UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| | x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2006
OR
| | ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File No. 000-27409
AKESIS PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| | |
| Nevada | | 84-1409219 |
(State or other jurisdiction of incorporation or organization) | | (I.R.S. Employer Identification No.) |
| |
888 Prospect Street Suite 320 La Jolla, California | | 92037 |
| (Address of principal executive offices) | | (Zip Code) |
(858) 454-4311
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act: Common Stock ($0.001 Par Value)
Indicate by check mark if the registrant is a well-known seasoned issuer as defined in Rule 405 of the Securities Act of 1933. Yes¨ Nox
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Exchange Act of 1934. Yes¨ Nox
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yesx No¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer (as defined in Rule 12b-2 of the Securities Exchange Act of 1934).
Large accelerated filer ¨ Accelerated filer ¨ Non-accelerated filer x
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Securities Exchange Act of 1934). Yes ¨ Nox
The aggregate market value of the common stock held by non-affiliates of the registrant as of June 30, 2006 was approximately $6.4 million. For purposes of this calculation, all executive officers and directors of the registrant and all beneficial owners of more than ten percent or more of the registrant’s common stock were considered affiliates.
As of January 31, 2007, the registrant had outstanding 22,580,884 shares of common stock.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive proxy statement to be delivered to stockholders in connection with our 2007 Annual Meeting of Stockholders are incorporated by reference in Part III of this Form 10-K. In addition, certain exhibits previously filed with the registrant’s prior Forms 10-K, Forms 10-Q and Forms 8-K are incorporated by reference into Part IV of Form 10-K.
AKESIS PHARMACEUTICAL, INC.
FORM 10-K
Year Ended December 31, 2006
INDEX
-i-
PART I
This document contains “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995 that are intended to be protected by the safe harbor created thereby. These statements, which are based on our current expectations regarding future events and circumstances, may contain words such as “will”, “may”, “expects”, “anticipates”, “intends”, “believes”, “estimates” or other words or phrases of similar meaning. Examples of such forward-looking statements include, but are not limited to, statements about:
| | • | | Our capital requirements and resources; |
| | • | | Development of new products; |
| | • | | Our intent to develop and sell products and services to companies in the pharmaceutical industry; |
| | • | | Technological change and uncertainty of new and emerging technologies; |
| | • | | Potential competitors or products; |
| | • | | Future employment of our key employees; |
| | • | | Future capital requirements; |
| | • | | Development of strategic relationships; |
| | • | | Statements about potential future dividends; |
| | • | | Statements about protection of our intellectual property; and |
| | • | | Possible changes in legislation. |
Such forward-looking statements are inherently subject to risks and uncertainties (including, but not limited to, those risks and uncertainties discussed in Item 1A below under the caption “Risk Factors” and in other sections of this document) and actual results and outcomes may differ materially from the results and outcomes discussed in or anticipated by the forward-looking statements. We assume no obligation to update any forward-looking statements to reflect the impact of events and circumstances occurring or arising after the date of this report.
Item 1.Business
The Company
One of our predecessors, Akesis Pharmaceuticals, Inc., a Delaware corporation (“Akesis Delaware”), was incorporated on April 27, 1998, for the purpose of direct marketing to consumers an established over-the-counter product for lowering blood glucose levels in the treatment of diabetes. Effective December 9, 2004, pursuant to the Agreement and Plan of Merger and Reorganization, dated as of September 27, 2004 (the “Merger Agreement”), among Akesis Delaware, another of our predecessors, Liberty Mint, Ltd., a Nevada corporation (“Liberty”), and Ann Arbor Acquisition Corporation, a wholly owned subsidiary of Liberty (“MergerSub”), MergerSub merged with and into Akesis Delaware, with Akesis Delaware as the surviving corporation and wholly owned subsidiary of Liberty. Effective January 11, 2005, the combined company (“we” or the “Company”) changed its name to Akesis Pharmaceuticals, Inc. and its trading symbol to AKES.OB.
Despite the legacy businesses of our predecessors, we are currently an early stage biopharmaceutical company engaged in the discovery, development and commercialization of complementary and alternative therapies for the treatment of three principal forms of carbohydrate intolerance – Type 2 diabetes, Syndrome X, and impaired glucose tolerance (“IGT”) – and their
-1-
associated complications. We have been granted patents and filed patent applications for a number of proprietary combination therapies, including combinations with existing diabetes medications, for use in the treatment of Type 2 diabetes. Our therapies are directed to combinations of vanadium, an insulin mimetic, and chromium, a glucose mobilizer, with current and future diabetes treatments, such as metformin. We intend to use our proprietary combinations to develop prescription treatments for Type 2 diabetes and related metabolic disorders. These products are in an early stage of development and no regulatory filings to commercialize our products have yet been made with the United States Food and Drug Administration, or the FDA, or any similar state or foreign authorities.
We have completed an initial clinical trial of one of our specific product combinations, which demonstrated a consistent improvement in glycated hemoglobin (“HbA1c”) levels compared to base line, after three months of treatment in a diabetic population. These combinations are covered by our issued patents as set forth below. This initial clinical trial was an 81-individual open-label study. Open-label studies are generally considered to be less reliable than double blind placebo controlled studies, and are not accepted by the FDA. The observed overall reduction in HbA1c (which is an established long-term measure of blood glucose) in this open-label study was approximately 1.7% for all treatment groups. This reduction implies an average improvement of almost 20% in blood glucose parameters in this patient population, including patients taking the initial product candidate as monotherapy, as well as with concomitant medications. We believe that this trial may suggest that our proprietary combinations show the potential for enhancing currently available oral antidiabetic therapeutic agents. We intend to conduct follow-on feasibility clinical trials with one or more of our combinations with a goal of confirming and extending the results of our initial clinical study. We believe that the successful completion of these feasibility trials could lead to partnering opportunities in the pharmaceutical industry. We are not currently in discussions with the FDA regarding the specific requirements for approval and we have not commenced any FDA-approved clinical trials.
Diabetes
Diabetes is the fifth leading cause of death by disease in the United States. Diabetes is characterized by poor control of glucose levels in the blood, and is often associated with severe long-term complications, such as heart, eye, kidney and peripheral vascular diseases.
It is estimated that over 194 million people worldwide have diabetes. Of that population, approximately 18 million have Type 1 diabetes, also known as juvenile-onset diabetes, and approximately 159 million have Type 2 diabetes, also known as adult-onset diabetes. In the United States alone, in 2005 there were approximately 15 million people diagnosed with Type 2 diabetes, and approximately 1.5 million new cases of diabetes are diagnosed each year. CDC estimates that in 2005 direct and indirect costs related to diabetes will be in excess of $150 billion.
For people suffering from diabetes, poor control of blood glucose concentrations has been shown to result in severe long-term complications. For instance, damage to small blood vessels due to diabetes may result in disorders such as retinopathy, nephropathy, neuropathy, and peripheral vascular disease.
Weight control and obesity are also major problems for patients with diabetes, particularly for those people using insulin as part of their treatment regimen. Other metabolic complications resulting from diabetes and associated metabolic disorders include high blood pressure and dyslipidemia, the
-2-
abnormal metabolism of fat. These undesired metabolic effects may result in additional complications involving large blood vessels, which can lead to heart attacks, strokes and amputations of lower extremities. Further, patients with diabetes frequently have wide fluctuations in blood sugar following meals. These fluctuations in blood sugar can significantly affect a patient’s quality of life. Collectively, these complications and associated metabolic disorders can lead to increased pain, suffering, reduced quality of life and early death.
The most widely accepted measure of long-term blood glucose relies on HbA1clevels. A person’s HbA1clevel is a recognized indicator of that individual’s average blood glucose concentrations over a 3- to 4-month period. Lower HbA1c,levels indicate better blood glucose control, on average. HbA1c levels in people without diabetes are usually less than 6%. The ADA’s Clinical Practice Recommendations suggest that people with diabetes should aim for an HbA1c level that is lower than 7%. Only a minority of people diagnosed with diabetes in the United States are able to achieve the ADA’s recommended target HbA1clevel, even with available drug therapies. Additionally, aggressive use of insulin and other available therapies to achieve target glucose control can be associated with an increased risk of hypoglycemia and weight gain. Consequently, there is a pressing need to develop new treatment strategies that improve the overall health profile of patients with diabetes and reduce the risk of complications without increased pain and suffering.
In 1993, a landmark study in patients with Type 1 diabetes, called the Diabetes Control and Complications Trial, showed that improved glucose control – as measured by any reduction in an individual’s HbA1c level – reduced the incidence of long-term complications. In 1998, a similar landmark study in patients with Type 2 diabetes, the United Kingdom Prospective Diabetes Study, reported similar conclusions for Type 2 diabetes. Unfortunately, both of these studies also showed that available therapies cannot mitigate the progressive nature of diabetes and that long-term complications are to be expected.
Description of Products
The initial product candidate we developed, HEALTHPROPAK, is a patented combination of micronutrients, the individual components of which have been used widely for many years. It is consumable in tablet form and includes components that are believed to address immediate health needs, longer-term health-maintenance issues, and aspects of general health and well-being. This product candidate has to date been sold as a dietary supplement under the 1994 Dietary Supplement Health and Education Act (“DSHEA”). Preliminary evidence suggests that it may be effective at promoting diabetic health and wellness when used as a stand-alone product, as well as when used as an adjunct to prescription anti-diabetic products, both oral and insulin.
It is contemplated that our new product combinations will be derived from our more recent patent issuances relating to AKP-111, AKP-201 and AKP-310. For instance, we plan to evaluate metformin in combination with vanadium and/or chromium for the treatment of diabetes. This combination product may be a prescription combination. The product will be taken orally, once or twice a day. Our second combination product candidate, which we intend to evaluate through a proposed feasibility study may be a combination of glimepride, or one of the thiazolidinediones with chromium and/or vanadium. This product will also be considered for prescription use and be taken orally in tablet form.
-3-
Our current development pipeline is illustrated in the following table:
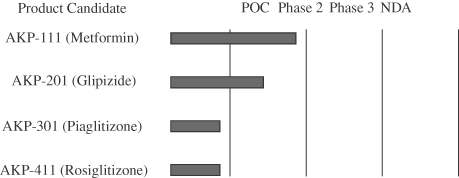
The product candidates under consideration are being developed in response to the increasing number of cases of diabetes and attempt to improve health and wellness without requiring substantial lifestyle changes. They do so by delivering a core micronutrient offering that appears to be important for maintaining good diabetic control.
The anchor components of HEALTHPROPAK are intended to mitigate insulin resistance, and include chromium (from its picolinate complex, although other forms are being considered), magnesium (as highly-bioavailable salts) and vanadium (as its sulfate, although other forms are being considered). These anchor components are supported by additional micronutrients that may support longer-term health and focus on cardiovascular benefits, specifically aspirin source and vitamin E (in its natural isomeric form).
The components of HEALTHPROPAK are presented below:
| | | | |
Ingredient
| | Amount
| | Daily Value
|
Biotin | | 300 µg | | 100% |
Calcium (from calcium carbonate/phosphate) | | 150 mg | | 15% |
Chromium (from polynicotinate complex) | | 333 µg | | 278% |
Copper (from copper chelate) | | 2 mg | | 100% |
Folic Acid | | 400 µg | | 100% |
Iodine (from sea kelp) | | 150 µg | | 100% |
Magnesium (from citrate/fumarate/malate/glutarate/succinate complex) | | 46 mg | | 12% |
Manganese (from manganese sulfate) | | 11 mg | | 550% |
Molybdenum (from citrate/fumarate/malate/glutarate/succinate complex) | | 75 µg | | 100% |
Niacinamide | | 20.1 mg | | 101% |
Pantothenic Acid (as calcium pantothenate) | | 10 mg | | 100% |
Phosphorous (from calcium phosphate) | | 115 mg | | 12% |
Riboflavin | | 3.6 mg | | 212% |
Selenium (from citrate/fumarate/malate/glutarate/succinate complex) | | 60 µg | | 86% |
Standardized Willow/Willow Bark Complex (aspirin source) | | 160 mg | | N/A |
Thiamine (mononitrate) | | 3 mg | | 200% |
Vandyl Sulfate (hydrate) | | 100 mg | | N/A |
Vitamin A | | 5000 IU | | 100% |
Vitamin B-6 (as pyridoxine•HCl) | | 23.1 mg | | 1155% |
Vitamin B-12 | | 48 µg | | 800% |
Vitamin C (ascorbic acid) | | 60 mg | | 100% |
Vitamin D-3 | | 400 IU | | 100% |
Vitamin E (natural) | | 400 IU | | 1333% |
Zinc | | 15 mg | | 100% |
-4-
Chromium and vanadium supplements in diabetes treatment
The first studies to suggest that chromium supplementation (with chromium picolinate) could have beneficial effects on body mass and glucose metabolism were published in the early 1960s. Chromium supplementation has been proposed to help with weight loss, glycemic control in diabetes, athletic performance, controlling hypercholesterolemia, corticosteroid-induced hyperglycemia and improving lean muscle mass. However many subsequent studies of chromium picolinate have failed to support these earlier findings.
There is now reasonable evidence to suggest that chromium deficiency may be associated with the development or progression of diabetes, and that supplementation with chromium can exert positive effects on insulin sensitivity, blood glucose levels and glycosylated hemoglobin levels in diabetic patients. However, there is as of yet no clear picture of whether populations susceptible to diabetes are chromium deficient, or what long term dosing is appropriate to treat or prevent diabetes, or what the long term side effects of chromium supplementation may arise.
The most commonly used form of chromium in health supplements is chromium picolinate, which is significantly more bioavailable than elemental chromium. It is rapidly absorbed in the stomach and subsequently absorbed into tissues, where it rapidly distributes within cells. Pharmacokinetic models predict that ingested chromium will accumulate and be retained in human tissues if the supplement is taken for extended periods of time; however this prediction has not been experimentally confirmed.
Accurate estimates of chromium levels in humans are difficult to determine, and tissue chromium levels do not necessarily correlate with serum chromium levels. The U.S. Food and Nutrition Board of the National Academy of Sciences concluded in 2001 that there was not enough existing evidence to set Recommended Daily Allowances (“RDAs”) for chromium and instead set Adequate Intakes (“AIs”) based on the amount of chromium that normal healthy people currently consume. Based on that data, the Institute of Medicine recommended AIs of chromium of 25-35 micrograms per day (µg or mcg).
The nutritional biochemistry and mechanism of action of chromium in the body is still poorly understood. No enzymes have been formally identified that require chromium for activity, and no chromium dependent co-factors have been biochemically characterized. Chromium has been shown to activate the tyrosine kinase activity of insulin activated insulin receptors and to activate a membrane phosphotyrosine phosphatase in adipocyte membranes. The physiological actions of chromium on insulin sensitivity and diabetes may also be mediated through the interaction of chromium on the transport, storage and intracellular uptake of iron. Chromium supplementation competes with iron for transport through transferrin and acts to reduce iron storage in the body. Excess iron accumulation has been linked to diabetes and insulin insensitivity in some studies, suggesting that the beneficial effects of chromium supplementation may be related to the short term reduction of iron accumulation, particularly in the elderly.
Chromium is considered safe up to doses of 1000 µg (1 mg) per day. However, the use of chromium picolinate has been associated with toxicity, especially at high dosesin vitro, possibly through the release of the picolinate ligand, which can independently act as an oxidant.
In March 2003, the Expert Group on Vitamins and Minerals of the U.K. Joint Food Standards and Safety Group requested that the health supplement industry voluntarily withdraw products containing
-5-
chromium picolinate while also consulting on a ban on the use and sale of chromium picolinate in the U.K. Currently the FDA, working with the U.S. National Academy of Sciences, is studying the potential regulation of chromium picolinate.
In summary, the long term biological effects of chromium accumulation in humans are poorly understood, and there have not yet been any long term studies on the effects of chromium supplementation. There is no conclusive proof, as evidenced by a large long-term controlled clinical trial, demonstrating that the benefits of high dose chromium supplementation for the treatment of diabetes significantly outweigh the risk of chromium toxicity. However, investigators at the Pennington Biomedical Research Institute in Baton Rouge, Louisiana recently showed that chromium picolinate enhanced the anti-diabetic properties of glipizide, a commonly used sulfonylurea. In the study, not only was serum glucose reduced but weight gain was attenutated in the chromium picolinate and glipizide group, as compared to glipizide only. To date no recommendations for supplementation yet exists in the U.S. for diabetes treatment.
Vanadium does not appear to be an essential element. There are no disorders in humans associated with vanadium deficiency, and the government has not established and RDA for vanadium consumption. The normal diet contains 10-30 micrograms (µg or mcg) of elemental vanadium per day. The reported Tolerable Upper Limit (“ULs”) for vanadium is approximately 1.8 mg/day for an adult.
Sodium vanadate was first reported to be effective for treating diabetes in 1899, almost 60 years before the discovery of chromium picolinate. Many subsequent studies have shown that a number of vanadium compounds have insulin enhancing actions bothin vitro andin vivo. Treatment with vanadium compounds such as vanadium sulfate resulted in the development of a modest increase in insulin sensitivity and decreased insulin requirements.
Vanadium has been proposed to act through at least three mechanisms: (1) a direct insulin-mimetic action, (2) an enhancement of insulin sensitivity, and (3), a prolongation of insulin biological response. The insulin mimetic action appears to be mediated by direct binding of vanadium, or vanadium complexes, with low molecular serum proteins to the insulin receptor. The synergistic enhancement of insulin sensitivity and prolongation of insulin response appear to be mediated via an inhibitory action of vanadate on phosphoprotein tyrosine phosphatases (“PTPs”) which would otherwise act to switch off the intracellular effects of insulin within the cell.
Vanadium salts such as vanadyl sulfate appear to be poorly absorbed through the gastrointestinal tract, with less than 5% of the absorbed dose being taken up. The use of enteric coated vanadyl sulfate capsules has been shown to approximately double the uptake of vanadate sulfate. Other salts, such as bis (ethylmaltolato) oxovanadium (IV) (BEVO), have increased absorption compared to other vanadium compounds. Absorbed vanadate has been shown to bind to transferrin and ferritin in plasma and other body fluids. Absorbed vanadium is mainly excreted in the urine in both high and low molecular weight complexes. Long term administration of vanadium results in the accumulation of vanadium in bone.
There is currently limited data on the long term toxicity of vanadium in humans. Vanadate appears to accumulate in bones and in clinical trials gastrointestinal side effects increase above doses of 75 mg / day. In one clinical trial, gastrointestinal side effects were experienced in 75% of the subjects in the first week, but well tolerated after that. In another study 12 subjects were given 13.5 mg daily for 2 weeks, followed by 22.5 mg daily for 5 months. Five developed gastrointestinal symptoms – nausea, vomiting, diarrhea, cramps – and five developed green tongues.
-6-
We believe, based on our initial studies, that supplementation with chromium and vanadium, either individually or combined, may provide synergistic effects when administered in combination with other diabetes therapeutics, potentially making those existing therapeutic strategies for treating diabetes more effective.
Target Markets
The initial product candidate is targeted at individuals with the three principal forms of carbohydrate intolerance – Type 2 diabetes, Syndrome X, and IGT – and their associated complications. Collectively, these conditions are found in one of every five or six individuals, and they are regarded by some analysts as significant growth engines for the human-health industry. All three of these market segments have clear needs and articulated demands for product offerings that deliver benefits such as those that may be available through our product candidates.
Type 2 diabetes is endemic in modern industrialized countries and, by many estimates, represents in excess of 95% of the diabetic population. There are an estimated 15 million or more individuals with Type 2 diabetes in the U.S. and in excess of 150 million individuals worldwide. General agreement exists that this population will more than triple during the next 25 years. Annual sales of oral anti-diabetic agents currently are on the order of $10 billion in the U.S. alone.
Syndrome X is an intermediate between Type 2 diabetes and IGT, and may represent a market of at least 30 million individuals in the U.S. Some recent studies have suggested that this condition is linked to 13 million or more cardiovascular disease cases, which would implicate Syndrome X in at least half of those reported.
Impaired glucose tolerance is an even larger market segment, as this condition affects at least 35 million individuals in the U.S. It has become an increasing area of focus by the ADA, which has advocated the identification and intensive management of the health and wellness of individuals with IGT.
At a more general level, the Akesis product line is tied to health-management trends that are (or are becoming) mainstream. It is aligned perfectly with an aging consumer population that cares more than ever about staying healthy and active.
Safety Findings
All individual micronutrient components in HEALTHPROPAK are found in food articles that either are part of standard diets or are found in widely-available dietary supplements that are legally marketed and sold, and that have extensive clinical and/or safety-in-use histories. Safe consumption of individual and combination micronutrients at levels equivalent to and in excess of those found in the product candidate is documented over decades and, in some cases, centuries. While there are some reports of mild adverse reactions (e.g. gastrointestinal distress and headache), no consistent reports specifically linking any of the micronutrient components of the product candidate to adverse events are found in reports deposited with FDA and other regulatory bodies responsible for maintaining such data.
-7-
For consumers with aspirin sensitivities or allergies, it should be noted that the initial product candidate contains a standardized willow/willow bark (“willow”) complex that is metabolized to acetylsalicylic acid, the active ingredient in aspirin. Willow is used in the initial product candidate at an equivalent of 20 mg aspirin to confer long-term cardioprotective benefits, which are advocated for people with diabetes by the ADA and the American Heart Association. We do not intend to use willow in new product candidates.
Reports of gastrointestinal distress and headaches have been made about individual micronutrients in the initial product candidate; however they generally are ameliorated or eliminated by concomitant food consumption or after a brief adaptation period.
We used WIL Laboratories, a contract toxicology organization, to perform a preliminary animal safety study with HEALTHPROPAK. The results from a good laboratory practices (“GLP”) 14-day, 70-rat toxicology study showed a clean profile when the product combination was administered at levels up to 20 times those levels intended for humans, a regimen recommended by the professional staff at WIL as providing an appropriate margin of safety. Specifically, the results from this study indicated that the product candidate was taken up by study animals and that no adverse observations were noted in a comprehensive examination of tissues, organs, and fluids. Furthermore, all histopathological examinations were completely normal.
Based on the foregoing, we do not anticipate any safety issues related to the individual components of the product candidate. However, the precise combination of micronutrients and active anti-diabetic drugs used (e.g., AKP-111, a metformin product) in the product candidate has not been tested in animals or humans. Accordingly, there can be no guarantee that toxicity issues will not arise or that results of human clinical trials of the product candidate will be consistent with the results obtained to date.
Preliminary Clinical Data
We performed an initial 81-individual, 12-week open-label human study of our initial product candidate, which provided preliminary but encouraging insight about its efficacy. This study was conducted over Thanksgiving and Christmas holidays, a period during which many diabetic individuals experience significant deteriorations of their diabetic control. Participants added the product candidate to their daily treatment regimen while making no changes to existing medications, diets, or exercise regimens. For these reasons, open-label studies can be used to make initial assessments about product efficacy, and are performed with prior physician and participant understanding that a product is being used that is expected to deliver health benefits. They differ from double-blind placebo-controlled trials, which are the industry-standard approach for testing prescription drugs and in which neither physicians nor patients know whether patients are taking a placebo or the product under investigation.
Open-label studies are generally considered to be less reliable than double blind placebo-controlled trials, and are not accepted by the FDA.
-8-
Participants for this study came from hospitals and physician practices in La Jolla, CA; Pittsburgh, PA; Las Vegas, NV; and Chicago, IL. Summary participant information is presented below:
| | |
Number of Participants | | 81 |
| |
Study Duration (Weeks) | | 12 |
| |
Average Age (Range) | | 61 (26 –81) |
| |
Gender (M/F) | | 41/40 |
| |
Average Disease Duration (Range) | | 7 (0 –24) |
| |
Type 2/Type 1 Diabetes | | 81/0 |
| |
Taking concomitant medications | | 88% |
The endpoint measured in the open-label study was HbA1c, which is the generally accepted standard against which diabetic control is evaluated. It provides a measure of average blood glucose readings throughout the previous 90 days. A generally accepted HbA1c range for the non-diabetic population is 4.5% to 7.5%. Values above 10.0% (correlating to an average blood glucose of 250 mg/dL) are observed in a large part of the diabetic population, and values above even 8.0% typically are precursors to serious micro-and macro-vascular complications.
The table below presents summary results of the improved diabetic control (as measured by reductions in HbA1c) that was delivered by our product candidate across various cross-sections of open-label study groups:
| | | |
Population
| | Change in HbA1c
| |
Entire | | -1.7 | % |
Those with starting HbA1c > 7.0 < 10.0% | | -2.2 | % |
Those taking only the Akesis product candidate | | -2.0 | % |
Those taking one concomitant prescription anti-diabetic | | -2.1 | % |
Those taking three concomitant prescription anti-diabetics | | -1.3 | % |
To provide context for these results, the following table compares the improvements in diabetic control delivered by our product candidate in the open-label study, when taken as the only product for diabetic control (i.e., as monotheraphy), with those delivered by prescription anti-diabetics taken as monotheraphy. Data for these prescriptions agents are taken from much larger and sometimes longer double-blind placebo-controlled studies. Accordingly, the data from these other studies is much more likely to be accurate than the data from our study. Further trials will need to be done to verify that the results with the product candidate are valid in larger populations and in longer-duration studies.
| | | | | |
Product
| | Study Duration (Weeks)
| | Change in HbA1c
| |
HEALTHPROPAK | | 12 | | -2.0 | % |
Glucophage® | | 29 | | -1.4 | % |
| | |
Rezulin® | | 26 | | -1.0 | % |
| | | 12 | | -0.8 | % |
| | |
Avandia® | | 26 | | -0.7 | % |
| | | 26 | | -0.6 | % |
| | |
Prandin™ | | 12 | | -0.6 | % |
-9-
The small size of the participant population for our study, combined with its being an open-label protocol, make it difficult to draw broad, statistically-significant conclusions. However, we believe the study involving our product candidate indicates the following:
| | 1. | Our product candidate may be efficacious, with activity that may be comparable to those delivered by prescription anti-diabetics. |
| | 2. | Our product candidate may be equally efficacious across all demographics (e.g., age, gender, weight, duration of condition, starting HbA1c, number/type of concomitant anti-diabetic medications). |
| | 3. | Our product candidate appears to be most effective in populations that are of greatest interest to the health- and managed-care industries – i.e., younger individuals, those with more recent onsets of diabetes, and individuals with higher baseline HbA1clevels. |
In order to test our product candidate under more demanding conditions, a randomized, double-blind, placebo-controlled, good clinical practices (“GCP”) human trial was conducted at Duke University on a pilot scale with 18 patients. The study evaluated the addition of our product candidate to an otherwise-unchanged diabetes-control program. Results from this trial were interpreted by Clinimetrics, an independent contract-research organization. Although this study was small by design, we believe the results may warrant undertaking larger similar studies.
Specifically, this blinded, placebo-controlled study made a stronger statement than trend-line information that generally is obtained from pilot trials. The treatment arm showed appreciably improved HbA1c values v. placebo (median improvement 1.0%, mean improvement 1.2%), although this improvement was not statistically significant (p = 0.07) due to the small sample size.
In summary, based on this preliminary clinical data, we believe that consumption of our initial product candidate may be an effective approach to improving the health of individuals with various manifestations of carbohydrate intolerance. However, there can be no assurance that the results of more extensive human studies with different combination products will be consistent with those obtained thus far.
Planned Feasibility Studies
In May 2006, we began the development of a clinical research protocol and began conducting other activities to support the initiation of new clinical trials related to our proposed pharmaceutical products. The cost of the clinical trial work during the year ended December 31, 2006 was approximately $98,000 compared to zero for 2005 and 2004. The Company is currently evaluating prospective suppliers and manufacturers for the raw materials and compounds that will be used for its clinical trial feasibility studies, but has not finalized any such arrangements.
Patents, Proprietary Rights, and Licenses
We believe that patents and other proprietary rights are important to our business. Our policy is to file patent applications to protect technology, inventions and improvements that may be important to the development of our business. We also rely upon trade secrets, know-how, continuing technological innovations and licensing opportunities to develop and maintain our competitive position. We plan to enforce our issued patents and our rights to proprietary information and technology.
-10-
Our core technology is covered by four issued U.S. patents, and we have one additional patent application pending. Each of these patents consist of method and composition claims. A summary of our intellectual property is as follows:
| | | | | | | | | | |
| Patent No. | | Title | | Application
Date | | Approval
Date | | Summary | | Target Market |
US 5,962,030 | | Dietary Supplement and method of treatment for diabetic control | | 11/05/97 | | 5/10/99 | | Claims dietary supplements for improving glucose metabolism comprising chromium, magnesium, and vanadium and at least one other ingredient | | Over the Counter Supplement market |
US 6,203,819 | | Dietary Supplement and method of treatment for diabetic control | | 3/19/99 | | 3/20/2001 | | Claims dietary supplements for improving glucose metabolism comprising chromium, vanadium and aspirin | | Over the Counter Supplement market |
US 6,376,549 | | Metformin-containing compositions for the treatment of diabetes | | 10/17/1998 | | 4/23/2002 | | Claims compositions for the treatment of diabetes comprising metformin, magnesium, chromium and vanadium | | Prescription diabetes market, particularly combination therapy approaches |
US 6,852,760 | | Compositions and methods for treatment of glucose metabolism disorders | | 10/17/99 | | 2/8/05 | | Claims compositions for the treatment of diabetes comprising a sulfonylurea class of drug, chromium and vanadium | | Prescription diabetes market, particularly combination therapy approaches |
Drugs. The FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries impose substantial requirements upon the clinical development, manufacture, marketing and distribution of drugs. These agencies and other federal, state and local entities regulate research and development activities and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion of our drug candidates.
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act and its implementing regulations. The process required by the FDA before our drug candidates may be marketed in the United States generally involves the following:
| | • | | completion of extensive preclinical laboratory tests, preclinical animal studies and formulation studies, all performed in accordance with GLP regulations; |
| | • | | submission to the FDA of an Investigational New Drug, or IND, application which must become effective before clinical trials may begin; |
-11-
| | • | | performance of adequate and well-controlled clinical trials to establish the safety and efficacy of the product candidate for each proposed indication; |
| | • | | submission of a New Drug Application, or NDA, to the FDA; |
| | • | | satisfactory completion of an FDA preapproval inspection of the manufacturing facilities at which the product is produced to assess compliance with current Good Manufacturing Practices, or cGMP, regulations; and |
| | • | | FDA review and approval of the NDA prior to any commercial marketing, sale or shipment of the drug. |
The testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our drug candidates will be granted on a timely basis, if at all.
Preclinical tests include laboratory evaluation of product chemistry, formulation and stability, as well as studies to evaluate toxicity in animals. The results of preclinical tests, together with manufacturing information and analytical data, are submitted as part of an IND application to the FDA.
An IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the conduct of the clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Our submission of an IND, or those of our collaborators, may not result in FDA authorization to commence a clinical trial. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development, and the FDA must grant permission before each clinical trial can begin. Further, an independent institutional review board, or IRB, for each medical center proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that center and it must monitor the study until completed. The FDA, the IRB, or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive GCP regulations and regulations for informed consent.
Clinical Trials. For purposes of NDA submission and approval, clinical trials are typically conducted in the following three sequential phases, which may overlap:
| | • | | Phase I: Studies are initially conducted in a limited population to test the drug candidate for safety, dose tolerance, absorption, metabolism, distribution and excretion in healthy humans or, on occasion, in patients. In some cases, a sponsor may decide to run what is referred to as a “Phase Ib” evaluation, which is a second safety-focused Phase I clinical trial typically designed to evaluate the impact of the drug candidate in combination with currently approved drugs. |
| | • | | Phase II: Studies are generally conducted in a limited patient population to identify possible adverse effects and safety risks, to determine the efficacy of the drug candidate for specific targeted indications and to determine dose tolerance and optimal dosage. Multiple Phase II clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive Phase III clinical trials. In some cases, a sponsor may decide to run what is referred to as a “Phase IIb” evaluation, which is a second, confirmatory Phase II clinical trial that could, if positive and accepted by the FDA, serve as a pivotal clinical trial in the approval of a drug candidate. |
-12-
| | • | | Phase III: These are commonly referred to as pivotal studies. When Phase II clinical trials demonstrate that a dose range of the drug candidate is effective and has an acceptable safety profile, Phase III clinical trials are undertaken in large patient populations to further evaluate dosage, to provide substantial evidence of clinical efficacy and to further test for safety in an expanded and diverse patient population at multiple, geographically dispersed clinical trial sites. |
In some cases, the FDA may condition approval of an NDA for a drug candidate on the sponsor’s agreement to conduct additional clinical trials to further assess the drug’s safety and effectiveness after NDA approval. Such post-approval trials are typically referred to as Phase IV clinical trials.
New Drug Application. The results of drug candidate development, preclinical testing and clinical trials are submitted to the FDA as part of a NDA. The NDA also must contain extensive manufacturing information. Once the submission has been accepted for filing, by law the FDA has 180 days to review the application and respond to the applicant. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA may refer the NDA to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. The FDA may deny approval of a NDA if the applicable regulatory criteria are not satisfied, or it may require additional clinical data or an additional pivotal Phase III clinical trial. Even if such data are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data from clinical trials is not always conclusive and the FDA may interpret data differently than we or our collaborators interpret data. Once issued, the FDA may withdraw drug approval if ongoing regulatory requirements are not met or if safety problems occur after the drug reaches the market. In addition, the FDA may require testing, including Phase IV clinical trials, and surveillance programs to monitor the effect of approved products which have been commercialized, and the FDA has the power to prevent or limit further marketing of a drug based on the results of these post-marketing programs. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are any modifications to the drug, including changes in indications, labeling, or manufacturing processes or facilities, we may be required to submit and obtain FDA approval of a new NDA or NDA supplement, which may require us to develop additional data or conduct additional preclinical studies and clinical trials.
Satisfaction of FDA regulations and requirements or similar requirements of state, local and foreign regulatory agencies typically takes several years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease. Typically, if a drug candidate is intended to treat a chronic disease, as is the case with some of the drug candidates we are developing, safety and efficacy data must be gathered over an extended period of time. Government regulation may delay or prevent marketing of drug candidates for a considerable period of time and impose costly procedures upon our activities. The FDA or any other regulatory agency may not grant approvals for new indications for our drug candidates on a timely basis, if at all. Even if a drug candidate receives regulatory approval, the approval may be significantly limited to specific disease states, patient populations and dosages. Further, even after regulatory approval is obtained, later discovery of previously unknown problems with a drug may result in restrictions on the drug or even complete withdrawal of the drug from the market. Delays in obtaining, or failures to obtain, regulatory approvals for any of our drug candidates would harm our business. In addition, we cannot predict what adverse governmental regulations may arise from future United States or foreign governmental action.
-13-
Other regulatory requirements. Any drugs manufactured or distributed by us or our collaborators pursuant to FDA approvals are subject to continuing regulation by the FDA, including recordkeeping requirements and reporting of adverse experiences associated with the drug. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including cGMPs, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. Failure to comply with the statutory and regulatory requirements can subject a manufacturer to possible legal or regulatory action, such as warning letters, suspension of manufacturing, seizure of product, injunctive action or possible civil penalties. We cannot be certain that we or our present or future third-party manufacturers or suppliers will be able to comply with the cGMP regulations and other ongoing FDA regulatory requirements. If our present or future third-party manufacturers or suppliers are not able to comply with these requirements, the FDA may halt our clinical trials, require us to recall a drug from distribution, or withdraw approval of the NDA for that drug.
The FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the Internet. A company can make only those claims relating to safety and efficacy that are approved by the FDA. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available drugs for uses that are not described in the drug’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, impose stringent restrictions on manufacturers’ communications regarding off-label use.
Dietary Supplements. The FDA regulates dietary supplements under DSHEA. DSHEA describes a dietary supplement as a product (other than tobacco) that is:
| | • | | intended to supplement the diet that bears or contains one or more of the following dietary ingredients: a vitamin, a mineral, an herb or other botanical, an amino acid, a dietary substance for use by man to supplement the diet by increasing the total daily intake, or a concentrate, metabolite, constituent, extract, or combinations of these ingredients; |
| | • | | intended for ingestion in pill, capsule, tablet, or liquid form; |
| | • | | not represented for use as a conventional food or as the sole item of a meal or diet; and |
| | • | | labeled as a “dietary supplement.” |
Under DSHEA, a manufacturer is not required to establish that a dietary supplement is safe or effective before the product can be marketed and the FDA does not have preapproval authority and does not scrutinize a dietary supplement before it enters the market. The FDA is permitted to restrict the sale of a dietary supplement or dietary ingredient if it poses a “significant and unreasonable risk” under the conditions of use on the label or as commonly consumed.
Dietary supplement manufacturers are not allowed to make claims that the product is intended for use in the diagnosis, cure, mitigation, treatment or prevention of disease. Claims that suggest such an
-14-
intended use subject the dietary supplement to regulation as a drug. Such a product becomes illegal if it fails to comply with all drug requirements, including the requirements for FDA approval of a NDA prior to marketing. Failure to comply with the requirements of DSHEA can subject a manufacturer to possible legal and regulatory action, such as warning letters, suspension of manufacturing, seizure of product, injunctive action or possible civil and criminal penalties.
Competition
Biotechnology and pharmaceutical companies are highly competitive. There are many pharmaceutical companies, biotechnology companies, public and private universities and research organizations actively engaged in the research and development of products that may compete with our products. A number of our largest competitors, including, but not limited to, Bristol-Myers Squibb Company, Sanofi-Aventis, GlaxoSmithKline plc, Eli Lilly and Company, Merck & Co., Novartis AG, Novo Nordisk A-S and Takeda Pharmaceuticals, are pursuing the development of, or are marketing, pharmaceuticals that target the same diseases that we are targeting, and it is possible that the number of companies seeking to develop products and therapies for the treatment of diabetes, obesity, and cardiovascular disease will increase. A number of supplement makers including Nutrition 21, have developed, or are developing similar products to ours. The government, through the National Center for Complementary and Alternative Medicine (“NCCAM”), funds a variety of private, and for-profit, and academic groups to conduct trials on chromium supplementation and related alternative approaches to treat diabetes.
Many of these and other existing or potential competitors have substantially greater financial, technical and human resources than we do and may be better equipped to develop, manufacture and market products. These companies may develop and introduce products and processes competitive with or superior to ours. In addition, other technologies or products may be developed that have an entirely different approach or means of accomplishing the intended purposes of our products, which might render our technology and products noncompetitive or obsolete.
If approved for marketing, our proprietary combinations may compete with established therapies for market share. In addition, many companies are pursuing the development of novel pharmaceuticals that target diabetes. These companies may develop and introduce products competitive with or superior to our proprietary combinations. Such competitive or potentially competitive products include: acarbose, nateglinide, metformin, miglitol, pioglitazone, repaglinide, rosiglitazone, sulfonyureas, and symlin.
Employees
As of January 31, 2007, we had 2 employees. Both of these employees are in general and administrative positions. All of our management employees and members of our Board of Directors have prior experience with pharmaceutical and biotechnology companies. None of our employees are covered by collective bargaining agreements, and our management considers relations with our employees to be good.
Corporate Website
We maintain a corporate website located athttp://www.akesis.com. The contents of that website are not intended to constitute, nor do they constitute, part of this report.
-15-
Item 1A.Risk Factors
Our future operating results may vary substantially from anticipated results due to a number of factors, many of which are beyond our control. The following discussion highlights some of these factors and the possible impact of these factors on future results of operations. You should carefully consider these factors before making an investment decision. If any of the following factors actually occur, our business, financial condition or results of operations could be harmed. In that case, the price of our common stock could decline, and you could experience losses on your investment.
We have a history of operating losses, anticipate future losses, may not generate revenues from product sales and may never become profitable.
We have experienced significant operating losses in each period since our inception. As of December 31, 2006, we have incurred total losses of $8.3 million. We expect these losses to continue and it is uncertain when, if ever, we will become profitable. These losses have resulted principally from costs incurred in conducting the initial open-label clinical trials, stock-based compensation for our executive officers and from general and administrative costs associated with operations. We expect to incur increasing operating losses in the future as a result of expenses associated with clinical trials (see the Liquidity and Capital Resources section of this report below) as well as general and administrative costs. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis.
We will require additional capital in the future to fund our expected operations, and if this additional capital is not available or not available on acceptable terms, we may have to reduce the size of our operations.
As of December 31, 2006, we have no long-term financial commitments. However, we are planning to proceed with additional feasibility clinical trials of our proposed products, which will require extensive additional funding (see the Liquidity and Capital Resources section of this report below). There can be no assurance that we will be able to raise additional financing. If we are unable to raise any additional required financing, our current cash resources would only enable us to continue operations based on our current level of commitments into the second half of 2008.
We may require substantial additional capital to finance future growth and fund ongoing operations beyond the second half of 2008. In particular, we may issue a substantial number of additional shares and warrants to raise additional financing and we have little control over the timing of any resales of such shares. As a result, the market price of our common stock may fall if a large portion of those shares is sold in the public market. We may raise additional funds through public or private financing, strategic relationships or other arrangements. We cannot be certain that the funding will be available on attractive terms, or at all. Furthermore, any additional equity financing may be dilutive to shareholders, and debt financing, if available, may involve restrictive covenants. Strategic arrangements, if necessary to raise additional funds, may require us to relinquish our rights to certain of our technologies or products. If we fail to raise capital when needed, our business will be negatively affected, which could cause the price of our common stock to decline.
We are currently assessing various prospective product combinations. We will require additional capital to fund the development and commercialization of our specific combinations. Our future capital requirements will depend on many factors, including:
| | • | | progress with our preclinical studies and toxicity studies; |
-16-
| | • | | the time and costs involved in obtaining regulatory approvals for the marketing of any of our specific combinations; |
| | • | | the costs of manufacturing any of our specific combinations; |
| | • | | our ability, and the ability of any partner, to effectively market, sell and distribute product, subject to obtaining regulatory approval; |
| | • | | our ability to establish one or more marketing, distribution or other commercialization arrangements; |
| | • | | the cost of any potential licenses or acquisitions; and |
| | • | | the costs involved in preparing, filing, prosecuting, maintaining and enforcing patents or defending ourselves against competing technological and market developments. |
If adequate funds are not available when required, we may have to delay, scale back or eliminate one or more of our development programs, or obtain funds by entering into more arrangements with collaborative partners or others that may require us to relinquish rights to certain of our specific combinations or technologies that we would not otherwise relinquish.
We may be unable to obtain regulatory clearance to market our proprietary combinations in the United States or foreign countries on a timely basis, or at all.
Our proprietary combinations are subject to extensive government regulations related to development, clinical trials, manufacturing and commercialization. The process of obtaining FDA and other regulatory approvals is costly, time consuming, uncertain and subject to unanticipated delays. The FDA may refuse to approve an application for approval of a specific combination if it believes that applicable regulatory criteria are not satisfied. The FDA may also require additional testing for safety and efficacy. Moreover, if the FDA grants regulatory approval of a product, the approval may be limited to specific indications or limited with respect to its distribution, which could limit our revenues. Foreign regulatory authorities may apply similar limitations or may refuse to grant any approval.
No diabetes product using our technologies has been approved for marketing. Consequently there is no precedent for the successful commercialization of products based on our technologies. This may impede our ability to obtain timely approvals from the FDA or foreign regulatory agencies. We will not be able to commercialize our proprietary products until we obtain regulatory approval, and consequently any delay in obtaining, or inability to obtain regulatory approvals could harm our business.
If we violate regulatory requirements at any stage, whether before or after marketing approval is obtained, we may be fined, forced to remove a product from the market or experience other adverse consequences, including delay, which would materially harm our financial results. Additionally, we may not be able to obtain the labeling claims necessary or desirable for product promotion.
Moreover, manufacturing facilities operated by the third-party manufacturers with whom we may contract to manufacture our proprietary combinations may not pass an FDA or other regulatory authority pre-approval inspection. Any failure or delay in obtaining these approvals could prohibit or delay us or any of our business partners from marketing our combinations.
-17-
Delays in the conduct or completion of our clinical trials, the analysis of the data from our clinical trials, or our manufacturing scale-up activities may result in delays in our planned filings for regulatory approvals, and may adversely affect our ability to enter into new collaborative arrangements.
We cannot predict whether we will encounter problems with any of our completed or planned clinical studies that will cause us or regulatory authorities to delay or suspend planned clinical studies.
Any of the following could delay the completion of our planned clinical studies:
| | • | | failure of the FDA or comparable foreign authorities to approve the scope or design of our clinical trials; |
| | • | | delays in enrolling volunteers; |
| | • | | insufficient supply or deficient quality of specific combination materials or other materials necessary for the performance of clinical trials; |
| | • | | negative results of clinical studies; or |
| | • | | serious side effects experienced by study participants relating to a specific combination. |
If the results of our planned clinical studies for our proprietary combinations are not available when we expect or if we encounter any delay in the analysis of data from our clinical studies or if we encounter delays in our ability to scale-up our manufacturing processes, we may have to delay our planned filings seeking regulatory approval of our proprietary combinations. Additionally we may not have the financial resources to continue research and development of any of our proprietary combinations; and we may not be able to enter into additional collaborative arrangements relating to any proprietary combinations subject to delay in clinical studies or delay in regulatory filings.
We have not commenced FDA trials and may not ever commence FDA trials.
We have not commenced FDA trials for any of our product combinations. There are a number of requirements that we must satisfy in order to begin FDA trials. These requirements will require substantial time, effort and financial resources. There can be no assurance that we will complete the steps necessary to reach FDA trials.
We currently have no internal sales and marketing resources and may have to rely on third parties in the event that we successfully develop our product candidates into commercial drug products.
To market any of our products in the United States or elsewhere, we must develop internally or obtain access to sales and marketing forces with technical expertise and with supporting distribution capability in the relevant geographic territory.
We may not be able to enter into marketing and distribution arrangements or find a corporate partner for our specific combination or our other specific combinations, and we are not likely to be able to market and distribute our products ourselves. If we are not able to enter into a marketing or distribution arrangement or find a corporate partner who can provide support for commercialization of
-18-
our specific combinations as we deem necessary, we may not be able to commercialize our products successfully. Moreover, any new marketer or distributor or corporate partner for our specific combinations, with whom we choose to contract may not establish adequate sales and distribution capabilities or gain market acceptance for our products, if any.
Our ability to generate revenues will be diminished if we fail to obtain acceptable prices or an adequate level of reimbursement for our products from third-party payors.
The requirements governing product licensing, pricing and reimbursement vary widely from country to country. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after product licensing approval is granted. As a result, we may obtain regulatory approval for a product in a particular country, but then be subject to price regulations that reduce our revenues from the sale of the product. Also, in some foreign markets, pricing of prescription pharmaceuticals is subject to continuing governmental control even after initial marketing approval. If we succeed in bringing a specific combination to market, we cannot be certain that the products will be considered cost effective and that reimbursement will be available or, if available, will be sufficient to allow us to sell the products on a competitive basis.
The continuing efforts of government and third-party payors to contain or reduce the costs of health care through various means, including efforts to increase the amount of patient co-pay obligations, may limit our commercial opportunity. For example, in some foreign markets, pricing and profitability of prescription pharmaceuticals are subject to government control. In the United States, we expect that there will continue to be a number of federal and state proposals to implement similar government control. In addition, increasing emphasis on managed care in the United States will continue to put pressure on the rate of adoption and pricing of pharmaceutical products. Cost control initiatives could decrease the price that any of our collaborators or we would receive for any products in the future. Further, cost control initiatives could adversely affect our collaborators’ ability to commercialize our products, our ability to realize revenues from this commercialization, and our ability to fund the development of future specific combinations.
Our ability to commercialize pharmaceutical products, alone or with collaborators, may depend in part on the extent to which adequate reimbursement for the products will be available from governmental and health administration authorities, private health insurers, and other third-party payors.
Significant uncertainty exists as to the reimbursement status of newly approved health care products. Third-party payors, including Medicare, are challenging the prices charged for medical products and services. Government and other third-party payors increasingly are attempting to contain health care costs by limiting both coverage and the level of reimbursement for new drugs and by refusing, in some cases, to provide coverage for uses of approved products for disease indications for which the FDA has not granted labeling approval. Third-party insurance coverage may not be available to patients for any products we discover and develop, alone or with collaborators. If government and other third-party payors do not provide adequate coverage and reimbursement levels for our products, the market acceptance of these products may be reduced.
We do not manufacture our own specific combinations and rely on third-party manufacturers to provide the components necessary for our specific combinations.
We do not manufacture our own specific combinations and may not be able to obtain adequate supplies, which could cause delays or reduce profit margins. The manufacturing of sufficient quantities
-19-
of new specific combinations is a time-consuming and complex process. We have no manufacturing capabilities. In order to continue to develop our proprietary combinations, apply for regulatory approvals and ultimately commercialize additional products, we need to contract or otherwise arrange for the necessary manufacturing.
If any of our existing or future manufacturers cease to manufacture or are otherwise unable to deliver any of the components of our specific combinations in either bulk or dosage form, or other product components, we may need to engage additional manufacturers. The cost and time to establish manufacturing facilities would be substantial. As a result, using a new manufacturer could disrupt our ability to supply our products and/or reduce our profit margins. Any delay or disruption in the manufacturing of bulk product, the dosage form of our products or other product components, including pens for delivery of our products, could harm our ability to generate product sales, harm our reputation and require us to raise additional funds.
We have not selected any third-party contract manufacturers for our proprietary combinations.
We have not yet selected manufacturers for our proprietary combinations and we cannot be certain that we will be able to obtain long-term supplies of those materials on acceptable terms. We do not currently have established quality control and quality assurance programs, including a set of standard operating procedures, analytical methods and specifications, designed to ensure that proprietary combinations are manufactured in accordance with current good manufacturing practices and other domestic and foreign regulations.
If our patents are determined to be unenforceable, or if we are unable to obtain new patents based on current patent applications or for future inventions, we may not be able to prevent others from using our intellectual property.
Our success will depend in part on our ability to obtain and expand patent protection for our specific combinations and technologies both in the United States and other countries. We cannot guarantee that any patents will issue from any pending or future patent applications owned by or licensed to us. Alternatively, a third party may successfully circumvent our patents. Our rights under any issued patents may not provide us with sufficient protection against competitive products or otherwise cover commercially valuable products or processes. In addition, because patent applications in the United States are maintained in secrecy for eighteen months after the filing of the applications, and publication of discoveries in the scientific or patent literature often lag behind actual discoveries, we cannot be sure that the inventors of subject matter covered by our patents and patent applications were the first to invent or the first to file patent applications for these inventions. In the event that a third party has also filed a patent on a similar invention, we may have to participate in interference proceedings declared by the U.S. Patent and Trademark Office to determine priority of invention, which could result in a loss of our patent position. Furthermore, we may not have identified all U.S. and foreign patents that pose a risk of infringement.
Litigation regarding patents and other proprietary rights may be expensive, cause delays in bringing products to market and harm our ability to operate.
Our success will depend in part on our ability to operate without infringing the proprietary rights of third parties. Legal standards relating to the validity of patents covering pharmaceutical and biotechnological inventions and the scope of claims made under these patents are still developing. As a result, our ability to obtain and enforce patents is uncertain and involves complex legal and factual
-20-
questions. Third parties may challenge or infringe upon existing or future patents. In the event that a third party challenges a patent, a court may invalidate the patent or determine that the patent is not enforceable. Proceedings involving our patents or patent applications or those of others could result in adverse decisions about:
| | • | | the patentability of our inventions and products relating to our specific combinations; and/or |
| | • | | the enforceability, validity or scope of protection offered by our patents relating to our specific combinations. |
The use of our technologies could potentially conflict with the rights of others.
The manufacture, use or sale of any of our proprietary combinations may infringe on the patent rights of others. If we are unable to avoid infringement of the patent rights of others, we may be required to seek a license, defend an infringement action or challenge the validity of the patents in court. Patent litigation is costly and time consuming. We may not have sufficient resources to bring these actions to a successful conclusion. In such case, we may be required to alter our products, pay licensing fees or cease activities. If our products conflict with patent rights of others, third parties could bring legal actions against us claiming damages and seeking to enjoin manufacturing and marketing of the affected products. If these legal actions are successful, in addition to any potential liability for damages, we could be required to obtain a license in order to continue to manufacture or market the affected products. We may not prevail in any legal action and a required license under the patent may not be available on acceptable terms, if at all.
We may be unable to adequately prevent disclosure of our trade secrets and other proprietary information.
In order to protect our proprietary technology and processes, we rely in part on confidentiality agreements with our corporate partners, employees, consultants, outside scientific collaborators and sponsored researchers and other advisors. These agreements may not effectively prevent disclosure of our confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of our confidential information. In addition, others may independently discover trade secrets and proprietary information. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
Competition in the biotechnology and pharmaceutical industries may result in competing products, superior marketing of other products and lower revenues or profits for us.
There are many companies that are seeking to develop products and therapies for the treatment of diabetes and other metabolic disorders. Our competitors include multinational pharmaceutical and chemical companies, specialized biotechnology firms and universities and other research institutions. A number of our largest competitors, including Bristol-Myers Squibb Company, Sanofi-Aventis, Eli Lilly and Company, GlaxoSmithKline, Merck & Co., Novartis AG, Novo Nordisk A-S and Takeda Pharmaceuticals, are pursuing the development or marketing of pharmaceuticals that target the same diseases that we are targeting, and it is possible that the number of companies seeking to develop products and therapies for the treatment of diabetes and other metabolic disorders will increase. The government, through NCCAM, funds a variety of private, and for-profit, and academic groups to conduct trials on chromium supplementation and related alternative approaches to treat diabetes.
-21-
Many of our competitors have substantially greater financial, technical, human and other resources than we do. In addition, many of these competitors have significantly greater experience than we do in undertaking preclinical testing and human clinical studies of new pharmaceutical products and in obtaining regulatory approvals of human therapeutic products. Accordingly, our competitors may succeed in obtaining FDA approval for products more rapidly than we do, which would provide these competitors with an advantage for the marketing of products with similar potential uses. Furthermore, if we are permitted to commence commercial sales of products, we may also be competing with respect to manufacturing and product distribution efficiency and sales and marketing capabilities, areas in which we have limited or no experience as an organization.
Our target patient population for our proprietary combinations is people with Type 2 diabetes. Other products are currently in development or exist in the market that may compete directly with the products that we are seeking to develop and market. Various products are available to treat Type 2 diabetes, including, sulfonyureas, metformin, insulin, glinides, alpha-glucosidase inhibitors, and thiazolidinediones.
In addition, several companies are developing various approaches to improve treatments for Type 1 and Type 2 diabetes. We cannot predict whether our proprietary combinations, even if successfully tested and developed, will have sufficient advantages over existing products to cause health care professionals to adopt them over other products or that our specific combinations will offer an economically feasible alternative to existing products.
We may not be able to keep up with the rapid technological change in the biotechnology and pharmaceutical industries, which could make our products obsolete and reduce our revenues.
Biotechnology and related pharmaceutical technologies have undergone and continue to be subject to rapid and significant change. Our future will depend in large part on our ability to maintain a competitive position with respect to these technologies. Any products that we develop may become obsolete before we recover expenses incurred in developing those products, which may require that we raise additional funds to continue our operations.
Our future success depends on our ability to retain our chief executive officer and other key executives.
Our success largely depends on the skills, experience and efforts of our key personnel, including our Chief Executive Officer, Jay B. Lichter, Ph.D. We have entered into a written employment agreement with Dr. Lichter that can be terminated at any time by us or by Dr. Lichter. The loss of Dr. Lichter, or our failure to retain other key personnel, would jeopardize our ability to execute our strategic plan and materially harm our business.
Our future success depends on our relationships with current and future consultants with specific areas of expertise that are critical to our business.
We currently have only two employees, and we have no immediate plans to hire additional employees. We currently are utilizing the services of eight consultants with expertise in such fields critical to our business as contract manufacturing, regulatory affairs, biostatistics, and diabetology. If we are unable to retain the services of those consultants, or obtain the services of additional consultants, our likelihood of success could decrease significantly.
-22-
Our business has a substantial risk of product liability claims, and insurance may be expensive or unavailable.
Our business exposes us to potential product liability risks that are inherent in the testing, manufacturing and marketing of human therapeutic products. Product liability claims could result in a recall of products or a change in the indications for which they may be used, either of which may have a material adverse effect on our business and results of operations.
We currently have limited product liability insurance, including clinical trial insurance, and will seek additional coverage prior to initiating clinical trials and marketing any of our specific combinations. However, we cannot assure you that our insurance will provide adequate coverage against potential liabilities. Furthermore, clinical trial and product liability insurance is becoming increasingly expensive. As a result, we may not be able to maintain current amounts of insurance coverage, obtain additional insurance or obtain insurance at a reasonable cost or in sufficient amounts to protect against losses that could have a material adverse effect on us.
Item 2.Properties
We lease an aggregate of approximately 300 square feet of office space located in La Jolla, California, and San Diego, California pursuant to two month-to-month leases. The La Jolla office space is sublet from Avalon Ventures. One of our directors, Kevin Kinsella, is a managing member of Avalon Ventures. The San Diego office space is sublet from Sirion Therapeutics, Inc. (“Sirion”). Jay Lichter, our Chief Executive Officer and a member of our Board of Directors, is a former officer and current consultant to Sirion, and Kevin Kinsella is a current member of the Board of Directors of Sirion. The Board of Directors has determined that the rent charged to us for both leases is fair and reasonable. We believe that our facilities are adequate to meet our operating needs for the foreseeable future.
Item 3.Legal Proceedings
We are not a party to any legal proceedings.
Item 4.Submission of Matters to a Vote of Security Holders
There were no matters submitted to a vote of security holders during the fourth quarter of 2006.
-23-
PART II
Item 5.Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Our common stock is traded on the OTC Bulletin Board Market under the symbol “AKES.OB”. The following table presents quarterly information on the price range of high and low sales prices for our common stock for the periods indicated since January 1, 2005.
| | | | | | |
| | | High
| | Low
|
2006 | | | | | | |
First Quarter | | $ | 2.49 | | $ | 1.30 |
Second Quarter | | $ | 1.60 | | $ | 0.55 |
Third Quarter | | $ | 1.25 | | $ | 0.55 |
Fourth Quarter | | $ | 1.50 | | $ | 0.60 |
| | |
2005 | | | | | | |
First Quarter | | $ | 8.75 | | $ | 4.75 |
Second Quarter | | $ | 9.48 | | $ | 5.35 |
Third Quarter | | $ | 9.60 | | $ | 5.40 |
Fourth Quarter | | $ | 7.50 | | $ | 1.65 |
As of January 31, 2007, there were approximately 408 stockholders of record of our common stock with approximately 22,580,884 shares outstanding. We have never declared or paid any dividends and do not expect to pay any dividends in the foreseeable future. We did not repurchase any securities of the Company in the fourth quarter of 2006.
The following table summarizes the securities authorized for issuance under our equity compensation plans as of December 31, 2006.
| | | | | | | |
Plan Category
| | Number of
Securities to be
Issued Upon
Exercise of
Outstanding
Options,
Warrants and
Rights
| | Weighted
Average
Exercise Price
of Outstanding
Options,
Warrants and
Rights
| | Number of
Securities
Remaining
Available for
Future
Issuance Under
Equity
Compensation
Plans
|
Equity compensation plans approved by stockholders | | 200,000 | | $ | 1.94 | | 1,100,000 |
Equity compensation arrangements not approved by stockholders | | 800,000 | | $ | 1.08 | | |
Warrants issued | | 1,726,300 | | $ | 0.94 | | |
| | |
| | | | |
|
Total | | 2,726,300 | | $ | 1.06 | | 1,100,000 |
| | |
| | | | |
|
-24-
Item 6.Selected Financial Data
The following table shows selected financial data. The selected financial data has been derived from our audited consolidated financial statements for fiscal years 2002 through 2006 and is qualified by reference to, and should be read in conjunction with, the Consolidated Financial Statements, and Notes thereto, and “Management’s Discussion and Analysis of Financial Condition and Results of Operations”, included in Item 7 of Part II of this report.
| | | | | | | | | | | | | | | | | | | | |
| | | Years Ended December 31,
| |
| | | 2006
| | | 2005
| | | 2004
| | | 2003
| | | 2002
| |
Consolidated Statement of Operations Data: | | | | | | | | | | | | | | | | | | | | |
Revenues | | $ | — | | | $ | — | | | $ | — | | | $ | 4,785 | | | $ | 26,621 | |
Operating expenses | | $ | 2,415,119 | | | $ | 3,110,246 | | | $ | 1,517,691 | | | $ | 28,480 | | | $ | 152,283 | |
Loss from operations | | $ | (2,415,119 | ) | | $ | (3,110,246 | ) | | $ | (1,517,691 | ) | | $ | (25,397 | ) | | $ | (130,726 | ) |
Net loss | | $ | (2,405,567 | ) | | $ | (3,105,826 | ) | | $ | (1,525,539 | ) | | $ | (31,291 | ) | | $ | (132,687 | ) |
Net loss per common share—basic and diluted | | $ | (0.15 | ) | | $ | (0.21 | ) | | $ | (0.24 | ) | | $ | (0.01 | ) | | $ | (0.02 | ) |
Weighted average common shares outstanding—basic and diluted | | | 16,020,232 | | | | 14,993,031 | | | | 6,341,604 | | | | 5,377,466 | | | | 5,377,466 | |
Consolidated Balance Sheet Data: | | | | | | | | | | | | | | | | | | | | |
Cash and cash equivalents | | $ | 4,111,070 | | | $ | 388,551 | | | $ | 1,234,250 | | | $ | — | | | $ | 1,747 | |
Working capital | | $ | 3,973,153 | | | $ | 320,278 | | | $ | 1,267,814 | | | $ | (29,621 | ) | | $ | (20,814 | ) |
Total assets | | $ | 4,199,083 | | | $ | 415,107 | | | $ | 1,350,250 | | | $ | — | | | $ | 5,697 | |
Stockholder loan | | $ | — | | | $ | — | | | $ | — | | | $ | 92,930 | | | $ | 70,716 | |
Stockholders’ equity (deficit) | | $ | 4,058,615 | | | $ | 337,670 | | | $ | 1,267,814 | | | $ | (122,551 | ) | | $ | (91,530 | ) |
| | | | | | | | | | | | | | | | |
| | | Quarters Ended (Unaudited)
| |
| Quarterly Statement of Operations Data For 2006: | | March 31,
| | | June 30,
| | | September 30,
| | | December 31,
| |
Total revenues | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
Net loss | | $ | (552,251 | ) | | $ | (446,313 | ) | | $ | (378,005 | ) | | $ | (1,028,998 | ) |
Basic and diluted net loss per share | | $ | (0.04 | ) | | $ | (0.03 | ) | | $ | (0.02 | ) | | $ | (0.06 | ) |
Basic and diluted weighted average number of shares of common stock | | | 15,228,552 | | | | 15,347,552 | | | | 15,347,552 | | | | 18,131,609 | |
| |
| | | Quarters Ended (Unaudited)
| |
| Quarterly Statement of Operations Data For 2005: | | March 31,
| | | June 30,
| | | September 30,
| | | December 31,
| |
Total revenues | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
Net loss | | $ | (892,500 | ) | | $ | (862,088 | ) | | $ | (777,725 | ) | | $ | (573,513 | ) |
Basic and diluted net loss per share | | $ | (0.06 | ) | | $ | (0.06 | ) | | $ | (0.05 | ) | | $ | (0.04 | ) |
Basic and diluted weighted average number of shares of common stock | | | 14,992,552 | | | | 14,992,552 | | | | 14,992,552 | | | | 14,994,454 | |
Item 7.Management’s Discussion and Analysis of Financial Condition and Results of Operations
This discussion and analysis should be read in conjunction with our audited financial statements and accompanying notes included elsewhere in this report. Operating results are not necessarily indicative of results that may occur in future periods.
Introduction
One of our predecessors, Akesis Delaware, was incorporated on April 27, 1998, for the purpose of direct marketing to consumers an established over-the-counter product for lowering blood glucose levels in the treatment of diabetes.
-25-
Effective December 9, 2004, pursuant to the Merger Agreement among Akesis Delaware, Liberty and MergerSub, MergerSub merged with and into Akesis Delaware, with Akesis Delaware as the surviving corporation and wholly owned subsidiary of Liberty.
Although we acquired Akesis Delaware as a result of the transaction, Akesis Delaware stockholders held a majority of our common stock following the transaction. Accordingly, for accounting purposes, the acquisition was a “reverse acquisition” and Akesis Delaware was the “accounting acquiror.” Therefore, the transaction was accounted for as a recapitalization of Akesis Delaware and recorded based on the fair value of Liberty’s net tangible assets acquired by Akesis Delaware. No goodwill or other intangible assets were recorded. Effective January 11, 2005, we changed our name to Akesis Pharmaceuticals, Inc. and the trading symbol was changed to AKES.OB.
Since Akesis Delaware was the surviving entity, the “Selected Financial Data” in Item 6 above and the “Financial Statements and Supplementary Data,” elsewhere in this Form 10-K reflect Akesis Delaware’s historical results of operations prior to its acquisition by us. The Management’s Discussion and Analysis of Financial Condition and Results of Operations that follows is a discussion and analysis of that financial data. The accounts of Liberty and Akesis Delaware have been consolidated as of December 9, 2004, the effective date of the acquisition.
The following discussion of results of operations, liquidity and capital resources contains forward-looking statements that involve risks and uncertainties. As described in Part I, our actual results may differ materially from the results discussed in the forward-looking statements. Factors that might cause or contribute to such differences include those discussed below and in the section entitled “Risk Factors” of this report.
Major Research and Development Projects
We are an early stage biopharmaceutical company engaged in the discovery, development and commercialization of complementary and alternative therapies for the treatment of three principal forms of carbohydrate intolerance – Type 2 diabetes, Syndrome X, and impaired glucose tolerance – and their associated complications. We have been granted patents and filed patent applications for a number of proprietary combination therapies, including combinations with existing diabetes medications, for use in the treatment of Type 2 diabetes. We intend to use our proprietary combinations to develop prescription treatments for diabetes and related metabolic disorders. These products are in an early stage of development and no regulatory filings to commercialize our products have yet been made with the FDA or any similar state or foreign authorities.
We have completed an initial clinical study of a specific combination product and demonstrated a consistent improvement in HbA1clevels, as compared to base line, after three months of treatment in a diabetic population. The observed overall reduction in HbA1c, (which is an established long-term measure of blood glucose), in this open-label study was approximately 1.7% for all treatment groups. This reduction implies an average improvement of almost 20% in blood glucose parameters in this patient population, including patients taking the initial product candidate as monotherapy, as well as with concomitant medications. We believe that these clinical studies may suggest that our combinations show the potential for enhancing currently available oral antidiabetic therapeutic agents. We intend to conduct follow-on feasibility clinical trials with one or more of our combination products with a goal of confirming and extending the results of our initial clinical studies. We believe that the successful completion of these feasibility trials could lead to partnering opportunities in the
-26-
pharmaceutical industry. We are not currently in discussions with the FDA regarding the specific requirements for approval of our products.
The risks and uncertainties associated with completing the development of our products on schedule, or at all, include the following, as well as the other risk factors described in this report:
| | • | | Our products may not be shown to be safe and efficacious in the clinical trials; |
| | • | | We may be unable to obtain regulatory approval of our products or be unable to obtain such approval on a timely basis; |
| | • | | We may be unable to recruit enough patients to complete the clinical trials in a timely manner; and |
| | • | | We may not have adequate funds to complete the development of our products even if we secure the additional amount of capital we have targeted if we have underestimated the cost of the clinical trials. |
If our products fail to achieve statistically significant results in the clinical trials, or we do not complete the clinical trials on a timely basis, our operations, financial position and liquidity could be severely impaired, including as follows:
| | • | | It could make it more difficult for us to consummate partnering opportunities in the pharmaceutical industry, or at all. |
| | • | | Our reputation among investors might be harmed, which could make it more difficult for us to obtain equity capital on attractive terms, or at all. |
Because of the many risks and uncertainties relating to the completion of clinical trials, consummation of partnering opportunities in the pharmaceutical industry, receipt of marketing approvals and acceptance in the marketplace, we cannot predict the period in which material cash inflows from our products will commence, if ever.
Results of Operations for the Years Ended December 31, 2006 and 2005
Year Ended December 31, 2006 Compared with December 31, 2005
Total operating expenses decreased to $2.4 million for the year ended December 31, 2006, from $3.1 million for the same time period in 2005. During the fourth quarter of 2005, we terminated one employee, and reduced the salaries of the three remaining employees to $14,400 annually. Effective March 15, 2006, we terminated a third employee. Salaries for the two remaining employees remained at $14,400 annually through November 15, 2006. At that time we increased the annual salary of our Chief Executive Officer and Chief Financial Officer to $150,000 and $100,000, respectively, and in addition paid performance bonuses to the Chief Executive Officer and Chief Financial Officer of $100,000 and $106,300, respectively. As a result, our cash-based payroll costs for 2006 decreased to approximately $268,000 from approximately $448,000 during 2005. Our non cash-based compensation costs for 2006 consisted of $1,150,961 of stock-based compensation charges determined in accordance with Statement of Financial Accounting Standards No. 123 (revised 2004), “Share Based Payment” (“SFAS No. 123(R)”) and warrants issued to two terminated employees with a value of $230,105
-27-
determined in accordance with a Black-Scholes model. Non cash-based compensation expense for 2005 consisted of stock-based compensation of approximately $1.8 million determined in accordance with SFAS No. 123(R). Also, we did not pay cash fees to our outside directors from October 1, 2005 through November 16, 2006. Director fees for the years ended December 31, 2006 and 2005 were $9,000 and $81,000, respectively. In addition, in May 2006, we began the development of a clinical research protocol and began conducting other activities to support the initiation of new clinical trials related to our proposed pharmaceutical products. The cost of the clinical trial work during the year ended December 31, 2006 was approximately $98,000 compared to zero for 2005. Finally, our legal fees decreased to approximately $154,000 during the year ended December 31, 2006 from approximately $277,000 for 2005. The primary reason for the decrease in legal fees during 2006 was that 2005 was the first full year we were a publicly traded company.
Year Ended December 31, 2005 Compared with December 31, 2004
Total operating expenses increased to $3.1 million for the year ended December 31, 2005, from $1.5 million for the same time period in 2004. Total operating expenses for the year ended December 31, 2005 included a non-cash stock-based compensation charge of approximately $1.8 million compared to $1.1 million for the year ended December 31, 2004. The stock-based compensation charges for the years ended December 31, 2005 and 2004 were determined in accordance with the provisions of SFAS No. 123(R), and recognized the expense for options to acquire our common stock that were issued to Edward B. Wilson, our former Chief Executive Officer, and John T. Hendrick, our current Chief Financial Officer, in December 2004 and to Kelly Joy, our former Vice President of Business Development, and Kevin Sayer, a member of our Board of Directors in December 2005. Additionally, we incurred approximately $448,000 in payroll related expenses during 2005 compared to approximately $32,000 during 2004 since we did not have any employees during 2004 prior to the completion of the acquisition of Akesis Delaware in December 2004. During the fourth quarter of 2005, in order to conserve cash, we terminated one employee and reduced the salaries of the remaining three employees to an amount approximating the minimum wage in the State of California. We also incurred approximately $456,000 in legal, audit and outside accounting fees, as well as printing and other charges, primarily related to being a publicly traded company during 2005 compared to $159,000 during 2004. Finally, our liability insurance premiums and outside director fees totaled approximately $198,000 during 2005 and we incurred no costs for those items during 2004. We did not pay fees to our outside directors during the fourth quarter of 2005.
Liquidity and Capital Resources
We have financed our operations primarily through the sale of equity securities and stockholder loans. We invest excess cash in investment securities that will be used to fund future operating costs. Cash, cash equivalents and investment securities totaled $4,111,070 at December 31, 2006, compared to $388,551 at December 31, 2005. We primarily fund current operations with our existing cash and investments. Cash used in operating activities for 2006 totaled $877,807. We have no long-term financial commitments.
We had no revenues or other income sources in 2006 to cover operating expenses, and we do not expect any revenues in the foreseeable future. During the fourth quarter of 2006, the salaries of our employees and other operating expenses increased significantly from the levels we incurred during the first nine months of 2006 and the fourth quarter of 2005. As explained in the following paragraphs, we sold common stock resulting in net proceeds of approximately $4.247 million in the fourth quarter of 2006. In addition, we have entered into a three year term loan credit facility that enables us to borrow
-28-
up to $1 million as more fully explained below. We believe our current financial resources should enable us to fund one clinical feasibility study as well as satisfy all of our general and administrative expenses for approximately the next fifteen to eighteen months based on our current level of commitments.
During the first quarter of 2006, we sold an aggregate of 180,000 shares of our common stock at a purchase price of $2.00 per share to certain accredited investors. In addition, we issued to those investors warrants to purchase an aggregate of up to 90,000 shares of our common stock in connection with the financing. The warrants are exercisable for shares of our common stock for three years from the date of issuance at an exercise price per share of $3.00. Our net proceeds in connection with those sales of common stock after deducting expenses related to the financing were approximately $350,000. All the shares and warrants issued in connection with the financing were exempt from registration by virtue of Section 4(2) of the Securities Act of 1933, as amended.
In connection with the financing described in the preceding paragraph, the Company (a) paid its placement agent a cash fee equal to one percent (1%) of all funds invested by investors introduced by such placement agent (excluding amounts paid by investors upon exercise of warrants), and (b) issued to its placement agent a warrant to purchase up to 11,550 shares of common stock.
During the fourth quarter of 2006, we sold an aggregate of 7,233,332 shares of our common stock at a purchase price of $0.60 per share to certain accredited investors. In addition, we issued to those investors warrants to purchase an aggregate of up to 1,085,000 shares of our common stock in connection with the financing. The warrants are exercisable for shares of our common stock for three years from the date of issuance at an exercise price per share of $0.60. Our net proceeds in connection with those sales of common stock after deducting expenses related to the financing were approximately $4.247 million. All the shares and warrants issued in connection with the financing were exempt from registration by virtue of Section 4(2) of the Securities Act of 1933, as amended.
In connection with the financing described in the preceding paragraph, the Company (a) paid its placement agent a cash fee equal to eight percent (8%) of all funds invested by investors introduced by such placement agent (excluding amounts paid by investors upon exercise of warrants), and (b) issued to its placement agent a warrant to purchase up to 83,000 shares of common stock.
In December 2006, we entered into a loan and security agreement with Square 1 Bank that provides for a loan of up to $1.0 million to finance general corporate purposes. The loan bears an interest rate equal to the lender’s prime rate plus 0.75% and matures in December 2009. We have granted a security interest in substantially all of our tangible assets as collateral for the loans under the loan and security agreement. The agreement imposes certain limitations on our ability to engage in certain transactions. At December 31, 2006, we had no borrowings under the loan and security agreement.
In order to finance additional feasibility trials beyond the trial scheduled for 2007-2008 to further validate our products, we will need to raise a significant amount of capital. We will also need to raise additional capital to finance our future operating cash needs. We may seek to raise capital through the sale of equity or debt securities or the development of other funding mechanisms. In addition, we may seek to form a strategic partnership for the development and commercialization of our products.
The first clinical feasibility study that we intend to initiate with the proceeds from the fourth quarter 2006 private placements of our common stock is related to our metformin combination product.
-29-
If we realize sufficient proceeds from future sources of capital, then we also plan to conduct clinical studies related to additional combination products including our thalidazione combination product.
Our actual capital requirements will depend upon numerous factors, including:
| | • | | the rate of progress and costs of our clinical trial and research and development activities; |
| | • | | actions taken by the FDA and other regulatory authorities; |
| | • | | the timing and amount of milestone or other payments we might receive from potential strategic partners; |
| | • | | our degree of success in commercializing our product candidates; |
| | • | | the emergence of competing technologies and products, and other adverse market developments; and |
| | • | | the costs of preparing, filing, prosecuting, maintaining and enforcing patent claims and other intellectual property rights. |
There can be no assurance that we will be able to obtain needed additional capital or enter into relationships with corporate partners on a timely basis, on favorable terms, or at all. Conditions in the capital markets in general, and the life science capital market specifically, may affect our potential financing sources and opportunities for strategic partnering.
Critical Accounting Policies
Basis of Revenue Recognition: To date, we have not had any significant ongoing revenue sources.
Stock-based compensation: We adopted SFAS No. 123(R) in 2004. Stock-based compensation for 2006, 2005 and 2004 was $1,150,961, $1,804,000 and $1,087,500, respectively.
Effect of New Accounting Standards
In March 2006, the Financial Accounting Standards Board (“FASB”) issued SFAS No. 156, “Accounting for Servicing of Financial Assets – an amendment of SFAS No. 140” (“SFAS No. 156”), which clarifies when servicing rights should be separately accounted for, requires companies to account for separately recognized servicing rights initially at fair value, and gives companies the option of subsequently accounting for those servicing rights at either fair value or under the amortization method. SFAS No. 156 is effective for fiscal years beginning after September 15, 2006. We do not expect the adoption of SFAS No. 156 to affect future reporting or disclosures.
In June 2006, the FASB issued Interpretation No. 48, “Accounting for Uncertainty in Income Taxes” (“FIN 48”). FIN 48 clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements in accordance with SFAS No. 109, “Accounting for Income Taxes”. This Interpretation prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. This Interpretation also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, and disclosure. FIN 48 is effective for fiscal years beginning after December 15, 2006. We are in the process of evaluating the adoption of FIN 48.
-30-
In September 2006, the FASB issued SFAS No. 157, “Fair Value Measurements” (“SFAS No. 157”). SFAS No. 157 provides guidance for using fair value to measure assets and liabilities and requires expanded information about the extent to which companies measure assets and liabilities at fair value, the information used to measure fair value, and the effect of fair value measurements on earnings. The standard applies whenever other standards require (or permit) assets or liabilities to be measured at fair value. The standard does not expand the use of fair value in any new circumstances. SFAS No. 157 is effective for fiscal years beginning after November 15, 2007. We are in the process of evaluating the adoption of SFAS No. 157.
In September 2006, the FASB issued SFAS No. 158, “Employers’ Accounting for Defined Benefit Pension and Other Postretirement Plans” (“SFAS No. 158”), an amendment of SFAS No. 87, 88, 106, and 132(R). SFAS No. 158 requires, among other things, that a company (1) recognize a net liability or asset to report the funded status of their defined benefit pensions and other postretirement plans on its balance sheet and (2) measure benefit plan assets and benefit obligations as of the company’s balance sheet date. SFAS No. 158 is effective for calendar year-end companies with publicly traded equity securities as of December 31, 2006. We do not expect the adoption of SFAS No. 158 to affect future reporting or disclosures.
Item 7A.Quantitative and Qualitative Disclosures About Market Risk
We do not use derivative financial instruments for trading or speculative purposes. However, we regularly invest excess cash in short-term investments that are subject to changes in short-term interest rates. We believe that the market risk arising from holding these financial instruments is minimal.
Because we have minimal debt, our exposure to market risks associated with changes in interest rates arise from increases or decreases in interest income earned on our investment portfolio. We attempt to ensure the safety and preservation of invested funds by limiting default risks, market risk, and reinvestment risk. We mitigate default risk by investing in short-term investments. A hypothetical 100 basis point decrease in interest rates along the entire interest rate yield curve would not materially affect the fair value of our interest sensitive financial instruments at December 31, 2006.
-31-
Item 8.Financial Statements and Supplementary Data
AKESIS PHARMACEUTICALS, INC.
INDEX TO FINANCIAL STATEMENTS
-32-
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
Akesis Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of Akesis Pharmaceuticals, Inc., a Nevada corporation, as of December 31, 2006 and 2005 and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2006. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of the Company as of December 31, 2006 and 2005 and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2006, in conformity with United States generally accepted accounting principles.
SWENSON ADVISORS, LLP
Independent Registered Public Accounting Firm
San Diego, California
February 7, 2007
-33-
Akesis Pharmaceuticals, Inc.
Consolidated Balance Sheets
As of December 31, 2006 and 2005
| | | | | | | | |
| | | 2006
| | | 2005
| |
Assets | | | | | | | | |
Current assets: | | | | | | | | |
Cash and cash equivalents | | $ | 4,111,070 | | | $ | 388,551 | |
Prepaid insurance and other current assets | | | 2,551 | | | | 9,164 | |
| | |
|
|
| |
|
|
|
Total current assets | | | 4,113,621 | | | | 397,715 | |
Property and equipment, net | | | — | | | | 17,392 | |
Debt issuance costs, net | | | 85,462 | | | | — | |
| | |
|
|
| |
|
|
|
Total assets | | $ | 4,199,083 | | | $ | 415,107 | |
| | |
|
|
| |
|
|
|
Liabilities and Stockholders’ Equity (Deficit) | | | | | | | | |
Current liabilities: | | | | | | | | |
Accounts payable | | $ | 140,468 | | | $ | 77,437 | |
| | |
|
|
| |
|
|
|
Total current liabilities | | | 140,468 | | | | 77,437 | |
| | |
|
|
| |
|
|
|
Total liabilities | | | 140,468 | | | | 77,437 | |
| | |
|
|
| |
|
|
|
Commitments and contingencies (Note 5) | | | — | | | | — | |
Stockholders’ equity: | | | | | | | | |
Convertible preferred stock, $0.001 par value, 10,000,000 shares authorized, and zero shares issued and outstanding as of December 31, 2006 and December 31, 2005 | | | — | | | | — | |
Common stock, $0.001 par value, 50,000,000 shares authorized: 22,580,884 and 15,167,552 shares issued and outstanding at December 31, 2006 and December 31, 2005, respectively | | | 22,581 | | | | 15,168 | |
Additional paid-in capital | | | 12,292,655 | | | | 6,173,556 | |
Accumulated deficit | | | (8,256,621 | ) | | | (5,851,054 | ) |
| | |
|
|
| |
|
|
|
Total stockholders’ equity | | | 4,058,615 | | | | 337,670 | |
| | |
|
|
| |
|
|
|
Total liabilities and stockholders’ equity | | $ | 4,199,083 | | | $ | 415,107 | |
| | |
|
|
| |
|
|
|
See accompanying notes.
-34-
Akesis Pharmaceuticals, Inc.
Consolidated Statements of Operations
For the Years Ended December 31, 2006, 2005, and 2004
| | | | | | | | | | | | |
| | | 2006
| | | 2005
| | | 2004
| |
Revenue | | $ | — | | | $ | — | | | $ | — | |
| | |
|
|
| |
|
|
| |
|
|
|
Operating costs and expenses: | | | | | | | | | | | | |
Selling, general and administrative | | | 2,316,935 | | | | 3,110,246 | | | | 1,517,691 | |
Research and development | | | 98,184 | | | | — | | | | — | |
| | |
|
|
| |
|
|
| |
|
|
|
Total expenses | | | 2,415,119 | | | | 3,110,246 | | | | 1,517,691 | |
| | |
|
|
| |
|
|
| |
|
|
|
Loss from operations | | | (2,415,119 | ) | | | (3,110,246 | ) | | | (1,517,691 | ) |
Interest income/(expense), net | | | 25,157 | | | | 7,620 | | | | (6,248 | ) |
Loss on disposal of assets | | | (13,805 | ) | | | — | | | | — | |
| | |
|
|
| |
|
|
| |
|
|
|
Loss before income taxes | | | (2,403,767 | ) | | | (3,102,626 | ) | | | (1,523,939 | ) |
Provision for income taxes | | | 1,800 | | | | 3,200 | | | | 1,600 | |
| | |
|
|
| |
|
|
| |
|
|
|
Net loss | | $ | (2,405,567 | ) | | $ | (3,105,826 | ) | | $ | (1,525,539 | ) |
| | |
|
|
| |
|
|
| |
|
|
|
Net loss per common share – basic and diluted | | $ | (0.15 | ) | | $ | (0.21 | ) | | $ | (0.24 | ) |
| | |
|
|
| |
|
|
| |
|
|
|
Weighted-average common shares outstanding – basic and diluted | | | 16,020,232 | | | | 14,993,031 | | | | 6,341,604 | |
| | |
|
|
| |
|
|
| |
|
|
|
See accompanying notes.
-35-
Akesis Pharmaceuticals, Inc.
Consolidated Statements of Stockholders’ Equity
For the Years Ended December 31, 2006, 2005, and 2004
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Convertible Preferred Stock
| | | | | | | Additional Paid-In Capital
| | Accumulated Deficit
| |
| | | Series A
| | | Series B
| | | Series C
| | | Common Stock
| | |
| | | Shares
| | | Amount
| | | Shares
| | | Amount
| | | Shares
| | | Amount
| | | Shares
| | Amount
| | |
Balance at December 31, 2003 | | 404,444 | | | $ | 404 | | | 166,022 | | | $ | 166 | | | — | | | $ | — | | | 5,377,466 | | $ | 1,633 | | $ | 1,094,935 | | $ | (1,219,689 | ) |
Issuance of preferred stock – series C | | — | | | | — | | | — | | | | — | | | 288,653 | | | | 289 | | | — | | | — | | | 129,326 | | | — | |
Issuance of common stock | | — | | | | — | | | — | | | | — | | | — | | | | — | | | 1,366,316 | | | 415 | | | 165,585 | | | — | |
Conversion of preferred stock – series A | | (404,444 | ) | | | (404 | ) | | — | | | | — | | | — | | | | — | | | 1,331,562 | | | 404 | | | — | | | — | |
Conversion of preferred stock – series B | | — | | | | — | | | (166,022 | ) | | | (166 | ) | | — | | | | — | | | 546,599 | | | 166 | | | — | | | — | |
Conversion of preferred stock – series C | | — | | | | — | | | — | | | | — | | | (288,653 | ) | | | (289 | ) | | 950,340 | | | 289 | | | — | | | — | |
Exercise of stock options | | — | | | | — | | | — | | | | — | | | — | | | | — | | | 927,702 | | | 282 | | | 134,072 | | | — | |
Merger with Liberty Mint, Ltd. | | — | | | | — | | | — | | | | — | | | — | | | | — | | | 4,492,567 | | | 11,804 | | | 1,386,631 | | | — | |
Stock-based compensation | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | — | | | 1,087,500 | | | — | |
Net loss | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | — | | | — | | | (1,525,539 | ) |
| | |
|
| |
|
|
| |
|
| |
|
|
| |
|
| |
|
|
| |
| |
|
| |
|
| |
|
|
|
Balance at December 31, 2004 | | — | | | | — | | | — | | | | — | | | — | | | | — | | | 14,992,552 | | | 14,993 | | | 3,998,049 | | | (2,745,228 | ) |
Issuance of common stock | | — | | | | — | | | — | | | | — | | | — | | | | — | | | 175,000 | | | 175 | | | 339,900 | | | — | |
Issuance of warrants for private | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | — | | | — | | | — | |
placement fees | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | — | | | 31,607 | | | — | |
Stock-based compensation | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | — | | | 1,804,000 | | | — | |
Net loss | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | — | | | — | | | (3,105,826 | ) |
| | |
|
| |
|
|
| |
|
| |
|
|
| |
|
| |
|
|
| |
| |
|
| |
|
| |
|
|
|
Balance at December 31, 2005 | | — | | | | — | | | — | | | | — | | | — | | | | — | | | 15,167,552 | | | 15,168 | | | 6,173,556 | | | (5,851,054 | ) |
Issuance of common stock | | — | | | | — | | | — | | | | — | | | — | | | | — | | | 7,413,332 | | | 7,413 | | | 4,589,813 | | | — | |
Sale of warrants to former employees | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | — | | | 3,000 | | | — | |
Issuance of warrants for employee termination costs | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | — | | | 230,105 | | | — | |
Issuance of warrants for private placement fees | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | — | | | 89,715 | | | — | |
Issuance of warrants for debt issuance costs | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | — | | | 55,505 | | | — | |
Stock-based compensation | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | — | | | 1,150,961 | | | — | |
Net loss | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | — | | | — | | | (2,405,567 | ) |
| | |
|
| |
|
|
| |
|
| |
|
|
| |
|
| |
|
|
| |
| |
|
| |
|
| |
|
|
|
Balance at December 31, 2006 | | — | | | $ | — | | | — | | | $ | — | | | — | | | $ | — | | | 22,580,884 | | $ | 22,581 | | $ | 12,292,655 | | $ | (8,256,621 | ) |
| | |
|
| |
|
|
| |
|
| |
|
|
| |
|
| |
|
|
| |
| |
|
| |
|
| |
|
|
|
See accompanying notes.
-36-
Akesis Pharmaceuticals, Inc.
Consolidated Statements of Cash Flows
For the Years Ended December 31, 2006, 2005, and 2004
| | | | | | | | | | | | |
| | | Year Ended December 31,
| |
| | | 2006
| | | 2005
| | | 2004
| |
Cash flows from operating activities: | | | | | | | | | | | | |
Net loss | | $ | (2,405,567 | ) | | $ | (3,105,826 | ) | | $ | (1,525,539 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | |
Depreciation and amortization | | | 5,929 | | | | 4,192 | | | | — | |
Loss on disposal of assets | | | 13,805 | | | | — | | | | — | |
Stock-based compensation | | | 1,150,961 | | | | 1,804,000 | | | | 1,087,500 | |
Warrants issued for private placement fees | | | 89,715 | | | | 31,607 | | | | — | |
Warrants issued for employee terminations | | | 230,105 | | | | — | | | | — | |
Changes in assets and liabilities: | | | | | | | | | | | | |
Other current assets | | | 6,613 | | | | 106,836 | | | | (116,000 | ) |
Debt issuance costs, net of warrants issued to lender | | | (32,399 | ) | | | — | | | | — | |
Accounts payable | | | 63,031 | | | | (4,999 | ) | | | 52,815 | |
| | |
|
|
| |
|
|
| |
|
|
|
Net cash used in operating activities | | | (877,807 | ) | | | (1,164,190 | ) | | | (501,224 | ) |
| | |
|
|
| |
|
|
| |
|
|
|
Cash flows from investing activities: | | | | | | | | | | | | |
Sale/(Purchase) of property and equipment | | | 100 | | | | (21,584 | ) | | | — | |
| | |
|
|
| |
|
|
| |
|
|
|
Net cash provided (used) in investing activities | | | 100 | | | | (21,584 | ) | | | — | |
| | |
|
|
| |
|
|
| |
|
|
|
Cash flows from financing activities: | | | | | | | | | | | | |
Proceeds from Series C preferred stock issuances | | | — | | | | — | | | | 129,615 | |
Proceeds from common stock issuances | | | 4,597,226 | | | | 340,075 | | | | 1,698,789 | |
Proceeds from sale of warrants to former employees | | | 3,000 | | | | — | | | | — | |
Payment of shareholders’ loans | | | — | | | | — | | | | (92,930 | ) |
| | |
|
|
| |
|
|
| |
|
|
|
Net cash provided by financing activities | | | 4,600,226 | | | | 340,075 | | | | 1,735,474 | |
| | |
|
|
| |
|
|
| |
|
|
|
Net increase (decrease) in cash and cash equivalents | | | 3,722,519 | | | | (845,699 | ) | | | 1,234,250 | |
Cash and cash equivalents at beginning of period | | | 388,551 | | | | 1,234,250 | | | | — | |
| | |
|
|
| |
|
|
| |
|
|
|
Cash and cash equivalents at end of year | | $ | 4,111,070 | | | $ | 388,551 | | | $ | 1,234,250 | |
| | |
|
|
| |
|
|
| |
|
|
|
Supplemental Disclosures of Cash Flow Information: | | | | | | | | | | | | |
Interest Expense | | $ | 2,442 | | | $ | — | | | $ | 6,248 | |
Income Taxes Paid | | $ | 1,800 | | | $ | 3,200 | | | $ | 1,600 | |
Supplemental Disclosures of Non-Cash Investing and Financing Activities: | | | | | | | | | | | | |
Conversion of shareholders’ loans to Series C convertible preferred stock | | $ | — | | | $ | — | | | $ | 129,615 | |
Conversion of Series A, B and C convertible preferred stock to common stock | | $ | — | | | $ | — | | | $ | 1,120,404 | |
See accompanying notes.
-37-
Akesis Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
As of December 31, 2006 and 2005 and For the Years Ended December 31, 2006, 2005 and 2004
| 1. | The Company and Recapitalization |
One of our predecessors, Akesis Delaware, was incorporated on April 27, 1998, for the purpose of direct marketing to consumers an established over-the-counter product for lowering blood glucose levels in the treatment of diabetes.
Effective December 9, 2004, pursuant to the Merger Agreement among Akesis Delaware, Liberty and MergerSub, MergerSub merged with and into Akesis Delaware, with Akesis Delaware as the surviving corporation and wholly owned subsidiary of Liberty.
Although Liberty acquired Akesis Delaware as a result of the transaction, Akesis Delaware stockholders held a majority of the Company’s common stock following the transaction. Accordingly, for accounting purposes, the acquisition was a “reverse acquisition” and Akesis Delaware was the “accounting acquiror.” Therefore, the transaction was accounted for as a recapitalization of Akesis Delaware and recorded based on the fair value of Liberty’s net tangible assets acquired by Akesis Delaware. No goodwill or other intangible assets were recorded. Effective January 11, 2005, Liberty changed its name to Akesis Pharmaceuticals, Inc. and the trading symbol was changed to AKES.OB.
| 2. | Summary of Significant Accounting Policies |
Principles of consolidation
The acquisition of Akesis Delaware by Liberty has been accounted for as a recapitalization of Akesis Delaware as described in Note 1. Since Akesis Delaware is the surviving entity, the accompanying consolidated financial statements reflect its historical results of operations prior to the acquisition. The accounts of Liberty and Akesis Delaware have been consolidated as of December 9, 2004, the effective date of the acquisition described in Note 1 above.
Use of estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amount of expenses during the reporting period. Actual results could differ from those estimates.
Business risk and concentrations of credit risk
Akesis Delaware’s business is in the health care industry and sells products that may not be successful in the marketplace. Financial instruments which potentially subject the Company to concentrations of credit risk consist primarily of cash and cash equivalents, including money market accounts. Substantially all of our cash and cash equivalents are maintained with one financial institution in the United States. Deposits held with that financial institution exceed the amount of insurance provided on such deposits. Those deposits may be redeemed upon demand and, therefore, bear minimal risk.
Fair value of financial instruments
The carrying amounts of cash and cash equivalents, prepaid assets and accounts payable approximate fair market value because of the short maturity of those instruments.
-38-
Cash and cash equivalents
Cash equivalents consist of highly liquid investment with original maturities of three months or less when purchased.
Property and equipment
Property and equipment are stated at cost and depreciated using the straight-line method over the estimated useful lives of the related assets ranging from 3 to 5 years. Maintenance and repairs are charged to expense as incurred, and improvements and betterments are capitalized. When assets are retired or otherwise disposed of, the cost and accumulated depreciation and amortization are removed from the accounts and any resulting gain or loss is reflected in operations in the period realized.
Income taxes
Income taxes are accounted for in accordance with Statement of Financial Accounting Standards No. 109, “Accounting for Income Taxes” (“SFAS No. 109”). Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities and net operating loss and credit carryforwards using enacted tax rates in effect for the year in which the differences are expected to reverse. Valuation allowances are established when necessary to reduce deferred tax assets to the amounts expected to be realized.
Revenue recognition
Product sales are recognized upon shipment to the customer and when payment is probable or collected immediately.
Research and development
Research and development costs are expensed as incurred. Such costs include consultants, supplies, and clinical trials.
Stock-based compensation
In December 2004, the FASB issued SFAS No. 123(R). SFAS No. 123(R) requires all share-based payments to employees, including grants of employee stock options, to be recognized in the financial statements based on their fair values and does not allow the previously permitted pro forma disclosure as an alternative to financial statement recognition. SFAS No. 123(R) supersedes Accounting Principles Board Opinion No. 25, “Accounting for Stock Issued to Employees” and related interpretations and amends SFAS No. 95, Statement of Cash Flows. SFAS No. 123(R) is required to be effective beginning in the first quarter of fiscal 2006. However, the Company decided to adopt SFAS No. 123(R) effective with the acquisition of Akesis Delaware by Liberty on December 9, 2004. In March 2005, the SEC issued Staff Accounting Bulletin No. 107 (“SAB 107”) relating to SFAS No. 123(R), which among other things, expanded the coverage of SFAS No. 123(R) to include share-based payments to outside directors. The Company has applied the provisions of SAB 107 in its adoption of SFAS No. 123(R).
Compensation costs for all share-based awards to employees and outside directors are measured based on the grant date fair value of those awards and is recognized over the period during which the employee or outside director is required to perform service in exchange for the award (generally over the vesting period of the award). The cost of share-based compensation awards is recognized during the period based on the value of the portion of share-based payment awards that is ultimately expected to vest during the period, and is amortized under the multiple option
-39-
methodology prescribed by SFAS No. 123(R). As share-based compensation expense recognized in the consolidated statement of operations for the years ended December 31, 2006, 2005 and 2004 is based on awards ultimately expected to vest, it has been reduced for estimated forfeitures. SFAS No. 123(R) requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
We have no awards with market or performance conditions. Excess tax benefits, as defined by SFAS No. 123(R), will be recognized as an addition to additional paid-in capital. The adoption of the SFAS No. 123(R) fair value method resulted in a non-cash stock-based compensation charge on the Company’s reported results of operations of $1,150,961, $1,804,000 and $1,077,000 for the years ended December 31, 2006, 2005 and 2004, respectively.
Prior to the adoption of SFAS No. 123(R), no stock options had been issued by Akesis Delaware to employees. However, stock options were issued prior to the recapitalization to non-employees and were recorded at their fair value in accordance with SFAS No. 123 and EITF 96-18, “Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling, Goods or Services”. Such stock options to non-employees were periodically re-measured as the stock options vested, and no re-measurement issues having a material impact on the financial statements were identified.
Stock offering costs
Expenses incurred in connection with common stock issuances are recorded as an offset to additional paid-in capital on the consolidated balance sheets. Such expenses consist of third-party related offering expenses.
Debt issuance costs
Debt issuance costs incurred to obtain debt financing are deferred and included on the consolidated balance sheets. The costs are amortized over the term of the debt. The amortization expense is included in interest expense on the consolidated statements of operations.
Net loss per share
Basic and diluted net loss per share is computed in accordance with Statement of Financial Accounting Standards No. 128, “Earnings per Share”. Basic loss per share includes no dilution and is computed by dividing net loss by the weighted-average number of shares of common stock outstanding for the period. Diluted loss per share reflects the potential dilution of securities that could share in the Company’s earnings, such as common stock equivalents which may be issued upon exercise of outstanding common stock options. Diluted loss per share is identical to basic loss per share for all periods reported because inclusion of common stock equivalents would be anti-dilutive.
For the years ended December 31, 2006, 2005, and 2004, the following options and warrants to purchase shares of common stock were excluded from the computation of diluted net loss per share, as the inclusion of such shares would be antidilutive:
| | | | | | |
| | | Years Ended December 31,
|
| | | 2006
| | 2005
| | 2004
|
Stock options | | 1,000,000 | | 1,462,499 | | 1,062,499 |
Stock warrants | | 1,726,300 | | 106,750 | | — |
-40-
Effect of new accounting standards
In March 2006, the FASB issued SFAS No. 156, which clarifies when servicing rights should be separately accounted for, requires companies to account for separately recognized servicing rights initially at fair value, and gives companies the option of subsequently accounting for those servicing rights at either fair value or under the amortization method. SFAS No. 156 is effective for fiscal years beginning after September 15, 2006. The Company does not expect the adoption of SFAS No. 156 to affect future reporting or disclosures.
In June 2006, the FASB issued FIN 48. FIN 48 clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements in accordance with SFAS No. 109, “Accounting for Income Taxes”. FIN 48 prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. FIN 48 also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, and disclosure. FIN 48 is effective for fiscal years beginning after December 15, 2006. The Company is in the process of evaluating the adoption of FIN 48.
In September 2006, the FASB issued SFAS No. 157. SFAS No. 157 provides guidance for using fair value to measure assets and liabilities and requires expanded information about the extent to which companies measure assets and liabilities at fair value, the information used to measure fair value, and the effect of fair value measurements on earnings. The standard applies whenever other standards require (or permit) assets or liabilities to be measured at fair value. The standard does not expand the use of fair value in any new circumstances. SFAS No. 157 is effective for fiscal years beginning after November 15, 2007. The Company is in the process of evaluating the adoption of SFAS No. 157.
In September 2006, the FASB issued SFAS No. 158. SFAS No. 158 requires, among other things, that a company (1) recognize a net liability or asset to report the funded status of their defined benefit pensions and other postretirement plans on its balance sheet and (2) measure benefit plan assets and benefit obligations as of the company’s balance sheet date. SFAS No. 158 is effective for calendar year-end companies with publicly traded equity securities as of December 31, 2006. The Company does not expect the adoption of SFAS No. 158 to affect future reporting or disclosures.
As of December 31, 2004, Akesis Delaware had no property and equipment, and depreciation expense for the year ended December 31, 2004 was zero. Property and equipment as of December 31, 2005 consisted of the following:
| | | | | | | | | | |
| | | Cost
| | Accumulated
Depreciation
| | | Net
|
Furniture and fixtures | | $ | 14,919 | | $ | (2,753 | ) | | $ | 12,166 |
Office equipment | | | 6,665 | | | (1,439 | ) | | | 5,226 |
| | |
|
| |
|
|
| |
|
|
Total property and equipment | | $ | 21,584 | | $ | (4,192 | ) | | $ | 17,392 |
| | |
|
| |
|
|
| |
|
|
On October 2, 2006, the Company sold its furniture, fixtures and office equipment to its former chief executive officer for $100 as part of a Separation Agreement and Release. Depreciation expense recognized in 2006 prior to the sale of the assets was $3,487 and the Company recorded a loss in the consolidated statement of operations of $13,805 on the disposal of the assets. As of December 31, 2006, the Company had no property and equipment.
-41-
Akesis Delaware received working capital contributions from two related-party lenders prior to its acquisition by Liberty in December 2004. Included in these balances was a convertible promissory note effective April 4, 2002 with the related-party lenders. The notes were due and payable to the related-party lenders at the earlier of 18 months from the effective date, Akesis Delaware closing a minimum financing by qualified investors in excess of $1,000,000, execution of a licensing agreement that would provide sufficient cash flow to repay the note, or a sale of all or substantially all of the Company’s assets. Interest accrues at 8% per annum based on a 360 day period. The note, including principal and accrued interest, could have been converted to stock of Akesis Delaware at any time, including Series B Preferred Stock with a conversion price of $5.00 or the same security from the closing of a minimum financing transaction by qualified investors in excess of $1,000,000. Akesis Delaware had the right to prepay the note with a 20 day written notice to the lender in either cash or stock. If no election was made by the lender within 10 days of notice, the prepayment would have been made in cash.
In September 2004, the holders of the loans and the Akesis Delaware Board of Directors agreed to convert stockholder loan principal balances of $115,461 into Series C Preferred Stock at $0.40 per share for a total of 288,653 shares. The principal balances converted into Series C Preferred Stock as of September 30, 2004. Additionally, Akesis Delaware recorded imputed interest of $14,154 as of September 30, 2004. However, interest was forgiven by the lenders, not paid upon conversion of the loans, and accordingly was recorded as additional paid-in capital.
| 5. | Commitments, Contingencies and Related Party Transactions |
Leases
The Company leases an aggregate of approximately 300 square feet of office space located in La Jolla, California, and San Diego, California, pursuant to two leases each on a month-to-month basis. The La Jolla office space is sublet from Avalon Ventures. One of the Company’s directors, Kevin Kinsella, is a managing member of Avalon Ventures. The San Diego office space is sublet from Sirion. Jay Lichter, the Company’s Chief Executive Officer and a member of the Board of Directors, is a former officer and current consultant to Sirion, and Kevin Kinsella is a current member of the Board of Directors of Sirion. In addition, from December 9, 2004 through July 31, 2006, the Company sublet approximately 900 square feet of office space in Carefree, Arizona from its former chief executive officer at his cost. The Board of Directors has determined that the rent charged to the Company for all three leases is fair and reasonable. The Company recorded rent expense during the years ended December 31, 2006, 2005 and 2004 of $25,200, $24,840 and $5,500, respectively.
Loan
In December 2006, the Company entered into a loan and security agreement with Square 1 Bank (the “Bank”) that provides for a loan of up to $1.0 million to finance general corporate purposes. The loan bears an interest rate equal to the Bank’s prime rate plus 0.75% and matures in December 2009. The agreement requires monthly interest payments on any outstanding principal balance, and any outstanding principal balance is amortized and payable over the term of the agreement beginning in June 2007. The Company has granted a security interest in substantially all of its tangible assets as collateral for the loans under the loan and security agreement, and the agreement imposes certain limitations on the Company’s ability to engage in certain transactions. At December 31, 2006, the Company had no borrowings under the loan and security agreement.
-42-
| 6. | Stock-based Compensation |
Stock-based compensation expense for the years ended December 31, 2006, 2005 and 2004, was $1,150,961, $1,804,000 and $1,087,500, respectively. Since we have a net operating loss carryforward as of December 31, 2006, no excess tax benefits for the tax deductions related to share-based awards were recognized in the consolidated statement of operations. At the present time, we intend to issue new common shares upon the exercise of stock options. None of the share-based awards are classified as a liability as of December 31, 2006.
Immediately following the acquisition of Akesis Delaware by Liberty in December 2004, two executive officers became entitled, through their respective employment offer letters, to nonstatutory stock options with a term of 10 years to acquire a total of 1,062,499 shares of common stock at an exercise price of $1.50 per share. Twenty percent of the shares of common stock subject to the options vested as of the effective date of the officers’ employment immediately following the acquisition of Akesis Delaware by Liberty and one forty-eighth (1/48th) of the remaining shares subject to the options were scheduled to vest each month following the effective date of the officers’ employment, subject to the officers’ continued employment with the Company on any such date. In addition, in the event of a change of control of the Company, then the officers would fully vest in and have the right to exercise the options as to all of the shares of common stock subject to the options. Effective September 30, 2006, one of the officers resigned and did not exercise the option to acquire the shares that had vested under the option agreement.
On October 2, 2006, the Company issued nonstatutory stock options with a term of 10 years to acquire a total of 600,000 shares of common stock at an exercise price of $0.94 per share to two executive officers and an outside director. The shares subject to the options vest one thirty-sixth (1/36th) each month for three years from the date of grant subject to the officers’ continued employment with the Company and the outside director’s continued service on the Company’s Board of Directors. In addition, in the event of a change of control of the Company, then the officers and outside director shall fully vest in and have the right to exercise the options as to all of the shares of common stock subject to the options.
The Company’s Board of Directors also authorized and reserved 1,500,000 shares of the Company’s common stock pursuant to a 2005 Stock Plan in January 2005 for option grants to the Company’s employees, directors and consultants. Options were granted pursuant to such 2005 Stock Plan to an officer and an outside director during the year ended December 31, 2005 for each of them to acquire 200,000 shares of our common stock. The stock options are nonqualified stock options with a term of 10 years and an exercise price of $1.94 per share. Twenty-five percent of the shares of common stock subject to the options were scheduled to vest as of the first anniversary of the officer’s and outside director’s service to the Company, and one forty-eighth (1/48th) of the shares subject to the options were scheduled to vest each month following the first anniversary of the commencement of the officer’s and outside director’s service to the Company, subject to the officer’s and outside director’s continued service to the Company on any such date. In addition, in the event of a change of control of the Company, then the officer and outside director would fully vest in and have the right to exercise the options as to all of the shares of common stock subject to the options as to which the officer and outside director would not otherwise be vested or exercisable. Both the officer and outside director joined the Company in January 2005. Effective March 15, 2006, the officer resigned and did not exercise the option to acquire the shares that had vested under the option agreement.
-43-
The fair value of each option award is estimated on the date of grant using the Black-Scholes method for option pricing that uses the assumptions noted in the following table. Expected volatilities are based on historical volatility of our common stock and other factors. The expected term of options granted is based on our management’s estimate since our operating history is too brief to have established historical rates for employee termination and option exercises. The risk-free interest rates are based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of the grant. Assumptions used in the Black-Scholes model for the years ended December 31, 2006, 2005 and 2004, respectively, were as follows:
| | | | | | |
| | | Years Ended December 31,
|
| | | 2006
| | 2005
| | 2004
|
Expected volatility | | 125% | | 125% | | 85% |
Annual expected termination rate | | 35% | | 25% | | 5% |
Risk-free interest rate (zero coupon U.S. Treasury Note) | | 4.68% | | 3.7% to 4.0% | | 1.9% to 3.4% |
Expected dividend yield | | 0% | | 0% | | 0% |
The fair value of options granted during the years ended December 31, 2006, 2005 and 2004 was $292,153, $428,584 and $4,505,500, respectively, and the fair value is amortized over the vesting period of the option using the multiple option methodology in accordance with the provisions of SFAS No. 123(R).
The following is a summary of the status of the 2006 and 2004 nonstatutory options and the options under the 2005 Stock Plan for the years ended December 31, 2006, 2005 and 2004:
| | | | | | | | | | | |
| | | Number
of Shares
| | | Weighted
Average
Exercise Price
Per Share
| | Weighted
Average
Remaining
Contractual
Term
(in years)
| | Aggregate
Intrinsic
Value
(in millions)
|
Balance at December 31, 2003 | | — | | | $ | — | | | | | |
Granted | | 1,062,499 | | | $ | 1.50 | | | | | |
Exercised | | — | | | | — | | | | | |
Cancelled | | — | | | | — | | | | | |
| | |
|
| | | | | | | | |
Balance at December 31, 2004 | | 1,062,499 | | | $ | 1.50 | | 9.92 | | $ | 4.5 |
Granted | | 400,000 | | | $ | 1.94 | | | | | |
Exercised | | — | | | | — | | | | | |
Cancelled | | — | | | | — | | | | | |
| | |
|
| | | | | | | | |
Balance at December 31, 2005 | | 1,462,499 | | | $ | 1.62 | | 9.21 | | $ | 4.9 |
Granted | | 600,000 | | | $ | 0.94 | | | | | |
Exercised | | — | | | | — | | | | | |
Expired | | (532,707 | ) | | $ | 1.55 | | | | | |
Cancelled | | (529,792 | ) | | $ | 1.62 | | | | | |
| | |
|
| | | | | | | | |
Balance at December 31, 2006 | | 1,000,000 | | | $ | 1.25 | | 9.06 | | $ | 1.4 |
| | |
|
| | | | | | | | |
Exercisable at December 31, 2006 | | 265,333 | | | $ | 1.55 | | 8.33 | | $ | 0.6 |
| | |
|
| | | | | | | | |
The grant-date fair values of options granted during the years ended December 31, 2006, 2005 and 2004 were $0.49 per share, $1.07 per share and $4.24 per share, respectively.
-44-
A summary of the status of our non-vested stock options as of December 31, 2006, 2005 and 2004 and changes during the years then ended are presented below.
| | | | | | |
| | | Number of Shares
| | | Average
Grant-Date Fair Value
Per Share
|
Nonvested at December 31, 2003 | | — | | | $ | — |
Granted | | 1,062,499 | | | $ | 4.24 |
Vested | | (212,500 | ) | | $ | 4.24 |
Cancelled | | — | | | $ | — |
| | |
|
| | | |
Nonvested at December 31, 2004 | | 849,999 | | | $ | 4.24 |
Granted | | 400,000 | | | $ | 1.07 |
Vested | | (212,500 | ) | | $ | 4.24 |
Cancelled | | — | | | $ | — |
| | |
|
| | | |
Nonvested at December 31, 2005 | | 1,037,499 | | | $ | 3.02 |
Granted | | 600,000 | | | $ | 0.49 |
Vested | | (373,040 | ) | | $ | 2.43 |
Cancelled | | (529,792 | ) | | $ | 3.39 |
| | |
|
| | | |
Nonvested at December 31, 2006 | | 734,667 | | | $ | 0.98 |
| | |
|
| | | |
As of December 31, 2006, there was $0.7 million of total unrecognized compensation cost, related to non-vested stock options, which is expected to be recognized over a weighted-average period of approximately 2.57 years. The total fair values of shares vested during the years ended December 31, 2006, 2005 and 2004 were $905,617, $901,000 and $901,000 respectively.
1998 Stock Option Plan
Akesis Delaware adopted the 1998 Incentive Stock Plan and terminated such immediately prior to the acquisition by Liberty of Akesis Delaware. All then outstanding options and rights were terminated immediately prior to the acquisition by Liberty of Akesis Delaware. Under the plan, nonstatutory stock options and stock purchase rights were granted to service providers, and incentive stock options were granted to employees. The fair market value of the shares was determined on the date of the option grant.
The term of each option was 10 years unless sooner terminated or amended by the Board. In the case of an incentive stock option granted to an optionee who, at the time of the grant, owned more than 10% of the voting power of all classes of stock, the term of the option was five years from the date of grant or as provided on the option agreement.
The exercise price of an option was determined by the Company’s administrator with the following exceptions: For an incentive stock option granted to an employee who owned more than 10% percent of the voting power of all classes of stock, the exercise price was no less than 110% of the fair market value per share on the date of grant. The exercise price for employees was no less than 100% of the fair market value on the date of grant. For nonstatutory stock options, the service provider who owned more than 10% percent of the voting power of all classes of stock, the exercise price was no less than 110% of the fair market value per share on the date of grant. The exercise price for other service providers was no less than 85% of the fair market value on the date of grant.
Options vested at a rate of no less than 20% per year over five years from the date of grant, except for options granted to officers, directors, and consultants.
-45-
The following is a summary of the status of options under the 1998 Incentive Stock Plan as of certain dates:
| | | | | | |
| | | Options
Outstanding
| | | Weighted-
Average
Exercise
Price
|
Balance at December 31, 2003 | | 878,592 | | | $ | 0.15 |
Granted | | 115,233 | | | $ | 0.12 |
Exercised | | (927,714 | ) | | $ | 0.14 |
Cancelled | | (66,111 | ) | | $ | 0.11 |
| | |
|
| | | |
Balance at December 31, 2004 | | — | | | $ | — |
| | |
|
| | | |
During the first quarter of 2006, the Company conducted a financing with certain accredited investors, including two members of its Board of Directors, where it sold an aggregate of 180,000 shares of its common stock at a purchase price of $2.00 per share. In addition, the Company issued warrants to the investors to purchase an aggregate of up to 90,000 shares of its common stock in connection with the financings. The warrants are exercisable for shares of the Company’s common stock for three years from the date of issuance at an exercise price per share of $3.00. The net proceeds from the financings after deducting expenses related to the financing were approximately $350,000. All the shares and warrants issued in connection with the financing were exempt from registration by virtue of Section 4(2) of the Securities Act of 1933, as amended.
In connection with the financing described in the preceding paragraph, the Company (a) paid its placement agent a cash fee equal to one percent (1%) of all funds invested by investors introduced by such placement agent (excluding amounts paid by investors upon exercise of warrants), and (b) issued to its placement agent a warrant to purchase up to 11,550 shares of its common stock. The warrant is exercisable for five years from the date of issuance at an exercise price per share of $2.00. The one percent cash commission totalling $2,100 paid to the placement agent was recorded as a reduction of paid in capital. The fair value of the warrant issued to the placement agent in connection with the financing was determined to be $18,965 using a Black-Scholes model, and that amount was recorded as a consulting fee. The warrant issued to the placement agents was exempt from registration by virtue of Section 4(2) of the Securities Act of 1933, as amended.
During the fourth quarter of 2006, the Company conducted an additional financing with certain accredited investors where it sold an aggregate of 7,233,332 shares of its common stock at a purchase price of $0.60 per share. In addition, the Company issued warrants to the investors to purchase an aggregate of up to 1,085,000 shares of its common stock in connection with the financing. The warrants are exercisable for shares of the Company’s common stock for three years from the date of closing at an exercise price per share of $0.60. The net proceeds from the financing after deducting expenses related to the financing were approximately $4.247 million. All the shares and warrants issued in connection with the financing were exempt from registration by virtue of Section 4(2) of the Securities Act of 1933, as amended.
In connection with the financing described in the preceding paragraph, an affiliate of Avalon Ventures purchased 5,000,000 shares of common stock for $3,000,000 and received a warrant to purchase up to 750,000 shares of common stock. Kevin Kinsella, a beneficial owner of the
-46-
Company’s common stock, a holder of warrants to purchase common stock and member of the Company’s board of directors, is a managing member of Avalon Ventures. Jay Lichter, the Company’s President and Chief Executive Officer and an optionholder and member of the Company’s board of directors, is a venture partner of Avalon Ventures.
Also in connection with the fourth quarter 2006 financing described above, the Company (a) paid its placement agent a cash fee equal to eight percent (8%) of all funds invested by investors introduced by such placement agent (excluding amounts paid by investors upon exercise of warrants), and (b) issued to its placement agent a warrant to purchase up to 83,000 shares of its common stock. The warrant is exercisable for five years from the date of issuance at an exercise price per share of $0.60. The eight percent cash commission totalling $66,400 paid to the placement agent was recorded as a reduction of paid in capital. The fair value of the warrant issued to the placement agent in connection with the financing was determined to be $70,750 using a Black-Scholes model, and that amount was recorded as a consulting fee. The warrant issued to the placement agent was exempt from registration by virtue of Section 4(2) of the Securities Act of 1933, as amended.
In December 2006, the Company entered into a loan and security agreement with the Bank that provides for a loan of up to $1.0 million to finance general corporate purposes as described in Note 5 above. In connection with the loan agreement, the Company issued to the Bank a warrant to purchase up to 50,000 shares of its common stock. The warrant is exercisable for seven years from the date of issuance at an exercise price per share of $0.60. The fair value of the warrants issued to the Bank in connection with the loan was determined to be $55,505 using a Black-Scholes model, and that amount was recorded as debt issuance costs. The warrant issued to the Bank was exempt from registration by virtue of Section 4(2) of the Securities Act of 1933, as amended.
In connection with separation agreements entered into with two former officers during 2006, the Company issued the former officers warrants to purchase up to 50,000 and 250,000 shares, respectively, of its common stock. The warrants are exercisable for shares of the Company’s common stock for three years from the effective date of the applicable separation agreement at an exercise price per share of $1.42 and $0.94, respectively. The officers purchased the warrants from the Company for $500 and $2,500, respectively. The fair value of these warrants was determined to be a total of $230,105 using a Black-Scholes model, and that amount was recorded as employee termination expense. The warrants were exempt from registration by virtue of Section 4(2) of the Securities Act of 1933, as amended.
Income taxes consisted of the following:
| | | | | | | | |
| | | 2006
| | | 2005
| |
Deferred (benefit) | | | | | | | | |
Federal | | $ | (277,000 | ) | | $ | (377,000 | ) |
State | | | (49,000 | ) | | | (67,000 | ) |
| | |
|
|
| |
|
|
|
| | | | (326,000 | ) | | | (444,000 | ) |
Change in valuation allowance | | | 326,000 | | | | 444,000 | |
| | |
|
|
| |
|
|
|
| | | $ | — | | | $ | — | |
| | |
|
|
| |
|
|
|
-47-
At December 31, 2006, Akesis Delaware has federal tax net operating loss carryforwards of approximately $4.6 million. The federal net operating loss carryforwards will begin to expire in 2018, unless previously utilized. Pursuant to Internal Revenue Code Section 382 and 383, use of Akesis Delaware’s net operating loss carryforwards may be limited if a cumulative change in ownership of more than 50% occurs within a three-year period. No assessment has been made as to whether such a change in ownership has occurred.
Significant components of Akesis Delaware’s net deferred tax assets at December 31, 2006 and 2005 are shown below. A valuation allowance of $1,605,000 and $1,279,000 has been established to offset the net deferred tax assets at December 31, 2006 and 2005, respectively, as realization of such assets is uncertain.
| | | | | | | | |
| | | December 31, 2006
| |
| | | 2006
| | | 2005
| |
Noncurrent Net Operating Loss Carryforwards | | $ | 1,565,000 | | | $ | 1,241,000 | |
Other noncurrent | | | 27,000 | | | | 25,000 | |
| | |
|
|
| |
|
|
|
Total noncurrent | | | 1,592,000 | | | | 1,266,000 | |
Other current | | | 13,000 | | | | 13,000 | |
| | |
|
|
| |
|
|
|
Total deferred tax assets | | | 1,605,000 | | | | 1,279,000 | |
Deferred tax asset valuation allowance | | | (1,605,000 | ) | | | (1,279,000 | ) |
| | |
|
|
| |
|
|
|
Net deferred taxes | | $ | — | | | $ | — | |
| | |
|
|
| |
|
|
|
A reconciliation of income taxes at the federal statutory rate to the effective tax rate is as follows:
| | | | | | | | |
| | | Years Ended December 31,
| |
| | | 2006
| | | 2005
| |
Income taxes (benefit) computed at the federal statutory rate | | $ | (842,000 | ) | | $ | (1,087,080 | ) |
Tax effect of change in valuation analysis | | | 326,000 | | | | 444,000 | |
Nondeductible compensation | | | 403,000 | | | | 632,000 | |
Deduction arising from exercise of stock options | | | — | | | | — | |
Nondeductible expenses arising from issuance of warrants | | | 113,000 | | | | 11,000 | |
| | |
|
|
| |
|
|
|
| | | $ | — | | | $ | — | |
| | |
|
|
| |
|
|
|
-48-
Item 9.Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
None.
Item 9A.Controls and Procedures
Evaluation of disclosure controls and procedures. Our Chief Executive Officer and our Chief Financial Officer evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. Based on this evaluation, our Chief Executive Officer and our Chief Financial Officer have concluded that the Company’s disclosure controls and procedures are effective to ensure that information we are required to disclose in reports that we file or submit under the Securities Exchange Act of 1934 is recorded, processed, summarized and reported within the time periods specified in Securities and Exchange Commission rules and forms.
Changes in internal control over financial reporting. Based on an evaluation performed by our Chief Executive Officer and Chief Financial Officer, we have concluded that there was no change in our internal control over financial reporting that occurred during the period covered by this Annual Report on Form 10-K that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Item 9B.Other Information
None.
-49-
PART III
Item 10.Directors, Executive Officers and Corporate Governance
The information regarding our directors and executive officers is incorporated by reference from our definitive proxy statement to be delivered to stockholders in connection with our 2007 Annual Meeting of Stockholders.
Item 11.Executive Compensation
The information required by this Item is incorporated by reference from our definitive proxy statement to be delivered to stockholders in connection with our 2007 Annual Meeting of Stockholders.
Item 12.Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this Item regarding security ownership of certain beneficial owners and management is incorporated by reference from our definitive proxy statement to be delivered to stockholders in connection with our 2007 Annual Meeting of Stockholders. The information required by this Item regarding equity compensation plans is incorporated by reference from Item 5 of this Annual Report on Form 10-K.
Item 13.Certain Relationships and Related Transactions, and Director Independence
The information required by this Item is incorporated by reference from our definitive proxy statement to be delivered to stockholders in connection with our 2007 Annual Meeting of Stockholders.
Item 14.Principal Accountant Fees and Services
The information required by this Item is incorporated by reference from our definitive proxy statement to be delivered to stockholders in connection with our 2007 Annual Meeting of Stockholders.
-50-
PART IV
Item 15.Exhibits and Financial Statement Schedules
| | (a) | The following documents are filed as part of this Form 10-K: |
| | (1) | Consolidated Financial Statements (included in Part II of this report): |
| | • | | Report of Swenson Advisors, LLP, Independent Registered Public Accounting Firm. |
| | • | | Consolidated Balance Sheets |
| | • | | Consolidated Statements of Operations |
| | • | | Consolidated Statement of Stockholders’ Equity |
| | • | | Consolidated Statements of Cash Flows |
| | • | | Notes to Consolidated Financial Statements |
| | (2) | Consolidated Financial Statement Schedules: |
All consolidated financial statement schedules are omitted because the information is inapplicable or presented in the notes to the financial statements.
| | |
Exhibit Number
| | Description
|
| 2.1(1) | | Agreement and Plan of Merger and Reorganization dated September 27, 2004 by and among the Registrant, Ann Arbor Acquisition Corporation, and Liberty Mint, Ltd. |
| 3.1(2) | | Articles of Incorporation, as amended, of the Registrant |
| 3.2(2) | | Bylaws of the Registrant |
| 4.1(3) | | Form of Warrant to Purchase Common Stock issued pursuant to that certain Common Stock and Warrant Purchase Agreement dated December 30, 2005 between the Company and the purchasers therein |
| 4.2(3) | | Form of Warrant to Purchase Common Stock issued pursuant to the Form of Finder Agreement |
| 4.3(7) | | Form of Warrant to Purchase Common Stock issued pursuant to that certain Securities Purchase Agreement dated November 21, 2006 between the Company and the purchasers therein |
| 4.4(7) | | Warrant to Purchase Common Stock dated November 21, 2006 between the Company and Globalvest Partners, LLC |
| 4.5(8) | | Form of Warrant issued to Investors in the Financing dated December 15, 2006 |
| 4.6(8) | | Warrant, dated as of December 15, 2006, issued to Globalvest Partners, LLC |
| 4.7(8) | | Warrant, dated as of December 15, 2006, issued to Square 1 Bank |
| 4.8(6) | | Warrant to Purchase Common Stock dated October 2, 2006 between the Company and Edward Wilson. |
| 10.1(2) | | Employment Offer Letter dated December 13, 2004 for Edward B. Wilson, President and CEO of the Registrant |
| 10.2(2) | | Employment Offer Letter dated December 13, 2004 for John T. Hendrick, CFO of the Registrant |
| 10.3(2) | | Stand-Alone Stock Option Agreement dated January 24, 2005 for Edward B. Wilson, President and CEO of the Registrant |
| 10.4(2) | | Stand-Alone Stock Option Agreement dated January 24, 2005 dated for John T. Hendrick, CFO of the Registrant |
| 10.5(2) | | 2005 Stock Plan of the Registrant |
| 10.6(3) | | Form of Finder Agreement |
| 10.7(5) | | Common Stock and Warrant Purchase Agreement dated March 31, 2006 |
-51-
| | |
Exhibit Number
| | Description
|
| 10.8(6) | | Separation Agreement and Release dated October 2, 2006 between the Company and the Edward Wilson |
| 10.9(6) | | Form of Stand-Alone Stock Option Agreement |
| 10.10(6) | | Offer Letter dated October 2, 2006 between the Company and Jay Lichter |
| 10.11(7) | | Securities Purchase Agreement dated November 21, 2006 between the Company and the purchasers therein |
| 10.12(7) | | Finders Agreement dated November 8, 2006 between the Company and Globalvest Partners, LLC |
| 10.13(8) | | Securities Purchase Agreement, dated as of December 15, 2006, by and among the Company and the Investors |
| 10.14(8) | | Loan and Security Agreement, dated as of December 15, 2006, by and between the Company and Square 1 Bank |
| 14.1(4) | | Code of Ethics of the Registrant |
| 21.1 | | Subsidiaries of the Registrant |
| 23.1 | | Consent of Swenson Advisors, LLP, Independent Registered Public Accounting Firm |
| 31.1 | | Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| 31.2 | | Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| 32.1 | | Certification of Chief Executive Officer and Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
(1) Incorporated | by reference to the Registrant’s Form 8-K as filed with the SEC on September 28, 2004. |
(2) | Incorporated by reference to the Registrant’s Form 10-K as filed with the SEC on March 25, 2005. |
(3) Incorporated | by reference to the Registrant’s Form 8-K as filed with the SEC on January 6, 2006. |
(4) Incorporated | by reference to the Registrant’s Amendment to Form 10-K as filed with the SEC on April 28, 2005. |
(5) Incorporated | by reference to the Registrant’s Form 8-K as filed with the SEC on April 4, 2006. |
(6) Incorporated | by reference to the Registrant’s Form 8-K as filed with the SEC on October 3, 2006. |
(7) Incorporated | by reference to the Registrant’s Form 8-K as filed with the SEC on November 27, 2006. |
(8) Incorporated | by reference to the Registrant’s Form 8-K as filed with the SEC on December 20, 2006. |
-52-
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities and Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | |
AKESIS PHARMACEUTICALS, INC. |
| |
By: | | /s/ Jay B. Lichter
|
| | | Jay B. Lichter, |
| | | President, Chief Executive Officer and Director |
Dated: February 8, 2007
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints Jay B. Lichter and John T. Hendrick and each of them acting individually, as his attorneys-in-fact, each with full power of substitution, for him in any and all capacities, to sign any and all amendments to this Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming our signatures as they may be signed by our said attorney to any and all amendments said Form 10-K.
Pursuant to the requirements of the Securities Act, this Form 10-K has been signed by the following persons in the capacities and on the dates indicated:
| | | | |
Signature
| | Title
| | Date
|
| | |
/s/ Jay B. Lichter
Jay B. Lichter | | President, Chief Executive Officer and Director (Principal Executive Officer) | | February 8, 2007 |
| | |
/s/ John T. Hendrick
John T. Hendrick | | Chief Financial Officer (Principal Financial and Accounting Officer) | | February 8, 2007 |
| | |
/s/ Kevin J. Kinsella
Kevin J. Kinsella | | Director | | February 8, 2007 |
| | |
/s/ Kevin R. Sayer
Kevin R. Sayer | | Director | | February 8, 2007 |
| | |
/s/ John F. Steel, IV
John F. Steel, IV | | Director | | February 8, 2007 |
-53-
EXHIBIT INDEX
| | |
Exhibit
Number
| | Description
|
| 2.1(1) | | Agreement and Plan of Merger and Reorganization dated September 27, 2004 by and among the Registrant, Ann Arbor Acquisition Corporation, and Liberty Mint, Ltd. |
| 3.1(2) | | Articles of Incorporation, as amended, of the Registrant |
| 3.2(2) | | Bylaws of the Registrant |
| 4.1(3) | | Form of Warrant to Purchase Common Stock issued pursuant to that certain Common Stock and Warrant Purchase Agreement dated December 30, 2005 between the Company and the purchasers therein |
| 4.2(3) | | Form of Warrant to Purchase Common Stock issued pursuant to the Form of Finder Agreement. |
| 4.3(7) | | Form of Warrant to Purchase Common Stock issued pursuant to that certain Securities Purchase Agreement dated November 21, 2006 between the Company and the purchasers therein |
| 4.4(7) | | Warrant to Purchase Common Stock dated November 21, 2006 between the Company and Globalvest Partners, LLC |
| 4.5(8) | | Form of Warrant issued to Investors in the Financing dated December 15, 2006 |
| 4.6(8) | | Warrant, dated as of December 15, 2006, issued to Globalvest Partners, LLC |
| 4.7(8) | | Warrant, dated as of December 15, 2006, issued to Square 1 Bank |
| 4.8(6) | | Warrant to Purchase Common Stock dated October 2, 2006 between the Company and Edward Wilson |
| 10.1(2) | | Employment Offer Letter dated December 13, 2004 for Edward B. Wilson, President and CEO of the Registrant |
| 10.2(2) | | Employment Offer Letter dated December 13, 2004 for John T. Hendrick, CFO of the Registrant |
| 10.3(2) | | Stand-Alone Stock Option Agreement dated January 24, 2005 for Edward B. Wilson, President and CEO of the Registrant |
| 10.4(2) | | Stand-Alone Stock Option Agreement dated January 24, 2005 dated for John T. Hendrick, CFO of the Registrant |
| 10.5(2) | | 2005 Stock Plan of the Registrant |
| 10.6(3) | | Form of Finder Agreement |
| 10.7(5) | | Common Stock and Warrant Purchase Agreement dated March 31, 2006 |
| 10.8(6) | | Separation Agreement and Release dated October 2, 2006 between the Company and the Edward Wilson |
| 10.9(6) | | Form of Stand-Alone Stock Option Agreement |
| 10.10(6) | | Offer Letter dated October 2, 2006 between the Company and Jay Lichter |
| 10.11(7) | | Securities Purchase Agreement dated November 21, 2006 between the Company and the purchasers therein |
| 10.12(7) | | Finders Agreement dated November 8, 2006 between the Company and Globalvest Partners, LLC. |
| 10.13(8) | | Securities Purchase Agreement, dated as of December 15, 2006, by and among the Company and the Investors |
| 10.14(8) | | Loan and Security Agreement, dated as of December 15, 2006, by and between the Company and Square 1 Bank |
| 14.1(4) | | Code of Ethics of the Registrant |
| 21.1 | | Subsidiaries of the Registrant |
| 23.1 | | Consent of Swenson Advisors, LLP, Independent Registered Public Accounting Firm |
| 31.1 | | Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| 31.2 | | Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| 32.1 | | Certification of Chief Executive Officer and Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
(1) | Incorporated by reference to the Registrant’s Form 8-K as filed with the SEC on September 28, 2004. |
(2) | Incorporated by reference to the Registrant’s Form 10-K as filed with the SEC on March 25, 2005. |
(3) | Incorporated by reference to the Registrant’s Form 8-K as filed with the SEC on January 6, 2006. |
(4) | Incorporated by reference to the Registrant’s Amendment to Form 10-K as filed with the SEC on April 28, 2005. |
(5) | Incorporated by reference to the Registrant’s Form 8-K as filed with the SEC on April 4, 2006. |
(6) | Incorporated by reference to the Registrant’s Form 8-K as filed with the SEC on October 3, 2006. |
(7) | Incorporated by reference to the Registrant’s Form 8-K as filed with the SEC on November 27, 2006. |
(8) | Incorporated by reference to the Registrant’s Form 8-K as filed with the SEC on December 20, 2006. |