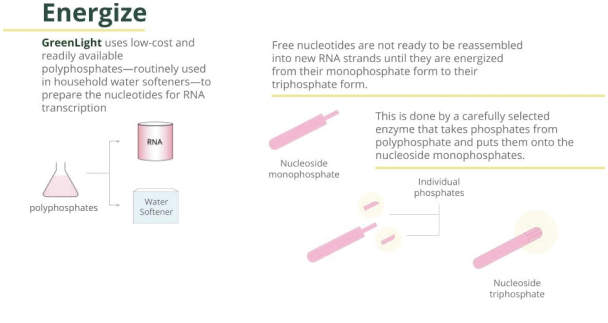
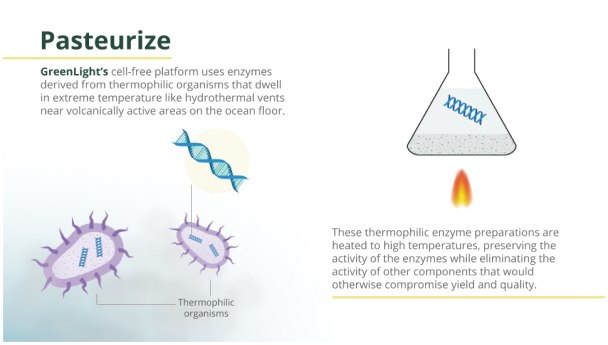
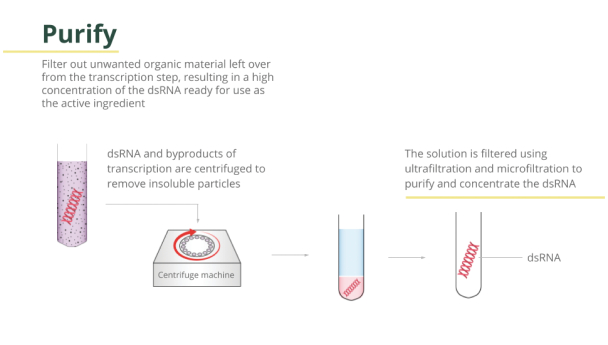
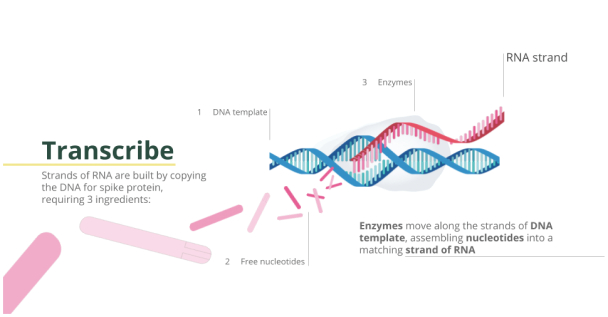
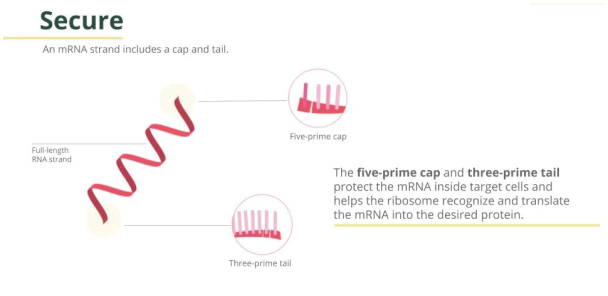
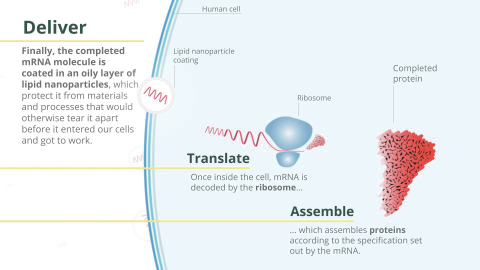
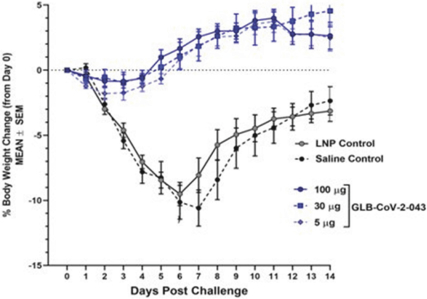
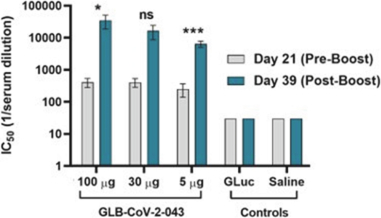
Filed Pursuant to Rule 424(b)(3)
Registration No. 333-262574
PROSPECTUS

GreenLight Biosciences Holdings, PBC
86,631,958 Shares of Common Stock
10,350,000 Shares of Common Stock Issuable Upon Exercise of Warrants
This prospectus relates to the resale, from time to time, of up to 86,631,958 shares of our common stock, par value $0.0001 per share (“Common Stock”), by the selling securityholders (including their pledgees, donees, transferees or other successors-in-interest) identified in this prospectus (the “Selling Securityholders”). This prospectus also relates to the issuance by us of up to 10,350,000 shares of Common Stock upon the exercise of outstanding warrants (the “Public Warrants”).
On February 2, 2022, we consummated the business combination, or the Business Combination, contemplated by the Business Combination Agreement (the “Business Combination Agreement”), dated August 9, 2021, by and among our company (formerly known as Environmental Impact Acquisition Corp. (“ENVI”)), GreenLight Biosciences, Inc. (“GreenLight”) and Honey Bee Merger Sub, Inc., pursuant to which Honey Bee Merger Sub, Inc. was merged with and into GreenLight, with GreenLight surviving the merger (the “Merger”). As a result of the Merger, and upon consummation of the Merger and the other transactions contemplated by the Business Combination Agreement, GreenLight became a wholly owned subsidiary of ENVI. Upon the closing of the Business Combination, we changed our name to GreenLight Biosciences Holdings, PBC (“New GreenLight”), with stockholders of GreenLight becoming stockholders of New GreenLight. See “Prospectus Summary—Background.”
We are registering 12,425,000 shares of Common Stock (the “PIPE Shares”) held by certain of the Selling Securityholders pursuant to the terms of subscription agreements (the “Subscription Agreements”), entered into with certain of the Selling Securityholders, or the PIPE Investors. Pursuant to the Subscription Agreements, the PIPE Investors purchased shares of our Common Stock in a private placement in connection with the Business Combination, or the PIPE Financing.
We are registering 65,644,695 shares of our Common Stock held by certain of the Selling Securityholders pursuant to the terms of an Investor Rights Agreement we entered into concurrently with the Business Combination Agreement, including (a) 58,407,195 shares of Common Stock issued to former securityholders of GreenLight pursuant to the Business Combination Agreement, (b) 5,175,000 shares of Common Stock issued to our initial securityholders and (c) 2,062,500 shares of Common Stock issuable upon the exercise of private placement warrants we issued to our initial securityholders (the “Private Placement Warrants”). Separately, we are registering 1,310,590 outstanding shares of Common Stock, and 7,251,673 shares of Common Stock issuable upon exercise of stock options, held by certain members of our Board of Directors and executive officers.
We are also registering the issuance of shares of Common Stock underlying the Public Warrants pursuant to the terms of a Warrant Agreement, dated January 13, 2021, between us and Continental Stock Transfer and Trust Company (the “Warrant Agreement”).
We will not receive any proceeds from the sale of the shares by the Selling Securityholders. We will receive the proceeds from any exercise of the warrants for cash.
We will bear all costs, expenses and fees in connection with the registration of the shares of Common Stock. The Selling Securityholders will bear all commissions and discounts, if any, attributable to their sales of the shares of Common Stock.
Our Common Stock is listed on the Nasdaq Capital Market (“Nasdaq”) under the symbol “GRNA” and our warrants are listed on Nasdaq under the symbol “GRNAW”. On February 14, 2022, the closing sale price of our Common Stock as reported on Nasdaq was $8.72, and the closing sale price of our warrants as reported on Nasdaq was $0.5499.
We are an “emerging growth company” under applicable Securities and Exchange Commission rules and, as such, have elected to comply with certain reduced public company disclosure requirements for this prospectus and future filings. See “Prospectus Summary—Implications of Being an Emerging Growth Company.”
Our business and investment in our Common Stock involve a high degree of risk. These risks are described in the section titled “Risk Factors” beginning on page 10 of this prospectus.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the accuracy or adequacy of this prospectus. Any representation to the contrary is a criminal offense.
The date of this prospectus is February 14, 2022.