|
| |
The Registrant has included in this Registration Statement a set of alternate pages after the back cover page of the Public Offering Prospectus (the “Alternate Pages”) to reflect the foregoing differences in the Resale Prospectus as compared to the Public Offering Prospectus. The Public Offering Prospectus will exclude the Alternate Pages and will be used for the public offering by the Registrant. The Resale Prospectus will be substantively identical to the Public Offering Prospectus except for the addition or substitution of the Alternate Pages and will be used for the resale offering by the selling stockholder.
(1) Assumes the underwriter’s over-allotment option has not been exercised.
The information in this preliminary prospectus is not complete and may be changed. We may not sell these securities nor may we accept offers to buy these securities until the registration statement filed with the Securities and Exchange Commission becomes effective. This preliminary prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
| PRELIMINARY PROSPECTUS | SUBJECT TO COMPLETION, DATED |
$9,960,340

SHUTTLE PHARMACEUTICALS HOLDINGS, INC.
Subscription Rights to Purchase Up to $4,500,000 in Units
1,225,888Units
Each Unit Consisting of One ShareUp to Shares of Common Stock, and
One WarrantWarrants to Purchase One ShareUp to Shares of Common Stockstock, and Up to 44% Interest, in total, in Shuttle Diagnostics, Inc.
This is an initial public offering of Shuttle Pharmaceuticals Holdings, Inc. We are offering a total of 1,225,888 units of securities, with each unit consisting of one share(the “Company,” “we,” “us” or “our”) is distributing to the holders (collectively, the “Holders”) of our common stock, par value $0.00001 per share (“common stock”(the “Common Stock”), and anon-transferable rights (the “Rights”) to purchase up to an aggregate of $4,500,000 in units, with each unit consisting of one share of common stock, one warrant to purchase one share of our common stock. The units are being offeredstock, exercisable at an assumed combined public offering price of $8.125 per unit and a combined effective offering price of $8.115$2.35 per share, of common stock and an offering priceinterest in our wholly-owned subsidiary, Shuttle Diagnostics, Inc., such that at the conclusion of $0.01 for each warrant. The holdersthis Rights Offering, the participants in the Rights Offering will hold 44% of the warrants may purchase our common stock atequity interests in Diagnostics and the Company will own 56% of the equity interests in Diagnostics, assuming the Rights Offering is fully subscribed. You will not be entitled to receive any Rights unless you are a Holder of record as of 5:00 p.m., New York City time, on [ ], 2024 (the “Record Date”). Holders, as of the Record Date, will receive one Right for every share of Common Stock owned. For purposes of this preliminary prospectus, we have assumed an exerciseestimated per Unit Price of $[ ] which is equal to 90% of the closing price of $0.01 per share. The shares included in the units are immediately separable, as a result of which we will be issuing an aggregate of 1,225,888 shares of our common stock on [ ], 2024 (the “Estimated Per Unit Purchase Price”). As noted elsewhere in this prospectus, the Estimated Per Unit Purchase Price is subject to change when calculated on the expiration date of this offering.
The Rights will expire if they are not exercised by 5:00 p.m., New York City time, on [ ], 2024, the expected expiration date of this Rights Offering. We, in our sole discretion, may extend the period for exercising the Rights. Rights which are not exercised by the expiration date of the Rights Offering will expire and warrantswill have no value. You should carefully consider whether or not to exercise your Rights before the expiration date. Once you have exercised your Rights, your exercise may not be revoked.
Rights may only be exercised in whole numbers of shares of Common Stock, and we will not issue fractional shares. Each subscription right will entitle you to purchase 1,225,888 sharesone Unit, which we refer to as the Basic Subscription Right, at a subscription price per Unit of our common stock. It is$[ ], which we refer to as the intentionSubscription Price. If you exercise your Basic Subscription Rights in full, and any portion of the underwritersUnits remain available under the Rights Offering, you will be entitled to immediatelyan over-subscription privilege to purchase a portion of the unsubscribed Units at the Subscription Price, subject to proration and ownership limitations, which we refer to as the Over-Subscription Privilege. Each subscription right consists of a Basic Subscription Right and an Over-Subscription Privilege, which we refer to as the Subscription Right.
SRO, LLC, an entity controlled by Keith Moore, Chairman of Boustead Securities, LLC (“BSL”), our placement agent, has entered into an agreement pursuant to which it is obligated to purchase $2,250,000 Units (the “Committed Investment”) in the Rights Offering and has the ability to act as a backstop purchaser should we be unable to raise the full $4,500,000 under the Rights Offerings, including the Committed Investment. However, there can be no assurance that they will participate up to the full $4,500,000, should we be unable to raise the full amount. Aside from the aforementioned Commitment Investment, the Rights Offering is being conducted on a “best efforts” basis.
If we fail to consummate this rights offering or obtain funds from other sources during this fiscal year, we may not be able to continue as a going concern and will need to explore restructuring and reorganization initiatives. See “Risk Factors - Risks Related to this Rights Offering - If the rights offering is not consummated or we are not able to obtain alternative financing during the fiscal year ending December 31, 2024, we will not have funds to meet our working capital requirements” and “Risk Factors - Risks Related to this Rights Offering - Even if the rights offering is completed, we will require additional capital to fund our operations, and if we fail to obtain financing when needed or on acceptable terms, we will not be able to meet our working capital requirements or fund business operations.”
You should carefully consider whether to exercise your subscription rights prior to the warrantsexpiration of the rights offering. All exercises of subscription rights are irrevocable, even if the rights offering is extended by our board of directors for additional periods (not to exceed 45 days).
All subscription payments will be deposited into an account maintained by the subscription agent for the benefit of the holders exercising their subscriptions under this rights offering, and if this rights offering is not completed for any reason all funds will be promptly returned to such subscribers in the amounts advanced in connection with their respective exercises. If we amend the rights offering to allow for an extension of the rights offering for additional periods aggregating to more than 45 days, or make a fundamental change to the terms of the rights offering set forth in this prospectus, you may cancel your subscription and receive a prompt refund of any money you have advanced. We may terminate the Rights Offering at any time prior to the expiration of the Rights Offering for any reason. In the event the Rights Offering is terminated, all subscription payments received will be returned within 10 business days, without interest or deduction. We expect the Rights Offering to expire on behalf[ ], 2024.
Rights under the Basic Subscription Right will be distributed in proportion to Holders’ holdings on the Record Date. If you exercise your Basic Subscription Right in full, and other Holders do not, you will be entitled to an Over-Subscription Right to purchase a portion of their clients simultaneously with the consummationunsubscribed shares at the subscription price, subject to the availability and pro rata allocation of Common Stock among persons exercising this Over-Subscription Right. See “Questions & Answers - What are the limitations of the Over-Subscription Right?”
Exercising the Rights and investing in our Common Stock involve significant risks. We urge you to read carefully the section titled “Risk Factors” beginning on page 21 of this public offering. In addition,prospectus, the selling shareholders (as defined herein) are offering 1,311,942 shares of common stocksection titled “Risk Factors” in our Annual Report on Form 10-K for the year ended December 31, 2023, and all other information included or incorporated by reference in this prospectus in its entirety before you decide whether to be sold pursuant to a separate resale prospectus. We will not receive any proceeds from the sale of the common stock to be sold by the selling stockholders.exercise your Rights.
Prior to this offering, no public market has existed for our common stock. No stockholder affiliated with managementOur Common Stock is selling shares in this offering. We have applied to list our common stock included in the units and our common stock issuable upon exercise of the warrants for tradinglisted on the Nasdaq Capital Market tier of The Nasdaq Stock Market LLC (“Nasdaq”) under the symbol “SHPH”. We do“SHPH.” On [ ], 2024, the last reported sale price of our Common Stock was $[ ]. The Rights being offered herein are non-transferrable, except that Rights will be transferable by operation of law (e.g., by death) or by such Holders that are closed-end funds to funds affiliated with such Holders. The Rights will not intend to applybe listed for listing of the warrantstrading on Nasdaq or on any other national securitiesstock exchange or trading system. Without an active trading market, the liquidity of the warrants will be limited.market. You are urged to obtain a current price quote for our Common Stock before exercising your Rights.
An investmentVstock Transfer, LLC will serve as the subscription agent (“Subscription Agent”), the information agent (“Information Agent”) and the escrow agent (the “Escrow Agent”) for this Rights Offering. The Subscription Agent will hold the funds we receive from subscribers until we complete, abandon or terminate the Rights Offering. If you want to participate in our common stock involves significant risks.this Rights Offering and you are the record holder of your securities, we recommend that you submit your subscription documents to the Subscription Agent well before the deadline. If you want to participate in this Rights Offering and you hold securities through your broker, dealer, bank, or other nominee, you should promptly contact your broker, dealer, bank, or other nominee and submit your subscription documents in accordance with the instructions and within the time period provided by your broker, dealer, bank, or other nominee. For a more detailed discussion, see “The Rights Offering - The Rights” beginning on page 32.
| Per Share | Total(2) | |||||||
| Subscription Price | $ | [ ] | $ | 4,500,000 | ||||
| Placement Agent Fees (1) | $ | [ ] | $ | 360,000 | ||||
| Proceeds to us, before expenses | $ | [ ] | $ | 4,140,000 | ||||
| (1) | In connection with this Rights Offering, we have agreed to pay to Boustead Securities, LLC, as placement agent, an aggregate cash fee equal to 8.0% of the gross proceeds received by us directly from exercises of the Subscription Rights. In addition, we agreed to pay BSL a commitment fee of $112,500, payable in cash or shares, upon the earlier of filing this registration statement or termination of this offering, and we also paid BSL $40,000 to cover its due diligence fees in advance of this Rights Offering. See “Plan of Distribution.” |
| (2) | Assumes the Rights Offering is fully subscribed but excludes proceeds from the exercise of Warrants included in the Units. |
If you have any questions or need further information about this Rights Offering, please contact the Information Agent toll-free at [ ] or via email at [ ]. It is anticipated that delivery of the shares of Common Stock purchased in this Rights Offering will be made on or about [ ], 2024 (the fifth business day following the expiration date), unless the expiration date is extended.
Our board of directors is making no recommendation regarding your exercise of the Subscription Rights. You should carefully consider whether to exercise your Subscription Rights before the risk factors beginning on page 13expiration date. You may not revoke or revise any exercises of this prospectus before you make your decision to invest in our common stock.Subscription Rights once made.
| Per Share | Total | |||||||
| Offering price | $ | 8.125 | $ | 9,960,340 | ||||
| Underwriting discounts and commissions (1) | $ | 0.568 | $ | 697,224 | ||||
| Proceeds, before expenses, to us | $ | 7.56 | $ | 9,263,116 | ||||
We have granted a 45-day option to the underwriter to purchase up to 183,883 additional units solely to cover over-allotments, if any.
We are an “emerging growth company” as defined in Section 2(a) of the Securities Act of 1933, as amended, and we have elected to comply with certain reduced public company reporting requirements.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The underwriter expects to deliver the shares of common stock against payment as set forth under “Underwriting” on or about , 2022.Placement Agent
Boustead Securities, LLC
The date of this prospectus is ____________, 2022.
TABLE OF CONTENTS
We have not, and the underwriter has not, authorized anyone to provide you with any information or to make any representation other than that contained in this prospectus, any amendment or supplement to this prospectus or in any free writing prospectus prepared we may authorize to be delivered or made available to you. We do not, and the underwriter does not, take any responsibility for, and can provide no assurance as to the reliability of, any information that others may provide to you. We are offering to sell, and seeking offers to buy, shares of our common stock only in jurisdictions where offers and sales are permitted. The information in this prospectus is accurate only as of the date on the front cover of this prospectus, regardless of the time of delivery of this prospectus or any sale of shares of our common stock.any units hereunder. Our business, financial condition, results of operations and prospects may have changed since that date. You should also read and consider the information in the documents to which we have referred you under the caption “Where You Can Find More Information” in this prospectus.
For investors outside the United States: Neither we nor the underwriterWe have not done anything that would permit a public offering of the securities or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of the securities and the distribution of this prospectus outside of the United States. You are required to inform yourselves about and to observe any restrictions relating to this offering and the distribution of this prospectus outside of the United States.
You should rely only on the information contained in or incorporated by reference in this prospectus. Neither we nor the underwriterprospectus and in any free writing prospectus prepared by or on behalf of us. We have not authorized any dealer, salesperson or other personanyone to provide you with information concerning us, except fordifferent from, or in addition to, that contained in or incorporated by reference in this prospectus or any related free writing prospectus. This prospectus is an offer to sell only the securities offered hereby and only under circumstances and in jurisdictions where it is lawful to do so. The information contained in or incorporated by reference in this prospectus.prospectus is current only as of its date. Our business, financial condition, results of operations and prospects may have changed since that date.
We are not offering to sell or seeking offers to purchase these securities in any jurisdiction where the offer or sale is not permitted. We have not done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus and any free writing prospectus related to this offering in jurisdictions outside the United States are required to inform themselves about and to observe any restrictions relating to this offering and the distribution of this prospectus and any such free writing prospectus applicable to that jurisdiction.
Unless otherwise indicated, any reference to Shuttle Pharmaceuticals Holdings, Inc., or as “we,” “us,” or “our” refers to Shuttle Pharmaceuticals Holdings, Inc. and its subsidiaries (“Shuttle,” “Shuttle Pharma” or the “Company”).
Unless otherwise indicated, information contained in this prospectus concerning our industry and the markets in which we operate or plan to operate, including our general expectations and market position, market opportunity and market share, is based on information from our own management estimates and research, as well as from industry and general publications and research, surveys and studies conducted by third parties. Management estimates are derived from publicly available information, our knowledge of our industry, and assumptions based on such information and knowledge which we believe to be reasonable. Our management estimates have not been verified by any independent source, and we have not independently verified any third-party information. In addition, assumptions and estimates of our company’s and our industry’s future performance are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in the section entitled “Risk Factors” beginning on page 13.21. These and other factors could cause our future performance to differ materially from our assumptions and estimates. See “Special Note Regarding Forward-Looking Statements” on page 3927 below.
| i |
PUBLIC OFFERINGPROSPECTUS SUMMARY
The following summary provides an overview of all material information contained in this prospectus. It does not contain all of the information you should consider before making a decision to purchase our shares of common stock in the offering. Prior to investing in our common stock, you should carefully and thoroughly read the more detailed information in this prospectus and review our financial statements and all other information that is included in this prospectus, including the section entitled “Risk Factors” beginning at page 13.21.
Unless the context otherwise requires, references in this prospectus to “Shuttle Pharma,” “Shuttle Pharmaceuticals,” “the Company,” “we,” “our” and “us” refers to Shuttle Pharmaceuticals Holdings, Inc. and its subsidiary, Shuttle Pharmaceuticals, Inc.
Our Company
Overview
Founded in 2012 by faculty members of the Georgetown University Medical Center, Shuttle PharmaceuticalsPharma is a discovery and developmentclinical stage specialty pharmaceutical company focused on improvingleveraging our proprietary technology to develop novel therapies designed to cure cancers. Our goal is to extend the outcomesbenefits of cancer patients treatedtreatments with surgery, radiation therapy, (RT). Our mission is to improve the lives of cancer patients by developing therapies that are designed to maximize the effectiveness of RT while limiting the late effects of radiation in cancer treatment. Although RT is a proven modality for treating cancers, by developing radiation sensitizers, we aim to increase cancer cure rates, prolong patient survival and improve quality of life when used as a primary treatment, or in combination with surgery, chemotherapy and immunotherapy. Radiation therapy (“RT”) is one of the most effective modalities for treating cancers. We currentlyare developing a pipeline of products designed to address the limitations of the current cancer therapies as well as to extend to the new applications of RT. We believe that our product candidates will enable us to deliver cancer treatments that are safer, more reliable and at a greater scale than that of the current standard of care.
Our product candidates include Ropidoxuridine, Extended Bio-availability Ropidoxuridine (IPdR/TPI), and a platform of HDAC inhibitors (SP-1-161, SP-2-225 and SP-1-303). In December 2023, we submitted an Investigational New Drug (“IND”) application with the U.S. Food and Drug Administration (“FDA”) to support the next phase of development of Ropidoxuridine. In January 2024, we received the ‘Safe to Proceed’ letter from the FDA for our IND application for the Phase II study of Ropidoxuridine (IPdR) as a radiation sensitizing agent during radiotherapy in patients with newly diagnosed IDH-wildtype glioblastoma with unmethylated MGMT promoter. Receipt of the letter allows us to commence the Phase II study of Ropidoxuridine (IPdR). We have no FDA approved products and we have not yet applied for a new drug application. To date, we have been funded by investments from private investors and government contracts obtained fromreceived FDA approval of Orphan designation for Ropidoxuridine and RT for treating brain cancer (glioblastoma). We believe our management team’s expertise in radiation therapy, combined modality cancer treatment and immuno-oncology will help drive the National Institutesdevelopment and, if approved, the commercialization of Health (NIH)these potentially curative therapies for performing research. We have no product revenue and our independent auditors, in their report dated June 3, 2022, expressed doubt about our ability to continue as a going concern.patients with aggressive cancers.
Historically,Radiation Oncology has gone through transformative technological innovation over the major advances in radiation oncology have focused on improving technologylast several years to increase the amountbetter define tumors, allow improved shaping of radiation that can be administered todelivery and support dose escalation with shorter courses of treatment. Furthermore, achieving higher dose distributions within tumor volumes has reached a tumor without damaging adjacent, normal tissues. Examples of other such technologies include intensity modulated radiation therapy (IMRT), stereotactic body radiation therapy (SBRT), stereotactic radiosurgery (SRS) and proton therapy – the backbones of state-of-the-art RT. All offer improvements in physical radiation dose shaping. The basic principle underlying the effectiveness of RT for curingpractical plateau, since cancers lies in the differential cancer cell kill achieved in tumors, as compared to the effects of RT on the normal surrounding tissues, which is achievedare frequently integrated with or surrounded by delivery of highly conformal RT doses – in other words, delivery of high-dose to volumes that are shaped to conform to the target cancers while minimizing the dose to surrounding normal tissues. The treated volumes frequently includemore sensitive normal tissues thereby limiting the magnitudesand further dose escalation increases risks of the prescribed RT doses. We suggest that technological innovations to define tumor volumes and shapetissue necrosis. To increase cancer cures at maximally tolerated radiation delivery have reached an effectiveness plateau and that further improvements in RT outcomes will requiredoses, pharmacological and immunological approachesbiological modifications of cells are needed to sensitize cancers, protect normal tissues, and engage the immune system.
At present, the drugs being used for sensitizing cancers to RT are chemotherapeutic agents possessing radiation sensitizing properties as secondary effects. With the exception of Cituximab, a growth factor targeting monoclonal antibody biologic, all other drugs used as radiation sensitizers are used “off-label” to address the clinical need for radiation sensitizers. For example, certain chemotherapeutic agents, such as 5-fluorouracil, capecitabine and cis-platinum, are approved as single agents for cancer treatment, but are used “off-label” as radiation sensitizers in combination with RT. Treatments with such agents are associated with inherent toxicities associated with the drug’s primary, single-agent mechanisms of action.
Shuttle Pharma’s platform of sensitizers offers a pipeline of product candidates designed to address the urgent clinical need and the current limitations of using “off-label” drugs with potential new sensitizer agents. Our pipeline includes Ropidoxuridine, our lead clinical sensitizer drug candidate, to sensitize rapidly growing cancer cells and selective histone deacetylase (HDAC) inhibitors to sensitize cancer cells and stimulate the immune system. Our novel technologies will be tested in combinations with radiation therapies (conventional X-ray and proton radiation therapies) and in combinations with immune-therapies. To date, Ropidoxuridine has completed a Phase I clinical trial. Our HDAC inhibitor platform drug candidates have been tested in preclinical models of solid tumor cancers. Ropidoxuridine and the selective HDAC6 inhibitor SP-2-225 are the clinical and preclinical candidate drug products we propose to develop using funding from this offering.
Our intellectual property for Ropidoxuridine includes novel formulations that show improved drug bioavailability (in a preclinical animal model) and for sensitizing cancers to proton and to conventional radiation therapies. Our HDAC inhibitor intellectual property includes new patent applications and granted patents for composition of matter and methods of use for treating cancers with HDAC inhibitors in combinations with radiation therapy.
To date, we have obtained funding for our research from private investors and Small Business Innovation Research (“SBIR”) contracts obtained through the National Institutes of Health (“NIH”) to support the development of the radiation sensitizer Ropidoxuridine in a Phase I clinical trial. We have also received awards for Phase I and II SBIR contracts for development of human cell cultures for health disparities studies and predictive biomarkers of radiation late effects through the NIH’s National Cancer Institute. The completed Phase I and II funded discovery work performed to establish “Cell-based Models for Prostate Cancer Health Disparity Research” and to develop “Predictive Biomarkers of Prostate Cancer Sensitivity for Radiation Late Effects” enables Shuttle Pharma to apply for NIH SBIR Phase II funding to develop these products for advancing basic science and clinical research.
Our Product Candidates
The U.S. Food and Drug Administration (the “FDA”) considers new molecular entities as drugs that use new and unique mechanisms of action for treating medical conditions. Our clinical stage agent, Ropidoxuridine (IPdR), increases DNA double strand breaks following radiation exposure and our inhibitors of histone deacetylases (HDACs) stimulate the immune system to produce T-lymphocytes targetingreact against antigens produced by irradiated, damaged cancer cells. Drugs that show sensitizing properties, or the ability to make cancer cells more sensitive to radiation, offer a solution to this problem. Currently, such drugs are chemotherapy agents used off-label, and many have inherent toxicities since they were designed for direct cancer treatments and not for sensitization.
Our objectiveWe are developing our products with the goal of addressing the unmet need in cancer treatment for a commercially marketable radiation response modifier solution that leads to greater sensitivity of cancer cells to ionizing radiation therapy. The goal of our products is to improveincrease the outcomestherapeutic index for patients receiving radiation and to decrease radiation-related toxicities in patients with solid tumors. Our products operate across three areas related to the treatment of cancer treatment through RT while reducing its side effects by:with RT:
| 2. | Activation of the DNA damage response pathway to kill cancer cells and protect adjacent normal cells. | |
| 3. | Activation of the immune system to kill any remaining cells after RT. |
Our platform technology allows for the creation of an inventory of products for radiation sensitizing, immune modulation, and protection of healthy tissue.
| 1 |
Our Pipeline
We are currently developing a pipeline of small molecule radiation sensitizers and immune response regulating drugs. Our most advanced product candidate is Ropidoxuridine, an orally available halogenated pyrimidine with strong cancer radiation sensitizing properties in preclinical studies. In addition, we have a pipeline of complimentary product candidates that we are developing to address a host of solid tumor cancer indications. Our pipeline is represented in the diagram below:
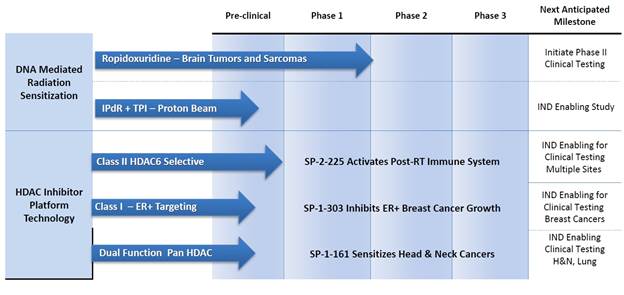
Timeline for clinical phase (Ropidoxuridine) and pre-clinical phase (HDAC inhibitors) pipeline.
Our lead product candidates include:
| ● |
| 2 |
| ● |
To our knowledge, no drug utilizing the mechanisms of our candidate small molecule drugs has received FDA approval as a radiation sensitizer. We have developed, to clinical stage, the small molecule strategies to sensitize growing cancer cells in tumors to conventional RT and to large fraction radiation therapy. The pre-clinical technology, HDAC inhibitor platform, is designed to target cancer cells while protecting healthy tissue/normal cells, thus enhancing the candidate radiation sensitizer product pipeline. The selective HDAC6 inhibitor (SP-2-225), discovered and developed by our scientists, has inhibited the growth of melanoma tumors and breast cancers in animal models by an immune stimulating mechanism.
We are focused on developing a clinical stage product candidate (Ropidoxuridine) and a pre-clinical product candidate, selective HDAC6 inhibitor (SP-2-225). We propose to develop these drug candidates as illustrated below:
Overview of Radiation Sensitizer Development

Ropidoxuridine, the clinical stage molecule, sensitizes rapidly growing cancers to radiation therapy by increasing reactive free radicals that increase DNA strand breaks. Ropidoxuridine development for treating glioblastoma will require Phase II clinical testing for use in treating brain tumors. The selective HDAC6 inhibitor, SP-2-225, a pre-clinical stage molecule, activates the innate immune system to target irradiated tumor cells by immune mechanisms.
Ropidoxuridine (IPdR)
Ropidoxuridine (IPdR) is an orally available halogenated pyrimidine (5-iodo-2-pyrimidinone-2-deoxyribose) with strong cancer radiation sensitizing properties. As a prodrug that does not become an active drug until after it is metabolized, IPdR is absorbed and metabolized to IUdR by enzymes in the liver and in cancer cells. IUdR, a halogenated pyrimidine, is incorporated into DNA by rapidly growing cancer cells. Cells that incorporate IUdR into their DNA then become more sensitive to the effects of RT. The Phase I clinical trial of Ropidoxuridine and RT, supported by an NIH SBIR contract to Shuttle Pharma, was sub-contracted to the Brown University Oncology Group (BrUOG) at the LifeSpan/Rhode Island Hospital. This Phase I clinical trial has been completed and the results were initially reported by the sub-contractor at the 30th EORTC-NCI-AACR Symposium in November 2018 and published in the medical journal Clinical Cancer Research in 2019. A maximum tolerated dose (MTD) of 1200 mg/day for 28 days was established for use in combination with radiation therapy to achieve therapeutic blood levels of IUdR.
The reported Phase I clinical trial of Ropidoxuridine in combination with RT provides the foundation for proposed Phase II clinical trials to establish the data necessary for the FDA to determine efficacy in treating brain tumors, sarcomas and pancreatic cancers, diseases that offer potential for orphan designations. The FDA granted approval of our application for orphan-drug designation for IPdR for the treatment of glioblastoma. Orphan designation protects the marketing position of Ropidoxuridine for up to seven years after marketing approval is received from the FDA. This approval integrates well into the overall intellectual property strategy for Ropidoxuridine which includes filed patent applications for “Method and Compositions for Cancer Therapies that Include Delivery of Halogenated Thymidines and Thymidine Phosphorylase Inhibitors in Combination with Radiation.” We believe that we are positioned to initiate Phase II clinical studies with Ropidoxuridine and RT in 2022.
Extended Bio-availability Ropidoxuridine (IPdR/TPI)
Ropidoxuridine and Tipiracil (IPdR/TPI) is a new combination drug formulation designed to increase the bio-availability and incorporation of IUdR into DNA. Shuttle Pharma’s preclinical studies of the combination of IPdR/TPI have shown up to 10-fold greater bioavailability of the active metabolite (IUdR) as compared to IPdR administered alone in controls. We have filed an application under the Patent Cooperation Treaty (or PCT) for the intellectual property. This new formulation will be tested in a Phase I clinical trial as a sensitizer of rectal cancers. Another nucleoside analogue, Trifluridine has been formulated in combination with Tipiracil (TAS-102) to enhance drug uptake by colon cancer cells to prolong survival in patients treated for metastatic colorectal cancers, as has been reported in the New England Journal of Medicine (N Engl J Med. 2015; 372:1909-1919). We anticipate testing for uptake of IPdR by colorectal cancer cells following administration of the IPdR/TPI drug formulation.
Proton radiation therapy is an advanced form of radiation therapy using charged proton particles (p+). Proton RT differs from conventional RT in that the radiation is delivered by a beam of protons to precisely target tumors and, due to the favorable physics of energy deposition by proton particles, there is no exit beam, resulting in less radiation to surrounding healthy tissues. The use of Proton RT is expanding rapidly in the U.S. and worldwide. According to the National Association of Proton Therapy, more than 30 facilities are currently in operation in the U.S. and an additional 30 facilities are planned for installation over the next five years. (See www.proton-therapy.org.) Much attention has been paid to proton therapy in the popular press, promoting its advantages, as well as addressing the increased health care costs. The role of a sensitizer that offers proton radiation sensitization presents an opportunity to enhance the value of proton radiation therapy as a cancer treatment modality. We believe the development of a proton therapy targeted radiation sensitizer, such as IPdR/TPI, is timely and consistent with current market needs to advance protons as a therapeutic modality.
We intend to perform clinical studies to support the development of the IPdR/TPI combination to advance this drug candidate with proton RT. The addressable market includes diseases such as brain tumors, cancers of the head and neck, GI cancers and lung cancers.
Selective HDAC Inhibitors
The roles of acetylation in the epigenetic regulation of chromatin structure and gene expression rests on the balance of activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs). Increased acetylation of histones leads to changes in chromatin structure and accessibility for key cellular proteins to specific target sites. Acetylations of non-histone proteins also modulates their enzymatic activities. We have discovered novel HDAC inhibitor molecules and testing in preclinical models has shown cancer radiation sensitizing properties, normal tissue protective properties and selective HDAC6 inhibitory properties. Our HDAC inhibitor platform, described below, will be evaluated in pre-clinical studies of radiation sensitization of solid tumors and activation of the immune response to irradiated cancer cells.
| ● | ||
| ● | SP-2-225 is Shuttle Pharma’s pre-clinical class IIb selective HDAC inhibitor that selectively affects histone deacetylase HDAC6. SP-2-225 has effects on the regulation of the immune system. The interactions of RT with the immune response for cancer treatment are of great current interest, offering insight into potential mechanisms for primary site and metastatic cancer treatment. For this reason, Shuttle Pharma has selected SP-2-225 as the candidate lead HDAC inhibitor for preclinical development. We have contracted with investigators at Georgetown University to perform preclinical studies of immune activation after radiation therapy in an animal tumor model. Requests for proposals for advancing drug manufacture and IND-enabling studies have been submitted and are under review to enable drug development to a Phase I clinical trial in 2024. With the introduction of check-point inhibitors, CAR-T therapies and personalized medicine in cancer, regulation of the immune response following RT is of significant clinical and commercial interest. | |
| ● | SP-1-303 is |
Our Approach
Drug Development Projects for Radiation Treatment of Cancers
To advance researchWe believe that is complementary to ourwe have established a leadership position in radiation sensitizer discovery and development. Over approximately seven years of research, we have identified two clinical phase product candidates and discovered new pre-clinical molecules using our proprietary platform technologies to increase the therapeutic index for patients receiving radiation for treatment of solid tumors. Our development projects,strategy has four key pillars: (1) to improve the efficacy of RT by demonstrating improved disease-free survival rates in patients who undergo radiation therapy, (2) reduce the amount of radiation needed for a favorable tumor response, thereby limiting the potential for radiation related toxicities to healthy cells, (3) decrease the extent of surgery needed to remove cancers and improve quality of life, and (4) leverage our next generation technologies to create drugs that regulate the immune response assisting immune checkpoint and CAR-T therapies and other personalized medicines targeting cancers.
We propose to perform Phase I and Phase II clinical trials to advance our clinical product candidates. In addition, candidate HDAC inhibitor molecules will be tested in animal models, and IND-enabling studies will be performed to prepare for Phase I clinical trials.
To date, we have been awarded three SBIR contracts from the NIH has awarded SBIR contracts to us to develop reagents for health disparities research and to develop biomarkers of radiation sensitivity for patients treated with radiation therapy. Our scientists have been engaged in developing model human cell systems for testing radiation sensitizers in tissue cultures. This project provides an efficient and low-cost screening technology to provide data for the FDA’s determination of drug efficacy and to identify candidate lead molecules for treating prostate cancers in African-Americans. First developed at Georgetown University, the conditional cellular reprogramming (CRC) technology offers the ability to establish new cell lines from biopsies of cancers. We have obtained a sub-license from Propagenix, Inc. to establish 100 normal and cancer cell lines from prostate biopsy samples for use in screening drug candidates and for health disparities research. A more detailed description of the Propagenix license is set forth on page 60 below.
In addition, to identify patients who may be more sensitive to radiation therapy and are at a higher risk to suffer treatment-related complications, collaborative research with Georgetown University has led to discovery of metabolite biomarkers, which are predictive of patient responses to radiation therapy. A patent for the intellectual property has been submitted by Georgetown University with Shuttle Pharma scientist (Scott Grindrod, PhD) as co-inventor. Developmental work in health disparities and predictive biomarker development has been supported by NIH SBIR contracts to Shuttle Pharmaceuticals for the following areas:to:
| ● | Develop | |
| ● | Develop prostate cancer cell cultures from African-American men, with donor matched normal prostate cells, establishing 50 pairs for accelerating research to reduce prostate cancer health disparities in African-American men. This project was funded under “Moonshot” designation and Shuttle Pharma is eligible to submit an application for additional SBIR (Phase IIb) funding to establish the infrastructure required to expand and distribute cells for research purposes. Cells from African-American patients are distributed to investigators who are conducting health disparities research. |
| 3 |
| ● | Develop predictive biomarkers |
All three SBIR funded projects have been completed. The NIH’sCompany is eligible to apply for SBIR Phase IIb funding to “bridge” the funding gap should Shuttle Pharma elect to advance the “Moonshot” health disparities or the predictive biomarker project. The NIH SBIR program is designed to encourage small businesses to engage in Federal Research/Research and Development (“R/R&D”) that has the potential for commercialization. Shuttle Pharma will apply for additional NIH SBIR grants and contracts to fund advancement of these projects.
Market Opportunity
The American Cancer Society (Cancer Facts & Figures 2020) estimates 1,806,590 new cancer cases and 606,520 cancer deaths each year in the United States and, according to the American Society for Radiation Oncology, more than 50% of patients undergo RT at some point in the treatment of their diseases. RT is used to treat cancers of the lung, breast, brain, esophagus, pancreas, rectum, head and neck, uterus, lymphomas and sarcomas. At present, we are developing drug candidates to address brain, pancreas, rectum, sarcomas and lymphomas, although we may test and seek approval for our drug candidates to treat other cancers in the future.
Currently, there is only one drug (the monoclonal antibody, Cetuximab) that has received FDA approval for the radiation sensitizer indication. Cetuximab is a recombinant monoclonal antibody that binds to epidermal growth factor receptor (EGFR) and inhibits the binding of epidermal growth factor (EGF). Cetuximab is administered via intravenous infusion and is used as monotherapy or in combination with other chemotherapies or radiation therapy. In clinical trials, cetuximab was associated with serious and fatal infusion reactions, cardiopulmonary arrest or sudden death, and serious dermatologic toxicities, toxicities that have created deterrents to its use as a radiation sensitizer. Present treatment utilizes “off-label” small molecule drugs, which are cytotoxic chemotherapy agents that also sensitize, but do not have radiation sensitization as an FDA approved indication. Moreover, since “off-label” drugs are cytotoxic, they are often associated with intrinsic acute and chronic side effects. Nevertheless, these drugs have shown clinically significant improvements in disease control and survival and are typically included in standard-of-care treatment recommendations for patients with cancers of the head and neck, brain, lung, esophagus, stomach, pancreas, liver, bladder, lymphomas and sarcomas. As a result, we believe that there is a significant market opportunity for our product candidates. Based on cancer incidence data published by the American Cancer Society, we have estimated the numbers of patients presenting with local/regional disease, suitable for treatment with RT.
Estimated RT Cases by Disease Site
| Cancer Type | Cases Diagnosed Annually | Estimated RT Cases | ||||||
| Brain | 23,890 | 21,979 | ||||||
| Pancreas | 57,600 | 32,832 | ||||||
| Sarcomas | 13,130 | 4,000 | ||||||
| Rectum | 43,340 | 26,437 | ||||||
Annual cancer cases for each disease site are estimated from American Cancer Society Facts & Figures 2020 publication. The fraction of patients optimally receiving RT for each disease site were obtained from published estimates of Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer. 2005 Sep 15;104(6):1129-37. doi: 10.1002/cncr.21324. The Estimated RT cases were obtained by multiplying Cases Diagnosed Annually by the fraction receiving RT for optimal utilization.
Our Development Strategy
Our goal is to maintain and build upon our leadership position in radiation sensitization. We plan to develop Ropidoxuridine and the HDAC6 inhibitor (SP-2-225) and, if approved by the FDA, to commercialize our product candidates for the treatment of cancers. While this process may require years to complete, we believe achieving this goal could result in new radiation sensitizer and immunotherapy products for cancer treatment.products. Key elements of our strategy include:
| ● | Capitalize on Ropidoxuridine as an orally available, small molecule radiation | |
| ● | Expand our leadership position within radiation sensitizers. In addition to our traditional radiation sensitizers, we plan to advance our near-term pipeline to include radiation sensitizers for proton therapy. Proton Therapy is growing worldwide as a form of radiation therapy due to its unique beam shaping characteristics. As a result, this new technology offers a major opportunity for Shuttle Pharma to strive to develop | |
| ● | Execute a disciplined business development strategy to strengthen our portfolio of product candidates. We have built our current product pipeline through in-house discovery, development, partnerships with leading academic institutions and through |
| ● | Invest in our HDAC platform technology | |
| ● | Enter into collaborations to realize the full potential of our |
We propose the following clinical development plan to identify, develop and commercialize drugs for use in cancer treatments in combination with RT:
Develop Ropidoxuridine (IPdR) for Orphan disease indications to take to market
Develop Ropidoxuridine and tipiracil (IPdR/TPI) for colorectal cancer indications to take to market
Develop HDAC Inhibitors for use in breast cancer for immune activation after RT
Management Team
Our management team has significant experience in radiation oncology and in progressing products from early-stage research through clinical trials. Our CEO, Anatoly Dritschilo, M.D., is an experienced clinician and researcher who has held senior academic and management positions including serving as Department Chairman, Hospital Medical Director and Cancer Center Director at Georgetown University Medical Center. Prior to co-founding our company, Dr. Dritschilo was a co-founder of Oncomed, Inc., a company that became public as NeoPharm, Inc. (Nasdaq: NEOL). He has experience in providing care for patients undergoing treatment for cancers of the prostate, breast, brain, lung, sarcomas and GI systems. Dr. Dritschilo has directed basic science research supported by grants from the National Cancer Institute (“NCI”) and performed clinical trials using drugs and radiation therapy. In addition, Dr. Dritschilo served as the principal investigator of pharmaceutical industry sponsored clinical evaluations of human interferon alpha-2 (Bristol-Myers) with radiation therapy and antisense raf oligonucleotides, LErafAON (NeoPharm) with radiation therapy. He serves as a Radiation Biology and Radiation Oncology expert on committees of the NIH to review Program Project (P01) grant applications, Specialized Program of Research Excellence (SPORE) grant applications and investigator-initiated research project (R01) applications.
| 4 |
Dr. Dritschilo is supported in our clinical development effort by Tyvin Rich, MD, our Chief Clinical Officer and Medical Director. Dr. Rich is the former Professor and Chairman of the Department of Therapeutic Radiology and Oncology at the University of Virginia Health Sciences Center and proton radiation therapy specialist at the Hampton Proton Therapy Center in Hampton, Virginia. Dr. Rich has served as principal investigator on multi-modality clinical trials for the treatment of gastrointestinal (GI) cancers and helped to develop treatment with 5-fluorouracil (5-FU) as a radiation sensitizer for use with RT in the treatment of GI cancers. He has extensive cancer clinical trial experience in developing radiation sensitizer applications through his participation in the Radiation Therapy Oncology Group (RTOG). Dr. Rich is a co-inventor with scientists at the University of Virginia of the Proton Activated Atomic Medicine technology.
Our administrative services are provided by Peter Dritschilo, MBA, who has served as our President and COO since 2012. Mr. Dritschilo’s experience in hospital administration and management of medical oncology clinical services and radiation therapy facilities, including management of day-to-day operations, human resources and financial oversight. Peter Dritschilo is the son of our Chairman and CEO, Dr. Anatoly Dritschilo. The addition of Michael Vander Hoek, as our CFO and Vice President for Operations and Regulatory expands our capability to provide the level of management needed for the proposed expansion of clinical trials. Mr. Vander Hoek has served as administrative director of the Lombardi Comprehensive Cancer Center (LCC) for the past 12 years and has extensive experience in negotiations, management and supervision of Contract Research Organizations (CROs) and research contracts in general. As the administrative director of the Lombardi Comprehensive Cancer Center, Mr. Vander Hoek also served as the chief financial officer. Taken together, we believe our leadership team of highly qualified specialists will help us achieve the proposed milestones for the development of radiation sensitizer products.
Pre-IPO Bridge Financing
In December 2021, we completed a $500,000 note offering pursuant to which we sold to two accredited investors (the “Investors”) units consisting of (i) a $250,000 promissory note bearing 10% interest, repayable at the time of this offering (the “Note”)Recent Financings – Our IPO and (ii) a warrant to purchase 250,000 shares (the “Warrant Shares”) of our common stock at an exercise price of $1.00 per share (the “Warrant”). Immediately before closing on the Offering, the Notes will be repaid and cancelled in exchange for the exercise of the Warrants and issuance of the 500,000 Warrant Shares to the Investors.Post-IPO Financings
In February and MarchOn September 2, 2022, we soldclosed on our IPO of 1,225,888 units (each a total of $365,000“Unit,” and $225,000, respectively, in convertible notes (“Convertible Notes”collectively, the “Units”) to certain accredited investors, which notes will automatically convert into units,, with each unitUnit consisting of one share of the Company’s common stock and aone warrant to purchase one share of common stock, upon effectiveness of this offering at a conversionpublic offering price of $3.00$8.125 per unit (the “Conversion Units”Unit. Our IPO, which was underwritten by Boustead Securities, LLC (“Boustead”). In addition, each, resulted in gross proceeds of the Convertible Note holders will be entitled to demand registration rights,$9,960,430, before deducting underwriting discounts and we are required to register the Conversioncommissions. On September 29, 2022, Boustead exercised its overallotment option, purchasing an additional 183,883 Units, within 180 daysresulting in gross proceeds of the closing$1,494,041, before deducting underwriter commissions and discounts. As a result, our IPO raised a total of this offering. We are using the net proceeds from the Convertible Notes offering to expand our current operations, including our technology$11,454,474, before deducting underwriting discounts, commissions and intellectual property portfolio, and to fund the costs of this offering. Any funds remaining will be used for working capital and other general corporate purposes.related IPO expenses.
In August 2022,On January 11, 2023, we completedentered into a $125,000 note offeringsecurities purchase agreement (the “SPA”) with Alto Opportunity Master Fund, SPC – Segregated Master Portfolio B, a Cayman entity (the “Investor”), pursuant to which wethe Company sold to three accredited investorsthe Investor a $4.3 million convertible note (the “Investors”) units consisting of (i) a total of $125,000 in 10% promissory notes bearing 10% interest, repayable at the time of this offering (the “August“Convertible Note”) and (ii) warrantswarrant (the “Warrant”) to purchase a total of 50,0001,018,079 shares of common stock exercisable at $2.50 per share. We anticipate that the notes will be repaid upon closing of the Offering.
Boustead Securities LLC (“Boustead”) acted as placement agentCompany, in both theexchange for gross proceeds of $4.0 million (the “Investment Amount”). The Convertible Note and Warrant offeringamortizes on a monthly basis and the Convertible Notes offering, pursuantCompany can make such monthly amortization payments in cash or, subject to which Boustead waived its cash compensation related to the Note and Warrant offerings and received cash compensation of $36,500 and $22,250certain equity conditions, in each of the February and March 2022 closings, respectively, and also received warrants to purchase 10% of the total number of Conversion Shares, exercisable at the conversion price of the Convertible Notes. For the August 2022 offering, Boustead received a warrant to purchase 5,000registered shares of common stock exercisable at $2.50 per share, andor a combination thereof. For equity repayment, the Convertible Note is entitled to receive $12,500 in cash compensation, which cash compensation has been deferred.
Our Resale Offering
Certain of our shareholders will be selling through a separate prospectus (the “Resale Prospectus”) (i) 486,942convertible into shares of common stock at price per share equal to the lower of (i) $2.35 (ii) 147,50090% of the three lowest daily VWAPs of the 15 trading days prior to the payment date or (iii) 90% of the VWAP of the trading day prior to payment date. The Convertible Note is repayable over 26 months and bears interest at the rate of 5% per annum. The Warrant is exercisable for four years from the date of closing and is exercisable at $2.35 per share. In the event the Investor exercises the Warrant in full, such exercise would result in additional gross proceeds to the Company of approximately $2.4 million.
On May 10, 2023, we entered into an amendment agreement (the “Amendment Agreement”) to the SPA. Under the Amendment Agreement, the Company and the Investor amended the transaction documents as follows: (i) amended and restated Section 2 of the Warrant so as to remove a provision that would have potentially required an adjustment to the number of warrant shares exercisable under the Warrant, (ii) stipulated that the Company would obtain majority shareholder approval to issue up to an additional $10 million in convertible notes (the “Subsequent Notes”) and warrants (the “Subsequent Warrants”) equal to 42.5% of the outstanding principal value of the Subsequent Notes, which Subsequent Note and Subsequent Warrant would be sold to the Investor on substantially the same terms as the existing Convertible Note and Warrant (each as amended by the Amendment Agreement) and upon conversion and/or exercise would cause the potential issuance of in excess of 19.9% of the Company’s issued and outstanding stock, (iii) that, upon obtaining majority stockholder approval, the Company would file a Schedule 14C related to such potential issuance of the shares of common stock related to the potential sale of the Subsequent Notes and warrantsSubsequent Warrants to the Investor within 30 calendar days of entry into the Amendment Agreement, and (iv) stipulated that the Investor would release $1,500,000 in cash collateral to the Company, with $1,000,000 to be released to the Company immediately upon singing of the Amendment Agreement and $500,000 to be released upon the Company’s filing of the Schedule 14C. The Company obtained majority stockholder consent to the potential sale of the Subsequent Notes and Subsequent Warrants to the Investor in advance of entry into the Amendment Agreement.
On June 4, 2023, we entered into amendment no. 1 (“Amendment No. 1”) to the Amendment Agreement dated May 11, 2023, for purposes of amending the terms of the SPA. Under Amendment No. 1 to the Amendment Agreement, the Company and the Investor agreed as follows: (i) that Section 15(q) to the Convertible Note, which required the Company to hold the Cash Collateral in a Controlled Account Agreement (as defined in the Convertible Note), would no longer be applicable, (ii) that the Investor would stipulate the release to the Company of the remaining Cash Collateral totaling $2,924,000 (thus releasing the full amount of the Cash Collateral to the Company), and (iii) that, should the Investor exercise its option to purchase 147,500 shares of common stock, which sharesthe Subsequent Notes and warrants willSubsequent Warrants, that such Subsequent Notes would omit Section 15(q) and that the Company would not be issuable upon conversion of convertible notes, which convertible notes will automatically convert into common stock and warrants upon the effectiveness of this offering and the date of this prospectus, and (iii) 530,000 shares of common stock issuable upon exercise of warrants held by certain warrant holders, as set forth in the Resale Prospectus. We will not receiverequired to maintain any proceeds from the sales by the selling stockholders of the securities set forth in thee Resale Prospectus.controlled accounts or otherwise be subject to any controlled account agreements.
The Resale Prospectus is substantively identical to this Prospectus, except for the following principal points:
Summary Risk Factors
Our business is subject to a number of risks you should be aware of before making an investment decision. These risks are discussed more fully in the “Risk Factors” section of this prospectus at page 1321 immediately following the section entitled “Questions and Answers Related to this prospectus summary.Rights Offering.” These risks include the following:
| ● | Our ability to continue as a going concern in the near term is dependent upon us successfully raising additional equity or debt financing to fund our operations. | |
| ● | Our success is primarily dependent on | |
| ● | ||
| product sales revenue. | ||
| ● | We face competition from entities that have |
| ● | If | |
| business. | ||
| ● |
| |
| ● | If we are not able to obtain and enforce patent protection for our technologies or product candidates, development and commercialization of our product candidates may be adversely affected. | |
| ● | We | |
| put our patents and other proprietary rights at risk. | ||
| ● | ||
| harmed. | ||
| ● | We may be unable to | |
| product candidates. | ||
| ● | ||
| unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, thereby harming our business. | ||
| ● | ||
| Our ability to obtain services, reimbursement or funding from the federal government may be impacted by possible reductions in federal spending. | ||
| ● | ||
| product candidates could be compromised. | ||
| ● | ||
| no guarantee that we will regain compliance without effectuating a reverse stock split. | ||
| ● | ||
| common stock could incur substantial losses. | ||
| ● | The | |
| equity or of debt securities that are convertible into common stock will dilute our share capital. | ||
| ● |
| 6 |
Implications of Being an Emerging Growth Company
| ● | As a smaller reporting company, and as a company with less than | ||
| ● | being permitted to provide only two years of audited financial statements, in addition to any required unaudited interim financial statements, with correspondingly reduced “Management’s Discussion and Analysis of Financial Condition and Results of Operations” disclosure; | ||
| ● | not being required to comply with the auditor attestation requirements in the assessment of our internal control over financial reporting; |
| ● | not being required to comply with any mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements; | |
| ● | reduced disclosure obligations regarding executive compensation; and | |
| ● | exemptions from the requirements of holding a non-binding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
We may take advantage of the above provisions for up to five years or until such earlier time that we are no longer an emerging growth company. We would cease to be an emerging growth company if we have more than $1.0$1.235 billion in annual revenues, have more than $700 million in market value of our capital stock held by non-affiliates or issue more than $1.0 billion of non-convertible debt over a three-year period. We may also choose to take advantage of some, but not all, of the available exemptions. We have taken advantage of some reduced reporting requirements in this prospectus. Accordingly, the information contained in this prospectus may be different than the information you receive from other public companies in which you hold stock.
In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This provision allows an emerging growth company to delay the adoption of some accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this extended transition period for adopting new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
Corporate Information
The Company was formed as a limited liability company in the state of Maryland in December 2012 and was converted to a C corporation, Shuttle Pharmaceuticals, Inc. (“Shuttle”), in August of 2016. In June 2018, Shuttle completed a share exchange with Shuttle Pharma Acquisition Corp. Inc. (“Acquisition Corp.”), pursuant to which Shuttle Pharmaceuticals, Inc. became a subsidiary of Acquisition Corp. and we subsequently changed the name of Acquisition Corp. to Shuttle Pharmaceuticals Holdings, Inc. Our executive offices are located at 1 Research Court, Suite 450, Rockville,401 Professional Drive, Gaithersburg, Maryland 2085020879 and our telephone number is (240) 403-4212. Our corporate website is www.shuttlepharma.com. Information appearing on our corporate website is not incorporated as part of this prospectus.
| 7 |
TheSummary of the Rights Offering
| Securities |
| |
| Size of Offering: | [ ] Units | |
| Subscription Price: | 90% of the VWAP of the Company’s common stock five days prior to the end of the Subscription Period. | |
| Percentage Interest in Diagnostics: | Each Unit will entitle the holder to a [ ]% equity interest in Shuttle Diagnostics such that, when all $4,500,000 in Units are sold, a total of 44% of the equity interest in Diagnostics will be distributed to the purchasers in the Rights Offering. | |
| Warrants: | Each Warrant will entitle the holder to purchase one share of our common stock at an exercise price of
| |
| 5:00 p.m., Eastern Time, , 2024 | ||
| Basic Subscription Right: | Your Basic Subscription Right will entitle you to purchase one Unit at the Subscription Price. You may exercise your Basic Subscription Right for some or all of your Subscription Rights, or you may choose not to exercise your Subscription Rights. If you choose to exercise your Subscription Rights, there is no minimum number of Units you must purchase. We | |
| Over-Subscription Privilege: | If you exercise your Basic Subscription Rights in full, you may also choose to exercise an over-subscription privilege to purchase a | |
| Expiration Date: | The Subscription Rights will expire at 5:00 p.m., Eastern Time, on , 2024. |
| 8 |
| Purchase Agreement, Committed Investor: | SRO, LLC, an entity controlled by Keith Moore, of Boustead Securities, LLC, has entered into a purchase agreement with us pursuant to which SRO, LLC has agreed to purchase at least $2.25 million of Units in In addition, the Rights Offering is backstopped by SRO, LLC, the Committed Investor such that the Committed Investor has also agreed to purchase up to See the “Purchase Agreement” and “Backstop Commitment” below. | |
Procedure for Exercising Subscription Rights: | To exercise your Subscription Rights, you must take the following steps: If you are a record holder, as of If as of the Record Date you are a beneficial owner of shares of common stock that are registered in the name of a broker, dealer, bank or other nominee, you should instruct your broker, dealer, bank or other nominee to exercise your Subscription Rights on your behalf. Please follow the instructions of your nominee, who may require that you meet a deadline earlier than 5:00 p.m., Eastern Time, on , 2024. | |
| Payment Adjustments: | If you send a payment that is insufficient to purchase | |
Delivery of Shares, Warrants and Equity Interests: | As soon as practicable after the expiration of the Rights Offering, and within five business days thereof, we expect to close on subscriptions and for the Subscription Agent to arrange for the issuance of the shares of common stock, Warrants and equity interests purchased pursuant to the Rights Offering. All shares, Warrants and equity interests that are purchased in the Rights Offering will be issued in book-entry, or uncertificated, form. If you hold your shares in the name of a bank, broker, dealer, or other nominee, DTC will credit your account with your nominee with the securities you purchased in the Rights Offering. However, since neither the Warrants nor the Equity Interests are registered, we will need to establish a separate account on your behalf with our transfer agent, Vstock Transfer LLC. |
| 9 |
| Non-transferability of Subscription Rights: | The Subscription Rights may not be sold, transferred, assigned or given away to anyone. The Subscription Rights will not be listed for trading on any stock exchange or market. | |
| Transferability of Warrants and Equity Interests: | The Warrants will be separately transferable following their issuance and for a period of three years after issuance; the Equity Interests will be separately transferable following their issuance. | |
| No Board Recommendation: | Our board of directors is not making a recommendation regarding your exercise of the Subscription Rights. You are urged to make your decision to invest based on your own assessment of our | |
| No Revocation: | Except as described below, all exercises of Subscription Rights are irrevocable, even if you later learn of information that you consider to be unfavorable to the exercise of your Subscription Rights. | |
| Dividend policy: | We have never paid cash dividends on our common stock and we do not anticipate paying any cash dividends in the foreseeable future. See the section entitled “Dividend Policy.” |
| Use of proceeds: |
$[_] million. We intend to use the net proceeds from | |
| Material U.S. Federal Income Tax Consequences: | For U.S. federal income tax purposes, we do not believe you should recognize income or loss upon receipt or exercise of a Subscription Right, but the receipt and exercise of the Subscription Rights is unclear in certain respects. You should consult your own tax advisor as to the tax consequences of the Rights Offering in light of your particular circumstances. See “Material U.S. Federal Income Tax Consequences.” | |
| Extension, Amendment and Termination: | Although we do not presently intend to do so, we may extend the Rights Offering for additional time in our sole discretion for any reason for up to an additional 45 days. For example, we may decide that changes in the market price of our common stock warrant an extension, or we may decide that the If we should make any fundamental changes to the terms set forth in this prospectus, we will (i) file a post-effective amendment to the registration statement of which this prospectus forms a part, (ii) offer potential purchasers who have subscribed for rights the opportunity to cancel such subscriptions and issue a refund of any money advanced by such stockholder or eligible warrant holder, and (iii) recirculate an updated prospectus after the post-effective amendment is declared effective with the SEC. | |
| Questions: | If you have any questions about the Rights Offering, please contact the Information Agent, VStock Transfer, LLC, by mail at 18 Lafayette Place, Woodmere, New York 11598 or by email at action@vstocktranser.com. | |
| Market for Common Stock: | Our common stock is listed on the Nasdaq Capital Market under the symbol “SHPH.” | |
| Market for Warrants and Equity Interests: | There is no established public trading market for either the Warrants or the Equity Interests, and we do not expect a market to develop. In addition, we do not intend to apply for listing of the | |
| (1) |
| ● | 1,446,155 warrants to purchase common stock; and | |
| ● | 2,221,820 shares |
| 10 |
INCORPORATION OF CERTAIN INFORMATION BY REFERENCE
The SEC allows us to “incorporate by reference” into this prospectus the information in other documents that we file with it. This means that we can disclose important information to you by referring you to those documents. The information incorporated by reference is considered to be a part of this prospectus, and information in documents that we file later with the SEC will automatically update and supersede information contained in documents filed earlier with the SEC or contained in this prospectus. We incorporate by reference in this prospectus the documents listed below and any future filings that we may make with the SEC under Sections 13(a), 13(c), 14, or 15(d) of the Exchange Act prior to the termination of the offering under this prospectus; provided, however, that we are not incorporating, in each case, any documents or information deemed to have been furnished and not filed in accordance with SEC rules:
| ● | ||
| ● |
Reverse Stock SplitAdditionally, all documents filed by us with the SEC under Sections 13(a), 13(c), 14 or 15(d) of the Exchange Act (i) prior to effectiveness of this registration statement, and (ii) after the effective date of this registration statement and before the termination or completion of any offering hereunder, shall be deemed to be incorporated by reference into this prospectus from the respective dates of filing of such documents, except that we do not incorporate any document or portion of a document that is “furnished” to the SEC, but not deemed “filed.”
Effective April 1, 2022,Any statement contained in this prospectus supplement, the accompanying prospectus or in a document incorporated or deemed to be incorporated by reference into this prospectus supplement and the accompanying prospectus will be deemed to be modified or superseded for purposes of this prospectus supplement and the accompanying prospectus to the extent that a statement contained in this prospectus supplement and the accompanying prospectus or any other subsequently filed document that is deemed to be incorporated by reference into this prospectus supplement and the accompanying prospectus modifies or supersedes the statement. Any statements so modified or superseded shall not be deemed, except as so modified or superseded, to constitute a part of this prospectus supplement and the accompanying prospectus.
We will furnish without charge to you, on written or oral request, a copy of any or all of the documents incorporated by reference in this prospectus, including exhibits to these documents. You should direct any requests for documents to Shuttle Pharmaceutical Holdings, Inc., 401 Professional Drive, Suite 260, Gaithersburg, MD 20879 or info@shuttlepharma.com.
You also may access these filings on our website at www.shuttlepharma.com. We do not incorporate the information on our website into this prospectus supplement or the accompanying prospectus and you should not consider any information on, or that can be accessed through, our website as part of this prospectus supplement or the accompanying prospectus (other than those filings with the SEC that we effectedspecifically incorporate by reference into this prospectus supplement and the accompanying prospectus)
| 11 |
WHERE YOU CAN FIND MORE INFORMATION
We have filed a 2-for-1 reverse stock splitregistration statement on Form S-1 with the SEC under the Securities Act of 1933, as amended. This prospectus is part of the registration statement, but the registration statement includes additional information and exhibits. We file annual, quarterly and current reports, proxy statements and other information with the SEC. The SEC maintains a web site that contains reports, proxy and information statements and other information regarding companies, such as ours, that file documents electronically with the SEC. The website address is www.sec.gov. The information on the SEC’s website is not part of this prospectus, and any references to this website or any other website are inactive textual references only.
| 12 |
QUESTIONS AND ANSWERS RELATING TO THE RIGHTS OFFERING
The following are examples of what we anticipate will be common questions about this Rights Offering. The answers are based on selected information included elsewhere in this prospectus. The following questions and answers do not contain all of the information that may be important to you and may not address all of the questions that you may have about the Rights Offering. This prospectus and the documents incorporated by reference into this prospectus contain more detailed descriptions of the terms and conditions of the Rights Offering and provides additional information about us and our business, including potential risks related to the Rights Offering, the Units offered hereby, and our business. We urge you to read this entire prospectus and the documents incorporated by reference into this prospectus.
Why are we conducting the Rights Offering?
We are conducting the Rights Offering to raise additional capital for general corporate purposes and to fund ongoing operations and expansion of our business.
What is a Unit?
Each Unit consists of one share of common stock, one warrant to purchase a share of common stock, and an equity interest in our wholly owned subsidiary, Shuttle Diagnostics, Inc. (“Diagnostics”), such that upon sale of all of the Units, the Unit Holders will hold a total of 44% of the equity interests in Diagnostics. Each Unit is being offered for at a subscription price of 90% of the VWAP of the Company’s common stock five days prior to the Record Date (or , 2024), which is $[ ] per Unit. No fractional Units will be issued. Each warrant entitles you to purchase one share of common stock at an exercise price of $2.35 per share, from the date of issuance through its expiration three years after the date of issuance. The shares of common stock, warrants and equity interests that comprise each Unit are immediately separable and will be issued separately in this Rights Offering, however, they may only be purchased as a Unit, and the Units will not trade as a separate security.
What is the Rights Offering?
We are distributing, at no charge, to holders of our common stock and outstanding warrants, as of the Record Date, non-transferable Subscription Rights to purchase Units at a price of $[ ] per Unit. The Subscription Rights will not be tradable. Each Unit consists of one share of our common stock, (the “Reverse Stock Split”one Warrant to purchase one share of our common stock, exercisable at $2.35 per share, and a [ ]% percent Equity Interest in our Diagnostics Subsidiary. See “Are there risks in exercising my Subscription Rights?” below. Upon expiration of the Rights Offering, the common stock, Warrants and Equity Interests comprising the Units will immediately separate and will be issued separately but may only be purchased as a Unit, and the Units will not trade as a separate security. There is no public trading market for the Warrants or Equity Interests and they will not be listed for trading on Nasdaq or any other securities exchange or market. The common stock to be issued upon exercise of the Warrants, like our existing shares of common stock, will be traded on the NASDAQ Capital Market under the symbol “SHPH.” You will receive one Subscription Right for every share of common stock or warrant that you owned as of 5:00 p.m., Eastern Time, on the Record Date. Each Subscription Right entitles the record holder to a Basic Subscription Right and an Over-Subscription Privilege. The Subscription Rights will expire if they are not exercised by 5:00 p.m., Eastern Time, on , 2024, unless we extend or earlier terminate the Rights Offering.
What are the Basic Subscription Rights?
For every share you owned (including each share of common stock issuable upon exercise of any outstanding warrants) as of the Record Date, you will receive one Basic Subscription Right, which gives you the opportunity to purchase one Unit, consisting of one share of our common stock, one Warrant to purchase one share of common stock, and a [ ]% Equity Interest in our subsidiary, Shuttle Diagnostics, Inc. (“Diagnostics”). All referencesFor example, if you owned 100 shares of common stock as of the Record Date, you will receive 100 Subscription Rights and will have the right to purchase 100 shares of our common stock, 100 Warrants and [ ]% Equity Interests in this prospectus refersDiagnostics for $[ ] per Unit (or a total payment of $[ ]), with the per Unit calculation to be equal to 90% of the five-day VWAP of the Company’s Common Stock immediately prior to the Record Date. You may exercise all or a portion of your Basic Subscription Rights or you may choose not to exercise any Basic Subscription Rights at all.
| 13 |
If you are a record holder of our common stock, or warrants to purchase our common stock, the number of shares you may purchase pursuant to your Basic Subscription Rights is indicated on the enclosed Rights Certificate. If you hold your common stock in the name of a broker, dealer, bank or other nominee who uses the services of the Depository Trust Company, or DTC, you will not receive a Rights Certificate. Instead, DTC will issue one Subscription Right to your nominee record holder for each share of our common stock that you beneficially own as of the Record Date. If you are not contacted by your nominee, you should contact your nominee as soon as possible.
What is the Over-Subscription Privilege?
If you exercise your Basic Subscription Rights in full, you may also choose to exercise your Over-Subscription Privilege to purchase a portion of any Units that are not purchased by other holders of common stock or warrant holders and remain available under the Rights Offering. You should indicate on your Rights Certificate, or the form provided by your nominee if your shares are held in the name of a nominee, how many additional Units you would like to purchase pursuant to your Over-Subscription Privilege, which we refer to as your Over-Subscription Request.
Subject to stock ownership limitations, if enough Units are available, we will seek to honor your Over-Subscription Request in full. If Over-Subscription Requests exceed the number of Units available, however, we will allocate the available Units pro-rata among the stockholders and warrant holders exercising the Over-Subscription Privilege in proportion to the number of shares of our common stock after giving effect(including each share of common stock issuable upon exercise of Participating Warrants) each of those stockholders and warrant holders owned on the Record Date, relative to the Reverse Stock Split (unless otherwise indicated).number of shares (including each share of common stock issuable upon exercise of outstanding warrants) owned on the Record Date by all record holders exercising the Over-Subscription Privilege. If this pro-rata allocation results in any stockholders or warrant holders receiving a greater number of Units than the stockholder or warrant holders subscribed for pursuant to the exercise of the Over-Subscription Privilege, then such stockholder or warrant holder will be allocated only that number of Units for which the stockholder or warrant holder oversubscribed, and the remaining Units will be allocated among all other stockholders and warrant holders exercising the Over-Subscription Privilege on the same pro rata basis described above. The proration process will be repeated until all Units have been allocated. See “The Rights Offering- Limitation on the Purchase of Units” for a description of certain stock ownership limitations.
To properly exercise your Over-Subscription Privilege, you must deliver to the Subscription Agent the subscription payment related to your Over- Subscription Privilege before the Rights Offering expires. Because we will not know the total number of unsubscribed Units before the expiration of the Rights Offering, if you wish to maximize the number of Units you purchase pursuant to your over-subscription privilege, you will need to deliver payment in an amount equal to the aggregate Subscription Price for the maximum number of Units available, assuming that no common stockholder or warrant holders other than you has purchased any Units pursuant to such stockholder’s or warrant holder’s basic subscription right and over-subscription privilege. See “The Rights Offering- The Subscription Rights-Over-Subscription Privilege.” To the extent you properly exercise your Over-Subscription Privilege for a number of Units that exceeds the number of unsubscribed Units available to you, any excess subscription payments will be returned to you within 10 business days after the expiration of the Rights Offering, without interest or deduction.
Vstock Transfer, LLC, our Subscription Agent for the Rights Offering, will determine the allocation of Over-Subscription Requests based on the formula described above.
May the Subscription Rights that I exercise be reduced for any reason?
Yes. While we are distributing to holders of our common stock and holders of the warrant holders, one Subscription Right for every share of common stock (including each share of common stock issuable upon exercise of warrants) owned on the Record Date, we are only seeking to raise $4.5 million dollars in gross proceeds in this Rights Offering. As a result, based on 16,894,893 shares of common stock outstanding as of April [_], 2024, and 1,466,153 shares of common stock issuable upon exercise of warrants, we would grant Subscription Rights to acquire [_] Units but will only accept subscriptions for [ ] Units. Accordingly, enough Units may not be available to honor your subscription in full. If exercises of Basic Subscription Rights exceed the number of Units available in the Rights Offering, we will allocate the available Units pro-rata among the record holders exercising the Basic Subscription Rights in proportion to the number of shares of our common stock (including each share of common stock issuable upon exercise of outstanding warrants) each of those record holders owned on the Record Date, relative to the number of shares owned on the Record Date by all record holders exercising the Basic Subscription Right. If this pro-rata allocation results in any record holders receiving a greater number of Units than the record holder subscribed for pursuant to the exercise of the Basic Subscription Rights, then such record holder will be allocated only that number of Units for which the record holder subscribed, and the remaining Units will be allocated among all other record holders exercising their Basic Subscription Rights on the same pro rata basis described above. The proration process will be repeated until all Units have been allocated. Please also see the discussion under “The Rights Offering-The Subscription Rights-Over-Subscription Privilege” and “The Rights Offering-Limitation on the Purchase of Units” for a description potential proration as to the Over-Subscription Privilege and certain stock ownership limitations.
| 14 |
If for any reason the number of Units allocated to you is less than you have subscribed for, then the excess funds held by the Subscription Agent on your behalf will be returned to you, without interest, as soon as practicable after the Rights Offering has expired and all prorating calculations and reductions contemplated by the terms of the Rights Offering have been effected, and we will have no further obligations to you.
SUMMARY FINANCIAL INFORMATIONWhat are the terms of the Warrants?
Each Warrant entitles the holder to purchase one share of our common stock at an exercise price of $2.35 per share from the date of issuance through its expiration three years from the date of issuance. The following summary financial data shouldWarrants will be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and the Financial Statements and notes thereto, included elsewhere in this prospectus.exercisable for cash.
Statements of Operations Data:Are the Warrants or Equity Interests listed?
There is no public trading market for the Warrants or Equity Interests and they will not be listed for trading on Nasdaq or any other securities exchange or market. The Warrants and Equity Interests will be issued in registered form under a warrant agent agreement with Vstock Transfer, LLC as agent for the Warrants and Equity Interests.
| For the Years Ended | For the Six Months Ended | |||||||||||||||
| December 31, | June 30, | |||||||||||||||
| 2021 | 2020 | 2022 | 2021 | |||||||||||||
| (As Restated) | ||||||||||||||||
| Revenue | $ | - | $ | - | $ | - | $ | - | ||||||||
| Operating expenses | $ | 1,742,992 | $ | 509,522 | $ | 992,022 | $ | 512,377 | ||||||||
| Loss from operations | $ | (1,742,992 | ) | $ | (509,522 | ) | $ | (992,002 | ) | $ | (512,337 | ) | ||||
| Other income (expense) | $ | 590,858 | $ | (296,210 | ) | $ | (287,092 | ) | $ | 36,212 | ||||||
| Net loss | $ | (1,152,134 | ) | $ | (805,732 | ) | $ | (1,279,114 | ) | $ | (476,125 | ) | ||||
| Weighted average common shares outstanding - basic and diluted | 9,301,750 | 9,291,526 | 9,312,583 | 9,291,526 | ||||||||||||
| Net loss per share - basic and diluted | $ | (0.12 | ) | $ | (0.09 | ) | $ | (0.14 | ) | $ | (0.05 | ) | ||||
Balance Sheet Data:Will fractional shares be issued upon exercise of Subscription Rights or the exercise of Warrants?
| As of June 30, 2022 | ||||||||||||
| Actual | Pro Forma | Pro Forma as Adjusted for Conversions and this Offering | ||||||||||
| Cash | $ | 50,437 | $ | 10,898,066 | $ | 10,974,541 | ||||||
| Total current assets | 63,952 | 10,911,581 | 10,988,056 | |||||||||
| Total assets | 173,480 | 11,021,109 | 11,097,584 | |||||||||
| Total current liabilities | 2,453,951 | 2,453,951 | 1,863,951 | |||||||||
| Total liabilities | 2,479,553 | 2,479,553 | 1,889,553 | |||||||||
| Accumulated deficit | (7,126,082 | ) | (7,126,082 | ) | (7,126,082 | ) | ||||||
| Total stockholders’ equity (deficit) | $ | (2,306,073 | ) | $ | 8,541,556 | $ | 9,208,031 | |||||
No. We will not issue fractional shares of common stock in the Rights Offering. We will only distribute Subscription Rights to acquire whole Units, rounded down to the nearest whole number of underlying common shares giving rise to such Subscription Rights. Any excess subscription payments received by the Subscription Agent will be returned within 10 business days after expiration of the Rights Offering, without interest or deduction.
The numberAdditionally, no fractional shares of common stock will be issued as a result of the exercise of Warrants. Instead, for any such fractional share that would otherwise have been issuable upon exercise of Warrants, we will round up to the next whole share.
What effect will the Rights Offering have on our outstanding common stock?
Assuming no other transactions by us involving our capital stock prior to the expiration of the Rights Offering, and if the Rights Offering is fully subscribed, upon consummation of the Rights Offering we will have shares of common stock issued and outstanding, actual, pro forma (reflecting the conversions of Series A preferred stock and convertible notes) and pro forma as adjusted in the table above excludes 3,000,000Warrants to purchase an additional shares reserved for issuance under our 2018 Equity Incentive Plan, of which 384,167 shares (on a post-reverse split basis) have been granted to date in the form of restricted stock units, some of which remain subject to certain vesting conditions.
An investment in our common stock involves a high degree of risk. You should carefully consider the following risk factors and all the other information in this prospectus before you decide to buy our common stock. If any of the following risks related to our business actually occurs, our business, financial condition, operating results, and prospects would be adversely affected. The market price of our common stock could decline due to anyissued and outstanding, and [ ] Equity Interests in Diagnostics based on [16,894,893] shares of these risks and uncertainties related to our business, or related to an investment in our common stock and you may lose part or all1,466,155 warrants outstanding as of your investment.[ ], 2024. The exact number of shares of common stock, Warrants and Equity Interests that we will issue in this offering will depend on the number of Units that are subscribed for in the Rights Offering.
How was the Subscription Price determined?
Risks Related to Our Business, Financial Condition and Capital Requirements
Our funds are limited and our independent auditing firm issued a going concern opinion related to our audit forIn determining the year ended December 31, 2021.
To date, we have been funded by investments from private investors and government contracts obtained fromSubscription Price, the National Institutes of Health (NIH) for performing research. While this has allowed us to complete a Phase I clinical trial for Ropidoxuridine and a pre-clinical trial for our HDAC inhibitor platform, we have not yet completed our clinical trials, will require additional funds to reach such milestone, and do not know if any of our products will ever achieve commercial viability. As such, to date we have no product revenue and our independent auditors, in their report dated June 3, 2022, have expressed doubt about our ability to continue as a going concern.
Our success is primarily dependent ondirectors considered, among other things, the successful development, regulatory approval and commercialization of our product candidates, all of which are in the early stages of development.
We currently have one clinical stage product candidate in the early stages of development. Ropidoxuridine has undergone an SBIR funded Phase 1 clinical trial at Lifespan/Rhode Island Hospital. We also have an HDAC inhibitor small molecule platform. The three lead drug candidate molecules are in preclinical phases of development. none of our product candidates have gained marketing approval for sale in the United States or any other country, and we cannot guarantee that we will ever have marketable products. To date, we have invested substantially all of our efforts and financial resources in the research and development and commercial planning for our current product candidate and our HDAC small molecule delivery platform. Our near-term prospects, including our ability to finance our Company and generate revenue, as well as our future growth, will depend heavily on the development, marketing approval and commercialization of our product candidates. The clinical and commercial success of product candidates will depend on a number of factors, including the following:
Many of the above-listed factors are beyond our control. Accordingly, we cannot assure you that we will ever be able to generate revenue through the sale of our product candidates. Any one of these factors or other factors discussed in this prospectus could affect our ability to commercialize product candidates, which could impact our ability to earn sufficient revenues to transition from a developmental stage company and continue our business. If we do not obtain marketing approval of and commercialization of our product candidates, or are significantly delayed in doing so, our business will be materially harmed. We have a limited operating history and have incurred significant losses since our inception, and we anticipate that we will continue to incur losses for the foreseeable future and may never achieve or maintain profitability.
We are a Phase I clinical stage pharmaceutical company with a limited operating history upon which you can evaluate our business and prospects. Specialty pharmaceutical product development is a highly speculative undertaking and involves a substantial degree of risk. We do not currently have any product candidates in advanced clinical trials or approved for sale, and we continue to incur significant research and development and general and administrative expenses related to our operations. In addition, we have limited experience and have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the specialty pharmaceutical industry. We have not generated any revenue and have incurred losses in each year since our founding in December 2012. Our accumulated deficit as of June 30, 2022 was $7,126,082. We expect to continue to incur significant losses for the foreseeable future. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
We currently have no source of product sales revenue.
We have not generated any revenues from commercial sales of our product candidates. Our ability to generate product revenue depends upon our ability to develop and commercialize products, including any of our current product candidates or other product candidates that we may develop, in-license or acquire in the future. We do not anticipate generating revenue from the sale of products for the foreseeable future. Our ability to generate future product revenue from our current or future product candidates also depends on a number of additional factors, including our ability to:
In addition, because of the numerous risks and uncertainties associated with clinical product development, including that our product candidates may not advance through development or achieve the endpoints of applicable clinical trials, we are unable to predict the timing or amount of any potential future product sales revenues. Our expenses also could increase beyond expectations if we decide to or are required by the FDA, or comparable foreign regulatory authorities, to perform studies or trials in addition to those that we currently anticipate. Even if we complete the development and regulatory processes described above, we anticipate incurring significant costs associated with launching and commercializing these products.
The market may not be receptive to our product candidates based on our novel therapeutic modality, and we may not generate any future revenue from the sale or licensing of product candidates.
Even if approval is obtained for a product candidate, we may not generate or sustain revenue from sales of the product due to factors such as whether the product can be sold at a competitive cost and otherwise accepted in the market. The product candidates that we are developing are based on new delivery platform therapeutic approaches (there currently is no drug which has FDA approval for indications of radiation sensitization). Market participants with significant influence over acceptance of new treatments, such as physicians and third-party payors, may not accept our delivery platform, and we may not be able to convince the medical community and third-party payors to accept and use, or to provide favorable reimbursement for, any product candidates developed by us. Market acceptance of our product candidates will depend on, among other factors:
| ● |
We will require substantial additional financing in order to obtain marketing approval of our product candidates and commercialize our product candidates; a failure to obtain this necessary capital when needed on acceptable terms, or at all, could force us to delay, limit, reduce or terminate our product development, other operations or commercialization efforts.
Since our inception, substantially all of our resources have been dedicated to the preclinical and clinical development of our HDAC small molecule delivery platform and our initial product candidate, Ropidoxuridine. Our capital needs to date have been met by contributions from existing shareholders, as well as through private offerings of our securities and our SBIR contracts. We believe that we will continue to expend substantial resources for the foreseeable future on the completion of clinical development and regulatory preparedness of our product candidates, preparations for a commercial launch of our product candidates, if approved, and development of any other current or future product candidates we may choose to further develop. These expenditures will include costs associated with research and development, conducting preclinical studies and clinical trials, obtaining marketing approvals, and, if we are not able to enter into planned collaborations, manufacturing and supply as well as marketing and selling any products approved for sale. In addition, other unanticipated costs may arise. Because the outcome of any drug development process is highly uncertain, we cannot reasonably estimate the actual amounts necessary to complete the development and commercialization of our current product candidates, if approved, or future product candidates, if any.
We estimate that our net proceeds from this Offering will be approximately $8,663,577, if all 1,225,888 units, with each unit consisting of (i) one share of common stock and (ii) a warrant to purchase one share of common stock, offered hereby, are offered and sold, less offering expenses payable by us. We believe that such proceeds together with our existing capital resources, will be sufficient to fund our operations through 2024. However, our operating plan may change as a result of factors currently unknown to us, and we may need to seek additional funds sooner than planned, through public or private equity or debt financings or other sources, such as strategic collaborations. Such financing may result in dilution to shareholders, imposition of debt covenants and repayment obligations, or other restrictions that may adversely affect our business. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans.
Our future capital requirements depend on many factors, including:
Additional funds may not be available when we need them, on terms that are acceptable to us, or at all. If adequate funds are not available to us on a timely basis, we may be required to:
Raising additional capital may cause dilution to our existing shareholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
We may seek additional capital through a combination of public and private equity offerings, debt financings, strategic collaborations and alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a shareholder. The incurrence of indebtedness would result in increased fixed payment obligations and could involve certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. If we raise additional funds through strategic collaborations and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates or grant licenses on terms unfavorable to us.
Unstable market and economic conditions caused by the ongoing conflict between the Ukraine and Russia, as well as the ongoing COVID-19 pandemic, may have serious adverse consequences on our business, financial condition and results of operations.
The global economy, including credit and financial markets, has experienced extreme volatility and disruptions as a result of the ongoing conflict between the Ukraine and Russia, as well as challenges arising from the ongoing COVID-19 pandemic, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in unemployment rates, increases in inflation rates and uncertainty about economic stability. Any such volatility and disruptions may have adverse consequences on us or the third parties upon whom we rely.
Our product candidates are in the early stages of development and may fail in development or suffer delays that materially adversely affect their commercial viability.
We have no products on the market and all of our product candidates are in the early stages of development. Our ability to achieve and sustain profitability depends on obtaining regulatory approvals, including institutional review board (“IRB”) approval, for and commercializing our product candidates, either alone or with third parties. Before obtaining regulatory approval for the commercial distribution of our product candidates, we or one of our collaborators must conduct extensive preclinical tests and clinical trials to demonstrate the safety and efficacy in humans of our product candidates, the final determination of which rests solely in the authority of the FDA. Preclinical testing and clinical trials are expensive, difficult to design and implement, can take many years to complete and are uncertain as to outcome. The start or end of a clinical study is often delayed or halted due to changing regulatory requirements, manufacturing challenges, required clinical trial administrative actions, slower than anticipated patient enrollment, changing standards of care, availability or prevalence of use of a comparative drug or required prior therapy, clinical outcomes or financial constraints. For instance, delays or difficulties in patient enrollment or difficulties in retaining trial participants can result in increased costs, longer development times or termination of a clinical trial. Clinical trials of a new product candidate require the enrollment of a sufficient number of patients, including patients who are suffering from the disease the product candidate is intended to treat and who meet other eligibility criteria. Rates of patient enrollment are affected by many factors, including the size of the patient population, the eligibility criteria for the clinical trial, the age and condition of the patients, the stage and severity of disease, the nature of the protocol, the proximity of patients to clinical sites and the availability of effective treatments for the relevant disease.
A product candidate can unexpectedly fail at any stage of preclinical and clinical development. The historical failure rate for product candidates is high due to scientific feasibility, lack of quality and effectiveness, changing standards of medical care and other variables. The results from preclinical testing or early clinical trials of a product candidate may not predict the results that will be obtained in later phase clinical trials of the product candidate. We, the FDA or other applicable regulatory authorities may suspend clinical trials of a product candidate at any time for various reasons, including a belief that subjects participating in such trials are being exposed to unacceptable health risks or adverse side effects. We may not have the financial resources to continue development of, or to enter into collaborations for, a product candidate if we experience any problems or other unforeseen events that delay or prevent regulatory approval of, or our ability to commercialize, product candidates, including:
If third parties on which we depend to conduct our preclinical studies, or any future clinical trials, do not perform as contractually required, fail to satisfy regulatory or legal requirements or miss expected deadlines, our development program could be delayed with materially adverse effects on our business, financial condition, results of operations and prospects.
We are relying on third party collaborators to conduct our efficacy clinical trials for Ropidoxuridine and plan to rely on third party clinical investigators, contract research organizations (“CROs”), clinical data management organizations and consultants to design, conduct, supervise and monitor preclinical studies of our product candidates and will do the same for any clinical trials. Because we plan to largely rely on third parties and do not have the ability to conduct preclinical studies or clinical trials independently, we have less control over the timing, quality and other aspects of preclinical studies and clinical trials than we would if we conducted them on our own. These investigators, CROs, and consultants are not our employees and we have limited control over the amount of time and resources that they dedicate to our programs. These third parties may have contractual relationships with other entities, some of which may be our competitors, which may draw time and resources from our programs. The third parties with whom we contract might not be diligent, careful or timely in conducting our preclinical studies or clinical trials, resulting in the preclinical studies or clinical trials being delayed or unsuccessful.
If we cannot contract with acceptable third parties on commercially reasonable terms, or at all, or if these third parties do not carry out their contractual duties, satisfy legal and regulatory requirements for the conduct of preclinical studies or clinical trials or meet expected deadlines, our clinical development programs could be delayed and otherwise adversely affected. In all events, we are responsible for ensuring that each of our preclinical studies and clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. The FDA requires clinical trials to be conducted in accordance with good clinical practices, including for conducting, recording and reporting the results of preclinical studies and clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of clinical trial participants are protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements. Any such event could have a material adverse effect on our business, financial condition, results of operations and/or prospects.
Because we rely on third party manufacturing and supply partners, our supply of research and development, preclinical and clinical development materials may become limited or interrupted or may not be of satisfactory quantity or quality.
We rely on third party supply and manufacturing partners to supply the materials and components for, and manufacture, our research and development, preclinical and clinical trial drug supplies. We do not own manufacturing facilities or supply sources for such components and materials. There can be no assurance that our supply of research and development, preclinical and clinical development drugs and other materials will not be limited, interrupted, restricted in certain geographic regions or of satisfactory quality or continue to be available at acceptable prices. In particular, any replacement of any drug product formulation manufacturer we may use could require significant effort and expertise in the event there are a limited number of qualified replacements for a particular product candidate.
The manufacturing process for a product candidate is subject to FDA and foreign regulatory authority review. Suppliers and manufacturers must meet applicable manufacturing requirements and undergo rigorous facility and process validation tests required by regulatory authorities in order to comply with regulatory standards, such as Current Good Manufacturing Practice (or CGMP). In the event that any of our suppliers or manufacturers fail to comply with such requirements or to perform its obligations to us in relation to quality, timing or otherwise, or if our supply of components or other materials becomes limited or interrupted for other reasons, we may be forced to manufacture the materials ourselves, for which we currently do not have the capabilities or resources, or enter into an agreement with another third party, which we may not be able to do on reasonable terms, if at all. In some cases, the technical skills or technology required to manufacture our product candidates may be unique or proprietary to the original manufacturer, and we may have difficulty, or there may be contractual restrictions prohibiting us from, transferring such skills or technology to another third party and a feasible alternative may not exist. These factors would increase our reliance on such manufacturer or require us to obtain a license from such manufacturer in order to have another third party manufacture our product candidates. If we are required to change manufacturers for any reason, we will be required to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. The delays associated with the verification of a new manufacturer could negatively affect our ability to develop product candidates in a timely manner or within budget.
We expect to continue to rely on third party manufacturers if we receive regulatory approval for any product candidate. To the extent that we have existing or future manufacturing arrangements with third parties, we will depend on these third parties to perform their obligations in a timely manner consistent with contractual and regulatory requirements, including those related to quality control and assurance. If we are unable to obtain or maintain third-party manufacturing for product candidates, or to do so on commercially reasonable terms, we may not be able to fully develop and commercialize our product candidates. Our or a third party’s failure to execute on our manufacturing requirements could adversely affect our business in a number of ways, including:
We may be unsuccessful in engaging in strategic transactions which could adversely affect our ability to develop and commercialize product candidates, impact our cash position, increase our expense and present significant distractions to our management.
From time to time, we may consider strategic transactions, such as collaborations, acquisitions of companies, asset purchases and out- or in- licensing of product candidates or technologies. In particular, we will evaluate and, if strategically attractive, seek to enter into additional collaborations, including with major biotechnology or pharmaceutical companies to complete development and marketing of our product candidates, if approved. The competition for collaborators is intense, and the negotiation process is time-consuming and complex. Any proposed collaboration may be on terms that are not optimal for us, and we may not be able to maintain any new or existing collaboration if, for example, development or approval of a product candidate is delayed, sales of an approved product candidate do not meet expectations or the collaborator terminates the collaboration. Any such collaboration, or other strategic transaction, may require us to incur non-recurring or other charges, increase our near- and long-term expenditures and pose significant integration or implementation challenges or disrupt our management or business. These transactions would entail numerous operational and financial risks, including exposure to unknown liabilities, disruption of our business and diversion of our management’s time and attention in order to manage a collaboration or develop acquired products, product candidates or technologies, incurrence of substantial debt or dilutive issuances of equity securities to pay transaction consideration or costs, higher than expected collaboration, acquisition or integration costs, write-downs of assets or goodwill or impairment charges, increased amortization expenses, difficulty and cost in facilitating the collaboration or combining the operations and personnel of any acquired business, impairment of relationships with key suppliers, manufacturers or customers of any acquired business due to changes in management and ownership and the inability to retain key employees of any acquired business. Accordingly, although there can be no assurance that we will undertake or successfully complete any transactions of the nature described above, any transactions that we do complete may be subject to the foregoing or other risks and have a material adverse effect on our business, results of operations, financial condition and prospects. Conversely, any failure to enter into any collaboration or other strategic transaction that would be beneficial to us could delay the development and potential commercialization of our product candidates and have a negative impact on the competitiveness of any product candidate that reaches market.
We face competition from entities that have developed or may develop product candidates for our target disease indications, including companies developing novel treatments and technology platforms based on modalities and technology similar to ours. If these companies develop technologies or product candidates more rapidly than we do or their technologies, including delivery technologies, are more effective, our ability to develop and commercialize product candidates may be adversely affected.
The development and commercialization of drugs is highly competitive. We compete with a variety of multinational pharmaceutical companies and specialized biotechnology companies, as well as with universities and other research institutions which are developing new technology. Our competitors have developed, are developing or will develop product candidates and processes competitive with our product candidates. Competitive therapeutic treatments include those that have already been approved and accepted by the medical community and any new treatments that enter the market. We believe that a significant number of products are currently under development, and may become commercially available in the future, for the treatment of conditions for which we may try to develop product candidates.
Many of our competitors have significantly greater financial, technical, manufacturing, marketing, sales and supply resources or experience than we have. If we obtain approval for any product candidate, we will face competition based on many different factors, including the quality and effectiveness of our products, the ease with which our products can be administered and the extent to which patients accept relatively new routes of administration, the timing and scope of regulatory approvals for these products, the availability and cost of manufacturing, marketing and sales capabilities, price, reimbursement coverage and patent position. Competing products could present superior treatment alternatives, including by being more effective, safer, less expensive or marketed and sold more effectively than any products we may develop. Competitive products may make any products we develop obsolete or noncompetitive before we recover the expense of developing and commercializing our product candidates. Such competitors could also recruit our employees, which could negatively impact our level of expertise and our ability to execute our business plan.
Any inability to attract and retain qualified key management and technical personnel would impair our ability to implement our business plan.
Our success largely depends on the continued service of certain key management and other specialized personnel, including Anatoly Dritschilo, M.D., our Chief Executive Officer, Mira Jung, Ph.D., our Chief Scientific Officer for Biology, Michael Vander Hoek, our Chief Financial Officer and Vice President Operations and Regulatory, and Peter Dritschilo, our President and Chief Operating Officer. The loss of one or more members of our management team or other key employees or advisors could delay our research and development programs and materially harm our business, financial condition, results of operations and prospects. The relationships that our key managers have cultivated within our industry make us particularly dependent upon their continued employment with us. We are dependent on the continued service of our technical personnel because of the highly technical nature of our product candidates and technologies and the specialized nature of the regulatory approval process. Because our management team and key employees are not obligated to provide us with continued service, they could terminate their employment with us at any time without penalty. We do not maintain key person life insurance policies on any of our management team members or key employees. Our future success will depend in large part on our continued ability to attract and retain other highly qualified scientific, technical and management personnel, as well as personnel with expertise in clinical testing, manufacturing, governmental regulation and commercialization. We face competition for personnel from other companies, universities, public and private research institutions, government entities and other organizations.
If our product candidates advance into Phase II and Phase III clinical trials, we may experience difficulties in managing our growth and expanding our operations.
We have limited experience in drug development and have not begun clinical trials for any of our product candidates, other than a Phase 1 clinical trial for Ropidoxuridine. As our product candidates enter and advance through preclinical studies and any clinical trials, we will need to expand our development, regulatory and manufacturing capabilities or contract with other organizations to provide these capabilities for us. In the future, we expect to have to manage additional relationships with collaborators or partners, suppliers and other organizations. Our ability to manage our operations and future growth will require us to continue to improve our operational, financial and management controls, reporting systems and procedures. We may not be able to implement improvements to our management information and control systems in an efficient or timely manner and may discover deficiencies in existing systems and controls.
If any of our product candidates are approved for marketing and commercialization and we are unable to develop sales, marketing and distribution capabilities on our own or enter into agreements with third parties to perform these functions on acceptable terms, we will be unable to commercialize any such future products.
We currently have no sales, marketing or distribution capabilities or experience. If any of our product candidates is approved, we plan to enter into collaborations with third parties to sell, market and distribute our products. In the alternative, we would have to develop internal sales, marketing and distribution capabilities to commercialize any approved product, which would be expensive and time-consuming, or, as is more likely, enter into collaborations with third parties to perform these services. If we rely on third parties with sales, marketing and distribution capabilities to market our products or decide to co-promote products with collaborators, we will need to establish and maintain marketing and distribution arrangements with third parties, and there can be no assurance that we will be able to enter into such arrangements on acceptable terms, if, at all. In entering into third-party marketing or distribution arrangements, any revenue we receive will depend upon the efforts of the third parties and there can be no assurance that such third parties will establish adequate sales and distribution capabilities or be successful in gaining market acceptance of any approved product. If we decide to market our products directly, we will need to commit significant financial and managerial resources to develop a marketing and sales force with technical expertise and supporting distribution, administration and compliance capabilities. If we are not able to commercialize any product approved in the future, either on our own or through third parties, our business, financial condition, results of operations and prospects could be materially adversely affected.
If we fail to comply with U.S. and foreign regulatory requirements, regulatory authorities could limit or withdraw any marketing or commercialization approvals we may receive and subject us to other penalties that could materially harm our business.
Even if we receive marketing and commercialization approval of a product candidate, there can be no assurance we will not be subject to future or continuing regulatory review, including in relation to adverse patient experiences with the product and clinical results that are reported after a product is made commercially available, both in the U.S. and any foreign jurisdiction in which we seek regulatory approval. The FDA has significant post-market authority, including the authority to require labeling changes based on new safety information and to require post-market studies or clinical trials to evaluate safety risks related to the use of a product or to require withdrawal of the product from the market. The FDA also has the authority to require a risk evaluation and mitigation strategies (“REMS”) plan after approval, which may impose further requirements or restrictions on the distribution or use of an approved drug. The manufacturer and manufacturing facilities we use to make a future product, if any, will also be subject to periodic review and inspection by the FDA and other regulatory agencies, including for continued compliance with CGMP requirements. The discovery of any new or previously unknown problems with our third-party manufacturers, manufacturing processes or facilities may result in restrictions on the product, manufacturer or facility, including withdrawal of the product from the market. If we rely on third-party manufacturers, we will not have control over compliance with applicable rules and regulations by such manufacturers. Any product promotion and advertising will also be subject to regulatory requirements and continuing regulatory review. If we or our collaborators, manufacturers or service providers fail to comply with applicable continuing regulatory requirements in the U.S. or foreign jurisdictions in which we seek to market our products, we or they may be subject to, among other things, fines, warning letters, holds on clinical trials, refusal by the FDA to approve pending applications or supplements to approved applications, suspension or withdrawal of regulatory approval, product recalls and seizures, refusal to permit the import or export of products, operating restrictions, injunction, civil penalties and criminal prosecution.
Our business entails a significant risk of product liability and our ability to obtain sufficient insurance coverage could have a material effect on our business, financial condition, results of operations or prospects.
Our business exposes us to significant product liability risks inherent in the development, testing, manufacturing and marketing of therapeutic treatments. Product liability claims could delay or prevent completion of our development programs. If we succeed in marketing products, such claims could result in an FDA investigation of the quality and effectiveness of our products, our manufacturing processes and facilities or our marketing programs and potentially a recall of our products or more serious enforcement action, limitations on the approved indications for which they may be used or suspension or withdrawal of approvals. Regardless of the merits or eventual outcome, liability claims may also result in decreased demand for our products, injury to our reputation, costs to defend the related litigation, a diversion of management’s time and our resources, substantial monetary awards to trial participants or patients and a decline in our stock price. We currently have product liability insurance that we believe is appropriate for our stage of development and may need to obtain higher levels prior to marketing any of our product candidates. Any insurance we have or may obtain may not provide sufficient coverage against potential liabilities. Furthermore, clinical trial and product liability insurance is becoming increasingly expensive. As a result, we may be unable to obtain sufficient insurance at a reasonable cost to protect us against losses caused by product liability claims that could have a material adverse effect on our business.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with FDA regulations, provide accurate information to the FDA, comply with manufacturing standards we may establish, comply with federal and state healthcare fraud and abuse laws and regulations, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. While we make an effort to maintain strict employee work processes and oversight, employee misconduct could expose us to liability through the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. Furthermore, it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Our internal computer systems, or those of our CROs or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs.
Despite the implementation of cyber security measures, our internal computer systems and those of our CROs and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. Such events could cause interruptions of our operations. For example, the loss of preclinical data or data from any future clinical trial involving our product candidates could result in delays in our development and regulatory filing efforts and significantly increase our costs. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the development of our product candidates could be delayed.
Our proprietary information, or that of our customers, suppliers and business partners, may be lost or we may suffer security breaches.
In the ordinary course of our business, we collect and store sensitive data, including intellectual property, clinical trial data, our proprietary business information and that of our customers, suppliers and business partners, and personally identifiable information of our customers, clinical trial subjects and employees, in our data centers and on our networks. The secure processing, maintenance and transmission of this information is critical to our operations. Despite our security measures, our information technology and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Although to our knowledge we have not experienced any such material security breach to date, any such breach could compromise our network, or the networks of our CROs or other third party service providers, and the information stored there could be accessed, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, regulatory penalties, disrupt our operations, damage our reputation, and cause a loss of confidence in our products and our ability to conduct clinical trials, which could adversely affect our business and reputation and lead to delays in gaining regulatory approvals for our drugs. Although we maintain business interruption insurance coverage, our insurance might not cover all losses from any future breaches of our systems.
Failure of our information technology systems could significantly disrupt the operation of our business.
Our business increasingly depends on the use of information technologies, which means that certain key areas such as research and development, production and sales are to a large extent dependent on our information systems or those of third-party providers. Our ability to execute our business plan and to comply with regulatory requirements with respect to data control and data integrity, depends, in part, on the continued and uninterrupted performance of our information technology systems, or IT systems and the IT systems supplied by third-party service providers. These systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and backup measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we and our third-party service providers have taken to prevent unanticipated problems that could affect our IT systems, sustained or repeated system failures or problems arising during the upgrade of any of our IT systems that interrupt our ability to generate and maintain data, and in particular to operate our proprietary technology platform, could adversely affect our ability to operate our business.
If we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected.
Our research, development and manufacturing involve the use of hazardous materials and various chemicals. We maintain quantities of various flammable and toxic chemicals in our facilities in Gaithersburg, Maryland that are required for our research, development and manufacturing activities. We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. We believe our procedures for storing, handling and disposing these materials in our Gaithersburg facilities comply with the relevant guidelines of Gaithersburg, the State of Maryland and the Occupational Safety and Health Administration of the U.S. Department of Labor. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards mandated by applicable regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. If an accident occurs, we could be held liable for resulting damages, which could be substantial. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of animals and biohazardous materials. Although we maintain workers’ compensation insurance to cover us for costs and expenses, we may incur due to injuries to our employees resulting from the use of these materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological or hazardous materials. Additional federal, state and local laws and regulations affecting our operations may be adopted in the future. We may incur substantial costs to comply with, and substantial fines or penalties if we violate any of these laws or regulations.
Our information technology systems could face serious disruptions that could adversely affect our business.
Our information technology and other internal infrastructure systems, including corporate firewalls, servers, leased lines and connection to the Internet, face the risk of systemic failure that could disrupt our operations. A significant disruption in the availability of our information technology and other internal infrastructure systems could cause interruptions in our collaborations with our partners and delays in our research and development work.
Changes in accounting rules and regulations, or interpretations thereof, could result in unfavorable accounting charges or require us to change our compensation policies.
Accounting methods and policies for pharmaceutical companies, including policies governing revenue recognition, research and development and related expenses and accounting for stock-based compensation are subject to review, interpretation and guidance from relevant accounting authorities, including the SEC. Changes to accounting methods or policies, or interpretations thereof, may require us to reclassify, restate or otherwise change or revise our financial statements, including those contained in this prospectus.
Risks Related to Our Intellectual Property
If we are not able to obtain and enforce patent protection for our technologies or product candidates, development and commercialization of our product candidates may be adversely affected.
Our success depends in part on our ability to obtain and maintain patents and other forms of intellectual property rights, including in-licenses of intellectual property rights of others, for our product candidates, methods used to manufacture our product candidates and methods for treating patients using our product candidates, as well as our ability to preserve our trade secrets, to prevent third parties from infringing upon our proprietary rights and to operate without infringing upon the proprietary rights of others. As of the date of this prospectus, we have filed five patent applications with the U.S. Patent and Trademark Office (the “USPTO”) with respect to various aspects of our HDAC inhibitor small molecule delivery platform and Ropidoxuridine, our lead product candidate. However, we may not be able to apply for patents on certain aspects of our product candidates or delivery technologies in a timely fashion or at all. To date, four US patents and two European patents have been granted. There is no guarantee that any of our pending patent applications will result in issued or granted patents, that any of our issued, granted or licensed patents will not later be found to be invalid or unenforceable or that any issued, granted or licensed patents will include claims that are sufficiently broad to cover our product candidates or delivery technologies or to provide meaningful protection from our competitors. Moreover, the patent position of specialty pharmaceutical companies can be highly uncertain because it involves complex legal and factual questions. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our current and future proprietary technology and product candidates are covered by valid and enforceable patents or are effectively maintained as trade secrets. If third parties disclose or misappropriate our proprietary rights, it may materially and adversely impact our position in the market.
The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other requirements during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case. The standards applied by the USPTO and foreign patent offices in granting patents are not always applied uniformly or predictably. For example, there is no uniform worldwide policy regarding patentable subject matter or the scope of claims allowable in pharmaceutical patents. As such, we do not know the degree of future protection that we will have on our proprietary products and technology. While we will endeavor to try to protect our product candidates with intellectual property rights such as patents, as appropriate, the process of obtaining patents is time-consuming, expensive and sometimes unpredictable.
We may decide for business reasons to no longer pursue or to abandon certain intellectual property rights in the US or elsewhere, including due to non-cooperation of inventors or owners of such intellectual property, prior art, or scope of protection, or for other reasons.
Once granted, patents may remain open to opposition, interference, re-examination, post-grant review, inter partes review, nullification or derivation action in court or before patent offices or similar proceedings for a given period after allowance or grant, during which time third parties can raise objections against such initial grant. In the course of such proceedings, which may continue for a protracted period of time, the patent owner may be compelled to limit the scope of the allowed or granted claims thus attacked, or may lose the allowed or granted claims altogether. In addition, there can be no assurance that:
We intend to license patent rights from third-party owners or licensees. If such owners or licensees do not properly or successfully obtain, maintain or enforce the patents underlying such licenses, or if they retain or license to others any competing rights, our competitive position and business prospects may be adversely affected. We may not be able to protect our intellectual property rights throughout the world.
Obtaining a valid and enforceable issued or granted patent covering our technology in the U.S. and worldwide can be extremely costly. In jurisdictions where we have not obtained patent protection, competitors may use our technology to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but where it is more difficult to enforce a patent as compared to the U.S. Competitor products may compete with our future products in jurisdictions where we do not have issued or granted patents or where our issued or granted patent claims or other intellectual property rights are not sufficient to prevent competitor activities in these jurisdictions. The legal systems of certain countries, particularly certain developing countries, make it difficult to enforce patents and such countries may not recognize other types of intellectual property protection, particularly that relating to biopharmaceuticals. This could make it difficult for us to prevent the infringement of patents or marketing of competing products in violation of our proprietary rights generally in certain jurisdictions. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
We generally file a provisional patent application first (a priority filing) at the USPTO. A U.S. utility application and international application under the Patent Cooperation Treaty (PCT) are usually filed within twelve months after the priority filing. Based on the PCT filing, national and regional patent applications may be filed in the European Union, Japan, Australia and Canada and other countries. We have so far not filed for patent protection in all national and regional jurisdictions where such protection may be available. In addition, we may decide to abandon national and regional patent applications before grant. Finally, the grant proceeding of each national or regional patent is an independent proceeding which may lead to situations in which applications might in some jurisdictions be refused by the relevant registration authorities, while granted by others. It is also quite common that depending on the country, various scopes of patent protection may be granted on the same product candidate or technology. The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws in the U.S., and many companies have encountered significant difficulties in protecting and defending such rights in such jurisdictions. If we or our licensors encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for our business in such jurisdictions, the value of these rights may be diminished, and we may face additional competition from others in those jurisdictions. Many countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we or any of our licensors are forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position in the relevant jurisdiction may be impaired and our business and results of operations may be adversely affected.
We or our licensors, or any future collaborators or a strategic partners may become subject to third party claims or litigation alleging infringement of patents or other proprietary rights or seeking to invalidate patents or other proprietary rights, and we may need to resort to litigation to protect or enforce our patents or other proprietary rights, all of which could be costly, time consuming, delay or prevent the development and commercialization of our product candidates, or put our patents and other proprietary rights at risk.
We or our licensors, or any future collaborators or strategic partners may be subject to third-party claims for infringement or misappropriation of patent or other proprietary rights. We are generally obligated under our license or collaboration agreements to indemnify and hold harmless our licensors or collaborator for damages arising from intellectual property infringement by us. If we or our licensors, or any future collaborators or strategic partners are found to infringe a third-party patent or other intellectual property rights, we could be required to pay damages, potentially including treble damages, if we are found to have willfully infringed. In addition, we or our licensors, collaborators or any future strategic partners may choose to seek, or be required to seek, a license from a third party, which may not be available on acceptable terms, if at all. Even if a license can be obtained on acceptable terms, the rights may be non-exclusive, which could give our competitors access to the same technology or intellectual property rights licensed to us. If we fail to obtain a required license, we or our collaborator, or any future collaborator, may be unable to effectively market product candidates based on our technology, which could limit our ability to generate revenue or achieve profitability and possibly prevent us from generating revenue sufficient to sustain our operations. In addition, we may find it necessary to pursue claims or initiate lawsuits to protect or enforce our patent or other intellectual property rights. The cost to us in defending or initiating any litigation or other proceeding relating to patent or other proprietary rights, even if resolved in our favor, could be substantial, and litigation would divert our management’s attention. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could delay our research and development efforts and limit our ability to continue our operations.
If we were to initiate legal proceedings against a third party to enforce a patent covering one of our products or our technology, the defendant could counterclaim that our patent is invalid or unenforceable. In patent litigation in the U.S., defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, for example, lack of novelty, obviousness or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. The outcome following legal assertions of invalidity and unenforceability during patent litigation is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on one or more of our products or certain aspects of our platform technology. Such a loss of patent protection could have a material adverse impact on our business. Patents and other intellectual property rights also will not protect our technology if competitors design around our protected technology without legally infringing our patents or other intellectual property rights.
Intellectual property rights of third parties could adversely affect our ability to commercialize our product candidates, and we might be required to litigate or obtain licenses from third parties in order to develop or market our product candidates. Such litigation or licenses could be costly or not available on commercially reasonable terms.
Our competitive position may suffer if patents issued to third parties or other third-party intellectual property rights cover our products or elements thereof, or our manufacture or uses relevant to our development plans. In such cases, we may not be in a position to develop or commercialize products or product candidates unless we successfully pursue litigation to nullify or invalidate the third-party intellectual property right concerned, or enter into a license agreement with the intellectual property right holder, if available on commercially reasonable terms.
Third party intellectual property right holders may also actively bring infringement claims against us. We cannot guarantee that we will be able to successfully settle or otherwise resolve such infringement claims. If we are unable to successfully settle future claims on terms acceptable to us, we may be required to engage in or continue costly, unpredictable and time-consuming litigation and may be prevented from or experience substantial delays in marketing our products. If we fail in any such dispute, in addition to being forced to pay damages, we may be temporarily or permanently prohibited from commercializing any of our product candidates that are held to be infringing. We might, if possible, also be forced to redesign product candidates so that we no longer infringe the third-party intellectual property rights. Any of these events, even if we were ultimately to prevail, could require us to divert substantial financial and management resources that we would otherwise be able to devote to our business.
If we fail to comply with our obligations under any license, collaboration or other agreements, we may be required to pay damages and could lose intellectual property rights that are necessary for developing and protecting our product candidates and delivery technologies or we could lose certain rights to grant sublicenses.
Our current licenses impose, and any future licenses we enter into are likely to impose, various development, commercialization, funding, milestone, royalty, diligence, sublicensing, insurance, patent prosecution and enforcement, and other obligations on us. If we breach any of these obligations, or use the intellectual property licensed to us in an unauthorized manner, we may be required to pay damages and the licensor may have the right to terminate the license, which could result in us being unable to develop, manufacture and sell products that are covered by the licensed technology or enable a competitor to gain access to the licensed technology. Moreover, our licensors may own or control intellectual property that has not been licensed to us and, as a result, we may be subject to claims, regardless of their merit, that we are infringing or otherwise violating the licensor’s rights. In addition, while we cannot currently determine the amount of the royalty obligations we would be required to pay on sales of future products, if any, the amounts may be significant. The amount of our future royalty obligations will depend on the technology and intellectual property we use in products that we aim to develop and commercialize, if any. Therefore, even if we are able to develop and commercialize products, we may be unable to achieve or maintain profitability.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patent protection for certain aspects of our product candidates and delivery technologies, we also consider trade secrets, including confidential and unpatented know-how important to the maintenance of our competitive position. We protect trade secrets and confidential and unpatented know-how, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to such knowledge, such as our employees, corporate collaborators, outside scientific collaborators, CROs, contract manufacturers, consultants, advisors and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants that obligate them to maintain confidentiality and assign their inventions to us. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time- consuming, and the outcome is unpredictable. In addition, some courts in the U.S. and certain foreign jurisdictions are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
We may be subject to claims that we or our employees or consultants have wrongfully used or disclosed alleged trade secrets of our employees’ or consultants’ former employers or their clients. These claims may be costly to defend and if we do not successfully do so, we may be required to pay monetary damages and may lose valuable intellectual property rights or personnel.
Many of our employees were previously employed at universities or biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper our ability to commercialize, or prevent us from commercializing, our product candidates, which could severely harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names or may be forced to stop using these names, which we need for name recognition by potential partners or customers in our markets of interest. If we are unable to establish name recognition based on our trademarks and trade names, we may not be able to effectively compete and our business may be adversely affected.
Risks Related to Government Regulation and Product Approvals
We may be unable to obtain U.S. or foreign regulatory approval and, as a result, unable to commercialize our product candidates.
Our product candidates are subject to extensive governmental regulations relating to, among other things, research, testing, development, manufacturing, safety, efficacy, approval, recordkeeping, reporting, labeling, storage, packaging, advertising and promotion, pricing, marketing and distribution of drugs. Rigorous preclinical testing and clinical trials and an extensive regulatory approval process are required to be completed in the U.S. and in many foreign jurisdictions before a new drug can be marketed. Satisfaction of these and other regulatory requirements is costly, time consuming, uncertain and subject to unanticipated delays. It is possible that none of the product candidates we may develop will obtain the regulatory approvals necessary for us or our collaborators to begin selling them.
We have very limited experience in conducting and managing the clinical trials necessary to obtain regulatory approvals, including approval by the FDA. The time required to obtain FDA and other approvals is unpredictable but typically takes many years following the commencement of clinical trials, depending upon the type, complexity and novelty of the product candidate. The standards that the FDA and its foreign counterparts use when regulating us are not always applied predictably or uniformly and can change. Any analysis we perform of data from preclinical and clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. We may also encounter unexpected delays or increased costs due to new government regulations, for example, from future legislation or administrative action, or from changes in FDA policy during the period of product development, clinical trials and FDA regulatory review. It is impossible to predict whether legislative changes will be enacted, or whether FDA or foreign regulations, guidance or interpretations will be changed, or what the impact of such changes, if any, may be.
Any delay or failure in obtaining required approvals could have a material adverse effect on our ability to generate revenues from the particular product candidate for which we are seeking approval. Furthermore, any regulatory approval to market a product may be subject to limitations on the approved uses for which we may market the product or the labeling or other restrictions. In addition, the FDA has the authority to require a Risk Evaluation and Mitigation Strategy (REMS) plan as part of an NDA or biologics license application (BLA) or after approval, which may impose further requirements or restrictions on the distribution or use of an approved drug or biologic, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria and requiring treated patients to enroll in a registry. These limitations and restrictions may limit the size of the market for the product and affect reimbursement by third-party payors.
If we or our collaborators, manufacturers or service providers fail to comply with healthcare laws and regulations, we or they could be subject to enforcement actions, which could affect our ability to develop, market and sell our products and may harm our reputation.
We and our collaborators are subject to federal, state, and foreign healthcare laws and regulations pertaining to fraud and abuse and patients’ rights. These laws and regulations include:
If our operations are found to be in violation of any such requirements, we may be subject to penalties, including civil or criminal penalties, monetary damages, the curtailment or restructuring of our operations, loss of eligibility to obtain approvals from the FDA, or exclusion from participation in government contracting, healthcare reimbursement or other government programs, including Medicare and Medicaid, any of which could adversely our financial results. Although effective compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, these risks cannot be entirely eliminated. Any action against us for an alleged or suspected violation could cause us to incur significant legal expenses and could divert our management’s attention from the operation of our business, even if our defense is successful. In addition, achieving and sustaining compliance with applicable laws and regulations may be costly to us in terms of money, time and resources.
If we or our collaborators, manufacturers or service providers fail to comply with applicable federal, state or foreign laws or regulations, we could be subject to enforcement actions, which could affect our ability to develop, market and sell our products successfully and could harm our reputation and lead to reduced acceptance of our products by the market. These enforcement actions include, among others:
Any drugs we develop may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, thereby harming our business.
The regulations that govern marketing approvals, pricing and reimbursement for new drugs vary widely from country to country. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. Although we intend to monitor these regulations, our programs are currently in the early stages of development and we will not be able to assess the impact of price regulations for a number of years. As a result, we might obtain regulatory approval for a product in a particular country, but then be subject to price regulations that delay our commercial launch of the product and negatively impact the revenues we are able to generate from the sale of the product in that country.
Our ability to commercialize any products also will depend in part on the extent to which reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Even if we succeed in bringing one or more products to the market, these products may not be considered cost-effective, and the amount reimbursed for any products may be insufficient to allow us to sell our products on a competitive basis. Because our programs are in the early stages of development, we are unable at this time to determine their cost effectiveness or the likely level or method of reimbursement. Increasingly, the third-party payors who reimburse patients or healthcare providers, such as government and private insurance plans, are requiring that drug companies provide them with predetermined discounts from listhistorical trading prices and are seeking to reduce the prices charged or the amounts reimbursed for pharmaceutical products. If the price we are able to charge for any products we develop, or the reimbursement provided for such products, is inadequate in light of our development and other costs, our return on investment could be adversely affected.
Our current product candidates will need to be administered under the supervision of a physician on an outpatient basis. Under currently applicable U.S. law, certain drugs that are not usually self-administered (including injectable drugs) may be eligible for coverage under the Medicare Part B program if:
There may be significant delays in obtaining coverage for newly-approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA. Moreover, eligibility for coverage does not imply that any drug will be reimbursed in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim payments for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement may be based on payments allowed for lower- cost drugs that are already reimbursed, may be incorporated into existing payments for other services and may reflect budgetary constraints or imperfections in Medicare data. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the U.S. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement rates. Our inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for new drugs that we develop and for which we obtain regulatory approval could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our financial condition.
We believe that the efforts of governments and third-party payors to contain or reduce the cost of healthcare and legislative and regulatory proposals to broaden the availability of healthcare will continue to affect the business and financial condition of pharmaceutical and biopharmaceutical companies. A number of legislative and regulatory changes in the healthcare system in the U.S. and other major healthcare markets have been proposed in recent years, and such efforts have expanded substantially in recent years. These developments have included prescription drug benefit legislation that was enacted and took effect in January 2006, healthcare reform legislation enacted by certain states, and Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (the “ACA”), a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending and enhance remedies against fraud and abuse. The ACA also contains provisions that will affect companies in the pharmaceutical industry and other healthcare related industries by imposing additional costs and changes to business practices. Provisions affecting pharmaceutical companies include the following:
Moreover, we cannot predict what healthcare reform initiatives may be adopted in the future. Further federal and state legislative and regulatory developments are likely, and we expect ongoing initiatives in the U.S. to increase pressure on drug pricing. Such reforms could have an adverse effect on anticipated revenues from product candidates that we may develop and for which we may obtain regulatory approval and may affect our overall financial condition and ability to develop product candidates.
Our ability to obtain services, reimbursement or funding from the federal government may be impacted by possible reductions in federal spending.
U.S. federal government agencies currently face potentially significant spending reductions. Under the Budget Control Act of 2011, the failure of Congress to enact deficit reduction measures of at least $1.2 trillion for the years 2013 through 2021 triggered automatic cuts to most federal programs. These cuts would include aggregate reductions to Medicare payments to providers of up to two percent per fiscal year, starting in 2013. Under the American Taxpayer Relief Act of 2012, which was enacted on January 1, 2013, the imposition of these automatic cuts was delayed until March 1, 2013. Certain of these automatic cuts have been implemented. The full impact on our business of these automatic cuts is uncertain. If federal spending is reduced, anticipated budgetary shortfalls may also impact the ability of relevant agencies, such as the FDA or the National Institutes of Health to continue to function at current levels. Amounts allocated to federal grants and contracts may be reduced or eliminated. These reductions may also impact the ability of relevant agencies to timely review and approve drug research and development, manufacturing, and marketing activities, which may delay our ability to develop, market and sell any products we may develop.
If any of our product candidates receives marketing approval and we or others later identify undesirable side effects caused by the product candidate, our ability to market and derive revenue from the product candidates could be compromised.
In the event that any of our product candidates receive regulatory approval and we or others identify undesirable side effects caused by one of our products, any of the following adverse events could occur, which could result in the loss of significant revenue to us and materially and adversely affect our results of operations and business:
Risks Related to our Common Stock and this Offering
Our internal controls may be inadequate, which could cause our financial reporting to be unreliable and lead to misinformation being disseminated to the public.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. As defined in Rule 13a-15(f) under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), internal control over financial reporting is a process designed by, or under the supervision of, the principal executive and principal financial officer and effected by the board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
We will be required to include a report of management on the effectiveness of our internal control over financial reporting. We expect to incur additional expenses and diversion of management’s time as a result of performing the system and process evaluation, testing and remediation required in order to comply with the management certification requirements.
We do not have a sufficient number of employees to segregate responsibilities and may be unable to afford increasing our staff or engaging outside consultants or professionals to overcome our lack of employees. During the course of our testing, we may identify other deficiencies that we may not be able to timely remediate. Moreover, effective internal controls, particularly those related to revenue recognition, are necessary for us to produce reliable financial reports and are important to help prevent financial fraud. If we cannot provide reliable financial reports or prevent fraud, our business and operating results could be harmed, investors could lose confidence in our reported financial information, and the trading price of our common stock, if a market ever develops, could drop significantly.
The Jobs Act has reduced the information that we are required to disclose.
Under the Jobs Act, the information that we will be required to disclose has been reduced in a number of ways.
As a company that had gross revenues of less than $1.0 billion during the Company’s last fiscal year, the Company is an “emerging growth company,” as defined in the Jobs Act (an “EGC”). We will retain that status until the earliest of (a) the last day of the fiscal year which we have total annual gross revenues of $1,000,000,000 (as indexed for inflation in the manner set forth in the Jobs Act) or more; (b) the last day of the fiscal year of following the fifth anniversary of the date of the first sale of the common stock pursuant to an effective registration statement under the Securities Act of 1933, as amended (the “Securities Act”); (c) the date on which we have, during the previous three year period, issued more than $1,000,000,000 in non-convertible debt; or (d) the date on which we are deemed to be a “large accelerated filer,” as defined in Rule 12b-2 under the Exchange Act or any successor thereto. As an EGC, the Company is relieved from the following:
Our stock price may be volatile, and purchasers of our common stock could incur substantial losses.
Our stock price is likely to be volatile. As a result of this volatility, investors may not be able to sell their common stock at or above the initial public offering price. The market price for our common stock may be influenced by many factors, including the other risks described in this section of the prospectus entitled “Risk Factors” and the following:
| ● |
| 15 |
| ● | the value of | |
| ● | the | |
| ● | ||
| ● | ||
| ● |
In addition,conjunction with the stock marketsreview of these factors, the board of directors also reviewed our history and prospects, including our past and present earnings and cash requirements, our prospects for the future, the outlook for our industry and our current financial condition. The board of directors also believed that the Subscription Price should be designed to provide an incentive to our current stockholders to participate in general,the Rights Offering and exercise their Basic Subscription Right and their Over-Subscription Privilege.
The Subscription Price does not necessarily bear any relationship to any established criteria for value. You should not consider the markets for pharmaceutical stocks, in particular, have experienced extreme volatility that has been often unrelated to the operating performanceSubscription Price as an indication of the issuer. These broad market and industry factors may seriously harm theactual value of our company or our common stock. The market price of our common stock regardless of our operating performance.
may decline during or after the Rights Offering. You will experience immediate and substantial dilution asshould obtain a result of this offering and may experience additional dilution in the future.
If you purchase common stock in this offering, you will incur immediate and substantial dilution of $3.32 per share, representing the difference between the assumed initial public offeringcurrent price of $4.00 per share and our pro forma net tangible book value per share after giving effect to this offering. In addition, we can offer no assurance that you will not experience substantial dilution in the future.
The future issuance of equity or of debt securities that are convertible into common stock will dilute our share capital.
We may choose to raise additional capital in the future, depending on market conditions, strategic considerations and operational requirements. To the extent that additional capital is raised through the issuance of shares or other securities convertible into shares of our common stock, our stockholders will be diluted. Future issuances of our common stock or other equity securities, or the perception that such sales may occur, could adversely affect the trading price ofquote for our common stock and impair our ability to raise capital through future offerings of shares or equity securities. No prediction can be made as to the effect, if any, that future sales of common stock or the availability of common stock for future sales will have on the trading priceperform an independent assessment of our common stock.
Holders of 1% or moreWarrants and Equity Interests before exercising your Subscription Rights and make your own assessment of our common stock priorbusiness and financial condition, our prospects for the future, the terms of the Rights Offering, the information in this prospectus and the other considerations relevant to your circumstances. Once made, all exercises of Subscription Rights are irrevocable. In addition, there is no established trading market for the Warrants or Equity Interests to be issued pursuant to this offering, willand the Warrants and Equity Interests may not be subjectwidely distributed.
Am I required to a six month lock-up agreement, andexercise all directors, officers and 10% shareholders will be subject to a one year lock-up. Nonetheless, at such time as such stock becomes available for sale, it is possible a significant number of our shares may cause the market price of our common stock to drop significantly.Basic Subscription Rights I receive in the Rights Offering?
Commencing six months after the date of this prospectus, 417,678 shares of our fully diluted common stock outstanding as of the date of this prospectus, including shares of common stock issuable upon conversion of convertible notes and Series A convertible preferred stock, will be eligible for sale in the public market from time to time thereafter pursuant to Rule 144 under the Securities Act, and 9,574,197 shares of our fully diluted common stock will be eligible for resale following a one-year lock-up period; some of such sharesNo. You may be subject to the volume and other restrictions of Rule 144. Further, we have 3,000,000 shares reserved for issuance under our 2018 Equity Incentive Plan, which shares may be issued from time to time by our management and which will be subject to vesting and other requirements. At such time as the lock-up periods end, it is possible that a significantexercise any number of such shares will be sold intoyour Basic Subscription Rights, or you may choose not to exercise any Basic Subscription Rights. If you do not exercise any Basic Subscription Rights, the market. At such time, the sale of a significant number of shares of our common stock you own (including each share of common stock issuable exercise of outstanding warrants) will not change. However, if you choose to not exercise your Basic Subscription Rights in full and other holders of Subscription Rights do exercise, your proportionate ownership interest in our company will decrease. If you do not exercise your Basic Subscription Rights in full, you will not be entitled to exercise your Over-Subscription Privilege.
How soon must I act to exercise my Subscription Rights?
If you received a Rights Certificate and elect to exercise any or all of your Subscription Rights, the Subscription Agent must receive your completed and signed Rights Certificate and payment for both your Basic Subscription Rights and any Over-Subscription Privilege you elect to exercise before the Rights Offering expires on , 2024, at 5:00 p.m., Eastern Time, unless we extend or earlier terminate the Rights Offering. If you hold your common shares in the public marketname of a broker, dealer, bank or other nominee, your nominee may establish a deadline before the perceptionexpiration of the Rights Offering by which you must provide it with your instructions to exercise your Subscription Rights, along with the required subscription payment.
May I transfer my Subscription Rights?
No. The Subscription Rights may be exercised only by the common stockholders and warrant holders to whom they are distributed, and they may not be sold, transferred, assigned or given away to anyone else, other than by operation of law. As a result, Rights Certificates may be completed only by the stockholder or warrant holder who receives the certificate. We do not intend to apply for the listing of the Subscription Rights on any securities exchange or recognized trading market.
| 16 |
Will our directors and executive officers participate in the Rights Offering?
To the extent they hold common stock as of the Record Date, our directors and executive officers will be entitled to participate in the Rights Offering on the same terms and conditions applicable to other Rights holders.
Are we requiring a minimum subscription to complete the Rights Offering?
We are not requiring a minimum per investor subscription. However, we are seeking to raise in aggregate the full $4.5 million being offered in this Rights Offering and may terminate the Rights Offering if we have inadequate participation. Should we be unable to raise the full $4.5 million in the Rights Offering and you have already subscribed, we will return your full subscription amount to you within 10 days of termination of the offering, without interest or deduction.
Purchase Agreement
SRO, LLC (“SRO”), an entity formed by Keith Moore, of Boustead Securities LLC, the entity acting as our Placement Agent in this Rights Offering, has entered into a securities purchase agreement (“SPA”) with us, effective February 7, 2024, pursuant to which SRO has committed to invest $2.25 million in the Rights Offering. As such, the total amount that such sales may occur could significantly reducewill be required from our existing shareholders will be $2.25 million in order to close the marketRights Offering, assuming SRO meets it commitment.
Backstop Commitment
Should we not receive adequate participation in the Rights Offering from our existing stockholders to meet the full $4.5 million offering amount, pursuant to the terms of the SPA between SRO and the Company, SRO has agreed to act as backstop participant in the Rights Offering and invest up to an additional $2.25 million to help us reach the full $4.5 offering amount in the Rights Offering.
Has the board of directors made a recommendation to stockholders regarding the Rights Offering?
No. Our board of directors is making no recommendation regarding your exercise of the Subscription Rights. Rights holders who exercise Subscription Rights will incur investment risk on new money invested. We cannot predict the price at which our shares of common stock will trade after the Rights Offering. On [ ], 2024, the last reported sale price of our common stock. In addition, pursuantstock on Nasdaq was $[_] per share. You should make your decision based on your assessment of our business and financial condition, our prospects for the future, the terms of the Rights Offering, the information contained in this prospectus and other considerations relevant to your circumstances. See “Risk Factors” starting at page 21 in this Prospectus and in our Annual Report on Form 10-K for the year ended December 31, 2023 for a separate prospectus accompanying this registration statement, we are registering for resale a totaldiscussion of 1,311,942.some of the risks involved in investing in our securities.
The offering price of the shares and the other terms of this Offering have been arbitrarily determined by the Company.How do I exercise my Subscription Rights?
The offering priceIf you are a common stockholder or a warrant holder of the shares and other terms of this offering have been arbitrarily determined by us and bear no relationship to our assets, book value, potential earnings or any other recognized criterion of value. In addition, no investment banker, appraiser, or other independent third party has been consulted concerning the offering price for the shares or the fairness of the offering price used for the shares.
The offering price of the primary offering and resale offering could differ.
The offering price ofrecord (meaning you hold your shares of our common stock or warrants in your name and not through a broker, dealer, bank or other nominee) and you wish to participate in the initial public offering has been determined by negotiations between the CompanyRights Offering, you must deliver a properly completed and the underwriter. The offering price in the initial public bears no relationship to our assets, earnings or book value, or any other objective standard of value. The selling shareholders may sell the resale shares at prevailing market prices or privately negotiated prices after closesigned Rights Certificate, together with payment of the offeringSubscription Price for both your Basic Subscription Rights and listing of our common stockany Over-Subscription Privilege you elect to exercise, to the Subscription Agent before 5:00 p.m., Eastern Time, on Nasdaq. Therefore,, 2024. If you are exercising your Subscription Rights through your broker, dealer, bank or other nominee, you should promptly contact your broker, dealer, bank or other nominee and submit your subscription documents and payment for the offering prices ofUnits subscribed for in accordance with the initial publicinstructions and resale offering could differ. As a result,within the purchasers in the resale offering could pay moretime period provided by your broker, dealer, bank or less than the offering price in the primary offering.other nominee.
The Resale by the Selling StockholdersWhat if my shares are held in our Resale Offering may cause the market price of our Common Stock to decline.“street name”?
The resale ofIf you hold your shares of our common stock by the selling stockholdersor warrants in the Resale Offering, as well asname of a broker, dealer, bank or other nominee, then your broker, dealer, bank or other nominee is the issuancerecord holder of common stockthe shares you beneficially own. The record holder must exercise the Subscription Rights on your behalf. Therefore, you will need to have your record holder act for you.
| 17 |
If you wish to participate in this Rights Offering could result in resalesand purchase Units, please promptly contact the record holder of our common stock by our current stockholders concerned aboutyour shares or Participating Warrants. We will ask the potential dilutionrecord holder of their holdings. In addition, the resale byyour shares or Participating Warrants, who may be your broker, dealer, bank or other unregistered stockholders after expirationnominee, to notify you of the lock-up period could have the effect of depressing the market price for our common stock.
this Rights Offering.
An active trading market for our common stock may not develop.What form of payment is required?
Prior to this offering, there has been no public marketYou must timely pay the full Subscription Price for our common stock. We intend to apply to have the shares of common stock listed on Nasdaq, subject to our sale of a sufficientfull number of shares inUnits you wish to acquire pursuant to the offeringexercise of Subscription Rights by delivering to meet the listing requirements of Nasdaq. There can be no assurance that an application for listing the shares on Nasdaq or on any other market will be approved. Accordingly, an active trading market for our shares may never develop or be sustained following this offering. If an active market for our common stock does not develop, it may be difficult for you to sell shares you purchase in this offering without depressing the market price for the shares or at all.
If we fail to meet or maintain applicable listing requirements, Nasdaq may delist our common stock from trading, in which case the liquidity and market price of our common stock could decline.
Assuming our common stock is listed on Nasdaq, we cannot assure you that we will be able to meet the continued listing standards of Nasdaq in the future. If we fail to comply with the applicable listing standards and Nasdaq delists our common stock, we and our stockholders could face significant material adverse consequences, including:Subscription Agent a:
| ● | personal check drawn on a | |
| ● | ||
| ● | ||
| ● |
If you send payment by personal uncertified check, payment will not be deemed to have been delivered to the Subscription Agent until the check has cleared. As such, any payments made by personal check should be delivered to the Subscription Agent no fewer than three business days prior to the expiration date.
If you send a payment that is insufficient to purchase the number of Units you requested, or if the number of Units you requested is not specified in the forms, the payment received will be applied to exercise your Subscription Rights to the fullest extent possible based on the amount of the payment received.
Will I receive interest on any funds I deposit with the Subscription Agent?
No. You will not be entitled to any interest on any funds that are deposited with the Subscription Agent pending completion or cancellation of the Rights Offering. If the Rights Offering is cancelled for any reason, the Subscription Agent will return this money to subscribers, without interest or penalty, as soon as practicable.
When will I receive my new shares of Common Stock, Warrants and Equity Interests?
As soon as practicable after the expiration of the Rights Offering, and within five business days thereof, we expect to close on subscriptions and for the Subscription Agent to arrange for the issuance of the shares of common stock, Warrants and Equity Interest purchased in the Rights Offering. At closing, all prorating calculations and reductions contemplated by the terms of the Rights Offering will have been effected and payment to us for the subscribed-for Units will have cleared. All shares common stock, Warrants and Equity Interests that you purchase in the Rights Offering will be issued in book-entry, or uncertificated, form meaning that you will receive a book entry account statement from our transfer agent reflecting ownership of these securities if you are a holder of record. If you hold your common stock in the name of a broker, dealer, bank or other nominee, DTC will credit your account with your nominee with the common stock you purchase in the Rights Offering, however, the Warrants and Rights will be reflected in book entry form only with the Company’s transfer agent. Vstock Transfer, LLC, is acting as the Warrant agent and agent for the Equity Interests in this offering.
After I send in my payment and Rights Certificate to the Subscription Agent, may I cancel my exercise of Subscription Rights?
No. Exercises of Subscription Rights are irrevocable, even if you later learn information that you consider to be unfavorable to the exercise of your Subscription Rights. You should not exercise your Subscription Rights unless you are certain that you wish to purchase Units at the Subscription Price.
How much will our company receive from the Rights Offering?
Assuming that all Units are sold in the Rights Offering, we estimate that the net proceeds from the Rights Offering will be approximately $[_] million, based on the Subscription Price of $[ ] per Unit, after deducting fees and expenses payable to the dealer-manager, and after deducting other estimated expenses payable by us and excluding any proceeds received upon exercise of any Warrants. If all Warrants included in the Units are exercised for cash at the exercise price of $2.35 per share, we will receive an additional $ million. We intend to use the net proceeds from the exercise of Subscription Rights to fund the operations and testing for Diagnostics, as well as for general corporate purposes and to fund ongoing operations. See “Use of Proceeds.”
| 18 |
Are there risks in exercising my Subscription Rights?
Yes. The exercise of your Subscription Rights involves risks. Exercising your Subscription Rights involves the purchase of shares of our common stock, Warrants to purchase common stock, and Equity Interests in Diagnostics and you should consider this investment as carefully as you would consider any other investment. In addition, our Warrants and Equity Interests will not be listed on Nasdaq and a market for the Warrants and Equity Interests does not exist. See “Risk Factors” for a discussion of additional risks involved in investing in our securities.
Can the board of directors terminate, extend or amend the Rights Offering?
Yes. Our board of directors may decide to terminate the Rights Offering at any time and for any reason before the expiration of the Rights Offering. We also have the right to extend the Rights Offering for additional periods in our sole discretion for up to an additional 45 days. We do not presently intend to extend the Rights Offering. We will notify stockholders and the public if the Rights Offering is terminated or extended by issuing a press release announcing the extension no later than 9:00 a.m., Eastern Time, on the next business day after the most recently announced expiration date of the Rights Offering. In the event that we decide to extend the Rights Offering and you have already exercised your Subscription Rights, your subscription payment will remain with the Subscription Agent until such time as the Rights Offering closes or is terminated.
Our board of directors also reserves the right to amend or modify the terms of the Rights Offering in its sole discretion. If we should make any fundamental changes to the terms of the Rights Offering set forth in this prospectus, we will file a post-effective amendment to the registration statement in which this prospectus is included, offer potential purchasers who have subscribed for rights the opportunity to cancel such subscriptions and issue a refund of any money advanced by such stockholder and recirculate an updated prospectus after the post-effective amendment is declared effective by the SEC. In addition, upon such event, we may extend the Expiration Date of the Rights Offering to allow holders of rights ample time to make new investment decisions and for us to recirculate updated documentation. Promptly following any such occurrence, we will issue a press release announcing any changes with respect to the Rights Offering and the new expiration date. The terms of the Rights Offering cannot be modified or amended after the Expiration Date of the Rights Offering. Although we do not presently intend to do so, we may choose to amend or modify the terms of the Rights Offering for any reason, including, without limitation, in order to increase participation in the Rights Offering. Such amendments or modifications may include a change in the subscription price, although no such change is presently contemplated. If we should make any fundamental changes to the terms set forth in this prospectus, we will (i) file a post-effective amendment to the registration statement of which this prospectus forms a part, (ii) offer potential purchasers who have subscribed for rights the opportunity to cancel such subscriptions, and (iii) issue a refund of any money advanced by such stockholder or eligible warrant holder and recirculate an updated prospectus after the post-effective amendment is declared effective with the SEC.
If the Rights Offering is not completed or is terminated, will my subscription payment be refunded to me?
Yes. The Subscription Agent will hold all funds it receives in a segregated bank account until completion of the Rights Offering. If we do not complete the Rights Offering, all subscription payments received by the Subscription Agent will be returned within 10 business days after the termination or expiration of the Rights Offering, without interest or deduction. If you own shares in “street name,” it may take longer for you to receive your subscription payment because the Subscription Agent will return payments through the record holder of your shares. In addition, if we should make any fundamental changes to the terms set forth in this prospectus, we will (i) file a post-effective amendment to the registration statement of which this prospectus forms a part, (ii) offer potential purchasers who have subscribed for rights the opportunity to cancel such subscriptions, and (iii) issue a refund of any money advanced by such stockholder or eligible warrant holder and recirculate an updated prospectus after the post-effective amendment is declared effective with the SEC.
| 19 |
How do I exercise my Rights if I live outside the United States?
The Subscription Agent will hold Rights Certificates for stockholders having addresses outside the United States. To exercise Subscription Rights, foreign stockholders must notify the Subscription Agent and timely follow other procedures described in the section entitled “The Rights Offering - Foreign Stockholders.”
What fees or charges apply if I purchase shares in the Rights Offering?
We are not charging any fee or sales commission to issue Subscription Rights to you or to issue shares of common stock, Warrants or Equity Interests to you if you exercise your Subscription Rights. If you exercise your Subscription Rights through a broker, dealer, bank or other nominee, you are responsible for paying any fees your broker, dealer, bank or other nominee may charge you.
What are the U.S. federal income tax consequences of receiving and/or exercising my Subscription Rights?
For U.S. federal income tax purposes, we do not believe you should recognize income or loss in connection with the receipt or exercise of Subscription Rights in the Rights Offering, but the receipt and exercise of the Subscription Rights is unclear in certain respects. You should consult your own tax advisor as to your tax consequences resulting from the receipt and exercise of Subscription Rights, including the receipt, ownership and disposition of our common stock, Warrants, Equity Interests and common stock received upon the exercise of Warrants. For further information, see “Material U.S. Federal Income Tax Consequences.”
To whom should I send my forms and payment?
If your shares of common stock or warrants are held in the name of a broker, dealer, bank or other nominee, then you should send your subscription documents and subscription payment to that broker, dealer, bank or other nominee. If you are the record holder, then you should send your subscription documents, Rights Certificate, and subscription payment to the Subscription Agent by hand delivery, first class mail or courier service to:
| By mail: | ||
Vstock Transfer LLC 18 Lafayette Place Woodmere, New York 11598 | ||
You or, if applicable, your nominee are solely responsible for completing delivery to the Subscription Agent of your subscription documents, Rights Certificate and payment. You should allow sufficient time for delivery of your subscription materials to the Subscription Agent and clearance of payment before the expiration of the Rights Offering at 5:00 p.m. Eastern Time on , 2024.
Whom should I contact if I have other questions?
If you have other questions or need assistance, please contact the Information Agent: Vstock Transfer, LLC, by telephone at (212) 828-8436, by mail at Vstock Transfer, LLC, 18 Lafeyette Place, Woodmere, New York 11598 or by email at action@vstocktransfer.com.
Who is the dealer-manager?
Boustead Securities LLC (“BSL”) is acting as the sole placement agent for the Rights Offering. Under the terms and subject to the conditions contained in the our agreement with BSL, BSL will use its best efforts to solicit the exercise of Subscription Rights. We have agreed to pay the placement agent certain fees for acting in such capacity and reimburse the placement agent for certain out-of-pocket expenses incurred in connection with this offering. The National Securities Markets Improvement Actplacement agent is not underwriting or placing any of 1996, which is a federal statute, preventsthe Subscription Rights or preempts the states from regulating the saleshares of certain securities, which are referred to as “covered securities.” Because we expect that our common stock, will be listed on Nasdaq,Warrants or Equity Interests being issued in the Rights Offering and is not making any recommendation with respect to such Subscription Rights (including with respect to the exercise or expiration of such Subscription Rights), shares of common stock, Warrants or Equity Interests.
| 20 |
Investing in our securities will be deemed covered securities. Althoughinvolves a high degree of risk. Before making an investment decision with respect to our securities, we urge you to carefully consider the states will be preempted from regulatingrisks described in the sale“Risk Factors” section of our securities,Annual Report on Form 10-K for the federal statute does allow statesyear ended December 31, 2023, which are incorporated by reference into this prospectus. These risk factors relate to investigate companies if there is a suspicionour business, intellectual property, regulatory matters, and ownership of fraudour common stock. In addition, the following risk factors present material risks and if there is a finding of fraudulent activity, thenuncertainties associated with the states can regulateRights Offering. The risks and uncertainties incorporated by reference into this prospectus or bardescribed below are not the sale of covered securities in a particular case. Further, ifonly ones we were no longer listed on Nasdaq, our securities wouldface. Additional risks and uncertainties not be covered securities and we would be subject to regulations in each state inpresently known or which we offerconsider immaterial as of the date hereof may also have an adverse effect on our business. If any of the matters discussed in the following risk factors were to occur, our business, financial condition, results of operations, cash flows or prospects could be materially adversely affected, the market price of our common stock could decline and you could lose all or part of your investment in our securities.
Risks Related to the Rights Offering
Because ourOur management will have broad discretion over the use of the net proceeds from this Offering,offering, you may not agree with how we use themthe proceeds and the proceeds may not be invested successfully.
We intend to use the net proceeds to us from this offering to fund offering to fund preclinical and clinical trials of product candidates, Ropidoxuridine and new formulations of Ropidoxuridine with Tipiracil, O-18 containing molecules for proton radiation sensitization, continued HDAC technology platform development, working capital and general corporate purposes, including the costs of operating as a public company, as well as potential acquisition or in-licensing activities. Therefore, ourOur management will have broad discretion as to the use of the net proceeds from this offering proceeds.and could use them for purposes other than those contemplated at the time of commencement of this offering. Accordingly, you will be relying on the judgment of our management with regard toregarding the use of these net proceeds, and you will not have the opportunity, as part of your investment decision, to assess whether the proceeds are being used appropriately. It is possible that, pending their use, we may invest the net proceeds will be invested in a way that does not yield a favorable, or any, return for us. The failure of our Company.management to use such funds effectively could have a material adverse effect on our business, financial condition, operating results and cash flows.
If securities or industry analystsYour interest in our company may be diluted as a result of this Rights Offering.
Stockholders and warrant holders who do not publish researchfully exercise their Subscription Rights should expect that they will, at the completion of this offering, own a smaller proportional interest in our company on a fully-diluted basis than would otherwise be the case had they fully exercised their Subscription Rights. Further, the shares issuable upon the exercise of the Warrants to be issued pursuant to the Rights Offering will dilute the ownership interest of stockholders not participating in this offering or reports aboutholders of warrants who have not exercised them.
Further, if you purchase Units in this offering at the Subscription Price, you may suffer immediate and substantial dilution in the net tangible book value of our business, orcommon stock. See “Dilution” in this prospectus for a more detailed discussion of the dilution which may incur in connection with this offering.
Completion of the Rights Offering is subject to us raising the full $4.5 million.
Completion of the Rights Offering is subject to us raising the full $4.5 million offering amount. As such, if they issue an adverse or misleading opinion regardinginadequate number of stockholders participate, and if SRO, LLC does not exercise its full backup commitment, we may be unable to complete this Rights Offering.
This Rights Offering may cause the trading price of our common stock our stock price and trading volume could decline.to decrease.
The trading market for our common stock will be influenced bySubscription Price, together with the research and reports that industry or securities analysts publish about us or our business. We do not currently have and may never obtain research coverage by securities and industry analysts. If no or few securities or industry analysts commence coveragenumber of us, the trading price for our stock would be negatively impacted. In the event we obtain securities or industry analyst coverage, if any of the analysts who cover us issue an adverse or misleading opinion regarding us, our business model, our intellectual property or our stock performance, or if our target studies and operating results fail to meet the expectations of analysts, our stock price would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline.
Our board of directors has the authority, without shareholder approval, to issue preferred stock with terms that may not be beneficial to holders of our common stock and such issuance could potentially affect adversely shareholder voting power and perpetuate their control over us.
Our Certificate of Incorporation, as amended to date, allows us to issue shares of preferred stock without any vote or further action by our shareholders. Our board of directors has the authority to fix and determine the relative rights and preferences of any preferred stock. As a result, our board of directors could authorize the issuance of a series of preferred stock that would grant to holders the preferred right to our assets upon liquidation, the right to receive dividend payments before dividends are distributed to the holders of common stock and the right to the redemptionissuable upon exercise of the shares, together with a premium, priorWarrants we propose to the redemption of shares of our common stock. These rightsissue and preferences could negatively affect the holders of our common stock.
The ability of our executive officers and directors, who are our principal stockholders, to control our business may limit or eliminate the ability of minority shareholders to influence corporate affairs.
Our executive officers and directors, who are our principal stockholders, own approximately 73% of our issued and outstanding common stock and, followingultimately will issue if this offering, will own approximately 63% of our issued and outstanding common stock. Accordingly, they will be able to effectively control the election of directors, as well as all other matters requiring shareholder approval. The interests of our principal stockholders may differ from the interests of other shareholders with respect to the issuance of shares, business transactions with or sales to other companies, selection of other directors and other business decisions. The minority shareholders have no way of overriding decisions made by our principal stockholders. This level of control may also have an adverse impact on the market value of our shares because our principal stockholders may institute or undertake transactions, policies or programs that result in losses and may not take any steps to increase our visibility in the financial community and/or may sell sufficient numbers of shares to significantly decrease our price per share.
Our Certificate of Incorporation and Bylaws, each as amended to date, provide for indemnification of officers and directors at the expense of the Company and limit their liability thatRights Offering is completed, may result in a major cost to us and hurt the interests of our shareholders because corporate resources may be expended for the benefit of officers and/or directors.
Our Certificate of Incorporation and Bylaws, each as amended to date, provide for the indemnification of our officers and directors. We have been advised that,an immediate decrease in the opinion of the SEC, indemnification for liabilities arising under federal securities laws is against public policy as expressed in the Securities Act and is therefore, unenforceable.
Our Certificate of Incorporation, as amended to date, provides that disputes must be resolved in the Court of Chancery of the State of Delaware, except for cases brought under the Securities Act or Exchange Act.
Our Certificate of Incorporation, as amended to date, provides that the Court of Chancery in the State of Delaware will be the exclusive forum for dispute resolution for certain enumerated actions, excluding any actions brought under the Securities Act or Exchange Act, or unless the Company consents in writing to an alternative jurisdiction. This exclusive forum selection clause may cause inconvenience of our shareholders or other stakeholders, should they need to bring suit against the Company for an action other than one arising under the Securities Act or Exchange Act.
We do not expect to pay cash dividends in the foreseeable future.
We have never paid cash dividends on our common stock. We do not expect to pay cash dividends on our common stock at any time in the foreseeable future. The future payment of dividends directly depends upon our future earnings, capital requirements, financial requirements and other factors that our board of directors will consider. Since we do not anticipate paying cash dividends on our common stock, return on your investment, if any, will depend solely on an increase, if any, in the market value of our common stock.
Provisions in our amended and restated certificate of incorporation, as amended, and bylaws, as amended, as well as Delaware law, might discourage, delay or prevent a change of control of our company or changes in our management and, therefore, depress the market price of our common stock.
Our Certificate This decrease may continue after the completion of Incorporationthis Rights Offering. If that occurs, you may have committed to buy shares of our common stock at a price greater than the prevailing market price. We cannot predict the effect, if any, that the availability of shares for future sale represented by the Warrants issued in connection with the Rights Offering will have on the market price of our common stock from time to time. Further, if a substantial number of Subscription Rights are exercised and Bylaws, each as amendedthe holders of the shares received upon exercise of those Subscription Rights or the related Warrants choose to date, and bylaws contain provisions thatsell some or all of the shares underlying the Subscription Rights or the related Warrants, the resulting sales could depress the market price of our common stock.
| 21 |
Our common stock is presently trading below $1.00 per share and if our stock price does not increase above $1.00 per share for at least 10 business days, we may be forced to effect a reverse stock split in order to bring the price of our common stock above $1.00 per share or face delisting from the Nasdaq Stock Market.
On August 31, 2023, we received a letter from the Nasdaq Listing Qualifications Staff of The Nasdaq Stock Market LLC (“Nasdaq”) stating that for the 30 consecutive business day period between July 20, 2023 to August 30, 2023 the Company’s common stock had failed to maintain a minimum closing bid price of $1.00 per share, as required for continued listing on The Nasdaq Capital Market pursuant to Nasdaq Listing Rule 5550(a)(2) (the “Minimum Bid Price Requirement”). Pursuant to Nasdaq Listing Rule 5810(c)(3)(A), the Company had an initial period of 180 calendar days, or until February 27, 2024 (the “Compliance Period”), to regain compliance with the Minimum Bid Price Requirement. To regain compliance, the closing bid price of the Company’s common stock must meet or exceed $1.00 per share for a minimum of 10 consecutive trading days, unless such period is extended by actingNasdaq. As the Company had not regained compliance with the Minimum Bid Price Requirement by February 27, 2024, the Company requested and was granted an additional 180-day period to discourage, delay,regain compliance (the “Additional Compliance Period”). To qualify, the Company was required to meet the continued listing requirement for market value of publicly held shares and all other initial listing standards for The Nasdaq Capital Market, with the exception of the Minimum Bid Price Requirement, and we would need to provide written notice of our intention to cure the bid price deficiency during the second compliance period, by effecting a reverse stock split, if necessary. Subsequently, Nasdaq staff determined that the Company was eligible for an additional 180 calendar day period and granted the Company an extension until August 26, 2024, to regain compliance.
The Company believes that it can either regain compliance organically through market forces or, preventif necessary, by effectuating a changereverse stock split prior to the end of the Additional Compliance Period. However, if the Company cannot regain compliance with the Minimum Bid Price Requirement during the Additional Compliance Period, Nasdaq will provide the Company with notice that our common stock will be subject to delisting. At that time, the Company may appeal Nasdaq’s delisting determination to a Nasdaq Hearings Panel. While Nasdaq’s notice to the Company of noncompliance has no immediate effect on the listing of our common stock and our common stock will continue to be listed on The Nasdaq Capital Market under the symbol “SHPH,” there can be no assurance that we will regain compliance with the Minimum Bid Price Requirement or maintain compliance with any of the other Nasdaq continued listing requirements. We will continue to monitor the closing bid price of our common stock and may, if appropriate, consider available options, including seeking stockholder approval to effectuate a reverse stock split, in controlorder to regain compliance with the Minimum Bid Price Requirement.
Our consolidated financial statements are prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business; our ability to continue as a going concern is dependent upon our ability to successfully conduct clinical trials, bring a drug candidate to commercialization, generate revenues, and to raise additional equity or debt financing to fund our operations.
Our consolidated financial statements are prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The Company has incurred losses since inception and had a net loss of approximately $6.6 million and no revenues for the year ended December 31, 2023, with working capital of approximately $4.6 million as of December 31, 2023. In addition, the convertible note payable outstanding at December 31, 2023 includes covenants and certain cash payment requirements. These conditions, and the Company’s ability to comply with such conditions, raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date that the consolidated financial statements are issued.
In September 2022, the Company completed its initial public offering of common stock, generating net proceeds of approximately $10.0 million. Additionally, in January 2023, the Company entered into a securities purchase agreement with an institutional investor through which the Company sold a convertible note with a principal value of $4.3 million, along with a four-year warrant to purchase 1,018,079 shares of common stock, exercisable at $2.35 per share, providing the Company with approximately $3.6 million in net proceeds. To date, the warrant has not yet been exercised. However, the Company’s existing cash resources, marketable securities and the cash received from the Company’s initial public offering and convertible note offering are not expected to provide sufficient funds to support the Company’s operations and clinical trials through the next twelve months.
| 22 |
The capital raises generated thus far have supported operations leading up to the manufacture of drug product and FDA approval of the IND for the Phase II clinical trial of Ropidoxuridine and radiation therapy in glioblastoma. The FDA recommended and the company agreed to an expansion of the clinical trial, necessitating additional capital expenditures to complete the trial. Management intends to initiate a rights offering for $4.5 million and has submitted SBIR applications for non-dilutive NIH funding for our pre-clinical project. The ability of the Company to continue as a going concern is dependent upon our ability to successfully conduct clinical trials, bring a drug candidate to commercialization, generate revenues, and to raise additional equity or debt financing to fund our operations.
Holders of our Warrants will have no rights as a common stockholder until such holders exercise their Warrants and acquire our common stock and the holders of Equity Interests will only have rights with respect to our subsidiary, Diagnostics.
Until holders of Warrants acquire shares of our common stock upon exercise of the Warrants, the holders of such securities will have no rights with respect to the shares of our common stock underlying such Warrants. Upon exercise of the Warrants, the holders thereof will be entitled to exercise the rights of a common stockholder only as to matters for which the record date occurs after the exercise date. The holders of Equity Interests will have no voting rights in the Company and will only have rights with respect to our subsidiary, Diagnostics, which is not publicly traded.
If we terminate this offering for any reason, we will have no obligation other than to return subscription monies within 10 business days.
We may decide, in our sole discretion and for any reason, to cancel or terminate the Rights Offering at any time prior to the expiration date. If this offering is cancelled or terminated, we will have no obligation with respect to Subscription Rights that have been exercised except to return within 10 business days, without interest or deduction, all subscription payments deposited with the Subscription Agent. If we terminate this offering and you have not exercised any Subscription Rights, such Subscription Rights will expire and be worthless.
The Subscription Price determined for this offering is not an indication of the fair value of our common stock.
In determining the Subscription Price, our board of directors considered a number of factors, including, but not limited to, our need to raise capital in the near term to continue our operations, the current and historical trading prices of our common stock, a price that would increase the likelihood of participation in the Rights Offering, the cost of capital from other sources, the value of the Warrants and Equity Interests being issued as components of the Unit, and comparable precedent transactions. The Subscription Price does not necessarily bear any relationship to any established criteria for value. No valuation consultant or investment banker has opined upon the fairness or adequacy of the Subscription Price. You should not consider the Subscription Price as an indication of the value of our company or our common stock.
If you do not act on a timely basis and follow subscription instructions, your exercise of Subscription Rights may be rejected.
Holders of Subscription Rights who desire to purchase shares of our common stock, Warrants and Equity Interests in this offering must act on a timely basis to ensure that all required forms and payments are actually received by the Subscription Agent prior to 5:00 p.m., New York City time, on the expiration date, unless extended. If you are a beneficial owner of shares of common stock or warrants and you wish to exercise your Subscription Rights, you must act promptly to ensure that your broker, dealer, bank, trustee or other nominee acts for you and that all required forms and payments are actually received by your broker, dealer, bank, trustee or other nominee in sufficient time to deliver such forms and payments to the Subscription Agent to exercise the Subscription Rights granted in this offering that you beneficially own prior to 5:00 p.m., New York City time on the expiration date, as may be extended. We will not be responsible if your broker, dealer, bank, trustee or other nominee fails to ensure that all required forms and payments are received by the Subscription Agent prior to 5:00 p.m., New York City time, on the expiration date.
If you fail to complete and sign the required subscription forms, send an incorrect payment amount, or otherwise fail to follow the subscription procedures that apply to your exercise in this Rights Offering, the Subscription Agent may, depending on the circumstances, reject your subscription or accept it only to the extent of the payment received. Neither we nor the Subscription Agent undertakes to contact you concerning an incomplete or incorrect subscription form or payment, nor are we under any obligation to correct such forms or payment. We have the sole discretion to determine whether a subscription exercise properly follows the subscription procedures.
| 23 |
You may not receive all of the Units for which you subscribe.
While we are distributing to holders of our common stock and warrants one Subscription Right for every share of common stock (including each share of common stock issuable upon exercise of warrants) owned on the Record Date, we are only seeking to raise $4.5 million dollars in gross proceeds in this Rights Offering. As a result, based on [16,894,893] shares of common stock outstanding as of [ ], 2024, and [1,446,155] shares of common stock issuable upon exercise of warrants, we would grant Subscription Rights to acquire [18,341,048] Units but will only accept subscriptions for [ ] Units. Accordingly, enough Units may not be available to honor your subscription in full. If excess Units are available after the exercise of Basic Subscription Rights, holders who fully exercise their Basic Subscription Rights will be entitled to subscribe for an additional number of Units. Over-Subscription Privileges will be allocated pro rata among Rights holders who over-subscribed, based on the number of over-subscription Units to which they have subscribed. We cannot guarantee that you will receive any or the entire number of Units for which you subscribed. If for any reason the number of Units allocated to you is less than you have subscribed for, then the excess funds held by the Subscription Agent on your behalf will be returned to you, without interest, as soon as practicable after the Rights Offering has expired and all prorating calculations and reductions contemplated by the terms of the Rights Offering have been effected, and we will have no further obligations to you.
Unless we otherwise agree in writing, a person or entity, together with related persons or entities, may not exercise Subscription Rights (including Over-Subscription Privileges) to purchase Units that, when aggregated with their existing ownership, would result in such person or entity, together with any related persons or entities, owning in excess of 9.99% of our issued and outstanding shares of common stock following the closing of the transactions contemplated by this Rights Offering. If the number of shares allocated to you is less than your subscription request, then the excess funds held by the Subscription Agent on your behalf will be returned to you, without interest, as soon as practicable after the Rights Offering has expired and all prorating calculations and reductions contemplated by the terms of the Rights Offering have been effected, and we will have no further obligations to you.
If you make payment of the Subscription Price by personal check, your check may not clear in sufficient time to enable you to purchase shares in this Rights Offering.
Any personal check used to pay for shares and Warrants to be issued in this Rights Offering must clear prior to the expiration date of this Rights Offering, and the clearing process may require five or more business days. If you choose to exercise your Subscription Rights, in whole or in part, and to pay for shares and Warrants by personal check and your check has not cleared prior to the expiration date of this Rights Offering, you will not have satisfied the conditions to exercise your Subscription Rights and will not receive the shares and Warrants you wish to purchase.
The receipt of Subscription Rights may be treated as a taxable distribution to you.
We believe the distribution of the Subscription Rights in this Rights Offering should be a non-taxable distribution to holders of shares of common stock and holders of warrants, under Section 305(a) of the Internal Revenue Code of 1986, as amended, or the Code. Please see the discussion under the heading “Material U.S. Federal Income Tax Consequences” below. This position is not binding on the IRS, or the courts, however. If this Rights Offering is deemed to be part of a “disproportionate distribution” under Section 305 of the Code, your receipt of Subscription Rights in this offering may be treated as the receipt of a taxable distribution to you equal to the fair market value of the Subscription Rights. Any such distribution would be treated as dividend income to the extent of our current and accumulated earnings and profits, if any, with any excess being treated as a return of capital to the extent thereof and then as capital gain. Each holder of shares of common stock and each holder of Participating Warrants is urged to consult his, her or its own tax advisor with respect to the particular tax consequences of this Rights Offering.
| 24 |
Proposed legislation in the U.S. Congress, including changes in our management thatU.S. tax law, may adversely impact the stockholdersCompany and the value of our company may deem advantageous. These provisions, among other things:
Subscription Rights, shares of our common stock, Warrants and Equity Interests.
Changes to U.S. tax laws (which changes may have retroactive application) could adversely affect the Company or holders of our Subscription Rights, shares of our common stock, Warrants and Equity Interests. In recent years, many changes to U.S. federal income tax laws have been proposed and made, and additional changes to U.S. federal income tax laws are likely to continue to occur in the future.
Exercising the Subscription Rights limits your ability to engage in certain hedging transactions that could provide you with financial benefits.
By exercising the Subscription Rights, you are representing to us that you have not entered into any short sale or similar transaction with respect to our common stock since the Record Date for the Rights Offering. In addition, the Subscription Rights provide that, upon exercise of the Subscription Right, you agree not to enter into any short sale or similar transaction with respect to our common stock for so long as you continue to hold common stock, Warrants or Equity Interests issued in connection with the exercise of the Subscription Right. These requirements prevent you from pursuing certain investment strategies that could provide you greater financial benefits than you might have realized if the Subscription Rights did not contain these requirements.
The Subscription Rights are not transferable, and there is no market for the Subscription Rights.
You may not sell, transfer, assign or give away your Subscription Rights. Because the Subscription Rights are non-transferable, there is no market or other means for you to directly realize any value associated with the Subscription Rights. You must exercise the Subscription Rights to realize any potential value from your Subscription Rights.
Absence of a public trading market for the Warrants may limit your ability to resell the Warrants.
There is no established trading market for the Warrants to be issued pursuant to this offering, and they will not be listed for trading on Nasdaq or any other securities exchange or market, and the Warrants may not be widely distributed. Purchasers of the Warrants may be unable to resell the Warrants or sell them only at an unfavorable price for an extended period of time, if at all.
There is no public market for the Equity Interests being sold in this offering.
There is no established public trading market for the Equity Interests in Diagnostics, and we do not expect a market to develop. In addition, we do not currently intend to apply for listing of the Equity Interests on any securities exchange or recognized trading system. Purchasers of the Equity Interests may be unable to resell their shares or interests in Diagnostics and may only be able to sell them at an unfavorable price for an extended period of time, if at all.
The market price of our common stock may never exceed the exercise price of the Warrants issued in connection with this offering.
The Warrants being issued in connection with this offering, which will be exercisable at a fixed price of $2.35 per share, become exercisable upon issuance and will expire three years from the date of issuance. The market price of our common stock may never exceed the exercise price of the Warrants prior to their date of expiration. Any Warrants not exercised by their date of expiration will expire worthless and we will be under no further obligation to the Warrant holder.
The Warrants contain features that may reduce your economic benefit from owning them.
For so long as you continue to hold Warrants, you will not be permitted to enter into any short sale or similar transaction with respect to our common stock. This could prevent you from pursuing investment strategies that could provide you greater financial benefits from owning the Warrant.
The placement agent is not underwriting the Subscription Rights or the securities underlying the Subscription Rights.
Boustead Securities, LLC (“Boustead”) is acting as sole placement agent for the Rights Offering. As provided in the advisory services agreement with Boustead, Boustead will be acting as an advisor and placement agent with respect to this Rights Offering. The placement agent will not be underwriting Units being issued in this offering and makes no guarantees as to the success of or ability to complete this Rights Offering. Boustead will not be subject to any liability to us in rendering the services contemplated by the advisory services agreement except for any act of bad faith, gross negligence or willful misconduct by Boustead. The Rights Offering may not be successful despite the services of the placement agent to us in this offering.
Since the Warrants are executory contracts, they may have no value in a bankruptcy or reorganization proceeding.
In addition, Section 203the event a bankruptcy or reorganization proceeding is commenced by or against us, a bankruptcy court may hold that any unexercised Warrants are executory contracts that are subject to rejection by us with the approval of the Delaware General Corporation Lawbankruptcy court. As a result, holders of the Warrants may, discourage, delayeven if we have sufficient funds, not be entitled to receive any consideration for their Warrants or preventmay receive an amount less than they would be entitled to if they had exercised their Warrants prior to the commencement of any such bankruptcy or reorganization proceeding.
We may amend or modify the terms of the Rights Offering at any time prior to the expiration of the Rights Offering in our sole discretion.
Our board of directors reserves the right to amend or modify the terms of the Rights Offering in its sole discretion. If we should make any fundamental changes to the terms of the Rights Offering set forth in this prospectus, we will file a post-effective amendment to the registration statement in which this prospectus is included, offer potential purchasers who have subscribed for rights the opportunity to cancel such subscriptions and issue a refund of any money advanced by such stockholder and recirculate an updated prospectus after the post-effective amendment is declared effective by the SEC. In addition, upon such event, we may extend the Expiration Date of the Rights Offering to allow holders of rights ample time to make new investment decisions and for us to recirculate updated documentation. Promptly following any such occurrence, we will issue a press release announcing any changes with respect to the Rights Offering and the new expiration date. The terms of the Rights Offering cannot be modified or amended after the Expiration Date of the Rights Offering. Although we do not presently intend to do so, we may choose to amend or modify the terms of the Rights Offering for any reason, including, without limitation, in order to increase participation in the Rights Offering. Such amendments or modifications may include a change in controlthe subscription price, although no such change is presently contemplated.
Risks Related to Our Business
Investors should carefully consider the risks and uncertainties and all other information contained or incorporated by reference in this prospectus, including the risks and uncertainties discussed under “Risk Factors” in our most recent Annual Report on Form 10-K, as may be amended from time to time, and in subsequent filings that are incorporated herein by reference. All these risk factors are incorporated by reference herein in their entirety. These risks and uncertainties are not the only ones facing us. Our business, financial condition or results of our company. Section 203 imposes certain restrictions on merger, business combinations and other transactions between us and holdersoperations could be materially adversely affected by any of 15% or morethese risks. The trading price of our common stock.stock could decline due to any of these risks, and you may lose all or part of your investment. This prospectus and the incorporated documents also contain forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of certain factors, including the risks mentioned in this prospectus.
| 26 |
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus containsand the documents incorporated herein by reference contain forward-looking statements that involve riskswithin the meaning of the Private Securities Litigation Reform Act of 1995. These statements are based on our management’s current beliefs, expectations and uncertainties. assumptions about future events, conditions and results and on information currently available to us. Discussions containing these forward-looking statements may be found, among other places, in the Sections entitled “Business,” “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” incorporated by reference from our most recent Annual Report on Form 10-K and our Quarterly Reports on Form 10-Q, as well as any amendments thereto, filed with the SEC.
All statements, other than statements of historical facts contained in this prospectusfact, included or incorporated herein regarding our strategy, future operations, financial position, future revenues, projected costs, plans, prospects and objectives are forward-looking statements. In some cases, you can identify forward-looking statements by terminologyWords such as “expect,” “anticipate,” “intend,” “plan,” “believe,” “seek,” “estimate,” “think,” “may,” “could,” “will,” “would,” “should,” “expect,” “plan,” “anticipate,” “believe,” “estimate,” “intend,” “predict,” “seek,” “contemplate,” “project,” “continue,” “potential,” “ongoing”“likely,” “opportunity” and similar expressions or the negativevariations of these terms or other comparable terminology. Thesesuch words are intended to identify forward-looking statements include, but are not limited to,the exclusive means of identifying forward-looking statements. Examples of our forward-looking statements about:include:
| ● | the initiation, timing, progress and results of our research and development programs, preclinical studies, any clinical trials and INDs, NDAs other regulatory submissions; | |
| ● | our expected dependence on third party collaborators for developing, obtaining regulatory approval for and commercializing product candidates; | |
| ● | our receipt and timing of any milestone payments or royalties under any research collaboration and license agreement we enter into; | |
| ● | our ability to identify and develop product candidates; | |
| ● | our or a collaborator’s ability to obtain and maintain regulatory approval of any of our product candidates; | |
| ● | the rate and degree of market acceptance of any approved products candidates; | |
| ● | the commercialization of any approved product candidates; | |
| ● | our ability to establish and maintain additional collaborations and retain commercial rights for our product candidates subject to collaborations; | |
| ● | the implementation of our business model and strategic plans for our business, technologies and product candidates; | |
| ● | our estimates of our expenses, ongoing losses, future revenue and capital requirements; | |
| ● | our ability to obtain additional funds for our operations; | |
| ● | our ability to obtain and maintain intellectual property protection for our technologies and product candidates and our ability to operate our business without infringing the intellectual property rights of others; | |
| ● | our reliance on third parties to conduct our preclinical studies or any future clinical trials; | |
| ● | our reliance on third party supply and manufacturing partners to supply the materials and components for, and manufacture, our research and development, preclinical and clinical trial drug supplies; | |
| ● | our ability to attract and retain qualified key management and technical personnel; | |
| ● | our use of net proceeds to us from this offering; | |
| ● | our expectations regarding the time during which we will be an emerging growth company under the JOBS Act; | |
| ● | our financial performance; and | |
| ● | developments relating to our competitors or our industry. |
TheseBecause forward-looking statements relateare inherently subject to risks and uncertainties, some of which cannot be predicted or quantified and some of which are beyond our control, you should not rely on these forward-looking statements as predictions of future events. The events and circumstances reflected in our forward-looking statements reflect our current view with respect to future events and may not be achieved or to our future financial performanceoccur and involve known and unknown risks, uncertainties and other factors that may cause our actual results performance or achievementscould differ materially from those projected in the forward-looking statements. Moreover, we operate in an evolving environment. New risk factors and uncertainties may emerge from time to be materially different from any future results, performance or achievements expressed or implied by these forward-looking statements.time, and it is not possible for management to predict all risk factors and uncertainties. Factors that may cause actual results to differ materially from current expectations include, among other things, those described in the section entitled “Risk Factors” and elsewhere in this prospectus.
Any forward-looking statement in this prospectus reflects our current view with respect to future events and is subject to these and other risks, uncertainties and assumptions relating to our operations, results of operations, industry and future growth. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Except as required by U.S. federal securities law, we assume no obligation to update or revise these forward-looking statements for any reason, even if new information becomes available in the future.
This prospectus and the documents incorporated herein by reference also refer to estimates and other statistical data made by independent parties and by us relating to market size and growth and other data about our industry. This data involves a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. In addition, projections, assumptions and estimates of our future performance and the future performance of the markets in which we operate are necessarily subject to a high degree of uncertainty and risk.
| 27 |
WeAssuming that all Units are subscribed for in the Rights Offering, we estimate that the net proceeds from this offeringthe Rights Offering will be approximately $8,663,577,$[_] million, after deducting underwriting discounts and commissions and estimatedexpenses relating to this offering expenses payable by us. Ifus estimated at approximately $ million, including placement agent fees and expenses in connection with the underwriter exercises its over-allotment option to purchase additional shares in full, we estimate that our netRights Offering, and excluding any proceeds will be approximately $10,157,577, after deducting underwriting discounts and commissions and estimated offering expenses payable by us.received upon exercise of any Warrants.
We intend to use the net proceeds from this offeringthe exercise of Subscription Rights to fund Phase II clinical trialsthe operations and IND-enabling studies of our product candidates, including radiation sensitizer Ropidoxuridine and the HDAC inhibitor small molecule technology platform, potential acquisition or in-licensing activities, and working capital andtesting for Diagnostics, as well as for general corporate purposes.purposes and to fund ongoing operations. We anticipate thatexpect to use any proceeds we receive from the exercise of Warrants for substantially the same purposes and in substantially the same manner. Pending these uses, we intend to invest the funds raised from this offering will allow us to complete Phase II clinical trial of 30 patients for Ropidoxuridine and complete IND-enabling studies to file for IND in preparation for a Phase I clinical trial for our selective HDAC6 inhibitor. The funds are expected to be used as follows:
The above estimates include the working capital necessary to fund each of the above listed projects; any additional funds raised in this offering will be used toward additional working capital.
Notwithstanding our plannedshort-term, investment grade, interest-bearing securities. It is possible that, pending their use, of funds, there is no guarantee that we will not require additional funds to complete the above clinical trials. In addition, following our completion of the above-listed clinical trials, we believe we will need approximately $22 million in additional funding to complete Phase III clinical trials for Ropidoxuridine and approximately $30 million in additional funding to complete Phase I through III clinical trials for our selective HDAC6 inhibitor. If such additional funds are required, we will have to secure additional funding from investors or through a joint development partner.
We believe thatmay invest the net proceeds from this offering, together with our existing cash on hand, cash equivalents and investments, will enable us to fund our operations and continued growth and development through at least the next 12 months. We have based this estimate on assumptionsin a way that may prove to be wrong and we may end up using our available capital resources more quickly than currently anticipated.does not yield a favorable, or any, return for us.
The expected use of the net proceeds from this offering represents our intentions based upon our current plans and business conditions, which could change in the future as our plans and business conditions evolve. The amounts and timing of our actual expenditures depend on numerous factors, including the progress of our preclinical development efforts, the results of any clinical trials and other studies and any unforeseen cash needs. All of our research and development programs are at an early stage and development of product candidates from these programs is highly uncertain and may not result in approved products. Completion dates and completion costs can vary significantly for each product candidate and are difficult to predict. Accordingly, we
Our management will have broad discretion inas to the useallocation of the net proceeds from this offering and could spenduse them for purposes other than those contemplated at the proceeds in ways that do not improve our resultstime of operations or enhance the valuecommencement of our stock.this offering.
We may seek additional financing to support the intended use of proceeds discussed above. If we secure additional equity funding, investors in this offering would be diluted. In all events, there can be no assurance that additional financing would be available when needed and, if available, on terms acceptable to us.
Pending the use of the proceeds from this offering, we intend to invest the net proceeds in short-term, interest-bearing, investment-grade securities, certificates of deposit or government securities.
We have not paid any dividends on our common stock since inception and we currently expect that, in the foreseeable future, all earnings (if any) will be retained for the development of our business and no dividends will be declared or paid. Any future dividends will be subject to the discretion of our board of directors and will depend upon, among other things, our earnings (if any), operating results, financial condition and capital requirements, general business conditions and other pertinent facts.
| 28 |
The following table sets forth our cash and cash equivalents and capitalization as of June 30, 2022:December 31, 2023:
| ● | on an actual basis; | |
| ● | on a pro forma as adjusted basis to reflect the |
You should read this table in conjunction with our financial statements and related notes and the sections titled “Use of Proceeds,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and “Description of Capital Stock” appearing elsewhereour audited financial statements for the year ended December 31, 2023, which is included in this prospectus.our Annual Report on Form 10-K filed with the SEC on March 21, 2024, which is incorporated by reference herein.
| Actual | Pro forma | Pro form As Adjusted | ||||||||||
| Cash and cash equivalents | $ | 50,437 | $ | 10,898,039 | $ | 10,974,514 | ||||||
| Capitalization: | ||||||||||||
| Notes payable | $ | 1,261,097 | $ | 1,261,097 | $ | 671,097 | ||||||
| Stockholders’ equity (deficit): | ||||||||||||
| Series A convertible preferred stock, $0.00001 par value; $1,000 per share liquidation value or aggregate of $1,212,500; 20,000,000 shares authorized; 1,213 shares issued and outstanding actual, 0 pro forma, and 0 pro forma as adjusted | - | - | - | |||||||||
| Common stock, $0.00001 par value; 100,000,000 shares authorized; 9,312,991 shares issued and outstanding at June 30, 2022 actual, 12,938,377 shares pro forma, and 13,283,377 shares pro forma as adjusted | 93 | 130 | 133 | |||||||||
| Additional paid-in capital | 4,819,916 | 15,667,481 | 16,333,953 | |||||||||
| Accumulated deficit | (7,126,082 | ) | (7,126,082 | ) | (7,126,082 | ) | ||||||
| Total stockholders’ equity (deficit) | (2,306,073 | ) | 8,541,529 | 9,208,004 | ||||||||
| Total capitalization | $ | (1,044,976 | ) | $ | 9,802,626 | $ | 9,879,101 | |||||
| Actual | Pro form As Adjusted | |||||||
| Cash and cash equivalents | $ | $ | ||||||
| Capitalization: | ||||||||
| Notes payable | $ | $ | ||||||
| Stockholders’ equity (deficit): | ||||||||
| Series A convertible preferred stock, $0.00001 par value; $1,000 per share liquidation value; 20,000,000 shares authorized; no shares outstanding | - | |||||||
| Common stock, $0.00001 par value; 100,000,000 shares authorized; 16,894,893 shares issued and outstanding at December 31, 2023 actual, [ ] shares pro forma as adjusted | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated deficit | ) | ) | ||||||
| Total stockholders’ equity (deficit) | ) | |||||||
| Total capitalization | $ | ) | $ |
The number of shares of common stock issued and outstanding actual, pro forma and pro forma as adjusted in the table above excludes 2,615,8832,321,820 shares reserved for issuance under our 2018 Equity Incentive Plan. To date, we have issued grants of 384,167 shares (on a post-reverse split basis), which were issued in the form of restricted stock units, some ofPlan or 1,446,155 warrants which remain subject to certain vesting conditions.exercise.
| 29 |
If you investPurchasers of Units in our common stock, your interestthe Rights Offering will be diluted to the extentexperience an immediate dilution of the difference between the public offering pricenet tangible book value per share of our 1,225,888 units, consistingcommon stock. Our net tangible book value as of 1,225,888December 31, 2023 was approximately $[ ], or $[ ] per share of our common stock (based upon 16,894,893 shares of our common stock purchased at an effective priceoutstanding). Net tangible book value per share is equal to our total tangible assets less our total liabilities, divided by the number of $8.115shares of our outstanding common stock.
Dilution per share of common stock and warrantsequals the difference between the amount paid by purchasers of Units in the Rights Offering (ascribing no value to purchase common stock, exercised at a price of $0.01 per share, that you pay upon purchase of the 1,225,888 units,Warrants contained in the Units) and the pro forma as adjusted net tangible book value per share of our common stock immediately after the Rights Offering.
Based on the sale by us in this Rights Offering which includes the conversion of 1,212.5 sharesa maximum of Series A convertible preferred stock (which will convert into approximately 336,805 shares of common stock upon completion of this offering, not including the 8.5% cumulative dividend, which is payableUnits at the timeSubscription Price of conversion in either cash or common stock at the Company’s determination), the$[ ] per Unit (assuming no exercise of approximately 336,805 shares of common stock issuable to the Series A convertible preferred stockholders upon exercise of warrants (which warrants will be issuable to the Series A convertible preferred stockholders upon completion of this offering, and which share calculation does not including the 8.5% cumulative dividend payable at the time of conversion in either cash or common stock at the Company’s determination), the exercise of 500,000 warrants into 500,000 shares of common stock (which shares are being registered in the Resale Offering)Warrants), and the conversion of $590,000 of convertible notes (which will convert into units consisting of a total of 147,500 shares of common stock at a conversion price of $4.00 per shareafter deducting estimated offering expenses and warrants to purchase 147,500 shares of common stock (the “Conversion Units”) upon completion of this Offeringdealer-manager fees and which Conversion Units are being registered in the Resale Offering). Net tangible book value per share is determinedexpenses payable by dividingus, our total tangible assets less our total liabilities by the number of shares of common stock outstanding. Our historicalpro forma net tangible book value as of June 30, 2022 was $(2,393,456) or $(0.26) per share, based on 9,312,991 shares of common stock, 500,000 warrants to purchase common stock, and 1,212.5 shares of Series A preferred stock and related warrants outstanding as of June 30, 2022.
Net tangible book value dilution per share represents the difference between the amount per share paid by new investors who purchase shares from us in this offering and the pro forma net tangible book value per share of common stock immediately after completion of this Offering. As of June 30, 2022, after giving effect to our sale in this Offering of 1,225,888 units, with each unit consisting of one share of common stock and one warrant to purchase one share of common stock, in this Offering at an initial offering price of $8.125 per unit, including the exercise of the warrants offered herein at an exercise price of $0.01 per share, the exercise of warrants to purchase 500,000 shares of common stock and the sale of such common stock by certain selling stockholders in the Resale Offering, the automatic conversion of our Series A convertible preferred stock into 673,610 shares of common stock and the automatic conversion of our convertible notes into the equivalent of 295,000 shares of common stock, after deducting estimated Offering expenses that we must pay, our pro forma as adjusted net tangible book valueDecember 31, 2023 would have been $9,120,648,approximately $ million, or $0.69$ per share. This represents an immediate increase in pro forma net tangible book value to existing stockholders of $0.95$ per share to existing shareholders, and an immediate dilution to purchasers in pro forma net tangible book valuethe Rights Offering of $3.37$ per share to new investors purchasing shares in this Offering. The table below illustrates this per share dilution as of June 30, 2022.
| Pro forma, | ||||||||
| Before | as adjusted | |||||||
| Offering | after offering | |||||||
| Assumed initial offering price per common share | $ | 4.06 | ||||||
| Net tangible book value per share as of June 30, 2022 | $ | (0.26 | ) | |||||
| Increase in pro forma net tangible book value per share attributable to new investors participating in this Offering and conversion of the Convertible Notes | $ | 0.954 | ||||||
| Pro forma as adjusted net tangible book value per share after this Offering and conversion of the Convertible Notes | $ | 0.69 | ||||||
| Dilution of pro forma net tangible book value per share to new investors | $ | 3.37 | ||||||
share. The following table sets forth, on a pro forma as adjusted basis as of June 30, 2022, the number of shares of common stock purchased or to be purchased from us, the total consideration paid or to be paid and the average price per share paid or to be paid by existing holders of common stock and by new investors, at a public offering price of $8.125 per unit, before deducting estimated Offering expenses that we must pay.
| Average | ||||||||||||||||||||
| Shares Purchased | Total Consideration | Price Per | ||||||||||||||||||
| Number | Percent | Amount | Percent | Share | ||||||||||||||||
| Existing stockholders | 9,312,991 | 79 | % | $ | 4,820,000 | 33 | % | $ | 0.52 | |||||||||||
| New investors | 2,451,776 | 21 | % | 9,960,340 | 67 | % | 4.06 | |||||||||||||
| Total | 11,764,767 | 100 | % | $ | 14,780,340 | 100 | % | $ | 1.26 | |||||||||||
*reflects both shares of common stock and warrants issued inillustrates this unit offering, and assumes the new investors’ exercise of warrants to purchase 1,225,888 shares of common stock, exercised at $0.01 per share.per-share dilution:
The foregoing discussion and tables are based on the number of shares of common stock outstanding as of June 30, 2022 but excluding 2,615,883 shares of common stock reserved for issuance under our 2018 Equity Incentive Plan. To date, 384,167 shares have been granted in the form of restricted stock units (“RSUs”), of which 357,390 RSUs have vested and 30,777 RSUs remain subject to vesting.
MANAGEMENT’S DISCUSSION AND ANALYSIS OFFINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following Management’s Discussion and Analysis should be read in conjunction with our financial statements and the related notes thereto included elsewhere in this prospectus. The Management’s Discussion and Analysis contains forward-looking statements that involve risks and uncertainties, such as statements of our plans, objectives, expectations and intentions. Any statements that are not statements of historical fact are forward-looking statements. When used, the words “believe,” “plan,” “intend,” “anticipate,” “target,” “estimate,” “expect,” and the like, and/or future-tense or conditional constructions (“will,” “may,” “could,” “should,” etc.), or similar expressions, identify certain of these forward-looking statements. These forward-looking statements are subject to risks and uncertainties that could cause actual results or events to differ materially from those expressed or implied by the forward-looking statements in this form. Our actual results and the timing of events could differ materially from those anticipated in these forward-looking statements as a result of several factors including, but not limited to, those noted under “Risk Factors” in this prospectus. In addition, any compensation disclosure contained in this prospectus regarding our officers and directors is likely to increase in the near term following completion of the offering, and therefore all such disclosures made within this prospectus reflect historical facts and will not reflect forward looking or anticipated compensation going forward. See the section entitled “Executive Compensation” below for a more detailed discussion.
We do not undertake any obligation to update forward-looking statements to reflect events or circumstances occurring after the date of this prospectus, except as required by U.S. federal securities laws.
Overview
Founded by Georgetown University Medical School faculty members, we are a discovery and development stage pharmaceutical company leveraging our proprietary technology to develop novel therapies that are designed to cure cancer. Originally formed as Shuttle Pharmaceuticals, LLC in 2012, our goal is to extend the benefits of cancer treatments by leveraging insights into cancer therapy with surgery, radiation therapy, chemotherapy and immunotherapy. While there are several therapies being developed with the goal of curing cancer, one of the most effective and proven approaches to this is radiation therapy (RT). We are developing a pipeline of products designed to address the limitations of this current standard of cancer therapies. We believe that our product candidates will enable us to deliver cancer treatments that are safer, more reliable and at a greater scale than that of the current standard of care.
Operations to date have focused on continuing our research and development efforts to advance Ropidoxuridine clinical testing and improved drug formulation, to advance HDAC6 inhibitor (SP-2-225) preclinical development, and complete SBIR contract work on predictive biomarkers of radiation response, as well as prostate cell lines for health disparities research. We have received SBIR contract funding from the NIH for the aforementioned projects. The clinical development of Ropidoxuridine has shown drug bioavailability and a maximum tolerated dose has been established for use in Phase II clinical trials. The radiation biomarker project and the health disparities project have been completed. Changes in operational, administrative, legal and professional expenses related to our operations are set forth in more detail in the discussion below.
Results of Operations
Comparison of the three months ended June 30, 2022 and 2021
The following table summarizes the results of our operations:
| Three Months Ended | ||||||||||||||||
| June 30, | Change | |||||||||||||||
| 2022 | 2021 | $ | % | |||||||||||||
| Revenue | $ | - | $ | - | $ | - | - | |||||||||
| Operating expenses: | ||||||||||||||||
| Research and development, net of contract expense reimbursements | 83,868 | 286,730 | (202,862 | ) | (71 | )% | ||||||||||
| General and administrative | 9,078 | 7,213 | 1,865 | 26 | % | |||||||||||
| Legal and professional | 260,680 | 42,308 | 218,372 | 516 | % | |||||||||||
| Total operating expenses | 353,626 | 336,251 | 17,375 | 5 | % | |||||||||||
| Other (income) expense: | ||||||||||||||||
| Interest expense - related party | 14,836 | 10,547 | 4,289 | 41 | % | |||||||||||
| Interest expense | 170,391 | 120 | 170,271 | n/a | ||||||||||||
| (Gain) Loss on change in fair value of warrant liability | 58,422 | (117 | ) | 58,539 | n/a | |||||||||||
| Total other expense | 243,649 | 10,550 | 233,099 | 2209 | % | |||||||||||
| Net loss | $ | 597,275 | $ | 346,801 | $ | 250,474 | 72 | % | ||||||||
Research and Development-Net of contract expense reimbursements. Research and development-net of contract expense reimbursements (“R&D”) was $83,368 for the three months ended June 30, 2022, as compared to $286,730 for three months ended June 30, 2021. For the three months ended June 30, 2022, the Company received $211,455 in reimbursement from the NIH contracts and incurred $295,323 in R&D expenses. For the three months ended June 30, 2021, reimbursement from the NIH totaled $0 and total expenses related to R&D was $286,731. Quarterly expenses increased by $8,592, or 3%, in the second quarter of 2022 as Topic 345 work was completed. Expenses were offset by a final payment of $211,455 received in April 2022 following the Company’s submission of a final report to the NIH for the period of performance ending March 15, 2022.
R&D expense reimbursements reflect the final payment of $211,455 received in April 2022 after the final report was filed with the NIH and reviewed. NIH requires that milestones included in the fixed price contract be met, therefore, compensation related expenses continued in 2022 under the no cost extension from the NIH. Compensation related expenses were $225,597 in the three months ended June 30,2021 as compared to $270,164 in the three months ended June 30, 2022. Compensation related expenses increased from 79% of total R&D in the three months ened June 30, 2021 to 91% for the three months ended June 30, 2022. Subcontract work made up 13% of total R&D expense in the three months ended June 30, 2021 and there were no subcontractor expenses during the three months ended June 30, 2022. All other R & D expenses were inconsequential.
Below is a breakdown of the actual costs and reimbursements received by the company for the three months ended June 30, 2022 and 2021, and a breakdown of how such cost and reimbursements were distributed across research projects.
For the three months ended June 30, 2022, total research and development costs were $295,323 for which all costs were funded by the Shuttle. For the three months ended June 30, 2021, total R & D costs were $286,730 for which 21.3% of the costs were allocated to the NIH funded project (Topic 345) and Shuttle funded the remaining costs of $225,597, or 78.7% of the total costs. For the three months ended June 30, 2022, total R&D costs were $295,323 for which $211, 455 was reimbursed by NIH and the remaining costs were funded by the Shuttle for a net R&D loss of $83,868. Company funded R&D activities increased in 2021 and decreased in 2022 due to NIH no cost extensions required to complete contracted work and file the final reports before receiving payment from the NIH.
Key Research and Development Projects
3 Month Periods ending June 30, 2021 and 2022 (Q2)
| Research & Development | NIH Topic 345 | NIH Topic 352* | Shuttle Funded | Total | ||||||||||||||||||||||||||||
| Revenue and Expenses | 2021 | 2022 | 2021 | 2022 | 2021 | 2022 | 2021 | 2022 | ||||||||||||||||||||||||
| NIH Reimbursement | - | 211,455 | - | - | - | - | - | 211,455.00 | ||||||||||||||||||||||||
| Compensation | - | - | - | 225,596.59 | 270,164.09 | 225,596.59 | 270,164.09 | |||||||||||||||||||||||||
| Subcontracts | 36,927.56 | - | - | - | - | - | 36,927.56 | - | ||||||||||||||||||||||||
| Supplies | 7,143.63 | - | - | - | - | 619.36 | 7,143.63 | 619.36 | ||||||||||||||||||||||||
| Other, Lab | 17,062.90 | - | - | - | - | 24,539.10 | 17,062.90 | 24,539.10 | ||||||||||||||||||||||||
| Expense total | 61,134.09 | - | - | - | 225,596.59 | 295,322.55 | 286,730.68 | 295,322.55 | ||||||||||||||||||||||||
| Research and Development, Net of Contracts | (61,134.09 | ) | 211,455 | - | - | (225,596.59 | ) | (295,322.55 | ) | (286,730.68 | ) | (83,867.55 | ) | |||||||||||||||||||
| 21.3 | % | 0.0 | % | 78.7 | % | 100.0 | % | |||||||||||||||||||||||||
| $ | ||||
| Net tangible book value per share as of December 31, 2023 | $ | |||
| Increase in | $ | |||
| Pro forma net tangible book value per share as of December 31, 2023, after giving effect to Rights Offering | $ | |||
| Dilution in net tangible book value per share to purchasers in the | $ |
In addition, the CEOThe information above is as of December 31, 2023 and CMO are actively involved in the research and development activities, but neither receives a salary from the Company. As such, research and development expenses are lower than might be incurred in the future.
The allocation of costs to each research project for the three months ended June 30, 2021 and 2022 are as follows:
Cost Allocation for the 3-month Period Ending June 30, 2021excludes:
| ● | ||
| ● | ||
| ● |
| 30 |
MARKET PRICE AND DIVIDEND POLICY
Our shares of common stock are currently quoted on The Nasdaq Capital Market under the symbol “SHPH”. On April [ ], 2024, the last reported sales price of our common stock on Nasdaq was $[_].
GeneralHolders of Record
As of April [ ], 2024, we had approximately 55 holders of record of our common stock. Because many of our shares of common stock are held by brokers and Administrative Expenses. Generalother institutions on behalf of stockholders, this number is not indicative of the total number of stockholders represented by these stockholders of record.
Dividends
We have not declared or paid dividends to stockholders since inception and Administrative expensesdo not plan to pay cash dividends in the three months ended June 30, 2022 increased by $1,865, from $7,213 in 2021foreseeable future. We currently intend to $9,078 in 2022.retain earnings, if any, to finance our growth.
Issuer Purchases of Equity Securities
None.
| 31 |
Legal and Professional Expenses. During the three months ended June 30, 2022, legal and professional expenses increased by $260,680 or 497%. This increase in legal and professional fees was primarily due to increases in our expenses related to obtaining pre-IPO financing and other expenses related to preparation for the IPO.The Subscription Rights
Other (Income) ExpenseWe are distributing to the record holders of our common stock and warrants, at no charge, non-transferable Subscription Rights to purchase one Unit at a subscription price of $[ ] per Unit. Each Basic Subscription Right will entitle you to purchase one share of our common stock, one warrant to purchase one share of common stock (a “Warrant”) and [ ]% equity interests (the “Equity Interest”) in our subsidiary Shuttle Diagnostics, Inc. (“Diagnostics”). Other expense was $243,649Assuming all Units are sold in the Rights Offering, at close, the Company will own 56% of the Equity Interests in Diagnostics and the participants in the Rights Offering will hold 44% of the Equity Interests in Diagnostic. Each Warrant will be exercisable for one share of our common stock at an exercise price of $2.35 per share from the date of issuance through the expiration three months ended June 30, 2022, which consistedyears from the date of $170,394 in interest expense on convertible loans, $14,836 in interest expense on related party loans,issuance. Each record holder of our common stock and holders of our warrants will receive one Subscription Right for every share of our common stock (including each share of common stock issuable upon exercise of warrants) owned by such record holder as of the Record Date. Each Subscription Right entitles the record holder to a loss on change in warrant liability of $58,422. Other income was $10,550 for the three months ended June 30, 2021, which consisted of $120 in interest expense, $10,547 in interest expense on related party loansBasic Subscription Right and a gain on change in warrant liability of $117.an Over-Subscription Privilege.
Comparison of the six months ended June 30, 2022 and 2021Basic Subscription Rights
The following table summarizesYour Basic Subscription Rights will entitle you to purchase Units, each comprised of one share of common stock, one Warrant and [ ]% Equity Interest in Diagnostics. For example, if you owned 100 shares of common stock as of the resultsRecord Date, you will receive 100 Subscription Rights and will have the right to purchase 100 shares of our operations:common stock, Warrants to purchase 100 shares of our common stock, exercisable at $2.35 per share, and [ ] Equity Interests in Diagnostics, at a price of $[ ] per Unit, or a total payment of $[ ]. You may exercise all or a portion of your Basic Subscription Rights, or you may choose not to exercise any of your Basic Subscription Rights. If you do not exercise your Basic Subscription Rights in full, you will not be entitled to exercise your Over-Subscription Privilege.
| Six Months Ended | ||||||||||||||||
| June 30, | Change | |||||||||||||||
| 2022 | 2021 | $ | % | |||||||||||||
| Revenue | $ | - | $ | - | $ | - | - | |||||||||
| Operating expenses: | ||||||||||||||||
| Research and development, net of contract expense reimbursements | 379,783 | 392,726 | (12,943 | ) | (3 | )% | ||||||||||
| General and administrative | 22,847 | 13,461 | 9,386 | 70 | % | |||||||||||
| Legal and professional | 589,392 | 106,150 | 483,242 | 455 | % | |||||||||||
| Total operating expenses | 992,022 | 512,337 | 479,685 | 94 | % | |||||||||||
| Other (income) expense: | ||||||||||||||||
| Interest expense - related party | 25,383 | 21,094 | 4,289 | 20 | % | |||||||||||
| Interest expense | 315,944 | 350 | 315,594 | n/a | ||||||||||||
| (Gain) Loss on change in fair value of warrant liability | 18,772 | (57,656 | ) | 76,428 | n/a | |||||||||||
| Gain on forgiveness of Paycheck Protection Program note payable | (73,007 | ) | - | (73,007 | ) | n/a | ||||||||||
| Total other (income) expense | 287,092 | (36,212 | ) | 323,304 | n/a | |||||||||||
| Net loss | $ | 1,279,114 | $ | 476,125 | $ | 802,989 | 169 | % | ||||||||
Additionally, sufficient Units may not be available to honor your Basic Subscription Right in full. While we are distributing one subscription right for every share of common stock owned or deemed owned on the record date, we are only seeking to raise $4.5 million dollars in gross proceeds in this Rights Offering. As a result, based on (1) [16,894,893] shares of common stock outstanding and (2) [1,446,155] shares of common stock issuable upon exercise of outstanding warrants, we would grant subscription rights to acquire [18,341,048] Units but will only accept subscriptions for [ ] Units.
Research and Development-NetIf exercises of contract expense reimbursements. Research and development-netBasic Subscription Rights exceed the number of contract expense reimbursements (“R&D”) was $379,783 forUnits available in the six months ended June 30, 2022, as compared to $392,726 for six months ended June 30, 2021. For six months ended June 30, 2022,Rights Offering, we will allocate the Company received $211,455available Units pro-rata among the record holders exercising the Basic Subscription Rights in reimbursement from the NIH contracts and incurred $591,237 in R&D expenses. For the six months ended June 30, 2021, reimbursement from NIH totaled $211,455 and total expenses related to R&D were $604,181. The decrease of $12,945, or 2%, is primarily relatedproportion to the Company reducing R&D spending asnumber of shares of our common stock each of those record holders owned or were deemed to own on the record date, relative to the number of shares owned or deemed owned on the record date by all record holders exercising the basic subscription right. If this pro-rata allocation results in any record holders receiving a resultgreater number of NIH contracts ending and new contract proposals being prepared but not yet started. The no cost extension fromUnits than the NIH ended on March 15, 2022,record holder subscribed for pursuant to the exercise of the basic subscription rights, then such record holder will be allocated only that number of Units for which the record holder subscribed, and the final reportremaining Units will be allocated among all other record holders exercising their Basic Subscription Rights on the same pro rata basis described above. The proration process will be repeated until all shares have been allocated.
If for any reason the number of Units allocated to you is less than you have subscribed for, then the NIH was filedexcess funds held by the Subscription Agent on your behalf will be returned to you, without interest, as soon as practicable after the Rights Offering has expired and accepted, resulting in a paymentall prorating calculations and reductions contemplated by the terms of $211,455 during the six months ended June 30, 2022.Rights Offering have been effected, and we will have no further obligations to you.
Over-Subscription Privilege
R&D expense reimbursements wereIf you exercise your Basic Subscription Rights in full, you may also choose to exercise your Over-Subscription Privilege. Subject to proration and the limitations described in this prospectus, we will seek to honor the Over-Subscription Requests in full. If Over-Subscription Requests exceed the number of Units available, however, we will allocate the available Units pro rata among the common stockholders and warrant holders as of the Record Date exercising the Over-Subscription Privilege in proportion to the number of shares of our common stock (including each share of common stock issuable upon exercise of warrants) each of those stockholders and/or warrant holders owned on the Record Date, relative to the number of shares owned on the Record Date by all stockholders and warrant holders as of the Record Date exercising the Over-Subscription Privilege. If this pro rata allocation results in any stockholder or warrant holder receiving a greater number of Units than the record holder subscribed for pursuant to the exercise of the Over-Subscription Privilege, then such record holder will be allocated only that number of Units for which the record holder oversubscribed, and the remaining Units will be allocated among all other stockholders or warrant holders exercising the Over-Subscription Privilege on the same duringpro rata basis described above. The proration process will be repeated until all Units have been allocated. Vstock Transfer, LLC, the six months ended June 30, 2021 and June 30, 2022. NIH requires that milestones included inSubscription Agent for the fixed price contract be met, therefore, compensation related expenses continued in 2022 underRights Offering, will determine the no cost extension fromover-subscription allocation based on the NIH. Compensation related expenses were $447,222 in the six months ended June 30, 2021 as compared to $532,961 in the six months ended June 30, 2022. Compensation related expenses increased from 74% of total R&D in the six months ended June 30, 2021 as compared to 90.1% in the six months ended June 30, 2022. Subcontract work made up 17% of total R&D expense in the six months ended June 30, 2021 and there were no subcontractor expenses during the six months ended June 30, 2022. All other R&D expenses were inconsequential.formula described above.
| 32 |
Below is a breakdownTo the extent the aggregate subscription payment of the actual costsnumber of unsubscribed Units available to you pursuant to the Over-Subscription Privilege is less than the amount you actually paid in connection with the exercise of the Over-Subscription Privilege, you will be allocated only the number of unsubscribed Units available to you, and reimbursements received byany excess subscription payments will be returned to you, without interest or deduction, with 10 business days after expiration of the Company for the six months ended June 30, 2022 and 2021, and a breakdown of how such cost and reimbursements were distributed across research projects.Rights Offering.
ForWe can provide no assurances that you will be entitled to purchase the six months ended June 30, 2022, total research and development costs were $591,236number of Units issuable upon the exercise of your Over-Subscription Privilege in full at the expiration of the Rights Offering. We will not be able to satisfy any requests for which $211,455 was paid by reimbursements received from the NIH, leaving a net of $392,726. For the six months ended June 30, 2021, total R&D costs were $604,181 for which $211,455 was paid by reimbursements received from the NIH, leaving a net of $379,783. Company funded R&D activities increased in the six months ended June 30, 2021 and remained relatively consistent during the six months ended June 30, 2022 dueUnits pursuant to the NIH no cost extensions required forOver-Subscription Privilege if all of our stockholders exercise their Basic Subscription Rights in full, and we will only honor an Over-Subscription Privilege to the Company to complete contracted work and fileextent sufficient Units are available following the final reports. A summaryexercise of the breakdown of costs is listed below.Basic Subscription Rights.
Key Research and Development Projects
6 Month Periods ending June 30, 2021 and 2022 (Q2)Limitation on the Purchase of Units
You may only purchase the number of Units purchasable upon exercise of the number of Basic Subscription Rights distributed to you in the Rights Offering, plus the Over-Subscription Privilege, if any. Accordingly, the number of Units that you may purchase in the Rights Offering is limited by the number of shares of our common stock (including each share of common stock issuable upon exercise of Participating Warrants) you held on the Record Date and by the extent to which other stockholders or warrant holders exercise their Basic Subscription Rights and Over-Subscription Privileges, all of which we cannot determine prior to completion of the Rights Offering. However, due to stock exchange restrictions, we will not issue Units in the Rights Offering to the extent that a holder would beneficially own, together with any other person with whom such holder’s securities may be aggregated under applicable law, more than 19.99% of our outstanding shares of common stock.
| Research & Development | NIH Topic 345 | NIH Topic 352* | Shuttle Funded | Total | ||||||||||||||||||||||||||||
| Revenue and Expenses | 2021 | 2022 | 2021 | 2022 | 2021 | 2022 | 2021 | 2022 | ||||||||||||||||||||||||
| NIH Reimbursement | - | 211,455 | - | - | - | - | 211,455.00 | 211,455.00 | ||||||||||||||||||||||||
| Compensation | - | - | - | - | 447,222.22 | 532,961.32 | 447,222.22 | 532,961.32 | ||||||||||||||||||||||||
| Subcontracts | 102,855.59 | - | - | - | - | - | 102,855.59 | - | ||||||||||||||||||||||||
| Supplies | 13,522.47 | - | - | - | - | 2,198.70 | 13,522.47 | 2,198.70 | ||||||||||||||||||||||||
| Other, Lab | 40,581.15 | - | - | - | - | 56,076.87 | 40,581.15 | 56,076.87 | ||||||||||||||||||||||||
| Expense total | 156,959.21 | - | - | - | 447,222.22 | 591,236.89 | 604,181.43 | 591,236.89 | ||||||||||||||||||||||||
| Research and Development, Net of Contracts | (156,959.21 | ) | 211,455 | - | - | (447,222.22 | ) | (591,236.89 | ) | (392,726.43 | ) | (379,781.89 | ) | |||||||||||||||||||
In addition, the CEO and CMO are actively involved in the research and development activities, but neither receives a salary from the Company. As such, research and development expenses are lower than might be incurred in the future.Subscription Price
The allocationSubscription Price is $[ ] per Unit. The Subscription Price does not necessarily bear any relationship to our past or expected future results of costsoperations, cash flows, current financial condition, or any other established criteria for value. No change will be made to each research project for the six months ended June 30, 2021 and 2022 are as follows:Subscription Price by reason of changes in the trading price of our common stock or other factor prior to the expiration of this Rights Offering.
Cost Allocation forDetermination of Subscription Price
In the six-month Period Ending June 30, 2022determining the Subscription Price, the board of directors considered a variety of factors including those listed below:
| ● | ||
General and Administrative Expenses. General and Administrative expenses increased by $9,386, from $13,461 in the six months ending June 30, 2021 to $22,847 in the six months ended June 30, 2022. Processing fees related to filing accounted for an increase of $6,215 and are related to pre-IPO activities. Website expenses during the six moths ended June 30, 2022 increased by $1,424 in order to maintain and update the company’s profile and prepare for the Company’s IPO.
Legal and Professional Expenses. Legal and professional expenses increased by $475,242, or 448%, primarily due to increases in fees related to obtaining pre-IPO financing and expenses incurred related to preparing for the IPO.
Other (Income) Expense. Other expense was $221,516 for the six months ended June 30, 2022, which consisted of $315,944 in interest expense on convertible loans, $25,383 in interest expense on related party loans, a gain on change in warrant liability of $46,804 and a $73,007 gain on the forgiveness of the Company’s Paycheck Protection Program loan. Other income was $36,212 for the six months ended June 30, 2021, which consisted of $350 in interest expense, $21,094 in interest expense on related party loans and a gain on change in warrant liability of $57,656.
Comparison of the Years ended December 31, 2021 (Restated) and 2020
The following table summarizes the results of our operations for the years ended December 31, 2021 (Restated) and 2020:
| Years Ended | ||||||||||||||||
| December 31, | ||||||||||||||||
| 2021 | 2020 | Change | % | |||||||||||||
| Revenue | $ | - | $ | - | $ | - | - | |||||||||
| Operating expenses: | ||||||||||||||||
| Research and development, net of contract expense reimbursements | 1,021,808 | 161,772 | 860,036 | 532 | % | |||||||||||
| General and administrative | 36,500 | 85,927 | (49,427 | ) | (58 | %) | ||||||||||
| Legal and professional | 684,684 | 261,823 | 422,861 | 162 | % | |||||||||||
| Total operating expenses | 1,742,992 | 509,522 | 1,233,470 | 242 | % | |||||||||||
| Other income (expense): | ||||||||||||||||
| Interest expense - related party | (46,947 | ) | (36,771 | ) | (10,176 | ) | 28 | % | ||||||||
| Interest expense | (3,841 | ) | (2,859 | ) | (982 | ) | 34 | % | ||||||||
| Change in fair value of warrant liability | 579,146 | (256,580 | ) | 835,726 | 326 | % | ||||||||||
| Gain on disposal of loan | 62,500 | - | 62,500 | 100 | % | |||||||||||
| Total other income (expense) | 590,858 | (296,210 | ) | 887,068 | 299 | % | ||||||||||
| Net loss | $ | (1,152,134 | ) | $ | (805,732 | ) | $ | (346,402 | ) | 43 | % | |||||
Research and Development-Net of contract expense reimbursements. Research and development-net of contract expense reimbursements was $1,021,808 for the year ended December 31, 2021, as compared to $161,772 for the year ended December 31, 2020. For the year ended December 31, 2021, the Company received $505,377 in reimbursement from NIH contracts and incurred $1,527,185 in research and development expenses. For the year ended December 31, 2020, reimbursement from the NIH totaled $1,258,141 and total expenses related to research and development was $1,419,913. The increase of $860,036 or 532% is primarily related to the Company receiving $752,764 less in contract reimbursements in 2021 as compared to 2020. The lower contract reimbursements are due to the completion of Topic 345-Predictive-Biomakers and Phase 1 of Topic 352-Prostate Health Disparity contracts in 2020. In 2021, the Company received $82,467 for Topic 352 Prostate Health Disparity Phase 2 and $422,910 Topic 345-Predictive-Biomakers and Phase 2.
The research and development expenses with the largest variances included compensation of $885,349, subcontractor expenses of $539,043, and lab supply costs of $30,181 for the year end December 31, 2021, as compared to compensation of $888,001, subcontractor expenses of $403,409, and lab supply costs of $57,355 for the year end December 31, 2020. Subcontractor expenses increased by $135,634, or 33.62%, between 2020 and 2021. These expenses increased as more subcontractor services were needed due to the Phase 2 NIH contracts and pending additional financing. All other research and development expense variances between 2020 and 2021 are immaterial.
Below is a breakdown of the actual costs and reimbursements received by the company for the years ended December 31, 2021 and 2020, and a breakdown of how such cost and reimbursements were distributed across research projects.
Key Research and Development Projects
Research and Development, Net of Contract Expense Reimbursements
Periods ending December 31, 2020 and 2021
| Research & Development Revenue and | NIH Topic 345 | NIH Topic 352* | Shuttle Funded | Total | ||||||||||||||||||||||||||||
| Expenses | 2020 | 2021 | 2020 | 2021 | 2020 | 2021 | 2020 | 2021 | ||||||||||||||||||||||||
| NIH Reimbursement | 845,820 | 422,910 | 412,321 | 82,467 | - | - | 1,258,141 | 505,377 | ||||||||||||||||||||||||
| Compensation | 183,183 | 198,426 | 174,026 | - | 530,791 | 686,923 | 888,000 | 885,349 | ||||||||||||||||||||||||
| Subcontracts | 236,633 | 539,043 | 163,979 | - | 2,797 | - | 403,409 | 539,043 | ||||||||||||||||||||||||
| Supplies | 24,670 | 30,181 | 32,655 | - | - | - | 57,355 | 30,181 | ||||||||||||||||||||||||
| Other, Lab | 36,969 | 72,611 | 34,179 | - | - | - | 71,148 | 72,611 | ||||||||||||||||||||||||
| Expense total | 481,485 | 840,261 | 404,840 | - | 533,588 | 686,923 | 1,419,913 | 1,527,185 | ||||||||||||||||||||||||
| Research and Development, Net of Contracts | 364,336 | (417,351 | ) | 7,481 | 82,467 | (533,588 | ) | (686,923 | ) | (161,772 | ) | (1,021,808 | ) | |||||||||||||||||||
Note: The Project 352 final reimbursement was received in 2021 and research costs were company funded through an NIH extension without cost
In addition, our CEO and CMO are actively involved in the research and development activities, but neither receives a salary from the Company. As such, research and development expenses are lower than might be incurred in the future.
General and Administrative Expenses. General and Administrative expenses decreased by $49,427, from $85,927 for the year ended December 31, 2020, to $36,500 for the year ended December 31, 2021. There were two changes that account for the decrease in expenses: (1) directors and officers insurance decreased by $34,322 from $45,629 in 2020 to $$11,308 in 2021; and (2) a decrease in travel and conference expenses by $16,686 from $16,887 in 2020 to $202 in 2021. The decrease in costs for insurance was due to a negotiated reduction in premiums for 2021 and the reduction in the number of members serving on our board of directors from seven directors in 2020 to five directors in 2021. Travel and conference fees decreased from 2020 to 2021 due to a shift to virtual investment conferences related to the ongoing Covid-19 pandemic and a change in company strategy from contacting investors through conferences in 2020 to contacting investors through introductions made through an agreement with Boustead Securities.
Legal and Professional Expenses. The 162% increase in legal and professional fees was primarily due to a non-cash payment of $420,000 for business consulting services resulting from the transfer of 210,000 shares (105,000 shares on a post-split basis) of common stock from a major shareholder, who is also the wife our Chairman and CEO, to a business consultant, and increases in accounting and consulting fees of $684,684 for the year ended December 31, 2021 as compared to $261,823 for the year ended December 31, 2020.
Other Income (Expense).Other income was $590,858 for the year ended December 31, 2021, which consisted of $3,841 for interest expense on convertible loans, $46,947 interest expense on a loan-related party, gain on change in warrant liability of $579,146 and $62,500 gain on the forgiveness of the PPP loan. Other expense was $296,210 for the year ended December 31, 2020, which consisted of $2,859 for interest expense on convertible loans, $36,771 interest expense on a loan-related party and loss on change in warrant liability of $256,580.
Liquidity and Capital Resources
Our capital needs to date have been met by contributions from existing shareholders, as well as through private offerings of our securities, SBIR contracts and other grants. In the six months ended June 30, 2022, we raised a total of $525,715 through the sale of convertible notes and warrants. In the year ended December 31, 2021, we raised a total of $687,932 through the sale of convertible notes and warrants and PPP loans. In addition, since inception, we have received a total of $5,531,722 in SBIR contracts and other grants received primarily through the National Institutes of Health.
We believe that we will continue to expend substantial resources for the foreseeable future on the completion of clinical development and regulatory preparedness of our product candidates, preparations for a commercial launch of our product candidates, if approved, and development of any other current or future product candidates we may choose to further develop. These expenditures will include costs associated with research and development, conducting preclinical studies and clinical trials, obtaining marketing approvals, and, if we are not able to enter into planned collaborations, manufacturing and supply as well as marketing and selling any products approved for sale. In addition, other unanticipated costs may arise. Because the outcome of any drug development process is highly uncertain, we cannot reasonably estimate the actual amounts necessary to complete the development and commercialization of our current product candidates, if approved, or future product candidates, if any.
There can be no assurance that additional financing will be available to us when needed, on favorable terms or otherwise. Moreover, any such additional financing may dilute the interests of existing shareholders. The absence of additional financing, when needed, could cause us to delay implementation of its business plan in whole or in part, curtail its business activities and seriously harm us and our prospects.
Balance Sheet Data:
| June 30, | December 31, | |||||||||||||||
| 2022 | 2021 | Change | % | |||||||||||||
| Current assets | $ | 63,952 | $ | 509,615 | $ | (445,663 | ) | (87 | )% | |||||||
| Current liabilities | 2,453,951 | 2,217,331 | 236,620 | 11 | % | |||||||||||
| Working capital deficiency | $ | (2,389,999 | ) | $ | (1,707,716 | ) | $ | (682,283 | ) | 40 | % | |||||
As of June 30, 2022, total current assets were $63,952. Total current liabilities as of June 30, 2022, were $2,453,951, resulting in a working capital deficiency of $2,389,999. As of December 31, 2021, total current assets were $509,615. Total current liabilities as of December 31, 2021, were $2,217,331, resulting in a working capital deficiency of $1,707,716. The current assets primarily resulted from $525,715 cash received from notes payable issued. The increase in current liabilities is due to the increase in notes payable issued, forgiveness of the PPP loan and payments on trades payable.
Cash Flows from Operating Activities
| Six Months Ended | ||||||||||||||||
| June 30, | ||||||||||||||||
| 2022 | 2021 | Change | % | |||||||||||||
| Cash used in operating activities | $ | (980,027 | ) | $ | (147,641 | ) | $ | (832,386 | ) | 564 | % | |||||
| Cash used in investing activities | $ | - | $ | - | $ | - | 0 | % | ||||||||
| Cash provided by financing activities | $ | 525,715 | $ | 73,007 | $ | 452,708 | 620 | % | ||||||||
| Cash on hand | $ | 50,437 | $ | 42,519 | $ | 7,918 | 19 | % | ||||||||
We have not generated positive cash flows from operating activities. For the six months ended June 30, 2022, net cash flows used in operating activities was $980,027, consisting of a net loss of $1,279,114, reduced by depreciation expense of $2,899, loss on change in warranty liability of $18,772 amortization of right of use assets of $29,599, amortization of debt discount of $278,531, stock-based compensation of $333,066, gain on forgiveness of the PPP of loan of $73,007 and a net change in working capital of $290,773. For the six months ended June 30, 2021, net cash flows used in operating activities was $147,641, consisting of a net loss of $476,125, adjusted for depreciation expense of $2,700, change in warranty liability of $57,656, amortization of right of use assets of $26,582, stock-based compensation of $245,034, and a net change in working capital of $111,824.
Cash Flows from Investing Activities
For the six months ended June 30, 2022 and 2021, we had no investing activities.
Cash Flows from Financing Activities
For the six months ended June 30, 2022, we received $525,715 from the issuance of convertible notes. For the six months ended June 30, 2021, we received, $73,007 from the Paycheck Protection Program.
Going Concern
The accompanying financial statements have been prepared in conformity with generally accepted accounting principles, which contemplate continuation of the Company as a going concern. The Company’s only revenue source since inception from 2014 through present has been awards from government contracts totaling $5,531,722, and the Company has incurred losses since inception, having accumulated a deficit of $7,126,082 as of June 30, 2022. We currently have limited liquidity and have not completed our efforts to establish a stabilized source of revenues sufficient to cover operating costs over an extended period. These factors, among others, raise substantial doubt about our ability to continue as a going concern.
We will need to raise capital to fund its operations. To address its financing requirements, the Company intends to seek financing through debt or equity financings with an aim to continue progress toward commercial viability of its products. We continue to submit Federal grant and contract applications which have historically been the primary source of revenue. The financial statements do not include any adjustments that might result from the outcome of the uncertainty of raising additional capital.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that are material to investors.
Critical Accounting Policies and Significant Judgments and Estimates
This discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with generally accepted accounting principles in the United States (“GAAP”). The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported expenses incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. While our significant accounting policies are described in more detail in the notes to our financial statements included elsewhere in this registration statement, we believe that the following accounting policies are critical to understanding our historical and future performance, as these policies relate to the more significant areas involving management’s judgments and estimates.
Our most critical accounting policies and estimates relate to the following:
Research and Development
Research and Development expenses are offset by contract receivable payments from an NIH SBIR contract that supports this scientific research. This is stated in the financials as research and development-net of contract expense reimbursements.
Operating Lease Right-of-use Assets and Operating Lease Liability
Operating lease right-of-use assets and liabilities are recognized at the present value of the future lease payments at the lease commencement date. The interest rate used to determine the present value is our incremental borrowing rate, estimated to be 10%, as the interest rate implicit in most of our leases is not readily determinable. Operating lease expense is recognized on a straight-line basis over the lease term.
Derivative Financial Instruments
We evaluate all of our agreements to determine if such instruments have derivatives or contain features that qualify as embedded derivatives. For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair value and is then re-valued at each reporting date, with changes in the fair value reported in the statements of operations. For stock-based derivative financial instruments, we use a Binomial Simulation model to value the derivative instruments at inception and on subsequent valuation dates. The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is evaluated at the end of each reporting period. Derivative instrument liabilities are classified in the balance sheet as current or non-current based on whether or not net-cash settlement of the derivative instrument could be required within 12 months of the balance sheet date. As of June 30, 2022, our only derivative financial instrument was an embedded warrant feature associated with warrants issuable to our Series A convertible preferred stockholders upon completion of our initial public offering or public listing due to certain provisions that allow for a change in the warrant value based on fluctuations of our fair value of common stock at the date of issuance of the warrant based on certain contingent call features.
We are a clinical stage pharmaceutical company leveraging our proprietary technology to develop novel therapies designed to cure cancers. Our goal is to extend the benefits of cancer treatments with surgery, radiation therapy, chemotherapy and immunotherapy. Radiation therapy (RT) is one of the most effective modalities for treating cancers. We are developing a pipeline of products designed to address limitations of the current cancer therapies as well as to extend to the new applications of radiation therapy. We believe that our product candidates will enable us to deliver cancer treatments that are safer, more reliable and at a greater scale than that of the current standard of care.
Our product candidates include Ropidoxuridine, Extended Bio-availability Ropidoxuridine (IPdR/TPI), and a platform of HDAC inhibitors (SP-1-161, SP-2-225 and SP-1-303). We have advanced Ropidoxuridine through a Phase I clinical trial using non-dilutive NIH SBIR contracts and are currently preparing a Phase II study that we intend to commence in 2022. We also plan to submit investigational new drug applications (INDs) for the extended Bio-availability Ropidoxuridine with the goals of initiating Phase I clinical trials in 2023, leveraging the outcomes of the Phase I clinical study results of Ropidoxuridine. We have applied for and received FDA approval of Orphan designation for Ropidoxuridine and RT for treating brain cancer (glioblastoma). In addition, we plan to continue to develop our pre-clinical products SP-1-161, SP-2-225 and SP-1-303 with the goal of submitting INDs in 2023 and 2024. We believe our management team’s expertise in radiation therapy, combined modality cancer treatment and immuno-oncology will help drive the development and, if approved, the commercialization of these potentially curative therapies for patients with aggressive cancers.
Radiation Oncology has gone through transformative technological innovation over the last several years to better define tumors, allow improved shaping of radiation delivery and support dose escalation with shorter courses of treatment. Furthermore, achieving higher dose distributions within tumor volumes has reached a practical plateau, since cancers are frequently integrated with or surrounded by more sensitive normal tissues and further dose increases risk of tissue necrosis. To increase cancer cures at maximally tolerated radiation doses, pharmacological and biological modifications of cells are needed to sensitize cancers, protect normal tissues, and stimulate the immune system to react against antigens produced by irradiated, damaged cancer cells. Drugs that show sensitizing properties, or the ability to make cancer cells more sensitive to radiation, offer a solution to this problem. Currently, such drugs are used off-label, and many have inherent toxicities since they were designed for direct cancer treatments and not for sensitization.
We are developing our products with the goal of addressing the unmet need in cancer treatment for a commercially marketable radiation response modifier solution that leads to greater sensitivity of cancer cells to ionizing radiation therapy. The goal of our products is to increase the therapeutic index for patients receiving radiation and to decrease radiation-related toxicities in patients with solid tumors. Our products operate across three areas related to the treatment of cancer with RT:
Our platform technology allows for the creation of an inventory of products for radiation sensitizing, immune modulation, and protection of healthy tissue.
Our Pipeline
We are currently developing a pipeline of small molecule radiation sensitizers and immune response regulating drugs. Our most advanced product candidate is Ropidoxuridine, an orally available halogenated pyrimidine with strong cancer radiation sensitizing properties in preclinical studies. In addition to our clinical study-ready candidate, we have a pipeline of complimentary product candidates that we are developing to address a host of solid tumor cancer indications. Our pipeline is represented in the diagram below:

Timeline for clinical phase (Ropidoxuridine) and pre-clinical phase (HDAC inhibitors) pipeline.
Our lead product candidates include:
| ● | ||
| ● | ||
| ● |
Our Approach
We believe that we have established a leadership position in radiation sensitizer discovery and development. Over approximately six years of research, we have identified two clinical phase product candidates and discovered new pre-clinical molecules using our proprietary platform technologies to increase the therapeutic index for patients receiving radiation for treatment of solid tumors. Our development strategy has four key pillars: (1) to improve the efficacy of RT by demonstrating improved disease-free survival rates in patients who undergo radiation therapy, (2) reduce the amount of radiation needed for a favorable tumor response, thereby limiting the potential for radiation related toxicities to healthy cells, (3) decrease the extent of surgery needed to remove cancers and improve quality of life, and (4) leverage our next generation technologies to create drugs that regulate the immune response assisting immune checkpoint and CAR-T therapies and other personalized medicines targeting cancers.
We propose to perform Phase I and Phase II clinical trials to advance our clinical product candidates. In addition, candidate HDAC inhibitor molecules will be tested, and IND-enabling studies will be performed to prepare for Phase I clinical trials.
To date, we have been awarded three SBIR contracts from the NIH to:
| ● | ||
| ● |
All three SBIR funded projects have been completed. The Company is eligible to apply for SBIR Phase IIb funding to “bridge” the funding gap should Shuttle Pharma elect to advance the “Moon shot” health disparities or the predictive biomarker project. The NIH SBIR program is designed to encourage small businesses to engage in Federal Research/Research and Development (“R/R&D”) that has the potential for commercialization.
Our Strategy
Our goal is to maintain and build upon our leadership position in radiation sensitization. We plan to develop Ropidoxuridine and the HDAC6 inhibitor (SP-2-225) and, if approved by the FDA, commercialize our product candidates for the treatment of cancers. While this process may require years to complete, we believe achieving this goal could result in new radiation sensitizer and immunotherapy products. Key elements of our strategy include:
| ● | ||
Radiation Therapy
The table below detailsSubscription Price does not necessarily bear any relationship to any established criteria for value. No valuation consultant or investment banker has opined upon the numberfairness or adequacy of the Subscription Price. You should not consider the Subscription Price as an indication of actual value of our company or our common stock. The market price of our common stock may decline during or after the Rights Offering. We cannot predict the price at which our shares of common stock will trade after the Rights Offering. You should obtain a current price quote for our common stock and rateperform an independent assessment of cancers occurring inour Warrants before exercising your Subscription Rights and make your own assessment of our business and financial condition, our prospects for the United States in 2018:future, and the terms of this Rights Offering. Once made, all exercises of Subscription Rights are irrevocable.
Estimated New Cancer Cases in the USNo Short-Sales
| Male | Female | |||||||||||||||||
| Prostate | 174,650 | 26 | % | Breast | 268,600 | 38 | % | |||||||||||
| Lung & bronchus | 116,440 | 17 | % | Lung & bronchus | 111,710 | 16 | % | |||||||||||
| Colon & rectum | 78,500 | 12 | % | Colon & rectum | 67,100 | 10 | % | |||||||||||
| Urinary bladder | 61,700 | 9 | % | Uterine corpus | 61,880 | 9 | % | |||||||||||
| Melanoma of the skin | 57,220 | 8 | % | Melanoma of the skin | 39,260 | 6 | % | |||||||||||
| Kidney & renal pelvis | 44,120 | 7 | % | Thyroid | 37,810 | 5 | % | |||||||||||
| Non-Hodgkin lymphoma | 41,090 | 6 | % | Non-Hodgkin lymphoma | 33,110 | 5 | % | |||||||||||
| Oral cavity & pharynx | 38,140 | 6 | % | Kidney & renal pelvis | 29,700 | 4 | % | |||||||||||
| Leukemia | 35,920 | 5 | % | Pancreas | 26,830 | 4 | % | |||||||||||
| Pancreas | 29,940 | 4 | % | Leukemia | 25,860 | 4 | % | |||||||||||
| All sites | 677,720 | All sites | 701,860 | |||||||||||||||
By exercising the Subscription Rights, you are representing to us that you have not entered into any short sale or similar transaction with respect to our common stock since the Record Date for the Rights Offering. In addition, the Subscription Rights provide that, upon exercise of the Subscription Right, you represent that you have not since the Record Date and, for so long as you continue to hold Warrants or Equity Interests issued in connection with the exercise of the Subscription Right, agree not to enter into any short sale or similar transaction with respect to our common stock. These requirements prevent you from pursuing certain investment strategies that could provide you greater financial benefits than you might have realized if the Subscription Rights did not contain these requirements.
2018 Clinical Cancer Advances Report, American College of Clinical Oncology, 2018
Colon Cancer Alliance. Colorectal Cancer Survival Rates from Facts and Figures, 2017. Chicago, IL; 2017
Engineered Radiation SensitizersNo Recombination
The market for radiation sensitizers in selected cancer types is defined bycommon stock, Warrants and Equity Interests comprising the need to improve local-regional tumor control. Treatment regimensUnits will separate upon the exercise of the Subscription Rights, and the Units will not trade as a separate security. Holders may not recombine shares of common stock, Warrants and Equity Interests comprising the Units after they have been developed to address patient needs for tumor control and quality of life. Since the initial applications of Ropidoxuridine and selective HDAC inhibitors are as adjuncts to the standard of care for the treatment of radiation responsive cancers, the unmet needs of the market lie in the potential for the following:
Various sources have estimated that more than 800,000 patients in the US are treated with radiation therapy for their cancers. According to the American Cancer Society about 50% are treated for curative purposes and the balance for palliative care. The market opportunity for radiation sensitizers lies with the 400,000 patients treated for curative purposes. The number of patients being treated with RT is expected to grow by more than 22% over the next five years. Based on a rough estimate of a course of radiation sensitizing brand drug therapy (off label at this time) of $12,000 per patient—the market size would be in excess of $4.0 billion. This would represent about 4% of the annual cost of cancer care in the US.
In the past two decades, developments in the field of oncology have resulted in an increase in the number of clinical trials of marketed products that exhibit radiation sensitizing properties. The following are a few examples of recently approved products that exhibit radiation sensitizing properties: topotecan (Hycamtin®) was approved for ovarian and small-cell lung cancer and also in cervical cancer when used in combination with cisplatin. Irinotecan (Camptosar®) is used for metastatic colorectal carcinoma, trastuzumab (Hercepetin®) for breast cancer, and gefitinib (Iressa®) for locally advanced non-small-cell lung cancer. However, the claims on radiation sensitization are anecdotal in the scientific literature.distributed.
In addition, clinical trials are in progress to develop novel molecules (such as poly (ADP-ribose) polymerase (PARP), histone deacetylase (HDAC) inhibitors (such Zolinza® (vorinostat) Non-Transferability of and heat-shock protein 90 (hsp90) inhibitors with potential to increase the therapeutic use of compounds with radiation sensitizing properties for other cancers. Several drugs with radiation sensitizing properties are currently in Phase III clinical trials, such as nimorazole (for head and neck cancer), motexafin gadolinium (for brain metastases), and cisplatin (for cervical cancer); though none are likely to apply for a radiosensitizing claim with the FDA since the radiosensitizing element in their clinical trials are not primary endpoints. While additional drugs with radiation sensitizing properties are expected to be launched in the future, thereby driving the radiation sensitizers market further, to date, there is no indication that any drug in development is expected to be approved specifically as a radiation sensitizer.Subscription Rights
The competitive environmentSubscription Rights are non-transferable (other than by operation of law) and, therefore, you may not sell, transfer, assign or give away your Subscription Rights to anyone. The Subscription Rights will not be listed for “off-label” radiation sensitizers for solid tumor cancers is anticipated to become predominantly generic. Avastin, Erbitux, Camptosar and Xeloda havetrading on any stock exchange or will lose patent protection in the next three years. Newer products under investigation or approved, such as Vectibix® (panitumumab) from Amgen will be promoted as having radiation sensitizing properties, along with indications for treatment for specific cancers. The high cost of these new therapies coupled with limited efficacy compared to current standard of care will be constrained by both public and private payors. Other new agents are in development but will face similar challenges.
We anticipate that new products launching into the cancer market with anecdotal claims for use as radiation sensitizers with improved effectiveness, quality and tolerability will initially be limited in their growth until they have been added to established clinical pathways and guidelines. If their effectiveness, quality and tolerability are demonstrated clinically, as determined by the FDA, it is anticipated the National Comprehensive Cancer Network (NCCN), the leading authority in oncology drug evaluation for treatment guidelines, would issue a recommendation and addition to standard of care within approximately six to twelve months after launch. An NCCN recommendation would positively impact the growth potential for a new product entering the market. Also, payors, both public and private, would add the new product to their approved drug lists and provide reimbursement giving providers incentive to use the product as neoadjuvant and adjuvant therapy to standard of care.
As with many cancer therapies, side effects can often have a distinct impact on quality of life and influence the potential for market growth. Patients increasingly have a stronger voice in the decision-making process for the appropriate therapies and costs to treat their cancers. As payors are increasingly placing more of the financial burden of the cost of therapy directly on patients, patients are voicing their opinions to their physicians and payors which have a direct effect on which products are selected. Many of the current therapies have significant side effects:
Private insurers are expected to have more restrictive formularies and medical benefits in which patients will be expected to carry more of the burden of the cost of drugs. Also, it is anticipated that increased application of third party developed treatment guidelines, such as those from the NCCN (National Comprehensive Cancer Network), are expected to be used by private payors to limit the access to products for specific conditions through prior authorizations and implementation of step therapy or increased out of pocket cost approaches. As many of the current drugs used as radiation sensitizers are expensive and not approved for use as radiation sensitizers (thus, such treatment is “off label”), and as many of the products in clinical trials are expected to be at the current or higher price levels, new products that may be specifically approved for an indication as the only approved product as a radiation sensitizer will have increased consideration for reimbursement.
CMS is increasingly moving many patients to private insurance through Medicare Advantage and ACOs. Medicare Advantage plans are capitation HMO and PPO plans offered through private insurers to Medicare patients. ACOs are being developed to increase quality of care for their patients. Most of the new ACOs are initially positioned for Medicare patients with over 400 approved by CMS. Several studies from the Center for Health Strategies, 2017, Journal of American Medical Association, 2018 and the Brookings Institute, 2015 estimated that almost 1000 ACOs for Medicare and non-Medicare patient populations would be approved by CMS or developed by a variety of healthcare entities to begin operating under the ACA in 2017. We expect the growth in ACOs to continue, regardless of any changes that may be made to the ACA going forward. In early 2017, Health Affairs, a magazine tracking ACOs, estimated that over 22 million patients are enrolled in Medicare and private ACOs. To address the quality of care measures designated by CMS and to gain additional incentives, use of clinical pathways or treatment guidelines is anticipated to be increasingly instituted to manage patient care. The impact on the uptake of new products in this environment can be profound if the new product is first in class and is included in national guidelines from organizations such as the NCCN and/or approval by the regional CMS contracting groups.
ROPIDOXURIDINE
The halogenated thymidine (TdR) analogs, bromodeoxyuridine (BUdR) and iododeoxyuridine (IUdR), are a class of pyrimidine analogs that have been recognized as potent radiosensitizing agents since the early 1960s. (See Kinsella TJ. An Approach to the Radiosensitization of Human Tumors. Cancer J Sci Am. Jul-Aug 1996:2(4); 184-193). Their cellular uptake and metabolism are dependent on the TdR salvage pathway where they are initially phosphorylated to the monophosphate derivative by the rate-limiting enzyme, thymidine kinase (TK). (See Shewach DS, Lawrence TS. Antimetabolite radiosensitizers. J Clin Oncol, Sep 10 2007; 25(26):4043-4050). After sequential phosphorylation to triphosphates, they are then used in DNA replication, in competition with deoxythymidine triphosphate (dTTP), by DNA polymerase. DNA incorporation is a prerequisite for radiosensitization of human tumors by the halogenated TdR analogs, and the extent of radiosensitization correlates directly with the percentage TdR replacement in DNA. (See Lawrence TS, Davis MA, Maybaum J, Stetson PL, Ensminger WD. The Dependence of Halogenated Pyrimidine Incorporation and Radiosensitization on the Duration of Drug Exposure. International Journal of radiation oncology, biology, physics. Jun 1990; 18(6);1393-1398). The molecular mechanisms of radiosensitization are most likely the result of increased susceptibility of TdR analog-substituted DNA to the generation of highly reactive uracil free radicals by ionizing radiation (IR), which may also damage unsubstituted complementary-strand DNA. Repair of IR damage may also be reduced by pre-IR exposure to these analogs.
The rationale for using Ropidoxuridine as a radiation sensitizer is based on prior clinical studies with the active metabolite IUdR; identified in NIH laboratories as a potent radiation sensitizer. Ropidoxuridine is an orally available prodrug of IUdR. In the body, Ropidoxuridine is metabolized in the liver into IUdR. IUdR is incorporated into the DNA of actively growing cells and when cells are exposed to ionizing radiation, DNA strand breaks are generated, resulting in more cell death and radiation sensitization. (See Gurkan E, Schupp JE, Aziz MA, Kinsella TJ, Loparo KA. Probabilistic modeling of DNA mismatch repair effects on cell cycle dynamics and iododeoxyuridine-DNA incorporation. Cancer Res. Nov 15 2007; 67(22):10993-11000).
Most of the clinical efficacy data were obtained from NIH supported studies performed with IUdR, the active metabolite of Ropidoxuridine. However, IUdR requires constant infusion over six weeks of therapy which creates a significant compliance issue for patients. Ropidoxuridine can be given as a capsule for oral administration, resulting in greater ease of medication delivery and potentially improved compliance and fewer complications.
Over the last 20 years, there has been renewed interest in these halogenated TdR analogs as experimental radiation sensitizers in selected cancer patient groups. These analogs are rapidly metabolized in both rodents and humans, principally with cleavage of deoxyribose and subsequent dehalogenation by hepatic and extrahepatic metabolism, when given as a bolus infusion with a plasma half-life of <5 min. Consequently, prolonged continuous or repeated intermittent drug infusions over several weeks before and during irradiation are necessary, based on in vivo human tumor kinetics, to maximize the proportion of tumor cells that incorporate these analogs into DNA during the S phase of the cell cycle. (See Fowler JF, Kinsella TJ. The Limiting Radiosensitization of Tumors by S-phase Sensitizers. Br J Cancer. 1996;74 (Suppl)(27):294-296). Phase I and Phase II trials using prolonged continuous or repeated intermittent intravenous infusions of BUdR or IUdR before and during radiation therapy (RT) have focused principally on patients with high-grade brain tumors. These clinically radiation resistant tumors can have a rapid proliferation rate (potential tumor doubling times of 5–15 days) and are surrounded by non-proliferating normal brain tissues that show little to no DNA incorporation of the TdR analogs. As such, high-grade brain tumors are ideal targets for this approach to radiation sensitization. In Phase I/Phase II clinical trials, prolonged survival outcomes were observed compared to RT alone in patients with anaplastic astrocytomas and in patients with glioblastoma multiforme IUdR continuous IV infusion (1000 mg/m2/ day/ 14 days), Total 39 patients (F. Sullivan, et al. Int J Radiat Oncol Biol Phys. 1994; 30(3):583-90.) A therapeutic gain in clinical radiation sensitization using these halogenated TdR analogs was proposed for other types of clinically poorly radiation responsive (radiation resistant) cancers, including locally advanced cervical cancer, head and neck cancers, unresectable hepatic metastases from colorectal cancers, and locally advanced sarcomas, based on the results of other Phase I/Phase II clinical trials.
Target Indication: Glioblastoma, Sarcomas and Rectal Cancers
After completion of the Phase I clinical trial of Ropidoxuridine and RT in advanced GI cancers, we proposed to perform Phase II efficacy clinical trials in brain tumors (glioblastoma), soft tissue sarcomas, and rectal cancers. Glioblastoma multiforme is a deadly malignancy of the brain with no known cure. Radiation therapy provides delay of disease progression and is standard of care following surgical resection or biopsy. Radiation therapy is combined with Temodar, a drug that has shown activity (~ four months survival benefit) in treating brain tumors. Preliminary data using radiation therapy in combination with IUdR resulted in a delay of disease progression of up to six months. We propose to test IPdR in combination with radiation therapy in the Phase II clinical trial. Similarly, delay in disease progression has been observed following treatment of sarcomas by the combination of IUdR and RT. Based on the Phase I data of our clinical trial we know that therapeutic levels of IUdR are reached by administering the orally available prodrug, IPdR.
Clinical DataExpiration Date; Extension
The Phase I resultssubscription period, during which you may exercise your Subscription Rights, expires at 5:00 p.m., Eastern Time, on , 2024, which is the expiration of the clinical trial supportedRights Offering. If you do not exercise your Subscription Rights before that time, your Subscription Rights will expire and will no longer be exercisable. We will not be required to issue shares to you if the Subscription Agent receives your Rights Certificate or your subscription payment after that time. We have the option to extend the Rights Offering in our sole discretion for any reason up to an additional 45 days, although we do not presently intend to do so. We may extend the Rights Offering by an SBIR contract to Shuttle Pharma and a sub-contractgiving oral or written notice to the Brown University Oncology Group (BrUOG) atSubscription Agent before the LifeSpan/Rhode Island Hospital were reported byRights Offering expires. If we elect to extend the subcontractor atRights Offering, we will issue a press release announcing the 30th EORTC-NCI-AACR Symposium in November 2018 andextension no later than 9:00 a.m., Eastern Time, on the next business day after the most recently announced expiration date of the Rights Offering.
If you hold your shares of common stock or warrants in the medical journal, Clinical Cancer Research,name of a broker, dealer, bank or other nominee, the nominee will exercise the Subscription Rights on your behalf in 2019. Eighteen patients completed dose escalation to 1800 mg/day for 30 days, establishingaccordance with your instructions. Please note that the maximum tolerated dose (MTD) of 1,200 mg/day in combination with RT. Therapeutic blood levels of IUdR were achieved. Four patients were scored as partial responses, nine patients had stable disease and one patient progressed innominee may establish a deadline that may be before 5:00 p.m., Eastern Time, on , 2024, which is the target lesions. These data support advancing IPdR and RT to clinical trialsexpiration date that we have established for the FDA to determine efficacy.Rights Offering.
| 34 |
Development PlanTermination
A keyWe may terminate the Rights Offering at any time and for any reason prior to driving the Ropidoxuridine product forward,expiration of the new formulationRights Offering. If we terminate the Rights Offering, we will issue a press release notifying stockholders and the public of IPdR/TPI, is the development of a clinical plan with aggressive timelines and support within the radiation oncology community to participate in clinical trials with the appropriate patients to ensure a comprehensive NDA dossier for each product. Initially, the plan is focusedtermination no later than 9:00 a.m., Eastern Time, on the Phase I and Phase II clinical trials. Uponnext business day after the most recently announced expiration date of the Rights Offering.
Return of Funds upon Completion or Termination
The Subscription Agent will hold funds received in payment for shares in a segregated account pending completion of these studies,the Rights Offering. The Subscription Agent will hold this money until the Rights Offering is completed or is terminated. To the extent you properly exercise your Over-Subscription Privilege for an amount of Units that exceeds the number of unsubscribed Units available to you, any excess subscription payments will be returned to you within 10 business days after the expiration of the Rights Offering, without interest or deduction. If the Rights Offering is terminated for any reason, all subscription payments received by the Subscription Agent will be returned within 10 business days, without interest or deduction.
Shares of Our Capital Stock, Warrants and Equity Interests in our Subsidiary Outstanding After the Rights Offering
Assuming no other transactions by us involving our capital stock prior to the expiration of the Rights Offering, and if the Rights Offering is fully subscribed, upon consummation of the Rights Offering we will determine whetherhave [ ] shares of common stock issued and outstanding, Warrants to extendpurchase additional shares of our common stock issued and outstanding, and Equity Interests in Diagnostics outstanding, based on 16,894,893 shares of our common stock outstanding as of [ ], 2024. Assuming the Phase II studyfull $4.5 million of Units are sold in the Rights Offering, a total of [ ] shares of common stock, [ ] warrants to a randomized Phase II, or to perform a randomized Phase III clinical trial. Such determinationpurchase common stock and [ ] Equity Interests in Diagnostics will be based,sold in part, on results of the initial clinical trials and the end of a Phase II meeting with the FDA. Shuttle Pharmaceuticals requested and received FDA orphan drug status for Ropidoxuridine as a clinical radiation sensitizer for treatment of glioblastoma and pre-operative treatment of soft tissue sarcomas.Rights Offering. As a result, we will have a total of [ ] shares of common stock outstanding following the applicationRights Offering, along with [ ] warrants to purchase common stock and we will have sold 44% of the Equity Interest in Diagnostics, with 56% of Diagnostics being held by the Company.
Methods for “orphan” designationExercising Subscription Rights
The exercise of Subscription Rights is irrevocable and may not be cancelled or modified. You may exercise your Subscription Rights as follows:
Subscription by Record Holders
If, as of the Record Date, you are a holder of record of common stock or warrants, the number of Units you may purchase pursuant to your Subscription Rights is indicated on the enclosed Rights Certificate. You may exercise your Subscription Rights by properly completing and executing the Rights Certificate and forwarding it, together with your full payment, to the Subscription Agent at the address given below under “Subscription Agent,” to be received before 5:00 p.m., Eastern Time, on , 2024.
Subscription by Beneficial Owners
If as of the Record Date you are a beneficial owner of shares of our common stock or warrants that are registered in the name of a broker, dealer, bank or other nominee, you will not receive a Rights Certificate. Instead, we will issue one Subscription Right to such nominee record holder for Ropidoxuridine with RTall shares of our common stock or warrants held by such nominee at the Record Date. If you are not contacted by your nominee, you should promptly contact your nominee in order to subscribe for glioblastoma has been approved. The applicationshares in the Rights Offering and follow the instructions provided by your nominee.
To properly exercise your Over-Subscription Privilege, you must deliver the subscription payment related to your Over-Subscription Privilege before the Rights Offering expires. Because we will not know the total number of unsubscribed Units before the Rights Offering expires, if you wish to maximize the number of shares you purchase pursuant to your Over-Subscription Privilege, you will need to deliver payment in an amount equal to the aggregate subscription payment for sarcomas, however, was not approved and will require addressing certain FDA comments and resubmission. The IPdR/TPI formulation clinical plan will focus on resectable stage II and III rectal cancer patients.the maximum number of Units that you wish to purchase.
| 35 |
Payment Method
Our clinical plan
Payments must be made in full in U.S. currency by personal check, certified check or bank draft, or by wire transfer, and payable to “Vstock Transfer, LLC, as Subscription Agent for Ropidoxuridine development includes:
Shuttle Pharmaceuticals Holdings, Inc.” You must timely pay the full subscription payment, including payment for the Over-Subscription Privilege, for the full number of shares of our common stock you wish to acquire pursuant to the exercise of Subscription Rights by delivering a:
| ● | ||
We believe the data obtained from the NIH/NCI SBIR funded Phase I clinical trial supports efforts to raise additional capital to enable performing the Phase II clinical trials of Ropidoxuridine. We aim to conduct and complete the Phase II clinical trial so that we may present data to the FDA for its determination of efficacy. We believe this will support our efforts to raise the additional required capital to fund Phase III clinical trials and seek FDA approval of an NDA with “orphan” designation.
The clinical plan for the IPdR/TPI formulation will focus on resectable Stage II and Stage III rectal cancer patients. Nonetheless, we cannot guarantee the successful completion of any of these trials. Our inability to meet any of the aforementioned milestones in the Phase II or Phase III clinical trials will cause us to be unable to proceed with our present efforts and will likely cause us to be unable to raise additional funds.
Our HDAC Small Molecule Delivery Platform
General
Since the founding of Shuttle Pharma, our discovery research and development efforts have been focused on our small molecule technology delivery platform which uses HDAC inhibitors, designed to target cancer cells, while protecting healthy tissue.
HDACs are a class of enzymes that regulate gene expression through chemical modification of histones and non-histone proteins. Increased HDAC activity leads to a more condensed chromatin (which is a protein complex consisting of DNA and other proteins), decreased gene expression and loss of key gene products, including tumor suppressor gene function. Inhibition of HDAC activity leads to a more open chromatin and increased expression of the key gene products. This chromatin modification underlies the epigenetic cellular regulatory system and is an area of intense investigation.
Our research and development efforts to date have focused on the discovery of novel, dual functional molecules for potential use in cancer treatment as radiation sensitizers of cancers, protectors of normal tissues, and activators of the immune responses to antigens expressed by irradiated cancer cells. To date, we have produced three candidate molecules:
SP-1-161 - A Dual Functional Agent
SP-1-161 is an HDAC inhibitor of the hydroxamate chemical class of compounds and an ATM activator of the indole chemical class. HDACs modify histones and non-histone proteins, which are key components of the chromatin structure, gene expression regulation, and cell growth. HDAC inhibitors inhibit cell proliferation, angiogenesis and immunity. Eighteen human HDACs have been identified, subdivided into four classes based on sequence and functional homology. In cancer cells, HDAC activity silences tumor suppressor genes important for cell growth regulation and to chromosomal instability. Abnormal HDAC activity is also associated with tumor cell growth, invasion, metastasis and resistance to therapy. Therefore, inhibitors of HDACs have emerged as anti-cancer agents for cancer therapy. Vorinostat and romidepsin have been approved by the FDA for treatment of patients with relapsed or refractory T-cell lymphomas. In addition, panobinostat received FDA approval for treatment of recurrent multiple myeloma in combination with bortezomib and dexamethasone.
In preclinical studies, SP-1-161 inhibited the activity of pan-HDACs and activated the ATM gene product. ATM is a critical protein for the activation of the cell stress response for cellular recovery from radiation exposure in normal cells, but not in cancer cells. ATM activates the P53 protein, referred to as the “guardian of the genome,” and serves as a tumor suppressor critical for normal cell function and activation of programmed cell death in cancer cells.
In preclinical studies, SP-1-161 protected normal breast epithelial cells (184A1) following exposure to ionizing radiation while increasing sensitivity of breast cancer cells (MCF7). SP-1-161 provides this dual function in a single molecule and this molecule is differentiated from other HDAC inhibitors by treatment of cancers while protecting normal cells.
SP-2-225
SP-2-225 is a selective HDAC inhibitor that affects histone deacetylase (HDAC6) and is a member of the class IIb HDAC family. Class II HDACs play important roles in cancer motility, invasion, neurological diseases, and immune checkpoint. HDAC6 inhibition has been most extensively studied for its role in the treatment of hematological cancers. HDAC6 is unique among HDAC enzymes in having two active catalytic domains and a unique physiological function. In addition to the modification of histones, HDAC6 targets specific substrates including α-tubulin and HSP90, and are involved in protein trafficking and degradation, cell shape and migration. Selective HDAC6 inhibitors are an emerging class of pharmaceuticals due to the involvement of HDAC6 in pathways related to neurodegenerative diseases, cancer and immunology. Specifically, its potential to affect regulation of the immune system and enhance the immune response in cancer is of great interest. With the introduction of check-point inhibitors, CAR-T therapies and personalized medicine in cancer, regulation of the immune response to this therapy is of significant clinical and commercial interest. (See Grindrod S, Brown M, Jung M. “Development of dual Function Small Molecules as Therapeutic Agents for Cancer Research,” Poster presentation #A178, American Association of Cancer Research Oct 2017).
Selective inhibition of HCAC6 reduces dose limiting side effects associated with non-selective HDAC inhibitors. Selective HDAC6 inhibitors may be combined with other cytotoxic agents. Shuttle’s discovery of selective HDAC inhibitors has yielded several HDAC6 selective candidate molecules including SP-2-225. HDAC6 inhibitors are under investigation for roles in the treatment of diseases such as multiple myeloma.
SP-1-303 - Target Indication: Breast Cancer
Histone deacetylase inhibitors sensitize cancers to the effects of radiation, protecte normal tissues from radiation injury and activate the immune system. SP-1-303 is a selective Class I HDAC inhibitor that inhibits HDAC1, 3 and 6 and has direct cellular toxicity in ER positive breast cancer cells. Furthermore, SP-1-303 increases the PD-L1 expression level in a time-dependent manner, support combination of SP-1-303 with an immune checkpoint blocker to enhance the therapeutic benefits. We are currently conducting preclinical efficacy studies of these molecules.
Development Plan
The HDAC inhibitor platform of candidate molecules will require pre-clinical evaluation, completion of IND-enabling studies and the lead drug candidates will be tested in Phase I clinical trials for pharmacokinetics and MTD determination. We have three lead candidates for potential development for the treatment of solid tumors, including breast cancer, lung cancer and multiple myeloma.
The results of Phase I and Phase II clinical trials will determine further drug development and Shuttle will seek to establish collaborative partnerships with other pharmaceutical companies to complete pre-clinical and clinical development, drug manufacturing and marketing of our product candidates. In the event we are unsuccessful in completing our clinical trials at any stage, or in the event we obtain negative results, we will likely be unable to raise additional funding related to our HDAC studies or will have to change direction of our research efforts regarding the HDAC inhibitor platform of candidate molecules.
Our Manufacturing Strategy
We have no manufacturing facilities that are company owned or operated. We have performed laboratory scale synthesis and testing in our research laboratories in Gaithersburg, Maryland. GMP synthesis of API, drug formulation and human dosage preparation will be performed under contracts with third-party manufacturers.
Strategic Agreements
We have developed important strategic agreements with academic institutions for access to resources such as intellectual property, core facilities and contracting relationships. In addition, we have established an agreement with Propagenix for intellectual property in-licensing. Our current and ongoing relationships include:
| ● |
Competition “Off-Label” Use
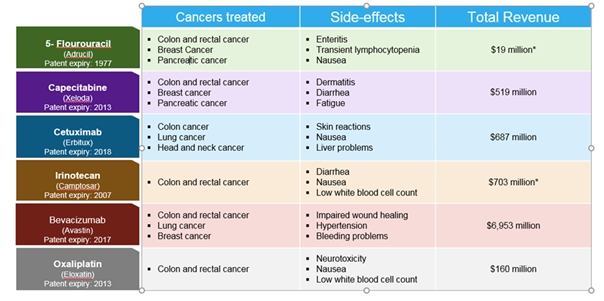
Drugs with radiation sensitizing properties.
Our Product Candidates
We are advancing a clinical stage product candidate, Ropidoxuridine, that we believe will target cancer cells while protecting healthy tissue when used in conjunction with RT.
Ropidoxuridine
Ropidoxuridine, an orally available halogenated pyrimidine with strong cancer radiation sensitizing properties, is our lead “clinical phase” product candidate. Halogenated pyrimidines are incorporated into DNA by rapidly growing cancer cells and become more sensitive to the effects of RT. We have received an SBIR contract from the NIH to fund a Phase I clinical trial in collaboration with Brown University at the Lifespan/Rhode Island Hospital to determine the maximum tolerated dose in patients with advanced gastrointestinal cancers. In connection with the trial, NCI has approved the Phase I clinical protocol and provided drug and clinical data management support to Rhode Island Hospital. The Phase I clinical trial has been completed and the results support advancing Ropidoxuridine to Phase II clinical trials of brain tumors, sarcomas and other tumors through contracted research organizations (or CROs).
The following tables provide data from reported clinical trials of Iododeoxyuridine and RT therapy in brain cancers (glioblastoma multiforme) and high-grade sarcomas. Our primary strategy for Ropidoxuridine and RT therapy is to provide oral drug delivery to effect radiation sensitization of cancers and validate effectiveness in glioblastoma and sarcoma, potential “Orphan” indications.
Brain Cancer Treatment
Efficacy compared to historical RT-alone controls for treatment
of high-grade primary brain tumors (RTOG*, NCI** trials)
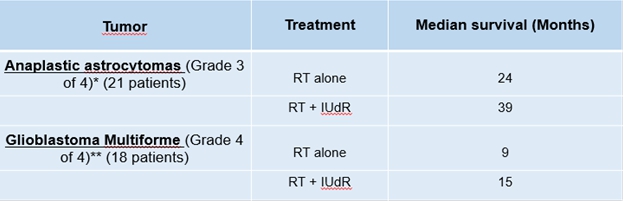
Sarcoma Treatment
Efficacy compared to historical RT-alone controls for treatment
of high-grade sarcomas (University of Michigan*** trials)

In addition to our primary product candidate, we are developing and planning to develop other cancer radiation sensitizers and radiation protectors, which target protecting normal tissue during the administration of RT, and other products utilizing our HDAC small molecule technology platform.
SBIR Contracts
The SBIR Program
The Small Business Innovation Research program, as developed by Congress under the Small Business Innovation Development Act of 1982, is designed to encourage domestic small businesses to engage in Federal Research/Research and Development (“R/R&D”) that has the potential for commercialization. Through a competitive awards-based program, SBIR enables small businesses to explore their technological potential and provides the incentive to profit from its commercialization. Some of the SBIR’s program goals include stimulating technological innovation, meeting Federal research and development needs and encouraging participation in innovation and entrepreneurship.
The SBIR program is a three-phase program. Phase 1 is to establish the technical merit and commercial potential of the proposed R/R&D efforts. Phase 2 is to continue the R/R&D efforts initiated in Phase 1 and funding is based on the results achieved in Phase 1. Phase 3 allows for the small business to pursue commercialization objectives resulting from the Phase 1 and 2 R/R&D activities. In addition, companies that have successfully completed Phases I and II are also eligible to apply for Phase IIb funding.
In addition to the SBIR contract to fund our Phase I clinical study on Ropidoxuridine in combination with RT for treatment of advanced gastrointestinal cancers, we have also received awards of SBIR contracts from the NIH to address prostate cancer health disparities and prostate cancer radiation biomarker development.
As of the date of this prospectus, all SBIR contracts received by the Company have been completed. The Company submitted a final report for SBIR contract # 75N81018C00031 on March 28, 2022. The following summary of terms for the three Phase II SBIR contracts is provided below.
Summary of SBIR Contracts
Prostate Cancer Studies to Address Health Disparities
Prostate cancer health disparities studies have shown that African-American men are at higher risk for developing prostate cancer, as well as at higher risk of cancer specific death rates as compared to Caucasian American men. The causes of disparities have been attributed to socioeconomic differences, environmental exposures and biological factors. Most disparities studies have been population based, in part, due to the lack of relevant in vitro and in vivo models to support biological studies.
Shuttle Pharma has been awarded Phase I and II SBIR contracts entitled “Cell-based models for prostate cancer health disparity research” to develop African-American prostate cancer cell lines with donor matched normal prostate epithelial cell lines from African American men.
The commercialization of the prostate cells will require additional support through the SBIR funding mechanism. Companies that have completed Phase I and II SBIR awards are eligible to apply for Phase IIb SBIR funding. These awards are intended to de-risk a project by providing up to $4 million of matching funding for product development to commercialization. We intend to apply for such government funding to advance laboratory facilities and to expand the availability of the cell cultures. We are not raising capital through this IPO for the health disparities project. Should we not be successful for SBIR IIb funding, we will pause and may have to terminate this project.
Prostate Cancer Biomarker Development
Patients treated for prostate cancer may experience treatment related late effects that adversely affect quality of life and may prove life-threatening. Shuttle has been awarded a Phase I SBIR contract entitled “Predictive biomarkers for prostate cancer patient sensitivity for radiation late effects” to determine the technical and commercial feasibility of a biomarker panel predictive of radiation mediated late effects in patients treated for prostate cancer.
Through collaboration with Georgetown University, patients treated with SBRT for prostate cancers will be analyzed for urinary and rectal symptoms and their blood will be analyzed by mass spectroscopy for predictive biomarkers. The discovery and validation of metabolite panels to serve as a predictive biomarker of patient outcomes following radiation therapy will support future development and commercialization of a diagnostic product through a Phase 2 SBIR effort.
The development to commercialization of the metabolite predictive biomarker panel will require additional support through the SBIR funding mechanism. We will be eligible to apply for Phase IIb SBIR funding the next round of solicitation. A Phase IIb will help de-risk the project by providing up to $4 million of matching funds for performing the clinical validation trial for product development to commercialization. We intend to apply for such government funding to advance this project. We do not intend to use the funds raised through this IPO for the health disparities project. Should we not be successful for SBIR IIb funding, we will terminate this project.
Collaborative Arrangements
While we intend to enter into collaborative arrangements to further develop our drug candidates in the future, at present we have not entered into any collaborative arrangements with third parties to develop our drug candidates as we are still completing clinical trials and, as a result, there can be no assurance that we will be able to do so on commercially reasonable terms or otherwise.
Intellectual Property
We invest significant amounts in research and development. Our research and development expenses before contract reimbursements were $591,237 and $604,181 for the six months ended June 30, 2022 and 2021 respectively. After reimbursements for contracts of $211,455 for the six months ended June 30, 2022 and 2021, net research and development expenses were $379,783 and $392,726, respectively.
We are seeking multifaceted protection for our intellectual property that includes licenses, confidentiality and non-disclosure agreements, copyrights, patents, trademarks and common law rights, such as trade secrets. We enter into confidentiality and proprietary rights agreements with our employees, consultants, collaborators, subcontractors and other third parties and generally control access to our documentation and proprietary information.
As of the date of this prospectus, we have filed four patent applications with the USPTO with respect to various aspects of our HDAC small molecule delivery platform and Ropidoxuridine, our lead product candidate. The following is the status of the patent applications Shuttle has filed to date:
Summary of Shuttle Pharma’s Intellectual Property Portfolio
| ||||||||
| ||||||||
Morgan, Lewis & Bockius LLP prepared patent applications related to Ropidoxuridine (IpdR) and HDAC inhibitors, and, in the fourth quarter of 2018, found no freedom to operate (FTO) issue for Ropidoxuridine used as radiosensitizer and used with tipiracil, and HDAC inhibitors SP-1-161 and SP-2-225.
Our strategy around protection of our proprietary technology, including any innovations and improvements, is to obtain worldwide patent coverage with a focus on jurisdictions that represent significant global pharmaceutical markets. Generally, patents have a term of twenty years from the earliest priority date, assuming that all maintenance fees are paid, no portion of the patent has been terminally disclaimed and the patent has not been invalidated. In certain jurisdictions, and in certain circumstances, patent terms can be extended or shortened. We are obtaining worldwide patent protection for at least novel molecules, composition of matter, pharmaceutical formulations, methods of use, including treatment of disease, methods of manufacture and other novel uses for the inventive molecules originating from our research and development efforts. We continuously assess whether it is strategically more favorable to maintain confidentiality for the “know-how” regarding a novel invention rather than pursue patent protection. For each patent application that is filed we strategically tailor our claims in accordance with the existing patent landscape around a particular technology.
There can be no assurance that an issued patent will remain valid and enforceable in a court of law through the entire patent term. Should the validity of a patent be challenged, the legal process associated with defending the patent can be costly and time consuming. Issued patents can be subject to oppositions, interferences and other third-party challenges that can result in the revocation of the patent limit patent claims such that patent coverage lacks sufficient breadth to protect subject matter that is commercially relevant. Competitors may be able to circumvent our patents. Development and commercialization of pharmaceutical products can be subject to substantial delays and it is possible that at the time of commercialization any patent covering the product has expired or will be in force for only a short period of time following commercialization. We cannot predict with any certainty if any third-party U.S. or foreign patent rights or other proprietary rights will be deemed infringed by the use of our technology. Nor can we predict with certainty which, if any, of these rights will or may be asserted against us by third parties. Should we need to defend ourselves and our partners against any such claims, substantial costs may be incurred. Furthermore, parties making such claims may be able to obtain injunctive or other equitable relief, which could effectively block our ability to develop or commercialize some or all of our products in the U.S. and abroad, and could result in the award of substantial damages. In the event of a claim of infringement, we or our partners may be required to obtain one or more licenses from a third party. There can be no assurance that we can obtain a license on a reasonable basis should we deem it necessary to obtain rights to an alternative technology that meets our needs. The failure to obtain a license may have a material adverse effect on our business, results of operations and financial condition.
We also rely on trade secret protection for our confidential and proprietary information. No assurance can be given that we can meaningfully protect our trade secrets on a continuing basis. Others may independently develop substantially equivalent confidential and proprietary information or otherwise gain access to our trade secrets.
It is our policy to require our employees and consultants, outside scientific collaborators, sponsored researchers and other advisors who receive confidential information from us to execute confidentiality agreements upon the commencement of employment or consulting relationships. These agreements provide that all confidential information developed or made known to these individuals during the course of the individual’s relationship with the company is to be kept confidential and is not to be disclosed to third parties except in specific circumstances. The agreements provide that all inventions conceived by an employee will be the property of our company. There can be no assurance, however, that these agreements will provide meaningful protection or adequate remedies for our trade secrets in the event of unauthorized use or disclosure of such information.
Our success will depend in part on our ability to obtain and maintain patent protection, preserve trade secrets, prevent third parties from infringing upon our proprietary rights and operate without infringing upon the proprietary rights of others, both in the U.S. and other territories worldwide.
Manufacturing and Supply
We do not currently own or operate manufacturing facilities for the production of preclinical, clinical or commercial quantities of any of our product candidates. We currently use a number of our suppliers for the raw materials and formulation to meet the preclinical and any clinical requirements of our product candidates. We do not have a long-term agreement with any of these parties and we believe alternative sources of supply exist.
We intend to enter into collaborations for the manufacture of our product candidates, with our collaborators assuming responsibility for such manufacturing. Manufacturing is subject to extensive regulations that impose various procedural and documentation requirements, which govern record keeping, manufacturing processes and controls, personnel, quality control and quality assurance, among others. Any collaborator or third-party contract manufacturer we use would need to be compliant with cGMP. cGMP is a regulatory standard for the production of pharmaceuticals that will be used in humans.
Competition
The development and commercialization of drugs is highly competitive. We compete with a variety of multinational pharmaceutical companies and specialized biotechnology companies, as well as technology being developed at universities and other research institutions. Our competitors have developed, are developing or will develop product candidates and processes competitive with our product candidates. Competitive therapeutic treatments include those that have already been approved and accepted by the medical community and any new treatments that enter the market. We believe that a significant number of products are currently under development, and may become commercially available in the future, for the treatment of conditions for which we may try to develop product candidates.
Many of our competitors have significantly greater financial, technical, manufacturing, marketing, sales and supply resources or experience than we have. If we are able to obtain approval for any product candidate, we will face competition based on many different factors, including the quality and effectiveness of our products, the ease with which our products can be administered and the extent to which patients accept relatively new routes of administration, the timing and scope of regulatory approvals for these products, the availability and cost of manufacturing, marketing and sales capabilities, price, reimbursement coverage and patent position. Competing products could present superior treatment alternatives, including by being more effective, safer, and less expensive or marketed and sold more effectively than any products we may develop. Competitive products may make any products we develop obsolete or noncompetitive before we recover the expense of developing and commercializing our product candidates. Such competitors could also recruit our employees, which could negatively impact our level of expertise and our ability to execute our business plan.
The following figure provides summary information about cytotoxic drugs that may be used with radiation therapy for their sensitizing properties that currently comprise the competition for Shuttle’s agents.

Fluorouracil (5-FU) is an anti-metabolite used to treat cancer, by injection, for colon cancer, esophageal cancer, stomach cancer, pancreatic cancer, breast cancer, and cervical cancer. Fluorouracil was patented in 1956 and is an effective and safe drug with radiation sensitizing properties. Capecitabine, an orally available formulation of 5-FU and was patented in 1992. It is used for the treatment of gastric, esophageal and other cancers for sensitization to radiation therapy.
Cetuximab is an epidermal growth factor receptor (EGFR) inhibitor used for the treatment of metastatic colorectal, lung cancer and head and neck cancers. This monoclonal antibody is administered by intravenous infusion and improves the 5-year survival of patients when used in combination with radiation therapy, compared with radiotherapy alone.
Platinum based compounds (cis-platin, carbo-platin and oxaloplatin) also exhibit radiation sensitizing properties. Platinum and radiation are used together for the treatment of locally advanced cervical cancer and for head and neck cancers. Cisplatin is believed to augment the effects of radiation by inhibiting the repair of radiation-induced sub-lethal damage.
Bevacizumab works as an anti-angiogenic agent. It was approved for medical use in the United States in 2004. The addition of bevacizumab to standard treatment can prolong the lives of breast and lung cancer patients by several months and may be used with radiation therapy.
Irinotecan is given by injection and is used to treat colon cancer and small cell lung cancer and can be combined with radiation therapy. For colon cancer it is used either alone or with fluorouracil.
Government Regulation and Product Approval
Governmental authorities in the U.S., at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, labeling, packaging, promotion, storage, advertising, distribution, marketing and export and import of products such as those we are developing. Our product candidates must be approved by the FDA through the NDA process before they may be legally marketed in the U.S. and will be subject to similar requirements in other countries prior to marketing in those countries. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
U.S. government regulation
NDA approval processes
In the U.S., the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act (the “FDCA”) and implementing regulations. Failure to comply with the applicable U.S. requirements at any time during the product development or approval process, or after approval, may subject an applicant to administrative or judicial sanctions, any of which could have a material adverse effect on us. These sanctions could include:
| ● | wire transfer of immediately available funds directly to the account maintained by Vstock Transfer, LLC, as Subscription Agent, for purposes of accepting subscriptions in this Rights Offering at: [ ] |
The process requiredIf you elect to exercise your Subscription Rights, you should consider using a wire transfer or certified check drawn on a U.S. bank to ensure that the Subscription Agent receives your funds before the Rights Offering expires. If you send a personal check, payment will not be deemed to have been received by the FDASubscription Agent until the check has cleared. The clearinghouse may require five or more business days to clear a personal check. Accordingly, holders who wish to pay the Subscription Price by means of a personal check should make payment sufficiently in advance of the expiration of the Rights Offering to ensure that the payment is received and clears by that date. If you send a certified check, payment will be deemed to have been received by the Subscription Agent immediately upon receipt of such instrument.
You should read the instruction letter accompanying the Rights Certificate carefully and strictly follow it. DO NOT SEND RIGHTS CERTIFICATES OR PAYMENTS DIRECTLY TO US. We will not consider your subscription received until the Subscription Agent has received delivery of a properly completed and duly executed Rights Certificate and payment of the full subscription payment.
The method of delivery of Rights Certificates and payment of the subscription payment to the Subscription Agent will be at the risk of the holders of Subscription Rights. If sent by mail, we recommend that you send those certificates and payments by registered mail, properly insured, with return receipt requested, or by overnight courier, and that you allow a sufficient number of days to ensure delivery to the Subscription Agent and clearance of payment before the Rights Offering expires.
Missing or Incomplete Subscription Forms or Payment
If you fail to complete and sign the Rights Certificate or otherwise fail to follow the subscription procedures that apply to the exercise of your Subscription Rights before the Rights Offering expires, the Subscription Agent will reject your subscription or accept it to the extent of the payment received. Neither we nor our Subscription Agent undertakes any responsibility or action to contact you concerning an incomplete or incorrect subscription form, nor are we under any obligation to correct such forms. We have the sole discretion to determine whether a drugsubscription exercise properly complies with the subscription procedures.
If you send a payment that is insufficient to purchase the number of shares you requested, or if the number of shares you requested is not specified in the forms, the payment received will be applied to exercise your Subscription Rights to the fullest extent possible based on the amount of the payment received. Any excess subscription payments received by the Subscription Agent will be returned, without interest or deduction, within 10 business days following the expiration of the Rights Offering.
| 36 |
Issuance of Common Stock, Warrants and Equity Interests
The shares of common stock, Warrants and Equity Interests that are purchased in the Rights Offering as part of the Units will be issued in book-entry, or uncertificated, form meaning that you will receive a DRS account statement from our transfer agent reflecting ownership of these securities if you are a holder of record. If you hold your shares of common stock and warrants in the name of a bank, broker, dealer, or other nominee, DTC will credit your account with your nominee with the securities you purchased in the Rights Offering.
Subscription, Information and Escrow Agent
The Subscription Agent, Information Agent and Escrow Agent for the Rights Offering is Vstock Transfer, LLC. The address to which Rights Certificates and payments should be mailed or delivered by overnight courier is provided below. If sent by mail, we recommend that you send documents and payments by registered mail, properly insured, with return receipt requested, and that you allow a sufficient number of days to ensure delivery to the Subscription Agent and clearance or payment before the Rights Offering expires. Do not send or deliver these materials to us.
If delivering by mail, hand or overnight courier:
VStock Transfer, LLC
Attn: Shay Galam
18 Lafayette Place
Woodmere, New York 11598
Phone: (212) 828-8436
If you deliver the Rights Certificates in a manner different than that described in this prospectus, we may not honor the exercise of your Subscription Rights.
You should direct any questions or requests for assistance concerning the method of subscribing for the shares of our common stock or for additional copies of this prospectus to the Information Agent, Vstock Transfer, LLC, at (212) 828-8436 or by mail at Vstock Transfer, LLC, 18 Lafayette Place, Woodmere, New York 11598.
Warrant Agent
The warrant agent for the Warrants is VStock Transfer LLC.
Equity Interest
The transfer agent for the Equity Interests in Diagnostics is VStock Transfer LLC.
No Fractional Shares
We will not issue fractional shares of common stock in the Rights Offering. We will only distribute Subscription Rights to acquire whole Units, rounded down to the nearest whole number of underlying common shares giving rise to such Subscription Rights. Any excess subscription payments received by the Subscription Agent will be returned within 10 business days after expiration of the Rights Offering, without interest or deduction. Similarly, no fractional shares of common stock will be issued in connection with the exercise of a Warrant. Instead, for any such fractional share that would otherwise have been issuable upon exercise of Warrants, we will round up such fraction to the next whole share.
Notice to Brokers and Nominees
If you are a broker, dealer, bank or other nominee holder that holds shares of our common stock or warrants for the account of others on the Record Date, you should notify the beneficial owners for whom you are the nominee of the Rights Offering as soon as possible to learn their intentions with respect to exercising their Subscription Rights. If a beneficial owner of our common stock or warrants so instructs, you should complete the Rights Certificate and submit it to the Subscription Agent with the proper subscription payment by the expiration date. You may exercise the number of Subscription Rights to which all beneficial owners in the aggregate otherwise would have been entitled had they been direct holders of our common stock on the Record Date, provided that you, as a nominee record holder, make a proper showing to the Subscription Agent by submitting the form entitled “Nominee Holder Certification,” which is provided with your Rights Offering materials. If you did not receive this form, you should contact our Subscription Agent to request a copy.
| 37 |
Validity of Subscriptions
We will resolve all questions regarding the validity and form of the exercise of your Subscription Rights, including time of receipt and eligibility to participate in the Rights Offering. Our determination will be final and binding. Once made, subscriptions are irrevocable; we will not accept any alternative, conditional, or contingent subscriptions. We reserve the absolute right to reject any subscriptions not properly submitted or the acceptance of which would be unlawful. You must resolve any irregularities in connection with your subscriptions before the expiration date of the Rights Offering, unless we waive them in our sole discretion. Neither we nor the Subscription Agent is under any duty to notify you or your representative of defects in your subscriptions. A subscription will be considered accepted, subject to our right to withdraw or terminate the Rights Offering, only when the Subscription Agent receives a properly completed and duly executed Rights Certificate and any other required documents and the full subscription payment including final clearance of any personal check. Our interpretations of the terms and conditions of the Rights Offering will be final and binding.
Stockholder Rights
Holders of Warrants issued in connection with the Rights Offering will not have rights as holders of our common stock until such Warrants are exercised and the shares of common stock underlying the Warrants are issued to the holder. In addition, holders of Equity Interests in Diagnostics will only have shareholder rights with respect to Diagnostics, which is a subsidiary of the Company and is not publicly trading, and will have no ownership rights with respect to the Company.
Foreign Stockholders
We will not mail this prospectus or Rights Certificates to stockholders with addresses that are outside the United States or that have an army post office or foreign post office address. The Subscription Agent will hold these Rights Certificates for their account. To exercise Subscription Rights, our foreign stockholders must notify the Subscription Agent prior to 5:00 p.m., Eastern Time, on , 2024, the third business day prior to the expiration date, of your exercise of Subscription Rights and provide evidence satisfactory to us, such as a legal opinion from local counsel, that the exercise of such Subscription Rights does not violate the laws of the jurisdiction in which such stockholder resides and payment by a U.S. bank in U.S. dollars before the expiration of the offer. If no notice is received by such time or the evidence presented is not satisfactory to us, the Subscription Rights represented thereby will expire.
No Revocation or Change
Once you submit the Rights Certificate or have instructed your nominee of your subscription request, you are not allowed to revoke or change the exercise or request a refund of monies paid. All exercises of Subscription Rights are irrevocable, even if you learn information about us that you consider to be unfavorable. You should not exercise your Subscription Rights unless you are certain that you wish to purchase shares at the Subscription Price. If we should make any fundamental changes to the terms set forth in this prospectus, we will (i) file a post-effective amendment to the registration statement of which this prospectus forms a part, (ii) offer potential purchasers who have subscribed for rights the opportunity to cancel such subscriptions, and (iii) issue a refund of any money advanced by such stockholder or eligible warrant holder and recirculate an updated prospectus after the post-effective amendment is declared effective with the SEC.
U.S. Federal Income Tax Treatment of Rights Distribution
For U.S. federal income tax purposes, we do not believe holders of shares of our common stock or warrants should recognize income or loss upon receipt or exercise of a Subscription Right, but the receipt and exercise of the Subscription Rights is unclear in certain respects. See “Material U.S. Federal Income Tax Consequences.”
No Recommendation to Rights Holders
Our board of directors is not making a recommendation regarding your exercise of the Subscription Rights. Stockholders who exercise Subscription Rights risk investment loss on money invested. We cannot predict the price at which our shares of common stock will trade after the Rights Offering. You should make your investment decision based on your assessment of our business and financial condition, our prospects for the future and the terms of this Rights Offering. Please see “Risk Factors” for a discussion of some of the risks involved in investing in our common stock.
| 38 |
Fees and Expenses
We will pay all fees charged by the Subscription Agent, the Information Agent and the Escrow Agent, and by the placement agent. You are responsible for paying any other commissions, fees, taxes or other expenses incurred in connection with the exercise of your Subscription Rights.
Listing
The Subscription Rights may not be sold, transferred, assigned or given away to anyone, and will not be listed for trading on any stock exchange or market. There is no public trading market for the Warrants or Equity Interests and they will not be listed for trading on Nasdaq or any other securities exchange or market. The shares of our common stock, including those issuable upon the exercise of the Warrants to be issued in the Rights Offering, are traded on Nasdaq under the symbol “SHPH.”
Important
Do not send Rights Certificates directly to us. You are responsible for choosing the payment and delivery method for your Rights Certificate and you bear the risks associated with such delivery. If you choose to deliver your Rights Certificate and payment by mail, we recommend that you use registered mail, properly insured, with return receipt requested. We also recommend that you allow a sufficient number of days to ensure delivery to the Subscription Agent and clearance of payment prior to the expiration time.
Distribution Arrangements
Boustead Securities LLC will act as placement agent for the Rights Offering. The placement agent will provide marketing assistance and advice to us in connection with the Rights Offering and will use its best efforts to solicit the exercise of Subscription Rights and participation in the Over-Subscription Privilege. The placement agent is not underwriting or placing any of the Subscription Rights or the shares of our common stock, Warrants or Equity Interests to be issued in the Rights Offering and does not make any recommendation with respect to such Subscription Rights (including with respect to the exercise or expiration of such Subscription Rights), shares or Warrants. We have agreed to pay the placement agent certain fees and to reimburse the dealer-manager for certain out-of-pocket expenses incurred in connection with this offering. See “Plan of Distribution.”
Other Matters
We are not making the Rights Offering in any state or other jurisdiction in which it is unlawful to do so, nor are we distributing or accepting any offers to purchase any units from subscription rights holders who are residents of those states or other jurisdictions or who are otherwise prohibited by federal or state laws or regulations from accepting or exercising the subscription rights. We may delay the commencement of the Rights Offering in those states or other jurisdictions, or change the terms of the Rights Offering, in whole or in part, in order to comply with the securities laws or other legal requirements of those states or other jurisdictions. Subject to state securities laws and regulations, we also have the discretion to delay allocation and distribution of any shares you may elect to purchase by exercise of your subscription privileges in order to comply with state securities laws. We may decline to make modifications to the terms of the Rights Offering requested by those states or other jurisdictions, in which case, if you are a resident in those states or jurisdictions or if you are otherwise prohibited by federal or state laws or regulations from accepting or exercising the subscription rights, you will not be eligible to participate in the Rights Offering. However, we are not currently aware of any states or jurisdictions that would preclude participation in the Rights Offering.
| 39 |
MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES
The following discussion describes the material U.S. federal income tax consequences of the receipt and exercise (or expiration) of the Subscription Rights acquired through the Rights Offering, the ownership and disposition of shares of our common stock, Warrants and Equity Interests received upon exercise of the Subscription Rights and the ownership and disposition of the shares of common stock exercise of the Warrants. This discussion does not purport to be a complete analysis of all potential tax consequences. The effects of other U.S. federal tax laws, such as estate and gift tax laws, alternative minimum tax laws, net investment income tax laws, and any applicable state, local or non-U.S. tax laws are not discussed. This discussion is based on the U.S. Internal Revenue Code of 1986, as amended, or the Code, U.S. Treasury Regulations promulgated thereunder, judicial decisions, and published rulings and administrative pronouncements of the U.S. Internal Revenue Service, or the IRS, in each case in effect as of the date hereof. These authorities may change or be subject to differing interpretations. Any such change or differing interpretation may be marketedapplied retroactively in a manner that could adversely affect a holder of the Subscription Rights, shares of our common stock, Warrants or Equity Interests. This summary does not discuss the potential effects, whether adverse or beneficial, of any proposed legislation that, if enacted, could be applied on a retroactive or prospective basis. We have not sought and will not seek any rulings from the IRS regarding the matters discussed below. There can be no assurance the IRS or a court will not take a contrary position to that discussed below regarding the tax consequences of the receipt of Subscription Rights through the Rights Offering by persons holding shares of our common stock, Warrants or Equity Interests, the exercise (or expiration) of the Subscription Rights, ownership and disposition (or expiration) of Warrants, and the acquisition, ownership and disposition of Equity Interests, each acquired upon exercise of the Subscription Rights, and the acquisition, ownership and disposition of shares of our common stock acquired upon exercise of the Warrants.
This discussion is limited to the Subscription Rights acquired through the Rights Offering, shares of our common stock, Warrants and Equity Interests acquired upon exercise of Subscription Rights and shares of our common stock acquired upon exercise of the Warrants, in each case, that are held as a “capital asset” within the meaning of Section 1221 of the Code (generally, property held for investment). This discussion does not address all U.S. generally involvesfederal income tax consequences relevant to a holder’s particular circumstances, including the following:impact of the alternative minimum tax or the unearned income Medicare contribution tax. In addition, it does not address consequences relevant to holders subject to particular rules, including, without limitation:
| ● |
| ● | U.S. holders (as defined below) that are subject to taxing jurisdictions other than, or in addition to, the United States or otherwise hold our common stock, the Subscription Rights, shares of | |
| ● | corporations organized outside the | |
| ● | persons holding our common stock, the Subscription Rights, Warrants or shares of | |
| ● | banks, insurance companies, underwriters, and other financial institutions; | |
| ● | brokers, dealers or traders in securities or currencies or traders that elect to mark-to-market their securities; | |
| ● | “controlled foreign corporations,” “passive foreign investment companies,” and corporations that accumulate earnings to avoid U.S. federal income tax; | |
| ● | partnerships or other entities or arrangements treated as partnerships or other pass-through entities for U.S. federal income tax purposes (and investors therein) and S corporations (and shareholders thereof); |
| 40 |
| ● | real estate investment trusts, regulated investment companies, grantor trusts, tax-exempt organizations or governmental organizations; | |
| ● | persons deemed to sell the Subscription Rights, shares of common stock, Warrants, Equity Interests or shares of our common stock under the constructive sale provisions of the | |
| ● | persons subject to special tax accounting rules as a result of any item of gross income being taken into account in an applicable financial statement (as defined in the Code); | |
| ● | persons for whom our stock constitutes “qualified small business stock” within the meaning of Section 1202 of the Code; | |
| ● | persons who received, hold or | |
| ● | tax-qualified retirement plans, individual retirement accounts, or other tax-deferred accounts; | |
| ● | persons subject to | |
| ● |
OnceIf an entity treated as a pharmaceutical candidate is identifiedpartnership for development, it entersU.S. federal income tax purposes holds shares of our common stock, the preclinicalSubscription Rights, or nonclinical testing stage. Nonclinical tests include laboratory evaluationsshares of product chemistry, toxicity and formulation, as well as animal studies. An IND sponsor must submit the resultsour common stock, Warrants or Equity Interests acquired upon exercise of Subscription Rights or shares of our common stock acquired upon exercise of the nonclinical tests, together with manufacturing information and analytical data, toWarrants, as the FDA as partcase may be, the tax treatment of a partner in the IND. Some nonclinical testing may continue even after the IND is submitted. In addition to including the results of the nonclinical studies, the INDpartnership will also include a protocol detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring quality and the effectiveness criteria to be evaluated if the first phase lends itself to an efficacy determination. The IND automatically becomes effective thirty (30) days after receipt by the FDA, unless the FDA, within the thirty (30) day time period, places the INDdepend on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin. A clinical hold may occur at any time during the life of an IND and may affect one or more specific studies or all studies conducted under the IND.
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with GCPs. They must be conducted under protocols detailing the objectives of the trial, dosing procedures, research subject selection and exclusion criteria and the quality and effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND, and progress reports detailing the status of the clinical trials must be submittedpartner, the activities of the partnership and certain determinations made at the partner level. Accordingly, partnerships and the partners in such partnerships should consult their own tax advisors regarding the U.S. federal income tax consequences to the FDA annually. Sponsors also must timely reportthem.
THIS DISCUSSION IS FOR INFORMATION PURPOSES ONLY AND IS NOT TAX ADVICE. INVESTORS SHOULD CONSULT THEIR OWN TAX ADVISORS WITH RESPECT TO THE APPLICATION OF THE U.S. FEDERAL INCOME TAX LAWS TO THEIR PARTICULAR SITUATIONS AS WELL AS ANY TAX CONSEQUENCES OF THE RECEIPT, OWNERSHIP AND EXERCISE OF SUBSCRIPTION RIGHTS, THE ACQUISITION, OWNERSHIP, AND DISPOSITION OF SHARES OF COMMON STOCK, WARRANTS AND EQUITY INTERESTS ACQUIRED UPON EXERCISE OF SUBSCRIPTION RIGHTS, AND SHARES OF OUR COMMON STOCK ACQUIRED UPON THE EXERCISE OF WARRANTS ARISING UNDER THE U.S. FEDERAL ESTATE OR GIFT TAX LAWS OR UNDER THE LAWS OF ANY STATE, LOCAL OR NON-U.S. TAXING JURISDICTION OR UNDER ANY APPLICABLE INCOME TAX TREATY.
Tax Considerations Applicable to FDA serious and unexpected adverse reactions, any clinically important increase in the rateU.S. Holders
Definition of a serious suspected adverse reaction over that listed in the protocol or investigation brochure or any findings from other studies or animal or in vitro testing that suggest a significant risk in humans exposed to the drug. An institutional review board (IRB) at each institution participating in the clinical trial must review and approve the protocol before a clinical trial commences at that institution and must also approve the information regarding the trial and the consent form that must be provided to each research subject or the subject’s legal representative, monitor the study until completed and otherwise comply with IRB regulations.U.S. Holder
Human clinical trials are typically conducted in three sequential phasesFor purposes of this discussion, a “U.S. holder” is any beneficial owner of shares of our common stock, our Subscription Rights, shares of our common stock, Warrants or Equity Interests acquired upon exercise of Subscription Rights or shares of our common stock acquired upon exercise of Warrants, as the case may be, that, may overlap or be combined.for U.S. federal income tax purposes, is:
| ● | ||
| ● | a | |
| ● |
Human clinical trials are inherently uncertain and Phase I, Phase II and Phase III testing may not achieve desired resultsits source; or otherwise be completed. The FDA or the sponsor may suspend a clinical trial at any time for a variety of reasons, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the product candidate has been associated with unexpected serious harm to patients.
During the development of a new product candidate, sponsors are given opportunities to meet with the FDA at certain points. These points may be prior to the submission of an IND, at the end of Phase II and before an NDA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date and for the FDA to provide advice on the next phase of development. Sponsors typically use the meeting at the end of Phase II to discuss their Phase II clinical results and present their plans for the pivotal Phase III clinical trial that they believe will support the approval of the new drug. If a Phase II clinical trial is the subject of discussion at the end of Phase II meeting with the FDA, a sponsor may be able to request a Special Protocol Assessment (“SPA”), the purpose of which is to reach agreement with the FDA on the Phase III clinical trial protocol design and analysis that will form the primary basis of an efficacy claim.
According to published guidance on the SPA process, a sponsor which meets the prerequisites may make a specific request for a SPA and provide information regarding the design and size of the proposed clinical trial. The FDA is supposed to evaluate the protocol within forty-five (45) days of the request to assess whether the proposed trial is adequate, which evaluation may result in discussions and a request for additional information. An SPA request must be made before the proposed trial begins, and all open issues must be resolved before the trial begins. If a written agreement is reached, it will be documented and made part of the record. The agreement will be binding on the FDA and may not be changed by the sponsor or the FDA after the trial begins except with the written agreement of the sponsor and the FDA or if the FDA determines that a substantial scientific issue essential to determining the safety or efficacy of the product candidate was identified after the testing began.
Concurrent with clinical trials, sponsors usually complete additional animal safety studies and also develop additional information about the chemistry and physical characteristics of the product candidate and finalize a process for manufacturing commercial quantities of the product candidate in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and the manufacturer must develop methods for testing the quality, purity and potency of the product candidate. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its proposed shelf-life.
The results of product development, nonclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests and other control mechanisms, proposed labeling and other relevant information are submitted to the FDA as part of an NDA requesting approval to market the product. The submission of an NDA is subject to the payment of user fees, but a waiver of such fees may be obtained under specified circumstances. The FDA reviews all NDAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. It may request additional information rather than accept an NDA for filing. In this event, the NDA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing.
Once the submission is accepted for filing, the FDA begins an in-depth review. NDAs receive either standard or priority review. A drug representing a significant improvement in treatment, prevention or diagnosis of disease may receive priority review. The FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical or other data. Even if such data are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP-compliant. The FDA may refer the NDA to an advisory committee for review and recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA, the FDA will inspect the facility or facilities where the product is manufactured and tested.
Expedited review and approval
The FDA has various programs, including Fast Track, priority review and accelerated approval, which are intended to expedite or simplify the process for reviewing product candidates, or provide for the approval of a product candidate on the basis of a surrogate endpoint. Even if a product candidate qualifies for one or more of these programs, the FDA may later decide that the product candidate no longer meets the conditions for qualification or that the time period for FDA review or approval will be shortened. Generally, product candidates that are eligible for these programs are those for serious or life-threatening conditions, those with the potential to address unmet medical needs and those that offer meaningful benefits over existing treatments. For example, Fast Track is a process designed to facilitate the development and expedite the review of product candidates to treat serious or life-threatening diseases or conditions and fill unmet medical needs. Priority review is designed to give product candidates that offer major advances in treatment or provide a treatment where no adequate therapy exists an initial review within six months as compared to a standard review time of ten (10) months.
Although Fast Track and priority review do not affect the standards for approval, the FDA will attempt to facilitate early and frequent meetings with a sponsor of a Fast Track designated product candidate and expedite review of the application for a product candidate designated for priority review. Accelerated approval, which is described in Subpart H of 21 CFR Part 314, provides for an earlier approval for a new product candidate that is intended to treat a serious or life-threatening disease or condition and that fills an unmet medical need based on a surrogate endpoint. A surrogate endpoint is a laboratory measurement or physical sign used as an indirect or substitute measurement representing a clinically meaningful outcome. As a condition of approval, the FDA may require that a sponsor of a product candidate receiving accelerated approval perform post-marketing clinical trials.
In the Food and Drug Administration Safety and Innovation Act (“FDASIA”), the U.S. Congress encouraged the FDA to utilize innovative and flexible approaches to the assessment of product candidates under accelerated approval. The law required the FDA to issue related draft guidance within a year after the law’s enactment and also promulgate confirming regulatory changes. In June 2013, the FDA published a draft Guidance for Industry titled “Expedited Programs for Serious Conditions—Drugs and Biologics,” which provides guidance on FDA programs that are intended to facilitate and expedite development and review of new product candidates as well as threshold criteria generally applicable to concluding that a product candidate is a candidate for these expedited development and review programs.
In addition to the Fast Track, accelerated approval and priority review programs discussed above, the FDA also provided guidance on a new program for Breakthrough Therapy designation. The FDA defines a Breakthrough Therapy as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. A drug designated as a Breakthrough Therapy is eligible for accelerated approval. The FDA must take certain actions, such as holding timely meetings and providing advice, intended to expedite the development and review of an application for approval of a Breakthrough Therapy. Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened. A request for Breakthrough Therapy designation should be submitted concurrently with, or as an amendment to an IND. FDA has already granted this designation to approximately thirty (30) new product candidates and has begun approving Breakthrough Therapy designated drugs.
Patent term restoration and marketing exclusivity
Depending upon the timing, duration and specifics of FDA approval of the use of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch- Waxman Act. The Hatch-Waxman Act permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of fourteen (14) years from the product candidate’s approval date. The patent term restoration period is generally one half of the time between the effective date of an IND and the submission date of an NDA, plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved product candidate is eligible for the extension and the application for extension must be made prior to expiration of the patent. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restorations of patent term for some of our currently owned or licensed patents to add patent life beyond their current expiration date, depending on the expected length of clinical trials and other factors involved in the submission of the relevant NDA.
Market exclusivity provisions under the FDCA also can delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the U.S. to the first applicant to gain approval of an NDA for a new chemical entity. A product candidate is a new chemical entity if the FDA has not previously approved any other new product candidate containing the same active moiety, which is the molecule or ion responsible for the action of the product candidate substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application (“ANDA”) or a 505(b)(2) NDA submitted by another company for another version of such product candidate where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b) (2) NDA, or supplement to an approved NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages or strengths of an existing product candidate. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for product candidates containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate quality and effectiveness.
Orphan drug designation
Under the Orphan Drug Act, the FDA may grant orphan drug designation to product candidates intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the U.S. or more than 200,000 individuals in the U.S. and for which there is no reasonable expectation that the cost of developing and making available in the U.S. a product candidate for this type of disease or condition will be recovered from sales in the U.S. for that product candidate. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the FDA publicly discloses the identity of the therapeutic agent and its potential orphan use. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product candidate that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product candidate is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same product candidate for the same indication, except in very limited circumstances, for seven years. Orphan drug exclusivity, however, could also block the approval of one of our product candidates for seven years if a competitor obtains approval of the same product candidate as defined by the FDA or if our product candidate is determined to be contained within the competitor’s product candidate for the same indication or disease.
Pediatric exclusivity and pediatric use
Under the Best Pharmaceuticals for Children Act (“BPCA”), certain product candidates may obtain an additional six months of exclusivity if the sponsor submits information requested in writing by the FDA (a “Written Request”) relating to the use of the active moiety of the product candidate in children. The FDA may not issue a Written Request for studies on unapproved or approved indications or where it determines that information relating to the use of a product candidate in a pediatric population, or part of the pediatric population, may not produce health benefits in that population.
In addition, the Pediatric Research Equity Act (“PREA”) requires a sponsor to conduct pediatric studies for most product candidates and biologics, for a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration. Under PREA, original NDAs, biologics license application and supplements thereto must contain a pediatric assessment unless the sponsor has received a deferral or waiver. The required assessment must assess the quality and effectiveness of the product candidate for the claimed indications in all relevant pediatric subpopulations and support dosing and administration for each pediatric subpopulation for which the product candidate is safe and effective. The sponsor or FDA may request a deferral of pediatric studies for some or all of the pediatric subpopulations. A deferral may be granted for several reasons, including a finding that the product candidate or biologic is ready for approval for use in adults before pediatric studies are complete, or that additional quality or effectiveness data needs to be collected before the pediatric studies begin. After April 2013, the FDA must send a noncompliance letter to any sponsor that fails to: submit the required assessment, keep a deferral current, or submit a request for approval of a pediatric formulation.
Post-approval requirements
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements is not maintained or if problems occur after the product candidate reaches the market. Later discovery of previously unknown problems with a product candidate may result in restrictions on the product candidate or even complete withdrawal of the product candidate from the market. After approval, some types of changes to the approved product candidate, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval. In addition, the FDA may require testing and surveillance programs to monitor the effect of approved product candidates that have been commercialized, and the FDA has the power to prevent or limit further marketing of a product candidate based on the results of these post-marketing programs.
Any product candidates manufactured or distributed by us pursuant to FDA approvals are subject to continuing regulation by the FDA, including, among other things:
| ● | a trust that (1) is subject to the primary supervision of |
| 41 |
Receipt of Subscription Rights
Although the rules and regulations governing transactions such as this Rights Offering are complex and do not speak directly to the consequences of certain aspects of this Rights Offering, including the inclusion of the right to purchase Warrants in the Subscription Rights (rather than the right to purchase shares of common stock or Equity Interests), the distribution of Subscription Rights to holders of warrants and the effects of the Over-Subscription Privilege, we do not believe a U.S. holder’s receipt of Subscription Rights pursuant to the Rights Offering should be treated as a taxable distribution with respect to its existing shares of common stock or warrants, as applicable, for U.S. federal income tax purposes. Section 305(a) of the Code generally provides that the receipt by a stockholder, or a holder of rights to acquire stock, of a right to acquire stock or warrants is not included in the taxable income of the stockholder; however, the general non-recognition rule in Section 305(a) of the Code is subject to exceptions described in Section 305(b) of the Code, which include “disproportionate distributions.” A disproportionate distribution is generally a distribution or a series of distributions, including deemed distributions, that has the effect of the receipt of cash or other property by some stockholders (including holders of rights to acquire stock and holders of debt instruments convertible into stock) and an increase in the proportionate interest of other stockholders (including holders of rights to acquire stock and holders of debt instruments convertible into stock) in a corporation’s assets or earnings and profits. During the last 36 months, we have not made any distributions of cash or property (other than stock or rights to acquire stock) with respect to: (i) our common stock or (ii) options or warrants to acquire our common stock. Currently we do not intend to make any future distributions of cash or property (other than stock or rights to acquire stock) with respect to: (i) our common stock or (ii) options or warrants to acquire our common stock; however, there is no guarantee that we will not make such distributions in the future.
The position regarding the non-taxable treatment of the Subscription Right distribution is not binding on the IRS, or the courts. If this position is finally determined by the IRS or a court to be incorrect, whether because, contrary to our expectations, distributions of cash or property (other than stock or rights to acquire stock) are made with respect to our common stock, options or warrants, because the issuance of the Subscription Rights is a “disproportionate distribution” or otherwise, the fair market value of the Subscription Rights would be taxable to U.S. holders of our common stock as a dividend to the extent of the U.S. holder’s pro rata share of our current and accumulated earnings and profits, if any, with any excess being treated as a return of capital to the extent thereof and then as capital gain. Although no assurance can be given, we anticipate that we will not have current and accumulated earnings and profits through the end of 2024.
The following discussion is based upon the treatment of the Subscription Right issuance as a non-taxable distribution with respect to a U.S. holder’s’ existing shares of common stock or warrants for U.S. federal income tax purposes.
Tax Basis in the Subscription Rights
If the fair market value of the Subscription Rights a U.S. holder receives is less than 15% of the fair market value of the U.S. holder’s existing shares of common stock or warrants (in each case, with respect to which the Subscription Rights are distributed) on the date the U.S. holder receives the Subscription Rights, the Subscription Rights will be allocated a zero tax basis for U.S. federal income tax purposes, unless the U.S. holder elects to allocate the tax basis in the holder’s existing shares of common stock or warrants between the existing shares of common stock or warrants and the Subscription Rights in proportion to the relative fair market values of the existing shares of common stock warrants and the Subscription Rights determined on the date of receipt of the Subscription Rights. If a U.S. holder chooses to allocate tax basis between the holder’s existing common stock or warrants and the Subscription Rights, the U.S. holder must make this election on a statement included with the holder’s timely filed U.S. federal income tax return (including extensions) for the taxable year in which the U.S. holder receives the Subscription Rights. Such an election is irrevocable.
| 42 |
However, if the fair market value of the Subscription Rights a U.S. holder receives is 15% or more of the fair market value of the holder’s existing shares of common stock or warrants on the date the U.S. holder receives the Subscription Rights, then the U.S. holder must allocate tax basis in the existing shares of common stock or warrants between those shares or warrants and the Subscription Rights the U.S. holder receives in proportion to their fair market values determined on the date the U.S. holder receives the Subscription Rights. Please refer to the discussion below regarding the U.S. tax treatment of a U.S. holder that, at the time of the receipt of the Subscription Right, no longer holds the common stock or warrants with respect to which the Subscription Right was distributed.
The fair market value of the Subscription Rights on the date that the Subscription Rights are distributed is uncertain and we have not obtained, and do not intend to obtain, an appraisal of the fair market value of the Subscription Rights on that date. In determining the fair market value of the Subscription Rights, U.S. holders should consider all relevant facts and circumstances, including without limitation any difference between the Subscription Price of the Subscription Rights and the trading price of our shares of common stock on the date that the Subscription Rights are distributed, the fair market value of the common stock and Equity Interests, the exercise price of the Warrants, the length of the period during which the Subscription Rights may be exercised and the fact that the Subscription Rights are non-transferable.
Exercise of Subscription Rights
A U.S. holder will not recognize gain or loss upon the exercise of a Subscription Right received in the Rights Offering. A U.S. holder’s adjusted tax basis, if any, in the Subscription Right plus the Subscription Price should be allocated between the share of common stock, the Warrants and the Equity Interests acquired upon exercise of the Subscription Right. We believe that the tax basis in the common stock or warrants upon which the Subscription Rights were issued which is allocated to the Subscription Rights under the prior section entitled “—Tax Basis in the Subscription Rights” should be further allocated between the shares of common stock, Warrants and Equity Interests acquired upon exercise of the Subscription Right in proportion to their relative fair market values on the date the Subscription Rights were distributed. The Subscription Price should be allocated between the shares of common stock, the Warrants and Equity Interests acquired upon exercise of the Subscription Right in proportion to their relative fair market values on the exercise date. These allocations will establish the U.S. holder’s initial tax basis for U.S. federal income tax purposes in the shares of common stock, Warrants and Equity Interests received upon exercise of such U.S. holder’s Subscription Right. The holding period of a share of a Warrant or Equity Interest acquired upon exercise of a Subscription Right in the Rights Offering will begin on the date of exercise.
If, at the time of the receipt or exercise of the Subscription Right, the U.S. holder no longer holds the common stock, or Warrant with respect to which the Subscription Right was distributed, then certain aspects of the tax treatment of the receipt and exercise of the Subscription Right are unclear, including (1) the allocation of the tax basis between the shares of our common stock or warrants previously sold and the Subscription Right, (2) the impact of such allocation on the amount and timing of gain or loss recognized with respect to the shares of our common stock or warrants previously sold, and (3) the impact of such allocation on the tax basis of the shares of our common stock, Warrants and Equity Interests acquired upon exercise of the Subscription Right. If a U.S. holder exercises a Subscription Right received in the Rights Offering after disposing of shares of our common stock or warrants with respect to which the Subscription Right is received, the U.S. holder should consult its own tax advisor.
Expiration of Subscription Rights
If a U.S. holder allows Subscription Rights received in the Rights Offering to expire, the U.S. holder should not recognize any gain or loss for U.S. federal income tax purposes, and the U.S. holder should re-allocate any portion of the tax basis in its existing common shares or warrants previously allocated to the Subscription Rights that have expired to such U.S. holder’s existing common shares and warrants, as applicable.
| 43 |
Sale or Other Disposition, Exercise or Expiration of Warrants
The exercise price of each Warrant that may be received upon exercise of a Subscription Right is $2.35 per share. The tax authorities that apply to warrants with an exercise price less than the fair market value of the underlying security which will be received upon exercise thereof are complex and subject to differing interpretations. Accordingly, the tax treatment of the Warrants which may be received upon exercise of a Subscription Right is uncertain. If the exercise price of a Warrant as of the date of receipt by a U.S. holder that exercises a Subscription Right is substantially less than the fair market value of the underlying share of our common stock which may be received upon exercise of such Warrant, the Warrant may be classified for U.S. federal income tax purposes as of the date of receipt by a U.S. holder as a share of our common stock. If a Warrant is properly classified as a share of common stock for U.S. federal income tax purposes, a U.S. holder will not be required to recognize income, gain or loss upon exercise of such Warrant and a U.S. holder’s tax basis in a share of our common stock will be equal to the sum of (1) the U.S. holder’s tax basis in the Warrants exchanged therefor and (2) the exercise price of such Warrants. A U.S. holder’s holding period in the shares of our common stock received upon exercise should generally include the holding period of such U.S. holder in the Warrants. For the U.S. federal income tax consequences of a sale or other taxable disposition (other than by exercise), or distributions (including constructive distributions) paid on the Warrants if they are properly classified as shares of our common stock for U.S. federal income tax purposes, see the discussions below under the headings “—Distributions on Common Stock and Equity Interests”, and “—Sale, Exchange or other Disposition of Common Stock or Equity Interests”. U.S. holders should consult their own tax advisors regarding the proper U.S. federal income tax classification of the Warrants. The remainder of this discussion assumes the Warrants are properly classified as warrants for U.S. federal income tax purposes.
Upon the sale or other taxable disposition of a Warrant (other than by exercise) received upon exercise of a Subscription Right, a U.S. holder will generally recognize capital gain or loss equal to the difference between the amount realized on the sale or other taxable disposition and the U.S. holder’s adjusted tax basis in the Warrant. A U.S. Holder’s adjusted tax basis in a Warrant will generally equal its initial tax basis (discussed above under “—Exercise of Subscription Rights”), as adjusted for any constructive dividends on the Warrant described below. This capital gain or loss will be long-term capital gain or loss if the U.S. holder’s holding period in such Warrant is more than one year at the time of the sale or other taxable disposition. The deductibility of capital losses is subject to certain limitations.
A U.S. holder will not be required to recognize income, gain or loss upon exercise of a Warrant received upon exercise of a Subscription Right. A U.S. holder’s tax basis in a share of our common stock received upon exercise of the Warrants for cash will be equal to the sum of (1) the U.S. holder’s tax basis in the Warrants exchanged therefor and (2) the exercise price of such Warrants. A U.S. holder’s holding period in the shares of our common stock received upon exercise will commence on the day after such U.S. holder exercises the Warrants.
In certain circumstances, the Warrants will be exercisable on a cashless basis. The U.S. federal income tax treatment of an exercise of a Warrant on a cashless basis is not clear, and could differ from the consequences described above. It is possible that a cashless exercise could be a taxable event. U.S. holders are urged to consult their own tax advisors as to the consequences of an exercise of a Warrant on a cashless basis, including with respect to whether the exercise is a taxable event, and their holding period and tax basis in the common stock received.
If a Warrant expires without being exercised, a U.S. holder will recognize a capital loss in an amount equal to such holder’s adjusted tax basis in the Warrant. Such loss will be long-term capital loss if, at the time of the expiration, the U.S. holder’s holding period in such Warrant is more than one year. The deductibility of capital losses is subject to certain limitations.
Distributions on Common Stock and Equity Interests
As described in the section entitled “Dividend Policy,” we do not anticipate declaring or paying dividends to holders of our common stock or Equity Interests in the foreseeable future. However, if we do make distributions of cash or property on our common stock or Equity Interests, such distributions will constitute dividends to the extent paid out of our current or accumulated earnings and profits, as determined for U.S. federal income tax purposes. Dividends received by a corporate U.S. holder may be eligible for a dividends received deduction, subject to applicable limitations. Dividends received by certain non-corporate U.S. holders, including individuals, are generally taxed at the lower applicable capital gains rate provided certain holding period and other requirements are satisfied. Distributions in excess of our current and accumulated earnings and profits will constitute a return of capital and first be applied against and reduce a U.S. holder’s adjusted tax basis in its common stock or Equity Interests but not below zero. Any excess will be treated as capital gain and will be treated as described below in the section relating to the sale or disposition of our common stock.
| 44 |
Sale, Exchange or Other Disposition Common Stock or Equity Interests
Upon a sale, exchange, or other taxable disposition of or our common stock or Equity Interests, a U.S. holder generally will recognize capital gain or loss equal to the difference between the amount realized (not including any amount attributable to declared and unpaid dividends, which will be taxable as described above to U.S. holders of record who have not previously included such dividends in income) and the U.S. holder’s adjusted tax basis in our common stock or Equity Interests. The U.S. holder’s adjusted tax basis in our Equity Interests generally will equal its initial tax basis (discussed above under “—Exercise of Subscription Rights”), as adjusted for applicable distributions, if any (including constructive dividends described below). A U.S. holder’s adjusted tax basis in our common stock generally will equal its initial tax basis in our common stock, as adjusted for applicable distributions (including constructive dividends described below). Such capital gain or loss generally will be long-term capital gain or loss if the U.S. holder’s holding period for our common stock or Equity Interests exceeded one year at the time of disposition. Long-term capital gains recognized by certain non-corporate U.S. holders, including individuals, generally are subject to reduced rates of taxation. The deductibility of capital losses is subject to limitations.
| 45 |
Information Reporting and Backup Withholding
A U.S. holder may be subject to information reporting and backup withholding when such holder receives dividend payments (including constructive dividends) or receives proceeds from the sale or other taxable disposition of the shares of common stock, Warrants or Equity Interests acquired through the exercise of Subscription Rights or shares of our common stock acquired through exercise of the Warrants. Certain U.S. holders are exempt from backup withholding, including certain corporations and certain tax-exempt organizations. A U.S. holder will be subject to backup withholding if such holder is not otherwise exempt (or fails to properly establish an exemption) and such holder:
| ● | fails to furnish the holder’s taxpayer identification number, which for an individual is ordinarily his or her social security number; | |
| ● | ||
| ● | is notified by the IRS that the holder previously failed to properly report payments of interest or dividends; or | |
| ● | fails to certify under penalties of perjury that the |
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules may be allowed as a refund or a credit against a U.S. holder’s U.S. federal income tax liability, provided the required information is timely furnished to the IRS. U.S. holders should consult their own tax advisors regarding their qualification for an exemption from backup withholding and the procedures for obtaining such an exemption.
Tax Considerations Applicable to Non-U.S. Holders
For purposes of this discussion, a “non-U.S. holder” is a beneficial owner of shares of our common stock or warrants, our Subscription Rights, shares of our common stock, Warrants and Equity Interests acquired upon exercise of Subscription Rights or shares of our common stock acquired upon exercise of Warrants, as the case may be, that is neither a U.S. holder nor an entity treated as a partnership for U.S. federal income tax purposes.
Receipt, Exercise and Expiration of the Subscription Rights
The discussion assumes that the receipt of Subscription Rights will be treated as a nontaxable distribution. See “—Tax Considerations Applicable to U.S. Holders—Receipt of Subscription Rights” above. In such case, non-U.S. holders will not be subject to U.S. federal income tax (or any withholding thereof) on the receipt, exercise or expiration of the Subscription Rights.
Exercise of Warrants
A non-U.S. holder will not be subject to U.S. federal income tax on the cash exercise of Warrants into shares of our common stock. As discussed above in “—Tax Considerations Applicable to U.S. Holders—Sale or Other Disposition, Exercise or Expiration of Warrants,” the U.S. federal income tax classification of the Warrants and the treatment of an exercise of a Warrant on a cashless basis is not clear. Non-U.S. holders are urged to consult their own tax advisors as to the proper U.S. federal income tax classification of the Warrants and the consequences of an exercise of a Warrant on a cashless basis, including with respect to whether the exercise is a taxable event, and their holding period and tax basis in the common stock received.
Constructive Dividends on Warrants
As described in the section entitled “Dividend Policy,” we do not anticipate declaring or paying dividends to holders of our common stock or Equity Interests in the foreseeable future. However, if at any time during the period in which a non-U.S. holder holds Warrants, if the exercise price is adjusted in certain other circumstances (or in certain circumstances, there is a failure to make appropriate adjustments), or there is an adjustment to the number of common shares that will be issued on exercise of the Warrants, such adjustments may result in the deemed payment of a taxable dividend to a non-U.S. holder. Any resulting withholding tax attributable to deemed dividends may be collected from other amounts payable or distributable to the non-U.S. holder. Non-U.S. holders should consult their own tax advisors regarding the proper treatment of any adjustments to the Warrants.
| 46 |
Distributions on Common Stock or Equity Interests
As described in the section entitled “Dividend Policy,” we do not anticipate declaring or paying dividends to holders of our common stock or Equity Interests in the foreseeable future. However, if we do make distributions of cash or property on our common stock or Equity Interests, such distributions will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. Amounts not treated as dividends for U.S. federal income tax purposes will constitute a return of capital and first be applied against and reduce a non-U.S. holder’s adjusted tax basis in its common stock or Equity Interests, as the case may be, but not below zero. Any excess will be treated as capital gain and will be treated as described below in the section relating to the sale or disposition of our common stock, Warrants, or Equity Interests. Because we may not know the extent to which a distribution is a dividend for U.S. federal income tax purposes at the time it is made, for purposes of the withholding rules discussed below we or the applicable withholding agent may treat the entire distribution as a dividend.
Subject to the discussion below on backup withholding and foreign accounts, dividends paid to a non-U.S. holder of our common stock or Equity Interests that are not effectively connected with the non-U.S. holder’s conduct of a trade or business within the United States will be subject to U.S. federal withholding tax at a rate of 30% of the gross amount of the dividends (or such lower rate specified by an applicable income tax treaty).
Non-U.S. holders will be entitled to a reduction in or an exemption from withholding on dividends as a result of either (1) an applicable income tax treaty or (2) the non-U.S. holder holding our common stock or Equity Interests in connection with the conduct of a trade or business within the United States and such dividends being effectively connected with that trade or business. To claim such a reduction in or exemption from withholding, the non-U.S. holder must provide the applicable withholding agent with a properly executed (a) IRS Form W-8BEN or W-8BEN-E (or other applicable documentation) claiming an exemption from or reduction of the withholding tax pursuant to the benefits of an income tax treaty between the United States and the country in which the non-U.S. holder resides or is established, or (b) IRS Form W-8ECI stating that the dividends are not subject to withholding tax because they are effectively connected with the conduct by the non-U.S. holder of a trade or business within the United States, as may be applicable. These certifications must be provided to the applicable withholding agent prior to the payment of dividends and must be updated periodically. Non-U.S. holders that do not timely provide the applicable withholding agent with the required certification, but that qualify for a reduced rate under an applicable income tax treaty, may obtain a refund of any excess amounts withheld by timely filing an appropriate claim for refund with the IRS.
If dividends paid to a non-U.S. holder are effectively connected with the non-U.S. holder’s conduct of a trade or business within the United States (and, if required by an applicable income tax treaty, the non-U.S. holder maintains a permanent establishment in the United States to which such dividends are attributable), then, although exempt from U.S. federal withholding tax (provided the non-U.S. holder provides appropriate certification, as described above, and subject to the discussion below on backup withholding and foreign accounts), the non-U.S. holder will be subject to U.S. federal income tax on such dividends on a net income basis at the regular graduated U.S. federal income tax rates. In addition, a non-U.S. holder that is a corporation may be subject to a branch profits tax at a rate of 30% (or such lower rate specified by an applicable income tax treaty) on its effectively connected earnings and profits for the taxable year that are attributable to such dividends, as adjusted for certain items. Non-U.S. holders should consult their own tax advisors regarding their entitlement to benefits under any applicable income tax treaty.
Sale or Other Disposition of Common Stock, Warrants or Equity Interests
Subject to the discussions below on backup withholding and foreign accounts, a non-U.S. holder will not be subject to U.S. federal income tax on any gain realized upon the sale or other taxable disposition of our common stock, Warrants or Equity Interests unless:
| ● | the gain is effectively connected with the non-U.S. holder’s conduct of |
| 47 |
| ● | the non-U.S. holder is a nonresident alien individual present in the United States for 183 days or more during the taxable year of the disposition and certain other requirements are met; or | |
| ● | our common stock, Warrants or Equity Interests constitutes a U.S. real property interest, or USRPI, by reason of our status as a U.S. real property holding corporation, or USRPHC, for U.S. federal income tax purposes. |
Drug manufacturers and other entities involvedGain described in the manufacture and distribution of approved product candidates are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and some state agencies for compliance with cGMP and other laws.
Regulation outside of the U.S.
In addition to regulations in the U.S., wefirst bullet point above generally will be subject to regulations of other countries governing any clinical trials and commercial sales and distribution of our product candidates. Whether or not we obtain FDA approval forU.S. federal income tax on a product, we must obtain approval bynet income basis at the comparable regulatory authorities of countries outside of the U.S. before we can commence clinical trials in such countries and approval of the regulators of such countries or economic areas, such as the European Union, before we may market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the timeregular graduated rates. A Non-U.S. holder that is a corporation also may be longer or shorter than that requiredsubject to a branch profits tax at a rate of 30% (or such lower rate specified by an applicable income tax treaty) on such effectively connected gain, as adjusted for FDA approval.certain items.
Under European Union regulatory systems, a company may submit marketing authorization applications either under a centralized or decentralized procedure. The centralized procedure, which is compulsory for medicines produced by biotechnology or those medicines intended to treat AIDS, cancer, neurodegenerative disorders or diabetes and is optional for those medicines which are highly innovative, provides for the grant of a single marketing authorization that is valid for all European Union member states. The decentralized procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the applications and assessments report, each member state must decide whether to recognize approval. If a member state does not recognize the marketing authorization, the disputed points are eventually referred to the European Commission, whose decision is binding on all member states.
AsGain described in the U.S., we may apply for designation of a product candidate as an orphan drug for the treatment of a specific indication in the European Union before the application for marketing authorization is made. Orphan drugs in Europe enjoy economic and marketing benefits, including up to ten years of market exclusivity for the approved indication unless another applicant can show that its product is safer, more effective or otherwise clinically superior to the orphan-designated product.
Reimbursement
Sales of our products will depend, in part, on the extent to which the costs of our productssecond bullet point above will be coveredsubject to U.S. federal income tax at a rate of 30% (or such lower rate specified by third-party payors, such as government health programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly challenging the prices charged for medical products and services. Additionally, the containment of healthcare costs has become a priority of federal and state governments and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictionsan applicable income tax treaty) on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. If these third-party payors do not consider our products to be cost-effective compared to other therapies, they may not cover our products after approved as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (“MMA”) imposed new requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities which will provide coverage of outpatient prescription drugs. Part D plans include both stand-alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutics committee. Government payment for some of the costs of prescription drugs may increase demand for our products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that resultsgain derived from the MMA may result in a similar reduction in payments from non-governmental payors.
The American Recovery and Reinvestment Act of 2009 provides funding for the federal government to compare the effectiveness of different treatments for the same illness. A plan for the research will be developed by the Department of Health and Human Services, the Agency for Healthcare Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures will be made to the U.S. Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payors, it is not clear what effect, if any, the research will have on the sales of any product, if any such product or the condition that it is intended to treat is the subject of a study. It is also possible that comparative effectiveness research demonstrating benefits in a competitor’s product could adversely affect the sales of our product candidates. If third-party payors do not consider our products to be cost-effective compared to other available therapies, they may not cover our products as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010, collectively referred to as the “ACA,” enacted in March 2010, had a significant impact on the health care industry by expanding coverage for the uninsured. With regard to pharmaceutical products, among other things, ACA is expanded and increased industry rebates for drugs covered under Medicaid programs and made changes to the coverage requirements under the Medicare Part D program. The administration and Congress which will take office in January 2017, has pledged to repeal and replace the ACA, largely because of significantly increasing health insurance premiums and decreasing participation by members of the insurance companies. We cannot predict the impact of any repeal, replacement or modificationsdisposition, which may be enacted.
In addition, in some non-U.S. jurisdictions, the proposed pricing for a product candidate must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitabilityoffset by U.S. source capital losses of the company placingnon-U.S. holder (even though the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our product candidates. Historically, product candidates launched in the European Union doindividual is not follow price structuresconsidered a resident of the U.S. and generally tend to be significantly lower.
Environment
Our third-party manufacturers are subject to inspections byUnited States), provided the FDA for compliance with cGMP and other U.S. regulatory requirements, includingnon-U.S. holder has timely filed U.S. federal state and local regulations regarding environmental protection and hazardous and controlled substance controls, among others. Environmental laws and regulations are complex, change frequently and have tended to become more stringent over time. We have incurred, and may continue to incur, significant expenditures to ensure we are in compliance with these laws and regulations. We would be subject to significant penalties for failure to comply with these laws and regulations.
Sales and Marketing
Our current focus is on the development of our existing portfolio, the completion of clinical trials and, if and where appropriate, the registration of our product candidates. We currently do not have marketing, sales and distribution capabilities. If we receive marketing and commercialization approval for any of our product candidates, we intend to market the product either directly or through collaborations, strategic alliances and distribution agreements with third parties. The ultimate implementation of our strategy for realizing the financial value of our product candidates is dependent on the results of clinical trials for our product candidates, the availability of funds and the ability to negotiate acceptable commercial terms with third parties.
Employees
As of the date of this prospectus, we have five employees, including our three executive officers, one engaged in research and development and one in administration. We consider our relationship with our employees to be good.
Facilities
Our corporate headquarters are located in Rockville, Maryland, where we lease shared access to office space and reception services. Our research and development activities are performed in approximately 1,727 square feet of laboratory and office space located in Gaithersburg, Maryland. All of such space is leased from a non-affiliated third party, pursuant to leases expiring in 2023, which provide for an aggregate monthly rental of $5,757.
We believe that our existing facilities are adequate for our current needs and have sufficient laboratory space to house additional scientists as we grow. When our lease expires, we may exercise our renewal options or look for additional or alternate space for our operations. We believe that suitable additional or alternative space will be available in the future on commercially reasonable terms.
Legal Proceedings
Currently, there are no legal proceedings pending or threatened against us.
Directors and Executive Officers
Our directors and executive officers and their respective ages and titles are as follows:
Set forth below is a description of the background and business experience of our directors and executive officers.
Anatoly Dritschilo, M.D. is a co-founder of the Company and has served as Chairman of the Board and Chief Executive Officer since the Company’s formation in December 2012. Dr. Dritschilo is a radiation oncologist by training and has held multiple leadership positions in health care. At Georgetown University Medical School in Washington, D.C., he served principally as Department Chair from 1980 to 2018; Chief of Radiation Oncology at MedStar-Georgetown University Hospital from 2005 to 2016; Medical Director of Georgetown University Hospital from 1994 to 1997; and Interim Director of the NCI-funded Lombardi Comprehensive Cancer Center from 2005 to 2007. He has also served on the Boards of Directors of MedStar-Georgetown University Hospital, the National Capital Rehabilitation Hospital and the MedStar Health Research Institute. His experience with Pharma includes Board of Directors membership of NeoPharm, Inc, and he was a founding director of Oncomed (Neopharm). His 200+ scientific publications and 12 issued patents have earned him election as a Fellow of the National Academy of Inventors. Dr. Dritschilo holds a Bachelor of Science degree in Chemical Engineering from the University of Pennsylvania, his medical degree from the College of Medicine of New Jersey and residency training from the Harvard, Joint Center for Radiation Therapy. His qualifications support his service as our Chairman of the Board of Directors.
Michael P. Vander Hoek serves as the Company’s Chief Financial Officer, a position he was appointed to in August 2019, and Vice President, Operations and Regulatory, a position he has held since 2019. From November 2019 until April 2021, Mr. Vander Hoek served as Director, Finance and Business Development at Georgetown Lombardi Comprehensive Cancer Center (“LCCC”), where he directed a new five-year $221.9 million institutional commitment for cancer center research under a new NCI-approved cancer consortium arrangement and recruited scientists to fulfill strategic objectives with senior leaders to improve cancer research and treatment. From 2007 until November 2019, Mr. Vander Hoek served as Associate Director, Administration, at Georgetown’s LCCC, where he was responsible for direct administrative operations for more than 400 faculty and staff in the department of oncology, radiation medicine, pathology and biostatistics, bioinformatics and biomathematics, including managing $216.9 million in institutional commitments to LCCC from Medstar Health, John Theurer Cancer Center (“JTCC”), and Georgetown University. and implementing an enterprise-wide clinical trial management system for Georgetown University and Medstar Health. From 2004 until 2007, Mr. Vander Hoek served as Chief Financial Officer at Georgetown’s LCCC. During his time at Georgetown, Mr. Vander Hoek negotiated a series of 12 research integration agreements between LCCC and the JTCC that resulted in the approval of an NCI recognized Consortium in 2019. From 2001 until 2004, Mr. Vander Hoek served as Vice-Chair, Planning and Administration, at MedStar Georgetown University Hospital, where he was responsible for managing administrative and financial operations for some 440 staff, physicians, residents and fellows in the departments of Medicine and Neurology. From 1996 until 2001, Mr. Vander Hoek served as Senior Associate Administrator, Finance and Information Systems, for the Department of Medicine, Georgetown University Medical Center, where he designed and managed the faculty compensation system, while managing the finances and information systems for the department. His financial management experience in publicly held companies includes Director of Managed Care Reimbursement for Critical Care America from 1990 to 1993 and Regional Controller for Laboratory Corporation of America (LabCorp) from 1993 to 1996. His responsibilities at both companies included extensive financial management related to mergers, acquisitions, and start-up operations. Mr. Vander Hoek holds a Master’s in Health Services Administration from The George Washington University and a Bachelor of Arts in Biology and Psychology from Hope College.
Peter Dritschilo has served as our President and Chief Operating Officer since Shuttle was formed in December 2012. He also served as our Chief Financial Officer from December 2012 until August 2019. Mr. Dritschilo has over 20 years of business management experience in medical services and cancer treatment. He has held administrative positions with Medstar-Rad America from 2001 to 2005, Georgetown University 2005 to 2006, Prince William Hospital and the Fauquier Hospital Cancer Center 2006 to 2011 and Inova Health System’s Schar Cancer Institute from 2011 to 2018. In 2014, Mr. Dritschilo filed for Chapter 7 bankruptcy protection due to the failure of a personal business venture. Mr. Dritschilo graduated from Georgetown University and received his MBA from the George Washington University.
Mira Jung, Ph.D., a co-founder of our company, presently serves as our Chief Scientific Officer for Biology and was a member of our board of directors from our formation in December 2012 until 2019. Since 2005, Dr. Jung has served as Professor of Radiation Medicine and Microbiology at Georgetown University Medical School, with over 20 years of experience in molecular radiation biology research. She is an expert in mechanisms of radiation resistance and on the roles of HDAC inhibitors in modifying the radiation response. Dr. Jung’s research has been funded by NIH and the DOD leading to 100 publications and six issued patents, including the first reports of HDAC inhibitor drug classes modifying cancer cell radiation resistance and protecting normal tissues from radiation damage. Dr. Jung holds an MA degree and a PhD in Microbiology and Molecular Virology from the University of Kansas.
Tyvin A. Rich, M.D. serves as our company’s Chief Medical Officer and is responsible for the clinical development of novel radiation sensitizers. Since 2010, Dr. Rich has served as a Staff Radiation Oncologist at the Hampton University Proton Therapy Institute in Hampton Virginia and Professor Emeritus at University of Virginia Health Sciences Center, Department of Radiation Oncology. From 1995 until 2010, Dr. Rich was a Professor and Chairman of the Department of Therapeutic Radiology and Oncology at the University of Virginia Health Sciences Center. And prior to that, from 1984 through 1995, Dr. Rich was a Professor of Radiotherapy and Director of Clinics in the Department of Radiotherapy of the University of Texas M. D. Anderson Cancer Center. He has served as the protocol chair for RTOG clinical trials that advanced the use of chemoradiation for the treatment of rectal and pancreatic cancers. He is an expert in the applications of infusional 5-Fluorouracil for chemoradiation therapy of gastro-intestinal cancers and has authored more than 200 scientific articles, reviews and book chapters. Dr. Rich received his undergraduate degree at Rutgers University, his medical degree at the University of Virginia, and completed residencies in internal medicine at Georgetown University Medical Center and radiation therapy at Massachusetts General, Harvard Medical School.
Joshua Schafer was appointed to be a member of our company’s board of directors in 2019. Since 2015, he has served as Senior Vice President and Head of Corporate Strategy and Business Development for Mallinckrodt Pharmaceuticals Incorporated. From 2009 until 2015, he served as Vice President and Oncology Therapeutic Area Head at Astellas Pharmaceuticals Incorporated, where he was responsible for building the company’s global oncology franchise. From 2000 until 2009, Mr. Schafer served in positions of increasing seniority at Takeda Pharmaceuticals North America, including Manager and Senior Manager, New Product and New Business Development; Senior Product Manager, Gastrointestinal Marketing; and Director, Oncology and Renal Marketing and Commercial Development. He began working in the healthcare and pharmaceutical industry in 1998, and has served in various positions including management consulting at Accenture (formerly Anderson Consulting), G.D. Searle & Co. (later acquired by Pfizer) and Cognia Corporation. He received his Bachelor of Arts in Biology and German at the University of Notre Dame, his MS in Biotechnology from Northwestern University and his MBA from Northwestern University. We believe Mr. Schafer’s extensive experience in pharmaceutical strategy, marketing and business development will assist our board’s oversight role as we build and develop our Company.
Steven Richards was appointed to be a member of our company’s board of directors in 2019. He is CEO and Founder of Endurance Media, a media finance company based in Santa Monica, California, that launched in 2014 with a strategic alliance with eOne Entertainment and a mandate to produce and finance commercially driven feature films. From 2006 to 2014, Mr. Richards served as Co-President and Chief Operating Officer of Silver Pictures where he oversaw all business activities and managed a team of more than 20 people responsible for film development, production and financial information. From 2000 to 2006, he served as Chief Financial Officer at Silver Pictures and from 1995 to 2000 as Vice President, Finance, at Silver Pictures. Mr. Richards holds an MBA in Finance from UCLA, a BBA in accounting from Temple University, and holds his CPA license. We believe his experience as a chief financial officer and in accounting will assist in providing our board guidance and oversight as we grow our company.
Milton Brown, M.D., Ph.D. is a co-founder of our company and has served as our Chief Scientific Officer for Chemistry and as a member of our board of directors since we were formed in December 2012. Since August 2022, Dr. Brown has also served as Vice Dean of Research at Eastern Virginia Medical School. Dr. Brown was a founder in 2004 of Rivanna Pharmaceuticals, a Virginia-based biopharmaceutical company engaged in the discovery and development of novel small molecule therapeutics for the treatment of neurological diseases and cancer. Since 2012, Dr. Brown has served as Director of the Drug Discovery Center at Georgetown University Medical School and since 2010, he has been the principal investigator of the NIH/NCI funded Chemical Diversity Center. He brings to Shuttle 15 years of experience in drug discovery with over 80 publications and eight issued patents, including discovery of novel HDAC inhibitors and has two drugs currently in clinical trials. He has served on government committees including the NIH Experimental Therapeutics Study Section, the NIH Drug Discovery and Molecular Pharmacology Study Section and was a scientific counselor to the U.S. Secretary of Health. Dr. Brown holds a Ph.D. in synthetic chemistry from University of Alabama, and an MD from the University of Virginia. His extensive experience and expertise in drug discovery makes him uniquely qualified to direct the company’s drug discovery program and serve as our director.
William H. Adkins was appointed to be a member of our company’s board of directors in 2019. From 2018 to present, Mr. Adkins has worked as a consultant to businesses, working with business owners to develop strategy, planning and problem solving, especially as concerns developing strategic goals, performance plans and marketing strategies. From 2017 until present, Mr. Adkins has served as the President and General Manager at Gen’R LLC, a business consulting firm. From 2014 until 2017 Mr. Adkins served as a Strategic Business Development Manager at automotiveMastermind Inc., where he was responsible for training dealer candidates and general management, including consulting with dealers and interviewing applicants for management instructor positions. While at automotiveMastermind Inc., he partnered with the company’s founders to develop automotiveMastermind Inc. into a major company that eventually merged with IHS Markit Ltd. From 2004 to 2014, Mr. Adkins was a management instructor for the National Automobile Dealers Association, where he trained managers and dealer successors how to effectively operate a retail automobile dealership with knowledge of the various departments in a traditional dealership (e.g. sales, service, parts, and accounting). From 1985 until 2003, Mr. Adkins was President and General Manager at Chevrolet dealerships in Ohio, California and New York, including Adkins Chevrolet Buick Oldsmobile, Bayview Lincoln Mercury and Palanker Chevrolet. Mr. Adkins attended the University of Maryland where he studied marketing and business management. Mr. Adkins studied marketing at the University of Cincinnati and law at Bryant and Stranton Business College. Mr. Adkins’ substantial business and marketing experience will help us as we develop the Company’s products and business.
Chris Senanayake, Ph.D. was appointed to be a member of the Company’s board of directors in 2021. He is CEO and Founder of TCG GreenChem, Inc., a US subsidiary of TCG Lifesciences Pvt. Ltd., a leading global Contract Research and Manufacturing Services (CRAMS) company in the area of drug discovery, development and commercialization. Dr. Senanayake has more than 30 years of pharmaceutical industry experience, making him an invaluable asset to Shuttle Pharma’s mission as the Company advances its pharmaceutical candidates in clinical trials. He is the Founder and Chief Executive Officer (CEO) of TCG GreenChem Inc. and Chief Scientific Officer of TCG Lifesciences, Pvt. Ltd. He has held positions of Senior Scientist at Dow Chemical, and Research Fellow at Merck & Co, Inc. Director and Executive Director of Process Research at Sepracor, Inc. (1996 to 2002), Director of Chemical Development and Vice President of Chemical Development for Boehringer Ingelheim Pharmaceuticals, Inc. In 2018, he was appointed as the CEO of Asta GreenChem, Inc in Richmond VA and Astatech (Chengdu) Biopharmaceuticals Corp. in China. He has a record of leading and delivering on high complexity APIs for manufacturing. Dr. Senanayake participated in development activities of many drugs, including multi-billion-dollar blockbuster drugs, such as Crixivan, Lunesta, Jardiance, Formotorol, Desvenlafaxine and other drug candidates. He is co-author of 425 scientific publications and is co-inventor of more than 150 patents. We believe he will provide value to us by introducing potential joint venture partners, as well as enhancing our oversight through his in-depth understanding of and experience in the pharmaceuticals industry.
Scientific Advisory Committee
Theodore L. Phillips, M.D. has served as the Chair of our Scientific Advisory Committee since 2018. He held the position of Chief Medical Officer and Clinical Director at Shuttle Pharmaceuticals from 2014 until 2018. Dr. Phillips’ distinguished career has included positions of Chair of the Department of Radiation Oncology (from1978 to 1998) and Associate Director (from 1996 to 1999) of the UCSF Cancer Center at the University of California at San Francisco. He is highly experienced in radiation oncology clinical trials of hypoxic radiation sensitizers. Dr. Phillips served as the principal investigator of the SBIR contract for the Phase I clinical trial of Ropidoxuridine. He previously served as Associate Director of the Northern California Oncology Group from 1983-1990, president of the American Society of Therapeutic Radiation Oncologists from 1984 to 1985, and is an elected member of the Institute of Medicine of the National Academy of Science. Dr. Phillips holds a BS degree from Dickinson College in Carlisle, Pennsylvania and a MD from the University of Pennsylvania. He provides advice to the leadership team to help design and implement clinical trials of radiation therapy and radiation response modifying drugs.
Ralph R. Weichselbaum, M.D. has served as Scientific Advisor to Shuttle Pharmaceuticals for translational research for the discovery and development of radiation response modifiers since 2013. Dr. Weichselbaum is the Daniel K. Ludwig Professor and Chairman of the Department of Radiation and Cellular Oncology, the University of Chicago, a position he has held since 1985. He is also an elected member of the Institute of Medicine, National Academy of Sciences. He has devoted his career to translational research in cancer with combined radiotherapy and chemotherapy. Dr. Weichselbaum and his colleagues conceived “genetic radiotherapy” and developed viral constructs for use in clinical tumor radiation sensitization. These were commercialized as TNFerade (GenVec, Inc.) and tested in a Phase I clinical trial in prostate cancer and a Phase III clinical trial for pancreatic cancer.
J. Martin Brown, Ph.D. has served as a Scientific Advisor to Shuttle Pharmaceuticals for translational research for the development of hypoxic radiation sensitizers since 2017. Dr. Brown received his Ph.D.in Cancer Biology from Oxford University in 1968 and was Director of the Division of Radiation and Cancer Biology at Stanford University from 1984 to 2004. He is an expert in the radiation biology of hypoxia in cancers and has more than 300 peer-reviewed published articles. He has received awards in recognition of his work, including the Gold Medal, American Society for Therapeutic Radiology and Oncology (1999, the Failla Memorial Award, Radiation Research Society (2000), the Weiss Medal, Association for Radiation Research (2001) and the Henry S. Kaplan Distinguished Scientist Award, International Association for Radiation Research (2007). He developed etanidazole, a hypoxic radiation sensitizer, and tirapazamine, a hypoxic cytotoxic drug, from bench to clinical trials.
Alejandro Villagra, Ph.D. has served as a Scientific Advisor to Shuttle Pharmaceuticals with expertise in cellular signaling pathways, epigenetics and immunology since 2017. Dr. Villagra received his Ph.D. in Molecular Biology from the University of Concepcion, in Chile in 2004 and completed post-graduate training at the H. Lee Moffitt Cancer Center and Research Institute in Tampa, Florida in Molecular Immunology in 2009, in the Laboratory of Eduardo Sotomayor, MD. He joined the faculty of the Moffitt Cancer Center and Research Institute, as a research scientist from 2009 through 2015 and advanced to Assistant Professor of Oncologic Sciences. He became an Assistant Professor in the Department of Biochemistry and Molecular Medicine at the George Washington University (GWU) School of Medicine and Health Sciences in 2015, as a member of the GWU Cancer Center. His research is focused on molecular and cellular roles of histone deacetylases (HDACs) in tumor immunology and as adjuvants for immunotherapy of cancers.
Joseph Armstrong, III, Ph.D. joined as a Scientific Advisor to Shuttle Pharmaceuticals in 2021, He received his Ph.D. from the University of Colorado in 1988, completed his post-doctoral work at the University of Virginia at Charlottesville and holds the position of Chief Operating Officer at and Global Head of Business Development TCG GreenChem, Inc. He provides industry experience in chemistry, drug development and process research, having previously held positions at Merck & Co. Inc. in Rahway, N.J and in the U.K. for two pharmaceutical companies in the areas of Pharmaceutical Research and Development. His primary areas of focus have been in the design and implementation of efficient synthesis of drug candidates amenable to large scale production. Dr. Armstrong led the development team that designed, developed and implemented the manufacturing process for the new treatment for Type II diabetes, Januvia TM. His team was awarded the Solvias Prize in 2004 (Basel, Switzerland), the IChemE Aztra-Zeneca Award for Green Chemistry and Engineering in 2005 (London, England), Dr. Armstrong has more than 40 publications and holds 10 patents.
Family Relationships
Dr. Anatoly Dritschilo and Peter Dritschilo are father and son. There are no other family relationships among our directors and executive officers.
Board of Directors
Our board of directors is responsible for overseeing the Company’s business consistent with its fiduciary duty to the stockholders. This significant responsibility requires highly skilled individuals with various qualities, attributes and professional experience. There are general requirements for service on the board that are applicable to directors and there are other skills and experience that should be represented on the board as a whole but not necessarily by each director. Our Corporate Governance and Nominating Committee, detailed below, considers the qualifications of director candidates individually and in the broader context of the board’s overall composition and the Company’s current and future needs.
Terms of Office
Our directors were initially appointed for staggered two and three-year terms as initial appointments. The Chairman of the Board is also the CEO and was appointed for an initial three-year term. Following this offering, we intend that all of our directors will be elected to one-year terms to hold office until the next annual meeting of our stockholders and until a successor is appointed and qualified, or until their removal, resignation, or death. Executive officers serve at the pleasure of the board of directors.
Director Independence
In order to qualify to list our shares of common stock for trading on Nasdaq, our board of directors must consist of a majority of “independent” directors, as defined under Nasdaq listing standards and Rule 10A-3(b)(1) under the Exchange Act. At present, four of the six directors serving on our board of directors qualify as “independent.” Our independent directors consist of Messrs. Adkins, Richards and Schafer and Dr. Senanayake.
Board Committees
General
Our board of directors has established three committees consisting of an audit committee, a compensation committee, and a nominating and corporate governance committee. The members of each committee qualify as “independent” as defined under Nasdaq listing standards and Rule 10A-3(b)(1). Moreover, at least one member of the audit committee qualifies as an “audit committee financial expert” as the term is defined under Nasdaq listing standards and applicable rules and regulations of the SEC, based on their respective business professional experience in the financial and accounting fields.
Audit Committee
The audit committee, which consists of Steve Richards, MBA, CPA (Chair), William Adkins and Chris Senanayake, MD, assists our board of directors in its oversight of the Company’s accounting and financial reporting processes and the audits of the Company’s financial statements, including (a) the quality and integrity of the Company’s financial statements (b) the Company’s compliance with legal and regulatory requirements, (c) the independent auditors’ qualifications and independence and (d) the performance of the Company’s internal audit functions and independent auditors, as well as other matters which may come before it as directed by the board of directors. Further, the audit committee, to the extent it deems necessary or appropriate, among its several other responsibilities, will:
Compensation Committee
The compensation committee, which consists of Steve Richards (Chair) and Joshua Schafer, aids our board of directors in meeting its responsibilities relating to the compensation of the Company’s executive officers and to administer all incentive compensation plans and equity-based plans of the Company, including the plans under which Company securities may be acquired by directors, executive officers, employees and consultants. Further, the compensation committee, to the extent it deems necessary or appropriate, among its several other responsibilities, will:
Nominating and Corporate Governance Committee
The nominating and corporate governance committee, which consists of Joshua Schafer (Chair) and Steve Richards, recommends to the board of directors individuals qualified to serve as directors and on committees of the board of directors to advise the board of directorsincome tax returns with respect to the board of directors composition, procedures and committees to develop and recommend to the board of directors a set of corporate governance principles applicable to the Company, and to oversee the evaluation of the board of directors and Shuttle’s management. In addition, the nominating and corporate governance committee will consider diversity of background including diversity of race, ethnicity, international background, gender and age when evaluating candidates for board membership.such losses.
Further, the nominating and corporate governance committee, to the extent it deems necessary or appropriate, among its several other responsibilities will:
Code of Ethics
We have adopted a code of ethics that applies to all of our executive officers, directors and employees. The code of ethics codifies the business and ethical principles that govern all aspects of our business. This document will be made available in print, free of charge, to any shareholder requesting a copy in writing from our Secretary at our executive offices in Rockville, Maryland. A copy of our code of ethics is available on our website at www.shuttlepharma.com.
Board of Directors Role in Risk Oversight
Members of the board of directors have periodic meetings with management and the Company’s independent auditors to perform risk oversight withWith respect to the Company’s internal control processes. The Company believes thatthird bullet point above, we believe we are not currently and do not anticipate becoming a USRPHC. Because the board’s role in risk oversight does not materially affect the leadership structuredetermination of the Company. The Company believes that its founders, leadership team and members of the board of directors exemplify diversity and inclusivity with respect to race, sex and ethnic origin. The board of directors presently has two diverse directors and is in the process of reviewing and vettingwhether we are a female candidate to serve as a director. As such, the Company anticipates being in full compliance with Nasdaq’s newly adopted diversity requirements by the end of its first year of listing.
Summary Compensation Table
The table below summarizes all compensation awarded to, earned by, or paid to our Chief Executive Officer and Chief Financial Officer and certain of our other executive officers for 2021 and 2020.
SUMMARY COMPENSATION TABLE
| Name and principal position | Year | Salary ($) | Bonus ($) | Stock Awards ($) | Option Awards ($) | Non-Equity Incentive Plan Compensation ($) | Nonqualified Deferred Compensation Earnings ($) | All Other Compensation ($) | Total ($) | |||||||||||||||||||||||||||
| Anatoly Dritschilo M.D., CEO | 2021 | 18,829 | - | - | - | - | - | - | 18,829 | |||||||||||||||||||||||||||
| 2020 | 35,144 | - | - | - | - | - | - | 35,144 | ||||||||||||||||||||||||||||
| Michael Vander Hoek, CFO, VP | 2021 | 18,338 | - | - | - | - | - | - | 18,138 | |||||||||||||||||||||||||||
| 2020 | 17,484 | - | - | - | - | - | - | 17,484 | ||||||||||||||||||||||||||||
| Peter Dritschilo, President and COO | 2021 | 31,534 | - | - | - | - | - | - | 31,534 | |||||||||||||||||||||||||||
| 2020 | 23,970 | - | - | - | - | - | - | 23,970 | ||||||||||||||||||||||||||||
Employment Agreements
Each of our executive officers has entered into an employment agreement with us. The employees each will receive compensationUSRPHC depends on an annual basis in cash, payable in monthly installments commencing at the completion of this offering, as well as restricted stock units subject to achieving certain key performance indicators. Certain of our executive officers are entitled to various target bonuses, upon achievement of certain milestones. The terms of the employment agreements are as follows:
Employment Agreement with Anatoly Dritschilo, MD
On June 28, 2019, we entered into an employment agreement with our Chief Executive Officer and Chairman of the Board, Anatoly Dritschilo, M.D. Under Dr. Dritschilo’s employment agreement, Dr. Dritschilo will receive base compensation of $274,000 per year. Dr. Dritschilo also received an initial restricted stock unit grant of 45,495 restricted stock units (“RSUs”) (22,747 on a post-reverse split basis) issuable under the Company’s 2018 Equity Incentive Plan, which RSUs vest over three years in substantially equal one-third installments on each one year anniversary of the agreement. Under his employment agreement, if Dr. Dritschilo terminates his employment for “Good Reason,” as defined in the agreement, Dr. Dritschilo will be entitled to his then applicable base salary for period of 12 months, subject to his continued compliance with certain requirements of his employment agreement.
Employment Agreement with Michael Vander Hoek
On September 1, 2019, we entered into an amended employment agreement with our Chief Financial Officer and Vice President for Operations and Regulatory, Michael Vander Hoek. Under Mr. Vander Hoek’s employment agreement, he will receive base compensation of $227,000 and is entitled to a target bonus of $72,000 upon achievement of certain milestones. Mr. Vander Hoek also received an initial restricted stock unit grant of 6,096 RSUs (on a post-reverse split basis) issuable under the Company’s 2018 Equity Incentive Plan, which RSUs vest over three years in substantially equal installments on each one year anniversary of the agreement. Under Mr. Vander Hoek’s employment agreement, if he terminates his employment for “Good Reason,” as defined in the agreement, he will be entitled to his then applicable base salary for period of 12 months, subject to his continued compliance with certain requirements of his employment agreement.
Employment Agreement with Peter Dritschilo
On May 30, 2019, we entered into an employment agreement with our President and Chief Operating Officer, Peter Dritschilo. Under Mr. Dritschilo’s employment agreement, Mr. Dritschilo will receive base compensation of $236,000 and is entitled to a target bonus of $72,000 upon achievement of certain milestones. Mr. Dritschilo also received an initial restricted stock unit grant of 20,760 RSUs (10,380 on a post-reverse split basis) issuable under the Company’s 2018 Equity Incentive Plan, which RSUs vest over three years in substantially equal installments on each one year anniversary of the agreement. Under Mr. Dritschilo’s employment agreement, if Mr. Dritschilo terminates his employment for “Good Reason,” as defined in the agreement, he will be entitled to his then applicable base salary for period of 12 months, subject to his continued compliance with certain requirements of his employment agreement.
Employment Agreement with Tyvin Rich, MD
On May 31, 2019, we entered into an employment agreement with our Chief Clinical Officer, Tyvin Rich, M.D. Under Dr. Rich’s employment agreement, Dr. Rich receives base compensation of $218,000 per year and is entitled to a target bonus of $43,000 upon achievement of certain milestones. Dr. Rich also received an initial restricted stock unit grant of 3,843 RSUs (on a post-reverse split basis) issuable under the Company’s 2018 Equity Incentive Plan, which RSUs vest over three years in substantially equal installments on each one year anniversary of the agreement. Under Dr. Rich’s employment agreement, if Dr. Rich terminates his employment for “Good Reason,” as defined in the agreement, he is entitled to his then applicable base salary for period of 12 months, subject to his continued compliance with certain provisions of his employment agreement.
Employment Agreement with Mira Jung, Ph.D.
On May 30, 2019, we entered into an employment agreement with our Chief Scientific Officer for Biology, Mira Jung, Ph.D. Under Dr. Jung’s employment agreement, Dr. Jung receives base compensation of $46,800 and is entitled to a target bonus of $14,200 upon achievement of certain milestones. Dr. Jung also received an initial restricted stock unit grant of 892 RSUs (on a post-reverse split basis) issuable under the Company’s 2018 Equity Incentive Plan, which RSUs vest over three years in substantially equal installments on each one year anniversary of the agreement. Under Dr. Jung’s employment agreement, if Dr. Jung terminates her employment for “Good Reason,” as defined in the agreement, Dr. Jung is then entitled to her then applicable base salary for period of 12 months, subject to her continued compliance with certain requirements of her employment agreement.
Outstanding Equity Awards at Fiscal Year-End
On a post-reverse split basis, a total of 384,167 RSUs have been granted to our executive officers under our 2018 Equity Incentive Plan (the “Plan”), of which 357,390 have vested to date yet remain unissued. After completion of this public offering, it is our intent to file a registration statement on Form S-8 to register the shares granted under our 2018 Equity Incentive Plan, at which time we will issue all such vested shares.
2018 Equity Incentive Stock Plan
Our 2018 Equity Incentive Plan provides for equity incentives to be granted to our employees, executive officers or directors and to key advisers and consultants. Equity incentives may be in the form of stock options with an exercise price of not less than the fair market value of the underlying shares as determined pursuantour USRPIs relative to the 2018fair market value of our other business assets and our non-U.S. real property interests, however, there can be no assurance we are not a USRPHC or will not become one in the future.
Non-U.S. holders should consult their own tax advisors regarding potentially applicable income tax treaties that may provide for different rules.
Information Reporting and Backup Withholding
Subject to the discussion below on foreign accounts, a non-U.S. holder will not be subject to backup withholding with respect to distributions on our common stock, Warrants or Equity Incentive Plan, restrictedInterests we make to the non-U.S. holder, provided the applicable withholding agent does not have actual knowledge or reason to know such holder is a United States person and the holder timely certifies its non-U.S. status, such as by providing a valid IRS Form W-8BEN, W- 8BEN-E or W-8ECI, or other applicable certification. However, information returns generally will be filed with the IRS in connection with any distributions (including deemed distributions) made on our common stock, awards, other stock- based awards,Warrants and Equity Interests to the non-U.S. holder, regardless of whether any tax was actually withheld. Copies of these information returns may also be made available under the provisions of a specific treaty or any combinationagreement to the tax authorities of the foregoing. The 2018 Equity Incentive Plancountry in which the non-U.S. holder resides or is administered byestablished.
| 48 |
Information reporting and backup withholding may apply to the Company’s compensation committeeproceeds of a sale or alternatively, if there is no compensation committee, the Company’s board of directors. We have reserved 3,000,000 sharesother taxable disposition of our common stock, for issuance underWarrants or Equity Interests within the 2018 Equity Incentive Plan (the “Plan”),United States, and information reporting may (although backup withholding generally will not) apply to the proceeds of which 384,167 shares have been granted under the Plan as of the date of this prospectus.
Director Compensation
Each of our non-employee directors, who were elected in 2019, receives compensation on an annual basis consisting of $25,000 in cash, payable in quarterly installments commencing 90 days after completion of the offering, and 2,702 restricted stock units. Pursuant to director offer letters entered into between each director and our company (the “Director Agreements”), the RSUs vest over a two-year period in one third increments, with one-third vesting immediately upon signing and then one-third vesting on each of the first and second anniversary of election. In addition, non-employee directors will also be reimbursed for out-of-pocket costs incurred in connection with attending meetings.
The following table sets forth, as of the date of this prospectus, the beneficial ownershipsale or other taxable disposition of our common stock, byWarrants or Equity Interests outside the United States conducted through certain U.S.-related financial intermediaries, in each director and executive officer, by each person known by uscase, unless the beneficial owner timely certifies under penalty of perjury that it is a non-U.S. holder on IRS Form W-8BEN or W-8BEN-E, or other applicable form (and the payor does not have actual knowledge or reason to beneficially own 5%know that the beneficial owner is a U.S. person) or moresuch owner otherwise timely establishes an exemption. Proceeds of a disposition of our common stock, and by directors and executive officersWarrants or Equity Interests conducted through a non-U.S. office of a non-U.S. broker generally will not be subject to backup withholding or information reporting.
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules may be allowed as a group. Unless otherwise stated,refund or a credit against a non-U.S. holder’s U.S. federal income tax liability, provided the address ofrequired information is timely furnished to the persons set forth in the table is c/o Shuttle Pharmaceuticals Holdings, Inc., One Research Court, Suite 450, Rockville, Maryland 20850.IRS.
Beneficial ownership is determined in accordance with SEC rules and generally includes voting or investment power with respectAdditional Withholding Tax on Payments Made to securities. Unless otherwise indicated, the stockholders listed in the table below have sole voting and investment power with respect to the shares indicated.Foreign Accounts
All share ownership figures include sharesWithholding taxes may be imposed under the Foreign Account Tax Compliance Act, or FATCA, on certain types of payments made to non-U.S. financial institutions and certain other non-U.S. entities. Specifically, a 30% withholding tax may be imposed on dividends (including deemed dividends) paid on our commonscommon stock, issuable upon securities convertibleWarrants or exchangeable into sharesEquity Interests, or gross proceeds from the sale or other disposition of our common stock, whetherWarrants or Equity Interests paid to a “foreign financial institution” or a “non-financial foreign entity” (each as defined in the Code), unless (1) the foreign financial institution undertakes certain diligence and reporting obligations, (2) the non-financial foreign entity either certifies it does not convertiblehave any “substantial United States owners” (as defined in the Code) or exchangeable within 60 daysfurnishes identifying information regarding each substantial United States owner, or (3) the foreign financial institution or non-financial foreign entity otherwise qualifies for an exemption from these rules. If the payee is a foreign financial institution and is subject to the diligence and reporting requirements in (1) above, it must enter into an agreement with the U.S. Department of the effective dateTreasury requiring, among other things, that it undertake to identify accounts held by certain “specified United States persons” or “United States-owned foreign entities” (each as defined in the Code), annually report certain information about such accounts, and withhold 30% on certain payments to non-compliant foreign financial institutions and certain other account holders. Non-U.S. holders typically will be required to furnish certifications (generally on the applicable IRS Form W-8) or other documentation to provide the information required by FATCA or to establish compliance with or an exemption from withholding under FATCA. FATCA withholding may apply where payments are made through a non-U.S. intermediary that is not FATCA compliant, even where the non-U.S. holder satisfies the holder’s own FATCA obligations. Foreign financial institutions located in jurisdictions that have an intergovernmental agreement with the United States governing FATCA may be subject to different rules.
Withholding under FATCA also potentially applies to payments of gross proceeds from the sale or other disposition of our common stock. Proposed regulations, however, would eliminate withholding under FATCA on such payments, and the U.S. Treasury Department has indicated that taxpayers may rely on this registration statement. Such sharesaspect of the proposed regulations until final regulations are deemed outstanding and beneficially owned by such person onlyissued. Because we may not know the extent to which a distribution is a dividend for U.S. federal income tax purposes at the time it is made, for purposes of computing histhese withholding rules we or her percentage ownership, but not for purposesthe applicable withholding agent may treat the entire distribution as a dividend. Prospective investors should consult their own tax advisors regarding the potential application of computing the percentage ownership for any other person.these withholding provisions.
As of August 15, 2022, there were issued and outstanding 9,312,991 shares of common stock, 336,805 shares of common stock issuable upon conversion of 1,212.5 shares of Series A convertible preferred stock, warrants to purchase up to 336,805 common stock issuable to the Series A convertible preferred stockholders upon completion of this initial public offering, 147,500 shares of common stock and warrants to purchase 147,500 shares of common stock issuable upon conversion of convertible notes, which conversion will occur immediately following completion of this initial public officer, and warrants to purchase 550,000 shares of common stock issuable upon exercise of such warrants.
| 49 |
| Names and addresses | Number of shares of common stock beneficially owned (#) | Percentage of shares of common stock beneficially owned before offering (%) | Number of shares of common stock beneficially owned after the Offering | Percentage of shares of common stock beneficially owned after offering (%) (1) | ||||||||||||
| Directors and Named Executive Officers: | ||||||||||||||||
| Anatoly Dritschilo, M.D.(2) | 4,427,979 | 47.5 | 4,427,979 | 33.3 | ||||||||||||
| Milton Brown, M.D., Ph.D.(3) | 1,073,826 | 11.5 | 1,073,826 | 7.6 | ||||||||||||
| Mira Jung, Ph.D.(4) | 1,071,716 | 11.5 | 1,071,716 | 7.5 | ||||||||||||
| Michael Vander Hoek(5) | 6,095 | - | 6,095 | - | ||||||||||||
| (5) | 10,380 | - | 10,380 | - | ||||||||||||
| Tyvin A. Rich, M.D. (5) | 3,843 | - | 3,843 | - | ||||||||||||
| Steve Richards(6) | 2,702 | - | 2,702 | - | ||||||||||||
| Joshua Schafer(6) | 2,702 | - | 2,702 | - | ||||||||||||
| Chris Senanayake(6) | 3,843 | - | 3,843 | - | ||||||||||||
| William H. Adkins(6)(7) | 280,480 | 3.0 | 280,480 | 2.1 | ||||||||||||
| All directors and officers as a group (eleven persons) | 6,957,384 | 73.5 | 6,957,384 | 50.5 | ||||||||||||
| Other 5% beneficial owners: | ||||||||||||||||
| Amir F. Heshmatpour(8) | 1,569,518 | 16.9 | 1,569,581 | 11.1 | ||||||||||||
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
Related Party Transactions
Unless described below, during the last two fiscal years, there were no transactions or series of similar transactions to which we were a party or will be a party, in which:
On January 25, 2018, Shuttle entered into a loan from Joy Dritschilo, the wife of our Chief Executive Officer, Anatoly Dritschilo, in the amount of $300,000 (the “January 2018 Loan”). The January 2018 Loan bears an interest rate of 7.5% per annum. The loan plus accrued interest was payable in full on January 25, 2019. On January 25, 2019, the Company amended the terms to extend the maturity date from January 25, 2019 to October 25, 2019.
On April 4, 2018, Shuttle entered into a loan from Mrs. Dritschilo in the amount of $50,000 (the “April 2018 Loan”). The April 2018 Loan bears an interest rate of 7.5% per annum. The loan plus accrued interest were payable in full on September 4, 2018. On October 31, 2018, the Company amended the terms to extend the maturity date of the April 2018 Loan from September 4, 2018 to April 4, 2019. On April 4, 2019, the Company amended the terms to extend the maturity date from April 4, 2019 to October 25, 2019.
On April 5, 2018, our predecessor in interest, Shuttle Pharma Acquisition Corp. Inc. (“Acquisition Corp.”), issued 3,600,000 shares to its founders, AFH Holding & Advisory, LLC and its affiliates (together, “AFH”). Such shares were issued at par value. AFH has also served as an advisor and consultant to the Company, and its owner, Amir Heshmatpour, has also served as a board member to our Company, a position he relinquished in advance of our commencement of the IPO process.
On May 31, 2018, the Company entered into a loan with our Chief Executive Officer in the amount of $25,000 (the “May 2018 Loan”). The May 2018 Loan bears interest at the rate of 7.5% per annum. The loan plus accrued interest were payable in full on July 15, 2018. On October 31, 2018, the Company amended the terms to extend the maturity date from July 15, 2018 to November 30, 2019.
On June 29, 2018, the Company entered into a loan with our Chief Executive Officer in the amount of $25,000. The loan bears an interest rate of 7.5% per annum. The loan plus accrued interest were payable in full on August 15, 2018. On December 6, 2018, the Company amended the terms to extend the maturity date from August 15, 2018 to February 15, 2019. On February 19, 2019, the Company paid off this note in full. The interest expense incurred on this loan was $1,223 for the year ended December 31, 2019.
On June 24, 2019, the Company entered into a loan from Mrs. Dritschilo in the amount of $70,000. The loan bears an interest rate of 7.5% per annum. The loans plus accrued interest are payable in full on June 23, 2020. This loan has since been satisfied in full.
In the fall of 2018 through to June 2019, we paid a total of $500,000 in cash to pay for a deposit on Acquisition Corp. in order to facilitate the process of taking the Company public.
On July 15, 2019, the Company issued 639,161 RSUs to our then consultant, AFH, to satisfy certain compensation owed to the consultant in relation to certain advisory services provided during 2018 and 2019. Such shares were issued pursuant to the Company’s 2018 Equity Incentive Plan.
On August 24, 2019, the Company entered into a loan with our Chief Executive Officer in the amount of $70,000. The loan bears interest at the rate of 7.5% per annum. The loan plus accrued interest is due and payable in full on August 24, 2020. This loan has since been satisfied in full.
On September 23, 2019, the Company entered into a loan with our Chief Executive Officer in the amount of $100,000 (the “September 2019 Loan”). The September 2019 Loan bear interest at the rate of 7.5% per annum and the loan plus accrued interest.
On December 1, 2020, the Company consolidated the January 2018 Loan and the April 2018 Loan into a single loan between Mrs. Dritschilo and the Company (the “2018 Consolidated Loan”) such that, with accrued interest, the 2018 Consolidated Loan had a principal balance of $424,005.65, bears interest at a rate of 7.5% per annum, and has a maturity date of December 31, 2021. The 2018 Consolidated Loan was extended until June 30, 2022, pursuant to an amendment to the 2018 Consolidated Loan agreement dated January 24, 2022. On July 29, 2022, the Company and Mrs. Dritschilo entered into an amendment to the 2018 Consolidated Loan, pursuant to which repayment was extended through June 30, 2023.
On December 1, 2020, the Company consolidated the May 2018 Loan and the September 2019 Loan with our Chief Executive Officer (the “2019 Consolidated Loan”), such that, with accrued interest, the 2019 Consolidated Loan had a principal balance of $138,448.20, bears interest at the rate of 7.5% per annum, and has a maturity date of December 31, 2021. The 2019 Consolidated Loan was extended until June 30, 2022, pursuant to an amendment to the 2019 Consolidated Loan agreement dated January 24, 2022. On July 29, 2022, the Company and our Chief Executive Officer entered into an amendment to the 2019 Consolidated Loan, pursuant to which repayment was extended through June 30, 2023.
On June 21, 2021, the Company entered into a loan agreement with Mrs. Dritschilo in the amount of $120,000 (principal), bearing interest at the rate of 7.5% per annum, with a single balloon payment due at maturity on June 21, 2022 (the “June 2021 Loan Agreement”). On July 29, 2022, the Company and Mrs. Dritschilo entered into an amendment to the June 2021 Loan Agreement, pursuant to which repayment was extended through June 30, 2023.
On September 22, 2021, Mrs. Dritschilo, who is one of our major shareholders, transferred 210,000 shares (105,000 shares post-split) of common stock of the Company in a private transaction to Steven Bayern, who had also been engaged by the Company to perform certain consulting services for the Company. Such shares, which represent approximately three percent of her total share ownership, were sold at par value pursuant to an exemption from registration under Section 4(a)(7) of the Securities Act. As a result of the transfer, the Company recognized $420,000 in non-cash stock compensation in legal and professional fees.
On August 1, 2022, in conjunction with our private placement of $125,000 of units consisting of 10% notes and warrants to purchase common stock, which were sold to three accredited investors in total, Mrs. Dritschilo purchased a $50,000 note and received warrants to purchase 20,000 shares of common stock at $2.50 per share. The notes and warrants were sold pursuant to an exemption from registration pursuant to Rule 506(b) of Regulation D of the Securities Act.
Review, Approval and Ratification of Related Party Transactions
All related party transactions are subject to the review, approval, or ratification of our board of directors or an appropriate committee thereof.
Capital Stock
Our authorized capital stock consists of 100,000,000 shares of common stock, par value $0.00001 per share, and 20,000,000 shares of preferred stock, par value $0.00001 per share.
Common Stock
As of the date of this prospectus, we had 9,312,99116,894,893 shares of common stock issued and outstanding. In addition, 500,000 shares of common stock will be issuable upon exercise of certain warrants at the effectiveness of this offering, and 147,500 shares will be issuable upon conversion of notes upon effectiveness of this offering. The shares of common stock and preferred stock presently outstanding are, and the shares of common stock in this offering, when issued and paid for as contemplated in this prospectus, will be, fully paid and non-assessable. Each holder of common stock is entitled to one vote for each share owned on all matters voted upon by shareholders, and a majority vote is required for all actions to be taken by shareholders. In the event we liquidate, dissolve or wind-up our operations, the holders of the common stock are entitled to share equally and ratably in our assets, if any, remaining after the payment of all our debts and liabilities and the liquidation preference of any shares of preferred stock that may then be outstanding. The common stock has no preemptive rights, no cumulative voting rights, and no redemption, sinking fund, or conversion provisions.
Holders of common stock are entitled to receive dividends, if and when declared by the board of directors, out of funds legally available for such purpose, subject to the dividend and liquidation rights of any preferred stock that may then be outstanding.
Warrants
General
The following is a summary of the material terms and provisions of the warrants that are being offered hereby. This summary is subject to and qualified in its entirety by the form of warrant, which has been filed as an exhibit to the registration statement of which this prospectus is a part. Prospective investors should carefully review the terms and provisions of the form of warrant for a complete description of the terms and conditions of the Warrants.
Duration and Exercise Price
Each Unit offered in this offering consists of one share of common stock and a warrant to purchase one share of common stock (each a “warrant”). Each whole warrant shall be exercisable into one share of common stock at an exercise price equal to $0.01 per share. The warrants will be immediately exercisable and will be exercisable for a five-year period after the date of issuance. It is the intention of the underwriters that the warrants will be immediately exercised on behalf of the holders of the units concurrently with completion of this offering. The exercise price and numbers of shares of common stock issuable upon exercise are subject to appropriate adjustment in the event of stock dividends, stock splits, reorganizations or similar events affecting our shares of common stock. The warrants will not trade.
Exercisability
The warrants will be exercisable, at the option of each holder, in whole or in part, by delivering to us a duly executed exercise notice accompanied by payment in full for the number of our shares of common stock purchased upon such exercise. A holder (together with its affiliates) may not exercise any portion of such holder’s warrants to the extent that the holder would own more than 4.99% of our outstanding shares of common stock immediately after exercise.
Transferability
A warrant may be transferred at the option of the holder upon surrender of the warrant to us together with the appropriate instruments of transfer.
Fractional Shares
No fractional shares of common stock will be issued upon the exercise of the warrants. Rather, the number of shares of common stock to be issued will, at our election, either be rounded up to the nearest whole number or we will pay a cash adjustment in respect of such final fraction in an amount equal to such fraction multiplied by the exercise price.
Trading Market
There is no established trading market for any of the warrants. Without an active trading market, the liquidity of the warrants will be limited.
Rights as a Shareholder
Except as otherwise provided in the warrants or by virtue of the holders’ ownership of shares of common stock, the holders of warrants do not have the rights or privileges of holders of our shares of common stock, including any voting rights, until such warrant holders exercise their warrants.
Waivers and Amendments
No term of the warrants may be amended or waived without the written consent of the holder of such warrant.
Exclusive Forum
We have agreed that any action, proceeding or claim against us arising out of or relating in any way to the warrants will be brought and enforced in the courts of the State of New York sitting in the City and County of New York or the United States District Court for the Southern District of New York, and we irrevocably submit to such jurisdiction, which jurisdiction will be the exclusive forum for any such action, proceeding or claim. See “Risk Factors”. This exclusive forum provision shall not apply to suits brought to enforce a duty or liability created by the Exchange Act, any other claim for which the federal courts have exclusive jurisdiction or any complaint asserting a cause of action arising under the Securities Act against us or any of our directors, officers, other employees or agents. Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder.
Preferred Stock
Our board of directors has the authority, without further action by the shareholders, to issue shares of preferred stock in one or more series and to fix the rights, preferences and the number of shares constituting any series or the designation of such series. While our Certificate of Incorporation and Bylaws, each as amended to date, do not contain any provisions that may delay, defer or prevent a change in control, the issuance of preferred stock may have the effect of delaying or preventing a change in control or make removal of our management more difficult. At present, our board of directors has authorized the issuance of up to 10,000 shares of Series A preferred stock, of which 1,212.5 shares arewere issued and outstanding assubsequently converted into common stock following completion of the date of this prospectus.our IPO.
Series A Convertible Preferred Stock
Our board of directors has designated and authorized the issuance of up to 10,000 shares of Series A Convertible Preferred Stock, par value $0.00001 per share (the “Series A Convertible Preferred Stock”), of which there are presentlywere 1,212.5 shares outstanding.issued, which were subsequently converted into common stock following completion of our IPO. A total of $1,212,500 was raised during 2018 and 2019 from the sale of our Series A Convertible Preferred Stock. The Series A Convertible Preferred Stock hashad a stated value of $1,000 per share, iswas entitled to receive a dividend at the rate of 8.5% per annum, which dividend iswas cumulative and will be payable at our option in shares of common stock or cash upon the date of conversion or redemption, as so determined by the Company. The Series A Convertible Preferred Stock, will bepursuant to its terms, were automatically convertible upon the earlier of (a) the closing of the sale of shares of common stock to the public at an offering price of at least $5.00 per share in a firm commitment underwritten public offering pursuant to an effective registration statement under the Securities Act, resulting in gross proceeds to us (before underwriter’s discounts, commissions and expenses) of an amount of at least $10 million (a “Qualified IPO”), or (b) listing of the common stock on the NYSE or Nasdaq (a “Qualified Listing”). All shares of Series A Convertible Preferred Stock will bewere then convertible at either (i) 90% of the gross public offering price per share of the Qualified IPO (before deducting underwriter’s discounts, commissions and expenses) or (ii) in the case of (b) above, $5.00 per share. Until such time asFollowing consummation of our IPO, the Series A Convertible Preferred Stock has beenwas converted into oura total of 336,810 shares of common stock, plus an additional 100,517 shares of common stock paid to the Series A Convertible Preferred stockholders have no voting rights other than to rights to vote on matters concerning the Series A Convertible Preferred Stock.Shareholders as dividends payable.
Series A Warrants
In conjunction with our sale of Series A Convertible Preferred Stock, our board of directors authorized the issuance of warrants to purchase up to approximately 269,444336,810 shares of common stock (the “Series A Warrants”), to the holders of Series A Preferred Stock. The Series A Warrants were issued following consummation of our IPO, are issuableexercisable for a period of three years following issuance, and have an exercise price of $4.00 per share.
| 50 |
January 2023 Note and Warrant Offering
On January 11, 2023, we entered into a securities purchase agreement (the “Purchase Agreement”) with an institutional investor (the “Investor”) and consummated the sale to such Investor of a senior secured convertible note (the “Convertible Note”) with an initial principal amount of $4,300,000, bearing interest at 5% per annum, and a warrant (the “Warrant”) to purchase 1,018,079 shares of our common stock at a fixed exercise price of $2.35 per share.
The Convertible Note was sold with an original issue discount of $300,000. As such, the Investor paid for the Convertible Note by delivering $4,000,000 in cash consideration to the Company. Boustead Securities LLC (“Boustead”) served as the sole placement agent for the private placement (the “Private Placement”) of the Convertible Note and Warrant. Boustead received a placement agent fee of $320,000 at the closing of the Private Placement, representing 7.0% of the gross cash proceeds at the closing plus 1% in non-accountable expenses. In addition, Boustead received a placement agent warrant to purchase 71,266 shares of common stock, representing 7.0% of the warrant shares issued in the Private Placement. After deducting the placement agent fee and our estimated expenses associated with the Private Placement, our estimated net cash proceeds at the closing were approximately $3,493,423.
Purchase Agreement
The Purchase Agreement contains certain representations and warranties, covenants and indemnities customary for similar transactions. Under the Purchase Agreement, we also agreed to the following additional covenants:
| ● | So long as the Convertible Note remains outstanding, we will not effect or enter into an agreement to effect any variable rate transaction other than a bona fide at-the-market offering or equity line of credit. |
| ● | While we obtained majority stockholder consent approving the offering and the issuance of common stock upon conversion of the Convertible Note and exercise of the Warrant, as is required under the rules of the Nasdaq Stock Market LLC (“Nasdaq”), we were required to file an information statement on Schedule 14C (the “Information Statement”) informing all stockholders of the action and mail such Information Statement to all stockholders within 70 days of closing. |
In addition, we granted the Investor participation rights in future equity and equity-linked offerings of securities, subject to certain limited exceptions, during the two years after the later of (a) the closing or (b) the date the Convertible Note no longer remains outstanding, in an amount of up to 30% of the securities being sold in such offerings.
Senior Secured Convertible Note
General
The Convertible Note was issued to the Investor on January 11, 2023 and matures on March 11, 2025 (subject to extension in certain circumstances, including bankruptcy and outstanding events of default).
Amortization
Starting on the earlier of (x) the date this Registration Statement is declared effective by the SEC and (y) February 28, 2023, then on the first trading day of the month for each month thereafter, and on the maturity date (each, an “Installment Date”), subject to certain exceptions and unless deferred as described below, the Company is required to make monthly amortization payments equal to 1/26th of the unrestricted principal (and related unrestricted original issue discount) and interest of the Convertible Note payable (the “Installment Amount”), which must be satisfied in cash at a redemption price equal to 105% of such Installment Amount (each, an “Installment Redemption”).
Notwithstanding the foregoing, the noteholder may, at its sole option, elect to defer any Installment Amount until a subsequent Installment Date selected by the noteholder, provided that any such deferred amounts shall not continue to accrue interest unless the deferral is at the request of the Company.
Interest
The Convertible Note bears interest at a rate of 5% per annum and, upon any conversion or redemption, shall include a make-whole of interest from such date of determination through the maturity date. After the occurrence and during the continuance of an Event of Default (as defined in the Convertible Note), the Convertible Note will accrue interest at the rate of 15% per annum. See “Events of Default” below.
| 51 |
Conversion; Alternate Conversion upon Event of Default
The Convertible Note is convertible, at the option of the noteholder, into shares of our common stock at the lower of (i) $2.35 per share, (ii) 90% of the three lowest VWAPs in the 15 trading days prior to the payment date or (iii) 90% of the VWAP on the trading day prior to the payment date. The conversion price is subject to full ratchet antidilution protection upon any subsequent transaction at a fixed price lower than the conversion price then in effect and standard adjustments in the event of any stock split, stock dividend, stock combination, recapitalization or other similar transaction. If we enter into any agreement to issue (or issue) any variable rate securities, other than a bona fide at-the-market offering or equity line of credit, the noteholder has the additional right to substitute such variable price (or formula) for the conversion price.
If an Event of Default has occurred under the Convertible Note, the noteholder may elect to alternatively convert the Convertible Note at a redemption premium of 115% of the conversion amount.
Conversion Limitation and Exchange Cap
The noteholder will not have the right to convert any portion of the Convertible Note, to the extent that, after giving effect to such conversion, the noteholder (together with certain related parties) would beneficially own in excess of 4.99% of the shares of our common stock outstanding immediately after giving effect to such conversion. The noteholder may from time to time increase this limit to 9.99%, provided that any such increase will not be effective until the 61st day after delivery of a notice to us of such increase.
In addition, until such time as we complete the Information Statement mailing to our stockholders notifying them that we have obtained the approval of a majority of our stockholders as required by Nasdaq, we are prohibited from issuing any shares of common stock upon conversion of the Convertible Note or otherwise pursuant to the terms of the Convertible Note, if the issuance of such shares of common stock would exceed 19.99% of our outstanding shares of common stock as of January 11, 2023 or otherwise exceed the aggregate number of shares of common stock which we may issue without breaching our obligations under the rules and regulations of Nasdaq.
Events of Default
The Convertible Note includes certain customary and other Events of Default, including, among other things, failure to maintain an effective registration statement with capacity to issue an offering amount equal to at least 300% of principal, the breach of certain financial covenants described below and maintaining our Nasdaq listing.
In connection with an Event of Default, the noteholder may require us to redeem in cash any or all of the Convertible Note. The redemption price will equal 115% of the outstanding principal of the Convertible Note to be redeemed, and accrued and unpaid interest and unpaid late charges thereon, or an amount equal to market value of the shares of our common stock underlying the Convertible Note, as determined in accordance with the Convertible Note, if greater.
Change of Control
In connection with a Change of Control (as defined in the Convertible Note), a noteholder may require us to redeem all or any portion of the Convertible Note. The redemption price per share will equal the greatest of (i) 115% of the outstanding principal of the Convertible Note to be redeemed, and accrued and unpaid interest and unpaid late charges thereon, (ii) 115% of the market value of the shares of our common stock underlying the Convertible Note, as determined in accordance with the terms of the Convertible Note, and (iii) 115% of the aggregate cash consideration that would have been payable in respect of the shares of our common stock underlying the Convertible Note, as determined in accordance with the terms of the Convertible Note.
| 52 |
Subsequent Placement Optional Redemption
At any time after the earlier of the closingdate a noteholder becomes aware of any placement by us of equity or equity-linked securities or the date of consummation of such a Qualified IPO or Qualified Listing,placement, subject to certain limited exceptions, the noteholder will have an exercisethe right to have us redeem a portion of each Convertible Note not in excess of 30% of the net proceeds from such placement at a redemption price per shareof 100% of the portion of the Convertible Note subject to redemption.
Covenants
We will be subject to certain customary affirmative and negative covenants regarding the incurrence of certain indebtedness, the existence of liens, the repayment of indebtedness, the payment of cash in respect of dividends, distributions or redemptions, and the transfer of assets, among other matters.
Control Account
In addition to customary affirmative and negative covenants, we are required to maintain the proceeds from the issuance and sale of the Convertible Note on deposit in a deposit account subject to a deposit account control agreement. The Company cannot make any withdrawals from such account or otherwise use the funds therein for any purpose other than for payment of its obligations to the noteholder. As such, as the Convertible Note is converted or repaid, the Company is permitted to withdraw $0.90 from such account for each $1.00 of the outstanding principal value of the Convertible Note that is converted into shares of common stock or is otherwise repaid.
Company Optional Redemption Rights
We may redeem the Convertible Notes at a price equal to 107.5% of the IPO’s initial per share offering priceoutstanding principal of the Convertible Notes (or, if greater, the market value of the shares underlying the Convertible Notes) to be redeemed and shall be exercisable at any time on or before expiration on the three-year anniversary following their issuance.
accrued and unpaid interest and unpaid late charges thereon.
Notes and Warrants
Warrant
On December 28, 2021,In addition to the Convertible Note, we completedissued a private placement offering (the “Note and Warrant Offering”) pursuant to which we sold to two accredited investors (the “Note and Warrant holders”)exercisable for four (4) years for the purchase of an aggregate of $500,000 of our 10% promissory notes, which are due upon completion of this Offering, and warrantsup to purchase $500,0001,018,079 shares of common stock (the “Warrant Shares”), at an exercise price of $1.00$2.35 per share. Such issuance was completed pursuantThe number of Warrant Shares and exercise price are each subject to an exemption from registrationadjustment as provided under Rule 506(b) of Regulation Dthe terms of the Securities Act. Boustead Securities, LLC actedWarrant. If, at the time of exercise of the Warrant, there is no effective registration statement registering, or no current prospectus available for, the issuance of the Warrant Shares to the Investor, and the registration statement is not subject to SEC review and the Company has otherwise affirmatively failed to maintain such registration statement’s effectiveness, then the Warrant may also be exercised, in whole or in part, by means of a “cashless exercise.” The Warrant may not be exercised if, after giving effect to the exercise the Investor, would beneficially own in excess of 4.99% of the number of shares of Common Stock outstanding immediately after giving effect to the issuance of the Warrant Shares. At the Investor’s option, the ownership limitation blocker may be raised or lowered to any other percentage not in excess of 9.99%, as placement agentapplicable, except that any raise will only be effective upon 61-days’ prior notice to the Company.
If the Company issues or sells, or the Company publicly announces the issuance or sale of, any shares of Common Stock, or convertible securities or options issuable or exchangeable into Common Stock (a “New Issuance”), under which such Common Stock is sold for a consideration per share less than the exercise price then in effect, the exercise price of the Warrant will be adjusted to the New Issuance price in accordance with the formulas provided in the Note andWarrant. Any such adjustment will not apply with respect to the issuance of Excluded Securities (as defined in the Warrant). Upon any adjustment to the exercise price, the number of Warrant offering but waived its compensationShares that may be purchased upon exercise of the Warrant will be increased or decreased proportionately, so that after such adjustment the aggregate exercise price payable for the adjusted number of Warrant Shares will be the same as the aggregate exercise price in relationeffect immediately prior to such offering. Upon completionadjustment. In addition, if the Company enters into a Fundamental Transaction (as defined in the Warrants) at any time that a Warrant is outstanding, then, upon any subsequent exercise of this Offering, the notes will be repaid andWarrant, the warrants will be exercised simultaneously, such that the Note and Warrant holdersInvestor will have the right to receive, for each Warrant Share that would have been repaid in full through their receiptissuable upon such exercise immediately prior to the occurrence of 500,000such Fundamental Transaction, the number of shares of Common Stock of the successor or acquiring corporation or of the Company, if it is the surviving corporation, and any additional consideration receivable as a result of such Fundamental Transaction by a holder of the number of shares of Common Stock for which the Warrant is exercisable immediately prior to such Fundamental Transaction, provided, further, that if holders of common stock all of which shares will then be registered for resaleare not offered or paid any consideration in the resale offering accompanying this Offering.
On August 1, 2022, we completed a private placement offering (the “2022 Note and Warrant Offering”) pursuant to which we sold to three accredited investors (the “Note and Warrant holders”) an aggregate of $125,000 of our 10% promissory notes, which are due upon completion of this Offering, and warrants to purchase 50,000 sharessuch Fundamental Transaction, such holder of common stock at an exercise price of $2.50 per share. Such issuance was completed pursuantwill be deemed to an exemption from registration under Rule 506(b) of Regulation Dhave received common stock of the Securities Act. Boustead Securities, LLC actedsuccessor entity (which entity may be the Company following such Fundamental Transaction) in such Fundamental Transaction.
Registration Rights Agreement
Pursuant to a Registration Rights Agreement, dated as placement agentof January 11, 2023, between the Investor and us, we have granted certain registration rights to the noteholder and warrantholder. The Registration Rights Agreement requires us to have the registration statement of which this prospectus is a part declared effective by the 60th day following the date of the Registration Rights Agreement (of March 11, 2023), or the 90th day (or April 10, 2023) following the date of the Registration Rights Agreement in the Note and Warrant offering but deferred its cash compensation in relationevent such registration statement is subject to such offering.SEC review. It also grants the Investor customary “piggyback” registration rights.
Additional Information
The foregoing is only a summary of the material terms of the Purchase Agreement, the Convertible Note, the Warrant, the Registration Rights Agreement and the other ancillary transaction documents (collectively, the “Transaction Documents”), and does not purport to be a complete description of the rights and obligations of the parties thereunder.
| 53 |
6% Convertible Notes
On February 8, 2022 and March 11, 2022, we completed private placement offerings pursuant to which we sold to several accredited investors an aggregate of $365,000 and $225,000, respectively, of our 6% convertible notes due three years from the date of issuance (the “Convertible Notes”), pursuant to an exemption from registration under Rule 506(b) of Regulation D of the Securities Act. Boustead Securities, LLC acted as placement agent in the Convertible Note and Warrant offering and received commissions and non-accountable reimbursements of 10% of the gross proceeds received and warrants to purchase 10% of the common stock.
Upon completion of this Offering, the Convertible Notes will automatically convert into units, comparable to those issued in this Offering, at a conversion price equal to 50% of the per share price in our initial public offering.
In connection with this Convertible Note offering, each purchaser of Convertible Notes entered into an investor rights and lock-up agreement (the “Investor Rights and Lock-up Agreement”) pursuant to which the Company has agreed, upon conversion of the Convertible Notes, to register the shares of common stock underlying the Convertible Notes and the Convertible Note holders have each agreed not to sell their shares of common stock for a period of 180 days following completion of this offering.
The Convertible Notes were registered in the resale offering prospectus accompanying our IPO prospectus and converted into shares of common stock upon completion of the IPO.
Registration RightsAugust 2022 Note and Warrant Offering
We are party toOn August 1, 2022, we completed a registration rights agreement with the holders of Series A Convertible Preferred Stockprivate placement offering (the “2022 Note and Warrant Offering”) pursuant to which we sold to three accredited investors (the “Note and Warrant holders”) an aggregate of $125,000 of our 10% promissory notes, which are obligateddue upon completion of this Offering, and warrants to register thepurchase 50,000 shares of common stock, underlying the Series A Convertible Preferred Stock and Warrants. Theat an exercise price of $2.50 per share. Such issuance was completed pursuant to an exemption from registration under Rule 506(b) of these sharesRegulation D of common stock would enable the holders to sell their shares without restriction under the Securities Act whenAct. Boustead Securities, LLC acted as placement agent in the registration statement is declared effective. We will pay forNote and Warrant offering but deferred its cash compensation in relation to such offering. At the registration expenses, other than underwriting discounts and commissions, of any shares subject to registration rights.
Generally, in an underwritten offering, the managing underwriter, if any, has the right, subject to specific conditions, to limit the number of shares such holders may include. As a resulttime of the underwriter objecting to the registrationIPO, 30,000 of the common stock underlying the Series A Convertible Preferred shareswarrants sold in this offering, we will be required2022 Note and Warrant Offering were registered for resale pursuant to file a separate resale registration statement registering the shares, with an obligation of having such registration statement declared effective within 180 days of the initial filing of such resale registration statement.
prospectus and were subsequently exercised.
Anti-Takeover Effects of Provisions of our Certificate of Incorporation, our Bylaws and Delaware Law
Some provisions of Delaware law, our certificate of incorporation and our bylaws contain provisions that could make the following transactions more difficult: acquisition of us by means of a tender offer; acquisition of us by means of a proxy contest or otherwise; or removal of our incumbent officers and directors. It is possible that these provisions could make it more difficult to accomplish or could deter transactions that stockholders may otherwise consider to be in their best interest or in our best interests, including transactions that might result in a premium over the market price for our shares.
These provisions, summarized below, are expected to discourage coercive takeover practices and inadequate takeover bids. These provisions are also designed to encourage persons seeking to acquire control of us to first negotiate with our Board of Directors. We believe that the benefits of increased protection of our potential ability to negotiate with the proponent of a non-friendly or unsolicited proposal to acquire or restructure us outweigh the disadvantages of discouraging these proposals because negotiation of these proposals could result in an improvement of their terms.
| 54 |
Delaware Anti-Takeover Statute
In general, Delaware corporations are subject to Section 203 of the DGCL, which prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years after the date that such stockholder became an interested stockholder, with the following exceptions:
| ● | before such date, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested holder; | |
| ● | upon completion of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction began, excluding for purposes of determining the voting stock outstanding (but not the outstanding voting stock owned by the interested stockholder) those shares owned (i) by persons who are directors and also officers and (ii) employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; |
| ● | on or after such date, the business combination is approved by the board of directors and authorized at an annual or special meeting of the stockholders, and not by written consent, by the affirmative vote of at least 662/3% of the outstanding voting stock that is not owned by the interested stockholder; or | |
| ● | the corporation does not have a class of voting stock that is: (i) listed on a national securities exchange; or (ii) held of record by more than 2,000 stockholders, unless any of the foregoing results from action taken, directly or indirectly, by an interested stockholder or from a transaction in which a person becomes an interested stockholder |
In general, Section 203 defines business combination to include the following:
| ● | any merger or consolidation involving the corporation and the interested stockholder; | |
| ● | any sale, transfer, pledge, or other disposition of 10% or more of the assets of the corporation involving the interested stockholder; | |
| ● | subject to certain exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; | |
| ● | any transaction involving the corporation that has the effect of increasing the proportionate share of the stock or any class or series of the corporation beneficially owned by the interested stockholder; or | |
| ● | the receipt by the interested stockholder of the benefit of any loss, advances, guarantees, pledges or other financial benefits by or through the corporation. |
Section 203 defines interested stockholder as an entity or person beneficially owning 15% or more of the outstanding voting stock of the corporation or any entity or person affiliated with or controlling or controlled by such entity or person.
While we are currently not subject to the restrictions contained in Section 203, we will become subject to these restrictions if our common stock is listed on a national securities exchange or we have more than 2,000 stockholders of record of our common stock.
| 55 |
Nasdaq ListingPotential Effects of Authorized but Unissued Stock
We have applied to list our shares of common stock and preferred stock available for future issuance without stockholder approval. We may utilize these additional shares for a variety of corporate purposes, including future public offerings to raise additional capital, to facilitate corporate acquisitions or payment as a dividend on the capital stock.
The Nasdaq Capital Market (“Nasdaq”)existence of unissued and unreserved common stock and preferred stock may enable our board of directors to issue shares to persons friendly to current management or to issue preferred stock with terms that could render more difficult or discourage a third-party attempt to obtain control of us by means of a merger, tender offer, proxy contest or otherwise, thereby protecting the continuity of our management. In addition, the board of directors has the discretion to determine designations, rights, preferences, privileges and restrictions, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences of each series of preferred stock, all to the fullest extent permissible under the symbol “SHPH”. There can be no assurance thatDelaware General Corporation Law and subject to any limitations set forth in our applicationcertificate of incorporation. The purpose of authorizing the board of directors to issue preferred stock and to determine the rights and preferences applicable to such preferred stock is to eliminate delays associated with a stockholder vote on specific issuances. The issuance of preferred stock, while providing desirable flexibility in connection with possible financings, acquisitions and other corporate purposes, could have the effect of making it more difficult for listing will be approved. We do not intenda third-party to apply for listingacquire, or could discourage a third-party from acquiring, a majority of the warrants included in the Units on Nasdaq or on any other national securities exchange or trading system. Without an active trading market, the liquidity of the warrants will be limited.
our outstanding voting stock.
Transfer Agent
The transfer agent and registrar of our common stock is VStock Transfer, LLC, of Woodmere, New York. Our transfer agent’s telephone number is (212) 828-8436.
| 56 |
SHARES ELIGIBLE FOR FUTURE SALEPLAN OF DISTRIBUTION
Prior to this offering, there has been no public market for our common stock, and we cannot assure you that a liquid trading market for our common stock will develop or be sustained after this offering.
Commencing 90 days after the date of this prospectus, subject to lock-up agreements of 365 days for directors, officer and affiliates and 180 days for holders of 1% or more of our comment stock (excluding the non-affiliate stockholders whose shares are being registered in the resale offering), the 9,312,991 shares of our common stock outstanding as of the date of this prospectus will be eligible for sale in the public market from time to time thereafter pursuant to Rule 144 under the Securities Act, and in some cases, subject to the volume and other restrictions of Rule 144. In addition, we have 1,212.5 share of Series A convertible preferred common stock, which shares will be convertible into approximately 336,805 shares of common stock upon the earlier of completion of the offering or the Company’s listing on a national securities exchange, as well as warrants to purchase up to 336,805 shares of common stock, which will be issuable upon closing of this offering. We also have 3,000,000 shares of common stock reserved for issuance under our 2018 Equity Incentive Plan, of which 384,167 shares have been granted to date. The sale of a significant number of shares of our common stock in the public market or the perception that such sales may occur could significantly reduce the market price of our common stock.
Rule 144
In general, under Rule 144 under the Securities Act, beginning 90 days afterPromptly following the effective date of the registration statement of which this prospectus form is a part, a person (or persons whosewe will distribute the Subscription Rights, Rights Certificates and copies of this prospectus to the holders of our common and warrants on the Record Date. Subscription Rights holders who wish to exercise their Subscription Rights and purchase Units must complete the Subscription Rights Certificate and return it with payment for the shares are aggregated) whoto the Subscription Agent at the following address:
| By hand or overnight courier: |
VStock Transfer, LLC
18 Lafayette Place
Woodmere, New York 11598
If you have any questions, you should contact our Information Agent for the Rights Offering: Vstock Transfer, LLC at (212) 828-8436, by email at action@vstocktransfer.com, or by mail at 18 Lafayette Place, Woodmere, New York, 11598.
Other than as described in this prospectus, we do not know of any existing agreements between any stockholder, broker, dealer, underwriter or agent relating to the sale or distribution of the underlying securities.
Boustead Securities LLC (“Boustead”) will act as placement agent for the Rights Offering. In such capacity, the placement agent will provide marketing assistance and financial advice (including determining the Subscription Price and the structure of the Rights Offering) to us in connection with this offering and will solicit the exercise of Subscription Rights and participation in the Over-Subscription Privilege. The placement agent will provide us with updated investor feedback and recommendations on pricing and structure through to the end of the subscription period. The placement agent is not deemed to have been an affiliate of ours atunderwriting other placing any time during the three months preceding a sale, and who has beneficially owned restricted securities within the meaning of Rule 144 for at least six months (including any period of consecutive ownership of preceding non-affiliated holders) would be entitled to sell those shares, subject only to the availability of current public information about us. A non-affiliated person who has beneficially owned restricted securities within the meaning of Rule 144 for at least one year would be entitled to sell those shares without regard to the provisions of Rule 144.
A person (or persons whose shares are aggregated) who is deemed to be an affiliate of ours and who has beneficially owned restricted securities within the meaning of Rule 144 for at least six months would be entitled to sell within any three-month period a number of shares that does not exceed the greater of one percent of the then outstandingSubscription Rights or the shares of our common stock, Warrants or the average weekly trading volume of our common stock reported through Nasdaq orEquity Interests being issued in this offering and does not make any recommendation with respect to such other market on which our shares of common stock are listed for trading during the four calendar weeks preceding such sale. Such sales are also subject to certain manner of sale provisions, notice requirements and the availability of current public information about us.
2018 Equity Incentive Plan Form S-8 Registration Statement
We intend to fileSubscription Rights (including with the SEC a registration statement on Form S-8 under the Securities Act covering the shares of common stock that we may issue upon exercise of awards which may be granted under our 2018 Equity Incentive Plan. Such registration statement is expected to be filed and become effective as soon as practicable after the effectiveness of this registration statement. Accordingly, shares registered under such registration statement will be available for sale in the open market following its effective date, subject to Rule 144 volume and manner of sale limitations, if applicable.
Selling Stockholder Resale Prospectus
As described in the Explanatory Noterespect to the registration statement of which this prospectus forms a part, the registration statement also contains the Resale Prospectus to be used in connection with the potential resale by certain selling stockholders of our common stock. These shares of common stock have been registered to permit public resaleexercise or expiration of such Subscription Rights), shares and the selling stockholders may offer the shares for resale from time to time pursuant to the Resale Prospectus. The selling stockholders may also sell, transfer or otherwise dispose of all or a portion of their shares in transactions exempt from the registration requirements of the Securities Act or pursuant to another effective registration statement covering those shares. Any shares sold by the selling stockholders until our common stock is listed or quoted on an established public trading market will take place at $4.00 per share, which is the low end of the public offering price range of the shares of common stock we are selling in our initial public offering. Thereafter, any sales will occur at prevailing market prices or in privately negotiated prices.
In connection with this offering,Rights Offering, we will enter into an underwriting agreement with Boustead Securities, LLC to serve as lead book-running manager of the offering and as representative of the several underwriters (if any) named below. Subject to the terms and conditions of the underwriting agreement, each underwriter will severally agree to purchase from us the number of shares of common stock set forth opposite its name below, at the public offering price, less the underwriting discount set forth on the cover page of this prospectus.
*Each unit consists of one share of common stock and one warrant to purchase one share of common stock, exercisable at $0.01 per share.
Subject to the terms and conditions set forth in the underwriting agreement, the underwriters have agreed to purchase the 1,225,888 units offered by this prospectus (other than those covered by the over-allotment option described below, if any are purchased).
The underwriters are offering the shares of common stock subject to various conditions and may reject all or part of any order. The underwriter has advised us that the underwriter proposes initially to offer the shares of common stock to the public at the public offering price set forth on the cover page of this prospectus and to dealers at a price less a concession not in excess of $0.56 per share of common stock to brokers and dealers. After the shares of common stock are released for sale to the public, the underwriter may change the offering price, the concession and other selling terms at various times.
The table below provides information regarding the amount of the discounts and commissions to be paid to the underwriters by us, before estimated expenses.
Over-Allotment Option
We have granted the underwriters an over-allotment option. This option, which is exercisable for up to 45 days after the date of this prospectus, permits the underwriter to purchase a maximum of 183,883 additional units from us to cover over-allotments. Such additional units consist of 183,883 additional shares of common stock and warrants to purchase 183,883 shares of common stock. If the underwriter exercises all or part of this option, it will purchase units covered by the option at the initial $8.125 public offering price per unit that appears on the cover page of this prospectus, less the underwriting discount. If this option is exercised in full, the total proceeds to us will be $11,454,389 before deduction of underwriting discounts and estimated offering expenses. The underwriters have agreed that, to the extent the over-allotment option is exercised, they will purchase a number of additional shares proportionate to the underwriter’s initial amount reflected in the foregoing table.
Underwriting Discount
The following table summarizes the compensation and estimated expenses we will pay. The information assumes either no exercise or full exercise by the underwriter of the over-allotment option.
| Per Share | Total | |||||||||||||||
| Without Over-allotment | With Over-allotment | Without Over-allotment | With Over-allotment | |||||||||||||
| Underwriting discount payable by us | $ | 0.56 | $ | 0.56 | $ | 697,200 | $ | 801,780 | ||||||||
| Non-accountable and accountable expenses payable by us | $ | 0.068 | $ | 0.068 | $ | 99,600 | $ | 114,500 | ||||||||
We have agreed to pay a non-accountable expense allowancefees to the underwriterBoustead as placement agent an aggregate cash fee equal to 1.0%8.0% of the gross proceeds (including proceeds subjectreceived by us directly from exercises of the Subscription Rights. We advanced $40,000 (the “Advance”) to the over-allotment option, ifBoustead as an advance against such non-accountable expense allowance upon their engagement as placement agent and to the extent it is exercised) for expenses in connection with this offering. We have also agreed to pay the underwriter an accountable expense reimbursementplacement agent a commitment fee of up to $255,000 for out-of-pocket expenses incurred by it with respect to this offering. We estimate that our total expenses of the offering, excluding the underwriting discounts and commissions,$112,500, which will be approximately $499,936.payable upon the filing of this registration statement (the “Filing Date”) and may be paid in cash or shares of SHPH common stock, with the price of such shares to be determined based the VWAP of SHPH common stock over the five trading days prior to the Filing Date.
We have also agreed to issue to the underwriter a warrant to purchase a number of shares of common stock equal to an aggregate of 7% of the aggregate number of the units sold in this offering. The warrant will be exercisable on a cashless basis at an exercise price equal to 150% of the offering price of the shares sold in this offering. The warrants are exercisable commencing six months after the date of effectiveness of the registration statement of which this prospectus forms a part, and will be exercisable for a period of five years from the effective date of the registration statement of which this prospectus forms a part. We have agreed to a one-time demand registration of the shares of common stock underlying the underwriter’s warrants for a period of five years from the effective date of the registration statement. The underwriter’s warrants also provide for immediate “piggyback” registration rights with respect to the underlying shares of common stock during the three-year period commencing from the effective date of the registration statement related to this offering. The warrants are not redeemable by us. The warrants and the shares of common stock issuable upon exercise of the warrants have been included on the registration statement of which this prospectus forms a part. Pursuant to applicable FINRA rules, and in particular Rule 5110, the warrants (and underlying shares) issued to the underwriter may not be sold, transferred, assigned, pledged, or hypothecated, or the subject of any hedging, short sale, derivative, put or call transaction that would result in the effective disposition of the securities by any person for a period of 180 days after the effective date of the registration statement related to this offering; provided, however, the warrants (and underlying shares) may be transferred to the underwriter’s officers, partners, registered persons or affiliates as long as the warrants (and underlying shares) remain subject to the lock-up.
We have agreed to indemnify the underwriterplacement agent and their respective affiliates against certain liabilities including liabilitiesarising under the Securities Act of 1933, as amended.
Pursuant to the underwriting agreement, we will provide the underwriter the right of first refusal for two years from the date of commencement of sales ofAct. The placement agent participation in this offering is subject to act as financial advisor or to act as joint financial advisor on at least equal economic terms on any public or private financing (debt or equity), merger, business combination, recapitalization or sale of some or all of the equity or assets of our company.
We have agreed to a six-month “lock-up” from the closing of this offering, during which, without the prior written consent of the underwriter, we will not issue, sell or register with the SEC (other than on Form S-8 or on any successor form) with respect to any of our equity securities (or any securities convertible into, exercisable for or exchangeable for any of our equity securities), except for (i) the issuance of the shares of common stock offered pursuant to this prospectus; and (ii) the issuance of shares of common stock pursuant to our existing stock option or bonus plan as describedcustomary conditions contained in the registration statement of which this prospectus forms a part.
Our executive officers, directorsplacement agent agreement between Boustead and certain of our significant stockholders have also agreedthe Company. The placement agent and their affiliates may provide to a 12-month “lock-up” period, and any stockholders who own in excess of 1% of the outstanding shares of our common stock will be subjectus from time to a six-month lock-up period, during which time without the prior written consent of the underwriter, they will not, directly or indirectly, (i) offer, pledge, assign, encumber, announce the intention to sell, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant to purchase, or otherwise transfer or dispose of, any shares of common stock or any securities convertible into or exercisable or exchangeable for common stock, owned either of record or beneficially (as defined in the Securities Exchange Act of 1934, as amended (the “Exchange Act”) by any signatory of the lock-up agreement on the date of the prospectus or thereafter acquired; (ii) enter into any swap or other agreement that transfers, in whole or in part, any of the economic consequences of ownership of the common stock or any securities convertible into or exercisable or exchangeable for common stock, whether any such transaction described in clauses (i) or (ii) above is to be settled by delivery of common stock or such other securities, in cash or otherwise, or publicly announce an intention to do any of the foregoing; and (iii) make any demand for or exercise any right with respect to, the registration of any shares of common stock or any security convertible into or exercisable or exchangeable for common stock. The foregoing will not apply to (i) common stock to be transferred as a gift or gifts (provided, that (a) any donee will execute and deliver to the underwriter, not later than one business day prior to such transfer, a lock-up agreement to the underwriter and (b) if the lock-up signatory is required to file a report under Section 16(a) of the Exchange Act, reporting a reduction in beneficial ownership of shares of common stock or beneficially owned shares or any securities convertible into or exercisable or exchangeable for common stock or beneficially owned shares during the six-month “lock-up,” the lock-up signatory will include a statement in such report to the effect that such transfer is being made as a gift), and (ii) the sale of the shares of common stock to be sold pursuant to this prospectus.
Rules of the SEC may limit the ability of the underwriter to bid for or purchase shares before the distribution of the shares is completed. However, the underwriter may engagefuture in the following activitiesordinary course of their business certain financial advisory, investment banking and other services in accordance with the rules:
Similar to other purchase transactions, the underwriter’s purchases to cover the syndicate short sales or to stabilize the market price of our common stock may have the effect of raising or maintaining the market price of our common stock or preventing or mitigating a decline in the market price of our common stock. As a result, the price of our common stock may be higher than the price that might otherwise exist in the open market. The imposition of a penalty bid might also have an effect on the priceterm of the common stock if it discourages resales of our shares of common stock.placement agent agreement.
Neither we nor the underwriters make any representation or prediction as to the effect that the transactions described above may have on the price of ourOur common stock. These transactions may occurstock is listed on the Nasdaq Capital Market, or otherwise. If such transactions are commenced, they may be discontinued without notice at any time.Nasdaq, under the symbol “SHPH.”
In December 2021, we completed a $500,000 unit offering private placement offering to two investors, which units consisted of a total of $500,000 of 10% promissory notes and warrants to purchase 500,000 shares of common stock, exercisable at a purchase price of $1.00 per share, which private placement was completed pursuant to an exemption from registration under Rule 506(b) of the Securities Act. Boustead acted as placement agent in the private placement, however, Boustead agreed to waive its placement agent fees and expenses.
In February and March 2022, we completed a $365,000 and a $225,000 convertible note offering, respectively, to certain accredited investors, which notes are convertible into shares of units, consisting of one share of common stock and a warrant to purchase one share of common stock, (the “Conversion Units”) at a conversion price of $4.00 per unit upon effectiveness of this offering. The convertible note offering was completed pursuant to an exemption from registration under Rule 506(b) of the Securities Act. Boustead acted as placement agent in each of the February and March 2022 private placements and received $36,500 and $22,250 cash compensation, respectively, and five-year warrants to purchase shares of common stock equal to 10% of the number of Conversion Shares, which warrants will be exercisable at the conversion price.
In August 2022, we completed a $125,000 unit offering private placement offering to three investors, which units consisted of a total of $125,000 of 10% promissory notes and warrants to purchase 50,000 shares of common stock, exercisable at a purchase price of $2.50 per share, which private placement was completed pursuant to an exemption from registration under Rule 506(b) of the Securities Act. Boustead acted as placement agent in the private placement and received a placement agent warrant to purchase 5,000 shares of our common stock, exercisable at $2.50 per share, however, Boustead agreed to waive its cash fees and expenses related to the private placement.
The warrants received in connection with the February, March 2022 and August 2022 private placements (the “Private Placement Warrants”) will not be exercisable or convertible more than five years from the commencement of this public offering. Pursuant to applicable FINRA rules and, in particular, Rule 5110(e)(1), the Private Placement Warrants may not be sold, transferred, assigned, pledged or hypothecated, or be the subject of any hedging, short sale, derivative, put or call transaction that would result in the effective economic disposition of the securities for a period of 180 days beginning on the date of commencement of sales of this public offering; provided, however, the Private Placement Warrants may be transferred to the underwriter’s officers, partners, registered persons or affiliates as long as the warrants remain subject to the lock-up restriction above.
A prospectus in electronic format may be delivered to potential investors by one or more of the underwriters participating in this offering. The prospectus in electronic format will be identical to the paper version of such preliminary prospectus. Other than the prospectus in electronic format, the information on any underwriter’s website and any information contained in any other website maintained by an underwriters are not part of the prospectus or the registration statement of which this prospectus forms a part.
Transfer Agent and Registrar
The transfer agent and registrar for shares of our common stock and preferred stock is VStock Transfer, LLC, Woodmere, New York. Our transfer agent’s telephone number is (212) 828-8436.
| 57 |
Notice to Non-US Investors
Canada
The securities may be sold in Canada only to purchasers purchasing, or deemed to be purchasing, as principal who are “accredited investors,” as defined in National Instrument 45-106 Prospectus Exemptions or subsection 73.3(1) of the Securities Act (Ontario), and are “permitted clients”, as defined in National Instrument 31-103 Registration Requirements, Exemptions and Ongoing Registrant Obligations. Any resale of the securities must be made in accordance with an exemption from, or in a transaction not subject to, the prospectus requirements of applicable securities laws. Securities legislation in certain provinces or territories of Canada may provide a purchaser with remedies for rescission or damages if this prospectus (including any amendment thereto) contains a misrepresentation, provided that the remedies for rescission or damages are exercised by the purchaser within the time limit prescribed by the securities legislation of the purchaser’s province or territory. The purchaser should refer to any applicable provisions of the securities legislation of the purchaser’s province or territory for particulars of these rights or consult with a legal advisor. Pursuant to section 3A.3 of National Instrument 33-105 Underwriting Conflicts (NI 33-105), the underwriters are not required to comply with the disclosure requirements of NI 33-105 regarding underwriter conflicts of interest in connection with this offering.
European Economic Area
In relation to each Member State of the European Economic Area which has implemented the Prospectus Directive, each, a Relevant Member State, with effect from and including the date on which the European Union Prospectus Directive, or the EU Prospectus Directive, was implemented in that Relevant Member State, or the Relevant Implementation Date, no offer of securities may be made to the public in that Relevant Member State other than:
provided that no such offer of securities shall require the Company or any underwriter to publish a prospectus pursuant to Article 3 of the Prospectus Directive and each person who initially acquires any securities or to whom any offer is made will be deemed to have represented, acknowledged and agreed to and with each of the underwriters and the Company that it is a “qualified investor” within the meaning of the law in that Relevant Member State implementing Article 2(1)(e) of the Prospectus Directive.
In the case of any securities being offered to a financial intermediary as that term is used in Article 3(2) of the Prospectus Directive, each such financial intermediary will be deemed to have represented, acknowledged and agreed that the securities acquired by it in the offer have not been acquired on a non-discretionary basis on behalf of, nor have they been acquired with a view to their offer or resale to, persons in circumstances which may give rise to an offer of any securities to the public other than their offer or resale in a Relevant Member State to qualified investors as so defined or in circumstances in which the prior consent of the representatives has been obtained to each such proposed offer or resale.
For the purposes of this provision, the expression an “offer of securities to the public” in relation to any securities in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and the securities to be offered so as to enable an investor to decide to purchase or subscribe for the securities, as the same may be varied in that Member State by any measure implementing the EU Prospectus Directive in that Member State. The expression “EU Prospectus Directive” means Directive 2003/71/EC (and any amendments thereto, including the 2010 PD Amending Directive, to the extent implemented in the Relevant Member State) and includes any relevant implementing measure in each Relevant Member State, and the expression “2010 PD Amending Directive” means Directive 2010/73/EU.
United Kingdom
In the United Kingdom, this document is being distributed only to, and is directed only at, and any offer subsequently made may only be directed at persons who are “qualified investors” (as defined in the Prospectus Directive) (i) who have professional experience in matters relating to investments falling within Article 19(5) of the Financial Services and Markets Act 2000 (Financial Promotion) Order 2005, as amended, or the Order, and/or (ii) who are high net worth companies (or persons to whom it may otherwise be lawfully communicated) falling within Article 49(2)(a) to (d) of the Order (all such persons together being referred to as “relevant persons”) or otherwise in circumstances which have not resulted and will not result in an offer to the public of the securities in the United Kingdom.
Any person in the United Kingdom who is not a relevant person should not act or rely on the information included in this document or use it as basis for taking any action. In the United Kingdom, any investment or investment activity that this document relates to may be made or taken exclusively by relevant persons.
The validity of the common stock being offered hereby has been passed upon by Michelman & Robinson, LLP, California and New York. The underwriter has been represented in connection with this offering by Olshan Frome Wolosky LLP, New York, New York.
The consolidated financial statements of Shuttle Pharmaceuticals Holdings, Inc. appearing in this prospectus(the “Company”) as of December 31, 2023 and related registration statement for the yearsfiscal year then ended, December 31, 2021 and 2020 have been audited by BF Borgers CPA PC, anFORVIS LLP, independent registered public accounting firm, as set forth in their report thereon, included in the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2023 (the “Annual Report on Form 10-K”), and are includedincorporated herein by reference. Such consolidated financial statements have been incorporated herein by reference in reliance upon such report given on the authority of BF Borgers CPA PCsuch firm as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed a registration statement on Form S-1 under the Securities Act with the SEC with respect to the shares The report of our common stock offered through this prospectus. This prospectus is filed as a part of that registration statement, but does not contain all of the information contained in the registration statement and exhibits. Statements made in the registration statement are summaries of the material terms of the referenced contracts, agreements or documents of the company. We refer you to our registration statement and each exhibit attached to it for a more detailed description of matters involving the company. You may inspect the registration statement, exhibits and schedules filed with the SEC at the SEC’s principal office in Washington, D.C. Copies of all or any part of the registration statement may be obtained from the Public Reference Section of the SEC, 100 F Street, N.E. Washington, D.C. 20549. Please Call the Commission at 1-800-SEC-0330 for further information on the operation of the public reference rooms. The SEC also maintains a web site at http://www.sec.gov thatFORVIS LLP contains reports, proxy Statements and informationan explanatory paragraph regarding registrants that files electronically with the SEC. Our registration statement and the referenced exhibits can also be found on this site.
DISCLOSURE OF SEC POSITION ON INDEMNIFICATION
FOR SECURITIES ACT LIABILITIES
In accordance with the provisions in our Certificate of Incorporation, as amended, we will indemnify an officer, director, or former officer or director, to the full extent permitted by law.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers and controlling persons pursuant to the foregoing provisions, or otherwise, we have been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable.
SHUTTLE PHARMACEUTICALS HOLDINGS, INC.
Consolidated Financial Statements
Contents
Shuttle Pharmaceuticals Holdings, Inc.
Condensed Consolidated Balance Sheets
(Unaudited)
| June 30, | December 31, | |||||||
| 2022 | 2021 | |||||||
| Assets | ||||||||
| Current assets | ||||||||
| Cash | $ | 50,437 | $ | 504,749 | ||||
| Prepaid expenses | 13,515 | 4,866 | ||||||
| Total current assets | 63,952 | 509,615 | ||||||
| Property and equipment, net | 15,665 | 18,564 | ||||||
| Other assets | 6,480 | 6,480 | ||||||
| Operating lease right-of-use asset | 87,383 | 116,982 | ||||||
| Total Assets | $ | 173,480 | $ | 651,641 | ||||
| Liabilities and Stockholders’ Deficit | ||||||||
| Current Liabilities | ||||||||
| Accounts payable and accrued expenses | $ | 515,786 | $ | 828,313 | ||||
| Accrued interest payable | 37,828 | 552 | ||||||
| Accrued interest payable - related parties | 72,330 | 46,947 | ||||||
| Dividends Payable | 382,595 | 331,059 | ||||||
| Notes payable to related parties | 685,473 | 685,473 | ||||||
| Notes payable | 575,624 | 91,021 | ||||||
| Paycheck Protection Program note payable | - | 73,007 | ||||||
| Derivative liability | 112,797 | 94,025 | ||||||
| Operating lease liability current portion | 71,518 | 66,934 | ||||||
| Total Current Liabilities | 2,453,951 | 2,217,331 | ||||||
| Operating lease liability non-current | 25,602 | 62,442 | ||||||
| Total Liabilities | 2,479,553 | 2,279,773 | ||||||
| Stockholders’ Deficit | ||||||||
| Series A convertible preferred stock, $0.00001 par value; $1,000 per share liquidation value or aggregate of $1,212,500; 20,000,000 shares authorized; 1,213 shares issued and outstanding at June 30, 2022 and December 31, 2021 | - | - | ||||||
| Common stock, $0.00001 par value; 100,000,000 shares authorized; 9,312,991 and 9,312,152 shares issued and outstanding at June 30, 2022 and December 31, 2021, respectively | 93 | 93 | ||||||
| Additional paid in capital | 4,819,916 | 4,150,867 | ||||||
| Common stock to be issued | - | 16,340 | ||||||
| Accumulated deficit | (7,126,082 | ) | (5,795,432 | ) | ||||
| Total Stockholders’ Deficit | (2,306,073 | ) | (1,628,132 | ) | ||||
| Total Liabilities and Stockholders’ Deficit | $ | 173,480 | $ | 651,641 | ||||
The accompanying footnotes are an integral part of these unaudited condensed consolidated financial statements.
Shuttle Pharmaceuticals Holdings, Inc.
Condensed Consolidated Statements of Operations
(Unaudited)
| Three Months Ended | Six Months Ended | |||||||||||||||
| June 30, | June 30, | |||||||||||||||
| 2022 | 2021 | 2022 | 2021 | |||||||||||||
| Revenue | $ | - | $ | - | $ | - | $ | - | ||||||||
| Operating expenses | ||||||||||||||||
| Research and development, net of contract expense reimbursements | 83,868 | 286,730 | 379,783 | 392,726 | ||||||||||||
| General and administrative | 9,078 | 7,213 | 22,847 | 13,461 | ||||||||||||
| Legal and professional | 260,680 | 42,308 | 589,392 | 106,150 | ||||||||||||
| Total operating expenses | 353,626 | 336,251 | 992,022 | 512,337 | ||||||||||||
| Net loss from operations | (353,626 | ) | (336,251 | ) | (992,022 | ) | (512,337 | ) | ||||||||
| Other income (expense) | ||||||||||||||||
| Interest expense - related parties | (14,836 | ) | (10,547 | ) | (25,383 | ) | (21,094 | ) | ||||||||
| Interest expense | (170,391 | ) | (120 | ) | (315,944 | ) | (350 | ) | ||||||||
| Change in fair value of derivative liability | (58,422 | ) | 117 | (18,772 | ) | 57,656 | ||||||||||
| Gain on forgiveness of Paycheck Protection Program note payable | - | - | 73,007 | - | ||||||||||||
| Total other income (expense) | (243,649 | ) | (10,550 | ) | (287,092 | ) | 36,212 | |||||||||
| Income (loss) before income taxes | (597,275 | ) | (346,801 | ) | (1,279,114 | ) | (476,125 | ) | ||||||||
| Provision for income taxes | - | - | - | - | ||||||||||||
| Net income (loss) | $ | (597,275 | ) | $ | (346,801 | ) | $ | (1,279,114 | ) | $ | (476,125 | ) | ||||
| Dividend on Series A Preferred Stock | (25,768 | ) | (25,768 | ) | (51,536 | ) | (51,536 | ) | ||||||||
| Net loss attributable to common stockholders | $ | (623,043 | ) | $ | (372,569 | ) | $ | (1,330,650 | ) | $ | (527,661 | ) | ||||
| Weighted average common shares outstanding - basic and diluted | 9,312,991 | 9,291,526 | 9,312,583 | 9,291,526 | ||||||||||||
| Net loss per shares - basic and diluted | $ | (0.06 | ) | $ | (0.04 | ) | $ | (0.14 | ) | $ | (0.05 | ) | ||||
The accompanying footnotes are an integral part of these unaudited condensed consolidated financial statements.
Shuttle Pharmaceuticals Holdings, Inc.
Condensed Consolidated Statements of Changes in Stockholders’ Deficit
(Unaudited)
Series A Preferred Stock | Common Stock | Additional Paid in | Common Stock to be | Accumulated | Total Stockholders’ | |||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Issued | Deficit | Deficit | |||||||||||||||||||||||||
| Balance - December 31, 2021 | 1,213 | $ | - | 9,312,152 | $ | 93 | $ | 4,150,867 | $ | 16,340 | $ | (5,795,432 | ) | $ | (1,628,132 | ) | ||||||||||||||||
| Warrants issued for financing costs | - | - | - | - | 319,643 | - | - | 319,643 | ||||||||||||||||||||||||
| Common stock issued for conversion of convertible debt and accrued interest | - | - | 839 | - | 16,340 | (16,340 | ) | - | - | |||||||||||||||||||||||
| Common stock issued for restricted stock units | - | - | - | - | 166,533 | - | - | 166,533 | ||||||||||||||||||||||||
| Dividends on Series A preferred stock | - | - | - | - | - | - | (25,768 | ) | (25,768 | ) | ||||||||||||||||||||||
| Net loss (Unaudited) | - | - | - | - | - | - | (681,839 | ) | (681,839 | ) | ||||||||||||||||||||||
| Balance - March 31, 2022 | 1,213 | $ | - | 9,312,991 | $ | 93 | $ | 4,653,383 | $ | - | $ | (6,503,039 | ) | $ | (1,849,563 | ) | ||||||||||||||||
| Warrants issued for financing costs | - | - | - | - | - | - | - | - | ||||||||||||||||||||||||
| Common stock issued for restricted stock units | - | - | - | - | 166,533 | - | - | 166,533 | ||||||||||||||||||||||||
| Dividends on Series A preferred stock | - | - | - | - | - | - | (25,768 | ) | (25,768 | ) | ||||||||||||||||||||||
| Net loss (Unaudited) | - | - | - | - | - | (597,275 | ) | (597,275 | ) | |||||||||||||||||||||||
| Balance - June 30, 2022 (Unaudited) | 1,213 | $ | - | 9,312,991 | $ | 93 | $ | 4,819,916 | $ | - | $ | (7,126,082 | ) | $ | (2,306,073 | ) | ||||||||||||||||
The accompanying footnotes are an integral part of these unaudited condensed consolidated financial statements.
Shuttle Pharmaceuticals Holdings, Inc.
Consolidated Statements of Changes in Stockholders’ Deficit
(Unaudited)
Series A Convertible Preferred Stock | Common Stock | Additional Paid-in | Common Stock to be | Accumulated | Total Stockholders’ | |||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Issued | Deficit | Deficit | |||||||||||||||||||||||||
| Balance, December 31, 2020 | 1,213 | $ | - | 9,291,526 | $ | 93 | $ | 2,833,507 | $ | 16,340 | $ | (4,540,236 | ) | $ | (1,690,296 | ) | ||||||||||||||||
| Warrants issued for financing costs | - | - | - | - | - | - | - | - | ||||||||||||||||||||||||
| Common stock issued for restricted stock units | - | - | - | - | 122,517 | - | - | 122,517 | ||||||||||||||||||||||||
| Dividends on Series A preferred stock | - | - | - | - | - | - | (25,768 | ) | (25,768 | ) | ||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (129,324 | ) | (129,324 | ) | ||||||||||||||||||||||
| Balance, March 31, 2021 | 1,213 | $ | - | 9,291,526 | $ | 93 | $ | 2,956,024 | $ | 16,340 | $ | (4,695,328 | ) | $ | (1,722,871 | ) | ||||||||||||||||
| Warrants issued for financing costs | - | - | - | - | - | - | - | - | ||||||||||||||||||||||||
| Common stock issued for restricted stock units | - | - | 7,738 | - | 122,517 | - | - | 122,517 | ||||||||||||||||||||||||
| Dividends on Series A preferred stock | - | - | - | - | - | - | (25,768 | ) | (25,768 | ) | ||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (346,801 | ) | (346,801 | ) | ||||||||||||||||||||||
| Balance, June 30, 2021 | 1,213 | $ | - | 9,299,264 | $ | 93 | $ | 3,078,541 | $ | 16,340 | $ | (5,067,897 | ) | $ | (1,972,923 | ) | ||||||||||||||||
The accompanying footnotes are an integral part of these unaudited condensed consolidated financial statements.
Shuttle Pharmaceuticals Holdings, Inc.
Consolidated Statements of Cash Flows
(Unaudited)
| Six Months Ended | ||||||||
| June 30, | ||||||||
| 2022 | 2021 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net income (loss) | $ | (1,279,114 | ) | $ | (476,125 | ) | ||
| Adjustments to reconcile net income (loss) to net cash used in operating activities: | ||||||||
| Depreciation | 2,899 | 2,700 | ||||||
| Change in fair value of derivative liability | 18,772 | (57,656 | ) | |||||
| Amortization of right-of-use asset | 29,599 | 26,582 | ||||||
| Amortization of debt discount | 278,531 | - | ||||||
| Gain on forgiveness of Paycheck Protection Program note payable | (73,007 | ) | - | |||||
| Stock-based compensation | 333,066 | 245,034 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Contracts receivable | - | 211,455 | ||||||
| Prepaid expenses | (8,649 | ) | 7,450 | |||||
| Accounts payable and accrued expenses | (312,527 | ) | (100,241 | ) | ||||
| Accrued interest payable | 37,276 | 210 | ||||||
| Accrued interest payable - related parties | 25,383 | 21,094 | ||||||
| Operating lease liability | (32,256 | ) | (28,144 | ) | ||||
| Net Cash used in Operating Activities | (980,027 | ) | (147,641 | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||
| Purchase of property and equipment | - | - | ||||||
| Net Cash used in Investing Activities | - | - | ||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Proceeds from PPP note payable | - | 73,007 | ||||||
| Proceeds from notes payable | 525,715 | - | ||||||
| Net Cash provided by Financing Activities | 525,715 | 73,007 | ||||||
| Net change in cash | (454,312 | ) | (74,634 | ) | ||||
| Cash, beginning of period | 504,749 | 117,153 | ||||||
| Cash, end of period | $ | 50,437 | $ | 42,519 | ||||
| Cash paid for: | ||||||||
| Interest | $ | - | $ | - | ||||
| Income taxes | $ | - | $ | - | ||||
| Supplemental non-cash financing activities: | ||||||||
| Shares issued for conversion of accrued interest | $ | 16,340 | $ | - | ||||
The accompanying footnotes are an integral part of these unaudited condensed consolidated financial statements.
Shuttle Pharmaceuticals Holdings, Inc.
Notes to Condensed Consolidated Financial Statements
For the Six Months Ended June 30, 2022 and 2021
(Unaudited)
Note 1 – Organization and Going Concern
Organization and Line of Business
The Company was formed as Shuttle Pharmaceuticals, LLC, in the State of Maryland on December 18, 2012. On August 12, 2016, the Company filed articles of conversion with the state of Maryland to convert from an LLC to a corporation and the Company changed its name to Shuttle Pharmaceuticals, Inc. (“Shuttle”). In connection with the conversion the Company issued 45,000,000 shares of common stock in exchange for 100% of the outstanding membership interests prior to conversion. On June 4, 2018, Shuttle completed a reverse merger with Shuttle Pharmaceuticals Holdings, Inc. (then known as Shuttle Pharma Acquisition Corp.), a Delaware corporation (the “Company”), pursuant to which Shuttle, our operating entity, became a wholly owned subsidiary of the Company.
The Company’s primary purpose is to develop and commercialize unique drugs for the sensitization of cancers and protection of normal tissues, with the goal of improving outcomes for cancer patients receiving radiation therapy. Shuttle has deployed its proprietary technology to develop novel cancer immunotherapies which has produced a pipeline of selective HDAC inhibitors for cancer and immunotherapy applications. The Company’s HDAC platform is designed to target candidate molecules with potential roles in therapeutics beyond cancer, including autoimmune, inflammatory, metabolic, neurological and infectious diseases. The Company’s Ropidoxuridine product, which is used with radiation therapy to sensitize cancer cells, was funded by a Small Business Innovation Research (SBIR) contract provided by the National Cancer Institute (NCI), a unit of the National Institutes of Health (NIH). Ropidoxuridine has been further developed though the Company’s collaborations with the University of Virginia for use in combination with proton therapy to improve patient survival. The Company is working on developing products through NIH grants, including a product to predict late effects of radiation with metabolite biomarkers and develop prostate cancer cell lines in health disparities research.
The production and marketing of the Company’s products and its ongoing research and development activities will be subject to extensive regulation by numerous governmental authorities in the United States. Prior to marketing in the United States, any combination product developed by the Company must undergo rigorous preclinical (animal) and clinical (human) testing and an extensive regulatory approval process implemented by the Food and Drug Administration (“FDA”) under the Food, Drug and Cosmetic Act. There can be no assurance that the Company will not encounter problems in clinical trials that will cause the Company or the FDA to delay or suspend clinical trials.
The Company’s success will depend in part on its ability to obtain patents and product license rights, maintain trade secrets, and operate without infringing on the proprietary rights of others, both in the United States and other countries. There can be no assurance that patents issued to or licensed by the Company will not be challenged, invalidated or circumvented, or that the rights granted thereunder will provide proprietary protection or competitive advantages to the Company now or in the future.
Reverse Stock Split
Effective April 1, 2022, we effected a 2-for-1 reverse stock split of our issued and outstanding common stock (the “Reverse Stock Split”). All references to shares of our common stock in this report refers to the number of shares of common stock after giving effect to the Reverse Stock Split (unless otherwise indicated).
Going Concern
The accompanying financial statements have been prepared in conformity with generally accepted accounting principles, which contemplate continuation of the Company as a going concern. The Company’s only revenue source since inception has been government awarded contracts totaling $5,531,722, and the Company has incurred losses since inception, having accumulated a deficit of $7,126,082 as of June 30, 2022. The Company currently has limited liquidity and has not completed its efforts to establish a stabilized source of revenues sufficient to cover operating costs over an extended period. These factors, among others, raise substantial doubt about the Company’s ability to continue as a going concern.
The Company will need to raise capital to fund its operations. To address its financing requirements, the Company intends to seek financing through debt or equity financings with an aim to continue progress toward commercial viability of its products. The Company continues to submit Federal grant and contract applications which have historically been the primary source of revenue. The financial statements do not include any adjustments that might result from the outcome of the uncertainty of raising additional capital.
Note 2 – Summary of Significant Accounting Policies
Basis of Presentation
The financial statements and related disclosures have been prepared pursuant to the rules and regulations of the Securities and Exchange Commission (“SEC”). The financial statements have been prepared using the accrual basis ofB.F Borgers CPA PC, independent registered public accounting in accordance with Generally Accepted Accounting Principles of the United States (“GAAP”).
Basis of Consolidation
The financial statements have been prepared on a consolidated basis with those offirm, audited the Company’s wholly-owned subsidiary, Shuttle Pharmaceuticals, Inc. All intercompany transactions and balances have been eliminated.
Use of Estimates
The preparation of consolidated financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. The Company regularly evaluates estimates and assumptions. The Company bases its estimates and assumptions on current facts, historical experience, and various other factors that it believes to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities and the accrual of costs and expenses that are not readily apparent from other sources. The actual results experienced by the Company may differ materially and adversely from the Company’s estimates. To the extent there are material differences between the estimates and the actual results, future results of operations will be affected. Significant estimates in the accompanying financial statements include useful lives of property and equipment, valuation of derivatives, and the valuation allowance on deferred tax assets.
Property and Equipment
Property and equipment are stated at cost. Expenditures for maintenance and repairs are charged to earnings as incurred; additions, renewals and betterments are capitalized. When property and equipment are retired or otherwise disposed of, the related cost and accumulated depreciation are removed from the respective accounts, and any gain or loss is included in operations. Depreciation of property and equipment is provided using the straight-line method for substantially all assets with estimated lives as follows:
Research and Development Expenses
Research and development expenses are charged to expense as incurred. Research and development expenses include, but are not limited to, product development, clinical and regulatory expenses, payroll and other personnel expenses, materials, supplies, related subcontract expenses, and consulting costs. The expenses assigned to NIH SBIR sponsored research are related to: (1) “Topic 352: Cell-Based Models for Prostate Cancer Health Disparity Research – Moonshot Project” and (2) “Topic 345: Predictive Biomarkers of Prostate Cancer Patient Sensitivity for Radiation Late Effects.”
The research expenses are assigned to the research projects to demonstrate proof of principle in patients with prostate cancer that may support development and commercialization of biomarker products and to gather prostate cancer cell lines in African American men to serve as the product for use in health disparities research. Costs that are not covered by the SBIR contract for performing the Phase I contract to determine commercialization feasibility included partial salary support of personnel and a consultant to develop a commercialization plan. Costs that are not covered in the Phase II contract include business development and partial salary support.
Research expenses related to new drug discovery include partial support of personnel, space, supplies, and legal costs.
During fiscal year 2022, the Company completed two SBIR contracts from the NIH to support research projects with potential for commercialization. The SBIR contract awards are fixed payments made by the NIH in response to quarterly Shuttle invoices and provide non-dilutive funds that do not include a repayment obligation. Details on the three contracts follow:
1. Contract #HHSN261201600027C/75N91018C00016 supported “Topic 345: Predictive Biomarkers of Prostate Cancer Patient Sensitivity for Radiation Late Effects.” This $299,502 Phase I award includes funded research from 9/19/2016 through 9/18/2017 and was advanced to Phase II of the awards with funding of $1,903,095 with a fixed price contract period of 9/17/2018 through 9/16/2020 and subsequent no cost extensions through 9/15/2021 and then 3/15/2022 (Reference 75N91019C00031). The Company received quarterly payments of $211,455 for a total of $845,820 in 2020; and 2 quarterly payments related to Topic 345 for a total of $422,910 in 2021. On April 6, 2022, the Company submitted the final invoice for “Topic 345: Predictive Biomarkers of Prostate Cancer Patient Sensitivity for Radiation Late Effects,” for $211,455, following the completion of the Final Quarterly Progress Report to NIH covering the performance period of 9/16/2019-3/15/2022. The invoice was paid in full on April 27, 2022. In Phase II of the SBIR effort, the Company will license the metabolite signatures (intellectual property) from Georgetown University, manufacture 500 “kits,” test and validate the metabolic kit performance and develop a multi-institutional clinical trial to be implemented in the Phase III effort. This contract includes a subcontract to Georgetown University (“Georgetown”) for use of Mass Spectrometry core facilities to analyze clinical samples. The contract was extended to complete the milestones which were delayed due to the impact of COVID-19.
On December 6, 2019, the Company engaged Georgetown to perform the $795,248 subcontract of its Phase II contract #HHSN75N91019C00031. The Company agreed to reimburse Georgetown for its allowable costs not to exceed the ceiling amount of $795,248. Georgetown invoiced the Company for a total of $791,017.12 as of June 30, 2022, leaving a balance of $4,230.88. Depending on the resources it uses, Georgetown may or may not invoice for the total subcontract amount. In the event Georgetown does not invoice for the total allowable amount, the Company is not obligated to pay the ceiling amount. As of April 2022, cumulative payments of $791,017.12 were made to Georgetown, including an additional invoice for $282,643 which was received but not paid until the second quarter of 2022. All invoices have now been paid.
2. The Phase II contract #HHSN261201800016C supports the discovery work following a Phase I contract # HHSN261600038C “Topic 352 – SBIR Phase II Cell-based Models for Prostate Cancer Health Disparity Research” and was awarded to provide $1,484,350 to fund research from 9/17/2018 through 9/16/2020 and was extended without cost through 11/16/2021 due to delays caused by the impact of COVID-19. For the entire contract period, the Company invoiced and received a total of $1,411,883. The final draft report was filed with the NIH along with the final invoice for $10,000, which payment was made on December 3, 2021, and no additional payments are expected. The Phase II contract also includes a subcontract to Georgetown University for $742,002 to establish prostate cancer cell lines from African American patients undergoing prostate surgery for cancers.
On December 5, 2018, the Company engaged Georgetown University to perform the $742,002 subcontract of its Phase II contract #HHSN261201800016C. Depending on the resources it uses, Georgetown may or may not invoice for the total subcontract amount. In the event Georgetown does not invoice for the total allowable amount, the Company is not obligated to pay the ceiling amount. The Company has been invoiced by Georgetown and has paid Georgetown a total of $305,866.35 as of June 30, 2022.
The Company recognizes the amounts received from the contract at fair value when there is reasonable assurance that the contract amount will be received, and it is probable that all attaching conditions will be complied with. The Company recognizes the amounts received in accordance with the contract as a reduction of research and development expenses over the periods necessary to match the contract on a systematic basis to the costs that it is intended to compensate. The Company records reimbursements on the balance sheet as contract receivables upon meeting the criteria discussed above until cash is received. During the quarter ended June 30, 2022, the Company recorded a net deficit of $83,868 with the Company funding the NIH no-cost extension along with other R&D activities. The NIH made the final payment of $211,455 in April 2022 for Topic 345.
Regarding the accounting treatment for reimbursements, GAAP provides limited guidance on the accounting for government grants received by for-profit companies. We understand there is more than one acceptable alternative for the accounting treatment – a reduction of costs, a deferred credit to be amortized, revenue, or other income. Due to the terms of the contracts, we have entered into the Company concluded that the reimbursements were more akin to a reduction of costs rather than any of the other alternatives that would match the contract reimbursements on a systematic basis to the costs that the contract is intended to compensate.
Derivative Financial Instruments
The Company evaluates all of its agreements to determine if such instruments have derivatives or contain features that qualify as embedded derivatives. For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair value and is then re-valued at each reporting date, with changes in the fair value reported in the statements of operations. For stock-based derivative financial instruments, the Company uses a Binomial Simulation model to value the derivative instruments at inception and on subsequent valuation dates. The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is evaluated at the end of each reporting period. Derivative instrument liabilities are classified in the balance sheet as current or non-current based on whether or not net-cash settlement of the derivative instrument could be required within 12 months of the balance sheet date. As of June 30, 2022, the Company’s only derivative financial instrument was an embedded warrant feature associated with Series A Convertible Preferred Stock due to certain provisions that allow for a change in the warrant value based on fluctuations of the Company’s fair value of common stock at the date of issuance of the warrant based on certain contingent call features.
Fair Value of Financial Instruments
For certain of the Company’s financial instruments, including cash, accounts receivable, accounts payable, accrued liabilities and short-term debt, the carrying amounts approximate their fair values due to their short maturities.
FASB ASC Topic 820, Fair Value Measurements and Disclosures, requires disclosure of the fair value of financial instruments held by the Company. FASB ASC Topic 825, Financial Instruments, defines fair value, and establishes a three-level valuation hierarchy for disclosures of fair value measurement that enhances disclosure requirements for fair value measures. The carrying amounts reported in the consolidated balance sheets for receivables and current liabilities each qualify as financial instruments and are a reasonable estimate of their fair values because of the short period of time between the origination of such instruments and their expected realization and their current market rate of interest. The three levels of valuation hierarchy are defined as follows:
The Company analyzes all financial instruments with features of both liabilities and equity under FASB ASC Topic 480, Distinguishing Liabilities from Equity, and FASB ASC Topic 815, Derivatives and Hedging.
For certain financial instruments, the carrying amounts reported in the balance sheets for cash and current liabilities, including convertible notes payable, each qualify as a financial instrument, and are a reasonable estimate of their fair values because of the short period of time between the origination of such instruments and their expected realization and their current market rate of interest.
An established trading market for the Company’s common stock does not exist. The fair value of the shares was determined based on the then most recent price per share at which we sold preferred stock to unrelated parties in a private placement during the six months then ended.
During the year ended December 31, 2020, the Company utilized $25.22 (post-share exchange) per share as the fair value of its common stock for accounting purposes based on preferred share transactions with investors from August 2018 through December 2019, with no transactions occurring in 2020 and $5.00 in 2021, $4.00 through March 31, 2022 and $6.00 through June 30, 2022.
At June 30, 2022, the Company identified the following liabilities that are required to be presented on the balance sheet at fair value:
| June 30, 2022 | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Liabilities | ||||||||||||||||
| Derivative Liabilities | $ | - | $ | - | $ | 112,797 | $ | 112,797 | ||||||||
At December 31, 2021, the Company identified the following liabilities that are required to be presented on the balance sheet at fair value:
| December 31, 2021 | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Liabilities | ||||||||||||||||
| Derivative Liabilities | $ | - | $ | - | $ | 94,025 | $ | 94,025 | ||||||||
Revenue Recognition
Revenue from providing research and development is recognized under Topic 606 in a manner that reasonably reflects the delivery of its services to customers in return for expected consideration and includes the following elements:
To satisfy these five elements, the Company records revenue for research and development services on a quarterly basis as services are provided. Revenue received from National Institutes of Health contracts is received in accordance with Federal grants and contracts policies. Research and development expenses are posted against revenue and recorded on the statement of operations as “Research and development, net of contract expense reimbursements.”
Basic and Diluted Earnings Per Share
Basic earnings per share (“EPS”) is computed based on the weighted average number of shares of common stock outstanding during the period. Diluted EPS is computed based on the weighted average number of shares of common stock plus the effect of dilutive potential common shares outstanding during the period using the treasury stock method and as if converted method. Dilutive potential common shares include outstanding warrants and Series A preferred stock.
For the six months ended June 30, 2022 and year ended December 31, 2021, the following common stock equivalents were excluded from the computation of diluted net loss per share as the result of the computation was anti-dilutive.
| June 30, | December 31, | |||||||
| 2022 | 2021 | |||||||
| Series A preferred stock | 97,062 | 97,062 | ||||||
| Warrants | 48,531 | 48,531 | ||||||
| 145,593 | 145,593 | |||||||
Recent Accounting Pronouncements
In August 2020, the FASB issued ASU 2020-06, ASC Subtopic 470-20 “Debt—Debt with “Conversion and Other Options” and ASC subtopic 815-40 “Hedging—Contracts in Entity’s Own Equity.” The standard reduced the number of accounting models for convertible debt instruments and convertible preferred stock. Convertible instruments that continue to be subject to separation models are (1) those with embedded conversion features that are not clearly and closely related to the host contract, that meet the definition of a derivative, and that do not qualify for a scope exception from derivative accounting; and (2) convertible debt instruments issued with substantial premiums for which the premiums are recorded as paid-in capital. The amendments in this update are effective for fiscal years beginning after December 15, 2021, including interim periods within those fiscal years. Early adoption is permitted. The Company adopted this standard on January 1, 2021.
Management does not believe that any recently issued, but not yet effective, accounting standards could have a material effect on the accompanying financial statements. As new accounting pronouncements are issued, we will adopt those that are applicable under the circumstances.
Note 3 – Property and Equipment, Net
Property and equipment consisted of the following:
| June 30, | December 31, | |||||||
| 2022 | 2021 | |||||||
| Office Furniture and equipment | $ | 8,861 | $ | 8,861 | ||||
| Laboratory equipment | 118,605 | 118,605 | ||||||
| 127,466 | 127,466 | |||||||
| Less accumulated depreciation | (111,801 | ) | (108,902 | ) | ||||
| Property and equipment, net | $ | 15,665 | $ | 18,564 | ||||
Depreciation expense for the six months ended June 30, 2022 and 2021, were $2,899 and $2,700 respectively.
Note 4 – Operating Lease Right-of-use Asset and Operating Lease Liability
Operating lease right-of-use assets and liabilities are recognized at the present value of the future lease payments at the lease commencement date. The interest rate used to determine the present value is our incremental borrowing rate, estimated to be 10%, as the interest rate implicit in most of our leases is not readily determinable. Operating lease expense is recognized on a straight-line basis over the lease term. During the six months ended June 30 2022, and 2021, the Company recorded $35,088 and $35,088, respectively, as operating lease expense.
The Company currently has a lease agreement which allows for the use of a laboratory facility for a monthly payment of $6,107, which monthly lease payment increases by 3% every year. The laboratory lease commenced October 1, 2018, with the first payment due January 1, 2019, and expires on October 31, 2023. A security deposit of $6,480 is being held for the duration of the lease term.
In adopting ASC Topic 842, Leases (Topic 842), the Company has elected the ‘package of practical expedients,’ which permits the Company to avoid reassessing its prior conclusions about lease identification, lease classification and initial direct costs under the new standard. The Company did not elect the use-of-hindsight or the practical expedient pertaining to land easements, as the latter is not applicable to the Company. In addition, the Company elected not to apply ASC Topic 842 to arrangements with lease terms of 12 month or less. On January 1, 2019, upon adoption of ASC Topic 842, the Company recorded a right-of-use asset.
The Right-of-use assets are summarized below:
| June 30, | December 31, | |||||||
| 2022 | 2021 | |||||||
| Office Lease | $ | 265,207 | $ | 265,207 | ||||
| Less accumulated amortization | (177,824 | ) | (148,225 | ) | ||||
| Right-of-use, net | $ | 87,383 | $ | 116,982 | ||||
Amortization on the right-of -use asset is included in rent expense on the statements of operations.
Operating lease liabilities are summarized below:
| June 30, | ||||
| 2022 | ||||
| Office Lease | $ | 97,120 | ||
| Less: current portion | (71,518 | ) | ||
| Long term portion | $ | 25,602 | ||
The Maturities of lease liabilities are summarized below:
| As of | ||||
| June 30, | ||||
| 2022 | ||||
| 2022 | $ | 38,502 | ||
| 2023 | 64,800 | |||
| Total future minimum lease payments | 103,302 | |||
| Less imputed interest | (6,182 | ) | ||
| PV of Payments | $ | 97,120 | ||
Note 5 – Notes Payable-Related Party
On December 1, 2020, the Company consolidated all of the outstanding loans owed to an officer of the Company and to his spouse, resulting in the following two loans: (i) a single loan from the spouse of an officer of the Company, dated December 1, 2020, with a principal balance of $426,243, bearing interest at the rate of 7.5% per annum, with a maturity date of December 31, 2021; and (ii) a single loan owed to an officer of the company in the principal amount of $139,229, bearing interest at the rate of 7.5% per annum, with a maturity date of December 31, 2021. In December of 2021 the maturity dates of these loans were extended to June 30, 2022. As of June 30, 2022, the accrued interest was $47,575 and $15,533, and the total balances with accrued interest of $473,818 and $154,762 respectively. Subsequent to the date of this report, in July of 2022 the notes were extended to June 30, 2023 (Note 8).
On June 21, 2021, the Company entered into a loan from the spouse of an officer of the Company in the amount of $120,000 (principal) with an interest rate of 7.5% per annum due June 21, 2022, due at maturity. As of June 30, 2022, the accrued interest was $9,222 and the total balances with accrued interest of $129,222. Subsequent to the date of this report, in July of 2022 the notes were extended to June 30, 2023 (Note 8).
Note 6 - Notes Payable
On March 9, 2021, the Company obtained a $73,007 term note issued under the Coronavirus Aid, Relief, and Economic Security Act’s Paycheck Protection Program (the “PPP”). The note bears an interest rate of 1% per annum, has a six-month deferral period with payments beginning the seventh month and all outstanding principal and interest is due within two years from the note’s inception date. All or a portion of the note may be forgiven in accordance with PPP requirements. No more than 25% of the amount forgiven can be attributable to non-payroll costs. As of December 31, 2021, a “Loan Forgiveness Application” was submitted to PNC Bank along with the requested documentation and during the quarter ended March 31, 2022 the note liability was reduced in its entirety.
On May 15, 2020, the Company obtained a $62,500 term note issued under the Coronavirus Aid, Relief, and Economic Security Act’s Paycheck Protection Program (the “PPP”). The note bears an interest rate of 1% per annum, has a six-month deferral period with payments beginning the seventh month and all outstanding principal and interest is due within two years from the note’s inception date. All or a portion of the note may be forgiven in accordance with PPP requirements. No more than 25% of the amount forgiven can be attributable to non-payroll costs. A “Loan Forgiveness Application” was submitted to PNC Bank along with the requested documentation and the note liability was reduced in its entirety during the year ended December 31, 2021.
| June 30, | December 31, | |||||||
| 2022 | 2021 | |||||||
| PPP Note payable | ||||||||
| PPP Note May 15, 2020 | $ | - | $ | 62,500 | ||||
| PPP Note March 9, 2021 | 73,007 | 73,007 | ||||||
| Loan Forgiveness | (73,007 | ) | (62,500 | ) | ||||
| $ | - | $ | 73,007 | |||||
On December 28, 2021, the Company issued $500,000 note units, consisting of two $250,000 notes, for a total of $500,000 10% unsecured promissory notes with a maturity date of December 28, 2022, and 500,000 warrants exercisable at $1.00 per share with an expiry date of December 28, 2026, and fees of $5,075. The value of the warrants was determined using a computed volatility of 85.5%, 0% dividend rate, and a risk free interest rate of 1.27% and was applied as a discount on the notes payable.
On February 8, 2022, and March 11, 2022, the Company sold $365,000 and $224,985, respectively, in 6% convertible notes (the “Notes”), which bear 6% interest, are repayable three years from the date of issuance, and will convert automatically into units, with each unit consisting of one share of common stock and a warrant to purchase one share of commons stock (the “Conversion Units”) at a conversion price equal to 50% of the per unit offering price upon closing of this offering. Boustead Securities LLC acted as placement agent for this offering and received compensation of $36,500 and $22,250, respectively, and warrants to purchase shares of common stock equal to 10% of the Conversion Shares, exercisable at the conversion price of the Convertible Notes. The value of the warrants was determined using computed volatility of 83.4%, 0% dividend rate, and a risk free interest rate of 1.27%, and computed volatility of 85.5% %, 0% dividend rate, and a risk free interest rate of 1.96%, and was applied as a discount on the notes payable.
| June 30, | December 31, | |||||||
| 2022 | 2021 | |||||||
| Promissory note issued on December 28, 2021 | $ | 500,000 | $ | 500,000 | ||||
| Promissory note issued on February 8, 2022 | 365,000 | - | ||||||
| Promissory note issued on March 11, 2022 | 225,000 | - | ||||||
| 1,090,000 | 500,000 | |||||||
| Less discount on notes payable | (514,376 | ) | (408,979 | ) | ||||
| $ | 575,624 | $ | 91,021 | |||||
Note 7 – Stockholders’ Equity
Pursuant to the Company’s amended and restated articles of incorporation, the Company is authorized to issue 100,000,000 shares of common stock, with a par value of $0.00001 per share, and 20,000,000 shares of preferred stock, with a par value of $0.00001 per share.
Series A Preferred Shares
The Series A Preferred Stock, in accordance with its terms, is automatically convertible into a number of shares of the Company’s common stock upon the closing of the sale of shares of common stock to the public in a qualified offering (as set forth in the Series A certificate of designation) or upon listing of the Company’s common stock on a national securities exchange.
As of June 30, 2022, and December 31, 2021, the Company had 1,213 shares of Series A Preferred Stock issued and outstanding.
As of June 30, 2022, and December 31, 2021, the Company accrued $51,536 for the 8.5% cumulative dividends on the Series A Preferred stock for the six months ended June 30, 2022 and $103,062 for the year ended December 31, 2021,2022, as set forth in their report thereon, and included in the Annual Report on Form 10-K, which is incorporated herein by reference.
In March 2023, the Audit Committee of our Board of Directors approved the engagement of FORVIS LLP as our independent registered public accounting firm for a totalthe fiscal year ended December 31, 2023, effective March 21, 2023. The dismissal of $382,595 and $331,059 respectively.BF Borgers CPA PC was effective March 21, 2023. Additional information regarding the change in our independent registered public accounting firm can be found in our Current Report on Form 8-K dated March 22, 2023, which is incorporated by reference into this prospectus.
Common Stock
As of June 30, 2022 and December 31, 2021, the Company had 9,312,991 and 9,312,152 shares of common stock issued and outstanding, respectively. The balance includes 20,626 and 21,530 shares of restricted stock issued in 2021 and 2020 respectively and 839 common shares issued to settle common stock to be issued.
Common Stock to be Issued
On June 4, 2018, $120,250 outstanding convertible notes were converted to 6,182 shares of common stock of the Company at a price of $19.44 per share. The Company recorded $16,340 of common stock to be issued for the accrued interest. As of June 30, 2022, 839 common shares were issued to settle the $16,340 of common stock to be issued.
The Series A Preferred Stock sold in the private placement offerings, included warrants to be issued upon the earlier of a closingvalidity of the sale of shares of common stock to the public at a prices per share of at least $13.88 or in a firm commitment underwritten public offering pursuant to an effective registration statement resulting in gross proceeds of at least $15,000,000. The warrants shall be exercisable for a period of three years after the date of issuance. The warrant exercise price is contingent on the terms of the public offering. If an initial public offering occurs at a price at or above $13.88 per share, then the exercise price shall be set to the issuance price of the common stock with the number of warrants determined based on a 10% discount to the per share common stock issuance price. In the scenario where the common stock is listed with the common stock issuance price below $13.88, the exercise pricesecurities offered hereby will be set to $20.82 with the number of warrants based on a fixed conversion price of $12.49, which represents a 10.0% discount to the $13.88 threshold. The warrants also have contingent call features based on the terms of the public offering. If an initial public offering occurs at a price at or above $13.88, then the warrants are callable if the 20-day VWAP of the common stock in at or above 150% of the variable exercise price. In the scenario where the common stock is listed with a common stock issuance price below $13.88, then the warrants are callable if the 20-day VWAP of the common stock is at or above the $20.82 exercise price. The detachable warrants contained terms and features that gave rise to derivative liability classification.
Current accounting principles that are provided in ASC 815 - Derivatives and Hedging require derivative financial instruments to be classified in liabilities and carried at fair value with changes recorded in income. The Company has selected the Binomial Option Pricing valuation technique to fair value the compound embedded derivative. Inherent in a binomial options pricing model are assumptions related to expected stock-price volatility, expected life, risk-free interest rate, and dividend yield. The Company estimates the volatility of its ordinary shares based on historical volatility of comparable companies that matches the expected remaining life of the warrants. The risk-free interest rate is based on the U.S. Treasury zero-coupon yield curve on the grant datepassed upon for a maturity similar to the expected remaining life of the warrants.
The derivative liability linked to the Series A Preferred Stock as of June 30, 2022, and December 31, 2021 was $47,221 and $94,025, respectively. For the period ended June 30, 2022 and 2021, the change in fair value of warrant liability was a gain of $46,804 and a gain of $57,656, respectively.
The estimated fair values of the liability measured on a recurring basis are as follows:
| June 30, | December 31, | June 30, | ||||||||||
| % | 2022 | 2021 | 2021 | |||||||||
| Expected average volatility | 87.4 | % | 85.5 | % | 86 | % | ||||||
| Dividend yield | 8.5 | % | 8.5 | % | 8.5 | % | ||||||
| Expected life | 1.84 Years | 2.33 Years | 2.84 Years | |||||||||
| Risk-free interest rate | 2.92 | % | 0.73 | % | 0.25 | % | ||||||
A continuity schedule of the Series A Preferred Stock warrants is set forth below:
| Number of Warrants | Weighted Average Exercise Price | Weighted Average Life (years) | ||||||||||
| Outstanding, December 31, 2020 | 48,532 | $ | 24.98 | 3.33 | ||||||||
| Granted | - | - | - | |||||||||
| Forfeited | - | - | - | |||||||||
| Exercised | - | - | - | |||||||||
| Outstanding and Exercisable, December 31, 2021 | 48,532 | $ | 24.98 | 2.33 | ||||||||
| Granted | - | - | - | |||||||||
| Forfeited | - | - | - | |||||||||
| Exercised | - | - | - | |||||||||
| Outstanding and Exercisable, June 30, 2022 | 48,532 | $ | 24.98 | 1.83 | ||||||||
Equity Incentive Plan
Our 2018 Equity Incentive Plan provides for equity incentives to be granted to our employees, executive officers or directors and to key advisers and consultants. Equity incentives may be in the form of stock options with an exercise price of not less than the fair market value of the underlying shares as determined pursuant to the 2018 Equity Incentive Plan, restricted stock awards, other stock-based awards, or any combination of the foregoing. The 2018 Equity Incentive Plan is administeredus by the Company’s compensation committee or, alternatively, if there is no compensation committee, the Company’s board of directors. We have reserved 3,000,000 shares of our common stock for issuance under the 2018 Equity Incentive Plan. As of June 30, 2022, 384,167 shares have been granted under the 2018 Equity Incentive Plan.
Restricted Stock Units. We may grant restricted stock units under our 2018 Plan. Restricted stock units are bookkeeping entries representing an amount equal to the fair market value of one share of our common stock. Subject to the provisions of our 2018 Plan, the administrator determines the terms and conditions of restricted stock units, including the vesting criteria and the form and timing of payment. The administrator, in its sole discretion, may pay earned restricted stock units in the form of cash, in shares or in some combination thereof. Notwithstanding the foregoing, the administrator, in its sole discretion, may accelerate the time at which any restrictions will lapse or be removed.
On August 16, 2019, five individuals were appointed to the Board of Directors of the Company to serve as directors. Each individual entered into an agreement pursuant to which they will serve as a director and pursuant to which they would each receive a grant of $75,000 worth of Restricted Stock Units (“RSUs”) issuable under the Company’s 2018 Equity Incentive Plan (the “2018 Plan”). The RSUs vest annually in one third increments from the date of appointment. Under the terms of the director agreements, the Company has also agreed to pay each director $25,000 per annum, payable in equal quarterly installments commencing 90 days following the Company becoming a publicly reporting company under the Securities Exchange Act of 1934, as amended.
During the six months ended June 30, 2022 and 2021, pursuant to the agreements with directors and officers compensation expense for the RSUs of $333,066 and $245,034 was included in wages, respectively.
As of June 30, 2022, there was $32,001 of total unrecognized compensation cost related to non-vested share-based compensation arrangements which is expected to be recognized within the current year.
A continuity schedule of the Restricted Stock Units (RSU) follows:
| Number of RSU | Weighted Average Exercise Price | Weighted Average Life (years) | ||||||||||
| Outstanding, December 31, 2020 | 61,884 | $ | 23.76 | 4.33 | ||||||||
| Granted | - | - | ||||||||||
| Forfeited | (2,702 | ) | 27.76 | - | ||||||||
| Exercised | - | - | - | |||||||||
| Outstanding, December 31, 2021 | 59,182 | $ | 23.57 | 3.33 | ||||||||
| Granted | - | - | - | |||||||||
| Forfeited | - | - | - | |||||||||
| Exercised | - | - | - | |||||||||
| Outstanding, June 30, 2022 | 59,182 | $ | 23.57 | 2.84 | ||||||||
| Exercisable, June 30, 2022 | 57,768 | $ | 23.74 | 2.84 | ||||||||
Note 8 – Subsequent Events
Management evaluated all additional events subsequent to the balance sheet date through August 12, 2022, the date the financial statements were available to be issued, and determined the following items:
In July of 2022, the Company signed extension agreements for the related party notes of $138,449, $424,036 and $120,000 that were due during June of 2022. The extensions updated the maturity date for the three notes to June 30, 2023.
In August of 2022, the Company closed on a bridge financing arrangement or $125,000 of 10% notes and warrants to purchase 50,000 shares of common stock exercisable at $2.50 per share. The notes are due 12 months from the date of execution or upon closing the initial public offering (“IPO”) and warrants are exercisable for five years.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the shareholders and the board of directors of Shuttle Pharmaceuticals Holdings, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Shuttle Pharmaceuticals Holdings, Inc. (the “Company”) as of December 31, 2021 and 2020, the related statements of operations, stockholders’ equity (deficit), and cash flows for the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021 and 2020, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States.
Restatement of December 31, 2021 Financial Statements
As discussed in Note 9 to the financial statements, the financial statements have been restated to correct certain misstatements.
Substantial Doubt about the Company’s Ability to Continue as a Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company’s significant operating losses raise substantial doubt about its ability to continue as a going concern. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud.
Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
/s/ BF Borgers CPA PC
BF Borgers CPA PC
Served as Auditor since 2021
Lakewood, CO
June 3, 2022, except for the effects of the restatement disclosed in Note 9, as to which the date is June 23, 2022
Shuttle Pharmaceuticals Holdings, Inc.
| December 31, | December 31, | |||||||
| 2021 | 2020 | |||||||
(As Restated) | ||||||||
| ASSETS | ||||||||
| Current Assets: | ||||||||
| Cash | $ | 504,749 | $ | 117,153 | ||||
| Prepaid expenses | 4,866 | 12,579 | ||||||
| Contracts receivable | - | 211,455 | ||||||
| Total current assets | 509,615 | 341,187 | ||||||
| Property and equipment, net | 18,564 | 24,782 | ||||||
| Other assets | 6,480 | 6,480 | ||||||
| Operating lease right-of-use asset | 116,982 | 171,598 | ||||||
| Total assets | $ | 651,641 | $ | 544,047 | ||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable and accrued expenses | $ | 828,313 | $ | 516,966 | ||||
| Accrued interest payable | 552 | 392 | ||||||
| Accrued interest payable - related parties | 46,947 | - | ||||||
| Dividends Payable | 331,059 | 227,997 | ||||||
| Notes payable - related parties | 685,473 | 565,473 | ||||||
| Notes payable | 91,021 | - | ||||||
| Paycheck Protection Program note payable, current portion | 73,007 | 62,500 | ||||||
| Derivative liability | 94,025 | 673,171 | ||||||
| Operating lease liability current portion | 66,934 | 58,468 | ||||||
| Total current liabilities | 2,217,331 | 2,104,967 | ||||||
| Operating lease liability non-current | 62,442 | 129,376 | ||||||
| Total liabilities | 2,279,773 | 2,234,343 | ||||||
| Stockholders’ Deficit: | ||||||||
| Series A convertible preferred stock, $0.00001 par value; $1,000 per share liquidation value or aggregate of $1,212,500; 20,000,000 shares authorized; 1,213 shares issued and outstanding at December 31, 2021 | - | - | ||||||
| Common stock, $0.00001 par value; 100,000,000 shares authorized; 9,312,152 and 9,291,526 shares issued and outstanding at December 31, 2021 and 2020, respectively | 93 | 93 | ||||||
| Additional paid in capital | 4,150,867 | 2,833,507 | ||||||
| Common stock to be issued | 16,340 | 16,340 | ||||||
| Accumulated deficit | (5,795,432 | ) | (4,540,236 | ) | ||||
| Total stockholders’ deficit | (1,628,132 | ) | (1,690,296 | ) | ||||
| Total liabilities and stockholders’ deficit | $ | 651,641 | $ | 544,047 | ||||
The accompanying footnotes are an integral part of these consolidated financial statements.
Shuttle Pharmaceuticals Holdings, Inc.
Consolidated Statements of Operations
| Year Ended | ||||||||
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| (As Restated) | ||||||||
| Revenue | $ | - | $ | - | ||||
| Operating expenses: | ||||||||
| Research and development, net of contract expense reimbursements | 1,021,808 | 161,772 | ||||||
| General and administrative | 36,500 | 85,927 | ||||||
| Legal and professional | 684,684 | 261,823 | ||||||
| Total operating expenses | 1,742,992 | 509,522 | ||||||
| Loss from operations | (1,742,992 | ) | (509,522 | ) | ||||
| Other income (expense): | ||||||||
| Loss on write off of deposit | - | - | ||||||
| Interest expense - related parties | (46,947 | ) | (36,771 | ) | ||||
| Interest expense | (3,841 | ) | (2,859 | ) | ||||
| Change in fair value of warrant liability | 579,146 | (256,580 | ) | |||||
| Gain on forgiveness of Paycheck Protection Program note payable | 62,500 | - | ||||||
| Total other income (expense) | 590,858 | (296,210 | ) | |||||
| Loss before income taxes | (1,152,134 | ) | $ | (805,732 | ) | |||
| Income tax provision | - | - | ||||||
| Net loss | $ | (1,152,134 | ) | $ | (805,732 | ) | ||
| Dividend on Series A preferred stock | (103,062 | ) | (103,062 | ) | ||||
| Net loss attributable to common stockholders | $ | (1,255,196 | ) | $ | (908,794 | ) | ||
| Weighted average common shares outstanding - basic and diluted | 9,301,750 | 9,291,526 | ||||||
| Diluted | 9,306,610 | 18,575,143 | ||||||
| Net loss per shares - basic and diluted | $ | (0.12 | ) | $ | (0.09 | ) | ||
The accompanying footnotes are an integral part of these consolidated financial statements.
Shuttle Pharmaceuticals Holdings, Inc.
Consolidated Statements of Changes in Stockholders’ Deficit
| Series A Convertible | Additional | Common | Total | |||||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Paid-in | Stock to | Accumulated | Stockholders’ | |||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | be Issued | Deficit | Deficit | |||||||||||||||||||||||||
| Balance, December 31, 2019 | 1,213 | $ | - | 9,269,996 | $ | 93 | $ | 2,373,918 | $ | 16,340 | $ | (3,631,442 | ) | $ | (1,241,091 | ) | ||||||||||||||||
| Common stock issued for restricted stock units | - | - | 21,530 | - | 459,589 | - | - | 459,589 | ||||||||||||||||||||||||
| Dividends on Series A preferred stock | - | - | - | - | - | - | (103,062 | ) | (103,062 | ) | ||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (805,732 | ) | (805,732 | ) | ||||||||||||||||||||||
| Balance, December 31, 2020 | 1,213 | $ | - | 9,291,526 | $ | 93 | $ | 2,833,507 | $ | 16,340 | $ | (4,540,236 | ) | $ | (1,690,296 | ) | ||||||||||||||||
| Stock based compensation (As Restated) | - | - | - | - | 420,000 | - | - | 420,000 | ||||||||||||||||||||||||
| Warrants issued for financing costs | - | - | - | - | 407,293 | - | - | 407,293 | ||||||||||||||||||||||||
| Common stock issued for restricted stock units | - | - | 20,626 | - | 490,067 | - | - | 490,067 | ||||||||||||||||||||||||
| Dividends on Series A preferred stock | - | - | - | - | - | - | (103,062 | ) | (103,062 | ) | ||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (1,152,134 | ) | (1,152,134 | ) | ||||||||||||||||||||||
| Balance, December 31, 2021 (As Restated) | 1,213 | $ | - | 9,312,152 | $ | 93 | $ | 4,150,867 | $ | 16,340 | $ | (5,795,432 | ) | $ | (1,628,132 | ) | ||||||||||||||||
The accompanying footnotes are an integral part of these consolidated financial statements.
Shuttle Pharmaceuticals Holdings, Inc.
Consolidated Statements of Cash Flows
| Year Ended | ||||||||
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| (As Restated) | ||||||||
| Cash Flows From Operating Activities: | ||||||||
| Net loss | $ | (1,152,134 | ) | $ | (805,732 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | 6,218 | 5,782 | ||||||
| Change in fair value of warrant liability | (579,146 | ) | 256,580 | |||||
| Amortization of right-of-use asset | 54,616 | 49,169 | ||||||
| Amortization of debt discount | 3,389 | - | ||||||
| Gain on forgiveness of Paycheck Protection Program note payable | (62,500 | ) | - | |||||
| Stock-based compensation | 490,067 | 459,589 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Contracts receivable | 211,455 | 164,927 | ||||||
| Prepaid expenses | 7,713 | (586 | ) | |||||
| Accounts payable and accrued expenses | 311,347 | (51,127 | ) | |||||
| Accrued interest payable | 160 | 392 | ||||||
| Accrued interest payable - related parties | 46,947 | (54,959 | ) | |||||
| Operating lease liability | (58,468 | ) | (50,866 | ) | ||||
| Net cash used in operating activities | (300,336 | ) | (26,831 | ) | ||||
| Cash Flows From Investing Activities: | ||||||||
| Purchase of property and equipment | - | (10,818 | ) | |||||
| Net cash used in investing activities | - | (10,818 | ) | |||||
| Cash Flows From Financing Activities: | ||||||||
| Proceeds from notes payable-related parties | 120,000 | 7,573 | ||||||
| Proceeds from PPP note payable | 73,007 | 62,500 | ||||||
| Proceeds from notes payable | 494,925 | - | ||||||
| Net cash provided by financing activities | 687,932 | 70,073 | ||||||
| Net change in cash | 387,596 | 32,424 | ||||||
| Cash, beginning of period | 117,153 | 84,729 | ||||||
| Cash, end of period | $ | 504,749 | $ | 117,153 | ||||
| Cash paid for: | ||||||||
| Interest | $ | 293 | $ | 1,886 | ||||
| Income taxes | $ | - | $ | - | ||||
The accompanying footnotes are an integral part of these consolidated financial statements.Dorsey & Whitney LLP, New York, New York.
Shuttle Pharmaceuticals Holdings, Inc.
Notes to Consolidated Financial Statements
Year Ended December 31, 2020 and 2021
Note 1 – Organization and Going Concern
Organization and Line of Business
The Company was formed as Shuttle Pharmaceuticals, LLC, in the State of Maryland on December 18, 2012. On August 12, 2016, the Company filed articles of conversion with the state of Maryland to convert from an LLC to a corporation and the Company changed its name to Shuttle Pharmaceuticals, Inc. (“Shuttle”). In connection with the conversion the Company issued 45,000,000 shares of common stock in exchange for 100% of the outstanding membership interests prior to conversion. On June 4, 2018, Shuttle completed a reverse merger with Shuttle Pharmaceuticals Holdings, Inc. (then known as Shuttle Pharma Acquisition Corp.), a Delaware corporation (the “Company”), pursuant to which Shuttle, our operating entity, became a wholly owned subsidiary of the Company.
The Company’s primary purpose is to develop and commercialize unique drugs for the sensitization of cancers and protection of normal tissues, with the goal of improving outcomes for cancer patients receiving radiation therapy. Shuttle has deployed its proprietary technology to develop novel cancer immunotherapies which has produced a pipeline of selective HDAC inhibitors for cancer and immunotherapy applications. The Company’s HDAC platform is designed to target candidate molecules with potential roles in therapeutics beyond cancer, including autoimmune, inflammatory, metabolic, neurological and infectious diseases. The Company’s Ropidoxuridine product, which is used with radiation therapy to sensitize cancer cells, was funded by a Small Business Innovation Research (SBIR) contract provided by the National Cancer Institute (NCI), a unit of the National Institutes of Health (NIH). Ropidoxuridine has been further developed though the Company’s collaborations with the University of Virginia for use in combination with proton therapy to improve patient survival. The Company is working on developing products through NIH grants, including a product to predict late effects of radiation with metabolite biomarkers and develop prostate cancer cell lines in health disparities research.
The production and marketing of the Company’s products and its ongoing research and development activities will be subject to extensive regulation by numerous governmental authorities in the United States. Prior to marketing in the United States, any combination product developed by the Company must undergo rigorous preclinical (animal) and clinical (human) testing and an extensive regulatory approval process implemented by the Food and Drug Administration (“FDA”) under the Food, Drug and Cosmetic Act. There can be no assurance that the Company will not encounter problems in clinical trials that will cause the Company or the FDA to delay or suspend clinical trials.
The Company’s success will depend in part on its ability to obtain patents and product license rights, maintain trade secrets, and operate without infringing on the proprietary rights of others, both in the United States and other countries. There can be no assurance that patents issued to or licensed by the Company will not be challenged, invalidated or circumvented, or that the rights granted thereunder will provide proprietary protection or competitive advantages to the Company now or in the future.
Reverse Stock Split
Effective April 1, 2022, we effected a 2-for-1 reverse stock split of our issued and outstanding common stock (the “Reverse Stock Split”). All references to shares of our common stock in this registration statement on Form S-1 refers to the number of shares of common stock after giving effect to the Reverse Stock Split (unless otherwise indicated).
Going Concern
The accompanying financial statements have been prepared in conformity with generally accepted accounting principles, which contemplate continuation of the Company as a going concern. The Company’s only revenue source since inception has been government awarded contracts totaling $5,531,722, and the Company has incurred losses since inception, having accumulated a deficit of $5,795,432 as of December 31, 2021. The Company currently has limited liquidity and has not completed its efforts to establish a stabilized source of revenues sufficient to cover operating costs over an extended period. These factors, among others, raise substantial doubt about the Company’s ability to continue as a going concern.
The Company will need to raise capital to fund its operations. To address its financing requirements, the Company intends to seek financing through debt or equity financings with an aim to continue progress toward commercial viability of its products. The Company continues to submit Federal grant and contract applications which have historically been the primary source of revenue. The financial statements do not include any adjustments that might result from the outcome of the uncertainty of raising additional capital.
Note 2 – Summary of Significant Accounting Policies
Basis of Presentation
The financial statements and related disclosures have been prepared pursuant to the rules and regulations of the Securities and Exchange Commission (“SEC”). The financial statements have been prepared using the accrual basis of accounting in accordance with Generally Accepted Accounting Principles of the United States (“GAAP”).
Basis of Consolidation
The financial statements have been prepared on a consolidated basis with those of the Company’s wholly-owned subsidiary, Shuttle Pharmaceuticals, Inc. All intercompany transactions and balances have been eliminated.
Use of Estimates
The preparation of consolidated financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. The Company regularly evaluates estimates and assumptions. The Company bases its estimates and assumptions on current facts, historical experience, and various other factors that it believes to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities and the accrual of costs and expenses that are not readily apparent from other sources. The actual results experienced by the Company may differ materially and adversely from the Company’s estimates. To the extent there are material differences between the estimates and the actual results, future results of operations will be affected. Significant estimates in the accompanying financial statements include useful lives of property and equipment, valuation of derivatives, and the valuation allowance on deferred tax assets.
Accounts Receivable
The Company’s accounts receivable consists of amounts due from the National Institutes of Health (“NIH”), a government agency under the Department of Health and Human Services. The Company submits quarterly reports of progress for tasks performed on research and development contracts it is awarded. The contracts are typically reimbursed on a fixed fee cost reimbursement basis and payments from the contracts are generally collected within 30 days of the Company’s submission for reimbursement. The Company currently does not provide an allowance for doubtful collections, which is based upon a review of outstanding receivables, historical collection information, and existing economic conditions. Normal receivable terms vary from 7-30 days after the issuance of the invoice and typically would be considered past due when the term expires. The Company’s allowance for doubtful accounts was $0 at December 31, 2020 and 2021.
Property and Equipment
Property and equipment are stated at cost. Expenditures for maintenance and repairs are charged to earnings as incurred; additions, renewals and betterments are capitalized. When property and equipment are retired or otherwise disposed of, the related cost and accumulated depreciation are removed from the respective accounts, and any gain or loss is included in operations. Depreciation of property and equipment is provided using the straight-line method for substantially all assets with estimated lives as follows:
Research and Development Expenses
Research and development expenses are charged to expense as incurred. Research and development expenses include, but are not limited to, product development, clinical and regulatory expenses, payroll and other personnel expenses, materials, supplies, related subcontract expenses, and consulting costs. The expenses assigned to NIH SBIR sponsored research are related to: (1) “Topic 352: Cell-Based Models for Prostate Cancer Health Disparity Research – Moonshot Project” and (2) “Topic 345: Predictive Biomarkers of Prostate Cancer Patient Sensitivity for Radiation Late Effects.”
The research expenses are assigned to the research projects to demonstrate proof of principle in patients with prostate cancer that may support development and commercialization of biomarker products and to gather prostate cancer cell lines in African American men to serve as the product for use in health disparities research. Costs that are not covered by the SBIR contract for performing the Phase I contract to determine commercialization feasibility included partial salary support of personnel and a consultant to develop a commercialization plan. Costs that are not covered in the Phase II contract include business development and partial salary support.
Research expenses related to new drug discovery include partial support of personnel, space, supplies, and legal costs.
During fiscal year 2021, the Company has made progress on completing two SBIR contracts from the NIH to support research projects with potential for commercialization. The SBIR contract awards are fixed payments made by the NIH in response to quarterly Shuttle invoices and provide non-dilutive funds that do not include a repayment obligation. Details on the three contracts follow:
1. Contract #HHSN261201600027C/75N91018C00016 supported “Topic 345: Predictive Biomarkers of Prostate Cancer Patient Sensitivity for Radiation Late Effects.” This $299,502 Phase I award includes funded research from 9/19/2016 through 9/18/2017 and was advanced to Phase II of the awards with funding of $1,903,095 with a fixed price contract period of 9/17/2018 through 9/16/2020 and subsequent no cost extensions through 9/15/2021 and then 3/15/2022 (Reference 75N91019C00031). The Company received quarterly payments of $211,455 for a total of $845,820 in 2020; and 2 quarterly payments related to Topic 345 for a total of $422,910 in 2021. In Phase II of the SBIR effort, the Company will license the metabolite signatures (intellectual property) from Georgetown University, manufacture 500 “kits,” test and validate the metabolic kit performance and develop a multi-institutional clinical trial to be implemented in the Phase III effort. This contract includes a subcontract to Georgetown University for use of Mass Spectrometry core facilities to analyze clinical samples. The contract was extended to complete the milestones which were delayed due to the impact of COVID-19.
On December 6, 2019, the Company engaged Georgetown University to perform the $795,248 subcontract of its Phase II contract #HHSN75N91019C00031. The Company will reimburse Georgetown for its allowable costs not to exceed the ceiling amount of $795,248. Depending on the resources it uses, Georgetown may or may not invoice for the total subcontract amount. In the event Georgetown does not invoice for the total allowable amount, the Company is not obligated to pay the ceiling amount. As of December 31, 2021, payments of $354,829 were made to Georgetown, and additional invoices for $436,188 were received but not yet paid.
2. The Phase II contract #HHSN261201800016C supports the discovery work following a Phase I contract # HHSN261600038C “Topic 352 – SBIR Phase II Cell-based Models for Prostate Cancer Health Disparity Research” and was awarded to provide $1,484,350 to fund research from 9/17/2018 through 9/16/2020 and was extended without cost through 11/16/2021 due to delays caused by the impact of COVID-19. For 2020, the Company received 2.5 quarterly payments for a total of $412,321 and $82,467 in 2021. The Phase II contract also includes a subcontract to Georgetown University for $742,002 to establish prostate cancer cell lines from African American patients undergoing prostate surgery for cancers.
On December 5, 2018, the Company engaged Georgetown University to perform the $742,002 subcontract of its Phase II contract #HHSN261201800016C. Depending on the resources it uses, Georgetown may or may not invoice for the total subcontract amount. In the event Georgetown does not invoice for the total allowable amount, the Company is not obligated to pay the ceiling amount. The Company has been invoiced by Georgetown and has paid Georgetown a total of $292,252 as of December 31, 2021.
The Company recognizes the amounts received from the contract at fair value when there is reasonable assurance that the contract amount will be received, and it is probable that all attaching conditions will be complied with. The Company recognizes the amounts received in accordance with the contract as a reduction of research and development expenses over the periods necessary to match the contract on a systematic basis to the costs that it is intended to compensate. The Company records reimbursements on the balance sheet as contract receivables upon meeting the criteria discussed above until cash is received. During the 12 months ended December 31, 2021, the Company recorded a net deficit of $1,021,808 from these two SBIR contracts which is the result of recording $1,527,185 in SBIR contract related expenses on revenues of $505,377.
Regarding the accounting treatment for reimbursements, GAAP provides limited guidance on the accounting for government grants received by for-profit companies. We understand there is more than one acceptable alternative for the accounting treatment – a reduction of costs, a deferred credit to be amortized, revenue, or other income. Due to the terms of the contracts, we have entered into the Company concluded that the reimbursements were more akin to a reduction of costs rather than any of the other alternatives that would match the contract reimbursements on a systematic basis to the costs that the contract is intended to compensate.
Derivative Financial Instruments
The Company evaluates all of its agreements to determine if such instruments have derivatives or contain features that qualify as embedded derivatives. For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair value and is then re-valued at each reporting date, with changes in the fair value reported in the statements of operations. For stock-based derivative financial instruments, the Company uses a Binomial Simulation model to value the derivative instruments at inception and on subsequent valuation dates. The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is evaluated at the end of each reporting period. Derivative instrument liabilities are classified in the balance sheet as current or non-current based on whether or not net-cash settlement of the derivative instrument could be required within 12 months of the balance sheet date. As of December 31, 2021, the Company’s only derivative financial instrument was an embedded warrant feature associated with Series A Convertible Preferred Stock due to certain provisions that allow for a change in the warrant value based on fluctuations of the Company’s fair value of common stock at the date of issuance of the warrant based on certain contingent call features.
Fair Value of Financial Instruments
For certain of the Company’s financial instruments, including cash, accounts receivable, accounts payable, accrued liabilities and short-term debt, the carrying amounts approximate their fair values due to their short maturities.
FASB ASC Topic 820, Fair Value Measurements and Disclosures, requires disclosure of the fair value of financial instruments held by the Company. FASB ASC Topic 825, Financial Instruments, defines fair value, and establishes a three-level valuation hierarchy for disclosures of fair value measurement that enhances disclosure requirements for fair value measures. The carrying amounts reported in the consolidated balance sheets for receivables and current liabilities each qualify as financial instruments and are a reasonable estimate of their fair values because of the short period of time between the origination of such instruments and their expected realization and their current market rate of interest. The three levels of valuation hierarchy are defined as follows:
The Company analyzes all financial instruments with features of both liabilities and equity under FASB ASC Topic 480, Distinguishing Liabilities from Equity, and FASB ASC Topic 815, Derivatives and Hedging.
For certain financial instruments, the carrying amounts reported in the balance sheets for cash and current liabilities, including convertible notes payable, each qualify as a financial instrument, and are a reasonable estimate of their fair values because of the short period of time between the origination of such instruments and their expected realization and their current market rate of interest.
An established trading market for the Company’s common stock does not exist. The fair value of the shares was determined based on the then most recent price per share at which we sold preferred stock to unrelated parties in a private placement during the six months then ended.
During the year ended December 31, 2020, the Company utilized $25.22 (post-share exchange) per share as the fair value of its common stock for accounting purposes based on preferred share transactions with investors from August 2018 through December 2019, with no transactions occurring in 2020 and $5.00 in 2021.
At December 31, 2021, the Company identified the following liabilities that are required to be presented on the balance sheet at fair value:
| Description | Level 1 | Level 2 | Level 3 | |||||||||
| Warrant Liability | $ | - | $ | - | $ | 94,025 | ||||||
At December 31, 2020, the Company identified the following liabilities that are required to be presented on the balance sheet at fair value:
| Description | Level 1 | Level 2 | Level 3 | |||||||||
| Warrant Liability | $ | - | $ | - | $ | 673,171 | ||||||
Revenue Recognition
Revenue from providing research and development is recognized under Topic 606 in a manner that reasonably reflects the delivery of its services to customers in return for expected consideration and includes the following elements:
To satisfy these five elements, the Company records revenue for research and development services on a quarterly basis as services are provided. Revenue received from National Institutes of Health contracts is received in accordance with Federal grants and contracts policies. Research and development expenses are posted against revenue and recorded on the statement of operations as “Research and development, net of contract expense reimbursements.”
Impairment of Long-Lived Assets
The Company reviews its long-lived assets for impairment whenever events or changes in business circumstances indicate that the carrying amount of the asset may not be fully recoverable. Recoverability of assets is measured by a comparison of the carrying amount of an asset to the estimated undiscounted cash flows expected to be generated by the asset. If the carrying amount of the asset exceeds its estimated future cash flows, an impairment charge will be recognized in the amount by which the carrying amount of the asset exceeds the fair value of the asset. There were no impairments of long-lived assets during the periods presented.
Income Taxes
The Company accounts for income taxes in accordance with ASC Topic 740, Income Taxes. ASC 740 requires a company to use the asset and liability method of accounting for income taxes, whereby deferred tax assets are recognized for deductible temporary differences, and deferred tax liabilities are recognized for taxable temporary differences. Temporary differences are the differences between the reported amounts of assets and liabilities and their tax bases. Deferred tax assets are reduced by a valuation allowance when, in the opinion of management, the Company does not foresee generating taxable income in the near future and utilizing its deferred tax asset, therefore, it is more likely than not that some portion, or all of, the deferred tax assets will not be realized. Deferred tax assets and liabilities are adjusted for the effects of changes in tax laws and rates on the date of enactment.
Under ASC 740, a tax position is recognized as a benefit only if it is “more likely than not” that the tax position would be sustained in a tax examination, with a tax examination being presumed to occur. The amount recognized is the largest amount of tax benefit that is greater than 50% likely of being realized on examination. For tax positions not meeting the “more likely than not” test, no tax benefit is recorded. The Company has no material uncertain tax positions for any of the reporting periods presented.
Basic and Diluted Earnings Per Share
Basic earnings per share (“EPS”) is computed based on the weighted average number of shares of common stock outstanding during the period. Diluted EPS is computed based on the weighted average number of shares of common stock plus the effect of dilutive potential common shares outstanding during the period using the treasury stock method and as if converted method. Dilutive potential common shares include outstanding warrants and Series A preferred stock.
For the years ended December 31, 2021, and 2020, the following common stock equivalents were excluded from the computation of diluted net loss per share as the result of the computation was anti-dilutive.
| December 31, | December 31, | |||||||
| 2021 | 2020 | |||||||
| Series A preferred stock | 97,062 | 97,062 | ||||||
| Warrants | 48,532 | 48,532 | ||||||
| Total | 145,594 | 145,594 | ||||||
Recent Accounting Pronouncements
In August 2020, the FASB issued ASU 2020-06, ASC Subtopic 470-20 “Debt—Debt with “Conversion and Other Options” and ASC subtopic 815-40 “Hedging—Contracts in Entity’s Own Equity.” The standard reduced the number of accounting models for convertible debt instruments and convertible preferred stock. Convertible instruments that continue to be subject to separation models are (1) those with embedded conversion features that are not clearly and closely related to the host contract, that meet the definition of a derivative, and that do not qualify for a scope exception from derivative accounting; and (2) convertible debt instruments issued with substantial premiums for which the premiums are recorded as paid-in capital. The amendments in this update are effective for fiscal years beginning after December 15, 2021, including interim periods within those fiscal years. Early adoption is permitted. The Company has adopted this standard on January 1, 2021.
Management does not believe that any recently issued, but not yet effective, accounting standards could have a material effect on the accompanying financial statements. As new accounting pronouncements are issued, we will adopt those that are applicable under the circumstances.
Note 3 – Property and Equipment, Net
Property and equipment consisted of the following as of December 31, 2021, and 2020:
| December 31, | December 31, | |||||||
| 2021 | 2020 | |||||||
| Office Furniture and equipment | $ | 8,861 | $ | 8,861 | ||||
| Laboratory equipment | 118,605 | 118,605 | ||||||
| 127,466 | 127,466 | |||||||
| Less accumulated depreciation | (108,902 | ) | (102,684 | ) | ||||
| Property and equipment, net | $ | 18,564 | $ | 24,782 | ||||
Depreciation expense for the twelve months ended December 31, 2021, and 2020 were $6,218 and $5,782 respectively.
Note 4 – Operating Lease Right-of-use Asset and Operating Lease Liability
Operating lease right-of-use assets and liabilities are recognized at the present value of the future lease payments at the lease commencement date. The interest rate used to determine the present value is our incremental borrowing rate, estimated to be 10%, as the interest rate implicit in most of our leases is not readily determinable. Operating lease expense is recognized on a straight-line basis over the lease term. During the twelve months ended December 31, 2021, and the year ended December 31, 2020, the Company recorded $74,028 and $71,872, respectively, as operating lease expense.
The Company currently has a lease agreement which allows for the use of a laboratory facility for a monthly payment of $6,107, which monthly lease payment increases by 3% every year. The laboratory lease commenced October 1, 2018, with the first payment due January 1, 2019, and expires on October 31, 2023. A security deposit of $6,480 is being held for the duration of the lease term.
In adopting ASC Topic 842, Leases (Topic 842), the Company has elected the ‘package of practical expedients,’ which permits the Company to avoid reassessing its prior conclusions about lease identification, lease classification and initial direct costs under the new standard. The Company did not elect the use-of-hindsight or the practical expedient pertaining to land easements, as the latter is not applicable to the Company. In addition, the Company elected not to apply ASC Topic 842 to arrangements with lease terms of 12 month or less. On January 1, 2019, upon adoption of ASC Topic 842, the Company recorded a right-of-use asset.
| December 31, | December 31, | |||||||
| 2021 | 2020 | |||||||
| Office Lease | $ | 265,207 | $ | 265,207 | ||||
| Less accumulated amortization | (148,225 | ) | (93,609 | ) | ||||
| Right-of-use, net | $ | 116,982 | $ | 171,598 | ||||
The Right-of-use assets are summarized below:
Amortization on the right-of -use asset is included in rent expense on the statements of operations.
Operating lease liabilities are summarized below:
| December 31, | ||||
| 2021 | ||||
| Office Lease | $ | 129,376 | ||
| Less: current portion | (66,934 | ) | ||
| Long term portion | $ | 62,442 | ||
The Maturities of lease liabilities are summarized below:
| As of | ||||
| December 31, | ||||
| 2021 | ||||
| 2022 | $ | 76,248 | ||
| 2023 | 64,800 | |||
| Total future minimum lease payments | 141,048 | |||
| Less imputed interest | (11,672 | ) | ||
| PV of Payments | $ | 129,376 | ||
Note 5 – Notes Payable-Related Party
On January 25, 2018, the Company entered into a loan from the spouse of an officer of the Company in the amount of $300,000. The loan bears interest at the rate of 7.5% per annum. The loan plus accrued interest was payable in full on January 25, 2019. On January 25, 2019, the Company amended the loan for purposes of extending the maturity date from January 25, 2019, to October 25, 2020. On December 1, 2020, the Company again amended the terms and extended the maturity date to December 31, 2021, bringing the total amount owed under the loan, together with accrued interest of $64,084, to a total of $364,084. This note was rolled into the note referenced below with a principal amount of $424,056.
On April 4, 2018, the Company entered into a loan from the spouse of an officer of the Company in the amount of $50,000. The loan bears an interest rate of 7.5% per annum. The loan plus accrued interest was payable in full on September 4, 2018. On October 31, 2018, the Company amended the terms to extend the maturity date from September 4, 2018, to April 4, 2019. On April 4, 2019, the Company amended the terms to extend the maturity date from April 4, 2019, to October 25, 2020. On December 1, 2020, the Company again amended the terms and extended the maturity date to December 31, 2021, bringing the total amount owed under the loan, together with accrued interest of $9,972, to a total of $59,972. This note was rolled into the note referenced below with a principal amount of $424,056.
On May 30, 2018, the Company entered into a loan with an officer of the Company in the amount of $25,000. The loan bears an interest rate of 7.5% per annum. The loan plus accrued interest was payable in full on July 15, 2018. On October 31, 2018, the Company amended the terms to extend the maturity date from July 15, 2018, to November 30, 2020. On December 1, 2020, the Company again amended the terms and extended the maturity date to December 31, 2021, bringing the total amount owed under the loan, together with accrued interest of $4,698, to a total of $29,698. This note was rolled into the note reference below with a principal amount of $138,448.
On June 24, 2019, the Company entered into a loan from the spouse of an officer of the Company in the amount of $70,000. The loan bore interest rate of 7.5% per annum and required monthly payments of $6,073. The loan plus accrued interest was paid in full on June 30, 2020.
On August 24, 2019, the Company entered into a loan from the spouse of an officer of the Company in the amount of $70,000. The loan bore an interest rate of 7.5% per annum and required monthly payments of $6,073. The loan plus accrued interest was paid in full on August 23, 2020.
On September 23, 2019, the Company entered into a loan from the spouse of an officer of the Company in the amount of $100,000. The loan bore an interest rate of 7.5% per annum and became payable in full on September 22, 2020. This note was rolled into the note reference below with a principal amount of $138,448.
On December 1, 2020, the Company consolidated all of the outstanding loans owed to an officer of the Company and to his spouse, resulting in the following two loans: (i) a single loan from the spouse of an officer of the Company, dated December 1, 2020, with a principal balance of $424,056, bearing interest at the rate of 7.5% per annum, with a maturity date of December 31, 2021; and (ii) a single loan owed to an officer of the company in the principal amount of $138,449, bearing interest at the rate of 7.5% per annum, with a maturity date of December 31, 2021. In December of 2021 the maturity dates of these loans were extended to June 30, 2022. As of December 31, 2021, the accrued interest was $31,804 and $10,384, and the total balances with accrued interest of $458,047 and $149,613 respectively.
On June 21, 2021, the Company entered into a loan from the spouse of an officer of the Company in the amount of $120,000 (principal) with an interest rate of 7.5% per annum due June 21, 2022, due at maturity. As of December 31, 2021, the accrued interest was $4,759 and the total balances with accrued interest of $124,759.
Note 6 - Notes Payable
On March 9, 2021, the Company obtained a $73,007 term note issued under the Coronavirus Aid, Relief, and Economic Security Act’s Paycheck Protection Program (the “PPP”). The note bears an interest rate of 1% per annum, has a six-month deferral period with payments beginning the seventh month and all outstanding principal and interest is due within two years from the note’s inception date. All or a portion of the note may be forgiven in accordance with PPP requirements. No more than 25% of the amount forgiven can be attributable to non-payroll costs. As of December 31, 2021, a “Loan Forgiveness Application” was submitted to PNC Bank along with the requested documentation and subsequent to the close of the December 31, 2021, reporting period the note liability was reduced in its entirety.
On May 15, 2020, the Company obtained a $62,500 term note issued under the Coronavirus Aid, Relief, and Economic Security Act’s Paycheck Protection Program (the “PPP”). The note bears an interest rate of 1% per annum, has a six-month deferral period with payments beginning the seventh month and all outstanding principal and interest is due within two years from the note’s inception date. All or a portion of the note may be forgiven in accordance with PPP requirements. No more than 25% of the amount forgiven can be attributable to non-payroll costs. A “Loan Forgiveness Application” was submitted to PNC Bank along with the requested documentation and the note liability was reduced in its entirety during the year ended December 31, 2021.
| December 31, | December 31, | |||||||
| 2021 | 2020 | |||||||
| PPP Note May 15, 2020 | $ | 62,500 | $ | 62,500 | ||||
| PPP Note March 9, 2021 | 73,007 | - | ||||||
| Loan Forgiveness | (62,500 | ) | - | |||||
| Total | $ | 73,007 | $ | 62,500 | ||||
On December 28, 2021, the Company issued $500,000 note units, consisting of two $250,000 notes, for a total of $500,000 10% unsecured promissory notes with a maturity date of December 28, 2022, and 500,000 warrants exercisable at $1.00 per share with an expiry date of December 28, 2026, and fees of $5,075. The value of the warrants was determined using a computed volatility of 85.5%, 0% dividend rate, and a risk free interest rate of 1.27% and was applied as a discount on the notes payable as follows:
| December 31, | ||||
| 2021 | ||||
| Promissory notes issued on December 28, 2021 | $ | 500,000 | ||
| Less unamortized discount on notes payable | (408,979 | ) | ||
| Total | $ | 91,021 | ||
Note 7 – Stockholders’ Equity
Pursuant to the Company’s amended and restated articles of incorporation, the Company is authorized to issue 100,000,000 shares of common stock, with a par value of $0.00001 per share, and 20,000,000 shares of preferred stock, with a par value of $0.00001 per share.
Series A Preferred Shares
The Series A Preferred Stock, in accordance with its terms, is automatically convertible into a number of shares of the Company’s common stock upon the closing of the sale of shares of common stock to the public in a qualified offering (as set forth in the Series A certificate of designation) or upon listing of the Company’s common stock on a national securities exchange.
As of December 31, 2021, and 2020, the Company had 1,213 shares of Series A Preferred Stock issued and outstanding.
As of December 31, 2021, and 2020, the Company had accrued the 8.5% cumulative dividends on the Series A Preferred stock of $331,059 and $227,997 respectively.
Common Stock
As of December 31, 2021, and 2020, the Company had 9,312,152 and 9,291,526 shares of common stock issued and outstanding, respectively. The balance includes 20,626 and 21,530 shares of restricted stock issued in 2021 and 2020 respectively.
Common Stock to be Issued
On June 4, 2018, $120,250 outstanding convertible notes were converted to 6,182 shares of common stock of the Company at a price of $19.44 per share. The Company has recorded $16,340 of common stock to be issued for the accrued interest. As of December 31, 2020 and 2021, the common stock has not been issued.
Warrants
The Series A Preferred Stock sold in the private placement offerings, included warrants to be issued upon the earlier of a closing of the sale of shares of common stock to the public at a prices per share of at least $13.88 or in a firm commitment underwritten public offering pursuant to an effective registration statement resulting in gross proceeds of at least $15,000,000. The warrants shall be exercisable for a period of three years after the date of issuance. The warrant exercise price is contingent on the terms of the public offering. If an initial public offering occurs at a price at or above $13.88 per share, then the exercise price shall be set to the issuance price of the common stock with the number of warrants determined based on a 10% discount to the per share common stock issuance price. In the scenario where the common stock is listed with the common stock issuance price below $13.88, the exercise price will be set to $20.82 with the number of warrants based on a fixed conversion price of $12.49, which represents a 10.0% discount to the $13.88 threshold. The warrants also have contingent call features based on the terms of the public offering. If an initial public offering occurs at a price at or above $13.88, then the warrants are callable if the 20-day VWAP of the common stock in at or above 150% of the variable exercise price. In the scenario where the common stock is listed with a common stock issuance price below $13.88, then the warrants are callable if the 20-day VWAP of the common stock is at or above the $20.82 exercise price. The detachable warrants contained terms and features that gave rise to derivative liability classification.
Current accounting principles that are provided in ASC 815 - Derivatives and Hedging require derivative financial instruments to be classified in liabilities and carried at fair value with changes recorded in income. The Company has selected the Binomial Option Pricing valuation technique to fair value the compound embedded derivative. Inherent in a binomial options pricing model are assumptions related to expected stock-price volatility, expected life, risk-free interest rate, and dividend yield. The Company estimates the volatility of its ordinary shares based on historical volatility of comparable companies that matches the expected remaining life of the warrants. The risk-free interest rate is based on the U.S. Treasury zero-coupon yield curve on the grant date for a maturity similar to the expected remaining life of the warrants.
The derivative liability linked to the Series A Preferred Stock as of December 31, 2021 and 2021 was $94,025 and $673,171, respectively. The change in fair value of warrant liability was a gain of $579,146 as of December 31, 2021 and a loss of $256,580 as of December 31, 2020.
For the year ended December 31, 2021, and 2020, the estimated fair values of the liability measured on a recurring basis are as follows:
| December 31, | December 31, | |||||||
| 2021 | 2020 | |||||||
| Expected average volatility | 85.5 | % | 69.8 | % | ||||
| Dividend yield | 8.5 | % | 8.5 | % | ||||
| Expected life | 2.33 Years | 3.33 Years | ||||||
| Risk-free interest rate | 0.73 | % | 0.17 | % | ||||
A continuity schedule of the Series A Preferred Stock warrants is set forth below:
| Number of Warrants | Weighted Average Exercise Price | Weighted Average Life (years) | Intrinsic Value | |||||||||||||
| Outstanding, December 31, 2019 | 48,532 | $ | 24.98 | 4.33 | $ | - | ||||||||||
| Granted | - | - | - | |||||||||||||
| Forfeited | - | - | - | |||||||||||||
| Exercised | - | - | - | |||||||||||||
| Outstanding, December 31, 2020 | 48,532 | 24.98 | 3.33 | - | ||||||||||||
| Granted | - | - | - | |||||||||||||
| Forfeited | - | - | - | |||||||||||||
| Exercised | - | - | - | |||||||||||||
| Outstanding and Exercisable, December 31, 2021 | 48,532 | $ | 24.98 | 2.33 | $ | - | ||||||||||
Equity Incentive Plan
Our 2018 Equity Incentive Plan provides for equity incentives to be granted to our employees, executive officers or directors and to key advisers and consultants. Equity incentives may be in the form of stock options with an exercise price of not less than the fair market value of the underlying shares as determined pursuant to the 2018 Equity Incentive Plan, restricted stock awards, other stock-based awards, or any combination of the foregoing. The 2018 Equity Incentive Plan is administered by the Company’s compensation committee or, alternatively, if there is no compensation committee, the Company’s board of directors. We have reserved 3,000,000 shares of our common stock for issuance under the 2018 Equity Incentive Plan. As of December 31, 2021, 384,167 shares have been granted under the 2018 Equity Incentive Plan.
Restricted Stock Units. We may grant restricted stock units under our 2018 Plan. Restricted stock units are bookkeeping entries representing an amount equal to the fair market value of one share of our common stock. Subject to the provisions of our 2018 Plan, the administrator determines the terms and conditions of restricted stock units, including the vesting criteria and the form and timing of payment. The administrator, in its sole discretion, may pay earned restricted stock units in the form of cash, in shares or in some combination thereof. Notwithstanding the foregoing, the administrator, in its sole discretion, may accelerate the time at which any restrictions will lapse or be removed.
On August 16, 2019, five individuals were appointed to the Board of Directors of the Company to serve as directors. Each individual entered into an agreement pursuant to which they will serve as a director and pursuant to which they would each receive a grant of $75,000 worth of Restricted Stock Units (“RSUs”) issuable under the Company’s 2018 Equity Incentive Plan (the “2018 Plan”). The RSUs vest annually in one third increments from the date of appointment. Under the terms of the director agreements, the Company has also agreed to pay each director $25,000 per annum, payable in equal quarterly installments commencing 90 days following the Company becoming a publicly reporting company under the Securities Exchange Act of 1934, as amended.
During the years ended December 31, 2021 and 2020, pursuant to the agreements with directors and officers 20,626 and 21,530 RSUs were issued with a value of $490,067 and $459,589 included in wages, respectively.
As of December 31, 2021, there was $390,067 of total unrecognized compensation cost related to non-vested share-based compensation arrangements which is expected to be recognized within the next two years.
A continuity schedule of the Restricted Stock Units (RSU) follows:
| Number of RSU | Weighted Average Exercise Price | Weighted Average Life (years) | ||||||||||
| Outstanding, December 31, 2019 | 60,169 | $ | 24.02 | - | ||||||||
| Granted | 4,417 | 22.64 | - | |||||||||
| Forfeited | (2,702 | ) | 27.76 | - | ||||||||
| Exercised | - | - | - | |||||||||
| Outstanding, December 31, 2020 | 61,884 | 23.76 | 4.33 | |||||||||
| Granted | - | - | ||||||||||
| Forfeited | (2,702 | ) | 27.76 | - | ||||||||
| Exercised | - | - | - | |||||||||
| Outstanding, December 31, 2021 | 59,182 | $ | 23.57 | 3.33 | ||||||||
| Exercisable, December 31, 2021 | 43,056 | $ | 23.93 | 3.33 | ||||||||
Note 8 – Income Taxes (Restated)
The Company has not made provision for income taxes for the years ended December 31, 2021, since the Company has the benefit of net operating losses in these periods.
The reconciliation of income tax benefit at the U.S. statutory rate of 21% to the Company’s tax expense is as follows:
| December 31, | December 31, | |||||||
| 2021 | 2020 | |||||||
| (Restated) | ||||||||
| Federal tax benefit at statutory rate | $ | (241,948 | ) | $ | (21,111 | ) | ||
| State income tax benefit, net of federal tax effect | (95,051 | ) | (8,293 | ) | ||||
| Permanent differences | 19,381 | 19,381 | ||||||
| Change in valuation allowance | 317,618 | 10,023 | ||||||
| $ | - | $ | - | |||||
The principal components[ ] Units consisting of deferred tax assets consist
[ ] shares of Common Stock
[ ] Warrants to purchase Common Stock and
A total of 44% of the following:
| December 31, | December 31, | |||||||
| 2021 | 2020 | |||||||
(Restated) | ||||||||
| Deferred income tax asset: | ||||||||
| Net operation loss carry forwards | $ | 890,970 | $ | 587,699 | ||||
| Fixed assets | 7,479 | 8,110 | ||||||
| Intangibles | 7,788 | 7,788 | ||||||
| Interest | 45,574 | 30,596 | ||||||
| R&D tax credits | 87,801 | 87,801 | ||||||
| Total deferred income tax asset | 1,039,612 | 721,994 | ||||||
| Less: valuation allowance | (1,039,612 | ) | (721,994 | ) | ||||
| Total deferred income tax asset | $ | - | $ | - | ||||
The Company has approximately $3,000,000Equity Interest of net operating losses (“NOL”) carried forward to offset taxable income, if any, in future. In assessing the realization of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. Management considers the scheduled reversal of deferred tax liabilities, projected future taxable income and tax planning strategies in making this assessment. Based on the assessment, management has established a full valuation allowance against all of the deferred tax asset relating to NOLs for every period because it is more likely than not that all of the deferred tax asset will not be realized.
Note 9 – Restatement of Financial Statementsour subsidiary, Shuttle Diagnostics, Inc.
The Company’s financial statements as of and for the year ended December 31, 2021, contained an understatement of legal and professional expenses of $420,000 for the valuation of a total of 210,000 shares (105,000 shares on a post-split basis) of incentive stock transferred from a significant shareholder of the Company , who is also the wife of our Chairman and CEO, to a consultant for the Company in a private transaction.
Originally Reported | Restatement Adjustment ($) | As Restated ($) | ||||||||||
| Stockholders’ Equity | ||||||||||||
| Additional paid-in capital | 3,730,867 | 420,000 | 4,150,867 | |||||||||
| Accumulated deficit | (5,375,432 | ) | (420,000 | ) | (5,795,432 | ) | ||||||
Year Ended December 31, 2021
| Originally Reported ($) | Restatement Adjustment ($) | As Restated ($) | ||||||||||
| OPERATING EXPENSES | ||||||||||||
| Legal and professional | 264,684 | 420,000 | 684,684 | |||||||||
| Total operating expenses | 1,322,992 | 420,000 | 1,742,992 | |||||||||
| Loss from operations | (1,322,992 | ) | (420,000 | ) | (1,742,992 | ) | ||||||
| Loss before Income Taxes | (732,134 | ) | (420,000 | ) | (1,152,134 | ) | ||||||
| Net loss | (732,134 | ) | (420,000 | ) | (1,152,134 | ) | ||||||
| Net loss attributable to common stockholders | (835,196 | ) | (420,000 | ) | (1,255,196 | ) | ||||||
| Net loss per shares - basic and diluted | (0.08 | ) | (0.04 | ) | (0.12 | ) | ||||||
| Originally Reported ($) | Restatement Adjustment ($) | As Restated ($) | ||||||||||
| Cash Flows From Operating Activities: | ||||||||||||
| Net loss | (732,134 | ) | (420,000 | ) | (1,152,134 | ) | ||||||
| Stock-based compensation | 490,067 | 420,000 | 910,067 | |||||||||
Note 10 – Subsequent Events
Management evaluated all additional events subsequent to the balance sheet date through June 3, 2022, the date the financial statements were available to be issued, and determined the following items:
On January 22, 2022, the Company received notice of forgiveness for the PPP loan entered into on March 9, 2021 for $73,007 and $641 in interest that reduced the note liability in its entirety.
On February 8, 2022, and March 11, 2022, the Company sold $365,000 and $224,985, respectively, in 6% convertible notes (the “Notes”), which bear 6% interest, are repayable three years from the date of issuance, and will convert automatically into shares of common stock (the “Conversion Shares”) at a conversion price of $2.50 per share upon closing of this offering. Boustead Securities LLC acted as placement agent for this offering and received compensation of $36,500 and $22,250, respectively, and warrants to purchase shares of common stock equal to 10% of the Conversion Shares, exercisable at the conversion price of the Convertible Notes.
Effective March 30, 2022, the Company issued 1,678 shares (839 shares post-reverse split) of common stock to correct an issuance error on completion of the 2018 share exchange which is valued at $16,340 common stock to be issued.
Effective April 1, 2022, pursuant to the consent of the Company’s board of directors and a majority of its common stockholders, the Company effectuated a two-for-one reverse split, pursuant to which each stockholder received one share of common stock for every two shares held.
On April 6, 2022, the Company submitted the final invoice for “Topic 345: Predictive Biomarkers of Prostate Cancer Patient Sensitivity for Radiation Late Effects,” for $211,455, following the completion of the Final Quarterly Progress Report to NIH covering the performance period of 9/16/2019-3/15/2022. The invoice was paid in full on April 27, 2022.
$9,960,340
1,225,888Units
Each Unit Consisting of One Share of Common Stock and
One Warrant to Purchase One Share of Common Stock
SHUTTLE PHARMACEUTICALS HOLDINGS, INC.


_______________
PROSPECTUS
______________
Boustead Securities, LLC
______________
_______, 2022
Through and including , 2022 (the 25th day after the date of this prospectus), all dealers effecting transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to a dealer’s obligation to deliver a prospectus when acting as an underwriter and with respect to an unsold allotment or subscription.
The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell and is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
SUBJECT TO COMPLETION, PRELIMINARY PROSPECTUS DATED AUGUST 23, 2022

SHUTTLE PHARMACEUTICALS HOLDINGS, INC.
1,311,942shares of common stock
This prospectus relates to the resale of 1,311,942 shares of common stock, par value $0.00001 per share of the Company, which includes (i) 496,942 shares of common stock, (ii) 147,500 shares of common stock and warrants to purchase 147,500 shares of common stock issuable upon conversion of certain convertible notes and (iii) exercise of warrants to purchase shares of common stock held by the selling stockholder named in this prospectus.
Prior to this offering, there has been no public market for our common stock. We have reserved the symbol “SHPH” for purposes of listing our Common Stock on The Nasdaq Capital Market and have applied to list our shares on Nasdaq. There is no guarantee or assurance that our shares of common stock will be approved for listing on Nasdaq. We will not receive any proceeds from the sale of shares by the selling stockholder.
Any shares sold by the selling stockholders until our common stock is listed or quoted on an established public trading market will take place at $8.125, which is the per share public offering price we are selling in our initial public offering. Thereafter, any sales will occur at prevailing market prices or in privately negotiated prices. The distribution of securities offered hereby may be effected in one or more transactions that may take place in ordinary brokers’ transactions, privately negotiated transactions or through sales to one or more dealers for resale of such securities as principals. Usual and customary or specifically negotiated brokerage fees or commissions may be paid by the selling shareholders. No sales of the shares covered by this prospectus shall occur until the common stock sold in our initial public offering begins trading on Nasdaq.
On ______, 2022, a registration statement under the Securities Act of 1933, as amended (the “Securities Act”) with respect to our initial public offering of shares of our common stock was declared effective by the U.S. Securities and Exchange Commission. We received approximately $___ million in net proceeds from the offering (assuming no exercise of the underwriters’ over-allotment option) after payment of underwriting discounts and commissions and estimated expenses of the offering.
Investing in our common stock involves a high degree of risk, including the risk of losing your entire investment. See “Risk Factors” beginning on page 13 of the primary offering prospectus contained in this Registration Statement to read about factors you should consider before buying our common stock.
Neither the Securities and Exchange Commission nor any state securities commission nor any other regulatory body has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this prospectus is , 2022
RESALE PROSPECTUS SUMMARY
The following summary provides an overview of all material information contained in this prospectus. It does not contain all of the information you should consider before making a decision to purchase our shares of common stock in the offering. Prior to investing in our common stock, you should carefully and thoroughly read the more detailed information in this prospectus and review our financial statements and all other information that is included in this prospectus, including the section entitled “Risk Factors” beginning at page 13.
Unless the context otherwise requires, references in this prospectus to “Shuttle Pharma,” “Shuttle Pharmaceuticals,” “the Company,” “we,” “our” and “us” refers to Shuttle Pharmaceuticals Holdings, Inc. and its subsidiary, Shuttle Pharmaceuticals, Inc.
Our Company
Overview
Founded in 2012 by faculty members of the Georgetown University Medical Center, Shuttle Pharmaceuticals is a discovery and development stage specialty pharmaceutical company focused on improving the outcomes of cancer patients treated with radiation therapy (RT). Our mission is to improve the lives of cancer patients by developing therapies that are designed to maximize the effectiveness of RT while limiting the late effects of radiation in cancer treatment. Although RT is a proven modality for treating cancers, by developing radiation sensitizers, we aim to increase cancer cure rates, prolong patient survival and improve quality of life when used as a primary treatment, or in combination with surgery, chemotherapy and immunotherapy. We currently have no FDA approved products and we have not yet applied for a new drug application. To date, we have been funded by investments from private investors and government contracts obtained from the National Institutes of Health (NIH) for performing research. We have no product revenue and our independent auditors, in their report dated June 3, 2022, expressed doubt about our ability to continue as a going concern.
Historically, the major advances in radiation oncology have focused on improving technology to increase the amount of radiation that can be administered to a tumor without damaging adjacent, normal tissues. Examples of other such technologies include intensity modulated radiation therapy (IMRT), stereotactic body radiation therapy (SBRT), stereotactic radiosurgery (SRS) and proton therapy – the backbones of state-of-the-art RT. All offer improvements in physical radiation dose shaping. The basic principle underlying the effectiveness of RT for curing cancers lies in the differential cancer cell kill achieved in tumors, as compared to the effects of RT on the normal surrounding tissues, which is achieved by delivery of highly conformal RT doses – in other words, delivery of high-dose to volumes that are shaped to conform to the target cancers while minimizing the dose to surrounding normal tissues. The treated volumes frequently include sensitive normal tissues, thereby limiting the magnitudes of the prescribed RT doses. We suggest that technological innovations to define tumor volumes and shape radiation delivery have reached an effectiveness plateau and that further improvements in RT outcomes will require pharmacological and immunological approaches to sensitize cancers, protect normal tissues and engage the immune system.
At present, the drugs being used for sensitizing cancers to RT are chemotherapeutic agents possessing radiation sensitizing properties as secondary effects. With the exception of Cituximab, a growth factor targeting monoclonal antibody biologic, all other drugs used as radiation sensitizers are used “off-label” to address the clinical need for radiation sensitizers. For example, certain chemotherapeutic agents, such as 5-fluorouracil, capecitabine and cis-platinum, are approved as single agents for cancer treatment, but are used “off-label” as radiation sensitizers in combination with RT. Treatments with such agents are associated with inherent toxicities associated with the drug’s primary, single-agent mechanisms of action.
Shuttle Pharma’s platform of sensitizers offers a pipeline of product candidates designed to address the urgent clinical need and the current limitations of using “off-label” drugs with potential new sensitizer agents. Our pipeline includes Ropidoxuridine, our lead clinical sensitizer drug candidate, to sensitize rapidly growing cancer cells and selective histone deacetylase (HDAC) inhibitors to sensitize cancer cells and stimulate the immune system. Our novel technologies will be tested in combinations with radiation therapies (conventional X-ray and proton radiation therapies) and in combinations with immune-therapies. To date, Ropidoxuridine has completed a Phase I clinical trial. Our HDAC inhibitor platform drug candidates have been tested in preclinical models of solid tumor cancers. Ropidoxuridine and the selective HDAC6 inhibitor SP-2-225 are the clinical and preclinical candidate drug products we propose to develop using funding from this offering.
Our intellectual property for Ropidoxuridine includes novel formulations that show improved drug bioavailability (in a preclinical animal model) and for sensitizing cancers to proton and to conventional radiation therapies. Our HDAC inhibitor intellectual property includes new patent applications and granted patents for composition of matter and methods of use for treating cancers with HDAC inhibitors in combinations with radiation therapy.
To date, we have obtained funding for our research from private investors and Small Business Innovation Research (“SBIR”) contracts obtained through the National Institutes of Health (“NIH”) to support the development of the radiation sensitizer Ropidoxuridine in a Phase I clinical trial. We have also received awards for Phase I and II SBIR contracts for development of human cell cultures for health disparities studies and predictive biomarkers of radiation late effects through the NIH’s National Cancer Institute. The completed Phase I and II funded discovery work performed to establish “Cell-based Models for Prostate Cancer Health Disparity Research” and to develop “Predictive Biomarkers of Prostate Cancer Sensitivity for Radiation Late Effects” enables Shuttle Pharma to apply for NIH SBIR Phase IIb funding to develop these products for advancing basic science and clinical research.
Our Product Candidates
The U.S. Food and Drug Administration (the “FDA”) considers new molecular entities as drugs that use new and unique mechanisms of action for treating medical conditions. Our clinical stage agent, Ropidoxuridine (IPdR), increases DNA double strand breaks following radiation exposure and our inhibitors of histone deacetylases (HDACs) stimulate the immune system to produce T-lymphocytes targeting cancer cells.
Our objective is to improve the outcomes of cancer treatment through RT while reducing its side effects by:
To our knowledge, no drug utilizing the mechanisms of our candidate small molecule drugs has received FDA approval as a radiation sensitizer. We have developed, to clinical stage, the small molecule strategies to sensitize growing cancer cells in tumors to conventional RT and to large fraction radiation therapy. The pre-clinical technology, HDAC inhibitor platform, is designed to target cancer cells while protecting healthy tissue/normal cells, thus enhancing the candidate radiation sensitizer product pipeline. The selective HDAC6 inhibitor (SP-2-225), discovered and developed by our scientists, has inhibited the growth of melanoma tumors and breast cancers in animal models by an immune stimulating mechanism.
We are focused on developing a clinical stage product candidate (Ropidoxuridine) and a pre-clinical product candidate, selective HDAC6 inhibitor (SP-2-225). We propose to develop these drug candidates as illustrated below:
Overview of Radiation Sensitizer Development
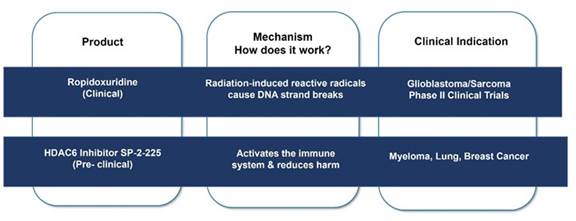
Ropidoxuridine, the clinical stage molecule, sensitizes rapidly growing cancers to radiation therapy by increasing reactive free radicals that increase DNA strand breaks. Ropidoxuridine development for treating glioblastoma will require Phase II clinical testing for use in treating brain tumors. The selective HDAC6 inhibitor, SP-2-225, a pre-clinical stage molecule, activates the innate immune system to target irradiated tumor cells by immune mechanisms.
Ropidoxuridine (IPdR)
Ropidoxuridine (IPdR) is an orally available halogenated pyrimidine (5-iodo-2-pyrimidinone-2-deoxyribose) with strong cancer radiation sensitizing properties. As a prodrug that does not become an active drug until after it is metabolized, IPdR is absorbed and metabolized to IUdR by enzymes in the liver and in cancer cells. IUdR, a halogenated pyrimidine, is incorporated into DNA by rapidly growing cancer cells. Cells that incorporate IUdR into their DNA then become more sensitive to the effects of RT. The Phase I clinical trial of Ropidoxuridine and RT, supported by an NIH SBIR contract to Shuttle Pharma, was sub-contracted to the Brown University Oncology Group (BrUOG) at the LifeSpan/Rhode Island Hospital. This Phase I clinical trial has been completed and the results were initially reported by the sub-contractor at the 30th EORTC-NCI-AACR Symposium in November 2018 and published in the medical journal Clinical Cancer Research in 2019. A maximum tolerated dose (MTD) of 1200 mg/day for 28 days was established for use in combination with radiation therapy to achieve therapeutic blood levels of IUdR.
The reported Phase I clinical trial of Ropidoxuridine in combination with RT provides the foundation for proposed Phase II clinical trials to establish the data necessary for the FDA to determine efficacy in treating brain tumors, sarcomas and pancreatic cancers, diseases that offer potential for orphan designations. The FDA granted approval of our application for orphan-drug designation for IPdR for the treatment of glioblastoma. Orphan designation protects the marketing position of Ropidoxuridine for up to seven years after marketing approval is received from the FDA. This approval integrates well into the overall intellectual property strategy for Ropidoxuridine which includes filed patent applications for “Method and Compositions for Cancer Therapies that Include Delivery of Halogenated Thymidines and Thymidine Phosphorylase Inhibitors in Combination with Radiation.” We believe that we are positioned to initiate Phase II clinical studies with Ropidoxuridine and RT in 2022.
Extended Bio-availability Ropidoxuridine (IPdR/TPI)
Ropidoxuridine and Tipiracil (IPdR/TPI) is a new combination drug formulation designed to increase the bio-availability and incorporation of IUdR into DNA. Shuttle Pharma’s preclinical studies of the combination of IPdR/TPI have shown up to 10-fold greater bioavailability of the active metabolite (IUdR) as compared to IPdR administered alone in controls. We have filed an application under the Patent Cooperation Treaty (or PCT) for the intellectual property. This new formulation will be tested in a Phase I clinical trial as a sensitizer of rectal cancers. Another nucleoside analogue, Trifluridine has been formulated in combination with Tipiracil (TAS-102) to enhance drug uptake by colon cancer cells to prolong survival in patients treated for metastatic colorectal cancers, as has been reported in the New England Journal of Medicine (N Engl J Med. 2015; 372:1909-1919). We anticipate testing for uptake of IPdR by colorectal cancer cells following administration of the IPdR/TPI drug formulation.
Proton radiation therapy is an advanced form of radiation therapy using charged proton particles (p+). Proton RT differs from conventional RT in that the radiation is delivered by a beam of protons to precisely target tumors and, due to the favorable physics of energy deposition by proton particles, there is no exit beam, resulting in less radiation to surrounding healthy tissues. The use of Proton RT is expanding rapidly in the U.S. and worldwide. According to the National Association of Proton Therapy, more than 30 facilities are currently in operation in the U.S. and an additional 30 facilities are planned for installation over the next five years. (See www.proton-therapy.org.) Much attention has been paid to proton therapy in the popular press, promoting its advantages, as well as addressing the increased health care costs. The role of a sensitizer that offers proton radiation sensitization presents an opportunity to enhance the value of proton radiation therapy as a cancer treatment modality. We believe the development of a proton therapy targeted radiation sensitizer, such as IPdR/TPI, is timely and consistent with current market needs to advance protons as a therapeutic modality.
We intend to perform clinical studies to support the development of the IPdR/TPI combination to advance this drug candidate with proton RT. The addressable market includes diseases such as brain tumors, cancers of the head and neck, GI cancers and lung cancers.
Selective HDAC Inhibitors
The roles of acetylation in the epigenetic regulation of chromatin structure and gene expression rests on the balance of activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs). Increased acetylation of histones leads to changes in chromatin structure and accessibility for key cellular proteins to specific target sites. Acetylations of non-histone proteins also modulates their enzymatic activities. We have discovered novel HDAC inhibitor molecules and testing in preclinical models has shown cancer radiation sensitizing properties, normal tissue protective properties and selective HDAC6 inhibitory properties. Our HDAC inhibitor platform, described below, will be evaluated in pre-clinical studies of radiation sensitization of solid tumors and activation of the immune response to irradiated cancer cells.
Drug Development Projects for Radiation Treatment of Cancers
To advance research that is complementary to our radiation sensitizer discovery and development projects, the NIH has awarded SBIR contracts to us to develop reagents for health disparities research and to develop biomarkers of radiation sensitivity for patients treated with radiation therapy. Our scientists have been engaged in developing model human cell systems for testing radiation sensitizers in tissue cultures. This project provides an efficient and low-cost screening technology to provide data for the FDA’s determination of drug efficacy and to identify candidate lead molecules for treating prostate cancers in African-Americans. First developed at Georgetown University, the conditional cellular reprogramming (CRC) technology offers the ability to establish new cell lines from biopsies of cancers. We have obtained a sub-license from Propagenix, Inc. to establish 100 normal and cancer cell lines from prostate biopsy samples for use in screening drug candidates and for health disparities research. A more detailed description of the Propagenix license is set forth on page 60 below.
In addition, to identify patients who may be more sensitive to radiation therapy and are at a higher risk to suffer treatment-related complications, collaborative research with Georgetown University has led to discovery of metabolite biomarkers, which are predictive of patient responses to radiation therapy. A patent for the intellectual property has been submitted by Georgetown University with Shuttle Pharma scientist (Scott Grindrod, PhD) as co-inventor. Developmental work in health disparities and predictive biomarker development has been supported by NIH SBIR contracts to Shuttle Pharmaceuticals for the following areas:
The NIH’s SBIR program is designed to encourage small businesses to engage in Federal Research/Research and Development (“R/R&D”) that has the potential for commercialization. Shuttle Pharma will apply for additional NIH SBIR grants and contracts to fund advancement of these projects.
Market Opportunity
The American Cancer Society (Cancer Facts & Figures 2020) estimates 1,806,590 new cancer cases and 606,520 cancer deaths each year in the United States and, according to the American Society for Radiation Oncology, more than 50% of patients undergo RT at some point in the treatment of their diseases. RT is used to treat cancers of the lung, breast, brain, esophagus, pancreas, rectum, head and neck, uterus, lymphomas and sarcomas. At present, we are developing drug candidates to address brain, pancreas, rectum, sarcomas and lymphomas, although we may test and seek approval for our drug candidates to treat other cancers in the future.
Currently, there is only one drug (the monoclonal antibody, Cetuximab) that has received FDA approval for the radiation sensitizer indication. Cetuximab is a recombinant monoclonal antibody that binds to epidermal growth factor receptor (EGFR) and inhibits the binding of epidermal growth factor (EGF). Cetuximab is administered via intravenous infusion and is used as monotherapy or in combination with other chemotherapies or radiation therapy. In clinical trials, cetuximab was associated with serious and fatal infusion reactions, cardiopulmonary arrest or sudden death, and serious dermatologic toxicities, toxicities that have created deterrents to its use as a radiation sensitizer. Present treatment utilizes “off-label” small molecule drugs, which are cytotoxic chemotherapy agents that also sensitize, but do not have radiation sensitization as an FDA approved indication. Moreover, since “off-label” drugs are cytotoxic, they are often associated with intrinsic acute and chronic side effects. Nevertheless, these drugs have shown clinically significant improvements in disease control and survival and are typically included in standard-of-care treatment recommendations for patients with cancers of the head and neck, brain, lung, esophagus, stomach, pancreas, liver, bladder, lymphomas and sarcomas. As a result, we believe that there is a significant market opportunity for our product candidates. Based on cancer incidence data published by the American Cancer Society, we have estimated the numbers of patients presenting with local/regional disease, suitable for treatment with RT.
Estimated RT Cases by Disease Site
| Cancer Type | Cases Diagnosed Annually | Estimated RT Cases | ||||||
| Brain | 23,890 | 21,979 | ||||||
| Pancreas | 57,600 | 32,832 | ||||||
| Sarcomas | 13,130 | 4,000 | ||||||
| Rectum | 43,340 | 26,437 | ||||||
Annual cancer cases for each disease site are estimated from American Cancer Society Facts & Figures 2020 publication. The fraction of patients optimally receiving RT for each disease site were obtained from published estimates of Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer. 2005 Sep 15;104(6):1129-37. doi: 10.1002/cncr.21324. The Estimated RT cases were obtained by multiplying Cases Diagnosed Annually by the fraction receiving RT for optimal utilization.
Our Development Strategy
Our goal is to maintain and build upon our leadership position in radiation sensitization. We plan to develop Ropidoxuridine and the HDAC6 inhibitor (SP-2-225) and, if approved by the FDA, to commercialize our product candidates for the treatment of cancers. While this process may require years to complete, we believe achieving this goal could result in radiation sensitizer and immunotherapy products for cancer treatment. Key elements of our strategy include:
We propose the following clinical development plan to identify, develop and commercialize drugs for use in cancer treatments in combination with RT:
Develop Ropidoxuridine (IPdR) for Orphan disease indications to take to market
Develop Ropidoxuridine and tipiracil (IPdR/TPI) for colorectal cancer indications to take to market
Develop HDAC Inhibitors for use in breast cancer for immune activation after RT
Management Team
Our management team has significant experience in radiation oncology and in progressing products from early-stage research through clinical trials. Our CEO, Anatoly Dritschilo, M.D., is an experienced clinician and researcher who has held senior academic and management positions including serving as Department Chairman, Hospital Medical Director and Cancer Center Director at Georgetown University Medical Center. Prior to co-founding our company, Dr. Dritschilo was a co-founder of Oncomed, Inc., a company that became public as NeoPharm, Inc. (Nasdaq: NEOL). He has experience in providing care for patients undergoing treatment for cancers of the prostate, breast, brain, lung, sarcomas and GI systems. Dr. Dritschilo has directed basic science research supported by grants from the National Cancer Institute (“NCI”) and performed clinical trials using drugs and radiation therapy. In addition, Dr. Dritschilo served as the principal investigator of pharmaceutical industry sponsored clinical evaluations of human interferon alpha-2 (Bristol-Myers) with radiation therapy and antisense raf oligonucleotides, LErafAON (NeoPharm) with radiation therapy. He serves as a Radiation Biology and Radiation Oncology expert on committees of the NIH to review Program Project (P01) grant applications, Specialized Program of Research Excellence (SPORE) grant applications and investigator-initiated research project (R01) applications.
Dr. Dritschilo is supported in our clinical development effort by Tyvin Rich, MD, our Chief Clinical Officer and Medical Director. Dr. Rich is the former Professor and Chairman of the Department of Therapeutic Radiology and Oncology at the University of Virginia Health Sciences Center and proton radiation therapy specialist at the Hampton Proton Therapy Center in Hampton, Virginia. Dr. Rich has served as principal investigator on multi-modality clinical trials for the treatment of gastrointestinal (GI) cancers and helped to develop treatment with 5-fluorouracil (5-FU) as a radiation sensitizer for use with RT in the treatment of GI cancers. He has extensive cancer clinical trial experience in developing radiation sensitizer applications through his participation in the Radiation Therapy Oncology Group (RTOG). Dr. Rich is a co-inventor with scientists at the University of Virginia of the Proton Activated Atomic Medicine technology.
Our administrative services are provided by Peter Dritschilo, MBA, who has served as our President and COO since 2012. Mr. Dritschilo’s experience in hospital administration and management of medical oncology clinical services and radiation therapy facilities, including management of day-to-day operations, human resources and financial oversight. Peter Dritschilo is the son of our Chairman and CEO, Dr. Anatoly Dritschilo. The addition of Michael Vander Hoek as our CFO and Vice President for Operations and Regulatory expands our capability to provide the level of management needed for the proposed expansion of clinical trials. Mr. Vander Hoek has served as administrative director of the Lombardi Comprehensive Cancer Center (LCC) for the past 12 years and has extensive experience in negotiations, management and supervision of Contract Research Organizations (CROs) and research contracts in general. As the administrative director of the Lombardi Comprehensive Cancer Center, Mr. Vander Hoek also served as the chief financial officer. Taken together, we believe our leadership team of highly qualified specialists will help us achieve the proposed milestones for the development of radiation sensitizer products.
Pre-IPO Bridge Financing
In December 2021, we completed a $500,000 note offering pursuant to which we sold to two accredited investors (the “Investors”) units consisting of (i) a $250,000 promissory note bearing 10% interest, repayable at the time of this offering (the “Note”) and (ii) a warrant to purchase 250,000 shares (the “Warrant Shares”) of our common stock at an exercise price of $1.00 per share (the “Warrant”). Immediately before closing on the Offering, the Notes will be repaid and cancelled in exchange for the exercise of the Warrants and issuance of the 500,000 Warrant Shares to the Investors.
In February and March 2022, we sold a total of $365,000 and $225,000, respectively, in convertible notes (“Convertible Notes”) to certain accredited investors, which notes will automatically convert upon effectiveness of this offering at a conversion price of 50% of the per unit offering price (the “Conversion Units”). In addition, each of the Convertible Note holders will be entitled to demand registration rights, and we are required to register the shares underlying the Convertible Notes within 180 days of the closing of this offering. We are using the net proceeds from the Convertible Notes offering to expand our current operations, including our technology and intellectual property portfolio, and to fund the costs of this offering. Any funds remaining will be used for working capital and other general corporate purposes.
In August 2022, we completed a $125,000 note and warrant offering pursuant to which we sold to three accredited investors (the “Investors”) units consisting in total of (i) $125,000 in promissory notes bearing 10% interest, repayable at the time of this offering (the “Note”) and (ii) warrant to purchase a total of 50,000 shares (the “Warrant Shares”) of our common stock at an exercise price of $2.50 per share (the “Warrants”).
Boustead Securities LLC (“Boustead”) acted as placement agent in both the Note and Warrant offering and the Convertible Notes offering, pursuant to which Boustead waived its cash compensation related to the Note and Warrant offering and received cash compensation of $36,500 and $22,250 in each of the February and March 2022 closings, respectively, and also received (i) warrants to purchase 5,000 shares of common stock, exercisable at $2.50 per share, and (ii) warrants to purchase 10% of the total number of Conversion Shares, exercisable at the conversion price of the Convertible Notes.
Summary Risk Factors
Our business is subject to a number of risks you should be aware of before making an investment decision. These risks are discussed more fully in the “Risk Factors” section of this prospectus at page 13 immediately following this prospectus summary. These risks include the following:
Implications of Being an Emerging Growth Company
We may take advantage of the above provisions for up to five years or until such earlier time that we are no longer an emerging growth company. We would cease to be an emerging growth company if we have more than $1.0 billion in annual revenues, have more than $700 million in market value of our capital stock held by non-affiliates or issue more than $1.0 billion of non-convertible debt over a three-year period. We may also choose to take advantage of some, but not all, of the available exemptions. We have taken advantage of some reduced reporting requirements in this prospectus. Accordingly, the information contained in this prospectus may be different than the information you receive from other public companies in which you hold stock.
In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This provision allows an emerging growth company to delay the adoption of some accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this extended transition period for adopting new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
Corporate Information
The Company was formed as a limited liability company in the state of Maryland in December 2012 and was converted to a C corporation, Shuttle Pharmaceuticals, Inc. (“Shuttle”), in August of 2016. In June 2018, Shuttle completed a share exchange with Shuttle Pharma Acquisition Corp. Inc. (“Acquisition Corp.”), pursuant to which Shuttle Pharmaceuticals, Inc. became a subsidiary of Acquisition Corp. and we subsequently changed the name of Acquisition Corp. to Shuttle Pharmaceuticals Holdings, Inc. Our executive offices are located at 1 Research Court, Suite 450, Rockville, Maryland 20850 and our telephone number is (240) 403-4212. Our corporate website is www.shuttlepharma.com. Information appearing on our corporate website is not incorporated as part of this prospectus.
THE RESALE OFFERING
USE OF PROCEEDS
We will not receive any of the proceeds from the sale of the common stock held by the selling stockholders named in this prospectus.
SELLING STOCKHOLDERS
The following table sets forth certain information with respect to the selling stockholders’ beneficial ownership of shares of common stock as of the date of this prospectus. Although there was no agreement between the Company and the stockholders to register these shares, the Company believes the registration of these shares is beneficial to the Company.
Percentage of beneficial ownership before this offering is based on 9,312,991 shares of our common stock outstanding as of August 15, 2022. We have determined beneficial ownership in accordance with the rules of the SEC. Beneficial ownership is based on information furnished by the selling stockholder. Unless otherwise indicated, the selling stockholder named in the following table has, to our knowledge, sole voting and investment power with respect to the shares it beneficially owns.
The selling stockholders may offer for sale from time to time any or all of the shares. The table below assumes that the selling stockholders will sell all of the shares offered for sale hereby. The selling stockholders, however, are under no obligation to sell any shares pursuant to this prospectus.
| Names and addresses | Number of shares of common stock beneficially owned before the Offering(#) | Number of shares being registered in the Offering | Number of shares of common stock beneficially owned after Offering | Percentage of class of common stock beneficially owned after Offering(*) | ||||||||||||
| Steven Bayern | 355,000 | (1) | 250,000 | 105,000 | 0.1 | % | ||||||||||
| Rui Wu | 250,000 | (2) | 250,000 | - | - | |||||||||||
| BaseStones, Inc. | 20,000 | (3) | 20,000 | - | - | |||||||||||
| Yuki Namamura | 10,000 | (3) | 10,000 | - | - | |||||||||||
| Gene Jung | 356,942 | 356,942 | - | - | ||||||||||||
| Lisa Dritschilo | 130,000 | 130,000 | ||||||||||||||
| Chris Etherington | 12,500 | (4) | 12,500 | - | - | |||||||||||
| Mel Feldman | 12,500 | (4) | 12,500 | - | - | |||||||||||
| Robert Frome (5) | 12,500 | (4) | 12,500 | - | - | |||||||||||
| Robert Gutman | 20,000 | (4) | 20,000 | - | - | |||||||||||
| One10 Food Sciences Pte. Ltd. | 50,000 | (4) | 50,000 | - | - | |||||||||||
| Jerry Katzman | 12,500 | (4) | 12,500 | - | - | |||||||||||
| John & Mary Lynch | 12,500 | (4) | 12,500 | - | - | |||||||||||
| Lawrence Shields | 37,500 | (4) | 37,500 | - | - | |||||||||||
| Kenneth Zuckerbrot | 12,500 | (4) | 12,500 | - | - | |||||||||||
| Dan Montzka | 50,000 | (4) | 50,000 | - | - | |||||||||||
| Daniel Hutt | 12,500 | (4) | 12,500 | - | - | |||||||||||
| Frank Bucciero | 12,500 | (4) | 12,500 | - | - | |||||||||||
| Tanya White | 12,500 | (4) | 12,500 | - | - | |||||||||||
| Lanier W. Moore II | 12,500 | (4) | 12,500 | - | - | |||||||||||
| Varkes Churukian | 12,500 | (4) | 12,500 | - | - | |||||||||||
SELLING STOCKHOLDER PLAN OF DISTRIBUTION
Since there is currently no public market established for our securities, the selling stockholders have represented to the Company that they will not offer or sell shares prior to the closing of the primary offering and listing of our common stock on the Nasdaq Capital Market (“Nasdaq”). After the primary offering closes, our common stock is listed on Nasdaq and there is an established market for these resale shares, the selling stockholder may sell the resale shares from time to time at the market price prevailing on the Nasdaq Capital Market at the time of offer and sale, or at prices related to such prevailing market prices or in negotiated transactions or a combination of such methods of sale directly or through brokers.
The selling stockholders may use any one or more of the following methods when disposing of shares or interests therein:
The selling stockholder may, from time to time, pledge or grant a security interest in some or all of the shares of common stock owned by them and, if they default in the performance of their secured obligations, the pledgees or secured parties may offer and sell the shares of common stock from time to time, under this prospectus, or under an amendment to this prospectus under Rule 424(b)(3) or other applicable provision of the Securities Act amending the list of selling stockholder to include the pledgee, transferee or other successors in interest as selling stockholder under this prospectus. The selling stockholder also may transfer the securities in other circumstances, in which case the transferees, pledgees or other successors in interest will be the selling beneficial owners for purposes of this prospectus.
In connection with the sale of our common stock or interests therein, the selling stockholder may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the common stock and deliver these securities to close out their short positions, or loan or pledge the common stock to broker-dealers that in turn may sell these securities. The selling stockholder may also enter into option or other transactions with broker-dealers or other financial institutions or the creation of one or more derivative securities which require the delivery to such broker-dealer or other financial institution of shares offered by this prospectus, which shares such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).
The aggregate proceeds to the selling stockholder from the sale of the common stock offered by them will be the purchase price of the common stock less discounts or commissions, if any. The selling stockholder reserves the right to accept and, together with their agents from time to time, to reject, in whole or in part, any proposed purchase of common stock to be made directly or through agents. We will not receive any of the proceeds from this offering.
Broker-dealers engaged by the selling stockholders may arrange for other broker-dealers to participate in sales. Broker-dealers may receive commissions or discounts from the selling stockholders (or, if any broker-dealer acts as agent for the purchase of shares, from the purchaser) in amounts to be negotiated. The selling stockholders do not expect these commissions and discounts to exceed what is customary in the types of transactions involved, and in no case will the maximum compensation received by any broker-dealer exceed seven percent (7%).
The selling stockholders also may resell all or a portion of the shares in open market transactions in reliance upon Rule 144 under the Securities Act, provided that they meet the criteria and conform to the requirements of that rule.
Any underwriters, agents, or broker-dealers, and any selling stockholders who are affiliates of broker-dealers, that participate in the sale of the Common Stock or interests therein may be “underwriters” within the meaning of Section 2(11) of the Securities Act. Any discounts, commissions, concessions or profit they earn on any resale of the shares may be underwriting discounts and commissions under the Securities Act. The selling stockholders who are “underwriters” within the meaning of Section 2(11) of the Securities Act will be subject to the prospectus delivery requirements of the Securities Act. We know of no existing arrangements between any of the selling stockholder and any other stockholder, broker, dealer, underwriter, or agent relating to the sale or distribution of the shares, nor can we presently estimate the amount, if any, of such compensation. See “Selling Stockholder” for description of any material relationship that a shareholder has with us and the description of such relationship.
To the extent required, shares of our common stock to be sold, the name of the selling stockholder, the respective purchase prices and public offering prices, the names of any agents, dealer or underwriter, any applicable commissions or discounts with respect to a particular offer will be set forth in an accompanying prospectus supplement or, if appropriate, a post-effective amendment to the registration statement that includes this prospectus.
In order to comply with the securities laws of some states, if applicable, the shares of common stock may be sold in these jurisdictions only through registered or licensed brokers or dealers. In addition, in some states the shares of common stock may not be sold unless it has been registered or qualified for sale or an exemption from registration or qualification requirements is available and is complied with.
We have advised the selling stockholder that the anti-manipulation rules of Regulation M under the Exchange Act may apply to sales of shares in the market and to the activities of the selling stockholder and its affiliates. In addition, we will make copies of this prospectus (as it may be supplemented or amended from time to time) available to the selling stockholder for the purpose of satisfying the prospectus delivery requirements of the Securities Act. The selling stockholder may indemnify any broker-dealer that participates in transactions involving the sale of the shares against certain liabilities, including liabilities arising under the Securities Act.
LEGAL MATTERS
Certain legal matters in connection with this offering with respect to the United States federal securities law and New York law will be passed upon for us by Michelman & Robinson LLP, Los Angeles, California and New York, New York.
1,311,942 shares of Common Stock

SHUTTLE PHARMACEUTICALS HOLDINGS, INC.
RESALE PROSPECTUS[ ], 2024
You should rely only on the information contained in this Resale Prospectus. No dealer, salesperson or other person is authorized to give information that is not contained in this Resale Prospectus. This Resale Prospectus is not an offer to sell nor is it seeking an offer to buy these securities in any jurisdiction where the offer or sale is not permitted. The information contained in this Resale Prospectus is correct only as of the date of this prospectus, regardless of the time of the delivery of this prospectus or the sale of these securities.
The date of this Resale Prospectus is , 2022
PART II
INFORMATION NOT REQUIRED IN THE PROSPECTUS
| ITEM 13. | OTHER EXPENSES OF ISSUANCE AND DISTRIBUTION |
| Financial Printing Fees | $ | 5,000 | ||
| SEC Filing Fees | $ | 1,711 | ||
| Nasdaq Application Fees | $ | 75,000 | ||
| FINRA Expenses | $ | 2,225 | ||
| Transfer Agent Fees | $ | 21,000 | ||
| Accounting Fees and Expenses | $ | 45,000 | ||
| Legal Fees and Expenses | $ | 350,000 | ||
| Miscellaneous Fees and Expenses | $ | - | ||
| Total | $ | 499,936 |
| Financial Printing Fees | $ | * | ||
| SEC Filing Fees | $ | * | ||
| FINRA Expenses | $ | * | ||
| Transfer Agent Fees | $ | * | ||
| Accounting Fees and Expenses | $ | * | ||
| Legal Fees and Expenses | $ | * | ||
| Miscellaneous Fees and Expenses | $ | * | ||
| Total | $ | * |
All amounts are estimates other than the Nasdaq Application FeesSEC and the SEC’s and FINRA’s registrationFINRA filing fees.
* To be completed upon amendment.
| ITEM 14. | INDEMNIFICATION OF DIRECTORS AND OFFICERS |
Section 145 of the Delaware General Corporation Law, as amended, authorizes us to indemnify any director or officer under certain prescribed circumstances and subject to certain limitations against certain costs and expenses, including attorney’s fees actually and reasonably incurred in connection with any action, suit or proceeding, whether civil, criminal, administrative or investigative, to which a person is party by reason of being one of our directors or officers if it is determined that such person acted in accordance with the applicable standard of conduct set forth in such statutory provisions. Our certificate of incorporation contains provisions relating to the indemnification of directors and officers and our by-laws extend such indemnities to the full extent permitted by Delaware law. We currently maintain insurance for the benefit of any director or officer which cover claims for which we could not indemnify such persons.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling us pursuant to the foregoing provisions, or otherwise, we have been advised that in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable.
| ITEM 15. | RECENT SALES OF UNREGISTERED SECURITIES |
During the past three years, we effected the following transactions in reliance upon exemptions from registration under the Securities Act, as amended:
During the period commencing October 29, 2018 through February 11, 2019, we issued 1,215.5 units (the “Units”), at a purchase price of $1,000 per Unit, which Units consist of a total of (i) 1,215.5 shares of Series A convertible preferred stock, par value $0.001 per share, which shares will convert into approximately 269,444 shares of common stock, and (ii) warrants to purchase up to 269,444 shares of common stock, which warrants will be issuable upon completion of the Company’s initial public offering (“IPO”). The Units were sold to a total of 21 accredited investors pursuant to an exemption from registration under Rule 506(b) of the Securities Act.
On July 15, 2019, the Company issued to AFH Holding & Advisory, LLC, as consultant (“AFH”), 639,161 restricted stock units (319,580 shares post-reverse split) under the Company’s 2018 Equity Incentive Plan.
On December 28, 2021, in conjunction with entering into two loan agreements for a total of $500,000, which arewere repayable at the time of our IPO, we issued warrants to purchase a total of 500,000 shares of our common stock, exercisable at $1.00 per share. Such warrants were sold to two accredited investors pursuant to an exemption from registration under Rule 506(b) of the Securities Act. Boustead Securities LLC acted as placement agent, but waived its cash compensation related to such offering and, to date, has received no warrant compensation.
| II-1 |
The above disclosures do not include 768,334 shares (384,167 shares on a post-reverse split basis) granted pursuant to the Shuttle Pharmaceuticals Holdings, Inc. 2018 Equity Incentive Plan, which were issued to certain employees, directors and consultants, and vest on a periodic basis in accordance with the grant agreements between such individuals and the Company.
On February 8, 2022 and March 11, 2022, the Company sold to certain accredited investors $365,000 and $224,985, respectively, in 6% convertible notes (the “Notes”), which bearbore 6% interest, arewere repayable three years from the date of issuance, and will convertconverted automatically into shares of common stock or, in the event that units are sold in the offering, units, at a conversion price of $3.00$4.00 per share or unit upon closing of this offering.our IPO. Such notes were sold to accredited investors pursuant to an exemption from registration under Rule 506(b) of the Securities Act. Boustead Securities LLC acted as placement agent and received compensation of (i) $36,500 in cash and warrants to purchase 10% of the total number of shares issuable upon conversion of the Convertible Notes, exercisable at the conversion price of the Convertible Notes for the February offering and (ii) $22,750 in cash and warrants to purchase 10% of the total number of shares issuable upon conversion of the Convertible Notes, exercisable at the conversion price of the Convertible Notes for the March offering.
Effective March 30, 2022, the Company issued a total of 1,678 shares (839 shares on a post-reverse split basis) of common stock (the “Issuance”) to some 23 existing shareholders in satisfaction of certain interest that had accrued as the result of an inaccurate conversion of convertible notes in our 2018 share exchange. The Issuance satisfied in full all interest owed or otherwise accruing as the result of the inaccurate conversion. Such issuance was made in accordance with Rule 506(b) of the Securities Act.
On August, 1 2022, in conjunction with entering into three loan agreements for a total of $125,000, which arewere repayable at the timefollowing consummation of our IPO, we issued warrants to purchase a total of 50,000 shares of our common stock, exercisable at $2.50 per share. Such warrants were sold to three accredited investors pursuant to an exemption from registration under Rule 506(b) of the Securities Act. Boustead Securities LLC acted as placement agent, and received warrants to purchase 5,000 shares of common stock exercisable at $2.50 per share, equal to 10% of the value of the note offering. To date, Boustead has deferred itsoffering, and $12,500 in cash compensation.
The above disclosures do not include 678,180 shares of common stock granted pursuant to the Shuttle Pharmaceuticals Holdings, Inc. 2018 Equity Incentive Plan, which were issued to certain employees, directors and consultants, and vest on a periodic basis in accordance with the grant agreements between such individuals and the Company.
| ITEM 16. | EXHIBITS |
| Exhibit No. | Description | |
ITEM 17. UNDERTAKINGS
The undersigned registrant hereby undertakes:
(a) (1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in this registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high and of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and
(iii) To include any material information with respect to the plan of distribution not previously disclosed in this registration statement or any material change to such information in this registration statement.
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time to be deemed the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(b) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
| II-3 |
| II-4 |
| II-5 |
#To be filed by amendment
*Filed herewith.
** Furnished herewith
| II-6 |
| ITEM 17. | UNDERTAKINGS |
| (a) | The undersigned registrant hereby undertakes: |
| (1) | To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement: |
| (i) | To include any prospectus required by section 10(a)(3) of the Securities Act of 1933; | |
| (ii) | To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and | |
| (iii) | To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement. |
| Provided, however, that paragraphs (a)(1)(i), (ii) and (iii) above do not apply if the information required to be included in a post-effective amendment by those paragraphs is contained in reports filed with or furnished to the Commission by the registrant pursuant to section 13 or section 15(d) of the Securities Exchange Act of 1934 that are incorporated by reference in this registration statement. |
| (2) | That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| (3) | To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering. |
| (4) | That, for the purpose of determining liability of the registrant under the Securities Act of 1933 to any purchaser in the initial distribution of the securities: The undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser: |
| (i) | Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424; | |
| (ii) | Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant; | |
| (iii) | The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and | |
| (iv) | Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser. |
| II-7 |
| (b) | The undersigned registrant hereby undertakes that, for purposes of determining any liability under the Securities Act of 1933, each filing of the registrant’s annual report pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 (and, where applicable, each filing of an employee benefit plan’s annual report pursuant to Section 15(d) of the Securities Exchange Act of 1934) that is incorporated by reference in the registration statement shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| (c) | Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue. |
| (d) | The undersigned registrant hereby undertakes that: |
| (1) | For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective; and |
| (2) | For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| II-8 |
SIGNATURES
In accordance with the requirements of the Securities Act of 1933, the registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing this registration statement on Form S-1 to be signed on its behalf by the undersigned, in Rockville,Gaithersburg, Maryland, on August 25, 2022.April 5, 2024.
| SHUTTLE PHARMACEUTICALS HOLDINGS, INC. | ||
| By: | /s/ Anatoly Dritschilo, M.D. | |
Anatoly Dritschilo, M.D., | ||
| Chief Executive Officer | ||
| (Principal Executive Officer) | ||
| By: | /s/ Michael Vander Hoek | |
| Michael Vander Hoek | ||
| Chief Financial Officer | ||
| (Principal Financial Officer) |
KNOW ALL MEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Anatoly Dritschilo, M.D. and Michael Vander Hoek, and each of them, as his or her true and lawful attorney-in-fact and agent, with full power of substitution and re- substitution, for each of them and in each name, place and stead, in any and all capacities, to sign any and all pre- or post-effective amendments to this registration statement, and to file the same with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent, full power and authority to do and perform each and every act and thing requisite or necessary to be done in and about the premises, as fully to all intents and purposes as each might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent or his substitute, may lawfully do or cause to be done by virtue hereof. In accordance with the requirements of the Securities Act of 1933, as amended, this registration statement was signed by the following persons in the capacities and on the dates stated.
| Signatures | Title(s) | Date | ||
| /s/ Anatoly Dritschilo | Chairman of the Board and | |||
| Anatoly Dritschilo, M.D. | Chief Executive Officer (Principal Executive Officer) | |||
| /s/ Michael Vander Hoek | Chief Financial Officer | |||
| Michael Vander Hoek | (Principal Financial and Accounting Officer) | |||
| /s/ Chris | Director | |||
| Chris Senanayake, Ph.D. | ||||
| /s/ Steven | Director | |||
| Steven Richards | ||||
| /s/ Josh | Director | |||
| Josh Schafer | ||||
| /s/ Milton | Director | |||
| Milton Brown, M.D., Ph.D. | ||||
| /s/ | Director | |||