PART I
Except where the context otherwise requires, the terms “we,” “us,” “our” or “the Company,” refer to the business of Protalix BioTherapeutics, Inc. and its consolidated subsidiaries, and “Protalix” or “Protalix Ltd.” refers to the business of Protalix Ltd., our wholly-owned subsidiary and sole operating unit.
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
AND RISK FACTORS SUMMARY
The statements set forth under the captions “Business,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Risk Factors,” and other statements included elsewhere in this Annual Report on Form 10-K, particularly with respect to our plans and strategy for our business and related financing, includes forward-looking statements within the meanings of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act, including statements regarding expectations, beliefs, intentions or strategies for the future. When used in this report, the terms “anticipate,” “believe,” “estimate,” “expect,” “can,” “continue,” “could,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “will,” “would” and other words or phrases of similar import, as they relate to our company or our subsidiaries or our management, are intended to identify forward-looking statements. We intend that all forward-looking statements be subject to the safe-harbor provisions of the Private Securities Litigation Reform Act of 1995. These forward-looking statements are only predictions and reflect our views as of the date they are made with respect to future events and financial performance, and we undertake no obligation to update or revise, nor do we have a policy of updating or revising, any forward-looking statement to reflect events or circumstances after the date on which the statement is made or to reflect the occurrence of unanticipated events, except as may be required under applicable law. Forward-looking statements are subject to many risks and uncertainties that could cause our actual results to differ materially from any future results expressed or implied by the forward-looking statements.
Examples of the risks and uncertainties include, but are not limited to, the following:
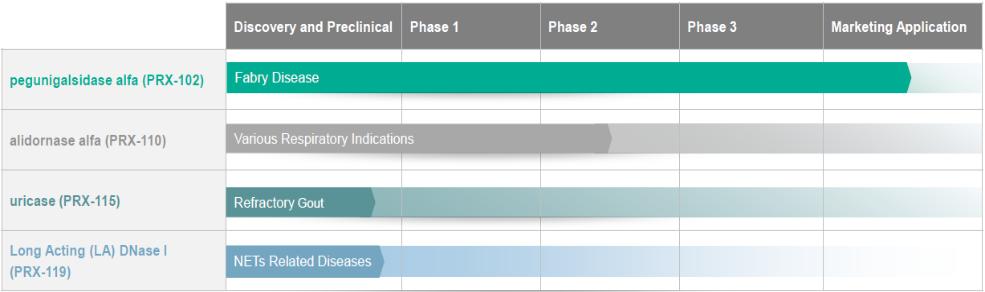
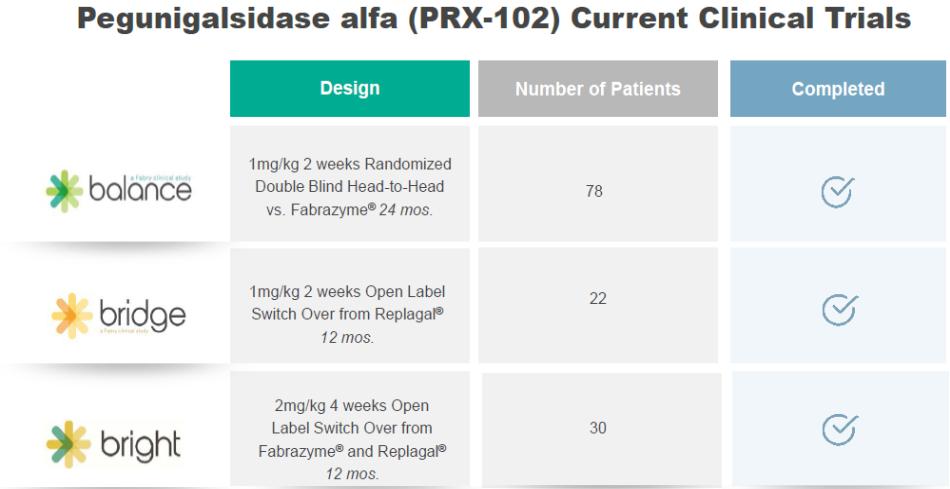
●risks related to the timing and progress of the preparation of an updated Biologics License Application, or BLA, for PRX-102 addressing the Complete Response Letter, or CRL;
●risks related to the timing, progress and likelihood of final approval by the U.S. Food and Drug Administration, or the FDA, and the European Medicines Agency, or the EMA, of a resubmitted Biologics License Application, or BLA, and of a Marketing Authorization Application, or an MAA, respectively, for PRX-102, and, if approved, whether the use of PRX-102 will be commercially successful;
●failure or delay in the commencement or completion of our preclinical studies and clinical trials, which may be caused by several factors, including: slower than expected rates of patient recruitment; unforeseen safety issues; determination of dosing issues; lack of effectiveness during clinical trials; inability or unwillingness of medical investigators and institutional review boards to follow our clinical protocols; inability to monitor patients adequately during or after treatment; and/or lack of sufficient funding to finance our clinical trials;
●the risk that the FDA, the European Medicines Agency, or EMA, or other foreign regulatory authorities may not accept or approve a marketing application we file for any of our product candidates, and other risks relating to the review process;
●risks associated with the novel coronavirus disease, or COVID-19, outbreak and variants, which may adversely impact our business;
●risks related to any transactions we may effect in the public or private equity markets to raise capital to finance future research and development activities, general and administrative expenses and working capital;
●risks relating to our evaluation and pursuit of strategic alternatives;
●the risk that the results of our clinical trials will not support the applicable claims of safety or efficacy and that our product candidates will not have the desired effects or will be associated with undesirable side effects or other unexpected characteristics;