As filed with the Securities and Exchange Commission on May 1, 2017August 7, 2023
Registration No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM S-3
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
Aeglea BioTherapeutics, Inc.
(Exact name of registrant as specified in its charter)
| | |
| Delaware | | 46-4312787 |
(State or other jurisdiction of incorporation or organization) | | (I.R.S. Employer Identification No.) |
901 S. MoPac Expressway221 Crescent Street
Barton Oaks Plaza OneBuilding 17, Suite 102B
Suite 250Waltham, MA 02453
Austin, TX 78746
(512) 942-2935
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
David G. Lowe, Ph.D.Jonathan D. Alspaugh
President and Chief ExecutiveFinancial Officer
Aeglea BioTherapeutics, Inc.
901 S. MoPac Expressway221 Crescent Street
Barton Oaks Plaza OneBuilding 17, Suite 102B
Suite 250Waltham, MA 02453
Austin, TX 78746
(512) 942-2935
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Ryan A. Murr
| | |
Robert A. Freedman, Esq.
Branden C. Berns Gibson, Dunn & Crutcher LLP 555 Mission Street, Suite 3000 San Francisco, California 94105 (415) 393-8373 Effie Toshav, Esq.
Fenwick & West LLP
801 California Street
Mountain View, California 94041
(650) 988-8500
| | Charles N. York II
Chief Financial Officer
Aeglea BioTherapeutics, Inc.
901 S. MoPac Expressway
Barton Oaks Plaza One
Suite 250
Austin, TX 78746
(512) 942-2935
|
Approximate date of commencement of proposed sale to the public: From time to time after the effective date of this Registration Statement.
If the only securities being registered on this Form are being offered pursuant to dividend or interest reinvestment plans, please check the following box: ☐
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, as amended, other than securities offered only in connection with dividend or interest reinvestment plans, check the following box: ☒
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering: ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earliest effective registration statement for the same offering: ☐
If this Form is a registration statement pursuant to General Instruction I.D. or a post-effective amendment thereto that shall become effective upon filing with the Commission pursuant to Rule 462(e) under the Securities Act, check the following box.box: ☐
If this Form is a post-effective amendment to a registration statement filed pursuant to General Instruction I. D. filed to register additional securities or additional classes of securities pursuant to Rule 413(b) under the Securities Act, check the following box.box: ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
| | | | | | | | |
| Large accelerated filer | | ☐ | | | | Accelerated filer | | ☐ |
| | | |
| Non-accelerated filer | | ☒ | | (Do not check if a smaller reporting company) | | Smaller reporting company | | ☐☒ |
| | | |
| | | | Emerging growth company | | ☒ | | | | | | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ☒☐
CALCULATION OF REGISTRATION FEE
| | | | | | | | |
|
Title of Each Class of Securities to be Registered (1) | | Amount to be
Registered (1)(2) | | Proposed Maximum
Offering Price Per Security (3) | | Proposed Maximum
Aggregate Offering Price (3) | | Amount of
Registration Fee (4) |
Common stock, $0.0001 par value per share | | | | | | | | |
Preferred stock, $0.0001 par value per share | | | | | | | | |
Debt securities | | | | | | | | |
Warrants | | | | | | | | |
Subscription rights | | | | | | | | |
Units | | | | | | | | |
Total | | | | | | $150,000,000 | | $17,385 |
|
|
(1) | There is being registered hereunder an indeterminate number of shares of (a) common stock, (b) preferred stock, (c) debt securities, (d) warrants to purchase common stock, preferred stock or debt securities of the Registrant, (e) subscription rights to purchase common stock, preferred stock or debt securities of the Registrant, and (f) units, consisting of some or all of these securities in any combination, as may be sold from time to time by the Registrant. Any securities registered hereunder may be sold separately or as units with other securities registered hereunder. There are also being registered hereunder an indeterminate number of shares of common stock, preferred stock and debt securities as shall be issuable upon conversion, exchange or exercise of any securities that provide for such issuance. In no event will the aggregate offering price of all types of securities issued by the Registrant pursuant to this registration statement exceed $150,000,000. |
(2) | Pursuant to Rule 416(a), this registration statement also covers any additional securities that may be offered or issued in connection with any stock split, stock dividend or similar transaction. |
(3) | The proposed maximum offering price per share and proposed maximum aggregate offering price for each type of security will be determined from time to time by the Registrant in connection with the issuance by the Registrant of the securities registered hereunder. |
(4) | Calculated pursuant to Rule 457(o) under the Securities Act of 1933, as amended. |
The Registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment whichthat specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until thethis registration statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
EXPLANATORY NOTE
This registration statement contains two prospectuses:
a base prospectus, which covers the offering, issuance and sale by us of up to a maximum aggregate offering price of $150,000,000 of our common stock, preferred stock, debt securities, warrants to purchase our common stock, preferred stock or debt securities, subscription rights to purchase our common stock, preferred stock or debt securities and/or units consisting of some or all of these securities; and
| • | | a sales agreement prospectus covering the offering, issuance and sale by us of up to a maximum aggregate offering price of $20,000,000 of our common stock that may be issued and sold under a Capital on DemandTM Sales Agreement dated May 1, 2017 with JonesTrading Institutional Services LLC. |
The base prospectus immediately follows this explanatory note. The specific terms of any securities to be offered pursuant to the base prospectus will be specified in a prospectus supplement to the base prospectus. The sales agreement prospectus immediately follows the base prospectus. The $20,000,000 of common stock that may be offered, issued and sold under the sales agreement prospectus is included in the $150,000,000 of securities that may be offered, issued and sold by us under the base prospectus.
The information in this prospectus is not complete and may be changed. WeThe selling stockholders named in this prospectus may not sell these securities until the registration statement filed with the Securities and Exchange Commission (the “SEC”) is effective. This prospectus is not an offer to sell these securities, and it is not soliciting an offer to buy these securities, in any statejurisdiction where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED MAY 1, 2017AUGUST 7, 2023
PROSPECTUS

$150,000,000
Aeglea BioTherapeutics, Inc.
1,099,284,385 Shares
Common Stock Preferred Stock,
Debt Securities, Warrants, Subscription Rights and UnitsOffered by the Selling Stockholders
From timeThis prospectus relates to time, we may offerthe proposed resale or other disposition by the selling stockholders identified herein (the “Selling Stockholders”), of up to $150,000,000 aggregate dollar amount of(i) 12,945,385 shares (“Merger Common Shares”) of our common stock, par value $0.0001 per share (“Common Stock”), (ii) 364,887,000 shares of Common Stock (“Merger Conversion Shares”), issuable upon the conversion of 364,887 shares (“Merger Preferred Shares”) of our Series A Preferred Stock, par value $0.0001 per share (“Series A Preferred Stock”), and (iii) 721,452,000 shares of Common Stock (“Private Placement Conversion Shares”), issuable upon the conversion of 721,452 shares (“Private Placement Preferred Shares”) of our Series A Preferred Stock. Subject to receiving the requisite stockholder approval and certain beneficial ownership limitations set by each preferred stockholder, each share of Series A Preferred Stock will automatically convert into an aggregate of approximately 1,000 shares of our Common Stock. The shares of Common Stock registered by this prospectus are referred to herein as the “Resale Shares.”
The Merger Common Shares and Merger Preferred Shares were issued and sold to former stockholders of Spyre Therapeutics, Inc., a Delaware corporation (“Spyre”), in connection with the acquisition (the “Asset Acquisition”) of Spyre, which closed on June 22, 2023. The Private Placement Preferred Shares were issued and sold to accredited investors in a private placement (the “PIPE” and, together with the Asset Acquisition, the “Transactions”), which closed on June 26, 2023. We are not selling any Resale Shares under this prospectus and will not receive any of the proceeds from the sale or preferred stock, debtother disposition of Resale Shares by the Selling Stockholders.
The Selling Stockholders may sell the Resale Shares on any national securities warrants to purchase our common stock, preferred stockexchange or debtquotation service on which the securities subscription rights to purchase our common stock, preferred stockmay be listed or debt securities and/or units consistingquoted at the time of some or all of these securities, in any combination, together or separately,sale, on the over-the-counter market, in one or more offerings, in amounts,transactions otherwise than on these exchanges or systems, such as privately negotiated transactions, or using a combination of these methods, and at fixed prices, and on the terms that we will determineat prevailing market prices at the time of the offering and which will be set forth in a prospectus supplement and any related free writing prospectus. The prospectus supplement and any related free writing prospectus may also add, updatesale, at varying prices determined at the time of sale, or change information containedat negotiated prices. See the disclosure under the heading “Plan of Distribution” elsewhere in this prospectus. prospectus for more information about how the Selling Stockholders may sell or otherwise dispose of their Resale Shares hereunder.
The totalSelling Stockholders may sell any, all or none of the securities offered by this prospectus and we do not know when or in what amount the Selling Stockholders may sell their Resale Shares hereunder following the effective date of these securities will have an initial aggregate offering pricethe registration statement of up to $150,000,000.which this prospectus forms a part.
You should carefully read this prospectus, the information incorporated, or deemed to be incorporated, by reference in this prospectus and any applicable prospectus supplement, and related free writing prospectus carefullyas well as any documents incorporated by reference herein or therein, before you invest.invest in any of the securities being offered.
Our common stockCommon Stock is traded on The NASDAQ GlobalNasdaq Capital Market under the symbol “AGLE.” On April 28, 2017August 4, 2023, the last reported sales price for our common stockCommon Stock was $7.15$0.48 per share. None of the other securities we may offer are currently traded on any securities exchange. The applicable prospectus supplement and any related free writing prospectus will contain information, where applicable, as to any other listing on The NASDAQ Global Market or any securities market or exchange of the securities covered by the prospectus supplement and any related free writing prospectus.
An investment in our securities involves a high degree of risk. You should carefully consider the information under the heading “Risk Factors” beginning on page 635 of this prospectus before investing in our securities.
Common stock, preferred stock, debt securities, warrants, subscription rights and/or units may be sold by us to or through underwriters or dealers, directly to purchasers or through agents designated from time to time. For additional information on the methods of sale, you should refer to the section entitled “Plan of Distribution” in this prospectus. If any underwriters, dealers or agents are involved in the sale of any securities with respect to which this prospectus is being delivered, the names of such underwriters or agents and any applicable fees, discounts or commissions, details regarding over-allotment options, if any,prospectus supplement, and under similar headings in the net proceeds to us will be set forth in a prospectus supplement. The price to the public of such securities and the net proceeds we expect to receive from such sale will also be set forth in a prospectus supplement.other documents that are incorporated by reference into this prospectus.
Neither the Securities and Exchange CommissionSEC nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this prospectus is , 20172023
i
ABOUT THIS PROSPECTUS
This prospectus is part of a registration statement on Form S-3that we filed with the Securities and Exchange Commission, or the SEC using a “shelf” registration process. Under this shelf registration process, the Selling Stockholders may, from time to time, we may sell any combination of the securities described in this prospectus in one or more offerings, up to a total dollar amount of $150,000,000. We have provided to you in thisofferings.
This prospectus a general description of the securities we may offer. Each time we sell securities under this shelf registration process, we will provide a prospectus supplementcontains and incorporates by reference information that will contain specific information about the terms of the offering. We may also add, update or change in the prospectus supplement any of the information contained in this prospectus. To the extent there is a conflict between the information contained in this prospectus and the prospectus supplement, you should rely onconsider when making your investment decision. Neither we, nor the information in the prospectus supplement;provided that, if any statement in one of these documents is inconsistent with a statement in another document having a later date—for example, a document incorporated by reference in this prospectus or any prospectus supplement—the statement in the document having the later date modifies or supersedes the earlier statement. You should read both this prospectus and any prospectus supplement together with additional information described under the next heading “Where You Can Find More Information.”
You should rely only on the information contained in or incorporated by reference into this prospectus or any applicable prospectus supplement. No dealer, salesperson or any other person isSelling Stockholders, have authorized anyone to give any information or to make any representation other than the information and representationsthose contained in or incorporated by reference intoin this prospectus or any applicable prospectus supplement. Ifprospectus. The Selling Stockholders are offering to sell, and seeking offers to buy, our securities only in jurisdictions where it is lawful to do so. We have not authorized anyone to provide you with different information is given or different representations are made, you may not rely on that information or those representations as having been authorized by us. You may not imply from the delivery of this prospectus and any applicable prospectus supplement, nor from a sale made under this prospectus and any applicable prospectus supplement, that our affairs are unchanged since the date of this prospectus and any applicable prospectus supplement or that the information contained in any document incorporated by reference is accurate as of any date other than the date of the document incorporated by reference, regardless of the time of delivery of this prospectus and any applicable prospectus supplement or any sale of a security.information. This prospectus and any applicableaccompanying prospectus supplement may only be used where it is legaldo not constitute an offer to sell or the securities.
THIS PROSPECTUS MAY NOT BE USED TO OFFER AND SELL SECURITIES UNLESS IT IS ACCOMPANIED BY AN ADDITIONAL PROSPECTUS OR A PROSPECTUS SUPPLEMENT.solicitation of an offer to buy any securities other than the securities described in any accompanying prospectus supplement or an offer to sell or the solicitation of an offer to buy such securities in any circumstances in which such offer or solicitation is unlawful. You should assume that the information appearing in this prospectus, any prospectus supplement, the documents incorporated by reference and any related free writing prospectus is accurate only as of their respective dates. Our business, financial condition, results of operations and prospects may have changed materially since those dates.
In this prospectus, unless the context otherwise requires, the terms “Aeglea,” the “Company,” “we,” “us,” and “our” refer to Aeglea BioTherapeutics, Inc., a Delaware corporation, and its consolidated subsidiaries.
This prospectus contains trade names, trademarks and service marks of others, which are the property of their respective owners. Solely for convenience, trademarks and trade names referred to in this prospectus may appear without the ® or TM symbols.
All references to “our product candidates,” “our programs” and “our pipeline” in this prospectus refer to the research programs with respect to which we have exercised the option to acquire intellectual property license rights to or have the option to acquire intellectual property license rights to pursuant to that certain antibody discovery and option agreement by and among Spyre, Paragon Therapeutics, Inc. (“Paragon”) and Parapyre Holding LLC (“Parapyre”) dated May 25, 2023 (the “Spyre Option Agreement”).
1
CAUTIONARY STATEMENT CONCERNING FORWARD-LOOKING STATEMENTS
This prospectus, including the documents that we incorporate by reference, contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). These forward-looking statements involve a number of risks and uncertainties. We caution readers that any forward-looking statement is not a guarantee of future performance and that actual results could differ materially from those contained in the forward-looking statement. These statements are based on current expectations of future events.
All statements, other than statements of historical facts contained in this prospectus, including, without limitation, statements regarding: stockholder approval of the conversion rights of the Series A Preferred Stock; any future payouts under the CVR (as defined herein); our ability to achieve the expected benefits or opportunities and related timing with respect to our acquisition of Spyre or to monetize any of our legacy assets, our future results of operations and financial position, business strategy, the length of time that we believe our existing cash resources will fund our operations, our market size, our potential growth opportunities, our preclinical and future clinical development activities, the efficacy and safety profile of our product candidates, the potential therapeutic benefits and economic value of our product candidates, the timing and results of preclinical studies and clinical trials, the expected impact of macroeconomic conditions, including inflation, increasing interest rates and volatile market conditions, current or potential bank failures, as well as global events, including the ongoing military conflict in Ukraine and geopolitical tensions in China on our operations, and the receipt and timing of potential regulatory designations, approvals and commercialization of product candidates. Forward-looking statements generally relate to future events or our future financial or operating performance. Forward-looking statements generally relate to future events or our future financial or operating performance. The words “believe,” “may,” “will,” “potentially,” “estimate,” “continue,” “anticipate,” “predict,” “target,” “intend,” “could,” “would,” “should,” “project,” “plan,” “expect,” and similar expressions that convey uncertainty of future events or outcomes are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words.
These forward-looking statements are subject to a number of risks, uncertainties and assumptions. Moreover, we operate in a very competitive and rapidly changing environment, and new risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. Factors that might cause such a difference include those discussed in our most recent Annual Report on Form 10-K, as supplemented and updated by subsequent Quarterly Reports on Form 10-Q and Current Reports on Form 8-K that we have filed or will file with the SEC, and in other documents which are incorporated by reference into this prospectus, as well as the risk factors and other information contained in or incorporated by reference into any accompanying prospectus supplement. You are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date of this prospectus or, in the case of documents referred to or incorporated by reference, the date of those documents.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based on information available to us as of the date of this prospectus. While we believe that such information provides a reasonable basis for these statements, such information may be limited or incomplete. Our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all relevant information. These statements are inherently uncertain, and investors are cautioned not to unduly rely on these statements.
All subsequent written or oral forward-looking statements attributable to us or any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section. We do not undertake any obligation to release publicly any revisions to these forward-looking statements to reflect events or circumstances after the date of this prospectus or to reflect the occurrence of unanticipated events, except as may be required under applicable U.S. securities laws. If we do update one or more forward-looking statements, no inference should be drawn that we will make additional updates with respect to those or other forward-looking statements.
2
PROSPECTUS SUMMARY
This summary may not contain all the information that you should consider before investing in securities. You should read the entire prospectus and the information incorporated by reference in this prospectus carefully, including “Risk Factors” and the financial data and related notes and other information incorporated by reference herein, before making an investment decision.
Company Overview
We areOn June 22, 2023, we completed the Asset Acquisition pursuant to Spyre Acquisition Agreement. Spyre was apre-clinical stage biotechnology company committedthat was incorporated on April 28, 2023 under the direction of Peter Harwin, a Managing Member of Fairmount, for the purpose of holding rights to certain intellectual property being developed by Paragon. Fairmount is a founder of Paragon.
Through the Asset Acquisition, we received the option to acquire the intellectual property rights related to four research programs (collectively, the “Option”) pursuant to the Spyre Option Agreement. On July 12, 2023, we exercised the Option with respect to one of these research programs to exclusively license intellectual property rights related to such research program directed to antibodies that selectively bind to a4b7 integrin and methods of using these antibodies, including methods of treating IBD using SPY001. If this research program is pursued non-provisionally and matures into issued patents, we would expect those patents to expire no earlier than 2044 subject to any disclaimers or extensions. The license agreement pertaining to such research program is currently being finalized. Furthermore, as of the date of this registration statement, the Option remains unexercised with respect to the intellectual property rights related to the three remaining research programs under the Spyre Option Agreement. For more information on the Spyre Option Agreement, see discussion under the heading “Spyre Option Agreement” below.
On July 27, 2023, we announced that we entered into an agreement to sell the global rights to pegzilarginase, an investigational treatment for the rare metabolic disease Arginase 1 Deficiency, to Immedica Pharma AB (“Immedica”) for $15.0 million in upfront cash proceeds and up to $100.0 million in contingent milestone payments (the “Immedica APA”). The sale of pegzilarginase to Immedica supersedes and terminates the license agreement between us and Immedica dated March 2021. See the section titled “Recent Developments” below for more information regarding the Immedica APA.
Following the Asset Acquisition and the entry into the Immedica APA, we have significantly reshaped the business into a preclinical stage biotechnology company focused on developing enzyme-basednext generation therapeutics for patients living with inflammatory bowel disease (“IBD”), including ulcerative colitis (“UC”) and Crohn’s disease (“CD”). Our portfolio of novel and proprietary monoclonal antibody product candidates has the potential to address unmet needs in the field of amino acid metabolismIBD care by improving efficacy, safety, and/or dosing convenience relative to treat rare genetic diseasesproducts currently available or product candidates in development. We have purposely engineered our product candidates to bind potently and cancer. Our engineered human enzymes are designed to degrade specific amino acids in the bloodselectively to target these diseases. In inborn errorsepitopes with extended half-lives. We plan to use combinations of metabolism, or IEM,our proprietary antibodies and patient enrichment strategies via companion diagnostics to enhance efficacy. We intend to deliver our product candidates through convenient, infrequently self-administered, subcutaneous (“SC”) injection as a subset of rare genetic diseases, we are seeking to reduce the toxic levels of amino acids in patients to the normal range. In oncology, we are seeking to reduce amino acid blood levels below the normal range where we believe we will be able to exploit the dependence of certain cancers on specific amino acids.pre-filled pen.
Our lead product candidate, AEB1102 (pegzilarginase),Strategy
Our goal is engineered to degrade the amino acid arginine and is being developed to treat two extremes of arginine metabolism, including arginine excess in patients with Arginase I deficiency, an IEM, as well as some cancers which have been shown to have a metabolic dependence on arginine. AEB1102 has demonstrated clinical proof of mechanism in both scenarios. In a Phase 1 clinical trialdevelop next-generation therapeutics for the treatment of patients with Arginase I deficiency,IBD, relying on three strategic pillars:
| • | | Advancing novel, potentially best-in-class, long-acting antibodies against validated IBD targets; |
| • | | Evaluating rational therapeutic combinations of potentially best-in-class antibodies; and |
3
| • | | Developing genetic- or biomarker-based companion diagnostics to match treatment targets to IBD sub-populations. |
Inflammatory Bowel Disease
IBD is a dose-proportional reduction in plasma arginine levels was observed inchronic condition characterized by inflammation within the gastrointestinal tract. It encompasses two patients. A reduction in blood arginine levels was also observed in Phase 1 clinical trials formain disorders: UC and CD. UC primarily affects the treatment of patients with advanced solid tumorscolon and the hematological malignancies relapsed refractory acute myeloid leukemia,rectum. Inflammation occurs in the innermost lining of the colon leading to ulcers. Symptoms include bloody diarrhea, abdominal pain, bowel urgency, and frequent bowel movements. CD can affect any part of the gastrointestinal tract, from the mouth to the anus. It is characterized by inflammation that extends through multiple layers of the bowel wall. Symptoms include abdominal pain, diarrhea, weight loss, fatigue, and complications such as strictures or AML,fistulas. Both conditions can significantly impact patients’ quality of life in terms of physical health, emotional well-being, and myelodysplastic syndrome,the unpredictability of symptom onset.
IBD affects millions of individuals worldwide, with increasing prevalence and incidence in both developed and developing countries. In the United States, it is estimated that approximately 1.7 million individuals currently have IBD, with approximately 70,000 patients newly diagnosed every year. The prevalence of UC in the United States is approximately 900,000 individuals, and the prevalence of CD in the United States is approximately 800,000 individuals. Based on research from the Crohn’s and Colitis Foundation of America, the market for IBD therapeutics is expected to experience steady growth, driven by rising disease prevalence, increasing diagnosis rates, and evolving treatment paradigms.
A range of pharmaceutical options exists, including anti-inflammatory drugs, immunosuppressants, and biologics. Treatment plans are often tailored to the individual patient’s disease severity, location, and response to therapy. In some cases, surgical interventions such as bowel resection or MDS. These preliminary results support AEB1102’s potential use as a therapeuticostomy formation may be necessary to manage complications or improve quality of both Arginase I deficiency and certain cancers associated with abnormal amino acid metabolism.life.
Despite available treatments, there remain substantial unmet needs in IBD management, including:
| • | | Inadequate response or loss of response to existing therapies; |
| • | | Side effects and safety concerns associated with long-term medication use; |
| • | | Limited options for patients with refractory or severe disease; and |
| • | | Adherence to frequent and/or inconvenient dosing regimens. |
Our Portfolio
We are conducting three clinical trials for AEB1102, consistingadvancing a broad pipeline of one Phase 1/2 clinical trial for the treatment of Arginase I deficiency and two Phase 1 clinical trials for the treatment of certain cancers.
Arginase I Deficiency. Following completion of dosing for the first two adult patientspotentially best-in-class monoclonal antibodies (“mAbs”) in our Phase 1 clinical trial for the treatment of patients with Arginase I deficiency, we submitted a protocol amendment in November 2016 to broaden the scope of our Phase 1 trial into a Phase 1/2 trial. The amended protocol includes dosing of pediatric patients (two and older) and weekly repeat dosing,connection with the intentresearch programs with respect to assesswhich we have exercised the safety, tolerability, pharmacokinetics, pharmacodynamics, and clinical response of AEB1102 in patients with this IEM. InOption to acquire intellectual property license rights to or have the first quarter of 2017, we received IRB approval forOption to acquire intellectual property license rights to pursuant to the Phase 1/2 protocol for the treatment of patients with Arginase I deficiency at multiple clinical trial sites. In March 2017, we received an information request from the FDA which included comments and recommendations on the protocol amendment and a request for supporting documents based on their review of our completed toxicology studies, our dose escalation plan and our information to support the inclusion of pediatric patients. As recommended by the FDA, we replied with supporting information and completed a follow-up meeting. At this time, we believe our Phase 1/2 protocol provides an appropriate path to evaluate the safety and tolerability of AEB1102 in pediatric patients, and pending FDA feedback, we plan to initiate dosing in pediatric patients in the middle of 2017. In March 2017, we announced results from the first two adult patients in our Phase 1 clinical trial for the treatment of Arginase I deficiency at the 2017 American College of Medical Genetics and Genomics Annual Clinical Genetics Meeting, or ACMG Annual Meeting. We intend to continue enrollment of adult patientsSpyre Option Agreement and plan to dose additional adult patientsdevelop a companion diagnostic for each program. The following table summarizes these programs*.
4
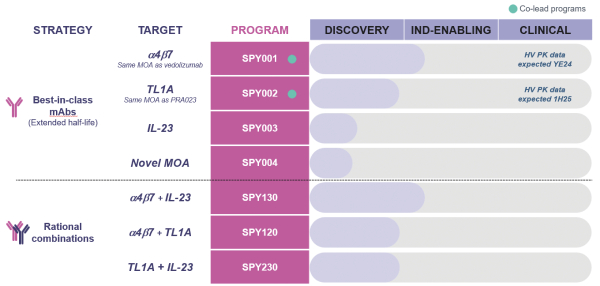
| * | We exercised the Option to acquire intellectual property license rights related to SPY001 on July 12, 2023. Although we hold the Option to acquire intellectual property license rights related to SPY002, SPY003 and SPY004, such Option remains unexercised as the date of this registration statement. See discussion under the heading “Spyre Option Agreement” in our next Quarterly Report on Form 10-Q, which will be incorporated by reference herein, for more information. |
SPY001 – anti-a4b7 mAb
Our most advanced product candidate, SPY001, is a highly potent, highly selective, and fully human monoclonal immunoglobulin G1 antibody designed to bind selectively to the a4b7 integrin. Thea4b7 integrin is a protein found on the surface of immune cells known as lymphocytes. This integrin regulates the migration of lymphocytes to the gut where they contribute to the inflammatory process in IBD. By binding to the a4b7 integrin, SPY001 is designed to prevent the interaction of these lymphocytes with MAdCAM-1, a molecule expressed on endothelial cells lining the blood vessels in the middlegut. This interaction is responsible for guiding lymphocytes from the bloodstream into the gut tissue, where they cause inflammation. By blocking the interaction between a4b7 integrin and MAdCAM-1, SPY001 aims to reduce the recruitment of 2017. Toplinelymphocytes to the gut, leading to a decrease in inflammation. Since it specifically targets the gut immune system, SPY001 is designed to help minimize systemic immunosuppressive effects unrelated to IBD pathology.
Vedolizumab is an anti-a4b7 integrin mAb marketed by Takeda as Entyvio. It is available in the United States as an intravenously administered product with an every 8-week maintenance dosing regimen. A SC version with an every 2-week dosing regimen is under review by the United States Food and Drug Administration (the “FDA”) for use in the maintenance setting and is approved in Europe and Japan. Vedolizumab is one of the leading therapies for moderate-to-severe IBD given its exquisite safety profile as a gut selective mechanism and its high efficacy rates in UC (particularly in the maintenance setting where it has the highest remission rates among approved biologics). In the induction setting, exposure-response data suggests that higher exposures may increase the likelihood of early remission in UC. In 2022, Takeda reported $5.3 billion in sales for Entyvio and updated its peak annual sales guidance to $7.5-9.0 billion. We believe that there is a significant opportunity for a SC regimen with a more infrequent dosing schedule (every eight to twelve weeks) in the maintenance settings, and potential efficacy upside in induction.
Our preclinical development program has demonstrated that SPY001 binds to the same epitope as vedolizumab with similar potency and selectivity. Both SPY001 and vedolizumab potently block MAdCAM-1 adhesion and
5
avoid interactions with a4b1 to prevent adhesion to VCAM-1, which can be associated with the risk of progressive multifocal leukoencephalopathy. Additionally, our preclinical studies have demonstrated that SPY001 binds to memory T-helper cells from this trialhuman donors with a comparable potency as vedolizumab.
SPY001 is engineered with half-life extension technology. This approach relies on modifying the Fc domain to improve the pharmacokinetic (“PK”) profile of the antibody and support infrequent dosing as a convenient SC injection. An evaluation of SPY001’s PK in non-human primates suggests an increase in half-life of approximately 2-fold relative to vedolizumab, potentially supporting a SC dosing administration of every eight to twelve weeks in humans.
We aim to achieve the following target product profile for SPY001 in IBD:
| • | | Less frequent administration: Lower the frequency of administration by incorporating half-life extension technology and deliver SPY001 through a single SC injection every eight to twelve weeks. |
| • | | High concentration, citrate-free formulation: Deliver at least 300 mg in a single 2 mL injection suitable for a pre-filled pen without the need for citrate to enhance patient comfort. |
| • | | Potential for increased induction efficacy in UC: Target greater exposure in the induction setting given exposure-response relationships observed with vedolizumab in UC. |
SPY001 is currently progressing through IND-enabling studies and is expected to enter first-in-human (“FIH”) studies in the first half of 2018.2024. We believe SPY001 has the potential to meaningfully transform the standard of care in IBD as a potent, low-frequency and self-administered SC injection.
SPY002 – anti-TL1A mAb
Our co-lead product candidate, SPY002, is a highly potent, highly selective, and fully human mAb designed to bind to tumor necrosis factor-like ligand 1A (“TL1A”). TL1A is a protein that plays a role in regulating the immune system and is found to be elevated in the gut tissue of individuals with IBD. TL1A interacts with its receptor, death receptor 3 (“DR3”), which is expressed in various immune cells, including T cells. This interaction triggers signaling pathways that contribute to inflammation and immune system activation, leading to IBD symptomology. SPY002 has been designed to block the interaction between TL1A and DR3 and thereby inhibit the downstream signaling events and dampen the inflammatory response. By neutralizing TL1A, SPY002 has the potential to modulate the immune response in IBD patients, potentially reducing disease activity and promoting mucosal healing.
TL1A is a clinically validated target that has been studied in multiple Phase 2 trials in UC and CD. Merck’s anti-TL1A molecule (PRA023) was studied in a randomized controlled Phase 2 trial where it showed a 25% placebo-adjusted clinical remission rate at week 12 and a placebo-adjusted endoscopic improvement rate of 31% for patients with UC. PRA023 was additionally studied in a Phase 2a open label trial in CD where it showed a 49% clinical remission rate at week 12 compared to a prespecified 16% historical placebo rate and a 26% endoscopic response compared to a prespecified 12% historical placebo rate. Roivant is also advancing an anti-TL1A molecule (RVT-3101) that showed a 21% placebo-adjusted clinical remission rate and a 21% placebo-adjusted endoscopic response in a Phase 2b study in UC patients. In the maintenance setting at week 56, RVT-3101 showed a compelling clinical remission rate of 36% (absolute) at the expected Phase 3 dose. Both PRA023 and RVT-3101 lack any half-life extension technologies and are dosed every four weeks in the maintenance setting.
Like SPY001, SPY002 is engineered with half-life extension technology to support infrequent, self-administration as a convenient SC injection. It is expected that these modifications, along with a high concentration formulation, will enable dosing every eight weeks or less frequently in humans.
6
Advanced Solid Tumors. In October 2015, we initiated enrollmentWe aim to achieve the following target product profile for a Phase 1 dose escalation trial for cancer patients with advanced solid tumors. In this ongoing trial, patients have demonstrated aSPY002 in IBD and other inflammatory diseases:
| | • | | reduction in blood arginine levels fromLess frequent administration: Lower the dosingfrequency of AEB1102, providing proof-of-mechanism. We expect to announce results of this Phase 1 dose escalation in patients with advanced solid tumorsadministration by incorporating half-life extension technology and anticipate initiating expansion arms in specific solid tumor types, potentially in combination with existingdeliver SPY002 through a single SC injection every eight weeks or emerging standards of care, in the fourth quarter of 2017 or the first quarter of 2018.less frequently.
|
Hematological Malignancies. In July 2016, we initiated | • | | High potency: High affinity to TL1A trimers, enabling lower doses. |
| • | | High concentration, citrate-free formulation: Deliver at least 300 mg in a single 2 mL injection suitable for a pre-filled pen. |
We expect to enter FIH studies in the second half of 2024. We believe SPY002 has the potential to be a Phase 1best-in-class TL1A antibody compared to others in development as a highly potent, highly selective and infrequently self-administered SC injection.
SPY003 – anti-IL-23 mAb
Our third program, SPY003, is a discovery stage program designed to bind to Interleukin 23 (“IL-23”). IL-23 is a cytokine that is produced by immune cells and is involved in immune response regulation. IL-23 promotes the differentiation and activation of Th17 cells. Th17 cells produce other inflammatory cytokines, such as IL-17, which contribute to the inflammation seen in IBD. IL-23 also helps in the recruitment and activation of other immune cells, such as neutrophils, which further contribute to tissue damage in the gut.
Several IL-23 mAbs have been approved or are in late-stage development for IBD including Skyrizi (AbbVie), Tremfya (Johnson & Johnson), and mirikizumab (Lilly).
We aim to achieve the following target product profile for SPY003 in IBD:
| • | | Less frequent administration: Lower the frequency of administration by incorporating half-life extension technology and deliver SPY003 through a single SC injection every eight weeks or less frequently. |
| • | | High concentration, citrate-free formulation: Deliver at least 300 mg in a single 2 mL injection suitable for a pre-filled pen. |
SPY004 – novel MOA mAb
SPY004 is an undisclosed novel mechanism of action (“MOA”) and incorporates half-life extension modifications.
Our combination programs – SPY120, SPY130, and SPY230
We aim to advance rational combinations of our therapeutic antibodies into clinical trialstudies. SPY120 combines SPY001 (a4b7) and SPY002 (TL1A), SPY130 combines SPY001(a4b7) and SPY003 (IL-23), and SPY230 combines SPY002 (TL1A) and SPY003 (IL23). We believe these combinations target orthogonal biology and could lead to greater remission rates in IBD.
Clinical proof-of-concept for the potential of combination therapy in IBD was demonstrated by Johnson & Johnson’s VEGA study which evaluated their anti-TNFa antibody, Simponi, in combination with their anti-IL-23 antibody, Tremfya, in patients with the hematological malignancies AML and MDSUC. The combination resulted in an absolute clinical remission rate of 47% in the United Statesinduction setting, which was nearly additive of the remission rates observed with either TNFa (25%) or IL-23 (24%) alone. We believe there is potential to build on this result by combining mechanisms that have shown greater efficacy rates in specific indications and Canada. Assettings and that exhibit a superior safety profile relative to the TNFa class.
7
Our precision immunology approach
We aim to develop genetic- or biomarker-based companion diagnostics across our portfolio of therapeutics to aid patients and physicians in selecting the optimal treatment regimen. Clinical proof-of-concept for the potential of companion diagnostic (“CDx”) approaches in IBD was demonstrated by both Prometheus (now Merck) and Roivant for their anti-TL1A programs. In Phase 2 studies, UC patients that were CDx positive were more likely to respond to anti-TL1A therapy as evidenced by a 7-13% increase in clinical remission rate at week 12 compared to all-comers. We are in discussions with several potential partners with access to large scale IBD biobanks to support CDx development across our portfolio.
Our Team and Investors
Our portfolio of potentially best-in-class antibodies were discovered and developed by Paragon. Spyre is the second company that was founded upon technology spun out of Paragon, a group of entrepreneurial scientists and investors with extensive mAb experience. Its scientific founders’ discoveries have also led to the creation of other successful biotechnology companies, including Cogent Biosciences, Inc., Viridian Therapeutics, Inc., Dianthus Therapeutics, Inc., and Apogee Therapeutics, Inc.
On June 22, 2023, we announced the completion of the Asset Acquisition with the sole focus on advancing a pipeline of next-generation antibodies for IBD with respect to which we have exercised the Option to acquire intellectual property license rights to or have the Option to acquire intellectual property license rights to pursuant to the Spyre Option Agreement. Concurrent with the closing of the acquisition, we completed a $210 million private placement of our Series A Preferred Stock with a syndicate of healthcare investors led by Fairmount Funds Management LLC, with participation from Fidelity Management & Research Company, Venrock Healthcare Capital Partners, Commodore Capital, Deep Track Capital, Perceptive Advisors, RTW Investments, Cormorant Asset Management, Driehaus Capital Management, Ecor1 Capital, RA Capital Management, Surveyor Capital (a Citadel company), and Wellington Management Company LLP, as well as additional institutional investors.
With respect to the Asset Acquisition, we determined that Aeglea was the acquiror for accounting purposes under ASC 805-10-25-4 and ASC 805-10-55-11. The primary factors considered were a) the relative voting rights in the trialcombined entity not resulting in a change of control, b) legacy members of our board of directors maintained control of the board, and c) the only change in the composition of senior management was the appointment of a new Chief Operating Officer. Next, we considered whether the Asset Acquisition should be defined as a business under ASC 805. ASC 805-10-55-5A through 55-5C describe a screen test to determine whether an acquired set of assets and activities is not a business. We determined that substantially all (greater than 90%) of the fair value of the assets acquired were concentrated in a single asset, Spyre’s Option to license intellectual property rights related to SPY001, SPY002, SPY003 and SPY004 pursuant to the Spyre Option Agreement. Accordingly, we treated the Asset Acquisition as an asset acquisition for accounting purposes. Even if the transaction would have failed the screen test, Spyre lacked the financial resources to have inputs, processes, and outputs to constitute a business under ASC 805. Following the Asset Acquisition, we will need to invest in infrastructure and the continuing research and development being conducted under the Spyre Option Agreement to be able to have any measurable outputs.
We have a strong management team and group of employees with diverse backgrounds and significant experience in developing novel treatments for patients at biopharmaceutical companies such as AbbVie, BridgeBio Pharma, Genentech, and Johnson & Johnson. Together, our team has a proven track record in the discovery, development, and commercialization of numerous approved therapeutics.
8
Recent Developments
On June 22, 2023, we acquired Spyre pursuant to an Agreement and Plan of Merger (the “Acquisition Agreement”), by and among the Company, Aspen Merger Sub I, Inc., a Delaware corporation and a wholly owned subsidiary of the Company (“First Merger Sub”), Sequoia Merger Sub II, LLC, a Delaware limited liability company and wholly owned subsidiary of the Company (“Second Merger Sub”), and Spyre. Pursuant to the Acquisition Agreement, First Merger Sub merged with advanced solid tumors,and into Spyre, pursuant to which Spyre was the first three cohortssurviving corporation and became a wholly owned subsidiary of this trial have demonstrated proof-of-mechanism.the Company (the “First Merger”). Immediately following the First Merger, Spyre merged with and into Second Merger Sub, pursuant to which Second Merger Sub became the surviving entity.
The Asset Acquisition was structured as a stock-for-stock transaction pursuant to which all of Spyre’s outstanding equity interests were exchanged based on a fixed exchange ratio of 1-for-13.73622, for consideration of a combination of 12,945,385 shares of Common Stock and 364,887 shares of Series A Preferred Stock (or 364,887,000 shares on an as-converted-to-common basis) in addition to the assumption of outstanding and unexercised stock options to purchase 68,365 shares of Common Stock from the Amended and Restated Spyre 2023 Equity Incentive Plan. Concurrently with the Asset Acquisition, we entered into a definitive agreement (the “Securities Purchase Agreement”) for a PIPE investment with existing and new investors (the “Investors”) to raise approximately $210 million in which the Investors were issued 721,452 shares of Series A Preferred Stock (or 721,452,000 shares on an as-converted-to-common basis) at a price of $291.08 per share (or $0.29108 per share on an as-converted-to-common basis). The Asset Acquisition was approved by our Board of Directors and the Board of Directors and stockholders of Spyre. The closings of the Transactions were not subject to the approval of Aeglea stockholders. Subject to Aeglea stockholder approval and certain beneficial ownership limitations set by each holder, each share of Series A Preferred Stock will automatically convert into 1,000 shares of Common Stock. We expect to announce resultsfile a proxy statement with the SEC to solicit such stockholder approval, among other matters, at a special meeting of Aeglea stockholders. Except as otherwise required by law (e.g. voting on a change to the authorized shares of Series A Non-Voting Preferred Stock or the rights of such shares as required by Delaware General Corporation Law) and Aeglea’s Certificate of Designation of Series A Non-Voting Convertible Preferred Stock (the “Certificate of Designation”), the Series A Preferred Stock does not have voting rights. For more information on the Series A Preferred Stock, please refer to the Certificate of Designation, which is incorporated by reference herein.
In connection with the execution of the Phase 1 dose escalation trialAcquisition Agreement, Aeglea and Spyre entered into stockholder support agreements (the “Support Agreements”) with certain of Aeglea’s officers and directors, which collectively own an aggregate of less than 1% of the outstanding shares of the Common Stock. The Support Agreements provide that, among other things, each of the parties thereto has agreed to vote or cause to be voted all of the shares of Common Stock owned by such stockholder in patientsfavor of the approval of the conversion of the Series A Preferred Stock into shares of Common Stock in accordance with AMLNasdaq Stock Market Rules at Aeglea’s stockholders’ meeting to be held in connection therewith.
Concurrently and MDS in the fourth quarter of 2017 or the first quarter of 2018.
Our pipeline of engineered human enzyme product candidates in preclinical development includes: AEB3103, an enzyme that degrades the amino acid cysteine, and its oxidized form cystine, to target a widely recognized, but previously underexploited vulnerability of cancer to oxidative stress; AEB2109, an enzyme that degrades the amino acid methionine to target methionine-dependent cancers and AEB4104, an engineered human enzyme to treat another IEM by degrading the amino acid homocysteine. We plan to continue preclinical development of AEB3103, AEB2109, AEB4104 and related variants of these candidatesconnection with the aim of submitting an IND for one or more of these development candidates in 2018.
We are a patient-focused organization consciousexecution of the fact that IEMAcquisition Agreement, certain Spyre stockholders as of immediately prior to the Asset Acquisition, and oncology patients have limited treatment options, and we recognize that their lives and well-being are highly dependent upon our efforts and the efforts of others to develop improved therapies. For this reason, we are passionate about discovering and developing therapeutics to address IEM and oncology indications where there is a significant unmet medical need. Our goal is to create a world-class company committed to efficiently developing a portfolio of product candidates to treat these diseases.
The Securities We May Offer
With this prospectus, we may offer common stock, preferred stock, debt securities, warrants, subscription rights to purchase our common stock, preferred stock or debt securities, and/or units consisting of some or all of these securities in any combination. The aggregate offering price of securities that we offer with this prospectus will not exceed $150,000,000. Each time we offer securities with this prospectus, we will provide offerees with a prospectus supplement that will contain the specific termscertain of the securities being offered. The following isdirectors and officers of Aeglea as of immediately prior to the Asset Acquisition entered into lock-up agreements with Aeglea and Spyre, pursuant to which each such stockholder will be subject to a summary of180-day lockup on the securities we may offer with this prospectus.
Common Stock
We may offer shares of our common stock, par value $0.0001 per share.
Preferred Stock
We may offer shares of our preferred stock, par value $0.0001 per share, in onesale or more series. Our board of directors or a committee designated by the board will determine the dividend, voting, conversion and other rights of the seriestransfer of shares of preferred stock being offered. Each seriesCommon Stock held by each such stockholder at the closing of preferred stockthe Asset Acquisition, including those shares received by such Spyre stockholders in the Asset Acquisition.
In connection with the Asset Acquisition, a non-transferrable contingent value right (a “CVR”) was distributed to Aeglea stockholders of record as of the close of business on July 3, 2023, but was not distributed to holders of shares of Common Stock or Series A Preferred Stock issued to the Investors or former stockholders of Spyre in
9
connection with the Transactions. Holders of the CVR will be entitled to receive certain stock and/or cash payments from proceeds received by us, if any, related to the disposition or monetization of Aeglea’s legacy assets for a period of one year following the closing of the Asset Acquisition.
On July 27, 2023, we announced that we entered the Immedica APA. The sale of pegzilarginase to Immedica supersedes and terminates the license agreement between the us and Immedica dated March 2021.
The milestone payments under the Immedica APA are contingent on formal reimbursement decisions by national authorities in key European markets and pegzilarginase approval by the FDA, among other events. The upfront payment and contingent milestone payments if paid, net of expenses and adjustments, will be distributed to holders of Aeglea’s CVR pursuant to the CVR Agreement that was entered into in connection with the Asset Acquisition.
For more fully described ininformation about the particular prospectus supplement that will accompany this prospectus, including redemption provisions, rights in the event of our liquidation, dissolutionTransactions or the winding up, voting rights and rights to convert into common stock.
Debt Securities
We may offer general obligations, which may be secured or unsecured, senior or subordinated and convertible into shares of our common stock or preferred stock. In this prospectus, we refer to the senior debt
securities and the subordinated debt securities together as the “debt securities.” Our board of directors will determine the terms of each series of debt securities being offered.
We will issue the debt securities under an indenture between us and a trustee. In this document, we have summarized general features of the debt securities from the indenture. We encourageImmedica APA, you to read the indenture, which is an exhibit to the registration statement of which this prospectus is a part.
Warrants
We may offer warrants for the purchase of debt securities, shares of preferred stock or shares of common stock. We may issue warrants independently or together with other securities. Our board of directors will determine the terms of the warrants.
Subscription Rights
We may offer subscription rights for the purchase of common stock, preferred stock or debt securities. We may issue subscription rights independently or together with other securities. Our board of directors will determine the terms of the subscription rights.
Units
We may offer units consisting of some or all of the securities described above, in any combination, including common stock, preferred stock, warrants and/or debt securities. The terms of these units will be set forth in a prospectus supplement. The description of the terms of these units in the related prospectus supplement will not be complete. You should refer to our Current Reports on Form 8-K filed with the applicable form of unitSEC on June 23, 2023 (excluding exhibit 99.1 furnished thereto) and unit agreement for complete information with respect to these units.July 27, 2023, respectively, which are incorporated by reference herein.
* * *Corporation Information
We were formed as a limited liability company under the laws of the State of Delaware in December 2013 and converted to a Delaware corporation in March 2015. In connection with our conversionOn June 22, 2023, we completed the Asset Acquisition, pursuant to which all of Spyre’s outstanding equity interests were exchanged based on a Delaware corporation, eachfixed exchange ratio of our outstanding13.73522 to 1 for consideration from Aeglea of 12,945,385 shares of the members of the limited liability company was converted intoCommon Stock and 364,887 shares of capital stock. On the date of conversion, each Series A convertible preferred share converted into a share of Series A convertible preferredPreferred Stock in addition to the assumption of outstanding and unexercised stock and each Common A share, Common A-1 share and Common B share converted intooptions to purchase 68,365 shares of common stock.Common Stock from the Amended and Restated Spyre 2023 Equity Incentive Plan. Our principal executive offices are located at 901 S. MoPac Expressway, Barton Oaks Plaza One,221 Crescent Street, Building 17, Suite 250, Austin, Texas 78746,102B, Waltham, MA 02453, and our telephone number is (512) 942-2935.
Our Relationship with Paragon and Parapyre
Paragon and Parapyre, a related party of Paragon, beneficially own more than 5% of our capital stock collectively through their holdings of our Common Stock and Series A Preferred Stock. Fairmount Funds Management LLC beneficially owns more than 5% of our capital stock, has two seats on our Board and beneficially owns more than 5% of Paragon, which is a joint venture between Fairmount Funds Management LLC and Fair Journey Biologics. Fairmount Funds Management LLC has appointed Paragon’s board of directors and has the contractual right to approve the appointment of any executive officers.
In connection with the Asset Acquisition, we assumed the rights and obligations of Spyre under the Spyre Option Agreement. Under the Spyre Option Agreement, we are obligated to compensate Paragon on a quarterly basis for its services performed under each research program based on the actual costs incurred. As of the date of the Asset Acquisition, Spyre had incurred total expenses of $19.3 million under the Spyre Option Agreement since inception, inclusive of a $3.0 million research initiation fee that was due upon signing of the Spyre Option Agreement and $16.3 million of reimbursable expenses under the Spyre Option Agreement for historical costs incurred by Paragon. As of the acquisition date, $19.3 million was unpaid and was assumed by us through the Asset Acquisition.
In July 2023, we exercised our option for SPY001 with the remaining three options for SPY002, SPY003, SPY004 remaining outstanding.
10
In connection with the Asset Acquisition, the Company assumed the Parapyre Option Obligation (as described under the section titled “Spyre Option Agreement” below)which provided for an annual equity grant of options to purchase 1% of the then outstanding shares of Spyre’s common stock, on a fully diluted basis, on the last business day of each calendar year, at the fair market value determined by the board of directors of Spyre. As a result of the Asset Acquisition, the Parapyre Option Obligation shall continue and Parapyre shall be entitled to receive the equivalent shares of the Company with the same terms.
See the section titled “Spyre Option Agreement” below for more information on the Spyre Option Agreement.
Spyre Option Agreement
In May 2023, Spyre entered into the Spyre Option Agreement with Paragon and Parapyre. In consideration for the Option granted under the Spyre Option Agreement, Spyre was obligated to pay Paragon an upfront cash amount of $3.0 million in research initiation fees. In addition, Spyre was obligated to pay incurred reimbursable research costs of $16.3 million to Paragon as of the closing of the Asset Acquisition. Furthermore, the Spyre Option Agreement provided for an annual equity grant of options to purchase 1% of the then outstanding shares of Spyre’s common stock, on a fully diluted basis, on the last business day of each calendar year, at the fair market value determined by the board of directors of Spyre (the “Parapyre Option Obligation”). As a result of the Asset Acquisition, we assumed the rights and obligations of Spyre under the Spyre Option Agreement, including the Parapyre Option Obligation. Pursuant to the Spyre Option Agreement, on a research program-by-research program basis following the finalization of the research plan for each respective research program, we are required to pay Paragon a nonrefundable fee in cash of $0.75 million. We are also obligated to compensate Paragon on a quarterly basis for its services performed under each research program based on the actual costs incurred. For the period from June 22, 2023 (Asset Acquisition date) to June 30, 2023, we did not make any payments to Paragon.
On July 12, 2023, we exercised our Option available under the Spyre Option Agreement with respect to the SPY001 research program and will enter into a SPY001 license agreement (the “SPY001 License Agreement”). Our Option available under the Spyre Option Agreement with respect to SPY002, SPY003 and SPY004 remains unexercised.
Following the execution of the SPY001 License Agreement, we will also obligated to pay Paragon up to $23.0 million upon the achievement of specific development and clinical milestones for the first product under the SPY001 License Agreement that achieves such specified milestones. Upon execution of the SPY001 License Agreement, we will pay Paragon a $1.5 million fee for nomination of a development candidate, and we are obligated to make a further milestone payment of $2.5 million upon the first dosing of a human patient in a Phase 1 trial. Subject to the execution of the Option with respect to SPY002, SPY003 or SPY004, we expect to be obligated to make similar payments upon and following the execution of license agreements with respect to SPY002, SPY003 and SPY004, respectively. For additional detail regarding our arrangements with Paragon, see the section titled “Our Relationship with Paragon and Parapyre” above.
Commercial
Should any of our product candidates be approved for commercialization, we intend to develop a plan to commercialize them in the United States and other key markets, through internal infrastructure and/or external partnerships in a manner that will enable us to realize the full commercial value of our programs. Given our stage of development, we have not yet established a commercial organization or distribution capabilities.
11
Manufacturing
We do not currently own or operate facilities for product manufacturing, testing, storage, and distribution. We are currently in the process of novating certain agreements with third parties for the performance of future clinical manufacturing and toxicology activities from Paragon to us. The initial forms of these agreements are generally non-specific master services agreements that allow an entity to begin the process of future manufacturing or toxicology services, respectively. As clinical development activities are commenced by us, the agreements will be revised to provide for the specific deliverables and associated costs that are needed under our development plan.
Competition
We expect to face intense competition from other biopharmaceutical companies that are developing agents for the treatment of inflammatory diseases. If approved for the treatment of patients with moderate-to-severe IBD, our portfolio of products would compete with TNF antibodies including Humira (AbbVie), Remicade (Johnson & Johnson), and Simponi (Johnson & Johnson); IL-12/23 and IL-23 antibodies including Stelara (Johnson & Johnson) and Skyrizi (AbbVie); a4b7 antibody Entyvio (Takeda); JAK inhibitors including Xeljanz (Pfizer), Rinvoq (AbbVie); and S1P1 receptor modulating therapies including Zeposia (Bristol Myers Squibb).
We are aware of several companies with product candidates in development for the treatment of patients with IBD, including Merck’s PRA023, Roivant’s RVT-3101, and Teva’s TEV-48574 TL1A antibodies; additional IL-23s including Tremfya (Johnson & Johnson) and mirikizumab (Lilly); additional S1P1 modulator etrasimod (Pfizer); and oral anti-integrin agents including Morphic Therapeutic’s MORF-057 and Gilead’s GS-1427, and a discovery program at Dice Therapeutics (Lilly).
Government Regulation
The FDA and other regulatory authorities at federal, state and local levels, as well as in foreign countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing, post-approval monitoring and post-approval reporting of biologics such as those we are developing. We, along with our third-party contractors, will be required to navigate the various preclinical, clinical and commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval or licensure of our product candidates. Failure to comply with the applicable United States requirements at any time during the product development process, approval process or post-market may subject an applicant to administrative or judicial sanctions. These sanctions could include, among other actions, the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, product recalls or market withdrawals, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement and civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
United States Biologics Regulation
In the United States, biological products are subject to regulation under the Federal Food, Drug, and Cosmetic Act (“FDCA”) and the Public Health Service Act (“PHSA”) and their implementing regulations, as well as other federal, state, local, and foreign statutes and regulations. The process required by the FDA before biologic product candidates may be marketed in the United States generally involves the following:
completion of preclinical laboratory tests and animal studies performed in accordance with applicable regulations, including the FDA’s current Good Laboratory Practices;
12
submission to the FDA of an investigational new drug application (“IND”), which must become effective before clinical trials may begin and must be updated annually or when significant changes are made;
approval by an independent institutional review board (“IRB”), or ethics committee at each clinical site before the trial may be commenced;
manufacture of the proposed biologic candidate in accordance with current Good Manufacturing Practices (“cGMPs”);
performance of adequate and well-controlled human clinical trials in accordance with applicable IND regulations, current Good Clinical Practice (“cGCP”) requirements and other clinical-trial related regulations to establish the safety, purity and potency of the proposed biologic product candidate for its intended purpose;
preparation of and submission to the FDA of a biologics license application (“BLA”), after completion of all pivotal clinical trials;
satisfactory completion of an FDA Advisory Committee review, if applicable;
a determination by the FDA within 60 days of its receipt of a BLA to file the application for review;
satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the proposed product is produced to assess compliance with cGMPs, and to assure that the facilities, methods and controls are adequate to preserve the biological product’s continued safety, purity and potency, and potential audit of selected clinical investigation sites to assess compliance with cGCPs;
payment of user fees for FDA review of the BLA, unless a waiver is applicable; and
FDA review and approval of a BLA to permit commercial marketing of the product for a particular indication(s) for use in the United States.
Preclinical and Clinical Development
Prior to beginning the first clinical trial with a product candidate, in the United States, we must submit an IND to the FDA. An IND is a request for authorization from the FDA to administer an investigational new drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol or protocols for preclinical studies and clinical trials. The IND also includes results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology and pharmacodynamic characteristics of the product, chemistry, manufacturing and controls information, and any available human data or literature to support the use of the investigational product. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day period, raises safety concerns or questions about the proposed clinical trial. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before the clinical trial can begin. Submission of an IND therefore may or may not result in FDA authorization to begin a clinical trial. Similar processes exist in countries outside the United States that we will be required to follow if we choose to execute trials in other countries.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with cGCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical study. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for
13
each successive clinical trial conducted during product development and for any subsequent protocol amendments. Furthermore, an independent IRB for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial begins at that site, and must monitor the study until completed.
Regulatory authorities, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk or that the trial is unlikely to meet its stated objectives. Some studies also include oversight by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board, which provides authorization for whether or not a study may move forward at designated check points based on access to certain data from the study and may recommend halting the clinical trial if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing preclinical studies and clinical trials and clinical study results to public registries. Sponsors of clinical trials of FDA-regulated products, including biological products, are required to register and disclose certain clinical trial information, which is publicly available at www.clinicaltrials.gov.
A sponsor who wishes to conduct a clinical trial outside of the United States may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. If a foreign clinical trial is not conducted under an IND, the sponsor may submit data from the clinical trial to the FDA in support of a BLA. The FDA will accept a well-designed and well-conducted foreign clinical trial not conducted under an IND if the trial was conducted in accordance with cGCP requirements and the FDA is able to validate the data through an onsite inspection if deemed necessary.
For the purposes of BLA approval, human clinical trials are typically conducted in three sequential phases that may overlap.
| • | | Phase 1. The investigational product is initially introduced into healthy human subjects or patients with the target disease or condition. These studies are designed to test the safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans and the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. |
| • | | Phase 2. The investigational product is administered to a limited patient population with a specified disease or condition to evaluate the preliminary efficacy, optimal dosages and dosing schedule and to identify possible adverse side effects and safety risks. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. |
| • | | Phase 3. The investigational product is administered to an expanded patient population to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the investigational product and to provide an adequate basis for product approval. |
In some cases, the FDA may require, or companies may voluntarily pursue, additional clinical trials after a product is approved to gain more information about the product. These so-called Phase 4 studies may be made a condition to approval of the BLA. Concurrent with clinical trials, companies may complete additional animal studies and develop additional information about the biological characteristics of the product candidate and must finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final product, or for biologics, the safety, purity and potency. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
14
During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data and clinical study investigators. Written IND safety reports must be promptly submitted to the FDA and the investigators for serious and unexpected suspected adverse events, any findings from other studies, tests in laboratory animals or in vitro testing that suggest a significant risk for human subjects, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information qualifies for reporting. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days after the sponsor’s initial receipt of the information.
BLA Submission and Review
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development, preclinical studies and clinical trials are submitted to the FDA as part of a BLA requesting approval to market the product for one or more indications. FDA approval of a BLA must be obtained before a biologic may be marketed in the United States. The BLA must include all relevant data available from pertinent preclinical studies and clinical trials, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls, and proposed labeling, among other things. Data can come from company-sponsored clinical studies intended to test the safety and effectiveness of the product, or from a number of alternative sources, including studies initiated and sponsored by investigators. The submission of a BLA requires payment of a substantial application user fee to the FDA, unless a waiver or exemption applies.
In addition, under the Pediatric Research Equity Act (“PREA”), a BLA or supplement to a BLA must contain data to assess the safety and effectiveness of the biological product candidate for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The Food and Drug Administration Safety and Innovation Act requires that a sponsor who is planning to submit a marketing application for a biological product that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration submit an initial pediatric study plan (“PSP”) within sixty days after an end-of-Phase 2 meeting or, if there is no such meeting, as early as practicable before the initiation of the Phase 3 or Phase 2/3 study as may be agreed between the sponsor and FDA. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. The FDA and the sponsor must reach an agreement on the PSP. A sponsor can submit amendments to an agreed-upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from preclinical studies, early phase clinical trials and/or other clinical development programs. Unless otherwise required by regulation, PREA does not apply to any biological product for an indication for which orphan designation has been granted.
Within 60 days following submission of the application, the FDA reviews the BLA to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the BLA must be resubmitted with additional information. Once a BLA has been accepted for filing, the FDA’s goal is to review standard applications within ten months after the filing date, or, if the application qualifies for priority review, six months after the FDA accepts the application for filing. In both standard and priority reviews, the review process may also be extended by FDA requests for additional information or clarification. The FDA reviews a BLA to determine, among other things, whether a product is safe, pure and potent and the facility in which it is manufactured, processed, packed or held meets standards designed to assure
15
the product’s continued safety, purity and potency. The FDA may convene an advisory committee to provide clinical insight on application review questions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving a BLA, the FDA will typically inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure compliance with cGCPs. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it typically will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
After the FDA evaluates a BLA and conducts inspections of manufacturing facilities where the investigational product and/or its drug substance will be produced, the FDA may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A Complete Response Letter will describe all of the deficiencies that the FDA has identified in the BLA, except that where the FDA determines that the data supporting the application are inadequate to support approval, the FDA may issue the Complete Response Letter without first conducting required inspections, testing submitted product lots and/or reviewing proposed labeling. In issuing the Complete Response Letter, the FDA may recommend actions that the applicant might take to place the BLA in condition for approval, including requests for additional information or clarification. If a Complete Response Letter is issued, the applicant may either resubmit the BLA, addressing all of the deficiencies identified in the letter, or withdraw the application or request an opportunity for a hearing. The FDA may delay or refuse approval of a BLA if applicable regulatory criteria are not satisfied, require additional testing or information and/or require post-marketing testing and surveillance to monitor safety or efficacy of a product.
If regulatory approval of a product is granted, such approval will be granted for particular indications and may entail limitations on the indicated uses for which such product may be marketed. For example, the FDA may approve the BLA with a Risk Evaluation and Mitigation Strategy (“REMS”) to ensure the benefits of the product outweigh its risks. A REMS is a safety strategy to manage a known or potential serious risk associated with a product and to enable patients to have continued access to such medicines by managing their safe use, and could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling or the development of adequate controls and specifications. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing requirements is not maintained or if problems occur after the product reaches the marketplace. The FDA may require one or more Phase 4 post-market studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization, and may limit further marketing of the product based on the results of these post-marketing studies.
Expedited Development and Review Programs
The FDA offers several expedited development and review programs for qualifying product candidates. The Fast Track program is intended to expedite or facilitate the process for reviewing new products that meet certain criteria. Specifically, new products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a Fast Track product has opportunities for more frequent interactions
16
with the review team during product development and, once a BLA is submitted, the product may be eligible for priority review. A Fast Track product may also be eligible for rolling review, where the FDA may consider for review sections of the BLA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the BLA, the FDA agrees to accept sections of the BLA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the BLA.
A product intended to treat a serious or life-threatening disease or condition may also be eligible for Breakthrough Therapy designation to expedite its development and review. A product can receive Breakthrough Therapy designation if preliminary clinical evidence indicates that the product, alone or in combination with one or more other drugs or biologics, may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The designation includes all of the Fast Track program features, as well as more intensive FDA interaction and guidance beginning as early as Phase 1 and an organizational commitment to expedite the development and review of the product, including involvement of senior managers.
Any marketing application for a biologic submitted to the FDA for approval, including a product with a Fast Track designation and/or Breakthrough Therapy designation, may be eligible for other types of FDA programs intended to expedite the FDA review and approval process, such as priority review and accelerated approval. A product is eligible for priority review if it has the potential to provide a significant improvement in the treatment, diagnosis or prevention of a serious disease or condition. For original BLAs, priority review designation means the FDA’s goal is to take action on the marketing application within six months of the 60-day filing date (as compared to ten months under standard review).
Additionally, products studied for their safety and effectiveness in treating serious or life-threatening diseases or conditions may receive accelerated approval upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. As a condition of accelerated approval, the FDA will generally require the sponsor to perform adequate and well-controlled post-marketing clinical studies with due diligence to verify and describe the anticipated effect on irreversible morbidity or mortality or other clinical benefit. Under the Food and Drug Omnibus Reform Act of 2022 (“FDORA”) the FDA may require, as appropriate, that such studies be underway prior to approval or within a specific time period after the date of approval for a product granted accelerated approval. Under FDORA, the FDA has increased authority for expedited procedures to withdraw approval of a product or indication approved under accelerated approval if the sponsor fails to conduct the required post-marketing studies or if such studies fail to verify the predicted clinical benefit. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product.
Fast Track designation, Breakthrough Therapy designation and priority review do not change the standards for approval but may expedite the development or approval process. Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened.
Post-Approval Requirements
Any products manufactured or distributed by us pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to record-keeping, reporting of adverse experiences, periodic reporting, product sampling and distribution, and advertising and
17
promotion of the product. As part of the manufacturing process, the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. After a BLA is approved for a biological product, the product also may be subject to official lot release. If the product is subject to official release by the FDA, the manufacturer submits samples of each lot of product to the FDA together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot. The FDA also may perform certain confirmatory tests on lots of some products before releasing the lots for distribution by the manufacturer. In addition, the FDA conducts laboratory research related to the regulatory standards on the safety, purity, potency and effectiveness of biologics. After approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing user fee requirements, under which the FDA assesses an annual program fee for each product identified in an approved BLA. Biologic manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMPs, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. Changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMPs and impose reporting requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMPs and other aspects of regulatory compliance.
The FDA may withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
restrictions on the marketing or manufacturing of a product, complete withdrawal of the product from the market or product recalls;
fines, warning letters or holds on post-approval clinical studies;
refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of existing product approvals;
product seizure or detention, or refusal of the FDA to permit the import or export of products;
consent decrees, corporate integrity agreements, debarment or exclusion from federal healthcare programs;
mandated modification of promotional materials and labeling and the issuance of corrective information;
the issuance of safety alerts, Dear Healthcare Provider letters, press releases and other communications containing warnings or other safety information about the product; or
injunctions or the imposition of civil or criminal penalties.
The FDA closely regulates the marketing, labeling, advertising and promotion of biologics. A company can make only those claims relating to safety and efficacy, purity and potency that are approved by the FDA and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. Failure to comply with these requirements can result in,
18
among other things, adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available products for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturer’s communications on the subject of off-label use of their products.
Biosimilars and Reference Product Exclusivity
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, the “ACA”), includes a subtitle called the Biologics Price Competition and Innovation Act of 2009 (“BPCIA”), which created an abbreviated approval pathway for biological products that are highly similar, or “biosimilar,” to or interchangeable with an FDA-approved reference biological product. The FDA has issued several guidance documents outlining an approach to review and approval of biosimilars.
Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency, is generally shown through analytical studies, animal studies, and a clinical study or studies. Interchangeability requires that a product is biosimilar to the reference product and the product must demonstrate that it can be expected to produce the same clinical results as the reference product in any given patient and, for products that are administered multiple times to an individual, the biologic and the reference biologic may be alternated or switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. A product shown to be biosimilar or interchangeable with an FDA-approved reference biological product may rely in part on the FDA’s previous determination of safety and effectiveness for the reference product for approval, which can potentially reduce the cost and time required to obtain approval to market the product. Complexities associated with the larger, and often more complex, structures of biological products, as well as the processes by which such products are manufactured, pose significant hurdles to implementation of the abbreviated approval pathway that are still being worked out by the FDA. In September 2021, the FDA issued two guidance documents intended to inform prospective applicants and facilitate the development of proposed biosimilars and interchangeable biosimilars, as well as to describe the FDA’s interpretation of certain statutory requirements added by the BPCIA.
Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing that applicant’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of its product. The BPCIA also created certain exclusivity periods for biosimilars approved as interchangeable products. FDA-approved interchangeable biosimilars may be substituted for the reference product without the intervention of the prescribing health care provider, subject to state laws, which differ by state.
A biological product can also obtain pediatric market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study.
The BPCIA is complex and continues to be interpreted and implemented by the FDA. In July 2018, the FDA announced an action plan to encourage the development and efficient review of biosimilars, including the
19
establishment of a new office within the agency that will focus on therapeutic biologics and biosimilars. On December 20, 2020, Congress amended the PHSA as part of the COVID-19 relief bill to further simplify the biosimilar review process by making it optional to show that conditions of use proposed in labeling have been previously approved for the reference product, which used to be a requirement of the application. In addition, government proposals have sought to reduce the 12-year reference product exclusivity period. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation. As a result, the ultimate impact, implementation, and impact of the BPCIA is subject to significant uncertainty.
As discussed below, the Inflation Reduction Act of 2022 (“IRA”) is a significant new law that intends to foster generic and biosimilar competition and to lower drug and biologic costs.
Other Healthcare Laws and Compliance Requirements
Pharmaceutical companies are subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which they conduct their business. Such laws include, without limitation: the federal Anti-Kickback Statute (“AKS”); the federal False Claims Act (“FCA”); the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) and similar foreign, federal and state fraud, abuse and transparency laws.
The AKS prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering or paying remuneration, to induce, or in return for, either the referral of an individual, or the purchase, lease, order, arrangement, or recommendation of an item or service for which payment may be made under any federal healthcare program. The term remuneration has been interpreted broadly to include anything of value. The AKS has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand, and prescribers and purchasers on the other. The government often takes the position that to violate the AKS, only one purpose of the remuneration need be to induce referrals, even if there are other legitimate purposes for the remuneration. There are a number of statutory exceptions and regulatory safe harbors protecting some common activities from AKS prosecution, but they are drawn narrowly and practices that involve remuneration, such as consulting agreements, that may be alleged to be intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exception or safe harbor. Our practices may not in all cases meet all of the criteria for protection under a statutory exception or regulatory safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the AKS. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances. Violations are subject to civil and criminal fines and penalties for each violation, plus up to three times the remuneration involved, imprisonment, and exclusion from government healthcare programs. In addition, the government may assert that a claim including items or services resulting from a violation of the AKS constitutes a false or fraudulent claim for purposes of the FCA or federal civil monetary penalties.
Civil and criminal false claims laws, including the FCA, and civil monetary penalty laws, which impose criminal and civil penalties and can be enforced through civil whistleblower or qui tam actions, prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment of federal government funds, including in federal healthcare programs, that are false or fraudulent; knowingly making, using or causing to be made or used, a false statement of record material to a false or fraudulent claim or obligation to pay or transmit money or property to the federal government or knowingly concealing or knowingly and improperly avoiding or decreasing an obligation to pay money to the federal government. Pharmaceutical and other healthcare companies have been prosecuted under these laws for engaging in a variety of different types of conduct that “caused” the submission of false claims to federal healthcare programs. Under the AKS, for example, a claim resulting from a violation of the AKS is deemed to be a false or fraudulent claim for purposes
20
of the FCA. The FCA also permits a private individual acting as a “whistleblower” to bring actions on behalf of the federal government alleging violations of the FCA and to share in any monetary recovery.
HIPAA created additional federal criminal statutes that prohibit, among other things, executing a scheme to defraud any healthcare benefit program, including private third-party payors, and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statements or representations relating to healthcare matters.
The FDCA addresses, among other things, the design, production, labeling, promotion, manufacturing, and testing of drugs, biologics and medical devices, and prohibits such acts as the introduction into interstate commerce of adulterated or misbranded drugs or devices. The PHSA also prohibits the introduction into interstate commerce of unlicensed or mislabeled biological products.
The United States federal Physician Payments Sunshine Act requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to annually report to the Centers for Medicaid & Medicare Services (“CMS”) information related to payments or other transfers of value made to various healthcare professionals including physicians, certain other licensed health care practitioners, and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Beginning on January 1, 2023, California Assembly Bill 1278 requires California physicians and surgeons to notify patients of the Open Payments database established under the federal Physician Payments Sunshine Act.
We are also subject to additional similar United States state and foreign law equivalents of each of the above federal laws, which, in some cases, differ from each other in significant ways, and may not have the same effect, thus complicating compliance efforts. If our operations are found to be in violation of any of such laws or any other governmental regulations that apply, we may be subject to penalties, including, without limitation, civil, criminal and administrative penalties, damages, fines, exclusion from government-funded healthcare programs, such as Medicare and Medicaid or similar programs in other countries or jurisdictions, integrity oversight and reporting obligations to resolve allegations of non-compliance, disgorgement, individual imprisonment, contractual damages, reputational harm, diminished profits and the curtailment or restructuring of our operations.
Data Privacy and Security
Numerous state, federal, and foreign laws govern the collection, dissemination, use, access to, confidentiality, and security of personal information, including health-related information. In the United States, numerous federal and state laws and regulations, including state data breach notification laws, state health information privacy laws, and federal and state consumer protection laws and regulations, govern the collection, use, disclosure, and protection of health-related and other personal information could apply to our operations or the operations of our partners. For example, HIPAA, as amended by the Health Information Technology for Economic and Clinical Health, and their respective implementing regulations imposes privacy, security, and breach notification obligations on certain health care providers, health plans, and health care clearinghouses, known as covered entities, as well as their business associates and their covered subcontractors that perform certain services that involve using, disclosing, creating, receiving, maintaining, or transmitting individually identifiable health information for or on behalf of such covered entities. Entities that are found to be in violation of HIPAA may be subject to significant civil, criminal, and administrative fines and penalties and/or additional reporting and oversight obligations if required to enter into a resolution agreement and corrective action plan with the U.S. Department of Health and Human Services (“HHS”) to settle allegations of HIPAA non-compliance. Further, entities that knowingly obtain, use, or disclose individually identifiable health information maintained by a HIPAA covered entity in a manner that is not authorized or permitted by HIPAA may be subject to criminal penalties.
21
Even when HIPAA does not apply, according to the FTC, violating consumers’ privacy rights or failing to take appropriate steps to keep consumers’ personal information secure may constitute unfair acts or practices in or affecting commerce in violation of Section 5(a) of the Federal Trade Commission Act.
In addition, state laws govern the privacy and security of personal information, including health-related information, in certain circumstances. Failure to comply with these laws, where applicable, can result in the imposition of significant civil and/or criminal penalties and private litigation. For example, the California Consumer Privacy Act, which went into effect on January 1, 2020, has created new data privacy obligations for covered companies and provided new privacy rights to California residents.
Coverage and Reimbursement
In the United States and markets in other countries, patients generally rely on third-party payors to reimburse all or part of the costs associated with their treatment. Adequate coverage and reimbursement from governmental healthcare programs, such as Medicare and Medicaid, and commercial payors is critical to new product acceptance. Our ability to successfully commercialize our product candidates will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize a sufficient return on our investment. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels.
Significant uncertainty exists as to the coverage and reimbursement status of any pharmaceutical or biological product for which we obtain regulatory approval. Sales of any product, if approved, depend, in part, on the extent to which such product will be covered by third-party payors, such as federal, state, and foreign government healthcare programs, commercial insurance and managed healthcare organizations, and the level of reimbursement, if any, for such product by third-party payors. Decisions regarding whether to cover any of our product candidates, if approved, the extent of coverage and amount of reimbursement to be provided are made on a plan-by-plan basis. Further, no uniform policy for coverage and reimbursement exists in the United States, and coverage and reimbursement can differ significantly from payor to payor. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement rates, but also have their own methods and approval process apart from Medicare determinations. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our product candidates to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. Factors payors consider in determining reimbursement are based on whether the product is:
a covered benefit under its health plan;
safe, effective and medically necessary;
appropriate for the specific patient;
neither experimental nor investigational.
Third-party payors are increasingly challenging the prices charged for medical products and services, examining the medical necessity and reviewing the cost effectiveness of pharmaceutical or biological products, medical devices and medical services, in addition to questioning safety and efficacy. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and
22
measures, could further limit sales of any product that receives approval. Decreases in third-party reimbursement for any product or a decision by a third-party not to cover a product could reduce physician usage and patient demand for the product.
For products administered under the supervision of a physician, obtaining coverage and adequate reimbursement may be particularly difficult because of the higher prices often associated with such drugs. Additionally, separate reimbursement for the product itself or the treatment or procedure in which the product is used may not be available, which may impact physician utilization. In addition, companion diagnostic tests require coverage and reimbursement separate and apart from the coverage and reimbursement for their companion pharmaceutical or biological products. Similar challenges to obtaining coverage and reimbursement, applicable to pharmaceutical or biological products, will apply to companion diagnostics.
In addition, net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. We cannot be sure that reimbursement will be available for any product candidate that we commercialize and, if reimbursement is available, the level of reimbursement. In addition, many pharmaceutical manufacturers must calculate and report certain price reporting metrics to the government, such as average sales price, and best price. Penalties may apply in some cases when such metrics are not submitted accurately and timely. Further, these prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs.
Finally, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union (“EU”) provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost effectiveness of a particular product candidate to currently available therapies. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of its product candidates. Historically, products launched in the EU do not follow price structures of the United States and generally prices tend to be significantly lower.
Healthcare reform
The United States and some foreign jurisdictions are considering or have enacted a number of reform proposals to change the healthcare system. There is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by federal and state legislative initiatives, including those designed to limit the pricing, coverage, and reimbursement of pharmaceutical and biopharmaceutical products, especially under government-funded health care programs, and increased governmental control of drug pricing.
The ACA, which was enacted in 2010, substantially changed the way healthcare is financed by both governmental and private insurers in the United States, and significantly affected the pharmaceutical industry.
23
The ACA contains a number of provisions of particular import to the pharmaceutical and biotechnology industries, including, but not limited to, those governing enrollment in federal healthcare programs. Since its enactment, there have been judicial and Congressional challenges to certain aspects of the ACA, and we expect there will be additional challenges and amendments to the ACA in the future.
Other legislative changes have been proposed and adopted since the ACA was enacted. For example, the Budget Control Act of 2011 and subsequent legislation, among other things, created measures for spending reductions by Congress that include aggregate reductions of Medicare payments to providers of 2% per fiscal year, which remain in effect through 2032. Due to the Statutory Pay-As-You-Go Act of 2010, estimated budget deficit increases resulting from the American Rescue Plan Act of 2021, and subsequent legislation, Medicare payments to providers will be further reduced starting in 2025 absent further legislation. The United States American Taxpayer Relief Act of 2012 further reduced Medicare payments to several types of providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
In addition, the Bipartisan Budget Act of 2018, among other things, amended the Medicare Act (as amended by the ACA) to increase the point-of-sale discounts that manufacturers must agree to offer under the Medicare Part D coverage discount program to 70% off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs being covered under Medicare Part D.
Moreover, there has recently been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted federal and state measures designed to, among other things, reduce the cost of prescription drugs, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. For example, in May 2019, CMS adopted a final rule allowing Medicare Advantage Plans the option to use step therapy for Part B drugs, permitting Medicare Part D plans to apply certain utilization controls to new starts of five of the six protected class drugs, and requiring the Explanation of Benefits for Part D beneficiaries to disclose drug price increases and lower cost therapeutic alternatives.
In addition, the United States government, state legislatures and foreign governments have continued implementing cost-containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. The IRA includes several provisions that may impact our business to varying degrees, including provisions that reduce the out-of-pocket spending cap for Medicare Part D beneficiaries from $7,050 to $2,000 starting in 2025, thereby effectively eliminating the coverage gap; impose new manufacturer financial liability on certain drugs under Medicare Part D, allow the United States government to negotiate Medicare Part B and Part D price caps for certain high-cost drugs and biologics without generic or biosimilar competition; require companies to pay rebates to Medicare for certain drug prices that increase faster than inflation; and delay until January 1, 2032 the implementation of the HHS rebate rule that would have limited the fees that pharmacy benefit managers can charge. Further, under the IRA, orphan drugs are exempted from the Medicare drug price negotiation program, but only if they have one rare disease designation and for which the only approved indication is for that disease or condition. If a product receives multiple rare disease designations or has multiple approved indications, it may not qualify for the orphan drug exemption. These provisions will take effect progressively starting in fiscal year 2023, although the Medicare drug price negotiation program is currently subject to legal challenges. The effects of the IRA on its business and the healthcare industry in general is not yet known.
President Biden has also issued multiple executive orders that have sought to reduce prescription drug costs. In February 2023, HHS also issued a proposal in response to an October 2022 executive order from President Biden that includes a proposed prescription drug pricing model that will test whether targeted Medicare payment
24
adjustments will sufficiently incentivize manufacturers to complete confirmatory trials for drugs approved through FDA’s accelerated approval pathway. Although a number of these and other proposed measures may require authorization through additional legislation to become effective, and the Biden administration may reverse or otherwise change these measures, both the Biden administration and Congress have indicated that they will continue to seek new legislative measures to control drug costs.
Notwithstanding the IRA and President Biden’s executive orders, continued legislative and enforcement interest exists in the United States with respect to specialty drug pricing practices. Specifically, we expect regulators to continue pushing for transparency to drug pricing, reducing the cost of prescription drugs under Medicare, reviewing the relationship between pricing and manufacturer patient programs, and reforming government program reimbursement methodologies for drugs.
Individual states in the United States have also become increasingly active in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain drug access and marketing cost disclosure and transparency measures, and designed to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions could harm our business, financial condition, results of operations and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce the ultimate demand for our drugs or put pressure on our drug pricing, which could negatively affect our business, financial condition, results of operations and prospects.
Other Government and Regulation Outside of the United States
In addition to regulations in the United States, we are subject to a variety of regulations in other jurisdictions governing, among other things, research and development, operationsclinical trials, testing, manufacturing, safety, efficacy, quality control, labeling, packaging, storage, record keeping, distribution, reporting, export and corporate officesimport, advertising, marketing and other promotional practices involving biological products as well as authorization, approval as well as post-approval monitoring and reporting of our products. Because biologically sourced raw materials are subject to unique contamination risks, their use may be restricted in Austin, TX.some countries.
RATIO OF EARNINGS TO FIXED CHARGESWhether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application much like an IND prior to the commencement of human clinical trials.
The requirements and process governing the conduct of clinical trials, including requirements to conduct additional clinical trials, product licensing, safety reporting, post-authorization requirements, marketing and promotion, interactions with healthcare professionals, pricing and reimbursement may vary widely from country to country. No action can be taken to market any product in a country until an appropriate approval application has been approved by the regulatory authorities in that country. The current approval process varies from country to country, and the time spent in gaining approval varies from that required for FDA approval. In certain countries, the sales price of a product must also be approved. The pricing review period often begins after market approval is granted. Even if a product is approved by a regulatory authority, satisfactory prices may not be approved for such product, which would make launch of such products commercially unfeasible in such countries.
25
Regulation in the European Union
The collection and use of personal health data and other personal data regarding individuals in the European Economic Area (“EEA”) is governed by the provisions of the European General Data Protection Regulation (EU) 2016/679 (“EU GDPR”) and related data protection laws in individual EEA member states, including additional requirements relating to health, genetic and biometric data implemented through national legislation. Similar processing of personal health data and other personal data regarding individuals in the United Kingdom (“UK”) is governed by the UK General Data Protection Regulation (“UK GDPR”) and the UK Data Protection Act 2018. In this document, “GDPR” refers to both the EU GDPR and the UK GDPR, unless specified otherwise. The GDPR imposes a number of strict obligations and restrictions on the ability to process, including collecting, analyzing and transferring, personal data of individuals, in particular with respect to health data from clinical trials and adverse event reporting. The GDPR includes requirements relating to the legal basis of the processing (such as consent of the individuals to whom the personal data relates), the information provided to the individuals prior to processing their personal data, the notification obligations to the national data protection authorities, and the security and confidentiality of the personal data.
In addition, the GDPR imposes specific restrictions on the transfer of personal data to countries outside of the EEA/UK that are not considered by the European Commission (“EC”) and the UK government as providing an adequate level of data protection (third countries), including the United States. Appropriate safeguards are required to enable such transfers. Among the appropriate safeguards that can be used, the data exporter may use the EC approved standard contractual clauses (“SCCs”) and the UK International Data Transfer Agreement/Addendum (“UK IDTA”). Where relying on the SCCs or UK IDTA for data transfers, we may also be required to carry out transfer impact assessments to assess whether the recipient is subject to local laws which allow public authority access to personal data. The international transfer obligations under the EEA/UK data protection regimes will require effort and cost and may result in us needing to make strategic considerations around where EEA/UK personal data is located and which service providers we can utilize for the processing of EEA/UK personal data. Although the UK is regarded as a third country under the EU GDPR, the EC has issued a decision recognizing the UK as providing adequate protection under the EU GDPR (“Adequacy Decision”) and, therefore, transfers of personal data originating in the EEA to the UK remain unrestricted. The UK government has confirmed that personal data transfers from the UK to the EEA remain free flowing. The UK government has also now introduced a Data Protection and Digital Information Bill (“UK Data Protection Bill”) into the UK legislative process with the intention for this bill to reform the UK’s data protection regime following table sets forthBrexit. If passed, the final version of the UK Data Protection Bill may have the effect of further altering the similarities between the UK and EU data protection regime. This may lead to additional compliance costs and could increase our dollar coverage deficiency.overall risk. The ratiorespective provisions and enforcement of earningsthe EU GDPR and UK GDPR may further diverge in the future and create additional regulatory challenges and uncertainties.
On March 25, 2022, the EC and the United States announced that they have agreed in principle on a new Trans-Atlantic Data Privacy Framework. Following this statement, on October 7, 2022, President Biden signed an Executive Order on ‘Enhancing Safeguards for United States Signals Intelligence Activities’, which implemented the agreement in principle. On that basis, the EC prepared a draft Adequacy Decision and launched its adoption procedure. While this new EU-United States privacy framework is expected to fixed chargesenter into force in 2023, there is not disclosed since itstill some uncertainty around the new framework.
Failure to comply with the requirements of the GDPR and the related national data protection laws of the EEA member states/UK may result in significant monetary fines for noncompliance of up to €20 million (£17.5 million for the UK) or 4% of the annual global revenues of the noncompliant company, whichever is greater, other administrative penalties and a number of criminal offenses (punishable by uncapped fines) for organizations and, in certain cases, their directors and officers, as well as civil liability claims from individuals whose personal data was processed. Data protection authorities from the different EEA member states/UK may still implement certain variations, enforce the GDPR and national data protection laws differently, and introduce
26
additional national regulations and guidelines, which adds to the complexity of processing personal data subject to the EEA/UK data protection regimes. Guidance developed at both the EU level and at the national level in individual EU member states concerning implementation and compliance practices are often updated or otherwise revised.
Compliance with the GDPR is a rigorous and time-intensive process that may increase our cost of doing business or require us to change our business practices, and despite those efforts, there is a risk that we may be subject to fines, penalties and litigation in connection with European activities, which could in turn have a negative numbereffect on our reputation and materially harm our business.
Furthermore, there is a growing trend towards the required public disclosure of clinical trial data in the EU, which adds to the complexity of obligations relating to processing health data from clinical trials. Such public disclosure obligations are provided in the new EU Clinical Trials Regulation (EU) No. 536/2014 (the “CTR”), European Medical Agency (“EMA”) disclosure initiatives and voluntary commitments by industry. Failure to comply with these obligations could lead to government enforcement actions and significant penalties against us, harm to our reputation, and adversely impact our business and operating results. The uncertainty regarding the interplay between different regulatory frameworks, such as the CTR and the GDPR, further adds to the complexity that we face with regard to data protection regulation.
Drug and Biologic Development Process
Regardless of where they are conducted, all clinical trials included in applications for marketing authorization for human medicines in the EU must have been carried out in accordance with EU regulations. This means that clinical trials conducted in the EU have to comply with EU clinical trial legislation but also that clinical trials conducted outside the EU have to comply with ethical principles equivalent to those set out in the EU, including adhering to international good clinical practice and the Declaration of Helsinki. The conduct of clinical trials in the EU is governed by the CTR, which entered into force on January 31, 2022. The CTR replaced the Clinical Trials Directive 2001/20/EC, (“Clinical Trials Directive”) and introduced a complete overhaul of the existing regulation of clinical trials for medicinal products in the EU.
Under the former regime, which will expire after a transition period of one or three years, respectively, as outlined below in more detail, before a clinical trial can be initiated it must be approved in each year and period shown below. Any time we offer debt securities pursuantEU member state where there is a site at which the clinical trial is to this prospectus, we will provide an updated table setting forth our ratio of earnings to fixed charges on a historical basisbe conducted. The approval must be obtained from two separate entities: the national competent authority in the applicable prospectus supplement,EU member state(s) and one or more ethics committees. The national competent authority of all EU member states in which the clinical trial will be conducted must authorize the conduct of the trial, and the independent ethics committee must grant a positive opinion in relation to the conduct of the clinical trial in the relevant EU member state before the commencement of the trial. Any substantial changes to the trial protocol or other information submitted with the clinical trial applications must be submitted to or approved by the relevant national competent authorities and ethics committees. Under the current regime all suspected unexpected serious adverse reactions to the investigated drug that occur during the clinical trial must be reported to the national competent authority and to the ethics committees of the EU member state where they occur.
A more unified procedure applies under the CTR. A sponsor can submit a single application for approval of a clinical trial through a centralized EU clinical trials portal (the “CTIS”). One national competent authority (the reporting EU member state proposed by the applicant) will take the lead in validating and evaluating the application, and will consult and coordinate with the other concerned EU member states. If an application is rejected, it may be amended and resubmitted through the EU clinical trials portal. If an approval is issued, the sponsor may start the clinical trial in all concerned EU member states. However, a concerned EU member state may in limited circumstances declare an “opt-out” from an approval and prevent the clinical trial from being
27
conducted in such member state. The CTR also aims to streamline and simplify the rules on safety reporting, and introduces enhanced transparency requirements such as mandatory submission of a summary of the clinical trial
results to the EU database. The CTR foresees a three-year transition period. On January 31, 2023, submission of initial clinical trial applications via CTIS became mandatory, and by January 31, 2025, all ongoing trials approved under the former Clinical Trials Directive will need to comply with the CTR and have to be transitioned to CTIS.
Under both the former regime and the CTR, national laws, regulations, and the applicable Good Clinical Practice and Good Laboratory Practice standards must also be respected during the conduct of the trials, including the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use guidelines on Good Clinical Practice and the ethical principles that have their origin in the Declaration of Helsinki.
During the development of a medicinal product, the EMA and national regulators within the EU provide the opportunity for dialogue and guidance on the development program. At the EMA level, this is usually done in the form of scientific advice, which is given by the Committee for Medicinal Products for Human Use (“CHMP”) on the recommendation of the Scientific Advice Working Party. A fee is incurred with each scientific advice procedure, but is significantly reduced for designated orphan medicines. Advice from the EMA is typically provided based on questions concerning, for example, quality (chemistry, manufacturing and controls testing), nonclinical testing and clinical studies, and pharmacovigilance plans and risk-management programs. Advice is not legally binding with regard to any future marketing authorization application (“MAA”) for the product concerned.
Drug Marketing Authorization
In the EU, medicinal products are subject to extensive pre- and post-market regulation by regulatory authorities at both the EU and national levels. To obtain regulatory approval of a product under the EU regulatory systems, we must submit an MAA under either the EU centralized procedure, or one of the national procedures in the EU.
Centralized Authorization Procedure
The centralized procedure provides for the grant of a single marketing authorization (“MA”) that is issued by the EC following the scientific assessment of the application by the EMA and that is valid for all EU member states as well as in the three additional EEA member states (Norway, Iceland and Liechtenstein). The centralized procedure is compulsory for certain types of medicinal products, including for medicines developed by means of certain biotechnological processes, products designated as orphan medicinal products, advanced therapy medicinal products (gene therapy, somatic cell therapy or tissue-engineered medicines) and medicinal products with a new active substance indicated for the treatment of certain diseases (HIV/AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and other immune dysfunctions and viral diseases). The centralized procedures is option for medicinal products containing a new active substance not yet authorized in the EU, or for products that constitute a significant therapeutic, scientific or technical innovation or for which the grant of an MA through the centralized procedure would be in the interest of public health at EU level.
Under the centralized procedure, the CHMP established at the EMA, is responsible for conducting the initial assessment of a drug. The CHMP is also responsible for several post-authorization and maintenance activities, such as the assessment of modifications or extensions to an existing marketing authorization. Under the centralized procedure, the timeframe for the evaluation of an MAA by the EMA’s CHMP is, in principle, 210 days from receipt of a valid MAA. However, this timeline excludes clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP, so the overall process typically takes a year or more, unless the application is eligible for an accelerated assessment. Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest, particularly from the point of view of therapeutic innovation.
28
Upon request, the CHMP can reduce the time frame to 150 days if required. Any time we offer shares of preferred stock pursuant to this prospectus, wethe applicant provides sufficient justification for an accelerated assessment. The CHMP will provide a table setting forthpositive opinion regarding the application only if it meets certain quality, safety and efficacy requirements. This opinion is then transmitted to the EC, which has the ultimate authority for granting the MA within 67 days after receipt of the CHMP opinion.
Decentralized and Mutual Recognition Procedures
Medicines that fall outside the mandatory scope of the centralized procedure can be authorized under a decentralized procedure where an applicant applies for simultaneous authorization in more than one EU member state, or they can be authorized in an EU member state in accordance with that state’s national procedures and then be authorized in other EU countries by a procedure whereby the countries concerned agree to recognize the validity of the original, national marketing authorization (mutual recognition procedure).
The decentralized procedure permits companies to file identical MA applications for a medicinal product to the competent authorities in various EU member states simultaneously if such medicinal product has not received marketing approval in any EU member state before. The competent authority of a single EU member state, the reference member state, is appointed to review the application and provide an assessment report. The competent authorities of the other EU member states, the concerned member states, are subsequently required to grant a marketing authorization for their territories on the basis of this assessment. The only exception to this is where the competent authority of an EU member state considers that there are concerns of potential serious risk to public health, the disputed points are subject to a dispute resolution mechanism and may eventually be referred to the EC, whose decision is binding for all EU member states.
Risk Management Plan
All new MAAs must include a Risk Management Plan (“RMP”) describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. RMPs are continually modified and updated throughout the lifetime of the medicine as new information becomes available. An updated RMP must be submitted: (i) at the request of EMA or a national competent authority, or (ii) whenever the risk-management system is modified, especially as the result of new information being received that may lead to a significant change to the benefit-risk profile or as a result of an important pharmacovigilance or risk-minimization milestone being reached. The regulatory authorities may also impose specific obligations as a condition of the MA. RMPs and Periodic Safety Update Reports (“PSURs”) are routinely available to third parties requesting access, subject to limited redactions.
MA Validity Period
In the EU, an MA has an initial duration of five years. After these five years, the authorization may subsequently be renewed on the basis of a reevaluation of the risk-benefit balance. Once renewed, the MA is valid for an unlimited period unless the EC or the national competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with only one additional five-year renewal. Applications for renewal must be made to the EMA at least nine months before the five-year period expires.
Exceptional Circumstances/Conditional Approval
Similar to accelerated approval regulations in the United States, conditional MAs can be granted in the EU for medicines intended for treating, preventing or diagnosing seriously debilitating or life-threatening diseases, or in a public health emergency. A conditional MA can be granted for medicinal products where, although comprehensive clinical data referring to the safety and efficacy of the medicinal product have not been supplied, the following criteria are fulfilled: (i) the benefit/risk balance of the product is positive, (ii) it is likely that the
29
applicant will be in a position to provide the comprehensive clinical data post-authorization, (iii) unmet medical needs will be fulfilled by the grant of the MA and (iv) the benefit to public health of the immediate availability on the market of the medicinal product concerned outweighs the risk inherent in the fact that additional data are still required. Once a conditional MA has been granted, the MA holder must fulfil specific obligations within defined timelines. A conditional MA must be renewed annually, but can be converted into a standard MA once the MA holder fulfils the obligations imposed and the complete data confirm that the medicine’s benefits continue to outweigh its risks.
Data and Market Exclusivity
As in the United States, it may be possible to obtain a period of market and/or data exclusivity in the EU that would have the effect of postponing the entry into the marketplace of a competitor’s generic, hybrid or biosimilar product (even if the pharmaceutical product has already received a MA) and prohibiting another applicant from relying on the MA holder’s pharmacological, toxicological and clinical data in support of another MA for the purposes of submitting an application, obtaining an MA or placing the product on the market. Innovative medicinal products (sometimes referred to as new chemical entities (“NCEs”)) approved in the EU generally qualify for eight years of data exclusivity and 10 years of marketing exclusivity.
If granted, the data exclusivity period begins on the date of the product’s first MA in the EU and prevents generic or biosimilar applicants from referencing the innovator’s preclinical and clinical trial data contained in the dossier of the reference product when applying for a generic or biosimilar marketing authorization in the EU. After eight years, a generic product application may be submitted and generic companies may rely on the MA holder’s data. However, a generic product cannot launch until two years later (or a total of 10 years after the first MA in the EU of the innovator product). An additional one-year period of marketing exclusivity is possible if, during the data exclusivity period (the first eight years of the 10-year marketing exclusivity period), the MA holder obtains an authorization for one or more new therapeutic indications that are deemed to bring a significant clinical benefit compared to existing therapies. Additionally, a standalone one-year period of data exclusivity can be granted where an application is made for a new indication for a well-established substance, provided that significant pre-clinical or clinical studies were carried out in relation to the new indication. Where a change of classification of a pharmaceutical product has been authorized on the basis of significant pre-trial tests or clinical trials, when examining an application by another applicant for or holder of an MA for a change of classification of the same substance the competent authority will not refer to the results of those tests or trials for one year after the initial change was authorized.
Products may not be granted data exclusivity since there is no guarantee that a product will be considered by the EU’s regulatory authorities to include a NCE. Even if a compound is considered to be a NCE and the MA applicant is able to gain the prescribed period of data exclusivity, another company nevertheless could also market another version of the medicinal product if such company can complete a full MAA with their own complete database of pharmaceutical tests, preclinical studies and clinical trials and obtain MA of its product.
Pediatric Development
In the EU, companies developing a new medicinal product are obligated to study their product in children and must therefore submit a PIP together with a request for agreement to the EMA, unless the EMA has granted a product-specific waiver, a class waiver, or a deferral for one or more of the measures included in the PIP. The EMA issues a decision on the PIP based on an opinion of the EMA’s Pediatric Committee. Companies must conduct pediatric clinical trials in accordance with the PIP approved by the EMA, unless a deferral (e.g. until enough information to demonstrate its effectiveness and safety in adults is available) or waiver (e.g. because the relevant disease or condition occurs only in adults) has been granted by the EMA. The MAA for the medicinal product must include the results of all pediatric clinical trials performed and details of all information collected in
30
compliance with the approved PIP, unless such a waiver or a deferral has been granted. Medicinal products that are granted an MA on the basis of the pediatric clinical trials conducted in accordance with the approved PIP are eligible for a six month extension of the protection under a supplementary protection certificate (“SPC”), provided an application for such extension is made at the same time as filing the SPC application for the product, or at any point up to two years before the SPC expires, or, in the case of orphan medicinal products, a two year extension of the orphan market exclusivity. This pediatric reward is subject to specific conditions and is not automatically available when data in compliance with the approved PIP are developed and submitted. An approved PIP is also required when an MA holder wants to add a new indication, medicinal form or route of administration for a medicine that is already authorized and covered by intellectual property rights.
PRIME Designation
In March 2016, the EMA launched an initiative to facilitate development of product candidates in indications, often rare, for which few or no therapies currently exist. The Priority Medicines (“PRIME”) scheme is intended to encourage drug development in areas of unmet medical need and provides accelerated assessment of products representing substantial innovation reviewed under the centralized procedure. Products from small- and medium-sized enterprises may qualify for earlier entry into the PRIME scheme than larger companies on the basis of compelling non-clinical data and tolerability data from initial clinical trials. Many benefits accrue to sponsors of product candidates with PRIME designation, including but not limited to, early and proactive regulatory dialogue with the EMA, frequent discussions on clinical trial designs and other development program elements, and potentially accelerated marketing authorization application assessment once a dossier has been submitted. Importantly, once a candidate medicine has been selected for the PRIME scheme, a dedicated contact point and rapporteur from the CHMP or from the Committee for Advanced Therapeutics (“CAT”) are appointed facilitating increased understanding of the product at EMA’s Committee level. A kick-off meeting with the CHMP/CAT rapporteur initiates these relationships and includes a team of multidisciplinary experts to provide guidance on the overall development plan and regulatory strategy. PRIME eligibility does not change the standards for product approval, and there is no assurance that any such designation or eligibility will result in expedited review or approval.
Post-Approval Regulation
Similar to the United States, both MA holders and manufacturers of medicinal products are subject to comprehensive regulatory oversight by the EMA, the EC and/or the competent regulatory authorities of the EU member states. This oversight applies both before and after grant of manufacturing licenses and marketing authorizations. It includes control of compliance with EU good manufacturing practices rules, manufacturing authorizations, pharmacovigilance rules and requirements governing advertising, promotion, sale, and distribution, recordkeeping, importing and exporting of medicinal products.
Failure by us or by any of our ratiothird-party partners, including suppliers, manufacturers and distributors to comply with EU laws and the related national laws of combined fixed chargesindividual EU member states governing the conduct of clinical trials, manufacturing approval, MA of medicinal products and preferred stock dividendsmarketing of such products, both before and after grant of MA, statutory health insurance, bribery and anti-corruption or other applicable regulatory requirements may result in administrative, civil or criminal penalties. These penalties could include delays or refusal to earnings, if required.authorize the conduct of clinical trials or to grant an MA, product withdrawals and recalls, product seizures, suspension, withdrawal or variation of the MA, total or partial suspension of production, distribution, manufacturing or clinical trials, operating restrictions, injunctions, suspension of licenses, fines and criminal penalties.
The holder of an MA for a medicinal product must also comply with EU pharmacovigilance legislation and its related regulations and guidelines, which entail many requirements for conducting pharmacovigilance, or the assessment and monitoring of the safety of medicinal products.
| | | | | | | | | | | | | | | | |
| | | Period From
December 16,
2013
(Inception) to
December 31,
2013 | | | Year Ended
December 31,
2014 | | | Year Ended
December 31,
2015 | | | Year Ended
December 31,
2016 | |
| | | (1) | | | (1) | | | (1) | | | (1) | |
| | | | | | | | | | | | | | | | |
31
MA holders must establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance, who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions and submission of PSURs in relation to medicinal products for which they hold MAs. The EMA reviews PSURs for medicinal products authorized through the centralized procedure. If the EMA has concerns that the risk benefit profile of a product has varied, it can adopt an opinion advising that the existing MA for the product be suspended, withdrawn or varied. The EMA can advise that the MA holder be obliged to conduct post-authorization Phase IV safety studies. If the EC agrees with the opinion, it can adopt a decision varying the existing MA. Failure by the MA holder to fulfill the obligations for which the EC’s decision provides can undermine the ongoing validity of the MA.
More generally, non-compliance with pharmacovigilance obligations can lead to the variation, suspension or withdrawal of the MA for the product or imposition of financial penalties or other enforcement measures.
The manufacturing process for pharmaceutical products in the EU is highly regulated and regulators may shut down manufacturing facilities that they believe do not comply with regulations. Manufacturing requires a manufacturing authorization, and the manufacturing authorization holder must comply with various requirements set out in the applicable EU laws, regulations and guidance, including Directive 2001/83/EC, Directive 2003/94/EC, Regulation (EC) No 726/2004 and the European Commission Guidelines for Good Manufacturing Practice (“GMP”). These requirements include compliance with GMP standards when manufacturing pharmaceutical products and active pharmaceutical ingredients, including the manufacture of active pharmaceutical ingredients outside of the EU with the intention to import the active pharmaceutical ingredients into the EU. Similarly, the distribution of pharmaceutical products into and within the EU is subject to compliance with the applicable EU laws, regulations and guidelines, including the requirement to hold appropriate authorizations for distribution granted by the competent authorities of the EU member states. The manufacturer or importer must have a qualified person who is responsible for certifying that each batch of product has been manufactured in accordance with GMP, before releasing the product for commercial distribution in the EU or for use in a clinical trial. Manufacturing facilities are subject to periodic inspections by the competent authorities for compliance with GMP.
Sales and Marketing Regulations
The advertising and promotion of our products is also subject to EU laws concerning promotion of medicinal products, interactions with physicians, misleading and comparative advertising and unfair commercial practices. In addition, other national legislation of individual EU member states may apply to the advertising and promotion of medicinal products and may differ from one country to another. These laws require that promotional materials and advertising in relation to medicinal products comply with the product’s Summary of Product Characteristics (“SmPC”) as approved by the national competent authorities. The SmPC is the document that provides information to physicians concerning the safe and effective use of the medicinal product. It forms an intrinsic and integral part of the marketing authorization granted for the medicinal product. Promotion of a medicinal product that does not comply with the SmPC is considered to constitute off-label promotion, which is prohibited in the EU. Direct-to-consumer advertising of prescription-only medicines is also prohibited in the EU. Violations of the rules governing the promotion of medicinal products in the EU could be penalized by administrative measures, fines and imprisonment.
Anti-Corruption Legislation
In the EU, interactions between pharmaceutical companies and physicians are also governed by strict laws, regulations, industry self-regulation codes of conduct and physicians’ codes of professional conduct both at EU level and in the individual EU member states. The provision of benefits or advantages to physicians to induce or encourage the prescription, recommendation, endorsement, purchase, supply, order or use of medicinal products
32
is prohibited in the EU. The provision of benefits or advantages to physicians is also governed by the national anti-bribery laws of the EU member states. Violation of these laws could result in substantial fines and imprisonment.
Payments made to physicians in certain EU member states also must be publicly disclosed. Moreover, agreements with physicians must often be the subject of prior notification and approval by the physician’s employer, his/her regulatory professional organization, and/or the competent authorities of the individual EU member states. These requirements are provided in the national laws, industry codes, or professional codes of conduct, applicable in the individual EU member states. Failure to comply with these requirements could result in reputational risk, public reprimands, administrative penalties, fines or imprisonment.
Other Markets
The UK formally left the EU on January 31, 2020 and the transition period, during which EU laws continued to apply to the UK, expired on December 31, 2020. This means EU laws now only apply to the UK in respect of Northern Ireland as laid out in the Northern Ireland Protocol. Following the end of the transition period, the EU and the UK concluded a trade and cooperation agreement (“TCA”), which applied provisionally from January 1, 2021 and entered into force on May 1, 2021.
The TCA includes specific provisions concerning pharmaceuticals, which include the mutual recognition of GMP, inspections of manufacturing facilities for medicinal products and GMP documents issued but does not provide for wholesale mutual recognition of UK and EU pharmaceutical regulations. At present, Great Britain has implemented EU legislation on the marketing, promotion and sale of medicinal products through the Human Medicines Regulations 2012 (as amended). Except in respect of the new CTR, the regulatory regime in Great Britain therefore largely aligns with current EU medicines regulations, however it is possible that these regimes will diverge more significantly in future now that Great Britain’s regulatory system is independent from the EU and the TCA does not provide for mutual recognition of UK and EU pharmaceutical legislation. However, notwithstanding that there is no wholesale recognition of EU pharmaceutical legislation under the TCA, under a new framework which will be put in place by the Medicines and Healthcare products Regulatory Agency (“MHRA”), from January 1, 2024, the MHRA has stated that it will take into account decisions on the approval of marketing authorizations from the EMA (and certain other regulators) when considering an application for a Great Britain marketing authorization.
On February 27, 2023, the UK government and the EC announced a political agreement in principle to replace the Northern Ireland Protocol with a new set of arrangements, known as the “Windsor Framework”. This new framework fundamentally changes the existing system under the Northern Ireland Protocol, including with respect to the regulation of medicinal products in the UK. In particular, the MHRA will be responsible for approving all medicinal products destined for the UK market (i.e., Great Britain and Northern Ireland), and the EMA will no longer have any role in approving medicinal products destined for Northern Ireland. A single UK-wide marketing authorization will be granted by the MHRA for all medicinal products to be sold in the UK, enabling products to be sold in a single pack and under a single authorization throughout the UK. The Windsor Framework was approved by the EU-UK Joint Committee on March 24, 2023, so the UK government and the EU will enact legislative measures to bring it into law.
For other countries outside of the EU, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials must be conducted in accordance with cGCPs and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
33
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension of clinical trials, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Employees and Human Capital Resources
As of June 30, 2023, we had 14 employees, all of whom were employed full time. None of our employees are represented by a labor union or covered under a collective bargaining agreement. We consider our relationship with our employees to be good.
Legal Proceedings
From time to time, we may be subject to legal proceedings. We are not currently a party to or aware of any proceedings that we believe will have, individually or in the aggregate, a material adverse effect on our business, financial condition or results of operations. Regardless of outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
(1) | Due to our net losses for the periods presented earnings were insufficient to cover fixed charges. For this reason, no ratios are provided. |
34
RISK FACTORS
An investment in our securities involves a high degree of risk. The prospectus supplement applicable to each offering of securities will contain a discussion of the risks applicable to an investment in our securities. Prior to making a decision about investing in our securities, you should carefully consider the specific factors discussed under the heading “Risk Factors” in the applicable prospectus supplement, together with all of the other information contained or incorporated by reference in the prospectus supplement or appearing or incorporated by reference in this prospectus. You should also carefully consider the risks, uncertainties and assumptions discussed under Part I, Item 1A, “Risk Factors,”other factors described in our most recent Annual Report on Form 10-K, for the year ended December 31, 2016, which is incorporated herein as supplemented and updated by reference,subsequent Quarterly Reports on Form 10-Q and may be amended, supplementedCurrent Reports on Form 8-K that we have filed or superseded from time to time by other reports wewill file with the SEC, and in other documents which are incorporated by reference into this prospectus, as well as the future.risk factors and other information contained in or incorporated by reference into any accompanying prospectus supplement before investing in any of our securities. The risks and uncertainties we have described are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also affect our operations.
35
FORWARD-LOOKING STATEMENTS
This prospectus and documents incorporated herein by reference contain “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. These forward-looking statements involve a number of risks and uncertainties. We caution readers that any forward-looking statement is not a guarantee of future performance and that actual results could differ materially from those contained in the forward-looking statement. These statements are based on current expectations of future events. Such statements include, but are not limited to, statements about future financial and operating results, plans, objectives, expectations and intentions, costs and expenses, outcome of contingencies, financial condition, results of operations, liquidity, cost savings, objectives of management, business strategies, debt financing, timing and plans for our nonclinical studies and clinical trials, the achievement of clinical and commercial milestones, the advancement of our technologies and our proprietary product candidates, and other statements that are not historical facts. You can find many of these statements by looking for words like “believes,” “expects,” “anticipates,” “estimates,” “may,” “might,” “should,” “will,” “could,” “plan,” “intend,” “project,” “seek” or similar expressions in this prospectus or in documents incorporated by reference into this prospectus. We intend that such forward-looking statements be subject to the safe harbors created thereby.
These forward-looking statements are based on the current beliefs and expectations of our management and are subject to significant risks and uncertainties. If underlying assumptions prove inaccurate or unknown risks or uncertainties materialize, actual results may differ materially from current expectations and projections. Factors that might cause such a difference include those discussed in Part I, Item 1A “Risk Factors” in our Annual Report on Form 10-K for the year ended December 31, 2016, as well as those discussed in this prospectus and in the documents incorporated by reference into this prospectus. You are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date of this prospectus or, in the case of documents referred to or incorporated by reference, the date of those documents.
All subsequent written or oral forward-looking statements attributable to us or any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section. We do not undertake any obligation to release publicly any revisions to these forward-looking statements to reflect events or circumstances after the date of this prospectus or to reflect the occurrence of unanticipated events, except as may be required under applicable U.S. securities law. If we do update one or more forward-looking statements, no inference should be drawn that we will make additional updates with respect to those or other forward-looking statements.
WHERE YOU CAN FIND MORE INFORMATION
We are subject to the informational requirements of the Securities Exchange Act of 1934, as amended, or the Exchange Act and are required to file annual, quarterly and other reports, proxy statements and other information with the SEC. You may inspect and copy these reports, proxy statements and other information at the public reference facilities maintained by the SEC in Washington, D.C., 100 F Street N.E., Washington, D.C. 20549. Copies of such materials can be obtained from the SEC’s public reference section at prescribed rates. You may obtain information on the operation of the public reference rooms by calling the SEC at (800) SEC-0330. Additionally, the SEC maintains an Internet site (http://www.sec.gov) that contains reports, proxy and information statements, and various other information about us. You may also inspect the documents described herein at our principal executive offices, 901 S. MoPac Expressway, Barton Oaks Plaza One, Suite 250, Austin, TX 78746, during normal business hours.
Information about us is also available at our website at http://www.aegleabio.com. However, the information on our website is not a part of this prospectus and is not incorporated by reference into this prospectus.
INCORPORATION OF INFORMATION BY REFERENCE
The SEC allows us to “incorporate by reference” information that we file with the SEC, which means that we can disclose important information to you by referring you to those other documents. The information incorporated by reference is an important part of this prospectus, and information we file later with the SEC will automatically update and supersede this information. A Current Report (or portion thereof) furnished, but not filed, on Form 8-K shall not be incorporated by reference into this prospectus. We incorporate by reference the documents listed below and any future filings we make with the SEC under Section 13(a), 13(c), 14, or 15(d) of the Exchange Act, including all filings made after the date of the filing of this registration statement and prior to the effectiveness of this registration statement, except, in each case, as to any portion of any future report or document that is not deemed filed under such provisions, prior to the termination of any offering of securities made by this prospectus:
our Annual Report on Form 10-K for the fiscal year ended December 31, 2016 filed with the SEC on March 23, 2017, including certain information incorporated by reference therein from our Definitive Proxy Statement for our 2017 annual meeting of stockholders filed with the SEC on April 21, 2017;
| • | | our Annual Report on Form 10-K for the fiscal year ended December 31, 2022, filed with the SEC on March 2, 2023, including certain information incorporated by reference therein from our Definitive Proxy Statement on Schedule 14A for our 2023 annual meeting of stockholders filed with the SEC on April 17, 2023; |
our Current Report on Form 8-K filed on February 16, 2017;
| • | | our Quarterly Report on Form 10-Q for the quarter ended March 31, 2023, filed with the SEC on May 11, 2023; |
the description of our common stock contained in our registration statement on Form 8-A filed with the SEC on March 28, 2016 under Section 12 of the Exchange Act, including any amendment or report filed for the purpose of updating such description; and
| • | | our Current Reports on Form 8-K filed on January 6, 2023, January 18, 2023, March 13, 2023, April 12, 2023, May 19, 2023, June 9, 2023, June 22, 2023, June 23, 2023 (excluding information in Item 7.01 and exhibit 99.1 furnished thereto), July 14, 2023 and July 27, 2023 (excluding information in Item 7.01 and exhibit 99.1 furnished thereto) (in each case, except for information contained therein which is furnished rather than filed); and |
filings we make with the SEC pursuant to the Exchange Act after the date of the initial registration statement, of which this prospectus is a part, and prior to the effectiveness of the registration statement.
| • | | the description of our Common Stock contained in our registration statement on Form 8-A filed with the SEC on March 28, 2016 under Section 12 of the Exchange Act, including any amendment or report filed for the purpose of updating such description. |
We will furnish without charge to you, on written or oral request, a copy of any or all of such documents that has been incorporated herein by reference (other than exhibits to such documents unless such exhibits are specifically incorporated by reference into the documents that this prospectus incorporates). Written or oral requests for copies should be directed Aeglea BioTherapeutics, Inc., Attn: Investor Relations, 901 S. MoPac Expressway, Barton Oaks Plaza One,221 Crescent Street, Building 17, Suite 250, Austin, Texas 78746,102B, Waltham, MA 02453, telephone number (512) 942-2935. See the section of this prospectus entitled “Where You Can Find More Information” for information concerning how to read and obtain copies of materials that we file with the SEC at the SEC’s public offices.SEC.
Any statement contained in this prospectus, or in a document all or a portion of which is incorporated by reference, shall be modified or superseded for purposes of this prospectus to the extent that a statement contained in this prospectus, any prospectus supplement or any document incorporated by reference modifies or supersedes such statement. Any such statement so modified or superseded shall not, except as so modified or superseded, constitute a part of this prospectus.
36
USE OF PROCEEDS
We are not selling any securities under this prospectus and we will retain broad discretion over the use of the netnot receive any proceeds to us from the sale of our securities under this prospectus. Unless otherwise provided in the applicable prospectus supplement, we intend to use theResale Shares covered hereby. The net proceeds from the sale of securitiesthe Resale Shares offered by this prospectus will be received by the Selling Stockholders.
Subject to limited exceptions, the Selling Stockholders will pay any underwriting discounts and commissions and expenses incurred by the Selling Stockholders for brokerage, accounting, tax or legal services or any other expenses incurred by the Selling Stockholders in disposing of any of the Resale Shares. We will bear the costs, fees and expenses incurred in effecting the registration of the Resale Shares covered by this prospectus, including all registration and filing fees, Nasdaq listing fees and fees and expenses of our counsel and our independent registered public accounting firm.
37
SELLING STOCKHOLDERS
This prospectus covers the resale or other disposition from time to time by the Selling Stockholders identified in the table below of up to an aggregate of 1,099,284,385 shares of our Common Stock. The Selling Stockholders may from time to time offer and sell any or all of the Resale Shares set forth below pursuant to this prospectus and any accompanying prospectus supplement.
On June 22, 2023, we entered into the Securities Purchase Agreement, pursuant to which we sold an aggregate of 721,452 shares of our Series A Preferred Stock, which are convertible into 1,000 shares of Common Stock pursuant to the Certificate of Designation, at an aggregate purchase price of approximately $210 million. Also on June 22, 2023, we completed our acquisition of Spyre in accordance with the Acquisition Agreement, pursuant to which we issued an aggregate of 12,945,385 shares of Common Stock and 364,887 shares of Series A Preferred Stock to certain of the Selling Stockholders. This prospectus covers the resale or other disposition by the Selling Stockholders or their pledgees, donees, transferees or other successors-in-interest that receive their shares after the date of this prospectus of up to the total number of shares issued to the Selling Stockholders pursuant to the Acquisition Agreement or issuable upon the conversion of the Series A Preferred Stock sold pursuant to the Securities Purchase Agreement or Acquisition Agreement. This prospectus covers the resale or other disposition by the Selling Stockholders or their transferees of up to the total number of Merger Common Shares, Merger Conversion Shares and Private Placement Conversion Shares issued to the Selling Stockholders pursuant to the Acquisition Agreement or the Securities Purchase Agreement. Throughout this prospectus, when we refer to the “Selling Stockholders,” we are referring to the purchasers under the Securities Purchase Agreement, certain of the securityholders under the Acquisition Agreement and certain of our officers listed in the table below.
We are registering the Resale Shares to permit the Selling Stockholders and their pledgees, donees, transferees or other successors-in interest that receive their shares after the date of this prospectus to resell or otherwise dispose of the shares in the manner contemplated under “Plan of Distribution” herein.
Except as otherwise disclosed herein, the Selling Stockholders do not have, and within the past three years have not had, any position, office or other material relationship with us.
The following table sets forth the names of the Selling Stockholders, the number of shares of our Common Stock owned by the Selling Stockholders, the number of shares of our Common Stock that may be offered under this prospectus and the number of shares of our Common Stock that will be owned after this offering by the Selling Stockholders assuming all of the shares registered for general corporate purposes, whichresale hereby are sold.
The Selling Stockholders may include funding researchsell some, all or none of their Resale Shares. We do not know how long the Selling Stockholders will hold the Resale Shares before selling them, and development, increasingwe currently have no agreements, arrangements or understandings with the Selling Stockholders regarding the sale or other disposition of any of the Resale Shares. The Resale Shares covered hereby may be offered from time to time by the Selling Stockholders, provided that Resale Shares issued upon conversion of Series A Preferred Stock may only be offered after such shares of Series A Preferred Stock are converted to Common Stock pursuant to the terms of the Certificate of Designation.
38
The information set forth below is based upon information obtained from the Selling Stockholders and upon information in our working capital, reducing indebtedness, acquisitions or investmentspossession regarding the issuance of the Series A Preferred Stock and Merger Common Shares in businesses, products or technologies thatconnection with the PIPE and the Asset Acquisition. The percentages of Common Stock owned after the offering by each Selling Stockholder below are complementarybased on 97,457,166 shares of Common Stock outstanding as of July 12, 2023, and, for each Selling Stockholder, assumes the conversion of only the Series A Preferred Stock owned by such Selling Stockholder but not the Series A Preferred Stock owned by any other Selling Stockholder.
| | | | | | | | | | | | | | | | |
Name of Selling
Stockholders(1) | | Common Stock
Beneficially
Owned
Before
Offering (2) | | | Common
Stock that May
Be Offered
Pursuant to
Prospectus | | | Common Stock
Beneficially
Owned After
Offering (2) | |
| | | | | | | | | Number | | | Percentage
(%) | |
Entities associated with Fairmount Funds Management LLC (3) | | | 532,384,540 | | | | 532,384,540 | | | | — | | | | * | |
Entities associated with Paragon Therapeutics, Inc. (4) | | | 78,010,483 | | | | 78,010,483 | | | | — | | | | * | |
Entities associated with Venrock Healthcare Capital Partners III, L.P. (5) | | | 52,192,000 | | | | 45,692,000 | | | | 6,500,000 | | | | 4.5 | |
Avoro Life Sciences Fund LLC (6) | | | 45,978,459 | | | | 45,692,000 | | | | 286,459 | | | | * | |
Commodore Capital Master LP (7) | | | 45,852,100 | | | | 42,944,000 | | | | 2,908,100 | | | | 2.1 | |
Deep Track Biotechnology Master Fund, Ltd. (8) | | | 42,944,000 | | | | 42,944,000 | | | | — | | | | * | |
Perceptive Life Sciences Master Fund, Ltd. (9) | | | 42,944,000 | | | | 42,944,000 | | | | — | | | | * | |
Entities associated with RTW Investments, LP (10) | | | 42,944,000 | | | | 42,944,000 | | | | — | | | | * | |
Entities associated with EcoR1 Capital (11) | | | 22,177,000 | | | | 17,177,000 | | | | 5,000,000 | | | | 4.4 | |
Entities associated with Driehaus Capital Management (USVI) LLC (12) | | | 21,820,357 | | | | 17,177,000 | | | | 4,643,357 | | | | 4.1 | |
Cameron Turtle (13) | | | 20,633,415 | | | | 18,662,699 | | | | 1,970,716 | | | | 1.7 | |
Entities associated with BVF Partners L.P. (14) | | | 17,177,000 | | | | 17,177,000 | | | | — | | | | * | |
Cormorant Global Healthcare Master Fund, LP (15) | | | 17,177,000 | | | | 17,177,000 | | | | — | | | | * | |
Entities associated with Franklin Biotechnology (16) | | | 17,177,000 | | | | 17,177,000 | | | | — | | | | * | |
RA Capital Healthcare Fund, L.P. (17) | | | 17,177,000 | | | | 17,177,000 | | | | — | | | | * | |
Entities associated with Logos Global (18) | | | 14,939,000 | | | | 8,589,000 | | | | 6,350,000 | | | | 6.0 | |
Citadel CEMF Investments Ltd. (19) | | | 10,306,000 | | | | 10,306,000 | | | | — | | | | * | |
Entities associated with Aisling Capital (20) | | | 10,280,151 | | | | 8,589,000 | | | | 1,691,151 | | | | 1.6 | |
Affinity Healthcare Fund, LP (21) | | | 7,477,000 | | | | 2,577,000 | | | | 4,900,000 | | | | 4.9 | |
Entities associated with FMR LLC (Fidelity) (22) | | | 5,400,000 | | | | 5,400,000 | | | | — | | | | * | |
Salthill Investors (Bermuda) L.P. (23) | | | 5,357,000 | | | | 5,357,000 | | | | — | | | | * | |
Salthill Partners, L.P. (23) | | | 4,949,000 | | | | 4,949,000 | | | | — | | | | * | |
Stempoint Capital (24) | | | 3,180,863 | | | | 2,654,117 | | | | 526,746 | | | | * | |
Andrew Spencer (25) | | | 2,519,299 | | | | 2,284,697 | | | | 234,602 | | | | * | |
Janet Gunzner-Toste (26) | | | 2,519,299 | | | | 2,284,697 | | | | 234,602 | | | | * | |
Justin LaFountaine (27) | | | 2,063,339 | | | | 1,866,269 | | | | 197,070 | | | | * | |
Wedbush Healthcare Partners 2023 Fund, LLC (28) | | | 1,718,000 | | | | 1,718,000 | | | | — | | | | * | |
Sessions LLC (29) | | | 859,000 | | | | 859,000 | | | | — | | | | * | |
Harry Turtle (30) | | | 344,000 | | | | 344,000 | | | | — | | | | * | |
| (1) | To our knowledge, unless otherwise indicated, all persons named in the table above have sole voting and investment power with respect to their shares of Common Stock. Unless an address is provided below, the address for the holder is 221 Crescent Street, Building 17, Suite 102B, Waltham MA 02453. |
| (2) | “Beneficial ownership” is a term broadly defined by the SEC in Rule 13d-3 under the Exchange Act, and includes more than the typical form of stock ownership, that is, stock held in the person’s name. The term |
39
| also includes what is referred to as “indirect ownership,” meaning ownership of shares as to which a person has or shares investment power. Notwithstanding the foregoing, the beneficial ownership amounts assume the sale of all Common Stock that may be offered pursuant to this prospectus without taking into account certain limitations, including that a holder of Series A Preferred Stock is prohibited from converting shares of Series A Preferred Stock into shares of Common Stock (i) prior to the affirmative vote of the holders of a majority of the then outstanding shares of the Series A Preferred Stock, or (ii) if, as a result of such conversion, such holder, together with its affiliates, would beneficially own more than a specified percentage (established by the holder between 0.00% and 19.99%) (the “Beneficial Ownership Limitation”) of the total number of shares of Common Stock issued and outstanding immediately after giving effect to such conversion. |
| (3) | Consists of (i) 424,362,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Fairmount Healthcare Fund II LP (“Fund II”), (ii) 9,184,458 shares of Common Stock held Fund II, (iii) 12,675,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Fairmount Healthcare Fund LP (“Fund I”), (iv) 276,082 shares of Common Stock held by Fund I and (v) 85,887,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Fairmount Healthcare Co-Invest LP (“Co-Invest”). Fairmount Funds Management LLC (“Fairmount Funds Management”) serves as investment manager for Fund I, Fund II and Co-Invest and may be deemed a beneficial owner of any securities of the Company held by Fund I, Fund II and Co-Invest. Fund I, Fund II and Co-Invest have delegated to Fairmount Funds Management the sole power to vote and the sole power to dispose of all securities held in Fund I’s, Fund II’s and Co-Invest’s portfolio. Because Fund I, Fund II and Co-Invest have divested themselves of voting and investment power over the securities they hold and may not revoke that delegation on less than 61 days’ notice, Fund I, Fund II and Co-Invest disclaim beneficial ownership of the securities they hold. The general partner of Fairmount Funds Management is Fairmount Funds Management GP LLC (“Fairmount GP”). As managing members of Fairmount GP, Peter Harwin and Tomas Kiselak may be deemed beneficial owners of any securities of the Company beneficially owned by Fairmount Funds Management. Fairmount GP, Fairmount Funds Management LLC, Mr. Harwin and Mr. Kiselak disclaim beneficial ownership of securities held by Fund I, Fund II and Co-Invest, except to the extent of their pecuniary interest therein. The address for the beneficial owners is 200 Barr Harbor Drive, Suite 400, West Conshohocken, PA 19428. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by each of Fund II, Fund I and Co-Invest are subject to a Beneficial Ownership Limitation of 0.00%, 0.00% and 19.99%, respectively. |
| (4) | Consists of (i) 33,157,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Paragon, (ii) 1,182,567 shares of Common Stock held Paragon, (iii) 33,157,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Parapyre, (iv) 1,182,567 shares of Common Stock held Parapyre, (v) 9,010,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Spyre Advisors LLC (“Spyre Advisors”), and (vi) 321,349 shares of Common Stock held Spyre Advisors. Parapyre and Spyre Advisors are owned and controlled by Paragon. Paragon is managed by a board of directors. |
| (5) | Consists of (i) 32,478,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Venrock Healthcare Capital Partners EG, L.P. (“VHCP EG”), (ii) 4,620,200 shares of Common Stock held by VHCP EG, (iii) 12,012,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Venrock Healthcare Capital Partners III, L.P. (“VHCP III”), (iv) 1,708,850 shares of Common Stock held by VHCP III, (v) 1,202,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by VHCP Co-Investment Holdings III, LLC (“VHCP III Co”), and (vi) 170,950 shares of Common Stock held by VHCP III Co. VHCP Management III, LLC (“VHCPM”) is the sole general partner of VHCP III and the sole manager of VHCP III Co. VHCP Management EG, LLC (“VHCPM EG”) is the sole general partner of VHCP EG. Dr. Bong Koh and Nimish Shah are the voting members of VHCPM and VHCPM EG. The address of each of these persons and entities is 7 Bryant Park, 23rd Floor, New York, NY 10018. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by VHCP EG, VHCP III, and VHCP III Co are subject to a Beneficial Ownership Limitation of 9.99%. |
40
| (6) | Consists of (i) 45,692,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Avoro Life Sciences Fund LLC and (ii) 286,459 shares of Common Stock held by Avoro Life Sciences Fund LLC. Avoro Capital Advisors LLC (“Avoro”) is the investment advisor for Avoro Life Sciences Fund LLC. Behzad Aghazadeh serves as the portfolio manager and controlling person of Avoro and may be deemed to have investment discretion and voting power over the shares held by Avoro. Mr. Aghazadeh disclaims beneficial ownership of these shares, except to the extent of his pecuniary interest in such shares, if any. The address of Avoro Life Sciences Fund LLC is 110 Greene Street, Suite 800, New York, NY 10012. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Avoro Life Sciences Fund LLC are subject to a Beneficial Ownership Limitation of 19.99% |
| (7) | Consists of (i) 42,944,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Commodore Capital Master LP and (ii) 2,908,100 shares of Common Stock held by Commodore Capital Master LP. Commodore Capital LP is the investment manager to Commodore Capital Master LP and may be deemed to beneficially own the shares held by Commodore Capital Master LP. Michael Kramarz and Robert Egen Atkinson are the managing partners of Commodore Capital LP and exercise investment discretion with respect to these shares. Commodore Capital LP and Commodore Capital Master LP have shared voting and dispositive power with respect to these shares. The address of Commodore Capital LP and Commodore Capital Master LP is 444 Madison Avenue, 35th Floor, New York, NY 10022. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Commodore Capital Master LP are subject to a Beneficial Ownership Limitation of 9.99%. |
| (8) | Consists of 42,944,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Deep Track Biotechnology Master Fund, Ltd. (“Deep Track Biotech”). Deep Track Capital, LP (“Deep Track Capital”) and David Kroin have shared voting power and shared dispositive power over the shares held by the Deep Track Biotech. Mr. Kroin may be considered a control person for Deep Track Capital. The address of Deep Track Capital and Mr. Kroin is 200 Greenwich Ave, 3rd Floor, Greenwich, CT 06830, and the address for Deep Track Biotech is 190 Elgin Avenue, George Town, KY1-9001, Cayman Islands. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Deep Track Biotech are subject to a Beneficial Ownership Limitation of 9.99%. |
| (9) | Consists of 42,944,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Perceptive Life Sciences Master Fund, Ltd. The address of Perceptive is 51 Astor Place, 10th Floor, New York, NY 10003. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Perceptive Life Sciences Master Fund, Ltd. are subject to a Beneficial Ownership Limitation of 19.99%. |
| (10) | Consists of an aggregate of 42,944,000 shares of Common Stock held by RTW Master Fund, Ltd., RTW Innovation Master Fund, Ltd. and RTW Biotech Opportunities Ltd (collectively, the “RTW Funds”). RTW Investments, LP (“RTW”), in its capacity as the investment manager of the RTW Funds, has the power to vote and the power to direct the disposition of the shares held by the RTW Funds. Accordingly, RTW may be deemed to be the beneficial owner of such securities. Roderick Wong, M.D., as the Managing Partner of RTW, has the power to direct the vote and disposition of the securities held by RTW. Dr. Wong disclaims beneficial ownership of the shares held by the RTW Funds, except to the extent of his pecuniary interest therein. The address and principal office of RTW Investments, LP is 40 10th Avenue, Floor 7, New York, NY 10014, and the address of Dr. Wong and each of the RTW Funds is c/o RTW Investments, LP, 40 10th Avenue, Floor 7, New York, NY 10014. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by the RTW Funds are subject to a Beneficial Ownership Limitation of 9.90%. |
| (11) | Consists of (i) 988,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by EcoR1 Capital Fund, L.P. (“EcoR1”), (ii) 16,189,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by EcoR1 Capital Fund Qualified, L.P. (“Qualified”), and (iii) 5,000,000 shares of Common Stock held by EcoR1 Capital, LLC (“EcoR1 Capital”). Oleg Nodelman is the control person of EcoR1 Capital. EcoR1 Capital is the general partner and investment advisor of EcoR1 and Qualified, which disclaims beneficial ownership of the shares. Mr. Nodelman, EcoR1, EcoR1 Capital and Qualified each disclaim beneficial ownership of the shares except to the extent of their pecuniary interest therein. The address of each of EcoR1, EcoR1 Capital, Qualified and Oleg Nodelman is |
41
| 357 Tehama Street #3, San Francisco, CA 94103. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by EcoR1 and Qualified are subject to a Beneficial Ownership Limitation of 9.99%. |
| (12) | Consists of (i) 12,693,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Driehaus Life Sciences Master Fund, L.P. (“Driehaus Master Fund”), (ii) 3,433,583 shares of Common Stock held by Driehaus Master Fund, (iii) 4,484,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Driehaus Life Sciences (QP) Fund, L.P. (“Driehaus QP Fund”), and (iv) 1,209,744 shares of Common Stock held by Driehaus QP Fund. Driehaus Capital Management (USVI) LLC is the general partner of both Driehaus Life Sciences Master Fund, L.P. and Driehaus Life Sciences (QP) Fund, L.P. By virtue of such relationship, Driehaus Capital Management (USVI) LLC may be deemed to have beneficial ownership over such securities. Driehaus Capital Management LLC is the investment advisor to both Driehaus Life Sciences Master Fund, L.P. and Driehaus Life Sciences (QP) Fund. Driehaus Capital Management LLC exercises voting and investment power through portfolio managers comprised of Michael Caldwell and Alex Munns, each of whom disclaims beneficial ownership of the shares held by Driehaus Life Sciences Master Fund, L.P. and Driehaus Life Sciences (QP) Fund, L.P.. The address for these entities is c/o Driehaus Capital Management LLC, 25 East Erie Street, Chicago, IL 60611. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Driehaus Master Fund, L.P. and Driehaus QP Fund are subject to a Beneficial Ownership Limitation of 19.99%. |
| (13) | Consists of (i) 18,086,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock, (ii) 576,699 shares of Common Stock and (iii) 1,970,716 shares of Common Stock issuable upon the exercise of options that are or will become exercisable within 60 days of the date hereof, held by Dr. Turtle. |
| (14) | Consists of (i) 9,177,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Biotechnology Value Fund, L.P. (“BVF LP”), (ii) 6,956,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Biotechnology Value Fund II, L.P. (“BVF2 LP”), (iii) 809,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Biotechnology Value Trading Fund OS LP (“BVF OS”), and (iv) 235,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by MSI BVF SPV, LLC (“MSI BVF” and together with BVF LP, BVF2 LP and BVF OS, the “BVF Entities”). BVF I GP LLC (“BVF GP”), as general partner of BVF LP, may be deemed to beneficially own the shares beneficially owned by BVF LP. BVF II GP LLC (“BVF2 GP”), as general partner of BVF2 LP, may be deemed to beneficially own the shares beneficially owned by BVF2 LP. BVF Partners OS Ltd. (“Partners OS”), as general partner of BVF OS, may be deemed to beneficially own the shares beneficially owned by BVF OS. BVF GP Holdings LLC (“BVF GPH”), as the sole member of BVF GP and BVF2 GP, may be deemed to beneficially own the shares beneficially owned in the aggregate by BVF GP and BVF2 GP. BVF Partners L.P. (“Partners”), as investment manager of BVF LP, BVF2 LP, BVF OS and MSI BVF, and the sole member of Partners OS, may be deemed to beneficially own the shares beneficially owned in the aggregate by the BVF Entities. BVF Inc., as the general partner of Partners, may be deemed to beneficially own the shares beneficially owned by Partners. Mark N. Lampert as director and officer of BVF Inc., may be deemed to beneficially own the shares beneficially owned by BVF Inc. Each of BVF GP, BVF2 GP, Partners OS, BVF GPH, Partners, BVF Inc. and Mark N. Lampert disclaims beneficial ownership of securities beneficially owned by the BVF Entities. The address of each of these entities is 44 Montgomery Street, Suite 4000, San Francisco, CA 94104. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by the BVF Entities are subject to a Beneficial Ownership Limitation of 9.90%. |
| (15) | Consists of 17,177,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Cormorant Global Healthcare Master Fund, LP (“Cormorant Global Healthcare”). Cormorant Global Healthcare GP, LLC (“Global Healthcare GP”) is the general partner of Cormorant Global Healthcare. Cormorant Asset Management, LP serves as the investment manager to Cormorant Global Healthcare. Bihua Chen serves as the managing member of both Global Healthcare GP and Cormorant Asset Management, LP and accordingly may be deemed to have voting and dispositive power with respect to shares held by Cormorant Global Healthcare. Each of Global Healthcare GP, Cormorant Asset Management, LP and Ms. Chen disclaims beneficial ownership of such shares except to the extent of any pecuniary interest therein. The principal address for the Cormorant Asset Management, LP entities is 200 |
42
| Clarendon Street 52nd Floor, Boston, MA 02116. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Cormorant Global Healthcare are subject to a Beneficial Ownership Limitation of 9.99%. |
| (16) | Consists of (i) 5,500,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Franklin Strategic Series – Franklin Biotechnology Discovery Fund (“Franklin Strategic”) and (ii) 11,677,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Franklin Templeton Investment Funds – Franklin Biotechnology Discovery Fund (“Franklin Templeton”). The address of the Franklin entities is c/o Franklin Advisers, Inc., One Franklin Parkway, San Mateo, CA 94403. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Franklin Strategic and Franklin Templeton are subject to a Beneficial Ownership Limitation of 19.99%. |
| (17) | Consists of 17,177,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by RA Capital Healthcare Fund, L.P. (“RACHF”). RA Capital Management, L.P. is the investment manager for RACHF. The general partner of RA Capital Management, L.P. is RA Capital Management GP, LLC, of which Peter Kolchinsky and Rajeev Shah are the managing members. Each of Mr. Kolchinsky and Mr. Shah may be deemed to have voting and investment power over the ordinary shares held by RACHF. Mr. Kolchinsky and Mr. Shah disclaim beneficial ownership of such shares, except to the extent of any pecuniary interest therein. The principal business address of these persons and entities is 200 Berkeley Street, 18th Floor, Boston, MA 02116. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by RACHF are subject to a Beneficial Ownership Limitation of 9.90%. |
| (18) | Consists of (i) 8,589,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Logos Opportunities Fund IV LP (“Logos Opportunities”) and (ii) 6,350,000 shares of Common Stock held by Logos Global Master Fund LP (“Global Fund”). Logos Opportunities IV GP LLC (“Logos Opportunities GP”) is the general partner of Logos Opportunities. Logos Global Management LP (“Logos Global”) is the investment advisor to Global Fund. Logos Global Management GP LLC (“Logos Global GP”) is the general partner of Logos Global. Arsani William and Graham Walmsley are the members of Logos Opportunities GP and Mr. William is a control person of Logos Global and Logos Global GP. Mr. William and Mr. Walmsley each disclaim beneficial ownership of these shares, except to the extent of each’s pecuniary interest in such shares, if any. The principal address of Logos Opportunities and Global Fund is 1 Letterman Drive, Building C, Suite C3-350, San Francisco, CA 94129. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Logos Opportunities are subject to a Beneficial Ownership Limitation of 9.99%. |
| (19) | Consists of 10,306,000 shares of Common Stock issuable upon conversion of Series A Preferred Stock held by Citadel CEMF Investments Ltd. (“Citadel CEMF”). Citadel Advisors LLC (“Citadel Advisors”) is the portfolio manager of Citadel CEMF. Citadel Advisors Holdings LP (“Citadel Holdings”) is the sole member of Citadel Advisors. Citadel GP LLC (“Citadel GP”) is the General Partner of Citadel Holdings. Kenneth Griffin owns a controlling interest in Citadel GP LLC. Mr. Griffin, as the owner of a controlling interest in Citadel GP LLC, may be deemed to have shared power to vote and/or shared power to dispose of the securities held by Citadel CEMF. This disclosure shall not be construed as an admission that Mr. Griffin or any of the Citadel related entities listed above is the beneficial owner of any of the Company’s securities other than the securities actually owned by such person (if any). The address of Citadel CEMF is Southeast Financial Center, 200 S. Biscayne Blvd., Suite 3300, Miami, FL 33131. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Citadel CEMF are subject to a Beneficial Ownership Limitation of 9.99%. |
| (20) | Consists of (i) 8,589,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held directly by Aisling Capital V, LP (“Aisling V”) and held indirectly by Aisling Capital Partners V, LP (“Aisling GP V”), as general partner of Aisling V, Aisling Capital Partners V LLC (“Aisling Partners V”), as general partner of Aisling GP V, and each of the individual managing members of Aisling Partners V and (ii) 1,691,151 shares of Common Stock held by Aisling Capital IV, LP (“Aisling IV”) and held indirectly by Aisling Capital Partners IV, LP (“Aisling GP IV”), as general partner of Aisling IV, Aisling Capital Partners IV LLC (“Aisling Partners IV”), as general partner of Aisling GP IV, and each of the individual managing members of Aisling Partners IV. The individual managing members (collectively, the “Managers”) of |
43
| Aisling Partners IV and Aisling Partners V are Dr. Andrew Schiff and Steve Elms. Aisling GP IV, Aisling GP V, Aisling Partners IV, Aisling Partners V and the Managers share voting and dispositive power over the shares directly held by Aisling V and Aisling IV. Each of Aisling GP IV, Aisling GP V, Aisling Partners IV, Aisling Partners V and the Managers may be deemed to be the beneficial owner of the securities listed above only to the extent of its pecuniary interest therein. The above information shall not be deemed an admission that any of Aisling GP IV, Aisling GP V, Aisling Partners IV, Aisling Partners V or any of the Managers is the beneficial owner of any securities reported herein. The address of the principal business offices of each of these entities and persons is 489 Fifth Avenue, 10th Floor, New York, NY 10017. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Aisling V are subject to a Beneficial Ownership Limitation of 19.99%. |
| (21) | Consists of (i) 2,577,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Affinity Healthcare Fund, LP (the “Affinity Fund”) and (ii) 4,900,000 shares of Common Stock held by Affinity Fund. Affinity Asset Advisors, LLC (the “Affinity Advisor”) is the investment manager of Affinity Fund and exercises investment discretion with regard to the shares of Common Stock owned by Affinity Fund. Affinity Fund and Affinity Advisor have the shared power to vote or to direct the vote and to dispose or direct the disposition of such shares of Common Stock owned by Affinity Fund. Affinity Advisor may be deemed to be the beneficial owner of such shares of Common Stock owned by Affinity Fund by virtue of its position as investment manager of Affinity Fund. The address of each of these entities is 19 Barn Lane, Bridgehampton, NY 11932. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Affinity Fund are subject to a Beneficial Ownership Limitation of 19.99%. |
| (22) | Consists of (i) 304,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Fidelity Select Portfolios: Pharmaceuticals Portfolio, (ii) 600,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Fidelity Advisor Series VII: Fidelity Advisor Biotechnology Fund, (iii) 4,900,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Fidelity Mt. Vernon Street Trust: Fidelity Series Growth Company Fund, (iv) 17,600,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Fidelity Mt. Vernon Street Trust: Fidelity Growth Company Fund, (v) 22,900,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Fidelity Growth Company Commingled Pool By: Fidelity Management Trust Company, as Trustee, and (vi) 5,400,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Fidelity Mt. Vernon Street Trust: Fidelity Growth Company K6 Fund. These funds and accounts are managed by direct or indirect subsidiaries of FMR LLC. Abigail P. Johnson is a Director, the Chairman and the Chief Executive Officer of FMR LLC. Members of the Johnson family, including Abigail P. Johnson, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR LLC. Neither FMR LLC nor Abigail P. Johnson has the sole power to vote or direct the voting of the shares owned directly by the various investment companies registered under the Investment Company Act of 1940 (“Fidelity Funds”) advised by Fidelity Management & Research Company LLC (“FMR Co. LLC”), a wholly owned subsidiary of FMR LLC, which power resides with the Fidelity Funds’ Boards of Trustees. FMR Co. LLC carries out the voting of the shares under written guidelines established by the Fidelity Funds’ Boards of Trustees. The address of these funds and accounts is 245 Summer Street, Boston, MA 02210. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by these funds and accounts are subject to a Beneficial Ownership Limitation of 14.90%. |
| (23) | Consists of (i) 5,357,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Salthill Investors (Bermuda) L.P. (“Salthill Investors”) and (ii) 4,949,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Salthill Partners, L.P. (“Salthill Partners”). Wellington Management Company LLP (“Wellington Management”), a registered investment adviser under |
44
| the Investment Advisers Act of 1940, as amended, is the investment adviser to Salthill Partners and Salthill Investors (collectively, the “Wellington Entities”). Wellington Management is an indirect subsidiary of Wellington Management Group LLP (“Wellington Group”). Wellington Group and Wellington Management may be deemed beneficial owners with shared voting and investment power over the shares held by the Wellington Entities. The address of these entities is c/o Wellington Management Company LLP, 280 Congress Street, Boston, MA 02210. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by the Wellington Entities are subject to a Beneficial Ownership Limitation of 19.99%. |
| (24) | Consists of (i) 2,654,117 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by StemPoint Capital Master Fund LP (“StemPoint Fund”) and (ii) 526,746 shares of Common Stock held by StemPoint Fund. StemPoint Capital LP is the investment advisor to StemPoint Fund and Jefferies Strategic Investments, LLC (“JSI”), and the sub-advisor to Titan Biotech Dislocation Fund SP (“TBDF”). StemPoint Capital LP exercises voting and investment power through its respective investment management agreement for both the StemPoint Fund and JSI, who disclaims beneficial ownership of the shares held by JSI. In aggregate, the total number of shares that StemPoint Capital LP as advisor exercises investment power over is 2,654,117 as of July 12, 2023. The total number of shares that StemPoint Capital LP as advisor exercises voting and investment power over is 526,746 as of July 12, 2023. The address of these funds and accounts is 520 Madison Avenue, 19th Floor, New York, NY 10022. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by StemPoint Fund are subject to a Beneficial Ownership Limitation of 19.99%. |
| (25) | Consists of (i) 2,206,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock and (ii) 78,697 shares of Common Stock and (iii) 234,602 shares of Common Stock issuable upon the exercise of options that are or will become exercisable within 60 days of the date hereof, held by Dr. Spencer. |
| (26) | Consists of (i) 2,206,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock and (ii) 78,697 shares of Common Stock and (iii) 234,602 shares of Common Stock issuable upon the exercise of options that are or will become exercisable within 60 days of the date hereof, held by Dr. Gunzner-Toste. |
| (27) | Consists of (i) 1,802,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock and (ii) 64,269 shares of Common Stock and (iii) 197,070 shares of Common Stock issuable upon the exercise of options that are or will become exercisable within 60 days of the date hereof, held by Mr. LaFountaine. |
| (28) | Consists of 1,718,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Wedbush Healthcare Partners 2023 Fund, LLC. The address for Wedbush Healthcare Partners 2023 Fund, LLC is Wedbush Center, 1000 Wilshire Blvd., Los Angeles, CA 90017. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Wedbush Healthcare Partners 2023 Fund, LLC are subject to a Beneficial Ownership Limitation of 9.99%. |
| (29) | Consists of 859,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Sessions LLC. The address for Sessions LLC is 96 Fairbanks Ave, Wellesley, MA 02481. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Sessions LLC are subject to a Beneficial Ownership Limitation of 19.99%. |
| (30) | Consists of 344,000 shares of Common Stock issuable upon the conversion of Series A Preferred Stock held by Harry Turtle. The shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Mr. Turtle are subject to a Beneficial Ownership Limitation of 19.99%. |
Relationship with the Selling Stockholders
In addition to the Securities Purchase Agreement, on June 22, 2023, in connection with the PIPE, we entered into a registration rights agreement (the “New RRA”) on June 22, 2023 with certain Selling Stockholders. Cameron Turtle is our Chief Operating Officer, Andrew Spencer is our SVP, Preclinical R&D, Janet Gunzner-Toste is our SVP, Operations and Justin LaFountaine is our VP, Corporate Development. Wedbush PacGrow served as lead financial advisor to our ownboard of directors with respect to the Asset Acquisition.
45
Registration Rights Agreement
Pursuant to the terms of our New RRA, we agreed to prepare and capital expenditures. We will set forthfile with the SEC a registration statement that permits the resale or other disposition of the Selling Stockholders’ shares of Common Stock issued upon conversion of the Series A Preferred Stock issued to such Selling Stockholder pursuant to the Securities Purchase Agreement and Acquisition Agreement and, subject to certain exceptions, use commercially reasonable efforts to keep the registration statement of which this prospectus forms a part effective under the Securities Act for so long as such securities registered for resale thereunder retain their character as Registrable Securities (as defined in the prospectus supplement our intended use forNew RRA).
We have also agreed, among other things, to indemnify the net proceeds receivedSelling Stockholders and each of their respective officers, directors, members, employees, partners, managers, stockholders, affiliates, investment advisors and agents, each person who controls any such Selling Stockholder and the officers, directors, members, employees, partners, managers, stockholders, affiliates, investment advisors and agents of each such controlling person from the sale ofcertain liabilities and pay all fees and expenses (excluding any securities. Pending the applicationlegal fees of the net proceeds, we intendselling holder(s), and any underwriting discounts and selling commissions) incident to investour obligations under the net proceeds in short-term or long-term, investment-grade, interest-bearing securities.
New RRA.46
PLAN OF DISTRIBUTION
We are registering the Resale Shares issued to the Selling Stockholders to permit the sale, transfer or other disposition of these shares by the Selling Stockholders or their donees, pledgees, transferees or other successors-in-interest from time to time after the date of this prospectus. We will not receive any of the proceeds from the sale by the Selling Stockholders of the Resale Shares. We will, or will procure to, bear all fees and expenses incident to our obligation to register the Resale Shares.
The Selling Stockholders may sell all or a portion of the securities coveredResale Shares beneficially owned by this prospectusthem and offered hereby from time to time, and in the case of the Private Placement Conversion Shares and Merger Conversion Shares, may only be offered after such shares are converted to Common Stock pursuant to the terms of the Certificate of Designation, directly or through one or more underwriters, broker-dealers or agents. If the Resale Shares are sold through underwriters or broker-dealers, the Selling Stockholders will be responsible for public offering and sale by them, andunderwriting discounts (it being understood that the Selling Stockholders shall not be deemed to be underwriters solely as a result of their participation in this offering) or commissions or agent’s commissions. The Resale Shares may also sellbe sold on any national securities exchange or quotation service on which the securities to investors directly or through agents. We will name any underwriter or agent involved in the offer and sale of securities in the applicable prospectus supplement. We have reserved the right to sell or exchange securities directly to investors on our own behalf in jurisdictions where we are authorized to do so. We may distribute the securities from time to time in one or more transactions:
at a fixed price or prices, which may be changed;
at market prices prevailing at the time of sale;
at prices related to such prevailing market prices;listed or
at negotiated prices.
We may directly solicit offers to purchase the securities being offered by this prospectus. We may also designate agents to solicit offers to purchase the securities from time to time. We will name in a prospectus supplement any agent involved in the offer or sale of our securities. Unless otherwise indicated in a prospectus supplement, an agent will be acting on a best efforts basis, and a dealer will purchase securities as a principal for resale at varying prices to be determined by the dealer.
If we utilize an underwriter in the sale of the securities being offered by this prospectus, we will execute an underwriting agreement with the underwriter quoted at the time of sale, in the over-the-counter market or in transactions otherwise than on these exchanges or systems or in the over-the-counter market and wein one or more transactions at fixed prices, at prevailing market prices at the time of the sale, at varying prices determined at the time of sale, or at negotiated prices. These sales may be effected in transactions, which may involve crosses or block transactions. The Selling Stockholders may use any one or more of the following methods when selling Resale Shares:
ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers;
block trades in which the broker-dealer will provideattempt to sell the nameshares as agent but may position and resell a portion of the block as principal to facilitate the transaction;
to or through underwriters or purchases by a broker-dealer as principal and resale by the broker-dealer for its account;
purchases by a broker-dealer as principal and resale by the broker-dealer for its account;
an exchange distribution in accordance with the rules of the applicable exchange;
privately negotiated transactions;
settlement of short sales entered into after the effective date of the registration statement of which this prospectus is a part;
broker-dealers may agree with the Selling Stockholders to sell a specified number of such shares at a stipulated price per share;
through the writing or settlement of options or other hedging transactions, whether such options are listed on an options exchange or otherwise;
a combination of any underwriter in the prospectus supplement that the underwriter will usesuch methods of sale; and
any other method permitted pursuant to make resalesapplicable law.
The Selling Stockholders also may resell all or a portion of the securitiesResale Shares in open market transactions in reliance upon Rule 144 under the Securities Act (“Rule 144”), as permitted by that rule, or Section 4(a)(1) under the Securities Act, if available, rather than under this prospectus, provided that they meet the criteria and conform to the public. In connection withrequirements of those provisions.
Broker-dealers engaged by the sale ofSelling Stockholders may arrange for other broker-dealers to participate in sales. If the securities, we,Selling Stockholders effect such transactions by selling Resale Shares to or the purchasers of securities for whom the underwriterthrough underwriters, broker-dealers or agents, such underwriters, broker-dealers or agents may act as agent, may compensate the underwriterreceive commissions in the form of underwriting discounts, or commissions. The underwriter may sell the securities to or through dealers, and those dealers may receive compensation in the form of discounts,
47
concessions or commissions from the underwritersSelling Stockholders or commissions from purchasers of the purchasersResale Shares for whom they may act as agent.
Weagent or to whom they may sell as principal. Such commissions will providebe in amounts to be negotiated, but, except as set forth in a supplement to this prospectus, in the applicablecase of an agency transaction will not be in excess of a customary brokerage commission in compliance with FINRA Rule 2121; and in the case of a principal transaction a markup or markdown in compliance with FINRA IM-2121.01.
In connection with sales of the Resale Shares or otherwise, the Selling Stockholders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the Resale Shares in the course of hedging in positions they assume. The Selling Stockholders may also sell Resale Shares short and if such short sale shall take place after the date that this registration statement is declared effective by the SEC, the Selling Stockholders may deliver Resale Shares covered by this prospectus supplement any compensation we pay to underwriters, dealers, or agentsclose out short positions and to return borrowed Resale Shares in connection with such short sales. The Selling Stockholders may also loan or pledge Resale Shares to broker-dealers that in turn may sell such shares, to the offeringextent permitted by applicable law. The Selling Stockholders may also enter into option or other transactions with broker-dealers or other financial institutions or the creation of one or more derivative securities which require the delivery to such broker-dealer or other financial institution of shares offered by this prospectus, which shares such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction). Notwithstanding the foregoing, the Selling Stockholders have been advised that they may not use Resale Shares to cover short sales of our Common Stock made prior to the date the registration statement, of which this prospectus forms a part, has been declared effective by the SEC.
The Selling Stockholders may, from time to time, pledge or grant a security interest in some or all of the securities,Resale Shares owned by them and, if they default in the performance of their secured obligations, the pledgees or secured parties may offer and sell the Resale Shares from time to time pursuant to this prospectus or any amendment to this prospectus under Rule 424(b)(3) or other applicable provision of the Securities Act, amending, if necessary, the list of Selling Stockholders to include the pledgee, transferee or other successors in interest as Selling Stockholders under this prospectus. The Selling Stockholders also may transfer and donate the Resale Shares in other circumstances in which case the transferees, donees, pledgees or other successors in interest will be the selling beneficial owners for purposes of this prospectus.
The Selling Stockholders and any discounts, concessionsbroker-dealer or commissions allowed by underwriters to participating dealers. Underwriters, dealers and agents participating in the distribution of the securitiesResale Shares may be deemed to be underwriters“underwriters” within the meaning of Section 2(11) of the Securities Act of 1933, as amended,in connection with such sales. In such event, any commissions paid, or the Securities Act, and any discounts and commissions received by themor concessions allowed to, any such broker-dealer or agent and any profit realized by them on the resale of the securitiesshares purchased by them may be deemed to be underwriting commissions or discounts under the Securities Act. Selling Stockholders who are “underwriters” within the meaning of Section 2(a)(11) of the Securities Act will be subject to the applicable prospectus delivery requirements of the Securities Act including Rule 172 thereunder and may be subject to certain statutory liabilities of, including but not limited to, Sections 11, 12 and 17 of the Securities Act and Rule 10b-5 under the Exchange Act.
Each Selling Stockholder has informed the Company that it is not a registered broker-dealer and does not have any written or oral agreement or understanding, directly or indirectly, with any person to distribute the Resale Shares. Upon the Company being notified in writing by a Selling Stockholder that any material arrangement has been entered into with a broker-dealer for the sale of Common Stock through a block trade, special offering, exchange distribution or secondary distribution or a purchase by a broker or dealer, a supplement to this prospectus will be filed, if required, pursuant to Rule 424(b) under the Securities Act, disclosing (i) the name of each such Selling Stockholder and of the participating broker-dealer(s), (ii) the number of Resale Shares involved, (iii) the price at which such the Resale Shares were sold, (iv) the commissions paid or discounts or concessions allowed to such broker-dealer(s), where applicable, (v) that such broker-dealer(s) did not conduct any investigation to verify the information set out or incorporated by reference in this prospectus, and (vi) other facts material to the transaction. In no event shall any broker-dealer receive fees, commissions and markups, which, in the aggregate, would exceed eight percent (8.0%).
48
Under the securities laws of some U.S. states, the Resale Shares may be sold in such states only through registered or licensed brokers or dealers. In addition, in some U.S. states the Resale Shares may not be sold unless such shares have been registered or qualified for sale in such state or an exemption from registration or qualification is available and is complied with.
There can be no assurance that any Selling Stockholder will sell any or all of the Resale Shares registered pursuant to the shelf registration statement, of which this prospectus forms a part.
Each Selling Stockholder and any other person participating in such distribution will be subject to applicable provisions of the Exchange Act and the rules and regulations thereunder, including, without limitation, to the extent applicable, Regulation M of the Exchange Act, which may limit the timing of purchases and sales of any of the Resale Shares by the Selling Stockholder and any other participating person. To the extent applicable, Regulation M may also restrict the ability of any person engaged in the distribution of the Resale Shares to engage in market-making activities with respect to the Resale Shares. All of the foregoing may affect the marketability of the Resale Shares and the ability of any person or entity to engage in market-making activities with respect to the Resale Shares.
We will pay all expenses of the registration of the Resale Shares pursuant to the New RRA, including, without limitation, SEC filing fees and expenses of compliance with state securities or “blue sky” laws; provided, however, that each Selling Stockholder will pay all underwriting discounts and commissions.selling commissions, if any and any related legal expenses incurred by it. We will indemnify the Selling Stockholders against certain liabilities, including some liabilities under the Securities Act, in accordance with the New RRA, or the Selling Stockholders will be entitled to contribution. We may enter into agreements to indemnify underwriters, dealers and agentsbe indemnified by the Selling Stockholders against certain civil liabilities set forth in the New RRA, including liabilities under the Securities Act, andthat may arise from any written information furnished to reimburse themus by the Selling Stockholders specifically for certain expenses. We may grant underwriters who participateuse in the distribution of our securities under this prospectus, an option to purchase additional securities to cover any over-allotments in connection with the distribution.
The securities we offer under this prospectus may or may not be listed through The NASDAQ Global Market or any other securities exchange. To facilitate the offering of securities, certain persons participating in the offering may engage in transactions that stabilize, maintain or otherwise affect the price of the securities. This may include short sales of the securities, which involves the sale by persons participating in the offering of more securities than we sold to them. In these circumstances, these persons would cover such short positions by making purchases in the open market or by exercising their option to purchase additional securities. In addition, these persons may stabilize or maintain the price of the securities by bidding for or purchasing securities in the open market or by imposing penalty bids, whereby selling concessions allowed to dealers participating in the offering may be reclaimed if securities sold by them are repurchased in connection with stabilization transactions. The effect of these transactions may be to stabilize or maintain the market price of the securities at a level above that which might otherwise prevail in the open market. These transactions may be discontinued at any time.
We may engage in at the market offerings into an existing trading market in accordance with Rule 415(a)(4) under the Securities Act. In addition,related registration rights agreements, or we may enter into derivative transactions with third parties, or sell securities not covered by this prospectusbe entitled to third parties in privately negotiated transactions. If the applicable prospectus supplement indicates, in connection with those derivatives, the third parties may sell securities covered by this prospectus and the applicable prospectus supplement, including short sale transactions. If so, the third party may use securities pledged by us or borrowed from us or others to settle those sales or to close out any related open borrowings of stock, and they may use securities received from us in settlement of those derivatives to close out any related open borrowings of stock. The third party in these sale transactions will be an underwriter and will be identified in the applicable prospectus supplement. In addition, we may otherwise loan or pledge securities to a financial institution or other third party that in turn may sell the securities short using this prospectus. The financial institution or other third party may transfer its economic short position to investors in our securities or in connection with a concurrent offering of other securities.
We will file a prospectus supplement to describe the terms of any offering of our securities covered by this prospectus. The prospectus supplement will disclose:contribution.
49
the terms of the offer;
the names of any underwriters, including any managing underwriters, as well as any dealers or agents;
the purchase price of the securities from us;
the net proceeds to us from the sale of the securities;
any delayed delivery arrangements;
any over-allotment or other options under which underwriters, if any, may purchase additional securities from us;
any underwriting discounts, commissions or other items constituting underwriters’ compensation, and any commissions paid to agents;
in a subscription rights offering, whether we have engaged dealer-managers to facilitate the offering or subscription, including their name or names and compensation;
any public offering price; and
other facts material to the transaction.
We will bear all or substantially all of the costs, expenses and fees in connection with the registration of our securities under this prospectus. The underwriters, dealers and agents may engage in transactions with us, or perform services for us, in the ordinary course of business.
In compliance with guidelines of the Financial Industry Regulatory Authority, or FINRA, the maximum consideration or discount to be received by any FINRA member or independent broker dealer may not exceed 8% of the aggregate amount of the securities offered pursuant to this prospectus and any applicable prospectus supplement.
DESCRIPTION OF CAPITAL STOCK
General
Our authorized capital stock consists of 500,000,000 shares of common stock,Common Stock, $0.0001 par value per share, and 10,000,000 shares of undesignated preferred stock (“Preferred Stock”), of which 1,086,341 shares have been designated as Series A Preferred Stock, $0.0001 par value per share.
As of July 12, 2023, there were 97,457,166 shares of our Common Stock and 1,086,339 shares of Series A Preferred Stock outstanding.
The following description summarizes the most important terms of our capital stock. Because it is only a summary, it does not contain all the information that may be important to you. For a complete description, you should refer to our restated certificateCertificate of incorporationIncorporation (the “Certificate of Incorporation”) and restated bylaws,Bylaws (the “Bylaws”), which are included as exhibits to the registration statement of which this prospectus forms a part,our most recent Annual Report on Form 10-K, and to the applicable provisions of Delaware law.
As of March 31, 2017, there were 13,452,260 shares of our common stock outstanding, and no shares of preferred stock outstanding.
Common Stock
Dividend rights
Subject to preferences that may apply to any shares of preferred stockPreferred Stock outstanding at the time, the holders of our common stockCommon Stock are entitled to receive dividends out of funds legally available if our board of directors, in its discretion, determines to issue dividends and then only at the times and in the amounts that our board of directors may determine. For more information about our dividend policy, see “Dividend Policy” in our Annual Report on Form 10-K for the year ended December 31, 2016, which is incorporated by reference in this prospectus.
Voting rights
Holders of our common stockCommon Stock are entitled to one vote for each share held on all matters submitted to a vote of stockholders. We have not provided for cumulative voting for the election of directors in our restated certificateCertificate of incorporation.Incorporation. Accordingly, pursuant to our restated certificateCertificate of incorporationIncorporation, holders of a majority of the shares of our common stockCommon Stock are able to elect all of our directors. Our restated certificateCertificate of incorporationIncorporation establishes a classified board of directors, divided into three classes with staggered three-year terms. Only one class of directors will be elected at each annual meeting of our stockholders, with the other classes continuing for the remainder of their respective three-year terms.
No preemptive or similar rights
Our common stockCommon Stock is not entitled to preemptive rights, and is not subject to conversion, redemption or sinking fund provisions.
Right to receive liquidation distributions
Upon our liquidation, dissolution or winding-up, the assets legally available for distribution to our stockholders would be distributable ratably among the holders of our common stockCommon Stock and any participating preferred stockPreferred Stock outstanding at that time, subject to prior satisfaction of all outstanding debt and liabilities and the preferential rights of and the payment of liquidation preferences, if any, on any outstanding shares of preferred stock.Preferred Stock.
Preferred Stock
Our board of directors is authorized, subject to limitations prescribed by Delaware law, to issue preferred stockPreferred Stock in one or more series, to establish from time to time the number of shares to be included in each series and to fix the designation, powers, preferences and rights of the shares of each series and any of their qualifications,
50
limitations or restrictions, in each case without further vote or action by our stockholders. OurSubject to the Certificate of Designation, our board of directors
can also increase or decrease the number of shares of any series of preferred stock,Preferred Stock, but not below the number of shares of that series then outstanding, without any further vote or action by our stockholders. Our board of directors may authorize the issuance of preferred stockPreferred Stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of our common stock.Common Stock. The issuance of preferred stock,Preferred Stock, while providing flexibility in connection with possible acquisitions and other corporate purposes, could, among other things, have the effect of delaying, deferring or preventing a change in control of our company and might adversely affect the market price of our common stockCommon Stock and the voting and other rights of the holders of our common stock.Common Stock. We have no current plan to issue any shares of preferred stock.Preferred Stock other than the shares of our Series A Preferred Stock that were issued in connection with the Transactions.
Series A Preferred Stock
Holders of Series A Preferred Stock are entitled to receive dividends on shares of Series A Preferred Stock equal to, on an as-if-converted-to-common-stock basis, and in the same form as dividends actually paid on shares of our Common Stock. Except as provided in the Certificate of Designation or as otherwise required by law, the Series A Preferred Stock does not have voting rights. However, as long as any shares of Series A Preferred Stock are outstanding, we will not, without the affirmative vote of the holders of a majority of the then outstanding shares of the Series A Preferred Stock: (a) alter or change adversely the powers, preferences or rights given to the Series A Preferred Stock, or alter or amend the Certificate of Designation, amend or repeal any provision of, or add any provision to, the Company’s Certificate of Incorporation or its Bylaws, or file any articles of amendment, certificate of designations, preferences, limitations and relative rights of any series of Preferred Stock, if such action would adversely alter or change the preferences, rights, privileges or powers of, or restrictions provided for the benefit of the Series A Preferred Stock, regardless of whether any of the foregoing actions will be by means of amendment to the Certificate of Incorporation or by merger, consolidation, recapitalization, reclassification, conversion or otherwise, (b) issue further shares of Series A Preferred Stock or increase or decrease (other than by conversion) the number of authorized shares of Series A Preferred Stock, (c) prior to the stockholder approval of the Conversion Proposal or at any time while at least 30% of the originally issued Series A Preferred Stock remains issued and outstanding, consummate (x) any Fundamental Transaction (as defined in the Certificate of Designation) or (y) any merger or consolidation of the Company with or into another entity or any stock sale to, or other business combination in which the stockholders of the Company immediately before such transaction do not hold at least a majority of the capital stock of the Company immediately after such transaction or (d) enter into any agreement with respect to any of the foregoing. The Series A Preferred Stock does not have a preference upon any liquidation, dissolution or winding-up of the Company.
Following stockholder approval of the Conversion Proposal, each share of Series A Preferred Stock will automatically convert into 1,000 shares of Common Stock, subject to certain limitations, including that a holder of Series A Preferred Stock is prohibited from converting shares of Series A Preferred Stock into shares of Common Stock if, as a result of such conversion, such holder, together with its affiliates, would beneficially own more than a specified percentage (established by the holder between 0% and 19.99%) of the total number of shares of Common Stock issued and outstanding immediately after giving effect to such conversion.
Registration Rights
Pursuant to the terms of our investors’registration rights agreement entered into on March 16, 2021 (the “Baker RRA”) with Baker Brothers Life Sciences, L.P. and 667, L.P. (together, the “Baker Funds”), following a request by the Baker Funds, we are obligated to file a resale registration statement on Form S-3, or other appropriate form, covering the shares of our Common Stock held by the Baker Funds (the “Baker Registrable Securities”). Under the Baker RRA, the Baker Funds also have the right to request up to two underwritten public offerings or block trades in March 2015,any twelve month period, but not more than three underwritten public offerings and eight block trades
51
in total, to effect the sale or distribution of the Baker Registrable Securities, subject to specified exceptions, conditions and limitations. The Baker RRA also includes customary indemnification obligations in connection with registrations conducted pursuant to the Baker RRA. The rights of the Baker Funds under the Baker RRA terminate automatically upon the earlier to occur of the following events: (i) all Baker Registrable Securities covered by the Baker RRA have been sold pursuant to an effective registration statement; (ii) all Baker Registrable Securities covered by the Baker RRA have been sold pursuant to Rule 144, or other similar rule; (iii) at any time after the Baker Funds are no longer our affiliate, all Baker Registrable Securities covered by the Baker RRA may be resold by the Baker Funds without limitations as to volume or manner of sale pursuant to Rule 144; or (iv) ten (10) years after the date of the Baker RRA.
In addition, certain of the holders of our common stock holdersSeries A Preferred Stock are entitled to certain rights with respect to the registration of their sharessuch securities as further provided under the Securities Act, as described below. We refer to these shares collectively as registrable securities.
heading “Demand registration rightsSelling Stockholders – Registration Rights Agreement
The holders of at least 62% of the shares of common stock issued upon the conversion of Series B convertible preferred stock in connection with our initial public offering may make a written request to us for the registration of any of the registrable securities under the Securities Act. We are only required to file two registration statements that are declared effective upon exercise of these demand registration rights. We may postpone the filing of a registration statement once during any 12-month period for a total cumulative period of not more than 90 days if our board of directors determines that the filing would be seriously detrimental to us and our stockholders, provided that we do not register any securities for our own account or any other stockholder during such 90-day period.
Form S-3 registration rights
The holders of at least 20% of the outstanding shares of common stock that were issued upon the conversion of shares of preferred stock in connection with our initial public offering can request that we register all or part of their shares on Form S-3 if we are eligible to file a registration statement on Form S-3 and if the aggregate price to the public of the shares offered is at least $1,000,000. We may postpone the filing of a registration statement on Form S-3 once during any 12-month period for a total cumulative period of not more than 90 days if our board of directors determines that the filing would be seriously detrimental to us and our stockholders, provided that we do not register any securities for our own account or any other stockholder during such 90-day period.
Piggyback registration rights
If we register any of our securities for public sale in an offering pursuant to this prospectus, holders of registrable securities will have the right to include their shares in the registration statement. However, this right does not apply to a registration relating to employee benefit plans or a registration on Form S-4 relating solely to a transaction under Rule 145 of the Securities Act. The underwriters of any underwritten offering will have the right to limit the number of shares registered by these holders if they determine that marketing factors require limitation, in which case the number of shares to be registered will be apportioned pro rata among these holders, according to the total amount of securities entitled to be included by each holder. However, the number of shares to be registered by these holders cannot be reduced below 25% of the total shares covered by the registration statement.
Expenses of registration rights
We generally will pay all expenses related to the registrations, other than underwriting discounts and commissions.
Expiration of registration rights
The registration rights described above will expire, with respect to any particular holder of these rights, on the earlier of the fifth anniversary of the closing of our initial public offering, or when that holder ceases to hold such registrable securities..”
Anti-Takeover Provisions
The provisions of Delaware law, our restated certificateCertificate of incorporationIncorporation and our restated bylawsBylaws could have the effect of delaying, deferring or discouraging another person from acquiring control of our company. These provisions, which are summarized below, may have the effect of discouraging takeover bids. They are also designed, in part, to encourage persons seeking to acquire control of us to negotiate first with our board of directors. We believe that the benefits of increased protection of our potential ability to negotiate with an unfriendly or unsolicited acquirer outweigh the disadvantages of discouraging a proposal to acquire us because negotiation of these proposals could result in an improvement of their terms.
Delaware law
We are subject to the provisions of Section 203 of the Delaware General Corporation Law (the “DGCL”) regulating corporate takeovers. In general, Section 203 prohibits a publicly-held Delaware corporation from engaging in a business combination with an interested stockholder for a period of three years following the date on which the person became an interested stockholder unless:
Priorprior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder;
Theupon completion of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding atupon consummation of the time the transaction, commenced, excluding for purposes of determining the voting stock outstanding, but not the outstanding voting stock owned by the interested stockholder, (1) shares owned by persons who are directors and also officers and (2) shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or
At or subsequent to the date of the transaction, the business combination is approved by the board of directors of the corporation and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 66.67%66 2/3% of the outstanding voting stock thatwhich is not owned by the interested stockholder.
Restated Certificate of Incorporation and Restated Bylaw Provisions
Our restated certificateCertificate of incorporationIncorporation and our restated bylawsBylaws include a number of provisions that could deter hostile takeovers or delay or prevent changes in control of our company, including the following:
| | • | | Board of Directors vacancies.vacancies. Our restated certificateCertificate of incorporationIncorporation and restated bylawsBylaws authorize our board of directors to fill vacant directorships, including newly created seats unless the board of directors |
52
| determines that any such vacancies shall be filled by the stockholders. In addition, the number of directors constituting our board of directors is permitted to be set only by a resolution adopted by a majority vote of our entire board of directors. These provisions prevent a stockholder from increasing the size of our board of directors and then gaining control of our board of directors by filling the resulting vacancies with its own nominees. This makes it more difficult to change the composition of our board of directors but promotes continuity of management. |
| | • | | Classified board.board. Our restated certificateCertificate of incorporationIncorporation and restated bylawsBylaws provide that our board is classified into three classes of directors, each with staggered three-year terms. A third party may bediscouraged from making a tender offer or otherwise attempting to obtain control of us as it is more difficult and time consuming for stockholders to replace a majority of the directors on a classified board of directors. |
| | • | | Stockholder action; special meetings of stockholders.Our restated certificateCertificate of incorporationIncorporation provides that our stockholders may not take action by written consent, but may only take action at annual or special meetings of our stockholders. As a result, a holder controlling a majority of our capital stock would not be able to amend our restated bylawsBylaws or remove directors without holding a meeting of our stockholders called in accordance with our restated bylaws.Bylaws. Further, our restated bylawsBylaws provide that special meetings of our stockholders may be called only by a majority of our board of directors, the chairperson of our board of directors, our Chief Executive Officer or our President, thus prohibiting a stockholder from calling a special meeting. These provisions might delay the ability of our stockholders to force consideration of a proposal or for stockholders controlling a majority of our capital stock to take any action, including the removal of directors. |
| | • | | Advance notice requirements for stockholder proposals and director nominations.nominations. Our restated bylawsBylaws provide advance notice procedures for stockholders seeking to bring business before our annual meeting of stockholders or to nominate candidates for election as directors at our annual meeting of stockholders. Our restated bylawsBylaws also specify certain requirements regarding the form and content of a stockholder’s notice. These provisions might preclude our stockholders from bringing matters before our annual meeting of stockholders or from making nominations for directors at our annual meeting of stockholders if the proper procedures are not followed. We expect that these provisions might also discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain control of our company. |
| | • | | No cumulative voting. The Delaware General Corporation LawDGCL provides that stockholders are not entitled to the right to cumulate votes in the election of directors unless a corporation’s certificate of incorporation provides otherwise. Our restated certificateCertificate of incorporationIncorporation and restated bylawsBylaws do not provide for cumulative voting. |
| | • | | Directors removed only for cause. Our restated certificateCertificate of incorporationIncorporation provides that stockholders may remove directors only for cause. |
| | • | | Amendment of charter provisions. Any amendment of the above provisions in our restated certificateCertificate of incorporationIncorporation requires approval by holders of at least two-thirds of our outstanding common stock,Common Stock, provided that if two-thirds of our board of directors approves such an amendment, then only the approval of a majority of holders is required. |
| | • | | Issuance of undesignated preferred stockPreferred Stock. Our board of directors has the authority, without further action by the stockholders, to issue up to 10,000,000 shares of undesignated preferred stockPreferred Stock with rights and preferences, including voting rights, designated from time to time by our board of directors. The existence of authorized but unissued shares of preferred stockPreferred Stock enables our board of directors to render more difficult or to discourage an attempt to obtain control of us by merger, tender offer, proxy contest or other means. |
| | • | | Choice of forum. Our restated certificateCertificate of incorporationIncorporation provides that the Court of Chancery of the State of Delaware is the sole and exclusive forum for any derivative action or proceeding brought on our behalf; any action asserting a breach of fiduciary duty; any action asserting a claim against us arising pursuant to the Delaware General Corporation Law,DGCL, our restated certificateCertificate of incorporationIncorporation or our restated bylaws;Bylaws; or any action asserting a claim against us that is governed by the internal affairs doctrine. In addition, our Bylaws also provide that the |
53
| federal district courts of the United States is the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act. |
Transfer Agent and Registrar
The transfer agent and registrar for our common stockCommon Stock is American Stock Transfer and Trust Company, LLC. The transfer agent’s address is 6201 15th15th Avenue, Brooklyn, New York 11219, and its telephone number is (800) 937-5449.
Exchange Listing
Our common stockCommon Stock is listed on The NASDAQ GlobalNasdaq Capital Market under the symbol “AGLE.”
54
DESCRIPTION OF DEBT SECURITIESLEGAL MATTERS
General
We will issueCertain legal matters, including the debtlegality of the securities offered, by this prospectus and any accompanying prospectus supplement under an indenture to be entered into between us and the trustee identified in the applicable prospectus supplement. The terms of the debt securities will include those stated in the indenture and those made part of the indenture by reference to the Trust Indenture Act of 1939, as in effect on the date of the indenture. We have filed a copy of the form of indenture as an exhibit to the registration statement in which this prospectus is included. The indenture will be subject to and governedpassed upon for us by the terms of the Trust Indenture Act of 1939.
We may offer under this prospectus up to an aggregate principal amount of $150,000,000 in debt securities, or if debt securities are issued at a discount, or in a foreign currency, foreign currency units or composite currency, the principal amount asGibson, Dunn & Crutcher LLP, San Francisco, California. Additional legal matters may be soldpassed upon for an aggregate public offering price of up to $150,000,000. Unless otherwise specified in the applicable prospectus supplement, the debt securities will represent our direct, unsecured obligations and will rank equally with all of our other unsecured indebtedness.
We may issue the debt securities in oneus or more series with the sameany underwriters, dealers or various maturities, at par, at a premium, or at a discount. We will describe the particular terms of each series of debt securities in a prospectus supplement relating toagents, by counsel that series, which we will file with the SEC. The prospectus supplement relating to the particular series of debt securities being offered will specify the particular amounts, prices and terms of those debt securities. These terms may include:
the title of the series;
the aggregate principal amount, and, if a series, the total amount authorized and the total amount outstanding;
the issue price or prices, expressed as a percentage of the aggregate principal amount of the debt securities;
any limit on the aggregate principal amount;
the date or dates on which principal is payable;
the interest rate or rates (which may be fixed or variable) or, if applicable, the method used to determine such rate or rates;
the date or dates from which interest, if any, will be payable and any regular record date for the interest payable;
the place or places where principal and, if applicable, premium and interest, is payable;
the terms and conditions upon which we may, or the holders may require us to, redeem or repurchase the debt securities;
the denominations in which such debt securities may be issuable, if other than denominations of $1,000 or any integral multiple of that number;
whether the debt securities are to be issuable in the form of certificated securities (as described below) or global securities (as described below);
the portion of principal amount that will be payable upon declaration of acceleration of the maturity date if other than the principal amount of the debt securities;
the currency of denomination;
the designation of the currency, currencies or currency units in which payment of principal and, if applicable, premium and interest, will be made;
if payments of principal and, if applicable, premium or interest, on the debt securities are to be made in one or more currencies or currency units other than the currency of denomination, the manner in which the exchange rate with respect to such payments will be determined;
if amounts of principal and, if applicable, premium and interest may be determined by reference to an index based on a currency or currencies or by reference to a commodity, commodity index, stock exchange index or financial index, then the manner in which such amounts will be determined;
the provisions, if any, relating to any collateral provided for such debt securities;
any addition to or change in the covenants and/or the acceleration provisions described in this prospectus or in the indenture;
any events of default, if not otherwise described below under “Events of Default”;
the terms and conditions, if any, for conversion into or exchange for shares of our common stock or preferred stock;
any depositaries, interest rate calculation agents, exchange rate calculation agents or other agents; and
the terms and conditions, if any, upon which the debt securities shall be subordinated in right of payment to our other indebtedness.
We may issue discount debt securities that provide for an amount less than the stated principal amount to be due and payable upon acceleration of the maturity of such debt securities in accordance with the terms of the indenture. We may also issue debt securities in bearer form, with or without coupons. If we issue discount debt securities or debt securities in bearer form, we will describe material U.S. federal income tax considerations and other material special considerations which apply to these debt securitiesname in the applicable prospectus supplement.
We may issue debt securities denominated in or payable in a foreign currency or currencies or a foreign currency unit or units. If we do, we will describe the restrictions, elections, and general tax considerations relating to the debt securities and the foreign currency or currencies or foreign currency unit or units in the applicable prospectus supplement.
Debt securities offered under this prospectus and any prospectus supplement will be subordinated in right of payment to certain of our outstanding senior indebtedness. In addition, we will seek the consent of the holders of any such senior indebtedness prior to issuing any debt securities under this prospectus to the extent required by the agreements evidencing such senior indebtedness.55
Registrar and Paying Agent
The debt securities may be presented for registration of transfer or for exchange at the corporate trust office of the security registrar or at any other office or agency that we maintain for those purposes. In addition, the debt securities may be presented for payment of principal, interest and any premium at the office of the paying agent or at any office or agency that we maintain for those purposes.
Conversion or Exchange Rights
Debt securities may be convertible into or exchangeable for shares of our common stock. The terms and conditions of conversion or exchange will be stated in the applicable prospectus supplement. The terms will include, among others, the following:
the conversion or exchange price;
the conversion or exchange period;
provisions regarding the convertibility or exchangeability of the debt securities, including who may convert or exchange;
events requiring adjustment to the conversion or exchange price;
provisions affecting conversion or exchange in the event of our redemption of the debt securities; and
any anti-dilution provisions, if applicable.
Registered Global Securities
If we decide to issue debt securities in the form of one or more global securities, then we will register the global securities in the name of the depositary for the global securities or the nominee of the depositary, and the global securities will be delivered by the trustee to the depositary for credit to the accounts of the holders of beneficial interests in the debt securities.
The prospectus supplement will describe the specific terms of the depositary arrangement for debt securities of a series that are issued in global form. None of us, the trustee, any payment agent or the security registrar will have any responsibility or liability for any aspect of the records relating to or payments made on account of beneficial ownership interests in a global debt security or for maintaining, supervising or reviewing any records relating to these beneficial ownership interests.
No Protection in the Event of Change of Control
The indenture does not have any covenants or other provisions providing for a put or increased interest or otherwise that would afford holders of our debt securities additional protection in the event of a recapitalization transaction, a change of control or a highly leveraged transaction. If we offer any covenants or provisions of this type with respect to any debt securities covered by this prospectus, we will describe them in the applicable prospectus supplement.
Covenants
Unless otherwise indicated in this prospectus or the applicable prospectus supplement, our debt securities will not have the benefit of any covenants that limit or restrict our business or operations, the pledging of our assets or the incurrence by us of indebtedness. We will describe in the applicable prospectus supplement any material covenants in respect of a series of debt securities.
Merger, Consolidation or Sale of Assets
The form of indenture provides that we will not consolidate with or merge into any other person or convey, transfer, sell or lease our properties and assets substantially as an entirety to any person, unless:
we are the surviving person of such merger or consolidation, or if we are not the surviving person, the person formed by the consolidation or into or with which we are merged or the person to which our properties and assets are conveyed, transferred, sold or leased, is a corporation organized and existing under the laws of the U.S., any state or the District of Columbia or a corporation or comparable legal entity organized under the laws of a foreign jurisdiction and has expressly assumed all of our obligations, including the payment of the principal of and, premium, if any, and interest on the debt securities and the performance of the other covenants under the indenture; and
immediately before and immediately after giving effect to the transaction on a pro forma basis, no event of default, and no event which, after notice or lapse of time or both, would become an event of default, has occurred and is continuing under the indenture.
Events of Default
Unless otherwise specified in the applicable prospectus supplement, the following events will be events of default under the indenture with respect to debt securities of any series:
we fail to pay any principal or premium, if any, when it becomes due;
we fail to pay any interest within 30 days after it becomes due;
we fail to observe or perform any other covenant in the debt securities or the indenture for 60 days after written notice specifying the failure from the trustee or the holders of not less than 25% in aggregate principal amount of the outstanding debt securities of that series; and
certain events involving bankruptcy, insolvency or reorganization of us or any of our significant subsidiaries.
The trustee may withhold notice to the holders of the debt securities of any series of any default, except in payment of principal of or premium, if any, or interest on the debt securities of a series, if the trustee considers it to be in the best interest of the holders of the debt securities of that series to do so.
If an event of default (other than an event of default resulting from certain events of bankruptcy, insolvency or reorganization) occurs, and is continuing, then the trustee or the holders of not less than 25% in aggregate principal amount of the outstanding debt securities of any series may accelerate the maturity of the debt securities. If this happens, the entire principal amount, plus the premium, if any, of all the outstanding debt securities of the affected series plus accrued interest to the date of acceleration will be immediately due and payable. At any time after the acceleration, but before a judgment or decree based on such acceleration is obtained by the trustee, the holders of a majority in aggregate principal amount of outstanding debt securities of such series may rescind and annul such acceleration if:
all events of default (other than nonpayment of accelerated principal, premium or interest) have been cured or waived;
all lawful interest on overdue interest and overdue principal has been paid; and
the rescission would not conflict with any judgment or decree.
In addition, if the acceleration occurs at any time when we have outstanding indebtedness that is senior to the debt securities, the payment of the principal amount of outstanding debt securities may be subordinated in right of payment to the prior payment of any amounts due under the senior indebtedness, in which case the holders of debt securities will be entitled to payment under the terms prescribed in the instruments evidencing the senior indebtedness and the indenture.
If an event of default resulting from certain events of bankruptcy, insolvency or reorganization occurs, the principal, premium and interest amount with respect to all of the debt securities of any series will be due and payable immediately without any declaration or other act on the part of the trustee or the holders of the debt securities of that series.
The holders of a majority in principal amount of the outstanding debt securities of a series will have the right to waive any existing default or compliance with any provision of the indenture or the debt securities of that series and to direct the time, method and place of conducting any proceeding for any remedy available to the trustee, subject to certain limitations specified in the indenture.
No holder of any debt security of a series will have any right to institute any proceeding with respect to the indenture or for any remedy under the indenture, unless:
the holder gives to the trustee written notice of a continuing event of default;
the holders of at least 25% in aggregate principal amount of the outstanding debt securities of the affected series make a written request and offer reasonable indemnity to the trustee to institute a proceeding as trustee;
the trustee fails to institute a proceeding within 60 days after such request; and
the holders of a majority in aggregate principal amount of the outstanding debt securities of the affected series do not give the trustee a direction inconsistent with such request during such 60-day period.
These limitations do not, however, apply to a suit instituted for payment on debt securities of any series on or after the due dates expressed in the debt securities.
We will periodically deliver certificates to the trustee regarding our compliance with our obligations under the indenture.
Modification and Waiver
From time to time, we and the trustee may, without the consent of holders of the debt securities of one or more series, amend the indenture or the debt securities of one or more series, or supplement the indenture, for certain specified purposes, including:
to provide that the surviving entity following a change of control permitted under the indenture will assume all of our obligations under the indenture and debt securities;
to provide for certificated debt securities in addition to uncertificated debt securities;
to comply with any requirements of the SEC under the Trust Indenture Act of 1939;
to provide for the issuance of and establish the form and terms and conditions of debt securities of any series as permitted by the indenture;
to cure any ambiguity, defect or inconsistency, or make any other change that does not materially and adversely affect the rights of any holder; and
to appoint a successor trustee under the indenture with respect to one or more series.
From time to time we and the trustee may, with the consent of holders of at least a majority in principal amount of an outstanding series of debt securities, amend or supplement the indenture or the debt securities series, or waive compliance in a particular instance by us with any provision of the indenture or the debt securities. We may not, however, without the consent of each holder affected by such action, modify or supplement the indenture or the debt securities or waive compliance with any provision of the indenture or the debt securities in order to:
reduce the amount of debt securities whose holders must consent to an amendment, supplement, or waiver to the indenture or such debt security;
reduce the rate of or change the time for payment of interest or reduce the amount of or postpone the date for payment of sinking fund or analogous obligations;
reduce the principal of or change the stated maturity of the debt securities;
make any debt security payable in money other than that stated in the debt security;
change the amount or time of any payment required or reduce the premium payable upon any redemption, or change the time before which no such redemption may be made;
waive a default in the payment of the principal of, premium, if any, or interest on the debt securities or a redemption payment;
waive a redemption payment with respect to any debt securities or change any provision with respect to redemption of debt securities; or
take any other action otherwise prohibited by the indenture to be taken without the consent of each holder affected by the action.
Defeasance of Debt Securities and Certain Covenants in Certain Circumstances
The indenture permits us, at any time, to elect to discharge our obligations with respect to one or more series of debt securities by following certain procedures described in the indenture. These procedures will allow us either:
to defease and be discharged from any and all of our obligations with respect to any debt securities except for the following obligations (which discharge is referred to as “legal defeasance”):
| 1. | to register the transfer or exchange of such debt securities; |
| 2. | to replace temporary or mutilated, destroyed, lost or stolen debt securities; |
| 3. | to compensate and indemnify the trustee; or |
| 4. | to maintain an office or agency in respect of the debt securities and to hold monies for payment in trust; or |
to be released from our obligations with respect to the debt securities under certain covenants contained in the indenture, as well as any additional covenants which may be contained in the applicable supplemental indenture (which release is referred to as “covenant defeasance”).
In order to exercise either defeasance option, we must irrevocably deposit with the trustee or other qualifying trustee, in trust for that purpose:
money;
U.S. Government Obligations (as described below) or Foreign Government Obligations (as described below) that through the scheduled payment of principal and interest in accordance with their terms will provide money; or
a combination of money and/or U.S. Government Obligations and/or Foreign Government Obligations sufficient in the written opinion of a nationally-recognized firm of independent accountants to provide money;
that, in each case specified above, provides a sufficient amount to pay the principal of, premium, if any, and interest, if any, on the debt securities of the series, on the scheduled due dates or on a selected date of redemption in accordance with the terms of the indenture.
In addition, defeasance may be effected only if, among other things:
in the case of either legal or covenant defeasance, we deliver to the trustee an opinion of counsel, as specified in the indenture, stating that as a result of the defeasance neither the trust nor the trustee will be required to register as an investment company under the Investment Company Act of 1940;
in the case of legal defeasance, we deliver to the trustee an opinion of counsel stating that we have received from, or there has been published by, the Internal Revenue Service a ruling to the effect that, or there has been a change in any applicable federal income tax law with the effect that (and the opinion shall confirm that), the holders of outstanding debt securities will not recognize income, gain or loss for U.S. federal income tax purposes solely as a result of such legal defeasance and will be subject to U.S. federal income tax on the same amounts, in the same manner, including as a result of prepayment, and at the same times as would have been the case if legal defeasance had not occurred;
in the case of covenant defeasance, we deliver to the trustee an opinion of counsel to the effect that the holders of the outstanding debt securities will not recognize income, gain or loss for U.S. federal income tax purposes as a result of covenant defeasance and will be subject to U.S. federal income tax on the same amounts, in the same manner and at the same times as would have been the case if covenant defeasance had not occurred; and
certain other conditions described in the indenture are satisfied.
If we fail to comply with our remaining obligations under the indenture and applicable supplemental indenture after a covenant defeasance of the indenture and applicable supplemental indenture, and the debt securities are declared due and payable because of the occurrence of any undefeased event of default, the amount of money and/or U.S. Government Obligations and/or Foreign Government Obligations on deposit with the trustee could be insufficient to pay amounts due under the debt securities of the affected series at the time of acceleration. We will, however, remain liable in respect of these payments.
The term “U.S. Government Obligations” as used in the above discussion means securities that are direct obligations of or non-callable obligations guaranteed by the United States of America for the payment of which obligation or guarantee the full faith and credit of the United States of America is pledged.
The term “Foreign Government Obligations” as used in the above discussion means, with respect to debt securities of any series that are denominated in a currency other than U.S. dollars, (1) direct obligations of the government that issued or caused to be issued such currency for the payment of which obligations its full faith and credit is pledged or (2) obligations of a person controlled or supervised by or acting as an agent or instrumentality of such government the timely payment of which is unconditionally guaranteed as a full faith and credit obligation by that government, which in either case under clauses (1) or (2), are not callable or redeemable at the option of the issuer.
Regarding the Trustee
We will identify the trustee with respect to any series of debt securities in the prospectus supplement relating to the applicable debt securities. You should note that if the trustee becomes a creditor of ours, the indenture and the Trust Indenture Act of 1939 limit the rights of the trustee to obtain payment of claims in certain cases, or to realize on certain property received in respect of any such claim, as security or otherwise. The trustee and its affiliates may engage in, and will be permitted to continue to engage in, other transactions with us and our affiliates. If, however, the trustee acquires any “conflicting interest” within the meaning of the Trust Indenture Act of 1939, it must eliminate such conflict or resign.
The holders of a majority in principal amount of the then outstanding debt securities of any series may direct the time, method and place of conducting any proceeding for exercising any remedy available to the trustee. If an event of default occurs and is continuing, the trustee, in the exercise of its rights and powers, must use the degree of care and skill of a prudent person in the conduct of his or her own affairs. Subject to that provision, the trustee will be under no obligation to exercise any of its rights or powers under the indenture at the request of any of the holders of the debt securities, unless they have offered to the trustee reasonable indemnity or security.
No Individual Liability of Incorporators, Stockholders, Officers or Directors
Each indenture provides that no incorporator and no past, present or future stockholder, officer or director of our company or any successor corporation in those capacities will have any individual liability for any of our obligations, covenants or agreements under the debt securities or such indenture.
Governing Law
The indentures and the debt securities will be governed by, and construed in accordance with, the laws of the State of New York.
DESCRIPTION OF WARRANTS
General
We may issue warrants for the purchase of our debt securities, preferred stock, common stock, or any combination thereof. Warrants may be issued independently or together with our debt securities, preferred stock or common stock and may be attached to or separate from any offered securities. Each series of warrants will be issued under a separate warrant agreement to be entered into between us and a bank or trust company, as warrant agent. The warrant agent will act solely as our agent in connection with the warrants. The warrant agent will not have any obligation or relationship of agency or trust for or with any holders or beneficial owners of warrants. This summary of certain provisions of the warrants is not complete. For the terms of a particular series of warrants, you should refer to the prospectus supplement for that series of warrants and the warrant agreement for that particular series.
Debt Warrants
The prospectus supplement relating to a particular issue of warrants to purchase debt securities will describe the terms of the debt warrants, including the following:
the title of the debt warrants;
the offering price for the debt warrants, if any;
the aggregate number of the debt warrants;
the designation and terms of the debt securities, including any conversion rights, purchasable upon exercise of the debt warrants;
if applicable, the date from and after which the debt warrants and any debt securities issued with them will be separately transferable;
the principal amount of debt securities that may be purchased upon exercise of a debt warrant and the exercise price for the warrants, which may be payable in cash, securities or other property;
the dates on which the right to exercise the debt warrants will commence and expire;
if applicable, the minimum or maximum amount of the debt warrants that may be exercised at any one time;
whether the debt warrants represented by the debt warrant certificates or debt securities that may be issued upon exercise of the debt warrants will be issued in registered or bearer form;
information with respect to book-entry procedures, if any;
the currency or currency units in which the offering price, if any, and the exercise price are payable;
if applicable, a discussion of material U.S. federal income tax considerations;
the antidilution provisions of the debt warrants, if any;
the redemption or call provisions, if any, applicable to the debt warrants;
any provisions with respect to the holder’s right to require us to repurchase the debt warrants upon a change in control or similar event; and
any additional terms of the debt warrants, including procedures and limitations relating to the exchange, exercise, and settlement of the debt warrants.
Debt warrant certificates will be exchangeable for new debt warrant certificates of different denominations. Debt warrants may be exercised at the corporate trust office of the warrant agent or any other office indicated in
the prospectus supplement. Prior to the exercise of their debt warrants, holders of debt warrants will not have any of the rights of holders of the debt securities purchasable upon exercise and will not be entitled to payment of principal or any premium, if any, or interest on the debt securities purchasable upon exercise.
Equity Warrants
The prospectus supplement relating to a particular series of warrants to purchase our common stock or preferred stock will describe the terms of the warrants, including the following:
the title of the warrants;
the offering price for the warrants, if any;
the aggregate number of warrants;
the designation and terms of the common stock or preferred stock that may be purchased upon exercise of the warrants;
if applicable, the designation and terms of the securities with which the warrants are issued and the number of warrants issued with each security;
if applicable, the date from and after which the warrants and any securities issued with the warrants will be separately transferable;
the number of shares of common stock or preferred stock that may be purchased upon exercise of a warrant and the exercise price for the warrants;
the dates on which the right to exercise the warrants shall commence and expire;
if applicable, the minimum or maximum amount of the warrants that may be exercised at any one time;
the currency or currency units in which the offering price, if any, and the exercise price are payable;
if applicable, a discussion of material U.S. federal income tax considerations;
the antidilution provisions of the warrants, if any;
the redemption or call provisions, if any, applicable to the warrants;
any provisions with respect to a holder’s right to require us to repurchase the warrants upon a change in control or similar event; and
any additional terms of the warrants, including procedures and limitations relating to the exchange, exercise and settlement of the warrants.
Holders of equity warrants will not be entitled:
to vote, consent, or receive dividends;
receive notice as stockholders with respect to any meeting of stockholders for the election of our directors or any other matter; or
exercise any rights as stockholders.
DESCRIPTION OF SUBSCRIPTION RIGHTS
We may issue subscription rights to purchase our common stock, preferred stock or debt securities. These subscription rights may be offered independently or together with any other security offered hereby and may or may not be transferable by the stockholder receiving the subscription rights in such offering. In connection with any offering of subscription rights, we may enter into a standby arrangement with one or more underwriters or other purchasers pursuant to which the underwriters or other purchasers may be required to purchase any securities remaining unsubscribed for after such offering.
The prospectus supplement relating to any subscription rights we offer, if any, will, to the extent applicable, include specific terms relating to the offering, including some or all of the following:
the price, if any, for the subscription rights;
the exercise price payable for our common stock, preferred stock or debt securities upon the exercise of the subscription rights;
the number of subscription rights to be issued to each stockholder;
the number and terms of our common stock, preferred stock or debt securities which may be purchased per each subscription right;
the extent to which the subscription rights are transferable;
any other terms of the subscription rights, including the terms, procedures and limitations relating to the exchange and exercise of the subscription rights;
the date on which the right to exercise the subscription rights shall commence, and the date on which the subscription rights shall expire;
the extent to which the subscription rights may include an over-subscription privilege with respect to unsubscribed securities or an over-allotment privilege to the extent the securities are fully subscribed; and
if applicable, the material terms of any standby underwriting or purchase arrangement which may be entered into by us in connection with the offering of subscription rights.
The description in the applicable prospectus supplement of any subscription rights we offer will not necessarily be complete and will be qualified in its entirety by reference to the applicable subscription rights certificate, which will be filed with the SEC if we offer subscription rights. We urge you to read the applicable subscription rights certificate and any applicable prospectus supplement in their entirety.
DESCRIPTION OF UNITS
We may issue units consisting of some or all of the securities described above, in any combination, including common stock, preferred stock, warrants and/or debt securities. The terms of these units will be set forth in a prospectus supplement. The description of the terms of these units in the related prospectus supplement will not be complete. You should refer to the applicable form of unit and unit agreement for complete information with respect to these units.
LEGAL MATTERS
Fenwick & West LLP, Mountain View, California, will issue an opinion about certain legal matters with respect to the securities. Any underwriters or agents will be advised about legal matters relating to any offering by their own counsel.
EXPERTS
The financial statements incorporated in this Prospectusprospectus by reference to the Annual Report on Form 10-K for the year ended December 31, 20162022 have been so incorporated in reliance on the report (which contains an explanatory paragraph relating to the Company’s ability to continue as a going concern as described in Note 1 to the financial statements) of PricewaterhouseCoopers LLP, an independent registered public accounting firm, given on the authority of said firm as experts in auditing and accounting.
The information in this prospectus and the accompanying prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus and accompanying prospectus are not an offer to sell these securities and they are not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
Subject to completion, dated May 1, 201756
PROSPECTUS
$20,000,000
COMMON STOCK
We have entered into a Capital on Demand™ Sales Agreement, or sales agreement, with JonesTrading Institutional Services LLC, or JonesTrading, relating to the sale of shares of our common stock offered by this prospectus and the accompanying prospectus. In accordance with the terms of the sales agreement, we may offer and sell shares of our common stock having an aggregate offering price of up to $20.0 million from time to time through JonesTrading acting as sales agent, at our discretion.
Our common stock is listed for trading on The Nasdaq Global Market under the symbol “AGLE.” On April 28, 2017, the last reported sale price of our common stock on The Nasdaq Global Market was $7.15 per share.
Sales of our common stock, if any, under this prospectus may be made in sales deemed to be “at the market offerings” as defined in Rule 415 promulgated under the Securities Act of 1933, as amended, or the Securities Act. There is no arrangement for funds to be received in any escrow, trust or similar arrangement.
We will pay JonesTrading commissions for its services in acting as agent in the sale of our common stock. JonesTrading will be entitled to compensation at a commission rate equal to up to 3.0% of the aggregate gross sales price of the shares sold. In connection with the sale of our common stock on our behalf, JonesTrading will be deemed to be an “underwriter” within the meaning of the Securities Act and the compensation of JonesTrading will be deemed to be underwriting commissions or discounts. We have also agreed to provide indemnification and contribution to JonesTrading with respect to certain liabilities, including liabilities under the Securities Act.
Investing in our common stock involves significant risks. Before buying shares of our common stock, you should carefully consider the risks described under the caption “Risk Factors” beginning on page S-5 of this prospectus and under similar headings in the accompanying prospectus and the documents incorporated by reference into this prospectus.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the accuracy or adequacy of this prospectus or the accompanying prospectus. Any representation to the contrary is a criminal offense.

The date of this prospectus is , 2017
Table Of Contents
Prospectus
About this Prospectus
This document is in two parts. The first part is this prospectus, including the documents incorporated by reference herein, which describes the specific terms of this offering and updates the information contained in the accompanying prospectus. The second part, the accompanying prospectus, including the documents incorporated by reference therein, provides general information. This prospectus and the accompanying prospectus are part of a registration statement that we filed with the Securities and Exchange Commission, or the SEC, utilizing a “shelf” registration process. Under this shelf registration process, we may from time to time sell shares of our common stock having an aggregate offering price of up to $150,000,000 under this prospectus at prices and on terms to be determined by market conditions at the time of the offering.
Before buying any of the common stock that we are offering, we urge you to carefully read this prospectus the accompanying prospectus and any free writing prospectus and all of the information incorporated by reference herein and therein, as well as the additional information described under the headings “Where You Can Find More Information” and “Incorporation of Documents by Reference.” These documents contain important information that you should consider when making your investment decision.
To the extent there is a conflict between the information contained in this prospectus, on the one hand, and the information contained in the accompanying prospectus or any document incorporated by reference herein filed prior to the date of this prospectus, on the other hand, you should rely on the information in this prospectus. If any statement in one of these documents is inconsistent with a statement in another document having a later date (for example, a document incorporated by reference in this prospectus), the statement in the document having the later date modifies or supersedes the earlier statement.
You should rely only on the information contained in or incorporated by reference in this prospectus, the accompanying prospectus, and any related free writing prospectus filed by us with the SEC. We have not, and JonesTrading has not, authorized anyone to provide you with different information. If anyone provides you with different or inconsistent information, you should not rely on it. This prospectus does not constitute an offer to sell or the solicitation of an offer to buy any securities other than the securities described in this prospectus or an offer to sell or the solicitation of an offer to buy such securities in any circumstances in which such offer or solicitation is unlawful. You should assume that the information appearing in this prospectus, the accompany prospectus, the documents incorporated by reference herein and any related free writing prospectus is accurate only as of their respective dates. Our business, financial condition, results of operations and prospects may have changed materially since those dates.
We further note that the representations, warranties and covenants made by us in any agreement that is filed as an exhibit to any document that is incorporated by reference in this prospectus and/or the accompanying prospectus were made solely for the benefit of the parties to such agreement, including, in some cases, for the purpose of allocating risk among the parties to such agreements, and should not be deemed to be a representation, warranty or covenant to you. Moreover, such representations, warranties or covenants were accurate only as of the date when made. Accordingly, such representations, warranties and covenants should not be relied on as accurately representing the current state of our affairs.
Unless the context indicates otherwise, as used in this prospectus, the terms “Company,” “Aeglea,” “Registrant,” “we,” “us” and “our” refer to Aeglea BioTherapeutics, Inc., a Delaware corporation, and its subsidiaries, taken as a whole, unless otherwise noted. “Aeglea” and all product candidate names are our common law trademarks. This prospectus and the information incorporated herein by reference contains additional trade names, trademarks and service marks of other companies, which are the property of their respective owners. We do not intend our use or display of other companies’ trade names, trademarks or service marks to imply a relationship with, or endorsement or sponsorship of us by, these other companies.
Forward-Looking Statements
This prospectus and documents incorporated herein by reference contain “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. These forward-looking statements involve a number of risks and uncertainties. We caution readers that any forward-looking statement is not a guarantee of future performance and that actual results could differ materially from those contained in the forward-looking statement. These statements are based on current expectations of future events. Such statements include, but are not limited to, statements about future financial and operating results, plans, objectives, expectations and intentions, costs and expenses, outcome of contingencies, financial condition, results of operations, liquidity, cost savings, objectives of management, business strategies, debt financing, timing and plans for our nonclinical studies and clinical trials, the achievement of clinical and commercial milestones, the advancement of our technologies and our proprietary product candidates, and other statements that are not historical facts. You can find many of these statements by looking for words like “believes,” “expects,” “anticipates,” “estimates,” “may,” “might,” “should,” “will,” “could,” “plan,” “intend,” “project,” “seek” or similar expressions in this prospectus, the documents incorporated by reference into this prospectus and any free writing prospectus. We intend that such forward-looking statements be subject to the safe harbors created thereby.
These forward-looking statements are based on the current beliefs and expectations of our management and are subject to significant risks and uncertainties. If underlying assumptions prove inaccurate or unknown risks or uncertainties materialize, actual results may differ materially from current expectations and projections. Factors that might cause such a difference include those discussed in Part I, Item 1A “Risk Factors” in our Annual Report on Form 10-K for the year ended December 31, 2016, as well as those discussed in this prospectus, the documents incorporated by reference into this prospectus and any free writing prospectus. You are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date of this prospectus or, in the case of documents referred to or incorporated by reference, the date of those documents.
All subsequent written or oral forward-looking statements attributable to us or any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section. We do not undertake any obligation to release publicly any revisions to these forward-looking statements to reflect events or circumstances after the date of this prospectus or to reflect the occurrence of unanticipated events, except as may be required under applicable U.S. securities law. If we do update one or more forward-looking statements, no inference should be drawn that we will make additional updates with respect to those or other forward-looking statements.
Prospectus SummaryWHERE YOU CAN FIND MORE INFORMATION
This summary highlights selected information contained elsewhere in this prospectus and the accompanying prospectus. It does not contain all of the information you should consider before making an investment decision. Before you decide to invest in our common stock, you should carefully read this entire prospectus and the accompanying prospectus, including the risk factors and the financial statements and related notes included or incorporated by reference herein and therein.
Overview
We are a biotechnology company committed to developing enzyme-based therapeutics in the field of amino acid metabolism to treat rare genetic diseases and cancer. Our engineered human enzymes are designed to degrade specific amino acids in the blood to target these diseases. In inborn errors of metabolism, or IEM, a subset of rare genetic diseases, we are seeking to reduce the toxic levels of amino acids in patients to the normal range. In oncology, we are seeking to reduce amino acid blood levels below the normal range where we believe we will be able to exploit the dependence of certain cancers on specific amino acids.
Our lead product candidate, AEB1102 (pegzilarginase), is engineered to degrade the amino acid arginine and is being developed to treat two extremes of arginine metabolism, including arginine excess in patients with Arginase I deficiency, an IEM, as well as some cancers which have been shown to have a metabolic dependence on arginine. AEB1102 has demonstrated clinical proof of mechanism in both scenarios. In a Phase 1 clinical trial for the treatment of patients with Arginase I deficiency, a dose-proportional reduction in plasma arginine levels was observed in two patients. A reduction in blood arginine levels was also observed in Phase 1 clinical trials for the treatment of patients with advanced solid tumors and the hematological malignancies relapsed refractory acute myeloid leukemia, or AML, and myelodysplastic syndrome, or MDS. These preliminary results support its potential use as a therapeutic of both Arginase I deficiency and certain cancers associated with abnormal amino acid metabolism.
Company Information
We were formed as a limited liability company under the laws of the State of Delaware in December 2013 and converted to a Delaware corporation in March 2015. Our principal executive offices are located at 901 S. MoPac Expressway, Barton Oaks Plaza One, Suite 250, Austin, Texas 78746, and our telephone number is (512) 942-2935. Our website address is www.aegleabio.com. The information contained on, or that can be accessed through, our website is not part of this prospectus, and you should not consider information on our website to be part of this prospectus.
The Offering
Issuer
| Aeglea BioTherapeutics, Inc. |
Common stock offered by us
| Shares of our common stock having an aggregate offering price of up to $20.0 million. |
Manner of offering
| Sales of our common stock, if any, under this prospectus may be made in sales deemed to be “at the market offerings” as defined in Rule 415 promulgated under the Securities Act. See “Plan of Distribution” on page S-10 of this prospectus. |
Use of proceeds
| We intend to use the net proceeds of this offering primarily to fund research and development of our product candidates, working capital, capital expenditures and other general corporate purposes. See “Use of Proceeds” on page S-7 of this prospectus. |
Risk factors
| Investing in our common stock involves significant risks. See “Risk Factors” beginning on page S-5 of this prospectus, as well as the other information included in or incorporated by reference in this prospectus and the accompanying prospectus, for a discussion of risks you should carefully consider before investing in our securities. |
Nasdaq Global Market listing
| Our common stock is listed on The Nasdaq Global Market under the symbol “AGLE.” |
Risk Factors
An investment in our common stock involves a high degree of risk. Before deciding whether to invest in our common stock, you should consider carefully the risks described below and discussed under the section captioned “Risk Factors” in Part I, Item 1A of our Annual Report on Form 10-K for the year ended December 31, 2016, which is incorporated by reference in this prospectus and the accompanying prospectus in its entirety, as well as any amendment or update to our risk factors reflected in subsequent filings with the SEC, together with other information in this prospectus, the accompanying prospectus, and the information and documents incorporated by reference that we have authorized for use in connection with this offering. The risks and uncertainties described below and incorporated by reference herein are not the only ones we face. Additional risks and uncertainties that we are unaware of or that we deem immaterial may also become important factors that adversely affect our business. If any of these risks actually occur, our business, financial condition, results of operations or cash flows could be seriously harmed. This could cause the trading price of our common stock to decline, resulting in a loss of all or part of your investment.
Risks Related to Our Common Stock and This Offering
The price of our common stock may be volatile and fluctuate substantially, which could result in substantial losses for purchasers of our common stock.
Our stock price is volatile. The stock market in general and the market for smaller biotechnology companies in particular have experienced extreme volatility that has often been unrelated or disproportionate to the operating performance of those companies. The market price for our common stock may be influenced by many factors, including:
the success or failure of competitive products or technologies;
results of ongoing or planned clinical trials of our product candidates or those of our competitors;
regulatory or legal developments in the United States and other countries;
developments or disputes concerning patent applications, issued patents or other proprietary rights;
the recruitment or departure of key personnel;
the level of expenses related to any of our product candidates or clinical development programs;
the results of our efforts to discover, develop, acquire or in-license additional product candidates or products;
actual or anticipated changes in estimates as to financial results, development timelines or recommendations by securities analysts;
operating results that fail to meet expectations of securities analysts that cover our company;
variations in our financial results or those of companies that are perceived to be similar to us;
changes in the structure of healthcare payment systems;
market conditions in the pharmaceutical and biotechnology sectors;
general economic and market conditions; and
the other factors described in this “Risk Factors” section and in the documents incorporated by reference herein.
If securities or industry analysts do not continue to publish research or reports about our business, or publish negative reports about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. We do not have any control over these analysts. There can be
no assurance that analysts will continue to cover us or provide favorable coverage. If one or more of the analysts who cover us downgrade our stock or change their opinion of our stock, our stock price would likely decline. If one or more of these analysts cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause our stock price or trading volume to decline.
We have broad discretion in the use of the net proceeds from this offering and may not use them effectively.
Our management has broad discretion in the application of the net proceeds from this offering, and you will not have the opportunity as part of your investment decision to assess whether the net proceeds are being used appropriately. Our management could spend the net proceeds from this offering in ways that do not improve our results of operations or enhance the value of our common stock. The failure by our management to apply these funds effectively could result in financial losses that could have a material adverse effect on our business, cause the price of our common stock to decline and delay the development of our product candidates. Pending their use, we may invest the net proceeds from this offering in a manner that does not produce income or that loses value.
If you purchase shares of common stock sold in this offering, you will incur immediate and substantial dilution.
If you purchase shares of our common stock in this offering, you will experience substantial and immediate dilution in the pro forma net tangible book value per share after giving effect to this offering, based on the assumed public offering price of $7.15 per share, which is the last reported sale price of our common stock on The NASDAQ Global Select Market on April 28, 2017, because the price that you pay will be substantially greater than the pro forma net tangible book value per share of the common stock that you acquire. This dilution is due in large part to the fact that certain of our earlier investors paid substantially less than the offering price when they purchased shares of our capital stock. You will experience additional dilution upon exercise of the outstanding stock options and other equity awards that may be granted under our equity incentive plans, and when we otherwise issue additional shares of our common stock. For more information, see “Dilution.”
Since we do not anticipate paying any cash dividends on our capital stock in the foreseeable future, stock price appreciation, if any, will be your sole source of gain.
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all of our future earnings, if any, to finance the growth and development of our business. In addition, the terms of any future debt agreements may preclude us from paying dividends. As a result, appreciation, if any, in the market price of our common stock will be your sole source of gain for the foreseeable future.
The actual number of shares we will issue under the sales agreement, at any one time or in total, is uncertain.
Subject to certain limitations in the sales agreement and compliance with applicable law, we have the discretion to deliver a placement notice to JonesTrading at any time throughout the term of the sales agreement. The number of shares that are sold by JonesTrading after delivering a placement notice will fluctuate based on the market price of the common shares during the sales period and limits we set with JonesTrading. Because the price per share of each share sold will fluctuate based on the market price of our common stock during the sales period, it is not possible at this stage to predict the number of shares that will be ultimately issued.
The common stock offered hereby will be sold in “at the market offerings”, and investors who buy shares at different times will likely pay different prices.
Investors who purchase shares in this offering at different times will likely pay different prices, and so may experience different outcomes in their investment results. We will have discretion, subject to market demand, to vary the timing, prices, and numbers of shares sold, and there is no minimum or maximum sales price. Investors may experience a decline in the value of their shares as a result of share sales made at prices lower than the prices they paid.
Use of Proceeds
We may issue and sell shares of our common stock having aggregate sale proceeds of up to $20,000,000 from time to time. There can be no assurance that we will be able to sell any shares under or fully utilize the sales agreement with JonesTrading as a source of financing. Because there is no minimum offering amount required as a condition to close this offering, the actual total public offering amount, commissions and proceeds to us, if any, are not determinable at this time. We currently intend to use any net proceeds from the sale of securities under this prospectus primarily to fund research and development of our product candidates, working capital, capital expenditures and other general corporate purposes. Additionally, we may use a portion of the net proceeds from this offering to expand our current business by in-licensing or acquiring, as the case may be, commercial products, product candidates, technologies, compounds, other assets or complementary businesses, using cash or shares of our common stock. However, we have no current commitments or obligations to do so.
The amounts and timing of our actual expenditures will depend on numerous factors, including the progress of our clinical trials and other development efforts and other factors described under “Risk Factors” in this prospectus and the documents incorporated by reference herein, as well as the amount of cash used in our operations. As a result, our management will have broad discretion over the uses of the net proceeds, if any, we receive in connection with securities offered pursuant to this prospectus and investors will be relying on the judgment of our management regarding the application of the proceeds. Pending these uses, we intend to invest the net proceeds in short-term, interest-bearing obligations, investment-grade instruments, certificates of deposit or guaranteed obligations of the U.S. government.
Dilution
If you invest in our common stock, your ownership interest will be diluted to the extent of the difference between the public offering price per share of our common stock and the as adjusted net tangible book value per share of our common stock immediately after this offering.
As of December 31, 2016, our net tangible book value was $63.0 million, or $4.69 per share of common stock. Net tangible book value per share represents the amount of our tangible assets less our liabilities divided by the total number of shares of our common stock outstanding.
Our as adjusted net tangible book value as of December 31, 2016 would be $82.1 million, or $5.06 per share of common stock. As adjusted net tangible book value per share reflects the sale by us of shares of our common stock in the full aggregate amount of $20,000,000 in this offering, at an assumed offering price of $7.15 per share, which was the last reported sale price of our common stock on The NASDAQ Global Market on April 28, 2017, after deducting the estimated commissions and estimated offering expenses payable by us. This represents an immediate increase in as adjusted net tangible book value of $0.37 per share to existing stockholders and immediate dilution of $2.09 per share to new investors purchasing shares in the offering.
The following table illustrates this per share dilution to new investors:
| | | | | | | | |
Assumed offering price per share | | | | | | $ | 7.15 | |
Net tangible book value per share as of December 31, 2016, before giving effect to this offering | | $ | 4.69 | | | | | |
Increase in as adjusted net tangible book value per share after giving effect to this offering | | | 0.37 | | | | | |
| | | | | | | | |
As adjusted net tangible book value per share, after giving effect to this offering | | | | | | | 5.06 | |
| | | | | | | | |
Dilution per share to investors purchasing shares in this offering | | | | | | $ | 2.09 | |
| | | | | | | | |
The shares sold in this offering, if any, will be sold from time to time at various prices. An increase of $1.00 per share in the price at which the shares are sold from the assumed offering price of $7.15 per share shown in the table above, assuming all of our common stock in the aggregate amount of $20,000,000 is sold at that price, would increase our as adjusted net tangible book value per share after the offering to $5.23 per share and would increase the dilution in net tangible book value per share to new investors to $2.92 per share, after deducting commissions and estimated aggregate offering expenses payable by us. A decrease of $1.00 per share in the price at which the shares are sold from the assumed offering price of $7.15 per share shown in the table above, assuming all of our common stock in the aggregate amount of $20,000,000 is sold at that price, would decrease our as adjusted net tangible book value per share after the offering to $4.89 per share and would decrease the dilution in net tangible book value per share to new investors to $1.26 per share, after deducting commissions and estimated aggregate offering expenses payable by us. This information is supplied for illustrative purposes only.
To the extent that outstanding options are exercised or outstanding restricted stock units vest, investors purchasing our common stock in this offering will experience further dilution. In addition, we may choose to raise additional capital due to market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the issuance of these securities could result in further dilution to our stockholders.
Information in the above table is based on 13,430,833 shares outstanding on December 31, 2016, and excludes:
1,146,935 shares of common stock issuable upon exercise of options outstanding as of December 31, 2016 at a weighted average exercise price of $5.64 per share; and
1,323,138 shares of common stock reserved and available for future issuance as of December 31, 2016 under our equity incentives plans, consisting of (1) 1,177,199 shares of common stock reserved and available for issuance under our 2016 Equity Incentive Plan and (2) 145,939 shares of common stock reserved for issuance under our 2016 Employee Stock Purchase Plan.
Plan of Distribution
We have entered into a Capital on DemandTM Sales Agreement, or sales agreement, with JonesTrading, under which we may issue and sell shares of our common stock having an aggregate gross sales price of up to $20,000,000 from time to time through JonesTrading acting as our sales agent. Sales of our common stock, if any, under this prospectus may be made in sales deemed to be “at the market offerings” as defined in Rule 415 promulgated under the Securities Act.
Each time we wish to issue and sell common stock, we will notify JonesTrading of the number of shares to be issued, the dates on which such sales are anticipated to be made, any minimum price below which sales may not be made and other sales parameters as we deem appropriate. Once we have so instructed JonesTrading, unless JonesTrading declines to accept the terms of the notice, JonesTrading has agreed, subject to the terms and conditions of the Sales Agreement, to use its commercially reasonable efforts consistent with its normal trading and sales practices to sell such shares up to the amount specified on such terms. We may instruct JonesTrading not to sell shares of common stock if the sales cannot be effected at or above the price designated by us in any such instruction. We or JonesTrading may suspend the offering of shares of common stock being made through JonesTrading under the sales agreement upon proper notice to the other party.
We will pay JonesTrading commissions for its services in acting as agent in the sale of our common stock. JonesTrading will be entitled to compensation at a commission rate equal to up to 3.0% of the aggregate gross sales price of the shares sold. JonesTrading may effect sales to or through dealers, and such dealers may receive compensation in the form of discounts, concessions or commissions from JonesTrading and/or purchasers of shares of common stock for whom they may act as agents or to whom they may sell as principal. Because there is no minimum offering amount required as a condition to close this offering, the actual total public offering amount, commissions and proceeds to us, if any, are not determinable at this time. We have also agreed to reimburse JonesTrading for certain specified expenses, including the fees and disbursements of its legal counsel in an amount not to exceed $35,000, as provided in the sales agreement. We estimate that the total expenses for the offering, excluding compensation and reimbursements payable to JonesTrading under the terms of the sales agreement, will be approximately $195,000.
Settlement for sales of common stock will occur on the third business day, or such shorter settlement cycle as may be in effect under Exchange Act Rule 15c6-1 from time to time, following the date on which any sales are made, or on some other date that is agreed upon by us and JonesTrading in connection with a particular transaction, in return for payment of the net proceeds to us. Sales of our common stock as contemplated in this prospectus will be settled through the facilities of our transfer agent, American Stock Transfer & Trust Company, LLC or by such other means as we and JonesTrading may agree upon. There is no arrangement for funds to be received in an escrow, trust or similar arrangement.
In connection with the sale of the common stock on our behalf, JonesTrading may be deemed to be an “underwriter” within the meaning of the Securities Act and the compensation of JonesTrading may be deemed to be underwriting commissions or discounts. We have agreed to provide indemnification and contribution to JonesTrading against certain civil liabilities, including liabilities under the Securities Act.
The offering of our common stock pursuant to the Sales Agreement will terminate upon the earlier of (i) the issuance and sale of all shares of our common stock subject to the sales agreement, or (ii) the termination of such sales agreement as permitted therein.
JonesTrading and its affiliates may in the future provide various investment banking and other financial services for us and our affiliates for which services they may in the future receive customary fees.
This summary of the material provisions of the sales agreement does not purport to be a complete statement of its terms and conditions. We are filing a copy of the sales agreement with this prospectus.
Legal Matters
The validity of the common stock offered hereby and certain legal matters in connection with this offering will be passed upon by Fenwick & West LLP, Mountain View, California. Duane Morris LLP, Newark, New Jersey, is counsel to JonesTrading in connection with this offering.
Experts
The financial statements incorporated in this prospectus by reference to the Annual Report on Form 10-K for the year ended December 31, 2016 have been so incorporated in reliance on the report of PricewaterhouseCoopers LLP, an independent registered public accounting firm, given on the authority of said firm as experts in auditing and accounting.
Where You Can Find More Information
We have filed with the SEC a registration statement on Form S-3 under the Securities Act with respect to the shares of common stock offered hereby. This prospectus, which constitutes a part of the registration statement, does not contain all of the information set forth in the registration statement or the exhibits filed therewith. For further information about us and the common stock offered hereby, reference is made to the accompanying prospectus and registration statement of which it is a part and the exhibits filed therewith. Statements contained in this prospectus regarding the contents of any contract or any other document that is filed as an exhibit to the accompanying prospectus and the registration statement of which it is a part are not necessarily complete, and in each instance we refer you to the copy of such contract or other document filed as an exhibit to the registration statement or the exhibits to the reports or other documents incorporated by reference in this prospectus for a copy of such contract or other document.
We are subject to the informational requirements of the Securities Exchange Act of 1934, as amended, or the Exchange Act and are required to file annual, quarterly and other reports, proxy statements and other information with the SEC. You may inspect and copy these reports, proxy statements and other information at the public reference facilities maintained by the SEC in Washington, D.C., 100 F Street N.E., Washington, D.C. 20549. Copies of such materials can be obtained from the SEC’s public reference section at prescribed rates. You may obtain information on the operation of the public reference rooms by calling the SEC at (800) SEC-0330. Additionally, theThe SEC maintains an Internet site (http:(http://www.sec.gov)www.sec.gov) that contains reports, proxy and information statements, and various other information about us. You may also inspect the documents described herein at our principal executive offices, 901 S. MoPac Expressway, Barton Oaks Plaza One, Suite 250, Austin, TX 78746, during normal business hours.
Information about us is also available at our website at http://www.aegleabio.com.www.aeglea.com. However, the information on our website is not a part of this prospectus and is not incorporated by reference into this prospectus.
Incorporation of Documents by Reference
The SEC allows us to “incorporate by reference” information that we file with the SEC, which means that we can disclose important information to you by referring you to those other documents. The information incorporated by reference is an important part of this prospectus and the accompanying prospectus, and information we file later with the SEC will automatically update and supersede this information. A Current Report (or portion thereof) furnished, but not filed, on Form 8-K shall not be incorporated by reference into this prospectus and the accompanying prospectus. We incorporate by reference the documents listed below and any57
future filings we make with the SEC under Section 13(a), 13(c), 14, or 15(d) of the Exchange Act prior to the termination of any offering of securities made by this prospectus and accompanying prospectus:
our Annual Report on Form 10-K for the fiscal year ended December 31, 2016 filed with the SEC on March 23, 2017, including certain information incorporated by reference therein from our Definitive Proxy Statement for our 2017 annual meeting of stockholders filed with the SEC on April 21, 2017;
our Current Report on Form 8-K filed on February 16, 2017; and
the description of our common stock contained in our registration statement on Form 8-A filed with the SEC on March 28, 2016 under Section 12 of the Exchange Act, including any amendment or report filed for the purpose of updating such description.
We will furnish without charge to you, on written or oral request, a copy of any or all of such documents that has been incorporated herein by reference (other than exhibits to such documents unless such exhibits are specifically incorporated by reference into the documents that this prospectus incorporates). Written or oral requests for copies should be directed Aeglea BioTherapeutics, Inc., Attn: Investor Relations, 901 S. MoPac Expressway, Barton Oaks Plaza One, Suite 250, Austin, Texas 78746, telephone number (512) 942-2935. See the section of this prospectus entitled “Where You Can Find More Information” for information concerning how to read and obtain copies of materials that we file with the SEC at the SEC’s public offices.
Any statement contained in this prospectus, or in a document all or a portion of which is incorporated by reference, shall be modified or superseded for purposes of this prospectus to the extent that a statement contained in this prospectus or any document incorporated by reference modifies or supersedes such statement. Any such statement so modified or superseded shall not, except as so modified or superseded, constitute a part of this prospectus.
$20,000,000
Common Stock
PROSPECTUS

, 2017
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
| Item 14. | Item 14. Other Expenses of Issuance and Distribution
|
The following table sets forth estimated expenses payable by us in connection with the issuance and distribution of the securities being registered:registered hereunder on Form S-3 (other than underwriting discounts and commissions, if any) are set forth below. The Selling Stockholders will not bear any portion of such expenses. Each item listed is estimated, except for the SEC registration fee:
| | | | |
SEC registration fee | | $ | 17,385 | |
FINRA filing fee | | | 23,000 | |
Printing and engraving* | | | * | |
Legal fees and expenses* | | | * | |
Accounting fees and expenses* | | | * | |
Transfer agent and registrar fees and expenses* | | | * | |
Miscellaneous expenses* | | | * | |
| | | | |
Total* | | $ | * | |
| | | | |
| | | | |
SEC registration fee | | $ | 60,570 | |
Printing and engraving | | | 25,000 | |
Legal fees and expenses | | | 50,000 | |
Accounting fees and expenses | | | 50,000 | |
Transfer agent and registrar fees and expenses | | | 0 | |
Miscellaneous expenses | | | 0 | |
| | | | |
Total | | $ | 185,570 | |
| | | | |
* | These fees are calculated based on the type of securities offered and the number of issuances and accordingly, cannot be estimated at this time. An estimate of the aggregate expenses in connection with the sale and distribution of securities being offered will be included in the applicable prospectus supplement. |
| Item 15. | Item 15. Indemnification of Officers and Directors
|
Section 145 of the Delaware General Corporation LawDGCL authorizes a court to award, or a corporation’s board of directors to grant, indemnity to directors and officers under certain circumstances and subject to certain limitations. The terms of Section 145 of the Delaware General Corporation LawDGCL are sufficiently broad to permit indemnification under certain circumstances for liabilities, including reimbursement of expenses incurred, arising under the Securities Act of 1933, as amended, or the Securities Act.
As permitted by the Delaware General Corporation Law,DGCL, the Registrant’s restated certificateCertificate of incorporationIncorporation contains provisions that eliminate the personal liability of its directors for monetary damages for any breach of fiduciary duties as a director, except liability for the following:
any breach of the director’s duty of loyalty to the Registrant or its stockholders;
acts or omissions not in good faith or that involve intentional misconduct or a knowing violation of law;
under Section 174 of the Delaware General Corporation LawDGCL (regarding unlawful dividends and stock purchases); or
any transaction from which the director derived an improper personal benefit.
As permitted by the Delaware General Corporation Law,DGCL, the Registrant’s restated bylawsBylaws provide that:
the Registrant is required to indemnify its directors and executive officers to the fullest extent permitted by the Delaware General Corporation Law,DGCL, subject to very limited exceptions;
the Registrant may indemnify its other employees and agents as set forth in the Delaware General Corporation Law;DGCL;
the Registrant is required to advance expenses, as incurred, to its directors and executive officers in connection with a legal proceeding to the fullest extent permitted by the Delaware General Corporation Law,DGCL, subject to very limited exceptions; and
the rights conferred in the restated bylawsBylaws are not exclusive.
II-1
The Registrant has entered, and intends to continue to enter, into separate indemnification agreements with its directors and executive officers to provide these directors and executive officers additional contractual assurances regarding the scope of the indemnification set forth in the Registrant’s restated certificateCertificate of incorporationIncorporation and restated bylawsBylaws and to provide additional procedural protections. At present, there is no pending litigation or
II-1
proceeding involving a director or executive officer of the Registrant regarding which indemnification is sought. The indemnification provisions in the Registrant’s restated certificateCertificate of incorporation, restated bylawsIncorporation, Bylaws and the indemnification agreements entered into or to be entered into between the Registrant and each of its directors and executive officers may be sufficiently broad to permit indemnification of the Registrant’s directors and executive officers for liabilities arising under the Securities Act.
The Registrant currently carries liability insurance for its directors and officers. OneTwo of the Registrant’s directors, Armen Shanafelt, isPeter Harwin and Tomas Kiselak, are also indemnified by histheir employer with regard to histheir service on the Registrant’s board of directors.
Reference is made to the following documents filed as exhibits to this Registration Statement regarding relevant indemnification provisions described above and elsewhere herein:
| | | | |
Exhibit
| | Number | |
Restated Certificate of Incorporation
| | | 3.1 | |
Restated Bylaws
| | | 3.2 | |
Amended and Restated Investors’ Rights Agreement, dated March 10, 2015, by and among the Registrant and certain of its stockholders, as amended
| | | 4.2 | |
Item 16. Exhibits
| | | | | | | | | | | | |
| | | Incorporated by Reference |
Exhibit
Number | | Exhibit Description | | Form | | File No. | | Exhibit | | Filing Date | | Filed
Herewith |
| | | | | | |
| 2.1 | | Agreement and Plan of Merger, dated June 22, 2023, by and among Aeglea BioTherapeutics, Inc. Aspen Merger Sub I, Inc., Sequoia Merger Sub II, LLC and Spyre Therapeutics, Inc. | | 8-K | | 001-37722 | | 2.1 | | 06/23/2023 | | |
| | | | | | |
| 3.1 | | Restated Certificate of Incorporation | | S-1/A | | 333-205001 | | 3.2 | | 9/14/2015 | | |
| | | | | | |
| 3.2 | | Amended and Restated Bylaws | | 8-K | | 001-37722 | | 3.1 | | 12/19/2022 | | |
| | | | | | |
| 3.3 | | Certificate of Designations of Series A Non-Voting Convertible Preferred Stock | | 8-K | | 001-37722 | | 3.1 | | 6/23/2023 | | |
| | | | | | |
| 4.1 | | Securities Purchase Agreement, dated June 22, 2023, by and among the Registrant and the purchasers named therein (incorporated by reference to Exhibit 10.1 to the Registrant’s current report on Form 8-K, filed on June 23, 2023) | | 8-K | | 001-37722 | | 10.1 | | 6/23/2023 | | |
| | | | | | |
| 4.2 | | Form of Common Stock Certificate | | S-1/A | | 333-205001 | | 4.1 | | 9/14/2015 | | |
| | | | | | |
| 4.3 | | Description of the Registrant’s securities | | 10-K | | 001-37722 | | 4.3 | | 3/18/2021 | | |
| | | | | | |
| 4.4 | | Registration Rights Agreement, dated March 16, 2021, by and among the Registrant and Baker Brothers Life Sciences, L.P. and 667, L.P. | | 10-K | | 001-37722 | | 4.5 | | 3/18/2021 | | |
| | | | | | |
| 4.5 | | Form of Registration Rights Agreement, by and among Aeglea BioTherapeutics, Inc. and certain purchasers | | 8-K | | 001-37722 | | 10.2 | | 6/23/2023 | | |
| | | | | | |
| 5.1 | | Opinion of Gibson, Dunn & Crutcher LLP | | | | | | | | | | X |
| | | | | | |
| 10.1* | | Biologics Master Services Agreement, effective June 20, 2022, by and between Paragon Therapeutics, Inc. and WuXi Biologics (Hong Kong) Limited | | | | | | | | | | |
| | | | | | |
| 10.2* | | Cell Line License Agreement, dated as of June 20, 2022, by and between Paragon Therapeutics, Inc. and WuXi Biologics (Hong Kong) Limited | | | | | | | | | | |
The exhibits listed in the accompanying Exhibit Index are filed (except where otherwise indicated) as part of this Registration Statement.II-2
Item 17.
| * | To be filed in an amendment. |
(a) The undersigned Registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) to include any prospectus required by Section 10(a)(3) of the Securities Act of 1933;Act;
(ii) to reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Securities and CommissionSEC pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table“Filing Fee Table” in the effective registration statement; and
(iii) to include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement;
provided, however, that subparagraphs (i),(ii), and (iii) do not apply if the information required to be included in a post-effective amendment by those paragraphs is contained in reports filed with or furnished to the Securities and CommissionSEC by the Registrant pursuant to Section 13 or Section 15(d) of the Securities
II-2
Exchange Act of 1934 that are incorporated by reference in the registration statement, or is contained in a form of prospectus filed pursuant to Rule 424(b) that is part of the registration statement.
(2) That, for the purpose of determining any liability under the Securities Act, of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initialbona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser:
(i) Each prospectus filed by the Registrant pursuant to Rule 424(b)(3) shall be deemed to be part of the registration statement as of the date the filed prospectus was deemed part of and included in the registration statement; and
II-3
(ii) Each prospectus required to be filed pursuant to Rule 424(b)(2), (b)(5), or (b)(7) as part of a registration statement in reliance on Rule 430B relating to an offering made pursuant to Rule 415(a)(1)(i), (vii), or (x) for the purpose of providing the information required by Section 10(a) of the Securities Act of 1933 shall be deemed to be part of and included in the registration statement as of the earlier of the date such form of prospectus is first used after effectiveness or the date of the first contract of sale of securities in the offering described in the prospectus. As provided in Rule 430B, for liability purposes of the issuer and any person that is at that date an underwriter, such date shall be deemed to be a new effective date of the registration statement relating to the securities in the registration statement to which that prospectus relates, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such effective date, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such effective date.
(5)(b) That, for the purpose of determining liability of the Registrant under the Securities Act of 1933 to any purchaser in the initial distribution of the securities, the undersigned Registrant undertakes that in a primary offering of securities of the undersigned Registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned Registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) any preliminary prospectus or prospectus of the undersigned Registrant relating to the offering required to be filed pursuant to Rule 424;
(ii) any free writing prospectus relating to the offering prepared by or on behalf of the undersigned Registrant or used or referred to by the undersigned Registrant;
(iii) the portion of any other free writing prospectus relating to the offering containing material information about the undersigned Registrant or its securities provided by or on behalf of the undersigned Registrant; and
(iv) any other communication that is an offer in the offering made by the undersigned Registrant to the purchaser.
(b) The undersigned Registrant hereby undertakes that, for purposes of determining any liability under the Securities Act, of 1933, each filing of the Registrant’s annual report pursuant to Section 13(a) or Section 15(d) of the Securities Exchange Act of 1934 (and, where applicable, each filing of an employee benefit plan’s annual
II-3
report pursuant to Section 15(d) of the Securities Exchange Act of 1934)Act) that is incorporated by reference in the registration statement shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(c) The undersigned Registrant hereby undertakes to deliver or cause to be delivered with the prospectus, to each person to whom the prospectus is sent or given, the latest annual report, to security holders that is incorporated by reference in the prospectus and furnished pursuant to and meeting the requirements of Rule 14a-3 or Rule 14c-3 under the Securities Exchange Act of 1934; and, where interim financial information required to be presented by Article 3 of Regulation S-X is not set forth in the prospectus, to deliver, or cause to be delivered to each person to whom the prospectus is sent or given, the latest quarterly report that is specifically incorporated by reference in the prospectus to provide such interim financial information.
(d) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers, and controlling persons of the Registrant pursuant to the foregoing provisions, or otherwise, the Registrant has been advised that in the opinion of the Securities and Exchange CommissionSEC such indemnification is against public policy as expressed in the Securities Act of 1933 and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer, or controlling person of the Registrant in the successful defense of any action, suit, or proceeding) is asserted by such director, officer, or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act of 1933 and will be governed by the final adjudication of such issue.
(e) If and when applicable, the Registrant hereby further undertakes to file an application for the purpose of determining the eligibility of the trustee to act under subsection (a) of Section 310 of the Trust Indenture Act in accordance with the rules and regulations prescribed by the Securities and Exchange Commission under Section 305(b)(2) of the Trust Indenture Act.
II-4
SIGNATURES
Pursuant to the requirements of the Securities Act, of 1933, the Registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form S-3 and has duly caused this Registration Statementregistration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Austin, StateWaltham, Commonwealth of Texas,Massachusetts, on May 1, 2017.August 7, 2023.
| | |
| AEGLEA BIOTHERAPEUTICS, INC. |
| |
| By: | | /s/ David G. Lowe, Ph.D.Jonathan D. Alspaugh |
| | David G. Lowe, Ph.D.Jonathan D. Alspaugh |
| | President and Chief ExecutiveFinancial Officer |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints David G. Lowe, Ph.D.Jonathan D. Alspaugh and Charles N. York II, and each of them,Dr. Cameron Turtle, as his or her true and lawful attorneys-in-fact, proxies and agents, each with full power of substitution, for him or her in any and all capacities, to sign any and all amendments to this registration statement (including post-effective amendments or any abbreviated registration statement and any amendments thereto filed pursuant to Rule 462(b) increasing the number of securities for which registration is sought), and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission,SEC, granting unto said attorneys-in-fact, proxies and agents full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully for all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact, proxies and agents, or their or his or her substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act, this Registration Statementregistration statement has been signed by the following persons in the capacities and on the dates indicated.
| | | | |
Signature | | Title | | Date |
| | |
/s/ David G. Lowe, Ph.D.Jonathan D. Alspaugh David G. Lowe, Ph.D.Jonathan D. Alspaugh
| | President Chief Executive Officer and Director (Principal Executive Officer)
| | May 1, 2017 |
| | |
/s/ Charles N. York II
Charles N. York II
| | Chief Financial Officer and Vice President ((Principal Executive Officer, Principal Accounting Officer and
Principal Financial Officer) | | May 1, 2017 |
| | |
/s/ Suzanne L. Bruhn, Ph.D.
Suzanne L. Bruhn, Ph.D.
| | Director
| | May 1, 2017August 7, 2023 |
| | |
/s/ Russell J. Cox Russell J. Cox | | Director | | May 1, 2017August 7, 2023 |
| | |
/s/ George Georgiou, Ph.D.Peter Harwin George Georgiou, Ph.D.Peter Harwin
| | Director | | May 1, 2017August 7, 2023 |
| | |
/s/ Sandesh Mahatme, LLMMichael Henderson, M.D.
Sandesh Mahatme, LLMMichael Henderson, M.D.
| | Director | | August 7, 2023 |
| | |
Director/s/ Tomas Kiselak
Tomas Kiselak | | May 1, 2017Director | | August 7, 2023 |
| | |
/s/ Alison Lawton
Alison Lawton | | Director | | August 7, 2023 |
II-5
| | | | |
| Signature | | Title | | Date |
Signature
/s/ Ivana Magovčević-Liebisch, Ph.D., J.D.
Ivana Magovčević-Liebisch, Ph.D., J.D. | | Title
Director | | Date
August 7, 2023 |
| | |
/s/ Anthony G. Quinn, M.B Ch.B, Ph.D.Hunter C. Smith, M.B.A.
Anthony G. Quinn, M.B Ch.B, Ph.D.Hunter C. Smith, M.B.A.
| | Director | | May 1, 2017 |
| | |
/s/ Armen Shanafelt
Armen Shanafelt, Ph.D.
| | Director
| | May 1, 2017August 7, 2023 |
***
II-6
EXHIBIT INDEX
| | | | | | | | | | | | |
| | | | | Incorporated by Reference |
Exhibit
Number | | Exhibit Description | | Form | | File No. | | Exhibit | | Filing Date | | Filed
Herewith |
| | | | | | |
| 1.1* | | Form of Underwriting Agreement | | | | | | | | | | |
| | | | | | |
| 1.2 | | Capital on DemandTM Sales Agreement, dated May 1, 2017, by and between the Registrant and JonesTrading Institutional Services LLC | | | | | | | | | | X |
| | | | | | |
| 3.1 | | Restated Certificate of Incorporation | | S-1/A | | 333-205001 | | 3.2 | | 9/14/2015 | | |
| | | | | | |
| 3.2 | | Restated Bylaws | | S-1/A | | 333-205001 | | 3.5 | | 9/14/2015 | | |
| | | | | | |
| 4.1 | | Form of Common Stock Certificate | | S-1/A | | 333-205001 | | 4.1 | | 9/14/2015 | | |
| | | | | | |
| 4.2 | | Amended and Restated Investors’ Rights Agreement, dated March 10, 2015, by and among the Registrant and certain of its stockholders, as amended | | S-1 | | 333-205001 | | 4.2 | | 6/16/2015 | | |
| | | | | | |
| 4.3 | | Form of Debt Security | | | | | | | | | | X |
| | | | | | |
| 4.4 | | Form of Indenture | | | | | | | | | | X |
| | | | | | |
| 4.5* | | Form of Warrant | | | | | | | | | | |
| | | | | | |
| 4.6* | | Form of Warrant Agreement | | | | | | | | | | |
| | | | | | |
| 4.7* | | Form of Preferred Stock Certificate | | | | | | | | | | |
| | | | | | |
| 4.8* | | Form of Subscription Rights Certificate | | | | | | | | | | |
| | | | | | |
| 4.9* | | Form of Unit | | | | | | | | | | |
| | | | | | |
| 4.10* | | Form of Unit Agreement | | | | | | | | | | |
| | | | | | |
| 5.1 | | Opinion of Fenwick & West LLP | | | | | | | | | | X |
| | | | | | |
| 12.1 | | Statement of Computation of Ratio of Earnings to Fixed Charges | | | | | | | | | | X |
| | | | | | |
| 23.1 | | Consent of PricewaterhouseCoopers LLP | | | | | | | | | | X |
| | | | | | |
| 23.2 | | Consent of Fenwick & West LLP (included in Exhibit 5.1) | | | | | | | | | | X |
| | | | | | |
| 24.1 | | Power of Attorney (included on the signature page hereto) | | | | | | | | | | X |
| | | | | | |
| 25.1** | | Form T-1 Statement of Eligibility of Trustee for Senior Indenture under the Trust Indenture Act of 1939. | | | | | | | | | | |
| | | | | | |
| 25.2** | | Form T-1 Statement of Eligibility of Trustee for Subordinated Indenture under the Trust Indenture Act of 1939. | | | | | | | | | | |
* | To be filed by amendment or as an exhibit to a report pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended and incorporated herein by reference. |
** | To be filed in accordance with the requirements of Section 305(b)(2) of the Trust Indenture Act of 1939 and Rule 5b-3 thereunder. |
II-7