QuickLinks -- Click here to rapidly navigate through this document
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF
THE SECURITIES EXCHANGE ACT OF 1934
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2007 COMMISSION FILE NUMBER: 000-21429
ARQULE, INC.
(EXACT NAME OF REGISTRANT AS SPECIFIED IN ITS CHARTER)
DELAWARE
(STATE OR OTHER JURISDICTION OF INCORPORATION OR ORGANIZATION) | | 04-3221586
(I.R.S. EMPLOYER
IDENTIFICATION NO.) |
19 PRESIDENTIAL WAY, WOBURN, MASSACHUSETTS 01801
(ADDRESS OF PRINCIPAL EXECUTIVE OFFICES INCLUDING ZIP CODE) |
REGISTRANT'S TELEPHONE NUMBER, INCLUDING AREA CODE:
(781) 994-0300 |
SECURITIES REGISTERED PURSUANT TO SECTION 12(b) OF THE ACT:
NONE |
(TITLE OF EACH CLASS)
| | NAME OF EACH EXCHANGE
ON WHICH REGISTERED
|
|---|
COMMON STOCK,
$.01 PAR VALUE | | The NASDAQ Stock Market LLC
(NASDAQ Global Market) |
SECURITIES REGISTERED PURSUANT TO SECTION 12(g) OF THE ACT:
NONE
Indicate by check mark if the registrant is a well-known issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of "accelerated filer and large accelerated filer" in Rule 12b-2 of the Exchange Act. (Check One)
| Large accelerated filer o | | Accelerated filer ý | | Non-accelerated filer o
(Do not check if a smaller reporting company) | | Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No ý
The aggregate market value of voting and non-voting common stock held by non-affiliates of the registrant as of June 30, 2007 was: $304,084,097
There were 43,772,003 shares of the registrant's Common Stock outstanding as of March 10, 2008.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive proxy statement for the Registrant's Annual Meeting of Shareholders to be held on May 16, 2008, which will be filed with the Securities and Exchange Commission not later that 120 days after the registrant's fiscal year end of December 31, 2007, are incorporated by reference into Part III of the Form 10-K.
IMPORTANT FACTORS REGARDING FORWARD-LOOKING STATEMENTS
You should carefully consider the risks described below together with all of the other information included in this Form 10-K before making an investment decision. An investment in our common stock involves a high degree of risk. We operate in a dynamic and rapidly changing industry that involves numerous uncertainties. The risks and uncertainties described below are not the only ones we face. Other risks and uncertainties, including those that we do not currently consider material, may impair our business. If any of the risks discussed below actually occur, our business, financial condition, operating results or cash flows could be materially adversely affected. This could cause the trading price of our common stock to decline, and you may lose all or part of your investment.
This Form 10-K, including information incorporated herein by reference, contains forward-looking statements as defined in the Private Securities Litigation Reform Act of 1995. All statements that are not descriptions of historical fact are forward-looking statements, based on estimates, assumptions and projections that are subject to risks and uncertainties. These statements can generally be identified by use of forward looking terminology such as "believes", "expects", "intends", "may", "will", "plans", "should", "anticipates" or similar terminology. Although we believe that the expectations reflected in such forward looking statements are reasonable as of the date thereof, such expectations are based on certain assumptions regarding the progress of product development efforts under collaborative agreements, the execution of new collaborative agreements and other factors relating to our growth. Such expectations may not materialize if product development efforts, including any necessary trials of our potential drug candidates, are delayed or suspended, if positive early results are not repeated in later studies or in humans, if planned acquisitions or negotiations with potential collaborators are delayed or unsuccessful, if we are unsuccessful at integrating acquired assets or technologies, or if other assumptions prove incorrect. The forward-looking statements contained herein represent the judgment of ArQule as of the date of this Form 10-K. ArQule disclaims any intent or obligation to update any forward-looking statement except to the extent required by law.
2
PART I
ITEM 1. BUSINESS
BUSINESS OVERVIEW
We are a clinical-stage biotechnology company organized as a Delaware corporation in 1993 and engaged in the research and development of innovative cancer therapeutics. Our mission is to introduce novel products that act selectively against cancer cells, target multiple tumor types and are well tolerated by patients. We believe our clinical stage products represent potential best-in-class or first-in-class small molecule candidates based on highly differentiated mechanisms of action.
Our lead products, which are in clinical-stage testing, consist of: ARQ 197, an orally administered inhibitor of the c-Met receptor tyrosine kinase; ARQ 501, an intravenously administered novel activator of the cell's DNA damage response mechanism mediated by the E2F-1 transcription factor; and ARQ 171, an intravenously administered second generation activator of E2F-1. Early-stage clinical trial results, which are available for ARQ 197 and ARQ 501, have demonstrated anti-cancer activity across multiple types of tumors.
We retain full worldwide commercial rights to ARQ 197 outside of Japan and certain other Asian countries, where we have granted commercial rights to Kyowa Hakko Kogyo Co., Ltd. ("Kyowa"). We are developing ARQ 501, ARQ 171 and ARQ 761 (a new chemical entity based on ARQ 501) pursuant to our collaboration with Hoffmann-La Roche ("Roche"). Our agreements with these partners provide for possible future milestone payments, royalties on product sales, and development funding, in addition to payments that we have already received.
Our pre-clinical programs are directed toward molecular targets that we believe play critical roles in the development of human cancers. The targets, mechanisms of action and chemistry related to compounds generated from these programs differ, offering the potential for multiple therapeutic opportunities. The most advanced of these programs are focused on the development of inhibitors of the Eg5 kinesin spindle protein and the B-RAF kinase. Toxicology testing is planned to begin in 2008 with a product candidate from the Eg5 program. Additional molecular targets are being explored in other pre-clinical programs.
Our products and research programs are based on our understanding of biological processes that lead to the proliferation and metastasis of cancer cells, combined with our ability to generate product candidates possessing certain pre-selected, drug-like properties and designed to act with specificity against cancer cells. We believe that these qualities, when present from the earliest stages of product development, increase the likelihood of producing safe, effective and marketable drugs. We believe that our combined expertise in chemistry and cancer biology differentiates us from other companies at a similar stage of evolution.
We will be focusing and advancing a significant portion of our drug discovery efforts based on our newly acquired understanding of the way ARQ 197 binds to c-Met. Insights into this novel binding mechanism form the basis of a discovery platform that we plan to leverage to generate a new type of selective kinase inhibitors. These compounds will be designed to inhibit a variety of kinases potently, selectively and without competing with ATP (adenosine triphosphate, an energy source for cells). We are currently assessing the potential of multiple kinases as targets for this drug discovery platform, and we are seeking to generate and validate compounds that inhibit these kinase targets with mechanisms similar to that of ARQ 197.
In May 2006, we terminated our chemistry services operations, which involved providing chemistry services to collaborators and customers in the pharmaceutical and biotechnology industries for their discovery programs. Our decision to terminate these operations was designed to ensure an operational focus on our oncology portfolio following our successful transition to an integrated research and
3
development company. We have maintained the know-how and trade secrets associated with our combinatorial chemistry expertise, developed and validated in the course of our chemistry services collaborations, and combined it with our biology expertise to form a powerful drug discovery engine.
CLINICAL STAGE PRODUCTS
ARQ 197
ARQ 197 is an orally available small molecule that inhibits c-Met, an enzyme belonging to a group known as receptor tyrosine kinases. C-Met is believed to play key roles in cancer cell growth, survival, angiogenesis, invasion and metastasis. The inappropriate expression of c-Met in many cancers and its role in controlling multiple signal transduction pathways involved in tumor growth and metastasis render it a highly compelling target for cancer therapy.
ARQ 197 is highly specific for c-Met and does not compete with ATP (adenosine triphosphate), an energy source for cells, for its binding to c-Met. Therefore, we believe ARQ 197 may offer an attractive therapeutic profile based on a combination of safety and anti-cancer activity. In clinical studies to date, treatment with ARQ 197 has been well tolerated and has resulted in tumor responses and prolonged stable disease across broad ranges of doses and tumors.
We initiated a Phase 1 clinical trial with ARQ 197 in late 2005. This open label, dose escalation trial included patients with multiple metastatic tumor types who had disease progression when treated with available therapy or for whom no standard systemic therapy existed. The primary objectives of the trial were to determine tolerability, safety and a recommended dosing regimen for Phase 2 trials. Additionally, the trial sought to define the pharmacokinetic and pharmacodynamic profiles of ARQ 197 and to assess its anti-tumor activity.
The trial employed a standard Phase 1 sequential dose-escalation design, with ten doses evaluated, from 10 milligrams (mg) twice daily through 180 mg twice daily. Sixty-three patients were enrolled with a broad range of solid tumors and confirmed, active metastatic disease. ARQ 197 was dose-escalated orally in two dosing schedules, the first one administered in cycles consisting of two weeks on treatment followed by one week off drug, and the second one in cycles consisting of three weeks on treatment with no time off drug.
At the 2007 Annual Meeting of the American Society of Clinical Oncology on June 2, 2007, we announced available data from this trial demonstrating that treatment with ARQ 197 was well tolerated over extended dosing periods, with approximately 60 percent of the evaluable patient population experiencing partial responses, minor responses or stable disease lasting eight weeks or longer. As measured by RECIST criteria (Response Evaluation Criteria in Solid Tumors), a partial response is at least a 30 percent decrease in target lesions, progressive disease is at least a 20 percent increase in target lesions, and stable disease is neither shrinkage sufficient to qualify for partial response nor increase sufficient to qualify for progressive disease. Minor response is not defined by RECIST criteria, but we define evidence of target lesion shrinkage of less than 30 percent as a minor response. Findings from this study resulted in a recommended Phase 2 dose of 120 mg twice daily (240 mg daily).
Treatment with ARQ 197 in this trial was well tolerated. Most adverse events were mild and transient. No drug-related grade three or four adverse events (as defined by the National Cancer Institute) were reported. No dose-limiting toxicity was observed on either dosing schedule. Substantial plasma exposure, at levels several times the predicted efficacious concentration, was maintained with
4
oral dosing. Patient compliance with dosing was high, and there were no treatment interruptions due to adverse events.
Thirty-nine patients were recruited into the intermittent, or two weeks out of three, dosing cohort. Of the 32 evaluable patients in this cohort, there were three patients with partial responses and 19 patients with stable disease. Eleven of these 19 patients with stable disease demonstrated evidence of tumor shrinkage and 8 of these patients achieved stable disease lasting six months or more. Partial responses per RECIST criteria were observed in patients with prostate, neuroendocrine and testicular tumors. Stable disease lasting more than four months was observed in a range of additional tumor types, including pancreatic, renal cell, non-small cell lung and papillary thyroid. To date, twenty-four patients have been recruited into the continuous, or three weeks out of three, dosing cohort. Of the 18 evaluable patients in this cohort 14 were evaluated as having stable disease. Future evaluations of active patients will occur periodically under the study protocol.
Data analysis from a review of patients in the Phase 1 trial with ARQ 197, based on independent readings of patients' original tumor scans, was presented at the 2007 AACR-NCI-EORTC International Conference on October 24, 2007. Forty-five patients who were evaluable and whose tumor image files were available for the independent review were included in an anti-metastatic analysis. Eighteen of 19 (94.7 percent) patients treated with ARQ 197 for 12 weeks or longer did not develop detectable new metastatic lesions. Five of 7 (71.4 percent) patients treated for 7 to 12 weeks did not develop detectable new metastatic lesions. In contrast, eleven of 19 (57.9 percent) patients treated for 6 weeks or less developed new metastatic lesions. We believe these findings support a viable hypothesis of anti-metastatic effect that merits additional investigation.
Investigators at the Royal Marsden Hospital in the United Kingdom are conducting a dose-escalation clinical trial, employing a continuous dosing schedule, designed to analyze c-Met inhibition by ARQ 197 in human tissue biopsies and to provide pharmacokinetic data. Initial findings demonstrate that a single oral 100 mg dose of ARQ 197 significantly reduced elevated levels of phospho-c-Met in tumor tissue biopsy samples. Initial pharmacokinetic data suggest that oral doses of ARQ 197 higher than the 120 mg twice daily dose currently used in our ongoing Phase 2 trials may result in increased systemic drug exposure.
In October 2007, we initiated two Phase 2 trials with ARQ 197, in MiT (Microphthalmia Transcription Factor) tumors and pancreatic adenocarcinoma. These trials are the first in a series of planned Phase 2 trials with ARQ 197, with additional planned trials to include non-small cell lung cancer (pending the successful completion of a Phase 1 trial evaluating the safety of combination therapy with ARQ 197 and erlotinib) and hormone-refractory prostate cancer. We expect to initiate these trials in 2008. We may consider additional trials based on the potential broad-spectrum impact of ARQ 197 on c-Met-mediated oncogenic processes in a variety of cancers.
MiT tumors include clear cell sarcoma (CCS), alveolar soft parts sarcoma (ASPS) and translocation-associated renal cell carcinoma (RCC). They are linked biologically through a common chromosomal abnormality that drives the over-expression of c-Met and the development of cancer. We have demonstrated the ability of ARQ 197 to kill clear cell sarcoma cellsin vitro.
5
Approximately 45 patients will be enrolled in a multi-center, single arm, two-stage trial. Eligible patients will receive 120 mg of ARQ 197 orally twice daily. During the first stage of the trial, approximately 23 patients will be treated. If a partial response or a complete response is observed in more than one patient in this stage, the study will continue to the second stage, where an additional 22 patients will be enrolled. Otherwise, the study may be stopped. The primary objective of the trial is to determine the overall response rate in patients treated with ARQ 197. Secondary objectives include the evaluation of progression-free survival time, as well as six-month and one-year overall survival in these patients.
In pancreatic cancer, between 78 and 88 percent of tumor tissue samples from patients are estimated to contain over-expressed c-Met, indicating that the c-Met signaling pathway may play a role in the development of this disease and that inhibition of this pathway may represent a viable therapeutic intervention. ARQ 197 has shown anti-cancer activity in animal models of human pancreatic cancer.
Approximately 72 patients from clinical sites in Eastern Europe will be enrolled in an open-label, randomized trial in which patients will be treated with either ARQ 197 or gemcitabine. Eligible patients will be randomized to receive either 120 mg of ARQ 197 orally twice daily or intravenous infusion of gemcitabine at a dose of 1000 mg/m2 (meter squared). Investigators will evaluate overall survival, progression-free survival and overall response rate.
ARQ 501, ARQ 171and ARQ 761: the E2F-1 Program
ARQ 501, ARQ 171, and ARQ 761 are designed to kill cancer cells selectively while sparing normal cells through the direct activation of DNA damage response/checkpoint pathways believed to be regulated by the E2F-1 regulatory protein, thereby restoring the ability of the cell to recognize DNA damage and initiating the process of apoptosis, or programmed cell death, in these cells. ARQ 501 is the first product generated in this program, while ARQ 171 and ARQ 761 are second-generation compounds.
We initiated a Phase 2 proof-of-principle program with ARQ 501 consisting of three separate clinical trials during 2006: monotherapy trials in leiomyosarcoma and in head and neck cancer, and a combination therapy trial with gemcitabine in pancreatic cancer. The primary endpoints for each of the Phase 2 trials with ARQ 501 was an objective response rate of 15 percent. Objective response rate was defined as the sum of complete responses and partial responses, and in the case of the leiomyosarcoma study, stable disease lasting more than 4 months was also considered a partial response.
Data from the pancreatic cancer trial showed a 16 percent objective response rate among evaluable patients treated with ARQ 501 and gemcitabine combination therapy, thus meeting the protocol-defined endpoint as agreed upon by Roche and us. Data from the leiomyosarcoma study showed a 17.5 percent objective response rate among evaluable patients to monotherapy treatment with ARQ 501, thus meeting the protocol-defined endpoint as agreed upon by Roche and us. Data from the head and neck cancer study showed a 2 percent objective response rate, thus failing to meet its endpoint.
We have also analyzed six-month survival data from the pancreatic cancer trial, comparing survival time among patients treated with ARQ 501 and gemcitabine with historical survival data from patients treated with gemcitabine monotherapy alone. We have shared this data with Roche under our agreement, and we expect to have analyzed twelve-month survival data from this trial in mid-2008.
6
As defined in our Roche collaboration agreement, Roche has an option to license worldwide rights for the development and commercialization of all products resulting from the E2F-1 program in the field of cancer therapy based on our delivery of a clinical data package from one of the Phase 2 monotherapy trials and the combination therapy trial with ARQ 501, as well as a recommended Phase 2 dose for a second-generation E2F-1 product.
We initiated patient recruitment in the Phase 1 trial with ARQ 171 in December 2006. ARQ 171, which is believed to have the same mechanism of action as ARQ 501, has been shown to have greater potency in preclinical tests. In November 2007, we announced that symptomatic and asymptomatic QTc prolongation, a potentially dangerous cardiac rhythm abnormality, has been observed in this trial. These observations were made based on electrocardiograms (EKGs) from patients who received doses of ARQ 171 from 380 to 760 mg/m2. We subsequently reviewed EKGs from all other patients treated with ARQ 171. We are completing this review and analysis of data, and we have decided not to enroll additional patients until we have fully assessed these observations.
We are also proceeding with the pre-clinical development of another second-generation E2F-1 compound, ARQ 761. Progress in the pre-clinical and Phase 1 clinical development of ARQ 761, as well as clarity regarding the status of ARQ 171, will inform our decisions relating to the choice of the second-generation E2F-1 compound for additional clinical development and the determination of the clinical pathway and indications for this compound. We believe this information will guide Roche's decision regarding their further participation in the E2F-1 program.
PRECLINICAL AND RESEARCH PIPELINE
We have a number of pre-clinical and research-stage programs based on product candidates directed toward molecular targets that we believe play critical roles in the development of human cancers and therefore may be attractive points for therapeutic intervention. The targets, mechanisms of action and chemistry related to compounds generated from these programs differ, offering the potential for multiple therapeutic opportunities. Such intervention may be designed to activate or to inhibit targeted molecules and cell signaling pathways, depending on their roles in biological processes related to cancer.
The targets of our most advanced research programs include the Eg5 kinesin spindle protein and the B-RAF Kinase. We have identified a candidate from our Eg5 program for which we plan to begin GLP toxicology testing in 2008. Pending the successful completion of such testing, we plan to file an Investigational New Drug application (IND) for this compound in late 2008.
KYOWA HAKKO KOGYO CO., LTD. ALLIANCE
On April 27, 2007, we announced an exclusive license agreement with Kyowa to develop and commercialize ARQ 197 in Japan and parts of Asia. The agreement includes $123 million in upfront and potential development milestone payments from Kyowa to ArQule, including $30 million in upfront licensing payments that we received in 2007. In addition, the agreement includes undisclosed sales milestone payments.
Upon commercialization, ArQule will receive double-digit royalties from Kyowa on net sales of ARQ 197. Kyowa will be responsible for clinical development costs and commercialization of the compound in certain Asian countries, consisting of Japan, China (including Hong Kong), South Korea and Taiwan.
In February 2008, we received a $3 million milestone payment from Kyowa marking the initiation by Kyowa of a Phase 1, dose escalation trial in Japan with ARQ 197. This payment was made under the terms of the exclusive license agreement signed between the two companies.
7
HOFFMANN-LA ROCHE ALLIANCE
On April 2, 2004, we announced an alliance with Roche to discover and develop drug candidates targeting the E2F biological pathway. Under the terms of the agreement, Roche obtained an option to license drugs resulting from our E2F program in the field of cancer therapy. Roche provided immediate research funding of $15 million and financial support for ongoing research and development. Through December 31, 2007, we have received approximately $31.8 million of $33 million in research and development support from Roche under this agreement.
We are responsible for advancing drug candidates from early stage development into Phase 2 trials. Roche has an option to license worldwide rights for the development and commercialization of products resulting from the E2F-1 program based on a clinical data package from one of the Phase 2 ARQ 501 monotherapy trials and the Phase 2 ARQ 501-gemcitabine combination therapy trial, as well as from a Phase 1 trial with a second-generation E2F compound such as ARQ 171 or ARQ 761. In order to license these rights, Roche must pay an option fee. We believe our completed analysis of QTc prolongation observed in the Phase 1 trial with ARQ 171 and our subsequent decision regarding further patient enrollment, as well as progress in the pre-clinical and Phase 1 clinical development of ARQ 761, will help guide Roche's decision regarding their further participation in the E2F-1 program.
BUSINESS STRATEGY
2008 Operational Goals
During 2008, we will invest in the clinical development of ARQ 197 through the continuing enrollment of patients in our ongoing Phase 2 trials in MiT tumors and pancreatic cancer. In addition, we have initiated a Phase 1 trial in non-small cell lung cancer evaluating the safety of combination therapy with ARQ 197 and erlotinib. Pending the successful completion of this trial, we plan to initiate a Phase 2 trial in this indication. Furthermore, we are exploring trial designs in prostate cancer that are intended to lead to the initiation of a Phase 2 trial late in the year.
With respect to our E2F-1 program, we will seek to identify a compound among ARQ 501, ARQ 171 and ARQ 761 for further clinical testing. We will continue to provide information related to the development of these compounds to Roche as part of our contractual obligation.
We plan to expand our clinical-stage pipeline pending the successful completion of GLP toxicology testing and IND submission with a product candidate from our Eg5 program. Our earlier-stage drug discovery efforts will be focused primarily upon the further development of our emerging kinase platform, leading to the generation of product candidates directed toward validated kinase targets.
Drug Discovery And Development Strategy
Our strategy for developing the Company and specific compounds into commercial products has the following components:
Grow organically and through business development. We plan to grow both organically and through business development activities. Organic growth will be based on our advancement of internally defined product candidates from pre-clinical through clinical development. These candidates will be based upon scientific platforms within the Company and targeted toward molecules with validated roles in oncogenic processes. Their design will be informed by our combined expertise in chemistry and cancer biology that we believe differentiates us from our competitors. Business development activities offer the opportunity to leverage the capabilities of a potential partner with resources complementary to ours in drug discovery and development.
We will be focusing and advancing a significant portion of our drug discovery efforts based on our newly acquired understanding of the way ARQ 197 binds to c-Met. Insights into this novel binding
8
mechanism form the basis of a discovery platform that we plan to leverage to generate a new type of selective kinase inhibitors. These compounds will be designed to inhibit a variety of kinases potently, selectively and without competing with ATP (adenosine triphosphate, an energy source for cells). We are currently assessing the potential of multiple kinases as targets for this drug discovery platform, and we are seeking to generate and validate compounds that inhibit these kinase targets with mechanisms similar to that of ARQ 197.
Simultaneously, we will consider a broad range of business development activities potentially encompassing product and technology acquisitions, licensing agreements and corporate combinations that will help expand the overall scope of product development and potentially accelerate the implementation of a commercialization infrastructure. We may also continue to invest in technology and personnel to enhance or expand our capabilities in drug discovery.
Focus on cancer, a market with a large unmet need. Cancer is the second most common cause of death in the western world. According to the American Cancer Society, approximately 560,000 cancer-related deaths were projected to occur and 1.4 million new cases were projected to be diagnosed in the U.S. during 2008. Demographic trends and improved screening are expected to increase the rate of cancer diagnoses, as 85 percent of cancers occur in the over-55 year old population. The National Cancer Institute estimates that the median age of cancer patients at death was 73, and the overall cost of cancer in the U.S. during 2007 was $219 billion.
Medical therapy for cancer has historically included surgery, cytotoxic (poisonous to cells) chemotherapy and radiation. While chemotherapies have evolved, many are still harmful to all rapidly dividing cells. More recently, a number of alternative therapies that are target specific have been introduced. We believe that targeted approaches to treating cancer, such as those we are pursuing, have the potential to be more selective for cancer cells than traditional chemotherapies and applicable to a broad spectrum of cancers.
Take advantage of available accelerated regulatory approval strategies as appropriate. Cancer compounds are eligible for potential accelerated regulatory approval and we will pursue opportunities for such approval as appropriate. Once on the market, with supportive data the agents may be approved for additional indications.
Benefit from the resources and strengths of collaborators. In April 2004, we entered into an agreement with Roche in which Roche acquired an option for certain compounds in our E2F program and to the E2F program in total for oncology indications. In April 2007, we announced that we entered into an exclusive license agreement with Kyowa to develop and commercialize ARQ 197 in Japan and parts of Asia. We benefit from the resources and expertise of these partners, and we intend to pursue future partnership arrangements as appropriate when the capabilities of a potential partner complement our strengths in oncology drug discovery and development.
Continue to exploit our strength in chemistry for oncology drug discovery and development. We have developed a chemistry-based drug discovery technology platform designed to create small molecules that possess drug-like characteristics. We believe that identifying drug-like characteristics early in discovery increases the likelihood that small molecules reaching preclinical development will have a greater potential to become medicines. Without such a technology platform, the traditional approach is to develop small molecules that have demonstrated activity toward biological targets, with little regard for whether the molecules otherwise would make good medicines. In our view, a drug that has the best set of drug-like characteristics for its indication (i.e., one that is the most effective and has the fewest side effects) will ultimately generate the most revenue in its category, even if it is not the first to become available on the market.
9
EXIT FROM CHEMISTRY SERVICES OPERATIONS
In 2005, we announced our plan to exit our chemistry services operations, which were the previous focus of the Company. These operations involved providing chemistry services to collaborators and customers for their discovery programs. Our decision, which followed our successful transition to an integrated research and development company was designed to ensure an operational focus on developing our oncology portfolio.
We terminated these operations, which included a major collaboration with Pfizer. Inc. ("Pfizer"), in May 2006. We had previously received notice from Pfizer on December 2, 2005 that, pursuant to the terms of our Collaboration Agreement with Pfizer dated December 19, 2001, Pfizer elected to terminate the agreement, effective May 22, 2006.
Following our decision to exit the chemistry services operations, we entered into agreements to sell and to license non-exclusively the majority of certain physical and intellectual property assets related to the chemistry services operations to Shanghai DESANO Pharmaceutical Holding Co. Ltd ("DESANO"). A purchase and sale agreement was executed, and the sale and licensing transactions were consummated in the fourth quarter of 2006. The assets conveyed were primarily chemical compound production and analysis equipment, production consumables and source code segments of production and information management software. The purchase price was $1,250,000. Chemistry services equipment not purchased by DESANO was sold at auction, generating net proceeds of $52,000.
Our collaboration with Pfizer was our largest chemistry services collaboration and accounted for virtually all of our compound development revenue in 2005 and 2006. Since the inception of this relationship in 1999, we have produced collections of chemical compounds exclusively for Pfizer using our automated high throughput system. As of December 31, 2006, we had received $289 million from Pfizer under this collaboration. Pfizer has made equity investments in our company of $10 million in 2001 and $8 million in 2003, based on the achievement of certain delivery milestones.
CHEMISTRY-BASED COLLABORATIONS (DISCONTINUED OPERATIONS)
We have received milestone payments from certain collaborators related to compounds we provided to them as part of our discontinued chemistry services operations. Should any of these compounds proceed further in the clinic, or become drugs, we will be eligible to receive various further milestone payments and royalties under the terms of the agreements.
BOSTON BIOMEDICAL, INC.
In January 2007, we entered into a $5.0 million, eight-month sponsored research agreement with the newly established Boston Biomedical, Inc. ("BBI"), an independent corporation. Dr. Chiang J. Li, our former chief scientific officer, transitioned to the position of chief executive officer of BBI from his previous role as chief scientific officer of ArQule. Our agreement with BBI included their completion of scientific research that included a number ofin vivo andin vitro studies, reports and publications related to mechanisms of action and biomarkers for our lead clinical-stage products. We retain all intellectual property and technology rights related to research conducted by BBI employees under their contract. ArQule does not have an equity position in BBI or any continuing interests other than that covered by the research agreement.
PATENTS AND PROPRIETY RIGHTS
We believe that patent and trade secret protection is crucial to our business and that our future will depend in part on our ability to obtain patents, maintain trade secret protection and operate without infringing the proprietary rights of others, both in the U.S. and other countries. As of February 12, 2008, we had 29 issued or allowed U.S. utility patents, one issued U.S. design patent,
10
numerous granted foreign patents, and numerous patent applications in the U.S. and other countries. While many patent applications have been filed in the U.S. and other countries with respect to our cancer programs, the majority of these have not yet been issued or allowed. The patent positions of companies in the biotechnology industry and the pharmaceutical industry are highly uncertain and involve complex legal and factual questions. Therefore, we cannot predict the breadth of claims, if any, that may be allowed under any of our patent applications, or the enforceability of any of our issued patents.
As needed, we obtain rights under patents owned by other parties through licenses. We have several exclusive and nonexclusive technology licenses from certain institutions which support our research programs. We anticipate that we will continue to seek licenses from universities and others where applicable technology complements our research and development efforts.
Our patent portfolio for ARQ 501 includes patents and pending patent applications in the U.S. and foreign countries. We have issued patents and pending applications that cover the formulations and syntheses of ARQ 501. For the uses of ARQ 501 in the treatment of cancer, we have patents and pending patent applications and have licensed rights under issued patents and pending applications from the Dana-Farber Cancer Institute. ARQ 501 is derived from a naturally occurring substance, and we do not have patents that cover the composition of this compound.
With respect to ARQ 197, we have pending U.S. and Patent Cooperation Treaty ("PCT") patent applications that cover the composition of this compound, pharmaceutical compositions containing this compound, and the uses of this compound in the treatment of cancer.
With respect to ARQ 171, we have pending U.S. and PCT patent applications that cover the composition of this compound, pharmaceutical compositions containing this compound, and the uses of this compound in the treatment of cancer.
Patents extend for varying periods according to the date of patent filing or grant and the legal term of patents in the various countries where patent protection is obtained. The actual protection afforded by a patent, which can vary from country to country, depends on the type of patent, the scope of its coverage and the availability of legal remedies in the country.
In an effort to maintain the confidentiality and ownership of our trade secrets and proprietary information, we require all of our employees and consultants to sign confidentiality agreements. Employees and consultants involved in scientific and technical endeavors also sign invention assignment agreements. We intend these confidentiality and assignment agreements to protect our proprietary information by controlling the disclosure and use of technology to which we have rights. These agreements also provide that we will own all the proprietary technology developed at ArQule or developed using our resources.
"ArQule", the ArQule logo, "Directed Array", "Mapping Array" and "AMAP" are trademarks of ArQule that are registered or entitled to be registered in the U.S. Patent and Trademark Office. The terms "AMAP", "ArQule Reactor", "Compass Array", "Custom Array", "MapMaker", "Optimal Chemical Entities", "OCEs", "Parallel Track", and "PrepQule" are trademarks of ArQule. The term "Activated Checkpoint Therapy" is a registered trademark of ArQule.
11
COMPETITION
The pharmaceutical and biotechnology industries are highly competitive. We face intense competition from organizations such as large pharmaceutical companies, biotechnology companies and academic and research organizations. The major pharmaceutical organizations competing with us have greater capital resources, larger overall research and development staff and facilities and considerably more experience in drug development and commercialization. Biotechnology companies competing with us may have these advantages as well. In addition to competition for collaborators and investors, these companies and institutions also compete with us in recruiting and retaining highly qualified scientific and management personnel.
With respect to our cancer drug discovery and development programs, other companies have potential drugs in preclinical and clinical trials that may result in effective, commercially successful treatments for the same cancers we target. We also experience competition for qualified subjects for our clinical studies of our drug candidates, which may result in longer and more costly clinical trials. In the area of small molecule anti-cancer therapeutics, we have identified a number of companies that have clinical development programs and focused research and development in small molecule approaches to cancer, including: Ariad Pharmaceuticals, Inc.; Array BioPharma Inc.; Cell Therapeutics, Inc.; Curis, Inc.; Exelixis, Inc.; Idera Pharmaceuticals, Inc.; Infinity Pharmaceuticals, Inc.; Kosan Biosciences, Inc.; Onyx Pharmaceuticals, Inc.; OSI Pharmaceuticals, Inc.; Oxigene, Inc.; Pharmacopeia; Telik, Inc.; and Vion Pharmaceuticals, Inc.
With respect to ARQ 197, we are aware of a number of companies that are or may be pursuing approaches to c-Met inhibition, including Exelixis, Inc., Bristol-Myers Squibb Company, Compugen Ltd., Amgen Inc., Pfizer, Merck & Co., Inc., Methylgene Inc., SGX Inc. and Supergen Inc.
We face competition in other areas of our business, including advancing a discovery and development portfolio of anti-cancer candidates that are selective for cancer cells and applicable across a broad spectrum of cancer types, and securing partners to co-develop and advance our drug candidates through later-stage clinical trials and beyond.
There can be no assurance that our competitors will not develop more effective or more affordable products or technology, or achieve earlier product development and commercialization than ArQule, thus rendering our technologies and/or products obsolete, uncompetitive or uneconomical.
GOVERNMENT REGULATION
Virtually all pharmaceutical and biotechnology products that our collaborative partners or we develop will require regulatory approval by governmental agencies prior to commercialization. The nature and the extent to which these regulations apply vary depending on the nature of the products. In particular, human pharmaceutical products are subject to rigorous preclinical and clinical testing and other approval procedures by the U.S. Food and Drug Administration ("FDA"). Various federal and, in some cases, state statutes and regulations also govern or influence the manufacturing, safety, labeling, storage, record keeping and marketing of these products. The process of obtaining these approvals and the subsequent compliance with appropriate federal and state statutes and regulations are time consuming and require substantial resources, and the outcome of these regulatory activities is uncertain.
Generally, in order to gain FDA approval, a company first must conduct preclinical studies in the laboratory and in animal models to gain preliminary information on a compound's activity and to identify potential safety problems. Preclinical studies must be conducted in accordance with FDA regulations. The results of these studies are submitted as a part of an IND application that the FDA must review before human clinical trials of an investigational drug can start. If the FDA does not respond with any questions, a drug developer can commence clinical trials thirty days after the submission of an IND.
12
In order to eventually commercialize any products, we or our collaborator will be required to initiate and oversee clinical studies under an IND to demonstrate the safety and efficacy that are necessary to obtain FDA marketing approval. Clinical trials are normally done in three phases and generally take several years, but may take longer to complete. Furthermore, the FDA may suspend clinical trials at any time if the FDA believes that the subjects participating in trials are being exposed to unacceptable risks or if the FDA finds deficiencies in the conduct of the trials or other problems with our product under development.
After completion of clinical trials of a new product, FDA marketing approval must be obtained. If the product is classified as a new pharmaceutical, our collaborator or we will be required to file a New Drug Application ("NDA"), and receive approval before commercial marketing of the drug. The NDA contains, among other things, the results of the non-clinical and clinical testing of the drug. NDAs submitted to the FDA can take several years to obtain approval and the FDA is not obligated to grant approval at all. FDA can condition NDA approval on the conduct of costly post-marketing follow-up studies or can place restrictions on the sale or marketing of the drug in order to manage risks.
Even if FDA regulatory clearances are obtained, a marketed product is subject to continual review and ongoing regulatory obligations. If and when the FDA approves any of our or our collaborators' products under development, the manufacture and marketing of these products will be subject to continuing regulation, including compliance with current Good Manufacturing Practices ("cGMP"), adverse event reporting requirements and prohibitions on promoting a product for unapproved uses or making false or misleading statements or omissions with respect to a drug in advertising or promotion. Later discovery of previously unknown problems or failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product or withdrawal of the product from the market as well as possible civil or criminal sanctions. Various federal and, in some cases, state statutes and regulations also govern or influence the manufacturing, safety, labeling, storage, record keeping and marketing of pharmaceutical products.
For marketing outside the U.S., we or our partners will be subject to foreign regulatory requirements governing human clinical trials, marketing approval and post-marketing activities for pharmaceutical products and biologics. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary widely from country to country.
EMPLOYEES
As of February 1, 2008, we employ 113 people in Woburn, Massachusetts. Of that total, 81 are engaged in research and development and 32 in general and administration, and 43 hold Ph.D.s and 23 hold Masters in the Sciences.
CERTAIN OTHER INFORMATION
We file annual, quarterly and current reports, proxy statements and other information with the Securities and Exchange Commission ("SEC"). You may read and copy any document we file at the SEC's Public Reference Room at 100 F Street, N.E., Washington D.C. 20549. Please call the SEC at 1-800-SEC-0330 for further information on the public reference room. The SEC maintains a website athttp://www.sec.gov that contains reports, proxy and information statements and other information concerning filers. We also maintain a web site athttp://www.ArQule.com that provides additional information about our company and links to documents we file with the SEC. The Company's Corporate Governance Guidelines; the charters of the Audit Committee, the Compensation Committee, and the Nominating and Governance Committee; and the Code of Conduct are also available on the Company's website.
13
EXECUTIVE OFFICERS
Set forth below is certain information regarding our current executive officers, including their respective ages, as of February 1, 2008. As announced on January 7, 2008, we have initiated a search for a successor to Dr. Stephen A. Hill, current president and chief executive officer, who has communicated to the board of directors his intention to leave the Company in the first quarter of 2008. Peter S. Lawrence, who was recently appointed to the position of chief operating officer, has assumed operational responsibility for leadership of the Company. A search committee has been appointed by the board and will consider Mr. Lawrence and external candidates for the chief executive officer position.
NAME
| | AGE
| | POSITION
|
|---|
| Dr. Stephen A. Hill | | 49 | | President, Chief Executive Officer and a Director |
| Peter S. Lawrence | | 44 | | Chief Operating Officer |
| Dr. Nigel J. Rulewski | | 53 | | Chief Medical Officer |
Stephen A. Hill, B.M. B.Ch., M.A., F.R.C.S.
President and Chief Executive Officer
Dr. Hill has served as ArQule's President and CEO since April 1999. Before joining ArQule, Dr. Hill was the Head of global Drug Development at F. Hoffmann-La Roche Ltd. from 1997-1999. Dr. Hill joined Roche in 1989 as Medical Adviser to Roche Products in the United Kingdom. He held several senior positions there that included Medical Director, responsible for clinical trials of compounds across a broad range of therapeutic areas, such as CNS, HIV, cardiovascular, metabolic and oncology products. Subsequently, he served as Head of International Drug Regulatory Affairs at Roche headquarters in Basel, Switzerland, where he led the regulatory submissions for seven major new chemical entities. Dr. Hill also was a member of Roche's Portfolio Management, Research, Development and Pharmaceutical Division Executive Boards. Prior to Roche, Dr. Hill served seven years with the National Health Service in the United Kingdom in General and Orthopedic Surgery. Dr. Hill is a Fellow of the Royal College of Surgeons of England and holds his scientific and medical degrees from St. Catherine's College at Oxford University.
Peter S. Lawrence
Executive Vice President and Chief Operating Officer
Mr. Lawrence joined ArQule in April 2006 from Pod Holding Ltd., an international venture capital firm which he co-founded in 2001 and where he most recently served as general partner. He helped drive the strategic growth of that firm, including deal sourcing and structuring, syndication and business expansion activities. Mr. Lawrence served as lead partner on investment activities for a number of companies, including the cancer company, Pintex Pharmaceuticals. Previously, Mr. Lawrence was an attorney and partner at Mintz, Levin, Cohn, Ferris Glovsky and Popeo, P.C., from 1991 to 2001. At Mintz Levin, he served as external corporate counsel to public and private companies, managed a transactional legal practice and provided strategic guidance to clients through periods of rapid growth and transformative corporate events. His public financing experiences include the initial public offering and numerous financings for America Online Inc. (AOL), as well as public financings for Biogen, Human Genome Sciences, Hybridon and many other companies. He worked on numerous mergers and acquisitions, including Roche/Compuchem, AOL/Time Warner, Steinway Piano, DEC/Intel, and Mitotix/GPC Biotech. Mr. Lawrence worked at Gaston & Snow from 1989 to 1991 in the firm's Corporate Law Department. He holds a Bachelor's degree from Amherst College and a J.D. from Boston University School of Law.
14
Nigel J. Rulewski, M.B., B.S., D.R.C.O.G., D.C.H.
Chief Medical Officer
Dr. Rulewski joined ArQule in August 2006 from BioAccelerate Holding Inc., a pharmaceutical development organization, where he was Senior Vice President. He brings to ArQule more than two decades of experience in clinical research, product development, regulatory affairs, commercialization, corporate planning and licensing activities. At BioAccelerate, Dr. Rulewski was responsible for all aspects of licensing and product development in the oncology area. Previously, as vice president, medical affairs and chief medical officer at Astra USA, Dr. Rulewski negotiated the approval of eight New Drug Applications (NDAs) and had responsibility for all issues pertaining to drug development in the U.S., including interactions with the U.S. Food and Drug Administration (FDA). He previously served as medical director at Serono Laboratories, where he managed three research groups, medical information and drug safety. He was also associate medical director and medical director, international operations, at Fisons Corporation. Dr. Rulewski practiced medicine in the United Kingdom following his graduation from St. Bartholomew's Hospital Medical School, University of London.
15
ITEM 1A. RISK FACTORS
RISKS RELATING TO OUR INDUSTRY AND BUSINESS STRATEGY
Development of our products is at an early stage and is based on scientific platforms that are unproven. We may not successfully develop a drug candidate that becomes a commercially viable drug.
The discovery and development of drugs is inherently risky and involves a high rate of failure. Discovery and development of commercial drugs are relatively new to us. Our drug candidates and drug research programs are in early stages and require significant, time-consuming and costly research and development, testing and regulatory approvals.
One of our clinical-stage product candidates, ARQ 197, is based on inhibition of the c-Met receptor tyrosine kinase. Two of our other product candidates in clinical trials, ARQ 501 and ARQ 171, are based on the DNA damage response mechanism mediated by the E2F-1 transcription factor. Although drugs have been approved that inhibit the activity of kinases and other enzymes, to our knowledge, no company has received regulatory approval for a drug based on an approach similar to our c-Metplatform or to our E2F-1 platform. Our approaches and scientific platforms may not lead to the development of approvable or marketable drugs.
In addition to our clinical-stage programs, we have a limited number of pre-clinical and research-stage programs in our pipeline. Our viability as a company depends, in part, on our ability to continue to create drug candidates for ourselves and our collaborators. Numerous significant factors will affect the success of our drug research and development efforts, including the biology and chemistry complexity involved, availability of appropriate technologies, the uncertainty of the scientific process and the capabilities and performance of our scientists. Our research and development capabilities may not be adequate to develop additional, viable drug candidates.
We must show the safety and efficacy of our product candidates through expensive, time consuming preclinical testing and clinical trials, the results of which are uncertain and governed by exacting regulations.
Our product candidates are in clinical or preclinical stages of development and may not prove to be sufficiently safe or effective in more advanced human clinical trials. We will need to conduct extensive further testing of all of our product candidates, expend significant additional resources and possibly partner with another company or companies to realize commercial value from any of our product candidates.
Before obtaining regulatory approvals for the commercial sale of our products, we must demonstrate through preclinical studies (laboratory or animal testing) and clinical trials (human testing) that our proposed products are safe and effective for use in each target indication. This testing is expensive and time-consuming, and failure can occur at any stage. If we terminate a preclinical or clinical program, we will have expended resources in an effort that will not provide a return on our investment and missed the opportunity to have allocated those resources to potentially more productive uses.
Clinical trials must meet FDA and foreign regulatory requirements. We have limited experience in designing, conducting and managing the preclinical studies and clinical trials necessary to obtain regulatory approval for our product candidates in any country. We or our collaborative partners may encounter problems in clinical trials that may cause us or the FDA or foreign regulatory agencies to delay, suspend or terminate our clinical trials at any phase. These problems could include our inability to manufacture or obtain sufficient quantities of materials produced in accordance with current Good Manufacturing Practice, or cGMP, for use in our clinical trials, conduct clinical trials at our preferred sites, enroll a sufficient number of patients for our clinical trials at one or more sites, or begin or successfully complete clinical trials in a timely fashion, if at all. Furthermore, we, the FDA or foreign
16
regulatory agencies may suspend clinical trials of our product candidates at any time if we or they believe the subjects participating in the trials are being exposed to unacceptable health risks as a result of adverse events occurring in our trials or if we or they find deficiencies in the clinical trial process or conduct of the investigation.
Acceptable results from initial preclinical studies and clinical trials of products under development are not necessarily indicative of results that will be obtained from subsequent or more extensive preclinical studies and clinical testing in humans. Clinical trials may not demonstrate sufficient safety and efficacy to obtain the required regulatory approvals or result in marketable products. Failure to adequately demonstrate the safety and efficacy of a product under development will delay and could prevent its regulatory approval.
A number of companies in the pharmaceutical industry, including biotechnology companies, have suffered significant setbacks in advanced clinical trials, even after generating promising results in earlier trials.
Though it is part of our strategy to pursue clinical development to take advantage of available accelerated regulatory approval processes, there is no guarantee that our product candidates will show the evidence predictive of clinical benefit necessary to qualify for such regulatory treatment.
Delays in clinical testing could result in increased costs to us and delay our ability to obtain regulatory approval and commercialize our product candidates.
Clinical trials typically take several years to complete. The duration and cost of clinical trials will vary greatly depending on the nature, complexity, and intended use of the drug being tested. We may not complete clinical testing within the time frame we have planned, or at all. At any time, a clinical trial can be placed on "clinical hold" or temporarily or permanently stopped for a variety of reasons, principally for safety concerns. We may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent us from receiving regulatory approval or commercializing our product candidates, including the following:
- •
- our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical and/or pre-clinical testing or to abandon programs;
- •
- trial results may not meet the level of statistical significance required by the FDA or other regulatory agencies;
- •
- enrollment in our clinical trials for our product candidates may be slower than we anticipate, resulting in significant delays;
- •
- we, or regulators, may suspend or terminate our clinical trials if the participating patients are being exposed to unacceptable health risks;
- •
- the effects of our product candidates on patients may not be the desired effects or may include undesirable side effects or other characteristics that may delay or preclude regulatory approval or limit their commercial use, if approved; and
- •
- the FDA or other regulatory agencies may lack experience in evaluating the safety and efficacy of drugs based on our c-Met or E2F-1 platforms, which could lengthen the regulatory review process.
Completion and duration of clinical trials depends on, among other things, our ability to enroll a sufficient number of patients, which is a function of many factors, including:
- •
- the incidence among the general population of diseases which contain therapeutic endpoints chosen for evaluation;
17
- •
- the eligibility criteria defined in the protocol;
- •
- the size of the patient population required for analysis of the trial's therapeutic endpoints;
- •
- our ability to recruit clinical trial investigators and sites with the appropriate competencies and experience;
- •
- our ability to obtain and maintain patient consents; and
- •
- competition for patients by clinical trial programs for other treatments.
We have limited clinical development and commercialization experience.
We have limited experience conducting clinical trials and have never obtained regulatory approvals for any drug. To date, we have filed three IND applications, and we have initiated five Phase 1 clinical trials, and five Phase 2 clinical trials. We have not conducted a Phase 3, or pivotal, clinical trial, filed an NDA or commercialized a drug. We have no experience as a company in the sale, marketing or distribution of pharmaceutical products and do not currently have a sales and marketing organization. Developing commercialization capabilities will be expensive and time-consuming, and could delay any product launch. We may not be able to develop a successful commercial organization. To the extent we are unable or determine not to acquire these resources internally, we will be forced to rely on third-party clinical investigators, clinical research organizations or marketing organizations. If we were unable to establish adequate capabilities independently or with others, our drug development and commercialization efforts could fail, and we may be unable to generate product revenues.
If our drug discovery and development programs do not progress as anticipated, our revenue and stock price could be negatively impacted.
We estimate the timing of a variety of preclinical, clinical, regulatory and other milestones for planning purposes, including when a drug candidate is expected to enter clinical trials, how soon patients will be recruited and enrolled in these trials, when a clinical trial will be completed and when an application for regulatory approval will be filed. We base our estimates on facts that are currently known to us and on a variety of assumptions, many of which are beyond our control. If we or our collaborators do not achieve milestones when anticipated, we will not receive the corresponding revenue, and our stock price could decline. In addition, our research and clinical testing may be delayed or abandoned if we or our competitors subsequently discover other compounds that we believe show significantly improved safety or efficacy compared to our product candidates, which could limit our ability to generate revenues, cause us to incur additional expense and cause the market price of our common stock to decline significantly.
RISKS RELATED TO OUR FINANCIAL CONDITION
We have incurred significant losses since our inception and anticipate that we will incur significant continued losses for the next several years, and our future profitability is uncertain.
From our inception in 1993 through December 31, 2007, we have incurred cumulative losses of approximately $282 million. These losses have resulted principally from the costs of our research activities, acquisitions, enhancements to our technology and early-stage clinical trials. In the past we derived our revenue primarily from license and technology transfer fees and payments for compound deliveries associated with our discontinued chemistry services operations; research and development funding paid under our agreements with collaboration partners; and to a limited extent, milestone payments.
We expect our expenses to increase significantly as we spend additional amounts to fund research, development, clinical testing and commercialization of our drug candidates. We currently have three
18
product candidates in various stages of clinical development, and we anticipate filing an IND application for an additional product candidate within the next twelve months. As a result, we will need to generate significant additional revenues to achieve profitability.
To attain profitability, we will need to develop clinical products successfully and market and sell them effectively, either by ourselves or with collaborators. We have never generated revenue from the commercialization of our product candidates, and there is no guarantee that we will be able to do so. Even if were to generate product revenues and achieve profitability, we may not be able to maintain or increase profitability. Because of the numerous risks and uncertainties associated with the development of drugs, we are unable to predict the extent of any future losses or when we will become profitable, if at all. If we fail to become profitable, or if we are unable to fund our continuing losses, we may be unable to continue our business.
We may need substantial additional funding and may be unable to raise capital when needed, or on terms favorable to us, which could force us to delay, reduce or eliminate our drug discovery, product development and commercialization activities.
Developing drugs, conducting clinical trials, and commercializing products are expensive. Our future funding requirements will depend on many factors, including:
- •
- the progress and cost of our ongoing and future collaborative and independent clinical trials and other research and development activities and our ability to share such costs of our clinical development efforts with third parties;
- •
- the costs and timing of obtaining regulatory approvals;
- •
- the costs of filing, prosecuting, maintaining, defending and enforcing any patent applications, claims, patents and other intellectual property rights;
- •
- the cost and timing of securing manufacturing capabilities for our clinical product candidates and commercial products, if any;
- •
- the costs and timing of commercializing our product candidates, including establishing or contracting for sales, marketing and distribution capabilities, if any such candidates receive regulatory approval for commercial sale; and
- •
- the costs of any acquisitions of or investments in businesses, products and technologies.
We may seek the capital necessary to fund our operations through public or private equity offerings, debt financings, and collaboration and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, our stockholders' ownership interests will be diluted and the terms of such securities may include liquidation or other preferences that adversely affect our stockholders' rights. Other debt-financing arrangements may require us to pledge certain assets and enter into covenants that would restrict certain business activities or our ability to incur further indebtedness. If we raise additional funds through collaboration and licensing arrangements with third parties, we may have to relinquish rights to certain product candidates that we might otherwise seek to develop or commercialize independently, or grant licenses on terms that are not favorable to us. There can be no assurance that sufficient funds will be available to us when required, on satisfactory terms, or at all. If we are unable to obtain additional funds when needed, we may have to delay, reduce the scope of or eliminate some of our development and commercialization programs, or obtain funds through other arrangements on unattractive terms, which could prevent us from successfully executing our business strategy.
19
Funds associated with certain of our auction rate securities may not be accessible for an undetermined period of time and our auction rate securities may experience an other than temporary decline in value, which would adversely affect our statement of operations.
Our marketable securities portfolio includes auction rate securities that are structured with short-term interest rate reset dates of generally less than ninety days, but with contractual maturities that can be well in excess of ten years. At the end of each reset period, which occurs every seven to twenty-eight days, investors can sell or continue to hold the securities at par. In the first quarter of 2008, certain auction rate securities failed auction due to sell orders exceeding buy orders. We will review our portfolio at the end of the quarter ending March 31, 2008, taking into consideration other-than-temporary impairment factors, to conclude whether ArQule has an impairment. If we determine there is an impairment, it would be recorded in the quarter ended March 31, 2008 within other comprehensive loss if related to a temporary impairment, and in the statement of operations if related to an other than temporary impairment related to these auction rate securities. Our marketable securities portfolio, which totals $124.2 million at December 31, 2007, includes $92.6 million (at cost) invested in auction rate securities, $67.7 million (at cost) are associated with auctions that failed subsequent to February 12, 2008. Subsequent to December 31, 2007, ArQule was able to sell at par $22.4 million of auction rate securities that were held at December 31, 2007. In addition, certain of our auction rate securities totaling $3.8 million were called for redemption and accordingly we expect to receive payment by the end of March 2008. The funds associated with failed auctions will not be accessible until a successful auction occurs, a buyer is found outside of the auction process or the underlying securities have matured. If conditions in the credit markets deteriorate further causing additional auctions to fail, the funds associated with these auction rate securities may also not be accessible for an undetermined period of time.
RISKS RELATED TO REGULATORY APPROVAL
Our product candidates are subject to a lengthy and uncertain regulatory process that may not result in the necessary regulatory approvals, which would adversely affect our ability to commercialize products. We have only limited experience in regulatory affairs.
Our product candidates, as well as the activities associated with their research, development and commercialization, are subject to extensive regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in other countries. Failure to obtain regulatory approval for a product candidate would prevent us from commercializing that product candidate. We have not received regulatory approval to market any of our product candidates in any jurisdiction and have only limited experience in preparing and filing the applications necessary to gain regulatory approvals. The process of obtaining regulatory approvals is expensive, often takes many years, if approval is obtained at all, and can vary substantially based upon the type, complexity and novelty of the product candidates involved.
Before a new drug application can be filed with the FDA, the product candidate must undergo extensive clinical trials. Any clinical trial may fail to produce results satisfactory to the FDA, typically for lack of safety or efficacy. For example, the FDA could determine that the design of a clinical trial is inadequate to produce reliable results. The regulatory process also requires preclinical testing, and data obtained from preclinical and clinical activities are susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. In addition, the results of later trials may not confirm the positive results of earlier preclinical studies or trials. Delays or rejections may be encountered based upon changes in regulatory policy for product approval during the period of product development and regulatory agency review. Changes in regulatory approval policy, regulations or statutes or the process for regulatory review during the development or approval periods of our product candidates may cause delays in the approval or rejection of an application. We are currently in Phase 2 clinical testing of ARQ 501 and Phases 1 and 2 clinical testing of ARQ 197 and Phase 1 testing of ARQ 171. We have
20
never conducted a Phase 3, or pivotal, clinical trial, nor have we filed or prosecuted the applications necessary to gain regulatory approvals.
Even if our drug candidates obtain regulatory approval, we and our collaborators will be subject to ongoing government regulation.
Even if regulatory authorities approve any of our drug candidates, the manufacture, marketing and sale of these drugs will be subject to strict and ongoing regulation. Compliance with such regulations may consume substantial financial and management resources and expose us and our collaborators to the potential for other adverse circumstances. For example, a regulatory authority can place restrictions on the sale or marketing of a drug in order to manage the risks identified during initial clinical trials or after the drug is on the market. A regulatory authority can condition the approval for a drug on costly post-marketing follow-up studies. Based on these studies, if a regulatory authority does not believe that the drug demonstrates a clinical benefit to patients, it could limit the indications for which a drug may be sold or revoke the drug's marketing approval. In addition, identification of certain side effects either during clinical trials or after a drug is on the market may result in reformulation of a drug, additional preclinical and clinical trials, labeling changes, or withdrawal of approval. Any of these events could delay or prevent us from generating revenue from the commercialization of these drugs and cause us to incur significant additional costs.
Even if we or our collaborators bring products to market, we may be unable to effectively price our products or obtain adequate reimbursement for sales of our products, which would have an adverse effect on our revenues.
Third party payors, such as government and private insurance plans, frequently require companies to provide rebates and predetermined discounts from list prices and are increasingly challenging the prices charged for pharmaceuticals and other medical products. Our products may not be considered cost-effective, and reimbursement to the patient may not be available or be sufficient to allow the sale of our products on a competitive basis. We, or our collaborators, may not be able to negotiate favorable reimbursement rates for our products. If we, or our collaborators, fail to obtain an adequate level of reimbursement for our products by third-party payors, sales of the drugs would be adversely affected or there may be no commercially viable market for the products.
We face potential liability related to the privacy of health information we obtain from research institutions.
Most health care providers, including research institutions from which we or our collaborators obtain patient information, are subject to privacy regulations promulgated under the Health Insurance Portability and Accountability Act of 1996, or HIPAA. Although we are not directly regulated by HIPAA, we could face substantial criminal penalties if we knowingly receive individually identifiable health information from a health care provider or research institution that has not satisfied HIPPA's disclosure standards. In addition, certain state privacy laws may apply directly to our operations and/or those of our collaborators and may impose restrictions on our use and dissemination of individuals' health information. Moreover, patients about whom we or our collaborators obtain information, as well as the providers who share this information with us, may have contractual rights that limit our ability to use and disclose the information. Claims that we have violated individuals' privacy rights or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm our business.
21
RISKS RELATED TO COLLABORATIONS
Part of our business strategy involves collaborative out-licensing of our drug candidates while retaining commercialization or co-promotional rights in parts of the world. We may not be able to find collaborators or successfully form suitable collaborations to further our drug development and commercialization efforts.
We may seek collaborators for our drug development and commercialization efforts. We may enter into these collaborations to obtain external financing for drug development and to obtain access to drug development and commercialization expertise. The availability of partners depends on the willingness of pharmaceutical and biotechnology companies to collaborate in drug discovery activities. Only a limited number of pharmaceutical and biotechnology companies would fit our requirements. The number could decline further through consolidation, or the number of collaborators with interest in our drugs could decline. If the number of our potential collaborators were to decline, the remaining collaborators may be able to negotiate terms less favorable to us.
We face significant competition in seeking drug development collaborations, both from other biotechnology companies and from the internal capabilities and compound pipelines of the pharmaceutical and biotechnology companies themselves. This competition is particularly intense in the oncology field. Our ability to interest such companies in forming co-development and commercialization arrangements with us will be influenced by, among other things:
- •
- the compatibility of technologies;
- •
- the potential partner's acceptance of our approach to drug discovery;
- •
- the novelty, quality and commercial potential of any drug candidate we may succeed in developing; and
- •
- our ability, and collaborators' perceptions of our ability, to achieve intended results in a timely fashion, with acceptable quality and cost.
Even if we are able to gain the interest of potential drug development partners, the negotiation, documentation and implementation of collaborative arrangements are complex and time-consuming. Collaborations may not be available on commercially acceptable terms and, if formed, may not be commercially successful or, if successful, may not realize sufficient benefit for us. If we are unable to form collaborations, we may not gain access to the financial resources and industry expertise necessary to develop and commercialize drug products or successfully market any products we develop on our own and, therefore, be unable to generate revenue from our products.
In fiscal year 2007, our collaborations with Roche and Kyowa accounted for substantially all of our research and development revenue. If Roche or Kyowa were to terminate its collaboration with us, our revenue may significantly decrease.
Our success depends in part on the efforts of our current and possible future collaborators, who will likely have substantial control and discretion over the continued development and commercialization of drug candidates which are the subjects of our collaborations.
Pursuant to our license agreement with Kyowa, Kyowa has, and if Roche exercises its option to acquire rights to compounds from the E2F-1 program, including ARQ 501, ARQ 171 and ARQ 761or if we were successful in establishing additional collaborations, Roche and any other collaborators would have significant discretion in determining the efforts and amount of resources that they dedicate to our collaborations. Our collaborators may determine not to proceed with clinical development or commercialization of a particular drug candidate for a number of reasons that are beyond our control, even under circumstances where we might have continued such a program. In addition, our rights to receive milestone payments and royalties from our collaborators will depend on our collaborators'
22
abilities to establish the safety and efficacy of our drug candidates, obtain regulatory approvals and achieve market acceptance of products developed from our drug candidates. We may also depend on our collaborators to manufacture clinical scale quantities of some of our drug candidates and, possibly, for commercial scale manufacture, distribution and direct sales. Our collaborators may not be successful in manufacturing our drug candidates on a commercial scale or in successfully commercializing them.
We face additional risks in connection with our existing and future collaborations, including the following:
- •
- our collaborators may develop and commercialize, either alone or with others, products that are similar to or competitive with the products that are the subject of the collaboration with us;
- •
- our collaborators may underfund or not commit sufficient resources to the testing, marketing, distribution or other development of our drug candidates;
- •
- our collaborators may not properly maintain or defend our intellectual property rights or they may utilize our proprietary information in such a way as to invite litigation that could jeopardize or potentially invalidate our intellectual property or proprietary information or expose us to potential liability;
- •
- our collaborators may encounter conflicts of interest, changes in business strategy or other business issues which could adversely affect their willingness or ability to fulfill their obligations to us (for example, pharmaceutical and biotechnology companies historically have re-evaluated their priorities following mergers and consolidations, which have been common in recent years in these industries); and
- •
- disputes may arise between us and our collaborators delaying or terminating the research, development or commercialization of our drug candidates, resulting in significant litigation or arbitration that could be time-consuming and expensive, or causing collaborators to act in their own self-interest and not in the interest of our stockholders.
We may not receive any further milestone, royalty or license payments under our current collaborations.
Although we have received license fees, milestone fees and other payments to date under our current drug development collaborations with Roche and Kyowa, we may not receive any royalty payments or additional license and milestone fees under such agreements. Our receipt of any future milestone, royalty or license payments depends on many factors, including whether our collaborators want or are able to continue to pursue potential drug candidates, intellectual property issues, unforeseen complications in the development or commercialization process, and the ultimate commercial success of the drugs.
RISKS RELATED TO RELATIONSHIPS WITH THIRD PARTY VENDORS
We rely heavily on third parties such as contract research organizations, to conduct clinical trials and perform research and analysis services for us. If third parties upon which we rely do not perform as contractually required or expected, we may not be able to develop further, obtain regulatory approval for or commercialize our product candidates.
We do not have the ability or the human resources to perform all of the testing or conduct all of the clinical trials that are necessary in connection with the development of our product candidates. We are using third-party clinical research organizations to oversee many of our ongoing clinical trials and expect to use the same or similar organizations for certain of our future clinical trials. Our reliance on these third parties reduces our control over these activities. We may face delays outside of our control if these parties do not perform their obligations in a timely or competent fashion or if we are forced to
23
change service providers or if the quality or accuracy of the data they obtain is compromised due to their failure to adhere to our clinical protocols or regulatory requirements or for other reasons. These risks are heightened if we conduct clinical trials outside of the United States, where it may be more difficult to ensure that studies are conducted in compliance with FDA requirements. Any third party that we hire to conduct clinical trials may also provide services to our competitors, which could compromise the performance of their obligations to us. If we experience significant delays in the progress of our clinical trials and in our plans to file NDAs, the commercial prospects for product candidates could be harmed and our ability to generate product revenue would be delayed or prevented.
We have limited manufacturing experience. We primarily rely on third parties to provide sufficient quantities of our product candidates to conduct pre-clinical and clinical studies. We have no control over our manufacturers' and suppliers' compliance with manufacturing regulations, and their failure to comply could interrupt our drug supply.
To date, our product candidates have been manufactured in relatively small quantities for preclinical and clinical trials. We have no experience in manufacturing any of our product candidates on a large scale and have contracted with third party manufacturers to provide material for clinical trials and to assist in the development and optimization of our manufacturing processes and methods. Our ability to conduct clinical trials and commercialize our product candidates will depend on the ability of such third parties to manufacture our products on a large scale at a competitive cost and in accordance with cGMP and other regulatory requirements. Significant scale-up of manufacturing may result in unanticipated technical challenges and may require additional validation studies that the FDA must review and approve. If we are not able to obtain contract cGMP manufacturing on commercially reasonable terms, obtain or develop the necessary materials and technologies for manufacturing, or obtain intellectual property rights necessary for manufacturing, we may not be able to conduct or complete clinical trials or commercialize our product candidates. There can be no assurance that we will be able to obtain such requisite terms, materials, technologies and intellectual property necessary to successfully manufacture our product candidates for clinical trials or commercialization. Our product candidates require precise, high-quality manufacturing. The failure to achieve and maintain these high manufacturing standards, including the incidence of manufacturing errors, could result in patient injury or death, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could seriously hurt our business.
The facilities used by our contract manufacturers must undergo inspections by the FDA for compliance with cGMP regulations before our product candidates produced there can receive marketing approval. If these facilities do not satisfy cGMP requirements in connection with the manufacture of our product candidates, we may need to conduct additional validation studies, or find alternative manufacturing facilities, either of which would result in significant cost to us as well as a delay of up to several years in obtaining approval for any affected product candidate. In addition, after approval of a product candidate for commercial use, our contract manufacturers and any alternative contract manufacturer we may utilize will be subject to ongoing periodic inspection by the FDA and corresponding state and foreign agencies for compliance with cGMP regulations, similar foreign regulations and other regulatory standards. We do not have control over our contract manufacturers' compliance with these regulations and standards. Any failure by our third-party manufacturers or suppliers to comply with applicable regulations could result in sanctions being imposed on them (including fines, injunctions and civil penalties), failure of regulatory authorities to grant marketing approval of our product candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or products, operating restrictions and criminal prosecution.
24
Materials necessary to manufacture our product candidates currently under development may not be available on commercially reasonable terms, or at all, which may delay our development and commercialization of these drugs.
Some of the materials necessary for the manufacture of our product candidates currently under development may, from time to time, be available either in limited quantities, or from a limited number of manufacturers, or both. We and/or our collaborators need to obtain these materials for our clinical trials and, potentially, for commercial distribution when and if we obtain marketing approval for these compounds. Suppliers may not sell us these materials at the time we need them or on commercially reasonable terms. If we are unable to obtain the materials needed for the conduct of our clinical trials, product testing and potential regulatory approval could be delayed, adversely impacting our ability to develop the product candidates. If it becomes necessary to change suppliers for any of these materials or if any of our suppliers experience a shutdown or disruption in the facilities used to produce these materials, due to technical, regulatory or other problems, it could significantly hinder or prevent manufacture of our drug candidates and any resulting products.
RISKS RELATED TO OUR COMMON STOCK
Our stock price may be extremely volatile.
The trading price of our common stock has been highly volatile. We believe the trading price of our common stock will remain highly volatile and may fluctuate substantially due to factors such as:
- •
- adverse results or delays in clinical trials;
- •
- announcement of FDA approval or non-approval, or delays in the FDA review process, of our or our collaborators' product candidates or those of our competitors or actions taken by regulatory agencies with respect to our, our collaborators' or our competitors' clinical trials;
- •
- announcement of new products by us or our competitors;
- •
- quarterly variations in our or our competitors' results of operations, including as a result of recognition of upfront licensing or other fees, the timing and amount of expenses incurred for clinical development, regulatory approval and commercialization of our product candidates;
- •
- litigation, including intellectual property infringement lawsuits, involving us;
- •
- financing transactions;
- •
- developments in the biotechnology and pharmaceutical industries;
- •
- the general performance of the equity markets and in particular the biopharmaceutical sector of the equity markets;
- •
- departures of key personnel or board members;
- •
- developments concerning current or future collaborations;
- •
- FDA or international regulatory actions affecting our industry generally; and
- •
- third-party reimbursement policies.
This volatility and general market declines in our industry over the past several years have affected the market prices of securities issued by many companies, often for reasons unrelated to their operating performance, and may adversely affect the price of our common stock. In the past, securities class action litigation has often been instituted following periods of volatility in the market price of a company's securities. A securities class action suit against us could result in potential liabilities, substantial costs and the diversion of management's attention and resources, regardless of the outcome of the action.
25
Some of our existing stockholders can exert control over us, and their interests could conflict with the best interests of our other stockholders.
Due to their combined stock holdings, our principal stockholders (stockholders holding more than 5% of our common stock), acting together, may be able to exert significant influence over all matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. In addition, this concentration of ownership may delay or prevent a change in control of our company, even when a change may be in the best interests of our stockholders. Furthermore, the interests of these stockholders may not always coincide with our interests as a company or the interests of other stockholders. Accordingly, these stockholders could cause us to enter into transactions or agreements that would not be widely viewed as beneficial.
If our officers, directors or principal stockholders sell substantial amounts of our common stock (including shares issued upon the exercise of options and warrants) in the public market, the market price of our common stock could fall. These sales also might make it more difficult for us to sell equity or equity-related securities in the future at a time and price that we deem appropriate.
Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent or deter attempts by our stockholders to replace or remove our current management.
Provisions in our corporate charter and bylaws and Delaware law may discourage, delay or prevent an acquisition of our company, a change in control, or attempts by our stockholders to replace or remove members of our current Board of Directors. Because our Board of Directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt by our stockholders to replace current members of our management team. These provisions include:
- •
- a Board of Directors having three classes of directors with a three-year term of office that expires as to one class each year, commonly referred to as a "staggered board";
- •
- a prohibition on actions by our stockholders by written consent;
- •
- the inability of our stockholders to call special meetings of stockholders;
- •
- the ability of our Board of Directors to issue preferred stock without stockholder approval, which could be used to institute a "poison pill" that would work to dilute the stock ownership of a potential hostile acquirer, effectively preventing acquisitions that have not been approved by our Board of Directors;
- •
- limitations on the removal of directors; and
- •
- advance notice requirements for director nominations and stockholder proposals.
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner. As a result, it is difficult for a third party to acquire control of us without the approval of our Board of Directors and, therefore, mergers with and acquisitions of us that our stockholders may consider in their best interests may not occur.
26
Because we do not intend to pay dividends, stockholders will benefit from an investment in our common stock only if it appreciates in value.
We have never declared or paid any cash dividends on our common stock. We currently intend to retain our future earnings, if any, to finance the expansion of our business and do not expect to pay any cash dividends in the foreseeable future. As a result, the success of an investment in our common stock will depend entirely upon any future appreciation. There is no guarantee that our common stock will appreciate in value or even maintain the price at which stockholders have purchased their shares.
RISKS RELATING TO COMPETITION
The drug research and development industry is highly competitive, and we compete with some companies that have a broader range of capabilities and better access to resources than we do.
The pharmaceutical and biotechnology industries are characterized by rapid and continuous technological innovation. We compete with companies worldwide that are engaged in the research and discovery, licensing, development and commercialization of drug candidates, including, in the area of small molecule anti-cancer therapeutics, Ariad Pharmaceuticals, Inc.; Array BioPharma Inc.; Cell Therapeutics, Inc.; Curis, Inc.; Exelixis, Inc.; Onyx Pharmaceuticals, Inc.; OSI Pharmaceuticals, Inc.; Oxigene, Inc.; Pharmacopeia, Inc.; SGX Pharmaceuticals; Telik, Inc.; Kosan Biosciences, Inc.; and Vion Pharmaceuticals, Inc. With respect to ARQ 197, we are aware of a number of companies that are pursuing approaches to c-Met inhibition, including Exelixis, Inc., Amgen Inc., Pfizer Inc, SGX Pharmaceuticals and Methylgene Inc.
Even if we are successful in bringing products to market, we face substantial competitive challenges in effectively marketing and distributing our products. Companies and research institutions, including large pharmaceutical companies with much greater financial resources and more experience in developing products, conducting clinical trials, obtaining FDA and foreign regulatory approvals and bringing new drugs to market are developing products within the field of oncology. Some of these entities already have competitive products on the market or product candidates in more advanced stages of development than we do. By virtue of having or introducing competitive products on the market before us, these entities may gain a competitive advantage. In addition, there may be product candidates of which we are not aware at an earlier stage of development that may compete with our product candidates. Some of our competitors have entered into collaborations with leading companies within our target markets.
We are in a rapidly evolving field of research. Consequently, our technology may be rendered non-competitive or obsolete by approaches and methodologies discovered by others, both before and after we have gone to market with our products. We also face competition from existing therapies that are currently accepted in the marketplace and from the impact of adverse events in our field that may affect regulatory approval or public perception.
We anticipate that we will face increased competition in the future as new companies enter the market and advanced technologies become available. If we are unable to successfully compete in our chosen field, we will not become profitable.
We may not be able to recruit and retain the scientists and management we need to compete.
Our success depends on our ability to attract, retain and motivate highly skilled scientific personnel and management, and our ability to develop and maintain important relationships with leading academic institutions, clinicians and scientists. We are highly dependent on our senior management and scientific staff, and the loss of the services of one or more of our other key employees could have an adverse effect on the successful completion of our clinical trials or the commercialization of our product candidates.
27
We compete intensely with pharmaceutical and biotechnology companies, including our collaborators, medicinal chemistry outsourcing companies, contract research and manufacturing organizations, and academic and research institutions in the recruitment of scientists and management. The shortage of personnel with experience in drug development could lead to increased recruiting, relocation and compensation costs, which may exceed our expectations and resources. If we cannot hire additional qualified personnel, the workload may increase for both existing and new personnel. If we are unsuccessful in our recruitment efforts, we may be unable to execute our strategy.
RISKS RELATED TO INTELLECTUAL PROPERTY
Our patents and other proprietary rights may fail to protect our business. If we are unable to adequately protect our intellectual property, third parties may be able to use our technology which could adversely affect our ability to compete in the market.
To be successful and compete, we must obtain and protect patents on our products and technology and protect our trade secrets. Where appropriate, we seek patent protection for certain aspects of the technology we are developing, but patent protection may not be available for some of our product candidates or their use, synthesis or formulations. The patent position of biotechnology firms is highly uncertain, involves complex legal and factual questions, and has recently been the subject of much litigation. No consistent policy has emerged from the U.S. Patent and Trademark Office or the courts regarding the breadth of claims allowed or the degree of protection afforded under many biotechnology patents. In addition, there is a substantial backlog of biotechnology patent applications at the U.S. Patent and Trademark Office. As a consequence of these factors, the approval or rejection of patent applications may take several years.
We do not know whether our patent applications will result in issued patents. In addition, the receipt of a patent might not provide much practical protection. If we receive a patent with a narrow scope it will be easier for competitors to design products that do not infringe our patent. We cannot be certain that we will receive any additional patents, that the claims of our patents will offer significant protection for our technology, or that our patents will not be challenged, narrowed, invalidated or circumvented.
Competitors may interfere with our patent protection in a variety of ways. Competitors may claim that they invented the claimed invention before us. Competitors may also claim that we are infringing on their patents and that, therefore, we cannot practice our technology as claimed under our patents. Competitors may also contest our patents by showing the patent examiner that the invention was not original, was not novel or was obvious. In litigation, a competitor could claim that our issued patents are not valid for a number of reasons. If a court agrees, our patents could be narrowed, invalidated or rendered unenforceable, or we may be forced to stop using the technology covered by these patents or to license the technology from third parties. As a company, we have no meaningful experience with competitors interfering with our patents or patent applications and therefore may not have the experience we would need to aggressively protect our patents should such action become necessary.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States. Many companies have encountered significant problems in protecting and defending such rights in foreign jurisdictions. Many countries, including certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of the patent. Compulsory licensing of life-saving drugs is also becoming increasingly popular in developing countries either through direct legislation or international initiatives. Such compulsory licenses could be extended to include some of our product candidates, which could limit our potential revenue opportunities. Moreover, the legal systems of
28
certain countries, particularly certain developing countries, do not favor the aggressive enforcement of patent and other intellectual property protection, which makes it difficult to stop infringement.
Drug candidates we develop that are approved for commercial marketing by the FDA would be subject to the provisions of the Drug Price Competition and Patent Term Restoration Act of 1984, known as the "Hatch-Waxman Act." The Hatch-Waxman Act provides companies with marketing exclusivity for varying time periods during which generic versions of a drug may not be marketed and allows companies to apply to extend patent protection for up to five additional years. It also provides a means for approving generic versions of a drug once the marketing exclusivity period has ended and all relevant patents have expired. The period of exclusive marketing, however, may be shortened if a patent is successfully challenged and defeated, which could reduce the amount of revenue we receive for such product.
Agreements we have with our employees, consultants and collaborators may not afford adequate protection for our trade secrets, confidential information and other proprietary information.
In addition to patent protection, we also rely on copyright and trademark protection, trade secrets, and know-how. It is unclear whether our trade secrets and know-how will prove to be adequately protected. To protect our trade secrets and know-how, we require our employees, consultants and advisors to execute agreements regarding the confidentiality and ownership of such proprietary information. We cannot guarantee, however, that these agreements will provide us with adequate protection against improper use or disclosure of confidential information and there may not be adequate remedies in the event of unauthorized use or disclosure. Our employees, consultants or advisors may unintentionally or willfully disclose our information to competitors. In addition, in some situations, these agreements may conflict with, or be subject to, the rights of third parties with whom our employees, consultants or advisors had or have previous employment or consulting relationships. Like patent litigation, enforcing a claim that a third party illegally obtained and is using our trade secrets is expensive and time-consuming and the outcome is unpredictable. In addition, courts outside the United States are sometimes less willing than our federal and state courts to protect trade secrets. Furthermore, others may independently develop substantially equivalent knowledge, methods and know-how. Our failure or inability to protect our proprietary information and techniques may inhibit or limit our ability to compete effectively or exclude certain competitors from the market.
Our success will depend partly on our ability to operate without infringing upon or misappropriating the proprietary rights of others.
There are many patents in our field of technology and we cannot guarantee that we do not infringe on those patents or that we will not infringe on patents granted in the future. If a patent holder believes a product of ours infringes on its patent, the patent holder may sue us even if we have received patent protection for our technology.
If we do not prevail in litigation or if other parties have filed, or in the future should file, patent applications covering products and technologies that we have developed or intend to develop, we may have to obtain licenses from third parties, which may not be available on commercially reasonable terms, or at all, and may require us to pay substantial royalties or grant a cross-license to some of our patents to another patent holder. Additionally, we may have to change the formulation of a product candidate so that we do not infringe third-party patents. Such reformulation may be impossible to achieve or which may require substantial time and expense. If we are unable to cost-effectively redesign our products so they do not infringe a patent, we may be unable to sell some of our products. Any of these occurrences will result in lost revenues and profits for us.
29
The drug research and development industry has a history of patent and other intellectual property litigation, and we may be involved in costly intellectual property lawsuits.
The drug research and development industry has a history of patent and other intellectual property litigation, and we believe these lawsuits are likely to continue. Legal proceedings relating to intellectual property would be expensive, take significant time and divert management's attention from other business concerns. We face potential patent infringement suits by companies that control patents for drugs or potential drugs similar to our product candidates or other suits alleging infringement of their intellectual property rights. There could be issued patents of which we are not aware that our products infringe or patents that we believe we do not infringe that we are ultimately found to infringe. The publication of discoveries in the scientific or patent literature frequently occurs substantially later than the date on which the underlying discoveries were made and patent applications were filed. Because patent applications can take many years to issue, there may be currently pending applications of which we are unaware that may later result in issued patents that we infringe with our drug candidates or resulting products. In addition, technology created under our research and development collaborations may infringe the intellectual property rights of third parties, in which case we may not receive milestone or royalty revenue from those collaborations.
If we do not prevail in an infringement lawsuit brought against us, we might have to pay substantial damages and we could be required to stop the infringing activity or obtain a license to use the patented technology or redesign our products so as not to infringe the patent. We may not be able to enter into licensing arrangements at a reasonable cost or effectively redesign our products. Any inability to secure licenses or alternative technology could delay the introduction of our products or prevent us from manufacturing or selling products.
RISKS RELATED TO EMPLOYEES AND FACILITIES
Our operations could be interrupted by damage to our laboratory facilities.
Our operations are dependent upon the continued use of our specialized laboratories and equipment in Woburn, Massachusetts. Catastrophic events, including fires or explosions, could damage our laboratories, equipment, scientific data, work in progress or inventories of chemical compounds and biological materials and may materially interrupt our business. We employ safety precautions in our laboratory activities in order to reduce the likelihood of the occurrence of these catastrophic events; however, we cannot eliminate the chance that such an event will occur. Rebuilding our facilities could be time consuming and result in substantial delays in our development of products and in fulfilling our agreements with our collaborators.
Security breaches may disrupt our operations and adversely affect our operating results.
Our network security and data recovery measures may not be adequate to protect against computer viruses, break-ins, and similar disruptions from unauthorized tampering with our computer systems. The misappropriation, theft, sabotage or any other type of security breach with respect to any of our proprietary and confidential information that is electronically stored, including research or clinical data, could have a material adverse impact on our business, operating results and financial condition. Additionally, any break-in or trespass of our facilities that results in the misappropriation, theft, sabotage or any other type of security breach with respect to our proprietary and confidential information, including research or clinical data, or that results in damage to our research and development equipment and assets could have a material adverse impact on our business, operating results, and financial condition.
30
RISKS RELATED TO PRODUCT LIABILITY
If our use of chemical and biological materials and hazardous materials violates applicable laws or causes personal injury, we may be liable for damages.
Our drug discovery activities, including the analysis and synthesis of chemical compounds, involve the controlled use of chemicals, including flammable, combustible, toxic and radioactive materials that are potentially hazardous if misused. Federal, state and local laws and regulations govern our use, storage, handling and disposal of these materials. These laws and regulations include the Resource Conservation and Recovery Act, the Occupational Safety and Health Act, local fire and building codes, regulations promulgated by the Department of Transportation, the Drug Enforcement Agency and the Department of Energy, the Department of Health and Human Services, and the laws of Massachusetts where we conduct our operations. We may incur significant costs to comply with these laws and regulations in the future and current or future environmental laws and regulations may impair our research, development and production efforts. Notwithstanding our extensive safety procedures for handling and disposing of materials, the risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of an accident, our business could be disrupted and we could be liable for damages. Our liability may exceed our insurance coverage and our total assets and have a negative impact on our financial condition and results of operations.
We may be exposed to potential liability related to the development, testing or manufacturing of compounds we develop and our insurance coverage may not be sufficient to cover losses.
We are developing, clinically testing and manufacturing potential therapeutic products for use in humans. In connection with these activities, we could be liable if persons are injured or die while using these drugs. We may have to pay substantial damages and/or incur legal costs to defend claims resulting from injury or death, and we may not receive expected royalty or milestone payments if commercialization of a drug is limited or ended as a result of such claims. We have product liability and clinical trial insurance that contains customary exclusions and provides coverage per occurrence at levels, in the aggregate, which we believe are customary and commercially reasonable in our industry given our current stage of drug development. Our product liability insurance does not cover every type of product liability claim that we may face or loss we may incur and may not adequately compensate us for the entire amount of covered claims or losses or for the harm to our business reputation. Also, we may be unable to maintain our current insurance policies or obtain and maintain necessary additional coverage at acceptable costs, or at all.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
In November 1999, we moved our main operations to a new facility in Woburn, Massachusetts, which includes approximately 128,000 square feet of laboratory and office space. This facility was designed to our specific requirements. In March 2001, we purchased this building and the land on which it sits and a developable adjacent parcel of land for $18.2 million and $2.3 million, respectively, in an arms-length transaction with the original developer.
On May 2, 2005, we completed a transaction to sell the Woburn facility and simultaneously lease the facility from the purchaser. The lease was subsequently amended on June 30, 2005. Under the terms of the transaction, the purchaser obtained two parcels of land and our headquarters building in exchange for a cash payment of approximately $40.1 million. We are leasing our existing facility and the associated land for a period of ten years at an average annual rental rate of $3.4 million. We also have options to extend the lease term for up to an additional ten years. See Note 7, "Property and
31
Equipment" in the Notes to Consolidated Financial Statements appearing in Item 8 in this Annual Report on Form 10-K.
In March 2002, we entered into an eight year lease with Pacific Shores Development LLC for approximately 34,000 square feet of laboratory and office space in Redwood City, California. We took occupancy in September 2002. Each base lease payment, the first of which was due and paid in September 2002, is $75,823 per month, subject to annual escalation provisions. In the third quarter of 2004, we entered into a sublease for the California facility. See Note 10, "Restructuring Actions" in the Notes to Consolidated Financial Statements appearing in Item 8 in this Annual Report on Form 10-K.
ITEM 3. LEGAL PROCEEDINGS
On January 16, 2002, we brought a complaint in the Superior Court of Middlesex County in the Commonwealth of Massachusetts for declaratory relief and damages against Cummings Properties, LLC ("Cummings") arising from a dispute over increased rent for lease of approximately 35,500 square feet of laboratory and office space in Medford, Massachusetts. On October 11, 2005, we and Cummings agreed to settle the lawsuit and file with the Court a stipulation of dismissal with prejudice.
In exchange for Cummings forgiving a portion of the rental payment obligations under the subject lease for the period from November 1, 2005 through July 30, 2006, we assigned sublease rent payments due to it for the leased premises during the same period to Cummings and guaranteed those payments. The total amount of those payments is approximately $0.3 million. As a result of this settlement, we saved approximately $0.6 million in rental payments. In connection with this settlement, on October 11, 2005, we and Cummings terminated the subject lease.
ITEM 4. SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS
No matters were submitted to stockholders for a vote during the fourth quarter of 2007.
32
PART II
ITEM 5. MARKET FOR THE REGISTRANT'S COMMON STOCK, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
STOCK PERFORMANCE GRAPH
The following graph shows the cumulative total stockholder return on our common stock over the period from December 31, 2002 to December 31, 2007, as compared with that of the NASDAQ Stock Market Index (U. S. Companies) and the NASDAQ Biotechnology Index, based on an initial investment of $100 in each on December 31, 2002. Total stockholder return is measured by dividing share price change plus dividends, if any, for each period by the share price at the beginning of the respective period, and assumes reinvestment of dividends. In the stock performance graph below we changed the comparison, for the years presented, from the NASDAQ Pharmaceuticals Index to the NASDAQ Biotechnology Index because we believe it is more reflective of our current business and peer group. The table below includes the NASDAQ Pharmaceuticals Index for historical comparison with the NADSDAQ Biotechnology Index.
COMPARISON OF CUMULATIVE TOTAL RETURN OF ARQULE, INC.,
NASDAQ STOCK MARKET (U.S. COMPANIES) INDEX
AND NASDAQ BIOTECHNOLOGY INDEX
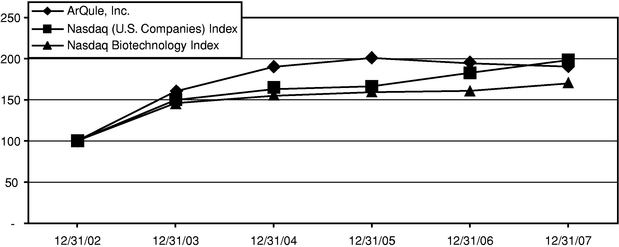
| | 12/31/02
| | 12/31/03
| | 12/31/04
| | 12/31/05
| | 12/31/06
| | 12/31/07
|
|---|
| ArQule, Inc. | | 100.00 | | 160.00 | | 189.84 | | 200.66 | | 194.10 | | 190.16 |
| NASDAQ Market (U.S. Companies) Index | | 100.00 | | 149.52 | | 162.72 | | 166.18 | | 182.58 | | 197.99 |
| NASDAQ Biotechnology Index | | 100.00 | | 145.75 | | 154.68 | | 159.06 | | 160.69 | | 169.71 |
| NASDAQ Pharmaceuticals Index | | 100.00 | | 146.59 | | 156.13 | | 171.93 | | 168.31 | | 176.97 |
ArQule's common stock is traded on the NASDAQ Global Market under the symbol "ARQL".
33
The following table sets forth, for the periods indicated, the range of the high and low sale prices for ArQule's common stock:
| | HIGH
| | LOW
|
|---|
| 2006 | | | | | | |
| First Quarter | | $ | 6.28 | | $ | 5.03 |
| Second Quarter | | | 6.41 | | | 4.01 |
| Third Quarter | | | 5.85 | | | 3.99 |
| Fourth Quarter | | | 7.09 | | | 3.83 |
2007 |
|
|
|
|
|
|
| First Quarter | | $ | 7.72 | | $ | 5.78 |
| Second Quarter | | | 10.59 | | | 6.85 |
| Third Quarter | | | 8.25 | | | 5.47 |
| Fourth Quarter | | | 8.34 | | | 5.60 |
2008 |
|
|
|
|
|
|
| First Quarter (through March 10, 2008) | | $ | 6.09 | | $ | 3.78 |
As of March 10, 2008, there were approximately 122 holders of record and approximately 6,485 beneficial shareholders of our common stock.
Dividend Policy
We have never paid cash dividends on our common stock and we do not anticipate paying any cash dividends in the foreseeable future. We currently intend to retain future earnings, if any, for use in our business.
34
ITEM 6. SELECTED FINANCIAL DATA
The following selected financial data have been derived from our audited historical consolidated financial statements, certain of which are included elsewhere in this Annual Report on Form 10-K. The following selected financial data should be read in conjunction with our consolidated financial statements and related notes and "Management's Discussion and Analysis of Financial Condition and Results of Operations" appearing elsewhere in this Annual Report on Form 10-K.
All current year and comparative prior period amounts have been restated to reflect our discontinued chemistry services operations. See Note 2 to our Consolidated Financial Statements for further information concerning discontinued operations.
This data is in thousands, except per share data.
| | YEAR ENDED DECEMBER 31,
| |
|---|
| | 2007*
| | 2006*
| | 2005
| | 2004
| | 2003
| |
|---|
| STATEMENT OF OPERATIONS DATA: | | | | | | | | | | | | | | | | |
| Revenue: | | | | | | | | | | | | | | | | |
| Research and development revenue(a)(b) | | $ | 9,165 | | $ | 6,626 | | $ | 6,628 | | $ | 5,012 | | $ | — | |
| Costs and expenses: | | | | | | | | | | | | | | | | |
| | Research and development | | | 53,727 | | | 47,428 | | | 24,646 | | | 20,181 | | | 18,836 | |
| | General and administrative | | | 15,069 | | | 11,560 | | | 8,688 | | | 8,982 | | | 9,560 | |
| | Restructuring charges/(credits)(c)(d) | | | — | | | — | | | — | | | (983 | ) | | 1,239 | |
| | Acquired in-process research and development(e) | | | — | | | — | | | — | | | — | | | 30,359 | |
| | |
| |
| |
| |
| |
| |
| | | Total costs and expenses | | | 68,796 | | | 58,988 | | | 33,334 | | | 28,180 | | | 59,994 | |
| | |
| |
| |
| |
| |
| |
| Loss from continuing operations | | | (59,631 | ) | | (52,362 | ) | | (26,706 | ) | | (23,168 | ) | | (59,994 | ) |
| Investment income, net | | | 6,259 | | | 5,139 | | | 3,331 | | | 1,086 | | | 610 | |
| Loss on investment(f) | | | — | | | — | | | (250 | ) | | — | | | (4,750 | ) |
| | |
| |
| |
| |
| |
| |
| Net loss from continuing operations | | | (53,372 | ) | | (47,223 | ) | | (23,625 | ) | | (22,082 | ) | | (64,134 | ) |
| Income from discontinued operations(g) | | | — | | | 15,783 | | | 16,105 | | | 17,161 | | | 29,383 | |
| | |
| |
| |
| |
| |
| |
| Net loss(h) | | $ | (53,372 | ) | $ | (31,440 | ) | $ | (7,520 | ) | $ | (4,921 | ) | $ | (34,751 | ) |
| | |
| |
| |
| |
| |
| |
| Basic and diluted income (loss) per share: | | | | | | | | | | | | | | | | |
| Net loss from continuing operations | | $ | (1.33 | ) | $ | (1.33 | ) | $ | (0.68 | ) | $ | (0.77 | ) | $ | (2.64 | ) |
| Income from discontinued operations(g) | | | — | | | 0.45 | | | 0.46 | | | 0.60 | | | 1.21 | |
| | |
| |
| |
| |
| |
| |
| | | $ | (1.33 | ) | $ | (0.88 | ) | $ | (0.22 | ) | $ | (0.17 | ) | $ | (1.43 | ) |
| | |
| |
| |
| |
| |
| |
| Weighted average common shares outstanding—basic and diluted | | | 40,040 | | | 35,539 | | | 34,619 | | | 28,819 | | | 24,333 | |
| | |
| |
| |
| |
| |
| |
- *
- As a result of the adoption of Statement of Financial Accounting Standards (SFAS) No. 123(R), "Share Based Payment," as of January 1, 2006, all share-based payments have been recognized in the statements of operations based on their fair values. The Company adopted the modified prospective transition method permitted under SFAS No. 123(R) and, consequently, has not adjusted results from prior years. Stock-based compensation expense related to SFAS 123(R) was
35
approximately $5.0 million and $3.2 million for the years ended December 31, 2007 and 2006, respectively.
| | DECEMBER 31,
|
|---|
| | 2007
| | 2006
| | 2005
| | 2004
| | 2003
|
|---|
| Cash, cash equivalents and marketable securities(i)(j) | | $ | 135,082 | | $ | 95,832 | | $ | 140,643 | | $ | 71,365 | | $ | 76,724 |
| Working capital | | | 111,797 | | | 80,557 | | | 105,646 | | | 54,782 | | | 59,446 |
| Total assets(k) | | | 142,210 | | | 104,820 | | | 156,684 | | | 120,218 | | | 128,424 |
| Long-term debt | | | — | | | — | | | — | | | 17 | | | 1,218 |
| Total stockholders' equity(i)(j) | | | 88,041 | | | 79,954 | | | 105,458 | | | 82,452 | | | 86,477 |
- (a)
- In April 2004, ArQule entered into an alliance with Roche to discover and develop drug candidates targeting the E2F biological pathway. Roche provided immediate research funding of $15 million, and is providing financial support for ongoing research and development.
- (b)
- In April 2007, ArQule entered into an exclusive license agreement with Kyowa to develop and commercialize ARQ 197 in Japan and parts of Asia. The agreement includes upfront licensing fees of $30 million, which were received in 2007. In addition the agreement provides for potential development milestones of $93 million, sales milestones and royalty payments upon commercialization.
- (c)
- In October 2003, we completed an agreement with InPharmatica Ltd. to sell certain assets of our former operations in the United Kingdom and to assign our facility obligation. As a result, we reversed $0.3 million of restructuring accrual to reflect a change in our original estimate of the remaining lease obligation and assumed sublease income in the United Kingdom. In December 2003, the adequacy of the restructuring accrual and assumed sublease income relative to the lease commitment in Redwood City, California was reassessed and, based on deteriorating market conditions, an additional provision of $1.5 million was recorded, to increase our restructuring accrual.
- (d)
- In the first quarter of 2004, we implemented a restructuring which necessitated a charge of $0.5 million for termination benefits. In the third quarter of 2004, we subleased our abandoned California facility. Since the terms of the sublease was more favorable than we had previously estimated, we recorded a restructuring credit of $1.5 million to reduce our restructuring accrual.
- (e)
- In September 2003, we acquired Cyclis Pharmaceuticals, Inc. for $25.9 million in a stock purchase transaction. In connection with the transaction, we immediately charged to income $30.4 million representing purchased in-process research and development that had not yet reached technological feasibility and had no future alternative use.
- (f)
- In the fourth quarter of 2003, the carrying value of an investment in a privately-held proteomic company was written down by $4.75 million to reflect the estimated fair value of the investment. Based on events affecting the financial condition of the Company in the second quarter of 2005, we recorded a non-cash loss of $0.25 million to write-off the remaining carrying value of the investment.
- (g)
- In the fourth quarter of 2006, we completed our exit from our chemistry services operations and disposed of the related assets. Pursuant to Statement of Financial Accounting Standards No. 144,Accounting for the Impairment or Disposal of Long-Lived Assets, we have reported the results of the chemistry services operations as discontinued operations in 2006, since the related cash flows of our chemistry services operations were eliminated from our ongoing operations and we do not have any significant continuing involvement in the operations of the component or the assets that were disposed.
36
- (h)
- Net loss for 2004 includes a $0.6 million fourth quarter adjustment for a loss on the sublease of our Medford facility. See Note 14, "Commitments and Contingencies" in the Notes to Consolidated Financial Statements appearing in Item 8 of this Annual Report on Form 10-K.
- (i)
- In January 2005, we completed a stock offering in which we sold 5.79 million shares of common stock at a price of $5.25 for net proceeds of $28.3 million after commissions and offering expenses.
- (j)
- In June 2007, we completed a stock offering in which we sold 7.0 million shares of common stock at a price of $7.75 for net proceeds of $50.5 million after commissions and offering expenses. In July 2007, we sold an additional 0.5 million shares of common stock upon exercise of a portion of the underwriters over allotment option at a price of $7.75 for net proceeds of $3.6 million after offering expenses.
- (k)
- In June 2005, we completed a transaction to sell our headquarters facility in Woburn, Massachusetts, and to simultaneously lease the facility from the purchaser. We received a cash payment of approximately $39.3 million, net of commissions and closing costs, and entered into a ten year lease at an average annual rental rate of $3.4 million. As a result of the transaction, we reduced our net fixed assets by $33.7 million, representing the net book value of the real estate sold, and realized a gain on the sale of $5.5 million, which was deferred and is being amortized over the initial ten-year term of the lease as a reduction in rent expense.
37
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS OVERVIEW
We are a clinical-stage biotechnology company organized as a Delaware corporation in 1993 and engaged in the research and development of innovative cancer therapeutics. Our mission is to introduce novel products that act selectively against cancer cells, target multiple tumor types and are well tolerated by patients. We believe our clinical stage products represent potential best-in-class or first-in-class small molecule candidates based on highly differentiated mechanisms of action.
Our lead products, which are in clinical-stage testing, consist of: ARQ 197, an orally administered inhibitor of the c-Met receptor tyrosine kinase; ARQ 501, an intravenously administered novel activator of the cell's DNA damage response mechanism mediated by the E2F-1 transcription factor; and ARQ 171, an intravenously administered second generation activator of E2F-1. Early-stage clinical trial results, which are available for ARQ 197 and ARQ 501, have demonstrated anti-cancer activity across multiple types of tumors.
We retain full worldwide commercial rights to ARQ 197 outside of Japan and certain other Asian countries, where we have granted commercial rights to Kyowa Hakko Kogyo Co., Ltd. ("Kyowa"). We are developing ARQ 501, ARQ 171 and ARQ 761 (a new chemical entity based on ARQ 501) pursuant to our collaboration with Hoffmann-La Roche ("Roche"). Our agreements with these partners provide for possible future milestone payments, royalties on product sales, and development funding, in addition to payments that we have already received.
Our pre-clinical programs are directed toward molecular targets that we believe play critical roles in the development of human cancers. The targets, mechanisms of action and chemistry related to compounds generated from these programs differ, offering the potential for multiple therapeutic opportunities. The most advanced of these programs are focused on the development of inhibitors of the Eg5 kinesin spindle protein and the B-RAF kinase. Toxicology testing is planned to begin in 2008 with a product candidate from the Eg5 program. Additional molecular targets being explored in other pre-clinical programs.
Our products and research programs are based on our understanding of biological processes that lead to the proliferation and metastasis of cancer cells, combined with our ability to generate product candidates possessing certain pre-selected, drug-like properties and designed to act with specificity against cancer cells. We believe that these qualities, when present from the earliest stages of product development, increase the likelihood of producing safe, effective and marketable drugs. We believe that our combined expertise in chemistry and cancer biology differentiates us from other companies with a similar stage of evolution.
We will be focusing and advancing a significant portion of our drug discovery efforts based on our newly acquired understanding of the way ARQ 197 binds to c-Met. Insights into this novel binding mechanism form the basis of a discovery platform that we plan to leverage to generate a new type of selective kinase inhibitors. These compounds will be designed to inhibit a variety of kinases potently, selectively and without competing with ATP (adenosine triphosphate, an energy source for cells). We are currently assessing the potential of multiple kinases as targets for this drug discovery platform, and we are seeking to generate and validate compounds that inhibit these kinase targets with mechanisms similar to that of ARQ 197.
In September 2005, we announced a strategic decision to exit our pre-existing chemistry services operations in order to focus operationally on developing our oncology portfolio. Revenue from our chemistry services operations terminated in 2006 as a result of our strategic decision to no longer provide these services and the subsequent decision by Pfizer to terminate its Collaborative Agreement ("Agreement") with us effective May 22, 2006. We did not incur any financial penalty as a result of termination. We continued to provide chemistry services to Pfizer pursuant to the Agreement through
38
the effective date of termination. Since December 2001, we produced for Pfizer annually an average of approximately 160,000 synthetic chemical compounds and received average annual cash payments of approximately $50 million for those compounds and related services. The Agreement provided for six months prior written notice by either party to the other for termination without cause and, in the event of termination by Pfizer, certain payments to us. In accordance with these provisions, we received approximately $19.8 million in December 2005 in connection with the termination.
We considered the chemistry services asset group to be a "component of an entity," as defined in Statement of Financial Accounting Standards No. 144,Accounting for the Impairment or Disposal of Long-Lived Assets ("SFAS 144"), since it comprised operations and cash flows that were clearly distinguished, operationally and for financial reporting purposes, from the remainder of the Company's operations. Pursuant to SFAS 144, we reported the results of the chemistry services component as discontinued operations in the year ended December 31, 2006 since their related cash flows were eliminated from our ongoing operations and we did not have any significant continuing involvement in the operations of the component or the assets that were disposed.
We have incurred a cumulative net loss of $282 million from inception through December 31, 2007. We expect research and development costs to increase in 2008, due to clinical testing of our lead product candidates. Although we have generated positive cash flow from operations for six consecutive years from 2000-2005, these cash flows were attributable to our discontinued chemistry services operations. We recorded a net loss for all but one of those years. We recorded a net loss for 2006 and 2007, and expect a net loss for 2008.
Our revenue consists primarily of development funding from our alliances with Roche and Kyowa. Revenue and expenses fluctuate from quarter to quarter based upon a number of factors, notably: the timing and extent of our cancer related research and development activities together with the length and outcome of our clinical trials.
On April 2, 2004, we announced an alliance with Roche to discover and develop drug candidates targeting the E2F biological pathway, including ARQ 501. Under the terms of the agreement, Roche obtained an option to license our E2F program in the field of cancer therapy. Roche provided immediate research funding of $15 million, and is providing financial support for ongoing research and development. Under this alliance, we are responsible for advancing drug candidates from early stage development to Phase 2 trials. Roche may opt to license worldwide rights for the development and commercialization of products resulting from this collaboration by paying an option fee. Assuming the successful development and commercialization of a compound under the program, we could receive up to $276 million in pre-determined payments, plus royalties based on net sales. Additionally, we have the option to co-promote products in the U.S.
On April 27, 2007, we entered into an exclusive license agreement with Kyowa to develop and commercialize ARQ 197, a small molecule, selective inhibitor of the c-Met receptor tyrosine kinase, in Japan and parts of Asia. A $3 million portion of an upfront licensing fee was received by the Company under this agreement in the first quarter of 2007 and an additional $27 million in upfront licensing fees was received on May 7, 2007. The agreement includes $123 million in upfront and potential development milestone payments from Kyowa to ArQule, including the $30 million cash upfront licensing payments. In addition, the agreement includes sales milestone payments. Upon commercialization, ArQule will receive double-digit royalties from Kyowa on net sales of ARQ 197. Kyowa will be responsible for all clinical development costs and commercialization of the compound in certain Asian countries, consisting of Japan, China (including Hong Kong), South Korea and Taiwan.
39
LIQUIDITY AND CAPITAL RESOURCES
| |
| |
| |
| | % increase (decrease)
| |
|---|
| | December 31,
| |
|---|
| | 2006 to 2007
| | 2005 to 2006
| |
|---|
| | 2007
| | 2006
| | 2005
| |
|---|
| | (in millions)
| |
| |
| |
|---|
| Cash, cash equivalents and marketable securities | | $ | 135.1 | | $ | 95.8 | | $ | 140.6 | | 41 | % | (32 | )% |
| Working capital | | | 111.8 | | | 80.6 | | | 105.6 | | 39 | % | (24 | )% |
| | 2007
| | 2006
| | 2005
| |
| |
| |
|---|
| | (in millions)
| |
| |
| |
|---|
| Cash flow from: | | | | | | | | | | | | | | |
| Operating activities | | $ | (16.0 | ) | $ | (47.8 | ) | $ | 3.5 | | | | | |
| Investing activities | | | (35.8 | ) | | 47.3 | | | (36.2 | ) | | | | |
| Financing activities | | | 56.3 | | | 2.0 | | | 30.3 | | | | | |
Cash flow from operating activities. Our uses of cash for operating activities have primarily consisted of salaries and wages for our employees, facility and facility-related costs for our offices and laboratories, fees paid in connection with preclinical and clinical studies, laboratory supplies and materials, and professional fees. The sources of our cash flow from operating activities have consisted primarily of payments from our collaborators for services performed or upfront payments for future services. In 2007, our net use of cash was primarily driven by the difference between cash receipts from our collaborators, and payments for operating expenses which resulted in a net cash outflow of $16.0 million.
Cash flow from investing activities. Our net cash used by investing activities of $35.8 million in 2007 was comprised of net purchases of marketable securities of $34.6 million and acquisitions of fixed assets of $1.2 million. The composition and mix of cash, cash equivalents and marketable securities may change frequently as a result of the Company's constant evaluation of conditions in financial markets, the maturity of specific investments, and our near term liquidity needs.
Our cash equivalents and marketable securities include money market funds, corporate bonds and US federal and state agency backed certificates, including auction rate securities and other investment grade debt securities that have strong credit ratings.
Our cash equivalents and our portfolio of marketable securities are subject to market risk due to changes in interest rates. Fixed rate interest securities may have their market value adversely impacted due to a rise in interest rates, while floating rate securities may produce less income than expected if interest rates fall. Due in part to these factors, our future investment income may fall short of expectation due to changes in interest rates or we may suffer losses in principal if we are forced to sell securities that decline in the market value due to changes in interest rates.
Auction rate securities are securities that are structured with short-term interest rate reset dates of generally less than ninety days but with contractual maturities that can be well in excess of ten years. At the end of each reset period, which occurs every seven to twenty-eight days, investors can sell or continue to hold the securities at par. These securities are subject to fluctuations in fair value depending on the supply and demand at each auction. In the first quarter of 2008, certain auction rate securities failed auction due to sell orders exceeding buy orders. We will review our portfolio at the end of the quarter ending March 31, 2008, taking into consideration other-than-temporary impairment factors, to conclude whether ArQule has an impairment. If we determine there is an impairment, it would be recorded in the quarter ended March 31, 2008 within other comprehensive loss if related to a temporary impairment, and in the statement of operations if related to an other than temporary impairment related to these auction rate securities. ArQule's marketable securities portfolio as of December 31, 2007 was $124.2 million. The portfolio included $92.6 million (at cost) invested in
40
auction rate securities of which, $67.7 million (at cost) are associated with auctions that failed subsequent to February 12, 2008. Subsequent to December 31, 2007, ArQule was able to sell at par $22.4 million of auction rate securities that were held at December 31, 2007. In addition, certain of auction rate securities totaling $3.8 million were called for redemption and accordingly we expect to receive payment by the end of March 2008. The funds associated with failed auctions will not be accessible until a successful auction occurs, a buyer is found outside of the auction process or the underlying securities have matured.
Although we were cash flow positive from operations from 1999 through 2005, we were not cash flow positive from operations in 2006 or 2007, nor do we expect to be cash flow positive in 2008, as a result of our decision to exit our chemistry services operations and the increased cost of developing our clinical candidates. Given the current liquidity constraints in the auction rate securities market, and depending on decisions we may make regarding our clinical trials, we expect that our available cash and marketable securities, together with cash from operations and investment income, will be sufficient to finance our working capital and capital requirements at least through the first quarter of 2009. Pending increased stability and liquidity in the auction rate securities market, we believe we can fund our operations through the end of 2009.
Cash flow from financing activities. Our net cash provided by financing activities of $56.3 million in the year ended December 31, 2007 was comprised primarily of the proceeds from our June 2007 stock offering, wherein we sold 7 million shares of common stock at $7.75 per share for aggregate net proceeds of $50.5 million after commissions and offering expenses. During July 2007, the Company received net proceeds of $3.6 million when the underwriters exercised a portion of their over-allotment option and purchased an additional 502,000 shares of common stock. Stock option exercises and employee stock plan purchases provided additional cash inflow of $2.2 million.
In January 2005, we completed a stock offering in which we sold 5.79 million shares of common stock at $5.25 per share for net proceeds of approximately $28.3 million after commissions and offering expenses. On May 2, 2005, we completed a transaction to sell our Woburn facility and simultaneously lease the facility from the purchaser. The lease was subsequently amended on June 30, 2005. Under the terms of the transaction, the purchaser obtained two parcels of land and our headquarters building in exchange for a cash payment of approximately $39.3 million, net of commissions and closing costs. We are leasing our existing facility and the associated land for a period of ten years at an average annual rental rate of $3.4 million.
Our cash requirements may vary materially from those now planned depending upon the results of our drug discovery and development strategies, our ability to enter into additional corporate collaborations and the terms of such collaborations, results of research and development, unanticipated required capital expenditures, competitive and technological advances, acquisitions and other factors. We cannot guarantee that we will be able to develop any of our drug candidates into a commercial product. It is likely we will need to raise additional capital or incur indebtedness to continue to fund our operations in the future. Our ability to raise additional funds will depend on financial, economic and market conditions and other factors, many of which are beyond our control. There can be no assurance that sufficient funds will be available to us when required, on satisfactory terms, or at all. If necessary funds are not available, we may have to delay, reduce the scope of, or eliminate some of our development programs, potentially delaying the time to market for any of our product candidates.
41
Our contractual obligations were comprised of the following as of December 31, 2007 (in thousands):
| | Payment due by period
|
|---|
Contractual Obligations
| | Total
| | Less than
1 year
| | 1-3 years
| | 3-5 years
| | More than
5 years
|
|---|
| Operating lease obligations | | $ | 25,735 | | $ | 3,854 | | $ | 7,458 | | $ | 7,096 | | $ | 7,327 |
| Purchase obligations | | | 9,316 | | | 9,316 | | | — | | | — | | | — |
| | |
| |
| |
| |
| |
|
| | Total | | $ | 35,051 | | $ | 13,170 | | $ | 7,458 | | $ | 7,096 | | $ | 7,327 |
| | |
| |
| |
| |
| |
|
Included in the total minimum payments for operating leases is approximately $1.3 million related to abandoned real estate in California, net of contractual sublease income. This net amount has been accrued as a liability as a part of the Company's restructuring charge in 2002 and subsequently adjusted in 2003 and 2004. Purchase obligations are comprised primarily of outsourced preclinical and clinical trial expenses and payments to license certain intellectual property to support the Company's research efforts.
DISCONTINUED OPERATIONS
On September 27, 2005, we announced our intention to exit our chemistry services operations. We received notice on December 2, 2005 that Pfizer had elected to terminate our Collaboration Agreement, pursuant to its terms, effective May 22, 2006. The Agreement provided for six months prior written notice by either party to the other for termination without cause and, in the event of termination by Pfizer, certain payments to us. In accordance with these provisions, we received approximately $19.8 million in December 2005 in connection with the termination. This amount was recorded as deferred revenue and was recognized as revenue when compounds were delivered through the termination date. We have fulfilled our compound production obligations under the Agreement, recognized the remaining deferred revenue, and ceased chemistry services operations in 2006.
The net book value of the assets associated with the chemistry services operations, which totaled $1.4 million, approximated the fair market value of the underlying assets. In December 2006, we completed the sale of the chemistry services assets, which consisted of commercially available laboratory instrumentation, for approximately $1.3 million, net of disposal costs.
We considered the chemistry services asset group to be a "component of an entity" (as defined in SFAS 144) since it comprised operations and cash flows that were clearly distinguished, operationally and for financial reporting purposes, from the remainder of the Company's operations. Pursuant to SFAS 144, we reported the results of the chemistry services component as discontinued operations in the year ended December 31, 2006, since their related cash flows were eliminated from our ongoing operations and we do not have any significant continuing involvement in the operations of the component or the assets that were disposed.
42
The following table presents operating results for the discontinued chemical services operations in, 2006 and 2005 (in thousands):
| | 2006
| | 2005
|
|---|
| Revenue | | $ | 26,718 | | $ | 46,296 |
| Costs and expenses: | | | | | | |
| | Cost of revenue | | | 8,375 | | | 30,191 |
| | Restructuring charge | | | 2,498 | | | — |
| | |
| |
|
| Total costs and expenses. | | | 10,873 | | | 30,191 |
| Loss from disposition of assets. | | | (62 | ) | | — |
| | |
| |
|
| Income from discontinued operations. | | $ | 15,783 | | $ | 16,105 |
| | |
| |
|
Historically, ArQule entered into various collaborative agreements with pharmaceutical and biotechnology companies under which ArQule produced and delivered compound arrays and provided research and development services. Revenue elements from collaborative agreements included non-refundable technology transfer fees, funding of compound development work, payments based upon delivery of specialized compounds meeting collaborators' specific criteria and certain milestones and royalties on product sales.
Pfizer notified us in December 2005 that, in accordance with the provisions of the Collaborative Agreement ("Agreement") with ArQule, it was terminating their collaboration with us effective May 22, 2006. In accordance with the terms of the Agreement we received $19,750 in December 2005 in connection with the termination. We were required to perform under the terms of the contract during the period from Pfizer's termination notification to us through the effective termination date of the contract, and we recognized revenue based on the total number of compounds delivered to Pfizer during that time. As of December 31, 2006, we have completed our compound production obligations under the terms of the Agreement and have ceased chemistry services operations.
Compound development revenue was derived from the following contractual elements in 2006 and 2005 (in thousands):
| | 2006
| | 2005
|
|---|
| Non-refundable technology transfer payments | | $ | 5 | | $ | 10 |
| Funding of compound development | | | — | | | 236 |
| Payments based on delivery of specialized compounds | | | 25,963 | | | 46,050 |
| Milestone payments | | | 750 | | | — |
| | |
| |
|
| Total compound development revenue | | $ | 26,718 | | $ | 46,296 |
| | |
| |
|
CRITICAL ACCOUNTING POLICIES AND ESTIMATES
A "critical accounting policy" is one which is both important to the portrayal of the Company's financial condition and results and requires management's most difficult, subjective or complex judgments, often as a result of the need to make estimates about the effect of matters that are inherently uncertain. Management believes the following are critical accounting policies. For additional information, please see the discussion of our significant accounting policies in Note 3 to the Consolidated Financial Statements included in Item 8 of this Form 10-K.
43
Research and Development Revenue Recognition
Pursuant to the terms of the Roche agreement, Roche obtained an option to license ArQule's E2F program in the field of cancer therapy in 2004. Roche provided immediate research funding of $15 million, and financial support for ongoing research and development. ArQule is responsible for advancing drug candidates from early stage development into Phase 2 trials. Roche may opt to license worldwide rights for the development and commercialization of products resulting from this collaboration by paying an option fee. Assuming the successful development and commercialization of a compound under the program, ArQule could receive up to $276 million in pre-determined milestone payments, plus royalties based on net sales. ArQule considers the development portion of the arrangement to be a single unit of accounting under EITF 00-21 for purposes of revenue recognition, and recognizes the initial and ongoing development payments as research and development revenue over the maximum estimated development period. We estimate the maximum development period could extend until December 2009. This period may ultimately be shorter depending upon the outcome of the development work, resulting in accelerated recognition of the development revenue. Milestone and royalty payments will be recognized as revenue when earned. The cost associated with satisfying the Roche contract is included in research and development expense in the Consolidated Statement of Operations.
On April 27, 2007, we entered into an exclusive license agreement with Kyowa to develop and commercialize ARQ 197, a small molecule, selective inhibitor of the c-Met receptor tyrosine kinase, in Japan and parts of Asia. A $3 million portion of an upfront licensing fee was received by the Company under this agreement in the first quarter of 2007 and an additional $27 million in upfront licensing fees was received on May 7, 2007. The agreement includes $123 million in upfront and potential development milestone payments from Kyowa to ArQule, including the $30 million cash upfront licensing payments. In addition, the agreement includes sales milestone payments. Upon commercialization, ArQule will receive double-digit royalties from Kyowa on net sales of ARQ 197. Kyowa will be responsible for all clinical development costs and commercialization of the compound in certain Asian countries, consisting of Japan, China (including Hong Kong), South Korea and Taiwan.
Pursuant to the Kyowa agreement, the initial license fee and any subsequent milestone payments, once earned, will be recognized as research and development revenue using the contingency-adjusted performance model. Under this model, when payments are earned, revenue is immediately recognized on a pro-rata basis in the period we achieve the milestone based on the time elapsed from inception of the Kyowa agreement to the time the milestone is earned over the estimated duration of the development period under the agreement. Thereafter, the remaining portion of the milestone payment is recognized on a straight-line basis over the remaining estimated development period under the agreement. We currently estimate the development period to be through April 2016. This period may ultimately be shorter or longer depending upon the outcome of the development work, resulting in accelerated or deferred recognition of the development revenue. Royalty payments will be recognized as revenue when earned. The cost associated with satisfying the Kyowa contract is included in research and development expense in the Consolidated Statement of Operations.
Restructuring Charges/Credits
Accruals for abandoned facilities under lease require significant management judgment and the use of estimates, including assumptions concerning the ability of a sublessee to fulfill its contractual sublease obligations. In the third quarter of 2004, we entered into a sublease for the Company's abandoned facility in Redwood City, California. The term of the sublease extends through 2010, the remaining term of the Company's primary lease. As a result of signing the sublease, we adjusted our accrual for abandoned facilities to reflect the full amount of the anticipated sublease income to be received. This assumption about the subleasee's ability to fulfill its contractual obligation is based on an analysis of their financial position and ability to generate future working capital. If the subleasee is
44
unable to meet its obligations, and the Company is unable to enter into another sublease for the facility, ArQule may be required to adjust its restructuring accrual and record additional restructuring expense of up to $2.2 million.
Investments in Non-Marketable Equity Securities
At December 31, 2003, we performed an assessment of the fair value of our investment in a privately held proteomics company. This assessment included analysis of that company's current financial condition, its prospects for generating additional cash flow from operating activities, the current market conditions for raising capital funding for companies in this industry and the likelihood that any funding raised would significantly dilute our ownership percentage. As a result of this initial analysis, it was our judgment that an impairment had occurred and that the fair value of our investment was $0.25 million, resulting in a non-cash loss on investment of $4.75 million. In the second quarter of 2005, events affecting the financial condition of the company caused us to conclude that the fair value of the investment had further declined, and as such, we recorded a non-cash loss on investment of $0.25 million to write-off the remaining carrying value of the investment.
Impairment or Disposal of Long-Lived Assets
We assess our long-lived assets for impairment whenever events or changes in circumstances (a "triggering event") indicate that the carrying value of a group of long-lived assets may not be recoverable in accordance with SFAS 144.
On September 27, 2005, we announced our intention to exit our chemistry services operations when we had completed our existing Agreement with Pfizer in 2008. We concluded that our intention to exit our chemistry services operations was a triggering event and that an impairment review was required. As a result of that review, we determined that the anticipated undiscounted future cash flows from our chemistry services operations exceeded the net carrying value of the group of long-lived assets attributed to those operations, and therefore there was no impairment in the quarter ended September 30, 2005.
On December 2, 2005, we received notice that Pfizer had elected to terminate the Agreement, pursuant to the Agreement's terms, effective May 22, 2006. We concluded that notification from Pfizer was also a triggering event and performed a second impairment review. As a result of this second review, we again determined that the anticipated undiscounted future cash flows from our chemistry services operations exceeded the net carrying value of the group of long-lived assets attributed to those operations, and therefore there was no impairment in the quarter ended December 31, 2005. Based on our decision to exit our chemistry services operations, in 2005 we adjusted the depreciable lives on fixed assets used exclusively in those operations in order to fully depreciate the remaining book value of those assets over the remaining period that we will provide services to Pfizer.
We were contractually required to perform under the terms of the Agreement until May 22, 2006 and, as such, the assets of the chemistry services operations were considered "held for use" at December 31, 2005. Although we were actively seeking a potential buyer for the chemistry services operations, the uncertainty of us successfully completing a sale transaction within one year, or deciding to abandon the assets, precluded us from classifying the assets of the chemistry services operations as "assets to be disposed of by sale" at December 31, 2005.
In the third quarter ended September 30, 2006, it became probable that we would sell the chemistry services operations, eliminate the associated cash flows, and have no continuing involvement in the chemistry services operations. Accordingly, the chemistry services operations was reported as "discontinued operations" in our statements of operations in accordance with SFAS 144.
45
The net book value of the assets associated with the chemistry services operations, which totaled $1.4 million, approximated the fair market value of the underlying assets. In December 2006, management completed the sale of the chemistry services assets, which consisted of commercially available laboratory instrumentation, for approximately $1.3 million, net of direct costs to sell such assets.
Sale Leaseback Accounting
On May 2, 2005, we completed a transaction to sell our Woburn headquarters facility and two parcels of land in exchange for a cash payment of $39.3 million, net of commissions and closing costs. Simultaneously with that sale, we entered into an agreement to lease back the entire facility and the associated land. The lease was subsequently amended on June 30, 2005. The amended lease has a term of ten years with an average annual rental rate of $3.4 million. We also have options to extend the lease term for up to an additional ten years. In accordance with Statement of Financial Accounting Standards No. 98,Accounting for Leases, we are applying sale leaseback accounting to the transaction and are treating the lease as an operating lease. As a result of this transaction, we reduced our net fixed assets by $33.7 million, representing the net book value of the assets sold on the date of the lease amendment, and realized a gain on the sale of $5.5 million, which has been deferred and will be amortized over the initial ten year lease term as a reduction in rent expense.
RESULTS OF OPERATIONS
The following results of operations for the years ended December 31, 2007, 2006 and 2005 exclude the effect of discontinued operations:
Revenue
| |
| |
| |
| | % increase (decrease)
| |
|---|
| | 2007
| | 2006
| | 2005
| | 2006 to 2007
| | 2005 to 2006
| |
|---|
| | (in millions)
| |
| |
| |
|---|
| Research and development revenue | | $ | 9.2 | | $ | 6.6 | | $ | 6.6 | | 38 | % | — | % |
2007 as compared to 2006: Research and development revenue is comprised of revenue from the Roche alliance agreement and from the Kyowa exclusive license agreement entered into on April 27, 2007.
2006 as compared to 2005: Research and development revenue which remained the same in both years is comprised of revenue from Roche in connection with the alliance agreement.
Research and development
| |
| |
| |
| | % increase (decrease)
| |
|---|
| | 2007
| | 2006
| | 2005
| | 2006 to 2007
| | 2005 to 2006
| |
|---|
| | (in millions)
| |
| |
| |
|---|
| Research and development | | $ | 53.7 | | $ | 47.4 | | $ | 24.6 | | 13 | % | 92 | % |
Our research and development expense consists primarily of salaries and related expenses for personnel, costs of contract manufacturing services, costs of facilities and equipment, fees paid to professional service providers in connection with our clinical trials, fees paid to research organizations in connection with preclinical animal studies, costs of materials used in research and development, consulting, license, and sponsored research fees paid to third parties and depreciation of associated
46
laboratory equipment. We expect our research and development expense to increase as we continue to develop our portfolio of oncology programs.
We have not accumulated and tracked our internal historical research and development costs or our personnel and personnel-related costs on a program-by-program basis. Our employee and infrastructure resources are allocated across several projects, and many of our costs are directed to broadly applicable research endeavors. As a result, we cannot state the costs incurred for each of our oncology programs on a program-by-program basis, or the cost to support our alliance agreement with Roche. The expenses incurred by us to third-parties for preclinical and clinical trials in 2007 and since inception of each program were as follows (in thousands):
Oncology program
| | Current status
| | 2007
| | Program-to-date
|
|---|
| E2F modulation—ARQ 501 | | Phase 2 | | $ | 7,666 | | $ | 27,387 |
| E2F modulation—ARQ 171 | | Phase 1 | | | 1,721 | | | 5,262 |
| cMet inhibition—ARQ 197 | | Phase 2 | | | 13,623 | | | 19,140 |
Our future research and development expenses in support of our current and future oncology programs will be subject to numerous uncertainties in timing and cost to completion. We test potential products in numerous preclinical studies for safety, toxicology, and efficacy. We then may conduct multiple clinical trials for each product. As we obtain results from trials, we may elect to discontinue or delay clinical trials for certain products in order to focus our resources on more promising products. Completion of clinical trials may take several years or more, but the length of time generally varies substantially according to the type, complexity, novelty, and intended use of a product. It is not unusual for the preclinical and clinical development of these types of products to each take nine years or more, and for total development costs to exceed $500 million for each product.
We estimate that clinical trials of the type generally needed to secure new drug approval are typically completed over the following timelines:
Clinical Phase
| | Estimated Completion Period
|
|---|
| Phase 1 | | 1-2 years |
| Phase 2 | | 2-3 years |
| Phase 3 | | 2-4 years |
The duration and the cost of clinical trials may vary significantly over the life of a project as a result of differences arising during clinical development, including, among others, the following:
- •
- the number of clinical sites included in the trials;
- •
- the length of time required to enroll suitable patient subjects;
- •
- the number of patients that ultimately participate in the trials;
- •
- the duration of patient follow-up to ensure the absence of long-term adverse safety events; and
- •
- the efficacy and safety profile of the product.
An element of our business strategy is to pursue the research and development of a broad pipeline of products. This is intended to allow us to diversify the risks associated with our research and development expenditures. As a result, we believe our future capital requirements and future financial success are not substantially dependent on any one product. To the extent we are unable to maintain a broad pipeline of products, our dependence on the success of one or a few products increases.
Our strategy includes entering into alliance arrangements with third parties to participate in the development and commercialization of our products, such as our collaboration agreements with Roche and Kyowa. In the event that third parties have control over the clinical trial process for a product, the
47
estimated completion date would largely be under control of that third party rather than under our control. We cannot forecast with any degree of certainty whether our products will be subject to future collaborative arrangements or how such arrangements would affect our development plans or capital requirements.
As a result of the uncertainties discussed above, we are unable to determine the duration and completion costs of our oncology programs or when and to what extent we will receive cash inflows from the commercialization and sale of a product. Our inability to complete our oncology programs in a timely manner or our failure to enter into collaborative agreements, when appropriate, could significantly increase our capital requirements and could adversely impact our liquidity. These uncertainties could force us to seek additional, external sources of financing from time-to-time in order to continue with our strategy. Our inability to raise additional capital, or to do so on terms reasonably acceptable to us, would jeopardize the future success of our business.
2007 as compared to 2006: The $6.3 million increase in research and development expense in 2007 is primarily due to (i) a $10.9 million increase in outsourced costs related to the phase 1 and 2 clinical programs for ARQ 197, (ii) an increase of $4.7 million in connection with the Company's sponsored research agreement with Boston Biomedical Institute (BBI), and (iii) a decrease of $7.5 million in outsourced costs related to the phase 1 and 2 clinical programs with ARQ 501. At December 31, 2007, we had 78 employees dedicated to our research and development program, down from 93 employees at December 31, 2006.
2006 as compared to 2005: The $22.8 million increase in research and development expense in 2006 is primarily due to: (i) an increase in outsourced preclinical, clinical and manufacturing costs of $16.4 million required to advance our oncology programs, principally ARQ 197, ARQ 501 and ARQ 171; (ii) an increase in personnel and related costs of $3.8 million reflecting the hiring of additional scientists and stock-based compensation charges recorded in 2006 but not 2005; (iii) increased professional fees of $0.8 million and (iv) increased facility and maintenance costs of $1.6 million, due to the absorption of these costs, formerly associated with the chemical services operations, by our research and development organization. At December 31, 2006, we had 93 employees dedicated to our research and development program, up from 86 employees at December 31, 2005.
General and administrative
| |
| |
| |
| | % increase (decrease)
| |
|---|
| | 2007
| | 2006
| | 2005
| | 2006 to 2007
| | 2005 to 2006
| |
|---|
| | (in millions)
| |
| |
| |
|---|
| General and administrative | | $ | 15.1 | | $ | 11.6 | | $ | 8.7 | | 30 | % | 33 | % |
2007 compared to 2006: General and administrative expense increased $3.5 million in 2007 primarily due to increased personnel-related costs of $2.6 million, including stock-based compensation expense of $1.6 million and $0.7 million of facility costs which are no longer absorbed by the chemical services operations. General and administrative headcount was 32 at December 31, 2007, compared to 33 at December 31, 2006.
2006 compared to 2005: General and administrative expense increased $2.9 million in 2006 primarily due to increased personnel-related expenses, including stock-based compensation expense of $1.3 million, and due to facility costs of $1.4 million which are no longer absorbed by the chemical services operations.
48
Restructuring
In December 2002, we announced a major restructuring of our operations in order to realign our workforce and expedite the transition towards becoming a drug discovery company. The restructuring actions included closing our facilities in Redwood City, California and Cambridge, United Kingdom.
The facility-related accrual, which represents the difference between our lease obligation for the California facility and the amount of sublease payments it will receive under its sublease agreement, will be paid out through 2010.
On January 19, 2006, our Board of Directors authorized termination benefits for employees in connection with a plan of termination for our discontinued chemistry services operations. The termination benefits, which affected 104 employees, consisted of cash payments and continuation of health care benefits. In 2006, a restructuring charge of $2.5 million was recorded pursuant to this action and is included in the 2006 Consolidated Statement of Operations as part of "Income from discontinued operations". As of December 31, 2006, all affected employees had been separated from the Company and the restructuring costs were fully paid.
Activities against the restructuring accrual in 2006 and 2007 were as follows (in thousands):
| | Balance as of
December 31, 2005
| | 2006
Provisions
| | 2006
Payments
| | Balance as of
December 31, 2006
|
|---|
| Termination benefits-discontinued operations | | $ | — | | $ | 2,383 | | $ | (2,383 | ) | $ | — |
| Other charges-discontinued operations | | | — | | | 115 | | | (115 | ) | | — |
| Facility-related | | | 2,706 | | | — | | | (662 | ) | | 2,044 |
| | |
| |
| |
| |
|
| | Total restructuring accrual | | $ | 2,706 | | $ | 2,498 | | $ | (3,160 | ) | $ | 2,044 |
| | |
| |
| |
| |
|
| | Balance as of
December 31, 2006
| | 2007
Provisions
| | 2007
Payments
| | Balance as of
December 31, 2007
|
|---|
| Facility-related | | $ | 2,044 | | $ | — | | $ | (678 | ) | $ | 1,366 |
| | |
| |
| |
| |
|
| | Total restructuring accrual | | $ | 2,044 | | $ | — | | $ | (678 | ) | $ | 1,366 |
| | |
| |
| |
| |
|
Investment income and interest expense
| |
| |
| |
| | % increase (decrease)
| |
|---|
| | 2007
| | 2006
| | 2005
| | 2006 to 2007
| | 2005 to 2006
| |
|---|
| | (in millions)
| |
| |
| |
|---|
| Investment income | | $ | 6.3 | | $ | 5.1 | | $ | 3.7 | | 22 | % | 39 | % |
| Interest expense | | | — | | | — | | | (0.4 | ) | — | | (100 | )% |
| | |
| |
| |
| |
| |
| |
| Investment income, net | | $ | 6.3 | | $ | 5.1 | | $ | 3.3 | | 22 | % | 54 | % |
| | |
| |
| |
| |
| |
| |
Investment income is derived from our portfolio of cash and short-term investments. Investment income increased year-to-year due to the increased average portfolio balance and to generally higher interest rates. Interest expense was zero in 2007 and 2006 because we had no debt in either year. Interest expense decreased in 2006 due to interest charges incurred in 2005 related to the sale of the Woburn facility. See "Critical Accounting Policies and Estimates" above for a discussion of our sale leaseback accounting.
49
Loss on investment
| | 2007
| | 2006
| | 2005
|
|---|
| | (in millions)
|
|---|
| Loss on investment | | $ | — | | $ | — | | $ | 0.25 |
The loss on investment in 2005 relates to impairment charges taken to write off our investment in a privately-held proteomics company.
RECENT ACCOUNTING PRONOUNCEMENTS
In September 2006, the Financial Accounting Standards Board ("FASB") issued SFAS No. 157Fair Value Measurements ("SFAS 157"). SFAS 157 defines fair value, establishes a framework for measuring fair value in generally accepted accounting principles and expands disclosures about fair value measurements. This accounting standard is effective for fiscal years beginning after November 15, 2007. The adoption of SFAS 157 is not anticipated to have a material effect on our financial position or results of operations.
In February 2007, the FASB issued SFAS No. 159,"The Fair Value Option for Financial Assets and Financial Liabilities" ("SFAS 159"), which is effective for financial statements issued for fiscal years beginning after November 15, 2007. Early adoption is permitted as of the beginning of a fiscal year that begins on or before November 15, 2007, provided the entity also elects to apply the provisions SFAS 157,Fair Value Measurements. SFAS 159 provides companies with an option to report selected financial assets and liabilities at fair value. The Statement also establishes presentation and disclosure requirements designed to facilitate comparisons between companies that choose different measurement attributes for similar types of assets and liabilities. SFAS 159 requires companies to provide additional information that will help investors and other users of financial statements to more easily understand the effect of the company's choice to use fair value on its earnings. It also requires entities to display the fair value of those assets and liabilities for which the company has chosen to use fair value on the face of the balance sheet. The implementation of SFAS 159 is not expected to have a material impact on the Company's financial statements.
In June 2007, the Emerging Issues Task Force ("EITF"), reached a consensus on EITF Issue No. 07-03,Accounting for Nonrefundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities. EITF 07-03 concludes that non-refundable advance payments for future research and development activities should be deferred and capitalized until the goods have been delivered or the related services have been performed. If an entity does not expect the goods to be delivered or services to be rendered, the capitalized advance payment should be charged to expense. This consensus is effective for fiscal years beginning after December 15, 2007. The initial adjustment to reflect the effect of applying the consensus as a change in accounting principle would be accounted for as a cumulative-effect adjustment to retained earnings as of the beginning of the year of adoption. We do not believe that our adoption of EITF 07-03 will have a material impact on our financial statements.
50
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We own financial instruments that are sensitive to market risk as part of our investment portfolio. We have implemented policies regarding the amount and credit ratings of investments. Our investment portfolio is used to preserve our capital until it is used to fund operations, including our research and development activities. None of these market-risk sensitive instruments are held for trading purposes. Our investments are evaluated quarterly to determine the fair value of the portfolio.
Our cash and marketable securities include money market funds, corporate bonds and US federal and state agency backed certificates, including auction rate securities and other investment grade debt securities that have strong credit ratings.
Our cash equivalents and our portfolio of marketable securities are subject to market risk due to changes in interest rates. Fixed rate interest securities may have their market value adversely impacted due to a rise in interest rates, while floating rate securities may produce less income than expected if interest rates fall. Due in part to these factors, our future investment income may fall short of expectation due to changes in interest rates or we may suffer losses in principal if we are forced to sell securities that decline in the market value due to changes in interest rates.
Auction rate securities are securities that are structured with short-term interest rate reset dates of generally less than ninety days but with contractual maturities that can be well in excess of ten years. At the end of each reset period, which occurs every seven to twenty-eight days, investors can sell or continue to hold the securities at par. These securities are subject to fluctuations in fair value depending on the supply and demand at each auction. In the first quarter of 2008, certain auction rate securities failed auction due to sell orders exceeding buy orders. We will review our portfolio at the end of the quarter ending March 31, 2008, taking into consideration other-than-temporary impairment factors, to conclude whether ArQule has an impairment. If we determine there is an impairment, it would be recorded in the quarter ended March 31, 2008 within other comprehensive loss if related to a temporary impairment, and in the statement of operations if related to an other than temporary impairment related to these auction rate securities. ArQule's marketable securities portfolio as of December 31, 2007 was $124.2 million. The portfolio included $92.6 million (at cost) invested in auction rate securities of which, $67.7 million (at cost) are associated with auctions that failed subsequent to February 12, 2008. Subsequent to December 31, 2007, ArQule was able to sell at par $22.4 million of auction rate securities that were held at December 31, 2007. In addition, certain of auction rate securities totaling $3.8 million were called for redemption and accordingly we expect to receive payment by the end of March 2008. The funds associated with failed auctions will not be accessible until a successful auction occurs, a buyer is found outside of the auction process or the underlying securities have matured.
51
ITEM 8. CONSOLIDATED FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| | Page
|
|---|
Report of Independent Registered Public Accounting Firm |
|
53 |
Consolidated Balance Sheets at December 31, 2007 and 2006 |
|
54 |
Consolidated Statements of Operations for the years ended December 31, 2007, 2006 and 2005 |
|
55 |
Consolidated Statements of Stockholders' Equity and Comprehensive Loss for the years ended December 31, 2007, 2006 and 2005 |
|
56 |
Consolidated Statements of Cash Flows for the years ended December 31, 2007, 2006 and 2005 |
|
57 |
Notes to Consolidated Financial Statements |
|
58 |
52
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of ArQule, Inc.:
In our opinion, the accompanying consolidated balance sheets and related consolidated statements of operations, of stockholders' equity and comprehensive income, and of cash flows present fairly, in all material respects, the financial position of ArQule, Inc. at December 31, 2007 and 2006, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2007 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2007 based on criteria established inInternal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company's management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management's Report on Internal Control Over Financial Reporting appearing under Item 9A. Our responsibility is to express opinions on these financial statements and on the Company's internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
As disclosed in Notes 3 and 13 to the consolidated financial statements, respectively, the Company changed the manner in which it accounts for stock-based compensation in 2006 and the manner for which it accounts for uncertain income tax positions in 2007.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
Boston, Massachusetts
March 17, 2008
53
ARQULE, INC.
CONSOLIDATED BALANCE SHEETS
| | December 31,
| |
|---|
| | 2007
| | 2006
| |
|---|
| | (IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
| |
|---|
| ASSETS | | | | | | | |
| Current assets: | | | | | | | |
| | Cash and cash equivalents | | $ | 10,835 | | $ | 6,242 | |
| | Marketable securities | | | 124,247 | | | 89,590 | |
| | Prepaid expenses and other current assets | | | 1,426 | | | 2,162 | |
| | |
| |
| |
| | | | Total current assets | | | 136,508 | | | 97,994 | |
| Property and equipment, net | | | 3,911 | | | 4,549 | |
| Other assets | | | 1,791 | | | 2,277 | |
| | |
| |
| |
| | | | Total assets | | $ | 142,210 | | $ | 104,820 | |
| | |
| |
| |
| LIABILITIES AND STOCKHOLDERS' EQUITY | | | | | | | |
| Current liabilities: | | | | | | | |
| | Accounts payable and accrued expenses | | $ | 14,162 | | $ | 10,276 | |
| | Current portion of deferred revenue | | | 9,997 | | | 6,609 | |
| | Current portion of deferred gain on sale leaseback | | | 552 | | | 552 | |
| | |
| |
| |
| | | | Total current liabilities | | | 24,711 | | | 17,437 | |
| Restructuring accrual, net of current portion | | | 738 | | | 1,366 | |
| Deferred revenue, net of current portion | | | 25,176 | | | 1,967 | |
| Deferred gain on sale leaseback, net of current portion | | | 3,544 | | | 4,096 | |
| | |
| |
| |
| | | | Total liabilities | | | 54,169 | | | 24,866 | |
| Commitments and contingencies (Note 14) | | | — | | | — | |
| Stockholders' equity: | | | | | | | |
| | | Preferred stock, $0.01 par value; 1,000,000 shares authorized; no shares issued or outstanding | | | — | | | — | |
| | | Common stock, $0.01 par value; 100,000,000 shares authorized; 43,761,113 and 35,811,709 shares issued and outstanding at December 31, 2007 and 2006, respectively | | | 438 | | | 358 | |
| Additional paid-in capital | | | 369,196 | | | 307,965 | |
| Accumulated other comprehensive loss | | | (4 | ) | | (152 | ) |
| Accumulated deficit | | | (281,589 | ) | | (228,217 | ) |
| | |
| |
| |
| | | | Total stockholders' equity | | | 88,041 | | | 79,954 | |
| | |
| |
| |
| | | | Total liabilities and stockholders' equity | | $ | 142,210 | | $ | 104,820 | |
| | |
| |
| |
The accompanying notes are an integral part of these consolidated financial statements.
54
ARQULE, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
| | YEAR ENDED DECEMBER 31,
| |
|---|
| | 2007
| | 2006
| | 2005
| |
|---|
| | (IN THOUSANDS, EXCEPT PER SHARE DATA)
| |
|---|
| Revenue: | | | | | | | | | | |
| | Research and development revenue | | $ | 9,165 | | $ | 6,626 | | $ | 6,628 | |
Costs and expenses: |
|
|
|
|
|
|
|
|
|
|
| | Research and development | | | 53,727 | | | 47,428 | | | 24,646 | |
| | General and administrative | | | 15,069 | | | 11,560 | | | 8,688 | |
| | |
| |
| |
| |
| | | | 68,796 | | | 58,988 | | | 33,334 | |
| | |
| |
| |
| |
| | | Loss from continuing operations | | | (59,631 | ) | | (52,362 | ) | | (26,706 | ) |
Investment income |
|
|
6,259 |
|
|
5,139 |
|
|
3,700 |
|
| Interest expense | | | — | | | — | | | (369 | ) |
| Loss on investment | | | — | | | — | | | (250 | ) |
| | |
| |
| |
| |
| Net loss from continuing operations | | | (53,372 | ) | | (47,223 | ) | | (23,625 | ) |
| Income from discontinued operations | | | — | | | 15,783 | | | 16,105 | |
| | |
| |
| |
| |
| | | Net loss | | $ | (53,372 | ) | $ | (31,440 | ) | $ | (7,520 | ) |
| | |
| |
| |
| |
Basic and diluted income (loss) per share: |
|
|
|
|
|
|
|
|
|
|
| | | Net loss from continuing operations | | $ | (1.33 | ) | $ | (1.33 | ) | $ | (0.68 | ) |
| | | Income from discontinued operations | | | — | | | 0.45 | | | 0.46 | |
| | |
| |
| |
| |
| Net loss per share | | $ | (1.33 | ) | $ | (0.88 | ) | $ | (0.22 | ) |
| | |
| |
| |
| |
Weighted average common shares outstanding-basic and diluted |
|
|
40,040 |
|
|
35,539 |
|
|
34,619 |
|
| | |
| |
| |
| |
The accompanying notes are an integral part of these consolidated financial statements.
55
ARQULE, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY AND COMPREHENSIVE LOSS
(IN THOUSANDS, EXCEPT SHARE DATA)
| | COMMON STOCK
| |
| |
| |
| |
| |
| |
|---|
| |
| | ACCUMULATED
OTHER
COMPREHENSIVE
INCOME/(LOSS)
| |
| |
| |
| |
|---|
| | SHARES
| | PAR
VALUE
| | ADDITIONAL
PAID-IN
CAPITAL
| | ACCUMULATED
DEFICIT
| | STOCKHOLDERS'
EQUITY
| | TOTAL
COMPREHENSIVE
LOSS
| |
|---|
| Balance at December 31, 2004 | | 28,982,774 | | $ | 290 | | $ | 271,805 | | $ | (386 | ) | $ | (189,257 | ) | $ | 82,452 | | | | |
| Stock option exercises | | 406,610 | | | 4 | | | 1,822 | | | | | | | | | 1,826 | | | | |
| Employee stock purchase plan | | 120,453 | | | 1 | | | 465 | | | | | | | | | 466 | | | | |
| Issuance of common stock from stock offering, net | | 5,788,095 | | | 58 | | | 28,291 | | | | | | | | | 28,349 | | | | |
| Stock based compensation expense | | | | | | | | 347 | | | | | | | | | 347 | | | | |
| Change in unrealized loss on marketable securities | | | | | | | | | | | (462 | ) | | | | | (462 | ) | $ | (462 | ) |
| Net loss | | | | | | | | | | | | | | (7,520 | ) | | (7,520 | ) | | (7,520 | ) |
| | |
| |
| |
| |
| |
| |
| | | | |
| Balance at December 31, 2005 | | 35,297,932 | | | 353 | | | 302,730 | | | (848 | ) | | (196,777 | ) | | 105,458 | | | | |
| | | | | | | | | | | | | | | | | | | |
| |
| 2005 Comprehensive loss | | | | | | | | | | | | | | | | | | | $ | (7,982 | ) |
| | | | | | | | | | | | | | | | | | | |
| |
| Stock option exercises and issuance of restricted stock | | 382,557 | | | 4 | | | 1,538 | | | | | | | | | 1,542 | | | | |
| Employee stock purchase plan | | 131,220 | | | 1 | | | 479 | | | | | | | | | 480 | | | | |
| Stock based compensation expense | | | | | | | | 3,218 | | | | | | | | | 3,218 | | | | |
| Change in unrealized loss on marketable securities | | | | | | | | | | | 696 | | | | | | 696 | | | 696 | |
| Net loss | | | | | | | | | | | | | | (31,440 | ) | | (31,440 | ) | | (31,440 | ) |
| | |
| |
| |
| |
| |
| |
| | | | |
| Balance at December 31, 2006 | | 35,811,709 | | | 358 | | | 307,965 | | | (152 | ) | | (228,217 | ) | | 79,954 | | | | |
| | | | | | | | | | | | | | | | | | | |
| |
| 2006 Comprehensive loss | | | | | | | | | | | | | | | | | | | $ | (30,744 | ) |
| | | | | | | | | | | | | | | | | | | |
| |
| Stock option exercises | | 351,621 | | | 4 | | | 1,757 | | | | | | | | | 1,761 | | | | |
| Employee stock purchase plan | | 95,783 | | | 1 | | | 474 | | | | | | | | | 475 | | | | |
| Stock based compensation expense | | | | | | | | 4,971 | | | | | | | | | 4,971 | | | | |
| Issuance of common stock from stock offering, net | | 7,502,000 | | | 75 | | | 54,029 | | | | | | | | | 54,104 | | | | |
| Change in unrealized loss on marketable securities | | | | | | | | | | | 148 | | | | | | 148 | | | 148 | |
| Cumulative translation adjustment | | | | | | | | | | | | | | | | | | | | | |
| Net loss | | | | | | | | | | | | | | (53,372 | ) | | (53,372 | ) | | (53,372 | ) |
| | |
| |
| |
| |
| |
| |
| | | | |
| Balance at December 31, 2007 | | 43,761,113 | | $ | 438 | | $ | 369,196 | | $ | (4 | ) | $ | (281,589 | ) | $ | 88,041 | | | | |
| | |
| |
| |
| |
| |
| |
| |
| |
| 2007 Comprehensive loss | | | | | | | | | | | | | | | | | | | $ | (53,224 | ) |
| | | | | | | | | | | | | | | | | | | |
| |
The accompanying notes are an integral part of these consolidated financial statements.
56
ARQULE, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| | YEAR ENDED DECEMBER 31,
| |
|---|
| | 2007
| | 2006
| | 2005
| |
|---|
| | (IN THOUSANDS)
| |
|---|
| Cash flows from operating activities: | | | | | | | | | | |
| | Net loss | | $ | (53,372 | ) | $ | (31,440 | ) | $ | (7,520 | ) |
| | Income from discontinued operations | | | — | | | (15,783 | ) | | (16,105 | ) |
| | Adjustments to reconcile net loss to net cash provided by (used in) operating activities: | | | | | | | | | | |
| | | Depreciation and amortization | | | 1,714 | | | 2,254 | | | 3,666 | |
| | | Amortization of premium/discount on marketable securities | | | 26 | | | 179 | | | 276 | |
| | | Amortization of deferred gain on sale leaseback | | | (552 | ) | | (552 | ) | | (277 | ) |
| | | Non-cash stock compensation. | | | 4,971 | | | 3,218 | | | 347 | |
| | | Loss on investment | | | — | | | — | | | 250 | |
| | | Loss on disposal of fixed assets. | | | 160 | | | 4 | | | 124 | |
| | Changes in operating assets and liabilities: | | | | | | | | | | |
| | | Accounts receivable | | | — | | | 6 | | | 63 | |
| | | Prepaid expenses and other current assets | | | 736 | | | (906 | ) | | 304 | |
| | | Other assets | | | 486 | | | (442 | ) | | (1,339 | ) |
| | | Accounts payable and accrued expenses | | | 3,886 | | | 4,961 | | | 1,040 | |
| | | Restructuring accrual, net of current portion. | | | (628 | ) | | (681 | ) | | (681 | ) |
| | | Deferred revenue | | | 26,597 | | | (1,610 | ) | | 143 | |
| | Net cash provided by (used in) operating activities from discontinued operations. | | | — | | | (7,046 | ) | | 23,203 | |
| | |
| |
| |
| |
| | | | Net cash provided by (used in) operating activities | | | (15,976 | ) | | (47,838 | ) | | 3,494 | |
| | |
| |
| |
| |
| Cash flows from investing activities: | | | | | | | | | | |
| | Purchases of marketable securities | | | (181,555 | ) | | (85,570 | ) | | (166,841 | ) |
| | Proceeds from sale or maturity of marketable securities | | | 147,020 | | | 132,335 | | | 94,500 | |
| | Additions to property and equipment | | | (1,236 | ) | | (814 | ) | | (2,663 | ) |
| | Net proceeds from sale of facility | | | — | | | — | | | 39,331 | |
| | Net cash provided by (used in) investing activities from discontinued operations | | | — | | | 1,302 | | | (484 | ) |
| | |
| |
| |
| |
| | | | Net cash provided by (used in) investing activities | | | (35,771 | ) | | 47,253 | | | (36,157 | ) |
| | |
| |
| |
| |
| Cash flows from financing activities: | | | | | | | | | | |
| | Principal payments of capital lease obligations | | | — | | | — | | | (135 | ) |
| | Principal payments of long-term debt | | | — | | | — | | | (169 | ) |
| | Proceeds from registered direct stock offering, net | | | 54,104 | | | — | | | 28,349 | |
| | Proceeds from issuance of common stock | | | 2,236 | | | 2,022 | | | 2,292 | |
| | |
| |
| |
| |
| | | | Net cash provided by financing activities | | | 56,340 | | | 2,022 | | | 30,337 | |
| | |
| |
| |
| |
| Net increase (decrease) in cash and cash equivalents. | | | 4,593 | | | 1,437 | | | (2,326 | ) |
| Cash and cash equivalents, beginning of period | | | 6,242 | | | 4,805 | | | 7,131 | |
| | |
| |
| |
| |
| Cash and cash equivalents, end of period | | $ | 10,835 | | $ | 6,242 | | $ | 4,805 | |
| | |
| |
| |
| |
SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION:
During 2007 and 2006 the Company paid no interest on debt and in 2005 paid approximately $369.
The accompanying notes are an integral part of these consolidated financial statements.
57
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
1. ORGANIZATION AND NATURE OF OPERATIONS
We are a clinical-stage biotechnology company organized as a Delaware corporation in 1993 and engaged in the research and development of innovative cancer therapeutics. Our mission is to introduce novel products that act selectively against cancer cells, target multiple tumor types and are well tolerated by patients.
Our lead products, which are in clinical-stage testing, consist of: ARQ 197, an orally administered inhibitor of the c-Met receptor tyrosine kinase; ARQ 501, an intravenously administered novel activator of the cell's DNA damage response mechanism mediated by the E2F-1 transcription factor; and ARQ 171, an intravenously administered second generation activator of E2F-1.
As part of our business since inception until 2006, we provided chemistry services to collaborators and customers for their discovery programs. In September 2005, we announced a strategic decision to exit our chemistry services operations in order to focus operationally on developing our oncology portfolio. On December 2, 2005, we received notice that our major collaborator and customer, Pfizer Inc. ("Pfizer") pursuant to the terms of the Collaborative Agreement ("Agreement") with ArQule, was terminating the Agreement effective on May 22, 2006. We continued to provide chemistry services to Pfizer through the effective date of termination.
2. DISCONTINUED OPERATIONS
On September 27, 2005, we announced our intention to exit our chemistry services operations. We received notice on December 2, 2005 that Pfizer had elected to terminate our Collaboration Agreement, pursuant to its terms, effective May 22, 2006. The Agreement provided for six months prior written notice by either party to the other for termination without cause and, in the event of termination by Pfizer, certain payments to us. In accordance with these provisions, we received approximately $19.8 million in December 2005 in connection with the termination. This amount was recorded as deferred revenue and was recognized as revenue when compounds were delivered through the termination date. We have fulfilled our compound production obligations under the Agreement, recognized the remaining deferred revenue, and ceased chemistry services operations in 2006.
The net book value of the assets associated with the chemistry services operations, which totaled $1.4 million, approximated the fair market value of the underlying assets. In December 2006, we completed the sale of the chemistry services assets, which consisted of commercially available laboratory instrumentation, for approximately $1.3 million, net of disposal costs.
We considered the chemistry services asset group to be a "component of an entity" as defined in Statement of Financial Accounting Standards No. 144,Accounting for the Impairment or Disposal of Long-Lived Assets ("SFAS 144"), since it comprised operations and cash flows that were clearly distinguished, operationally and for financial reporting purposes, from the remainder of the Company's operations. Pursuant to SFAS 144, we reported the results of the chemistry services component as discontinued operations in the year ended December 31, 2006, since its related cash flows were eliminated from our ongoing operations and we do not have any significant continuing involvement in the operations of the component or the assets that were disposed.
58
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
2. DISCONTINUED OPERATIONS (Continued)
The following table presents operating results for the discontinued chemical services operations in 2006 and 2005:
| | 2006
| | 2005
|
|---|
| Revenue | | $ | 26,718 | | $ | 46,296 |
| Costs and expenses: | | | | | | |
| | Cost of revenue | | | 8,375 | | | 30,191 |
| | Restructuring charge | | | 2,498 | | | — |
| | |
| |
|
| Total costs and expenses | | | 10,873 | | | 30,191 |
| Loss from disposition of assets | | | (62 | ) | | — |
| | |
| |
|
| Income from discontinued operations | | $ | 15,783 | | $ | 16,105 |
| | |
| |
|
Revenue Recognition—Compound Development Revenue (Discontinued Operations)
Historically, ArQule entered into various collaborative agreements with pharmaceutical and biotechnology companies under which ArQule produced and delivered compound arrays and provided research and development services. Revenue elements from collaborative agreements included non-refundable technology transfer fees, funding of compound development work, payments based upon delivery of specialized compounds meeting collaborators' specific criteria and certain milestones and royalties on product sales.
Pfizer notified us in December 2005 that, in accordance with the provisions of the Collaborative Agreement ("Agreement") with ArQule, it was terminating their collaboration with us effective May 22, 2006. In accordance with the terms of the Agreement we received $19,750 in December 2005 in connection with the termination. We were required to perform under the terms of the contract during the period from Pfizer's termination notification to us through the effective termination date of the contract, and we recognized revenue based on the total number of compounds delivered to Pfizer during that time.
Compound development revenue was derived from the following contractual elements in 2006 and 2005:
| | 2006
| | 2005
|
|---|
| Non-refundable technology transfer payments | | $ | 5 | | $ | 10 |
| Funding of compound development | | | — | | | 236 |
| Payments based on delivery of specialized compounds | | | 25,963 | | | 46,050 |
| Milestone payments | | | 750 | | | — |
| | |
| |
|
| Total compound development revenue | | $ | 26,718 | | $ | 46,296 |
| | |
| |
|
Cost of Compound Development Revenue (Discontinued Operations)
Cost of compound development revenue represents the actual costs incurred in connection with performance pursuant to our chemistry-based collaborative agreements and the costs incurred to
59
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
2. DISCONTINUED OPERATIONS (Continued)
develop and produce compounds under these agreements. These costs consist primarily of payroll and payroll-related costs, chemicals, supplies and overhead expenses.
3. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Significant accounting policies followed in the preparation of these financial statements are as follows:
Basis of Consolidation
The consolidated financial statements include the accounts of ArQule, Inc. and its wholly-owned subsidiary ArQule U.K. Ltd., (collectively, "we", "us", "our" and the "Company"). All intercompany transactions and balances have been eliminated. In February 2005, ArQule U.K. Ltd. was formally dissolved.
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from these estimates.
Cash Equivalents and Marketable Securities
We consider all highly liquid investments purchased within three months of original maturity date to be cash equivalents. We invest our available cash primarily in money market mutual funds, U.S. federal and state agency backed certificates, including auction rate certificates, corporate bonds and other investment grade debt securities that have strong credit ratings. Auction rate securities are structured with short-term interest reset dates of generally less than 90 days, but with contractual maturities that can be well in excess of ten years. At the end of each reset period, which occurs every seven to twenty-eight days, investors can sell or continue to hold the securities at par value. We account for our short-term marketable securities in accordance with SFAS No. 115, "Accounting for Certain Investments in Debt and Equity Securities" ("SFAS No. 115"). We classify our marketable securities as available-for-sale at the time of purchase and re-evaluate such designation as of each consolidated balance sheet date.
We determine on a quarterly basis the fair value of our investment portfolio. Our securities are recorded on our balance sheet at fair value. Unrealized gains and losses on securities are included in stockholders' equity, net of related tax effects. We evaluate our investments periodically for possible other-than-temporary impairment by reviewing factors such as the length of time and extent to which fair value has been below the cost basis, the financial condition of the issuer and our ability and intent to hold the investment for a period of time which may be sufficient for anticipated recovery of market value. We record an impairment charge to the extent that the carrying value of our available for sale securities exceeds the estimated fair value of the securities and the decline in value is determined to be other-than-temporary.
60
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
3. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Fair Value of Financial Instruments
At December 31, 2007 and 2006, our financial instruments consist of cash, cash equivalents, marketable securities, accounts receivable, accounts payable, and accrued expenses. The carrying amounts of these instruments approximate their fair values.
Investments in Non-Marketable Equity Securities
Investments in non-marketable equity securities are accounted for under the cost method if ArQule owns less than 20 percent of the outstanding stock of the investee and our management determines we do not exert significant influence over the management of the investee. We assess the fair value of investments in non-marketable equity securities quarterly, or whenever events or changes in circumstances indicate the carrying value may not be recoverable. In the event fair value is determined to be less than the carrying value of an investment, the carrying value is written down to fair value if the decline in value is significant and is deemed to be other than temporary. Since there is no readily available market information concerning the fair value of these investments, such assessments require significant management judgment in analyzing the investee's financial position and projected future financial results and cash flows. Although our best estimates of fair value are based upon available information, the use of different estimates could yield different conclusions concerning the recoverability of the carrying value of investments.
Property and Equipment
Property and equipment are recorded at cost and depreciated using the straight-line method over their estimated useful lives. Assets under capital leases and leasehold improvements are amortized over the shorter of their estimated useful lives or the term of the respective leases by use of the straight-line method. Maintenance and repair costs are expensed as incurred.
Revenue Recognition—Research and Development Revenue
On April 2, 2004, ArQule announced an alliance with Hoffmann-La Roche ("Roche") to discover and develop drug candidates targeting the E2F biological pathway. The alliance includes the compounds ARQ 501 and ARQ 171 which are currently in phase 2 and 1 clinical development. Under the terms of the agreement, Roche obtained an option to license ArQule's E2F program in the field of cancer therapy. Roche provided immediate research funding of $15,000, and financial support for ongoing research and development. ArQule is responsible for advancing drug candidates from early stage development into Phase 2 trials. Roche has an option to further develop and commercialize products resulting from this collaboration by paying an option fee for the worldwide licensing rights. Assuming the successful development and commercialization of a compound under the program, ArQule could receive up to $276,000 in pre-determined payments, plus royalties based on net sales. Additionally, ArQule has the option to co-promote products in the U.S.
ArQule considers the development portion of the arrangement to be a single unit of accounting under EITF No. 00-21,Accounting for Revenue Arrangements with Multiple Deliverables ("EITF 00-21") for purposes of revenue recognition, and will recognize the initial and ongoing development payments as research and development revenue over the maximum estimated development period. We estimate
61
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
3. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
the maximum development period could extend until December 2009, although this period may ultimately be shorter depending upon the outcome of the development work, which would result in accelerated recognition of the development revenue. Milestone and royalty payments will be recognized as revenue when earned. The cost associated with satisfying the Roche contract is included in research and development expense in the Consolidated Statement of Operations as incurred.
On April 27, 2007, we entered into an exclusive license agreement with Kyowa Hakko Kogyo Co., Ltd. ("Kyowa") to develop and commercialize ARQ 197, a small molecule, selective inhibitor of the c-Met receptor tyrosine kinase, in Japan and parts of Asia. A $3 million portion of an upfront licensing fee was received by the Company under this agreement in the first quarter of 2007 and an additional $27 million in upfront licensing fees was received on May 7, 2007. The agreement includes $123 million in upfront and potential development milestone payments from Kyowa to ArQule, including the $30 million cash upfront licensing payments. In addition, the agreement includes sales milestone payments. Upon commercialization, ArQule will receive double-digit royalties from Kyowa on net sales of ARQ 197. Kyowa will be responsible for all clinical development costs and commercialization of the compound in certain Asian countries, consisting of Japan, China (including Hong Kong), South Korea and Taiwan.
Under the Kyowa agreement, the initial license fee and any subsequent milestone payments, once earned, will be recognized as research and development revenue using the contingency-adjusted performance model. Under this model, when payments are earned, revenue is immediately recognized on a pro-rata basis in the period we achieve the milestone based on the time elapsed from inception of the Kyowa agreement to the time the milestone is earned over the estimated duration of the development period under the agreement. Thereafter, the remaining portion of the milestone payment is recognized on a straight-line basis over the remaining estimated development period under the agreement. We currently estimate the development period to be through April 2016. This period may ultimately be shorter or longer depending upon the outcome of the development work, resulting in accelerated or deferred recognition of the development revenue. Royalty payments will be recognized as revenue when earned. The cost associated with satisfying the Kyowa contract is included in research and development expense in the Consolidated Statement of Operations.
Research and Development Costs
Costs of internal research and development, which are expensed as incurred, are comprised of the following types of costs incurred in performing research and development activities and those incurred in connection with research and development revenue: salaries and benefits, allocated overhead and occupancy costs, clinical trial and related clinical manufacturing costs, contract services, and other outside costs. We incurred research and development expenses of $53,727, $47,428, and $24,646 in 2007, 2006 and 2005, respectively.
Restructuring Charges/Credits
The Company accounts for restructuring charges/credits in accordance with Statement of Financial Accounting Standards No. 146,Accounting for Costs Associated with Exit or Disposal Activities. Accruals are established for one-time employee termination benefits in the same period that the appropriate level of management and the Board of Directors approve and commit the Company to a termination
62
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
3. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
that meets the following criteria and has been communicated to employees: (i) specifically identifies the number, location and job level of employees to be terminated, (ii) specifies the benefits terminated employees are to receive, and (iii) assures that employees will be terminated within sixty days. Accruals are established for property and equipment and facility-related costs for facilities that have been abandoned and which have no future economic benefit to the Company at the time the Company ceases to occupy the facility.
Accruals for property and equipment and facility related costs of abandoned facilities require significant management judgment and the use of estimates, including assumptions concerning our ability to sublease certain operating leases for abandoned real estate and the ability of a sublessee to fulfill its contractual sublease obligation. Estimates of the time required to sublease facilities and sublease rates the Company will receive are based on management's analysis of the local real estate markets and general economic conditions in the regions of the abandoned facilities. If either the time it takes to sublease these facilities or the actual sublease rates achieved differ from the Company's assumptions, we may be required to adjust our restructuring accrual and record a restructuring charge or credit. When abandoned facilities are subleased, the Company must estimate the ability of the sublessee to satisfy the contractual lease obligation based on its financial position and projected ability to generate future working capital. If the sublessee's actual performance on the sublease is different from the Company estimates, we may be required to adjust our restructuring accrual and record a restructuring charge or credit.
Impairment or Disposal of Long-Lived Assets
We assess our long-lived assets for impairment whenever events or changes in circumstances (a "triggering event") indicate that the carrying value of a group of long-lived assets may not be recoverable in accordance with SFAS 144.
On September 27, 2005, we announced our intention to exit our chemistry services operations when the Agreement with Pfizer ended in 2008. We concluded that our intention to exit our chemistry services operations was a triggering event and that an impairment review was required. As a result of that review, we determined that the anticipated undiscounted future cash flows from our chemistry services operations exceeded the net carrying value of the group of long-lived assets attributed to those operations, and therefore there was no impairment in the quarter ended September 30, 2005.
On December 2, 2005, we received notice that Pfizer had elected to terminate the Agreement, pursuant to the Agreement terms, effective May 22, 2006. We concluded that notification from Pfizer was also a triggering event and performed a second impairment review. As a result of this second review, we again determined that the anticipated undiscounted future cash flows from our chemistry services operations exceeded the net carrying value of the group of long-lived assets attributed to those operations, and therefore there was no impairment in the quarter ended December 31, 2005.
63
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
3. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
We were contractually required to perform under the terms of the Agreement until May 22, 2006 and, as such, the assets of the chemistry services operations were considered "held for use" at December 31, 2005. Although we were actively seeking a potential buyer for the chemistry services operations, the uncertainty of us successfully completing a sale transaction within one year, or deciding to abandon the assets, precluded us from classifying the assets of the chemistry services operations as "assets to be disposed of by sale" at December 31, 2005.
In the third quarter ended September 30, 2006, it became probable that we would sell the chemistry services operations, eliminate the associated cash flows, and have no continuing involvement in the chemistry services operations. Accordingly, the chemistry services operations was reported as "discontinued operations" in our statements of operations in accordance with SFAS 144.
The net book value of the assets associated with the chemistry services operations, which totaled $1.4 million, approximated the fair market value of the underlying assets. In December 2006, management completed the sale of the chemistry services assets, which consisted of commercially available laboratory instrumentation, for approximately $1.3 million, net of direct costs to sell such assets.
Segment Data
Management uses consolidated financial information in determining how to allocate resources and assess performance. For this reason, we have determined that we are principally engaged in one operating segment. See Note 15 with respect to significant customers. Substantially all of our revenue since inception has been generated in the United States and substantially all of our long-lived assets are located in the United States.
Income Taxes
Income taxes have been accounted for using the asset and liability method. Under the asset and liability method, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. A valuation allowance against deferred tax assets is recorded if, based upon the weight of all available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
Earnings (Loss) Per Share
The computations of basic and diluted earnings (loss) per common share from continuing and discontinued operations are based upon the weighted average number of common shares outstanding and potentially dilutive securities. Potentially dilutive securities include stock options. Options to purchase 4,084,265, 3,872,946 and 4,477,862 shares of common stock were not included in the 2005, 2006 and 2007 computations of diluted net loss per share, respectively, because inclusion of such shares would have an anti-dilutive effect.
64
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
3. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Stock-Based Compensation
Effective January 1, 2006, we adopted the provisions of Statement of Financial Accounting Standards No. 123(R) ("SFAS 123(R)"), "Share-Based Payment", which establishes accounting for equity instruments exchanged for employee services. Under the provisions of SFAS 123(R), stock-based compensation cost is measured at the grant date, based on the calculated fair value of the award, and is recognized as an expense over the employees' requisite service period (generally the vesting period of the equity grant). Before January 1, 2006, we accounted for stock-based compensation to employees in accordance with Accounting Principles Board Opinion No. 25, "Accounting for Stock Issued to Employees," and related interpretations. We also followed the disclosure requirements of SFAS No. 123, "Accounting for Stock-Based Compensation." We elected to adopt the modified prospective transition method as provided by SFAS 123(R) beginning January 1, 2006 and, accordingly, financial statement amounts for the periods beginning before January 1, 2006 presented in this Form 10-K have not been restated to reflect the fair value method of expensing stock-based compensation.
The following table presents stock-based compensation expense included in our Consolidated Statements of Operations (in thousands):
| | Year ended
December 31, 2007
| | Year ended
December 31, 2006
|
|---|
| Research and development | | $ | 2,118 | | $ | 1,556 |
| General and administrative | | | 2,853 | | | 1,325 |
| Discontinued operations | | | — | | | 337 |
| | |
| |
|
| | Total compensation expense | | $ | 4,971 | | $ | 3,218 |
| | |
| |
|
In the years ended December 31, 2007 and 2006, no stock-based compensation expense was capitalized and there were no recognized tax benefits associated with the stock-based compensation charge. The stock-based compensation charge reduced basic and diluted net loss in the years ended December 31, 2007 and 2006 by $0.12 and $0.09 per share, respectively.
We estimate the fair value of stock options using the Black-Scholes valuation model. Key input assumptions used to estimate the fair value of stock options include the exercise price of the award, expected option term, expected volatility of our stock over the option's expected term, risk-free interest rate over the option's expected term, and the expected annual dividend yield. We believe that the valuation technique and approach utilized to develop the underlying assumptions are appropriate in calculating the fair values of our stock options granted in the year ended December 31, 2007 and 2006.
65
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
3. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
The fair value of stock options and employee stock purchase plan shares granted in the years ended December 31, 2007and 2006 respectively were estimated on the grant date using the Black-Scholes option-pricing model with the following assumptions:
| | 2007
| | 2006
|
|---|
| Dividend yield(1) | | 0.0% | | 0.0% |
| Expected volatility factor(2) | | 54.7—62.3% | | 63—90% |
| Risk free interest(3) | | 3.3—4.9% | | 4.3—4.9% |
| Expected term, excluding options issued pursuant to the Employee Stock Purchase Plan(4) | | 4.0—5.9 years | | 3.9—4.9 years |
| Expected term—Employee Stock Purchase Plan(5) | | 6 months | | 6 months |
- (1)
- We have historically not paid dividends on our common stock and we do not anticipate paying any dividends in the foreseeable future.
- (2)
- Measured using an average of historical daily price changes of our stock. The weighted average expected volatility in 2007 and 2006 was approximately 61% and 90%, respectively.
- (3)
- The risk-free interest rate for periods equal to the expected term of share option based on the U.S. Treasury yield in effect at the time of grant.
- (4)
- The expected term is the number of years that we estimate, based on historical experience, that options will be outstanding before exercise or cancellation. The range in expected term is the result of certain groups of employees exhibiting different exercising behavior.
- (5)
- The expected term of options issued in connection with our Employee Stock Purchase Plan is 6 months based on the terms of the plan.
We recognized employee stock-based compensation cost of $289 for the year ended December 31, 2005. If compensation cost had been determined based on the fair value at the grant dates, our net loss for the years ended December 31, 2005 would have been the pro forma amounts indicated in the table below (in thousands, except for per share data):
| | 2005
| |
|---|
| Net loss as reported | | $ | (7,520 | ) |
| Add: Stock-based employee compensation expense included in reported net loss | | | 289 | |
| Less: Stock-based employee compensation under the fair-value method for all awards | | | (4,615 | ) |
| | |
| |
| Pro forma net loss | | $ | (11,846 | ) |
| | |
| |
| Basic and diluted net loss per share | | | | |
| As reported | | $ | (0.22 | ) |
| Pro forma | | $ | (0.34 | ) |
66
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
3. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
The fair value of stock options and employee stock purchase plan shares granted in 2005 were estimated on the grant date using the Black-Scholes option-pricing model with the following assumptions:
| | 2005
|
|---|
| Dividend yield | | 0.0% |
| Expected volatility factor | | 80% |
| Risk-free interest rate | | 4.4% |
| Expected term, excluding options issued pursuant the Employee Stock Purchase Plan | | 4.5 years |
| Expected term—Employee Stock Purchase Plan | | 6—18 months |
Comprehensive Income (Loss)
Other comprehensive income (loss) refers to revenues, expenses, gains and losses that under accounting principles generally accepted in the United States of America are included in comprehensive income (loss) but are excluded from net income (loss) as these amounts are recorded directly as an adjustment to stockholders' equity, net of tax. Our other comprehensive income (losses) were $148, $696 and ($462) in 2007, 2006 and 2005 respectively, composed of unrealized gains and losses on marketable securities.
Recent Accounting Pronouncements
In September 2006, the Financial Accounting Standards Board ("FASB") issued SFAS No. 157Fair Value Measurements ("SFAS 157"). SFAS 157 defines fair value, establishes a framework for measuring fair value in generally accepted accounting principles and expands disclosures about fair value measurements. This accounting standard is effective for fiscal years beginning after November 15, 2007. The adoption of SFAS 157 is not anticipated to have a material effect on our financial position or results of operations.
In February 2007, the FASB issued SFAS No. 159,"The Fair Value Option for Financial Assets and Financial Liabilities", ("SFAS 159"), which is effective for financial statements issued for fiscal years beginning after November 15, 2007. Early adoption is permitted as of the beginning of a fiscal year that begins on or before November 15, 2007, provided the entity also elects to apply the provisions SFAS 157,Fair Value Measurements. SFAS 159 provides companies with an option to report selected financial assets and liabilities at fair value. The Statement also establishes presentation and disclosure requirements designed to facilitate comparisons between companies that choose different measurement attributes for similar types of assets and liabilities. SFAS 159 requires companies to provide additional information that will help investors and other users of financial statements to more easily understand the effect of the company's choice to use fair value on its earnings. It also requires entities to display the fair value of those assets and liabilities for which the company has chosen to use fair value on the face of the balance sheet. The implementation of SFAS 159 is not expected to have a material impact on the Company's financial statements.
In June 2007, the EITF, reached a consensus on EITF Issue No. 07-03,Accounting for Nonrefundable Advance Payments for Goods or Services to Be Used in Future Research and Development
67
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
3. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Activities ("EITF 07-03"). EITF 07-03 concludes that non-refundable advance payments for future research and development activities should be deferred and capitalized until the goods have been delivered or the related services have been performed. If an entity does not expect the goods to be delivered or services to be rendered, the capitalized advance payment should be charged to expense. This consensus is effective for fiscal years beginning after December 15, 2007. The initial adjustment to reflect the effect of applying the consensus as a change in accounting principle would be accounted for as a cumulative-effect adjustment to retained earnings as of the beginning of the year of adoption. We do not believe that our adoption of EITF 07-03 will have a material impact on our financial statements.
4. RELATED PARTIES
In January 2007, we entered into a $5.0 million, eight-month sponsored research agreement with the newly established Boston Biomedical, Inc. ("BBI"), an independent corporation led by our former chief scientific officer. BBI conducts scientific research under the agreement that includes a number ofin vivo andin vitro studies, reports and publications related to mechanisms of action and biomarkers for our lead products, which are in human clinical trials. See Note 16 to the consolidated financial statements for further terms of the agreement.
5. COLLABORATIONS AND ALLIANCES
Research and Development Alliance
On April 2, 2004, we announced an alliance with Hoffmann-La Roche ("Roche") to discover and develop drug candidates targeting the E2F biological pathway, including ARQ 501, which is currently in Phase 2 clinical testing, and ARQ 171, which is currently in Phase 1 clinical testing. Under the terms of the agreement, Roche obtained an option to license drugs resulting from our E2F program in the field of cancer therapy. Roche provided immediate research funding of $15 million and financial support for ongoing research and development. To date, we have received approximately $31.8 million in research and development support from Roche pursuant to this agreement and have recognized revenue of approximately $24.8 million. We have recognized revenue from Roche of approximately $6.6 million in each of the years ended December 31, 2007, 2006 and 2005.
We are responsible for advancing drug candidates from early stage development into Phase 2 trials. Roche has an option to license worldwide rights for the development and commercialization of products resulting from the E2F-1 program based on a clinical data package from one of the ongoing Phase 2 ARQ 501 monotherapy trials and the Phase 2 ARQ 501-gemcitabine combination therapy trial, as well as from the Phase 1 trial with ARQ 171. In order to license these rights, Roche must pay an option fee.
Assuming the successful development and commercialization of a compound under the program, we could receive up to $276 million in predetermined payments, plus royalties based on net sales. Additionally, we have the option to co-promote products in the U.S. Revenue from the Roche alliance is included in research and development revenue in the consolidated statements of operations.
68
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
5. COLLABORATIONS AND ALLIANCES (Continued)
Kyowa Licensing Agreement
On April 27, 2007, we entered into an exclusive license agreement with Kyowa to develop and commercialize ARQ 197, a small molecule, selective inhibitor of the c-Met receptor tyrosine kinase, in Japan and parts of Asia. A $3 million portion of an upfront licensing fee was received by the Company under this agreement in the first quarter of 2007 and an additional $27 million in upfront licensing fees was received on May 7, 2007. The agreement includes $123 million in upfront and potential development milestone payments from Kyowa to ArQule, including the $30 million cash upfront licensing payments. In addition, the agreement includes sales milestone payments. Upon commercialization, ArQule will receive double-digit royalties from Kyowa on net sales of ARQ 197. Kyowa will be responsible for all clinical development costs and commercialization of the compound in certain Asian countries, consisting of Japan, China (including Hong Kong), South Korea and Taiwan.
For the year ended December 31, 2007, $2.3 million was recognized as revenue. At December 31, 2007 $28.2 million remains in deferred revenue. In February 2008, we received a $3 million milestone payment from Kyowa.
Chemistry-Based Collaborations (Discontinued Operations)
In the past, we have entered into chemistry-based collaborations with a number of companies, including Pfizer, Bayer AG, GlaxoSmithKline plc, Sankyo Company, Ltd., Wyeth Pharmaceuticals, Solvay, Johnson & Johnson, and the Novartis Institute for BioMedical Research, Inc., an affiliate of Novartis AG. These collaborations have generally involved the production of chemical compounds and compound screening. We have fulfilled our obligations under these collaborations. Some of these collaborations contain trailing obligations of our collaborators to make, under specified circumstances, development milestone and royalty payments to us in the event products developed under these collaborations are commercialized.
6. MARKETABLE SECURITIES
We account for our short-term marketable securities in accordance with SFAS No. 115. We classify our marketable securities as available-for-sale at the time of purchase and re-evaluate such designation as of each consolidated balance sheet date. Our marketable securities are classified as cash equivalents if the original maturity, from the date of purchase, is ninety days or less and as short-term investments if the original maturity, from the date of purchase, is in excess of ninety days since we intend to convert them into cash as necessary to meet our liquidity requirements.
Our marketable securities are reported at fair value with the related unrealized gains and losses included in accumulated other comprehensive income (loss), a component of stockholders' equity, net of tax. Realized gains or losses on the sale of marketable securities are determined using the specific-identification method and were not material for fiscal year 2007, 2006 and 2005. We evaluate our investments periodically for possible other-than-temporary impairment by reviewing factors such as the length of time and extent to which fair value has been below cost basis, the financial condition of the issuer and our ability and intent to hold the investment for a period of time which may be sufficient for anticipated recovery of market value. We record an impairment charge to the extent that the carrying
69
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
6. MARKETABLE SECURITIES (Continued)
value of our available for sale securities exceeds the estimated fair value of the securities and the decline in value is determined to be other-than-temporary.
The following is a summary of the fair value of available-for-sale marketable securities we held at December 31, 2007 and 2006:
December 31, 2007
| | Amortized
Cost
| | Gross
Unrealized
Gains
| | Gross
Unrealized
Losses
| | Fair
Value
|
|---|
| Security type | | | | | | | | | | | | |
| | Time deposits | | $ | 6,169 | | $ | — | | $ | (1 | ) | $ | 6,168 |
| | Corporate bonds and notes | | | 25,190 | | | 14 | | | (17 | ) | | 25,187 |
| | Auction rate securities | | | 92,892 | | | — | | | — | | | 92,892 |
| | |
| |
| |
| |
|
| Total marketable securities | | $ | 124,251 | | $ | 14 | | $ | (18 | ) | $ | 124,247 |
| | |
| |
| |
| |
|
December 31, 2006
| | Amortized
Cost
| | Gross
Unrealized
Gain
| | Gross
Unrealized
Losses
| | Fair
Value
|
|---|
| Security type | | | | | | | | | | | | |
| | Time deposits | | $ | 5,004 | | $ | — | | $ | (9 | ) | $ | 4,995 |
| | Corporate bonds and notes | | | 32,775 | | | 11 | | | (96 | ) | | 32,690 |
| | US federal agency backed securities | | | 12,179 | | | — | | | (58 | ) | | 12,121 |
| | Auction rate securities | | | 39,784 | | | — | | | — | | | 39,784 |
| | |
| |
| |
| |
|
| Total marketable securities | | $ | 89,742 | | $ | 11 | | $ | (163 | ) | $ | 89,590 |
| | |
| |
| |
| |
|
We invest our available cash primarily in money market mutual funds, U.S. federal and state agency backed certificates, including auction rate certificates, corporate bonds and other investment grade debt securities that have strong credit ratings. Auction rate securities are structured with short-term interest reset dates of generally less than 90 days, but with contractual maturities that can be well in excess of ten years. At the end of each reset period, which occurs every seven to twenty-eight days, investors can sell or continue to hold the securities at par value. If any of our auction rate securities were to fail an auction, due to sell orders exceeding buy orders, the funds associated with a failed auction would not be accessible until a successful auction occurred, a buyer was found outside the auction process or the underlying securities matured.
In the first quarter of 2008, certain auction rate securities failed auction due to sell orders exceeding buy orders. We will review our portfolio at the end of the quarter ending March 31, 2008, taking into consideration other-than-temporary impairment factors, to conclude whether ArQule has an impairment. If we determine there is an impairment, it would be recorded in the quarter ended March 31, 2008 within other comprehensive loss if related to a temporary impairment, and in the statement of operations if related to an other than temporary impairment related to these auction rate securities. ArQule's marketable securities portfolio as of December 31, 2007 was $124.2 million. The portfolio included $92.6 million (at cost) invested in auction rate securities of which, $67.7 million (at cost) are associated with auctions that failed subsequent to February 12, 2008. Subsequent to December 31, 2007, ArQule was able to sell at par $22.4 million of auction rate securities that were
70
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
6. MARKETABLE SECURITIES (Continued)
held at December 31, 2007. In addition, certain of our auction rate securities totaling $3.8 million were called for redemption and accordingly we expect to receive payment by the end of March 2008. The funds associated with failed auctions will not be accessible until a successful auction occurs, a buyer is found outside of the auction process or the underlying securities have matured.
At December 31, 2007 and 2006, marketable securities are carried at fair value and are classified as current as the funds will be needed to meet working capital needs and to fund current operations. The net unrealized losses on marketable securities at December 31, 2007 and 2006 were $4 and $152, respectively.
The following table summarizes our investments with gross unrealized losses, aggregated by investment category and length of time that individual securities have been in a continuous unrealized loss position, at December 31, 2007:
| | Less than 12 Months
| | 12 months or more
| | Total
|
|---|
| | Fair Value
| | Unrealized Losses
| | Fair Value
| | Unrealized Losses
| | Fair Value
| | Unrealized Losses
|
|---|
| Time deposits | | $ | 6,168 | | $ | 1 | | — | | — | | $ | 6,168 | | $ | 1 |
| Corporate bonds | | | 25,187 | | | 17 | | — | | — | | | 25,187 | | | 17 |
| | |
| |
| |
| |
| |
| |
|
| Total temporarily impaired securities | | $ | 31,355 | | $ | 18 | | — | | — | | $ | 31,355 | | $ | 18 |
| | |
| |
| |
| |
| |
| |
|
The securities summarized above represent a total of 21 investments purchased by the Company in order to maximize its return on liquid assets in excess of its immediate needs. The temporary impairments relate to unfavorable market interest rate fluctuations that have decreased the fair value of the investments below the original investment cost. The Company believes these fluctuations are temporary and therefore has not realized an impairment loss on these investments at December 31, 2007.
7. PROPERTY AND EQUIPMENT
Property and equipment consist of the following at December 31, 2007 and 2006:
| | USEFUL LIFE
ESTIMATED
(YEARS)
| | 2007
| | 2006
|
|---|
| Machinery and equipment | | 5 | | $ | 12,971 | | $ | 12,189 |
| Leasehold improvements | | 3-10 | | | 2,257 | | | 2,143 |
| Furniture and fixtures | | 7 | | | 1,209 | | | 1,209 |
| Computer equipment | | 3 | | | 5,729 | | | 5,634 |
| Construction-in-progress | | — | | | 124 | | | 113 |
| | | | |
| |
|
| | | | | | 22,290 | | | 21,288 |
| Less: Accumulated depreciation and amortization | | | | | 18,379 | | | 16,739 |
| | | | |
| |
|
| | | | | $ | 3,911 | | $ | 4,549 |
| | | | |
| |
|
71
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
7. PROPERTY AND EQUIPMENT (Continued)
On May 2, 2005, we completed a transaction to sell our Woburn headquarters facility and two parcels of land in exchange for a cash payment, net of commissions and closing costs, of $39,331. Simultaneous with that sale, we entered into an agreement to lease back the entire facility and the associated land. The lease was subsequently amended on June 30, 2005. The amended lease has a term of ten years with an average annual rental rate of $3,409. We also have options to extend the lease term for up to an additional ten years. In accordance with Statement of Financial Accounting Standards No. 98,Accounting for Leases, we are applying sale leaseback accounting to the transaction and are treating the lease as an operating lease. As a result of this transaction, we reduced our net fixed assets by $33,709, representing the net book value of the assets sold on the date of the lease amendment, and realized a gain on the sale of $5,477, which was deferred and is being amortized over the initial ten year lease term as a reduction in rent expense.
In December 2006, we completed the sale of the assets, which consisted of commercially available laboratory instrumentation, from our discontinued chemistry service operations,. These assets had a net book value of $1,364 and were sold for $1,302 net of direct costs to sell such assets, resulting in a $62 loss on disposal.
8. OTHER ASSETS
Other assets include the following at December 31, 2007 and 2006:
| | 2007
| | 2006
|
|---|
| Security deposits | | $ | 976 | | $ | 1,002 |
| Prepaid rent, net of current portion | | | 769 | | | 962 |
| Other long-term prepaid assets | | | 46 | | | 313 |
| | |
| |
|
| Total other assets | | $ | 1,791 | | $ | 2,277 |
| | |
| |
|
In July 2001, we purchased approximately 1.8 million preferred shares of a privately owned proteomics company for $5,000. This represented approximately an 8% ownership interest. At December 31, 2003, we performed an assessment based on an analysis of the investment's current financial condition, its prospects of generating additional cash flow from operating activities, the current market conditions for raising capital funding for companies in this industry and the likelihood that any funding raised would significantly dilute our ownership percentage. As a result of this analysis it was our judgment that a permanent impairment had occurred and that the fair value of our investment was $250, resulting in a non-cash loss on investment of $4,750. In the second quarter of 2005, events affecting the financial condition of the investment caused us to conclude that the fair value of the investment had further declined, and as such, we recorded a non-cash loss on investment of $250 to write-off the remaining carrying value of this investment.
72
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
9. ACCOUNTS PAYABLE AND ACCRUED EXPENSES
Accounts payable and accrued expenses include the following at December 31, 2007 and 2006:
| | 2007
| | 2006
|
|---|
| Accounts payable | | $ | 548 | | $ | 208 |
| Accrued payroll | | | 1,954 | | | 1,726 |
| Accrued outsourced pre-clinical and clinical fees | | | 9,307 | | | 6,197 |
| Accrued professional fees | | | 496 | | | 434 |
| Accrued restructuring-current portion | | | 629 | | | 678 |
| Other accrued expenses | | | 1,228 | | | 1,033 |
| | |
| |
|
| | | $ | 14,162 | | $ | 10,276 |
| | |
| |
|
10. RESTRUCTURING ACTIONS
In December 2002, we announced a major restructuring of our operations in order to realign our workforce and expedite the transition towards becoming a drug discovery company. The restructuring actions included closing our facilities in Redwood City, California and Cambridge, United Kingdom.
The facility-related accrual, which primarily represents the difference between the Company's lease and other facility related obligations for its California facility and the amount of sublease and other payments it will receive under its sublease agreement, will be paid out through 2010. The portions of the restructuring accrual that are expected to be paid out within one year and longer than one year are included in the Consolidated Balance Sheet under "Accounts payable and accrued expenses" and "Restructuring accrual, net of current portion," respectively.
Accruals for abandoned facilities under lease requires significant management judgment and the use of estimates, including assumptions concerning the ability of a sublessee to fulfill its contractual sublease obligation. As a result of signing the sublease for the California facility, we adjusted our accrual for abandoned facilities to reflect the full amount of the anticipated sublease income to be received. This assumption about the subleasee's ability to fulfill its contractual obligation is based on an analysis of their financial position and ability to generate future working capital. If the subleasee is unable to meet its obligations, and the Company is unable to enter into another sublease for the facility, ArQule may be required to adjust its restructuring accrual and record additional restructuring expense of up to $2,153.
On January 19, 2006, our Board of Directors authorized termination benefits for employees in connection with a plan of termination for our chemistry services operations. The termination benefits, which affected 104 employees, consisted of cash payments and continuation of health care benefits. In 2006, a restructuring charge of $2.5 million was recorded pursuant to this action and is included in the 2006 Consolidated Statement of Operations as part of "Income from discontinued operations". As of December 31, 2006, all affected employees had been separated from the Company and the restructuring costs were fully paid.
73
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
10. RESTRUCTURING ACTIONS (Continued)
Activities against the restructuring accrual in 2006 and 2007 were as follows:
| | Balance as of
December 31, 2005
| | 2006
Provisions
| | 2006
Payments
| | Balance as of
December 31, 2006
|
|---|
| Termination benefits-discontinued operations | | $ | — | | $ | 2,383 | | $ | (2,383 | ) | $ | — |
| Other charges-discontinued operations | | | — | | | 115 | | | (115 | ) | | — |
| Facility-related | | | 2,706 | | | — | | | (662 | ) | | 2,044 |
| | |
| |
| |
| |
|
| | Total restructuring accrual | | $ | 2,706 | | $ | 2,498 | | $ | (3,160 | ) | $ | 2,044 |
| | |
| |
| |
| |
|
| | Balance as of
December 31, 2006
| | 2007
Provisions
| | 2007
Payments
| | Balance as of
December 31, 2007
|
|---|
| Facility-related | | $ | 2,044 | | $ | — | | $ | (678 | ) | $ | 1,366 |
| | |
| |
| |
| |
|
| | Total restructuring accrual | | $ | 2,044 | | $ | — | | $ | (678 | ) | $ | 1,366 |
| | |
| |
| |
| |
|
11. STOCKHOLDERS' EQUITY
Preferred Stock
We are authorized to issue up to one million shares of preferred stock. As of December 31, 2007 and 2006, there were no outstanding shares of preferred stock. Our Board of Directors will determine the terms of the preferred stock if and when the shares are issued.
Common Stock
Our amended Certificate of Incorporation authorizes the issuance of up to 100 million shares of $0.01 par value common stock.
In June 2007, we completed a stock offering in which we sold 7.0 million shares of common stock at a price of $7.75 for net proceeds of $50.5 million after commissions and offering expenses. In July 2007, we sold an additional 0.5 million shares of common stock upon exercise of a portion of the underwriters over allotment option at a price of $7.75 for net proceeds of $3.6 million after offering expenses..
At December 31, 2007, we have 2,816,549 common shares reserved for future issuance under the Employee Stock Purchase Plan ("Purchase Plan") and for the exercise of common stock options pursuant to the 1994 Amended and Restated Equity Incentive Plan ("Equity Incentive Plan") and the 1996 Amended and Restated Director Stock Option Plan ("Directors Plan").
In January 2005, we completed a stock offering whereby we sold 5.79 million shares of common stock at $5.25 per share for aggregate net proceeds of $28.3 million after commissions and offering expenses.
12. STOCK OPTION PLANS
During 2005, our shareholders approved an amendment to the Equity Incentive Plan to increase the number of shares available to 9,600,000. All shares are awarded at the discretion of our Board of
74
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
12. STOCK OPTION PLANS (Continued)
Directors in a variety of stock based forms including stock options and restricted stock. Pursuant to the Equity Incentive Plan, incentive stock options may not be granted at less than the fair market value of our common stock at the date of the grant, and the option term may not exceed ten years. Stock options issued pursuant to the Equity Incentive Plan generally vest over four years. For holders of 10% or more of our voting stock, options may not be granted at less than 110% of the fair market value of the common stock at the date of the grant, and the option term may not exceed five years. Stock appreciation rights granted in tandem with an option shall have an exercise price not less than the exercise price of the related option. As of December 31, 2007, no stock appreciation rights have been issued. At December 31, 2007, there were 2,016,409 shares available for future grant under the Equity Incentive Plan.
During 2005, our Shareholders approved an amendment to the Director Plan to increase the number of shares available to 500,500. In May 2006, our shareholders approved an amendment to the Director Plan to increase the number of options granted to the Chairman of the Board and Directors. Under the terms of the Director Plan, options to purchase shares of common stock are automatically granted (A) to the Chairman of the Board of Directors (1) upon his or her initial election or appointment in the amount of 25,000 and vesting over three years and (2) upon his or her re-election or continuation on our board immediately after each annual meeting of stockholders in the amount of 15,000 and vesting immediately, and (B) to each other Director (1) upon his or her initial election to our board in the amount of 20,000 and vesting over three years and (2) upon his or her re-election or continuation on our board in the amount of 10,000 and vesting immediately. All options granted pursuant to the Director Plan have a term of ten years with exercise prices equal to fair market value on the date of grant. In May 2007, our shareholders approved an amendment to the Director Plan to increase the number of shares available from 500,500 to 750,500. Through December 31, 2007, options to purchase 496,949 shares of common stock have been granted under this plan of which 382,501 shares are currently exercisable. As of December 31, 2007, 315,051 shares are available for future grant.
As of December 31, 2007 and 2006, no stock-based compensation expense was capitalized and there were no recognized tax benefits associated with the stock-based compensation charge. For the year ended December 31, 2007, stock-based compensation expense of $637, included in research and development, was related to Boston Biomedical, Inc. transition costs (see Note 16, Boston Biomedical, Inc. Collaboration in this Form 10-K). Additionally in the year ended December 31, 2007, $438 of stock-based compensation expense was incurred in conjunction with the acceleration of vesting of 103,798 stock options and the extension of the post-employment exercise period of 395,942 stock options held by certain employees who will meet eligibility criteria for a retirement benefit within the next four years. On October 4, 2007, the exercise period associated with 1,115,000 stock options was extended and the vesting of 165,625 stock options was accelerated in conjunction with an amendment to the CEO's employment agreement. The amount of stock option expense associated with this amendment was $703 in the year ended December 31, 2007 and will be $1,406 in the first quarter of 2008.
75
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
12. STOCK OPTION PLANS (Continued)
During 2007, we issued 27,500 fully-vested options to certain members of our Scientific Advisory Board under the Equity Incentive Plan. In 2006 and 2005, we issued 15,000 and 13,500 of such grants, respectively. Compensation expense in 2007, 2006 and 2005 was $121, $74 and $58, respectively. In 2005, we amended the terms of certain options awarded to employees whose positions were terminated, resulting in a non-cash charge of $289.
Option activity under the Plans for the years ended December 31, 2005, 2006 and 2007 was as follows:
Stock Options
| | Number
of Shares
| | Weighted Average
Exercise Price
|
|---|
| Outstanding as of December 31, 2004 | | 4,228,511 | | $ | 8.10 |
| Granted | | 1,199,705 | | | 6.42 |
| Exercised | | (406,610 | ) | | 4.50 |
| Cancelled | | (937,341 | ) | | 10.53 |
| | |
| |
|
| Outstanding as of December 31, 2005 | | 4,084,265 | | | 7.41 |
| Granted | | 1,464,260 | | | 5.70 |
| Exercised | | (348,403 | ) | | 4.40 |
| Cancelled | | (1,327,176 | ) | | 8.50 |
| | |
| |
|
| Outstanding as of December 31, 2006 | | 3,872,946 | | | 6.66 |
| Granted | | 1,334,825 | | | 6.60 |
| Exercised | | (355,029 | ) | | 5.14 |
| Cancelled | | (374,880 | ) | | 6.46 |
| | |
| |
|
| Outstanding as of December 31, 2007 | | 4,477,862 | | $ | 6.78 |
| | |
| |
|
| Exercisable as of December 31, 2007 | | 2,297,283 | | $ | 7.35 |
| | |
| |
|
| Weighted average grant-date fair value of options granted during the year ended December 31, 2007 | | | | $ | 3.46 |
| | | | |
|
The following table summarizes information about options outstanding at December 31, 2007:
| | Options Outstanding
| | Options Exercisable
|
|---|
Range of Exercise Prices
| | Number
Outstanding at
December 31, 2007
| | Weighted
Average
Remaining
Contractual Life
| | Weighted
Average
Exercise
Price
| | Exercisable as of
December 31, 2007
| | Weighted
Average
Exercise
Price
|
|---|
| $ 2.80 — 5.60 | | 1,256,202 | | 5.0 | | $ | 4.72 | | 991,302 | | $ | 4.61 |
| 5.60 — 8.40 | | 2,664,935 | | 7.9 | | | 6.27 | | 785,256 | | | 6.29 |
| 8.40 — 11.20 | | 252,510 | | 6.0 | | | 9.50 | | 216,510 | | | 9.53 |
| 11.20 — 14.00 | | 123,328 | | 4.0 | | | 13.32 | | 123,328 | | | 13.32 |
| 14.00 — 16.80 | | 14,250 | | 0.4 | | | 16.60 | | 14,250 | | | 16.60 |
| 16.80 — 19.60 | | 53,137 | | 2.1 | | | 18.22 | | 53,137 | | | 18.22 |
| 19.60 — 22.40 | | 86,000 | | 2.3 | | | 20.04 | | 86,000 | | | 20.04 |
| 22.40 — 25.20 | | 7,500 | | 2.9 | | | 23.13 | | 7,500 | | | 23.13 |
| 25.20 — 28.00 | | 20,000 | | 3.1 | | | 28.00 | | 20,000 | | | 28.00 |
| | |
| |
| |
| |
| |
|
| | | 4,477,862 | | 6.6 | | $ | 6.78 | | 2,297,283 | | $ | 7.35 |
| | |
| |
| |
| |
| |
|
76
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
12. STOCK OPTION PLANS (Continued)
The aggregate intrinsic value of options outstanding at December 31, 2007 was $1.4 million, of which $1.2 million related to exercisable options. The weighted average fair value of options granted in year ended December 31, 2007, 2006 and 2005 was $3.46, $4.03 and $4.52 per share, respectively. The intrinsic value of options exercised in the year ended December 31, 2007, 2006 and 2005 was $1,173, $317 and $805, respectively.
Shares vested, expected to vest and exercisable as of December 31, 2007 are as follows:
| | Shares
| | Weighted-Average
Exercise Price
| | Weighted-Average
Remaining
Contractual
Term (in years)
| | Aggregate
Intrinsic Value
|
|---|
| Vested and unvested expected to vest at December 31, 2007 | | 4,267,457 | | $ | 6.78 | | 6.1 | | $ | 1,371 |
Exercisable at December 31, 2007 |
|
2,297,283 |
|
$ |
7.35 |
|
4.5 |
|
$ |
1,185 |
The total compensation cost not yet recognized as of December 31, 2007 related to non-vested option awards was $6.1 million, which will be recognized over a weighted-average period of 2.7 years. During the year ended December 31, 2007, there were 305,603 shares forfeited with a weighted average grant date fair values of $3.99 per share. The weighted average remaining contractual life for options exercisable at December 31, 2007 was 4.8 years.
On January 19, 2006, we granted 40,860 shares of restricted stock to employees of our chemistry services business, which vested upon their separation from ArQule pursuant to a plan of termination (See Note 10, Restructuring Actions). Through December 31, 2006, 3,880 shares were forfeited, and the remaining 36,980 shares were fully vested. The shares of restricted stock were issued at no cost to the recipients. The fair value of the restricted stock at the time of grant was $5.73 per share, and was expensed ratably over the vesting period. We recognized share-based compensation expense related to the restricted stock of $212 for the year ended December 31, 2006.
In 1996, the stockholders adopted the Purchase Plan. This plan enables eligible employees to exercise rights to purchase our common stock at 85% of the fair market value of the stock on the date the right was granted or the date the right is exercised, whichever is lower. Rights to purchase shares under the Purchase Plan are granted by the Board of Directors. The rights are exercisable during a period determined by the Board of Directors; however, in no event will the period be longer than twenty-seven months. The Purchase Plan is available to substantially all employees, subject to certain limitations. In May 2005, our shareholders approved an amendment to the Purchase Plan to increase the aggregate number of shares of the Company's common stock that may be issued from 1,020,000 shares to 1,230,000 shares. In May 2007, our shareholders approved an amendment to the Purchase Plan to increase the aggregate number of shares of the Company's common stock that may be issued from 1,230,000 shares to 1,600,000. As of December 31, 2007, 1,114,911 shares have been purchased and 485,089 shares are available for future sale under the Purchase Plan.
13. INCOME TAXES
There was no current or deferred tax expense for the year ended December 31, 2007, 2006, or 2005.
77
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
13. INCOME TAXES (Continued)
The following is a reconciliation between the U.S. federal statutory rate and the effective tax rate for continuing operations for the years ended December 31, 2007, 2006 and 2005:
| | 2007
| | 2006
| | 2005
| |
|---|
| Income tax (benefit) expense at statutory rate | | $ | (18,147 | ) | $ | (16,056 | ) | $ | (8,033 | ) |
| State tax (benefit) expense, net of Federal tax (benefit) expense | | | (3,118 | ) | | (2,799 | ) | | (1,431 | ) |
| Permanent items | | | 982 | | | 688 | | | 108 | |
| Effect of change in valuation allowance | | | 21,776 | | | 19,662 | | | 10,133 | |
| Tax credits | | | (2,202 | ) | | (2,001 | ) | | (893 | ) |
| Other | | | 709 | | | 506 | | | 116 | |
| | |
| |
| |
| |
| Tax expense | | $ | — | | $ | — | | $ | — | |
| | |
| |
| |
| |
The income tax effect of temporary differences comprising the deferred tax assets and deferred tax liabilities on the accompanying balance sheets is a result of the following at December 31, 2007 and 2006:
| | 2007
| | 2006
| |
|---|
| Deferred tax assets: | | | | | | | |
| | Pre-operating costs capitalized for tax purposes | | $ | 21 | | $ | 41 | |
| | Net operating loss carryforwards | | | 62,045 | | | 41,534 | |
| | Tax credit carryforwards | | | 14,345 | | | 12,303 | |
| | Equity based compensation | | | 1,085 | | | 518 | |
| | Book depreciation in excess of tax | | | 3,053 | | | 3,674 | |
| | Reserves and accruals | | | 291 | | | 456 | |
| | Deferred revenue | | | 1,564 | | | 2,109 | |
| | Loss on investment | | | 2,013 | | | 2,013 | |
| | Other | | | 48 | | | 41 | |
| | |
| |
| |
| | | | 84,465 | | | 62,689 | |
| | Valuation allowance | | | (84,465 | ) | | (62,689 | ) |
| Deferred tax liabilities | | | — | | | — | |
| | |
| |
| |
| | Net deferred tax assets | | $ | — | | $ | — | |
| | |
| |
| |
Total valuation allowance increased by $21,776 for the year ended December 31, 2007. As required by Statement of Financial Accounting Standards No. 109,Accounting for Income Taxes, we have evaluated positive and negative evidence bearing upon the realizability of our deferred tax assets, which are comprised principally of federal net operating loss ("NOL"), net capital loss and research & development credit carryforwards. We have determined that it is more likely than not that we will not recognize the benefits of our federal and state deferred tax assets and, as a result, we have established a full valuation allowance against our net deferred tax assets as of December 31, 2007.
As of December 31, 2007, we had federal NOL, state NOL, and research and development credit carryforwards of approximately $179,629, $114,057 and $16,038 respectively, which can be used to offset future federal and state income tax liabilities and expire at various dates through 2027. Federal net
78
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
13. INCOME TAXES (Continued)
capital loss carryforwards of approximately $5,000 can be used to offset future federal capital gains and expire at various dates through 2008. Approximately $17,394 of our federal NOL and $1,679 of our state NOL were generated from excess tax deductions from share-based awards, the tax benefit of which will be credited to additional paid-in-capital when the deductions reduce current taxes payable.
We adopted the provisions of FASB Interpretation No. 48 ("FIN 48")Accounting for Uncertainty in Income Taxes, an interpretation of FASB Statement No. 109 ("SFAS 109") on January 1, 2007. As a result of the implementation of FIN 48, we recorded no adjustment for unrecognized income tax benefits. At the adoption date of January 1, 2007 and also at December 31, 2007, we had no unrecognized tax benefits. We do not expect that the total amount of unrecognized tax benefits will significantly increase in the next twelve months.
We recognize interest and penalties related to uncertain tax positions in income tax expense. As of December 31, 2007, we had no accrued interest or penalties related to uncertain tax positions. The tax years 2003 through 2006 remain open to examination by the major taxing jurisdictions to which we are subject, which is primarily the U.S.
Utilization of NOL and R&D credit carryforwards may be subject to a substantial annual limitation due to ownership change limitations that have occurred previously or that could occur in the future provided by Section 382 of the Internal Revenue Code of 1986, as amended, as well as similar state provisions. These ownership changes may limit the amount of NOL and R&D credit carryforwards that can be utilized annually to offset future taxable income and tax, respectively. In general, an ownership change, as defined by Section 382, results from transactions increasing the ownership of certain shareholders or public groups in the stock of a corporation by more than 50 percentage points over a three-year period. Since the Company's formation, the Company has raised capital through the issuance of capital stock on several occasions which, combined with the purchasing shareholders' subsequent disposition of those shares, may have resulted in a change of control, as defined by Section 382, or could result in a change of control in the future upon subsequent disposition. The Company has not currently completed a study to assess whether a change of control has occurred or whether there have been multiple changes of control since the Company's formation due to the significant complexity and cost associated with such study and that there could be additional changes in control in the future. If we have experienced a change of control at any time since Company formation, utilization of our NOL or R&D credit carryforwards would be subject to an annual limitation under Section 382. Any limitation may result in expiration of a portion of the NOL or R&D credit carryforwards before utilization. Further, until a study is completed and any limitation known, no amounts are being presented as an uncertain tax position under FIN 48.
79
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
14. COMMITMENTS AND CONTINGENCIES
Leases
We lease facilities under non-cancelable operating leases. At December 31, 2007, the minimum lease commitments for all leased facilities, net of sublease income, are as follows:
YEAR ENDING DECEMBER 31,
| | OPERATING LEASES
|
|---|
| 2008 | | $ | 3,854 |
| 2009 | | | 3,982 |
| 2010 | | | 3,476 |
| 2011 | | | 3,523 |
| 2012 | | | 3,573 |
| Thereafter | | | 7,327 |
| | |
|
| Total minimum lease payments | | $ | 25,735 |
| | |
|
Included in the total minimum payments for operating leases is approximately $1.3 million related to unoccupied real estate in California, net of contractual sublease income, which is accrued as a net liability as a part of the Company's restructuring accrual. (See Note 10).
Rent expense under non-cancelable operating leases was approximately $2,935, $3,142 and $2,341 for the years ended December 31, 2007, 2006 and 2005, respectively. Sublease income, which is recorded as a reduction of rent expense, was approximately $425, $402 and $316 for the years ended December 31, 2007, 2006 and 2005 respectively.
On January 16, 2002, we brought a complaint in the Superior Court of Middlesex County in the Commonwealth of Massachusetts for declaratory relief and damages against Cummings Properties, LLC ("Cummings") arising from a dispute over increased lease rates related to approximately 35,500 square feet of laboratory and office space in Medford, Massachusetts. As a result of developments in the pre-trial phase of our litigation, in the fourth quarter of 2004, we recorded an expense of $637 to accrue the difference between our contractual lease obligations for a portion of the Medford facility and the amount of contractual sublease income we expected to receive over the term of the lease ("accrued loss on sublease"). On October 11, 2005, the parties agreed to settle the lawsuit and file with the Court a stipulation of dismissal of the lawsuit with prejudice. In exchange for Cummings forgiving a portion of the rental payment obligations for the period from November 1, 2005 through July 30, 2006, we paid Cummings $262 and assigned our sublease rent payments during that period to Cummings and guaranteed those payments. There are no remaining sublease payments due at December 31, 2007.
15. CONCENTRATION OF CREDIT RISK
Revenue from one customer represented approximately 72% of total revenue during 2007 and 100% in 2006 and 2005. In 2007, revenue from another customer represented approximately 25% of total revenue. One customer accounted for 100% of our accounts receivable balance at December 31, 2005. There was no accounts receivable balance at December 31, 2006 or 2007.
80
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
16. BOSTON BIOMEDICAL, INC. COLLABORATION
In January 2007, we entered into a $5.0 million, eight-month sponsored research agreement with the newly established Boston Biomedical, Inc. ("BBI"), an independent corporation led by our former chief scientific officer. Approximately 26 former employees of ArQule have joined BBI.
BBI conducts scientific research under the agreement that includes a number ofin vivo andin vitro studies, reports and publications related to mechanisms of action and biomarkers for our clinical-stage products. These products include ARQ 197, ARQ 501 and ARQ 171. We retain all intellectual property and technology rights related to research conducted by BBI employees under the contract. ArQule has no equity position in BBI.
In connection with the foregoing events, on January 26, 2007, our former chief scientific officer entered into a separation agreement and general release with us and was paid a lump sum severance payment comprised of (i) one year's salary in the amount of $321 (ii) the average of his cash bonuses over the last two years in the amount of $110 and (iii) the amount of $113 to which he was entitled under our Annual Incentive Program for fiscal year 2006.
In addition, he was granted an option to purchase 64,375 shares of our common stock, which is fully vested and exercisable on the date of grant and will expire on December 31, 2008. His previously vested option grants covering 216,250 shares were amended to extend the exercise period through December 31, 2007. In connection with his appointment as Chairman of our Scientific Advisory Board, he was granted an additional option to purchase 12,500 shares, which is fully vested and exercisable on the date of grant and will expire ten years after the date of grant. As a result of his separation from service, all his unvested options have lapsed.
Approximately 26 of our former employees joined BBI in January 2007 and each employee who transitioned to BBI executed and delivered a Separation Agreement and General Release. In consideration for entering into such agreement, each employee received a fully-vested option to purchase shares of our common stock with an exercise period terminating on December 31, 2008, as well as an amendment to their previously vested stock options to extend the exercise period through December 31, 2007. The total number of fully vested stock options issued to these employees was 87,500, and the total number of stock options that were amended to extend the exercise was 92,504. As a result of separation of service all unvested options of such employees have lapsed.
In the first quarter of 2007, we expensed approximately: $431 related to lump sum cash payments under the separation and general release agreement with our former chief scientific officer, as well as certain non-cash charges for stock based compensation, including $201 for stock options granted to him; and $168 arising from the extension of the exercise period of his vested options. Additionally, in the first quarter of 2007, we expensed approximately $197 for stock options granted to other employees related to their separation agreements and releases, and $71 arising from the extension of the exercise period of their vested options.
Through December 31, 2007, in connection with the BBI sponsored research agreement, we incurred $4,669 of research and development expense. We made payments of $4,351 to BBI and the remaining $318 is reported in other accrued expenses.
81
ARQULE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
17. SELECTED QUARTERLY FINANCIAL DATA (UNAUDITED)
| | FIRST
QUARTER
| | SECOND
QUARTER
| | THIRD
QUARTER
| | FOURTH
QUARTER
| |
|---|
| 2007 | | | | | | | | | | | | | |
| Net revenues | | $ | 1,652 | | $ | 2,235 | | $ | 2,736 | | $ | 2,542 | |
| Net loss from continuing operations | | | (14,504 | ) | | (13,361 | ) | | (11,118 | ) | | (14,389 | ) |
| Income (loss) from discontinued operations | | | — | | | — | | | — | | | — | |
| | |
| |
| |
| |
| |
| Net income (loss) | | $ | (14,504 | ) | $ | (13,361 | ) | $ | (11,118 | ) | $ | (14,389 | ) |
Basic and diluted income (loss) per share: |
|
|
|
|
|
|
|
|
|
|
|
|
|
| Net loss from continuing operations | | $ | (0.40 | ) | $ | (0.36 | ) | $ | (0.26 | ) | $ | (0.33 | ) |
| Net income from discontinued operations | | | — | | | — | | | — | | | — | |
| | |
| |
| |
| |
| |
| Net income (loss) per share | | $ | (0.40 | ) | $ | (0.36 | ) | $ | (0.26 | ) | $ | (0.33 | ) |
| | FIRST
QUARTER
| | SECOND
QUARTER
| | THIRD
QUARTER
| | FOURTH
QUARTER
| |
|---|
| 2006 | | | | | | | | | | | | | |
| Net revenues | | $ | 1,652 | | $ | 1,652 | | $ | 1,652 | | $ | 1,670 | |
| Net loss from continuing operations | | | (9,719 | ) | | (9,415 | ) | | (15,321 | ) | | (12,768 | ) |
| Income (loss) from discontinued operations | | | 14,005 | | | 1,840 | | | — | | | (62 | ) |
| | |
| |
| |
| |
| |
| Net income (loss) | | | 4,286 | | | (7,575 | ) | | (15,321 | ) | | (12,830 | ) |
Basic and diluted income (loss) per share: |
|
|
|
|
|
|
|
|
|
|
|
|
|
| Net loss from continuing operations | | $ | (0.28 | ) | $ | (0.26 | ) | $ | (0.43 | ) | $ | (0.36 | ) |
| Net income from discontinued operations | | | 0.40 | | | 0.05 | | | — | | | — | |
| | |
| |
| |
| |
| |
| Net income (loss) per share | | $ | 0.12 | | $ | (0.21 | ) | $ | (0.43 | ) | $ | (0.36 | ) |
82
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Business Officer (Principal Financial Officer), evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2007. The term "disclosure controls and procedures," as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended ("Exchange Act"), means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC's rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company's management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Our management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and our management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2007, our Chief Executive Officer and Chief Operating Officer (Principal Financial Officer) concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Management's Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under the framework in Internal Control—Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2007.
The effectiveness of our internal control over financial reporting as of December 31, 2007 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report which is included herein.
Changes in Internal Control
There has been no change in our internal control over financial reporting during the quarter ended December 31, 2007 that has materially effected or is likely to materially affect our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
None.
83
PART III
Except as otherwise indicated, the following information required by the Instructions to Form 10-K is incorporated herein by reference from various sections of the ArQule, Inc. Proxy Statement for the annual meeting of shareholders to be held on May 16, 2008, as summarized below:
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS, AND CORPORATE GOVERNANCE
"Election of Directors;" "Section 16(a) Beneficial Ownership Reporting Compliance;" "Corporate Governance;" and "Board Committees and Meetings."
Information regarding the executive officers of the Company is incorporated by reference from "Executive Officers of the Registrant" at the end of Item 1 of this report.
ITEM 11. EXECUTIVE COMPENSATION
"Compensation Discussion and Analysis;" "Executive Compensation;" "Director Compensation;" "Compensation, Nominating and Governance Committee Interlocks and Insider Participation;" and "Compensation Committee Report."
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
"Share Ownership of Certain Beneficial Owners" and "Securities Authorized for Issuance Under Equity Compensation Plans."
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
"Certain Relationships and Related Transactions" and "Director Independence."
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
Fees paid to the Company's independent registered public accounting firm are disclosed under the caption "Ratification of the Selection of an Independent Registered Public Accountants."
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENTS SCHEDULES
The financial statements are listed under Item 8 of this report.
The financial statement schedules are omitted from this report because they are not applicable or required information and are shown in the financial statements of the footnotes thereto.
84
EXHIBIT
NO.
| | DESCRIPTION
|
|---|
| 3.1 | | Amended and Restated Certificate of Incorporation of the Company. Filed as Exhibit 3.1 to the Company's Registration Statement on Form S-1 (File No. 333-22945) and incorporated herein by reference. |
3.2 |
|
Certificate of Amendment to Amended and Restated Certificate of Incorporation filed as Exhibit 3.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2006 (File No. 000-21429) and incorporated herein by reference. |
3.3 |
|
Amended and Restated By-laws of the Company. Filed as Exhibit 3.1 to the Company's Current Report on Form 8-K filed on November 19, 2007 (File No. 000-21429) and incorporated herein by reference. |
4.1 |
|
Specimen Common Stock Certificate. Filed as Exhibit 4.1 to the Company's Registration Statement on Form S-1 (File No. 333-11105) and incorporated herein by reference. |
10.1* |
|
Amended and Restated 1994 Equity Incentive Plan, as amended through May 11, 2005. Filed as Exhibit 4 to the Company's Registration Statement on Form S-8 filed on September 30, 2005 (File No. 333-128740) and incorporated herein by reference. |
10.2* |
|
Amended and Restated 1996 Employee Stock Purchase Plan. Filed as Annex B to the Company's Definitive Proxy Statement filed on April 16, 2007 (File No. 000-21429) and incorporated herein by reference. |
10.3* |
|
Amended and Restated 1996 Director Stock Option Plan. Filed as Annex A to the Company's Definitive Proxy Statement filed on April 16, 2007 (File No. 000-21429) and incorporated herein by reference. |
10.4* |
|
2005 Director Stock Compensation Plan. Filed as Exhibit 4 to the Company's Registration Statement on Form S-8 filed on December 6, 2005 (File No. 333-130159) and incorporated herein by reference. |
10.5 |
|
Lease by and between Pacific Shores Center LLC and the Company, dated March 1, 2002. Filed as Exhibit 10.40 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2002 (File No. 000-21429) and incorporated herein by reference. |
10.6* |
|
Employment Agreement between the Company and Stephen A Hill, dated January 1, 2004. Filed as Exhibit 10.45 to the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2003 filed with the Commission on March 12, 2004 (File No. 000-21429) and incorporated herein by reference. |
10.7+ |
|
Strategic Alliance Agreement by and between F. Hoffmann–La Roche Ltd., Hoffmann–La Roche Inc. and the Company dated April 1, 2004. Filed as Exhibit 10.49+ to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31,2004 filed with the Commission on May 7, 2004 (File No. 000-21429) and incorporated herein by reference. |
10.8 |
|
Form of Agreement of Purchase and Sale between ARE-MA Region No. 20, LLC and the Company, dated April 28, 2005. Filed as Exhibit 10.1 to the Company's Current Report on Form 8-K filed with the Commission on May 6, 2005 (File No. 000-21429) and incorporated herein by reference. |
85
10.9 |
|
Amended and Restated Lease by and between ARE-MA Region No. 20, LLC and the Company, dated June 30, 2005. Filed as Exhibit 10.21 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2005 filed with the Commission on August 5, 2005 (file No. 000-21429) and incorporated herein by reference. |
10.10* |
|
Employment Agreement between the Company and Peter S. Lawrence, dated April 13, 2006. Filed as Exhibit 10.1 to the Company's Current Report on Form 8-K dated April 18, 2006 (File No. 000-21429) and incorporated herein by reference. |
10.11* |
|
Employment Agreement between the Company and Nigel J. Rulewski, MD, dated August 1, 2006. Filed as Exhibit 10.1 to the Company's Current Report on Form 8-K dated August 1, 2006 (File No. 000-21429) and incorporated herein by reference. |
10.12+ |
|
Exclusive License Agreement, by and between the Company and Kyowa Hakko Kogyo Co., Ltd. Filed as Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2007 filed with the Commission on August 7, 2007. |
10.13* |
|
Amendment to Employment Agreement, dated as of October 4, 2007, by and between the Company and Peter S. Lawrence. Filed as Exhibit 10.1 to the Company's Current Report on Form 8-K filed on October 10, 2007 (File No. 000-21429) , and incorporated herein by reference. |
10.14* |
|
Amendment to Employment Agreement, dated as of October 4, 2007, by and between the Company and Stephen A. Hill. Filed as Exhibit 10.3 to the Company's Current Report on Form 8-K filed on October 10, 2007 (File No. 000-21429), and incorporated herein by reference. |
10.15* |
|
Amendment to Employment Agreement, effective as of January 7, 2008, by and between the Company and Stephen A. Hill. Filed as Exhibit 10.1 to the Company's Current Report on Form 8-K filed on January 8, 2008 (File No. 000-21429), and incorporated herein by reference. |
10.16* |
|
Form of Incentive Stock Option Agreement. Filed herewith. |
10.17* |
|
Form of Non-Statutory Stock Option Agreement. Filed herewith. |
23.1 |
|
Consent of PricewaterhouseCoopers LLP, an Independent Registered Public Accounting Firm, filed herewith. |
31.1 |
|
Rule 13a-14(a) Certificate of Chief Executive Officer, filed herewith. |
31.2 |
|
Rule 13a-14(a) Certificate of Principal Financial Officer, filed herewith. |
32 |
|
Rule 13a-14(b) Certificate of Chief Executive Officer and Principal Financial Officer, filed herewith. |
- *
- Indicates a management contract or compensatory plan.
- +
- Certain confidential material contained in the document has been omitted and filed separately with the Securities and Exchange Commission pursuant to Rule 406 of the Securities Act of 1933, as amended or Rule 24b-2 of the Securities and Exchange Act of 1934, as amended.
86
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | ARQULE, INC. |
|
|
By: |
|
/s/ STEPHEN A. HILL
Stephen A. Hill
President and Chief Executive Officer |
|
|
Date: March 17, 2008 |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
SIGNATURE
| | TITLE
| | DATE
|
|---|
|
|
|
|
|
/s/ STEPHEN A. HILL
Stephen A. Hill | | President, Chief Executive Officer and Director
(Principal Executive Officer) | | March 17, 2008 |
/s/ PETER S. LAWRENCE
Peter S. Lawrence |
|
Chief Operating Officer
(Principal Financial Officer) |
|
March 17, 2008 |
/s/ ROBERT J. WEISKOPF
Robert J. Weiskopf |
|
Vice President of Finance,
Corporate Controller and Treasurer
(Principal Accounting Officer) |
|
March 17, 2008 |
/s/ PATRICK J. ZENNER
Patrick J. Zenner |
|
Director—Chairman of the Board |
|
March 17, 2008 |
/s/ TIMOTHY C. BARABE
Timothy C. Barabe |
|
Director |
|
March 17, 2008 |
/s/ RONALD M. LINDSAY
Ronald M. Lindsay |
|
Director |
|
March 17, 2008 |
/s/ MICHAEL D. LOBERG
Michael D. Loberg |
|
Director |
|
March 17, 2008 |
/s/ WILLIAM G. MESSENGER
William G. Messenger |
|
Director |
|
March 17, 2008 |
/s/ NANCY A. SIMONIAN
Nancy A. Simonian |
|
Director |
|
March 17, 2008 |
87
QuickLinks
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 FOR THE FISCAL YEAR ENDED DECEMBER 31, 2007 COMMISSION FILE NUMBER: 000-21429 ARQULE, INC. (EXACT NAME OF REGISTRANT AS SPECIFIED IN ITS CHARTER)DOCUMENTS INCORPORATED BY REFERENCEIMPORTANT FACTORS REGARDING FORWARD-LOOKING STATEMENTSPART IPART IIINDEX TO CONSOLIDATED FINANCIAL STATEMENTSReport of Independent Registered Public Accounting FirmARQULE, INC. CONSOLIDATED BALANCE SHEETSARQULE, INC. CONSOLIDATED STATEMENTS OF OPERATIONSARQULE, INC. CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY AND COMPREHENSIVE LOSS (IN THOUSANDS, EXCEPT SHARE DATA)ARQULE, INC. CONSOLIDATED STATEMENTS OF CASH FLOWSARQULE, INC. NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)PART IIIPART IVSIGNATURES