UNITED STATES SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| |
þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2008
OR
| |
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File No. 000-30334
Angiotech Pharmaceuticals, Inc.
(Exact Name of Registrant as Specified in its Charter)
| | |
British Columbia, Canada | | 98-0226269 |
(State or Other Jurisdiction of Incorporation or Organization) | | (I.R.S. Employer Identification No.) |
| | |
1618 Station Street, Vancouver, BC, Canada | | V6A 1B6 |
(Address of principal executive offices) | | (Zip Code) |
Registrant’s telephone number, including area code: (604) 221-7676
Securities registered pursuant to Section 12(b) of the Act:
| | |
Title of each class | | Name of each exchange on which registered |
Common shares, without par value | | The NASDAQ Global Select Market |
Class I preference shares, without par value | | The NASDAQ Global Select Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes o No þ
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes o No þ
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months and (2) has been subject to such filing requirements for the past 90 days. Yes þ No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ¨
Accelerated filer x
Non-accelerated filer ¨ (Do not check if a smaller reporting company)
Smaller reporting company¨
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes o No þ
The aggregate market value of the common shares held by non-affiliates as of June 30, 2008 of the registrant (based on the last reported sale price of the common shares of U.S. $2.98, as reported on The NASDAQ Global Select Market) was approximately U.S. $253,663,509.
As of March 12, 2009 the registrant had 85,121,983 outstanding common shares.
DOCUMENTS INCORPORATED BY REFERENCE
Part III of this Annual Report on Form 10-K incorporates by reference information from the definitive Proxy Statement for the registrant’s 2009 annual general meeting of shareholders.
ANGIOTECH PHARMACEUTICALS INC.
TABLE OF CONTENTS
ANNUAL REPORT ON FORM 10K FOR THE YEAR ENDED DECEMBER 31, 2008
| | | |
PART I |
| |
| Item 1. | BUSINESS | 4 |
| Item 1A. | RISK FACTORS | 14 |
| Item 1B. | UNRESOLVED STAFF COMMENTS | 29 |
| Item 2 | PROPERTIES | 30 |
| Item 3. | LEGAL PROCEEDINGS | 31 |
| Item 4. | SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS | 32 |
| |
| |
PART II |
| |
| Item 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER | |
| | MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES | 33 |
| Item 6. | SELECTED FINANCIAL DATA | 35 |
| Item 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND | |
| | RESULTS OF OPERATIONS | 38 |
| Item 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK | 58 |
| Item 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA | 59 |
| Item 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND | |
| | FINANCIAL DISCLOSURE | 59 |
| Item 9A. | CONTROLS AND PROCEDURES | 59 |
| Item 9B. | OTHER INFORMATION | 60 |
| |
| |
PART III |
| |
| Item 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE | 61 |
| Item 11. | EXECUTIVE COMPENSATION | 61 |
| Item 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND | |
| | RELATED STOCKHOLDER MATTERS | 61 |
| Item 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR | |
| | INDEPENDENCE | 61 |
| Item 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES | 61 |
| |
| |
PART IV |
| |
| Item 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES | 62 |
| |
| |
| SIGNATURES | 63 |
- 2 -
In this Annual Report on Form 10-K, references to “the Company”, “Angiotech”, “us” or “we” are to Angiotech Pharmaceuticals, Inc. and all of its subsidiaries as a whole, except where it is clear that these terms only refer to Angiotech Pharmaceuticals, Inc.
Accounting Standards
In this Annual Report on Form 10-K, all dollar amounts are in U.S. dollars, except where otherwise stated and financial reporting is made in accordance with U.S. generally accepted accounting principles (“U.S. GAAP”).
Currency and Exchange Rates
We use the U.S. dollar as our reporting currency, while the Canadian dollar is the functional currency for Angiotech Pharmaceuticals, Inc. and its Canadian subsidiaries, the U.K pound is the functional currency of our U.K subsidiary, Pearsalls Limited, the Danish Kroner is the functional currency of our Danish subsidiary, PBN Medicals Denmark A/S, and the U.S. dollar is the functional currency for all other subsidiaries.
NOTICE REGARDING WEBSITE ACCESS TO COMPANY REPORTS
We file electronically with the Securities and Exchange Commission our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q and Current Reports on Form 8-K pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 (“the Exchange Act”). You may obtain a free copy of our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q and Current Reports on Form 8-K, and any amendments to those reports, on the day of filing with the SEC and on our website at: www.angiotech.com. Our website and the information on our website is not part of this Annual Report on Form 10-K.
- 3 -
PART I
Item 1.
BUSINESS
Summary
We are a specialty pharmaceutical and medical device company that discovers, develops and markets innovative technologies primarily focused on acute and surgical applications. We generate our revenue through our sales of medical products and components, as well as from royalties derived from sales by our partners of products utilizing certain of our proprietary technologies. For the year ended December 31, 2008, we recorded $190.8 million in direct sales of our various medical products and $92.5 million in royalties and license fees received from our partners.
Our research and development efforts focus on understanding and characterizing biological conditions that often occur concurrent with medical device implantation, surgery or acute trauma, including scar formation and inflammation, cell proliferation, bleeding and coagulation, infection, and tumor tissue overgrowth. Our strategy is to utilize our various technologies in the areas of drugs, drug delivery, surface modification, biomaterials and medical devices to create and commercialize novel, proprietary medical products that reduce surgical procedure side effects, improve surgical outcomes, shorten hospital stays, or are easier or safer for a physician to use.
We develop our products using a proprietary and systematic discovery approach. We use our drug screening capabilities to identify new uses for known pharmaceutical compounds. We look for compounds that address the underlying biological causes of conditions that can occur with medical device implantation, surgery or acute trauma. Once appropriate drugs have been identified, we work to formulate the drug, or a combination of drugs, with our portfolio of drug, drug delivery and surface modification technologies and biomaterials to develop a novel surgical implant or medical device. We have patent protected, or have filed patent applications for, certain of our technology and many of our products and potential product candidates.
In March 2007, due to declines in the amount of royalties received from sales of TAXUS by Angiotech's partners, the high levels of cash interest payable on Angiotech's outstanding debt and the capital-intensive nature of Angiotech’s business, Angiotech determined that its balance sheet was not supported by its existing business model and that a restructuring was necessary. Although Angiotech believes it has developed planned courses of action and identified other opportunities to mitigate these operating and liquidity risks, there can be no assurance that Angiotech will be able to achieve any or all of these plans.
In 2008, TAXUS royalties declined more significantly than anticipated and Angiotech’s stock price declined precipitously, such that the market value of the company became significantly less than the outstanding principal amount of its indebtedness. During this time, Angiotech continued to unsuccessfully explore various financial and strategic alternatives. In September 2008, Angiotech announced a restructuring effort to address its liquidity concerns, which included efforts to reduce operating costs through significant cuts to its operating budget and efforts to focus its business on its more promising Medical Devices product segment and away from its Pharmaceutical Technologies segment.
In March 2009, we announced that it had entered into a new secured credit facility, which is intended to provide the company additional near-term liquidity and enable the company to continue the evaluation and exploration of various financial and strategic alternatives that may reduce our total outstanding debt obligations and provide the Company with greater financial flexibility.
Business Strategy
Our strategy is to apply our various technologies to develop and commercialize novel, proprietary medical device, surgical implant and pharmaceutical products that address the most frequent problems or complications observed in connection with medical device use or surgical procedures. Specific elements of our strategy include:
•
Identifying and Prioritizing Market Opportunities. We begin our product development process by identifying medical devices or surgical procedures where problems or complications arise soon after device implantation or the initial procedure, and where re-intervention is expensive, potentially harmful for patients or difficult to perform. We target areas where we have previously developed successful technologies, such as the treatment of scar formation and cell proliferation with paclitaxel and its analogues or derivatives, or where we have particular scientific focus or expertise. For example, numerous human trials have indicated that paclitaxel dramatically improves the clinical performance of stents used to treat patients with coronary artery disease, which has led to improved patient outcomes, premium pricing and growth of the stent market. We believe other medical devices and surgical implants could be similarly affected through the proprietary addition of locally-delivered therapeutics.
- 4 -
•
Developing Novel, Proprietary Product Candidates. After prioritizing opportunities, we identify the biology that may contribute to complications or failures of a device or procedure. We then screen and select a potential drug or drugs to combine with our biomaterial or drug delivery technologies to create proprietary, locally-delivered drug formulation or drug-device combination product candidates. We continually assess and study applications of our technology, including analyzing the biology pertaining to the failure of certain medical devices and surgical implants, and determining the therapeutic drug selection, concentration, total dose and drug release characteristics required to enhance medical device and surgical implant performance. We believe this approach may allow us to create additional novel drug-eluting medical devices and surgical implants that achieve better clinical results than drugs, medical devices or surgical implants may achieve independently.
•
Establishing and Developing Intellectual Property Portfolio. After identifying potentially useful technologies or developing novel product candidates, we incorporate these elements into new patent filings or our existing patent portfolio, and we attempt to develop and establish new intellectual property in jurisdictions throughout the world. We believe we are among the first companies to develop an extensive intellectual property portfolio of products combining approved pharmaceutical agents, such as paclitaxel, with medical devices and surgical implants. Recognizing the importance of intellectual property in our industry, we plan to continue to pursue patent protection in the United States, the European Union and other significant markets, as well as to protect trade secrets and know-how as our research and development activities uncover additional important medical product and therapeutic opportunities.
•
Selecting Commercialization Path. Once we reach a certain stage with a technology or product candidate, we select a development and commercialization path. Where we have a distribution channel, we may elect to fully develop a product ourselves, or in selected cases where product opportunities may not fit our commercial capabilities, we will license certain of our products, technologies or product candidates to be developed, manufactured or commercialized by partners.
•
Pursuing Selective Strategic Acquisitions and Licenses. To support our product development and commercialization activities, we have pursued, and will continue to selectively pursue, acquisitions or licenses to obtain proprietary pharmaceutical compounds or compound classes, formulation technologies, medical device technologies and intellectual property or other commercial assets.
Business Overview
We currently operate in two segments: Pharmaceutical Technologies and Medical Products.
Pharmaceutical Technologies
Our Pharmaceutical Technologies segment focuses primarily on establishing product development and marketing partnerships with major medical device, pharmaceutical or biomaterials companies and to date has derived the majority of its revenue from royalties due from partners that develop, market and sell products incorporating our technologies. Currently our principal revenues in this segment are from royalties derived from sales by Boston Scientific Corporation (“BSC”) of TAXUS® coronary stent systems incorporating the drug paclitaxel.
TAXUS Drug-Eluting Stents
The most significant commercial products containing technology developed and licensed by our Pharmaceutical Technologies segment are the TAXUS Express and TAXUS Liberté coronary stent systems. These products are sold by BSC, our exclusive licensee in the coronary paclitaxel-eluting stent field. We derive royalty revenue from sales by BSC of TAXUS coronary stent systems. Royalty revenue received from BSC was $84.1 million for the year ended December 31, 2008.
BSC commenced sales of the TAXUS Express paclitaxel-eluting coronary stent system in January 2003 in the European Union and other countries outside of the United States, and in March 2004 in the United States. BSC announced the completion of the initial launch of the TAXUS Liberté paclitaxel-eluting coronary stent system in 18 countries in January 2005 and in Europe in September 2005, and commenced sales of TAXUS Liberté in the United States in October of 2008. The TAXUS Liberté stent system represents BSC’s next generation product incorporating our research, technology and intellectual property related to the use of paclitaxel to treat restenosis and other local inflammatory diseases. The TAXUS Liberté stent has been designed by BSC to further enhance coronary stent deliverability and blood vessel conformability, particularly in challenging coronary lesions. In addition, in September 2008 BSC received approval to mark et and sell the Taxus Express2 Atom™ Paclitaxel-Eluting Coronary Stent System in the United States. The TAXUS Express 2 Atom stent system is the only drug-eluting stent available that is specifically designed to treat lesions with diameters as small as 2.25 millimeters. We are entitled to receive royalties based on the commercial sale of all of BSC’s various TAXUS products.
- 5 -
BSC has concluded several clinical trials of paclitaxel-eluting coronary stents that have demonstrated positive results, including six separate clinical studies, with a total of over 31,000 patients studied over a five-year period, designed to evaluate the near and long-term safety and efficacy of both the slow and moderate drug release versions of the TAXUS Express coronary stent system. These various studies have universally indicated that paclitaxel-eluting coronary stents significantly reduce the incidence of restenosis and the need for repeat surgical procedures at the treated lesion site in coronary artery disease patients.
Certain clinicians have suggested that the use of drug-eluting stents may cause an increased rate of late thrombosis (the formation of blood clots in the stent long after the initial stent implantation) and in turn, potentially lead to an increased rate of myocardial infarctions (heart attacks) or deaths as compared to bare metal stents. These suggestions led to a significant decrease in the use of drug-eluting stents, beginning in the second half of 2006 and continuing through 2008. A meta-analysis of a majority of the previous clinical trials of TAXUS is comprised of five-year follow up data. The overall thrombosis rate indicated by this analysis is no different between the drug-eluting stent arm of the studies and the bare metal stent arm of the studies. Death rates and myocardial infarction rates also show no difference at the end of five years, while the target lesion revascularization rate continues to maintain a statistically sign ificant benefit for the patients receiving the drug-eluting paclitaxel stent as compared to those patients who received a bare metal stent (TLR 12.1% vs 21.0%, p<0.001).
CoSealTM Surgical Sealant
CoSeal surgical sealant is a commercially approved, surgical biomaterial product and was obtained as part of our acquisition of Angiotech BioMaterials Corp. (formerly known as Cohesion Technologies, Inc.) in 2003. CoSeal is the first fully synthetic vascular sealing agent approved by the United States Food and Drug Administration (“FDA”), and has been sold in the United States since February 2002. CoSeal is approved for use in the United States and the European Union to achieve adjunctive hemostasis in vascular reconstruction by mechanically sealing areas of leakage. CoSeal is designed to rapidly seal tissue surfaces, suture lines and synthetic grafts during surgery. A premixed configuration of CoSeal received both FDA and European CE Mark approval in February 2003 and April 2003 respectively, and received Health Canada approval in October 2003. The premixed configuration of CoSeal is simpler to use, can be stored at room temperature and has a two-hour lifespan once activated.
On February 25, 2003, we entered into a strategic alliance with Baxter Healthcare Corporation (“Baxter”), which provides Baxter with worldwide (excluding Japan) sales, marketing and distribution rights of our CoSeal surgical sealant product. This alliance also gives Baxter an option for distribution rights in Japan. In April 2003, we broadened the strategic alliance to provide Baxter with worldwide manufacturing rights for CoSeal. As a result of these transactions, we receive royalties on the end-user sales of CoSeal by Baxter. We have retained all development and commercial rights for drug-loaded versions of CoSeal.
Medical Products
Our Medical Products segment manufactures and markets a wide range of single-use specialty medical products, primarily medical device products and medical device components. These products are sold directly to end users or other third party medical device manufacturers. This segment contains two specialized direct sales and distribution organizations as well as significant manufacturing capabilities. Many of our medical products are made using our proprietary manufacturing processes, or are protected by our intellectual property. Our Medical Products segment may apply certain of our proprietary technologies to its products to create novel, next generation medical products to market directly to end users or medical products distributors.
Interventional Radiology and Biopsy Products
We offer a variety of products targeted at the interventional radiology and biopsy markets. We currently sell the majority of these products in finished form, either directly to or through distributors. We also sell selected biopsy needles and related components to other third-party medical device suppliers that compete in various general surgery markets. Some of our more significant commercial products in this area include:
·
SKATERTM Drainage Catheters – the SKATER catheter line is used to facilitate drainage of fluid from wounds, infectious tissues or surgical sites, and features larger lumens and drainage holes, kink resistance, resistance to encrustation and high radiopacity.
·
V+PadTMHemostatic Pad – the V+Pad product is a hydrophilic wound dressing that is intended for use in the local management of bleeding wounds such as vascular access sites (percutaneous catheters or tubes), surgical debridement and lacerations. The V+Pad promotes rapid control of bleeding in patients on anticoagulation therapy and in patients following hemodialysis. We are the exclusive distributor of V+Pad in the United States.
·
Soft Tissue Biopsy Needles – we offer a wide range of soft tissue biopsy needles, both disposable and re-usable for use in different types of breast, lung, spinal and bone marrow biopsies. Some key features of our biopsy needles include: an echogenic tip for placement under ultrasound guidance, numerically ordered centimeter markings to facilitate precise depth placement, and adjustable needle stops allowing the restriction of forward movement and localizing the needle to the biopsy site.
- 6 -
·
BioPinceTM Full Core Biopsy Devices – BioPince is our biopsy instrument product line featuring a proprietary tri-axial “Cut and Trap” cannula system. This system allows the device to deliver cylindrical, full-length biopsy specimens that are complete and largely undamaged, which we believe significantly improves the diagnostic value of the sample.
·
T-LockTM Bone Marrow Biopsy Device – our bone marrow biopsy device, the T-Lock, has an ergonomically designed twist-lock handle, which improves a clinician’s ability to penetrate hard bone to obtain the biopsy sample. An adjustable needle stop to control the depth of penetration allows the clinician to safely aspirate at the sternum of the patient.
·
HemoStreamTM Dialysis Catheter –our HemoStream product is used for short-term vascular access for patients undergoing hemodialysis. HemoStream has a novel triple outflow lumen that is designed to prevent the common complication of “sidewalling”, which occurs when the catheter is drawn next to the vessel wall during dialysis. This is a common problem with dialysis catheters. We are the exclusive distributor of the HemoStream dialysis catheter line.
·
Bio-Seal™Lung Biopsy Tract Plug System – Bio-Seal is our biopsy system containing a proprietary hydrogel plug designed to prevent air leaks in patients having lung biopsies. On contact with moist tissue, the hydrogel plug absorbs fluids and expands to fill the void created by the biopsy needle puncture. The plug is absorbed into the body after healing of the puncture site has occurred. We are the exclusive worldwide manufacturer and distributor of the Bio-Seal Lung Biopsy Tract Plug System, which has received CE Mark approval and is currently marketed and sold in the European Union. BioSeal has undergone a human clinical trial in the United States. and is not yet commercially available in the United States or other markets outside of the European Union.
·
Other Selected Products – other selected products for the interventional radiology and biopsy markets include fixation pins, tunneling stylets and surgical tunnelers and isotope seed spacers and needles for prostate cancer treatment.
General Surgery Products
We offer a variety of products targeted at various surgical disciplines for wound closure, wound healing and general surgery. We currently sell the majority of these products in finished form, either directly or through distributors. We also sell selected surgical needles, suture products and wound closure product components to other third-party medical device suppliers that compete in various general surgery markets. Our most significant commercial products in this market area include:
·
QuillTM SRS – our Quill SRS product is a suture material that contains proprietary patterns of tiny barbs, which can eliminate the need for surgeons to tie knots when closing certain wound types. We believe that use of Quill SRS may lead to faster surgical times, improved wound cosmesis and lower wound infection and complication rates.
·
SharpointTM Specialty Microsurgical Products – our Sharpoint microsurgical sutures include specialized suture configurations and materials that are complex to manufacture, as well as needles that are manufactured from specialty stainless steel wire that has been tempered to a specific balance of hardness and tensile strength, which allows a fine edge that resists bending as the surgeon applies pressure.
·
Silk and Polyester Braided and Monofilament Suture Materials – we manufacture a wide array of configurations of non-absorbable and absorbable sutures. Our suture configurations vary in diameter size, length, and material.
·
Drilled End and Channel Needles – we manufacture a wide array of surgical needles for our own surgical product lines, as well as for other manufacturers of wound closure products. Our product line includes drilled end suture needles, which have a precisely drilled butt end for maximum suture holding strength and are adequately tempered for securing strong attachment of the suture, and channel needles, which have a channel with an underlying receptacle for attachment of the suture.
·
Lifespan™ ePTFE Vascular Grafts – we manufacture reinforced ePTFE synthetic vascular graft for use in bypass or reconstruction of diseased or occluded blood vessels or to provide arteriovenous shunts for blood access in hemodialysis patients. Our ePTFE product line has over 75 product iterations and includes vascular grafts of various sizes and lengths.
·
Surgical Blades Used in Cataract Surgery – we have a wide range of surgical blades for cataract surgery, including clear corneal knives, standard implant knives, pilot tip implant knives, precision depth knives, sharptone crescent knives, spoon knives, stab knives, vitrectomy knives to create small, precise incisions, sub-2mm series knives, and a variety of slit and specialty knives.
Research and Product Development
We, or our partners, are evaluating several new product candidates that incorporate our proprietary technologies and capabilities, including a significant number of product candidates undergoing human clinical testing conducted both by us and by our partners. We also maintain several pre-clinical, or research stage, programs, as well as conduct various product and process improvement initaitives with respect to our currently marketed products and within our manufacturing facilities. The following discussion outlines our most advanced product candidates and their stage of development, and summarizes our most recently concluded or terminated clinical development programs.
- 7 -
Ongoing Clinical Programs
·
TAXUS Element™ Platinum Chromium Paclitaxel-Eluting Coronary Stent System. The TAXUS Element paclitaxel-eluting coronary stent system is the third generation BSC coronary stent platform that incorporates our research, technology and intellectual property related to the use of paclitaxel. The TAXUS Element stent features BSC’s proprietary platinum chromium alloy, which is designed to enable thinner stent struts, increased flexibility and a lower stent profile while improving radial strength, recoil and radiopacity. In addition, the TAXUS Element stent platform incorporates new balloon technology intended to improve upon BSC’s market-leading Maverick® Balloon Catheter technology.
On October 10, 2008, we announced that BSC had completed enrollment in the PERSEUS clinical trial, designed to evaluate the third-generation TAXUS Element paclitaxel-eluting coronary stent. The PERSEUS clinical program has enrolled nearly 1,500 patients at 100 U.S. and international centers since July 2007, and will compare the TAXUS Element stent to the prior-generation TAXUS Express2 stent marketed in this United States since 2004.
·
TAXUS Petal™ Bifurcation Paclitaxel-Eluting Coronary Stent System. The TAXUS Petal bifurcation paclitaxel-eluting coronary stent system, which is under evaluation in clinical trials being conducted by BSC, represents a novel BSC coronary stent product candidate that incorporates our research, technology and intellectual property related to the use of paclitaxel. Conventional coronary stents were designed to treat tubular arteries, and are considered less than optimal for the y-shaped anatomy of a bifurcated area of the coronary arteries. The TAXUS Petal is a specialized coronary stent designed to treat both the main branch and the side branch of a bifurcation by incorporating an innovative side structure (the Petal strut) in the middle of the stent that opens into a side branch.
On July 18, 2007, BSC initiated the TAXUS PETAL I First Human Use (FHU) trial, which is expected to enroll a total of 45 patients in New Zealand, France and Germany. The trial is a non-randomized study with an initial assessment of acute performance and safety (including rates of death, myocardial infarction and target vessel revascularization) at 30 days and six months, with continued annual follow-up to occur for five years. Upon successful completion of this study, BSC has indicated that it intends to begin a pivotal trial which if successful would provide a basis for United States and international approvals for the commercialization of the TAXUS Petal stent.
·
ZILVER PTX Paclitaxel-Eluting Peripheral Vascular Stent System. The ZILVER PTX paclitaxel-eluting peripheral vascular stent, which is under evaluation in clinical trials being conducted by our partner Cook Medical, Inc (“Cook”) is a specialized stent product incorporating our proprietary paclitaxel technology and is designed for placement in diseased arteries in the limbs to restore blood flow. Cook is a co-exclusive licensee, together with BSC, of our proprietary paclitaxel technology to reduce restenosis following stent placement in peripheral artery disease. The ZILVER PTX paclitaxel-eluting peripheral stent is designed to reduce restenosis following placement of a stent in PAD patients.
The ZILVER PTX is currently undergoing multiple human clinical trials in the United States, Japan, the European Union and selected other countries to assess product safety and efficacy. In January 2007, Cook released nine-month data from its EU clinical study. The preliminary data presented by Cook on the first 60 patients in the randomized trial, which is examining the safety of using Cook's ZILVER PTX paclitaxel-eluting stent to treat blockages, or lesions, of the superficial femoral artery (“SFA”) above the knee, indicated that the ZILVER PTX stent showed an equal adverse event rate to conventional angioplasty for treating SFA lesions. The ZILVER PTX stent also displayed a zero-percent fracture rate for 41 lesions at six months and 18 lesions at one year.
On June 11, 2008, Cook reported positive interim results from the registry arm of a clinical study designed to measure the efficacy of the ZILVER PTX in treating PAD patients, specifically in the treatment of blockages in the femoropopliteal artery. The results were reported by trial investigators at the 2008 SVS Vascular Annual Meeting, and revealed clinical improvement, excellent durability and fracture resistance, high rates of event-free survival (“EFS”) and freedom from target lesion revascularization (“TLR”). Interim data was compiled at six and 12 months using 435 patients and 200 patients, respectively. The corresponding EFS rates were 94 percent and 84 percent, and freedom from TLR was 96 percent and 88 percent. Evaluation of stent x-rays is ongoing, and currently suggests stent fractures in approximately one percent of cases at six months and less than two percent of cases at 12 months. In addition, the ZILVER PTX stent exhibited no safety concerns and results were better than expected for Trans-Atlantic Inter-Society Consensus class C and D lesions, occlusions, in-stent restenosis and lesions greater than seven centimeters. Follow-up to the registry arm of the study will continue through two years.
On July 16, 2007 Cook announced that the first U.S. patients in a randomized pivotal human clinical trial of ZILVER PTX were treated at Tri-City Medical Center in Oceanside, CA. The trial is designed to randomize patients to receive either the ZILVER PTX stent or balloon angioplasty. Data from this clinical trial is intended to be used to support submission to the FDA for approval in the U.S. to market the device. In addition, data collected on Japanese and United States patients is expected to be combined for the final evaluation of the device and used for regulatory submissions in both markets for approval.
- 8 -
On September 10, 2008 Cook announced it had completed enrollment in its pivotal human clinical trial for the ZILVER PTX. The 420 patients enrolled in Cook’s randomized trial include peripheral artery disease patients treated in Germany, the United States and Japan. Based on a release by Cook on September 10, 2008, Cook had enrolled an additional 780 patients in the European Union, Canada and Korea in a clinical registry to evaluate the safety of the ZILVER PTX device. The data from such trials have been used for submission in Europe for CE Mark approval to market the device there, with additional regulatory submissions pending in additional markets.
·
Bio-Seal™ Lung Biopsy Tract Plug System. Bio-Seal is a novel technology designed to prevent air leaks in patients having lung biopsies by plugging the biopsy track with an expanding hydrogel plug. On contact with moist tissue, the hydrogel plug absorbs fluids and expands to fill the void created by the biopsy needle puncture. The seal is airtight and the plug is absorbed into the body after healing of the puncture site has occurred.
Bio-Seal has undergone a human clinical trial in the United States which was designed to assess the safety and efficacy of Bio-Seal, with the primary endpoint being reduction in rates of pneumothorax in patients undergoing lung biopsy procedures. The clinical trial was a prospective randomized multi-centered safety and efficacy evaluation. The trial enrolled its first patient in October 2005 and completed enrolment in June 2008. The study was designed to provide a basis for U.S. clearance for the commercialization of Bio-Seal. Data from this clinical trial study has been submitted to the FDA. The FDA has responded to our submission with additional questions about the study. We have responded to the FDA, and upon further review by the FDA we may either receive 510(k) clearance to market Bio-Seal in the United States or be required to respond to additional questions or conduct additional clinical studies f or this product candidate. Upon receiving such further information from the FDA, we will determine the timing of product launch or any further development work necessary to achieve approval should we choose to continue the development of this product candidate. The complete data for the Bio-Seal study was presented at the 2009 Society of Interventional Radiology in San Diego, CA on March 9, 2009. The trial hit its primary end point with clinical success in 85% of the treatment patients compared to 69% for the control patients (p=0.002). The product has already received CE Mark approval.
·
Option™ Vena Cava Filter. The Option™ Vena Cava Filter, which we licensed in March 2008 from our partner Rex Medical L.P., is under evaluation in a pivotal human clinical trial. We believe this vena cava filter may have a number of potential benefits, which include unique filter apex and retention anchors, insertion through either the femoral or jugular route, and non-thrombogenic material. The purpose of the United States multi-center prospective clinical trial is to evaluate the device’s safety and efficacy in preventing pulmonary emboli, and to assess the ability to retrieve the device from the body up to 175 days following implantation. Interim results of the pivotal trial were presented at the AIM/Veith Meeting in New York in November 2008. The complete results, representing a total of 100 patients, were presented at the the 2009 Society of Interventional Radiology in San Diego, CA. The clinical data from this trial has been submitted to the FDA. The FDA has responded to our submission with additional questions about the study and we are preparing a response.
·
Anti- Infective HemoStreamTM Dialysis Catheter. We have an FDA-approved dialysis catheter, HemoStream, that we sell through our direct sales force. A dialysis catheter is used for exchanging blood between a hemodialysis machine and a patient. Some common malfunctions of dialysis catheters include clotting, infection, and kinking. Through our proprietary drug identification strategy, we have identified 5-Fluorouracil (“5-FU”), a drug previously approved by the FDA for treatment of various types of cancer, as a compound that may help to prevent certain types of infection in patients receiving selected types of implantable devices, including certain dialysis catheters. We are currently developing this 5-FU anti-infective technology for our HemoStream dialysis catheter line, and we currently anticipate f iling in 2009 for a 510(k) clearance to market this product candidate in the U.S.
·
MultiStem® Stem Cell Therapy. The MultiStem stem cell therapy is under evaluation in clinical trials being conducted together with our partner Athersys, Inc. (“Athersys”) for the treatment of acute myocardial infarction. MultiStem stem cells are proprietary adult stem cells derived from bone marrow, which have demonstrated the ability in laboratory experiments to form a wide range of cell types. MultiStem may work through several mechanisms, but a primary mechanism appears to be the production of multiple therapeutic molecules produced in response to inflammation and tissue damage. We and Athersys believe that MultiStem may represent a unique “off the shelf” stem cell product candidate, based on its potential ability to be used without tissue matching or immunosupression, and its potential capacity for large scale production. We entered an agreement with Athersys in May 2006 to co-develo p and commercialize MultiStem for use in the indications of acute myocardial infarction and peripheral vascular disease. On December 20, 2007, we and Athersys announced we had received authorization from the FDA to commence a phase I human clinical trial to evaluate the safety of MultiStem in the treatment of acute myocardial infarction. Upon completion of a phase I human clinical trial currently being conducted by Athersys, we may assume lead responsibility for further clinical development. We currently own marketing and commercialization rights with respect to this product candidate. On September 22, 2008, as part of certain cost reduction initiatives, we announced a potential amendment of, and reduction in, cash outlays related to our collaboration with Athersys. The final terms of such amendment to our collaboration with Athersys may have an impact on our expected future expenditures for research and development of MultiStem, and the extent of our future financial and commercial commitments a nd rights relating to this product candidate.
- 9 -
Completed or Suspended Clinical Programs
·
Anti-infective Central Venous Catheter. Central venous catheters (“CVC”) are usually inserted into critically ill patients for extended periods of time to administer fluids, drugs, and nutrition, as well as facilitate frequent blood draws. We have elected to evaluate 5-FU as a compound that may help to prevent certain types of infection in patients receiving a CVC. We recently completed a human clinical trial in the United States designed to assess the safety and efficacy of our 5-FU-eluting CVC in preventing various types of catheter related infections. The study was a randomized, single-blind, 960-patient, 25-center study and was designed to evaluate whether our 5-FU-eluting CVC prevents bacterial colonization at least as well as the market leading anti-infective CVC. On July 10, 2007, we announced that we had comp leted enrolment of the study, and on October 9, 2007 we announced this study had met its primary statistical endpoint of non-inferiority as compared to the market leading anti-infective CVC (a chlorhexidine / silver sulfadiazine (CH-SS) coated CVC) and indicated an excellent safety profile. In March 2008, we presented the clinical trial data at the 28th International Symposium on Intensive Care and Emergency Medicine (ISICEM) in Brussels, Belgium. Based on the clinical trial data, the investigators concluded that our 5-FU CVC met the primary endpoint of the study, specifically that our 5-FU CVC product candidate was non-inferior in its ability to prevent bacterial colonization of the catheter tip when compared to catheters coated with CH-SS. There were no statistically significant differences in the rate of adverse events related to the study devices, or in the rates of catheter-related bloodstream infections. Additionally, there was no evidence for acquired resistance to 5-FU in clinical isolate s exposed to the drug for a second time. Based on the positive results achieved in the study, in December 2007 we filed a request for 510(k) clearance from the FDA to market and sell the CVC in the United States and on April 17, 2008, we announced that we had received 510(k) clearance from the FDA to market our 5-FU CVC in the United States but we have not commercially launched the product.
·
TAXUS Liberté™ paclitaxel-eluting coronary stent system. The TAXUS Liberté paclitaxel-eluting coronary stent system is BSC’s second generation coronary stent system platform that incorporates our research, technology and intellectual property related to the use of paclitaxel to prevent restenosis. The TAXUS Liberté stent system has been designed to further enhance coronary stent deliverability and blood vessel conformability, particularly in challenging coronary lesions. To date, BSC has only commenced sales of the TAXUS Liberté in countries outside of the United States. On October 10, 2008, we announced that BSC had received approval from the FDA to market and sell the TAXUS Liberte in the United States.
·
Vascular Wrap™ Our paclitaxel-eluting mesh surgical implant, or Vascular Wrap, is designed to treat complications, including graft stenosis or restenosis, that may occur in connection with vascular graft implants in hemodialysis patients or in patients that have peripheral artery disease. Vascular grafts are implanted in patients in order to bypass diseased blood vessels, or to provide access to the vascular system of kidney failure patients in order to facilitate the process of hemodialysis. In many cases, these vascular grafts fail due to proliferation of cells or scar into the graft (graft stenosis or restenosis), which can negatively impact blood flow through the vascular graft.
In April 2008, we elected to suspend enrollment in our U.S. and EU human clinical trials for our Vascular Wrap product candidate in patients undergoing surgery for hemodialysis access, pending a safety review to evaluate an imbalance of infections observed between the two study groups. As a result of these observations, we elected to notify physicians to suspend further enrolment in the trials, pending a full review of the potential cause of the implant site infections. There are currently no plans to resume enrollment in these clinical trials, and we are continuing to evaluate alternatives for this program,including potential collaborations, partnerships, divestitures or future clinical development initiatives.
Patents, Proprietary Rights and Licenses
Patents and other proprietary rights are essential to our business. We file patent applications to protect technology, inventions and improvements to inventions that are considered important to our business. We also rely upon trade secrets, continuing technological innovations and licensing opportunities to develop and maintain our competitive position. Furthermore, we endeavor to extend our intellectual property portfolio by exclusively licensing patents and patent applications from others and initiating research collaborations with outside sources.
Our patents and patent applications relate to, among other things:
•
the use of paclitaxel and other agents for the treatment of chronic inflammatory diseases, such as restenosis (including as a stent component);
•
compositions of and use of pharmacologic agents as medical device coatings and drug-eluting biomaterials;
•
hybrid polymer systems designed for localized delivery of bioactive agents and controlled elution of drugs from medical device surfaces;
- 10 -
•
non-reactive hydrophilic/hydrophobic polymer matrix, which can be varied to optimize performance characteristics such as lubricity, flexibility and hydration;
•
biocompatible surface treatments used in ultrasound imaging technology to cause medical devices to shine brightly in ultrasound images providing greater echogenicity, or visibility;
•
hemostatic and sealant compositions, as well as broad intellectual property rights relating to collagen;
•
drug delivery compositions, such as our proprietary systemic formulation, medical device coatings and other local drug delivery systems; and
•
methods of administration.
As part of our patent strategy, we have filed a variety of patent applications internationally. Oppositions have been filed against various granted patents that we either own or license and which are related to certain of our technologies. See “― Legal Proceedings” elsewhere in this Annual Report on Form 10-K for a discussion of the proceedings related to certain of such oppositions. An adverse decision by an Opposition Division in any country, or subsequently, by a Board of Appeal, could result in revocation of our patent or a narrowing of the scope of protection afforded by the patent. The ultimate outcomes of the pending oppositions, including appeals, are uncertain at this time. We do not know whether the patents that we have received or licensed, or those we may be able to obtain or license in the future, would be held valid or enforceable by a court or whether our patents would be found to infringe a competitor’s patents related to its technology or product. Further, we have no assurance that third parties will not (whether pursuant to or in breach of the terms) modify or terminate any license they have granted to us.
Regulatory
The research and development, manufacture, labeling, advertising, sale and marketing of our drug and medical device products and those of our collaborators are subject to extensive regulation by the FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries. Because we focus on combining pharmaceutical compounds with medical devices and biomaterials, we are subject to both drug and device regulations depending upon the categorization of the commercial product or potential product candidate.
Drug and device licensing laws require registration of manufacturing facilities, carefully controlled research and testing of products, government review and approval or clearance of results prior to marketing of products, adherence to current Good Manufacturing Practices (“cGMPs”), during production and processing of products, and compliance with comprehensive post-marketing requirements including restrictions on advertising and promotion and requirements for reporting adverse events to the FDA in the United States and other regulatory authorities abroad. In the United States, these activities are subject to rigorous regulation by the FDA. Similar regulations apply in the European Union and other markets.
Our success is ultimately dependent upon our and our collaborators obtaining marketing approval or clearance for potential product candidates currently under development and will depend on our and our collaborators’ ability to comply with FDA and other market regulations governing the manufacturing, quality control, pre-clinical evaluation, and clinical testing of those candidates. If we or our present or future collaborators are not able to comply with the continuing FDA regulations that accompany FDA approval, the FDA may halt our clinical trials, require us to recall a drug from distribution, or withdraw approval of the new drug application for the relevant drug or the 510(k) or Premarket Approval for the relevant device. Depending on the circumstances surrounding the clinical evaluation of a product, we may sponsor clinical trials, contract clinical trial activities to contract research organizations (“CROs”) or rely upon our corporate collab orators to sponsor such clinical development. There can be no assurance that any of our and our collaborators’ clinical trials or product development activities will provide favorable data or that the data will be acceptable to any government regulatory agency to provide us with approval to market and sell our products or product candidates. Moreover, even if we and our collaborators believe the results of our product development activities are favorable, regulatory authorities may interpret the results differently than we do.
We have significant medical device component engineering and manufacturing, as well as finished goods and packaging manufacturing operations. The manufacturing processes used in the production of finished medical devices are subject to FDA regulatory inspection, and must comply with FDA regulations, including its Quality Systems Regulation, (“QSR”), which sets forth the cGMP requirements for medical devices. The QSR requires manufacturers of finished medical devices to follow elaborate design, testing, control, documentation and other quality assurance procedures during the finished device manufacturing process. Our FDA registered facilities are subject to FDA inspection at any time for compliance with the QSR and other FDA regulatory requirements. Failure to comply with these regulatory requirements and similar foreign requirements may result in civil and criminal enforcement actions, including financial penalties, seizures, injunctions or other meas ures. In some cases, failure to comply with the QSR could prevent us or our customers from marketing products or gaining clearance to market additional products. Our products must also comply with state and foreign regulatory requirements.
- 11 -
Manufacturing and Distribution
We have significant manufacturing capabilities for our medical devices. Our medical device manufacturing capabilities include polymer handling and extrusion, metals, plastics, textiles and packaging and assembly. Our suture manufacturing capabilities include the ability to make and handle various approved polymer materials in multiple lengths, diameters and configurations. Our metal manufacturing capabilities include bending, grinding, drilling, polishing, chemical etching, thermal curing and wire winding. Our plastics manufacturing capabilities include injection molding, insert molding and wire coating. Our textile manufacturing capabilities include braiding and embroidery of selected suture materials, including silk. Our assembly and packaging capabilities include small lot assembly, suture attachment and kit assembly. We also maintain manufacturing capabilities for certain of our biomaterials and coating technologies, including dedicated resources and te chnology to process and manufacture certain types of drug-eluting medical devices.
We devote resources to develop procedures and process controls for our products and product candidates to ensure successful technology transfer for commercial scale manufacturing. We expect that process and assay development performed during pre-clinical and clinical stages of manufacturing will result in products with the desired use and stability. These activities include scaling up production methods, developing quality control systems, optimizing batch-to-batch reproducibility, testing sterilization methods and establishing reliable sources of raw materials for synthesizing proprietary products.
Sales and Marketing
We market and sell various medical device products through two specialty direct sales organizations. In order to augment these capabilities in certain areas and to allow our direct sales forces to focus on our most critical growth opportunities, we also sell certain of our medical devices and medical device components through a network of independent sales representatives and independent distributors.
Our direct sales teams comprised of about 120 individuals as of December 2008. The majority of our direct sales teams are based in the United States and focus primarily on selling certain of our product lines to interventional radiologists, general surgeons, orthopaedic surgeons, aesthetic surgeons and ophthalmologists.
These sales forces target hospital-based physician customers, as well as alternate site health care providers such as ambulatory surgery centers or urgent care centers. The primary goal of our sales team is to increase market share of our highest margin products into markets that we believe are not as well served by our larger competitors. These competitors often sell their products to larger hospitals or large group purchasing organizations based on contracts for a range of products and in some cases do not employ direct sales professionals targeting our key market segments.
Competition
We expect to face competition from companies utilizing and developing technologies that target the same diseases and clinical indications that our technologies target. Some of the companies developing these technologies have substantially greater product development capabilities and financial, scientific, manufacturing, sales and marketing resources and experience than we do.
The medical device, biotechnology and pharmaceutical industries are subject to rapid and substantial technological change. There can be no assurance that developments by others will not render obsolete our products or technologies or that we will be able to keep pace with technological developments. Some competitive products have an entirely different approach or means to accomplish the desired therapeutic effect than our product candidates. These competitive products, including any enhancements or modifications made to such products in the future, could be more effective and/or less costly than our products, technologies or our product candidates.
There are a number of companies that are marketing or developing competing drug-eluting coronary stents or other treatments for restenosis that may be considered to be directly competitive with our technology. Certain of our competitors have developed and commercialized coronary drug- eluting stents that compete with BSC’s TAXUS stents and which have been approved for use in the United States, European Union and certain other countries. The launch of these competing products has had a significant impact on BSC’s sales of TAXUS, and has resulted in a decline in our royalty revenue received from BSC. The continued successful commercialization of any of these or other technologies to treat restenosis following angioplasty or stent placement could have a material adverse impact on our business.
We compete with a number of additional large companies across various of our product lines. These competitors have substantially greater business and financial resources than we do. These competitors include the Ethicon division Johnson & Johnson and the U.S. Surgical division of Covidien, Inc. in surgical sutures and wound closure devices; C.R. Bard Inc., the Allegiance division of Cardinal Health, Inc. and the Sherwood, Davis & Geck division of Covidien, Inc. in biopsy needle devices; and Alcon, Inc., Allergan, Inc. and the Allegiance division of Cardinal Health, Inc. in ophthalmology products.
- 12 -
Employees
As of December 31, 2008, on a worldwide basis, we have approximately 1,450 employees, including 900 in manufacturing, 100 in R&D, 200 in sales and marketing and 250 in administration. In addition, we have contractual arrangements with scientists at various research and academic institutions. All personnel or their management companies execute confidentiality agreements with us. While we will continue to seek to hire highly qualified employees, we believe that we have employed sufficiently qualified personnel.
Reimbursement
In the United States, health care providers generally rely on third-party payers, including both private health insurance plans and governmental payers, such as Medicare and Medicaid, to cover and reimburse all or part of the cost of a medical device and the procedures in medical devices that are used. Our ability to commercialize human therapeutic products and product candidates successfully will depend in part on the extent to which coverage and reimbursement for such products and related treatments will be available from government health administration authorities, private health insurers and other third-party payers or supported by the market for these products. Third-party payers are increasingly challenging the price of medical products and services and instituting cost containment measures to control or significantly influence the purchase of medical products and services. These cost containment measures, if instituted in a manner affecting the coverage for or paymen t of our products, could have a material adverse effect on our ability to operate profitably.
There can be no assurance that third-party payers’ coverage and reimbursement will be available or sufficient for our current products or the products we might develop. We believe that recommendations and endorsements by physicians are essential for market acceptance of our products, and we do not know whether these recommendations or endorsements will be obtained. We also believe that surgeons will not use these products unless they determine, based on clinical data and other factors, that the clinical benefits to patients and cost savings achieved through use of these products outweigh their cost. Acceptance among physicians may also depend upon the ability to train surgeons and other potential users of our products and the willingness of such users to learn new techniques.
In the United States, there have been and we expect there will continue to be a number of legislative and regulatory proposals to change the healthcare system in ways that could significantly affect our business. Efforts by governmental and third-party payers to reduce healthcare costs or the announcement of legislative proposals or reforms to implement government controls could cause a reduction in sales or in the selling price of our products, which could seriously harm our business. We cannot predict the impact on our business of any legislation or regulations related to the healthcare system that may be enacted or adopted in the future.
Environmental
Our business is subject to a broad range of government regulation and requirements, including federal, state, local and foreign environmental laws and regulations governing, among other matters, air emissions, wastewater discharges, workplace health and safety as well as the use, handling, storage and disposal of hazardous materials and remediation of contamination associated with the release of these materials at or from our facilities or off-site disposal locations. Some of these laws can impose liability for remediation costs on a party regardless of fault or the lawfulness of its conduct. Many of our manufacturing processes involve the use and subsequent regulated disposal of hazardous materials. To date, such matters have not had a material adverse impact on our business or financial condition. We cannot assure you, however, that such matters will not have such an effect in the future.
- 13 -
Item 1A.
RISK FACTORS
You should consider carefully the following information about these risks, together with all of the other information contained within this document. Additional risks and uncertainties not currently known to us or that we currently deem immaterial may impair our business operations. If any of the following risks actually occur, our business, results of operations and financial condition could be harmed.
Risks Related to Our Business
We were not profitable for the quarter and the year ended December 31, 2008 and may not be able to regain and maintain profitability.
We began operations in 1992 and have incurred a loss from operations in each of the years of our existence except for fiscal years 2004 and 2006. As of December 31, 2008, our accumulated deficit was $843.7 million. Our ability to become profitable again will depend on, among other things, the amounts of royalty revenue we receive from our corporate partners; our ability to restructure our existing indebtedness; our ability to maintain and improve sales of our existing product lines; our ability to successfully market and sell certain new products and technologies; our ability to research, develop and successfully launch new products and technologies; our ability to improve our gross profit margins through realization of lower manufacturing costs and efficiencies or improved product sales mix; our ability to effectively control our various operating costs; and foreign currency fluctuations.
Our working capital and funding needs may vary significantly depending upon a number of factors including, but not limited to, the level of royalty revenue we receive from corporate partners; our levels of sales and gross profit; costs associated with our manufacturing operations, including capital expenditures, labour and raw materials costs, and our ability to realize manufacturing efficiencies from our various operations; fluctuations in certain working capital items, including inventory and accounts receivable, that may be necessary to support the growth of our business or new product introductions; progress of our research and development programs and costs associated with completing clinical studies and the regulatory process; collaborative and license arrangements with third parties; the cost of filing, prosecuting and enforcing our patent claims and other intellectual property rights; expenses associated with litigation; opportunities to in-license complementary te chnologies or potential acquisitions; potential milestone or other payments we may make to licensors or corporate partners; and technological and market developments that impact our royalty revenue, sales levels or competitive position in the marketplace.
The sharp reduction in TAXUS royalty payments from BSC, the large amount of our outstanding indebtedness and the related cash interest payments due on such indebtedness, the current economic conditions affecting our and our partners’ financial stability, as well as the capital expenditures required to develop the medical products segment of our business, among other factors, have magnified our working capital needs and funding shortages. As described below, on March 2, 2009, we announced that we had entered into a senior secured credit facility to provide enhanced liquidity in the near term and to provide us with the flexibility and time to explore longer-term options for our overall capital structure and working capital needs. See “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources—New Senior Secured Credit Facilities” in this Annual Report on Form 10-K. These longer-term options include one or more financ ial and strategic alternatives, including a capital influx from one or more new investors and the restructuring of our outstanding indebtedness, but such efforts have not been successful to date due to our significant debt burden. Due to numerous factors that may impact our future cash position, working capital and liquidity as discussed below and the significant cash that will be necessary to continue to service our current level of debt obligations, there can be no assurances that we will have adequate liquidity and capital resources to satisfy our financial obligations beyond 2009. If our cash flows are worse than expected, we may require additional funds in order to meet the funding requirements of our commercial operations for our research and development programs, to satisfy certain contractual obligations, for other operating and capital requirements, for potential acquisitions or in-licensing of technologies, to satisfy milestone or other payment obligations due to licensors or corporate partners, or to repay or refinance our indebtedness. Financing in addition to our new credit facility may not be available, and even if available, may not be available on attractive or acceptable terms due to difficult credit markets and other factors.
Our obligation to pay cash interest on our existing debt has had, and we expect will continue to have, an adverse effect on our liquidity.
We currently have outstanding $325 million aggregate principal amount of Senior Floating Rate Notes due 2013 (the “Floating Rate Notes”) and $250 million aggregate principal amount of Senior Subordinated Notes due 2014 (the “Subordinated Notes”). We are obligated to make periodic cash interest payments on both the Floating Rate Notes and the Subordinated Notes. Using current interest rates, the annual combined cash interest cost of these notes is $43.1 million.
As a result of the cash interest payments we are obligated to make on our outstanding notes, we have had significant liquidity issues. If our cash flows are worse than expected, an inability to access additional sources of liquidity to fund our cash needs beyond 2009, or to refinance our outstanding notes, could further adversely affect our financial condition or results of operations and our ability to make payments on our debt, and could force us to seek the protection of the bankruptcy laws.
- 14 -
During 2008, we commenced certain cost reductions with the goal of achieving positive consolidated free cash flow (after the incurrence of net interest expense). These efforts may not be sufficient to achieve our goal. If further cost reductions beyond those that are currently scheduled become necessary, our future prospects may be adversely impacted.
In September 2008 we announced that we were pursuing various initiatives to reduce operating costs and focus our business efforts on our most promising near term product opportunities. We believe certain cost reduction measures, in addition to a potential financial or strategic transaction of significant magnitude, are concurrently necessary to address potential liquidity issues likely to arise in the near term relating to our current balance sheet structure.
We have implemented, and we expect to continue to implement, operating cost reductions across all functions in the company, including in research and development and general and administrative functions, with more limited reduction initiatives in sales and marketing. The cost reduction efforts are designed to reduce certain expenses while maintaining support for the sales our existing marketed product lines. Our remaining resources subsequent to these changes will be focused primarily on our existing Medical Products business, and on selected new products that have recently launched or are expected to be launched in the near future, including Quill SRS, the HemoStream™ Chronic Dialysis Catheter and the Bio-Seal™ Lung Biopsy System. Selected actions that have been taken, or that we expect to take, with respect to the reorganization include postponement of the scheduled launch of our 5-fluorouracil-eluting central venous catheter (5-FU CVC); closure of our resear ch and manufacturing facility in Rochester, New York; postponement of certain pre-clinical-stage research activities, pending the completion of partnering or other funding activities that would offset direct costs and personnel costs associated with such programs; reduction of certain financial and personnel contributions relating to our joint venture with Genzyme Corporation; potential amendment of and reduction in cash outlays related to our collaboration with Athersys, Inc.; rationalization or elimination of office and laboratory space in Vancouver, British Columbia, North Bend, Washington and Herndon, Virginia; rationalization of selected pending and issued intellectual property; elimination of certain expenses and reductions in personnel in all general and administrative and in research and development departments; selective reduction in certain sales and marketing investments personnel and in medical affairs; and postponement of selected planned capital expenditures.
Failure to achieve cost reductions through the above or other measures at the rate or levels we expect could adversely affect our ability to achieve our previously stated goal of achieving positive consolidated free cash flow (after the incurrence of net interest expense). If we are required to make further reductions to our expense levels beyond those that are ongoing or currently contemplated, our future business prospects may be adversely impacted.
We depend on BSC for a significant amount of our future revenues and development of TAXUS.
Although the acquisition of our Medical Products segment has diversified our revenue, we anticipate that a significant amount of our revenue for the next few years will be derived from and dependent upon royalty revenues from BSC. We do not have control over the sales and marketing efforts, stent pricing, production volumes, distribution or regulatory environment related to BSC’s paclitaxel-eluting coronary stent program. Our involvement is limited to the terms of our 1997 License Agreement (as amended) with BSC and Cook, which provides for the receipt of royalty revenue based on the net sales of TAXUS and specifies the applicable royalty rates.
Royalty revenue from BSC for the quarter and year ended December 31, 2008 decreased by 46% and 24% respectively from the same periods in 2007, which BSC has attributed to a decline in the number of angioplasty procedures in the United States and is expected to decline further during the year ending December 31, 2009. If BSC is impaired in its ability to market and distribute TAXUS, whether for this reason or due to a failure to comply with applicable regulatory requirements, discovery of a defect in the device, increased incidence of adverse events or identification of other safety issues, or previously-unknown problems with the manufacturing operations for TAXUS (any of which could, under certain circumstances, result in a manufacturing injunction), our revenues could be further significantly reduced. BSC’s failure to resolve these issues in a timely manner and to the satisfaction of the FDA and other regulatory authorities, or the occurrence of similar problem s in the future, could delay the launch of TAXUS Liberté in the United States and could have a significant impact on our royalty revenue from sales of TAXUS.
Additionally, BSC may terminate our 1997 License Agreement under certain circumstances, including, if BSC is unable to acquire a supply of paclitaxel at a commercially reasonable price, if BSC reasonably determines that the paclitaxel-eluting coronary stent is no longer commercially viable, or if our license agreement with the National Institutes of Health (“NIH”), certain rights under which are sublicensed to BSC, terminates. During the year ended December 31, 2008, revenue from BSC represented approximately 30% of our total revenue from continuing operations, compared to 37% in 2007.
The amounts payable by BSC to us vary from 1% to 9% of net sales depending on various factors, including volume of sales from time to time and patent protection laws in the country of sale. From these amounts, we must pay certain royalties to our licensors, including the NIH and the University of British Columbia (“UBC”), under license agreements. For the year ended December 31, 2008, the average gross royalty rate earned was 7.1% for sales in the United States and 6.4% for sales in other countries. For the year ended December 31, 2007, the average gross royalty rate earned was 7.6% for sales in the United States and 5.6% for sales in other countries. There is no guarantee that royalty payments under our 1997 License Agreement will continue, and demand for BSC’s paclitaxel-eluting coronary stent products could continue to decline as a result of the factors stated above, as well as competition, technological change, reimbursement or other factors. Also, the royalty rate payable by BSC could decline if and when patent protection expires, or no longer exists as defined by our 1997 License Agreement, in certain jurisdictions.
- 15 -
Boston Scientific may be enjoined from the selling, or otherwise become subject to limitations applicable to its ability to sell, TAXUS in the United States.
Our royalty revenue derived from the sale of paclitaxel-eluting coronary stents depends on BSC’s ability to continue to sell its TAXUS Express 2 stent and to launch next generation paclitaxel-eluting stents, including the TAXUS Liberté stent, in the United States. Historically, stent manufacture and sale is the subject of a substantial amount of U.S. patent litigation, and we anticipate that our licensees, including BSC and others, may be involved in material legal proceedings related to paclitaxel-eluting stents.
Many of the products we are depending on to grow our business are not yet ready for sale or have only recently been introduced for sale.
Many of the products we are depending on to drive future growth are not yet ready for sale or have only recently been introduced for sale. For example, our Option IVC filter has not yet been approved for sale in the U.S, our 5-FU CVC has been approved for sale but has not yet been commercially launched, and our Quill SRS and HemoStream Chronic Dialysis Catheter products have only recently become available for sale. If any of these or our other products are not approved for sale or do not achieve market acceptance, our ability to generate revenues will be adversely affected.
If our products are alleged to be harmful, we may not be able to sell them, we may be subject to product liability claims not covered by insurance and our reputation could be damaged.
The nature of our business exposes us to potential liability risks inherent in the testing, manufacturing and marketing of pharmaceutical products and medical devices. Using our drug candidates or devices in clinical trials may expose us to product liability claims. These risks will expand with respect to drugs or devices, if any, that receive regulatory approval for commercial sale. In addition, some of the products we manufacture and sell are designed to be implanted in the human body for varying periods of time. Even if a drug or device were approved for commercial use by an appropriate governmental agency, there can be no assurance that users will not claim that effects other than those intended may have resulted from our products. Component failures, manufacturing flaws, quality system failures, design defects, inadequate disclosure of product-related risks or product-related information or other safety issues with respect to these or other products we manufacture or sell could result in an unsafe condition or injury to, or death of, a patient. In addition, although many of our products are subject to review and approval by the FDA or other regulatory agencies, under the current state of law, any approval of our products by such agencies will not prohibit products liability lawsuits from being brought against us in the event that our products are alleged to be defective, even if such products have been used for their approved indications and appropriate labels have been included.
In the event that anyone alleges that any of our products are harmful, we may experience reduced consumer demand for our products or our products may be recalled from the market. In addition, we may be forced to defend individual or class action lawsuits and, if unsuccessful, to pay a substantial amount in damages. A recall of some of our products could result in exposure to additional product liability claims, lost sales and significant expense to perform the recall. The outcome of litigation, particularly class action lawsuits, is difficult to assess or quantify. Plaintiffs in these types of lawsuits often seek recovery of very large or indeterminate amounts, including not only actual damages, but also punitive damages. The magnitude of the potential loss relating to these types of lawsuits may remain unknown for substantial periods of time. In addition, the cost to defend against any future litigation may be significant.
We do not have insurance covering our costs and losses as a result of any recall of products or devices incorporating our technologies whether such recall is instituted by a device manufacturer or us as required by a regulatory agency. Insurance to cover costs and losses associated with product recalls is expensive. If we seek insurance covering product recalls in the future it may not be available on acceptable terms. Even if obtained, insurance may not fully protect us against potential liability or cover our losses. Some manufacturers that suffered such claims in the past have been forced to cease operations or declare bankruptcy.
We do have insurance covering product liability. However, our insurance may not fully protect us from potential product liability claims. If a product liability claim or a series of claims is brought against us in excess of our insurance coverage, our business could suffer. Some manufacturers that suffered such claims in the past have been forced to cease operations or even to declare bankruptcy.
Our success depends on the successful commercialization of our technology.
The successful commercialization of our technology is crucial for our success. Successful product development in the pharmaceutical industry is highly uncertain and very few research and development projects produce a commercial product. Medical devices, pharmaceutical applications and surgical implants utilizing our technology are in various stages of clinical and commercial development and face a variety of risks and uncertainties. Principally, these risks and uncertainties include the following:
o
Future clinical trial results may show that some or all of our technology, or the technology of our strategic collaborators that incorporate our technology, is not safe or effective.
- 16 -
o
Even if our technology is shown to be safe and effective, we and our strategic collaborators may face significant or unforeseen difficulties in manufacturing our medical devices or the medical devices and surgical implants that use our technology. These difficulties may become apparent when we or our strategic collaborators manufacture the medical devices or surgical implants on a small scale for clinical trials and regulatory approval or may only become apparent when scaling-up the manufacturing to commercial scale.
o
Even if our technology-based products are successfully developed, receive all necessary regulatory approvals and are commercially produced, there is no guarantee that there will be market acceptance of them or that they will not cause unanticipated side effects in patients. For example, if drug-eluting stents are found to cause, or are perceived to be the cause of, blood clots in patients, then sales of our drug-eluting stent products may be adversely affected. In addition, there is no guarantee that there will be market acceptance of our products. Our ability to achieve market acceptance for any of our products will depend on a number of factors, including whether or not competitors may develop technologies which are superior to or less costly than our technology-based products, and whether governmental and private third-party payers provide adequate coverage and reimbursement for our products, with the result that our technology-based products, e ven if they are successfully developed, manufactured and approved, may not generate significant revenues.
If we are unsuccessful in dealing with any of these risks, or if we are unable to successfully commercialize our technology for some other reason, it would likely seriously harm our ability to generate revenue.
We depend on our strategic collaborators for the development, regulatory approval, testing, manufacturing and the potential commercialization of our products.
Historically, our strategy has been to enter into various arrangements with corporate and academic collaborators, licensors, licensees and others for the research, development, clinical testing, regulatory approval, manufacturing, marketing and commercialization of our product candidates. For instance, we collaborate with BSC and Cook to develop and market paclitaxel-eluting coronary and peripheral stents, and with Baxter to manufacture and market our CoSealTM product for use as both a sealant and adhesion prevention product. Strategic collaborators, both existing (particularly BSC) and those that we may collaborate with in the future, are or may be essential to the development of our technology and potential revenue and we have little control over or access to information regarding our collaborators’ activities with respect to our products.
Our strategic collaborators may fail to successfully develop or commercialize our technology to which they have rights for a number of reasons, including:
o
failure of a strategic collaborator to continue, or delays in, its funding, research, development and commercialization activities;
o
the pursuit or development by a strategic collaborator of alternative technologies, either on its own or with others, including our competitors, as a means for developing treatments for the diseases targeted by our programs;
o
the preclusion of a strategic collaborator from developing or commercializing any product, through, for example, litigation or other legal action; and
o
the failure of a strategic collaborator to make required milestone payments, meet contractual milestone obligations or exercise options which may result in our terminating applicable licensing arrangements.
We have and we expect that we will continue to enter into licensing agreements with third parties to give us access to technologies that we may use to develop products through our strategic collaboration and partnership arrangements. The technologies governed by these license agreements may be critical to our ability to maintain our competitive advantage in our existing products and to develop future products. For example, through licenses with NIH and UBC, we have been granted access to technologies that have contributed to the development of the TAXUS paclitaxel-eluting coronary stent.
Pursuant to terms of existing license agreements, licensors have the ability to terminate their respective licenses upon the occurrence of certain specified circumstances. Events which may allow licensors to exercise these termination provisions include our bankruptcy, sub-licensing without the licensor’s consent, a transaction which results in a change of control of us, our failure to use the required level of diligence efforts to develop, market and sell products based on the licensed technology, our failure to maintain adequate levels of insurance with respect to the licensed technologies or other acts or omissions that may constitute a breach by us of our license agreement. In addition, any failure to continue to have access to these technologies may materially affect the benefits that we currently derive from the collaboration and partnership arrangements and may negatively impact our results and operations.
If our process related to product development does not result in an approved and commercially successful product, our business could be adversely affected.
We focus our research and development activities on areas in which we have particular strengths. The outcome of any development program is highly uncertain, notwithstanding how promising a particular program may seem. Success in pre-clinical and early-stage clinical trials may not necessarily translate into success in large scale clinical trials. Further, to be successful in clinical trials, increased investment will be necessary, which will adversely affect our short-term profitability.
- 17 -
In addition, we will need to obtain and maintain regulatory approval in order to market new products. Notwithstanding the outcome of clinical trials for new products, regulatory approval may not be achieved. The results of clinical trials are susceptible to varying interpretations that may delay, limit or prevent approval or result in the need for post-marketing studies. In addition, changes in regulatory policy for product approval during the period of product development and review by regulators of a new application may cause delays or rejection. Even if we receive regulatory approval, this approval may include limitations on the indications for which we can market the product. There is no guarantee that we will be able to satisfy the applicable regulatory requirements, and we may suffer a significant variation from planned revenue as a result.
Our current and planned clinical trials may not begin on time, or at all, and may not be completed on schedule, or at all.
The commencement or completion of any of our clinical trials may be delayed or halted for numerous reasons, including, but not limited to, the following:
o
the FDA or other regulatory authorities do not approve a clinical trial protocol or a clinical trial, or place a clinical trial on hold;
o
the data and safety monitoring committee of a clinical trial recommends that a trial be placed on hold or suspended;
o
patients do not enroll in clinical trials at the rate we expect;
o
patients are not followed-up at the rate we expect;
o
patients experience adverse side effects or events related to our products;
o
patients die or suffer adverse medical effects during a clinical trial for a variety of reasons, including the advanced stage of their disease and medical problems, which may or may not be related to our product candidates;
o
regulatory inspections of our clinical trials or manufacturing facilities, which may, among other things, require us to undertake corrective action or suspend or terminate our clinical trials if investigators find us not to be in compliance with regulatory requirements;
o
the failure of our manufacturing process to produce finished products which conform to design and performance specifications;
o
changes in governmental regulations or administrative actions;
o
the interim results of the clinical trial are inconclusive or negative;
o
pre-clinical or clinical data is interpreted by third parties in different ways;
o
our clinical trial expenditures are constrained by our budgetary considerations; or
o
our trial design, although approved, is inadequate to demonstrate safety and/or efficacy.
Clinical trials may require the enrollment of large numbers of patients, and suitable patients may be difficult to identify and recruit. Patient enrollment in clinical trials and completion of patient follow-up in clinical trials depend on many factors, including the size of the patient population, the nature of the trial protocol, the proximity of patients to clinical sites and the eligibility criteria for the study and patient compliance. For example, patients may be discouraged from enrolling in our clinical trials if the trial protocol requires them to undergo extensive post-treatment procedures to assess the safety and effectiveness of our products, or they may be persuaded to participate in contemporaneous trials of competitive products. Delays in patient enrollment or failure of patients to continue to participate in a study may cause an increase in costs and delays or may result in the failure of the trial.
Our clinical trial costs will increase if we have material delays in our clinical trials or if we need to perform more or larger clinical trials than planned. Adverse events during a clinical trial could cause us to repeat a trial, terminate a trial or cancel an entire program.
Pre-clinical development is a long, expensive and uncertain process, and we may terminate one or more of our pre-clinical development programs.
We may determine that certain pre-clinical product candidates or programs do not have sufficient potential to warrant the allocation of resources. Accordingly, we may elect to terminate our programs for such product candidates. If we terminate a pre-clinical program in which we have invested significant resources, our prospects will suffer, as we will have expended resources on a program that will not provide a return on our investment and we will have missed the opportunity to have allocated those resources to potentially more productive uses.
We may not be able to protect our intellectual property or obtain necessary intellectual property rights from third parties, which could adversely affect our business.
Our success depends, in part, on ensuring that our intellectual property rights are covered by valid and enforceable patents or effectively maintained as trade secrets and our ability to detect violations of our intellectual property rights and enforce such rights against others.
- 18 -
The validity of our patent claims depends, in part, on whether prior art references described or rendered obvious our inventions as of the filing date of our patent applications. We may not have identified all prior art, such as U.S. and foreign patents, published applications or published scientific literature, that could adversely affect the validity of our issued patents or the patentability of our pending patent applications. For example, patent applications in the United States are maintained in confidence for up to 18 months after their filing. In some cases, however, patent applications remain confidential in the U.S. Patent and Trademark Office, which we refer to as the U.S. Patent Office, for the entire time prior to issuance as a U.S. patent. Patent applications filed in countries outside the United States are not typically published until at least 18 months from their first filing date. Similarly, publication of discoveries in scientific or patent literature oft en lags behind actual discoveries. Therefore, we cannot be certain that we were the first to invent, or the first to file patent applications related to, our technology. In the event that a third party has also filed a U.S. patent application covering a similar invention, we may have to participate in an adversarial proceeding, known as an interference, declared by the U.S. Patent Office to determine priority of invention in the United States. It is possible that we may be unsuccessful in the interference, resulting in a loss of some portion or all of our U.S. patent positions. The laws in some foreign jurisdictions do not protect intellectual property rights to the same extent as in the United States, and many companies have encountered significant difficulties in protecting and defending such rights in foreign jurisdictions. If we encounter such difficulties or we are otherwise precluded from effectively protecting our intellectual property rights in foreign jurisdictions, our business prospects could be s ubstantially harmed.
We frequently seek patents to protect our intellectual property. It should be recognized that we may not be able to obtain patent protection for key elements of our technology, as the patent positions of pharmaceutical, biotechnology and medical device companies are uncertain and involve complex legal and factual questions for which important legal issues are largely unresolved. For example, no consistent policy has emerged regarding the scope of health-related patent claims that are granted by the U.S. Patent Office or enforced by the U.S. federal courts. Rights under any of our issued patents may not provide us with commercially meaningful protection for our products or afford us a commercial advantage against our competitors or their competitive products or processes. In addition, even if a patent is issued, the coverage claimed in a patent application may be significantly reduced in the patent as granted.
There can be no assurance that:
o
patent applications will result in the issuance of patents;
o
additional proprietary products developed will be patentable;
o
licenses we have obtained from third parties that we use in connection with our technology will not be terminated;
o
patents issued will provide adequate protection or any competitive advantages;
o
patents will not be successfully challenged by any third parties; or
o
the patents of others will not impede our or our collaborators’ ability to commercialize our technology.
For example, the drug paclitaxel is itself not covered by composition of matter patents. Therefore, although we are developing an intellectual property portfolio around the use of paclitaxel for intended commercial applications, others may be able to engage in off-label use of paclitaxel for the same indications, causing us to lose potential revenue. Furthermore, others may independently develop similar products or technologies or, if patents are issued to us, design around any patented technology developed by us, which could affect our potential to generate revenues and harm our results of operations.
Patent protection for our technology may not be available based on prior art. The publication of discoveries in scientific or patent literature often lags behind actual discoveries. As a consequence, there may be uncertainty as to whether we or a third party were the first creator of inventions covered by issued patents or pending patent applications or that we or a third party were the first to file patent applications for such inventions. Moreover, we might have to participate in interference proceedings declared by the U.S. Patent Office, or other proceedings outside the United States, including oppositions, to determine priority of invention or patentability, which could result in substantial cost to us even if the outcome were favorable. An unfavorable outcome in an interference or opposition proceeding could preclude us, our collaborators and our licensees from making, using or selling products using the technology or require us to obtain license rights from prevaili ng third parties. We do not know whether any prevailing party would offer us a license on commercially acceptable terms, if at all. We may also be forced to pay damages or royalties for our past use of such intellectual property rights, as well as royalties for any continued usage.
As part of our patent strategy, we have filed a variety of patent applications internationally. Oppositions have been filed against various granted patents that we either own or license and which are related to certain of our technologies. See “Legal Proceedings” elsewhere in this Annual Report on Form 10-K for a discussion of the proceedings related to certain of such oppositions.
Our future success and competitive position depend in part on our ability to obtain and maintain certain proprietary intellectual property rights used in our approved products and principal product candidates. Any such success depends in part on effectively prosecuting claims against others who we believe are infringing our rights and by effectively defending claims of intellectual property infringement brought by our competitors and others. The stent-related markets have experienced rapid technological change and obsolescence in the recent past, and our competitors have strong incentives to attempt to stop or delay us from introducing new products and technologies. See “—We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.”
- 19 -
We do not know whether the patents that we have obtained or licensed, or may be able to obtain or license in the future, would be held valid or enforceable by a court or whether a competitor’s technology or product would be found to infringe such patents. Further, we have no assurance that third parties will not properly or improperly modify or terminate any license they have granted to us.
We have obtained licenses from third parties with respect to their intellectual property that we use in connection with our technology. However, we may need to obtain additional licenses for the development of our current or future products. Licenses may not be available on satisfactory terms or at all. If available, these licenses may obligate us to exercise diligence in bringing our technology to market and may obligate us to make minimum guarantee or milestone payments. This diligence and these milestone payments may be costly and could adversely affect our business. We may also be obligated to make royalty payments on the sales, if any, of products resulting from licensed technology and may be responsible for the costs of filing and prosecuting patent applications. These costs could affect our results of operations and decrease our earnings.
Certain of our key technologies include trade secrets and know-how that may not be protected by patents. There can be no assurance that we will be able to protect our trade secrets. To help protect our rights, we undertake to require employees, consultants, advisors and collaborators to enter into confidentiality agreements. We cannot assure you that all employees, consultants, advisors and collaborators have signed such agreements, or that these agreements will adequately protect our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure. Furthermore, we may not have adequate remedies for any such breach. Any disclosure of confidential data into the public domain or to third parties could allow our competitors to learn our trade secrets and use the information in competition against us.
If certain single-source suppliers fail to deliver key product components in a timely manner, our manufacturing ability would be impaired and our product sales could suffer.
We depend on certain single-source suppliers that supply components used in the manufacture of certain of our products, including our Quill SRS product. If we need alternative sources for key component parts for any reason, these component parts may not be immediately available to us. If alternative suppliers are not immediately available, we will have to identify and qualify alternative suppliers, and production of these components may be delayed. We may not be able to find an adequate alternative supplier in a reasonable time period or on commercially acceptable terms, if at all. Shipments of affected products have been limited or delayed as a result of such problems in the past, and similar problems could occur in the future. Our inability to obtain our key source supplies for the manufacture of our products may require us to delay shipments of products, harm customer relationships or force us to curtail or cease operations.
If physicians do not recommend and endorse our products or products that use our technology, or if our working relationships with physicians deteriorate, our products or products that use our technology may not be accepted in the marketplace, which could adversely affect our sales and royalty revenues.
In order for us to sell our products or continue to receive royalty revenues from the sale of products that use our technologies, physicians must recommend and endorse them. We believe that recommendations and endorsements by physicians will be essential for market acceptance of our products, and we do not know whether we will obtain the necessary recommendations or endorsements from physicians. Acceptance of our products or of products that use our technology depends on educating the medical community as to the distinctive characteristics, perceived benefits, safety, clinical efficacy and cost-effectiveness of these products compared to products of competitors, and on training physicians in the proper application of these products. If we are not successful in obtaining the recommendations or endorsements of physicians for our products or our collaborators are not successful in doing the same for their products that use our technology, our sales and royalty revenues may no t increase or may decline.
In addition, if we fail to maintain our working relationships with physicians, many of our products may not be developed and marketed in line with the needs and expectations of professionals who use and support our products. The research, development, marketing and sales of many of our new and improved products is dependent upon our maintaining working relationships with physicians. We rely on these professionals to provide us with considerable knowledge and experience regarding our products and the marketing of our products. Physicians assist us as researchers, marketing consultants, product consultants, inventors and as public speakers. If we are unable to maintain our strong relationships with these professionals and continue to receive their advice and input, the development and marketing of our products could suffer, which could adversely affect the acceptance of our products in the marketplace and our sales.
- 20 -
If we are unable to license new technologies to utilize in the development of products, or our existing license agreements are terminated, our ability to maintain our competitive advantage in our existing products and to develop future products may be adversely affected.
We have entered into, and we expect that we will continue to enter into, licensing agreements with third parties to give us access to technologies that we may use to develop products through our strategic collaboration and partnership arrangements. The technologies governed by these license agreements may be critical to our ability to maintain our competitive advantage in our existing products and to develop future products. For example, through licenses with the NIH and UBC, we have been granted access to technologies that have contributed to the developments of the Taxus Paclitaxel-Eluting Coronary Stent.
Pursuant to terms of our existing license agreements, licensors have the right under certain specified circumstances to terminate their respective licenses. Events that may allow licensors to exercise these termination provisions include:
o
our bankruptcy;
o
sub-licensing without the licensor’s consent;
o
a transaction which results in a change of control of us;
o
our failure to use the required level of diligence to develop, market and sell products based on the licensed technology;
o
our failure to maintain adequate levels of insurance with respect to the licensed technologies; or
o
other acts or omissions that may constitute a breach by us of our license agreement.
In addition, any failure to continue to have access to these technologies may materially adversely affect the benefits that we currently derive from our collaboration and partnership arrangements and may adversely affect our results and operations.
Compulsory licensing and/or generic competition may affect our business in certain countries.
In a number of countries, governmental authorities and other groups have suggested that companies which manufacture medical products (i.e., pharmaceuticals and medical devices) should make products available at a low cost. In some cases, governmental authorities have held that where a pharmaceutical or medical device company does not do so, their patents might not be enforceable to prevent generic competition. Alternatively, some governmental authorities could require that we grant compulsory licenses to allow competitors to manufacture and sell their own versions of our products, thereby reducing our sales or the sales of our licensee(s). In all of these situations, the results of our operations in these countries could be adversely affected.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.
In connection with maintaining the value of our various intellectual property and exclusivity rights, we regularly evaluate the activities of others worldwide. Our success will depend, in part, on our ability to obtain patents, or licenses to patents, maintain trade secret protection and enforce our rights against others. Should it become necessary to protect those rights, we intend to pursue all cost-efficient strategies, including, when appropriate, negotiation or litigation in any relevant jurisdiction. For a summary of certain of our current legal proceedings, see “Legal Proceedings” elsewhere in this Annual Report on Form 10-K.
We intend to pursue and to defend vigorously any and all actions of third parties related to our material patents and pioneering technology. Any failure to obtain and protect intellectual property could adversely affect our business and our ability to operate could be hindered by the proprietary rights of others.
Our involvement in intellectual property litigation could result in significant expense, adversely affecting the development of product candidates or sales of the challenged product or intellectual property and diverting the efforts of our technical and management personnel, whether or not such litigation is resolved in our favor. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources and intellectual property litigation may be used against us as a means of gaining a competitive advantage. Competing parties frequently file multiple suits to leverage patent portfolios across product lines, technologies and geographies and to balance risk and exposure between the parties. Uncertainties resulting from the initiation and continuation of any litigation could affect our ability to continue our operations. In the event of an adverse outcome as a defendant in any such liti gation, we may, among other things, be required to:
o
pay substantial damages or back royalties;
o
cease the development, manufacture, use or sale of product candidates or products that infringe upon the intellectual property of others;
o
expend significant resources to design around a patent or to develop or acquire non-infringing intellectual property;
o
discontinue processes incorporating infringing technology; or
o
obtain licenses to the infringed intellectual property.
We cannot be assured that we will be successful in developing or acquiring non-infringing intellectual property or that necessary licenses will be available upon reasonable terms, if at all. Any such development, acquisition or license could require the expenditure of substantial time and other resources and could adversely affect our business and financial results. If we cannot develop or acquire such intellectual property or obtain such licenses, we could encounter delays in any introduction of products or could find that the development, manufacture or sale of products requiring such licenses could be prohibited.
- 21 -
If third parties file patent applications, or are issued patents claiming technology also claimed by us in pending applications, we may be required to participate in interference proceedings with the U.S. Patent Office, or other proceedings outside the United States, including oppositions, to determine priority of invention or patentability, which could result in substantial cost to us even if the eventual outcome were favorable.
Our ability to operate could be hindered by the proprietary rights of others.
A number of pharmaceutical, biotechnology and medical device companies as well as research and academic institutions have developed technologies, filed patent applications or received patents on various technologies that may be related to our business. Some of these technologies, applications or patents may conflict with or adversely affect our technologies or intellectual property rights, including those that we license from others. We are aware of other parties holding intellectual property rights that may represent prior art or other potentially conflicting intellectual property, including stents coated with agents intended to reduce restenosis. Any conflicts with the intellectual property of others could limit the scope of the patents, if any, that we may be able to obtain or result in the denial of our current or future patent applications altogether.
If patents that cover our activities are issued to other persons or companies, we could be charged with infringement. In the event that other parties’ patents cover any portion of our activities, we may be forced to develop alternatives or negotiate a license for such technology. We do not know whether we would be successful in either developing alternative technologies or acquiring licenses upon reasonable terms, if at all. Obtaining any such licenses could require the expenditure of substantial time and other resources and could harm our business and decrease our earnings. If we do not obtain such licenses, we could encounter delays in the introduction of our products or could find that the development, manufacture or sale of products requiring such licenses is prohibited.
Technological advances and evolving industry standards could reduce our future product sales, which could cause our revenues to grow more slowly or decline.
The markets for our products are characterized by rapidly changing technology, changing customer needs, evolving industry standards and frequent new product introductions and enhancements. The emergence of new industry standards in related fields may adversely affect the demand for our products. This could happen, for example, if new standards and technologies emerged that were incompatible with customer deployments of our applications. In addition, any compounds, products or processes that we develop may become obsolete or uneconomical before we recover any of the expenses incurred in connection with their development. We cannot assure you that we will succeed in developing and marketing product enhancements or new products that respond to technological change, new industry standards, changed customer requirements or competitive products on a timely and cost-effective basis. Additionally, even if we are able to develop new products and product enhancements, we cannot assu re you that they will achieve market acceptance.
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no such claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper or prevent our ability to commercialize certain product candidates, which could severely harm our business.
We may incur significant costs complying with environmental laws and regulations.
Our research and development processes and manufacturing operations involve the use of hazardous materials. We are subject to federal, state, provincial, local and other laws and regulations in the countries in which we operate or sell our products, which govern the use, manufacture, storage, handling and disposal of such materials and certain waste products. The risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of an accident or the discovery of pre-existing contamination at one or more of our facilities, we could be held liable for any damages that result and any such liability could exceed our resources. We may not be specifically insured with respect to this liability, and we do not know whether we will be required to incur significant costs to comply with environmental laws and regulations in the future, or whether our operations, business or assets will be harmed by current or future environmental laws or re gulations.
- 22 -
We face and will continue to face significant competition.
Competition from pharmaceutical companies, medical device companies, biotechnology companies and academic and research institutions is intense and is expected to increase. Many of our competitors and potential competitors have substantially greater product development capabilities, experience conducting clinical trials and financial, scientific, manufacturing, sales and marketing resources and experience than our company. Some of these competitors include JNJ, Guidant Corporation, Genzyme Corporation, Baxter, Abbott Laboratories, BSC, Medtronic, Inc., Wyeth, Inc., Novartis AG, C.R. Bard, the Allegiance division of Cardinal Health, Inc., Bausch & Lomb, and Covidien Ltd., among others. We also face competition from non-medical device companies, such as pharmaceutical companies, which may offer non-surgical alternative therapies for disease states which are currently or intended to be treated using our products. Other companies may:
o
develop and obtain patent protection for products earlier than us;
o
design around patented technology developed by us;
o
obtain regulatory approvals for such products more rapidly;
o
have greater manufacturing capabilities and other resources;
o
have larger or more experienced sales forces;
o
develop more effective or less expensive products; or
o
have greater success in obtaining adequate third-party payer coverage and reimbursement for their competing products.
While we intend to expand our technological capabilities in order to remain competitive, there is a risk that:
o
research and development by others will render our technology or product candidates obsolete or non-competitive;
o
treatments or cures developed by others will be superior to any therapy developed by us; and
o
any therapy developed by us will not be preferred to any existing or newly-developed technologies.
The commercial potential of our products and product candidates will be significantly limited if we are not able to obtain adequate levels of reimbursement or market acceptance for them.
Our ability to commercialize human therapeutic products and product candidates successfully will depend in part on the extent to which coverage and reimbursement for such products and related treatments will be available from government health administration authorities, private health insurers and other third-party payers or supported by the market for these products. There can be no assurance that third-party payers’ coverage and reimbursement will be available or sufficient for the products we might develop.
Third party payers are increasingly challenging the price of medical products and services and instituting cost containment measures to control or significantly influence the purchase of medical products and services. These cost containment measures, if instituted in a manner affecting the coverage of or payment for our products, could have a material adverse effect on our ability to operate profitably. In some countries in the European Union and in the United States, significant uncertainty exists as to the reimbursement status of newly-approved healthcare products, and we do not know whether adequate third-party coverage and reimbursement will be available for us to realize an appropriate return on our investment in product development, which could seriously harm our business. In the United States, while reimbursement amounts previously approved appear to have provided a reasonable rate of return, there can be no assurance that our products will continue to be reimbursed at current rates or that third-party payers will continue to consider our products cost-effective and provide coverage and reimbursement for our products, in whole or in part.
We cannot be certain that our products will gain commercial acceptance among physicians, patients and third party payers, even if necessary international and United States marketing approvals are maintained. We believe that recommendations and endorsements by physicians will be essential for market acceptance of our products, and we do not know whether these recommendations or endorsements will be obtained. We also believe that surgeons will not use these products unless they determine, based on clinical data and other factors, that the clinical benefits to patients and cost savings achieved through use of these products outweigh their cost. Acceptance among physicians may also depend upon the ability to train surgeons and other potential users of our products and the willingness of such users to learn these relatively new techniques.
Future legislation or regulatory changes to, or consolidation in, the healthcare system may affect our ability to sell our product profitably.
There have been, and we expect there will continue to be, a number of legislative and regulatory proposals to change the healthcare system, and some could involve changes that could significantly affect our business. Efforts by governmental and third-party payers to reduce health care costs or the announcement of legislative proposals or reforms to implement government controls could cause a reduction in sales or in the selling price of our products, which would seriously harm our business. Additionally, initiatives to reduce the cost of healthcare have resulted in a consolidation trend in the healthcare industry, including hospitals. This in turn has resulted in greater pricing pressures and the exclusion of certain suppliers from certain market segments as consolidated groups such as group purchasing organizations, independent delivery networks and large single accounts continue to consolidate purchasing decisions for some of our hospital customers. We expect that market demand, government regulation, and third-party reimbursement policies will continue to change the worldwide healthcare industry, resulting in further business consolidations and alliances among our customers and competitors, which may reduce competition, exert further downward pressure on the prices of our products and may adversely impact our business, financial condition or results of operations.
- 23 -
We must receive regulatory approval for each of our product candidates before they can be sold commercially in Canada, the United States or internationally, which can take significant time and be very costly.
The development, manufacture and sale of medical devices and human therapeutic products in Canada, the United States and internationally is governed by a variety of statutes and regulations. These laws require, among other things:
o
regulatory approval of manufacturing facilities and practices;
o
adequate and well-controlled research and testing of products in pre-clinical and clinical trials;
o
review and approval of submissions containing manufacturing, pre-clinical and clinical data in order to obtain marketing approval based on establishing the safety and efficacy of the product for each use sought, including adherence to cGMPs during production and storage; and
o
control of marketing activities, including advertising and labelling.
The product candidates currently under development by us or our collaborators will require significant research, development, pre-clinical and clinical testing, pre-market review and approval, and investment of significant funds prior to their commercialization. In many instances, we are dependent on our collaborators for regulatory approval and compliance, and have little or no control over these matters. The process of completing clinical testing and obtaining such approvals is likely to take many years and require the expenditure of substantial resources, and we do not know whether any clinical studies by us or our collaborators will be successful, that regulatory approvals will be received, or that regulatory approvals will be obtained in a timely manner. Despite the time and resources expended by us, regulatory approval is never guaranteed. Even if regulatory approval is obtained, regulatory agencies may limit the approval to certain diseases, conditions or categories of patients who can use them.
If any of our development programs are not successfully completed in a timely fashion, required regulatory approvals are not obtained in a timely fashion, or products for which approvals are obtained are not commercially successful, it could seriously harm our business.
Our products and manufacturing facilities that have, or may receive, regulatory approval, are or will be subject to ongoing regulation. In addition, we have little or no control over the manufacturing facilities of our collaborators in which certain of our products are manufactured.
Our products and manufacturing operations are subject to extensive regulation in the United States by the FDA and by similar regulatory agencies abroad. Ongoing regulation includes compliance with an array of manufacturing and design controls and testing, quality control, storage and documentation procedures. Regulatory agencies may also require expensive post-approval studies. Any adverse events associated with our products must also be reported to regulatory authorities. If deficiencies in our or our collaborators’ manufacturing and laboratory facilities are discovered, or we or our collaborators fail to comply with applicable post-market regulatory requirements, a regulatory agency may close the facility or suspend manufacturing.
With respect to products manufactured by third-party contractors, we are, and we expect to continue to be, dependent on our collaborators for continuing regulatory compliance and we may have little or no control over these matters. Our ability to control third-party compliance with FDA and other regulatory requirements will be limited to contractual remedies and rights of inspection. Our failure or the failure of third-party manufacturers to comply with regulatory requirements applicable to our products may result in legal or regulatory action by those regulatory authorities. There can be no assurance that our or our collaborators’ manufacturing processes will satisfy regulatory, cGMP or International Standards Organization (“ISO”) requirements.
In addition, there may be uncertainty as to whether or not we or others who are involved in the manufacturing process will be able to make the transition to commercial production of some of our newly developed products. A failure to achieve regulatory approval for manufacturing facilities or a failure to make the transition to commercial production for our products will adversely affect our prospects, business, financial condition and results of operations.
If we are unable to fully comply with federal and state “fraud and abuse laws”, we could face substantial penalties, which may adversely affect our business, financial condition and results of operations.
We are subject to various laws pertaining to health care fraud and abuse, including the federal Anti-Kickback Statute, physician self-referral laws, the federal False Claims Act, the federal Health Insurance Portability and Accountability Act of 1996, the federal False Statements Statute, and state law equivalents to these federal laws, which may not be limited to government-reimbursed items and may not contain identical exceptions. Violations of these laws are punishable by criminal and civil sanctions, including, in some instances, civil and criminal penalties, damages, fines, exclusion from participation in federal and state healthcare programs, including Medicare and Medicaid, and the curtailment or restructuring of operations. Any action against us for violation of these laws could have a significant impact on our business. In addition, we are subject to the U.S. Foreign Corrupt Practices Act. We have a network of approximately 160 distributors. Any action against us for violation by us or our distributors of this act could have a significant impact on our business.
- 24 -
We may be unsuccessful in marketing, selling and distributing certain of our products.
We distribute a number of our products worldwide. In order to achieve commercial success for our approved products, we have previously expanded our sales and marketing force in the United States, Europe and other parts of the world. If our distribution personnel or methods are not sufficient to ensure we have supply to meet demand for our products or if there is a quality control failure with our products, it could harm our prospects, business, financial condition and results of operations.
To the extent that we enter into co-promotion or other marketing and sales arrangements with other companies, any revenues received will be dependent on the efforts of others, and we do not know whether these efforts will be successful. Failure to develop a direct sales and marketing force or enter into appropriate arrangements with other companies to market and sell our products will reduce our ability to generate revenues.
We may encounter unanticipated costs or loss of business associated with terminating or relocating facilities and operations.
We have consolidated our Syracuse, New York and Puerto Rico manufacturing facilities into two facilities in Puerto Rico. There is a risk that there may be further unanticipated costs associated with this consolidation.
Consolidation in the healthcare industry could have an adverse effect on our revenues and results of operations.
Many healthcare industry companies, including medical device companies, are consolidating to create new companies with greater market power. As the healthcare industry consolidates, competition to provide goods and services to industry participants will become more intense. These industry participants may try to use their market power to negotiate price concessions or reductions for medical devices that incorporate components produced by us. If we are forced to reduce our prices because of consolidation in the healthcare industry, our revenues would decrease and our consolidated earnings, financial condition or cash flows would suffer.
We may incur losses associated with foreign currency fluctuations.
We report our operating results and financial position in U.S. dollars in order to more accurately represent the currency of the economic environment in which we operate.
Our operations are in some instances conducted in currencies other than the U.S. dollar and fluctuations in the value of foreign currencies relative to the U.S. dollar could cause us to incur currency exchange losses. In addition to the U.S. dollar, we currently conduct operations in Canadian dollars, Euros, Swiss francs, Danish kroner, and U.K. pounds sterling. Exchange rate fluctuations may reduce our future operating results and comprehensive income. For the year ended December 31, 2008, we reported $0.5 million of net foreign exchange gains due to foreign currency fluctuations compared to $0.3 million of foreign exchange losses for the year ended December 31, 2007.
We have not entered into any forward currency contracts or other financial derivatives to hedge foreign exchange risk, and therefore we are subject to foreign currency transaction and translation gains and losses. We purchase goods and services in U.S. and Canadian dollars, Swiss francs, Danish krone, Euros, and U.K. pounds sterling, and earn a significant portion of our license and milestone revenues in U.S. dollars. Foreign exchange risk is managed primarily by satisfying foreign denominated expenditures with cash flows or assets denominated in the same currency.
Acquisition of companies or technologies may result in disruptions to our business.
As part of our business strategy, we may acquire additional assets and businesses principally relating to or complementary to our current operations. Any acquisitions or mergers by us will be accompanied by the risks commonly encountered in acquisitions of companies. These risks include, among other things, higher than anticipated acquisition costs and expenses, the difficulty and expense of integrating the operations and personnel of the companies and the loss of key employees and customers as a result of changes in management.
In addition, geographic distances may make integration of acquired businesses more difficult. We may not be successful in overcoming these risks or any other problems encountered in connection with any acquisitions.
If significant acquisitions are made for cash consideration, we may be required to use a substantial portion of our available cash, cash equivalents and short-term investments. Future acquisitions by us may cause large one-time expenses or create goodwill or other intangible assets that could result in significant asset impairment charges in the future. Acquisition financing may not be available on acceptable terms, if at all.
- 25 -
If we fail to hire and retain key management, scientific and technical personnel, we may be unable to successfully implement our business plan.
We are highly dependent on our senior management and scientific and technical personnel. The competition for qualified personnel in the healthcare field is intense, and we rely heavily on our ability to attract and retain qualified managerial, scientific and technical personnel. Our ability to manage growth effectively will require continued implementation and improvement of our management systems and the ability to recruit and train new employees. We may not be able to successfully attract and retain skilled and experienced personnel, which could harm our ability to develop our product candidates and generate revenues.
Risks Relating to our Indebtedness, Shares, and Organization and Structure
Our existing and future permitted debt could adversely affect our operations and we may need to restructure our existing debt to ensure that our cash flows are adequate to service our debt.
As of December 31, 2008, we had outstanding $575 million of indebtedness, excluding accrued interest. Excluding intercompany transactions, our subsidiaries that are not guarantors of the Floating Rate Notes or Subordinated Notes accounted for approximately $85.1 million or 30% of our total revenues for the year ended December 31, 2008 ($53.0 million or 18% respectively for the year ended December 31, 2007), and approximately $0.3 million of our total assets and $0.1 million of our total liabilities as of December 31, 2008 ($488 million and $116 million respectively for the year ended December 31, 2007). The Floating Rate Notes and the Subordinated Notes are guaranteed by the same group of our subsidiaries.
The amount and terms of our indebtedness and other financial obligations have adversely affected our operations. For example, it:
o
increases our vulnerability to general adverse economic and industry conditions;
o
limits our ability to obtain additional financing in the future for working capital, capital expenditures, acquisitions, general corporate purposes or other purposes;
o
requires us to dedicate a substantial portion of our cash flow from operations to the payment of principal and interest on our indebtedness, thereby reducing the funds available to us for operations and any future business opportunities, including acquisitions permitted by our Subordinated Notes and Floating Rate Notes;
o
limits our planning flexibility for, or ability to react to, changes in our business and the industry as discussed under “Management’s Discussion and Analysis of Financial Condition and Results of Operations —Business Overview —Financial and Strategic Alternatives Process” in this Annual Report on Form 10-K; and
o
places us at a competitive disadvantage with competitors who may have less indebtedness and other obligations or greater access to financing.
The Floating Rate Notes bear interest at rates that fluctuate with changes in certain prevailing benchmarks. If interest rates increase, we may be unable to meet our debt service obligations under the Floating Rate Notes and Subordinated Notes and other indebtedness.
On March 2, 2009, we announced that we had completed a financing transaction with Wells Fargo Foothill, LLC, consisting of a delayed draw term loan facility of up to $10 million and a new secured revolving credit facility providing up to an additional $22.5 million in available credit. See “Management’s Discussion and Analysis of Financial Condition and Results of Operations —Liquidity and Capital Resources —New Senior Secured Credit Facilities” in this Annual Report on Form 10-K. Although this new credit facility provides enhanced liquidity in the near term, if our cash flows are worse than expected, we may need to refinance or restructure all or a portion of our indebtedness on or before maturity. We cannot assure you that we will be able to repay or refinance any of our debt on commercially reasonable terms or at all. If we are unable to refinance our debt or obtain new financing under these circumstances, we would have to consider other options, such as:
·
sales of certain assets to meet our debt service obligations;
·
sales of equity; and
·
negotiations with our lenders and the holders of our Floating Rate Notes and our Subordinated Notes to restructure the applicable debt.
We may not be able to implement one or more of these alternatives on terms acceptable to us or at all. Our financing arrangements and the indentures governing our Floating Rate Notes and Subordinated Notes may restrict, or market or business conditions may limit, our ability to do some of these things. Moreover, if we are unable to obtain sufficient financing when we need it or on terms satisfactory to us, our development activities could have to be delayed, curtailed or eliminated and our financial results could be adversely affected.
- 26 -
We and our subsidiaries are permitted to incur substantially more debt, which could further exacerbate the risks associated with our leverage.
The terms of the indentures governing the Floating Rate Notes and Subordinated Notes expressly permit the incurrence of additional amounts of debt for specified purposes. For example, on March 2, 2009, we announced that we had entered into a new revolving credit facility, and all borrowings under that facility rank senior to the Subordinated Notes and the related guarantees, to the extent of the value of the assets securing such borrowings. Moreover, the indentures governing the Floating Rate Notes and Subordinated Notes do not impose any limitation on our incurrence of liabilities that are not defined as “Indebtedness” under such indentures (such as trade payables). If new debt or other liabilities are added to our and our subsidiaries’ current levels of debt, the related risks that we and they now face could be exacerbated.
If our cash flows prove inadequate to service our debt and provide for our other obligations, we may be required to refinance all or a portion of our existing debt or future debt at terms unfavorable to us.
Our ability to make payments on and refinance our debt, including the Floating Rate Notes, the Subordinated Notes and other financial obligations, and to fund our capital expenditures and acquisitions will depend on our ability to generate substantial operating cash flow. This will depend on our future performance, which will be subject to prevailing economic conditions and to financial, business and other factors beyond our control. Although we have recently entered into a new senior secured credit facility, if our cash flows are worse than expected, and the amounts we are able to draw on that facility prove inadequate to meet our debt service and other obligations in the future, we may be required to refinance all or a portion of our existing or future debt, including the Floating Rate Notes and Subordinated Notes, on or before maturity, to sell assets or to obtain additional financing. We cannot assure you that we will be able to refinance any of our indebtedness, inclu ding the Floating Rate Notes and Subordinated Notes, sell any such assets or obtain such additional financing on commercially reasonable terms or at all. Additionally, because the indentures governing the Floating Rate Notes and Subordinated Notes require that, upon the occurrence of a “change of control,” as defined in the indentures, we must make an offer to repurchase the Floating Rate Notes and Subordinated Notes, respectively, at a price equal to 101% of the principal amount thereof, plus accrued and unpaid interest, if any, to the date of repurchase. In the event that we were required to repurchase the Floating Rate Notes and Subordinated Notes pursuant to our offer, we would not have enough cash available to make such repurchase.
For additional risks related to the refinancing of our existing debt, see “—Our existing and future permitted debt could adversely affect our operations, and we need to restructure our existing debt to ensure that our cash flows are adequate to service our debt.”
The indentures governing the Floating Rate Notes and Subordinated Notes contain covenants that may limit our ability to take advantage of certain business opportunities advantageous to us that may arise.
The indentures governing the Floating Rate Notes and Subordinated Notes contain certain covenants that, among other things, limit our ability and the ability of certain of our subsidiaries to:
o
incur, assume or guarantee additional indebtedness or issue preferred stock;
o
pay dividends or make other equity distributions to our shareholders;
o
purchase or redeem our capital stock;
o
make certain investments;
o
create liens;
o
sell or otherwise dispose of assets;
o
engage in transactions with our affiliates; and
o
merge or consolidate with another entity or transfer all or substantially all of our assets.
These restrictions could limit our ability to obtain future financing, make acquisitions or needed capital expenditures, withstand economic downturns in our business, industry or the economy in general, conduct operations or otherwise take advantage of business opportunities that may arise.
Although the indentures for the Floating Rate Notes and Subordinated Notes contain a fixed charge coverage test that limits our ability to incur indebtedness, this limitation is subject to a number of significant exceptions and qualifications. Moreover, the indentures do not impose any limitation on our incurrence of liabilities that are not considered “Indebtedness” under the indentures (such as operating leases), nor do they impose any limitation on the amount of liabilities incurred by subsidiaries, if any, that might be designated as “Unrestricted Subsidiaries” under the indentures. Despite current indebtedness levels, we and our subsidiaries may still be able to incur substantially more debt. This could further exacerbate the risks associated with our leverage. Also, although the indentures limit our ability to make restricted payments, these restrictions are subject to significant exceptions and qualifications.
- 27 -
The current global credit and financial market conditions may exacerbate certain risks affecting our business.
Sales of our products are dependent, in large part, on reimbursement from government health administration authorities, private health insurers, distribution partners and other organizations. As a result of the current global credit and financial market conditions, these organizations may be unable to satisfy their reimbursement obligations or may delay payment. In addition, federal and state health authorities may reduce Medicare and Medicaid reimbursements, and private insurers may increase their scrutiny of claims. A reduction in the availability or extent of reimbursement could negatively affect our product sales and revenue.
Due to the recent tightening of global credit, there may be a disruption or delay in the performance of our third-party contractors, suppliers or collaborators. We rely on third parties for several important aspects of our business, including royalty revenue, portions of our product manufacturing, clinical development of future collaboration products, conduct of clinical trials and raw materials. If such third parties are unable to satisfy their commitments to us, our business would be adversely affected.
Certain of our products are used in elective medical procedures which are not covered by insurance. Adverse changes in the economy or other conditions or events have had and may continue to have an adverse effect on consumer spending and may reduce the demand for these procedures. Any such changes, conditions or events could have an adverse effect on our sales and results of operations.
The NASDAQ and/or the Toronto Stock Exchange may delist our common shares from quotation on its exchange, which could limit investors’ ability to make transactions in our common shares and subject us to additional trading restrictions.
The NASDAQ rules provide that the exchange can delist a company’s shares for failing to maintain a share price above a dollar.Our common shares are currently trading at less than a dollar on the NASDAQ. The NASDAQ hascurrently placed a moratorium on delistings for failing to maintain a share price above a dollar. We cannot assure you that the price of our common shares will rise above a dollar or that our common shares will continue to be traded on the NASDAQ in the future.
The Toronto Stock Exchange may delist a company’s shares if, in the opinion of the Toronto Stock Exchange, the financial condition and/or the operating results of the company appear to be unsatisfactory or appear not to warrant continuation of the securities on the trading list. The Toronto Stock Exchange may consider a number of factors when determining whether to delist a company’s shares, including the company’s ability to meet its obligations as they become due, working capital position, quick asset position, total assets, capitalization, cash flow, earnings, annual revenues, public distribution of the listed shares, share price and trading activity. We cannot assure you that we will continue to meet the listing requirements of the Toronto Stock Exchange or that our common shares will continue to be traded on the Toronto Stock Exchange in the future.
The threat of delisting and/or a delisting of our common shares could have material adverse effects by, among other things:
·
reducing the liquidity and market price of our common shares;
·
reducing the number of investors willing to hold or acquire our commons shares, thereby further restricting our ability to obtain equity financing;
·
reducing the amount of news and analyst coverage of our company; and
·
reducing our ability to retain, attract and motivate our directors, officers and employees.
United States investors may not be able to obtain enforcement of civil liabilities against us.
We were formed under the laws of British Columbia, Canada. A substantial portion of our assets are located outside the United States. In addition, a majority of the members of our board of directors and our officers are residents of countries other than the United States. As a result, it may be impossible for United States investors to affect service of process within the United States upon us or these persons or to enforce against us or these persons any judgments in civil and commercial matters, including judgments under United States federal or state securities laws. In addition, a Canadian court may not permit United States investors to bring an original action in Canada or to enforce in Canada a judgment of a state or federal court in the United States.
Laws and provisions in our notice of articles, articles, shareholder rights plan and stock option plan could delay or deter a change in control.
Our notice of articles and articles allow for the issuance of Class I preference shares. The board of directors may set the rights and restrictions of any series of preference shares in its sole discretion without the approval of the holders of our common shares. The rights and restrictions of our preference shares may be superior to those of the common shares. Accordingly, the issuance of preference shares also could have the effect of delaying or preventing a change of control of our company. There are at present, no preference shares outstanding.
In addition, under theBusiness Corporations Act (British Columbia) and our articles, some business combinations, including the sale, lease or other disposition of all or substantially all of our undertaking, must be approved by at least three-quarters of the votes cast by our shareholders in aggregate or, in some cases, approved by at least three-quarters of the votes cast by holders of each class of shares. In some cases, a business combination must be approved by a court. Shareholders may also have a right to dissent from the transaction, in which case, we would be required to pay dissenting shareholders the fair value of their common shares provided they have followed the required procedures.
- 28 -
In addition, our shareholders adopted a shareholder rights plan which provides for substantial dilution to an acquiror of 20% or more of our common shares, except in certain circumstances, including a) the acquiror makes a bid to all shareholders, which, among other things, is held open for at least 60 days and is accepted by independent shareholders holding at least 50% of the outstanding common shares, or b) the bid is otherwise approved by our board of directors. The shareholder rights plan was amended and restated on October 30, 2008 and must be reconfirmed by the shareholders every three years.
Furthermore, all of our executive officers have contractual rights under employment agreements that provide for 12 to 24 months severance pay in the event of a change of control of our company. Under our stock option plan, following a change of control, all outstanding stock options vest immediately. In the event that an offer is made to our shareholders generally or to a class of our shareholders, that if accepted would result in the offeror becoming a control person (generally meaning a person holding 20% or more of a company’s voting shares), our board of directors has the discretion under our stock option plan to determine to accelerate the vesting and expiry date of all outstanding options.
Limitations on the ability to acquire and hold our common shares may be imposed by the Competition Act (Canada). This legislation permits the Commissioner of Competition to review any acquisition of a significant interest in our company. This legislation grants the Commissioner jurisdiction to challenge such an acquisition before the Competition Tribunal if the Commissioner believes that it would, or would be likely to, result in a substantial lessening or prevention of competition in any market in Canada. The Investment Canada Act (Canada) subjects an acquisition of control of a company by a non-Canadian to government review if the value of our assets as calculated pursuant to the legislation exceeds a threshold amount which, for an investor from a World Trade Organization member country, is CDN$312 million in 2009. A reviewable acquisition may not proceed unless the relevant minister is satisfied or is deemed to be satisfied that there is likely to be a net benefit to Ca nada from the transaction.
Each of these matters could delay or deter a change in control that would be attractive to, and provide liquidity for, shareholders, and could limit the price that investors are willing to pay in the future for our common shares.
Item 1B.
UNRESOLVED STAFF COMMENTS
None.
- 29 -
Item 2.
PROPERTIES
We have 16 facilities located in six different countries, which include Canada, the United States, Puerto Rico, the United Kingdom, Denmark and Switzerland. Of the 16 facilities, 11 are primarily used to manufacture and distribute medical devices or materials for medical device products. Our other 5 facilities are primarily used for research, development and administrative activities. Collectively, these facilities have over 540,000 square feet of modern technical manufacturing, research and administrative operations. The following chart summarizes the facilities where our domestic and international operations occur:
| Location1 | Primary Purpose | Segment | Owned or Leased |
| Chicago, IL | Administration | Medical Products | Leased |
| Lausanne, Switzerland | Administration | Medical Products | Leased |
| Gainesville, FL | Manufacturing | Medical Products | Owned |
| Henrietta, NY | Manufacturing | Medical Products | Leased |
| Laguna Hills, CA | Manufacturing | Medical Products | Leased |
| North Bend, WA | Administration | Pharmaceutical | Leased |
| | | Technologies | |
| Reading, PA | Manufacturing | Medical Products | Owned |
| Aguadilla, Puerto Rico | Manufacturing | Medical Products | Leased |
| Rincon, Puerto Rico | Manufacturing | Medical Products | Leased |
| Stenlose, Denmark (3 facilities) | Manufacturing | Medical Products | 2 Leased, 1 Owned |
| Taunton, United Kingdom | Manufacturing | Medical Products | Owned |
| Vancouver, B.C. (2 facilities). | Administration and Research | Pharmaceutical | Leased |
| | | Technologies | |
| Wheeling, IL | Manufacturing | Medical Products | Owned |
| 1Excluded from this list are our two properties that are vacant and are held for sale. One facility is in Syracuse, NY with��77,000 square feet and the other is in Rincon, Puerto Rico with 31,000 square feet. |
We have several important, and in some cases proprietary, manufacturing capabilities and processes. These include specific capabilities in the areas of various metal bending, grinding and chemical etching, electroplating and electropolishing, plastic injection molding, wire coating, braiding and weaving of surgical textiles and kit assembly and packaging.
- 30 -
Item 3.
LEGAL PROCEEDINGS
i)
On April 4, 2005, the Company together with BSC commenced a legal action in the Netherlands against Sahajanand Medical Technologies Pvt. Ltd. for patent infringement. On May 3, 2006, the Dutch trial court ruled in favor of Angiotech, finding that Angiotech’s EP (NL) 0 706 376 patent was valid, and that SMT’s Infinnium™ stent infringed the patent. On March 13, 2008, a Dutch Court of Appeal held a hearing to review the correctness of the trial court’s decision. The Court of Appeal released its judgment on January 27, 2009, ruling against Sahajanand (finding the Angiotech patent novel, inventive, and sufficiently disclosed). The Court of Appeal however requested amendment of claim 12 before rendering its decision on infringement. This amendment is due to the court on April 7th, 2009. A date for the court’s decision on infringement has not yet been set. The decisio n of the Court of Appeal is appealable to the Supreme Court of the Netherlands.
ii)
On March 23, 2006, RoundTable Healthcare Partners, LP as Seller Representative, Angiotech as Buyer, and LaSalle Bank as Escrow Agent, executed an Escrow Agreement for certain representations and warranties under which Angiotech deposited $20 million with LaSalle. On April 4, 2007, LaSalle Bank received an Escrow Claim Notice issued by Angiotech, which directed LaSalle to remit the $20 million to Angiotech as Buyer. On or about Apri1 16, 2007, LaSalle received from RoundTable a Notice of Objection to Angiotech's Escrow Claim Notice. On July 3, 2007, LaSalle filed an action in the Circuit Court of Cook County, IL, asking the court to resolve this dispute. After various hearings and discussions, Angiotech executed a Joint Letter of Direction allowing the release of $6.5 million to RoundTable, thereby leaving the amount in dispute being approximately $13.5 million. On March 21, 2008, this action was moved to the US District Court S outhern District of New York. We are now in the discovery phase of this litigation.
iii)
In July 2004, Dr. Gregory Baran initiated legal action, alleging infringement by our subsidiary, Medical Device Technologies Inc (“MDT”) of two U.S. patents owned by Dr. Baran. These patents allegedly cover MDT’s BioPince™ automated biopsy device. On September 25, 2007, the judge issued her decision pursuant to the Markman hearing of December 2005. We have submitted a Motion for Summary Judgment to the court based upon the judges’ Markman decision. No hearing date has yet been set by the court.
iv)
At the EPO, various patents either owned or licensed by or to the Company are in opposition proceedings including the following:
·
In EP0774964 (which is licensed from the Massachusetts Institute of Technology) the patent was revoked after a hearing held July 17, 2007. An appeal was filed on October 2, 2007. The parties have been summoned to attend Oral Proceedings at the EPO on March 19, 2009.
·
In EP0784490, the proceedings are ongoing and the parties are awaiting communication from the EPO.
·
In EP0809515 (which is licensed from (and to) BSC), the EPO held an oral hearing on January 30, 2008 and thereafter revoked this patent. An appeal was filed on April 22, 2008. The parties have been summoned to attend Oral Proceedings at the EPO on June 19, 2009.
·
In EP0830110 (which is licensed from Edwards LifeSciences Corporation) an amended form of this patent was found valid after an oral hearing on September 28, 2006; however, the opponent appealed the decision on December 21, 2006 and briefs are being exchanged.
·
In EP0876166 the EPO held an oral hearing on September 24, 2008, and thereafter revoked this patent. Our deadline to file an appeal is March 23, 2009 and we have determined that we will not file an appeal.
·
In EP0975340 (which is licensed from (and to) BSC), Oral Proceedings were held on December 4, 2008. The EPO issued an Interlocutory Decision on December 23, 2008 stating the patent was found to have met the requirements of the convention. The decision may be appealed, but no appeal has yet been filed.
·
In EP1118325 (which is licensed from the NIH), the EPO has set a hearing date of April 7, 2009.
·
In EP1155689, briefs are being exchanged.
·
In EP1407786 (which is licensed from (and to) BSC), the patent was revoked in a decision from the EPO on December 9, 2008. An appeal was filed on January 30, 2009.
·
In EP1429664, briefs are being exchanged.
·
In EP1159974, briefs are being exchanged.
·
In EP1075843, a response to the Notice of Appeal was filed. We are awaiting further action from the EPO.
·
In EP0991359, our response to the opposition is to be filed prior to the deadline.
·
In EP1322235, briefs have been filed and we are awaiting further action from the EPO.
·
In EP1216042, an oral hearing has been scheduled for April 23, 2009.
·
In EP0876165, the parties have been summoned to attend an Oral Proceeding at the EPO on June 24, 2009.
- 31 -
Item 4.
SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS
(a) On October 30, 2008, we held its 2008 annual and special general meeting of shareholders.
(b) Proxies were solicited by our management pursuant to Regulation 14A of the Securities Exchange Act of 1934, as amended. Those directors nominated (Proposal 1) in the proxy statement are shown under (c) below. There was no solicitation opposing management’s nominees for directors and all such nominees were elected pursuant to the vote of the shareholders.
(c) Those matters voted upon and the results were as follows:
(1)
Fixing the size of the Board of Directors at seven and the election of directors (Proposal 1);
(2)
Appointment of auditors for the ensuing year and the authorization of the directors to fix the remuneration to be paid to the auditors (Proposal 2);
(3)
Reconfirmation of our Shareholder Rights Plan with minor technical amendments (Proposal 3); and
(4)
Amendment of our articles to increase the quorum for meetings of shareholders to comply with NASDAQ’s quorum requirements (Proposal 4).
| | | | | |
Proposal | For | Against | Withheld | Abstentions | Broker Non-Votes |
Proposal 1 | 53,939,257 | 1,105,027 | - | - | 1 |
William L. Hunter | 53,893,391 | - | 1,150,892 | - | 2 |
David T. Howard | 53,406,923 | - | 1,637,360 | - | 2 |
Hartley T. Richardson | 33,196,622 | - | 21,822,518 | - | 2 |
Edward M. Brown | 53,907,578 | - | 1,136,705 | - | 2 |
Arthur H. Willms | 53,918,873 | - | 1,125,410 | - | 2 |
Laura Brege | 53,880,741 | - | 1,163,542 | - | 2 |
Henry A. McKinnell Jr. | 53,919,391 | - | 1,124,892 | - | 2 |
Proposal 2 | 54,920,220 | - | 124,064 | - | 1 |
Proposal 3 | 30,978,013 | 961,921 | - | - | 23,104,351 |
Proposal 4 | 53,775,536 | 1,268,748 | - | - | 1 |
- 32 -
PART II
Item 5.
MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Trading Price and Volume
Our common shares are publicly traded on the Toronto Stock Exchange under the trading symbol “ANP” and on the NASDAQ Global Select Market under the trading symbol “ANPI”. The following table summarizes the high and low prices for our common shares during 2008 and 2007 on quarterly basis. The prices listed below are in Canadian dollars for trading on the Toronto Stock Exchange and in U.S dollars for trading on the NASDAQ Global Select Market.
| | | | |
Quarter | Canadian Dollars
(TSX) | U.S. Dollars
(NASDAQ) |
2008: | High | Low | High | Low |
Q1 | $3.68 | $1.50 | $3.68 | $1.50 |
Q2 | 3.55 | 2.07 | 3.49 | 2.10 |
Q3 | 3.10 | 0.55 | 3.10 | 0.53 |
Q4 | 0.88 | 0.11 | 0.79 | 0.10 |
| | | | |
Quarter | Canadian Dollars
(TSX) | U.S. Dollars
(NASDAQ) |
2007: | High | Low | High | Low |
Q1 | $11.15 | $6.16 | $9.47 | $5.33 |
Q2 | 8.00 | 6.02 | 7.40 | 5.28 |
Q3 | 7.89 | 5.75 | 7.56 | 5.40 |
Q4 | 7.69 | 3.05 | 7.90 | 3.10 |
Comparative Shareholder Return Performance Graph
The following graph compares cumulative total Shareholder return on $100 invested in Common shares of the Company on December 31, 2003 with the cumulative total return of the S&P/TSX Composite Index (“S&P/TSX”) and the NASDAQ Biotechnology Index, assuming the re-investment of dividends. The Common shares began trading on the TSX on December 18, 1997.
- 33 -
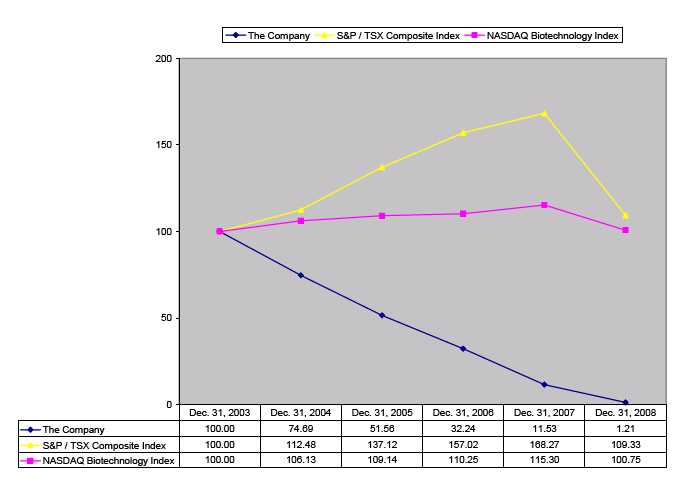
Based on information provided by our transfer agent, as of March 5, 2009, we had 600 shareholders of record. We believe we have approximately 14,300 beneficial owners of our common shares in total. This belief is based on the preliminary search for beneficial holders conducted in order to estimate the number of proxy statements and annual reports required by intermediaries for our 2009 Annual General Meeting.
Dividend Policy
We have not declared or paid any dividends on its common shares since inception. The declaration of dividend payments is at the sole discretion of our Board of Directors. The Board of Directors may declare dividends in the future depending upon numerous factors that ordinarily affect dividend policy, including the results of our operations, our financial position and general business conditions.
- 34 -
Item 6.
SELECTED FINANCIAL DATA
The following table sets forth selected consolidated financial information for each of our five most recently completed financial years. This data should be read in conjunction with our Management's Discussion and Analysis of Financial Condition and Results of Operations, the audited consolidated financial statements, including the notes to the financial statements, and the risk factors set out in this Annual Report on Form 10-K.
CONSOLIDATED STATEMENTS OF INCOME
| | | | | | | | | | | | | | | |
| | | Year ended | | | Year ended | | | Year ended | | | Year ended | | | Year ended | |
| | | December 31, | | | December 31, | | | December 31, | | | December 31, | | | December 31, | |
| (in thousands of U.S. $) | | 2008 | | | 2007 | | | 2006 | | | 2005 | | | 2004 | |
| REVENUE | | | | | | | | | | | | | | | |
| Royalty revenue | $ | 91,546 | | $ | 116,659 | | $ | 175,254 | | $ | 189,203 | | $ | 100,638 | |
| Product sales, net (6) | | 190,816 | | | 170,193 | | | 138,590 | | | 5,334 | | | 8,281 | |
| License fees | | 910 | | | 842 | | | 1,231 | | | 5,111 | | | 17,312 | |
| |
| | | 283,272 | | | 287,694 | | | 315,075 | | | 199,648 | | | 126,231 | |
| EXPENSES | | | | | | | | | | | | | | | |
| License and royalty fees | | 14,258 | | | 18,652 | | | 25,986 | | | 28,345 | | | 18,072 | |
| Cost of products sold | | 101,052 | | | 94,949 | | | 69,543 | | | 5,653 | | | 5,632 | |
| Research and development | | 53,192 | | | 53,963 | | | 45,393 | | | 31,988 | | | 26,659 | |
| Selling, general and administration | | 98,483 | | | 99,315 | | | 78,933 | | | 37,837 | | | 21,180 | |
| Depreciation and amortization | | 33,998 | | | 33,429 | | | 36,014 | | | 9,540 | | | 9,235 | |
| In-process research and development | | 2,500 | | | 8,125 | | | 1,042 | | | 54,957 | | | 6,375 | |
| |
| | | 303,483 | | | 308,433 | | | 256,911 | | | 168,320 | | | 87,153 | |
| |
| Operating (loss) income | | (20,211 | ) | | (20,739 | ) | | 58,164 | | | 31,328 | | | 39,078 | |
| Other (expenses) income: | | | | | | | | | | | | | | | |
| Foreign exchange (loss) gain | | 540 | | | (341 | ) | | 515 | | | 1,092 | | | 2,050 | |
| Investment and other income | | 1,192 | | | 10,393 | | | 6,235 | | | 10,006 | | | 5,668 | |
| Interest expense on long-term debt | | (44,490 | ) | | (51,748 | ) | | (35,502 | ) | | - | | | - | |
| Write-down and other deferred financing charges (1) | | (16,544 | ) | | - | | | (9,297 | ) | | - | | | - | |
| Write-down / loss on redemption of investments (2) | | (23,587 | ) | | (8,157 | ) | | - | | | (5,967 | ) | | - | |
| Write-down of assets held for sale (3) | | (1,283 | ) | | - | | | - | | | - | | | - | |
| Write-down of goodwill (4) | | (649,685 | ) | | - | | | - | | | - | | | - | |
| |
| Total other (expenses) income | | (733,857 | ) | | (49,853 | ) | | (38,049 | ) | | 5,131 | | | 7,718 | |
| (Loss) income from continuing operations before | | | | | | | | | | | | | | | |
| income taxes and cumulative effect of change in | | | | | | | | | | | | | | | |
| accounting policy | | (754,068 | ) | | (70,592 | ) | | 20,115 | | | 36,459 | | | 46,796 | |
| |
| Income tax (recovery) expense | | (12,892 | ) | | (14,545 | ) | | 2,092 | | | 28,055 | | | (6,183 | ) |
| (Loss) income from continuing operations before | | | | | | | | | | | | | | | |
| cumulative effect of change in accounting policy | | (741,176 | ) | | (56,047 | ) | | 18,023 | | | 8,404 | | | 52,979 | |
| |
| Loss from discontinued operations, net of income | | - | | | (9,893 | ) | | (7,708 | ) | | (9,591 | ) | | (527 | ) |
| taxes (5) | | | | | | | | | | | | | | | |
| Cumulative effect of change in accounting policy | | - | | | - | | | 399 | | | - | | | - | |
| |
| Net (loss) income | $ | (741,176 | ) | $ | (65,940 | ) | $ | 10,714 | | $ | (1,187 | ) | $ | 52,452 | |
| |
| Basic net income (loss) per common share: | | | | | | | | | | | | | | | |
| Continuing operations | $ | (8.71 | ) | $ | (0.66 | ) | $ | 0.21 | | $ | 0.10 | | $ | 0.63 | |
| Discontinued operations | | - | | | (0.12 | ) | | (0.09 | ) | | (0.11 | ) | | - | |
| Total | $ | (8.71 | ) | $ | (0.78 | ) | $ | 0.12 | | $ | (0.01 | ) | $ | 0.63 | |
| Diluted net income (loss) per common share: | | | | | | | | | | | | | | | |
| Continuing operations | $ | (8.71 | ) | $ | (0.66 | ) | $ | 0.21 | | $ | 0.10 | | $ | 0.62 | |
| Discontinued operations | | - | | | (0.12 | ) | | (0.09 | ) | | (0.11 | ) | | (0.01 | ) |
| Total | $ | (8.71 | ) | $ | (0.78 | ) | $ | 0.12 | | $ | (0.01 | ) | $ | 0.61 | |
- 35 -
| | | | | | |
| Basic weighted average number of common | | | | | |
| shares outstanding (in thousands) | 85,118 | 85,015 | 84,752 | 84,121 | 83,678 |
| Diluted weighted average number of common | | | | | |
| shares outstanding (in thousands) | 85,118 | 85,015 | 85,437 | 85,724 | 85,697 |
BALANCE SHEET INFORMATION
| | | | | | | | | | | | | | | | |
| As at | December 31, |
| (in thousands of U.S.$) | | 2008 | | | 2007 | | | 2006 | | | 2005 | | | 2004 | |
| Cash, cash equivalents and short-term investments | $ | 39,800 | | $ | 91,326 | | $ | 108,617 | | $ | 195,442 | | $ | 271,484 | |
| Working capital | | 47,737 | | | 97,745 | | | 115,892 | | | 181,317 | | | 268,300 | |
| Total assets | | 385,197 | | | 1,150,108 | | | 1,224,624 | | | 494,694 | | | 479,077 | |
| Total long-term obligations | | 633,655 | | | 645,096 | | | 658,366 | | | 4,459 | | | 12,882 | |
| Deficit | | (843,673 | ) | | (102,497 | ) | | (34,893 | ) | | (45,607 | ) | | (44,420 | ) |
| Total shareholders’ (deficit) equity | | (299,873 | ) | | 442,072 | | | 498,692 | | | 462,680 | | | 441,826 | |
QUARTERLY RESULTS
The following tables present our unaudited consolidated quarterly results of operations for each of our last eight quarters:
| | | | | | | | | | | | | |
| | Quarter ended |
| (in thousands of U.S.$, except per share data) | | December 31, | | | September 30, | | | June 30, | | | March 31, | |
| | | 2008 | | | 2008 | | | 2008 | | | 2008 | |
| Revenue | | | | | | | | | | | | |
| Royalty and license revenue | $ | 16,027 | | $ | 21,858 | | $ | 25,589 | | $ | 28,982 | |
| Product sales | | 46,054 | | | 46,502 | | | 50,533 | | | 47,727 | |
| Total revenues | | 62,081 | | | 68,360 | | | 76,122 | | | 76,709 | |
| Gross Margin: | | | | | | | | | | | | |
| Pharmaceutical Technologies | | 13,254 | | | 18,406 | | | 21,928 | | | 24,611 | |
| Medical Products | | 22,431 | | | 21,730 | | | 23,724 | | | 21,878 | |
| Total Gross Margin | | 35,685 | | | 40,136 | | | 45,652 | | | 46,489 | |
| Operating loss | | (1,839 | ) | | (2,452 | ) | | (7,284 | ) | | (8,636 | ) |
| Net loss | $ | (76,964 | ) | $ | (622,378 | ) | $ | (26,071 | ) | $ | (15,763 | ) |
| |
| Basic loss per share | $ | (0.90 | ) | $ | (7.31 | ) | $ | (0.31 | ) | $ | (0.19 | ) |
| |
| Diluted loss per share | $ | (0.90 | ) | $ | (7.31 | ) | $ | (0.31 | ) | $ | (0.19 | ) |
| | | | | | | | | | | | | |
| | Quarter ended |
| (in thousands of U.S.$, except per share data) | | December 31, | | | September 30, | | | June 30, | | | March 31, | |
| | | 2007 | | | 2007 | | | 2007 | | | 2007 | |
| Revenue | | | | | | | | | | | | |
| Royalty and license revenue | $ | 27,423 | | $ | 26,674 | | $ | 29,932 | | $ | 33,472 | |
| Product sales | | 43,935 | | | 41,352 | | | 42,420 | | | 42,486 | |
| Total revenues | | 71,358 | | | 68,026 | | | 72,352 | | | 75,958 | |
| Gross Margin: | | | | | | | | | | | | |
| Pharmaceutical Technologies | | 23,007 | | | 22,148 | | | 25,663 | | | 28,031 | |
| Medical Products | | 20,247 | | | 17,967 | | | 17,336 | | | 19,694 | |
| Total Gross Margin | | 43,254 | | | 40,115 | | | 42,999 | | | 47,725 | |
| Operating (loss) income | | (6,034 | ) | | (5,907 | ) | | (11,150 | ) | | 2,352 | |
| Net (loss) income from continuing operations | | (23,498 | ) | | (10,832 | ) | | (15,045 | ) | | (6,672 | ) |
| Net (loss) income | | (26,444 | ) | | (11,988 | ) | | (15,215 | ) | | (12,293 | ) |
| |
| Basic loss per share: | | | | | | | | | | | | |
| Continuing operations | $ | (0.28 | ) | $ | (0.13 | ) | $ | (0.18 | ) | $ | (0.08 | ) |
| Discontinued operations | | (0.03 | ) | | (0.01 | ) | | - | | | (0.07 | ) |
| Total | $ | (0.31 | ) | $ | (0.14 | ) | $ | (0.18 | ) | $ | (0.15 | ) |
| |
| Diluted loss per share: | | | | | | | | | | | | |
| Continuing operations | $ | (0.28 | ) | $ | (0.13 | ) | $ | (0.18 | ) | $ | (0.08 | ) |
| Discontinued operations | | (0.03 | ) | | (0.01 | ) | | - | | | (0.07 | ) |
| Total | $ | (0.31 | ) | $ | (0.14 | ) | $ | (0.18 | ) | $ | (0.15 | ) |
1) We expensed the deferred financing charge of $13.5 million related to the suspension of the note purchase agreement in the third quarter of 2008 and a further $3.0 million in the fourth quarter of 2008. In 2006, we repaid a credit facility of $425 million and as a result, expensed the unamortized balance of the deferred financing charges related to this facility.
- 36 -
2) In 2008, we wrote-down and realized a loss on investments of $10.7 million, $1.9 million and $11.0 million in the second, third and fourth quarters respectively. In 2007, we wrote down and realized a loss on investments of $8.2 million in the first quarter.
3) We recorded an impairment charge of $1.3 million in the fourth quarter of 2008 on the two properties we intend to sell and classified these properties and one other as available for sale.
4) We initially wrote-down our goodwill carrying value associated with our Medical Products segment by $599.4 million at the end of the third quarter of 2008 and wrote off the remaining balance of $26.8 million in the fourth quarter of 2008. We also determined that the goodwill associated with our Pharmaceuticals Technology segment was impaired as at December 31, 2008 and accordingly, wrote off the remaining balance of $23.5 million. As a result, our goodwill balance is nil for the Medical Products and Pharmaceutical Technology segments as at December 31, 2008.
5) We recorded an impairment charge of $7.7 million in the fourth quarter of December 2006 related to our decision to discontinue the following subsidiaries: American Medical Instruments Inc. (Dartmouth), Point Technologies, Inc and Point Technologies SA. In the first quarter of 2007, we recorded a further impairment of $8.9 million related to the discontinuance of these operations.
6) We recorded $2.6 million against our product sales revenue in the second quarter of 2007 related to our decision to accept returns of Contour Threads brand name as part of our consolidation and discontinuation of the Contour Threads brand name, coincident with our launch of our Quill SRS brand name.
On March 23, 2006, we completed the acquisition of 100% of the outstanding share of privately held AMI, a leading independant manufacturer of specialty, single use medical devices.
- 37 -
Item 7.
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
ANGIOTECH PHARMACEUTICALS, INC.
For the year ended December 31, 2008
(All amounts following are expressed in U.S. dollars unless otherwise indicated.)
The following management’s discussion and analysis (“MD&A”), dated December 31, 2008, should be read in conjunction with our audited consolidated financial statements for the year ended December 31, 2008 prepared in accordance with U.S. generally accepted accounting principles (“U.S. GAAP”) and the applicable rules and regulations of the United States Securities and Exchange Commission (“SEC”) for the presentation of annual financial information. Additional information relating to our Company, including our 2007 Audited Annual Financial Statements and 2007 Annual Information Form (“AIF”), is available by accessing the SEDAR website at www.sedar.com or the SEC’s IDEA website at idea.sec.gov
Forward-Looking Statements and Cautionary Factors That May Affect Future Results
Statements contained in this Annual Report on Form 10-K that are not based on historical fact, including without limitation statements containing the words “believes,” “may,” “plans,” “will,” “estimates,” “continues,” “anticipates,” “intends,” “expects” and similar expressions, constitute “forward-looking statements” within the meaning of the U.S. Private Securities Litigation Reform Act of 1995 and constitute “forward-looking information” within the meaning of applicable Canadian securities laws. All such statements are made pursuant to the “safe harbor” provisions of applicable securities legislation. Forward-looking statements may involve, but are not limited to, comments with respect to our objectives and priorities for 2009 and beyond, our strategies or future actions, our targets, expectations for our financial condition and t he results of, or outlook for, our operations, research and development and product and drug development. Such forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause the actual results, events or developments to be materially different from any future results, events or developments expressed or implied by such forward-looking statements.
Many such known risks, uncertainties and other factors are taken into account as part of our assumptions underlying these forward-looking statements and include, among others, the following: general economic and business conditions, both nationally and in the regions in which we operate; market demand; technological changes that could impact our existing products or our ability to develop and commercialize future products; competition; existing governmental regulations and changes in, or the failure to comply with, governmental regulations; availability of financial reimbursement coverage from governmental and third-party payers for products and related treatments; adverse results or unexpected delays in pre-clinical and clinical product development processes; adverse findings related to the safety and/or efficacy of our products or products sold by our partners; decisions, and the timing of decisions, made by health regulatory agencies regarding app roval of our technology and products; the requirement for substantial funding to conduct research and development, to expand manufacturing and commercialization activities or to consummate acquisitions; and any other factors that may affect our performance.
In addition, our business is subject to certain operating risks that may cause any results expressed or implied by the forward-looking statements in this Annual Report on Form 10-K to differ materially from our actual results. These operating risks include: our ability to attract and retain qualified personnel; our ability to successfully complete pre-clinical and clinical development of our products; changes in our business strategy or development plans; our failure to obtain patent protection for discoveries; loss of patent protection resulting from third-party challenges to our patents; commercialization limitations imposed by patents owned or controlled by third parties; our ability to obtain rights to technology from licensors; liability for patent claims and other claims asserted against us; our ability to obtain and enforce timely patent and other intellectual property protection for our technology and products; the ability to enter into, and to maintain, corporate alliances relating to the development and commercialization of our technology and products; market acceptance of our technology and products; our ability to successfully manufacture, market and sell our products; the availability of capital to finance our activities; our ability to restructure and to service our debt obligations; and any other factors referenced in our other filings with the applicable Canadian securities regulatory authorities or the SEC.
- 38 -
For a more thorough discussion of the risks associated with our business, see the section entitled “Risk Factors” in this Annual Report on Form 10-K.
Given these uncertainties, assumptions and risk factors, investors are cautioned not to place undue reliance on such forward-looking statements. Except as required by law, we disclaim any obligation to update any such factors or to publicly announce the result of any revisions to any of the forward-looking statements contained in this Annual Report on Form 10-K to reflect future results, events or developments.
This Annual Report on Form 10-K contains forward-looking information that constitutes "financial outlooks" within the meaning of applicable Canadian securities laws. We have provided this information to give shareholders general guidance on management's current expectations of certain factors affecting our business, including our financial results. Given the uncertainties, assumptions and risk factors associated with this type of information, including those described above, investors are cautioned that the information may not be appropriate for other purposes.
Business Overview
We are a specialty pharmaceutical and medical device company that discovers, develops and markets innovative technologies primarily focused on acute and surgical applications. We generate our revenue through our sales of medical products and components, as well as from royalties derived from sales by our partners of products utilizing certain of our proprietary technologies. For the year ended December 31, 2008, we recorded $190.8 million in direct sales of our various medical products and $92.5 million in royalties and license fees received from our partners.
Our research and development efforts focus on understanding and characterizing biological conditions that often occur concurrent with medical device implantation, surgery or acute trauma, including scar formation and inflammation, cell proliferation, bleeding and coagulation, infection, and tumor tissue overgrowth. Our strategy is to utilize our various technologies in the areas of drugs, drug delivery, surface modification, biomaterials and medical devices to create and commercialize novel, proprietary medical products that reduce surgical procedure side effects, improve surgical outcomes, shorten hospital stays, or are easier or safer for a physician to use.
We develop our products using a proprietary and systematic discovery approach. We use our drug screening capabilities to identify new uses for known pharmaceutical compounds. We look for compounds that address the underlying biological causes of conditions that can occur with medical device implantation, surgery or acute trauma. Once appropriate drugs have been identified, we work to formulate the drug, or a combination of drugs, with our portfolio of drug, drug delivery and surface modification technologies and biomaterials to develop a novel surgical implant or medical device. We have patent protected, or have filed patent applications for, certain of our technology and many of our products and potential product candidates.
We currently operate in two segments: Pharmaceutical Technologies and Medical Products.
Pharmaceutical Technologies
Our Pharmaceutical Technologies segment focuses primarily on establishing product development and marketing partnerships with major medical device, pharmaceutical or biomaterials companies and to date has derived the majority of its revenue from royalties due from partners that develop, market and sell products incorporating our technologies. Currently, our principal revenues in this segment are from royalties derived from sales by Boston Scientific Corporation (“BSC”) of TAXUS® coronary stent systems incorporating the drug paclitaxel.
Medical Products
Our Medical Products segment manufactures and markets a wide range of single-use specialty medical products, primarily medical device products and medical device components. These products are sold directly to end users or other third party medical device manufacturers. This segment contains two specialized direct sales and distribution organizations as well as significant manufacturing capabilities. Many of our medical products are made using our proprietary manufacturing processes, or are protected by our intellectual property. Our Medical Products segment may apply certain of our proprietary technologies to its products to create novel, next generation medical products to market directly to end users or medical products distributors.
Financial and Strategic Alternatives Process.During approximately the last eight calendar quarters, revenue in our Pharmaceutical Technologies segment has declined significantly, primarily due to lower than expected royalties derived from sales by BSC of TAXUS® coronary stent systems. This decline in royalty revenue has significantly impacted our ability to fund our operations and service our debt obligations, and has materially impacted our liquidity position. During 2008, our management and Board of Directors determined to explore and pursue various restructuring and cost reduction actions in order to conserve our liquidity, as well as various financial and strategic transactions that could potentially reduce or eliminate our existing debt obligations and improve our working capital position.
- 39 -
Our management and Board of Directors continue to believe a transaction of significant size and scope is necessary to meaningfully address the working capital needs of our business initiatives, as well as to address liquidity issues related to our current balance sheet structure, likely to arise in the near term due to several factors that have impacted our business and cash flows from operations. In particular, we continue to expect to receive lower revenues from BSC over the next several quarters, mainly due to the impact of new competitive entrants into the market for drug-eluting stents and other factors.
On July 7, 2008, we announced that our Board of Directors had authorized a transaction to create a new subsidiary, Angiotech Pharmaceutical Interventions Inc. (“API”), and that we would contribute certain business assets and intellectual property to API, primarily consisting of business assets of Angiotech other than the intellectual property and royalty revenue related to TAXUS. In connection with this transaction, we were to receive $200 to $300 million of new financing from new investors to establish API, which financing was targeted to reduce substantial amounts of our existing debt obligations with equivalent amounts of convertible debt bearing interest in additional equity of the newly created subsidiary in kind, as opposed to cash. This transaction, if approved by our shareholders and completed, would have substantially reduced our annual cash pay interest obligations.
On September 22, 2008, we announced that we had postponed the planned shareholder vote regarding the proposed API transaction. As of that date, given the time required for more extended discussions to address concerns of certain of our shareholders and bondholders, and given various other factors impacting our business and cash position (including lower expected revenues derived from BSC), we did not believe we would be able to satisfy the transaction condition with respect to the minimum level of cash and cash equivalents required to be held at the time of the transaction’s close.
As a result of the uncertainty regarding our proposed transaction, we also announced on September 22, 2008 that management and the Board of Directors would continue to explore alternatives to our balance sheet and current capital structure, including but not limited to whether we could consummate the previously announced transaction or other similar transaction alternatives.
On November 12, 2008, the API transaction was terminated. On November 21, 2008, we announced that we had engaged The Blackstone Group to assist us in evaluating various alternatives for our business and capital structure, including, but not limited to, securing interim senior secured financing for working capital and liquidity purposes, evaluating various restructuring alternatives to pursue with the holders of our Senior Floating Rate Notes due 2013 (the “Floating Rate Notes”) and our 7.75% Senior Subordinated Notes due 2014 (the “Subordinated Notes”), and assisting us in evaluating proposals or potential proposals from various financial parties regarding a significant investment of capital.On March 2, 2009, we announced that we had completed a financing transaction with Wells Fargo Foothill, LLC, consisting of a delayed draw secured term loan facility of up to $10 million and a new secured revolving credit facility providing up to an additional $22.5 million in available credit. See “—Liquidity and Capital Resources—New Senior Secured Credit Facilities”.
Cost Reduction Initiatives. On September 22, 2008, we announced that, in connection with the update to our financial and strategic alternatives process as described above, we would pursue various initiatives to further reduce operating costs and focus our business efforts, including:
·
Postponement of the scheduled launch of our 5-flourouracil-eluting central venous catheter (5-FU CVC);
·
Closure of our research and manufacturing facility in Rochester, NY, and rationalization or elimination of office and laboratory space in Vancouver, BC, North Bend, WA and Herndon, VA.;
·
Postponement of certain pre-clinical-stage research activities, pending the completion of partnering or other funding activities that would offset direct costs and personnel costs associated with such programs;
·
Reduction of certain financial and personnel contributions relating to our joint venture with Genzyme Corporation;
·
Potential amendment of and reduction in cash outlays related to our collaboration with Athersys, Inc.;
·
Rationalization of selected pending and issued intellectual property;
·
Elimination of certain expenses and reductions in personnel in all general and administrative departments;
·
Selective reduction in certain sales and marketing investments and in medical affairs; and
·
Postponement of selected planned capital expenditures.
Our remaining resources subsequent to these changes have been focused primarily on our existing medical device products business, with particular emphasis on our portfolio of Promoted Brands, and on selected new products that have recently launched including Quill™ SRS, and the HemoStream™ Chronic Dialysis Catheter.
FDA Approval Received by our Partner BSC for TAXUS Liberté® and TAXUS Express Atom™ Stents. On October 10, 2008, we announced that BSC had received approval from the United States Food and Drug Administration (“FDA”) to market and sell the second-generation TAXUSLiberté® Paclitaxel-Eluting Coronary Stent System in the United States. On September 25, 2008, we announced that BSC had received approval from the FDA to market and sell the TAXUS Express2 Atom™ Paclitaxel-Eluting Coronary Stent System in the United States. We are entitled to receive royalties based on the commercial sale of these products by BSC.
- 40 -
Enrollment Completed for TAXUS PERSEUS Clinical Trial. On October 8, 2008, we announced that BSC had completed enrollment in the PERSEUS clinical trial, designed to evaluate the third-generation TAXUS Element paclitaxel-eluting coronary stent. The PERSEUS clinical program has enrolled nearly 1,500 patients at 100 U.S. and international centers since July 2007, and will compare the TAXUS Element stent to the prior-generation TAXUS Express2 stent marketed in the United States since 2004.
Clinical Trial Enrollment and CE Mark Filings Completed by our Partner Cook Medical Inc. for the ZILVER® PTX Paclitaxel-Eluting Peripheral Stent Product Candidate. On September 10, 2008 we announced that Cook Medical Inc. (“Cook”), a multinational medical device manufacturer, had completed enrollment in the first international clinical trial of a drug-eluting stent designed to treat arterial blockages outside the coronary arteries. The 420 patients enrolled in Cook’s randomized trial of its ZILVER PTX Drug-Eluting Peripheral Stent include peripheral artery disease (“PAD”) patients treated in Germany, the United States and Japan. Cook also already has enrolled 780 patients in the European Union, Canada and Korea in a clinical registry to evaluate the safety of the ZILVER PTX device. The data from such trials have been used for submission in Europefor CE Mark approval to market the device there, with additional regulatory submissions pending in additional markets. We are entitled to receive royalties based on the commercial sale of this product by Cook.
CE Mark Approval Received of HemoStreamTM Chronic Dialysis Catheter. On August 12, 2008, we announced that we had received CE Mark approval to market and sell our HemoStream™ chronic dialysis catheter in Europe. We received FDA clearance to market and sell this product in the United States in August 2007, but have not yet commercially launched this product.
Ongoing Clinical Programs
The following discussion describes our product candidates, or certain of our partners’ product candidates, that are being evaluated in ongoing human clinical trials and their stage of development:
TAXUS Element™ Platinum Chromium Paclitaxel-Eluting Coronary Stent System. The TAXUS Element paclitaxel-eluting coronary stent system is the third generation BSC coronary stent platform that incorporates our research, technology and intellectual property related to the use of paclitaxel. The TAXUS Element stent features BSC’s proprietary platinum chromium alloy, which is designed to enable thinner stent struts, increased flexibility and a lower stent profile while improving radial strength, recoil and radiopacity. In addition, the TAXUS Element stent platform incorporates new balloon technology intended to improve upon BSC’s market-leading Maverick® Balloon Catheter technology.
TAXUS Petal™ Bifurcation Paclitaxel-Eluting Coronary Stent System. The TAXUS PETAL bifurcation paclitaxel-eluting coronary stent system, which is under evaluation in clinical trials being conducted by BSC, represents a novel BSC coronary stent product candidate that incorporates our research, technology and intellectual property related to the use of paclitaxel. Conventional coronary stents were designed to treat tubular arteries, and are considered less than optimal for the y-shaped anatomy of a bifurcated area of the coronary arteries. The TAXUS PETAL is a specialized coronary stent designed to treat both the main branch and the side branch of a bifurcation by incorporating an innovative side structure (the Petal strut) in the middle of the stent that opens into a side branch.
On July 18, 2007, BSC initiated the TAXUS PETAL I First Human Use (FHU) trial, which is expected to enroll a total of 45 patients in New Zealand, France and Germany. The trial is a non-randomized study with an initial assessment of acute performance and safety (including rates of death, myocardial infarction and target vessel revascularization) at 30 days and six months, with continued annual follow-up to occur for five years. Upon successful completion of this study, BSC has indicated that it intends to begin a pivotal trial which if successful would provide a basis for U.S. and international approvals for the commercialization of the TAXUS Petal stent.
ZILVER PTX Paclitaxel-Eluting Peripheral Vascular Stent System. The ZILVER PTX paclitaxel-eluting peripheral vascular stent, which is under evaluation in clinical trials being conducted by our partner Cook, is a specialized stent product incorporating our proprietary paclitaxel technology and is designed for placement in diseased arteries in the limbs to restore blood flow. Cook is a co-exclusive licensee, together with BSC, of our proprietary paclitaxel technology to reduce restenosis following stent placement in peripheral artery disease. The ZILVER PTX paclitaxel-eluting peripheral stent is designed to reduce restenosis following placement of a stent in PAD patients.
The ZILVER PTX is currently undergoing multiple human clinical trials in the United States, Japan, the European Union and selected other countries to assess product safety and efficacy. In January 2007, Cook released nine-month data from its EU clinical study. The preliminary data presented by Cook on the first 60 patients in the randomized trial, which is examining the safety of using Cook's ZILVER PTX paclitaxel-eluting stent to treat blockages, or lesions, of the superficial femoral artery (“SFA”) above the knee, indicated that the ZILVER PTX stent showed an equal adverse event rate to conventional angioplasty for treating SFA lesions. The ZILVER PTX stent also displayed a zero-percent fracture rate for 41 lesions at six months and 18 lesions at one year.
- 41 -
On June 11, 2008, Cook reported positive interim results from the registry arm of a clinical study designed to measure the efficacy of the Zilver PTX in treating PAD patients, specifically in the treatment of blockages in the femoropopliteal artery. The results were reported by trial investigators at the 2008 SVS Vascular Annual Meeting, and revealed clinical improvement, excellent durability and fracture resistance, high rates of event-free survival (“EFS”) and freedom from target lesion revascularization (“TLR”). Interim data was compiled at six and 12 months using 435 patients and 200 patients, respectively. The corresponding EFS rates were 94 percent and 84 percent, and freedom from TLR was 96 percent and 88 percent. Evaluation of stent x-rays is ongoing, and currently suggests stent fractures in approximately one percent of cases at six months and less than two percent of cases at 12 months. In addition, the Zilver PTX stent exhibit ed no safety concerns and results were better than expected for Trans-Atlantic Inter-Society Consensus (“TASC”) class C and D lesions, occlusions, in-stent restenosis and lesions greater than seven centimeters. Follow-up to the registry arm of the study will continue through two years.
On July 16, 2007 Cook announced that the first U.S. patients in a randomized pivotal human clinical trial of ZILVER PTX were treated at Tri-City Medical Center in Oceanside, CA. The trial is designed to randomize patients to receive either the ZILVER PTX stent or balloon angioplasty. Data from this clinical trial is intended to be used to support submission to the FDA for approval in the United States to market the device. In addition, data collected on Japanese and U.S. patients is expected to be combined for the final evaluation of the device and used for regulatory submissions in both markets for approval.
On September 10, 2008 Cook announced it had completed enrollment in its pivotal human clinical trial for the ZILVER PTX. The 420 patients enrolled in Cook’s randomized trial include peripheral artery disease patients treated in Germany, the United States and Japan. On the same date, Cook announced that it had enrolled an additional 780 patients in the European Union, Canada and Korea in a clinical registry to evaluate the safety of the ZILVER PTX device. Those data have been used for submission in Europe for CE Mark approval to market the device there, with additional regulatory submissions pending in additional markets.
Bio-Seal™ Lung Biopsy Tract Plug System. Bio-Seal™ is a novel technology designed to prevent air leaks in patients having lung biopsies by plugging the biopsy track with an expanding hydrogel plug. On contact with moist tissue, the hydrogel plug absorbs fluids and expands to fill the void created by the biopsy needle puncture. The seal is airtight and the plug is absorbed into the body after healing of the puncture site has occurred.
Bio-Seal has undergone a human clinical trial in the U.S. which was designed to assess the safety and efficacy of Bio-Seal, with the primary endpoint being reduction in rates of pneumothorax in patients undergoing lung biopsy procedures. The clinical trial was a prospective randomized multi-centered safety and efficacy evaluation. The trial enrolled its first patient in October 2005 and completed enrolment in June 2008. The study was designed to provide a basis for U.S. clearance for the commercialization of Bio-Seal. Data from this clinical trial study has been submitted to the FDA. The FDA has responded to our submission with additional questions about the study. We have responded to the FDA, and upon further review by the FDA we may either receive 510(k) clearance to market Bio-Seal in the United States or be required to respond to additional questions or conduct additional clinical studies for this product candidate. Upon receiving such further informat ion from the FDA, we will determine the timing of product launch or any further development work necessary to achieve approval should we choose to continue the development of this product candidate. The complete data for the Bio-Seal study was presented at the 2009 Society of Interventional Radiology in San Diego, CA on March 9, 2009. The trial hit its primary end point with clinical success in 85% of the treatment patients compared to 69% for the control patients (p=0.002). The product has already received CE Mark approval.
Option™ Vena Cava Filter. The Option™ Vena Cava Filter, which we licensed in March 2008 from our partner Rex Medical L.P., is under evaluation in a pivotal human clinical trial. We believe this vena cava filter may have a number of potential benefits, which include unique filter apex and retention anchors, insertion through either the femoral or jugular route, and non-thrombogenic material. The purpose of the U.S. multi-center prospective clinical trial is to evaluate the device’s safety and efficacy in preventing pulmonary emboli, and to assess the ability to retrieve the device from the body up to 175 days following implantation. Interim results of the pivotal trial were presented at the AIM/Veith Meeting in New York in November 2008. The complete results, representing a total of 100 patients, were presented at the 2009 Society of Interventional Radiology in San Diego, CA on March 9, 2009. The clinical data from this trial has been submi tted to the FDA. The FDA has responded to our submission with additional questions about the study and we are preparing a response.
- 42 -
MultiStem® Stem Cell Therapy. The MultiStem stem cell therapy is under evaluation in clinical trials being conducted together with our partner Athersys, Inc. (“Athersys”) for the treatment of acute myocardial infarction. MultiStem stem cells are proprietary adult stem cells derived from bone marrow, which have demonstrated the ability in laboratory experiments to form a wide range of cell types. MultiStem may work through several mechanisms, but a primary mechanism appears to be the production of multiple therapeutic molecules produced in response to inflammation and tissue damage. We and Athersys believe that MultiStem may represent a unique “off the shelf” stem cell product candidate, based on its potential ability to be used without tissue matching or immunosupression, and its potential capacity for large scale production. We entered into an agreement with Athersys in May 2006 to co-develop and commercialize MultiStem for use in the indications o f acute myocardial infarction and peripheral vascular disease. On December 20, 2007, we and Athersys announced we had received authorization from the FDA to commence a phase I human clinical trial to evaluate the safety of MultiStem in the treatment of acute myocardial infarction. Upon completion of a phase I human clinical trial currently being conducted by Athersys, we may assume lead responsibility for further clinical development. We currently own marketing and commercialization rights with respect to this product candidate. On September 22, 2008, as part of certain cost reduction initiatives, we announced a potential amendment of, and reduction in, cash outlays related to our collaboration with Athersys. The final terms of such amendment to our collaboration with Athersys may have an impact on our expected future expenditures for research and development of MultiStem, and the extent of our future financial and commercial commitments and rights relating to this product candidate.
Completed or Suspended Clinical Programs
5-FU-Eluting Central Venous Catheter. Central venous catheters (“CVC”) are usually inserted into critically ill patients for extended periods of time to administer fluids, drugs, and nutrition, as well as facilitate frequent blood draws. Through our proprietary drug identification strategy, we have elected to evaluate 5-Fluorouracil (“5-FU”), a drug previously approved by the FDA for treatment of various types of cancer, as a compound that may help to prevent certain types of infection in patients receiving a CVC. We recently completed a human clinical trial in the United States designed to assess the safety and efficacy of our 5-FU-eluting CVC in preventing various types of catheter related infections. The study was a randomized, single-blind, 960-patient, 25-center study and was designed to evaluate whether our 5-FU-eluting CVC prevents bacterial colonization at least as well as the market leading anti-infective CVC. On July 10, 2007, we announce d that we had completed enrollment of the study, and on October 9, 2007, we announced this study had met its primary statistical endpoint of non-inferiority as compared to the market leading anti-infective CVC (a chlorhexidine / silver sulfadiazine (CH-SS) coated CVC) and indicated an excellent safety profile. In March 2008, we presented the clinical trial data at the 28th International Symposium on Intensive Care and Emergency Medicine (ISICEM) in Brussels, Belgium Based on the clinical trial data, the investigators concluded that our 5-FU CVC met the primary endpoint of the study; specifically that our 5-FU CVC product candidate was non-inferior in its ability to prevent bacterial colonization of the catheter tip when compared to catheters coated with CH-SS. There were no statistically significant differences in the rate of adverse events related to the study devices, or in the rates of catheter-related bloodstream infections. Additionally, there was no evidence for acquired resistance to 5-FU in clinical isolates exposed to the drug for a second time. Based on the positive results achieved in the study, in December 2007 we filed a request for 510(k) clearance from the FDA to market and sell the CVC in the United States and on April 17, 2008, we announced that we had received 510(k) clearance from the FDA to market our 5-FU CVC in the United States but have not yet commercially launched the product.
TAXUS Liberté™ paclitaxel-eluting coronary stent system. The TAXUS Liberté paclitaxel-eluting coronary stent system is BSC’s second generation coronary stent system platform that incorporates our research, technology and intellectual property related to the use of paclitaxel to prevent restenosis. The TAXUS Liberté stent system has been designed to further enhance coronary stent deliverability and blood vessel conformability, particularly in challenging coronary lesions. To date, BSC has only commenced sales of the TAXUS Liberté in countries outside of the United States. On October 10, 2008, we announced that BSC had received approval from the FDA to market and sell the TAXUS Liberté in the United States.
Vascular Wrap™ Our paclitaxel-eluting mesh surgical implant, or Vascular Wrap, is designed to treat complications, including graft stenosis or restenosis, that may occur in connection with vascular graft implants in hemodialysis patients or in patients that have peripheral artery disease. Vascular grafts are implanted in patients in order to bypass diseased blood vessels, or to provide access to the vascular system of kidney failure patients in order to facilitate the process of hemodialysis. In many cases, these vascular grafts fail due to proliferation of cells or scar into the graft (graft stenosis or restenosis), which can negatively impact blood flow through the vascular graft.
In April 2008, we elected to suspend enrollment in our U.S. and EU human clinical trials for our Vascular Wrap product candidate in patients undergoing surgery for hemodialysis access, pending a safety review to evaluate an imbalance of infections observed between the two study groups. As a result of these observations, we elected to notify physicians to suspend further enrollment in the trials, pending a full review of the potential cause of the implant site infections. There are currently no plans to resume enrollment in these clinical trials, and we are continuing to evaluate alternatives for this program, including potential collaborations, partnerships, divestitures or future clinical development initiatives.
Acquisitions
·
Quill Medical, Inc. (“Quill”).On June 26, 2006, we completed the acquisition of 100% of the equity of Quill for $40 million cash consideration. Through this transaction, we acquired the rights, in all possible fields of use, to develop and market applications of Quill’s proprietary self-anchoring wound closure technology. Unlike conventional sutures which are smooth, the Quill products have tiny teeth-like barbs or cogs along the surface. This “self-anchoring” wound closure technology may be used to close certain wounds or surgical incisions without the need for suture knots. Eliminating knot-tying can save surgical time, may reduce the risk of infection, and may reduce wound leakage.
- 43 -
We are currently working to develop a portfolio of next-generation products using the Quill technology. In January 2007, we launched the first of these new products, the Quill SRS for various wound closure and tissue approximation applications in general and aesthetic surgery. In 2007 and throughout 2008 we launched several new Quill SRS product lines.
The launch of the Quill SRS for various indications in January 2007 triggered a development milestone of $10.0 million that was paid to the former shareholders of Quill in August 2007. This milestone payment is creditable against any future contingent payments that we may be required to make based upon the achievement of significant incremental revenue growth of products incorporating the Quill technology over an approximately five-year period from the date of the acquisition. This $10.0 million payment was recorded as an increase to goodwill during the first quarter of 2007.
·
American Medical Instruments Holdings, Inc. (“AMI”). On March 23, 2006, we completed the acquisition of 100% of the equity of AMI for $787.9 million cash consideration. AMI provided us with the majority of the assets and products comprising our Medical Products business segment.
Collaboration, License and Sales and Distribution Agreements
In connection with our research and development efforts, we have entered into various arrangements with corporate and academic collaborators, licensors, licensees and others for the research, development, clinical testing, regulatory approval, manufacturing, marketing and commercialization of our product candidates. Terms of the various license agreements may require us, or our collaborators, to make milestone payments upon achievement of certain product development and commercialization objectives and pay royalties on future sales of commercial products, if any, resulting from the collaborations. During 2008, we did not enter into any significant collaboration, license or sales and distribution agreements.
On September 22, 2008, we announced a reduction of certain financial and personnel contributions relating to our collaboration with Genzyme Corporation, and a potential amendment of, and reduction in, cash outlays related to our collaboration with Athersys, Inc.
Our most significant collaborations, licences, sales and distribution agreements are listed in the exhibits index to this Annual Report on Form 10-K and may be found at the locations specified therein.
Critical Accounting Policies and Estimates
Our consolidated financial statements are prepared in accordance with U.S. GAAP. These accounting principles require management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenue and expenses. We believe that the estimates and assumptions upon which we rely are reasonable and are based upon information available to us at the time the estimates and assumptions were made. Actual results could differ materially from our estimates.
We believe the following policies to be critical to understanding our financial condition, results of operations, and expectations for 2009 because these policies require management to make significant estimates, assumptions and judgments about matters that are inherently uncertain.
Revenue Recognition
(i) Royalty revenue
We recognize royalty revenue when we have fulfilled the terms in accordance with the contractual agreement, have no future obligations, the amount of the royalty fee is determinable and collection is reasonably assured. We record royalty revenue from BSC on a cash basis due to our inability to accurately estimate the BSC royalty before we receive the reports and payments from BSC. This results in a one quarter lag between the time we record royalty revenue and the time the associated sales were recorded by BSC.
(ii) Product sales
We recognize revenue from product sales, including shipments to distributors, when the product is shipped from our facilities to the customer provided that we have not retained any significant risks of ownership or future obligations with respect to products shipped. We recognize revenue from product sales net of provisions for future returns. These provisions are established in the same period as the related product sales are recorded and are based on estimates derived from our historical experience.
We consider revenue to be realized or realizable and earned when all of the following criteria are met: persuasive evidence of a sales arrangement exists; delivery has occurred or services have been rendered; the price is fixed or determinable; and collectibility is reasonably assured. These criteria are generally met at the time of shipment when the risk of loss and title passes to the customer or distributor.
- 44 -
We include amounts billed to customers for shipping and handling in revenue. The corresponding costs for shipping and handling are included in cost of products sold.
(iii) License fees
License fees are comprised of initial fees and milestone payments derived from collaborative and other licensing arrangements. We recognize non-refundable milestone payments upon the achievement of specified milestones when the milestone payment is substantive in nature, the achievement of the milestone was not reasonably assured at the inception of the agreement and we have no further significant involvement or obligation to perform under the arrangement. Initial fees and non-refundable milestone payments received which require our ongoing involvement are deferred and amortized into income on a straight-line basis over the period of our ongoing involvement.
Income tax expense
Income taxes are accounted for under the liability method. Deferred tax assets and liabilities are recognized for the differences between the financial statement and income tax bases of assets and liabilities, and for operating losses and tax credit carry forwards. The carrying value of our net deferred tax assets assumes that we will be able to generate sufficient future taxable income in certain tax jurisdictions to realize the value of these assets. Management evaluates the realizability of the deferred tax assets and assesses the need for any valuation allowance adjustment. A valuation allowance is provided for the portion of deferred tax assets that is more likely than not to be unrealized. Deferred tax assets and liabilities are measured using the enacted tax rates and laws.
Significant estimates are required in determining our provision for income taxes including, but not limited to, accruals for tax contingencies and valuation allowances for deferred income tax assets. Some of these estimates are based on interpretations of existing tax laws or regulations. Our effective tax rate may change from period to period based on the mix of income among the different foreign jurisdictions in which we operate, changes in tax laws in these jurisdictions, and changes in the amount of valuation allowance recorded.
Effective January 1, 2007, we adopted Financial Accounting Standards Board (“FASB”) Interpretation No. 48, Accounting for Uncertainty in Income Taxes – an Interpretation of FASB Statement No. 109 (“FIN 48”). FIN 48 is designed to reduce diversity and provide consistent accounting practices and criteria for how companies should recognize, measure, present, and disclose in their financial statements all significant uncertain tax positions.
Stock-based compensation
We account for stock-based compensation in accordance with Statement of Financial Accounting Standards (“SFAS”) 123(R) Share-Based Payment, a revision to SFAS 123, Accounting for Stock-Based Compensation. SFAS 123(R) requires us to recognize the grant date fair value of share-based compensation awards granted to employees over the requisite service period. We use the Black-Scholes option pricing model to calculate stock option values, which requires certain assumptions including the future stock price volatility and expected time to exercise. Changes to any of these assumptions, or the use of a different option pricing model (such as the binomial model), could produce a different fair value for stock-based compensation, which could have a material impact on our earnings.
Cash equivalents, short and long-term investments
We invest our excess cash balances in short-term securities, principally investment grade commercial debt and government agency notes. At December 31, 2008, we held one equity security which was classified as available-for-sale, and accordingly, was recorded at fair market value. The security has been trading below carrying value for an extended period of time and management does not intend to hold it for a sufficient period of time to reasonably expect a recovery to initial carrying value. As such, the unrealized loss on this security has been reported in other income and expenses.
As part of our strategic product development efforts, we also invest in equity securities of certain companies with which we have collaborative agreements. The equity securities of some of these companies are not publicly traded and so fair value is not readily available. These investments are recorded using the cost method of accounting and are tested for impairment by reference to anticipated undiscounted cash flows expected to result from the investment, the results of operations and financial position of the investee, and other evidence supporting the net realizable value of the investment.
Goodwill
- 45 -
We test goodwill for possible impairment at least annually and whenever changes in circumstances occur that would indicate an impairment in the value of goodwill. When the carrying value of a reporting unit’s goodwill exceeds the implied fair value of the goodwill, an impairment loss is recognized in an amount equal to the excess. Circumstances that could trigger an impairment include adverse changes or outcomes in legal or regulatory matters, technological advances, decreases in anticipated demand, unanticipated competition, and significant declines in our share price. We estimate fair value based on a discounted projection of future cash flows which are subject to significant uncertainty and estimates. If future cash flows are less than those projected, an impairment charge may become necessary that could have a material impact on our financial position and results of operations. We tested our goodwill for impairment as at September 30, 2008 and determined that due to a significant and sustained decline in our public stock market capitalization our goodwill was impaired and as a consequence we recorded an impairment charge of $599.4 million during the three months ended September 30, 2008.
Given the continued decline in our market value in the fourth quarter of 2008 and the further negative indicators of the economy as a whole, we updated our impairment tests of goodwill and acquired intangible assets and concluded a further impairment had occurred in the carrying amounts of goodwill associated with our Medical Products segment as well as an impairment in the carrying amounts of goodwill associated with our Pharmaceuticals Technologies segment. Accordingly, we recorded a further impairment charge of $50.3 million in the fourth quarter of 2008 leaving no goodwill on our books as of December 31, 2008.
Intangible assets
Our identifiable intangible assets are primarily comprised of technologies acquired through our business combinations. Intangible assets also include in-licensed proven medical technologies. We amortize intangible assets on a straight-line basis over the estimated life of the technologies, which range from two to twelve years depending on the circumstances and the intended use of the technology. We determine the estimated useful lives for intangible assets based on a number of factors such as legal, regulatory or contractual limitations; known technological advances; anticipated demand for our products; and the existence or absence of competition. We review the carrying value of our intangible assets for impairment indicators at least annually and whenever there has been a significant change in any of these factors listed above. A significant change in these factors may warrant a revision of the expected remaining useful life of the intangible asset, resulting in accelerated amortization or an impairment charge, which would impact earnings. We tested our intangible assets for impairment as at September 30, 2008 and determined that there was no impairment. Given the continued decline in our market value in the fourth quarter of 2008 and the further negative indicators of the economy as a whole, we updated our impairment tests of intangible assets as at December 31, 2008 and determined that there was no impairment.
Results of Operations
Overview
The following discussion and analysis of results from our operations excludes the financial results from our discontinued operations (see “Results of Operations - Discontinued Operations”) for the comparative periods, unless otherwise noted.
We completed our acquisition of AMI on March 23, 2006. Accordingly, the results of AMI are not included in our results for the period from January 1, 2006 to the date of acquisition on March 23, 2006.
| | | | | | | | | | |
| (in thousands of U.S.$, except per share data) | Years ended December 31, |
| | | 2008 | | | 2007 | | | 2006 | |
| Revenues | | | | | | | | | |
| Pharmaceutical Technologies | $ | 92,456 | | $ | 117,501 | | $ | 176,485 | |
| Medical Products | | 190,816 | | | 170,193 | | | 138,590 | |
| Total revenues | | 283,272 | | | 287,694 | | | 315,075 | |
| Operating (loss) income | | (20,211 | ) | | (20,739 | ) | | 58,164 | |
| |
| Other (expense) income | | (733,857 | ) | | (49,853 | ) | | (38,049 | ) |
| |
| (Loss) income from continuing operations before income | | | | | | | | | |
| taxes and cumulative effect of change in accounting policy | | (754,068 | ) | | (70,592 | ) | | 20,115 | |
| Income tax (recovery) expense | | (12,892 | ) | | (14,545 | ) | | 2,092 | |
| Net (loss) income from continuing operations | $ | (741,176 | ) | $ | (56,047 | ) | $ | 18,023 | |
| |
| Basic and diluted net (loss) income per common share, | | | | | | | | | |
| continuing operations | $ | (8.71 | ) | $ | (0.66 | ) | $ | 0.21 | |
For the year ended December 31, 2008, we recorded a net loss from continuing operations of $741.2 million ($8.71 basic net loss per share) compared to net loss from continuing operations of $56.0 million ($0.66 basic net loss per share) for the year ended December 31, 2007.
- 46 -
The increase in the net loss of $685.2 million is due primarily to the write-down of an aggregate of $649.7 million of goodwill that we recorded in the third and fourth quarters of 2008. Also increasing the net loss were (i) a reduction of $25.1 million in royalty revenue, as BSC’s sales of paclitaxel-eluting coronary stent systems declined substantially in 2008 as compared to prior comparable periods; (ii) $16.5 million of transaction costs accrued related to the abandoned proposed API transaction; (iii) an $18.6 million write-down or loss on redemption of two available-for-sale equity securities, one of which we disposed of, and the other of which was trading below cost for an extended period of time; (iv) a $5.0 million write-down of a long term investment for which we could not obtain sufficient assurance we would be able to recover our carrying value; and (v) a $2.5 million payment for in-process research and development expense made during the first quarter of 2008 (as compared to $8.0 million in 2007). Partially offsetting the increase in the net loss were a $14.5 million increase in gross profit relating to higher medical products sales and a $7.2 million reduction in interest expense, as the interest rate applied to our Floating Rate Notes declined.
The $56.0 million we incurred in net loss for the year ended December 31, 2007 as compared to net income of $18.0 million for the year ended December 31, 2006 is primarily due to (i) a reduction of $49.0 million in royalty revenue derived from BSC’s sales of paclitaxel-eluting coronary stent systems; (ii) a decrease of $9.6 million in other royalty revenue due to a non-recurring sale to Orthovita, Inc. in 2006 of profit sharing rights for certain of their products for $9.0 million; (iii) a higher volume of lower margin Original Equipment Manufactured (“OEM”) product lines, reducing gross profit margins from 49.8% to 44.2%; (iv) an increase of $12.9 million in sales and marketing salaries as we invested significantly in our sales infrastructure with additions to our direct sales and marketing support personnel in the United States and the European Union; (v) an increase of $7.1 million for in-process research and development (“IPR&D”) expense, m ainly due to a $7.0 million payment we made to CombinatoRx for the extension of our collaboration agreement; (vi) non-recurring charges of $5.2 million for reorganization activities and personnel reductions relating to the announced plan to close and consolidate our Syracuse, NY manufacturing facility; and (vii) an additional $16.2 million in interest expense related to debt incurred to partially fund the AMI acquisition on March 23, 2006. Partially offsetting these factors was an income tax recovery of $14.5 million in 2007 compared to an income tax expense of $2.1 million in 2006.
Revenues
| |
| (in thousands of U.S.$) | Years ended December 31, |
| | | 2008 | | 2007 | | 2006 |
| Pharmaceutical Technologies: | | | | | | |
| Royalty revenue – paclitaxel-eluting stents | $ | 84,079 | $ | 110,477 | $ | 159,487 |
| Royalty revenue – other | | 7,467 | | 6,182 | | 15,767 |
| License fees | | 910 | | 842 | | 1,231 |
| | | 92,456 | | 117,501 | | 176,485 |
| Medical Products: | | | | | | |
| Product sales | | 190,816 | | 170,193 | | 138,590 |
| Total revenues | $ | 283,272 | $ | 287,694 | $ | 315,075 |
We operate in two reportable segments:
Pharmaceutical Technologies
Our Pharmaceutical Technologies segment includes royalty revenue generated from licensing our proprietary paclitaxel technology to various partners, as well as revenue derived from the licensing of certain of our biomaterials and other technologies.
Royalty revenue derived from sales of paclitaxel-eluting coronary stent systems by BSC for the year ended December 31, 2008 decreased by 24% as compared to the year ended December 31, 2007. The decrease in royalty revenues was primarily a result of lower sales of paclitaxel-eluting stents by BSC. Royalty revenue for the year ended December 31, 2008 was based on BSC’s net sales for the period October 1, 2007 to September 30, 2008 of $1.2 billion, of which $637 million was in the United States, compared to net sales of $1.6 billion, of which $1.0 billion was in the United States, for the comparable period in the prior year. The average gross royalty rate earned in the year ended December 31, 2008 on BSC’s net sales was 7.1% for sales in the United States and 6.4% for sales in other countries, compared to an average rate of 7.6% for sales in the United States and 5.6% for sales in other countries for the year ended December 31, 2007.
The average gross royalty rate for countries other than the United States improved in the year ended December 31, 2008 primarily due to the contribution of royalty revenue from BSC’s sales in Japan during the period, where the average gross royalty rate we receive is higher, as compared to minimal royalty revenue received relating to sales in Japan in the comparable period in the prior year.
- 47 -
Royalty revenue derived from sales of paclitaxel-eluting coronary stent systems by BSC for the year ended December 31, 2007 decreased by 31% as compared to the year ended December 31, 2006. The decrease in royalty revenues was primarily a result of lower sales of paclitaxel-eluting stents by BSC. Royalty revenue for the year ended December 31, 2007 was based on BSC’s net sales for the period October 1, 2006 to September 30, 2007 of $1.6 billion, of which $1.0 billion was in the United States, compared to net sales of $2.2 billion, of which $1.5 billion was in the United States for the comparable periods in the prior year. The average gross royalty rate earned in the year ended December 31, 2007 on BSC’s net sales was 7.6% for sales in the United States and 5.6% for sales in other countries compared to an average rate of 7.9% for sales in the United States and 6.0% for sales in other countries for the year ended December 31, 2006.
Other royalty revenue increased by $1.3 million for the year ended December 31, 2008 as compared to the year ended December 31, 2007. Other royalty revenue decreased by $9.6 million to $6.2 million for the year ended December 31, 2007 as compared to $15.8 million for the year ended December 31, 2006. The majority of this decrease was due to the non-recurring $9.0 million payment received from Orthovita, Inc in December 2006, which was classified as royalty revenue in 2006, under an agreement whereby Orthovita purchased our profit-sharing royalty rights for certain of its products.
We expect revenues in our Pharmaceutical Technologies segment will decrease into 2009 as compared to 2008 and 2007, primarily as a result of new competitors entering into the drug-eluting coronary stent market in the United States in the second half of 2008 and other factors. Our royalties derived from sales by BSC of TAXUS declined from $50.8 million in the first half of 2008 to $33.3 million in the second half of 2008.
Medical Products
Our Medical Products segment manufactures and markets a range of single use, specialty medical devices. The Medical Products segment also manufactures finished medical devices and medical device components for third party medical device manufacturers and marketers.
Revenue from our Medical Products segment for the year ended December 31, 2008 was $190.8 million, an increase of 12% compared to $170.2 million in the year ended December 31, 2007. The increase was due to several factors, including increased sales of various of our Interventional product lines, increased sales of certain of our Surgical product lines, the impact of certain new product launches during the year and improvement of sales of medical devices and device components to other third party medical device manufacturers. This increase also reflects the effect of the one-time $3.0 million charge against revenue in 2007 relating to the discontinuation of the Contour Threads brand and the accrual of a reserve against potential product that may be returned in that year. Excluding the impact of the $3.0 million accrual in 2007 relating to the Contour Threads brand discontinuation, revenue growth as compared to 2007 was 10%.
Revenue from our Medical Products segment for the year ended December 31, 2007 was $170.2 million compared to $138.6 million in the year ended December 31, 2006. Product revenue in our Medical Products segment for the year ended December 31, 2007 is not directly comparable to 2006, because the results for 2006 only include revenue in this segment from March 23, 2006 (the date we completed our acquisition of AMI).
We expect that revenues in our Medical Products segment will continue to increase during 2009 as compared to 2008 and 2007, reflecting the potential for growth of certain existing and newly launched product lines. This expected growth may be offset partially by the impact of the difficult economic environment and increasingly difficult credit market and liquidity environment on our customer base, particularly relating to certain third party medical device company customers that purchase medical devices and components from our various manufacturing operations.
Expenditures
| | | | | | | |
| (in thousands of U.S.$) | Years ended December 31, |
| | | 2008 | | 2007 | | 2006 |
| License and royalty fees | $ | 14,258 | $ | 18,652 | $ | 25,986 |
| Cost of products sold | | 101,052 | | 94,949 | | 69,543 |
| Research and development | | 53,192 | | 53,963 | | 45,393 |
| Selling, general and administrative | | 98,483 | | 99,315 | | 78,933 |
| Depreciation and amortization | | 33,998 | | 33,429 | | 36,014 |
| In-process research and development | | 2,500 | | 8,125 | | 1,042 |
| | $ | 303,483 | $ | 308,433 | $ | 256,911 |
License and royalty fees on royalty revenue
License and royalty fee expenses include license and royalty payments due to certain of our licensors, primarily relating to paclitaxel-eluting coronary stent system royalty revenue received from BSC. The decrease in this expense in 2008 and 2007 reflects the reduction in our royalty revenue. We expect license and royalty fee expense to continue to be a significant cost in 2009, commensurate with the amount of royalty revenue we earn.
- 48 -
Cost of products sold
Cost of products sold is comprised of costs and expenses related to the production of our various medical device, device component and biomaterial products and technologies, including direct labor, raw materials, depreciation and certain fixed overhead costs related to our various manufacturing facilities and operations.
Cost of products sold increased by $6.2 million to $101.1 million for the year ended December 31, 2008 compared to $94.9 million for the year ended December 31, 2007, primarily due to increased sales. Gross margins for our product sales were 47.1% for the year ended December 31, 2008 compared to 44.2% for the year ended December 31, 2007. Gross margins in 2008 compared to 2007 were positively impacted by an increase in sales of higher margin product lines, the launch of certain new, higher margin product lines for which there were no material sales in the comparable prior year period, and the effect of higher sales volumes on the absorption of fixed overhead and labour costs.
Cost of products sold increased by $25.4 million to $94.9 million for the year ended December 31, 2007 compared to $69.5 million for the year ended December 31, 2006, primarily because the 2006 results did not include a full year of the results of the operations acquired through the AMI acquisition which occurred on March 23, 2006.
Gross margins in our Medical Products segment were 44.2% for the year ended December 31, 2007 compared to 49.8% for the year ended December 31, 2006. Gross margins in 2007 were impacted by (i) the one-time charge against revenue of $3.0 million, with no corresponding reduction in cost of products sold, for actual and estimated potential returns of the Contour Threads brand product relating to a marketing incentive program offered to customers in support of the Quill SRS product launch and the concurrent discontinuation of the Contour Threads brand marketing and training support for certain indications of use; (ii) the overall product sales mix reflecting certain lower margin product lines; and (iii) certain non-recurring costs related to the closure and consolidation of our Syracuse, NY manufacturing facility.
We expect that cost of products sold will continue to be significant, and that gross margins may continue to improve in 2009, primarily as a result of improved sales mix, including potential increases in sales of selected product lines that provide higher relative contribution margins, the impact of anticipated higher levels of product sales on the absorption of fixed overhead costs and the reduction in fixed overhead and labor costs relative to certain product sales due to the completion of the consolidation of our Syracuse, NY operations, which will result in certain products being manufactured in a lower cost location in Puerto Rico as well as greater production volume concentrated over fixed overhead costs in our Puerto Rico location. These improvements may be offset by the impact of certain sales and pricing initiatives for selected product lines designed to improve sales volume, market share and overall operating cash flow derived from ou r fixed assets.
Research and development
Our research and development expense is comprised of costs incurred in performing research and development activities, including salaries and benefits, clinical trial and related clinical manufacturing costs, contract research costs, patent procurement costs, materials and supplies, and operating and occupancy costs. Our research and development activities occur in two main areas:
(i) Discovery and pre-clinical research - Our discovery and pre-clinical research efforts are divided into several distinct areas of activity, including screening and pre-clinical evaluation of pharmaceuticals and various biomaterials and drug delivery technologies, evaluation of mechanism of action of pharmaceuticals, mechanical engineering and pursuing patent protection for our discoveries.
(ii) Clinical research and development - Clinical research and development refers to internal and external activities associated with clinical studies of product candidates in humans, and advancing clinical product candidates towards a goal of obtaining regulatory approval to manufacture and market these product candidates in various geographies.
Research and development expenses, organized by stage of development, for the periods indicated were as follows:
| | | | | | | | | |
| (in thousands of U.S.$) | Years ended December 31, |
| |
| | | 2008 | | | 2007 | | | 2006 | |
| |
| Discovery and pre-clinical research | $ | 25,808 | | $ | 22,744 | | $ | 23,066 | |
| Ongoing clinical programs | | 3,367 | | | - | | | - | |
| Completed or Suspended Clinical Programs: | | | | | | | | | |
| Vascular WrapTMPaclitaxel-Eluting Mesh | | 13,552 | | | 11,743 | | | 9,399 | |
| Other clinical programs | | 3,337 | | | 7,249 | | | 7,835 | |
| | | 46,064 | | | 41,736 | | | 40,300 | |
| Medical Products | | 7,809 | | | 12,362 | | | 5,169 | |
| Stock-based compensation | | 795 | | | 2,056 | | | 2,340 | |
| Less: Depreciation, amortization and inter- | | | | | | | | | |
| company charges allocated to projects above | | (1,476 | ) | | (2,191 | ) | | (1,997 | ) |
| Total research and development | | 53,192 | | | 53,963 | | | 45,812 | |
| Less: Research and development relating to | | | | | | | | | |
| discontinued operations | | - | | | - | | | (419 | ) |
| Total research and development relating to | | | | | | | | | |
| continuing operations | $ | 53,192 | | $ | 53,963 | | $ | 45,393 | |
- 49 -
Research and development program expenses include all direct costs, as well as certain indirect expenses based on time allocated by certain research, clinical and administrative staff to each program.
Research and development expenditures were $53.2 million in 2008 as compared to $54.0 million in 2007. Research and development expenditures were up in the first half of 2008, primarily relating to our Vascular Wrap clinical program, and due to severance costs of $1.9 million relating to a significant reduction in staffing within our clinical and research departments completed in the second quarter. This was offset in the second half of 2008 by a $0.6 million decrease in clinical trial costs for our suspended Vascular Wrap clinical program and a $1.4 million reduction related to our completed 5-FU CVC clinical program (for which significant clinical trial activities have materially concluded). Ongoing clinical programs added $0.7 million in costs as compared to 2007. Also adding to research and development expenditures in the second half of 2008 were severance costs of $1.6 million due to a significant additional reduction in staffi ng within our clinical and research departments completed primarily during the third quarter.
Research and development expenditures increased by $8.6 million to $54.0 million for the year ended December 31, 2007 as compared to $45.4 million for 2006. The increase was due to a $3.1 million increase in clinical trial activity mainly associated with our Vascular Wrap and Bio-Seal programs, the addition of discovery and pre-clinical research personnel, an initial payment of $0.8 million for a new early-stage research collaboration, a one-time payment of $0.9 million to terminate a development agreement, and the incurrence of research and development expenses of our Medical Products segment for a full twelve month period in 2007.
We currently expect our research and development expenditures may decrease through 2009 as compared to 2008 and 2007, reflecting the suspension of enrolment in our Vascular Wrap clinical trial and the reduction or elimination of staffing and other direct costs during 2008 relating to certain other research programs and activities. Even after these expected declines, we anticipate we will continue to incur significant research and development expenditures in 2009.
Selling, general and administrative expenses
Our selling, general and administrative expenses are comprised of direct selling and marketing costs related to the sale of our various medical products, including salaries, benefits and sales commissions, and our various management and administrative support functions, including salaries, commissions, benefits and other operating and occupancy costs.
Selling, general and administrative expenditures were $98.5 million in 2008 as compared to $99.3 million in 2007. Higher expenditures in salaries, benefits and related costs, accounting and tax consulting fees and travel were offset by lower expenditures for other operating costs and lower litigation expenses in 2008 as compared to 2007. Salaries, benefits and related costs increased by $6.5 million in 2008 primarily due to the increase in direct sales and marketing personnel implemented in the United States and Europe during the second half of 2007. Litigation costs decreased $1.7 million as compared to the prior year period, due primarily to reaching agreement in the third quarter of 2007 with Conor MedSystems, Inc. (“Conor”) and its parent company Johnson & Johnson to settle all outstanding patent litigation with respect to Conor’s CoStar® paclitaxel-eluting stent. Other operating costs decreased by $4.6 m illion in 2008 as compared to 2007 due to certain personnel and other cost reduction initiatives undertaken in our general and administrative areas, primarily during the third quarter.
Selling, general and administrative expenditures for the year ended December 31, 2007 increased by $20.4 million to $99.3 million compared to $78.9 million in the year ended December 31, 2006. The higher expenditures were primarily due to the fact that 2007 results included a full year of Medical Products selling, marketing, general and administrative costs, as compared to only approximately nine months in 2006. Also contributing to the increase were additional salaries, benefits, recruiting, and travel costs relating to additions to our direct sales and marketing support personnel in the United States and the European Union, and $3.2 million for severance charges related to the announced plan to close and consolidate our Syracuse, NY manufacturing facility. Partially offsetting the increase was a $5.5 million reduction in legal litigation expense, due primarily to reaching a favorable agreement with Conor and its parent company Johnson & amp; Johnson to settle all outstanding patent litigation with respect to Conor’s CoStar® paclitaxel-eluting stent.
- 50 -
During 2009, we expect that selling, general and administrative expenses may be lower than in 2008 and 2007 due to staff reductions implemented primarily in the third quarter of 2008. Expenditures could materially increase or fluctuate depending on product sales levels, the timing of launch of certain new products and growth of new product sales, or as a result of litigation or other legal expenses that may be incurred to support and defend our intellectual property portfolio or other aspects of our business.
Depreciation and amortization
Depreciation and amortization expense was $34.0 million for the year ended December 31, 2008, compared to $33.4 million for the year ended December 31, 2007, and is comprised of amortization of licensed technologies and identifiable intangible assets purchased through business combinations of $30.2 million and $29.9 million, respectively, and depreciation of property, plant and equipment of $3.8 million and $3.5 million, respectively.
Depreciation and amortization expense was $33.4 million for the year ended December 31, 2007, compared to $36.0 million for the year ended December 31, 2006, and is comprised of amortization of licensed technologies and identifiable intangible assets purchased through business combinations of $29.9 million and $32.7 million, respectively, and depreciation of property, plant and equipment of $3.5 million and $3.3 million, respectively. The decrease in amortization expense in 2007 is primarily due to a one-time expense of $2.9 million in 2006 for accelerated amortization of the intangible assets related to the monetization of the profit-sharing royalty stream from Orthovita, Inc.
We expect depreciation and amortization expense to remain consistent in 2009 as compared to 2008.
In-process research and development (“IPR&D”)
We record IPR&D expense relating to acquired or in-licensed technologies that are at an early stage of development and have no alternative future use.
For the year ended December 31, 2008, we recorded IPR&D expense of $2.5 million for an initial license payment made to Rex Medical L.P. to obtain the marketing rights for the Option inferior vena cava filter. For the year ended December 31, 2007, we recorded IPR&D expense of $8.1 million, of which $7.0 million relates to the extension of our collaboration with CombinatoRx Inc. and $1.0 million relates to a collaboration agreement with Rex Medical L.P. For the year ended December 31, 2006, we recorded IPR&D expense of $1.0 million as a result of license milestone payments made to Poly-Med, Inc. in accordance with a license agreement.
We may incur further IPR&D expenditures in future periods in the event we in-license or acquire additional early stage technologies.
Other Income (Expense)
| | | | | | | | | | |
| (in thousands of U.S.$) | Years ended December 31, |
| | | 2008 | | | 2007 | | | 2006 | |
| Foreign exchange (loss) gain | $ | 540 | | $ | (341 | ) | $ | 515 | |
| Investment and other income | | 1,192 | | | 10,393 | | | 6,235 | |
| Interest expense on long term-debt | | (44,490 | ) | | (51,748 | ) | | (35,502 | ) |
| Write-down and other deferred financing charges | | (16,544 | ) | | - | | | (9,297 | ) |
| Write-down of assets held for sale | | (1,283 | ) | | - | | | - | |
| Write-down or net loss on redemption of investments | | (23,587 | ) | | (8,157 | ) | | - | |
| Write-down of goodwill | | (649,685 | ) | | - | | | - | |
| | $ | (733,857 | ) | $ | (49,853 | ) | $ | (38,049 | ) |
Net foreign exchange gains and losses were primarily the result of changes in the relationship of the U.S. to Canadian dollar and other foreign currency exchange rates when translating our foreign currency denominated cash, cash equivalents and short-term investments to U.S. dollars for reporting purposes at period end. We continue to hold foreign currency denominated cash and cash equivalents to meet our anticipated operating and capital expenditure needs in future periods in jurisdictions outside of the United States. We do not use derivatives to hedge against exposures to foreign currency arising from our balance sheet financial instruments and therefore are exposed to future fluctuations in the U.S. dollar to foreign currency exchange rates.
Investment and other income for the year ended December 31, 2008 decreased by $9.2 million when compared to 2007, primarily due to the fact that the 2007 comparative period included a gain of $7.5 million realized on the recovery of investments owned by Cohesion Technologies, Inc. which we acquired in 2003. In addition, we earned lower interest income in 2008 due primarily to lower cash balances available to invest and lower interest rates.
- 51 -
Investment and other income for the year ended December 31, 2007 increased by $4.2 million when compared to 2006, primarily due to a gain in the first quarter of 2007 of $7.5 million realized on the recovery of investments owned by Cohesion Technologies, Inc. which we acquired in 2003, offset partially by a reduction in investment income due to a lower cash balance available to invest because of the use of cash resources for the acquisitions of AMI and Quill and the write-off of certain capitalized tax assets totalling $1.9 million related to the AMI acquisition.
During the year ended December 31, 2008, we incurred interest expense of $44.5 million on our outstanding long-term debt obligations, as compared to $51.7 million for 2007. The decrease of $7.1 million has resulted from a decline, during 2008, in the interest rate applicable on our Floating Rate Notes due to a decline in the LIBOR rates. The interest rate decline resulted in an average interest rate of 7.7% for 2008 as compared to 9.0% in 2007. Interest expense in 2008 also includes $2.3 million for amortization of deferred financing costs.
During the year ended December 31, 2007, we incurred interest expense of $51.7 million on our outstanding long-term debt obligations, as compared to $35.5 million for 2006. The increase is primarily because our debt obligations that were issued in connection with our acquisition of AMI on March 23, 2006 were not outstanding for the full twelve month comparative period of 2006. Also contributing to the increase, the interest rate on our Floating Rate Notes is higher than the rate on the credit facility that they replaced. Interest expense for 2007 also includes $2.2 million for amortization of deferred financing costs.
During the year ended December 31, 2006, we incurred interest expense of $35.5 million on our outstanding long-term debt obligations. The senior secured term loan was extinguished in December 2006 and replaced with the Floating Rate Notes. During March 2006 to December 2006, the period, the senior secured term loan was in place, interest rates ranged between 6.3% and 8.8% and the interest rate on the Subordinated Notes remained fixed at 7.75%. Interest expense in 2006 also includes $2.0 million for amortization of deferred financing costs.
In 2008, we recognized costs of $16.5 million related to our exploration of proposed financing and strategic alternatives.
During the year ended December 31, 2006, we recognized a write-down of $9.3 million of deferred financing costs related to the extinguishment of the senior secured term loan.
In connection with our plans for capacity rationalization and consolidation in the Medical Products segment, we reclassified two of its long-lived assets as current assets held for sale in accordance with guidance in SFAS 144, Accounting for the Impairment or Disposal of Long-Lived Assets. These properties have a carrying value of approximately $3.1 million, which represents the lower of cost and fair value less cost to sell. An impairment charge of $1.3 million has been recorded against income from continuing operations.
In addition, and also in connection with our capacity rationalization in the Pharmaceutical Technology segment, we reclassified a property that is no longer used and has a carrying value of approximately $5.3 million as held for sale.
The write-down or net loss on redemption of investments for the year ended December 31, 2008 of $23.6 million is comprised of a loss of $8.8 million realized on the sale of our common stock holdings in CombinatoRx, Inc., an unrealized loss of $9.7 million on an available-for-sale equity security that has been trading below carrying value for an extended period and for which management does not intend to hold for a sufficient period of time to reasonably expect a recovery to initial carrying value and an unrealized loss of $5.0 million on the write-down in carrying value of shares held in a private biotechnology company classified as long term investments.
The write-down or net loss on redemption of investments for the year ended December 31, 2007 of $8.2 million is comprised of a loss of $9.6 million realized on the sale of our common stock holdings in Orthovita, Inc., partially offset by a gain of $1.4 million realized on the sale of our common stock holdings in NuVasive, Inc.
As a result of the significant and sustained decline in our public market capitalization, we performed an interim impairment test of goodwill and acquired intangible assets. Upon conclusion of such testing, we determined that the carrying amounts of goodwill associated with our Medical Products segment were impaired and we recorded an impairment charge of $599.4 million in the third quarter of 2008. The goodwill impairment charge was determined by comparing the carrying value of goodwill assigned to our Medical Products segment with the implied fair value of the goodwill. We utilized the income approach in determining the implied fair value of the goodwill, which requires estimates of future operating results and cash flows discounted using estimated discount rates.
Given the continued decline in our market value in the fourth quarter of 2008, we updated our impairment tests of goodwill and acquired intangible assets and concluded a further impairment had occurred in the carrying amounts of goodwill associated with our Medical Products segment as well as an impairment in the carrying amounts of goodwill associated with our Pharmaceutical Technologies segment. Accordingly, we recorded an additional impairment charge of $50.3 million in the fourth quarter of 2008, leaving no goodwill on our balance sheet as of December 31, 2008.
- 52 -
Income Tax
Income tax recovery for the year ended December 31, 2008 was $12.9 million compared to income tax recovery of $14.5 million for the year ended December 31, 2007. The income tax recovery for 2008 is primarily due to a net loss from operations, the amortization of identifiable intangible assets, tax deductions relating to international financing structures, and a recovery of taxes paid in prior years as a result of the carryback of current year losses. The income tax recovery for 2008 also includes a recovery of $3.8 million relating to the settlement of an outstanding Quebec income tax reassessment and a charge of $6.1 million related to an accrual under FIN 48.
The effective tax rate for 2008 was 1.7% comparedto an effective tax rate of 20.6% for 2007. The effective tax rate for 2008 is lower than the statutory Canadian tax rate of 31.0%, primarily due to valuation allowances on net operating losses, tax deductions related to international financing structures, provincial income tax credits, the net effect of lower tax rates on earnings in foreign jurisdictions, and goodwill impairment charges.
For the year ended December 31, 2008, income tax recovery of $12.9 million consisted of a current and deferred income tax recovery of $6.2 million on a net loss from Canadian operations and a current and deferred income tax recovery of $6.7 million on net losses from U.S. and foreign operations.
Discontinued Operations
In September 2006, we determined that certain operating subsidiaries acquired through the AMI acquisition were not aligned with our business strategy and we began actively looking to dispose of these subsidiaries. The operations were categorized as discontinued and the net loss for these operations has been shown separately on the statements of operations of these subsidiaries. On July 31, 2007, we completed the sale of 100% of the issued and outstanding shares of Point Technologies, Inc. and its subsidiary Point Technologies S.A. for proceeds of $2.6 million. On August 30, 2007, we sold all of the assets and liabilities of American Medical Instruments (Dartmouth), Inc. for proceeds of $2.2 million.
The operating results of discontinued operations are summarized as follows:
| | | | | | | |
| (in thousands of U.S.$) | | | | | | |
| | Years ended December 31, |
| | | 2007 | | | 2006 | |
| Revenue | $ | 7,580 | | $ | 10,092 | |
| Operating loss | | (632 | ) | | (4,045 | ) |
| Other income and expense | | (1,991 | ) | | 4 | |
| Impairment charge | | (8,879 | ) | | (7,700 | ) |
| Loss before income taxes | | (11,502 | ) | | (11,741 | ) |
| Income tax recovery | | (1,609 | ) | | (4,033 | ) |
| Net loss from discontinued operations | ($ | 9,893 | ) | ($ | 7,708 | ) |
Liquidity and Capital Resources
At December 31, 2008, we had working capital of $47.7 million and cash resources of $39.0 million, consisting of cash and cash equivalents. In aggregate, our working capital decreased by $62.0 million as compared to December 31, 2007, primarily relating to the decline in our royalty revenues derived from sales of paclitaxel-eluting coronary stent systems by BSC (as described previously) and our loss on redemption and write-down of equity securities. These cash resources, in addition to cash generated from operations, are used to support our continuing clinical studies, research and development initiatives, sales and marketing initiatives, working capital requirements, debt servicing requirements and for general corporate purposes. We may also use our cash resources to fund acquisitions of, or investments in, businesses, products or technologies that expand, complement or are otherwise related to our business.
On March 2, 2009 we announced we had completed a financing transaction with Wells Fargo Foothill, LLC. The financing includes a delayed draw secured term loan facility of up to $10 million and a secured revolving credit facility, with a borrowing base comprised of certain of our finished goods inventory and accounts receivable, providing up to an additional $22.5 million in available credit, subject to certain terms and conditions. At any time, the amount of financing available under the revolving credit facility may be significantly less than $22.5 million and is expected to fluctuate from month to month with changes in levels of finished goods and accounts receivable. As of February 23, 2009, the amount of financing available under the revolving credit facility was approximately (unaudited) $9.5 million. Any borrowings outstanding under the term loan and revolving credit facility bear interest ranging from LIBOR + 3.25% to LIBOR +3.75%, with a minimum Base LIBOR Rate of 2.25%. The term loan and revolving credit facility include certain covenants and restrictions with respect to our operations and require us to maintain certain levels of EBITDA and interest coverage ratios, among other terms and conditions. Repayment of any amounts drawn under the term loan and revolving credit facility can be made at certain points in time with ultimate maturity being February 27, 2013. Amounts repaid under the secured term loan, and prepayments made under the revolving credit facility in certain circumstances, cannot be re-borrowed by us. The purpose of this financing is to provide additional liquidity and capital resources for working capital and general corporate purposes.
As the minimum Base LIBOR Rate under the term loan and revolving credit facility is 2.25% and the LIBOR rate was 1.425% on December 31, 2008, a .5% increase or decrease in the LIBOR rate as of December 31, 2008 would have no impact on interest payable under the credit facilities.
- 53 -
On March 23, 2006 we completed an offering of $250.0 million in aggregate principal amount of Subordinated Notes in a private placement transaction, and entered into a $425.0 million senior secured credit facility consisting of a $350.0 million senior term loan facility maturing in 2013 and a $75.0 million senior secured revolving credit facility maturing in 2011. None of the $75.0 million revolving credit facility was drawn. The net proceeds from the sale of the $250.0 million Senior Subordinated Notes and the $350.0 million term loan, as well as cash on hand, were used to finance the acquisition of AMI. In December 2006, we repaid the term loan with the proceeds from the issuance of Senior Floating Rate Notes due 2013 in the aggregate principal amount of $325.0 million and cash on hand. We also terminated the $75 million revolving credit facility.
The significant terms relating to our Subordinated Notes and Floating Rate Notes are described below (see “Senior Floating Rate Notes” and “Senior Subordinated Notes”).
Due to numerous factors that may impact our future cash position, working capital and liquidity as discussed below and the significant cash that will be necessary to continue to service our current level of debt obligations, there can be no assurance that we will have adequate liquidity and capital resources to satisfy our financial obligations beyond 2009.
Our cash inflows and the amounts of expenditures that will be necessary to execute our business plan are subject to numerous uncertainties, including but not limited to changes in drug-eluting coronary stent markets, including the impact of new competitive entrants into such markets and research relating to the efficacy of drug-eluting stents, the sales achieved in such markets by our partner BSC, the timing and success of product sales and marketing initiatives and new product launches, the timing and success of our research, product development and clinical trial activities, the timing of completing certain operational initiatives including facility closures, our ability to effect reductions in certain aspects of our budgets in an efficient and timely manner, and changes in interest rates. These and other uncertainties may adversely affect our liquidity and capital resources to a significant extent and may force us to further reduce our expen ditures on research and development or on our various new product and sales and marketing initiatives in order for us to continue to service our debt obligations. Such further reductions in our budgeted expenditures may have an adverse effect on our new product development and sales growth initiatives and reduce our ability to achieve the revenue growth targets, product launch or new product development timelines in our current operating plan. There can also be no assurance that such reductions in expenditures will be adequate to provide enough cash flow to continue to service our current level of debt obligations.
In particular, should our royalties received from BSC decline more significantly than we expect in future periods as a result of new competitive entrants into the U.S. drug-eluting stent market and due to negative research or publications relating to the efficacy of drug-eluting stents, our liquidity has been and may continue to be adversely affected, and we have been forced to explore alternative funding sources through debt, equity or other public or private securities offerings, or to pursue certain reorganization, restructuring or other strategic alternatives. We may not be able to complete any restructuring, reorganization or strategic activities on terms that would be favourable for our shareholders. Capital markets conditions deteriorated significantly during 2008 and early 2009, including a significant and material decline in the level of corporate lending activity, combined with a significant increase in the cost of any such lend ing. Current market conditions may have a material impact on our ability to complete any of the activities as described on favourable terms, if at all.
As a result of uncertainty and volatility relating to the market and competitive environment for drug-eluting stents and to address our liquidity needs, we commenced the exploration of a broad range strategic and financial alternatives in the second half of 2007 See “Business Overview —Financial and Strategic Alternatives Process”.
Cash Flow Highlights
| | | | | | | | | | |
| (in thousands of U.S.$) | Years ended December 31, |
| | | 2008 | | | 2007 | | | 2006 | |
| Cash and cash equivalents, beginning of year | $ | 91,326 | | $ | 99,332 | | $ | 62,163 | |
| |
| Net (loss) income excluding non-cash items | | (16,866 | ) | | (14,697 | ) | | 58,662 | |
| Working capital requirements | | (21,141 | ) | | 9,135 | | | (2,791 | ) |
| Cash (used in) provided by operating activities | | (38,007 | ) | | (5,562 | ) | | 55,871 | |
| Cash (used in) provided by investing activities | | (8,239 | ) | | 259 | | | (575,208 | ) |
| Cash (used in) provided by financing activities | | (4,378 | ) | | (1,637 | ) | | 556,926 | |
| Effect of exchange rate changes on cash | | (1,750 | ) | | (1,066 | ) | | (420 | ) |
| Net (decrease) increase in cash and cash equivalents | | (52,374 | ) | | (8,006 | ) | | 37,169 | |
| |
| Cash and cash equivalents, end of year | $ | 38,952 | | $ | 91,326 | | $ | 99,332 | |
Cash Flows from Operating Activities
- 54 -
Cash used in operating activities for the year ended December 31, 2008 was $38.0 million compared to cash used of $5.6 million for 2007. Net loss for the current year, excluding non-cash items, resulted in cash outflows of $16.9 million compared to cash outflows of $14.7 million for 2007. The increase in cash used in operating activities was due to factors consistent with those that impacted net loss, as described above under “Results of Operations – Overview”. Working capital requirements resulted in net cash outflows of $21.1 million during 2008 compared to net cash inflows of $9.1 for 2007. The net cash outflows in 2008 primarily resulted from a $5.5 million increase during the year in trade accounts receivable, consistent with the increase in medical product sales as compared to 2007, an increase of $5.1 million during the year of inventory balances built in order to meet production requirements for certain of our medical products in the United States and Europe and a decrease of $12.5 million in accounts payable and accrued liabilities due to lower spending.
Cash used in operating activities for the year ended December 31, 2007 was $5.6 million compared to cash provided of $55.9 million for 2006. Net loss for the current year, excluding non-cash items, resulted in cash outflows of $14.7 million compared to cash inflows of $58.7 million for 2006. The decrease in cash provided by operating activities was due to factors consistent with those that impacted net loss, as described above under “Results of Operations – Overview”. Working capital requirements resulted in cash inflows of $9.1 million during 2007 compared to cash outflows of $2.8 million for 2006. Cash inflows related to working capital for 2007 primarily resulted from a $10.6 million decrease in trade accounts receivable due to focused collection efforts and slightly higher trade and tax payables balances. Partially offsetting these net inflows was a decrease in other accounts payable as a $5.0 million milestone payable accrued at December 31, 2006 was paid during 2007.
Cash Flows from Investing Activities
Net cash used in investing activities for the year ended December 31, 2008 was $8.2 million compared to net cash provided of $0.3 million for 2007. For 2008, the net cash used in investing activities was primarily related to capital expenditures of $7.7 million. We also paid a $0.8 million licensing milestone to a partner upon our successful completion of enrollment in our Bio-Seal human clinical trial and invested $2.5 million in IPR&D for an initial license payment to Rex Medical L.P. These cash outflows were offset by proceeds on sale of short-term investments of $2.8 million.
Net cash provided by investing activities for the year ended December 31, 2007 was $0.3 million compared to net cash used of $575.2 million for 2006. For 2007, cash provided by investing activities was primarily from the proceeds on the sale of long term assets, and the funds were used for the acquisition of R&D assets including intangible assets, IPR&D and equipment. For 2006, cash used in investing activities was primarily related to cash used to fund the acquisitions of AMI and Quill, partly offset by net redemptions of short-term investments.
We invest our excess cash balances in short-term marketable securities, principally investment grade commercial debt and government agency notes. The primary objectives of our marketable securities portfolio are liquidity and safety of principal. Investments are made with the objective of achieving the highest rate of return while meeting our two primary objectives. Our investment policy limits investments to certain types of instruments issued by institutions with investment grade credit ratings and places restrictions on maturities and concentration by type and issuer. At December 31, 2008, we were not holding any short-term marketable securities.
At December 31, 2008 and December 31, 2007, we retained the following cash and cash equivalents denominated in foreign currencies in order to meet our anticipated foreign operating and capital expenditures in future periods.
| | | | |
| | | December 31, | | December 31, |
| (in thousands of U.S.$) | | 2008 | | 2007 |
| |
| Canadian dollars | $ | 3,942 | $ | 15,488 |
| Swiss franc | | 3,053 | | 3,870 |
| Euro | | 5,263 | | 1,395 |
| Danish krone | | 2,022 | | 1,756 |
| Other | | 2,806 | | - |
Cash Flows from Financing Activities
Net cash used in financing activities for the year ended December 31, 2008 was $4.4 million, primarily for expenditures related to the proposed API financing transaction as described above as well as expenditures related to the credit facility announced on March 2, 2009 (see “Liquidity and Capital Resources” above and “Subsequent Events” in note 27 to audited consolidated financial statements for year ended December 31, 2008). The expenditures related to the proposed financing transaction had been capitalized as part of deferred financing costs related to the transaction, but were expensed in late 2008 based upon our September 22, 2008 announcement that we may not be able to satisfy certain closing conditions of the proposed transaction. These cash outflows were offset with a small amount of cash received on the exercise of employee stock options.
- 55 -
Net cash used in financing activities for the year ended December 31, 2007 of $1.6 million was related to long-term debt financing costs of $1.9 million, partially offset by proceeds from exercise of stock options of $0.2 million. Net cash provided by financing activities for 2006 of $556.9 million was mainly due to the proceeds received from the senior secured credit facility and Subordinated Notes used to fund the AMI acquisition.
Senior Floating Rate Notes
On December 11, 2006, we issued Floating Rate Notes in the aggregate principal amount of $325 million. The Floating Rate Notes bear interest at an annual rate of LIBOR (London Interbank Offered Rate) plus 3.75%, which is reset quarterly. Interest is payable quarterly in arrears on March 1, June 1, September 1, and December 1 of each year through to maturity. The Floating Rate Notes are unsecured senior obligations, are guaranteed by certain of our subsidiaries and rank equally in right of payment to all of our existing and future senior indebtedness. At December 31, 2008, the interest rate on these notes was 5.97%. We may redeem all or a part of the notes at specified redemption prices.
Senior Subordinated Notes
On March 23, 2006, we issued Subordinated Notes in the aggregate principal amount of $250.0 million. These Subordinated Notes bear interest at an annual rate of 7.75% payable semi-annually in arrears on April 1 and October 1 of each year through to maturity. The Subordinated Notes are unsecured obligations. The Subordinated Notes and related note guarantees provided by us and certain of our subsidiaries are subordinated to Floating Rate Notes described above and our existing and future senior indebtedness.
Prior to April 1, 2009, we may redeem at a specified redemption price up to 35% of the aggregate principal amount of the notes using net proceeds from certain equity and convertible debt offerings or we may redeem all, or a portion, of the aggregate principal amount of the notes at any time by paying a make-whole redemption price. On or after April 1, 2009, we may redeem all or a part of the notes at specified redemption prices.
Debt Covenants
The terms of the indentures governing our Floating Rate Notes and our Subordinated Notes include various covenants that impose restrictions on the operation of our business and the business of our subsidiaries, including the incurrence of certain liens and other indebtedness. As of December 31, 2008, we are in material compliance with all covenants and are not in breach of any provision of the indentures governing the Subordinated Notes and Floating Rate Notes that would cause an event of default to occur.
In addition, the terms of our senior secured credit facility that we announced on March 2, 2009, include customary financial covenants, including maintaining certain levels of EBITDA and interest coverage ratios, as well as covenants that limit our ability to, among other things, incur indebtedness, create liens, merge or consolidate, sell or dispose of assets, change the nature of their business, make distributions and make advances, loans or investments.
Contractual Obligations
Our significant contractual obligations for the next five years and thereafter include:
| | | | | | |
| (in thousands of U.S.$) | Payments due by period | | | |
| |
| | Total | Less than 1 year | 1 to 3 years | 3 to 5 years | More than 5 |
| | | | | | years |
| |
| Long-term debt repayments | 575,000 | - | - | 325,000 | 250,000 |
| Long-term debt interest | | | | | |
| obligations | 185,955 | 36,851 | 72,854 | 71,406 | 4,844 |
| Operating leases | 24,708 | 3,271 | 5,605 | 4,996 | 10,836 |
| License, research and technology | | | | | |
| development agreements | 1,563 | 1,563 | - | - | - |
| Total obligations | 787,226 | 41,685 | 78,459 | 401,402 | 265,680 |
Long-term debt includes $325.0 million of Floating Rate Notes and $250.0 million of Subordinated Notes. Repayments are based on contractual commitments as defined in the indentures governing the notes. Long-term debt interest obligations on variable (floating) rate debt are estimated using the current interest rates in effect at December 31, 2008. Long-term debt repayments and interest obligations assume no early repayment of principal.
We have entered into operating leases in the ordinary course of business for office and laboratory space with various third parties, which expire through July 2019.
- 56 -
Included in the above schedule are our commitments arising from our acquisition of Quill, to spend a further $1.6 million over the period of January 1, 2009 to June 30, 2009 in relation to the technology, including sales and marketing, research and development, and corporate support.
The table above does not include any cost sharing or milestone payments in connection with research and development collaborations with third parties as these payments are contingent on the achievement of specific developmental, regulatory or commercial activities and milestones. In addition, we may have to make royalty payments based on a percentage of future sales of certain products in the event regulatory approval for marketing is obtained. We have a contingent obligation of $10.0 million to former Afmedica equity holders should we reach certain development and regulatory milestones with respect to any Afmedica product. In addition, we may be required to make additional contingent payments of up to $150.0 million to the former shareholders of Quill should we achieve certain revenue and development milestones. These payments to the former Quill shareholders are primarily contingent upon the achievement of significant incremental revenue growth over a five year period from the close of the acquisition, subject to certain conditions. We may also have to make royalty payments based on a percentage of future sales of certain products associated with certain collaborators and licensors in the event regulatory approval for marketing is obtained.
Contingencies
We are party to various legal proceedings, including patent infringement litigation and other matters. See Part I, Item 3 and Note 19(b) “Contingencies”, in the Notes to the Consolidated Financial Statements of Part II, Item 8 of this Annual Report on Form 10-K for more information.
Inflation
The effects of inflation or changing prices has not had a material impact on our net sales, revenues or income from continuing operations for the last three years.
Off-Balance Sheet Arrangements
As of December 31, 2008, we do not have any off-balance sheet arrangements, as defined by applicable securities regulators in Canada and the United States, that have, or are reasonably likely to have, a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that would be material to investors.
Recent Accounting Pronouncements
In December 2007, the FASB issued SFAS No. 141 (Revised 2007), Business Combinations, or SFAS No. 141(R). SFAS No. 141(R) will change the accounting for business combinations. Under SFAS No. 141(R), an acquiring entity will be required to recognize all the assets acquired and liabilities assumed in a transaction at the acquisition-date fair value with limited exceptions. SFAS No. 141(R) will change the accounting treatment and disclosure for certain specific items in a business combination. SFAS No. 141(R) applies prospectively to business combinations for which the acquisition date is on or after the beginning of the first annual reporting period beginning on or after December 15, 2008. Accordingly, any business combinations we engage in will be recorded and disclosed following existing GAAP until December 31, 2008. We expect SFAS No. 141(R) will have an impact on accounting for business combinations once adopted but the effect is dependent upon acquisitions at that time.
In December 2007, the FASB issued SFAS No. 160, Non-controlling Interests in Consolidated Financial Statements—An Amendment of ARB No. 51, or SFAS No. 160. SFAS No. 160 establishes new accounting and reporting standards for the noncontrolling interest in a subsidiary and for the deconsolidation of a subsidiary. SFAS No. 160 is effective for fiscal years beginning on or after December 15, 2008. We do not expect the adoption of SFAS No. 160 to have a material impact on its consolidated balance sheets, results of operations or cash flows.
In November 2007, the Emerging Issues Task Force issued EITF Issue 07-01, Accounting for Collaborative Arrangements or EITF No. 07-01. EITF No. 07-01 requires collaborators to present the results of activities for which they act as the principal on a gross basis and report any payments received from (made to) other collaborators based on other applicable GAAP or, in the absence of other applicable GAAP, based on analogy to authoritative accounting literature or a reasonable, rational, and consistently applied accounting policy election. Further, EITF No. 07-01 clarified that the determination of whether transactions within a collaborative arrangement are part of a vendor-customer (or analogous) relationship subject to Issue 01-9, Accounting for Consideration Given by a Vendor to a Customer. EITF No. 07-01 is effective for fiscal years beginning December 15, 2008. We have not yet completed its evaluation of EITF 07-01, but does not believe that it wi ll have a material impact on its consolidated financial position, results of operations or cash flows.
- 57 -
In March 2008, the FASB issued SFAS No. 161, Disclosures about Derivative Instruments and Hedging Activities, or SFAS No. 161. SFAS No. 161 amends and expands the disclosure requirements of SFAS No. 133, Accounting for Derivative Instruments and Hedging Activities. It requires qualitative disclosures about objectives and strategies for using derivatives, quantitative disclosures about fair value amounts of gains and losses on derivative instruments, and disclosures about credit-risk-related contingent features in derivative agreements. This statement is effective for financial statements issued for fiscal years beginning after November 15, 2008. We are still assessing the impact of this pronouncement.
In April 2008, the FASB issued FSP FAS 142-3, Determination of the Useful Life of Intangible Assets, or FSP FAS 142-3. FSP FAS 142-3 amends the factors that should be considered in developing renewal or extension assumptions used to determine the useful life of a recognized intangible asset under SFAS No. 142, Goodwill and Other Intangible Assets. The intent of the position is to improve the consistency between the useful life of a recognized intangible asset under SFAS No. 142 and the period of expected cash flows used to measure the fair value of the intangible asset. FSP FAS 142-3 is effective for fiscal years beginning after December 15, 2008. We are assessing the potential impact that the adoption of FSP FAS 142-3 may have on its consolidated financial position, results of operations or cash flows.
In June 2008, the Emerging Issues Task Force issued EITF 08-3, Accounting for Lessees for Maintenance Deposits Under Lease Arrangements or EITF 08-3. EITF 08-3 provides guidance for accounting for nonrefundable maintenance deposits. It also provides revenue recognition accounting guidance for the lessor. EITF 08-3 is effective for fiscal years beginning after December 15, 2008. We have not yet completed its evaluation of EITF 8-03, but does not believe that it will have a material impact on its consolidated financial position, results of operations or cash flows.
In November 2008, the Emerging Issues Task Force issued EITF 08-6, Equity method Investment Accounting Considerations, or EITF 08-6. EITF 08-6 addresses a number of matters associated with the impact of SFAS No. 141(R) and SFAS No. 160 on the accounting for equity method investments including initial recognition and measurement and subsequent measurement issues. EITF 08-6 is effective, on a prospective basis, for fiscal years beginning after December 15, 2008 and interim periods within those fiscal years. We have not yet completed its evaluation of EITF 8-03, but does not believe that it will have a material impact on its consolidated financial position, results of operations or cash flows.
In November 2008, the Emerging Issues Task Force issued EITF 08-07, Accounting for Defensive Intangible Assets, or ETIF 08-7. EITF 08-7 provides guidance for accounting for defensive intangible assets subsequent to their acquisition in accordance with SFAS No. 141(R) and SFAS No. 157 including the estimated useful life that should be assigned to such assets. EITF 08-7 is effective for intangible assets acquired on or after the beginning of the first annual reporting period beginning on or after December 15, 2008. We do not expect EITF 08-7 will have an impact on accounting for business combinations once adopted but the effect is dependent upon acquisitions at that time.
Outstanding Share Data
As of December 31, 2008, there were 85,121,983 common shares issued and outstanding for a total of $472.7 million in share capital. At December 31, 2008, we had 7,644,632 CDN dollar stock options outstanding under the Angiotech Pharmaceuticals, Inc. stock option plan (of which 5,676,629 were exercisable) at a weighted average exercise price of CDN$12.82. We also had 1,790,968 U.S. dollar stock options outstanding under this plan at December 31, 2008, (of which 370,478 were exercisable) at a weighted average exercise price of US$3.19 per option. Each CDN dollar stock option and U.S. dollar stock option is exercisable for one common share of Angiotech Pharmaceuticals, Inc.
As of December 31, 2008, there were 94 stock options outstanding in the AMI stock option plan (of which 13 were exercisable). Each AMI stock option is exercisable for approximately 3,852 common shares of Angiotech Pharmaceuticals, Inc. upon exercise at a weighted average exercise price of US$15.44 per option.
Item 7A.
QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
The primary objective of our investment activities is to preserve our capital to fund operations. We also seek to maximize income from our investments without assuming significant risk. To achieve our objectives, we maintain a portfolio of cash equivalents and investments in a variety of securities of high credit quality. As of December 31, 2008 we had cash and cash equivalents of $39.0 million.
Interest Rate Risk
As the issuer of the Floating Rate Notes, we are exposed to interest rate risk. The interest rate on the Floating Rate Notes is reset quarterly to 3-month LIBOR plus 3.75%. The aggregate principal amount of the Floating Rate Notes is $325million and the notes bear interest at a rate of approximately 6.0% at December 31, 2008 (8.9% at December 31, 2007). Based upon the average floating rate debt levels of the Company during the year, a 100 basis point increase in interest rates would have impacted our interest expense by approximately $3.3 million for each of the years ended December 31, 2008 and 2007. We do not use derivatives to hedge against interest rate risk.
- 58 -
Foreign Currency Risk
We operate internationally and enter into transactions denominated in foreign currencies. As such, our financial results are subject to the variability that arises from exchange rate movements in relation to the U.S. dollar. Our foreign currency exposures are primarily limited to the Canadian dollar, the Swiss franc, the Danish kroner, the Euro and the U.K. pound sterling. We incurred foreign exchange gain of $0.5 million for the year ended December 31, 2008 ($0.3 million foreign exchange loss for the year ended December 31, 2007) primarily as a result of changes in the relationship of the U.S. to Canadian dollar and other foreign currency exchange rates when translating our foreign currency-denominated cash, cash equivalents and short-term investments to U.S. dollars for reporting purposes at period end. We have not entered into any forward currency contracts or other financial derivatives to hedge foreign exchange risk, and therefore we are subject to foreig n currency transaction and translation gains and losses. We purchase goods and services in U.S. and Canadian dollars, Swiss francs, Danish kroner, Euros, and U.K. pounds sterling, and earn a significant portion of our license and milestone revenues in U.S. dollars. Foreign exchange risk is managed primarily by satisfying foreign denominated expenditures with cash flows or assets denominated in the same currency.
Since we operate internationally and approximately 13.4% of our net revenue for the year ended December 31, 2008 was generated in other than the U.S. dollar, foreign currency exchange rate fluctuations could significantly impact our financial position, results of operations, cash flows and competitive position.
For purposes of specific risk analysis, we used a sensitivity analysis to measure the potential impact to our consolidated financial statements for a hypothetical 10% strengthening of the U.S. dollar compared with the Canadian dollar, the Swiss franc, the Danish kroner, the Euro and the U.K. pounds sterling for the year ended December 31, 2008. Assuming a 10% strengthening of the U.S. dollar, our product net revenue would have been negatively impacted by approximately $3.5 million.
Item 8.
FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Management’s Report on Internal Control over Financial Reporting
See index to financial statements included under Item 15 in this Annual Report on Form 10-K.
Item 9.
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
Item 9A.
CONTROLS AND PROCEDURES
(a) Evaluation of Disclosure Controls and Procedures.
Based upon an evaluation of the effectiveness of disclosure controls and procedures, our Chief Executive Officer (“CEO”) and Chief Financial Officer (“CFO”) have concluded that as of the end of the period covered by this Form 10-K our disclosure controls and procedures (as defined in Rules 13a-15(e) or 15d-15(e) under the Exchange Act) were effective to provide reasonable assurance that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified by the rules and forms of the SEC and is accumulated and communicated to management, including the CEO and CFO, as appropriate to allow timely decisions regarding required disclosure.
It should be noted that the design of any system of controls is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions, regardless of how remote. However, our CEO and CFO have concluded that our disclosure controls and procedures are effective under circumstances where our disclosure controls and procedures should reasonably be expected to operate effectively.
(b) Management's Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act) to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Due to its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate due to changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
- 59 -
Under the supervision and with the participation of our management, including our CEO and CFO, we conducted an evaluation of the effectiveness of our internal control over financial reporting using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control—Integrated Framework. Based on its evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2008.
The effectiveness of our internal control over financial reporting as at December 31, 2008 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report which appears herein.
(c) Changes in Internal Control over Financial Reporting
No change was made to our internal control over financial reporting during the fiscal quarter ended December 31, 2008 that has materially affected, or is reasonably likely to materially affect, such internal control over financial reporting.
Item 9B.
OTHER INFORMATION
None
- 60 -
PART III
Item 10.
DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this Item is set forth under the captions “Election of Directors” and “Corporate Governance” in our definitive Proxy Statement, which will be filed with the SEC pursuant to Regulation 14A under the Exchange Act and is incorporated herein by reference.
Item 11.
EXECUTIVE COMPENSATION
The information required by this Item is set forth under the captions “Executive Compensation”, and “Corporate Governance” in our definitive Proxy Statement, which will be filed with the SEC pursuant to Regulation 14A under the Exchange Act and is incorporated herein by reference.
Item 12.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this Item is set forth under the caption “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” in our definitive Proxy Statement, which will be filed with the SEC pursuant to Regulation 14A under the Exchange Act and is incorporated herein by reference.
Item 13.
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this Item is set forth under the captions “Certain Relationships and Related Transactions” and “Corporate Governance” in our definitive Proxy Statement, which will be filed with the SEC pursuant to Regulation 14A under the Exchange Act and is incorporated herein by reference.
Item 14.
PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this Item is set forth under the caption “Appointment of Auditors” in our definitive Proxy Statement, which will be filed with the SEC pursuant to Regulation 14A under the Exchange Act and is incorporated herein by reference.
- 61 -
PART IV
Item 15.
EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a)
The following documents are included as part of this Annual Report on Form 10-K.
(1)
Financial Statements:
| | | Page† |
| | Report of Independent Registerd Public Accounting Firm | 13-1 |
| | Consolidated Balance Sheets as of December 31, 2008 and 2007 | 13-2 |
| | Consolidated Statements of Operations for the years ended December 31, 2008, 2007 and 2006. | 13-3 |
| | Consolidated Statements of Shareholders’ (Deficit) Equity as of December 31, 2008, 2007 and 2006 | 13-4 |
| | Consolidated Statements of Cash Flows for the years ended December 31, 2008, 2007 and 2006 | 13-5 |
| | Notes to Consolidated Financial Statements | 13-6 |
| | †Page numbers refer to Exhibit 13 of this Annual Report on Form 10-K. | |
(2)
Financial Statement Schedule:
| | | Page† |
| | Report of Independent Registerd Public Accounting Firm | 13-44 |
| | Schedule II – Valuation of Qualifying Accounts for the years ended December 31, 2008, 2007 and 2006 | 13-45 |
| | †Page numbers refer to Exhibit 13 of this Annual Report on Form 10-K. | |
All other schedules are omitted as the information required is inapplicable or the information is presented in the consolidated financial statements or related notes.
(3)
Exhibits:
The exhibits listed below are filed as part of this Annual Report on Form 10-K. References under the caption “Location” to exhibits or other filings indicate that the exhibit or other filing has been filed, that the indexed exhibit and the exhibit or other filing referred to are the same and that the exhibit or other filing referred to is incorporated by reference. Management contracts and compensatory plans or arrangements filed as exhibits to this Annual Report on Form 10-K are identified by an asterisk. The Commission file number for our Exchange Act filings referenced below is 000-30334.
| | | | | |
| Exhibit | | Description | | Location |
| 3.1 | | Notice of Articles of Angiotech Pharmaceuticals, Inc. | | Filed herewith |
| |
| 3.2 | | Articles of Angiotech Pharmaceuticals, Inc. | | Exhibit 3, Form 6-K filed March 31, 2005 |
| |
| 4.1 | | Indenture dated March 23, 2006 among Angiotech Pharmaceuticals, Inc., certain of its subsidiaries and Wells Fargo Bank N.A., for 7.75% Senior Subordinated Notes due 2014 | | Exhibit 99.2, Form 6-K filed April 3, 2006 |
| |
| 4.2 | | Indenture dated December 11, 2006 among Angiotech Pharmaceuticals, Inc., certain of its subsidiaries and Wells Fargo Bank N.A., for Senior Floating Rate Notes due 2013 | | Exhibit 2, Form 6-K filed December 15, 2006 |
| |
| 4.3 | | Shareholder Rights Plan Agreement between Angiotech Pharmaceuticals, Inc. and Computershare Trust Company of Canada as Rights Agent, Amended and Restated as of October 30, 2008 | | Filed herewith |
| |
| 10.1 | | Credit Agreement, dated as of February 27, 2009, by and among Angiotech Pharmaceuticals, Inc., as Parent, the subsidiaries of Parent listed as borrowers on the signature pages thereto, as Borrowers, the lenders signatory thereto, as Lenders, and Wells Fargo Foothill, LLC, as Arranger and Administrative Agent. | | Exhibit 10.1, Form 8-K filed March 5, 2009 |
| |
| 10.2 | | License Agreement by and among Angiotech Pharmaceuticals, Inc., Boston Scientific Corporation and Cook Incorporated, dated as of July 9, 1997 | | Exhibit, Form 20-FR filed October 5, 1999 |
| | | | | |
| 10.3 | | September 24, 2004 Amendment Between Angiotech Pharmaceuticals, Inc. and Cook Incorporated Modifying July 9, 1997 License Agreement Among Angiotech Pharmaceuticals, Inc., Boston Scientific Corporation, and Cook Incorporated** | | Filed herewith |
| |
| 10.4 | | Amendment Between Angiotech Pharmaceuticals, Inc. and Boston Scientific Corporation Modifying July 9, 1997 License Agreement Among Angiotech Pharmaceuticals, Inc., Boston Scientific Corporation, and Cook Incorporated | | Exhibit 10.2, Form 6-K filed March 31, 2005 |
| |
| 10.5 | | Distribution and License Agreement by and among Angiodevice International GmbH (as assignee of Angiotech Pharmaceuticals, Inc., Angiotech International, GmbH, and Cohesion Technologies, Inc.), Baxter Healthcare Corporation and Baxter Healthcare, S.A., dated April 1, 2003** | | Filed herewith |
| |
| 10.6 | | Manufacturing and Supply Agreement, by and among Angiodevice International GmbH (as assignee of Angiotech Pharmaceuticals, Inc., Angiotech International, GmbH, and Cohesion Technologies, Inc.), Baxter Healthcare Corporation and Baxter Healthcare, S.A., dated April 1, 2003** | | Filed herewith |
| |
| 10.7 | | Amendment No. 1 dated December 23, 2004 to Distribution and License Agreement, by and among Angiodevice International GmbH (as assignee of Angiotech Pharmaceuticals, Inc., Angiotech International, GmbH, and Cohesion Technologies, Inc.), Baxter Healthcare Corporation and Baxter Healthcare, S.A., dated April 1, 2003 | | Exhibit 1, Form 6-K filed March 28, 2008 |
| |
| 10.8 | | Amendment No. 1 dated January 21, 2005 to Manufacturing and Supply Agreement, by and among Angiodevice International GmbH (as assignee of Angiotech Pharmaceuticals, Inc., Angiotech International, GmbH, and Cohesion Technologies, Inc.), Baxter Healthcare Corporation and Baxter Healthcare, S.A., dated April 1, 2003 | | Exhibit 2, Form 6-K filed March 28, 2008 |
| |
| 10.9 | | Amendment No. 2 dated October 8, 2007 to Distribution and License Agreement, by and among Angiodevice International GmbH (as assignee of Angiotech Pharmaceuticals, Inc., Angiotech International, GmbH, and Cohesion Technologies, Inc.), Baxter Healthcare Corporation and Baxter Healthcare, S.A., dated April 1, 2003** | | Filed herewith |
| |
| 10.10 | | Agreement and Plan of Merger among Angiotech Pharmaceuticals, Inc., Angiotech Pharmaceuticals (US), Inc., Quaich Acquisition, Inc. and Quill Medical, Inc. dated May 25, 2006 ** | | Filed herewith |
| |
| 10.11 | | License Agreement between Public Health Service (through the Office of Technology Transfer, National Institutes of Health) and Angiotech Pharmaceuticals, Inc., dated November 19, 1997** | | Filed herewith |
| |
| 10.12 | | First Amendment between Public Health Service (through the Office of Technology Transfer, National Institutes of Health) and Angiotech Pharmaceuticals, Inc., dated March 28, 2002 modifying the License Agreement between Public Health Service (through the Office of Technology Transfer, National Institutes of Health) and Angiotech Pharmaceuticals, Inc., dated November 19, 1997** | | Filed herewith |
| |
| 10.13 | | Second Amendment between Public Health Service (through the Office of Technology Transfer, National Institutes of Health) and Angiotech Pharmaceuticals, Inc., dated May 23, 2007 modifying the License Agreement between Public Health Service (through the Office of Technology Transfer, National Institutes of Health) and Angiotech Pharmaceuticals, Inc., dated November 19, 1997** | | Filed herewith |
| |
| 10.14 | | License Agreement between Angiotech Pharmaceuticals, Inc. and The University of British Columbia, dated August 1, 1997** | | Filed herewith |
| |
| 10.15 | | Amendment to Licence Agreement between Angiotech Pharmaceuticals, Inc. and The University of British Columbia, dated February 27, 2004, modifying the License Agreement between Angiotech Pharmaceuticals, Inc. and The University of British Columbia, dated August 1, 1997** | | Filed herewith |
| | | | | |
| 10.16* | | Form of Indemnification Agreement for Officers – US | | Filed herewith |
| | | | | |
| 10.17* | | Form of Indemnification Agreement for Officers – Canada | | Filed herewith |
| | | | | |
| 10.18* | | Form of Indemnification Agreement for Directors | | Filed herewith |
| | | | | |
| 10.19* | | Executive Employment Agreement between Angiotech Pharmaceuticals, Inc. and William L. Hunter, dated April 23, 2004 | | Filed herewith |
| | | | | |
| 10.20* | | Executive Employment Agreement between Angiotech Pharmaceuticals, Inc. and K. Thomas Bailey, dated August 8, 2007 ** | | Filed herewith |
| | | | | |
| 10.21* | | Executive Employment Agreement between Angiotech Pharmaceuticals, Inc. and David D. McMasters, dated October 30, 2007 ** | | Filed herewith |
| | | | | |
| 10.22* | | Executive Employment Agreement between Angiotech Pharmaceuticals, Inc. and Rui Avelar, dated October 15, 2007 | | Filed herewith |
| | | | | |
| 10.23* | | Executive Employment Agreement between Angiotech Pharmaceuticals, Inc. and Jeffrey Walker, dated October 15, 2007 ** | | Filed herewith |
| | | | | |
| 10.24* | | Executive Employment Agreement between Angiotech Pharmaceuticals, Inc. and Chris J.W. Dennis, dated December 17, 2007 (as amended)** | | Filed herewith |
| | | | | |
| 10.25* | | Executive Employment Agreement between Angiotech Pharmaceuticals, Inc. and Gary Ingenito, dated November 26, 2007 ** | | Filed herewith |
| | | | | |
| 10.26* | | Angiotech Pharmaceuticals, Inc. 2001 Stock Option Plan | | Exhibit to Form S-8, filed September 27, 2001 |
| | | | | |
| 10.27* | | Angiotech Pharmaceuticals, Inc. 2004 Stock Option Plan | | Filed herewith |
| | | | | |
| 10.28* | | Angiotech Pharmaceuticals, Inc. 2006 Stock Option Plan | | Filed herewith |
| | | | | |
| 10.29* | | American Medical Instruments Holdings, Inc. 2003 Stock Option Plan | | Filed herewith |
| | | | | |
| 13 | | Disclosure incorporated by reference into Part II of this Annual Report on Form 10-K. | | Filed herewith |
| | | | | |
| 14.1 | | Code of Ethics for Chief Executive Officer | | Filed herewith |
| | | | | |
| 14.2 | | Code of Ethics for Chief Financial Officer | | Filed herewith |
| | | | | |
| 14.3 | | Code of Ethics for Directors and Officers | | Filed herewith |
| | | | | |
| 14.4 | | Guide of Standards and Business Conduct | | Exhibit 1, Form 6-K filed April 18, 2007 |
| | | | | |
| 21 | | List of Worldwide Subsidiaries | | Filed herewith |
| | | | | |
| 31.1 | | Certification of CEO Pursuant to Securities Exchange Act Rules 13a-14 and 15d-14 as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | | Filed herewith |
| |
| 31.2 | | Certification of CFO Pursuant to Securities Exchange Act Rules 13a-14 and 15d-14 as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | | Filed herewith |
| |
| 32.1 | | Certification of CEO and CFO Pursuant to 18 U.S.C. Section 1350 as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | | Filed herewith |
** portions of this exhibit have been omitted and filed separately with the Commission pursuant to a request for confidential treatment
- 62 -
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | |
| | | | |
| Angiotech Pharmaceuticals, Inc. | |
Date: March 16, 2009 | By: | /s/ K. Thomas Bailey |
| | K. Thomas Bailey Chief Financial Officer |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS that each person whose signature appears below constitute and appoint William L. Hunter, Chief Executive Officer, and K. Thomas Bailey, Chief Financial Officer, and each of them, the lawful attorney and agent or attorneys and agents with power and authority to do any and all acts and things and to execute all instruments which said attorneys and agents, or either of them, determine may be necessary or advisable or required to enable Angiotech Pharmaceuticals, Inc. to comply with the Securities Exchange Act of 1934, as amended, and any rules or regulations or requirements of the Securities and Exchange Commission in connection with this Annual Report on Form 10-K. Without limiting the generality of the foregoing power and authority, the powers granted include the power and authority to sign the names of the undersigned officers and directors in the capacities indicated below to this Annual Report on Form 10-K or amendments or supplements thereto, and ea ch of the undersigned hereby ratifies and confirms all that said attorneys and agents or either of them, shall do or cause to be done by virtue hereof. This Power of Attorney may be signed in several counterparts.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | | | |
| | | | |
Signatures | | Title | | Date |
| | | | |
/s/ William L. Hunter William L. Hunter, M.D. | | President, Chief Executive Officer and Director (Principal Executive Officer) | | March 16, 2009 |
| | | | |
/s/ K. Thomas Bailey K. Thomas Bailey | | Chief Financial Officer (Principal Financial Officer) | | March 16, 2009 |
| | | | |
/s/ Jay Dent Jay Dent | | Senior Vice President, Finance (Principal Accounting Officer) | | March 16, 2009 |
| | | | |
/s/ David Howard David Howard | | Chairman of the Board of Directors | | March 16, 2009 |
| | | | |
/s/ Laura Brege Laura Brege | | Director | | March 16, 2009 |
| | | | |
/s/ Edward Brown Edward Brown | | Director | | March 16, 2009 |
| | | | |
/s/ Hartley T. Richardson Hartley T. Richardson | | Director | | March 16, 2009 |
| | | | |
/s/ Arthur H. Willms Arthur Willms | | Director | | March 16, 2009 |
| | | | |
/s/ Henry A, McKinnell Jr. Henry A, McKinnell Jr. | | Director | | March 16, 2009 |
| | | | |
- 63 -