
As filed with the Securities and Exchange Commission on July 25, 2016March 28, 2024
Registration No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DCD.C. 20549
FORM S-3
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
GALENA BIOPHARMA, INC.SELLAS Life Sciences Group, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 20-8099512 | |
(State or other jurisdiction
| ( Identification |
2000 Crow Canyon Place,
7 Times Square, Suite 3802503
San Ramon, California 94583New York, New York 10036
(855) 855-4253(646) 200-5278
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Mark W. Schwartz, Ph.D.Angelos M. Stergiou, M.D., ScD h.c.
President and Chief Executive Officer
Galena Biopharma, Inc.7 Times Square, Suite 2503
2000 Crow Canyon Place, Suite 380New York, New York 10036
San Ramon, California 94583
(855) 855-4253(646) 200-5278
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Joel I. Papernik, Esq.
Daniel A. Bagliebter, Esq.
Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C.
919 Third Avenue
New York, New York 10022
(212) 935-3000
Approximate date of commencement of proposed sale to the public: From time to time after the effective date of this registration statement.
If the only securities being registered on this Form are being offered pursuant to dividend or interest reinvestment plans, please check the following box.box: ¨
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, other than securities offered only in connection with dividend or interest reinvestment plans, check the following box. box: x
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a registration statement pursuant to General Instruction I.D. or a post-effective amendment thereto that shall become effective upon filing with the Commission pursuant to Rule 462(e) under the Securities Act, check the following box. ¨
If this Form is a post-effective amendment to a registration statement filed pursuant to General Instruction I.D. filed to register additional securities or additional classes of securities pursuant to Rule 413(b) under the Securities Act, check the following box. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer” andfiler,” “smaller reporting company” and “emerging growth company” inRule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | ¨ | Accelerated filer | ||||
| Non-accelerated filer | x | Smaller reporting company | x | |||
| Emerging growth company | ¨ |
CALCULATION OF REGISTRATION FEE
| ||||||||
Title of each class of securities to be registered | Amount to be registered(1) | Proposed maximum offering price per share(2) | Proposed maximum aggregate offering price(2) | Amount of registration fee | ||||
Common stock, par value $0.0001 per share | 3,658,012 shares | $0.4075 | $1,490,640.20 | $150.11 | ||||
| ||||||||
| ||||||||
The Registrant hereby amends this registration statement on such dateIf an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or dates as may be necessaryrevised financial accounting standards provided pursuant to delay its effective date until the Registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a)7(a)(2)(B) of the Securities Act of 1933 or until the registration statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.Act. ¨
THE REGISTRANT HEREBY AMENDS THIS REGISTRATION STATEMENT ON SUCH DATE OR DATES AS MAY BE NECESSARY TO DELAY ITS EFFECTIVE DATE UNTIL THE REGISTRANT SHALL FILE A FURTHER AMENDMENT WHICH SPECIFICALLY STATES THAT THIS REGISTRATION STATEMENT SHALL THEREAFTER BECOME EFFECTIVE IN ACCORDANCE WITH SECTION 8(a) OF THE SECURITIES ACT OF 1933, AS AMENDED, OR UNTIL THE REGISTRATION STATEMENT SHALL BECOME EFFECTIVE ON SUCH DATE AS THE SECURITIES AND EXCHANGE COMMISSION, ACTING PURSUANT TO SAID SECTION 8(a), MAY DETERMINE.
The information in this prospectus is not complete and may be changed. WeA registration statement relating to these securities has been filed with the Securities and Exchange Commission. The selling stockholders may not sell these securities until the registration statement filed with the Securities and Exchange Commission isdeclares the registration statement effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED JULY 25, 2016MARCH 28, 2024
PROSPECTUS
3,658,01213,029,316 Shares of Common Stock
ThisThe selling stockholders of SELLAS Life Sciences Group, Inc. (“SELLAS,” “we,” “us” or the “Company”) listed beginning on page 11 of this prospectus relates to themay offer and saleresell under this prospectus up to 13,029,316 shares of our Common Stock issuable upon exercise of warrants acquired by certain of the selling stockholders identified inunder the Purchase Agreement (defined below) (the “Warrants”). The selling stockholders acquired the Warrants from us pursuant to a Securities Purchase Agreement (the “Purchase Agreement”), dated March 15, 2024, by and among the Company and the investors listed therein (the “Investors”).
We are registering the resale of the shares of Common Stock covered by this prospectus as required by the Purchase Agreement. The selling stockholders will receive all of up to 3,658,012the proceeds from any sales of the shares of our common stock.offered hereby. We are not selling any securities under this prospectus and will not receive any of the proceeds, frombut we will incur expenses in connection with the saleoffering. To the extent the Warrants are exercised for cash, if at all, we will receive the exercise price of shares by the selling stockholders.Warrants.
The selling stockholders may sell these shares through public or private transactions at market prices prevailing at the time of sale or at negotiated prices. The timing and amount of any sale are within the sole discretion of the selling stockholders. Our registration of the shares of common stock described inCommon Stock covered by this prospectus in a numberdoes not mean that the selling stockholders will offer or sell any of different ways and at varying prices. Seethe shares. For further information regarding the possible methods by which the shares may be distributed, see “Plan of Distribution” for more information about how the selling stockholders may sell the sharesbeginning on page 13 of common stock being offered pursuant to this prospectus.
We will pay the expenses incurred in registering the shares, including legal and accounting fees. See “Plan of Distribution”.
Our common stockCommon Stock is tradedlisted on The NASDAQNasdaq Capital Market under the symbol “GALE.“SLS.” On July 22, 2016, the closingThe last reported sale price of our common stockCommon Stock on The NASDAQ Capital MarketMarch 27, 2024 was $0.45.$1.03 per share.
An investmentInvesting in our common stockCommon Stock is highly speculative and involves a highsignificant degree of risk. See “Risk Factors”Please consider carefully the specific factors set forth under “Risk Factors” beginning on page 116 of this prospectus.
NEITHER THE SECURITIES AND EXCHANGE COMMISSION NOR ANY STATE SECURITIES COMMISSION HAS APPROVED OR DISAPPROVED OF THESE SECURITIES OR PASSED UPON THE ADEQUACY OR ACCURACY OF THIS PROSPECTUS. ANY REPRESENTATION TO THE CONTRARY IS A CRIMINAL OFFENSE.prospectus and in our filings with the Securities and Exchange Commission.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the accuracy or adequacy of the disclosures in this prospectus. Any representation to the contrary is a criminal offense.
The date of this prospectus is , 2016.2024
TABLEOF CONTENTSTable of Contents
This prospectus is part of thea registration statement on Form S-3 that we have filed with the Securities and Exchange Commission (the “SEC”) pursuant to which the selling stockholder named herein may, from time to time, offer and sell or the SEC. As permitted by the rules and regulationsotherwise dispose of the SEC, the registration statement filedshares of our Common Stock (“Shares”) covered by us includes additional information not contained in this prospectus.
If information in this prospectus is inconsistent with any document incorporated by reference that was filed with the SEC before the date of this prospectus, you should rely on this prospectus. This prospectus and the documents incorporated by reference into this prospectus include important information about us, the securities being offered and other information you should know before investing in our securities. You should also read and consider information in the documents to which we have referred you in the section of this prospectus entitled “Where You Can Find More Information.”
You should rely only on this prospectus and the information incorporated or deemed to be incorporated by reference in this prospectus. We have not authorized anyone to provide you with information that is in addition to or different from that contained or incorporated by reference in this prospectus. If anyone provides you with different or inconsistent information, you should not rely on it. This prospectus does not constitute an offer to sell or the solicitation of an offer to buy securities in any jurisdiction to any person to whom it is unlawful to make such offer or solicitation in such jurisdiction. You should not assume that the information contained or incorporated by reference in this prospectus is accurate as ofon any date other than as ofsubsequent to the date set forth on the front cover of this prospectus or that any information we have incorporated by reference is correct on any date subsequent to the date of the document incorporated by reference, even though this prospectus is delivered or Shares are sold or otherwise disposed of on a later date.
This prospectus does not contain all of the information included in the caseregistration statement. For a more complete understanding of the offering of the Shares, you should refer to the registration statement including the exhibits. Copies of some of the documents referred to herein have been filed, will be filed or will be incorporated by reference as exhibits to the dateregistration statement of such documents regardless of the time of delivery ofwhich this prospectus or any saleis a part, and you may obtain copies of our shares of common stock. Our business, financial condition, liquidity, results of operations and prospects may have changed since those dates.
documents as described below under the heading “Where You Can Find More Information.” We further note that the representations, warranties and covenants made by us in any agreement that is filed as an exhibit to any document that is incorporated by reference in thisthe accompanying prospectus were made solely for the benefit of the parties to such agreement, including in some cases, for the purpose of allocating risk among the parties to such agreements, and should not be deemed to be a representation, warranty or covenant to you. Moreover, such representations, warranties or covenants were accurate only as of the date when made. Accordingly, such representations, warranties and covenants should not be relied on as accurately representing the current state of our affairs. It is important for you to read and consider all information contained in this prospectus, including the documents incorporated by reference therein, in making your investment decision. You should also read and consider the information in the documents to which we have referred you under “Where You Can Find More Information” and “Incorporation of Certain Documents by Reference” in this prospectus.
Unless otherwise indicated,
The selling stockholders named in this prospectus may sell up to 13,029,316 shares of our Common Stock issuable upon exercise of warrants to purchase shares of our Common Stock from time to time. This prospectus also covers any shares of Common Stock that may become issuable as a result of share splits, share dividends, or similar transactions. We have agreed to pay the expenses incurred in registering these shares, including legal and accounting fees.
We and the selling stockholders have not authorized anyone to give any information or to make any representation to you other than those contained or incorporated by reference in this prospectus. You must not rely upon any information or representation not contained or incorporated by reference in this prospectus. This prospectus concerningdoes not constitute an offer to sell or the solicitation of an offer to buy any of our industry,shares of Common Stock other than the Shares covered hereby, nor does this prospectus constitute an offer to sell or the solicitation of an offer to buy any securities in any jurisdiction to any person to whom it is unlawful to make such offer or solicitation in such jurisdiction. Persons who come into possession of this prospectus in jurisdictions outside the United States are required to inform themselves about, and to observe, any restrictions as to the offering and the distribution of this prospectus applicable to those jurisdictions.
This prospectus, including our general expectations and market opportunity, isthe documents incorporated by reference herein, include statements that are based on information from our own management estimates and research, as well as from industry and general publications and research, surveys and studies conducted by third parties. Management estimates are derived from publicly available information, our knowledge of our industry and assumptions based on such information and knowledge, which we believe to be reasonable. In addition,various assumptions and estimates that are subject to numerous known and unknown risks and uncertainties. Some of ourthese risks and our industry’s future performanceuncertainties are necessarily uncertain due to a variety of factors, including those described in the section entitled “Risk Factors” beginning on page 116 of this prospectus.prospectus and as described in Part I, Item 1A (Risk Factors) of our most recent Annual Report on Form 10-K for the year ended December 31, 2023 filed with the SEC on March 28, 2024, as updated by our subsequent filings with the SEC under the Securities Exchange Act of 1934, as amended (the “Exchange Act”). These and other important factors could cause our future performanceresults to differbe materially different from ourthe results expected as a result of, or implied by, these assumptions and estimates.
Except as otherwise indicated herein or as the context otherwise requires, references in this prospectus supplement, the accompanying prospectus and the information incorporated by referenced herein or therein to “SELLAS,” “the Company,” “we,” “us,” “our” and similar terms refer to SELLAS Life Sciences Group, Inc. and, where appropriate, our subsidiaries.
| 1 |
This summary description about us and our business highlights selected information appearingcontained elsewhere in this prospectus or incorporated by reference into this prospectus andprospectus. It does not contain all of the information that may be important to you or that you should consider before investing in our common stock. This prospectus includes or incorporatessecurities. Important information is incorporated by reference information about the securities we areinto this prospectus. To understand this offering as well as information regarding our business and detailed financial data. Before making an investment decision,fully, you should read thiscarefully the entire prospectus, and the information incorporated by reference herein in their entirety, including “Risk Factors” beginningand “Cautionary Note Regarding Forward-Looking Statements,” together with the additional information described under “Information Incorporated by Reference.”
Overview
We are a late-stage clinical biopharmaceutical company focused on page 11the development of this prospectus.novel therapeutics for a broad range of cancer indications. Our product candidates currently include galinpepimut-S, or GPS, a peptide immunotherapy directed against the Wilms tumor 1, or WT1, antigen, and SLS009 (formerly, GFH009), a highly selective small molecule cyclin-dependent kinase 9, or CDK9, inhibitor.
About Galena
OverviewGalinpepimut-S
Galena Biopharma, Inc. (“we,” “us,” “our,” “Galena” or the “company”)
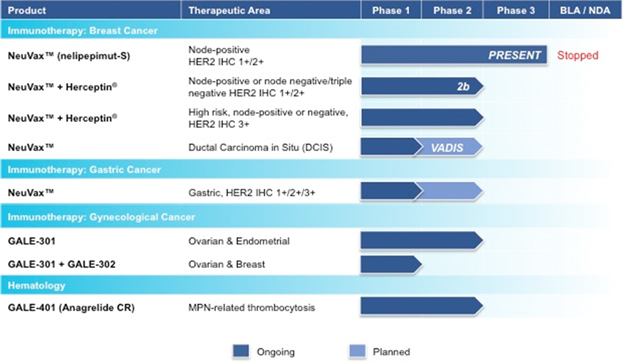
Our lead product candidate, GPS, is a biopharmaceutical company committed tocancer immunotherapeutic agent licensed from Memorial Sloan Kettering Cancer Center, or MSK, that targets the development and commercializationWT1 protein, which is present in 20 or more cancer types. Based on its mechanism of targeted oncology therapeutics that address major unmet medical needs. Galena’s development portfolio is focused primarily on addressing the rapidly growing patient populations of cancer survivors by harnessing the power of the immune system to prevent cancer recurrence. The Company’s pipeline consists of multiple mid- to late-stage clinical assets, including novel cancer immunotherapy programs led by NeuVax™ (nelipepimut-S), GALE-301 and GALE-302. The company is also developing a hematology asset, GALE-401, a controlled release version of the approved drug anagrelide for the treatment of patients with thrombocythemia, secondary to myeloproliferative disorders, to reduce the elevated platelet count. NeuVax is currently in several concurrent Phase 2 trials ongoing bothaction as a singledirectly immunizing agent, andGPS has potential as a monotherapy or in combination with other therapies. GALE-301 is inimmunotherapeutic agents to address a Phase 2a clinical trial in ovarian and endometrialbroad spectrum of hematologic, or blood, cancers, and in a Phase 1b clinical trial given sequentially with GALE-302.GALE-401 has completed a Phase 2 trial.
We are seeking to build value for shareholders through pursuit of the following objectives:
The chart below summarizes the current status of our clinical development pipeline:indications.
Novel Cancer Immunotherapies
Our targeted cancer immunotherapy approach is currently based upon two key areas: preventing secondary recurrence of cancer, which is becoming increasingly important as the number of cancer survivors continues to grow; and, primary prevention intended to treat breast cancer earlier in the treatment spectrum. Once a patient’s tumor becomes metastatic, the outcome is often fatal, making the prevention of recurrence a potentially critical component of overall patient care. Our programs primarily target patients in the adjuvant (after-surgery) setting who have relatively healthy immune systems, but may still have residual disease. Minimal residual disease, or single cancer cells (occult cancer cells) or micrometastasis, that are undetectable by current radiographic scanning technologies, can result in disease recurrence.
Our therapies utilize an immunodominant peptide combined with the immune adjuvant, recombinant human granulocyte macrophage-colony stimulating factor (rhGM-CSF), and work by harnessing the patient’s own immune system to seek out and attack any residual cancer cells. Using peptide immunogens has many potential clinical advantages, including a favorable safety profile, since these drugs may lack the toxicities typical of most cancer therapies. They also have the potential to evoke long-lasting protection through activation of the immune system and a convenient, intradermal mode of delivery. We are currently engaged in multiple clinical trials with NeuVax™ (nelipepimut-S), GALE-301, and GALE-302, targeting the prevention of recurrence in breast, gastric, ovarian and endometrial cancers.
NeuVax™ (nelipepimut-S)
NeuVax™ (nelipepimut-S) is a cancer immunotherapy targeting human epidermal growth factor receptor (HER2) expressing cancers. NeuVax is the immunodominant nonapeptide derived from the extracellular domain of the HER2 protein, a well-established and validated target for therapeutic intervention in breast and gastric carcinomas. The NeuVax vaccine is combined with GM-CSF for injection under the skin, or intradermal administration. Data has shown that an increased presence of circulating tumor cells (CTCs) may predict Disease Free Survival (DFS) and Overall Survival (OS) suggesting a presence of isolated micrometastases, not detectable clinically, but, over time, can lead to recurrence, most often in distant sites. After binding to the specific HLA molecules on antigen presenting cells, the nelipepimut-S sequence stimulates specific cytotoxic T lymphocyte, or CTLs, causing significant clonal expansion. These activated CTLs recognize, neutralize and destroy, through cell lysis, HER2 expressing cancer cells, including occult cancer cells and micrometastatic foci. The nelipepimut immune response can also generate CTLs to other immunogenic peptides through inter- and intra-antigenic epitope spreading.
Breast Cancer: According to the National Cancer Institute (NCI), over 230,000 women in the U.S. are diagnosed with breast cancer annually. While improved diagnostics and targeted therapies have decreased breast cancer mortality in the U.S., metastatic breast cancer remains incurable. Approximately 75% to 80% of breast cancer patients have tissue test positive for some increased amount of the HER2 receptor, which is associated with disease progression and decreased survival. Only approximately 20% to 30% of all breast cancer patients—those with HER2 immunohistochemistry (IHC) 3+ disease, or IHC 2+ and fluorescence in situ hybridization (FISH) amplified—have a HER2 directed, approved treatment option available after their initial standard of care. This leaves the majority of breast cancer patients with low-to-intermediate HER2 expression (IHC 1+/2+) ineligible for therapy and without an effective targeted treatment option to prevent cancer recurrence.
We have multiple trials currently ongoing with NeuVax. On June 24, 2016, the assembled Independent Data Monitoring Committee, or IDMC, met to conduct a pre-planned safety and futility analysis of the Phase 3 PRESENT (Prevention of Recurrence in Early-Stage, Node- Positive Breast Cancer with Low to Intermediate HER2 Expression with NeuVax Treatment) clinical trial. On June 27, 2016, the IDMC recommended that the Phase 3 trial “be stopped for futility unless it is determined that there has been a systematic reversal in the study drug treatments in the two arms, in which case the IDMC should reevaluate the clinical evidence.” We have stopped the PRESENT trial, and initiated an investigation into the causes of the recommendation. We currently have two additional Phase 2 breast cancer trials ongoing with NeuVax in combination with trastuzumab (Herceptin®; Genentech/Roche) targeting the prevention of recurrence in expanded indications.
We also recently announced our intent to initiate a Phase 2 trial with NeuVax as a single agent in patients with ductal carcinoma in situ, or DCIS, in collaboration with the NCI, potentially positioning NeuVax as a treatment for earlier stage disease. The trial will have an immunological endpoint evaluating NeuVax peptide-specific cytotoxic T lymphocyte (CTL; CD8+ T-cell) response in vaccinated patients. DCIS is defined by the NCI as a noninvasive condition in which abnormal cells are found in the lining of a breast duct, and is the most common type of breast cancer. The abnormal cells have not spread outside the duct to other tissues in the breast. In some cases, DCIS may become invasive cancer and spread to other tissues, and at this time, there is no way to know which lesions could become invasive. Current treatment options for DCIS include breast-conserving surgery and radiation therapy with or without tamoxifen, breast-conserving surgery without radiation therapy, or total mastectomy with or without tamoxifen. According to the American Cancer Society, in 2015 there were over 60,000 diagnoses of DCIS.
Gastric Cancer: According to the NCI, gastric (stomach) cancer is a disease in which malignant (cancer) cells form in the lining of the stomach. Almost all gastric cancers are adenocarcinomas (cancers that begin in cells that make and release mucus and other fluids). Other types of gastric cancer are gastrointestinal carcinoid tumors, gastrointestinal stromal tumors, and lymphomas. Infection with bacteria called Helicobacter pylori (H. pylori) is a common cause of gastric cancer and age, diet, and stomach disease can affect the risk of developing gastric cancer. Gastric cancer is often diagnosed at an advanced stage because there are no early signs or symptoms. Gastric, or stomach cancer, is the second-most common cancer among males and third-most common among females in Asia and worldwide with over 63,000 new cases a year in India, where an initial clinical trial of NeuVax will be run. Overexpression of the HER2 receptor occurs in approximately 20% of gastric and gastro-esophageal junction adenocarcinomas, predominantly those of the intestinal type. Overall, without regard to the stage of cancer, only approximately 28% of patients with stomach cancer live at least five years following diagnosis and new adjuvant treatments are needed to prevent disease recurrence.
We currently have a number of ongoing or planned clinical trials designed to expand the clinical and geographical footprint of NeuVax:
GALE-301 and GALE-302
Our second immunotherapy franchise targets folate binding protein (FBP) receptor-alpha. FBP is a well-validated therapeutic target that is highly over-expressed in ovarian, endometrial and breast cancers, and is the source of immunogenic peptides that can stimulate cytotoxic T lymphocytes to recognize and destroy FBP-expressing cancer cells. Current treatments after surgery for these diseases are principally with platinum based chemotherapeutic agents. These patients suffer a high recurrence rate and most relapse with an extremely poor prognosis. GALE-301 and GALE-302 are immunogenic peptides that consist of a peptide derived from FBP combined with GM-CSF for the prevention of cancer recurrence in the adjuvant setting. GALE-301 is the E39 peptide, while GALE-302 is an attenuated version of this peptide, known as E39. Two trials are ongoing with our FBP peptides: theGALE-301 Phase 2a portion of the Phase 1/2a clinical trial is ongoing in ovarian and endometrial adenocarcinomas, and the GALE-301 plus GALE-302 Phase 1b clinical trial is ongoing in breast and ovarian cancers.
Ovarian Cancer: According to the NCI Surveillance, Epidemiology, and End Results (SEER) Program, new cases of ovarian cancer occur at an annual rate of 11.9 per 100,000 womenJanuary 2020, we commenced in the United States with an estimated 22,280 cases for 2016. Although ovarian cancer represents about 1.3% of all cancers, it represents about 2.4% of all cancer deaths, or an estimated 14,180 deaths in 2015. Approximately 1.3% of women will be diagnosed with ovarian cancer at some point during their lifetime (2010 - 2012 data). The prevalence of ovarian cancer in the U.S. is about 192,000 women, and the five-year survivorship for women with ovarian cancer is 45.6%. Due to the lack of specific symptoms, the majority of ovarian cancer patients are diagnosed at later stages of the disease, with an estimated 80% of women presenting with advanced-stage (III or IV) disease. These patients have their tumors routinely surgically debulked to minimal residual disease, and then are treated with platinum- and/or taxane-based chemotherapy. While many patients respond to this treatment regimen and become clinically free-of-disease, the majority of these patients will relapse. Depending upon their level of residual disease, the risk for recurrence after completion of primary therapy is approximately 70%. Unfortunately for these women, once the disease recurs, treatment options are limited and the disease is most likely incurable. According to the NCI SEER Program, new cases of endometrial cancer occur at an annual rate of 25.1 per 100,000 women in the U.S., with an estimated 54,870 cases for 2015. Although endometrial cancer represents about 3.3% of all cancers, it represents about 1.7% of all cancer deaths, or an estimated 10,170 deaths in 2015. Approximately 2.8% of women will be diagnosed with endometrial cancer at some point during their lifetime (2010 - 2012 data). The prevalence of endometrial cancer in the U.S. is about 620,000 women.
Hematology
GALE-401 (anagrelide controlled release (CR))
GALE-401 (Anagrelide Controlled Release) contains the active ingredient anagrelide, an FDA-approved product, for the treatment of patients with myeloproliferative neoplasms (MPNs) to lower abnormally elevated platelet levels. The currently available immediate release (IR) version of anagrelide causes adverse events that are believed to be dose and plasma concentration dependent. These adverse events may limit the use of the IR version of the drug. Therefore, reducing the maximum concentration (Cmax) is hypothesized to reduce the side effects, but preserve efficacy, potentially allowing a broader use of the drug. Multiple Phase 1 studies in 98 healthy subjects have shown GALE-401 reduces the Cmax of anagrelide following oral administration, appears to be well tolerated at the doses administered, and to be capable of reducing platelet levels. The Phase 1 program provided the desired PK/PD (pharmacokinetic/pharmacodynamic) profile to enable the initiation of a Phase 2 proof-of-concept trial. The Phase 2, open label single arm, proof-of-conceptrandomized Phase 3 clinical trial, enrolled 18 patients in the United StatesREGAL study, for the treatment of thrombocytosis, or elevated platelet countsGPS monotherapy in patients with MPNs. Final safety and efficacy data from this pilot Phase 2 trial was presented in December 2015 and demonstrated a prolonged clinical benefit with a potentially improved safety profile. The follow-up portion of the Phase 2 is now complete and we plan to submit a final Phase 2 manuscript at the end of 2016. We continue to review our data and are analyzing the treatment landscape for MPNs, with a current focus on Essential Thrombocythemia (ET). We are planning to meet with the FDA later this year to discuss our Phase 2 data, our development opportunities in MPN patients, and confirm the 505(b)2 regulatory pathway for approval.
MPNs are a closely related group of hematological malignancies in which the bone marrow cells that produce the body’s blood cells develop and function abnormally. The main MPNs are ET, Polycythemia Vera (PV), Primary Myelofibrosis (PMF), and Chronic Myelogenous Leukemia (CML), all of which are associated with high platelet counts. ET is an acquired disease of the bone marrow and is associated with vascular complications including thrombosis and bleeding events. The MPNs are progressive blood cancers that can strike anyone at any age.
Alliance Partners in Therapeutic Areas
We are actively looking to leverage our technology platforms by seeking to work with pharmaceutical and biotechnology partners in a number of therapeutic areas in oncology. Our team has experience targeting products in multiple indications, and based on this experience, we believe we can expand the clinical utility of our current development candidates as well as discover more drug candidates by working with partners than we can develop with our own resources. We are seeking to work with partnersacute myeloid leukemia, or AML, in the discovery and developmentmaintenance setting after achievement of drugs insecond complete remission, or CR2, following successful completion of second-line antileukemic therapy. Patients are randomized to receive either GPS or best available treatment, or BAT. We expect this study will be used as the basis for submission of a number of therapeutic areas and technology platforms.
Intellectual Property
Patents and other intellectual property rights are crucial to our success. It is our policy to protect our intellectual property rights through available means, including filing and prosecuting patent applications in the U.S. and other countries, protecting trade secrets, and utilizing regulatory protections such as data exclusivity. We also include restrictions regarding use and disclosure of our proprietary information in our contracts with third parties, and utilize customary confidentiality agreements with our employees, consultants, clinical investigators and scientific advisors to protect our confidential information and know-how. Together with our licensors, we also rely on trade secrets to protect our combined technology especially where we do not believe patent protection is appropriateBiologics License Application, or obtainable. It is our policy to operate without infringing on, or misappropriating, the proprietary rights of others. The following chart summarizes our intellectual property rights. The patents for the products are for the U.S. and certain foreign countries:
|
|
|
| |||
Out-License Agreements
Teva Pharmaceuticals
Effective December 3, 2012, we entered into a license and supply agreement with ABIC Marketing Limited, a subsidiary of Teva Pharmaceuticals (“ABIC”). Under the agreement, we granted ABIC exclusive rights to seek marketing approval in Israel for our NeuVax product candidate for the treatment of breast cancer following its approval by the FDA or the European Medicines Agency, and to market, sell and distribute NeuVax in Israel assuming such approval is obtained. ABIC’s rights also include a right of first refusal in Israel for all future indications for which NeuVax may be approved.
Under the license and supply agreement, ABIC will assume responsibility for regulatory registration of NeuVax in Israel, provide financial support for local development, and commercialize the product in the region in exchange for making royalty payments to us based on future sales of NeuVax. ABIC also agrees in the license and supply agreement to purchase all supplies of NeuVax from us at a price determined accordingBLA, subject to a specified formula.
Dr. Reddy’s Laboratories Ltd.
Effective January 14, 2014, we entered into a strategic developmentstatistically significant and commercialization partnership with Dr. Reddy’s Laboratories Ltd. (“Dr. Reddy’s”), under which we licensed commercial rights in India to Dr. Reddy’s for NeuVax in breastclinically meaningful data outcome and gastric cancers. Under the agreement, Dr. Reddy’s will lead the Phase 2 development of NeuVax in India in gastric cancer, significantly expanding the potential patient population addressable with NeuVax.
Kwangdong Pharmaceutical Co., Ltd.
Effective April 30, 2009, we entered into a license agreement with Kwangdong Pharmaceutical Co, Ltd (Kwangdong). Under the agreement, we granted Kwangdong exclusive rights to seek marketing approval in The Republic of Korea (South Korea) for our NeuVax product candidate for the treatment of breast cancer following its approval by the FDA or the European Medicines Agency, and to market, sell and distribute NeuVax in South Korea assuming such approval is obtained.
Recent Developments (in reverse chronological order)
Discontinued NeuVax™ (nelipepimut-S) Phase 3, PRESENT Interim Analysis based on Independent Data Monitoring Committee Recommendation
On June 24, 2016, the assembled IDMC met to conduct a pre-planned safety and futility analysis of the Phase 3 PRESENT (Prevention ofRecurrence in Early-Stage, Node- Positive Breast Cancer with Low to Intermediate HER2 Expression with NeuVax Treatment) Trial. On June 27, 2016, the IDMC recommended that the study be stopped for futility unless it is determined that there has been a systematic reversal in the study drug treatments in the two arms, in which case the IDMC should reevaluate the clinical evidence. We have stopped the PRESENT Trial, and initiated an investigation into the causes of the recommendation.
Derivative and Securities Litigation – The U.S. District Court for the District of Oregon granted final approval of the settlements previously reported.
On December 3, 2015, we reached an agreement in principle to settle the consolidated shareholder derivative action, In reGalena Biopharma, Inc. Derivative Litigation, Civil Action No. 3:14-cv-00382-SI against us and certain of our current and former officers and directors. Following the hearing on June 23, 2016, on June 24, 2016, the U.S. District Court for the District of Oregon entered a final order and judgment in In re Galena Biopharma, Inc. Derivative Litigation, granting final approval to the settlement. On the same day, the Court also issued an opinion and order awarding attorney’s fees of $4.5 million plus costs, which will be paid by our insurance carriers. The settlement includes a payment of $15 million in cash by our insurance carriers, which we will use to fund a portion of the class action settlement, and cancellation of 1,200,000 outstanding director stock options. The settlement also requires that we adopt and implement certain corporate governance measures. The settlement does not include any admission of wrongdoing or liability on the part of us or the individual defendants and includes a full release of us and the current and former officers and directors in connection with the allegations made in the consolidated federal derivative actions and state court derivative actions.
On December 3, 2015, we also agreed in principal to resolve and settle the securities putative class action lawsuit,In re Galena Biopharma, Inc. Securities Litigation, Civil Action No. 3:14-cv-00367-SI, against us, certain of our current and former officers and directors and other defendants in the United States District Court for the District of Oregon. Following the hearing on June 23, 2016, on June 24, 2016, the U.S. District Court for the District of Oregon entered a final order and partial judgment in In re Galena Biopharma, Inc. Securities Litigation, granting final approval of the settlement. On the same day, the Court also issued an opinion and order awarding attorney’s fees of $4.5 million plus costs, which is paid out of the settlement funds. The agreement provides for a settlement payment of $20 million to the class and the dismissal of all claims against us and the other defendants in connection with the consolidated federal securities class actions. Of the $20 million settlement payment to the class, $16.7 million was paid by our insurance carriers and $2.3 million in cash was paid by us on July 1, 2016, along with $1 million in shares of our common stock (480,053 shares) paid by us on July 6, 2016. We will be responsible for defense costs and any settlements or judgments incurred for any related opt-out lawsuits.
Securities Litigation Opt Out Claims
We have resolved claims brought by shareholders and others that relate to the securities litigation mentioned above in one case for $150,000 plus $150,000 in shares (291,262) of our common stock, and in another case for $1.5 million in shares of our common stock (3,366,750 shares). The shares issued in connection with such settlements are included in the offering covered by this prospectus. The settlements do not include any admission of wrongdoing or liability on the part of us or any of the current or former directors and officers and includes a full release of us and the current and former directors and officers in connection with the allegations made. We are not aware of any other claims made by shareholders who have opted out of the securities litigation.
Presented GALE-401 Combined Safety Data
On June 13, 2016 we presented combined safety data from our GALE-401 clinical trials at the European Hematology Association 21stCongress. A total of six trials have been run with GALE-401, five Phase 1 trials in healthy volunteers (N=98), and one Phase 2 single arm, open label pilot study in MPN patients (N=18). The poster, entitled, “Anagrelide Controlled Release (GALE-401) Safety Profile Consistently Well Tolerated in Myeloproliferative Neoplasms Patients and Healthy Volunteers” was designed to characterize the safety profile of GALE-401 in all subjects treated to date. The results demonstrated that GALE-401 is well tolerated in MPN patients as well as in healthy volunteers and we observed predominantly mild to moderate toxicities that did not reveal any unexpected AEs.
Received Two Orphan Drug Designations for GALE-301 and GALE-301/GALE-302
On June 10, 2016, we announced that the U.S. Food and Drug Administration, or the FDA. The primary endpoint of the REGAL study is overall survival, or OS. We planned to enroll approximately 125 to 140 patients at approximately 95 clinical sites in North America, Europe and Asia with a planned interim safety, efficacy and futility analysis after 60 events (deaths). In March 2024, we announced the completion of enrollment. Under our current assumptions with respect to enrollment and the estimated survival times for both the treated and control groups in the study, we believe, after discussions with our external statisticians and experts, that the planned interim analysis after 60 events (deaths) per the protocol will occur in the first half of 2024 and the final analysis after 80 events will occur by the end of 2024. Because these analyses are event driven, they are difficult to predict with any certainty and may occur at a different time than currently expected.
In December 2020, we entered into an exclusive license agreement with 3D Medicines Inc., or 3D Medicines, a China-based biopharmaceutical company developing next-generation immuno-oncology drugs, for the development and commercialization of GPS, as well as the Company’s next generation heptavalent immunotherapeutic GPS+, which is at preclinical stage, across all therapeutic and diagnostic uses in mainland China, Hong Kong, Macau and Taiwan, which we refer to as Greater China. We have retained sole rights to GPS and GPS+ outside of Greater China. In November 2022, we announced that we had agreed with 3D Medicines for 3D Medicines to participate in the REGAL study through the inclusion of approximately 20 patients from mainland China. Although the REGAL study has completed enrollment as announced in March 2024, in accordance with the predetermined statistical analysis plan, 3D Medicines may still enroll patients in mainland China. The timing of such participation and patient enrollment by 3D Medicines, if at all, cannot be predicted with certainty. In December 2023, we announced that we had commenced a binding arbitration proceeding against 3D Medicines to resolve a dispute regarding, among other things, the trigger and payment of relevant milestone payments due to us under the 3D Medicines Agreement. As of March 15, 2024, we have received an aggregate of $10.5 million in upfront and milestone payments under our license agreement with 3D Medicines, or the 3D Medicines Agreement, and a total of $191.5 million in potential future development, regulatory and sales milestones, not including future royalties, remains under the license agreement, which milestones are variable in nature and not under our control.
| 2 |
In December 2018, pursuant to a Clinical Trial Collaboration and Supply Agreement, we initiated a Phase 1/2 multi-arm "basket" type clinical study of GPS in combination with Merck & Co., Inc.’s anti-PD-1 therapy, pembrolizumab (Keytruda®). In 2020, we, together with Merck, determined to focus on ovarian cancer (second or third line). In November 2022, we reported topline clinical and initial immune response data from this study, which showed that treatment with the combination of GPS and pembrolizumab compared favorably to treatment with anti-PD-1 therapy alone in a similar patient population. In November 2023, additional immunobiological and clinical data from the study was presented at the International Gynecologic Cancer Society 2023 Annual Global Meeting which showed a correlation between immune response and progression free survival, or PFS.
In February 2020, a Phase 1 open-label investigator-sponsored clinical trial of GPS, in combination with Bristol-Myers Squibb’s anti-PD-1 therapy, nivolumab (Opdivo®), in patients with malignant pleural mesothelioma, or MPM, who harbor relapsed or refractory disease after having received frontline standard of care multimodality therapy was commenced at MSK. Enrollment of a target total of 10 evaluable patients was completed at the end of 2022. We reported positive topline safety and efficacy data from this study in June 2023 and positive follow-up immune response and survival data in December 2023.
GPS was granted Orphan Drug Designations, or ODD, from the FDA, granted two orphan-drugas well as orphan medicines designations from the European Medicines Agency, or EMA, for GPS in AML, MPM, and multiple myeloma, or MM, as well as Fast Track designations for Galena’s two cancer immunotherapy peptides derivedAML, MPM, and MM from Folate Binding Protein (FBP)the FDA.
SLS009
On March 31, 2022, we entered into an exclusive license agreement, or the GenFleet Agreement, with GenFleet Therapeutics (Shanghai), Inc., or GenFleet, a clinical-stage biotechnology company developing cutting-edge therapeutics in oncology and immunology, that grants rights to us for the treatment (including preventiondevelopment and commercialization of recurrence)SLS009, a highly selective small molecule CDK9 inhibitor, across all therapeutic and diagnostic uses worldwide, except for Greater China.
CDK9 activity has been shown to correlate negatively with OS in a number of ovarian cancer: onecancer types, including hematologic cancers, such as AML and lymphomas, as well as solid cancers, such as osteosarcoma, pediatric soft tissue sarcomas, melanoma, endometrial, lung, prostate, breast and ovarian. As demonstrated in preclinical and clinical data, to date, SLS009’s high selectivity has the potential to reduce toxicity as compared to older CDK9 inhibitors and other next-generation CDK9 inhibitors currently in clinical development and to potentially be more efficacious.
We completed a Phase 1 dose-escalating clinical trial in the United States and China for GALE-301 (E39),SLS009 in mid-2023 and onereported positive safety and efficacy data for GALE-301 (E39)both patient cohorts, that is relapsed and/or refractory AML and GALE-302 (E39’).refractory lymphoma. We also established in the trial a recommended Phase 2 dose, or RP2D, of 60 mg for AML and 100 mg for lymphomas.
Presented GALE-301
In the second quarter of 2023, we commenced an open label, single arm, multi-center Phase 1/2a Primary Analysis
On June 6, 2016, we presented the primary analysis from the Company’s GALE-301 Phase 1/2a clinical trial of SLS009 in combination with venetoclax and azacitidine, or aza/ven, in AML patients who failed or did not respond to treatment with venetoclax-based therapies. The Phase 2a trial is evaluating safety, tolerability and efficacy at two dose levels, 45 mg once weekly, and 60 mg once weekly or 30 mg twice a week.
In the American Societyfourth quarter of Clinical Oncology Annual Meeting 2016. The poster, entitled, “The primary analysis2023, we announced the dosing of the first patient in a phase I/IIa dose findingPhase 1b/2 open-label, single arm trial in relapsed/refractory, or r/r, peripheral T-cell lymphoma, or PTCL, which will enroll up to 95 patients to evaluate safety and efficacy and, based on results, may serve as a registrational study. This study is fully funded by GenFleet and is being conducted in China.
In March 2024, we announced positive topline data from the Phase 2a clinical trial of SLS009 in combination with aza/ven in r/r/ AML. A total of 21 patients were enrolled in the study as of March 15, 2024: 10 in the 45 mg safety cohort and 11 in the 60 mg cohort (30 mg twice a folate binding protein vaccine, E39 + GM-CSFweek or 60 mg once a week). Response rates observed in ovarianthe three cohorts were 10% in the 45 mg once a week safety dose cohort (dose level below the RP2D), 20% in the 60 mg once a week dose cohort, and endometrial cancer50% in the 30 mg twice a week dose cohort. Additionally, we observed strong anti-leukemic activity, which is defined as 50% or more bone marrow blast reduction in 67% of patients to prevent recurrence,” demonstrated thatacross all dose levels. Median OS has not been reached in any of the vaccine is well toleratedcohorts and immunogenic. Inthe first patient enrolled in the study who achieved a CR continues on the study and remains leukemia-free 9 months after enrollment. During the trial, we identified potential biomarkers currently undergoing testing as predictive markers in the most recent portion of the study. Patients with the identified biomarkers exhibited significantly higher response rates: 100% response rate at the optimal dose group,level (30 mg twice a week) and 57% response rate across all dose levels. Furthermore, we have clarified the results demonstrate potential clinical benefitproposed biological basis and mechanism of action for GALE-301SLS009 activity in patients with these biomarkers. The relevant biomarkers are present in multiple hematologic and solid cancer indications, with a substantial proportion of patients exhibiting them in additional indications, ranging up to prevent recurrence~50% of patients in these patients,some indications.
SLS009 was granted ODD for AML and that boosters may sustain this effect.PTCL and Fast Track designations for r/r AML and r/r PTCL by the FDA.
| 3 |
Recent Developments
Received Fast Track Designation for NeuVax™ (nelipepimut-S) PRESENT Clinical TrialJanuary 2024 Registered Direct Offering
On June 1, 2016,January 4, 2024, we announced thatentered into Securities Purchase Agreements with certain investors (the “January Investors”), pursuant to which we agreed to issue and sell, in a registered direct offering by the FDA has designated NeuVax™ (nelipepimut-S)Company directly to the January Investors (the “January Registered Offering”), combined GM-CSF, as a Fast Track development program for the treatment(i) an aggregate of patients with early stage, node positive breast cancer with low to intermediate HER2 expression, otherwise known as HER2 1+ or 2+, following standard of care.
Recent Operating Results and Financial Condition
We had cash and cash equivalents of approximately $19.6 million as of June 30, 2016. In addition, we had approximately $24 million of restricted cash that is reserved for a lender who has the option to redeem all, or part, of such amount within 30 days of our public announcement on June 29, 2016, of the discontinuation of the Phase 3 PRESENT Trial upon the IDMC’s recommendation dated June 27, 2016. As of the date of this prospectus, the lender has not redeemed the approximately $24 million in full or in part.
We had no revenue for the quarter ending June 30, 2016, and our cash burn from operations for the quarter ending June 30, 2016 was approximately $13 million. In addition, we paid off our loan with Oxford Finance LLC for a total of $3.1 million. The Company has stopped the PRESENT Trial and is running an investigation that is estimated to cost between $250,000 and $500,000 over the next three months. Depending on the outcome of the investigation, the estimated cost to close out the PRESENT Trial will be between $4 million to $7 million. On July 1, 2016, we paid $2.3 million for the securities class action settlement. We believe that our existing cash and cash equivalents together with the net proceeds of $11.7 million we received on July 13, 2016 from selling 28,000,00010,130,000 shares of common stock and 14,000,000(ii) an aggregate of 1,870,000 pre-funded warrants exercisable for shares of common stock (the “January Pre-Funded Warrants”), together with warrants (the “July Financing”“January Common Warrants”) should be sufficient to fund our operations for at least six months. This projection is based on our current planned operations, stopping the PRESENT Trial and investigationpurchase up to 12,000,000 shares of the causes of the failure of such clinical trial, anticipated payments for defense costs for the cooperation and discussions with the staff in the SEC investigation and other governmental investigations, and is subject to changes in our plans and uncertainties inherent in our business. We will need to seek to replenish our existing cash and cash equivalents priorcommon stock, to the endJanuary Investors. Each share of 2016. We also have funding available under our purchase agreement with Lincoln Park Capital Fund, LLCcommon stock and sales agreements with MLV & Co.accompanying January Common Warrant was sold at a combined offering price of $0.75, and Maxim Group LLC described ineach January Pre-Funded Warrant and accompanying January Common Warrant was sold at a combined offering price of $0.7499. Each January Common Warrant has an exercise price of $0.75 and was exercisable immediately on the previously filed prospectuses, but there is no guarantee that such fundingissuance date and will be availableexpire five years from the issuance date. The aggregate gross proceeds to us on favorable terms or will be sufficient to meet all of our future funding needs. Additionally, in connection with the July Financing, we have agreed not to issue any shares of our common stock (including under our purchase agreement with Lincoln Park Capital Fund, LLC and under our sales agreements with MLV & Co. and Maxim Group LLC) for a period of 75 days from the date ofJanuary Registered Offering were approximately $9.0 million before deducting the closing of the July Financing. At our annual meeting of stockholders adjourned on July 15, 2016, our stockholders approved an increase in our authorized common stock from 275,000,000 to 350,000,000. We are not able to predict whether these additional shares will be sufficient based upon our current stock price to meet the Company’s ongoing financing requirements to maintain the Company’s operations.
If we fail to obtain additional future funding when needed, we could be forced to scale back or terminate our operations, or to seek to merge with or to be acquired by another company. We may not be able to meet our obligations as they come due, raising substantial doubts as to our ability to continue as a going concern. Any such inability to continue as a going concern may result in our common stock holders losing their entire investment. There is no guaranty that we will become profitable or secure additional financing.
Senior Secured DebenturesMarch 2024 Registered Direct Offering and Concurrent Private Placement
On May 10, 2016,March 15, 2024, we entered into a Securities Purchase Agreement (the “Purchase Agreement”) with JGB (Cayman) Newton Ltd.institutional investors (the “Investors”), or JGB, pursuant to which we sold, atagreed to issue and sell, in a 6.375% original issue discount, a total of $25,530,000 Senior Secured Debentures, orregistered direct offering by the Debentures, and warrants to purchase up to 2.0 million shares of our common stock. Net proceeds to us from sale of the Debentures, after payment of commissions and legal fees, were approximately $23,400,000. The Debentures mature November 10, 2018, and accrue interest at 9% per year and contain no conversion features to shares of our common stock.
The Debentures carry an interest only period of six months following which the holder shall have the rights, at its option, to require us to redeem up to $1,100,000 of the outstanding principal amount of these Debentures. Interest is payable at the end of each month based on the outstanding principal. We are required to promptly, but in any event no more than three trading days after the holder delivers a redemption notice to us, to pay the applicable redemption amount in cash or, at our election and subject to certain conditions, in shares of our common stock. If we elect to pay the redemption amount in shares of our common stock, then the shares will be delivered at the lesser of A) 7.5% discountCompany directly to the averageInvestors (the “Registered Offering”), (i) an aggregate of the 3 lowest volume weighted average prices over the prior 20 trading days or B) a 7.5% discount to the prior trading day’s volume weighted average price. We may only opt for payment in11,000,000 shares of common stock if certain equity conditions are met.
As noted above, because the interim analysisand (ii) an aggregate of the PRESENT Trial resulted in a discontinuation2,029,316 pre-funded warrants exercisable for shares of the study, JGB has the right to require us to prepay in cash all, or any portion, of the outstanding principal amount of this Debenture funded in cash by JGB on the closing date, plus all accrued and unpaid interest. If JGB elects such prepayment of the Debentures, then the number of shares subjectcommon stock (the “Pre-Funded Warrants”) to the warrants issuedInvestors. Each share of common stock was sold at an offering price of $1.535, and each Pre-Funded Warrant was sold at an offering price of $1.5349. In a concurrent private placement (the “Private Placement” and together with the Registered Offering, the “Offerings”), we agreed to issue to the holder will be reduced in proportionInvestors warrants exercisable for up to the percentagean aggregate of principal required and accrued interest to be prepaid by us. In addition to the Debenture, JGB received a Series A warrant to purchase 1 million13,029,316 shares upon the closing on the sale of the Debenturescommon stock (the “Common Warrants”) at an exercise price of $1.51. The Series A warrant is$1.41 per share. Each Common Warrant was exercisable beginning November 10, 2016immediately on the issuance date and for a period ofwill expire five years thereafter. Additionally,and six months from the issuance date. The aggregate gross proceeds to us from the Offerings were approximately $20.0 million before deducting the placement agent’s fees and related offering expenses.
Smaller reporting company
We are a “smaller reporting company” as defined in accordance with the termsSecurities Exchange Act of 1934, as amended, or the Exchange Act. We may take advantage of certain of the Securities Purchase Agreement, we issuedscaled disclosures available to JGB a Series B warrantsmaller reporting companies and will be able to purchase 1take advantage of these scaled disclosures for so long as our voting and non-voting Common Stock held by non-affiliates is less than $250.0 million shares uponmeasured on the last business day of our announcementsecond fiscal quarter, or our annual revenue is less than $100.0 million during the most recently completed fiscal year and our voting and non-voting Common Stock held by non-affiliates is less than $700.0 million measured on June 29, 2016,the last business day of the interim analysis conducted by the Independent Data Monitoring Committee of the PRESENT Trial at an exercise price of $0.43. The Series B warrant is exercisable beginning November 10, 2016 and for a period of five years thereafter.our second fiscal quarter.
Risks Associated with Our Business
Our obligations under the Debentures can be acceleratedbusiness and our ability to implement our business strategy are subject to numerous risks, as more fully described in the event we undergo a changesection entitled “Risk Factors” in controlthis prospectus and other customary events of default. In the event of default and acceleration of our obligations, we would be required to pay an amount equal to 103.5% of all amounts of principal and interest then outstanding under the Debentures in cash. Our obligations under the Debentures are secured under a Security Agreement by a senior lien on all of our assets, including all of our interests in our consolidated subsidiaries.most recent Annual Report on Form 10-K incorporated herein by reference. You should read these risks before you invest in our securities. We must also have maintained a minimum of $24.0 million in cash through the date of the public announcement of the interim analysis of the PRESENT Trial. If the trial had not been discontinued as a result of the interim analysis then the minimum cash requirement would have been reducedmay be unable, for many reasons, including those that are beyond our control, to $12.0 million.implement our business strategy.
Armentum Partners, LLC, or Armentum, acted as the placement agent in the offering of the Debentures and we agreed to pay Armentum a fee equal to 2% of the funds received from the sale of the Debentures. We paid half of the placement fee upon funding with the remaining payable unless we prepay the loan, or any portion, of all outstanding principal amounts of the Debentures if the PRESENT trial is discontinued for certain specified reasons.
Corporate Information
We were incorporated on April 3, 2006 in Delaware as Argonaut Pharmaceuticals, Inc. On November 28, 2006, we changed our name to RXi Pharmaceuticals Corporation and began operations January 2007. On September 26, 2011, we changed our name to Galena Biopharma, Inc. In December 2017, we completed a business combination with SELLAS Life Sciences Group, Ltd., and changed our name to “SELLAS Life Sciences Group, Inc.”
Our principal executive offices are located at 2000 Crow Canyon Place,7 Times Square, Suite 380, San Ramon, California 94583,2503, New York, NY 10036, and our phone number is (855) 855-4253.(646) 200-5278. Our website address is www.galenabiopharma.com. We dowww.sellaslifesciences.com. The information contained on, or that can be accessed through, our website is not incorporate the information on our websitepart of, and is not incorporated by reference into, this prospectus and you should not consider such informationbe considered to be part of this prospectus.
We were incorporated as Argonaut Pharmaceuticals, Inc. in Delaware on April 3, 2006 and changed our name to RXi Pharmaceuticals Corporation on November 28, 2006. On September 26, 2011, we changed our company name from RXi Pharmaceuticals Corporation to Galena Biopharma, Inc.
| 4 |
| Shares of Common | ||
| Common Stock to be Outstanding Immediately after this Offering, Assuming Cash Exercise of the Warrants | 69,296,986 shares of Common Stock. | |
| Use of | We will not receive | |
| Nasdaq Capital Market | SLS | |
| Risk | Investing in our | |
The number of shares of common stockour Common Stock shown above to beissued and outstanding after this offeringin the table above is based on 181,837,11756,267,670 shares of our Common Stock outstanding as of March 31, 201625, 2024 and excludes as of such date:excludes:
| · | 2,267,670 shares of Common Stock issuable upon exercise of outstanding options; |
| · | 766,641 shares of Common Stock issuable upon vesting of outstanding restricted stock units; and |
| · | warrants outstanding for the purchase of an aggregate of 41,801,843 shares of Common Stock. |
Throughout this prospectus, when we refer to the shares of our common stock subjectCommon Stock being registered on behalf of the selling stockholders for offer and sale, we are referring to outstanding options having a weighted-average exercise price of $2.59 per share;
The number of shares of common stock issuable upon the exercise of our outstanding warrants and the exercise prices thereof are subject to adjustment in certain circumstances.
NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the other documents we have filed with the SEC that are incorporated herein by reference contain forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995 that involve risks and uncertainties,selling stockholders, as well as assumptions that, if they never materialize or prove incorrect, could cause our results to differ materially from those expressed or implied by such forward-looking statements. All statements other than statementsthe shares of historical fact are statements that could be deemed forward-looking statements, including any projections of financing needs, revenue, expenses, earnings or losses from operations, or other financial items, any statementsCommon Stock issuable upon exercise of the plans, strategiesWarrants, each as described under “The Offering” and objectives of management for future operations, any statements concerning product research, development and commercialization plans and timelines, any statements regarding safety and efficacy of product candidates, any statements of expectation or belief and any statements of assumptions underlying any of“Selling Stockholders.” When we refer to the foregoing. In addition, forward-looking statements may containselling stockholders in this prospectus, we are referring to the words “believe,” “anticipate,” “expect,” “estimate,” “intend,” “plan,” “project,” “will be,” “will continue,” “will result,” “seek,” “could,” “may,” “might,” or any variations of such words or other words with similar meanings. All forward-looking statements attributable to us or to persons acting on our behalf are expressly qualified in their entirety by the cautionary statements and risk factors set forth under “Risk Factors” and elsewhereselling stockholders identified in this prospectus and, set forthas applicable, their donees, pledgees, transferees or other successors-in-interest selling shares of Common Stock or interests in our Form 10-K forshares of Common Stock received after the year ended December 31, 2015 and subsequent Quarterly Reports on Form 10-Q filed with the SEC. See “Where You Can Find More Information” and “Incorporationdate of Certain Documents by Reference” in this prospectus for information on how to access or obtain our reports filed with the SEC.
Given their inherent uncertainty, you should not place undue reliance on these forward-looking statements. You should read this prospectus and the documents that we reference in this prospectus with the understanding that our actual future results may be materially different from what we expect. Except as required by law, we do not undertake any obligation to update or revise any forward-looking statements contained in this prospectus, whethera selling stockholder as a result of new information, future eventsgift, pledge, partnership distribution or otherwise.other transfer.
| 5 |
Investing in our securities involves significant risks. Before making an investment decision,a high degree of risk. You should carefully consider and evaluate all of the information contained in this prospectus and in the documents we incorporate by reference into this prospectus before you decide to purchase our securities. In particular, you should carefully consider and evaluate the risks and uncertainties described below, together withunder the information underheading “Risk Factors” in our most recent Annual Report on Form 10-K10-K. Each of the risks described in these sections and subsequent Quarterly Reports on Form 10-Qdocuments could materially and the other information incorporated by reference in this prospectus. Some of these factors relate principally to our business and the industry in which we operate. Other factors relate principally to your investment in our common stock. If any of these risks were to occur,adversely affect our business, financial condition, results of operations cash flowsand prospects, and could result in a partial or prospects could be materially and adversely affected. In such case, you may lose all or partcomplete loss of your investment.
| 6 |
On March 15, 2024, we entered into the Purchase Agreement with the Investors, pursuant to which we issued and sold, in a registered direct offering directly to the Investors (the “Registered Offering”), (i) an aggregate of 11,000,000 shares of our common stock at an offering price of $1.535 per share and (ii) an aggregate of 2,029,316 pre-funded warrants exercisable for shares of common stock (the “Pre-Funded Warrants”) at an offering price of $1.5349 per Pre-Funded Warrant, for net proceeds of approximately $18.4 million after deducting the placement agent’s fee and estimated offering expenses payable by us.
In a concurrent private placement (the “Private Placement” and together with the Registered Offering, the “Offerings”), we agreed to issue to the Investors who participated in the Registered Offering Warrants exercisable for an aggregate of 13,029,316 shares of Common Stock at an exercise price of $1.41 per share. Each Warrant was exercisable immediately on the issuance date and will expire five years and six months from the issuance date.
In connection with the Offerings, we entered into a placement agency agreement (the “Placement Agent Agreement”) with A.G.P./Alliance Global Partners (the “Placement Agent”), pursuant to which the Placement Agent acted as the exclusive placement agent in connection with the Offerings. Pursuant to the Placement Agent Agreement, we agreed to pay the Placement Agent a fee equal to 7.0% of the aggregate gross proceeds from the Offerings.
Pursuant to the terms of the Purchase Agreement, we agreed to use commercially reasonable efforts to cause a registration statement providing for the resale by holders of shares of our Common Stock issuable upon the exercise of the Warrants, to become effective 30 days (or, in the event of a “full review” by the SEC, within 60 days) following the closing of the Offerings and to keep such registration statement effective at all times.
The foregoing descriptions of the form of Purchase Agreement, the Placement Agent Agreement and the form of Warrant are not complete and are subject to and qualified in their entirety by reference to the form of Purchase Agreement, the form of Placement Agent Agreement and the form of Warrant, respectively, copies of which are attached as Exhibits 1.1, 4.2 and 10.1, respectively, to the Current Report on Form 8-K dated March 15, 2024, and are incorporated herein by reference.
| 7 |
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the documents incorporated by reference into this prospectus contain forward-looking statements about us and our industry that involve substantial risks and uncertainties. All statements other than statements of historical facts contained in this prospectus, the applicable prospectus supplement, and the documented incorporated by reference, including statements regarding our future financial condition, business strategy and plans, and objectives of management for future operations, are forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as “aim,” “anticipate,” “assume,” “believe,” “contemplate,” “continue,” “could,” “design,” “due,” “estimate,” “expect,” “goal,” “intend,” “may,” “objective,” “plan,” “predict,” “positioned,” “potential,” “seek,” “should,” “target,” “will,” “would” and other similar expressions that are predictions of or indicate future events and future trends, or the negative of these terms or other comparable terminology.
We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our financial condition, results of operations, business strategy and financial needs. These forward-looking statements are subject to a number of known and unknown risks, uncertainties and assumptions, including risks described below andin the section titled “Risk Factors” contained in our most recent Annual Report on Form 10-K and subsequent Quarterly Reports on Form 10-Q are notand incorporated by reference in this prospectus, as the only ones facing us. Additionalsame may be amended, supplemented or superseded by the risks and uncertainties described under similar headings in the other documents that are filed after the date hereof and incorporated by reference into this prospectus, regarding, among other things:
| · | our future financial and business performance; |
| · | strategic plans for our business and product candidates; |
| · | our ability to develop or commercialize products; |
| · | the expected results and timing of clinical trials and nonclinical studies; |
| · | our ability to comply with the terms of our license agreements; |
| · | developments and projections relating to our competitors and industry; |
| · | our expectations regarding our ability to obtain, develop and maintain intellectual property protection and not infringe on the rights of others; |
| · | our ability to retain and attract highly-skilled executive officers and employees; |
| · | our future capital requirements and the timing of those requirements and sources and uses of cash; |
| · | our ability to obtain funding for our operations; |
| · | changes in applicable laws or regulations; |
| · | risks associated with preclinical or clinical development and trials; |
| · | changes in the assumptions underlying our expectations regarding our future business or business model; |
| · | our ability to develop, manufacture and commercialize product candidates; |
| · | general economic, financial, legal, political and business conditions and changes in domestic and foreign markets; |
| · | changes in applicable laws or regulations; |
| 8 |
| · | the impact of natural disasters, including climate change, and the impact of health epidemics, on our business; |
| · | the size and growth potential of the markets for our products, and our ability to serve those markets; |
| · | market acceptance of our planned products; |
| · | our ability to raise capital; |
| · | the possibility that we may be adversely affected by other economic, business, and/or competitive factors; and |
| · | other risks and uncertainties set forth herein in the section entitled “Risk Factors.” |
These risks are not presently known to usexhaustive. Other sections of this prospectus, the applicable prospectus supplement, or the documents incorporated herein by reference may include additional factors that we currently deem immaterial may also materially and adversely affectcould harm our business and operations.
Risks Relatingfinancial performance. Moreover, we operate in a very competitive and rapidly changing environment. New risk factors emerge from time to Our Former Commercial Operations
We are subject to U.S. federaltime, and state health care fraud and abuse and false claims laws and regulations, and we recently have been subpoenaed in connection with marketing and promotional practices related to Abstral. Prosecutions under such laws have increased in recent years and we may become subject to such prosecutions or related litigation under these laws. If we haveit is not fully complied with such laws, we could face substantial penalties.
Our former commercial operations are subject to various U.S. federal and state fraud and abuse laws, including, without limitation, the federal False Claims Act, federal Anti-Kickback Statute, and the federal Sunshine Act.
A federal investigation of two of the high-prescribing physicianspossible for Abstral has resulted in the criminal prosecution of the two physicians for alleged violations of the federal False Claims Act and other federal statutes. The criminal trial is set for October 2016. We have received a trial subpoena for documents in connection with that investigation and we have been in contact with the U.S. Attorney’s Office for the Southern District of Alabama, which is handling the criminal trial, and are cooperating in the production of documents. On April 28, 2016, a second superseding indictment was filed in the criminal case, which added additional information about the defendant physicians and provided information regarding the facts and circumstances involving a rebate agreement between the Company and the defendant physicians’ pharmacy as well as their ownership of our stock. Certain former employees have received trial subpoenas to appear at the trial and provide oral testimony. We have agreed to reimburse those former employees’ attorney’s fees. To our knowledge, we are not a target or subject of that investigation.
There also have been federal and state investigations of a company that has a product that competes with Abstral in the same therapeutic class, and we have learned that the FDA and other governmental agencies are investigating our Abstral promotion practices. On December 16, 2015, we received a subpoena issued by the U.S. Attorney’s Office in District of New Jersey requesting the production of a broad range of documents pertaining to our marketing and promotional practices for Abstral. We have been in contact with the U.S. Attorney’s Office for the District of New Jersey and are cooperating in the production of the requested documents. We are unablemanagement to predict whetherall risk factors, nor can we could become subject to legal or administrative actions as a result of these matters, orassess the impact of such matters. If we are found to be in violation of the False Claims Act, Anti-Kickback Statute, Patient Protection and Affordable Care Act, or any other applicable state or any federal fraud and abuse laws, we may be subject to penalties, such as civil and criminal penalties, damages, fines, or an administrative action of exclusion from government health care reimbursement programs. We can make no assurances as to the time or resources that will need to be devoted to these matters or their outcome, or the impact, if any, that these matters or any resulting legal or administrative proceedings may haveall factors on our business or financial condition.the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in, or implied by, any forward-looking statements.
The federal False Claims Act prohibits persons from knowingly filing,
You should not rely upon forward-looking statements as predictions of future events. We cannot assure you that the events and circumstances reflected in the forward-looking statements will be achieved or causingoccur. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. Except as required by law, we undertake no obligation to be filed, a false claimupdate publicly any forward-looking statements for any reason after the date of this prospectus or to or the knowing use of falseconform these statements to obtain payment from,actual results or to changes in our expectations.
You should carefully read this prospectus, and the federal government. Qui tam suits filedapplicable prospectus supplement, together with the information incorporated herein by reference as described under the False Claims Act can be broughtheading “Incorporation by any individual on behalf of the government and such individuals, commonly known as “relators” or “whistleblowers,Reference,” may share in any amounts paid by the entity to the government in fines or settlement. The frequency of filing qui tam actions has increased significantly in recent years, causing greater numbers of health care companies to have to defend such qui tam actions and pay substantial sums to settle such actions.
The federal Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering, receiving, or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing or arranging for a good or service, for which payment may be made under a federal health care program such as the Medicare and Medicaid programs. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal health care covered business, the statute has been violated. The Anti-Kickback Statute is broad, and despite a series of narrow safe harbors, prohibits many arrangements and practices that are lawful in businesses outside of the health care industry. Penalties for violations of the federal Anti-Kickback Statute include criminal penalties and civil and administrative sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other federal health care programs. An alleged violation of the Anti-Kickback Statute may be used as a predicate offense to establish liability pursuant to other federal laws and regulations such as the federal False Claims Act. Many states have also adopted laws similar to the federal Anti-Kickback Statute, some of which apply to the referral of patients for health care items or services reimbursed by any source, not only Medicare and Medicaid programs.
The federal Patient Protection and Affordable Care Act includes provisions expanding the ability of certain relators to bring actions that would have been dismissed under prior law. When an entity is determined to have violated the federal False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties for each separate false claim. The Deficit Reduction Act of 2005 encouraged states to enact or modify their state false claims acts to be at least as effective as the federal False Claims Act by granting states a portion of any federal Medicaid funds recovered through Medicaid-related actions. Most states have enacted state false claims laws, and many of those states included laws including qui tam provisions. The federal Patient Protection and Affordable Care Act includes provisions known as the Physician Payments Sunshine Act, which requires manufacturers of drugs, biologics, devices and medical supplies covered under Medicare and Medicaid to record any transfers of value to physicians and teaching hospitals and to report this data beginning in 2013 to the Centers for Medicare and Medicaid Services for subsequent public disclosures. Manufacturers must also disclose investment interests held by physicians and their family members. Failure to submit the required information may result in civil monetary penalties of up to $1 million per year for knowing violations and may result in liability under other federal laws or regulations. Similar reporting requirements have also been enacted on the state level in the U.S., and an increasing number of countries worldwide either have adopted or are considering similar laws requiring transparency of interactions with health care professionals. In addition, some states such as Massachusetts and Vermont imposed an outright ban on certain gifts to physicians. These laws could affect our product promotional activities by limiting the kinds of interactions we could have with hospitals, physicians or other potential purchasers or users of our system. Both the disclosure laws and gift bans also will impose administrative, cost and compliance burdens on us.
We face product liability exposure and, if successful claims are brought against us, we may incur substantial liability if our insurance coverage for those claims is inadequate.
The commercial sale of our products after they are approved as well as the usedocuments filed as exhibits to the registration statement of which this prospectus is a part with the understanding that our products in clinical trials exposes us to possible product liability claims. This risk exists even if a product is approved for commercial sale by the FDAactual future results, levels of activity, performance and manufactured in facilities licensed and regulated by the FDA, if our products were sold to third parties, or if our products are provided in clinical trials. Our products are designed to affect important bodily functions and processes. Any side effects, manufacturing defects, misuse or abuse associated with our products could result in injury to a patient or even death. For example, because the placebo may have performed better than NeuVax in the PRESENT Trial, the use of NeuVax may have worsened the patient’s condition.
Product liability claimsachievements may be brought against us by consumers, health care providers, pharmaceutical companies or others selling or otherwise coming into contact with our products or generic versions of our products. Ifmaterially different from what we cannot successfully defend ourselves against product liability claims we could incur substantial liabilities. Because we have sold Abstral and Zuplenz and provided NeuVax as a study drug in the Present Trial, regardless of merit or eventual outcome, product liability claims may result in:
expect. We have obtained product liability insurance coverage for commercial product sales with a $10 million per occurrence and a $10 million annual aggregate coverage limit. Our insurance coverage may not be sufficient to coverqualify all of our product liability related expensesforward-looking statements by these cautionary statements.
Except as required by law, we assume no obligation to update these forward-looking statements publicly, or losses and may not cover us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive and,to update the reasons actual results could differ materially from those anticipated in these forward-looking statements, even if new information becomes available in the future, wefuture.
| 9 |
We will not receive any of the proceeds from the sale of Common Stock by the selling stockholders named in this prospectus, and the selling stockholders will receive all of the proceeds from this offering.
We may not be ablereceive up to maintain insurance coverage at a reasonable cost,approximately $18.4 million in sufficient amounts or upon adequate terms to protect us against losses due to product liability. If we determine that it is prudent to increase our product liability coverageaggregate gross proceeds from cash exercises of the Warrants, based on salesthe per share exercise price of the Warrants. Any proceeds we receive from the exercise of the Warrants will be used to advance our products,clinical programs and for working capital and general corporate purposes. The holders of the Warrants are not obligated to exercise their Warrants, and we may be unablecannot predict whether holders of the Warrants will choose to obtainexercise all or any of their Warrants.
| 10 |
This prospectus relates to the sale or other disposition of up to 13,029,316 shares of Common Stock issuable to the selling stockholders upon exercise of the Warrants by the selling stockholders named below, and their donees, pledgees, transferees or other successors-in-interest selling shares of Common Stock or interests in shares of Common Stock received after the date of this increased product liability insuranceprospectus from a selling stockholder as a gift, pledge, partnership distribution or other transfer. The shares of Common Stock covered hereby were issued by us in the Private Placement. See “The Private Placement” beginning on commercially reasonable terms or at all. Large judgments have been awarded in class action or individual lawsuits based on drugs that had unanticipated side effects, including side effectspage 7 of this prospectus.
The table below sets forth information as of March 25, 2024, to our knowledge, for the selling stockholders and other information regarding the beneficial ownership (as determined under Section 13(d) of the Exchange Act and the rules and regulations thereunder) of the shares of Common Stock held by the selling stockholders. The second column lists the number of shares of Common Stock and percentage beneficially owned by the selling stockholders as of March 25, 2024. The third column lists the maximum number of shares of Common Stock that may be less severe than thosesold or otherwise disposed of our products. A successful product liability claim or seriesby the selling stockholders pursuant to the registration statement of claims brought against us could cause our stock price to decline and, if judgments exceed our insurance coverage, could decrease our cash and havewhich this prospectus forms a material adverse effect on our business, results of operations, financial condition and prospects.
Our business involves the use of hazardous materials and we and our third-party manufacturers and suppliers must comply with environmental laws and regulations, which can be expensive and restrict how we do business.
Our third-party manufacturers and suppliers activities involve the controlled storage, use and disposal of hazardous materials. We and our manufacturers and suppliers are subject to laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials even after wepart. The selling stockholders may sell or otherwise dispose of the products. In some, cases, these hazardous materialsall or none of their shares. Pursuant to Rules 13d-3 and various wastes resulting from their use will be stored at our contractors or manufacturers’ facilities pending use and disposal. We cannot completely eliminate the risk of contamination, which could cause injury to our employees and others, environmental damage resulting in costly clean-up and liabilities under applicable laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. Although we expect that the safety procedures utilized by our third-party contractors and manufacturers for handling and disposing of these materials will generally comply with the standards prescribed by these laws and regulations, we cannot guarantee that this will be the case or eliminate the risk of accidental contamination or injury from these materials. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources. We do not currently carry biological or hazardous waste insurance coverage and our property and casualty and general liability insurance policies specifically exclude coverage for damages and fines arising from biological or hazardous waste exposure or contamination.
We will continue to be responsible for certain liabilities and obligations related to Abstral and Zuplenz, and if unknown liabilities were to arise it could have a material adverse effect on us.
Under our respective asset purchase agreements with Sentynl and Midatech, our future obligations under our former agreements with Orexo AB and MonoSol have been assumed by Sentynl and Midatech, respectively, except that we will continue to be responsible for chargebacks, rebates, patient assistance and certain other product distribution channel liabilities related to Abstral and Zuplenz for a specified period of time post-closing. We also will be responsible for any pre-closing liabilities and obligations related to Abstral and Zuplenz, including unknown liabilities, and have agreed in the respective asset purchase agreements to indemnify Sentynl and Midatech for any breach of our representations, warranties and covenants in the respective asset purchase agreements up to a certain agreed to amount. We cannot quantify these responsibilities to Sentanyl and Midatech, but if substantial unknown liabilities were to arise, it could have a material adverse effect on our financial condition.
Risks Relating to Our Development Programs
Our drug candidates may not receive regulatory approval or be successfully commercialized.
Before they can be marketed, our products in development must be approved by the FDA or similar foreign governmental agencies. The process for obtaining FDA approval is both time-consuming and costly, with no certainty of a successful outcome. Before obtaining regulatory approval for the sale of any drug candidate, we must conduct extensive preclinical tests and clinical trials to demonstrate the safety and efficacy in humans of our product candidates. Although our drug candidates have exhibited no serious
adverse events, or SAEs, in the Phase 1 and 1/2 clinical trial, SAEs or other unexpected side effects may arising during further testing and development. A failure of any preclinical study or clinical trial can occur at any stage of testing. The results of preclinical and initial clinical testing of these products may not necessarily indicate the results that will be obtained from later or more extensive testing. It also is possible to suffer significant setbacks in advanced clinical trials, even after obtaining promising results in earlier trials.
Our Phase 3 PRESENT clinical trial has been stopped due to futility and though we are conducting an investigation13d-5 of the causes for the failure of the clinical trial, we are not certain that the investigation will result in the reason(s) for the failure.
On June 27, 2016, the Independent Data Monitoring Committee conducting the pre-planned interim analysis of the PRESENT Trial recommended that we stop the clinical trial because of futility. We are conducting an investigation of the causes of the failure of the trial, which may take a number of months. Once the investigation has been concluded, we may or may not know the reasons for the failure of the clinical trial. As we are conducting the investigation, we have stopped any further dosing of the patients. The investigation may cause us to unblind the data of the clinical trial. Because we will be investigating the causes of the failure of the clinical trial, the PRESENT Trial may no longer likely be considered a registrational clinical trial. Even if we determine the causes of the failure, NeuVax may never be approved for the treatment of patients with Node positive HER2 negative breast cancer.
Our products, GALE 301 and GALE 302 have the same mechanism of actions NeuVax and may not be a viable product to prevent the recurrence of ovarian cancer or other types of cancers.
GALE 301/302, which have a similar mechanism of action as NeuVax, may no longer be a viable product to prevent the recurrence of ovarian cancer or other types of cancer, depending upon the reasons for the causes of the failure of NeuVax.
A number of different factors could prevent us from obtaining regulatory approval or commercializing our product candidates on a timely basis, or at all.
We, the FDA or other applicable regulatory authorities, an Independent Data Safety Monitoring Board, or IDSMB, governing our clinical trials, or an institutional review board, or IRB, which is an independent committee registered with and overseen by the U.S. Department of Health and Human Services, or HHS, that functions to approve, monitor and review biomedical and behavioral research involving humans, may suspend clinical trials of a drug candidate at any time for various reasons, including if we or it believe the subjects or patients participating in such trials are being exposed to unacceptable health risks. Among other reasons, adverse side effects of a drug candidate on subjects or patients in a clinical trial could result in the FDA or other regulatory authorities suspending or terminating the trial and refusing to approve a particular drug candidate for any or all indications of use.
Clinical trials of a new drug candidate require the enrollment of a sufficient number of patients, including patients who are suffering from the disease the drug candidate is intended to treat and who meet other eligibility criteria. Rates of patient enrollment are affected by many factors, and delays in patient enrollment can result in increased costs and longer development times than we expect at present. Patients who are enrolled at the outset of this standard of care also may eventually choose for personal reasons not to participate in the study. We also compete for eligible patients with other breast cancer trials underway from time to time, and we may experience delays in patient enrollment due to the dependency of other large trials underway in the same patient population.
Clinical trials also require the review and oversight of IRBs, which approve and continually review clinical investigations to protect the rights and welfare of human subjects. An inability or delay in obtaining IRB approval could prevent or delay the initiation and completion of clinical trials, and the FDA may decide not to consider any data or information derived from a clinical investigation not subject to initial and continuing IRB review and approval.
In addition, cancer vaccines are a relatively new form of therapeutic treatment and a very limited number of such products have received regulatory approval. Therefore, the FDA or other regulatory authority may apply standards for approval of a new cancer vaccine that is different from past experience.
Numerous factors could affect the timing, cost or outcome of our drug development efforts, including the following:
It is possible that none of the product candidates that we develop will obtain the appropriate regulatory approvals necessary for us to begin selling them or that any regulatory approval to market a product may be subject to limitations on the indicated uses for which we may market the product. The time required to obtain FDA and other approvals is unpredictable but often can take years following the commencement of clinical trials, depending upon the complexity of the drug candidate. Any analysis we perform of data from clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. Any delay or failure in obtaining required approvals could have a material adverse effect on our ability to generate revenue from the particular drug candidate.
In addition, the length of time to develop the product candidates as well as any regulatory delays in the development and regulatory approval process could cause the patent exclusivity to be unavailable or greatly reduced for each product candidate. The lack of patent exclusivity could have a material adverse effect on our ability to generate revenue from the particular drug candidate.
We are also subject to numerous foreign regulatory requirements governing the conduct of clinical trials, manufacturing and marketing authorization, pricing and third-party reimbursement. The foreign regulatory approval processExchange Act, beneficial ownership includes all of the risks associated with the FDA approval described above as well as risks attributable to the satisfaction of local regulations in foreign jurisdictions. Approval by the FDA does not assure approval by regulatory authorities outside of the U.S.
We are dependent upon contract manufacturers for clinical supplies of our product candidates.
We do not have the facilities or expertise to manufacture supplies of any of our product candidates for clinical trials. Accordingly, we are dependent upon contract manufacturers for these supplies. There can be no assurance that we will be able to secure needed supply arrangements on reasonable terms, or at all. Our failure to secure these arrangements as needed could have a materially adverse effect on our ability to complete the development of our product candidates or, if we obtain regulatory approval for our product candidates, to commercialize them.
Our current plans call for the manufacture of our compounds by contract manufacturers offering research grade, Good Laboratory Practices grade and Good Manufacturing Practices grade materials for preclinical studies (e.g., toxicology studies) and for clinical use. Certain of our product candidates are complex molecules requiring many synthesis steps, which may lead to challenges with purification and scale-up. These challenges could result in increased costs and delays in manufacturing.
NeuVax is administered in combination with Leukine, a “GM-CSF” available in both liquid and lyopholyzed forms exclusively from Genzyme Corporation, or “Genzyme,” a subsidiary of Sanofi-Aventis. We will continue to be dependent on Genzyme for our supply of Leukine in connection with the ongoing NeuVax and GALE-301/GALE-302 trials and the potential commercial manufacture of these programs. Any temporary interruptions or discontinuation of the availability of Leukine, or any determination by us to change the GM-CSF used with NeuVax or GALE-301/GALE-302, may have a material adverse effect on our clinical trials and any commercialization of the assets.
We may not be able to establish or maintain the third-party relationships that are necessary to develop or potentially commercialize some or all of our product candidates.
We expect to depend on collaborators, partners, licensees, clinical research organizations and other third parties to support our discovery efforts, to formulate product candidates, to manufacture our product candidates, and to conduct clinical trials for some or all of our product candidates. We cannot guarantee that we will be able to successfully negotiate agreements for or maintain relationships with collaborators, partners, licensees, clinical investigators, vendors and other third parties on favorable terms, if at all. Our ability to successfully negotiate such agreements will depend on, among other things, potential partners’ evaluation of the superiority of our technology over competing technologies and the quality of the preclinical and clinical data that we have generated, and the perceived risks specific to developing our product candidates. If we are unable to obtain or maintain these agreements, we may not be able to clinically develop, formulate, manufacture, obtain regulatory approvals for or commercialize our product candidates. Under certain license agreements that we have already entered into, we have minimum dollar amounts per year that we are obligated to spend on the development of the technology we have licensed from our contract partners and other obligations to maintain certain licenses. If we fail to meet this requirement under any of our licenses that contain such requirements or any other obligations under these licenses, we may be in breach of our obligations under such agreement, which may result in the loss of the technology licensed. We cannot necessarily control the amount or timing of resources that our contract partners will devote to our research and development programs, product candidates or potential product candidates, and we cannot guarantee that these parties will fulfill its obligations to us under these arrangements in a timely fashion. We may not be able to readily terminate any such agreements with contract partners even if such contract partners do not fulfill its obligations to us.
In addition, we may receive notices from third parties from time to time alleging that our technology or product candidates infringe upon the intellectual property rights of those third parties. Any assertion by third parties that our activities or product candidates infringe upon its intellectual property rights may adversely affect our ability to secure strategic partners or licensees for our technology or product candidates or our ability to secure or maintain manufacturers for our compounds.
We are subject to competition and may not be able to compete successfully.
The biotechnology industry, including the cancer immunotherapy market, is intensely competitive and involves a high degree of risk. We compete with other companies that have far greater experience and financial, research and technical resources than us. Potential competitors in the U.S. and worldwide are numerous and include pharmaceutical and biotechnology companies, educational institutions and research foundations, many of which have substantially greater capital resources, marketing experience, research and development staffs and facilities than us. Some of our competitors may develop and commercialize products that compete directly with those incorporating our technology, introduce products to market earlier than our products or on a more cost effective basis. In addition, our technology may be subject to competition from other technology or methods developed using techniques other than those developed by traditional biotechnology methods. Our competitors compete with us in recruiting and retaining qualified scientific and management personnel as well as in acquiring technologies complementary to our technology. We and our collaborators may face competition with respect to product efficacy and safety, ease of use and adaptability to various modes of administration, acceptance by physicians, the timing and scope of regulatory approvals, availability of resources, reimbursement coverage, price and patent position, including the potentially dominant patent positions of others. An inability to successfully complete our product development could lead to us having limited prospects for establishing market share or generating revenue from our technology.
For patients with early stage breast cancer, adjuvant therapy is often given to prevent recurrence and increase the chance of long-term disease free survival. Adjuvant therapy for breast cancer can include chemotherapy, hormonal therapy, radiation therapy, or combinations thereof. In addition, the HER2 targeted drug trastuzumab (Herceptin®) may be given to patients with tumors with high expression of HER2 (IHC 3+), in the adjuvant setting which may be useful in treating breast cancer.
There are a number of cancer vaccines in development for breast cancer, including but not limited to Lapuleucel-T (Dendreon), and AE-37 (Antigen Express). While these development candidates are aimed at a number of different targets, and AE-37 has published data in the HER2 breast cancer patient population, there is no guarantee that any of the these compounds will not in the future be indicated for treatment of low to intermediate HER2 breast cancer patients and become directly competitive with NeuVax.
We are dependent on technologies we license, and if we lose the right to license such technologies or we fail to license new technologies in the future, our ability to develop new products would be harmed.
We currently are dependent on licenses from third parties for technologies relating to our product candidates. Our current licenses impose, and any future licenses we enter into are likely to impose, various development, funding, royalty, diligence, sublicensing, insurance and other obligations on us. If our license with respect to any of these technologies is terminated for any reason, the development of the products contemplated by the licenses would be delayed, or suspended altogether, while we seek to license similar technology or develop new non-infringing technology. The costs of obtaining new licenses are high.
Risks associated with operating in foreign countries could materially adversely affect our product development.
We may conduct future studies in countries outside of the U.S. Consequently, we may be subject to risks related to operating in foreign countries. Risks associated with conducting operations in foreign countries include:
In addition, there may be political instability, including war, terrorism, riots, civil insurrection or social unrest, and natural or man-made disasters, including famine, flood, fire, earthquake, storm or disease, which could seriously harm the progress of our clinical trials at sites in particular foreign countries or regions.
Risks Relating to Our Financial Position and Capital Requirements
We may not be able to obtain sufficient financing, and may not be able to develop our product candidates.
We had cash and cash equivalents of approximately $19.6 million as of June 30, 2016. In addition, we had approximately $24 million of restricted cash that is reserved for a lender who has the option to redeem all, or part, of such amount within 30 trading days of our public announcement on June 29, 2016, of the discontinuation of the Phase 3 PRESENT Trial upon the IDMC’s recommendation on June 27, 2016. As of the date of this prospectus, the lender has not redeemed the approximately $24 million in full or in part.
We had no revenue for the quarter ending June 30, 2016, and our cash burn from operations for the quarter ending June 30, 2016 was approximately $13 million. In addition, we paid off our loan with Oxford Finance LLC for a total of $3.1 million. The Company has stopped the PRESENT Trial and is running an investigation that is estimated to cost between $250,000 and $500,000 over the next three months. Depending on the outcome of the investigation, the estimated cost to close out the PRESENT Trial will be between $4 million to $7 million. On July 1, 2016, we paid $2.3 million for the securities class action settlement. We believe that our existing cash and cash equivalents together with the net proceeds we received on July 13, 2016 from selling 28,000,000 shares of common stock and 14,000,000 warrants (the “July Financing”) should be sufficient to fund our operations for at least six months. This projection is based on our current planned operations, stopping the PRESENT Trial and investigation of the causes of the failure of such clinical trial, anticipated payments for defense costs for the cooperation and discussions with the staff in the SEC investigation and other governmental investigations, and is subject to changes in our plans and uncertainties inherent in our business. We will need to seek to replenish our existing cash and cash equivalents prior to the end of 2016. We also have funding available under our purchase agreement with Lincoln Park Capital Fund, LLC and sales agreements with MLV & Co. and Maxim Group LLC described in the previously filed prospectuses, but there is no guarantee that such funding will be available to us on favorable terms or will be sufficient to meet all of our future funding needs. Additionally, in connection with the July Financing, we have agreed not to issue any shares of our common stock (including under our purchase agreement with Lincoln Park Capital Fund, LLC and under our sales agreements with MLV & Co. and Maxim Group LLC) for a period of 75 days from the date of the closing of the July Financing. At our annual meeting of stockholders adjourned on July 15, 2016, our stockholders approved an increase in our authorized common stock from 275,000,000 to 350,000,000. We are not able to predict whether these additional shares will be sufficient based upon our current stock price to meet the Company’s ongoing financing requirements to maintain the Company’s operations.
If we fail to obtain additional future funding when needed, we could be forced to scale back or terminate our operations, or to seek to merge with or to be acquired by another company. We may not be able to meet our obligations as they come due, raising substantial doubtsCommon Stock as to our ability to continue aswhich a going concern. Any such inability to continue as a going concern may result in our common stock holders losing their entire investment. There is no guaranty that we will become profitablestockholder has sole or secure additional financing.
We expect to continue to incur significant researchshared voting power or investment power, and development expenses, which may make it difficult for us to attain profitability, and may lead to uncertainty about our ability to continue as a going concern.
Substantial funds were expended to develop our technologies and product candidates, and additional substantial funds will be required for further preclinical testing and clinical trialsalso any shares of our product candidates, and to manufacture and market any products that are approved for commercial sale. BecauseCommon Stock which the successful development of our products is uncertain, we are unable to precisely estimate the actual funds we will require to develop and potentially commercialize them. In addition, we may not be able to generate enough revenue, even if we are able to commercialize any of our product candidates, to become profitable.
In the event that we are unable to achieve or sustain profitability or to secure additional financing, we may not be able to meet our obligations as they come due, raising substantial doubts as to our ability to continue as a going concern. Any such inability to continue as a going concern may result in our common stock holders losing their entire investment. There is no guaranty that we will become profitable or secure additional financing. Our financial statements contemplate that we will continue as a going concern and do not contain any adjustments that might result if we were unable to continue as a going concern. Changes in our operating plans, our existing and anticipated working capital needs, the acceleration or modification of our expansion plans, increased expenses, potential acquisitions or other events will all affect our ability to continue as a going concern. Future financing may be obtained through, and future development efforts may be paid for by, the issuance of debt or equity, which may have an adverse effect on our security holders or may otherwise adversely affect our business.
If we raise funds through the issuance of debt or equity, any debt securities or preferred stock issued will have rights, preferences and privileges senior to those of holders of our common stock in the event of a liquidation. In such event, there is a possibility that once all senior claims are settled, there may be no assets remaining to pay out to the holders of common stock. In addition, if we raise funds through the issuance of additional equity, whether through private placements or additional public offerings, such an issuance would dilute your ownership in us.
The terms of debt securities may also impose restrictions on our operations, which may include limiting our ability to incur additional indebtedness, to pay dividends on or repurchase our capital stock, or to make certain acquisitions or investments. In addition, we may be subject to covenants requiring us to satisfy certain financial tests and ratios, and our ability to satisfy such covenants may be affected by events outside of our control.
You may have difficulty evaluating our business, and our historical financial information may not be representative of our future results.
We recently announced the recommendation to stop the PRESENT Trial for futility and we are investigating the reasons for such failure. As a result, we will focus our resources on our pipeline of other product candidates. As a result, we will have no recurring revenues unless and until we are able to obtain marketing approval of one or more of our other product candidates and our historical financial information may not be representative of our future results.
We may be unable to comply with our reporting and other requirements under federal securities laws.
As a publicly traded company, we are subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, or the “Exchange Act,” and the Sarbanes-Oxley Act of 2002, or the “Sarbanes-Oxley Act.” In addition, the Exchange Act requires that we file annual, quarterly and current reports. Our failure to prepare and disclose this information in a timely manner could subject us to penalties under federal securities laws, expose us to lawsuits and restrict our ability to access financing. The Sarbanes-Oxley Act requires that we, among other things, establish and maintain effective internal controls and procedures for financial reporting. From time to time we evaluate our existing internal controls in light of the standards adopted by the Public Company Accounting Oversight Board. It is possible that we or our independent registered public accounting firm may identify significant deficiencies or material weaknesses in our internal control over financial reporting in the future. Any failure or difficulties in implementing and maintaining these controls could cause us to fail to meet the periodic reporting obligations or result in material misstatements in our financial statements.
Section 404 of the Sarbanes-Oxley Act requires annual management and independent auditor assessments of the effectiveness of our internal control over financial reporting. Our failure to satisfy the requirements of Section 404 on a timely basis could result in the loss of investor confidence in the reliability of our financial statements, which in turn could have a material adverse effect on our business and our common stock.
Risks Related to Our Intellectual Property
We may not be able to obtain and enforce patent rights or other intellectual property rights that cover our product candidates and that are of sufficient breadth to prevent third parties from competing against us.
Our success with respect to our product candidates will depend in part on our ability to obtain and maintain patent protection in the U.S. and abroad, to preserve our trade secrets, and to prevent third parties from infringing upon our proprietary rights. Our patents and patent applications, however, may not be sufficient to provide protection for NeuVax or our other products and product candidates against commercial competition.
The active peptide found in NeuVax, the E75 peptide,stockholder has been known and studied for many years. We have one issued U.S. patent, US 6,514,942, covering the composition of matter of the E75 peptide, which expired in mid-2015, prior to any potential commercialization of NeuVax. We do not have and will not be able to obtain any composition of matter patent protection for E75, the active peptide in NeuVax. We also have a license from The Henry M. Jackson Foundation to issued U.S., European, Japanese, Korean, Mexican and Australian method of use patents, which expire in 2028, that are directed to a method of inducing immunity against breast cancer recurrence by administering a composition comprising the E75 peptide to patients who have both an immunohistochemistry (IHC) rating of 1+ or 2+ for HER2/neu protein expression, as well as a fluorescence in situ hybridization (FISH) rating of less than about 2.0 for HER2/neu gene expression. The license further includes an issued U.S. method of use patent directed to a method of inducing immunity against recurrence of any HER2/neu expressing tumors by administering the E75 peptide to patients with tumors having a FISH rating of less than about 2.0 for HER2/neu gene expression; an allowed U.S application which includes claims to the use of E75 to reduce the risk of cancer recurrence, including bone only recurrence; and pending applications with similar claims in a number of foreign jurisdiction, all of which expire in 2028. Also included in the license is a method of use patent, which expires in 2026, that is directed to the use of NeuVax in combination with Herceptin® to treat any HER2/neu expressing cancer. Thus, our method of use patents may not prevent competitors from seeking to develop and market NeuVax for use in cancer patients who do not meet these criteria. If any such alternative uses were approved, this could lead to off-label use and price erosion for our NeuVax product. We may seek FDA approval for use of NeuVax to treat cancer patients who fall outside the claimed IHC and FISH ranges and for other cancers as well. Although we are pursuing additional patent protection for NeuVax through pending patent applications, we may not be able to obtain additional patent protection that would provide us with a significant commercial advantage.
Anagrelide hydrochloride, the sole active pharmaceutical ingredient, or API, in GALE-401, has been approved for many years and, thus, it is not possible to obtain composition of matter patents that cover anagrelide hydrochloride. As a result, competitors who obtain the requisite regulatory approval can offer products with the same 43 API as GALE-401, so long as the competitors do not infringe any formulation patents that we may have or may obtain or license, if any. The only patent protection that we have or are likely to obtain covering GALE-401 are patents relating to specific formulations, methods using these formulations, and methods of manufacturing and packaging. We have an issued U.S. Patent, which expires in 2020, covering methods of using anagrelide to reduce platelet count in patients subject to veno-occlusive events. We have granted patents in the U.S., United Kingdom and Japan, which expire in 2029, covering controlled release formulations of anagrelide and methods of use. We also are prosecuting pending patent applications in other territories including, but not limited to, the U.S. Europe, India and Japan, which may not issue prior to any potential commercialization of GALE-401. We may seek FDA approval for use of GALE-401 to treat patients with myeloproliferative neoplasms that include several hermatological disorders. Although we are pursuing additional patent protection for GALE-401 through pending patent applications, we may not be able to obtain additional patent protection that would provide us with a significant commercial advantage.
The active peptides found in GALE-301 and GALE-302 are derived from Folate Binding Protein. One of the active peptides, E39, has been known and studied for many years. The other active peptide, GALE-302, is a derivative of E39. We have a license from The Henry M. Jackson Foundation to issued and granted patents in the U.S., Europe, Canada, and Japan, covering composition of matter for the E39 derivative peptides, including GALE-302, alone and in combination with E39, as well as the use of these compositions for the treatment of cancer. These patents are expected to expire in 2022, prior to any potential commercialization of GALE-301. We do not have and will not be able to obtain any composition of matter patent protection for the E39 peptide in any territory. The license we have from The Henry M. Jackson Foundation grants us the right to develop and market GALE-301 for any use, including methodsacquire within 60 days of treating cancer, our patents may not prevent competitors from seeking to develop and market the E39 peptide alone. If any such alternative usesMarch 25, 2024. The percentage of compositions containing the E39 peptide were approved, this could lead to off label use and price erosion for GALE-301. We may seek FDA approval for use of GALE-301, alone or in combination with GALE-302, to treat cancer patients with ovarian and endometrial cancers and for other cancers, as well. Although we are pursuing additional patent protection for GALE-301 and the combination of GALE-301 and GALE-302 through pending patent applications, we may not be able to obtain additional patent protection that would provide us with a significant commercial advantage.
Our ability to obtain, maintain and enforce patents is uncertain and involves complex legal and factual questions. Accordingly, rights under any patents we have or may obtain or license may not provide us with sufficient protection for our commercial product and product candidates to afford a commercial advantage against competitive products or processes, including those from branded and generic pharmaceutical companies. In addition, we cannot guarantee that any patents will issue from any pending or future patent applications owned by or licensed to us. Nor can we guarantee that the claims of these patents will be held valid or enforceable by the courts or will provide us with any significant protection against competitive products or otherwise be commercially valuable to us.
Changes in either the patent laws or in the interpretations of patent laws in the U.S. or abroad may diminish the value of our intellectual property. In addition, on September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to the U.S. patent law. These include provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act, in particular the first-to-file provision and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement of or defense of our issued patents, all of which could have a material adverse effect on our business and financial condition. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents.
While we intend to take actions reasonably necessary to enforce our patent rights, we may not be able to detect infringement of our own or in-licensed patents, which may be especially difficult for methods of manufacturing or formulation products, and we depend, in part, on our licensors and collaborators to protect a substantial portion of our proprietary rights. In addition, third parties may challenge our in-licensed patents and any of our own patents that we may obtain, which could result in the invalidation or unenforceability of some or all of the relevant patent claims. Litigation or other proceedings to enforce or defend intellectual property rights is very complex, expensive, and may divert our management’s attention from our core business and may result in unfavorable results that could adversely affect our ability to prevent third parties from competing with us.
If another party has reason to assert a substantial new question of patentability against any of our claims in our own and in-licensed patents, the third party can request that the patent claims be reexamined, which may result in a loss of scope of some claims or a loss of the entire patent. In addition to potential infringement suits and, interference and reexamination proceedings, we may become a party to patent opposition proceedings where either the patentability of the inventions subject of our patents are challenged, or we are challenging the patents of others. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful. As the medical device, biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that others may assert our commercial product and/or product candidates infringe their patent rights. If a third-party is patents were found to cover our commercial product and product candidates, proprietary technologies or its uses, we or our collaborators could be enjoined by a court and required to pay damages and could be unable to continue to commercialize our products or use our proprietary technologies unless we or it obtained a license to the patent. A license may not be available to us or our collaborators on acceptable terms, if at all. In addition, during litigation, the patent holder could obtain a preliminary injunction or other equitable relief, which could prohibit us from making, using or selling our commercial product and product candidates pending a trial on the merits, which could be years away.
Proprietary trade secrets and unpatented know-how are also very important to our business. Although we have taken steps to protect our trade secrets and unpatented know-how, by entering into confidentiality agreements with third parties, and proprietary information and invention agreements with certain employees, consultants and advisors, third parties may still obtain this information or we may be unable to protect our rights. We also have limited control over the protection of trade secrets used by our licensors, collaborators and suppliers. There can be no assurance that binding agreements will not be breached, that we would have adequate remedies for any breach, or that our trade secrets and unpatented know-how will not otherwise become known or be independently discovered by our competitors. If trade secrets are independently discovered, we would not be able to prevent their use. Enforcing a claim that a third party illegally obtained and is using our trade secrets or unpatented know-how is expensive and time consuming, and the outcome is unpredictable.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed to us alleged trade secrets of their other clients or former employers. As is common in the biotechnology and pharmaceutical industry, certain of our employees were formerly employed by other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Moreover, we engage the services of consultants to assist us in the development of our commercial product and product candidates, many of whom were previously employed at or may have previously been or are currently providing consulting services to, other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We may be subject to claims that these employees and consultants or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers or their former or current customers. Litigation may be necessary to defend against these types of claims. Even if we are successful in defending against any such claims, any such litigation would likely be protracted, expensive, a distraction to our management team, not viewed favorably by investors and other third parties, and may potentially result in an unfavorable outcome.
Our product candidates may face competition sooner than expected after the expiration of our composition of matter patent protection for such products.
Our composition of matter patents for many of our product candidates have expired or will expire prior to any product approval. We intend to seek data exclusivity or market exclusivity for our NeuVax as well as our GALE-301 and GALE-302 product candidates provided under the Federal Food, Drug and Cosmetic Act, or FDCA, and similar laws in other countries. We believe that these product candidates will qualify for 12 years of data exclusivity under the Biologics Price Competition and Innovation Act of 2009, or BPCIA, which was enacted as part of the Patient Protection and Affordable Care Act and the Health Care and Education Affordability Reconciliation Act of 2010 (collectively, the Affordable Care Act or ACA) enacted in March 2010. Under the BPCIA, an application for a biosimilar product or biologics license application (BLA) cannot be submitted to the FDA until four years, or if approved by the FDA, until 12 years, after the original brand product identified as the reference product is approved under a BLA. The BPCIA provides an abbreviated pathwaybeneficial ownership for the approval of biosimilar and interchangeable biological products. The new abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable”selling stockholders is based on its similarity to an existing brand product. The new law is complex and is only beginning to be interpreted and implemented by the FDA. While it is uncertain when any such processes may be fully adopted by the FDA, any such processes could have a material adverse effect on the future commercial prospects for our biological product candidates. There is also a risk that the U.S. Congress could amend the BPCIA to shorten this exclusivity period as proposed by President Obama, potentially creating the opportunity for biosimilar competition sooner than anticipated after the expiration of our patent protection. Moreover, the extent to which a biosimilar, once approved, will be substituted for any reference product in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
If our product candidates are not considered biologics that would qualify for exclusivity under the BPCIA, they may be eligible for market exclusivity as drugs under the FDCA. The FDCA provides a five-year period of non-patent marketing exclusivity within the U.S. to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA, submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by FDA to be essential to the approval of the application, for example, for new indications, dosages, or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent.
Even if, as we expect, GALE-301 and GALE-302 are considered to be reference products eligible for 12 years of exclusivity under the BPCIA or five years of exclusivity under the FDCA, another company could market competing products if the FDA approves a full BLA or full NDA for such product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of the products.
In some countries outside of the U.S., peptide vaccines, such as GALE-301 and GALE-302, are regulated as chemical drugs rather than as biologics and may or may not be eligible for non-patent exclusivity.
Although we have received orphan drug designation for both GALE-301, as well as GALE-301and GALE-302, there is no guarantee that the drugs will be successfully approved by the FDA, that they will be commercially successful in the marketplace, or that another drug will not be approved for the same indication ahead of our drugs.
Risks Relating to Ownership of Our Common Stock
The market price and trading volume of our common stock may be volatile.
The market price of our common stock has exhibited substantial volatility recently. Between July 13, 2015 and July 13, 2016, the sale price of our common stock as reported on The NASDAQ Capital Market ranged from a low of $0.28 to a high of $2.49. The market price of our common stock could continue to fluctuate significantly for many reasons, including the following factors:
Factors beyond our control may also have an impact on the price of our stock. For example, to the extent that other companies within our industry experience declines in their stock prices, our stock price may decline as well.
We are, and in the future may be, subject to legal or administrative actions that could adversely affect our financial condition and our business.
On December 3, 2015, we agreed in principle to resolve and settle the consolidated shareholder derivative action, In re Galena Biopharma, Inc. Derivative Litigation, Civil Action No. 3:14-cv-00382-SI against us and certain of our current and former officers and directors. Following the hearing on June 23, 2016, on June 24, 2016, the U.S. District Court for the District of Oregon entered a final order and judgment in In re Galena Biopharma, Inc. Derivative Litigation, granting final approval to the settlement. On the same day, the court also issued an opinion and order awarding attorney’s fees of $4.5 million plus costs, which will be paid by our insurance carriers. The settlement includes a payment of $15 million in cash by our insurance carriers, which we will use to fund a portion of the class action settlement, and cancellation of 1,200,000 outstanding director stock options. The settlement also requires that we adopt and implement certain corporate governance measures. The settlement does not include any admission of wrongdoing or liability on the part of us or the individual defendants and includes a full release of us and the individual defendants in connection with the allegations made in the consolidated federal derivative actions and state court derivative actions.
On December 3, 2015, we also agreed in principal to resolve and settle the securities putative class action lawsuit,In re Galena Biopharma, Inc. Securities Litigation, Civil Action No. 3:14-cv-00367-SI, against us, certain of our current and former officers and directors and other defendants in the United States District Court for the District of Oregon. Following the hearing on June 23, 2016, on June 24, 2016, the U.S. District Court for the District of Oregon entered a final order and partial judgment in In re Galena Biopharma, Inc. Securities Litigation, granting final approval of the settlement. On the same day, the Court also issued an opinion and order awarding attorney’s fees of $4.5 million plus costs, which is paid out of the settlement funds. The agreement provides for a settlement payment of $20 million to the class and the dismissal of all claims against us and the other defendants in connection with the consolidated federal securities class actions. Of the $20 million settlement payment to the class, $16.7 million was paid by our insurance carriers and $2.3 million in cash was paid by us on July 1, 2016, along with $1 million in56,267,670 shares of our common stock (480,053 shares) was paid by us on July 6, 2016. We will be responsible for defense costs and any settlements or judgments incurred for any related opt-out lawsuits.
We are aware that the SEC is investigating certain matters relating to the useCommon Stock outstanding as of certain outside investor-relations professionals by us and other public companies. We have been in contact with the SEC staff through our counsel and are cooperating with the investigation and in discussions with the SEC staff.
A federal investigation of two of the high-prescribing physicians for Abstral has resulted in the criminal prosecution of the two physicians for alleged violations of the federal False Claims Act and other federal statutes. The criminal trial is set for October 2016. We have received a trial subpoena for documents in connection with that investigation and we have been in contact with the U.S. Attorney’s Office for the Southern District of Alabama, which is handling the criminal trial, and are cooperating in the production of documents. On April 28, 2016, a second superseding indictment was filed in the criminal case, which added additional information about the defendant physicians and provided information regarding the facts and circumstances involving a rebate agreement between the CompanyMarch 25, 2024, and the defendant physicians’ pharmacy as well as their ownership of our stock. Certain former employees have received trial subpoenas to appear at the trial and provide oral testimony. We have agreed to reimburse those former employees’ attorney’s fees. To our knowledge, we are not a target or subject of that investigation.
There also have been federal and state investigations of a company that has a product that competes with Abstral in the same therapeutic class, and we have learned that the FDA and other governmental agencies are investigating our Abstral promotion practices. On December 16, 2015, we received a subpoena issued by the U.S. Attorney’s Office in District of New Jersey requesting the production of a broad range of documents pertaining to our marketing and promotional practices for Abstral. We have been in contact with the U.S. Attorney’s Office for the District of New Jersey and are cooperating in the production of the requested documents. We are unable to predict whether we could become subject to legal or administrative actions as a result of these matters, or the impact of such matters. If we are found to be in violation of the False Claims Act, Anti-Kickback Statute, Patient Protection and Affordable Care Act, or any other applicable state or any federal fraud and abuse laws, we may be subject to penalties, such as civil and criminal penalties, damages, fines, or an administrative action of exclusion from government health care reimbursement programs. We can make no assurances as to the time or resources that will need to be devoted to these matters or their outcome, or the impact, if any, that these matters or any resulting legal or administrative proceedings may have on our business or financial condition.
Litigation is inherently uncertain. We have incurred and may continue to incur substantial unreimbursed legal fees and other expenses in connection with these or other legal and regulatory proceedings that may not qualify for coverage under, or may exceed the limits of, our applicable directors and officers liability insurance policies and could have a material adverse effect on our financial condition, liquidity, and results of operations. These matters also may distract the time and attention of our officers and directors or divert our other resources away from our ongoing development programs. An unfavorable outcome in any of these matters could damage our business and reputation or result in additional claims or proceedings against us.
Our common stock is currently trading at prices less than $1.00, which is the minimum bid price requirement under NASDAQ’s continued listing standards. If our common stock continues to trade at such prices, our common stock may be subject to delisting from the NASDAQ Capital Market.
The continued listing requirements of the NASDAQ Capital Market require that the closing bid price of our common stock not be less than $1.00. Following our announcement on June 29, 2016, that we had stopped our PRESENT trial, the closing bid price of our common stock has been less than $1.00. If the closing bid price of our common stock remains under $1.00 for a period of at least 30 consecutive trading days, we expect to receive a letter from NASDAQ advising us that we are not in compliance with its continued listing requirements under NASDAQ Listing Rule 5502(a)(2). In accordance with Nasdaq Listing Rule 5810(c)(3)(A), we would then have 180 calendar days to regain compliance with the $1.00 minimum bid price requirement. We can regain compliance with the $1.00 minimum bid listing requirement if the closing bid price of our is at least $1.00 per share for a minimum of ten consecutive trading days during this initial 180-day compliance period. If compliance is not achieved within the 180-day period, NASDAQ would provide written notification to us that our common stock is subject to delisting.
In the event that we fail to regain compliance with NASDAQ continued listing standards by the expiration of the applicable cure period or any extension period, NASDAQ will commence suspension and delisting procedures with respect to our common stock, which could impair the value of your investment. If our common stock is delisted from NASDAQ Capital Market in the future, such securities may be traded over-the-counter on the “pink sheets.” Such alternative market, however, is generally considered to be less efficient than, and not as broad as, NASDAQ. Accordingly, delisting of our common stock from NASDAQ could have a significant negative effect on the trading volume, liquidity and market price of our common stock. In addition, the delisting of our common stock could adversely affect our ability to raise capital on terms acceptable to us or at all and could reduce the number of investors willing to hold or acquire our common stock.
Future sales of substantial amounts of our common stock, or the possibility that such sales could occur, could adversely affect the market price of our common stock.
Future sales in the public market of shares of our common stock, including shares referred to in the foregoing risk factors or shares issuedCommon Stock issuable upon exercise or conversion of convertible securities that are currently exercisable or convertible or are exercisable or convertible within 60 days of March 25, 2024, beneficially owned by the applicable selling stockholder. Except as described below, to our knowledge, none of the selling stockholders has been an officer or director of ours or of our outstanding stock options,affiliates within the past three years or has any material relationship with us or our affiliates within the perception by the market that these sales could occur, could lower the market price of our common stock or make it difficult for us to raise additional capital.
As of March 31, 2016, we had reserved for issuance 12,000,552 shares of our common stock issuable upon the exercise of outstanding stock options at a weighted-average exercise price of $2.59 per share and 35,775,656 shares of our common stock issuable upon the exercise of outstanding warrants at a weighted-average exercise price of $1.84 per share. Upon exercise of these options and warrants, the underlying shares may be resold into the public market. In the case of outstanding options and warrants that have exercise prices that are below the market price of our common stock from time to time, our stockholders would experience dilution upon the exercise of these options.
past three years. Our outstanding warrants may result in dilution to our stockholders.
Our outstanding April 2011 warrants to purchase a total of 615,398 shares of common stock as of March 31, 2016 at a current exercise price of $0.65 per share contain so-called full-ratchet anti-dilution provisions. Our outstanding March 2010 and December 2012 warrants to purchase 25,000 shares and 3,031,311 shares, respectively, of common stock as of March 31, 2016 at exercise prices of $1.92 and $1.75, respectively, per share contain so-called weighted-average anti-dilution provisions. These anti-dilution provisions may be triggered by the issuance of the shares being offered hereby or upon any future issuance by us of shares of our common stock or common stock equivalents at a price per share below the then-exercise price of the warrants, subject to some exceptions.
To the extent that these anti-dilution provisions are triggered in the future, we would be required to reduce the exercise price of all of the warrantsknowledge is based on either a full-ratchet or weighted-average basis, which would have a dilutive effect on our stockholders.
We may issue preferred stock in the future, and the terms of the preferred stock may reduce the value of our common stock.
We are authorized to issue up to 5 million shares of preferred stock in one or more series. Our board of directors may determine the terms of future preferred stock offerings without further action by our stockholders. If we issue preferred stock, it could affect stockholder rights or reduce the market value of our outstanding common stock. In particular, specific rights granted to future holders of preferred stock may include voting rights, preferences as to dividends and liquidation, conversion and redemption rights, sinking fund provisions, and restrictions on our ability to merge with or sell our assets to a third party.
Anti-takeover provisions of our amended and restated certificate of incorporation and amended and restated bylaws and provisions of Delaware law could delay or prevent a change of control.
Anti-takeover provisions of our amended and restated certificate of incorporation and amended and restated bylaws may discourage, delay or prevent a merger or other change of control that stockholders may consider favorable or may impede the ability of the holders of our common stock to change our management and may be constrained by other contractual agreements with third parties. These provisions of our amended and restated certificate of incorporation and amended and restated bylaws, among other things:
In addition, Section 203 of the Delaware General Corporation Law provides that, subject to limited exceptions, persons that acquire, or are affiliated with a person that acquires, more than 15% of the outstanding voting stock of a Delaware corporation such as our company shall not engage in any business combination with that corporation, including by merger, consolidation or acquisitions of additional shares for a three-year period following the date on which that person or its affiliate crosses the 15% stock ownership threshold. Section 203 could operate to delay or prevent a change of control of our company.
We have never declared or paid cash dividends on our capital stock and we do not anticipate paying cash dividends in the foreseeable future.
Our business requires significant funding. We currently plan to invest all available funds and future earnings in the development and growth of our business and do not anticipate paying any cash dividends on our common stock in the foreseeable future, and are prohibited by the terms of our outstanding indebtedness from paying dividends on any common stock, except with the prior consent of our lenders. As a result, capital appreciation, if any, of our common stock will be our stockholders’ sole source of potential gain for the foreseeable future.
The terms of our outstanding indebtedness may inhibit potential acquirers.
We are prohibited by the terms of our outstanding indebtedness from disposing of any of our business or property, except with the consent of our lenders or if we were to prepay the outstanding indebtedness and related fees in accordance with the loan security agreement. Our outstanding indebtedness may inhibit potential acquirers or other interested parties from seeking to acquire all or a part of our business or assets, and there is no assurance that our lenders would consent to any proposed future transaction that might be beneficial to our stockholders.
This prospectus relates to shares of our common stock that may be offered and sold from time to time by the selling stockholders. We will receive no proceeds from the sale of shares of common stockinformation provided by the selling stockholders in connection with the filing of this offering.prospectus, as well as information obtained from relevant Schedule 13D and 13G filings.
The selling stockholders are offering under this prospectus up to 3,658,012 shares of our common stock. The selling stockholdersCommon Stock being covered hereby may be sold or otherwise disposed of from time to time offer and sell pursuant toduring the period the registration statement of which this prospectus anyis a part remains effective, by or allfor the account of the shares offered hereby. Theselling stockholders. After the date of effectiveness of such registration statement, the selling stockholders may sell or transfer, in transactions covered by this prospectus or in transactions exempt from the registration requirements of the Securities Act, some or all or none of its shares. We do not know how longtheir Common Stock.
Under the terms of the Warrants, the selling stockholders will holdmay not exercise the Warrants to the extent such exercise would cause such selling stockholders, together with their respective affiliates and attribution parties, to beneficially own a number of shares before selling them, and we currentlyof Common Stock which would exceed 4.99% (or for certain holders, 9.99%) of our then outstanding Common Stock following such exercise, excluding for purposes of such determination shares of Common Stock issuable upon exercise of the Warrants which have no agreements, arrangements or understandings withnot been exercised. The number of shares in the second column does not reflect this limitation.
Information about the selling stockholders regardingmay change over time. Any changed information will be set forth in an amendment to the saleregistration statement or supplement to this prospectus, to the extent required by law.
| Shares of Common Stock Beneficially Owned Prior to this Offering | Number of Shares of Common Stock Being Offered Hereby | Shares of Common Stock Beneficially Owned After this Offering | ||||||||||||||||||
| Selling Stockholder | Number (1) | % (2) | Number (3) | % (3) | ||||||||||||||||
| Highbridge Tactical Credit Master Fund, L.P. (4) | 10,816,161 | 16.1 | 5,322,222 | 5,493,939 | 7.9 | |||||||||||||||
| Highbridge Tactical Credit Institutional Fund, Ltd.(5) | 2,448,551 | 4.1 | 1,192,436 | 1,256,115 | 1.8 | |||||||||||||||
| Anson Investments Master Fund LP(6) | 10,862,866 | 16.2 | 5,081,433 | 5,781,433 | 8.3 | |||||||||||||||
| Anson East Master Fund LP (7) | 3,041,450 | 5.1 | 1.433,225 | 1,608,225 | 2.3 | |||||||||||||||
| 11 |
| * | Less than one percent |
| (1) | The shares of Common Stock underlying warrants are convertible or exercisable within 60 days of March 25, 2024. |
| (2) | Based on a denominator equal to the sum of (i) 56,267,670 shares of our Common Stock outstanding on March 25, 2024, and (ii) the number of shares of our Common Stock issuable upon exercise or conversion of convertible securities that are currently exercisable or convertible or are exercisable or convertible within 60 days of March 25, 2024 beneficially owned by the applicable selling stockholder. |
| (3) | Assumes that (i) all of the shares of Common Stock to be registered by the registration statement of which this prospectus is a part are sold in this offering and (ii) the selling stockholders do not acquire additional shares of our Common Stock after the date of this prospectus and prior to completion of this offering. The percentage of beneficial ownership after the offering is based on 69,296,986 shares of Common Stock, consisting of (a) 56,267,670 shares of our Common Stock outstanding on March 25, 2024, and (b) the 13,029,316 shares of our Common Stock underlying the Warrants offered under this prospectus. |
| (4) | Consists of 10,816,161 shares of Common Stock issuable upon the exercise of warrants. Highbridge Capital Management, LLC is the trading manager of Highbridge Tactical Credit Master Fund, L.P. Highbridge Tactical Credit Master Fund, L.P. disclaims beneficial ownership over these shares of Common Stock. The address of Highbridge Capital Management, LLC is 277 Park Avenue, 23rd Floor, New York, NY 10172, and the address of Highbridge Tactical Credit Master Fund, L.P. is c/o Maples Corporate Services Limited #309 Ugland House, South Church Street, George Town, Grand Cayman KY1-1104, Cayman Islands. |
| (5) | Consists of 2,448,551 shares of Common Stock issuable upon the exercise of warrants. Highbridge Capital Management, LLC is the trading manager of Highbridge Tactical Credit Institutional Fund, Ltd. Highbridge Tactical Credit Institutional Fund, Ltd. disclaims beneficial ownership over these shares of Common Stock. The address of Highbridge Capital Management, LLC is 277 Park Avenue, 23rd Floor, New York, NY 10172, and the address of Highbridge Tactical Credit Institutional Fund, Ltd. is c/o Maples Corporate Services Limited #309 Ugland House, South Church Street, George Town, Grand Cayman KY1-1104, Cayman Islands. |
| (6) | Consists of (i) 4,290,000 shares of Common Stock and (ii) 6,572,866 shares of Common Stock issuable upon the exercise of warrants. Anson Advisors Inc and Anson Funds Management LP, the Co-Investment Advisers of Anson Investments Master Fund LP (“Anson”), hold voting and dispositive power over the shares of Common Stock held by Anson. Tony Moore is the managing member of Anson Management GP LLC, which is the general partner of Anson Funds Management LP. Moez Kassam and Amin Nathoo are directors of Anson Advisors Inc. Mr. Moore, Mr. Kassam and Mr. Nathoo each disclaim beneficial ownership of these shares of Common Stock except to the extent of their pecuniary interest therein. The principal business address of Anson is Maple Corporate Services Limited, PO Box 309, Ugland House, Grand Cayman, KYI-1104, Cayman Islands. |
| (7) | Consists of (i) 1,210,000 shares of Common Stock and (ii) 1,831,450 shares of Common Stock issuable upon the exercise of warrants. Anson Advisors Inc and Anson Funds Management LP, the Co-Investment Advisers of Anson East Master Fund LP (“Anson East”), hold voting and dispositive power over the shares of Common Stock held by Anson East. Tony Moore is the managing member of Anson Management GP LLC, which is the general partner of Anson Funds Management LP. Moez Kassam and Amin Nathoo are directors of Anson Advisors Inc. Mr. Moore, Mr. Kassam and Mr. Nathoo each disclaim beneficial ownership of these shares of Common Stock except to the extent of their pecuniary interest therein. The principal business address of Anson East is Maple Corporate Services Limited, PO Box 309, Ugland House, Grand Cayman, KYI-1104, Cayman Islands. |
| 12 |
We are registering the shares of anyCommon Stock issuable upon exercise of the Warrants to permit the resale of these shares of Common Stock by the holders thereof and holders of the shares or the exercise of the warrants.
The following table presents information regarding the selling stockholdersCommon Stock and the shares that it may offer and sellWarrants from time to time underafter the date of this prospectus. The table is prepared based on information suppliedWe will receive proceeds in connection with the applicable exercise price of the Warrants to uspurchase shares of our Common Stock. Other than such exercise price, we will not receive any of the proceeds from the sale by the selling stockholders and reflects their holdings as of July 22, 2016. None of the selling stockholders, nor any affiliate of a selling stockholder, has held a position or office, or had any other material relationship, with us or any of our predecessors or affiliates. Beneficial ownership is determined in accordance with Rule 13d-3(d) promulgated by the SEC under the Exchange Act. The percentage of shares beneficially owned prior to the offering is based on 210,843,953 shares of Common Stock. We will bear all fees and expenses incident to our common stock actually outstanding asobligation to register the shares of July 22, 2016.Common Stock.
Selling Stockholder | Shares Beneficially Owned Before this Offering | Percentage of Outstanding Shares Beneficially Owned Before this Offering | No. of Outstanding Shares Offered by Selling Stockholder | Percentage of Outstanding Shares Beneficially Owned After this Offering | ||||||||||||
Peter Barber | 975,684 | * | 975,684 | * | ||||||||||||
Daniel DiPietro | 975,684 | * | 975,684 | * | ||||||||||||
Matthew Wyckoff | 859,868 | * | 859,868 | * | ||||||||||||
John Liatos | 505,013 | * | 505,013 | * | ||||||||||||
Paul Glidden | 50,501 | * | 50,501 | * | ||||||||||||
Hosie Kenneth Riley | 291,262 | * | 291,262 | * | ||||||||||||
|
| |||||||||||||||
TOTALS | 3,658,012 | |||||||||||||||
Each selling stockholder of the shares offered herebysecurities and any of their pledgees, assignees and successors-in-interest may, from time to time, sell any or all of their securities covered hereby on the NASDAQThe Nasdaq Capital Market or any other stock exchange, market or trading facility on which the securities are traded or in private transactions. These sales may be at fixed, prevailing market prices at the time of sale, prices related to such prevailing market prices, varying prices determined at the time of sale or negotiated prices. A Selling Stockholderselling stockholder may use any one or more of the following methods when selling securities:
| · | ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers; |
| · | block trades in which the broker-dealer will attempt to sell the securities as agent but may position and resell a portion of the block as principal to facilitate the transaction; |
| · | purchases by a broker-dealer as principal and resale by the broker-dealer for its account; |
| · | an exchange distribution in accordance with the rules of the applicable exchange; |
| · | privately negotiated transactions; |
| · | settlement of short sales; |
| · | in transactions through broker-dealers that agree with the Selling Stockholders to sell a specified number of such securities at a stipulated price per security; |
| · | through the writing or settlement of options or other hedging transactions, whether through an options exchange or otherwise; |
| · | a combination of any such methods of sale; or |
| · | any other method permitted pursuant to applicable law. |
The selling stockholders may also sell securities under Rule 144 or any other exemption from registration under the Securities Act of 1933, as amended (the “Securities Act”), if available, rather than under this prospectus.
Broker-dealers engaged by the selling stockholders may arrange for otherbrokers-dealers to participate insales. Broker-dealers may receive commissions or discounts from the selling stockholders (or, if anybroker-dealer acts as agent for the purchaser of securities, from the purchaser) in amounts to be negotiated, but, except as set forth in a supplement to this Prospectus, in the case of an agency transaction not in excess of a customary brokerage commission in compliance with FINRA Rule 2440;2121; and in the case of a principal transaction a markup or markdown in compliance with FINRA IM-2440.
In connection with the sale of the securities or interests therein, the selling stockholders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the securities in the course of hedging the positions they assume. The selling stockholders may also sell securities short and deliver these securities to close out their short positions, or loan or pledge the securities to broker-dealers that in turn may sell these securities. The selling stockholders may also enter into option or other transactions with broker-dealers or other financial institutions or create one or more derivative securities which require the delivery to such broker-dealer or other financial institution of securities offered by this prospectus, which securities such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).
The selling stockholders and any broker-dealers or agents that are involved in selling the securities may be deemed to be “underwriters” within the meaning of the Securities Act in connection with such sales. In such event, any commissions received by such broker-dealers or agents and any profit on the resale of the securities purchased by them may be deemed to be underwriting commissions or discounts under the Securities Act. Each selling stockholdersstockholder has informed the Company that it does not have any written or oral agreement or understanding, directly or indirectly, with any person to distribute the securities.
| 13 |
The Selling Stockholders also may transfer the securities in other circumstances, in which case the transferees, pledgees or other successors-in-interest will be the selling beneficial owners for purposes of this prospectus. Upon being notified by either of the Selling Stockholders that a donee, pledgee, transferee, other successor- in-interest intends to sell our Shares, we will, to the extent required, promptly file a supplement to this prospectus to name specifically such person as a selling stockholder.
The Selling Stockholders will act independently of us in making decisions with respect to the timing, manner, and size of each resale or other transfer. There can be no assurance that the Selling Stockholders will sell any or all of the Shares under this prospectus. Further, we cannot assure you that the Selling Stockholders will not transfer, distribute, devise or gift the Shares by other means not described in this prospectus.
The underwriters, broker-dealers and agents that may engage in transactions with us or the Selling Stockholders, may have banking, lending or other relationships with us or perform services for us or the Selling Stockholders, in the ordinary course of business.
The Company is required to pay certain fees and expenses incurred by the Company incident to the registration of the securities. The Company has agreed to indemnify the selling stockholders against certain losses, claims, damages and liabilities, including liabilities under the Securities Act.
Because selling stockholders may be deemed to be “underwriters” within the meaning of the Securities Act, they will be subject to the prospectus delivery requirements of the Securities Act including Rule 172 thereunder. In addition, any securities covered by this prospectus which qualify for sale pursuant to Rule 144 under the Securities Act may be sold under Rule 144 rather than under this prospectus. The selling stockholders have advised us that there is no underwriter or coordinating broker acting in connection with the proposed sale of the resale securities by the selling stockholders.
We agreed to keep this prospectus effective until the earlier of (i) the date on which the securities may be resold by the selling stockholders without registration and without regard to any volume or manner-of-sale limitations by reason of Rule 144, without the requirement for the Company to be in compliance with the current public information under Rule 144 under the Securities Act or any other rule of similar effect or (ii) all of the securities have been sold pursuant to this prospectus or Rule 144 under the Securities Act or any other rule of similar effect. The resale securities will be sold only through registered or licensed brokers or dealers if required under applicable state securities laws. In addition, in certain states, the resale securities covered hereby may not be sold unless they have been registered or qualified for sale in the applicable state or an exemption from the registration or qualification requirement is available and is complied with.
Under applicable rules and regulations under the Exchange Act, any person engaged in the distribution of the resale securities may not simultaneously engage in market making activities with respect to the common stockCommon Stock for the applicable restricted period, as defined in Regulation M, prior to the commencement of the distribution. In addition, the selling stockholders will be subject to applicable provisions of the Exchange Act and the rules and regulations thereunder, including Regulation M, which may limit the timing of purchases and sales of the common stockCommon Stock by the selling stockholders or any other person. We will make copies of this prospectus available to the selling stockholders and have informed them of the need to deliver a copy of this prospectus to each purchaser at or prior to the time of the sale (including by compliance with Rule 172 under the Securities Act).
| 14 |
Our business requires significant funding. We currently plan to invest all available funds and any future earnings in our business and do not anticipate paying any cash dividends on our common stock in the foreseeable future. We currently are prohibited by the terms of our outstanding indebtedness from paying dividends on our common stock, except with the prior consent of our lenders.
The following summary of the terms of our securities is subject to and qualified by reference to our amended and restated certificate of incorporation and our restated bylaws, copies of which are on file with the SEC as exhibits to previous SEC filings. Please refer to “Where You Can Find More Information” below for directions on obtaining these documents.
General
Our authorized capital stock consists of 355,000,000 shares, which includes:
As of July 22, 2016, there were 210,843,953 shares of common stock outstanding and no shares of preferred stock outstanding.
Common Stock
Each holder of our common stock is entitled to one vote for each share of common stock held on all matters submitted to a vote of stockholders. Common stockholders are not be entitled to cumulative voting in the election of directors by our certificate of incorporation. This means that the holders of a majorityvalidity of the shares voted will be able to elect allof Common Stock offered in this prospectus has been passed upon for us by Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C., New York, New York.
The consolidated financial statements of SELLAS Life Sciences, Group, Inc. (the “Company”) incorporated in this Prospectus by reference from the Annual Report on Form 10-K of the directors then standingCompany for election. Subjectthe year ended December 31, 2023, have been audited by Moss Adams LLP, an independent registered public accounting firm, as stated in their report (which report expresses an unqualified opinion and includes an explanatory paragraph related to preferences that may apply to shares of preferred stock outstanding ata going concern emphasis), which is incorporated herein by reference. Such consolidated financial statements are incorporated by reference in reliance upon the time, the holders of outstanding shares of our common stock are entitled to receive dividends out of assets legally available at the times and in the amounts that our board of directors may determine from time to time.
Upon our liquidation, dissolution or winding-up, the holders of our common stock will be entitled to share ratably in all assets remaining after payment of all liabilities and the liquidation preferences of any series of capital stock ranking senior to the common stock upon liquidation. Holders of common stock have no preemptive or conversion rights or other subscription rights. There will be no redemption or sinking fund provisions applicable to the common stock. All outstanding shares of common stock are fully paid and nonassessable, and the shares of common stock to be issued under this prospectus, when they are paid for, will be fully paid and nonassessable.
Preferred Stock
Our board of directors is authorized, subject to any limitations prescribed by law, without further vote or action by the stockholders, to issue from time to time any of the authorized shares of preferred stock in one or more series without stockholder approval. Each such series of preferred stock shall have such number of shares, designations, preferences, voting powers, qualifications, and special or relative rights or privileges as shall be determined by our board of directors, which may include, among others, dividend rights, voting rights, liquidation preferences, conversion rights and preemptive rights.
The authority possessed by our board to issue preferred stock could potentially be used to discourage attempts by third parties to obtain control of our company through a merger, tender offer, proxy contest or otherwise by making such attempts more difficult or more costly. Our board may issue preferred stock with voting rights or conversion rights that, if exercised, could adversely affect the voting power of the holders of common stock. There are no current agreements or understandings with respect to the issuance of preferred stock.
Anti-Takeover Effects of Delaware Law and Our Restated Certificate of Incorporation and Bylaws
Certain provisions of Delaware law, our amended and restated certificate of incorporation and our amended and restated bylaws contain provisions that could have the effect of delaying, deferring or discouraging another party from acquiring control of us. These provisions, which are summarized below, are expected to discourage certain types of coercive takeover practices and inadequate takeover bids. These provisions are also designed, in part, to encourage persons seeking to acquire control of us to first negotiate with our board of directors. We believe that the benefits of increased protection of our potential ability to negotiate with an unfriendly or unsolicited acquirer outweigh the disadvantages of discouraging such proposals, including proposals that are priced above the then-current market value of our common stock, because, among other reasons, the negotiationreport of such proposals could resultfirm given their authority as experts in an improvement of their terms.
Certificate of Incorporationaccounting and Bylaws
Our amended and restated certificate of incorporation and amended and restated bylaws include provisions that:auditing.
WHERE YOU CAN FIND ADDITIONAL INFORMATION
Delaware Anti-Takeover Statute
We are subject to the provisions of Section 203information requirements of the Delaware General Corporation Law regulating corporate takeovers. In general, Section 203 prohibits a publicly-held Delaware corporation from engaging, under certain circumstances, in a business combination with an interested stockholder for a period of three years following the date the person became an interested stockholder unless:
Generally, a “business combination” includes a merger, asset or stock sale, or other transaction resulting in a financial benefit to the “interested stockholder.” Generally, an “interested stockholder” is a person who, together with affiliates and associates, owns or, within three years prior to the determination of interested stockholder status, did own 15% or more of a corporation’s outstanding voting stock. We expect the existence of this provision to have an anti-takeover effect with respect to transactions our board of directors does not approve in advance. We also anticipate that Section 203 may discourage business combinations or other attempts that might result in a premium over the market price for the shares of common stock held by our stockholders.
The provisions of Delaware law, our restated certificate of incorporation and our amended and restated bylaws could have the effect of discouraging others from attempting hostile takeovers and, as a consequence, they may also inhibit temporary fluctuations in the market price of our common stock that often result from actual or rumored hostile takeover attempts. These provisions may also have the effect of preventing changes in our management. It is possible that these provisions could make it more difficult to accomplish transactions that stockholders may otherwise deem to be in their best interests.
Listing
Our common stock is listed on The NASDAQ Capital Market under the symbol “GALE.”
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is Computershare Trust Company, N.A.
Fredrikson & Byron P.A., Minneapolis, Minnesota, has rendered an opinion with respect to the validity of the shares of common stock offered by this prospectus.
The consolidated financial statements of the Company as of December 31, 2015 and 2014, and for each of the three years in the period ended December 31, 2015, and the effectiveness of internal control over financial reporting of the Company as of December 31, 2015, have been incorporated by reference herein in reliance upon the reports of Moss Adams LLP, independent registered public accounting firm, and upon the authority of said firm as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
Wewe therefore file annual, quarterly and currentperiodic reports, proxy statements and other information with the SEC. Our SEC filings are availablerelating to the public over the Internetour business, financial statements and other matters. The reports, proxy statements and other information we file may be inspected and copied at prescribed rates at the SEC’sSEC's Public Reference Room located at 100 F Street, N.E., Washington, D.C. 20549. You may obtain information on the operation of the SEC's Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC also maintains a website at http://www.sec.gov. The SEC’s websitethat contains reports, proxy and information statements and other information regarding issuers such aslike us that file electronically with the SEC. The address of the SEC's website is http://www.sec.gov.
This prospectus constitutes part of a registration statement filed under the Securities Act with respect to the shares of Common Stock covered hereby. As permitted by the SEC's rules, this prospectus omits some of the information, exhibits and undertakings included in the registration statement. You may also read and copy any documentthe information omitted from this prospectus but contained in the registration statement, as well as the periodic reports and other information we file with the SEC, at the SEC’s Public Reference Room at 100 F Street, N.E., Room 1580, Washington D.C. 20549.public reference room and website of the SEC referred to above. You may also obtain copiesaccess our filings with the SEC on our website, which is located at http://www.sellaslifesciences.com. The information contained on our website is not part of these documents at prescribed rates by writingthis prospectus.
Statements contained in this prospectus as to the SEC. Please callcontents of any contract or other document are not necessarily complete, and in each instance we refer you to the SEC at 1-800-SEC-0330 for further information oncopy of the operation of its Public Reference Room.contract or other document filed or incorporated by reference as an exhibit to the registration statement or as an exhibit to our Exchange Act filings, each such statement being qualified in all respects by such reference.
| 15 |
INCORPORATION OF CERTAIN DOCUMENTSINFORMATION INCORPORATED BY REFERENCE
The SEC allows us to “incorporateincorporate by reference”reference the information from other documents that we file with it, which means that we can disclose important information to you by referring you to those documents. Theanother document that we have filed separately with the SEC. You should read the information incorporated by reference because it is considered to bean important part of this prospectus.prospectus and the applicable prospectus supplement. Information in this prospectus supersedes information incorporated by reference that we filed with the SEC prior to the date of this prospectus, while information that we file later with the SEC will automatically update and supersede the information in this prospectus.prospectus and the applicable prospectus supplement. We incorporate by reference into this prospectus and the registration statement of which this prospectus is a part the information or documents listed below that we have filed with the SEC:SEC (Commission File No. 001-33958):
| · | our Annual Report on Form 10-K for the fiscal year ended December 31, 2023, filed with the SEC on March 28, 2024; and |
| · | the description of our Common Stock set forth in our registration statement on Form 8-A, filed with the SEC on February 8, 2008, as amended on February 12, 2008, including any further amendments thereto or reports filed for the purposes of updating this description. |
We also incorporate by reference into this prospectus all additional documentsany future filings (other than the portions of current reports furnished under Item 2.02 or Item 7.01 of Form 8-K and exhibits filed on such form that we fileare related to such items unless such Form 8-K expressly provides to the contrary) made with the SEC under the terms of Sectionpursuant to Sections 13(a), 13(c), 14 or 15(d) of the Securities Exchange Act, of 1934 that areincluding those made after the date of the initial filing of the registration statement of which this prospectus is a part and beforeprior to effectiveness of such registration statement, until we file a post-effective amendment that indicates the termination of the offering of securities offeredthe Common Stock made by this prospectus. Weprospectus and will become a part of this prospectus from the date that such documents are not, however, incorporating,filed with the SEC. Information in each case,such future filings updates and supplements the information provided in this prospectus and any documents or information that we areapplicable prospectus supplement. Any statements in any such future filings will automatically be deemed to modify and supersede any information in any document we previously filed with the SEC that is incorporated or deemed to be incorporated herein by reference to the extent that statements in the later filed document modify or replace such earlier statements.
We will furnish without charge to each person, including any beneficial owner, to whom a prospectus and not file in accordance with SEC rules.
You mayapplicable prospectus supplement is delivered, upon written or oral request, a copy of any or all of the documents incorporated by reference into this prospectus at no cost, by writing or telephoning us atbut not delivered with the following address: Galena Biopharma, Inc., 2000 Crow Canyon Place, Suite 380, San Ramon, California 94583, Attention: Investor Relations, Phone: (855) 855-4253. We will not sendprospectus and applicable prospectus supplement, including exhibits to any documents, unless the exhibitsthat are specifically incorporated by reference into the document.
such documents. You should direct any requests for documents to SELLAS Life Sciences Group, Inc., Attention: Corporate Secretary, 7 Times Square, Suite 2503, New York, NY 10036. Our phone number is (646) 200-5278.
PROSPECTUSYou should rely only on information contained in, or incorporated by reference into, this prospectus and any prospectus supplement. We have not authorized anyone to provide you with information different from that contained in this prospectus or incorporated by reference into this prospectus. We are not making offers to sell the securities in any jurisdiction in which such an offer or solicitation is not authorized or in which the person making such offer or solicitation is not qualified to do so or to anyone to whom it is unlawful to make such offer or solicitation.
| 16 |

3,658,012 Shares of Common Stock
The date of this prospectus is , 2016.
PART II
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
| Other Expenses of Issuance and |
The following table sets forth all costs and expenses payable by the expenses to be paidRegistrant, in connection with the offeringsale of the securities described inbeing registered under this registration statement. All amounts shown are estimates except for the Securities and Exchange Commission, or SEC, registration fee.
Securities and Exchange Commission registration fee | $ | 150 | ||
Printing and engraving expenses | 5,000 | |||
Legal fees and expenses | 10,000 | |||
Accounting fees and expenses | 20,000 | |||
Transfer agent and registrar fees | 2,500 | |||
|
| |||
TOTAL | $ | 37,650 | ||
|
|
| Amount | |||||
| SEC registration fee | $ | 2,048.13 | |||
| Legal fees and expenses | $ | 25,000.00 | |||
| Accounting fees and expenses | $ | 7,500 | |||
| Total | $ | 34,548.13 | |||
| Indemnification of Directors and Officers. |
As permitted by Section 145102 of the Delaware General Corporation Law, provides that a corporation may indemnify any person who waswe have adopted provisions in our amended and restated certificate of incorporation and amended and restated bylaws, each as amended, which limit or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative, other than an action by or in the right of the corporation, by reason of the fact that the person is or was a director, officer, employee or agent of the corporation or is or was serving at the corporation’s request as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses, including attorneys’ fees, judgments, fines and amounts paid in settlement actually and reasonably incurred by the person in connection with the action, suit or proceeding if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe the person’s conduct was unlawful. The power to indemnify applies to actions brought by or in the right of the corporation as well, but only to the extent of expenses, including attorneys’ fees but excluding judgments, fines and amounts paid in settlement, actually and reasonably incurred by the person in connection with the defense or settlement of the action or suit if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interests of the corporation and except that no indemnification shall be made in respect of any claim, issue or matter as to which such person shall have been adjudged to be liable to the corporation unless and only to the extent that a court of competent jurisdiction shall determine that such indemnity is proper.
Section 145(g) of the Delaware General Corporation Law provides that a corporation shall have the power to purchase and maintain insurance on behalf of its officers, directors, employees and agents, against any liability asserted against and incurred by such persons in any such capacity.
Section 102(b)(7) of the General Corporation Law of the State of Delaware provides that a corporation may eliminate or limit the personal liability of our directors for a breach of their fiduciary duty of care as a director. The duty of care generally requires that, when acting on behalf of the corporation, directors exercise an informed business judgment based on all material information reasonably available to them. Consequently, a director to the corporation or its stockholders for monetary damages for breach of fiduciary duty as a director, provided that such provision shallwill not eliminate or limit the liability of a director (i) for any breach of the director’s duty of loyalty to the corporation or its stockholders, (ii) for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law, (iii) under Section 174 of the General Corporation Law of the State of Delaware, or (iv) for any transaction from which the director derived an improper personal benefit. No such provision shall eliminate or limit the liability of a director for any act or omission occurring prior to the date when such provision becomes effective.
Our Amended and Restated Certificate of Incorporation provides that our directors shall not be personally liable to us or our stockholders for monetary damages for breach of fiduciary duty as a director, except for liability for:
| · | any breach of the director’s duty of loyalty to us or our stockholders; |
| · | any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; |
| · | any act related to unlawful stock repurchases, redemptions or other distributions or payment of dividends; or |
| · | any transaction from which the director derived an improper personal benefit. |
These limitations of liability do not affect the availability of equitable remedies such as injunctive relief or rescission. Our amended and restated certificate of incorporation also authorizes us to indemnify our officers, directors and other agents to the fullest extent that the exculpation from liabilities is not permitted under Delaware law.
As permitted by Section 145 of the Delaware General Corporation Law, our amended and restated bylaws, as in effect at the time such liability is determined. In addition, our Amended and Restated Certificate of Incorporation provides that we shall, to the maximumamended, provide that:
| · | we may indemnify our directors, officers and employees to the fullest extent permitted by the Delaware General Corporation Law, subject to limited exceptions; |
| · | we may advance expenses to our directors, officers and employees in connection with a legal proceeding to the fullest extent permitted by the Delaware General Corporation Law, subject to limited exceptions; and |
| · | the rights provided in our bylaws are not exclusive. |
Our amended and restated certificate of incorporation and bylaws, each as amended, which are filed as Exhibits 3.1 and 3.2 to our Annual Report on Form 10-K, provide for the indemnification provisions described above and elsewhere herein. We have entered into separate indemnification agreements with our directors and officers that may be broader than the specific indemnification provisions contained in the Delaware General Corporation Law of the State of Delaware,Law. These indemnification agreements generally require us, among other things, to indemnify each person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative,our officers and directors against liabilities that may arise by reason of their status or service as directors or officers, other than liabilities arising from willful misconduct. These indemnification agreements also generally require us to advance any expenses incurred by the fact that he isdirectors or was, or has agreed to become, our director or officer, or is or was serving, or has agreed to serve, at our request,officers as a director, officer, employee or trustee of, or in a similar capacity with, another corporation, partnership, joint venture, trust or other enterprise, including any employee benefit plan (all such persons being referred to hereafter as an “Indemnitee”), or by reasonresult of any action allegedproceeding against them as to which they could be indemnified. In addition, we have been taken or omitted in such capacity, against all expenses (including attorneys’ fees), judgments, finespurchased a policy of directors’ and amounts paid in settlement actually and reasonably incurred by him or on his behalf in connection with such action, suit or proceeding and any appeal therefrom, if he acted in good faith and in a manner he reasonably believed to be in, or not opposed to, our best interests, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful, andofficers’ liability insurance that we shall not indemnify an Indemnitee seeking indemnification in connection with any action, suit, proceeding, claim or counterclaim, or part thereof, initiated by the Indemnitee unless the initiation thereof was approved by our board of directors.
II-1
Our Amended and Restated Certificate of Incorporation also provides that any indemnification under the preceding paragraph (unless ordered by a court) shall be made by us only as authorized in the specific case upon a determination that indemnification is proper in the circumstances because such person has either met the applicable standard of conduct set forth in this paragraph and that the amount requested has been actually and reasonably incurred. Such determination shall be made:
1. by a majority vote of the directors who are not parties to such action, suit or proceeding, even though less than a quorum; or
2. by a committee of such directors designated by a majority vote of such directors, even though less than a quorum; or
3. if there are no such directors, or if such directors so direct, by independent legal counsel in a written opinion; or
4. by the holders of our common stock.
Our Amended and Restated Certificate of Incorporation provides that we may purchase and maintain insurance policies on behalf ofinsures our directors and officers against specifiedthe cost of defense, settlement or payment of a judgment in some circumstances. These indemnification provisions and the indemnification agreements may be sufficiently broad to permit indemnification of our officers and directors for liabilities, for actions taken in its capacities as such, including liabilitiesreimbursement of expenses incurred, arising under the Securities Act.
| II-1 |
We have obtained directors’ and officers’ liability insurance to cover liabilitiesentered into indemnification agreements with our directors and executive officers, may incur in connection with their services to us.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to our directors, officers and controlling persons pursuantaddition to the foregoing provisions, or otherwise, we have been advised thatindemnification provided for in our amended and restated certificate of incorporation and amended and restated bylaws, and intend to enter into indemnification agreements with any new directors and executive officers in the opinionfuture.
We have purchased and currently intend to maintain insurance on behalf of the SECeach and every person who is or was a director or officer of our company against any loss arising from any claim asserted against him or her and incurred by him or her in any such indemnification is against public policy as expressed in the Securities Act of 1933 and is, therefore, unenforceable.capacity, subject to certain exclusions.
The Exhibit Index that follows the signature pagefollowing exhibits are filed as part of this registration statement lists the exhibits thatRegistration Statement on Form S-3 or are filed with this registration statement, and such Exhibit Index is incorporated herein by reference.
(a) The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:* Filed herewith.
(i) To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement; notwithstanding the foregoing, any increase or decrease in the volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Securities and Exchange Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20 percentItem 17. Undertakings.
| (a) | The undersigned registrant hereby undertakes: |
| (1) | To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement: |
| (i) | To include any prospectus required by section 10(a)(3) of the Securities Act of 1933; |
| (ii) | To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective
|
| II-2 |
| (iii) | To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement; |
provided, however, that paragraphs (a)(1)(i), (a)(1)(ii) and (a)(1)(iii) above do not apply if the information required to be included in a post-effective amendment by those paragraphs is contained in reports filed with or furnished to the Securities and Exchange Commission by the registrant pursuant to Sectionsection 13 or section 15(d) of the Securities Exchange Act of 1934 that are incorporated by reference in the registration statement, or is contained in a form of prospectus filed pursuant to Rule 424(b) that is part of the registration statement.
(2)
| (2) | That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| (3) | To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering. |
| (4) | That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser: |
| (i) | Each prospectus filed by the registrant pursuant to Rule 424(b)(3) shall be deemed to be part of the registration statement as of the date the filed prospectus was deemed part of and included in the registration statement; and |
| (ii) | Each prospectus required to be filed pursuant to Rule 424(b)(2), (b)(5), or (b)(7) as part of a registration statement in reliance on Rule 430B relating to an offering made pursuant to Rule 415(a)(1)(i), (vii), or (x) for the purpose of providing the information required by section 10(a) of the Securities Act of 1933 shall be deemed to be part of and included in the registration statement as of the earlier of the date such form of prospectus is first used after effectiveness or the date of the first contract of sale of securities in the offering described in the prospectus. As provided in Rule 430B, for liability purposes of the issuer and any person that is at that date an underwriter, such date shall be deemed to be a new effective date of the registration statement relating to the securities in the registration statement to which that prospectus relates, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such effective date, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such effective date. |
| (5) | That, for the purpose of determining liability of the registrant under the Securities Act to any purchaser in the initial distribution of the securities, the undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser: |
| (i) | Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424; |
| (ii) | Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant; |
| (iii) | The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and |
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
| II-3 |
| (iv) | Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser. |
| (b) | The undersigned registrant hereby undertakes that, for purposes of determining any liability under the Securities Act of 1933, each filing of the registrant’s annual report pursuant to section 13(a) or section 15(d) of the Securities Exchange Act of 1934 (and, where applicable, each filing of an employee benefit plan’s annual report pursuant to section 15(d) of the Securities Exchange Act of 1934) that is incorporated by reference in the registration statement shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| (c) | Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue. |
| II-4 |
II-2
(4) That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness; provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(5) The undersigned registrant hereby undertakes that, for purposes of determining any liability under the Securities Act of 1933, each filing of the registrant’s annual report pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 and (and, where applicable, each filing of an employee benefit plan’s annual report pursuant to Section 15(d) of the Securities Exchange Act of 1934) that is incorporated by reference in the registration statement shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(b) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act of 1933 and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act of 1933 and will be governed by the final adjudication of such issue.
II-3
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form S-3 and has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of San Ramon, State of California,New York, New York, on July 25, 2016.March 28, 2024.
| By: | /s/ Angelos M. Stergiou | ||
| |||
| President and Chief Executive Officer | |||
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby severally constitutes and appoints jointlyAngelos M. Stergiou and severally, Mark W. Schwartz, Ph.D.John T. Burns, and each of them singly, as hissuch person’s true and lawful attorney-in-factattorneys-in-fact and agent,agents, with full power of substitution and resubstitution, for himsuch person and in hissuch person’s name, place and stead, in any and all capacities, to sign any and all amendments (including, without limitation, post-effective amendments) to this registration statement on Form S-3, and to sign(or any registration statement for the same offering covered by this registration statement that is to be effective upon filing pursuant to Rule 462(b) promulgated under the Securities Act of 1933, and all post-effective amendments thereto,1933), and to file the same, and all prospectus supplements, with all exhibits thereto, and all documents in connection therewith, with the Securities and Exchange Commission, granting unto each said attorney-in-fact and agent, full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as hesuch person might or could do in person, hereby ratifying and confirming all that any said attorney-in-fact and agent, or hisany substitute or substitutes of any of them, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities and on the dates indicated:indicated.
| Signature | Title | Date | ||
|
|
| ||
| President, Chief Executive Officer and Director (Principal Executive Officer) | |||
| March 28, 2024 | ||||
/s/ John T. Burns John T. Burns |
(Principal Financial and Accounting Officer) | |||
/s/ Jane Wasman | Chair of the Board of Directors | March 28, 2024 | ||
/s/ David Scheinberg, M.D., Ph.D. David Scheinberg, M.D., Ph.D. | Director | |||
/s/ Robert Van Nostrand | Director | |||
/s/ John Varian | Director | |||
/s/ Katherine Kalin | Director | |||
| ||||
| ||||
| ||||
| March 28, 2024 | ||||
II-4
EXHIBIT INDEX
The following exhibits are filed with this registration statement or are incorporated by reference as part of this registration statement:
|
| |
II-5