Preliminary Offering Circular Dated September 21, 2015
An offering statement pursuant to Regulation A relating to these securities has been filed with the Securities and Exchange Commission, which we refer to as the Commission. Information contained in this Preliminary Offering Circular is subject to completion or amendment. These securities may not be sold nor may offers to buy be accepted before the offering statement filed with the Commission is qualified. This Preliminary Offering Circular shall not constitute an offer to sell or the solicitation of an offer to buy nor may there be any sales of these securities in any state in which such offer, solicitation or sale would be unlawful before registration or qualification under the laws of any such state. We may elect to satisfy our obligation to deliver a Final Offering Circular by sending you a notice within two business days after the completion of our sale to you that contains the URL where the Final Offering Circular or the offering statement in which such Final Offering Circular was filed may be obtained.

APERION BIOLOGICS, INC.
Best Efforts Offering of Shares of Common Stock
This Offering Circular relates to the offer and sale of up to shares of common stock of Aperion Biologics, Inc., a Delaware corporation. We anticipate that the offering price will be between $ and $ per share.
| | Price to Public | Underwriting Discounts and Commissions | Proceeds to Issuer |
| Per share: | $ | $ | $ |
| Total Minimum: | $ | $ | $ |
| Total Maximum: | $ | $ | $ |
Acting as Underwriter
See “Risk Factors” on page [ ] to read about factors you should consider before buying shares of our common stock.
The underwriter has agreed to use its best efforts to procure potential purchasers for the shares of common stock offered pursuant to this Offering Circular.
The shares are being offered on an all or none basis. The offering will commence on the date of this Offering Circular. All investor funds received from the date of this Offering Circular to the closing date of this offering, which shall take place on , 2015, will be deposited into an escrow account until closing. The closing date is also the termination date of this offering. If, on the closing date, investor funds are not received for the full amount of shares to be sold in this offering, the offering will terminate and any funds received will be returned promptly, without interest.
Following this offering, our common stock will be quoted on OTCQB under the symbol “ .”
The Company is following the “Offering Circular” format disclosure under Form 1-A.
GENERALLY NO SALE MAY BE MADE TO YOU IN THIS OFFERING IF THE AGGREGATE PURCHASE PRICE YOU PAY IS MORE THAN 10% OF THE GREATER OF YOUR ANNUAL INCOME OR NET WORTH. DIFFERENT RULES APPLY TO ACCREDITED INVESTORS AND NON-NATURAL PERSONS. BEFORE MAKING ANY REPRESENTATION THAT YOUR INVESTMENT DOES NOT EXCEED APPLICABLE THRESHOLDS, WE ENCOURAGE YOU TO REVIEW RULE 251(d)(2)(i)(C) OF REGULATION A. FOR GENERAL INFORMATION ON INVESTING, WE ENCOURAGE YOU TO REFER TO WWW.INVESTOR.GOV.
THE UNITED STATES SECURITIES AND EXCHANGE COMMISSION DOES NOT PASS UPON THE MERITS OF OR GIVE ITS APPROVAL TO ANY SECURITIES OFFERED OR THE TERMS OF THE OFFERING, NOR DOES IT PASS UPON THE ACCURACY OR COMPLETENESS OF ANY OFFERING CIRCULAR OR OTHER SELLING LITERATURE. ANY REPRESENTATION TO THE CONTRARY IS UNLAWFUL.
THE SECURITIES HAVE NOT BEEN REGISTERED UNDER THE SECURITIES ACT OF 1933, AS AMENDED (THE “SECURITIES ACT”) OR APPLICABLE STATE SECURITIES LAWS, AND THESE SECURITIES ARE OFFERED PURSUANT TO AN EXEMPTION FROM REGISTRATION WITH THE COMMISSION. HOWEVER, THE COMMISSION HAS NOT MADE AN INDEPENDENT DETERMINATION THAT THE SECURITIES OFFERED HEREUNDER ARE EXEMPT FROM REGISTRATION.
THIS OFFERING CIRCULAR CONTAINS ALL OF THE REPRESENTATIONS BY THE COMPANY CONCERNING THIS OFFERING, AND NO PERSON SHALL MAKE DIFFERENT OR BROADER STATEMENTS THAN THOSE CONTAINED HEREIN. INVESTORS ARE CAUTIONED NOT TO RELY UPON ANY INFORMATION NOT EXPRESSLY SET FORTH IN THIS OFFERING CIRCULAR.
TABLE OF CONTENTS
Page
Offering Circular Summary | 4 |
Risk Factors | 9 |
Dilution | 21 |
Underwriting | 24 |
Use of Proceeds | 26 |
Description of Business | 28 |
Management’s Discussion and Analysis of Financial Condition and Results of Operations | 51 |
Directors, Executive Officers and Significant Employees | 54 |
Compensation of Directors and Executive Officers | 57 |
Security Ownership of Management and Certain Securityholders | 58 |
Interest of Management and Others in Certain Transactions | 59 |
Securities Being Offered | 63 |
Financial Statements | 69 |
PART III—EXHIBITS | 70 |
SIGNATURES | 72 |
OFFERING CIRCULAR SUMMARY
This summary highlights information contained elsewhere in this Offering Circular and does not contain all of the information that you should consider in making your investment decision. Before investing in our common stock, you should carefully read this entire Offering Circular, including our consolidated financial statements and the related notes and the information set forth under the headings “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” in each case included elsewhere in this Offering Circular. Unless otherwise stated, all references to “us,” “our,” “we,” the “Company” and similar designations refer to Aperion Biologics, Inc.
Overview
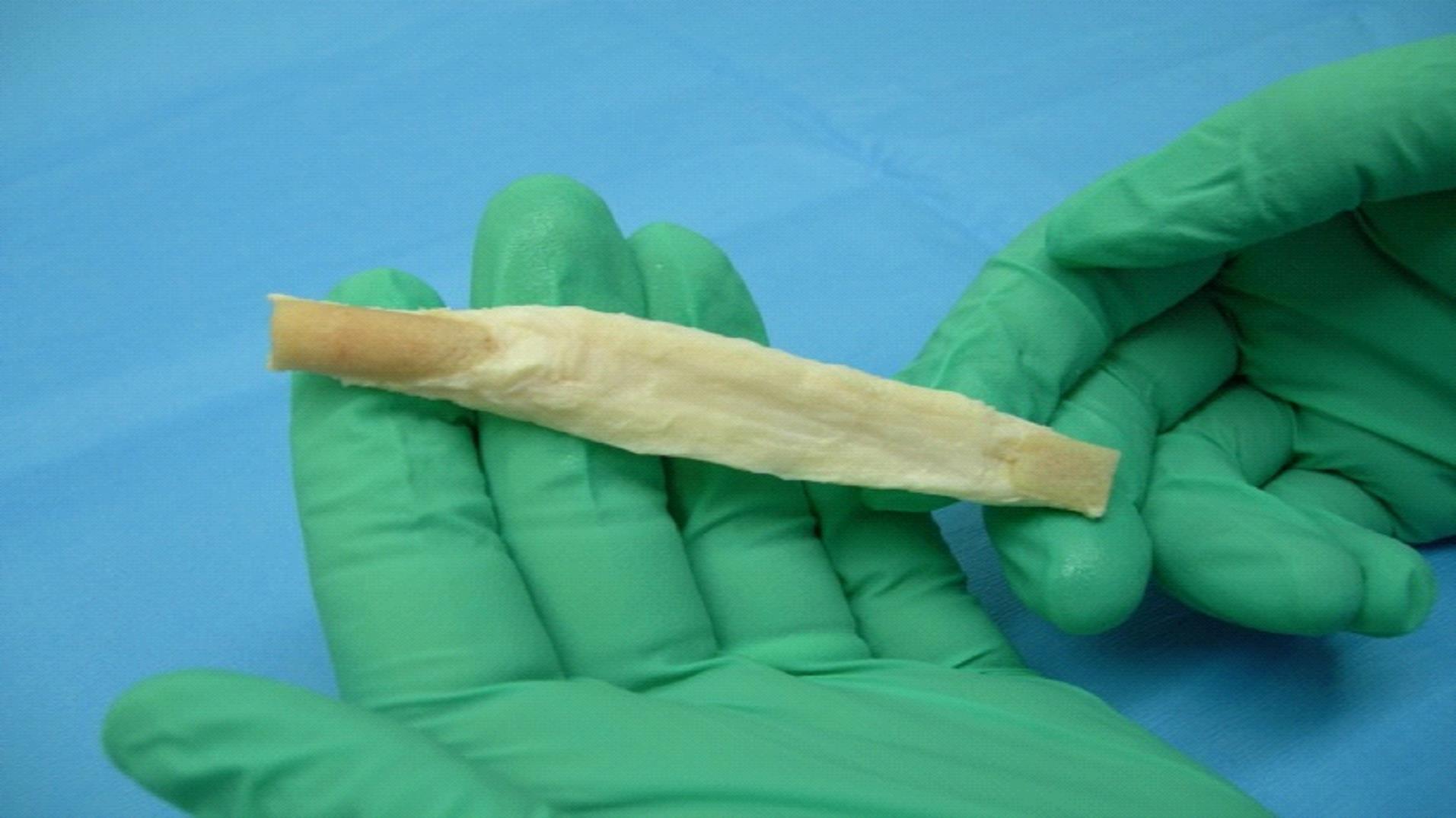
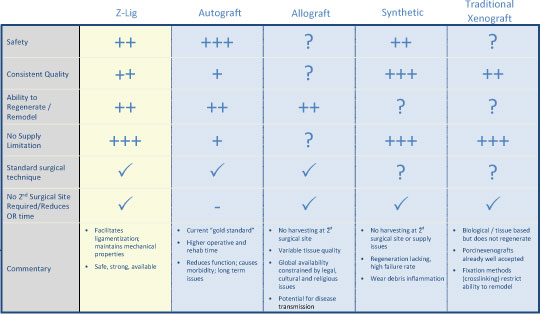
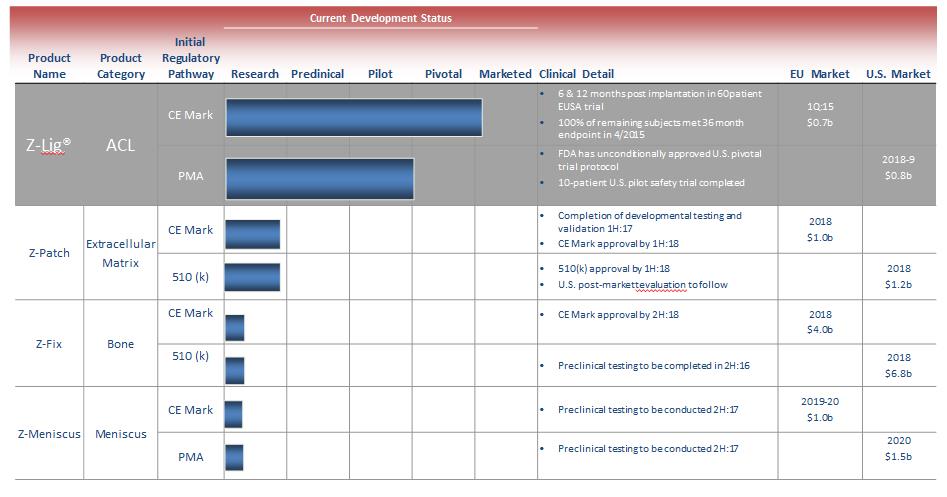
We are a commercial-stage medical device company addressing the significant need for an alternative to human-based sources of tissues to be used in surgical procedures. Our lead product, the Z-Lig®, is produced by a patent-protected process for porcine tendons and its safety and performance was demonstrated in multicenter, prospective randomized trials in Europe and South Africa. In 2014, we received the CE Mark (the regulatory permission to market the product in Europe) for use initially as a knee joint anterior cruciate ligament (“ACL”) replacement in revision and multiligament procedures, which allows us to distribute the Z-Lig in any market which recognizes this approval. We have been building the appropriate distribution channels, primarily focusing on establishing a network of distributors that have strong relationships with sports medicine doctors.
Z-Lig is the only known biological alternative to human tissue for ACL replacement. Since Z-Lig is a biological implant that maintains a scaffold, it can become populated and remodeled with the patient’s own cells (similar to human-sourced tissue), referred to in the industry as “ligamentization”. This ligamentization is one of the keys to Z-Lig’s proven ability to be highly functional in patients many years post-implantation. Our propriety process is protected by an extensive portfolio of patents and patent applications, including 23 issued patents in the U.S. and internationally.
Our Solution
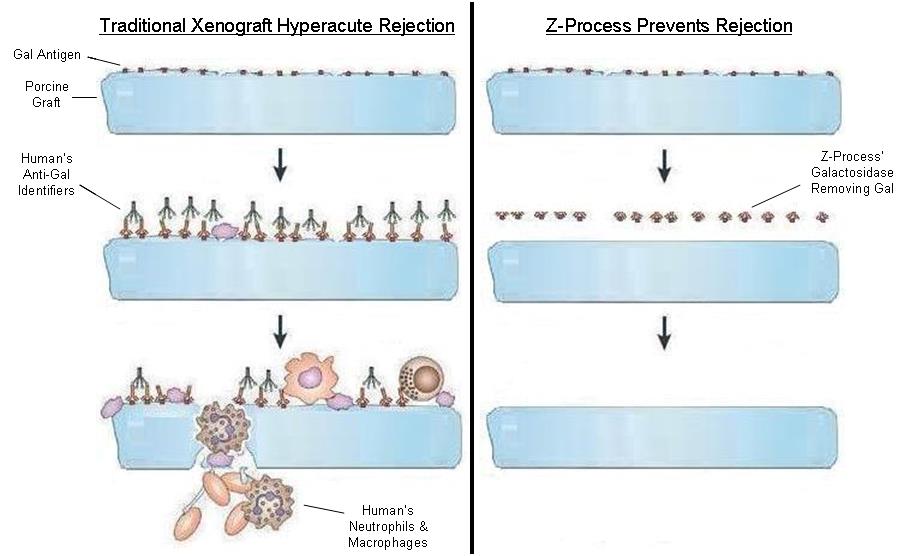
To solve for xenotransplant rejection problems, we developed the Z-Process, which is a proprietary process that immunochemically modifies animal tissue so that it is compatible with the human immune system. The Z-Process addresses both α-gal and non-gal antigens on the xenograft to create porcine tendons that can be safely used in humans while still maintaining their biological scaffold activity. Using the Z-Process, we developed the Z-Lig® device as an immunocompatible, porcine-derived ACL reconstruction alternative that provides a readily available, off-the-shelf solution and is strong, sterile, cost-effective and consistent.
Our Product Candidate Pipeline
In addition to the ACL, our processing technology represents a platform which can also potentially be applied to opportunities involving cartilage, soft tissue patches (extracellular matrices), bone, heart valves, vascular grafts, collagen and other tissues throughout the body.
The Company has several product families in clinical and preclinical development and research stages that fall into four general groupings:
| • | Z-Lig – family of devices designed for use in a range of ligament reconstruction procedures, in addition to the initial application for the ACL. |
| • | Z-Patch – an extracellular matrix product used in soft tissue repair and augmentation procedures. |
| • | Z-Fix – a bone-based product for biologic fixation in ACL reconstruction procedures which would be complementary to the Z-Lig device permitting an all-biologic ACL reconstruction. |
| • | Z-Meniscus – a meniscus device which can be use in the repair or reconstruction of meniscal injuries and defects. |
Our Strategies
Our contact with clinicians confirmed that the initial market need for the Z-Lig is comprised of those revision and multiligament knee joint cases where a graft option is required to achieve the desired clinical result. The first phase of our European roll-out calls for the targeting of a select number of markets where human or synthetic graft options are accepted; in particular those markets whose sites participated in our European and South African (EUSA) clinical trials. Thus, the initial target markets for Z-Lig distribution include Italy, South Africa, Poland, Spain, UK, and the Benelux countries (Netherlands, Belgium, Luxembourg). We have identified and signed with a select group of distributors that have met our selection criteria, which include existing complementary product lines, market share, financial stability, and willingness to invest in the product line. In these markets, we are introducing the product through a targeted approach by focusing on clinicians chosen by our investigative surgeons and our distribution partners.
Supporting our indirect distribution efforts will be Aperion direct personnel specifically tasked with sales management and also clinical marketing specialists for training and technical/scientific field education.
The next distribution phase is expected to expand our application of use to include all ACL indications and furthermore to add European and Asian markets that accept the CE Mark, with a specific focus on high-volume clinical centers or those specializing in knee injuries. The third and final phase would include pan-European expansion into markets in the EU and continued introduction in markets outside the United States that accept the CE Mark and potential distribution into non-European nations whose regulatory bodies require additional regulatory approval.
We intend to build a sizable revenue base with the Z-Lig in overseas (outside U.S.) markets over the next several years. Our marketing efforts in the U.S. are contingent on obtaining FDA approval for the Z-Lig and other devices. The FDA has granted us an unconditional approval for a clinical trial of the Z-Lig in the United States. We plan to market our products in the U.S. after the trial is completed and FDA approval is obtained.
Our Risks
An investment in our common stock involves a high degree of risk. You should carefully consider the risks summarized below. These risks are discussed more fully in the “Risk Factors”
section immediately following this prospectus summary. These risks include, but are not limited to, the following:
| · | We have incurred significant operating losses since inception and may continue to incur losses for the foreseeable future; |
| · | We only recently received approval in Europe to commercialize our Z-Lig products, and we have no significant experience or capability to sell our products on a commercial scale; |
| · | Our Z-Lig products may not gain market acceptance among surgeons, physicians, patients, healthcare payors and the medical community; |
| · | We may not be able to generate long-term or additional positive clinical data to support or expand our commercialization efforts; |
| · | We may not be able to protect our intellectual property; and, |
| · | CrossCart LLC, our controlling stockholder, controls all aspects of our business. |
Company and Other Information
We were initially incorporated under the laws of the State of California as CrossCart, Inc., in 1996. In June 2008, CrossCart, Inc. was reincorporated under the laws of the State of Delaware, and in May 2009, CrossCart, Inc. changed its name to “Aperion Biologics, Inc.” Our principal executive office is located at 11969 Starcrest Dr. San Antonio, TX 78247, and our telephone number is (210) 858-7056. Our website address is www.aperionbiologics.com. We do not incorporate the information on or accessible through our website into this Offering Circular, and you should not consider any information on, or that can be accessed through, our website a part of this Offering Circular.
We own various U.S. federal trademark registrations and applications, and unregistered trademarks, including the following marks referred to in this Offering Circular: “Z-Lig” and “Aperion Biologics”. All other trademarks or trade names referred to in this Offering Circular are the property of their respective owners. Solely for convenience, the trademarks and trade names in this Offering Circular are referred to without the symbols ® and ™, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto.
This Offering Circular summary highlights information contained elsewhere and does not contain all of the information that you should consider in making your investment decision. Before investing in our common stock, you should carefully read this entire Offering Circular, including our financial statements and the related notes included elsewhere in this Offering Circular. You should also consider, among other things, the matters described under “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in each case appearing elsewhere in this Offering Circular.
The Offering
Common stock offered by us | Shares |
Common stock to be outstanding immediately after this offering | Shares |
Use of proceeds | We intend to use approximately $700,000 of net proceeds from this offering to pay certain accounts payable. We also intend to use a portion of the net proceeds to repay outstanding principal amounts and interest on certain demand notes and line of credit notes issued to CrossCart LLC in the amount of approximately $186,720, including accrued interest. We also intend to use approximately $300,000 of net proceeds to pay certain compensation owed to our employees as a result of the reduction of their salaries and fees. We intend to use the remaining net proceeds to launch our commercialization plan for Z-Lig products in Europe, South Africa and other countries that accept our approved CE Mark; fund post-market registry studies of Z-Lig; conduct pre-clinical and R&D activities for additional product candidates; and initiate clinical trials for Z-Lig products in the U.S. See “Use of Proceeds” on page [ ]. |
Risk factors | You should carefully read “Risk Factors” on page [ ] in this Offering Circular for a discussion of factors that you should consider before deciding to invest in our common stock. |
The number of shares of our common stock to be outstanding after this offering is based on 72,963,279 shares of our common stock outstanding as of June 30, 2015 and excludes:
| · | 6,447,747 shares of common stock issuable upon the exercise of stock options outstanding as of June 30, 2015 at a weighted average exercise price of $0.08 per share; |
| · | 12,142,842 shares of common stock issuable upon the exercise of warrants outstanding as of June 30, 2015 at a weighted average exercise price of $0.08 per share, which warrants prior to the completion of this offering are exercisable to purchase common stock or convertible preferred stock, assuming such warrants will not be exercised prior to the completion of this offering; |
| · | 1,888,775 shares of common stock reserved for future issuance under our 2008 Stock Option/Stock Issuance Plan; |
Unless otherwise indicated, all information in this Offering Circular reflects or assumes the following:
| · | the filing of our amended and restated certificate of incorporation and the adoption of our amended and restated bylaws, which will occur immediately prior to the completion of this offering; |
| · | the conversion of all of our outstanding shares of convertible preferred stock into an aggregate of 37,504,969 shares of common stock upon the completion of this offering, including shares of common stock to be issued as cumulative dividends accrued under such preferred stock; |
| · | the issuance of 31,366,725 shares of common stock upon the conversion of approximately an aggregate of $9,838,058 in outstanding principal and accrued interest on our convertible promissory notes (which is currently convertible into shares of our preferred stock but will be amended to be convertible into shares of our common stock in connection with the offering), upon the completion of this offering, at an average conversion price of $0.31 per share, assuming that the offering is completed on ; |
| · | the issuance of additional shares of common stock as a result of anti-dilution adjustments provided in our certificate of incorporation; and |
| · | a one-for- reverse split of our common stock, which became effective on . |
Risk Factors
Investing in our common stock involves a high degree of risk. You should carefully consider the risks described below along with all of the other information contained in this Offering Circular, including our financial statements and the related notes, before deciding whether to purchase our common stock. If any of the adverse events described in the following risk factors, as well as other factors which are beyond our control, actually occurs, our business, results of operations and financial condition may suffer significantly. As a result, the trading price of our common stock could decline, and you may lose all or part of your investment in our common stock.
Risks Related to Our Business and Finance
We have incurred significant operating losses since inception, anticipate that we will continue to incur losses for the foreseeable future and will need to raise additional capital.
We have generated operating losses since we began operations in 2008, and our net losses attributable to common stockholders for the fiscal year ended September 30, 2014 and 2013 were $3.29 million and $5.45 million respectively. We expect to continue to incur additional operating losses for the foreseeable future as we plan the commercialization of Z-Lig products and continue our clinical and pre-clinical studies. Our recurring operating losses and our need for additional sources of capital to fund our ongoing operations raise substantial doubt about our ability to continue as a going concern. As a result, our independent registered public accounting firm included an explanatory paragraph in its report on our financial statements as of and for the year ended September 30, 2014 with respect to this uncertainty. If the time required to generate material revenues and achieve profitability is longer than we currently anticipate, or if we are unable to raise new capital through equity or obtain debt financing or other sources of funding, we may be forced to curtail or suspend our operations, which would have a material adverse effect on the value of your investment.
We expect capital outlays and operating expenditures to increase over the next several years as we establish and expand our marketing infrastructure to commercialize our Z-Lig products. Following the completion of this offering, we believe our financial resources will be adequate to sustain our current operations at least through . However, we will need to raise additional capital. We cannot be certain that we will be able to obtain financing on terms acceptable to us, or at all. Our failure to obtain adequate and timely funding will materially adversely affect our business and our ability to develop our products and would have a material adverse effect on the value of your investment.
We only recently received approval in Europe to commercialize our Z-Lig products, and we have no significant experience or capability to sell our products on a commercial scale.
Our future is significantly dependent on the commercial success of our Z-Lig family of products. We have only recently received a CE Mark in April 2014 that allowed us to conduct limited sales in select European markets beginning in February 2015. As a result, we have no significant history or experience in selling Z-Lig or any other products in Europe or in any other markets, and we have limited relationships with surgeons, physicians, clinicians and hospitals that may purchase or use our products. In order for us to commercialize our products successfully, we will need to develop, or obtain through outsourcing arrangements, the capability to market and
sell our products on a commercial scale. We may not have the ability and sufficient resources to establish the infrastructure and organizations needed to execute these functions, which can be complex and costly. Our effort to commercialize Z-Lig products is also subject to a number of additional risks, which could have a material adverse effect on the value of your investment, including:
| · | competitors’ established relationships with our potential customers; |
| · | limitations in our ability to demonstrate the advantages of our products compared to competing products and the relative safety, efficacy and ease of use of our products; |
| · | our inability to convince doctors and hospitals to use our products; and |
| · | the introduction and market acceptance of competing products and technologies. |
Our Z-Lig products may not gain market acceptance among surgeons, physicians, patients, healthcare payors and the medical community.
A critical element in our commercialization strategy is to persuade and educate the medical community on the safe and effective use of our products and how Z-Lig products differentiate from the existing supplies of surgical issues. Surgeons, physicians and hospitals may not perceive the benefits of our products and may be unwilling to change from the devices they are currently using. A number of factors may limit the market acceptance of our Z-Lig products, including the following:
| · | rate of adoption by healthcare practitioners; |
| · | rate of a product’s acceptance by the target population; |
| · | timing of market entry relative to competitive products; |
| · | availability of third-party reimbursement; |
| · | government review and approval requirements; |
| · | extent of marketing efforts by us and third-party distributors or agents retained by us; and |
| · | side effects or unfavorable publicity concerning our products or similar products. |
Therefore, even after we have demonstrated the effectiveness of the Z-Lig products, we may not be able to commercialize these products successfully if we cannot achieve an adequate level of market acceptance. Our inability to successfully commercialize our products will have a material adverse effect on the value of your investment.
We may experience defects and manufacturing issues in our supplies of animal issues or raw materials.
As we continue to expand our clinical development and commercialization activities, we expect to scale up the manufacturing and supplies of animal tissues that are the main component of our Z-Lig products. We may not be able to control or maintain a consistent and high quality of
raw materials, such as porcine tissues, or uncover defects prior to implantation in humans. In the past, we observed and experienced contamination in porcine tissues resulting from the processing and handling of these materials during clinical trials, which led to bacterial infections associated with the grafts in patients implanted with Z-Lig in the trial. While we have implemented stringent validation, safety and corrective procedures and intend to conduct additional post-market studies to confirm the effectiveness of such preventive measures, there is no guarantee that we can eliminate this risk in the future. In addition, external factors outside of our control may affect the quality of our supplies of animal or porcine issues. For example, swine diseases or pandemics may severely reduce the supply of porcine tissues required to manufacture our Z-Lig products. Mishandling or lack of quality control at our third-party suppliers may also negatively impact our ability to obtain a sufficient and acceptable level of porcine tissues to meet our needs. Our failure to eliminate contamination and deficiency in the animal and porcine issues used in our products may require us to incur additional costs to implement preventive measures, or cause significant delays and disruptions in the development and commercialization of our products, which would have an adverse effect on our business operations and financial condition.
The safety and efficacy of our products is supported by limited clinical data, and we may not be able to generate long-term or additional positive clinical data to support or expand our commercialization efforts.
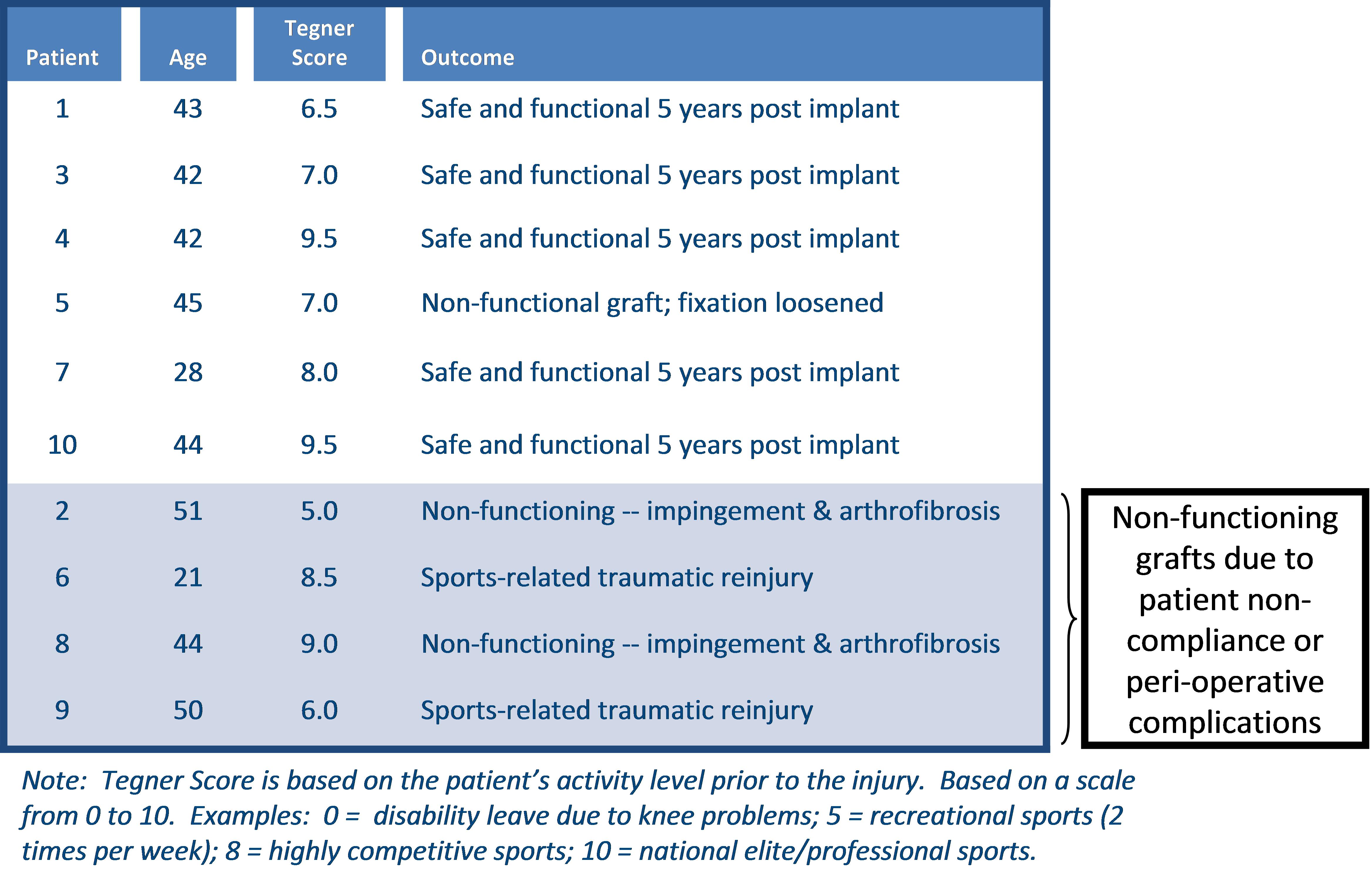
We have obtained a CE Mark to market and sell our Z-Lig products in Europe based on the 6- and 12-month data collected from our clinical studies in 2013 and the longer term (12 years) safety data from the U.S. pilot safety trial. We have committed to continue these clinical studies to support post-market commercial activities in Europe and to meet certain post-market approval requirements by European regulatory agencies. In addition, we intend to conduct additional clinical studies to support further commercialization efforts and expand the markets for our Z-Lig products. For example, the CE Mark we received in April 2014 is limited to revision and multiligament ACL surgeries and does not cover primary ACL surgeries. We have established post-market clinical plans to generate additional data required to expand the indication of Z-Lig products to cover primary ACL surgeries, which would allow us to reach a broader ACL reconstruction market. There is no guarantee that we can duplicate the positive results from our earlier trials for future studies or generate the required data to support the expansion of Z-Lig indication. Our failure to do so may result in the loss of CE Mark approval or other regulatory approvals, delays, failures in the adoption of our products by surgeons and physicians, damage to our reputation and legal claims against us.
In addition, any negative data from our post-market clinical studies may adversely impact our ability to conduct clinical trials and seek regulatory approval of our products from the FDA to market our products in the United States. Furthermore, we are developing several products based on our Z-process that would allow a commercialization path based on FDA’s Section 510(K) pre-market clearance procedures. This procedure is shorter and typically requires the submission of less supporting documentation than other FDA approval processes and does not require long-term clinical studies. As result, we may encounter difficulties and delays in marketing these products which otherwise may not be the case if the products have been proven safe and effective in more extensive and long-term clinical trials.
Our clinical studies of our current or future products may not produce results necessary to support regulatory clearance or approval in the United States or elsewhere.
We plan to seek FDA approval of Z-Lig by conducting pivotal clinical trials in the United States. While we have conducted a limited safety pilot study in the United States showing that Z-Lig was safe, and we have received a CE Mark based on the clinical studies in Europe and South Africa, there is no guarantee that the same results will be duplicated in a clinical trial with a larger patient population in the United States. Our inability to achieve acceptable results in future clinical studies would have a material adverse effect on our business operations and financial condition and the value of your investment.
If third-party payors fail to provide appropriate levels of reimbursement for the use of our products, our revenues could be adversely affected.
Sales of our products depend on the availability of adequate reimbursement from third-party payors. In each market in which we do business, our inability to obtain reimbursement approval or the failure of third-party payors to reimburse health care providers at a level which justifies the use of our products instead of cheaper alternatives will hurt our business.
Moreover, we are unable to predict what changes will be made to the reimbursement methodologies used by third-party payors in the future. We cannot be certain that under current and future payment systems, in which healthcare providers may be reimbursed a set amount based on the type of procedure performed, such as those utilized by Medicare and in many privately managed care systems, the cost of our products will be justified and incorporated into the overall cost of the procedure. A failure by third-party payors to provide appropriate levels of reimbursement for the use of our products will have a material adverse effect on our business operations and financial condition and the value of your investment.
As we expand into multiple international markets, we will face similar risks relating to adverse changes in coverage and reimbursement procedures and policies in those markets. Reimbursement and healthcare payment systems vary significantly among markets in different jurisdictions. Our inability to obtain international coverage and reimbursement approval, or any adverse changes in coverage and the reimbursement policies of foreign third-party payors, could negatively affect our ability to sell our products.
We rely on third party suppliers for our raw materials, including porcine tissues, and their inability to supply us with an adequate supply of materials could harm our business.
We rely on third-party suppliers to supply and manufacture the raw materials, including porcine tissues, for the development of our products. To be successful, our suppliers must be able to provide us with products and components in substantial quantities, in compliance with regulatory requirements (including ISO 22442), in accordance with agreed upon specifications, at acceptable cost and on a timely basis. Among other factors, our anticipated growth could strain the ability of suppliers to deliver an increasingly large supply of products, materials and components. If we are unable to obtain sufficient quantities of high quality components to meet customer demand on a timely basis, we could lose customers, our reputation may be harmed and our business could suffer.
In addition, we currently use one supplier for the supply of porcine tissues required to manufacture our Z-Lig products, biologics, and components. Our dependence on one supplier involves several risks, including limited control over pricing, availability, quality and delivery schedules. If any one or more of our suppliers cease to provide us with sufficient quantities of our components in a timely manner or on terms acceptable to us, we would have to seek alternative sources of supplies. We could incur significant delays while we locate and engage
alternative qualified suppliers, including delays associated with qualifying the new supplier to meet all applicable regulatory requirements in the United States, Europe and other markets in which we develop and sell our products. Even if we are able to identify an alternative supplier, we might not be able to negotiate or receive the same or more favorable terms as those provided by our existing suppliers. Any such disruption or increased expenses could harm our commercialization efforts and adversely affect our ability to generate revenue. Any inability to meet our customers’ demands for these products could lead to decreased sales and harm our reputation and result in the loss of customers, which would have a material adverse effect on our business operations and financial condition and the value of your investment.
Failure to attract, retain, and motivate skilled personnel may delay our commercialization plans and research and development efforts.
Our commercial success depends on our continued ability to attract, retain, and motivate highly qualified management and scientific personnel. Competition for skilled and qualified personnel in the medical device industry is intense. If we lose the services of personnel with the necessary skills, including the members of our senior management team, it could significantly impede our commercialization and research and development objectives. Replacing key personnel and consultants may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to develop, gain regulatory approval of and commercialize products successfully.
Our international operations subject us to certain risks.
We intend to market our Z-Lig products initially in select European countries and in South Africa, including Denmark, United Kingdom, Netherlands, Belgium, Spain, Italy and Poland. We also have plans to expand our commercialization activities to other non-European countries that accept the CE Mark, such as Canada, Turkey, Saudi Arabia, Australia and Korea. We will also pursue various clinical and pre-clinical activities in the United States and Europe. Our international operations will subject us to rules, regulations and customs of multiple jurisdictions, and compliance with these rules and regulations is costly and exposes us to penalties for non-compliance. Other laws and regulations that can significantly impact us include various anti-bribery laws, including the U.S. Foreign Corrupt Practices Act and anti-boycott laws, as well as export controls laws. Any failure to comply with applicable legal and regulatory obligations could impact us in a variety of ways that include, but are not limited to, significant criminal, civil and administrative penalties, denial of export privileges, seizure of shipments and restrictions on certain business activities. Also, the failure to comply with applicable legal and regulatory obligations could result in the disruption of our sales activities.
Our international operations expose us to additional risks, including:
| · | difficulties in enforcing or defending intellectual property rights; |
| · | pricing pressure that we may experience internationally; |
| · | third-party reimbursement policies that may require some of the patients who receive our products to directly absorb medical costs or that may necessitate the reduction of the selling prices of our products; |
| · | competitive disadvantage to competition with established business and customer relationships; |
| · | the imposition of additional U.S. and foreign governmental controls or regulations; changes in duties and tariffs, license obligations and other non-tariff barriers to trade; |
| · | foreign currency exchange rate fluctuations; |
| · | difficulties in communicating with our employees, partners and collaborators, and maintaining consistency with our internal guidelines; |
| · | difficulties in enforcing agreements and collecting receivables through certain foreign legal systems; and |
| · | difficulties in establishing and maintaining an effective internal control system to ensure timely and accurate financial reporting. |
If we experience any of these risks, our sales in international countries may be harmed and our results of operations would suffer.
We may face product liability claims that could result in costly litigation and significant liabilities.
Manufacturing and marketing of our products, and clinical testing of our products under development, may expose us to product liability and other tort claims. Although we have, and intend to maintain, liability insurance, the coverage limits of our insurance policies may not be adequate and one or more successful claims brought against us may have a material adverse effect on our business and results of operations. Additionally, product liability claims could negatively affect our reputation, product sales, and our ability to obtain and maintain regulatory approval for our products.
Risks Relating to Our Intellectual Property
If we are unable to protect our intellectual property, our business will be negatively affected.
The market for medical devices is subject to frequent litigation regarding patent and other intellectual property rights. It is possible that our patents or licenses may not withstand challenges made by others or protect our rights adequately.
Our success depends in large part on our ability to secure effective patent protection for our products and processes in Europe and the United States. We have filed and intend to continue to file patent applications to protect our proprietary technology. However, we face the risks that:
| · | we may fail to secure necessary patents prior to or after obtaining regulatory clearances, thereby permitting competitors to market competing products; and |
| · | our already-granted patents may be re-examined, invalidated or not extended. |
We also own trade secrets and confidential information that we try to protect by entering into confidentiality agreements with our employees and other parties. However, the confidentiality agreements may not be honored or, if breached, we may not have sufficient remedies to protect our confidential information. Further, our competitors may independently learn our trade secrets or develop similar or superior technologies. To the extent that our consultants, key employees
or others apply technological information to our projects that they develop independently or others develop, disputes may arise regarding the ownership of proprietary rights to such information, and such disputes may not be resolved in our favor. If we are unable to protect our intellectual property adequately, our business and commercial prospects will suffer.
Because it is difficult and costly to protect our proprietary rights, and third parties have filed patent applications that are similar to ours, we cannot ensure the proprietary protection of our technologies and products.
Our commercial success will depend in part on obtaining patent protection of our technology and successfully defending any of our patents that may be challenged. The patent positions of medical device companies can be highly uncertain and can involve complex legal and factual questions, and we cannot predict the breadth of claims allowed in patents we own or license.
We are a party to certain license agreements that give us rights under specified patents and patent applications, including a license agreement with the University of Missouri. Our current licenses, and our future licenses frequently will, contain performance obligations. If we fail to meet those obligations, the licenses could be terminated. If we are unable to continue to license these technologies on commercially reasonable terms, or at all, we may be forced to delay or terminate our product development and research activities.
We are unable to exercise the same degree of control over intellectual property that we license from third parties as we exercise over our internally developed intellectual property. We do not control the prosecution of certain of the patent applications that we license from third parties; therefore, the patent applications may not be prosecuted as we desire or in a timely manner.
The degree of future protection for our proprietary rights is uncertain, and we cannot ensure that:
| · | we or our licensors will be the first to file patent applications for these inventions; |
| · | the patents of others will not have an adverse effect on our ability to do business; |
| · | others will not independently develop similar or alternative technologies or reverse engineer any of our products, processes or technologies; |
| · | any of our pending patent applications will result in issued patents; |
| · | any patents issued or licensed to us or our collaborators or strategic partners will provide a basis for commercially viable products or will provide us with any competitive advantages; |
| · | any patents issued or licensed to us will not be challenged and invalidated by third parties; or |
| · | we will develop additional products, processes or technologies that are patentable. |
Others have filed and in the future are likely to file patent applications that are similar to ours. The costs of litigating any infringement claim could be substantial. Moreover, we cannot predict whether we, our collaborators, or strategic partners would prevail in any actions. In addition, if the relevant patent claims were upheld as valid and enforceable and our products or processes
were found to infringe the patent or patents, we could be prevented from making, using, or selling the relevant product or process unless we could obtain a license or were able to design around the patent claims. We can give no assurance that such a license would be available on commercially reasonable terms, or at all, or that we would be able to successfully design around the relevant patent claims. There may be significant litigation in our industry regarding patent and other intellectual property rights, which could subject us to litigation. If we become involved in litigation, it could consume a substantial portion of our managerial and financial resources.
We rely on trade secrets to protect technology where we believe patent protection is not appropriate or obtainable. Trade secrets, however, are difficult to protect. While we require employees, and consultants to enter into confidentiality agreements, we may not be able to adequately protect our trade secrets or other proprietary information or enforce these confidentiality agreements.
We may infringe the intellectual property rights of others, which may prevent or delay our product development efforts.
Our success will depend in part on our ability to operate without infringing the proprietary rights of third parties. We cannot guarantee that our products, or manufacture or use of our product candidates, will not infringe third-party patents. Furthermore, a third party may claim that we are using inventions covered by the third party’s patent rights and may go to court to stop us from engaging in our normal operations and activities, including making or selling our product candidates. These lawsuits are costly and could affect our results of operations and divert the attention of managerial and scientific personnel. Some of these third parties may be better capitalized and have more resources than us. There is a risk that a court would decide that we are infringing the third party’s patents and would order us to stop the activities covered by the patents. In that event, we may not have a viable way around the patent and may need to halt commercialization of the relevant product candidate. In addition, there is a risk that a court will order us to pay the other party damages for having violated the other party’s patents. In addition, we may be obligated to indemnify our licensors and collaborators against certain intellectual property infringement claims brought by third parties, which could require us to expend additional resources. The pharmaceutical and biotechnology industries have produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform.
Patent litigation is costly and time consuming, even in cases where the claims against us are without merit. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, if we do not obtain a license, develop or obtain non-infringing technology, fail to defend an infringement action successfully or have infringed patents declared invalid, we may incur substantial monetary damages, encounter significant delays in bringing our product candidates to market and be precluded from manufacturing or selling our product candidates.
International patent protection is particularly uncertain, and if we are involved in opposition proceedings in foreign countries, we may have to expend substantial sums and management resources.
The laws of some foreign countries may not protect our intellectual property rights to the same extent as the laws of the United States. For example, certain countries do not grant patent claims that are directed to the treatment of humans. We may participate in opposition
proceedings to determine the validity of our foreign patents or our competitors’ foreign patents, which could result in substantial costs and diversion of our efforts.
The medical device industry is characterized by extensive patent litigation, and we could become subject to litigation that could be costly, result in the diversion of management’s attention, and harm our business and financial condition.
Our success depends in part on not infringing the patents or violating the other proprietary rights of others. Significant litigation regarding patent rights occurs in the medical industry, including among companies focused on regenerative medicine. It is possible that U.S. and foreign patents and pending patent applications controlled by third parties may be alleged to cover our Z-Process. Our competitors in Europe, the United States and abroad, many of which have substantially greater resources and have made substantial investments in patent portfolios and competing technologies, may have applied for or obtained or may in the future apply for and obtain, patents that will prevent, limit or otherwise interfere with our ability to make, use and sell our products. At any given time, we may be involved as either a plaintiff or a defendant in a number of patent infringement actions, the outcomes of which may not be known for prolonged periods of time.
The large number of patents, the rapid rate of new patent applications and issuances, the complexities of the technologies involved and the uncertainty of litigation significantly increase the risks related to any patent litigation. Any potential intellectual property litigation also could force us to do one or more of the following:
| · | stop selling, making, or using products that use the disputed intellectual property; |
| · | obtain a license from the intellectual property owner to continue selling, making, licensing, or using products, which license may require substantial royalty payments and may not be available on reasonable terms, or at all; |
| · | incur significant legal expenses; |
| · | pay substantial damages or royalties to the party whose intellectual property rights we may be found to be infringing; |
| · | pay the attorney fees and costs of litigation to the party whose intellectual property rights we may be found to be infringing; or |
| · | redesign those products that contain the allegedly infringing intellectual property, which could be costly, disruptive and/or infeasible. |
If any of the foregoing occurs, we may have to withdraw existing products from the market or may be unable to commercialize one or more of our products, all of which could have a material adverse effect on our business, results of operations and financial condition. Any litigation or claim against us, even those without merit, may cause us to incur substantial costs, and could place a significant strain on our financial resources, divert the attention of management from our core business and harm our reputation. Further, as the number of participants in the regenerative tissue industry grows, the possibility of intellectual property infringement claims against us increases.
Risks Related to Our Industry
We are in a highly competitive market segment, and if our competitors are better able to develop and market products that are safer, more effective, less costly, easier to use or otherwise superior than any products that we may develop, our business will be adversely impacted.
The medical device industry is highly competitive and subject to technological change. Our success depends, in part, upon our ability to maintain a competitive position in the development of technologies and products for use in the ACL knee reconstruction graft and other orthopaedic markets. Any product we develop that achieves regulatory clearance or approval will have to compete for market acceptance and market share. We believe that the primary competitive factors in ACL replacement graft markets are clinical effectiveness, product safety, reliability and durability, and eligible patient populations, physician experience and comfort with use of a particular device, ease of use, product support and service, sales force experience and relationships and price. We face significant competition in the United States and internationally, and we expect the intensity of competition will increase over time. We compete with companies that provide allograft and traditional xenograft solutions for orthopaedic surgical procedures. Many of the companies developing or marketing competing products enjoy several advantages to us, including:
| · | greater financial and human resources for product development, sales and marketing; |
| · | greater name recognition; |
| · | long established relationships with physicians and hospitals; |
| · | longer-term clinical trial data due to earlier regulatory approval; |
| · | the ability to offer rebates or bundle multiple product offerings to offer greater discounts or incentives; and |
| · | more established sales and marketing programs and distribution networks; and |
Our competitors may develop and patent processes or products competitive with us, obtain regulatory clearance or approvals for competing products more rapidly than us or develop more effective or less expensive products or technologies that render our technology or products obsolete or less competitive. We also face fierce competition in recruiting and retaining qualified sales, scientific, and management personnel, establishing clinical trial sites and enrolling patients in clinical studies. Our inability to successfully compete with existing and future market competitors will have a material adverse effect on our business operations and financial condition and the value of your investment.
Our business is subject to extensive governmental regulation that could make it more expensive and time consuming for us to introduce new or improved products.
Our products must comply with regulatory requirements imposed by various regulatory agencies. These requirements involve lengthy and detailed laboratory and clinical testing procedures, sampling activities, an extensive agency review process, and other costly and time-consuming procedures. It often takes several years to satisfy these requirements, depending on the complexity and novelty of the product. We also are subject to numerous additional licensing
and regulatory requirements relating to safe working conditions, manufacturing practices, environmental protection, fire hazard control, and disposal of hazardous or potentially hazardous substances. Some of the most important requirements we face include:
| · | European Union CE mark requirements; |
| · | Medical Device Quality Management System Requirements (ISO 13485:2003); and |
| · | Occupational Safety and Health Administration requirements. |
Government regulation may impede our ability to conduct clinical studies and to manufacture our existing and future products. Government regulation also could delay our marketing of new products for a considerable period of time and impose costly procedures on our activities. The regulatory agencies may not approve any of our future products on a timely basis, if at all. Any delay in obtaining, or failure to obtain, such approvals could negatively impact our marketing of any future products and reduce our product revenues.
Our products remain subject to strict regulatory controls on manufacturing, marketing and use. We may be forced to modify or recall a product after release in response to regulatory action or unanticipated difficulties encountered in general use. Any such action could have a material effect on the reputation of our products and on our business and financial position.
Further, regulations may change, and any additional regulation could limit or restrict our ability to use any of our technologies, which could harm our business. We could also be subject to new international, federal, state or local regulations that could affect our research and development programs and harm our business in unforeseen ways. If this happens, we may have to incur significant costs to comply with such laws and regulations, which will harm our results of operations.
Risks Related to Our Common Stock and Corporate Structure
CrossCart LLC controls all aspects of our business.
CrossCart LLC (“CrossCart”) is our largest and controlling stockholder and currently beneficially owns approximately 78.5% of our outstanding shares of common stock on a fully-diluted basis assuming the conversion of all of our outstanding preferred stock and convertible notes. Following the completion of this offering, CrossCart will beneficially own approximately % of our outstanding shares of common stock. In addition, Dr. Kevin R. Stone, Chairman of our Board of Directors, is the controlling member and manager of CrossCart. In addition, CrossCart is our most significant creditor and historically we have depended on the funding of CrossCart and Dr. Stone to maintain our operations. As a result, CrossCart and Dr. Stone are and will be able to exercise significant control over our business operations and on all matters requiring stockholders’ approval, including the election of directors and approval of significant corporate transactions. Due to CrossCart’s and Dr. Stone’s controlling position, we may take actions with respect to our business that may conflict with the desire of other stockholders.
An active trading market for our common stock may not develop and you may not be able to resell your shares at or above the initial offering price.
Prior to this offering, there has been no public market for shares of our common stock. In the absence of an active trading market for our common stock, investors may not be able to sell their common stock at or above the initial public offering price or at the time that they would like to sell. In addition, we intend to submit a listing application for our common stock to be listed on a national securities exchange, including the NASDAQ Capital Market (“NASDAQ”), and there is no guarantee that we can meet the listing standards or that such listing application will be accepted. Even if such application is accepted and our common stock is listed on NASDAQ or other exchanges, an active trading market for our common stock may never develop, which will adversely impact your ability to sell our shares. If shares of common stock are not listed on NASDAQ or another national securities exchange, we anticipate that our common stock will be quoted at over-the-counter, or OTC markets, following the completion of this offering. There is no assurance that an active trading market for our common stock will develop on the OTC market.
Our stock price may be influenced by public perception of alternative tissue replacement and government regulation of such technology.
External events in the field of ligament reconstruction or the use of animal-derived tissue in human transplantation may have a significant negative impact on the public perception and stock price of certain companies involved in this kind of work. Potential adverse events in this field may occur in the future that could result in increased governmental regulation of our Z-Lig device. These external events may have a negative impact on public perceptions of our business, which could cause our stock price to decline.
We do not intend to pay dividends on our common stock.
We have never paid dividends on our common stock and we currently intend to retain any future earnings and do not expect to pay any cash dividends on our common stock in the foreseeable future. The declaration and payment of all future dividends, if any, will be at the sole discretion of our board of directors, which retains the right to change our dividend policy at any time, and may be limited by our debt arrangements in place from time to time. Consequently, stockholders must rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize any future gains on their investment.
Anti-takeover provisions in our certificate of incorporation and Delaware law could make an acquisition of the Company more difficult and could prevent attempts by our stockholders to remove or replace current management.
Provisions of Delaware law and our amended and restated certificate of incorporation and amended and restated bylaws, which will be effective upon the completion of this offering, may discourage, delay or prevent a merger, acquisition or other change in control that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares. These provisions may also prevent or delay attempts by stockholders to replace or remove our current management or members of our board of directors. These provisions include:
| · | advance notice requirements for stockholder proposals and nominations; |
| · | the inability of stockholders to act by written consent or to call special meetings; |
| · | the ability of our board of directors to make, alter or repeal our amended and restated bylaws: and |
| · | the authority of our board of directors to issue preferred stock with such terms as our board of directors may determine. |
The affirmative vote of the holders of at least 75% of our shares of capital stock entitled to vote, and not less than 75% of the outstanding shares of each class entitled to vote thereon as a class, is generally necessary to amend or repeal the above provisions that are contained in our amended and restated certificate of incorporation. In addition, absent approval of our board of directors, our amended and restated bylaws may only be amended or repealed by the affirmative vote of the holders of at least 75% of our shares of capital stock entitled to vote.
In addition, upon the completion of this offering, we will be subject to the provisions of Section 203 of the Delaware General Corporation Law, which limits business combination transactions with stockholders of 15% or more of our outstanding voting stock that our board of directors has not approved. These provisions and other similar provisions make it more difficult for stockholders or potential acquirers to acquire us without negotiation. These provisions may apply even if some stockholders may consider the transaction beneficial to them.
As a result, these provisions could limit the price that investors are willing to pay in the future for shares of our common stock. These provisions might also discourage a potential acquisition proposal or tender offer, even if the acquisition proposal or tender offer is at a premium over the then-current market price for our common stock.
Dilution
If you invest in our common stock in this offering, your interest will be diluted to the extent of the difference between the public offering price per share of our common stock and the pro forma as adjusted net tangible book value per share of our common stock immediately after this offering.
Our historical net tangible book value as of June 30, 2015 was ($28,398,078), or $($6.94) per share of our common stock. Historical net tangible book value per share represents our total tangible assets less total liabilities divided by the number of shares of our common stock outstanding. Our pro forma net tangible book value (deficit) as of June 30, 2015 was approximately ($28,398,078), or ($0.39) per share of common stock. Pro forma tangible book value per share represents our total tangible assets less total liabilities divided by the number of shares of common stock outstanding as of June 30, 2015 after giving effect to (i) the automatic conversion of all of the outstanding shares of our convertible preferred stock into an aggregate of 37,504,969 shares of our common stock upon the completion of this offering, including shares of common stock to be issued as cumulative dividends accrued under such preferred stock and (ii) approximately an aggregate of $9,838,058 in outstanding principal and accrued interest on our convertible promissory notes (which is currently convertible into shares of our preferred stock but will be amended to be convertible into shares of our common stock in connection with the offering), upon the completion of this offering, at an average conversion price of $0.31, assuming that the offering is completed on 2015. Pro forma as adjusted net tangible book value per share gives further effect to the issuance of shares of our common stock at an assumed initial public offering price of $ per share, the midpoint of the price range set forth on the cover page of this Offering Circular, and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. Our pro forma, as
adjusted net tangible book value as of June 30, 2015 would have been $ million, or $ per share of common stock. This represents an immediate increase in pro forma net tangible book value of $ per share to existing stockholders and an immediate dilution in pro forma net tangible book value of $ per share to investors purchasing common stock in this offering.
The following table illustrates this per share dilution:
Assumed initial public offering price per share | | | $ | |
Historical net tangible book value per share as of June 30, 2015 | $____ | | | |
Decrease attributable to the conversion of outstanding convertible preferred stock and convertible promissory notes | $____ | | | |
Pro forma net tangible book value per share as of June 30, 2015 | | | | |
Increase per share attributable to new investors | $____ | | | |
| | | | | |
Pro forma, as adjusted net tangible book value per share after this offering | | | | |
| | | | | |
Dilution per share to investors in this offering | | | | |
| | | | | |
The following table summarizes on an as adjusted basis as of June 30, 2015, the difference between the number of shares of common stock purchased from us, the total consideration paid and the average price per share paid by existing stockholders and by new investors, assuming an initial public offering price of $ per share, the midpoint of the price range set forth on the cover page of this Offering Circular, and before deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us:
| | Shares Purchased | | | Total Consideration(1) | | | Average
Price per
Share | |
| | Number | | Percent | | | Amount | | | Percent | | |
Existing stockholders | | | | | | % | | $ | | | | | | % | | $ | | |
New investors | | | | | | | | | | | | | | | | $ | | |
| | | | | | | | | | | | | | | | | | | |
Total | | | | | 100 | % | | $ | | | | | 100 | % | | | | |
| | | | | | | | | | | | | | | | | | | |
| (1) | A $1.00 increase (decrease) in the assumed initial public offering price of $ per share, the midpoint of the price range set forth on the cover of this Offering Circular, would increase (decrease) the total consideration paid to us by new investors and total consideration paid to us by all stockholders by $ million, assuming that the number of shares offered by us, as set forth on the cover page of this Offering Circular, remains the same and after deducting estimated placement agent fees and estimated offering expenses payable by us. An increase (decrease) of 1.0 million shares in the number of shares offered by us would increase (decrease) the total consideration paid |
to us by new investors and total consideration paid to us by all stockholders by $ million, assuming the assumed initial public offering price of $ per share remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
As of June 30, 2015, options to purchase 6,447,747 shares of common stock were outstanding at a weighted average exercise price of $0.08 per share and warrants to purchase 12,142,842 shares of common stock and preferred stock were outstanding at a weighted average exercise price of $0.08 per share. Assuming all of our outstanding options and warrants are exercised, new investors will own approximately % of our outstanding shares while contributing approximately % of the total amount paid to fund our company.
Underwriting
We and WR Hambrecht + Co, LLC (the underwriter) have entered into an underwriting agreement with respect to the shares being offered. Subject to certain conditions, the underwriter has agreed to use its best efforts to procure potential purchasers for the shares of our common stock offered hereby. This offering is being undertaken on a best efforts only basis. The underwriter is not required to take or pay for any specific number or dollar amount of our common stock.
The shares are being offered on an all or none basis. Investor funds will be deposited into an escrow account for the benefit of investors set up by at . The offering will not be completed unless we sell the number of shares specified on the cover page of this Offering Circular. All investor funds received prior to the closing will be deposited into the escrow account until closing. The escrow account will be opened on the date of this Offering Circular and will remain open until the closing date. All funds received into the escrow account after the pricing of the offering will be held in a non-interest bearing account in accordance with Rule 15c2-4 under the Exchange Act. The escrow account will not be opened until the date of this Offering Circular. All funds must be transmitted directly by wire to the specified bank account at per the instructions disseminated at pricing by the underwriter. will not accept or handle any funds. On the closing date, the escrow agent will notify the underwriter whether the full amount necessary to purchase the shares to be sold in this offering has been received. If, on the closing date, investor funds are not received in respect of the full amount of shares to be sold in this offering, then all investor funds that were deposited into the escrow account will be returned promptly to investors, and the offering will terminate. The following table shows the per share and total underwriting discounts and commissions to be paid to the underwriter.
| | | Per
Share | | | Total | |
| Public offering price | | $ | | | | $ | | |
| Underwriting discounts and commissions payable by us | | $ | | | | $ | | |
| Proceeds, before expenses, to us | | $ | | | | $ | | |
Certain expenses of the underwriter (excluding discounts and commissions) of up to $ are payable by us. Shares sold to the public will initially be offered at the initial public offering price set forth on the cover of this Offering Circular. Any shares sold to securities dealers may be sold at a discount of up to $ per share from the initial public offering price. After the initial offering of the shares, the offering price and the other selling terms may be subject to change. The offering of the shares is subject to receipt and acceptance and subject to the right to reject any order in whole or in part.
We and our officers, directors, and holders of five percent or more of our common stock have agreed, or will agree, with the underwriter, subject to certain exceptions, that, without the prior written consent of the underwriter, we and they will not, directly or indirectly, during the period ending 90 days after the date of the Offering Circular:
| · | offer, pledge, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant for the sale of, or otherwise dispose of or transfer any shares of the our common stock or any securities convertible into or exchangeable or exercisable for our common stock, whether now owned or |
hereafter acquired by the undersigned or with respect to which the undersigned has or hereafter acquires the power of disposition; or
| · | enter into any swap or any other agreement or any transaction that transfers, in whole or in part, the economic consequence of ownership of our common stock, whether any such swap or transaction is to be settled by delivery of our common stock or other securities, in cash or otherwise |
This agreement does not apply, in our case, to securities issued pursuant to existing employee benefit plans or securities issued upon exercise of options and other exceptions, and in the case of our officers, directors and other holders of our securities, exercise of stock options issued pursuant to a stock option or similar plans, and other exceptions.
Prior to the offering, there has been no public market for the shares. The initial public offering price will be determined by negotiation between us and the underwriter. The principal factors to be considered in determining the initial public offering price will include:
| · | the information set forth in this Offering Circular and otherwise available to the underwriter; |
| · | our history and prospects and the history of and prospects for the industry in which we compete; |
| · | our past and present financial performance; |
| · | our prospects for future earnings and the present state of our development; |
| · | the general condition of the securities markets at the time of this offering; |
| · | the recent market prices of, and demand for, publicly traded common stock of generally comparable companies; and |
| · | other factors deemed relevant by the underwriter and us. |
We intend to submit an application with The NASDAQ Stock Market LLC (“NASDAQ”) to list shares of our common under the symbol “ZLIG.” In order to meet one of the requirements for listing our common stock on NASDAQ, the underwriter intends to sell lots of 100 or more shares to a minimum of 300 beneficial holders. If our common stock is not listed on NASDAQ, we expect that our common stock will be quoted on OTCQB immediately following this offering under the symbol “ “.
Any underwriter who is a qualified market maker on NASDAQ may engage in passive market making transactions on NASDAQ in accordance with Rule 103 of Regulation M of the Exchange Act, during the business day prior to the pricing of the offering, before the commencement of offers or sales. Passive market makers must comply with applicable volume and price limitations and must be identified as passive market makers. In general, a passive market maker must display its bid at a price not in excess of the highest independent bid for such security; if all independent bids are lowered below the passive market maker’s bid, however, the passive market maker’s bid must then be lowered when certain purchase limits are exceeded.
We have agreed to indemnify the underwriter against certain liabilities, including liabilities under the Securities Act. The underwriter and its affiliates are engaged in various activities, which may include securities trading, commercial and investment banking, financial advisory, investment management, investment research, principal investment, hedging, financing and brokerage activities. The underwriter and its affiliates may in the future perform various financial advisory and investment banking services for us, for which they received or will receive customary fees and expenses.
In the ordinary course of their various business activities, the underwriter and its affiliates may make or hold a broad array of investments and actively trade debt and equity securities (or related derivative securities) and financial instruments (including bank loans) for their own account and for the accounts of their customers, and such investment and securities activities may involve securities and/or instruments of the issuer. The underwriter and its affiliates may also make investment recommendations and/or publish or express independent research views in respect of such securities or instruments, or recommend to clients that they acquire, long and/or short positions in such securities and instruments.
Selling Restrictions
Switzerland
The shares may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange (SIX) or on any other stock exchange or regulated trading facility in Switzerland. This Offering Circular has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering or marketing material relating to the shares or the offering may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering or marketing material relating to the offering, the Company, the shares have been or will be filed with or approved by any Swiss regulatory authority. In particular, this document will not be filed with, and the offer of shares will not be supervised by, the Swiss Financial Market Supervisory Authority FINMA (FINMA), and the offer of shares has not been and will not be authorized under the Swiss Federal Act on Collective Investment Schemes (CISA). The investor protection afforded to acquirers of interests in collective investment schemes under the CISA does not extend to acquirers of shares.
Use of Proceeds
We estimate that the net proceeds to us from the sale of shares of common stock in this offering will be approximately $ based upon an assumed initial offering price of $ per share, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us.
We anticipate that we will use approximately $700,000 of net proceeds from the offering to repay certain outstanding accounts payable, including trade payables, consulting and professional fees. We also intend to repay outstanding principal amounts and accrued interest owed under our demand notes (the “Demand Notes”) and a line of credit note (the “LOC Note”) issued to CrossCart LLC. The outstanding principal of each Demand Note carries an interest rate of 8% per annum, which may be increased to 10% if an event of default occurs. As of June 30, 2015, the total amount outstanding under the Demand Notes was $125,093,
including accrued interest. The LOC Note bears interest at a rate of 10% per annum, and all principal and accrued interest will be due and payable on May 1, 2016. The LOC Note contains customary events of default. If the Company fails to pay all amounts due by May 1, 2016, the interest on the remaining outstanding balance will accrue at a rate of 12% per annum. As of June 30, 2015, the total amount outstanding under the LOC Note was $61,627, including accrued interest.
We also intend to use approximately $300,000 of the net proceeds to pay certain compensation owed to our employees as a result of the reduction of their salaries, including approximately $210,000 of reduced compensation owed to our Chief Executive Officer and Chief Financial Officer. For a description of the salary reduction, see Compensation of Directors and Executive Officers on page [ ].
We anticipate that we will use the remaining net proceeds, approximately $ , from this offering to:
| · | launch our commercialization plan for Z-Lig products in Europe, South Africa and other countries that accept our approved CE Mark, including the establishment of sales, marketing and distribution infrastructure, expansion of our processing and manufacturing capacity to a commercial scale and hiring of additional personnel; |
| · | fund post-market registry studies of Z-Lig in Europe, South Africa and other select markets; |
| · | conduct pre-clinical and R&D activities for product candidates; and |
| · | initiate clinical trials for Z-Lig products in the U.S. |
We anticipate using the remaining amounts, if any, for general corporate purposes.
The expected use of net proceeds from this offering represents our intentions based upon our current plans and business conditions, which could change in the future as our plans and business conditions evolve. The amounts and timing of our actual expenditures may vary significantly depending on numerous factors, including the progress of our commercialization effort, the results from clinical trials and preclinical studies, the amount of cash available from other sources and any unforeseen cash needs. As a result, our management will retain broad discretion over the allocation of the net proceeds from this offering.
Description of Business
Overview
Aperion Biologics, Inc. (“ABI”), headquartered in San Antonio, Texas, is a commercial-stage medical device company addressing the significant need for an alternative to human-based sources of tissues to be used in surgical procedures. Our lead product, the Z-Lig, is a patent-protected porcine tendon currently in limited commercial release for use initially as an anterior cruciate ligament (ACL) replacement in revision and multiligament knee joint procedures (see Figure 1). Z-Lig is the only known biological alternative to human tissue for ACL replacement. Since the Z-Lig is a biological implant that maintains a scaffold, it can become populated and remodeled with the patient’s own cells (similar to human-sourced tissue), referred to in the industry as “ligamentization”. This ligamentization is one of the keys to Z-Lig’s proven ability to be highly functional in patients for many years after implantation. Our propriety process is protected by an extensive portfolio of patents and patent applications, including 23 issued patents in the U.S. and internationally.
Figure 1: ABI’s Z-Lig® (in product packaging on right) for use in ACL replacement surgery.
With the exception of synthetic ACL replacements, which are deemed inadequate by most orthopaedic surgeons , the only current ACL replacement choices include using tendon harvested from another part of the patient’s own body or tendon from a human cadaver (autograft and allograft, respectively). These existing choices have a number of shortcomings. As for autografts, the main clinical issue is the need for a second surgical site for harvesting. This results in longer operation times, increased patient pain and morbidity, longer rehabilitation time and reduction of function at the harvest site. Many clinicians believe autograft harvesting actually causes “harm” to patients by sacrificing one part of the human body to fix another.
As for allografts, the main clinical issue is the lack of supply of tissue of adequate quality. Allografts are harvested from cadaveric donors. The quality of allografts can be quite variable due to the age, sex, size, genetics, and medical condition of the donors and how the allograft tissue is processed. Because tissue donation is not commonplace, the supply of allograft tissue can be quite variable. Even in the U.S. with over an estimated 300,000 ACL replacements performed annually, less than 25% of these procedures were performed using allograft. The remaining patients must have the graft harvested from their own bodies.
Figure 2: Torn ACLs can be replaced with human-based or synthetic grafts.
The need for an alternative to human-sourced grafts has been recognized in the medical community. The only non-human commercialized grafts are synthetics which tend to quickly wear, degenerate and fail. Synthetic grafts are not approved for use in the U.S. and we are not aware of any in clinical development in the U.S. In Europe, synthetic grafts have extremely limited commercial acceptance and are primarily used for patients who wish to return to sport in the shortest time period possible. We believe that the lack of durability of a synthetic graft can mainly be attributed to its inability to regenerate (as tissue-based implants do) after wear from use. The only synthetic graft in development in Europe is a resorbable polymer based scaffold graft currently in clinical safety trials and will require an additional clinical study prior to commercial availability.
Given the issues with synthetic grafts, we believe that animal tissue is the only viable alternative to human-sourced ACL grafts. No other company sells or has in human clinical development an animal-based (xenograft) ACL product.
The xenograft market is a well-accepted segment of the medical device industry. Xenografts from porcine, bovine and equine sources have been used as heart valves, extracellular matrices (patches), injectable collagen and other replacement/augmentation tissues for many years. Our products are different from currently commercialized xenografts in that they (i) act as biological scaffolds (unlike, for example, heart valves) and (ii) do not illicit an immunological reaction (unlike, for example, extracellular matrices and injectable collagen).
We were originally incorporated in the State of Delaware under the name “CrossCart, Inc.” CrossCart Inc. was established in 1996 to develop xenograft replacement tissues including cartilage, meniscus cartilage, soft tissue devices, and ligaments. Its efforts to do so were then focused on ligaments due to the ability to measure objectively the outcomes, the presence of an FDA ligament guidance document, and the large unmet needs. The Company developed the Z-Lig process to be flexibly applied to each of these tissue devices. Due to the limitations of the CrossCart name, the Company was renamed Aperion Biologics Inc. in 2008 and established a manufacturing facility in San Antonio.
CrossCart, Inc. is a separate and distinct entity from CrossCart LLC, which was formed in 2013 as an investment vehicle to purchase outstanding securities the Company held by a previous controlling stockholder. In addition, Dr. Kevin R. Stone, a member of our Board of Directors, is the controlling member and manager of CrossCart LLC.
Industry and Market Opportunities
ABI believes that regenerative medicine is one of the markets with the highest growth potential within the medical device industry. The potential size of the tissue engineering market totals $74 billion according to industry research. We estimate that the market expenditures for ACL autografts and allografts total over $2 billion worldwide, including $750 million in the U.S., $625 million in Europe and $750 million in the rest of the world. These figures include both the cost of allografts (approximately $2,500 per procedure based on our calculation) and the direct surgical costs of the autograft harvesting and subsequent direct incremental rehabilitation and medication costs related to the second surgical site (approximately $2,500 per procedure in total based on our calculation).
We believe that the global supply of quality allografts does not sufficiently meet the demand for knee ligament procedures. Furthermore, the demand is expected to be driven higher by the expanding aging and active population suffering from degenerative ailments. Over 250,000 ACL reconstruction surgeries are performed in Europe annually. The vast majority of such surgeries were performed using autografts. Throughout most of Europe, allografts are available on a very limited basis and the need for an alternative option is more acute.
We have also conducted a study to estimate of the size of the potential markets applicable to our earlier stage pipeline, including Z-Patch, Z-Fix, and Z-Meniscus. Based on this study, we believe the total addressable U.S. and European markets for extracellular matrices, wound healing, bone replacement and meniscus replacement are $3.6 billion, $5.0 billion, $2.1 billion, and $300.0 million, respectively.
Current Treatment and Limitations
The major problems with the existing ACL replacement graft market are (i) autografts require a second surgical site which causes significant additional cost and morbidity; (ii) allografts are in short supply, are highly inconsistent in quality and can transmit human disease; (iii) synthetic grafts are unable to regenerate and therefore fail due to the continuous abrasion from regular use; and (iv) traditional xenografts have been plagued with rejection or lose their ability to be populated with the host’s cells.
Below are the specific issues within each category of graft:
Problems with Human-Sourced Grafts (Autografts and Allografts)
Autografts
As shown in Figure [ ] below, during an autograft procedure, a second surgical site is chosen on the patient’s body and tendon (patellar tendon or hamstring) is harvested. Consequently, the patient suffers from additional pain, morbidity and scarring at the harvest site. The requirement for a second surgical site prolongs operating time and causes a more complex and extended recovery period. Most patients have impaired function, including muscle weakness and many even develop arthritis at the autograft harvest site. The direct surgical costs of the autograft harvesting and the subsequent direct incremental rehabilitation and medication costs are high and it is approximately $3,500 per procedure. In addition, patients’ own tendons available for harvesting are of limited supply and do not grow back. Given that many active patients reinjure their repaired ACL or injure their other ACL, autografts may not be a viable follow-on procedure.
Figure 3
Allografts
Supply of cadaver tissue remains extremely constrained and demand for high quality tissue significantly outstrips the supply. One allograft tendon is needed for each ACL replacement. Only half of the 25,000 cadavers available in the U.S. annually provide tissue meeting the specific requirements of orthopaedic tissue donation and, of those that meet the requirements, the yield is only about six harvested grafts suitable for ACL replacement per cadaver. Therefore, of the over 300,000 ACL replacements performed annually in the U.S., approximately 20-25% of these procedures were performed using allograft. Outside the U.S., the availability of allografts is even more limited on a country by country basis. Of these procedures performed with an allograft, we believe a significant portion were performed with a graft that would generally be considered by most surgeons to be less than ideal, typically because the graft was not: (i) the preferred type (bone-tendon-bone, bone-tendon or soft tissue); (ii) the preferred length; or (iii) from a young enough donor.
Allografts have been shown to have inconsistent characteristics due to the broad range of ages of the tissue donors. Published studies document the biomechanical properties of human allografts based on age. Tensile testing has been performed on allografts from different aged donors. Tensile testing to failure from different allograft donors, as shown in the chart below, demonstrate that the effect of donor age on the strength of allografts can be significant. For example, an allograft from a 66-75 year old donor can only withstand (i) approximately 60% of the stress load that an 18-35 year old allograft can withstand or (ii) approximately 40% of the stress load that a Z-Lig can withstand. Surgeons and certain patients are aware of the importance of the age of the cadaver from which the allograft is derived and given the preference for young donors, the supply shortage of quality allografts is more pronounced.
Allografts are produced in tissue banks which are regulated differently than medical devices by the FDA. Product from tissue banks is not subject to the FDA approval process and is therefore not subject to the same controls and regulations as is a medical device. Despite a range of improvements in industry monitoring and oversight made beginning predominantly in 2005, soft tissue allograft infections in patients continue to be reported and have resulted in recalls of allografts from several tissue banks. While allografts are generally safe, the risk of human disease transmission continues to exist.
In addition to the disadvantage discussed above, the availability of allograft is even more restricted in Europe and other international markets. These patients are often left with little to no choice but to use autografts because the allograft market in these markets is severely underdeveloped due to legal, cultural and religious issues. Allografts in some international markets are only available from hospital-based or regional tissue banks. The quality, processing and consistency of these grafts have even greater variability and thus limits allograft acceptance.
Problems with Non-Human Grafts (Synthetic Grafts and Traditional Xenografts)
Synthetic Grafts
Numerous attempts have been made to develop artificial ligament devices. Ligaments made from Dacron, carbon fiber, Gore-Tex and other chemicals/fibers have been tested by competitors. However, we believe that no synthetic device to date has possessed the necessary biological and biomechanical properties to achieve long-term success because they not have supported meaningful cellular remodeling by the human’s own cells. The only graft that has survived the test of time in the ACL indication are biologic grafts. Regardless of these limitations, a limited number of synthetic devices are available outside the U.S. but the acceptance of such devices by surgeons has been very low. Combination devices, which include a synthetic device augmented with a human allograft, have seen only limited technical success and they do not address most of the issues discussed above regarding human allograft sourcing and use.
Xenografts
Specific types of antibodies in humans,known as anti-gal antibodies, bind to the α-gal antigen
on animal-based grafts and initiate a rapid immune rejection process. The α-gal-mediated rejection may be prevented by: (i) removing or deactivating the antigens found on the xenograft or (ii) sourcing the xenograft from an animal that has been genetically modified to remove or eliminate α-gal antigens.
Deactivation of antigens involves crosslinking -- a chemical manipulation of the xenograft to make it acceptable to the human body. However, too much crosslinking results in a xenograft that is incapable of acting as a biological scaffold and thereby incapable of becoming populated with the patient’s own cells (no ligamentization occurs). The ability of a xenograft to become remodeled and incorporated into the host has been shown to be vital to the long-term performance of the implant. Too little crosslinking results in the patient’s immune system attacking the xenograft and rejecting it either acutely or over the longer-term (see Figure 4 below). Historical attempts by competitors to use xenografts have ended mostly with failure. Companies have attempted to develop animal-based products for the ACL market using high concentrations of glutaraldehyde to attenuate recognition of antigens on the graft, previously successfully used to “fix” porcine heart valves, but were abandoned due to the frequent failure of subsequent implants.
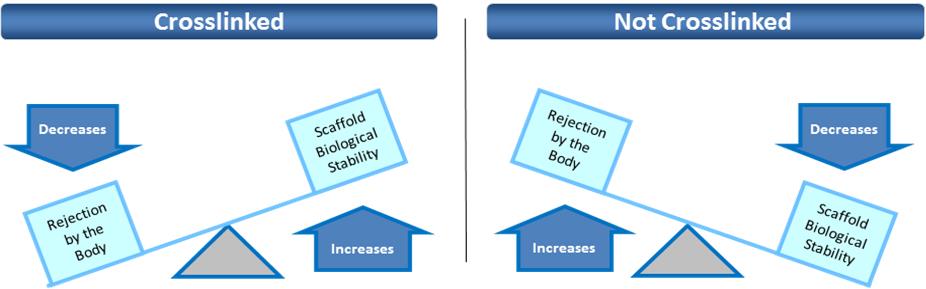
Figure 4
We believe that previous failure of knee-related xenograft was mainly due to local toxicity and synovitis caused by residuals of the chemical crosslinking agent leaching into the surrounding joint areas and not rapidly dissipating. Knee capsules, unlike heart valves and rotator cuffs, generally have very little turnover of surrounding fluid, and, as a result, residual crosslinking agent does not have the opportunity to meaningfully dissipate or be removed.
An alternative method to prevent α-gal-mediated rejection is by using genetically altered animals. Genetic alteration has been shown to be technically feasible in pigs. However, scaled-up production of such pigs has not occurred because genetically modified pigs have not been found to be completely void of the α-gal antigen nor reliably reproduce offspring free of the α-gal antigen. We believe the resultant production cost and regulatory hurdles will be very significant, are we are not aware of any company that is attempting to scale-up production of genetically modified pigs for commercial tissue use.
Our proprietary approach has been shown to solve both the rejection as well as the compatibility issues. Currently, we believe we are the only company that is pursuing an animal-derived ACL implant. See “Our Solutions and Advantages” section for additional details.
Our Solutions and Advantages
To solve the xenotransplant rejection problems mentioned above, we developed the Z-Process®. The Z-Process is a proprietary process that immunochemically modifies animal tissue so that it is compatible with the human immune system (see Figure [ ] below). The Z-Process addresses both α-gal and non-gal antigens on the xenograft. Our Z-Process also creates porcine tendons for safe use in humans while still maintaining biological scaffold activity of the implant. Using our Z-Process, we developed the Z-Lig device as an immunocompatible, porcine-derived ACL reconstruction alternative which provides a readily available, off-the-shelf solution that is strong, sterile, and reproducible. The process and resultant product is protected by our extensive portfolio of patents and patent applications. See “—Intellectual Property”
Figure 5
We uses α (alpha) -galactosidase enzyme to cleave the terminal galactose of the gal antigen so that the carbohydrate chain remaining on the animal tissue is the same in its structure as the carbohydrate chains present in humans. Importantly, our technology uses α-galactosidase enzyme to remove α-gal antigens from non-primate animal tissues, including ligaments, cartilage, bone, heart valves, vascular tissue, injectable collagen and other tissues. We are not aware of any competing technology that acts as a replacement for α-galactosidase for cleaving the gal antigen while still maintaining the tissue’s biological scaffold. Therefore, we believe that
competitors are prevented from deactivating the α-gal antigen from specific animal tissues without either (i) infringing ABI’s patents or (ii) making the xenograft non-functional or inert and incapable of incorporating the host’s own cells.
While the α-galactosidase enzyme treatment solves the α-gal rejection issue, it has no known effect on the non-gal immune response. To address the non-gal response, low level chemical crosslinking is utilized to control the response due to the non-gal antigens. This multi-step process was developed by our team and refined over many years to the final, highly specific and reproducible process used in the clinical trials. Our process patents cover the crosslinking treatment of tissues for non-gal antigens and subsequent sterilization using forms of e-beam irradiation.
Outside of autografts and allografts, Z-Lig represents a new and unique method in the industry in that it incorporates the host’s cells. When first implanted, Z-Lig contains no live cells. Z-Lig is gradually populated and remodeled by the patient’s own cells, eventually yielding a mature human ligament through the process of ligamentization. We believe that Z-Lig remodels at a similar rate as human allografts, with substantial ligamentization occurring within 6 to 12 months and complete remodeling within two to three years. This remodeling is key to Z-Lig’s long-term functionality.
In regenerative medicine much of the work with stem cells, growth factors and bioactive agents is also focused on the development of an ideal scaffold. Many different techniques have been employed to replicate the architecture and organization of normal tissue and organs, i.e. non-woven and woven materials, development of porous materials, 3-D printing, etc. All of these efforts may have shortcomings in terms of the architecture, mechanical properties and biological-materials interactions, among others. We believe scaffolds produced using the Z-Process may offer optimal properties in terms of the natural architecture, target mechanical properties, and inherent material properties of a biological substrate. The use of scaffolds produced using the Z-Process could represent a sizeable advance in the development of regenerative therapies.
Product Candidates and Pipeline
The Company’s product pipeline is summarized in Figure 6 below:
Figure 6
The Company currently has four product candidates in clinical and preclinical development and research stages. Below is the current status, clinical results and regulatory strategy for the product candidates in the portfolio:
Z-Lig® Family - The Z-Lig family of devices are designed for use in a range of ligament reconstruction procedures, especially the initial application for the anterior cruciate ligament (ACL). The device is sourced from animal tissue but is sized and has the requisite biological and mechanical properties for use in these type procedures. A key advantage of this device is that little training is required for its use. The use of the device does not require the surgeon to change his surgical technique, his preferred fixation system, nor his rehabilitation protocols.
We received the CE Mark for the BT+ (bone-tendon) and BTB (bone-tendon-bone) versions of the Z-Lig Device Family for revision and multiligament ACL reconstruction (ACLR) procedures. We have developed a portfolio of data supportive of clinical safety and performance to enable the opportunity to market the device in the EU. We also have plans to provide additional clinical data required to expand our approved indication to the full ACL indication to cover all ACL procedures, including primary ACL surgeries. The Z-Lig has thus been evaluated to demonstrate its suitability as a biocompatible graft alternative to allograft and/or autograft through bench testing and extensive in vivo testing. We plan to expand indications as well as introduce a ST (soft tissue) version to complete the spectrum of ligament reconstruction devices for the Z-Lig Device Family.
Z-Patch – an extracellular matrix product used in soft tissue repair and augmentation procedures, such as hernia and Achilles tendon repair. Sourced from porcine dermis, this product has potential applications in orthopaedics, general, reconstructive, plastic and wound care indications. In the U.S., this product may be developed and approved through the 510(k) clearance route under the FDA process.
Z-Fix – a xenograft bone based product which is a biologic fixation device for ACL reconstruction and would be complementary to the Z-Lig device (all biologic ACL reconstruction). In the U.S., this product may be developed and approved through the 510(k) clearance route. Designs using bone could be expanded to include indications such as bone graft repair and spacer implants used in spine surgery.
Z-Meniscus – a xenograft meniscus device which can be use in the repair or reconstruction of meniscal injuries and defects. In the U.S. and markets outside the U.S., this device would require additional clinical data prior to regulatory approval and commercialization.
Our Current Status – Clinical Results with the Z-Lig
After extensive review of our regulatory dossier, we were granted the CE Mark for the Z-Lig device for multiligament and revision ACL procedures in April 2014. The dossier was an extensive summary of the science, preclinical, validation data and included the data collected from the European and South African multicenter study. We are also committed to plans for continued clinical and medical evaluation of this device family to insure the processing related issues have been addressed (post-market controls, reporting / vigilance, and long-term safety and performance). The additional data on 20 to 30 subjects at one year period will be submitted to support the indication expansion to the full ACL indication. In addition, we have committed to post-market safety evaluations and plans to continue Z-Lig Subject follow-up for an extended period of time (up to 60 month follow-up) for post-market surveillance purposes, which will generate the additional clinical data for market expansion opportunities both in the existing markets covered by the CE Mark and in the U.S.
In 2012, we initiated our multicenter, prospective, controlled (allograft), randomized and blinded clinical study in Europe and South Africa and subjects enrolled in the trial have surpassed the 36 month long-term follow-up period. This study enrolled sixty-six subjects (n=66). All time points to complete the endpoint analysis for Primary Performance and Efficacy have been achieved, therefore the study continues for the purpose of collecting additional post-market safety data. Generally, the failure of ACL grafts occurs within the early post-implantation window (3 months after the operation), when the ligamentization process has initiated and graft strength is most vulnerable. Our assessment with the Primary Performance measure (primary endpoint for CE Marking) supported the conclusion of non-inferiority of Z-Lig versus allograft, with 100% success in all subjects at 6 months follow-up in the PP population. Assessment of clinically relevant success outcomes such as patient activity, patient satisfaction, stability, functional and effusion resulted in compelling data showing comparable performance to the allograft control.
Surgeons using the device (our study Principal Investigators) have reported consistent, comparable or superior quality of the Z-Lig graft materials in comparison to allograft controls. Surgeons have reported that the Z-Lig graft has a superior bone plug material, is easily used and managed during surgery, and requires no change to their standard surgical procedure.
Within the initial 12 months, bacterial infections associated with the graft were reported in 6 subjects implanted with Z-Lig in this study. We conducted a proactive and extensive safety investigation which concluded that the infections were caused by contamination of incoming tissue and processing deficiencies. Excepting these cases of bacterial infection, it was concluded that the reported complications are consistent with those found in the medical and scientific literature and were unrelated to the graft. We have implemented a robust corrective action plan to address the sources of bacterial contamination, and these validation activities
have been completed to verify that the corrective actions were sufficient and effective and that sterility can be assured. Our corrective action plan and validation data were reviewed by the Notified Body for CE Marking and they required no additional action by us. We plan to execute additional post-market study to confirm the effectiveness of the corrective actions.
U.S. Human Clinical Results
Z-Lig is currently under development in the U.S. under an Investigational Device Exemption (IDE) with the FDA. Z-Lig’s development trials follow the FDA guidance document on Intra-Articular Knee Ligament Reconstruction Devices. The safety testing modules conducted for CE Mark approval can also support FDA approval of Z-Lig as well as 510(k) approvals of follow-on products.
The results of the U.S. safety pilot study demonstrated that the Z-Lig was safe (see Figure 7 below). Data from the human clinical study demonstrated the long-term safety and feasibility of the Z-Lig device. The study included 10 subjects followed for 2 years under the protocol. This group represented a highly active subject population, including subjects with high pre-injury Tegner activity levels (competitive athletes with Tegner ratings 8-10), subjects with clinically significant comorbidities, and subjects undergoing revision ACL surgeries. These are all known risk factors to outcomes following ACL surgery. The device performed as intended during the surgical procedure and post-surgically through progressive weight bearing and return to normal activities. The grafts were well-tolerated without observation of a deleterious immunogenic response. In 4 of the 10 subjects, non-device related adverse events were experienced either due to vigorous sports activities (e.g., sports injury, rupture) or events related to the initial procedure (e.g., fixation, procedure error). No systemic diseases or secondary infections resulted from the implantation of the device and there was no acute immune response or long-term rejection response exhibited by any of the subjects. The 12-month results of the pilot study described below were submitted to the FDA, leading to unconditional acceptance by the FDA of a pivotal clinical trial protocol. As assessed by the operating surgeon and two non-affiliated orthopaedic knee specialists, five of six evaluable subjects presented with functional grafts at the 24-month post-operative time point and satisfied all success criteria, including effusion, knee laxity (KT 1000), pivot shift, Lachmans and Anterior Drawer tests. Assessment by MRI also showed significant remodeling and maturation of the functioning grafts. One of six evaluable patients that presented without a functional graft had a tibial bone fixation that loosened.
Figure 7
The above figure describes the outcomes of subjects five years postsurgery. Recently, we completed 12-year follow-up on the five evaluable patients with functional grafts. Safety of the devices continued to be exhibited throughout the 12-year interval, and all evaluable patients continued to demonstrate graft function with knee stability. The nature and rate of complications and adverse events seen during this feasibility study were similar to those cited in the literature. In April 2014, we received unconditional approval from FDA to move forward with its pivotal efficacy trial.
The pivotal efficacy trial will involve 326 patients at up to 10 clinical sites. The study will be controlled, randomized and evaluator-blinded. Half of the patients will receive Z-Lig while the other half will receive an allograft control. The study’s primary endpoint is non-inferiority of function of Z-Lig (relative to allograft) at 12 months and 24 months post-implant. We plan to continue discussions with the FDA regarding ways to optimize the study through randomization schemes, the number of clinical sites and discussions surrounding the data gathered during the European trial.
The body of data in support of the safety and performance of the Z-Lig Device Family is thus consistent and extensive, with multiple studies concluding that the Z-Lig Device Family performs in accordance with design criteria and has demonstrated adequate safety and performance in support of its intended use. Studies for long-term efficacy analysis have been completed (reported herein) and post-market surveillance studies are in effect for ongoing assessment.
Preclinical Studies
Prior to the clinical experience, we conducted controlled studies in 20 Rhesus monkeys to evaluate Z-Lig’s safety and performance prior to use in humans. Our studies showed that knee joints of monkeys implanted with a Z-Lig displayed normal motion and laxity within a few weeks post implantation. This normal motion and laxity was maintained for the entire one-year period of the study. Serological studies of these monkeys showed a transient production of non-gal
antibodies that was deemed to have substantially decreased at approximately nine months post-implantation due to the gradual replacement of the pig tissue and matrix by the recipient’s own cells and tissue. Biomechanical analysis of the implants after one year in monkeys demonstrated characteristics similar to those of the control (monkey allograft) and a significant return to normal leg function (as also seen in the allograft-implanted monkey). Histological studies of the implants one year post-implantation revealed effective replacement of the porcine tissue components with those of the recipient, demonstrating that the device served as a scaffold that supported gradual remodeling with the monkey’s own cells. In addition to these animal studies, we also successfully completed initial safety and performance studies, including cytotoxicity, acute systemic toxicity, pyrogenicity, residual risk assessment, viral safety, viral inactivation, tensile strength and dynamic abrasion.
Our Marketing & Distribution Strategy
Markets and Methods of Distribution
The orthopaedic surgery marketplace is fragmented with many clinicians focused on certain types of surgical procedures, such as trauma, total joints, spine, sports medicine. Our marketing efforts will be focused on developing awareness of the Z-Lig to stakeholders treating sports or activity related injuries. The scope of our marketing and distribution efforts includes the patients who desire their specific expertise and suppliers who service their needs. The identification of this market segment and the key opinion leaders enable a more efficient, focused sales and marketing effort. Additionally since the Z-Lig device does not require changes to surgical technique and thus avoids additional costs for training, marketing activities are focused on educating on the science, technology and the previous clinical experiences.
The following describes our plans to initiate marketing and distribution activities as an independent organization through a distribution model. With the CE Mark approval, we intend to introduce the Z-Lig ACLR device through a focused, phased market introduction. We believe the initial market need for the Z-Lig are revision and multiligament cases where a graft option is typically required to achieve the desired clinical result. Initially we intend to target a select number of markets where human or synthetic graft options are accepted, in particular those nations whose sites participated in our European and South African (EUSA) clinical trials, which includes Italy, South Africa, Poland, Spain, UK, and the Benelux countries (Netherlands, Belgium, Luxemburg). We will utilize indirect distribution channels who have met our selection criteria (i.e. existing/complementary product lines, market share, financial, willing to invest in the product line, among others). In these select markets, we will introduce the product through a targeted approach, clinicians chosen by our investigative surgeons and/or our distribution partners. Supporting the efforts of our indirect distribution channel will be direct personnel specifically tasked with sales management and clinical marketing specialists for training and technical/scientific field education. The next distribution phase will expand our distribution reach to additional European and Asian markets that accept the CE Mark, with a specific focus on high-volume clinical centers or those specializing in knee injuries. The third and final phase would include pan-European expansion into markets in the EU and continued global introduction in markets outside the United States who accept the CE Mark and potential distribution into non-European nations whose regulatory bodies require additional regulatory approval.
The following describes the main elements our proposed structured rollout in each region:
| • | Identify Targets in Each Country |
| • | Adapt Brand Messaging and Marketing Communication |
| o | Marketing Materials adapted for local markets |
| o | Product Training material: develop Sales and Surgeon training (product and technology) presentations. |
| o | Sales Operations: Ordering and shipping processes |
| • | Seed and Expansion stage |
| § | Sales training meetings in each market with distributors |
| § | Distributors identify 4-10 “launch” clinicians |
| • | Tradeshows – national/in-country – European Society of Sports Traumatology, Knee Surgery, & Arthroscopy (ESSKA), International Society of Arthroscopy, Knee Surgery and Orthopaedic Sports Medicine (ISAKOS), European Federation of National Associations of Orthopaedics and Traumatology (EFFORT), and others. |
| • | “Surgeons selling surgeons” - Company sponsored symposia |
| • | Post Sale Monitoring and Partnering |
Phase 1 (launch to 12-18 months)
Distribution efforts will be in 6 to 10 markets which have accepted options for ACL reconstruction (allograft and synthetic), have reimbursement pathways for graft alternatives, and/or are countries where we have conducted our clinical evaluations. Some of these initial markets may be characterized as:
| · | Major markets which utilize graft options such as allograft, xenograft or synthetic; clinicians who are receptive to the use of alternatives. |
| · | Markets with minimal coding restraints. |
| · | Markets where we have conducted our clinical evaluations to take advantage of our clinical successes and leverage our study clinicians and key opinion leaders (KOLs) to introduce and penetrate the local markets. |
| · | Collaboration with our distributors or clinical investigators; initial distribution in each market will focus on a select number of thought leader clinicians to effect an orderly launch of our product and gain clinical experience and feedback, verify current sizing and surgical techniques, and help insure success and traction in the marketplace. |
| · | This limited release will enable us to gather first hand data, establish successes and help fine tune training and marketing activities. |
| · | In some markets release will take the form of a registry to establish a clinical database for the local market and obtain product performance feedback. The data will also be used for broader marketing and reimbursement activities in the future. |
| · | Presence at key international medical events with exhibit supplemented with symposia or presentations. |
Phase 2 (12-18 months to 24-36 months)
Efforts will focus on expanding our customer base in existing countries and extend our distribution to additional European markets with a specific focus on high-volume clinical centers or those specializing in knee injuries. This phase will also involve markets outside of Europe that accept the CE Mark (i.e. Canada, Turkey, Saudi Arabia, Australia and Korea). Working with these centers may require implementation of additional “mini-studies” for clinical acceptance and reimbursement. Continue our presence at international meetings and assist our distributors at national events. Continue to add and train distributors is expansion markets.
Phase 3 (24 months and beyond)
Efforts will continue in the expansion of customer base in markets with established distribution. In addition to pan-European expansion in the EU markets, continued introduction in markets outside the United States who accept the CE Mark. These non-European markets would include those whose regulatory bodies require additional regulatory submissions and approvals. Developing access, advocacy and adoption in the non–European markets.
Our Distributors
We require that each indirect distributor purchase stock of our products and store grafts and provide local distribution, billing, and collection for its designated markets. Initially distributors will be directed to identify a select group of qualified clinicians to become users and potential key opinion leaders of the Z-Lig within their territory.
Choosing the right distribution partners as we move into the various international markets is key. Some of the issues that have to be explored and expectations established with any potential business partner before distribution can begin are:
| · | A current presence in the geographic region in the orthopedic, sports medicine or sports traumatology market with existing products |
| · | The experience, ability, or the desire to handle a regenerative medicine/tissue-based product (store, monitor, distribute, invoice, collect, etc) |
| · | Presence in the target market including accounts and relationships with clinicians |
| · | Presence at local, regional, and national logical meetings |
| · | A willingness to invest in training their sales force and customers, and a willingness to invest in marketing, training and promotional activities that will drive distribution and acceptance of their products |
| · | A willingness to invest in activities needed to establish products in their markets, including those related to establishing reimbursement. Local knowledge of regulatory barriers and time to market conditions are valuable. |
| · | Distributors with adequate financial resources |
Quality of the product, safety, efficacy, and price are important, but the value of surgeon relationships and customer service are equally important factors for success.
Our Management, Direct Sales and Marketing Team
As we continue to advance our commercialization efforts and expand our operations, we expect to hire additional management members to execute our strategies. These additional hires may include senior management positions overseeing our business development and regulatory affairs. We also intend to hire additional personnel to enhance our financial reporting, accounting and auditing capabilities, which will ensure that we comply with any ongoing reporting requirements under the rules and regulations of the SEC.
We will initially use an indirect distribution model. We intend to increase our headcounts by hiring additional sales and marketing employees, including a Vice President of European, Middle East and Africa Distribution Management responsible for establishing our distribution network, initiating and coordinating our distribution activities within the distribution network, and driving distribution objectives; and Clinical Marketing Specialist(s) who will be focused on both surgeon and sales representative education on the science and technology surrounding the Z-Process, Z-Lig product, and our company.
Marketing Approach
The introduction of an innovative technology, especially one for ligament reconstruction, will meet an unmet market need but will also encounter the challenges of clinical acceptance. Our efforts to execute a Level I clinical trial with KOLs in Europe represent a significant step towards gaining scientific and clinical credibility. The study design includes post-marketing surveillance out to 5 years as a means of providing continual product performance feedback. We will focus on peer-to-peer marketing activities as a rapid and effective means of establishing credibility and market acceptance of our new technology.
Direct marketing efforts at logical meetings, symposiums and others are highly effective, especially those with clinician-to-clinician interactions. For example, we have conducted lunchtime symposiums or have had invited lectures at the ISAKOS (International Society of Arthroscopy, Knee Surgery and Orthopaedic Sports Medicine) and ESSKA (European Society for Sports Traumatology, Knee Surgery and Arthroscopy) meetings to introduce surgeons to the Z-Lig in a peer-to-peer environment. We plan to repeat this successful format at additional international meetings. Independent surgeon training symposia with KOLs will be encouraged/supported. Exhibiting at major international meetings is planned and supporting local distributors to exhibit at in-country meetings will be encouraged. Marketing efforts will also be expected by the local distribution channels to augment company efforts.
Use of Initial Market Feedback
In some markets, post launch market feedback will be collected from clinicians who have had the opportunity to use the product. Data will be used to confirm safety and performance expectations as well as provide information for product enhancements, market reception and education or training opportunities. We anticipate surveys to be sent to approximately 20-24 clinicians who have used the product in a minimum of 6 cases and can provide follow up assessments 6 and 12 months post-operative. These surveys will be conducted with surgeons who have been targeted in our launch activities.
The Advantage of Clinical Registries
In addition to the ongoing multicenter trial (post-market surveillance), performance of smaller, local market studies as registries will be encouraged. These additional studies have been proposed to supplement our CE Mark approval and may be needed to encourage clinical Z-Lig use in new markets (those outside of trial countries). These studies can explore issues such as cost (reimbursement), performance versus autograft, overall operative time savings, and other advantages.
Competition
As has been mentioned elsewhere in the business section, the principal competition for our Z-Lig device arises from other types of ACL graft materials: allograft (from cadaveric tissue) and synthetic (polymers that are designed to replicate the physical, strength characteristics of native human ligaments. We do not believe that there are other xenograft (from other species) or biologic ligaments either in the market or under development by the established orthopaedic companies. In some global markets, synthetic devices such as those produced by LARS, Xiros (Neoligaments) and Telos have limited clinical use due to their less than satisfactory clinical history. Currently, the only synthetic device under clinical testing that we are aware of is a resorbable polymer-based scaffold that is being developed by Soft Tissue Regeneration.
Intellectual Property
Our product candidates and Z-Process technology are protected by an extensive intellectual property portfolio. As of June 30, 2015, we held 23 issued patents in the U.S. and internationally, consisted of 17 issued patents in the U.S. and 6 issued patents in Europe and Japan. In addition, we currently hold 3 patent applications pending in the U.S. and Europe. These issued patents will expire between 2015 and 2024. ABI’s patent portfolio includes claims relating to treatment and application of specific tissues for a variety of medical and surgical uses. The intellectual property portfolio includes combined patents of process and material composition that support deantigenation, sterilization and viral inactivation of a wide range of biological tissues. ABI has exclusive rights to the only known method for humanizing non-primate animal grafts for replacement of ligaments, bone, etc. in humans without making the graft inert.
Patents regarding Z-Process include coverage of key immunochemical modification and sterilization techniques as applied to a variety of animal-derived tissues, including connective tissue grafts (tendon, ligament, articular cartilage and fibrocartilage); cardiovascular tissues (heart valve and pericardium); and calcified and de-calcified bone (granular bone matrix, cortical and cancellous structural bone for struts, cages and machined implants). The patent portfolio also includes immunochemical modifications of solubilized and homogenized dermal- and tendon-derived injectable collagen formulations.
In January 2009, we entered into a licensing agreement with the Curators of University of Missouri pursuant to which we obtained an exclusive right and license to use five U.S. Patents covering an alpha-galactosidase. The license was obtained to ensure a viable source of alpha-galactosidase to use the Z-Process. Under this license agreement, we are required to pay royalty in an amount equal to a percentage in the low single digits of net sales of licensed products. In addition, we are required to pay an annual minimum royalty payment of $35,000. All payments under this agreement will be terminated if the aggregate amount paid under the agreement (including both sale royalties and minimum royalty payments) equals to a specified amount.
Manufacturing
Z-Lig raw materials are sourced from suppliers that meet ABI’s strict quality and performance requirements and are compliant with ISO 22442 standards. Through simple, predictable ramp-up of the breeding and processing of the pigs and supply chain redundancies, we believe we can readily meet all of the expected market demand. Accordingly, we have not entered into any formal supply agreements with our suppliers to provide a guaranteed volume of raw materials. In addition, by utilizing varying ages and strains of pig, we established a flexible manufacturing process that permits us to adjust the size of our Z-Lig products to accommodate the demand of customers.
Government Regulation
Our products are medical devices that are subject to extensive regulation by government authorities in the European Union (the “EU”), the United States and in other countries and jurisdictions, including South Africa, and non-EU European countries. These governmental authorities regulate the marketing and distribution of medical devices in their respective geographies. The regulations cover the entire life cycle of the product, including the research, development, testing, manufacture, quality control, packaging, storage, labeling, advertising and
promotion of the devices. In addition, post-approval monitoring and reporting, as well as import and export of medical devices, are subject to regulatory requirements.
Review and approval of medical devices in the EU
The EU has a coordinated system for the authorization of medical devices. The EU Medical Devices Directive (Council Directive 93/42/EEC, as amended) sets out the basic regulatory framework for medical devices in the European Union. In the EU our medical devices must comply with the Essential Requirements in Annex I to the EU Medical Devices Directive, which we refer to as the Essential Requirements. Compliance with these requirements is a prerequisite to be able to affix the Certificate of Conformity mark, or CE Mark, to our medical devices, without which they cannot be marketed or sold in the European Economic Area, or EEA. To demonstrate compliance with the Essential Requirements we must undergo a conformity assessment procedure, which varies according to the type of medical device and its classification. Except for low risk medical devices (Class I with no measuring function and which are not sterile), where the manufacturer can issue a CE Declaration of Conformity based on a self-assessment of the conformity of its products with the Essential Requirements, a conformity assessment procedure requires the intervention of a third-party organization designated by competent authorities of an EU country to conduct conformity assessments, which is referred to as a Notified Body. The Notified Body would typically audit and examine products' technical file and the quality system for the manufacture, design and final inspection of the devices before issuing a CE Certificate of Conformity demonstrating compliance with the relevant Essential Requirements. To date, we have used the CE Marking process to satisfy the conformity standards required to market and sell our Z-Lig products in the EU.
With respect to implantable devices or devices classified as Class III in the EU, the manufacturer must conduct clinical studies to obtain the required clinical data, unless reliance on existing clinical data from similar devices can be justified. As part of the conformity assessment process, depending on the type of devices, the Notified Body will review the manufacturer's clinical evaluation process, assess the clinical evaluation data of a representative sample of the devices' subcategory or generic group, or assess all the clinical evaluation data, verify the manufacturer's assessment of that data and assess the validity of the clinical evaluation report and the conclusions drawn by the manufacturer. To date, we have conducted clinical studies in Europe and South Africa to obtain clinical data as part of the clinical evaluation process.
Even after we receive a CE Certificate of Conformity enabling us to affix the CE Mark on a product and to sell our product in the EEA countries, a Notified Body or a competent authority may require post-marketing studies of our product. Failure to comply with such requirements in a timely manner could result in the withdrawal of our CE Certificate of Conformity and the recall or withdrawal of our product from the market in the EU, which would prevent us from generating revenue from sales of that product in the EEA. Moreover, each CE Certificate of Conformity is valid for a maximum of five years, but more commonly three years. Our current CE Certificates of Conformity are valid through 2019 for our Z-Lig products. At the end of each period of validity we are required to apply to the Notified Body for a renewal of the CE Certificate of Conformity. There may be delays in the renewal of the CE Certificate of Conformity or the Notified Body may require modifications to our products or to the related Technical Files before it agrees to issue the new CE Certificate of Conformity.
In addition, we must inform the Notified Body that carried out the conformity assessment of the medical devices we market or sell in the EEA of any planned substantial changes to our devices
that could affect compliance with the Essential Requirements or the devices' intended purpose. The Notified Body will then assess the changes and verify whether they affect the products' conformity with the Essential Requirements or the conditions for the use of the devices. If the assessment is favorable, the Notified Body will issue a new CE Certificate of Conformity or an addendum to the existing CE Certificate of Conformity attesting compliance with the Essential Requirements. If it is not, we may not be able to continue to market and sell the product in the EEA.
On September 26, 2012, the European Commission adopted a package of legislative proposals designed to replace the existing regulatory framework for medical devices in the EU. These proposals provide for a revision of the current regulatory framework for medical devices in the EU to strengthen patient safety, transparency and product traceability. The proposals, for instance, include reinforced rules governing clinical evaluation throughout the life of the device, improved traceability of devices in the supply chain, including a phased and risk-based introduction of unique device identification, or UDI, improved market surveillance and vigilance, as well as better co-ordination between national regulators, increased powers for Notified Bodies to undertake unannounced inspections and strengthened supervision of Notified Bodies by member states. The European Commission's proposals may undergo significant amendments as they are reviewed by the European Council and European Parliament as part of the EU legislative process. If and when adopted, the proposed new legislation may prevent or delay the EU approval or clearance of our products under development or may impact our ability to modify our currently EU approved or cleared products on a timely basis and impose additional costs relating to clinical evaluation, vigilance and product traceability.
Marketing and sales considerations in the EU
In the EU, medical devices may be promoted only for the intended purpose for which the devices have been CE Marked. Failure to comply with this requirement could lead to the imposition of penalties by the competent authorities of the EU Member States. The penalties could include warnings, orders to discontinue the promotion of the medical device, seizure of the promotional materials and fines. Promotional materials must also comply with various laws and codes of conduct developed by medical device industry bodies in the EU governing promotional claims, comparative advertising, advertising of medical devices reimbursed by the national health insurance systems and advertising to the general public.
Post-approval monitoring in the EU
Additionally, all manufacturers placing medical devices into the market in the EU are legally bound to report any serious or potentially serious incidents involving devices they produce or sell to the competent authority in whose jurisdiction the incident occurred. In the EU, manufacturers must comply with the EU Medical Device Vigilance System. Under this system, incidents must be reported to the relevant authorities of the EU countries, and manufacturers are required to take field safety corrective actions to reduce a risk of death or serious deterioration in the state of health associated with the use of a medical device that is already placed on the market.
Third-party reimbursement
In the United States and most other major joint implant markets, third-party payors, including government health programs, commercial health insurers and managed care organizations, reimburse hospitals and other medical facilities an aggregate amount for all elements of a joint
replacement procedure, including operating room time, patient care and the joint replacement product. As a result, our products generally are not reimbursed separately, but instead are subject to the limits imposed by third-party payors on the coverage and reimbursement of procedures that utilize our products.
Sales of our products will depend, in part, on the extent to which the costs of such procedures involving the use of our products cleared by the EU and other jurisdictions will be covered by third-party payors, including government health programs in the United States, such as Medicare and Medicaid, commercial health insurers and managed care organizations. The process for determining whether a payor will provide coverage for a particular procedure may be separate from the process for setting the price or reimbursement rate that the payor will pay for the procedure once coverage is approved. Third party payors may limit coverage to particular procedures on an approved list, or formulary, which might not include all of the approved procedures involving the use of our products for a particular indication.
In the EU, pricing and reimbursement schemes vary widely from country to country. In many foreign markets, pricing of medical devices is subject to governmental control. In the United States, there have been, and we expect that there will continue to be, a number of federal and state proposals to limit payments by governmental payors for medical devices, and the procedures in which medical devices are used. While we cannot predict whether such legislative or regulatory proposals will be adopted, the adoption of such proposals could have a material adverse effect on our business, financial condition and profitability.
Review, approval and clearance of medical devices in the United States
Medical devices in the United States are strictly regulated by the FDA. Unless an exemption applies, a medical device may not be marketed in the United States unless it has been cleared through filing of a 510(k) premarket notification, or 510(k), or approved by the FDA pursuant to a premarket approval application, or PMA. The information that must be submitted to the FDA in order to obtain clearance or approval to market a new medical device varies depending on how the medical device is classified by the FDA. Medical devices are classified into one of three classes depending on the level of control necessary to assure the safety and effectiveness of the device. Class I devices have the lowest level or risk associated with them, and are subject to general controls, including labeling, premarket notification and adherence to the Quality System Regulations (“QSRs”). Class II devices are subject to general controls and special controls, including performance standards. Class III devices, which have the highest level of risk associated with them, are subject to most of the aforementioned requirements as well as to premarket approval. Most Class I devices and some Class II devices are exempt from the 510(k) requirement, although manufacturers of these devices are still subject to registration, listing, labeling and QSR requirements.
The PMA process is more complex, costly and time consuming than the 510(k) clearance procedure. A PMA must be supported by extensive data, including technical, preclinical, clinical, manufacturing, control and labeling information, that demonstrate the safety and effectiveness of the device for its intended use. After a PMA is submitted, the FDA has 45 days to determine whether it is sufficiently complete to permit a substantive review. If the PMA is complete, the FDA will file the PMA. The FDA is subject to performance goal review times for PMAs and may issue a decision letter as a first action on a PMA.
In evaluating a 510(k), the FDA will determine whether the device has the same intended use as the predicate device, and (a) has the same technological characteristics as the predicate
device, or (b) has different technological characteristics, and (1) the data supporting substantial equivalence contains information, including appropriate clinical or scientific data, if deemed necessary by the FDA, that demonstrates that the device is as safe and as effective as a legally marketed device, and (2) does not raise different questions of safety and effectiveness than the predicate device. Most 510(k)s do not require clinical data for clearance, but the FDA may request such data. The FDA seeks to review and act on a 510(k) within 90 days of submission, but it may take longer if the agency finds that it requires more information to review the 510(k).
Our Z-Lig products are classified as Class III medical devices. We intend to obtain regulatory approval of our Z-Lig products by filing a PMA with the FDA. The PMA process for the Z-Lig may be subject to additional clinical evaluation under an Investigational Device Exemption (IDE) to collect more human clinical data prior to filing a PMA. We intend to rely on the 510(k) premarket notification process to obtain FDA clearance for our product candidates which may pursue this regulatory pathway.
Investigational device exemption
A clinical trial to support safety and effectiveness of the device is typically required for a PMA and, in a small percentage of cases, the FDA may require a clinical study in support of a 510(k) submission. A manufacturer that wishes to conduct a clinical study involving the device is subject to the FDA's Investigational Device Exemption, or IDE, regulation. The IDE regulation distinguishes between significant and nonsignificant risk device studies and the procedures for obtaining approval to begin the study differ accordingly. Also, some types of studies are exempt from the IDE regulations. A significant risk device presents a potential for serious risk to the health, safety, or welfare of a subject. Significant risk devices are devices that are substantially important in diagnosing, curing, mitigating or treating disease or in preventing impairment to human health. Studies of devices that pose a significant risk require both FDA approval and an approval of an independent institutional review board, or IRB, prior to initiation of a clinical study. Nonsignificant risk devices are devices that do not pose a significant risk to the human subjects. A nonsignificant risk device study requires only IRB approval prior to initiation of a clinical study.
An IDE application is considered approved 30 days after it has been received by the FDA, unless the FDA otherwise informs the sponsor prior to 30 calendar days from the date of receipt, that the IDE is granted (“approved”), granted with conditions, or disapproved. The clinical trial must be conducted in accordance with applicable regulations, including but not limited to the FDA's IDE regulations and current good clinical practices. A clinical trial may be suspended by the FDA, the IRB or the sponsor at any time for various reasons, including a belief that the risks to the study participants outweigh the benefits of participation in the trial.
Post-marketing restrictions and enforcement
After a device is placed on the market, numerous regulatory requirements apply. These include: compliance with the QSR and other compliance requirements; labeling regulations, which prohibit the promotion of products for uncleared or unapproved or "off-label" uses and impose other restrictions on labeling; and medical device reporting obligations, which require that manufacturers investigate and report to the FDA adverse events, including deaths, or serious injuries that may have been or were caused by a medical device and malfunctions in the device that would likely cause or contribute to a death or serious injury if it were to recur.
Facilities
We lease a 4,000 square foot facility in San Antonio, Texas, under a lease agreement that was recently renewed and will expire on December 31, 2018, at our option. The facility has a 1,000-square foot Class 8 clean room area which contains four 100-square foot modular clean rooms of Class 6 to Class 7 certification. We believe that we have adequate capacity to support Z-Lig and other production requirements for clinical trials through commercial launch in Europe and the U.S. The remainder of ABI’s facility is used for product development, distribution and general and administrative purposes
Legal Proceedings
We may from time to time be involved in various claims and legal proceedings of a nature we believe are normal and incidental to a medical device business. These matters may include product liability, intellectual property, employment, and other general claims. We accrue for contingent liabilities when it is probable that a liability has been incurred and the amount can be reasonably estimated. We are not presently a party to any legal proceedings that, in the opinion of our management, are likely to have a material adverse effect on our business. Regardless of outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Management’s Discussion and Analysis of
Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of our operations together with our financial statements and related notes appearing at the end of this Offering Circular. This discussion contains forward-looking statements reflecting our current expectations that involve risks and uncertainties. Actual results and the timing of events may differ materially from those contained in these forward-looking statements due to a number of factors, including those discussed in the section entitled "Risk Factors" and elsewhere in this Offering Circular.
Aperion Biologics, Inc. (“we” or the “Company”) is a commercial-stage medical device company addressing the significant unmet need for an alternative to human-based sources of tissues to be used in surgical procedures. Our core technology platform is an enzymatic stripping of the key carbohydrate antigens followed by a unique conversion process that both “humanizes” and sterilizes animal tissues without unduly affecting their biomechanical or biological properties. The Company’s lead product, the Z-Lig, is a patent-protected porcine tendon currently in limited commercial release for use initially as an anterior cruciate ligament (ACL) replacement in revision and multiligament knee joint procedures. Z-Lig is the only known biological alternative to human tissue for ACL replacement. Since the Z-Lig is a biological implant that maintains a scaffold, it can become populated and remodeled with the patient’s own cells (similar to human-sourced tissue), referred to in the industry as “ligamentization.” This ligamentization is one of the keys to Z-Lig’s proven ability to be highly functional in patients for many years after implantation. Our propriety process is protected by an extensive portfolio of patents and patent applications, including 23 issued patents in the U.S. and internationally.
The Company’s primary activities since its inception have been the development and implementation of its business plans, performing research on its platform technology, conducting clinical trials on its products in both animals and humans, pursuing regulatory approvals to market its product in U.S. and international markets, and raising capital. In April 2014 the Company received the CE Mark to allow commercial sales of the Z-Lig in foreign markets that accept the CE Mark (primarily Europe). The first commercial sale took place in February 2015.
The Company was founded in October 1996 under the name CrossCart, Inc. The Company changed its name to Aperion Biologics, Inc. (a Delaware corporation) in May 2009. To date, the Company has funded its business primarily with the proceeds from private placements of equity and debt securities.
Results of Operations
The Nine Months ended June 30, 2015 as compared to June 30, 2014
As mentioned above, the Company commenced its first commercial sale of Z-Lig in February 2015 and recognized $27,000 of sales during the nine months ended June 30, 2015. There were no sales during the nine months ended June 30, 2014.
Costs related to clinical studies decreased by $38,833 from $165,896 for the nine months ended June 30, 2014 to $127,063 for the nine months ended June 30, 2015. The decrease was primarily driven by lower expenses associated with the Company’s clinical trials outside of the United States.
Research and development expenses decreased by $16,429 from $56,345 for the nine months ended June 30, 2014 to $39,916 for the nine months ended June 30, 2015, primarily due to completion of an R&D consulting contract which reduced expenses $15,174 between the periods.
Sales, general and administrative expenses increased by $19,421 from $1,099,724 for the nine months ended June 30, 2014 to $1,119,146 for the nine months ended June 30, 2015. Sales and marketing activities related to the launch of the Z-Lig product increased $103,451. In addition, consulting activities related to fundraising resulted in an increase of $100,734 for the period. Offsetting these increases were a decrease of $55,262 in salary and personnel costs in operations, a decrease of $48,567 in legal fees and a decrease of $84,469 and in regulatory expenses due to the completion of the CE Mark application process.
Other expenses increased for the period primarily as a result of higher interest expense related to the Company’s borrowings to support its activities, as interest expense increased $91,564 between the periods. This increase was driven by the increase in the total amount of average interest bearing debt from $6,400,014 for the nine months ended June 30, 2014 to $7,956,327 for the nine months ended June 30, 2015.
Fiscal Year ended September 30, 2014 as compared to September 30, 2013
Operating expenses decreased by $1,537,299 from $3,389,580 in fiscal year 2013 to $1,852,281 in fiscal year 2014. The decrease was primarily due to the completion of the Company’s clinical trials outside of the U.S. and related follow-up examinations, data gathering and compilation activities and reduced salary and personnel expenses.
Research and development expenses decreased by $358,599 from $417,844 in fiscal year 2013 to $59,245 in fiscal year 2014, primarily due to a reduction of salary and personnel expenses and reduced use of consultants.
Sales, general and administrative expenses decreased by $604,906 from $2,101,434 in fiscal year 2013 to $1,496,528 in fiscal year 2014. The decrease was primarily attributed to a $226,098 reduction in spending in our Quality Assurance department related to the completion of significant activities relating to our CE Mark application and the adverse event investigation in our clinical trials, a $133,404 decrease in salary and personnel expenses in the Sales department, and a $178,832 decrease in salary and personnel expenses in our Operations Department as a result of the completion of process validation production, which was offset by an increase of $57,411 in fees related to fundraising activities and an increase of $62,499 in fees to Board members due to increasing the size of the Board of Directors.
Other expenses increased for the period primarily as a result of higher interest expense related to the Company’s borrowings to support its activities, as interest expense increased $234,931 between the periods. This increase was driven by the increase in the total amount of average interest bearing debt from $4,537,000 for fiscal year 2013 to $6,600,014 for fiscal year 2014.
Liquidity and Capital Resources
We had cash of $124,769 at September 30, 2014 and $258,564 at June 30, 2015, sourced primarily from the proceeds received in private placements of convertible promissory notes.
As of September 30, 2014, the Company had working capital and a stockholders’ deficiency of $25,345,307 and $25,990,881,respectively. During the year ended September 30, 2014, the Company incurred a net loss attributable to common stockholders of $3,291,838. The Company has not generated any material revenues and has incurred net losses since inception. Our recurring operating losses and our need for additional sources of capital to fund our ongoing operations raise substantial doubt about our ability to continue as a going concern. As a result, our independent registered public accounting firm included an explanatory paragraph in its report on our financial statements as of and for the year ended September 30, 2014 with respect to this uncertainty.
Since August 2013, the Company has relied primarily on the issuance of convertible promissory notes to fund its continuing operations. Most of this funding has been provided by CrossCart, LLC, which is the Company’s largest shareholder and controlled by Dr. Kevin R. Stone, who is the Chairman of the Board of Directors of the Company. As of June 30, 2015 the Company had borrowed $6,825,615 from CrossCart, LLC and other related parties. As of June 30, 2015, we had a total of $8,156,027 in outstanding principal amount of convertible promissory notes, the majority of which have matured and are due and payable, and we have not been able to make payments due to the lack of funding. However, we expect to enter into an agreement with each noteholder, including CrossCart LLC, to (i) waive any default resulting from such non-payment of notes and (ii) provide that all outstanding amount due under the convertible promissory notes will be converted into shares of our common stock immediately following the completion of the offering at the initial conversion price.
We believe that the net proceeds from this offering of approximately $ will be sufficient to fund our current operations at least through the end of calendar year , and thereafter we may need additional capital to continue our operations. We do not expect to be able to satisfy our cash requirements solely through sales of the Z-Lig in the near future, therefore we expect to rely on equity and debt financing to fund our operations. The sale of additional equity securities could result in additional dilution to our stockholders and those securities may have rights senior to those of our common stock. The incurrence of indebtedness would result in increased debt service obligations and could result in operating and financing covenants that would restrict our operations.
We cannot assure that we will have sufficient capital to finance our growth and business operations or that such capital will be available on terms that are favorable to us or at all. We are currently incurring operating deficits that are expected to continue for the foreseeable future, and as we begin to prosecute our marketing plan, we expect our operating deficit will continue to grow initially until we begin to generate a sufficient level of revenue from our commercialization efforts.
Plan of Operations
Since the receipt of the CE Mark in April 2014, the Company has developed a detailed marketing plan that will initially focus on distribution efforts of its Z-Lig products in 6 to 10 markets which have accepted options for ACL reconstruction (allograft and synthetic), have reimbursement pathways for graft alternatives, and/or are countries where we have conducted our clinical trials. We intend to use an indirect distribution model and focus on peer-to-peer marketing activities as a rapid and effective means of establishing credibility and market acceptance of our new technology. We intend to increase our headcounts by hiring additional sales and marketing employees, including a Vice President of EMEA Distribution Management responsible for establishing our distribution network, initiating and coordinating our distribution
activities within the distribution network, and driving distribution objectives. We also intend to hire Clinical Marketing Specialist(s) who will be focused on both surgeon and sales representative education on the science and technology surrounding the Z-Process, Z-Lig product, and our Company. In later years the Company intend to expand into other countries to broaden the reach of Z-Lig.
The Company recognizes that following the completion of this offering, it will need to raise additional capital in order to meet its obligations and execute its business plan within the next two years. If the Company is unable to raise sufficient additional funds through this offering, it will be required to develop and implement an alternative plan to further extend payables, reduce overhead or scale back its business plan until sufficient additional capital is raised to support further operations. There can be no assurance that such a plan will be successful.
Directors, Executive Officers and Significant Employees
The following table sets forth information regarding our executive officers, directors and significant employees, including their ages as of June 30, 2015:
| Name and Principal Position | Age | Term of Office | Approximate hours per week for part-time employees |
| Executive Officers | | | |
| Daniel Lee, Chief Executive Officer and Director | 54 | Since August 2008 | _ |
| David Cocke, Chief Financial Officer | 50 | Since September 2008 | _ |
| | | | |
| Directors | | | |
| David W. Anderson | 62 | Since February 2012 | _ |
| France Dixon Helfer | 62 | Since March 2014 | _ |
| Alfred G. Holcomb | 63 | Since 2008 | _ |
| Kevin R. Stone, M.D. | 60 | Since our inception | _ |
| Mike Ward | 44 | Since March 2014 | _ |
| Significant Employee | | | |
| Lance Johnson, Vice President Quality | 49 | Since November 2010 | -- |
| | | | |
Executive Officers
Daniel R. Lee Mr. Lee has been serving as our Chief Executive Officer since August 2008. Prior to joining us, from July 2006 to August 2008, Mr. Lee was Director of Marketing at Smith & Nephew Endoscopy, a sports medicine technology company, responsible for its TRUREPAIR business unit. From November 2003 to July 2006, Mr. Lee was Director of Marketing at OsteoBiologics, Inc., a medical supply company providing off-the-shelf bio-absorbable implant for articular cartilage, which was acquired by Smith & Nephew Endoscopy in 2006. Prior to joining OsteoBiologics, Inc., Mr. Lee was the Director of Marketing for Regeneration Technologies, Inc (“RTI”), a leading allograft tissue processor for orthopaedic surgical applications, from 1999 to 2003. Prior to joining RTI, he was the Director of the Sports Medicine Research and Development group at Surgical Dynamics, a subsidiary of U.S. Surgical Corporation. He is a member of the Society for Biomaterials and the American Association of
Tissue Banks where he holds a Certified Tissue Bank Specialist certification. He currently holds thirteen (13) patents on implants and instruments used in orthopaedic and general surgery. Mr. Lee holds an M.S. in Biomedical Engineering from the University of Alabama at Birmingham and a B.A. in Materials Science and Engineering from the Johns Hopkins University.
David Cocke Mr. Cocke has been serving as our Chief Financial Officer since September 2008. Since 1997, Mr. Cocke has been serving as a General Manager of NuPak Medical, Ltd., an ISO 13485-certified contract manufacturing medical device company. He was responsible for all aspects of management, including sales, finance and operations. Prior to that, from November 1993 to May 1996, Mr. Cocke was Chief Financial Officer of NuTech, Inc., a technology incubator subsidiary for KCI, a leader in tissue-based products for surgical procedures and wound healing. From 1991 to 1993, he was Director at the Corporate Development department at KCI. Prior to KCI, David was employed by GE Capital in its Corporate Finance Group and Salomon Brothers Inc in its Investment Banking Group. Mr. Cocke holds an MBA from the University of Virginia’s Darden Graduate School of Business Administration and a BBA with High Honors from the University of Texas at Austin.
Directors
David W. Anderson. Mr. Anderson has been our director since February 2012. Since September 2014, Mr. Anderson has been serving as the Chief Executive Officer of Orteq Ltd., an orthopedic medical device and regenerative medicine company focused on sports medicine. He also serves as Chairman of the Board of Gentis, Inc. a clinical stage spinal implant company which he joined in 2004. From 1999 to 2004, he was the founder and a director of Replication Medical, Inc., an orthopedic medical device company focused on spine surgeries. From 1994 to 1999, he served as the Chief Executive Officer of Bionx Implants, Inc., a leading manufacturer and marketer of reinforced polymer implants. From 1986 to 1989, he was the founder and Executive Vice President of Osteotech, Inc., a global leader in providing biologic solutions for regenerative medicine, which was acquired by Medtronic Inc. in 2010. Mr. Anderson also served as the Chief Executive Officer of Kensey Nash Corporation, a medical device company, from 1992 to 1994, and Sterilox Technologies, a manufacturer of non-toxic disinfectants, from 2000 to 2004. Mr. Anderson holds a B.S. in Chemical Engineering from Cornell University. Mr. Anderson brings to the Board over 25 years of experiences in entrepreneurship, strategic transactions and financing in the medical device industry.
France Dixon Helfer. Ms. Helfer has been our director since February 2014. Ms. Helfer is currently the President and Chief Executive Officer of TinyKicks LLC, a University of California healthcare technology company based in Irvine, California, since January 2015. From September 2011 to September 2014, Ms. Helfer was the President, Chief Executive Officer and Director of Halo Healthcare Inc., a medical device and diagnostics company focused on the detection and screening of breast cancer. Prior to that, from August 2004 to January 2008, Ms. Helfer was the President and Founder of Pegasus Biologics, Inc., which was acquired by Baxter Healthcare Inc. Prior to that, Ms. Helfer held a number of executive positions at various biotechnology companies,including Medtronic, Xenotech Labs, SORIN Bimedica, Eclipse Surgical and MMDDataDirect. Ms. Helfer serves on the boards of various business organizations and academic institutions, including Chapman University’s Crean College of Health and Behavioral Sciences. Ms. Helfer holds a B.A. in Biologic Sciences from California State University, Fullerton. Ms. Helfer brings to the Board over 20 years of experience as a senior level executive in the medical device industry.
Alfred G. Holcomb. Mr. Holcomb has been our director since 2008. Since 2004, Mr. Holcomb has been serving as the Vice President of Acquisition and Divestutures at Lewis Energy Corp., an oil drilling company,where he led the completion of numerous strategic transactions. From 1978 to 2004, Mr. Holcomb was a partner at the law firm of Schoenbum, Curphy & Scanlan. Mr. Holcomb has been an active speaker on the Eagle Ford in conferences around the world. Mr. Holcomb holds his B.A. in Finance in from University of Texas, a J.D. from St. Mary's University School of Law and a LL.M. in Taxation from New York University. Mr. Holcomb brings to the Board extensive experience and knowledge on strategic, legal and financial matters.
Kevin R. Stone, M.D. Dr. Stone is the founding scientist of our company and served as our Chief Executive Officer from 1996 to 2008, as a director since our inception and as our Chairman of the Board since 2014. Dr. Stone is a renowned orthopaedic surgeon and founded the Stone Clinic in 1988, an orthopedic clinic focused on biologic approaches to treating joint injuries. He is also the Founder and Chairman of Stone Research Foundation, an independent research institution investigating new techniques for joint health, arthritis and human performance, since 1996. Dr. Stone is the controlling member and manager of CrossCart LLC, our controlling stockholder. Dr. Stone is also an attending staff at the California Pacific Medical Center and the San Francisco Surgery Center. From 1999 to 2003, Dr. Stone served as the Chief Executive Officer and Chairman of the Board of Joint Juice, Inc. (now Post Holdings, Inc.). Dr. Stone is also the founder and Chief Executive Officer of Rescue Reel, LLC since 2006 and the founder of ProprioSense Holdings, LLC since 2012. Dr. Stone holds an A.B. cum laude in biology from Harvard College and an M.D. from the University of North Carolina School of Medicine, residency training in general surgery at Stanford University and orthopaedics at Harvard and Fellowship trained in knee surgery and sports medicine. Dr. Stone is recognized internationally as a leading expert and authority of advanced orthopedic surgical and rehabilitation techniques to repair damaged cartilage and ligaments. He brings to the Board extensive expertise, knowledge and experience in the field of orthobiologic medicine and provides us with significant management, operational and financial support.
Mike Ward. Mr. Ward has been our director since March 2014. Mr. Ward has been serving as the Vice President, Corporate Development at BioNano Genomics, Inc., a genomic mapping company, since March 2014. Prior to joining BioNano Genomics, Inc., from 2009 to 2013, Mr. Ward was a Vice President at Lurie Investment Fund, LLC, the venture capital arm of Ann and Robert H. Lurie Foundation, where he was responsible for managing the life sciences venture capital and private equity investments of the foundation. Prior to 2009, Mr. Ward worked for more than 15 years in the investment banking industry, holding various positions at Leerink Partners, Credit Suisse, Dresdner Kleinwort Wasserstein, BMO and Vector Securities. Mr. Ward also serves on the boards of the following healthcare companies: Nanosphere, Inc., a publicly traded molecular diagnostics company and CytoPherx, Inc., an acute renal failure-focused therapeutic medical device company. Mr. Ward holds a B.S. in finance from the University of Illinois. Mr. Ward brings to the Board over 20 years of life-sciences dedicated investment banking, venture capital and private equity experiences.
Significant Employee
Lance Johnson. Mr. Johnson has been serving as our Vice President, Quality since November 2010. Prior to joining us, he was the Quality Manager at Zimmer Spine, the orthopaedic spine division of Zimmer, Inc., from March 2009 to October 2010. From March 2005 to November 2009, Mr. Johnson was the Associate Quality Engineering Manager for Abbott Spine, an orthopaedic spine company, working specifically with new product development teams. In addition, he served as an investigator with the FDA from 1989 to 2005. From 1994 to 2005, he
was the resident investigator in charge of the Austin field office and as contributor to the FDA international cadre. During his time at the FDA, he audited medical device firms in the US, Europe and Canada. Mr. Johnson received his Medical Device Level II certification in 1998 and received his Level II Auditor certification in 2003. Lance trained and certified FDA, Texas, and BSI auditors during his career with the FDA. Mr. Johnson holds a B.S. in Biotechnology from Oklahoma State University.
Compensation of Directors and Executive Officers
The following table represents information regarding the total compensation earned by the three highest paid individuals who served as an executive officer or director of the Company as of June 30, 2015:
| Name and Principal Position | Cash Compensation ($) | Other Compensation ($)(2) | Total Compensation ($) |
| Daniel R. Lee, Chief Executive Officer | $235,000 | $23,349 | $258,349 |
David Cocke, Chief Financial Officer(1) | $60,000 | $4,239 | $64,239 |
| Kevin R. Stone, Chairman of the Board | $45,833 | $0 | $45,833 |
(1) Mr. Cocke is performing his services as Chief Financial Officer pursuant to the Independent Consulting Agreement, dated September 1, 2008.
(2) Represents stock-based compensation of stock options issued to the Chief Executive Officer and Chief Financial Officer.
The aggregate cash compensation of all of our directors for the fiscal year ended September 30, 2014 was $142,499. In addition, we have granted, in the aggregate, stock options to purchase to a total of 330,000 shares of our common stock to our directors (excluding Mr. Lee), during the fiscal year ended September 30, 2014.
As part of our effort to preserve cash, since July 2013, we have reduced, in full or partially, the salaries of our Chief Executive Officer, Chief Financial Officer and other full time employees until the Company has received financing of at least $2.0 million (beyond any funding received from CrossCart LLC) or the signing of a license or distribution agreement with at least $2.0 million of upfront payments or milestone fees achievable within 12 months of signing. Following the completion of this offering, we expect to pay the amount of such salaries to our Chief Executive Officer, Chief Financial Officer and other employees (the “Reduced Salary”). As of June 30, 2015, the total amount of the Reduced Salary for the Chief Executive Officer and Chief Financial Officer was $210,604. Since July 2013, we have reduced a total of $289,999 in cash compensation to our Board members as of June 30, 2015. We expect to enter an agreement with the directors pursuant to which we will agree to pay 70% of such compensation by issuing an aggregate of 751,849 shares of our common stock to these directors, and that the remaining 30% of cash compensation owed to such directors will be paid in cash.
On December 16, 2013, our Board approved the Management Retention Plan, pursuant to which our management members, including the Chief Executive Officer and Chief Financial Officer, will receive a retention bonus upon an external financing of at least $2.0 million (beyond any funding received from CrossCart LLC) or the signing of a license or distribution agreement with at least $2.0 million of upfront payments or milestone fees achievable within 12 months of signing. As of June 30, 2015, and assuming the completion of this offering, the total amount of the management retention bonus for all eligible management members will be approximately
$85,000, and we intend to pay this amount by issuing shares of common stock having the same value based on the public offering price.
Security Ownership of Management and Certain Securityholders
The following table sets forth certain information known to us regarding beneficial ownership of our capital stock as of June 30, 2015 for (i) all executive officers and directors as a group and (ii) each person, or group of affiliated persons, known by us to be the beneficial owner of more than ten percent (10%) of our capital stock. The percentage of beneficial ownership in the table below is based on 72,963,279 shares of common stock deemed to be outstanding as of June 30, 2015 and gives effect to the conversion of all outstanding shares of our convertible preferred stock into an aggregate of 37,504,969 shares of our common stock following the completion of this offering, including shares of common stock to be issued as cumulative dividends accrued under such preferred stock and (ii) the issuance of 31,366,725 shares of common stock upon conversion of approximately an aggregate of $9,838,058 in outstanding principal and accrued interest on our convertible promissory notes (which is currently convertible into shares of our preferred stock but will be amended to be convertible into shares of our common stock in connection with the offering), upon the completion of this offering, assuming that the offering is completed on . In addition, shares of common stock that may be acquired by the stockholder within 60 days of June 30, 2015, pursuant to the exercise of stock options are deemed to be outstanding for the purpose of computing the percentage ownership of such shareholder, but are not deemed to be outstanding for the purpose of computing the percentage ownership of any other person shown in the table.
| Name and Address of Beneficial Owner | Number of Shares
Beneficially Owned | Number of Shares Acquirable | Percent of Class |
| | | | |
| CrossCart LLC (1) | 57,304,590 | 0(3) | 78.5% |
| Dr. Kevin R. Stone (1) (2) | 60,777,692 | 100,000(4) | 83.2% |
| All executive officers and directors as a group | 61,532,732 | 3,602,833(5) | 85.1% |
____________________________________________________________________________
| (1) | Dr. Kevin R. Stone, a director of the Company, is the controlling member and manager of CrossCart LLC. As such, Dr. Stone may be deemed to have voting and dispositive power of all securities beneficially owned by CrossCart LLC reported herein. Dr. Stone disclaims beneficial ownership of all shares held by CrossCart LLC. The mailing address of CrossCart LLC and Dr. Stone is 3727 Buchanan Street, San Francisco, CA 94123. |
| (2) | All shares of common stock, warrants and convertible notes reported herein are held directly by the Stone Family Revocable Trust dated 9/20/90 (the “Trust”) for which Dr. Stone and Susan Stone serve as co-trustees. As the co-trustee of the Trust, Dr. Stone is deemed to have beneficial ownership of all securities held by the Trust. |
| (3) | Excludes 5,327,224 shares of common stock issuable upon exercises of warrants to purchase shares of our common stock and convertible preferred stock and the subsequent conversion of such shares of convertible preferred stock to shares of common stock. |
| (4) | Includes 100,000 shares issuable upon exercise of stock options within 60 days of June 30, 2015 and excludes 2,388,238 shares of common stock issuable upon exercises of warrants to purchase shares |
of our common stock and convertible preferred stock and the subsequent conversion of such shares of convertible preferred stock into shares of common stock.
(5) Includes an aggregate of 3,602,833 shares of common stock issuable upon exercise of stock options within 60 days of June 30, 2015.
Interest of Management and Others in Certain Transactions
Transactions with CrossCart and Dr. Kevin Stone
Convertible Note Financing
Dr. Kevin Stone, a director of the Company, is a controlling member and the manager of CrossCart LLC, a California limited liability company (“CrossCart”). Between August 19, 2013 and May 23, 2014, CrossCart loaned an aggregate of $1,520,015 to the Company in exchange for the issuance of eight (8) separate and identical convertible promissory notes in the principal amount of $200,000, $200,000, $150,000, $250,000, $250,000, $150,000, $150,000,$150,000, and $20,015 respectively (the “CrossCart Series D Notes”). Each CrossCart Series D Note bears an interest of 8% per annum, which rate increases to 10% per annum upon maturity or if an event of default occurs. In general, all principal and interest accrued under each CrossCart Series D Note is due and payable, upon demand at the sole option of CrossCart, following each such note’s maturity dates, all of which have occurred with the exception of the CrossCart Series D Note with a principal amount of $20,015 which matures on May 23, 2016. The CrossCart Series D Notes contain customary events of default. The principal and any accrued interest under the CrossCart Series D Notes are convertible, at CrossCart’s discretion, into shares of Series D Preferred Stock of the Company at an initial conversion price of $0.25 per share. Under the terms of the CrossCart Series D Notes, the Company is required to use reasonable efforts to cause a sufficient number of shares of Series D Preferred Stock to be authorized and reserved for the conversion of such notes. As of June 30, 2015 the total outstanding amount due under the CrossCart Series D Notes was $3,260,851, including accrued interest. CrossCart has informed us that it does not intend to demand repayment of the CrossCart Series D Notes prior to the completion of this offering. In addition, we expect to enter into an agreement with CrossCart to provide that upon completion of this offering, all outstanding amounts due under the 2013 CrossCart Series D Notes will be automatically converted into shares of our common stock at the initial conversion price of $0.25 per share.
Demand Notes
On December 18, 2014, the Company issued a demand note to CrossCart for an aggregate principal amount of $120,000 (the “Demand Note”). The outstanding principal of each Demand Note carries an interest rate of 8% per annum, which may be increased to 10% if an event of default occurs. All principal and accrued interest under the Demand Note is due and payable, upon demand, at the sole option and discretion of CrossCart. The Demand Note includes customary events of default. The outstanding balance on the Demand Note as of June 30, 2015 was $125,093, including accrued interest.
In addition, as inducement to enter into the Demand Note, the Company granted CrossCart certain royalty rights (the “Royalty Right”), pursuant to which the Company agrees to pay a royalty equal to 5% of any net sales from products manufactured using the Company’s Z-Process on the first $12,000,000 of net sales. The Royalty Right will expire on December 31,
2016 or when $12,000,000 in net sales has been achieved. The Royalty Right was further amended in connection with the execution of the Line of Credit Note as described below. Furthermore, as inducement to enter into the Demand Note, the Company agreed to pay CrossCart a transaction bonus equal to 3% of the gross proceeds received from any control change transaction of the Company (“Transaction Bonus”). In September 2015, the Company and CrossCart entered into an agreement to provide that CrossCart’s right to receive the Transaction Bonus will terminate and expire upon completion of this offering. As of June 30, 2015, the total amount outstanding under the Demand Note was $125,093.
Line of Credit Note
On May 1, 2015, the Company entered into a Line of Credit Note with CrossCart that provides the Company with a credit line up to $100,000. During the term of this note, the Company may request borrowings, with a minimum amount of $10,000 for each request, from CrossCart, and such request may be granted or denied at CrossCart’s sole discretion. The Company may repay the outstanding amount of this note at its discretion. The note bears interest at rate of 10% per annum, and all principal amount and accrued interest will be due and payable on May 1, 2016. The note contains customary events of default. If the Company fails to pay all amounts due by May 1, 2016, the interest on the remaining outstanding balance will accrue at a rate of 12% per annum. As of June 30, 2015, the total amount outstanding under the Line of Credit Note was $61,627, including accrued interest. In addition, as inducement to enter into the Line of Credit Note, the Company amended the Royalty Right such that CrossCart will be entitled to receive a 5% royalty on all net sales in excess of $12,000,000 without an expiration date (the “Additional Royalty Rights”). In September 2015, the Company and CrossCart entered into an agreement to provide that the Additional Royalty Rights will terminate and expire upon completion of this offering.
Extension of Warrants and Stock Options
On or about December 23, 2013, the Company entered into a letter agreement (the “Letter Agreement”) with CrossCart and Dr. Stone to extend the expiration dates of the following warrants held by CrossCart (the “Series C Warrants”):
| · | A warrant to purchase 10,625 shares of Series C Preferred Stock issued on July 25, 2007 having an exercise price of $0.48 per share and an expiration date of July 25, 2012. |
| · | A warrant to purchase 6,250 shares of Series C Preferred Stock issued on August 14, 2007 having an exercise price of $0.48 per share and an expiration date of August 14, 2012. |
The expiration dates of the two Series C Warrants were extended five (5) years from the initial expiration dates, such that the expiration dates are now July 25, 2017 and August 14, 2017, respectively.
In addition, the Letter Agreement extended the expiration date of stock options to purchase 100,000 shares of common stock of the Company, which was issued to Dr. Stone on February 11, 2003 with an exercise price of $0.45 per share. The expiration date of such stock option was extended to February 11, 2018.
Transaction between CrossCart and Lurie Investment
On August 19, 2013, CrossCart, Dr. Stone, Lurie Investment Fund, L.L.C. (“Lurie”) and Alfa-Tech, L.L.C. (“Alfa-Tech” and, together with “Lurie”, the “Lurie Entities”) entered into a Stock and Note Purchase Agreement (the “Lurie Purchase Agreement”), pursuant to which CrossCart purchased from the Lurie Entities the following securities of the Company held by Lurie: (i) an aggregate of 937,602 shares of Series B Preferred Stock; (ii) an aggregate of 7,083,334 shares of Series C Preferred Stock; (iii) an aggregate of 12,952,917 shares of Series C-2 Preferred Stock; (iv) warrants to purchase an aggregate of 937,500 shares of Series C-1 Preferred Stock at an exercise price of $0.48 per share; (v) warrants to purchase an aggregate of 1,171,875 shares of Series C-2 Preferred Stock at an exercise price of $0.48 per share; (vi) warrants to purchase an aggregate of 3,217,849 shares of common stock at an exercise price of $0.01 per share; and (vi) convertible promissory notes in the aggregate principal amount of $5,000,000 (collectively, the “Lurie Securities”). In exchange for the Lurie Securities, CrossCart agreed to pay the Lurie Entities cash consideration of $1.00 and certain contingent consideration upon the consummation of a sale of the Company, including the occurrence of a “deemed liquation event” as defined in the Company’s Certificate of Incorporation (the “Lurie Contingent Consideration”). In general, the Lurie Contingent Consideration shall equal to 20% of the gross proceeds received by CrossCart from the Company in such sale to the extent the proceeds exceed CrossCart’s capital contribution made by the purchaser to the Company.
Change of Control Bonus
On March 3, 2012, the Company awarded Dr. Stone a bonus (the “Stone Bonus Agreement”) in the event of an acquisition of the Company. An acquisition is defined as a sale, merger, consolidation, transfer or other disposition of all or substantially all (more than 80%) of the Company to another entity (the “Acquisition”). An initial public offering of the Company’s Common Stock pursuant to an effective registration statement filed with the Securities and Exchange Commission under the Securities Act of 1933, as amended (the “Securities Act”), does not constitute an Acquisition. Dr. Stone is entitled to receive a bonus award in an amount equal to 1% of the proceeds received in the Acquisition that occurred on or prior to December 16, 2013. On December 16, 2013, the Board amended and extended the Stone Bonus Agreement to cover any Acquisition that occurs on or before the completion of an initial public offering of the Company’s stock pursuant to an effective registration statement filed with the Securities and Exchange Commission under the Securities Act. In September 2015 the Company and Dr. Stone entered into an amendment to the Stone Bonus Agreement, pursuant to which the Stone Bonus Agreement will be terminated, and no bonus will be paid to Dr. Stone, upon the completion of this offering.
Transactions with Bruce Stone
Between May 22, 2014 and June 2, 2015, the Company issued three convertible promissory notes in the amount of $50,000, $35,000 and $40,000, respectively, to Mr. Bruce Stone, who is the brother of Dr. Kevin Stone, pursuant to Note Purchase Agreements dated April 17, 2014, November 12, 2014 and May 20, 2015, respectively (collectively, the “Bruce Stone Notes”). Each note bears interest at a rate of 8% per annum, which may be increased to 10% per annum if an event of default occurs. In general, all principal and interest accrued under each Bruce Stone Note are due and payable, upon demand at the sole option of Mr. Stone, on or after two years after the date of issuance. The notes contain customary events of default. The principal and any accrued interest under the Bruce Stone Notes are convertible, at Mr. Stone’s sole discretion, into shares of Series D Preferred Stock of the Company at an initial conversion price of $0.25 per share. Under the terms of the Bruce Stone Notes, the Company is required to use reasonable efforts to cause a sufficient number of shares of Series D Preferred Stock
authorized and reserved for issuance upon conversion of the notes. As of June 30, 2015 the total amount outstanding under the Bruce Stone Notes was $131,407, including accrued interest. In addition, we expect to enter into an agreement with Mr. Stone to provide that upon completion of this offering, all outstanding amount due under the Bruce Stone Notes will be automatically converted into shares of our common stock at the initial conversion price.
Management Retention Plan
On December 16, 2013, the Board approved the Management Retention Plan (the “Plan”), pursuant to which certain key employees, including Mr. Lee and Mr. Cocke, are entitled to receive a bonus upon the occurrence of specified events, provided that the participant must continue as an employee of the Company through the occurrence of the triggering event. Under the Plan, upon an external financing (beyond any financing from CrossCart) of at least $2.0 million or a signing of a license or distribution agreement with at least $2.0 million of upfront payments or milestone payments achievable within 12 months of signing, Mr. Lee is entitled to receive a bonus equal to $35,000 plus a salary delta amount (the “Salary Delta Amount”), and Mr. Cocke is entitled to receive a bonus equal to $15,000 plus the Salary Delta Amount. The Salary Delta Amount is calculated as the difference between the salary actually received after July 1, 2013 and the amount of salary such person would have received if the salary in effect during June 2013 had continued from July 2013 until the bonus is earned. We expect to amend the Plan to provide that all bonuses under the Plan shall be paid in shares of common stock based on the public offering price in this offering.
In addition, the Plan also provides that upon closing of a deemed liquidation events specified in the Company’s certificate of incorporation or a sale of the Company’s securities representing 50% or more of total voting power of outstanding securities (the “Acquisition”), Mr. Lee and Mr. Cocke are entitled to receive certain bonus payments based, in part, on the amount of proceeds received by the Company in the Acquisition (the “Acquisition Bonus”). We expect the Board to approve an amendment to the Plan to provide that the Acquisition Bonus will terminate and expire upon the completion of this offering.
Transactions with Directors
In December 2013, the Company entered into a letter agreement with each of David W. Anderson and Alfred G. Holcomb, two of our directors, to provide certain acquisition bonus payments (the “Director Bonus Agreement”), Under the Director Bonus Agreement, in the event of an acquisition of the Company or the occurrence of a “deemed liquidation” event under our certificate of incorporation (the “Acquisition”), each director will be eligible to receive the following cash bonus payments (the “Director Acquisition Bonuses”):
| · | if the proceeds of the Acquisition is at least $15 million and less than $25 million, a cash bonus of $100,000; |
| · | if the proceeds of the Acquisition is at least $25 million and less than $100,000 million, a cash bonus of $250,000; and |
| · | if the proceeds of the Acquisition is at least $100 million, a cash bonus equal to 10% of 4% of such proceeds. |
We expect to enter into an agreement with each of Mr. Anderson and Mr. Holcom to provide that the Director Acquisition Bonuses will terminate and expire immediately upon
completion of this offering.
Securities Being Offered
The following descriptions are summaries of the material terms of our amended and restated certificate of incorporation and amended and restated bylaws, which will be effective upon completion of this offering. The descriptions of the common stock and preferred stock give effect to changes to our capital structure that will occur immediately prior to the completion of this offering. We refer in this section to our amended and restated certificate of incorporation as our certificate of incorporation, and we refer to our amended and restated bylaws as our bylaws.
General
Upon completion of this offering, our authorized capital stock will consist of shares of common stock, par value $0.001 per share, and shares of preferred stock, par value $0.001 per share, all of which shares of preferred stock will be undesignated.
As of June 30, 2015, 72,963,279 shares of our common stock were outstanding and held by 61 stockholders of record. This amount assumes (i) the conversion of all outstanding shares of our convertible preferred stock into common stock, which will occur immediately prior to the completion of this offering and (ii) the issuance of shares of common stock upon the conversion of all principal amount and accrued interests under our Notes (as discussed below) upon the completion of this offering, assuming that the offering is completed on , 2015. In addition, as of June 30, 2015, we had outstanding options to purchase 6,447,747 shares of our common stock under our 2008 Stock Option Plan, and a predecessor plan, at a weighted average exercise price of $0.08 per share, of which options to purchase an aggregate of 6,043,809 were exercisable.
Common Stock
The holders of our common stock are entitled to one vote for each share held on all matters submitted to a vote of the stockholders. The holders of our common stock do not have any cumulative voting rights. Holders of our common stock are entitled to receive ratably any dividends declared by the board of directors out of funds legally available for that purpose, subject to any preferential dividend rights of any outstanding preferred stock. Our common stock has no preemptive rights, conversion rights or other subscription rights or redemption or sinking fund provisions.
In the event of our liquidation, dissolution or winding up, holders of our common stock will be entitled to share ratably in all assets remaining after payment of all debts and other liabilities and any liquidation preference of any outstanding preferred stock. The shares to be issued by us in this offering will be, when issued and paid for, validly issued, fully paid and non-assessable.
Preferred Stock
Immediately prior to the completion of this offering, we expect that all outstanding shares of our convertible preferred stock will be converted into shares of our common stock. Immediately prior to the completion of this offering, our amended and restated certificate of incorporation will be amended and restated to delete all references to such shares of convertible preferred stock. Upon the completion of this offering, our board of directors will have the authority, without
further action by our stockholders, to issue up to shares of preferred stock in one or more series and to fix the rights, preferences, privileges and restrictions thereof. These rights, preferences and privileges could include dividend rights, conversion rights, voting rights, terms of redemption, liquidation preferences, sinking fund terms and the number of shares constituting, or the designation of, such series, any or all of which may be greater than the rights of common stock. The issuance of our preferred stock could adversely affect the voting power of holders of common stock and the likelihood that such holders will receive dividend payments and payments upon our liquidation. In addition, the issuance of preferred stock could have the effect of delaying, deferring or preventing a change in control of our company or other corporate action. Immediately after completion of this offering, no shares of preferred stock will be outstanding, and we have no present plan to issue any shares of preferred stock.
Convertible Promissory Notes
In fiscal 2012, we borrowed $3,250,000 through the issuance of certain convertible promissory notes to existing stockholders. These notes accrue interest at 8% per year and are convertible into shares of the Company’s Series C-2 Preferred Stock at a conversion price of $0.48 per share (the “Series C-2 Notes”). In fiscal 2013 and 2014, we borrowed $4,126,027 through the issuance of certain convertible promissory notes to new and existing investors. These notes accrue interest at 8% and are due upon demand of the holder. The holders may, in their sole discretion, convert all principal and accrued interest under these notes into shares of the Company’s Series D Preferred Stock at a conversion price of $0.25 per share (the “Series D Notes”). In 2015, we borrowed $780,000 through the issuance of certain convertible promissory notes to existing and new investors. These notes accrue interest at a rate of 8% per year and are convertible into shares of the Company’s Series D or Series E Preferred Stock at a conversion price of $0.25 per share (the “Series E Notes”, and together with the Series C-2 Notes and Series D Notes, the “Notes”).
We intend to enter into agreements with each holder of the Notes to provide that upon the completion of this offering, subject to certain limitations on the conversion of the Notes, all principal and accrued interest under the Notes will convert into an aggregate of shares of common stock, assuming that the offering is completed on 2015. For more information on the Notes, see “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources—Convertible Note Financings.”
Warrants
As of June 30, 2015, we had outstanding warrants to purchase 954,375 shares of Series C convertible preferred stock and 1,171,875 shares of Series C-2 convertible preferred stock, together which are exercisable for an aggregate of 2,126,250 shares of common stock upon the completion of this offering. In addition, as of June 30, 2015, we had outstanding warrants to purchase an aggregate of 10,016,592 shares of our common stock.
On August 19, 2013, our largest stockholder, CrossCart LLC purchased preferred stock, promissory notes and warrants for common stock and preferred stock previously held by Lurie Investment Fund, LLC, another stockholder of the Company. The warrants have a net exercise provision and contain provisions for the adjustment of the exercise price and the number of shares issuable upon the exercise of the warrant in the event of certain stock dividends, stock splits, recapitalizations, reclassifications and consolidations. The warrants for preferred stock are exercisable until their expiration on either (i) the earlier of certain dates in 2020 or a sale or merger of the Company, or (ii) the earlier of certain dates in 2022 or the conversion of the
promissory notes as noted above. The warrants for common stock are exercisable until their expiration on June 19, 2018.
Registration Rights
Upon the completion of this offering, the holders of shares of our common stock, including shares issuable upon the conversion of our convertible preferred stock and Notes, or their permitted transferees, are entitled to rights with respect to the registration of these securities under the Securities Act, which we refer to as our registrable securities. These rights are provided under the terms of an investor rights agreement between us and certain holders of our common stock, Series A convertible preferred stock, Series B convertible preferred stock, and Series C convertible preferred stock. The investor rights agreement includes demand registration rights, short-form registration rights and piggyback registration rights. All fees, costs and expenses of underwritten registrations under these agreements will be borne by us and all selling expenses, including underwriting discounts and selling commissions, will be borne by the holders of the shares being registered.
Demand Registration Rights
Beginning 180 days after the completion of this offering, the holders of at least 60% of our Series C convertible preferred stock are entitled to demand registration rights. Under the terms of the investor rights agreement, upon the written request of such holders to sell registrable securities with an anticipated aggregate offering price (net of underwriting discounts and commissions) of at least $5.0 million, we will be required to use our best efforts to file a registration statement covering the offering and sale of such securities and use reasonable, diligent efforts to effect the registration of all or a portion of these securities for public resale. We are required to effect only two registrations pursuant to this provision of the investor rights agreement. In the event we register securities in connection with an underwritten offering, the underwriters will have the right to limit the number of shares included in such offering.
Short-Form Registration Rights
Upon the completion of this offering, the holders of at least 30% of our registrable securities are also entitled to short form registration rights. Pursuant to the investor rights agreement, if we are eligible to file a registration statement on Form S-3, upon the written request of such holders to sell registrable securities, we will be required to use our best efforts to effect a registration of such securities. In the event we register securities in connection with an underwritten offering, the underwriters will have the right to limit the number of shares included in such offering.
Piggyback Registration Rights
Upon the completion of this offering, the holders of our registrable securities are entitled to piggyback registration rights. If we register any of our securities either for our own account or for the account of other security holders, such holders are entitled to include their shares in the registration. In the event we register securities in connection with an underwritten offering, the underwriters will have the right to limit the number of shares included in such offering.
Our investor rights agreement contains customary cross-indemnification provisions, under which we are obligated to indemnify holders of registrable securities in the event of material
misstatements or omissions in the registration statement attributable to us, and they are obligated to indemnify us for material misstatements or omissions attributable to them.
Expiration of Registration Rights
The registration rights granted under the investor rights agreement will terminate after the earlier of (i) the fifth anniversary of the Company’s first underwritten public offering of its common stock under the Securities Act, (ii) with respect to any holder of registrable securities, the date on which all of such holder’s shares can be sold during a three-month period without registration in reliance on Rule 144 under the Securities Act, (iii) the closing of a deemed liquidation event or (iv) termination of the agreement upon consent of the parties.
Anti-Takeover Effects of Our Certificate of Incorporation and Bylaws and Delaware Law
Our certificate of incorporation and bylaws include a number of provisions that may have the effect of delaying, deferring or preventing another party from acquiring control of us and encouraging persons considering unsolicited tender offers or other unilateral takeover proposals to negotiate with our board of directors rather than pursue non-negotiated takeover attempts. These provisions include the items described below.
Board Composition and Filling Vacancies
Our certificate of incorporation provides for the division of our board of directors into three classes serving staggered three-year terms, with one class being elected each year. Our certificate of incorporation also provides that directors may be removed only for cause and then only by the affirmative vote of the holders of 75% or more of the shares then entitled to vote at an election of directors. Furthermore, any vacancy on our board of directors, however occurring, including a vacancy resulting from an increase in the size of our board, may only be filled by the affirmative vote of a majority of our directors then in office even if less than a quorum. The classification of directors, together with the limitations on removal of directors and treatment of vacancies, has the effect of making it more difficult for stockholders to change the composition of our board of directors.
No Written Consent of Stockholders
Our certificate of incorporation provides that all stockholder actions are required to be taken by a vote of the stockholders at an annual or special meeting, and that stockholders may not take any action by written consent in lieu of a meeting. This limit may lengthen the amount of time required to take stockholder actions and would prevent the amendment of our bylaws or removal of directors by our stockholders without holding a meeting of stockholders.
Meetings of Stockholders
Our certificate of incorporation and bylaws provide that only a majority of the members of our board of directors then in office may call special meetings of stockholders and only those matters set forth in the notice of the special meeting may be considered or acted upon at a special meeting of stockholders. Our bylaws limit the business that may be conducted at an annual meeting of stockholders to those matters properly brought before the meeting.
Advance Notice Requirements
Our bylaws establish advance notice procedures with regard to stockholder proposals relating to the nomination of candidates for election as directors or new business to be brought before meetings of our stockholders. These procedures provide that notice of stockholder proposals must be timely given in writing to our corporate secretary prior to the meeting at which the action is to be taken. Generally, to be timely, notice must be received at our principal executive offices not less than 90 days nor more than 120 days prior to the first anniversary date of the annual meeting for the preceding year. Our bylaws specify the requirements as to form and content of all stockholders’ notices. These requirements may preclude stockholders from bringing matters before the stockholders at an annual or special meeting.
Amendment to Certificate of Incorporation and Bylaws
Any amendment of our certificate of incorporation must first be approved by a majority of our board of directors, and if required by law or our certificate of incorporation, must thereafter be approved by a majority of the outstanding shares entitled to vote on the amendment and a majority of the outstanding shares of each class entitled to vote thereon as a class, except that the amendment of the provisions relating to stockholder action, board composition, limitation of liability and the amendment of our bylaws and certificate of incorporation must be approved by not less than 75% of the outstanding shares entitled to vote on the amendment, and not less than 75% of the outstanding shares of each class entitled to vote thereon as a class. Our bylaws may be amended by the affirmative vote of a majority of the directors then in office, subject to any limitations set forth in the bylaws; and may also be amended by the affirmative vote of at least 75% of the outstanding shares entitled to vote on the amendment, or, if our board of directors recommends that the stockholders approve the amendment, by the affirmative vote of the majority of the outstanding shares entitled to vote on the amendment, in each case voting together as a single class.
Undesignated Preferred Stock
Our certificate of incorporation provides for authorized shares of preferred stock. The existence of authorized but unissued shares of preferred stock may enable our board of directors to discourage an attempt to obtain control of us by means of a merger, tender offer, proxy contest or otherwise. For example, if in the due exercise of its fiduciary obligations, our board of directors were to determine that a takeover proposal is not in the best interests of our stockholders, our board of directors could cause shares of preferred stock to be issued without stockholder approval in one or more private offerings or other transactions that might dilute the voting or other rights of the proposed acquirer or insurgent stockholder or stockholder group. In this regard, our certificate of incorporation grants our board of directors broad power to establish the rights and preferences of authorized and unissued shares of preferred stock. The issuance of shares of preferred stock could decrease the amount of earnings and assets available for distribution to holders of shares of common stock. The issuance may also adversely affect the rights and powers, including voting rights, of these holders and may have the effect of delaying, deterring or preventing a change in control of us.
Section 203 of the Delaware General Corporation Law
Upon completion of this offering, we will be subject to the provisions of Section 203 of the Delaware General Corporation Law. In general, Section 203 prohibits a publicly held Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a three-year period following the time that this stockholder becomes an interested stockholder, unless the business combination is approved in a prescribed manner. Under Section 203, a
business combination between a corporation and an interested stockholder is prohibited unless it satisfies one of the following conditions:
| · | before the stockholder became interested, our board of directors approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder; |
| · | upon consummation of the transaction which resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the voting stock outstanding shares owned by persons who are directors and also officers, and employee stock plans, in some instances, but not the outstanding voting stock owned by the interested stockholder; or |
| · | at or after the time the stockholder became interested, the business combination was approved by our board of directors and authorized at an annual or special meeting of the stockholders by the affirmative vote of at least two-thirds of the outstanding voting stock which is not owned by the interested stockholder. |
Section 203 defines a business combination to include:
| · | any merger or consolidation involving the corporation and the interested stockholder; |
| · | any sale, transfer, lease, pledge or other disposition involving the interested stockholder of 10% or more of the assets of the corporation; |
| · | subject to exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; |
| · | subject to exceptions, any transaction involving the corporation that has the effect of increasing the proportionate share of the stock of any class or series of the corporation beneficially owned by the interested stockholder; and |
| · | the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation. |
In general, Section 203 defines an interested stockholder as any entity or person beneficially owning 15% or more of the outstanding voting stock of the corporation and any entity or person affiliated with or controlling or controlled by the entity or person.
Financial Statements
[To be provided by Company]
PART III—EXHIBITS
Exhibit | Description of Document |
1.1 | Form of Underwriting Agreement* |
2.1 | Amended and Restated Certificate of Incorporation of the Issuer, as currently in effect |
2.2 | Second Amended and Restated Certificate of Incorporation, as in effect following the offering |
2.3 | Bylaws of the Issuer, as currently in effect* |
2.4 | Amended and Restated Bylaws, as in effect following the offering* |
3.1 | Third Amended and Restated Investors’ Rights Agreement |
3.2 | Form of Demand Promissory Note issued to CrossCart LLC |
3.3 | Form of Convertible Promissory Note |
3.4 | Line of Credit Note issued to CrossCart LLC |
3.5 | Form of Warrant to purchase Preferred Stock of the Company |
3.6 | Form of Warrant to purchase Common Stock of the Company |
6.1† | License Agreement dated January 8, 2009 between the Company and The Curators of the University of Missouri |
6.2 | Letter Agreement dated December 23, 2013 by and among the Company, CrossCart LLC and Dr. Kevin R. Stone |
6.3 | Aperion Management Retention Plan |
6.4 | Aperion Amended and Restated Management Retention Plan |
6.5 | Lease Agreement dated December 17, 2008 by and among the Company and Titan Mac Fund I, LP, as amended on June 21, 2012. |
6.6 | Independent Consultant Agreement effective September 1, 2008 between the Company and David Cocke |
6.7 | 2008 Stock Option/Stock Issuance Plan |
8.1 | Escrow Agreement* |
10.1 | Power of Attorney |
11.1 | Consent of Marcum LLP |
12.1 | Opinion Of Morgan, Lewis & Bockius LLP* |
13.1 | Testing the water materials |
* To be filed by amendment.
| † | Confidential treatment has been requested for certain information contained in this document. Such information has been omitted and filed separately with the Securities and Exchange Commission |
SIGNATURES
Pursuant to the requirements of Regulation A, the issuer certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form 1-A and has duly caused this offering statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the city of San Antonio, State of Texas, on September 18, 2015.
| | APERION BIOLOGICS, INC. |
| | By: | /s/ Daniel R. Lee
Daniel R. Lee Chief Executive Officer |
KNOW ALL MEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Daniel R. Lee and David Cocke, jointly and severally, as his or her true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him or her, and in his or her name, place and stead, in any and all capacities, to sign the Offering Statement on Form 1-A of Aperion Biologics, Inc. and any or all amendments (including post-effective amendments) thereto, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents full power and authority to do and perform each and every act and thing requisite or necessary to be done in and about the premises hereby ratifying and confirming all that said attorneys-in-fact and agents, or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, as amended, this Offering Statement has been signed by the following persons in the capacities indicated.
Signature | Title | Date |
| | | |
/s/ Daniel R. Lee | Chief Executive Officer and Director | September 21, 2015 |
| Daniel R. Lee | (Principal Executive Officer) | |
| | | |
| | | |
/s/ David Cocke | Chief Financial Officer | September 21, 2015 |
| David Cocke | (Principal Financial Officer and Principal Accounting Officer) |
| | | |
/s/ David W. Anderson | Director | September 21, 2015 |
| | | |
| | | |
| | | |
| Director | September 21, 2015 |
| France Dixon Helfer | | |
| | | |
| | | |
| Director | September 21, 2015 |
| Alfred Holcomb | | |
| | | |
| | | |
/s/ Kevin R. Stone, M.D. | Director | September 21, 2015 |
| Kevin R. Stone, M.D. | | |
| | | |
| | | |
/s/ Mike Ward | Director | September 21, 2015 |
| Mike Ward | | |