As Filed with the Securities and Exchange Commission on February 23, 2021
Registration No. 333-252454
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
AMENDMENT NO. 1
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
CANNABICS PHARMACEUTICALS LTD.
(Exact name of registrant as specified in its charter)
| Nevada | 20-3373669 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification Number) |
# 3 Bethesda Metro Center, Suite 700
Bethesda, MD 20814
(877) 424-2429
(Address, including zip code, and telephone number, including area code,
of registrant’s principal executive offices)
Law Offices of David E. Price
# 3 Bethesda Metro Center, Suite 700
Bethesda, MD 20814
(202) 536-5191
(Address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
SRK Law Offices
7 Oppenheimer St.
Rabin Science Park
Rehovot, Israel
Telephone No.: (011) (972) 8-936-0999
Facsimile No.: (011) (972) 8-936-6000
Approximate date of commencement of proposed sale to the public: As soon as practicable after this Registration Statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box. o
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer o | Accelerated filer o |
| Non-accelerated filer o | Smaller reporting company x |
| Emerging growth company o |
CALCULATION OF REGISTRATION FEE
Title of each class of securities to be registered | Amount to be registered (1) | Proposed maximum offering price per share (2) | Proposed maximum aggregate offering price (2) | Amount of registration fee | |||||||||
| Common stock, par value $0.0001 per share | 20,777,779 | $0.395 | $8,207,223 | $895.41 | |||||||||
| (1) | The amount is comprised of (i) 15,277,779 shares of the registrant’s common stock which represent 300% of the shares issuable upon the conversion of convertible notes in the aggregate amount of $1,375,000 at an exercise price of $0.27, and (ii) 5,500,000 shares of the registrant’s common stock issuable upon the exercise of a warrant. The convertible notes and warrant are held by the selling stockholder named in the prospectus contained herein and any supplements thereto. The registrant is not selling any shares of common stock in this offering and therefore will not receive any proceeds from this offering. Pursuant to Rule 416 under the Securities Act of 1933, as amended (the “Securities Act”), there is also being registered hereby such indeterminate number of shares of the registrant’s common stock as may be issued or issuable with respect to the shares being registered hereunder to prevent dilution by reason of any stock dividend, stock split, recapitalization, or other similar transaction. |
| (2) | Estimated solely for the purpose of calculating the amount of the registration fee pursuant to Rule 457(c) of the Securities Act. The proposed maximum offering price per share and proposed maximum aggregate offering price are based upon the average of the high ($0.45) and low ($0.34) sales prices of the registrant’s common stock on February 12, 2021, as reported on the OTCQB. It is not known how many shares will be sold under this registration statement nor at what price or prices such shares will be sold.
|
| (3) | Calculated pursuant to Rule 457(o) based on an estimate of the proposed maximum aggregate offering price. |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission acting pursuant to said Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. The selling stockholder may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and we and the selling stockholder are not soliciting offers to buy these securities in any state where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED _________________, 2021
PROSPECTUS

CANNABICS PHARMACEUTICALS, INC.
20,777,779 SHARES
COMMON STOCK
This prospectus relates to the resale by the selling stockholder named in this prospectus of up to (i) 15,279,779 shares of our common stock, $0.0001 par value per share (“Common Stock”) which represent 300% of the shares issuable upon the conversion of convertible notes in the aggregate amount of $1,375,000 (the “Notes”) at an exercise price of $0.27, and (ii) 5,500,000 shares of Common Stock issuable upon the exercise of a warrant (“Warrant”), acquired by the selling stockholder in a private placement transaction (the shares issuable upon conversion of the Notes and upon exercise of the Warrant, together with the Pre-Delivery Shares, the “Shares”).
The selling stockholder is the holder named in the table under the section entitled “Selling Stockholder” beginning on page 37 of this prospectus or named in a supplement to this prospectus. The description of the issuances of the securities to the Selling Stockholder is set forth below in the section entitled “Prospectus Summary” beginning on page 1 of this prospectus, where the private placement is described.
The selling stockholder may sell the Shares directly, or through underwriters, agents, brokers, or dealers designated from time to time, on terms to be determined at the time of sale. Underwriters, agents, brokers or dealers may receive discounts, commissions, or concessions from the selling stockholders, from the purchasers in connection with sales of the Shares, or from both. The prices at which the selling stockholder may sell the Shares will be determined by the prevailing market price for the Shares or in negotiated transactions. Additional information relating to the distribution of the Shares by the selling stockholder is set forth below in the section entitled “Plan of Distribution.” If underwriters or dealers are involved in the sale of any securities offered by this prospectus, their names, and any applicable purchase price, fee, commission or discount arrangement between or among them, will be set forth, or will be calculable from the information set forth, in a supplement to this prospectus.
We will not receive any of the proceeds from the sale of the Shares by the selling stockholder, although we may receive proceeds from the exercise of the Warrant for shares of our Common Stock. We are registering the Shares on behalf of the selling stockholder, and we will bear the cost of the registration of these Shares.
Our Common Stock is quoted on the OTC Markets OTCQB (“OTCQB”) under the symbol “CNBX.” On February 12, 2021, the last sale of our Common Stock as reported on the OTCQB was $0.38 per share.
Investing in our common stock involves substantial risks. See the section entitled “Risk Factors” beginning on page 10 for a discussion of information and factors that should be considered before investing in our securities.
Neither the Securities and Exchange Commission (the “SEC”) nor any other regulatory body has approved or disapproved of these securities or passed upon the accuracy or adequacy of this prospectus. Any representation to the contrary is a criminal offense.
The date of this prospectus is __________, 2021.
TABLE OF CONTENTS
You should rely only on the information contained or incorporated by reference in this prospectus and in any prospectus supplement hereto. We have not authorized, and the selling stockholder may not authorize, any other person to provide you with different information. If anyone provides you with different or inconsistent information, you should not rely on it. We are not, and the selling stockholder is not, making an offer to sell these securities in any jurisdiction where the offer or sale is not permitted. You should assume that the information appearing in this prospectus is accurate only as of the date on the front cover of this prospectus. Our business, financial condition, results of operations and prospectus may have changed since that date.
| i |
This prospectus is part of a registration statement on Form S-1 that we filed with the Securities and Exchange Commission, or SEC, on behalf of the selling stockholder, who is named in the table under the section entitled “Selling Stockholder” beginning on page 37 of this prospectus.
This prospectus provides you with a general description of the securities that the selling stockholder may offer. To the extent required, the number of shares of our Common Stock to be sold, the purchase price, the public offering price, the names of any agent or dealer, and any applicable commission or discount with respect to a particular offering by the selling stockholder may be set forth in an accompanying prospectus supplement. A prospectus supplement may also add, update, or change information contained in this prospectus. You should read both this prospectus and any prospectus supplement together with the additional information described in the section below entitled “Where You Can Find More Information.”
To the extent permitted by applicable law, rules, or regulations, we may add, update, or change the information contained in this prospectus by means of a prospectus supplement or post-effective amendments to the registration statement of which this prospectus forms a part, through filings we make with the SEC that are incorporated by reference into this prospectus or by another method as may then be permitted under applicable law, rules or regulations.
You should rely only on the information contained in this prospectus or any related prospectus supplement, including the content of all documents now or in the future incorporated by reference into the registration statement of which this prospectus forms a part.
PRIOR TO MAKING A DECISION ABOUT INVESTING IN OUR COMMON STOCK, YOU SHOULD CAREFULLY CONSIDER THE SPECIFIC RISKS CONTAINED IN THE SECTION ENTITLED “RISK FACTORS” IN THIS PROSPECTUS, AND ANY APPLICABLE PROSPECTUS SUPPLEMENT, TOGETHER WITH ALL OF THE OTHER INFORMATION CONTAINED IN THIS PROSPECTUS AND ANY PROSPECTUS SUPPLEMENT OR APPEARING IN THE REGISTRATION STATEMENT OF WHICH THIS PROSPECTUS FORMS A PART.
In this prospectus, we refer to Cannabics Pharmaceuticals, Inc. as “we,” “us,” “our,” the “Company” or “Cannabics.” References to “selling stockholder” refer to the holder of our Common Stock listed herein under “Selling Stockholder,” who may sell Shares from time to time as described in this prospectus. All trade names used in this prospectus are either our registered trademarks or trademarks of their respective holders.
| ii |
This prospectus relates to the distribution of securities of an entity that is involved in the cannabis industry. Cannabis is classified as an illegal Schedule 1 drug and illegal under the Controlled Substances Act, 21 U.S.C. § 811 (hereafter referred to as the “CSA”). On December 20, 2018, President Donald J. Trump signed into law the Agriculture Improvement Act of 2018, otherwise known as the “Farm Bill.” Prior to its passage, hemp, a member of the cannabis family, was classified as a Schedule 1 controlled substance and also was illegal under the CSA.
The Federal Agricultural Improvement Act of 2018 (the “2018 Farm Bill”) legalized hemp and cannabinoids extracted from hemp in the U.S., but such extracts remain subject to state laws and the regulation by other U.S. federal agencies, such as the Food and Drug Administration (“FDA”) and the U.S. Department of Agriculture (“USDA”). With the passage of the Farm Bill, hemp cultivation is broadly permitted. The Farm Bill explicitly allows the transfer of hemp-derived products across state lines for commercial or other purposes. It also puts no restrictions on the sale, transport, or possession of hemp-derived products, so long as those items are produced in a manner consistent with the law. The hemp plant and the marijuana plant are both part of the same cannabis genus of plant, except that hemp has not more than 0.3% dry weight content of delta-9-tetrahydrocannabinol (“THC”). The same plant, with a higher THC content, is marijuana, which is legal under certain state laws, but which is currently not legal under U.S. federal law. Despite the passage of the Farm Bill, many aspects of the cannabis industry, particularly those not defined as hemp within the Farm Bill, remain illegal under U.S. federal Law.
At this time, we do not manufacture, distribute, dispense, or possess any controlled substances, including cannabis or cannabis-based preparations, in the United States, and we are not engaged in any business that falls outside of what is permissible under the Farm Bill.
In the future, we may become involved in business activities that would fall outside of the Farm Bill, such as the research and development, growth, cultivation and/or processing of cannabis that are not covered under the Farm Bill. Currently, 33 states plus the District of Columbia and Guam, have laws and/or regulations that recognize, in one form or another, legitimate medical and adult uses for cannabis and consumer use of cannabis in connection with medical treatment or for recreational use. Many other states are considering similar legislation. Conversely, under the CSA, the policies and regulations of the federal government and its agencies are that cannabis has no medical benefit and a range of activities including cultivation and the personal use of cannabis is prohibited. Unless and until Congress amends the CSA with respect to cannabis, there is a risk that federal authorities may enforce current federal law against our licensees and distributors, who may be deemed to be producing, cultivating, dispensing and/or aiding or abetting the possession and distribution of cannabis in violation of federal law. Active enforcement of the current CSA on cannabis may thus directly and adversely affect our potential revenues and profits. The risk of strict enforcement of the CSA in light of Congressional activity, judicial holdings, and stated federal policy remains uncertain. See the sections entitled “Risk Factors” and “U.S. Government Regulation of Cannabis.”
| iii |
This summary highlights, and is qualified in its entirety by, the more detailed information and financial statements included elsewhere in this prospectus. This summary does not contain all of the information that may be important to you in making your investment decision. You should read this entire prospectus carefully, especially the “Risk Factors” section beginning on page 10 and our financial statements and the related notes appearing at the end of this prospectus, before deciding to invest in our common stock.
As used in this prospectus, unless the context otherwise requires, references to “we,” “us,” “our” the “Company” and “Cannabics” refer to Cannabics Pharmaceuticals, Inc.
Overview
The Company is a biopharmaceutical corporation specializing in the discovery, development and commercialization of novel cannabinoid-based products and innovative technologies for the treatment of cancer. We combine the power of our proprietary technologies with the expertise of our leading scientists to unlock the medicinal properties of cannabis and its diversity of bioactive compounds. We have conducted thousands of tests on biopsies and cell lines in order to identify the physiologic impact of cannabinoids on cell cycle and cell death. This scientific workflow has generated an ongoing stream of biological data through which we have accumulated in-depth knowledge of the various therapeutic effects of cannabinoids and identified cannabinoid ratios demonstrating anti-tumor potential. We believe that our cannabinoid research coupled with our proprietary technologies and intellectual property positions the Company to play an important role in the rapidly growing medical cannabis marketplace.
Our core technology is a continuously evolving bioinformatics platform that utilizes high-throughput screening technology, advanced data analytics, and proprietary methodologies to rapidly examine the physiologic impact of multiple cannabinoid compounds on tumor cells. This technology enables us to screen thousands of cannabinoid combinations weekly, generating multiple datasets on the anti-tumor properties of different cannabis cultivars, formulations and ratios. We conduct a broad range of preclinical research on cannabinoids through our bioinformatics platform, which informs the development of our product candidates.
Through our research and development activities, we are building a portfolio of intellectual property assets comprised of patents, proprietary technologies, and bioinformatics that have a variety of research, analytic and therapeutic applications.
Our business model is based on technology development and the out-licensing of our intellectual property to strategic partners and to global pharmaceutical and biotechnology companies, but always in accordance with the law of each applicable jurisdiction. Cannabics does not itself manufacture, distribute, dispense, or possess any controlled substances, including cannabis or cannabis-based preparations, in the United States.
U.S. Government Regulation of Cannabis
The United States federal government regulates drugs through the Controlled Substances Act (21 U.S.C. § 811), which places controlled substances, including cannabis, in a schedule. Cannabis is classified as a Schedule I drug, which is viewed as highly addictive and having no medical value. The United States Federal Drug Administration has not approved the sale of cannabis for any medical application. Doctors may not prescribe cannabis for medical use under federal law; however, they can recommend its use under the First Amendment. In 2010, the United States Veterans Affairs Department clarified that veterans using medicinal cannabis will not be denied services or other medications that are denied to those using illegal drugs.
As of October 23, 2019, 33 states, the District of Columbia and Guam have laws legalized cannabis in some form for either medicinal or recreational use governed by state specific laws and regulations.
| 1 |
These state laws are in conflict with the federal Controlled Substances Act, which makes cannabis use and possession illegal on a national level. However, on August 29, 2013, the U.S. Department of Justice issued a memorandum providing that where states and local governments enact laws authorizing cannabis-related use, and implement strong and effective regulatory and enforcement systems, the federal government will rely upon states and local enforcement agencies to address cannabis activity through the enforcement of their own state and local narcotics laws. The memorandum further stated that the U.S Justice Department’s limited investigative and prosecutorial resources will be focused on eight priorities to prevent unintended consequences of the state laws, including distribution of cannabis to minors, preventing the distribution of cannabis from states where it is legal to states where it is not, and preventing money laundering, violence and drugged driving.
On December 11, 2014, the U.S. Department of Justice issued another memorandum with regard to its position and enforcement protocol with regard to Indian Country, stating that the eight priorities in the previous federal memo would guide the United States Attorneys' cannabis enforcement efforts in Indian Country. On December 16, 2014, as a component of the federal spending bill, the Obama administration enacted regulations that prohibits the Department of Justice from using funds to prosecute state-based legal medical cannabis programs.
On January 4, 2018, Attorney General Jeff Sessions issued a memorandum for all United States Attorneys concerning cannabis enforcement. Mr. Sessions rescinded all previous prosecutorial guidance issued by the Department of Justice regarding cannabis, including the August 29, 2013 memorandum. Mr. Sessions stated that U.S. Attorneys must decide whether or not to pursue prosecution of cannabis activity based upon factors including: the seriousness of the crime, the deterrent effect of criminal prosecution, and the cumulative impact of particular crimes on the community. Mr. Sessions reiterated that the cultivation, distribution and possession of cannabis continues to be a crime under the U.S. Controlled Substances Act.
U.S. Government Regulation of Hemp
On December 20, 2018, President Donald J. Trump signed into law the Agriculture Improvement Act of 2018, otherwise known as the “Farm Bill”. Prior to its passage, hemp, a member of the cannabis family, was classified as a Schedule 1 controlled substance, and so illegal under the Controlled Substances Act. The hemp plant and the marijuana plant are both part of the same cannabis genus of plant, except that hemp has not more than 0.3% dry weight content of delta-9-tetrahydrocannabinol (“THC”). The same plant, with a higher THC content, is marijuana, which is legal under certain state laws, but which is currently not legal under U.S. federal law.
With the passage of the Farm Bill, hemp cultivation is broadly permitted. The Farm Bill explicitly allows the transfer of hemp-derived products across state lines for commercial or other purposes. It also puts no restrictions on the sale, transport, or possession of hemp-derived products, so long as those items are produced in a manner consistent with the law.
Additionally, there will be significant, shared state-federal regulatory power over hemp cultivation and production. Under Section 10113 of the Farm Bill, state departments of agriculture must consult with the state’s governor and chief law enforcement officer to devise a plan that must be submitted to the United States Department of Agriculture (“USDA”). A state’s plan to license and regulate hemp can only commence once the USDA approves that state’s plan. In states opting not to devise a hemp regulatory program, USDA will construct a regulatory program under which hemp cultivators in those states must apply for licenses and comply with a federally run program. This system of shared regulatory programming is similar to options states had in other policy areas such as health insurance marketplaces under Affordable Care Act, or workplace safety plans under Occupational Health and Safety Act—both of which had federally-run systems for states opting not to set up their own systems.
The Farm Bill outlines actions that are considered violations of federal hemp law (including such activities as cultivating without a license or producing hemp with more than 0.3 percent THC, the psychoactive agent in cannabis).
| 2 |
The 2018 Farm Bill extends the protections for hemp research and the conditions under which such research can and should be conducted. Further, Section 7501 of the Farm Bill extends hemp research by including hemp under the Critical Agricultural Materials Act. This provision recognizes the importance, diversity, and opportunity of the plant and the products that can be derived from it, but also recognizes that there is a still a lot to learn about hemp and its products from commercial and market perspectives.
About this Prospectus
This prospectus relates to the resale by the selling stockholder of up to 20,777,779 Shares, which are comprised of 15,277,779 shares of our Common Stock which represent 300% of the shares issuable upon the conversion of the Notes and 5,500,000 shares of our Common Stock issuable upon the exercise of the Warrant. An initial Note in the amount of $825,000 (the “Initial Note”) and the Warrant were issued at the initial closing of a private placement transaction held on December 21, 2020, pursuant to a Securities Purchase Agreement entered into with us on December 16, 2020 (the “Private Placement”). A second Note in the amount of $550,000 was issued on February 22, 2021 (the “Second Note”). The number of Shares being registered pursuant to this registration statement is based on a good faith estimate of the number of shares issuable to the selling stockholder predicated on the following facts and assumptions:
| Face value of the Initial Note and the Second Note: | $1,375,000 |
| Per share market price as of February 12, 2021: | $0.38 |
| Per share conversion price as of February 12, 2021: | $0.27 |
| Total number of shares underlying the Initial Note and the Second Note: | 5,092,593 |
| 300% of the number of shares issuable upon conversion: | 15,277,779 |
| Number of shares issuable upon exercise of the Warrant: | 5,500,000 |
| Total number of shares being registered: | 20,777,779 |
The conversion price for the Notes is equal to the lower of (a) $0.35 per share or (b) eighty percent (80%) of the average of the two lowest daily volume-weighted average price for the Company’s Common Stock during the ten (10) consecutive trading days preceding the conversion date.
The Private Placement
On December 16, 2020, we entered into a securities purchase agreement with an institutional investor to sell a new series of senior secured convertible notes in a three tranche private placement to the investor, with an aggregate principal amount of $2,750,000 having an aggregate original issue discount of 10%, and ranking senior to all outstanding and future indebtedness. Pursuant to the securities purchase agreement, one note (the “Initial Note”) in an aggregate original principal amount of $825,000 was issued to the investor in the first tranche closing on December 21, 2020. On February 22, 2021, we entered into an amendment and restated securities purchase agreement pursuant to which a second Note in an aggregate principal amount of $550,000 was issued (the “Second Note”). Both Notes were issued in reliance upon the exemption from securities registration afforded by Section 4(a)(2) of the Securities Act of 1933, and Rule 506(b) of Regulation D as promulgated by the United States Securities and Exchange Commission under the Securities Act of 1933, together with the issuance of the warrant to acquire our common stock, as described below. The Initial Note has a face amount of $825,000 for which we received cash proceeds of $750,000. The Second Note has a face amount of $550,000 for which we received cash proceeds of $500,000. Subject to the satisfaction of customary closing and equity conditions, at any time prior to December 31, 2021 (or such later date as the parties shall mutually agree), we have the right to require an additional closing of an additional note in the aggregate principal amount of $1,375,000 (the “Additional Note”, and together with the Initial Note and the Second Note, the “Notes”) on the twentieth trading day after the date this registration statement is declared effective by the Securities and Exchange Commission. The Convertible Notes are being sold with an original issue discount of 10% and do not bear interest except upon the occurrence of an event of default.
Our Business
We are a biopharmaceutical company that focuses on the discovery, development, and commercialization of innovative cannabinoid-based products and services for the treatment of cancer. We have developed a proprietary cannabinoid bioinformatics platform that we believe enables us to unlock the medicinal properties of cannabis and its diverse bioactive compounds to create personalized natural therapeutics that may be tailored to specific cancers and the genetics of individual patients for improved clinical outcomes.
The Company’s main focus is the development and marketing of various new and innovative anti-cancer and palliative products, therapies, and biotechnological tools aimed at treating cancer and providing relief from diverse ailments that respond to the active ingredients sourced from the cannabis plant. These advanced tools include innovative delivery systems for cannabinoids, personalized medicine therapies and procedures based on cannabis originated compounds, and bioinformatics tools.
| 3 |
Our core technology is a continuously evolving bioinformatics platform utilizing high throughput screening technology, advanced data analytics, and proprietary methodologies to rapidly examine the physiologic impact of multiple cannabinoid compounds on tumor cells. This technology enables us to screen thousands of compounds simultaneously to measure their therapeutic effects, generating mega-data with respect to the anti-tumor properties of different cannabis chemovars, formulations, and ratios. The data produced by our platform informs our development of novel cannabinoid preparations, drug candidates, and related technologies that may be optimized to treat cancers for which current treatment options are ineffective, costly, or cause unwanted side effects.
Our lead product candidate is RCC-33, an oral capsule for the treatment of colorectal cancer, or CRC. RCC-33 contains high concentrations of the cannabinoids CBDV and CBGA, which have demonstrated in our laboratory testing complex synergistic anti-tumor activity in vitro with minimal psychoactive effects. We are currently in the early planning stage of a clinical development pathway for RCC-33. We plan to conduct further preclinical studies to establish the safety and efficacy of RCC-33 in an in vivo murine model of colorectal cancer.
Cannabics SR is a lipid-based capsule containing a standardized formulation of cannabinoids that we are developing as a product candidate for the treatment of cancer anorexia-cachexia syndrome, or CACS. With a rapid onset of action and sustained effects for up to 6-8 hours, we believe that the convenience of once or twice daily oral dosing of Cannabics SR may improve quality of life and increase patient compliance with treatment regimens, leading to better health outcomes. A two-year pilot study of Cannabics SR led by Dr. Gil Bar-Sela of the Rambam Hospital Health Care Campus, Division of Oncology, in Haifa, Israel, demonstrated a clinically significant weight increase in CACS patients treated with Cannabics SR capsules (Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6785913/). In the second half of 2021, we intend to commence an additional pilot study in Israel to assess the pharmacokinetics and pharmacodynamics of Cannabics SR in humans. Data from the study will inform our clinical development plan for Cannabics SR.
Another product candidate we are developing is Cannabics CDx, a drug sensitivity test designed to provide innovative decision support to healthcare providers interested in personalizing cannabinoid-based cancer therapies. Cannabics CDx applies data analytics and high-content drug sensitivity screening integrated with our proprietary database to measure the effectiveness of cannabinoid compounds on a patient’s biopsy, suggest preferred alternatives, and alert healthcare providers to cannabinoids that may be contraindicated. We believe that Cannabics CDx will meet a significant unmet need of the growing population of cancer patients being treated with cannabis by enabling healthcare providers to more precisely tailor cannabinoid treatments to a patient’s cancer and clinical profile. We are currently seeking strategic partners for a clinical validation study expected to commence in 2022 to assess the sensitivity and specificity of Cannabics CDx with a view towards commercializing Cannabics CDx in Europe, the United States, and other territories.
Business Strategy
Our goal is to capitalize on our pioneering work in the field of plant-derived cannabinoid therapeutics to become a leading developer of personalized cannabinoid-based medicines and diagnostics. Our business model focuses on the research and development of anti-cancer drugs by leveraging the expertise of our multiplex database to provide innovative biopharmaceutical products that offer the potential for superior efficacy and safety over existing therapies. To achieve this goal, the key elements of our strategy include:
| · | Leveraging Our Bioinformatics Platform. | |
| · | In-House Pre-Clinical Research. | |
| · | Commercialization Partnerships. | |
| · | Strategic Out-Licensing. |
| 4 |
Corporate Information
The Company was originally incorporated as Thrust Energy Corp. on September 15, 2004, under the laws of the State of Nevada, for the purpose of acquiring oil and gas exploration properties and non-operating interests. In April 2011, the Company expanded its business to include the acquisition of mineral exploration rights. On May 5, 2011, the name of the Company was changed to American Mining Corporation. On April 25, 2014, Cannabics Inc., a Delaware corporation, acquired 99.1% voting control of the Company. On June 19, 2014, the Company changed its name to Cannabics Pharmaceuticals Inc., and redirected its business focus towards its current operations.
All of our research and development in Israel is conducted by our wholly-owned subsidiary, G.R.I.N. Ultra Ltd., which was incorporated under the laws of Israel on August 25, 2014.
On November 15, 2020, we established a majority-owned subsidiary, Digestix Bioscience, Inc., incorporated under the laws of Delaware, to engage in the development of medical devices and pharmaceutical compositions for the treatment of precancerous and early-stage neoplastic local tumors, and in particular, adenomatous colorectal polyps. The other shareholders of Digestix are Professor Eitan Scapa, Dr. Erez Scapa, and Gabriel Yariv. Mr. Yariv, who currently serves as Director and COO of Cannabics, also serves as interim Chairman and CEO of Digestix. We do not have any other subsidiaries.
Our principal executive offices are located at #3 Bethesda Metro Center, Suite 700, Bethesda, Maryland, 20814, and our telephone number is (877) 424-2429. Our website address is http://www.cannabics.com. The information on, or that can be accessed through, our website is not incorporated by reference into this prospectus and should not be considered to be a part of this prospectus. We have included our website address as an inactive textual reference only. Any website references (URL’s) in this Registration Statement are inactive textual references only and are not active hyperlinks.
| 5 |
The Offering
The following is a brief summary of some of the terms of the offering and is qualified in its entirety by reference to the more detailed information appearing elsewhere in this prospectus. For a more complete description of the terms of our common stock, see “Description of Our Capital Stock – Common Stock” on page 91.
| Issuer | Cannabics Pharmaceuticals, Inc. | |
| Common Stock outstanding prior to offering | 135,237,584 shares of Common Stock (1) | |
| Common Stock to be outstanding after the offering | 156,015,363 shares of Common Stock (2) | |
| Securities offered by us | Shares issuable upon the conversion of up to $1,375,000 in original principal amount of Notes, and upon exercise of the Warrant (3) | |
| Maturity of Notes | Unless earlier converted or redeemed, the Notes will mature on the one year anniversary of the date on which they are issued. | |
| Original Issue Discount; Default Interest | The Notes will be issued with a 10% original issue discount. The Notes shall not bear interest except upon the occurrence (and during the continuance) of an event of default. After the occurrence and during the continuance of an event of default, the notes will accrue interest at the rate of 18.0% per annum. | |
| Conversion Rights | All amounts due under the Notes are convertible at any time, in whole or in part, at the option of the holders into shares of our common stock at a conversion price equal to the lower of (i) an initial conversion price of $0.35 per share, which conversion price is subject to adjustment pursuant to the terms of the Notes, or (ii) eighty percent (80%) of the average of the two lowest daily volume-weighted average price for the Company’s Common Stock during the ten (10) consecutive trading days preceding the conversion date. The conversion price is subject to customary adjustments upon an event of default or upon any stock dividend, stock split, stock combination, reclassification, or similar transaction that proportionately decreases or increases the price of our shares of common stock. | |
| Events of Default | The Notes include standard customary events of default, subject to any cure periods set forth in the Notes, where applicable.
If an event of default occurs, a holder may force us to redeem, within a specified time as further described in the Notes (regardless of whether such event of default has been timely cured), all or any portion of its note at a price equal to 125% of the greater of (i) the conversion amount being redeemed and (ii) the market value of the shares of our common stock underlying such conversion amount being redeemed, as determined in accordance with the Notes. See “Description of Notes” for additional information. |
| 6 |
| Use of proceeds | We will not receive any proceeds from the sale of the Shares offered by the selling stockholder under this prospectus. However, we will receive up to $2,750,000 in the aggregate from the selling stockholder if it exercises in full, on a cash basis, all of its unexercised Warrant. We will use such proceeds from the exercise of the Warrant for working capital and other corporate purposes. | |
| Risk Factors | You should carefully read the “Risk Factors” beginning on page 10 and the other information included in this prospectus for a discussion of factors you should consider carefully before deciding to invest in our common stock. | |
| No Listing of Notes | We do not intend to apply for listing of the notes on any securities exchange. | |
| OTCQB Symbol | “CNBX” |
_______________
(1) As of February 21, 2021. Does not include the Pre-Delivery Shares.
(2) Assumes the full conversion of the Notes held by the selling stockholder to acquire 15,277,779 shares of our Common Stock, the full exercise of the unexercised Warrant held by the selling stockholder to acquire 5,500,000 shares of our Common Stock, and that all other outstanding warrants and options are not exercised. We pre-delivered 3,913,663 shares of our Common Stock of the conversion shares under the Notes on the issuance date of the Initial Note (the “Pre-Delivery Shares”).
(3) The Initial Note in the amount of $825,000 was issued on December 21, 2020, and the Second Note in the amount of $550,000 was issued on February 22, 2021.
The selling stockholder may sell the Shares subject to this prospectus from time to time and may also decide not to sell all the Shares they are allowed to sell under this prospectus. The selling stockholder will act independently of us in making decisions with respect to the timing, manner, and size of each sale. Furthermore, the selling stockholder may effectuate such transactions by selling the securities to or through broker-dealers, and such broker-dealers may receive compensation in the form of discounts, concessions or commissions from the selling stockholder and/or the purchasers of the securities for whom such broker-dealers may act as agent or to whom they sell as principal, or both. The selling stockholder may be deemed to be an “underwriter,” as defined in the Securities Act. If any broker-dealers are used by the selling stockholder, any commissions paid to broker-dealers and, if broker-dealers purchase the selling stockholder’s securities as principals, any profits received by such broker-dealers on the resale of the selling stockholder’s securities may be deemed to be underwriting discounts or commissions under the Securities Act. In addition, any profits realized by the selling stockholder may be deemed to be underwriting commissions.
All costs, expenses and fees in connection with the registration of the selling stockholder’s securities offered by selling stockholder will be borne by us. Brokerage commission, if any, attributable to the sale of the selling stockholders’ securities will be borne by the selling stockholder. Based on information provided to us by the selling stockholder, to our knowledge, the selling stockholders does not have an existing short position in our common stock as of the date of this prospectus.
See the section entitled “Plan of Distribution” beginning on page 38.
Our common stock is quoted on the OTCQB under the symbol “CNBX.”
| 7 |
Summary Financial Data
The following tables set forth a summary of our financial data as of, and for the periods ended on, the dates indicated. We have derived the summary statement of operations data for the years ended August 31, 2020 and 2019, and balance sheet data as of August 31, 2020 and August 31, 2019, from our audited financial statements appearing at the end of this prospectus. We have derived the summary statement of operations data for the quarter ended November 30, 2020, and balance sheet data as of November 30, 2020, from our unaudited financial statements appearing at the end of this prospectus. Our historical results are not necessarily indicative of the results that should be expected in the future. You should read the following summary financial data together with our financial statements and the related notes appearing at the end of this prospectus and the “Selected Financial Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” sections of this prospectus.
| November 30, | August 31, | August 31, | ||||||||||
| 2020 | 2020 | 2019 | ||||||||||
| Unaudited | Audited | Audited | ||||||||||
| ASSETS | ||||||||||||
| Current assets: | ||||||||||||
| Cash and cash equivalents | $ | 184,031 | $ | 777,611 | $ | 265,982 | ||||||
| Prepaid expenses and other receivables | 168,574 | 152,299 | 284,496 | |||||||||
| Held for trading Investments | – | – | 3,256,456 | |||||||||
| Current royalties | – | – | 500,000 | |||||||||
| Total current assets | 352,605 | 929,910 | 4,306,934 | |||||||||
| Available for sale Investment | 539,609 | 426,522 | 6,010,946 | |||||||||
| Long term royalties | – | – | 3,863,000 | |||||||||
| Equipment, net | 806,317 | 862,879 | 1,002,286 | |||||||||
| Total assets | $ | 1,698,531 | $ | 2,219,311 | $ | 15,183,166 | ||||||
| LIABILITIES AND STOCKHOLDERS' EQUITY | ||||||||||||
| Current liabilities: | ||||||||||||
| Accounts payable and accrued liabilities | $ | 219,042 | $ | 231,141 | $ | 215,229 | ||||||
| Due to a related party | 223,645 | 223,645 | 223,645 | |||||||||
| Total current liabilities | 442,687 | 454,786 | 438,874 | |||||||||
| Stockholders' equity (deficit): | ||||||||||||
| Preferred stock, $.0001 par value, 5,000,000 shares authorized no shares issued and outstanding. | – | – | – | |||||||||
| Common stock, $.0001 par value, 900,000,000 shares authorized, 135,237,584, 135,080,441 and 134,498,775 shares issued and outstanding at November 30, 2020, August 31, 2020 and August 31, 2019 respectively. | 13,524 | 13,508 | 13,450 | |||||||||
| Additional paid-in capital | 15,405,295 | 15,372,311 | 15,300,250 | |||||||||
| issuance of warrants | 2,784,387 | 2,784,387 | 2,784,387 | |||||||||
| Other comprehensive income | (2,661,324 | ) | (2,774,411 | ) | 2,810,013 | |||||||
| Accumulated deficit | (14,286,038 | ) | (13,631,271 | ) | (6,163,807 | ) | ||||||
| Total stockholders' equity (deficit) | 1,255,844 | 1,764,524 | 14,744,292 | |||||||||
| Total liabilities and stockholders' equity | $ | 1,698,531 | $ | 2,219,311 | $ | 15,183,166 | ||||||
| 8 |
Balance Sheet Data
Quarter Ended November 30, 2020 | Year Ended August 31, 2020 | Year Ended August 31, 2019 | ||||||||||
| Unaudited | Audited | Audited | ||||||||||
| Cash and cash equivalents | $ | 184,031 | $ | 777,611 | $ | 265,982 | ||||||
| Working capital (deficit) | $ | (90,082 | ) | $ | 475,123 | $ | 3,868,060 | |||||
| Total assets | $ | 1,698,531 | $ | 2,219,311 | $ | 15,183,166 | ||||||
| Total current liabilities | $ | 442,687 | $ | 454,787 | $ | 438,874 | ||||||
| Total long-term liabilities | $ | – | $ | – | $ | – | ||||||
| Shareholders’ equity | $ | 1,255,844 | $ | 1,764,524 | $ | 14,744,292 | ||||||
| Number of Ordinary Shares outstanding | 135,237,584 | 135,080,441 | 134,498,775 | |||||||||
| 9 |
An investment in our common stock involves a high degree of risk. Before investing in our common stock, you should consider carefully the specific risks and uncertainties detailed in this “Risk Factors” section, together with all of the other information contained in this prospectus. If any of these risks occurs, our business, results of operations and financial condition could be harmed, the price of our common stock could decline, and you may lose all or part of your investment. This prospectus also contains forward-looking statements that involve risks and uncertainties. See “Cautionary Statement Regarding Forward-Looking Statements.” Our actual results could differ materially and adversely from those anticipated in these forward-looking statements as a result of certain factors, including those set forth below.
Risks Related to Our Company and Business
1. Our independent auditors have expressed substantial doubt about our ability to continue operating as a going concern, which could prevent us from obtaining new financing on reasonable terms or at all.
Our independent registered public accountants have expressed substantial doubt about our ability to continue as a going concern. This opinion could materially limit our ability to raise additional funds by issuing new debt or equity securities or otherwise. If we fail to raise sufficient capital when needed, we will not be able to complete our proposed business. As a result we may have to liquidate our business and investors may lose their investments. Our ability to continue as a going concern is dependent upon our ability to successfully accomplish our plan of operations described in this prospectus, obtain financing and eventually attain profitable operations. Investors should consider our independent registered public accountant’s comments when deciding whether to invest in the Company.
2. We have not generated any significant revenue since our inception and we may never achieve profitability.
We are an early stage biotechnology company and have not generated any significant revenue since we commenced our present operations in April 2014. At the present time, Cannabics SR is the only product that we have commercialized. To date, we have financed our operations primarily through private placements of common stock, warrants, and direct equity investments. As we continue our research and development of cannabinoid-based treatments, our expenses are expected to increase significantly. Accordingly, we will need to generate significant revenue to achieve profitability. Even as we begin to commercialize our technologies, we expect our losses to continue as a result of ongoing research and development expenses. These losses, among other things, have had and will continue to have an adverse effect on our working capital, total assets and stockholders’ equity. Because of the numerous risks and uncertainties associated with product development and commercialization efforts, we are unable to predict at what stage the Company will become profitable. We may never become profitable. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. If we are unable to achieve and then maintain profitability, our business, financial condition and results of operations will be negatively affected and the market value of our common stock will decline.
3. Since we have a limited operating history in our business, it is difficult for potential investors to evaluate our business.
We commenced operations as a biotechnology company in April 2014, and therefore have a relatively short operating history upon which an evaluation of our future success or failure can objectively be made. Our business is a highly speculative undertaking and involves a substantial degree of risk. We have not demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by early-stage companies in new and rapidly evolving competitive fields, including under-capitalization, cash shortages, limitations with respect to personnel, financial, and other resources and lack of revenue. The likelihood of our success must be considered in light of the early stage of our operations. There is no assurance that our business will ever be successful or that we will be able to attain profitability. Any failure by the Company to report profits may adversely affect the price of our common stock.
| 10 |
4. We will need to raise additional capital to meet our business requirements in the future, which may be costly or difficult to obtain and could dilute our stockholders’ ownership interests.
The Company has not yet generated significant revenue and will require additional capital to continue its research and development activities, conduct clinical trials, commercialize its products and otherwise fund its operations. Our ability to secure required financing will depend in part upon investor perception of our ability to create a successful business. Capital market conditions and other factors beyond our control may also play important roles in our ability to raise capital. There can be no assurance that debt or equity financing will be available or sufficient for our requirements or for other corporate purposes, or if debt or equity financing is available, that it will be on terms acceptable to us. Moreover, future activities may require us to alter our capitalization significantly. Our inability to access sufficient capital for our operations could have a material adverse effect on our financial condition, results of operations and prospects. If we are unable to obtain additional funding as needed, we may be required to reduce the scope of our research and development activities, which could harm our business plan, financial condition and operating results, or we may be required to cease our operations entirely, in which case, our investors will lose all of their investment.
Any additional capital raised through the sale of equity or equity-backed securities may dilute our stockholders’ ownership percentages and could also result in a decrease in the market value of our equity securities. The terms of any securities issued by us in future capital transactions may be more favorable to new investors, and may include preferences, superior voting rights and the issuance of warrants or other derivative securities, which may have a further dilutive effect on the holders of our securities then outstanding. Any debt financing secured in the future could involve restrictive covenants relating to capital raising activities and other financial and operational matters, which may make it more difficult for us to obtain additional capital and to pursue business opportunities.
In addition, we may incur substantial costs in pursuing future capital financing, including investment banking fees, legal fees, accounting fees, securities law compliance fees, printing and distribution expenses and other costs. We may also be required to recognize non-cash expenses in connection with certain securities we issue, such as convertible notes and warrants, which may have an adverse impact on our financial condition.
5. We are highly dependent on the success of cannabinoid technology, and we may not be able to develop the technology, successfully obtain regulatory or marketing approval for, or successfully commercialize, our products or product candidates.
Our business is focused entirely upon the research, development and commercialization of cannabinoid-based technologies and products for the treatment of cancer. Our success is dependent upon the viability of this technology and the development of cancer therapies.
Neither we nor any other company has received regulatory approval from the United States Food and Drug Administration (the “FDA”) to market any therapeutics based on botanical cannabinoids, though the FDA has approved two drugs that contain a synthetic substance that acts similarly to cannabis compounds but is not present in the cannabis plant.
The scientific evidence underlying the feasibility of developing cannabinoid-based technologies for the treatment of cancer is both preliminary and limited. In 2017, an ad hoc committee of the National Academies of Sciences, Engineering, and Medicine determined that while there is conclusive or substantial evidence that oral cannabinoids are effective antiemetics in the treatment of chemotherapy-induced nausea and vomiting, there was insufficient evidence to make any statement about the efficacy of cannabinoids as a treatment for cancer. The ad hoc committee went on to state that further clinical research into the anti-cancer effects of cannabinoids needs to be conducted.
If our cannabinoid technology is found to be ineffective or unsafe in humans, or if it never receives regulatory approval for commercialization, we may never be able bring our product candidates to market and may never become profitable. Further, our current business strategy, including all of our research and development, is focused on utilizing cannabinoid technology to treat cancer. This lack of diversification increases the risk associated with the ownership of our common stock. If we are unsuccessful in developing and commercializing our cannabinoid-based technology and its application to the treatment of cancer, we may be required to alter our scope and direction and steer away from the intellectual property we have developed as well as the core capabilities of our management team and advisory board. Without successful commercialization of our products and product candidates, we may never become profitable, which would have a material adverse effect on our business, results of operations and financial condition.
| 11 |
6. Our success depends upon our ability to retain our senior management and our ability to attract, retain and motivate other qualified personnel.
We are an early stage biopharmaceutical company. As of the date of this prospectus, we had nine employees and several consultants. Our success materially depends upon the efforts of our management and other key personnel, including but not limited to Dr. Eyal Ballan, our Chief Technology Officer. If we lose the services of Dr. Ballan or any other executive officers or significant employees, our business would likely be materially and adversely affected. At this time, we do not currently have “key man” life insurance for Dr. Ballan or any other executive officer.
Because of the specialized scientific and managerial nature of our business, we rely heavily on our ability to attract and retain qualified scientific, technical and managerial personnel. The competition for qualified personnel in the biotechnology industry is intense. Due to this intense competition, we may be unable to continue to attract and retain qualified personnel necessary for the development of our business or to recruit suitable replacement personnel. Any difficulties in obtaining and retaining qualified officers, employees and consultants could have a material adverse effect on our operations.
7. The relative lack of public company experience by our management team may put us at a competitive disadvantage.
As a company with a class of securities registered under the United States Securities Exchange Act of 1934, as amended (the “Exchange Act”), we are subject to reporting and other legal, accounting, corporate governance, and regulatory requirements imposed by the Exchange Act and rules and regulations promulgated under the Exchange Act. With the exception of our CFO, Uri Ben-Or, our management team lacks significant public company experience, which could impair our ability to comply with these legal, accounting, and regulatory requirements. Such responsibilities include complying with federal securities laws and making required disclosures on a timely basis. Our senior management may not be able to implement and effect programs and policies in an effective and timely manner that adequately respond to such increased legal and regulatory compliance and reporting requirements. Our failure to do so could lead to the imposition of fines and penalties and further result in the deterioration of our business.
8. If we are unable to enter into acceptable sales, marketing and distribution arrangements with third parties or establish sales, marketing and distribution capabilities, we may not be successful in commercializing any product candidate that we develop if and when a product candidate is approved.
We do not have any sales, marketing or distribution infrastructure and have no experience in the commercialization of biotechnology. To achieve commercial success for any product, we must develop a sales and marketing organization, outsource these functions to third parties or license our products to others.
In the United States, we intend to commercialize our products solely by licensing the right to produce and distribute them to organizations having greater resources and experience than we do. There can be no assurance that our licensing efforts will be successful, or that we will be able to license any future products on satisfactory terms, or at all. We do not presently have any other agreement or arrangement for the commercialization of our products in the United States or elsewhere.
While we generally intend to adopt a licensing model for the commercialization of our products, we may also seek one or more strategic partners for commercialization of our products outside the United States. As a result of entering into arrangements with third parties to perform sales, marketing and distribution services, our product revenue or the profitability of our product revenue may be lower, perhaps substantially lower, than if we were to directly market and sell products in those markets. Furthermore, we may be unsuccessful in entering into the necessary arrangements with third parties or may be unable to do so on terms that are favorable to us. In addition, we may have little or no control over such third parties and any of them may fail to devote the necessary resources and attention to sell and market our product candidates effectively.
| 12 |
If we do not license our products or outsource our commercialization efforts, we will be required to develop our own sales, marketing and distribution capabilities, which will require substantial resources and will be time-consuming, and could delay any product launch. Moreover, we may not be able to hire or retain a sales force that is sufficient in size or has adequate expertise in the consumer health markets that we plan to target. If we are unable to establish or retain a sales force and marketing and distribution capabilities, our operating results may be adversely affected.
If we do not successfully license our products or establish sales and marketing capabilities, either on our own or in collaboration with third parties, it is likely that we will be unable to commercialize any of our products.
9. We face intense competition, often from companies with greater resources and experience than we have, which may result in others developing or commercializing competing products before us or more successfully.
The market for cancer therapies is intensely competitive, subject to rapid change and significantly affected by new product introductions and other market activities of industry participants. Our competitors include large multinational corporations and their operating units, including GW Pharms, Abbott Laboratories Inc., Cepheid Inc., Philips, GE Healthcare, Siemens, Gen-Probe Incorporated, MDxHealth SA, EpiGenomics AG, Roche Diagnostics, Exact Sciences Corporation, Sequenom, Inc. and several others. We also compete against pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide, as well as smaller and other early-stage companies. Other potential competitors include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
Many of our competitors and potential competitors have or will have substantially greater financial, technological, managerial and research and development resources and experience than we have, and many have been engaged in the biotechnology industry for a much longer time than we have. Many of our competitors spend significantly more funds on research, development, promotion and sale of new and existing products than we do, and may therefore be able to react more quickly to new or emerging technologies, shifting market conditions and regulatory changes.
There can be no assurance that any of our current or future products and technologies will have a competitive advantage in the marketplace, or that they will remain competitive following the introduction of competing products or technologies. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, more convenient or less expensive. There can be no assurance that we will be successful in the face of increasing competition from new technologies or products introduced by existing companies in the industry or by new companies entering the market.
If we are unable to compete successfully, there may be a material and adverse effect on our business, financial condition and results of operations.
10. If the marketplace does not accept the products in our development pipeline or any other products we might develop, we may be unable to generate sufficient revenue to sustain and grow our business.
Even if we are able to successfully develop and obtain regulatory approval of a product candidate, our ability to generate significant revenue will depend on the acceptance of our products by physicians and patients. Physicians, hospitals, clinical laboratories, researchers or others in the healthcare industry may not use our current or future product candidates unless they are determined to be an effective and cost-efficient means of treating cancer. Market acceptance of our current or future therapeutic products will depend on a number of factors, including the indication statement and warnings approved by regulatory authorities in the product label, continued demonstration of efficacy and safety in commercial use, physicians’ willingness to prescribe the product, reimbursement from third-party payers such as government healthcare systems and insurance companies, the price of the product, the nature of any post-approval risk management plans mandated by regulatory authorities, competition, marketing and distribution support. In addition, we will need to expend a significant amount of resources on marketing and educational efforts to create awareness of our products and to encourage their acceptance and adoption. If the market for our products does not develop sufficiently or the products are not accepted, our revenue potential will be harmed.
| 13 |
11. We do not presently have any product liability insurance coverage and there is no assurance that we will be able to obtain such insurance at an affordable price or that it will be sufficient to cover all liabilities that we may incur.
We are exposed to potential product liability risks that are inherent in the testing, manufacturing and marketing of cancer treatments, pharmaceuticals and dietary supplements. While we do not presently carry any product liability insurance coverage, we intend to obtain such insurance in amounts we believe to be commercially reasonable for our current level of activity and exposure. There is no assurance, however, that we will be able to obtain or maintain insurance coverage that will be adequate to cover our potential liabilities, or that premiums will be commercially justifiable. Furthermore, insurance that might otherwise be readily available, may be more difficult for us to find and more expensive because we work with medicinal cannabis. If we are the subject of a successful product liability claim that exceeds the limits of, or is not otherwise covered by our insurance, or if we incur such liability at a time when we are not able to obtain liability insurance, we may incur substantial charges that adversely affect our earnings and require the commitment of capital resources that might otherwise be available for the development and commercial launch of our product programs.
12. If we fail to protect our intellectual property rights, our ability to pursue the development of our technologies and products would be negatively affected.
Our success will depend in part on our ability to protect our intellectual property. This is done, in part, by obtaining patents and trademarks and then maintaining adequate protection of our technologies, tradenames and products. If we do not adequately protect our intellectual property, competitors may be able to use our technologies to produce and market products in direct competition with us and erode our competitive advantage. Some foreign countries lack rules and methods for defending intellectual property rights and do not protect proprietary rights to the same extent as the United States. Many companies have had difficulty protecting their proprietary rights in these foreign countries. We may not be able to prevent misappropriation of our proprietary rights.
We are currently seeking patent protection for several processes and finished products. However, the patent process is subject to numerous risks and uncertainties, and there can be no assurance that we will be successful in protecting our products by obtaining and defending patents. These risks and uncertainties include the following:
| · | patents that may be issued or licensed may be challenged, invalidated, or circumvented, or otherwise may not provide any competitive advantage; |
| · | our competitors, many of which have substantially greater resources than us and many of which have made significant investments in competing technologies, may seek, or may already have obtained, patents that will limit, interfere with, or eliminate our ability to make, use, and sell our products and product candidates either in the United States or in international markets; |
| · | there may be significant pressure on the United States government and other international governmental bodies to limit the scope of patent protection both inside and outside the United States for treatments that prove successful as a matter of public policy regarding worldwide health concerns; |
| · | countries other than the United States may have less restrictive patent laws than those upheld by United States courts, allowing foreign competitors the ability to exploit these laws to create, develop, and market competing products. |
| 14 |
Any patents issued to us may not provide us with meaningful protection, and third parties may challenge, circumvent or narrow them. Third parties may also independently develop products similar to our products or product candidates, duplicate our unpatented product or product candidates, and design around any patents on product candidates we may develop.
Additionally, extensive time is required for development, testing and regulatory review of product candidates. While extension of a patent term due to regulatory delays may be available, it is possible that before any of our product candidates can be commercialized, any related patent, even with an extension, may expire or remain in force for only a short period following commercialization, thereby reducing any advantages of the patent.
In addition, the United States Patent and Trademark Office (the “USPTO”), and patent offices in other jurisdictions have often required that patent applications concerning biotechnology-related inventions be limited or narrowed substantially to cover only the specific innovations exemplified in the patent application, thereby limiting the scope of protection against competitive challenges. Thus, even if we or our licensors are able to obtain patents, the patents may be substantially narrower than anticipated.
In addition to patents, we rely on a combination of trade secrets, confidentiality, nondisclosure and other contractual provisions, and security measures to protect our confidential and proprietary information. These measures may not adequately protect our trade secrets or other proprietary information. If they do not adequately protect our rights, third parties could use our technology, and we could lose any competitive advantage we may have. In addition, others may independently develop similar proprietary information or techniques or otherwise gain access to our trade secrets, which could impair any competitive advantage we may have.
13. Costly litigation may be necessary to protect our intellectual property rights and we may be subject to claims alleging the violation of the intellectual property rights of others.
We may face significant expense and liability as a result of litigation or other proceedings relating to patents and other intellectual property rights of others. If another party has also filed a patent application or been issued a patent relating to an invention or technology claimed by us in pending applications, we may be required to participate in an interference proceeding declared by the USPTO to determine priority of invention, which could result in substantial uncertainties and costs, even if the eventual outcome were favorable to us. We could also be required to participate in interference proceedings involving issued patents and pending applications of another entity. An adverse outcome in an interference proceeding could require us to cease using the technology or to license rights from prevailing third parties.
The cost to us of any patent litigation or other proceeding relating to our patents or patent applications, even if resolved in our favor, could be substantial. Our ability to enforce our patent protection could be limited by our financial resources, and may be subject to lengthy delays.
A third party might claim that we are using inventions claimed by their patents and might go to court to stop us from engaging in our normal operations and activities, such as research, development and the sale of any future products. Such lawsuits are expensive and would consume time and other resources. There is a risk that the court will decide that we are infringing the third party’s patents and will order us to stop the activities claimed by the patents, redesign our products or processes to avoid infringement or obtain licenses (which may not be available on commercially reasonable terms). In addition, there is a risk that a court will order us to pay the other party damages for having infringed their patents.
There is no guarantee that any prevailing patent owner would offer us a license so that we could continue to engage in activities claimed by the patent, or that such a license, if made available to us, could be acquired on commercially acceptable terms. In addition, third parties may, in the future, assert other intellectual property infringement claims against us with respect to our products, technologies or other matters.
| 15 |
14. Failure in our information technology or storage systems could significantly disrupt our operations and our research and development efforts, which could adversely impact our revenues, as well as our research, development and commercialization efforts.
Our ability to execute our business strategy depends, in part, on the continued and uninterrupted performance of our information technology (“IT”), systems, which support our operations and our research and development efforts, as well as our storage systems. Due to the sophisticated nature of the technology we use in our products and service offerings, we are substantially dependent on our IT systems. IT systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and back-up measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we have taken to prevent unanticipated problems that could affect our IT systems, sustained or repeated system failures that interrupt our ability to generate and maintain data, could adversely affect our ability to operate our business.
15. We will need to grow the size of our organization, and we may experience difficulties in managing any growth we may achieve.
As of the date of this prospectus, we have nine full-time employees. As our development and commercialization plans and strategies progress, we expect to need additional research, development, managerial, operational, sales, marketing, financial, accounting, legal and other resources. Future growth would impose significant added responsibilities on our management, which may not be able to accommodate those added responsibilities. If we fail to effectively manage our future growth, it could delay the execution of our business plan and disrupt our operations.
16. We are subject to financial reporting and other requirements that place significant demands on our resources.
We are subject to reporting and other obligations under the Securities Exchange Act of 1934, as amended, including the requirements of Section 404 of the Sarbanes-Oxley Act of 2002. Section 404 requires us to conduct an annual management assessment of the effectiveness of our internal controls over financial reporting. These reporting and other obligations place significant demands on our management, administrative, operational, internal audit and accounting resources. The costs of preparing and filing annual and quarterly reports, proxy statements and other information with the SEC and furnishing audit reports to stockholders causes our expenses to be higher than they would be if we had remained a privately-held company. The increased costs associated with operating as a public company may decrease our net income or increase our net loss, and may cause us to reduce costs in other areas of our business or increase the prices of our product to offset the effect of such increased costs. Additionally, if these requirements divert our management’s attention from other business concerns, they could have a material adverse effect on our business, financial condition and results of operations.
17. Our disclosure controls and procedures and internal controls over financial reporting were determined not to be effective for the prior fiscal year ended August 31, 2020, and may not be effective in future periods.
Effective internal controls are necessary for us to provide reasonable assurance with respect to our financial reports and to effectively prevent fraud. If we cannot provide reasonable assurance with respect to our financial reports and effectively prevent fraud, our reputation and operating results could be harmed. Pursuant to the Sarbanes-Oxley Act of 2002, we are required to furnish a report by management on internal control over financial reporting, including management’s assessment of the effectiveness of such control. Internal control over financial reporting may not prevent or detect misstatements because of its inherent limitations, including the possibility of human error, the circumvention or overriding of controls, or fraud. Therefore, even effective internal controls can provide only reasonable assurance with respect to the preparation and fair presentation of financial statements. In addition, projections of any evaluation of effectiveness of internal control over financial reporting to future periods are subject to the risk that the control may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. If we fail to maintain the adequacy of our internal controls, including any failure to implement required new or improved controls, or if we experience difficulties in their implementation, our business and operating results could be adversely impacted, we could fail to meet our reporting obligations, and our business and stock price could be adversely affected.
| 16 |
At August 31, 2020, our Chief Executive Officer and Chief Financial Officer evaluated the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) and concluded that, subject to the inherent limitations identified in Item 9A of Part II of our Annual Report on Form 10-K for the fiscal year ended August 31, 2019, our disclosure controls and procedures were not effective due to the existence of material weaknesses in our internal control over financial reporting arising from inadequate segregation of duties over authorization, review and recording of transactions, as well as the financial reporting of such transactions, the lack of an audit committee, insufficient documentation of review procedures and insufficient information technology procedures.
We believe we have taken appropriate and reasonable steps to make the necessary improvements to remediate these deficiencies, however we cannot be certain that our remediation efforts will ensure that our management designs, implements and maintains adequate controls over our financial processes and reporting in the future or that the changes made will be sufficient to address and eliminate the material weaknesses previously identified. Our inability to remedy any additional deficiencies or material weaknesses that may be identified in the future could, among other things, have a material adverse effect on our business, results of operations and financial condition, as well as impair our ability to meet our quarterly, annual and other reporting requirements under the Exchange Act in a timely manner, and require us to incur additional costs or to divert management resources.
18. An occurrence of an uncontrollable event such as the COVID-19 pandemic is likely to negatively affect, and has to date negatively affected, our operations.
The occurrence of an uncontrollable event such as the COVID-19 pandemic has negatively affected our operations. A pandemic typically results in social distancing, travel bans and quarantine, and the effects of, and response to, the COVID-19 pandemic has limited access to our facilities, management, support staff and professional advisors. These, in turn, have negatively impacted our operations, financial condition, and ability to advance our business. The full effect on our business and operations is currently unknown.
Risks Related to Cannabis
19. Our failure to comply with controlled substance legislation could restrict or harm our ability to develop and commercialize our products.
Our business is, and will be, subject to wide-ranging laws and regulations of Israel, the United States (federal and state), the European Community and other governments in each of the countries where we may develop and market our products. We must comply with all regulatory requirements in each jurisdiction if we expect to be successful.
Most countries are parties to the Single Convention on Narcotic Drugs of 1961 as amended by the 1972 Protocol, which governs international trade and domestic control of narcotic substances, including cannabis extracts. Countries may interpret and implement their treaty obligations in a way that creates a legal obstacle to us obtaining marketing approval in those countries for any cannabinoid-based products we develop. These countries may not be willing or able to amend or otherwise modify their laws and regulations to permit our products to be marketed, or achieving such amendments to the laws and regulations may take a prolonged period of time. In the case of countries with similar obstacles, we would be unable to market our product candidates in countries in the near future or perhaps at all if the laws and regulations in those countries do not change.
Any cannabinoid-based product candidate that we may develop for use in the United States, will be subject to U.S. controlled substance laws and regulations that will require us, along with our collaborators and licensees, to expend time, money and effort in all areas of regulatory compliance, including, if applicable, manufacturing, production, quality control and assurance and clinical trials. Any failure to comply with these laws and regulations, or the cost of compliance with these laws and regulations, could adversely affect the results of our business operations and our financial condition.
| 17 |
The constant evolution of laws and regulations affecting the research and development of cannabis-based therapies could detrimentally affect our business. Laws and regulations related to the therapeutic uses of cannabis are subject to changing interpretations. These changes may require us to incur substantial costs associated with legal and compliance fees and ultimately require us to alter our business plan. Furthermore, violations or alleged violation of these laws could disrupt our business and result in a material adverse effect on our operations, including our ability to conduct clinical trials that are prerequisite to our ability to commercialize our cannabis-based medical products and therapies. We cannot predict the nature of any future laws, regulations, interpretations or applications of laws and regulations and it is possible that new laws and regulations may be enacted in the future that will be directly applicable to our business.
20. Cannabis remains illegal under U.S. federal law, and any change in the enforcement priorities of the federal government could render our current and planned future operations in the U.S. unprofitable or even prohibit such operations.
We are a biopharmaceutical company focused on the research and development of cannabinoid-based treatments and palliative therapies. The commercial viability of our products and technologies in the United States depends, in part, on state laws and regulations; however, Cannabis remains illegal under federal law.
The United States federal government regulates drugs through the Controlled Substances Act, which places controlled substances, including cannabis, on one of five schedules. Cannabis is currently classified as a Schedule I controlled substance, which is viewed as having a high potential for abuse and no currently accepted medical use in treatment in the United States. No prescriptions may be written for Schedule I substances, and such substances are subject to production quotas imposed by the United States Drug Enforcement Administration (the “DEA”). Because of this, doctors may not prescribe cannabis for medical use under federal law, although they can recommend its use under the First Amendment.
Currently, thirty-three U.S. states and the District of Columbia allow the use of medical cannabis. Eight states and the District of Columbia also allow its recreational use. Because cannabis is a Schedule I controlled substance, however, the development of a legal cannabis industry under the laws of these states is in conflict with the Federal Controlled Substances Act, which makes cannabis use and possession illegal on a national level. The United States Supreme Court has confirmed that the federal government has the right to regulate and criminalize cannabis, including for medical purposes, and that federal law criminalizing the use of cannabis pre-empts state laws that legalize its use.
In 2014, the United States House of Representatives passed an amendment (the “Rohrabacher-Farr Amendment”) to the Commerce, Justice, Science, and Related Agencies Appropriations Bill, which funds the United States Department of Justice (the “DOJ”). The Rohrabacher-Farr Amendment prohibits the DOJ from using funds to prevent states with medical cannabis laws from implementing such laws. In August 2016, a Ninth Circuit federal appeals court ruled in United States v. McIntosh that the Rohrabacher-Farr Amendment bars the DOJ from spending funds on the prosecution of conduct that is allowed by state medical cannabis laws, provided that such conduct is in strict compliance with applicable state law. In March 2015, bipartisan legislation titled the Compassionate Access, Research Expansion, and Respect States Act (the “CARERS Act”) was introduced, proposing to allow states to regulate the medical use of cannabis by changing applicable federal law, including by reclassifying cannabis under the Controlled Substances Act to a Schedule II controlled substance and thereby changing the plant from a federally-criminalized substance to one that has recognized medical uses.
Although these developments have been met with a certain amount of optimism in the scientific community, the CARERS Act has not yet been adopted, and the Rohrabacher-Farr Amendment, being an amendment to an appropriations bill, must be renewed annually. The currently enacted Commerce, Justice, Science, and Related Agencies Act, which includes the Rohrabacher-Farr Amendment, is effective, by passage of a short-term continuing resolution, through April 28, 2017. The federal government could at any time change its enforcement priorities against the cannabis industry. We do not grow or distribute cannabis, but our current and planned business operations involve licensing cannabinoid-based products and technology. Any change in enforcement priorities could render such operations unprofitable or even prohibit such operations.
| 18 |
21. Our ability to earn revenue through licensing our product in the United States is dependent on additional states legalizing medical marijuana.
We are engaged in the business developing and commercializing cannabinoid-based products for the treatment of cancer. Our ability to commercialize our products in the United States is dependent upon the continued progress of legislative authorization of cannabis at the state level for medical purposes and, in certain states, based on the specifics of the legislation passed in that state. Any number of factors could slow or halt the progress. Furthermore, progress, while encouraging, is not assured. The legislative process normally encounters setbacks before achieving success. While there may be ample public support for legislative proposals, there must be political will in the legislative committee or a bill may never advance to a vote. Numerous factors impact the legislative process. Any one of these factors could slow or halt the progress and adoption of cannabis for medical purposes, which would limit the market for our products and negatively impact our business and revenues.
22. Changes in consumer preferences and acceptance of medical cannabis, or any negative trends, will adversely affect our business.
Our business is substantially dependent on market acceptance of medical cannabis. Market perception of medical cannabis can be significantly influenced by a number of social, political and economic factors that are beyond our control, including scientific research or findings, regulatory investigations, litigation, media attention and other publicity regarding such products and treatments. There can be no assurance that future scientific research, findings, regulatory proceedings, litigation, media attention or other research findings or publicity will be favorable to the market for any of our current or future cannabinoid-based therapies. Future research reports, findings, regulatory proceedings, litigation, media attention or other publicity that are perceived as less favorable than, or that question, earlier research reports, findings or publicity could have a material adverse effect on the demand for our products, as well as our business, results of operations, financial condition and cash flows.
We believe that as cannabis-based biotechnology becomes more widely accepted by the U.S. medical community and the public at large, the stigma associated with medical cannabis will moderate and, as a result, consumer demand will likely continue to grow. There is, however, no assurance that such increase in demand will occur, that we will benefit from any demand increase or that our business will ever become profitable. We cannot predict the future growth rate and size of the market, assuming that the regulatory climate permits, of which there can be no assurance. Any negative outlook on medical cannabis will adversely affect our business prospects.
We also believe that large, well-funded pharmaceutical and other related businesses and industries may have economic reasons to oppose cannabinoid-based therapies. The pharmaceutical industry is well-funded with a strong and experienced lobby presence at both the federal and state levels, as well as internationally, that surpasses financial resources of the current group of medical cannabis research and development companies. Any effort by the pharmaceutical lobby to halt or delay cannabinoid-based medical products and therapies could have a detrimental impact on our business.
Risks Related to Product Development
23. If we fail to successfully develop and commercialize pharmaceutical or therapies, we may be unable to execute our plan of operations.
Our current business strategy focuses on discovering, developing and commercializing cannabinoid-based cancer treatments and palliative therapies. To date, we have commercialized only Cannabics SR, our non-pharmaceutical sustained release capsules for palliative therapy. The success of our business will depend upon our ability to fully develop and commercialize the product candidates in our current development pipeline as well as to continue the discovery and development of other products and technology.
| 19 |
Prior to commercializing our product candidates, we will be required to undertake time-consuming and costly development activities with uncertain outcomes, including conducting clinical studies and obtaining regulatory clearance or approval in Israel, the United States, the European Union and other countries where we may develop and market our product candidates. Delays in obtaining approvals and clearances could have material adverse effects on us and our ability to fully carry out our plan of operations. We have limited experience in taking products through these processes and there are considerable risks involved in these activities. The science and methods that we are employing are innovative and complex, and it is possible that our development programs will ultimately not yield products suitable for commercialization or government approval. Product candidates that appear promising in early development may fail to be validated in subsequent studies, and even if we achieve positive results, we may still fail to obtain the necessary regulatory clearances or approvals. Few research and development projects result in commercial products, and perceived viability in early clinical studies often is not replicated in later studies. At any point, we may abandon development of a product candidate, or we may be required to expend considerable resources obtaining additional clinical and nonclinical data, which would adversely impact the timing for generating potential revenue from those products. Further, our ability to develop and launch product candidates is dependent on our receipt of substantial additional funding. If our discovery and development programs yield fewer commercial product candidates than we expect, we may be unable to execute our business plan, and our business, financial condition and results of operations may be adversely affected.
If we fail to maintain or establish satisfactory arrangements for the supply of raw materials or the manufacture of our product candidates for preclinical or clinical trials, or if we experience an interruption of supply, we might not have sufficient quantities of our product candidates at an acceptable cost, which could delay, prevent or impair our development or commercialization efforts
We do not ourselves produce medical cannabis, and therefore our ability to research, develop and commercialize our cannabinoid-based product candidates is dependent upon a sufficient supply of medical cannabis strains. Any significant interruption or negative change in the availability or economics of the supply chain for medical cannabis could materially impact our business, financial condition and operating results. Some strains of medical cannabis may only be available from a single supplier or a limited group of suppliers. If a sole source supplier were to go out of business, we might be unable to find a replacement source in a timely manner or at all. If a sole source supplier were to be acquired by a competitor, that competitor might elect not to supply us. Any inability to secure required supplies of medical cannabis or to do so on appropriate terms could have a materially adverse impact on our business, financial condition and operating results.
24. Our clinical claims may never be validated.
The FDA regulates the sale and distribution, in interstate commerce, of in vitro test and treatment kits, reagents and instruments used to perform testing and treatment. To the extent that any product we develop is regarded as an in vitro test rather than as a Laboratory Developed Test (“LDT”), we will be subject to increased FDA regulation that will delay and add to the cost of commercialization of our product candidates, which will have a material adverse effect on our business, results of operations and financial condition.
We are also subject to the United States Clinical Laboratory Improvement Amendments of 1988 (“CLIA”), federal regulatory standards that apply to all clinical laboratories that perform testing on specimens derived from humans in the United States for the purpose of providing information for the diagnosis, prevention or treatment of disease. CLIA is intended to ensure the quality and reliability of clinical laboratories in the United States by mandating specific standards in the areas of personnel qualifications, administration, and participation in proficiency testing, patient test management, quality control, quality assurance and inspections. Accreditation by the College of American Pathologists (“CAP”), one of six CLIA-approved accreditation organizations, is sufficient to satisfy the requirements of CLIA.
The validation for CLIA or CAP is a two-step process. The first step is optimization of all of the steps of the test protocol to show that the test is able to produce repeatable and consistent results. The second step is the clinical validation, in which statistically significant sensitivity and specificity of the test on the appropriate human samples are determined. Overall, the purpose of the validation process is to determine the accuracy, precision, sensitivity and specificity of the test. The time and cost to complete the validation process can vary widely, and it is possible that we would be unable to complete the validation process along the timeline and within the budget as planned.
| 20 |
As of the date of this prospectus, our clinical claims have not yet been validated for commercialization in a CLIA or CAP laboratory, and we have not yet begun the validation process. We may be unable to enter into an agreement with a CLIA or CAP laboratory on favorable terms, or at all. Although we may be able to validate the tests, they might have sensitivity and specificity that is insufficient to bring the product to market. Any delays or incurrence of greater costs than budgeted in validating these tests may have a material adverse effect on our business, results of operations and financial condition.
25. The Federal Food and Drug Administration may impose additional regulatory obligations and costs upon the development of our products.
On October 3, 2014, the FDA issued draft guidance regarding oversight of LDTs, titled “Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs).” According to this guidance, the FDA plans to take a phased-in risk-based approach to regulating LDTs. The FDA plans to phase in enforcement of LTD premarket review, quality system oversight and adverse event reporting over a number of years. The FDA would require that laboratories providing LDTs, subject to certain limited exemptions, within six months after the guidance documents are finalized to comply with (i) either a new notification procedure in which the laboratory must provide the FDA with certain basic information about each LDT offered by their laboratory or the FDA’s device registration and listing requirements, and (ii) the medical device reporting, or MDR, requirements for LDTs offered by that laboratory. Under this new risk-based approach, it is possible that some level of pre-market review may be required for our LDTs, which may require us to obtain additional clinical data.
The FDA draft guidance was subject to public comment until February 2, 2015. On January 13, 2017, the FDA issued a discussion paper on LDTs that does not represent the formal position of FDA and is not enforceable, but is intended to advance public discussion on future LDT oversight. At the present time, we cannot assess what the additional costs and regulatory burdens of any FDA final guidance or FDA enforcement will be, or the impact it may have on our business and operations.
If the FDA requires us to seek clearance or approval for any of our products (as opposed to simply licensing our technology to a CLIA lab), we may not be able to obtain such approvals on a timely basis, or at all. The cost of conducting clinical trials and otherwise developing data and information to support any applications may be significant. Failure to comply with applicable regulatory requirements of the FDA could result in enforcement action, including receiving untitled or warning letters, fines, injunctions, or civil or criminal penalties. In addition, we could be subject to a recall or seizure of products, operating restrictions, partial suspension or total shutdown of production. Any such enforcement action would have a material adverse effect on our business, financial condition and operations.
26. Changes in laws and regulations concerning clinical tests may adversely affect our business, financial condition and results of operations.
The clinical laboratory testing industry is highly regulated, and failure to comply with applicable regulatory, supervisory or licensing requirements may adversely affect our business, financial condition and results of operations. In particular, the laws and regulations governing the marketing and research of clinical testing are extremely complex, and in many instances there are no clear regulatory or judicial interpretations of these laws and regulations, which increase the risk that we may be found to be in violation of these laws.
The regulatory environment in which we operate may change significantly and adversely in the future. The molecular therapeutics industry as a whole is a growing industry and regulatory agencies such as the FDA may also apply heightened scrutiny to new developments in the field of molecular therapeutics. Should we be deemed to not be in compliance with regulatory requirements or any changes thereto, we may be subject to sanctions which could include required changes to our operations, adverse publicity, substantial financial penalties and criminal proceedings. Any change in the laws and the regulations relating to our business, whether in the form of new or amended laws or regulations or regulatory policies, or the application of any of the above, may adversely affect our business, financial condition and results of operations by increasing our costs to comply with the new laws or constraining our ability to develop, market and commercialize our products.
| 21 |
For example, a development affecting our industry is the increased enforcement of the federal False Claims Act and, in particular, actions brought pursuant to the False Claims Act's "whistleblower" or " qui tam " provisions. The False Claims Act imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal governmental payer program. The qui tam provisions of the False Claims Act allow a private individual to bring civil actions on behalf of the federal government for violations of the False Claims Act and permit such individuals to share in any amounts paid by the defendant to the government in fines or settlement. When an entity is determined to have violated the False Claims Act, it is subject to mandatory damages of three times the actual damages sustained by the government, plus mandatory civil penalties ranging from $5,500 to $11,000 for each false claim. In addition, various states have enacted false claim laws analogous to the federal False Claims Act, and in some cases go even further because many of these state laws apply where a claim is submitted to any third-party payer and not merely a governmental payer program.
In addition, there has been a recent trend of increased U.S. federal and state regulation of payments made to physicians, which are governed by laws and regulations including the Stark Law. Among other requirements, the Stark Law requires laboratories to track, and places a cap on, non-monetary compensation provided to referring physicians. While we have a compliance plan to address compliance with applicable fraud and abuse laws and regulations, the evolving commercial compliance environment and the need to build and maintain robust and expandable systems to comply with multiple jurisdictions with different compliance and reporting requirements increases the possibility that we could violate one or more of these requirements.
27. All of our product candidates are in clinical and preclinical development, the validation of which may not be successful and may be subject to delays, which would have a material adverse effect on our business, results of operation and financial condition.
To date, we have devoted our resources towards developing the technology upon which we are building our clinical product candidates. Our clinical claims have yet to be validated and our clinical product candidates are currently in a preclinical development phase. As of the date of this prospectus, only Cannabics SR, our non-pharmaceutical palliative therapy, has been commercialized.
We may be unable to successfully complete the clinical validation process for our product candidates due to several factors, including our ability to acquire enough samples for full validation and the procurement of materials necessary to conduct testing.
We may not be able to successfully complete the preclinical testing necessary to advance our therapeutic product candidates into clinical development, including animal pharmacology and toxicity studies. The results of any preclinical work may indicate that our product candidates do not have the safety or efficacy necessary to file an Investigational New Drug (“IND”) application with the FDA in order to move our product on to the clinical development process.
Once we initiate the clinical development of our product candidates, it may be difficult to identify and qualify patients to participate in future clinical trials for our product candidates, and the timing of our clinical trials depends on the speed at which we can recruit patients to participate in testing as well as completion of required follow-up periods. If patients are unwilling to participate in our clinical trials due to concerns over the safety of the product candidate or for other reasons, the timeline for conducting the trials and obtaining regulatory approval may be delayed. Furthermore, we may also compete for patients with other companies conducting similar clinical trials. Any delays in our future clinical trials could result in increased costs, delays in product development or termination of the clinical trials altogether.
Any of these events could have a material adverse effect on our business, results of operations and financial condition.
| 22 |
28. We may fail to demonstrate the safety and efficacy of our product candidates in accordance with regulatory standards and may incur delays and substantial costs in our clinical trials.
In order to commercialize our product candidates, we must conduct extensive clinical trials demonstrating the safety and efficacy of our product candidates in humans. The clinical testing process is expensive, difficult to design and implement, takes many years to complete and is unpredictable in both its duration and outcome. A failure of one or more clinical trials can occur at any stage of testing. There is a high failure rate for drugs and biological products proceeding through clinical trials. The research, testing, manufacturing, labeling, packaging, storage, approval, sale, marketing, advertising and promotion, pricing, export, import and distribution of drug products are subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries, which regulations differ from country to country. We are not permitted to market our product candidates as a prescription pharmaceutical product in the United States until we receive approval of a New Drug Application (“NDA”), from the FDA, or in any foreign countries until we receive the requisite approval from such countries. In the United States, the FDA generally requires the completion of pre-clinical testing and clinical trials of each drug to establish its safety and efficacy and extensive pharmaceutical development to ensure its quality before an NDA is approved. Regulatory authorities in other jurisdictions impose similar requirements. Of the large number of drugs in development, only a small percentage result in the submission of an NDA to the FDA and even fewer are eventually approved for commercialization. We have not submitted an NDA to the FDA or comparable applications to other regulatory authorities. Preclinical and clinical data is often susceptible to varying interpretations and types of analyses and regulatory authorities may fail to approve our product. In addition, even if we successfully complete early clinical trials, such results may not be indicative of the success or results of our later clinical trials.
Our successful completion of clinical trials may be materially adversely affected by many factors, including:
| · | ineffective trial design and disagreement with the FDA on final trial design; |
| · | imposition of a clinical hold following an inspection of our clinical trial operations by the FDA or other regulatory authorities; |
| · | difficulties or delays in reaching an agreement with a contract research organization, and clinical trial sites; |
| · | delays in obtaining required institutional review board approval for each trial site; |
| · | data collected from clinical trials may not be sufficient to support the submission of a NDA or other submission or to obtain regulatory approval in the United States or elsewhere; |
| · | delays or difficulties in recruiting suitable patients to participate in clinical trials; |
| · | delays in manufacturing or delivering products and materials to clinical trial sites; |
| · | delays or difficulties caused by lack of patient adherence to treatment or post-treatment follow-up; |
| · | delays caused by patients dropping out of a trial and the need for recruiting additional patients; and |
| · | delays caused by clinical sites dropping out of the trial and the time required to recruit a new site. |
| 23 |
Any of these delays or difficulties could cause us to be delayed in obtaining marketing approval from regulatory authorities, if at all, or allow us to obtain approval for specific indications or patient populations that are not as broad as currently targeted. In addition, such delays or difficulties may cause our development costs or our time to bring our product candidates to market to increase, may weaken our competitive positioning in the market and may have a material adverse effect on our business, results of operations and financial condition.
29. We cannot predict if or when we will receive regulatory approval to commercialize a product candidate.
We cannot commercialize a product candidate until the appropriate regulatory authorities, such as the FDA or a state regulating authority, have reviewed and approved the product candidate. Even if our product candidates demonstrate safety and efficacy in clinical trials, regulatory agencies may not complete their review processes in a timely manner, and we may not be able to obtain timely regulatory approval. We may never be able to receive regulatory approval for our product candidates at all. Additional delays may result if an FDA advisory committee or other regulatory authority recommends non-approval or restrictions on approval. In addition, we may experience delays or rejections based upon additional government regulation from future legislation or administrative action, or changes in regulatory agency policy during the period of product development, clinical trials and the review process. Regulatory agencies may also approve a product candidate for fewer or more limited indications than requested or may grant approval subject to the performance of post-marketing studies. In addition, regulatory agencies may not approve the labeling claims that are necessary or desirable for the successful commercialization of our product candidates. Delays or failure to obtain necessary regulatory approvals could have a material adverse effect on our business, results of operations and financial condition.
30. Even if we obtain regulatory approval for a product candidate, we will remain subject to extensive regulatory scrutiny.
Even if we obtain regulatory approval in the United States for our product candidates, the FDA and other appropriate regulatory agencies may still impose significant restrictions or delays, including restriction of patient population or indications or additional costly studies. Any changes to the approved product or its labeling or manufacturing process would require FDA approval. Any advertisements or promotions must comply with FDA regulations and are subject to FDA review as well as state and federal laws. Drug product manufacturers are subject to continual review and inspection by the FDA and other regulatory authorities to comply with Current Good Manufacturing Practice standards. If the FDA or other regulatory authority finds previously undiscovered compliance issues with products, such as unanticipated adverse effects or issues with the manufacturing facility, the FDA or other regulatory authority may:
| · | issue a warning letter asserting that we are in violation of law; |
| · | seek an injunction; |
| · | impose civil or criminal penalties or monetary fines; |
| · | suspend or withdraw regulatory approval; |
| · | suspend currently ongoing clinical trials; |
| · | refuse any pending applications; |
| · | seize product; or |
| · | prohibit us from entering into beneficial or necessary contracts such as supply or government contracts. |
| 24 |
Any government investigation of alleged violations of law could require us to expend significant time and resources in response, could result in litigation and litigation-related expense and could generate negative publicity. The occurrence of any event or penalty described above may inhibit our ability to commercialize our product candidates and generate revenue, which would have a material adverse effect on our business, results of operations and financial condition.
In addition, even if we obtain regulatory approvals, the timing or scope of any approvals may prohibit or reduce our ability to commercialize our product candidates successfully. For example, if the approval process takes too long, we may miss market opportunities and give other companies the ability to develop competing products or establish market dominance. Any regulatory approval that we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render our products not commercially viable. For example, regulatory authorities may approve our product candidates for fewer or more limited indications than we request, may not approve the price we intend to charge for our product candidates, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve our product candidates with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that indication. Further, the FDA may place conditions on approvals including potential requirements or risk management plans and the requirement for a Risk Evaluation and Mitigation Strategy (“REMS”) to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the NDA must submit a proposed REMS; the FDA will not approve the NDA without an approved REMS, if required. A REMS could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of our product candidates. Moreover, product approvals may be withdrawn for non-compliance with regulatory standards or if problems occur following the initial marketing of the product. Any of the foregoing scenarios could materially harm the commercial success of our product candidates and have a material adverse effect on our business, results of operations and financial condition.
31. We may fail to obtain orphan drug status for our product candidates.
We intend to seek orphan drug status from the FDA for those anti-cancer product candidates we are presently developing to the extent such product candidates are eligible for orphan drug status under the Orphan Drug Act of 1983. Orphan drug status gives the manufacturer specific financial incentives to develop a pharmacological agent. If a product that has an orphan drug designation receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications to market the same medication for the same indication, except in very limited circumstances, for seven years. Failure to obtain an orphan drug designation for our product candidates may have a material adverse effect on our business, results of operations and financial condition.
32. Any of our product candidates may cause adverse effects or have properties that could delay or prevent their regulatory approval or limit the scope of any specific indications or market acceptance.
Adverse events caused by our product candidates could cause interruptions, delays or the halting of our clinical trials. If adverse effects are observed in any clinical trials for our product candidates, we may be unable to obtain timely, or any, regulatory approval of our product candidates. Adverse effects caused by our product candidates could also subject us to litigation and liability, which could have a material adverse effect on our business, results of operations and financial condition.
In addition, if any of our product candidates are approved for commercialization and are found to cause serious or unpredicted side effects, serious consequences may result, including but not limited to, the withdrawal of marketing approval by regulatory authorities, restrictions on distribution by regulatory authorities, the need to conduct additional clinical trials, litigation and potential liability for personal injury to patients and damage to our reputation. Furthermore, our ability to achieve and maintain profitability may be permanently impaired. Any of these events could have a material adverse effect on our business, results of operations and financial condition.
| 25 |
33. Our products are subject to government regulation, both in the United States and internationally, which could increase our costs significantly and limit or prevent the sale of our dietary supplements.
The manufacture, packaging, labeling, advertising, promotion, distribution and sale of Cannabics SR, and any other cannabinoid-based products that we may develop and commercialize, are subject to regulation by numerous national and local governmental agencies in the United States and other countries, including the FDA and Federal Trade Commission in the United States, and the Ministry of Health in Israel. Failure to comply with these regulatory requirements may result in various types of penalties or fines. These include injunctions, product withdrawals, recalls, product seizures, fines and criminal prosecutions. Individual states also regulate dietary supplements. A U.S. state may interpret claims or products presumptively valid under federal law as illegal under that state’s regulations. In markets outside the United States, we will likely be required to obtain approvals, licenses, or certifications from a country’s ministry of health or comparable agency, as well as labeling and packaging regulations, all of which vary from country to country. Approvals or licensing may be conditioned on reformulation of products or may be unavailable with respect to certain products or product ingredients. Any of these government agencies, as well as legislative bodies, can change existing regulations, or impose new ones, or could take aggressive measures, causing or contributing to a variety of negative consequences, including:
| · | requirements for the reformulation of certain or all products to meet new standards; |
| · | the recall or discontinuance of certain or all products; |
| · | additional record keeping; |
| · | expanded documentation of the properties of certain or all products; |
| · | expanded or different labeling; |
| · | adverse event tracking and reporting; and |
| · | additional scientific substantiation. |
Any or all of these requirements could have a material adverse effect on us. There can be no assurance that the regulatory environment in which we operate will not change or that such regulatory environment, or any specific action taken against us, will not result in a material adverse effect on us.
34. Changes in legislation or regulation in the health care systems in the United States and foreign jurisdictions may affect us.
Our ability to successfully commercialize our cannabinoid-based products may depend on how the healthcare systems of the United States, the European Union and other governments provide coverage or reimbursement. Reimbursement and healthcare payment systems in international markets vary significantly by country, and include both government sponsored healthcare and private insurance. To obtain reimbursement or pricing approval in some countries, we may be required to produce clinical data, which may involve one or more clinical trials, that compares the cost-effectiveness of our products to other available therapies. We may not obtain international reimbursement or pricing approvals in a timely manner, if at all. Our failure to receive international reimbursement or pricing approvals would negatively impact market acceptance of our products in the international markets in which those approvals are sought.
| 26 |
We believe that future reimbursement may be subject to increased restrictions in the United States, the European Union and in other international markets. There is increasing pressure by governments worldwide to contain health care costs by limiting both the coverage and the level of reimbursement for therapeutic products and by refusing, in some cases, to provide any coverage for products that have not been approved by the relevant regulatory agency. Future legislation, regulation or reimbursement policies of third party payers may adversely affect the demand for our product candidates currently under development and limit our ability to sell our product candidates on a profitable basis. In addition, third party payers continually attempt to contain or reduce the costs of healthcare by challenging the prices charged for healthcare products and services. If reimbursement for our products is unavailable or limited in scope or amount or if pricing is set at unsatisfactory levels, market acceptance of our products candidates will be impaired and future revenues, if any, will be adversely affected.
Risks Related to Our Dependence on Third Parties
35. We rely and expect to continue to rely heavily on third parties to conduct our preclinical studies and clinical trials, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such studies and trials.
Although we have in-house research facilities, we do not have the resources to conduct clinical trial and must rely on third parties to conduct our clinical trials. We expect to continue to rely heavily on third parties, such as contract research organizations, clinical data management organizations, medical institutions, clinical investigators and others to conduct our clinical trials. Our agreements with these third parties generally allow the third party to terminate our agreement with them at any time. If we are required to enter into alternative arrangements because of any such termination, the introduction of our product candidates to market could be delayed.
Our reliance on third parties for research and development will reduce our control over such activities but will not relieve us of our responsibilities. Likewise, our reliance on third parties whom we do not control does not relieve us of our responsibility to comply with regulatory requirements to use current Good Clinical Practice (“GCP”) standards when conducting, recording and reporting the results of clinical trials in order to ensure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. We are also required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database of regulatory agencies within specified timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
The third parties on whom we rely may also have relationships with other entities, some of whom may be our competitors. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical trials in accordance with the requirements of a regulatory agency or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for our product candidates and will not be able to, or may be delayed in our efforts to, successfully commercialize our product candidates.
36. Collaboration agreements that we may enter into in the future may not be successful, which could adversely affect our ability to develop and commercialize our product candidates.
We may enter into collaboration agreements with pharmaceutical companies and biotechnology institutions and laboratories for the development or commercialization of our cannabinoid-based product candidates, which agreements may contain provisions based upon, among other things, the merits of retaining certain rights. We will face significant competition in seeking appropriate collaborators and in negotiating agreements at acceptable terms, if at all. We may not be successful in our efforts to enter, implement and maintain collaboration agreements. Disagreements stemming from collaboration agreements concerning development, intellectual property, regulatory or commercialization matters can lead to delays and, in some cases, termination of our collaboration agreements or otherwise result in the potentially significant costs and fees in seeking to enforce or protect our rights, if any. Any such disagreements can be difficult if, in fact, neither of the parties has final decision-making authority. The resulting outcome of any disputes or disagreements would in all likelihood adversely affect our business.
| 27 |
37. Data provided by collaborators and others upon which we rely that has not been independently verified could turn out to be false, misleading, or incomplete.
We rely on third-party vendors, scientists, and collaborators to provide us with significant data and other information related to our projects, clinical trials, and our business. If such third parties provide inaccurate, misleading, or incomplete data, our business, prospects, and results of operations could be materially adversely affected.
38. Our business model is substantially dependent on third party licensees to market and sell our products, which will subject us to a number of risks.
We depend on third party licensees to sell, market, and service our products and current and future products in our intended markets. We are subject to a number of risks associated with reliance upon third party licensees, including:
| · | lack of day-to-day control over the activities of licensees; |
| · | third party licensees may not commit the necessary resources to market and sell our current and future products to our level of expectations; |
| · | third party licensees may terminate their arrangements with us on limited or no notice or may change the terms of these arrangements in a manner unfavorable to us; and |
| · | disagreements with our future licensees could result in costly and time-consuming litigation or arbitration which we could be required to conduct in jurisdictions with which we are not familiar. |
If we fail to establish and maintain satisfactory relationships with our future third party licensees, our revenue and market share may not grow as anticipated, and we could be subject to unexpected costs which could harm our results of operations and financial condition.
Risks Related To Operating In Israel
39. Failure to secure the necessary Israeli licenses to use cannabis for medical research could limit our ability to execute our research and development activities, delay the launch of our products and adversely affect the results of our business operations.
To date, we have conducted our research only in Israel, and, in fact, have limited our operations to Israel. The biotechnologies that we are developing contain cannabis, a “controlled substance” as defined in the Israeli Dangerous Drugs Ordinance [New Version], 5733 - 1973. In Israel, licenses to cultivate, possess and to use cannabis for medical research are granted by the Ministry of Health, Israel Medical Cannabis Unit (“IMCU”), on an ad hoc basis. We have obtained all IMCU licenses that are necessary for us to carry out our research. Even though we have an established track record of successfully obtaining the requisite licenses as required, there can be no assurance that we will continue to be able to secure licenses in the future. If we fail to comply with Israeli rules and regulations related to the licensing of cannabis, we may not be able to research and develop our product candidates as we intend or at all.
| 28 |
40. We may become subject to claims for remuneration or royalties for assigned service invention rights by our employees, which could result in litigation and adversely affect our business.
A significant portion of our intellectual property has been developed by our Israeli employees in the course of their employment for us. Under the Israeli Patent Law, 5727-1967 (the “Israeli Patent Law”), inventions conceived of by an employee during the term and as part of the scope of his or her employment with a company are regarded as “service inventions,” which belong to the employer, absent a specific agreement between the employee and employer giving the employee service invention rights. The Israeli Patent Law also provides that if there is no such agreement between an employer and an employee, the Israeli Compensation and Royalties Committee (the “C&R Committee”), a body constituted under the Israeli Patent Law, shall determine whether the employee is entitled to remuneration for his or her inventions. The C&R Committee (decisions of which have been upheld by the Israeli Supreme Court) has held that employees may be entitled to remuneration for their service inventions despite having specifically waived any such rights. Further, the C&R Committee has not yet set specific guidelines regarding the method for calculating this remuneration or the criteria or circumstances under which an employee’s waiver of his or her right to remuneration will be disregarded. We generally enter into intellectual property assignment agreements with our employees pursuant to which such employees assign to us all rights to any inventions created in the scope of their employment or engagement with us. Although our employees have agreed to assign to us service invention rights and have specifically waived their right to receive any special remuneration for such assignment beyond their regular salary and benefits, we may face claims demanding remuneration in consideration for assigned inventions. As a consequence of such claims, we could be required to pay additional remuneration or royalties to our current or former employees, or be forced to litigate such claims, which could negatively affect our business.
41. We expect that our results of operations will be subject to fluctuations in currency exchange rates because a substantial portion of our anticipated revenue will be generated in U.S. dollars and Euros while a significant portion of our expenses will be incurred in New Israeli Shekels.
We expect a substantial portion of our revenue will be generated in U.S. dollars and Euros, while a significant portion of our expenses, principally salaries and related personnel expenses, is paid in New Israeli Shekels, or NIS. As a result, we are exposed to the risk that the rate of inflation in Israel will exceed the rate of devaluation of the NIS in relation to the Euro or the U.S. dollar, or that the timing of this devaluation will lag behind inflation in Israel. Because inflation has the effect of increasing the U.S. dollar and Euro costs of our operations, it would therefore have an adverse effect on our dollar-measured results of operations. The value of the NIS, against the Euro, the U.S. dollar, and other currencies may fluctuate and is affected by, among other things, changes in Israel’s political and economic conditions. Any significant revaluation of the NIS may materially and adversely affect our cash flows, revenues and financial condition. Fluctuations in the NIS exchange rate, or even the appearance of instability in such exchange rate, could adversely affect our ability to operate our business.
42. We may not be able to enforce covenants not-to-compete under current Israeli law.
We have non-competition agreements with most of our employees, all of which are governed by Israeli law. These agreements generally prohibit our employees from competing with us or working for our competitors for a specified period following termination of their employment. However, Israeli courts are reluctant to enforce non-compete undertakings of former employees and tend, if at all, to enforce those provisions for relatively brief periods of time in restricted geographical areas and only when the employee has unique value specific to that employer’s business and not just regarding the professional development of the employee. Any such inability to enforce non-compete covenants may cause us to lose any competitive advantage arising from confidential information known to such former employees.
| 29 |
43. It may be difficult for investors in the United States to enforce any judgments obtained against us or some of our directors or officers.
The majority of our assets are located outside the United States. In addition, certain of our officers are nationals or residents of countries other than the United States, and all or a substantial portion of such persons’ assets are located outside the United States. As a result, it may be difficult for investors to enforce within the United States any judgments obtained against us or any of our non-U.S. officers, including judgments predicated upon the civil liability provisions of the securities laws of the United States or any state thereof. It may also be difficult to assert claims under United States securities law in actions originally instituted outside of the United States. Moreover, Israeli courts may refuse to hear a United States securities law claim because Israeli courts may not be the most appropriate forums in which to bring such a claim. Even if an Israeli court agrees to hear a claim, it may determine that Israeli law, and not U.S. law, is applicable to the claim. Further, if U.S. law is found to be applicable, certain content of applicable U.S. law must be proved as a fact, which can be a time-consuming and costly process, and certain matters of procedure would still be governed by Israeli law. Consequently, our investors may be effectively prevented from pursuing remedies under U.S. federal and state securities laws against us or any of our non-U.S. directors or officers.
44. If there are significant shifts in the political, economic and military conditions in Israel and its neighbors, it could have a material adverse effect on our business relationships and profitability.
All of our research facilities and certain of our key personnel are located in Israel. Our business is directly affected by the political, economic and military conditions in Israel and its neighbors. Since the establishment of the State of Israel in 1948, a number of armed conflicts have occurred between Israel and its Arab neighbors. A state of hostility, varying in degree and intensity, has caused security and economic problems in Israel. Although Israel has entered into peace treaties with Egypt and Jordan, and various agreements with the Palestinian Authority, there has been a marked increase in violence, civil unrest and hostility, including armed clashes, between the State of Israel and the Palestinians since September 2000. The establishment in 2006 of a government in the Gaza Strip by representatives of the Hamas militant group has created heightened unrest and uncertainty in the region. In mid-2006, Israel engaged in an armed conflict with Hezbollah, a Shiite Islamist militia group based in Lebanon, and in June 2007, there was an escalation in violence in the Gaza Strip. From December 2008 through January 2009 and again in November and December 2012, Israel engaged in an armed conflict with Hamas, which involved missile strikes against civilian targets in various parts of Israel and negatively affected business conditions in Israel. In July 2014, Israel launched an additional operation against Hamas operatives in the Gaza strip in response to Palestinian groups launching rockets at Israel. Recent political uprisings and social unrest in Syria are affecting its political stability, which has led to the deterioration of the political relationship between Syria and Israel and have raised new concerns regarding security in the region and the potential for armed conflict. Similar civil unrest and political turbulence is currently ongoing in many countries in the region. The continued political instability and hostilities between Israel and its neighbors and any future armed conflict, terrorist activity or political instability in the region could adversely affect our operations in Israel and adversely affect the market price of our shares of common stock. In addition, several countries restrict doing business with Israel and Israeli companies have been and are today subjected to economic boycotts. The interruption or curtailment of trade between Israel and its present trading partners could adversely affect our business, financial condition and results of operations.
Risks Related to Our Stock
45. There can be no assurance of an active, liquid and orderly trading market for our common stock or that investors will be able to sell their shares of common stock.
At present, our common stock is quoted on the OTCQB tier of the marketplace maintained by OTC Markets Group Inc., under the symbol “CNBX.” There is only a limited, liquid public trading market for our common stock. There can be no assurance that a liquid market for our common stock will continue. Market liquidity will depend on the perception of our business and any steps that our management might take to bring public awareness of our business to the investing public within the parameters of the federal securities laws. There is no assurance that any such awareness will be generated or sustained. Therefore, investors may not be able to liquidate their investment or liquidate it at a price paid by investors equal to or greater than their initial investment in our common stock. Moreover, holders of our common stock may not find purchasers for their shares should they to decide to sell the common stock held by them at any particular time if ever. Our common stock should be purchased only by investors who have no immediate need for liquidity in their investment and who can hold our common stock, possibly for a prolonged period of time.
| 30 |
46. The price of our common stock is volatile, and the value of your investment could decline.
The market price of our common stock has been highly volatile. Between September 1, 2019, and August 31, 2020, the sales price of our stock on the OTCQB ranged from a low of $0.08 per share to a high of $0.57 per share. Accordingly, it is difficult to forecast the future performance of our common stock. The market price of our common stock may be higher or lower than the price you pay, depending on many factors, some of which are beyond our control and may not be related to our operating performance. These fluctuations could cause you to lose all or part of your investment in our common stock. Factors that could cause fluctuations in the trading price of our common stock include the following:
| · | technological innovations or new products and services by us or our competitors; |
| · | regulatory developments at the federal, state or local level; |
| · | additions or departures of key personnel; |
| · | our ability to execute our business plan; |
| · | operating results that fall below expectations; |
| · | loss of any strategic relationship; |
| · | industry developments; |
| · | economic, political and other external factors; and |
| · | period-to-period fluctuations in our financial results. |
The stock market generally and in particular, the market for stocks of biotechnology companies with lower market capitalizations, like us, have from time to time experienced, and likely will again experience significant price and volume fluctuations that are unrelated to the operating performance of a particular company. The trading price of our common stock might decline in reaction to events that affect other companies in our industry, even if these events do not directly affect us.
Periods of volatility in the market price of a company’s securities have often been followed by securities class action litigation against that company. If our stock price continues to be volatile, we may become the target of securities litigation, which could result in substantial costs and divert our management’s attention and resources from our business. This could have a material adverse effect on our business, operating results and financial condition.
47. If we continue to experience losses, we could experience difficulty meeting our business plan and our stock price could be negatively affected.
If we are unable to earn revenues from our products, we will experience continuing operating losses and negative cash flow from our operations. Any failure to achieve or maintain profitability could negatively impact the market price of our common stock. We anticipate that we will continue to incur product development, sales and marketing and administrative expenses. As a result, we will need to generate significant quarterly revenues if we are to achieve and maintain profitability. A substantial failure to achieve profitability could make it difficult or impossible for us to grow our business. Our business strategy may not be successful, and we may not generate significant revenues or achieve profitability. Any failure to significantly increase revenues would also harm our ability to achieve and maintain profitability. If we do achieve profitability in the future, we may not be able to sustain or increase profitability on a quarterly or annual basis.
| 31 |
48. We may never pay any dividends to our shareholders.
We currently intend to retain any future earnings for use in the operation and expansion of our business. Accordingly, we do not expect to pay any dividends in the foreseeable future, but will review this policy as circumstances dictate. The declaration and payment of all future dividends, if any, will be at the sole discretion of our board of directors, which retains the right to change our dividend policy at any time. Consequently, our stockholders must rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize any future gains on their investment.
49. Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
As at December 21, 2020, Cannabics Inc., a Delaware corporation, owns approximately 65% of our common stock. Both of our Directors are also holders of shares in Cannabics Inc., and therefore have influence over it. Accordingly, the Company (and our management) may be able to control the outcome of stockholder votes, including votes concerning the election of directors, amendment of our organizational documents, approval of mergers, sales of assets and other significant corporate transactions. This concentration of ownership in Cannabics Inc. (and our management) may have the effect of delaying or preventing a change in our management and voting control of Cannabics Inc., including preventing or discouraging unsolicited acquisition proposals or offers for our common stock that some of our stockholders may believe is in their best interest.
50. We may issue shares of preferred stock with greater rights than our common stock, which may entrench management and result in dilution of our stockholders' investment.
Our Articles of Incorporation authorize the issuance of up to 100 million shares of preferred stock, par value $0.0001 per share. The authorized but unissued preferred stock may be issued by our board of directors from time to time on any number of occasions, without stockholder approval, as one or more separate series of shares comprised of any number of the authorized but unissued shares of preferred stock, designated by resolution of our board of directors stating the name and number of shares of each series and setting forth separately for such series the relative rights, privileges and preferences thereof, including, if any, the: (i) rate of dividends payable thereon; (ii) price, terms and conditions of redemption; (iii) voluntary and involuntary liquidation preferences; (iv) provisions of a sinking fund for redemption or repurchase; (v) terms of conversion to common stock, including conversion price, and (vi) voting rights. Such preferred stock may enable our board of directors to hinder or discourage any attempt to gain control of the Company by a merger, tender offer at a control premium price, proxy contest or otherwise. Consequently, the preferred stock could entrench our management. The market price of our common stock could be depressed by the existence of the preferred stock.
51. Nevada law and certain provisions of our Articles of Incorporation and bylaws may discourage mergers and other transactions.
Provisions of Nevada law, such as its business combination statute, and certain provisions of our Articles of Incorporation and by-laws could make it more difficult for someone to acquire control of the Company and limit the price that certain investors might be willing to pay for shares of our common stock. These provisions may make it more difficult for stockholders to take certain corporate actions and could delay or prevent someone from acquiring our business. The provisions could be beneficial to our management and the board of directors in a hostile tender offer, and could have an adverse impact on stockholders who might want to participate in such tender offer, or who might want to replace some or all of the members of the board of directors.
| 32 |
52. Our common stock may be subject to penny stock rules, which may make it more difficult for our investors to sell their common stock.
Our common stock is presently considered to be a "penny stock" and is subject to SEC rules and regulations that impose limitations upon the manner in which such shares may be publicly traded, and regulate broker-dealer practices in connection with transactions in "penny stocks." Penny stocks generally are equity securities with a price of less than $5.00 (other than securities registered on certain national securities exchanges or quoted on the NASDAQ system, provided that current price and volume information with respect to transactions in such securities is provided by the exchange or system). The penny stock rules require a broker-dealer, prior to a transaction in a penny stock not otherwise exempt from the rules, to deliver a standardized risk disclosure document that provides information about penny stocks and the risks in the penny stock market. The broker-dealer must also provide the customer with current bid and offer quotations for the penny stock, the compensation of the broker-dealer and its salesperson in the transaction, and monthly account statements showing the market value of each penny stock held in the customer's account. In addition, the penny stock rules generally require that prior to a transaction in a penny stock, the broker-dealer make a special written determination that the penny stock is a suitable investment for the purchaser and receive the purchaser's written agreement to the transaction. These disclosure requirements may have the effect of reducing the level of trading activity in the secondary market for a stock that becomes subject to the penny stock rules which may increase the difficulty investors may experience in attempting to liquidate such securities. These requirements could also hamper our ability to raise funds in the primary market for our shares of common stock.
53. You may experience additional dilution upon the exercise of outstanding warrants.
In addition to the shares underlying the warrants that are being registered pursuant to this prospectus, as of December 21, 2020, we have 5,000,000 shares of Common Stock issuable upon exercise of outstanding warrants. If the holders of the warrants exercise their rights, you may experience additional dilution in the net tangible book value of your Common Stock.
54. You may experience dilution of your ownership interest due to the issuance of additional shares of Common Stock upon the conversion of our convertible debt, especially since our convertible debt has fluctuating conversion rates that are set at a discount to market prices of our shares of Common Stock during the period immediately preceding conversion.
We have raised approximately $1,375,000 in financing through the issuance of the Initial Note and the Second Note, and intend to obtain additional convertible debt financing of up to an aggregate of $2,750,000. The lenders may choose to convert their loans, which are convertible into shares of Common Stock of the Company at a conversion price equal to the lower of (i) $0.35 or (ii) 80% of the average of the two lowest closing bid price of the Company’s shares of Common Stock in the ten trading days prior to the conversion. This could result in material dilution to existing shareholders of the Company. Because the conversion price is based upon the trading prices of our shares at the time of conversion, the number of shares into which the convertible debt may be converted may increase without an upper bound. If the trading prices of our shares are low when the conversion price of the convertible debt is determined, we would be required to issue a greater number of shares to the converting debtholder, which could cause substantial dilution to our shareholders. In addition, if any or all of the holders of our convertible debt convert and then sell our common stock, this could result in an imbalance of supply and demand for our common stock and reduce our stock price. The further our stock price declines, the further the adjustment of the conversion price will fall and the greater the number of shares we will have to issue upon conversion, resulting in further dilution to our shareholders. Because a market price-based conversion formula can lead to dramatic stock price reductions and corresponding negative effects on both a company and its shareholders, convertible security financings with market price-based conversion ratios have colloquially been called “floorless,” “toxic,” “death spiral,” and “ratchet” convertibles.
55. Future sales of our common stock by our stockholders could negatively affect our stock price after this offering.
Sales of a substantial number of shares of our common stock in the public market by our stockholders after this offering, or the perception that these sales might occur, could depress the market price of our common stock and could impair our ability to raise capital through the sale of additional equity securities. All of the shares of common stock sold in this offering will be freely tradable without restrictions or further registration under the Securities Act of 1933, as amended, (the “Securities Act”), and thus the price of our common stock may decline.
| 33 |
56. Future sales of our common stock by us could adversely affect its price and our future capital-raising activities could involve the issuance of equity securities, which would dilute your investment and could result in a decline in the trading price of our common stock.
We may in the future sell securities in the public or private equity markets if and when conditions are favorable, even if we do not have an immediate need for additional capital at that time. Sales of substantial amounts of common stock, or the perception that such sales could occur, could adversely affect the prevailing market price of our common stock and our ability to raise capital. We may issue additional common stock in future financing transactions or as incentive compensation for our executive management and other key personnel, consultants and advisors. Issuing any equity securities would be dilutive to the equity interests represented by our then-outstanding shares of common stock. The market price for our common stock could decrease as the market takes into account the dilutive effect of any of these issuances. Furthermore, we may enter into financing transactions at prices that represent a substantial discount to the market price of our common stock. A negative reaction by investors and securities analysts to any discounted sale of our equity securities could result in a decline in the trading price of our common stock.
57. We will have broad discretion in using the net proceeds from the Private Placement, and the benefits from our use of the proceeds may not meet investors' expectations.
Our management will have broad discretion over the allocation of our net proceeds from the Private Placement as well as over the timing of their use without stockholder approval. We have not yet determined how the net proceeds will be used, other than for working capital and other general corporate purposes. As a result, investors will be relying upon management's judgment with only limited information about our specific intentions for the use of our net proceeds from the Private Placement. Our failure to apply these proceeds effectively could cause our business to suffer.
58. If securities analysts do not publish research or if securities analysts or other third parties publish inaccurate or unfavorable research about us, the price of our common stock could decline.
The trading market for our common stock will rely in part on the research and reports that securities analysts and other third parties choose to publish about us. We do not control these analysts or other third parties. The price of our common stock could decline if one or more securities analysts downgrade our common stock or if one or more securities analysts or other third parties publish inaccurate or unfavorable research about us or cease publishing reports about us.
| 34 |
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements that can involve substantial risks and uncertainties. All statements other than statements of historical facts contained in this prospectus, including statements regarding our future results of operations and financial position, business strategy, prospective products, product approvals, research and development costs, future revenue, timing and likelihood of success, plans and objectives of management for future operations, future results of anticipated products and prospects, plans and objectives of management are forward-looking statements. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements.
In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “expect,” “plan,” “anticipate,” “could,” “intend,” “target,” “project,” “contemplate,” “believe,” “estimate,” “predict,” “potential,” “would” or “continue” or the negative of these terms or other similar expressions. The forward-looking statements in this prospectus are only predictions. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our business, financial condition and results of operations. These forward-looking statements speak only as of the date of this prospectus and are subject to a number of risks, uncertainties and assumptions described under the sections in this prospectus entitled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere in this prospectus. Because forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified and some of which are beyond our control, you should not rely on these forward-looking statements as predictions of future events. The events and circumstances reflected in our forward-looking statements may not be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. Moreover, we operate in an evolving environment. New risk factors and uncertainties may emerge from time to time, and it is not possible for management to predict all risk factors and uncertainties. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein, whether as a result of any new information, future events, changed circumstances or otherwise.
We obtained the industry, market and competitive position data in this prospectus from our own internal estimates and research as well as from industry and general publications and research, surveys and studies conducted by third parties. Industry publications, studies and surveys generally state that they have been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. While we believe that each of these studies and publications is reliable, we have not independently verified market and industry data from third-party sources. While we believe our internal company research as to such matters is reliable and the market definitions are appropriate, neither such research nor these definitions have been verified by any independent source.
All of the Shares being offered under this prospectus are being sold by or for the account of the selling stockholder. We will not receive any proceeds from the sale of Shares offered by this prospectus. However, with respect to the 5,500,000 shares of Common Stock underlying the Warrant that is being offered by the selling stockholder under this prospectus, we will receive up to $2,750,000 in the aggregate from the selling stockholder if it exercises in full, on a cash basis, all of its unexercised Warrant to purchase these shares of Common Stock. We will use such proceeds from the exercise of the Warrant for working capital and other corporate purposes.
The holder of the Warrant may exercise its Warrant at any time until its expiration. There can be no assurance that the Warrant will be exercised. Because the holder of the Warrant may exercise the Warrant in its own discretion, if at all, we cannot plan on specific uses of proceeds beyond application of proceeds to general corporate purposes.
We have agreed to bear the expenses (other than any underwriting discounts or commissions or agent’s commissions) in connection with the registration of the Shares being offered hereby by the selling stockholder.
| 35 |
MARKET PRICE OF OUR COMMON STOCK
Our common stock has been quoted on the OTCQB under the symbol “CNBX” since August 10, 2015. On February 12, 2021, the last reported sales price of our common stock on the OTCQB was $0.38 per share.
As of February 12, 2021, the Company’s common stock was held by 76 shareholders of record, which does not include shares that are held in street or nominee name.
The closing share prices presented below represent prices between broker-dealers and do not include retail mark-ups and mark-downs or any commission to the dealer.
| QUARTER ENDED | HIGH | LOW | ||||||
| November 30, 2020 | $ | 0.30 | $ | 0.15 | ||||
| August 31, 2020 | $ | 0.31 | $ | 0.19 | ||||
| May 31, 2020 | $ | 0.36 | $ | 0.16 | ||||
| February 29, 2020 | $ | 0.57 | $ | 0.08 | ||||
| November 30, 2019 | $ | 0.26 | $ | 0.12 | ||||
| August 31, 2019 | $ | 0.34 | $ | 0.25 | ||||
| May 31, 2019 | $ | 0.40 | $ | 0.30 | ||||
| February 28, 2019 | $ | 0.48 | $ | 0.28 | ||||
| November 30, 2018 | $ | 1.23 | $ | 0.40 | ||||
| August 31, 2018 | $ | 1.05 | $ | 0.73 | ||||
We have never declared or paid dividends on our capital stock. We do not expect to pay dividends on our common stock for the foreseeable future. Instead, we anticipate that all of our earnings will be used for the operation and growth of our business. Any future determination to declare cash dividends would be subject to the discretion of our board of directors and would depend upon various factors, including our results of operations, financial condition and liquidity requirements, restrictions that may be imposed by applicable law and our contracts and other factors deemed relevant by our board of directors.
DETERMINATION OF THE OFFERING PRICE
The selling stockholder will determine at what price it may sell the Shares offered by this prospectus, and such sales may be made at prevailing market prices, or at privately negotiated prices. For additional information, please see the section entitled "Plan of Distribution" below.
| 36 |
The shares of common stock being offered by the selling stockholder are those issuable to the selling stockholder upon conversion of the notes and exercise of the warrants. For additional information regarding the issuance of the notes and the warrants, see “Private Placement of Notes and Warrant” below. We are registering the shares of common stock in order to permit the selling stockholder to offer the shares for resale from time to time. Except for the ownership of the notes and the warrants issued pursuant to the Private Placement, the selling stockholder has not had any material relationship with us within the past three years.
The table below lists the selling stockholder and other information regarding the beneficial ownership (as determined under Section 13(d) of the Securities Exchange Act of 1934, as amended, and the rules and regulations thereunder) of the shares of common stock held by the selling stockholder. The second column lists the number of shares of common stock beneficially owned by the selling stockholder, based on its ownership of shares of common stock, notes and warrants, as of February 12, 2021, assuming conversion of the notes and exercise of the warrants held by the selling stockholder on that date but taking account of any limitations on conversion and exercise set forth therein.
The third column lists the shares of common stock being offered by this prospectus by the selling stockholder and does not take in account any limitations on (i) conversion of the notes set forth therein or (ii) exercise of the warrants set forth therein.
In accordance with the terms of a registration rights agreement with the holders of the notes and the warrants, this prospectus generally covers the resale of the sum of (i) 300% of the maximum number of shares of common stock issued or issuable pursuant to the Notes at an exercise price of $0.27, and (ii) the maximum number of shares of common stock issued or issuable upon exercise of the Warrant, in each case, determined as if the outstanding notes and warrants were converted or exercised (as the case may be) in full (without regard to any limitations on conversion or exercise contained therein solely for the purpose of such calculation) at a conversion price calculated based on the average of the two lowest closing bid price of the Company’s shares of Common Stock in the ten trading days prior to February 12, 2021. Because the conversion price of the notes and the exercise price of the warrants may be adjusted, the number of shares that will actually be issued may be more or less than the number of shares being offered by this prospectus. The fourth column assumes the sale of all of the shares offered by the selling shareholder pursuant to this prospectus.
Under the terms of the notes and the warrants, the selling stockholder may not convert the notes or exercise the warrants to the extent (but only to the extent) the selling stockholder or any of its affiliates would beneficially own a number of shares of our common stock which would exceed 4.99% of the outstanding shares of the Company. The number of shares in the second column reflects these limitations. The selling stockholders may sell all, some or none of their shares in this offering. See “Plan of Distribution.”
Name of Selling Stockholder | Number of Shares of Common Stock Owned Prior to Offering (1) | Maximum Number of Shares of Common Stock to be Sold Pursuant to this Prospectus (2) | Number of Shares of Common Stock Owned After Offering (3) | |||||||||
| 3i, LP (4) | 7,466,733 | 20,777,779 | 0 | |||||||||
| (1) | This column lists the number of shares of our common stock beneficially owned by the Selling Stockholder, as of February 12, 2021, (i) including 3,913,633 Pre-Delivery Shares that were pre-delivered to the Selling Stockholder on the issuance date of the Initial Note, and (ii) after giving effect to the 4.99% limitation on conversion of the Initial Note a nd the Second Note and exercise of the Warrant, as set forth in the Initial Note and Warrant, respectively, up to an aggregate additional 3,553,100 shares of our common stock either issuable upon conversion of the Initial Note and the Second Note and/or exercise of the Warrant, as applicable. Without regard to such 4.99% limitation on conversion of the Initial Note and the Second Note and exercise of the Warrant, respectively, (A) an additional 1,178,960 shares of our common stock (in addition to the 3,913,633 shares of our common stock that were pre-delivered to the Selling Stockholder on the issuance date of the Initial Note) are issuable upon conversion of the Initial Note and the Second Note, and (B) 5,500,000 shares of our common stock are issuable upon exercise of the Warrant. |
| 37 |
| (2) | In accordance with the terms of a registration rights agreement with the holder of the Notes and the Warrant, this prospectus generally covers the resale of (i) 300% of the maximum number of shares of common stock issuable upon conversion of the Notes, namely, 15,277,779 shares of our common stock (includes the 3,913,633 shares of our common stock that were pre-delivered to the Selling Stockholder on the issuance date of the Initial Note) issuable upon conversion of the Notes (y) assuming the full conversion of all the Notes issued and issuable under the securities purchase agreement and (z) without regard to any limitations on conversion in the Notes, in each case, solely for the purpose of such calculation), and (ii) 5,500,000 shares of our common stock issuable upon exercise of the Warrant ((x) assuming the exercise in full of the Warrant and (y) without regard to any limitations on exercise set forth in the Warrant, in each case, solely for the purpose of such calculation). Because the conversion price of the Notes is subject to adjustment in accordance with its terms, the number of shares that will actually be issued may be more or less than the number of shares being offered by this prospectus. |
| (3) | The ownership of shares after the offering assumes the issuance of all of the shares underlying the Notes and the Warrant that are offered for resale hereby, and the sale by the selling stockholder of all of the shares offered for resale hereby. |
| (4) | The business address of 3i, LP (referred to herein as the “Selling Stockholder”) is 140 Broadway, 38th Floor, New York, NY 10005. 3i, LP’s principal business is that of a private investor. Maier Joshua Tarlow is the manager of 3i Management, LLC, the general partner of 3i LP, and has sole voting control and investment discretion over securities beneficially owned directly indirectly by 3i Management, LLC and 3i, LP. We have been advised that none of Mr. Tarlow, 3i Management, LLC or 3i, LP is a member of the Financial Industry Regulatory Authority, or FINRA, or an independent broker-dealer, or an affiliate or associated person of a FINRA member or independent broker-dealer. Mr. Tarlow disclaims any beneficial ownership of the securities beneficially owned directly by 3i, LP and indirectly by 3i Management, LLC. |
RELATIONSHIP OF SELLING STOCKHOLDER TO THE COMPANY
The selling stockholder has not had any material transaction or relationship with us or any of our predecessors or affiliates within the past three years.
We are registering the shares of common stock issuable upon conversion of the notes and exercise of the warrants to permit the resale of these shares of common stock by the holders of the notes and warrants from time to time after the date of this prospectus. We will not receive any of the proceeds from the sale by the selling stockholder of the shares of common stock, although we will receive the exercise price of any Warrants not exercised by the selling stockholder on a cashless exercise basis. We will bear all fees and expenses incident to our obligation to register the shares of common stock.
| 38 |
The selling stockholder may sell all or a portion of the shares of common stock held by it and offered hereby from time to time directly or through one or more underwriters, broker-dealers or agents. If the shares of common stock are sold through underwriters or broker-dealers, the selling stockholder will be responsible for underwriting discounts or commissions or agent’s commissions. The shares of common stock may be sold in one or more transactions at fixed prices, at prevailing market prices at the time of the sale, at varying prices determined at the time of sale or at negotiated prices. These sales may be effected in transactions, which may involve crosses or block transactions, pursuant to one or more of the following methods:
| · | on any national securities exchange or quotation service on which the securities may be listed or quoted at the time of sale; |
| · | in the over-the-counter market; |
| · | in transactions otherwise than on these exchanges or systems or in the over-the-counter market; |
| · | through the writing or settlement of options, whether such options are listed on an options exchange or otherwise; |
| · | ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers; |
| · | block trades in which the broker-dealer will attempt to sell the shares as agent but may position and resell a portion of the block as principal to facilitate the transaction; |
| · | purchases by a broker-dealer as principal and resale by the broker-dealer for its account; |
| · | an exchange distribution in accordance with the rules of the applicable exchange; |
| · | privately negotiated transactions; |
| · | short sales made after the date the Registration Statement is declared effective by the SEC; |
| · | broker-dealers may agree with a selling security holder to sell a specified number of such shares at a stipulated price per share; |
| · | a combination of any such methods of sale; and |
| · | any other method permitted pursuant to applicable law. |
The selling stockholder may also sell shares of common stock under Rule 144 promulgated under the Securities Act of 1933, as amended, if available, rather than under this prospectus. In addition, the selling stockholder may transfer the shares of common stock by other means not described in this prospectus. If the selling stockholder effects such transactions by selling shares of common stock to or through underwriters, broker-dealers or agents, such underwriters, broker-dealers or agents may receive commissions in the form of discounts, concessions or commissions from the selling stockholder or commissions from purchasers of the shares of common stock for whom they may act as agent or to whom they may sell as principal (which discounts, concessions or commissions as to particular underwriters, broker-dealers or agents may be in excess of those customary in the types of transactions involved). In connection with sales of the shares of common stock or otherwise, the selling stockholder may enter into hedging transactions with broker-dealers, which may in turn engage in short sales of the shares of common stock in the course of hedging in positions they assume. The selling stockholder may also sell shares of common stock short and deliver shares of common stock covered by this prospectus to close out short positions and to return borrowed shares in connection with such short sales. The selling stockholder may also loan or pledge shares of common stock to broker-dealers that in turn may sell such shares.
The selling stockholder may pledge or grant a security interest in some or all of the notes, warrants or shares of common stock owned by it and, if it defaults in the performance of its secured obligations, the pledgees or secured parties may offer and sell the shares of common stock from time to time pursuant to this prospectus or any amendment to this prospectus under Rule 424(b)(3) or other applicable provision of the Securities Act amending, if necessary, the list of selling stockholders to include the pledgee, transferee or other successors in interest as selling stockholders under this prospectus. The selling stockholder also may transfer and donate the shares of common stock in other circumstances in which case the transferees, donees, pledgees or other successors in interest will be the selling beneficial owners for purposes of this prospectus.
| 39 |
To the extent required by the Securities Act and the rules and regulations thereunder, the selling stockholder and any broker-dealer participating in the distribution of the shares of common stock may be deemed to be “underwriters” within the meaning of the Securities Act, and any commission paid, or any discounts or concessions allowed to, any such broker-dealer may be deemed to be underwriting commissions or discounts under the Securities Act. At the time a particular offering of the shares of common stock is made, a prospectus supplement, if required, will be distributed, which will set forth the aggregate amount of shares of common stock being offered and the terms of the offering, including the name or names of any broker-dealers or agents, any discounts, commissions and other terms constituting compensation from the selling stockholders and any discounts, commissions or concessions allowed or re-allowed or paid to broker-dealers.
Under the securities laws of some states, the shares of common stock may be sold in such states only through registered or licensed brokers or dealers. In addition, in some states the shares of common stock may not be sold unless such shares have been registered or qualified for sale in such state or an exemption from registration or qualification is available and is complied with.
There can be no assurance that the selling stockholder will sell any or all of the shares of common stock registered pursuant to the registration statement, of which this prospectus forms a part.
The selling stockholder and any other person participating in such distribution will be subject to applicable provisions of the Securities Exchange Act of 1934, as amended, and the rules and regulations thereunder, including, without limitation, to the extent applicable, Regulation M of the Exchange Act, which may limit the timing of purchases and sales of any of the shares of common stock by the selling stockholder and any other participating person. To the extent applicable, Regulation M may also restrict the ability of any person engaged in the distribution of the shares of common stock to engage in market-making activities with respect to the shares of common stock. All of the foregoing may affect the marketability of the shares of common stock and the ability of any person or entity to engage in market-making activities with respect to the shares of common stock.
We will pay all expenses of the registration of the shares of common stock pursuant to the registration rights agreement, estimated to be approximately $72,000 in total, including, without limitation, Securities and Exchange Commission filing fees and expenses of compliance with state securities or “blue sky” laws; provided, however, a selling stockholder will pay all underwriting discounts and selling commissions, if any. We will indemnify the selling stockholder against liabilities, including some liabilities under the Securities Act in accordance with the registration rights agreements or the selling stockholder will be entitled to contribution. We may be indemnified by the selling stockholder against civil liabilities, including liabilities under the Securities Act that may arise from any written information furnished to us by the selling stockholder specifically for use in this prospectus, in accordance with the related registration rights agreements or we may be entitled to contribution.
Once sold under the registration statement, of which this prospectus forms a part, the shares of common stock will be freely tradable in the hands of persons other than our affiliates.
History
Cannabics Pharmaceuticals Inc. was incorporated in the State of Nevada as Thrust Energy Corp. on September 15, 2004, for the purposes of acquiring oil and gas exploration properties and non-operating interests. On April 25, 2014, Cannabics Inc., a Delaware research and development company, acquired a majority controlling interest in the Company. As a result of the change of control, we redirected our business focus towards our current operations and changed the name of the Company to Cannabics Pharmaceuticals Inc.
| 40 |
On August 25, 2014, the Company incorporated G.R.I.N. Ultra Ltd. under the laws of the State of Israel as a wholly owned subsidiary to conduct our research and development activities.
On October 26, 2014, G.R.I.N. Ultra was licensed by the Israeli Ministry of Health to possess and use cannabis for medical research. Cannabis is a controlled substance under Israeli law.
On July 24, 2017, we completed development of G.R.I.N. Ultra’s research laboratory in Rehovot, Israel.
On November 28, 2017, G.R.I.N. Ultra Ltd. was granted a further license by the Israeli Ministry of Health to conduct research into the characterization of anti-tumor activity of cannabinoids.
On August 10, 2018, we entered into a convertible loan agreement with Eroll Grow-Tech Ltd., an Israeli corporation that produces fully-automated, self-contained hydroponics systems. Under the terms of the agreement, we obtained the right to acquire up to 20% of the issued and outstanding common stock of Eroll Grow-Tech in exchange for an aggregate purchase price of up to $2 million. Pursuant to the terms of the agreement, the Company provided Eroll Grow-Tech with a loan in the amount of $2,000,000, and the Company has converted the loan into 3,580,567 shares of Eroll Grow-Tech. We are also entitled to receive aggregate royalties from Eroll Grow-Tech of up to $8 million. In 2019, we received royalties in the amount of $500,000.
On February 7, 2019, we entered into a joint venture agreement with Wize Pharma, Inc., a clinical-stage biopharmaceutical company with operations in Israel focused on the treatment of ophthalmic disorders, for the purpose of researching, developing, and administering cannabinoid formulations to treat ophthalmic conditions. Under the terms of the agreement, Wize Pharma issued 900,000 shares of its common stock to the Company and we issued 2,263,944 shares of the Company’s common stock to Wize Pharma.
On November 15, 2020, we established a majority-owned subsidiary, Digestix Bioscience, Inc., incorporated under the laws of Delaware, to engage in the development of medical devices and pharmaceutical compositions for the treatment of precancerous and early-stage neoplastic local tumors, and in particular, adenomatous colorectal polyps. The other shareholders of Digestix are Professor Eitan Scapa, Dr. Erez Scapa, and Gabriel Yariv. Mr. Yariv, who currently serves as Director and COO of Cannabics, also serves as interim Chairman and CEO of Digestix.
Overview
The Company is a biopharmaceutical corporation specializing in the discovery, development and commercialization of novel cannabinoid-based products and innovative technologies for the treatment of cancer. We combine the power of our proprietary technologies with the expertise of our leading scientists to unlock the medicinal properties of cannabis and its diversity of bioactive compounds. We have conducted thousands of tests on biopsies and cell lines in order to identify the physiologic impact of cannabinoids on cell cycle and cell death. This scientific workflow has generated an ongoing stream of biological data through which we have accumulated in-depth knowledge of the various therapeutic effects of cannabinoids and identified cannabinoid ratios demonstrating anti-tumor potential. We believe that our cannabinoid research coupled with our proprietary technologies and intellectual property positions the Company to play an important role in the rapidly growing medical cannabis marketplace.
Our core technology is a continuously evolving bioinformatics platform that utilizes high-throughput screening technology, advanced data analytics, and proprietary methodologies to rapidly examine the physiologic impact of multiple cannabinoid compounds on tumor cells. This technology enables us to screen thousands of cannabinoid combinations weekly, generating multiple datasets on the anti-tumor properties of different cannabis cultivars, formulations and ratios. We conduct a broad range of preclinical research on cannabinoids through our bioinformatics platform, which informs the development of our product candidates.
| 41 |
Our lead product candidate is RCC-33, an oral capsule for the treatment of colorectal cancer (“CRC”). RCC-33 contains high concentrations of the cannabinoids CBDV and CBGA, which have demonstrated in our laboratory testing complex synergistic anti-tumor activity in our in vitro studies, with minimal psychoactive effects. We are currently in the early planning stage of a clinical development pathway for RCC-33. We plan to conduct further preclinical studies to establish the safety and efficacy of RCC-33 in an in vivo murine model of colorectal cancer. Subject to the results of our preclinical studies, we intend to proceed with first-in-human clinical testing in the second half of 2022.
Cannabics SR is a lipid-based capsule containing a standardized formulation of cannabinoids that we are developing as a product candidate for the treatment of cancer anorexia-cachexia syndrome (“CACS”). With a rapid onset of action and sustained effects for up to 6-8 hours, we believe that the convenience of once or twice daily oral dosing of Cannabics SR may improve quality of life and increase patient compliance with treatment regimens, leading to better health outcomes. A two-year pilot study of Cannabics SR led by Dr. Gil Bar-Sela of the Rambam Hospital Health Care Campus, Division of Oncology, in Haifa, Israel, demonstrated a clinically significant weight increase in CACS patients treated with Cannabics SR capsules (Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6785913/). In the second half of 2021, we intend to commence an additional pilot study in Israel to assess the pharmacokinetics and pharmacodynamics of Cannabics SR in humans. Data from the study will inform our clinical development plan for Cannabics SR. In the meantime, we anticipate that the results of these studies may enable us to obtain a permit from the Israeli Ministry of Health to commercialize Cannabics SR in Israel.
Another product candidate we are developing is Cannabics CDx, a drug sensitivity test designed to provide innovative decision support to healthcare providers interested in personalizing cannabinoid-based cancer therapies. Cannabics CDx applies data analytics and high-content drug sensitivity screening integrated with our proprietary database to measure the effectiveness of cannabinoid compounds on a patient’s biopsy, suggest preferred alternatives, and alert healthcare providers to cannabinoids that may be contraindicated. We believe that Cannabics CDx will meet a significant unmet need of the growing population of cancer patients being treated with cannabis by enabling healthcare providers to more precisely tailor cannabinoid treatments to a patient’s cancer and clinical profile. We are currently seeking strategic partners for a clinical validation study expected to commence in 2022 to assess the sensitivity and specificity of Cannabics CDx with a view towards commercializing Cannabics CDx in Europe, the United States, and other territories.
Figure 1: Products currently under development by Cannabics Pharmaceuticals Inc.
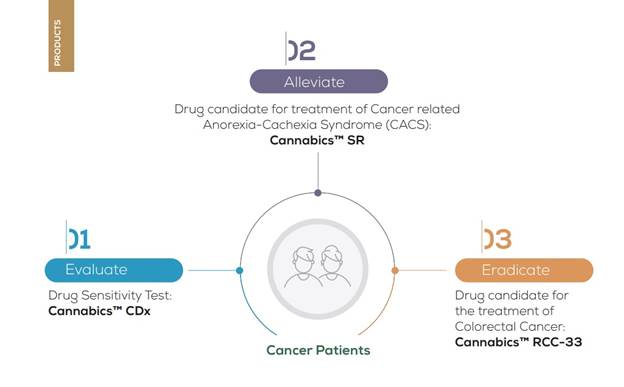
| 42 |
In addition to facilitating the discovery of our technologies and product candidates, we believe that our bioinformatics platform may be of benefit to other biopharmaceutical companies across a range of other applications, representing collaborative opportunities, market potential, and downstream value-creation for the Company. In particular, we believe that by identifying susceptible patient populations in advance of clinical trials, our platform can improve success rates, shorten trial periods, and reduce development costs.
As of the date of this prospectus, we have entered into several research collaborations with cannabis producers around the world, using our bioinformatics platform to examine the anti-tumor properties of distinct cannabinoid cultivars on cancer cell lines, with a view towards expanding our cannabinoid compound library and developing future cultivars that may be useful in the treatment of cancer. See “Description of Business - Strategic Partnerships” elsewhere in this prospectus.
We are led by a forward-looking management team and board of directors that have a strong record of innovation and value creation, with experience in business and product development, research, commercialization, and finance. Complementing our management is a distinguished advisory board of experts in the fields of pharmaceutical development, regulation, clinical research, and finance. Our scientists are leaders in the fields of cannabinoid research, oncology, and molecular genetics.
All of our pre-clinical research and development is conducted in our laboratory in Rehovot, Israel, under license from the Israeli Ministry of Health. Israel has had a favorable regulatory environment for clinical studies using cannabis since 1996, and has become a global leader in medical cannabis research and innovation. The location of our research facilities in Israel enables us to expand our expertise in the development of cannabinoid profiles and therapeutic formulations, establish collaborative arrangements, and attract leading minds in the field.
We currently conduct all of our preclinical research and discovery efforts internally, but will collaborate from time to time with a broad network of leading scientists conducting clinical research into cancer and the medical application of cannabinoids. We see great potential in scientific collaborations with like-minded companies interested in cannabinoid medicines and diagnostics. Past collaborations have included Rambam Medical Center (Israel), the Technion (Israel), and Simfo Gmbh (Germany).
Through our research and development activities, we are building a portfolio of intellectual property assets comprised of patents, proprietary technologies, and bioinformatics with a variety of research, analytic, and therapeutic applications. As of the date of this prospectus, our intellectual property portfolio includes two issued worldwide patents and 19 pending patent applications directed to plant extracts, pharmaceutical formulations, drug delivery, and the therapeutic uses of cannabinoids, together with know-how and trade secrets. We continue to strengthen our intellectual property portfolio while actualizing our cannabinoid technologies in our laboratory facilities and developing the scientific data to back our achievements. We anticipate that we will file additional patent applications in conjunction with the research, testing, and development of our technologies.
We intend to adopt an opportunistic approach towards commercializing any product we may develop based on optimal returns to the Company. We plan to advance our drug candidates through early human trials, at which point we expect to partner with established pharmaceutical companies to complete clinical development, obtain regulatory approvals, and commercialize our products. For our other non-drug product candidates, we may seek strategic partnerships earlier in the clinical development process. We may also out-license our product candidates and technologies to collaborative partners in respect of indications that are outside our development focus at the time.
Our operations strictly adhere to all applicable laws with respect to cannabis. Our use and possession of cannabis is strictly limited to medical research in Israel, in accordance with Israeli law. The Company does not, and will not possess, manufacture, distribute or dispense cannabis in the United States or in any other jurisdiction where such activities are not permitted.
| 43 |
Our goal is to capitalize on our work in the field of cannabinoid therapeutics to become a leading developer of cannabinoid-based anti-cancer medicines and technologies for global markets with significant unmet medical needs. The key elements of our strategy include:
● Leveraging Our Bioinformatics Platform. We believe that our proprietary bioinformatics platform, together with our in-house development expertise, generates data and analytical insights that enable us to discover, develop, and commercialize novel first-in-class product candidates based on proprietary cannabinoid formulations. We intend to leverage the power of our bioinformatics platform in collaborative relationships with pharmaceutical companies, academic institutions, and cannabis producers that may yield valuable data for the Company along with potential commercial opportunities of mutual interest and benefit.
● In-House Pre-Clinical Research. We conduct all of our pre-clinical research in-house at our laboratory facility in Israel. As we develop our product candidates, we may supplement our in-house clinical and regulatory capabilities in the design and implementation of clinical trials by engaging external consultants, collaborators, and leading contract research organizations to investigate the therapeutic effects of cannabis strains, to identify patterns of cannabinoid ratios that are useful in treating various indications, and to assess the effectiveness of our novel treatments and technologies in diverse indications. We may also enter into collaborations with leading academic and other research centers to augment our core expertise in specific programs.
● Commercialization Partnerships. We generally intend to advance our drug candidates through Phase 1 and Phase 2 clinical trials as appropriate to establish their clinical and commercial potential, at which point we will determine a commercialization path based on optimal returns to the Company. We expect to enter into strategic partnerships with established pharmaceutical and biotechnology companies to continue the development of our drug candidates beyond Phase 2 clinical trials, seek regulatory approvals, and ultimately market and sell any products we may develop. For other product candidates, such as Cannabics CDx, we may seek strategic partnerships earlier in their clinical development to accelerate the approval process and facilitate commercialization. We believe this strategy mitigates the risks inherent in late-stage clinical development by eliminating the need for us to raise the significant capital required to perform the large multi-center trials required for regulatory approval of any drug or other product we may develop and to build the resources necessary to market them.
● Strategic Out-Licensing. Our current research focus is on the development of cannabinoid therapies and other technologies for the treatment of gastrointestinal cancers. We intend to out-license our product candidates and technologies to collaborative partners for the development of therapies of low strategic interest to the Company. We believe that these partnerships will enhance the value of our intellectual property and allow the Company to retain selected interests in such therapies without having to acquire the resources needed for in-house development.
● Expanding and Protecting our Intellectual Property Portfolio. We seek patent protection for the technology, inventions, and improvements that we consider important for the development of our business, but only in those cases where we believe that the costs of obtaining patent protection is justified by the scientific and commercial potential of the technology, and typically only in those jurisdictions that we believe present significant commercial opportunities. We also seek to protect our know-how and trade secrets through an active program of legal mechanisms including assignments, confidentiality agreements, material transfer agreements, research collaborations, and licenses. See “Description of Business – Intellectual Property” elsewhere in this prospectus.
| 44 |
Cancer is a general term used to describe a group of more than 100 related diseases characterized by uncontrolled growth and spread of abnormal cells, leading to the development of a mass commonly known as a tumor, followed by invasion of the surrounding tissues and subsequent spread, or metastasis, to other parts of the body. Despite enormous investment in research and the introduction of new treatments, cancer remains a critical area of unmet medical need. According to the World Health Organization, cancer is the second leading cause of mortality worldwide, responsible for an estimated 9.6 million deaths in 2018. As of January 1, 2019, there were more than 16.9 million people with a history of cancer living in the United States, with 1.8 million new cases and 606,520 cancer deaths expected in 2020 (Source: American Cancer Society. Cancer Facts & Figures 2020).
Over the past decade, there has been growing interest in the therapeutic value of cannabinoid compounds in oncology. Cannabis has long been suggested as a well-tolerated, safe, and effective option to help patients cope with cancer related symptoms by reducing nausea and vomiting, alleviating cancer pain, stimulating appetite, and improving quality of life. Beyond their palliative benefits, however, cannabinoids have also been receiving increased attention for their anti-cancer potential, which we believe may one day revolutionize cancer therapy.
Cannabinoids are a diverse class of chemical compounds that occur naturally within cannabis plants and are pharmacologically similar to cannabinoids produced by the human body, known as endocannabinoids. Endocannabinoids form part of the human endocannabinoid system (ECS), a complex biological network that also includes cannabinoid receptors and enzymes involved in cannabinoid formation, transport, and degradation. The ECS is regarded as an important endogenous system implicated in regulation of the most vital biological processes to maintain homeostasis, assisting the body to remain stable and balanced despite external, or environmental, fluctuations (Source: Current Pharmaceutical Design, 2016;22(12):1756-1766).
Dysregulation of the ECS owing to variation in the expression and function of cannabinoid receptors or enzymes or the concentration of endocannabinoids has been associated with several diseases, including cancer (Source: International Journal of Molecular Sciences, 2020;21(3):747). Indeed, the mechanisms involved in the regulation of the ECS as well as the processes that it regulates include practically every pathway important in cancer biology. Expression of the ECS is altered in numerous types of tumors, compared to healthy tissue, and this aberrant expression has been related to cancer prognosis and disease outcome, depending on the origin of the cancer (Source: British Journal of Pharmacology, 2018;175(13):2566-2580). Recent studies suggest that endocannabinoids contribute to maintaining balance in cell proliferation and that targeting the ECS can affect cancer growth (Source: Canadian Urological Association Journal, 2017;11(3-4):E138-E142).
Cannabinoids can interact with the cannabinoid receptors in the ECS, sometimes with a higher affinity than endocannabinoids. As a consequence, all the processes regulated by endocannabinoids are susceptible to interference by cannabinoids. The ability to use cannabinoids to modulate the ECS encompasses several attractive pharmacotherapeutic targets for systemic anti-cancer treatment and has sparked considerable research examining cannabinoid action on cancer cells (Source: Pharmacological Reviews, 2006;58(3):389-462).
Cannabinoids have demonstrated selective anti-tumor properties in preclinical studies, exerting anti-proliferative, proapoptotic, anti-angiogenic, and anti-metastatic and anti-inflammatory effects depending on tumor type and specific setting (Source: Cancer Medicine, 2018:7(3):765-775). These effects appear to be more pronounced when cannabinoids are used together versus being administered separately, a mechanism known as the entourage effect. We believe, therefore, that cannabinoid combinations may hold promise for an improved anti-proliferative strategy for cancer management.
In addition to their potential role as anti-cancer agents, cannabinoids have been observed to act synergistically with some conventional antineoplastic drugs, such as chemotherapeutic agents, enhancing their effectiveness (Source: Cancer Medicine, 2018;7(3)765-775). This raises the potential for combinational therapies that may increase the range of chemotherapeutic options available to patients and enable targeting of tumor progression at different levels while also permitting dosages of cytotoxic drugs to be dramatically reduced without compromising efficacy.
| 45 |
Figure 2: Our pre-clinical data showing synergistic effects of cannabis extracts and chemotherapies on cancer biopsy after treatment with the same extract and three different chemotherapy combinations

As of the date of this prospectus, we are not aware of any cannabinoid-based therapies approved for the treatment of cancer.
We have developed a continuously evolving preclinical bioinformatics platform that enables us to evaluate and classify the physiological impact of multiple cannabinoid compounds on various cancer cells. Utilizing state-of-the-art high-throughput screening and flow cytometry, our platform is capable of testing thousands of compounds weekly, allowing us to rapidly and effectively examine their interactions with a growing library of human cancer cell lines and biopsies. Through the large body of data generated by our platform, we are accumulating in-depth knowledge of the various therapeutic effects of cannabinoids and patterns of cannabinoid ratios that demonstrate meaningful physiologic impact on cancer.
Our bioinformatics platform includes the following:
| Ø | high-throughput screening, high content screening, flow cytometry, machine learning, robotics, and proprietary methodologies; |
| Ø | a library of human cancer cell lines and thousands of different combinations and ratios of cannabinoid compounds in a costumed matrix; |
| Ø | a growing database of biological response data; |
| Ø | in-house extraction, processing methodologies, and analytical techniques that yield well-characterized and standardized extracts; |
| Ø | collaborations with regulated cannabis producers that may expand our cannabinoid compound library and provide us with access for future proprietary cultivars; |
| Ø | fully integrated in-house research and development; and |
| Ø | regulatory expertise. |
| 46 |
Once a series of potentially active cannabinoids is identified for a specific cancer type, we then test and confirm their activity through in vitro and ex-vivo evaluation studies to determine their potential activity. Through this process, we are able to assess their therapeutic potential. The results of our pre-clinical experiments provide starting points for our clinical development programs.
Biopharmaceutical Collaboration
As medical cannabis becomes increasingly recognized for its therapeutic potential in the age of personalized medicine and genomics, we believe that there is a growing global demand by biopharmaceutical companies for research and diagnostic tools that both facilitate and accelerate the generation of biological information for the development of cannabinoid drugs and formulations. We believe that our bioinformatics platform will be of benefit to such companies and may therefore represent collaborative opportunities, market potential and downstream value-creation for the Company.
Finding novel ways to treat and cure diseases is a fundamental challenge in biomedical research. Unsuccessful clinical trials are the most expensive obstacle for drug development because of the immense costs and the low success rate. Only 1 out of 10 drugs successfully pass through clinical development, with 80% of drugs excluded before Phase 3 clinical trials (Source: Biotechnology Innovation Organization, “Clinical Development Success Rates, 2006-2015). The low clinical target validation success rate reflects a lack of reliable drug target prediction methods. This is particularly true in the case of research on cancer, which is increasingly being understood as not just many but thousands of different diseases, requiring more well-defined targets and biomarkers. A 2018 study by MIT found that trials using biomarkers for patient stratification have higher success rates, especially in the area of oncology, where clinical trials using biomarkers exhibited almost twice the overall probability of success compared to trials without biomarkers (Source: Biostatistics, 2019;20(2):273-286).
We believe that our bioinformatics platform could make the development of cannabinoid-based drugs more successful by providing a more accurate and reliable drug target prediction method. Our proprietary analytics may benefit biopharmaceutical companies across a range of applications, including patient selection and recruitment for clinical trials and identification of new targets for drug development.
We are currently developing a portfolio of proprietary technologies and formulations with a variety of research, analytic, and therapeutic applications. Our most advanced development programs include the following:
| Product Candidate | Indication/Description | Current Development Status | Expected Next Steps | Partner(s) |
| RCC-33 | Colorectal Cancer | Pre-clinical | Pre-IND meeting with FDA in the second half of 2021.
Phase 1/2a clinical trial expected to commence in the second half of 2022 | None |
| Cannabics SR | Cancer Anorexia-Cachexia Syndrome | Phase 0 | Additional pilot studies expected to commence in the second half of 2021 | None |
| Cannabics CDx | Drug sensitivity test for cannabinoid-based cancer therapies. | Pre-clinical | Clinical validation study to commence in 2022 | To be determined |
We continue to conduct research and seek collaborations for new advances in biotechnology that may lead to the development of additional product candidates.
| 47 |
Overview
Our lead product candidate is RCC-33, which we are developing as a treatment for CRC. RCC-33 is an oral capsule containing a proprietary formulation of cannabinoids that have demonstrated synergistic efficacy in reducing the viability of human colon cancer cell lines in preclinical studies.
Colorectal Cancer
CRC is one of the more common forms of cancer worldwide, representing a significant challenge to the global healthcare system. According to the World Health Organization, CRC is the third most diagnosed cancer in the world and the second-leading cause of cancer-related mortality. In the United States, there were approximately 1,348,087 people living with CRC in 2017 (Source: National Cancer Institute. “Cancer Stat Facts: Colorectal Cancer”). It is estimated that 147,950 Americans will be diagnosed with CRC in 2020, representing 8.2% of all new cancer cases, and 53,200 Americans will die from the disease (Source: American Cancer Society. “Cancer Facts & Figures 2020”).
Most CRCs begin as a noncancerous growth called a polyp that develops on the inner lining of the colon or rectum. The most common kind of polyp is called an adenomatous polyp or adenoma. According to the American Cancer Society, an estimated one-third to one-half of all individuals will eventually develop one or more adenomas. Although all adenomas have the capacity to become cancerous, fewer than 10% are estimated to progress to invasive cancer. The likelihood that an adenoma will evolve into cancer increases as it becomes larger or when it acquires certain histopathological characteristics. Adenomas that become cancerous, called adenocarcinomas, comprise nearly 96% of all CRCs (Source: American Cancer Society. “Colorectal Cancer Facts & Figures 2017-2019”). Adenocarcinomas may grow into blood vessels or lymph vessels, increasing the chance of metastasis to other anatomical sites.
CRC usually develops slowly, over a period of 10 to 20 years. The complex sequence of events occurring during initiation, development and propagation of adenocarcinomas is likely the result of a lifelong accumulation of mutations caused by both genetic and environmental factors known as the adenoma to carcinoma sequence. While the specific cause of any particular case of CRC is often unknown, more than one-half of all cases and deaths are attributable to lifestyle and environmental factors, such as smoking, unhealthy diet, high alcohol consumption, physical inactivity, and excess body weight (Source: American Cancer Society. “Cancer Facts & Figures 2020”).
CRC does not usually cause symptoms until the disease is advanced, therefore early detection of adenomas by screening is vital. If not treated or removed, an adenoma can become a potentially life-threatening cancer.
Current Standard of Care
Treatment options for CRC patients depend on several factors, including the type and stage of cancer, possible side effects, and the patient’s preferences and overall health. Surgical removal of the tumor is the most common form of treatment, particularly in the early stages of malignancy. Patients with more advanced stages of CRC may be given adjuvant chemotherapy to kill any cancer cells remaining after surgery, though standard chemotherapy is associated with severe side effects and provides marginal benefit to the majority of patients. While radiation therapy is often used to treat rectal cancer, it is not generally recommended for colon cancer patients except in the later stages of the disease (Source: American Cancer Society. “Treating Colorectal Cancer”).
CRC is a heterogeneous disease with distinct clinical, molecular, and pathophysiological characteristics. As a result, the response to treatment is variable between patients, even when they are diagnosed at the same clinical stage. Such heterogeneity remains an obstacle to the optimization of treatment for each individual. Researchers are continuing to investigate new treatment options, such as immunotherapy and targeted therapy, that focus upon the genes, proteins, and other factors in a particular tumor (Source: American Cancer Society. “Advances in Colorectal Research”).
| 48 |
Immunotherapy uses the body’s own immune system to kill cancer cells. There are already several FDA-approved immunotherapy options for CRC, such as pembrolizumab (Keytruda®), nivolumab (Opdivo®), and ipilimumab (Yervoy®). Many immunotherapies that have shown promise in addressing other types of cancer are also being tested for CRC. While immunotherapy has had some encouraging results, significant limitations remain. Its efficacy is often unpredictable and the treatment can lead to the body becoming resistant or result in off-target toxicities where the body’s immune system attacks healthy tissue. Immunotherapy may take longer than other protocols and it is substantially more expensive than classical treatments (Source: Pharmacy & Therapeutics, 2017;42(8):514-521).
Targeted therapy uses drugs to target specific molecules inside cancer cells or on their surface to slow the growth of cancer, destroy cancer cells, and relieve cancer symptoms. There are different types of targeted therapy drugs, each working differently depending on what molecule the drug is targeting. A treatment is chosen based on the types of molecules expressed on the patient’s tumor cells, which allows doctors to tailor cancer treatment for each person. Several targeted therapy drugs, such as bevacizumab (Avasin®) and cetuximab (Erbitux®), are already used to treat advanced CRC. Despite showing clinical promise, targeted therapy has challenges, such as tumor heterogeneity, off-target toxicity, and acquired resistance (Source: Medical Research Journal, 2019;4(2):99-105). The lack of biomarkers by which to identify patients having a high probability of response is also a particularly significant obstacle. As with immunotherapy, the cost of targeted therapy is substantially higher than classical treatments.
We believe that there is no “magic bullet” to cure cancer and that a personalized combination of cancer treatments may be the best course for long term survival benefits in each case. To that end, the development of more prevention strategies and novel agents will be essential.
Cannabinoids and Colorectal Cancer
One area of increasing interest in the treatment of CRC lies in the development and use of cannabinoid therapeutics. The ECS is regarded as an important regulatory system in the gastrointestinal tract, being involved in several important functions such as motility, secretion, sensation, inflammation, and carcinogenesis. Recent studies advocate that the ECS plays a critical role in the development of CRC and should therefore be considered as an appropriate target for CRC inhibition (Source: Frontiers in Pharmacology, 2016;7:361). The expression of ECS components in CRC has been found to be increased and associated with poorer prognosis and advanced stages of disease (Source: Cannabis and Cannabinoid Research, 2018, 3(1):272-281). For example, cannabinoid receptors have been found to be overexpressed in tumor cells of the colon and this up-regulation has been postulated to be an indicator of cancer outcome (Source: British Journal of Pharmacology, 2018; 175(13): 2566-2580).
Research on the effects of cannabinoid compounds on CRC has demonstrated an ability to reduce the viability of CRC cell lines in vitro (Source: Cancer Medicine, 2018;7(3):765-775), while there is also convincing scientific evidence that cannabinoids are able to prevent or reduce carcinogenesis in different animal models of colon cancer (Source: Expert Review of Gastroenterology & Hepatology, 11:10, 871-873).
We believe that cannabinoids are a promising therapeutic agent for the treatment of CRC. We have conducted several in vitro unpublished studies using our bioinformatics platform to confirm that cannabinoids cause necrosis in colon cancer cells. While many cannabinoids demonstrate levels of toxicity on cancer cells, we have found that certain cannabinoid extracts and combinations show increased levels of toxicity relative to other isolated or combined cannabinoids. These findings have spurred the development of RCC-33, our product candidate for the treatment of CRC.
Figure 3: Our pre-clinical data showing synergistic effects of different cannabinoid combinations on viability of a colon cancer cell line.
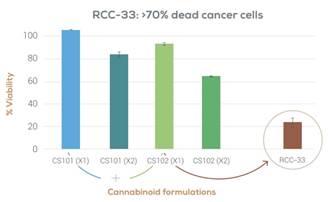
| 49 |
RCC-33
We are developing RCC-33 as an oral capsule containing high concentrations of the cannabinoids CBDV and CBGA in a novel formulation, which we believe may be effective in the treatment of adenocarcinomas of the colon. The cannabinoids in RCC-33 have demonstrated complex synergistic anti-tumor effects in combination, with no psychoactive effect. In our preclinical in vitro studies evaluating the influence of 15 different cannabinoids on human colon cancer cell lines (RKO, HCT116), alone and in combination, RCC-33 demonstrated clear efficacy in reducing the viability of colon cancer cells versus alternative cannabinoid combinations.
Development Plan
We are currently in the early planning stage of a clinical development pathway for RCC-33. We plan to conduct further preclinical studies to establish the safety and efficacy of RCC-33 before proceeding with first-in-human clinical testing.
Preclinical Studies
We intend to conduct a proof of concept non-clinical study in a murine model for colon adenocarcinoma to validate the results obtained in our cell-based assays. In addition, we plan to conduct non-clinical safety studies following Good Laboratory Practice (GLP) to evaluate the systemic and local toxicity of escalating doses of RCC-33 and establish dosing parameters. The results of these preclinical studies, which are expected in the fourth quarter of 2020, will guide our planned Phase 1/2a clinical trial. The non-clinical requirements to support the development program will be verified with the FDA at a pre-IND meeting expected to take place in the second half of 2021. Such studies may include repeated dose toxicity studies, male and female fertility studies, embryofetal development studies, animal abuse related studies, pharmacokinetics studies, drug-drug interaction studies, and others.
Clinical Trials
We plan to evaluate the safety, tolerability, and pharmacokinetic properties of RCC-33 in a Phase 1/2A ascending dose clinical trial in CRC patients, commencing in the second half of 2022. The clinical trial will examine the tolerability, pharmacokinetics, pharmacodynamics, and efficacy of multiple doses of RCC-33 in CRC patients. We are currently identifying potential contract research organizations and clinical trial centers to conduct the Phase 1/2a human proof of concept study, which is estimated to cost $1,000,000. We believe that the Company’s currently available funds will be sufficient to obtain all regulatory approvals necessary to conduct the Phase 1/2a trial. As at the date of this prospectus, however, the Company does not have sufficient funds to complete the Phase 1/2a study.
Subject to the results from our Phase 1 trials, we plan to submit an IND to the FDA for RCC-33 with the clinical protocol for a Phase 2 double-blind placebo controlled clinical trial evaluating RCC-33 in patients with CRC at various dosing levels versus placebo. The outcomes from the planned Phase 2 human proof of concept trial will inform our decision regarding further steps in the clinical development of RCC-33.
At this time, we do not expect to independently develop RCC-33 up to regulatory approval. Instead, we plan to seek a pharmaceutical partner or partners to continue our commercialization efforts. However, we may also seek to further advance the RCC-33 program with additional human clinical trials prior to finding a suitable pharmaceutical partner or partners. We estimate that it will take more than five years to bring RCC-33 to market, if at all, at a cost of more than $10 million.
| 50 |
Cannabics SR for Cancer Anorexia-Cachexia Syndrome (CACS)
Overview
We are developing Cannabics SR as a product candidate for the treatment of CACS. Cannabics SR is a sustained-release oral capsule containing a standardized compound of cannabinoids that has demonstrated a clinically significant weight increase in CACS patients in a peer-reviewed pilot study conducted by Dr. Gil Bar-Sela of the Rambam Hospital Health Care Campus, Division of Oncology, in Haifa, Israel. Our patent-pending technology provides for a convenient, once or twice daily administration, with rapid onset and a steady state of therapeutic effect for a 6 to 8-hour duration.
Cancer Anorexia-Cachexia Syndrome
CACS is a common complication of cancer associated with high morbidity and mortality. It is a complex metabolic syndrome in which a persistently elevated basal metabolic rate is not compensated for by adequate calorie or protein intake, causing involuntary and progressive weight loss leading to increasing functional impairment in cancer patients, especially in advanced stages of the disease. Once established, CACS cannot presently be reversed using available pharmacological or nutritional support techniques.
Unlike starvation, body-weight loss in CACS patients arises mainly from loss of muscle mass, characterized by increased catabolism of skeletal muscle and decreased protein synthesis. This weight loss is associated with important clinical outcomes such as increased morbidity, diminished effectiveness of chemotherapy, muscle wasting, inflammation, fatigue, and reduced survival expectations. The impact of CACS on the patient is not, however, limited to the effect of weight loss. Quality of life, functional abilities, symptoms, psychological outcomes, and social aspects are all affected by CACS.
According to the National Cancer Institute, nearly one-third of cancer deaths can be attributed to the severe weight loss and “metabolic mutiny” associated with CACS, and more than 50% of patients with cancer die with cachexia being present. The overall prevalence of CACS is currently estimated to range from 40% at cancer diagnosis to 70-80% in advanced phases of the disease (Source: Critical Reviews in Oncology/Hematology, 2013;88(3):625-636), while the overall prevalence of weight loss in cancer patients may be as high as 86% in the last 1-2 weeks of life (Source: Journal of Pain and Symptom Management 2007;34:94–104).
The cause and subsequent development of CACS is still poorly understood, but several factors and biological pathways are known to be involved, including inflammation, decreased secretion of anabolic hormones, and altered metabolic response. While there have been important advances in the study of CACS over the past decade, including progress in understanding its mechanisms and the development of promising pharmacologic and supportive care interventions, there is presently no effective pharmacologic therapy for CACS.
Current treatments for CACS are generally based on nutritional support and CACS pathophysiology-modulating drugs, with the most common being the progestogens, megestrol and medroxyprogesterone, and corticosteroids. Progestogens appear to stimulate appetite and improvements in body weight by increasing adipose tissue, but have not been confirmed to augment lean body mass. Megestrol also carries an increased risk of mortality and thromboembolism. Nonetheless, megestrol is the only FDA approved treatment option for CACS and no drug to date has been shown to be superior to it in efficacy and tolerability. Corticosteroids are also considered effective in stimulating appetite and reducing fatigue but should only be used for short periods and in selected cases because of side effects from longer term use, such as insulin resistance, fluid retention, steroidal myopathy, skin fragility, adrenal insufficiency, and sleep and cognitive disorders. Other drugs are being investigated or are in development. Given the dearth of approved therapies, we believe that CACS remains a significant area of unmet medical need.
| 51 |
Cannabinoid Therapies for CACS
Cannabis has long been suggested as a well-tolerated, safe, and effective option to help patients cope with cancer related symptoms with fewer serious side effects than most prescription drugs currently used as anti-emetics, analgesics, and the like. As such, cannabinoids are finding application in palliative care for reducing nausea and vomiting, alleviating cancer pain, and stimulating appetite, as well as improving quality of life in cancer patients. Dronabinol (Marinol®) and nabilone (Cesamet®), two drugs based on synthetic cannabinoids, have each been approved by the FDA for the treatment of chemotherapy-related nausea in patients who do not respond to conventional antiemetic therapy. Another drug, nabiximols (Sativex®), a specific cannabis extract, is approved in Canada and the United Kingdom for symptomatic relief of pain in advanced cancer patients.
Despite interest in cannabinoid-based therapies as a treatment for CACS, their use has been limited by impediments beyond the legal status of cannabis. The most significant obstacle is the lack of clinical research demonstrating their efficacy. While there is evidence that cannabinoids improve appetite, body weight, body fat level, caloric intake, mood, and quality of life in cancer patients, the few studies on these effects have yielded mixed and inconclusive findings. In addition, some of these studies have suffered from methodological constraints that limit any ability to draw firm conclusions.
The therapeutic use of cannabinoids has also been inhibited by limitations associated with traditional administration routes that reduce their effectiveness. Smoking and ingestion of cannabis suffer from wide variability in potency due to a lack of standardized and reproducible formulations. The ingestion of unformulated cannabis has also been associated with poor absorption and low bioavailability versus other administration routes, requiring higher doses and a greater risk of negative side effects. Additionally, the lack of available information on cannabinoid strains has made it difficult for healthcare providers to establish dosing rates. In our experience, however, the principal concern of patients with respect to medical cannabis lies in the undesirable side effects, such as disorientation and dizziness, which result from significant variability in peak blood levels of active cannabinoids soon after administration. We further believe that these side effects, which are common among immediate release methods, are a significant factor in the failure of patients to adhere to recommended treatment regimens and are therefore a pervasive threat to their health and wellbeing.
Cannabics SR
Cannabics SR is an oral composition in the form of a hydroxypropylmethylcellulose (HPMC) capsule containing a patent-pending formulation of cannabinoid extracts suspended in a lipid emulsion. It provides a relatively rapid onset of action, typically within 30-40 minutes, followed by a gradual and sustained release of active cannabinoids, resulting in a steady state level of beneficial effects for up to 6 to 8 hours with each capsule. Cannabics SR provides a consistent, predictable concentration of cannabinoids with an absorption profile and bioavailability of active ingredients that we believe to be superior to other oral cannabinoid administrations. We believe that the multifactorial benefits of the active pharmaceutical ingredients in Cannabics SR address an unmet medical need for a safe and effective treatment of CACS, leading to improved patient adherence and better health outcomes.
Cannabics SR capsules contain only food grade materials without any artificial additives. The active ingredients of each capsule are standardized in composition, formulation, and dose, and are comprised of only pure, natural extracts of active cannabinoids from selected strains of medical cannabis. All excipients are recognized by the FDA as Generally Regarded as Safe.
In addition to the therapeutic potential of Cannabics SR as a treatment for CACS, we believe that our SR technology may be formulated to serve the unique needs of patients suffering from other indications for which a sustained release of a cannabinoid formulation may be beneficial.
| 52 |
Clinical Development
In 2016, we commenced a two-year pilot study to evaluate the influence of Cannabics SR capsules on CACS, and, in particular, on weight loss in advanced cancer patients. The study was led by Professor Gil Bar-Sela, the former Deputy Director of the Division of Oncology at Rambam Health Care Campus, Head of the Palliative and Supportive Oncology Unit, and Head of Service for Melanoma and Sarcoma Patients.
Patients were administered 2 × 10 mg of Cannabics SR per 24 hours for six months. During the study, after some patients reported several psychoactive side effects, the dosage of each capsule was reduced to 5 mg. Almost no side effects were reported with the 5 mg dosage. Participants were weighed at each physician visit. The primary objective of the study was a weight gain of ≥10% from baseline. Of 24 patients who agreed to participate in the study, 17 started the Cannabics SR treatment, but only 11 received the capsules for more than two weeks. Three of six patients who completed the study period met the primary end-point. The remaining three patients had stable weights. In quality of life questionnaires patients reported less appetite loss after the Cannabics SR treatment (p=0.05). According to patients’ self-reports, improvement in appetite and mood as well as a reduction in pain and fatigue was demonstrated.
Despite various limitations, the preliminary study demonstrated a weight increase of ≥10% in 3 out of 17 (17.6%) of patients with doses of 5 mg × 1 or 5 mg × 2 capsules daily, without significant side effects. The remaining patients had stable weights. Also, all patients who remained in the study for at least 4.5 months reported an increase in appetite, as did 83% of the patients who completed the study. For 50% of the patients who completed the study, there were reports of pain reduction and sleep improvement. Additional results showed a significant decrease of appetite loss complaints among 83% of the patients who completed the study. (See Bar-Sela, Gil et al. “The Effects of Dosage-Controlled Cannabis Capsules on Cancer-Related Cachexia and Anorexia Syndrome in Advanced Cancer Patients: Pilot Study.” Integrative Cancer Therapies vol. 18 (2019): 1534735419881498. doi:10.1177/1534735419881498.)
Figure 4: Appetite loss among the six patients who completed Cannabics SR treatment, as reported on European Organization of Research and Treatment of Cancer Quality of Life Questionnaire (EORTC QLC-C30)
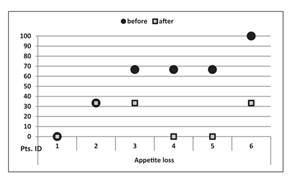
We intend to conduct additional pilot studies in Israel to assess the pharmacokinetics and pharmacodynamics of Cannabics SR in humans. These studies are expected to commence in 2021 at an anticipated cost of $250,000. Data from the pilot studies will guide our decisions regarding further clinical development and may better inform the design of our anticipated Phase 1 trials.
| 53 |
Commercialization
The results of our planned pilot studies may permit us to commercialize Cannabics SR in Israel under license by the Israeli Ministry of Health. If we are granted such a permit, we intend to engage a GMP manufacturer in Israel to produce Cannabics SR capsules for national distribution.
On May 13, 2020, the Israeli Ministry of Economy signed a Free Export Order, authorizing the export of GMP certified medical cannabis products from Israel. We are currently evaluating our export opportunities and optimal commercialization path for Cannabics SR across all available international markets, particularly with regard to the European Union, Canada, and Australia.
In 2019, we signed a letter of intent with NewCanna Hub to establish a joint venture for the production and marketing of Cannabics SR capsules in Colombia. NewCanna Hub specializes in genetic registration, large scale cultivation, research and development, manufacturing, and distribution. Under the joint venture, Cannabics SR capsules are expected to be produced in various formulations at NewCanna's Good Manufacturing Practice (GMP) certified facility in Columbia. The intention of the joint venture will be to seek international distribution agreements for Cannabics SR in relevant regulated territories.
Cannabics CDx Drug Sensitivity Test
Overview
Cannabics CDx is an ex-vivo drug sensitivity test that we are developing as a product candidate to provide healthcare providers with clinical decision support data from which they can identify, for a particular cancer patient undergoing cannabinoid therapy, which cannabinoids or cannabinoid combinations may have the most beneficial anti-cancer effects and which cannabinoids may be contraindicated.
Cancer and Personalized Medicine
The normal behavior of each cell in the human body is controlled by its genetic material, comprised of chains of deoxyribonucleic acid (DNA), units arranged in a particular order and packaged into condensed structures called chromosomes, inside the cell’s nucleus. The order of the DNA units as well as their three-dimensional structure dictates which protein and how much of it is made by each cell. Alterations in the DNA sequence, referred to as mutations, can disrupt normal protein function and are the leading cause of cancer development. Each person’s cancer has a unique combination of mutations, and as a cancer progresses, additional mutations accumulate. The number of cells within a growing tumor that carry a given mutation depends on when the mutation was acquired during tumor growth. Thus, even within the same tumor, different cancer cells often have different genetic mutations. This variation, or heterogeneity, within a tumor or between a primary and metastatic tumor, is a leading cause of resistance to treatment and thereby disease progression.
Comprehensive analyses of human cancer genomes over the past decade have revealed numerous genetic mutations that are associated with a variety of cancers. These discoveries have led to the development of molecular therapies targeted at rectifying the cellular changes that arise due to the mutations. While such therapeutics have improved patient outcomes, tumor heterogeneity among patients has limited the efficacy of these drugs to specific patient subtypes and contributed to relapse. In addition, intra-tumor heterogeneity leads to the emergence of drug resistant sub-populations of cancer cells, particularly while under sudden selective pharmacological pressure (such as chemotherapy) and further limits the efficiency of cancer treatment.
The diversity of cancer patients, including their biological characteristics and lifestyle factors, as well as the complex milieu of tumor cells within a single patient, has led to a novel, more personalized approach toward drug development and diagnosis. Personalized medicine aims to tailor each person’s health care to the prevention and treatment strategies most likely to be of benefit, sparing each person the cost of and potential harms from interventions and treatments that are unlikely to be of benefit. It has the potential to transform medical interventions by providing effective, tailored therapeutic strategies based on the genomic, epigenomic, and proteomic profile of a patient in the context of their personal situation. Personalized medicine may also improve health outcomes by reducing healthcare costs, drug-development costs, and time.
| 54 |
Cannabinoids and Personalized Medicine
While preclinical research during the last decade has stimulated interest in the therapeutic potential of cannabinoid compounds in oncology, one of the challenges facing healthcare providers and patients in selecting a cannabinoid based therapy has been the diversity of the cannabis plant, which encompasses thousands of distinct profiles, each with its own chemical composition and effects. After decades of highly restrictive regulation, there is presently a lack of clinically relevant information as to which cannabinoids are best suited to the unique medical needs of a patient across multiple lines of therapy. The result has left healthcare providers and patients at a loss as to which cannabinoids may be best suited to treat the unique cancer profiles of individual patients.
Cannabics CDx
We believe that the success of personalized medicine depends on the development of accurate and reliable diagnostics. Our goal is to expand the scope of personalized medicine across the cancer care continuum to include cannabinoid-based therapies and enable clinicians to make better informed decisions leading to improved clinical outcomes and lower healthcare costs. To that end, we are developing Cannabics CDx as a product candidate to provide clinical decision support data to healthcare providers interested in personalizing cannabinoid-based cancer therapies. We believe that by making cannabinoid therapy selection more accurate and accessible, Cannabics CDx may play a significant role in ushering medical cannabinoids into the mainstream of oncology.
Cannabics CDx is an innovative drug screening system that measures the effectiveness of cannabinoid compounds on a patient’s biopsy, identifies alternatives, and alerts healthcare providers to cannabinoids that may be contraindicated. Biopsied samples are delivered by courier to our laboratory, where we perform novel cannabinoid sensitivity tests on them using our high-content drug sensitivity screening integrated with our bioinformatics platform. We then apply advanced analytics to the test results and other relevant biological and clinical information provided by each patient’s healthcare provider to derive clinical support data from which healthcare providers can make better informed treatment decisions.
Figure 5: Sample personalized patient report produced by Cannabics CDx
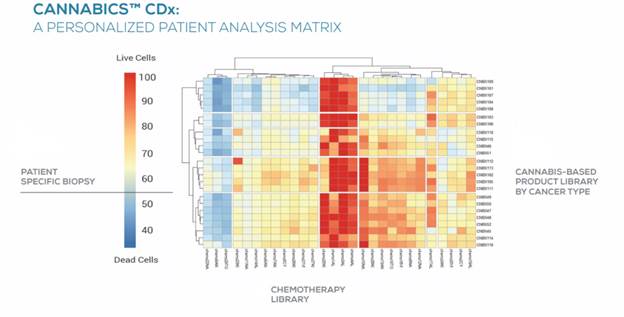
By enabling healthcare providers to more precisely tailor cannabinoid therapies to a patient’s cancer and clinical profile, we believe that Cannabics CDx will meet a significant unmet need of the growing population of cancer patients being treated with cannabis.
Validation
We are currently planning a clinical validation study expected to commence in 2022 to assess the sensitivity and specificity of Cannabics CDx. We are currently seeking strategic partners to collaborate on the validation and commercialization process.
| 55 |
Commercialization
Upon completion of our planned validation study, we will evaluate our options for commercializing Cannabics CDx with a view towards maximizing our return while expanding our collaborative network and opportunities in the rapidly emerging sector of pharmaceutical development. Consistent with our policies, we will not offer Cannabics CDx in any jurisdiction where it is not permitted or where it might otherwise be construed as a violation of law. In particular, we will not offer Cannabics CDx in the United States while cannabis is listed by the DEA as a Schedule I controlled substance.
Other Research and Development Programs
Our bioinformatics platform enables us to conduct a broad range of preclinical research and development activities to explore other uses of cannabinoids in the treatment of human diseases with unmet medical needs. While our current research focus is on the development of cannabinoid therapies and other technologies for the treatment of cancer, particularly cancers of the gastrointestinal tract, other areas of research include Alzheimer’s disease and auto immune diseases. These other programs are at various early stages of development and their continued progress is subject to available resources and our ability to secure necessary funding. We will determine which programs to continue based on several strategic factors, including economic potential and available resources. We may choose to partner with external parties for some or all of these programs.
We continue to explore and establish strategic partnerships with prominent companies and leading-edge research institutions in areas where we believe such relationships will benefit the further development of our product candidates and technologies. We may also out-license our product candidates and technologies to collaborative partners for the development of therapies of low strategic interest to the Company. We believe that these partnerships will enhance the value of our intellectual property and allow us to retain selected interests in such therapies without having to acquire the resources needed for in-house development.
Maripharm
On September 1, 2020, we entered into a memorandum of understanding with Maripharm Production B.V., a Dutch company that develops and grows unique cannabis strains, for a collaboration on a proof of concept evaluation of certain oil extracts from Maripharm’s cannabis strains whereby we will perform certain HTS screening to determine the necrolic and apoptotic effects of these strains upon cancer cell lines.
NewCanna Hub
On February 24, 2020, we entered into a collaboration agreement with NewCanna Hub for the purpose of examining the potential anti-tumor properties of five cannabinoid strains found in indigenous Colombian landraces on various gastrointestinal cancer cell lines. NewCanna has amassed one of the world's most extensive assortments of legally registered landraces and hybrid cannabis cultivars, including several landraces, unique to Colombia. The cannabis landraces to be studied as part of the collaboration are native plant populations that have grown for centuries, adapting to the environmental conditions of their geographical location, resulting in the development of unique characteristics over time.
RCKMC Ltd.
On February 5, 2020, we entered into a memorandum of understanding with RCKMC Ltd., an Israeli company specializing in the breeding of tailor-made strains of medical cannabis and the production of reliably homogeneous cannabis hybrid-seeds. The memorandum contemplates a research collaboration whereby RCKMC will provide raw cannabis flowers containing approximately 10-15 separate cannabinoid profiles to the Company, from which we will extract the resin and perform high-throughput screening for necrotic and apoptotic effects of extracts on gastrointestinal cancer cell lines. Subject to positive results from such research, the memorandum contemplates that the parties will enter into a joint venture agreement for the development of specific chemovars of medical cannabis specifically targeting gastro-intestinal cancers.
| 56 |
On March 11, 2020, we announced that our analysis of the cannabinoid profiles provided by RCKMC identified two specific cultivars demonstrating increased necrotic effects on gastric adenocarcinoma cells. The results will be used to further breed the selected cultivars as a possible source of active pharmaceutical ingredients in the development of pharmaco product candidates for the treatment of gastrointestinal and other forms of cancer.
Cannomed Medical Cannabis Industries Ltd.
On June 9, 2020, we entered into a memorandum of understanding with Cannomed Medical Cannabis Industries Ltd., a publicly traded Israeli company engaged in the production and distribution of medical cannabis. The memorandum contemplates a research collaboration whereby Cannomed Medical Cannabis Industries will provide the Company with raw cannabis plants representing 17 unique strains from which we will extract the resin and perform full high-throughput screening to determine their necrotic and apoptotic effects on cancer cell lines. If both parties are satisfied with the screening results, the memorandum further contemplates that they will enter into a joint venture agreement for the development of specific strains of medical cannabis with elevated chemovars specifically targeting cancers.
Commercial Operations
We have not established a sales, marketing, or product distribution infrastructure. We plan to commercialize any drugs we develop through licensing arrangements and strategic partnerships with established companies in the pharmaceutical industry having strong marketing capabilities and distribution networks. We generally intend to advance our drug candidates through Phase 1 and Phase 2 clinical trials as appropriate in order to establish their clinical and commercial potential before negotiating the terms of any licensing or collaboration. For other product candidates, such as Cannabics CDx, we may seek strategic partnerships earlier in their clinical development to accelerate the approval process, facilitate commercialization and mitigate risk. We believe that this approach will achieve the fullest marketing and distribution potential of any drugs or other products that we may develop in the short term.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. We believe that our scientific knowledge, experience, technology and development capabilities provide us with competitive advantages, but we face competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions, governmental agencies and public and private research institutions. Many of our current and potential competitors have longer operating histories and substantially greater financial, scientific, technical, intellectual property, regulatory and human resources than we have, as well as greater experience in developing and commercializing products, including obtaining FDA and other regulatory approvals.
As the medical use of cannabis increasingly receives government approval worldwide, we face growing competition from many new and existing companies seeking to develop cannabinoid-based therapies. Companies currently known to be developing cannabinoid-based human therapeutics include GW Pharmaceuticals PLC, Cannabis Science Inc., InMed Pharmaceuticals Inc., Emerald Bioscience Inc., Corbus Pharmaceuticals Holdings Inc., Zynerba Pharmaceuticals Inc., PharmaCyte Biotech, Inc., Tetra Bio-Pharma Inc., and Cure Pharmaceutical Holding Corp.
Many of our competitors are conducting research targeting the same technologies, applications, and markets as we are. Consequently, they may develop products for the same indications we are pursuing or may pursue in the future that are more effective, better tolerated, more widely-prescribed or accepted, more useful, and less costly. Any products that we successfully develop and commercialize will compete with existing products, as well as those currently in development or that may become available in the future.
In addition to competing for market position, we will also compete in terms of recruiting and retaining qualified personnel, acquiring intellectual property, establishing clinical trial sites, and enrolling patients for clinical trials and in obtaining funding.
| 57 |
Given the rapid changes affecting the global, national, and regional economies in general and cannabis-related medical research and development in particular, we may not be able to create and maintain a competitive advantage in the marketplace. Time-to-market is a critical factor in our industry and our success will depend on our ability to timely develop innovative technologies that will be accepted by patients. Our competitors may be better able to react to market changes, respond more rapidly to new regulations, or allocate greater resources to the development of their products than we can, which may result in our technologies and products becoming obsolete before we are able to enter the market, recover the expenses incurred to develop them, or generate significant revenue. Our success will depend, in part, upon our ability to develop our product candidates in a timely manner, keep our future products current with advancing technologies, achieve market acceptance of our future products, gain name recognition and a positive reputation in the healthcare industry, and establish successful marketing, sales, and distribution efforts. We cannot be certain that we will be able to compete against current or future competitors or that competitive pressure will not seriously harm our business prospects.
Research and Development
During the quarter ended November 30, 2020, we incurred research and development expenses of $433,730. During the years ended August 31, 2020, and August 31, 2019, we incurred research and development expenses of $1,682,462, and $1,543,759, respectively. All of our research and development expenses are directly related to the activities of our wholly-owned subsidiary, G.R.I.N. Ultra Ltd. See “Management’s Discussion and Analysis of Financial Condition and Results of Operations” elsewhere in this prospectus.
Patents
The proprietary nature of, and protection for, our technologies, processes, and know-how are vitally important to our business. Our success will substantially depend upon our ability to protect the proprietary nature of our technologies and know-how, to protect our technologies from infringement, misappropriation, discovery and duplication, to defend against any challenge or opposition and to operate without infringing on the proprietary rights of others.
We seek patent protection for the technologies, inventions and improvements that we consider important to the development of our business, but only in those cases where we believe that the costs of obtaining patent protection is justified by the scientific and commercial potential of the technology, and typically only in those jurisdictions that we believe present significant commercial opportunities. We also rely upon unpatented trade secrets, know-how, and continuing technological innovation to develop and maintain our competitive position. We seek to protect our ownership of know-how and trade secrets through an active program of legal mechanism including assignments, confidentiality agreements, material transfer agreements, research collaborations, and licenses.
While we cannot patent the naturally occurring individual cannabinoids used in our product candidates, there are a number of other approaches to protect our inventions, such as patenting cannabinoid combinations that provide novel compositions and methods for treating diseases, formulations designed specifically to increase the safety and efficacy of drug treatments, cannabinoid delivery technology, screening and manufacturing processes. We intend to design these patent methodologies so as to thoroughly protect our multi-faceted approach to the development of novel cannabinoid therapies. We do not intend to file for patent protection of our bioinformatics platform and data but instead plan to protect this proprietary asset as internal know-how.
We cannot be sure that any of our pending patent applications will be granted, or that any of the patents we own or obtain in the future will fully protect our position. Our patent rights and the patent rights of biotechnology and pharmaceutical companies in general, are highly uncertain and include complex legal and factual issues. We believe that our existing technology and the patents we hold or for which we have applied do not infringe any other patent rights. We believe our patent rights will provide meaningful protection against others duplicating our proprietary technologies. We cannot be sure of this, however, because of the complexity of the legal and scientific issues that could arise in litigation over these issues.
Despite these measures, any of our intellectual property and proprietary rights could be challenged, invalidated, circumvented, infringed, or misappropriated, or such intellectual property and proprietary rights may not be sufficient to permit us to take advantage of current market trends or otherwise to provide competitive advantages.
| 58 |
As of the date of this prospectus, our intellectual property portfolio includes the patent applications described in the following table, which have been filed with the U.S. Patent and Trademark Office, as well as know-how and trade secrets:
| Title | Application Type | Country | Status | Filing Date | ||||
| System and Method for High-throughput Screening of Cancer Cells | Non-provisional Utility patent application (national Phase) | United States | Published, Pending | 17/09/2017 | ||||
| System and Method for High-throughput Screening of Cancer Cells | National Phase Patent application | China | Published, Pending | 27/11/2017 | ||||
| System and Method for High-throughput Screening of Cancer Cells | National Phase Patent application | Japan | Published, Pending | 27/11/2017 | ||||
| System and Method for High-throughput Screening of Cancer Cells | National Phase Patent application | Canada | Pending, (not Published) | 13/09/2017 | ||||
| System and Method for High-throughput Screening of Cancer Cells | National Phase Patent application | India | Published, Pending | 15/09/2017 | ||||
| System and Method for High-throughput Screening of Cancer Cells | National Phase Patent | Israel | Granted | 18/09/2017 | ||||
| System and Method for High-throughput Screening of Cancer Cells | National Phase Patent application | Australia | Pending, (not Published) | 19/09/2017 | ||||
| System and Method for High-throughput Screening of Cancer Cells | National Phase Patent application | Europe | Published, Pending | 04/10/2017 | ||||
| System and Method for High-throughput Screening of Cancer Cells | National Phase Patent application | Brazil | Published, Pending | 10/10/2017 | ||||
| System and Method for High-throughput Screening of Cancer Cells | National Phase Patent application | Mexico | Pending, (not Published) | 10/10/2017 | ||||
| System and Method for High-throughput Screening of Cancer Cells | National Phase Patent | South Africa | Granted | 13/10/2017 | ||||
| System and Method for High-throughput Screening of Cancer Cells | National Phase Patent application | Hong Kong | Pending, (not Published) | 11/10/2018 | ||||
| Cannabinoid Compositions, Methods of Manufacture and Use Thereof | National Phase Patent application | Israel | Published, Pending | 24/02/2016 | ||||
| Cannabinoid Compositions, Methods of Manufacture and Use Thereof | Patent Co-operation Treaty, International patent application | Israel | Published, Pending | 23/02/2017 | ||||
| Cannabinoid Compositions, Methods of Manufacture and Use Thereof | Non-provisional Utility patent application (national Phase) | United States | Pending, (not Published) | 23/08/2018 | ||||
| The Effects of Dosage-Controlled Cannabis Capsules on Cancer- Related Cachexia and Anorexia Syndrome Among Advanced Cancer Patients | Provisional patent application | United States | Filed, Pending, (not Published) | 25/10/2018 | ||||
| Method for Sensitivity Testing of Cannabinoids on Patient-Derived Tumor Biopsies and CTCs | Patent Co-operation Treaty, International patent application | Israel | Filed, Pending, (not Published) | 02/01/2018 | ||||
| Novel System and Method for Microbiome Profiling and Modulation by Means of Cannabis Administration | Non-provisional Utility patent application (national Phase) | United States | Pending , (not Published) | 07/11/2018 | ||||
| A System and Method for Providing or Growing a Personalized Cannabis Product | Provisional patent application | United States | Filed, Pending, (not Published) | 17/09/2018 | ||||
| A System and Method for Providing Magnetic Targeting of Cannabinoids to Cancer Patients | Provisional patent application | United States | Filed, Pending, (not Published) | 28/11/2018 | ||||
| Personalized Cannabimetic Compositions Modeling and Production System and Methods Thereof | Provisional patent application | United States | Filed, Pending, (not Published) | 28/11/2018 | ||||
| A System and Method for Providing a Personalized Cannabis Derived Product for an Alcohol Withdrawal Syndrome (AWS) Patient | Provisional patent application | United States | Filed, Pending, (not Published) | 27/12/2018 | ||||
| A System and Method for Providing a Personalized Cannabis Derived Product for a Psychogenic Non-Epileptic Seizures (PNES) Patient | Provisional patent application | United States | Filed, Pending, (not Published) | 07/01/2019 |
We anticipate that we will file additional patent applications in conjunction with the research, testing, and development of our technologies.
| 59 |
Trademark
We have obtained trademark protection for CANNABICS in the United States. We intend to seek trademark protection in the United States and outside the United States where available and when appropriate. We intend to use these registered marks in connection with our biotechnological research and development, as well as with any products we may develop.
Licenses
All of our research and development activities are conducted in and limited to Israel. The product candidates and technologies we are developing contain or utilize cannabis, which is classified as a controlled substance under the Israeli Dangerous Drugs Ordinance [New Version], 5733 - 1973. In Israel, licenses to cultivate, possess, and use cannabis for medical research are granted by the Israeli Ministry of Health on an ad hoc basis.
We have been licensed by the Ministry of Health to possess and use cannabis for medical research since October 26, 2014. On November 28, 2017, we were issued a new research license for the characterization of anti-tumor activity of cannabinoids, which enables us to continue development of diagnostic tools and services for cancer patients that are receiving treatment with cannabis. While we have thus far been successful in obtaining all the licenses necessary to carry out our medical research, there can be no assurance that we will be able to acquire or maintain such licenses as we require them or at all.
Specialized Skill and Knowledge
The research and development of cannabinoid-based compounds and technologies requires specialized scientific and medical skill and knowledge. We believe that our current management offers a good combination of expertise in drug discovery, development, pre-clinical and clinical trials, as well as the licensing and commercialization of new investigative drugs. See “Management” and “Risk Factors”, elsewhere in this prospectus.
In addition to our management team, we have established an advisory board consisting of industry experts to supplement our internal capabilities with their expertise in the areas of finance, business development, cannabinoid science, formulation development and clinical practice. Our advisors generally meet twice a year as a group to assist us in formulating our research, development, clinical and sales and marketing strategies. Some individual advisors consult with and meet informally with us on a more frequent basis. Our advisors are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us. In addition, our advisors may have arrangements with other companies to assist those companies in developing products or technologies that may compete with ours.
As of the date of this prospectus, the following persons comprise our advisory board:
Dr. Gil Feiler – Advisory Board Chairman and Business Development Advisor. Dr. Feiler has served on the boards of Advanced Vision Technology Ltd. and Safra Bank Mutual Fund Division, as well as serving as Administrative Advisor for the government of Ras Al Khaimah, UAE. Dr. Feiler has published extensively on business opportunities and strategies and was a frequent speaker in world events, including the World Economic Forum in Davos.
| 60 |
Dr. Sigalit Ariely-Portnoy - Strategic Regulatory Advisor, Clinical, Quality and Validation. Dr. Sigalit Ariely-Portnoy has more than 17 years’ experience in the pharmaceutical industry. She has managed pharmaceutical and chemical plants at Taro Pharmaceutical Industries Ltd. as Operation Group Vice President and at Teva Pharmaceutical Industries Ltd. as Kfar-Saba OSD Plant Manager. Dr. Ariely-Portnoy managed Teva's largest plant worldwide (9 billion tablets per annum and more than $2B revenues). During her career, she has led more than 50 inspections by the FDA, the European Medicines Agency, the Israeli Ministry of Health and others. Dr. Ariely-Portnoy spearheaded the construction of a 200,000 sq. ft. pharmaceutical plant, several chemical plants and bio-warehouses, as well as many significant plant expansions for manufacturers of semisolids, liquids and oral solid dosage forms. Between the years 2003-2006, Dr. Ariely-Portnoy was the president of the Israel chapter of the Parenteral Drug Association. For the last five years, Dr. Ariely-Portnoy has managed Gsap, the leading consulting firm in Israel for providing professional services to pharmaceutical, medical device, biotechnology and cell therapy companies. Dr. Ariely-Portnoy received her B.Sc., M.Sc., and D.Sc. in the fields of chemical engineering and biomedical engineering from the Technion Institute of Technology in Haifa, Israel.
Dr. Danna Ben-Ami Shor, MD – Strategic Design and Implementation of Clinical Validation. Dr. Ben-Ami Shor is a recognized expert in invasive endoscopy and gastroenterology in the Sourasky Medical Center in Tel-Aviv, Israel. Dr. Ben-Ami Shor earned her M.D in 2009, graduating cum laude from the Sackler Faculty of Medicine, Tel Aviv University. She specialized in internal medicine and gastroenterology at the Sheba Medical Center, Israel. She also successfully completed an advanced endoscopy (ASGE) accredited fellowship within the Center for Interventional Endoscopy at AdventHealth, in Florida. Additionally, Dr. Ben-Ami Shor is proficient in both diagnostic and therapeutic endoscopic ultrasound (EUS), and endoscopic retrograde cholangiopancreatography (ERCP).
Prof. Zamir Halpern - Medical Advisor. Prof. Halpern, is a senior physician at the Gastroenterology Institute of the Sourasky Medical Center in Tel-Aviv, Israel, and current Chairman of the National Gastro Nutrition and Liver Diseases Council at the Israeli Ministry of Health. He has also served as Chairman of the Israeli Association of the Study of Liver, Chairman of the Israeli Gastroenterology Association and Chairman of the National Council for Food and Agriculture.
Dr. Tal Mofkadi - Financial Advisor. Dr. Mofkadi holds a Ph.D. in financial economics from Tel Aviv University. He is a lecturer at Tel Aviv University, the Interdisciplinary Center in Herzliya and universities abroad. He is the author of The Handbook of Corporate Valuation. Dr. Mofkadi provides expert opinions in financing, economic, and legal proceedings, writing valuations and optimal pricing policies as well as economic analysis of competition and regulatory aspects, risk assessment and more.
Dr. Erez Scapa, MD - Medical Advisor. Dr. Scapa earned his M.D. in 2000 at the Technion Institute of Technology in Haifa, Israel, cum laude. He later held a Research Fellowship in Hepatology at the Brigham and Women's Hospital, Harvard University, Boston, Massachusetts, as well as a fellowship in Endoscopic Submucosal Dissection at the NTT Medical Center in Tokyo, Japan. He is an expert in invasive endoscopy and has extensive experience in preforming colonoscopies and gastroscopies. He is proficient in both diagnostic as well as invasive endoscopic ultrasound and has served as the head of the Endoscopic Submucosal Dissection program in Tel-Aviv Sourasky Medical Center since 2019.
Prof. Amos Toren, MD - Medical Advisor. Prof. Toren has been the Director of the Pediatric Hemato-Oncology and Bone Marrow Transplant Department at the Sheba Medical Center since 2001, and is a senior lecturer at the Sackler School of Medicine, Tel-Aviv University. Prof. Toren is a specialist in pediatrics, general hematology and pediatric hemato-oncology. He has a Ph.D. in genetics and is qualified as a Master of Health Administration (MHA) at the Recannati Business School, Tel-Aviv University. Professor Toren runs numerous clinical research trials, including those that are investigator initiated, company initiated, unicenter and multicenter.
Government Regulation
Regulation of Pharmaceutical Products
Government authorities in the United States, at the federal, state and local level, and in other countries, extensively regulate, among other things, the research, development, testing, manufacture, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, import, and export of pharmaceutical products such as those we are developing. The processes for obtaining regulatory approvals in the United States and in foreign countries, along with subsequent compliance with applicable statutes and regulations, require the expenditure of substantial time and financial resources. A failure to comply with such laws and regulations or prevail in any enforcement action or litigation related to noncompliance could have a material adverse impact on our business, financial condition and results of operations and could cause the market value of our common stock to decline.
\
| 61 |
FDA Approval Process
In the United States, pharmaceutical products are subject to extensive regulation by the FDA. The Food, Drug, and Cosmetic Act and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as imposition of clinical holds, FDA refusal to approve a pending NDA, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement, civil penalties and criminal prosecution.
Pharmaceutical product development in the U.S. typically involves pre-clinical laboratory and animal tests and submission to the FDA of an IND, which must become effective before clinical testing may commence. For commercial approval, the sponsor must submit adequate tests by all methods reasonably applicable to show that the drug is safe for use under the conditions prescribed, recommended or suggested in the proposed labeling. The sponsor must also submit substantial evidence, generally consisting of adequate, well-controlled clinical trials to establish that the drug will have the effect it purports or is represented to have under the conditions of use prescribed, recommended or suggested in the proposed labeling. In certain cases, the FDA may determine that a drug is effective based on one clinical study plus confirmatory evidence. Satisfaction of FDA regulatory requirements for marketing approval typically takes many years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease.
Pre-clinical studies include in vitro and animal studies to assess the potential safety and efficacy of the product. Chemistry, manufacturing and controls tests include laboratory evaluation of product chemistry, formulation, quality attributes and the relationship between manufacturing process parameters and product characteristics. The conduct of pre-clinical tests must comply with federal regulations and requirements, including the FDA’s GLP regulations and the U.S. Department of Agriculture’s regulations implementing the Animal Welfare Act. The results of pre-clinical testing are submitted to the FDA as part of an IND along with other information, including information about product chemistry, manufacturing and controls, a proposed clinical trial protocol as well as results of previous clinical studies. Long-term pre-clinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has not imposed a clinical hold on the IND or otherwise commented or questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin.
Clinical trials involve the administration of the investigational new drug to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted: (i) in compliance with federal regulations, (ii) in compliance with GCP, an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators and monitors, and (iii) under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to the FDA as part of the IND.
The FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time or impose other sanctions if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The trial protocol and informed consent information for patients in clinical trials must also be submitted to an institutional review board (IRB) for approval. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements or may impose other conditions.
| 62 |
Clinical trials to support NDAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap. In Phase 1, the initial introduction of the drug into healthy human subjects or patients, the drug is tested to assess metabolism, pharmacokinetics, pharmacological actions, side effects associated with increasing doses, and, if possible, early evidence on effectiveness. Phase 2 usually involves trials in a limited patient population to determine the effectiveness of the drug for a particular indication, dosage tolerance, and optimum dosage, and to identify common adverse effects and safety risks. If a compound demonstrates evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to obtain the additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug and to provide adequate information for the labeling of the drug. In most cases, the FDA requires two adequate and well-controlled Phase 3 clinical trials to demonstrate the efficacy of the drug. A single Phase 3 trial with other confirmatory evidence may be sufficient in rare instances where the study is a large multi-center trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible.
Pursuant to the 21st Century Cures Act, the manufacturer of an investigational drug for a serious or life-threatening disease is required to make available, such as by posting on its website, its policy on evaluating and responding to requests for expanded access.
After completion of the required clinical testing, an NDA is prepared and submitted to the FDA. FDA approval of the NDA is required before marketing of the product may begin in the U.S. The NDA must include the results of all pre-clinical, clinical, and other testing and a compilation of data relating to the product’s pharmacology, chemistry, manufacture, and controls. The cost of preparing and submitting an NDA is substantial.
The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of NDAs. While most such applications for standard review drug products are reviewed within ten to twelve months; most applications for priority review drugs are reviewed in six to eight months. Priority review can be applied to drugs that the FDA determines offer major advances in treatment, or provide a treatment where no adequate therapy exists. The review process for both standard and priority review may be extended by the FDA for three additional months to consider certain late-submitted information or information intended to clarify information already provided in the submission.
The FDA may also refer applications for novel drug products, or drug products that present difficult questions of safety or efficacy, to an advisory committee (typically a panel that includes clinicians and other experts) for review, evaluation, and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the product unless compliance with current good manufacturing practice (cGMP), is satisfactory and the NDA contains data providing substantial evidence that the drug is safe and effective in the indication studied.
After the FDA evaluates the NDA and the manufacturing facilities, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing, or information, in order for the FDA to reconsider the application. If, or when, those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included.
An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require a risk evaluation and mitigation strategy (REMS) to help ensure that the benefits of the drug outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use (ETASU). ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the drug. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
| 63 |
Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented. An NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing NDAs.
Orphan Drugs
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition - generally a disease or condition that affects fewer than 200,000 individuals in the U.S (or affects more than 200,000 in the U.S. and for which there is no reasonable expectation that the cost of developing and making available in the U.S. a drug for such disease or condition will be recovered from sales in the U.S. of such drug). Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the generic identity of the drug and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The first NDA applicant to receive FDA approval for a particular active ingredient to treat a particular disease with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the U.S. for that product, for that indication. During the seven-year exclusivity period, the FDA may not approve any other applications to market the same drug for the same disease, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. If the FDA designates an orphan drug based on a finding of clinical superiority, the FDA must provide a written notification to the sponsor that states the basis for orphan designation, including “any plausible hypothesis” relied upon by the FDA. The FDA must also publish a summary of its clinical superiority findings upon granting orphan drug exclusivity based on clinical superiority. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA application user fee.
Pediatric Information
Under the Pediatric Research Equity Act (PREA), NDAs or supplements to NDAs must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may grant full or partial waivers, or deferrals, for submission of data. Unless otherwise required by regulation, PREA generally does not apply to a drug for an indication for which orphan designation has been granted; however, as of August 18, 2020, PREA will apply to NDAs for orphan-designated drugs if the drug is molecularly targeted cancer product intended for the treatment of an adult cancer and is directed at a molecular target that the FDA has determined is substantially relevant to the growth or progression of a pediatric cancer.
The Best Pharmaceuticals for Children Act (BPCA) provides NDA holders a six-month extension of any exclusivity (patent or non-patent) for a drug if certain conditions are met. Conditions for exclusivity include the FDA’s determination that information relating to the use of a new drug in the pediatric population may produce health benefits in that population, the FDA making a written request for pediatric studies, and the applicant agreeing to perform, and reporting on, the requested studies within the statutory timeframe. Applications under the BPCA are treated as priority applications, with all of the benefits that designation confers.
Fast Track Designation and Accelerated Approval
The FDA is required to facilitate the development and expedite the review of drugs that are intended for the treatment of a serious or life-threatening disease or condition for which there is no effective treatment and which demonstrate the potential to address unmet medical needs for the condition. Under the fast track program, the sponsor of a new product candidate may request that the FDA designate the product candidate for a specific indication as a fast track drug concurrent with, or after, the filing of the IND for the product candidate. The FDA must determine if the product candidate qualifies for fast track designation within 60 days of receipt of the sponsor’s request.
| 64 |
Under the fast track program, the FDA may designate a drug for fast-track status if it is intended to treat a serious or life-threatening illness and nonclinical or clinical data demonstrate the potential to address an unmet medical need. Similarly, the agency may designate a drug for accelerated approval if it treats a serious condition and generally provides meaningful therapeutic benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. In clinical trials, a surrogate endpoint is a measurement of laboratory or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient feels, functions or survives. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A product candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, will allow the FDA to withdraw the drug from the market on an expedited basis. All promotional materials for product candidates approved under accelerated regulations are subject to prior review by the FDA.
In addition to other benefits such as the ability to use surrogate endpoints and engage in more frequent interactions with the FDA, the FDA may initiate review of sections of a fast track drug’s NDA before the application is complete. This rolling review is available if the applicant provides, and the FDA approves, a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, the FDA’s time period goal for reviewing an application does not begin until the last section of the NDA is submitted. Additionally, the fast track designation may be withdrawn by the FDA if it believes that the designation is no longer supported by data emerging in the clinical trial process.
Special Protocol Assessment
A company may reach an agreement with the FDA under the special protocol assessment (SPA), process as to the required design and size of clinical trials intended to form the primary basis of an efficacy claim. According to its performance goals, the FDA is supposed to evaluate the protocol within 45 days of the request to assess whether the proposed trial is adequate, which evaluation may result in discussions and a request for additional information. An SPA request must be made before the proposed trial begins, and all open issues must be resolved before the trial begins. If a written agreement is reached, it will be documented and made part of the administrative record. Under the Food, Drug, and Cosmetic Act and FDA guidance implementing the statutory requirement, an SPA is generally binding upon the FDA except in limited circumstances, such as if the FDA identifies a substantial scientific issue essential to determining safety or efficacy after the study begins, public health concerns emerge that were unrecognized at the time of the protocol assessment, the sponsor and the FDA agree to the change in writing, or if the study sponsor fails to follow the protocol that was agreed upon with the FDA.
Breakthrough Therapy Designation
The FDA is required to expedite the development and review of an application for approval of a drug that is intended to treat a serious or life-threatening disease or condition where preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. Under the breakthrough therapy program, the sponsor of a new product candidate may request that the FDA designate the product candidate for a specific indication as a breakthrough therapy concurrent with, or after, the filing of the IND for the product candidate. The FDA must determine if the product candidate qualifies for breakthrough therapy designation within 60 days of receipt of the sponsor’s request.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of FDA-regulated products, including drugs, are required to register and disclose certain clinical trial information. Information related to the product, patient population, phase of investigation, trial sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to discuss the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed in certain circumstances for up to two years after the date of completion of the trial. Competitors may use this publicly-available information to gain knowledge regarding the progress of development programs.
| 65 |
Advertising and Promotion
Once an NDA is approved, a product will be subject to certain post-approval requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved labeling. Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented. An NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing NDAs.
Adverse Event Reporting and GMP Compliance
Adverse event reporting and submission of periodic reports is required following FDA approval of an NDA. The FDA also may require post-marketing testing, known as Phase 4 testing, REMS and surveillance to monitor the effects of an approved product, or the FDA may place conditions on an approval that could restrict the distribution or use of the product. In addition, quality-control, drug manufacture, packaging and labeling procedures must continue to conform to cGMPs after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies. Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects manufacturing facilities to assess compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality-control to maintain compliance with cGMPs. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered.
The Hatch-Waxman Act
Orange Book Listing
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent whose claims cover the applicant’s product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential generic competitors in support of approval of an abbreviated new drug application (ANDA). An ANDA provides for marketing of a drug product that has the same active ingredients in the same strengths and dosage form as the listed drug and has been shown through bioequivalence testing to be therapeutically equivalent to the listed drug. Other than the requirement for bioequivalence testing, ANDA applicants are not required to conduct, or submit results of, pre-clinical or clinical tests to prove the safety or effectiveness of their drug product. Drugs approved in this way are commonly referred to as “generic equivalents” to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA applicant is required to certify to the FDA concerning any patents listed for the approved product in the Orange Book. Specifically, the applicant must certify that (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new product. The ANDA applicant may also elect to submit a Section VIII statement certifying that its proposed ANDA label does not contain (or carve out) any language regarding the patented method-of-use rather than certify to a listed method-of-use patent. If the applicant does not challenge the listed patents, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired. A certification that the new product will not infringe the already approved product’s listed patents, or that such patents are invalid, is called a Paragraph IV certification. If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit, or a decision in the infringement case that is favorable to the ANDA applicant.
| 66 |
The ANDA application also will not be approved until any applicable non-patent exclusivity listed in the Orange Book for the referenced product has expired.
Exclusivity
Upon NDA approval of a new chemical entity (NCE), which is a drug that contains no active moiety that has been approved by the FDA in any other NDA, that drug receives five years of marketing exclusivity during which the FDA cannot receive any ANDA seeking approval of a generic version of that drug. Certain changes to a drug, such as the addition of a new indication to the package insert, are associated with a three-year period of exclusivity during which the FDA cannot approval an ANDA for a generic drug that includes the change. An ANDA may be submitted one year before NCE exclusivity expires if a Paragraph IV certification is filed. If there is no listed patent in the Orange Book, there may not be a Paragraph IV certification, and, thus, no ANDA may be filed before the expiration of the exclusivity period.
Patent Term Extension
After NDA approval, owners of relevant drug patents may apply for up to a five-year patent extension. The allowable patent term extension is calculated as half of the drug’s testing phase (the time between IND application and NDA submission) and all of the review phase (the time between NDA submission and approval up to a maximum of five years). The time can be shortened if the FDA determines that the applicant did not pursue approval with due diligence. The total patent term after the extension may not exceed 14 years and only one patent may be extended.
For patents that might expire during the application phase, the patent owner may request an interim patent extension. An interim patent extension increases the patent term by one year and may be renewed up to four times. For each interim patent extension granted, the post-approval patent extension is reduced by one year. The director of the United States Patent and Trademark Office must determine that approval of the drug covered by the patent for which a patent extension is being sought is likely. Interim patent extensions are not available for a drug for which an NDA has not been submitted.
International Drug Review and Approval
In addition to regulations in the United States, we will be subject to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products. The approval process varies from country to country, and the time may be longer or shorter that that required for FDA approval. If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension of clinical trials, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Some countries outside of the United States have a similar process that requires the submission of a clinical trial application (CTA), much like the IND prior to the commencement of human clinical trials. In the European Union, for example, a CTA must be submitted to the national health authority of each European Union (E.U.) Member State in which the clinical trial is to be conducted and an independent ethics committee, much like the FDA and IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed.
| 67 |
To obtain regulatory approval to commercialize a new drug under European Union regulatory systems, we must submit a marketing authorization application (MAA). In the European Union, marketing authorization for a medicinal product can be obtained through a centralized, mutual recognition, decentralized procedure, or the national procedure of an individual E.U. Member State. In accordance with the centralized procedure, the applicant can submit a single application for marketing authorization to the European Medicines Agency (EMA), to be assessed by the Committee of Medicinal Products for Human Use. The agency will provide a positive opinion regarding the application if it meets certain quality, safety, and efficacy requirements. Following the opinion of the EMA, the European Commission makes a final decision to grant a centralized marketing authorization that permits the marketing of a product in all 27 E.U. Member States and three of the four European Free Trade Association States, Iceland, Liechtenstein and Norway. The centralized procedure is mandatory for certain medicinal products, including orphan medicinal products, medicinal products derived from certain biotechnological processes, advanced therapy medicinal products and certain other medicinal products containing a new active substance for the treatment of certain diseases. This route is optional for certain other products, including medicinal products that are a significant therapeutic, scientific or technical innovation, or whose authorization would be in the interest of public or animal health.
Unlike the centralized authorization procedure, the decentralized marketing authorization procedure requires a separate application to, and leads to separate approval by, the competent authorities of each E.U. Member State in which the product is to be marketed. This application process is identical to the application that would be submitted to the EMA for authorization through the centralized procedure. The reference E.U. Member State prepares a draft assessment and drafts of the related materials within 120 days after receipt of a valid application. The resulting assessment report is submitted to the concerned E.U. Member States who, within 90 days of receipt, must decide whether to approve the assessment report and related materials. If a concerned E.U. Member State cannot approve the assessment report and related materials due to concerns relating to a potential serious risk to public health, disputed elements may be referred to the European Commission, whose decision is binding on all E.U. Member States.
The mutual recognition procedure is similarly based on the acceptance by the competent authorities of the E.U. Member States of the marketing authorization of a medicinal product by the competent authorities of other E.U. Member States. The holder of a national marketing authorization may submit an application to the competent authority of an E.U. Member State requesting that this authority recognize the marketing authorization delivered by the competent authority of another E.U. Member State.
For other countries outside of the European Union, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In Israel and internationally, clinical trials are generally required to be conducted in accordance with GCP, applicable regulatory requirements of each jurisdiction and the medical ethics principles that have their origin in the Declaration of Helsinki.
Data Exclusivity
In the European Union, marketing authorization applications for generic medicinal products are not required to include the results of pre-clinical and clinical trials, but instead can refer to the data included in the marketing authorization of a reference product for which regulatory data exclusivity has expired. If a marketing authorization is granted for a medicinal product containing a new active substance, that product benefits from eight years of data exclusivity, during which generic marketing authorization applications referring to the data of that product may not be accepted by the regulatory authorities, and a further two years of market exclusivity, during which such generic products may not be placed on the market. The two-year period may be extended to three years if during the first eight years a new therapeutic indication with significant clinical benefit over existing therapies is approved.
| 68 |
Orphan Medicinal Products
The EMA’s Committee for Orphan Medicinal Products (COMP), may recommend orphan medicinal product designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than 5 in 10,000 persons in the European Union. Additionally, designation is granted for products intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the product in the European Union would be sufficient to justify the necessary investment in developing the medicinal product. The COMP may only recommend orphan medicinal product designation when the product in question offers a significant clinical benefit over existing approved products for the relevant indication. Following a positive opinion by the COMP, the European Commission adopts a decision granting orphan status. The COMP will reassess orphan status in parallel with EMA review of a marketing authorization application and orphan status may be withdrawn at that stage if it no longer fulfills the orphan criteria (for instance because in the meantime a new product was approved for the indication and no convincing data are available to demonstrate a significant benefit over that product). Orphan medicinal product designation entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity is granted following marketing authorization. During this period, the competent authorities may not accept or approve any similar medicinal product, unless it offers a significant clinical benefit. This period may be reduced to six years if the orphan medicinal product designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Pediatric Development
In the European Union, companies developing a new medicinal product must agree to a Pediatric Investigation Plan (PIP), with the EMA and must conduct pediatric clinical trials in accordance with that PIP unless a waiver applies, for example, because the relevant disease or condition occurs only in adults. The marketing authorization application for the product must include the results of pediatric clinical trials conducted in accordance with the PIP, unless a waiver applies, or a deferral has been granted, in which case the pediatric clinical trials must be completed at a later date. Products that are granted a marketing authorization on the basis of the pediatric clinical trials conducted in accordance with the PIP are eligible for a six-month extension of the protection under a supplementary protection certificate (if the product covered by it qualifies for one at the time of approval). This pediatric reward is subject to specific conditions and is not automatically available when data in compliance with the PIP are developed and submitted.
U.S. Regulation of Controlled Substances
The federal Controlled Substances Act of 1970 (CSA), and its implementing regulations establish a “closed system” of regulations for controlled substances. The CSA imposes registration, security, recordkeeping and reporting, storage, manufacturing, distribution, importation and other requirements under the oversight of the DEA. The DEA is the federal agency responsible for regulating controlled substances, and requires those individuals or entities that manufacture, import, export, distribute, research, or dispense controlled substances to comply with the regulatory requirements in order to prevent the diversion of controlled substances to illicit channels of commerce.
The DEA categorizes controlled substances into one of five schedules (Schedule I, II, III, IV or V) with varying qualifications for listing in each schedule. Schedule I substances by definition have a high potential for abuse, have no currently accepted medical use in treatment in the U.S. and lack accepted safety for use under medical supervision. Pharmaceutical products having a currently accepted medical use that are otherwise approved for marketing may be listed as Schedule II, III, IV or V substances, with Schedule II substances presenting the highest potential for abuse and physical or psychological dependence, and Schedule V substances presenting the lowest relative potential for abuse and dependence.
Facilities that manufacture, distribute, import or export any controlled substance must register annually with the DEA. The DEA registration is specific to the particular location, activities and controlled substance schedules. For example, separate registrations are required for importation and manufacturing activities, and each registration authorizes which schedules of controlled substances the registrant may handle. However, certain coincident activities are permitted without obtaining a separate DEA registration, such as distribution of controlled substances by the manufacturer that produces them.
| 69 |
The DEA inspects all manufacturing facilities to review security, recordkeeping, reporting and handling prior to issuing a controlled substance registration. The specific security requirements vary by the type of business activity and the schedule and quantity of controlled substances handled. The most stringent requirements apply to manufacturers of Schedule I and Schedule II substances. Required security measures commonly include background checks on employees and physical control of controlled substances through storage in approved vaults, safes and cages, and through use of alarm systems and surveillance cameras. An application for a manufacturing registration as a bulk manufacturer (not a dosage form manufacturer or a repacker/relabeler) for a Schedule I or II substance must be published in the Federal Register, and is open for 60 days to permit interested persons to submit comments, objections or requests for a hearing. A copy of the notice of the Federal Register publication is simultaneously forwarded by DEA to all those registered, or applicants for registration, as bulk manufacturers of that substance. Once registered, manufacturing facilities must maintain records documenting the manufacture, receipt and distribution of all controlled substances. Manufacturers must submit periodic reports to the DEA of the distribution of Schedule I and II controlled substances, Schedule III narcotic substances, and other designated substances. Registrants must also report any controlled substance thefts or significant losses, and must obtain authorization to destroy or dispose of controlled substances. As with applications for registration as a bulk manufacturer, an application for an importer registration for a Schedule I or II substance must also be published in the Federal Register, which remains open for 30 days for comments. Imports of Schedule I and II controlled substances for commercial purposes are generally restricted to substances not already available from a domestic supplier or where there is not adequate competition among domestic suppliers. In addition to an importer or exporter registration, importers and exporters must obtain a permit for every import or export of a Schedule I and II substance or Schedule III, IV and V narcotic, and submit import or export declarations for Schedule III, IV and V non-narcotics. In some cases, Schedule III non-narcotic substances may be subject to the import and export permit requirements, if necessary to ensure that the U.S. complies with its obligations under international drug control treaties.
For drugs manufactured in the U.S., the DEA establishes annually an aggregate quota for the amount of substances within Schedules I and II that may be manufactured or produced in the U.S. based on the DEA’s estimate of the quantity needed to meet legitimate medical, scientific, research and industrial needs. This limited aggregate amount of cannabis that the DEA allows to be produced in the U.S. each year is allocated among individual companies, which, in turn, must annually apply to the DEA for individual manufacturing and procurement quotas. The quotas apply equally to the manufacturing of the active pharmaceutical ingredient and production of dosage forms. The DEA may adjust aggregate production quotas a few times per year, and individual manufacturing or procurement quotas from time to time during the year, although the DEA has substantial discretion in whether or not to make such adjustments for individual companies.
The U.S. states also maintain separate controlled substance laws and regulations, including licensing, recordkeeping, security, distribution, and dispensing requirements. State authorities, including Boards of Pharmacy, regulate use of controlled substances in each state. Failure to maintain compliance with applicable requirements, particularly as manifested in the loss or diversion of controlled substances, can result in enforcement action that could have a material adverse effect on our business, operations and financial condition. The DEA may seek civil penalties, refuse to renew necessary registrations, or initiate proceedings to revoke those registrations. In certain circumstances, violations could lead to criminal prosecution.
All of our operations are currently conducted in Israel, under license from the Israeli Ministry of Health. The Company does not and will not possess, manufacture, distribute or dispense cannabis in the United States while such activities are not permitted under U.S. federal law or in any other jurisdiction where such activities are not permitted. The regulation of cannabis, cannabis extracts and some cannabinoids may, however, limit our ability and that of our strategic partners, to conduct clinical trials, market our product candidates and to become profitable.
The Single Convention on Narcotic Drugs, 1961
Many countries, including the United States, are parties to the 1961 Single Convention on Narcotic Drugs, which governs international trade and domestic control of narcotic substances, including cannabis and cannabis extracts. Countries may interpret and implement their treaty obligations in a way that creates a legal obstacle to the Company obtaining marketing approval for our products in those countries. These countries may not be willing or able to amend or otherwise modify their laws and regulations to permit our products to be marketed or achieving such amendments to the laws and regulations may take a prolonged period of time. In that case, we would be unable to market our products in those countries in the near future or perhaps at all.
| 70 |
E.U. Regulation of In vitro Diagnostics
We expect that Cannabics CDx will be subject to regulation in the E.U. as an in vitro medical device. In the E.U., in vitro medical devices are currently required to conform with the essential requirements of the E.U. Directive on in vitro diagnostic medical devices (Directive No 98/79/EC, as amended). To demonstrate compliance with the essential requirements, the manufacturer must undergo a conformity assessment procedure. The conformity assessment varies according to the type of medical device and its classification. The conformity assessment of in vitro diagnostic medical devices can require the intervention of an accredited E.U. Notified Body. If successful, the conformity assessment concludes with the drawing up by the manufacturer of an EC Declaration of Conformity entitling the manufacturer to affix the CE mark to its products and to sell them throughout the E.U.. On April 5, 2017, the E.U. adopted the new In vitro Device Regulation (EU) 2017/746 (IVDR), which repeals and replaces Directive No 98/79/EC. Unlike directives, which must be implemented into the national laws of the E.U. member states, a regulation is directly applicable, i.e., without the need for adoption of E.U. member state laws implementing them, in all E.U. member states. The IVDR, among other things, is intended to establish a uniform, transparent, predictable and sustainable regulatory framework across the E.U. for in vitro diagnostic medical devices and ensure a high level of safety and health while supporting innovation. The IVDR will not become fully applicable until five years following its entry into force. Once applicable, the IVDR will among other things:
| · | strengthen the rules on placing devices on the market and reinforce surveillance once they are available; |
| · | establish explicit provisions on manufacturers’ responsibilities for the follow-up of the quality, performance and safety of devices placed on the market; |
| · | improve the traceability of medical devices throughout the supply chain to the end-user or patient through a unique identification number; and |
| · | set up a central database to provide patients, healthcare professionals and the public with comprehensive information on products available in the E.U. |
Healthcare Reform
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively the Affordable Care Act, was intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Among the provisions of the Affordable Care Act that have been implemented since enactment and are of importance to the pharmaceutical industry are the following:
| · | an annual, non-deductible fee on any entity that manufactures or imports specified branded prescription drugs or biologic agents; |
| · | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program; |
| · | expansion of healthcare fraud and abuse laws, including the False Claims Act and the Anti-Kickback Statute, new government investigative powers, and enhanced penalties for noncompliance; |
| · | a Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for a manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| · | extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| 71 |
| · | expansion of eligibility criteria for Medicaid programs; |
| · | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; |
| · | requirements to report certain financial arrangements with physicians and teaching hospitals; |
| · | a requirement to annually report certain information regarding drug samples that manufacturers and distributors provide to physicians; and |
| · | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
In addition, other legislative changes have been proposed and adopted since passage of the Affordable Care Act. The Budget Control Act of 2011, among other things, created the Joint Select Committee on Deficit Reduction to recommend proposals in spending reductions to Congress. The Joint Select Committee did not achieve its targeted deficit reduction of an amount greater than $1.2 trillion for the fiscal years 2012 through 2021, triggering the legislation’s automatic reductions to several government programs. These reductions included aggregate reductions to Medicare payments to healthcare providers of up to 2.0% per fiscal year, which went into effect in April 2013. Subsequent litigation extended the 2% reduction, on average, to 2025.
There have been significant ongoing efforts to modify or eliminate the Affordable Care Act. For example, the Tax Cuts and Jobs Act enacted on December 22, 2017 repealed the shared responsibility payment for individuals who fail to maintain minimum essential coverage under section 5000A of the Internal Revenue Code, commonly referred to as the individual mandate, beginning in 2019. The Joint Committee on Taxation estimates that the repeal will result in over 13 million fewer Americans maintaining their health insurance coverage over the next ten years and is likely to lead to increases in insurance premiums.
On January 20, 2017, the President signed an executive order directing federal agencies to exercise existing authorities to reduce burdens associated with the Affordable Care Act pending further action by Congress. In April 2018, the Centers for Medicare & Medicaid Services (CMS) issued a final rule and guidance documents which changed requirements for health plans sold through the Affordable Care Act marketplaces for 2019. These changes include, for example, (i) turning over responsibility for ensuring that marketplace plans have enough health care providers in their networks to the states that rely on the federal HealthCare.gov exchange; (ii) allowing states to alter aspects of the essential health benefits required of health plans sold through the federal and state insurance marketplaces; (iii) eliminating certain Small Business Health Options Program regulatory requirements; and (iv) outlining criteria by which insurers may reduce the percentage of income allocated to patient care. The U.S. Department of Labor issued a final rule in June 2018 to expand the availability of association health plans available to small business owners and self-employed individuals, beginning on September 1, 2018. These association health plans will not be required to provide the essential health benefits mandated by the Affordable Care Act. These and other regulations may impact coverage of certain health care services.
In 2018, Congress proposed further legislation to repeal or revise the Affordable Care Act, which if enacted, may have a significant impact on the health care system. We expect that further changes to the Affordable Care Act, as well as other healthcare reform measures that have been and may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product, and could seriously harm our future revenue. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may compromise our ability to generate revenue, attain profitability or commercialize our product candidates.
| 72 |
Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any products for which we obtain regulatory approval. In the United States and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of reimbursement from third-party payors. Third-party payors include government health administrative authorities, managed care providers, private health insurers and other organizations. The process for determining whether a payor will provide coverage for a drug product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the drug product. Third-party payors may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA approved drugs for a particular indication. Third-party payors are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain FDA approvals. Our product candidates, if approved, may not be considered medically necessary or cost-effective. A payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003 changed the way Medicare covers and pays for pharmaceutical products, including creating the Medicare Part D prescription drug benefit, which became effective at the beginning of 2006. Government payment for some of the costs of prescription drugs may increase demand for any products for which we receive marketing approval. However, to obtain payments under this program, we would be required to sell products to Medicare recipients through prescription drug plans operating pursuant to this legislation. These plans will likely negotiate discounted prices for our products. In addition, we will be subject to the rules and regulations issued by CMS from time to time for Medicare Part D, such as the requirement, effective January 1, 2021, to include drug price increases and lower cost therapeutic alternatives on its Part D Explanation of Benefits that Medicare Part D sends members to inform Medicare beneficiaries about possible ways to lower their out of pocket costs by considering a lower cost medication.
Existing federal law requires pharmaceutical manufacturers to pay rebates to state governments, based on a statutory formula, on covered outpatient drugs reimbursed by the Medicaid program as a condition of having their drugs paid for by Medicaid. Rebate amounts for a product are determined by a statutory formula that is based on prices defined in the statute: average manufactured price (AMP), which must be calculated for all products that are covered outpatient drugs under the Medicaid program, and best price, which must be calculated only for those covered outpatient drugs that are a single source drug or innovator multiple source drug, such as biologic products. Manufacturers are required to report AMP and best price for each of their covered outpatient drugs to the government on a regular basis. Additionally, some state Medicaid programs have imposed a requirement for supplemental rebates over and above the formula set forth in federal law, as a condition for coverage. In addition to the Medicaid rebate program, federal law also requires that if a pharmaceutical manufacturer wishes to have its outpatient drugs covered under Medicaid as well as under Medicare Part B, it must sign a “Master Agreement” obligating it to provide a formulaic discount of approximately 24% known as the federal ceiling price for drugs sold to the U.S. Departments of Defense (including the TRICARE retail pharmacy program), Veterans Affairs, the Public Health Service and the Coast Guard, and also provide discounts through a drug pricing agreement meeting the requirements of Section 340B of the Public Health Service Act, for outpatient drugs sold to certain specified eligible healthcare organizations. The formula for determining the discounted purchase price under the 340B drug pricing program is defined by statute and is based on the AMP and rebate amount for a particular product as calculated under the Medicaid drug rebate program, discussed above.
Different pricing and reimbursement schemes exist in other countries. In the European Union, each E.U. Member States can restrict the range of medicinal products for which its national health insurance system provides reimbursement and can control the prices of medicinal products for human use marketed on its territory. As a result, following receipt of marketing authorization in an E.U. Member State, through any application route, the applicant is required to engage in pricing discussions and negotiations with the competent pricing authority in the individual E.U. Member State. The governments of the E.U. Member States influence the price of pharmaceutical products through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. Some E.U. Member States operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed upon. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies. Other E.U. Member States allow companies to fix their own prices for medicines, but monitor and control company profits. Others adopt a system of reference pricing, basing the price or reimbursement level in their territories either on the pricing and reimbursement levels in other countries or on the pricing and reimbursement levels of medicinal products intended for the same therapeutic indication. Further, some E.U. Member States approve a specific price for the medicinal product or may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. The downward pressure on healthcare costs in general, particularly prescription drugs, has become more intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, we may face competition for our product candidates from lower-priced products in foreign countries that have placed price controls on pharmaceutical products. In addition, in some countries, cross-border imports from low-priced markets exert a commercial pressure on pricing within a country.
| 73 |
Health Technology Assessment (HTA), of medicinal products, however, is becoming an increasingly common part of the pricing and reimbursement procedures in some E.U. Member States. These E.U. Member States include the United Kingdom, France, Germany, Ireland, Italy and Sweden. HTA is the procedure according to which the assessment of the public health impact, therapeutic impact and the economic and societal impact of use of a given medicinal product in the national healthcare systems of the individual country is conducted. HTA generally focuses on the clinical efficacy and effectiveness, safety, cost, and cost-effectiveness of individual medicinal products as well as their potential implications for the healthcare system. Those elements of medicinal products are compared with other treatment options available on the market.
The outcome of HTA regarding specific medicinal products will often influence the pricing and reimbursement status granted to these medicinal products by the competent authorities of individual E.U. Member States. The extent to which pricing and reimbursement decisions are influenced by the HTA of the specific medicinal product varies between E.U. Member States.
In addition, pursuant to Directive 2011/24/EU on the application of patients’ rights in cross-border healthcare, a voluntary network of national authorities or bodies responsible for HTA in the individual E.U. Member States was established. The purpose of the network is to facilitate and support the exchange of scientific information concerning HTAs. This may lead to harmonization of the criteria taken into account in the conduct of HTAs between E.U. Member States and in pricing and reimbursement decisions and may negatively affect price in at least some E.U. Member States.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, an increasing emphasis on managed care in the United States has increased and will continue to increase the pressure on pharmaceutical pricing. With few exceptions (e.g., limitations on Medicare Part D sponsors concerning certain formulary changes), coverage policies and third-party reimbursement rates may change at any time.
Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Other Healthcare Laws and Compliance Requirements
In the U.S., our planned activities are potentially subject to additional regulation, particularly if third-party reimbursement becomes available for one or more of any products we may develop, by various federal, state and local authorities in addition to the FDA, including CMS, other divisions of the Department of Health and Human Services (HHS), the DOJ and individual U.S. Attorney offices within the DOJ, and state and local governments.
A variety of federal and state laws prohibit fraud and abuse. These laws are interpreted broadly and enforced aggressively by various state and federal agencies, including CMS, the Department of Justice, the Office of Inspector General, for HHS, and various state agencies. In addition, the Medicare and Medicaid programs increasingly use a variety of contractors to review claims data and to identify improper payments as well as fraud and abuse. These contractors include Recovery Audit Contractors, Medicaid Integrity Contractors and Zone Program Integrity Contractors. In addition, CMS conducts Comprehensive Error Rate Testing audits, the purpose of which is to detect improper Medicare payments. Any overpayments identified must be repaid unless a favorable decision is obtained on appeal. In some cases, these overpayments can be used as the basis for an extrapolation, by which the error rate is applied to a larger universe of claims, and which can result in even higher repayments.
The Health Insurance Portability and Accountability Act (HIPAA), as amended by the Health Information Technology for Economic and Clinical Health Act (HITECH), imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program, including private third-party payors and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement or representation, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry in connection with the delivery of or payment for healthcare benefits, items or services. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from federal health care programs, such as the Medicare and Medicaid programs.
| 74 |
The federal False Claims Act prohibits any person from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds or knowingly making, using or causing to be made or used, a false record or statement material to an obligation to pay money to the government, or knowingly and improperly avoiding, decreasing or concealing an obligation to pay money to the federal government. Pharmaceutical and other healthcare companies have been investigated and reached substantial financial settlements under these laws for, among other things, allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies’ marketing of the product for unapproved, and thus non-reimbursable, uses. Pharmaceutical and other healthcare companies are also subject to other federal false claims laws, including, among others, federal criminal healthcare fraud and false statement statutes that extend to non-government health benefit programs.
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting, receiving, or providing remuneration, directly or indirectly, to induce or in return for either the referral of an individual, or the furnishing, recommending, or arranging for the purchase, lease or order of any health care item or service reimbursable, in whole or in part, under a federal health care program. The definition of “remuneration” has been broadly interpreted to include anything of value, including gifts, discounts, credit arrangements, payments of cash, ownership interests and providing anything at less than its fair market value. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, and formulary managers on the other. Although there are a number of statutory exemptions and regulatory safe harbors protecting some business arrangements from prosecution, the exemptions and safe harbors are drawn narrowly and practices that involve remuneration intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exemption or safe harbor. The reach of the Anti-Kickback Statute was broadened by the Affordable Care Act, which, among other things, amends the intent requirement of the federal Anti-Kickback Statute. Pursuant to the statutory amendment, a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. In addition, the Affordable Care Act provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act or the Civil Monetary Penalties Law, which imposes penalties against any person who is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent.
The federal Physician Payment Sunshine Act, being implemented as the Open Payments Program, requires certain pharmaceutical and biological manufacturers to engage in extensive tracking of payments or transfers of value to physicians and teaching hospitals and public reporting of the payment data. Pharmaceutical and biological manufacturers with products for which payment is available under Medicare, Medicaid or the State Children’s Health Insurance Program are required to track such payments, and must submit a report on or before the 90th day of each calendar year disclosing reportable payments made in the previous calendar year.
Many states have similar fraud and abuse statutes or regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Some state laws also require pharmaceutical companies to report expenses relating to the marketing and promotion of pharmaceutical products and to report gifts and payments to certain health care providers in those states. Some of these states also prohibit certain marketing-related activities including the provision of gifts, meals, or other items to certain health care providers. In addition, California, Connecticut, Nevada and Massachusetts require pharmaceutical companies to implement compliance programs or marketing codes of conduct.
Because of the breadth of these laws and the narrowness of available statutory and regulatory exemptions, it is possible that some of our business activities could be subject to challenge under one or more of such laws. If our operations are found to be in violation of any of the federal and state laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including criminal and significant civil monetary penalties, damages, fines, imprisonment, exclusion from participation in government programs, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre-marketing product approvals, private “qui tam” actions brought by individual whistleblowers in the name of the government or refusal to allow us to enter into supply contracts, including government contracts, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. To the extent that any of our products are sold in a foreign country, we may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws, and implementation of corporate compliance programs and reporting of payments or transfers of value to healthcare professionals.
| 75 |
In addition, at the federal level, the Drug Supply Chain Security Act (DSCA), regulates the distribution and tracing of prescription drugs. The DSCA imposes requirements to ensure accountability in prescription drug distribution, for example, it requires manufacturers to affix a product identifier to each package and case of a prescription drug product intended for sale. A product identifier is an electronically-readable graphic that contains information including the product’s unique numerical identifier, lot number, and expiration date. The DSCA also requires relevant parties and to identify and remove illegitimate products from the market, including products that are counterfeit, stolen, intentionally contaminated, or otherwise harmful. The Prescription Drug Marketing Act, its implementing regulations and state laws also regulate the distribution of prescription drug product samples.
In order to distribute products commercially, we must also comply with state law requirements for registration of manufacturers and wholesale distributors of pharmaceutical products, including, in some states, manufacturers and distributors who ship products into the state even if such manufacturers or distributors have no place of business within the state. Several states, and more recently some large cities, have enacted legislation requiring pharmaceutical companies to, among other things, establish marketing compliance programs, file periodic reports with the state, register their sales representatives, and/or limit other specified sales and marketing practices. All of our activities are potentially subject to federal and state consumer protection and unfair competition laws.
The Foreign Corrupt Practices Act
The Foreign Corrupt Practices Act (FCPA), prohibits any U.S. individual or business from paying, offering, or authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the United States to comply with accounting provisions requiring such companies to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations.
In Europe, and throughout the world, other countries have enacted anti-bribery laws and/or regulations similar to the FCPA. Violations of any of these anti-bribery laws, or allegations of such violations, could have a negative impact on our business, results of operations and reputation.
Data Privacy and Security Laws
We expect that the Company may also be subject to data protection laws and regulations (i.e., laws and regulations that address privacy and data security). In the U.S., numerous federal and state laws and regulations, including state data breach notification laws, state health information privacy laws, and federal and state consumer protection laws (e.g., Section 5 of the FTC Act), govern the collection, use, disclosure and protection of health-related and other personal information. Failure to comply with data protection laws and regulations could result in government enforcement actions and create liability for the Company (which could include civil and/or criminal penalties), private litigation and/or adverse publicity that could negatively affect our operating results and business. HIPAA, as amended by HITECH, and its implementing regulations, among other things, imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information.
To the extent that we conduct clinical trials or seek to commercialize our products outside of the United States, we will also be subject to a variety of foreign data protection laws and regulations. E.U. member states, Israel and other countries have adopted data protection laws and regulations that impose significant compliance obligations. In the European Union, the collection and use of personal health data is governed by the provisions of the General Data Protection Regulation (GDPR). The GDPR became effective on May 25, 2018, repealing its predecessor directive and increasing responsibility and liability of pharmaceutical companies in relation to the processing of personal data of E.U. subjects. The GDPR, together with the national legislation of the E.U. member states governing the processing of personal data, impose strict obligations and restrictions on the ability to collect, analyze and transfer personal data, including health data from clinical trials and adverse event reporting. In particular, these obligations and restrictions concern the consent of the individuals to whom the personal data relates, the information provided to the individuals, the transfer of personal data out of the European Union, security breach notifications, security and confidentiality of the personal data and imposition of substantial potential fines for breaches of the data protection obligations. Data protection authorities from the different E.U. member states may interpret the GDPR and national laws differently and impose additional requirements, which add to the complexity of processing personal data in the European Union. Guidance on implementation and compliance practices are often updated or otherwise revised. For our clinical trials in Israel, to the extent that the sites for our trials include certain university, company or government agencies, we may be subject to restrictions and data protection obligations under the Protection of Privacy Law, 5741-1981, and the Protection of Privacy Regulations (Data Security), 5777-2017. All of these laws may impact our business. Our failure to comply with these privacy laws or significant changes in the laws restricting our ability to obtain required patient information could significantly impact our business and our future business plans.
| 76 |
Israel Laboratory Licensing and Regulation
We are required to maintain licensure under the law of Israel for our laboratory in Rehovot, Israel. The Israeli law includes standards for the day-to-day operation of a clinical reference laboratory, including the training and skills required of personnel and quality control. In addition, Israeli laws mandate proficiency testing, which involves testing of specimens that have been specifically prepared for the laboratory. Our research laboratory was certified by Israel in 2017. If we do not meet the requirements of Israeli laws, the Israeli Ministry of Health may suspend, restrict or revoke our license to operate our laboratory, assess substantial civil money penalties, or impose specific corrective action plans.
Our laboratory will be subject to Israeli regulations relating to the handling and disposal of regulated medical waste, hazardous waste and biohazardous waste, including chemical, biological agents and compounds, blood samples and other human tissue. Typically, we will use outside vendors who are contractually obligated to comply with applicable laws and regulations to dispose of such waste. These vendors will be licensed or otherwise qualified to handle and dispose of such waste.
The Israeli Institute for Occupational Safety & Health has established extensive requirements relating to workplace safety for health care employers, including requirements to develop and implement programs to protect workers from exposure to blood-borne pathogens by preventing or minimizing any exposure through needle stick or similar penetrating injuries.
Information Systems
Information systems are used extensively in virtually all aspects of our businesses. In our clinical laboratory services business, our information systems are critical with respect to laboratory testing, billing, accounts receivable, customer service, logistics, and management of medical data. Our success depends, in part, on the continued and uninterrupted performance of our information technology systems. Computer systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters.
Moreover, despite network security measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. We have invested heavily in the upgrade of our information and telecommunications systems to improve the quality, efficiency and security of our businesses.
Despite the precautionary measures that we have taken to prevent unanticipated problems that could affect our information technology systems, sustained or repeated system failures that interrupt our ability to process test orders, deliver test results or perform tests in a timely manner could adversely affect our reputation and result in a loss of customers and net revenues.
Employees
As at the date of this prospectus, we have nine full-time employees and three part-time employees on a consolidated basis, of which six are responsible for oversight of research and development, clinical trials, and regulatory affairs, and six are responsible for corporate and business development, administration, investor relations, information technology, accounting and finance.
Our scientific staff, which includes five individuals with post graduate degrees, possess a wide range of experience and expertise in the areas of molecular cell biology, biochemistry, assay development, endo-cannabinoid biology and high-throughput drug screening.
In addition to our permanent workforce, we regularly utilize outside consultants to provide advice on our research and development, clinical development planning and preclinical research programs on a project-by-project basis. We also retain, from time to time, independent contractors and consultants to perform specialized services.
| 77 |
Our employees and consultants have entered into non-disclosure and invention assignment agreements with us regarding our intellectual properties, trade secrets and other confidential information. In addition, we have entered into non-competition agreements with each of our key employees and consultants.
We have no collective bargaining agreements with our employees and none are represented by labor unions. We consider our relationship with our employees to be good.
We expect to increase the number of our employees and independent contractors as we expand our operations. See “Risk Factors” elsewhere in this prospectus.
Insurance
We currently maintain director and officer insurance, as well as property and general liability insurance. As our needs grow, we will secure insurance coverage as necessary. We do not have key person insurance. See “Risk Factors” elsewhere in this prospectus.
Legal Proceedings
On March 8, 2020, we joined Cannabics Inc., our largest shareholder, in a suit in Tel Aviv, Israel against Seach Sarid Ltd., Seach Medical Group Ltd., and Shay Sarid in Tel Aviv, Israel, claiming that the defendants pursued certain business arrangements which rightfully inured to Cannabics Inc. and us. The Tel Aviv court has referred the dispute to arbitration in Tel Aviv. Other that this arbitration proceeding, we are not currently a party to any legal proceedings.
Property and Facilities
We do not own any real property. We have no policies with respect to investments in real estate or interests in real estate.
Our head office is located in a shared office suite at # 3 Bethesda Metro Center, Suite 700 Bethesda, Maryland.
Our research and development facilities and in-house laboratory are located in Rehovot, Israel, where we currently occupy approximately 230 square meters at an annual rental rate of NIS 150,480 ($41,294) under a two-year lease agreement that expires on January 31, 2021. Lease payments are linked to the Israeli Consumer Price Index, or CPI, based on the CPI published on February 1, 2018. We have an option to extend this lease for an additional two years.
The administrative offices for our Israeli operations are located in Tel Aviv, Israel, where we currently occupy approximately 134 square meters at an annual rental rate of NIS 147,600 (approximately $42,171) under a one-year lease agreement that expires on January 4, 2021.
We believe that our current facilities will be sufficient to meet our anticipated needs for the foreseeable future and are suitable for the conduct of our business. Even so, we believe that suitable additional or alternative space will be available in the future on commercially reasonable terms, if required.
| 78 |
MANAGEMENT’S DISCUSSION AND ANALYSIS AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and related notes appearing at the end of this prospectus. This discussion and other parts of this prospectus contain forward-looking statements that involve risks and uncertainties, such as statements regarding our plans, objectives, expectations, intentions and projections. Our actual results could differ materially from those discussed in these forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, those discussed in the “Risk Factors” section of this prospectus.
Overview
The Company was incorporated in the State of Nevada, on September 15, 2004, as Thrust Energy Corp. On May 5, 2011, the Company changed its name to American Mining Company. Our principal offices are in Bethesda, Maryland. On May 21, 2014, the Company changed its name to its current Cannabics Pharmaceuticals Inc.
The Company was originally engaged in the exploration, development and production of oil and gas projects within North America but was unable to operate profitably. In May 2011, the Company suspended its oil and gas operations and changed its business to toll milling and refining and mine development. As of April 2014, the Company has changed its course of business to Biotechnology Pharmaceutical development. As such, the Company has divested itself of its former mining properties.
Financing
We will require additional financing to implement our business plan, which may include joint venture projects and debt or equity financings. The nature of this enterprise and lack of positive cash flow places debt financing beyond the credit-worthiness required by most banks or typical investors of corporate debt until such time as an economically viable profits and losses can be demonstrated. Therefore any debt financing of our activities may be costly and result in substantial dilution to our stockholders.
Future financing through equity investments is likely to be dilutive to existing stockholders. Also, the terms of securities we may issue in future capital transactions may be more favorable for our new investors. Newly issued securities may include preferences, superior voting rights, and the issuance of warrants or other derivative securities, which may have additional dilutive effects. Further, we may incur substantial costs in pursuing future capital and financing, including investment banking fees, legal fees, accounting fees, and other costs. We may also be required to recognize non-cash expenses in connection with certain securities we may issue, such as convertible notes and warrants, which will adversely impact our financial condition.
Our ability to obtain needed financing may be impaired by such factors as the capital markets, both generally and specifically in the bio-pharma industry, and the fact that we have not been profitable to date, which could impact the availability or cost of future financings. If the amount of capital we are able to raise from financing activities, together with our revenue from operations, is not sufficient to satisfy our capital needs, even to the extent that we reduce our operations accordingly, we may be required to cease operations.
There is no assurance that we will be able to obtain financing on terms satisfactory to us, or at all. We do not have any arrangements in place for any future financing. If we are unable to secure additional funding, we may cease or suspend operations. We have no plans, arrangements or contingencies in place in the event that we cease operations.
| 79 |
Results of Operations
Quarter ended November 30, 2020 compared to quarter ended November 30, 2019
Revenues
We have not received royalties during the three months ended November 30, 2020 due to the termination of the royalties agreement, compared to $1,754 for the three months ended November 30, 2019. The reason for the decrease in revenues is due to the termination of our relationship with the licensee.
Operating Expenses
For the three months ended November 30, 2020 our total operating expenses were $659,732 compared to $817,610 for the three months ended November 30, 2019 resulting in a decrease of $157,878. The decrease is attributable to decrease of $108,009 in general administration expenses and a total decrease of $49,869 in research and development expenses, and sales and marketing expenses.
We incurred a financial income of $4,965 for the three months ended November 30, 2020, compared to financial expense of $4,326,339 for the three months ended November 30, 2019. The decrease in financial expense was mainly attributable to revaluation of a financial asset of $4,363,000. This revaluation relates to our Company’s investment in Seedo Corp., which is obligated by the afore-mentioned sum in future royalties. Subsequent to November 30th, 2019, but prior to the date of this filing, the Company was made aware of a Petition for Debt Settlement filing by E-Roll Grow Tech Ltd., the fully owned subsidiary of Seedo Corp. Said filing calls into question the ability of Seedo Corp. to fulfill its royalty obligations to us. This accounting treatment does not release Seedo from its obligations under the agreement. As such, the Company has removed its anticipated Royalties for accounting purposes from Seedo at this time. As a result, the net loss was $654,767 for the three months ended November 30, 2020 compared to $5,141,195 for the three months ended November 30, 2019.
Net loss
Net loss decreased by $4,487,428 to $654,767 for the three months ended November 30, 2020, compared to a net loss of $5,142,195 for the three months ended November 30, 2019.
Year ended August 31, 2020 compared to the year ended August 31, 2019
Revenues
The revenues for the year ended August 31, 2020 totaled to $7,157 compared to $9,843 for the year ended August 31, 2019.
Operating and Other Expenses
For the year ended August 31, 2020, our total operating expenses were $3,050,609 compared to $3,313,450 for the year ended August 31, 2019. The decrease is attributable mainly to a decrease in sales and marketing expenses of $228,166, a decrease in general and administrative expenses of $173,378, and an increase in research and development expenditures of $138,702. The decrease in the sales and marketing expenses is attributable mainly to a decrease in company PR services of $21,665, and the fact that the Company did not have any travel abroad or conferences expenses for the year ended August 31, 2020, compared to $125,672 of company PR Services and $51,817 of travel and conference expenses for the year ended August 31, 2019.
| 80 |
Net Loss
The net loss for the year ended August 31, 2020 was $7,467,463 compared to net income of $1,132,970 for the year ended August 31, 2019.
Year ended August 31, 2019 compared to the year ended August 31, 2018
Revenues
The revenues for the year ended August 31, 2019 totaled to $9,843 compared to $9,601 for the year ended August 31, 2018.
Operating and Other Expenses
For the year ended August 31, 2019 our total operating expenses were $3,313,450 compared to $3,818,328 for the year ended August 31, 2018. The decrease is attributable mainly to, general and administrative of $910,870, decrease in marketing expenses of $133,852, and increase in research and development expenditures of $539,843. The decrease in the general and administrative expenses attributed mainly to the company share based compensation in total of $247,307 for the year ended August 31, 2019 comparing to $910,870 for the year ended August 31, 2018.
The net income for the year ended August 31, 2019 was $1,132,970 compared to net loss $3,776,617 for the year ended August 31, 2018.
Liquidity and Capital Resources
Overview
For the quarter ended November 30, 2020, the year ended August 31, 2020, as well as for the year ended August 31, 2019, we funded our operations through the issuance of common stock and advances from our majority shareholder. Our principal use of funds during the quarter ended November 30, 2020, the year ended August 31, 2020 has been for laboratory and clinical research relating to our proprietary materials and normative corporate operating expenses.
Quarter ended November 30, 2020 compared to quarter ended November 30, 2019
As of November 30, 2020, we had $184,031 in cash compared to $319,344 on November 30, 2019. We expect to incur a minimum of $1,000,000 in expenses during the next twelve months of operations. We estimate that these expenses will be comprised primarily of general expenses including overhead, legal and accounting fees, research and development expenses, and fees payable to outside medical centers for clinical studies.
We used cash in operations of $592,637 for the three months ended November 30, 2020 compared to cash used in operations of $761,687 for the three months ended November 30, 2019. The negative cash flow from operating activities for the three months ended November 30, 2020 is primarily attributable to the Company's net loss from operations of $654,767, a decrease in accounts payables and accrued liabilities of $12,100 and an increase of $16,275 in account receivables and prepaid expenses. Offset by depreciation of $57,505 and shares issued for services of $33,000.
| 81 |
We had cash flow from investing activities of $943 during the three months ended November 30, 2020, compared to $815,049 cash flow from investing activities for the three months ended November 30, 2019. The reason for the decrease in cash flow from investing activities is primarily due to the purchase of fixed assets in an aggregate amount of $934, for the period ended November 30, 2020, and realization of held for trading shares in the aggregate amount of $824,849 for the period ended November 30, 2019.
We will have to raise funds to pay for our expenses. We may have to borrow money from shareholders, issue equity or enter into a strategic arrangement with a third party. There can be no assurance that additional capital will be available to us. We currently have no arrangements or understandings with any person to obtain funds through bank loans, lines of credit or any other sources. Since we have no such arrangements or plans currently in effect, our inability to raise funds for our operations will have a severe negative impact on our ability to remain a viable company.
Year ended August 31, 2020 compared to the year ended August 31, 2019
As of August 31, 2020, we had $777,611 in cash compared to $265,982 as of August 31, 2019. The Company used cash in operations of $2,774,954 for the year ended August 31, 2020 compared to cash used in operations of $3,093,782 for the year ended August 31, 2019.
During the year ended August 31, 2020, the Company's net cash used for investing activities totaled $3,286,584. This compares to net cash used for investing activities in the year ended August 31, 2019 in the amount of $5,251,115. The difference reflects primarily the realization of some held for trading investments and the purchase of fixed assets.
During the year ended August 31, 2020, the Company had no financing activities. This compares to net cash earned from financing activities in the year ended August 31, 2020 in the amount of $7,217,270.
Year ended August 31, 2019 compared to the year ended August 31, 2018
For the year ended August 31, 2019, as well as the year ended August 31, 2018, we funded our operations through issuance of common stock and advances from our majority shareholder. Our principal use of funds during the year ended August 31, 2019 has been for laboratory and clinical research relating to our proprietary materials normative corporate operating expenses.
As of August 31, 2019, we had cash $265,982 compared to $1,393,608 as of August 31, 2018. The Company used cash in operations of $3,093,782 for the year ended August 31, 2019 compared to cash used in operations of $1,974,692 for the year ended August 31, 2018.
During the year ended August 31, 2019, the Company's investing activities totaled $5,251,115. This compares to net cash used for investing activities in the year ended August 31, 2018 in the amount of $1,442,394. The difference reflects primarily of held for trading and available for sale investments and purchase of fixed assets.
During the year ended August 31, 2019, the Company's financing activities earned $7,217,270. This compares to net cash earned for financing activities in the year ended August 31, 2018 in the amount of $1,443,000. During the year ended August 31,2019.
| 82 |
Cash Flows
The following table summarizes our sources and uses of cash for each of the periods presented:
| For the Nine Months ended November 30, | Year Ended August 31, | |||||||||||
| 2020 | 2020 | 2019 | ||||||||||
| Unaudited | Audited | |||||||||||
| Cash provided by (used) in operating activities | $ | (592,637 | ) | $ | (2,774,955 | ) | $ | (3,093,782 | ) | |||
| Cash from (used) in investing activities | (943 | ) | 3,286,584 | (5,251,115 | ) | |||||||
| Cash provided by financing activities | – | – | 7,217,270 | |||||||||
| Net increase (decrease) in cash and cash equivalents | $ | (593,580 | ) | $ | 511,629 | $ | (1,127,626 | ) | ||||
Going Concern
Due to the uncertainty of our ability to meet our current operating and capital expenses, our independent auditors included an explanatory paragraph in their report on the audited financial statements for the year ended August 31, 2020 regarding concerns about our ability to continue as a going concern. Our financial statements contain additional note disclosures describing the circumstances that lead to this disclosure by our independent auditors.
Our financial statements have been prepared on a going concern basis, which assumes the realization of assets and settlement of liabilities in the normal course of business. Our ability to continue as a going concern is dependent upon our ability to generate profitable operations in the future and/ or to obtain the necessary financing to meet our obligations and repay our liabilities arising from normal business operations when they become due. The outcome of these matters cannot be predicted with any certainty at this time and raise substantial doubt that we will be able to continue as a going concern. Our financial statements do not include any adjustments to the amount and classification of assets and liabilities that may be necessary should we be unable to continue as a going concern.
There is no assurance that our operations will be profitable. The Company has conducted private placements of its common stock, which have generated funds to satisfy the initial cash requirements of its planned Nevada exploration ventures. Our continued existence and plans for future growth depend on our ability to obtain the additional capital necessary to operate either through the generation of revenue or the issuance of additional debt or equity.
| 83 |
Contractual Obligations and Commitments
The following table summarizes our contractual obligations as of November 30, 2020 and August 31, 2020 and the effects that such obligations are expected to have on our liquidity and cash flows in future periods:
| Payments Due by Period | ||||||||||||||||||||
| As of August 31, 2020 | Total | Less Than 1 Year | 1 to 3 Years | 4 to 5 Years | More than 5 Years | |||||||||||||||
| Operating lease commitments (1) | $ | 95,700 | $ | 78,000 | $ | 17,700 | $ | – | $ | – | ||||||||||
| Loan payable (2) | $ | 223,645 | $ | – | $ | 223,645 | $ | – | $ | – | ||||||||||
| Total | $ | 319,345 | $ | 78,000 | $ | 241,345 | $ | – | $ | – | ||||||||||
| Payments Due by Period | ||||||||||||||||||||
| As of November 30, 2020 | Total | Less Than 1 Year | 1 to 3 Years | 4 to 5 Years | More than 5 Years | |||||||||||||||
| Operating lease commitments (1) | $ | 58,500 | $ | 58,500 | $ | – | $ | – | $ | – | ||||||||||
| Loan payable (2) | $ | 223,645 | $ | – | $ | 223,645 | $ | – | $ | – | ||||||||||
| Total | $ | 282,345 | $ | 58,500 | $ | 223,645 | $ | – | $ | – |
| |||||||||
| (1) | Amounts in the table reflect payments due for our lease of cars, office space, and laboratory facilities in Israel. The operating lease for our laboratory facilities in Israel expires on January 31, 2021, and we intend to exercise our option to renew the lease. |
| (2) | The balance due to Cannabics Inc. (our parent company) at November 30, 2020, at August 31, 2020, and at August 31, 2019 was $223,645. The loan is due on demand and bears no interest. |
Off-Balance Sheet Arrangements
We currently have no off-balance sheet arrangements that have or are reasonably likely to have a current or future material effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources.
Recently Issued and Adopted Accounting Pronouncements
We have reviewed all recently issued standards and have determined that, other than as disclosed in Note 2 to our financial statements appearing at the end of this prospectus, such standards are not expected to have a material impact on our financial statements or do not otherwise apply to our operations.
Quantitative and Qualitative Disclosures About Market Risk.
We are a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and are not required to provide the information required under this item.
| 84 |
Directors and Executive Officers
The following individuals serves as Directors and Executive Officers of the Company as of the date of this prospectus. Directors of the Company hold office until the next annual meeting of our shareholders or until their successors have been elected and qualified. Executive officers of the Company are appointed by our board of directors and hold office until their death, resignation or removal from office.
| Name | Position | Age | Held Position Since | |||
| Eyal Barad | Director, CEO | 55 | November 13, 2017 (Director) | |||
| Dr. Eyal Ballan | CTO | 46 | April 29, 2014 | |||
| Uri Ben-Or | CFO | 49 | December 9, 2016 | |||
| Gabriel Yariv | Director, COO | 43 | October 2, 2019 (Director) |
Eyal Barad, 55, is a co-founder of Cannabics Inc. He was named Director & COO on November 13, 2017, and was appointed CEO on January 29, 2018. Mr. Barad brings over 20 years of executive managerial experience in successful technology ventures. He has a BA in Economics & International Relations from the Hebrew University in Jerusalem, and an MBA with Honors from Haifa University.
Dr. Eyal Ballan, 46, is a co-founder of Cannabics Inc. and is our CTO. Dr. Ballan holds a Ph.D. in Neurophysiology, EEG, Brain Wave Analysis and Cortical Connectivity from Haifa University. After obtaining his Ph.D. he was an entrepreneur in the field of Biofeedback Studies and developed a Resonating Neuro-Feedback system. Dr. Ballan holds a M.Sc. from Tel-Aviv University - Magna Cum Laude - in anticancer drug development. Dr. Ballan was part of the renowned research team which developed Salirasib (Treatment for Non-Small Cell Lung Cancer). He is an expert in molecular biology, cell cultures and genomics with a focus towards identification of anticancer compounds and delivery systems to tumors and a member of the American Neurology Association.
Uri Ben-Or, 49 CPA, – was appointed CFO on December 9, 2016. He has more than 15 years of experience as CFO of public and private companies in the life science and Medical Device Industry. Uri has significant expertise in public Life Science companies traded on the TASE. In addition, Uri has strong finance, operation, and business development background in both startups and public global companies in the US, Europe, and Israel including developing strategic policy and guidance with respect to corporate structure and fundraising. Mr. Ben-Or earned a MBA from Bar Ilan University.
Gabriel Yariv, 43, has been a Director since October 2019 and was appointed COO on November 6, 2019. He brings over 20 years of successful executive experience in the medical industry. Mr. Yariv was part of the founding group of BreathID, an Oridion Medical business unit (now Medtronic) and its subsequent spinoff company, Exalenz Bioscience, which develops and manufactures advanced non-invasive diagnostic medical devices for gastrointestinal and liver conditions. Mr. Yariv also co-founded SimuTec, a medical simulation and training company in Brazil that develops and commercializes advanced personalized Virtual Reality training programs for physicians. Mr. Yariv is actively engaged in non-profit and philanthropic activities including ongoing business mentoring of entrepreneurs, founder of the Yariv Foundation for Leadership, and current member of the Friends of the Israel Museum society. Mr. Yariv holds a BA (Cum Laude) in History, Philosophy & Political science from Boston University, and a Certificate Course in Cyberlaw from Harvard University.
��
All directors serve for one-year terms and are subject to re-election at our annual meeting of shareholders, unless they earlier resign.
| 85 |
There are no material proceedings to which any of our directors, officers or affiliates, any owner of record or beneficially of more than five percent of any class of our voting securities, or any associate of any such director, officer, affiliate, or security holder is a party adverse to us or any of our subsidiaries or has a material interest adverse to us or any of our subsidiaries.
We have attempted and will continue to attempt to ensure that any transactions between we and our officers, directors, principal shareholders, or other affiliates have been and will be on terms no less favorable to us than could be obtained from unaffiliated third parties on an arm’s length basis.
Director Independence
Our board of directors is not independent, as both of our Directors are employed by the Company. There are no family relationships among any of our directors or executive officers.
Involvement in Certain Legal Proceedings
Except as noted herein or below, during the last ten (10) years none of our directors or officers have:
(1) had any bankruptcy petition filed by or against any business of which such person was a general partner or executive officer either at the time of the bankruptcy or within two years prior to that time;
(2) been convicted in a criminal proceeding or subject to a pending criminal proceeding;
(3) been subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining, barring, suspending or otherwise limiting his involvement in any type of business, securities or banking activities; or
(4) been found by a court of competent jurisdiction in a civil action, the Commission or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated.
All of these filing requirements were satisfied by the Company’s officers, directors, and ten-percent holders.
In making these statements, we have relied on the written representation of our Directors and Officers or copies of the reports that they have filed with the Commission.
Committees of the Board
All proceedings of the board of directors for the fiscal year ended August 31, 2020 were conducted by resolutions consented to in writing by our board of directors and filed with the minutes of the proceedings of our board of directors. Our Company currently does not have nominating, compensation or audit committees or committees performing similar functions nor does our company have a written nominating, compensation or audit committee charter. Our board of directors does not believe that it is necessary to have such committees because it believes that the functions of such committees can be adequately performed by the board of directors.
The Company does not have any defined policy or procedure requirements for shareholders to submit recommendations or nominations for directors. The Company’s board of directors believes that, given the stage of our development, a specific nominating policy would be premature and of little assistance until our business operations develop to a more advanced level. The Company does not currently have any specific or minimum criteria for the election of nominees to the board of directors and we do not have any specific process or procedure for evaluating such nominees. The board of directors will assess all candidates, whether submitted by management or shareholders, and make recommendations for election or appointment.
| 86 |
A shareholder who wishes to communicate with the Company’s board of directors may do so by directing a written request addressed to any of our Directors at the address appearing on the first page of this registration statement.
Audit Committee Financial Expert
We do not have a standing audit committee. Our directors perform the functions usually designated to an audit committee. Our board of directors has determined that we do not have a board member that qualifies as an "audit committee financial expert" as defined in Item 407(d)(5) of Regulation S-K, nor do we have a board member that qualifies as "independent" as the term is used in Item 7(d)(3)(iv)(B) of Schedule 14A under the Securities Exchange Act of 1934, as amended, and as defined by Rule 4200(a)(14) of the NASD Rules.
We believe that our board of directors is capable of analyzing and evaluating our financial statements and understanding internal controls and procedures for financial reporting. Our board of directors does not believe that it is necessary to have an audit committee because management believes that the functions of an audit committees can be adequately performed by the board of directors. In addition, we believe that retaining an independent director who would qualify as an "audit committee financial expert" would be overly costly and burdensome and is not warranted in our circumstances given the stage of our development and the fact that we have not generated positive cash flow to date.
Potential Conflict of Interest
Since we do not have an audit or compensation committee comprised of independent Directors, the functions that would have been performed by such committees are performed by our Board of Directors. Thus, there is a potential conflict of interest in that our Directors have the authority to determine issues concerning management compensation, in essence their own, and audit issues that may affect management decisions. We are not aware of any other conflicts of interest with any of our executives or Directors.
Board Leadership Structure
We do not have a formal chairman of the board.
Board’s Role in Risk Oversight
The Board assesses on an ongoing basis the risks faced by the Company. These risks include financial, technological, competitive, and operational risks. The Board dedicates time at each of its meetings to review and consider the relevant risks faced by the Company at that time. In addition, since the Company does not have an Audit Committee, the Board is also responsible for the assessment and oversight of the Company’s financial risk exposures.
Code of Ethics
The Company has adopted a Code of Ethics for Senior Financial Officers that is applicable to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. Our Code of Ethics is available on our website at www.cannabics.com.
Indemnification
Under our Articles of Incorporation and Bylaws, we may indemnify an officer or director who is made a party to any proceeding, including a law suit, because of his position, if he acted in good faith and in a manner he reasonably believed to be in our best interest. We may advance expenses incurred in defending a proceeding. To the extent that the officer or director is successful on the merits in a proceeding as to which he is to be indemnified, we must indemnify him against all expenses incurred, including attorney's fees. With respect to a derivative action, indemnity may be made only for expenses actually and reasonably incurred in defending the proceeding, and if the officer or director is judged liable, only by a court order. The indemnification is intended to be to the fullest extent permitted by the laws of the State of Nevada.
Regarding indemnification for liabilities arising under the Securities Act of 1933, which may be permitted to directors or officers under Nevada law, we have been advised that, in the opinion of the Securities and Exchange Commission, indemnification is against public policy, as expressed in the Securities Act, and is, therefore, unenforceable.
| 87 |
We pay a monthly salary to our two Directors who are also employees of the Company. We also pay a monthly salary to our CTO and CFO.
There are no stock option plans, retirement, pension, or profit-sharing plans for the benefit of our officers or directors. We do not have any long-term incentive plans that provide compensation intended to serve as incentive for performance.
Summary Compensation Table
| Stock | All Other | |||||||||||||||||||||
| Name and Principal Position | Year | Salary | Bonus | Awards | Compensation | Total | ||||||||||||||||
| Eyal Ballan, CTO | 2020 | $ | 222,000 | $ | – | $ | – | $ | 9,347 | $ | 231,347 | |||||||||||
| 2019 | $ | 257,832 | $ | – | $ | – | $ | 6,030 | $ | 263,862 | ||||||||||||
| Eyal Barad, Director, CEO | 2020 | $ | 222,000 | $ | – | $ | – | $ | 19,120 | $ | 241,120 | |||||||||||
| 2019 | $ | 257,723 | $ | – | $ | – | $ | 18,037 | $ | 275,760 | ||||||||||||
| Gabriel Yariv, Director, COO | 2020 | $ | 151,000 | $ | -- | $ | -- | $ | -- | $ | 151,000 | |||||||||||
| 2019 | $ | 32,300 | $ | -- | $ | -- | $ | -- | $ | 32,300 | ||||||||||||
| Uri Ben-Or, CFO | 2020 | $ | 75,000 | $ | 75,000 | |||||||||||||||||
| 2019 | $ | 55,000 | $ | 55,000 | ||||||||||||||||||
Eyal Ballan and Eyal Barad have entered into employment agreements with us, but their compensation is paid by our Israeli subsidiary, Grin Ultra. Gabriel Yariv has entered into an employment agreement with our Israeli subsidiary, Grin Ultra, and is not formally an employee of the Company. Uri Ben-Or is not an employee of the Company and has executed a consulting services agreement with Grin Ultra. These agreements contain provisions regarding non-competition, confidentiality of information, and assignment of inventions. The enforceability of covenants not to compete in Israel and the United States is subject to limitations. For example, Israeli courts have recently required employers seeking to enforce non-compete undertakings of a former employee to demonstrate that the competitive activities of the former employee will harm one of a limited number of material interests of the employer which have been recognized by the courts, such as the secrecy of a company’s confidential commercial information or its intellectual property.
The material terms and conditions of these agreements are described below.
Eyal Ballan:
| ● | Term: At will, no fixed term. |
| ● | Salary: $250,000 per year |
| ● | Bonus: 3 months salary, subject to the discretion of the Board |
| ● | Profit Participation: Two and one-half percent (2.5%) of the Company’s annual consolidated profits (after tax) above US1,000,000 (one million US Dollars), but no more than the sum equaling 4 times the annual base salary. | |
●
| Notice Period: 4 months |
| 88 |
Eyal Barad:
| ● | Term: At will, no fixed term. |
| ● | Salary: $250,000 per year |
| ● | Bonus: 3 months salary, subject to the discretion of the Board |
| ● | Profit Participation: Two and one-half percent (2.5%) of the Company’s annual consolidated profits (after tax) above US1,000,000 (one million US Dollars), but no more than the sum equaling 4 times the annual base salary. | |
● | Notice Period: 4 months |
Gabriel Yariv:
| ● | Term: Initial term of 3 years from November 1, 2019, with automatic renewal for an additional 2 years. |
| ● | Salary: NIS 50,000 per month |
| ● | Options: Upon the adoption of a stock option plan, the Company will grant Gabriel Yariv options to purchase up to 850,000 shares. |
| ● | Notice Period: 5 months |
Uri Ben-Or:
| ● | Term: One year term from November 1, 2016, with renewal upon mutual agreement of the parties. |
| ● | Consulting Fees: NIS 15,000 per month, plus VAT. |
| ● | Bonus: 1% of funds raised in a private or public offering that Mr. Ben-Or leads. |
| ● | Notice Period: 60 days |
Director Compensation
Both of our directors are employees, either of Cannabics or of our Israeli subsidiary, Grin Ultra, and receive salaries as employees. They do not receive any compensation for their service as directors.
| 89 |
Option/SAR Grants
We have not adopted a stock option plan, and we have not granted any options or any stock appreciation rights (SARs) to any executive officer or Director No stock options have been granted or exercised by any of the officers or Directors since we were founded. Nevertheless, the Company has undertaken to grant Gabriel Yariv stock options to purchase up to 850,000 shares once it has formally adopted a stock option plan.
Long-Term Incentive Plans and Awards
We do not have any long-term incentive plans that provide compensation intended to serve as incentive for performance. No individual grants or agreements regarding future payouts under non-stock price-based plans have been made to any executive officer or any Director or any employee or consultant since our inception; accordingly, no future payouts under non-stock price-based plans or agreements have been granted or entered into or exercised by any of the officers or Directors or employees or consultants since we were founded.
Employment Contracts, Termination of Employment, Change-in-Control Arrangements
Other than standard severance payments mandated by Israeli law (one month’s salary for each year worked), there are no compensation plans or arrangements, including payments to be made by us, with respect to our officers, Directors or consultants that would result from the resignation, retirement or any other termination of such Directors, officers or consultants from us. There are no arrangements for Directors, officers, employees or consultants that would result from a change-in-control.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth, as of February 12, 2021, information concerning ownership of our securities by (i) each director, (ii) each executive officer, (iii) all directors and executive officers as a group; and (iv) each person known to us to be the beneficial owner of more than five percent of each class. That table is based on 135,237,584 issued and outstanding shares, and does not include the Pre-Delivery Shares.
The number and percentage of shares beneficially owned includes any shares as to which the named person has sole or shared voting power or investment power and any shares that the named person has the right to acquire within 60 days.
The mailing address for all directors, executive officers, and beneficial owners of more than 5% of our common stock is #3 Bethesda Metro Center, Suite 700, Bethesda, Maryland, 20814.
| Beneficial Ownership | ||||||||
| Name of Beneficial Owner | Shares of Common Stock | Percentage | ||||||
| Cannabics Inc.* | 88,289,594 | 65.28% | ||||||
____________________
*Both of our Directors, Eyal Barad and Gabriel Yariv, are shareholders of Cannabics, Inc.
| 90 |
The shareholders of Cannabics, Inc. who are directors or officers of the Company, or who hold more than five percent of the shares of Cannabics, Inc. are listed below:
| Shareholder of Cannabics, Inc. | Position in Cannabics Pharmaceuticals, Inc. | Common Stock | % of Cannabics Inc. | |||||||
| Eyal Barad | Director, CEO | 316 | 26.25% | |||||||
| Eyal Ballan | CTO | 141 | 11.71% | |||||||
| Gabriel Yariv | Director, COO | 4 | 0.33% | |||||||
| Shay Avraham Sarid | 223 | 18.52% | ||||||||
| Itamar Borochov | 222 | 18.44% | ||||||||
| J Reiger Ltd | 128 | 10.63% | ||||||||
Unless otherwise noted, we believe that all persons or entities named in the table have sole voting and investment power with respect to all shares of common stock beneficially owned by them.
We are unaware of any contract or other arrangement the operation of which may at a subsequent date result in a change in control of our Company
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
No material related party transactions between the Company and its officers, directors or control persons occurred during the fiscal years ended August 31, 2020 and 2019.
Employment Agreements
We have entered into employment agreements with our named executive officers and a consulting agreement with our CFO. For more information regarding the arrangements with our named executive officers, see “Executive and Director Compensation—Executive Compensation Arrangements.”
The following summary describes our capital stock and the material provisions of our amended and restated articles of incorporation (the “Articles of Incorporation”) and our amended and restated bylaws (“Bylaws”), and of Chapter 78 of the Nevada Revised Statutes (“NRS”). Because the following is only a summary, it does not contain all of the information that may be important to you. For a complete description, you should refer to our Articles of Incorporation, and our Bylaws, copies of which are incorporated by reference as exhibits to the registration statement of which this prospectus is a part.
General
Our authorized capital stock consists of 900,000,000 shares of common stock, par value $0.0001, and 5,000,000 shares of preferred stock, par value $0.0001. As of February 12, 2021, there were 135,237,584 shares of our common stock issued and outstanding (not including the Pre-Delivery Shares), 5,000,000 shares of our common stock subject to outstanding warrants, and no shares of preferred stock issued or outstanding.
As of February 12,2021, there were 76 holders of record of our common stock. This number does not include beneficial owners whose shares are held by nominees in street name.
| 91 |
Common Stock
Voting Rights
Each outstanding share of our common stock entitles the holder thereof to one vote per share on all matters submitted to a stockholder vote. Our common stock does not carry any cumulative voting rights. As a result, holders of a majority of the shares of our common stock voting for the election of directors can elect all of our directors. At all meetings of stockholders, except where otherwise provided by statute or by our Articles of Incorporation or our Bylaws, the presence in person or by proxy duly authorized by holders of not less than a majority of the stockholding voting power shall constitute a quorum for the transaction of business. A vote by the holders of a majority of our outstanding shares is required to effect certain fundamental corporate changes such as liquidation, merger, or an amendment to our Articles of Incorporation.
Dividends
Holders of our common stock are entitled to share in all dividends that our board of directors, in its discretion, declares from legally available funds.
Liquidation
In the event of liquidation, dissolution or winding up, each outstanding share entitles its holder to participate pro rata in all assets that remain after payment of liabilities and after providing for each class of stock, if any, having preference over our common stock.
Other Rights and Preferences
Holders of our common stock have no pre-emptive, subscription, or conversion rights, and there are no redemption or sinking fund provisions applicable to our common stock. The rights, preferences, and privileges of holders of common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of preferred stock that we may designate and issue in the future.
Quotation
Our common stock is quoted on the OTCQB tier of the marketplace maintained by OTC Markets Group Inc., under the symbol “CNBX.”
Indemnification
Our Articles of Incorporation limits the liability of our directors and officers to the full extent permitted by the NRS and provides that we will indemnify each of our directors and officers to the full extent permitted by the NRS. Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers, or persons controlling us under the foregoing provisions, we have been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is ClearTrust, LLC, whose address is 16540 Pointe Village Dr., Suite 210, Lutz, Florida 33558 (telephone: 813-235-4490; e-mail: inbox@cleartrusttransfer.com).
| 92 |
Preferred Stock
Our board of directors has the authority, without action by the stockholders, to designate and issue up to an aggregate of 100,000,000 shares of preferred stock in one or more series. The board of directors can fix the rights, preferences, and privileges of the shares of each series and any of its qualifications, limitations, or restrictions. Our board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of common stock. The issuance of preferred stock, while providing flexibility in connection with possible future financings and acquisitions and other corporate purposes could, under certain circumstances, have the effect of delaying or preventing a change in control of Cannabics. In addition, the issuance of preferred stock, depending on the rights and preferences associated with such stock, might harm the market price of our common stock.
We are authorized to issue shares of preferred stock in one or more series as may be determined by our board of directors, who may establish, from time to time, the number of shares to be included in each series, may fix the designation, powers, preferences, and rights of the shares of each such series, and any qualifications, limitations, or restrictions thereof without obtaining the affirmative vote or the written consent of our stockholders. Any preferred stock so issued by our board of directors may rank senior to our common stock with respect to the payment of dividends or amounts upon liquidation, dissolution, or winding up of Cannabics, or both. Under certain circumstances, the issuance of preferred stock or the existence of unissued preferred stock might tend to discourage or render more difficult a merger or other change of control. No shares of preferred stock are currently outstanding.
Our board of directors is empowered to authorize and direct the payment of dividends to the holders of our preferred stock in shares of any class or series of our capital stock, including shares of common stock, as it may determine, without obtaining the affirmative vote or the written consent of our stockholders.
The issuance of shares of preferred stock, while providing desirable flexibility in connection with possible acquisitions and other corporate purposes, could have the effect of making it more difficult for a third party to acquire, or of discouraging a third party from acquiring, a majority of our outstanding voting stock.
Registration Rights
We are party to various subscription agreements which grant the holders of warrants for the purchase of 5,000,000 shares of our common stock the right to demand that we file a registration statement for their shares of our common stock issued upon exercise of these warrants or request that their shares of our common stock issued upon exercise of these warrants be covered by a registration statement that we are otherwise filing. In addition, we have granted the holders of the notes and warrant issued in the Private Placement registration rights with regard to the shares issuable upon the conversion of the notes and the exercise of the warrant. These shares that are subject to registration rights are referred to in this prospectus as “Registrable Securities.”
Demand Registration Rights
Two holders of the warrants for the purchase of 5,000,000 shares of our common stock have the right to demand that we file a registration statement to register all of their Registrable Securities. The holders of the notes and warrants issued in the Private Placement have the right to demand that we file a registration statement to register all of their Registrable Securities.
Piggyback Registration Rights
If we propose to register any of our securities for sale to the public under the Securities Act by filing a registration statement, the holders of all of the Registrable Securities are entitled to receive notice of such registration and to request that we include their Registrable Securities for resale in such registration statement.
| 93 |
Expenses of Registration; Indemnification
We are generally required to bear all registration expenses incurred in connection with any offerings pursuant to the demand and piggyback registration rights described above, other than underwriting commissions and discounts. The relevant subscription agreements contain customary indemnification provisions with respect to registration rights.
Anti-takeover Effects of Our Articles of Incorporation and Bylaws
Our Articles of Incorporation and Bylaws contain certain provisions that may have anti-takeover effects, making it more difficult for or preventing a third party from acquiring control of Cannabics or changing our board of directors and management.
According to our Articles of Incorporation and Bylaws, neither the holders of our common stock nor the holders of our preferred stock have cumulative voting rights in the election of our directors. The present ownership by a single stockholder of a significant portion of our issued and outstanding common stock and lack of cumulative voting make it more difficult for other stockholders to replace our board of directors or for a third party to obtain control of Cannabics by replacing our board of directors.
The authorization of classes of common stock or preferred stock with either specified voting rights or rights providing for the approval of extraordinary corporate action could be used to create voting impediments or to frustrate persons seeking to effect a merger or to otherwise gain control of Cannabics by diluting their stock ownership. In addition, the ability of our directors to distribute shares of any class or series (within limits imposed by applicable law) as a dividend in respect of issued shares of preferred stock could be used to dilute the stock ownership or voting rights of a person seeking to obtain control of Cannabics and effectively delay or prevent a change in control without further action by the stockholders.
Nevada Anti-Takeover laws
Business Combinations
The “business combination” provisions of NRS 78.411 to 78.444, inclusive, prohibit a Nevada corporation with at least 200 stockholders from engaging in various “combination” transactions with any interested stockholder for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the transaction is approved by the board of directors prior to the date the interested stockholder obtained such status; or after the expiration of the three-year period, unless:
| · | the transaction is approved by the board of directors or a majority of the voting power held by disinterested stockholders; or |
| · | if the consideration to be paid by the interested stockholder is at least equal to the highest of: (a) the highest price per share paid by the interested stockholder within the three years immediately preceding the date of the announcement of the combination or in the transaction in which it became an interested stockholder, whichever is higher, (b) the market value per share of common stock on the date of announcement of the combination and the date the interested stockholder acquired the shares, whichever is higher, or (c) for holders of preferred stock, the highest liquidation value of the preferred stock, if it is higher. |
A “combination” is defined to include mergers or consolidations or any sale, lease exchange, mortgage, pledge, transfer, or other disposition, in one transaction or a series of transactions, with an “interested stockholder” having: (a) an aggregate market value equal to 5% or more of the aggregate market value of the assets of the corporation, (b) an aggregate market value equal to 5% or more of the aggregate market value of all outstanding shares of the corporation, or (c) 10% or more of the earning power or net income of the corporation.
| 94 |
In general, an “interested stockholder” is a person who, together with affiliates and associates, owns (or within three years, did own) 10% or more of a corporation's voting stock. The statute could prohibit or delay mergers or other takeover or change in control attempts and, accordingly, may discourage attempts to acquire Cannabics even though such a transaction may offer our stockholders the opportunity to sell their stock at a price above the prevailing market price.
Control Share Acquisitions
The “control share” provisions of NRS 78.378 to 78.3793, inclusive, which apply only to Nevada corporations with at least 200 stockholders, including at least 100 stockholders of record who are Nevada residents, and which conduct business directly or indirectly in Nevada, prohibit an acquirer, under certain circumstances, from voting its shares of a target corporation's stock after crossing certain ownership threshold percentages, unless the acquirer obtains approval of the target corporation's disinterested stockholders. The statute specifies three thresholds: one-fifth or more but less than one-third, one-third but less than a majority, and a majority or more, of the outstanding voting power. Once an acquirer crosses one of the above thresholds, those shares in an offer or acquisition and acquired within 90 days thereof become “control shares” and such control shares are deprived of the right to vote until disinterested stockholders restore the right. These provisions also provide that if control shares are accorded full voting rights and the acquiring person has acquired a majority or more of all voting power, all other stockholders who do not vote in favor of authorizing voting rights to the control shares are entitled to demand payment for the fair value of their shares in accordance with statutory procedures established for dissenters’ rights.
DESCRIPTION OF THE PRIVATE PLACEMENT, NOTES AND WARRANT
Description of the Private Placement
On December 16, 2020, we entered into a securities purchase agreement with an institutional investor to sell a new series of senior secured convertible notes in a four tranche private placement (the “Private Placement”) to the investor, with an aggregate principal amount of $2,750,000 having an aggregate original issue discount of 10%, and ranking senior to all outstanding and future indebtedness. Pursuant to the securities purchase agreement, one note (the “Initial Note”) in an aggregate original principal amount of $825,000 was issued to the investor in the first tranche closing on December 21, 2020. On February 22, 2021, we entered into an amended and restated securities purchase agreement pursuant to which a second Note in an aggregate principal amount of $550,000 was issued (the “Second Note”). Both Notes were issued in reliance upon the exemption from securities registration afforded by Section 4(a)(2) of the Securities Act of 1933, and Rule 506(b) of Regulation D as promulgated by the United States Securities and Exchange Commission under the Securities Act of 1933, together with the issuance of the warrant to acquire our common stock, as described below. The Initial Note has a face amount of $825,000 for which we received cash proceeds of $750,000. The Second Note has a face amount of $550,000 for which we received cash proceeds of $500,000. Subject to the satisfaction of customary closing and equity conditions, at any time prior to December 31, 2021 (or such later date as the parties shall mutually agree), we have the right to require an additional closing of an additional note in the principal amount of $1,375,000 (the “Additional Notes”, and together with the Initial Note, the “Notes”) on the twentieth trading day after the date this registration statement is declared effective by the Securities and Exchange Commission. The Convertible Notes are being sold with an original issue discount of 10% and do not bear interest except upon the occurrence of an event of default.
The following is intended to provide a summary of the terms of the agreements and securities described above. This summary is qualified in its entirety by reference to the full text of the agreements, each of which is attached as an exhibit to our Current Report on Form 8-K dated December 21, 2020. The amendment and restated securities purchase agreement is attached as an exhibit to this registration statement. Readers should review those agreements for a complete understanding of the terms and conditions associated with these transactions.
Notes
Terms not defined in the following description of the particular terms of the Notes have the meanings given to them or the Notes.
| 95 |
Maturity and Repayment Dates
Each Note matures on the one year anniversary of the date on which each respective Note is issued (the “Issuance Date”). The Notes must be paid in cash and we may not voluntarily prepay except as described in “Redemptions at Our Election” below.
Interest
The Notes are being sold with an original issue discount and do not bear interest except upon the occurrence of an Event of Default (described below), in which event the applicable rate will be 18.00% per annum.
Conversion
The Notes are convertible at any time or times after the Issuance Date in whole or in part, at the option of the holders thereof, into shares of our common stock at a rate equal to the amount of principal, interest (if any) and unpaid late charges (if any), divided by a conversion price of $0.35 (subject to adjustment as provided in the Notes, the “Conversion Price”). The Conversion Price has full ratchet antidilution protection upon any subsequent placement below the Conversion Price then in effect and is subject to standard adjustments in the event of any stock split, stock dividend, stock combination, recapitalization or other similar transaction. If we enter into any agreement to issue (or issue) any variable price securities, the holder has the additional right to substitute such variable price (or formula) for the Conversion Price.
Alternate Conversion
The holders of the Notes may alternatively convert all or any portion of the Notes at any time at an alternate conversion price equal to the lower of (i) the conversion price then in effect, and (ii) 80% of the price computed as the quotient of (i) the sum of the VWAP of our common stock for each of the two (2) trading days with the lowest volume weighted average price (“VWAP”) of our common stock during the ten (10) consecutive trading day period ending and including the trading day immediately preceding the delivery or deemed delivery of the applicable notice of conversion divided by two (2) (the “Alternate Conversion Price”).
In connection with the occurrence of an Event of Default, the holders of the Notes will be entitled to convert all or any portion of the Notes at the Alternate Conversion Price.
Conversion Limitation and Exchange Cap
The holders of the Notes will not have the right to convert any portion of the Notes, to the extent that, after giving effect to such conversion, such holder (together with certain related parties) would beneficially own in excess of 4.99% of the shares of our common stock outstanding immediately after giving effect to such conversion. A holder may from time to time increase this limit to 9.99%, provided that any such increase will not be effective until the 61st day after delivery of a notice to us of such increase.
Events of Default
The Notes include certain customary Events of Default as described in the form of Note attached to our Current Report on Form 8-K filed with the SEC on December 21,2020. In connection with an Event of Default, the holders of the Notes may require us to redeem any portion or all of the Notes. The redemption price will equal the greater of (i) the product of (A) the Conversion Amount to be redeemed multiplied by (B) 125% and (ii) the product of (X) the Conversion Rate with respect to the Conversion Amount in effect at such time as the holder delivers an Event of Default Redemption Notice multiplied by (Y) the product of (1) 125% multiplied by (2) the greatest Closing Sale Price of our common stock on any trading day during the period commencing on the date immediately preceding such Event of Default and ending on the date we make the entire payment required, as determined in accordance with the Notes.
| 96 |
Upon the occurrence of a Bankruptcy Event of Default (as defined in the Notes), the Notes will automatically become immediately due and payable in cash in an amount equal to all outstanding principal, interest, and late charges multiplied by a redemption premium of 125%.
Change of Control
In connection with a Change of Control (as defined in the Notes), the holders of the Notes may require us to redeem all or any portion of the Notes. The redemption price per share will equal the greatest of (i) 125% of the outstanding principal of the Notes to be redeemed, and accrued and unpaid interest and unpaid late charges thereon, (ii) 125% of the market value of the shares of our common stock underlying the Notes, as determined in accordance with the Notes, and (iii) 125% of the aggregate cash consideration that would have been payable in respect of the shares of our common stock underlying the Notes, as determined in accordance with the Notes.
Other Corporate Events
We cannot enter a Fundamental Transaction (as defined in the Notes), unless the successor entity assumes all of the obligations under the Notes pursuant to written agreements satisfactory to the holder of the Notes, and the successor entity is a publicly traded corporation whose shares of common stock are quoted or listed on a national securities exchange. If at any time we grant any Purchase Rights (as defined in the Notes) or make any distribution of assets pro rata to all or substantially all of the holders of any class of our common stock, then the holders of the Notes will be entitled to acquire the aggregate Purchase Rights or assets which such holder could have acquired if such holder had held the number of shares of our common stock acquirable upon complete conversion of the Notes (without taking into account any limitations on conversion) held by such holder immediately prior to the date as of which the record holders are to be determined for such grant of purchase rights or distributions. To the extent any such grant of rights or distribution would result in the holders exceeding the maximum percentage described in first paragraph of “—Conversion Limitation and Exchange Cap” above, such rights shall be held in abeyance for up to ninety trading days.
Redemptions at Our Election
At any time after the Issuance Date, we have the right to redeem all, or any part, of the Conversion Amount then remaining under the Notes at cash price equal to 115% of the greater of (i) the Conversion Amount being redeemed and (ii) the product of (1) the Conversion Rate with respect to the Conversion Amount being redeemed multiplied by (2) the greatest Closing Sale Price of our common stock on any trading day during the period commencing on the date immediately preceding such redemption notice date and ending on the Trading Day immediately prior to the date the Company makes the entire payment required to be made for the redemption.
Covenants
The Notes require our compliance with certain customary affirmative and negative covenants regarding the incurrence of indebtedness, the existence of liens, the repayment of indebtedness, the payment of cash in respect of dividends, distributions or redemptions, and the transfer of assets, among other matters.
Warrant
Pursuant to the terms of the Securities Purchase Agreement, at the Initial Closing we issued to the selling stockholder a warrant to acquire up to 5,500,000 shares of our common stock, which we refer to herein as the “Warrant”.
Terms not defined in the following description of the particular terms of the Warrant have the meanings given to them in the Warrant.
| 97 |
Expiration and Exercise
The Warrant, for the purchase of an aggregate of 5,500,000 shares of our common stock (subject to standard adjustments in the event of any stock split, stock dividend, stock combination, recapitalization or other similar transaction), expires on the third anniversary of the Issuance Date (the “Expiration Date”). Prior to or on the Expiration Date, the Warrants are exercisable at any time after the Issuance Date, in whole or in part, at the option of the holders.
The exercise price for the Warrant is $0.50 (subject to adjustment as provided in the Warrant, the “Exercise Price”). The Exercise Price has full ratchet antidilution protection upon any subsequent placement below the Exercise Price then in effect and is subject to standard adjustments in the event of any stock split, stock dividend, stock combination, recapitalization or other similar transaction. If we enter into any agreement to issue (or issue) any variable price securities, the holder has the additional right to substitute such variable price (or formula) for the Exercise Price.
Cashless exercise is available to the holder of the Warrant.
Exercise Limitation and Exchange Cap
The holder of the Warrant will not have the right to convert any portion of the Warrant to the extent that, after giving effect to such conversion, such holder (together with certain related parties) would beneficially own in excess of 4.99% of the shares of our common stock outstanding immediately after giving effect to such exercise. The holder may from time to time increase this limit to 9.99%, provided that any such increase will not be effective until the 61st day after delivery of a notice to us of such increase.
Events of Default
Events of Default are cross-referenced to the definition contained in the Notes.
At any time after the occurrence of an Event of Default, at the request of the holder, we or the successor entity (as the case may be) shall purchase the Warrant from the holder on the date of such request by paying to the holder cash in an amount equal to the Event of Default Black Scholes Value (as defined in the Warrant).
Other Corporate Events
We cannot enter a Fundamental Transaction (as defined in the Warrant), unless the successor entity assumes all of the obligations under the Warrant pursuant to written agreements satisfactory to the holder of the Warrant, and the successor entity is a publicly traded corporation whose shares of common stock are quoted or listed on a national securities exchange. If at any time we grant any Purchase Rights (as defined in the Warrant) or make any distribution of assets pro rata to all or substantially all of the holders of any class of its common stock, then the holder of the Warrant will be entitled to acquire the aggregate Purchase Rights or assets which such holder could have acquired if such holder had held the number of shares of the our common stock acquirable upon complete exercise of the Warrant held by such holder immediately prior to the date as of which the record holders are to be determined for such grant of purchase rights or distributions. To the extent any such grant of rights or distribution would result in the holders exceeding the maximum percentage described in first paragraph of “Exercise Limitation and Exchange Cap” above, such rights shall be held in abeyance for the benefit of the holders until such time or times, if ever, as such holders’ rights to participate would not result in the holders exceeding the maximum percentage.
| 98 |
Notwithstanding the foregoing, at the request of the holder delivered at any time commencing on the earliest to occur of (x) the public disclosure of any Fundamental Transaction, (y) the consummation of any Fundamental Transaction and (z) the holder first becoming aware of any Fundamental Transaction through the date that is ninety (90) days after the public disclosure of the consummation of such Fundamental Transaction by us pursuant to a Current Report on Form 8-K filed with the SEC, we or the successor entity (as the case may be) shall purchase the Warrant from the holder on the date of such request by paying to the holder cash in an amount equal to the Black Scholes Value (as defined in the Warrant). Payment of such amounts shall be made by us (or at our direction) to the holder on or prior to the later of (x) the second (2nd) trading day after the date of such request and (y) the date of consummation of such Fundamental Transaction.
Securities Purchase Agreement
The securities purchase agreement contains certain representations and warranties, covenants and indemnities customary for similar transactions. Under the Purchase Agreement, we also agreed that as long as the Notes remain outstanding, we will not effect or enter into an agreement to effect any variable rate transaction other than a bona fide at-the-market offering or equity line of credit.
Registration Rights Agreement
We entered into a registration rights agreement with the holder as of the initial closing date of the securities purchase agreement. Under the registration rights agreement, we have agreed to register 300% of the shares issuable under the notes and 100% of the shares issuable under the warrant, with filing to occur no later than 30 days of the closing and with effectiveness to occur no later than 90 days of the closing. If we are unable to meet either of these deadlines, we may be required to pay certain cash damages under the registration rights agreement or, with the passage of additional time, an event of default under the notes may occur.
Dollar Value of Underlying Securities and Potential Profits on Conversion
The following table sets forth the potential profit to be realized upon conversion by the selling stockholder based on the conversion price on February 12, 2021 and the closing price of our common stock on February 12, 2021.
Potential Profit from Conversion of the Notes at the Option of the Selling Stockholder
| Per share market price as of February 12, 2021 | $ | 0.38 | ||
| Per share conversion price as of February 12, 2021 | $ | 0.27 | ||
| Total number of shares underlying notes based on conversion price (1) | 5,092,593 | |||
| Aggregate market value of underlying shares based on per share market price as of February 12, 2021 | $ | 1,935,185 | ||
| Aggregate gross cash purchase price for the convertible notes | $ | 1,375,000 | ||
| Aggregate original issue discount | $ | 125,000 | ||
| Aggregate net cash purchase price for the convertible notes | $ | 1,250,000 | ||
| Total premium to market price of underlying shares | 41% |
(1) The aggregate principal amount of the Notes is $1,375,000.
Since the warrant exercise price is greater than the current market price for the Company’s shares of common stock, the selling stockholder will not realize any profit if it exercises the Warrant at this time.
| 99 |
Payments to Selling Stockholder and Affiliates
In connection with the notes, we are or may be required to make the following payments to the selling stockholder and its affiliates:
| Payee | Maximum Early Redemption Premiums (1) | Maximum Registration Penalties (2) | Total Maximum Payments During First 12 Months (3) | |||||||
| Selling Stockholder | $ | 849,063 | $ | 275,000 | $1,124,063 | |||||
(1) Represents the cash amount that would be payable by us if we were required to redeem the notes (assuming all as a result of an event of default or change of control assuming (a) that the applicable premium to be applied upon the event of default or change of control is 125%, (b) that the event of default or change of control occurs on February 21, 2021, and (c) that the required payments continue until December 20, 2021. The default interest rate is 18% per annum upon the occurrence and continuance of an event of default.
(2) Represents the maximum monetary penalties and interest that would be payable if we failed to timely file, obtain a declaration of effectiveness or maintain the effectiveness with respect to the registration statement required under the below-described registration rights agreement. Assumes that (a) the monetary penalties begin to accrue on February 21, 2021, (b) the monetary penalties continue to accrue until December 20, 2021, and (c) the monetary penalties will not be paid until December 20, 2021 (which results in the payment of interest on unpaid amounts at a rate of 2% per month from February 21, 2021 to December 20, 2021).
(3) Represents the maximum amounts payable in cash under the other columns in this table during the first 12 months after the sale of the notes.
Net Proceeds from Private Placement of Notes
The following table sets forth the gross proceeds received from the private placement of the notes and calculates the net proceeds thereof after deduction of the anticipated payments pursuant to the notes and related documents. The net proceeds do not include the payment of any contingent payments, such as liquidated damages or repayment premiums in the case of default or a change of control. The net proceeds assumes that all interest and principal will be paid in cash, notwithstanding that we may pay (and are expected to pay) interest and principal in shares of its common stock under specified circumstances, as described above. The table assumes that none of the notes are converted prior to maturity. Based on the foregoing assumptions, the net proceeds represent approximately 85.7% of the gross proceeds.
| Gross Proceeds | $ | 1,375,000 | ||
| Approximate Aggregate Payments (including Original Issue Discount) | $ | 125,000 | ||
| Approximate Transaction Costs (including Costs and Expenses of this Offering) | $ | 72,000 | ||
| Net Proceeds | $ | 1,178,000 |
Comparison of Issuer Proceeds to Potential Investor Profit
As discussed above, we plan to use the proceeds from the sale of the convertible notes for general corporate purposes. The following table summarizes the potential proceeds we will receive pursuant to the securities purchase agreement, the notes and the warrant. For purposes of this table, we have assumed that the selling stockholder will exercise all of the warrant on a cash basis. We have also assumed that the notes will be held by the selling stockholder through the maturity date thereof.
| 100 |
| Total Gross Proceeds Payable to us (1) | $ | 4,125,000 | ||
| Payments that have been made or may be required to be made by us until maturity (2) | $ | 125,000 | ||
| Net proceeds to us assuming maximum payments made by selling stockholder (3) | $ | 4,000,000 | ||
| Total possible profit to the selling stockholder (4) | $ | 685,185 | ||
| Percentage of payments and profit over net proceeds (5) | 17% |
(1) Includes gross proceeds payable to us on the sale of the notes in the amount of $1,375,000 and assumes full exercise of the warrant to yield an aggregate exercise price of $2,750,000. However, there is no assurance that the warrant will actually be exercised or if it is exercised, whether it will be exercised for cash.
(2) Total possible payments (excluding repayment of principal but including the original issue discount) payable by us to the selling stockholder or its affiliates assuming the notes remain outstanding until the maturity date, namely, the original issue discount. Assumes that no liquidated damages are incurred and that no redemption premium on the notes will be applicable.
(3) Total net proceeds to us calculated by subtracting the result in footnote (2) from the result in footnote (1) (but does not include costs and expenses of this Offering).
(4) This number represents the total possible profit to the selling stockholder based on the aggregate discount to market price of the shares underlying the notes and the original issue discount as indicated in the above table entitled “Potential Profit from Conversion of the Notes at the Option of the Selling Stockholder.”
(5) Percentage of the profit calculated in footnote (4) compared to the net proceeds disclosed in footnote (3).
Comparison of Registered Shares to Outstanding Shares
The following table compares the number of shares held by persons other than the selling stockholder, company affiliates, and affiliates of selling stockholder with the number of shares registered for resale and sold by such parties in prior transactions as well as in the current transactions involving the notes and warrant:
| Shares Outstanding Prior to the Private Placement Held by Persons Other than the Selling Stockholder, or Affiliates of us or Selling Stockholder | 48,141,201 | |||
| Shares Registered for Resale by Selling Stockholder or Affiliates of Selling Stockholder in Prior Registration Statements | 0 | |||
| Shares Registered for Resale by Selling Stockholder or Affiliates of the Selling Stockholder that Continue to be Held by Such Persons | 0 | |||
| Shares Sold in Registered Resale Transactions by the Selling Stockholder or Affiliates Thereof | 0 | |||
| Shares Registered for Resale on behalf of the Selling Stockholder or Affiliates Thereof in Connection with the private placement (including reserves) | 20,777,779 |
Other Information
As of the date of this prospectus, we do not believe that we will have the financial ability to make all payments on the notes in cash when due. Accordingly, we do intend, as of the date of this prospectus, to make such payments in shares of our common stock to the greatest extent possible.
| 101 |
The selling stockholder has advised us that it may enter into short sales in the ordinary course of its business of investing and trading securities. The selling stockholder has advised us that no short sales were entered into during the period beginning when such selling stockholder obtained knowledge that we were contemplating a private placement and ending upon the public announcement of any such private placement.
We have not had any material relationships or arrangements with the selling stockholder, its affiliates, or any person with whom the selling stockholder has a contractual relationship regarding this private placement (or any predecessors of those persons).
We believe that the closing of the remaining tranches of this offering will provide sufficient financial resources to fund our operations, working capital, and capital expenditures through December 31, 2022.
Subsequent sources of outside funding will be required to fund our working capital, capital expenditures and rate operations beyond 2022. No assurances can be given that we will be successful in complying with certain of the terms and conditions in the issuance of the notes or in arranging further funding, if needed, to continue the execution of our business plan, or if successful, on what terms. Failure to obtain such funding will require management to substantially curtail, if not cease operations, which will result in a material adverse effect on the financial position and our results of operations.
SHARES ELIGIBLE FOR FUTURE SALE
Upon the completion of this offering, a total of 192,784,269 shares of common stock will be outstanding, assuming conversion of the Notes and exercise of the Warrant for the shares being registered in this prospectus. Of these shares, the shares of common stock of the selling stockholder registered in this offering will be freely tradable in the public market without restriction or further registration under the Securities Act.
Rule 144
Of the 135,237,584 shares of our common stock issued and outstanding as of December 21, 2020 (not including the Pre-Delivery Shares), a total of 87,708,055 shares are deemed “restricted securities” within the meaning of Rule 144. The Pre-Delivery Shares are restricted securities. Restricted securities are eligible for public sale only if they are registered under the Securities Act or if they qualify for an exemption from registration under Rules 144.
In general, subject to the satisfaction of certain conditions, including the availability of current information, Rule 144 permits a person who presently is not and who has not been an affiliate of ours for at least three months immediately preceding the sale and who has beneficially owned the shares of common stock for at least six months to sell such shares without regard to any of the volume limitations set forth in Rule 144. This provision may already apply with respect to the shares issued in connection with the Private Placement, assuming that at such time the relevant criteria of Rule 144 are satisfied by both the selling stockholder owning the restricted shares and ourselves.
Under Rule 144, subject to the satisfaction of certain other conditions, a person deemed to be one of our affiliates, who has beneficially owned restricted shares of our Common Stock for at least six months is permitted to sell in a brokerage transaction, within any three-month period, a number of shares that does not exceed the greater of 1% of the total number of outstanding shares of the same class, or, if our common stock is quoted on a stock exchange, the average weekly trading volume during the four calendar weeks preceding the sale, if greater.
The possibility that substantial amounts of our common stock may be sold under Rule 144 into the public market may adversely affect prevailing market prices for the Common Stock and could impair our ability to raise capital in the future through the sale of equity securities.
| 102 |
The validity of the common stock being offered hereby will be passed upon for us by SRK Kronengold Law Offices.
The financial statements of Cannabics Pharmaceuticals, Inc. as of August 31, 2020 and 2019, and appearing in this prospectus in the registration statements have been audited by Weinstein International CPA, independent registered public accounting firm, given upon the authority of said firm as experts in accounting and auditing. The unaudited financial statements of Cannabics Pharmaceuticals, Inc. as of November 30, 2020 have been reviewed by Weinstein International CPA.
The audit reports covering the August 31, 2020 and 2019 financial statements contains an explanatory paragraph that states that the Company’s recurring losses from operations and negative operating cash flows raise substantial doubt about the entity’s ability to continue as a going concern. The financial statements do not include any adjustments that might result from the outcome of that uncertainty.
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
Weinstein International CPA is our auditor. There have not been any changes in or disagreements with our accountants on accounting and financial disclosure or any other matter.
WHERE YOU CAN FIND MORE INFORMATION
We electronically file annual, quarterly and special reports, proxy and information statements and other information with the SEC. The public may read and copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of that site is http://www.sec.gov. Our website address is www.cannabics.com. Information contained in, or accessible through, our website is not a part of this prospectus.
| 103 |
| F-1 |
CANNABICS PHARMACEUTICALS INC.
| November 30, | August 31, | |||||||
| 2020 | 2020 | |||||||
| Unaudited | Audited | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 184,031 | $ | 777,611 | ||||
| Prepaid expenses and other receivables | 168,574 | 152,299 | ||||||
| Total current assets | 352,605 | 929,910 | ||||||
| Available for sale Investment | 539,609 | 426,522 | ||||||
| Equipment, net | 806,317 | 862,879 | ||||||
| Total assets | $ | 1,698,531 | $ | 2,219,311 | ||||
| LIABILITIES AND STOCKHOLDERS' EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable and accrued liabilities | $ | 219,042 | $ | 231,142 | ||||
| Due to a related party | 223,645 | 223,645 | ||||||
| Total current liabilities | 442,687 | 454,787 | ||||||
| Stockholders' equity (deficit): | ||||||||
| Preferred stock, $.0001 par value, 5,000,000 shares authorized, no shares issued and outstanding. | – | – | ||||||
| Common stock, $.0001 par value, 900,000,000 shares authorized, 135,237,584 shares issued and outstanding at November 30 2020 And 135,080,441 shares issued and outstanding at August 31, 2020. | 13,524 | 13,508 | ||||||
| Additional paid-in capital | 15,405,295 | 15,372,311 | ||||||
| Issuance of warrants | 2,784,387 | 2,784,387 | ||||||
| Other comprehensive loss | (2,661,324 | ) | (2,774,411 | ) | ||||
| Accumulated deficit | (14,286,038 | ) | (13,631,271 | ) | ||||
| Total stockholders' equity (deficit) | 1,255,844 | 1,764,524 | ||||||
| Total liabilities and stockholders' equity | $ | 1,698,531 | $ | 2,219,311 | ||||
See accompanying notes to consolidated financial statements.
| F-2 |
CANNABICS PHARMACEUTICALS INC.
Consolidated Statements of Operations and Comprehensive Loss
(Unaudited)
| For the Three Months Ended | ||||||||
| November 30, | November 30, | |||||||
| 2020 | 2019 | |||||||
| Unaudited | ||||||||
| Net revenue | – | 1,754 | ||||||
| Operating expenses: | ||||||||
| Research and development expense | $ | 433,730 | $ | 466,033 | ||||
| Sales and marketing expenses | 7,551 | 24,117 | ||||||
| General and administrative expenses | 218,451 | 326,460 | ||||||
| Total operating expenses | 659,732 | 817,610 | ||||||
| Loss from operations | (659,732 | ) | (815,856 | ) | ||||
| Other income | ||||||||
| Financial (Loss) Income | 4,965 | (4,326,339 | ) | |||||
| Net loss | (654,767 | ) | (5,142,195 | ) | ||||
| Income (Loss) from available for sale assets | 113,087 | (3,550,813 | ) | |||||
| Total comprehensive (income) loss | $ | (541,680 | ) | $ | (8,693,008 | ) | ||
| Net loss per share - basic and diluted: | $ | (0.005 | ) | $ | (0.038 | ) | ||
| Weighted average number of shares outstanding - Basic and Diluted | 135,105,839 | 133,653,941 | ||||||
See accompanying notes to consolidated financial statements.
| F-3 |
CANNABICS PHARMACEUTICALS INC.
Consolidated Statements of Stockholders' Equity (Deficit)
Unaudited
| Common Stock | Additional Paid In | Other Comprehensive | Accumulated | Total Stockholders’ Equity | ||||||||||||||||||||||||
| Shares | Amount | Capital | Warrants | Gain | Deficit | (Deficit) | ||||||||||||||||||||||
| Balance, August 31, 2020 | 135,080,441 | $ | 13,508 | $ | 15,372,311 | $ | 2,784,387 | $ | (2,774,411 | ) | $ | (13,631,271 | ) | $ | 1,764,524 | |||||||||||||
| Issuance of common stock for services. | 157,143 | 16 | 32,984 | – | – | – | 33,000 | |||||||||||||||||||||
| Other comprehensive loss | – | – | – | – | 113,087 | – | 113,087 | |||||||||||||||||||||
| Net loss | – | – | – | – | – | (654,767 | ) | (654,767 | ) | |||||||||||||||||||
| Balance, November 30, 2020 | 135,237,584 | $ | 13,524 | $ | 15,405,295 | $ | 2,784,387 | $ | (2,661,324 | ) | $ | (14,286,038 | ) | $ | 1,255,844 | |||||||||||||
| Common Stock | Additional Paid In | Other Comprehensive | Accumulated | Total Stockholders’ Equity | ||||||||||||||||||||||||
| Shares | Amount | Capital | Warrants | Gain | Deficit | (Deficit) | ||||||||||||||||||||||
| Balance, August 31, 2019 | 134,418,775 | $ | 13,450 | $ | 15,300,250 | $ | 2,784,387 | $ | 2,810,013 | $ | (6,163,807 | ) | $ | 14,744,292 | ||||||||||||||
| Issuance of common stock for services. | 80,000 | – | (30,000 | ) | – | – | – | (30,000 | ) | |||||||||||||||||||
| Other comprehensive loss | – | – | – | – | (3,550,813 | ) | – | (3,550,813 | ) | |||||||||||||||||||
| Net loss | – | – | – | – | – | (5,142,195 | ) | (5,142,195 | ) | |||||||||||||||||||
| Balance, November 30, 2019 | 134,498,775 | $ | 13,450 | $ | 15,270,250 | $ | 2,784,387 | $ | (740,800 | ) | $ | (11,306,003 | ) | $ | 6,021,284 | |||||||||||||
The accompanying notes are an integral part of the financial statements.
| F-4 |
CANNABICS PHARMACEUTICALS INC.
Consolidated Statements of Cash Flows
(Unaudited)
| For the Three Months Ended | ||||||||
| November 30, | November 30, | |||||||
| 2020 | 2019 | |||||||
| Unaudited | ||||||||
| Cash flows from operating activities: | ||||||||
| Net Loss | $ | (654,767 | ) | $ | (5,142,194 | ) | ||
| Adjustments required to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | 57,505 | 51,578 | ||||||
| Royalties receivables valuation | – | 4,363,000 | ||||||
| Stock issued for services | 33,000 | (30,000 | ) | |||||
| Profit from held for trading investments | – | (58,730 | ) | |||||
| Changes in operating assets and liabilities: | ||||||||
| Decrease (increase) Accounts Receivable and prepaid expenses | (16,275 | ) | 53,076 | |||||
| Increase (decrease) Accounts payable and accrued liabilities | (12,100 | ) | 1,583 | |||||
| Net cash used in operating activities | (592,637 | ) | (761,687 | ) | ||||
| Cash flows from investing activities: | ||||||||
| Realization of Held for trading Investments | – | 824,849 | ||||||
| Acquisition of equipment | (943 | ) | (9,800 | ) | ||||
| Net cash used in investing activities | (943 | ) | 815,049 | |||||
| Net increase (Decrease) in cash | (593,580 | ) | 53,362 | |||||
| Cash and cash equivalents at beginning of the Period | 777,611 | 265,982 | ||||||
| Cash and cash equivalents at end of the Period | $ | 184,031 | $ | 319,344 | ||||
See accompanying notes to consolidated financial statements.
| F-5 |
CANNABICS PHARMACEUTICALS INC.
Notes to Consolidated Financial Statements
(unaudited)
Note 1– Nature of Business, Presentation and Going Concern
Organization
Cannabics Pharmaceuticals Inc. (the “Company”), was incorporated in the State of Nevada, on September 15, 2004, under the name of Thrust Energy Corp.
On April 25, 2014, the Company experienced a change in control. Cannabics, Inc. (“Cannabics”) acquired a majority of the issued and outstanding common stock of the Company in accordance with stock purchase agreements. On the closing date, April 25, 2014, pursuant to the terms of the Stock Purchase Agreement, Cannabics purchased 41,000,000 shares of the Company’s outstanding restricted common stock for $198,000, representing 51%.
On May 21, 2014, the Company changed its name, via merger in the state of Nevada, to Cannabics Pharmaceuticals Inc. The Company’s principal offices are in Bethesda, Maryland. The Company changed its course of business to laboratory research and development.
On June 3, 2014, the Company’s Board of Directors declared a two-to-one forward stock split of all outstanding shares of common stock. The stock split was approved by FINRA on June 25, 2014.
On August 25, 2014, the Company organized G.R.I.N. Ultra Ltd. (“GRIN”), an Israeli corporation, as a wholly-owned subsidiary. GRIN will provide research and development activities for the Company’s products in Israel.
On July 24, 2017, the Company announced its establishment of a genetics laboratory to develop diagnostic tools based on human genome, tumor genetics and specific cannabinoids.
On August 20th, 2020, the Company announced the creation of a new Division for its Anti-Tumor drug candidate RCC-33, for the treatment of colorectal cancer. The emanates from the Company’s focus on a clinical validation path, including in-vivo experiments, collaborations with key medical centers, and the preparation of a product dossier with which the company plans to schedule a Pre IND-Meeting with the US FDA.
| F-6 |
Basis of Presentation
The accompanying unaudited financial statements have been prepared in accordance with accounting principles generally accepted in the United States (“U.S. GAAP”) for interim financial statement presentation and in accordance with Form 10-Q. Accordingly, they do not include all of the information and footnotes required in annual financial statements. In the opinion of management, the unaudited financial statements contain all adjustments (consisting only of normal recurring accruals) necessary to present fairly the financial position and results of operations and cash flows. The results of operations presented are not necessarily indicative of the results to be expected for any other interim period or for the entire year.
These unaudited financial statements should be read in conjunction with our August 31, 2020 annual financial statements included in our Form 10-K, filed with the U.S. Securities and Exchange Commission (“SEC”) on November 4, 2020.
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and GRIN. All significant inter-company balances and transactions have been eliminated in consolidation.
Going Concern
The accompanying unaudited financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The Company has incurred a net loss of $654,767 for the three months ended November 30, 2020, and has incurred cumulative losses since inception of $14,286,038. These conditions raise substantial doubt about the ability of the Company to continue as a going concern.
The ability of the Company to continue as a going concern is dependent upon its abilities to generate revenues, to continue to raise investment capital, and develop and implement its business plan. No assurance can be given that the Company will be successful in these efforts.
Research and Development Costs
The Company accounts for research and development costs in accordance with Accounting Standards Codification 730 “Research and Development” (“ASC 730”). ASC 730 requires that research and development costs be charged to expense when incurred. Research and development costs charged to expense were $433,730 and $466,033 for the three months ended November 30, 2020 and 2019, respectively.
Reclassifications
Certain amounts in the prior period financial statements have been reclassified to conform to the current period presentation. These reclassifications had no effect on reported losses, total assets, or stockholders’ equity as previously reported.
Note 2 – Related Party Transactions
During the three months ending November 30, 2020, the Company paid $105,000 in salaries, including socials benefits, to two directors, compared to $125,000 for the three months ending November 30, 2019.
The Company had a balance outstanding at November 30, 2020 and at November 30, 2019 of $223,645 payable to Cannabics, Inc. The advance is due on demand and bears no interest.
| F-7 |
Note 3 – Stockholders’ Equity (Deficit)
Authorized Shares
The Company is authorized to issue up to 900,000,000 shares of common stock, par value $0.0001 per share. Each outstanding share of common stock entitles the holder to one vote per share on all matters submitted to a stockholder vote. All shares of common stock are non-assessable and non-cumulative, with no pre-emptive rights.
Note 4 – Commitments and Contingencies
The company leases its office in Tel Aviv. In May, 2019, the Company extended its lease agreement for an additional twelve-month extension which expires on April 1st, 2021. The monthly lease is $1,390.00
Effective June 1, 2018, the Company entered into a 36-month car lease for one of its executive officers.
As security for its obligation under a property lease agreement, car lease and credit cards, the Company’s subsidiary provided a bank guarantee in the amount of $50,000.
Note 5 – Subsequent Events
On October 27th, 2020, the Company announced that it had received an "Intention to Grant a European Patent" by the European Patent Office for its System and Method for High Throughput Screening of Cancer Cells patent application relating to its “CDx” diagnostic drug sensitivity test.
On November 16th, 2020, the Company established a subsidiary named Digestix Bioscience Inc., specifically dedicated to development of medical devices and pharmaceutical compositions for precancerous neoplastic tumors, as noted in the 8-K filed November 26th, 2020.
On December 16, 2020, the Company executed a Securities Purchase Agreement with 3i LP for a series of convertible notes for an aggregate funding total of $2,750,000, to be carried out over four tranches, the first of which having been received. Per the agreements between the companies, the underlying shares for said notes are to be registered with the SEC. Until such time, the Company has issued 3,913,663 shares on December 18, 2020. Any other shares are to be registered in the future. For complete and detailed information on the Agreements with 3i LP, please see our 8-K for this event, filed December 21, 2020.
On December 21st, 2020, the Company announced that it had initiated is In-Vivo Animal Studies for an FDA Pre-IND meeting. The purpose of these studies is for the Company’s “RCC-33” rug candidate on mice transplanted with colorectal cancer tumor cell lines.
The Company has evaluated subsequent events through the date the financial statements were issued and filed with the SEC and has determined that there are no other such events that warrant disclosure or recognition in the financial statements.
| F-8 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and the Board of Directors of Cannabics Pharmaceuticals Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Cannabics Pharmaceuticals Inc. ("the Company") as of August 31, 2020 and 2019 and the related statements of operations, changes in stockholders' deficit and cash flows, for each of the years ended August 31, 2020 and 2019, and the related notes and schedules (collectively referred to as the "financial statements"). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of August 31, 2020 and 2019, and the results of its operations and its cash flows for each of the periods ended August 31, 2020 and 2019, in conformity with generally accepted accounting principles in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Going Concern
The accompanying financial statements have been prepared assuming the Company will continue as a going concern. As more fully described in Note 1, the Company has incurred significant losses and needs to raise additional funds to meet its obligations and sustain its operations. These conditions raise substantial doubt about the Company's ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 1. These financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ Weinstein International. C.P.A.
We have served as the Company's auditor since 2019.
Tel - Aviv, Israel
November 4, 2020
| F-9 |
CANNABICS PHARMACEUTICALS INC.
Audited Consolidated Balance Sheets
| August 31, | August 31, | |||||||
| 2020 | 2019 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 777,611 | $ | 265,982 | ||||
| Prepaid expenses and other receivables | 152,299 | 284,496 | ||||||
| Held for trading Investments | – | 3,256,456 | ||||||
| Current royalties | – | 500,000 | ||||||
| Total current assets | 929,910 | 4,306,934 | ||||||
| Available for sale Investment | 426,522 | 6,010,946 | ||||||
| Long term royalties | – | 3,863,000 | ||||||
| Equipment, net | 862,879 | 1,002,286 | ||||||
| Total assets | $ | 2,219,311 | $ | 15,183,166 | ||||
| LIABILITIES AND STOCKHOLDERS' EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable and accrued liabilities | $ | 231,142 | $ | 215,229 | ||||
| Due to a related party | 223,645 | 223,645 | ||||||
| Total current liabilities | 454,787 | 438,874 | ||||||
| Stockholders' equity (deficit): | ||||||||
| Preferred stock, $.0001 par value, 5,000,000 shares authorized no shares issued and outstanding. | – | – | ||||||
| Common stock, $.0001 par value, 900,000,000 shares authorized, 135,080,441 and 134,498,775 shares issued and outstanding at August 31, 2020 and August 31, 2019 respectively. | 13,508 | 13,450 | ||||||
| Additional paid-in capital | 15,372,311 | 15,300,250 | ||||||
| issuance of warrants | 2,784,387 | 2,784,387 | ||||||
| Other comprehensive income | (2,774,411 | ) | 2,810,013 | |||||
| Accumulated deficit | (13,631,271 | ) | (6,163,807 | ) | ||||
| Total stockholders' equity (deficit) | 1,764,524 | 14,744,292 | ||||||
| Total liabilities and stockholders' equity | $ | 2,219,311 | $ | 15,183,166 | ||||
The accompanying notes are an integral part of the financial statements
| F-10 |
CANNABICS PHARMACEUTICALS INC.
Audited Consolidated Statements of Operations
| For the Year Ended August 31, | ||||||||
| 2020 | 2019 | |||||||
| Net revenue | $ | 7,157 | $ | 9,843 | ||||
| Operating expenses: | ||||||||
| Research and development expense | 1,682,462 | 1,543,759 | ||||||
| Sales and marketing expenses | 59,997 | 288,163 | ||||||
| General and administrative expenses | 1,308,150 | 1,481,528 | ||||||
| Total operating expenses | 3,050,609 | 3,313,450 | ||||||
| Loss from operations | (3,043,451 | ) | (3,303,607 | ) | ||||
| Other income | ||||||||
| Financial (loss) income, net | (4,424,012 | ) | 4,436,576 | |||||
| Net (loss) income | $ | (7,467,464 | ) | $ | 1,132,970 | |||
| (Loss) Profit from available for sale assets | (5,584,424 | ) | 2,810,013 | |||||
| Total comprehensive (loss) income | (13,051,887 | ) | 3,942,983 | |||||
| Net (loss) per share - basic and diluted: | $ | (0.10 | ) | $ | 0.02 | |||
| Weighted average number of shares outstanding - Basic and Diluted | 134,551,721 | 132,450,953 | ||||||
The accompanying notes are an integral part of the financial statements.
| F-11 |
CANNABICS PHARMACEUTICALS INC.
Audited Consolidated Statements of Cash Flows
| For the year Ended August 31, | ||||||||
| 2020 | 2019 | |||||||
| Cash flows from operating activities: | ||||||||
| Net (Loss) Profit | $ | (7,467,464 | ) | $ | 1,132,970 | |||
| Adjustments required to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | 215,155 | 188,928 | ||||||
| Royalties receivables valuation | 4,363,000 | (4,363,000 | ) | |||||
| Stock issued for services | 72,120 | 247,307 | ||||||
| Profit from held for trading investments | (105,876 | ) | (142,226 | ) | ||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts Receivable and prepaid expenses | 132,197 | (57,252 | ) | |||||
| Accounts payable and accrued liabilities | 15,913 | (100,509 | ) | |||||
| Net cash used in operating activities | (2,774,955 | ) | (3,093,782 | ) | ||||
| Cash flows from investing activities: | ||||||||
| Available for sale investments | – | (1,920,000 | ) | |||||
| Held for trading Investments | 3,362,332 | (3,114,233 | ) | |||||
| Acquisition of equipment | (75,748 | ) | (216,882 | ) | ||||
| Net cash used in investing activities | 3,286,584 | (5,251,115 | ) | |||||
| Cash flows from financing activities: | ||||||||
| Proceeds from sale of common stock | – | 7,294,259 | ||||||
| Costs of raising capital | – | (76,989 | ) | |||||
| Net cash provided by financing activities | – | 7,217,270 | ||||||
| Net increase (decrease) in cash | 511,629 | (1,127,626 | ) | |||||
| Cash and cash equivalents at beginning of year | 265,982 | 1,393,608 | ||||||
| Cash and cash equivalents at end of the year | $ | 777,611 | $ | 265,982 | ||||
| Significant non-cash transactions: | ||||||||
| Issuance of Shares | $ | – | $ | 939,933 | ||||
The accompanying notes are an integral part of the financial statements.
| F-12 |
CANNABICS PHARMACEUTICALS INC.
Audited Consolidated Statements of Stockholders' Equity (Deficit)
| Common stock | Additional paid in | Accumulated | Total stockholders' equity | |||||||||||||||||||||
| Shares | Amount | capital | Warrants | deficit | (deficit) | |||||||||||||||||||
| Balance, August 31, 2018 | 121,575,388 | $ | 12,158 | $ | 9,840,420 | $ | 89,722 | $ | (7,296,777 | ) | $ | 2,645,523 | ||||||||||||
| Issuance of shares of common stock for cash | 10,277,777 | 1,028 | 4,441,384 | 2,784,387 | – | 7,226,799 | ||||||||||||||||||
| Issuance of common stock for services | 2,645,610 | 264 | 1,018,445 | – | – | 1,018,709 | ||||||||||||||||||
| Expiration of warrants | – | – | – | (89,722 | ) | – | – | |||||||||||||||||
| Other comprehensive profit | – | – | – | – | – | 2,810,013 | ||||||||||||||||||
| Net profit for the year ended August 31, 2020 | – | – | – | – | 1,132,970 | 1,132,970 | ||||||||||||||||||
| Balance, August 31, 2019 | 134,498,775 | $ | 13,450 | $ | 15,300,249 | $ | 2,784,387 | $ | (6,163,807 | ) | $ | 14,744,292 | ||||||||||||
| Issuance of common stock for services | 581,666 | 58 | 72,062 | – | – | 72,120 | ||||||||||||||||||
| Other comprehensive profit | – | – | – | – | – | (5,584,424 | ) | |||||||||||||||||
| Net loss for the year ended August 31, 2020 | – | – | – | – | (7,467,464 | ) | (7,467,464 | ) | ||||||||||||||||
| Balance, August 31, 2020 | 135,080,441 | $ | 13,508 | $ | 15,372,311 | $ | 2,784,387 | $ | (13,631,271 | ) | $ | 1,764,524 | ||||||||||||
The accompanying notes are an integral part of the financial statements.
| F-13 |
CANNABICS PHARMACEUTICALS INC.
Notes to Consolidated Financial Statements
As of August 31, 2020
Note 1 – Nature of Business, Presentation and Going Concern
Organization
Cannabics Pharmaceuticals Inc. (the "Company"), was incorporated in the State of Nevada, on September 15, 2004, under the name of Thrust Energy Corp. The Company was originally engaged in the exploration, exploitation, development and production of oil and gas projects within North America, but was unable to operate profitably.
In May 2011, the Company changed its name to American Mining Corporation, suspending its oil and gas operations and changing its business to toll milling and refining, mineral exploration and mine development.
On April 25, 2014, the Company experienced a change in control. Cannabics, Inc. (“Cannabics”) acquired a majority of the issued and outstanding common stock of the Company in accordance with stock purchase agreements by and between Cannabics and Thomas Mills (“Mills”). On the closing date, April 25, 2014, pursuant to the terms of the Stock Purchase Agreement, Cannabics purchased from Mills 41,000,000 shares of the Company’s outstanding restricted common stock for $198,000, representing 51%.
On May 21, 2014, the Company changed its name, via merger in the state of Nevada, to Cannabics Pharmaceuticals Inc. The Company’s principle offices are in Bethesda, Maryland. As of May 21, 2014, the Company has changed its course of business to laboratory research and development.
On August 25, 2014, the Company organized G.R.I.N. Ultra Ltd. (“GRIN”), an Israeli corporation, as a wholly-owned subsidiary. GRIN will provide research and development activities for the Company’s products in Israel.
Stock Split
On June 3, 2014, the Company’s Board of Directors declared a two-to-one forward stock split of all outstanding shares of common stock. The stock split was approved by FINRA on June 25, 2014. The effect of the stock split increased the number of shares of common stock outstanding from 40,880,203 to 81,760,406. All common share and per common share data in these financial statements and related notes hereto have been retroactively adjusted to account for the effect of the stock split for all periods presented prior to June 3, 2014. The total number of authorized common shares and the par value thereof was not changed by the split.
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America and the rules and regulations of the Securities and Exchange Commission (“SEC”).
| F-14 |
Note 1 – Nature of Business, Presentation and Going Concern (Continued)
Going Concern
The accompanying financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The Company has incurred a net loss of $7,467,463 for the year ended August 31, 2020 and has incurred cumulative losses since inception of $13,631,271. These conditions raise substantial doubt about the ability of the Company to continue as a going concern.
The Company’s continuation as a going concern is dependent upon its ability to generate revenues, its ability to continue to raise investment capital, and implementing its business plan. No assurance can be given that the Company will be successful in these efforts.
These financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might be necessary should the Company be unable to continue as a going concern. Management believes that actions presently being taken to obtain additional funding and implement its strategic plans provide the opportunity for the Company to continue as a going concern. No assurance can be given that the Company will be successful in these efforts.
Note 2 – Summary of Significant Accounting Policies
Functional currency
The currency of the primary economic environment in which the operations of the Company and its Subsidiary are conducted is the U.S. dollar (“$” or “dollar”). Therefore, the functional currency of the Company and its Subsidiary is the dollar.
Transactions and balances denominated in dollars are presented at their original amounts. Non-dollar transactions and balances have been re-measured to dollars in accordance with the provisions of ASC 830-10 (formerly Statement of Financial Accounting Standard 52), "Foreign Currency Translation". All transaction gains and losses from re-measurement of monetary balance sheet items denominated in non-dollar currencies are reflected in the statement of operations as financial income or expenses, as appropriate.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. Significant estimates in the accompanying financial statements include the amortization period for intangible assets, impairment valuation of intangible assets, valuation of share-based payments and the valuation allowance on deferred tax assets.
Principles of Consolidation
The consolidated financial statements include the accounts of Cannabics Pharmaceutical Inc. and its wholly-owned subsidiary, G.R.I.N. Ultra Ltd. All significant inter-company balances and transactions have been eliminated in consolidation.
| F-15 |
Note 2 – Summary of Significant Accounting Policies (Continued)
Cash and Cash Equivalents
The Company considers all highly liquid temporary cash investments with an original maturity of three months or less to be cash equivalents. At August 31, 2020 and 2019, cash equivalents consisted of bank accounts held at financial institutions.
Concentration of Credit Risk
The Company places its cash and cash equivalents with high credit quality financial institutions. There is Federal Deposit Insurance on the Company’s U.S. bank accounts.
Equipment, net
Equipment at August 31, 2020 consists of computer equipment, office equipment and cars recorded at cost. Expenditures for major additions and betterments are capitalized. Maintenance and repairs are charged to operations as incurred. Depreciation of property and equipment is computed by the straight-line method (after taking into account their respective estimated residual values) over the assets estimated useful lives of 3 years for computer equipment, 14 years for office equipment and 7 years for cars. Upon sale or retirement of property and equipment, the related cost and accumulated depreciation are removed from the accounts and any gain or loss is reflected in the consolidated statements of operations.
Depreciation expense was $215,155 and $188,928 for the years ended August 31, 2020 and 2019, respectively.
Revenue recognition
Revenue is recognized when delivery has occurred, evidence of an arrangement exists, title and risks and rewards for the products are transferred to the customer, collection is reasonably assured and product returns can be reliably estimated.
Revenue from license agreements is recognized over the periods from which the Company is entitled to the respective payments.
The Company’s revenues are concentrated in a small number of customers. For the year ended August 31, 2020 the revenues were for license fees.
Impairment or Disposal of Long-Lived Assets
The Company accounts for the impairment or disposal of long-lived assets according to the Financial Accounting Standards Board’s (“FASB”) Accounting Standards Codification (“ASC”) 360 “Property, Plant and Equipment”. ASC 360 clarifies the accounting for the impairment of long-lived assets and for long-lived assets to be disposed of, including the disposal of business segments and major lines of business. Long-lived assets are reviewed when facts and circumstances indicate that the carrying value of the asset may not be recoverable. When necessary, impaired assets are written down to estimated fair value based on the best information available. Estimated fair value is generally based on either appraised value or measured by discounting estimated future cash flows. Considerable management judgment is necessary to estimate discounted future cash flows. Accordingly, actual results could vary significantly from such estimates.
| F-16 |
Note 2 – Summary of Significant Accounting Policies (Continued)
Fair Value of Financial Instruments
The following provides an analysis of financial instruments that are measured subsequent to initial recognition at fair value, grouped into Levels 1 to 3 based on the degree to which fair value is observable:
Level 1 - fair value measurements are those derived from quoted prices (unadjusted) in active markets for identical assets or liabilities;
Level 2 - fair value measurements are those derived from inputs other than quoted prices included within Level 1 that are observable for the asset or liability, either directly (i.e. as prices) or indirectly (i.e. derived from prices); and
Level 3 - fair value measurements are those derived from valuation techniques that include inputs for the asset or liability that are not based on observable market data (unobservable inputs).
Fair value estimates discussed herein are based upon certain market assumptions and pertinent information available to management as of August 31, 2020 and 2019. The respective carrying value of certain on-balance-sheet financial instruments approximated their fair values due to the short-term nature of these instruments.
The Company applied ASC 820 for all non-financial assets and liabilities measured at fair value on a non-recurring basis. The adoption of ASC 820 for non-financial assets and liabilities did not have a significant impact on the Company’s financial statements.
As of August 31, 2020, the fair values of the Company’s level 1 financial instruments are in total of $426,522. And none of level 3 investments. As of August 31, 2020, the fair values of the Company’s financial instruments approximate their historical carrying amount.
Research and development, net
Research and development expenses include costs directly attributable to the conduct of research and development programs, including the cost of salaries, employee benefits, the cost of supplies, the cost of services provided by outside contractors, including services related to the Company’s clinical trials, clinical trial expenses and the full cost of manufacturing product for use in research and preclinical development. All costs associated with research and developments are expensed as incurred.
Clinical trial costs are a significant component of research and development expenses and include costs associated with third-party contractors. The Company outsources a substantial portion of its clinical trial activities, utilizing external entities such as Contract Research Organizations, independent clinical investigators, and other third-party service providers to assist the Company with the execution of its clinical studies. For each clinical trial that the Company conducts, clinical trial costs are expensed immediately.
| F-17 |
Note 2 – Summary of Significant Accounting Policies (Continued)
Stock Based Compensation
The Company accounts for Stock-Based Compensation under ASC 718 “Compensation – Stock Compensation”, which addresses the accounting for transactions in which an entity exchanges its equity instruments for goods or services, with a primary focus on transactions in which an entity obtains employee services in share-based payment transactions. ASC 718-10 requires measurement of the cost of employee services received in exchange for an award of equity instruments based on the grant-date fair value of the award. Incremental compensation costs arising from subsequent modifications of awards after the grant date must be recognized.
The Company accounts for stock-based compensation awards to non-employees in accordance with ASC 505-50, Equity-Based Payments to Non-Employees. Under ASC 505-50, the Company determines the fair value of the warrants or stock-based compensation awards granted as either the fair value of the consideration received or the fair value of the equity instruments issued, whichever is more reliably measurable. Any stock options or warrants issued to non-employees are recorded in expense and additional paid-in capital in shareholders' deficit over the applicable service periods using variable accounting through the vesting dates based on the fair value of the options or warrants at the end of each period.
The Company issues stock to consultants for various services. The costs for these transactions are measured at the fair value of the consideration received or the fair value of the equity instruments issued, whichever is more reliably measurable. The value of the common stock is measured at the earlier of (i) the date at which a firm commitment for performance by the counterparty to earn the equity instruments is reached or (ii) the date at which the counterparty's performance is complete. The Company recognized consulting expense and a corresponding increase to additional paid-in-capital related to stock issued for services.
Income Taxes
Income taxes are accounted for under the liability method of accounting for income taxes. Under the liability method, future tax liabilities and assets are recognized for the estimated future tax consequences attributable to differences between the amounts reported in the financial statement carrying amounts of assets and liabilities and their respective tax bases. Future tax assets and liabilities are measured using enacted or substantially enacted income tax rates expected to apply when the asset is realized or the liability settled. The effect of a change in income tax rates on future income tax liabilities and assets is recognized in income in the period that the change occurs. Future income tax assets are recognized to the extent that they are considered more likely than not to be realized.
The FASB has issued ASC 740 “Income Taxes”. ASC 740 clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements. This standard requires a company to determine whether it is more likely than not that a tax position will be sustained upon examination based upon the technical merits of the position.
If the more-likely-than-not threshold is met, a company must measure the tax position to determine the amount to recognize in the financial statements.
As a result of the implementation of this standard, the Company performed a review of its material tax positions in accordance with recognition and measurement standards established by ASC 740 and concluded that the tax position of the Company has not met the more-likely-than-not threshold as of August 31, 2020.
| F-18 |
Note 2 – Summary of Significant Accounting Policies (Continued)
Comprehensive Income
The Company adopted ASC 220, Comprehensive Income which establishes standards for reporting and display of comprehensive income, its components and accumulated balances. The Company is disclosing this information on its Statement of Stockholders' Equity. Comprehensive income comprises equity except those resulting from investments by owners and distributions to owners. The Company has no elements of “other comprehensive income” for the years ended August 31, 2020 and 2019.
Basic and Diluted Loss per Share
The Company computes income (loss) per share in accordance with ASC 260, "Earnings per Share", which requires presentation of both basic and diluted earnings per share (“EPS”) on the face of the statement of operations. Basic EPS is computed by dividing income (loss) available to common shareholders by the weighted average number of shares outstanding during the period. Diluted EPS gives effect to all dilutive potential shares of common stock outstanding during the period using the treasury stock method and convertible preferred stock using the if-converted method. In computing diluted EPS, the average stock price for the period is used in determining the number of shares assumed to be purchased from the exercise of stock options or warrants. Diluted EPS excludes all dilutive potential shares if their effect is anti-dilutive. As of August 31, 2020, and 2019, the potentially dilutive shares were anti-dilutive.
Segment Information
In accordance with the provisions of ASC 280-10, “Disclosures about Segments of an Enterprise and Related Information”, the Company is required to report financial and descriptive information about its reportable operating segments. The Company does not consider itself to have any operating segments as of August 31, 2020 and 2019.
Reclassification
Certain amounts in the prior period financial statements have been reclassified to conform to the current period presentation.
Note 3 – Recent Accounting Pronouncements
In February 2015, the Financial Accounting Standards Board (the “FASB”) issued Accounting Standards Update (“ASU”) 2015-02, “Consolidation (Topic 810): Amendments to the Consolidation Analysis”, which provides guidance in evaluating entities for inclusion in consolidations. ASU 2015-02 is effective for fiscal years beginning after December 15, 2015. The Company does not believe the adoption of ASU 2015-02 will have a material effect on its consolidated financial statements.
In June 2014, the FASB issued Accounting Standards Update (“ASU”) No. 2014-10 (“ASU 2014-10”), Development Stage Entities (Topic 915), Elimination of Certain Financial Reporting Requirements, Including an Amendment to Variable Interest Entities Guidance in Topic 810, Consolidation. The objective of the amendments in this Update is to improve financial reporting by reducing the cost and complexity associated with incremental reporting requirements for development stage entities. The amendments in this Update also eliminate an exception provided to development stage entities in Topic 810, Consolidation, for determining whether an entity is a variable interest entity on the basis of the amount of investment equity at risk. The amendments related to the elimination of inception-to-date information and the other remaining disclosure requirements of Topic 915 should be applied retrospectively except for the clarification to Topic 275, which shall be applied prospectively. These amendments are effective for annual reporting periods beginning after December 15, 2014, and interim periods therein.
| F-19 |
Note 3 – Recent Accounting Pronouncements (Continued)
The amendment eliminating the exception to the sufficiency-of-equity-at-risk criterion for development stage entities in paragraph 810-10-15-16 should be applied retrospectively for annual reporting periods beginning after December 15, 2015 and interim periods therein. Early application of each of the amendments is permitted for any annual reporting period or interim period for which the entity’s financial statements have not yet been issued. The Company has adopted ASU 2014-10 in the fourth quarter of 2014 and does not expect this adoption to have a material impact on its consolidated financial condition, results of operations or cash flows.
In August 2014, the FASB issued ASU No. 2014-15, Presentation of Financial Statements-Going Concern (Subtopic 205-40), Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern. The objective of the amendments in this Update is to provide guidance on determining when and how to disclose going-concern uncertainties in the financial statements. The new standard requires management to perform interim and annual assessments of an entity’s ability to continue as a going concern within one year of the date the financial statements are issued. An entity must provide certain disclosures if “conditions or events raise substantial doubt about the entity’s ability to continue as a going concern.” The ASU applies to all entities and is effective for annual periods ending after December 15, 2017, and interim periods thereafter, with early adoption permitted. The Company is evaluating the impact of ASU 2014-15 on its consolidated financial condition, results of operations and cash flows.
In September 2015, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update No. 2015-16 (ASU 2015-16) "Simplifying the Accounting for Measurement Period Adjustments". ASU 2015-16 require that an acquirer recognize adjustments to provisional amounts that are identified during the measurement period in the reporting period in which the adjustment amounts are determined. The amendments in ASU 2015-16 require that the acquirer record, in the same period’s financial statements, the effect on earnings of changes in depreciation, amortization, or other income effects, if any, as a result of the change to the provisional amounts, calculated as if the accounting had been completed at the acquisition date. The amendments in ASU 2015-16 require an entity to present separately on the face of the income statement or disclose in the notes the portion of the amount recorded in current-period earnings by line item that would have been recorded in previous reporting periods if the adjustment to the provisional amounts had been recognized as of the acquisition date. For public business entities, the amendments in ASU 2015-16 are effective for fiscal years beginning after December 15, 2015, including interim periods within those fiscal years. The amendments in ASU 2015-16 should be applied prospectively to adjustments to provisional amounts that occur after the effective date of this update with earlier application permitted for financial statements that have not been issued. For all other entities, the amendments in ASU 2015-16 are effective for fiscal years beginning after December 15, 2017, and interim periods within fiscal years beginning after December 15, 2017. The amendments in ASU 2015-16 should be applied prospectively to adjustments to provisional amounts that occur after the effective date of this update with earlier application permitted for financial statements that have not yet been made available for issuance.
Note 4 – Related Party Transactions
During the year ended August 31, 2020 and August 31 2019, the Company paid approximately of $595,000 and $515,555 as salary to three of its directors.
Cannabics Inc. (the parent company) balance at August 31, 2020 and at August 31, 2019 was $223,645. The advance is due on demand and bears no interest.
Note 5 – Commitments and Contingencies
As security for its obligation under a property lease agreement, cars lease and credit cards of the Company’s subsidiary provided a bank guarantee in the amount of $45,000.
Note 6 – Stockholders’ Equity (Deficit)
Authorized Shares
The Company is authorized to issue up to 900,000,000 shares of common stock par value $0.0001 per share. Each outstanding share of common stock entitles the holder to one vote per share on all matters submitted to a stockholder vote. All shares of common stock are non-assessable and non-cumulative, with no pre-emptive rights. The Company’s initial Articled authorized 5,000,000 preferred shares at .0001 par value, no other attributes have been assigned and no such shares have ever been issued.
Common Stock
During the year ended August 31, 2020, the Company issued 581,666 shares of its common stock to 2 consultants and an advisor for services rendered at a fair value of $72,120 or an average of $0.124 per share.
| F-20 |
Note 7 – Warrants
Since August 31st, 2019, the company has issued no Warrants. The only Warrants outstanding are 5,000,000 Warrants to two entities from 2018, with a strike price of $1.00 per Warrant and are exercisable until August 24th, 2023.
| 1. | On September 24, 2018, as part of a securities purchase agreement the company issued 5,000,000 Warrants, to purchase common shares of the Company at $1.00 per share; said Warrants are valid for five years, expiring on August 24th, 2023. | |
| The fair value of each warrant is approximately $0.556 and the total value of the 5,000,000 warrants is $2,784,387 | ||
| The fair value of the warrants are estimated using the Black Scholes option-pricing model with the following assumptions: |
| PV of exercise Share price | $1 | |
| Expected Volatility | 102.0% | |
| Risk Free Interest Rate | 1.58% | |
| Expected Term (years) | 5 | |
| Expected Dividend Yield | 0% |
| 2. | On December 12, 2018 the company granted 50,000 Warrants to an advisor, to purchase 50,000 common shares of the Company at $1 per share; said Warrants are valid for one year, expiring on December 12, 2019. | |
| The fair value of each warrant is approximately $0.0.0874 and the total value of the 50,000 warrants issued was $4,370. | ||
| The fair value of the warrants are estimated using the Black Scholes option-pricing model with the following assumptions: |
| PV of exercise Share price | $1 | |
| Expected Volatility | 112.72 % | |
| Risk Free Interest Rate | 2.7% | |
| Expected Term (years) | 1 | |
| Expected Dividend Yield | 0% |
| The said Warrants expired on December 12, 2019. | ||
| F-21 |
Note 7 – Warrants (Continued)
| 3. | On June 19, 2019 the company granted 125,000 Warrants each to two of its directors totaling 250,000 Warrants, to purchase common shares of the Company at $0.30 per share; said Warrants are valid for one year, expiring on June 19, 2020. | |
| The fair value of each warrant is approximately $0.1224 and the total value of the 250,000 warrants issued was $30,600. | ||
| The fair value of the warrants are estimated using the Black Scholes option-pricing model with the following assumptions: |
| PV of exercise Share price | $0.3 | |
| Expected Volatility | 100.34% | |
| Risk Free Interest Rate | 1.96% | |
| Expected Term (years) | 1 | |
| Expected Dividend Yield | 0% |
| The said Warrants expired on June 19, 2020. |
The following table presents the warrant activity for the years ended August 31, 2020 and 2019.
| 2020 | 2019 | 2018 | ||||||||||||||||||||||
| Weighted Average Exercise | Weighted Average Exercise | Weighted Average Exercise | ||||||||||||||||||||||
| Warrants | Price | Warrants | Price | Warrants | Price | |||||||||||||||||||
| Warrants outstanding as of September 1 | 5,300,000 | $ | 1.00 | 500,000 | $ | 1.00 | 1,566,671 | $ | 0.02 | |||||||||||||||
| Issued | – | $ | – | 5,300,000 | $ | 0.97 | 1,000,000 | $ | 2.00 | |||||||||||||||
| Exercised | – | $ | – | – | $ | – | (500,000 | ) | $ | 0.02 | ||||||||||||||
| Expired | (300,000 | ) | $ | 0.42 | (1,000,000 | ) | $ | 2.00 | (1,066,671 | ) | $ | 0.20 | ||||||||||||
| Warrants outstanding as of August 31 | 5,000,000 | $ | 1.00 | 5,300,000 | $ | 0.97 | 1,000,000 | $ | 2.00 | |||||||||||||||
| Warrants exercisable as of August 31 | 5,000,000 | $ | 1.00 | 5,300,000 | $ | 0.97 | 1,000,000 | $ | 2.00 | |||||||||||||||
The weighted average remaining contractual life for the options outstanding as of August 31, 2020 was 3 years.
Note 8 – Income Taxes
Taxes on income included in the consolidated statements of operations represent current taxes due to taxable income of the Company and its Subsidiary.
Corporate taxation in the U.S.
The applicable corporate tax rate for the Company is 21%.
No provision for income tax was made for the period from September 15, 2004 (Inception) to August 31, 2020 as the Company had cumulative operating losses. For the years ended August 31, 2020 and 2019, the Company incurred net losses for tax purposes of $2,229,392 and $1,114,659, respectively. Under U.S. tax laws, subject to certain limitations, carry forward tax losses expire 20 years after the year in which incurred. In the case of the Company, subject to potential limitations in accordance with the relevant law, the net loss carry forward will expire in the years 2032 through 2036.
| F-22 |
Note 8 – Income Taxes (continued)
Corporate taxation in Israel:
The Subsidiary is taxed in accordance with Israeli tax laws. The corporate tax rate applicable to 2020 is 23%.
In December 2016, the Israeli Parliament approved the Economic Efficiency Law (Legislative Amendments for Applying the Economic Policy for the 2020 and 2019 Budget Years), which further reduces the corporate income tax rate to 24% (instead of 25%) effective from January 1, 2017 and to 23% effective from January 1, 2020.
As of August 31, 2020, the Subsidiary has an accumulated tax loss carry forward of approximately $7,833,000 (as of August 31, 2019, approximately $5,600,000). Under the Israeli tax laws, carry forward tax losses have no expiration date.
The income tax expense (benefit) differs from the amount computed by applying the United States Statutory corporate income tax rate as follows:
| For the Year Ended August 31, | ||||||||
| 2020 | 2019 | |||||||
| United States statutory corporate income tax rate | 21.0 | % | 21.0 | % | ||||
| Change in valuation allowance on deferred tax assets | -21.0 | % | -21.0 | % | ||||
| Provision for income tax | – | % | – | % | ||||
Deferred income taxes reflect the tax effect of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for tax purposes. The components of the net deferred income tax assets are approximately as follows:
| August 31, | ||||||||
| 2020 | 2019 | |||||||
| US Deferred income tax assets: | ||||||||
| Net operating loss carry forwards benefit | $ | 1,800,098 | $ | 785,628 | ||||
| Valuation allowance | (1,800,098 | ) | (785,628 | ) | ||||
| Net deferred income tax assets | $ | – | $ | – | ||||
| Outside US Deferred income tax assets: | ||||||||
| Net operating loss carry forwards benefit | $ | 1,845,645 | $ | 1,209,215 | ||||
| Valuation allowance | (1,845,645 | ) | (1,209,215 | ) | ||||
| $ | – | $ | – | |||||
| August 31, | ||||||||
| 2020 | 2019 | |||||||
| Consolidated Deferred income tax assets: | ||||||||
| Net operating loss carry forwards benefit | $ | 3,615,744 | $ | 1,994,843 | ||||
| Valuation allowance | (3,615,744 | ) | (1,994,843 | ) | ||||
| Net deferred income tax assets | $ | – | $ | – | ||||
| F-23 |
Note 8 – Income Taxes (continued)
The amount taken into income as deferred income tax assets must reflect that portion of the income tax loss carry forwards that is more likely than not to be realized from future operations. The Company has established a full valuation allowance on its net deferred tax assets because of a lack of sufficient positive evidence to support its realization. The valuation allowance increased by $1,670,901 and decreased $234,549 for the years ended August 31, 2020 and 2019, respectively.
No provision for income taxes has been provided in these financial statements due to the net loss for the years ended August 31, 2020 and 2019. At August 31, 2020, the Company has net operating loss carry forwards of approximately $13,631,270 which expire commencing 2032. The potential tax benefit of these losses may be limited due to certain change in ownership provisions under Section 382 of the Internal Revenue Code (“IRS”) and similar state provisions.
IRS Section 382 places limitations (the “Section 382 Limitation”) on the amount of taxable income which can be offset by net operating loss carry forwards after a change in control (generally greater than 50% change in ownership) of a loss corporation. Generally, after a change in control, a loss corporation cannot deduct operating loss carry forwards in excess of the Section 382 Limitation. Due to these “change in ownership” provisions, utilization of the net operating loss and tax credit carry forwards may be subject to an annual limitation regarding their utilization against taxable income in future periods. The Company has not concluded its analysis of Section 382 through August 31, 2020, but believes the provisions will not limit the availability of losses to offset future income.
The Company is subject to income taxes in the U.S. federal jurisdiction and is subject to examination for a period of three years for current filings and indefinitely for any delinquent filings. The tax regulations within each jurisdiction are subject to interpretation of related tax laws and regulations and require significant judgment to apply. The Company estimates that the amount of penalties, if any, will not have a material effect on the results of operations, cash flows or financial position. No provisions have been made in the financial statements for such penalties, if any.
Note 9 – Subsequent Events
On September 2nd, 2020, the Board dismissed Itamar Borochov as Director and Chair of the company.
The Company has evaluated subsequent events through the date the financial statements were issued and filed with the Securities and Exchange Commission and has determined that there are no other such events that warrant disclosure or recognition in the financial statements.
Note 10 – General & administrate expenses
| For the year Ended August 31, 2020 | For the year Ended August 31, 2019 | |||||||
| Salaries and related expenses | $ | 220,586 | $ | 97,060 | ||||
| Legal and professional fees | 828,003 | 676,096 | ||||||
| Consulting – Stock based compensation | 72,120 | 247,307 | ||||||
| Insurance | 103,389 | 131,853 | ||||||
| Other expenses | 84,051 | 329,212 | ||||||
| $ | 1,308,150 | $ | 1,481,528 | |||||
Note 11 – Financial (Income) expenses
| For the year Ended August 31, 2020 | For the year Ended August 31, 2019 | |||||||
| Interest and bank charges | $ | 8,133 | $ | 18,348 | ||||
| Other financial expense | 60,000 | – | ||||||
| Capital gain from selling of held for trading investments | (69,974 | ) | (19,621 | ) | ||||
| Loss (Gain) from Held for trading Investments | – | (54,077 | ) | |||||
| Loss (Profit) from royalties valuation | 4,363,000 | (4,363,000 | ) | |||||
| Currency exchange differences loss (profit) | 62,852 | (18,226 | ) | |||||
| $ | 4,424,011 | $ | (4,436,576 | ) | ||||
| F-24 |
PART II
INFORMATION NOT REQUIRED IN THE PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution
The following table lists the costs and expenses payable by the Registrant in connection with the sale of the common stock being registered under this registration statement. All amounts shown are estimates except for the SEC registration fee.
| SEC registration fee | $ | 1,632.37 | ||
| Legal fees and expenses | $ | 70,000.00 | ||
| Accounting fees and expenses | $ | – | ||
| Miscellaneous fees and expenses | $ | 500.00 | ||
| Total | $ | 72,132.37 |
Item 14. Indemnification of Directors and Officers
Neither our Articles of Incorporation nor Bylaws prevent us from indemnifying our officers, directors, and agents to the extent permitted under the NRS. NRS 78.7502 provides that a corporation shall indemnify any director, officer, employee, or agent of a corporation against expenses, including attorneys' fees, actually and reasonably incurred by him in connection with any the defense to the extent that a director, officer, employee, or agent of a corporation has been successful on the merits or otherwise in defense of any action, suit, or proceeding referred to NRS 78.7502(1) or 78.7502(2), or in defense of any claim, issue, or matter therein.
NRS 78.7502(1) provides that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending, or completed action, suit, or proceeding, whether civil, criminal, administrative, or investigative, except an action by or in the right of the corporation, by reason of the fact that he is or was a director, officer, employee, or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee, or agent of another corporation, partnership, joint venture, trust, or other enterprise, against expenses, including attorneys' fees, judgments, fines, and amounts paid in settlement actually and reasonably incurred by him in connection with the action, suit, or proceeding if he: (a) is not liable pursuant to NRS 78.138; or (b) acted in good faith and in a manner which he reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful.
NRS 78.7502(2) provides that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending, or completed action or suit by or in the right of the corporation to procure a judgment in its favor by reason of the fact that he is or was a director, officer, employee, or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee, or agent of another corporation, partnership, joint venture, trust, or other enterprise against expenses, including amounts paid in settlement and attorneys' fees actually and reasonably incurred by him in connection with the defense or settlement of the action or suit if he: (a) is not liable pursuant to NRS 78.138; or (b) acted in good faith and in a manner which he reasonably believed to be in or not opposed to the best interests of the corporation. Indemnification may not be made for any claim, issue, or matter as to which such a person has been adjudged by a court of competent jurisdiction, after exhaustion of all appeals there from, to be liable to the corporation or for amounts paid in settlement to the corporation, unless and only to the extent that the court in which the action or suit was brought or other court of competent jurisdiction determines upon application that in view of all the circumstances of the case, the person is fairly and reasonably entitled to indemnity for such expenses as the court deems proper.
NRS 78.747 provides that except as otherwise provided by specific statute, no director or officer of a corporation is individually liable for a debt or liability of the corporation, unless the director or officer acts as the alter ego of the corporation. The court as a matter of law must determine the question of whether a director or officer acts as the alter ego of a corporation.
| II-1 |
Article 5.1 of our Articles of Incorporation provides that a director or officer of Cannabics shall not be personally liable to Cannabics or its stockholders for damages for breach of fiduciary duty as a director or officer, unless such liability results from (i) acts or omissions which involve intentional misconduct, fraud, or a knowing violation of law, or (ii) the unlawful payment of distributions.
Article 6.1 of our Articles of Incorporation further provides that every person who was or is a party or is threatened to be made a party to or is involved in any action, suit, or proceedings, whether civil, criminal, administrative, or investigative, by reason of the fact that he is or was a director or officer of Cannabics, shall be indemnified and held harmless to the fullest extent legally permissible under the law of the State of Nevada from time to time against all expenses, liability, and loss (including attorney's fees, judgments, fines and amounts paid or to be paid in settlement) reasonably incurred or suffered by such person in connection therewith.
Our Bylaws provide that Cannabics shall indemnify its officers and directors to the fullest extent not prohibited by the laws of Nevada or any other applicable law. Cannabics is permitted by the Bylaws to purchase and maintain insurance on behalf of its officers and directors against any liability and expense incurred in such capacity, whether or not Cannabics would have the power to indemnify such person against such liability.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers or persons controlling the registrant pursuant to the foregoing provisions, the registrant has been informed that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is therefore unenforceable.
| Item 15. | Recent Sales of Unregistered Securities |
The following sets forth information regarding all unregistered securities sold within the past three years through December 21, 2020 and provides the share number, per share price, and aggregate consideration as of the date of such sale:
Private Placement of Notes and Warrant
On December 16, 2020, we entered into a Securities Purchase Agreement (“SPA”) with an institutional investor for a private placement of senior secured convertible notes totaling up to an aggregate of $2,750,000 to be issued in three tranches subject to the achievement of certain milestones. The convertible notes include a conversion right, at the Investor’s option, to convert the convertible notes into shares of our Common Stock at a conversion price equal to the lower of (i) $0.35 per share or (ii) eighty percent (80%) of the average of the two lowest daily volume-weighted average price for the Company’s Common Stock during the ten (10) consecutive trading days preceding the conversion date (the “Notes”). The investor has the right to have the conversion price reduced if we issue Common Stock or convertible notes at a lower conversion price than $0.35 during the period that the Notes are outstanding. The Notes will be due one year from issuance. The Notes will be interest free, but in the event of a default, they will bear annual interest at a rate of 18.00%. The SPA and the Notes contain events of default, including, among other things, failure to repay the Notes by the maturity date, and bankruptcy and insolvency events, that would result in the imposition of the default interest rate.
On December 21, 2020, we closed the first tranche (the “Initial Closing”) and issued a Note in the amount of $825,000 (the “Initial Note”). On February 22, 2021, we closed the second tranche and issued a second Note in the amount of $550,000 (the “Second Note”). The Initial Note was issued at a discount of $75,000, and the Second Note was issued at a discount of $50,000. Each additional Note to be issued will be issued with an original issue discount of ten percent (10%). In addition, we issued to the Investor 3,913,663 shares of Common Stock as pre-delivery shares in accordance with the terms of the SPA (the “Pre-Delivery Shares”), which shares will be deducted from the total number of shares to be issued to the Investor upon conversion of the Initial Note. We have the right to require an additional closing of an additional Note in the amount of $1,375,000 on the twentieth trading day after the date this registration statement is declared effective by the Securities and Exchange Commission.
| II-2 |
In addition, the SPA granted the Investor a right to receive 100% warrant coverage, and we issued a warrant to the investor for up to 5,500,000 shares of our Common Stock, which expires three years from the issuance date of the warrant, with an exercise price of $0.50 per share (the “Warrant”). The Warrant may be exercised and converted to Common Stock at the investor’s option at any time until the warrant expiration date.
These securities were issued in reliance on the exemption from registration provided by Section 4(a)(2) of the Securities Act, and Regulation D promulgated thereunder, as these securities were sold to “accredited investors” within the meaning of Regulation D.
| Item 16. | Exhibits. |
A list of exhibits filed with this registration statement on Form S-1 is set forth on the attached Exhibit Index and is incorporated herein by reference.
| II-3 |
| Exhibit 10.9 | Employment Agreement between Eyal Ballan and the Company, dated June 10, 2018. |
| Exhibit 10.10 | Amended Employment Agreement between Gabriel Yariv and Grin Ultra Ltd., dated September 10, 2020. |
| Exhibit 10.11 | Consulting Agreement between CFO Direct and Grin Ultra Ltd., dated November 1, 2016. |
| Exhibit 10.12 | |
| Exhibit 10.13 | Form of Convertible Note with 3i, LP, incorporated by reference from Form 8-K of December 21, 2020. |
| Exhibit 10.14 | Form of Warrant with 3i, LP, incorporated by reference from Form 8-K of December 21, 2020. |
| Exhibit 10.15 | |
| Exhibit 10.16 | |
| Exhibit 10.17 | |
| Exhibit 10.18 | Amended and Restated Securities Purchase Agreement |
| Exhibit 21 | Subsidiaries |
| Exhibit 23.1 | Consent of Independent Registered Public Accounting Firm |
| Exhibit 23.2 | Consent of SRK Kronengold Law Offices (included in the opinion filed as Exhibit 5.1) |
| Exhibit 24.1 | Power of Attorney (contained on the signature page of the registration statement)* |
| 101.INS** | XBRL Instance Document |
| 101.SCH** | XBRL Taxonomy Extension Schema Document |
| 101.CAL** | XBRL Taxonomy Extension Calculation Linkbase Document |
| 101.DEF** | XBRL Taxonomy Extension Definition Linkbase Document |
| 101.LAB** | XBRL Taxonomy Extension Label Linkbase Document |
| 101.PRE** | XBRL Taxonomy Extension Presentation Linkbase Document |
| * Filed herewith. | |
| ** Previously filed | |
| II-4 |
Item 17. Undertakings
The undersigned Registrant hereby undertakes to:
(a) file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement to:
(i) include any prospectus required by section 10(a)(3) of the Securities Act;
(ii) reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or together, represent a fundamental change in the information in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the SEC pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and
(iii) include any material information with respect to the plan of distribution not previously disclosed in this registration statement or any material change to such information in the registration statement.
(b) that, for the purpose of determining any liability under the Securities Act, each post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(c) to file a post-effective amendment to remove from registration any of the securities that remain unsold at the end of the offering.
(d) that insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the Registrant, the Registrant has been advised that in the opinion of the SEC, such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registration of expenses incurred or paid by a director, officer or controlling person to the Registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question of whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
(e) that, for the purpose of determining liability under the Securities Act to any purchaser, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(f) that, for the purpose of determining liability of the Registrant under the Securities Act to any purchaser in the initial distribution of the securities, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the Registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) any preliminary prospectus or prospectus of the Registrant relating to the offering filed pursuant to Rule 424;
(ii) any free writing prospectus relating to the offering prepared by or on behalf of the Registrant or used or referred to by the Registrant;
(iii) the portion of any other free writing prospectus relating to the offering containing material information about the Registrant or its securities provided by or on behalf of the Registrant; and
(iv) any other communication that is an offer in the offering made by the Registrant to the purchaser.
| II-5 |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, as amended, the Registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form S-1 and has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Bethesda, State of Maryland, on the 23rd day of February, 2021.
| CANNABICS PHARMACEUTICALS INC. | |
| By: /s/ Eyal Barad | |
| Eyal Barad | |
| Chief Executive Officer and Director | |
| (Principal Executive Officer) |
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints Eyal Barad as his true and lawful attorney-in-fact and agent with full power of substitution, for him in any and all capacities, to sign any and all amendments to this registration statement (including post-effective amendments) and any registration statement related thereto filed pursuant to Rule 462(b) increasing the number of securities for which registration is sought, and to file the same, with all exhibits thereto and other documents in connection therewith, with the United States Securities and Exchange Commission, granting unto said attorney-in-fact and agent full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully for all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent, or his substitute, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities and on the dates indicated.
| Signature | Title | Date |
| /s/ Eyal Barad | Chief Executive Officer and a Director (Principal Executive Officer) | February 23, 2021 |
| Eyal Barad | ||
| /s/ Gabriel Yariv | Chief Operating Officer and Director | February 23, 2021 |
| Gabriel Yariv | ||
| /s/ Uri Ben-Or | Chief Financial Officer | February 23, 2021 |
Uri Ben-Or | (Principal Accounting Officer) |
|
| II-6 |